WO2008026602A1 - Dérivé hétéroacène, dérivé tétrahalotérphényle, et leurs procédés de production - Google Patents
Dérivé hétéroacène, dérivé tétrahalotérphényle, et leurs procédés de production Download PDFInfo
- Publication number
- WO2008026602A1 WO2008026602A1 PCT/JP2007/066684 JP2007066684W WO2008026602A1 WO 2008026602 A1 WO2008026602 A1 WO 2008026602A1 JP 2007066684 W JP2007066684 W JP 2007066684W WO 2008026602 A1 WO2008026602 A1 WO 2008026602A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- general formula
- represented
- group
- substituent
- derivative
- Prior art date
Links
- 238000004519 manufacturing process Methods 0.000 title claims description 33
- 125000001424 substituent group Chemical group 0.000 claims abstract description 96
- 239000000126 substance Substances 0.000 claims abstract description 54
- 239000004065 semiconductor Substances 0.000 claims abstract description 50
- 230000003647 oxidation Effects 0.000 claims abstract description 48
- 238000007254 oxidation reaction Methods 0.000 claims abstract description 48
- 239000000463 material Substances 0.000 claims abstract description 36
- 239000010409 thin film Substances 0.000 claims abstract description 31
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims abstract description 28
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 28
- 239000011593 sulfur Substances 0.000 claims abstract description 28
- 229910052796 boron Inorganic materials 0.000 claims abstract description 27
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 25
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 claims abstract description 24
- 125000001309 chloro group Chemical group Cl* 0.000 claims abstract description 24
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims abstract description 22
- 229910052698 phosphorus Inorganic materials 0.000 claims abstract description 22
- 229910052801 chlorine Inorganic materials 0.000 claims abstract description 21
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 21
- 239000001301 oxygen Substances 0.000 claims abstract description 21
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 claims abstract description 20
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 20
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 20
- 229910052711 selenium Inorganic materials 0.000 claims abstract description 20
- 239000011669 selenium Substances 0.000 claims abstract description 20
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 claims abstract description 17
- 239000011574 phosphorus Substances 0.000 claims abstract description 17
- 229910052714 tellurium Inorganic materials 0.000 claims abstract description 16
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical compound [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 claims abstract description 16
- 239000003153 chemical reaction reagent Substances 0.000 claims abstract description 14
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 13
- 229910052782 aluminium Inorganic materials 0.000 claims abstract description 11
- 125000003118 aryl group Chemical group 0.000 claims abstract description 11
- 229910052731 fluorine Inorganic materials 0.000 claims abstract description 11
- 125000001153 fluoro group Chemical group F* 0.000 claims abstract description 11
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 claims abstract description 10
- 238000006243 chemical reaction Methods 0.000 claims description 54
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 36
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 30
- 239000000758 substrate Substances 0.000 claims description 23
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 claims description 20
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 19
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 18
- 229910052740 iodine Inorganic materials 0.000 claims description 18
- 229910052763 palladium Inorganic materials 0.000 claims description 18
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 claims description 18
- 229910052751 metal Inorganic materials 0.000 claims description 15
- 239000002184 metal Substances 0.000 claims description 15
- 239000003054 catalyst Substances 0.000 claims description 12
- 239000000376 reactant Substances 0.000 claims description 12
- YJTKZCDBKVTVBY-UHFFFAOYSA-N 1,3-Diphenylbenzene Chemical group C1=CC=CC=C1C1=CC=CC(C=2C=CC=CC=2)=C1 YJTKZCDBKVTVBY-UHFFFAOYSA-N 0.000 claims description 9
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 8
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 claims description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 8
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 8
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 8
- 229910052799 carbon Inorganic materials 0.000 claims description 8
- 229910052710 silicon Inorganic materials 0.000 claims description 8
- 239000010703 silicon Substances 0.000 claims description 8
- 239000011135 tin Substances 0.000 claims description 8
- 229910052725 zinc Inorganic materials 0.000 claims description 8
- 239000011701 zinc Substances 0.000 claims description 8
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 claims description 7
- 150000004703 alkoxides Chemical class 0.000 claims description 7
- 150000004820 halides Chemical class 0.000 claims description 7
- 239000011777 magnesium Substances 0.000 claims description 7
- 229910052749 magnesium Inorganic materials 0.000 claims description 7
- 229910052718 tin Inorganic materials 0.000 claims description 7
- 125000004391 aryl sulfonyl group Chemical group 0.000 claims description 5
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 claims description 4
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 claims description 4
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 3
- 238000000576 coating method Methods 0.000 abstract description 26
- 238000006263 metalation reaction Methods 0.000 abstract description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical group CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 124
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 69
- 239000000243 solution Substances 0.000 description 65
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 54
- 238000003786 synthesis reaction Methods 0.000 description 43
- 230000015572 biosynthetic process Effects 0.000 description 42
- 239000002904 solvent Substances 0.000 description 35
- -1 p- (n-hexyl) phenyl group Chemical group 0.000 description 34
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 33
- 238000001819 mass spectrum Methods 0.000 description 33
- 239000007787 solid Substances 0.000 description 33
- 150000001875 compounds Chemical class 0.000 description 31
- 239000000203 mixture Substances 0.000 description 31
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 29
- 239000012074 organic phase Substances 0.000 description 27
- 239000012299 nitrogen atmosphere Substances 0.000 description 24
- 238000000034 method Methods 0.000 description 23
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 22
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 22
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 21
- 239000012071 phase Substances 0.000 description 20
- 229910052757 nitrogen Inorganic materials 0.000 description 18
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 18
- 238000003756 stirring Methods 0.000 description 17
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- 238000005259 measurement Methods 0.000 description 14
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 14
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 11
- 239000013078 crystal Substances 0.000 description 11
- 239000011248 coating agent Substances 0.000 description 10
- 238000004817 gas chromatography Methods 0.000 description 10
- WGOPGODQLGJZGL-UHFFFAOYSA-N lithium;butane Chemical compound [Li+].CC[CH-]C WGOPGODQLGJZGL-UHFFFAOYSA-N 0.000 description 10
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 125000001246 bromo group Chemical group Br* 0.000 description 9
- 238000001816 cooling Methods 0.000 description 9
- 125000003963 dichloro group Chemical group Cl* 0.000 description 9
- 239000002243 precursor Substances 0.000 description 9
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 8
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- 125000001624 naphthyl group Chemical group 0.000 description 8
- 239000000047 product Substances 0.000 description 8
- SLIUAWYAILUBJU-UHFFFAOYSA-N pentacene Chemical compound C1=CC=CC2=CC3=CC4=CC5=CC=CC=C5C=C4C=C3C=C21 SLIUAWYAILUBJU-UHFFFAOYSA-N 0.000 description 7
- 239000011592 zinc chloride Substances 0.000 description 7
- 235000005074 zinc chloride Nutrition 0.000 description 7
- RFFLAFLAYFXFSW-UHFFFAOYSA-N 1,2-dichlorobenzene Chemical compound ClC1=CC=CC=C1Cl RFFLAFLAYFXFSW-UHFFFAOYSA-N 0.000 description 6
- 238000005160 1H NMR spectroscopy Methods 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- 239000011521 glass Substances 0.000 description 6
- 229910052736 halogen Inorganic materials 0.000 description 6
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 6
- 229910052759 nickel Inorganic materials 0.000 description 6
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 6
- 239000011541 reaction mixture Substances 0.000 description 6
- 150000003839 salts Chemical class 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- UORVGPXVDQYIDP-UHFFFAOYSA-N trihydridoboron Substances B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 6
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Natural products P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 5
- 230000032683 aging Effects 0.000 description 5
- 239000012267 brine Substances 0.000 description 5
- 230000008859 change Effects 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 5
- 150000002367 halogens Chemical class 0.000 description 5
- 229910052744 lithium Inorganic materials 0.000 description 5
- LVKCSZQWLOVUGB-UHFFFAOYSA-M magnesium;propane;bromide Chemical compound [Mg+2].[Br-].C[CH-]C LVKCSZQWLOVUGB-UHFFFAOYSA-M 0.000 description 5
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 5
- 238000001953 recrystallisation Methods 0.000 description 5
- 238000000859 sublimation Methods 0.000 description 5
- 230000008022 sublimation Effects 0.000 description 5
- 239000006228 supernatant Substances 0.000 description 5
- OIAQMFOKAXHPNH-UHFFFAOYSA-N 1,2-diphenylbenzene Chemical group C1=CC=CC=C1C1=CC=CC=C1C1=CC=CC=C1 OIAQMFOKAXHPNH-UHFFFAOYSA-N 0.000 description 4
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 4
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 4
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical group CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 4
- 241000233855 Orchidaceae Species 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 229910052786 argon Inorganic materials 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 229910000085 borane Inorganic materials 0.000 description 4
- 229910052802 copper Inorganic materials 0.000 description 4
- 239000010949 copper Substances 0.000 description 4
- IMDXZWRLUZPMDH-UHFFFAOYSA-N dichlorophenylphosphine Chemical compound ClP(Cl)C1=CC=CC=C1 IMDXZWRLUZPMDH-UHFFFAOYSA-N 0.000 description 4
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 4
- 238000005191 phase separation Methods 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N silicon dioxide Inorganic materials O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 239000002002 slurry Substances 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- WJECKFZULSWXPN-UHFFFAOYSA-N 1,2-didodecylbenzene Chemical compound CCCCCCCCCCCCC1=CC=CC=C1CCCCCCCCCCCC WJECKFZULSWXPN-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- YVKOUANNKYWVHL-UHFFFAOYSA-N C1=CC=CC2=CC=CC=C12.[I] Chemical compound C1=CC=CC2=CC=CC=C12.[I] YVKOUANNKYWVHL-UHFFFAOYSA-N 0.000 description 3
- 239000005749 Copper compound Substances 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 3
- 150000008064 anhydrides Chemical class 0.000 description 3
- YQUSJUJNDKUWAM-UHFFFAOYSA-N benzenesulfonylsulfanylsulfonylbenzene Chemical compound C=1C=CC=CC=1S(=O)(=O)SS(=O)(=O)C1=CC=CC=C1 YQUSJUJNDKUWAM-UHFFFAOYSA-N 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 150000001880 copper compounds Chemical class 0.000 description 3
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 238000004992 fast atom bombardment mass spectroscopy Methods 0.000 description 3
- 230000008014 freezing Effects 0.000 description 3
- 238000007710 freezing Methods 0.000 description 3
- 150000004795 grignard reagents Chemical class 0.000 description 3
- 229910001623 magnesium bromide Inorganic materials 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 239000011368 organic material Substances 0.000 description 3
- UFCVADNIXDUEFZ-UHFFFAOYSA-N pentacene-6,13-dione Chemical compound C1=CC=C2C=C3C(=O)C4=CC5=CC=CC=C5C=C4C(=O)C3=CC2=C1 UFCVADNIXDUEFZ-UHFFFAOYSA-N 0.000 description 3
- 125000005007 perfluorooctyl group Chemical group FC(C(C(C(C(C(C(C(F)(F)F)(F)F)(F)F)(F)F)(F)F)(F)F)(F)F)(F)* 0.000 description 3
- 239000003444 phase transfer catalyst Substances 0.000 description 3
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 3
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000007639 printing Methods 0.000 description 3
- 239000002994 raw material Substances 0.000 description 3
- 230000035484 reaction time Effects 0.000 description 3
- 230000009257 reactivity Effects 0.000 description 3
- 229910052938 sodium sulfate Inorganic materials 0.000 description 3
- 235000011152 sodium sulphate Nutrition 0.000 description 3
- 238000010257 thawing Methods 0.000 description 3
- 150000003568 thioethers Chemical class 0.000 description 3
- 238000010792 warming Methods 0.000 description 3
- 239000008096 xylene Substances 0.000 description 3
- QPFMBZIOSGYJDE-UHFFFAOYSA-N 1,1,2,2-tetrachloroethane Chemical compound ClC(Cl)C(Cl)Cl QPFMBZIOSGYJDE-UHFFFAOYSA-N 0.000 description 2
- ONUFSRWQCKNVSL-UHFFFAOYSA-N 1,2,3,4,5-pentafluoro-6-(2,3,4,5,6-pentafluorophenyl)benzene Chemical group FC1=C(F)C(F)=C(F)C(F)=C1C1=C(F)C(F)=C(F)C(F)=C1F ONUFSRWQCKNVSL-UHFFFAOYSA-N 0.000 description 2
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 2
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 2
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- JPVYNHNXODAKFH-UHFFFAOYSA-N Cu2+ Chemical compound [Cu+2] JPVYNHNXODAKFH-UHFFFAOYSA-N 0.000 description 2
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 239000007818 Grignard reagent Substances 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- WZHAVYGMKGPNFN-UHFFFAOYSA-M [Br-].FC1=CC=C([Mg+])C=C1F Chemical compound [Br-].FC1=CC=C([Mg+])C=C1F WZHAVYGMKGPNFN-UHFFFAOYSA-M 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 2
- 125000006267 biphenyl group Chemical group 0.000 description 2
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 2
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 2
- 239000002178 crystalline material Substances 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 230000006837 decompression Effects 0.000 description 2
- CDHICTNQMQYRSM-UHFFFAOYSA-N di(propan-2-yl)alumane Chemical compound CC(C)[AlH]C(C)C CDHICTNQMQYRSM-UHFFFAOYSA-N 0.000 description 2
- 125000000950 dibromo group Chemical group Br* 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- 125000001891 dimethoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 239000012776 electronic material Substances 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 229910052737 gold Inorganic materials 0.000 description 2
- 239000010931 gold Substances 0.000 description 2
- 239000011261 inert gas Substances 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 2
- OTCKOJUMXQWKQG-UHFFFAOYSA-L magnesium bromide Chemical compound [Mg+2].[Br-].[Br-] OTCKOJUMXQWKQG-UHFFFAOYSA-L 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000012046 mixed solvent Substances 0.000 description 2
- LDZCNNKHPNYYSX-UHFFFAOYSA-N n,n,2-triphenylaniline Chemical group C1=CC=CC=C1N(C=1C(=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 LDZCNNKHPNYYSX-UHFFFAOYSA-N 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000001979 organolithium group Chemical group 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 125000000538 pentafluorophenyl group Chemical group FC1=C(F)C(F)=C(*)C(F)=C1F 0.000 description 2
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 2
- 150000002978 peroxides Chemical class 0.000 description 2
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 2
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 2
- 150000003003 phosphines Chemical class 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 2
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 150000003216 pyrazines Chemical group 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- ZGNPLWZYVAFUNZ-UHFFFAOYSA-N tert-butylphosphane Chemical compound CC(C)(C)P ZGNPLWZYVAFUNZ-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- GNLJBJNONOOOQC-UHFFFAOYSA-N $l^{3}-carbane;magnesium Chemical compound [Mg]C GNLJBJNONOOOQC-UHFFFAOYSA-N 0.000 description 1
- CXWYTOFTBOZXGQ-UHFFFAOYSA-N (1,2-dichloro-2-diphenylphosphanylethyl)-diphenylphosphane;nickel Chemical compound [Ni].C=1C=CC=CC=1P(C=1C=CC=CC=1)C(Cl)C(Cl)P(C=1C=CC=CC=1)C1=CC=CC=C1 CXWYTOFTBOZXGQ-UHFFFAOYSA-N 0.000 description 1
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- LHLHWDNRHALZNF-BQYQJAHWSA-N (e)-2,3-di(propan-2-yl)but-2-enedioic acid Chemical compound CC(C)C(\C(O)=O)=C(\C(C)C)C(O)=O LHLHWDNRHALZNF-BQYQJAHWSA-N 0.000 description 1
- LHLHWDNRHALZNF-FPLPWBNLSA-N (z)-2,3-di(propan-2-yl)but-2-enedioic acid Chemical compound CC(C)C(\C(O)=O)=C(/C(C)C)C(O)=O LHLHWDNRHALZNF-FPLPWBNLSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- PKDUEAKQNWATCJ-UHFFFAOYSA-N 1,2-bis(2-ethylhexyl)naphthalene Chemical group C1=CC=CC2=C(CC(CC)CCCC)C(CC(CC)CCCC)=CC=C21 PKDUEAKQNWATCJ-UHFFFAOYSA-N 0.000 description 1
- WQONPSCCEXUXTQ-UHFFFAOYSA-N 1,2-dibromobenzene Chemical compound BrC1=CC=CC=C1Br WQONPSCCEXUXTQ-UHFFFAOYSA-N 0.000 description 1
- GJDURYMLURTLIK-UHFFFAOYSA-N 1,2-didodecylanthracene-9,10-dione Chemical compound C(CCCCCCCCCCC)C1=C(C=2C(C3=CC=CC=C3C(C2C=C1)=O)=O)CCCCCCCCCCCC GJDURYMLURTLIK-UHFFFAOYSA-N 0.000 description 1
- GOYDNIKZWGIXJT-UHFFFAOYSA-N 1,2-difluorobenzene Chemical compound FC1=CC=CC=C1F GOYDNIKZWGIXJT-UHFFFAOYSA-N 0.000 description 1
- VOMPMOXLDNRRRY-UHFFFAOYSA-N 1,2-dihexylnaphthalene Chemical group C1=CC=CC2=C(CCCCCC)C(CCCCCC)=CC=C21 VOMPMOXLDNRRRY-UHFFFAOYSA-N 0.000 description 1
- QNLZIZAQLLYXTC-UHFFFAOYSA-N 1,2-dimethylnaphthalene Chemical group C1=CC=CC2=C(C)C(C)=CC=C21 QNLZIZAQLLYXTC-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- SWJPEBQEEAHIGZ-UHFFFAOYSA-N 1,4-dibromobenzene Chemical compound BrC1=CC=C(Br)C=C1 SWJPEBQEEAHIGZ-UHFFFAOYSA-N 0.000 description 1
- UKJZVGGDYYQVOT-UHFFFAOYSA-N 1-dodecylanthracene Chemical compound C1=CC=C2C=C3C(CCCCCCCCCCCC)=CC=CC3=CC2=C1 UKJZVGGDYYQVOT-UHFFFAOYSA-N 0.000 description 1
- QPUYECUOLPXSFR-UHFFFAOYSA-N 1-methylnaphthalene Chemical group C1=CC=C2C(C)=CC=CC2=C1 QPUYECUOLPXSFR-UHFFFAOYSA-N 0.000 description 1
- IYDMICQAKLQHLA-UHFFFAOYSA-N 1-phenylnaphthalene Chemical group C1=CC=CC=C1C1=CC=CC2=CC=CC=C12 IYDMICQAKLQHLA-UHFFFAOYSA-N 0.000 description 1
- TWZSIAFEFBKCNN-UHFFFAOYSA-N 2,3-dibromo-1-benzothiophene Chemical compound C1=CC=C2C(Br)=C(Br)SC2=C1 TWZSIAFEFBKCNN-UHFFFAOYSA-N 0.000 description 1
- IQHSSYROJYPFDV-UHFFFAOYSA-N 2-bromo-1,3-dichloro-5-(trifluoromethyl)benzene Chemical group FC(F)(F)C1=CC(Cl)=C(Br)C(Cl)=C1 IQHSSYROJYPFDV-UHFFFAOYSA-N 0.000 description 1
- BEJFSZRYERIHNZ-UHFFFAOYSA-N 2-bromo-6,7-didodecyl-3-iodoanthracene-9,10-dione Chemical compound O=C1C2=CC(Br)=C(I)C=C2C(=O)C2=C1C=C(CCCCCCCCCCCC)C(CCCCCCCCCCCC)=C2 BEJFSZRYERIHNZ-UHFFFAOYSA-N 0.000 description 1
- LDJXFZUGZASGIW-UHFFFAOYSA-L 2-diphenylphosphanylethyl(diphenyl)phosphane;palladium(2+);dichloride Chemical compound Cl[Pd]Cl.C=1C=CC=CC=1P(C=1C=CC=CC=1)CCP(C=1C=CC=CC=1)C1=CC=CC=C1 LDJXFZUGZASGIW-UHFFFAOYSA-L 0.000 description 1
- ABMULKFGWTYIIK-UHFFFAOYSA-N 2-hexylphenol Chemical compound CCCCCCC1=CC=CC=C1O ABMULKFGWTYIIK-UHFFFAOYSA-N 0.000 description 1
- LZQRFVUKPVNFJO-UHFFFAOYSA-N 2-hexylpyrazine Chemical group CCCCCCC1=CN=CC=N1 LZQRFVUKPVNFJO-UHFFFAOYSA-N 0.000 description 1
- NZLJDTKLZIMONR-UHFFFAOYSA-N 2-hexylpyridine Chemical group CCCCCCC1=CC=CC=N1 NZLJDTKLZIMONR-UHFFFAOYSA-N 0.000 description 1
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical group CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 1
- VQGHOUODWALEFC-UHFFFAOYSA-N 2-phenylpyridine Chemical group C1=CC=CC=C1C1=CC=CC=N1 VQGHOUODWALEFC-UHFFFAOYSA-N 0.000 description 1
- OBAQODNMTCJDSX-UHFFFAOYSA-N 3,4-dichloro-2-phenylphenol Chemical compound OC1=CC=C(Cl)C(Cl)=C1C1=CC=CC=C1 OBAQODNMTCJDSX-UHFFFAOYSA-N 0.000 description 1
- BCKVHOUUJMYIAN-UHFFFAOYSA-N 5-bromo-2-benzofuran-1,3-dione Chemical compound BrC1=CC=C2C(=O)OC(=O)C2=C1 BCKVHOUUJMYIAN-UHFFFAOYSA-N 0.000 description 1
- PSYIWEUNIZFSNC-UHFFFAOYSA-N 5-bromo-6-iodo-2-benzofuran-1,3-dione Chemical compound C1=C(I)C(Br)=CC2=C1C(=O)OC2=O PSYIWEUNIZFSNC-UHFFFAOYSA-N 0.000 description 1
- NUUCWIXGBYNWQZ-UHFFFAOYSA-N 9,10-didodecylanthracene Chemical compound C1=CC=C2C(CCCCCCCCCCCC)=C(C=CC=C3)C3=C(CCCCCCCCCCCC)C2=C1 NUUCWIXGBYNWQZ-UHFFFAOYSA-N 0.000 description 1
- 229920003026 Acene Polymers 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 101100346155 Caenorhabditis elegans oma-2 gene Proteins 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- HGXHHGLLQBGDPK-UHFFFAOYSA-N ClC1(C(=C(C(=C1Cl)Cl)Cl)Cl)Cl.C1=CC=CC2=CC=CC=C12 Chemical compound ClC1(C(=C(C(=C1Cl)Cl)Cl)Cl)Cl.C1=CC=CC2=CC=CC=C12 HGXHHGLLQBGDPK-UHFFFAOYSA-N 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical group COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- LTEQMZWBSYACLV-UHFFFAOYSA-N Hexylbenzene Chemical group CCCCCCC1=CC=CC=C1 LTEQMZWBSYACLV-UHFFFAOYSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- VAYOSLLFUXYJDT-RDTXWAMCSA-N Lysergic acid diethylamide Chemical compound C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N(CC)CC)C2)=C3C2=CNC3=C1 VAYOSLLFUXYJDT-RDTXWAMCSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000004642 Polyimide Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- QMELDNSRILFMRS-UHFFFAOYSA-N S.Br.Br Chemical compound S.Br.Br QMELDNSRILFMRS-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 241000705989 Tetrax Species 0.000 description 1
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical group C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- 239000005858 Triflumizole Substances 0.000 description 1
- 229910052770 Uranium Inorganic materials 0.000 description 1
- ZWVMFEHQWMPYMM-UHFFFAOYSA-N [Ni].ClNCCNCl Chemical compound [Ni].ClNCCNCl ZWVMFEHQWMPYMM-UHFFFAOYSA-N 0.000 description 1
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 125000000746 allylic group Chemical group 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229910021417 amorphous silicon Inorganic materials 0.000 description 1
- 125000005577 anthracene group Chemical group 0.000 description 1
- PYKYMHQGRFAEBM-UHFFFAOYSA-N anthraquinone Natural products CCC(=O)c1c(O)c2C(=O)C3C(C=CC=C3O)C(=O)c2cc1CC(=O)OC PYKYMHQGRFAEBM-UHFFFAOYSA-N 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- ODWXUNBKCRECNW-UHFFFAOYSA-M bromocopper(1+) Chemical compound Br[Cu+] ODWXUNBKCRECNW-UHFFFAOYSA-M 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- YMKDRGPMQRFJGP-UHFFFAOYSA-M cetylpyridinium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCC[N+]1=CC=CC=C1 YMKDRGPMQRFJGP-UHFFFAOYSA-M 0.000 description 1
- 229960001927 cetylpyridinium chloride Drugs 0.000 description 1
- 239000002800 charge carrier Substances 0.000 description 1
- SWAKCLHCWHYEOW-UHFFFAOYSA-N chloro selenohypochlorite Chemical compound Cl[Se]Cl SWAKCLHCWHYEOW-UHFFFAOYSA-N 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 239000011651 chromium Substances 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- AQEFLFZSWDEAIP-UHFFFAOYSA-N di-tert-butyl ether Chemical compound CC(C)(C)OC(C)(C)C AQEFLFZSWDEAIP-UHFFFAOYSA-N 0.000 description 1
- FXNXUCCZCNRKFM-UHFFFAOYSA-N dichloro(hexyl)borane Chemical compound CCCCCCB(Cl)Cl FXNXUCCZCNRKFM-UHFFFAOYSA-N 0.000 description 1
- NCQDQONETMHUMY-UHFFFAOYSA-N dichloro(phenyl)borane Chemical compound ClB(Cl)C1=CC=CC=C1 NCQDQONETMHUMY-UHFFFAOYSA-N 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- LMZLQYYLELWCCW-UHFFFAOYSA-N dimethoxy(phenyl)phosphane Chemical compound COP(OC)C1=CC=CC=C1 LMZLQYYLELWCCW-UHFFFAOYSA-N 0.000 description 1
- NQKXFODBPINZFK-UHFFFAOYSA-N dioxotantalum Chemical compound O=[Ta]=O NQKXFODBPINZFK-UHFFFAOYSA-N 0.000 description 1
- 238000003618 dip coating Methods 0.000 description 1
- 239000012769 display material Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- PXJJSXABGXMUSU-UHFFFAOYSA-N disulfur dichloride Chemical compound ClSSCl PXJJSXABGXMUSU-UHFFFAOYSA-N 0.000 description 1
- VHJLVAABSRFDPM-ZXZARUISSA-N dithioerythritol Chemical compound SC[C@H](O)[C@H](O)CS VHJLVAABSRFDPM-ZXZARUISSA-N 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- 239000003759 ester based solvent Substances 0.000 description 1
- 239000004210 ether based solvent Substances 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 238000007646 gravure printing Methods 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- YCSJJCRCOUGEPI-UHFFFAOYSA-N hexyl(dimethoxy)alumane Chemical compound CCCCCC[Al](OC)OC YCSJJCRCOUGEPI-UHFFFAOYSA-N 0.000 description 1
- NEGKMWMOGLVJDA-UHFFFAOYSA-N hexyl(dimethoxy)borane Chemical compound CCCCCCB(OC)OC NEGKMWMOGLVJDA-UHFFFAOYSA-N 0.000 description 1
- MWWVTUYEKHQJHW-UHFFFAOYSA-N hexyl(dimethoxy)phosphane Chemical compound CCCCCCP(OC)OC MWWVTUYEKHQJHW-UHFFFAOYSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- AMGQUBHHOARCQH-UHFFFAOYSA-N indium;oxotin Chemical compound [In].[Sn]=O AMGQUBHHOARCQH-UHFFFAOYSA-N 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 238000007641 inkjet printing Methods 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 239000011147 inorganic material Substances 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 238000003475 lamination Methods 0.000 description 1
- 239000004973 liquid crystal related substance Substances 0.000 description 1
- AFRJJFRNGGLMDW-UHFFFAOYSA-N lithium amide Chemical compound [Li+].[NH2-] AFRJJFRNGGLMDW-UHFFFAOYSA-N 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 1
- CETVQRFGPOGIQJ-UHFFFAOYSA-N lithium;hexane Chemical compound [Li+].CCCCC[CH2-] CETVQRFGPOGIQJ-UHFFFAOYSA-N 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- CWTPEXDGZPTZSH-UHFFFAOYSA-M magnesium;decane;bromide Chemical compound [Mg+2].[Br-].CCCCCCCCC[CH2-] CWTPEXDGZPTZSH-UHFFFAOYSA-M 0.000 description 1
- MBHQEXPGNCWWBP-UHFFFAOYSA-M magnesium;dodecane;bromide Chemical compound [Mg+2].[Br-].CCCCCCCCCCC[CH2-] MBHQEXPGNCWWBP-UHFFFAOYSA-M 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- AUHZEENZYGFFBQ-UHFFFAOYSA-N mesitylene Substances CC1=CC(C)=CC(C)=C1 AUHZEENZYGFFBQ-UHFFFAOYSA-N 0.000 description 1
- 125000001827 mesitylenyl group Chemical group [H]C1=C(C(*)=C(C([H])=C1C([H])([H])[H])C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- BZLVMXJERCGZMT-UHFFFAOYSA-N methyl tert-butyl ether Substances COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 1
- DVSDBMFJEQPWNO-UHFFFAOYSA-N methyllithium Chemical compound C[Li] DVSDBMFJEQPWNO-UHFFFAOYSA-N 0.000 description 1
- XKBGEWXEAPTVCK-UHFFFAOYSA-M methyltrioctylammonium chloride Chemical compound [Cl-].CCCCCCCC[N+](C)(CCCCCCCC)CCCCCCCC XKBGEWXEAPTVCK-UHFFFAOYSA-M 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- SGNSAMGUMVFALJ-UHFFFAOYSA-N n,n'-dichloroethane-1,2-diamine Chemical compound ClNCCNCl SGNSAMGUMVFALJ-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- QCYXGORGJYUYMT-UHFFFAOYSA-N nickel;triphenylphosphane Chemical compound [Ni].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 QCYXGORGJYUYMT-UHFFFAOYSA-N 0.000 description 1
- RBLGTYCOUOIUNY-UHFFFAOYSA-L octylaluminum(2+);dichloride Chemical compound CCCCCCCC[Al](Cl)Cl RBLGTYCOUOIUNY-UHFFFAOYSA-L 0.000 description 1
- CDKDZKXSXLNROY-UHFFFAOYSA-N octylbenzene Chemical group CCCCCCCCC1=CC=CC=C1 CDKDZKXSXLNROY-UHFFFAOYSA-N 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 125000002524 organometallic group Chemical group 0.000 description 1
- TWLXDPFBEPBAQB-UHFFFAOYSA-N orthoperiodic acid Chemical compound OI(O)(O)(O)(O)=O TWLXDPFBEPBAQB-UHFFFAOYSA-N 0.000 description 1
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 1
- BPUBBGLMJRNUCC-UHFFFAOYSA-N oxygen(2-);tantalum(5+) Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[Ta+5].[Ta+5] BPUBBGLMJRNUCC-UHFFFAOYSA-N 0.000 description 1
- 150000002941 palladium compounds Chemical class 0.000 description 1
- 125000005459 perfluorocyclohexyl group Chemical group 0.000 description 1
- 125000005005 perfluorohexyl group Chemical group FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)* 0.000 description 1
- BPQPJXCUBLCZIB-UHFFFAOYSA-L phenylaluminum(2+);dichloride Chemical compound [Cl-].[Cl-].[Al+2]C1=CC=CC=C1 BPQPJXCUBLCZIB-UHFFFAOYSA-L 0.000 description 1
- NHKJPPKXDNZFBJ-UHFFFAOYSA-N phenyllithium Chemical compound [Li]C1=CC=CC=C1 NHKJPPKXDNZFBJ-UHFFFAOYSA-N 0.000 description 1
- 239000004038 photonic crystal Substances 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229920000301 poly(3-hexylthiophene-2,5-diyl) polymer Polymers 0.000 description 1
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920000139 polyethylene terephthalate Polymers 0.000 description 1
- 239000005020 polyethylene terephthalate Substances 0.000 description 1
- 229920001721 polyimide Polymers 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 229920000098 polyolefin Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 239000011698 potassium fluoride Substances 0.000 description 1
- 235000003270 potassium fluoride Nutrition 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 229910052705 radium Inorganic materials 0.000 description 1
- HCWPIIXVSYCSAN-UHFFFAOYSA-N radium atom Chemical compound [Ra] HCWPIIXVSYCSAN-UHFFFAOYSA-N 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 238000007650 screen-printing Methods 0.000 description 1
- 150000004756 silanes Chemical group 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 229910052814 silicon oxide Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 238000004528 spin coating Methods 0.000 description 1
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- HIFJUMGIHIZEPX-UHFFFAOYSA-N sulfuric acid;sulfur trioxide Chemical compound O=S(=O)=O.OS(O)(=O)=O HIFJUMGIHIZEPX-UHFFFAOYSA-N 0.000 description 1
- 229910052715 tantalum Inorganic materials 0.000 description 1
- GUVRBAGPIYLISA-UHFFFAOYSA-N tantalum atom Chemical compound [Ta] GUVRBAGPIYLISA-UHFFFAOYSA-N 0.000 description 1
- PBCFLUZVCVVTBY-UHFFFAOYSA-N tantalum pentoxide Inorganic materials O=[Ta](=O)O[Ta](=O)=O PBCFLUZVCVVTBY-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 125000005628 tolylene group Chemical group 0.000 description 1
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- PYJJCSYBSYXGQQ-UHFFFAOYSA-N trichloro(octadecyl)silane Chemical compound CCCCCCCCCCCCCCCCCC[Si](Cl)(Cl)Cl PYJJCSYBSYXGQQ-UHFFFAOYSA-N 0.000 description 1
- 150000008648 triflates Chemical class 0.000 description 1
- HSMVPDGQOIQYSR-KGENOOAVSA-N triflumizole Chemical compound C1=CN=CN1C(/COCCC)=N/C1=CC=C(Cl)C=C1C(F)(F)F HSMVPDGQOIQYSR-KGENOOAVSA-N 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- WRECIMRULFAWHA-UHFFFAOYSA-N trimethyl borate Chemical compound COB(OC)OC WRECIMRULFAWHA-UHFFFAOYSA-N 0.000 description 1
- 125000005580 triphenylene group Chemical group 0.000 description 1
- NHDIQVFFNDKAQU-UHFFFAOYSA-N tripropan-2-yl borate Chemical compound CC(C)OB(OC(C)C)OC(C)C NHDIQVFFNDKAQU-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 238000001771 vacuum deposition Methods 0.000 description 1
- 230000008016 vaporization Effects 0.000 description 1
- 229910052724 xenon Inorganic materials 0.000 description 1
- FHNFHKCVQCLJFQ-UHFFFAOYSA-N xenon atom Chemical compound [Xe] FHNFHKCVQCLJFQ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6564—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms
- C07F9/6568—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus atoms as the only ring hetero atoms
- C07F9/65683—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having phosphorus atoms, with or without nitrogen, oxygen, sulfur, selenium or tellurium atoms, as ring hetero atoms having phosphorus atoms as the only ring hetero atoms the ring phosphorus atom being part of a phosphine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D329/00—Heterocyclic compounds containing rings having oxygen and selenium or oxygen and tellurium atoms as the only ring hetero atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/093—Preparation of halogenated hydrocarbons by replacement by halogens
- C07C17/10—Preparation of halogenated hydrocarbons by replacement by halogens of hydrogen atoms
- C07C17/12—Preparation of halogenated hydrocarbons by replacement by halogens of hydrogen atoms in the ring of aromatic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/26—Preparation of halogenated hydrocarbons by reactions involving an increase in the number of carbon atoms in the skeleton
- C07C17/263—Preparation of halogenated hydrocarbons by reactions involving an increase in the number of carbon atoms in the skeleton by condensation reactions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/26—Preparation of halogenated hydrocarbons by reactions involving an increase in the number of carbon atoms in the skeleton
- C07C17/263—Preparation of halogenated hydrocarbons by reactions involving an increase in the number of carbon atoms in the skeleton by condensation reactions
- C07C17/2632—Preparation of halogenated hydrocarbons by reactions involving an increase in the number of carbon atoms in the skeleton by condensation reactions involving an organo-magnesium compound, e.g. Grignard synthesis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/35—Preparation of halogenated hydrocarbons by reactions not affecting the number of carbon or of halogen atoms in the reaction
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/361—Preparation of halogenated hydrocarbons by reactions involving a decrease in the number of carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C25/00—Compounds containing at least one halogen atom bound to a six-membered aromatic ring
- C07C25/18—Polycyclic aromatic halogenated hydrocarbons
- C07C25/22—Polycyclic aromatic halogenated hydrocarbons with condensed rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C46/00—Preparation of quinones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/54—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/62—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D495/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/22—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains four or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D517/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having selenium, tellurium, or halogen atoms as ring hetero atoms
- C07D517/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having selenium, tellurium, or halogen atoms as ring hetero atoms in which the condensed system contains two hetero rings
- C07D517/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/027—Organoboranes and organoborohydrides
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/657—Polycyclic condensed heteroaromatic hydrocarbons
- H10K85/6576—Polycyclic condensed heteroaromatic hydrocarbons comprising only sulfur in the heteroaromatic polycondensed ring system, e.g. benzothiophene
Definitions
- the present invention relates to a heterocene derivative that can be developed into an electronic material such as an organic semiconductor, its use, and a production method thereof. Furthermore, the present invention relates to a tetrahaloterphenyl derivative that is a precursor compound of the heterocene derivative and a method for producing the same.
- Organic semiconductor devices typified by organic thin-film transistors have recently attracted attention because they have features not found in inorganic semiconductor devices such as energy saving, low cost, and flexibility.
- This organic semiconductor device is composed of several kinds of materials such as an organic semiconductor active phase, a substrate, an insulating phase, and an electrode.
- the organic semiconductor active phase responsible for charge carrier movement has a central role of the device.
- the organic semiconductor device performance is affected by the carrier mobility of the organic material constituting the organic semiconductor active phase.
- the organic semiconductor active phase is produced by vaporizing the organic material under a high temperature vacuum. A vacuum deposition method and a coating method in which an organic material is dissolved in an appropriate solvent and the solution is applied are known.
- the coating can be performed using a printing technique without using high-temperature and high-vacuum conditions. Therefore, the coating method is an economically preferable process because printing can greatly reduce the manufacturing cost of manufacturing the device.
- conventionally there has been a problem that it becomes difficult to form an organic semiconductor active phase by a coating method as a material having higher performance as an organic semiconductor device.
- Pentacenoacene with a condensed thiophene ring has improved oxidation resistance compared to pentacene! /, Has low carrier mobility! / And requires multiple steps for its synthesis ( For example, see Non-Patent Document 3) Practically preferred! / Not a material.
- Non-Patent Document 1 “Journal of Applied Physics” (USA), 2002, 92, 5259-5263
- Patent Document 1 WO2003 / 016599
- the present invention has excellent oxidation resistance in view of the problems of the above-described prior art.
- Another object of the present invention is to provide a heterocacene derivative capable of forming an organic semiconductor active phase by a coating method, an oxidation-resistant organic semiconductor material using the same, and an organic thin film.
- Another object of the present invention is to provide a tetrahaloterphenyl derivative useful as a precursor of the heterocene derivative and a method for producing the same.
- the present inventors have found a novel heterocene derivative of the present invention.
- the heterocene derivative is excellent in oxidation resistance and can be applied by a coating method, a crystalline thin film can be easily and stably produced. Therefore, the oxidation resistant organic semiconductor containing the heterocene derivative can be used.
- the present inventors have found a material and its organic thin film and have completed the present invention.
- the present inventors have found a novel precursor compound capable of efficiently producing the heteroacene derivative, that is, a specific tetrahaloterphenyl derivative, and a method for efficiently producing such a tetrahaloterphenyl derivative. The present invention has been completed.
- the present invention has the following configuration.
- T 1 and T 2 are the same or different and represent sulfur, selenium, tellurium, oxygen, phosphorus, boron, aluminum, and 1 and m are each an integer of 0 or 1
- rings A and B are the same or different and have a structure represented by the following general formula (A-1) or (A-2).
- substituents R 5 to R U are the same or different and are a hydrogen atom, a fluorine atom, a chlorine atom, an aryl group having 4 to 30 carbon atoms, an alkyl group having 3 to 20 carbon atoms, a carbon number of 1 shows the 20 halogenated alkyl group.
- substituents R 5 to R 6 and R 8 to R U are each, any two or more substituents are bonded to each other in each substituent group, the substituent Can form a benzene ring, an optionally substituted pyridine ring, and an optionally substituted pyrazine ring.
- T 3 represents sulfur, selenium, tellurium, oxygen, phosphorus, boron, n is an integer of 0 or 1, provided that when T 1 and T 2 are sulfur, rings A and B are (A- 1) or a ring represented by (A-2) having a substituent.
- heteroacene derivative according to any one of 1 to 3 above, wherein n is 0 and T 3 is sulfur, selenium, tellurium or oxygen.
- substituents xi to X 4 represent a bromine atom, an iodine atom, or a chlorine atom
- substituents R 1 and R 2 and rings A and B are represented by the general formula (1) described in 1 above. And the same meaning as the substituent and ring.
- rings A and B are a ring represented by (A-1).
- the tetrahaloterphenyl derivative represented by the general formula (2) described in any one of 5 to 7 above is tetrametalated using a metallizing agent, and the following general formula (3) and the following general formula (4) 5.
- M 1 represents a halide of magnesium, boron, zinc, tin, or silicon; no, idoxide; alkoxide; an alkylate; and the substituent X 1 and ring A are the same as those described in 8 above. It has the same meaning as the substituent and ring represented by formula (2).
- M 2 represents a halide of magnesium, boron, zinc, tin, or silicon; a nodoxide; an alkoxide; an alkylated product, and the substituent X 4 and the B ring are represented by the general formula (2 ) The same meaning as the substituent and ring shown by).
- X 5 and X 6 are iodine atoms, and X 2 and X 3 are bromine atoms and / or chlorine atoms.
- An oxidation-resistant organic semiconductor material comprising the heteroacene derivative described in any one of 1 to 4 above.
- the present invention provides a heterocene derivative having excellent oxidation resistance and capable of forming an organic semiconductor active phase by a coating method and use thereof. Furthermore, a tetrahaloterphenyl derivative that is a precursor compound of the heterocene derivative and a method for producing the same are also provided.
- the present invention is described in detail below.
- the description describes a heterophacene derivative and a production method thereof, a tetrahaloterphenyl derivative that is a precursor of the heterocacene derivative, a production method thereof, an oxidation-resistant organic semiconductor material containing the heterophacene derivative, and an organic thin film thereof.
- the heteroacene derivative of the present invention is represented by the following general formula (1).
- substituents I ⁇ to R 4 are the same or different and are a hydrogen atom, a fluorine atom, a chlorine atom, an aryl group having 4 to 30 carbon atoms, an alkyl group having 3 to 20 carbon atoms, or a carbon number.
- 1 to 20 halogenated alkyl groups, T 1 and T 2 are the same or different and represent sulfur, selenium, tellurium, oxygen, phosphorus, boron, aluminum, and 1 and m are each an integer of 0 or 1
- rings A and B are the same or different and have a structure represented by the following general formula (A-1) or (A-2).
- T 3 represents sulfur, selenium, tellurium, oxygen, phosphorus, boron, n is an integer of 0 or 1, provided that when T 1 and T 2 are sulfur, rings A and B are (A- 1) or a ring represented by (A-2) having a substituent.
- the aryl group having 4 to 30 carbon atoms in the substituents Ri to R 4 is not particularly limited, for example, a phenyl group, a p trinole group, a p- (n-hexyl) phenyl group, a p- (n octinore) phenyl group, p — (Cyclohexenole) phenyl group, m— (n-octinole) phenyl group, p-fluorophenenole group, pentafluorophenyl group, p (trifluoromethyl) phenyl group, p— (n-fluorooctyl) ) Phenyl group, 2 Chenyl group, 5— (n-hexyl) -2 Che Ninole group, 2, 2, 1 Biphenyl 2 group, Biphenyl group, Perfluorobiphenyl group, 1 Naphthyl group, 2-naphthyl
- halogenated alkyl group having 1 to 20 carbon atoms in the substituent I ⁇ to R 4 is not particularly limited, for example triflates Ruo Russia methyl, triflumizole Ruo Roe methyl group, per full O Roo Chi group, par Furuorododeshiru Group, perfluorooctadecyl group, perfluorocyclohexyl group, Perfluoroalkyl groups such as perfluorocyclooctyl group; or halogenated alkyl groups in which part of hydrogen such as pentadecafluorooctyl group and octadecafluorodecyl group is substituted with fluorine. And preferably a perfluoroalkyl group, particularly preferably a perfluorooctyl group or a perfluorododecyl group.
- the substituents T 1 and T 2 are sulfur, selenium, tellurium, oxygen, phosphorus, boron, and aluminum. Among them, sulfur, selenium, phosphorus, and boron are preferable, and sulfur, phosphorus, and boron are more preferable. Further, when T 1 and T 2 are sulfur, it is preferable that the rings A and B are (A-1) or a ring represented by (A-2) having a substituent! /.
- 1 and m are each an integer of 0 or 1. However, 1 and m are 0 when the substituents 1 and T 2 are sulfur, selenium, tenol, and oxygen, and 1 when the substituents 1 and T 2 are phosphorus, boron, and aluminum. , M is 1.
- rings A and B represented by general formulas (A-1) and (A-2) will be described.
- the heteroacene derivative of the present invention is a derivative having rings A and B, and rings A and B have a structure represented by the general formula (A-1) or (A-2).
- the aryl group having 4 to 30 carbon atoms in the substituents R 5 to R U is not particularly limited, for example, a pheninore group, a p trinole group, a p- (n-hexyl) phenyl group, a p- (n octinore) phenyl group, p— (Cyclohexenole) phenyl group, m— (n-octinole) phenyl group, p-fluorophenenole group, pentafluorophenyl group, p (trifluoromethyl) phenyl group, p— (n-nofluoro Octyl) phenyl group, 2 chenyl group, 5— (n-hexyl) -2 che ninole group, 2, 2, 1 biphenyl bis group, biphenyl group, perfluorobiphenyl group, 1 naphthyl group , 2-naph
- the alkyl group having 3 to 20 carbon atoms in the substituents R 5 to R U is not particularly limited, for example, propyl group, n butyl group, isobutyl group, t butyl group, neopentyl group, hexynole group, octyl group, dodecyl group, Examples include a cyclohexyl group, a cyclooctyl group, and a 2-ethylhexino group.
- the halogenated alkyl group having 1 to 20 carbon atoms in the substituents R 5 to R U is not particularly limited, and examples thereof include a trifluoromethyl group, a trifnoleoloetinole group, a perfluorooctyl group, a single group.
- a fluorocyclohexyl group, a perfluorocyclooctyl group, etc. can be mentioned, Preferably it is a perfluorooctyl group.
- ring A and substituent group R 5 to R 6 and R 8 to R U of B are each, any two or more substituents are bonded to each other in each substituent group, substituted ! /, May! /, Benzene ring, with substituent, may! /, Pyridine ring, with substituent! /, May! /, Forms a pyrazine ring It is preferably a benzene ring which may have a substituent.
- the benzene ring which may have a substituent is not particularly limited, for example, benzene ring, methylbenzene ring, (n-hexyl) benzene ring, (n-octyl) benzene ring, dimethylbenzene ring, di (n-hexyl).
- pyridine ring di (n-octadecafluorododecyl) naphthalene ring, phenylnaphthalene ring, anthracene ring, triphenylene ring, quinoline ring, etc., which may have a substituent
- the pyridine ring include, but are not limited to, a pyridine ring, a methylpyridine ring, an (n-hexyl) pyridine ring, a phenyl pyridine ring, and the like.
- Examples thereof include a pyrazine ring, a methylbiazine ring, a dimethylbiazine ring, an (n-hexyl) pyrazine ring, and a phenylvirazine ring, and a benzene ring which may have a substituent is preferred.
- n Dodecyl) naphthalene ring and di (n-perfluorododecyl) naphthalene ring are particularly preferred.
- the substituents R 8 to R U , R 9 And R 1U are preferably bonded to each other and have a substituent! /, May! /, To form a benzene ring.
- substituents R 5 to R 6 and R 8 to R U in particular a hydrogen atom, a fluorine atom, a hydrogen atom gesture et preferred is benzene ring which may have a substituent, a fluorine atom, a benzene ring preferable.
- Substituent T 3 is sulfur, selenium, tellurium, oxygen, phosphorus, or boron, preferably sulfur, selenium, phosphorus, or boron, and more preferably sulfur, phosphorus, or boron.
- n is an integer of 0 or 1, and when n is 0, T 3 is sulfur, selenium, tellurium, oxygen, and when n is a, T 3 is phosphorus, boron.
- the heteroacene derivative represented by the general formula (1) of the present invention includes the heteroacene derivative, an oxidation-resistant organic semiconductor material containing the heteroacene derivative, and an organic thin film thereof having high oxidation resistance and carrier.
- the following compounds are preferred because of their mobility.
- substituents 1 , T 2 , R 4 and symbols 1 and m have the same meaning as the substituent and symbol represented by the general formula (1), and the substituent L 1 L 2 is a chlorine atom, bromine atom, iodine atom, A group, acetoxy group, and a phenylsulfonyl group; p and q each represents an integer of 0 or 2; )
- a tetra-metalation means that substitution in the metal Xi ⁇ X 4 respectively in Formula (2).
- the amount of the metallizing agent used is preferably 3 to 20 equivalents relative to 1 equivalent of the tetrahaloterphenyl derivative of the general formula (2), particularly preferably 4 to; 15 equivalents, more preferably 5 to 10 equivalents. Can be used in a range.
- the tetrametalation is preferably carried out in a solvent.
- the solvent used is not particularly limited.
- THF tetrahydrofuran
- jetyl ether methyl-tert-butinoleethenole
- ethyleneglycolenoresmechinoleatenore di-xane, tonolene, hexane, cyclohexane, etc. Yes, particularly preferably THF.
- These solvents may be used alone or as a mixture of two or more.
- the temperature for the tetrametalation is from ⁇ 100 to 50 ° C. Force S, particularly preferably from 1 to 20 ° C.
- the reaction time is preferably 1 to 120 minutes, particularly preferably 5 to 60 minutes.
- the progress of tetrametalation can be monitored by taking out a part of the reaction solution, stopping the reaction with water, and analyzing by gas chromatography.
- the tetrametal salt produced by the tetrametalation is then reacted with a reactant represented by general formula (3) and general formula (4) to obtain a heterocacene derivative represented by general formula (1). is there.
- the reaction with the reactive agent is carried out by reacting the reaction mixture containing the tetrametal salt generated by the tetrametalation with the reactive agent directly, after isolating the generated tetrametal salt, Any method of reacting may be used.
- the substituents 1 , T 2 , R 4 and symbols 1 and m have the same meanings as the substituents and symbols represented by the general formula (1).
- general formula (3) and general formula (4) bis (phenylsulfonole) sulfide, dichlorophenylphosphine, dichlorophenylphenol and the like are preferred.
- Substituent L 2 represents a chlorine atom, a bromine atom, an iodine atom, an oxy group having 1 to 20 carbon atoms, an acetoxy group, or an arylsulfonyl group, preferably a chlorine atom, bromine atom, or carbon number;! To 20 And an arylsulfonyl group.
- the oxy group having 1 to 20 carbon atoms is not particularly limited, for example, methoxy group, ethoxy group, ⁇ -butoxy group, phenoxy group, (2-metho Xyl) phenoxy group and the like, and the arylsulfonyl group is not particularly limited. Examples thereof include a phenylsulfonyl group and a p-tolylsulfonyl group. Of these, a phenylsulfonyl group is particularly preferred.
- the reactant represented by the general formula (4) for example 2 sulfur chloride; dibromide sulfur; bis (phenylpropyl sulfo Nino Les) Sunorefuido, bis (p - Bis (arylsulfonyl) sulfides such as Torinolesnorehoninore) sulfide; Sulfur; Selenium dichloride; Selenium; Dichlorinated tenoler; Tenoler; Dichlorophenylphosphine, Dimethoxyphenylphosphine, Diphenoxyphenylphosphine, Dichloro ⁇ 4— (n-octinole) phenylene phosphine ⁇ arylenophosphines; dichloro (n-hexinole) phosphine, dichloro (n-octinole) phosphine, dimethoxy (n-hexyl) phosphine,
- the heterocene derivative represented by the general formula (1) of the present invention obtained by the above method can be further refined.
- the purification method is not particularly limited, and examples thereof include column chromatography, recrystallization, or sublimation.
- the tetrahaloterphenyl derivative represented by the general formula (2) of the present invention includes a tetrahalobenzene represented by the following general formula (5) and a 2-haloaryl represented by the following general formula (6) and the following general formula (7). It can be produced by reacting metal reagents in the presence of palladium and / or nickel catalysts.
- the reactants represented by general formula (6) and general formula (7) may be the same compound.
- the substituents X 5 and X 6 in the general formula (5) represent a bromine atom, an iodine atom or a chlorine atom, preferably a bromine atom or an iodine atom, and more preferably an iodine atom.
- R 2 , X 2 and X 3 have the same meaning as the substituent represented by the general formula (2).
- M 2 is a halide of magnesium, boron, zinc, tin, or silicon; a hydride oxide; an alkoxide; an alkylated product, and can be eliminated by the palladium and / or nickel catalyst described above to replace palladium and / or nickel.
- MgCl MgBr B (OH) B (OMe) tetra
- S preferably ZnCl ⁇ ( ⁇ ).
- Specific compounds represented by general formula (6) and general formula (7) include, for example, 3-bromobenzocenyl-2 zinc chloride, 2 bromo-4,5 difluorophenyl magnesium bromide, 2 Bromonaphthyl-3 Magnesium bromide, 2 Bromophenylporonic acid and the like.
- the 2-haloaryl metal reagents represented by the general formula (6) and the general formula (7) are, for example, allylic dihalogen substitutes as raw materials thereof such as Grignard reagents such as isopropyl magnesium bromide or ⁇ butyl lithium.
- Grignard reagents such as isopropyl magnesium bromide or ⁇ butyl lithium.
- an organolithium reagent it can be suitably prepared by reacting with zinc chloride, trimethoxyborane or the like.
- the halogen / metal exchange reaction using the Grignard reagent can be carried out using the method described in “Janore Taiga, Taiga Noreganic Garry”, 2000, Vol. 65, pages 4618-4634 ”, using an organolithium reagent.
- the halogen / metal exchange reaction for example, the method described in “Journal of Chemicanol Research Synopsis”, 1981, p. 185 can be used.
- the catalyst used in the reaction of the roaryl metal reagent is not particularly limited as long as it is a palladium and / or nickel catalyst.
- tetrakis (triphenylphosphine) palladium tris (dibenzylideneacetone) dipalladium / triphenylphosphine mixture
- dichlorobis (triphenylphosphine) ) Palladium bis (tree tert butylphosphine) palladium, diacetatobis (triphenylphosphine) palladium, dichloro (1,2-bis (diphenylphosphino) ethane) palladium, palladium acetate / triphenylphosphine mixture, paradium acetate / tree tert Butylphosphine mixture, palladium acetate / 2- (dicyclohexylphosphino) 1,1, -biphenyl mixture, dichloro (ethylenediamine) pall
- -Nickel catalysts such as tetramethylethylenediamine) nickel / triphenylphosphine mixture, bis (1,5-cyclocyclogen) nickel / triphenylphosphine mixture, and the like.
- a preferred catalyst is a zero-valent palladium compound, and a particularly preferred catalyst is tetrakis (triphenylphosphine) palladium.
- These catalysts may be used alone or as a mixture of two or more.
- the solvent to be used is not particularly limited, for example, THF, cetinoleethenole, methinole tert-butenoleethenore, dixane, ethylene glycol dimethyl ether, toluene, xylene, hexane, cyclohexane, ethanol, water, N, N-dimethylformamide, N-methylpyrrolidone, triethylamine, piperidine, piprine lysine, diisopropylamine, etc. may be mentioned, and these solvents may be used alone or in a mixture of two or more. For example, it can be used in a two-component system such as toluene / water or toluene / ethanol / water.
- the amount of nickel catalyst is against the tetrahalobenzene 1 mole of the general formula (5), 0.;! ⁇ 20 mol 0/0 preferably tool particularly preferably 1; 10 mol 0/0 It is a range.
- the amount of the 2-haloaryl metal reagent represented by the general formula (6) or (7) is preferably 0.8 to 3.2 equivalents relative to 1 equivalent of the tetrahalobenzene of the general formula (5), particularly preferably 1 0 to 2.8 equivalents, more preferably 1.;! To 2.5 equivalents can be used.
- the temperature during the reaction is preferably 10 to 120 ° C, particularly preferably 30 to 100 ° C, more preferably 40 to 90 ° C, and the reaction time is preferably! To 48 hours. Particularly preferably, it can be suitably carried out in the range of 2 to 30 hours.
- a base can also be present in the reaction system.
- the type of base is not particularly limited, for example, sodium carbonate, sodium bicarbonate, potassium carbonate, cesium carbonate.
- Inorganic bases such as potassium, potassium phosphate, sodium phosphate, sodium tert butoxide, potassium fluoride; triethylamine, trimethylamine, tributylamine, ethylenediamine, N, N, N ', N'-tetramethylethylenediamine
- organic bases such as diisopropylamine and pyridine can be cited as suitable.
- the amount of these bases used is preferably 0.5 to 10.0 equivalents, more preferably 2.0 to 8.0 equivalents per 1 equivalent of the tetrahalobenzene of the general formula (5). Can do.
- a phase transfer catalyst can be used in combination with these bases.
- the type of the phase transfer catalyst is not particularly limited. For example, trioctylmethylammonium chloride, tetraptylammonium chloride, cetylpyridinium chloride and the like can be mentioned as suitable ones.
- These phase transfer catalysts are used in an amount of 0 ⁇ ;! to 1 ⁇ 5 equivalents, particularly preferably in the range of 0.2 to 0.8 equivalents per equivalent of tetrahalobenzene of the general formula (5). It is.
- phosphines such as triphenylphosphine can be present in the reaction system.
- the amount of these phosphines used is preferably 0.9 to 8.0 equivalents, more preferably 1.0 to 3.0 equivalents per equivalent of the palladium and / or nickel catalyst. be able to.
- a copper compound can also be present in the reaction system.
- the copper compound is not particularly limited, for example, copper chloride (1), copper bromide (1), copper iodide (1), monovalent copper such as copper acetate (I); copper chloride (II), bromide Examples include divalent copper such as copper (II), copper (II) iodide, copper (II) acetate, and acetyl cetate copper (II). Of these, monovalent copper is preferred, and copper (I) iodide is particularly preferred.
- the amount of these copper compounds used is preferably 0.3 to 10.0 equivalents, more preferably 0.6 to 6.0 equivalents per 1 equivalent of the palladium and / or nickel catalyst. Touch with S.
- Tetroha mouth terphenyl derivatives of general formula (2) can be produced, for example, by using X 5 and X 6 in the general formula (5) as iodine atoms and X 2 and X 3 as bromine atoms and / or chlorine atoms.
- the oxidation-resistant organic semiconductor material is excellent in solubility in a solvent and oxidation resistance, and has suitable coating properties.
- the oxidation-resistant organic semiconductor material can be produced by dissolving the heteroacene derivative represented by the general formula (1) of the present invention in a solvent.
- an oxidation-resistant organic semiconductor material containing the heteroacene derivative represented by the general formula (1) is obtained.
- the temperature at the time of mixing and stirring is preferably 10 to 200 ° C, particularly preferably 20 to 190 ° C.
- the concentration of the heteroacene derivative represented by the general formula (1) during mixing and stirring can vary depending on the solvent and the temperature, and is preferably 0.0;! To 10.0% by weight.
- the solution can be prepared in air, preferably in an inert atmosphere such as nitrogen or argon.
- the oxidation resistance of the oxidation-resistant organic semiconductor material containing the heteroacene derivative represented by the general formula (1) can be evaluated by a method in which the solution is brought into contact with air for a predetermined time.
- the solvent to be used is degassed in advance to remove dissolved oxygen.
- the contact time with air can be appropriately selected depending on the temperature, and is preferably 0.5 minutes to 3 hours.
- Oxidation can proceed by changing the color of the solution and detecting the oxide by gas chromatography and gas chromatography (GC) -mass spectrum (GCMS) analysis.
- GC gas chromatography and gas chromatography
- GCMS gas chromatography-mass spectrum
- the oxidation-resistant organic semiconductor material containing the heteroacene derivative represented by the general formula (1) of the present invention has a relatively low temperature because the heteroacene derivative itself represented by the general formula (1) used has moderate aggregation properties. Therefore, it can be suitably applied to the production of an organic thin film by a coating method. That is, since it is not necessary to strictly remove air from the atmosphere, the coating process can be simplified.
- the coating can be carried out in air, but is preferably carried out under a nitrogen stream in consideration of solvent drying.
- the viscosity of the oxidation-resistant organic semiconductor material containing the heteroacene derivative represented by the general formula (1) of the present invention is preferably in the range of 0.005 to 20 poise. .
- an organic thin film using an oxidation-resistant organic semiconductor material containing a heterocacene derivative represented by the general formula (1) of the present invention will be described.
- Such an organic thin film can be produced by recrystallization of the above-mentioned oxidation resistant organic semiconductor material (solution) or application to a substrate, and it is particularly preferred to produce the organic thin film by application to a substrate. Then, an organic thin film is formed on the substrate by being manufactured by coating on the substrate.
- a thin film by recrystallization can be formed by cooling the oxidation-resistant organic semiconductor material.
- the atmosphere for producing the organic thin film is preferably carried out under an inert gas such as nitrogen or argon or under air, particularly preferably under an inert gas such as nitrogen or argon.
- the concentration of the heteroacene derivative represented by the general formula (1) in the solution is not particularly limited, for example, 0.01 to 10.0% by weight.
- Cooling is preferably performed by cooling between a temperature of 60-200 ° C and a temperature of 20-60 ° C, particularly preferably between 10-40 ° C.
- the crystalline organic thin film thus produced can be laminated on an appropriate substrate, that is, it can be produced on a substrate by lamination or the like.
- the substrate can also be produced using printing techniques such as screen printing I inkjet printing I gravure printing.
- printing techniques such as screen printing I inkjet printing I gravure printing.
- the material of the substrate used, and various crystalline and non-crystalline materials can be used.
- Specific examples of the substrate include, for example, polyethylene terephthalate, polymethyl methacrylate, polyethylene, polypropylene, polystyrene, cyclic polyolefin, polyimide, polycarbonate, polyvinyl alcohol, polybutyl alcohol, poly (diisopropyl fumaric acid), and poly (jet fumaric acid).
- Plastic substrates such as poly (diisopropylmaleic acid); inorganic material substrates such as glass, quartz, aluminum oxide, silicon, silicon oxide, tantalum dioxide, tantalum pentoxide, and indium tin oxide; gold, copper, chromium, titanium It is possible to use a metal substrate such as Further, the surfaces of these substrates can be used even if they are modified with silanes such as octadecyltrichlorosilane and octadecinoletrimethoxysilane; and silinoleamines such as hexamethinoresilazane. Furthermore, the substrate may be a material having an insulating property or an inductive property.
- the heterocene derivative is dissolved in a solvent such as toluene or black benzene, and is not easily oxidized by air even in a solution state. Therefore, a semiconductor thin film can be easily formed by a coating method. Therefore, the heteroacene derivative represented by the general formula (1) of the present invention is an electron paper. 1.
- Organic semiconductors for transistors such as organic EL displays, liquid crystal displays, IC tags, active phase applications; organic EL display materials; organic semiconductor laser materials; organic thin film solar cell materials; and electronic materials such as photonic crystal materials be able to.
- the vessel was cooled with water and the reaction was stopped by adding 4 ml of 1N hydrochloric acid. Toluene is added and the resulting suspension is filtered. The solid on the filter plate was washed with toluene and water. The solid was dried under reduced pressure to obtain 292 mg of a white solid.
- the filtrate was phase-separated and the organic phase was washed with water. The organic phase was concentrated under reduced pressure and the solvent was distilled off. The resulting solid was washed with hexane (10 ml) and the residue was recrystallized from toluene. The precipitated crystals were dried under reduced pressure to obtain 206 mg of a white solid. Combined with the white solid after the previous filtration, the target product was obtained in a yield of 75%.
- Example 9 Synthesis of Tetraphenyl Dichenoacene (Heterocene Derivative) 4, 5, 4 ", 5" — Tetraphenylolene synthesized in Example 8 in a lOOmh Nulenk reaction vessel under nitrogen atmosphere 2,5,2 "-Tetrabromo-1, 1 ', 4, 1" -Terphenyl 416 mg (0.489 mmol) and THF 30 ml were added.
- This suspension was cooled to 80 ° C, and 3.9 ml (3.9 mmol) of cyclohexane / hexane solution of sec-butyllithium (manufactured by Kanto Chemical Co., Ltd., 1.0 M) was added dropwise as a metallizing agent. Went. After stirring for 20 minutes, 507 mg (l.61 mmol) (compounds of the general formulas (3) and (4)) of bis (phenylsulfonyl) sulfide (manufactured by Across) was charged at ⁇ 75 ° C. all at once. The temperature was gradually raised, and the reaction temperature was raised to room temperature overnight.
- Example 12 (3, 2, 5, 5, 3 "tetrabromo-6, 7, 6", 7 "(tetradodecyl) 2, 1 ', 4 ,, 2 "—Synthesis of Dinaphthoterphenyl (Tetrahaloterphenyl Derivative)) 2-Bromo-3-chloro 6, 7-di synthesized in Synthesis Example 4 in a 100 ml Schlenk reaction vessel under nitrogen atmosphere 461 mg (0.640 mmol) of dodecylanthracene and 8 ml of THF were added, the solution was cooled to ⁇ 40 ° C., and 1.0 ml (0.665 mmol) of a THF solution of isopropylmagnesium bromide (manufactured by Kanto Chemical Co., Ltd., 0.665 M) was added dropwise.
- Example 13 Synthesis of tetradodecyldinaphthodichenoacene (heterocene derivative) 3, 2 ', 5', 3 "-tetrabromo-6 synthesized in Example 12 in a 100 ml Schlenk reaction vessel under nitrogen atmosphere 7,6 ", 7"-(tetradodecyl) 2,1 ', 4', 2 "dinaphthophenyl 122 mg (0.086 mmol) and THF 6 ml were added.
- Example 14 Synthesis of tetradodecyldinaphthodichenoacene (heterocene derivative) Under a nitrogen atmosphere, a 100-m tunlenk reaction vessel was cooled to 75 ° C, THF 6 ml and sec butyllithium as a metallizing agent (Kanto) Chemical 1.0M) cyclohexane / hexane solution 1. Oml (1.Ommol) was added.
- a concave glass substrate was heated to 70 ° C., and the above solution was applied onto the substrate using a dropper and dried under normal pressure to produce an organic thin film having a thickness of 280 nm.
- a concave glass substrate was heated to 70 ° C., and the above solution was applied onto the substrate using a dropper and dried under normal pressure to produce an organic thin film having a thickness of 220 nm.
- the oxidation resistance was evaluated using pentacene.
- the present invention it is possible to provide a heteroacene derivative having excellent oxidation resistance and capable of forming an organic semiconductor active phase by a coating method and use thereof. Further, the present invention provides a tetrahaloterphenyl derivative that is a precursor compound of the heterocene derivative and a method for producing the same.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Physics & Mathematics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
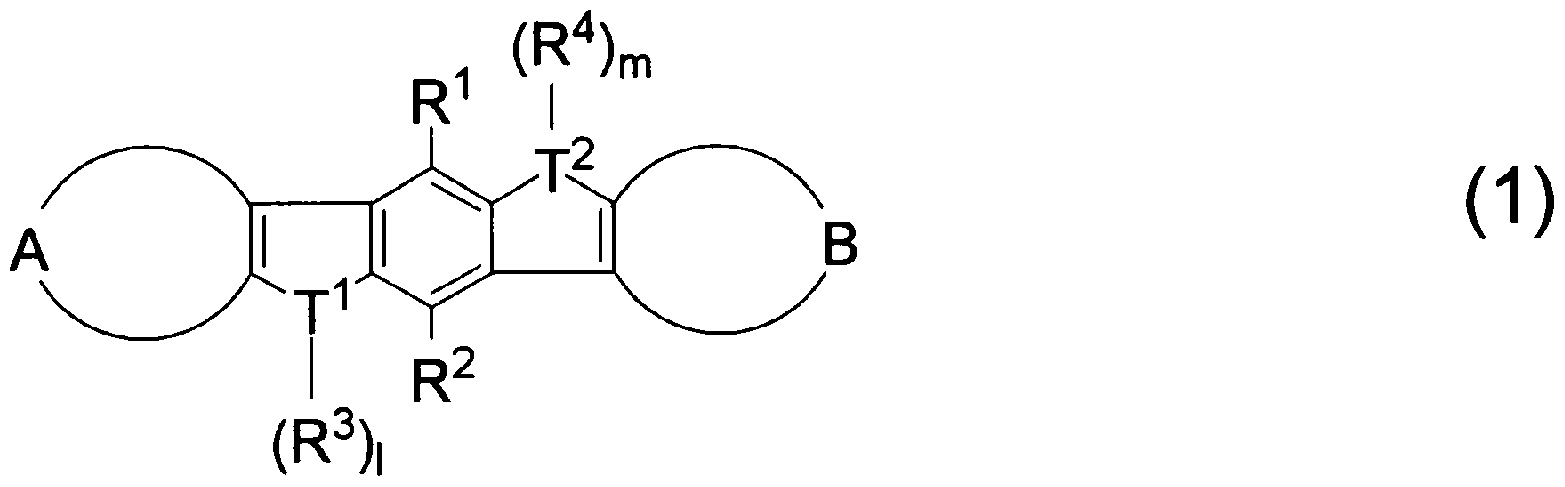
































Claims






Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/439,341 US8138355B2 (en) | 2006-08-28 | 2007-08-28 | Heteroacene derivative, tetrahaloterphenyl derivative, and processes for producing the same |
| KR1020097004243A KR101430529B1 (ko) | 2006-08-28 | 2007-08-28 | 헤테로아센 유도체, 테트라할로터페닐 유도체, 및 그것들의제조방법 |
| EP07806161.1A EP2067782B2 (en) | 2006-08-28 | 2007-08-28 | Heteroacene derivative, tetrahaloterphenyl derivative, and their production methods |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006231082 | 2006-08-28 | ||
| JP2006-231082 | 2006-08-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008026602A1 true WO2008026602A1 (fr) | 2008-03-06 |
Family
ID=39135884
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2007/066684 WO2008026602A1 (fr) | 2006-08-28 | 2007-08-28 | Dérivé hétéroacène, dérivé tétrahalotérphényle, et leurs procédés de production |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8138355B2 (ja) |
| EP (1) | EP2067782B2 (ja) |
| JP (2) | JP5585680B2 (ja) |
| KR (1) | KR101430529B1 (ja) |
| WO (1) | WO2008026602A1 (ja) |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009099658A (ja) * | 2007-10-15 | 2009-05-07 | Mitsui Chemicals Inc | 有機トランジスタ |
| WO2010000670A1 (en) * | 2008-07-02 | 2010-01-07 | Basf Se | High performance solution processable seminconductor based on dithieno [2,3-d:2',3'-d']benzo[1,2-b:4,5-b'] dithiophene |
| WO2010027106A1 (ja) * | 2008-09-08 | 2010-03-11 | 住友化学株式会社 | 新規化合物及び有機半導体材料 |
| WO2011016648A1 (en) * | 2009-08-04 | 2011-02-10 | Rohm And Haas Electronic Materials Korea Ltd. | Novel organic electroluminescent compounds and organic electroluminescent device using the same |
| WO2011030918A1 (en) * | 2009-09-11 | 2011-03-17 | Ricoh Company, Ltd. | Leaving substituent-containing compound, organic semiconductor material, organic semiconductor film containing the material, organic electronic device containing the film, method for producing film-like product, pi-electron conjugated compound and method for producing the pi-electron conjugated compound |
| WO2011108765A1 (ja) * | 2010-03-05 | 2011-09-09 | 住友化学株式会社 | 多環式化合物 |
| CN102227484A (zh) * | 2008-11-28 | 2011-10-26 | 剑桥显示技术有限公司 | 有机半导体 |
| JP2011213705A (ja) * | 2010-01-12 | 2011-10-27 | Ricoh Co Ltd | 置換基脱離化合物および有機半導体材料およびその膜およびそれを用いた有機トランジスタ |
| JP2012020987A (ja) * | 2010-06-15 | 2012-02-02 | Ricoh Co Ltd | 置換基脱離化合物とそれから得られる有機半導体材料、それを用いた有機電子デバイス、有機薄膜トランジスタおよびディスプレイ装置 |
| CN102511089A (zh) * | 2009-07-22 | 2012-06-20 | 株式会社理光 | 新型有机半导体材料以及使用其的电子器件 |
| CN102639591A (zh) * | 2009-12-02 | 2012-08-15 | 巴斯夫欧洲公司 | 二噻吩并苯并噻吩并[3,2-b]噻吩共聚物及其作为高性能可溶液加工半导体聚合物的用途 |
| WO2014027581A1 (ja) * | 2012-08-14 | 2014-02-20 | 国立大学法人九州大学 | 複素環化合物及びその利用 |
| CN103641832A (zh) * | 2009-03-20 | 2014-03-19 | 罗门哈斯电子材料韩国有限公司 | 新的有机电致发光化合物和使用该化合物的有机电致发光设备 |
| WO2014136827A1 (ja) | 2013-03-05 | 2014-09-12 | Jnc株式会社 | カルコゲン含有有機化合物およびその用途 |
| US9260451B2 (en) | 2012-08-24 | 2016-02-16 | Nippon Kayaku Kabushiki Kaisha | Method for producing aromatic compound |
| US9988472B2 (en) | 2015-08-31 | 2018-06-05 | Samsung Electronics Co., Ltd. | Composition, electronic device, and thin film transistor |
| US10358578B2 (en) | 2015-05-29 | 2019-07-23 | Samsung Electronics Co., Ltd. | Insulating ink and insulator and thin film transistor and electronic device |
| US10522771B2 (en) | 2014-12-01 | 2019-12-31 | Samsung Electronics Co., Ltd. | Composition, electronic device, and thin film transistor |
| US11038116B2 (en) | 2018-11-26 | 2021-06-15 | Samsung Electronics Co., Ltd. | Compound and organic thin film and thin film transistor and electronic device |
| US11069863B2 (en) | 2018-12-03 | 2021-07-20 | Samsung Electronics Co., Ltd. | Organic thin film and organic thin film transistor and electronic device |
| US11242357B2 (en) | 2017-10-18 | 2022-02-08 | Samsung Electronics Co., Ltd. | Fused polycyclic heteroaromatic compound and organic thin film and electronic device |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101430529B1 (ko) * | 2006-08-28 | 2014-08-18 | 도소 가부시키가이샤 | 헤테로아센 유도체, 테트라할로터페닐 유도체, 및 그것들의제조방법 |
| US8049411B2 (en) | 2008-06-05 | 2011-11-01 | Idemitsu Kosan Co., Ltd. | Material for organic electroluminescence device and organic electroluminescence device using the same |
| DE102009052428A1 (de) | 2009-11-10 | 2011-05-12 | Merck Patent Gmbh | Verbindung für elektronische Vorrichtungen |
| EP2818464A4 (en) | 2012-02-22 | 2015-11-18 | Jnc Corp | NOVEL CHALCO-CONTAINING ORGANIC COMPOUND AND USE THEREOF |
| JP5896935B2 (ja) | 2012-08-27 | 2016-03-30 | 富士フイルム株式会社 | 有機薄膜トランジスタ、有機半導体薄膜および有機半導体材料 |
| JP5896863B2 (ja) | 2012-08-27 | 2016-03-30 | 富士フイルム株式会社 | 有機薄膜トランジスタ、有機半導体薄膜および有機半導体材料 |
| KR102178662B1 (ko) * | 2013-02-08 | 2020-11-13 | 미쯔비시 가스 케미칼 컴파니, 인코포레이티드 | 화합물, 리소그래피용 하층막 형성재료, 리소그래피용 하층막 및 패턴 형성방법 |
| WO2014123102A1 (ja) | 2013-02-08 | 2014-08-14 | 三菱瓦斯化学株式会社 | 化合物、リソグラフィー用下層膜形成材料、リソグラフィー用下層膜及びパターン形成方法 |
| EP2955575B1 (en) | 2013-02-08 | 2020-07-29 | Mitsubishi Gas Chemical Company, Inc. | Resist composition, resist pattern formation method, and polyphenol derivative used in same |
| JP6247568B2 (ja) * | 2014-03-03 | 2017-12-13 | 富士フイルム株式会社 | 有機薄膜トランジスタ、非発光性有機半導体デバイス用有機半導体材料およびその応用 |
| EP3165550A4 (en) * | 2014-07-03 | 2018-04-25 | Sumitomo Chemical Company Limited | Polymeric compound and light-emitting element using same |
| JP6482821B2 (ja) * | 2014-11-04 | 2019-03-13 | 山本化成株式会社 | 有機トランジスタ |
| US10745372B2 (en) | 2014-12-25 | 2020-08-18 | Mitsubishi Gas Chemical Company, Inc. | Compound, resin, material for forming underlayer film for lithography, underlayer film for lithography, pattern forming method, and purification method |
| EP3050887B1 (en) * | 2015-01-29 | 2017-06-28 | Samsung Electronics Co., Ltd. | Fused polycyclic heteroaromatic compound, organic thin film including compound and electronic device including organic thin film |
| CN107533291B (zh) | 2015-03-31 | 2021-06-11 | 三菱瓦斯化学株式会社 | 化合物、抗蚀剂组合物及使用其的抗蚀图案形成方法 |
| JP6766803B2 (ja) | 2015-03-31 | 2020-10-14 | 三菱瓦斯化学株式会社 | レジスト組成物、レジストパターン形成方法、及びそれに用いるポリフェノール化合物 |
| CN107924131B (zh) | 2015-08-31 | 2020-12-25 | 三菱瓦斯化学株式会社 | 光刻用下层膜形成材料及其组合物用于形成光刻用下层膜的用途、以及抗蚀图案形成方法 |
| US11143962B2 (en) | 2015-08-31 | 2021-10-12 | Mitsubishi Gas Chemical Company, Inc. | Material for forming underlayer film for lithography, composition for forming underlayer film for lithography, underlayer film for lithography and production method thereof, pattern forming method, resin, and purification method |
| JP6848869B2 (ja) | 2015-09-10 | 2021-03-24 | 三菱瓦斯化学株式会社 | 化合物、樹脂、レジスト組成物又は感放射線性組成物、レジストパターン形成方法、アモルファス膜の製造方法、リソグラフィー用下層膜形成材料、リソグラフィー用下層膜形成用組成物、回路パターンの形成方法、及び、精製方法 |
| US10686145B2 (en) | 2017-07-28 | 2020-06-16 | Samsung Electronics Co., Ltd. | Organic compound, organic thin film, and electronic device |
| US11066418B2 (en) | 2018-11-26 | 2021-07-20 | Samsung Electronics Co., Ltd. | Compound and thin film transistor and electronic device |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4836439B1 (ja) * | 1969-08-05 | 1973-11-05 | ||
| WO2003016599A1 (fr) | 2001-08-09 | 2003-02-27 | Asahi Kasei Kabushiki Kaisha | Element a semi-conducteur organique |
| US20040076853A1 (en) * | 2002-04-24 | 2004-04-22 | Eastman Kodak Company | Organic light-emitting diode devices with improved operational stability |
| JP2005120379A (ja) * | 2003-10-15 | 2005-05-12 | Merck Patent Gmbh | ポリ(ベンゾジチオフェン) |
| JP2005154371A (ja) * | 2003-11-27 | 2005-06-16 | Japan Science & Technology Agency | 新規なベンゾジカルコゲノフェン誘導体、その製造方法およびそれを用いた有機半導体デバイス |
| JP2006231082A (ja) | 2006-05-19 | 2006-09-07 | Tohoku Teikku:Kk | 温浴施設 |
| JP2007019294A (ja) * | 2005-07-08 | 2007-01-25 | Konica Minolta Holdings Inc | 有機半導体材料、有機半導体膜、有機半導体素子及び有機薄膜トランジスタ |
| WO2007068618A1 (en) * | 2005-12-12 | 2007-06-21 | Ciba Holding Inc. | Organic semiconductors and their manufacture |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4836439A (ja) | 1971-09-09 | 1973-05-29 | ||
| JP4836439B2 (ja) | 2004-11-25 | 2011-12-14 | 株式会社東芝 | 非水電解質電池用電極材料、非水電解質電池用電極および非水電解質電池 |
| US7985885B2 (en) * | 2005-04-08 | 2011-07-26 | Tosoh Corporation | Terphenylene derivative, tetrahaloterphenyl derivative, and processes for producing both |
| DE102005023437A1 (de) * | 2005-05-20 | 2006-11-30 | Merck Patent Gmbh | Verbindungen für organische elektronische Vorrichtungen |
| JP2007088222A (ja) * | 2005-09-22 | 2007-04-05 | Konica Minolta Holdings Inc | 有機半導体材料、有機半導体膜、有機半導体デバイス及び有機薄膜トランジスタ |
| JP5007988B2 (ja) * | 2005-10-27 | 2012-08-22 | 国立大学法人名古屋大学 | 多環縮環化合物およびそれらの製造法ならびに多環縮環化合物を用いる有機電界発光素子 |
| JP2007288033A (ja) * | 2006-04-19 | 2007-11-01 | Konica Minolta Holdings Inc | 有機半導体材料、有機半導体膜、有機半導体デバイス及び有機薄膜トランジスタ |
| JP5499422B2 (ja) * | 2006-06-28 | 2014-05-21 | コニカミノルタ株式会社 | 有機半導体材料、有機半導体膜、有機薄膜トランジスタ及び有機薄膜トランジスタの製造方法 |
| JP5272345B2 (ja) * | 2006-08-28 | 2013-08-28 | 東ソー株式会社 | ヘテロアセン誘導体、テトラハロターフェニル誘導体及びそれらの製造方法 |
| KR101430529B1 (ko) * | 2006-08-28 | 2014-08-18 | 도소 가부시키가이샤 | 헤테로아센 유도체, 테트라할로터페닐 유도체, 및 그것들의제조방법 |
-
2007
- 2007-08-28 KR KR1020097004243A patent/KR101430529B1/ko active IP Right Grant
- 2007-08-28 WO PCT/JP2007/066684 patent/WO2008026602A1/ja active Application Filing
- 2007-08-28 US US12/439,341 patent/US8138355B2/en active Active
- 2007-08-28 EP EP07806161.1A patent/EP2067782B2/en active Active
-
2013
- 2013-03-26 JP JP2013064929A patent/JP5585680B2/ja active Active
- 2013-03-26 JP JP2013064928A patent/JP5716781B2/ja active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4836439B1 (ja) * | 1969-08-05 | 1973-11-05 | ||
| WO2003016599A1 (fr) | 2001-08-09 | 2003-02-27 | Asahi Kasei Kabushiki Kaisha | Element a semi-conducteur organique |
| US20040076853A1 (en) * | 2002-04-24 | 2004-04-22 | Eastman Kodak Company | Organic light-emitting diode devices with improved operational stability |
| JP2005120379A (ja) * | 2003-10-15 | 2005-05-12 | Merck Patent Gmbh | ポリ(ベンゾジチオフェン) |
| JP2005154371A (ja) * | 2003-11-27 | 2005-06-16 | Japan Science & Technology Agency | 新規なベンゾジカルコゲノフェン誘導体、その製造方法およびそれを用いた有機半導体デバイス |
| JP2007019294A (ja) * | 2005-07-08 | 2007-01-25 | Konica Minolta Holdings Inc | 有機半導体材料、有機半導体膜、有機半導体素子及び有機薄膜トランジスタ |
| WO2007068618A1 (en) * | 2005-12-12 | 2007-06-21 | Ciba Holding Inc. | Organic semiconductors and their manufacture |
| JP2006231082A (ja) | 2006-05-19 | 2006-09-07 | Tohoku Teikku:Kk | 温浴施設 |
Non-Patent Citations (15)
| Title |
|---|
| "BERICHTE" (GERMANY), vol. 66B, 1933, pages 1876 - 1891 |
| "JOURNAL OF ORGANIC CHEMISTRY" (USA), vol. 16, 1951, pages 1577 - 1581 |
| JOURNAL OF AMERICAN CHEMICAL SOCIETY, vol. 119, 1997, pages 4578 - 4593 |
| JOURNAL OF AMERICAN CHEMICAL SOCIETY, vol. 127, 2005, pages 13281 - 13286 |
| JOURNAL OF APPLIED PHYSICS", (USA), vol. 92, 2002, pages 5259 - 5263 |
| JOURNAL OF CHEMICAL RESEARCH SYNOPSIS, 1981, pages 185 |
| JOURNAL OF ORGANIC CHEMISTRY, vol. 65, 2006, pages 4618 - 4634 |
| OSMAN A.-M.: "Reactions between chloro-p-benzoquinones and beta-naphthol", JOURNAL OF ORGANIC CHEMISTRY, vol. 22, 1957, pages 342 - 344, XP003021333 * |
| SCIENCE", (USA), vol. 280, 1998, pages 1741 - 1744 |
| See also references of EP2067782A4 |
| SIRRINGHAUS H. ET AL.: "Dibenzothienobisbenzothiophene-a novel fused-ring oligomer with high field-effect mobility", JOURNAL OF MATERIALS CHEMISTRY, vol. 9, no. 9, 1999, pages 2095 - 2101, XP002244281 * |
| SYNLETT, 2003, pages 29 - 34 |
| SYNTHESIS, 1993, pages 387 - 390 |
| SYNTHETIC COMMUNICATIONS, vol. 33, 2003, pages 2751 - 2756 |
| WANG C.-H. ET AL.: "Linear C2-symmetric polycyclic benzodithiophene: Efficient, highly diversified approaches and the optical properties", TETRAHEDRON LETTERS, vol. 46, no. 47, 10 October 2005 (2005-10-10), pages 8153 - 8157, XP005123855 * |
Cited By (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009099658A (ja) * | 2007-10-15 | 2009-05-07 | Mitsui Chemicals Inc | 有機トランジスタ |
| US8367717B2 (en) | 2008-07-02 | 2013-02-05 | Basf Se | High performance solution processable semiconductor based on dithieno [2,3-D:2′, 3′-D′]benzo[1,2-B:4,5-B′] dithiophene |
| WO2010000670A1 (en) * | 2008-07-02 | 2010-01-07 | Basf Se | High performance solution processable seminconductor based on dithieno [2,3-d:2',3'-d']benzo[1,2-b:4,5-b'] dithiophene |
| CN102083838A (zh) * | 2008-07-02 | 2011-06-01 | 巴斯夫欧洲公司 | 基于二噻吩并[2,3-d:2′,3′-d′]苯并[1,2-b:4,5-b′]二噻吩的高性能可溶液加工半导体 |
| CN102083838B (zh) * | 2008-07-02 | 2016-05-25 | 巴斯夫欧洲公司 | 基于二噻吩并[2,3-d:2′,3′-d′]苯并[1,2-b:4,5-b′]二噻吩的高性能可溶液加工半导体 |
| WO2010027106A1 (ja) * | 2008-09-08 | 2010-03-11 | 住友化学株式会社 | 新規化合物及び有機半導体材料 |
| CN102209720A (zh) * | 2008-09-08 | 2011-10-05 | 住友化学株式会社 | 新型化合物及有机半导体材料 |
| CN102227484A (zh) * | 2008-11-28 | 2011-10-26 | 剑桥显示技术有限公司 | 有机半导体 |
| CN103641832A (zh) * | 2009-03-20 | 2014-03-19 | 罗门哈斯电子材料韩国有限公司 | 新的有机电致发光化合物和使用该化合物的有机电致发光设备 |
| US9133211B2 (en) | 2009-07-22 | 2015-09-15 | Ricoh Company, Ltd. | Dithienobenzodithiophene semiconductive material and electronic device using the same |
| CN102511089A (zh) * | 2009-07-22 | 2012-06-20 | 株式会社理光 | 新型有机半导体材料以及使用其的电子器件 |
| WO2011016648A1 (en) * | 2009-08-04 | 2011-02-10 | Rohm And Haas Electronic Materials Korea Ltd. | Novel organic electroluminescent compounds and organic electroluminescent device using the same |
| WO2011030918A1 (en) * | 2009-09-11 | 2011-03-17 | Ricoh Company, Ltd. | Leaving substituent-containing compound, organic semiconductor material, organic semiconductor film containing the material, organic electronic device containing the film, method for producing film-like product, pi-electron conjugated compound and method for producing the pi-electron conjugated compound |
| US9224959B2 (en) | 2009-09-11 | 2015-12-29 | Ricoh Company, Ltd. | Method for producing a pi-electron conjugated compound |
| US8680296B2 (en) | 2009-09-11 | 2014-03-25 | Ricoh Company, Ltd. | Leaving substituent-containing compound, products produced using the same, and methods for producing the products |
| US9123896B2 (en) | 2009-09-11 | 2015-09-01 | Ricoh Company, Ltd. | Organic electronic device containing an organic semiconductor material film which contains a leaving substituent-containing compound |
| CN102639591A (zh) * | 2009-12-02 | 2012-08-15 | 巴斯夫欧洲公司 | 二噻吩并苯并噻吩并[3,2-b]噻吩共聚物及其作为高性能可溶液加工半导体聚合物的用途 |
| CN102639591B (zh) * | 2009-12-02 | 2015-05-13 | 巴斯夫欧洲公司 | 二噻吩并苯并噻吩并[3,2-b]噻吩共聚物及其作为高性能可溶液加工半导体聚合物的用途 |
| JP2011213705A (ja) * | 2010-01-12 | 2011-10-27 | Ricoh Co Ltd | 置換基脱離化合物および有機半導体材料およびその膜およびそれを用いた有機トランジスタ |
| WO2011108765A1 (ja) * | 2010-03-05 | 2011-09-09 | 住友化学株式会社 | 多環式化合物 |
| JP2012020987A (ja) * | 2010-06-15 | 2012-02-02 | Ricoh Co Ltd | 置換基脱離化合物とそれから得られる有機半導体材料、それを用いた有機電子デバイス、有機薄膜トランジスタおよびディスプレイ装置 |
| KR101981367B1 (ko) | 2012-08-14 | 2019-05-22 | 닛뽄 가야쿠 가부시키가이샤 | 헤테로시클릭 화합물 및 이의 용도 |
| WO2014027581A1 (ja) * | 2012-08-14 | 2014-02-20 | 国立大学法人九州大学 | 複素環化合物及びその利用 |
| KR20150042257A (ko) * | 2012-08-14 | 2015-04-20 | 고쿠리쓰다이가쿠호진 규슈다이가쿠 | 헤테로시클릭 화합물 및 이의 용도 |
| US9187493B2 (en) | 2012-08-14 | 2015-11-17 | Nippon Kayaku Kabushiki Kaisha | Heterocyclic compound and use thereof |
| JPWO2014027581A1 (ja) * | 2012-08-14 | 2016-07-25 | 日本化薬株式会社 | 複素環化合物及びその利用 |
| US9260451B2 (en) | 2012-08-24 | 2016-02-16 | Nippon Kayaku Kabushiki Kaisha | Method for producing aromatic compound |
| JP6008158B2 (ja) * | 2013-03-05 | 2016-10-19 | Jnc株式会社 | カルコゲン含有有機化合物およびその用途 |
| US20160013425A1 (en) * | 2013-03-05 | 2016-01-14 | Jnc Corporation | A chalcogen-containing organic compound and a use thereof |
| KR20150126898A (ko) | 2013-03-05 | 2015-11-13 | 제이엔씨 주식회사 | 칼코겐 함유 유기 화합물 및 그의 용도 |
| US9853225B2 (en) | 2013-03-05 | 2017-12-26 | Jnc Corporation | Chalcogen-containing organic compound and a use thereof |
| WO2014136827A1 (ja) | 2013-03-05 | 2014-09-12 | Jnc株式会社 | カルコゲン含有有機化合物およびその用途 |
| US10522771B2 (en) | 2014-12-01 | 2019-12-31 | Samsung Electronics Co., Ltd. | Composition, electronic device, and thin film transistor |
| US10879475B2 (en) | 2014-12-01 | 2020-12-29 | Samsung Electronics Co., Ltd. | Composition, electronic device, and thin film transistor |
| US10358578B2 (en) | 2015-05-29 | 2019-07-23 | Samsung Electronics Co., Ltd. | Insulating ink and insulator and thin film transistor and electronic device |
| US9988472B2 (en) | 2015-08-31 | 2018-06-05 | Samsung Electronics Co., Ltd. | Composition, electronic device, and thin film transistor |
| US11242357B2 (en) | 2017-10-18 | 2022-02-08 | Samsung Electronics Co., Ltd. | Fused polycyclic heteroaromatic compound and organic thin film and electronic device |
| US11038116B2 (en) | 2018-11-26 | 2021-06-15 | Samsung Electronics Co., Ltd. | Compound and organic thin film and thin film transistor and electronic device |
| US11069863B2 (en) | 2018-12-03 | 2021-07-20 | Samsung Electronics Co., Ltd. | Organic thin film and organic thin film transistor and electronic device |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20090042289A (ko) | 2009-04-29 |
| US20090261300A1 (en) | 2009-10-22 |
| JP2013189434A (ja) | 2013-09-26 |
| JP5585680B2 (ja) | 2014-09-10 |
| EP2067782B1 (en) | 2014-08-13 |
| KR101430529B1 (ko) | 2014-08-18 |
| JP2013173748A (ja) | 2013-09-05 |
| US8138355B2 (en) | 2012-03-20 |
| EP2067782A4 (en) | 2010-08-04 |
| EP2067782B2 (en) | 2018-06-27 |
| JP5716781B2 (ja) | 2015-05-13 |
| EP2067782A1 (en) | 2009-06-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008026602A1 (fr) | Dérivé hétéroacène, dérivé tétrahalotérphényle, et leurs procédés de production | |
| JP5272345B2 (ja) | ヘテロアセン誘導体、テトラハロターフェニル誘導体及びそれらの製造方法 | |
| JP5716821B2 (ja) | ヘテロアセン誘導体、その前駆化合物及びそれらの製造方法 | |
| JP5061896B2 (ja) | ターフェニレン誘導体、テトラハロターフェニル誘導体及びそれらの製造方法 | |
| JP5549829B2 (ja) | ヘテロアセン誘導体及びその用途 | |
| JP4792796B2 (ja) | 2,3−ジハロビフェニレン誘導体、その前駆化合物及び製造方法 | |
| JP5481815B2 (ja) | ビフェニレン誘導体、その用途、及びその製造方法 | |
| JP2009203183A (ja) | ヘテロアセン誘導体の製造方法 | |
| JP6318863B2 (ja) | ジチエノベンゾジチオフェン誘導体の製造方法 | |
| JP5076466B2 (ja) | ビフェニレン誘導体、その用途、及びその製造方法 | |
| JP4978003B2 (ja) | ジベンゾシロール誘導体、その前駆化合物、及びそれらの製造方法 | |
| JP6344063B2 (ja) | ジチエノベンゾジチオフェン誘導体の製造方法 | |
| JP2010070473A (ja) | ターフェニレン誘導体の製造方法 | |
| JP5076599B2 (ja) | ターフェニレン誘導体の製造方法 | |
| JP5617296B2 (ja) | ビ(アントラカルコゲノフェニル)誘導体、その前駆化合物及びそれらの製造方法 | |
| JP5169191B2 (ja) | ヘテロアセン誘導体、(テトラハロ)ジアリールチエノチオフェン誘導体及びそれらの製造方法 | |
| JP4935130B2 (ja) | ジベンゾシロール誘導体、その前駆化合物及び製造方法 | |
| JP2007210964A (ja) | テトラフェニレン誘導体、その用途、及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07806161 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12439341 Country of ref document: US Ref document number: 1020097004243 Country of ref document: KR Ref document number: 2007806161 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |