WO2015080184A1 - 樹脂組成物およびその用途 - Google Patents
樹脂組成物およびその用途 Download PDFInfo
- Publication number
- WO2015080184A1 WO2015080184A1 PCT/JP2014/081325 JP2014081325W WO2015080184A1 WO 2015080184 A1 WO2015080184 A1 WO 2015080184A1 JP 2014081325 W JP2014081325 W JP 2014081325W WO 2015080184 A1 WO2015080184 A1 WO 2015080184A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin
- resin composition
- copper salt
- general formula
- neutralizing
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/49—Phosphorus-containing compounds
- C08K5/51—Phosphorus bound to oxygen
- C08K5/53—Phosphorus bound to oxygen bound to oxygen and to carbon only
- C08K5/5317—Phosphonic compounds, e.g. R—P(:O)(OR')2
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/49—Phosphorus-containing compounds
- C08K5/51—Phosphorus bound to oxygen
- C08K5/52—Phosphorus bound to oxygen only
- C08K5/521—Esters of phosphoric acids, e.g. of H3PO4
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
Definitions
- the present invention relates to a resin composition and its use, and more particularly to a resin composition comprising a near infrared absorber, a neutralizing dispersant, and a resin, and its use.
- laminated glass is used in various applications such as vehicles such as automobiles, buildings, and solar cells.
- an interlayer film for laminated glass a polyvinyl butyral resin film, an ionomer resin film, and the like are known.
- the sun rays include ultraviolet rays, infrared rays and the like in addition to visible rays.
- infrared rays infrared rays having a wavelength close to visible light are called near infrared rays.
- Near-infrared rays are also called heat rays and are one of the causes of temperature rise inside vehicles and buildings.
- Patent Document 1 a copper salt composition containing a phosphonic acid copper salt, a polysiloxane component, a plasticizer, and a dispersant is known (see, for example, Patent Document 1).
- Patent Document 1 provides an infrared absorption film in which the resin composition containing the copper salt composition and the resin is excellent in visible light transmission and stability even when exposed to high temperatures. It is disclosed that it is possible.
- the present invention has been made in view of the above prior art, and an object of the present invention is to provide a resin composition containing a near-infrared absorber having excellent light resistance.
- the resin composition of the present invention is a resin composition comprising a near-infrared absorber, a neutralizing dispersant, and a resin.
- the near-infrared absorber includes at least a copper salt and the following general formula (1).
- the neutralizing dispersant is neutralized with an alkali metal salt at least one phosphate ester compound selected from phosphate monoesters and phosphate diesters. It is a neutralizing dispersant obtained by doing.
- R 1 is a monovalent group represented by —CH 2 CH 2 —R 11
- R 11 is a hydrogen atom, an alkyl group having 1 to 20 carbon atoms, or 1 carbon atom. Represents -20 fluorinated alkyl groups.
- the alkali metal salt is preferably at least one salt selected from a sodium salt, a potassium salt, and a cesium salt, and more preferably a sodium salt.
- the phosphoric acid monoester is a phosphoric acid monoester represented by the following general formula (2)
- the phosphoric acid diester is a phosphoric acid diester represented by the following general formula (3).
- R 2 , R 3 and R 4 are monovalent groups represented by — (CH 2 CH 2 O) n R 12 , and n is from 2 to 65 R 12 is an integer, and R 12 represents an alkyl group having 6 to 35 carbon atoms or an alkylphenyl group having 6 to 35 carbon atoms. However, R 2 , R 3 and R 4 may be the same or different.
- the resin is at least one selected from polyvinyl acetal resin, ethylene-vinyl acetate copolymer, (meth) acrylic acid resin, polyester resin, polyurethane resin, vinyl chloride resin, polyolefin resin, polycarbonate resin, and norbornene resin.
- a resin is preferable, and a polyvinyl butyral resin or an ethylene-vinyl acetate copolymer is more preferable. It is preferable to contain 0.05 to 50 parts by mass of a near infrared absorber per 100 parts by mass of the resin.
- the interlayer film for laminated glass of the present invention is formed from the resin composition.
- the laminated glass of the present invention has the interlayer film for laminated glass.
- the resin composition containing the near infrared absorber of the present invention is excellent in light resistance.
- the resin composition of the present invention is a resin composition comprising a near infrared absorber, a neutralizing dispersant, and a resin, and the near infrared absorber is represented by at least a copper salt and the general formula (1). It is obtained by reacting with a phosphonic acid compound, and the neutralizing dispersant neutralizes at least one phosphoric ester compound selected from phosphoric monoester and phosphoric diester with an alkali metal salt. It is a neutralizing dispersant obtained.
- the resin composition of the present invention is also referred to as a copper salt fine particle dispersed resin.
- the neutralizing dispersant used in the present invention is a neutralizing dispersant obtained by neutralizing at least one phosphate ester compound selected from phosphate monoester and phosphate diester with an alkali metal salt. .
- the neutralizing dispersant used in the present invention may be one kind or two or more kinds.
- alkali metal salt examples include alkali metal oxides, hydroxides, halides, hydrides, and organic acid salts such as hydrogen carbonate, carbonate, nitrate, hydrogen sulfate, sulfate, formate, and acetate.
- alkali metal hydroxides, bicarbonates and carbonates are preferred.
- the metal species constituting the alkali metal salt include lithium, sodium, potassium, rubidium, cesium, and francium.
- Sodium, potassium, and cesium are preferable, and sodium is more preferable.
- the alkali metal salt is preferably at least one salt selected from a sodium salt, a potassium salt, and a cesium salt, and more preferably a sodium salt.
- alkali metal salt 1 type may be used or 2 or more types may be used.
- the at least one phosphate ester compound selected from the phosphate monoester and phosphate diester is not particularly limited, but the phosphate monoester is a phosphorus represented by the following general formula (2). It is preferable that it is an acid monoester, and it is preferable that the said phosphoric acid diester is a phosphoric acid diester represented by following General formula (3).
- R 2 , R 3 and R 4 are monovalent groups represented by — (CH 2 CH 2 O) n R 12 , and n is from 2 to 65 R 12 is an integer, and R 12 represents an alkyl group having 6 to 35 carbon atoms or an alkylphenyl group having 6 to 35 carbon atoms. However, R 2 , R 3 and R 4 may be the same or different.
- n is an integer of 2 to 65, preferably an integer of 2 to 35. When n is less than 2, transparency may be insufficient when a laminated glass or the like is produced. Moreover, when n exceeds the said range, the quantity of a phosphoric acid ester compound required in order to obtain the laminated glass etc. which have sufficient transparency will increase, and there exists a tendency which becomes a cause of high cost.
- R 12 is an alkyl group having 6 to 35 carbon atoms or an alkylphenyl group having 6 to 35 carbon atoms, preferably an alkyl group having 6 to 25 carbon atoms or an alkylphenyl group having 6 to 25 carbon atoms, An alkyl group having 6 to 25 carbon atoms is preferable, and an alkyl group having 10 to 20 carbon atoms is more preferable. If R 12 is a group having less than 6 carbon atoms, transparency may be insufficient when a laminated glass or the like is produced. Further, if R 12 is a group having more than 35 carbon atoms, the amount of the phosphoric acid ester compound necessary for obtaining a laminated glass having sufficient transparency tends to increase, resulting in high costs. .
- the neutralizing dispersant it is preferable that at least one of the phosphoric acid monoester represented by the general formula (2) and the phosphoric acid diester represented by the general formula (3) is used. It is more preferable to use both the phosphoric acid monoester represented by the general formula (2) and the phosphoric acid diester represented by the general formula (3).
- the phosphoric acid monoester represented by the general formula (2) and the phosphoric acid diester represented by the general formula (3) are used, it tends to be excellent in transparency and heat resistance of laminated glass or the like.
- the phosphoric acid monoester represented by the general formula (2) When both the phosphoric acid monoester represented by the general formula (2) and the phosphoric acid diester represented by the general formula (3) are used, the phosphoric acid monoester represented by the general formula (2)
- the ratio of the phosphoric acid diester represented by the general formula (3) is not particularly limited, but is usually 10:90 to 90:10 in a molar ratio (general formula (2): general formula (3)). is there.
- phosphoric acid monoester represented by the said General formula (2) it may be used individually by 1 type, or 2 or more types may be used, As phosphoric acid diester represented by the said General formula (3), These may be used alone or in combination of two or more.
- a commercially available phosphate ester Compounds such as DDP-2, DDP-4, DDP-6, DDP-8, DDP-10, TDP-2, TDP-6, TDP-8, TDP-10, TLP-4, TCP-5, TOP-0V (Prisurf A212C, Plysurf A215C, Plysurf AL12H, Plysurf AL, Plysurf A208F, Plysurf A208N, Plysurf A208B, Plysurf A219B, Plysurf A210D, Plysurf DBS (manufactured by Daiichi Kogyo Seiyaku Co., Ltd.) or the like can also be used.
- the commercially available phosphate ester compounds include components other than the phosphate monoester represented by the general formula (2) and the phosphate diester represented by the general formula (3), for example, polyoxyalkylene mono
- alkyl ethers and phosphoric acid triesters represented by the following general formula (4) are contained. In the present invention, even if such a polyoxyalkylene monoalkyl ether or a phosphate triester is present in the phosphate compound, it can be used without any problem.
- the total amount of phosphoric acid monoester represented by general formula (2) and phosphoric diester represented by general formula (3) is 100 parts by mass.
- the polyoxyalkylene monoalkyl ether is preferably 1 to 300 parts by mass.
- polyoxyalkylene monoalkyl ether As polyoxyalkylene monoalkyl ether, HO— (CH 2 CH 2 O) n R 12 , (n is an integer of 2 to 65, preferably 2 to 35, and R 12 is a carbon number of 6 -35, preferably 6-25 alkyl groups or C6-C35, preferably 6-25 alkylphenyl groups).
- the polyoxyalkylene monoalkyl ether is considered to be derived from a raw material used in producing a phosphate ester.
- the phosphoric acid triester represented by General formula (4) is contained, the phosphoric acid monoester represented by General formula (2) and the phosphoric acid represented by the said General formula (3)
- the phosphoric acid triester represented by the general formula (4) is preferably 1 to 300 parts by mass with respect to 100 parts by mass in total of the diesters.
- R 5 , R 6 and R 7 are monovalent groups represented by — (CH 2 CH 2 O) n R 12 , and n is an integer of 2 to 65, preferably R 12 represents an integer of 2 to 35, and R 12 represents an alkyl group having 6 to 35 carbon atoms, preferably 6 to 25 carbon atoms or an alkylphenyl group having 6 to 35 carbon atoms, preferably 6 to 25 carbon atoms.
- R 5 , R 6 and R 7 may be the same or different.
- n and R 12 in the polyoxyalkylene monoalkyl ether and general formula (4) are preferably the same as n and R 12 in general formulas (2) and (3).
- At least one phosphate ester compound selected from the phosphate monoester and phosphate diester can be neutralized with an alkali metal salt.
- the method of preparing the aqueous solution of a phosphate ester compound and the aqueous solution of an alkali metal salt, respectively, adding the aqueous solution of an alkali metal salt in the aqueous solution of a phosphate ester compound, and neutralizing is mentioned.
- the solvent used for neutralization include alcohols such as methanol, ethanol, and propanol, and mixed solvents of these alcohols and water, in addition to water.
- the first neutralization point where one of the two hydroxyl groups of the phosphoric acid monoester is neutralized, and the two are neutralized. There will be a second neutralization point.
- the neutralizing dispersant used in the present invention is preferably neutralized to the second neutralization point from the viewpoint of further reducing the addition amount of the neutralizing dispersant.
- the solution is neutralized to the first neutralization point by neutralizing to pH 5.0 to 6.9, and neutralized to pH 8.5 to 11.5. By doing so, it is neutralized to the second neutralization point.
- the neutralization is preferably carried out at least until the pH is 5.3 to 6.6, which is the first neutralization point, and the pH which is the second neutralization point is 9.0 to 11. More preferably, it is carried out until it becomes zero.
- the neutralizing dispersant may be obtained as a solid such as wax by removing the solvent such as water after neutralization, and dissolved in the solution without removing the solvent such as water. Alternatively, it may be obtained in a dispersed state.
- At least one phosphate ester compound selected from the phosphate monoester represented by the general formula (2) and the phosphate diester represented by the general formula (3) is neutralized with an alkali metal salt. What is marketed in the state obtained may be used as the neutralizing dispersant.
- Commercially available neutralizing dispersants include DLP-10 (sodium salt of phosphate ester compound), DOP-8NV (sodium salt of phosphate ester compound) (both manufactured by Nikko Chemicals Co., Ltd.) and the like.
- the neutralized dispersant is further neutralized. It may be used as
- the resin composition of the present invention contains the neutralizing dispersant, coloring and a decrease in visible light transmittance due to irradiation with light such as sunlight are suppressed.
- the near-infrared absorber used in the present invention is a near-infrared absorber obtained by reacting at least a copper salt with a phosphonic acid compound represented by the following general formula (1).
- R 1 is a monovalent group represented by —CH 2 CH 2 —R 11
- R 11 is a hydrogen atom, an alkyl group having 1 to 20 carbon atoms, or 1 carbon atom. Represents -20 fluorinated alkyl groups.
- the near-infrared absorber used in the present invention may be obtained by reacting only a copper salt and a phosphonic acid compound represented by the general formula (1) as raw materials.
