WO2011111423A1 - ヨウ素化芳香族化合物の製造方法 - Google Patents
ヨウ素化芳香族化合物の製造方法 Download PDFInfo
- Publication number
- WO2011111423A1 WO2011111423A1 PCT/JP2011/051078 JP2011051078W WO2011111423A1 WO 2011111423 A1 WO2011111423 A1 WO 2011111423A1 JP 2011051078 W JP2011051078 W JP 2011051078W WO 2011111423 A1 WO2011111423 A1 WO 2011111423A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- general formula
- ring
- compound
- aromatic compound
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/91—Dibenzofurans; Hydrogenated dibenzofurans
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/093—Preparation of halogenated hydrocarbons by replacement by halogens
- C07C17/20—Preparation of halogenated hydrocarbons by replacement by halogens of halogen atoms by other halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/093—Preparation of halogenated hydrocarbons by replacement by halogens
- C07C17/20—Preparation of halogenated hydrocarbons by replacement by halogens of halogen atoms by other halogen atoms
- C07C17/202—Preparation of halogenated hydrocarbons by replacement by halogens of halogen atoms by other halogen atoms two or more compounds being involved in the reaction
- C07C17/208—Preparation of halogenated hydrocarbons by replacement by halogens of halogen atoms by other halogen atoms two or more compounds being involved in the reaction the other compound being MX
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/82—Carbazoles; Hydrogenated carbazoles
- C07D209/88—Carbazoles; Hydrogenated carbazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/24—Benzimidazoles; Hydrogenated benzimidazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
Definitions
- the present invention relates to a method for producing an iodinated aromatic compound useful as an intermediate of an organic synthetic compound and a material for organic electroluminescence.
- An iodinated aromatic compound in which an iodine atom is bonded to an aromatic compound has a wide demand as an important intermediate in the production of organic photoreceptors, dyes, agricultural chemicals, pharmaceuticals and the like used for materials for organic electroluminescence.
- an iodinated aromatic compound can be iodinated directly or a corresponding aromatic amino compound can be synthesized by a Sandmeyer reaction.
- an oxidizing agent and an acid catalyst are used when the aromatic compound and the iodine compound are reacted.
- an oxidizing agent to be used a method using peracetic acid (see Non-Patent Document 1), a method using iodic acid (see Non-Patent Document 2), a method using periodic acid (see Non-Patent Document 3), and the like are known.
- these oxidizing agents are expensive and require treatment of the waste liquid, which is industrially expensive and economically disadvantageous.
- a halogen exchange reaction is known in which chlorine atoms of chlorinated aromatic compounds and bromine atoms of brominated aromatic compounds are exchanged for iodine atoms.
- halogen exchange using sodium iodide, copper iodide and diamine (ligand) in an alcohol solvent or dioxane (see Non-Patent Documents 4, 5, and Patent Document 3).
- the cost is high because an expensive ligand is required, and the purification operation is not easy, so that it is not an industrially satisfactory method.
- Non-Patent Document 6 a method of performing halogen exchange using potassium iodide and copper iodide in hexamethylphosphoric triamide (see Non-Patent Document 6) has also been reported.
- hexamethylphosphoric triamide as a reaction solvent has a very high carcinogenicity and is harmful to the human body, and therefore has a problem as an industrial production method.
- the reaction rate of the halogen exchange reaction is very slow.
- the reaction point is Since it is sterically crowded, the reactivity is low and the reaction hardly proceeds.
- the halogen exchange reaction may proceed by increasing the temperature or increasing the amount of the catalyst. It was very difficult to obtain a fluorinated aromatic compound in a high yield, and there was no satisfactory industrial production method.
- the present invention has been made in view of the above problems, and an object of the present invention is to produce an iodinated aromatic compound efficiently and safely without using a ligand and without using a solvent harmful to the human body. It is to provide a method that can. Furthermore, even when the substrate has a sterically bulky structure such as a condensed ring structure, it is an object of the present invention to provide a production method capable of suppressing the side reaction and obtaining the target iodinated aromatic compound in a high yield.
- a compound represented by the following general formula [1] and an alkali metal iodide salt are reacted in the presence of copper or a copper ion and a compound represented by the following general formula [2].
- Compound production method Compound production method.
- Ar represents an aromatic hydrocarbon ring group or an aromatic heterocyclic group
- X represents a chlorine atom or a bromine atom
- n represents an integer of 1 or more.
- R 1 , R 2 , R 3 and R 4 each independently represents an alkyl group or an aryl group, and R 1 and R 3 , R 2 and R 4, and R 3 and R 4 may be bonded to each other to form a ring.
- R 1 and R 2 each independently represents an alkyl group or an aryl group.
- the present invention it is possible to provide a method capable of efficiently and safely producing an iodinated aromatic compound without using a ligand and without using a solvent harmful to the human body. Furthermore, even when the substrate has a three-dimensionally bulky structure such as a condensed ring structure, it is possible to provide a production method in which side reactions are suppressed and the target iodinated aromatic compound can be obtained in high yield.
- the production method of an iodinated aromatic compound useful as an intermediate of an organic synthetic compound and a material for organic electroluminescence is a method of the present invention, which suppresses side reactions without using a solvent harmful to the human body,
- the aromatic compound is obtained in high yield and has an excellent effect.
- Ar represents an aromatic hydrocarbon ring group or an aromatic heterocyclic group.
- aromatic hydrocarbon ring group examples include a benzene ring group, a biphenyl ring group, a naphthalene ring group, an azulene ring group, an anthracene ring group, a phenanthrene ring group, a pyrene ring group, a chrysene ring group, a naphthacene ring group, and a triphenylene ring.
- o-terphenyl ring group o-terphenyl ring group, m-terphenyl ring group, p-terphenyl ring group, acenaphthene ring group, coronene ring group, fluorene ring group, fluoranthrene ring group, pentacene ring group, perylene ring group, pentaphen
- examples thereof include a ring group, a picene ring group, a pyranthrene ring group, an anthraanthrene ring group, a dibenzofuran ring group, a dibenzodiophene ring group, and a carbazole ring group.
- aromatic heterocyclic group examples include a furan ring group, a thiophene ring group, a pyridine ring group, a pyridazine ring group, a pyrimidine ring group, a pyrazine ring group, a triazine ring group, an oxadiazole ring group, a triazole ring group, and an imidazole.
- Ring group pyrazole ring group, thiazole ring group, indole ring group, benzimidazole ring group, benzothiazole ring group, benzoxazole ring group, quinoxaline ring group, quinazoline ring group, phthalazine ring group, benzofuran ring group, dibenzofuran ring group,
- Examples include a benzothiophene ring group, a dibenzothiophene ring group, and a carbazole ring group.
- These rings may have a substituent, and specific examples of the substituent include alkyl groups having 1 to 25 carbon atoms (methyl group, ethyl group, propyl group, isopropyl group, tert-butyl group, pentyl group).
- Preferred as the aromatic hydrocarbon ring group and aromatic heterocyclic group represented by Ar are benzene ring group, naphthalene ring group, azulene ring group, anthracene ring group, phenanthrene ring group, benzothiazole ring group, benzoxazole ring Group, benzofuran ring group, dibenzofuran ring group, benzothiophene ring group, dibenzothiophene ring group, and carbazole ring group. Particularly preferred is a dibenzofuran ring group.
- X represents a chlorine atom or a bromine atom.
- n represents an integer of 1 or more.
- n is an integer of 1 or more and is not particularly limited because it depends on the structure of the compound represented by the general formula [1], but n is preferably 1 to 10, and more preferably 1 to 3.
- R 1 , R 2 , R 3 and R 4 each independently represents an alkyl group or an aryl group
- R 1 and R 3 , R 2 and R 4 and R 3 and R 4 are They may combine with each other to form a ring.
- Specific examples of the alkyl group include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, sec-butyl group, tert-butyl group, pentyl group, hexyl group, octyl group, decyl group and the like.
- Specific examples of the aryl group include a phenyl group, a naphthyl group, and an anthracenyl group.
- R 1 and R 3 and R 2 and R 4 may be bonded to each other to form a ring, and R 1 and R 3 and R 2 and R 4 may be bonded together with an adjacent nitrogen atom to form a ring.
- Examples thereof include 5- to 7-membered rings. Preferably, it is a 5-membered or 6-membered ring.
- Examples of the ring formed by bonding R 3 and R 4 together with the adjacent urea bond include a 5- to 7-membered ring.
- the ring formed by bonding R 3 and R 4 together with the adjacent urea bond may be a single ring or a condensed polycycle.
- R 1 and R 2 are preferably a methyl group, an ethyl group, or an isopropyl group, and most preferably a methyl group.
- R 3 and R 4 are preferably bonded together with an adjacent urea bond to form a ring, and the formed ring is more preferably a 5- or 6-membered ring, A structure represented by the general formula [3] is more preferable.
- R 1 and R 2 each independently represents an alkyl group or an aryl group. Specific examples of the alkyl group or aryl group represented by R 1 and R 2 include the groups cited as specific examples of the alkyl group or aryl group represented by R 1 and R 2 in the general formula [2] .
- the addition amount is based on 1 mol of the halide represented by the general formula [1]. 10 mol or more is preferable.
- the reaction solvent include dimethylformamide, dimethyl sulfoxide, dimethylacetamide, N-methylpyrrolidone, tetrahydrofuran, ethyl acetate, toluene and the like.
- the amount of the compound represented by the general formula [2] or the general formula [3] may be increased and used as a solvent. The amount used is 5 to 10 times the amount of 1 g of halide. preferable.
- 1,3-dimethyl-2-imidazolidinone in which R 1 and R 2 are methyl groups has no carcinogenicity and is less harmful to the human body even when used as a solvent And is most preferred in the present invention.
- X represents a chlorine atom or a bromine atom
- Y represents an oxygen atom
- S represents an alkyl group or an aryl group
- W represents a substituent.
- alkyl group represented by R 5 examples include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, pentyl group, hexyl group, octyl group, decyl group and the like.
- aryl group examples include a phenyl group and a naphthyl group.
- Y is preferably an oxygen atom.
- substituent represented by W include alkyl groups having 1 to 25 carbon atoms, cycloalkyl groups, aryl groups, heteroaryl groups, halogen atoms, alkoxy groups, aryloxy groups, amino groups, cyano groups, carboxyl groups. Group, hydroxyl group and the like. When there are a plurality of Ws, each W may be the same or different.
- Examples of the copper or copper ion used in the production method of the present invention include copper chloride (I), copper bromide (I), copper iodide (I), copper oxide (I), copper chloride (II), odor Examples thereof include copper (II) chloride, copper (II) sulfate, copper nitrate (II), copper acetate (II), copper hydroxide (II), etc., preferably copper (I) chloride, copper (I) bromide Copper (I) iodide, more preferably copper iodide.
- the alkali metal iodide salt is used in an amount of 1 mol to 20 mol, preferably 1 mol to 15 mol, and copper or copper ions from 1 mol to 1 mol of the halide represented by the general formula [1]. It is preferable to use in the range of 8 mol.
- the reaction temperature is usually preferably 130 to 160 ° C, particularly preferably 140 to 150 ° C.
- exemplary compound 1-44 5.0 g (20 mmol), copper iodide 19.0 (100 mmol), potassium iodide 49.8 g (300 mmol), exemplary compound 2-1 (1,3-dimethyl-2- 50 ml of imidazolidinone (hereinafter abbreviated as DMI) was added and stirred at 140 to 150 ° C. for 12 hours. Next, tetrahydrofuran and saturated saline were added, the aqueous layer was removed, and the extracted organic layer was concentrated under reduced pressure. When the obtained product was analyzed by HPLC, 5.5 (yield 92%) of the desired 4-iododibenzofuran and 0.1 g (yield 3%) of by-product (H form) were obtained.
- DMI imidazolidinone
- exemplary compound 1-47 7.4 g (20 mmol), copper iodide 1.9 (10 mmol), sodium iodide 6.0 g (40 mmol), (1R, 2R)-( ⁇ )-N, N ′ -2.9 g (20 mmol) of dimethylcyclohexane-1,2-diamine and 50 ml of dioxane were added and stirred at 110 ° C. for 20 hours.
- tetrahydrofuran and saturated saline were added, the aqueous layer was removed, and the extracted organic layer was concentrated under reduced pressure.
- the obtained product was analyzed by HPLC. As a result, the desired 2,6-diiododibenzofuran was 1.7 g (yield 20%), and the by-product (H form) was 0.79 g (yield 2.3%). The rest was unreacted raw material.
- the by-product (H-form) from which iodine was eliminated was generated in the method of the comparative example, and the yield of the target iodo-form was decreased, but the by-product (H-form) was observed in the method of the present invention. Is hardly produced, and the target product is obtained in a high yield, which is superior to the comparative example.
- a solvent harmful to the human body such as HMPA or dioxane is used, but it can be seen that the present invention is superior in that a safe solvent can be used in the present invention.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Furan Compounds (AREA)
- Indole Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
本発明は、配位子が不要で、人体に有害な溶媒を使用することなく、ヨウ素化芳香族化合物を効率的かつ安全に製造できる方法を提供する。更に、基質が縮環構造のような立体的に嵩高い構造である場合においても、副反応を抑え、目的のヨウ素化芳香族化合物が高収率で得られる製造方法を提供する。このヨウ素化芳香族化合物の製造方法は、下記一般式[1]で表される化合物とヨウ化アルカリ金属塩を、銅もしくは銅イオン及び下記一般式[2]で表される化合物の存在下で反応させることを特徴とする。
Description
本発明は、有機合成化合物の中間体及び有機エレクトロルミネッセンス用材料として有用なヨウ素化芳香族化合物の製造方法に関する。
芳香族化合物にヨウ素原子が結合したヨウ素化芳香族化合物は、有機エレクトロルミネッセンス用材料等に使用される有機感光体や染料、農薬、医薬品等の製造において、重要な中間体として幅広い需要がある。
ヨウ素化芳香族化合物は、一般的には、芳香族化合物を直接ヨウ素化するか、対応する芳香族アミノ化合物をサンドマイヤー反応により合成できる。芳香族化合物を直接ヨウ素化する方法においては、芳香族化合物とヨウ素化合物とを反応させる際に、酸化剤と酸触媒が使用される。使用される酸化剤として、過酢酸を用いる方法(非特許文献1参照)、ヨウ素酸を用いる方法(非特許文献2参照)、過ヨウ素酸を用いる方法(非特許文献3参照)等が知られているが、これらの酸化剤は高価であり、また廃液の処理も必要となるため工業的に高コストとなり経済的に不利である。他にも、過硫酸塩(特許文献1参照)や過酸化水素を用いる反応(特許文献2参照)も報告されているが、これらの酸化剤は取り扱いに注意を要する上に、反応の収率も低い。サンドマイヤー反応(非特許文献4参照)の場合には、反応の段階数が多く、反応後の後処理も煩雑であり、工業的な製造方法としては効率が悪く、好ましくない。
ヨウ素化芳香族化合物の別の合成法として、塩素化芳香族化合物の塩素原子や臭素化芳香族化合物の臭素原子をヨウ素原子に交換するハロゲン交換反応が知られている。例えば、アルコール系溶媒やジオキサン中、ヨウ化ナトリウム、ヨウ化銅及びジアミン(配位子)を用いてハロゲン交換する方法(非特許文献4、5、特許文献3参照)等が知られているが、高価な配位子が必要であるためコストが高く、また、精製操作も容易ではないため、工業的に満足できる方法ではない。同様に銅触媒を用いる反応で配位子を添加しない系として、ヘキサメチルリン酸トリアミド中、ヨウ化カリウム及びヨウ化銅を用いてハロゲン交換を行う方法(非特許文献6参照)も報告されているが、反応溶媒のヘキサメチルリン酸トリアミドは発がん性が非常に高く、人体に有害であるため、工業的な製造法としては問題がある。
更に、基質が立体的に嵩高い場合、例えば、本発明に係る下記一般式[4]のような構造である場合には、ハロゲン交換反応の反応速度は非常に遅い。特に、本発明に係る下記一般式[5]で表されるような化合物のように、縮環の隣接炭素に置換している塩素原子または臭素原子をヨウ素に交換する反応においては、反応点が立体的に混み合っているため反応性が低く、反応はほとんど進行しない。また、反応速度を上げるために、温度を上げたり触媒量を増やしたりすることでハロゲン交換反応が進行する場合もあるが、交換されたヨウ素の脱離反応が競争的に生じるため、目的のヨウ素化芳香族化合物を高収率で得ることは非常に困難であり、工業的な製造法として満足できるものはなかった。
Journal of American Chemical Society,1968,90,p6187
Liebigs Annalen,1960,634,p84
Organic Syntheses,Collective Vol.VI,1988,p700
Organic Syntheses,Collective Vol.II,1943,p351
Journal of American Chemical Society,2002,124,p14844
Chemistry Letters,1985,p411
本発明は、上記課題に鑑みなされたものであり、本発明の目的は、配位子が不要で、人体に有害な溶媒を使用することなく、ヨウ素化芳香族化合物を効率的かつ安全に製造できる方法を提供することにある。更に、基質が縮環構造のような立体的に嵩高い構造である場合においても、副反応を抑え、目的のヨウ素化芳香族化合物が高収率で得られる製造方法を提供することにある。
本発明の上記目的は、下記の構成により達成される。
1.下記一般式[1]で表される化合物とヨウ化アルカリ金属塩を、銅もしくは銅イオン及び下記一般式[2]で表される化合物の存在下で反応させることを特徴とするヨウ素化芳香族化合物の製造方法。

(式中、Arは芳香族炭化水素環基または芳香族複素環基を表す。Xは塩素原子または臭素原子を表し、nは1以上の整数を表す。R1、R2、R3及びR4はそれぞれ独立に、アルキル基またはアリール基を表し、R1とR3、R2とR4及びR3とR4は互いに結合して環を形成してもよい。)
2.前記一般式[2]が下記一般式[3]で表されることを特徴とする前記1に記載のヨウ素化芳香族化合物の製造方法。
2.前記一般式[2]が下記一般式[3]で表されることを特徴とする前記1に記載のヨウ素化芳香族化合物の製造方法。

(式中、R1及びR2はそれぞれ独立に、アルキル基またはアリール基を表す。)
3.前記一般式[1]が下記一般式[4]で表されることを特徴とする前記1または2に記載のヨウ素化芳香族化合物の製造方法。
3.前記一般式[1]が下記一般式[4]で表されることを特徴とする前記1または2に記載のヨウ素化芳香族化合物の製造方法。

(式中、Xは塩素原子または臭素原子を表し、Yは酸素原子、硫黄原子またはN-R5を表し、R5はアルキル基またはアリール基を表す。Wは置換基を表し、m及びnは1から4の整数を表す。)
4.前記一般式[4]が下記一般式[5]で表されることを特徴とする前記3に記載のヨウ素化芳香族化合物の製造方法。
4.前記一般式[4]が下記一般式[5]で表されることを特徴とする前記3に記載のヨウ素化芳香族化合物の製造方法。

(式中、Xは塩素原子または臭素原子を表し、Yは酸素原子、硫黄原子またはN-R5を表し、R5はアルキル基またはアリール基を表す。Wは置換基を表し、mは1から4の整数を表す。)
5.前記一般式[5]において、Yが酸素原子であることを特徴とする前記4に記載のヨウ素化芳香族化合物の製造方法。
5.前記一般式[5]において、Yが酸素原子であることを特徴とする前記4に記載のヨウ素化芳香族化合物の製造方法。
本発明によれば、配位子が不要で、人体に有害な溶媒を使用することなく、ヨウ素化芳香族化合物を効率的かつ安全に製造できる方法を提供することができる。更に、基質が縮環構造のような立体的に嵩高い構造である場合においても、副反応を抑え、目的のヨウ素化芳香族化合物が高収率で得られる製造方法を提供することができる。
有機合成化合物の中間体及び有機エレクトロルミネッセンス用材料として有用なヨウ素化芳香族化合物の製造方法は、本発明の方法により、人体に有害な溶媒を使用することなく、副反応を抑え、目的のヨウ素化芳香族化合物が高収率で得られ、優れた効果を有する。
以下、本発明を実施するための形態について説明するが、本発明はこれらに限定されない。
以下、本発明を更に詳細に述べる。
前記一般式[1]において、Arは芳香族炭化水素環基または芳香族複素環基を表す。
前記芳香族炭化水素環基としては、例えば、ベンゼン環基、ビフェニル環基、ナフタレン環基、アズレン環基、アントラセン環基、フェナントレン環基、ピレン環基、クリセン環基、ナフタセン環基、トリフェニレン環基、o-テルフェニル環基、m-テルフェニル環基、p-テルフェニル環基、アセナフテン環基、コロネン環基、フルオレン環基、フルオラントレン環基、ペンタセン環基、ペリレン環基、ペンタフェン環基、ピセン環基、ピラントレン環基、アンスラアントレン環基、ジベンゾフラン環基、ジベンゾジオフェン環基、カルバゾール環基等が挙げられる。
前記芳香族複素環基としては、例えば、フラン環基、チオフェン環基、ピリジン環基、ピリダジン環基、ピリミジン環基、ピラジン環基、トリアジン環基、オキサジアゾール環基、トリアゾール環基、イミダゾール環基、ピラゾール環基、チアゾール環基、インドール環基、ベンゾイミダゾール環基、ベンゾチアゾール環基、ベンゾオキサゾール環基、キノキサリン環基、キナゾリン環基、フタラジン環基、ベンゾフラン環基、ジベンゾフラン環基、ベンゾチオフェン環基、ジベンゾチオフェン環基、カルバゾール環基等が挙げられる。
これらの環は置換基を有していてもよく、置換基の具体例としては、炭素数1~25のアルキル基(メチル基、エチル基、プロピル基、イソプロピル基、tert-ブチル基、ペンチル基、ヘキシル基、オクチル基、デシル基、ドデシル基、オクタデシル基等)、シクロアルキル基(シクロヘキシル基、シクロペンチル基等)、アリール基(フェニル基等)、ヘテロアリール基(ピリジル基、チアゾリル基、オキサゾリル基、イミダゾリル基、フリル基、ピロリル基等)、ハロゲン原子(塩素原子、臭素原子、ヨウ素原子、フッ素原子等)、アルコキシ基(メトキシ基、エトキシ基、プロピルオキシ基等)、アリールオキシ基(フェノキシ基等)、アルコキシカルボニル基(メチルオキシカルボニル基、エチルオキシカルボニル基等)、スルホンアミド基(メタンスルホンアミド基、エタンスルホンアミド基、ブタンスルホンアミド基、ヘキサンスルホンアミド基、シクロヘキサンスルホンアミド基、ベンゼンスルホンアミド基等)、スルファモイル基(アミノスルホニル基、メチルアミノスルホニル基、ジメチルアミノスルホニル基、ブチルアミノスルホニル基、ヘキシルアミノスルホニル基、シクロヘキシルアミノスルホニル基、フェニルアミノスルホニル基、2-ピリジルアミノスルホニル基等)、カルバモイル基(アミノカルボニル基、メチルアミノカルボニル基、ジメチルアミノカルボニル基、プロピルアミノカルボニル基等)、アミド基(アセトアミド基、プロピオンアミド基、ベンズアミド基等)、スルホニル基(メチルスルホニル基、エチルスルホニル基、フェニルスルホニル基等)、アミノ基(アミノ基、エチルアミノ基、ジメチルアミノ基等)、シアノ基、カルボキシル基、ヒドロキシル基等を挙げることができる。
Arで表される芳香族炭化水素環基、芳香族複素環基として好ましいものは、ベンゼン環基、ナフタレン環基、アズレン環基、アントラセン環基、フェナントレン環基、ベンゾチアゾール環基、ベンゾオキサゾール環基、ベンゾフラン環基、ジベンゾフラン環基、ベンゾチオフェン環基、ジベンゾチオフェン環基、カルバゾール環基である。特に好ましくはジベンゾフラン環基である。
前記一般式[1]において、Xは塩素原子または臭素原子を表す。nは1以上の整数を表す。nは1以上の整数であり、一般式[1]で表される化合物の構造によるので特に限定はされないが、nは1~10であることが好ましく、1~3であることがより好ましい。
前記一般式[2]において、R1、R2、R3及びR4はそれぞれ独立に、アルキル基またはアリール基を表し、R1とR3、R2とR4及びR3とR4は互いに結合して環を形成してもよい。アルキル基の具体例としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、sec-ブチル基、tert-ブチル基、ペンチル基、ヘキシル基、オクチル基、デシル基等が挙げられる。アリール基の具体例としては、フェニル基、ナフチル基、アントラセニル基等が挙げられる。
R1とR3及びR2とR4は互いに結合して環を形成してもよく、R1とR3及びR2とR4が隣接する窒素原子と共に互いに結合して形成される環として、例えば、5~7員の環が挙げられる。好ましくは、5員または6員の環である。
R3とR4が隣接する尿素結合と共に互いに結合して形成される環は、例えば、5~7員の環が挙げられる。またR3及びR4が隣接する尿素結合と共に互いに結合して形成される環は単環であっても縮合多環であってもよい。
R1及びR2として好ましくはメチル基、エチル基、イソプロピル基であり、最も好ましくはメチル基である。
R3及びR4としては、R3及びR4が隣接する尿素結合と共に互いに結合して環を形成することが好ましく、形成される環が5または6員環であることがより好ましく、特に、一般式[3]で表される構造であることが更に好ましい。一般式[3]において、R1及びR2はそれぞれ独立に、アルキル基またはアリール基を表す。R1及びR2で表されるアルキル基またはアリール基の具体例としては、一般式[2]においてR1及びR2で表されるアルキル基またはアリール基の具体例として挙げた基が挙げられる。
本発明において、一般式[2]または一般式[3]で表される化合物を反応の際の添加剤として使用する場合、添加量は一般式[1]で表されるハロゲン化物1molに対して10mol以上が好ましい。反応の溶媒として、例えば、ジメチルホルムアミド、ジメチルスルホキシド、ジメチルアセトアミド、N-メチルピロリドン、テトラヒドロフラン、酢酸エチル、トルエン等が挙げられる。また、一般式[2]または一般式[3]で表される化合物の添加量を増やして溶媒として使用してもよく、使用量としては、ハロゲン化物1gに対して5倍から10倍量が好ましい。
一般式[3]において、特に、R1及びR2がメチル基である1,3-ジメチル-2-イミダゾリジノンは、発がん性がなく、溶媒として使用しても人体に対する有害性が少ない化合物であり、本発明において最も好ましい。
前記一般式[4]及び一般式[5]において、Xは塩素原子または臭素原子を表し、Yは酸素原子、硫黄原子またはN-R5を表し、R5はアルキル基またはアリール基を表す。Wは置換基を表す。
R5で表されるアルキル基の具体例としては、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、ペンチル基、ヘキシル基、オクチル基、デシル基等が挙げられる。アリール基の具体例としては、フェニル基、ナフチル基等が挙げられる。
Yとして好ましくは、酸素原子である。
Wで表される置換基の具体例としては、炭素数1~25のアルキル基、シクロアルキル基、アリール基、ヘテロアリール基、ハロゲン原子、アルコキシ基、アリールオキシ基、アミノ基、シアノ基、カルボキシル基、ヒドロキシル基等を挙げることができる。Wが複数のときは、それぞれのWが同じでも異なっていてもよい。
以下に、本発明に係る一般式[1]、[4]、[5]で表される化合物の代表的具体例を示すが、本発明はこれらに限定されるものではない。

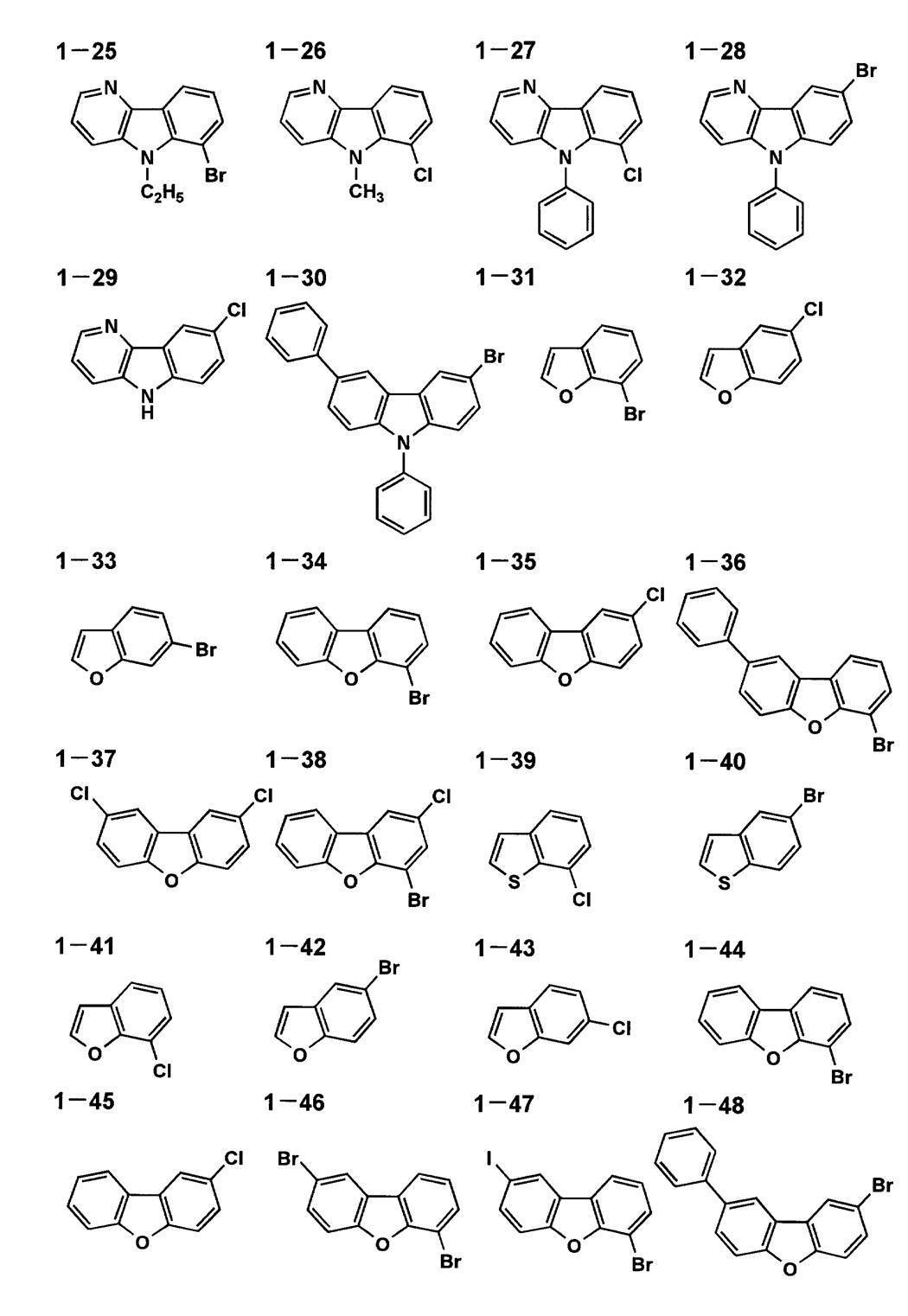

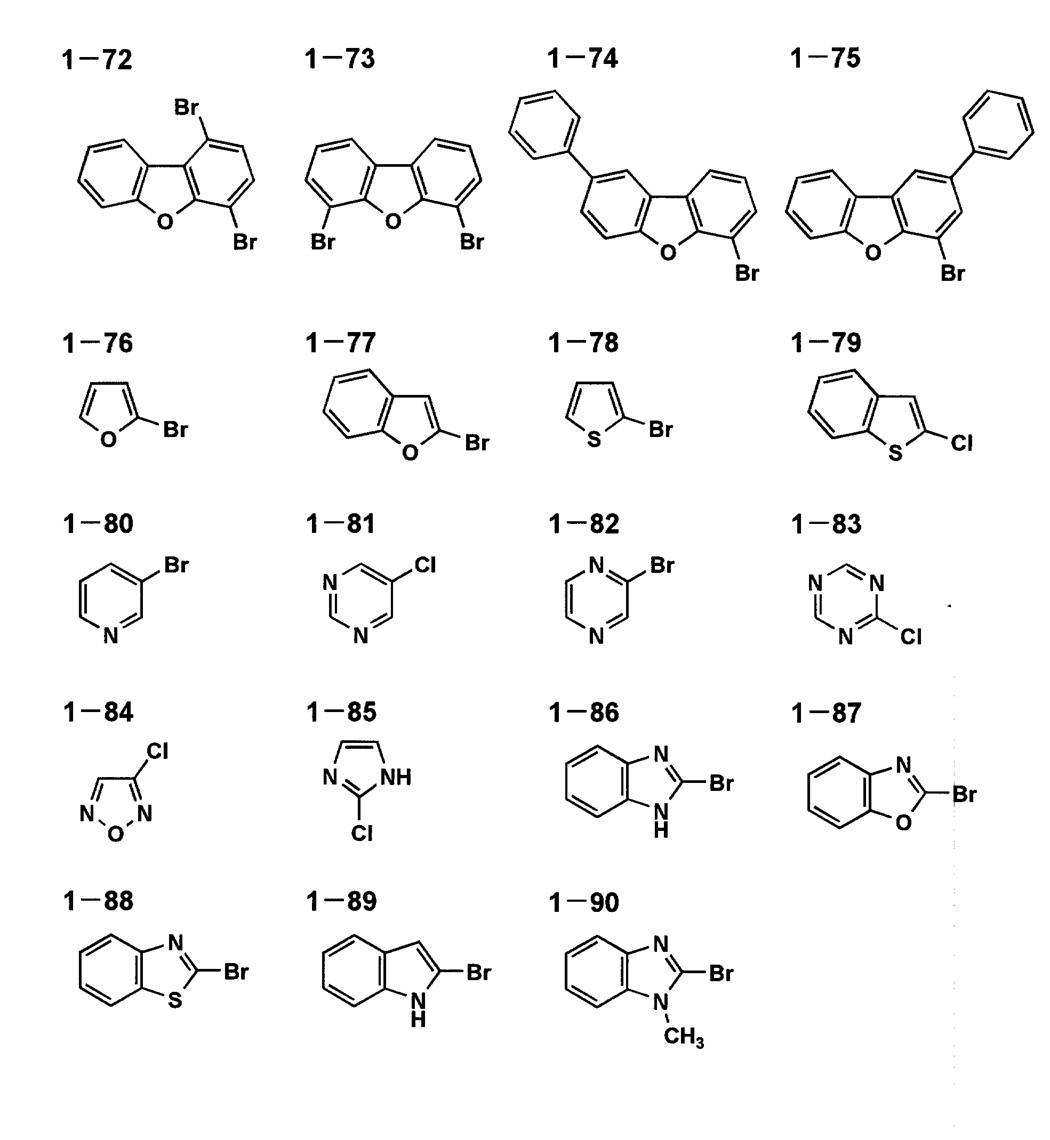
以下に、本発明に係る一般式[2]及び[3]で表される化合物の代表的具体例を示すが、本発明はこれらに限定されるものではない。

本発明の製造方法において使用するヨウ化アルカリ金属塩としては、例えば、ヨウ化リチウム、ヨウ化ナトリウム、ヨウ化カリウム、ヨウ化セシウム等が挙げられ、好ましくはヨウ化ナトリウム、ヨウ化カリウム、より好ましくはヨウ化カリウムである。
本発明の製造方法において使用する銅もしくは銅イオンとしては、例えば、塩化銅(I)、臭化銅(I)、ヨウ化銅(I)、酸化銅(I)、塩化銅(II)、臭化銅(II)、硫酸銅(II)、硝酸銅(II)、酢酸銅(II)、水酸化銅(II)等が挙げられ、好ましくは、塩化銅(I)、臭化銅(I)、ヨウ化銅(I)、より好ましくはヨウ化銅である。
本発明の製造方法においては、上記ヨウ化アルカリ金属塩を一般式[1]で表されるハロゲン化物1molに対して1molから20mol、好ましくは、1molから15mol、また、銅もしくは銅イオンを1molから8molの範囲で使用することが好ましい。反応温度は、通常130~160℃が好ましく、140~150℃が特に好ましい。
以下に実施例を挙げて本発明を具体的に説明するが、本発明の実施態様はこれらに限定されるものではない。
実施例1
〔4-ヨードジベンゾフランの合成〕(比較例)
〔4-ヨードジベンゾフランの合成〕(比較例)

窒素気流下、例示化合物1-44 5.0g(20mmol)、ヨウ化銅19.0(100mmol)、ヨウ化カリウム49.8g(300mmol)、ヘキサメチルリン酸トリアミド(HMPA)50mlを投入し、140~150℃で12時間攪拌した。次に、テトラヒドロフラン及び飽和食塩水を投入し、水層を除去した後、抽出した有機層を減圧濃縮した。得られた生成物をHPLCで解析したところ、目的の4-ヨードジベンゾフランは0.6g(収率10%)、副生物(H体)は2.9g(収率85%)得られた。
実施例2
〔4-ヨードジベンゾフランの合成〕(本発明)
〔4-ヨードジベンゾフランの合成〕(本発明)

窒素気流下、例示化合物1-44 5.0g(20mmol)、ヨウ化銅19.0(100mmol)、ヨウ化カリウム49.8g(300mmol)、例示化合物2-1(1,3-ジメチル-2-イミダゾリジノン;以下略称DMI)50mlを投入し、140~150℃で12時間攪拌した。次に、テトラヒドロフラン及び飽和食塩水を投入し、水層を除去した後、抽出した有機層を減圧濃縮した。得られた生成物をHPLCで解析したところ、目的の4-ヨードジベンゾフランは5.5(収率92%)、副生物(H体)は0.1g(収率3%)得られた。
1H-NMR(400MHz,CDCl3):δ=7.94(t,2H),7.83(d,1H),7.67(d,1H),7.51(dt,1H),7.39(t,1H),7.13(t,1H)
実施例3
〔2,6-ジヨードジベンゾフラン合成〕(比較例)
実施例3
〔2,6-ジヨードジベンゾフラン合成〕(比較例)

窒素気流下、例示化合物1-47 7.4g(20mmol)、ヨウ化銅1.9(10mmol)、ヨウ化ナトリウム6.0g(40mmol)、(1R,2R)-(-)-N,N′-ジメチルシクロヘキサン-1,2-ジアミン2.9g(20mmol)、ジオキサン50ml投入し、110℃で20時間攪拌した。次に、テトラヒドロフラン及び飽和食塩水を投入し、水層を除去した後、抽出した有機層を減圧濃縮した。得られた生成物をHPLCで解析したところ、目的の2,6-ジヨードジベンゾフランは1.7g(収率20%)、副生物(H体)は0.79g(収率2.3%)得られ、残りは未反応の原料であった。
実施例4
〔2,6-ジヨードジベンゾフランの合成〕(本発明)
〔2,6-ジヨードジベンゾフランの合成〕(本発明)

窒素気流下、例示化合物1-47 7.4g(20mmol)、ヨウ化銅19.0(100mmol)、ヨウ化カリウム49.8g(300mmol)、例示化合物2-1(DMI)50mlを投入し、140~150℃で12時間攪拌した。次に、テトラヒドロフラン及び飽和食塩水を投入し、水層を除去した後、抽出した有機層を減圧濃縮した。得られた生成物をHPLCで解析したところ、目的の2,6-ジヨードジベンゾフランは8.2g(収率95%)、副生物(H体)は0.7g(収率2%)得られた。
1H-NMR(400MHz,CDCl3):δ=8.26(d,1H),7.87(t,1H),7.86(t,1H),7.78(dd,1H),7.45(d,1H),7.14(t,1H)
実施例5
〔ヨードベンゼンの合成〕(本発明)
実施例5
〔ヨードベンゼンの合成〕(本発明)

窒素気流下、例示化合物1-1 2.2g(20mmol)、ヨウ化銅19.0(100mmol)、ヨウ化カリウム49.8g(300mmol)、例示化合物2-1(DMI) 50mlを投入し、140~150℃で12時間攪拌した。次に、テトラヒドロフラン及び飽和食塩水を投入し、水層を除去した後、抽出した有機層を減圧濃縮した。得られた生成物をHPLCで解析したところ、目的のヨードベンゼンは3.8g(収率94%)得られた。
Massスペクトル;(API法、m/e(相対強度)) 205((60) MH+)
実施例6
〔N-フェニル-3-ヨードカルバゾールの合成〕(本発明)
実施例6
〔N-フェニル-3-ヨードカルバゾールの合成〕(本発明)

窒素気流下、例示化合物1-20 6.4g(20mmol)、ヨウ化銅19.0(100mmol)、ヨウ化カリウム49.8g(300mmol)、例示化合物2-1(DMI) 50mlを投入し、140~150℃で12時間攪拌した。次に、テトラヒドロフラン及び飽和食塩水を投入し、水層を除去した後、抽出した有機層を減圧濃縮した。得られた生成物をHPLCで解析したところ、目的のN-フェニル-3-ヨードカルバゾールは6.9g(収率93%)得られた。
Massスペクトル;(API法、m/e(相対強度)) 370((55) MH+)
実施例7
〔2-ヨード-N-メチル-ベンゾイミダゾールの合成〕(本発明)
実施例7
〔2-ヨード-N-メチル-ベンゾイミダゾールの合成〕(本発明)

窒素気流下、例示化合物1-90 4.2g(20mmol)、ヨウ化銅19.0(100mmol)、ヨウ化カリウム49.8g(300mmol)、例示化合物2-1(DMI) 50mlを投入し、140~150℃で12時間攪拌した。次に、テトラヒドロフラン及び飽和食塩水を投入し、水層を除去した後、抽出した有機層を減圧濃縮した。得られた生成物をHPLCで解析したところ、目的のN-フェニル-3-ヨードカルバゾールは4.8g(収率94%)得られた。
Massスペクトル;(API法、m/e(相対強度)) 259((45) MH+)
実施例中の各化合物の同定はMASSスペクトル及びNMRスペクトルで行い、それぞれ目的化合物及び副生物(H体)であることを確認した。その他の例示化合物も上記の方法に準じて合成することができる。
実施例中の各化合物の同定はMASSスペクトル及びNMRスペクトルで行い、それぞれ目的化合物及び副生物(H体)であることを確認した。その他の例示化合物も上記の方法に準じて合成することができる。
以上の結果から、比較例の方法ではヨウ素が脱離した副生物(H体)が生成し、目的のヨード体の収率の低下が見られるが、本発明の方法では副生物(H体)はほとんど生成せず、しかも、目的物が高収率で得られ、比較例に比べて優れていることが判る。また、比較例では、HMPAやジオキサン等の人体に有害な溶媒を使用するが、本発明では安全な溶媒を使用できるという点においても、本発明が優れていることが判る。
Claims (5)
- 下記一般式[1]で表される化合物とヨウ化アルカリ金属塩を、銅もしくは銅イオン及び下記一般式[2]で表される化合物の存在下で反応させることを特徴とするヨウ素化芳香族化合物の製造方法。

(式中、Arは芳香族炭化水素環基または芳香族複素環基を表す。Xは塩素原子または臭素原子を表し、nは1以上の整数を表す。R1、R2、R3及びR4はそれぞれ独立に、アルキル基またはアリール基を表し、R1とR3、R2とR4及びR3とR4は互いに結合して環を形成してもよい。) - 前記一般式[2]が下記一般式[3]で表されることを特徴とする請求項1に記載のヨウ素化芳香族化合物の製造方法。

(式中、R1及びR2はそれぞれ独立に、アルキル基またはアリール基を表す。) - 前記一般式[1]が下記一般式[4]で表されることを特徴とする請求項1または2に記載のヨウ素化芳香族化合物の製造方法。

(式中、Xは塩素原子または臭素原子を表し、Yは酸素原子、硫黄原子またはN-R5を表し、R5はアルキル基またはアリール基を表す。Wは置換基を表し、m及びnは1から4の整数を表す。) - 前記一般式[4]が下記一般式[5]で表されることを特徴とする請求項3に記載のヨウ素化芳香族化合物の製造方法。

(式中、Xは塩素原子または臭素原子を表し、Yは酸素原子、硫黄原子またはN-R5を表し、R5はアルキル基またはアリール基を表す。Wは置換基を表し、mは1から4の整数を表す。) - 前記一般式[5]において、Yが酸素原子であることを特徴とする請求項4に記載のヨウ素化芳香族化合物の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012504349A JP5598533B2 (ja) | 2010-03-08 | 2011-01-21 | ヨウ素化芳香族化合物の製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010050332 | 2010-03-08 | ||
| JP2010-050332 | 2010-03-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011111423A1 true WO2011111423A1 (ja) | 2011-09-15 |
Family
ID=44563252
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/051078 WO2011111423A1 (ja) | 2010-03-08 | 2011-01-21 | ヨウ素化芳香族化合物の製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP5598533B2 (ja) |
| WO (1) | WO2011111423A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012130709A1 (en) | 2011-03-25 | 2012-10-04 | Basf Se | 4h-imidazo[1,2-a]imidazoles for electronic applications |
| JP2019199457A (ja) * | 2018-05-18 | 2019-11-21 | 国立大学法人千葉大学 | フェナントリジン化合物の製造方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005037797A1 (en) * | 2003-10-21 | 2005-04-28 | Pharmacia Corporation | Substituted pyrazole urea compounds for the treatment of inflammation |
| JP2005306869A (ja) * | 2004-04-20 | 2005-11-04 | Xerox Corp | ヨード芳香族化合物を調製する方法及びヨード芳香族化合物の使用 |
| JP2005534700A (ja) * | 2002-08-02 | 2005-11-17 | マサチューセッツ インスティテュート オブ テクノロジー | 銅触媒による炭素−ヘテロ原子および炭素−炭素結合の形成 |
-
2011
- 2011-01-21 JP JP2012504349A patent/JP5598533B2/ja not_active Expired - Fee Related
- 2011-01-21 WO PCT/JP2011/051078 patent/WO2011111423A1/ja active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005534700A (ja) * | 2002-08-02 | 2005-11-17 | マサチューセッツ インスティテュート オブ テクノロジー | 銅触媒による炭素−ヘテロ原子および炭素−炭素結合の形成 |
| WO2005037797A1 (en) * | 2003-10-21 | 2005-04-28 | Pharmacia Corporation | Substituted pyrazole urea compounds for the treatment of inflammation |
| JP2005306869A (ja) * | 2004-04-20 | 2005-11-04 | Xerox Corp | ヨード芳香族化合物を調製する方法及びヨード芳香族化合物の使用 |
Non-Patent Citations (3)
| Title |
|---|
| ARTIS K. ET AL.: "Copper-Catalyzed Halogen Exchange in Aryl Halides: An Aromatic Finkelstein Reaction", J. AM. CHEM. SOC., vol. 124, 2002, pages 14844 - 14845, XP002337383, DOI: doi:10.1021/ja028865v * |
| HITOMI S. ET AL.: "Preparation of Aromatic Iodides from Bromides via The Reverse Halogen Exchange", CHEMISTRY LETTERS, 1985, pages 411 - 412 * |
| NALAN T. ET AL.: "Synthesis and structure- activity relationships of indole and benzimidazole piperazines as histamine H4 receptor antagonists", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 14, 2004, pages 5251 - 5256, XP004580509, DOI: doi:10.1016/j.bmcl.2004.08.035 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012130709A1 (en) | 2011-03-25 | 2012-10-04 | Basf Se | 4h-imidazo[1,2-a]imidazoles for electronic applications |
| EP3034508A1 (en) | 2011-03-25 | 2016-06-22 | Basf Se | 4h-imidazo[1,2-a]imidazoles for electronic applications |
| EP3640252A1 (en) | 2011-03-25 | 2020-04-22 | UDC Ireland Limited | 4h-imidazo[1,2-a]imidazoles for electronic applications |
| JP2019199457A (ja) * | 2018-05-18 | 2019-11-21 | 国立大学法人千葉大学 | フェナントリジン化合物の製造方法 |
| JP7061772B2 (ja) | 2018-05-18 | 2022-05-02 | 国立大学法人千葉大学 | フェナントリジン化合物の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2011111423A1 (ja) | 2013-06-27 |
| JP5598533B2 (ja) | 2014-10-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101676054B1 (ko) | 폴리(펜타플루오로설파닐)방향족 화합물을 제조하기 위한 공정 | |
| WO2009096202A1 (ja) | ハロ多環芳香族化合物及びその製造方法 | |
| JP2019214584A (ja) | ハロゲン化化合物およびフラーレン誘導体の製造方法 | |
| TW201813959A (zh) | 雙呋喃二鹵化物,用於製造雙呋喃二鹵化物的方法,及利用雙呋喃二鹵化物製造雙呋喃二酸、雙呋喃二醇或雙呋喃二胺的方法 | |
| US9580604B2 (en) | Pentafluorosulfanyl phthalocyanine derivatives and intermediates thereof | |
| JP5598533B2 (ja) | ヨウ素化芳香族化合物の製造方法 | |
| JP7049604B2 (ja) | ペンタフルオロスルファニル芳香族化合物の製造方法 | |
| JP2019081748A (ja) | ハロゲン化合物の製造方法 | |
| JP2009046408A (ja) | ジハロ多環芳香族化合物、ピロリル多環芳香族化合物、及びそれらの製造方法 | |
| JPWO2018025892A1 (ja) | 重合性トリプチセン誘導体化合物 | |
| JP5741091B2 (ja) | 含窒素縮合複素環化合物の製造方法 | |
| WO2017209297A1 (ja) | トリアリーレン化合物及びその製造方法 | |
| WO2018025554A1 (ja) | 含窒素複素環化合物の製造方法 | |
| JP5546872B2 (ja) | N,n,n’,n’−テトラアリールベンジジン誘導体の製造方法 | |
| JP6902275B2 (ja) | 芳香族複素多環式ハロゲン化合物の製造方法 | |
| JP2005502701A (ja) | 3−ブロモメチル安息香酸の製造方法 | |
| JP7220450B2 (ja) | ジスルフィドを触媒とする芳香族ヨウ素化合物の製造方法 | |
| JP2011148751A (ja) | 含窒素縮合複素環化合物の製造方法 | |
| JP5396997B2 (ja) | 含窒素縮合複素環化合物の製造方法 | |
| JP5900675B2 (ja) | ピロリル多環芳香族化合物の製造方法 | |
| JP5968214B2 (ja) | ジハロ多環芳香族化合物の製造方法 | |
| WO2023048244A1 (ja) | テトラフルオロスルファニル基含有アリール化合物の製造方法 | |
| KR20230105943A (ko) | 1,2,4-셀레나다이아졸 화합물의 제조방법 | |
| JP4153834B2 (ja) | 種々の置換基を有する1,1’−ビインデニリデン誘導体の新規合成方法 | |
| JP2004010599A (ja) | ハロゲン化縮環芳香族化合物の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11753085 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012504349 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11753085 Country of ref document: EP Kind code of ref document: A1 |