WO2006100289A1 - Verfahren zur herstellung von alkoxylierten 2,5-dihydrofuran- oder tetra-1,1,4,4-alkoxylierten but-2-enderivaten - Google Patents
Verfahren zur herstellung von alkoxylierten 2,5-dihydrofuran- oder tetra-1,1,4,4-alkoxylierten but-2-enderivaten Download PDFInfo
- Publication number
- WO2006100289A1 WO2006100289A1 PCT/EP2006/060989 EP2006060989W WO2006100289A1 WO 2006100289 A1 WO2006100289 A1 WO 2006100289A1 EP 2006060989 W EP2006060989 W EP 2006060989W WO 2006100289 A1 WO2006100289 A1 WO 2006100289A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- derivatives
- general formula
- alkyl
- butene
- alkoxy
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/26—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D307/30—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D307/32—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/87—Benzo [c] furans; Hydrogenated benzo [c] furans
- C07D307/88—Benzo [c] furans; Hydrogenated benzo [c] furans with one oxygen atom directly attached in position 1 or 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/87—Benzo [c] furans; Hydrogenated benzo [c] furans
- C07D307/89—Benzo [c] furans; Hydrogenated benzo [c] furans with two oxygen atoms directly attached in positions 1 and 3
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B3/00—Electrolytic production of organic compounds
- C25B3/20—Processes
- C25B3/23—Oxidation
Definitions
- the invention relates to a novel process for the preparation of substituted in 3 or 4-position-2,5-Dihydrofuranderivaten each carrying a CrCe alkoxy in the 2- or in the 5-position or at both positions, or in 3- or 4-position substituted 1, 1, 4,4-tetraalkoxy-but-2-enes (DHF alkoxy derivatives).
- DE-A-27 10 420 and DE-A-848 501 describe the anodic oxidation of furans in the presence of sodium or ammonium bromide as conductive salts.
- EP-A-078 004 discloses the anodic oxidation of furans with alkoxides, halides and sulfonates as conducting salts while WO 2004/85710 describes the direct anodic oxidation of furans on specific boron-doped diamond electrodes.
- the object was therefore to provide an electrochemical process for the preparation of alkoxylated 2,5-dihydrofuran or tetra-1,1,4,4-alkoxybut-2-ene derivatives which is economical and the desired products in high yields and with good selectivity provides.
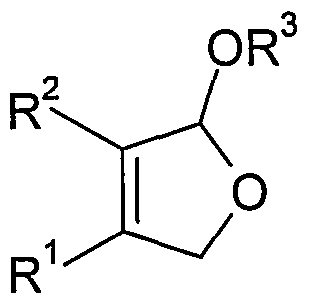
- radicals R 1 and R 2 are independently hydrogen, C 1 - to C 6 -alkyl, C 6 to C 12 aryl such as phenyl or C 5 - to C 12 cycloalkyl or R 1 and R 2 together with the double bond to which they are attached, a C 6 - to C 12 -aryl radical such as, for example, phenyl mono- or poly-C 1 - to C 6 -alkyl, halogen- or alkoxy-substituted phenyl, or a mono- or polyunsaturated one C 5 - to C 12 - form cycloalkyl,
- C 1 - to Ce-monoalkyl alcohol methanol or isopropanol is preferably used.
- the process according to the invention is particularly preferably used for the production of
- R 1 , R 2 independently of one another are hydrogen, C 1 - to C 6 -alkyl, C 6 - to C 12 -aryl or C 5 - to C 12 -cycloalkyl,
- R 1 and R 2 together with the double bond to which they are attached form a C 6 - to C 12 -aryl radical or a mono- or polyunsaturated C 5 - to C 12 -cycloalkyl radical
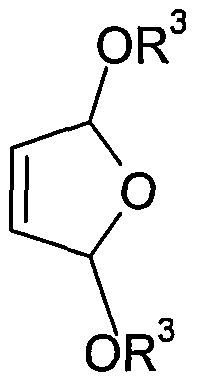
- R 3 C 1 - to C 6 -alkyl means, by electrochemical oxidation in the presence of a C 1 - to C 6 -Monoalkylalkohols prepares from 2-butene-diol derivatives of the formula (I).
- radicals R 1 , R 2 and R 3 have the same meaning as in the general formula (II) from 2-butene-diol derivatives of the formula (I) or a mixture thereof with DHF-alkoxy derivatives of the general formula (II)
- R 3 is C 1 - to C 6 -alkyl, from buten-1,4-diol of the general formula (I), where R 1 and R 2 in formula (I) are hydrogen.
- 2-butene-1,4-diol is considerably less expensive. Due to a higher boiling point of the 2-butene-1, 4-diol also reduces the cooling effort during the reaction and higher reaction temperatures are possible. An essential further advantage of this educt is its significantly lower toxicity. Cis-butene-1,4-diol or at least 20% by weight of cis-butene-1,4-diol-containing diastereomer mixtures are preferably used in the process according to the invention. Particularly suitable is the process according to the invention for the preparation of DHF-alkoxy derivatives of the general formula (IIIb),
- radicals R 4 , R 5 , R 6 and R 7 are hydrogen, C 1 - to C 4 -alkyl, C 1 - to C 6 - alkoxy or halogen, and R 3 in the general formula (II) indicated Meaning,
- radicals R 4 , R 5 , R 6 and R 7 are hydrogen, C 1 - to C 4 -alkyl, C 1 - to C 6 - alkoxy or halogen,
- radicals R 4 , R 5 , R 6 and R 7 are hydrogen, C 1 - to C 4 -alkyl, C 1 - to C 6 - alkoxy or halogen, and R 3 in the general formula (II) indicated Meaning,
- the radicals R 4 , R 5 , R 6 and R 7 are hydrogen.
- the desired target products are a compound of the general formula (III) or (IV), starting from 2-butene-1,4-diol derivatives of the general formula (I).
- the unwanted compound of general formula (II) is returned to the electrolysis cell and then used together with the corresponding 2-butene-1, 4-diol derivative of the general formula (I) as starting material for the preparation of the target products with the desired higher number of alkoxy radicals.
- the C 1 - to C 6 monoalcohol based on the 2-butene-1, 4-diol derivative of the general formula (I), equimolar or used in excess of up to 1:20 and then serves at the same time as a solvent or diluent for the compound of general formula (II) and the compound of general formula (I) formed.
- a solvent or diluent for the compound of general formula (II) and the compound of general formula (I) formed Preference is given to using a C 1 -C 6 -monoalkyl alcohol and very particularly preferably methanol.
- the electrolysis solution is added to customary cosolvents.
- these are the inert solvents commonly used in organic chemistry. tel with a high oxidation potential. Examples include dimethylformamide, dimethyl carbonate or propylene carbonate.
- Conducting salts which are contained in the electrolysis solution are generally at least one compound selected from the group consisting of potassium, sodium, lithium, iron, alkali, alkaline earth metal, Te ⁇ a (C 1 - to C 6- alkyl) ammonium, preferably tri (C 1 - to C 6 alkyl) -methylammonium salts.
- Suitable counterions are sulfate, bisulfate, alkyl sulfates, aryl sulfates, halides, phosphates, carbonates, alkyl phosphates, alkyl carbonates, nitrate, alcoholates, tetrafluoroborate or perchlorate.
- acids derived from the abovementioned anions are suitable as conductive salts.
- MTBS methyltributylammonium methylsulfate
- methyltriethylammonium methylsulfate methyltri-propylmethylammonium methylsulfates.
- suitable electrolyte salts are ionic liquids. Suitable ionic liquids are described in "Lonic Liquids in Synthesis”, ed. Peter Wasserscheid, Tom Welton, Verlag Wiley VCH, 2003, Chap. 3.6, pages 103 - 126.
- the pH of the electrolyte is by addition of organic and inorganic acids such as citric acid, tartaric acid, sulfuric acid, phosphoric acid, sulfonic acids, C 1 - to C 6 carboxylic acids such as formic acid, acetic acid, propionic acid or by using per se known buffer systems on a pH in the range of 2 to 7, preferably 2.5 to 5 set.
- organic and inorganic acids such as citric acid, tartaric acid, sulfuric acid, phosphoric acid, sulfonic acids, C 1 - to C 6 carboxylic acids such as formic acid, acetic acid, propionic acid or by using per se known buffer systems on a pH in the range of 2 to 7, preferably 2.5 to 5 set.
- the process according to the invention can be carried out in all customary types of electrolytic cell. Preferably, one works continuously with undivided flow cells.
- bipolar switched capillary gap cells or Plattenstapelzellen in which the electrodes are designed as plates and are arranged plane-parallel (see Ullmann's Encyclopedia of Industrial Chemistry, 1999 electronic release, Sixth Edition, VCH Verlag Weinheim, Volume Eiectrochemistry, Chapter 3.5 Special Cell Designs and Chapter 5, Organic Ectrochemistry, Subchapter 5.4.3.2 Cell Design).
- electrolysis cells are e.g. also described in DE-A-19533773.
- the current densities at which the process is carried out are generally 1 to 20, preferably 3 to 5 mA / cm 2 .
- the temperatures are usually -20 to 55 0 C, preferably 20 to 40 0 C. In general, working at atmospheric pressure. Higher pressures are preferably used when working at higher temperatures. tet is to be avoided in order to avoid boiling of the starting compounds or cosolvents.
- Suitable anode materials are, for example, noble metals such as platinum or metal oxides such as ruthenium or chromium oxide or mixed oxides of the type RuO x TiO x . Preference is given to graphite or carbon electrodes. Furthermore, anodes with diamond surfaces are preferred.
- cathode materials for example, iron, steel, stainless steel, nickel or precious metals such as platinum and graphite or carbon materials into consideration, with graphite is preferred. Furthermore, cathodes with diamond surfaces are preferred.
- the system is graphite as the anode and cathode and graphite as the anode and nickel, stainless steel or steel as the cathode. Furthermore, anodes with diamond surfaces are preferred.
- the electrolysis solution is worked up by general separation methods.
- the electrolysis solution is generally first brought to a pH of 8 to 9, then distilled and fertilize the individual compounds are obtained separately in the form of different fractions. Further purification can be carried out, for example, by crystallization, distillation or by chromatography. If 2,5-dimethoxytetrahydrofuran is to be prepared from 2,5-dihydro-2,5-dimethoxyfuran, purification is not necessary and the crude product obtained by the process according to the invention can be used.
- MTBS methyltributylammonium methylsulfate
- the electrolyte was pumped through the cell for 19 h at a flow rate of 200 l / h via a heat exchanger.
- Electrolyte 35.0 g of 1,2-benzenedimethanol
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- Materials Engineering (AREA)
- Metallurgy (AREA)
- Electrolytic Production Of Non-Metals, Compounds, Apparatuses Therefor (AREA)
Abstract
Description









Claims




Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008502421A JP2008536007A (ja) | 2005-03-24 | 2006-03-23 | アルコキシル化された2,5−ジヒドロフラン誘導体又はテトラ−1,1,4,4−アルコキシル化されたブタ−2−エン誘導体の製造方法 |
| CA002602077A CA2602077A1 (en) | 2005-03-24 | 2006-03-23 | Method for producing alkoxylated 2,5-dihydrofuran but-2-ene derivatives or tetra-1,1,4,4-alkoxylated but-2-ene derivatives |
| EP06725267A EP1863781A1 (de) | 2005-03-24 | 2006-03-23 | Verfahren zur herstellung von alkoxylierten 2,5-dihydrofuran- oder tetra-1,1,4,4-alkoxylierten but-2-enderivaten |
| US11/908,506 US20080110763A1 (en) | 2005-03-24 | 2006-03-23 | Method For Producing Alkoxylated 2,5-Dihydrofuran But-2-Ene Derivatives Or Tetra-1,1,4,4-Alkoxylated But-2-Ene Derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102005013631A DE102005013631A1 (de) | 2005-03-24 | 2005-03-24 | Verfahren zur Herstellung von alkoxylierten 2,5-Dihydrofuran- oder tetra-1,1,4,4-alkoxylierten But-2-enderivaten |
| DE102005013631.1 | 2005-03-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006100289A1 true WO2006100289A1 (de) | 2006-09-28 |
Family
ID=36649870
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/060989 WO2006100289A1 (de) | 2005-03-24 | 2006-03-23 | Verfahren zur herstellung von alkoxylierten 2,5-dihydrofuran- oder tetra-1,1,4,4-alkoxylierten but-2-enderivaten |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20080110763A1 (de) |
| EP (1) | EP1863781A1 (de) |
| JP (1) | JP2008536007A (de) |
| CN (1) | CN101137635A (de) |
| CA (1) | CA2602077A1 (de) |
| DE (1) | DE102005013631A1 (de) |
| WO (1) | WO2006100289A1 (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023242064A1 (en) | 2022-06-15 | 2023-12-21 | Dsm Ip Assets B.V. | Process for the preparation of alkoxylated 2,5-dihydrofuran |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2472719T3 (es) * | 2010-03-25 | 2014-07-02 | Basf Se | Lavadora, procedimiento, combinación de lavado textil electroqu�mico y bola de blanqueo electrol�tico |
| CN102633754B (zh) * | 2012-03-28 | 2014-02-05 | 南开大学 | 用改性纳米氧化铝催化剂制备高纯度2,5-二氢呋喃的方法 |
| EP3545120A1 (de) * | 2016-11-24 | 2019-10-02 | Avantium Knowledge Centre B.v. | Verfahren zur behandlung einer furan-2,5-dicarbonsäure-zusammensetzung |
| CN109518211B (zh) * | 2019-01-08 | 2020-11-06 | 合肥工业大学 | 一种芳香偶酰类化合物的电化学合成方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0078004A1 (de) * | 1981-10-28 | 1983-05-04 | BASF Aktiengesellschaft | Elektrochemisches Verfahren zur Herstellung von 2,5-Dialkoxy-2,5-dihydrofuranen |
| WO2004106316A1 (de) * | 2003-05-28 | 2004-12-09 | Basf Aktiengesellschaft | Verfahren zur herstellung von alkoxylierten 2,5-dihydrofuran- oder tetra-1,1,4,4- alkoxylierten but-2-enderivaten |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19962102A1 (de) * | 1999-12-22 | 2001-06-28 | Basf Ag | Verfahren zur elektrochemischen Oxidation von organischen Verbindungen |
-
2005
- 2005-03-24 DE DE102005013631A patent/DE102005013631A1/de not_active Withdrawn
-
2006
- 2006-03-23 EP EP06725267A patent/EP1863781A1/de not_active Withdrawn
- 2006-03-23 JP JP2008502421A patent/JP2008536007A/ja not_active Withdrawn
- 2006-03-23 WO PCT/EP2006/060989 patent/WO2006100289A1/de not_active Application Discontinuation
- 2006-03-23 US US11/908,506 patent/US20080110763A1/en not_active Abandoned
- 2006-03-23 CN CNA2006800080086A patent/CN101137635A/zh active Pending
- 2006-03-23 CA CA002602077A patent/CA2602077A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0078004A1 (de) * | 1981-10-28 | 1983-05-04 | BASF Aktiengesellschaft | Elektrochemisches Verfahren zur Herstellung von 2,5-Dialkoxy-2,5-dihydrofuranen |
| WO2004106316A1 (de) * | 2003-05-28 | 2004-12-09 | Basf Aktiengesellschaft | Verfahren zur herstellung von alkoxylierten 2,5-dihydrofuran- oder tetra-1,1,4,4- alkoxylierten but-2-enderivaten |
Non-Patent Citations (1)
| Title |
|---|
| NAITO, KATSUYUKI ET AL: "Isobenzofuran: new approaches from 1,3-dihydro-1-methoxyisobenzofuran", JOURNAL OF ORGANIC CHEMISTRY , 45(20), 4061-2 CODEN: JOCEAH; ISSN: 0022-3263, 1980, XP002390843 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023242064A1 (en) | 2022-06-15 | 2023-12-21 | Dsm Ip Assets B.V. | Process for the preparation of alkoxylated 2,5-dihydrofuran |
Also Published As
| Publication number | Publication date |
|---|---|
| US20080110763A1 (en) | 2008-05-15 |
| CN101137635A (zh) | 2008-03-05 |
| CA2602077A1 (en) | 2006-09-28 |
| JP2008536007A (ja) | 2008-09-04 |
| EP1863781A1 (de) | 2007-12-12 |
| DE102005013631A1 (de) | 2006-09-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2748353B1 (de) | Verfahren zur elektrochemischen darstellung von gamma-hydroxycarbonsäureestern und gamma-lactonen | |
| WO2010023258A1 (de) | Verfahren zur anodischen dehydrodimerisierung von substituierten arylalkoholen | |
| WO2006100289A1 (de) | Verfahren zur herstellung von alkoxylierten 2,5-dihydrofuran- oder tetra-1,1,4,4-alkoxylierten but-2-enderivaten | |
| EP1619273B1 (de) | Verfahren zur Herstellung von 2-Alkin-1-acetalen | |
| EP2411564B1 (de) | Elektrochemisches verfahern zur herstellung von 3-tert.-butylbenzaldehyd-dimethylacetal | |
| EP2041336B1 (de) | Elektrochemische herstellung sterisch gehinderter amine | |
| EP0902846A1 (de) | Verfahren zur herstellung von phthaliden | |
| EP1769103B1 (de) | Elektrochemisches verfahren zur herstellung cyclopropylbenzylaminen | |
| WO2008145627A1 (de) | Elektrochemische oxidation an allylgruppen | |
| EP1913178A1 (de) | Verfahren zur herstellung von 1,1,4,4-tetraalkoxy-but-2-enderivaten | |
| DE848501C (de) | Verfahren zur Herstellung von 2, 5-substituierten 2, 5-Dihydrofuranderivaten | |
| EP0638665A1 (de) | Verfahren zur Herstellung von Bezaldehyddialkylacetalen | |
| WO2004106316A1 (de) | Verfahren zur herstellung von alkoxylierten 2,5-dihydrofuran- oder tetra-1,1,4,4- alkoxylierten but-2-enderivaten | |
| EP0078004B1 (de) | Elektrochemisches Verfahren zur Herstellung von 2,5-Dialkoxy-2,5-dihydrofuranen | |
| WO2002020446A1 (de) | Verfahren zur herstellung von orthocarbonsäuretrialkylestern | |
| EP1430165B1 (de) | Verfahren zur herstellung von orthocarbonsäuretrialkylestern | |
| DE2710420C2 (de) | Verfahren zur elektrolytischen Herstellung von 2,5-Dialkoxy-2,5-dihydrofuranen | |
| DE2428878C2 (de) | Verfahren zur Herstellung von p-Hydroxymethyl-benzoesäureestern | |
| EP0326855A1 (de) | Verfahren zur Herstellung von Fluormalonsäure und ihren Derivaten | |
| EP0085158B1 (de) | Verfahren zur Herstellung von Cycloalkenonderivaten | |
| EP0621352A2 (de) | Verfahren zur Herstellung von Terephthalaldehydtetraalkylacetalen | |
| DE4407986A1 (de) | Verfahren zur Herstellung von o-Phthalaldehydtetraalkylacetalen | |
| EP2171128B1 (de) | Verfahren zur herstellung von isocyanaten durch anodische oxidation vom formamiden | |
| DE102004045029A1 (de) | Verfahren zur Herstellung von Glyoxalsäurealkylesterdialkylacetal | |
| DE10143161A1 (de) | Verfahren zur Herstellung von Oxo-cyclohexyl-oder Oxo-cyclohexylenderivaten |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006725267 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2602077 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680008008.6 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11908506 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008502421 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006725267 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 11908506 Country of ref document: US |