WO2016153064A1 - ジアミンおよびその利用 - Google Patents
ジアミンおよびその利用 Download PDFInfo
- Publication number
- WO2016153064A1 WO2016153064A1 PCT/JP2016/059755 JP2016059755W WO2016153064A1 WO 2016153064 A1 WO2016153064 A1 WO 2016153064A1 JP 2016059755 W JP2016059755 W JP 2016059755W WO 2016153064 A1 WO2016153064 A1 WO 2016153064A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- polyimide
- thin film
- diamine
- carbon atoms
- Prior art date
Links
- 0 CCC(C)=C*(N)=C Chemical compound CCC(C)=C*(N)=C 0.000 description 5
- KAWCEDACAFGLKU-UHFFFAOYSA-N Cc(cc(cc1)N)c1-c(cc1)c(C(F)(F)F)cc1N Chemical compound Cc(cc(cc1)N)c1-c(cc1)c(C(F)(F)F)cc1N KAWCEDACAFGLKU-UHFFFAOYSA-N 0.000 description 1
- MEEACOHBVRZXQZ-UHFFFAOYSA-N O=C1OCC(C(C2)C3)C1C2C3(CCC1(CC(C2)C3C(O4)=O)C2C3C4=O)C1=O Chemical compound O=C1OCC(C(C2)C3)C1C2C3(CCC1(CC(C2)C3C(O4)=O)C2C3C4=O)C1=O MEEACOHBVRZXQZ-UHFFFAOYSA-N 0.000 description 1
- KUYUMJCLIMPLRQ-UHFFFAOYSA-N OC(C(C1C2C(O3)=O)C2C3=O)OC1=O Chemical compound OC(C(C1C2C(O3)=O)C2C3=O)OC1=O KUYUMJCLIMPLRQ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/49—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by carboxyl groups
- C07C205/57—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by carboxyl groups having nitro groups and carboxyl groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C227/00—Preparation of compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C227/04—Formation of amino groups in compounds containing carboxyl groups
- C07C227/06—Formation of amino groups in compounds containing carboxyl groups by addition or substitution reactions, without increasing the number of carbon atoms in the carbon skeleton of the acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C229/00—Compounds containing amino and carboxyl groups bound to the same carbon skeleton
- C07C229/52—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton
- C07C229/54—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton with amino and carboxyl groups bound to carbon atoms of the same non-condensed six-membered aromatic ring
- C07C229/60—Compounds containing amino and carboxyl groups bound to the same carbon skeleton having amino and carboxyl groups bound to carbon atoms of six-membered aromatic rings of the same carbon skeleton with amino and carboxyl groups bound to carbon atoms of the same non-condensed six-membered aromatic ring with amino and carboxyl groups bound in meta- or para- positions
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K1/00—Printed circuits
- H05K1/02—Details
- H05K1/03—Use of materials for the substrate
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
Definitions
- the present invention relates to a diamine and use thereof.
- retardation is the product of birefringence (difference between two orthogonal refractive indexes) and film thickness, and this numerical value, particularly retardation in the thickness direction, is important because it affects viewing angle characteristics. Since it is a numerical value and a large retardation value may cause a decrease in display quality of the display (see, for example, Patent Document 3), these flexible display substrates have these in addition to high flexibility (flexibility). These characteristics are also required.
- the present invention has been made in view of such circumstances, and an object of the present invention is to provide a diamine that not only is excellent in flexibility and transparency, but also has a characteristic of low retardation.
- a polyimide that is soluble in an organic solvent can be obtained by copolymerizing with a fluorine-containing aromatic diamine and an alicyclic tetracarboxylic dianhydride such as tetracyclobutanoic dianhydride, and the polyimide can be used as an organic solvent. It was found that a thin film having not only excellent flexibility and transparency but also low retardation can be obtained from the composition obtained by dissolving in the present invention.
- the present invention relates to a diamine represented by the formula (1-1) as a first aspect.
- R 1 , R 2 , R 3 , R 4 and R 5 each independently represents a halogen atom, an alkyl group having 1 to 5 carbon atoms or an alkoxy group having 1 to 5 carbon atoms
- R 6 and R 7 each independently represent a hydrogen atom, a halogen atom, an alkyl group having 1 to 5 carbon atoms or an alkoxy group having 1 to 5 carbon atoms
- a, b, d and e each independently represents an integer of 0 to 4
- c represents an integer of 0 to 2.
- the present invention relates to the diamine according to the first aspect, which is a diamine represented by the formula (1-2).
- a third aspect relates to the diamine according to the second aspect, which is a diamine represented by formula (1-3) or formula (1-4).
- a 4th viewpoint it is related with the polyamic acid which is a reaction product of the diamine component containing the diamine as described in any one of a 1st viewpoint thru
- the said diamine component is related with the polyamic acid as described in a 4th viewpoint which further contains the diamine represented by a formula (A1).
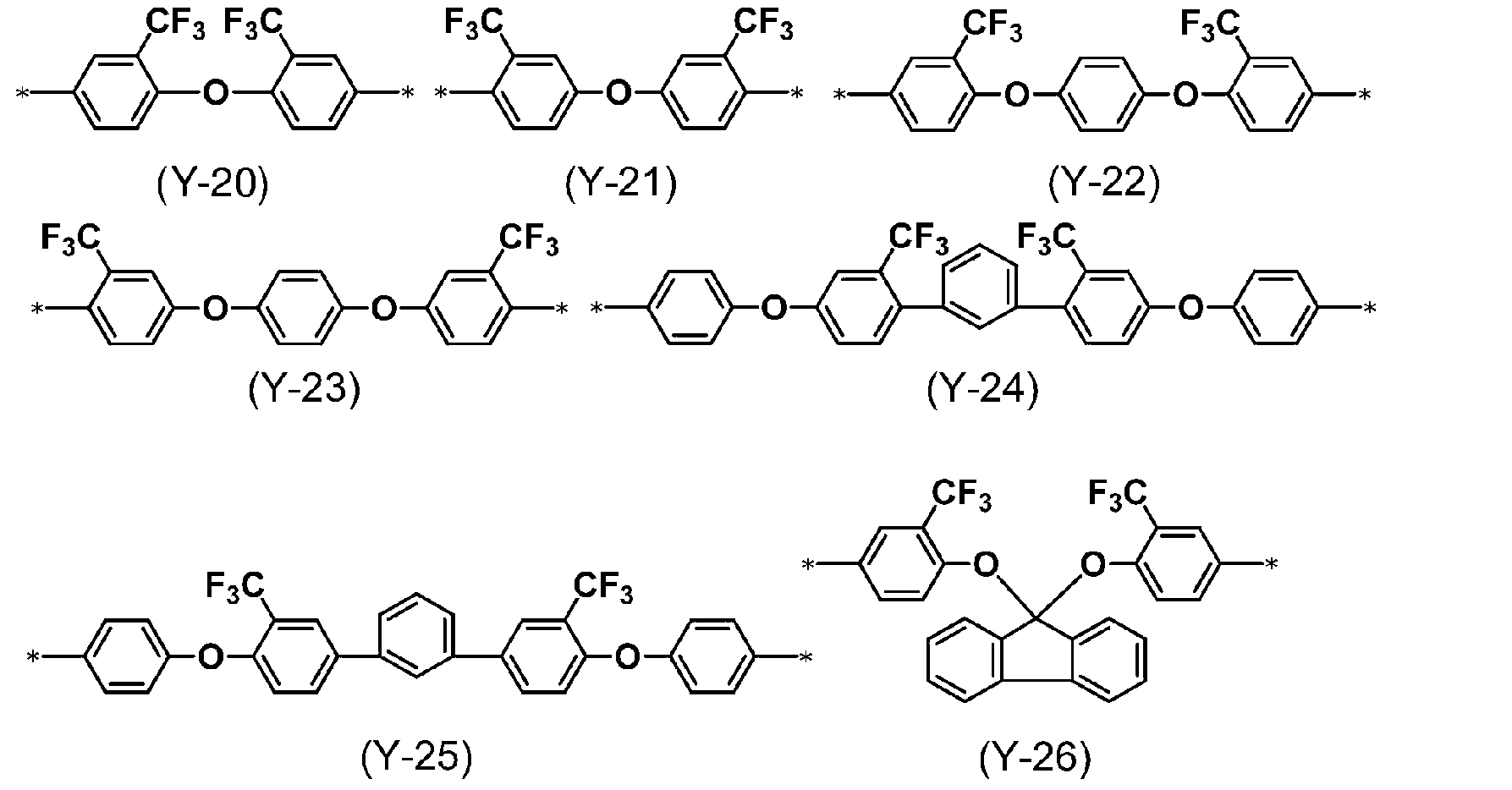
- B 2 represents a divalent group selected from the group consisting of formulas (Y-1) to (Y-34)).
- * represents a bond.
- the said acid dianhydride component is related with the polyamic acid as described in a 4th viewpoint or a 5th viewpoint containing the acid dianhydride represented by Formula (C1).
- B 1 represents a tetravalent group selected from the group consisting of formulas (X-1) to (X-12).
- the present invention relates to a polyimide obtained by imidizing the polyamic acid according to any one of the fourth aspect to the sixth aspect.
- a composition for forming a thin film comprising the polyimide according to the seventh aspect, an organic solvent, and silicon dioxide particles having an average particle diameter calculated from a specific surface area value measured by a nitrogen adsorption method of 100 nm or less Related to things.
- the present invention relates to the thin film forming composition according to the eighth aspect, wherein the mass ratio of the polyimide and the silicon dioxide particles is 1:10 to 10: 1.
- a 10th viewpoint it is related with the composition for thin film formation as described in an 8th viewpoint or a 9th viewpoint whose said average particle diameter is 60 nm or less.
- an 11th viewpoint it is related with the thin film formed from the composition for thin film formation as described in any one of 8th viewpoint thru
- a 12th viewpoint it is related with the board
- a 13th viewpoint it is related with the film forming composition containing the polyimide as described in a 7th viewpoint, and an organic solvent.
- the present invention relates to a dinitro compound characterized by being represented by the formula (2-1).
- R 1 , R 2 , R 3 , R 4 and R 5 each independently represents a halogen atom, an alkyl group having 1 to 5 carbon atoms or an alkoxy group having 1 to 5 carbon atoms
- R 6 and R 7 each independently represent a hydrogen atom, a halogen atom, an alkyl group having 1 to 5 carbon atoms or an alkoxy group having 1 to 5 carbon atoms
- a, b, d and e each independently represents an integer of 0 to 4
- c represents an integer of 0 to 2.
- the present invention relates to the dinitro compound according to the fifteenth aspect, which is a dinitro compound represented by the formula (2-2).
- the seventeenth aspect relates to the dinitro compound according to the sixteenth aspect, which is a dinitro compound represented by formula (2-3) or formula (2-4).
- An eighteenth aspect is a method for producing a diamine represented by formula (1-1), (Wherein R 1 , R 2 , R 3 , R 4 and R 5 each independently represents a halogen atom, an alkyl group having 1 to 5 carbon atoms or an alkoxy group having 1 to 5 carbon atoms, R 6 and R 7 each independently represent a hydrogen atom, a halogen atom, an alkyl group having 1 to 5 carbon atoms or an alkoxy group having 1 to 5 carbon atoms, a, b, d and e each independently represents an integer of 0 to 4, and c represents an integer of 0 to 2.
- the present invention relates to a production method including a step of obtaining a diamine represented by formula (1-1) by reducing a nitro group of a dinitro compound represented by formula (2-1).
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , a, b, c, d and e have the same meaning as described above.
- the novel diamine compound of the present invention can obtain a polyimide soluble in an organic solvent by copolymerizing with a alicyclic tetracarboxylic dianhydride together with a conventionally known fluorine-containing aromatic diamine.
- the polyimide obtained from the diamine compound of the present invention is excellent in flexibility and transparency, and can form a thin film capable of realizing a lower retardation.
- the resin film is also a flexible device, In particular, it can be suitably used as a substrate for a flexible display.
- membrane formed using the polyimide of this invention shows high transparency (high light transmittance, low yellowness) and low retardation, it can be used suitably as a board
- the diamine according to the present invention is a diamine represented by the formula (1-1), and particularly preferred is a diamine represented by the formula (1-2). Among them, a thin film having excellent flexibility and transparency and low retardation. Considering that the above can be obtained with good reproducibility, a diamine represented by formula (1-3) or formula (1-4) is preferable.
- R 1 , R 2 , R 3 , R 4 and R 5 are each independently a halogen atom, an alkyl group having 1 to 5 carbon atoms, or 1 to 5 carbon atoms.
- R 6 and R 7 each independently represents a hydrogen atom, a halogen atom, an alkyl group having 1 to 5 carbon atoms or an alkoxy group having 1 to 5 carbon atoms, and a, b, d and e each independently represents an integer of 0 to 4, and c represents an integer of 0 to 2.
- halogen atom examples include a fluorine atom, a chlorine atom, and a bromine atom.
- alkyl group having 1 to 5 carbon atoms include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, sec-butyl group, tert-butyl group, and n-pentyl group. , Isoamyl group, neopentyl group, tert-amyl group, sec-isoamyl group, cyclopentyl group, n-hexyl group and the like.
- alkoxy group having 1 to 5 carbon atoms examples include methoxy group, ethoxy group, n-propoxy group, isopropoxy group, n-butoxy group, isobutoxy group, sec-butoxy group, tert-butoxy group, and n-pentoxy group. , Isopentoxy group, neopentoxy group, tert-pentoxy group and the like.
- the diamines represented by the above formulas (1-1) to (1-4) of the present invention are obtained by reducing the nitro groups of the dinitro compounds represented by the following formulas (2-1) to (2-4), respectively. Obtainable. (In the formula, R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , a, b, c, d and e have the same meaning as described above.)
- the diamine represented by the formula (1-1) is 9,10- [1,2] benzenoanthracene-1,4-in an organic solvent as shown in the following scheme as an example.
- a diol compound hereinafter also referred to as a benzenoanthracenediol compound
- a nitrobenzoyl halide compound are reacted in the presence of a base catalyst to produce an intermediate (9,10- [1,2] benzenoanthracene-1,4 -Diyl bis (nitrobenzoate) compound) (compound represented by formula (2-1)) can be obtained (first stage), and the nitro group of this intermediate can be reduced (second stage). .
- the dinitro compounds represented by the above formulas (2-1) to (2-4) which are intermediates are also the object of the present invention.
- X represents a halogen atom
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , a, b, c, d and e have the same meaning as described above.
- the charging ratio of the benzenoanthracenediol compound and the nitrobenzoyl halide compound is preferably 2 to 4 mol of the nitrobenzoyl halide compound with respect to 1 mol of the benzenoanthracenediol compound.
- the nitrobenzoyl halide compound is low in stability in the reaction solution, it is preferable to add the necessary amount in several portions instead of adding the necessary amount at once.
- Base catalysts include trimethylamine, triethylamine, diisopropylamine, diisopropylethylamine, N-methylpiperidine, 2,2,6,6-tetramethyl-N-methylpiperidine, pyridine, 4-dimethylaminopyridine, N-methylmorpholine, etc.
- Organic bases such as organic amines are preferably used.
- the amount of the base catalyst used is not particularly limited as long as it is 2 mol or more per 1 mol of the benzenoanthracenediol compound, but it is usually about 2 to 10 mol.
- an acid absorbent may be used to neutralize an acid such as hydrochloric acid by-produced in the reaction.
- the acid absorbent examples include epoxides such as propylene oxide.
- the amount of the acid absorbent used is not particularly limited as long as it is 2 mol or more with respect to 1 mol of the benzenoanthracenediol compound, but it is usually about 2 to 10 mol.
- the organic solvent is not particularly limited as long as it does not affect the reaction, but is an aromatic hydrocarbon such as benzene, toluene, xylene; N, N-dimethylformamide (hereinafter referred to as DMF), Amides such as N, N-dimethylacetamide (hereinafter referred to as DMAc) and N-methyl-2-pyrrolidone (hereinafter referred to as NMP); diethyl ether, tetrahydrofuran, 1,4-dioxane, 1,2-dimethoxyethane, cyclopentyl Ethers such as methyl ether, ketones such as 2-butanone and 4-methyl-2-pentanone, nitriles such as acetonitrile, dimethyl sulfoxide (hereinafter referred to as DMSO) and the like can be used.
- DMF N, N-dimethylformamide
- NMP N-methyl-2-pyrrolidone
- the reaction temperature can be about 0 to 200 ° C., preferably 20 to 150 ° C.
- the solvent is distilled off, and the crude product is used in the next step as it is or after purification.
- the purification method is arbitrary and may be appropriately selected from known methods such as recrystallization, distillation, silica gel column chromatography.
- the method for reducing the nitro group of the intermediate to an amino group may be a known method and is not particularly limited.
- palladium-carbon, platinum oxide, Raney nickel, platinum- There is a method in which carbon, rhodium-alumina, platinum carbon sulfide, reduced iron, iron chloride, tin, tin chloride, zinc or the like is used as a catalyst, and hydrogen gas, hydrazine, hydrogen chloride, ammonium chloride or the like is used.
- catalytic hydrogenation is preferred because side reactions due to the ester sites of the intermediate are unlikely to occur and the desired product can be easily obtained.
- Examples of the hydrogen atom source for catalytic hydrogenation include hydrogen gas, hydrazine, hydrogen chloride, ammonium chloride, and ammonium formate.
- Examples of the catalyst used for the catalytic hydrogenation include powders of metals such as platinum, palladium, ruthenium, rhodium, nickel, iron, zinc, tin and the like, and the metal powder may be supported on an active material.
- the type of the catalyst is appropriately determined according to the type of the hydrogen source and the reaction conditions, and is not particularly limited, but may be any catalyst that can reduce only the nitro group, preferably palladium-carbon, platinum oxide, Raney nickel, Examples include platinum-carbon, rhodium-alumina, and platinum sulfide carbon.
- the amount of the catalyst used is not particularly limited because it is appropriately determined according to the type of hydrogen source and the reaction conditions, but is usually 0.01 mol% in terms of metal with respect to the raw dinitro compound (intermediate). 50 mol%, preferably 0.1 mol% to 20 mol%.
- the reaction solvent a solvent that does not affect the reaction can be used.
- ester solvents such as ethyl acetate and methyl acetate
- aromatic hydrocarbon solvents such as toluene and xylene
- aliphatic hydrocarbon solvents such as n-hexane, n-heptane and cyclohexane, 1,2-dimethoxyethane, tetrahydrofuran
- Ether solvents such as dioxane
- alcohol solvents such as methanol and ethanol
- ketone solvents such as 2-butanone and 4-methyl-2-pentanone
- Examples include aprotic polar solvents such as -2-pyrrolidone and dimethyl sulfoxide, and water.
- the reaction can be carried out at a temperature at which the reaction proceeds efficiently as long as it is not higher than the boiling point of the solvent used without decomposition of the raw materials and products.
- a temperature from ⁇ 78 ° C. to the boiling point of the solvent is preferable, and from the viewpoint of ease of synthesis, a temperature from 0 ° C. to the boiling point of the solvent is more preferable, more preferably from 0 to 100 ° C., and even more preferable.
- the catalytic hydrogenation may be performed under pressure conditions such as using an autoclave.
- the target diamine After the reaction, after distilling off the solvent, the target diamine can be obtained by purification using a known method such as recrystallization, distillation, silica gel column chromatography or the like. If the solvent contains a large amount of oxygen, the produced diamine compound may be colored. Therefore, the solvent used for the reaction and purification is preferably degassed before use. Moreover, in order to prevent coloring more, it is preferable to deaerate also the reaction liquid after the solvent distillation after the reaction.
- the benzenoanthracene diol compound used in the present invention for example, as shown in the following scheme as an example, is a Diels-Alder reaction of an anthracene compound and a 1,4-benzoquinone compound in an organic solvent according to a known method.
- 9,10- [1,2] benzenoanthracene-13,16 (9H, 10H) -dione compound obtained by treatment under heating conditions in acetic acid solvent in the presence of 47% hydrogen bromide. it can.
- X represents a halogen atom
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , a, b, c, d and e have the same meaning as described above.
- Polyamic acid and polyimide The diamine of the present invention described above can be converted into a polyamic acid by a polycondensation reaction with an acid dianhydride and then converted into a corresponding polyimide by a dehydration ring-closing reaction using heat or a catalyst. Both the polyamic acid and the polyimide are objects of the present invention.
- the diamine component used in the production of the polyamic acid of the present invention is not only excellent in flexibility and transparency, but also from the viewpoint of obtaining a polyamic acid and a polyimide that give a thin film having a characteristic of low retardation with good reproducibility.
- a fluorine-containing aromatic diamine is preferably contained, and a diamine represented by the following formula (A1) is more preferably contained.
- B 2 represents a divalent group selected from the group consisting of formulas (Y-1) to (Y-34)).
- * represents a bond.
- B 2 in the formula is the formula (Y-12), (Y-13), (Y-14), (Y-15), (Y-18)
- a diamine represented by (Y-27), (Y-28), (Y-30), (Y-33) is preferred, and the B 2 is represented by the formula (Y-12), (Y-13), Diamines represented by (Y-14), (Y-15), and (Y-33) are particularly preferred.
- the diamine component other diamine compounds other than the diamine represented by the above formula (1-1) and the diamine represented by the above formula (A1) are used. May be.
- the diamine represented by the formula (1-1) of the present invention when the fluorine-containing aromatic diamine is used together with the diamine represented by the formula (1-1) of the present invention, the diamine represented by the formula (1-1) and the fluorine-containing atom aromatic are used.
- the acid dianhydride component used in the production of the polyamic acid of the present invention is not only excellent in flexibility and transparency, but also has a reproducibility of polyamic acid and polyimide that give a thin film having a characteristic of low retardation.
- alicyclic tetracarboxylic dianhydride more preferably acid dianhydride represented by the following formula (C1).
- B 1 represents a tetravalent group selected from the group consisting of formulas (X-1) to (X-12).
- a plurality of R's independently represent a hydrogen atom or a methyl group, and * represents a bond.
- B 1 in the formula is represented by the formula (X-1), (X-2), (X-4), (X-5), (X -6), (X-7), (X-8), (X-9), (X-11), and (X-12) are preferred acid dianhydrides, wherein B 1 is the above formula Acid dianhydrides represented by (X-1), (X-2), (X-6), (X-11), and (X-12) are particularly preferred.
- the alicyclic tetracarboxylic acid dicarboxylic acid in the acid dianhydride component used in the production of the polyamic acid of the present invention is used.
- the anhydride content is preferably 50 mol% or more, more preferably 60 mol% or more, even more preferably 70 mol% or more, still more preferably 80 mol% or more, still more preferably 90 mol% or more, and most preferably 100 mol%. Mol%.
- the diamine represented by the above formula (1-1) and the diamine represented by the above formula (A1) are used as the diamine component, and the acid dianhydride component represented by the above (C1) is used as the acid dianhydride component.
- the polyamic acid has a monomer unit represented by the following formula (4-1) and a monomer unit represented by the following formula (4-2).
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , a, b, c, d, e, B 1 and B 2 represent the same meaning as described above.
- the method for obtaining the polyamic acid of the present invention is not particularly limited, and the aforementioned acid dianhydride component and diamine component may be reacted and polymerized by a known method.
- Examples of the solvent used for polyamic acid synthesis include m-cresol, N-methyl-2-pyrrolidone (NMP), N, N-dimethylformamide (DMF), N, N-dimethylacetamide (DMAc), and N-methyl.
- Examples include caprolactam, dimethyl sulfoxide (DMSO), tetramethyl urea, pyridine, dimethyl sulfone, hexamethyl phosphoramide, and ⁇ -butyrolactone. These may be used alone or in combination. Furthermore, even if it is a solvent which does not melt
- the temperature of the polycondensation reaction can be selected from -20 to 150 ° C, preferably -5 to 100 ° C.
- the polyamic acid solution obtained by the polymerization reaction of the polyamic acid described above can be used as a film-forming composition for forming a polyimide film, which will be described later, as it is, or after dilution or concentration.
- a poor solvent such as methanol or ethanol is added to the polyamic acid to precipitate a polyimide, and the polyamic acid is isolated.
- the isolated polyamic acid is redissolved in an appropriate solvent, and this is a film-forming composition described later. It can also be used as a product.
- the solvent for re-dissolution is not particularly limited as long as the polyamic acid obtained is dissolved, and examples thereof include m-cresol, 2-pyrrolidone, NMP, N-ethyl-2-pyrrolidone, N-vinyl- Examples include 2-pyrrolidone, DMAc, DMF, and ⁇ -butyrolactone.
- the solvent alone does not dissolve the polyamic acid, it can be used in addition to the above solvent as long as the polyamic acid does not precipitate.
- Specific examples thereof include ethyl cellosolve, butyl cellosolve, ethyl carbitol, butyl carbitol, ethyl carbitol acetate, ethylene glycol, 1-methoxy-2-propanol, 1-ethoxy-2-propanol, and 1-butoxy-2-propanol.
- the polyimide of the present invention can be obtained by subjecting the above-mentioned polyamic acid to dehydration ring closure (thermal imidization) by heating, or chemically ring closure using a known dehydration ring closure catalyst.
- the method by heating can be performed at an arbitrary temperature of 100 to 300 ° C., preferably 120 to 250 ° C.
- the chemical ring closure method can be carried out, for example, in the presence of pyridine, triethylamine, 1-ethylpiperidine, etc., and acetic anhydride. You can choose.
- the polyimide obtained from the polyamic acid having the monomer unit represented by the above formula (4-1) and the monomer unit represented by the above formula (4-2) thus obtained is represented by the following formula (5-1). And a monomer unit represented by the following formula (5-2).
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , a, b, c, d, e, B 1 and B 2 represent the same meaning as described above.
- the polyimide solution obtained by the ring-closing reaction of the polyamic acid described above can be used as it is or after diluting or concentrating, as a film-forming composition described later.
- a poor solvent such as methanol or ethanol is added to the polyimide solution to precipitate the polyimide, and the polyimide is isolated.
- the isolated polyimide is redissolved in an appropriate solvent, and this is used as a film forming composition described later. Can be used.
- these film forming compositions can be used for the preparation of a thin film forming composition containing polyimide and silicon dioxide particles described later.
- the solvent for re-dissolution is not particularly limited as long as it can dissolve the obtained polyimide.
- the solvent alone does not dissolve the polyimide, it can be used in addition to the above solvent as long as the polyimide does not precipitate.
- Specific examples thereof include ethyl cellosolve, butyl cellosolve, ethyl carbitol, butyl carbitol, ethyl carbitol acetate, ethylene glycol, 1-methoxy-2-propanol, 1-ethoxy-2-propanol, and 1-butoxy-2-propanol.
- the number average molecular weight of the polyamic acid (polyimide) is preferably 5,000 or more, more preferably 10,000 or more, and still more preferably from the viewpoint of improving the flexibility, strength, etc. of the thin film obtained. 15,000 or more, more preferably 20,000 or more, and preferably 200,000 or less, more preferably 100,000 or less, and still more preferably 50,000 or more from the viewpoint of ensuring the solubility of the resulting polyimide. 000 or less.
- a number average molecular weight is a value which is measured by a GPC (gel permeation chromatography) apparatus and calculated as polyethylene glycol and polyethylene oxide equivalent values.
- composition for forming a thin film comprising the polyimide of the present invention, an organic solvent, and silicon dioxide particles is also an object of the present invention.
- the silicon dioxide (silica) used in the present invention is not particularly limited, but silicon dioxide in the form of particles, for example, the average particle diameter is 100 nm or less, for example, 5 nm to 100 nm, preferably 5 nm to 60 nm, more preferably 5 nm to 55 nm, and more From the viewpoint of obtaining a highly transparent thin film with good reproducibility, the thickness is preferably 5 nm to 50 nm, more preferably 5 nm to 45 nm, still more preferably 5 nm to 35 nm, and still more preferably 5 nm to 30 nm.
- the average particle diameter of silicon dioxide particles is an average particle diameter value calculated from specific surface area values measured by a nitrogen adsorption method using silicon dioxide particles.
- colloidal silica having the above average particle size can be suitably used, and silica sol can be used as the colloidal silica.
- silica sol there can be used an aqueous silica sol produced by a known method using a sodium silicate aqueous solution as a raw material, and an organosilica sol obtained by substituting water as a dispersion medium of the aqueous silica sol with an organic solvent.
- alkoxysilanes such as methyl silicate and ethyl silicate are obtained by hydrolysis and condensation in an organic solvent such as alcohol in the presence of a catalyst (for example, an alkali catalyst such as ammonia, an organic amine compound, or sodium hydroxide).
- a silica sol obtained by replacing the silica sol with another organic solvent can be used.
- the present invention preferably uses an organosilica sol whose dispersion medium is an organic solvent.
- Examples of the organic solvent in the above-described organosilica sol include: lower alcohols such as methyl alcohol, ethyl alcohol and isopropanol; linear amides such as N, N-dimethylformamide and N, N-dimethylacetamide; N-methyl-2- Examples include cyclic amides such as pyrrolidone; ethers such as ⁇ -butyrolactone; glycols such as ethyl cellosolve and ethylene glycol, acetonitrile, and the like. This substitution can be performed by a usual method such as a distillation method or an ultrafiltration method.
- the viscosity of the organosilica sol is about 0.6 mPa ⁇ s to 100 mPa ⁇ s at 20 ° C.
- organosilica sols examples include, for example, trade name MA-ST-S (methanol-dispersed silica sol, manufactured by Nissan Chemical Industries, Ltd.), trade name MT-ST (methanol-dispersed silica sol, manufactured by Nissan Chemical Industries, Ltd.).
- Product name XBA-ST xylene / n-butanol mixed solvent dispersed silica sol, manufactured by Nissan Chemical Industries, Ltd.
- product name EAC-ST ethyl acetate dispersed silica sol, manufactured by Nissan Chemical Industries, Ltd.
- product Name PMA-ST propylene glycol monomethyl ether acetate dispersed silica sol, Nissan Chemical Industries, Ltd.
- Trade name MEK-ST methyl ethyl ketone dispersed silica sol, manufactured by Nissan Chemical Industries, Ltd.
- trade name MEK-ST-UP methyl ethyl ketone dispersed silica sol, manufactured by Nissan Chemical Industries, Ltd.
- trade name MEK-ST-L examples thereof include, but are not limited to, methyl ethyl ketone-dispersed silica sol, manufactured by Nissan Chemical Industries, Ltd., and trade name MIBK-ST (methyl isobutyl ketone-dispersed silica sol, manufactured by Nissan Chemical Industries
- the thin film forming composition of the present invention contains an organic solvent in addition to the polyimide and silicon dioxide.
- This organic solvent is not specifically limited, For example, the thing similar to the specific example of the reaction solvent used at the time of preparation of the said polyamic acid and a polyimide is mentioned. More specifically, N, N-dimethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidone, 1,3-dimethyl-2-imidazolidinone, N-ethyl-2-pyrrolidone, ⁇ - Examples include butyrolactone.
- an organic solvent may be used individually by 1 type, and may be used in combination of 2 or more type. Among these, N, N-dimethylacetamide, N-methyl-2-pyrrolidone, and ⁇ -butyrolactone are preferable in view of obtaining a thin film with high flatness with good reproducibility.
- the present invention is a composition for forming a thin film comprising the polyimide, silicon dioxide, and an organic solvent.
- the composition for forming a thin film of the present invention is uniform and phase separation is not observed.
- the blending amount of the solid content in the thin film forming composition of the present invention is usually about 0.5 to 30% by mass, preferably about 5 to 25% by mass.
- solid content concentration When the solid content concentration is less than 0.5% by mass, the film-forming efficiency is lowered in producing a thin film, and the viscosity of the composition for forming a thin film is lowered, so that it is difficult to obtain a coating film having a uniform surface. On the other hand, if the solid content concentration exceeds 30% by mass, the viscosity of the composition for forming a thin film becomes too high, and there is a possibility that the film forming efficiency is deteriorated and the surface uniformity of the coating film is lacking.
- solid content here means the total mass of components other than an organic solvent, and even if it is a liquid monomer etc., it shall be included in a weight as solid content.
- the viscosity of the composition for forming a thin film is appropriately set in consideration of the thickness of the thin film to be produced, etc. It is about 500 to 50,000 mPa ⁇ s at 25 ° C., preferably about 1,000 to 20,000 mPa ⁇ s.
- various organic or inorganic low-molecular or high-molecular compounds may be blended in the thin film-forming composition of the present invention in order to impart processing characteristics and various functionalities.
- a catalyst an antifoaming agent, a leveling agent, a surfactant, a dye, a plasticizer, fine particles, a coupling agent, a sensitizer, and the like can be used.
- the catalyst can be added for the purpose of reducing the retardation of the thin film and the linear expansion coefficient.
- the composition for thin film formation which contains a catalyst in addition to the said polyimide, silicon dioxide, and an organic solvent can also be made into the object of this invention.
- composition for forming a thin film of the present invention can be obtained by dissolving the polyimide and silicon dioxide obtained by the above-described method in the above-mentioned organic solvent, and adding silicon dioxide to the reaction solution after preparation of the polyimide, If desired, the organic solvent may be further added.
- the organic solvent is removed by applying the thin film forming composition of the present invention described above to a substrate, drying and heating, high heat resistance, high transparency, moderate flexibility, and moderate linear expansion. And a thin film having a small retardation. And the said thin film, ie, the thin film containing the said polyimide and the said inorganic silica compound, is also the object of this invention.
- the base material used for the production of the thin film examples include plastics (polycarbonate, polymethacrylate, polystyrene, polyester, polyolefin, epoxy, melamine, triacetylcellulose, ABS, AS, norbornene resin, etc.), metal, stainless steel (SUS). Wood, paper, glass, silicon wafer, slate and the like.
- the base material to be applied is glass or a silicon wafer from the viewpoint that existing equipment can be used, and the obtained thin film has good peelability. Of these, glass is more preferable.
- the linear expansion coefficient of the substrate to be applied is preferably 35 ppm / ° C.
- the coating method of the composition for forming a thin film on the substrate is not particularly limited.
- cast coating method spin coating method, blade coating method, dip coating method, roll coating method, bar coating method, die coating method.
- Method ink jet method, printing method (letter plate, intaglio plate, planographic plate, screen printing, etc.) and the like, and these can be appropriately used according to the purpose.
- the heating temperature is preferably 300 ° C. or lower. If it exceeds 300 ° C., the resulting thin film becomes brittle, and a thin film particularly suitable for display substrate use may not be obtained.
- the heating temperature is increased stepwise as it is, Finally, it is desirable to heat at over 175 ° C. to 280 ° C. for 30 minutes to 2 hours.
- the low thermal expansion characteristic can be expressed by heating at a temperature of two or more stages of drying the solvent and promoting molecular orientation.
- the applied thin film-forming composition is heated at 40 ° C. to 100 ° C.
- Heating for ⁇ 2 hours is preferred.
- the appliance used for heating include a hot plate and an oven.
- the heating atmosphere may be under air or under an inert gas such as nitrogen, and may be under normal pressure or under reduced pressure, and different pressures are applied at each stage of heating. May be.
- the thickness of the thin film is usually about 1 to 60 ⁇ m, preferably about 5 to 50 ⁇ m.
- a thin film having a desired thickness can be obtained by adjusting the thickness of the coating before heating. Form.
- there is no limitation in particular as a method of peeling the thin film formed in this way from a base material The thin film is cooled with the base material, the thin film is cut and peeled, and a tension is applied through a roll to separate. And the like.
- the polyamic acid-containing solution and the polyimide-containing solution can be suitably used as a film-forming composition for forming a polyimide film. That is, the polyamic acid-containing solution applied on the substrate is heated to cause an imidization reaction while evaporating the solvent, or the polyimide-containing solution applied on the substrate is heated to evaporate the solvent. Thereby, the film
- the heating temperature is usually about 40 to 500 ° C., and for example, it may be heated stepwise in the range of 40 to 150 ° C., 180 to 350 ° C., and further 380 to 450 ° C.
- a known additive such as a coupling agent may be added to the polyamic acid solution or the polyimide solution.
- the film-forming composition and a film formed using the composition are also objects of the present invention.
- the known additives that can be blended in the film-forming composition and the various conditions relating to the formation of the polyimide film are the additives that can be blended in the thin-film-forming composition described in detail above, and the composition. Various conditions relating to the production of the thin film formed from the above can be appropriately employed.
- Toluene (900 g), anthracene (90 g) and 1,4-benzoquinone (63.32 g) were placed in the flask, and the flask was evacuated and purged with nitrogen, and then heated to dissolve the solid. The resulting mixture was stirred for 20 hours under reflux conditions (110 ° C.). During heating and stirring, precipitation of the product was confirmed as the reaction progressed. Thereafter, the reaction mixture was cooled to room temperature, and the precipitate was collected by filtration and washed with toluene (540 g). Finally, the washed filtrate (135.64 g) was dried at 60 ° C.
- Acetic acid (693.5 g) was placed in the fluffco, and TB (95 g) obtained in Synthesis Example 1 was added and dissolved therein. Then, the temperature was raised to 70 ° C., and a 47% HBr aqueous solution (7.8 g) was added dropwise over 3 minutes, and then the resulting mixture was stirred at 70-80 ° C. for 1 hour. Thereafter, the reaction mixture was cooled to 30 ° C., and the precipitate was collected by filtration and washed successively with acetic acid (135.76 g) and toluene (221.79 g). Finally, the washed product (99.39 g) was dried at 70 ° C.
- THDNB 9,10-dihydro-9,10- [1,2] benzenoanthracene-1,4-diyl bis (4-nitrobenzoate)
- THDAB 9,10-dihydro-9,10- [1,2] benzenoanthracene-1,4-diyl bis (4-aminobenzoate)
- TH (20 g) obtained in Synthesis Example 2 was dissolved in DMF (1000 g) at 25 ° C., and triethylamine (29.8 g) was added.
- DMF 1000 g
- triethylamine 29.8 g
- about 3-5 g of 3-nitrobenzoyl chloride was added to the solution at 21 ° C., and the mixture was stirred at 21 ° C. to 27 ° C. for 5 minutes. This operation was repeated a total of 8 times, and a total of 29.8 g of 3-nitrobenzoyl chloride was added.
- water (1000 g) was added to the reaction solution at 25 ° C., and further stirred at 25 ° C. for 1 hour.
- the used Pd—C was washed with N, N-dimethylformamide (81 g), and the dimethylformamide used for washing was recovered together with the previous filtrate.
- Water (2300 g) was added dropwise to the collected filtrate at 25 ° C., and then the precipitate was collected by filtration, and the filtered product was washed with water (500 g).
- the obtained filtered product (66.2 g) was dried under reduced pressure at 70 ° C. to obtain a m-THDAB crude product (35.2 g).
- This m-THDAB recrystallized product (30.8 g) was dissolved in degassed tetrahydrafuran (308 g), and after adding 30 mg of 79% hydrazine monohydrate, special white birch activated carbon (3.08 g) was added, After stirring for 1 hour, it was filtered. The obtained filtrate was dried at 70 ° C. under reduced pressure to obtain m-THDAB activated carbon treated product (28.6 g). Furthermore, this m-THDAB activated carbon treatment product was added to degassed hexane (858 g), and the mixture was stirred for 1 hour under reflux conditions.
- reaction mixture was cooled to 50 ° C., and then further 1,2,3,4-cyclobutanetetracarboxylic dianhydride (CBDA) 0. 637g was added and it stirred as it was overnight. Thereafter, the reaction mixture is diluted with ⁇ -butyrolactone so that the solid concentration is 8% by mass, and 2.65 g of acetic anhydride and 1.542 g of pyridine are added to the diluted reaction mixture, and then the reaction mixture is added under a nitrogen atmosphere. Stir at 4 ° C. for 4 hours. The resulting reaction mixture was then added dropwise to 100 g of methanol and stirred for 30 minutes, The precipitate was collected by filtration. This operation was repeated three times. Finally, the obtained residue was dried under reduced pressure at 150 ° C. for 8 hours to obtain polyimide (3.248 g yield: 87.2%).
- CBDA 1,2,3,4-cyclobutanetetracarboxylic dianhydride
- THDA 9,10-dihydro-9,10- [1,2] benzenoanthracene skeleton in the molecule
- polyimide was obtained in the same manner as in Example 1 (3.22 g, 86.2%).
- THDA was synthesized according to the method described in Journal of Polymer Science Part A: Polymer Chemistry, Vol. 49, 3109-3120 (2011).
- Example 2 Preparation of polyimide solution (varnish)
- the polyimide obtained in Example 1 was dissolved in ⁇ -butyrolactone so as to have a concentration of 12% by mass to obtain a polyimide solution.
- Example 3 Preparation of polyimide film
- the polyimide solution obtained in Example 2 was subjected to pressure filtration using a 5 ⁇ m filter. Thereafter, the filtered polyimide solution was applied onto a glass substrate in the air, and heated sequentially at 50 ° C. for 30 minutes, 140 ° C. for 30 minutes, and 200 ° C. for 60 minutes to obtain a polyimide film. Then, a rectangular cut was made in the obtained polyimide film, and the film was peeled off to obtain an evaluation sample.
- Example 3 A polyimide film was obtained by the same procedure and method as in Example 3 except that the polyimide solution obtained in Comparative Example 2 was used instead of the polyimide solution obtained in Example 2. Then, a rectangular cut was made in the obtained polyimide film, and the film was peeled off to obtain an evaluation sample.
- Example 5 Preparation of thin film forming composition
- the average particle diameter calculated from the specific surface area value measured by the nitrogen adsorption method was 22 nm.
- the specific surface area of the dry powder of silica sol was measured using a specific surface area measuring device Monosorb MS-16 manufactured by Yuasa Ionics Co., and D was measured using the measured specific surface area S (m 2 / g).
- Example 6 The thin film-forming composition obtained in Example 5 was applied to a glass substrate, and the coating film was sequentially heated under a vacuum of ⁇ 97 kPa at 50 ° C. for 30 minutes, 140 ° C. for 30 minutes, and 200 ° C. for 60 minutes. A thin film was obtained. For the heating, three ovens set in advance to a desired temperature were used. The obtained thin film was peeled off by mechanical cutting and subjected to subsequent evaluation.
- Td 5% The 5% weight loss temperature (Td 5% [° C.]) is measured by using a TGA Q500 manufactured by TA Instruments and raising the temperature of about 5 to 10 mg of a thin film to 50 to 800 ° C. at 10 ° C./min in nitrogen. I asked for it.
- thickness direction retardation ( Rth ) and in-plane retardation ( R0 ) are calculated by the following formula
- R th [(Nx + Ny) / 2 ⁇ Nz]
- ⁇ d [( ⁇ Nxz ⁇ d) + ( ⁇ Nyz ⁇ d)] / 2 Nx
- Ny Two in-plane orthogonal refractive indexes (Nx> Ny, Nx is also called the slow axis, and Ny is also called the fast axis)
- Nz Refractive index in the thickness (perpendicular) direction (perpendicular) to the surface d: Film thickness ⁇ Nxy: Difference between two in-plane refractive indices (Nx ⁇ Ny) (birefringence)
- ⁇ Nxz difference between in-plane refractive index Nx and thickness direction refractive index Nz (birefringence)
- the membrane produced using the diamine of the present invention is more linearly expanded than the membrane produced using a known diamine structurally similar to the diamine of the present invention (Comparative Example 3).
- the coefficient was low and could have a low value of approximately 30 ppm / ° C.
- the transmittance was higher than that of the comparative example, the heat resistance was further improved, and the yellowness (CIE b * ) was also low.
- the retardation R th in the thickness direction was also less than 700 nm, and the result was as low as the comparative example.
- the thin film (Example 6) manufactured using the composition for forming a thin film containing the polyimide manufactured using the diamine of the present invention and the silicon dioxide particles contains the silicon dioxide particles
- the light transmittance The coefficient of linear expansion at 50 ° C. to 200 ° C. is about 15 ppm / ° C., which is lower than that of the film of Example 3, that is, excellent in dimensional stability during heating and 5% weight reduction.
- the heat resistance evaluated by temperature was also improved.
- the thin film is obtained by multiplying two birefringences (differences between two in-plane refractive indices and a refractive index in the thickness direction) when viewed from a cross section in the thickness direction by the film thickness.
- the thin film produced using the diamine of the present invention has characteristics such as a low linear expansion coefficient, high transparency (high light transmittance, low yellowness), and low retardation, that is, the base of the flexible display substrate. It can be expected that the film satisfies the necessary requirements as a film and can be particularly suitably used as a base film of a flexible display substrate.
- the reaction mixture was then cooled to 50 ° C., 0.637 g (3.25 mmol) of CBDA was added, and the reaction was allowed to proceed overnight under a nitrogen atmosphere. The next day, dilute the reaction mixture with GBL to a solids concentration of 8% by weight, add 2.654 g (0.026 mol) of acetic acid and 1.542 g (19.5 mmol) of pyridine, at 100 ° C. Stir for 4 hours. Next, the obtained reaction mixture was dropped into 100 g of methanol and stirred for 30 minutes, stirred for 30 minutes, and solid polyimide was separated by filtration. This operation was repeated three times. Methanol in the polyimide was removed by drying in a vacuum oven at 150 ° C. for 8 hours to obtain 3.2438 g (yield 81.17%) of polyimide I finally dried. Next, powdered polyimide I was dissolved in GBL so as to have a concentration of 12% to obtain a polyimide I solution.
- Example 13 Preparation of polyimide film-forming composition and preparation of polyimide film
- Example 13 At room temperature, 1 g of the polyimide I obtained in Example 7 was dissolved in a GBL solvent so that the polyimide concentration was 12% by mass, and this solution was slowly filtered through a 5 ⁇ m filter to obtain a thin film forming composition. It was. Next, the obtained thin film forming composition was applied to a glass substrate and heated in air at 50 ° C. for 30 minutes, 140 ° C. for 30 minutes and 200 ° C. for 60 minutes to obtain a transparent film PI-I. . The obtained thin film was peeled off from the glass substrate by mechanical cutting. The optical and thermal properties are shown in Table 2.
- Example 14 Preparation of polyimide film-forming composition and preparation of polyimide film
- Example 14 At room temperature, 1 g of the polyimide II obtained in Example 8 was dissolved in a GBL solvent so that the polyimide concentration was 12% by mass, and this solution was slowly filtered through a 5 ⁇ m filter to obtain a thin film forming composition. It was. Next, the obtained thin film-forming composition was applied to a glass substrate and heated in air at 50 ° C. for 30 minutes, 140 ° C. for 30 minutes and 200 ° C. for 60 minutes to obtain a transparent film PI-II. . The obtained thin film was peeled off from the glass substrate by mechanical cutting. The optical and thermal properties are shown in Table 2.
- Example 15 Preparation of polyimide film forming composition and creation of polyimide film
- Example 15 At room temperature, 1 g of the polyimide III obtained in Example 9 was dissolved in a GBL solvent so that the polyimide concentration was 12% by mass, and this solution was slowly filtered through a 5 ⁇ m filter to obtain a thin film forming composition. It was. Next, the obtained thin film-forming composition was applied to a glass substrate and heated in air at 50 ° C. for 30 minutes, 140 ° C. for 30 minutes and 200 ° C. for 60 minutes to obtain a transparent film PI-III. . The obtained thin film was peeled off from the glass substrate by mechanical cutting. The optical and thermal properties are shown in Table 2.
- This thin film forming composition was applied to a glass substrate and heated under reduced pressure of ⁇ 97 kPa at 50 ° C. for 30 minutes, 140 ° C. for 30 minutes and 200 ° C. for 60 minutes to obtain a transparent film PI-III-A. .
- the obtained thin film was peeled off from the glass substrate by mechanical cutting.
- the optical and thermal properties are shown in Table 2.
- Example 17 Preparation of polyimide film forming composition and creation of polyimide film
- Example 17 At room temperature, 1 g of polyimide IV obtained in Example 4 was dissolved in a GBL solvent so that the polyimide concentration was 12% by mass, and this solution was slowly filtered under pressure through a 5 ⁇ m filter to obtain a composition for forming a thin film. It was. Next, the obtained thin film-forming composition was applied to a glass substrate, heated in air at 50 ° C. for 30 minutes, 140 ° C. for 30 minutes and 200 ° C. for 60 minutes, and under a reduced pressure of ⁇ 100 kPa, Heating at 280 ° C. for 60 minutes gave a transparent film PI-IV. The obtained thin film was peeled off from the glass substrate by mechanical cutting. The optical and thermal properties are shown in Table 3.
- Example 18 Preparation of polyimide film-forming composition and preparation of polyimide film
- Example 18 At room temperature, 1 g of the polyimide V obtained in Example 11 was dissolved in a GBL solvent so that the polyimide concentration was 12% by mass, and this solution was slowly filtered under pressure through a 5 ⁇ m filter to obtain a thin film forming composition. It was. Next, the obtained thin film-forming composition was applied to a glass substrate, heated in air at 50 ° C. for 30 minutes, 140 ° C. for 30 minutes and 200 ° C. for 60 minutes, and under a reduced pressure of ⁇ 100 kPa, Heating was performed at 280 ° C. for 60 minutes to obtain a transparent film PI-V. The obtained thin film was peeled off from the glass substrate by mechanical cutting. The optical and thermal properties are shown in Table 3.
- Example 19 Preparation of polyimide film-forming composition and preparation of polyimide film
- Example 19 At room temperature, 1 g of the polyimide V obtained in Example 6 was dissolved in a GBL solvent so as to have a polyimide concentration of 12% by mass, and this solution was slowly filtered through a 5 ⁇ m filter to obtain a composition for forming a thin film. It was.
- the obtained thin film-forming composition was applied to a glass substrate, heated in air at 50 ° C. for 30 minutes, 140 ° C. for 30 minutes and 200 ° C. for 60 minutes, and under a reduced pressure of ⁇ 100 kPa, Heating was performed at 280 ° C. for 60 minutes to obtain a transparent film PI-VI.
- the obtained thin film was peeled off from the glass substrate by mechanical cutting.
- the optical and thermal properties are shown in Table 2.
- the thermal decomposition temperature (Td point) was carried out using a TGA Q500 manufactured by TA Instruments under a nitrogen stream at a heating rate of 10 ° C./min.
- a weight loss of 5% was specified at 150 ° C.
- the number average molecular weight (Mn) and the weight average molecular weight (Mw) were determined using Showdex GPC-101 manufactured by Showa Denko K.K.
- a PTFE 0.45 ⁇ m filter for polymer filtration was used, and standard polystyrene was used for the calibration curve.
- Film formation was performed using the Kortest Instruments free automatic film applicator PFA-2010-1, and film baking was performed using a circular oven DO45 from Deng YNG. The film thickness was measured with a thickness gauge manufactured by Teclock Co., Ltd.
- the membranes (Examples 13 to 15 and Examples 17 to 19) produced using the diamine of the present invention have a low coefficient of linear expansion and transmission.
- the rate was high, the heat resistance was good, and the yellowness (CIE b * ) was low. Also were preferable results with respect to the retardation R th in the thickness direction.
- the thin film (Example 16) manufactured using the composition for forming a thin film containing the polyimide manufactured using the diamine of the present invention and the silicon dioxide particles contains the silicon dioxide particles, it has a light transmittance.
- the linear expansion coefficient at 50 ° C. to 200 ° C. was as low as about 16 ppm / ° C.
- the results showed that the dimensional stability during heating was excellent, and the heat resistance evaluated at a 5% weight loss temperature was also improved.
- the thin film is obtained by multiplying two birefringences (differences between two in-plane refractive indices and a refractive index in the thickness direction) when viewed from a cross section in the thickness direction by the film thickness.
- thickness direction retardation R th expressed as an average value of the phase difference is as low as less than 150 nm, was an extremely low value of birefringence ⁇ n also 0.004.
- the thin film produced using the diamine of the present invention has characteristics such as a low linear expansion coefficient, high transparency (high light transmittance, low yellowness), and low retardation, that is, the base of the flexible display substrate. It can be expected that the film satisfies the necessary requirements as a film and can be particularly suitably used as a base film of a flexible display substrate.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Engineering & Computer Science (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description
これらのデバイスにおいては、ガラス基板上に様々な電子素子、例えば、薄膜トランジスタや透明電極等が形成されているが、このガラス材料を柔軟かつ軽量な樹脂材料に替えることで、デバイス自体の薄型化や軽量化、フレキシブル化が期待される。
そして、そのような樹脂材料の候補としてはポリイミドが注目を集めており、ポリイミドフィルムに関する種々の報告が従来よりなされている(例えば特許文献1,2参照)。
すなわち、リタデーション(位相差)とは、複屈折(直交する2つの屈折率の差)と膜厚との積をいうが、この数値、特に厚さ方向のリタデーションは視野角特性に影響する重要な数値であり、大きなリタデーション値は、ディスプレイの表示品質の低下を招く原因となり得ることから(例えば特許文献3参照)、フレキシブルディスプレイ基板にあっても、高い柔軟性(可撓性)以外に、これらの特性も求められている。

R6及びR7は、それぞれ独立して、水素原子、ハロゲン原子、炭素原子数1乃至5のアルキル基又は炭素原子数1乃至5のアルコキシ基を表し、
a、b、d及びeは、それぞれ独立して、0~4の整数を表し、そして
cは0~2の整数を表す。)
第2観点として、式(1-2)で表されるジアミンである、第1観点に記載のジアミンに関する。


第5観点として、前記ジアミン成分が、式(A1)で表されるジアミンをさらに含む、第4観点に記載のポリアミック酸に関する。





第6観点として、前記酸二無水物成分が、式(C1)で表される酸二無水物を含む、第4観点又は第5観点に記載のポリアミック酸に関する。


第7観点として、第4観点乃至第6観点のうちいずれか一項に記載のポリアミック酸をイミド化して得られるポリイミドに関する。
第8観点として、第7観点に記載のポリイミドと、有機溶媒と、窒素吸着法により測定された比表面積値から算出される平均粒子径が100nm以下である二酸化ケイ素粒子を含む、薄膜形成用組成物に関する。
第9観点として、前記ポリイミドと前記二酸化ケイ素粒子の質量比が、1:10~10:1である、第8観点に記載の薄膜形成用組成物に関する。
第10観点として、前記平均粒子径が、60nm以下である、第8観点又は第9観点に記載の薄膜形成用組成物に関する。
第11観点として、第8観点乃至第10観点のうちいずれか一項に記載の薄膜形成用組成物から形成される薄膜に関する。
第12観点として、第11観点に記載の薄膜からなるフレキシブルデバイス用基板に関する。
第13観点として、第7観点に記載のポリイミドと、有機溶媒とを含む膜形成用組成物に関する。
第14観点として、第13観点に記載の膜形成用組成物から形成される膜からなるフレキシブルデバイス用基板に関する。
第15観点として、式(2-1)で表されることを特徴とするジニトロ化合物に関する。

R6及びR7は、それぞれ独立して、水素原子、ハロゲン原子、炭素原子数1乃至5のアルキル基又は炭素原子数1乃至5のアルコキシ基を表し、
a、b、d及びeは、それぞれ独立して、0~4の整数を表し、そして
cは0~2の整数を表す。)
第16観点として、式(2-2)で表されるジニトロ化合物である、第15観点に記載のジニトロ化合物に関する。



R6及びR7は、それぞれ独立して、水素原子、ハロゲン原子、炭素原子数1乃至5のアルキル基又は炭素原子数1乃至5のアルコキシ基を表し、
a、b、d及びeは、それぞれ独立して、0~4の整数を表し、そして
cは0~2の整数を表す。)
式(2-1)で表されるジニトロ化合物のニトロ基を還元して式(1-1)で表されるジアミンを得る段階を含む、製造方法に関する。

また本発明のジアミン化合物から得られるポリイミドは、柔軟性及び透明性に優れ、さらに低いリタデーションを実現できる薄膜を形成できる。
さらに本発明のポリイミドを含む薄膜形成用組成物より得られる薄膜は、柔軟性及び透明性に優れることに加え、特に低い線膨張係数、低いリタデーションを示すことから、該樹脂膜についてもフレキシブルデバイス、特にフレキシブルディスプレイの基板として好適に用いることができる。
そして、本発明のポリイミドを用いて形成される膜は、高い透明性(高い光線透過率、低い黄色度)、低いリタデーションを示すことから、フレキシブルデバイス、特にフレキシブルディスプレイの基板として好適に用いることができる。
以下、本発明についてさらに詳しく説明する。
本発明に係るジアミンは、式(1-1)で表されるジアミンであり、特に式(1-2)で表されるジアミンが好ましく、中でも、柔軟性及び透明性に優れ、低リタデーションの薄膜等を再現性よく得ることを考慮すると、好ましくは式(1-3)又は式(1-4)で表されるジアミンである。

上記炭素原子数1乃至5のアルキル基としては、例えばメチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、n-ペンチル基、イソアミル基、ネオペンチル基、tert-アミル基、sec-イソアミル基、シクロペンチル基、n-ヘキシル基等が挙げられる。
また炭素原子数1乃至5のアルコキシ基としては、メトキシ基、エトキシ基、n-プロポキシ基、イソプロポキシ基、n-ブトキシ基、イソブトキシ基、sec-ブトキシ基、tert-ブトキシ基、n-ペントキシ基、イソペントキシ基、ネオペントキシ基、tert-ペントキシ基等が挙げられる。


塩基触媒としては、トリメチルアミン、トリエチルアミン、ジイソプロピルアミン、ジイソプロピルエチルアミン、N-メチルピペリジン、2,2,6,6-テトラメチル-N-メチルピペリジン、ピリジン、4-ジメチルアミノピリジン、N-メチルモルホリン等の有機アミン類等の有機塩基が好適に用いられる。また、塩基触媒の使用量は、ベンゼノアントラセンジオール化合物1モルに対して2モル以上であれば特に限定されるものではないが、通常2~10モル程度である。
また、反応で副生する塩酸等の酸を中和するために、酸吸収剤を用いてもよい。酸吸収剤としては、プロピレンオキシド等のエポキシド類が挙げられる。酸吸収剤の使用量は、ベンゼノアントラセンジオール化合物1モルに対して2モル以上であれば特に限定されるものではないが、通常2~10モル程度である。
有機溶媒としては、反応に影響を及ぼさない溶媒であれば特に限定されるものではないが、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;N,N-ジメチルホルムアミド(以下、DMFという)、N,N-ジメチルアセトアミド(以下、DMAcという)、N-メチル-2-ピロリドン(以下、NMPという)等のアミド類;ジエチルエーテル、テトラヒドロフラン、1,4-ジオキサン、1,2-ジメトキシエタン、シクロペンチルメチルエーテル等のエーテル類、2-ブタノン、4-メチル-2-ペンタノンなどのケトン類、アセトニトリル等のニトリル類、ジメチルスルホキシド(以下、DMSOという)などを用いることができる。これらの溶媒は、単独で用いても、2種以上を組み合わせて用いてもよい。なお、溶媒中に水分が多く含まれると、エステルの加水分解が起こることから、溶媒は脱水溶媒を使用する、もしくは、脱水してから使用することが好ましい。
反応温度は、0~200℃程度とすることができるが、20~150℃が好ましい。
反応後は、溶媒を留去し、粗生成物のまま、あるいは精製して次工程に用いる。精製法は任意であり、再結晶、蒸留、シリカゲルカラムクロマトグラフィ等公知の手法から適宜選択すればよい。
接触水素化の水素原子源としては、水素ガスやヒドラジン、塩化水素、塩化アンモニウム、ギ酸アンモニウム等が挙げられる。
接触水素化に用いる触媒としては、白金、パラジウム、ルテニウム、ロジウム、ニッケル、鉄、亜鉛、スズ等の金属の粉末が挙げられ、金属の粉末が活性体に担持されたものであってもよい。触媒の種類は、水素源の種類や反応条件に応じて適宜決定されるため、特に限定されないが、ニトロ基のみを還元できる触媒であればよく、好ましくは、パラジウム-炭素、酸化白金、ラネーニッケル、白金-炭素、ロジウム-アルミナ、硫化白金炭素が挙げられる。また、触媒の使用量は、水素源の種類や反応条件に応じて適宜決定されるため、特に限定されないが、原料のジニトロ体(中間体)に対して金属換算で通常0.01モル%から50モル%、好ましくは0.1モル%から20モル%である。
反応溶媒としては、反応に影響を及ぼさない溶媒を用いることができる。例えば、酢酸エチル、酢酸メチルなどのエステル系溶媒、トルエン、キシレンなどの芳香族炭化水素溶媒、n-ヘキサン、n-ヘプタン、シクロヘキサンなどの脂肪族炭化水素溶媒、1,2-ジメトキシエタン、テトラヒドロフラン、ジオキサンなどのエーテル系溶媒、メタノール、エタノールなどのアルコール系溶媒、2-ブタノン、4-メチル-2-ペンタノンなどのケトン系溶媒、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチル-2-ピロリドン、ジメチルスルホキシドなどの非プロトン性極性溶媒、水などが挙げられる。これらの溶媒は、単独、又は、2種類以上混合して使用することができる。
反応温度は、原料や生成物が分解することなく、用いる溶媒の沸点以下であれば、反応が効率よく進行する温度で行なうことができる。具体的には、-78℃から溶媒の沸点以下の温度が好ましく、合成の簡便性の観点から、0℃から溶媒の沸点以下の温度がより好ましく、さらに好ましくは0~100℃、さらにより好ましくは10~50℃である。
また、接触水素化は、オートクレーブを用いる等して、加圧条件の下で行ってもよい。
反応後は、溶媒を留去後、再結晶、蒸留、シリカゲルカラムクロマトグラフィ等公知の手法を用いて精製し、目的物のジアミンを得ることができる。なお、溶媒中に酸素が多く含まれると、生成したジアミン化合物の着色が起こる場合があるため、反応および精製に使用する溶媒は脱気してから使用することが好ましい。また、より着色を防ぐために、反応後の溶媒留去前、溶媒留去後の反応液も脱気することが好ましい。

以上説明した本発明のジアミンは、酸二無水物との重縮合反応によりポリアミック酸とした後、熱または触媒を用いた脱水閉環反応により、対応するポリイミドとすることができる。該ポリアミック酸及びポリイミドともに本発明の対象である。





また、本発明の効果を損なわない範囲において、前記ジアミン成分には、上記式(1-1)で表されるジアミン、上記式(A1)で表されるジアミン以外の、その他のジアミン化合物を用いてもよい。



ポリアミック酸を合成する際の酸二無水物成分のモル数とジアミン成分のモル数との比は、酸二無水物成分/ジアミン成分=0.8~1.2である。
重縮合反応の温度は、-20~150℃、好ましくは-5~100℃の任意の温度を選択することができる。
再溶解用溶媒は、得られたポリアミック酸を溶解させるものであれば特に限定されるものではなく、例えば、m-クレゾール、2-ピロリドン、NMP、N-エチル-2-ピロリドン、N-ビニル-2-ピロリドン、DMAc、DMF、γ-ブチロラクトンなどが挙げられる。
加熱による方法は、100~300℃、好ましくは120~250℃の任意の温度で行うことができる。
化学的に閉環する方法は、例えば、ピリジンやトリエチルアミン、1-エチルピペリジンなどと、無水酢酸などとの存在下で行うことができ、この際の温度は、-20~200℃の任意の温度を選択することができる。

再溶解用溶媒は、得られたポリイミドを溶解させるものであれば特に限定されるものではなく、例えば、m-クレゾール、2-ピロリドン、NMP、N-エチル-2-ピロリドン、N-ビニル-2-ピロリドン、DMAc、DMF、γ-ブチロラクトンなどが挙げられる。
上述の本発明のポリイミドと、有機溶媒と、二酸化ケイ素粒子を含む薄膜形成用組成物も本発明の対象である。
本発明に用いる二酸化ケイ素(シリカ)は特に限定されないが、粒子形態の二酸化ケイ素、例えば平均粒子径が100nm以下、例えば5nm~100nm、好ましくは5nm~60nm、より好ましくは5nm~55nmであり、より高透明の薄膜を再現性よく得る観点から、好ましくは5nm~50nm、より好ましくは5nm~45nm、より一層好ましくは5nm~35nm、さらに好ましくは5nm~30nmである。
本発明において二酸化ケイ素粒子の平均粒子径とは、二酸化ケイ素粒子を用いて窒素吸着法により測定された比表面積値から算出される平均粒子径値である。
また、メチルシリケートやエチルシリケート等のアルコキシシランを、アルコール等の有機溶媒中で触媒(例えば、アンモニア、有機アミン化合物、水酸化ナトリウム等のアルカリ触媒)の存在下において加水分解し、縮合して得られるシリカゾル、又はそのシリカゾルを他の有機溶媒に溶媒置換したオルガノシリカゾルも用いることができる。
これらの中でも本発明は分散媒が有機溶媒であるオルガノシリカゾルを用いることが好ましい。
上記のオルガノシリカゾルの粘度は、20℃で、0.6mPa・s~100mPa・s程度である。
本発明において二酸化ケイ素、例えばオルガノシリカゾルとして使用される上記製品に挙げたような二酸化ケイ素は、二種以上を混合して用いてもよい。
本発明の薄膜形成用組成物は、前記ポリイミド及び二酸化ケイ素に加えて、有機溶媒を含む。該有機溶媒は、特に限定されるものではなく、例えば、上記ポリアミック酸及びポリイミドの調製時に用いた反応溶媒の具体例と同様のものが挙げられる。より具体的には、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチル-2-ピロリドン、1,3-ジメチル-2-イミダゾリジノン、N-エチル-2-ピロリドン、γ-ブチロラクトンなどが挙げられる。なお、有機溶媒は、1種を単独で使用してもよく、2種以上を組み合わせて使用してもよい。
これらの中でも、平坦性の高い薄膜を再現性よく得ることを考慮すると、N,N-ジメチルアセトアミド、N-メチル-2-ピロリドン、γ-ブチロラクトンが好ましい。
本発明は、前記ポリイミドと二酸化ケイ素と有機溶媒とを含有する薄膜形成用組成物である。ここで本発明の薄膜形成用組成物は、均一なものであって、相分離は認められないものである。
本発明の薄膜形成用組成物において、前記ポリイミドと前記二酸化ケイ素の配合比は、質量比で、ポリイミド:二酸化ケイ素=10:1~1:10であることが好ましく、より好ましくは8:2~2:8、例えば7:3~3:7である。
また本発明の薄膜形成用組成物における固形分量の配合量は、通常0.5~30質量%程度、好ましくは5~25質量%程度である。固形分濃度が0.5質量%未満であると薄膜を作製する上において製膜効率が低くなり、また薄膜形成用組成物の粘度が低くなるため、表面が均一な塗膜を得られにくい。また固形分濃度が30質量%を超えると、薄膜形成用組成物の粘度が高くなりすぎて、やはり成膜効率の悪化や塗膜の表面均一性に欠ける虞がある。なおここでいう固形分量とは、有機溶媒以外の成分の総質量を意味し、液状のモノマー等であっても固形分として重量に含めるものとする。
なお薄膜形成用組成物の粘度は、作製する薄膜の厚み等を勘案し適宜設定するものではあるが、特に5~50μm程度の厚さの薄膜を再現性よく得ること目的とする場合、通常、25℃で500~50,000mPa・s程度、好ましくは1,000~20,000mPa・s程度である。
本発明の薄膜形成用組成物は、上述の方法で得られたポリイミド並びに二酸化ケイ素を上述の有機溶媒に溶解して得ることができるし、ポリイミドの調製後の反応溶液に二酸化ケイ素を添加し、所望により前記有機溶媒を更に加えたものとしてもよい。
以上説明した本発明の薄膜形成用組成物を基材に塗布して乾燥・加熱することで有機溶媒を除去し、高い耐熱性と、高い透明性と、適度な柔軟性と、適度な線膨張係数とを有し、しかもリタデーションの小さい薄膜を得ることができる。
そして上記薄膜、すなわち上記ポリイミドと、上記無機シリカ化合物とを含有する薄膜も本発明の対象である。
特に、電子デバイスの基板材料として適用する場合においては、既存設備を利用することができるという観点から、適用する基材がガラス、シリコンウェハであることが好ましく、また得られる薄膜が良好な剥離性を示すことからガラスであることがさらに好ましい。なお、適用する基材の線膨張係数としては塗工後の基材の反りの観点から、好ましくは35ppm/℃以下、より好ましくは30ppm/℃以下、より一層好ましくは25ppm/℃以下、さらに好ましくは、20ppm/℃以下である。
また、得られる薄膜の耐熱性と線膨張係数特性を考慮すると、塗布した薄膜形成用組成物を40℃~100℃で5分間~2時間加熱した後に、そのまま段階的に加熱温度を上昇させ、最終的に175℃超~280℃で30分~2時間加熱することが望ましい。このように、溶媒を乾燥させる段階と分子配向を促進する段階の2段階以上の温度で加熱することにより、低熱膨張特性を発現させることができる。
特に、塗布した薄膜形成用組成物は、40℃~100℃で5分間~2時間加熱した後に、100℃超~175℃で5分間~2時間、次いで、175℃超~280℃で5分~2時間加熱することが好ましい。
加熱に用いる器具は、例えばホットプレート、オーブン等が挙げられる。加熱雰囲気は、空気下であっても窒素等の不活性ガス下であってもよく、また、常圧下であっても減圧下であってもよく、また加熱の各段階において異なる圧力を適用してもよい。
なおこのようにして形成された薄膜を基材から剥離する方法としては特に限定はなく、該薄膜を基材ごと冷却し、薄膜に切れ目を入れ剥離する方法やロールを介して張力を与えて剥離する方法等が挙げられる。
上述したように、前記ポリアミック酸含有溶液や前記ポリイミド含有溶液はポリイミドの膜を形成するための膜形成用組成物として好適に用いることができる。
すなわち、基材上に塗布した上記ポリアミック酸含有溶液を加熱し、溶媒を蒸発させつつイミド化反応をさせることで、あるいは、基材上に塗布した上記ポリイミド含有溶液を加熱し、溶媒を蒸発させることで、本発明のポリイミドを含む膜を得ることができる。この際、加熱温度は、通常40~500℃程度であり、例えば、40~150℃の範囲、180~350℃の範囲、さらに380~450℃の範囲で段階的に加熱してもよい。
なお、ポリイミドの膜と基材との密着性を更に向上させる目的で、ポリアミック酸溶液やポリイミド溶液に、カップリング剤等の公知の添加剤を加えてもよい。
上記膜形成用組成物並びに該組成物を用いて形成される膜も本発明の対象である。
なお、膜形成用組成物に配合され得る公知の添加剤や、ポリイミドの膜の形成等に係る諸条件は、先に詳述した薄膜形成用組成物に配合され得る添加剤や、該組成物から形成される薄膜の製造等に係る諸条件を適宜採用することができる。
カラム:Inertsil ODS-3、5μm、4.6×250mm
オーブン:40℃、 検出波長:254nm、 流速:1.0mL/分
溶離液:
TB:アセトニトリル/0.5%リン酸水溶液=70/30 サンプル注入量:2μL
TH:アセトニトリル/0.5%リン酸水溶液=70/30 サンプル注入量:1μL
THDNB:アセトニトリル/0.5%リン酸水溶液=80/20 サンプル注入量:10μL
THDAB:アセトニトリル/水=80/20 サンプル注入量:5μL
<HPLC分析(2)>
カラム:Inertsil ODS-3、5μm、4.6×250mm
オーブン:40℃、 検出波長:200nm,254nm、 流速:1.0mL/分
溶離液:
m-THDNB:アセトニトリル/0.5%リン酸水溶液=70/30 サンプル注入量:10μL
m-THDAB:アセトニトリル/0.5%リン酸水溶液=70/30 サンプル注入量:10μL
装置:フーリエ変換型超伝導核磁気共鳴装置(FT-NMR)(INOVA-400(Varian社)400MHz、
溶媒:DMSO-d6、CDCl3
内標準物質:テトラメチルシラン(TMS)
<数平均分子量(Mn)及び重量平均分子量(Mw)の測定>
装置:昭和電工(株)製、Showdex GPC-101
カラム:KD803およびKD805
カラム温度:50℃
溶出溶媒:DMF、流量:1.5ml/分
検量線:標準ポリスチレン
[合成例1] 9,10-[1,2]ベンゼノアントラセン-13,16(9H,10H)-ジオン(以下、TBという)の合成

その後、反応混合物を室温まで冷却し、析出物をろ過によって回収し、トルエン(540g)で洗浄した。
最後に、洗浄したろ取物(135.64g)を減圧下、60℃で乾燥することで、TBを122.73g得た(収率;84.9%、HPLC面百値(保持時間;6.3min);98.8%)。
1HNMR(CDCl3、δppm):7.4(m,1H)、7.2(m,2H )、7.1(m,1H)、6.3(s,1H)、4.9(s,1H)、3.1(t,1H).

その後、反応混合物を30℃まで冷却し、析出物をろ過によって回収し、酢酸(135.76g)、トルエン(221.79g)で順次洗浄した。
最後に、洗浄した取物(99.39g)を減圧下、70℃で乾燥することで、THを89.89g得た(収率;94.6%、HPLC面百値(保持時間;4.1min);99.8%)。
1HNMR(DMSO-d6、δppm):8.8(s,2H)、7.4(m,4H )、7.0(m,4H)、6.3(s,2H)、5.8(s,2H).

次いで、得られた混合物へ、約5-10gの4-ニトロベンゾイルクロリドを加え、5分間攪拌した。この4-ニトロベンゾイルクロリドの添加および5分間の撹拌を、計9回繰り繰り返し、合計59.62gのニトロベンゾイルクロリドを加えた。
その後、50℃まで昇温して2時間攪拌し、得られた反応混合物に240gの水を加えて20-30℃に冷却し、更に16時間攪拌した。
撹拌後、ろ過によって析出物を回収し、水(750g)、メタノール(750g)で順次を洗浄し、未乾燥の粗物を得た(粗物1)。
以上の操作を、THの使用量を35gとして用いた以外は同一の条件で繰り返して行い、更に未乾燥の粗物(粗物2)を得、合計211.18gの未乾燥の粗物(粗物1+2)を得た。
この未乾燥の粗物(粗物1+粗物2)を減圧下、70℃で十分乾燥し、乾燥したTHDNB粗物を157.37g得た。
乾燥したTHDNB粗物60gとDMF(3L)とを混合することで得られた懸濁液を120℃で1時間30分間攪拌し、撹拌後20℃まで冷却した。そして、析出物をろ過によって回収し、メタノール(400g)で洗浄した後、洗浄したろ取物を減圧下、70℃にて乾燥し、THDNBを52.09g得た(収率;86.8%、HPLC面百値(保持時間;10.0min);99.2%)。
なお、上記得られた結果物であるTHDNBは、汎用の重溶媒に溶解しなかったため、NMRで同定できなかったが、後記の様に、当該結果物を還元することでTHDABが得られた事実から、当該結果物はTHDNBであることが確認された。

HPLCを用いて反応が終了したことを確認した後、ろ過によって反応混合物からPd-Cを取り除き、このPd-Cをジメチルホルムアミド(38g)で洗浄し、洗浄に用いたジメチルホルムアミドをろ液とともに回収した(ろ液1)。
以上の操作をもう一度行って、ろ液を回収し(ろ液2)、ろ液1とろ液2を合わせた。
次いで、このろ液(ろ液1+ろ液2)を水中(1623g)に滴下した後、析出物をろ過によって回収し、水(395g)で洗浄した。そして、得られたろ取物にメタノール(300g)を加え、26℃でスラリー洗浄を行った。
最後に、混合物をろ過し、ろ物を減圧下、70℃で乾燥することでTHDAB(15.46g)を得た(収率;86.2%、HPLC面百値(保持時間;4.7min);99.5%)。
1HNMR(DMSO-d6、δppm):8.0(m,4H)、7.4(m,4H )、7.0(m,4H)、6.9(s,2H)、6.7(m,4H)、6.3(s,4H)、5.6(s,2H).
反応容器内を窒素置換したオートクレーブ内に、合成例3で得たTHDNB(10g)、5%Pd-C(STDタイプ、wet品、エヌ・イー ケムキャット(株)製、1g)およびジメチルホルムアミド(70g)を入れて反応容器内を水素置換した後、水素圧常圧の条件下、30℃で92.5時間攪拌した。
HPLCを用いて反応が終了したことを確認した後、ろ過によって反応混合物からPd-Cを取り除き、このPd-Cをジメチルホルムアミド(18g)で洗浄し、洗浄に用いたジメチルホルムアミドをろ液とともに回収した。
次いで、このろ液を水中(679g)に滴下した後、析出物ろ過によって回収し、水(200g)で洗浄した。そして、次に得られたろ取物にメタノール(148g)を加え、23℃でスラリー洗浄を行った。
最後に、混合物をろ過し、ろ物を減圧下、70℃で乾燥することでTHDAB(8.57g)を得た(収率;95.5%、HPLC面百値(保持時間;4.7min);99.4%)。
1HNMR(DMSO-d6、δppm):8.0(m,4H)、7.4(m,4H )、7.0(m,4H)、6.9(s,2H)、6.7(m,4H)、6.3(s,4H)、5.6(s,2H).

次いでその溶液に21℃にて約3-5gの3-ニトロベンゾイルクロリドを添加し、21℃から27℃にて5分間撹拌した。この操作を計8回繰り返し、計29.8gの3-ニトロベンゾイルクロリドを添加した。23℃から27℃にて18時間撹拌後、25℃にて反応液に水(1000g)を添加し、更に25℃にて1時間撹拌した。
ろ過によって析出物を回収し、水(200g)でろ取物を洗浄し、m-THDNB粗物の未乾燥品を得た。ここで得られたm-THDNB粗物のすべてをメタノール(400g)に加え、25℃にて1時間撹拌後、ろ過し、ろ取物をメタノール(200g)で洗浄した。得られたろ取物(88.5g)を70℃にて減圧乾燥し、m-THDNBを43.1g得た(収率;99.3%、HPLC面百値(保持時間;10.0min);96.8%)。
なお、上記得られた結果物であるm-THDNBは、汎用の重溶媒に溶解しなかったため、NMRで同定できなかったが、後記の様に、当該結果物を還元することでm-THDABが得られた事実から、当該結果物はm-THDNBであることが確認された。

HPLCを用いて反応が完了したことを確認した後、ろ過によって反応混合物からPd-Cを取り除き、ろ液を得た。また使用したPd-CをN,N-ジメチルホルムアミド(81g)で洗浄し、洗浄に用いたジメチルホルムアミドを、先のろ液とともに回収した。回収したろ液に水(2300g)を25℃にて滴下した後、析出物をろ過によって回収し、ろ取物を水(500g)で洗浄した。そして得られたろ取物(66.2g)を70℃にて減圧乾燥することでm-THDAB粗物(35.2g)を得た。
このm-THDAB粗物(35.2g)を、脱気したテトラヒドロフラン(106g)に溶解し、79%ヒドラジン1水和物(30mg)を添加後、5℃に冷却した。この溶液に脱気した2-プロパノール(317g)を滴下し、1時間撹拌した。析出物をろ過し、ろ取物を脱気した2-プロパノール(70g)で2回洗浄し、得られたろ取物のすべてを70℃にて減圧乾燥することでm-THDAB再結晶物(30.8g)を得た。
このm-THDAB再結晶物(30.8g)を、脱気したテトラヒドラフラン(308g)に溶解し、79%ヒドラジン1水和物を30mg添加後、特製白鷺活性炭(3.08g)を加え、1時間撹拌後ろ過した。得られたろ液を70℃にて減圧乾燥することで、m-THDAB活性炭処理物(28.6g)を得た。
さらにこのm-THDAB活性炭処理物を、脱気したヘキサン(858g)に加え、還流条件下で1時間撹拌した。室温に冷却後、ろ過して得られたろ出物を脱気したヘキサン(143g)で3回洗浄した。得られたろ取物(27.5g)を70℃にて減圧乾燥することで、m-THDABの結晶を26.4g得た(収率;70.6%、HPLC面百値(保持時間;6.7min);99.7%)。
この結晶は、1HNMR分析結果から、m-THDABであることを確認した。
1HNMR(DMSO-d6、δppm):7.5(dd,2H)、7.4(m,6H )、7.3(dd,2H)、7.0(m,4H)、7.0(s,2H)、7.0(ddd,2H)、5.6(s,2H)、5.6(br,4H).
[実施例1]
窒素置換したフラスコ内に、2,2’-ジ(トリフルオロメチル)ベンジジン(TFMB)1.46g及びTHDAB 0.898gを入れた。そこへγ-ブチロラクトン 14.9gを加え、撹拌してTFMB及びTHDABが溶解したことを確認した後、更に2,3,5-トリカルボキシシクロペンチル酢酸-1,4:2,3-二無水物(TCA)0.728gを加えた。そして、得られた混合物を窒素雰囲気下、90℃で4時間撹拌し、反応混合物を50℃まで冷却した後、更に1,2,3,4-シクロブタンテトラカルボン酸二無水物(CBDA) 0.637gを加え、そのまま一晩撹拌した。
その後、固形物濃度が8質量%となるようにγ-ブチロラクトンを用いて反応混合物を希釈し、希釈した反応混合物に無水酢酸2.65gおよびピリジン1.542gを加えた後、窒素雰囲気下、100℃で4時間撹拌した。
次いで、得られた反応混合物を100gのメタノール中に滴下して30分間撹拌し、
ろ過によって析出物を回収した。この操作を3回繰り返した。
最後に、得られたろ物を減圧下、150℃で8時間乾燥し、ポリイミドを得た(3.248g 収率:87.2%)。
THDABの代わりに、本発明のジアミンと同様に分子内に9,10-ジヒドロ-9,10-[1,2]ベンゼノアントラセン骨格を有する以下の既知ジアミン(以下THDA)0.913gを用いた以外は、実施例1と同様の方法でポリイミドを得た(3.22g 86.2%)。
なお、THDAは、Journal of Polymer Science Part A: Polymer Chemistry, Vol. 49, 3109-3120 (2011)記載の方法に従って合成した。

[実施例2]
実施例1で得られたポリイミドを、濃度が12質量%となるようにγ-ブチロラクトンに溶解させ、ポリイミド溶液を得た。
実施例1で得られたポリイミドの代わりに、比較例1で得られたポリイミドを用いた以外は、実施例2と同様の方法でポリイミド溶液を得た。
[実施例3]
まず、実施例2で得られたポリイミド溶液を、5μmのフィルターを用いて加圧ろ過した。
その後、大気下で、ろ過したポリイミド溶液をガラス基板上に塗布し、50℃で30分間、140℃で30分間、200℃で60分間、順次加熱し、ポリイミドの膜を得た。そして、得られたポリイミドの膜に四角形の切込みを入れて膜を剥がし、評価試料とした。
実施例2で得られたポリイミド溶液の代わりに、比較例2で得られたポリイミド溶液を用いた以外は、実施例3と同様の手順・方法でポリイミドの膜を得た。そして、得られたポリイミドの膜に四角形の切込みを入れて膜を剥がし、評価試料とした。
[実施例4]
窒素注入/排出口を有しメカニカルスターラーと冷却器が取り付けられた100mLの三口フラスコ内に、TFMB 10.087g(31.5mmol)、THDAB 1.611g(3.5mmol)を仕込んだ。続いてγ-ブチロラクトン 45.9gを加え、撹拌を開始した。その後すぐにノルボルナン-2-スピロ-α-シクロペンタノン-α’-スピロ-2”-ノルボルナン-5,5”,6,6”-テトラカルボン酸二無水物(CpODA)6.726g(17.5mmol)を加え、さらにγ-ブチロラクトン 9.836gを加え、窒素雰囲気下にて90℃に加熱し、20分間撹拌した。その後、CBDA 3.431g(17.5mmol)と、1-エチルピペリジン 0.655gを加え、さらに、γ-ブチロラクトン 9.836gを加え、窒素雰囲気下にて6時間、180℃で撹拌した。次いで得られた反応混合物に対して、メタノールを用いた析出物の回収・精製を行い、得られたろ物を乾燥させ、ポリイミドを収率86.2%にて得た(Mn:49,646,Mw:119,613)。
[実施例5]
室温で、実施例4で得られたポリイミド 3gを、以下の[参考例]で調製したGBL-M:γ-ブチロラクトン分散シリカゾル(シリカ固形分濃度:25.25質量%)に加えて30分間混合した後、撹拌した混合物を一晩静置状態で放置することで、薄膜形成用組成物(固形分濃度:18.97質量%、ポリイミド:二酸化珪素粒子=3:7(質量比))を得た。
[参考例]シリカゾルの調製例
1000mLの丸底フラスコに、日産化学工業(株)製メタノール分散シリカゾル:MA-ST-M 350g(シリカ固形分濃度:40.4質量%)とγ-ブチルラクトン419gを入れた。そして、そのフラスコを真空エバポレーターと繋いでフラスコ内を減圧にし、約35℃の温水浴に20~50分間浸すことで、溶媒がメタノールからγ-ブチルラクトンに置換されたシリカゾル(GBL-M)約560.3gを得た(シリカ固形分濃度:25.25質量%)。
なお、上記シリカゾルにおいて、窒素吸着法により測定された比表面積値から算出される平均粒子径は22nmであった。なお具体的には、シリカゾルの乾燥粉末の比表面積をユアサアイオニクス社製、比表面積測定装置モノソーブMS-16を用いて測定し、測定された比表面積S(m2/g)を用いてD(nm)=2720/Sの式で平均一次粒子径を算出した。
[実施例6]
実施例5で得られた薄膜形成用組成物をガラス基板に塗布し、塗膜を-97kPaの真空下で、50℃で30分間、140℃で30分間、200℃で60分間、順次加熱して薄膜を得た。なお、加熱には、予め所望の温度に設定をした3つのオーブンを使用した。
得られた薄膜を機械的切断にて剥がし、その後の評価に供した。
上述の手順にて作製した各薄膜(評価試料)の耐熱性及び光学特性、すなわち、50℃乃至200℃における線膨張係数(CTE)、5%重量減少温度(Td5%)、光線透過率(T400nm、T550nm)及びCIE b*値(黄色評価)、リタデーション(Rth、R0)並びに複屈折(Δn)に関して、下記手順に従いそれぞれ評価した。結果を表1に示す。
1)線膨張係数(CTE)
TAインスツルメンツ社製 TMA Q400を用いて、薄膜を幅5mm、長さ16mmのサイズにカットし、まず10℃/minで昇温して50乃至300℃まで加熱(第一加熱)し、次いで10℃/minで降温して50℃まで冷却した後に、10℃/minで昇温して50乃至420℃まで加熱(第二加熱)した際の、第二加熱の50℃乃至200℃における線膨張係数(CTE[ppm/℃])の値を測定することで求めた。なお、第一加熱、冷却および第二加熱を通じて、荷重0.05Nを加えた。
2)5%重量減少温度(Td5%)
5%重量減少温度(Td5%[℃])は、TAインスツルメンツ社製 TGA Q500を用い、窒素中、薄膜約5乃至10mgを50乃至800℃まで10℃/minで昇温して測定することで求めた。
3)光線透過率(透明性)(T400nm、T550nm)及びCIE b値(CIE b*)
波長400nm及び550nmの光線透過率(T400nm、T550nm[%])及びCIE b値(CIE b*)は、日本電色工業(株)製 SA4000スペクトロメーターを用いて、室温にて、リファレンスを空気として、測定を行った。
4)リタデーション(Rth、R0)
厚さ方向リタデーション(Rth)及び面内リタデーション(R0)を、王子計測機器(株)製、KOBURA 2100ADHを用いて、室温にて測定した。
なお、厚さ方向リタデーション(Rth)及び面内リタデーション(R0)は以下の式にて算出される。
R0=(Nx-Ny)×d=ΔNxy×d
Rth=[(Nx+Ny)/2-Nz]×d=[(ΔNxz×d)+(ΔNyz×d)]/2
Nx、Ny:面内の直交する2つの屈折率(Nx>Ny、Nxを遅相軸、Nyを進相軸とも称する)
Nz:面に対して厚さ(垂直)方向(垂直)の屈折率
d:膜厚
ΔNxy:面内の2つの屈折率の差(Nx-Ny)(複屈折)
ΔNxz:面内の屈折率Nxと厚さ方向の屈折率Nzの差(複屈折)
ΔNyz:面内の屈折率Nyと厚さ方向の屈折率Nzの差(複屈折)
5)膜厚(d)
得られた薄膜の膜厚は、(株)テクロック製 シックネスゲージにて測定した。
6)複屈折(Δn)
前述の<4)リタデーション>により得られた厚さ方向リタデーション(Rth)の値を用い、以下の式にて算出した。
ΔN=[Rth/d(フィルム膜厚)]/1000

さらに、本発明のジアミンを用いて製造したポリイミドと、二酸化ケイ素粒子とを含む薄膜形成用組成物を用いて製造した薄膜(実施例6)は、二酸化ケイ素粒子を含むにも関わらず光線透過率が高く、50℃乃至200℃における線膨張係数がおよそ15ppm/℃であり、実施例3の膜と比べてさらに低い線膨張係数を示し、すなわち加熱時の寸法安定に優れ、また5%重量減少温度で評価される耐熱性も改善されるという結果となった。特に該薄膜は、厚さ方向の断面からみたときの2つの複屈折(面内の2つの屈折率と厚さ方向の屈折率との夫々の差)にそれぞれ膜厚を掛けて得られる2つの位相差の平均値として表される厚さ方向リタデーションRthが150nm未満と極めて低く、複屈折Δnも0.004という極めて低い値となった。
このように、本発明のジアミンを用いて製造した薄膜等は、低線膨張係数、高い透明性(高い光線透過率、低い黄色度)、低いリタデーションという特性を有し、すなわちフレキシブルディスプレイ基板のベースフィルムとして必要な要件を満たすものであり、フレキシブルディスプレイ基板のベースフィルムとして特に好適に用いることができることが期待できる。
[実施例7]

[実施例8]

[実施例9]

[実施例10]

[実施例11]

[実施例12]

[実施例13]
室温で、実施例7で得られたポリイミドI 1gをポリイミド濃度12質量%となる様にGBL溶媒に溶解し、この溶液を5μmフィルタに通してゆっくり加圧濾過して薄膜形成用組成物を得た。次いで、得られた薄膜形成用組成物をガラス基板に塗布し、大気下にて、50℃で30分間、140℃で30分間及び200℃で60分間加熱し透明な膜PI-Iを得た。得られた薄膜を機械的切断にてガラス基板から剥がした。光学的及び熱的性質を表2に示す。
[実施例14]
室温で、実施例8で得られたポリイミドII 1gをポリイミド濃度12質量%となる様にGBL溶媒に溶解し、この溶液を5μmフィルタに通してゆっくり加圧濾過して薄膜形成用組成物を得た。次いで、得られた薄膜形成用組成物をガラス基板に塗布し、大気下にて、50℃で30分間、140℃で30分間及び200℃で60分間加熱し透明な膜PI-IIを得た。得られた薄膜を機械的切断にてガラス基板から剥がした。光学的及び熱的性質を表2に示す。
[実施例15]
室温で、実施例9で得られたポリイミドIII 1gをポリイミド濃度12質量%となる様にGBL溶媒に溶解し、この溶液を5μmフィルタに通してゆっくり加圧濾過して薄膜形成用組成物を得た。次いで、得られた薄膜形成用組成物をガラス基板に塗布し、大気下にて、50℃で30分間、140℃で30分間及び200℃で60分間加熱し透明な膜PI-IIIを得た。得られた薄膜を機械的切断にてガラス基板から剥がした。光学的及び熱的性質を表2に示す。
[実施例16]
室温で、実施例9で得られたポリイミドIII 3gをポリイミド濃度12質量%となる様にGBL溶媒に溶解し、この溶液を5μmフィルタに通してゆっくり加圧濾過し、得られた溶液を実施例5に記載されたGBL-M:γ-ブチルラクトン分散シリカゾル(γ-ブチルラクトン中に分散された18乃至23nmのサイズのSiO2 25.25%) 3.326gに加え、30分間混合した後、静置状態で放置することで薄膜形成用組成物を得た。この薄膜形成用組成物をガラス基板に塗布し、-97kPa減圧下にて、50℃で30分間、140℃で30分間及び200℃で60分間加熱し透明な膜PI-III-Aを得た。得られた薄膜を機械的切断にてガラス基板から剥がした。光学的及び熱的性質を表2に示す。
[実施例17]
室温で、実施例4で得られたポリイミドIV 1gをポリイミド濃度12質量%となる様にGBL溶媒に溶解し、この溶液を5μmフィルタに通してゆっくり加圧濾過して薄膜形成用組成物を得た。次いで、得られた薄膜形成用組成物をガラス基板に塗布し、大気下にて、50℃で30分間、140℃で30分間及び200℃で60分間加熱し、そして-100kPa減圧下にて、280℃で60分間加熱し、透明な膜PI-IVを得た。得られた薄膜を機械的切断にてガラス基板から剥がした。光学的及び熱的性質を表3に示す。
[実施例18]
室温で、実施例11で得られたポリイミドV 1gをポリイミド濃度12質量%となる様にGBL溶媒に溶解し、この溶液を5μmフィルタに通してゆっくり加圧濾過して薄膜形成用組成物を得た。次いで、得られた薄膜形成用組成物をガラス基板に塗布し、大気下にて、50℃で30分間、140℃で30分間及び200℃で60分間加熱し、そして-100kPa減圧下にて、280℃で60分間加熱し、透明な膜PI-Vを得た。得られた薄膜を機械的切断にてガラス基板から剥がした。光学的及び熱的性質を表3に示す。
[実施例19]
室温で、実施例6で得られたポリイミドV 1gをポリイミド濃度12質量%となる様にGBL溶媒に溶解し、この溶液を5μmフィルタに通してゆっくり加圧濾過して薄膜形成用組成物を得た。次いで、得られた薄膜形成用組成物をガラス基板に塗布し、大気下にて、50℃で30分間、140℃で30分間及び200℃で60分間加熱し、そして-100kPa減圧下にて、280℃で60分間加熱し、透明な膜PI-VIを得た。得られた薄膜を機械的切断にてガラス基板から剥がした。光学的及び熱的性質を表2に示す。
実施例13乃至実施例19で作製した各ポリイミド膜の光学的及び熱的性質を以下の機器を用いて測定した。
ポリイミド膜の光透過率(T400nm、T550nm)及び黄色度(CIE b*)は、室温で日本電色SA4000分光計を使用して計測した。
厚さ芳香リタデーション(Rth)及び内面リタデーション(Ro)を、王子計測機器(株)製KOBURA 2100 ADHを用いて、室温にて測定した。
線膨張係数(CTE)は、ティー・エイ・インスツルメント社のTMA Q400を用いて、窒素気流下で、10℃/分の加熱速度で行い、50℃から200℃の温度範囲で測定した。
熱分解温度(Td点)は窒素気流下で、10℃/分の加熱速度で、ティー・エイ・インスツルメント社のTGA Q500を使用して実施した。150℃で5%の重量損失を規定した。
数平均分子量(Mn)及び重量平均分子量(Mw)は、昭和電工(株)製のShowdex GPC-101を用いて決定された。高分子濾過のためのPTFEの0.45μmのフィルターを使用し、検量線は、標準ポリスチレンを用いた。を用いた
膜形成は、コーテスト インスツルメント フリー オートマティック フィルム アプリケ-タ- PFA-2010-1を使用して実施し、フィルムベーキングは、Deng YNG社の円形オーブンDO45を使用して実施した。
膜厚は(株)テクロック製、シックネスゲージにて測定した。


さらに、本発明のジアミンを用いて製造したポリイミドと、二酸化ケイ素粒子とを含む薄膜形成用組成物を用いて製造した薄膜(実施例16)は、二酸化ケイ素粒子を含むにも関わらず光線透過率が高く、50℃乃至200℃における線膨張係数がおよそ16ppm/℃という非常に低い数値を示した。すなわち加熱時の寸法安定に優れ、また5%重量減少温度で評価される耐熱性も改善されるという結果となった。特に該薄膜は、厚さ方向の断面からみたときの2つの複屈折(面内の2つの屈折率と厚さ方向の屈折率との夫々の差)にそれぞれ膜厚を掛けて得られる2つの位相差の平均値として表される厚さ方向リタデーションRthが150nm未満と極めて低く、複屈折Δnも0.004という極めて低い値となった。
このように、本発明のジアミンを用いて製造した薄膜等は、低線膨張係数、高い透明性(高い光線透過率、低い黄色度)、低いリタデーションという特性を有し、すなわちフレキシブルディスプレイ基板のベースフィルムとして必要な要件を満たすものであり、フレキシブルディスプレイ基板のベースフィルムとして特に好適に用いることができることが期待できる。
Claims (18)
- 式(1-1)で表されることを特徴とするジアミン。

R6及びR7は、それぞれ独立して、水素原子、ハロゲン原子、炭素原子数1乃至5のアルキル基又は炭素原子数1乃至5のアルコキシ基を表し、
a、b、d及びeは、それぞれ独立して、0~4の整数を表し、そして
cは0~2の整数を表す。) - 式(1-2)で表されるジアミンである、請求項1に記載のジアミン。

- 式(1-3)又は式(1-4)で表されるジアミンである、請求項2に記載のジアミン。

- 請求項1乃至請求項3のうちいずれか一項に記載のジアミンを含むジアミン成分と、酸二無水物成分との反応物であるポリアミック酸。
- 前記ジアミン成分が、式(A1)で表されるジアミンをさらに含む、請求項4に記載のポリアミック酸。





- 前記酸二無水物成分が、式(C1)で表される酸二無水物を含む、請求項4又は請求項5に記載のポリアミック酸。

- 請求項4乃至請求項6のうちいずれか一項に記載のポリアミック酸をイミド化して得られるポリイミド。
- 請求項7に記載のポリイミドと、有機溶媒と、窒素吸着法により測定された比表面積値から算出される平均粒子径が100nm以下である二酸化ケイ素粒子を含む、薄膜形成用組成物。
- 前記ポリイミドと前記二酸化ケイ素粒子の質量比が、1:10~10:1である、請求項8に記載の薄膜形成用組成物。
- 前記平均粒子径が、60nm以下である、請求項8又は請求項9に記載の薄膜形成用組成物。
- 請求項8乃至請求項10のうちいずれか一項に記載の薄膜形成用組成物から形成される薄膜。
- 請求項11に記載の薄膜からなるフレキシブルデバイス用基板。
- 請求項7に記載のポリイミドと、有機溶媒とを含む膜形成用組成物。
- 請求項13に記載の膜形成用組成物から形成される膜からなるフレキシブルデバイス用基板。
- 式(2-1)で表されることを特徴とするジニトロ化合物。

R6及びR7は、それぞれ独立して、水素原子、ハロゲン原子、炭素原子数1乃至5のアルキル基又は炭素原子数1乃至5のアルコキシ基を表し、
a、b、d及びeは、それぞれ独立して、0~4の整数を表し、そして
cは0~2の整数を表す。) - 式(2-2)で表されるジニトロ化合物である、請求項15に記載のジニトロ化合物。

- 式(2-3)又は式(2-4)で表されるジニトロ化合物である、請求項16に記載のジニトロ化合物。

- 式(1-1)で表されるジアミンを製造する方法であって、

R6及びR7は、それぞれ独立して、水素原子、ハロゲン原子、炭素原子数1乃至5のアルキル基又は炭素原子数1乃至5のアルコキシ基を表し、
a、b、d及びeは、それぞれ独立して、0~4の整数を表し、そして
cは0~2の整数を表す。)
式(2-1)で表されるジニトロ化合物のニトロ基を還元して式(1-1)で表されるジアミンを得る段階を含む、製造方法。

Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017507647A JP6748378B2 (ja) | 2015-03-25 | 2016-03-25 | ジアミンおよびその利用 |
| CN201680029316.0A CN107614483B (zh) | 2015-03-25 | 2016-03-25 | 二胺及其利用 |
| KR1020177030061A KR102557784B1 (ko) | 2015-03-25 | 2016-03-25 | 디아민 및 그의 이용 |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-062790 | 2015-03-25 | ||
| JP2015062790 | 2015-03-25 | ||
| JP2015185776 | 2015-09-18 | ||
| JP2015-185776 | 2015-09-18 | ||
| JP2016-012560 | 2016-01-26 | ||
| JP2016012560 | 2016-01-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016153064A1 true WO2016153064A1 (ja) | 2016-09-29 |
Family
ID=56977556
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/059755 WO2016153064A1 (ja) | 2015-03-25 | 2016-03-25 | ジアミンおよびその利用 |
Country Status (5)
| Country | Link |
|---|---|
| JP (1) | JP6748378B2 (ja) |
| KR (1) | KR102557784B1 (ja) |
| CN (1) | CN107614483B (ja) |
| TW (1) | TWI684580B (ja) |
| WO (1) | WO2016153064A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017122730A1 (ja) * | 2016-01-14 | 2017-07-20 | 日産化学工業株式会社 | ジアミンおよびその利用 |
| WO2018062422A1 (ja) * | 2016-09-28 | 2018-04-05 | 日産化学工業株式会社 | ジアミンおよびその利用 |
| JPWO2017098936A1 (ja) * | 2015-12-09 | 2018-09-27 | 株式会社カネカ | ポリアミド酸、ポリイミド、ポリアミド酸溶液、ポリイミド積層体、フレキシブルデバイス基板、及びそれらの製造方法 |
| WO2019131896A1 (ja) * | 2017-12-28 | 2019-07-04 | 宇部興産株式会社 | ポリイミド、ポリイミド溶液組成物、ポリイミドフィルム、及び基板 |
| WO2019246233A1 (en) * | 2018-06-20 | 2019-12-26 | Dupont Electronics, Inc. | Polymers for use in electronic devices |
| CN113490543A (zh) * | 2019-02-28 | 2021-10-08 | 富士胶片株式会社 | 聚合物及其制造方法、使用该聚合物的气体分离膜、气体分离模块、及气体分离装置、以及间苯二胺化合物 |
| US11806661B2 (en) | 2019-02-28 | 2023-11-07 | Fujifilm Corporation | Polymer and method for producing the same, gas separation membrane, gas separation module, and gas separation apparatus using the polymer, and m-phenylenediamine compound |
| US11964966B2 (en) | 2018-08-08 | 2024-04-23 | Dupont Electronics, Inc. | Polymers for use in electronic devices |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020018617A1 (en) * | 2018-07-20 | 2020-01-23 | Dupont Electronics, Inc. | Polymers for use in electronic devices |
| CN111960960B (zh) * | 2019-05-20 | 2022-12-30 | 广东生益科技股份有限公司 | 二胺化合物、其制备方法、热固性树脂组合物及其应用 |
| CN113387817A (zh) * | 2021-07-27 | 2021-09-14 | 中国科学院长春应用化学研究所 | 一种含氟芳香二胺化合物及其制备方法和无色透明聚酰亚胺薄膜 |
| CN115869788B (zh) * | 2021-09-27 | 2023-07-14 | 中国石油化工股份有限公司 | 具有三蝶烯基结构的聚酰亚胺无规共聚物及其制备方法和应用 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004359599A (ja) * | 2003-06-04 | 2004-12-24 | Hitachi Chem Co Ltd | トリプチセンジアミンの製造方法 |
| JP2006001968A (ja) * | 2004-06-15 | 2006-01-05 | Hitachi Chem Co Ltd | トリプチセン骨格を有するポリアミド酸、ポリイミド樹脂及びこれを用いた光学部品 |
| JP2008308433A (ja) * | 2007-06-14 | 2008-12-25 | Idemitsu Kosan Co Ltd | トリプチセン構造を有する化合物、フォトレジスト基材及びフォトレジスト組成物 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS58208322A (ja) | 1982-05-31 | 1983-12-05 | Japan Synthetic Rubber Co Ltd | ポリイミド化合物の製造方法 |
| JPS60188427A (ja) | 1984-03-09 | 1985-09-25 | Nissan Chem Ind Ltd | 新規なポリイミド樹脂及びその製造方法 |
| TWI390007B (zh) * | 2007-08-03 | 2013-03-21 | Chien Hong Cheng | 三蝶烯衍生物及其在有機電子元件的應用 |
| US20120259036A1 (en) | 2009-12-22 | 2012-10-11 | Akebono Brake Industry Co., Ltd. | Friction material and method for producing friction material |
-
2016
- 2016-03-25 KR KR1020177030061A patent/KR102557784B1/ko active IP Right Grant
- 2016-03-25 CN CN201680029316.0A patent/CN107614483B/zh active Active
- 2016-03-25 JP JP2017507647A patent/JP6748378B2/ja active Active
- 2016-03-25 TW TW105109816A patent/TWI684580B/zh active
- 2016-03-25 WO PCT/JP2016/059755 patent/WO2016153064A1/ja active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004359599A (ja) * | 2003-06-04 | 2004-12-24 | Hitachi Chem Co Ltd | トリプチセンジアミンの製造方法 |
| JP2006001968A (ja) * | 2004-06-15 | 2006-01-05 | Hitachi Chem Co Ltd | トリプチセン骨格を有するポリアミド酸、ポリイミド樹脂及びこれを用いた光学部品 |
| JP2008308433A (ja) * | 2007-06-14 | 2008-12-25 | Idemitsu Kosan Co Ltd | トリプチセン構造を有する化合物、フォトレジスト基材及びフォトレジスト組成物 |
Non-Patent Citations (5)
| Title |
|---|
| GUNG, BENJAMIN W. ET AL.: "A Threshold for Charge Transfer in Aromatic Interactions? A Quantitative Study of pi-Stacking Interactions", J. ORG. CHEM., vol. 70, no. 25, December 2005 (2005-12-01), pages 10532 - 10537, XP055320295 * |
| GUNG, BENJAMIN W. ET AL.: "The Strength of Parallel-Displaced Arene-Arene Interactions in Chloroform", J. ORG. CHEM., vol. 70, no. 9, April 2005 (2005-04-01), pages 3641 - 3644, XP055320293 * |
| RAFIEE, ZAHRA ET AL.: "Synthesis and Study of New Poly(ester-imide)s Containing Triptycene Groups", POLYMER ENGINEERING AND SCIENCE, vol. 54, no. 10, October 2014 (2014-10-01), pages 2252 - 2257, XP055320291 * |
| RAFIEE, ZHARA ET AL.: "Preparation and characterization of heat-resistant polyimide/ titanium dioxide nanocomposite films containing triptycene side units by sol-gel processes", HIGH PERFORMANCE POLYMERS, vol. 26, 2014, pages 373 - 380 * |
| YANG, JYE-SHANE ET AL.: "Solid-State Molecular Folding and Supramolecular Structures of Triptycene-Derived Secondary Dicarboxamides", J. ORG. CHEM., vol. 67, no. 21, October 2002 (2002-10-01), pages 7343 - 7354, XP055320289 * |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2017098936A1 (ja) * | 2015-12-09 | 2018-09-27 | 株式会社カネカ | ポリアミド酸、ポリイミド、ポリアミド酸溶液、ポリイミド積層体、フレキシブルデバイス基板、及びそれらの製造方法 |
| JP7122437B2 (ja) | 2015-12-09 | 2022-08-19 | 株式会社カネカ | ポリイミド、ポリイミド積層体及びフレキシブルデバイス基板 |
| JP2021152173A (ja) * | 2015-12-09 | 2021-09-30 | 株式会社カネカ | ポリイミド、ポリイミド積層体及びフレキシブルデバイス基板 |
| WO2017122730A1 (ja) * | 2016-01-14 | 2017-07-20 | 日産化学工業株式会社 | ジアミンおよびその利用 |
| JPWO2017122730A1 (ja) * | 2016-01-14 | 2018-11-08 | 日産化学株式会社 | ジアミンおよびその利用 |
| WO2018062422A1 (ja) * | 2016-09-28 | 2018-04-05 | 日産化学工業株式会社 | ジアミンおよびその利用 |
| CN111770949A (zh) * | 2017-12-28 | 2020-10-13 | 宇部兴产株式会社 | 聚酰亚胺、聚酰亚胺溶液组合物、聚酰亚胺膜和基板 |
| JPWO2019131896A1 (ja) * | 2017-12-28 | 2021-01-14 | 宇部興産株式会社 | ポリイミド、ポリイミド溶液組成物、ポリイミドフィルム、及び基板 |
| JP7069478B2 (ja) | 2017-12-28 | 2022-05-18 | Ube株式会社 | ポリイミド、ポリイミド溶液組成物、ポリイミドフィルム、及び基板 |
| WO2019131896A1 (ja) * | 2017-12-28 | 2019-07-04 | 宇部興産株式会社 | ポリイミド、ポリイミド溶液組成物、ポリイミドフィルム、及び基板 |
| CN111770949B (zh) * | 2017-12-28 | 2024-01-16 | Ube株式会社 | 聚酰亚胺、聚酰亚胺溶液组合物、聚酰亚胺膜和基板 |
| WO2019246233A1 (en) * | 2018-06-20 | 2019-12-26 | Dupont Electronics, Inc. | Polymers for use in electronic devices |
| US11964966B2 (en) | 2018-08-08 | 2024-04-23 | Dupont Electronics, Inc. | Polymers for use in electronic devices |
| CN113490543A (zh) * | 2019-02-28 | 2021-10-08 | 富士胶片株式会社 | 聚合物及其制造方法、使用该聚合物的气体分离膜、气体分离模块、及气体分离装置、以及间苯二胺化合物 |
| US11806661B2 (en) | 2019-02-28 | 2023-11-07 | Fujifilm Corporation | Polymer and method for producing the same, gas separation membrane, gas separation module, and gas separation apparatus using the polymer, and m-phenylenediamine compound |
Also Published As
| Publication number | Publication date |
|---|---|
| CN107614483A (zh) | 2018-01-19 |
| CN107614483B (zh) | 2020-07-31 |
| KR20170131514A (ko) | 2017-11-29 |
| KR102557784B1 (ko) | 2023-07-20 |
| TWI684580B (zh) | 2020-02-11 |
| JPWO2016153064A1 (ja) | 2018-01-25 |
| TW201710234A (zh) | 2017-03-16 |
| JP6748378B2 (ja) | 2020-09-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6748378B2 (ja) | ジアミンおよびその利用 | |
| JP6631804B2 (ja) | 樹脂薄膜の製造方法および樹脂薄膜形成用組成物 | |
| JP6990354B2 (ja) | 樹脂薄膜形成用組成物 | |
| JP7047852B2 (ja) | ポリイミド前駆体、ポリイミド、ポリイミドフィルム、ワニス、及び基板 | |
| WO2014077253A1 (ja) | ポリイミド樹脂フィルム及びポリイミド樹脂フィルムからなる電子デバイス用基板 | |
| JP6806089B2 (ja) | ジアミンおよびその利用 | |
| JP7036012B2 (ja) | ポリイミド樹脂フィルム及びポリイミド樹脂フィルムの製造方法 | |
| JP7037122B2 (ja) | ジアミンおよびその利用 | |
| JP6966725B2 (ja) | 酸二無水物およびその利用 | |
| JP7054064B2 (ja) | フレキシブルデバイス基板形成用組成物 | |
| KR20210123333A (ko) | 폴리이미드계 수지 분체의 제조 방법 | |
| WO2008007629A1 (fr) | Acide polyamique et polyimide | |
| JP6966726B2 (ja) | 酸二無水物およびその利用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16768967 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017507647 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20177030061 Country of ref document: KR Kind code of ref document: A |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16768967 Country of ref document: EP Kind code of ref document: A1 |