WO2017078096A1 - アミノグリコシド系抗生物質の製造方法 - Google Patents
アミノグリコシド系抗生物質の製造方法 Download PDFInfo
- Publication number
- WO2017078096A1 WO2017078096A1 PCT/JP2016/082666 JP2016082666W WO2017078096A1 WO 2017078096 A1 WO2017078096 A1 WO 2017078096A1 JP 2016082666 W JP2016082666 W JP 2016082666W WO 2017078096 A1 WO2017078096 A1 WO 2017078096A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- salt
- compound
- formula
- group
- optionally substituted
- Prior art date
Links
- 0 CO[C@](C(CO)O[C@](C[C@@](C(*)C([C@](*)C1O[C@]2OC(C*)CCC2N)O)C1O)C1O)C1N Chemical compound CO[C@](C(CO)O[C@](C[C@@](C(*)C([C@](*)C1O[C@]2OC(C*)CCC2N)O)C1O)C1O)C1N 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/20—Carbocyclic rings
- C07H15/22—Cyclohexane rings, substituted by nitrogen atoms
- C07H15/222—Cyclohexane rings substituted by at least two nitrogen atoms
- C07H15/226—Cyclohexane rings substituted by at least two nitrogen atoms with at least two saccharide radicals directly attached to the cyclohexane rings
- C07H15/234—Cyclohexane rings substituted by at least two nitrogen atoms with at least two saccharide radicals directly attached to the cyclohexane rings attached to non-adjacent ring carbon atoms of the cyclohexane rings, e.g. kanamycins, tobramycin, nebramycin, gentamicin A2
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to a novel method for producing aminoglycoside antibiotics.
- MRSA microporous swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine-resistant swine, and the development of therapeutic drugs is actively being carried out.
- aminoglycoside antibiotics have a broad antibacterial spectrum from Gram-positive bacteria to Gram-negative bacteria and have an excellent bactericidal activity, and thus are promising to overcome various resistant bacteria including MRSA. Expected to be a drug, research into its derivatives is ongoing.
- (2S) -2-hydroxyarbekacin is a part of the present inventors as an aminoglycoside antibiotic that exhibits a broad antibacterial spectrum and excellent antibacterial activity, and can avoid serious nephrotoxicity. Is disclosed.
- (2S) -2-hydroxyarbekacin is obtained from (2S) -2-hydroxydibekacin by the procedure shown in Scheme 1 below.
- the object of the present invention is to provide a new method for efficiently producing (2S) -2-hydroxyarbekacin from (2S) -2-hydroxydibekacin.
- a synthetic intermediate comprising a compound of formula III or a salt thereof for producing a compound of formula II or a salt thereof from a compound of formula I or a salt thereof: [Wherein R 1 represents an optionally substituted C1-6 alkyloxycarbonyl group or an optionally substituted arylalkyloxycarbonyl group].
- the protecting groups R 3 and R 1 are respectively introduced into the 1-position and 3-position amino groups of the compound of formula III or a salt thereof to obtain the compound of formula VI or a salt thereof, [Wherein, the protective group R 3 represents an optionally substituted alkylcarbonyl group]
- the compound of Formula VI or a salt thereof is deprotected at the 1-position, and the amino group at the 1-position is reacted with the compound of Formula IV or a salt thereof to obtain a compound of Formula VII or a salt thereof.
- a protecting group R 3 is introduced into the 1-position and 3-position amino groups of the compound described in Formula III or a salt thereof to obtain the compound described in Formula VIII or a salt thereof, [Wherein, the protective group R 3 represents an optionally substituted alkyl group] Deprotecting the amino group at position 1 in the compound according to formula VIII or a salt thereof, reacting the amino group at position 1 with the compound according to formula IV or a salt thereof, and then reacting the compound according to formula IX or the salt thereof Get salt, [Wherein R 2 represents an optionally substituted arylalkyloxycarbonyl group, F represents a hydrogen atom or a carboxylic acid activating group, and the configuration of the carbon atom of * represents R or S] The method according to (3) or (4), comprising deprotecting the amino group in the compound according to formula IX or a salt thereof to obtain the compound according to formula II or a salt thereof.
- alkyl as a group or a part of the group means a linear, branched or cyclic alkyl group.
- aryl means phenyl or naphthyl.
- Arylalkyl means alkyl in which one or more hydrogen atoms are substituted with an aryl group.
- salt of a compound may absorb moisture and adsorb water or become a hydrate when left in the air or by recrystallization.
- the invention also includes such various hydrates, solvates and polymorphic compounds.
- the compound described in Formula III used as a synthetic intermediate has a protective group R 1 introduced in any amino group other than the 1-position and the 3-position, and R 1 may be substituted. It represents an alkyloxycarbonyl group or an arylalkyloxycarbonyl group which may be substituted.
- the alkyloxycarbonyl group represented by R 1 is preferably a C1-6 alkyloxycarbonyl group, more preferably a C1-4 alkyloxycarbonyl group, still more preferably a methoxycarbonyl group or a tert-butoxycarbonyl group. More preferably a tert-butoxycarbonyl group.
- One or more hydrogen atoms of the alkyloxycarbonyl group represented by R 1 may be substituted.
- substituents include halogen atoms such as chlorine, bromine, and fluorine, methoxy groups, trifluoroalkyl groups, and nitro groups. Can be mentioned.
- the arylalkyloxycarbonyl group represented by R 1 is preferably an aryl C1-4 alkyloxycarbonyl group, more preferably an aryl C1-2 alkyloxycarbonyl group, still more preferably a benzyloxycarbonyl group. .
- One or more hydrogen atoms of the arylalkyloxycarbonyl group represented by R 1 may be substituted.
- substituents include halogen atoms such as chlorine, bromine and fluorine, methoxy groups, trifluoromethoxy groups, and nitro groups. Etc.
- the compound described by Formula III can exist as a salt. Although it does not specifically limit as the salt, for example, the acid addition salt of an amino group is mentioned. Such salts can be composed of pharmaceutically acceptable salts. Specific examples of the salt include hydrohalides such as hydrofluoride, hydrochloride, hydrobromide or hydroiodide, sulfate, nitrate, phosphate, perchlorate.
- inorganic acid salts such as carbonate, acetate, trichloroacetate, trifluoroacetate, hydroxyacetate, lactate, citrate, tartrate, oxalate, benzoate, mandelate, butyrate, Carboxylate such as maleate, propionate, formate or malate, amino acid salt such as arginate, aspartate or glutamate, or sulfone such as methanesulfonate or p-toluenesulfonate Examples include acid salts.
- the above-mentioned salts are the same in other compounds in the present invention.
- the compound described in Formula III can be provided as an agent for producing the target compound.
- (2S) -2-Hydroxyarbekacin compound described in formula II or a salt thereof
- (2S) -2 is prepared by a production method using a compound described in formula III or a salt thereof as a synthetic intermediate.
- -Hydroxyarbekacin compound described in Formula II
- the compound according to formula II is represented by the following formula II-a.
- the protecting group R 1 is introduced into amino groups other than the 1-position and 3-position in the compound described in Formula I or a salt thereof to obtain the compound described in Formula III or a salt thereof (Step a)
- the compound of formula III is reacted with the compound of formula IV to obtain a compound of formula V or a salt thereof (step b), and the amino group is deprotected to convert the compound of formula II Obtain (step c).
- Step a of Route A as described above, a protecting group R 1 is introduced into amino groups other than the 1-position and 3-position in the compound of Formula I or a salt thereof in the presence of a copper ion. Presence of copper cations, is a surprising fact that the compounds according to Formula I or R 1 groups to the amino groups other than the 1- and 3-position of its salts is introduced with high selectivity.
- the copper ion is not particularly limited as long as the effect of the present invention is not hindered, but is preferably a divalent copper ion.
- generates this copper ion Preferably it is an organic salt or inorganic salt of a cupric, More preferably, it is a copper acetate.
- step A of route A the compound described in formula IV is used as an acylating agent.
- the arylalkyloxycarbonyl group represented by R 2 in the compound described in Formula IV is preferably an aryl C1-4 alkyloxycarbonyl group, more preferably an aryl C1-2 alkyloxycarbonyl group, and still more preferably A benzyloxycarbonyl group;
- One or more hydrogen atoms of the arylalkyloxycarbonyl group represented by R 2 may be substituted.
- substituents include halogen atoms such as chlorine, bromine and fluorine, methoxy groups, trifluoromethoxy groups, A nitro group etc. are mentioned, Preferably it is a methoxy group.
- F represents a hydrogen atom or a carboxylic acid activating group.
- the ester with the carboxylic acid activating group is preferably a succinimide ester.
- a specific example of a compound according to Formula IV is 4-benzyloxycarbonylamino-2-hydroxybutyric acid succinimide ester.
- the configuration of the carbon atom of * in the compound described in Formula IV represents R or S, and preferably represents S.
- route A As shown in Examples 1 to 4 described later, the yield (about 34%) is about twice that of the method described in Patent Document 1. Therefore, route A is advantageous in industrial production of (2S) -2-hydroxyarbekacin as compared with the conventional method.
- the protecting groups R 3 and R 1 are introduced into the amino groups at the 1-position and the 3-position respectively in the compound of formula III obtained in step a or a salt thereof, A compound or salt thereof is obtained (step d and step e), the amino group at position 1 in the compound or salt thereof according to formula VI is deprotected (step f), the amino group at position 1 and the formula IV Reacting the compound or a salt thereof to give a compound of formula VII or a salt thereof (step g), deprotecting the amino group in the compound of formula VII or a salt thereof, The salt is obtained (step h).
- Step d of Route B it has been clarified by the present inventors that when the protecting group R 3 is used, the amino group at the 1-position can be selectively protected while leaving the amino group at the 3-position. Further, as shown in Step e and Step f of Route B, it was found that only the R 3 group of the amino group at the 1-position can be selectively deprotected after protecting the amino group at the 3-position of the compound with the R 1 group. . According to such route B, since the amino group can be selectively protected or deprotected, generation of by-products can be prevented and the target product can be produced efficiently.
- the protecting group R 3 is introduced into the amino groups at the 1-position and 3-position of the compound represented by formula III or a salt thereof obtained in step a to obtain the compound represented by formula VIII or a salt thereof.
- Step i deprotecting the amino group at position 1 in the compound of formula VIII or salt thereof (step j), and reacting the amino group at position 1 with the compound of formula IV or salt thereof To obtain a compound of formula IX or a salt thereof (step k) and deprotect the amino group in the compound of formula IX or a salt thereof to obtain a compound of formula II or a salt thereof (steps l and Step m).
- Step i of Route C two R 3 groups are introduced into the amino groups at the 1-position and 3-position in the compound described in Formula III.
- the R 3 protector is subjected to a deprotection reaction, it has been revealed by the inventors that only the R 3 group at the 1-position is selectively removed.
- acylation at the 1-position step k can also be selectively performed, so that generation of by-products can be prevented and the target product can be produced efficiently.
- Example 10 Preparation of (2S) -2-hydroxyarbekacin
- the title compound of Example 9 was treated in the same manner as in Example 4 using 1.06 g (1.22 mmol) of the title compound: 319 mg (46%) of the title compound: (2S) -2-hydroxyarbekacin.
- Example 13 Preparation of 2 ', 6', 3 "-tri-Nt-butoxycarbonyl-4 '''-Np-methoxybenzyloxycarbonyl- (2S) -2-hydroxyarbekacin 171 g (0.198 mol) of the title compound of Example 12 was dissolved in 1200 ml of THF, and 228 g (0.6%) of (S) -4- (4-methoxybenzyloxy) carbonylamino-2-hydroxybutyric acid ester was added to this solution. Mole) in THF (3500 ml) and triethylamine (117 ml) were added, and the mixture was stirred overnight at room temperature.
- Example 14 250 g of the title compound of Example 13 was dissolved in 2000 ml of methanol, cooled to 5 ° C., 2000 ml of 6 molar hydrochloric acid was added, and the mixture was stirred at room temperature for 6 hours. The reaction solution was neutralized with concentrated aqueous ammonia, and the precipitate was removed and concentrated to 900 ml. This solution was diluted with 2000 ml of ethyl acetate and extracted with 500 ml of water. The aqueous layer was neutralized with concentrated aqueous ammonia and purified with an ion exchange resin in the same manner as in Example 2 to obtain 53.7 g (40%) of the title compound of Example 14.
- DIW 4.8L and 2-OH-DKB 1.2Kg were put into a plastic container and dissolved.
- 490 g of copper acetate monohydrate was added and washed with 4.8 L of MeOH to prepare a chelate solution.
- MeOH 18L was charged into the reaction kettle, and then the prepared chelate solution was added and rinsed with MeOH 18L.
- the internal temperature was cooled to 15 ° C. or lower, 2.41 kg of (Boc) 2 O was added so that the internal temperature did not exceed 20 ° C., washed with 2.4 L of MeOH, and the internal temperature was stirred at 20-25 ° C. overnight. .
- EXAMPLE 15 1.03 L of TEA was added to an EtOH solution of the compound and washed with 1.2 L of EtOH. To this solution, 2.92 L of TFAOEt was further added, washed with 1.2 L of EtOH, and stirred at 40 ° C. overnight. After confirming the end point of the reaction, this solution was directly used in the next step.
- Example 17 THF (3.6 L) was added to the compound, followed by the addition of (S) -4- (4-methoxybenzyloxy) carbonylamino-2-hydroxybutyric acid succinimide ester in THF and washing with 3.6 L THF.
- the mixture was cooled to 15 ° C. or lower, 1.37 L of TEA was added so that the internal temperature did not exceed 20 ° C., and the mixture was washed with 1.2 L of THF.
- the reaction temperature was raised to 20 ° C. and stirred at that temperature overnight. After confirming the end point of the reaction, 24.6 L of 1N NaOH aqueous solution was added at an internal temperature of 30 ° C. or lower.
- Example 19 Preparation of (2S) -2-hydroxyarbekacin 24 L of MeOH was added to the solution of Example 18, and 24 L of 6N hydrochloric acid was added to this solution so that the internal temperature did not exceed 30 ° C., and the mixture was stirred at an internal temperature of 25-30 ° C. for 4.5 hours to confirm the end point of the reaction. . While maintaining the internal temperature at about 25 ° C., the pH was adjusted to around 5.8 with concentrated aqueous ammonia. This solution was washed with 24 L of ethyl acetate, and the obtained aqueous layer was quantified to confirm that 1.052 kg of crude (2S) -2-hydroxyarbekacin was produced.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Saccharide Compounds (AREA)
Abstract
式IIIに記載の化合物またはその塩を合成中間体として用いることを特徴とする、式Iに記載の(2S)-2-ヒドロキシジベカシンまたはその塩から、式IIに記載の化合物(2S)-2-ヒドロキシアルベカシンまたはその塩を効率的に製造する方法を提供する。[式中、R1は、置換されていてもよいアルキルオキシカルボニル基または置換されていてもよいアリールアルキルオキシカルボニル基を表す]
Description
本特許出願は、先に出願された日本国における特許出願である特願2015-216137号(出願日:2015年11月2日)に基づく優先権の主張を伴うものである。この先の特許出願における全開示内容は、引用することにより本明細書の一部とされる。
本発明は、アミノグリコシド系抗生物質の新規な製造方法に関する。
近年、感染症の治療に用いられる抗菌剤に対して抵抗性を示す薬剤耐性菌が出現し、その耐性菌を起因菌とする感染症の治療が医療現場で大きな問題となっている。特にMRSAは、院内感染により急速に伝播し、臨床上重篤な感染症を引き起こす主要な薬剤耐性菌のひとつであることが知られており、その治療薬剤の開発が盛んに行われている。
このような技術状況下、アミノグリコシド系抗生物質は、グラム陽性菌からグラム陰性菌までの幅広い抗菌スペクトラムを有しかつ優れた殺菌力を有することから、MRSAを含めた各種耐性菌を克服する有望な薬剤になるものと期待され、その誘導体の研究が継続的に行われている。
特許文献1には、幅広い抗菌スペクトラムと優れた抗菌活性とを発揮し、かつ重篤な腎毒性を回避しうるアミノグリコシド系抗生物質として(2S)-2-ヒドロキシアルベカシンが本発明者らの一部により開示されている。特許文献1では、以下のスキーム1に示される手順によって、(2S)-2-ヒドロキシジベカシンから(2S)-2-ヒドロキシアルベカシンが取得されている。

しかしながら、特許文献1に記載の方法を用いて(2S)-2-ヒドロキシジベカシンから2S-ヒドロキシアルベカシンを製造すると、脱保護反応またはアシル化反応において複数の副生成物が発生することが本発明者らの検討から明らかとなった。(2S)-2-ヒドロキシアルベカシンの工業的生産の観点からは、副生成物を低減することが望ましい。したがって、副生成物を低減して、(2S)-2-ヒドロキシジベカシンから(2S)-2-ヒドロキシアルベカシンを効率的に取得するための新たな方法を創出することが望まれているといえる。
本発明は、(2S)-2-ヒドロキシジベカシンから(2S)-2-ヒドロキシアルベカシンを効率的に製造するための新たな方法を提供することをその目的としている。
本発明者らは、今般、鋭意検討した結果、特定の合成中間体を介した製造方法により、下式に示される通り、(2S)-2-ヒドロキシジベカシン(式I)から(2S)-2-ヒドロキシアルベカシン(式II)を効率的に取得しうることを見出した。本発明は、かかる知見に基づくものである。
本発明によれば、以下の発明を提供される。
(1)式Iに記載の化合物またはその塩から式IIに記載の化合物またはその塩を製造するための、式IIIに記載の化合物またはその塩からなる、合成中間体:



[式中、R1は、置換されていてもよいC1~6アルキルオキシカルボニル基または置換されていてもよいアリールアルキルオキシカルボニル基を表す]。
(1)式Iに記載の化合物またはその塩から式IIに記載の化合物またはその塩を製造するための、式IIIに記載の化合物またはその塩からなる、合成中間体:
(2)式Iに記載の化合物またはその塩から式IIに記載の化合物またはその塩を製造するための、式IIIに記載の化合物またはその塩の使用:



[式中、R1は、置換されていてもよいC1~6アルキルオキシカルボニル基または置換されていてもよいアリールアルキルオキシカルボニル基を表す]。
(3)式IIIに記載の化合物またはその塩を合成中間体として用いることを特徴とする、式Iに記載の化合物またはその塩から式IIに記載の化合物またはその塩を製造する方法:



[式中、R1は、置換されていてもよいアルキルオキシカルボニル基またはまたは置換されていてもよいアリールアルキルオキシカルボニル基を表す]。
(4)式Iに記載の化合物またはその塩における1位および3位以外のアミノ基に保護基R1を銅イオンの存在下で導入して、式IIIに記載の化合物またはその塩を得ることを含んでなる、(3)に記載の方法。
(5)式IIIに記載の化合物またはその塩と、式IVに記載の化合物またはその塩とを反応させて、式Vに記載の化合物またはその塩を得、

[式中、R2は置換されていてもよいアリールアルキルオキシカルボニル基を表し、Fは水素原子またはカルボン酸活性化基を表し、*の炭素原子の立体配置はRまたはSを表す]
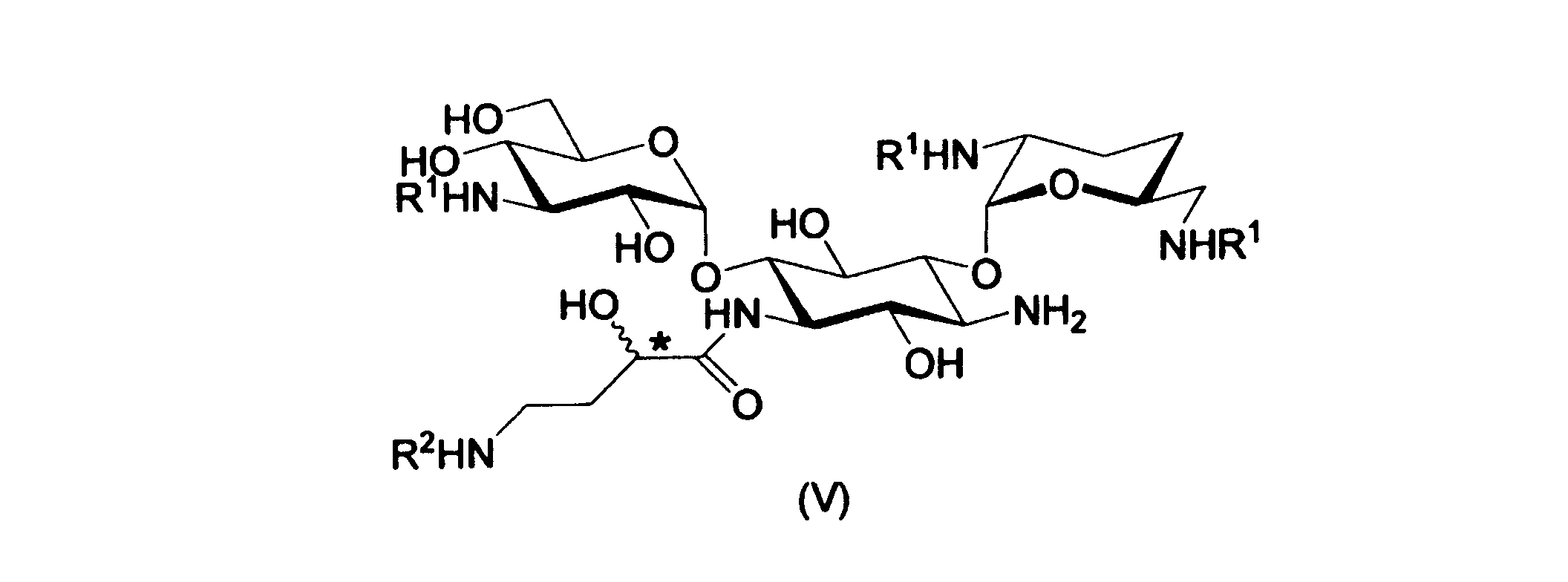
式Vに記載の化合物またはその塩におけるアミノ基を脱保護し、式IIに記載の化合物またはその塩を得ること
を含んでなる、(3)または(4)に記載の方法。
を含んでなる、(3)または(4)に記載の方法。
(6)式IIIに記載の化合物またはその塩における1位および3位のアミノ基にそれぞれ、保護基R3およびR1を導入して、式VIに記載の化合物またはその塩を得、

[式中、保護基R3は、置換されていてもよいアルキルカルボニル基を表す]
式VIに記載の化合物またはその塩における1位を脱保護し、該1位のアミノ基と式IVに記載の化合物またはその塩とを反応させて、式VIIに記載の化合物またはその塩を得、

[式中、R2は置換されていてもよいアリールアルキルオキシカルボニル基を表し、Fは水素原子またはカルボン酸活性化基を表し、*の炭素原子の立体配置はRまたはSを表す]

式VIIに記載の化合物またはその塩におけるアミノ基を脱保護し、式IIに記載の化合物またはその塩を得ることを含んでなる、(3)または(4)に記載の方法。
式VIに記載の化合物またはその塩における1位を脱保護し、該1位のアミノ基と式IVに記載の化合物またはその塩とを反応させて、式VIIに記載の化合物またはその塩を得、
(7)式IIIに記載の化合物またはその塩における1位および3位のアミノ基に保護基R3を導入して、式VIIIに記載の化合物またはその塩を得、

[式中、保護基R3は、置換されていてもよいアルキル基を表す]
式VIIIに記載の化合物またはその塩における1位のアミノ基を脱保護し、該1位のアミノ基と式IVに記載の化合物またはその塩とを反応させて、式IXに記載の化合物またはその塩を得、

[式中、R2は置換されていてもよいアリールアルキルオキシカルボニル基を表し、Fは水素原子またはカルボン酸活性化基を表し、*の炭素原子の立体配置はRまたはSを表す]

式IXに記載の化合物またはその塩におけるアミノ基を脱保護し、式IIに記載の化合物またはその塩を得ること
を含んでなる、(3)または(4)に記載の方法。
式VIIIに記載の化合物またはその塩における1位のアミノ基を脱保護し、該1位のアミノ基と式IVに記載の化合物またはその塩とを反応させて、式IXに記載の化合物またはその塩を得、
を含んでなる、(3)または(4)に記載の方法。
本発明によれば、従来の製造方法に比べて、(2S)-2-ヒドロキシジベカシン(式I)から顕著に高い収率の(2S)-2-ヒドロキシアルベカシン(式II)を効率的に取得することができる。
本明細書において、特に断らない限り、基または基の一部としての「アルキル」とは、基が直鎖状、分枝鎖状、または環状のアルキルを意味する。また、特に断らない限り、「アリール」とは、フェニルまたはナフチルを意味する。また、「アリールアルキル」とは、1以上の水素がアリール基で置換されたアルキルを意味する。
本明細書において、化合物の「塩」とは、大気中に放置したりまたは再結晶をすることにより、水分を吸収し、吸着水が付いたり、水和物となったりする場合があり、本発明には、そのような各種の水和物、溶媒和物および結晶多形の化合物も包含する。
式IIIに記載の化合物またはその塩(合成中間体)
本発明において、合成中間体として用いられる式IIIに記載の化合物は、1位および3位以外のアミノ基にいずれも保護基R1が導入されており、R1は、置換されていてもよいアルキルオキシカルボニル基または置換されていてもよいアリールアルキルオキシカルボニル基を表すことを特徴とする。
本発明において、合成中間体として用いられる式IIIに記載の化合物は、1位および3位以外のアミノ基にいずれも保護基R1が導入されており、R1は、置換されていてもよいアルキルオキシカルボニル基または置換されていてもよいアリールアルキルオキシカルボニル基を表すことを特徴とする。
R1で表されるアルキルオキシカルボニル基は、好ましくはC1~6アルキルオキシカルボニル基であり、より好ましくはC1~4アルキルオキシカルボニル基であり、さらに好ましくはメトキシカルボニル基またはtert-ブトキシカルボニル基であり、さらに好ましくはtert-ブトキシカルボニル基である。
R1で表されるアルキルオキシカルボニル基の1以上の水素原子は置換されていてもよく、かかる置換基としては塩素、臭素、フッ素等のハロゲン原子、メトキシ基、トリフルオロアルキル基またはニトロ基が挙げられる。
また、R1で表されるアリールアルキルオキシカルボニル基は、好ましくはアリールC1~4アルキルオキシカルボニル基あり、より好ましくはアリールC1~2アルキルオキシカルボニル基であり、さらに好ましくはベンジルオキシカルボニル基である。
R1で表されるアリールアルキルオキシカルボニル基の1以上の水素原子は置換されていてもよく、かかる置換基としては塩素、臭素、フッ素等のハロゲン原子、メトキシ基、トリフルオロメトキシ基またはニトロ基等が挙げられる。
また、式IIIに記載の化合物は、塩として存在することができる。その塩としては、特に限定されないが、例えば、アミノ基の酸付加塩が挙げられる。かかる塩は、薬学的に許容可能な塩により構成することができる。また、塩の具体例としては、フッ化水素酸塩、塩酸塩、臭化水素酸塩またはヨウ化水素酸塩等のハロゲン化水素酸塩、硫酸塩、硝酸塩、リン酸塩、過塩素酸塩または炭酸塩等の無機酸塩、酢酸塩、トリクロロ酢酸塩、トリフルオロ酢酸塩、ヒドロキシ酢酸塩、乳酸塩、クエン酸塩、酒石酸塩、シュウ酸塩、安息香酸塩、マンデル酸塩、酪酸塩、マレイン酸塩、プロピオン酸塩、ギ酸塩またはリンゴ酸塩等のカルボン酸塩、アルギニン酸塩、アスパラギン酸塩またはグルタミン酸塩等のアミノ酸塩、あるいはメタンスルホン酸塩またはp―トルエンスルホン酸塩等のスルホン酸塩等が挙げられる。上述の塩は、本発明における他の化合物においても同様である。
また、式IIIに記載の化合物は目的化合物の製造用用剤として提供することができる。
また、式IIIに記載の化合物は目的化合物の製造用用剤として提供することができる。
(2S)-2-ヒドロキシアルベカシン(式IIに記載の化合物)またはその塩
本発明によれば、式IIIに記載の化合物またはその塩を合成中間体として用いた製造方法により、(2S)-2-ヒドロキシアルベカシン(式IIに記載の化合物)またはその塩を効率的に取得することができる。本発明の1つの態様によれば、式IIに記載の化合物は、以下の式II-aで表される。

本発明によれば、式IIIに記載の化合物またはその塩を合成中間体として用いた製造方法により、(2S)-2-ヒドロキシアルベカシン(式IIに記載の化合物)またはその塩を効率的に取得することができる。本発明の1つの態様によれば、式IIに記載の化合物は、以下の式II-aで表される。
(2S)-2-ヒドロキシアルベカシン(式IIに記載の化合物)の製造方法
本発明によれば、式IIIに記載の化合物を合成中間体として用いて、(2S)-2-ヒドロキシジベカシン(式Iに記載の化合物)から(2S)-2-ヒドロキシアルベカシン(式IIに記載の化合物)を、以下の3つのルートA~Cにより有利に取得することができる。なお、ルートA~Cに記載の方法において、温度、溶媒および試薬の種類・量等の反応条件の詳細は特段の記載のない限り、当業者によって適宜決定することができる。
本発明によれば、式IIIに記載の化合物を合成中間体として用いて、(2S)-2-ヒドロキシジベカシン(式Iに記載の化合物)から(2S)-2-ヒドロキシアルベカシン(式IIに記載の化合物)を、以下の3つのルートA~Cにより有利に取得することができる。なお、ルートA~Cに記載の方法において、温度、溶媒および試薬の種類・量等の反応条件の詳細は特段の記載のない限り、当業者によって適宜決定することができる。

ルートAによれば、式Iに記載の化合物またはその塩における1位および3位以外のアミノ基に保護基R1を導入して式IIIに記載の化合物またはその塩を取得し(ステップa)、次に、式IIIに記載の化合物と式IVに記載の化合物とを反応させて式Vに記載の化合物またはその塩を得(ステップb)、アミノ基を脱保護して式IIの化合物を得る(ステップc)。
ルートAのステップaでは、上述の通り、銅イオンの存在下、式Iに記載の化合物またはその塩における1位および3位以外のアミノ基に保護基R1を導入する。銅カチオンの存在下、式Iに記載の化合物またはその塩の1位および3位以外のアミノ基にR1基が高選択的に導入されることは意外な事実である。
銅イオンは、本発明の効果を妨げない限り特に限定されないが、2価の銅イオンであることが好ましい。かかる銅イオンを生成する試薬としては、好ましくは第二銅の有機塩または無機塩であり、より好ましくは酢酸銅である。
また、ルートAのステップbでは、アシル化剤として式IVに記載の化合物が用いられる。
式IVに記載の化合物におけるR2で表されるアリールアルキルオキシカルボニル基は、好ましくはアリールC1~4アルキルオキシカルボニル基であり、より好ましくはアリールC1~2アルキルオキシカルボニル基であり、さらに好ましくはベンジルオキシカルボニル基である。
式IVに記載の化合物におけるR2で表されるアリールアルキルオキシカルボニル基は、好ましくはアリールC1~4アルキルオキシカルボニル基であり、より好ましくはアリールC1~2アルキルオキシカルボニル基であり、さらに好ましくはベンジルオキシカルボニル基である。
また、R2で表されるアリールアルキルオキシカルボニル基の1以上の水素原子は置換されていてもよく、かかる置換基としては塩素、臭素、フッ素等のハロゲン原子、メトキシ基、トリフルオロメトキシ基またはニトロ基等が挙げられ、好ましくはメトキシ基である。
また、式IVに記載の化合物において、Fは水素原子またはカルボン酸活性化基を表す。
カルボン酸活性化基とのエステルは、好ましくはスクシンイミドエステル体である。式IVに記載の化合物の具体例としては、4-ベンジルオキシカルボニルアミノ-2-ヒドロキシ酪酸スクシンイミドエステルが挙げられる。
カルボン酸活性化基とのエステルは、好ましくはスクシンイミドエステル体である。式IVに記載の化合物の具体例としては、4-ベンジルオキシカルボニルアミノ-2-ヒドロキシ酪酸スクシンイミドエステルが挙げられる。
また、式IVに記載の化合物の*の炭素原子の立体配置はRまたはSを表し、好ましくはSを表す。
さらに、式IVに記載の化合物は、好ましくは以下の式IV-aで表される。

ルートAによれば、後述する例1~4に示される通り、特許文献1に記載の方法と比較して約2倍の収率(約34%)が達成される。したがって、ルートAは、従来の方法と比較して、(2S)-2-ヒドロキシアルベカシンの工業生産上有利である。

ルートBによれば、ステップaで得られた式IIIに記載の化合物またはその塩における1位および3位のアミノ基にそれぞれ、保護基R3およびR1を導入して、式VIに記載の化合物またはその塩を得(ステップdおよびステップe)、式VIに記載の化合物またはその塩における1位のアミノ基を脱保護し(ステップf)、該1位のアミノ基と式IVに記載の化合物またはその塩とを反応させて、式VIIに記載の化合物またはその塩を得(ステップg)、式VIIに記載の化合物またはその塩におけるアミノ基を脱保護し、式IIに記載の化合物またはその塩を得る(ステップh)。
ルートBのステップdでは、保護基R3を用いると、3位のアミノ基を残したまま1位のアミノ基を選択的に保護しうることが本発明者らの検討により明らかとなった。さらにルートBのステップeおよびステップfに示される通り、化合物の3位のアミノ基をR1基で保護した後に、1位のアミノ基のR3基のみを選択的に脱保護できることを見出した。
このようなルートBによれば、アミノ基を選択的に保護または脱保護しうることから、副生成物の発生を防ぎ、効率的に目的物を製造することが可能となる。
このようなルートBによれば、アミノ基を選択的に保護または脱保護しうることから、副生成物の発生を防ぎ、効率的に目的物を製造することが可能となる。
また、後述する例5~7および8~10によれば、ルートAに比べ3工程増えているにもかかわらず、ルートBの収率(約46%)はルートAよりも向上している。したがって、ルートBは、式IIに記載の化合物の工業生産上一層有利であるといえる。

ルートCによれば、ステップaで得られた式IIIに記載の化合物またはその塩における1位および3位のアミノ基に保護基R3を導入して、式VIIIに記載の化合物またはその塩を得(ステップi)、式VIIIに記載の化合物またはその塩における1位のアミノ基を脱保護し(ステップj)、該1位のアミノ基と式IVに記載の化合物またはその塩とを反応させて、式IXに記載の化合物またはその塩を得(ステップk)、式IXに記載の化合物またはその塩におけるアミノ基を脱保護し、式IIに記載の化合物またはその塩を得る(ステップlおよびステップm)。
ルートCのステップiでは、式IIIに記載の化合物における1位および3位のアミノ基に2個のR3基を導入している。このR3保護体を脱保護反応にかけると、1位のR3基のみが選択的に脱離することが本発明者らの検討により明らかとなった。このようなルートCによれば、1位のアシル化(ステップk)も選択的に実施しうることから、副生成物の発生を防ぎ、効率的に目的物を製造することが可能となる。
後述する例11~19によれば、ルートAに比べ工程数が増えているにもかかわらず、ルートCの収率(約55%)は、ルートAおよびルートBと比較しても向上している。したがって、ルートCは、式IIに記載の化合物の工業生産上一層有利であるといえる。
本発明を下記実施例により具体的に説明するが、本発明はこれら実施例により限定されるものではない。
ルートA(R
1
=ベンジルオキシカルボニル基(Cbz))例1:2’,6’,3’’-トリ-N-ベンジルオキシカルボニル-(2S)-2-ヒドロキシジベカシンの製造

WO2007/142150(特許文献1)に記載の方法に準じて得た(2S)-2-ヒドロキシジベカシン(遊離塩基)1g(2.13ミリモル)と酢酸銅1水和物469mg(2.34ミリモル)の混合物にメタノール20mlを加えて均一溶液になるまで撹拌した。この溶液にN-(ベンジルオキシカルボニルオキシ)スクシンシミド1.75g(7.03ミリモル)を室温で加え、同温で16時間放置した。反応液を濃縮しクロロホルムを加えた後、濃アンモニア水で2回洗浄した。これを濃縮することにより表題化合物1.84g(99%)を得た。
ESIMS : m/z 892 [M+Na]+
1H-NMR (Pyridine-d5) : δ7.15-7.50 (m, 15H), 5.75 (d, 1H, J=3Hz), 5.67(d, 1H, J=3Hz), 5.15-5.45 (m, 6H)
1H-NMR (Pyridine-d5) : δ7.15-7.50 (m, 15H), 5.75 (d, 1H, J=3Hz), 5.67(d, 1H, J=3Hz), 5.15-5.45 (m, 6H)
例2:(2S)-2-ヒドロキシアルベカシンの製造

例1の表題化合物1.74g(2ミリモル)をピリジン3mlに溶解し、この溶液にKawaguchiらの報告(Journal of Antibiotics. Vol.
25. pp695-708(1972))に従って合成した(S)-4-ベンジルオキシカルボニルアミノ-2-ヒドロキシ酪酸スクシンイミドエステル980mg(2.8ミリモル)を-10~-5℃で5分間かけて加え、1時間後室温に戻し16時間撹拌した。反応液に濃アンモニア水0.5mlを加え室温で1時間撹拌後、濃縮し得られた残渣をクロロホルムに溶解し2回水洗後、濃縮した。残渣を1,4-ジオキサン-酢酸-水(4:1:1)の混合溶媒20mlに溶解し、パラジウムブラックを加え常温、常圧で6時間水素雰囲気下、接触還元を行った。反応液からパラジウムブラックを除去後、濃縮を行い、得られた残渣を水20mlに溶解した。この水溶液をアンバーライトCG-50アンモニアタイプの陽イオン交換樹脂カラム100mlに吸着し水洗後、0.1Nから1Nのアンモニア水溶液で溶離を行った。相当する画分を濃縮後凍結乾燥を行い表題化合物:(2S)-2-ヒドロキシアルベカシン376mg(33%)を得た。
25. pp695-708(1972))に従って合成した(S)-4-ベンジルオキシカルボニルアミノ-2-ヒドロキシ酪酸スクシンイミドエステル980mg(2.8ミリモル)を-10~-5℃で5分間かけて加え、1時間後室温に戻し16時間撹拌した。反応液に濃アンモニア水0.5mlを加え室温で1時間撹拌後、濃縮し得られた残渣をクロロホルムに溶解し2回水洗後、濃縮した。残渣を1,4-ジオキサン-酢酸-水(4:1:1)の混合溶媒20mlに溶解し、パラジウムブラックを加え常温、常圧で6時間水素雰囲気下、接触還元を行った。反応液からパラジウムブラックを除去後、濃縮を行い、得られた残渣を水20mlに溶解した。この水溶液をアンバーライトCG-50アンモニアタイプの陽イオン交換樹脂カラム100mlに吸着し水洗後、0.1Nから1Nのアンモニア水溶液で溶離を行った。相当する画分を濃縮後凍結乾燥を行い表題化合物:(2S)-2-ヒドロキシアルベカシン376mg(33%)を得た。
ESIMS : m/z 569 [M+H]+
1H-NMR (26%ND4OD) : δ5.13 (d, 1H, J=3.5Hz), 5.03 (d, 1H, J=4 Hz), 4.16 (dd, 1H, J=4, 9.5Hz), 3.98 (m, 1H), 3.83-3.90 (m, 2H), 3.65-3.77 (m, 2H), 3.37 (t, 1H, J=10, 10Hz), 2.82 (m, 1H), 2.70-2.78 (m, 2H), 2.65 (dd,1H, J=7, 13Hz), 2.62 (dd, 1H, J=7, 13Hz), 1.86-1.96 (m, 1H), 1.69-1.80 (m, 3H), 1.61 (dq, 1H, J= 4, 13Hz), 1.37 (dq,1H, J=4, 13Hz)
1H-NMR (26%ND4OD) : δ5.13 (d, 1H, J=3.5Hz), 5.03 (d, 1H, J=4 Hz), 4.16 (dd, 1H, J=4, 9.5Hz), 3.98 (m, 1H), 3.83-3.90 (m, 2H), 3.65-3.77 (m, 2H), 3.37 (t, 1H, J=10, 10Hz), 2.82 (m, 1H), 2.70-2.78 (m, 2H), 2.65 (dd,1H, J=7, 13Hz), 2.62 (dd, 1H, J=7, 13Hz), 1.86-1.96 (m, 1H), 1.69-1.80 (m, 3H), 1.61 (dq, 1H, J= 4, 13Hz), 1.37 (dq,1H, J=4, 13Hz)
ルートA(R
1
=tert-ブトキシカルボニル基(Boc))例3:2’,6’,3’’-トリ-N-t-ブトキシカルボニル-(2S)-2-ヒドロキシジベカシンの製造

WO2007/142150(特許文献1)に記載の方法に準じて得た(2S)-2-ヒドロキシジベカシン(遊離塩基)1g(2.13ミリモル)と酢酸銅1水和物469mg(2.34ミリモル)の混合物にメタノール20mlを加えて均一溶液になるまで撹拌した。この溶液に二炭酸ジ-t-ブチル1.53g(7.03ミリモル)を室温で加え、同温で16時間放置した。反応液を濃縮しクロロホルムを加えた後、濃アンモニア水で2回洗浄した。これを濃縮することにより表題化合物1.86g(定量)を得た。
ESIMS : m/z 790 [M+Na]+
1H-NMR (Pyridine-d5) : 5.79 (d, 1H, J=3Hz), 5.68 (d, 1H, J=3Hz), 1.48(s, 27H)
1H-NMR (Pyridine-d5) : 5.79 (d, 1H, J=3Hz), 5.68 (d, 1H, J=3Hz), 1.48(s, 27H)
例4:(2S)-2-ヒドロキシアルベカシンの製造

