WO2014166386A1 - 抗新生血管的化合物、其中间体及其用途 - Google Patents
抗新生血管的化合物、其中间体及其用途 Download PDFInfo
- Publication number
- WO2014166386A1 WO2014166386A1 PCT/CN2014/074977 CN2014074977W WO2014166386A1 WO 2014166386 A1 WO2014166386 A1 WO 2014166386A1 CN 2014074977 W CN2014074977 W CN 2014074977W WO 2014166386 A1 WO2014166386 A1 WO 2014166386A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- halogen
- neovascularization
- preparation
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 175
- 230000003527 anti-angiogenesis Effects 0.000 title 1
- 201000010099 disease Diseases 0.000 claims abstract description 40
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 40
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 claims abstract description 25
- 230000002159 abnormal effect Effects 0.000 claims abstract description 24
- 102000001253 Protein Kinase Human genes 0.000 claims abstract description 23
- 108060006633 protein kinase Proteins 0.000 claims abstract description 22
- 230000002401 inhibitory effect Effects 0.000 claims abstract description 18
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 claims abstract description 17
- 230000035755 proliferation Effects 0.000 claims abstract description 17
- 206010064930 age-related macular degeneration Diseases 0.000 claims abstract description 15
- 208000000208 Wet Macular Degeneration Diseases 0.000 claims abstract description 14
- 102100023600 Fibroblast growth factor receptor 2 Human genes 0.000 claims abstract description 11
- 101710182389 Fibroblast growth factor receptor 2 Proteins 0.000 claims abstract description 11
- 238000002360 preparation method Methods 0.000 claims description 46
- 206010029113 Neovascularisation Diseases 0.000 claims description 31
- 239000000203 mixture Substances 0.000 claims description 28
- 239000003814 drug Substances 0.000 claims description 27
- 229940079593 drug Drugs 0.000 claims description 23
- 150000003839 salts Chemical class 0.000 claims description 23
- 208000005590 Choroidal Neovascularization Diseases 0.000 claims description 16
- 206010060823 Choroidal neovascularisation Diseases 0.000 claims description 16
- 239000000651 prodrug Substances 0.000 claims description 16
- 229940002612 prodrug Drugs 0.000 claims description 16
- 210000004204 blood vessel Anatomy 0.000 claims description 15
- 125000000217 alkyl group Chemical group 0.000 claims description 13
- 229910052736 halogen Inorganic materials 0.000 claims description 13
- 150000002367 halogens Chemical class 0.000 claims description 12
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 8
- 108010051742 Platelet-Derived Growth Factor beta Receptor Proteins 0.000 claims description 8
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 claims description 8
- -1 amino, hydroxyl Chemical group 0.000 claims description 8
- 208000010412 Glaucoma Diseases 0.000 claims description 7
- 201000003142 neovascular glaucoma Diseases 0.000 claims description 7
- 206010012689 Diabetic retinopathy Diseases 0.000 claims description 6
- 102000001332 SRC Human genes 0.000 claims description 6
- 108060006706 SRC Proteins 0.000 claims description 6
- 229910052739 hydrogen Inorganic materials 0.000 claims description 6
- 239000012528 membrane Substances 0.000 claims description 6
- 125000001424 substituent group Chemical group 0.000 claims description 6
- 229910052799 carbon Inorganic materials 0.000 claims description 5
- 125000004432 carbon atom Chemical group C* 0.000 claims description 5
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 4
- 125000005842 heteroatom Chemical group 0.000 claims description 4
- 229940043355 kinase inhibitor Drugs 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 4
- 239000003909 protein kinase inhibitor Substances 0.000 claims description 4
- 101000997832 Homo sapiens Tyrosine-protein kinase JAK2 Proteins 0.000 claims description 3
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 claims description 3
- 239000004037 angiogenesis inhibitor Substances 0.000 claims description 3
- 150000003573 thiols Chemical class 0.000 claims description 3
- 229940091171 VEGFR-2 tyrosine kinase inhibitor Drugs 0.000 claims 1
- 230000000694 effects Effects 0.000 abstract description 21
- 201000011510 cancer Diseases 0.000 abstract description 8
- 206010061218 Inflammation Diseases 0.000 abstract description 7
- 230000033115 angiogenesis Effects 0.000 abstract description 7
- 230000004054 inflammatory process Effects 0.000 abstract description 7
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 162
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 90
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 60
- 239000011541 reaction mixture Substances 0.000 description 50
- 235000019439 ethyl acetate Nutrition 0.000 description 44
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 35
- 238000006243 chemical reaction Methods 0.000 description 35
- 239000012043 crude product Substances 0.000 description 34
- 230000005764 inhibitory process Effects 0.000 description 27
- 239000011734 sodium Substances 0.000 description 27
- 239000012267 brine Substances 0.000 description 23
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 23
- 239000012074 organic phase Substances 0.000 description 21
- 238000012360 testing method Methods 0.000 description 17
- 241000252212 Danio rerio Species 0.000 description 16
- HKSZLNNOFSGOKW-UHFFFAOYSA-N ent-staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 HKSZLNNOFSGOKW-UHFFFAOYSA-N 0.000 description 16
- 239000000243 solution Substances 0.000 description 16
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical compound C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 16
- CGPUWJWCVCFERF-UHFFFAOYSA-N staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(OC)O1 CGPUWJWCVCFERF-UHFFFAOYSA-N 0.000 description 16
- 210000001508 eye Anatomy 0.000 description 15
- 238000000034 method Methods 0.000 description 14
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- 239000007787 solid Substances 0.000 description 13
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- 230000002062 proliferating effect Effects 0.000 description 12
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- MWKFXSUHUHTGQN-UHFFFAOYSA-N decan-1-ol Chemical compound CCCCCCCCCCO MWKFXSUHUHTGQN-UHFFFAOYSA-N 0.000 description 10
- 108090000623 proteins and genes Proteins 0.000 description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 9
- 239000000543 intermediate Substances 0.000 description 9
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 8
- 238000002073 fluorescence micrograph Methods 0.000 description 8
- 239000007924 injection Substances 0.000 description 8
- 238000002347 injection Methods 0.000 description 8
- 239000013642 negative control Substances 0.000 description 8
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 8
- 235000018102 proteins Nutrition 0.000 description 8
- 102000004169 proteins and genes Human genes 0.000 description 8
- 206010028980 Neoplasm Diseases 0.000 description 7
- 238000011161 development Methods 0.000 description 7
- 230000018109 developmental process Effects 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- 239000012299 nitrogen atmosphere Substances 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 208000009043 Chemical Burns Diseases 0.000 description 6
- 108091000080 Phosphotransferase Proteins 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 230000005856 abnormality Effects 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 102000020233 phosphotransferase Human genes 0.000 description 6
- 206010055665 Corneal neovascularisation Diseases 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- 238000010171 animal model Methods 0.000 description 5
- 229940125773 compound 10 Drugs 0.000 description 5
- 201000000159 corneal neovascularization Diseases 0.000 description 5
- 239000003112 inhibitor Substances 0.000 description 5
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 5
- 208000002780 macular degeneration Diseases 0.000 description 5
- 229910000029 sodium carbonate Inorganic materials 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- 230000002792 vascular Effects 0.000 description 5
- 230000006459 vascular development Effects 0.000 description 5
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- 208000018380 Chemical injury Diseases 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- 241000699666 Mus <mouse, genus> Species 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 230000005754 cellular signaling Effects 0.000 description 4
- 229940125898 compound 5 Drugs 0.000 description 4
- 230000003511 endothelial effect Effects 0.000 description 4
- FJKIXWOMBXYWOQ-UHFFFAOYSA-N ethenoxyethane Chemical compound CCOC=C FJKIXWOMBXYWOQ-UHFFFAOYSA-N 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 238000001819 mass spectrum Methods 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 239000002904 solvent Substances 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 210000005166 vasculature Anatomy 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 241000251468 Actinopterygii Species 0.000 description 3
- 208000024827 Alzheimer disease Diseases 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 206010058314 Dysplasia Diseases 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-diisopropylethylamine Substances CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 3
- 108091008606 PDGF receptors Proteins 0.000 description 3
- 102000011653 Platelet-Derived Growth Factor Receptors Human genes 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 230000001772 anti-angiogenic effect Effects 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 210000004087 cornea Anatomy 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 238000013100 final test Methods 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 230000000649 photocoagulation Effects 0.000 description 3
- 239000013641 positive control Substances 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 238000010898 silica gel chromatography Methods 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 125000000547 substituted alkyl group Chemical group 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 230000009261 transgenic effect Effects 0.000 description 3
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 description 2
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- 206010002091 Anaesthesia Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 108091008794 FGF receptors Proteins 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 208000032843 Hemorrhage Diseases 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 2
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 108091008605 VEGF receptors Proteins 0.000 description 2
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 2
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 2
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 230000037005 anaesthesia Effects 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940125758 compound 15 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 239000003889 eye drop Substances 0.000 description 2
- 239000003885 eye ointment Substances 0.000 description 2
- NIHNNTQXNPWCJQ-UHFFFAOYSA-N fluorene Chemical compound C1=CC=C2CC3=CC=CC=C3C2=C1 NIHNNTQXNPWCJQ-UHFFFAOYSA-N 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 208000026278 immune system disease Diseases 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- XTEGVFVZDVNBPF-UHFFFAOYSA-N naphthalene-1,5-disulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1S(O)(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-N 0.000 description 2
- BDJRBEYXGGNYIS-UHFFFAOYSA-N nonanedioic acid Chemical compound OC(=O)CCCCCCCC(O)=O BDJRBEYXGGNYIS-UHFFFAOYSA-N 0.000 description 2
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 238000002428 photodynamic therapy Methods 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- WLJVNTCWHIRURA-UHFFFAOYSA-N pimelic acid Chemical compound OC(=O)CCCCCC(O)=O WLJVNTCWHIRURA-UHFFFAOYSA-N 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 2
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 210000001525 retina Anatomy 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- SQGYOTSLMSWVJD-UHFFFAOYSA-N silver(1+) nitrate Chemical compound [Ag+].[O-]N(=O)=O SQGYOTSLMSWVJD-UHFFFAOYSA-N 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical compound OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 229940124676 vascular endothelial growth factor receptor Drugs 0.000 description 2
- 230000032665 vasculature development Effects 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- SAIZARLJIMKKAT-UHFFFAOYSA-N 2-(2-ethoxy-2-oxoethyl)-2-hydroxy-4-oxo-4-propoxybutanoic acid Chemical compound CCCOC(=O)CC(CC(=O)OCC)(C(=O)O)O SAIZARLJIMKKAT-UHFFFAOYSA-N 0.000 description 1
- OQMYZVWIXPPDDE-UHFFFAOYSA-N 2-(cyclohexylazaniumyl)acetate Chemical compound OC(=O)CNC1CCCCC1 OQMYZVWIXPPDDE-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-PPJXEINESA-N 2-phenylacetic acid Chemical compound O[14C](=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-PPJXEINESA-N 0.000 description 1
- VIUDTWATMPPKEL-UHFFFAOYSA-N 3-(trifluoromethyl)aniline Chemical compound NC1=CC=CC(C(F)(F)F)=C1 VIUDTWATMPPKEL-UHFFFAOYSA-N 0.000 description 1
- CYWHLOXWVAWMFO-UHFFFAOYSA-N 3-sulfanyl-1h-pyridine-2-thione Chemical compound SC1=CC=CN=C1S CYWHLOXWVAWMFO-UHFFFAOYSA-N 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 1
- ULBNVDPBWRTOPN-UHFFFAOYSA-N 4-sulfanylbenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(S)C=C1 ULBNVDPBWRTOPN-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 102100032528 C-type lectin domain family 11 member A Human genes 0.000 description 1
- 101710167766 C-type lectin domain family 11 member A Proteins 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 239000005632 Capric acid (CAS 334-48-5) Substances 0.000 description 1
- 239000005635 Caprylic acid (CAS 124-07-2) Substances 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical class NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- ZEOWTGPWHLSLOG-UHFFFAOYSA-N Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F Chemical compound Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F ZEOWTGPWHLSLOG-UHFFFAOYSA-N 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 208000028006 Corneal injury Diseases 0.000 description 1
- 241000252210 Cyprinidae Species 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010014733 Endometrial cancer Diseases 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- 241000283070 Equus zebra Species 0.000 description 1
- KIWBPDUYBMNFTB-UHFFFAOYSA-N Ethyl hydrogen sulfate Chemical compound CCOS(O)(=O)=O KIWBPDUYBMNFTB-UHFFFAOYSA-N 0.000 description 1
- 102000044168 Fibroblast Growth Factor Receptor Human genes 0.000 description 1
- 102100023593 Fibroblast growth factor receptor 1 Human genes 0.000 description 1
- 101710182386 Fibroblast growth factor receptor 1 Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 108010072039 Histidine kinase Proteins 0.000 description 1
- 101000916644 Homo sapiens Macrophage colony-stimulating factor 1 receptor Proteins 0.000 description 1
- 101000669917 Homo sapiens Rho-associated protein kinase 1 Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101000808011 Homo sapiens Vascular endothelial growth factor A Proteins 0.000 description 1
- 101000851018 Homo sapiens Vascular endothelial growth factor receptor 1 Proteins 0.000 description 1
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 208000032984 Intraoperative Complications Diseases 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- 239000003798 L01XE11 - Pazopanib Substances 0.000 description 1
- 102000004856 Lectins Human genes 0.000 description 1
- 108090001090 Lectins Proteins 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 102100028198 Macrophage colony-stimulating factor 1 receptor Human genes 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 101150038994 PDGFRA gene Proteins 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 208000034038 Pathologic Neovascularization Diseases 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 102100024924 Protein kinase C alpha type Human genes 0.000 description 1
- 101710109947 Protein kinase C alpha type Proteins 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 102100039313 Rho-associated protein kinase 1 Human genes 0.000 description 1
- 102000038012 SFKs Human genes 0.000 description 1
- 108091008118 SFKs Proteins 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 206010039509 Scab Diseases 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 229940100514 Syk tyrosine kinase inhibitor Drugs 0.000 description 1
- 239000006180 TBST buffer Substances 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- BGDKAVGWHJFAGW-UHFFFAOYSA-N Tropicamide Chemical compound C=1C=CC=CC=1C(CO)C(=O)N(CC)CC1=CC=NC=C1 BGDKAVGWHJFAGW-UHFFFAOYSA-N 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 102100033178 Vascular endothelial growth factor receptor 1 Human genes 0.000 description 1
- 206010047571 Visual impairment Diseases 0.000 description 1
- XMGZWGBXVLJOKE-UHFFFAOYSA-N acetic acid;toluene Chemical compound CC(O)=O.CC1=CC=CC=C1 XMGZWGBXVLJOKE-UHFFFAOYSA-N 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- JIMXXGFJRDUSRO-UHFFFAOYSA-N adamantane-1-carboxylic acid Chemical compound C1C(C2)CC3CC2CC1(C(=O)O)C3 JIMXXGFJRDUSRO-UHFFFAOYSA-N 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229960004909 aminosalicylic acid Drugs 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 230000002491 angiogenic effect Effects 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 229940125644 antibody drug Drugs 0.000 description 1
- 229940045988 antineoplastic drug protein kinase inhibitors Drugs 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 210000005252 bulbus oculi Anatomy 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 210000000748 cardiovascular system Anatomy 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 210000000795 conjunctiva Anatomy 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 230000000235 effect on cancer Effects 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- 210000002257 embryonic structure Anatomy 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 230000010595 endothelial cell migration Effects 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- FPIQZBQZKBKLEI-UHFFFAOYSA-N ethyl 1-[[2-chloroethyl(nitroso)carbamoyl]amino]cyclohexane-1-carboxylate Chemical compound ClCCN(N=O)C(=O)NC1(C(=O)OCC)CCCCC1 FPIQZBQZKBKLEI-UHFFFAOYSA-N 0.000 description 1
- XBPOBCXHALHJFP-UHFFFAOYSA-N ethyl 4-bromobutanoate Chemical compound CCOC(=O)CCCBr XBPOBCXHALHJFP-UHFFFAOYSA-N 0.000 description 1
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 230000004720 fertilization Effects 0.000 description 1
- 102000052178 fibroblast growth factor receptor activity proteins Human genes 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 230000023597 hemostasis Effects 0.000 description 1
- 102000058223 human VEGFA Human genes 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 150000001261 hydroxy acids Chemical class 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 206010023332 keratitis Diseases 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 229940076783 lucentis Drugs 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 210000001161 mammalian embryo Anatomy 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 206010027191 meningioma Diseases 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- JJWRKQIITDNSSJ-UHFFFAOYSA-N n,n-didecylaniline Chemical compound CCCCCCCCCCN(CCCCCCCCCC)C1=CC=CC=C1 JJWRKQIITDNSSJ-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 201000011519 neuroendocrine tumor Diseases 0.000 description 1
- 238000011587 new zealand white rabbit Methods 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 229960002446 octanoic acid Drugs 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 102000002574 p38 Mitogen-Activated Protein Kinases Human genes 0.000 description 1
- 108010068338 p38 Mitogen-Activated Protein Kinases Proteins 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 231100000915 pathological change Toxicity 0.000 description 1
- 230000036285 pathological change Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- CUIHSIWYWATEQL-UHFFFAOYSA-N pazopanib Chemical compound C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 CUIHSIWYWATEQL-UHFFFAOYSA-N 0.000 description 1
- 229960000639 pazopanib Drugs 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-M perchlorate Inorganic materials [O-]Cl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-M 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- DDBREPKUVSBGFI-UHFFFAOYSA-N phenobarbital Chemical compound C=1C=CC=CC=1C1(CC)C(=O)NC(=O)NC1=O DDBREPKUVSBGFI-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 230000009822 protein phosphorylation Effects 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 229960003876 ranibizumab Drugs 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000003938 response to stress Effects 0.000 description 1
- 230000002207 retinal effect Effects 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229910001961 silver nitrate Inorganic materials 0.000 description 1
- 235000020183 skimmed milk Nutrition 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 239000012085 test solution Substances 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- YFDSDPIBEUFTMI-UHFFFAOYSA-N tribromoethanol Chemical compound OCC(Br)(Br)Br YFDSDPIBEUFTMI-UHFFFAOYSA-N 0.000 description 1
- 229950004616 tribromoethanol Drugs 0.000 description 1
- 239000003656 tris buffered saline Substances 0.000 description 1
- 229960004791 tropicamide Drugs 0.000 description 1
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 210000003606 umbilical vein Anatomy 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 208000029257 vision disease Diseases 0.000 description 1
- 230000004393 visual impairment Effects 0.000 description 1
- QDLHCMPXEPAAMD-QAIWCSMKSA-N wortmannin Chemical compound C1([C@]2(C)C3=C(C4=O)OC=C3C(=O)O[C@@H]2COC)=C4[C@@H]2CCC(=O)[C@@]2(C)C[C@H]1OC(C)=O QDLHCMPXEPAAMD-QAIWCSMKSA-N 0.000 description 1
- QDLHCMPXEPAAMD-UHFFFAOYSA-N wortmannin Natural products COCC1OC(=O)C2=COC(C3=O)=C2C1(C)C1=C3C2CCC(=O)C2(C)CC1OC(C)=O QDLHCMPXEPAAMD-UHFFFAOYSA-N 0.000 description 1
- 108700024526 zebrafish sox32 Proteins 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
- C07D215/22—Oxygen atoms attached in position 2 or 4
- C07D215/227—Oxygen atoms attached in position 2 or 4 only one oxygen atom which is attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- Anti-angiogenic compound intermediate thereof and use thereof
- the present invention relates to a neovascular inhibitor and/or protein kinase inhibitor-like compound and the use of said compound. Background technique
- Neovascularization is associated with a variety of major human diseases, such as malignant tumors. It has been found that ocular neovascular diseases, including age-related macular degeneration (AMD), diabetic retinopathy, neovascular glaucoma, etc., are common features of these diseases in the abnormal proliferation of ocular neovascularization (Jin Xiao, et al. , Research progress in the application and mechanism of anti-VEGF drugs in ophthalmic diseases, Chinese and foreign medical treatment, 201 2 years).
- AMD age-related macular degeneration
- neovascular glaucoma etc.
- macular degeneration mainly has two kinds of conditions, namely, wet and macular degeneration (AMD), which is characterized by choroidal neovascularization into the subretinal and subsequent pathological changes such as hemorrhage, exudation and edema.
- AMD macular degeneration
- Wet macular degeneration will rapidly lose vision and be more severe than dryness.
- photodynamic therapy Although photodynamic therapy has improved the efficacy, it is still not ideal.
- Lucentis vascular endothelial factor antagonist
- VEGF subtype monoclonal antibody fragment that reduces neovascularization.
- the drug was approved by the US FDA for the treatment of wet macular degeneration, and its efficacy was good.
- this type of anti-VEGF drug has therapeutic effects on diabetic retinopathy and neovascular glaucoma.
- ranibizumab is an antibody drug, the price is extremely high, and it cannot be universalized throughout the world. Therefore, the study of small-molecule angiogenesis inhibitors with good efficacy and low price is the focus of intense competition in the international pharmaceutical industry today.
- Protein kinases also known as protein phosphakins, are a class of enzymes that catalyze the phosphorylation of proteins. It can transfer the ⁇ -phosphate on adenosine triphosphate (ATP) to the amino acid residues of the protein shield molecule, such as the hydroxyl group of certain serine, threonine or tyrosine residues, thereby changing the protein and enzyme. Conformation and activity. Phosphorylation of proteins is an important link in many signaling pathways. Most important life processes in cells are inseparable from protein phosphorylation.
- ATP adenosine triphosphate
- Protein kinases are classified into five classes: protein serine/threonine kinase, protein tyrosine kinase, protein histidine kinase, protein tryptophan kinase, and protein aspartate/glutathione kinase. Protein kinases play an important role in the regulation and maintenance of cellular processes, and abnormalities in kinase activity have been observed in many disease states, including: malignant neoplasms, immune diseases, cardiovascular diseases, diabetes, infectious diseases. Arthritis and other immune disorders, neurological diseases such as Alzheimer's disease, Alzheimer's disease (AD), etc., have been associated with more than 400 human diseases associated with protein kinases.
- AD Alzheimer's disease
- VEGFR vascular endothelial growth factor receptor family members
- VEGFR1, VEGFR2, etc. receptor tyrosine kinases, such as VEGFR1, VEGFR2, etc., which are involved in the growth and metastasis of malignant tumors and vascular proliferative diseases (such as macular degeneration, Tumors and other diseases have important effects in the development of diseases.
- PDGFR platelet-derived growth factor receptor
- receptor tyrosine kinases such as PDGFRa And PDGFRp
- colony stimulating factor 1 receptor stem cell growth factor receptor KIT, etc.
- abnormal expression of PDGFR has been found in melanoma, meningiomas, neuroendocrine tumors, ovarian cancer, prostate cancer, lung cancer and pancreatic cancer.
- Abnormal activation of KIT is a direct cause of many tumorigenesis and development.
- FGFR family members (fibroblast growth factor receptors), including FGFR1, FGFR2, etc., are closely related to cancer. For example, abnormal activation of FGFR2 has been found in endometrial cancer, cervical cancer, breast cancer, lung cancer, and gastric cancer.
- the SRC kinase family is a protein with tyrosine protein kinase activity. It was originally discovered as an oncogene protein with the ROUS sarcoma retrovirus. It has been found to inhibit SRC and may have a therapeutic or ameliorating effect on cancer or other diseases.
- the p38 mitogen-activated protein kinase (MAPK) pathway is an intracellular stress response signaling pathway that is closely related to inflammatory responses.
- the present invention provides a compound of formula I, and a pharmaceutically acceptable salt or prodrug thereof, having the structural formula:
- H amino, hydroxy or decyl
- R 2 and R 3 together with the atom to which they are attached form a substituted or unsubstituted five- or six-membered ring containing from 1 to 2 heteroatoms, the hetero atom being N, fluorene or S, the substituent being C1 -6 alkyl.
- R 2 and R 3 together with the carbon atom to which they are bonded form a substituted or unsubstituted five- or six-membered ring containing one N, and the substituent is a C 1-3 alkyl group.
- R 2 is selected from an amino group or -(CH 2 ) n NHR 8 ; or R 2 and R 3 together with the carbon atom to which they are bonded constitute a substituted or unsubstituted five- or six-membered ring containing one N,
- the substituent is an alkyl group of C1 to 3.
- halogen is F or CL
- at least one of R 2 and R 3 is an amino group, the balance being H; , and the same and selected from F or C1;
- the preparation method of the compound of the present invention may be any suitable method.
- the compound of the present invention can be preferably produced by an intermediate compound represented by the formula II.
- R 5 and R 6 are each independently selected from the group consisting of H, halogen, a C 1-6 pit group or a halogen-substituted alkyl group; and R 7 is selected from the group consisting of H, C1-6 alkyl or halogen.
- R 4 , R 5 and R 6 are each independently selected from the group consisting of a C, an alkyl group of C 1-2 or a substituted alkyl group; and R 7 is selected from H or a halogen.
- the preparation method of the intermediate compound represented by the formula II may include the following steps: w 0 > I
- R4,, R 6 and R 7 are as defined above.
- R, R 2 and R 3 are as defined above,
- the preparation method of the compound of the preferred embodiment of the present invention comprises the following reaction steps: (1) Synthesis of the intermediate compound represented by Formula 7:
- R 5 have the same definitions as above;
- the present invention also provides the use of the above compound, a pharmaceutically acceptable salt or prodrug thereof for the preparation of a medicament for inhibiting neovascularization of abnormal proliferation.
- the drug for inhibiting abnormal proliferation of neovascularization is a vascular endothelial growth receptor 2 (VEGFR2) inhibitor.
- VEGFR2 vascular endothelial growth receptor 2
- the drug for inhibiting abnormally proliferating neovascularization is a drug against ocular neovascularization. Further, the drug for inhibiting abnormally proliferating neovascularization is a choroidal neovascularization inhibitor. Wherein the drug for inhibiting abnormally proliferating neovascularization is a medicament for treating or preventing wet macular degeneration, diabetic retinopathy or neovascular glaucoma.
- the drug is an ophthalmic preparation.
- the ophthalmic preparation is an eye drop, an eye ointment or an ophthalmic injection.
- the ophthalmic injection is an intravitreal injection.
- the present invention also provides the use of the above compound, a pharmaceutically acceptable salt or prodrug thereof for the preparation of a medicament for the treatment of a disease associated with dysplastic neovascularization.
- the disease associated with abnormally proliferating neovascularization is a disease caused by abnormal vascular endothelial growth receptor 2 (VEGFR2).
- VEGFR2 abnormal vascular endothelial growth receptor 2
- the disease associated with abnormally proliferating neovascularization is a disease associated with ocular neovascularization.
- the disease associated with abnormally proliferating neovascularization is a disease associated with choroidal neovascularization.
- the disease associated with abnormally proliferating neovascularization is wet macular degeneration, diabetic retinal lesion or neovascular glaucoma.
- the present invention also provides a method for inhibiting abnormally proliferating neovascularization or a disease associated with neovascularization, comprising administering an effective amount of a compound of the present invention, a pharmaceutically acceptable salt or a prodrug thereof to a patient in need of treatment, Or a pharmaceutical composition as described below.
- the neovascularization which inhibits abnormal proliferation refers to a neovascularization which inhibits abnormal ocular hyperplasia; the disease associated with abnormally proliferating neovascularization is a disease associated with ocular neovascularization.
- the neovascularization which inhibits abnormal proliferation refers to inhibition of choroidal neovascularization; and the disease associated with abnormally proliferating neovascularization is a disease associated with choroidal neovascularization.
- the method for treating a neoplastic blood vessel which inhibits abnormal proliferation or a disease associated with dysplastic neovascularization specifically refers to a method for treating or preventing wet macular degeneration, diabetic retinopathy or neovascular glaucoma.
- the administration means direct administration to the eye or intravitreal or subconjunctival injection.
- the present invention also provides the above compound, a pharmaceutically acceptable salt or prodrug thereof for preparing protein kinase Use in the preparation of drugs.
- the present invention also provides the use of the above compound, a pharmaceutically acceptable salt or prodrug thereof for the preparation of a medicament for treating a disease caused by abnormal protein kinase.
- the present invention also provides a method for treating a disease caused by a protein kinase abnormality, comprising administering an effective amount of a compound of the present invention, a pharmaceutically acceptable salt or a prodrug thereof, or a drug described below to a patient in need of treatment. combination.
- the protein kinase is VEGFR2, PDGF- ⁇ , KIT, AURORA-B, FGFR2, SRC, JAK2 or P38-a, preferably VEGFR2, KIT or PDGFR-p.
- the disease caused by the abnormality of the protein kinase refers to an inflammation or a malignant tumor.
- the present invention also provides a pharmaceutical composition comprising an effective amount of the above compound and a pharmaceutically acceptable salt or prodrug thereof. Further, the composition is an ophthalmic preparation. In addition to the above-mentioned compounds provided by the present invention, the ophthalmic preparation may further comprise other drugs known to have similar therapeutic uses.
- the ophthalmic preparation is an eye drop, an eye ointment or an ophthalmic injection.
- the ophthalmic injection is an intravitreal or subconjunctival injection.
- Salts of the compounds of the invention can be prepared by methods known in the art. Treatment with an acid, or with a suitable anion exchanger, can form a salt with the above compounds.
- the pharmaceutically acceptable salt of the compound of the present invention may be an organic or inorganic acid addition salt having a basic nitrogen atom from the above compound.
- suitable inorganic acids include, but are not limited to, hydrogen (e.g., hydrochloric acid), sulfuric acid, or citric acid.
- suitable organic acids include, but are not limited to, carboxylic acids, phosphoric acids, sulfonic acids or aminocarboxylic acids such as acetic acid, propionic acid, caprylic acid, capric acid, dodecanoic acid, glycolic acid, lactic acid, fumaric acid, Succinic acid, adipic acid, pimelic acid, suberic acid, azelaic acid, malic acid, tartaric acid, citrate, amino acid, such as glutamic acid or aspartic acid, maleic acid, hydroxy acid, thiol Acid, cyclohexanecarboxylic acid, adamantanecarboxylic acid, benzoic acid, salicylic acid, 4 aminosalicylic acid, phthalic acid, phenylacetic acid, mandelic acid, cinnamic acid, methane or ethanesulfonic acid Acid, 2-hydroxyethanesulfonic acid, ethane-1,2-disulfonic acid
- it may be used in the isolation or purification of pharmaceutically unacceptable salts such as picrate or perchlorate.
- pharmaceutically acceptable salts such as picrate or perchlorate.
- it can only be a pharmaceutically acceptable salt or free compound, in the form of a suitable pharmaceutical preparation.
- the pharmaceutically acceptable prodrug of the present invention refers to a compound obtained by chemically modifying the compound to release an active ingredient by enzymatic or non-enzymatic conversion in vivo to exert a pharmacological effect.
- the above-mentioned compound or a pharmaceutically acceptable salt thereof is also isotopically labeled, and the isotopically labeled compound means the same as the compound listed herein, but one or more of the atoms are Another atomic substitution, the atomic mass or mass of the atom is different from the atomic mass or mass number that is common in nature.
- Isotopes which may be introduced into the compound include hydrogen, carbon, nitrogen, oxygen, sulfur, i.e., 2 H, 3 H, 13 C, 14 C, 15 N, 17 0> 18 0, 35 S.
- Compounds containing the above isotopes and/or other atomic isotopes and stereoisomers thereof, as well as pharmaceutically acceptable salts of the compounds, stereoisomers, are intended to be encompassed within the scope of the invention.
- the key intermediates and compounds in the present invention are isolated and purified in the manner of organic chemistry. Separation and purification methods are used and examples of the methods include filtration, extraction, drying, spin drying, and various types of chromatography. Alternatively, the intermediate can be subjected to the next reaction without purification.
- the compound of the invention has good anti-angiogenic angiogenesis, and the compound is inhibited by
- VEGFR2 (aka KDR) produces activity.
- KDR neovascularization
- FGFR2 FGFR2
- protein kinase abnormalities such as wet macular degeneration, inflammation, malignant tumors and the like.
- Figure 1 is a mass spectrum of Compound 2-1.
- Figure 2 is a mass map of Compound 2-2.
- Figure 3 is a nuclear magnetic diagram of Compound 2-2.
- Figure 4 is a mass spectrum of Compound 2-3.
- Figure 5 is a mass spectrum of Compound 2-4.
- Figure 6 is a mass spectrum of Compound 2-5.
- Figure 7 shows a fluorescence micrograph of zebrafish ridge vasculature development, in which 7A is a fluorescence micrograph of zebrafish ridge vasculature normal (negative control); 7B is treated with 1 uM compound 2-2, zebra Fluorescence micrographs of fish ridges that were inhibited by vascular development (100%).
- Fig. 8 is a graph showing the results of inhibition of VEGFR2 by in vitro compounds 2-1, 2-2 and 2-4 (i.e., DR2 in the figure).
- Figure 9 shows a Zeiss fluorescence micrograph of choroidal neovascularization, in which 9A is a Zeiss fluorescence micrograph of Compound 2-2 significantly inhibiting choroidal neovascularization at luM concentration; 9B is a Zeiss with PBS as a negative control Fluorescence micrograph.
- Figure 10 is a dose effect curve of the compound of the present invention, wherein 1 OA is a positive control for Staurosporine; 10B is Compound 2-1; and 10C is Compound 2-2.
- Figure 11 is a photograph showing the effect of Compound 2-2 on corneal neovascularization in mice, wherein 11A is the right eye of the mouse treated with Compound 2-2; 11B is the left eye of the mouse treated with PBS as a control .
- Figure 12 is a photograph showing the effect of Compound 2-2 on corneal neovascularization in rabbit eyes, wherein 12A is treated with Compound 2-2; 12B is treated with PBS as a control; and 12C is treated with Compound 2-1. detailed description
- step 1
- step 1
- Test Example 1 Zebrafish vascular development inhibition test
- the zebrafish (Danio rerio, also known as Zebra fish) is a hard-bone fish of the genus Cyprinidae (Danio).
- the similarity between the gene and the human gene is as high as 85%, and the female can produce 200-300 at a time.
- the fertilization and embryo development process are carried out in vitro, and the formation can be formed within 24 hours, and the embryo is transparent and easy to observe. Changes in organ tissue. Many characteristics make it one of the five fish experimental animals recognized by the International Organization for Standardization.
- zebrafish are widely used in human disease research, and there are many studies on cardiovascular systems. Zebrafish can be used to screen for the effects of small molecule compounds on angiogenesis.
- This experiment uses an animal model of FLK1-GFP transgenic zebrafish, which is commonly used as a screening compound for angiogenesis, and can observe blood vessels in vivo under a fluorescence microscope (Suk-Won Jin, 2005, Development).
- the post-natal FLK1-GFP transgenic zebrafish embryos are selected, placed in culture and placed in a 28 ° C incubator for 3-5 days.
- the compound of the present invention, Pazopanib (130B, positive control) was directly added to the culture medium of zebrafish bred for 3-5 days; 40 uM DMSO was used as a negative control.
- the development of the spinal vasculature was examined 24 hours later and photographed with a fluorescence microscope.
- the inhibition rate of each compound on the zebrafish ridge vasculature development is shown in Table 1.
- the vascular development status of the negative control group was set to an inhibition rate of 0%, and the condition that the blood vessel was completely undeveloped was set to an inhibition rate of 100%.
- Figure 7A and Figure 7B show fluorescence micrographs of zebrafish after treatment with 40 uM DMSO in the negative control group and fluorescence micrographs of zebrafish after treatment with luM compound 2-2, respectively, showing the zebrafish of the negative control group
- the spinal vasculature develops normally, and after treatment with compound 2-2, the development of spinal vasculature is 100% inhibited.
- the cells were grown to 70-80%, and the compounds 2-1, 2-2 and 2-4 of the present invention were added (the concentration of each compound was ⁇ , ⁇ and ⁇ ), and VEGF was used as a control, and incubated for 30 minutes;
- the mouse was C57 BL (Jax Lab, USA) and was experimented with a laser-induced animal model of Choroidal Neovascularization (CNV).
- CNV Choroidal Neovascularization
- This model is currently the most widely used animal model for studying the effects of drugs on CNV formation in wet macular degeneration. All mice were 2-3 months old. After anesthesia with Avertin, 1% tropicamide (Alcon) was dilated, and 4 photocoagulation burns were applied to each eye using IRJDEX OcuLight GL532nm laser photocoagulation (IRIDEX) and slit lamp delivery system. Point, the parameter is power 120mW, spot size 75 ⁇ , and duration 0.1 seconds.
- compound 2-2 (luM) can significantly inhibit choroidal neovascularization and effectively treat or alleviate wet macular degeneration.
- the compounds of the present invention have a good antiangiogenic effect against dysplasia, and such compounds inhibit the activity of VEGFR2 production. Such compounds are useful in the treatment of diseases caused by neovascular abnormalities, such as wet macular degeneration.
- Test Example 4 Effect of the compound of the present invention on kinase
- the compounds of the present invention 2-1 and 2-2 have a good inhibitory activity against KDR and PDGFR- ⁇ , and the effect of the compound 2-2 is superior to that of the positive reference staurosporine.
- kinase activity inhibition assays were tested on compounds 2-1 and 2-2 at a test concentration of 5000 nM, repeated twice.
- the test compound was first dissolved in 100% DMSO at a concentration of 100 times the final test concentration, and the DMSO concentration in the test solution at all final tests was 1%.
- SB-202191 was used as a control
- ⁇ 3 ⁇ - ⁇ was treated with wortmannin as a control
- Staurosporine was used as a control.
- the concentration at the time of the test was 10 uM.
- both compounds 2-1 and 2-2 can selectively and selectively inhibit protein kinase KDR, compound 2-1 inhibition rate is greater than 97%, and compound 2-2 inhibition rate is 100%. These compounds also inhibit certain other protein kinases associated with diseases such as inflammation and tumors. Among them, the inhibition rate of 2-1 for KIT is about 72%, and the inhibition rate for PDGFR- ⁇ is about 71%. To some extent, AURORA-B (inhibition rate of about 31%), FGFR2 (about 36%), SRC (about 21%), and JAK2 (about 21%) were inhibited, and most other kinase inhibition rates were less than 5%.
- Compound 2-2 has an inhibition rate of FGFR2 of about 65%, a PDGFR- ⁇ inhibition rate of about 97%, a KIT inhibition rate of about 92%, a P38- ⁇ inhibition rate of about 80%, and an SRC inhibition rate of about 25%. It is 65%, but most of the remaining protein kinase inhibitory activity is less than 5%.
- the compound of the present invention has a high specificity and high inhibition effect on DR compared with the previously developed small molecule kinase inhibitor, and is closely related to tumor and inflammation to FGFR2 and PDGFR- ⁇ . Protein kinase also has a certain inhibitory effect, which indicates that compounds 2-1 and 2-2 have potential therapeutic activities against various tumors, inflammation and macular degeneration associated with abnormal activation of several protein kinases such as KDR and FGFR2.
- Test Example 5 Study of corneal neovascularization
- the animal model of chemical corneal injury is a widely used pharmacodynamic study disease model.
- mice were C57/BL (USA, Jax Lab).
- chemical burns were induced with 75% silver nitrate solution and 25% nitrate oblique rod. mold.
- 0.02 mi of compound 2-2 concentration lOOuM
- 0.02 mi of compound 2-2 was injected into the conjunctiva of the right eye, and then 0.2 ml of compound 2-2 was administered twice a day (concentration lOOuM), and the left eye was treated with PBS as a control, and treated in the same manner as the right eye.
- the cornea was photographed after 7 days. See Fig. 11 for the scab, where 11A is the result of treatment with compound 2-2 and 11B is the result of treatment with PBS as a negative control.
- the compounds of the present invention have a good angiogenic effect against dysplasia, which is produced by inhibiting VEGFR2 (aka K R ).
- VEGFR2 aka K R
- Such compounds can be used for the treatment of abnormally proliferating neovascularization, KDR, FGFR2 and other protein kinase abnormalities, such as wet macular degeneration, inflammation, malignant tumors and the like.
Abstract
Description









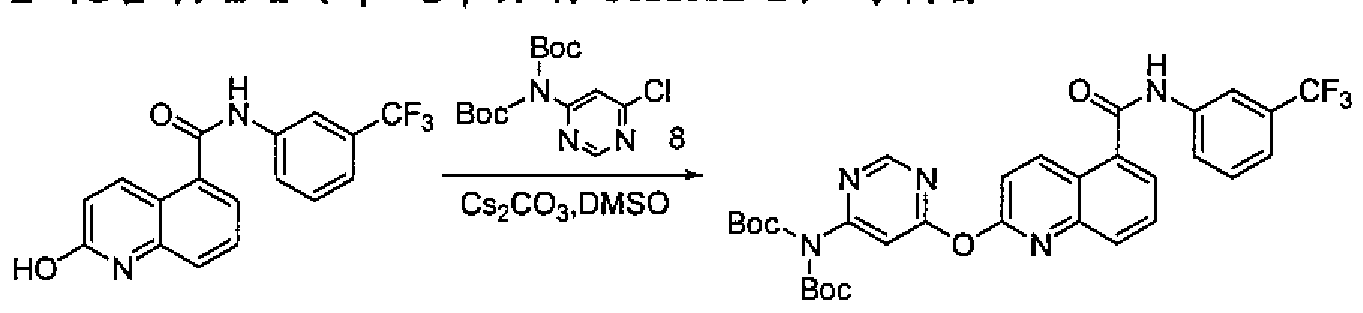

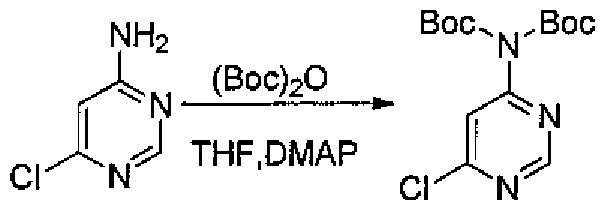














Claims




Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2014252584A AU2014252584B2 (en) | 2013-04-09 | 2014-04-09 | Anti-angiogenesis compound, intermediate and use thereof |
| ES14782609T ES2899726T3 (es) | 2013-04-09 | 2014-04-09 | Compuesto antiangiogénesis, intermediario y uso del mismo |
| CN201480000555.4A CN104334543B (zh) | 2013-04-09 | 2014-04-09 | 抗新生血管的化合物、其中间体及其用途 |
| KR1020157032078A KR101854203B1 (ko) | 2013-04-09 | 2014-04-09 | 신생혈관형성 억제 화합물, 그의 중간체 및 용도 |
| JP2016506767A JP6152921B2 (ja) | 2013-04-09 | 2014-04-09 | 抗血管新生化合物、中間体ならびにその使用 |
| CA2908849A CA2908849C (en) | 2013-04-09 | 2014-04-09 | Anti-angiogenesis compound, intermediate and use thereof |
| US14/392,101 US9540381B2 (en) | 2013-04-09 | 2014-04-09 | Anti-angiogenesis compound, intermediate and use thereof |
| EP14782609.3A EP2985283B1 (en) | 2013-04-09 | 2014-04-09 | Anti-angiogenesis compound, intermediate and use thereof |
| SG11201508330WA SG11201508330WA (en) | 2013-04-09 | 2014-04-09 | Anti-angiogenesis compound, intermediate and use thereof |
| NZ713187A NZ713187A (en) | 2013-04-09 | 2014-04-09 | Anti-angiogenesis compound, intermediate and use thereof |
| HK16103268.1A HK1215566A1 (zh) | 2013-04-09 | 2016-03-21 | 抗新生血管的化合物、其中間體及其用途 |
| US15/298,545 US10064864B2 (en) | 2013-04-09 | 2016-10-20 | Anti-angiogenesis compound, intermediate and use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201310122138.4 | 2013-04-09 | ||
| CN201310122138 | 2013-04-09 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/392,101 A-371-Of-International US9540381B2 (en) | 2013-04-09 | 2014-04-09 | Anti-angiogenesis compound, intermediate and use thereof |
| US15/298,545 Continuation US10064864B2 (en) | 2013-04-09 | 2016-10-20 | Anti-angiogenesis compound, intermediate and use thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014166386A1 true WO2014166386A1 (zh) | 2014-10-16 |
Family
ID=51688942
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2014/074977 WO2014166386A1 (zh) | 2013-04-09 | 2014-04-09 | 抗新生血管的化合物、其中间体及其用途 |
Country Status (12)
| Country | Link |
|---|---|
| US (2) | US9540381B2 (zh) |
| EP (1) | EP2985283B1 (zh) |
| JP (1) | JP6152921B2 (zh) |
| KR (1) | KR101854203B1 (zh) |
| CN (2) | CN107245071A (zh) |
| AU (1) | AU2014252584B2 (zh) |
| CA (1) | CA2908849C (zh) |
| ES (1) | ES2899726T3 (zh) |
| HK (1) | HK1215566A1 (zh) |
| NZ (1) | NZ713187A (zh) |
| SG (1) | SG11201508330WA (zh) |
| WO (1) | WO2014166386A1 (zh) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107812006A (zh) * | 2017-11-13 | 2018-03-20 | 全椒先奇医药科技有限公司 | 一种治疗急性肾衰药物组合物及其应用 |
| US10064864B2 (en) | 2013-04-09 | 2018-09-04 | Guangzhou Kangrui Biological Pharmaceutical Technology Co., Ltd. | Anti-angiogenesis compound, intermediate and use thereof |
| CN111743885A (zh) * | 2020-08-17 | 2020-10-09 | 山东省科学院生物研究所 | 一种对羟基苯乙酸在预防和/或治疗心血管疾病中的应用 |
| WO2023143611A1 (zh) * | 2022-01-30 | 2023-08-03 | 兰泰克生物技术公司 | 一种治疗癌症的方法 |
| WO2023226902A1 (zh) * | 2022-05-27 | 2023-11-30 | 苏州泽璟生物制药股份有限公司 | 一种kras g12c抑制剂的制备方法及其中间体 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102499780B1 (ko) * | 2016-05-27 | 2023-02-16 | 항저우 인노게이트 파마 컴퍼니 리미티드 | Fgfr4 저해제인 헤테로 고리 화합물 |
| CN115484957A (zh) * | 2020-04-24 | 2022-12-16 | 广州康睿生物医药科技股份有限公司 | 用于治疗眼部状况的制剂 |
| CN112194598B (zh) * | 2020-10-15 | 2021-12-21 | 郑州猫眼农业科技有限公司 | 3-(叔丁氧基羰基-r氧基羰基甲基-氨基)-丙酸酯的制备方法 |
| CN114634421A (zh) * | 2022-02-24 | 2022-06-17 | 济宁晟泰药业有限公司 | 一种达沙替尼中间体的制备方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1346271A (zh) * | 1999-02-10 | 2002-04-24 | 阿斯特拉曾尼卡有限公司 | 用作血管生成抑制剂的喹唑啉衍生物 |
| US20080194605A1 (en) * | 2005-04-12 | 2008-08-14 | Timo Heinrich | (1H-Ind0l-7-Yl)-(Pyrimidin-2-Ylamino)Methanone Derivatives and Related Compounds as Igf-R1 Inhibitors for the Treatment of Cancer |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4757743A (en) * | 1987-04-29 | 1988-07-19 | Vickers, Incorporated | Power transmission |
| JP4932495B2 (ja) * | 2004-01-23 | 2012-05-16 | アムゲン インコーポレイテッド | 化合物及び使用方法 |
| GT200600411A (es) * | 2005-09-13 | 2007-05-21 | Novartis Ag | Combinaciones que comprenden un inhibidor del receptor del factor de crecimiento endotelial vascular |
| RU2008127486A (ru) * | 2005-12-08 | 2010-01-20 | Милленниум Фармасьютикалз, Инк. (Us) | Бициклические соединения с ингибиторной активностью в отношении киназы |
| GB0604937D0 (en) * | 2006-03-10 | 2006-04-19 | Novartis Ag | Organic compounds |
| GB0605120D0 (en) * | 2006-03-14 | 2006-04-26 | Novartis Ag | Organic Compounds |
| ATE528002T1 (de) * | 2006-05-19 | 2011-10-15 | Bayer Pharma AG | Pyridoncarboxamidderivate zur behandlung hyperproliferativer und angiogenese-vermittelter leiden |
| WO2014154173A1 (zh) * | 2013-03-28 | 2014-10-02 | 广州康睿生物医药科技有限公司 | 一种抗血管新生化合物 |
| CA2908849C (en) | 2013-04-09 | 2017-11-07 | Guangzhou Hui Bo Rui Biological Pharmaceutical Technology Co., Ltd. | Anti-angiogenesis compound, intermediate and use thereof |
-
2014
- 2014-04-09 CA CA2908849A patent/CA2908849C/en active Active
- 2014-04-09 SG SG11201508330WA patent/SG11201508330WA/en unknown
- 2014-04-09 EP EP14782609.3A patent/EP2985283B1/en active Active
- 2014-04-09 NZ NZ713187A patent/NZ713187A/en unknown
- 2014-04-09 ES ES14782609T patent/ES2899726T3/es active Active
- 2014-04-09 JP JP2016506767A patent/JP6152921B2/ja active Active
- 2014-04-09 CN CN201710702483.3A patent/CN107245071A/zh active Pending
- 2014-04-09 AU AU2014252584A patent/AU2014252584B2/en active Active
- 2014-04-09 KR KR1020157032078A patent/KR101854203B1/ko active IP Right Grant
- 2014-04-09 CN CN201480000555.4A patent/CN104334543B/zh active Active
- 2014-04-09 WO PCT/CN2014/074977 patent/WO2014166386A1/zh active Application Filing
- 2014-04-09 US US14/392,101 patent/US9540381B2/en active Active
-
2016
- 2016-03-21 HK HK16103268.1A patent/HK1215566A1/zh unknown
- 2016-10-20 US US15/298,545 patent/US10064864B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1346271A (zh) * | 1999-02-10 | 2002-04-24 | 阿斯特拉曾尼卡有限公司 | 用作血管生成抑制剂的喹唑啉衍生物 |
| US20080194605A1 (en) * | 2005-04-12 | 2008-08-14 | Timo Heinrich | (1H-Ind0l-7-Yl)-(Pyrimidin-2-Ylamino)Methanone Derivatives and Related Compounds as Igf-R1 Inhibitors for the Treatment of Cancer |
Non-Patent Citations (2)
| Title |
|---|
| See also references of EP2985283A4 |
| XIAO JIN ET AL.: "Research Progress on the Clinical Application and Basic Mechanism of Anti-VEGF Drug", CHINA FOREIGN MEDICAL TREATMENT, 2012 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10064864B2 (en) | 2013-04-09 | 2018-09-04 | Guangzhou Kangrui Biological Pharmaceutical Technology Co., Ltd. | Anti-angiogenesis compound, intermediate and use thereof |
| CN107812006A (zh) * | 2017-11-13 | 2018-03-20 | 全椒先奇医药科技有限公司 | 一种治疗急性肾衰药物组合物及其应用 |
| CN111743885A (zh) * | 2020-08-17 | 2020-10-09 | 山东省科学院生物研究所 | 一种对羟基苯乙酸在预防和/或治疗心血管疾病中的应用 |
| CN111743885B (zh) * | 2020-08-17 | 2022-03-15 | 山东省科学院生物研究所 | 一种对羟基苯乙酸在预防和/或治疗心血管疾病中的应用 |
| WO2023143611A1 (zh) * | 2022-01-30 | 2023-08-03 | 兰泰克生物技术公司 | 一种治疗癌症的方法 |
| WO2023226902A1 (zh) * | 2022-05-27 | 2023-11-30 | 苏州泽璟生物制药股份有限公司 | 一种kras g12c抑制剂的制备方法及其中间体 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2985283B1 (en) | 2021-10-06 |
| AU2014252584A8 (en) | 2015-11-12 |
| US9540381B2 (en) | 2017-01-10 |
| CN104334543B (zh) | 2017-08-25 |
| KR20150139962A (ko) | 2015-12-14 |
| CN107245071A (zh) | 2017-10-13 |
| JP6152921B2 (ja) | 2017-06-28 |
| HK1215566A1 (zh) | 2016-09-02 |
| US20170119768A1 (en) | 2017-05-04 |
| US10064864B2 (en) | 2018-09-04 |
| SG11201508330WA (en) | 2015-11-27 |
| CN104334543A (zh) | 2015-02-04 |
| EP2985283A1 (en) | 2016-02-17 |
| CA2908849A1 (en) | 2014-10-16 |
| US20160075708A1 (en) | 2016-03-17 |
| EP2985283A4 (en) | 2016-09-14 |
| KR101854203B1 (ko) | 2018-06-20 |
| AU2014252584B2 (en) | 2016-10-13 |
| NZ713187A (en) | 2017-04-28 |
| CA2908849C (en) | 2017-11-07 |
| AU2014252584A1 (en) | 2015-11-05 |
| JP2016515636A (ja) | 2016-05-30 |
| ES2899726T3 (es) | 2022-03-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6152921B2 (ja) | 抗血管新生化合物、中間体ならびにその使用 | |
| TWI250151B (en) | Phthalazine derivatives and pharmaceutical composition comprising same | |
| TW539675B (en) | Novel 2,3-disubstituted-4(3H)-quinazolinones | |
| JP2013532689A (ja) | プロドラッグ形態のキナーゼ阻害剤化合物を使用して眼疾患を治療する方法 | |
| JP6579714B2 (ja) | 化合物 | |
| JP2012507556A (ja) | サーチュインモジュレーターとしてのピリジン、二環式ピリジンおよび関連するアナログ | |
| WO2008061796A2 (en) | Compounds for the treatment of diseases associated with amyloid or amyloid-like proteins | |
| EA013748B1 (ru) | Производные n-(гетероарил)-1-гетероарилалкил-1h-индол-2-карбоксамидов, их получение и их применение в терапии | |
| CN106715422A (zh) | 新型茚衍生物、其制备方法以及包含其作为活性成分的用于预防或治疗视网膜疾病的药物组合物 | |
| CN106146473B (zh) | 一种抗血管新生化合物 | |
| JP6291066B2 (ja) | ナフチル尿素誘導体およびその医療適用 | |
| JP2011201777A (ja) | 光学活性なヘテロシクリデン−n−アリールアセトアミド誘導体 | |
| JPH06509318A (ja) | トロポロン誘導体ならびに虚血性疾患を予防および治療するためのその薬剤組成物 | |
| CN101291913A (zh) | 作为5-羟色胺-6配体的吖嗪基-3-磺酰基吲唑衍生物 | |
| WO2023276828A1 (ja) | 腎障害の予防および/または治療用医薬組成物、並びにオートファジー活性化剤 | |
| JP2017516867A (ja) | 抗増殖性化合物としての1h−1,8−ナフチリジン−2−オン | |
| KR102619332B1 (ko) | Bet 단백질을 저해하는 신규한 카르복스아마이드 리독스 유도체 및 이를 이용한 안과질환 예방 및 치료용 조성물 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14782609 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2908849 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2016506767 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14392101 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014782609 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2014252584 Country of ref document: AU Date of ref document: 20140409 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20157032078 Country of ref document: KR Kind code of ref document: A |