- the copper salt and the general formula In addition to the phosphonic acid compound represented by 1), other components may be used as raw materials, and these may be obtained by reaction.
- the near-infrared absorber is a copper salt represented by the following general formula (1 ′), and the general formula (1)
- the near-infrared absorber contains a copper salt represented by the following general formula (1 ′).
- R 1 is a monovalent group represented by —CH 2 CH 2 —R 11
- R 11 is a hydrogen atom, an alkyl group having 1 to 20 carbon atoms, or a carbon number 1 to 20 fluorinated alkyl groups are shown.
- R 11 in the general formulas (1) and (1 ′) is preferably a hydrogen atom or an alkyl group having 1 to 20 carbon atoms.
- R 11 hydrogen atom, methyl group, ethyl group, propyl group, butyl group, pentyl group, hexyl group, heptyl group, octyl group, nonyl group, decyl group, undecyl group, dodecyl group, tridecyl group Tetradecyl group, pentadecyl group, hexadecyl group, heptadecyl group, octadecyl group and the like are preferable.
- a phosphonic acid compound represented by General formula (1) it may be used individually by 1 type, or 2 or more types may be used.
- the method for producing the near infrared absorber used in the present invention is not particularly limited, and for example, it can be produced by the following method.
- a copper salt and a phosphonic acid compound represented by the general formula (1) are mixed and reacted in a solvent to obtain a reaction mixture (hereinafter also referred to as a reaction step). ), A method having a step (hereinafter also referred to as a solvent removal step) of obtaining a near-infrared absorber as fine particles comprising a phosphonic acid copper salt by removing the solvent in the reaction mixture.
- the phosphonic acid compound represented by the general formula (1) those in which R 11 is a hydrogen atom or an alkyl group having 1 to 20 carbon atoms as described above are preferable.
- the phosphonic acid compound represented by the general formula (1) include ethylphosphonic acid, propylphosphonic acid, butylphosphonic acid, pentylphosphonic acid, hexylphosphonic acid, heptylphosphonic acid, octylphosphonic acid, nonylphosphonic acid, and decylphosphonic acid.
- alkylphosphonic acids such as acid, undecylphosphonic acid, dodecylphosphonic acid, tridecylphosphonic acid, tetradecylphosphonic acid, pentadecylphosphonic acid, hexadecylphosphonic acid, heptadecylphosphonic acid, and octadecylphosphonic acid.
- the copper salt a copper salt capable of supplying divalent copper ions is usually used.
- the copper salt may be a copper salt other than the phosphonic acid copper salt represented by the general formula (1 ').
- Examples of the copper salt include copper acetate anhydride, copper formate anhydride, copper stearate anhydride, copper benzoate anhydride, copper ethyl acetoacetate anhydride, copper pyrophosphate anhydride, copper naphthenate anhydride, copper citrate Copper salt of organic acid such as anhydride, hydrate or hydrate of copper salt of organic acid; copper salt of inorganic acid such as copper oxide, copper chloride, copper sulfate, copper nitrate, basic copper carbonate, inorganic Hydrates or hydrates of acid copper salts; copper hydroxide.
- you may use individually by 1 type, or may use 2 or more types.
- copper salt copper acetate anhydride and copper acetate monohydrate are preferably used in terms of solubility and removal of by-products.
- a dispersant When producing a near infrared absorber, a dispersant may be used in the reaction step.
- the dispersant include at least one phosphate ester compound selected from a phosphate monoester represented by the general formula (2) and a phosphate diester represented by the general formula (3).
- the neutralization dispersing agent which the resin composition of this invention contains.
- the phosphoric acid monoester represented by the general formula (2) which is a preferred embodiment of at least one phosphoric acid ester compound selected from phosphoric acid monoesters and phosphoric acid diesters used in obtaining a neutralizing dispersant.
- the ester and the phosphoric acid diester represented by the general formula (3) are also preferable as a dispersant used in producing a near-infrared absorber.
- the same kind may be used for the phosphate ester used when obtaining a neutralization dispersing agent, and the phosphate ester used when obtaining a near-infrared absorber, and another thing may be used.
- the dispersant is at least one phosphate ester compound selected from a phosphate monoester represented by the general formula (2) and a phosphate diester represented by the following general formula (3) Further, 5 to 100 parts by mass are preferably used per 100 parts by mass of the copper salt, and more preferably 10 to 50 parts by mass.
- the solvent examples include alcohols such as methanol, ethanol, isopropyl alcohol, n-butyl alcohol, methanol / ethanol mixed solvent, isopropyl alcohol / ethanol mixed solvent, tetrahydrofuran (THF), dimethylformamide (DMF), water, and the like. From the viewpoint of performing the reaction satisfactorily, methanol, ethanol, a methanol / ethanol mixed solvent, and an isopropyl alcohol / ethanol mixed solvent are preferable.
- the reaction step is preferably performed at a temperature of 0 to 80 ° C., more preferably 10 to 60 ° C., preferably 0.5 to 80 hours, more preferably 1 to 50 hours.
- the phosphonic acid compound represented by the general formula (1) reacts with the copper salt, and fine phosphonic acid copper salt that does not dissolve in the solvent is generated by the reaction.
- At least one phosphate compound selected from the phosphoric acid monoester represented by the general formula (2) and the phosphoric acid diester represented by the following general formula (3) acts as a good dispersant during the reaction. Therefore, the phosphonic acid copper salt can maintain high dispersibility and suppress aggregation.
- the reaction step not only the reaction between the phosphonic acid compound represented by the general formula (1) and the copper salt, but also, for example, the phosphoric acid monoester represented by the general formula (2) and the following general formula (3) At least one phosphate ester compound selected from the phosphoric acid diesters represented by formula (1) and a part of the copper salt may react. Further, a part of the raw material may remain without reacting.
- fine particles comprising a phosphonic acid copper salt are usually obtained by removing at least a part of the solvent from the reaction mixture.
- the solvent removal step at least a part of the solvent is removed from the reaction mixture.
- the liquid components in the reaction mixture may be removed together.
- the solvent removal step at least a part of the solvent is usually removed by heating the reaction mixture, and the heating condition is usually room temperature to 70 ° C., preferably 40 to 60 ° C.
- the solvent removal step may be performed under normal pressure or under reduced pressure. When the solvent removal step is performed under reduced pressure, heating may not be performed or the heating temperature may be low.
- the reaction mixture is allowed to stand to precipitate fine particles composed of copper phosphonate, and the supernatant (solvent) is removed, or the reaction mixture is centrifuged.
- fine-particles which consist of a phosphonic acid copper salt, and removing a supernatant liquid (solvent) is mentioned.
- a step of removing the dispersion medium after the near infrared absorber is dispersed in the dispersion medium may be provided.
- the near-infrared absorber fine particles made of a phosphonic acid copper salt having an average particle diameter of 1 to 1000 nm are usually used.
- the average particle size is more preferably 5 to 300 nm in order to ensure dispersibility in the monomer and transparency of the resin composition.
- a resin is used.
- the resin used in the present invention is not particularly limited as long as it can disperse the above-described near-infrared absorber.
- the following resins can be used.
- the resin used in the present invention is selected from polyvinyl acetal resin, ethylene-vinyl acetate copolymer, (meth) acrylic acid resin, polyester resin, polyurethane resin, vinyl chloride resin, polyolefin resin, polycarbonate resin, and norbornene resin. It is preferable that at least one type of resin can disperse the near-infrared absorber well and is excellent in visible light transmittance.
- the resin used in the present invention is more preferably at least one resin selected from polyvinyl acetal resin and ethylene-vinyl acetate copolymer, polyvinyl butyral resin (PVB), and ethylene-vinyl acetate copolymer Particularly preferred is at least one resin selected from a coalescence, and most preferred is a polyvinyl butyral resin or an ethylene-vinyl acetate copolymer.
- the polyvinyl acetal resin is used, the dispersibility of the above-mentioned near-infrared absorber is excellent.
- the resin of the present invention is excellent in adhesion to glass or the like.
- the composition is flexible and deformation of the near-infrared absorber due to a change in temperature hardly occurs.
- polyvinyl acetal resin it is preferable to use polyvinyl butyral resin (PVB) from the viewpoints of glass adhesion, dispersibility, transparency, heat resistance, light resistance, and the like.
- the polyvinyl acetal resin may be a blend of two or more kinds depending on the required physical properties, or may be a polyvinyl acetal resin obtained by acetalizing a combination of aldehydes during acetalization.
- the molecular weight, molecular weight distribution, and degree of acetalization of the polyvinyl acetal resin are not particularly limited, but the degree of acetalization is generally 40 to 85%, with a preferred lower limit being 60% and an upper limit being 75%.
- the polyvinyl acetal resin can be obtained by acetalizing a polyvinyl alcohol resin with an aldehyde.
- the polyvinyl alcohol resin is generally obtained by saponifying polyvinyl acetate, and a polyvinyl alcohol resin having a saponification degree of 80 to 99.8 mol% is generally used.
- the preferable lower limit of the viscosity average polymerization degree of the polyvinyl alcohol resin is 200, and the upper limit is 3000. If it is less than 200, the penetration resistance of the resulting laminated glass may be lowered. When it exceeds 3000, the moldability of the resin composition may be deteriorated, and the rigidity of the resin composition is excessively increased, resulting in poor processability.
- a more preferred lower limit is 500 and an upper limit is 2200.
- the viscosity average degree of polymerization and the degree of saponification of the polyvinyl alcohol resin can be measured based on, for example, JISK 6726 “Testing method for polyvinyl alcohol”.
- the aldehyde is not particularly limited, and examples thereof include aldehydes having 1 to 10 carbon atoms, and more specifically, for example, n-butyraldehyde, isobutyraldehyde, n-valeraldehyde, 2-ethylbutylartaldehyde. N-hexylaldehyde, n-octylaldehyde, n-nonylaldehyde, n-decylaldehyde, formaldehyde, acetaldehyde, benzaldehyde and the like. Of these, n-butyraldehyde, n-hexylaldehyde, n-valeraldehyde and the like are preferable. More preferred is butyraldehyde having 4 carbon atoms.
- an ethylene-vinyl acetate copolymer is preferable from the viewpoints of excellent dispersibility of the above-mentioned near-infrared absorber and glass adhesion, dispersibility, transparency, heat resistance, light resistance, and the like.
- the resin composition of the present invention is a resin composition comprising a near-infrared absorber, a neutralizing dispersant, and a resin.
- the resin composition of the present invention is excellent in light resistance.
- the resin composition of the present invention is not particularly limited as long as it is a composition comprising a near-infrared absorber, a neutralizing dispersant, and a resin as described above.
- Examples of the method for producing the resin composition of the present invention include, for example, a solvent such as toluene, ethanol / toluene mixed solvent, methanol / toluene mixed solvent, methylene chloride, chloroform, resin, near infrared absorber dispersion, neutralizing dispersant. And a method of dissolving the resin by stirring, ultrasonic irradiation or the like to obtain a dispersion and removing the solvent from the dispersion.
- the resin is dissolved by stirring, ultrasonic irradiation, etc.
- a method of removing the solvent from the dispersion by adding a near-infrared absorber dispersion and a neutralizing dispersant to the solution in which the resin is dissolved, followed by stirring, ultrasonic irradiation, etc. to obtain a dispersion.
- a solvent such as toluene, ethanol / toluene mixed solvent, methanol / toluene mixed solvent, methylene chloride, chloroform, etc.
- the dispersion is separately prepared by, for example, adding the dispersion to the solution, or adding the solution to the dispersion, if necessary, stirring and irradiating with ultrasonic waves, mixing both, The method of removing is mentioned.
- the said near-infrared absorber dispersion liquid can be prepared by disperse
- the resin composition of the present invention preferably contains 0.05 to 50 parts by mass, more preferably 0.1 to 25 parts by mass of a near infrared absorber per 100 parts by mass of the resin. If the amount is less than 0.05 parts by mass, sufficient near infrared absorption characteristics may not be obtained. If the amount is more than 50 parts by mass, the transparency and adhesiveness of the resin may be significantly reduced.
- the resin composition of the present invention preferably contains 0.01 to 10 parts by mass, more preferably 0.1 to 5.0 parts by mass of the neutralizing dispersant per 100 parts by mass of the resin. Within the said range, since transparency of resin and adhesiveness with glass can be kept favorable, it is preferable.
- the resin composition of the present invention has excellent near-infrared absorptivity, and is suitable as an intermediate film for structural materials such as laminated glass because coloring when irradiated with light and reduction in visible light transmittance are suppressed. Can be used.
- additives may be contained in the resin composition of the present invention.
- the additives include plasticizers, other dispersants, crosslinking agents, chelating agents, antioxidants, ultraviolet absorbers, light stabilizers, color tone correction agents, and the like. These additives may be added when the resin composition of the present invention is produced, or may be added when the above-described near infrared absorber, neutralizing dispersant, and resin are produced.
- the resin composition of the present invention is usually used for applications where it is desired to absorb near infrared rays.
- the resin film formed from the resin composition of the present invention has excellent near-infrared absorption ability and excellent light resistance, and therefore can be suitably used as an intermediate film for structural materials such as an intermediate film for laminated glass.
- the laminated glass of the present invention has the interlayer film for laminated glass.
- glass which comprises the laminated glass of this invention A conventionally well-known thing can be used.
- the phosphoric acid ester compound (A) used in the following production examples is a phosphoric acid monoester represented by the general formula (2), a phosphoric acid diester represented by the general formula (3), and a polyoxyalkylene mono A mixture of an alkyl ether (C 12 H 25 (OCH 2 CH 2 ) n OH) and a phosphoric acid triester represented by the general formula (4), wherein R in the above formulas (2) to (4) 2 , R 3 , R 4 , R 5 , R 6 and R 7 , and n of the polyoxyalkylene monoalkyl ether is 20, and R 12 is an alkyl group having 12 carbon atoms.
- the abundance ratio (molar ratio) of the monoester, diester, and polyoxyalkylene monoalkyl ether in the phosphate ester compound (A) is approximately 1: 1: 1.
- the phosphate ester compound (A) contains a small amount (about 5% by mass) of phosphate triester.
- a blue-green solid (copper salt, near infrared absorber) with a yield of 1.40 g (yield 100%) was obtained. To this was added 30 g of toluene, and ultrasonic irradiation was performed for 1.5 hours to prepare an n-butylphosphonic acid copper salt toluene dispersion (1).
- a thin light blue solid (copper salt, near-infrared absorber) with a yield of 16.7 g (yield 100%) was obtained.
- Example 1 Preparation of copper salt fine particle dispersed resin
- PVB polyvinyl butyral
- the resin sheet (1) was further preheated at 200 ° C. and 3 MPa for 1 minute using a 0.8 mm-thick mold and a compression molding machine manufactured by Shindo Metal Industry Co., Ltd., and then at 10 MPa for 5 minutes.
- the resin sheet (2) was obtained by pressing.
- the both sides of the resin sheet (2) were sandwiched between slide glasses (thickness 1.2 to 1.5 mm), and laminated glass (1) on a 70 ° C. plate.
- the laminated glass (1) was heated in an autoclave under a nitrogen atmosphere at a pressure of 1.5 MPa and 130 ° C. for 0.5 hours to obtain a measurement sample (1) in which slide glasses were disposed on both surfaces of the resin sheet. .
- the Tvis (visible light transmittance) of the measurement sample (1) was determined using a spectrophotometer (U-4000, manufactured by Hitachi, Ltd.).
- the measurement sample (1) is put into a super xenon weather meter (manufactured by Suga Test Instruments Co., Ltd.), the intensity of the irradiated light is 180 W / m 2 , and it is stored for 400 hours under the conditions of no rain, and then the spectrum is measured. Tvis (visible light transmittance) was determined.
- Tvis after 400 hours was 55.2%, and the Tvis of the measurement sample (1) before the light resistance test was 82.3%. Therefore, if the difference in Tvis before and after the test is ⁇ Tvis, ⁇ Tvis is 27 It was 1%.
- Example 2 Preparation of copper salt fine particle dispersed resin
- PVB resin (2) resin composition (2) in which n-butylphosphonic acid copper salt fine particles were dispersed was carried out in the same manner as in Example 1 except that the addition amount of the neutralizing dispersant (1) was changed to 58.0 mg. )).
- Example 3 Preparation of copper salt fine particle dispersed resin
- PVB polyvinyl butyral
- Example 4 Preparation of copper salt fine particle dispersed resin
- PVB resin (4) resin composition (4) in which n-butylphosphonic acid copper salt fine particles were dispersed was carried out in the same manner as in Example 3 except that the addition amount of the neutralizing dispersant (1) was changed to 92.8 mg. )).
- Example 5 Preparation of copper salt fine particle dispersed resin
- PVB in which n-butylphosphonic acid copper salt fine particles were dispersed was carried out in the same manner as in Example 3 except that 36.8 mg of neutralizing dispersant (2) was added instead of 34.8 mg of neutralizing dispersant (1).
- Resin (5) (resin composition (5)) was obtained.
- Example 6 Preparation of copper salt fine particle dispersed resin
- PVB in which n-butylphosphonic acid copper salt fine particles were dispersed was carried out in the same manner as in Example 3 except that 61.2 mg of neutralizing dispersant (2) was added instead of 34.8 mg of neutralizing dispersant (1).
- Resin (6) (resin composition (6)) was obtained.
- the measurement sample (6) was prepared in the same manner as in the production of the resin sheet of Example 1, except that the PVB resin (1) in which the copper salt fine particles were dispersed was replaced with the PVB resin (6) in which the copper salt fine particles were dispersed. Got.
- Example 7 Preparation of copper salt fine particle dispersed resin
- PVB in which n-butylphosphonic acid copper salt fine particles were dispersed was carried out in the same manner as in Example 3 except that 11.6 mg of neutralizing dispersant (2) was added instead of 34.8 mg of neutralizing dispersant (1).
- Resin (7) (resin composition (7)) was obtained.
- Example 8 Preparation of copper salt fine particle dispersed resin
- PVB in which n-butylphosphonic acid copper salt fine particles were dispersed was carried out in the same manner as in Example 3 except that 23.2 mg of neutralizing dispersant (2) was added instead of 34.8 mg of neutralizing dispersant (1).
- Resin (8) (resin composition (8)) was obtained.
- the measurement sample (8) was prepared in the same manner as in the production of the resin sheet of Example 1, except that the PVB resin (1) in which the copper salt fine particles were dispersed was replaced with the PVB resin (8) in which the copper salt fine particles were dispersed. Got.
- Example 9 Preparation of copper salt fine particle dispersed resin
- PVB polyvinyl butyral
- Liquid D was added to liquid C, stirred at 25 ° C. for 1 hour, and then irradiated with ultrasonic waves for 1 hour to uniformly dissolve PVB. This dispersion was spread on a Teflon (registered trademark) vat and air-dried at 25 ° C. for 20 hours. Further, vacuum drying was performed at 40 ° C. for 5 hours and at 70 ° C. for 3.5 hours to completely remove the solvent, thereby obtaining a PVB resin (9) in which copper salt fine particles were dispersed.
- Teflon registered trademark
- a measurement sample (9) was prepared in the same manner as in the production of the resin sheet of Example 1, except that the PVB resin (1) in which the copper salt fine particles were dispersed was replaced with the PVB resin (9) in which the copper salt fine particles were dispersed.
- Table 1 shows the evaluation results of light resistance in Examples 1 to 9 and Comparative Examples 1 and 2.
- Examples 1, 2 and Comparative Example 1 using the n-butylphosphonic acid copper salt toluene dispersion (1) are compared, it can be seen that the examples are excellent in light resistance.
- Examples 3 to 9 and Comparative Example 2 using n-butylphosphonic acid copper salt toluene dispersion (2) were compared, it was found that the examples were excellent in light resistance.
- the water was distilled off with an evaporator, then 100 ml of toluene was added, and the water and toluene were removed by distilling off with an evaporator to obtain 11.7 g of a white wax-like solid (neutralizing dispersant (6)).
- the phosphate ester compound (B) used in Production Example 9 is a phosphate monoester represented by the general formula (2), a phosphate diester represented by the general formula (3), and a polyoxyalkylene mono An alkyl ether (R 12 (OCH 2 CH 2 ) n OH), wherein R 2 , R 3 and R 4 in the above formulas (2) and (3), and n of the polyoxyalkylene monoalkyl ether are And R 12 is an alkyl group having 12 to 15 carbon atoms (mixture having different carbon numbers).
- the abundance ratio (molar ratio) of the monoester, the diester, and the polyoxyalkylene monoalkyl ether in the phosphoric ester compound (B) is approximately 1: 2: 1.
- a sodium salt of a phosphate ester compound (DLP-10, manufactured by Nikko Chemicals Co., Ltd.) was used.
- DLP-10 sodium salt of a phosphate ester compound
- Example 10 Preparation of copper salt fine particle dispersed resin
- PVB polyvinyl butyral
- Example 11 Preparation of copper salt fine particle dispersed resin
- PVB polyvinyl butyral