例3の表題化合物1.61g(2.1ミリモル)をピリジン3mlに溶解し、この溶液に(S)-4-(4-メトキシベンジルオキシ)カルボニルアミノ-2-ヒドロキシ酪酸スクシンイミドエステル1.12g(2.94ミリモル)を-10~-5℃で5分間かけて加え、1時間後室温に戻し16時間撹拌した。濃アンモニア水0.5mlを加え室温で1時間撹拌後、濃縮し得られた残渣をクロロホルムに溶解し2回水洗後、濃縮した。残渣にトリフルオロ酢酸5mlを加え室温で1時間撹拌した。濃縮を後得られた残渣を水20mlに溶解し濃アンモニア水で中和した。この水溶液を例1と同様の処理を行い表題化合物:(2S)-2-ヒドロキシアルベカシン407mg(34%)を得た。
ESIMS : m/z 569 [M+H]+
1H-NMR (26%ND4OD) : δ5.13 (d, 1H, J=3.5Hz), 5.03 (d, 1H, J=4Hz), 4.16 (dd, 1H, J=4, 9.5Hz),3.98 (m, 1H), 3.83-3.90 (m, 2H), 3.65-3.77 (m, 2H), 3.37 (t, 1H, J=10, 10Hz), 2.82 (m, 1H), 2.70-2.78 (m, 2H), 2.65 (dd, 1H, J=7, 13Hz), 2.62 (dd, 1H, J=7, 13Hz), 1.86-1.96 (m, 1H), 1.69-1.80 (m, 3H), 1.61 (dq, 1H, J=4, 13Hz), 1.37 (dq, 1H, J=4, 13Hz)
1H-NMR (26%ND4OD) : δ5.13 (d, 1H, J=3.5Hz), 5.03 (d, 1H, J=4Hz), 4.16 (dd, 1H, J=4, 9.5Hz),3.98 (m, 1H), 3.83-3.90 (m, 2H), 3.65-3.77 (m, 2H), 3.37 (t, 1H, J=10, 10Hz), 2.82 (m, 1H), 2.70-2.78 (m, 2H), 2.65 (dd, 1H, J=7, 13Hz), 2.62 (dd, 1H, J=7, 13Hz), 1.86-1.96 (m, 1H), 1.69-1.80 (m, 3H), 1.61 (dq, 1H, J=4, 13Hz), 1.37 (dq, 1H, J=4, 13Hz)
ルートB
例5:2’,6’,3’’-トリ-N-ベンジルオキシカルボニル-1-N-トリフルオロアセチル-(2S)-2-ヒドロキシジベカシンの製造

例1の表題化合物1g(1.15ミリモル)をメタノール2mlに溶解し、この溶液にトリフルオロ酢酸エチル0.16ml(1.38ミリモル)を-10~-5℃で加え、室温に戻し1時間撹拌し、濃縮して、例5の表題化合物1.10g(99%)を得た。
例5:2’,6’,3’’-トリ-N-ベンジルオキシカルボニル-1-N-トリフルオロアセチル-(2S)-2-ヒドロキシジベカシンの製造
ESIMS : m/z 988 [M+Na]+
δ7.15-7.50 (m, 15H), 5.75 (d, 1H, J=3Hz), 5.67 (d, 1H, J=3Hz), 5.15-5.45 (6H, m)
δ7.15-7.50 (m, 15H), 5.75 (d, 1H, J=3Hz), 5.67 (d, 1H, J=3Hz), 5.15-5.45 (6H, m)
例6:3,2’,6’,3’’-テトラ-N-ベンジルオキシカルボニル-(2S)-2-ヒドロキシジベカシンの製造

例5の表題化合物1.05g(1.09ミリモル)を1,4-ジオキサン2mlに溶解し、この溶液にトリエチルアミン0.2mlとN-(ベンジルオキシカルボニルオキシ)スクシンイミド298mg(1.2ミリモル)を室温で加え、同温で1時間撹拌した。反応液に濃アンモニア水1mlを加え室温で16時間撹拌した。反応液を濃縮後クロロホルムに溶解し2回水洗後に濃縮し、例6の表題化合物1.09g(定量)を得た。
ESIMS : m/z 1026 [M+Na]+
1H-NMR (Pyridine-d5) : δ7.15-7.55 (m, 20H), 5.86 (d, 1H, J=3Hz), 5.66 (d, 1H, J=3Hz), 5.25-5.45 (m, 8H)
1H-NMR (Pyridine-d5) : δ7.15-7.55 (m, 20H), 5.86 (d, 1H, J=3Hz), 5.66 (d, 1H, J=3Hz), 5.25-5.45 (m, 8H)
例7:(2S)-2-ヒドロキシアルベカシンの製造

例6の表題化合物990mg(0.99ミリモル)を用い例2と同様に処理し表題化合物:(2S)-2-ヒドロキシアルベカシン253mg(45%)を得た。
ESIMS : m/z 569 [M+H]+
1H-NMR (26%ND4OD) : δ5.13 (d, 1H, J=3.5Hz), 5.03 (d, 1H, J=4Hz), 4.16 (dd, 1H, J=4, 9.5Hz),3.98 (m, 1H), 3.83-3.90 (m, 2H), 3.65-3.77 (m, 2H), 3.37 (t, 1H, J=10, 10Hz), 2.82 (m, 1H), 2.70-2.78 (m, 2H), 2.65 (dd, 1H, J=7, 13Hz), 2.62 (dd, 1H, J=7, 13Hz), 1.86-1.96 (m, 1H), 1.69-1.80 (m, 3H), 1.61 (dq, 1H, J=4, 13Hz), 1.37 (dq, 1H, J=4, 13Hz)
1H-NMR (26%ND4OD) : δ5.13 (d, 1H, J=3.5Hz), 5.03 (d, 1H, J=4Hz), 4.16 (dd, 1H, J=4, 9.5Hz),3.98 (m, 1H), 3.83-3.90 (m, 2H), 3.65-3.77 (m, 2H), 3.37 (t, 1H, J=10, 10Hz), 2.82 (m, 1H), 2.70-2.78 (m, 2H), 2.65 (dd, 1H, J=7, 13Hz), 2.62 (dd, 1H, J=7, 13Hz), 1.86-1.96 (m, 1H), 1.69-1.80 (m, 3H), 1.61 (dq, 1H, J=4, 13Hz), 1.37 (dq, 1H, J=4, 13Hz)
例8:2’,6’,3’’-トリ-N-t-ブトキシカルボニル-1-N-トリフルオロアセチル-(2S)-2-ヒドロキシジベカシンの製造

例3の表題化合物1g(1.3ミリモル)をメタノール2mlに溶解し、この溶液にトリフルオロ酢酸エチル0.18ml(1.56ミリモル)を-10~-5℃で加え、室温に戻し1時間撹拌した。濃縮し例8の表題化合物1.12g(定量)を得た。
ESIMS : m/z 886 [M+Na]+
1H-NMR (Pyridine-d5) : 5.76 (d, 1H, J=3Hz), 5.66 (d, 1H, J=3Hz), 1.47 (s, 27H)
1H-NMR (Pyridine-d5) : 5.76 (d, 1H, J=3Hz), 5.66 (d, 1H, J=3Hz), 1.47 (s, 27H)
例9:3,2’,6’,3’’-テトラ-N-t-ブトキシカルボニル-(2S)-2-ヒドロキシジベカシンの製造

例8の表題化合物1.1g(1.27ミリモル)を1,4-ジオキサン2mlに溶解し、この溶液にトリエチルアミン0.2mlと二炭酸ジ-t-ブチル330mg(1.52ミリモル)を室温で加え、同温で1時間撹拌した。反応液に濃アンモニア水1mlを加え室温で16時間撹拌した。反応液を濃縮後クロロホルムに溶解し2回水洗後、濃縮して、例9の表題化合物1.11g(定量)を得た。
ESIMS : m/z 890 [M+Na]+
1H-NMR (Pyridine-d5) : 5.84 (d, 1H, J=3Hz), 5.73 (d, 1H, J=3Hz), 1.48 (s, 36H)
1H-NMR (Pyridine-d5) : 5.84 (d, 1H, J=3Hz), 5.73 (d, 1H, J=3Hz), 1.48 (s, 36H)
例10:(2S)-2-ヒドロキシアルベカシンの製造

例9の表題化合物1.06g(1.22ミリモル)を用い例4と同様に処理し、表題化合物:(2S)-2-ヒドロキシアルベカシン319mg(46%)を得た。
ESIMS : m/z 569 [M+H]+
1H-NMR (26%ND4OD) : δ5.13 (d, 1H, J=3.5Hz), 5.03 (d, 1H, J=4Hz), 4.16 (dd, 1H, J=4, 9.5Hz),3.98 (m, 1H), 3.83-3.90 (m, 2H), 3.65-3.77 (m, 2H), 3.37 (t, 1H, J=10, 10Hz), 2.82 (m, 1H), 2.70-2.78 (m, 2H), 2.65 (dd, 1H, J=7, 13Hz), 2.62 (dd, 1H, J=7, 13Hz), 1.86-1.96 (m, 1H), 1.69-1.80 (m, 3H), 1.61 (dq, 1H, J=4, 13Hz), 1.37 (dq, 1H, J=4, 13Hz)
1H-NMR (26%ND4OD) : δ5.13 (d, 1H, J=3.5Hz), 5.03 (d, 1H, J=4Hz), 4.16 (dd, 1H, J=4, 9.5Hz),3.98 (m, 1H), 3.83-3.90 (m, 2H), 3.65-3.77 (m, 2H), 3.37 (t, 1H, J=10, 10Hz), 2.82 (m, 1H), 2.70-2.78 (m, 2H), 2.65 (dd, 1H, J=7, 13Hz), 2.62 (dd, 1H, J=7, 13Hz), 1.86-1.96 (m, 1H), 1.69-1.80 (m, 3H), 1.61 (dq, 1H, J=4, 13Hz), 1.37 (dq, 1H, J=4, 13Hz)
ルートC(1)
例11:2’,6’,3’’-トリ-N-t-ブトキシカルボニル-1、3-ジ-N-トリフルオロアセチル-(2S)-2-ヒドロキシジベカシンの製造

例11:2’,6’,3’’-トリ-N-t-ブトキシカルボニル-1、3-ジ-N-トリフルオロアセチル-(2S)-2-ヒドロキシジベカシンの製造
例3の表題化合物208g(0.27モル)をメタノール1600 mlに溶解し、この溶液にトリエチルアミン88mlとトリフルオロ酢酸エチル175 ml(1.48モル)を加え、室温で終夜撹拌した。反応液を酢酸エチル3200 mlで希釈後、水3200 ml及び5%重曹水で洗浄した。有機層を硫酸マグネシウム脱水後に濃縮して、例11の表題化合物208g(80%)を得た。
例12:2’,6’,3’’-トリ-N-t-ブトキシカルボニル-3-N-トリフルオロアセチル-(2S)-2-ヒドロキシジベカシンの製造

例11の表題化合物208g(0.22モル)をメタノール4000mlに溶解し、この溶液に1モル水酸化ナトリウム水溶液420mlを加え、室温で終夜撹拌した。反応液をクロロホルム8000mlで希釈後、水4000mlで洗浄した。水層をクロロホルム4000 mlで抽出後、有機層を合わせ硫酸マグネシウム脱水後に濃縮して、例12の表題化合物171g(90%)を得た。
例13:2’,6’,3’’-トリ-N-t-ブトキシカルボニル-4’’’-N-p-メトキシベンジルオキシカルボニル-(2S)-2-ヒドロキシアルベカシンの製造

例12の表題化合物171 g(0.198モル)をTHF1200 mlに溶解し、この溶液に(S)-4-(4-メトキシベンジルオキシ)カルボニルアミノ-2-ヒドロキシ酪酸スクシンイミドエステル228g(0.6モル)のTHF溶液3500mlとトリエチルアミン117mlを加え、室温で終夜撹拌した。反応液に1モル水酸化ナトリウム水溶液2100mlとベンジルトリエチルアンモニウムクロリド(BTEAC)47.8gを加え、室温で7時間撹拌した。反応液をクロロホルム4800mlで希釈後、水4800 mlで洗浄した。水層をクロロホルム2400mlで抽出後、有機層を合わせ硫酸マグネシウム脱水後に濃縮して、例13の表題化合物250g(120 %)を得た。
例14:(2S)-2-ヒドロキシアルベカシンの製造

例13の表題化合物250gをメタノール2000mlに溶解し、5℃に冷却し6モル塩酸2000 mlを加え室温で6時間撹拌した。反応液を濃アンモニア水で中和後、析出物を除去後900mlまで濃縮した。この溶液を酢酸エチル2000mlで希釈後、水500ml抽出を行った。水層を濃アンモニア水で中和後、例2と同様にイオン交換樹脂で精製を行い、例14の表題化合物53.7g(40%)を得た。
ESIMS : m/z 569 [M+H]+
1H-NMR (26%ND4OD) : δ5.13 (d, 1H, J=3.5Hz), 5.03 (d, 1H, J=4Hz), 4.16 (dd, 1H, J=4, 9.5Hz),3.98 (m, 1H), 3.83-3.90 (m, 2H), 3.65-3.77 (m, 2H), 3.37 (t, 1H, J=10, 10Hz), 2.82 (m, 1H), 2.70-2.78 (m, 2H), 2.65 (dd, 1H, J=7, 13Hz), 2.62 (dd, 1H, J=7, 13Hz), 1.86-1.96 (m, 1H), 1.69-1.80 (m, 3H), 1.61 (dq, 1H, J=4, 13Hz), 1.37 (dq, 1H, J=4, 13Hz)
1H-NMR (26%ND4OD) : δ5.13 (d, 1H, J=3.5Hz), 5.03 (d, 1H, J=4Hz), 4.16 (dd, 1H, J=4, 9.5Hz),3.98 (m, 1H), 3.83-3.90 (m, 2H), 3.65-3.77 (m, 2H), 3.37 (t, 1H, J=10, 10Hz), 2.82 (m, 1H), 2.70-2.78 (m, 2H), 2.65 (dd, 1H, J=7, 13Hz), 2.62 (dd, 1H, J=7, 13Hz), 1.86-1.96 (m, 1H), 1.69-1.80 (m, 3H), 1.61 (dq, 1H, J=4, 13Hz), 1.37 (dq, 1H, J=4, 13Hz)
ルートC(2)-ラージスケール
例15
2’,6’,3’’-トリ-N-t-ブトキシカルボニル-(2S)-2-ヒドロキシジベカシンの製造

例15
2’,6’,3’’-トリ-N-t-ブトキシカルボニル-(2S)-2-ヒドロキシジベカシンの製造
DIW 4.8Lと2-OH-DKB 1.2Kgをポリ容器に投入し、溶解した。この溶液に酢酸銅一水和物490gを加え、MeOH 4.8Lで洗いこみ、キレート溶液を調製した。MeOH 18Lを反応釜に投入し、続いて調製したキレート溶液を加え、MeOH 18Lで洗いこんだ。内温を15℃以下に冷却し、(Boc)2O 2.41Kgを内温が20℃を超えないように加え、MeOH 2.4Lで洗いこみ、内温を20~25℃で終夜撹拌した。反応の終点を確認した後、濃アンモニア水を2.51L加え、30分間撹拌した。0.1N EDTA水溶液を49.1 L加えた。この混合液にクロロホルム 24 Lを加え、有機層を分け取った。水層からクロロホルム 24Lを用いて抽出し、得られた有機層を併せ定量を行い、2.29 Kgの標記化合物が得られたことを確認した。EtOHで12Lまで置換濃縮し、次工程に使用した。
LC Mass : m/s 768.5 [M+H]+
LC Mass : m/s 768.5 [M+H]+
例16:2’,6’,3’’-トリ-N-t-ブトキシカルボニル-1,3-ジ-N-トリフルオロアセチル-(2S)-2-ヒドロキシジベカシンの製造

例15化合物のEtOH溶液にTEA 1.03Lを投入しEtOH 1.2Lで洗浄した。この溶液に更にTFAOEt 2.92Lを加えEtOH 1.2Lで洗浄し、40℃で終夜撹拌した。反応の終点を確認した後、この溶液をそのまま次工程に使用した。
LC Mass : m/s 960.4 [M+H]+
LC Mass : m/s 960.4 [M+H]+
例17:2’,6’,3’’-トリ-N-t-ブトキシカルボニル-3-N-トリフルオロアセチル-(2S)-2-ヒドロキシジベカシンの製造

例16のEtOH溶液に0.5N NaOH水溶液24Lを、pHが9.5を超えないように注意しながら加えた。この混合液に0.3N NaOH水溶液を加えpHを10.90に調整した。0.3N NaOH水溶液は22 L要した。30分ごとにpHをチェックし、反応時間2時間まではpHが10.70以下ならば0.3N NaOHでpHを10.90に調整し、た。2時間以降はpHが10.65以下であったら.3N NaOHでpHを10.80に調整し、反応終点を3.5時間後に確認した。加温を停止し、即座に1N HClを用いてpHを9.0に調整した。この混合液にクロロホルム24Lを加え、有機層を分け取った。水層からクロロホルム24Lを用いて抽出し、得られた有機層を併せ定量を行い、2.07Kgの標記化合物が得られたことを確認した。DMFで12Lまで置換濃縮し、次工程に使用した。
LC Mass : m/s 864.4 [M+H]+
LC Mass : m/s 864.4 [M+H]+
例18

例17化合物にTHF 3.6Lを加え、続いて(S)-4-(4-メトキシベンジルオキシ)カルボニルアミノ-2-ヒドロキシ酪酸スクシンイミドエステルのTHF溶液を加え、THF 3.6Lで洗いこんだ。混合液を15℃以下に冷却し、内温が20℃を超えないようにTEA 1.37Lを加え、THF 1.2Lで洗いこんだ。反応温度を20℃に昇温し、その温度で終夜撹拌した。反応の終点を確認した後、1N NaOH水溶液 24.6Lを内温30℃以下で加えた。この混合液にDMF 12Lを加え、30℃で3時間撹拌した。5N NaOH水溶液 2.46Lを1時間ごとに混合液に加えた。反応の終点を確認した後、6N塩酸水でpHを10.0に調整し、クロロホルム24Lを加え有機層を分け取り、得られた水層をクロロホルム24Lで抽出した。有機層を併せた後、DMFで12Lまで置換濃縮し、この溶液を次工程に使用した。
LC Mass : m/s 1033.5 [M+H]+
LC Mass : m/s 1033.5 [M+H]+
例19:(2S)-2-ヒドロキシアルベカシンの製造

例18の溶液にMeOH 24Lを加え、この溶液に6N塩酸水24Lを内温が30℃を上回らないように加え、内温25~30℃で4.5時間撹拌し、反応の終点を確認した。内温約25℃を維持しながら濃アンモニア水でpHを5.8付近に調整した。この溶液を酢酸エチル24Lを用いて洗浄後、得られた水層を定量し、1.052Kgの粗(2S)-2-ヒドロキシアルベカシンが生成していることを確認した。
ESIMS : m/z 569 [M+H]+
さらに、レジン(WK-100,FPC-3500)を用い、アンモニア水で溶出した後に硫酸塩化し、(2S)-2-ヒドロキシアルベカシン硫酸塩として568g得た。
ESIMS : m/z 569 [M+H]+
さらに、レジン(WK-100,FPC-3500)を用い、アンモニア水で溶出した後に硫酸塩化し、(2S)-2-ヒドロキシアルベカシン硫酸塩として568g得た。
Claims (7)
- 式Iに記載の化合物またはその塩から式IIに記載の化合物またはその塩を製造するための、式IIIに記載の化合物またはその塩からなる、合成中間体:



- 式Iに記載の化合物またはその塩から式IIに記載の化合物またはその塩を製造するための、式IIIに記載の化合物またはその塩の使用:



- 式IIIに記載の化合物またはその塩を合成中間体として用いることを特徴とする、式Iに記載の化合物またはその塩から式IIに記載の化合物またはその塩を製造する方法:



- 式Iに記載の化合物またはその塩における1位および3位以外のアミノ基に保護基R1を銅イオンの存在下で導入して、式IIIに記載の化合物またはその塩を得ることを含んでなる、請求項3に記載の方法。
- 式IIIに記載の化合物またはその塩と、式IVに記載の化合物またはその塩とを反応させて、式Vに記載の化合物またはその塩を得、


を含んでなる、請求項3または4に記載の方法。 - 式IIIに記載の化合物またはその塩における1位および3位のアミノ基にそれぞれ、保護基R3およびR1を導入して、式VIに記載の化合物またはその塩を得、

式VIに記載の化合物またはその塩における1位のアミノ基を脱保護し、該1位のアミノ基と式IVに記載の化合物またはその塩とを反応させて、式VIIに記載の化合物またはその塩を得、


- 式IIIに記載の化合物またはその塩における1位および3位のアミノ基に保護基R3を導入して、式VIIIに記載の化合物またはその塩を得、

式VIIIに記載の化合物またはその塩における1位を脱保護し、該1位のアミノ基と式IVに記載の化合物またはその塩とを反応させて、式IXに記載の化合物またはその塩を得、


を含んでなる、請求項3または4に記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-216137 | 2015-11-02 | ||
| JP2015216137A JP2019001716A (ja) | 2015-11-02 | 2015-11-02 | アミノグリコシド系抗生物質の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017078096A1 true WO2017078096A1 (ja) | 2017-05-11 |
Family
ID=58662175
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/082666 WO2017078096A1 (ja) | 2015-11-02 | 2016-11-02 | アミノグリコシド系抗生物質の製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2019001716A (ja) |
| WO (1) | WO2017078096A1 (ja) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007142150A1 (ja) * | 2006-06-02 | 2007-12-13 | Meiji Seika Kaisha, Ltd. | 新規アミノグリコシド系抗生物質 |
-
2015
- 2015-11-02 JP JP2015216137A patent/JP2019001716A/ja active Pending
-
2016
- 2016-11-02 WO PCT/JP2016/082666 patent/WO2017078096A1/ja active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007142150A1 (ja) * | 2006-06-02 | 2007-12-13 | Meiji Seika Kaisha, Ltd. | 新規アミノグリコシド系抗生物質 |
Non-Patent Citations (1)
| Title |
|---|
| PROTECTIVE GROUPS IN ORGANIC SYNTHESIS, 1999, pages 503 - 547 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2019001716A (ja) | 2019-01-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA3059135C (en) | Synthesis of polycyclic-carbamoylpyridone compounds | |
| JP4104463B2 (ja) | 4’’−置換された−9−デオキソ−9a−アザ−9a−ホモエリスロマイシン誘導体を製造するための方法 | |
| CA2739992C (en) | Process for preparing 4-[2-(2-fluorophenoxymethyl)phenyl]piperidine compounds | |
| CN106459087A (zh) | 活化的凝血因子X (FXa)抑制剂的制备方法 | |
| CN106749242B (zh) | 阿维巴坦中间体的制备方法 | |
| ES2688597T3 (es) | Compuesto de benzoxaborol tricíclico, método de preparación y uso del mismo | |
| JP2019210273A (ja) | エドキサバンの製造方法 | |
| AU2016260693B2 (en) | Process for preparation of nitrogen mustard derivatives | |
| WO2015198505A1 (ja) | 合成ペンタペプチドの製造法 | |
| JP2019147763A (ja) | プロリンアミド化合物の製造方法 | |
| US20170267633A1 (en) | Novel crystalline arylalkylamine compound and process for producing the same | |
| CN111072660B (zh) | 一种瑞来巴坦的简便制备方法 | |
| WO2017078096A1 (ja) | アミノグリコシド系抗生物質の製造方法 | |
| FI100531B (fi) | Menetelmä 8-kloorikinolonijohdannaisten valmistamiseksi | |
| WO2014081047A1 (en) | Process for the preparation of (1s,4s,5s)-4-bromo-6-oxabicyclo[3.2.1] octan-7-one | |
| JP4294121B2 (ja) | ピリドンカルボン酸誘導体の製造方法およびその中間体 | |
| WO1994010185A1 (en) | Erythromycin derivative | |
| KR20150066777A (ko) | 광학활성 인돌린 유도체 및 이의 제조방법 | |
| TWI816708B (zh) | 用於合成環狀縮肽的方法 | |
| JP6239751B2 (ja) | ラコサミドの製造方法 | |
| JPS6396186A (ja) | 7−[(メタ−置換)フェニルグリシン]1−カルバ−1−デチアセファロスポリン類 | |
| JPWO2009142194A1 (ja) | 光学活性アミノアルコール誘導体の製造方法 | |
| ES2297415T3 (es) | Compuesto intermedio que se utiliza para la preparacion de pioglitazona. | |
| CN105859780A (zh) | 磷酸特地唑胺的制备方法 | |
| CN111247127B (zh) | 用于合成药物的中间体化合物的生产方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16862156 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16862156 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |