WO2015174214A1 - α-ブロモアセトフェノン化合物の製造方法 - Google Patents
α-ブロモアセトフェノン化合物の製造方法 Download PDFInfo
- Publication number
- WO2015174214A1 WO2015174214A1 PCT/JP2015/061873 JP2015061873W WO2015174214A1 WO 2015174214 A1 WO2015174214 A1 WO 2015174214A1 JP 2015061873 W JP2015061873 W JP 2015061873W WO 2015174214 A1 WO2015174214 A1 WO 2015174214A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- ether
- acid ester
- organic acid
- hydroxyl group
- Prior art date
Links
- KIPLQQRDGUKNJI-UHFFFAOYSA-N CC(C)(C(c(c(C)c1)ccc1OCCOCCO)=O)Br Chemical compound CC(C)(C(c(c(C)c1)ccc1OCCOCCO)=O)Br KIPLQQRDGUKNJI-UHFFFAOYSA-N 0.000 description 1
- FIBKKXYBZPPQJU-UHFFFAOYSA-N CC(C)(C(c(c(OC)c1)ccc1OCCOCCO)=O)Br Chemical compound CC(C)(C(c(c(OC)c1)ccc1OCCOCCO)=O)Br FIBKKXYBZPPQJU-UHFFFAOYSA-N 0.000 description 1
- FBWPUBXQIMAFAZ-UHFFFAOYSA-N CC(C)(C(c(cc1)cc(Br)c1OCCOCCOCCOC(C)=O)=O)Br Chemical compound CC(C)(C(c(cc1)cc(Br)c1OCCOCCOCCOC(C)=O)=O)Br FBWPUBXQIMAFAZ-UHFFFAOYSA-N 0.000 description 1
- KOJAWQIKRZGDAX-UHFFFAOYSA-N CC(C)(C(c(cc1)ccc1OCCOCCOCCOC(C)=O)=O)Br Chemical compound CC(C)(C(c(cc1)ccc1OCCOCCOCCOC(C)=O)=O)Br KOJAWQIKRZGDAX-UHFFFAOYSA-N 0.000 description 1
- ZLNJUVAKEQOXHG-UHFFFAOYSA-N CC(C)C(c(cc1)ccc1OCCOCCOCCOC(C)=O)=O Chemical compound CC(C)C(c(cc1)ccc1OCCOCCOCCOC(C)=O)=O ZLNJUVAKEQOXHG-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/28—Preparation of carboxylic acid esters by modifying the hydroxylic moiety of the ester, such modification not being an introduction of an ester group
- C07C67/287—Preparation of carboxylic acid esters by modifying the hydroxylic moiety of the ester, such modification not being an introduction of an ester group by introduction of halogen; by substitution of halogen atoms by other halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/63—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by introduction of halogen; by substitution of halogen atoms by other halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Definitions
- the present invention relates to a method for producing an ⁇ -bromoacetophenone compound.
- an ⁇ -hydroxyacetophenone photopolymerization initiator As an initiator for a polymerizable compound having an ethylenically unsaturated bond, an ⁇ -hydroxyacetophenone photopolymerization initiator is known.
- ⁇ -Hydroxyacetophenone-based photopolymerization initiators are generally used after acylating a phenyl compound in the presence of a Lewis acid using a carboxylic acid halide such as isobutyric acid chloride and brominating the ⁇ carbon of this acyl group.
- the bromine atom can be substituted with a hydroxyl group.
- Patent Document 1 describes that the ⁇ -carbon of the isobutyryl group of 4- (2-acetoxyethoxy) -phenyl-2-propylketone or 4-hydroxyphenyl-2-propylketone is brominated.
- Patent Document 2 describes bromination of the ⁇ -carbon of the isobutyryl group of 4-methoxyisobutyrophenone.
- photopolymerization initiators there are various uses for photopolymerization initiators. In addition to the use as a curing agent component for paints, adhesives, optical films, solder resist materials, etc., applications that require solubility in solvents containing water are also expanding. For example, when a photopolymerization initiator is used as a curable ink component used in an inkjet printer, it is required to increase the polarity and impart hydrophilicity.
- the photopolymerization is carried out by introducing a polar group having a specific chain length such as an ethylene oxide chain as a substituent onto the benzene ring of the phenyl compound used as a synthesis raw material.
- the hydrophilicity of the initiator can be increased.
- the ⁇ -halogenoacetophenone compound, which is a synthetic intermediate to be obtained, or the ⁇ -hydroxyacetophenone compound, which is a target photopolymerization initiator becomes liquid with a lower melting point. In that case, purification by the recrystallization method cannot be performed.
- the present invention provides a method for producing an ⁇ -bromoacetophenone compound which is useful as a synthesis intermediate of a photopolymerization initiator having a specific polar group and which is liquid at 5 to 30 ° C. with excellent reaction purity. Is an issue.
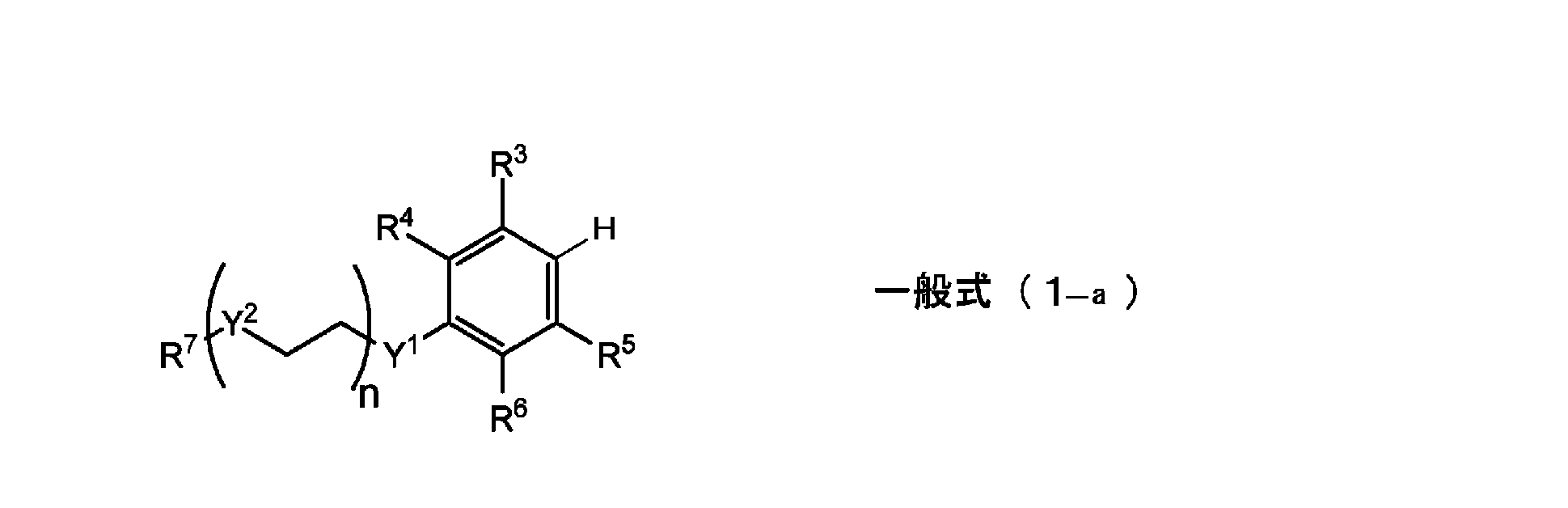
- R 1 and R 2 each independently represents an alkyl group.
- R 3 to R 6 each independently represent a hydrogen atom or a substituent. However, at least one of R 3 to R 6 is a hydrogen atom.
- Y 1 and Y 2 each independently represent an oxygen atom or a sulfur atom, and n is 2 to 3.
- R 7 represents a hydrogen atom, an alkyl group, an acyl group, or a trialkylsilyl group.
- R 1 ⁇ R 7, Y 1, Y 2 and n are respectively the same as R 1 ⁇ R 7, Y 1 , Y 2 and n in the general formula (1).
- the solvent selected from an organic acid ester compound and an ether compound having no hydroxyl group is one or more selected from methyl acetate, ethyl acetate, butyl acetate, propyl acetate, isopropyl acetate, and 1,4-dioxane. [1] or [2].
- Bromination is a mixed liquid containing bromine or a solvent selected from bromine and an organic acid ester compound and an ether compound having no hydroxyl group, and selected from a phenyl compound, an organic acid ester compound and an ether compound having no hydroxyl group.
- a liquid ⁇ -bromoacetophenone compound can be obtained at a temperature of 5 ° C. to 30 ° C. with a high reaction purity, and usually does not require a purification operation such as recrystallization.
- the production method of the present invention (hereinafter simply referred to as “the production method of the present invention”) will be described in detail below.
- the production method of the present invention uses a phenyl compound represented by the following general formula (1) as a starting material.
- R 1 and R 2 each independently represents an alkyl group. This alkyl group may be linear or branched.
- R 1 and R 2 are preferably an alkyl group having 1 to 10 carbon atoms, more preferably 1 to 5 carbon atoms, still more preferably 1 to 3 carbon atoms, still more preferably methyl or ethyl, and most preferably methyl. It is.
- R 1 and R 2 may be connected to each other to form a ring.
- the ring structure group formed by linking R 1 and R 2 is preferably a cycloalkyl group.
- the cycloalkyl group is more preferably a cycloalkyl group having 3 to 10 carbon atoms, more preferably 4 to 8 carbon atoms, and more specifically preferably cyclopropyl, cyclobutyl, cycloheptyl or cyclohexyl. .
- R 3 to R 6 each independently represents a hydrogen atom or a substituent. However, at least one of R 3 to R 6 is a hydrogen atom. In addition, two or more of R 3 to R 6 (preferably two or more including R 4 and R 6 ) are preferably hydrogen atoms, and preferably three or more (preferably three including R 4 and R 6 ). The above is more preferably a hydrogen atom, and more preferably all of R 3 to R 6 are hydrogen atoms.
- an alkyl group preferably an alkyl group having 1 to 5, more preferably 1 to 3, more preferably 1 or 2 carbon atoms
- halogen as the substituent Atom (specifically, fluorine atom, chlorine atom, bromine atom or iodine atom), amino group, hydroxy group, cyano group, nitro group, carboxy group, sulfo group, sulfonyl group, phosphonyl group, boric acid group, alkoxy group, Amide group etc. are mentioned. Of these, a methyl, ethyl, or halogen atom is preferred.
- R 7 represents a hydrogen atom, an alkyl group, an acyl group, or a trialkylsilyl group.
- R 7 is more preferably an acyl group.
- R 7 When R 7 is an alkyl group, it may be linear or branched and may have a substituent.
- the alkyl group preferably has 1 to 10 carbon atoms, more preferably 1 to 6 carbon atoms, and still more preferably 1 to 5 carbon atoms.
- substituents of the alkyl group may be linked to form a ring structure. Examples of the alkyl group having such a ring structure include 2-tetrahydropyranyl.
- R 7 when R 7 is an alkyl group, it is also preferably an alkoxymethyl group (preferably an alkoxymethyl group having 1 to 10 carbon atoms, more preferably 1 to 5 carbon atoms).
- R 7 is an alkyl group
- R 7 is an alkyl group
- R 7 is an alkyl group
- R 7 is an alkyl group
- examples when R 7 is an alkyl group include, for example, methyl, ethyl, propyl, isopropyl, t-butyl, methoxymethyl, ethoxymethyl, benzyl, p-methoxybenzyl, phenethyl and 2-tetrahydropyranyl. More preferably, methyl, methoxymethyl, benzyl or 2-tetrahydropyranyl.
- R 7 is an acyl group
- the carbon number thereof is preferably 2 to 12, more preferably 2 to 10, and further preferably 2 to 6.
- R 7 is more preferably acetyl, pivaloyl, acryloyl or benzoyl, and even more preferably acetyl.
- R 7 is a trialkylsilyl group
- the alkyl group of the trialkylsilyl group may be linear or branched.
- the alkyl group preferably has 1 to 10 carbon atoms, more preferably 1 to 6 carbon atoms, and still more preferably 1 to 4 carbon atoms. More specifically, examples include trimethylsilyl, triethylsilyl, t-butyldimethylsilyl, and triisopropylsilyl, and trimethylsilyl is preferable.
- Y 1 and Y 2 each independently represent an oxygen atom or a sulfur atom, and more preferably Y 1 and Y 2 are both oxygen atoms.
- n is 2 to 3, preferably 2.
- the compound of the general formula (2) which is a reaction product of bromination (bromination reaction) usually becomes liquid at 5 ° C. to 30 ° C.
- n is 0 to 1
- the regioselectivity of the bromination site in the bromination reaction is lowered.
- the method for synthesizing the phenyl compound represented by the general formula (1) is not particularly limited and can be obtained by a conventional method.
- the compound represented by the following general formula (1-a) is reacted with an acylating agent represented by the following general formula (1-b) in the presence of a Lewis acid such as aluminum chloride (III).
- a Lewis acid such as aluminum chloride (III).
- X represents a halogen atom, preferably a chlorine atom or a bromine atom.
- the phenyl compound represented by the general formula (1) is usually a liquid at 5 to 30 ° C. as described above.
- the compound of the following general formula (2) obtained by acylating the phenyl compound of the general formula (1) is converted to 5 ° C. to 30 ° C. It can be liquid at 0C.
- the compound being liquid at 5 ° C. to 30 ° C. means that crystals do not precipitate even when the compound is placed in an atmosphere at 5 ° C. to 30 ° C. for 1 week under normal pressure.
- the phenyl compound represented by the general formula (1) and bromine are reacted at a specific reaction temperature using a specific solvent.
- an ⁇ -bromoacetophenone compound represented by the following general formula (2) and liquid at 5 ° C. to 30 ° C. can be obtained with a high purity of about 95% to 100% without any special purification operation.
- the ⁇ -bromoacetophenone compound represented by the following general formula (2) obtained by the production method of the present invention is a liquid which is difficult to purify by a general recrystallization method because it is liquid at a low temperature of 30 ° C. or less. is there.
- the bromination reaction of the phenyl compound of the general formula (1) in the bromination reaction of the phenyl compound of the general formula (1), the bromination reaction of the carbon atoms constituting the benzene ring can be greatly suppressed. Therefore, the ⁇ -bromoacetophenone compound of the following general formula (2) can be obtained with high purity without subjecting the reaction product obtained by the bromination reaction to a purification operation such as recrystallization.
- R 1 ⁇ R 7, Y 1, Y 2 and n are the same as R 1 ⁇ R 7, Y 1 , Y 2 and n, respectively, in the general formula (1), preferred embodiments Is the same.
- the phenyl compound represented by the general formula (1) is reacted with bromine in a solvent containing at least one compound selected from an organic acid ester compound and an ether compound having no hydroxyl group.
- the solvent used in this bromination reaction is a compound other than an organic acid ester compound and an ether compound not having a hydroxyl group, as long as at least one compound selected from an organic acid ester compound and an ether compound having no hydroxyl group is used as the solvent. May be contained in a solvent.
- the bromination reaction (side reaction) of the benzene ring of the phenyl compound of the general formula (1) can be significantly suppressed.
- the target compound can be obtained with excellent purity. The reason for this is not clear, but is estimated as follows.
- bromination reaction of the phenyl compound of the general formula (1) bromination of the benzene ring, which is a side reaction, is considered to be caused by the reaction of ⁇ + of bromine (corresponding to a bromo cation) and the benzene ring.
- the solvent is bromine ⁇ + via an oxygen atom constituting an ester bond or an ether bond. It is presumed that the bromination reaction of the benzene ring is less likely to occur when ⁇ + is trapped in the solvent, for example, by coordination to. Even if the solvent has an oxygen atom in the molecular structure, the side reaction cannot be effectively suppressed in the case of a protic solvent such as alcohol or acetic acid. This is presumably because the solvent is less likely to coordinate with ⁇ + of bromine by coordinating to the proton generated from the solvent via its own oxygen atom.
- organic acid ester compound that can be used as a solvent in the production method of the present invention.
- the “organic acid ester compound” that can be used as a solvent in the present invention may contain an ether bond in its molecular structure. That is, a solvent having both an ester bond and an ether bond of an organic acid in its molecular structure is included in the “organic acid ester compound” in the present invention. That is, in the present invention, the ether compound is a solvent that contains an ether bond and does not contain an organic acid ester bond.
- the ether compound having no hydroxyl group that can be used as a solvent is not particularly limited.
- butyl ether and 1,4-dioxane are preferably used, and 1,4-dioxane is more preferably used.
- the solvent used in the bromination reaction in the present invention is methyl acetate, ethyl acetate, butyl acetate, propyl acetate, isopropyl acetate, methyl propionate, ethyl propionate, and 1, from the viewpoint of workability such as volatility and liquid separation.
- One or more selected from 4-dioxane is preferable, and one or more selected from methyl acetate, ethyl acetate, butyl acetate, propyl acetate, isopropyl acetate, and 1,4-dioxane is more preferable.
- the solvent used in the bromination reaction in the present invention does not have an organic acid ester compound and a hydroxyl group as long as it includes a solvent selected from an organic acid ester compound and an ether compound not having a hydroxyl group.
- a solvent other than the ether compound may be included.
- a solvent selected from ketones, alcohols, dialkylformamides, N-alkylpyrrolidones, and toluene can be used, but the present invention is not limited to these embodiments, and a solvent that can be generally used for bromination reaction is widely used. It comes out.
- the total content of the solvent selected from the organic acid ester compound and the ether compound having no hydroxyl group is preferably 50 to 100% by mass, and 70 to 100% by mass. Is more preferable, and 80 to 100% by mass is even more preferable.
- the phenyl compound represented by the general formula (1) is subjected to a bromination reaction in the coexistence state with the specific solvent.
- the amount of the solvent selected from the organic acid ester compound and the ether compound having no hydroxyl group in the reaction solution of the bromination reaction is , Preferably 0.1 mL or more per 1 g of the phenyl compound mixed in the reaction solution of bromination reaction, more preferably 0.3 to 120 mL, also preferably 0.5 to 60 mL, 0 It is also preferably 5 to 10 mL, and more preferably 0.5 to 5 mL.
- the bromination reaction (side reaction) of the phenyl compound represented by the general formula (1) to the benzene ring is more effectively suppressed. And the bromination reaction to the target site ( ⁇ carbon of the acyl group) proceeds more efficiently.
- the amount ratio of the phenyl compound represented by the general formula (1) and bromine used in the bromination reaction is determined by the stoichiometric ratio, but the molar ratio is [bromine] / [general
- the phenyl compound of formula (1)] is preferably 0.95 to 1.3, more preferably 1.0 to 1.2, and still more preferably 1.0 to 1.1.
- the reaction temperature of the bromination reaction is not limited, but is preferably 10 ° C. or higher, more preferably 20 ° C. or higher, further preferably 30 ° C. or higher, more preferably 35 ° C. or higher, and most preferably 40 ° C. °C or more.
- the reaction temperature of the bromination reaction is preferably a temperature near or below the boiling point of bromine, specifically 60 ° C. or less, more preferably 55 ° C. or less.
- part increases and the purity of the compound of General formula (2) obtained improves significantly.
- the desired reaction at the ⁇ -position of the acyl group is promoted, and excess bromine is reduced in the reaction system. For this reason, it is presumed that the side reaction with the benzene ring hardly proceeds and the selectivity of the desired reaction at the ⁇ -position of the acyl group is improved.
- the reaction time of the bromination reaction is not particularly limited, but preferably 15 minutes or more from the start of the reaction (that is, from the start of mixing of bromine with the phenyl compound of the general formula (1)). 30 minutes or more is more preferable.
- the reaction time for the bromination reaction is usually within 5 hours, preferably within 3 hours.
- the bromination is preferably carried out by a drop reaction. This dropping reaction will be described below.
- bromine is charged into a dropping funnel, and the bromine is mixed with a phenyl compound represented by the above general formula (1) and a solvent selected from an organic acid ester compound and an ether compound having no hydroxyl group (preferably, the bromination reaction is carried out by dropping into a solution in which the phenyl compound represented by the general formula (1) is dissolved in a solvent containing a solvent selected from an organic acid ester compound and an ether compound not having a hydroxyl group, the same applies hereinafter)
- a mixed liquid containing bromine and a solvent selected from an organic acid ester compound and an ether compound having no hydroxyl group preferably bromine is selected from an organic acid ester compound and an ether compound having no hydroxyl group
- a solution dissolved in a solvent containing a solvent the same shall apply hereinafter
- a mode in which the bromination reaction is carried out by dripping in the mixture (III) A mixed solution containing bromine and a solvent selected from an organic acid ester compound and an ether compound having no hydroxyl group is charged into a dropping funnel, and this mixed solution is An embodiment in which a bromination reaction is carried out by dropping into a mixed solution containing a phenyl compound represented by the general formula (1) and a solvent selected from an organic acid ester compound and an ether compound having no hydroxyl group.
- the above-described embodiment (I) or (III) is preferable, and the embodiment (III) is more preferable.
- the solvent mixed with bromine and the solvent mixed with the compound represented by the general formula (1) may be the same or different.
- the above-described reaction temperature of the bromination reaction in the present invention is the temperature of the liquid on the dropping side (the liquid containing the compound represented by the general formula (1)).
- a compound represented by the general formula (1) or a mixed solution of this and a solvent is charged in a reaction vessel such as a flask or a reaction kettle, and a dropping funnel is installed to drop bromine or a mixed solution of bromine and a solvent.
- the temperature of the liquid in the reaction vessel is the reaction temperature of the bromination reaction in the present invention.
- the dropping time is preferably 1 minute or longer, more preferably 15 minutes or longer, and further preferably 30 minutes or longer. From the same viewpoint, the dropping time is preferably within 180 minutes, more preferably within 120 minutes, further preferably within 90 minutes, and further preferably within 60 minutes.
- the dropping is preferably performed while stirring the reaction solution. After completion of the dropwise addition, the reaction is usually matured by continuing stirring for about 30 minutes to 5 hours. The stirring time after completion of the dropping is more preferably 1 to 3 hours.
- the total amount of solvent for 1 g of the reaction substrate (phenyl compound) of the general formula (1) is preferably 0.7 mL or more and 120 mL or less, more preferably 1.0 mL or more and 120 mL or less, further preferably 1.5 mL or more and 120 mL or less, and most preferably 2 mL or more and 120 mL or less. preferable.
- the ⁇ -bromoacetophenone compound represented by the general formula (2) is particularly suitable as a synthesis intermediate for the photopolymerization initiator. That is, a compound represented by the following general formula (3) can be obtained by reacting an ⁇ -bromoacetophenone compound represented by the general formula (2) with a base. This reaction is preferably performed by adding water.
- the compound represented by the general formula (3) can be suitably used as a photopolymerization initiator, particularly a radical polymerization initiator, or a precursor thereof.
- R 1 ⁇ R 6, Y 1, Y 2 and n are the same as R 1 ⁇ R 6, Y 1 , Y 2 and n in each of the above general formula (1), preferred embodiments Is the same.
- R 9 represents a hydrogen atom, an alkyl group (preferably an alkyl group having 1 to 10 carbon atoms, more preferably 1 to 5 carbon atoms, and still more preferably 1 to 3 carbon atoms), an acyl group (preferably 2 to 10 carbon atoms, More preferably, the number of carbon atoms is 2 to 6, more preferably 2 to 4 carbon atoms) or a trialkylsilyl group (one alkyl group of the trialkylsilyl group preferably has 1 to 10 carbon atoms, more preferably 1 carbon atom).
- Z represents a hydroxy group, an alkoxy group (preferably an alkoxy group having 1 to 10 carbon atoms, more preferably 1 to 5 carbon atoms, and still more preferably 1 to 3 carbon atoms) or an alkylamino group (including a dialkylamino group).
- the alkylamino group preferably has 1 to 10 carbon atoms, more preferably 1 to 5, more preferably 1 to 3 carbon atoms per alkyl group.
- Examples of the base include sodium hydroxide, potassium hydroxide, calcium hydroxide, lithium hydroxide, cesium hydroxide, rubidium hydroxide, sodium alkoxide (for example, sodium methoxide, sodium ethoxide), and alkylamine.
- sodium hydroxide is used as the base and an aqueous sodium hydroxide solution is added to react with the ⁇ -bromoacetophenone compound, Z in the general formula (3) can be converted into a hydroxy group.
- R 7 in the general formula (2) is an acyl group, it is hydrolyzed and R 9 in the general formula (3) becomes a hydrogen atom.
- Z in General formula (3) can be made into an alkoxy group.
- Z in General formula (3) can be made into an alkylamino group.
- the amount of the base used is preferably 1 or more and 50 or less, and more preferably 5 or more and 20 or less, with respect to the ⁇ -bromoacetophenone compound 1.
- a mixed solvent of water and a water-soluble organic solvent examples include alkanediols (polyhydric alcohols) such as glycerin, 1,2,6-hexanetriol, trimethylolpropane, ethylene glycol, propylene glycol; sugar alcohols; ethanol, methanol, butanol, propanol, C1-C4 alkyl alcohols such as isopropanol; ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, ethylene glycol monobutyl ether, ethylene glycol monomethyl ether acetate, diethylene glycol monomethyl ether, diethylene glycol monoethyl ether, diethylene glycol mono-n-propyl Ether, ethylene glycol mono-isopropyl ether, diethylene glycol mono-iso Propyl ether,
- alkanediols polyhydric alcohols
- glycerin 1,2,6-hexanetriol
- the reaction between the ⁇ -bromoacetophenone compound represented by the general formula (2) and the base is preferably carried out at 10 to 40 ° C.
- the reaction time for this reaction is preferably 0.5 to 5 hours.
- [Mol% of reaction substrate] 100 ⁇ [peak area of 7.95 ppm] / ⁇ [peak area of 7.95 ppm ⁇ + [peak area of 8.21 ppm] + [peak area of 8.41 ppm] ⁇
- [Mole% of target product] 100 ⁇ [8.21 ppm peak area] / ⁇ [7.95 ppm peak area ⁇ + [8.21 ppm peak area] + [8.41 ppm peak area] ⁇
- [Mole% of by-product] 100 ⁇ [peak area of 8.41 ppm] / ⁇ [peak area of 7.95 ppm ⁇ + [peak area of 8.21 ppm] + [peak area of 8.41 ppm] ⁇ The results are shown in the table below.
- Examples 2 to 38, 43 to 45 In the synthesis of compound 2a of Example 1, the reaction conditions were changed to the reaction conditions shown in the table below, and the compounds 2a of Examples 2-38 and 43-45 were synthesized in the same manner as the synthesis of compound 2a of Example 1. Got. The purity of the obtained compound 2a was measured in the same manner as in Example 1. The results are shown in the table below.
- Compound 2f was obtained in the same manner as in Example 1 except that compound 1f was used instead of compound 1a in the synthesis of compound 2a of Example 1 and the reaction conditions were changed to the reaction conditions shown in the following table. .
- Compound 1f was synthesized in the same manner as in Reference Example 1 except that 2-ethylhexanoyl chloride was used instead of isobutyric acid chloride in Reference Example 1 above.
- Compounds 1f and 2f were both liquid at 5-30 ° C.
- the purity of the obtained compound 2f was calculated based on the peak ratio of 1 H-NMR (CDCl 3 ) in the same manner as in Example 1. The results are shown in the table below.
Abstract
α-ブロモアセトフェノン化合物の製造方法は、少なくとも有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる化合物を溶媒とし、特定のフェニル化合物と臭素とを反応させてフェニル化合物を臭素化することを含み、5℃から30℃で液体であるα-ブロモアセトフェノン化合物を得る、製造方法である。
Description
本発明は、α-ブロモアセトフェノン化合物の製造方法に関する。
エチレン性不飽和結合を有する重合性化合物の重合開始剤として、α-ヒドロキシアセトフェノン系の光重合開始剤が知られている。α-ヒドロキシアセトフェノン系の光重合開始剤は一般的に、イソ酪酸クロリド等のカルボン酸ハライドを用いて、ルイス酸の存在下でフェニル化合物をアシル化し、このアシル基のα炭素を臭素化した後、この臭素原子をヒドロキシル基に置換して得ることができる。上記臭素化反応に関し、特許文献1には、4-(2-アセトキシエトキシ)-フェニル-2-プロピルケトンや4-ヒドロキシフェニル-2-プロピルケトンのイソブチリル基のα炭素を臭素化したことが記載されている。また、特許文献2には、4-メトキシイソブチロフェノンのイソブチリル基のα炭素を臭素化したことが記載されている。
しかし、特許文献1及び2に記載される臭素化方法では、アシル基のα炭素の他にベンゼン環の環構成炭素原子も一定程度臭素化されてしまう。そのため、反応後の精製工程が必要となり、アシル基のα炭素のみが臭素化されたα-ブロモアセトフェノン化合物を、精製操作を経ずに高純度に得ることは困難であった。また、目的の化合物(すなわちα-ブロモアセトフェノン化合物あるいはこれから導かれるα-ヒドロキシアセトフェノン化合物等)が融点の低い液体状化合物である場合には、再結晶による精製が困難であった。このように、目的化合物の精製、高純度化にも制約がある。
光重合開始剤の用途は多岐に渡る。塗料、接着剤、光学フィルム、ソルダーレジスト材料等の硬化剤成分としての使用に加え、水を含む溶媒に対する溶解性が求められる用途も拡大している。例えば、光重合開始剤をインクジェットプリンタに用いる硬化性インク成分として用いる場合には、極性を高めて親水性を付与することが求められる。光重合開始剤としてα-ヒドロキシアセトフェノン系の光重合開始剤を用いる場合、合成原料とするフェニル化合物のベンゼン環上にエチレンオキシド鎖等の特定鎖長の極性基を置換基として導入することで光重合開始剤の親水性を高めることができる。しかし、エチレンオキシド鎖等を導入すると、得られる合成中間体であるα-ハロゲノアセトフェノン化合物ないしは目的の光重合開始剤であるα-ヒドロキシアセトフェノン化合物は、その融点が低下して液状となる。その場合、再結晶法による精製ができない。
本発明は、特定の極性基を有する光重合開始剤の合成中間体として有用な、5℃から30℃で液体状のα-ブロモアセトフェノン化合物を、優れた反応純度で製造する方法を提供することを課題とする。
本発明の上記課題は下記の手段により解決された。
〔1〕
有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる少なくとも1種の化合物を含む溶媒中で、下記一般式(1)で表されるフェニル化合物と臭素とを反応させてフェニル化合物を臭素化することを含む、下記一般式(2)で表されるα-ブロモアセトフェノン化合物の製造方法であって、得られるα-ブロモアセトフェノン化合物が5℃~30℃で液体である、製造方法。
〔1〕
有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる少なくとも1種の化合物を含む溶媒中で、下記一般式(1)で表されるフェニル化合物と臭素とを反応させてフェニル化合物を臭素化することを含む、下記一般式(2)で表されるα-ブロモアセトフェノン化合物の製造方法であって、得られるα-ブロモアセトフェノン化合物が5℃~30℃で液体である、製造方法。

一般式(1)中、R1及びR2は各々独立にアルキル基を示す。R3~R6は各々独立に水素原子又は置換基を示す。但し、R3~R6の少なくとも1つは水素原子である。Y1及びY2は各々独立に酸素原子又は硫黄原子を示し、nは2~3である。R7は水素原子、アルキル基、アシル基又はトリアルキルシリル基を示す。

一般式(2)中、R1~R7、Y1、Y2及びnはそれぞれ一般式(1)におけるR1~R7、Y1、Y2及びnと同義である。
〔2〕
臭素化の反応温度が20℃以上である、〔1〕に記載の製造方法。
〔3〕
有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒が、酢酸メチル、酢酸エチル、酢酸ブチル、酢酸プロピル、酢酸イソプロピル、及び1,4-ジオキサンから選ばれる1種又は2種以上である、〔1〕又は〔2〕に記載の製造方法。
〔4〕
臭素化の反応温度が40℃以上である、〔1〕~〔3〕のいずれかに記載の製造方法。
〔5〕
臭素化に用いる溶媒の総量中、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒の総含有量が50~100質量%である、〔1〕~〔4〕のいずれかに記載の製造方法。
〔6〕
臭素化の反応液中の有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒の量が、この反応液中に混合されたフェニル化合物1g当たり0.5~60mLである、〔1〕~〔5〕のいずれかに記載の製造方法。
〔7〕
臭素化は、臭素もしくは、臭素と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液を、フェニル化合物と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液中に滴下して行うか、又は、
臭素と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液を、フェニル化合物中もしくは、フェニル化合物と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液中に滴下して行う、〔1〕~〔6〕のいずれかに記載の製造方法。
〔8〕
臭素化は、臭素と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液を、フェニル化合物と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液中に滴下して行う、〔7〕に記載の製造方法。
〔9〕
臭素と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液中の有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒の量Aと、フェニル化合物と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液中の有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒の量Bの比率が、体積比で、B:A=90:10~30:70である、〔8〕に記載の製造方法。
〔2〕
臭素化の反応温度が20℃以上である、〔1〕に記載の製造方法。
〔3〕
有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒が、酢酸メチル、酢酸エチル、酢酸ブチル、酢酸プロピル、酢酸イソプロピル、及び1,4-ジオキサンから選ばれる1種又は2種以上である、〔1〕又は〔2〕に記載の製造方法。
〔4〕
臭素化の反応温度が40℃以上である、〔1〕~〔3〕のいずれかに記載の製造方法。
〔5〕
臭素化に用いる溶媒の総量中、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒の総含有量が50~100質量%である、〔1〕~〔4〕のいずれかに記載の製造方法。
〔6〕
臭素化の反応液中の有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒の量が、この反応液中に混合されたフェニル化合物1g当たり0.5~60mLである、〔1〕~〔5〕のいずれかに記載の製造方法。
〔7〕
臭素化は、臭素もしくは、臭素と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液を、フェニル化合物と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液中に滴下して行うか、又は、
臭素と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液を、フェニル化合物中もしくは、フェニル化合物と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液中に滴下して行う、〔1〕~〔6〕のいずれかに記載の製造方法。
〔8〕
臭素化は、臭素と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液を、フェニル化合物と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液中に滴下して行う、〔7〕に記載の製造方法。
〔9〕
臭素と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液中の有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒の量Aと、フェニル化合物と有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液中の有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒の量Bの比率が、体積比で、B:A=90:10~30:70である、〔8〕に記載の製造方法。
本発明の製造方法によれば、5℃~30℃で液体状のα-ブロモアセトフェノン化合物を、高い反応純度で得ることができ、通常、再結晶等の精製操作を必要としない。
本発明のα-ブロモアセトフェノン化合物の製造方法(以下、単に「本発明の製造方法という。)について、以下に詳細に説明する。
本発明の製造方法は、出発原料として下記一般式(1)で表されるフェニル化合物を用いる。

一般式(1)中、R1及びR2は、各々独立にアルキル基を示す。このアルキル基は直鎖でも、分岐していてもよい。R1及びR2は好ましくは炭素数1~10、より好ましくは炭素数1~5、さらに好ましくは炭素数1~3のアルキル基であり、さらに好ましくはメチル又はエチルであり、最も好ましくはメチルである。
また、R1及びR2は互いに連結して環を形成していてもよい。R1及びR2が連結して形成される環構造の基はシクロアルキル基であることが好ましい。このシクロアルキル基はより好ましくは炭素数3~10、さらに好ましくは炭素数4~8のシクロアルキル基であり、より具体的にはシクロプロピル、シクロブチル、シクロヘプチル又はシクロへキシルであることが好ましい。
また、R1及びR2は互いに連結して環を形成していてもよい。R1及びR2が連結して形成される環構造の基はシクロアルキル基であることが好ましい。このシクロアルキル基はより好ましくは炭素数3~10、さらに好ましくは炭素数4~8のシクロアルキル基であり、より具体的にはシクロプロピル、シクロブチル、シクロヘプチル又はシクロへキシルであることが好ましい。
一般式(1)中、R3~R6は、各々独立に水素原子又は置換基を表す。但し、R3~R6の少なくとも1つは水素原子である。また、R3~R6のうち2つ以上(好ましくはR4及びR6を含む2つ以上)が水素原子であることが好ましく、3つ以上(好ましくはR4及びR6を含む3つ以上)が水素原子であることがより好ましく、さらに好ましくはR3~R6のすべてが水素原子である。
R3~R6が置換基である場合、この置換基としてアルキル基(好ましくは炭素数1~5、より好ましくは炭素数1~3、さらに好ましくは炭素数1又は2のアルキル基)、ハロゲン原子(具体的にはフッ素原子、塩素原子、臭素原子又はヨウ素原子)、アミノ基、ヒドロキシ基、シアノ基、ニトロ基、カルボキシ基、スルホ基、スルホニル基、ホスホニル基、ホウ酸基、アルコキシ基、アミド基等が挙げられる。なかでもメチル、エチル、又はハロゲン原子が好ましい。
一般式(1)中、R7は水素原子、アルキル基、アシル基又はトリアルキルシリル基を示す。R7はアシル基であることがより好ましい。
R7がアルキル基の場合、直鎖でも、分岐していてもよく、置換基を有してもよい。このアルキル基は炭素数1~10であることが好ましく、より好ましくは炭素数1~6であり、さらに好ましくは炭素数1~5である。また、R7がアルキル基の場合、このアルキル基が有する置換基が連結して環構造を形成してもよい。このように環構造を形成したアルキル基としては、例えば2-テトラヒドロピラニルが挙げられる。また、R7がアルキル基の場合、アルコキシメチル基(好ましくは炭素数1~10、より好ましくは炭素数1~5のアルコキシメチル基)であることも好ましい。R7がアルキル基の場合の好ましい例としては、例えば、メチル、エチル、プロピル、イソプロピル、t-ブチル、メトキシメチル、エトキシメチル、ベンジル、p-メトキシベンジル、フェネチル及び2-テトラヒドロピラニルが挙げられ、メチル、メトキシメチル、ベンジル又は2-テトラヒドロピラニルであることがより好ましい。
R7がアシル基の場合、その炭素数は2~12であることが好ましく、2~10であることがより好ましく、2~6であることがさらに好ましい。R7はより好ましくは、アセチル、ピバロイル、アクリロイル又はベンゾイルであり、さらに好ましくはアセチルである。
R7がトリアルキルシリル基の場合、トリアルキルシリル基のアルキル基は、直鎖でも、分岐していてもよい。このアルキル基は炭素数1~10であることが好ましく、より好ましくは炭素数1~6であり、さらに好ましくは炭素数1~4である。より具体的には、例えば、トリメチルシリル、トリエチルシリル、t-ブチルジメチルシリル及びトリイソプロピルシリルが挙げられ、好ましくはトリメチルシリルである。
一般式(1)中、Y1及びY2は、各々独立に酸素原子又は硫黄原子を示し、より好ましくはY1及びY2がいずれも酸素原子である。
一般式(1)中、nは2~3であり、好ましくは2である。nが2又は3であると、臭素化(臭素化反応)の反応生成物である一般式(2)の化合物は、通常5℃~30℃で液体状となる。また、nが0~1であると、臭素化反応における臭素化部位の位置選択性が低下する。また、反応生成物である一般式(2)の化合物の物性を本発明の規定内とすることが困難となる。
一般式(1)で表されるフェニル化合物の合成方法に特に制限はなく常法により得ることができる。例えば、下記一般式(1-a)で表される化合物に、塩化アルミニウム(III)等のルイス酸の存在下で、下記一般式(1-b)で表されるアシル化剤を反応させて得ることができる。

一般式(1-a)及び(1-b)中、R1~R7、Y1、Y2及びnは、それぞれ一般式(1)におけるR1~R7、Y1、Y2及びnと同義であり、好ましい形態も同じである。Xはハロゲン原子を表し、好ましくは塩素原子又は臭素原子である。
上記一般式(1)で表されるフェニル化合物は上述のとおり、通常は5℃~30℃で液体である。上記一般式(1)の化合物が5℃~30℃で液体状であることで、上記一般式(1)のフェニル化合物をアシル化して得られる下記一般式(2)の化合物を5℃~30℃で液体状とすることができる。本発明において、化合物が5℃~30℃で液体であるとは、当該化合物を、常圧下、5℃~30℃の雰囲気中に1週間置いても結晶が析出しないことを意味する。
上記一般式(1)で表されるフェニル化合物の具体例を以下に示すが、本発明はこれらに限定されない。



本発明の製造方法では、上記一般式(1)で表されるフェニル化合物と臭素とを、特定の溶媒を用いて、特定の反応温度で反応させる。これにより、下記一般式(2)で表され、5℃~30℃で液体であるα-ブロモアセトフェノン化合物を、特別の精製操作を経ることなく、通常95~100%程度の高純度で得ることができる。
本発明の製造方法で得られる下記一般式(2)で表されるα-ブロモアセトフェノン化合物は、30℃以下の低温において液体状であるため、一般的な再結晶法による精製が困難な化合物である。しかし、本発明の製造方法によれば、一般式(1)のフェニル化合物の臭素化反応において、ベンゼン環の環構成炭素原子の臭素化反応を大幅に抑えることができる。したがって、上記臭素化反応により得られる反応生成物を再結晶等の精製操作に付さなくても、下記一般式(2)のα-ブロモアセトフェノン化合物を高純度に得ることができる。
本発明の製造方法で得られる下記一般式(2)で表されるα-ブロモアセトフェノン化合物は、30℃以下の低温において液体状であるため、一般的な再結晶法による精製が困難な化合物である。しかし、本発明の製造方法によれば、一般式(1)のフェニル化合物の臭素化反応において、ベンゼン環の環構成炭素原子の臭素化反応を大幅に抑えることができる。したがって、上記臭素化反応により得られる反応生成物を再結晶等の精製操作に付さなくても、下記一般式(2)のα-ブロモアセトフェノン化合物を高純度に得ることができる。

上記一般式(2)中、R1~R7、Y1、Y2及びnは、それぞれ一般式(1)におけるR1~R7、Y1、Y2及びnと同義であり、好ましい形態も同じである。
[溶媒]
本発明の製造方法では、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる少なくとも1種の化合物を含む溶媒中で、上記一般式(1)で表されるフェニル化合物と臭素とを反応させる。この臭素化反応に用いる溶媒は、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる少なくとも1種の化合物を溶媒として用いれば、有機酸エステル化合物及び水酸基を有さないエーテル化合物以外の化合物を溶媒に含んでいてもよい。
有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒を用いることで、一般式(1)のフェニル化合物が有するベンゼン環の臭素化反応(副反応)を大幅に抑制することができ、目的の化合物を優れた純度で得ることができる。この理由は定かではないが、以下のように推定される。
一般式(1)のフェニル化合物の臭素化反応において、副反応であるベンゼン環の臭素化は、臭素のδ+(ブロモカチオンに相当)とベンゼン環が反応することにより生じると考えられる。しかし、本発明により、臭素化反応における溶媒として有機酸エステル化合物あるいは水酸基を有さないエーテル化合物を用いた場合には、エステル結合あるいはエーテル結合を構成する酸素原子を介して溶媒が臭素のδ+に配位するなどし、δ+が溶媒にトラップされることで、ベンゼン環の臭素化反応が生じにくくなると推定される。分子構造中に酸素原子を有する溶媒であっても、アルコールや酢酸などのプロトン性溶媒の場合には上記副反応を効果的に抑えることはできない。これは、溶媒が、自身の酸素原子を介して溶媒から生じるプロトンに配位することにより、臭素のδ+に対して配位しにくくなるためと考えられる。
本発明の製造方法では、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる少なくとも1種の化合物を含む溶媒中で、上記一般式(1)で表されるフェニル化合物と臭素とを反応させる。この臭素化反応に用いる溶媒は、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる少なくとも1種の化合物を溶媒として用いれば、有機酸エステル化合物及び水酸基を有さないエーテル化合物以外の化合物を溶媒に含んでいてもよい。
有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒を用いることで、一般式(1)のフェニル化合物が有するベンゼン環の臭素化反応(副反応)を大幅に抑制することができ、目的の化合物を優れた純度で得ることができる。この理由は定かではないが、以下のように推定される。
一般式(1)のフェニル化合物の臭素化反応において、副反応であるベンゼン環の臭素化は、臭素のδ+(ブロモカチオンに相当)とベンゼン環が反応することにより生じると考えられる。しかし、本発明により、臭素化反応における溶媒として有機酸エステル化合物あるいは水酸基を有さないエーテル化合物を用いた場合には、エステル結合あるいはエーテル結合を構成する酸素原子を介して溶媒が臭素のδ+に配位するなどし、δ+が溶媒にトラップされることで、ベンゼン環の臭素化反応が生じにくくなると推定される。分子構造中に酸素原子を有する溶媒であっても、アルコールや酢酸などのプロトン性溶媒の場合には上記副反応を効果的に抑えることはできない。これは、溶媒が、自身の酸素原子を介して溶媒から生じるプロトンに配位することにより、臭素のδ+に対して配位しにくくなるためと考えられる。
本発明の製造方法に溶媒として用いうる有機酸エステル化合物に特に制限はなく、例えば、酢酸メチル、酢酸エチル、酢酸プロピル、酢酸イソプロピル、酢酸ブチル、酢酸へキシル、プロピオン酸メチル、プロピオン酸エチル、プロピオン酸プロピル、プロピオン酸イソプロピル、プロピオン酸ブチル、プロピオン酸へキシル、酪酸メチル、酪酸エチル、酪酸プロピル、イソ酪酸メチル、イソ酪酸エチル、イソ酪酸プロピル、γ―ブチロラクトン、ε―カプロラクトン、エチレンカーボネート、プロピレンカーボネート、2-アセトキシ-1-メトキシプロパン、メチル-3-メトキシプロピオネート、酢酸セロソルブ、カルビトールアセテート、トリエチレングリコールジアセテート、及びジメチルアジピン酸が挙げられ、これらの1種又は2種以上を用いることができる。なかでも、酢酸メチル、酢酸エチル、酢酸ブチル、酢酸プロピル、及び酢酸イソプロピルから選ばれる1種又は2種以上を用いることが好ましい。
なお、本発明に溶媒として用いうる「有機酸エステル化合物」は、その分子構造中にエーテル結合を含む場合がある。すなわち、分子構造中に有機酸のエステル結合とエーテル結合の両者を有する溶媒は、本発明においては「有機酸エステル化合物」に包含される。つまり、本発明においてエーテル化合物は、エーテル結合を含み、有機酸のエステル結合を含まない溶媒である。
なお、本発明に溶媒として用いうる「有機酸エステル化合物」は、その分子構造中にエーテル結合を含む場合がある。すなわち、分子構造中に有機酸のエステル結合とエーテル結合の両者を有する溶媒は、本発明においては「有機酸エステル化合物」に包含される。つまり、本発明においてエーテル化合物は、エーテル結合を含み、有機酸のエステル結合を含まない溶媒である。
本発明の製造方法において、溶媒として用いうる水酸基を有さないエーテル化合物に特に制限はなく、例えば、ジエチルエーテル、ジエチレングリコールジメチルエーテル、ジプロピルエーテル、ジブチルエーテル、ジへキシルエーテル、エチレングリコールジエチルエーテル、ジエチレングリコールジメチルエーテル、イソアミルエーテル、ジエチレングリコールジエチルエーテル、トリエチレングリコールジメチルエーテル、テトラエチレングリコールジメチルエーテル、ジエチレングリコールメチルエチルエーテル、メチルターシャリーブチルエーテル、ジエチレングリコールジブチルエーテル、1,4-ジオキサン、及びテトラヒドロフランが挙げられ、これらの1種又は2種以上を用いることができる。なかでもジエチレングリコールメチルエーテル、ジプロピルエーテル、ジブチルエーテル、ジへキシルエーテル、エチレングリコールジエチルエーテル、ジエチレングリコールジメチルエーテル、イソアミルエーテル、ジエチレングリコールジエチルエーテル、トリエチレングリコールジメチルエーテル、テトラエチレングリコールジメチルエーテル、ジエチレングリコールメチルエチルエーテル、ジエチレングリコールジブチルエーテル、及び1,4-ジオキサンから選ばれる1種又は2種以上を用いることが好ましく、1,4-ジオキサンを用いることがより好ましい。
本発明において臭素化反応に用いる溶媒は、揮発性、分液性といった作業性の観点から、酢酸メチル、酢酸エチル、酢酸ブチル、酢酸プロピル、酢酸イソプロピル、プロピオン酸メチル、プロピオン酸エチル、及び1,4-ジオキサンから選ばれる1種又は2種以上が好ましく、酢酸メチル、酢酸エチル、酢酸ブチル、酢酸プロピル、酢酸イソプロピル、及び1,4-ジオキサンから選ばれる1種又は2種以上がより好ましい。
また、上述したように、本発明において臭素化反応の際に用いる溶媒は、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒を含む限り、有機酸エステル化合物及び水酸基を有さないエーテル化合物以外の溶媒を含んでもよい。有機酸エステル化合物及び水酸基を有さないエーテル化合物以外の溶媒に特に制限はない。例えば、ケトン、アルコール、ジアルキルホルムアミド、N-アルキルピロリドン、及びトルエンから選ばれる溶媒を用いることができるが、本発明はこれらの態様に限定されず、臭素化反応に通常使用されうる溶媒を広く用いることがでる。本発明において、臭素化反応の際に用いる溶媒の総量中、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒の総含有量は50~100質量%が好ましく、70~100質量%がより好ましく、80~100質量%がさらに好ましい。
本発明の製造方法において、上記一般式(1)で表されるフェニル化合物は、上記特定の溶媒との共存状態で臭素化反応に付される。臭素化反応の反応液中(すなわち、溶媒と臭素と反応基質であるフェニル化合物のすべてが混合された反応液中)の有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒の量は、臭素化反応の反応液中に混合されたフェニル化合物1g当たり0.1mL以上とすることが好ましく、0.3~120mLとすることがより好ましく、0.5~60mLとすることも好ましく、0.5~10mLとすることも好ましく、0.5~5mLとすることも好ましい。上記溶媒と上記フェニル化合物の量比を上記好ましい比率とすることで、上記一般式(1)で表されるフェニル化合物のベンゼン環への臭素化反応(副反応)をより効果的に抑制することができ、且つ、目的部位(アシル基のα炭素)への臭素化反応がより効率的に進行する。
また、本発明の製造方法において、臭素化反応に用いる上記一般式(1)で表されるフェニル化合物と臭素の量比は化学量論比で定まるが、モル比で、[臭素]/[一般式(1)のフェニル化合物]を0.95~1.3とすることが好ましく、1.0~1.2とすることがより好ましく、1.0~1.1とすることがさらに好ましい。
本発明の製造方法において、臭素化反応の反応温度に制限は無いが、好ましくは10℃以上、より好ましくは20℃以上、さらに好ましくは30℃以上、さらに好ましくは35℃以上、最も好ましくは40℃以上とする。また、臭素化反応の反応温度は臭素の沸点付近の温度あるいはそれ以下の温度とすることが好ましく、具体的には60℃以下とすることが好ましく、より好ましくは55℃以下である。これにより、臭素化部位の選択性が高まり、得られる一般式(2)の化合物の純度が大きく向上する。
反応温度を上げることでアシル基のα位の所望の反応が促進され、反応系中に余剰の臭素が少なくなる。そのため、ベンゼン環との副反応が進行しにくくなり、アシル基のα位の所望の反応の選択性が向上すると推定される。
反応温度を上げることでアシル基のα位の所望の反応が促進され、反応系中に余剰の臭素が少なくなる。そのため、ベンゼン環との副反応が進行しにくくなり、アシル基のα位の所望の反応の選択性が向上すると推定される。
本発明の製造方法において、臭素化反応の反応時間に特に制限はないが、反応開始から(すなわち、臭素と一般式(1)のフェニル化合物との混合を開始してから)15分以上が好ましく、30分以上がより好ましい。また、臭素化反応の反応時間は通常は5時間以内であり、3時間以内とするのが好ましい。
臭素化は滴下反応で実施することが好ましい。この滴下反応について以下に説明する。
臭素化は滴下反応で実施することが好ましい。この滴下反応について以下に説明する。
本発明の製造方法における臭素化反応を滴下反応で実施する場合、好ましい態様として以下の態様が挙げられる。
(I)臭素を滴下ロートに仕込み、この臭素を、上記一般式(1)で表されるフェニル化合物と、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液(好ましくは上記一般式(1)で表されるフェニル化合物が有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒を含む溶媒に溶解した液、以下同様)中に滴下して臭素化反応を実施する態様
(II)臭素と、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液(好ましくは臭素を有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒を含む溶媒に溶解した液、以下同様)を滴下ロートに仕込み、この混合液を、上記一般式(1)で表されるフェニル化合物(液体)中に滴下して臭素化反応を実施する態様
(III)臭素と、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液を滴下ロートに仕込み、この混合液を、上記一般式(1)で表されるフェニル化合物と、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液中に滴下して臭素化反応を実施する態様
(I)臭素を滴下ロートに仕込み、この臭素を、上記一般式(1)で表されるフェニル化合物と、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液(好ましくは上記一般式(1)で表されるフェニル化合物が有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒を含む溶媒に溶解した液、以下同様)中に滴下して臭素化反応を実施する態様
(II)臭素と、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液(好ましくは臭素を有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒を含む溶媒に溶解した液、以下同様)を滴下ロートに仕込み、この混合液を、上記一般式(1)で表されるフェニル化合物(液体)中に滴下して臭素化反応を実施する態様
(III)臭素と、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液を滴下ロートに仕込み、この混合液を、上記一般式(1)で表されるフェニル化合物と、有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒とを含む混合液中に滴下して臭素化反応を実施する態様
なかでも、上記(I)又は(III)の態様が好ましく、(III)の態様がより好ましい。上記(III)の態様において、臭素と混合する溶媒と、上記一般式(1)で表される化合物と混合する溶媒は同一でも異なっていてもよい。
上記の滴下反応においては、本発明における臭素化反応の上述した反応温度は、滴下される側の液(一般式(1)で表される化合物を含む液)の温度とする。例えば、フラスコや反応釜等の反応容器に一般式(1)で表される化合物又はこれと溶媒との混合液を仕込み、滴下ロートを設置して臭素又は臭素と溶媒との混合液を滴下する反応系では、反応容器内の液の温度が本発明における臭素化反応の反応温度である。
また、上記の滴下反応において、滴下時間に特に制限はなく、滴下される臭素の消費に合わせて適宜に調節される。臭素化部位の選択性をより高める観点から、滴下時間は1分以上が好ましく、15分以上がより好ましく、30分以上がさらに好ましい。また、同様の観点から、滴下時間は180分以内が好ましく、120分以内がより好ましく、90分以内がさらに好ましく、60分以内がさらに好ましい。滴下は反応液を撹拌しながら行うことが好ましい。
滴下終了後も通常は30分~5時間程度撹拌を続けて反応を熟成させる。滴下終了後の撹拌時間は1時間~3時間とすることがより好ましい。
滴下終了後も通常は30分~5時間程度撹拌を続けて反応を熟成させる。滴下終了後の撹拌時間は1時間~3時間とすることがより好ましい。
上記の滴下反応の滴下開始時点において、臭素を含む滴下液中の有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる溶媒量Aと、一般式(1)のフェニル化合物を含む液中の有機酸エステル化合物及びエーテル化合物から選ばれる溶媒量Bの比に特に制限はないが、B:A(体積比)を100:0~10:90とすることが好ましく、100:0~20:80とすることがより好ましく、90:10~30:70とすることがさらに好ましく、80:20~40:60とすることがさらに好ましく、70:30~50:50とすることがさらに好ましい。
上記の滴下反応に用いる、一般式(1)の反応基質(フェニル化合物)1gに対する全溶媒量に特に制限はないが、反応基質に対する全溶媒量が少なくなると副反応が起こりやすくなる。全溶媒量が多くなることに関しては反応上特に問題は無いが、製造コスト上多すぎるのは好ましくない。一般式(1)の反応基質1gに対する全溶媒量としては0.7mL以上120mL以下が好ましく、1.0mL以上120mL以下がより好ましく、1.5mL以上120mL以下がさらに好ましく、2mL以上120mL以下が最も好ましい。
上記一般式(2)で表される5℃から30℃で液体である化合物の具体例を以下に示すが、本発明はこれらに限定されない。



一般式(2)で表されるα-ブロモアセトフェノン化合物は、特に光重合開始剤の合成中間体として好適である。
すなわち、一般式(2)で表されるα-ブロモアセトフェノン化合物と、塩基とを反応させることで、下記一般式(3)で表される化合物を得ることができる。この反応は水を添加して行うことが好ましい。この一般式(3)で表される化合物は光重合開始剤、特にラジカル重合開始剤、又はその前駆体として好適に用いることができる。
すなわち、一般式(2)で表されるα-ブロモアセトフェノン化合物と、塩基とを反応させることで、下記一般式(3)で表される化合物を得ることができる。この反応は水を添加して行うことが好ましい。この一般式(3)で表される化合物は光重合開始剤、特にラジカル重合開始剤、又はその前駆体として好適に用いることができる。

一般式(3)中、R1~R6、Y1、Y2及びnは、それぞれ上記一般式(1)におけるR1~R6、Y1、Y2及びnと同義であり、好ましい形態も同じである。R9は水素原子、アルキル基(好ましくは炭素数1~10、より好ましくは炭素数1~5、さらに好ましくは炭素数1~3のアルキル基)、アシル基(好ましくは炭素数2~10、より好ましくは炭素数2~6、さらに好ましくは炭素数2~4のアシル基)又はトリアルキルシリル基(トリアルキルシリル基のアルキル基1つにつき炭素数が好ましくは1~10、より好ましくは1~5、さらに好ましくは1又は2のトリアルキルシリル基)を表す。Zはヒドロキシ基、アルコキシ基(好ましくは炭素数1~10、より好ましくは炭素数1~5、さらに好ましくは炭素数1~3のアルコキシ基)又はアルキルアミノ基(ジアルキルアミノ基を含む。アルキルアミノ基のアルキル基1つにつき炭素数が好ましくは1~10、より好ましくは1~5、さらに好ましくは1~3のアルキルアミノ基)を表す。
上記塩基としては、例えば、水酸化ナトリウム、水酸化カリウム、水酸化カルシウム、水酸化リチウム、水酸化セシウム、水酸化ルビジウム、ナトリウムアルコキシド(例えば、ナトリウムメトキシド、ナトリウムエトキシド)、アルキルアミンが挙げられる。
例えば、塩基として水酸化ナトリウムを用い、水酸化ナトリウム水溶液を添加してα-ブロモアセトフェノン化合物と反応を行えば、一般式(3)中のZをヒドロキシ基とすることができる。また、この場合、一般式(2)のR7がアシル基であれば、加水分解されて一般式(3)のR9は水素原子となる。
また、塩基としてナトリウムアルコキシドを用いれば、一般式(3)中のZをアルコキシ基とすることができる。
また、塩基としてアルキルアミンを用いれば、一般式(3)中のZをアルキルアミノ基とすることができる。
塩基の使用量は、α-ブロモアセトフェノン化合物1に対して、モル比で1以上50以下とすることが好ましく、5以上20以下とすることがより好ましい。
例えば、塩基として水酸化ナトリウムを用い、水酸化ナトリウム水溶液を添加してα-ブロモアセトフェノン化合物と反応を行えば、一般式(3)中のZをヒドロキシ基とすることができる。また、この場合、一般式(2)のR7がアシル基であれば、加水分解されて一般式(3)のR9は水素原子となる。
また、塩基としてナトリウムアルコキシドを用いれば、一般式(3)中のZをアルコキシ基とすることができる。
また、塩基としてアルキルアミンを用いれば、一般式(3)中のZをアルキルアミノ基とすることができる。
塩基の使用量は、α-ブロモアセトフェノン化合物1に対して、モル比で1以上50以下とすることが好ましく、5以上20以下とすることがより好ましい。
一般式(2)で表されるα-ブロモアセトフェノン化合物と塩基との反応は、水と水溶性有機溶媒との混合溶媒を用いることが好ましい。この水溶性有機溶媒としてはグリセリン、1,2,6-ヘキサントリオール、トリメチロールプロパン、エチレングリコール、プロピレングリコール等のアルカンジオール(多価アルコール類);糖アルコール類;エタノール、メタノール、ブタノール、プロパノール、イソプロパノールなどの炭素数1~4のアルキルアルコール類;エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、エチレングリコールモノブチルエーテル、エチレングリコールモノメチルエーテルアセテート、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールモノ-n-プロピルエーテル、エチレングリコールモノ-イソプロピルエーテル、ジエチレングリコールモノ-イソプロピルエーテル、エチレングリコールモノ-n-ブチルエーテル、エチレングリコールモノ-t-ブチルエーテル、ジエチレングリコールモノ-t-ブチルエーテル、トリエチレングリコールモノエチルエーテル、1-メチル-1-メトキシブタノール、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、プロピレングリコールモノ-t-ブチルエーテル、プロピレングリコールモノ-n-プロピルエーテル、プロピレングリコールモノ-イソプロピルエーテル、ジプロピレングリコール、ジプロピレングリコールモノメチルエーテル、ジプロピレングリコールモノエチルエーテル、ジプロピレングリコールモノ-n-プロピルエーテル、ジプロピレングリコールモノ-イソプロピルエーテル、トリプロピレングリコールモノメチルエーテルなどのグリコールエーテル類等が挙げられる。なかでも揮発除去性の観点から、炭素数1~4のアルコール(例えばエタノール、メタノール、ブタノール、プロパノール及びイソプロパノールから選ばれる1種又は2種以上)を用いることが好ましい。
一般式(2)で表されるα-ブロモアセトフェノン化合物と塩基との反応は、好ましくは10~40℃で行われる。この反応の反応時間は0.5~5時間とすることが好ましい。
以下、本発明を実施例に基づきさらに詳細に説明するが、本発明はこれらの実施例に限定されない。
[参考例1]
一般式(1)で表される化合物として下記化合物1aを下記スキーム1により合成した。
一般式(1)で表される化合物として下記化合物1aを下記スキーム1により合成した。

(化合物aの合成)
90℃に加熱した170.0gのフェニルジグリコール(PhDG、日本乳化剤製)(0.93mol)に、97.2gの無水酢酸(0.95mol)を滴下し、120℃で6時間加熱攪拌した。その後、減圧下で濃縮し、化合物aを204.4g得た(収率98%)。
90℃に加熱した170.0gのフェニルジグリコール(PhDG、日本乳化剤製)(0.93mol)に、97.2gの無水酢酸(0.95mol)を滴下し、120℃で6時間加熱攪拌した。その後、減圧下で濃縮し、化合物aを204.4g得た(収率98%)。
1H-NMR(CDCl3)
δ:2.10(3H,s),3.78(2H,m),3.87(2H,m),4.15(2H,m),4.26(2H,m),6.90-6.98(3H,m),7.25-7.32(2H,m)
δ:2.10(3H,s),3.78(2H,m),3.87(2H,m),4.15(2H,m),4.26(2H,m),6.90-6.98(3H,m),7.25-7.32(2H,m)
(化合物1aの合成)
10.0gの化合物a(44.59mmol)をクロロベンゼン40mLに溶解し、氷浴中で5℃まで冷却した後、17.84gの塩化アルミニウム(III)(133.78mmol)を添加した。その後、イソ酪酸クロリド5.59mL(53.51mmol)を滴下し、1時間攪拌した。次いで、氷80gに反応液を注ぎ、生成物を酢酸エチル40mLで抽出し、有機層を重曹水80mL、塩水40mLで順次洗浄した後、硫酸マグネシウム10gを用いて乾燥した。これをろ過後、ろ液を減圧濃縮することにより化合物1aを11.87g得た(収率90%)。化合物1aは30℃で液体である。
10.0gの化合物a(44.59mmol)をクロロベンゼン40mLに溶解し、氷浴中で5℃まで冷却した後、17.84gの塩化アルミニウム(III)(133.78mmol)を添加した。その後、イソ酪酸クロリド5.59mL(53.51mmol)を滴下し、1時間攪拌した。次いで、氷80gに反応液を注ぎ、生成物を酢酸エチル40mLで抽出し、有機層を重曹水80mL、塩水40mLで順次洗浄した後、硫酸マグネシウム10gを用いて乾燥した。これをろ過後、ろ液を減圧濃縮することにより化合物1aを11.87g得た(収率90%)。化合物1aは30℃で液体である。
1H-NMR(CDCl3)
δ:1.20(3H,s),1.21(3H,s),2.10(3H,s),3.52(1H,m),3.78(2H,m),3.89(2H,m),4.20(2H,m),4.27(2H,m),6.97(2H,d),7.95(2H,d)
δ:1.20(3H,s),1.21(3H,s),2.10(3H,s),3.52(1H,m),3.78(2H,m),3.89(2H,m),4.20(2H,m),4.27(2H,m),6.97(2H,d),7.95(2H,d)
[実施例1]
(化合物2aの合成)
(化合物2aの合成)

滴下ロートと温度計を設置した100mL容量の三口フラスコに10.0gの化合物1a(0.034mol)と酢酸エチル12.5mLを加え、三口フラスコ内の液温度を45℃に調節して攪拌した。酢酸エチル12.5mLと臭素5.7g(0.036mol)を混合し、混合液を滴下ロートに加え、30分かけて滴下した後、さらに2時間攪拌した。この間、三口フラスコ内の反応液の温度は45℃に保ち続けた。次いで、3wt%亜硫酸水素ナトリウム水溶液を攪拌後の溶液に7mL滴下し、有機層を炭酸水素ナトリウム水溶液、飽和食塩水で順次洗浄した後、硫酸マグネシウムを用いて乾燥した。これをろ過後、ろ液を減圧濃縮することにより、化合物2a(反応生成物)を11.6g得た(収率91%)。化合物1a及び2aは5℃~30℃でいずれも液体であった。
1H-NMR(CDCl3)
δ:2.04(6H,s),2.08(3H,s),3.79(2H,m),3.85(2H,m),4.21(2H,m),4.26(2H,m),6.94(2H,d),8.21(2H,d)
上記で得られた化合物2aの純度を調べるため、反応生成物中に存在する化合物1a(反応基質)、化合物2a(目的生成物)、化合物3a(副生成物)のモル%を、それぞれ1H-NMR(CDCl3)の7.95ppm、8.21ppm、8.41ppmのピーク面積に基づき、下記式により算出した。
[反応基質のモル%]=100×[7.95ppmのピーク面積]/{[7.95ppmのピーク面積}+[8.21ppmのピーク面積]+[8.41ppmのピーク面積]}
[目的生成物のモル%]=100×[8.21ppmのピーク面積]/{[7.95ppmのピーク面積}+[8.21ppmのピーク面積]+[8.41ppmのピーク面積]}
[副生成物のモル%]=100×[8.41ppmのピーク面積]/{[7.95ppmのピーク面積}+[8.21ppmのピーク面積]+[8.41ppmのピーク面積]}
結果を下表に示す。
1H-NMR(CDCl3)
δ:2.04(6H,s),2.08(3H,s),3.79(2H,m),3.85(2H,m),4.21(2H,m),4.26(2H,m),6.94(2H,d),8.21(2H,d)
上記で得られた化合物2aの純度を調べるため、反応生成物中に存在する化合物1a(反応基質)、化合物2a(目的生成物)、化合物3a(副生成物)のモル%を、それぞれ1H-NMR(CDCl3)の7.95ppm、8.21ppm、8.41ppmのピーク面積に基づき、下記式により算出した。
[反応基質のモル%]=100×[7.95ppmのピーク面積]/{[7.95ppmのピーク面積}+[8.21ppmのピーク面積]+[8.41ppmのピーク面積]}
[目的生成物のモル%]=100×[8.21ppmのピーク面積]/{[7.95ppmのピーク面積}+[8.21ppmのピーク面積]+[8.41ppmのピーク面積]}
[副生成物のモル%]=100×[8.41ppmのピーク面積]/{[7.95ppmのピーク面積}+[8.21ppmのピーク面積]+[8.41ppmのピーク面積]}
結果を下表に示す。
[実施例2~38、43~45]
実施例1の化合物2aの合成において、反応条件を下表に示す反応条件に変更したこと以外は実施例1の化合物2aの合成と同様にして、実施例2~38および43~45の化合物2aを得た。得られた化合物2aの純度を実施例1と同様にして測定した。結果を下表に示す。
実施例1の化合物2aの合成において、反応条件を下表に示す反応条件に変更したこと以外は実施例1の化合物2aの合成と同様にして、実施例2~38および43~45の化合物2aを得た。得られた化合物2aの純度を実施例1と同様にして測定した。結果を下表に示す。
[実施例39]

実施例1の化合物2aの合成において、化合物1aに代えて化合物1dを用い、且つ、反応条件を下表に示す反応条件に変更したこと以外は実施例1の化合物2aの合成と同様にして、化合物2dを得た。なお、化合物1dは、上記参考例1において、PhDGに代えてフェニルトリグリコール(Tetrahedron 1988,44,5,p.1553-1558に記載の方法で合成した)を用いたこと以外は参考例1と同様にして合成した。化合物1d及び2dは5℃~30℃でいずれも液体であった。
得られた化合物2dの純度を、実施例1と同様に1H-NMR(CDCl3)のピーク比に基づき算出した。結果を下表に示す。
得られた化合物2dの純度を、実施例1と同様に1H-NMR(CDCl3)のピーク比に基づき算出した。結果を下表に示す。
[実施例40]

実施例1の化合物2aの合成において、化合物1aに代えて化合物1eを用い、且つ、反応条件を下表に示す反応条件に変更したこと以外は実施例1の化合物2aの合成と同様にして、化合物2eを得た。なお、化合物1eは、上記参考例1において、イソ酪酸クロリドに代えてシクロプロパンカルボニルクロリドを用いたこと以外は参考例1と同様にして合成した。化合物1e及び2eは5℃~30℃でいずれも液体であった。
得られた化合物2eの純度を、実施例1と同様に1H-NMR(CDCl3)のピーク比に基づき算出した。結果を下表に示す。
得られた化合物2eの純度を、実施例1と同様に1H-NMR(CDCl3)のピーク比に基づき算出した。結果を下表に示す。
[実施例41]

実施例1の化合物2aの合成において、化合物1aに代えて化合物1fを用い、且つ、反応条件を下表に示す反応条件に変更したこと以外は実施例1と同様にして、化合物2fを得た。なお、化合物1fは、上記参考例1において、イソ酪酸クロリドに代えて2-エチルヘキサノイルクロリドを用いたこと以外は参考例1と同様にして合成した。化合物1f及び2fは5℃~30℃でいずれも液体であった。
得られた化合物2fの純度を、実施例1と同様に1H-NMR(CDCl3)のピーク比に基づき算出した。結果を下表に示す。
得られた化合物2fの純度を、実施例1と同様に1H-NMR(CDCl3)のピーク比に基づき算出した。結果を下表に示す。
[実施例42]

実施例1の化合物2aの合成において、化合物1aに代えて化合物1gを用い、且つ、反応条件を下表に示す反応条件に変更したこと以外は実施例1の化合物2aの合成と同様にして、化合物2gを得た。なお、化合物1gは、上記参考例1において、イソ酪酸クロリドに代えてシクロヘキサンカルボニルクロリドを用いたこと以外は参考例1と同様にして合成した。化合物1g及び2gは5℃~30℃でいずれも液体であった。
得られた化合物2gの純度を、実施例1と同様に1H-NMR(CDCl3)のピーク比に基づき算出した。結果を下表に示す。
得られた化合物2gの純度を、実施例1と同様に1H-NMR(CDCl3)のピーク比に基づき算出した。結果を下表に示す。
[比較例1]
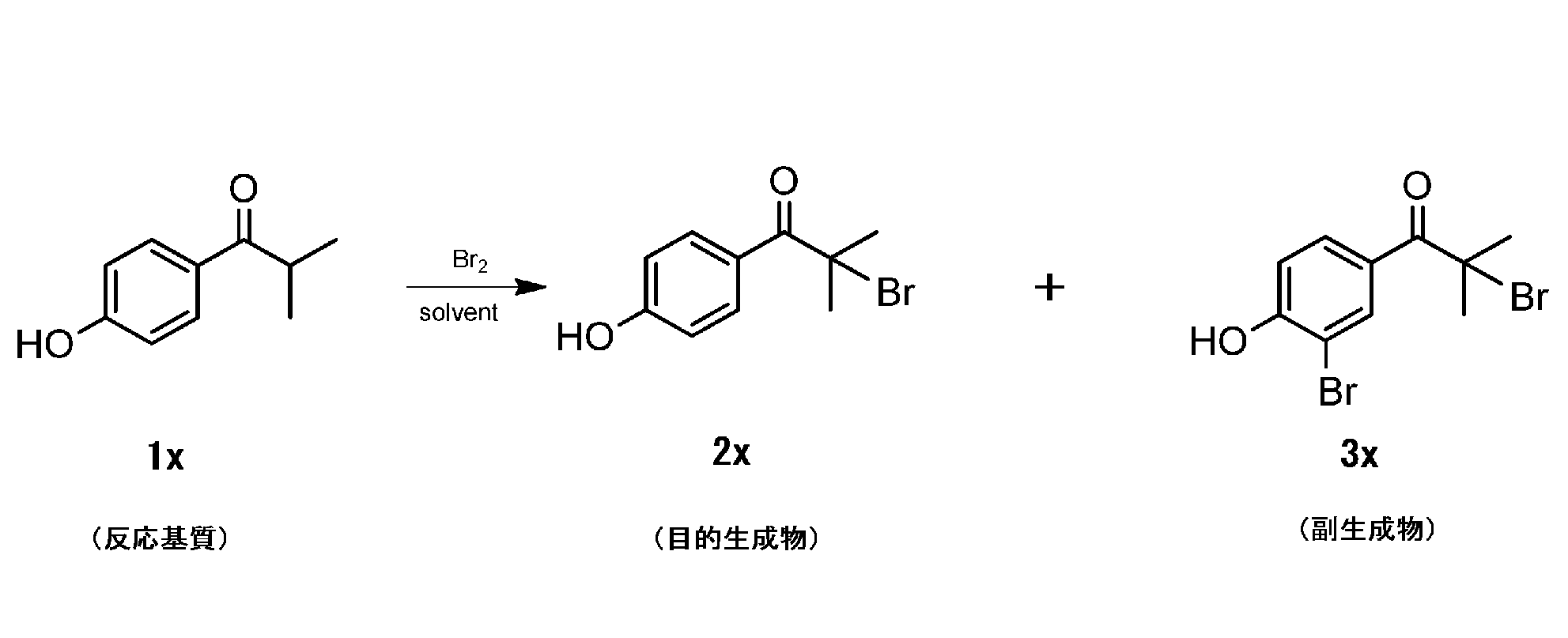
実施例1の化合物2aの合成において、化合物1aに代えて化合物1xを用い、且つ、反応条件を下表に示す反応条件に変更したこと以外は実施例1の化合物2aの合成と同様にして、化合物2xを得た。なお、化合物1xは、欧州特許第1073702号明細書に記載の方法と同様にして合成した。化合物1x及び2xは5℃~30℃でいずれも固体であった。化合物2xの融点は、56℃である。
得られた化合物2xの純度を、実施例1と同様に1H-NMR(CDCl3)のピーク比に基づき算出した。結果を下表に示す。
得られた化合物2xの純度を、実施例1と同様に1H-NMR(CDCl3)のピーク比に基づき算出した。結果を下表に示す。
[比較例2~15]
実施例1の化合物2aの合成において、反応条件を下表に示す反応条件に変更したこと以外は実施例1の化合物2aの合成と同様にして化合物2aを得た。得られた化合物2aの純度を実施例1と同様にして測定した。結果を下表に示す。






上記比較例1の結果から、反応基質として、一般式(1)におけるnが本発明で規定するよりも小さい化合物を用いた場合、本発明で規定する溶媒を用いても、臭素化部位の選択性が低く、得られる目的のα-ブロモアセトフェノン化合物の純度が低い結果となった。
また、上記比較例2~15の結果から、使用する溶媒が水酸基を有さないエーテル化合物及び有機酸エステル化合物以外であると、やはり臭素化部位の選択性が低く、得られる目的のα-ブロモアセトフェノン化合物の純度が低い結果となった。
また、上記比較例2~15の結果から、使用する溶媒が水酸基を有さないエーテル化合物及び有機酸エステル化合物以外であると、やはり臭素化部位の選択性が低く、得られる目的のα-ブロモアセトフェノン化合物の純度が低い結果となった。
これに対し、本発明で規定する要件のすべてを満たす実施例1~45では、臭素化反応における臭素化部位の選択性が大幅に高まり、再結晶化が困難なα-ブロモアセトフェノン化合物が高い反応純度で得られることがわかった。
Claims (9)
- 有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる少なくとも1種の化合物を含む溶媒中で、下記一般式(1)で表されるフェニル化合物と臭素とを反応させて該フェニル化合物を臭素化することを含む、下記一般式(2)で表されるα-ブロモアセトフェノン化合物の製造方法であって、得られる前記α-ブロモアセトフェノン化合物が5℃~30℃で液体である、α-ブロモアセトフェノン化合物の製造方法。


- 前記臭素化の反応温度が20℃以上である、請求項1に記載のα-ブロモアセトフェノン化合物の製造方法。
- 前記有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる前記溶媒が、酢酸メチル、酢酸エチル、酢酸ブチル、酢酸プロピル、酢酸イソプロピル、及び1,4-ジオキサンから選ばれる1種又は2種以上である、請求項1又は2に記載のα-ブロモアセトフェノン化合物の製造方法。
- 前記臭素化の反応温度が40℃以上である、請求項1~3のいずれか1項に記載のα-ブロモアセトフェノン化合物の製造方法。
- 前記臭素化に用いる溶媒の総量中、前記有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる前記溶媒の総含有量が50~100質量%である、請求項1~4のいずれか1項に記載のα-ブロモアセトフェノン化合物の製造方法。
- 前記臭素化の反応液中の前記有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる前記溶媒の量が、該反応液中に混合された前記フェニル化合物1g当たり0.5~60mLである、請求項1~5のいずれか1項に記載のα-ブロモアセトフェノン化合物の製造方法。
- 前記臭素化は、前記臭素もしくは、前記臭素と前記有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる前記溶媒とを含む混合液を、前記フェニル化合物と前記有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる前記溶媒とを含む混合液中に滴下して行うか、又は、
前記臭素と前記有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる前記溶媒とを含む混合液を、前記フェニル化合物中もしくは、前記フェニル化合物と前記有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる前記溶媒とを含む混合液中に滴下して行う、請求項1~6のいずれか1項に記載のα-ブロモアセトフェノン化合物の製造方法。 - 前記臭素化は、前記臭素と前記有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる前記溶媒とを含む混合液を、前記フェニル化合物と前記有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる前記溶媒とを含む前記混合液中に滴下して行う、請求項7に記載のα-ブロモアセトフェノン化合物の製造方法。
- 前記臭素と前記有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる前記溶媒とを含む前記混合液中の当該有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる当該溶媒の量Aと、前記フェニル化合物と前記有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる前記溶媒とを含む前記混合液中の当該有機酸エステル化合物及び水酸基を有さないエーテル化合物から選ばれる当該溶媒の量Bの比率が、体積比で、B:A=90:10~30:70である、請求項8に記載のα-ブロモアセトフェノン化合物の製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201580020362.XA CN106255676B (zh) | 2014-05-15 | 2015-04-17 | α-溴代苯乙酮化合物的制造方法 |
| JP2016519179A JP6204583B2 (ja) | 2014-05-15 | 2015-04-17 | α−ブロモアセトフェノン化合物の製造方法 |
| US15/292,151 US10081589B2 (en) | 2014-05-15 | 2016-10-13 | Method for manufacturing α-bromoacetophenone compound |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-101626 | 2014-05-15 | ||
| JP2014101626 | 2014-05-15 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/292,151 Continuation US10081589B2 (en) | 2014-05-15 | 2016-10-13 | Method for manufacturing α-bromoacetophenone compound |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015174214A1 true WO2015174214A1 (ja) | 2015-11-19 |
Family
ID=54479758
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/061873 WO2015174214A1 (ja) | 2014-05-15 | 2015-04-17 | α-ブロモアセトフェノン化合物の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10081589B2 (ja) |
| JP (1) | JP6204583B2 (ja) |
| CN (1) | CN106255676B (ja) |
| TW (1) | TW201612145A (ja) |
| WO (1) | WO2015174214A1 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113801005B (zh) * | 2021-06-16 | 2023-06-13 | 浙江师范大学 | 一种α-溴代苯乙酮类化合物的制备方法 |
| CN114573534A (zh) * | 2022-03-30 | 2022-06-03 | 八叶草健康产业研究院(厦门)有限公司 | 一种5-溴苯并呋喃酮的制备方法 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59167546A (ja) * | 1983-02-18 | 1984-09-21 | ヂバ−ガイギ−・アクチエンゲゼルシヤフト | 光硬化性着色組成物のための光開始剤 |
| JPS62502403A (ja) * | 1985-04-03 | 1987-09-17 | チバ―ガイギー アクチェンゲゼルシャフト | 新規ケトン誘導体 |
| JPH11511753A (ja) * | 1996-05-31 | 1999-10-12 | メルク エンド カンパニー インコーポレーテッド | Cox―2阻害剤として有用なフェニルヘテロ環の製造方法 |
| WO2003027059A1 (fr) * | 2001-09-18 | 2003-04-03 | Ishihara Sangyo Kaisha, Ltd. | Derives d'amides acides, procedes de production et agent antiparasitaire contenant ces derives |
| JP2012001451A (ja) * | 2010-06-14 | 2012-01-05 | Fujifilm Corp | 新規化合物 |
| WO2015045857A1 (ja) * | 2013-09-30 | 2015-04-02 | 富士フイルム株式会社 | α-ハロゲノアセトフェノン化合物の製造方法、及びα-ブロモアセトフェノン化合物 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2640358A1 (de) * | 1976-09-08 | 1978-03-16 | Hoechst Ag | Thiazolidinderivate und verfahren zu ihrer herstellung |
| JP5136035B2 (ja) * | 2007-12-11 | 2013-02-06 | コニカミノルタホールディングス株式会社 | インクジェットインク及びインクジェット記録方法 |
| CN101811951A (zh) * | 2010-05-07 | 2010-08-25 | 甘肃金盾化工有限责任公司 | 一种2-羟基-1-{4-(2-羟乙氧基)苯基}-2-甲基-1-丙酮的制备方法 |
-
2015
- 2015-04-17 WO PCT/JP2015/061873 patent/WO2015174214A1/ja active Application Filing
- 2015-04-17 JP JP2016519179A patent/JP6204583B2/ja not_active Expired - Fee Related
- 2015-04-17 CN CN201580020362.XA patent/CN106255676B/zh not_active Expired - Fee Related
- 2015-04-29 TW TW104113625A patent/TW201612145A/zh unknown
-
2016
- 2016-10-13 US US15/292,151 patent/US10081589B2/en not_active Expired - Fee Related
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS59167546A (ja) * | 1983-02-18 | 1984-09-21 | ヂバ−ガイギ−・アクチエンゲゼルシヤフト | 光硬化性着色組成物のための光開始剤 |
| JPS62502403A (ja) * | 1985-04-03 | 1987-09-17 | チバ―ガイギー アクチェンゲゼルシャフト | 新規ケトン誘導体 |
| JPH11511753A (ja) * | 1996-05-31 | 1999-10-12 | メルク エンド カンパニー インコーポレーテッド | Cox―2阻害剤として有用なフェニルヘテロ環の製造方法 |
| WO2003027059A1 (fr) * | 2001-09-18 | 2003-04-03 | Ishihara Sangyo Kaisha, Ltd. | Derives d'amides acides, procedes de production et agent antiparasitaire contenant ces derives |
| JP2012001451A (ja) * | 2010-06-14 | 2012-01-05 | Fujifilm Corp | 新規化合物 |
| WO2015045857A1 (ja) * | 2013-09-30 | 2015-04-02 | 富士フイルム株式会社 | α-ハロゲノアセトフェノン化合物の製造方法、及びα-ブロモアセトフェノン化合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN106255676B (zh) | 2019-04-30 |
| US20170029356A1 (en) | 2017-02-02 |
| JPWO2015174214A1 (ja) | 2017-04-20 |
| US10081589B2 (en) | 2018-09-25 |
| TW201612145A (en) | 2016-04-01 |
| JP6204583B2 (ja) | 2017-09-27 |
| CN106255676A (zh) | 2016-12-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2016098699A1 (ja) | カルボン酸エステルの製造方法 | |
| JP6204583B2 (ja) | α−ブロモアセトフェノン化合物の製造方法 | |
| JP5309318B2 (ja) | エステル、カルボン酸及びアミドの製造方法 | |
| WO2015186787A1 (ja) | カルボン酸無水物の製造方法およびカルボン酸エステルの製造方法 | |
| JP2016011292A (ja) | 混合酸無水物の製造方法 | |
| JP6089110B2 (ja) | α−ハロゲノアセトフェノン化合物の製造方法、及びα−ブロモアセトフェノン化合物 | |
| US10562834B2 (en) | Process for preparing substituted crotonic acids | |
| WO2006109570A1 (ja) | 2-イソプロペニル-5-メチル-4-ヘキセン-1-イル 3-メチル-2-ブテノアートの製造方法 | |
| WO2015159672A1 (ja) | ハロゲン化合物の製造方法、カリウム塩の製造方法、及びカリウム塩 | |
| JP5205971B2 (ja) | テトラヒドロピラン化合物の製造方法 | |
| JP2007056024A (ja) | ノルボルネン誘導体の製造方法 | |
| JP2010116363A (ja) | 2−アリール−2,2−ジフルオロ酢酸エステルの製造方法 | |
| JP2006312644A (ja) | β−ケトニトリル類の製法 | |
| JP2008105955A (ja) | (メタ)アクリル酸エステルの製造方法 | |
| JP2008120759A (ja) | エーテル基を有するβ−ジケトン化合物の製造法 | |
| JP4710698B2 (ja) | シリルエーテル基を有するβ−ジケトン化合物の製造法 | |
| JP6766459B2 (ja) | カルボン酸エステルの製造方法 | |
| JP4800933B2 (ja) | シクロプロパンモノアセタール誘導体の製造方法およびその中間体 | |
| JP4483183B2 (ja) | 6,7−ジヒドロキシクマリン−3−カルボン酸誘導体の製法及びその中間体 | |
| JPH07252183A (ja) | フェノール誘導体の製造方法 | |
| JP2024045205A (ja) | 2-ヒドロキシ-2-(パーフルオロアルキル)マロン酸エステル誘導体の製造方法、並びに2-(トリメチルシリルオキシ)-2-(パーフルオロアルキル)マロン酸エステル誘導体及び5-ヒドロキシ-5-(パーフルオロアルキル)ピリミジン-2,4,6(1h,3h,5h)-トリオンとそれらの製造方法 | |
| JP2019034903A (ja) | ビス(ジフルオロメチル)亜鉛反応剤 | |
| JP4154567B2 (ja) | 4−ジフルオロメトキシ−3−ヒドロキシベンズアルデヒドの製造方法 | |
| JP2006282611A (ja) | シリルエーテル基を有するβ−ジケトン化合物の製法 | |
| JP2008266183A (ja) | 3,4−ジアルコキシ−2−(アルキルチオ)フェノールの製法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15792318 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2016519179 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15792318 Country of ref document: EP Kind code of ref document: A1 |