RU2239455C2 - Фармацевтические композиции, содержащие агонист или антагонист аденозинового рецептора - Google Patents
Фармацевтические композиции, содержащие агонист или антагонист аденозинового рецептора Download PDFInfo
- Publication number
- RU2239455C2 RU2239455C2 RU2002109228/15A RU2002109228A RU2239455C2 RU 2239455 C2 RU2239455 C2 RU 2239455C2 RU 2002109228/15 A RU2002109228/15 A RU 2002109228/15A RU 2002109228 A RU2002109228 A RU 2002109228A RU 2239455 C2 RU2239455 C2 RU 2239455C2
- Authority
- RU
- Russia
- Prior art keywords
- group
- adenosine
- active ingredient
- iodobenzyl
- alkyl
- Prior art date
Links
- 0 CCN[C@@](C)CCC*C Chemical compound CCN[C@@](C)CCC*C 0.000 description 1
- PWTBZOIUWZOPFT-UHFFFAOYSA-N Nc1nc(NCCc(cc2)ccc2O)nc2nc(-c3ccc[o]3)n[n]12 Chemical compound Nc1nc(NCCc(cc2)ccc2O)nc2nc(-c3ccc[o]3)n[n]12 PWTBZOIUWZOPFT-UHFFFAOYSA-N 0.000 description 1
Images


















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
- A61K31/708—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid having oxo groups directly attached to the purine ring system, e.g. guanosine, guanylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Abstract
Предложено: новые фармацевтические композиции, содержащие агонисты аденозиновых рецепторов, в частности агонисты, связывающиеся с аденозиновым рецептором A3. Указанные композиции используют для индукции продуцирования или секреции G-CSF в организме, предупреждения или лечения токсичного побочного действия лекарственного средства или для лечения лейкопении, в частности лейкопении, вызываемой лекарственными средствами; и для ингибирования аномальных роста и пролиферации клеток. Изобретение расширяет арсенал средств заявленного назначения. 7 с. и 29 з.п. ф-лы, 20 ил.
Description
Область техники, к которой относится изобретение
Настоящее изобретение относится к области раковых заболеваний и касается лечения рака или лечения, предназначенного противодействовать побочному действию лечения рака.
Уровень техники
Далее приводится перечень работ, относящихся к описанию уровня техники в области, к которой относится изобретение. Упоминание данных ссылок в описании осуществляется путем указания в скобках номера ссылки из приведенного ниже перечня.
1. Linden J. The FASEB J. 5:2668-2676 (1991);
2. Stiles G.L. Clin. Res. 38:10-18 (1990);
3. Stolfi R.L., et al. Cancer Res. 43:561-566 (1983);
4. Belandinelli L. et al. Prog. Cardiovasc. Dis. 32:73-97 (1989);
5. Collis M.G. Pharmacol. Ther. 41:143-162 (1989);
6. Clark В. and Coupe M. Int. J. Cardiol. 23:1-10 (1989);
7. Dubey R.K. et al. Circulation 96:2656-2666 (1997);
8. Soderback U. et al. Clin. Sci. 81:691-694 (1994);
9. Gilbertsen R.B. Agents actions 22:91-98 (1987);
10. Bouma M.G. et al. J. Immunol. 153:4159-4168 (1994);
11. Rozengurt E. Exp. Cell Res. 139:71-78 (1982);
12. Gonzales F.A. et al., PNAS USA 87:9717-9721 (1990);
13. Sandberg G. and Fredholm B.B. Thymus 3:63-75 (1981);
14. Pastan I.H. et al. Annu. Rev. Biochem. 44:491-495 (1975);
15. WO 99/02143;
16. Fishman P., et al. Cancer Res. 58:3181-3187 (1998);
17. Djaldetti M. et al. Clin. Exp. Metastasis 14:189-196 (1996);
18. Fishman P. et al. Cancer Research 58:3181-3187 (1998).
Предпосылки создания изобретения
Миелотоксичность является распространенным тяжелым осложнением химиотерапии и одним из факторов, ограничивающих дозу химиотерапевтического лекарственного средства, которую можно вводить. Она вызывает угрожающие жизни заболевания у проходящих лечение пациентов и большую фактическую смертность, чем любое другое побочное действие химиотерапии, и может привести к более длительному пребыванию пациента в стационаре. Кроме того, вызываемая лекарственным средством миелосупрессия ограничивает назначение больших, возможно, более эффективных доз химиотерапии больным со злокачественными заболеваниями. Некоторые подходы к решению вопроса о таком побочном эффекте включают применение лития, простагландина Е, интерферона, лактоферрина и факторов роста - гранулярно-макрофагиального колониестимулирующего фактора (GM-CSF) и гранулоцитарного колониестимулирующего фактора (G-CSF). В настоящее время применение факторов роста, таких как G-CSF, является обычной терапией для раковых пациентов с нейтропенией. Такая терапия стимулирует пролиферацию и дифференциацию гематопоэтических предшественников и также регулирует функциональную активность нейтрофилов и макрофагов. Однако лечение G-CSF является дорогостоящим, так как G-CSF является рекомбинантным белком, и сопровождается побочными действиями.
Эндогенный пуриновый нуклеозид аденозин является ubiquitous в типах клеток млекопитающих. Аденозин, присутствующий в плазме и других внеклеточных жидкостях, опосредует многие свои физиологические действия через рецепторы клеточной поверхности и является важным регуляторным белком. Он высвобождается во внеклеточную окружающую среду из метаболически активных или подвергнутых действию внешней среды клеток. Известно, что аденозин действует через связывание со специфическими ассоциированными с G-протеином мембранными рецепторами A1, A2 и А3(1-2). Взаимодействие аденозина со своими рецепторами инициирует пути сигнальной трансдукции, главным образом, аденилатциклазную эффекторную систему, утилизирующую сАМР в качестве второго переносчика. В то время как рецепторы A1 и A2, сочетающиеся с Gi-протеинами, ингибируют аденилатциклазу и приводят к снижению уровня внутриклеточного сАМР, рецептор A2, сочетающийся с Gs-протеинами, активирует аденилатциклазу, причем посредством этого повышается уровень сАМР(3).
Так как специфические для аденозина рецепторы поверхности обнаруживаются чуть ли не во всех клетках, почти все системы органов организма до некоторой степени регулируются его локальным высвобождением. Сюда относятся регуляция электрофизиологических свойств сердца, успокоение и подавление высвобождения нейротрансмиттеров и регуляция высвобождения реннина и сосудистого тонуса в почках(4-7). Аденозин проявляет различное действие на иммунную систему, включая противовоспалительную активность, через ингибирование высвобождения цитокинов, ингибирование агрегации тромбоцитов, индукцию продуцирования эритропоэтина и модуляцию функции лимфоцитов(8-10). Кроме того, обнаружено, что аденозин играет некую роль в модуляции некоторых функций центральной нервной системы (ЦНС), при заживлении ран, при диурезе и борьбе с болью. Также показано, что аденозин способен индуцировать пролиферацию самых разных типов здоровых клеток(11-14). Такая модуляция клеточного роста, вероятно, опосредуется через аденилатциклазную эффекторную систему, описанную выше.
В исследованиях, проводимых в последнее время, обнаружено, что аденозин действует как хемозащитное средство, и такая активность связана, вероятно, с его способностью стимулировать пролиферацию клеток костного мозга. Также обнаружено, что аденозин проявляет ингибирующее действие на пролиферацию опухолевых клеток, по-видимому, через остановку клеточного цикла GO/G1 и уменьшение теломерного сигнала(17-18). Такое двойственное действие аденозина является привлекательным для лечения рака.
Краткое изложение сущности изобретения
Согласно настоящему изобретению обнаружено, что агонисты аденозинового рецетора A3 (A3RAg) обладают двойным действием, проявляющимся в том, что они ингибируют пролиферацию злокачественных клеток, с одной стороны, и, с другой стороны, противодействуют токсичному побочному действию химиотерапевтических лекарственных средств. Конкретно, соединения A3RAg ингибируют пролиферацию и рост опухолевых клеток, проявляют синергизм с противоопухолевым цитотоксичным лекарственным средством по уменьшению опухолевой нагрузки, индуцируют пролиферацию и дифференциацию клеток костного мозга и клеток белой крови и противодействуют токсичному побочному действию других лекарственных средств, в частности химиотерапевтических лекарственных средств. Кроме того, согласно изобретению обнаружено, что A3RAg проявляет такие активности при различных формах введения, включая парентеральное введение и, в частности, пероральное введение. Также обнаружено согласно изобретению, что некоторые активности A3RAg можно имитировать с помощью других агонистов и антагонистов аденозиновых рецепторов A1 или А2: агонисты аденозинового рецептора А1 (A1RAg) делят с A3RAg его способность индуцировать секрецию G-CSF; агонист аденозинового рецептора А2 (A2RAg) делит с A3RAg его способность ингибировать пролиферацию злокачественных клеток; и антагонист аденозинового рецептора А2 (A2RAn) делит с A3RAg его способность противодействовать токсичному побочному действию лекарственных препаратов, например, при лечении или предупреждении лейкопении.
Изобретение, в самом широком смысле, относится к применению активного ингредиента с получением одного из следующих терапевтических/биологических эффектов: индуцирование продуцирования или секреции G-CSF в организме; предупреждение или лечение токсичного побочного действия лекарственного средства или предупреждение или лечение лейкопении, в частности лейкопении, вызванной лекарственным средством; и ингибирование аномального роста и пролиферации клеток. Активный ингредиент может представлять собой A3RAg или агониста, или антагониста системы аденозиновых рецепторов, которые могут дать один из указанных терапевтических эффектов, достигаемых применением A3RAg.
Возможно несколько вариантов воплощения изобретения. Первый вариант, называемый в данном описании "вариантом для индуцирования G-CSF’’, включает применение активного ингредиента, который может представлять собой A3Rag или A1Rag, с получением секреции G-CSF в организме обработанного субъекта. Известно, что G-CSF стимулирует пролиферацию и дифференциацию гематопоэтических предшественников и регулирует функциональную активность нейтрофилов и макрофагов. Таким образом, G-CSF-индуцирующий агент, такой, какие указывались выше, может иметь большую лечебную ценность, например, при противодействии (т.е. предупреждении, снижении или уменьшении интенсивности) миелотоксичности.
Согласно такому варианту своего воплощения изобретение относится к способу индуцирования секреции G-CSF в организме субъекта, включающему введение указанному субъекту эффективного количества активного ингредиента, выбранного из группы, состоящей из A3RAg, A1RAg и сочетания A3RAg и A1RAg. Согласно такому варианту изобретение также относится к способу лечения, включающему введение нуждающемуся в этом субъекту эффективного количества указанного активного ингредиента для достижения лечебного действия, включающего индукцию продуцирования или секреции G-CSF. К этому варианту относится также использование указанного активного ингредиента для получения фармацевтической композиции для индуцирования G-CSF секреции. К такому варианту также относится фармацевтическая композиция для индуцирования продуцирования или секреции G-CSF в организме, содержащая эффективное количество указанного активного ингредиента в фармацевтически приемлемом носителе.
Согласно другому варианту воплощения изобретения, называемому в описании "вариантом для предупреждения лейкопении" или конкретнее - "вариантом для предупреждения нейтропении", активный ингредиент, который может представлять собой A3RAg или A2RAn, применяют для предупреждения или лечения лейкопении, которая может быть результатом миелотоксичности.
Согласно такому варианту своего воплощения изобретение относится к способу индуцирования пролиферации или дифференциации клеток костного мозга или белой крови у субъекта, включающему введение указанному субъекту эффективного количества активного ингредиента, выбранного из группы, состоящей из A3RAg, A2RAn аденозина и сочетания A3RAg или A2RAn. Указанный вариант также относится к способу предупреждения или лечения лейкопении, включающему введение нуждающемуся в этом субъекту эффективного количества указанного активного ингредиента. Согласно такому варианту изобретение также относится к применению указанного активного ингредиента для изготовления фармацевтической композиции для индуцирования пролиферации или дифференциации клеток костного мозга или белой крови. Еще согласно такому варианту изобретение также относится к применению указанного активного ингредиента для изготовления фармацевтической композиции для предупреждения или лечения лейкопении. Фармацевтическую композицию можно применять, в частности, для предупреждения или лечения лейкопении.
Согласно родственному варианту, называемому в данном описании "вариантом для предупреждения токсичности", вышеуказанный активный ингредиент (а именно один из A3RAg или A2RAn, а также их сочетания) применяют для противодействия токсичному побочному действию лекарственных средств, таких как химиотерапевтические лекарственные средства или немолептические (nemoleptic) лекарственные средства.
Согласно указанному последнему варианту изобретение относится, таким образом, к способу предупреждения или лечения токсичных побочных действий лекарственного средства, включающему введение нуждающемуся в этом субъекту эффективного количества активного ингредиента, выбранного из группы, состоящей из A3RAg, A2RAn и сочетания A3RAg и A2RAn. Также указанный вариант относится к применению указанного активного ингредиента для изготовления фармацевтической композиции для предупреждения или лечения токсичности, вызываемой лекарственными средствами. Такой вариант также относится к фармацевтической композиции для предупреждения или лечения токсичных побочных действий лекарственного средства, содержащей эффективное количество указанного активного ингредиента и фармацевтически приемлемый носитель.
Вообще, для цели противодействия лейкопении, вызываемой лекарственными средствами, или токсичному побочному действию, вызываемому лекарственными средствами, иногда желательно составлять композицию из лекарственного средства, имеющего такое токсичное побочное действие, вместе с указанным активным ингредиентом для комбинированного введения обоих. Таким образом, изобретение также относится к фармацевтической композиции, содержащей в сочетании лекарственное средство, которое может вызвать токсичное побочное действие у субъекта, которого лечат посредством него, и указанный активный ингредиент; а также к применению указанного активного ингредиента для изготовления такой фармацевтической композиции. Указанные активные ингредиенты, включенные в указанную композицию, находятся в количестве, эффективном для предупреждения или лечения токсичного побочного действия.
Согласно еще одному варианту воплощения изобретения, называемому в описании "вариантом для ингибирования пролиферации", активный ингредиент, который может представлять собой A3RAg, A2RAg или их сочетание, применяют для селективного ингибирования аномального роста клеток, например роста опухолевых клеток.
Согласно такому варианту изобретение относится к способу ингибирования аномального роста клеток у субъекта, включающему введение субъекту терапевтически эффективного количества активного ингредиента, выбранного из группы, состоящей из A3RAg, A2RAg и сочетания A3RAg и A2RAg. Согласно такому варианту изобретение также относится к применению указанного активного ингредиента для изготовления фармацевтической композиции для ингибирования аномального роста клеток. Еще согласно такому варианту изобретение также относится к фармацевтической композиции для ингибирования аномального роста клеток, содержащей указанный активный ингредиент и фармацевтически приемлемый носитель.
В одном из вариантов воплощения изобретения введение активного ингредиента предназначается для достижения двойного терапевтического эффекта: ингибирования аномального роста клеток и уменьшения токсичного побочного действия лекарственного средства, вызывающего такое действие.
Предпочтительным активным ингредиентом согласно изобретению является A3RAg. Предпочтительным способом введения активного ингредиента согласно изобретению является пероральный способ. Однако указанное предпочтение не исключает ни другие активные ингредиенты, ни другие способы введения активных ингредиентов.
Дозировка активного ингредиента, в частности, в случае, когда активный ингредиент представляет собой A3RAg, предпочтительно составляет менее 100 мкг на кг массы тела, как правило менее 50 мкг и, желательно, находится в интервале 1-10 мкг на кг массы тела.
Подробное описание изобретения
Согласно изобретению предлагается новое лечебное применение некоторых активных веществ, в частности агонистов и антагонистов аденозиновых рецепторов. По одному из вариантов воплощения изобретения - варианту для индуцирования G-CSF некоторые такие вещества используют для опосредования продуцирования и секреции G-CSF из клеток. Согласно другому варианту для предупреждения токсичности некоторые такие вещества используют для противодействия токсичному побочному действию лекарственных средств, например химиотерапевтических или немолептических лекарственных средств. Еще в одном из вариантов - варианте для предупреждения лейкопении некоторые такие вещества используют для противодействия лейкопении, в частности лейкопении, вызываемой лекарственными средствами. Согласно еще одному из вариантов - варианту для ингибирования пролиферации некоторые такие вещества используют для селективного ингибирования аномального роста клеток.
Термин "лейкопения", используемый в данном описании, относится к уменьшению в кровотоке числа клеток белой крови. Хотя, как правило, лейкопения обычно характеризуется пониженным числом нейтрофилов в крови (нейтропения), время от времени может обнаруживаться пониженное число лимфоцитов, моноцитов, эозинофилов или базофилов.
Лейкопения, которая может явиться результатом пониженного продуцирования или избыточной селезеночной секвестрации нейтрофилов, может происходить из-за наследственных или врожденных заболеваний. Однако она наблюдается, главным образом, после лечения лекарственными средствами, такими как цитостатические противораковые лекарственные средства, антитиреоидные лекарственные средства, фенотиазины, противосудорожные средства, пенициллины, сульфонамиды и хлорамфеникол. Некоторые антибластомные средства вызывают лейкопению как предсказуемое побочное действие.
В дальнейшем в данном описании снижение числа лейкоцитов или числа нейтрофилов лекарственными средствами будет называться "лейкопенией, вызываемой лекарственными средствами" или "нейтропенией, вызываемой лекарственными средствами". Кроме того, когда упоминается лейкопения, этот термин следует понимать как относящийся, в частности, к "нейтропении".
Далее, термин "предупреждение или лечение лейкопении" следует понимать как обозначение процедуры, посредством которой снижение числа лейкоцитов, которое может происходить в противном случае, уменьшается, полностью предотвращается или, если такое снижение имеет место, процедуры, которая приводит к повышению числа лейкоцитов. Лейкопения проявляется посредством многих побочных действий, таких как повышенная возможность заражения инфекционными факторами, и других. Термин "предупреждение или лечение лейкопении" также следует понимать как обозначение улучшения таких параметров, которые могут иметь место в результате лейкопении.
Фармацевтически или терапевтически "эффективное количество" для целей, о которых идет речь в данном описании, определяется такими соображениями, которые могут быть известны в технике. Количество должно быть эффективным для достижения нужного терапевтического действия, которое зависит от типа и способа лечения. Как ясно специалисту, количество должно быть эффективным для получения улучшения коэффициента выживаемости, для получения более быстрого восстановления, для получения ослабления или устранения симптомов или любых других показателей, выбранных в качестве соответствующих единиц измерений специалистами в данной области. Когда, например, указанный активный ингредиент вводят для индуцирования продуцирования G-CSF, эффективное количество активного ингредиента может представлять собой количество, которое приводит к продуцированию и секреции G-CSF из мононуклеарных клеток периферической крови, эндотелиальных клеток или фибробластов, в которых продуцируется G-CSF, посредством, например, стимуляции созревания предшественников гранулоцитов до зрелых нейтрофилов. Когда активный ингредиент вводят для противодействия лейкопении, вызываемой лекарственными средствами, эффективное количество активного ингредиента может представлять собой количество, которое защищает индивидуума от вызываемого лекарственным средством снижения числа лейкоцитов, в частности нейтрофилов; количество активного ингредиента, которое может вызвать повышение уже пониженного содержания таких клеток, например восстановить содержание до нормального уровня, а иногда даже более высокого; и т.п. Когда активный ингредиент вводят для того, чтобы уменьшить токсичное побочное действие лекарственного средства, количество активного ингредиента может представлять собой, например, количество, эффективное для снижения потери массы, происходящей из-за вводимого лекарственного средства. Когда активный ингредиент вводят для того, чтобы ингибировать аномальный рост клеток, что подробнее определяется далее, эффективное количество может представлять собой количество, ингибирующее пролиферацию таких клеток у субъекта, которого лечат, и даже устранить опухоль. Когда активный ингредиент вводят для того, чтобы усилить действие противоракового химиотерапевтического лекарственного средства, эффективное количество может представлять собой количество, которое или повышает специфическую противораковую токсичность химиотерапевтического лечения; или количество, эффективное для снижения количества химиотерапевтического лекарственного средства или сочетания лекарственных средств, необходимых для достижения нужного действия химиотерапевтического лекарственного средства или сочетания лекарственных средств, т.е. снижения опухолевой нагрузки; и т.п. Примером эффективного количества является введение A3RAg в количестве менее 100 мкг на кг массы тела в сутки, как правило менее 50 мкг на кг массы тела и, необязательно, даже менее 10 мкг на кг массы тела, например 3-6 мкг на кг массы тела. Такое количество A3RAg, как правило, вводят в виде однократной суточной дозы, хотя иногда суточную дозу можно разделить на несколько доз, вводимых в течение суток, или иногда несколько суточных доз можно объединить в одну дозу, которую дают пациенту один раз в несколько дней, в частности, если вводят композицию с отсроченным высвобождением.
Активный ингредиент по изобретению представляет собой предпочтительно A3RAg. A3RAg является агонистом, который связывается с рецепторами A3 и затем активирует их с получением лечебного действия. Следует отметить, что иногда A3RAg также может взаимодействовать с другими рецепторами, например с рецепторами А1 и А2. Однако A3RAg, используемый согласно изобретению, проявляет свое основное действие через рецептор A3 (а именно могут также иметь место незначительные действия, проявляемые через взаимодействие с другими аденозиновыми рецепторами).
По одному из вариантов воплощения изобретения активный ингредиент по изобретению представляет собой нуклеозидное производное. Термином "нуклеозид" обозначается любое соединение, содержащее сахар, предпочтительно рибозу или дезоксирибозу, или пуриновое или пиримидиновое основание, или сочетание сахара с пуриновым или пиримидиновым основанием, предпочтительно за счет N-гликозильной связи. Термин "нуклеозидное производное" будет использоваться для обозначения в данном описании нуклеозида, встречающегося в природе, как определено выше, синтетического нуклеозида или нуклеозида, подвергнутого химическим модификациям путем вставки/вставок, делеции/ий или экзоциклического(их) и эндоциклического(их) замещения(ий) его группы/групп, или конформационным модификациям, дающим производное с нужным биологическим действием.
Согласно одному из предпочтительных вариантов воплощения изобретения активный ингредиент представляет собой A3RAg.
Согласно одному из вариантов воплощения изобретения активный ингредиент представляет собой нуклеозидное производное общей формулы (I)

где R1 представляет собой (C1-С10)-алкил, (C1-С10)-гидроксиалкил, (C1-С10)-карбоксиалкил или (C1-С10)-цианоалкил, или группу общей формулы (II)

где
- Y представляет собой кислород, серу или -СН2-;
- X1 представляет собой Н, (C1-С10)-алкил, RaRbNC(=O)- или HORc-, где Ra и Rb могут быть одинаковыми или разными и выбраны из группы, состоящей из водорода, (C1-С10)-алкила, амино, (C1-С10)-галогеналкила, (C1-С10)-аминоалкила, (C1-С10)-BOC-аминоалкила и (C3-С10)-циклоалкила, или соединяются вместе с образованием гетероциклического кольца, содержащего два-пять атомов углерода, и Rc выбирают из группы, состоящей из (C1-С10)-алкилена, -NH-, (C1-С10)-галогеналкилена, (C1-С10)-аминоалкилена, (C1-С10)-ВОС-аминоалкилена и (C3-С10)-циклоалкилена;
- Х2 представляет собой Н, гидроксил, (C1-С10)-алкиламино, (C1-С10)-алкиламидо или (C1-С10)-гидроксиалкил;
- Х3 и X4, каждый независимо, представляет собой водород, гидроксил, амино, амидо, азидо, галоген, алкил, алкокси, карбокси, нитрило, нитро, трифтор, арил, алкарил, тио, сложную тиоэфирную группу, простую тиоэфирную группу, -OCOPh, -OC(=S)OPh, или Х3 и Х4 - оба представляют собой кислород, связанный с >C=S с образованием 5-членного цикла, или Х2 и Х3 образуют цикл формулы (III)

где R’ и R’’ представляют собой, независимо, (C1-С10)-алкил;
- R2 выбирают из группы, состоящей из водорода, галогена, простой (C1-С10)-алкилэфирной группы, амино, гидразидо, (C1-С10)-алкиламино, (C1-С10)-алкокси, (C1-С10)-тиоалкокси, пиридилтио, (C2-С10)-алкенила, (C2-С10)-алкинила, тио и (C1-С10)-алкилтио; и
- R3 представляет собой группу -NR4R5, причем R4 представляет собой водород или группу, выбранную из алкила, замещенного алкила или арил-NH-C(Z)-, причем Z представляет собой О, S или NRa, причем Ra имеет указанные выше значения,
- и R5, когда R4 представляет собой водород, выбирают из группы, состоящей из R- и S-1-фенилэтильной, бензильной, фенилэтильной или анилидной групп, незамещенных или замещенных в одном или нескольких положениях заместителем, выбранным из группы, состоящей из (C1-С10)-алкила, амино, галогена, (C1-С10)-галогеналкила, нитро, гидроксила, ацетамидо, (C1-С10)-алкокси и сульфоновой кислоты или ее соли; или R4 представляет собой бензодиоксанметил, фурфурил, L-пропилаланиламинобензил, β-аланиламинобензил, Т-ВОС-β-аланиламинобензил, фениламино, карбамоил, фенокси или (C3-С10)-циклоалкил, или R5 представляет собой группу формулы

или подходящую соль соединения, определение которому дается выше, например его триэтиламмониевую соль; или
- когда R4 представляет собой группу, выбранную из алкила, замещенного алкила или арил-NН-С(Z)-, тогда R5 выбирают из группы, состоящей из замещенного или незамещенного гетероарил-NRa-C(Z)-, гетероарил-С(Z)-, аралкил-NRa-С(Z)-, аралкил-С(Z)-, арил-NR-C(Z)- и арил-С(Z)-;
где Z имеет значения, указанные выше.
Согласно данному варианту воплощения изобретения активный ингредиент предпочтительно является нуклеозидным производным общей формулы (IV)

где X1, R2 и R4 имеют значения, указанные выше.
Предпочтительные активные ингредиенты по данному варианту воплощения изобретения можно, как правило, отнести к N6-бензиладенозин-5'-уронамидам, и обнаружено, что их производные являются агонистами, селективными в отношении аденозинового рецептора A3. Примерами таких производных являются N6-2-(4-аминофенил)этиладенозин (APNEA), N6-(4-амино-3-иодбензил)-аденозин-5'-(N-метилуронамид) (АВ-МЕСА) и 1-дезокси-1-{6-[({3-иодфенил}метил)амино]-9Н-пурин-9-ил}-N-метил-β-D-рибофурануронамид, последний также называют N6-3-иодбензил-5’-метилкарбоксамидоаденозином, N6-(3-иодбензил)-аденозин-5’-N-метилуронамидом, и обозначается выше и далее в описании аббревиатурой IB-MECA, или хлорированное производное IB-MECA (R2=Сl), называемое Cl-IB-MECA. В настоящее время особенно предпочтительны IB-MECA и Cl-IB-MECA.
Согласно другому варианту воплощения изобретения активный ингредиент может представлять собой производное аденозина, как правило, имеют в виду N6-бензиладенозин-5'-алкилуронамид-N1-оксид или N6-бензиладенозин-5'-N-диалкилуронамид-N1-оксид.
Кроме того, активный ингредиент может представлять собой ксантин-7-рибозидное производное общей формулы (V)

где Х представляет собой О или S;
- R6 представляет собой RaRbNC(=O)- или HORc-,
где
- Ra и Rb могут быть одинаковыми или разными и выбраны из группы, состоящей из водорода, (С1-С10)-алкила, амино, (С1-С10)-галогеналкила, (С1-С10)-аминоалкила и (С3-С10)-циклоалкила, или соединяются вместе с образованием гетероциклического кольца, содержащего два-пять атомов углерода, и
- Rc выбирают среди (С1-С10)-алкила, амино, (С1-С10)-галогеналкила, (С1-С10)-аминоалкила, (С1-С10)-ВОС-аминоалкила и (С1-С10)-циклоалкила;
- R7 и R8 могут быть одинаковыми или разными, и их выбирают из группы, состоящей из (С1-С10)-алкила, (С1-С10) -циклоалкила, R- или S-1-фенилэтильной, незамещенной бензильной или анилидной группы и простого фенилового эфира бензильной группы, замещенной в одном или нескольких положениях заместителем, выбранным из группы, состоящей из (С1-С10)-алкила, амино, галогена, (С1-С10)-галогеналкила, нитро, гидроксила, ацетамидо, (С1-С10)-алкокси и сульфоновой кислоты;
- R9 выбирают из группы, состоящей из галогена, бензила, фенила, (С3-С10)-циклоалкила и (С1-С10)-алкокси;
или соль такого соединения, например его триэтиламмониевую соль.
Некоторые из описанных выше соединений и методику их синтеза можно найти в US 5688774; US 5773423, US 6048865, WO 95/02604, WO 99/20284 и WO 99/06053, включенных в настоящее описание в качестве ссылок.
Активный ингредиент в случае варианта для индуцирования GSF также может представлять собой A1RAg. Он является, как правило, производным аденозина приведенной далее формулы

где R1 представляет собой низший алкил, циклоалкил, предпочтительно (С3-С8)-циклоалкил (включая хорошо известные циклогексил- и циклопентилсодержащие производные, известные как СРА и СНА соответственно), циклоалкильная группа может быть замещена, например гидроксилом или низшим алкилом; R1 также представляет гидроксил или гидроксиалкил; фенил, анилид или низший алкилфенил, которые все, необязательно, замещены одним или несколькими заместителями, например галогеном, низшим алкилом, галогеналкилом, таким как трифторметил, нитро, циано, -(CH2)mCO2Ra, -(CH2)mCONR2RaRb, -(CH2)mCORa, причем m равен целому числу от 0 до 6; -SORc, -SO2Rc, -SО3Н, -SO2NRaRb, -ORa, -SRa, -NHSO2Rc, -NHCORa, -NRaRb или -NHRaCO2Rb; где
- Ra и Rb представляют, независимо, водород, низший алкил, алканоил, фенил или нафтил (последний может быть частично насыщенным), причем алкильная группа, необязательно, замещена замещенной или незамещенной фенильной или феноксигруппой; или, когда R1 представляет -NRaRb, указанные Ra и Rb образуют вместе с атомом азота 5- или 6-членное гетероциклическое кольцо, необязательно содержащее второй гетероатом, выбранный среди кислорода или азота, и указанный второй гетероатом может быть, необязательно, также замещен водородом или низшим алкилом; или -NRaRb представляет собой группу общей формулы (VII) или (VIII)


где n равен целому числу от 1 до 4;
- Z представляет собой водород, низший алкил или гидроксил;
- Y представляет собой водород, низший алкил или OR', где R' представляет собой водород, низший алкил или низший алканоил;
- А представляет собой связь или низший алкилен, предпочтительно (C1-C4)-алкенил; и
- Х и X' представляют собой, каждый независимо, водород, низший алкил, низший алкокси, гидрокси, низший алканоил, нитро, галогеналкил, такой как трифторметил, галоген, амино, моно- или ди(низший алкил)амино, или Х и X', взятые вместе, представляют метилендиоксигруппу;
- Rc представляет собой низший алкил;
- R2 представляет водород; галоген; замещенную или незамещенную низшую алкильную или алкенильную группу; низший галогеналкил или галогеналкенил; циано; ацетамидо; низший алкокси; низший алкиламино; NRdRe, где Rd и Re представляют собой, независимо, водород, низший алкил, фенил или фенил, замещенный низшим алкилом, низшим алкокси, галогеном или галогеналкилом, таким как трифторметил, или алкоксилом; или -SRf где Rf представляет собой водород, низший алкил, низший алканоил, бензоил или фенил;
- W представляет группу -ОСН2-, -NHCH2-, -SCH2- или -NН(С=O);
- R3, R4 и R5 представляют, независимо, водород, низший алкил или низший алкенил, разветвленный или неразветвленный (C1-C12)-алканоил, бензоил или бензоил, замещенный низшим алкилом, низшим алкокси, галогеном, или R4 и R5 вместе образуют пятичленный цикл, необязательно замещенный низшим алкилом или алкенилом; R3 также представляет, независимо, фосфат, вторичный фосфат или первичный фосфат, или щелочной металл или аммоний, или его двухосновный или диаммониевый ион;
- R6 представляет водород, атом галогена; или
- одна из групп R (т.е. R1-R6) представляет собой серосодержащий углеводородный радикал формулы Rg-SО3-Rh, где Rg представляет группу, выбранную из (C1-С10)-алифатической, фенильной и замещенной низшим алкилом ароматической группы, которая может быть замещенной или незамещенной, и R11 представляет одновалентный катион. Подходящими одновалентными катионами являются ионы лития, натрия, калия и аммония или триалкиламмоний, которые делают возможной диссоциацию для замены в физиологических условиях. Остальные группы R представляют собой водород или атом галогена, незамещенный углеводородный остаток или любую другую не содержащую серы группу, имеющую указанные выше значения.
В данном случае углеводородные цепи могут быть линейными или разветвленными. В частности, термины "алкил" или "алкенил", используемые в данном описании, обозначают алкильные или алкенильные группы с линейными или разветвленными цепями. Термины "низший алкил" или "низший алкенил" обозначают соответственно (C1-С10)-алкильные или (C2-С10)-алкенильные группы, и предпочтительно - (C1-С6)-алкильные и (C2-С6)-алкенильные группы.
Предпочтительными производными аденозина формулы (VI) являются производные N6-циклопентиладенозин (СРА), 2-хлор-СРА (ССРА) и N6-циклогексиладенозин (СНА), получение которых хорошо известно специалистам в данной области. Другие производные аденозина, о которых известно, что они селективны в отношении рецептора А1, представляют собой производные, где R1 является анилидной группой, и последняя может быть незамещенной или замещена, например, гидроксилом, алкилом, алкокси или группой -СН2С(О)R’’, причем R’’ представляет собой гидроксильную группу, -NHCH3, -NHCH2CO2C2H5 (этилглицинат), толуоидид (также где метильная группа замещена галогеналкильной группой), или группой -CH2C(O)NHC6H4CH2C(O)R’’’, где R’’’ представляет группу, образующую метилэфирный заместитель (-ОСН3), амидный заместитель (например, R’’’ представляет группу -NНСН3), или R’’’ представляет собой гидразид, этилендиамин, -NHC2H5NHC(O)СН3, 4-(гидроксифенил)пропионил, биотинилированный этилендиамин или любой другой подходящий углеводород, который представляет соединение агонист A1.
С другой стороны, N6-замещенные производные аденозина, применяемые согласно настоящему изобретению в качестве активных ингредиентов, могут представлять собой производные, содержащие эпоксидную группу и, конкретнее, представляющие собой циклоалкилэпоксисодержащие производные аденозина (например, оксабицикло, такие как норборнанил, или оксатрицикло, такие как адамантанил). Некоторые такие соединения можно определить общей формулой (I),
где R1 представляет собой группу общей формулы (IХа) и (IXb)


где М представляет собой низшую алкильную группу, имеющую значения, указанные выше.
Варианты соединений агонистов, содержащих эпоксид-N6-норборнильную группу, включают эндо- и экзо-изомеры и, конкретнее, могут представлять собой один из четырех изомеров: форму 2R-экзо, 2R-эндо, 2S-экзо и 2S-эндо.
Другой вариант N6-нopбopнильнoгo производного может включать атом кислорода в положение N1 пуринового цикла. Такое соединение называется N6-(5,6-эпоксинорборн-2-ил)аденозин-1-оксидом.
Иногда A1RAg может представлять собой производное аденозина, в котором β-D-рибофуранозильная группа аденозина заменена водородом или фенильной группой.
A2RAn, которые можно использовать согласно изобретению, представляют собой 8-стирильные производные 1,3,7-замещенных ксантинов формулы (X)

где R1 и R3 представляют собой (C1-C4) -алкил, аллил или пропаргил;
R7 представляет собой Н, метил или (C2-C8) -алкил; n равен 1-3;
и Х представляет собой галоген, трифторалкил, алкокси, гидрокси, нитро, амино, диалкиламино, диазоний, изотиоцианат, бензилокси, аминоалкокси, алкоксикарбониламино, ацетокси, ацетиламино, сукциниламино, 4-(4-NН2-транс-CH2CH=CHCH2O-3,5-(MeO)2, 4-(4-АсNН-транс-СН2СН=СНСН2O)-3,5-(МеО)2, 4-(4-трет-ВОС-NН-транс-СН2СН=СНСН2O)-3,5-(МеО)2.
Конкретным примером соединения формулы (X) является (3,7-диметил-1-пропаргилксантан).
A2RAn также могут представлять собой соединения приведенных далее формул

или

Как очевидно, изобретение не может ограничиваться конкретными соединениями A3RAg, A2RAg или A2RAn, указанными выше.
Активный игредиент согласно изобретению может быть таким, какой описан выше, или может находиться в форме своих солей или сольватов, в частности, своих физиологически приемлемых солей и сольватов. Кроме того, когда активный ингредиент содержит один или несколько асимметричных атомов углерода, он может включать изомеры и диастереоизомеры описанных выше активных ингредиентов или их смеси.
Фармацевтически приемлемые соли описанных выше активных ингредиентов включают соли, образованные фармацевтически приемлемыми неорганическими и органическими кислотами. Примерами подходящих кислот являются хлороводородная, бромоводородная, серная, азотная, перхлорная, фумаровая, малеиновая, фосфорная, гликолевая, молочная, салициловая, янтарная, толуол-п-сульфоновая, винная, уксусная, лимонная, метансульфоновая, муравьиная, бензойная, малоновая, нафталин-2-сульфоновая и бензолсульфоновая кислоты.
Активный ингредиент можно вводить в виде неактивного вещества (например, пролекарства), и он становится активным только после дальнейшей(их) модификации(ий) естественным способом в определенном участке организма субъекта. В любом случае, производное будет таким, чтобы сохранялась терапевтическая функциональность фармацевтической композиции изобретения. Такие пролекарства также охватываются термином "активный ингредиент", используемым в данном описании. Подобным образом, термины "A3RAg", "A1RAg", "A1RAn", "A2RAg" и ″A2Ran″ следует понимать как охватывающие пролекарства, которые хотя a priori не обладают агонистической или антагонистической активностью (такой случай может быть), становятся активными in vivo.
A3RAg согласно изобретению можно выбрать путем отбора таких соединений, которые качественно обладают активностью, схожей с активностью IB-MECA. Например, такие соединения для применения в соответствии с вариантом для ингибирования лейкопении можно выбрать, основываясь на их способности стимулировать пролиферацию клеток костного мозга или белой крови, и затем, основываясь на их способности проявлять свою активность in vivo. Для применения в варианте для ингибирования пролиферации соединения можно отобрать по их способности ингибировать пролиферацию опухолевых клеток и также затем по проявлению их активности in vivo.
A1RAn и A2RAn можно испытать на их активность и отобрать для применения при лечении таким же способом, какой описан для A3RAg, с соответствующими изменениями.
Фармацевтическая композиция изобретения может содержать активный ингредиент как таковой, но может содержать и другие ингредиенты, которые могут представлять собой фармацевтически приемлемый носитель, разбавитель, эксципиент, добавку и/или адьювант, известные специалистам, например, для целей придания фармацевтической композиции вкуса, запаха, окраски, смазывающих свойств или подобных свойств. Очевидно, фармацевтически приемлемый(е) носитель(и), разбавитель(и), эксципиент(ы), добавка(и), используемые согласно изобретению, как правило, должны относиться к инертным нетоксичным твердым или жидким наполнителям, разбавителям или инкапсулирующим материалам, которые предпочтительно не взаимодействуют с соединениями в композиции изобретения.
Далее, активный ингредиент также можно вводить в сочетании с химиотерапевтическим лекарственным средством, в частности, в случае варианта для предупреждения лейкопении. Таким образом, фармацевтическая композиция по изобретению может содержать, кроме указанного активного ингредиента, химиотерапевтическое лекарственное средство. Согласно одному из вариантов воплощения изобретения, химиотерапевтическое лекарственное средство представляет собой противораковое химиотерапевтическое лекарственное средство. Следует иметь в виду, что таким термином обозначается любое цитотоксичное лекарственное средство или смесь, содержащая комбинацию двух или нескольких цитотоксичных лекарственных средств, назначаемых пациенту для цели снижения у него массы опухоли.
Один из выводов согласно изобретению состоит в том, что А3RАg является биологически доступным перорально и проявляет свою двойственную активность (снижение клеточной пролиферации и предупреждение или ослабление лейкопении) при пероральном введении. Таким образом, согласно одному из предпочтительных вариантов фармацевтическую композицию изобретения получают для перорального введения. Такая пероральная композиция может также содержать фармацевтически приемлемый носитель, разбавитель, эксципиент, добавку или адъювант, подходящие для перорального введения.
В объеме варианта воплощения изобретения для индуцирования G-CSF описываемую фармацевтическую композицию применяют, в частности, для повышения содержания G-CSF, секретируемого клетками. Такие композиции можно применять для ускорения восстановления нейтрофилов после химиотерапии и пересадки костного мозга или для ингибирования аномального роста клеток. В настоящее время такое лечение включает введение самих факторов роста, которые, как известно, обладают нежелательным побочным действием. Более того, известно, что средняя стоимость G-CSF терапии очень высокая.
В пределах объема варианта воплощения настоящего изобретения для предупреждения лейкопении или варианта для предупреждения токсичности описываемую фармацевтическую композицию используют, в частности, для повышения у субъекта уровня лейкоцитов в кровотоке или противодействия другим токсичным действиям, таким как потеря массы. Данный аспект изобретения применим в различных клинических ситуациях. Очевидно, что пониженный уровень лейкоцитов в кровотоке, и в частности нейтрофилов, может привести к ослаблению иммунной системы. Примером ослабления иммунной системы, которое можно лечить согласно такому аспекту изобретения, является таковое, часто встречающееся на ранних стадиях рака, или ослабление, являющееся результатом лейкопении, вызываемой лекарственными средствами, или нейтропении, вызываемой лекарственными средствами.
Вариант для ингибирования пролиферации полезен для лечения различных аномалий, связанных с аномальным ростом клеток, таких как рак, псориаз и некоторые аутоиммунные заболевания. В частности, композицию изобретения используют для ингибирования пролиферации опухолевых клеток, предпочтительно в рамках противораковой терапии.
В случае обработки A3RAg клеток лимфомы ингибирование пролиферации указанных клеток выражено более явно, чем при воздействии аденозина или агонистов ‘A1’ или ‘A2’, хотя некоторая активность наблюдается также с A2RAg (см., например, фиг.5А). Такие результаты показывают, что ингибирование пролиферации опухолевых клеток следует приписывать главным образом связыванию A3RAg со своим соответствующим рецептором, но это явление до некоторой степени также может имитироваться A2RAg. Вышеуказанные неожиданные результаты также предлагают новую лечебную мишень для будущих противораковых цитостатических лекарственных средств.
Также обнаружено, что A3RAg является сильным ингибитором роста других опухолевых клеток, кроме клеток лимфомы, например меланомы или карциномы ободочной кишки (см., например, фиг.6). Опытный специалист в данной области отчетливо представит преимущество лечения субъекта неспецифическим противораковым лекарственным средством, способным ингибировать рост аномально делящихся клеток, причем в то же время способным восстанавливать иммунную систему субъекта путем индуцирования пролиферации клеток костного мозга.
Фиг.7А-7В, например, показывают дифференцирующее действие A3RAg. В данном конкретном случае оценивают действие IB-MECA на опухолевые и здоровые клетки. Приведенные данные также ясно представляют более выраженное действие, полученное с использованием A3RAg по сравнению с аденозином. Терапевтическое действие A3RAg также полностью меняется, когда используют антагонист рецептора A3 MRS-1220.
Исследования in vivo подтверждают результаты, полученные in vitro, и показывают хемозащитное действие A3RAg на мышах, обрабатываемых одновременно A3RAg и цитотоксичным средством, при сравнении с мышами, обработанными только цитотоксичным лекарственным средством (см., например, фиг.8). Кроме того, наблюдают снижение числа очагов у мышей, обработанных A3RAg, что указывает на химиотерапевтическую активность A3RAg (см., например, фиг.9). Фигуры 10А-10В, а также 19А и 19В, например, показывают, что мыши, имеющие опухоль, обрабатываемые только цитотоксичным лекарственным средством, обнаруживают падение числа лейкоцитов и нейтрофилов периферической крови, в то время как введение А3RАg после химиотерапии приводит к восстановлению общего числа клеток белой крови, проявляющемуся в повышении процентного содержания нейтрофилов.
Таким образом, можно сделать вывод, что А3RАg обладает двойственной терапевтической функцией, так как действует и как химиотерапевтическое средство, и как хемозащитное средство. Понятно, что применение A3RAg для достижения такого двойного действия также входит в объем настоящего изобретения.
В любом случае фармацевтические композиции изобретения вводят и дозируют согласно принятой медицинской практике с учетом клинического состояния отдельного пациента, места и способа введения, графика введения, возраста, пола, массы тела пациента и других факторов, известных практикующим врачам.
Композицию изобретения можно вводить разными способами. Ее можно вводить перорально, подкожно или парентерально, в том числе внутривенно, интраартериально, внутримышечно, интраперитонеально, или посредством интраназального введения, а также внутриоболочечно и методами инфузии, известными специалистам в данной области.
Как известно, курс лечения у людей, как правило, продолжительнее, чем у животных, т.е. мышей, которых используют в приведенных примерах. Лечение имеет длительность, пропорциональную длительности болезненного процесса и эффективности активного вещества. Схема лечения включает однократные или многократные дозы в течение периода в несколько дней или более длительного. Лечение, как правило, имеет длительность, зависящую от хода болезненного процесса, эффективности активного вещества и вида пациента, которого лечат.
Когда композицию настоящего изобретения вводят парентерально, как правило, ее получают в стандартной лекарственной форме для инъекций (раствор, суспензия, эмульсия). Фармацевтическая композиция, подходящая для инъекции, включает стерильные водные растворы или дисперсии и стерильные порошки для восстановления в стерильные растворы или суспензии для инъекции. Используемый носитель может представлять собой растворитель или дисперсионную среду, содержащие, например, воду, этанол, полиол (например, глицерин, пропиленгликоль, липидполиэтиленгликоль и т.п.), их подходящие смеси и растительные масла.
Неводные носители, такие как хлопковое масло, кунжутное масло, оливковое масло, соевое масло, кукурузное масло, подсолнечное масло или арахисовое масло, и сложный эфир, такой как изопропилмиристат, также иногда можно использовать в качестве системы растворителей для активного ингредиента.
Кроме того, можно добавлять различные добавки, повышающие устойчивость, стерильность и изотоничность композиций, в том числе антимикробные консерванты, антиоксиданты, хелатообразователи и буферные вещества. Предупреждение действия микроорганизмов можно обеспечить с помощью различных антимикробных и противогрибковых веществ, например парабенов, хлорбутанола, фенола, сорбиновой кислоты и т.п.
Для цели перорального введения активный ингредиент можно ввести в применяемые формы таблеток, суспензий, растворов, эмульсий, капсул, порошков, сиропов и т.п., которые можно получить методами, хорошо известными фармацевтам.
Настоящее изобретение определяется формулой изобретения, содержание которой должно прочитываться в описании, и теперь будет поясняться примерами со ссылкой на прилагаемые фигуры. Следует иметь в виду, что используемая терминология предназначается для описания, а не для ограничения.
Хотя в приведенном выше описании подробно описывается только несколько конкретных вариантов осуществления изобретения, специалистам в данной области следует иметь в виду, что изобретение ими не ограничивается, и что возможны другие изменения в форме и деталях без отхода от объема и сущности описываемого изобретения.
Краткое описание фигур
Для того, чтобы понять изобретение и представить, как его можно осуществить на практике, теперь будут описаны предпочтительные варианты воплощения изобретения с помощью примеров, не являющихся ограничительными, со ссылкой на прилагаемые чертежи, на которых изображено следующее.
Фиг.1 представляет гистограмму, показывающую результаты анализа in vivo, при котором демонстрируется влияние аденозина (Ad), DPCPX (A1RAn), СРА и ССРА (оба A1RAg) или IB-MECA (A3RAg) на продуцирование G-CSF. Культуры, обработанные модифицированной RPMI, служат в качестве контроля. Результаты приводятся в виде процента от контроля (контроль = 100%).
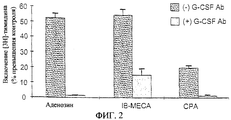
Фиг.2 представляет гистограмму, показывающую результаты анализа с включением [3Н]-тимидина - эксперимента, при котором проверяют стимуляцию пролиферации клеток костного мозга аденозином, СРА или IB-MECA с антителами против G-CSF ((+) G-CSF Аb - слабозаштрихованные столбики) или без них ((-) G-CSF Аb - густо заштрихованные столбики). Результаты показывают нейтрализующее действие антител против G-CSF. Результаты приводятся в виде процента повышения по сравнению с контролем (контроль = 0%).
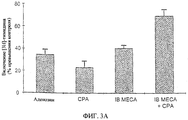
Обе фигуры - фиг.3А и фиг.3В представляют собой гистограммы, показывающие результаты анализа с включением [3Н]-тимидина - эксперимента, при котором проверяют пролиферацию клеток костного мозга в присутствии аденозина, агонистов аденозиновых рецепторов (фиг.3А) или аденозина в сочетании с антагонистами аденозиновых рецепторов (фиг.3В). Испытываемыми рецепторными агонистами (фиг.3А) являются СРА (A1RAg) и IB-MECA (A3RAg); испытываемыми рецепторными антагонистами (фиг.3В) являются DPCPX (A1RAn), DMPX (A2RAn) и MRS (A3RAn). Результаты приводятся в виде процента повышения включения тимидина по сравнению с контролем (контроль = 0%).
Фиг.4 представляет собой гистограмму, показывающую результаты эксперимента in vitro, при котором проверяют пролиферацию клеток костного мозга при трех различных концентрациях IB-MECA (0,01 мкМ, 0,1 мкМ и 1,0 мкМ). Результаты приводятся в виде процента повышения включения [3Н]-тимидина по сравнению с контролем (контроль = 0%). Цифры под столбиками показывают концентрации IB-MECA (в мкМ).
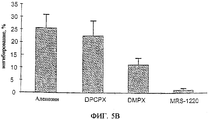
Фиг.5А и 5В представляют собой гистограммы, показывающие результаты двух экспериментов, оба in vitro и основанные на анализе числа клеток, при которых проверяют влияние аденозина и его антагониста на рост клеток лимфомы (Nb2-11C). В эксперименте, отраженном на фиг.5А, проверяют влияние на рост клеток лимфомы аденозина, СРА (A1RAg), DMPA (A2RAg) или IB-МЕСА (A3RAg). В эксперименте, отраженном на фиг.5В, проверяют влияние на рост клеток лимфомы аденозина, DPCPX (A1RAn), DPMX (A2RAn) или MRS-1220 (A3RAn). В качестве контроля служат клетки лимфомы, обработанные RPMI. Результаты приводятся в виде % ингибирования роста клеток относительно контроля (контроль = 0%).
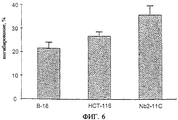
Фиг.6 представляет собой гистограмму, показывающую результаты анализа in vitro, при котором рост клеток опухолей разных типов (меланомы В16, карциномы ободочной кишки НТС-116, лимфомы Nb2-11C) ингибируют в присутствии A3RAg IB-MECA. В качестве контроля служат клетки, обработанные RPMI. Результаты приводятся в виде % ингибирования роста клеток относительно контроля (контроль = 0%).
Фиг.7А и 7В представляют собой гистограммы, показывающие результаты анализа in vitro, при котором проверяют влияние аденозина или A3RAg IB-MECA на рост опухолевых клеток (лимфомы Nb2-11C, фиг.7А) или клеток костного мозга (фиг.7В). Результаты на фиг.7А и 7В приводятся в виде процента ингибирования и процента стимуляции соответственно относительно контроля (контроль = 0%).
Фиг.8 представляет собой гистограмму, показывающую результаты эксперимента in vivo, при котором проверяют число клеток периферической белой крови (WBC) после 5 и 9 дней обработки химиотерапевтическим лекарственным средством (циклофосфамидом). Циклофосфамид вводят или один (слабозаштрихованные столбики), или в сочетании с IB-MECA, вводимым перорально (в 1 мл раствора) ежедневно, начиная с 24 часов после введения химиотерапевтического лекарственного средства. В качестве контроля служат мыши, обработанные PBS. Число WBC (числа WBC) приводится в виде процента, превышающего контроль (контроль = 0%).
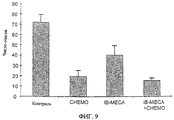
Фиг.9 представляет собой гистограмму, показывающую результаты эксперимента in vivo, при котором проверяют число очагов меланомы, развивающейся у мышей после инокуляции 2×105 клеток меланомы мышам, обработанным химиотерапевтическим средством циклофосфамидом (СНЕМО), A3RAg IB-MECA, сочетанием IB-MECA и СНЕМО или забуференным фосфатом физиологическим раствором (PBS), что служит в качестве контроля.
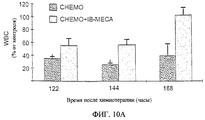
Фиг.10А и 10В представляют собой гистограммы, показывающие результаты эксперимента in vivo, демонстрирующего химиотерапевтическую активность IB-MECA. Показывается содержание клеток белой крови (WBC, фиг.10А) и нейтрофилов (фиг.10В) как функция времени (часы после введения химиотерапевтического лекарственного средства циклофосфамида (СНЕМО) с IB-MECA (СНЕМО + IB-MECA) и без IB-MECA). В качестве контроля служат мыши, обработанные PBS. Число нейтрофилов приводится в виде %, превышающего контроль (контроль = 0%).
Фиг.11 показывает массу "голых" мышей на 7, 10 и 14 дни после начала обработки (введение 5-FU, Cl-IB-MECA или сочетания 5-FU и Cl-IB-MECA) в виде % от контроля (необработанные мыши = 100%). Обработка заключается во введении 5-FU (густо заштрихованные столбики), введении 5-FU в сочетании с Cl-IB-MECA (A3RAg) - слабозаштрихованные столбики) и одного Cl-IB-MECA (незаштрихованные столбики).
Фиг.12А и 12В показывают результаты эксперимента, при котором проверяют влияние Cl-IB-MECA на снижение миелотоксичности, вызванной доксорубицином. Эксперимент осуществляют на мышах ICR. Фиг.12А показывает число клеток белой крови (WBC), в то время как фиг.12В показывает число ядросодержащих клеток костного мозга. На фиг.12А приводятся результаты двух различных обработок в четыре разные момента времени, причем уровень для контроля показан пунктирной линией, в то время как на фиг.12В приводятся результаты в два разные момента времени, и контрольный уровень представлен столбиком с левой стороны.
Фиг.13 показывает действие антител против G-CSF на число клеток белой крови (WBC) у контрольных мышей, мышей, обработанных химиотерапевтическим лекарственным средством, и мышей, обработанных химиотерапевтическим лекарственным средством и Cl-IB-MECA, вводимым перорально (6 мкг на кг массы тела, в 0,2 мл PBS). Число WBC после инъекции антител против G-CSF представляют слабозаштрихованные столбики. Все результаты приводятся в процентах от контроля (контроль = 100%).
Фиг.14 показывает размер опухоли со временем, развивающейся у "голых" мышей после инъекции клеток карциномы ободочной кишки человека НСТ-116, в контрольной группе и в обработанной группе (пероральное введение Cl-IB-MECA).
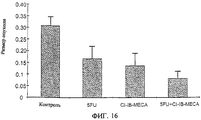
Фиг.15 показывает результаты эксперимента, подобного отраженному на фиг.14, где измеряют размер опухоли, развивающейся у мышей после инъекции клеток карциномы ободочной кишки человека НСТ-116. Испытывают четыре группы: контрольную группу, группу, получающую химиотерапевтическое лекарственное средство 5-FU, группу, которой вводят перорально Cl-IB-MECA, и группу, получающую комбинированную обработку 5-FU и Cl-IB-MECA.
Фиг.16 представляет собой гистограмму, показывающую размер опухоли на 30 день эксперимента, отраженного на фиг.15.
Фиг.17 представляет собой гистограмму, показывающую результаты эксперимента, где определяют пролиферацию клеток костного мозга, индуцированную Cl-IB-MECA, при разных концентрациях (0,05 мкг/мл и 0,5 мкг/мл) антител против G-CSF (0 - отсутствие антител). Пролиферацию определяют с помощью анализа с включением [3Н]-тимидина.
Фиг.18 показывает результаты эксперимента in vitro, где определяют пролиферацию или клеток меланомы В-16, или клеток костного мозга. Пролиферацию определяют с помощью анализа с включением [3Н]-тимидина. Клетки подвергают воздействию или 0,01 мкМ или 0,1 мкМ Cl-IB-MECA с A3RAg MRS-1523 (слабозаштрихованные столбики) или без A3RAg MRS-1523 (густозаштрихованные столбики). Результаты приводятся в виде процентов от контроля (контроль = 100%).
Фиг.19А и 19В показывают результаты эксперимента, подобного отраженному на фиг.10А и 10В соответственно, осуществленного с Cl-IB-MECA.
Фиг.20 показывает результаты эксперимента in vitro, где определяют пролиферацию клеток костного мозга, индуцированную IB-MECA или Cl-IB-MECA. Указанные два A3RAg добавляют к культуре клеток костного мозга в концентрации или 1 нМ, или 10 нМ, с добавлением A3RAn MRS-1523 в концентрации 10 нМ (слабозаштрихованные столбики - "(+) антагонисты") или без добавления (густозаштрихованные столбики - "(-) антагонисты"). Пролиферацию определяют с помощью анализа с включением [3Н]-тимидина. Результаты приводятся в виде процента стимулирования относительно контроля (необработанные клетки костного мозга, контроль = 0%).
Экспериментальные результаты
Опухолевые клетки
Использовали мышиные опухолевые клеточные линии (меланомы В-16 и лимфомы крыс Nb2 11c). Клетки меланомы В-16 получены из Американской коллекции типовых культур (АТСС), Роквилл, Мэриленд. Клетки лимфомы крыс Nb2-11C [Pines М., and Gertler A., J. of Cellular Biochem., 37; 119-129 (1988)] любезно предоставлены д-ром A. Gertler, Hebrew университет, Израиль.
Использовали также клетки карциномы ободочной кишки (НСТ-116), полученные из АТСС.
Клетки обычным образом хранят в среде RPMI, содержащей 10% сыворотки плода коровы (FBS, Biological Industries, Beit Haemek, Израиль). Дважды в неделю клетки переносят в свежеприготовленную среду.
Здоровые клетки
Использовали клетки костного мозга, полученные из бедренной кости мышей C57BL/6J. Клетки получали так, как описано ранее [17].
Лекарственные средства, соединения
Использовали лекарственные средства: аденозин; агонисты аденозинового рецептора А1: ССРА [2-хлор-N6-циклопентиладенозин], СРА (N-циклопентиладенозин); A1Ran: DPCPX (1,3-дипропил-8-циклопентилксантин); агонист аденозинового рецептора A2: DMPA (N6-[2-(3,5-диметоксифенил)-2-(2-метилфенил)этил]аденозин); A2RAn: DMPX (3,7-диметил-1-пропаргилксантан); A3Rag: IB-MECA (1-дезокси-1-{6-[({3-иодфенил}метил)амино]-9Н-пурин-9-ил}-N-метил-β-D-рибофурануронамид), CE-IB-MECA (2-хлор-N6-3-иодбензил)аденозин-5'-N-метилуронамид); и антагонисты аденозинового рецептора A3: MRS-1523 (5-пропил-2-этил-4-пропил-3-этилсульфанилкарбонил)-6-фенилпиридин-5-карбоксилат) и MRS-1200 (9-хлор-2-(2-фуранил)-5-[(фенилацетил)амино][1,2,4]-триазоло[1,5-с]хиназолин).
Использовали антитела против мышиного G-CSF (кроличья антисыворотка, очищенная хроматографией для протеина А, Cytolab LTD, Вейсманновский научно-исследовательский институт, Израиль).
Циклофосфамид закуплен у Taro Pharmaceutical Industries Ltd., Хайфа-Бэй, Израиль.
Мыши
Использовали самок мышей ICR, C57BL/6J или мышей происхождения BALB/C в возрасте 3 месяцев со средней массой 25 г. Мышей закупали в Harlan Laboratories, Иерусалим, Израиль. Давали стандартный гранулированный рацион и водопроводную воду.
Пример 1. Влияние аденозина и антагонистов аденозиновых рецепторов на продуцирование G-CSF и пролиферацию клеток костного мозга
Для того чтобы проверить предположение о том, что аденозин проявляет свое биологическое действие через стимуляцию продуцирования G-CSF, выращивают здоровые клетки в присутствии аденозина, агониста или антагониста аденозина.
Для этой цели клетки костного мозга, полученные из бедренной кости мышей C5BL/6J или ICR, сначала разъединяют, пропуская через иглу 25G. Затем клетки (3×105 клеток на лунку в 96-луночных микротитрационных планшетах) инкубируют со средой RPMI, содержащей 10% сыворотки плода коровы (FBS), в присутствии аденозина (25 мкМ). К культурам клеток костного мозга в отсутствие аденозина добавляют аденозин или агонистов аденозиновых рецепторов А1 и A3 СРА (A1RAg, 0,01 мкМ), ССРА (A1RAg, 0,01 мкМ) или IB-MECA (A3RAg, 0,01 мкМ); антагониста аденозиновых рецепторов A1 DPCPX (0,1 мкМ) добавляют к культуре клеток костного мозга в присутствии аденозина (25 мкМ).
В качестве контроля в описанном выше эксперименте служат культуры, содержащие клетки, суспендированные в среде RPMI и 5% FBS.
Для оценки пролиферации клеток костного мозга используют анализ с включением [3Н]-тимидина. Для указанной цели после 30-часовой инкубации каждую лунку "заряжают" 1 мкКи [3Н]-тимидина. После инкубации в целом в течение 48 часов клетки собирают и определяют поглощение [3Н]-тимидина в жидкостном сцинтилляционном счетчике LKB (LKB, Piscataway, Нью-Джерси, США). Результаты такого анализа отображены на фиг.1, которая показывает, что A1RAg или A3RAg обладают влиянием на продуцирование G-CSF, которое подобно действию, полученному в случае аденозина.
Для того чтобы подтвердить, что аденозин и его агонисты проявляют свое действие через стимуляцию продуцирования G-CSF, проводят другой анализ, где антитела против G-CSF (62,5 нг/мл) добавляют к культуре клеток костного мозга в присутствии аденозина (25 мкМ), СРА (0,01 мкМ) или IB-MECA (0,01 мкМ). Пролиферацию клеток оценивают так, как описано выше. Результаты такого эксперимента отображены на фиг.2, которая показывает, что антитела к G-CSF ингибируют стимулирующее действие аденозина и его агонистов на пролиферацию клеток костного мозга. Такие результаты позволяют предположить, что по меньшей мере некоторые из активностей, связанных с взаимодействием с аденозиновыми рецепторами, опосредуются через индукцию G-CSF.
Оценивают кумулятивное действие на пролиферацию клеток костного мозга при использовании сочетания A1RAg и A3RAg (СРА и IB-MECA). Анализ осуществляют подобно анализу в эксперименте, результаты которого приводятся на фиг.1. Клетки, являющиеся разъединенными, инкубируют в присутствии или аденозина (25 мкМ), СРА (0,01 мкМ), IB-MECA (0,01 мкМ), или сочетания IB-MECA и СРА (каждого в концентрации 0,01 мкМ), и затем обрабатывают так, как описано выше. Результаты приводятся на фиг.3А, которая показывает возросшее комбинированное действие IB-MECA и СРА.
Для того чтобы произвести сравнительную оценку влияния агониста аденозиновых рецепторов на пролиферацию клеток костного мозга, следуя той же методологии, какая описана выше, клетки инкубируют с одним аденозином или с сочетанием с DMPX (A2RAn), DPCPX (A1RAn), MRS-1220 (A3RAn) или с сочетанием DPCPX и MRS-1220. Результаты приводятся на фиг.2В. Как можно видеть, блокирование рецептора А2 DMPX также приводит к возросшей пролиферации клеток костного мозга, которая даже выше, чем в случае одного аденозина. Для сравнения пролиферация с DPCPX или MRS-1220 снижает повышение примерно на 50% по сравнению с использованием одного аденозина, в то время как DPCPX в сочетании с MRS-1220 вовсе ингибирует пролиферацию.
Клетки, предварительно обработанные так, как описано выше, инкубируют при различных концентрациях IB-MECA (1 мкМ, 0,1 мкМ или 0,01 мкМ). Процент стимуляции определяют с помощью анализа с включением [3Н]-тимидина, и результаты отображают на фиг.3, которая показывает, что IB-MECA стимулирует пролиферацию клеток костного мозга в зависимости от дозы.
Пример 2. Модуляция роста опухолевых клеток аденозином и его агонистами
Клетки лимфомы крыс Nb2-11C (1,2×l04 клеток/мл) инкубируют в течение 48 часов в 96-луночных микротитрационных планшетах с 1 мл среды RPMI, содержащей 5% сыворотки плода коровы. Добавляют или 25 мкМ аденозина, или 0,01 мкМ агонистов аденозиновых рецепторов (A1RAg CPA; A2RAg DPMA или A3RAg IB-MECA), или 0,1 мкМ антагонистов аденозиновых рецепторов (A1RAn DPCPX; A2RAn DMPX или A3RAn MRS-1220) в сочетании с аденозином (25 мкМ).
В качестве контроля в описанном выше эксперименте служат культуры, содержащие клетки, суспендированные в среде RPMI с 5% сыворотки плода коровы. Степень пролиферации определяют анализом с подсчетом числа клеток.
Результаты приводятся на фиг.5А и 5В, которые можно сравнить с ингибированием аденозином. Как можно видеть, пролиферация клеток Nb2-11C заметно ингибируется после инкубации с A3RAg IB-MECA. Незаметно ингибирования роста в присутствии A1RAg CPA, и меньшее ингибирование роста наблюдают в присутствии A2RAn DMPX. Неудача CPA при ингибировании пролиферации указанных двух (типов) опухолевых клеток позволяет предположить, что аденозиновый рецептор А1 в такого рода активности не участвует. Однако игибирующая активность как DMPA, так и IB-MECA позволяет сделать предположение о роли аденозиновых рецепторов А2 и A3 соответственно в таком ингибирующем действии.
Кроме того, можно видеть, что A1RAn DPCDX, по существу, не оказывает действия, в то время как в присутствии A3RAn MRS-1220 влияние аденозина на пролиферацию клеток Nb2-11C, по существу, устраняется. Небольшое, однако, все еще заметное действие проявляет A2RAn DMPX. Такие результаты приводят к заключению, что рост опухолевых клеток можно эффективно ингибировать A3RAg или A2RAn.
Таким же способом, какой описан выше, оценивают игибирование A3RAg IB-MECA роста клеток меланомы В-16, карциномы ободочной кишки НСТ-116 и лимфомы Nb2-11C. Результаты приводятся на фиг.6 в виде процента ингибирования пролиферации.
Пример 3. Агонисты аденозинового рецептора A3 проявляют дифферецирующее действие на опухолевые и здоровые клетки
Влияние аденозина, A3RAn и A3RAg на рост опухолевых клеток проверяют, следуя экспериментальной процедуре, описанной выше.
Вкратце, клетки лимфомы Nb2-11C или костного мозга инкубируют в присутствии или аденозина или IB-MECA. Двойственное действие A3RAg, проявляющееся в ингибировании роста опухолевых клеток и в то же время в пролиферации клеток костного мозга отражается на фиг.7А и 7В.
Пример 4. Исследования in vivo
Разделяют на 4 группы 40 мышей C57BL6/J и каждую группу обрабатывают согласно одной из описанных далее схем.
1. Контрольная группа: ежедневная интраперитонеальная (i.p.) инъекция 1 мл физиологического раствора на мышь со дня инокуляции опухоли до умерщвления мышей.
2. Химиотерапевтическая группа: одна i.p. инъекция циклофосфамида через 24 часа после инокуляции опухолевых клеток и ежедневная i.p. инъекция 1 мл физиологического раствора на мышь со дня инокуляции опухоли до умерщвления мышей.
3. Группа агониста аденозинового рецептора A3 (A3RAg):
ежедневное пероральное введение IB-MECA со дня инокуляции опухоли до умерщвления мышей.
4. Группа A3RAg + химиотерапия: одна i.p. инъекция циклофосфамида через 24 часа после инокуляции опухоли и ежедневное введение 3 мкг на кг массы тела IB-MECA.
На 5 и 9 день у мышей берут кровь из хвостовой вены и получают образцы крови для подсчета клеток белой крови (WBC). Результаты отображены на фиг.8.
Кроме того, в последующие 18 дней мышей умерщвляют и подсчитывают число опухолевых очагов в легких. Результаты отображены на фиг.9.
Проводят другой эксперимент, для того чтобы оценить хемозащитное действие A3RAg. Мышей обрабатывают циклофосфамидом (50 мг на кг массы тела в 0,3 мл PBS). Через 48 и 72 часа после введения цитотоксического лекарственного средства мышам дают i.p. инъекцию аденозина (25 мкг на кг массы тела) или IB-MECA (3 или 6 мкг на кг массы тела в 0,2 мл PBS). Проверяют число клеток белой крови (WBC) и нейтрофилов. Результаты приводятся на фиг.10А (WBC) и 10В (нейтрофилы) соответственно.
Как можно видеть, у мышей, обработанных только циклофосфамидом, обнаруживают падение числа лейкоцитов и нейтрофилов периферической крови, если сравнивать с группой, обработанной только IB-MECA. Когда вводят аденозин или IB-MECA, общее число клеток белой крови восстанавливается, причем последний имеет более выраженный эффект, причем полное восстановление получают через 168 часов (7 дней).
Пример 5. Агонисты аденозинового рецептора A3 предупреждают потерю массы у мышей, обработанных химиотерапевтическим лекарственным средством
Обрабатывают 4 группы "голых" мышей (происхождение BALB/C), по 10 в каждой, так, как описано далее.
Группа 1: мышей не обрабатывают [подтверждается как угодно]
Группа 2: мышам дают интраперитонеально (i.p.) инъекцию 5-фторурацила (5-FU, 30 мг на кг массы тела в PBS) в течение последующих пяти дней.
Группа 3: мышам дают i.p. инъекцию 5-FU как в группе 2, но начиная со 2 дня, и затем через день мышам дают перорально Cl-IB-MECA (6 мкг на кг массы тела в 0,2 мл PBS).
Группа 4: мыши получают Cl-IB-MECA, как описано выше.
Массу мышей измеряют на 7, 10 и 14 день. Результаты приводятся на фиг.11.
Как можно видеть, 5-FU сильно влияет на массу мышей по сравнению с контролем, в то время как Cl-IB-MECA, вводимый вместе с 5-FU, до некоторой степени предотвращает такую потерю массы. Сам по себе Cl-IB-MECA, по существу, не вызывает какой-либо потери массы.
Данный эксперимент демонстрирует, что агонисты аденозинового рецептора A3 обладают общим защитным действием от некоторых токсичных действий химиотерапии.
Пример 6. Cl-IB-MECA защищает мышей от миелотоксичного действия химиотерапевтического лекарственного средства доксорубицина
Мышей ICR обрабатывают дсксорубицином (i.p. инъекция 10 мг/кг в 0,5 мл PBS). Через 24, 48 и 72 часа после введения цитотоксичного лекарственного средства мышам вводят перорально Cl-IB-MECA (6 мкг на кг массы тела). В точках 72 часа, 96 часов, 120 часов и 144 часа мышей умерщвляют и берут образцы крови. Кроме того, из бедренных костей мышей отсасывают клетки костного мозга и определяют число ядросодержащих клеток в полученных отсосанных препаратах после окрашивания препарата кумасси синим.
Испытывают три группы мышей.
Группа 1 (контрольная): мышам вводят только PBS.
Группа 2: мышей обрабатывают только доксорубицином.
Группа 3: введение доксорубицина, как описано выше, сочетают с введением Cl-IB-MECA.
Результаты подсчета числа клеток белой крови показаны на фиг.12А, а числа ядросодержащих клеток костного мозга - на фиг.12В. Полученные результаты ясно показывают, что после введения Cl-IB-MECA имеется заметное возрастание числа клеток периферической белой крови, а также числа ядросодержащих клеток костного мозга. Это доказывает защитное действие A3RAg против миелотоксичного действия доксорубицина.
Пример 7. Антитела против G-CSF нейтрализуют миелозащитное действие Cl-IB-MECA
Мышей ICR делят на 6 групп, описанных далее.
Группа 1: контроль, введение только одного носителя.
Группа 2: контроль, с антителами против G-CSF (5 мкг/мышь).
Группа 3: химиотерапия, введение циклофосфамида (CYP, 50 мг на кг массы тела).
Группа 4: химиотерапия (50 мг на кг массы тела CYP) + антитела против G-CSF (5 мкг/мышь).
Группа 5: химиотерапия (50 мг на кг массы тела CYP) + Cl-IB-MECA (6 мкг на кг массы тела) + антитела против G-CSF (5 мкг/мышь).
Группа 6: химиотерапия (50 мг на кг массы тела CYP) + Cl-IB-MECA (6 мкг на кг массы тела) + антитела против G-CSF (5 мкг/мышь).
Каждая группа состоит из 10 мышей, и эксперимент повторяют дважды.
CYP инъецируют интраперитонеально в 0,2 мл PBS, который служит в качестве носителя.
Cl-IB-MECA дают перорально (в 0,2 мл PBS) через 48 часов и 72 часа после введения циклофосфамида.
Антитела против G-CSF инъецируют внутривенно (в 0,2 мл PBS) через 72 часа после введения химиотерапевтических лекарственных средств.
Образцы крови берут 124 часа после химиотерапии. Число клеток белой крови (WBC) определяют с помощью счетчика Coulter и подсчет дифференцированных клеток осуществляют на препаратах-мазках, окрашенных раствором May-Grundvald-Giesma.
Результаты подсчета WBC приводятся на фиг.13. Как можно видеть, мыши, обработанные только циклофосфамидом, показывают падение числа WBC периферической крови. В группе, обработанной Cl-IB-MECA, число WBC и процент нейтрофилов значительно выше по сравнению с группой, обработанной химиотерапевтическим средством (результаты, касающиеся нейтрофилов, не приводятся). Когда антитела против G-CSF вводят контрольной или обработанной химиотерапевтическим средством группам, наблюдают ожидаемое падение числа WBC. Введение антител против G-CSF мышам, обработанным сочетанием химиотерапевтического лекарственного средства и Cl-IB-MECA, аннулирует защитное действие Cl-IB-MECA, что можно четко видеть на фиг.13. Полученные результаты приводят к выводу, что защитное действие C1-IB-MECA на миелоидную систему опосредуется через способность C1-IB-MECA промотировать продуцирование и секрецию G-CSF.
Пример 8. Cl-IB-MECA ингибирует развитие НСТ-116 карциномы ободочной кишки человека у "голых" мышей
Опухоли создают подкожной инъекцией 1×106 клеток карциномы ободочной кишки человека НСТ-116 "голым" мышам (просхождение BALB/C) (Harlan, Иерусалим, Израиль). Мышей обрабатывают перорально 6 мкг Cl-IB-MECA на кг массы тела (в 0,2 мл PBS) через день. Мыши, которых обрабатывают только носителем (PBS), составляют контрольную группу. Каждая группа состоит из 10 мышей. Скорость роста опухоли определяют, измеряя два ортогональных диаметра каждой опухоли дважды в неделю, а размер опухоли оценивают по формуле ¶/6[D1D2]. Результаты отображены на фиг.14. Как можно видеть, в обработанной группе имеется значительное ингибирование роста опухоли.
В отдельной группе экспериментов проверяют комбинированную терапию Cl-IB-MECA и 5-фторурацилом (5-FU). "Голым" мышам инъецируют подкожно 1×106 клеток НСТ-116. Днем позже инъецируют интраперитонеально 5-FU (30 мг на кг массы тела, в 0,2 мл PBS) и еще подкожно в 4 последующих дня. Через день мышам вводят перорально 5 мкг на кг массы C1-IB-MECA (в 0,2 мл PBS). Мыши, которых обрабатывают или только носителем (PBS), или только 5-FU, служат в качестве контроля. Каждая группа состоит из 10 мышей. Скорость роста опухоли определяют, измеряя два ортогональных диаметра каждой опухоли дважды в неделю, а размер опухоли оценивают по формуле ¶/6[D1D2].
Результаты отображены на фиг.15 и 16. Заметное ингибирование роста опухоли наблюдают в группах, обработанных 5-FU, Cl-IB-MECA и комбинированной терапией Cl-IB-MECA и 5-FU. Через 20 дней видно отчетливое синергичное действие между Cl-IB-MECA и 5-FU при записи массы опухоли, как отображено, в частности, на фиг.16 (результаты, представленные на фиг.16, являются результатами в день 30).
Пример 9. Cl-IB-MECA стимулирует пролиферацию клеток костного мозга через индукцию продуцирования G-SCF
Клетки костного мозга (3×106 клеток/мл) инкубируют в лунках 96-луночных микротитрационных планшетов. Добавляют Сl-IB-MECA при конечной концентрации 10 нМ с антителами против G-SCF при конечной концентрации 0,05 и 0,5 мкг/мл или без них. Пролиферацию клеток измеряют с помощью анализа с включением [3Н]-тимидина. Результаты приводятся на фиг.17.
Как можно видеть, антитела против G-SCF ингибируют пролиферацию клеток костного мозга в зависимости от дозы. Такой эксперимент также показывает, что действие Cl-IB-MECA опосредуется через путь G-SCF (включая секрецию G-SCF из клеток).
Пример 10. Cl-IB-MECA ингибирует рост опухолевых клеток и стимулирует пролиферацию и дифференциацию клеток костного мозга
Клетки меланомы В-16 (5×105 клеток/мл) и клетки костного мозга (3×106 клеток/мл) инкубируют в лунках 96-луночного микротитрационного планшета. Добавляют культуру, состоящую из среды RPMI с добавлением 10% FTS, и Cl-IB-MECA при концентрации 0,01 мкМ или 0,1 мкМ, с антагонистом аденозинового рецептора A3 MRS-1523 или без него. Пролиферацию клеток измеряют с помощью анализа с включением [3Н]-тимидина, упомянутого ранее. Результаты приводятся на фиг.18. Как можно видеть, в присутствии MRS-1523 пролиферация как клеток меланомы, так и клеток костного мозга не меняется при сравнении с контролем. Напротив, Cl-IB-MECA проявляет ингибирующее действие на пролиферацию клеток меланомы и стимулирующее пролиферацию действие на клетки костного мозга.
Приведенные результаты демонстрируют двойственное действие агонистов аденозинового рецептора A3.
Пример 11. Cl-IB-MECA действует как хемозащитное средство
Выполняют эксперимент с Cl-IB-MECA, подобный описанному в примере 4, и результаты приводятся на фиг.19А и 19В, которые показывают хемозащитную активность Cl-IB-MECA.
Пример 12. Влияние IB-MECA и Cl-IB-MECA на пролиферацию клеток костного мозга
Клетки мышиного костного мозга культивируют так, как описано выше. К культурам добавляют IB-MECA или Cl-IB-MECA при концентрации 1 или 10 нМ в присутствии или в отсутствие А3RАn MRS-1523. Антагонист добавляют в концентрации 10 нМ. Результаты приводятся на фиг.20.
Как можно видеть на фиг.20, влияние как IB-MECA, так и Cl-IB-MECA зависит от дозы. Кроме того, также можно видеть, что указанное влияние в большой степени ингибируется A3RAn.
Claims (38)
1. Применение агониста аденозинового рецептора A3 (A3RAg) в качестве активного ингредиента фармацевтической композиции для индуцирования секреции или продуцирования гранулоцитарного колониестимулирующего фактора (G-CSF)y субъекта, нуждающегося в этом, где агонист аденозинового рецептора A3 (A3RAg) проявляет свое основное действие через рецептор аденозина A3.
2. Применение по п.1, где указанный активный ингредиент представляет собой производное нуклеотида общей формулы (I)

где R1 представляет собой (C1-С10)-алкил, (C1-С10)-гидроксиалкил, (C1-С10)-карбоксиалкил или (C1-С10)-цианоалкил или группу общей формулы (II)

где Y представляет собой кислород, серу или СН2;
X1 представляет собой Н, (C1-С10)-алкил, RaRbNC(=O) - или HORc-, где R3 и R13 могут быть одинаковыми или разными и выбраны из группы, состоящей из водорода, (C1-С10)-алкила, амино, (C1-С10)-галогеналкила, (C1-С10)-аминоалкила, (C1-С10)-BOC-аминоалкила и (C3-С10)-циклоалкила, или соединены вместе с образованием гетероциклического кольца, содержащего два-пять атомов углерода, и Rc выбирают из группы, состоящей из (C1-С10)-алкилена, NH-, (C1-С10)-галогеналкилена, (C1-С10)-аминоалкилена, (C1-С10)-ВОС-аминоалкилена и (C1-С10)-циклоалкилена;
Х2 представляет собой Н, гидроксил, (C1-С10)-алкиламино, (C1-С10)-алкиламидо или (C1-С10)-гидроксиалкил;
Х3 и Х4 каждый независимо представляет собой водород, гидроксил, амино, амидо, азидо, галоген, алкил, алкокси, карбокси, нитрило, нитро, трифтор, арил, алкарил, тио, сложную тиоэфирную группу, простую тиоэфирную группу, -OCOPh, OC(=S)OPh, или Х3 и Х4 оба представляют собой кислород, связанный с >C=S с образованием 5-членного цикла, или Х2 и Х3 образуют цикл формулы (III)

где R′ и R′′ представляют собой независимо (C1-С10)-алкил;
R2 выбирают из группы, состоящей из водорода, галогена, простой (C1-С10)-алкилэфирной группы, амино, гидразидо, (C1-С10)-алкиламино, (C1-С10)-алкокси, (C1-С10)-тиоалкокси, пиридилтио, (C2-С10)-алкенила, (C2-С10)-алкинила, тио и (C1-С10) -алкилтио;
R3 представляет собой группу -NR4R5, причем R4 представляет собой водород или группу, выбранную из алкила, замещенного алкила или арил-NH-C(Z)-, Z представляет собой О, S или NRa, причем Ra имеет указанные выше значения, R4 представляет собой водород, R5 выбирают из группы, состоящей из R- и S-1-фенилэтильной, бензильной, фенилэтильной или анилидной групп, незамещенных или замещенных в одном или нескольких положениях заместителем, выбранным из группы, состоящей из (C1-С10)-алкила, амино, галогена, (C1-С10)-галогеналкила, нитро, гидроксила, ацетамидо, (C1-С10)-алкокси и сульфоновой кислоты или ее соли, или R5 представляет собой бензодиоксанметил, фурфурил, L-пропилаланиламинобензил, β-аланиламинобензил, Т-ВОС-β-аланиламинобензил, фениламино, карбамоил, фенокси или (C3-С10)-циклоалкил, или R5 представляет собой группу формулы

или приемлемую соль соединения, определенного выше, например его триэтиламмониевую соль; или когда R4 представляет собой группу, выбранную из алкила, замещенного алкила или арил-NH-C(Z)-, тогда R5 выбирают из группы, состоящей из замещенного или незамещенного гетероарил-NRa-C(Z)-, гетероарил-С(Z)-, алкарил-Nra-C(Z)-, алкарил-С(Z)-, арил-NR-C(Z)- и арил-C(Z)-, где Z имеет значения, указанные выше.
3. Применение по п.1 или 2, где указанный активный ингредиент представляет собой производное нуклеозида общей формулы (IV)

где X1, R2 и R5 имеют значения, указанные в п.2.
4. Применение по п.3, где указанный активный ингредиент представляет собой производное N6-бензиладенозин-5'-уронамида.
5. Применение по п.4, где указанный активный ингредиент выбирают из группы, состоящей из N6-2-(4-аминофенил)-этиладенозина (APNEA), N6-(4-амино-3-иодбензил)аденозин-5'-(N-метилуронамида) (АВ-МЕСА) и N6-(3-иодбензил)-аденозин-5'-N-метилуронамида (IB-MECA) и 2-хлор-N6-(3-иодбензил)аденозин-5'-N-метилуронамида (CI-IB-MECA).
6. Применение по п.1, где активный ингредиент представляет собой N6-бeнзилaдeнoзин-5'-aлкилypoнaмид-Nl-oкcид или N6-бензиладенозин-5'-диалилуронамид-N1-оксид.
7. Применение по п.1, где активный ингредиент представляет собой производное ксантин-7-рибозида общей формулы (V)

где Х представляет собой О или S;
R6 представляет собой RaRbNC(=O)- или HORc-, где Ra и Rb могут быть одинаковыми или разными и выбраны из группы, состоящей из водорода, (C1-С10)-алкила, амино, (C1-C10)-галогеналкила, (C1-C10)-аминоалкила и (С3-С10)-циклоалкила, или соединены вместе с образованием гетероциклического кольца, содержащего два-пять атомов углерода, и Rc выбирают из (C1-С10) -алкила, амино, (C1-С10)-галогеналкила, (C1-С10)-аминоалкила, (C1-С10)-ВОС-аминоалкила и (C3-С10)-циклоалкила;
R7 и R8 могут быть одинаковыми или разными и выбраны из группы, состоящей из (C1-С10)-алкила, (C3-С10)-циклоалкила, R- или S-1-фенилэтильной, незамещенной бензильной или анилидной группы и простого фенилового эфира бензильной группы, замещенной в одном или нескольких положениях заместителем, выбранным из группы, состоящей из (C1-С10)-алкила, амино, галогена, (C1-С10)-галогеналкила, нитро, гидроксила, ацетамидо, (C1-С10)-алкокси и сульфоновой кислоты;
R9 выбирают из группы, состоящей из галогена, бензила, фенила,(С3-С10)-циклоалкила и (C1-С10)-алкокси,
или соль такого соединения, например, его триэтиламмониевую соль.
8. Применение агониста аденозинового рецептора A3 (A3RAg) в качестве активного ингредиента фармацевтической композиции для противодействия миелотоксичности, индуцированной лекарственным средством, где A3RAg проявляет свое основное действие через рецептор A3.
9. Применение по п.8, где указанное лекарственное средство представляет собой химиотерапевтическое лекарственное средство, назначаемое субъекту в рамках противоракового лечения.
10. Применение по п.8, где указанный активный ингредиент представляет собой активный ингредиент по любому из пп.2-7.
11. Применение по любому из пп.1-10, где указанный активный ингредиент включен в препаративную форму для перорального введения.
12. Применение A3RAg в качестве активного ингредиента фармацевтической композиции для индуцирования пролиферации или дифференциации клеток костного мозга или белой крови, где A3RAg проявляет свое основное действие через рецептор A3.
13. Применение по п.12, где указанный активный ингредиент представляет собой активный ингредиент по любому из пп.2-7.
14. Применение по п.12 или 13, где указанный активный ингредиент включен в препаративную форму для перорального введения.
15. Применение A3RAg в качестве активного ингредиента фармацевтической композиции для предупреждения или лечения лейкопении, где A3RAg проявляет свое основное действие через рецептор A3.
16. Применение по п.15 для предупреждения или лечения лейкопении, вызываемой лекарственным средством.
17. Применение по п.15, где указанный активный ингредиент представляет собой активный ингредиент по любому из пп.2-7.
18. Применение по любому из пп.15-17, где указанный активный ингредиент включен в препаративную форму для перорального введения.
19. Применение по п.16 в комбинации с лекарственным средством, которое может вызвать лейкопению у субъекта, подвергаемого лечению.
20. Применение по п.19, где указанное лекарственное средство является химиотерапевтическим лекарственным средством.
21. Применение A3RAg в качестве активного ингредиента фармацевтической композиции для ингибирования аномального клеточного роста, где A3RAg проявляет свое основное действие через A3 рецептора.
22. Применение по п.21 для ингибирования роста или пролиферации опухолевых клеток.
23. Применение по п.21 или 22, где указанный активный ингредиент представляет собой активный ингредиент по любому из пп.2-7.
24. Применение по любому из пп.21-23, где указанный активный ингредиент включен в препаративную форму для перорального введения.
25. Применение A3RAg в качестве активного ингредиента фармацевтической композиции для лечения рака, причем A3RAg оказывает свое основное действие через рецептор A3 и оказывает двойное действие как для ингибирования пролиферации раковых клеток, так и для противодействия токсическому побочному действию при лечении того же субъекта химиотерапевтическим лекарственным средством.
26. Применение по п.25, где указанный активный ингредиент действует синергически с указанным лекарственным средством для получения более сильного противоопухолевого действия.
27. Применение по п.25 или 26, где активный ингредиент представляет собой активный ингредиент по любому из пп.2-7.
28. Применение по любому из пп.25-27, где указанный активный ингредиент включен в препаративную форму для перорального введения.
29. Применение по любому из пп.1, 8, 12, 15, 21 или 25, где дозировка A3RAg составляет менее 100 мкг/кг массы тела.
30. Применение по п.29, где дозировка A3RAg составляет менее 50 мкг/кг массы тела.
31. Применение по п.30, где дозировка A3RAg составляет от 1 до 10 мкг/кг массы тела.
32. Применение по любому из пп.1, 2, 8, 12, 15, 21, 25, 29, 30 или 31, где указанный активный агент выбирают из группы, состоящей из
N6-(3-иодбензил)-9-метиладенина;
N6-(3-иодбензил)-9-гидроксиэтиладенина;
R-N6-(3-иодбензил)-9-(2,3-дигидроксипропил)аденина;
S-N6-(3-иодбензил)-9-(2,3-дигидроксипропил)аденина;
N6-(3-иодбензиладенин-9-ил)уксусной кислоты;
N6-(3-иодбензил)-9-(3-цианопропил)аденина;
2-хлор-N6-(3-иодбензил)-9-метиладенина;
2-амино-N6-(3-иодбензил)-9-метиладенина;
2-гидразидо-N6-(3-иодбензил)-9-метиладенина;
N6-(3-иодбензил)-2-метиламино-9-метиладенина;
2-диметиламино-N6-(3-иодбензил)-9-метиладенина;
N6-(3-иодбензил)-9-метил-2-пропиламиноаденина;
2-гексиламино-N6-(3-иодбензил)-9-метиладенина;
N6-(3-иодбензил)-2-метокси-9-метиладенина;
N6-(3-иодбензил)-9-метил-2-метилтиоаденина;
N6-(3-иодбензил)-9-метил-2-(4-пиридилтио)аденина;
(1S, 2R, 3S, 4R)-4-(6-амино-2-фенилэтиламино-9Н-пурин-9-ил)циклопентан-1, 2, 3-триола;
(1S, 2R, 3S, 4R)-4-(6-амино-2-хлор-9Н-пурин-9-ил)циклопентан-1, 2, 3-триола;
(±)-9-[2α,3α-дигидрокси-4β-(N-метилкарбамоил)циклопент-1β-ил)]N6-(3-иодбензил)аденина;
2-хлор-9-(2'-амино-2',3'-дидезокси-β-D-5'-метиларабино-фуронамидо)-N6-(3-иодбензил)аденина;
2-хлор-9-(2',3'-дидезокси-2'-фтор-λ-D-5'-метиларабино-фуронамидо)-N6-(3-иодбензил)аденина;
9-(2-ацетил-3-дезокси-β-D-5-метилрибофуронамидо)-2-хлор-N6-(3-иодбензил)аденина;
2-хлор-9-(3-дезокси-2-метансульфонил-β-D-5-метилрибо-фуронамидо)-N6-(3-иодбензил)аденина;
2-хлор-9-(3-дезокси-β-D-5-метилрибофуронамидо)-N6-(3-иодбензил)аденина;
2-хлор-9-(3,5-1,1,3,3-тетраизопропилдисилоксил-β-D-5-рибофуранозил)-N6-(3-иодбензил)аденина;
2-хлор-9-[2',3'-O-тиокарбонил-β-D-5-метилрибофуронамидо)N6-(3-иодбензил)аденина;
9-(2-фенокситиокарбонил-3-дезокси-β-D-5-метилрибофуронамидо)-2-хлор-N6-(3-иодбензил)аденина;
1-(6-бензиламино-9Н-пурин-9-ил)-1-дeзoкcи-N,4-диметил-β-D-рибофуранозидуронамида;
2-хлор-9-(2,3-дидезокси-β-D-5-метилрибофуронамидо)-N6-бензиладенина;
2-хлор-9-(2'-азидо-2',3'-дидезокси-β-D-5'-метиларабинофуронамидо)-N6-бензиладенина;
2-хлор-9-(β-D-эритрофуранозид)-N6-(3-иодбензил)аденина;
N6-(бензодиоксанметил)аденозина;
1-(6-фурфуриламино-9Н-пурин-9-ил)-1-дезокси-N-метил-β-D-рибофуранозидуронамида;
N6-[3-(L-пролиламино)бензил]аденозин-5'-N-метилуронамида;
N6-[3-(β-аланиламино)бензил]аденозин-5'-N-метилуронамида;
N6-[3-(N-Т-Вос-β-аланиламино)бензил]аденозин-5'-N-метилуронамида;
N6-[N'-фенилгидразинил)пурин-9-β-рибофуранозид-5'-N-метилуронамида;
6-(O-фенилгидроксиламино)пурин-9-β-рибофуранозид-5'-N-метилуронамида;
9-(β-D-2',3'-дидезоксиэритрофуранозил)-N6-(3-λ-аланиламино)бензил]аденозина;
9-(β-D-эритрофуранозид)-2-метиламино-N6-(3-иодбензил]аденина;
2-хлор-N-(3-иодбензил)-9-(2-тетрагидрофурил)-9Н-пурин-6-амина;
2-хлор-(2’-дезокси-6’-тио-L-арабинозил)аденина;
2-хлор-(6'-тио-L-арабинозил)аденина;
N6-(4-бифенилкарбониламино)аденозин-5'-N-этилуронамида;
N6-(2,4-дихлорбензилкарбониламино)аденозин-5'-N-этилуронамида;
N6-(4-метоксифенилкарбониламино)аденозин-5'-N-этилуронамида;
N6-(4-хлорфенилкарбониламино)аденозин-5'-N-этилуронамида;
N6-(фенилкарбониламино)аденозин-5'-N-этилуронамида;
N6-(бензилкарбамоиламино)аденозин-5’-N-этилуронамида;
N6-(4-сульфонамидофенилкарбамоил)аденозин-5'-N-этилуронамида;
N6-(4-ацетилфенилкарбамоил)аденозин-5'-N-этилуронамида;
N6-((R)-α-фенилэтилкарбамоил)аденозин-5'-N-этилуронамида;
N6-((S)-α-фенилэтилкарбамоил)аденозин-5'-N-этилуронамида;
N6-(5-метилизоксазол-3-ил-карбамоил)аденозин-5'-N-этилуронамида;
N6-(1,3,4-тиадиазол-2-ил-карбамоил)аденозин-5'-N-этилуронамида;
N6-(4-н-пропоксифенилкарбамоил)аденозин-5'-N-этилуронамида;
N6-биc-(4-нитрофенилкарбамоил)аденозин-5'-N-этилуронамида; и
N6-бис-(5-хлорпиридин-2-ил-карбамоил)аденозин-5'-N-этилуронамида.
33. Применение по любому из пп.1, 2, 3, 8, 12, 15, 21, 25 или 29, 30 и 31, где указанный активный ингредиент выбран из группы, состоящей из соединений формулы (IV), где X1 представляет собой RaRbNC(=O), где Ra и Rb могут быть одинаковыми или разными и выбраны из группы, состоящей из водорода, (C1-С10)алкила, амино, (C1-С10) галогеналкила, (C1-С10)-аминоалкила, и (C3-С10)циклоалкила; R2 выбран из группы, состоящей из водорода, галогена, (C1-С10)алкокси, амино, (C2-С10)алкенила и (C2-С10)алкинила, и R5 выбран из группы, состоящей из R- и S-1-фенилэтила, незамещенной бензильной группы и бензильной группы, замещенной в одном или более положениях заместителем, выбранным из группы, состоящей из (C1-С10)-алкила, амино, галогена, (C1-С10)галогеналкила, нитро, гидрокси, ацетамидо, (C1-С10)алкокси и сульфо.
34. Применение по п.33, где указанный активный ингредиент выбран из группы, состоящей из соединений формулы (IV), где Ra и Rb могут быть одинаковыми или разными и выбраны из группы, состоящей из водорода, (C1-С10)-алкила, и R2 является водородом или галогеном; Ra является водородом, R2 является водородом и R5 является незамещенным бензилом; Rb представляет (C1-С10) алкил или (C3-С10) циклоалкил и R5 в R или S-1-фенилэтиле или бензиле замещен в одном или более положениях заместителем, выбранным из группы, состоящей из галогена, амино, ацетамидо, (C1-С10)галогеналкила и сульфогруппы, где сульфопроизводным является соль; R2 представляет (C2-С10) алкилен формулы Rd-C=C-, где Rd представляет (C1-С8) алкил, или R2 представляет галоген, (C1-С10) алкиламино, или (C1-С10) алкилтио; Ra является водородом, Rb представляет (C1-С10)алкил и R5 - замещенный бензил.
35. Применение по любому из пп.1, 7, 8, 12, 15, 21, 25 или 29, 30 и 31, где указанный активный ингредиент выбран из группы, состоящей из соединений формулы (V), где X1 представляет собой О, R6 - RaRbNC(=O), где Ra и Rb могут быть одинаковыми или разными и выбраны из группы, состоящей из водорода, (C1-С10)алкила, амино, (C1-С10)галогеналкила, (C1-С10) -аминоалкила, (C3-С10) циклоалкила, и R7 и R8 могут быть одинаковыми или разными и выбраны из группы, состоящей из (C1-С10)алкила, R- и S-1-фенилэтильной, незамещенной бензильной группы и бензильной группы, замещенной в одном или нескольких положениях заместителем, выбранным из группы, состоящей из (C1-С10)алкила, амино, галогена, (C1-С10)галогеналкила, нитро, гидрокси, ацетамидо, (C1-С10)алкокси и сульфогруппы, и R9 выбран из группы, состоящей из галогена, бензила, фенила и (C3-С10)циклоалкила.
36. Применение по любому из пп.1-31 или 32-35, где активный ингредиент представлен в форме соли триэтиламмония.
Приоритет по пунктам:
23.12.1999 по пп.1-11, 13-14, 17, 19-20, 23, 25-36;
10.09.1999 по пп.12, 15-16, 18, 21-22 и 24.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IL131864 | 1999-09-10 | ||
| IL13186499A IL131864A0 (en) | 1999-09-10 | 1999-09-10 | Pharmaceutical compositions comprising an adenosine receptor agonist or antagonist |
| IL13368099A IL133680A0 (en) | 1999-09-10 | 1999-12-23 | Pharmaceutical compositions comprising an adenosine receptor agonist or antagonist |
| IL133680 | 1999-12-23 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2002109228A RU2002109228A (ru) | 2004-01-10 |
| RU2239455C2 true RU2239455C2 (ru) | 2004-11-10 |
Family
ID=26323880
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2002109228/15A RU2239455C2 (ru) | 1999-09-10 | 2000-09-08 | Фармацевтические композиции, содержащие агонист или антагонист аденозинового рецептора |
Country Status (19)
| Country | Link |
|---|---|
| US (2) | US7064112B1 (ru) |
| EP (1) | EP1261322B1 (ru) |
| JP (2) | JP4980530B2 (ru) |
| KR (2) | KR100674529B1 (ru) |
| CN (1) | CN100358512C (ru) |
| AU (1) | AU782826B2 (ru) |
| BR (1) | BR0013905A (ru) |
| CA (1) | CA2384111C (ru) |
| DE (1) | DE60037277T2 (ru) |
| DK (1) | DK1261322T3 (ru) |
| ES (1) | ES2296637T3 (ru) |
| HK (1) | HK1052653B (ru) |
| HU (1) | HU226913B1 (ru) |
| IL (1) | IL133680A0 (ru) |
| MX (1) | MXPA02002546A (ru) |
| PL (1) | PL199852B1 (ru) |
| PT (1) | PT1261322E (ru) |
| RU (1) | RU2239455C2 (ru) |
| WO (1) | WO2001019360A2 (ru) |
Families Citing this family (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL121272A (en) * | 1997-07-10 | 2000-06-01 | Can Fite Technologies Ltd | Pharmaceutical compositions comprising adenosine and their use for treating or preventing leukopenia |
| CN1234358C (zh) * | 2000-06-21 | 2006-01-04 | 弗·哈夫曼-拉罗切有限公司 | 苯并噻唑衍生物 |
| AU2002219497B2 (en) * | 2001-01-16 | 2004-08-26 | Can-Fite Biopharma Ltd. | Use of an adenosine A3 receptor agonist for inhibition of viral replication |
| US20020115635A1 (en) * | 2001-02-21 | 2002-08-22 | Pnina Fishman | Modulation of GSK-3beta activity and its different uses |
| US20040204481A1 (en) * | 2001-04-12 | 2004-10-14 | Pnina Fishman | Activation of natural killer cells by adenosine A3 receptor agonists |
| US7049303B2 (en) * | 2001-11-07 | 2006-05-23 | Medical Research Council | Inhibition of viruses |
| US7414036B2 (en) | 2002-01-25 | 2008-08-19 | Muscagen Limited | Compounds useful as A3 adenosine receptor agonists |
| US20040137477A1 (en) * | 2002-10-22 | 2004-07-15 | Can-Fite Biopharma, Ltd. | A3AR as a marker for a diseased state |
| US7087761B2 (en) | 2003-01-07 | 2006-08-08 | Hoffmann-La Roche Inc. | Cyclization process for substituted benzothiazole derivatives |
| EA011279B1 (ru) | 2004-05-24 | 2009-02-27 | Ф. Хоффманн-Ля Рош Аг | (4-метокси-7-морфолин-4-илбензотиазол-2-ил)-амид 4-гидрокси-4-метилпиперидин-1-карбоновой кислоты |
| KR101167342B1 (ko) | 2004-05-26 | 2012-07-19 | 이노텍 파마슈티컬스 코포레이션 | 아데노신 a1 수용체 아고니스트로서의 퓨린 유도체 및 이것의 사용 방법 |
| US7825102B2 (en) | 2004-07-28 | 2010-11-02 | Can-Fite Biopharma Ltd. | Treatment of dry eye conditions |
| AU2005286946B2 (en) | 2004-09-20 | 2012-03-15 | Inotek Pharmaceuticals Corporation | Purine derivatives and methods of use thereof |
| JP4633123B2 (ja) | 2004-11-05 | 2011-02-16 | エフ.ホフマン−ラ ロシュ アーゲー | イソニコチン酸誘導体の製造方法 |
| MX2007005525A (es) * | 2004-11-08 | 2007-07-05 | Can Fite Biopharma Ltd | Tratamiento terapeutico de resorcion ose acelerada. |
| JP2008520746A (ja) * | 2004-11-22 | 2008-06-19 | キング・ファーマシューティカルズ・リサーチ・アンド・デベロプメント・インコーポレイティッド | アデノシンa3受容体アンタゴニストを用いる癌及びhif−1が介在する疾患の促進的治療 |
| KR100968989B1 (ko) | 2005-03-23 | 2010-07-09 | 에프. 호프만-라 로슈 아게 | mGluR2 길항제로서 아세틸렌일-피라졸로-피리미딘 유도체 |
| CN101273040B (zh) | 2005-09-27 | 2011-11-09 | 弗·哈夫曼-拉罗切有限公司 | 作为mglur2拮抗剂的*二唑基吡唑并嘧啶类化合物 |
| AU2006320578B2 (en) | 2005-11-30 | 2013-01-31 | Inotek Pharmaceuticals Corporation | Purine derivatives and methods of use thereof |
| CN101365430B (zh) * | 2006-01-27 | 2011-09-21 | 坎-菲特生物药物有限公司 | 用于治疗干眼病的腺苷a3受体激动剂 |
| WO2008023362A2 (en) * | 2006-08-21 | 2008-02-28 | Can-Fite Biopharma Ltd. | Use of a combination of methotrexate and an a3ar agonist for the treatment of cancer |
| JP2010505848A (ja) * | 2006-10-06 | 2010-02-25 | ザ トラスティーズ オヴ ザ ユニヴァーシティー オヴ ペンシルバニア | 眼内圧を低下させるための種交差性a3アデノシン受容体アンタゴニストの効果的送達 |
| GB0625100D0 (en) * | 2006-12-15 | 2007-01-24 | Univ Murcia | Epigallocatechin-3-gallate compositions for cancer therapy and chemoprotection |
| US20090088403A1 (en) * | 2007-05-07 | 2009-04-02 | Randy Blakely | A3 adenosine receptors as targets for the modulation of central serotonergic signaling |
| TW200914048A (en) * | 2007-07-17 | 2009-04-01 | Combinatorx Inc | Combinations for the treatment of B-cell proliferative disorders |
| US20090053168A1 (en) * | 2007-07-17 | 2009-02-26 | Richard Rickles | Treatments of b-cell proliferative disorders |
| WO2009151569A2 (en) * | 2008-06-09 | 2009-12-17 | Combinatorx, Incorporated | Beta adrenergic receptor agonists for the treatment of b-cell proliferative disorders |
| WO2010027935A1 (en) | 2008-09-08 | 2010-03-11 | Merck Sharp & Dohme Corp. | Ahcy hydrolase inhibitors for treatment of hyper homocysteinemia |
| CA2761499A1 (en) | 2009-05-17 | 2010-11-25 | Can-Fite Biopharma Ltd. | A3 adenosine receptor agonists for the reduction of intraocular pressure |
| WO2011010306A1 (en) | 2009-07-21 | 2011-01-27 | Ramot At Tel-Aviv University Ltd. | A3 adenosine receptor ligands for modulation of pigmentation |
| US20120322815A1 (en) * | 2009-12-17 | 2012-12-20 | Ewha University-Industry Collaboration Foundation | Pharmaceutical composition containing a3 adenosine receptor agonist |
| WO2011085361A1 (en) | 2010-01-11 | 2011-07-14 | Inotek Pharmaceuticals Corporation | Combination, kit and method of reducing intraocular pressure |
| CA2790869A1 (en) | 2010-03-03 | 2011-09-09 | Government Of The Usa, Represented By The Secretary, Department Of Healt H And Human Services | A3ar agonists for the treatment of uveitis |
| US8476247B2 (en) | 2010-03-26 | 2013-07-02 | Inotek Pharmaceuticals Corporation | Method of reducing intraocular pressure in humans |
| US20130109645A1 (en) * | 2010-03-31 | 2013-05-02 | The united States of America,as represented by Secretary,Dept.,of Health and Human Services | Adenosine receptor agonists for the treatment and prevention of vascular or joint capsule calcification disorders |
| UY34536A (es) | 2011-12-22 | 2013-07-31 | Alios Biopharma Inc | Nucleósidos sustituidos, nucleótidos y análogos de los mismos |
| WO2013111132A1 (en) * | 2012-01-23 | 2013-08-01 | Can-Fite Biopharma Ltd. | Treatment of liver conditions |
| AU2013211957B2 (en) | 2012-01-26 | 2017-08-10 | Inotek Pharmaceuticals Corporation | Anhydrous polymorphs of (2R,3S,4R,5R)-5-(6-(cyclopentylamino)-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl) } methyl nitrate and processes of preparation thereof |
| US9441007B2 (en) | 2012-03-21 | 2016-09-13 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| USRE48171E1 (en) | 2012-03-21 | 2020-08-25 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US8822460B2 (en) | 2012-04-06 | 2014-09-02 | Janssen Pharmaceutica Nv | Fused cyclopentyl antagonists of CCR2 |
| CN105358551A (zh) | 2012-07-19 | 2016-02-24 | 詹森药业有限公司 | Ccr2的八氢环戊并吡咯基拮抗剂 |
| MX2015001644A (es) | 2012-08-09 | 2015-08-14 | Can Fite Biopharma Ltd | Ligandos del receptor de adenosina a3 para su uso en el tratamiento de una disfuncion sexual. |
| CA2903114A1 (en) | 2013-03-15 | 2014-09-25 | Inotek Pharmaceuticals Corporation | Ophthalmic formulations |
| IL242723B (en) | 2015-11-23 | 2019-12-31 | Can Fite Biopharma Ltd | A3 adenosine receptor ligand for the treatment of ectopic fat accumulation |
| WO2017185061A1 (en) | 2016-04-21 | 2017-10-26 | Astrocyte Pharmaceuticals, Inc. | Compounds and methods for treating neurological and cardiovascular conditions |
| IL254535A0 (en) | 2017-09-17 | 2017-11-30 | Can Fite Biopharma Ltd | Adenosine a3 receptor ligand for use in the management of cytokine release syndrome |
| JP7311855B2 (ja) | 2018-02-09 | 2023-07-20 | アストロサイト ファーマシューティカルズ, インコーポレイテッド | 嗜癖および関連する障害を処置するための化合物および方法 |
| CN112930349A (zh) | 2018-09-26 | 2021-06-08 | 阿斯特罗赛特制药公司 | 多晶形化合物及其用途 |
| IL264112A (en) | 2019-01-06 | 2020-07-30 | Fishman Pnina | Adenosine a3 receptor ligand for use in lowering adipocyte levels |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5443836A (en) * | 1993-03-15 | 1995-08-22 | Gensia, Inc. | Methods for protecting tissues and organs from ischemic damage |
| US5688774A (en) * | 1993-07-13 | 1997-11-18 | The United States Of America As Represented By The Department Of Health And Human Services | A3 adenosine receptor agonists |
| AU7331094A (en) | 1993-07-13 | 1995-02-13 | United States Of America, As Represented By The Secretary, Department Of Health And Human Services, The | A3 adenosine receptor agonists |
| GB2289218A (en) * | 1994-05-06 | 1995-11-15 | Merck & Co Inc | Inhibition of TNFalpha production with agonists of the A2b subtype of the adenosine receptor |
| RU2071770C1 (ru) | 1994-12-14 | 1997-01-20 | Владислав Алексеевич Шматко | Способ лечения рака шейки матки и яичников |
| RU2071771C1 (ru) | 1995-01-11 | 1997-01-20 | Владислав Алексеевич Шматко | Способ лечения рака молочной железы |
| US6211165B1 (en) * | 1997-05-09 | 2001-04-03 | The Trustees Of The University Of Pennsylvania | Methods and compositions for reducing ischemic injury of the heart by administering adenosine receptor agonists and antagonists |
| IL121272A (en) * | 1997-07-10 | 2000-06-01 | Can Fite Technologies Ltd | Pharmaceutical compositions comprising adenosine and their use for treating or preventing leukopenia |
| EP1019427A4 (en) * | 1997-07-29 | 2000-07-19 | Medco Res Inc | N? 6 -SUBSTITUES-ADENOSINE-5'-URONAMIDES USEFUL AS MODULATORS OF ADENOSINE RECEPTORS |
| US6048865A (en) | 1997-07-29 | 2000-04-11 | Medco Research, Inc. | N6 -substituted-adenosine-5'-uronamides as adenosine receptor modulator |
| US6329349B1 (en) | 1997-10-23 | 2001-12-11 | Trustees Of The University Of Pennsylvania | Methods for reducing ischemic injury of the heart via the sequential administration of monophosphoryl lipid A and adenosine receptor agents |
| CA2316994A1 (en) * | 1998-06-08 | 1999-12-16 | Epigenesis Pharmaceuticals, Inc. | Composition and method for prevention and treatment of cardiopulmonary and renal failure or damage associated with ischemia, endotoxin release, ards or brought about by administration of certain drugs |
| US6448253B1 (en) | 1998-09-16 | 2002-09-10 | King Pharmaceuticals Research And Development, Inc. | Adenosine A3 receptor modulators |
| IL127947A0 (en) * | 1999-01-07 | 1999-11-30 | Can Fite Technologies Ltd | Pharmaceutical use of adenosine agonists |
| UA72912C2 (ru) * | 1999-02-01 | 2005-05-16 | Юніверсіті Оф Вірджінія Патент Фаундейшн | Translated By PlajКОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНОЙ РЕАКЦИИ |
| IL131096A (en) | 1999-07-26 | 2004-09-27 | Can Fite Technologies Ltd | Method for preparing a composition derived from muscle tissue |
-
1999
- 1999-12-23 IL IL13368099A patent/IL133680A0/xx not_active IP Right Cessation
-
2000
- 2000-09-08 RU RU2002109228/15A patent/RU2239455C2/ru active
- 2000-09-08 AU AU68634/00A patent/AU782826B2/en not_active Expired
- 2000-09-08 DE DE60037277T patent/DE60037277T2/de not_active Expired - Lifetime
- 2000-09-08 JP JP2001522994A patent/JP4980530B2/ja not_active Expired - Lifetime
- 2000-09-08 KR KR1020067006473A patent/KR100674529B1/ko active IP Right Grant
- 2000-09-08 PL PL356469A patent/PL199852B1/pl unknown
- 2000-09-08 DK DK00956773T patent/DK1261322T3/da active
- 2000-09-08 CN CNB008148007A patent/CN100358512C/zh not_active Expired - Lifetime
- 2000-09-08 KR KR1020027003164A patent/KR100584797B1/ko active IP Right Grant
- 2000-09-08 ES ES00956773T patent/ES2296637T3/es not_active Expired - Lifetime
- 2000-09-08 EP EP00956773A patent/EP1261322B1/en not_active Expired - Lifetime
- 2000-09-08 WO PCT/IL2000/000550 patent/WO2001019360A2/en active IP Right Grant
- 2000-09-08 CA CA002384111A patent/CA2384111C/en not_active Expired - Lifetime
- 2000-09-08 PT PT00956773T patent/PT1261322E/pt unknown
- 2000-09-08 US US09/700,751 patent/US7064112B1/en not_active Expired - Lifetime
- 2000-09-08 HU HU0203870A patent/HU226913B1/hu unknown
- 2000-09-08 MX MXPA02002546A patent/MXPA02002546A/es active IP Right Grant
- 2000-09-08 BR BR0013905-0A patent/BR0013905A/pt not_active Application Discontinuation
-
2003
- 2003-07-11 HK HK03105018.4A patent/HK1052653B/zh not_active IP Right Cessation
-
2005
- 2005-11-25 US US11/286,376 patent/US20060084626A1/en not_active Abandoned
-
2007
- 2007-05-14 JP JP2007128137A patent/JP2007204496A/ja active Pending
Non-Patent Citations (3)
| Title |
|---|
| Противоопухолевая терапия. Справочник/Под ред. Н.И.Переводчиковой - М., 1993 с.192 и 202. * |
| Реферат из АБД Medline: Ezeamuzie CI et al. Adenosine A3 receptors on human eosinophils mediate inhibition of degranulation and superoxide anion release. Br J Pharmacol. - 1999, May; 127(1): 188-94. Справочник по онкологии. - М.: Медицина 1964 с.63. * |
| Реферат из АБД Medline: Yao Y et al. Adenosine A3 receptor agonists protect HL-60 and U-937 cells from apoptosis induced by A3 antagonists Biochem Biophys Res Commun. 1997 Mar 17; 232(2):317-22. * |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2239455C2 (ru) | Фармацевтические композиции, содержащие агонист или антагонист аденозинового рецептора | |
| US6579857B1 (en) | Combination cancer therapy comprising adenosine and deaminase enzyme inhibitors | |
| AU2012321106B2 (en) | Administration of NEDD8-activating enzyme inhibitor and hypomethylating agent | |
| DE60202278T2 (de) | Aktivierung natürlicher killerzellen durch adenosin-a3-rezeptoragonisten | |
| PT1385498E (pt) | Ácidos gordos como factores de sobrevivência e activação de neutrófilos | |
| JP2013166789A (ja) | 骨関節炎の治療におけるa3アデノシン受容体アゴニストの使用 | |
| EP1272897A2 (en) | Adenosine a2a receptor antagonists for treating and preventing hepatic fibrosis, cirrhosis and fatty liver | |
| AU2001238124A1 (en) | Adenosine a2a receptor antagonists for treating and preventing hepatic fibrosis,cirrhosis and fatty liver | |
| US20100069291A1 (en) | Combination comprising cndac (2'-cyano-2'-deoxy-n4-palmitoyl-1-beta-d-arabinofuranosyl-cytosine) and a cytotoxic agent | |
| US20080090848A1 (en) | Substituted Purinyl Derivatives With Immunomodulator And Chemoprotective Activity And Use Alone Or With Medium-Chain Length Fatty Acids Or Glycerides | |
| CN112165945A (zh) | 治疗淋巴样恶性疾病之方法 | |
| AU1888400A (en) | Use of adenosine agonists in cancer therapy | |
| JP6895688B2 (ja) | 血液がんの新規治療法及び新規治療剤 | |
| KR20040078123A (ko) | 에포틸론 및 대사길항물질을 포함하는 조합물 | |
| US20010031742A1 (en) | Pharmaceutical use of adenosine agonists | |
| JP2005526086A (ja) | 癌の治療用のcdk阻害剤と5−fuの組合せ | |
| US11957701B2 (en) | Therapy and new therapeutic agent for blood cancer | |
| US20020037871A1 (en) | Pharmaceutical use of adenosine agonists | |
| Alberts et al. | Phase I trial of gemcitabine and CPT-11 given weekly for four weeks every six weeks | |
| TW202203942A (zh) | 血液癌之新穎治療法及新穎治療劑 |