WO2013104703A1 - Substituierte annellierte pyrimidine und triazine und ihre verwendung - Google Patents
Substituierte annellierte pyrimidine und triazine und ihre verwendung Download PDFInfo
- Publication number
- WO2013104703A1 WO2013104703A1 PCT/EP2013/050381 EP2013050381W WO2013104703A1 WO 2013104703 A1 WO2013104703 A1 WO 2013104703A1 EP 2013050381 W EP2013050381 W EP 2013050381W WO 2013104703 A1 WO2013104703 A1 WO 2013104703A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrogen
- methyl
- alkyl
- group
- formula
- Prior art date
Links
- 150000003230 pyrimidines Chemical class 0.000 title abstract description 5
- 150000003918 triazines Chemical class 0.000 title abstract description 5
- 238000000034 method Methods 0.000 claims abstract description 66
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 62
- 238000011282 treatment Methods 0.000 claims abstract description 42
- 201000010099 disease Diseases 0.000 claims abstract description 36
- 238000011321 prophylaxis Methods 0.000 claims abstract description 35
- 239000003814 drug Substances 0.000 claims abstract description 9
- 238000004519 manufacturing process Methods 0.000 claims abstract description 5
- 150000001875 compounds Chemical class 0.000 claims description 245
- 229910052739 hydrogen Inorganic materials 0.000 claims description 229
- 239000001257 hydrogen Substances 0.000 claims description 228
- -1 hydroxy, hydroxycarbonyl Chemical group 0.000 claims description 187
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 167
- 229910052731 fluorine Inorganic materials 0.000 claims description 145
- 239000011737 fluorine Substances 0.000 claims description 145
- 150000002431 hydrogen Chemical class 0.000 claims description 145
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 137
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 120
- 125000001424 substituent group Chemical group 0.000 claims description 108
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 104
- 150000003839 salts Chemical class 0.000 claims description 104
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 95
- 239000012453 solvate Substances 0.000 claims description 95
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 81
- 238000006243 chemical reaction Methods 0.000 claims description 81
- 125000000623 heterocyclic group Chemical group 0.000 claims description 81
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 80
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 71
- 239000002904 solvent Substances 0.000 claims description 60
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 58
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 50
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 claims description 48
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 47
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 45
- 125000000217 alkyl group Chemical group 0.000 claims description 44
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 38
- 239000000203 mixture Substances 0.000 claims description 38
- 229910052757 nitrogen Inorganic materials 0.000 claims description 38
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 38
- 239000012442 inert solvent Substances 0.000 claims description 37
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 35
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims description 34
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 34
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 34
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 33
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 33
- 239000002585 base Substances 0.000 claims description 33
- 125000004043 oxo group Chemical group O=* 0.000 claims description 32
- 125000004076 pyridyl group Chemical group 0.000 claims description 31
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 30
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 27
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical group C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 claims description 26
- 208000035475 disorder Diseases 0.000 claims description 25
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 24
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims description 23
- 125000004399 C1-C4 alkenyl group Chemical group 0.000 claims description 23
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 claims description 22
- 125000006650 (C2-C4) alkynyl group Chemical group 0.000 claims description 20
- VOPWNXZWBYDODV-UHFFFAOYSA-N Chlorodifluoromethane Chemical compound FC(F)Cl VOPWNXZWBYDODV-UHFFFAOYSA-N 0.000 claims description 20
- 229910052799 carbon Inorganic materials 0.000 claims description 20
- 239000000460 chlorine Substances 0.000 claims description 20
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 claims description 19
- 150000001412 amines Chemical class 0.000 claims description 19
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 claims description 19
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 19
- 125000001113 thiadiazolyl group Chemical group 0.000 claims description 19
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims description 19
- 229920002554 vinyl polymer Polymers 0.000 claims description 19
- 125000004429 atom Chemical group 0.000 claims description 18
- 229910052801 chlorine Inorganic materials 0.000 claims description 18
- 239000003112 inhibitor Substances 0.000 claims description 18
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 18
- 125000001153 fluoro group Chemical group F* 0.000 claims description 17
- 229910052736 halogen Inorganic materials 0.000 claims description 17
- 150000002367 halogens Chemical class 0.000 claims description 17
- 125000001715 oxadiazolyl group Chemical group 0.000 claims description 17
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 17
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 17
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims description 17
- 125000000335 thiazolyl group Chemical group 0.000 claims description 17
- 125000003545 alkoxy group Chemical group 0.000 claims description 16
- 125000002393 azetidinyl group Chemical group 0.000 claims description 16
- 239000003054 catalyst Substances 0.000 claims description 16
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 claims description 16
- 125000001425 triazolyl group Chemical group 0.000 claims description 16
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 15
- 206010019280 Heart failures Diseases 0.000 claims description 15
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 15
- 125000004432 carbon atom Chemical group C* 0.000 claims description 15
- 125000004497 pyrazol-5-yl group Chemical group N1N=CC=C1* 0.000 claims description 15
- AMFYRKOUWBAGHV-UHFFFAOYSA-N 1h-pyrazolo[4,3-b]pyridine Chemical compound C1=CN=C2C=NNC2=C1 AMFYRKOUWBAGHV-UHFFFAOYSA-N 0.000 claims description 14
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 14
- 230000002829 reductive effect Effects 0.000 claims description 14
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 13
- PIGFYZPCRLYGLF-UHFFFAOYSA-N Aluminum nitride Chemical compound [Al]#N PIGFYZPCRLYGLF-UHFFFAOYSA-N 0.000 claims description 12
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 12
- 230000003176 fibrotic effect Effects 0.000 claims description 12
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 claims description 12
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 claims description 11
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 11
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 11
- 229910052794 bromium Inorganic materials 0.000 claims description 11
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 claims description 11
- 238000002360 preparation method Methods 0.000 claims description 11
- 208000002815 pulmonary hypertension Diseases 0.000 claims description 11
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 11
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 claims description 10
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 claims description 10
- 206010020772 Hypertension Diseases 0.000 claims description 10
- 208000001647 Renal Insufficiency Diseases 0.000 claims description 10
- 201000006370 kidney failure Diseases 0.000 claims description 10
- 229910052723 transition metal Inorganic materials 0.000 claims description 10
- 150000003624 transition metals Chemical class 0.000 claims description 10
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 9
- 206010002383 Angina Pectoris Diseases 0.000 claims description 9
- 150000001204 N-oxides Chemical class 0.000 claims description 9
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 claims description 9
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 9
- 125000006555 (C3-C5) cycloalkyl group Chemical group 0.000 claims description 8
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims description 8
- 229910052805 deuterium Inorganic materials 0.000 claims description 8
- ORTFAQDWJHRMNX-UHFFFAOYSA-N hydroxidooxidocarbon(.) Chemical group O[C]=O ORTFAQDWJHRMNX-UHFFFAOYSA-N 0.000 claims description 8
- 208000028867 ischemia Diseases 0.000 claims description 8
- 125000002971 oxazolyl group Chemical group 0.000 claims description 8
- 230000009424 thromboembolic effect Effects 0.000 claims description 8
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 claims description 8
- 208000019553 vascular disease Diseases 0.000 claims description 8
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 7
- 206010003210 Arteriosclerosis Diseases 0.000 claims description 7
- 239000002253 acid Substances 0.000 claims description 7
- 208000011775 arteriosclerosis disease Diseases 0.000 claims description 7
- 125000004850 cyclobutylmethyl group Chemical group C1(CCC1)C* 0.000 claims description 7
- 230000008569 process Effects 0.000 claims description 7
- 125000006239 protecting group Chemical group 0.000 claims description 7
- 125000004289 pyrazol-3-yl group Chemical group [H]N1N=C(*)C([H])=C1[H] 0.000 claims description 7
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 claims description 6
- 125000004008 6 membered carbocyclic group Chemical group 0.000 claims description 6
- 239000003146 anticoagulant agent Substances 0.000 claims description 6
- 239000003795 chemical substances by application Substances 0.000 claims description 6
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 claims description 6
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 claims description 6
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 claims description 6
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 6
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 claims description 5
- OWFXIOWLTKNBAP-UHFFFAOYSA-N isoamyl nitrite Chemical compound CC(C)CCON=O OWFXIOWLTKNBAP-UHFFFAOYSA-N 0.000 claims description 5
- 125000002757 morpholinyl group Chemical group 0.000 claims description 5
- 125000003566 oxetanyl group Chemical group 0.000 claims description 5
- 239000008194 pharmaceutical composition Substances 0.000 claims description 5
- 125000003386 piperidinyl group Chemical group 0.000 claims description 5
- 125000000464 thioxo group Chemical group S=* 0.000 claims description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 4
- 239000004480 active ingredient Substances 0.000 claims description 4
- 229910021529 ammonia Inorganic materials 0.000 claims description 4
- CPPKAGUPTKIMNP-UHFFFAOYSA-N cyanogen fluoride Chemical compound FC#N CPPKAGUPTKIMNP-UHFFFAOYSA-N 0.000 claims description 4
- 229910052500 inorganic mineral Inorganic materials 0.000 claims description 4
- 239000011707 mineral Substances 0.000 claims description 4
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 4
- 125000004193 piperazinyl group Chemical group 0.000 claims description 4
- 125000004522 1,3,4-thiadiazol-5-yl group Chemical group S1C=NN=C1* 0.000 claims description 3
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 claims description 3
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 3
- 150000007513 acids Chemical class 0.000 claims description 3
- 229940030600 antihypertensive agent Drugs 0.000 claims description 3
- 239000002220 antihypertensive agent Substances 0.000 claims description 3
- 229960004676 antithrombotic agent Drugs 0.000 claims description 3
- XMPZTFVPEKAKFH-UHFFFAOYSA-P ceric ammonium nitrate Chemical compound [NH4+].[NH4+].[Ce+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O XMPZTFVPEKAKFH-UHFFFAOYSA-P 0.000 claims description 3
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 3
- 238000005984 hydrogenation reaction Methods 0.000 claims description 3
- 231100000252 nontoxic Toxicity 0.000 claims description 3
- 230000003000 nontoxic effect Effects 0.000 claims description 3
- 229940082615 organic nitrates used in cardiac disease Drugs 0.000 claims description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 claims description 3
- 241001465754 Metazoa Species 0.000 claims description 2
- 208000001435 Thromboembolism Diseases 0.000 claims description 2
- 238000006114 decarboxylation reaction Methods 0.000 claims description 2
- 230000007062 hydrolysis Effects 0.000 claims description 2
- 238000006460 hydrolysis reaction Methods 0.000 claims description 2
- 150000002632 lipids Chemical class 0.000 claims description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 claims description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims 3
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 claims 1
- 208000024172 Cardiovascular disease Diseases 0.000 abstract description 5
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 171
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 90
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 75
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 64
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 64
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 62
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 60
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 59
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 54
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 54
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 49
- 229940083542 sodium Drugs 0.000 description 49
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 48
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 41
- 239000011734 sodium Substances 0.000 description 39
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 36
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 32
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Chemical class OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 30
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 30
- 239000000243 solution Substances 0.000 description 30
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 30
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 27
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 27
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 26
- 229910052708 sodium Inorganic materials 0.000 description 26
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 25
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 25
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 24
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 24
- 238000005481 NMR spectroscopy Methods 0.000 description 21
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 20
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 20
- UIIMBOGNXHQVGW-UHFFFAOYSA-M sodium bicarbonate Substances [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 20
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 18
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 18
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 18
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 18
- 239000012074 organic phase Substances 0.000 description 18
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical class Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 17
- 239000003480 eluent Substances 0.000 description 17
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 16
- 239000011591 potassium Substances 0.000 description 16
- 229960003975 potassium Drugs 0.000 description 16
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 15
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical class COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 15
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical class CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 15
- 150000002170 ethers Chemical class 0.000 description 15
- 235000019253 formic acid Nutrition 0.000 description 15
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 15
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 14
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 14
- 230000015572 biosynthetic process Effects 0.000 description 14
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 14
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 14
- 150000003254 radicals Chemical class 0.000 description 14
- 229920006395 saturated elastomer Polymers 0.000 description 14
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 13
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 13
- 150000001721 carbon Chemical group 0.000 description 13
- 239000011541 reaction mixture Substances 0.000 description 13
- PAMIQIKDUOTOBW-UHFFFAOYSA-N 1-methylpiperidine Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 12
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 12
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 12
- 229910000024 caesium carbonate Inorganic materials 0.000 description 12
- WMFOQBRAJBCJND-UHFFFAOYSA-M lithium hydroxide Inorganic materials [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 12
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 12
- 235000017557 sodium bicarbonate Nutrition 0.000 description 12
- 102000007637 Soluble Guanylyl Cyclase Human genes 0.000 description 11
- 108010007205 Soluble Guanylyl Cyclase Proteins 0.000 description 11
- 239000003208 petroleum Substances 0.000 description 11
- 230000009467 reduction Effects 0.000 description 11
- 239000000725 suspension Substances 0.000 description 11
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 10
- 239000003513 alkali Substances 0.000 description 10
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 10
- 229930195733 hydrocarbon Natural products 0.000 description 10
- 150000002430 hydrocarbons Chemical class 0.000 description 10
- 229910052763 palladium Inorganic materials 0.000 description 10
- 229910000029 sodium carbonate Inorganic materials 0.000 description 10
- 239000011780 sodium chloride Substances 0.000 description 10
- 239000003643 water by type Substances 0.000 description 10
- 239000008096 xylene Substances 0.000 description 10
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 9
- 102000011016 Type 5 Cyclic Nucleotide Phosphodiesterases Human genes 0.000 description 9
- 108010037581 Type 5 Cyclic Nucleotide Phosphodiesterases Proteins 0.000 description 9
- 229910052783 alkali metal Inorganic materials 0.000 description 9
- 239000005557 antagonist Substances 0.000 description 9
- 238000001816 cooling Methods 0.000 description 9
- 229910052808 lithium carbonate Inorganic materials 0.000 description 9
- 229910000027 potassium carbonate Inorganic materials 0.000 description 9
- 238000000746 purification Methods 0.000 description 9
- 238000010992 reflux Methods 0.000 description 9
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 9
- 125000004847 2-fluorobenzyl group Chemical group [H]C1=C([H])C(F)=C(C([H])=C1[H])C([H])([H])* 0.000 description 8
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 8
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 8
- 208000017169 kidney disease Diseases 0.000 description 8
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 8
- 229910052938 sodium sulfate Inorganic materials 0.000 description 8
- 235000011152 sodium sulphate Nutrition 0.000 description 8
- 238000003786 synthesis reaction Methods 0.000 description 8
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 8
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 8
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 7
- 230000001154 acute effect Effects 0.000 description 7
- 150000001298 alcohols Chemical class 0.000 description 7
- 150000001340 alkali metals Chemical class 0.000 description 7
- 239000011575 calcium Substances 0.000 description 7
- 239000012071 phase Substances 0.000 description 7
- 229910052700 potassium Inorganic materials 0.000 description 7
- 239000007787 solid Substances 0.000 description 7
- 238000000825 ultraviolet detection Methods 0.000 description 7
- HSINOMROUCMIEA-FGVHQWLLSA-N (2s,4r)-4-[(3r,5s,6r,7r,8s,9s,10s,13r,14s,17r)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid Chemical compound C([C@@]12C)C[C@@H](O)C[C@H]1[C@@H](CC)[C@@H](O)[C@@H]1[C@@H]2CC[C@]2(C)[C@@H]([C@H](C)C[C@H](C)C(O)=O)CC[C@H]21 HSINOMROUCMIEA-FGVHQWLLSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 6
- 206010012289 Dementia Diseases 0.000 description 6
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 6
- 239000003613 bile acid Substances 0.000 description 6
- 229960005069 calcium Drugs 0.000 description 6
- 238000004587 chromatography analysis Methods 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 238000004128 high performance liquid chromatography Methods 0.000 description 6
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 6
- RPDAUEIUDPHABB-UHFFFAOYSA-N potassium ethoxide Chemical compound [K+].CC[O-] RPDAUEIUDPHABB-UHFFFAOYSA-N 0.000 description 6
- BDAWXSQJJCIFIK-UHFFFAOYSA-N potassium methoxide Chemical compound [K+].[O-]C BDAWXSQJJCIFIK-UHFFFAOYSA-N 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 208000011580 syndromic disease Diseases 0.000 description 6
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 5
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 5
- 108010078321 Guanylate Cyclase Proteins 0.000 description 5
- 102000014469 Guanylate cyclase Human genes 0.000 description 5
- 208000006011 Stroke Diseases 0.000 description 5
- 229960000583 acetic acid Drugs 0.000 description 5
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 5
- 229910000019 calcium carbonate Inorganic materials 0.000 description 5
- 229910002091 carbon monoxide Inorganic materials 0.000 description 5
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 5
- 239000012973 diazabicyclooctane Substances 0.000 description 5
- 238000005755 formation reaction Methods 0.000 description 5
- 150000007529 inorganic bases Chemical class 0.000 description 5
- 230000000155 isotopic effect Effects 0.000 description 5
- 229910052744 lithium Inorganic materials 0.000 description 5
- 229960001078 lithium Drugs 0.000 description 5
- 150000007530 organic bases Chemical class 0.000 description 5
- 229910000104 sodium hydride Inorganic materials 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 4
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 4
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 4
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical class OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 229940083712 aldosterone antagonist Drugs 0.000 description 4
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 4
- 150000008041 alkali metal carbonates Chemical class 0.000 description 4
- 125000003282 alkyl amino group Chemical group 0.000 description 4
- 150000001408 amides Chemical class 0.000 description 4
- 150000001540 azides Chemical class 0.000 description 4
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 4
- 230000001684 chronic effect Effects 0.000 description 4
- 229910052802 copper Inorganic materials 0.000 description 4
- 239000010949 copper Substances 0.000 description 4
- BERDEBHAJNAUOM-UHFFFAOYSA-N copper(i) oxide Chemical compound [Cu]O[Cu] BERDEBHAJNAUOM-UHFFFAOYSA-N 0.000 description 4
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 4
- 239000012043 crude product Substances 0.000 description 4
- 230000006378 damage Effects 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 150000003278 haem Chemical class 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- 229910052740 iodine Inorganic materials 0.000 description 4
- 239000011630 iodine Substances 0.000 description 4
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 4
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 4
- 230000002093 peripheral effect Effects 0.000 description 4
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 4
- 235000015497 potassium bicarbonate Nutrition 0.000 description 4
- 239000011736 potassium bicarbonate Substances 0.000 description 4
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 4
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 230000002265 prevention Effects 0.000 description 4
- JPJALAQPGMAKDF-UHFFFAOYSA-N selenium dioxide Chemical compound O=[Se]=O JPJALAQPGMAKDF-UHFFFAOYSA-N 0.000 description 4
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 4
- ODZPKZBBUMBTMG-UHFFFAOYSA-N sodium amide Chemical compound [NH2-].[Na+] ODZPKZBBUMBTMG-UHFFFAOYSA-N 0.000 description 4
- 230000000638 stimulation Effects 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 4
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 4
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 4
- YYGNTYWPHWGJRM-UHFFFAOYSA-N (6E,10E,14E,18E)-2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene Chemical compound CC(C)=CCCC(C)=CCCC(C)=CCCC=C(C)CCC=C(C)CCC=C(C)C YYGNTYWPHWGJRM-UHFFFAOYSA-N 0.000 description 3
- SGUVLZREKBPKCE-UHFFFAOYSA-N 1,5-diazabicyclo[4.3.0]-non-5-ene Chemical compound C1CCN=C2CCCN21 SGUVLZREKBPKCE-UHFFFAOYSA-N 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 3
- QGHFTIKMLOOOSM-UHFFFAOYSA-N 5-fluoro-1-[(2-fluorophenyl)methyl]-3-iodopyrazolo[3,4-b]pyridine Chemical compound N1=C(I)C2=CC(F)=CN=C2N1CC1=CC=CC=C1F QGHFTIKMLOOOSM-UHFFFAOYSA-N 0.000 description 3
- GPEUANAWZCBBHU-UHFFFAOYSA-N 5-fluoro-1-[(2-fluorophenyl)methyl]pyrazolo[3,4-b]pyridine-3-carbonitrile Chemical compound N1=C(C#N)C2=CC(F)=CN=C2N1CC1=CC=CC=C1F GPEUANAWZCBBHU-UHFFFAOYSA-N 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- 239000005541 ACE inhibitor Substances 0.000 description 3
- 102000015427 Angiotensins Human genes 0.000 description 3
- 108010064733 Angiotensins Proteins 0.000 description 3
- 102100040214 Apolipoprotein(a) Human genes 0.000 description 3
- 229940127291 Calcium channel antagonist Drugs 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 3
- 108010033266 Lipoprotein(a) Proteins 0.000 description 3
- 102100031545 Microsomal triglyceride transfer protein large subunit Human genes 0.000 description 3
- 208000009525 Myocarditis Diseases 0.000 description 3
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 3
- 206010030113 Oedema Diseases 0.000 description 3
- 206010064911 Pulmonary arterial hypertension Diseases 0.000 description 3
- 239000007868 Raney catalyst Substances 0.000 description 3
- 229910000564 Raney nickel Inorganic materials 0.000 description 3
- BHEOSNUKNHRBNM-UHFFFAOYSA-N Tetramethylsqualene Natural products CC(=C)C(C)CCC(=C)C(C)CCC(C)=CCCC=C(C)CCC(C)C(=C)CCC(C)C(C)=C BHEOSNUKNHRBNM-UHFFFAOYSA-N 0.000 description 3
- 208000009729 Ventricular Premature Complexes Diseases 0.000 description 3
- 208000027418 Wounds and injury Diseases 0.000 description 3
- 229910000102 alkali metal hydride Inorganic materials 0.000 description 3
- 150000008046 alkali metal hydrides Chemical class 0.000 description 3
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 230000006399 behavior Effects 0.000 description 3
- 239000002876 beta blocker Substances 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical class OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 3
- XJHCXCQVJFPJIK-UHFFFAOYSA-M cesium fluoride Substances [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 3
- 239000003354 cholesterol ester transfer protein inhibitor Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical class OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 3
- 206010012601 diabetes mellitus Diseases 0.000 description 3
- NZZFYRREKKOMAT-UHFFFAOYSA-N diiodomethane Chemical compound ICI NZZFYRREKKOMAT-UHFFFAOYSA-N 0.000 description 3
- PRAKJMSDJKAYCZ-UHFFFAOYSA-N dodecahydrosqualene Natural products CC(C)CCCC(C)CCCC(C)CCCCC(C)CCCC(C)CCCC(C)C PRAKJMSDJKAYCZ-UHFFFAOYSA-N 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 230000009977 dual effect Effects 0.000 description 3
- 229940088598 enzyme Drugs 0.000 description 3
- 239000010408 film Substances 0.000 description 3
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 3
- 229910052737 gold Inorganic materials 0.000 description 3
- 239000010931 gold Substances 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 208000014674 injury Diseases 0.000 description 3
- 229910052742 iron Inorganic materials 0.000 description 3
- 238000002955 isolation Methods 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- PQUSVJVVRXWKDG-UHFFFAOYSA-N methyl 2-bromo-2-methylpropanoate Chemical compound COC(=O)C(C)(C)Br PQUSVJVVRXWKDG-UHFFFAOYSA-N 0.000 description 3
- 235000010755 mineral Nutrition 0.000 description 3
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 3
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 3
- 229910000105 potassium hydride Inorganic materials 0.000 description 3
- 238000002953 preparative HPLC Methods 0.000 description 3
- 230000002685 pulmonary effect Effects 0.000 description 3
- 208000005069 pulmonary fibrosis Diseases 0.000 description 3
- 125000002098 pyridazinyl group Chemical group 0.000 description 3
- HMOPBGFCZBTBMX-UHFFFAOYSA-N pyrrolo[2,3-d]pyrimidin-6-one Chemical compound C1=NC=NC2=NC(=O)C=C21 HMOPBGFCZBTBMX-UHFFFAOYSA-N 0.000 description 3
- 229940044601 receptor agonist Drugs 0.000 description 3
- 239000000018 receptor agonist Substances 0.000 description 3
- 239000002461 renin inhibitor Substances 0.000 description 3
- 229940086526 renin-inhibitors Drugs 0.000 description 3
- QEVHRUUCFGRFIF-MDEJGZGSSA-N reserpine Chemical compound O([C@H]1[C@@H]([C@H]([C@H]2C[C@@H]3C4=C(C5=CC=C(OC)C=C5N4)CCN3C[C@H]2C1)C(=O)OC)OC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 QEVHRUUCFGRFIF-MDEJGZGSSA-N 0.000 description 3
- 125000006413 ring segment Chemical group 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 229940031439 squalene Drugs 0.000 description 3
- TUHBEKDERLKLEC-UHFFFAOYSA-N squalene Natural products CC(=CCCC(=CCCC(=CCCC=C(/C)CCC=C(/C)CC=C(C)C)C)C)C TUHBEKDERLKLEC-UHFFFAOYSA-N 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 3
- 102000004217 thyroid hormone receptors Human genes 0.000 description 3
- 108090000721 thyroid hormone receptors Proteins 0.000 description 3
- 125000004306 triazinyl group Chemical group 0.000 description 3
- QVCUKHQDEZNNOC-UHFFFAOYSA-N 1,2-diazabicyclo[2.2.2]octane Chemical compound C1CC2CCN1NC2 QVCUKHQDEZNNOC-UHFFFAOYSA-N 0.000 description 2
- RQFCJASXJCIDSX-UHFFFAOYSA-N 14C-Guanosin-5'-monophosphat Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(COP(O)(O)=O)C(O)C1O RQFCJASXJCIDSX-UHFFFAOYSA-N 0.000 description 2
- ZVYNUGSPFZCYEV-UHFFFAOYSA-N 2,6-dichloro-5-fluoropyridine-3-carboxamide Chemical compound NC(=O)C1=CC(F)=C(Cl)N=C1Cl ZVYNUGSPFZCYEV-UHFFFAOYSA-N 0.000 description 2
- OFJRNBWSFXEHSA-UHFFFAOYSA-N 2-(3-amino-1,2-benzoxazol-5-yl)-n-[4-[2-[(dimethylamino)methyl]imidazol-1-yl]-2-fluorophenyl]-5-(trifluoromethyl)pyrazole-3-carboxamide Chemical compound CN(C)CC1=NC=CN1C(C=C1F)=CC=C1NC(=O)C1=CC(C(F)(F)F)=NN1C1=CC=C(ON=C2N)C2=C1 OFJRNBWSFXEHSA-UHFFFAOYSA-N 0.000 description 2
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 2
- SUBZTZVHVGYOPM-UHFFFAOYSA-N 2-chloro-5-fluoropyridine-3-carbonitrile Chemical compound FC1=CN=C(Cl)C(C#N)=C1 SUBZTZVHVGYOPM-UHFFFAOYSA-N 0.000 description 2
- KHRZSLCUROLAAR-UHFFFAOYSA-N 2-chloro-5-fluoropyridine-3-carboxamide Chemical compound NC(=O)C1=CC(F)=CN=C1Cl KHRZSLCUROLAAR-UHFFFAOYSA-N 0.000 description 2
- UFONWAPOQPJJLL-UHFFFAOYSA-N 5-fluoro-2h-pyrazolo[3,4-b]pyridin-3-amine Chemical compound C1=C(F)C=C2C(N)=NNC2=N1 UFONWAPOQPJJLL-UHFFFAOYSA-N 0.000 description 2
- 208000004476 Acute Coronary Syndrome Diseases 0.000 description 2
- 208000009304 Acute Kidney Injury Diseases 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 201000001320 Atherosclerosis Diseases 0.000 description 2
- ATOAHNRJAXSBOR-UHFFFAOYSA-N BAY 41-2272 Chemical compound NC1=NC(C=2C3=CC=CN=C3N(CC=3C(=CC=CC=3)F)N=2)=NC=C1C1CC1 ATOAHNRJAXSBOR-UHFFFAOYSA-N 0.000 description 2
- 229940127280 BAY 41-2272 Drugs 0.000 description 2
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 2
- 206010007556 Cardiac failure acute Diseases 0.000 description 2
- 206010007558 Cardiac failure chronic Diseases 0.000 description 2
- 229940127328 Cholesterol Synthesis Inhibitors Drugs 0.000 description 2
- 101710178035 Chorismate synthase 2 Proteins 0.000 description 2
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 2
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 2
- 101710152694 Cysteine synthase 2 Proteins 0.000 description 2
- 201000003883 Cystic fibrosis Diseases 0.000 description 2
- 206010012689 Diabetic retinopathy Diseases 0.000 description 2
- 206010013801 Duchenne Muscular Dystrophy Diseases 0.000 description 2
- 206010014561 Emphysema Diseases 0.000 description 2
- 206010048554 Endothelial dysfunction Diseases 0.000 description 2
- 102000002045 Endothelin Human genes 0.000 description 2
- 108050009340 Endothelin Proteins 0.000 description 2
- 208000010228 Erectile Dysfunction Diseases 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 206010016654 Fibrosis Diseases 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical class OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 208000010412 Glaucoma Diseases 0.000 description 2
- 206010018364 Glomerulonephritis Diseases 0.000 description 2
- XKMLYUALXHKNFT-UUOKFMHZSA-N Guanosine-5'-triphosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XKMLYUALXHKNFT-UUOKFMHZSA-N 0.000 description 2
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 206010020853 Hypertonic bladder Diseases 0.000 description 2
- 102100021711 Ileal sodium/bile acid cotransporter Human genes 0.000 description 2
- 229940127470 Lipase Inhibitors Drugs 0.000 description 2
- 208000026139 Memory disease Diseases 0.000 description 2
- 206010027525 Microalbuminuria Diseases 0.000 description 2
- 208000034486 Multi-organ failure Diseases 0.000 description 2
- 208000010718 Multiple Organ Failure Diseases 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- 208000008589 Obesity Diseases 0.000 description 2
- 208000009722 Overactive Urinary Bladder Diseases 0.000 description 2
- 108010016731 PPAR gamma Proteins 0.000 description 2
- 102100038831 Peroxisome proliferator-activated receptor alpha Human genes 0.000 description 2
- 239000005922 Phosphane Substances 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical class OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 108010022233 Plasminogen Activator Inhibitor 1 Proteins 0.000 description 2
- 102100039418 Plasminogen activator inhibitor 1 Human genes 0.000 description 2
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 2
- 206010037423 Pulmonary oedema Diseases 0.000 description 2
- 208000033626 Renal failure acute Diseases 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- 201000001880 Sexual dysfunction Diseases 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- 208000030886 Traumatic Brain injury Diseases 0.000 description 2
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical group ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 2
- SECKRCOLJRRGGV-UHFFFAOYSA-N Vardenafil Chemical compound CCCC1=NC(C)=C(C(N=2)=O)N1NC=2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(CC)CC1 SECKRCOLJRRGGV-UHFFFAOYSA-N 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- RYXZOQOZERSHHQ-UHFFFAOYSA-N [2-(2-diphenylphosphanylphenoxy)phenyl]-diphenylphosphane Chemical compound C=1C=CC=C(P(C=2C=CC=CC=2)C=2C=CC=CC=2)C=1OC1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RYXZOQOZERSHHQ-UHFFFAOYSA-N 0.000 description 2
- 230000009102 absorption Effects 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- RBYGDVHOECIAFC-UHFFFAOYSA-L acetonitrile;palladium(2+);dichloride Chemical compound [Cl-].[Cl-].[Pd+2].CC#N.CC#N RBYGDVHOECIAFC-UHFFFAOYSA-L 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 206010069351 acute lung injury Diseases 0.000 description 2
- 239000003463 adsorbent Substances 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 125000003342 alkenyl group Chemical group 0.000 description 2
- 208000006682 alpha 1-Antitrypsin Deficiency Diseases 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 238000002399 angioplasty Methods 0.000 description 2
- 229940127219 anticoagulant drug Drugs 0.000 description 2
- 229940127218 antiplatelet drug Drugs 0.000 description 2
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical class OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 229950005499 carbon tetrachloride Drugs 0.000 description 2
- 230000000747 cardiac effect Effects 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 238000000451 chemical ionisation Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 229960001701 chloroform Drugs 0.000 description 2
- 230000001906 cholesterol absorption Effects 0.000 description 2
- 208000020832 chronic kidney disease Diseases 0.000 description 2
- 230000004087 circulation Effects 0.000 description 2
- 208000019425 cirrhosis of liver Diseases 0.000 description 2
- 208000010877 cognitive disease Diseases 0.000 description 2
- 230000001276 controlling effect Effects 0.000 description 2
- NKNDPYCGAZPOFS-UHFFFAOYSA-M copper(i) bromide Chemical compound Br[Cu] NKNDPYCGAZPOFS-UHFFFAOYSA-M 0.000 description 2
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 2
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N coumarin Chemical compound C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 2
- 229940109239 creatinine Drugs 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- 230000007850 degeneration Effects 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- OQOCQBJWOCRPQY-UHFFFAOYSA-N diethyl 2-methyl-3-oxosuccinate Chemical compound CCOC(=O)C(C)C(=O)C(=O)OCC OQOCQBJWOCRPQY-UHFFFAOYSA-N 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- YWEUIGNSBFLMFL-UHFFFAOYSA-N diphosphonate Chemical compound O=P(=O)OP(=O)=O YWEUIGNSBFLMFL-UHFFFAOYSA-N 0.000 description 2
- CNXMDTWQWLGCPE-UHFFFAOYSA-N ditert-butyl-(2-phenylphenyl)phosphane Chemical compound CC(C)(C)P(C(C)(C)C)C1=CC=CC=C1C1=CC=CC=C1 CNXMDTWQWLGCPE-UHFFFAOYSA-N 0.000 description 2
- 239000002934 diuretic Substances 0.000 description 2
- 229940030606 diuretics Drugs 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 230000008694 endothelial dysfunction Effects 0.000 description 2
- ZUBDGKVDJUIMQQ-UBFCDGJISA-N endothelin-1 Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(O)=O)NC(=O)[C@H]1NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](C(C)C)NC(=O)[C@H]2CSSC[C@@H](C(N[C@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N2)=O)NC(=O)[C@@H](CO)NC(=O)[C@H](N)CSSC1)C1=CNC=N1 ZUBDGKVDJUIMQQ-UBFCDGJISA-N 0.000 description 2
- 239000000066 endothelium dependent relaxing factor Substances 0.000 description 2
- 229960001208 eplerenone Drugs 0.000 description 2
- JUKPWJGBANNWMW-VWBFHTRKSA-N eplerenone Chemical compound C([C@@H]1[C@]2(C)C[C@H]3O[C@]33[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)C(=O)OC)C[C@@]21CCC(=O)O1 JUKPWJGBANNWMW-VWBFHTRKSA-N 0.000 description 2
- 210000005225 erectile tissue Anatomy 0.000 description 2
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 2
- FUBBWDWIGBTUPQ-UHFFFAOYSA-N ethyl 2-[4-[3-[(4-fluorophenyl)-hydroxymethyl]-4-hydroxyphenoxy]-3,5-dimethylanilino]-2-oxoacetate Chemical compound CC1=CC(NC(=O)C(=O)OCC)=CC(C)=C1OC1=CC=C(O)C(C(O)C=2C=CC(F)=CC=2)=C1 FUBBWDWIGBTUPQ-UHFFFAOYSA-N 0.000 description 2
- FMVJYQGSRWVMQV-UHFFFAOYSA-N ethyl propiolate Chemical compound CCOC(=O)C#C FMVJYQGSRWVMQV-UHFFFAOYSA-N 0.000 description 2
- KTWOOEGAPBSYNW-UHFFFAOYSA-N ferrocene Chemical compound [Fe+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 KTWOOEGAPBSYNW-UHFFFAOYSA-N 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 229940052308 general anesthetics halogenated hydrocarbons Drugs 0.000 description 2
- RQFCJASXJCIDSX-UUOKFMHZSA-N guanosine 5'-monophosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O RQFCJASXJCIDSX-UUOKFMHZSA-N 0.000 description 2
- 150000008282 halocarbons Chemical class 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 210000002216 heart Anatomy 0.000 description 2
- 208000019622 heart disease Diseases 0.000 description 2
- 210000003709 heart valve Anatomy 0.000 description 2
- 239000001307 helium Substances 0.000 description 2
- 229910052734 helium Inorganic materials 0.000 description 2
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 2
- 239000005457 ice water Substances 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 201000001881 impotence Diseases 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000001786 isothiazolyl group Chemical group 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical class CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 230000003902 lesion Effects 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 239000002394 mineralocorticoid antagonist Substances 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 201000006938 muscular dystrophy Diseases 0.000 description 2
- 208000010125 myocardial infarction Diseases 0.000 description 2
- 201000009925 nephrosclerosis Diseases 0.000 description 2
- 235000020824 obesity Nutrition 0.000 description 2
- 208000020629 overactive bladder Diseases 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 108091008725 peroxisome proliferator-activated receptors alpha Proteins 0.000 description 2
- JPHSWUBDHJSNLZ-UHFFFAOYSA-N phenyl-[2-(2-phenylphosphanylphenoxy)phenyl]phosphane Chemical compound C=1C=CC=C(PC=2C=CC=CC=2)C=1OC1=CC=CC=C1PC1=CC=CC=C1 JPHSWUBDHJSNLZ-UHFFFAOYSA-N 0.000 description 2
- 229910000064 phosphane Inorganic materials 0.000 description 2
- RLOWWWKZYUNIDI-UHFFFAOYSA-N phosphinic chloride Chemical compound ClP=O RLOWWWKZYUNIDI-UHFFFAOYSA-N 0.000 description 2
- 239000002590 phosphodiesterase V inhibitor Substances 0.000 description 2
- DLYUQMMRRRQYAE-UHFFFAOYSA-N phosphorus pentoxide Inorganic materials O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 2
- 230000000019 pro-fibrinolytic effect Effects 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 208000005333 pulmonary edema Diseases 0.000 description 2
- 150000005229 pyrazolopyridines Chemical class 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 230000009103 reabsorption Effects 0.000 description 2
- 239000003087 receptor blocking agent Substances 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000000452 restraining effect Effects 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- 201000000306 sarcoidosis Diseases 0.000 description 2
- 230000037390 scarring Effects 0.000 description 2
- 231100000872 sexual dysfunction Toxicity 0.000 description 2
- 230000035939 shock Effects 0.000 description 2
- 229960003310 sildenafil Drugs 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- 229940127296 soluble guanylate cyclase stimulator Drugs 0.000 description 2
- 229960002256 spironolactone Drugs 0.000 description 2
- LXMSZDCAJNLERA-ZHYRCANASA-N spironolactone Chemical compound C([C@@H]1[C@]2(C)CC[C@@H]3[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)SC(=O)C)C[C@@]21CCC(=O)O1 LXMSZDCAJNLERA-ZHYRCANASA-N 0.000 description 2
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- IEHKWSGCTWLXFU-IIBYNOLFSA-N tadalafil Chemical compound C1=C2OCOC2=CC([C@@H]2C3=C([C]4C=CC=CC4=N3)C[C@H]3N2C(=O)CN(C3=O)C)=C1 IEHKWSGCTWLXFU-IIBYNOLFSA-N 0.000 description 2
- 229960000835 tadalafil Drugs 0.000 description 2
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Chemical class OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 238000000844 transformation Methods 0.000 description 2
- 125000003258 trimethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])[*:1] 0.000 description 2
- 210000002229 urogenital system Anatomy 0.000 description 2
- 229960002381 vardenafil Drugs 0.000 description 2
- 235000012431 wafers Nutrition 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- CEMAWMOMDPGJMB-UHFFFAOYSA-N (+-)-Oxprenolol Chemical compound CC(C)NCC(O)COC1=CC=CC=C1OCC=C CEMAWMOMDPGJMB-UHFFFAOYSA-N 0.000 description 1
- TXUICONDJPYNPY-UHFFFAOYSA-N (1,10,13-trimethyl-3-oxo-4,5,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl) heptanoate Chemical compound C1CC2CC(=O)C=C(C)C2(C)C2C1C1CCC(OC(=O)CCCCCC)C1(C)CC2 TXUICONDJPYNPY-UHFFFAOYSA-N 0.000 description 1
- NXWGWUVGUSFQJC-GFCCVEGCSA-N (2r)-1-[(2-methyl-1h-indol-4-yl)oxy]-3-(propan-2-ylamino)propan-2-ol Chemical compound CC(C)NC[C@@H](O)COC1=CC=CC2=C1C=C(C)N2 NXWGWUVGUSFQJC-GFCCVEGCSA-N 0.000 description 1
- DMYZJLOWGSRVKP-RTBURBONSA-N (2r,4r)-1-n-(4-chlorophenyl)-4-hydroxy-2-n-[4-(3-oxomorpholin-4-yl)phenyl]pyrrolidine-1,2-dicarboxamide Chemical compound N1([C@H](C[C@H](C1)O)C(=O)NC=1C=CC(=CC=1)N1C(COCC1)=O)C(=O)NC1=CC=C(Cl)C=C1 DMYZJLOWGSRVKP-RTBURBONSA-N 0.000 description 1
- AMNXBQPRODZJQR-DITALETJSA-N (2s)-2-cyclopentyl-2-[3-[(2,4-dimethylpyrido[2,3-b]indol-9-yl)methyl]phenyl]-n-[(1r)-2-hydroxy-1-phenylethyl]acetamide Chemical compound C1([C@@H](C=2C=CC=C(C=2)CN2C3=CC=CC=C3C3=C(C)C=C(N=C32)C)C(=O)N[C@@H](CO)C=2C=CC=CC=2)CCCC1 AMNXBQPRODZJQR-DITALETJSA-N 0.000 description 1
- ZXEIEKDGPVTZLD-NDEPHWFRSA-N (2s)-2-dodecylsulfanyl-n-(4-hydroxy-2,3,5-trimethylphenyl)-2-phenylacetamide Chemical compound O=C([C@@H](SCCCCCCCCCCCC)C=1C=CC=CC=1)NC1=CC(C)=C(O)C(C)=C1C ZXEIEKDGPVTZLD-NDEPHWFRSA-N 0.000 description 1
- LJCBAPRMNYSDOP-LVCYMWGESA-N (2s)-3-(7-carbamimidoylnaphthalen-2-yl)-2-[4-[(3s)-1-ethanimidoylpyrrolidin-3-yl]oxyphenyl]propanoic acid;hydron;chloride;pentahydrate Chemical compound O.O.O.O.O.Cl.C1N(C(=N)C)CC[C@@H]1OC1=CC=C([C@H](CC=2C=C3C=C(C=CC3=CC=2)C(N)=N)C(O)=O)C=C1 LJCBAPRMNYSDOP-LVCYMWGESA-N 0.000 description 1
- AJBMORBNKXNZSF-COSHMZDQSA-N (2s,3s,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3s,4s,5r,6r)-2-carboxy-4,5-dimethoxy-6-[(2r,3r,4s,5r,6s)-6-methoxy-4,5-disulfooxy-2-(sulfooxymethyl)oxan-3-yl]oxyoxan-3-yl]oxy-4,5-disulfooxy-2-(sulfooxymethyl)oxan-3-yl]oxy-4,5-dimethoxy-3-[(2r,3r,4s,5r,6r)-3,4 Chemical compound OS(=O)(=O)O[C@@H]1[C@@H](OS(O)(=O)=O)[C@@H](OC)O[C@H](COS(O)(=O)=O)[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@@H]2[C@@H]([C@@H](OS(O)(=O)=O)[C@H](O[C@H]3[C@@H]([C@@H](OC)[C@H](O[C@@H]4[C@@H]([C@@H](OC)[C@H](OC)[C@@H](COS(O)(=O)=O)O4)OC)[C@H](O3)C(O)=O)OC)[C@@H](COS(O)(=O)=O)O2)OS(O)(=O)=O)[C@H](C(O)=O)O1 AJBMORBNKXNZSF-COSHMZDQSA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- FJLGEFLZQAZZCD-MCBHFWOFSA-N (3R,5S)-fluvastatin Chemical compound C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)=C1C1=CC=C(F)C=C1 FJLGEFLZQAZZCD-MCBHFWOFSA-N 0.000 description 1
- CABVTRNMFUVUDM-VRHQGPGLSA-N (3S)-3-hydroxy-3-methylglutaryl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)C[C@@](O)(CC(O)=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 CABVTRNMFUVUDM-VRHQGPGLSA-N 0.000 description 1
- BHKIPHICFOJGLD-HOFKKMOUSA-N (5s)-4-cyclohexyl-2-cyclopentyl-3-[(s)-fluoro-[4-(trifluoromethyl)phenyl]methyl]-7,7-dimethyl-6,8-dihydro-5h-quinolin-5-ol Chemical compound C1([C@@H](F)C=2C=CC(=CC=2)C(F)(F)F)=C(C2CCCCC2)C([C@@H](O)CC(C2)(C)C)=C2N=C1C1CCCC1 BHKIPHICFOJGLD-HOFKKMOUSA-N 0.000 description 1
- RWIUTHWKQHRQNP-ZDVGBALWSA-N (9e,12e)-n-(1-phenylethyl)octadeca-9,12-dienamide Chemical compound CCCCC\C=C\C\C=C\CCCCCCCC(=O)NC(C)C1=CC=CC=C1 RWIUTHWKQHRQNP-ZDVGBALWSA-N 0.000 description 1
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 1
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical class OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- TWBNMYSKRDRHAT-RCWTXCDDSA-N (S)-timolol hemihydrate Chemical compound O.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1 TWBNMYSKRDRHAT-RCWTXCDDSA-N 0.000 description 1
- KOHIRBRYDXPAMZ-YHBROIRLSA-N (S,R,R,R)-nebivolol Chemical compound C1CC2=CC(F)=CC=C2O[C@H]1[C@H](O)CNC[C@@H](O)[C@H]1OC2=CC=C(F)C=C2CC1 KOHIRBRYDXPAMZ-YHBROIRLSA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- PCTZLSCYMRXUGW-UHFFFAOYSA-N 1,1,1,2,2-pentafluorobutane Chemical group [CH2]CC(F)(F)C(F)(F)F PCTZLSCYMRXUGW-UHFFFAOYSA-N 0.000 description 1
- FFWQLZFIMNTUCZ-UHFFFAOYSA-N 1-(bromomethyl)-2-fluorobenzene Chemical compound FC1=CC=CC=C1CBr FFWQLZFIMNTUCZ-UHFFFAOYSA-N 0.000 description 1
- MOHYOXXOKFQHDC-UHFFFAOYSA-N 1-(chloromethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCl)C=C1 MOHYOXXOKFQHDC-UHFFFAOYSA-N 0.000 description 1
- ZUCFGSAENWHFPO-UHFFFAOYSA-N 1-[4-amino-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-4-[3-(3-hydroxypropoxy)phenyl]-2-oxo-1,8-naphthyridin-3-yl]urea;hydrate;hydrochloride Chemical compound O.Cl.CC(C)C=1C=C(N)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C1=CC=CC(OCCCO)=C1 ZUCFGSAENWHFPO-UHFFFAOYSA-N 0.000 description 1
- PMGZJNCIQHGNLT-UHFFFAOYSA-N 1-[bis(2,2-dimethylpropanoyloxymethoxy)phosphoryl]-4-(3-phenoxyphenyl)butane-1-sulfonic acid Chemical compound CC(C)(C)C(=O)OCOP(=O)(OCOC(=O)C(C)(C)C)C(S(O)(=O)=O)CCCC1=CC=CC(OC=2C=CC=CC=2)=C1 PMGZJNCIQHGNLT-UHFFFAOYSA-N 0.000 description 1
- 125000006218 1-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- NZROMXGKFPXZPG-UHFFFAOYSA-N 1-o,2-o-diethyl 3-o-methyl 2-methyl-1-oxopropane-1,2,3-tricarboxylate Chemical compound CCOC(=O)C(=O)C(C)(CC(=O)OC)C(=O)OCC NZROMXGKFPXZPG-UHFFFAOYSA-N 0.000 description 1
- XEZNGIUYQVAUSS-UHFFFAOYSA-N 18-crown-6 Chemical compound C1COCCOCCOCCOCCOCCO1 XEZNGIUYQVAUSS-UHFFFAOYSA-N 0.000 description 1
- GUSVHVVOABZHAH-OPZWKQDFSA-N 1aw8p77hkj Chemical compound O([C@@H]1[C@@H](CO)O[C@H]([C@@H]([C@H]1O)O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4C[C@H]5[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@@H]([C@]1(OC[C@H](C)CC1)O5)C)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O GUSVHVVOABZHAH-OPZWKQDFSA-N 0.000 description 1
- GOIWZZQXWJVDOG-UHFFFAOYSA-N 2,2,2-trifluoroethyl trichloromethanesulfonate Chemical compound FC(F)(F)COS(=O)(=O)C(Cl)(Cl)Cl GOIWZZQXWJVDOG-UHFFFAOYSA-N 0.000 description 1
- DEDKKOOGYIMMBC-UHFFFAOYSA-N 2,6-dichloro-5-fluoropyridine-3-carbonitrile Chemical compound FC1=CC(C#N)=C(Cl)N=C1Cl DEDKKOOGYIMMBC-UHFFFAOYSA-N 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- DEMLYXMVPJAVFU-UHFFFAOYSA-N 2-(chloromethyl)oxirane;2-methyl-1h-imidazole Chemical compound ClCC1CO1.CC1=NC=CN1 DEMLYXMVPJAVFU-UHFFFAOYSA-N 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical class CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- HQSRVYUCBOCBLY-XOOFNSLWSA-N 2-[(2r)-butan-2-yl]-4-[4-[4-[4-[[(2s,4r)-2-(4-chlorophenyl)-2-[(4-methyl-1,2,4-triazol-3-yl)sulfanylmethyl]-1,3-dioxolan-4-yl]methoxy]phenyl]piperazin-1-yl]phenyl]-1,2,4-triazol-3-one Chemical compound O=C1N([C@H](C)CC)N=CN1C1=CC=C(N2CCN(CC2)C=2C=CC(OC[C@H]3O[C@@](CSC=4N(C=NN=4)C)(OC3)C=3C=CC(Cl)=CC=3)=CC=2)C=C1 HQSRVYUCBOCBLY-XOOFNSLWSA-N 0.000 description 1
- CMLUGNQVANVZHY-POURPWNDSA-N 2-[1-[2-[(3r,5s)-1-(3-acetyloxy-2,2-dimethylpropyl)-7-chloro-5-(2,3-dimethoxyphenyl)-2-oxo-5h-4,1-benzoxazepin-3-yl]acetyl]piperidin-4-yl]acetic acid Chemical compound COC1=CC=CC([C@@H]2C3=CC(Cl)=CC=C3N(CC(C)(C)COC(C)=O)C(=O)[C@@H](CC(=O)N3CCC(CC(O)=O)CC3)O2)=C1OC CMLUGNQVANVZHY-POURPWNDSA-N 0.000 description 1
- FBMYKMYQHCBIGU-UHFFFAOYSA-N 2-[2-hydroxy-3-[[1-(1h-indol-3-yl)-2-methylpropan-2-yl]amino]propoxy]benzonitrile Chemical compound C=1NC2=CC=CC=C2C=1CC(C)(C)NCC(O)COC1=CC=CC=C1C#N FBMYKMYQHCBIGU-UHFFFAOYSA-N 0.000 description 1
- UOJMJBUYXYEPFX-UHFFFAOYSA-N 2-[4-(4-hydroxy-3-propan-2-ylphenoxy)-3,5-dimethylanilino]-2-oxoacetic acid Chemical compound C1=C(O)C(C(C)C)=CC(OC=2C(=CC(NC(=O)C(O)=O)=CC=2C)C)=C1 UOJMJBUYXYEPFX-UHFFFAOYSA-N 0.000 description 1
- TXIIZHHIOHVWJD-UHFFFAOYSA-N 2-[7-(2,2-dimethylpropanoylamino)-4,6-dimethyl-1-octyl-2,3-dihydroindol-5-yl]acetic acid Chemical compound CC(C)(C)C(=O)NC1=C(C)C(CC(O)=O)=C(C)C2=C1N(CCCCCCCC)CC2 TXIIZHHIOHVWJD-UHFFFAOYSA-N 0.000 description 1
- QHVBWSIFLCIXBD-UHFFFAOYSA-N 2-[[2-[3-(diaminomethylidene)-6-oxocyclohexa-1,4-dien-1-yl]oxy-3,5-difluoro-6-[3-(1-methyl-4,5-dihydroimidazol-2-yl)phenoxy]pyridin-4-yl]-methylamino]acetic acid Chemical compound N=1C(OC=2C=C(C=CC=2)C=2N(CCN=2)C)=C(F)C(N(CC(O)=O)C)=C(F)C=1OC1=CC(=C(N)N)C=CC1=O QHVBWSIFLCIXBD-UHFFFAOYSA-N 0.000 description 1
- JIVPVXMEBJLZRO-CQSZACIVSA-N 2-chloro-5-[(1r)-1-hydroxy-3-oxo-2h-isoindol-1-yl]benzenesulfonamide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC([C@@]2(O)C3=CC=CC=C3C(=O)N2)=C1 JIVPVXMEBJLZRO-CQSZACIVSA-N 0.000 description 1
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- SGUAFYQXFOLMHL-UHFFFAOYSA-N 2-hydroxy-5-{1-hydroxy-2-[(4-phenylbutan-2-yl)amino]ethyl}benzamide Chemical compound C=1C=C(O)C(C(N)=O)=CC=1C(O)CNC(C)CCC1=CC=CC=C1 SGUAFYQXFOLMHL-UHFFFAOYSA-N 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- YZQLWPMZQVHJED-UHFFFAOYSA-N 2-methylpropanethioic acid S-[2-[[[1-(2-ethylbutyl)cyclohexyl]-oxomethyl]amino]phenyl] ester Chemical compound C=1C=CC=C(SC(=O)C(C)C)C=1NC(=O)C1(CC(CC)CC)CCCCC1 YZQLWPMZQVHJED-UHFFFAOYSA-N 0.000 description 1
- AUYYCJSJGJYCDS-UHFFFAOYSA-N 2/3/6893 Natural products IC1=CC(CC(N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-UHFFFAOYSA-N 0.000 description 1
- QMQZXWWFXNWPMP-UHFFFAOYSA-N 3-(dimethylamino)-2-fluoroprop-2-enal Chemical compound CN(C)C=C(F)C=O QMQZXWWFXNWPMP-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical class NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- RXXCIBALSKQCAE-UHFFFAOYSA-N 3-methylbutoxymethylbenzene Chemical compound CC(C)CCOCC1=CC=CC=C1 RXXCIBALSKQCAE-UHFFFAOYSA-N 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- NCGICGYLBXGBGN-UHFFFAOYSA-N 3-morpholin-4-yl-1-oxa-3-azonia-2-azanidacyclopent-3-en-5-imine;hydrochloride Chemical compound Cl.[N-]1OC(=N)C=[N+]1N1CCOCC1 NCGICGYLBXGBGN-UHFFFAOYSA-N 0.000 description 1
- PWNLEGTUORZMTM-UHFFFAOYSA-N 3-pyrimidin-2-yl-1h-pyrazolo[4,3-b]pyridine Chemical class N1=CC=CN=C1C1=NNC2=CC=CN=C12 PWNLEGTUORZMTM-UHFFFAOYSA-N 0.000 description 1
- NOBZETMXGVAWIM-UHFFFAOYSA-N 4-[(2-carbamimidoyl-3,4-dihydro-1h-isoquinolin-7-yl)oxymethyl]-1-pyridin-4-ylpiperidine-4-carboxylic acid;methanesulfonic acid Chemical compound CS(O)(=O)=O.C1=C2CN(C(=N)N)CCC2=CC=C1OCC(CC1)(C(O)=O)CCN1C1=CC=NC=C1 NOBZETMXGVAWIM-UHFFFAOYSA-N 0.000 description 1
- 150000005007 4-aminopyrimidines Chemical class 0.000 description 1
- CDIIZULDSLKBKV-UHFFFAOYSA-N 4-chlorobutanoyl chloride Chemical compound ClCCCC(Cl)=O CDIIZULDSLKBKV-UHFFFAOYSA-N 0.000 description 1
- KEWSCDNULKOKTG-UHFFFAOYSA-N 4-cyano-4-ethylsulfanylcarbothioylsulfanylpentanoic acid Chemical compound CCSC(=S)SC(C)(C#N)CCC(O)=O KEWSCDNULKOKTG-UHFFFAOYSA-N 0.000 description 1
- PFVRWSTVUQUXKP-UHFFFAOYSA-N 5-fluoro-1-[(2-fluorophenyl)methyl]pyrazolo[3,4-b]pyridine-3-carboxamide Chemical compound C12=NC=C(F)C=C2C(C(=O)N)=NN1CC1=CC=CC=C1F PFVRWSTVUQUXKP-UHFFFAOYSA-N 0.000 description 1
- HSJHOTUQSLQXLL-UHFFFAOYSA-N 5-fluoro-3-iodo-2h-pyrazolo[3,4-b]pyridine Chemical compound FC1=CN=C2NN=C(I)C2=C1 HSJHOTUQSLQXLL-UHFFFAOYSA-N 0.000 description 1
- KKJUPNGICOCCDW-UHFFFAOYSA-N 7-N,N-Dimethylamino-1,2,3,4,5-pentathiocyclooctane Chemical compound CN(C)C1CSSSSSC1 KKJUPNGICOCCDW-UHFFFAOYSA-N 0.000 description 1
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 201000010053 Alcoholic Cardiomyopathy Diseases 0.000 description 1
- UXOWGYHJODZGMF-QORCZRPOSA-N Aliskiren Chemical compound COCCCOC1=CC(C[C@@H](C[C@H](N)[C@@H](O)C[C@@H](C(C)C)C(=O)NCC(C)(C)C(N)=O)C(C)C)=CC=C1OC UXOWGYHJODZGMF-QORCZRPOSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 208000000044 Amnesia Diseases 0.000 description 1
- 206010002199 Anaphylactic shock Diseases 0.000 description 1
- 206010002329 Aneurysm Diseases 0.000 description 1
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical group NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 208000003017 Aortic Valve Stenosis Diseases 0.000 description 1
- 206010002915 Aortic valve incompetence Diseases 0.000 description 1
- 206010002921 Aortitis Diseases 0.000 description 1
- QNZCBYKSOIHPEH-UHFFFAOYSA-N Apixaban Chemical compound C1=CC(OC)=CC=C1N1C(C(=O)N(CC2)C=3C=CC(=CC=3)N3C(CCCC3)=O)=C2C(C(N)=O)=N1 QNZCBYKSOIHPEH-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 206010003130 Arrhythmia supraventricular Diseases 0.000 description 1
- 206010003178 Arterial thrombosis Diseases 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 206010003658 Atrial Fibrillation Diseases 0.000 description 1
- 208000002102 Atrial Premature Complexes Diseases 0.000 description 1
- 206010003662 Atrial flutter Diseases 0.000 description 1
- 208000006808 Atrioventricular Nodal Reentry Tachycardia Diseases 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 206010061666 Autonomic neuropathy Diseases 0.000 description 1
- PTQXTEKSNBVPQJ-UHFFFAOYSA-N Avasimibe Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1CC(=O)NS(=O)(=O)OC1=C(C(C)C)C=CC=C1C(C)C PTQXTEKSNBVPQJ-UHFFFAOYSA-N 0.000 description 1
- 208000037157 Azotemia Diseases 0.000 description 1
- 239000005552 B01AC04 - Clopidogrel Substances 0.000 description 1
- 239000005528 B01AC05 - Ticlopidine Substances 0.000 description 1
- AQYFUZRYBJBAGZ-UHFFFAOYSA-N BAY-41-8543 Chemical compound NC1=NC(C=2C3=CC=CN=C3N(CC=3C(=CC=CC=3)F)N=2)=NC(N)=C1N1CCOCC1 AQYFUZRYBJBAGZ-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Chemical class 0.000 description 1
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 1
- 206010048962 Brain oedema Diseases 0.000 description 1
- BMTAFVWTTFSTOG-UHFFFAOYSA-N Butylate Chemical group CCSC(=O)N(CC(C)C)CC(C)C BMTAFVWTTFSTOG-UHFFFAOYSA-N 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 1
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 1
- KSFOVUSSGSKXFI-GAQDCDSVSA-N CC1=C/2NC(\C=C3/N=C(/C=C4\N\C(=C/C5=N/C(=C\2)/C(C=C)=C5C)C(C=C)=C4C)C(C)=C3CCC(O)=O)=C1CCC(O)=O Chemical compound CC1=C/2NC(\C=C3/N=C(/C=C4\N\C(=C/C5=N/C(=C\2)/C(C=C)=C5C)C(C=C)=C4C)C(C)=C3CCC(O)=O)=C1CCC(O)=O KSFOVUSSGSKXFI-GAQDCDSVSA-N 0.000 description 1
- 201000002829 CREST Syndrome Diseases 0.000 description 1
- 101100296726 Caenorhabditis elegans pde-5 gene Proteins 0.000 description 1
- 206010007572 Cardiac hypertrophy Diseases 0.000 description 1
- 208000006029 Cardiomegaly Diseases 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- 206010007637 Cardiomyopathy alcoholic Diseases 0.000 description 1
- JOATXPAWOHTVSZ-UHFFFAOYSA-N Celiprolol Chemical compound CCN(CC)C(=O)NC1=CC=C(OCC(O)CNC(C)(C)C)C(C(C)=O)=C1 JOATXPAWOHTVSZ-UHFFFAOYSA-N 0.000 description 1
- 206010008120 Cerebral ischaemia Diseases 0.000 description 1
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 description 1
- 229940122502 Cholesterol absorption inhibitor Drugs 0.000 description 1
- 102100037637 Cholesteryl ester transfer protein Human genes 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- 208000026151 Chronic thromboembolic pulmonary hypertension Diseases 0.000 description 1
- 229920002905 Colesevelam Polymers 0.000 description 1
- 229920002911 Colestipol Polymers 0.000 description 1
- 208000031288 Combined hyperlipidaemia Diseases 0.000 description 1
- 208000014526 Conduction disease Diseases 0.000 description 1
- 208000002330 Congenital Heart Defects Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 206010056370 Congestive cardiomyopathy Diseases 0.000 description 1
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 1
- 208000011990 Corticobasal Degeneration Diseases 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- NOTFZGFABLVTIG-UHFFFAOYSA-N Cyclohexylethyl acetate Chemical compound CC(=O)OCCC1CCCCC1 NOTFZGFABLVTIG-UHFFFAOYSA-N 0.000 description 1
- 208000026292 Cystic Kidney disease Diseases 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- XUIIKFGFIJCVMT-GFCCVEGCSA-N D-thyroxine Chemical compound IC1=CC(C[C@@H](N)C(O)=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-GFCCVEGCSA-N 0.000 description 1
- 206010067889 Dementia with Lewy bodies Diseases 0.000 description 1
- 101000783577 Dendroaspis angusticeps Thrombostatin Proteins 0.000 description 1
- 101000783578 Dendroaspis jamesoni kaimosae Dendroaspin Proteins 0.000 description 1
- 206010012335 Dependence Diseases 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical class OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 1
- 206010056340 Diabetic ulcer Diseases 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical compound C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 201000010046 Dilated cardiomyopathy Diseases 0.000 description 1
- 206010052804 Drug tolerance Diseases 0.000 description 1
- 208000032928 Dyslipidaemia Diseases 0.000 description 1
- 206010014418 Electrolyte imbalance Diseases 0.000 description 1
- 208000005189 Embolism Diseases 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- YARKMNAWFIMDKV-UHFFFAOYSA-N Epanolol Chemical compound C=1C=CC=C(C#N)C=1OCC(O)CNCCNC(=O)CC1=CC=C(O)C=C1 YARKMNAWFIMDKV-UHFFFAOYSA-N 0.000 description 1
- JIGUQPWFLRLWPJ-UHFFFAOYSA-N Ethyl acrylate Chemical compound CCOC(=O)C=C JIGUQPWFLRLWPJ-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 206010015856 Extrasystoles Diseases 0.000 description 1
- 229940123583 Factor Xa inhibitor Drugs 0.000 description 1
- 206010057671 Female sexual dysfunction Diseases 0.000 description 1
- 108010049003 Fibrinogen Proteins 0.000 description 1
- 102000008946 Fibrinogen Human genes 0.000 description 1
- 208000022461 Glomerular disease Diseases 0.000 description 1
- 206010018366 Glomerulonephritis acute Diseases 0.000 description 1
- 238000003747 Grignard reaction Methods 0.000 description 1
- 206010019196 Head injury Diseases 0.000 description 1
- 101000880514 Homo sapiens Cholesteryl ester transfer protein Proteins 0.000 description 1
- 208000023105 Huntington disease Diseases 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- 208000002682 Hyperkalemia Diseases 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- 206010021036 Hyponatraemia Diseases 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- 101710156096 Ileal sodium/bile acid cotransporter Proteins 0.000 description 1
- 206010021518 Impaired gastric emptying Diseases 0.000 description 1
- 206010021639 Incontinence Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 102100025306 Integrin alpha-IIb Human genes 0.000 description 1
- 101710149643 Integrin alpha-IIb Proteins 0.000 description 1
- 206010022562 Intermittent claudication Diseases 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- 206010048858 Ischaemic cardiomyopathy Diseases 0.000 description 1
- 208000002260 Keloid Diseases 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 102000007330 LDL Lipoproteins Human genes 0.000 description 1
- 108010007622 LDL Lipoproteins Proteins 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 244000123592 Laurus azorica Species 0.000 description 1
- 208000020358 Learning disease Diseases 0.000 description 1
- 206010024119 Left ventricular failure Diseases 0.000 description 1
- 201000002832 Lewy body dementia Diseases 0.000 description 1
- 238000005684 Liebig rearrangement reaction Methods 0.000 description 1
- 229940086609 Lipase inhibitor Drugs 0.000 description 1
- 208000017170 Lipid metabolism disease Diseases 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- 208000000185 Localized scleroderma Diseases 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000001145 Metabolic Syndrome Diseases 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 206010027982 Morphoea Diseases 0.000 description 1
- 208000007101 Muscle Cramp Diseases 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- AHVYPIQETPWLSZ-UHFFFAOYSA-N N-methyl-pyrrolidine Natural products CN1CC=CC1 AHVYPIQETPWLSZ-UHFFFAOYSA-N 0.000 description 1
- 108020001621 Natriuretic Peptide Proteins 0.000 description 1
- 102000004571 Natriuretic peptide Human genes 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 206010029164 Nephrotic syndrome Diseases 0.000 description 1
- 208000007256 Nevus Diseases 0.000 description 1
- SNIOPGDIGTZGOP-UHFFFAOYSA-N Nitroglycerin Chemical compound [O-][N+](=O)OCC(O[N+]([O-])=O)CO[N+]([O-])=O SNIOPGDIGTZGOP-UHFFFAOYSA-N 0.000 description 1
- 239000000006 Nitroglycerin Substances 0.000 description 1
- 208000036576 Obstructive uropathy Diseases 0.000 description 1
- 208000022873 Ocular disease Diseases 0.000 description 1
- 208000010195 Onychomycosis Diseases 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- UXRZLDREKITWRO-UHFFFAOYSA-N P(c1ccccc1)c1ccccc1.CC1(C)c2ccccc2Oc2ccccc12 Chemical compound P(c1ccccc1)c1ccccc1.CC1(C)c2ccccc2Oc2ccccc12 UXRZLDREKITWRO-UHFFFAOYSA-N 0.000 description 1
- 108010015181 PPAR delta Proteins 0.000 description 1
- 102000000536 PPAR gamma Human genes 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 206010033799 Paralysis Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 101150003085 Pdcl gene Proteins 0.000 description 1
- 208000000450 Pelvic Pain Diseases 0.000 description 1
- 102100038824 Peroxisome proliferator-activated receptor delta Human genes 0.000 description 1
- 102100038825 Peroxisome proliferator-activated receptor gamma Human genes 0.000 description 1
- 229940080774 Peroxisome proliferator-activated receptor gamma agonist Drugs 0.000 description 1
- 229940123333 Phosphodiesterase 5 inhibitor Drugs 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 206010063985 Phytosterolaemia Diseases 0.000 description 1
- UJEWTUDSLQGTOA-UHFFFAOYSA-N Piretanide Chemical compound C=1C=CC=CC=1OC=1C(S(=O)(=O)N)=CC(C(O)=O)=CC=1N1CCCC1 UJEWTUDSLQGTOA-UHFFFAOYSA-N 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 208000000418 Premature Cardiac Complexes Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Chemical class OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 206010037596 Pyelonephritis Diseases 0.000 description 1
- 208000003782 Raynaud disease Diseases 0.000 description 1
- 206010038423 Renal cyst Diseases 0.000 description 1
- 206010063897 Renal ischaemia Diseases 0.000 description 1
- 206010063837 Reperfusion injury Diseases 0.000 description 1
- 206010038748 Restrictive cardiomyopathy Diseases 0.000 description 1
- 206010039163 Right ventricular failure Diseases 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 108091006614 SLC10A2 Proteins 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 208000034189 Sclerosis Diseases 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- 206010040639 Sick sinus syndrome Diseases 0.000 description 1
- 208000002227 Sitosterolemia Diseases 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 208000005392 Spasm Diseases 0.000 description 1
- 208000007718 Stable Angina Diseases 0.000 description 1
- 206010066218 Stress Urinary Incontinence Diseases 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 208000003734 Supraventricular Tachycardia Diseases 0.000 description 1
- 206010065342 Supraventricular tachyarrhythmia Diseases 0.000 description 1
- 206010051379 Systemic Inflammatory Response Syndrome Diseases 0.000 description 1
- 208000008253 Systolic Heart Failure Diseases 0.000 description 1
- 208000001871 Tachycardia Diseases 0.000 description 1
- 208000001163 Tangier disease Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Chemical class [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 101000712605 Theromyzon tessulatum Theromin Proteins 0.000 description 1
- 229940122388 Thrombin inhibitor Drugs 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical compound IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- 208000009205 Tinnitus Diseases 0.000 description 1
- 208000018452 Torsade de pointes Diseases 0.000 description 1
- 208000002363 Torsades de Pointes Diseases 0.000 description 1
- NGBFQHCMQULJNZ-UHFFFAOYSA-N Torsemide Chemical compound CC(C)NC(=O)NS(=O)(=O)C1=CN=CC=C1NC1=CC=CC(C)=C1 NGBFQHCMQULJNZ-UHFFFAOYSA-N 0.000 description 1
- VXFJYXUZANRPDJ-WTNASJBWSA-N Trandopril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@H]2CCCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 VXFJYXUZANRPDJ-WTNASJBWSA-N 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- FNYLWPVRPXGIIP-UHFFFAOYSA-N Triamterene Chemical compound NC1=NC2=NC(N)=NC(N)=C2N=C1C1=CC=CC=C1 FNYLWPVRPXGIIP-UHFFFAOYSA-N 0.000 description 1
- 201000001943 Tricuspid Valve Insufficiency Diseases 0.000 description 1
- 206010044640 Tricuspid valve incompetence Diseases 0.000 description 1
- 206010044642 Tricuspid valve stenosis Diseases 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 208000007814 Unstable Angina Diseases 0.000 description 1
- 208000012931 Urologic disease Diseases 0.000 description 1
- 201000004810 Vascular dementia Diseases 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- 206010047249 Venous thrombosis Diseases 0.000 description 1
- 206010047289 Ventricular extrasystoles Diseases 0.000 description 1
- 206010065341 Ventricular tachyarrhythmia Diseases 0.000 description 1
- 201000008485 Wernicke-Korsakoff syndrome Diseases 0.000 description 1
- 201000008803 Wolff-Parkinson-white syndrome Diseases 0.000 description 1
- 206010048215 Xanthomatosis Diseases 0.000 description 1
- DQASIYVADJWVLY-UHFFFAOYSA-N [1-[(2-fluorophenyl)methyl]pyrazolo[3,4-b]pyridine-3-carboximidoyl]azanium;chloride Chemical compound Cl.C12=NC=CC=C2C(C(=N)N)=NN1CC1=CC=CC=C1F DQASIYVADJWVLY-UHFFFAOYSA-N 0.000 description 1
- 229960000446 abciximab Drugs 0.000 description 1
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 1
- PBOVUCXZHRXDEY-UHFFFAOYSA-N acetic acid;5-fluoro-1-[(2-fluorophenyl)methyl]pyrazolo[3,4-b]pyridine-3-carboximidamide Chemical compound CC(O)=O.C12=NC=C(F)C=C2C(C(=N)N)=NN1CC1=CC=CC=C1F PBOVUCXZHRXDEY-UHFFFAOYSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 231100000851 acute glomerulonephritis Toxicity 0.000 description 1
- 201000011040 acute kidney failure Diseases 0.000 description 1
- 201000005180 acute myocarditis Diseases 0.000 description 1
- 208000012998 acute renal failure Diseases 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 1
- 229950000221 adaprolol Drugs 0.000 description 1
- IPGLIOFIFLXLKR-AXYNENQYSA-N adaprolol Chemical compound C1=CC(OCC(O)CNC(C)C)=CC=C1CC(=O)OCCC1(C2)C[C@@H](C3)C[C@H]2C[C@@H]3C1 IPGLIOFIFLXLKR-AXYNENQYSA-N 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000002170 aldosterone antagonist Substances 0.000 description 1
- 229960004601 aliskiren Drugs 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Chemical class OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229960002213 alprenolol Drugs 0.000 description 1
- PAZJSJFMUHDSTF-UHFFFAOYSA-N alprenolol Chemical compound CC(C)NCC(O)COC1=CC=CC=C1CC=C PAZJSJFMUHDSTF-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229960002414 ambrisentan Drugs 0.000 description 1
- OUJTZYPIHDYQMC-LJQANCHMSA-N ambrisentan Chemical compound O([C@@H](C(OC)(C=1C=CC=CC=1)C=1C=CC=CC=1)C(O)=O)C1=NC(C)=CC(C)=N1 OUJTZYPIHDYQMC-LJQANCHMSA-N 0.000 description 1
- XSDQTOBWRPYKKA-UHFFFAOYSA-N amiloride Chemical compound NC(=N)NC(=O)C1=NC(Cl)=C(N)N=C1N XSDQTOBWRPYKKA-UHFFFAOYSA-N 0.000 description 1
- 229960002576 amiloride Drugs 0.000 description 1
- 238000005576 amination reaction Methods 0.000 description 1
- 150000005005 aminopyrimidines Chemical class 0.000 description 1
- 229960000528 amlodipine Drugs 0.000 description 1
- ZPBWCRDSRKPIDG-UHFFFAOYSA-N amlodipine benzenesulfonate Chemical compound OS(=O)(=O)C1=CC=CC=C1.CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl ZPBWCRDSRKPIDG-UHFFFAOYSA-N 0.000 description 1
- 239000001099 ammonium carbonate Substances 0.000 description 1
- 235000012501 ammonium carbonate Nutrition 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- MZZLGJHLQGUVPN-HAWMADMCSA-N anacetrapib Chemical compound COC1=CC(F)=C(C(C)C)C=C1C1=CC=C(C(F)(F)F)C=C1CN1C(=O)O[C@H](C=2C=C(C=C(C=2)C(F)(F)F)C(F)(F)F)[C@@H]1C MZZLGJHLQGUVPN-HAWMADMCSA-N 0.000 description 1
- 229950000285 anacetrapib Drugs 0.000 description 1
- 208000003455 anaphylaxis Diseases 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000003705 antithrombocytic agent Substances 0.000 description 1
- 239000003698 antivitamin K Substances 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 206010002906 aortic stenosis Diseases 0.000 description 1
- 201000002064 aortic valve insufficiency Diseases 0.000 description 1
- 229960003886 apixaban Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 229940114079 arachidonic acid Drugs 0.000 description 1
- 235000021342 arachidonic acid Nutrition 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 230000006793 arrhythmia Effects 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 229960002274 atenolol Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- 230000001746 atrial effect Effects 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 229950010046 avasimibe Drugs 0.000 description 1
- 229950003799 axitirome Drugs 0.000 description 1
- PCCNIENXBRUYFK-UHFFFAOYSA-O azanium;cerium(4+);pentanitrate Chemical compound [NH4+].[Ce+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O PCCNIENXBRUYFK-UHFFFAOYSA-O 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 1
- 229960004324 betaxolol Drugs 0.000 description 1
- CHDPSNLJFOQTRK-UHFFFAOYSA-N betaxolol hydrochloride Chemical compound [Cl-].C1=CC(OCC(O)C[NH2+]C(C)C)=CC=C1CCOCC1CC1 CHDPSNLJFOQTRK-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 1
- 229960002781 bisoprolol Drugs 0.000 description 1
- VHYCDWMUTMEGQY-UHFFFAOYSA-N bisoprolol Chemical compound CC(C)NCC(O)COC1=CC=C(COCCOC(C)C)C=C1 VHYCDWMUTMEGQY-UHFFFAOYSA-N 0.000 description 1
- OIRCOABEOLEUMC-GEJPAHFPSA-N bivalirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)CNC(=O)CNC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 OIRCOABEOLEUMC-GEJPAHFPSA-N 0.000 description 1
- 229960001500 bivalirudin Drugs 0.000 description 1
- 108010055460 bivalirudin Proteins 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 230000036770 blood supply Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 230000004097 bone metabolism Effects 0.000 description 1
- 229960003065 bosentan Drugs 0.000 description 1
- SXTRWVVIEPWAKM-UHFFFAOYSA-N bosentan hydrate Chemical compound O.COC1=CC=CC=C1OC(C(=NC(=N1)C=2N=CC=CN=2)OCCO)=C1NS(=O)(=O)C1=CC=C(C(C)(C)C)C=C1 SXTRWVVIEPWAKM-UHFFFAOYSA-N 0.000 description 1
- 208000006752 brain edema Diseases 0.000 description 1
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 1
- 229940124630 bronchodilator Drugs 0.000 description 1
- 239000000168 bronchodilator agent Substances 0.000 description 1
- 239000006189 buccal tablet Substances 0.000 description 1
- 229950005341 bucindolol Drugs 0.000 description 1
- 229960004064 bumetanide Drugs 0.000 description 1
- MAEIEVLCKWDQJH-UHFFFAOYSA-N bumetanide Chemical compound CCCCNC1=CC(C(O)=O)=CC(S(N)(=O)=O)=C1OC1=CC=CC=C1 MAEIEVLCKWDQJH-UHFFFAOYSA-N 0.000 description 1
- 229960000330 bupranolol Drugs 0.000 description 1
- HQIRNZOQPUAHHV-UHFFFAOYSA-N bupranolol Chemical compound CC1=CC=C(Cl)C(OCC(O)CNC(C)(C)C)=C1 HQIRNZOQPUAHHV-UHFFFAOYSA-N 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 230000003222 cGMP degradation Effects 0.000 description 1
- XQPRBTXUXXVTKB-UHFFFAOYSA-M caesium iodide Chemical compound [I-].[Cs+] XQPRBTXUXXVTKB-UHFFFAOYSA-M 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- ZJKZKKPIKDNHDM-UHFFFAOYSA-L calcium;6-(5-carboxylato-5-methylhexoxy)-2,2-dimethylhexanoate Chemical compound [Ca+2].[O-]C(=O)C(C)(C)CCCCOCCCCC(C)(C)C([O-])=O ZJKZKKPIKDNHDM-UHFFFAOYSA-L 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- SGZAIDDFHDDFJU-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SGZAIDDFHDDFJU-UHFFFAOYSA-N 0.000 description 1
- 229960000932 candesartan Drugs 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 230000023852 carbohydrate metabolic process Effects 0.000 description 1
- 235000021256 carbohydrate metabolism Nutrition 0.000 description 1
- 125000005521 carbonamide group Chemical group 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 229960001222 carteolol Drugs 0.000 description 1
- LWAFSWPYPHEXKX-UHFFFAOYSA-N carteolol Chemical compound N1C(=O)CCC2=C1C=CC=C2OCC(O)CNC(C)(C)C LWAFSWPYPHEXKX-UHFFFAOYSA-N 0.000 description 1
- 229960004195 carvedilol Drugs 0.000 description 1
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 description 1
- 229960002320 celiprolol Drugs 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 208000015114 central nervous system disease Diseases 0.000 description 1
- 230000003727 cerebral blood flow Effects 0.000 description 1
- 206010008118 cerebral infarction Diseases 0.000 description 1
- 229960001523 chlortalidone Drugs 0.000 description 1
- 229940125881 cholesteryl ester transfer protein inhibitor Drugs 0.000 description 1
- 208000022831 chronic renal failure syndrome Diseases 0.000 description 1
- 235000019504 cigarettes Nutrition 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 208000024980 claudication Diseases 0.000 description 1
- GKTWGGQPFAXNFI-HNNXBMFYSA-N clopidogrel Chemical compound C1([C@H](N2CC=3C=CSC=3CC2)C(=O)OC)=CC=CC=C1Cl GKTWGGQPFAXNFI-HNNXBMFYSA-N 0.000 description 1
- 229960003009 clopidogrel Drugs 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- GMRWGQCZJGVHKL-UHFFFAOYSA-N colestipol Chemical compound ClCC1CO1.NCCNCCNCCNCCN GMRWGQCZJGVHKL-UHFFFAOYSA-N 0.000 description 1
- 229960002604 colestipol Drugs 0.000 description 1
- 208000028831 congenital heart disease Diseases 0.000 description 1
- 208000018631 connective tissue disease Diseases 0.000 description 1
- 239000002872 contrast media Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000007887 coronary angioplasty Methods 0.000 description 1
- 208000029078 coronary artery disease Diseases 0.000 description 1
- 210000004351 coronary vessel Anatomy 0.000 description 1
- 210000005226 corpus cavernosum Anatomy 0.000 description 1
- 229960000956 coumarin Drugs 0.000 description 1
- 235000001671 coumarin Nutrition 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 1
- ZOOSILUVXHVRJE-UHFFFAOYSA-N cyclopropanecarbonyl chloride Chemical compound ClC(=O)C1CC1 ZOOSILUVXHVRJE-UHFFFAOYSA-N 0.000 description 1
- PFWWSGFPICCWGU-UHFFFAOYSA-N cyclopropanesulfonyl chloride Chemical compound ClS(=O)(=O)C1CC1 PFWWSGFPICCWGU-UHFFFAOYSA-N 0.000 description 1
- YBSJFWOBGCMAKL-UHFFFAOYSA-N dabigatran Chemical compound N=1C2=CC(C(=O)N(CCC(O)=O)C=3N=CC=CC=3)=CC=C2N(C)C=1CNC1=CC=C(C(N)=N)C=C1 YBSJFWOBGCMAKL-UHFFFAOYSA-N 0.000 description 1
- 229960003850 dabigatran Drugs 0.000 description 1
- 229950004181 dalcetrapib Drugs 0.000 description 1
- FEJVSJIALLTFRP-LJQANCHMSA-N darusentan Chemical compound COC1=CC(OC)=NC(O[C@H](C(O)=O)C(OC)(C=2C=CC=CC=2)C=2C=CC=CC=2)=N1 FEJVSJIALLTFRP-LJQANCHMSA-N 0.000 description 1
- 229950008833 darusentan Drugs 0.000 description 1
- 229960002887 deanol Drugs 0.000 description 1
- 238000007257 deesterification reaction Methods 0.000 description 1
- 238000005661 deetherification reaction Methods 0.000 description 1
- 229960005227 delapril Drugs 0.000 description 1
- WOUOLAUOZXOLJQ-MBSDFSHPSA-N delapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N(CC(O)=O)C1CC2=CC=CC=C2C1)CC1=CC=CC=C1 WOUOLAUOZXOLJQ-MBSDFSHPSA-N 0.000 description 1
- 239000000841 delta opiate receptor agonist Substances 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 229960001767 dextrothyroxine Drugs 0.000 description 1
- 201000009101 diabetic angiopathy Diseases 0.000 description 1
- 208000033679 diabetic kidney disease Diseases 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 239000012954 diazonium Substances 0.000 description 1
- 150000001989 diazonium salts Chemical class 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- YJNFSFXBSVRTSG-UHFFFAOYSA-N diethyl 2-(cyclopropylmethyl)-2-methyl-3-oxobutanedioate Chemical compound CCOC(=O)C(=O)C(C)(C(=O)OCC)CC1CC1 YJNFSFXBSVRTSG-UHFFFAOYSA-N 0.000 description 1
- FPUQGCOBYOXAED-UHFFFAOYSA-N diethyl 2-[[2-[3-(dimethylcarbamoyl)-4-[[2-[4-(trifluoromethyl)phenyl]benzoyl]amino]phenyl]acetyl]oxymethyl]-2-phenylpropanedioate Chemical compound C=1C=CC=CC=1C(C(=O)OCC)(C(=O)OCC)COC(=O)CC(C=C1C(=O)N(C)C)=CC=C1NC(=O)C1=CC=CC=C1C1=CC=C(C(F)(F)F)C=C1 FPUQGCOBYOXAED-UHFFFAOYSA-N 0.000 description 1
- LHHLYOVYBZWIGM-UHFFFAOYSA-N diethyl 2-formylbutanedioate Chemical compound CCOC(=O)CC(C=O)C(=O)OCC LHHLYOVYBZWIGM-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 229960002768 dipyridamole Drugs 0.000 description 1
- IZEKFCXSFNUWAM-UHFFFAOYSA-N dipyridamole Chemical compound C=12N=C(N(CCO)CCO)N=C(N3CCCCC3)C2=NC(N(CCO)CCO)=NC=1N1CCCCC1 IZEKFCXSFNUWAM-UHFFFAOYSA-N 0.000 description 1
- SPBWMYPZWNFWES-UHFFFAOYSA-N disodium;azanylidyneoxidanium;iron(2+);pentacyanide;dihydrate Chemical compound O.O.[Na+].[Na+].[Fe+2].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-].[O+]#N SPBWMYPZWNFWES-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000009513 drug distribution Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 229960000622 edoxaban Drugs 0.000 description 1
- PSMMNJNZVZZNOI-SJILXJHISA-N edoxaban tosylate hydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.N([C@H]1CC[C@@H](C[C@H]1NC(=O)C=1SC=2CN(C)CCC=2N=1)C(=O)N(C)C)C(=O)C(=O)NC1=CC=C(Cl)C=N1 PSMMNJNZVZZNOI-SJILXJHISA-N 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 229950005925 eflucimibe Drugs 0.000 description 1
- 238000007336 electrophilic substitution reaction Methods 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- XFLQIRAKKLNXRQ-UUWRZZSWSA-N elobixibat Chemical compound C12=CC(SC)=C(OCC(=O)N[C@@H](C(=O)NCC(O)=O)C=3C=CC=CC=3)C=C2S(=O)(=O)CC(CCCC)(CCCC)CN1C1=CC=CC=C1 XFLQIRAKKLNXRQ-UUWRZZSWSA-N 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 206010014665 endocarditis Diseases 0.000 description 1
- 201000010048 endomyocardial fibrosis Diseases 0.000 description 1
- 239000002308 endothelin receptor antagonist Substances 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 229960002711 epanolol Drugs 0.000 description 1
- AQNDDEOPVVGCPG-UHFFFAOYSA-N esmolol Chemical compound COC(=O)CCC1=CC=C(OCC(O)CNC(C)C)C=C1 AQNDDEOPVVGCPG-UHFFFAOYSA-N 0.000 description 1
- 229960003745 esmolol Drugs 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical class CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- IDGUHHHQCWSQLU-UHFFFAOYSA-N ethanol;hydrate Chemical compound O.CCO IDGUHHHQCWSQLU-UHFFFAOYSA-N 0.000 description 1
- 238000006266 etherification reaction Methods 0.000 description 1
- GETWUUTZCWTXBD-UHFFFAOYSA-N ethyl 2-[4-azido-2-[1-[(2-fluorophenyl)methyl]pyrazolo[3,4-b]pyridin-3-yl]pyrimidin-5-yl]acetate Chemical compound CCOC(=O)Cc1cnc(nc1N=[N+]=[N-])-c1nn(Cc2ccccc2F)c2ncccc12 GETWUUTZCWTXBD-UHFFFAOYSA-N 0.000 description 1
- ZIUSEGSNTOUIPT-UHFFFAOYSA-N ethyl 2-cyanoacetate Chemical compound CCOC(=O)CC#N ZIUSEGSNTOUIPT-UHFFFAOYSA-N 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- OLNTVTPDXPETLC-XPWALMASSA-N ezetimibe Chemical compound N1([C@@H]([C@H](C1=O)CC[C@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 OLNTVTPDXPETLC-XPWALMASSA-N 0.000 description 1
- 229960000815 ezetimibe Drugs 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 229940012952 fibrinogen Drugs 0.000 description 1
- 229950010034 fidexaban Drugs 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229960003765 fluvastatin Drugs 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 229960001318 fondaparinux Drugs 0.000 description 1
- KANJSNBRCNMZMV-ABRZTLGGSA-N fondaparinux Chemical compound O[C@@H]1[C@@H](NS(O)(=O)=O)[C@@H](OC)O[C@H](COS(O)(=O)=O)[C@H]1O[C@H]1[C@H](OS(O)(=O)=O)[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](OS(O)(=O)=O)[C@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O[C@@H]4[C@@H]([C@@H](O)[C@H](O)[C@@H](COS(O)(=O)=O)O4)NS(O)(=O)=O)[C@H](O3)C(O)=O)O)[C@@H](COS(O)(=O)=O)O2)NS(O)(=O)=O)[C@H](C(O)=O)O1 KANJSNBRCNMZMV-ABRZTLGGSA-N 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 230000002431 foraging effect Effects 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 210000001652 frontal lobe Anatomy 0.000 description 1
- 239000001530 fumaric acid Chemical class 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 229960003883 furosemide Drugs 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 208000001288 gastroparesis Diseases 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 230000001434 glomerular Effects 0.000 description 1
- 206010061989 glomerulosclerosis Diseases 0.000 description 1
- 229960003711 glyceryl trinitrate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 239000002628 heparin derivative Substances 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 238000004896 high resolution mass spectrometry Methods 0.000 description 1
- 230000003054 hormonal effect Effects 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 229960002003 hydrochlorothiazide Drugs 0.000 description 1
- 201000005991 hyperphosphatemia Diseases 0.000 description 1
- 230000001631 hypertensive effect Effects 0.000 description 1
- 208000006575 hypertriglyceridemia Diseases 0.000 description 1
- 206010020871 hypertrophic cardiomyopathy Diseases 0.000 description 1
- 230000001969 hypertrophic effect Effects 0.000 description 1
- 208000026621 hypolipoproteinemia Diseases 0.000 description 1
- 230000036543 hypotension Effects 0.000 description 1
- 208000022368 idiopathic cardiomyopathy Diseases 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 229950005809 implitapide Drugs 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- NDDAHWYSQHTHNT-UHFFFAOYSA-N indapamide Chemical compound CC1CC2=CC=CC=C2N1NC(=O)C1=CC=C(Cl)C(S(N)(=O)=O)=C1 NDDAHWYSQHTHNT-UHFFFAOYSA-N 0.000 description 1
- 229960004569 indapamide Drugs 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 239000001023 inorganic pigment Substances 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N iron oxide Inorganic materials [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 1
- 235000013980 iron oxide Nutrition 0.000 description 1
- VBMVTYDPPZVILR-UHFFFAOYSA-N iron(2+);oxygen(2-) Chemical class [O-2].[Fe+2] VBMVTYDPPZVILR-UHFFFAOYSA-N 0.000 description 1
- 208000002551 irritable bowel syndrome Diseases 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- MOYKHGMNXAOIAT-JGWLITMVSA-N isosorbide dinitrate Chemical compound [O-][N+](=O)O[C@H]1CO[C@@H]2[C@H](O[N+](=O)[O-])CO[C@@H]21 MOYKHGMNXAOIAT-JGWLITMVSA-N 0.000 description 1
- 229960000201 isosorbide dinitrate Drugs 0.000 description 1
- YWXYYJSYQOXTPL-SLPGGIOYSA-N isosorbide mononitrate Chemical compound [O-][N+](=O)O[C@@H]1CO[C@@H]2[C@@H](O)CO[C@@H]21 YWXYYJSYQOXTPL-SLPGGIOYSA-N 0.000 description 1
- 229960003827 isosorbide mononitrate Drugs 0.000 description 1
- 210000001117 keloid Anatomy 0.000 description 1
- 229960001632 labetalol Drugs 0.000 description 1
- 239000004310 lactic acid Chemical class 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229950005241 landiolol Drugs 0.000 description 1
- WMDSZGFJQKSLLH-RBBKRZOGSA-N landiolol Chemical compound O1C(C)(C)OC[C@H]1COC(=O)CCC(C=C1)=CC=C1OC[C@@H](O)CNCCNC(=O)N1CCOCC1 WMDSZGFJQKSLLH-RBBKRZOGSA-N 0.000 description 1
- 201000003723 learning disability Diseases 0.000 description 1
- 230000007786 learning performance Effects 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- CZRQXSDBMCMPNJ-ZUIPZQNBSA-N lisinopril dihydrate Chemical compound O.O.C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 CZRQXSDBMCMPNJ-ZUIPZQNBSA-N 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 229960003566 lomitapide Drugs 0.000 description 1
- QKVKOFVWUHNEBX-UHFFFAOYSA-N lomitapide mesylate Chemical compound CS(O)(=O)=O.C12=CC=CC=C2C2=CC=CC=C2C1(C(=O)NCC(F)(F)F)CCCCN(CC1)CCC1NC(=O)C1=CC=CC=C1C1=CC=C(C(F)(F)F)C=C1 QKVKOFVWUHNEBX-UHFFFAOYSA-N 0.000 description 1
- 239000002171 loop diuretic Substances 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- NXPHGHWWQRMDIA-UHFFFAOYSA-M magnesium;carbanide;bromide Chemical compound [CH3-].[Mg+2].[Br-] NXPHGHWWQRMDIA-UHFFFAOYSA-M 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical class OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Chemical class 0.000 description 1
- 239000001630 malic acid Chemical class 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- CUONGYYJJVDODC-UHFFFAOYSA-N malononitrile Chemical compound N#CCC#N CUONGYYJJVDODC-UHFFFAOYSA-N 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- DKWNMCUOEDMMIN-PKOBYXMFSA-N melagatran Chemical compound C1=CC(C(=N)N)=CC=C1CNC(=O)[C@H]1N(C(=O)[C@H](NCC(O)=O)C2CCCCC2)CC1 DKWNMCUOEDMMIN-PKOBYXMFSA-N 0.000 description 1
- 229960002137 melagatran Drugs 0.000 description 1
- 229950008446 melinamide Drugs 0.000 description 1
- 230000006984 memory degeneration Effects 0.000 description 1
- 208000023060 memory loss Diseases 0.000 description 1
- 230000007334 memory performance Effects 0.000 description 1
- 229960003134 mepindolol Drugs 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- YDCHPLOFQATIDS-UHFFFAOYSA-N methyl 2-bromoacetate Chemical compound COC(=O)CBr YDCHPLOFQATIDS-UHFFFAOYSA-N 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 229960002704 metipranolol Drugs 0.000 description 1
- BLWNYSZZZWQCKO-UHFFFAOYSA-N metipranolol hydrochloride Chemical compound [Cl-].CC(C)[NH2+]CC(O)COC1=CC(C)=C(OC(C)=O)C(C)=C1C BLWNYSZZZWQCKO-UHFFFAOYSA-N 0.000 description 1
- 229960002237 metoprolol Drugs 0.000 description 1
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 206010027599 migraine Diseases 0.000 description 1
- 208000027061 mild cognitive impairment Diseases 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 208000005907 mitral valve insufficiency Diseases 0.000 description 1
- 208000006887 mitral valve stenosis Diseases 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- XLFWDASMENKTKL-UHFFFAOYSA-N molsidomine Chemical compound O1C(N=C([O-])OCC)=C[N+](N2CCOCC2)=N1 XLFWDASMENKTKL-UHFFFAOYSA-N 0.000 description 1
- 229960004027 molsidomine Drugs 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 208000029744 multiple organ dysfunction syndrome Diseases 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 206010028537 myelofibrosis Diseases 0.000 description 1
- 230000002107 myocardial effect Effects 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- XWNYUWKYRSXKGN-UHFFFAOYSA-N n'-amino-1-[(2-fluorophenyl)methyl]pyrazolo[3,4-b]pyridine-3-carboximidamide Chemical compound C12=NC=CC=C2C(C(N)=NN)=NN1CC1=CC=CC=C1F XWNYUWKYRSXKGN-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001298 n-hexoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229960004255 nadolol Drugs 0.000 description 1
- VWPOSFSPZNDTMJ-UCWKZMIHSA-N nadolol Chemical compound C1[C@@H](O)[C@@H](O)CC2=C1C=CC=C2OCC(O)CNC(C)(C)C VWPOSFSPZNDTMJ-UCWKZMIHSA-N 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical class C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 239000000692 natriuretic peptide Substances 0.000 description 1
- 229920005615 natural polymer Polymers 0.000 description 1
- 229960000619 nebivolol Drugs 0.000 description 1
- 201000008383 nephritis Diseases 0.000 description 1
- 230000002580 nephropathic effect Effects 0.000 description 1
- 230000007372 neural signaling Effects 0.000 description 1
- 230000001272 neurogenic effect Effects 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- AHLBNYSZXLDEJQ-FWEHEUNISA-N orlistat Chemical compound CCCCCCCCCCC[C@H](OC(=O)[C@H](CC(C)C)NC=O)C[C@@H]1OC(=O)[C@H]1CCCCCC AHLBNYSZXLDEJQ-FWEHEUNISA-N 0.000 description 1
- 229960001243 orlistat Drugs 0.000 description 1
- 229950009478 otamixaban Drugs 0.000 description 1
- PFGVNLZDWRZPJW-OPAMFIHVSA-N otamixaban Chemical compound C([C@@H](C(=O)OC)[C@@H](C)NC(=O)C=1C=CC(=CC=1)C=1C=C[N+]([O-])=CC=1)C1=CC=CC(C(N)=N)=C1 PFGVNLZDWRZPJW-OPAMFIHVSA-N 0.000 description 1
- 208000024449 overflow incontinence Diseases 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229960004570 oxprenolol Drugs 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 229950003510 pactimibe Drugs 0.000 description 1
- 230000036407 pain Effects 0.000 description 1
- 208000021090 palsy Diseases 0.000 description 1
- VWMZIGBYZQUQOA-QEEMJVPDSA-N pamaqueside Chemical compound O([C@@H]1[C@@H](CO)O[C@H]([C@@H]([C@H]1O)O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4C[C@H]5[C@@H]([C@]4(CC(=O)[C@@H]3[C@@]2(C)CC1)C)[C@@H]([C@]1(OC[C@H](C)CC1)O5)C)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VWMZIGBYZQUQOA-QEEMJVPDSA-N 0.000 description 1
- 229950005482 pamaqueside Drugs 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000004963 pathophysiological condition Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 229960002035 penbutolol Drugs 0.000 description 1
- KQXKVJAGOJTNJS-HNNXBMFYSA-N penbutolol Chemical compound CC(C)(C)NC[C@H](O)COC1=CC=CC=C1C1CCCC1 KQXKVJAGOJTNJS-HNNXBMFYSA-N 0.000 description 1
- 210000003899 penis Anatomy 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- PNJWIWWMYCMZRO-UHFFFAOYSA-N pent‐4‐en‐2‐one Natural products CC(=O)CC=C PNJWIWWMYCMZRO-UHFFFAOYSA-N 0.000 description 1
- 230000008447 perception Effects 0.000 description 1
- 208000008494 pericarditis Diseases 0.000 description 1
- 229960002582 perindopril Drugs 0.000 description 1
- IPVQLZZIHOAWMC-QXKUPLGCSA-N perindopril Chemical compound C1CCC[C@H]2C[C@@H](C(O)=O)N(C(=O)[C@H](C)N[C@@H](CCC)C(=O)OCC)[C@H]21 IPVQLZZIHOAWMC-QXKUPLGCSA-N 0.000 description 1
- 208000027232 peripheral nervous system disease Diseases 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 206010034674 peritonitis Diseases 0.000 description 1
- 108091008765 peroxisome proliferator-activated receptors β/δ Proteins 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 150000004031 phenylhydrazines Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229960002508 pindolol Drugs 0.000 description 1
- PHUTUTUABXHXLW-UHFFFAOYSA-N pindolol Chemical compound CC(C)NCC(O)COC1=CC=CC2=NC=C[C]12 PHUTUTUABXHXLW-UHFFFAOYSA-N 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 229960001085 piretanide Drugs 0.000 description 1
- 229960002797 pitavastatin Drugs 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 230000010118 platelet activation Effects 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 230000002980 postoperative effect Effects 0.000 description 1
- 229960000206 potassium canrenoate Drugs 0.000 description 1
- JTZQCHFUGHIPDF-RYVBEKKQSA-M potassium canrenoate Chemical compound [K+].O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@](CC4)(O)CCC([O-])=O)[C@@H]4[C@@H]3C=CC2=C1 JTZQCHFUGHIPDF-RYVBEKKQSA-M 0.000 description 1
- 229940086066 potassium hydrogencarbonate Drugs 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 239000003286 potassium sparing diuretic agent Substances 0.000 description 1
- 229940097241 potassium-sparing diuretic Drugs 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- IENZQIKPVFGBNW-UHFFFAOYSA-N prazosin Chemical compound N=1C(N)=C2C=C(OC)C(OC)=CC2=NC=1N(CC1)CCN1C(=O)C1=CC=CO1 IENZQIKPVFGBNW-UHFFFAOYSA-N 0.000 description 1
- 229960001289 prazosin Drugs 0.000 description 1
- 201000011461 pre-eclampsia Diseases 0.000 description 1
- 208000003476 primary myelofibrosis Diseases 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 229950003776 protoporphyrin Drugs 0.000 description 1
- 210000001147 pulmonary artery Anatomy 0.000 description 1
- 208000009138 pulmonary valve stenosis Diseases 0.000 description 1
- 150000003217 pyrazoles Chemical class 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical class O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 229960003401 ramipril Drugs 0.000 description 1
- HDACQVRGBOVJII-JBDAPHQKSA-N ramipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@@H]2CCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 HDACQVRGBOVJII-JBDAPHQKSA-N 0.000 description 1
- 229950010535 razaxaban Drugs 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 201000002793 renal fibrosis Diseases 0.000 description 1
- 208000015658 resistant hypertension Diseases 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- WXXSNCNJFUAIDG-UHFFFAOYSA-N riociguat Chemical compound N1=C(N)C(N(C)C(=O)OC)=C(N)N=C1C(C1=CC=CN=C11)=NN1CC1=CC=CC=C1F WXXSNCNJFUAIDG-UHFFFAOYSA-N 0.000 description 1
- 229960001148 rivaroxaban Drugs 0.000 description 1
- KGFYHTZWPPHNLQ-AWEZNQCLSA-N rivaroxaban Chemical compound S1C(Cl)=CC=C1C(=O)NC[C@@H]1OC(=O)N(C=2C=CC(=CC=2)N2C(COCC2)=O)C1 KGFYHTZWPPHNLQ-AWEZNQCLSA-N 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229960000672 rosuvastatin Drugs 0.000 description 1
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000036303 septic shock Effects 0.000 description 1
- 208000007056 sickle cell anemia Diseases 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 229960002578 sitaxentan Drugs 0.000 description 1
- PHWXUGHIIBDVKD-UHFFFAOYSA-N sitaxentan Chemical compound CC1=NOC(NS(=O)(=O)C2=C(SC=C2)C(=O)CC=2C(=CC=3OCOC=3C=2)C)=C1Cl PHWXUGHIIBDVKD-UHFFFAOYSA-N 0.000 description 1
- 208000019116 sleep disease Diseases 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 229940083618 sodium nitroprusside Drugs 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 229960002370 sotalol Drugs 0.000 description 1
- ZBMZVLHSJCTVON-UHFFFAOYSA-N sotalol Chemical compound CC(C)NCC(O)C1=CC=C(NS(C)(=O)=O)C=C1 ZBMZVLHSJCTVON-UHFFFAOYSA-N 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000001119 stannous chloride Substances 0.000 description 1
- 235000011150 stannous chloride Nutrition 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- 208000022170 stress incontinence Diseases 0.000 description 1
- 239000006190 sub-lingual tablet Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000011477 surgical intervention Methods 0.000 description 1
- 206010042772 syncope Diseases 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 230000006794 tachycardia Effects 0.000 description 1
- 239000011975 tartaric acid Chemical class 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- 125000006318 tert-butyl amino group Chemical group [H]N(*)C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 230000000542 thalamic effect Effects 0.000 description 1
- 239000003451 thiazide diuretic agent Substances 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 239000003868 thrombin inhibitor Substances 0.000 description 1
- 230000002537 thrombolytic effect Effects 0.000 description 1
- PHWBOXQYWZNQIN-UHFFFAOYSA-N ticlopidine Chemical compound ClC1=CC=CC=C1CN1CC(C=CS2)=C2CC1 PHWBOXQYWZNQIN-UHFFFAOYSA-N 0.000 description 1
- 229960005001 ticlopidine Drugs 0.000 description 1
- 229960004605 timolol Drugs 0.000 description 1
- 201000005882 tinea unguium Diseases 0.000 description 1
- 231100000886 tinnitus Toxicity 0.000 description 1
- 229950004437 tiqueside Drugs 0.000 description 1
- COKMIXFXJJXBQG-NRFANRHFSA-N tirofiban Chemical compound C1=CC(C[C@H](NS(=O)(=O)CCCC)C(O)=O)=CC=C1OCCCCC1CCNCC1 COKMIXFXJJXBQG-NRFANRHFSA-N 0.000 description 1
- 229960003425 tirofiban Drugs 0.000 description 1
- 229960005461 torasemide Drugs 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 238000007832 transition metal-catalyzed coupling reaction Methods 0.000 description 1
- 230000009529 traumatic brain injury Effects 0.000 description 1
- 229960001288 triamterene Drugs 0.000 description 1
- MWKJTNBSKNUMFN-UHFFFAOYSA-N trifluoromethyltrimethylsilane Chemical compound C[Si](C)(C)C(F)(F)F MWKJTNBSKNUMFN-UHFFFAOYSA-N 0.000 description 1
- MTPVUVINMAGMJL-UHFFFAOYSA-N trimethyl(1,1,2,2,2-pentafluoroethyl)silane Chemical compound C[Si](C)(C)C(F)(F)C(F)(F)F MTPVUVINMAGMJL-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 238000000870 ultraviolet spectroscopy Methods 0.000 description 1
- 208000009852 uremia Diseases 0.000 description 1
- 230000002485 urinary effect Effects 0.000 description 1
- 208000014001 urinary system disease Diseases 0.000 description 1
- 210000001635 urinary tract Anatomy 0.000 description 1
- 201000002327 urinary tract obstruction Diseases 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 1
- 230000001196 vasorelaxation Effects 0.000 description 1
- 208000003663 ventricular fibrillation Diseases 0.000 description 1
- 206010047302 ventricular tachycardia Diseases 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- 206010047470 viral myocarditis Diseases 0.000 description 1
- 210000001835 viscera Anatomy 0.000 description 1
- 229940019333 vitamin k antagonists Drugs 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- ZXIBCJHYVWYIKI-PZJWPPBQSA-N ximelagatran Chemical compound C1([C@@H](NCC(=O)OCC)C(=O)N2[C@@H](CC2)C(=O)NCC=2C=CC(=CC=2)C(\N)=N\O)CCCCC1 ZXIBCJHYVWYIKI-PZJWPPBQSA-N 0.000 description 1
- 229960001522 ximelagatran Drugs 0.000 description 1
- MTZBBNMLMNBNJL-UHFFFAOYSA-N xipamide Chemical compound CC1=CC=CC(C)=C1NC(=O)C1=CC(S(N)(=O)=O)=C(Cl)C=C1O MTZBBNMLMNBNJL-UHFFFAOYSA-N 0.000 description 1
- 229960000537 xipamide Drugs 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 239000004246 zinc acetate Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- the present application relates to novel substituted fused pyrimidines and triazines, processes for their preparation, their use alone or in combinations for the treatment and / or prophylaxis of diseases and their use for the preparation of medicaments for the treatment and / or prophylaxis of diseases, in particular Treatment and / or prophylaxis of cardiovascular diseases.
- cyclic guanosine monophosphate cGMP
- NO nitric oxide
- the guanylate cyclases catalyze the biosynthesis of cGMP from guanosine triphosphate (GTP).
- GTP guanosine triphosphate
- the previously known members of this family can be divided into two groups according to both structural features and the nature of the ligands: the particulate guanylate cyclases stimulable by natriuretic peptides and the soluble guanylate cyclases stimulable by NO.
- the soluble guanylate cyclases consist of two subunits and most likely contain one heme per heterodimer that is part of the regulatory center. This is central to the activation mechanism. NO can bind to the iron atom of the heme and thus significantly increase the activity of the enzyme. On the other hand, heme-free preparations can not be stimulated by NO. Also, carbon monoxide (CO) is able to bind to the central iron atom of the heme, with stimulation by CO being significantly less than by NO.
- CO carbon monoxide
- guanylate cyclase plays a crucial role in various physiological processes, in particular in the relaxation and proliferation of smooth muscle cells, platelet aggregation and adhesion, neuronal signaling and diseases based on a disturbance of the above operations.
- the NO / cGMP system may be suppressed, which may, for example, lead to hypertension, platelet activation, increased cell proliferation, endothelial dysfunction, arteriosclerosis, angina pectoris, heart failure, myocardial infarction, thrombosis, stroke and sexual dysfunction.
- a NO-independent treatment option for such diseases which is aimed at influencing the cGMP pathway in organisms, is a promising approach on account of the expected high efficiency and low side effects.
- the dual principle is met for the purposes of the present invention, when the compounds of the invention show an effect on recombinant guanylate cyclase reporter cell lines according to the investigation under B-2 as a minimal effective concentration (MEC) of ⁇ 3 ⁇ and inhibition of human phosphodiesterase 5 (PDE5 ) according to the study under B-6 as IC50 ⁇ 100 nM.
- MEC minimal effective concentration
- PDE5 human phosphodiesterase 5
- Phosphodiesterase-5 is the name given to one of the enzymes that cleaves the phosphoric acid ester bond in cGMP to give 5'-guanosine monophosphate (5'-GMP).
- phosphodiesterase-5 occurs predominantly in the smooth muscle of the penile erectile tissue (corpus cavernosum penis) and the pulmonary arteries.
- Blocking of cGMP degradation by inhibition of PDE5 leads to increased signals of the relaxation signal pathways and especially to increased blood supply to the penile erectile tissue and pressure reduction in the blood vessels of the lung. They are used to treat erectile dysfunction and pulmonary arterial hypertension.
- WO 2004/009590 describes Pyrazolopyridines with substituted 4-aminopyrimidines for the treatment of CNS diseases.
- WO 2010/065275 and WO 2011/149921 disclose substituted pyrrolo and dihydropyridopyrimidines as sGC activators.
- the sGC stimulators described in WO 2012/004259 are fused aminopyrimidines and in WO 2012/004258, WO 2012/143510 and WO 2012/152629 fused pyrimidines and triazines.
- WO 2012/28647 discloses pyrazolopyridines with various azaheterocycles for the treatment of cardiovascular diseases.
- the object of the present invention was to provide new substances which act as stimulators of soluble guanylate cyclase and as stimulators of soluble guanylate cyclase and phosphodiesterase-5 inhibitors (dual principle) and have a similar or improved therapeutic profile compared to the compounds known from the prior art , such as for their in vivo properties, such as their pharmacokinetic and pharmacodynamic behavior and / or their metabolism profile and / or their dose-response relationship.
- the present invention relates to compounds of the general formula (I)
- Pv 5 is hydrogen, deuterium, halogen, difluoromethyl, trifluoromethyl, (G-C4) -alkyl,
- # l is the point of attachment to the carbonyl group, the point of attachment to the pyrimidine or triazine ring is, m is a number 0, 1 or 2,
- R 6A is hydrogen, fluorine, (C 1 -C 4) -alkyl, hydroxyl or amino, in which (C 1 -C 4) -alkyl having 1 to 3 substituents independently of one another selected from the group fluorine, trifluoromethyl, hydroxy, (C 1 -C 4) Alkoxy, Ffydroxycarbonyl, (C 1 -C 4) alkoxycarbonyl and amino may be substituted,
- R 6B represents hydrogen, fluorine, difluoromethyl, trifluoromethyl, (C 1 -C 6) -alkyl, cyano, (C 3 -C 7) -cycloalkyl, difluoromethoxy, trifluoromethoxy or a group of the formula -MR 12 , where (C 1 -C 6) -alkyl may be substituted with 1 to 3 substituents independently selected from the group fluorine, cyano, trifluoromethyl, (C3-C7) -cycloalkyl, difluoromethoxy and trifluoromethoxy, and wherein
- M is a bond or (C 1 -C 4) -alkanediyl
- R 8 and R 9 together with the atom (s) to which they are respectively attached form a 4- to 7-membered heterocycle wherein the 4- to 7-membered heterocycle in turn is independently selected with 1 or 2 substituents 10 from the group cyano, trifluoromethyl, (G-C6) -alkyl,
- R 9 and R 10 together with the atom (s) to which they are respectively attached form a 4- to 7-membered heterocycle, wherein the 4- to 7-membered heterocycle in turn has 1 or 2 substituents independently selected from the group cyano, trifluoromethyl, (C 1 -C 6) -alkyl, 20 hydroxy, oxo, (C 1 -C 6) -alkoxy, trifluoromethoxy, (GG) -
- Alkoxycarbonyl, amino, mono- (Ci-C6) -alkylamino and di- (Ci-C6) -alkylamino may be substituted
- R 11 is (C 1 -C 6 ) -alkyl or (C 3 -C 7 ) -cycloalkyl, or
- R 8 and R 11 together with the atom (s) to which they are respectively attached form a 4- to 7-membered heterocycle, wherein the 4- to 7-membered heterocycle in turn has 1 or 2 substituents independently selected from the group cyano, trifluoromethyl, (G-C6) -alkyl, 30 hydroxy, oxo, (GG) -alkoxy, trifluoromethoxy, (GG) - Alkoxycarbonyl, amino, mono- (C 1 -C 6) -alkylamino and di- (C 1 -C 6) -alkylamino, and wherein 4- to 7-membered heterocyclyl, phenyl and 5- or 6-membered heteroaryl in turn with 1 to 3 substituents independently of one another selected from the group halogen, cyano, difluoromethyl, trifluoromethyl, (C 1 -C 6 ) -alkyl, (C 3 -C 7 ) -cycloalkyl, hydroxy
- R 6A and R 6B together with the carbon atom to which they are attached, a
- R 7A is hydrogen, fluorine, (C 1 -C 4 ) -alkyl, (C 1 -C 4 ) -alkoxycarbonyl or hydroxyl,
- R is hydrogen, fluorine, (C 1 -C 4 ) -alkyl or trifluoromethyl, represents hydrogen, cyano, (C 1 -C 4 ) -alkyl, (C 1 -C 4 ) -alkoxy, trifluoromethyl, difluoromethyl, (C 3 -C 6) - Cycloalkyl or halogen, is benzyl, where benzyl is substituted by 1 to 3 substituents independently of one another selected from the group consisting of fluorine, chlorine, (C 1 -C 4) -alkyl, cyclopropyl or (C 1 -C 4) -alkoxy,
- R 3 is hydrogen, cyano, (C 1 -C 4 ) -alkyl, (C 1 -C 4 ) -alkoxy, trifluoromethyl, difluoromethyl or (C 3 -C 6 ) -cycloalkyl,
- R 4 is hydrogen, cyano, (C 1 -C 4 ) -alkyl, (C 1 -C 4 ) -alkoxy, trifluoromethyl, difluoromethyl or (C 3 -C 6 ) -cycloalkyl, and also their N-oxides, salts, solvates, salts of N-oxides and solvates of N-oxides and salts.
- the present invention relates to compounds of the general formula (I)
- A is nitrogen or CR 5 ,
- (C 1 -C 6) -alkyl may be substituted by a group -NR 13 R 14 , in which
- R 13 is hydrogen, methyl or ethyl, in which
- (Ci-C4) -alkyl may be substituted by 1 to 3 substituents fluorine, and wherein
- R 15 is (C 1 -C 4 ) -alkyl or (C 3 -C 5 ) -cycloalkyl
- R 16 is (C 1 -C 4 ) -alkyl or (C 3 -C 5 ) -cycloalkyl, or
- Form 4- to 7-membered heterocycle wherein the 4- to 7-membered heterocycle in turn may be substituted with 1 or 2 substituents independently selected from the group trifluoromethyl, (Ci-C4) alkyl, hydroxy and oxo, wherein
- R 21 is hydrogen or (C 1 -C 4 ) -alkyl
- R 22 is hydrogen or (C 1 -C 4 ) -alkyl, in which (C 1 -C 4 ) -alkyl may in each case in each case be substituted by hydroxyl or fluorine, or
- R 21 and R 22 together with the atom to which they are respectively attached, form a 4- to 7-membered heterocycle, for a group # 1 -CR 6A R 6B - (CR 7A R 7B) m - # 2; in which
- # l is the point of attachment to the carbonyl group, the point of attachment to the pyrimidine or triazine ring is, m is a number 0, 1 or 2,
- R 6A is hydrogen, fluorine, (C 1 -C 4) -alkyl, hydroxyl or amino, in which (C 1 -C 4) -alkyl having 1 to 3 substituents independently of one another selected from the group fluorine, trifluoromethyl, hydroxy, (C 1 -C 4) Alkoxy, hydroxycarbonyl, (C 1 -C 4) -alkoxycarbonyl and amino may be substituted,
- R 6B is hydrogen, fluorine, difluoromethyl, trifluoromethyl, (C 1 -C 6 ) -alkyl, (C 2 -C 6 ) -alkenyl, cyano, (C 3 -C 7 ) -cycloalkyl, difluoromethoxy, trifluoromethoxy or a group of the formula -MR 12 , wherein (Ci-C6) alkyl having 1 to 3 substituents independently of one another selected from the group (Ci-C4) alkoxy, fluoro, cyano, trifluoromethyl, (C3-C7) cycloalkyl, difluoromethoxy and trifluoromethoxy may be substituted in which (C 1 -C 4) -alkoxy may be substituted by phenyl, and in which
- M is a bond or (C 1 -C 4) -alkanediyl
- R 8 , R 9 and R 10 are each independently hydrogen
- R 8 and R 9 together with the atom (s) to which they are respectively attached form a 4- to 7-membered heterocycle wherein the 4- to 7-membered heterocycle in turn is independently selected with 1 or 2 substituents from the group consisting of cyano, trifluoromethyl, (Ci-Ce) alkyl, hydroxy, oxo, (Ci-Ce) alkoxy, trifluoromethoxy, (CI-C ⁇ ) - alkoxycarbonyl, amino, mono- (Ci-C6) alkylamino and Di (Ci-C6) alkylamino may be substituted, or
- R 9 and R 10 together with the atom (s) to which they are respectively attached, form a 4- to 7-membered heterocycle, wherein the 4- to 7-membered heterocycle in turn is independently selected with 1 or 2 substituents from the group consisting of cyano, trifluoromethyl, (Ci-Ce) alkyl, hydroxy, oxo, (Ci-Ce) alkoxy, trifluoromethoxy, (CI-C ⁇ ) - alkoxycarbonyl, amino, mono- (Ci-C6) alkylamino and Di- (Ci-C6) -alkylamino may be substituted,
- R 11 is (C 1 -C 6 ) -alkyl or (C 3 -C 7 ) -cycloalkyl, or
- R 8 and R 11 together with the atom (s) to which they are respectively attached form a 4- to 7-membered heterocycle, wherein the 4- to 7-membered heterocycle in turn having 1 or 2 substituents independently selected from the group cyano, trifluoromethyl, (G-CeJ-alkyl, hydroxy, oxo, (Ci-Ce) alkoxy, trifluoromethoxy, (G-C ⁇ ) - alkoxycarbonyl, amino, mono- (Ci-C6) -alkylamino and di- (Ci-C6) -alkylamino, and wherein 4-7-membered heterocyclyl, phenyl and 5- or 6-membered heteroaryl in turn with 1 to 3 substituents independently of one another are selected from the group consisting of halogen, cyano, difluoromethyl, trifluoromethyl, (C 1 -C 6) -alkyl, (C 3 -C 7) -cycloalkyl,
- R TM is hydrogen, fluorine, (C 1 -C 4 ) -alkyl or trifluoromethyl
- R 1 is hydrogen, fluorine, cyano, (C 1 -C 4) -alkyl, (C 1 -C 4) -alkoxy, trifluoromethyl, difluoromethyl, (C 3 -C 6) -cycloalkyl or halogen
- R 2 is benzyl, 3,3,3 Trifluoroprop-1-yl, 4,4,4-trifluorobut-1-yl or 3,3,4,4,4-pentafluorobut-1-yl, wherein benzyl having 1 to 3 substituents independently selected from the group fluorine , Chlorine, (C 1 -C 4) -alkyl, cyclopropyl or (C 1 -C 4) -alkoxy,
- R 3 is hydrogen, cyano, (C 1 -C 4 ) -alkyl, (C 1 -C 4 ) -alkoxy, trifluoromethyl, difluoromethyl or (C 3 -C 6 ) -cycloalkyl,
- R 4 is hydrogen, cyano, (C 1 -C 4 ) -alkyl, (C 1 -C 4 ) -alkoxy, trifluoromethyl, difluoromethyl or (C 3 -C 6 ) -cycloalkyl, and also their N-oxides, salts, solvates, salts of N-oxides and solvates of N-oxides and salts.
- Compounds according to the invention are the compounds of the formula (I) and their salts, solvates and solvates of the salts comprising the compounds of the formulas below and their salts, solvates and solvates of the salts and of the formula (I) encompassed by formula (I), hereinafter referred to as exemplary compounds and their salts, solvates and solvates of the salts, as far as the compounds of formula (I), the compounds mentioned below are not already salts, solvates and solvates of the salts.
- Salts used in the context of the present invention are physiologically acceptable salts of the compounds according to the invention. Also included are salts which are themselves unsuitable for pharmaceutical applications but can be used, for example, for the isolation or purification of the compounds of the invention.
- Physiologically acceptable salts of the compounds according to the invention include acid addition salts of mineral acids, carboxylic acids and sulfonic acids, for example salts of hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, methanesulfonic acid, ethanesulfonic acid, toluenesulfonic acid, benzenesulfonic acid, naphthalenedisulfonic acid, formic acid, acetic acid, trifluoroacetic acid, propionic acid, lactic acid, tartaric acid, Malic acid, citric acid, fumaric acid, maleic acid and benzoic acid.
- Physiologically acceptable salts of the compounds according to the invention also include salts of customary bases, such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts) and ammonium salts derived from ammonia or organic amines having 1 to 16 carbon atoms, such as, by way of example and by way of illustration, ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, arginine, lysine, ethylenediamine and N-methylpiperidine.
- customary bases such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts
- solvates are those forms of the compounds according to the invention which form a complex in the solid or liquid state by coordination with solvent molecules. Hydrates are a special form of solvates that coordinate with water. As solvates, hydrates are preferred in the context of the present invention.
- the compounds of the invention may exist in different stereoisomeric forms depending on their structure, i. in the form of configurational isomers or, if appropriate, also as conformational isomers (enantiomers and / or diastereomers, including those of atropisomers).
- the present invention therefore includes the enantiomers and diastereomers and their respective mixtures. From such mixtures of enantiomers and / or diastereomers, the stereoisomerically uniform components can be isolated in a known manner; Preferably, chromatographic methods are used for this, in particular HPLC chromatography on achiral or chiral phase.
- the present invention encompasses all tautomeric forms.
- the present invention also includes all suitable isotopic variants of the compounds of the invention.
- An isotopic variant of a compound according to the invention is understood to mean a compound in which at least one atom within the compound according to the invention is exchanged for another atom of the same atomic number but with a different atomic mass than the atomic mass that usually or predominantly occurs in nature.
- isotopes which can be incorporated into a compound of the invention are those of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, chlorine, bromine and iodine, such as 2 H (deuterium), 3 H (tritium), 13 C, 14 C, 15 N, 17 0, 18 0, 32 P, 33 P, 33 S, 34 S, 35 S, 36 S, 18 F, 36 Cl, 82 Br, 123 I, 124 I, 129 I and 131 I.
- Certain isotopic variants of a compound according to the invention such as in particular those in which one or more radioactive isotopes are incorporated, may be of use, for example for the investigation of the active compound.
- isotopes such as deuterium may result in certain therapeutic benefits as a result of greater metabolic stability of the compound, such as prolonging the body's half-life or reducing the required effective dose; Such modifications of the compounds of the invention may therefore optionally also constitute a preferred embodiment of the present invention.
- Isotopic variants of the compounds according to the invention can be prepared by the processes known to the person skilled in the art, for example by the methods described below and the rules given in the exemplary embodiments, by using appropriate isotopic modifications of the respective reagents and / or starting compounds.
- the present invention also includes prodrugs of the compounds of the invention.
- prodrugs refers to compounds which themselves may be biologically active or inactive, but are converted during their residence time in the body to compounds of the invention (for example metabolically or hydrolytically).
- alkyl is a linear or branched alkyl radical having in each case the number of carbon atoms specified.
- alkyl is a linear or branched alkyl radical having in each case the number of carbon atoms specified.
- Alkanediyl in the context of the invention is a linear or branched divalent alkyl radical having 1 to 4 carbon atoms. Examples which may be mentioned are: methylene, ethane-1,2-diyl, ethane-1,1-diyl, propane-1,3-diyl, propane-1,1-diyl, propane-1,2-diyl, propane 2,2-diyl, butane-1,4-diyl, butane-1,2-diyl, butane-1,3-diyl and butane-2,3-diyl.
- Cycloalkyl or carbocycle in the context of the invention is a monocyclic, saturated alkyl radical having the particular number of carbon atoms indicated. Examples which may be mentioned by way of example include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
- Alkenyl in the context of the invention represents a linear or branched alkenyl radical having 2 to 4 carbon atoms and one double bond. By way of example and by way of preference: vinyl, allyl, isopropenyl and n-but-2-en-1-yl.
- Alkynyl in the context of the invention represents an alkynyl radical having 2 to 4 carbon atoms with a triple bond. Examples which may be mentioned are ethynyl, propynyl and butynyl.
- Alkoxy in the context of the invention is a linear or branched alkoxy radical having 1 to 6 or 1 to 4 carbon atoms.
- Examples include: methoxy, ethoxy, n-propoxy, isopropoxy, 1-methylpropoxy, n-butoxy, iso-butoxy, tert-butoxy, n-pentoxy, iso-pentoxy, 1-ethylpropoxy, 1-methylbutoxy, 2-methylbutoxy , 3-methylbutoxy and n-hexoxy.
- Preference is given to a linear or branched alkoxy radical having 1 to 4 carbon atoms.
- Alkoxycarbonyl in the context of the invention is a linear or branched alkoxy radical having 1 to 4 carbon atoms and an oxygen-bonded carbonyl group. Examples which may be mentioned by way of example include methoxycarbonyl, ethoxycarbonyl, n-propoxycarbonyl, isopropoxycarbonyl and tert-butoxycarbonyl.
- Mono-alkylamino in the context of the invention represents an amino group having a linear or branched alkyl substituent which has 1 to 6 carbon atoms. Examples which may be mentioned are: methylamino, ethylamino, n-propylamino, isopropylamino and tert-butylamino.
- Di-alkylamino in the context of the invention represents an amino group having two identical or different linear or branched alkyl substituents, each having 1 to 6 carbon atoms. Examples which may be mentioned are: N, N-dimethylamino, N, N-diethylamino, N-ethyl-N-methylamino, N-methyl-Nn-propylamino, N-isopropyl-Nn-propylamino, N-tert-butyl-N - methylamino, N-ethyl-Nn-pentylamino and Nn-hexyl-N-methylamino.
- Heterocyclyl or heterocycle in the context of the invention is a saturated heterocycle having a total of 4 to 7 ring atoms which contains one or two ring heteroatoms from the series ⁇ , O, S, SO and / or SO 2 and is linked via a ring carbon atom , Examples which may be mentioned are: azetidinyl, oxetanyl, pyrrolidinyl, pyrazolidinyl, tetrahydrofuranyl, piperidinyl, piperazinyl, tetrahydropyranyl, morpholinyl, thiomorpholinyl and Dioxidothiomorpholinyl.
- C-linked 5- or 6-membered heteroaryl is in the context of the invention for a monocyclic aromatic heterocycle (heteroaromatic) with a total of 5 or 6 ring atoms, up to three identical or different ring heteroatoms from the series N, O and / or S. contains and is linked via a ring carbon atom.
- Examples which may be mentioned are: furyl, pyrrolyl, thienyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isoxazolyl, isothiazolyl, triazolyl, oxadiazolyl, thiadiazolyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl and triazinyl.
- Preference is given to pyrazol-3-yl, pyrazol-5-yl, pyrazol-5-yl, thiazolyl, oxadiazolyl, thiadiazolyl, pyridyl and pyrimidinyl.
- 5- or 6-membered heteroaryl is in the context of the invention for a monocyclic aromatic heterocycle (heteroaromatic) with a total of 5 or 6 ring atoms, which contains up to three identical or different ring heteroatoms from the series N, O and / or S. a ring carbon atom or optionally linked via a ring nitrogen atom.
- Examples which may be mentioned are: furyl, pyrrolyl, thienyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isoxazolyl, isothiazolyl, triazolyl, oxadiazolyl, thiadiazolyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl and triazinyl. Preference is given to pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl and triazinyl.
- Halogen is in the context of the invention for fluorine, chlorine, bromine and iodine. Preference is given to bromine and iodine.
- An oxo group in the context of the invention is an oxygen atom which is bonded to a carbon atom via a double bond.
- a thiooxo group in the context of the invention represents a sulfur atom which is bonded via a double bond to a carbon atom.
- the end point of the line where the symbol # 1 , or * stands is not a carbon atom or a CH 2 group, but is part of the bond to the respectively designated atom to which L or R 2 is bonded.
- treatment includes inhibiting, delaying, arresting, alleviating, attenuating, restraining, reducing, suppressing, restraining or curing a disease, a disease, a disease, an injury or a medical condition , the unfolding, the course or progression of such conditions and / or the symptoms of such conditions.
- therapy is understood to be synonymous with the term “treatment”.
- prevention means the avoidance or reduction of the risk, a disease, a disease, a disease, an injury or a health disorder, a development or a Progression of such conditions and / or to get, experience, suffer or have the symptoms of such conditions.
- the treatment or the prevention of a disease, a disease, a disease, an injury or a health disorder can be partial or complete.
- A is nitrogen or CR 5 , where
- R 5 is hydrogen, deuterium, fluorine, iodine, difluoromethyl, trifluoromethyl, (C 1 -C 4) -alkyl, vinyl, allyl, ethynyl, cyclopropyl, cyclobutyl, hydroxy, phenyl, pyrazol-3-yl, pyrazol-4-yl, pyrazole -5-yl or pyridyl, wherein (Ci-C4) alkyl, vinyl, allyl, ethynyl, phenyl, pyrazol-3-yl, pyrazol-4-yl, pyrazol-5-yl and pyridyl with 1 or 2 substituents independently selected from the group consisting of fluorine, difluoromethyl, trifluoromethyl, methyl, ethyl, methoxy, ethoxy, cyclopropyl and cyclobutyl,
- L is a group # 1 -CR 6A R 6B - (CR 7A R 7B) m - # 2, where # l is the point of attachment to the carbonyl group,
- # is the point of attachment to the pyrimidine or triazine ring
- m is a number 0 or 1
- R 6A is hydrogen, fluorine, methyl, ethyl, hydroxy or amino
- R represents hydrogen, fluorine, difluoromethyl, trifluoromethyl, (C 1 -C 4 ) -alkyl, cyano, cyclopropyl, cyclobutyl, cyclopentyl or a group of the formula -MR 12 , where (C 1 -C 4 ) -alkyl having 1 to 3 substituents independently of one another selected from the group fluorine, cyano, trifluoromethyl, cyclopropyl, cyclobutyl, Cyclopentyl, difluoromethoxy, trifluoromethoxy, methoxy and ethoxy may be substituted, and wherein
- M is a bond, methylene, ethane-1, 2-diyl or propane-1, 3-diyl,
- R 8 and R 9 are each independently hydrogen, methyl
- Ethyl iso -propyl, cyclopropyl, cyclobutyl, cyclopentyl, oxetanyl, azetidinyl, tetrahydrofuranyl, pyrrolidinyl, tetrahydropyranyl, piperidinyl, piperazinyl, morpholinyl, phenyl, pyrazolyl or pyridyl, wherein methyl, ethyl and iso-propyl are further independently 1 or 2 substituents selected from the group consisting of fluorine, difluoromethyl, trifluoromethyl, cyclopropyl, cyclobutyl, cyclopentyl, hydroxy, difluoromethoxy, trifluoromethoxy, methoxy, ethoxy, hydroxycarbonyl, methoxycarbonyl, ethoxycarbonyl and amino may be substituted,
- R 11 is methyl, ethyl, iso-propyl, cyclopropyl, cyclobutyl, cyclopentyl, and wherein oxadiazolonyl, oxadiazolethionyl, phenyl, oxazolyl, thiazolyl, pyrazolyl, triazolyl, oxadiazolyl, thiadiazolyl, pyridyl, pyrimidinyl and pyrazinyl are themselves independent with 1 or 2 substituents from one another selected from the group consisting of fluorine, chlorine, cyano, difluoromethyl, trifluoromethyl, methyl, ethyl, isopropyl, 2,2,2-trifluoroethyl, 1,1,2,2,2-pentafluoroethyl, cyclopropyl, cyclobutyl, Cyclopropylmetyl, cyclobutylmethyl, hydroxy, methoxy and ethoxy
- R 7A is hydrogen, fluorine, methyl, ethyl or hydroxy
- R TM is hydrogen, fluorine, methyl, ethyl or trifluoromethyl
- R 1 is hydrogen or fluorine
- R 2 is benzyl, where benzyl is substituted by 1 to 3 substituents independently of one another selected from the group consisting of fluorine, methyl or methoxy,
- R 3 is hydrogen or methyl
- R 4 is hydrogen
- R 13 is hydrogen, methyl or ethyl, in which
- (C 1 -C 4 ) -Alkyl may be substituted by 1 to 3 substituents fluorine, and wherein
- R 15 is (C 1 -C 4 ) -alkyl or (C 3 -C 5 ) -cycloalkyl
- R 16 is (C 1 -C 4 ) -alkyl or (C 3 -C 5 ) -cycloalkyl, or together with the Nitrogen atom to which they are attached, a
- 4- to 6-membered heterocycle form wherein the 4- to 6-membered heterocycle in turn may be substituted with 1 or 2 substituents independently selected from the group trifluoromethyl, (Ci-C4) alkyl and oxo, and wherein 2 1 is hydrogen or (C 1 -C 4 ) -alkyl,
- R 22 is hydrogen or (C 1 -C 4 ) -alkyl, in which (C 1 -C 4 ) -alkyl may in each case in each case be substituted by hydroxyl or fluorine, or
- R 21 and R 22 together with the atom to which they are respectively attached, form a 4- to 7-membered heterocycle, for a group # 1 -CR 6A R 6B - (CR 7A R 7B) m - # 2; in which
- # 1 is the point of attachment to the carbonyl group, the point of attachment to the pyrimidine or triazine ring is, m is a number 0 or 1,
- R 6A is hydrogen, fluorine, methyl, ethyl, hydroxy or amino
- R 6B is hydrogen, fluorine, difluoromethyl, trifluoromethyl, (C 1 -C 4 ) -alkyl, (C 2 -C 6 ) -
- -NR 8 - (C O) -OR n , -NR ⁇ C ⁇ -NR ⁇ 10 , oxadiazolonyl, oxadiazolothionyl, phenyl, oxazolyl, thiazolyl, pyrazolyl, triazolyl, oxadiazolyl, thiadiazolyl, pyridyl, pyrimidinyl or pyrazinyl, wherein r the number 0 or 1 means
- R 8 and R 9 are each independently hydrogen
- R 11 is methyl, ethyl, iso-propyl, cyclopropyl, cyclobutyl, cyclopentyl, and wherein oxadiazolonyl, oxadiazolethionyl, phenyl, oxazolyl, thiazolyl, pyrazolyl, triazolyl, oxadiazolyl, thiadiazolyl, pyridyl, pyrimidinyl and pyrazinyl are themselves independent with 1 or 2 substituents selected from the group consisting of fluorine, chlorine, cyano, difluoromethyl, trifluoromethyl, methyl, ethyl, isopropyl, 2,2,2-trifluoroethyl, 1,1,2,2,2-pentafluoroethyl, cyclopropyl, Cyclobutyl, cyclopropylmetyl, cyclobutylmethyl, hydroxy, methoxy and ethoxy, or
- R 6A and R 6B together with the carbon atom to which they are attached, a
- Form pyrrolidinyl or tetrahydropyranyl ring in which the cyclopropyl, cyclobutyl, cyclopentyl, azetidinyl, tetrahydrofuranyl, pyrrolidinyl and tetrahydropyranyl ring may be substituted with 1 or 2 substituents independently selected from the group of fluorine and methyl,
- R 7A is hydrogen, fluorine, methyl, ethyl or hydroxy
- R TM is hydrogen, fluorine, methyl, ethyl, trifluoromethyl or (C 1 -C 4) -alkoxycarbonyl
- R 1 is hydrogen or fluorine
- R 2 is benzyl, 3,3,3-trifluoroprop-1-yl, 4,4,4-trifluorobut-1-yl or 3,3,4,4,4-pentafluorobutyl yl, where benzyl is substituted by 1 to 3 substituents independently of one another selected from the group fluorine, methyl or methoxy,
- R 3 is hydrogen, methyl or trifluoromethyl
- R 4 is hydrogen, and their salts, solvates and solvates of the salts.
- # l represents the point of attachment to the carbonyl group
- # 2 represents the point of attachment to the pyrimidine or triazine ring
- m is a number
- R 6A is hydrogen, fluorine, methyl, ethyl, hydroxy or amino
- R 6B is hydrogen, fluorine, difluoromethyl, trifluoromethyl, methyl, ethyl, cyclopropyl, cyclobutyl, cyclopentyl or a group of the formula -MR 12 , wherein methyl and ethyl having 1 to 3 substituents independently selected from the group fluorine, cyano, trifluoromethyl , Cyclopropyl, cyclobutyl, difluoromethoxy, trifluoromethoxy, methoxy and ethoxy, and wherein
- R 8 and R 9 are each independently hydrogen or
- Cyclopropyl and in which phenyl, thiazolyl, triazolyl, oxadiazolyl, thiadiazolyl and pyrimidinyl, in turn, having 1 or 2 substituents selected independently of one another from the group fluorine, difluoromethyl, trifluoromethyl, methyl, ethyl, isopropyl, 2,2,2-trifluoroethyl, 1,1, 2,2,2-pentafluoroethyl, cyclopropyl, cyclobutyl, cyclopropylmetyl and cyclobutylmethyl may be substituted, or
- R 13 is fluorine
- R 14 is hydrogen, fluorine, methyl or methoxy
- R 15 is hydrogen or fluorine
- R 3 is hydrogen or methyl
- R 4 is hydrogen, and their salts, solvates and solvates of the salts.
- R 21 is hydrogen or (C 1 -C 4 ) -alkyl
- R 22 is hydrogen or (C 1 -C 4 ) -alkyl, or a group -CH 2 -NR 13 R 14 or -CH 2 -CH 2 - NR 13 R 14 is in which
- R 13 is hydrogen, methyl or ethyl
- Ethyl may be substituted by 1 to 3 substituents fluorine, and in which
- R 15 is methyl, ethyl or cyclopropyl
- R 16 is methyl, ethyl or cyclopropyl
- Form a 5- or 6-membered heterocycle, wherein the 5- or 6-membered heterocycle in turn may be substituted with oxo, is a group # 1 -CR 6A R 6B - (CR 7A R 7B ) m - # 2 , wherein
- # l represents the point of attachment to the carbonyl group, the point of attachment to the pyrimidine or triazine ring is, m is a number 0,
- R 6A is hydrogen, methyl, ethyl, hydroxy or amino
- R 6B represents hydrogen, fluorine, difluoromethyl, trifluoromethyl, 1,1,2,2,2-pentafluoroeth-1-yl, methyl, ethyl, allyl, but-3-en-1-yl, or a group of the formula -MR 12 , wherein methyl and ethyl may be substituted with 1 to 3 substituents independently selected from the group of fluorine, cyano, trifluoromethyl, cyclopropyl, cyclobutyl, difluoromethoxy, trifluoromethoxy, methoxy and ethoxy, wherein methoxy and ethoxy may be substituted with phenyl, and wherein
- R 8 and R 9 are each independently hydrogen or
- Cyclopropyl, and wherein l, 3,4-thiadiazol-5-yl may be substituted with 1 or 2 substituents independently selected from the group of fluorine, trifluoromethyl, methyl or ethyl, or
- R 17 , R 18 , R 19 and R 20 independently represent hydrogen, fluorine, methyl or
- R 2 is 3,3,3-trifluoroprop-1-yl or 3,3,4,4,4-pentafluorobut-1-yl
- R 3 is hydrogen, methyl or trifluoromethyl
- R 4 is hydrogen, and theirs Salts, solvates and solvates of salts.
- R 13 is hydrogen or methyl
- Ethyl may be substituted with 1 to 3 substituents fluorine, wherein
- R 15 is methyl, ethyl or cyclopropyl
- R 16 is methyl, ethyl or cyclopropyl
- R 6A is hydrogen, methyl, ethyl, hydroxy or amino
- R 6B is hydrogen, trifluoromethyl, 1,1,2,2,2-pentafluoroeth-1-yl, methyl, ethyl, allyl, but-3-en-1-yl or a group of the formula -MR 12 wherein methyl and ethyl having 1 to 2 substituents independently selected from the group consisting of fluorine, trifluoromethyl, cyclopropyl, cyclobutyl, methoxy and ethoxy, in which methoxy and ethoxy may be substituted with phenyl, and wherein
- M is a bond
- R 8 and R 9 are each independently hydrogen or
- Cyclobutyl or cyclopentyl ring form represents hydrogen or fluorine, a group of the formula
- R 17 , R 18 , R 19 and R 20 independently of one another are hydrogen, fluorine, methyl or methoxy, with the proviso that at least one of the radicals R 17 , R 18 , R 19 or R 20 is different from hydrogen, with the In that at least one of the radicals R 17 , R 18 , R 19 or R 20 is hydrogen, and with the proviso that only one of the radicals R 17 , R 18 , R 19 or R 20 is methyl or methoxy, or
- R 2 is 3,3,3-trifluoroprop-1-yl or 3,3,4,4,4-pentafluorobut-1-yl,
- R 3 is hydrogen, methyl or trifluoromethyl
- R 4 is hydrogen, and their salts, solvates and solvates of the salts.
- A is nitrogen or CR 5 , where
- R 5 is methyl or pyrazol-5-yl, wherein methyl is substituted with a group -NR R, wherein
- R ] 13 is hydrogen
- R ] 14 is hydrogen, methyl, ethyl or -S (O) 2 -R 16 wherein Ethyl may be substituted with 1 to 3 substituents fluorine, and wherein
- R 16 is methyl or cyclopropyl
- 5-membered heterocycle form in which the 5-membered heterocycle may in turn be substituted with oxo
- L is a group # 1 -CR 6A R 6B - (CR 7A R 7B) m - # 2, where
- R 6A is methyl
- R 6B is methyl, R 1 is hydrogen or fluorine, R 2 is a group of the formula
- R 17 , R 18 , R 19 and R 20 independently of one another are hydrogen, fluorine, methyl or methoxy, with the proviso that at least one of the radicals R 17 , R 18 , R 19 or R 20 is different from hydrogen, with the In that at least one of the radicals R 17 , R 18 , R 19 or R 20 is hydrogen, and with the proviso that in each case only one of the radicals R 17 , R 18 , R 19 or R 20 is methyl or methoxy,
- R 3 is hydrogen, methyl or trifluoromethyl
- R 4 is hydrogen, and their salts, solvates and solvates of the salts.
- A is nitrogen or CR 5 , where
- R 5 L stands for hydrogen, for a group # 1 -CR 6A R 6B - (CR 7A R 7B) m - # 2, where
- # 1 represents the point of attachment to the carbonyl group, the point of attachment being to the pyrimidine or triazine ring, m is a number 0, R 6A is amino,
- R 6B is hydrogen, fluorine, difluoromethyl, trifluoromethyl, methyl, ethyl, cyclopropyl, cyclobutyl, cyclopentyl or a group of the formula -MR 12 , wherein methyl and ethyl may be substituted with 1 to 3 substituents independently selected from the group of fluorine, cyano, trifluoromethyl, cyclopropyl, cyclobutyl, difluoromethoxy, trifluoromethoxy, methoxy and ethoxy, and wherein
- R 8 and R 9 are each independently hydrogen or
- Cyclopropyl and in which phenyl, thiazolyl, triazolyl, oxadiazolyl, thiadiazolyl and pyrimidinyl themselves with 1 or 2 substituents independently of one another selected from the group fluorine, difluoromethyl, trifluoromethyl, methyl, ethyl, iso-propyl, 2,2,2-trifluoroethyl, 1,1,2,2,2-pentafluoroethyl, cyclopropyl, cyclobutyl, cyclopropylmetyl and cyclobutylmethyl, is hydrogen or fluorine, a group of the formula
- R 13 is fluorine
- R 14 is hydrogen, fluorine, methyl or methoxy
- R 15 is hydrogen or fluorine
- R 3 is hydrogen or methyl
- R 4 is hydrogen
- A is nitrogen or CR 5 , where
- R 5 L stands for hydrogen, for a group # 1 -CR 6A R 6B - (CR 7A R 7B) m - # 2, where
- # ! represents the point of attachment to the carbonyl group
- # is the point of attachment to the pyrimidine or triazine ring
- m is a number 0
- R 6A is methyl or hydroxy
- R 6B is pentafluoroethyl or cyclopropylmethyl, R 1 is hydrogen or fluorine, R 2 is a group of the formula stands, where
- Pv 14 is hydrogen, fluorine, methyl or methoxy
- Pv 15 is hydrogen or fluorine
- P 3 is hydrogen or methyl
- R 4 is hydrogen
- A is nitrogen or CR 5 , where
- R 5 L stands for hydrogen, for a group # 1 -CR 6A R 6B - (CR 7A R 7B) m - # 2, where
- # is the point of attachment to the pyrimidine or triazine ring
- m is a number
- R 6A stands for amino, R is hydrogen, fluorine, difluoromethyl, trifluoromethyl, methyl, ethyl, cyclopropyl, cyclobutyl, cyclopentyl or a group of the formula -MR 12 , where methyl and ethyl having 1 to 3 substituents independently of one another selected from the group fluorine, cyano, trifluoromethyl, Cyclopropyl, cyclobutyl, difluoromethoxy, trifluoromethoxy, methoxy and ethoxy may be substituted, and wherein
- R 8 and R 9 are each independently hydrogen or
- Cyclopropyl and in which phenyl, thiazolyl, triazolyl, oxadiazolyl, thiadiazolyl and pyrimidinyl themselves with 1 or 2 substituents independently of one another selected from the group fluorine, difluoromethyl, trifluoromethyl, methyl, ethyl, iso-propyl, 2,2,2-trifluoroethyl, 1,1,2,2,2-pentafluoroethyl, cyclopropyl, cyclobutyl, cyclopropylmetyl and cyclobutylmethyl, is hydrogen or fluorine, a group of the formula
- Pv 17 , R 18 , R 19 and R 20 are independently hydrogen, fluoro, methyl or
- R 20 are different from hydrogen, with the proviso that at least one of R 17 , R 18 , R 19 or R 20 is hydrogen, and with the proviso that in each case only one of R 17 , R 18 , R 19 or
- R 20 is methyl or methoxy
- R 3 is hydrogen or methyl
- R 4 is hydrogen, and their salts, solvates and solvates of the salts.
- R 4 is hydrogen, and their salts, solvates and solvates of the salts.
- A is nitrogen or CR 5 , where
- R 5 L is hydrogen, a group -CR 6A R 6B # I - (CR 7A R 7B) m - # 2, where
- # ! is the point of attachment to the carbonyl group, for the attachment point to the to the pyrimidine or triazine ring, m is a number 0, R 6A is hydroxy,
- R 6B is 2,2,2-trifluoroethyl, pentafluoroethyl or (C 1 -C 4 ) -alkyl, in which (C 1 -C 4 ) -alkyl having 1 substituent selected from the group consisting of (C 3 -C 7) -cycloalkyl, difluoromethoxy, trifluoromethoxy and (C 1 -C 4) -alkoxy, R 1 is hydrogen or fluorine, a group of the formula
- R 13 is fluorine
- R 14 is hydrogen, fluorine, methyl or methoxy
- R 15 is hydrogen or fluorine
- R 3 is hydrogen or methyl
- R 4 is hydrogen, and their salts, solvates and solvates of the salts.
- A is nitrogen or CR 5 , where R 5 is hydrogen
- L is a group # 1 -CR 6A R 6B - (CR 7A R 7B) m - # 2, in which
- # is the point of attachment to the pyrimidine or triazine ring
- m is a number
- R 6A is hydroxyl
- R 6B is 2,2,2-trifluoroethyl, pentafluoroethyl or (C 1 -C 4 ) -alkyl, in which (C 1 -C 4 ) -alkyl having 1 substituent selected from the group consisting of (C 3 -C 7) -cycloalkyl, difluoromethoxy, trifluoromethoxy and (C 1 -C 4) -alkoxy is substituted,
- R 1 is hydrogen or fluorine
- R 2 is a group of the formula
- R 17 , R 18 , R 19 and R 20 are independently hydrogen, fluoro, methyl or
- radicals R 17 , R 18 , R 19 or R 20 are different from hydrogen, with the proviso that at least one of R 17 , R 18 , R 19 or R 20 is hydrogen , and with the proviso that in each case only one of the radicals R 17 , R 18 , R 19 or R 20 is methyl or methoxy,
- R 3 is hydrogen or methyl
- R 4 is hydrogen, and their salts, solvates and solvates of the salts.
- A is CR 5 , where
- R 5 is (C 1 -C 4 ) -alkyl, (C 2 -C 4 ) -alkenyl, (C 2 -C 4 ) -alkynyl, cyclopropyl, cyclobutyl, phenyl or C-linked 5- or 6-membered heteroaryl, wherein (Ci-C 4 ) alkyl, (C 2 -C 4 ) alkenyl, (C 2 -C 4 ) alkynyl, phenyl and 5- or 6-membered heteroaryl having 1 to 3 substituents independently selected from the group with fluorine, difluoromethyl, trifluoromethyl, (GC 4 ) alkyl, difluoromethoxy, trifluoromethoxy, (Ci-C 4 ) alkoxy, (Ci-C 4 ) alkoxycarbonyl, cyclopropyl and cyclobutyl are substituted, and their salts, solvates and solvates of salts.
- A is nitrogen
- L is a group # 1 -CR 6A R 6B - (CR 7A R 7B) m - # 2, which is the point of attachment to the carbonyl group is the point of attachment to the triazine ring, represents a number 0, represents amino , is hydrogen, fluorine, difluoromethyl, trifluoromethyl, (C 1 -C 6) -alkyl, cyano, (C 3 -C 7) -cycloalkyl, difluoromethoxy, trifluoromethoxy or a group of the formula -MR 12 , wherein (Ci-Ce) alkyl having 1 to 3 substituents independently selected from the group of fluorine, cyano, trifluoromethyl, (C3-C7) -cycloalkyl, difluoromethoxy and trifluoromethoxy may be substituted, and wherein
- M is a bond or (C 1 -C 4) -alkanediyl
- R 8 , R 9 and R 10 are each independently hydrogen
- R 8 and R 9 together with the AtonV (s) to which they are respectively attached form a 4- to 7-membered heterocycle wherein the 4- to 7-membered heterocycle is in turn selected from 1 or 2 substituents independently the group cyano, trifluoromethyl, (G-C6) alkyl, hydroxy, oxo, (Ci-Ce) alkoxy, trifluoromethoxy, (CI-C ⁇ ) - alkoxycarbonyl, amino, mono- (Ci-C6) -alkylamino and di - (Ci-C6) -alkylamino, or
- R 9 and R 10 together with the atom (s) to which they are respectively attached form a 4- to 7-membered heterocycle, in which the 4- to 7-membered heterocycle in turn contains 1 or 2 substituents independently of one another selected from the group cyano, trifluoromethyl, (G-CeJ-alkyl, hydroxy, oxo, (Ci-Ce) -alkoxy, trifluoromethoxy, (GC ⁇ ) Alkoxycarbonyl, amino, mono- (C 1 -C 6) -alkylamino and di- (C 1 -C 6) -alkylamino may be substituted,
- R 11 is (C 1 -C 6 ) -alkyl or (C 3 -C 7 ) -cycloalkyl, or
- R 8 and R 11 together with the AtonV (s) to which they are respectively attached, form a 4- to 7-membered heterocycle, wherein the 4- to 7-membered heterocycle is in turn selected from 1 or 2 substituents independently the group cyano, trifluoromethyl, (G-CeJ-alkyl, hydroxy, oxo, (Ci-Ce) alkoxy, trifluoromethoxy, (GC ⁇ ) - alkoxycarbonyl, amino, mono- (Ci-C6) alkylamino and di- (Ci -C6) -alkylamino, and wherein 4- to 7-membered heterocyclyl, phenyl and 5- or 6-membered heteroaryl in turn having 1 to 3 substituents independently selected from the group halogen, cyano, difluoromethyl, trifluoromethyl, (Ci -C 6 ) alkyl, (C 3 -C 7 ) cycloalkyl, hydroxy, ox
- L is a group # 1 -CR 6A R 6B - (CR 7A R 7B) m - # 2, where
- # ' represents the point of attachment to the carbonyl group, for the attachment site to the triazine ring, m is a number 0,
- R 6A stands for amino
- R 6B is difluoromethyl, trifluoromethyl or (C 1 -C 6 ) -alkyl, in which (C 1 -C 6 ) -alkyl having 1 to 3 substituents independently of one another selected from the group consisting of fluorine, trifluoromethyl, (C 3 -C 7) -cycloalkyl, difluoromethoxy, Trifluoromethoxy and (C 1 -C 4) -alkoxy may be substituted,
- R 1 , R 2 , R 3 and R 4 each have the meanings given above, and their salts, solvates and solvates of the salts.
- R 2 is 2-fluoro-3-methylbenzyl, 2-fluoro-4-methylbenzyl, 2-fluoro-3-methoxybenzyl or 4-methoxybenzyl, and their salts, solvates and solvates of the salts.
- L represents a group * -CR 6A R 6B - (CR 7A R 7B ) p - #, where
- # represents the point of attachment to the pyrimidine or triazine ring
- p stands for a number
- R 6A stands for amino
- R 6B is pentafluoroethyl, ethoxyethyl, methoxycarbonyl, ethoxycarbonyl, (benzyloxy) methyl or cyclopropylmethyl, and their salts, solvates and solvates of the salts.
- * represents the point of attachment to the pyrimidine or triazine ring
- p stands for a number
- R 6A is methyl
- R 6B is pentafluoroethyl, ethoxyethyl, methoxycarbonyl, ethoxycarbonyl, benzyloxy) methyl or cyclopropylmethyl, and their salts, solvates and solvates of the salts.
- R 6B is pentafluoroethyl, ethoxyethyl, methoxycarbonyl, ethoxycarbonyl, benzyloxy) methyl or cyclopropylmethyl, and their salts, solvates and solvates of the salts.
- T formula (T) in which
- L represents a group * -CR 6A R 6B - (CR 7A R 7B ) p - #, where
- * represents the point of attachment to the carbonyl group
- # represents the point of attachment to the pyrimidine or triazine ring
- p stands for a number
- R 6A is methyl
- R 6B is methyl, and their salts, solvates and solvates of the salts.
- R 6B is methyl, and their salts, solvates and solvates of the salts.
- particular preference is also given to compounds of the formula (I) in which
- A is nitrogen
- L represents a group * -CR 6A R 6B - (CR 7A R 7B ) p - #, in which
- # represents the point of attachment to the pyrimidine or triazine ring
- R ⁇ 6A is methyl
- R 6B is methyl, and their salts, solvates and solvates of the salts.
- R 21 is hydrogen or (C 1 -C 4) -alkyl
- R 22 is hydrogen or (C 1 -C 4) -alkyl, and wherein methyl and ethyl may be substituted with a group -NR 13 R 14 , wherein
- R 13 is hydrogen or methyl
- Ethyl may be substituted with 1 to 5 substituents fluorine, and wherein
- R 15 is methyl, ethyl or cyclopropyl
- R 16 is methyl, ethyl or cyclopropyl
- R 13 and R 14 together with the nitrogen atom to which they are attached form a 5-membered heterocycle in which the 5-membered heterocycle may in turn be substituted by oxo, and their salts, solvates and solvates of the salts.
- A is CR 5 , where
- R 5 is methyl or pyrazol-5-yl, wherein methyl is substituted with a group -NR 13 R 14 , wherein
- R 13 and R 14 independently of one another represent hydrogen, methyl, ethyl or -S (O) 2 -R 16 , wherein
- Ethyl may be substituted with 1 to 3 substituents fluorine, and wherein
- R ] 16 is methyl or cyclopropyl, or
- R 13 and R] 14 together with the nitrogen atom to which they are attached form a 5-membered heterocycle wherein 5-membered heterocycle can in turn be substituted with oxo, and their salts, solvates and solvates of the salts.
- radicals are substituted in the compounds according to the invention, the radicals can, unless otherwise specified, be monosubstituted or polysubstituted. In the context of the present invention, the meaning is independent of each other for all radicals which occur repeatedly. Substitution with one, two or three identical or different substituents is preferred.
- the invention further provides a process for the preparation of the compounds of the formula (I) according to the invention which comprises reacting a compound of the formula (II)
- T 1 is (C 1 -C 4 ) -alkyl, to give a compound of the formula (IV)
- T 3 is (C 1 -C 4 ) -alkyl
- T 4 is (C 1 -C 4 ) -alkyl
- R 5A in (XV-A) is (C 2 -C 4 ) -alkenyl or (C 2 -C 4 ) -alkynyl,
- X 3 is bromine or iodine, to give a compound of formula (ID)
- n, L, R 1 , R 2 , R 3 , R 4 and R 5A each have the meanings given above,
- Y is chlorine, in an inert solvent in the presence of a suitable base to give a compound of the formula (XVII)
- R 5A in (XV-A) is (C 2 -C 4 ) -alkenyl or (C 2 -C 4 ) -alkynyl,
- R and R independently of one another represent cyano or (C 1 -C 4) -alkoxycarbonyl, and in the following the protective group R is split off as described in [E], whereby compound (1-E) is formed by hydrolysis and decarboxylation,
- Inert solvents for process step (II) + (III) - (IV) are, for example, alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers such as diethyl ether, dioxane, dimethoxyethane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone (II), pyridine, Acetonitrile, sulfolane or even water. It is likewise possible to use mixtures of the solvents mentioned. Preference is given to
- Suitable bases for process step (II) + (III) - (IV) are alkali metal hydroxides such as lithium, sodium or potassium hydroxide, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, alkali hydrogen carbonates such as sodium or potassium hydrogen carbonate, Alkali alcoholates such as sodium or potassium methoxide, sodium or potassium ethoxide or potassium tert-butoxide, or organic amines such as triethylamine, diisopropylethylamine, pyridine, l, 8-diazabicyclo [5.4.0] undec-7-ene (DBU) or l, 5-diazabicyclo [4.3.0] non-5-ene (DBN). Preference is given to potassium tert-butoxide or sodium methoxide.
- alkali metal hydroxides such as lithium, sodium or potassium hydroxide
- alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate
- alkali hydrogen carbonates such as sodium
- the reaction (II) + (III) - (IV) is generally carried out in a temperature range from + 20 ° C to + 150 ° C, preferably at + 75 ° C to + 100 ° C, optionally in a microwave.
- the Reaction can be carried out at normal, elevated or at reduced pressure (for example from 0.5 to 5 bar). Generally, one works at normal pressure.
- halogen sources in the reaction (IV) - (V) are diiodomethane, a mixture of cesium iodide, iodine and copper (I) iodide or copper (II) bromide.
- the process step (IV) - (V) is carried out in the case of diiodomethane as the halogen source with a molar ratio of 10 to 30 moles of isopentyl nitrite and 10 to 30 moles of the iodine equivalent based on 1 mole of the compound of formula (IV).
- the process step (IV) - (V) is carried out with or without solvent.
- Suitable solvents are all organic solvents which are inert under the reaction conditions.
- Preferred solvent is dimethoxyethane.
- the reaction (IV) - (V) is generally carried out in a temperature range of + 20 ° C to + 100 ° C, preferably in the range of + 50 ° C to + 100 ° C, optionally in a microwave.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- Inert solvents for process step (V) - (IA) are alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol, tert-butanol or 1,2-ethanediol, ethers such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or Diethylene glycol dimethyl ether, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), NN'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile or water. It is likewise possible to use mixtures of the solvents mentioned. Preferred is DMF.
- the reduction (V) - (I-A) is carried out with hydrogen in conjunction with transition metal catalysts such as palladium (10% on activated carbon), Raney nickel or palladium hydroxide.
- transition metal catalysts such as palladium (10% on activated carbon), Raney nickel or palladium hydroxide.
- the reaction (V) - (I-A) is generally carried out in a temperature range of + 20 ° C to + 50 ° C.
- the reaction may be carried out at normal or elevated pressure (e.g., in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- Inert solvents for process step (II) + (VI) - (VII) are, for example, alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers, such as diethyl ether, dioxane, dimethoxyethane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), NN'-dimethyl propyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine or acetonitrile. It is likewise possible to use mixtures of the solvents mentioned. Preference is given to methanol or ethanol.
- Suitable bases for process step (II) + (VI) - (VII) are alkali metal hydroxides such as lithium, sodium or potassium hydroxide, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, alkali hydrogen carbonates such as sodium or potassium bicarbonate, Alkali alcoholates such as sodium or potassium methoxide, sodium or potassium ethoxide or potassium tert-butoxide, or organic amines such as triethylamine, diisopropylethylamine, pyridine, l, 8-diazabicyclo [5.4.0] undec-7-ene (DBU) or l, 5-diazabicyclo [4.3.0] non-5-ene (DBN).
- alkali metal hydroxides such as lithium, sodium or potassium hydroxide
- alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate
- alkali hydrogen carbonates such as sodium or potassium bicarbonate
- Alkali alcoholates such as sodium or potassium meth
- reaction (II) + (VI) - (VII) is generally carried out in a temperature range from + 50 ° C to + 120 ° C, preferably from + 50 ° C to + 100 ° C, optionally in a microwave.
- the reaction may be carried out at normal or elevated pressure (e.g., in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- the reactions (VII) - (VTII) and (XII) - (XIII) can be carried out in a solvent which is inert under the reaction conditions or without solvent.
- Preferred solvent is sulfolane.
- the reactions (VII) - (VIII) and (XII) - ( ⁇ ) are generally carried out in a temperature range from + 70 ° C to + 150 ° C, preferably from + 80 ° C to + 130 ° C, optionally in a microwave ,
- the reaction may be carried out at normal or elevated pressure (e.g., in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- the reaction takes place (XII) - (XIII) without solvent in a temperature range from 0 ° C to + 50 ° C at atmospheric pressure.
- the process step (VIII) - (IX) is carried out by reaction with sodium azide with intermediate formation of the azide derivatives, which are further reduced directly to the corresponding amines.
- Inert solvents for the azide formation are, for example, ethers, such as diethyl ether, dioxane, dimethoxyethane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons, such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents, such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), NN '-Dimethylpropylen- urea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile or sulfolane.
- ethers such as diethyl ether, dioxane, dimethoxye
- the azide is generally formed in a temperature range from + 50 ° C to + 100 ° C, preferably from + 60 ° C to + 80 ° C, at atmospheric pressure.
- the reduction is carried out in an inert solvent such as, for example, alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol, tert-butanol or 1,2-ethanediol, ethers, such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether. or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), NN'-dimethyl propyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile or even water. It is likewise possible to use mixtures of the solvents mentioned. Preferred is DMF.
- alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol, tert-butanol or 1,2-ethanedi
- the reduction takes place at + 10 ° C to + 30 ° C with hydrogen in conjunction with transition metal catalysts such as palladium (10% on activated carbon), platinum dioxide or palladium hydroxide, or without hydrogen with stannous chloride and hydrochloric acid.
- transition metal catalysts such as palladium (10% on activated carbon), platinum dioxide or palladium hydroxide, or without hydrogen with stannous chloride and hydrochloric acid.
- reaction (VIII) - (IX) can also be carried out in a step analogous to process step (XIII) - (XIV).
- Process (XIII) - (XIV) takes place in a solvent which is inert under the reaction conditions.
- Suitable solvents are, for example, ethers, such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, or other solvents, such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile or else Water. It is likewise possible to use mixtures of the solvents mentioned. Preference is given to acetonitrile.
- the reaction (XIII) - (XIV) is generally carried out in a temperature range from + 20 ° C to + 100 ° C, preferably from + 40 ° C to + 70 ° C, optionally in a microwave.
- the reaction may be carried out at normal or elevated pressure (e.g., in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- the cyclizations ( ⁇ ) - (IB) and (XIV) - (IC) are carried out in a reaction inert solvent such as alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers such as Diethyl ether, dioxane, dimethoxyethane, tetrahydrofuran (THF), glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethylsulfoxide (DMSO), N, N'-dimethylpropyleneurea ( DMPU), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile or sulfolane.
- a reaction inert solvent such
- Suitable bases for process steps (IX) - (IB) and (XIV) - (IC) are alkali metal hydroxides such as lithium, sodium or potassium hydroxide, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, alkali metal bicarbonates such as sodium or Potassium bicarbonate, alkali metal alcoholates such as sodium or potassium methoxide, sodium or potassium ethoxide or potassium tert-butoxide, or organic amines such as triethylamine, diisopropylethylamine, pyridine, 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) or l, 5-diazabicyclo [4.3.0] non-5-ene (DBN). Preference is given to potassium tert-butoxide.
- alkali metal hydroxides such as lithium, sodium or potassium hydroxide
- alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate
- the reactions (DQ - (IB) and (XIV) - (IC) are generally carried out in a temperature range from 0 ° C to + 50 ° C, preferably from + 10 ° C to + 30 ° C, optionally in a microwave Reaction can be carried out at normal or elevated pressure (eg in the range of 0.5 to 5 bar) . Generally, one works at atmospheric pressure.
- the cyclization to (I-B) or (I-C) directly in the reduction of the azide to the corresponding amine (IX) or in the reaction ( ⁇ ) - (XIV) without addition of other reagents.
- Inert solvents for process step (X) + (XI) - (XII) are, for example, alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers such as diethyl ether, dioxane, dimethoxyethane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), NN'-dimethyl propyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine or acetonitrile. It is likewise possible to use mixtures of the solvents mentioned. Preference is given to methanol or ethanol.
- the reaction (X) + (XI) - (XII) is generally carried out in a temperature range from + 50 ° C to + 120 ° C, preferably from + 50 ° C to + 100 ° C, optionally in a microwave.
- the reaction can be carried out at normal or elevated pressure (for example in the range from 0.5 to 5 bar). Generally, one works at normal pressure.
- Inert solvents for process step (II) - (X) are, for example, alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers, such as diethyl ether, dioxane, Dimethoxyethane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine or acetonitrile. It is likewise possible to use mixtures of the solvents mentioned. Preferred is ethanol.
- Suitable bases for process step (II) - (X) are alkali metal hydroxides such as lithium, sodium or potassium hydroxide, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, alkali hydrogen carbonates such as sodium or potassium bicarbonate, alkali metal alcoholates such as sodium or potassium methoxide, sodium or potassium ethoxide or potassium tert-butoxide, or organic amines such as triethylamine, diisopropylethylamine, pyridine, 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) or 1,5-diazabicyclo [ 4.3.0] non-5-en (DBN).
- DBU 1,8-diazabicyclo [5.4.0] undec-7-ene
- DBN 1,5-diazabicyclo [ 4.3.0] non-5-en
- triethylamine Preferably triethylamine.
- the reaction (II) - (X) is generally carried out in a temperature range from 0 ° C to + 60 ° C, preferably from + 10 ° C to + 30 ° C.
- the reaction may be carried out at normal or elevated pressure (e.g., in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- Process step (V) + (XV-A) or (XV-B) or (XV-C) or (XV-D) - (I-D) takes place in a solvent which is inert under the reaction conditions.
- Suitable solvents are, for example, ethers, such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, or other solvents, such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone (II), pyridine, acetonitrile or water. It is likewise possible to use mixtures of the solvents mentioned. Preference is given to acetonitrile, dioxane and tetrahydrofuran.
- the reaction (V) + (XV-A) or (XV-B) or (XV-C) or (XV-D) - (ID) can be carried out in the presence of a suitable palladium and / or copper catalyst.
- a suitable palladium and / or copper catalyst is, for example, palladium on activated carbon, palladium (II) acetate, tetrakis (triphenylphosphine) palladium (0), bis (triphenylphospliin) palladium (II) chloride, bis (acetonitrile) - palladium (II ) -chloride, [l, l'-bis (diphenylphospliino) ferrocene] dichloropalladium (n) and corresponding dichloromethane complex, optionally in combination with additional phosphane ligands such as (2-biphenyl) di-tert-butylphosphine, dicyclohexyl [
- Suitable copper catalysts are, for example, copper bronze, copper (I) oxide, copper (I) iodide or copper (I) bromide.
- alkali metal hydroxides such as lithium, sodium or potassium hydroxide, alkali metal or alkaline earth metal carbonates such as lithium, sodium, potassium, calcium or cesium carbonate, alkali alcoholates such as sodium or potassium, sodium or potassium or sodium or Potassium tert-butoxide, alkali metal hydrides such as sodium or potassium hydride, amides such as sodium amide, lithium, sodium or potassium bis (trimethylsilyl) amide or lithium diisopropylamide, or organic amines such as triethylamine, N-methylmorpholine, N-methylpiperidine, N, N-diisopropylethylamine, pyridine, 1,5-diazabicyclo [4.3.0] non-5-ene (DBN), 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) or 1,4- diazabicyclo [2.2.2] octane (DABCO ®).
- DBN 1,5-diazabicyclo
- the reaction (V) + (XV-A) or (XV-B) or (XV-C) or (XV-D) - (ID) is generally in a temperature range of 0 ° C to + 200 ° C, preferably carried out at + 10 ° C to + 150 ° C.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., from 0.5 to 5 bar). Generally, one works at normal pressure.
- radical R 5A is unsaturated, this can then be completely or partially saturated.
- the reduction is carried out with hydrogen in conjunction with transition metal catalysts such as palladium (10% on activated carbon), Raney nickel or palladium hydroxide.
- transition metal catalysts such as palladium (10% on activated carbon), Raney nickel or palladium hydroxide.
- the reduction is generally carried out in a temperature range from + 20 ° C to + 50 ° C.
- the reaction can be carried out at normal or elevated pressure (for example in the range from 1 to 150 bar). In general, it works at 1 to 3 bar.
- the process step (V) + (R 25 -Y) - (XVII) takes place in a solvent which is inert under the reaction conditions.
- Inert solvents for process step (V) + (R 25 -Y) - (XVII) are, for example, halogenated hydrocarbons such as dichloromethane, trichloromethane, tetrachloromethane, trichlorethylene or chlorobenzene, ethers such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene , Toluene, xylene, hexane, cyclohexane or petroleum fractions, or other solvents such as acetone, methyl ethyl ketone, ethyl acetate, acetonitrile, N, N-dimethylformamide, N, N-dimethylacet
- alkali metal hydroxides such as, for example, lithium, sodium or potassium hydroxide, alkali metal or alkaline earth metal carbonates such as lithium, sodium, potassium, calcium or cesium carbonate, alkali alcoholates such as sodium or potassium methoxide, sodium or potassium ethanolate or sodium or cesium carbonate, Alkali hydrides such as sodium or potassium hydride, amides such as sodium amide, lithium, sodium or potassium bis (trimethylsilyl) amide or lithium diisopropylamide, or organic amines such as triethylamine, N-methylmorpholine, N-methylpiperidine, N, N-diisopropylethylamine, pyridine , l, 5-diazabicyclo [4.3.0] non-5-ene (DBN), l, 8-diazabicyclo [5.4.0] undec-7-ene (DBU) or l, 4-diazabicyclo [2.2.2] octane (DABCO)
- the reaction (V) + (R 25 -Y) - (XVII) is generally carried out in a temperature range of -20 ° C to + 200 ° C, preferably at + 10 ° C to + 100 ° C.
- the reaction can be carried out at normal, elevated or at reduced pressure (for example from 0.5 to 5 bar). Generally, one works at normal pressure.
- Process step (XVII) + (XV-A) or (XV-B) or (XV-C) or (XV-D) - (XVIII) takes place in a solvent which is inert under the reaction conditions.
- Suitable solvents are, for example, ethers, such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, or other solvents, such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone (II), toluene , Acetonitrile or water. It is likewise possible to use mixtures of the solvents mentioned. Preferred are dioxane and tetrahydrofuran.
- reaction (XVII) + (XV-A) or (XV-B) or (XV-C) or (XV-D) - (XVTII) can be carried out in the presence of a suitable palladium and / or copper catalyst.
- Suitable palladium catalysts are palladium on activated carbon, palladium (II) acetate, tetrakis (triphenylphosphine) palladium (0), bis (triphenylphosphine) palladium (II) chloride, bis (acetonitrile) palladium (II ) -chloride, [l, l'-bis (diphenylphospliino) ferrocene] dichloropalladium (n) and corresponding dichloromethane complex, optionally in combination with additional phosphane ligands such as (2-biphenyl) di-tert-butylphosphine, dicyclohexyl [2 ' , 4 ', 6'-tris (1-methylethyl) biphenyl-2-yl] phosphine (XPHOS), bis (2-phenylphosphinophenyl) ether (DPEphos) or 4,5-bis (dipheny
- Suitable copper catalysts are, for example, copper bronze, copper (I) oxide, copper (I) iodide or copper (I) bromide.
- the reaction (XVII) + (XV-A) or (XV-B) or (XV-C) or (XV-D) - (XVIII) takes place in the presence of a suitable base.
- Suitable bases for this reaction are the usual inorganic or organic bases.
- alkali metal or alkaline earth metal carbonates such as lithium, sodium, potassium, calcium or cesium carbonate or organic amines such as triethylamine, N-methylmorpholine, N-methylpiperidine, N, N-diisopropylethylamine, pyridine, 1,5-diazabicyclo [4.3 .0] non-5-ene (DBN), l, 8-diazabicyclo [5.4.0] undec-7-ene (DBU) or 1,4-diazabicyclo [2.2.2] octane (DABCO ®).
- triethylamine or sodium bicarbonate is used.
- the reaction (XVII) + (XV-A) or (XV-B) or (XV-C) or (XV-D) - (XVIII) is generally carried out in a temperature range of 0 ° C to + 200 ° C, preferably carried out at + 10 ° C to + 150 ° C.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., from 0.5 to 5 bar). Generally, one works at normal pressure.
- the process step (XVIII) - (ID) is carried out in the case of PMB by reaction with a mixture of trifluoromethanesulfonic anhydride and trifluoroacetic acid or trifluoroacetic acid and trifluoromethanesulfonic acid or cerium (IV) ammonium nitrate in suitable solvents such as acetonitrile, DMF or NMP and in the case of SEM as a protective group by reaction first with trifluoroacetic acid in suitable solvents we dichloromethane and then with aqueous mineral acid in suitable solvents such as ethanol, THF or dioxane.
- suitable solvents such as acetonitrile, DMF or NMP
- SEM as a protective group
- the reaction (XVIII) - (I-D) is generally carried out in a temperature range of 0 ° C to + 200 ° C, preferably at + 10 ° C to + 150 ° C, optionally in the microwave.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., from 0.5 to 20 bar). In general, it works at 0.5 to 10 bar.
- Suitable solvents are, for example, ethers, such as diethyl ether, dioxane, dimethoxyethane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons, such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents, such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropylene urea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile or sulfolane. It is likewise possible to use mixtures of the solvents mentioned. Preferred is DMF.
- Suitable bases for this reaction are the usual inorganic or organic bases. These include preferably alkali metal or alkaline earth metal carbonates such as lithium, sodium, potassium, Calcium or cesium carbonate, alkali alcoholates such as sodium or potassium methoxide, sodium or potassium ethoxide or sodium or potassium tert-butoxide, alkali metal hydrides such as sodium or potassium hydride, amides such as sodium amide, lithium, sodium or potassium bis (trimethylsilyl) amide or lithium diisopropylamide, or organic amines such as triethylamine, N-methylmorpholine, N-methylpiperidine, N, N-diisopropylethylamine, pyridine, l, 5-diazabicyclo [4.3.0] non-5-ene (DBN), 1,8-diazabicyclo [5.4.0] undec-7
- alkali metal or alkaline earth metal carbonates such as lithium, sodium, potassium, Calcium or cesium carbonate
- the reaction (XVIII) + (XIX) - (XX) is generally carried out in a temperature range from 0 ° C to + 200 ° C, preferably at + 20 ° C to + 100 ° C.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., from 0.5 to 5 bar). Generally, one works at normal pressure.
- Process step (V) + copper cyanide - (XXI) takes place in a solvent which is inert under the reaction conditions.
- suitable solvents are, for example, ethers, such as diethyl ether, dioxane, dimethoxyethane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons, such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents, such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile or sulfolane.
- ethers such as diethyl ether, dioxane, dimethoxyethane, tetrahydrofuran, glycol dimethyl ether or diethylene glyco
- reaction (V) + Kupfercaynid- (XXI) is generally carried out in a temperature range of 0 ° C to + 200 ° C, preferably at + 40 ° C to + 180 ° C.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., from 0.5 to 5 bar). Generally, one works at normal pressure.
- the reduction (XXI) - (I-F) is carried out with hydrogen in conjunction with transition metal catalysts such as palladium (10% on activated carbon), Raney nickel or palladium hydroxide.
- transition metal catalysts such as palladium (10% on activated carbon), Raney nickel or palladium hydroxide.
- the reaction (XXI) - (IF) is generally carried out in a temperature range of + 20 ° C to + 100 ° C.
- the reaction can be carried out at normal or elevated pressure (for example in the range from 0.5 to 100 bar). In general, it works at 1 to 3 bar.
- Process step (IF) + (XXII) - (IG) takes place in a solvent which is inert under the reaction conditions.
- Suitable solvents are, for example, halogenated hydrocarbons such as dichloromethane, trichloromethane, carbon tetrachloride, trichlorethylene or chlorobenzene, ethers such as Diethyl ether, dioxane, dimethoxyethane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU ), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile or sulfolane. It is likewise possible to use mixtures of the solvents mentioned. Preferred is DMF or a mixture of DMF and dichloromethane
- Suitable bases for this reaction are the usual inorganic or organic bases. These include preferably alkali metal or alkaline earth metal carbonates such as lithium, sodium, potassium, calcium or cesium carbonate, alkali alcoholates such as sodium or potassium, sodium or potassium or sodium or potassium tert-butoxide, alkali metal hydrides such as sodium or potassium , Amides such as sodium amide, lithium, sodium or potassium bis (trimethylsilyl) amide or lithium diisopropylamide, or organic amines such as triethylamine, N-methylmorpholine, N-methylpiperidine, N, N-diisopropylethylamine, pyridine, $ -dimethylamino-pyridine, 1.5 - diazabicyclo [4.3.0] non-5-ene (DBN), l, 8-diazabicyclo [5.4.0] undec-7-
- alkali metal or alkaline earth metal carbonates such as lithium, sodium, potassium, calcium or cesium carbonate
- the reaction (I-F) + (XXII) - (I-G) is generally carried out in a temperature range of 0 ° C to + 200 ° C, preferably at + 10 ° C to + 50 ° C.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., from 0.5 to 5 bar). Generally, one works at normal pressure
- the preparation of the compounds of the formula (I) according to the invention can be carried out by reversing the reaction steps using protective group chemistry, as shown by way of example in the following synthesis scheme (Scheme 9):
- transformations are carried out by conventional methods known to those skilled in the art and include, for example, reactions such as nucleophilic and electrophilic substitutions, oxidations, reductions, hydrogenations, transition metal-catalyzed coupling reactions, Grignard reactions, eliminations, alkylation, acylation, amination, esterification, ester cleavage, etherification, Ether cleavage, formation of carbonamides, and introduction and removal of temporary protecting groups.
- reactions such as nucleophilic and electrophilic substitutions, oxidations, reductions, hydrogenations, transition metal-catalyzed coupling reactions, Grignard reactions, eliminations, alkylation, acylation, amination, esterification, ester cleavage, etherification, Ether cleavage, formation of carbonamides, and introduction and removal of temporary protecting groups.
- reactions such as nucleophilic and electrophilic substitutions, oxidations, reductions, hydrogenations, transition metal-catalyzed coupling reactions, Grignard reactions, eliminations
- the compounds of the formula (II) are known from the literature (see, for example, WO 2011/147809, WO 03/095451, Example 6A) or can be prepared in analogy to processes known from the literature.
- the compounds of the formula (VI), (XI), (XV-A), (XV-B), (XV-C), (XV-D), (XVI), (XIX) and (XXII) are commercially available , known from the literature or can be prepared in analogy to known literature.
- the compounds of the invention act as potent stimulators of soluble guanylate cyclase and inhibitors of phosphodiesterase-5, have valuable pharmacological properties, and have an improved therapeutic profile, such as their in vivo properties and / or their pharmacokinetic behavior and / or metabolic profile. They are therefore suitable for the treatment and / or prophylaxis of diseases in humans and animals.
- the compounds of the invention cause vasorelaxation and inhibition of platelet aggregation and lead to a reduction in blood pressure and to an increase in coronary blood flow. These effects are mediated by direct stimulation of soluble guanylate cyclase and intracellular cGMP increase.
- the compounds according to the invention enhance the action of substances which increase cGMP levels, such as, for example, endothelium-derived relaxing factor (EDRF), NO donors, protoporphyrin IX, arachidonic acid or phenylhydrazine derivatives.
- EDRF endothelium-derived relaxing factor
- NO donors NO donors
- protoporphyrin IX arachidonic acid or phenylhydrazine derivatives.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of cardiovascular, pulmonary, thromboembolic and fibrotic disorders.
- the compounds according to the invention can therefore be used in medicaments for the treatment and / or prophylaxis of cardiovascular diseases such as hypertension, resistant hypertension, acute and chronic heart failure, coronary heart disease, stable and unstable angina pectoris, peripheral and cardiac vascular diseases, arrhythmias, atrial arrhythmias and the ventricles as well as conduction disorders such as atrio-ventricular blockades grade I-III (AB block I-III), supraventricular tachyarrhythmia, atrial fibrillation, atrial flutter, ventricular fibrillation, ventricular tachyarrhythmia, torsades de pointes tachycardia, atrial and ventricular extrasystoles , AV junctional extrasystoles, sick sinus syndrome, syncope, AV nodal reentrant tachycardia, Wolff-Parkinson-White syndrome, acute coronary syndrome (ACS), autoimmune heart disease (pericarditis, endocarditis, valvolitis, aor
- cardiac failure includes both acute and chronic manifestations of cardiac insufficiency, as well as more specific or related forms of disease such as acute decompensated heart failure, right heart failure, left heart failure, global insufficiency, ischemic cardiomyopathy, dilated cardiomyopathy, hypertrophic cardiomyopathy, idiopathic cardiomyopathy, congenital heart defects.
- Cardiac insufficiency in cardiac valve defects mitral valve stenosis, mitral valve insufficiency, aortic valve stenosis, aortic valve insufficiency, tricuspid stenosis, tricuspid insufficiency, pulmonary valve stenosis, pulmonary valvular insufficiency, combined heart valve defects, myocarditis, chronic myocarditis, acute myocarditis, viral myocarditis, diabetic cardiac insufficiency, alcoholic cardiomyopathy, cardiac storage disorders, dystolic heart failure and systolic heart failure and acute phase n worsening of existing chronic heart failure.
- the compounds according to the invention may also be used for the treatment and / or prophylaxis of arteriosclerosis, lipid metabolism disorders, hypolipoproteinemias, dyslipidemias, hypertriglyceridemias, hyperlipidemias, hypercholesterolemias, abetelipoproteinaemia, sitosterolemia, xanthomatosis, Tangier's disease, obesity (obesity) and combined hyperlipidemias and the metabolic syndrome.
- the compounds of the invention may be used for the treatment and / or prophylaxis of primary and secondary Raynaud's phenomenon, microcirculatory disorders, claudication, peripheral and autonomic neuropathies, diabetic microangiopathies, diabetic retinopathy, diabetic ulcers on the extremities, gangrenous, CREST syndrome, erythematosis, onychomycosis
- the compounds according to the invention are also suitable for the treatment of muscular dystrophy, such as the muscular dystrophy Becker-Kiener (BMD) and Duchenne muscular dystrophy (DMD).
- the compounds according to the invention are suitable for the treatment of urological diseases such as benign prostatic syndrome (BPS), benign prostatic hyperplasia (BPH), benign prostate enlargement (BPE), bladder emptying disorder (BOO), lower urinary tract syndromes (LUTS, including Feiine's urological syndrome ( FUS)), diseases of the Urogenital system including neurogenic overactive bladder (OAB) and (IC), incontinence (UI) such as mixed, urgency, stress, or overflow incontinence (MUI, UUI, SUI, OUI), pelvic pain, benign and malignant diseases the organs of the male and female urogenital system.
- BPS benign prostatic syndrome
- BPH benign prostatic hyperplasia
- BPE benign prostate enlargement
- BOO bladder emptying disorder
- LUTS lower urinary tract syndromes
- FUS lower urinary tract syndromes
- UI incontinence
- MUI mixed, urgency, stress, or overflow incontinence
- MUI UUI, SUI, OUI
- kidney diseases in particular of acute and chronic renal insufficiency, as well as of acute and chronic renal failure.
- renal insufficiency includes both acute and chronic manifestations of renal insufficiency, as well as underlying or related renal diseases such as renal hypoperfusion, intradialytic hypotension, obstructive uropathy, glomerulopathies, glomerulonephritis, acute glomerulonephritis, glomerulosclerosis, tubulo-interstitial disorders, nephropathic disorders such as primary and congenital kidney disease, nephritis, immunological kidney diseases such as renal transplant rejection, immune complex-induced kidney disease, nephropathy induced by toxic substances, contrast agent-induced nephropathy, diabetic and non-diabetic nephropathy, pyelonephritis, renal cysts, nephrosclerosis, hyperten
- the present invention also encompasses the use of the compounds of the invention for the treatment and / or prophylaxis of sequelae of renal insufficiency, such as pulmonary edema, heart failure, uremia, anemia, electrolyte imbalances (eg, hyperkalemia, hyponatremia) and disorders in bone and carbohydrate metabolism.
- sequelae of renal insufficiency such as pulmonary edema, heart failure, uremia, anemia, electrolyte imbalances (eg, hyperkalemia, hyponatremia) and disorders in bone and carbohydrate metabolism.
- the compounds according to the invention are also suitable for the treatment and / or prophylaxis of asthmatic diseases, pulmonary arterial hypertension (PAH) and other forms of pulmonary hypertension (PH), including left heart disease, HIV, sickle cell anemia, thromboembolism (CTEPH), sarcoidosis, COPD or Pulmonary fibrosis-associated pulmonary hypertension, chronic obstructive pulmonary disease (COPD), acute respiratory tract syndrome (ARDS), acute lung injury (ALI), alpha-1-antitrypsin deficiency (AATD), pulmonary fibrosis, pulmonary emphysema (eg, cigarette smoke-induced Pulmonary emphysema) and cystic fibrosis (CF).
- PAH pulmonary arterial hypertension
- PH pulmonary hypertension
- COPD chronic obstructive pulmonary disease
- ARDS acute respiratory tract syndrome
- ALI acute lung injury
- AATD alpha-1-antitrypsin deficiency
- CF
- the compounds described in the present invention are also agents for controlling diseases in the central nervous system, which are characterized by disorders of the NO / cGMP system.
- they are suitable for improving the perception, concentration performance, learning performance or memory performance after cognitive disorders such as occur in situations / diseases / syndromes such as mild cognitive impairment, age-associated learning and memory disorders, age-associated memory loss, vascular dementia, cranial brain -Trauma, stroke, post-stroke dementia, post-traumatic traumatic brain injury, generalized concentration disorder, difficulty concentrating in children with learning and memory problems, Alzheimer's disease, dementia with Lewy bodies , Dementia with degeneration of the frontal lobes including Pick's syndrome, Parkinson's disease, progressive nuclear palsy, dementia with corticobasal degeneration, amyolateral sclerosis (ALS), Huntington's disease, demyelinization, multiple sclerosis, thalamic degeneration, Creutzfeld-Jacob dementia, HIV dementia, schizophrenia with dementia or Korsakoff's psychosis.
- the compounds according to the invention are also suitable for the treatment and / or prophylaxis of diseases of the central nervous system such as states of anxiety, tension and depression, central nervous conditional sexual dysfunctions and sleep disorders as well as for the regulation of pathological disorders of food, pleasure and addiction.
- the compounds according to the invention are also suitable for regulating cerebral blood flow and are effective agents for combating migraine. They are also suitable for the prophylaxis and control of the consequences of cerebral infarct events (Apoplexia cerebri) such as stroke, cerebral ischaemias and craniocerebral trauma , Likewise, the compounds of the invention can be used to combat pain and tinnitus.
- the compounds according to the invention have antiinflammatory activity and can therefore be used as antiinflammatory agents for the treatment and / or prophylaxis of sepsis (SIRS), multiple organ failure (MODS, MOF), inflammatory diseases of the kidney, chronic intestinal inflammation (IBD, Crohn's Disease, UC). , Pancreatitis, peritonitis, rheumatoid diseases, inflammatory skin diseases and inflammatory ocular diseases.
- SIRS sepsis
- MODS multiple organ failure
- IBD chronic intestinal inflammation
- UC chronic intestinal inflammation
- Pancreatitis peritonitis
- rheumatoid diseases inflammatory skin diseases and inflammatory ocular diseases.
- the compounds of the invention can also be used for the treatment and / or prophylaxis of autoimmune diseases.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of fibrotic disorders of the internal organs such as, for example, the lung, the heart, the kidney, the bone marrow and in particular the liver, as well as dermatological fibroses and fibrotic disorders of the eye.
- fibrotic disorders includes in particular the following terms: liver fibrosis, cirrhosis, pulmonary fibrosis, endomyocardial fibrosis, nephropathy, glomerulonephritis, interstitial renal fibrosis, fibrotic damage as a result of diabetes, bone marrow fibrosis and similar fibrotic disorders, scleroderma, morphea, keloids, hypertrophic scarring (also after surgical interventions), nevi, diabetic retinopathy, proliferative vitroretinopathy and connective tissue disorders (eg sarcoidosis).
- the compounds of the invention are useful for controlling postoperative scarring, e.g. as a result of glaucoma surgery.
- the compounds according to the invention can likewise be used cosmetically for aging and keratinizing skin.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of hepatitis, neoplasm, osteoporosis, glaucoma and gastroparesis.
- Another object of the present invention is the use of the compounds of the invention for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- Another object of the present invention is the use of the compounds of the invention for the treatment and / or prophylaxis of heart failure, angina pectoris, hypertension, pulmonary hypertension, ischaemia, vascular disease, renal insufficiency, thromboembolic disorders, fibrotic diseases and arteriosclerosis.
- the present invention furthermore relates to the compounds according to the invention for use in a method for the treatment and / or prophylaxis of cardiac insufficiency, angina pectoris, hypertension, pulmonary hypertension, ischaemias, vascular disorders, renal insufficiency, thromboembolic disorders, fibrotic disorders and atherosclerosis.
- Another object of the present invention is the use of the compounds of the invention for the manufacture of a medicament for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- Another object of the present invention is the use of the compounds of the invention for the manufacture of a medicament for the treatment and / or prophylaxis of heart failure, angina pectoris, hypertension, pulmonary hypertension, ischemia, vascular diseases, renal insufficiency, thromboembolic disorders, fibrotic diseases and arteriosclerosis.
- Another object of the present invention is a method for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases, using an effective amount of at least one of the compounds of the invention.
- the present invention further provides a method for the treatment and / or prophylaxis of cardiac insufficiency, angina pectoris, hypertension, pulmonary hypertension, ischaemias, vascular diseases, renal insufficiency, thromboembolic disorders, fibrotic diseases and atherosclerosis, using an effective amount of at least one of the compounds according to the invention ,
- the compounds of the invention may be used alone or as needed in combination with other agents.
- Another object of the present invention are pharmaceutical compositions containing at least one of the compounds of the invention and one or more other active ingredients, in particular for the treatment and / or prophylaxis of the aforementioned diseases.
- suitable combination active ingredients may be mentioned by way of example and preferably:
- organic nitrates and NO donors such as sodium nitroprusside, nitroglycerin, isosorbide mononitrate, isosorbide dinitrate, molsidomine or SIN-1, and inhaled NO;
- cGMP cyclic guanosine monophosphate
- PDE phosphodiesterases
- Antithrombotic agents by way of example and preferably from the group of thrombocyte aggregation inhibitors, anticoagulants or profibrinolytic substances;
- Antihypertensive agents by way of example and preferably from the group of calcium antagonists, angiotensin AII antagonists, ACE inhibitors, endothelin antagonists, renin inhibitors, alpha-receptor blockers, beta-receptor blockers, mineralocorticoid receptor Antagonists and diuretics; and / or ⁇ fat metabolism-altering agents, by way of example and preferably from the group of thyroid receptor agonists, cholesterol synthesis inhibitors, as exemplified and preferably HMG co-eductase or squalene synthesis inhibitors, ACAT inhibitors, CETP inhibitors, MTP inhibitors, PPAR-alpha, PPAR-gamma and / or PPAR-delta agonists, cholesterol absorption inhibitors, lipase inhibitors, polymeric bile acid adsorbents, bile acid reabsorption inhibitors, and lipoprotein (a) antagonists.
- Antithrombotic agents are preferably understood as meaning compounds from
- the compounds according to the invention are administered in combination with a platelet aggregation inhibitor, such as, by way of example and by way of preference, aspirin, clopidogrel, ticlopidine or dipyridamole.
- a platelet aggregation inhibitor such as, by way of example and by way of preference, aspirin, clopidogrel, ticlopidine or dipyridamole.
- the compounds according to the invention are administered in combination with a thrombin inhibitor, such as, by way of example and by way of preference, ximelagatran, dabigatran, melagatran, bivalirudin or Clexane.
- a thrombin inhibitor such as, by way of example and by way of preference, ximelagatran, dabigatran, melagatran, bivalirudin or Clexane.
- the compounds according to the invention are administered in combination with a GPIIb / IIIa antagonist, such as, by way of example and by way of preference, tirofiban or abciximab.
- a GPIIb / IIIa antagonist such as, by way of example and by way of preference, tirofiban or abciximab.
- the compounds according to the invention are used in combination with a factor Xa inhibitor, such as by way of example and preferably rivaroxaban, DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux, PMD-3112, YM-150, KFA -1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 or SSR-128428.
- a factor Xa inhibitor such as by way of example and preferably rivaroxaban, DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux, PMD-3112, YM-150, KFA -1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC
- the compounds according to the invention are administered in combination with heparin or a low molecular weight (LMW) heparin derivative.
- LMW low molecular weight
- the compounds according to the invention are administered in combination with a vitamin K antagonist, such as by way of example and preferably coumarin.
- antihypertensive agents are preferably compounds from the group of calcium antagonists, angiotensin AII antagonists, ACE inhibitors, endothelin antagonists, renin inhibitors, alpha-receptor blocker, beta-receptor blocker, mineralocorticoid receptor Antagonists and diuretics understood.
- the compounds according to the invention are administered in combination with a calcium antagonist, such as, by way of example and by way of preference, nifedipine, amlodipine, verapamil or diltiazem.
- the compounds according to the invention are administered in combination with an alpha-1-receptor blocker, such as by way of example and preferably prazosin.
- the compounds according to the invention are used in combination with a beta-receptor blocker, such as by way of example and preferably propranolol, atenolol, timolol, pindolol, alprenolol, oxprenolol, penbutolol, bupranolol, metipranolol, nadolol, mepindolol, carazalol, sotalol, Metoprolol, betaxolol, celiprolol, bisoprolol, carteolol, esmolol, labetalol, carvedilol, adaprolol, landiolol, nebivolol, epanolol or bucindolol.
- a beta-receptor blocker such as by way of example and preferably propranolol, atenolol, timolol
- the compounds according to the invention are administered in combination with an angiotensin AII antagonist, such as by way of example and preferably losartan, candesartan, valsartan, telmisartan or embursatan.
- an ACE inhibitor such as, by way of example and by way of preference, enalapril, captopril, lisinopril, ramipril, delapril, fosinopril, quinopril, perindopril or trandopril.
- the compounds according to the invention are administered in combination with an endothelin antagonist such as, by way of example and by way of preference, bosentan, darusentan, ambrisentan or sitaxsentan.
- an endothelin antagonist such as, by way of example and by way of preference, bosentan, darusentan, ambrisentan or sitaxsentan.
- the compounds of the invention are administered in combination with a renin inhibitor, such as by way of example and preferably aliskiren, SPP-600 or SPP-800.
- a renin inhibitor such as by way of example and preferably aliskiren, SPP-600 or SPP-800.
- the compounds according to the invention are administered in combination with a mineralocorticoid receptor antagonist, such as by way of example and preferably spironolactone or eplerenone.
- a mineralocorticoid receptor antagonist such as by way of example and preferably spironolactone or eplerenone.
- the compounds of the invention are used in combination with a loop diuretic such as furosemide, torasemide, bumetanide and piretanide with potassium sparing diuretics such as amiloride and triamterene, with aldosterone antagonists such as spironolactone, potassium canrenoate and Eplerenone and thiazide diuretics, such as hydrochlorothiazide, chlorthalidone, xipamide, and indapamide administered.
- a loop diuretic such as furosemide, torasemide, bumetanide and piretanide
- potassium sparing diuretics such as amiloride and triamterene
- aldosterone antagonists such as spironolactone, potassium canrenoate and Eplerenone and thiazide diuretics, such as hydrochlorothiazide, chlorthalidone, xipamide, and indapamide administered.
- lipid metabolizing agents are preferably compounds from the group of CETP inhibitors, thyroid receptor agonists, cholesterol synthesis inhibitors such as HMG-CoA eduktase or squalene synthesis inhibitors, the ACAT inhibitors, MTP inhibitors, PPAR-alpha, PPAR gamma and / or PPAR delta agonists, cholesterol absorption inhibitors, polymeric bile acid adsorbers, bile acid reabsorption inhibitors, lipase inhibitors, and lipoprotein (a) antagonists.
- CETP inhibitors such as HMG-CoA eduktase or squalene synthesis inhibitors
- ACAT inhibitors such as HMG-CoA eduktase or squalene synthesis inhibitors
- MTP inhibitors MTP inhibitors
- PPAR-alpha PPAR-alpha
- PPAR gamma and / or PPAR delta agonists cholesterol absorption inhibitors
- the compounds according to the invention are administered in combination with a CETP inhibitor, such as, for example and preferably, dalcetrapib, BAY 60-5521, anacetrapib or CETP vaccine (CETi-1).
- a CETP inhibitor such as, for example and preferably, dalcetrapib, BAY 60-5521, anacetrapib or CETP vaccine (CETi-1).
- the compounds of the invention are administered in combination with a thyroid receptor agonist such as, by way of example and by way of preference, D-thyroxine, 3,5,3'-triiodothyronine (T3), CGS 23425 or axitirome (CGS 26214).
- a thyroid receptor agonist such as, by way of example and by way of preference, D-thyroxine, 3,5,3'-triiodothyronine (T3), CGS 23425 or axitirome (CGS 26214).
- T3 3,5,3'-triiodothyronine
- CGS 23425 CGS 23425
- axitirome CGS 26214
- the compounds according to the invention are administered in combination with an HMG-CoA reductase inhibitor from the class of statins, such as by way of example and preferably lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastat
- the compounds according to the invention are administered in combination with a squalene synthesis inhibitor, such as by way of example and preferably BMS-188494 or TAK-475.
- a squalene synthesis inhibitor such as by way of example and preferably BMS-188494 or TAK-475.
- the compounds according to the invention are administered in combination with an ACAT inhibitor, such as by way of example and preferably avasimibe, melinamide, pactimibe, eflucimibe or SMP-797.
- an MTP inhibitor such as, for example and preferably, implitapide, BMS-201038, R-103757 or JTT-130.
- the compounds of the invention are administered in combination with a PPAR gamma agonist such as, by way of example and by way of preference, pioglitazone or rosiglitazone.
- a PPAR gamma agonist such as, by way of example and by way of preference, pioglitazone or rosiglitazone.
- the compounds according to the invention are administered in combination with a PPA-delta agonist, such as by way of example and preferably GW 501516 or BAY 68-5042.
- the compounds according to the invention are administered in combination with a cholesterol absorption inhibitor, such as by way of example and preferably ezetimibe, tiqueside or pamaqueside.
- a cholesterol absorption inhibitor such as by way of example and preferably ezetimibe, tiqueside or pamaqueside.
- the compounds according to the invention are administered in combination with a lipase inhibitor, such as, for example and preferably, orlistat.
- a lipase inhibitor such as, for example and preferably, orlistat.
- the compounds of the invention are administered in combination with a polymeric bile acid adsorbent such as, by way of example and by way of preference, cholestyramine, colestipol, colesolvam, cholesta gel or colestimide.
- a polymeric bile acid adsorbent such as, by way of example and by way of preference, cholestyramine, colestipol, colesolvam, cholesta gel or colestimide.
- ASBT IBAT
- the compounds of the invention are administered in combination with a lipoprotein (a) antagonist such as, by way of example and by way of preference, gemcabene calcium (CI-1027) or nicotinic acid.
- a lipoprotein (a) antagonist such as, by way of example and by way of preference, gemcabene calcium (CI-1027) or nicotinic acid.
- compositions containing at least one compound of the invention usually together with one or more inert, non-toxic, pharmaceutically suitable excipients, and their use for the purposes mentioned above.
- the compounds according to the invention can act systemically and / or locally.
- they may be applied in a suitable manner, e.g. oral, parenteral, pulmonary, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctival, otic or as an implant or stent.
- the compounds according to the invention can be administered in suitable administration forms.
- the compounds according to the invention quickly and / or modified donating application forms, the Compounds according to the invention in crystalline and / or amorphised and / or dissolved form, such as tablets (uncoated or coated tablets, for example, with enteric or delayed-dissolving or insoluble coatings, which control the release of the compound of the invention) in the oral cavity quickly disintegrating tablets or films / wafers, films / lyophilisates, capsules (for example hard or soft gelatin capsules), dragées, granules, pellets, powders, emulsions, suspensions, aerosols or solutions.
- Parenteral administration can be accomplished by bypassing a resorption step (e.g., intravenously, intraarterially, intracardially, intraspinal, or intralumbar) or by resorting to absorption (e.g., intramuscularly, subcutaneously, intracutaneously, percutaneously, or intraperitoneally).
- a resorption step e.g., intravenously, intraarterially, intracardially, intraspinal, or intralumbar
- absorption e.g., intramuscularly, subcutaneously, intracutaneously, percutaneously, or intraperitoneally.
- parenteral administration are suitable as application forms u.a. Injection and infusion preparations in the form of solutions, suspensions, emulsions, lyophilisates or sterile powders.
- Inhalation medicaments including powder inhalers, nebulizers
- nasal drops solutions or sprays
- lingual, sublingual or buccal tablets films / wafers or capsules
- suppositories ear or eye preparations
- vaginal capsules aqueous suspensions (lotions, shake mixtures)
- lipophilic suspensions ointments
- creams transdermal therapeutic systems (eg plasters)
- milk pastes, foams, powdered powders, implants or stents.
- compositions according to the invention can be converted into the stated administration forms. This can be done in a conventional manner by mixing with inert, non-toxic, pharmaceutically suitable excipients.
- adjuvants include, among others.
- Excipients for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecyl sulfate, polyoxysorbitol oleate
- binders for example polyvinylpyrrolidone
- synthetic and natural polymers for example albumin
- stabilizers For example, antioxidants such as ascorbic acid
- dyes eg, inorganic pigments such as iron oxides
- flavor and / or odoriferous for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecyl sulfate, polyoxysorbitol oleate
- binders for example polyvinylpyrrolidone
- synthetic and natural polymers for example albumin
- stabilizers for example, antioxidants such as
- the dosage is about 0.001 to 2 mg / kg, preferably about 0.001 to 1 mg kg of body weight.
- Device Type MS Waters ZQ
- Device Type HPLC Agilent 1100 Series
- UV DAD Column: Thermo Hypersil GOLD 3 ⁇ 20 mm x 4 mm
- Eluent A 1 l of water + 0.5 ml of 50% formic acid
- eluent B 1 l of acetonitrile + 0.5 ml of 50% formic acid
- UV detection 210 nm.
- Instrument Thermo DFS, Trace GC Ultra; Column: Restek RTX-35, 15 m x 200 ⁇ x 0.33 ⁇ ; constant flow with helium: 1.20 ml / min; Oven: 60 ° C; Inlet: 220 ° C; Gradient: 60 ° C, 30 ° C / min 300 ° C (hold for 3.33 min).
- Method 9 Instrument: Waters ACQUITY SQD UPLC System; Column: Waters Acquity UPLC HSS T3 1.8 ⁇ 50 x 1 mm; Eluent A: 1 l of water + 0.25 ml of 99% formic acid, eluent B: 1 l of acetonitrile + 0.25 ml of 99% formic acid; Gradient: 0.0 min 95% A 6.0 min 5% A 7.5 min 5% A Oven: 50 ° C; Flow: 0.35 ml / min; UV detection: 210 - 400 nm.
- Method 10 Instrument: Micromass GCT, GC6890; Column: Restek RTX-35, 15 mx 200 ⁇ x 0.33 ⁇ ; constant flow with helium: 0.88 ml / min; Oven: 70 ° C; Inlet: 250 ° C; Gradient: 70 ° C, 30 ° C / min -> 310 ° C (hold for 3 min).
- Instrument Micromass GCT, GC6890; Column: Restek RTX-35, 15 mx 200 ⁇ x 0.33 ⁇ ; constant flow with helium: 0.88 ml / min; Oven: 70 ° C; Inlet: 250 ° C; Gradient: 70 ° C, 30 ° C / min -> 310 ° C (hold for 3 min).
- Example 1A Example 1A
- Example 3A 55.00 g (135 mmol) of Example 3A were initially charged in sulfolane (220 ml) and admixed with 41.40 g (270 mmol) of phosphoryl chloride. Thereafter, it was heated to 120 ° C for 1 h. After cooling, it was added to warm water (1500 ml) and then neutralized with solid sodium bicarbonate. The precipitate which formed was filtered off with suction and washed with water. Thereafter, it was further purified by chromatography on silica gel (mobile phase: cyclohexane / ethyl acetate 3: 2). After drying under high vacuum, 43.0 g of the title compound were obtained (73% of theory).
- Example 4A 10.00 g (23,482 mmol) of Example 4A were initially charged in DMF (200 ml) and treated with 2,290 g (35,223 mmol) of sodium azide. Thereafter, it was heated to 60 ° C for 1 h. After cooling, the reaction mixture was added to water and extracted three times with ethyl acetate. The organic phases were combined and washed once with saturated aqueous sodium chloride solution, then dried over sodium sulfate, filtered and concentrated. The residue was used without further purification in the next step.
- Example 6A Ethyl ⁇ 4-amino-2- [1- (2-fluorobenzyl) -1H-pyrazolo [3,4-b] pyridin-3-yl] pyrirnidin-5-yl ⁇ acetate
- Example 7A 2.00 g (5.550 mmol) of Example 7A were initially charged in dioxane (200 ml) and treated with 3.079 g (27.751 mmol) of selenium dioxide and heated to reflux for 2 h. After cooling, the mixture was filtered and the filtrate was concentrated and purified by chromatography on silica gel (mobile phase: cyclohexane / ethyl acetate 1: 1). 890 mg of the title compound were obtained (42% of theory).
- Example 12A 1.45 g (about 4.950 mmol) of Example 12A were initially charged in ethanol (20 ml), treated dropwise with a suspension of 1.37 g (about 3.300 mmol) Example 11 A in 20 ml of ethanol and then heated to reflux overnight. After cooling, it was filtered from a precipitate and washed with ethanol. The filtrate was concentrated and the residue treated with diethyl ether. It was again filtered from a precipitate and the filtrate was concentrated and then purified by preparative HPLC (methanol: water gradient). There were obtained 297 mg of the title compound (18% of theory).
- Example 3 337 mg (0.682 mmol) of Example 3 were reacted in analogy to Example 10A. There were obtained 236 mg of the title compound (67% of theory).
- the diazonium salt thus prepared was added in portions to a 0 ° C cold solution of 12.81 g (85.45 mmol) of sodium iodide in acetone (329 ml) and the mixture stirred for 30 min at RT.
- the reaction mixture was added to ice water (1.8 L) and extracted twice with ethyl acetate (487 mL each).
- the collected organic phases were washed with saturated aqueous sodium chloride solution (244 ml), dried, filtered and concentrated. 12.1 g (86% purity, 60% of theory) of the title compound were obtained as a solid.
- the crude product was reacted without further purification.
- Example 21A 1.00 g (3.152 mmol) of Example 21A were stirred in 10 ml of a 7N solution of ammonia in methanol at RT for three days. It was then concentrated in vacuo. 908 mg (99% of theory) of the title compound were obtained.
- the aqueous phase was separated and extracted twice more with ethyl acetate (200 ml each time).
- the combined organic Phases were washed twice with 10% aqueous sodium chloride solution (100 ml each), dried and concentrated in vacuo.
- the crude product was reacted without further purification.
- Variant B 900 mg (3.122 mmol) of the compound obtained under Example 22A were dissolved in THF (14 ml) and treated with 0.646 ml (7.993 mmol) of pyridine. Then, with stirring, 1.129 ml (7.993 mmol) of trifluoroacetic anhydride were slowly added dropwise and then stirred at RT overnight. Thereafter, the reaction mixture was poured into water and extracted three times with ethyl acetate. The combined organic phases were extracted with saturated aqueous sodium bicarbonate solution and 1N hydrochloric acid and then washed with saturated aqueous sodium chloride solution. The organic phase was dried over sodium sulfate, filtered and concentrated. 850 mg (99% of theory) of the title compound were obtained.
- Example 24A 23,000 g (66.22 mmol) of Example 24A were dissolved in 322 ml of ethanol and treated at 0 ° C with 26,804 g (264.88 mmol) of triethylamine and 6.027 g (66.22 mmol) hydrazine hydrate (55% solution in water). The mixture was stirred overnight at rt and then added to 1.715 l of a 10% aqueous sodium chloride solution and extracted twice with ethyl acetate. The combined organic phases were washed with 10% aqueous sodium chloride solution, dried over sodium sulfate and concentrated on a rotary evaporator. The residue was purified on silica gel (eluent: dichloromethane / methanol, 95: 5). There were obtained 15,000 g (75% of theory) of the title compound.
- Example 26A 1.272 g (about 4.962 mmol) of Example 26A were initially charged in 10 ml of ethanol and heated to reflux. For this purpose, a suspension of 1.00 g (3.308 mmol) of Example 25 A in 40 ml of ethanol was added dropwise. After heating overnight, an additional 2.24 g of Example 26A was added and refluxed for a further night. After cooling, a solid was filtered off with suction, washed with a little ethanol and the filtrate was concentrated. The residue was purified by preparative HPLC purified (acetonitrile: water gradient). 270 mg (16% of theory) of the title compound were obtained.
- Example 30A 1,143 g (4,962 mmol) of Example 30A were reacted in analogy to Example 27A. The residue was purified by preparative HPLC (acetonitrile: water (+ 1% trifluoroacetic acid) gradient). There were obtained 334 mg (21% of theory) of the title compound.
- Example 19A 10.00 g (38.021 mmol) of Example 19A were reacted with 4-methoxybenzyl chloride in analogy to the procedure of Example 20A. Chromatography on silica gel (eluent: cyclohexane-ethyl acetate mixture) gave 8.94 g (61% of theory) of the title compound.
- Example 32A 8.94 g (23.332 mmol) of Example 32A were reacted in analogy to the procedure under Example 23A, variant A. The resulting crude product was reacted without further purification.
- Example 33A 6.52 g (23,098 mmol) of Example 33A were reacted in analogy to the procedure in Example 24A. Yield: 6.16 g (74% of theory)
- Example 34 A 6.16 g (17.141 mmol) of Example 34 A were reacted in analogy to the procedure under Example 25A. A purification on silica gel was not carried out. There were obtained 4.90 g (90% of theory) of the title compound, which was reacted without further purification.
- Example 40A 0.150 g (0.349 mmol) of the compound from Example 40A were reacted in analogy to the procedure of Example 41A with 1- (bromomethyl) -2-fluoro-3-methylbenzene. After filtration, it was purified by preparative HPLC (acetonitrile: water (+0.05% formic acid) gradient). 83 mg of the title compound (44% of theory) were obtained.
- Example 44A 0.50 g (1.654 mmol) of the compound from Example 44A were reacted with 907 mg (3.308 mmol) of Example 25A in analogy to the procedure of Example 27A. 42 mg (5% of theory) of the title compound were obtained.
- Example 2A 5,887 g (19,256 mmol) of Example 2A were initially charged in tert-butanol (50 ml) and treated with 2,593 g (23.107 mmol) of potassium tert-butoxide. Subsequently, 3.2 g (19.256 mmol) of Example 1A in tert-butanol (25 ml) were added dropwise and the mixture heated to reflux overnight. The next day another 0.64 g (3.851 mmol) of Example 1 A was added and it was heated to reflux for a further day. After cooling, a precipitate was filtered off, which was washed with diethyl ether. It was then slurried in water and filtered once more and washed with diethyl ether. After drying under high vacuum, 6.65 g of the title compound were obtained (85% of theory).
- Example 46A 5.00 g (12.394 mmol) of Example 46A were initially charged in iso-pentyl nitrite (35.87 ml) and diiodomethane (1.16 mol, 93.71 ml) and heated to 85 ° C. for 12 h. After cooling, it was filtered off from solids, concentrated and then purified by chromatography on silica gel (mobile phase: first cyclohexane-dichloromethane gradient, then dichloromethane-methanol gradient). There were obtained 5.50 g of the title compound (67% of theory).
- Example 48A 28 g (168,513 mmol) of Example 48A were reacted in analogy to the procedure of Example 19A. After chromatography on silica gel (cyclohexane: ethyl acetate 9: 1), 14.9 g (31% of theory) of the title compound were obtained.
- Example 49A 13 g (46,925 mmol) of Example 49A were reacted in analogy to the procedure under Example 20A. Chromatography on silica gel (cyclohexane: ethyl acetate gradient) gave 8.4 g (43% of theory) of the title compound.
- Example 50A 9.3 g (24,146 mmol) of Example 50A were reacted in analogy to the procedure under Example 23A, variant A. After chromatography on silica gel (cyclohexane: ethyl acetate gradient), 5.7 g (80% of theory, 95% strength by weight) of the title compound were obtained.
- Example 51A were reacted in analogy to the procedure under Example 24A. This gave 6.6 g (96% of theory) of the title compound.
- Example 52A 500 mg (1.384 mmol) of Example 52A were reacted in analogy to the procedure under Example 25A. 365 mg (83% of theory) of the title compound were obtained.
- Example 53A 365 mg (1154 mmol) of Example 53A were reacted analogously to the procedure of Example 13A with 325 mg (1.731 mmol) of dimethyl 2,2-dimethyl-3-oxobutanedioate. 589 mg (92% of theory, 82% purity) of the title compound were obtained.
- Example 52A 1g (2.767 mmol) of Example 52A were reacted in analogy to the procedure under Example 46A. 971 mg (80% of theory) of the title compound were obtained.
- Example 55A 960 mg (2,205 mmol) of Example 55A were reacted in analogy to the procedure under Example 47A. 749 mg (62% of theory, 84% strength) of the title compound were obtained.
- the filtrate was concentrated in vacuo.
- the residue was admixed with ethanol and the insoluble solid was filtered off and washed with ethanol. After drying under high vacuum, 0.61 g (9% of theory) of the title compound were obtained.
- the filtrate was concentrated in vacuo and the residue was dried under high vacuum. 3.14 g (43% of theory, 43% strength by weight) of the title compound were obtained.
- Example 63 A 100 mg (0.27 mmol) of Example 63 A in 3 ml of THF were initially taken under argon. At 0 ° C., 56 ⁇ l (0.32 mmol) of ⁇ , ⁇ -diisopropylethylamine were added, and 0.16 ml (0.32 mmol) of ammonia (2M in ethanol) were added dropwise. The mixture was then stirred at RT overnight. Now was partitioned between dichloromethane and IN aqueous hydrochloric acid, the aqueous phase with dichloromethane extracted, the combined organic phases dried over sodium sulfate and concentrated in vacuo.
- Example 65A 84.37 g (348.23 mmol) of Example 65A were initially charged in 1.10 l of ethanol. Under reflux, 55 g (0.174 mol, purity 90%) of Example I IA were added in portions and it was heated further at reflux overnight. Then, with a test batch starting from 11.1 g (35.1 mmol), Example 65 A was combined. It was then cooled to 5 ° C, the precipitated solid was filtered off and washed with tert-butyl methyl ether. The solid was discarded. The filtrate was concentrated in vacuo, the residue treated with 500 ml of tert-butyl methyl ether and stirred for 1 h at room temperature.
- Example 69A 100 mg (0.16 mmol) of Example 69A in 3.3 ml of a mixture of DMF, water and triethylamine (25: 4: 4) were initially charged under argon, with 170 ⁇ (1.55 mmol) of ethyl acrylate, 25 mg (0.03 mmol) of palladium (II). acetate and 115 mg (0.31 mmol) of tetra-n-butylammonium iodide and stirred at 60 ° C for 9 h.
- Example 2A Under an argon atmosphere, 4.69 g (15.32 mmol) of Example 2A in 120 ml of tert-butanol were initially charged and 3.07 g (30.66 mmol) of potassium bicarbonate and 4.2 g (17.63 mmol) of Example 74A were added at room temperature. It was stirred for 5 h at 85 ° C bath temperature. After cooling, water was added and stirred for 30 min at room temperature. The precipitated solid was filtered off and washed with water and diethyl ether. After drying under high vacuum, 6.2 g (88% of theory) of the title compound were obtained.
- Example 33 Under an argon atmosphere, 600 mg (0.77 mmol, purity 57%) of Example 33 were initially charged and 2.50 ml (64.12 mmol) of 80% hydrazine hydrate were added. It was stirred for 30 min at 80 ° C, cooled, concentrated by rotary evaporation and dried under high vacuum. 566 mg of the title compound were obtained as crude product.
- Example 76A To 55 mg (0.13 mmol) of Example 76A in 1 ml of acetonitrile was added 1 ml (26.88 mmol) of formic acid. It was stirred for 1.5 h at 80 ° C bath temperature, cooled and concentrated. The residue was combined with ethyl acetate and washed once each with saturated aqueous sodium bicarbonate solution and saturated aqueous sodium chloride solution and then dried over sodium sulfate and concentrated. 29.7 mg (50% of theory) of the title compound were obtained.
- Example 78A Under argon atmosphere, 500 mg (1.36 mmol) of Example 78A in 10 ml of t-butanol were initially charged and treated with 272.6 mg (2.72 mmol) of potassium bicarbonate and 373 mg (1.57 mmol) Example 74A. After 5h stirring at 85 ° C was cooled and treated with water. After stirring for 30 minutes at room temperature, the precipitated solid was filtered off and washed with water and a little ether. After drying under high vacuum, 458 mg (63% of theory) of the title compound were obtained.
- Example 25A 4.12 g (13.62 mmol) of Example 25A were reacted in analogy to Example 66A. There were obtained 2.03 g (22% of theory, purity 70%) of the title compound.
- Etyl-5-amino-1- (2-fluorobenzyl) -4-formyl-1H-pyrazole-3-carboxylate was prepared analogously to compounds known from the literature from 2-fluorobenzyl bromide and sodium 1,4-diethoxy-1,4-dioxobutyl 2-en-2-olate (see Kelley et al J. Med Chem 1995, 38, 3884-3888, Toche et al J. Het Chem 2010, 47, 287-291 and Patent US4833246 , Column 24. a) Preparation of (2-fluorobenzyl) hydrazine:
- Example 90 A Emyl-3 2- [l 2-fluorobenzyl) -1H-pyrazolo [3,4-b] pyrene
- Example 69A Under argon were added 500 mg (0.78 mmol) of Example 69A, 14.8 mg (0.08 mmol) of copper (T) iodide, 127 mg (1.51 mmol) of sodium bicarbonate, 0.31 ml (304.4 mg, 3.1 mmol) of ethyl propiolate and 54.6 mg (0.08 mmol) of dichlorobistriphenylphosphine Palladium (II) in 7 ml of DMF overnight at 60 ° C stirred. Aqueous ammonium chloride solution was added and the mixture extracted with ethyl acetate. The organic phases were dried, concentrated under reduced pressure and the residue was purified by column chromatography on silica gel with cyclohexane / ethyl acetate.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Urology & Nephrology (AREA)
- Hospice & Palliative Care (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description
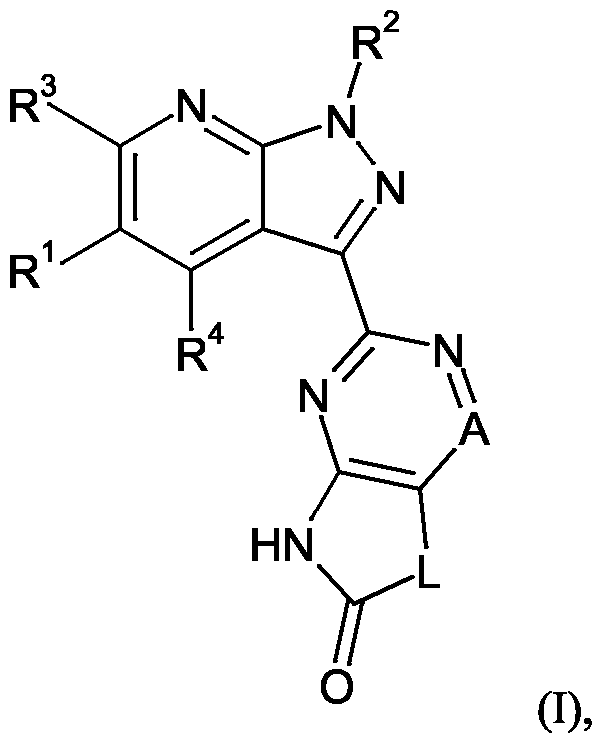


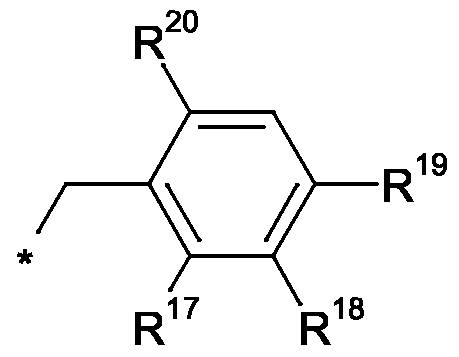








































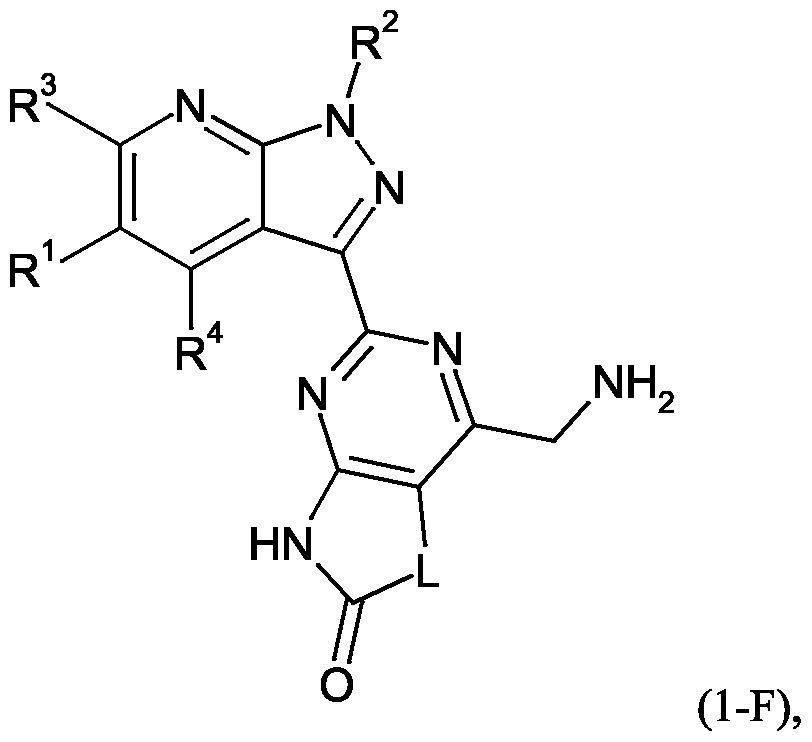

















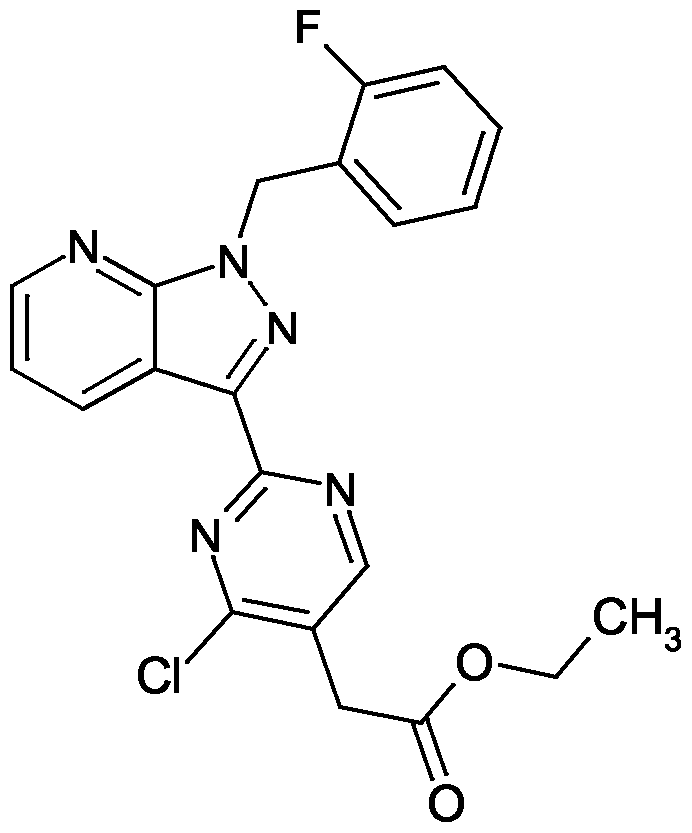

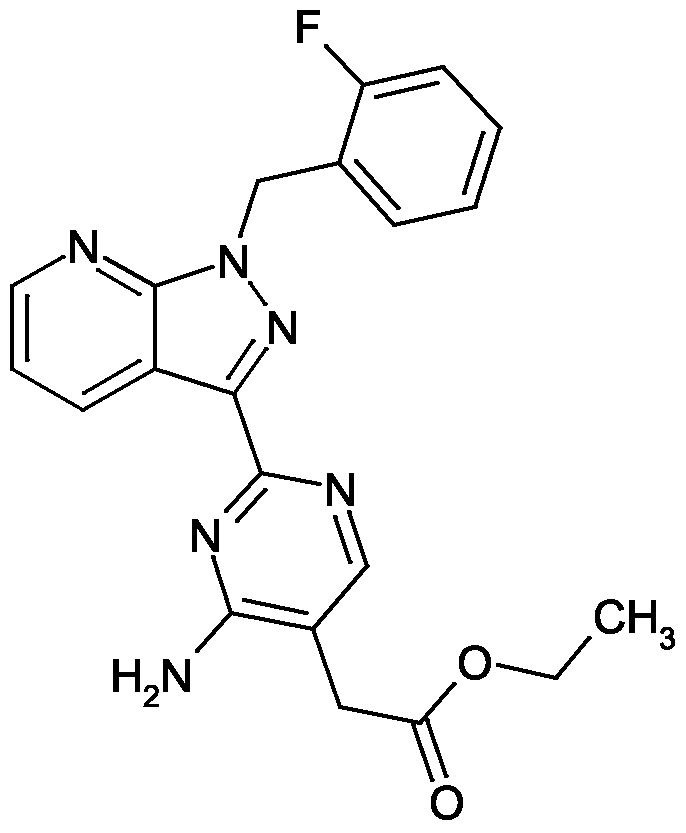



























































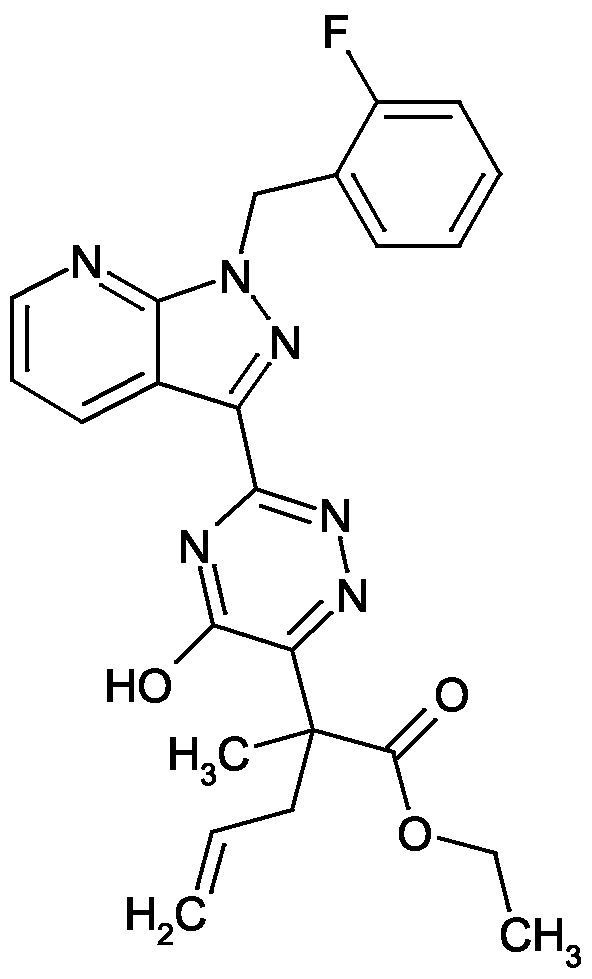



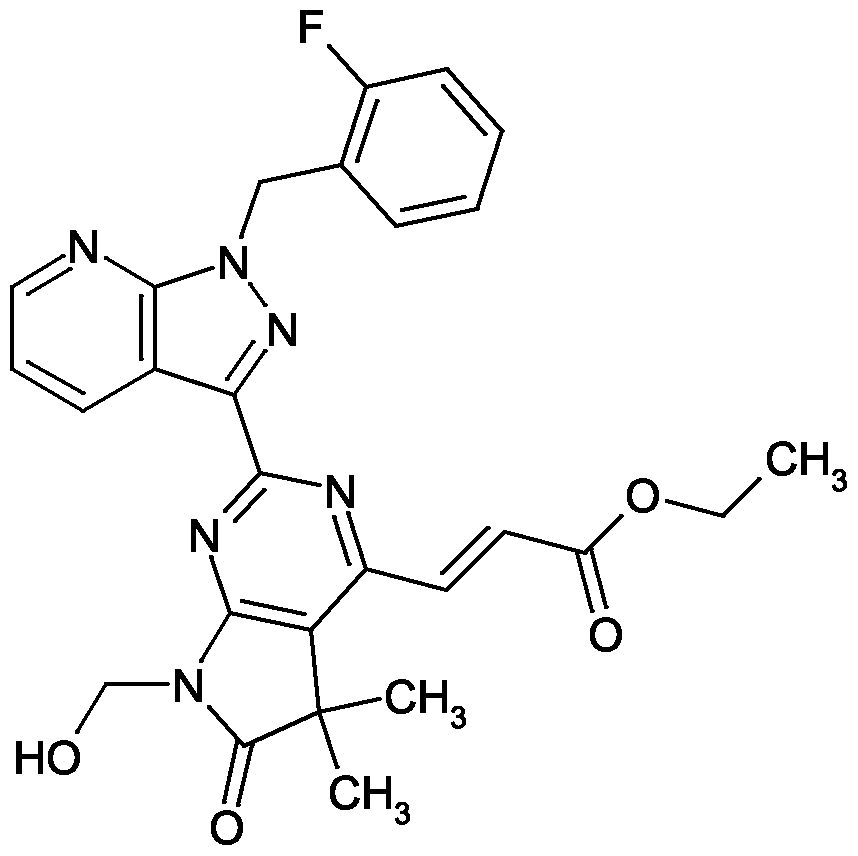

















































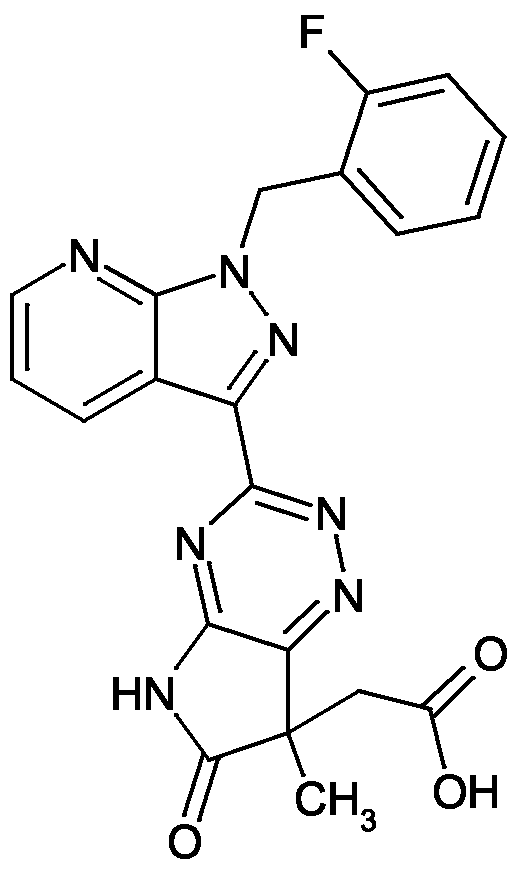







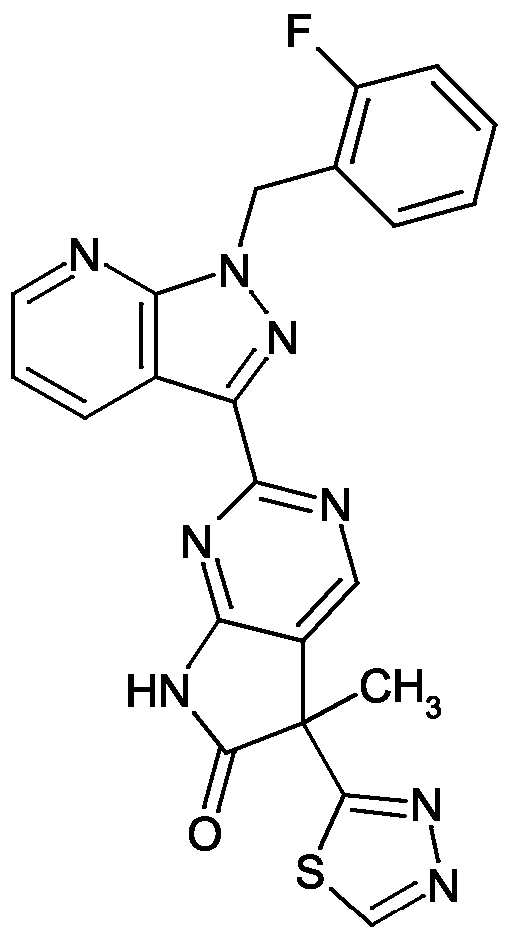



























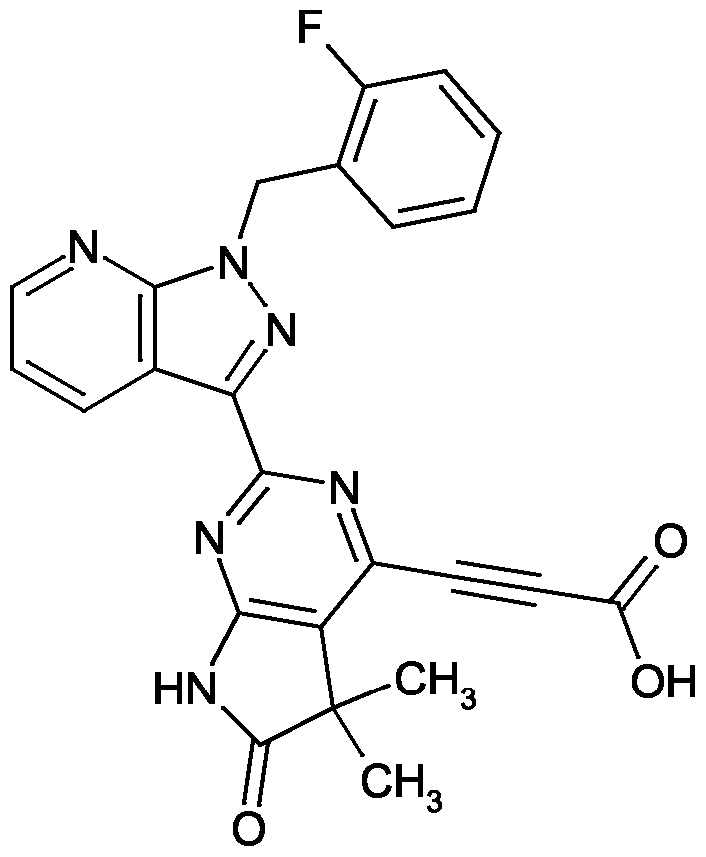



Claims

















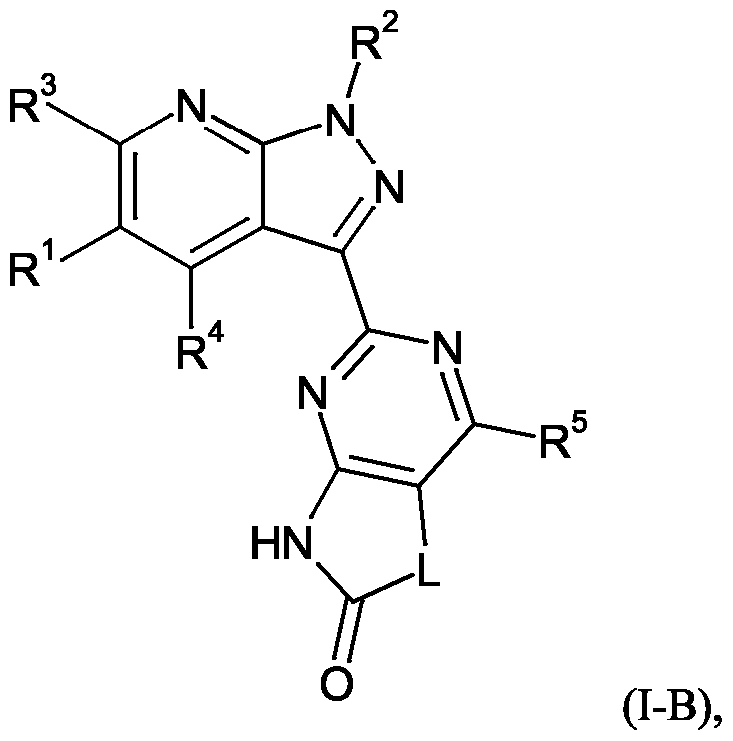

















Priority Applications (22)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020147021969A KR20140114416A (ko) | 2012-01-11 | 2013-01-10 | 치환된 융합 피리미딘 및 트리아진, 및 그의 용도 |
| CA2860855A CA2860855A1 (en) | 2012-01-11 | 2013-01-10 | Substituted annulated pyrimidines and triazines, and use thereof |
| EA201491339A EA025837B1 (ru) | 2012-01-11 | 2013-01-10 | Замещенные аннеллированные пиримидины и триазины |
| NZ626394A NZ626394A (en) | 2012-01-11 | 2013-01-10 | Substituted annulated pyrimidines and triazines, and use thereof |
| EP13700288.7A EP2802592A1 (de) | 2012-01-11 | 2013-01-10 | Substituierte annellierte pyrimidine und triazine und ihre verwendung |
| BR112014016971A BR112014016971A8 (pt) | 2012-01-11 | 2013-01-10 | pirimidinas e triazinas fundidas substituídas e seus usos |
| JP2014551609A JP6251183B2 (ja) | 2012-01-11 | 2013-01-10 | 置換された縮合ピリミジンおよびトリアジン化合物ならびにその使用 |
| CN201380013679.1A CN104812762B (zh) | 2012-01-11 | 2013-01-10 | 取代的环状嘧啶和三嗪以及它们的用途 |
| US14/371,046 US20140357637A1 (en) | 2012-01-11 | 2013-01-10 | Substituted annulated pyrimidines and triazines, and use thereof |
| MX2014008201A MX2014008201A (es) | 2012-01-11 | 2013-01-10 | Pirimidinas y triazinas fusionadas sustituidas y su uso. |
| AU2013208968A AU2013208968A1 (en) | 2012-01-11 | 2013-01-10 | Substituted annulated pyrimidines and triazines, and use thereof |
| SG11201403345UA SG11201403345UA (en) | 2012-01-11 | 2013-01-10 | Substituted annulated pyrimidines and triazines, and use thereof |
| AP2014007844A AP2014007844A0 (en) | 2012-01-11 | 2013-01-10 | Substituted annulated pyrimidines and triazines, and use thereof |
| IL233460A IL233460A0 (en) | 2012-01-11 | 2014-06-30 | Cyclic triazines and pyrimidines are converted and their use |
| CU2014000082A CU20140082A7 (es) | 2012-01-11 | 2014-07-07 | Pirimidinas y triazinas fusionadas sustituidas y sus uso |
| MA37178A MA35844B1 (fr) | 2012-01-11 | 2014-07-07 | Pyrimidines et triazines annelées substituées et leur utilisation |
| CR20140327A CR20140327A (es) | 2012-01-11 | 2014-07-08 | Pirimidinas y triazinas fusionadas sustituidas y su uso |
| TNP2014000298A TN2014000298A1 (en) | 2012-01-11 | 2014-07-09 | Substituted annulated pyrimidines and triazines, and use thereof |
| PH12014501595A PH12014501595A1 (en) | 2012-01-11 | 2014-07-10 | Substituted annulated pyrimidines and triazines, and use thereof |
| ZA2014/05042A ZA201405042B (en) | 2012-01-11 | 2014-07-10 | Substituted annulated pyrimidines and triazines, and use thereof |
| US14/738,148 US9505786B2 (en) | 2012-01-11 | 2015-06-12 | Substituted annulated triazines and use thereof |
| HK15112419.1A HK1211585A1 (en) | 2012-01-11 | 2015-12-17 | Substituted annulated pyrimidines and triazines, and use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102012200349.5 | 2012-01-11 | ||
| DE102012200349A DE102012200349A1 (de) | 2012-01-11 | 2012-01-11 | Substituierte annellierte Pyrimidine und Triazine und ihre Verwendung |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/371,046 A-371-Of-International US20140357637A1 (en) | 2012-01-11 | 2013-01-10 | Substituted annulated pyrimidines and triazines, and use thereof |
| US14/738,148 Division US9505786B2 (en) | 2012-01-11 | 2015-06-12 | Substituted annulated triazines and use thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013104703A1 true WO2013104703A1 (de) | 2013-07-18 |
Family
ID=47559479
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2013/050381 WO2013104703A1 (de) | 2012-01-11 | 2013-01-10 | Substituierte annellierte pyrimidine und triazine und ihre verwendung |
Country Status (29)
| Country | Link |
|---|---|
| US (2) | US20140357637A1 (de) |
| EP (1) | EP2802592A1 (de) |
| JP (1) | JP6251183B2 (de) |
| KR (1) | KR20140114416A (de) |
| CN (1) | CN104812762B (de) |
| AP (1) | AP2014007844A0 (de) |
| AU (1) | AU2013208968A1 (de) |
| BR (1) | BR112014016971A8 (de) |
| CA (1) | CA2860855A1 (de) |
| CL (1) | CL2014001779A1 (de) |
| CO (1) | CO7000776A2 (de) |
| CR (1) | CR20140327A (de) |
| CU (1) | CU20140082A7 (de) |
| DE (1) | DE102012200349A1 (de) |
| DO (1) | DOP2014000160A (de) |
| EA (1) | EA025837B1 (de) |
| EC (1) | ECSP14008556A (de) |
| GT (1) | GT201400147A (de) |
| HK (1) | HK1211585A1 (de) |
| IL (1) | IL233460A0 (de) |
| MA (1) | MA35844B1 (de) |
| MX (1) | MX2014008201A (de) |
| NZ (1) | NZ626394A (de) |
| PE (1) | PE20142293A1 (de) |
| PH (1) | PH12014501595A1 (de) |
| SG (2) | SG10201605655UA (de) |
| TN (1) | TN2014000298A1 (de) |
| WO (1) | WO2013104703A1 (de) |
| ZA (1) | ZA201405042B (de) |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014084312A1 (ja) | 2012-11-30 | 2014-06-05 | アステラス製薬株式会社 | イミダゾピリジン化合物 |
| WO2015004105A1 (de) * | 2013-07-10 | 2015-01-15 | Bayer Pharma Aktiengesellschaft | Benzyl-1h-pyrazolo[3,4-b]pyridine und ihre verwendung |
| JP2015509515A (ja) * | 2012-03-06 | 2015-03-30 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | 置換アザ二環およびその使用 |
| WO2015089182A1 (en) | 2013-12-11 | 2015-06-18 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2015106268A1 (en) | 2014-01-13 | 2015-07-16 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF NEUROMUSCULAR DISORDERS |
| WO2015124544A1 (de) * | 2014-02-19 | 2015-08-27 | Bayer Pharma Aktiengesellschaft | 3-(pyrimidin-2-yl)imidazo[1,2-a]pyridine |
| WO2016030354A1 (de) | 2014-08-29 | 2016-03-03 | Bayer Pharma Aktiengesellschaft | Amino-substituierte annellierte pyrimidine und ihre verwendung |
| WO2016030362A1 (de) | 2014-08-29 | 2016-03-03 | Bayer Pharma Aktiengesellschaft | Substituierte annellierte pyrimidine und ihre verwendung |
| WO2016177660A1 (en) | 2015-05-06 | 2016-11-10 | Bayer Pharma Aktiengesellschaft | The use of sgc stimulators, sgc activators, alone and combinations with pde5 inhibitors for the treatment of digital ulcers (du) concomitant to systemic sclerosis (ssc) |
| WO2017013010A1 (de) | 2015-07-23 | 2017-01-26 | Bayer Pharma Aktiengesellschaft | Stimulatoren und/oder aktivatoren der löslichen guanylatzyklase (sgc) in kombination mit einem inhibitor der neutralen endopeptidase (nep inhibitor) und/oder einem angiotensin aii-antagonisten und ihre verwendung |
| WO2017106175A2 (en) | 2015-12-14 | 2017-06-22 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF GASTROINTESTINAL SPHINCTER DYSFUNCTION |
| KR101753652B1 (ko) * | 2015-10-21 | 2017-07-05 | 한국화학연구원 | N-아릴-1h-피라졸로피리딘-3-아민 유도체 또는 이의 약학적으로 허용가능한 염, 이의 제조방법 및 이를 유효성분으로 포함하는 melk 관련 질환의 예방 또는 치료용 약학적 조성물 |
| US9776997B2 (en) | 2013-06-04 | 2017-10-03 | Bayer Pharma Aktiengesellschaft | 3-aryl-substituted imidazo[1,2-A]pyridines and their use |
| WO2018069126A1 (de) | 2016-10-11 | 2018-04-19 | Bayer Pharma Aktiengesellschaft | Kombination enthaltend sgc stimulatoren und mineralocorticoid-rezeptor-antagonisten |
| WO2018111795A2 (en) | 2016-12-13 | 2018-06-21 | Ironwood Pharmaceuticals, Inc. | Use of sgc stimulators for the treatment of esophageal motility disorders |
| CN108640923A (zh) * | 2018-07-09 | 2018-10-12 | 湖南天地恒制药有限公司 | 一种托法替布关键中间体的制备方法 |
| US10292970B2 (en) | 2014-12-02 | 2019-05-21 | Bayer Pharma Aktiengesellschaft | Heteroaryl-substituted imidazo[1,2-A]pyridines and their use |
| WO2019219672A1 (en) | 2018-05-15 | 2019-11-21 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| WO2020014504A1 (en) | 2018-07-11 | 2020-01-16 | Cyclerion Therapeutics, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF MITOCHONRIAL DISORDERS |
| WO2020165010A1 (en) | 2019-02-13 | 2020-08-20 | Bayer Aktiengesellschaft | Process for the preparation of porous microparticles |
| US11331308B2 (en) | 2016-10-11 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Combination containing sGC activators and mineralocorticoid receptor antagonists |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PE20130779A1 (es) | 2010-07-09 | 2013-06-21 | Bayer Ip Gmbh | Pirimidinas y triazinas condensadas y su uso |
| CA2833698A1 (en) * | 2011-04-21 | 2012-10-26 | Bayer Intellectual Property Gmbh | Fluoroalkyl-substituted pyrazolopyridines and use thereof |
| DE102012200349A1 (de) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituierte annellierte Pyrimidine und Triazine und ihre Verwendung |
| DE102012200360A1 (de) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituierte Triazine und ihre Verwendung |
| DE102012200352A1 (de) * | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituierte, annellierte Imidazole und Pyrazole und ihre Verwendung |
| KR20150121007A (ko) | 2013-03-01 | 2015-10-28 | 바이엘 파마 악티엔게젤샤프트 | 트리플루오르메틸-치환된 고리-융합된 피리미딘 및 그의 용도 |
| ES2616042T3 (es) * | 2013-03-01 | 2017-06-09 | Bayer Pharma Aktiengesellschaft | Pirazolopiridinas sustituidas con bencilo y su uso |
| EP3079700B1 (de) | 2013-12-11 | 2020-11-25 | Merck Sharp & Dohme Corp. | Lösliche guanylat-cyclase-aktivatoren |
| EP3079701B1 (de) | 2013-12-11 | 2021-08-11 | Merck Sharp & Dohme Corp. | Lösliche guanylat-cyclase-aktivatoren |
| US9796733B2 (en) | 2014-06-04 | 2017-10-24 | Merck Sharp & Dohme Corp. | Imidazo-pyrazine derivatives useful as soluble guanylate cyclase activators |
| TW201625635A (zh) | 2014-11-21 | 2016-07-16 | 默沙東藥廠 | 作為可溶性鳥苷酸環化酶活化劑之三唑并吡基衍生物 |
| WO2016191334A1 (en) | 2015-05-27 | 2016-12-01 | Merck Sharp & Dohme Corp. | Imidazo-pyrazinyl derivatives useful as soluble guanylate cyclase activators |
| WO2016191335A1 (en) | 2015-05-28 | 2016-12-01 | Merck Sharp & Dohme Corp. | Imidazo-pyrazinyl derivatives useful as soluble guanylate cyclase activators |
| WO2017107052A1 (en) | 2015-12-22 | 2017-06-29 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase stimulators |
| WO2017197555A1 (en) | 2016-05-16 | 2017-11-23 | Merck Sharp & Dohme Corp. | Fused pyrazine derivatives useful as soluble guanylate cyclase stimulators |
| WO2017200857A1 (en) | 2016-05-18 | 2017-11-23 | Merck Sharp & Dohme Corp. | Methods for using triazolo-pyrazinyl soluble guanylate cyclase activators in fibrotic disorders |
| IL292968A (en) | 2016-09-02 | 2022-07-01 | Cyclerion Therapeutics Inc | scg motors in converging cycles |
| EP3609883B1 (de) | 2017-04-11 | 2022-06-29 | Sunshine Lake Pharma Co., Ltd. | Fluorsubstituierte indazolverbindungen und verwendungen davon |
| CN113368240B (zh) * | 2021-06-13 | 2022-08-02 | 重庆医科大学 | 一种二茂铁基金属有机框架纳米粒及其制备方法 |
Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4833246A (en) | 1986-12-02 | 1989-05-23 | Fuji Photo Film Co., Ltd. | Novel pyrazolone dye |
| WO2000006569A1 (de) | 1998-07-29 | 2000-02-10 | Bayer Aktiengesellschaft | Mit sechsgliedrigen heterocyclischen ringen kondensierte substituierte pyrazolderivate |
| WO2000006568A1 (de) | 1998-07-29 | 2000-02-10 | Bayer Aktiengesellschaft | Substituierte pyrazolderivate |
| WO2003095451A1 (de) | 2002-05-08 | 2003-11-20 | Bayer Healthcare Ag | Carbamat-substituierte pyrazolopyridine |
| WO2004009590A1 (de) | 2002-07-18 | 2004-01-29 | Bayer Healthcare Ag | 4-aminosubstituierte pyrimidinderivate |
| WO2005073234A2 (en) | 2004-01-31 | 2005-08-11 | Actimis Pharmaceuticals, Inc. | Imidazo[1,2-c]pyrimidinylacetic acid derivatives |
| WO2006130673A1 (en) | 2005-05-31 | 2006-12-07 | Janssen Pharmaceutica, N.V. | 3-benzoimidazolyl-pyrazolopyridines useful in treating kinase disorders |
| WO2007041052A2 (en) | 2005-09-29 | 2007-04-12 | Merck & Co., Inc. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| WO2010065275A1 (en) | 2008-11-25 | 2010-06-10 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2011014992A1 (zh) | 2009-08-07 | 2011-02-10 | 上海贝尔股份有限公司 | 一种用于减小切换VoIP通话时的中断时间的方法和装置 |
| WO2011149921A1 (en) | 2010-05-27 | 2011-12-01 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2011147809A1 (de) | 2010-05-26 | 2011-12-01 | Bayer Pharma Aktiengesellschaft | Substituierte 5-fluor-1h-pyrazolopyridine und ihre verwendung |
| WO2012004258A1 (de) | 2010-07-09 | 2012-01-12 | Bayer Pharma Aktiengesellschaft | Annellierte pyrimidine und triazine und ihre verwendung zur behandlung bzw. prophylaxe von herz-kreislauf-erkrankungen |
| WO2012004259A1 (de) | 2010-07-09 | 2012-01-12 | Bayer Pharma Aktiengesellschaft | Annellierte 4 -aminopyrimidine und ihre verwendung als stimulatoren der löslichen guanylatcyclase |
| WO2012028647A1 (de) | 2010-09-03 | 2012-03-08 | Bayer Pharma Aktiengesellschaft | Bicyclische aza-heterocyclen und ihre verwendung |
| WO2012143510A1 (de) | 2011-04-21 | 2012-10-26 | Bayer Intellectual Property Gmbh | Fluoralkyl-substituierte pyrazolopyridine und ihre verwendung |
| WO2012152629A1 (de) | 2011-05-06 | 2012-11-15 | Bayer Intellectual Property Gmbh | Substituierte imidazopyridine und imidazopyridazine und ihre verwendung |
Family Cites Families (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES537947A0 (es) | 1984-11-26 | 1985-11-01 | Fordonal Sa | Procedimiento de preparacion de un derivado de piracina. |
| SE8704248D0 (sv) | 1987-10-30 | 1987-10-30 | Haessle Ab | Medical use |
| GB9314412D0 (en) | 1993-07-13 | 1993-08-25 | Rhone Poulenc Agriculture | New compositions of matter |
| AU5348396A (en) | 1995-05-01 | 1996-11-21 | Fujisawa Pharmaceutical Co., Ltd. | Imidazo 1,2-a pyridine and imidazo 1,2-a pyridezine derivati ves and their use as bone resorption inhibitors |
| EP0743066A3 (de) | 1995-05-16 | 1998-09-30 | Mitsui Pharmaceuticals, Inc. | Wundheilmittel |
| DE19642255A1 (de) | 1996-10-14 | 1998-04-16 | Bayer Ag | Verwendung von 1-Benzyl-3-(substituierten-hetaryl) -kondensierten Pyrazol-Derivaten |
| US6166027A (en) | 1996-10-14 | 2000-12-26 | Bayer Aktiengesellschaft | Heterocyclylmethyl-substituted pyrazole derivatives and their use for treating cardiovascular diseases |
| US6451805B1 (en) | 1997-11-14 | 2002-09-17 | Bayer Aktiengesellschaft | Substituted pyrazole derivatives for the treatment of cardiocirculatory diseases |
| DE10021069A1 (de) | 2000-04-28 | 2001-10-31 | Bayer Ag | Substituiertes Pyrazolderivat |
| EP1333833B1 (de) | 2000-10-23 | 2011-08-24 | GlaxoSmithKline LLC | Neues trisubstitutiertes 8H-Pyrido[2,3-d]pyrimidin-7-onderivat zur Behandlung von durch CSBP/p38kinase vermittelten Krankheiten |
| AU2002221827A1 (en) | 2000-11-22 | 2002-06-03 | Bayer Aktiengesellschaft | Novel lactame-substituted pyrazolopyridine derivatives |
| DE10132416A1 (de) | 2001-07-04 | 2003-01-16 | Bayer Ag | Neue Morpholin-überbrückte Pyrazolopyridinderivate |
| CA2521695C (en) | 2003-05-09 | 2011-08-16 | Asahi Glass Company, Limited | Method for producing 3-substituted 2-chloro-5-fluoro-pyridine or its salt |
| CN100355732C (zh) | 2003-11-03 | 2007-12-19 | 上海药明康德新药开发有限公司 | 2-氯-5-氟-烟酸酯及酸的制备方法 |
| CN101146796A (zh) | 2005-01-26 | 2008-03-19 | 先灵公司 | 作为蛋白激酶抑制剂用于治疗癌症的3-(吲唑-5-基)-(1,2,4)三嗪衍生物及相关化合物 |
| DE102006043443A1 (de) | 2006-09-15 | 2008-03-27 | Bayer Healthcare Ag | Neue aza-bicyclische Verbindungen und ihre Verwendung |
| AU2008282156B2 (en) | 2007-07-31 | 2014-07-17 | Vertex Pharmaceuticals Incorporated | Process for preparing 5-fluoro-1H-pyrazolo [3, 4-b] pyridin-3-amine and derivatives thereof |
| JP2011513483A (ja) | 2008-03-10 | 2011-04-28 | バーテックス ファーマシューティカルズ インコーポレイテッド | タンパク質キナーゼの阻害剤として有用なピリミジンおよびピリジン |
| DE102009004245A1 (de) | 2009-01-09 | 2010-07-15 | Bayer Schering Pharma Aktiengesellschaft | Neue anellierte, Heteroatom-verbrückte Pyrazol- und Imidazol-Derivate und ihre Verwendung |
| CA2762680C (en) | 2009-05-21 | 2018-04-17 | Chlorion Pharma, Inc. | Methyl sulfanyl pyrmidmes useful as antiinflammatories, analgesics, and antiepileptics |
| US20130178475A1 (en) | 2010-03-17 | 2013-07-11 | Ironwood Pharmaceuticals, Inc. | sGC STIMULATORS |
| CA2803688A1 (en) | 2010-06-25 | 2011-12-29 | Bayer Intellectual Property Gmbh | Use of stimulators and activators of soluble guanylate cyclase for treating sickle-cell anemia and conserving blood substitutes |
| DE102010040234A1 (de) | 2010-09-03 | 2012-03-08 | Bayer Schering Pharma Aktiengesellschaft | Verfahren zur Herstellung von 5-Flour-1H-pyrazolo[3,4-b]pyridin-3-carbonitril |
| DE102010043380A1 (de) | 2010-11-04 | 2012-05-10 | Bayer Schering Pharma Aktiengesellschaft | Benzyl-substituierte Carbamate und ihre Verwendung |
| EA201391769A1 (ru) | 2011-05-30 | 2014-04-30 | Астеллас Фарма Инк. | Имидазопиридиновые соединения |
| MX2014000029A (es) | 2011-07-06 | 2014-02-17 | Bayer Ip Gmbh | Pirazolopiridinas sustituidas con heteroarilo y uso de las mismas como estimuladores de guanilato ciclasas solubles. |
| JP6096778B2 (ja) | 2011-09-01 | 2017-03-15 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | ピロロピラジンキナーゼ阻害剤 |
| CN104039784B (zh) | 2011-09-02 | 2017-08-22 | 拜耳知识产权有限责任公司 | 取代的增环嘧啶及其用途 |
| DE102012200349A1 (de) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituierte annellierte Pyrimidine und Triazine und ihre Verwendung |
| DE102012200360A1 (de) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituierte Triazine und ihre Verwendung |
| DE102012200352A1 (de) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituierte, annellierte Imidazole und Pyrazole und ihre Verwendung |
| EP2822951B1 (de) | 2012-03-06 | 2017-07-26 | Bayer Intellectual Property GmbH | Substituierte azabicyclen und ihre verwendung |
| ES2616042T3 (es) | 2013-03-01 | 2017-06-09 | Bayer Pharma Aktiengesellschaft | Pirazolopiridinas sustituidas con bencilo y su uso |
| KR20150121007A (ko) | 2013-03-01 | 2015-10-28 | 바이엘 파마 악티엔게젤샤프트 | 트리플루오르메틸-치환된 고리-융합된 피리미딘 및 그의 용도 |
| MX2016000258A (es) | 2013-07-10 | 2016-04-28 | Bayer Pharma AG | Bencil-1h-pirazolo[3,4-b]piridinas y su uso. |
-
2012
- 2012-01-11 DE DE102012200349A patent/DE102012200349A1/de not_active Withdrawn
-
2013
- 2013-01-10 SG SG10201605655UA patent/SG10201605655UA/en unknown
- 2013-01-10 JP JP2014551609A patent/JP6251183B2/ja not_active Expired - Fee Related
- 2013-01-10 AU AU2013208968A patent/AU2013208968A1/en not_active Abandoned
- 2013-01-10 KR KR1020147021969A patent/KR20140114416A/ko not_active Application Discontinuation
- 2013-01-10 NZ NZ626394A patent/NZ626394A/en not_active IP Right Cessation
- 2013-01-10 US US14/371,046 patent/US20140357637A1/en not_active Abandoned
- 2013-01-10 SG SG11201403345UA patent/SG11201403345UA/en unknown
- 2013-01-10 BR BR112014016971A patent/BR112014016971A8/pt not_active IP Right Cessation
- 2013-01-10 EA EA201491339A patent/EA025837B1/ru not_active IP Right Cessation
- 2013-01-10 EP EP13700288.7A patent/EP2802592A1/de not_active Withdrawn
- 2013-01-10 WO PCT/EP2013/050381 patent/WO2013104703A1/de active Application Filing
- 2013-01-10 PE PE2014001063A patent/PE20142293A1/es not_active Application Discontinuation
- 2013-01-10 MX MX2014008201A patent/MX2014008201A/es not_active Application Discontinuation
- 2013-01-10 CA CA2860855A patent/CA2860855A1/en not_active Abandoned
- 2013-01-10 AP AP2014007844A patent/AP2014007844A0/xx unknown
- 2013-01-10 CN CN201380013679.1A patent/CN104812762B/zh not_active Expired - Fee Related
-
2014
- 2014-06-30 IL IL233460A patent/IL233460A0/en unknown
- 2014-07-03 CL CL2014001779A patent/CL2014001779A1/es unknown
- 2014-07-07 MA MA37178A patent/MA35844B1/fr unknown
- 2014-07-07 CU CU2014000082A patent/CU20140082A7/es unknown
- 2014-07-08 CR CR20140327A patent/CR20140327A/es unknown
- 2014-07-08 DO DO2014000160A patent/DOP2014000160A/es unknown
- 2014-07-08 CO CO14146937A patent/CO7000776A2/es unknown
- 2014-07-09 TN TNP2014000298A patent/TN2014000298A1/fr unknown
- 2014-07-09 EC ECIEPI20148556A patent/ECSP14008556A/es unknown
- 2014-07-10 ZA ZA2014/05042A patent/ZA201405042B/en unknown
- 2014-07-10 PH PH12014501595A patent/PH12014501595A1/en unknown
- 2014-07-10 GT GT201400147A patent/GT201400147A/es unknown
-
2015
- 2015-06-12 US US14/738,148 patent/US9505786B2/en not_active Expired - Fee Related
- 2015-12-17 HK HK15112419.1A patent/HK1211585A1/xx unknown
Patent Citations (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4833246A (en) | 1986-12-02 | 1989-05-23 | Fuji Photo Film Co., Ltd. | Novel pyrazolone dye |
| WO2000006569A1 (de) | 1998-07-29 | 2000-02-10 | Bayer Aktiengesellschaft | Mit sechsgliedrigen heterocyclischen ringen kondensierte substituierte pyrazolderivate |
| WO2000006568A1 (de) | 1998-07-29 | 2000-02-10 | Bayer Aktiengesellschaft | Substituierte pyrazolderivate |
| WO2003095451A1 (de) | 2002-05-08 | 2003-11-20 | Bayer Healthcare Ag | Carbamat-substituierte pyrazolopyridine |
| WO2004009590A1 (de) | 2002-07-18 | 2004-01-29 | Bayer Healthcare Ag | 4-aminosubstituierte pyrimidinderivate |
| WO2005073234A2 (en) | 2004-01-31 | 2005-08-11 | Actimis Pharmaceuticals, Inc. | Imidazo[1,2-c]pyrimidinylacetic acid derivatives |
| WO2006130673A1 (en) | 2005-05-31 | 2006-12-07 | Janssen Pharmaceutica, N.V. | 3-benzoimidazolyl-pyrazolopyridines useful in treating kinase disorders |
| WO2007041052A2 (en) | 2005-09-29 | 2007-04-12 | Merck & Co., Inc. | Acylated spiropiperidine derivatives as melanocortin-4 receptor modulators |
| WO2010065275A1 (en) | 2008-11-25 | 2010-06-10 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2011014992A1 (zh) | 2009-08-07 | 2011-02-10 | 上海贝尔股份有限公司 | 一种用于减小切换VoIP通话时的中断时间的方法和装置 |
| WO2011147809A1 (de) | 2010-05-26 | 2011-12-01 | Bayer Pharma Aktiengesellschaft | Substituierte 5-fluor-1h-pyrazolopyridine und ihre verwendung |
| WO2011149921A1 (en) | 2010-05-27 | 2011-12-01 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2012004258A1 (de) | 2010-07-09 | 2012-01-12 | Bayer Pharma Aktiengesellschaft | Annellierte pyrimidine und triazine und ihre verwendung zur behandlung bzw. prophylaxe von herz-kreislauf-erkrankungen |
| WO2012004259A1 (de) | 2010-07-09 | 2012-01-12 | Bayer Pharma Aktiengesellschaft | Annellierte 4 -aminopyrimidine und ihre verwendung als stimulatoren der löslichen guanylatcyclase |
| WO2012028647A1 (de) | 2010-09-03 | 2012-03-08 | Bayer Pharma Aktiengesellschaft | Bicyclische aza-heterocyclen und ihre verwendung |
| WO2012143510A1 (de) | 2011-04-21 | 2012-10-26 | Bayer Intellectual Property Gmbh | Fluoralkyl-substituierte pyrazolopyridine und ihre verwendung |
| WO2012152629A1 (de) | 2011-05-06 | 2012-11-15 | Bayer Intellectual Property Gmbh | Substituierte imidazopyridine und imidazopyridazine und ihre verwendung |
Non-Patent Citations (20)
| Title |
|---|
| DALEY, J. AM. CHEM. SOC., vol. 124, 2002, pages 3680 - 3691 |
| E. M. BECKER ET AL., BMC PHARMACOLOGY, vol. 1, no. 13, 2001 |
| F. WUNDER ET AL., ANAL. BIOCHEM., vol. 339, 2005, pages 104 - 112 |
| HASSAN J. ET AL., CHEM. REV., vol. 102, 2002, pages 1359 - 1469 |
| HASSAN J. ET AL., CHEM. REV., vol. 102, pages 1359 - 1469 |
| J. AM. CHEM. SOC., vol. 124, no. 14, 2002, pages 3680 - 3691 |
| JUSTUS, LIEBIGS ANNALEN DER CHEMIE, 1970, pages 99 - 107 |
| KELLEY ET AL., J. MED. CHEM., vol. 38, 1995, pages 3884 - 3888 |
| KLAUS WITTE; KAI HU; JOHANNA SWIATEK; CLAUDIA MÜSSIG; GEORG ERTL; BJÖRN LEMMER: "Experimental heart failure in rats: effects on cardiovascular circadian rhythms and on myocardial ßadrenergic signaling", CARDIOVASC RES, vol. 47, no. 2, 2000, pages 203 - 405 |
| KOZO OKAMOTO: "Spontaneous hypertension in rats", INT REV EXP PATHOL, vol. 7, 1969, pages 227 - 270 |
| MAARTEN VAN DEN BUUSE: "Circadian Rhythms of Blood Pressure, Heart Rate, and Locomotor Activity in Spontaneously Hypertensive Rats as Measured With Radio-Telemetry", PHYSIOLOGY & BEHAVIOR, vol. 55, no. 4, 1994, pages 783 - 787 |
| MÜLSCH ET AL., BRIT. J. PHARMACOL., vol. 120, 1997, pages 681 |
| OUDOUT ET AL., EUR. UROL., vol. 60, 2011, pages 1020 - 1026 |
| SHARKOVSKA Y; KALK P; LAWRENZ B; GODES M; HOFFMANN LS; WELLKISCH K; GESCHKA S; RELLE K; HOCHER B; STASCH JP: "NO-independent stimulation of soluble guanylate cyclase reduces target organ damage in low- and high-renin models of hypertension", J. HYPERTENSION, vol. 28, 2010, pages 1666 - 1675 |
| STASCH J.-P. ET AL., CHEMMEDCHEM, vol. 4, 2009, pages 853 - 865 |
| STASCH J.-P. ET AL., CIRCULATION, 2011 |
| STASCH J.-P. ET AL., CIRCULATION, vol. 123, 2011, pages 2263 - 2273 |
| STASCH J.-P. ET AL., NAT. REV. DRUG DISC., vol. 5, 2006, pages 755 - 768 |
| TOCHE ET AL., J. HET. CHEM., vol. 47, 2010, pages 287 - 291 |
| WU ET AL., BLOOD, vol. 84, 1994, pages 4226 |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015509515A (ja) * | 2012-03-06 | 2015-03-30 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | 置換アザ二環およびその使用 |
| WO2014084312A1 (ja) | 2012-11-30 | 2014-06-05 | アステラス製薬株式会社 | イミダゾピリジン化合物 |
| US9776997B2 (en) | 2013-06-04 | 2017-10-03 | Bayer Pharma Aktiengesellschaft | 3-aryl-substituted imidazo[1,2-A]pyridines and their use |
| CN105745215A (zh) * | 2013-07-10 | 2016-07-06 | 拜耳制药股份公司 | 苯甲基-1H-吡唑并[3,4-b]吡啶及其用途 |
| WO2015004105A1 (de) * | 2013-07-10 | 2015-01-15 | Bayer Pharma Aktiengesellschaft | Benzyl-1h-pyrazolo[3,4-b]pyridine und ihre verwendung |
| US9605008B2 (en) | 2013-07-10 | 2017-03-28 | Bayer Pharma Aktiengesellschaft | Benzyl-1H-pyrazolo[3,4-b]pyridines and use thereof |
| JP2016523944A (ja) * | 2013-07-10 | 2016-08-12 | バイエル・ファルマ・アクティエンゲゼルシャフト | ベンジル−1H−ピラゾロ[3,4−b]ピリジンおよびその使用 |
| WO2015089182A1 (en) | 2013-12-11 | 2015-06-18 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2015106268A1 (en) | 2014-01-13 | 2015-07-16 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF NEUROMUSCULAR DISORDERS |
| WO2015124544A1 (de) * | 2014-02-19 | 2015-08-27 | Bayer Pharma Aktiengesellschaft | 3-(pyrimidin-2-yl)imidazo[1,2-a]pyridine |
| CN106459090A (zh) * | 2014-02-19 | 2017-02-22 | 拜耳制药股份公司 | 3‑(嘧啶‑2‑基)咪唑并[1,2‑a]吡啶 |
| US9688699B2 (en) | 2014-02-19 | 2017-06-27 | Bayer Pharma Aktiengesellschaft | 3-(pyrimidine-2-yl)imidazo[1,2-a]pyridines |
| WO2016030362A1 (de) | 2014-08-29 | 2016-03-03 | Bayer Pharma Aktiengesellschaft | Substituierte annellierte pyrimidine und ihre verwendung |
| WO2016030354A1 (de) | 2014-08-29 | 2016-03-03 | Bayer Pharma Aktiengesellschaft | Amino-substituierte annellierte pyrimidine und ihre verwendung |
| US10292970B2 (en) | 2014-12-02 | 2019-05-21 | Bayer Pharma Aktiengesellschaft | Heteroaryl-substituted imidazo[1,2-A]pyridines and their use |
| WO2016177660A1 (en) | 2015-05-06 | 2016-11-10 | Bayer Pharma Aktiengesellschaft | The use of sgc stimulators, sgc activators, alone and combinations with pde5 inhibitors for the treatment of digital ulcers (du) concomitant to systemic sclerosis (ssc) |
| WO2017013010A1 (de) | 2015-07-23 | 2017-01-26 | Bayer Pharma Aktiengesellschaft | Stimulatoren und/oder aktivatoren der löslichen guanylatzyklase (sgc) in kombination mit einem inhibitor der neutralen endopeptidase (nep inhibitor) und/oder einem angiotensin aii-antagonisten und ihre verwendung |
| US11166932B2 (en) | 2015-07-23 | 2021-11-09 | Bayer Pharma Aktiengesellschaft | Stimulators and/or activators of soluble guanylate cyclase (sGC) in combination with an inhibitor of neutral endopeptidase (NEP inhibitor) and/or an angiotensin AII antagonist and the use thereof |
| KR101753652B1 (ko) * | 2015-10-21 | 2017-07-05 | 한국화학연구원 | N-아릴-1h-피라졸로피리딘-3-아민 유도체 또는 이의 약학적으로 허용가능한 염, 이의 제조방법 및 이를 유효성분으로 포함하는 melk 관련 질환의 예방 또는 치료용 약학적 조성물 |
| WO2017106175A2 (en) | 2015-12-14 | 2017-06-22 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF GASTROINTESTINAL SPHINCTER DYSFUNCTION |
| WO2018069126A1 (de) | 2016-10-11 | 2018-04-19 | Bayer Pharma Aktiengesellschaft | Kombination enthaltend sgc stimulatoren und mineralocorticoid-rezeptor-antagonisten |
| US10918639B2 (en) | 2016-10-11 | 2021-02-16 | Bayer Pharma Aktiengesellschaft | Combination containing SGC stimulators and mineralocorticoid receptor antagonists |
| US11331308B2 (en) | 2016-10-11 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Combination containing sGC activators and mineralocorticoid receptor antagonists |
| US11684621B2 (en) | 2016-10-11 | 2023-06-27 | Bayer Pharma Aktiengesellschaft | Combination containing sGC stimulators and mineralocorticoid receptor antagonists |
| WO2018111795A2 (en) | 2016-12-13 | 2018-06-21 | Ironwood Pharmaceuticals, Inc. | Use of sgc stimulators for the treatment of esophageal motility disorders |
| WO2019219672A1 (en) | 2018-05-15 | 2019-11-21 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| CN108640923A (zh) * | 2018-07-09 | 2018-10-12 | 湖南天地恒制药有限公司 | 一种托法替布关键中间体的制备方法 |
| WO2020014504A1 (en) | 2018-07-11 | 2020-01-16 | Cyclerion Therapeutics, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF MITOCHONRIAL DISORDERS |
| WO2020165010A1 (en) | 2019-02-13 | 2020-08-20 | Bayer Aktiengesellschaft | Process for the preparation of porous microparticles |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2802592A1 (de) | Substituierte annellierte pyrimidine und triazine und ihre verwendung | |
| EP2802587B1 (de) | Substituierte, annellierte imidazole und pyrazole und ihre verwendung | |
| EP2611802B1 (de) | Bicyclische aza-heterocyclen und ihre verwendung | |
| EP2751106B1 (de) | Substituierte annellierte pyrimidine und ihre verwendung | |
| EP2822951B1 (de) | Substituierte azabicyclen und ihre verwendung | |
| WO2012004258A9 (de) | Annellierte pyrimidine und triazine und ihre verwendung zur behandlung bzw. prophylaxe von herz-kreislauf-erkrankungen | |
| WO2013004785A1 (de) | Heteroaryl-substituierte pyrazolopyridine und ihre verwendung als stimulatoren der löslichen guanylatcyclase | |
| EP2705037A1 (de) | Substituierte imidazopyridine und imidazopyridazine und ihre verwendung | |
| WO2012004259A1 (de) | Annellierte 4 -aminopyrimidine und ihre verwendung als stimulatoren der löslichen guanylatcyclase | |
| EP3186251A1 (de) | Substituierte annellierte pyrimidine und ihre verwendung | |
| DE102012200351A1 (de) | Substituierte annellierte Pyrimidine und ihre Verwendung | |
| DE102011003315A1 (de) | Annellierte Pyrimindine und Triazine und ihre Verwendung | |
| DE102010031149A1 (de) | Annellierte Pyrimidine und Triazine und ihre Verwendung | |
| DE102012200354A1 (de) | Heteroaryl-substituierte Pyrazolopyridine und ihre Verwendung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13700288 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013700288 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 233460 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014001779 Country of ref document: CL Ref document number: 001063-2014 Country of ref document: PE Ref document number: MX/A/2014/008201 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2860855 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14146937 Country of ref document: CO Ref document number: 14371046 Country of ref document: US Ref document number: CR2014-000327 Country of ref document: CR |
|
| ENP | Entry into the national phase |
Ref document number: 2014551609 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P745/2014 Country of ref document: AE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: IDP00201404259 Country of ref document: ID |
|
| ENP | Entry into the national phase |
Ref document number: 2013208968 Country of ref document: AU Date of ref document: 20130110 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201408869 Country of ref document: UA |
|
| ENP | Entry into the national phase |
Ref document number: 20147021969 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201491339 Country of ref document: EA |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014016971 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112014016971 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140709 |