- Example 12 Preparation of copper salt fine particle dispersed resin
- PVB polyvinyl butyral
- a measurement sample (12) was prepared in the same manner as in the production of the resin sheet of Example 1, except that the PVB resin (1) in which the copper salt fine particles were dispersed was replaced with the PVB resin (12) in which the copper salt fine particles were dispersed.
- Example 13 Preparation of copper salt fine particle dispersed resin
- PVB polyvinyl butyral
- the measurement sample (13) was obtained in the same manner as in the production of the resin sheet of Example 1, except that the PVB resin (1) in which the copper salt fine particles were dispersed was replaced with the PVB resin (13) in which the copper salt fine particles were dispersed.
- Example 14 Preparation of copper salt fine particle dispersed resin
- PVB polyvinyl butyral
- the measurement sample (14) was obtained in the same manner as in the production of the resin sheet of Example 1, except that the PVB resin (1) in which the copper salt fine particles were dispersed was replaced with the PVB resin (14) in which the copper salt fine particles were dispersed.
- Example 15 Preparation of copper salt fine particle dispersed resin
- PVB polyvinyl butyral
- Table 2 shows the evaluation results of light resistance in Examples 10 to 15. When Examples 10 to 15 using n-butylphosphonic acid copper salt toluene dispersion (2) and Comparative Example 2 described above are compared, it can be seen that the examples are excellent in light resistance.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Joining Of Glass To Other Materials (AREA)
Abstract
本発明は、耐光性に優れる、近赤外線吸収剤を含有する樹脂組成物を提供することを目的とし、本発明の樹脂組成物は、近赤外線吸収剤と、中和分散剤と、樹脂とからなる樹脂組成物であり、前記近赤外線吸収剤が、少なくとも銅塩と、下記一般式(1)[式中、R1は、-CH2CH2-R11で表される1価の基であり、R11は水素原子、炭素数1~20のアルキル基、または炭素数1~20のフッ素化アルキル基を示す。]で表わされるホスホン酸化合物とを反応させることにより得られ、前記中和分散剤が、リン酸モノエステルおよびリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物を、アルカリ金属塩で中和することにより得られる中和分散剤であることを特徴とする。
Description
本発明は、樹脂組成物およびその用途に関し、詳しくは近赤外線吸収剤と、中和分散剤と、樹脂とからなる樹脂組成物およびその用途に関する。
従来から、自動車等の車両、建築物、太陽電池等の各種用途で、合わせガラスが用いられている。合わせガラス用中間膜としては、ポリビニルブチラール樹脂膜、アイオノマー樹脂膜等が知られている。
ところで、太陽光線には、可視光線の他に紫外線、赤外線等が含まれている。赤外線の中でも波長が可視光に近い赤外線は、近赤外線と呼ばれる。近赤外線は熱線とも呼ばれ車両や建築物内部の温度上昇の原因の一つである。
該温度上昇を抑制するために、車両や建築物に用いられる合わせガラスに、可視光線の透過性を保持したまま、熱線吸収性を付与することが考えられる。例えば、ホスホン酸銅塩と、ポリシロキサン成分と、可塑剤と、分散剤とを含有する銅塩組成物が知られている(例えば、特許文献1参照)。一般に金属塩を、樹脂と混合して得られた樹脂組成物は高温にさらされた場合には、可視光線の透過性が低下する場合や、黄変する場合があった。
しかしながら、特許文献1には、前記銅塩組成物と樹脂とを含有する樹脂組成物は、高温にさらされた場合であっても可視光の透過性および安定性に優れる赤外線吸収膜を提供することが可能である旨が開示されている。
しかしながら、特許文献1には、前記銅塩組成物と樹脂とを含有する樹脂組成物は、高温にさらされた場合であっても可視光の透過性および安定性に優れる赤外線吸収膜を提供することが可能である旨が開示されている。
特許文献1に記載された樹脂組成物は、高温にさらされた場合における黄変が、ホスホン酸銅塩および樹脂のみからなる樹脂組成物と比べると抑制されているが、シラン化合物を脱水縮合する工程が必要となるため、工程数が増えるという問題があった。
また、合わせガラス等に用いられる樹脂組成物としては、太陽光によって着色や、可視光透過率の低下が起こらないこと、すなわち耐光性を有することが望まれるが、特許文献1に開示された発明では耐光性についての検討は行われていなかった。
本発明は、上記従来技術を鑑みてされたものであり、耐光性に優れる、近赤外線吸収剤を含有する樹脂組成物を提供することを目的とする。
本発明者らは上記課題を達成するため鋭意研究を重ねた結果、特定の近赤外線吸収剤と、特定の中和分散剤と、樹脂とからなる樹脂組成物は、上記課題を解決することができることを見出し、本発明を完成させた。
すなわち、本発明の樹脂組成物は、近赤外線吸収剤と、中和分散剤と、樹脂とからなる樹脂組成物であり、前記近赤外線吸収剤が、少なくとも銅塩と、下記一般式(1)で表わされるホスホン酸化合物とを反応させることにより得られ、前記中和分散剤が、リン酸モノエステルおよびリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物を、アルカリ金属塩で中和することにより得られる中和分散剤であることを特徴とする。

[一般式(1)中、R1は、-CH2CH2-R11で表される1価の基であり、R11は水素原子、炭素数1~20のアルキル基、または炭素数1~20のフッ素化アルキル基を示す。]
前記アルカリ金属塩が、ナトリウム塩、カリウム塩、およびセシウム塩から選択される少なくとも1種の塩であることが好ましく、ナトリウム塩であることがより好ましい。
前記アルカリ金属塩が、ナトリウム塩、カリウム塩、およびセシウム塩から選択される少なくとも1種の塩であることが好ましく、ナトリウム塩であることがより好ましい。
前記リン酸モノエステルが、下記一般式(2)で表されるリン酸モノエステルであり、前記リン酸ジエステルが、下記一般式(3)で表されるリン酸ジエステルであることが好ましい。

[一般式(2)および(3)中、R2、R3およびR4は、-(CH2CH2O)nR12で表される1価の基であり、nは2~65の整数であり、R12は、炭素数6~35のアルキル基または炭素数6~35のアルキルフェニル基を示す。ただし、R2、R3およびR4は、それぞれ同一でも異なっていてもよい。]
前記樹脂が、ポリビニルアセタール樹脂、エチレン‐酢酸ビニル共重合体、(メタ)アクリル酸樹脂、ポリエステル樹脂、ポリウレタン樹脂、塩化ビニル樹脂、ポリオレフィン樹脂、ポリカーボネート樹脂、およびノルボルネン樹脂から選択される少なくとも1種の樹脂であることが好ましく、ポリビニルブチラール樹脂、またはエチレン‐酢酸ビニル共重合体であることがより好ましい。
前記樹脂100質量部あたり、近赤外線吸収剤を0.05~50質量部含有することが好ましい。
前記樹脂が、ポリビニルアセタール樹脂、エチレン‐酢酸ビニル共重合体、(メタ)アクリル酸樹脂、ポリエステル樹脂、ポリウレタン樹脂、塩化ビニル樹脂、ポリオレフィン樹脂、ポリカーボネート樹脂、およびノルボルネン樹脂から選択される少なくとも1種の樹脂であることが好ましく、ポリビニルブチラール樹脂、またはエチレン‐酢酸ビニル共重合体であることがより好ましい。
前記樹脂100質量部あたり、近赤外線吸収剤を0.05~50質量部含有することが好ましい。
前記樹脂100質量部あたり、中和分散剤を0.01~10質量部含有することが好ましい。
本発明の合わせガラス用中間膜は、前記樹脂組成物から形成される。
本発明の合わせガラスは、前記合わせガラス用中間膜を有する。
本発明の近赤外線吸収剤を含有する樹脂組成物は、耐光性に優れる。
次に本発明について具体的に説明する。
本発明の樹脂組成物は、近赤外線吸収剤と、中和分散剤と、樹脂とからなる樹脂組成物であり、前記近赤外線吸収剤が、少なくとも銅塩と、一般式(1)で表わされるホスホン酸化合物とを反応させることにより得られ、前記中和分散剤が、リン酸モノエステルおよびリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物を、アルカリ金属塩で中和することにより得られる中和分散剤であることを特徴とする。
なお、本発明の樹脂組成物を、銅塩微粒子分散樹脂とも記す。
[中和分散剤]
本発明に用いられる中和分散剤は、リン酸モノエステルおよびリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物を、アルカリ金属塩で中和することにより得られる中和分散剤である。
本発明に用いられる中和分散剤は、リン酸モノエステルおよびリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物を、アルカリ金属塩で中和することにより得られる中和分散剤である。
本発明に用いられる中和分散剤としては一種を用いても、二種以上を用いてもよい。
前記アルカリ金属塩としては、アルカリ金属の酸化物、水酸化物、ハロゲン化物、水素化物や、炭酸水素塩、炭酸塩、硝酸塩、硫酸水素塩、硫酸塩、ギ酸塩、酢酸塩などの有機酸塩等が挙げられ、アルカリ金属の水酸化物、炭酸水素塩、炭酸塩が好ましい。
また、アルカリ金属塩を構成する金属種としては、リチウム、ナトリウム、カリウム、ルビジウム、セシウム、フランシウムが挙げられるが、ナトリウム、カリウム、セシウムが好ましく、ナトリウムがより好ましい。すなわち、前記アルカリ金属塩としてはナトリウム塩、カリウム塩、およびセシウム塩から選択される少なくとも1種の塩が好ましく、ナトリウム塩がより好ましい。
なお、アルカリ金属塩としては一種を用いても、二種以上を用いてもよい。
また、前記リン酸モノエステルおよびリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物としては、特に限定はないが、前記リン酸モノエステルが、下記一般式(2)で表されるリン酸モノエステルであることが好ましく、前記リン酸ジエステルが、下記一般式(3)で表されるリン酸ジエステルであることが好ましい。

[一般式(2)および(3)中、R2、R3およびR4は、-(CH2CH2O)nR12で表される1価の基であり、nは2~65の整数であり、R12は、炭素数6~35のアルキル基または炭素数6~35のアルキルフェニル基を示す。ただし、R2、R3およびR4は、それぞれ同一でも異なっていてもよい。]
nは2~65の整数であり、好ましくは2~35の整数である。nが2未満である場合には、合わせガラス等を製造した際に透明性が不充分となる場合がある。また、nが前記範囲を超えると、充分な透明性を有する合わせガラス等を得るために必要な、リン酸エステル化合物の量が増え、コスト高の原因となる傾向がある。
nは2~65の整数であり、好ましくは2~35の整数である。nが2未満である場合には、合わせガラス等を製造した際に透明性が不充分となる場合がある。また、nが前記範囲を超えると、充分な透明性を有する合わせガラス等を得るために必要な、リン酸エステル化合物の量が増え、コスト高の原因となる傾向がある。
また、R12は、炭素数6~35のアルキル基または炭素数6~35のアルキルフェニル基であり、好ましくは炭素数6~25のアルキル基または炭素数6~25のアルキルフェニル基であり、炭素数6~25のアルキル基であることが好ましく、10~20のアルキル基であることがより好ましい。R12が、炭素数6未満の基であると、合わせガラス等を製造した際に透明性が不充分となる場合がある。また、R12が、炭素数35を超える基であると、充分な透明性を有する合わせガラス等を得るために必要な、リン酸エステル化合物の量が増え、コスト高の原因となる傾向がある。
中和分散剤を得る際には、前記一般式(2)で表されるリン酸モノエステルおよび前記一般式(3)で表されるリン酸ジエステルの少なくとも一方が用いられることが好ましいが、前記一般式(2)で表されるリン酸モノエステルおよび前記一般式(3)で表されるリン酸ジエステルの両方を用いることがより好ましい。前記一般式(2)で表されるリン酸モノエステルおよび前記一般式(3)で表されるリン酸ジエステルを用いると、合わせガラス等の透明性、耐熱性に優れる傾向があり好ましい。前記一般式(2)で表されるリン酸モノエステルおよび前記一般式(3)で表されるリン酸ジエステルの両方を用いる場合には、前記一般式(2)で表されるリン酸モノエステルと前記一般式(3)で表されるリン酸ジエステルとの割合は、特に限定されないが、通常はモル比(一般式(2):一般式(3))で10:90~90:10である。
また、前記一般式(2)で表されるリン酸モノエステルとしては、一種単独で用いても、二種以上を用いてもよく、前記一般式(3)で表されるリン酸ジエステルとしては、一種単独で用いても、二種以上を用いてもよい。
前記一般式(2)で表されるリン酸モノエステルおよび前記一般式(3)で表されるリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物としては、市販されているリン酸エステル化合物、例えばDDP-2,DDP-4、DDP-6、DDP-8、DDP-10、TDP-2、TDP-6、TDP-8、TDP-10、TLP-4、TCP-5、TOP-0V(以上、日光ケミカルズ(株)製)や、プライサーフA212C、プライサーフA215C、プライサーフAL12H、プライサーフAL、プライサーフA208F、プライサーフA208N、プライサーフA208B、プライサーフA219B、プライサーフA210D、プライサーフDBS(以上、第一工業製薬(株)製)等を用いることもできる。
なお、市販されているリン酸エステル化合物には、一般式(2)で表されるリン酸モノエステルおよび前記一般式(3)で表されるリン酸ジエステル以外の成分、例えば、ポリオキシアルキレンモノアルキルエーテルや、下記一般式(4)で表されるリン酸トリエステルが含有されている場合がある。本発明では、リン酸エステル化合物中に、このようなポリオキシアルキレンモノアルキルエーテルや、リン酸トリエステルが存在しても、問題なく使用することができる。
なお、ポリオキシアルキレンモノアルキルエーテルが含まれている場合には、一般式(2)で表されるリン酸モノエステルおよび前記一般式(3)で表されるリン酸ジエステルの合計100質量部に対して、ポリオキシアルキレンモノアルキルエーテルが、1~300質量部であることが好ましい。
なお、ポリオキシアルキレンモノアルキルエーテルとしては、HO-(CH2CH2O)nR12、(nは2~65の整数、好ましくは2~35の整数であり、R12は、炭素数6~35、好ましくは6~25のアルキル基または炭素数6~35、好ましくは6~25のアルキルフェニル基を示す。)が挙げられる。該ポリオキシアルキレンモノアルキルエーテルは、リン酸エステルを製造する際に用いられる原料に由来すると考えられる。
なお、一般式(4)で表されるリン酸トリエステルが含まれている場合には、一般式(2)で表されるリン酸モノエステルおよび前記一般式(3)で表されるリン酸ジエステルの合計100質量部に対して、一般式(4)で表されるリン酸トリエステルが、1~300質量部であることが好ましい。

[一般式(4)中、R5、R6およびR7は、-(CH2CH2O)nR12で表される1価の基であり、nは2~65の整数、好ましくは2~35の整数であり、R12は、炭素数6~35、好ましくは6~25のアルキル基または炭素数6~35、好ましくは6~25のアルキルフェニル基を示す。ただし、R5、R6およびR7は、それぞれ同一でも異なっていてもよい。]
なお、前記ポリオキシアルキレンモノアルキルエーテルや一般式(4)におけるnやR12としては、一般式(2)および(3)におけるnやR12と同様のものが好ましい。
なお、前記ポリオキシアルキレンモノアルキルエーテルや一般式(4)におけるnやR12としては、一般式(2)および(3)におけるnやR12と同様のものが好ましい。
本発明に用いる中和分散剤を得る方法としては、前記リン酸モノエステルおよびリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物を、アルカリ金属塩で中和することができればよく、特に限定されないが、例えばリン酸エステル化合物の水溶液および、アルカリ金属塩の水溶液をそれぞれ調製し、リン酸エステル化合物の水溶液中にアルカリ金属塩の水溶液を添加し、中和を行う方法が挙げられる。中和に用いる溶媒としては、水の他に、メタノール、エタノール、プロパノールなどのアルコールや、これらアルコールと水との混合溶媒が挙げられる。
なお、リン酸エステル化合物として、リン酸モノエステルが含まれている場合には、リン酸モノエステルが有する二つの水酸基のうち一つが中和される第一中和点、および二つが中和される第二中和点が存在することになる。
本発明に用いる中和分散剤としては、中和分散剤の添加量をより削減する観点から、第二中和点まで中和されていることが好ましい。
水溶液中で中和分散剤を得る際に、pHが5.0~6.9まで中和することにより、第一中和点まで中和され、pHが8.5~11.5まで中和することにより、第二中和点まで中和される。本発明では、前記中和は少なくとも第一中和点である、pHが5.3~6.6を超えるまで行われることが好ましく、第二中和点であるpHが9.0~11.0となるまで行われることがより好ましい。
なお、中和分散剤としては、中和を行った後、水等の溶媒を除去することにより、ワックス等の固体として得てもよく、水等の溶媒の除去を行わず、溶液中に溶解あるいは分散した状態で得てもよい。
また、前記一般式(2)で表されるリン酸モノエステルおよび前記一般式(3)で表されるリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物を、アルカリ金属塩で中和した状態で市販されているものを、中和分散剤として用いてもよい。市販されている中和分散剤としては、DLP-10(リン酸エステル化合物のナトリウム塩)、DOP-8NV(リン酸エステル化合物のナトリウム塩)(共に、日光ケミカルズ(株)製)等が挙げられる。なお、市販されている中和分散剤中に、リン酸エステルの水酸基が残存している場合、すなわち未中和の水酸基が存在する場合には、さらに中和を行った後に、中和分散剤として用いてもよい。
本発明の樹脂組成物は、前記中和分散剤を含有するため、太陽光等の光が照射されることによる着色や可視光透過率の低下が抑制されている。
[近赤外線吸収剤]
本発明に用いられる近赤外線吸収剤は、少なくとも銅塩と、下記一般式(1)で表わされるホスホン酸化合物とを反応させることにより得られる近赤外線吸収剤である。
本発明に用いられる近赤外線吸収剤は、少なくとも銅塩と、下記一般式(1)で表わされるホスホン酸化合物とを反応させることにより得られる近赤外線吸収剤である。
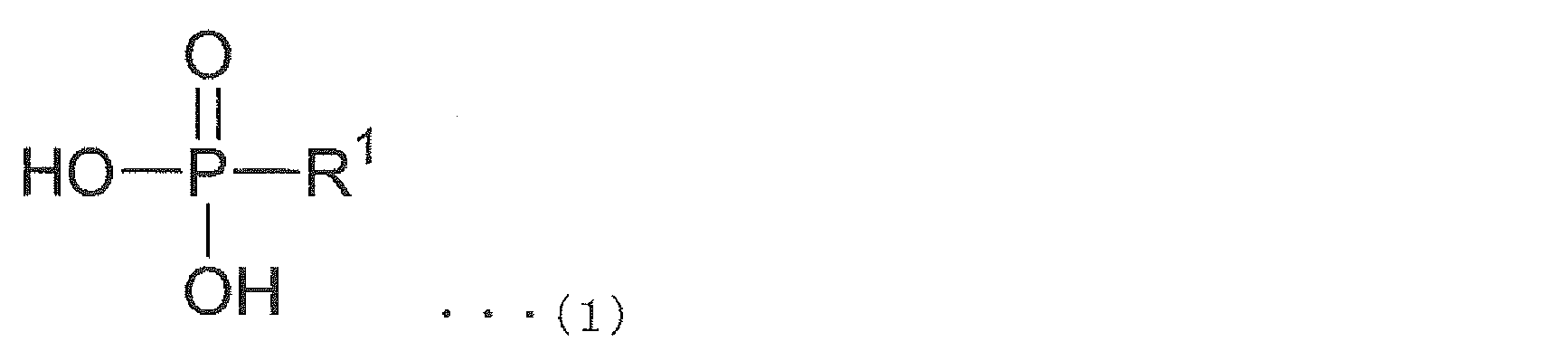
[一般式(1)中、R1は、-CH2CH2-R11で表される1価の基であり、R11は水素原子、炭素数1~20のアルキル基、または炭素数1~20のフッ素化アルキル基を示す。]
本発明に用いられる近赤外線吸収剤は、銅塩および、一般式(1)で表わされるホスホン酸化合物のみを原料とし、これらを反応させることにより得られるものでもよく、銅塩および、一般式(1)で表わされるホスホン酸化合物に加えて、他の成分を原料として用い、これらを反応させることにより得られるものでもよい。
本発明に用いられる近赤外線吸収剤は、銅塩および、一般式(1)で表わされるホスホン酸化合物のみを原料とし、これらを反応させることにより得られるものでもよく、銅塩および、一般式(1)で表わされるホスホン酸化合物に加えて、他の成分を原料として用い、これらを反応させることにより得られるものでもよい。
なお、銅塩および一般式(1)で表わされるホスホン酸化合物のみを原料とした場合には、近赤外線吸収剤は、下記一般式(1’)で表される銅塩であり、一般式(1)で表わされるホスホン酸化合物に加えて、他の成分を原料とした場合には、近赤外線吸収剤は、下記一般式(1’)で表される銅塩を含有する。

[一般式(1’)中、R1は、-CH2CH2-R11で表される1価の基であり、R11は水素原子、炭素数1~20のアルキル基、または炭素数1~20のフッ素化アルキル基を示す。]
前記一般式(1)および一般式(1’)におけるR11としては、水素原子または炭素数1~20のアルキル基であることが好ましい。具体的にはR11としては、水素原子、メチル基、エチル基、プロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基等が好ましい。
前記一般式(1)および一般式(1’)におけるR11としては、水素原子または炭素数1~20のアルキル基であることが好ましい。具体的にはR11としては、水素原子、メチル基、エチル基、プロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基、ドデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基等が好ましい。
なお、一般式(1)で表わされるホスホン酸化合物としては、一種単独で用いても、二種以上を用いてもよい。
本発明に用いる、近赤外線吸収剤の製造方法としては、特に限定はないが、例えば以下の方法で製造することができる。
近赤外線吸収剤の製造方法としては、溶媒中で、銅塩と、上記一般式(1)で表わされるホスホン酸化合物とを混合し、反応させ、反応混合物を得る工程(以下、反応工程とも記す)、該反応混合物中の溶媒を除去することにより近赤外線吸収剤を、ホスホン酸銅塩からなる微粒子として得る工程(以下、溶媒除去工程とも記す)を有する方法が挙げられる。
前記一般式(1)で表わされるホスホン酸化合物としては、前述のようにR11が水素原子または炭素数1~20のアルキル基であるものが好ましい。一般式(1)で表されるホスホン酸化合物としては例えば、エチルホスホン酸、プロピルホスホン酸、ブチルホスホン酸、ペンチルホスホン酸、ヘキシルホスホン酸、ヘプチルホスホン酸、オクチルホスホン酸、ノニルホスホン酸、デシルホスホン酸、ウンデシルホスホン酸、ドデシルホスホン酸、トリデシルホスホン酸、テトラデシルホスホン酸、ペンタデシルホスホン酸、ヘキサデシルホスホン酸、ヘプタデシルホスホン酸、オクタデシルホスホン酸等のアルキルホスホン酸が挙げられる。
前記銅塩としては、2価の銅イオンを供給することが可能な銅塩が通常用いられる。前記銅塩としては、前記一般式(1’)で表わされるホスホン酸銅塩以外の銅塩であればよい。前記銅塩としては例えば、酢酸銅無水物、蟻酸銅無水物、ステアリン酸銅無水物、安息香酸銅無水物、エチルアセト酢酸銅無水物、ピロリン酸銅無水物、ナフテン酸銅無水物、クエン酸銅無水物等の有機酸の銅塩、該有機酸の銅塩の水和物もしくは水化物;酸化銅、塩化銅、硫酸銅、硝酸銅、塩基性炭酸銅等の無機酸の銅塩、該無機酸の銅塩の水和物もしくは水化物;水酸化銅が挙げられる。なお、銅塩としては、一種単独で用いても、二種以上を用いてもよい。
銅塩としては、酢酸銅無水物、酢酸銅一水和物が、溶解性や副生成物の除去の点から好ましく用いられる。
近赤外線吸収剤を製造する際には、前記反応工程において分散剤を用いてもよい。前記分散剤としては、例えば一般式(2)で表されるリン酸モノエステルおよび一般式(3)で表されるリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物が挙げられる。なお、近赤外線吸収剤を製造する際には、本発明の樹脂組成物が含有する中和分散剤を用いないことが好ましい。
すなわち、中和分散剤を得る際に用いられるリン酸モノエステルおよびリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物の好適態様である、一般式(2)で表されるリン酸モノエステルおよび一般式(3)で表されるリン酸ジエステルは、近赤外線吸収剤を製造する際に用いられる分散剤としても好ましい。
なお、中和分散剤を得る際に用いるリン酸エステルと、近赤外線吸収剤を得る際に用いるリン酸エステルとは、同種のものを用いてもよく、別のものを用いてもよい。
なお、ホスホン酸銅塩からなる微粒子を製造する際には、前記銅塩1モルあたり、一般式(1)で表わされるホスホン酸化合物を0.5~1.5モル用いることが好ましく、0.8~1.2モル用いることがより好ましい。また、分散剤が、一般式(2)で表されるリン酸モノエステルおよび下記一般式(3)で表されるリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物である場合には、前記銅塩100質量部あたり、5~100質量部用いることが好ましく、10~50質量部用いることがより好ましい。
前記溶媒としては、メタノール、エタノール、イソプロピルアルコール、n-ブチルアルコール、メタノール/エタノール混合溶媒、イソプロピルアルコール/エタノール混合溶媒等のアルコール、テトラヒドロフラン(THF)、ジメチルホルムアミド(DMF)、水等が挙げられ、良好に反応を行う観点から、メタノール、エタノール、メタノール/エタノール混合溶媒、イソプロピルアルコール/エタノール混合溶媒が好ましい。また、反応工程は、好ましくは0~80℃、より好ましくは10~60℃の温度条件で、好ましくは0.5~80時間、より好ましくは1~50時間行われる。
前記反応工程では、前記一般式(1)で表わされるホスホン酸化合物と、前記銅塩とが反応し、該反応によって、前記溶媒に溶解しない微粒子状のホスホン酸銅塩が生成する。前記一般式(2)で表されるリン酸モノエステルおよび下記一般式(3)で表されるリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物は、反応時に良好な分散剤として作用することができるため、前記ホスホン酸銅塩は分散性が高く保たれ、凝集を抑制することができる。
なお、前記反応工程では、前記一般式(1)で表わされるホスホン酸化合物と銅塩との反応のみではなく、例えば一般式(2)で表されるリン酸モノエステルおよび下記一般式(3)で表されるリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物と、銅塩の一部とが反応してもよい。また、原料の一部が反応せずに残存していてもよい。
なお、前記近赤外線吸収剤の製造方法では、通常、前記反応混合物から、少なくとも前記溶媒の一部を除去することにより、ホスホン酸銅塩からなる微粒子を得る。
溶媒除去工程では、反応混合物中から、少なくとも前記溶媒の一部を除去する。溶媒除去工程では、溶媒以外にも、反応混合物中の液体成分を合わせて除去してもよい。
溶媒除去工程では、通常反応混合物を加熱することにより、少なくとも前記溶媒の一部を除去するが、加熱条件は通常、室温~70℃であり、好ましくは40~60℃である。また、溶媒除去工程は、常圧下で行ってもよく、減圧下で行ってもよい。減圧下で溶媒除去工程を行う場合には、加熱を行わなくてもよい場合や、加熱温度が低くてもよい場合がある。
また、溶媒を除去する別の方法としては、反応混合物を静置することにより、ホスホン酸銅塩からなる微粒子を沈殿させ、上澄み液(溶媒)を除去する方法や、反応混合物を遠心分離処理することにより、ホスホン酸銅塩からなる微粒子を沈殿させ、上澄み液(溶媒)を除去する方法が挙げられる。
なお、溶媒を除去する方法としては、これらの方法を組み合わせて行ってもよい。
また、溶媒除去工程を行った後に、近赤外線吸収剤中に含まれる不純物の除去を目的として、近赤外線吸収剤を、分散媒に分散した後に、該分散媒を除去する工程を設けてもよい。
前記近赤外線吸収剤としては通常、平均粒径が1~1000nmのホスホン酸銅塩からなる微粒子が用いられる。平均粒径としては、モノマーへの分散性や樹脂組成物の透明性を確保するため、5~300nmであることがより好ましい。
[樹脂]
本発明には、樹脂が用いられる。本発明に用いられる樹脂としては、前述の近赤外線吸収剤を分散することが可能であればよく特に限定はないが、例えば以下の樹脂を用いることができる。
本発明には、樹脂が用いられる。本発明に用いられる樹脂としては、前述の近赤外線吸収剤を分散することが可能であればよく特に限定はないが、例えば以下の樹脂を用いることができる。
本発明に用いる樹脂としては、ポリビニルアセタール樹脂、エチレン‐酢酸ビニル共重合体、(メタ)アクリル酸樹脂、ポリエステル樹脂、ポリウレタン樹脂、塩化ビニル樹脂、ポリオレフィン樹脂、ポリカーボネート樹脂、およびノルボルネン樹脂から選択される少なくとも1種の樹脂が、近赤外線吸収剤を良好に分散することが可能であり、かつ可視光の透過性に優れることが好ましい。
本発明に用いる樹脂としては、ポリビニルアセタール樹脂、およびエチレン‐酢酸ビニル共重合体から選択される少なくとも1種の樹脂であることがより好ましく、ポリビニルブチラール樹脂(PVB)、およびエチレン‐酢酸ビニル共重合体から選択される少なくとも1種の樹脂であることが特に好ましく、ポリビニルブチラール樹脂、またはエチレン‐酢酸ビニル共重合体が最も好ましい。ポリビニルアセタール樹脂を用いると、前述の近赤外線吸収剤の分散性に優れ、本発明の樹脂組成物を用いて、光学材料を製造する際に、ガラス等への密着性に優れ、本発明の樹脂組成物が柔軟であり、かつ温度変化に伴う近赤外線吸収剤の変形が起こり難いため好ましい。また、ポリビニルアセタール樹脂としては、ポリビニルブチラール樹脂(PVB)を用いることが、ガラス密着性、分散性、透明性、耐熱性、耐光性などの観点から好ましい。
ポリビニルアセタール樹脂は、必要な物性に応じて、二種以上を組み合わせたブレンド物であってもよく、アセタール化時にアルデヒドを組み合わせてアセタール化することにより得られるポリビニルアセタール樹脂であってもよい。上記ポリビニルアセタール樹脂の分子量、分子量分布およびアセタール化度は特に限定されないが、アセタール化度は、一般に40~85%であり、好ましい下限は60%、上限は75%である。
ポリビニルアセタール樹脂は、ポリビニルアルコール樹脂をアルデヒドによりアセタール化することにより得ることができる。上記ポリビニルアルコール樹脂は、一般にポリ酢酸ビニルを鹸化することにより得られるものであり、鹸化度80~99.8モル%のポリビニルアルコール樹脂が一般的に用いられる。上記ポリビニルアルコール樹脂の粘度平均重合度は好ましい下限は200、上限は3000である。200未満であると、得られる合わせガラスの耐貫通性が低下する場合がある。3000を超えると、樹脂組成物の成形性が悪くなる場合があり、しかも樹脂組成物の剛性が大きくなり過ぎ、加工性が悪くなる。より好ましい下限は500、上限は2200である。なお、ポリビニルアルコール樹脂の粘度平均重合度、および鹸化度は、例えば、JISK 6726「ポリビニルアルコール試験方法」に基づいて測定することができる。
アルデヒドとしては特に限定されず、例えば、炭素数が1~10のアルデヒド等が挙げられ、より具体的には、例えば、n-ブチルアルデヒド、イソブチルアルデヒド、n-バレルアルデヒド、2-エチルブチルアルテヒド、n-へキシルアルデヒド、n-オクチルアルデヒド、n-ノニルアルデヒド、n-デシルアルデヒド、ホルムアルデヒド、アセトアルデヒド、ベンズアルデヒド等が挙げられる。なかでも、n-ブチルアルデヒド、n-へキシルアルデヒド、n-バレルアルデヒド等が好ましい。より好ましくは、炭素数が4のブチルアルデヒドである。
また、エチレン‐酢酸ビニル共重合体を用いると、前述の近赤外線吸収剤の分散性に優れ、ガラス密着性、分散性、透明性、耐熱性、耐光性などの観点から好ましい。
〔樹脂組成物〕
本発明の樹脂組成物は、前述のように近赤外線吸収剤と、中和分散剤と、樹脂とからなる樹脂組成物である。本発明の樹脂組成物は、耐光性に優れる。
本発明の樹脂組成物は、前述のように近赤外線吸収剤と、中和分散剤と、樹脂とからなる樹脂組成物である。本発明の樹脂組成物は、耐光性に優れる。
本発明の樹脂組成物は、前述のように近赤外線吸収剤と、中和分散剤と、樹脂とからなる組成物であればよく、その製造方法としては特に限定されない。本発明の樹脂組成物の製造方法としては、例えば、トルエン、エタノール/トルエン混合溶媒、メタノール/トルエン混合溶媒、塩化メチレン、クロロホルム等の溶剤に、樹脂、近赤外線吸収剤分散液、中和分散剤を添加した後、撹拌、超音波照射等によって、樹脂を溶解させ、分散液を得て、該分散液から溶剤を除去する方法が挙げられる。また、別の方法としては、トルエン、エタノール/トルエン混合溶媒、メタノール/トルエン混合溶媒、塩化メチレン、クロロホルム等の溶剤に、樹脂を添加した後、撹拌、超音波照射等によって、樹脂を溶解させ、次いで樹脂が溶解した溶液中に、近赤外線吸収剤分散液、中和分散剤を添加した後、撹拌、超音波照射等を行い、分散液を得て、該分散液から溶剤を除去する方法が挙げられる。さらに別の方法としては、トルエン、エタノール/トルエン混合溶媒、メタノール/トルエン混合溶媒、塩化メチレン、クロロホルム等の溶剤に、樹脂を添加した後、撹拌、超音波照射等によって、樹脂を溶解させた溶液と、トルエン、メタノール、エタノール/トルエン混合溶媒、メタノール/トルエン混合溶媒、塩化メチレン、クロロホルム等の溶剤に、近赤外線吸収剤分散液と、中和分散剤とを添加した後、撹拌、超音波照射等によって、分散させた分散液とを別に調整し、該溶液に該分散液を加え、あるいは該分散液に該溶液を加え、必要により撹拌、超音波照射を行い、両者を混合した後に、溶剤を除去する方法が挙げられる。
なお、前記近赤外線吸収剤分散液は、近赤外線吸収剤を、トルエン、メタノール、塩化メチレン、クロロホルム等に分散することにより調製することができる。
本発明の樹脂組成物は、前記樹脂100質量部あたり、近赤外線吸収剤を0.05~50質量部含有することが好ましく、0.1~25質量部含有することがより好ましい。0.05質量部より少ないと充分な近赤外線吸収特性が得られない可能性があり、50質量部より多すぎると樹脂の透明性や接着性が大幅に低下するおそれがある。
本発明の樹脂組成物は、前記樹脂100質量部あたり、前記中和分散剤を0.01~10質量部含有することが好ましく、0.1~5.0質量部含有することがより好ましい。前記範囲内では、樹脂の透明性やガラスとの接着性が良好に保てるため好ましい。
本発明の樹脂組成物は、近赤外線吸収能に優れ、光照射がされた場合の着色や、可視光透過率の低下が抑制されているため、合わせガラス等の構造材料用の中間膜として好適に使用することが可能である。
また、本発明の樹脂組成物には、各種添加剤が含有されていてもよい。添加剤としては、例えば可塑剤、他の分散剤、架橋剤、キレート剤、酸化防止剤、紫外線吸収剤、光安定剤、色調補正剤等が挙げられる。これらの添加剤は、本発明の樹脂組成物を製造する際に、添加されてもよく、前述の近赤外線吸収剤、中和分散剤、樹脂それぞれを製造する際に添加されてもよい。
〔樹脂組成物の用途〕
本発明の樹脂組成物は、近赤外線を吸収することが望まれる用途に通常は用いられる。
本発明の樹脂組成物は、近赤外線を吸収することが望まれる用途に通常は用いられる。
本発明の樹脂組成物から形成される樹脂膜は、近赤外線吸収能に優れ、耐光性に優れるため合わせガラス用中間膜等の構造材料用中間膜として好適に用いることが可能である。
また、本発明の合わせガラスは、前記合わせガラス用中間膜を有している。本発明の合わせガラスを構成するガラスとしては特に限定はなく、従来公知のものを用いることができる。
次に本発明について実施例を示してさらに詳細に説明するが、本発明はこれらによって限定されるものではない。
下記製造例において用いたリン酸エステル化合物(A)は、前記一般式(2)で表されるリン酸モノエステルと、前記一般式(3)で表されるリン酸ジエステルと、ポリオキシアルキレンモノアルキルエーテル(C12H25(OCH2CH2)nOH)と、前記一般式(4)で表されるリン酸トリエステルとの混合物であり、前記式(2)~(4)中におけるR2、R3、R4、R5、R6およびR7、並びにポリオキシアルキレンモノアルキルエーテルのnが20であり、R12が炭素数12のアルキル基であるものである。なお、リン酸エステル化合物(A)中のモノエステルとジエステルとポリオキシアルキレンモノアルキルエーテルとの存在比(モル比)は、ほぼ1:1:1である。また、リン酸エステル化合物(A)中に、リン酸トリエステルは少量(約5質量%)含まれている。
〔製造例1〕
(銅塩分散液の調製)
200mlナスフラスコに、酢酸銅一水和物1.164g(5.83mmol)、エタノール35gを加え、25℃で1時間攪拌し、溶液(A液)を得た。
(銅塩分散液の調製)
200mlナスフラスコに、酢酸銅一水和物1.164g(5.83mmol)、エタノール35gを加え、25℃で1時間攪拌し、溶液(A液)を得た。
別の容器に、リン酸エステル化合物(A)0.232g、n-ブチルホスホン酸0.806gをエタノール20gに溶解し、溶液(B液)を作成した。
B液を、A液に対して2時間かけて滴下した。この反応液を25℃で20時間撹拌した。
その後、エバポレーターで反応液から溶媒を留去した。溶媒が留去された固形分にトルエン30gを加え、恒量になり、酢酸臭がしなくなるまでエバポレーターで留去した。
収量1.40g(収率100%)の青緑色固体(銅塩、近赤外線吸収剤)が得られた。これにトルエン30gを加え、超音波照射を1.5時間行い、n-ブチルホスホン酸銅塩トルエン分散液(1)を作成した。
〔製造例2〕
(銅塩分散液の調製)
1Lナスフラスコに、酢酸銅一水和物14.0g(70.13mmol)、メタノール490gを加え、25℃で1時間攪拌し、溶液(A液)を得た。
(銅塩分散液の調製)
1Lナスフラスコに、酢酸銅一水和物14.0g(70.13mmol)、メタノール490gを加え、25℃で1時間攪拌し、溶液(A液)を得た。
別の容器に、リン酸エステル化合物(A)2.8g、n-ブチルホスホン酸9.59gをメタノール96gに溶解し、溶液(B液)を作成した。
B液を、A液に対して3時間かけて滴下した。この反応液を25℃で20時間撹拌した。
その後、エバポレーターで反応液から溶媒を留去した。溶媒が留去された固形分にトルエン100gを加え、恒量になり、酢酸臭がしなくなるまでエバポレーターで留去した。
収量16.7g(収率100%)の薄い水色固体(銅塩、近赤外線吸収剤)が得られた。これにトルエン140gを加え、超音波照射を6時間行い、n-ブチルホスホン酸銅塩トルエン分散液(2)を作成した。
〔製造例3〕
(中和分散剤の作成)
リン酸エステル化合物(A)10gを純水100mlに溶解し、1MのNaOH水溶液で中和した。pHメーターで水溶液を測定し、pH7となったところで中和を終了した。
(中和分散剤の作成)
リン酸エステル化合物(A)10gを純水100mlに溶解し、1MのNaOH水溶液で中和した。pHメーターで水溶液を測定し、pH7となったところで中和を終了した。
エバポレーターで水を留去し、次いでトルエン100mlを添加し、エバポレーターで留去して水分およびトルエンを除去し、白色のワックス状固体(中和分散剤(1))を10.2g得た。
〔製造例4〕
(中和分散剤の作成)
リン酸エステル化合物(A)10gを純水100mlに溶解し、1MのNaOH水溶液で中和した。pHメーターで水溶液を測定し、pH10.6となったところで中和を終了した。
(中和分散剤の作成)
リン酸エステル化合物(A)10gを純水100mlに溶解し、1MのNaOH水溶液で中和した。pHメーターで水溶液を測定し、pH10.6となったところで中和を終了した。
エバポレーターで水を留去し、次いでトルエン100mlを添加し、エバポレーターで留去して水分およびトルエンを除去し、白色のワックス状固体(中和分散剤(2))を10.3g得た。
〔実施例1〕
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン250ml、ポリビニルブチラール(PVB)5.00gを加えた。
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン250ml、ポリビニルブチラール(PVB)5.00gを加えた。
これに、前記n-ブチルホスホン酸銅塩トルエン分散液(1)3.14g(銅塩を0.583mmol含む)を添加した。
これに、前記中和分散剤(1)を92.8mg加えた。25℃で10時間撹拌後、1時間超音波照射し、PVBを均一に溶解させた。この分散液をテフロン(登録商標)製バットに広げ、20時間25℃で風乾した。さらに40℃で5時間、70℃で3.5時間真空乾燥を行って溶媒を完全に除去し、銅塩微粒子が分散したPVB樹脂(1)(樹脂組成物(1))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
前記銅塩微粒子が分散したPVB樹脂(1)を、厚さ0.8mmの型枠および(株)神藤金属工業所製の圧縮成形機を用い、120℃、3MPaで予熱1分間を行った後、15MPaで3分間プレスし、樹脂シート(1)を得た。
<樹脂シートおよび測定サンプルの作製>
前記銅塩微粒子が分散したPVB樹脂(1)を、厚さ0.8mmの型枠および(株)神藤金属工業所製の圧縮成形機を用い、120℃、3MPaで予熱1分間を行った後、15MPaで3分間プレスし、樹脂シート(1)を得た。
前記樹脂シート(1)を、さらに厚さ0.8mmの型枠および(株)神藤金属工業所製の圧縮成形機を用い、200℃、3MPaで予熱1分間を行った後、10MPaで5分間プレスし、樹脂シート(2)を得た。
前記樹脂シート(2)の両面を、スライドガラス(厚み1.2~1.5mm)で挟み、70℃のプレート上で合わせガラス(1)とした。
該合わせガラス(1)をオートクレーブ内で、窒素雰囲気下、圧力1.5MPa、130℃で0.5時間加熱し、樹脂シートの両面にスライドガラスが配設された測定サンプル(1)を得た。
<耐光性の評価>
耐光性試験は以下の方法で行った。
耐光性試験は以下の方法で行った。
測定サンプル(1)のTvis(可視光透過率)を、分光光度計(U-4000形、(株)日立製作所製)を使用し求めた。
次いで、スーパーキセノンウェザーメーター(スガ試験機(株)製)に測定サンプル(1)を入れ、照射する光の強度は180W/m2、降雨なしの条件で400時間保管した後、分光を測定してTvis(可視光透過率)を求めた。
400時間後のTvisは55.2%であり、耐光性試験前の測定サンプル(1)のTvisは82.3%であったので、試験前後のTvisの差をΔTvisとすると、ΔTvisは、27.1%であった。
〔実施例2〕
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)の添加量を58.0mgに変更したこと以外は実施例1と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(2)(樹脂組成物(2))を得た。
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)の添加量を58.0mgに変更したこと以外は実施例1と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(2)(樹脂組成物(2))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(2)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(2)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(2)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(2)を得た。
<耐光性の評価>
前記測定サンプル(2)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは41.3%であり、耐光性試験前の測定サンプル(2)のTvisは83.3%であったので、ΔTvisは、42.0%であった。
前記測定サンプル(2)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは41.3%であり、耐光性試験前の測定サンプル(2)のTvisは83.3%であったので、ΔTvisは、42.0%であった。
〔比較例1〕
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)を添加しないこと以外は実施例1と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(c1)(樹脂組成物(c1))を得た。
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)を添加しないこと以外は実施例1と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(c1)(樹脂組成物(c1))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(c1)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(c1)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(c1)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(c1)を得た。
<耐光性の評価>
前記測定サンプル(c1)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは29.8%であり、耐光性試験前の測定サンプル(c1)のTvisは85.2%であったので、ΔTvisは、55.4%であった。
前記測定サンプル(c1)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは29.8%であり、耐光性試験前の測定サンプル(c1)のTvisは85.2%であったので、ΔTvisは、55.4%であった。
〔実施例3〕
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.9g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.0gを加えた。
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.9g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.0gを加えた。
これに、前記n-ブチルホスホン酸銅塩トルエン分散液(2)1.30g(銅塩を0.583mmol含む)を添加した。
これに、前記中和分散剤(1)を34.8mg加えた。25℃で10時間撹拌後、1時間超音波照射し、PVBを均一に溶解させた。この分散液をテフロン(登録商標)製バットに広げ、20時間25℃で風乾した。さらに40℃で5時間、70℃で3.5時間真空乾燥を行って溶媒を完全に除去し、銅塩微粒子が分散したPVB樹脂(3)(樹脂組成物(3))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(3)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(3)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(3)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(3)を得た。
<耐光性の評価>
前記測定サンプル(3)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは22.2%であり、耐光性試験前の測定サンプル(3)のTvisは84.5%であったので、ΔTvisは、62.3%であった。
前記測定サンプル(3)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは22.2%であり、耐光性試験前の測定サンプル(3)のTvisは84.5%であったので、ΔTvisは、62.3%であった。
〔実施例4〕
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)の添加量を92.8mgに変更したこと以外は実施例3と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(4)(樹脂組成物(4))を得た。
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)の添加量を92.8mgに変更したこと以外は実施例3と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(4)(樹脂組成物(4))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(4)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(4)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(4)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(4)を得た。
<耐光性の評価>
前記測定サンプル(4)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは28.9%であり、耐光性試験前の測定サンプル(4)のTvisは84.2%であったので、ΔTvisは、55.3%であった。
前記測定サンプル(4)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは28.9%であり、耐光性試験前の測定サンプル(4)のTvisは84.2%であったので、ΔTvisは、55.3%であった。
〔実施例5〕
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)34.8mgの代わりに、中和分散剤(2)36.8mgを添加したこと以外は実施例3と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(5)(樹脂組成物(5))を得た。
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)34.8mgの代わりに、中和分散剤(2)36.8mgを添加したこと以外は実施例3と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(5)(樹脂組成物(5))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(5)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(5)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(5)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(5)を得た。
<耐光性の評価>
前記測定サンプル(5)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは34.8%であり、耐光性試験前の測定サンプル(5)のTvisは83.5%であったので、ΔTvisは、48.7%であった。
前記測定サンプル(5)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは34.8%であり、耐光性試験前の測定サンプル(5)のTvisは83.5%であったので、ΔTvisは、48.7%であった。
〔実施例6〕
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)34.8mgの代わりに、中和分散剤(2)61.2mgを添加したこと以外は実施例3と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(6)(樹脂組成物(6))を得た。
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)34.8mgの代わりに、中和分散剤(2)61.2mgを添加したこと以外は実施例3と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(6)(樹脂組成物(6))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(6)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(6)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(6)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(6)を得た。
<耐光性の評価>
前記測定サンプル(6)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは24.4%であり、耐光性試験前の測定サンプル(6)のTvisは82.9%であったので、ΔTvisは、58.5%であった。
前記測定サンプル(6)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは24.4%であり、耐光性試験前の測定サンプル(6)のTvisは82.9%であったので、ΔTvisは、58.5%であった。
〔実施例7〕
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)34.8mgの代わりに、中和分散剤(2)11.6mgを添加したこと以外は実施例3と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(7)(樹脂組成物(7))を得た。
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)34.8mgの代わりに、中和分散剤(2)11.6mgを添加したこと以外は実施例3と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(7)(樹脂組成物(7))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(7)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(7)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(7)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(7)を得た。
<耐光性の評価>
前記測定サンプル(7)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは29.7%であり、耐光性試験前の測定サンプル(7)のTvisは81.2%であったので、ΔTvisは、51.5%であった。
前記測定サンプル(7)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは29.7%であり、耐光性試験前の測定サンプル(7)のTvisは81.2%であったので、ΔTvisは、51.5%であった。
〔実施例8〕
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)34.8mgの代わりに、中和分散剤(2)23.2mgを添加したこと以外は実施例3と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(8)(樹脂組成物(8))を得た。
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)34.8mgの代わりに、中和分散剤(2)23.2mgを添加したこと以外は実施例3と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(8)(樹脂組成物(8))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(8)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(8)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(8)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(8)を得た。
<耐光性の評価>
前記測定サンプル(8)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは33.8%であり、耐光性試験前の測定サンプル(8)のTvisは81.1%であったので、ΔTvisは、47.3%であった。
前記測定サンプル(8)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは33.8%であり、耐光性試験前の測定サンプル(8)のTvisは81.1%であったので、ΔTvisは、47.3%であった。
〔実施例9〕
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.9g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.0gを加え、25℃で10時間撹拌し、溶液(C液)を得た。
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.9g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.0gを加え、25℃で10時間撹拌し、溶液(C液)を得た。
50mlナスフラスコに、前記n-ブチルホスホン酸銅塩トルエン分散液(2)1.30g(銅塩を0.583mmol含む)、トルエン5g、前記中和分散剤(1)92.8mgを加え、25℃で3時間撹拌し、分散液(D液)を得た。
D液をC液に加え、25℃で1時間撹拌後、1時間超音波照射し、PVBを均一に溶解させた。この分散液をテフロン(登録商標)製バットに広げ、20時間25℃で風乾した。さらに40℃で5時間、70℃で3.5時間真空乾燥を行って溶媒を完全に除去し、銅塩微粒子が分散したPVB樹脂(9)を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(9)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(9)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(9)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(9)を得た。
<耐光性の評価>
前記測定サンプル(9)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは41.3%であり、耐光性試験前の測定サンプル(9)のTvisは82.8%であったので、ΔTvisは、41.5%であった。
前記測定サンプル(9)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは41.3%であり、耐光性試験前の測定サンプル(9)のTvisは82.8%であったので、ΔTvisは、41.5%であった。
〔比較例2〕
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)を添加しないこと以外は実施例3と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(c2)(樹脂組成物(c2))を得た。
(銅塩微粒子分散樹脂の調製)
中和分散剤(1)を添加しないこと以外は実施例3と同様に行い、n-ブチルホスホン酸銅塩微粒子が分散したPVB樹脂(c2)(樹脂組成物(c2))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(c2)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(c2)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(c2)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(c2)を得た。
<耐光性の評価>
前記測定サンプル(c2)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは17.6%であり、耐光性試験前の測定サンプル(c2)のTvisは86.3%であったので、ΔTvisは、68.7%であった。
前記測定サンプル(c2)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは17.6%であり、耐光性試験前の測定サンプル(c2)のTvisは86.3%であったので、ΔTvisは、68.7%であった。
実施例1~9、比較例1、2における耐光性の評価結果を表1に示す。n-ブチルホスホン酸銅塩トルエン分散液(1)を用いた実施例1、2および比較例1を比較すると、実施例は、耐光性に優れることがわかる。また、n-ブチルホスホン酸銅塩トルエン分散液(2)を用いた実施例3~9および比較例2を比較すると、実施例は、耐光性に優れることがわかる。

〔製造例5〕
(中和分散剤の作成)
リン酸エステル化合物(A)10gを純水100mlに溶解し、0.91MのKOH水溶液で中和した。pHメーターで水溶液を測定し、pH6.1となったところで中和を終了した。
(中和分散剤の作成)
リン酸エステル化合物(A)10gを純水100mlに溶解し、0.91MのKOH水溶液で中和した。pHメーターで水溶液を測定し、pH6.1となったところで中和を終了した。
エバポレーターで水を留去し、次いでトルエン100mlを添加し、エバポレーターで留去して水分およびトルエンを除去し、白色のワックス状固体(中和分散剤(3))を10.5g得た。
〔製造例6〕
(中和分散剤の作成)
リン酸エステル化合物(A)10gを純水100mlに溶解し、0.91MのKOH水溶液で中和した。pHメーターで水溶液を測定し、pH10.3となったところで中和を終了した。
(中和分散剤の作成)
リン酸エステル化合物(A)10gを純水100mlに溶解し、0.91MのKOH水溶液で中和した。pHメーターで水溶液を測定し、pH10.3となったところで中和を終了した。
エバポレーターで水を留去し、次いでトルエン100mlを添加し、エバポレーターで留去して水分およびトルエンを除去し、白色のワックス状固体(中和分散剤(4))を10.8g得た。
〔製造例7〕
(中和分散剤の作成)
リン酸エステル化合物(A)10gを純水100mlに溶解し、0.87MのCsOH水溶液で中和した。pHメーターで水溶液を測定し、pH5.4となったところで中和を終了した。
(中和分散剤の作成)
リン酸エステル化合物(A)10gを純水100mlに溶解し、0.87MのCsOH水溶液で中和した。pHメーターで水溶液を測定し、pH5.4となったところで中和を終了した。
エバポレーターで水を留去し、次いでトルエン100mlを添加し、エバポレーターで留去して水分およびトルエンを除去し、白色のワックス状固体(中和分散剤(5))を11.1g得た。
〔製造例8〕
(中和分散剤の作成)
リン酸エステル化合物(A)10gを純水100mlに溶解し、0.87MのCsOH水溶液で中和した。pHメーターで水溶液を測定し、pH10.5となったところで中和を終了した。
(中和分散剤の作成)
リン酸エステル化合物(A)10gを純水100mlに溶解し、0.87MのCsOH水溶液で中和した。pHメーターで水溶液を測定し、pH10.5となったところで中和を終了した。
エバポレーターで水を留去し、次いでトルエン100mlを添加し、エバポレーターで留去して水分およびトルエンを除去し、白色のワックス状固体(中和分散剤(6))を11.7g得た。
〔製造例9〕
(中和分散剤の作成)
リン酸エステル化合物(B)10gを純水100mlに溶解し、1MのNaOH水溶液で中和した。pHメーターで水溶液を測定し、pH5.4となったところで中和を終了した。
(中和分散剤の作成)
リン酸エステル化合物(B)10gを純水100mlに溶解し、1MのNaOH水溶液で中和した。pHメーターで水溶液を測定し、pH5.4となったところで中和を終了した。
エバポレーターで水を留去し、次いでトルエン100mlを添加し、エバポレーターで留去して水分およびトルエンを除去し、白色のワックス状固体(中和分散剤(7))を10.3g得た。
製造例9において用いたリン酸エステル化合物(B)は、前記一般式(2)で表されるリン酸モノエステルと、前記一般式(3)で表されるリン酸ジエステルと、ポリオキシアルキレンモノアルキルエーテル(R12(OCH2CH2)nOH)との混合物であり、前記式(2)、(3)中におけるR2、R3およびR4、並びにポリオキシアルキレンモノアルキルエーテルのnが10であり、R12が炭素数12~15のアルキル基(炭素数が異なる混合物)であるものである。なお、リン酸エステル化合物(B)中のモノエステルとジエステルとポリオキシアルキレンモノアルキルエーテルとの存在比(モル比)は、ほぼ1:2:1である。
中和分散剤(8)として、リン酸エステル化合物のナトリウム塩(DLP-10、日光ケミカルズ(株)製)を使用した。リン酸エステル化合物のナトリウム塩10gを純水100mlに溶解した水溶液を、pHメーターで測定したところpHは5.4であった。
〔実施例10〕
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.00gを加えた。
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.00gを加えた。
これに、前記n-ブチルホスホン酸銅塩トルエン分散液(2)1.30g(銅塩を0.583mmol含む)を添加した。
これに、前記中和分散剤(3)を58.2mg加えた。25℃で10時間撹拌後、1時間超音波照射し、PVBを均一に溶解させた。この分散液をテフロン(登録商標)製バットに広げ、20時間25℃で風乾した。さらに40℃で5時間、70℃で3.5時間真空乾燥を行って溶媒を完全に除去し、銅塩微粒子が分散したPVB樹脂(10)(樹脂組成物(10))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(10)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(10)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(10)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(10)を得た。
<耐光性の評価>
前記測定サンプル(10)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは41.1%であり、耐光性試験前の測定サンプル(10)のTvisは80.9%であったので、ΔTvisは、39.8%であった。
前記測定サンプル(10)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは41.1%であり、耐光性試験前の測定サンプル(10)のTvisは80.9%であったので、ΔTvisは、39.8%であった。
〔実施例11〕
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.00gを加えた。
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.00gを加えた。
これに、前記n-ブチルホスホン酸銅塩トルエン分散液(2)1.30g(銅塩を0.583mmol含む)を添加した。
これに、前記中和分散剤(4)を24.3mg加えた。25℃で10時間撹拌後、1時間超音波照射し、PVBを均一に溶解させた。この分散液をテフロン(登録商標)製バットに広げ、20時間25℃で風乾した。さらに40℃で5時間、70℃で3.5時間真空乾燥を行って溶媒を完全に除去し、銅塩微粒子が分散したPVB樹脂(11)(樹脂組成物(11))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(11)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(11)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(11)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(11)を得た。
<耐光性の評価>
前記測定サンプル(11)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは49.9%であり、耐光性試験前の測定サンプル(11)のTvisは82.7%であったので、ΔTvisは、32.8%であった。
前記測定サンプル(11)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは49.9%であり、耐光性試験前の測定サンプル(11)のTvisは82.7%であったので、ΔTvisは、32.8%であった。
〔実施例12〕
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.00gを加えた。
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.00gを加えた。
これに、前記n-ブチルホスホン酸銅塩トルエン分散液(2)1.30g(銅塩を0.583mmol含む)を添加した。
これに、前記中和分散剤(5)を58.6mg加えた。25℃で10時間撹拌後、1時間超音波照射し、PVBを均一に溶解させた。この分散液をテフロン(登録商標)製バットに広げ、20時間25℃で風乾した。さらに40℃で5時間、70℃で3.5時間真空乾燥を行って溶媒を完全に除去し、銅塩微粒子が分散したPVB樹脂(12)(樹脂組成物(12))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(12)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(12)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(12)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(12)を得た。
<耐光性の評価>
前記測定サンプル(12)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは36.9%であり、耐光性試験前の測定サンプル(12)のTvisは81.1%であったので、ΔTvisは、44.2%であった。
前記測定サンプル(12)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは36.9%であり、耐光性試験前の測定サンプル(12)のTvisは81.1%であったので、ΔTvisは、44.2%であった。
〔実施例13〕
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.00gを加えた。
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.00gを加えた。
これに、前記n-ブチルホスホン酸銅塩トルエン分散液(2)1.30g(銅塩を0.583mmol含む)を添加した。
これに、前記中和分散剤(6)を23.8mg加えた。25℃で10時間撹拌後、1時間超音波照射し、PVBを均一に溶解させた。この分散液をテフロン(登録商標)製バットに広げ、20時間25℃で風乾した。さらに40℃で5時間、70℃で3.5時間真空乾燥を行って溶媒を完全に除去し、銅塩微粒子が分散したPVB樹脂(13)(樹脂組成物(13))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(13)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(13)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(13)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(13)を得た。
<耐光性の評価>
前記測定サンプル(13)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは44.0%であり、耐光性試験前の測定サンプル(13)のTvisは81.7%であったので、ΔTvisは、37.7%であった。
前記測定サンプル(13)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは44.0%であり、耐光性試験前の測定サンプル(13)のTvisは81.7%であったので、ΔTvisは、37.7%であった。
〔実施例14〕
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.00gを加えた。
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.00gを加えた。
これに、前記n-ブチルホスホン酸銅塩トルエン分散液(2)1.30g(銅塩を0.583mmol含む)を添加した。
これに、前記中和分散剤(7)を93.3mg加えた。25℃で10時間撹拌後、1時間超音波照射し、PVBを均一に溶解させた。この分散液をテフロン(登録商標)製バットに広げ、20時間25℃で風乾した。さらに40℃で5時間、70℃で3.5時間真空乾燥を行って溶媒を完全に除去し、銅塩微粒子が分散したPVB樹脂(14)(樹脂組成物(14))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(14)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(14)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(14)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(14)を得た。
<耐光性の評価>
前記測定サンプル(14)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは47.2%であり、耐光性試験前の測定サンプル(14)のTvisは81.8%であったので、ΔTvisは、34.6%であった。
前記測定サンプル(14)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは47.2%であり、耐光性試験前の測定サンプル(14)のTvisは81.8%であったので、ΔTvisは、34.6%であった。
〔実施例15〕
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.00gを加えた。
(銅塩微粒子分散樹脂の調製)
300ml三角フラスコに、トリエチレングリコールビス(2-エチルヘキサノエート)1.90g、トルエン100g、エタノール40g、ポリビニルブチラール(PVB)5.00gを加えた。
これに、前記n-ブチルホスホン酸銅塩トルエン分散液(2)1.30g(銅塩を0.583mmol含む)を添加した。
これに、前記中和分散剤(8)を93.1mg加えた。25℃で10時間撹拌後、1時間超音波照射し、PVBを均一に溶解させた。この分散液をテフロン(登録商標)製バットに広げ、20時間25℃で風乾した。さらに40℃で5時間、70℃で3.5時間真空乾燥を行って溶媒を完全に除去し、銅塩微粒子が分散したPVB樹脂(15)(樹脂組成物(15))を得た。
(評価)
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(15)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(15)を得た。
<樹脂シートおよび測定サンプルの作製>
銅塩微粒子が分散したPVB樹脂(1)を、銅塩微粒子が分散したPVB樹脂(15)に代えた以外は、実施例1の樹脂シートの作製の項と同様に行い、測定サンプル(15)を得た。
<耐光性の評価>
前記測定サンプル(15)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは37.5%であり、耐光性試験前の測定サンプル(15)のTvisは79.8%であったので、ΔTvisは、42.3%であった。
前記測定サンプル(15)を使用し、実施例1に示した方法と同様の方法で耐光性試験を行った。400時間後のTvisは37.5%であり、耐光性試験前の測定サンプル(15)のTvisは79.8%であったので、ΔTvisは、42.3%であった。
実施例10~15における耐光性の評価結果を表2に示す。n-ブチルホスホン酸銅塩トルエン分散液(2)を用いた実施例10~15および前述の比較例2を比較すると、実施例は、耐光性に優れることがわかる。

Claims (10)
- 近赤外線吸収剤と、中和分散剤と、樹脂とからなる樹脂組成物であり、
前記近赤外線吸収剤が、少なくとも銅塩と、下記一般式(1)で表わされるホスホン酸化合物とを反応させることにより得られ、
前記中和分散剤が、リン酸モノエステルおよびリン酸ジエステルから選択される少なくとも1種のリン酸エステル化合物を、アルカリ金属塩で中和することにより得られる中和分散剤であることを特徴とする樹脂組成物。

- 前記アルカリ金属塩が、ナトリウム塩、カリウム塩、およびセシウム塩から選択される少なくとも1種の塩である請求項1に記載の樹脂組成物。
- 前記アルカリ金属塩が、ナトリウム塩である請求項1に記載の樹脂組成物。
- 前記リン酸モノエステルが、下記一般式(2)で表されるリン酸モノエステルであり、前記リン酸ジエステルが、下記一般式(3)で表されるリン酸ジエステルである請求項1~3のいずれか一項に記載の樹脂組成物。

- 前記樹脂が、ポリビニルアセタール樹脂、エチレン‐酢酸ビニル共重合体、(メタ)アクリル酸樹脂、ポリエステル樹脂、ポリウレタン樹脂、塩化ビニル樹脂、ポリオレフィン樹脂、ポリカーボネート樹脂、およびノルボルネン樹脂から選択される少なくとも1種の樹脂である請求項1~4のいずれか一項に記載の樹脂組成物。
- 前記樹脂が、ポリビニルブチラール樹脂、またはエチレン‐酢酸ビニル共重合体である請求項1~4のいずれか一項に記載の樹脂組成物。
- 前記樹脂100質量部あたり、近赤外線吸収剤を0.05~50質量部含有する請求項1~6のいずれか一項に記載の樹脂組成物。
- 前記樹脂100質量部あたり、中和分散剤を0.01~10質量部含有する請求項1~7のいずれか一項に記載の樹脂組成物。
- 前記請求項1~8のいずれか一項に記載の樹脂組成物から形成される合わせガラス用中間膜。
- 前記請求項9に記載の合わせガラス用中間膜を有する合わせガラス。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013247431 | 2013-11-29 | ||
| JP2013-247431 | 2013-11-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015080184A1 true WO2015080184A1 (ja) | 2015-06-04 |
Family
ID=53199123
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/081325 WO2015080184A1 (ja) | 2013-11-29 | 2014-11-27 | 樹脂組成物およびその用途 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2015080184A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018090809A (ja) * | 2016-04-21 | 2018-06-14 | 日本板硝子株式会社 | 赤外線吸収性組成物、赤外線カットフィルタ、及び撮像光学系 |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002069305A (ja) * | 2000-08-25 | 2002-03-08 | Kureha Chem Ind Co Ltd | 光学材料及びその製造方法 |
| JP2006240893A (ja) * | 2005-02-28 | 2006-09-14 | Sekisui Chem Co Ltd | 合わせガラス用着色中間膜および合わせガラス |
| WO2009123016A1 (ja) * | 2008-03-31 | 2009-10-08 | 株式会社クレハ | ホスホン酸銅化合物、並びにこれを含む赤外吸収材料及び積層体 |
| JP2009242650A (ja) * | 2008-03-31 | 2009-10-22 | Kureha Corp | 銅塩組成物、並びに、これを用いた樹脂組成物、赤外吸収膜及び光学部材 |
| JP2011099038A (ja) * | 2009-11-05 | 2011-05-19 | Kureha Corp | 近赤外線吸収剤及びその製造方法、並びに光学材料 |
| WO2012105150A1 (ja) * | 2011-02-02 | 2012-08-09 | 株式会社クレハ | 樹脂組成物およびその用途 |
| JP2012201865A (ja) * | 2011-03-28 | 2012-10-22 | Kureha Corp | 銅塩微粒子分散樹脂の製造方法、および銅塩微粒子分散樹脂 |
| WO2014119618A1 (ja) * | 2013-01-31 | 2014-08-07 | 株式会社クレハ | 近赤外線吸収剤の製造方法、近赤外線吸収剤およびその用途 |
| WO2014175231A1 (ja) * | 2013-04-26 | 2014-10-30 | 株式会社クレハ | 樹脂組成物およびその用途 |
| JP2014218597A (ja) * | 2013-05-09 | 2014-11-20 | 株式会社クレハ | 樹脂組成物およびその用途 |
-
2014
- 2014-11-27 WO PCT/JP2014/081325 patent/WO2015080184A1/ja active Application Filing
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002069305A (ja) * | 2000-08-25 | 2002-03-08 | Kureha Chem Ind Co Ltd | 光学材料及びその製造方法 |
| JP2006240893A (ja) * | 2005-02-28 | 2006-09-14 | Sekisui Chem Co Ltd | 合わせガラス用着色中間膜および合わせガラス |
| WO2009123016A1 (ja) * | 2008-03-31 | 2009-10-08 | 株式会社クレハ | ホスホン酸銅化合物、並びにこれを含む赤外吸収材料及び積層体 |
| JP2009242650A (ja) * | 2008-03-31 | 2009-10-22 | Kureha Corp | 銅塩組成物、並びに、これを用いた樹脂組成物、赤外吸収膜及び光学部材 |
| JP2011099038A (ja) * | 2009-11-05 | 2011-05-19 | Kureha Corp | 近赤外線吸収剤及びその製造方法、並びに光学材料 |
| WO2012105150A1 (ja) * | 2011-02-02 | 2012-08-09 | 株式会社クレハ | 樹脂組成物およびその用途 |
| JP2012201865A (ja) * | 2011-03-28 | 2012-10-22 | Kureha Corp | 銅塩微粒子分散樹脂の製造方法、および銅塩微粒子分散樹脂 |
| WO2014119618A1 (ja) * | 2013-01-31 | 2014-08-07 | 株式会社クレハ | 近赤外線吸収剤の製造方法、近赤外線吸収剤およびその用途 |
| WO2014175231A1 (ja) * | 2013-04-26 | 2014-10-30 | 株式会社クレハ | 樹脂組成物およびその用途 |
| JP2014218597A (ja) * | 2013-05-09 | 2014-11-20 | 株式会社クレハ | 樹脂組成物およびその用途 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018090809A (ja) * | 2016-04-21 | 2018-06-14 | 日本板硝子株式会社 | 赤外線吸収性組成物、赤外線カットフィルタ、及び撮像光学系 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5919401B2 (ja) | 合わせガラス用中間膜及び合わせガラス | |
| JP4891455B2 (ja) | 赤外線遮断性樹脂 | |
| RU2405744C2 (ru) | Промежуточная пленка для многослойного стекла и многослойное стекло | |
| JP2018066015A (ja) | シート | |
| JP5738031B2 (ja) | 銅塩微粒子分散樹脂の製造方法、および銅塩微粒子分散樹脂 | |
| JP5766218B2 (ja) | 樹脂組成物およびその用途 | |
| CN1833014A (zh) | 红外线吸收组合物、树脂组合物、夹层玻璃用中间膜、层合体、夹层玻璃和建材 | |
| JP4912591B2 (ja) | ローカラーpvbシートおよびその製造方法 | |
| JP2016124903A (ja) | 近赤外線吸収剤微粒子分散液の製造方法、近赤外線吸収剤微粒子分散液およびその用途 | |
| JP2015134893A (ja) | 近赤外線吸収剤分散液の製造方法、近赤外線吸収剤分散液およびその用途 | |
| CN115427502A (zh) | 树脂组合物、夹层玻璃用中间膜及夹层玻璃 | |
| WO2015080184A1 (ja) | 樹脂組成物およびその用途 | |
| WO2014119618A1 (ja) | 近赤外線吸収剤の製造方法、近赤外線吸収剤およびその用途 | |
| JP2012087243A (ja) | 銅塩微粒子分散樹脂の製造方法、銅塩微粒子分散樹脂およびマスターバッチ | |
| WO2014027639A1 (ja) | 樹脂組成物およびその用途 | |
| JP2014218597A (ja) | 樹脂組成物およびその用途 | |
| WO2014171215A1 (ja) | 樹脂組成物およびその用途 | |
| WO2014175231A1 (ja) | 樹脂組成物およびその用途 | |
| JP5401232B2 (ja) | 合わせガラス用中間膜及び合わせガラス | |
| CN109863444A (zh) | 发光显示系统和平视显示器 | |
| JP2010150099A (ja) | 合わせガラス用中間膜およびそれを用いた合わせガラス | |
| JP2016060871A (ja) | 樹脂組成物およびその用途 | |
| JP2016060903A (ja) | 近赤外線吸収剤微粒子分散液の製造方法、近赤外線吸収剤微粒子分散液およびその用途 | |
| JP2016094512A (ja) | 近赤外線吸収剤分散液の製造方法および近赤外線吸収剤分散液並びにその用途 | |
| JP2010059009A (ja) | 合わせガラス用中間膜及び合わせガラス |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14865262 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14865262 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |