KR20200052349A - 엔독시펜을 제조 및 사용하는 방법 - Google Patents
엔독시펜을 제조 및 사용하는 방법 Download PDFInfo
- Publication number
- KR20200052349A KR20200052349A KR1020207010196A KR20207010196A KR20200052349A KR 20200052349 A KR20200052349 A KR 20200052349A KR 1020207010196 A KR1020207010196 A KR 1020207010196A KR 20207010196 A KR20207010196 A KR 20207010196A KR 20200052349 A KR20200052349 A KR 20200052349A
- Authority
- KR
- South Korea
- Prior art keywords
- composition
- endoxifene
- endoxifen
- formula
- compound
- Prior art date
Links
- MHJBZVSGOZTKRH-IZHYLOQSSA-N 4-Hydroxy-N-desmethyltamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCNC)=CC=1)/C1=CC=C(O)C=C1 MHJBZVSGOZTKRH-IZHYLOQSSA-N 0.000 title claims description 400
- 238000004519 manufacturing process Methods 0.000 title claims description 20
- 239000000203 mixture Substances 0.000 claims abstract description 699
- 238000000034 method Methods 0.000 claims abstract description 297
- 150000003839 salts Chemical class 0.000 claims abstract description 171
- 230000001419 dependent effect Effects 0.000 claims abstract description 156
- 229940088597 hormone Drugs 0.000 claims abstract description 155
- 239000005556 hormone Substances 0.000 claims abstract description 155
- 208000020207 Reproductive tract disease Diseases 0.000 claims abstract description 73
- 210000000481 breast Anatomy 0.000 claims abstract description 49
- 150000001875 compounds Chemical class 0.000 claims description 255
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 claims description 185
- 235000002639 sodium chloride Nutrition 0.000 claims description 183
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical group CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 130
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 110
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 105
- 208000030270 breast disease Diseases 0.000 claims description 103
- 239000002904 solvent Substances 0.000 claims description 97
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 claims description 96
- 239000002775 capsule Substances 0.000 claims description 90
- 229960001603 tamoxifen Drugs 0.000 claims description 89
- 206010006187 Breast cancer Diseases 0.000 claims description 86
- 208000026310 Breast neoplasm Diseases 0.000 claims description 86
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 80
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical group [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 75
- 230000036470 plasma concentration Effects 0.000 claims description 70
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 66
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 65
- 239000003960 organic solvent Substances 0.000 claims description 56
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 55
- 239000007787 solid Substances 0.000 claims description 55
- 238000011282 treatment Methods 0.000 claims description 54
- 239000012458 free base Substances 0.000 claims description 53
- 239000007894 caplet Substances 0.000 claims description 52
- 239000003814 drug Substances 0.000 claims description 50
- 235000019439 ethyl acetate Nutrition 0.000 claims description 49
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 48
- 239000012452 mother liquor Substances 0.000 claims description 46
- 239000003826 tablet Substances 0.000 claims description 46
- -1 titanium halide Chemical class 0.000 claims description 46
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 40
- 230000003111 delayed effect Effects 0.000 claims description 39
- 201000000079 gynecomastia Diseases 0.000 claims description 39
- 206010006256 Breast hyperplasia Diseases 0.000 claims description 37
- 208000037396 Intraductal Noninfiltrating Carcinoma Diseases 0.000 claims description 37
- 239000003638 chemical reducing agent Substances 0.000 claims description 36
- 150000003608 titanium Chemical class 0.000 claims description 36
- 238000006243 chemical reaction Methods 0.000 claims description 33
- 206010020718 hyperplasia Diseases 0.000 claims description 30
- 239000002253 acid Substances 0.000 claims description 29
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 27
- 239000012649 demethylating agent Substances 0.000 claims description 27
- 229940079593 drug Drugs 0.000 claims description 27
- 235000019441 ethanol Nutrition 0.000 claims description 27
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 26
- MHJBZVSGOZTKRH-OCOZRVBESA-N 4-[(e)-1-[4-[2-(methylamino)ethoxy]phenyl]-2-phenylbut-1-enyl]phenol Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCNC)=CC=1)\C1=CC=C(O)C=C1 MHJBZVSGOZTKRH-OCOZRVBESA-N 0.000 claims description 25
- 238000000605 extraction Methods 0.000 claims description 25
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 24
- 230000003472 neutralizing effect Effects 0.000 claims description 24
- 239000011701 zinc Substances 0.000 claims description 24
- 238000004821 distillation Methods 0.000 claims description 23
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 22
- 208000028176 atypical lobular breast hyperplasia Diseases 0.000 claims description 22
- 239000003795 chemical substances by application Substances 0.000 claims description 22
- 208000028715 ductal breast carcinoma in situ Diseases 0.000 claims description 22
- 239000008194 pharmaceutical composition Substances 0.000 claims description 22
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical group ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 21
- RGHNJXZEOKUKBD-KLVWXMOXSA-N L-gluconic acid Chemical compound OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)C(O)=O RGHNJXZEOKUKBD-KLVWXMOXSA-N 0.000 claims description 21
- 206010073099 Lobular breast carcinoma in situ Diseases 0.000 claims description 21
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 claims description 21
- QOPVNWQGBQYBBP-UHFFFAOYSA-N chloroethyl chloroformate Chemical compound CC(Cl)OC(Cl)=O QOPVNWQGBQYBBP-UHFFFAOYSA-N 0.000 claims description 20
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 18
- 206010033128 Ovarian cancer Diseases 0.000 claims description 18
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 18
- 206010060862 Prostate cancer Diseases 0.000 claims description 18
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 18
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 18
- 208000002495 Uterine Neoplasms Diseases 0.000 claims description 18
- 206010047741 Vulval cancer Diseases 0.000 claims description 18
- 208000004354 Vulvar Neoplasms Diseases 0.000 claims description 18
- 201000010881 cervical cancer Diseases 0.000 claims description 18
- 206010046766 uterine cancer Diseases 0.000 claims description 18
- 206010046885 vaginal cancer Diseases 0.000 claims description 18
- 208000013139 vaginal neoplasm Diseases 0.000 claims description 18
- 201000005102 vulva cancer Diseases 0.000 claims description 18
- 238000005406 washing Methods 0.000 claims description 18
- 239000007950 delayed release tablet Substances 0.000 claims description 17
- 238000010992 reflux Methods 0.000 claims description 17
- 208000011580 syndromic disease Diseases 0.000 claims description 17
- 206010014733 Endometrial cancer Diseases 0.000 claims description 16
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 16
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 claims description 16
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 16
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 claims description 16
- KRIOVPPHQSLHCZ-UHFFFAOYSA-N propiophenone Chemical compound CCC(=O)C1=CC=CC=C1 KRIOVPPHQSLHCZ-UHFFFAOYSA-N 0.000 claims description 16
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 claims description 15
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 claims description 15
- 210000000936 intestine Anatomy 0.000 claims description 15
- 206010036596 premature ejaculation Diseases 0.000 claims description 15
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 claims description 15
- 235000011121 sodium hydroxide Nutrition 0.000 claims description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 15
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 14
- 239000011780 sodium chloride Substances 0.000 claims description 13
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 claims description 12
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 10
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical group [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 10
- 230000002265 prevention Effects 0.000 claims description 10
- 229940124834 selective serotonin reuptake inhibitor Drugs 0.000 claims description 10
- 239000012896 selective serotonin reuptake inhibitor Substances 0.000 claims description 10
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 9
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 9
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 claims description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 9
- AHOUBRCZNHFOSL-YOEHRIQHSA-N (+)-Casbol Chemical compound C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-YOEHRIQHSA-N 0.000 claims description 8
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 claims description 8
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 claims description 8
- 229910000147 aluminium phosphate Inorganic materials 0.000 claims description 8
- 238000002425 crystallisation Methods 0.000 claims description 8
- 230000008025 crystallization Effects 0.000 claims description 8
- 235000019253 formic acid Nutrition 0.000 claims description 8
- 229960002296 paroxetine Drugs 0.000 claims description 8
- 238000000746 purification Methods 0.000 claims description 8
- 239000000377 silicon dioxide Substances 0.000 claims description 8
- 229910052725 zinc Inorganic materials 0.000 claims description 8
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical group CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 claims description 7
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 claims description 7
- 238000001640 fractional crystallisation Methods 0.000 claims description 7
- 238000010438 heat treatment Methods 0.000 claims description 7
- 229910052700 potassium Inorganic materials 0.000 claims description 7
- 239000011591 potassium Substances 0.000 claims description 7
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 7
- 238000002360 preparation method Methods 0.000 claims description 7
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 claims description 6
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 claims description 6
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 claims description 6
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 claims description 6
- URLKBWYHVLBVBO-UHFFFAOYSA-N Para-Xylene Chemical group CC1=CC=C(C)C=C1 URLKBWYHVLBVBO-UHFFFAOYSA-N 0.000 claims description 6
- AHOUBRCZNHFOSL-UHFFFAOYSA-N Paroxetine hydrochloride Natural products C1=CC(F)=CC=C1C1C(COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-UHFFFAOYSA-N 0.000 claims description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 claims description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 6
- 150000001298 alcohols Chemical class 0.000 claims description 6
- 229910052782 aluminium Inorganic materials 0.000 claims description 6
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 claims description 6
- 235000019270 ammonium chloride Nutrition 0.000 claims description 6
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 claims description 6
- DIOQZVSQGTUSAI-UHFFFAOYSA-N decane Chemical compound CCCCCCCCCC DIOQZVSQGTUSAI-UHFFFAOYSA-N 0.000 claims description 6
- 229910052744 lithium Inorganic materials 0.000 claims description 6
- 229910052749 magnesium Inorganic materials 0.000 claims description 6
- 239000011777 magnesium Substances 0.000 claims description 6
- 239000006187 pill Substances 0.000 claims description 6
- VGKDLMBJGBXTGI-SJCJKPOMSA-N sertraline Chemical compound C1([C@@H]2CC[C@@H](C3=CC=CC=C32)NC)=CC=C(Cl)C(Cl)=C1 VGKDLMBJGBXTGI-SJCJKPOMSA-N 0.000 claims description 6
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 claims description 6
- 230000000699 topical effect Effects 0.000 claims description 6
- 230000002792 vascular Effects 0.000 claims description 6
- 230000003442 weekly effect Effects 0.000 claims description 6
- WSEQXVZVJXJVFP-HXUWFJFHSA-N (R)-citalopram Chemical compound C1([C@@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-HXUWFJFHSA-N 0.000 claims description 5
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 claims description 5
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 claims description 5
- 229910052784 alkaline earth metal Inorganic materials 0.000 claims description 5
- 229960001653 citalopram Drugs 0.000 claims description 5
- 230000008878 coupling Effects 0.000 claims description 5
- 238000010168 coupling process Methods 0.000 claims description 5
- 238000005859 coupling reaction Methods 0.000 claims description 5
- WSEQXVZVJXJVFP-FQEVSTJZSA-N escitalopram Chemical compound C1([C@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 WSEQXVZVJXJVFP-FQEVSTJZSA-N 0.000 claims description 5
- 229960004341 escitalopram Drugs 0.000 claims description 5
- 229960002464 fluoxetine Drugs 0.000 claims description 5
- 239000012535 impurity Substances 0.000 claims description 5
- 230000001404 mediated effect Effects 0.000 claims description 5
- 229960002073 sertraline Drugs 0.000 claims description 5
- 235000012239 silicon dioxide Nutrition 0.000 claims description 5
- 238000003756 stirring Methods 0.000 claims description 5
- LJCZNYWLQZZIOS-UHFFFAOYSA-N 2,2,2-trichlorethoxycarbonyl chloride Chemical compound ClC(=O)OCC(Cl)(Cl)Cl LJCZNYWLQZZIOS-UHFFFAOYSA-N 0.000 claims description 4
- LNGQDBJPFPIRBF-UHFFFAOYSA-N 2,2-dichloroethyl carbonochloridate Chemical compound ClC(Cl)COC(Cl)=O LNGQDBJPFPIRBF-UHFFFAOYSA-N 0.000 claims description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 claims description 4
- VBBSPGVSLKBZRW-UHFFFAOYSA-N [4-[2-(dimethylamino)ethoxy]phenyl]-(4-hydroxyphenyl)methanone Chemical compound C1=CC(OCCN(C)C)=CC=C1C(=O)C1=CC=C(O)C=C1 VBBSPGVSLKBZRW-UHFFFAOYSA-N 0.000 claims description 4
- 230000001335 demethylating effect Effects 0.000 claims description 4
- 238000001035 drying Methods 0.000 claims description 4
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 claims description 4
- IVSZLXZYQVIEFR-UHFFFAOYSA-N m-xylene Chemical group CC1=CC=CC(C)=C1 IVSZLXZYQVIEFR-UHFFFAOYSA-N 0.000 claims description 4
- BKIMMITUMNQMOS-UHFFFAOYSA-N nonane Chemical compound CCCCCCCCC BKIMMITUMNQMOS-UHFFFAOYSA-N 0.000 claims description 4
- 235000015497 potassium bicarbonate Nutrition 0.000 claims description 4
- 239000010936 titanium Substances 0.000 claims description 4
- 229910052719 titanium Inorganic materials 0.000 claims description 4
- VXUYXOFXAQZZMF-UHFFFAOYSA-N titanium(IV) isopropoxide Chemical compound CC(C)O[Ti](OC(C)C)(OC(C)C)OC(C)C VXUYXOFXAQZZMF-UHFFFAOYSA-N 0.000 claims description 4
- PLIYGCKUMSKVCU-UHFFFAOYSA-N (4-hydroxyphenyl)-[4-[2-(methylamino)ethoxy]phenyl]methanone Chemical compound C1=CC(OCCNC)=CC=C1C(=O)C1=CC=C(O)C=C1 PLIYGCKUMSKVCU-UHFFFAOYSA-N 0.000 claims description 3
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 claims description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 claims description 3
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 claims description 3
- 239000003513 alkali Substances 0.000 claims description 3
- 150000001342 alkaline earth metals Chemical class 0.000 claims description 3
- CBTVGIZVANVGBH-UHFFFAOYSA-N aminomethyl propanol Chemical compound CC(C)(N)CO CBTVGIZVANVGBH-UHFFFAOYSA-N 0.000 claims description 3
- 239000000908 ammonium hydroxide Substances 0.000 claims description 3
- 229910052786 argon Inorganic materials 0.000 claims description 3
- 238000001816 cooling Methods 0.000 claims description 3
- ATDGTVJJHBUTRL-UHFFFAOYSA-N cyanogen bromide Chemical compound BrC#N ATDGTVJJHBUTRL-UHFFFAOYSA-N 0.000 claims description 3
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 claims description 3
- 239000003085 diluting agent Substances 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- PQLFROTZSIMBKR-UHFFFAOYSA-N ethenyl carbonochloridate Chemical compound ClC(=O)OC=C PQLFROTZSIMBKR-UHFFFAOYSA-N 0.000 claims description 3
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 claims description 3
- 238000010575 fractional recrystallization Methods 0.000 claims description 3
- WSFSSNUMVMOOMR-BJUDXGSMSA-N methanone Chemical compound O=[11CH2] WSFSSNUMVMOOMR-BJUDXGSMSA-N 0.000 claims description 3
- KADXVMRYQRCLAH-UHFFFAOYSA-N n'-iodobutanediamide Chemical group NC(=O)CCC(=O)NI KADXVMRYQRCLAH-UHFFFAOYSA-N 0.000 claims description 3
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 claims description 3
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 claims description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 3
- 235000011181 potassium carbonates Nutrition 0.000 claims description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 3
- 235000017550 sodium carbonate Nutrition 0.000 claims description 3
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 claims description 3
- XROWMBWRMNHXMF-UHFFFAOYSA-J titanium tetrafluoride Chemical compound [F-].[F-].[F-].[F-].[Ti+4] XROWMBWRMNHXMF-UHFFFAOYSA-J 0.000 claims description 3
- YONPGGFAJWQGJC-UHFFFAOYSA-K titanium(iii) chloride Chemical compound Cl[Ti](Cl)Cl YONPGGFAJWQGJC-UHFFFAOYSA-K 0.000 claims description 3
- 239000008096 xylene Substances 0.000 claims description 3
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 claims description 2
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 2
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 claims description 2
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 claims description 2
- MPCVYAVDVXEPTK-UHFFFAOYSA-N butane Chemical compound CCC[CH2+] MPCVYAVDVXEPTK-UHFFFAOYSA-N 0.000 claims description 2
- 150000004649 carbonic acid derivatives Chemical group 0.000 claims description 2
- 238000004440 column chromatography Methods 0.000 claims description 2
- TVZPLCNGKSPOJA-UHFFFAOYSA-N copper zinc Chemical compound [Cu].[Zn] TVZPLCNGKSPOJA-UHFFFAOYSA-N 0.000 claims description 2
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 claims description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 claims description 2
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 claims description 2
- 239000012022 methylating agents Substances 0.000 claims description 2
- 229910052750 molybdenum Inorganic materials 0.000 claims description 2
- 239000011733 molybdenum Substances 0.000 claims description 2
- 229910052758 niobium Inorganic materials 0.000 claims description 2
- 239000010955 niobium Substances 0.000 claims description 2
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 claims description 2
- 229940078552 o-xylene Drugs 0.000 claims description 2
- 229910000028 potassium bicarbonate Inorganic materials 0.000 claims description 2
- 239000011736 potassium bicarbonate Substances 0.000 claims description 2
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 claims description 2
- 239000008213 purified water Substances 0.000 claims description 2
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 claims description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 claims description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 claims description 2
- 239000008247 solid mixture Substances 0.000 claims description 2
- UBZYKBZMAMTNKW-UHFFFAOYSA-J titanium tetrabromide Chemical compound Br[Ti](Br)(Br)Br UBZYKBZMAMTNKW-UHFFFAOYSA-J 0.000 claims description 2
- NLLZTRMHNHVXJJ-UHFFFAOYSA-J titanium tetraiodide Chemical compound I[Ti](I)(I)I NLLZTRMHNHVXJJ-UHFFFAOYSA-J 0.000 claims description 2
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 claims description 2
- 229910052721 tungsten Inorganic materials 0.000 claims description 2
- 239000010937 tungsten Substances 0.000 claims description 2
- 229910052720 vanadium Inorganic materials 0.000 claims description 2
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 claims description 2
- 229910052726 zirconium Inorganic materials 0.000 claims description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 claims 1
- 208000005107 Premature Birth Diseases 0.000 claims 1
- 206010036590 Premature baby Diseases 0.000 claims 1
- 229910001115 Zinc-copper couple Inorganic materials 0.000 claims 1
- 239000012280 lithium aluminium hydride Substances 0.000 claims 1
- 229910052698 phosphorus Inorganic materials 0.000 claims 1
- 239000011574 phosphorus Substances 0.000 claims 1
- OGHBATFHNDZKSO-UHFFFAOYSA-N propan-2-olate Chemical compound CC(C)[O-] OGHBATFHNDZKSO-UHFFFAOYSA-N 0.000 claims 1
- 210000002381 plasma Anatomy 0.000 description 68
- 102100028085 Glycylpeptide N-tetradecanoyltransferase 1 Human genes 0.000 description 50
- 229940050410 gluconate Drugs 0.000 description 45
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 30
- 206010028980 Neoplasm Diseases 0.000 description 27
- 239000000546 pharmaceutical excipient Substances 0.000 description 27
- 201000011510 cancer Diseases 0.000 description 26
- 239000011541 reaction mixture Substances 0.000 description 24
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 23
- 239000000523 sample Substances 0.000 description 23
- 238000012360 testing method Methods 0.000 description 21
- 208000035475 disorder Diseases 0.000 description 19
- 238000009472 formulation Methods 0.000 description 17
- 239000006186 oral dosage form Substances 0.000 description 17
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical compound BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 description 16
- 239000002207 metabolite Substances 0.000 description 16
- 208000037819 metastatic cancer Diseases 0.000 description 16
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 16
- 239000000126 substance Substances 0.000 description 16
- 229940124597 therapeutic agent Drugs 0.000 description 16
- 201000009030 Carcinoma Diseases 0.000 description 15
- 239000000843 powder Substances 0.000 description 14
- 239000000370 acceptor Substances 0.000 description 13
- 238000010520 demethylation reaction Methods 0.000 description 13
- 229920000642 polymer Polymers 0.000 description 13
- 210000002966 serum Anatomy 0.000 description 13
- 210000001519 tissue Anatomy 0.000 description 13
- 239000007884 disintegrant Substances 0.000 description 12
- 229960004756 ethanol Drugs 0.000 description 12
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 11
- 239000002246 antineoplastic agent Substances 0.000 description 11
- 201000010099 disease Diseases 0.000 description 11
- 239000002552 dosage form Substances 0.000 description 11
- 239000003112 inhibitor Substances 0.000 description 11
- 229920002472 Starch Polymers 0.000 description 10
- 239000002585 base Substances 0.000 description 10
- 239000008107 starch Substances 0.000 description 10
- 229940032147 starch Drugs 0.000 description 10
- 235000019698 starch Nutrition 0.000 description 10
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 9
- 238000009098 adjuvant therapy Methods 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 9
- 238000001959 radiotherapy Methods 0.000 description 9
- 238000002560 therapeutic procedure Methods 0.000 description 9
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 8
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 8
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 8
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 8
- 239000004480 active ingredient Substances 0.000 description 8
- HJJPJSXJAXAIPN-UHFFFAOYSA-N arecoline Chemical compound COC(=O)C1=CCCN(C)C1 HJJPJSXJAXAIPN-UHFFFAOYSA-N 0.000 description 8
- 239000000090 biomarker Substances 0.000 description 8
- 210000001072 colon Anatomy 0.000 description 8
- 238000002648 combination therapy Methods 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 238000009093 first-line therapy Methods 0.000 description 8
- 229940102396 methyl bromide Drugs 0.000 description 8
- 239000004014 plasticizer Substances 0.000 description 8
- 238000001356 surgical procedure Methods 0.000 description 8
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 8
- RPFIMPDXTABYCN-BJFQDICYSA-N 4-[(z)-1-[4-[2-(methylamino)ethoxy]phenyl]-2-phenylbut-1-enyl]phenol;hydrochloride Chemical compound Cl.C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCNC)=CC=1)/C1=CC=C(O)C=C1 RPFIMPDXTABYCN-BJFQDICYSA-N 0.000 description 7
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 7
- 108010001237 Cytochrome P-450 CYP2D6 Proteins 0.000 description 7
- 102100021704 Cytochrome P450 2D6 Human genes 0.000 description 7
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 7
- 239000013543 active substance Substances 0.000 description 7
- 239000011230 binding agent Substances 0.000 description 7
- 210000004369 blood Anatomy 0.000 description 7
- 239000008280 blood Substances 0.000 description 7
- 102000015694 estrogen receptors Human genes 0.000 description 7
- 108010038795 estrogen receptors Proteins 0.000 description 7
- 239000000945 filler Substances 0.000 description 7
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 7
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 7
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 7
- 239000000314 lubricant Substances 0.000 description 7
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 7
- 229920001223 polyethylene glycol Polymers 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- 241000269627 Amphiuma means Species 0.000 description 6
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 6
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 6
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 6
- 208000005726 Inflammatory Breast Neoplasms Diseases 0.000 description 6
- 206010021980 Inflammatory carcinoma of the breast Diseases 0.000 description 6
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 6
- 239000002202 Polyethylene glycol Substances 0.000 description 6
- 238000010521 absorption reaction Methods 0.000 description 6
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 238000002512 chemotherapy Methods 0.000 description 6
- 230000017858 demethylation Effects 0.000 description 6
- 230000008030 elimination Effects 0.000 description 6
- 238000003379 elimination reaction Methods 0.000 description 6
- 239000002702 enteric coating Substances 0.000 description 6
- 238000009505 enteric coating Methods 0.000 description 6
- 229960002949 fluorouracil Drugs 0.000 description 6
- 201000004653 inflammatory breast carcinoma Diseases 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 239000008184 oral solid dosage form Substances 0.000 description 6
- 201000010198 papillary carcinoma Diseases 0.000 description 6
- 229940068196 placebo Drugs 0.000 description 6
- 239000000902 placebo Substances 0.000 description 6
- 229960003975 potassium Drugs 0.000 description 6
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 5
- AEQDJSLRWYMAQI-UHFFFAOYSA-N 2,3,9,10-tetramethoxy-6,8,13,13a-tetrahydro-5H-isoquinolino[2,1-b]isoquinoline Chemical compound C1CN2CC(C(=C(OC)C=C3)OC)=C3CC2C2=C1C=C(OC)C(OC)=C2 AEQDJSLRWYMAQI-UHFFFAOYSA-N 0.000 description 5
- 229920000623 Cellulose acetate phthalate Polymers 0.000 description 5
- 102000004328 Cytochrome P-450 CYP3A Human genes 0.000 description 5
- 108010081668 Cytochrome P-450 CYP3A Proteins 0.000 description 5
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 5
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 5
- 229940081735 acetylcellulose Drugs 0.000 description 5
- 239000008186 active pharmaceutical agent Substances 0.000 description 5
- 235000010443 alginic acid Nutrition 0.000 description 5
- 229920000615 alginic acid Polymers 0.000 description 5
- 229940041181 antineoplastic drug Drugs 0.000 description 5
- 229960000997 bicalutamide Drugs 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 229920002301 cellulose acetate Polymers 0.000 description 5
- 229940081734 cellulose acetate phthalate Drugs 0.000 description 5
- 229950006191 gluconic acid Drugs 0.000 description 5
- 235000011187 glycerol Nutrition 0.000 description 5
- 208000030776 invasive breast carcinoma Diseases 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 229940091250 magnesium supplement Drugs 0.000 description 5
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 5
- 239000008108 microcrystalline cellulose Substances 0.000 description 5
- 229940016286 microcrystalline cellulose Drugs 0.000 description 5
- 230000000683 nonmetastatic effect Effects 0.000 description 5
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 210000000813 small intestine Anatomy 0.000 description 5
- 239000000176 sodium gluconate Substances 0.000 description 5
- 235000012207 sodium gluconate Nutrition 0.000 description 5
- 229940005574 sodium gluconate Drugs 0.000 description 5
- 235000010356 sorbitol Nutrition 0.000 description 5
- 239000000600 sorbitol Substances 0.000 description 5
- 239000003765 sweetening agent Substances 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- 239000001993 wax Substances 0.000 description 5
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 4
- MPPPKRYCTPRNTB-UHFFFAOYSA-N 1-bromobutane Chemical compound CCCCBr MPPPKRYCTPRNTB-UHFFFAOYSA-N 0.000 description 4
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 4
- 108010012934 Albumin-Bound Paclitaxel Proteins 0.000 description 4
- 102100022595 Broad substrate specificity ATP-binding cassette transporter ABCG2 Human genes 0.000 description 4
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 4
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 4
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- 208000006402 Ductal Carcinoma Diseases 0.000 description 4
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 4
- 101000823298 Homo sapiens Broad substrate specificity ATP-binding cassette transporter ABCG2 Proteins 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 4
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 4
- 241000124008 Mammalia Species 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 4
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 4
- 229910002651 NO3 Inorganic materials 0.000 description 4
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 4
- 229920002253 Tannate Polymers 0.000 description 4
- 229940034982 antineoplastic agent Drugs 0.000 description 4
- 230000001580 bacterial effect Effects 0.000 description 4
- 229910052788 barium Inorganic materials 0.000 description 4
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 4
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 4
- 229910052797 bismuth Inorganic materials 0.000 description 4
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 4
- 230000005907 cancer growth Effects 0.000 description 4
- 239000001913 cellulose Substances 0.000 description 4
- 235000010980 cellulose Nutrition 0.000 description 4
- 229920002678 cellulose Polymers 0.000 description 4
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 4
- 229960002023 chloroprocaine Drugs 0.000 description 4
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 4
- 229960001231 choline Drugs 0.000 description 4
- 230000001276 controlling effect Effects 0.000 description 4
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 4
- 229940043237 diethanolamine Drugs 0.000 description 4
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 4
- 239000001177 diphosphate Substances 0.000 description 4
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 4
- 235000011180 diphosphates Nutrition 0.000 description 4
- 229950005627 embonate Drugs 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 229960004671 enzalutamide Drugs 0.000 description 4
- WXCXUHSOUPDCQV-UHFFFAOYSA-N enzalutamide Chemical compound C1=C(F)C(C(=O)NC)=CC=C1N1C(C)(C)C(=O)N(C=2C=C(C(C#N)=CC=2)C(F)(F)F)C1=S WXCXUHSOUPDCQV-UHFFFAOYSA-N 0.000 description 4
- UFNVPOGXISZXJD-JBQZKEIOSA-N eribulin Chemical compound C([C@H]1CC[C@@H]2O[C@@H]3[C@H]4O[C@@H]5C[C@](O[C@H]4[C@H]2O1)(O[C@@H]53)CC[C@@H]1O[C@H](C(C1)=C)CC1)C(=O)C[C@@H]2[C@@H](OC)[C@@H](C[C@H](O)CN)O[C@H]2C[C@@H]2C(=C)[C@H](C)C[C@H]1O2 UFNVPOGXISZXJD-JBQZKEIOSA-N 0.000 description 4
- 229940011871 estrogen Drugs 0.000 description 4
- 239000000262 estrogen Substances 0.000 description 4
- 229940012017 ethylenediamine Drugs 0.000 description 4
- 239000010408 film Substances 0.000 description 4
- 239000000796 flavoring agent Substances 0.000 description 4
- 235000013355 food flavoring agent Nutrition 0.000 description 4
- 235000003599 food sweetener Nutrition 0.000 description 4
- 235000012208 gluconic acid Nutrition 0.000 description 4
- 229930195712 glutamate Natural products 0.000 description 4
- 229940074045 glyceryl distearate Drugs 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 229910001385 heavy metal Inorganic materials 0.000 description 4
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 4
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 4
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 4
- FABUFPQFXZVHFB-PVYNADRNSA-N ixabepilone Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)N1)O)C)=C\C1=CSC(C)=N1 FABUFPQFXZVHFB-PVYNADRNSA-N 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 229940049920 malate Drugs 0.000 description 4
- BJEPYKJPYRNKOW-UHFFFAOYSA-L malate(2-) Chemical compound [O-]C(=O)C(O)CC([O-])=O BJEPYKJPYRNKOW-UHFFFAOYSA-L 0.000 description 4
- 238000009607 mammography Methods 0.000 description 4
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 4
- 229960003194 meglumine Drugs 0.000 description 4
- LRMHVVPPGGOAJQ-UHFFFAOYSA-N methyl nitrate Chemical compound CO[N+]([O-])=O LRMHVVPPGGOAJQ-UHFFFAOYSA-N 0.000 description 4
- OSWPMRLSEDHDFF-UHFFFAOYSA-N methyl salicylate Chemical compound COC(=O)C1=CC=CC=C1O OSWPMRLSEDHDFF-UHFFFAOYSA-N 0.000 description 4
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 230000035772 mutation Effects 0.000 description 4
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 4
- 229940014662 pantothenate Drugs 0.000 description 4
- 235000019161 pantothenic acid Nutrition 0.000 description 4
- 239000011713 pantothenic acid Substances 0.000 description 4
- 239000012188 paraffin wax Substances 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 4
- 239000010452 phosphate Substances 0.000 description 4
- 235000021317 phosphate Nutrition 0.000 description 4
- 229960005141 piperazine Drugs 0.000 description 4
- 229940100467 polyvinyl acetate phthalate Drugs 0.000 description 4
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 4
- 229960004919 procaine Drugs 0.000 description 4
- 230000000069 prophylactic effect Effects 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 238000001953 recrystallisation Methods 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 4
- 229960001860 salicylate Drugs 0.000 description 4
- 235000010413 sodium alginate Nutrition 0.000 description 4
- 239000000661 sodium alginate Substances 0.000 description 4
- 229940114926 stearate Drugs 0.000 description 4
- 210000002784 stomach Anatomy 0.000 description 4
- 229960000575 trastuzumab Drugs 0.000 description 4
- MWWSFMDVAYGXBV-MYPASOLCSA-N (7r,9s)-7-[(2r,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;hydrochloride Chemical compound Cl.O([C@@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-MYPASOLCSA-N 0.000 description 3
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 3
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 3
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 3
- 208000009458 Carcinoma in Situ Diseases 0.000 description 3
- 206010010356 Congenital anomaly Diseases 0.000 description 3
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 3
- 101150029707 ERBB2 gene Proteins 0.000 description 3
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 3
- 229920003134 Eudragit® polymer Polymers 0.000 description 3
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 3
- 206010020850 Hyperthyroidism Diseases 0.000 description 3
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 3
- 208000000265 Lobular Carcinoma Diseases 0.000 description 3
- 229930195725 Mannitol Natural products 0.000 description 3
- 229920000881 Modified starch Polymers 0.000 description 3
- NYDCDZSEEAUOHN-IZHYLOQSSA-N N-Desmethyltamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCNC)=CC=1)/C1=CC=CC=C1 NYDCDZSEEAUOHN-IZHYLOQSSA-N 0.000 description 3
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 3
- 229920001800 Shellac Polymers 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 description 3
- 230000005856 abnormality Effects 0.000 description 3
- 229940028652 abraxane Drugs 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- TXUZVZSFRXZGTL-QPLCGJKRSA-N afimoxifene Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=C(O)C=C1 TXUZVZSFRXZGTL-QPLCGJKRSA-N 0.000 description 3
- 125000000129 anionic group Chemical group 0.000 description 3
- 201000003714 breast lobular carcinoma Diseases 0.000 description 3
- 229910000019 calcium carbonate Inorganic materials 0.000 description 3
- 235000010216 calcium carbonate Nutrition 0.000 description 3
- 229960004117 capecitabine Drugs 0.000 description 3
- 229960004562 carboplatin Drugs 0.000 description 3
- 239000001768 carboxy methyl cellulose Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- UHZZMRAGKVHANO-UHFFFAOYSA-M chlormequat chloride Chemical compound [Cl-].C[N+](C)(C)CCCl UHZZMRAGKVHANO-UHFFFAOYSA-M 0.000 description 3
- 229960004316 cisplatin Drugs 0.000 description 3
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 229960004397 cyclophosphamide Drugs 0.000 description 3
- 238000003745 diagnosis Methods 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- 229960003668 docetaxel Drugs 0.000 description 3
- PRAKJMSDJKAYCZ-UHFFFAOYSA-N dodecahydrosqualene Natural products CC(C)CCCC(C)CCCC(C)CCCCC(C)CCCC(C)CCCC(C)C PRAKJMSDJKAYCZ-UHFFFAOYSA-N 0.000 description 3
- 229960001904 epirubicin Drugs 0.000 description 3
- 201000007281 estrogen-receptor positive breast cancer Diseases 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 3
- 229960005277 gemcitabine Drugs 0.000 description 3
- 229940075507 glyceryl monostearate Drugs 0.000 description 3
- 229940118951 halaven Drugs 0.000 description 3
- 230000003054 hormonal effect Effects 0.000 description 3
- 229920003132 hydroxypropyl methylcellulose phthalate Polymers 0.000 description 3
- 229940031704 hydroxypropyl methylcellulose phthalate Drugs 0.000 description 3
- 229960003943 hypromellose Drugs 0.000 description 3
- 238000009169 immunotherapy Methods 0.000 description 3
- 206010073096 invasive lobular breast carcinoma Diseases 0.000 description 3
- 229960002014 ixabepilone Drugs 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 230000003902 lesion Effects 0.000 description 3
- 229960001078 lithium Drugs 0.000 description 3
- 208000019423 liver disease Diseases 0.000 description 3
- 239000000594 mannitol Substances 0.000 description 3
- 235000010355 mannitol Nutrition 0.000 description 3
- 229920003145 methacrylic acid copolymer Polymers 0.000 description 3
- 229960000485 methotrexate Drugs 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 230000000813 microbial effect Effects 0.000 description 3
- 210000003097 mucus Anatomy 0.000 description 3
- OSFCMRGOZNQUSW-UHFFFAOYSA-N n-[4-[2-(6,7-dimethoxy-3,4-dihydro-1h-isoquinolin-2-yl)ethyl]phenyl]-5-methoxy-9-oxo-10h-acridine-4-carboxamide Chemical compound N1C2=C(OC)C=CC=C2C(=O)C2=C1C(C(=O)NC1=CC=C(C=C1)CCN1CCC=3C=C(C(=CC=3C1)OC)OC)=CC=C2 OSFCMRGOZNQUSW-UHFFFAOYSA-N 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 235000019198 oils Nutrition 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- 229940063179 platinol Drugs 0.000 description 3
- 239000010734 process oil Substances 0.000 description 3
- 235000018102 proteins Nutrition 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 239000000376 reactant Substances 0.000 description 3
- 230000001850 reproductive effect Effects 0.000 description 3
- 210000005000 reproductive tract Anatomy 0.000 description 3
- 235000013874 shellac Nutrition 0.000 description 3
- 239000004208 shellac Substances 0.000 description 3
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 3
- 229940113147 shellac Drugs 0.000 description 3
- 229940005550 sodium alginate Drugs 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- 230000002459 sustained effect Effects 0.000 description 3
- 229940063683 taxotere Drugs 0.000 description 3
- 208000022679 triple-negative breast carcinoma Diseases 0.000 description 3
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 3
- 229960002066 vinorelbine Drugs 0.000 description 3
- MEJYDZQQVZJMPP-ULAWRXDQSA-N (3s,3ar,6r,6ar)-3,6-dimethoxy-2,3,3a,5,6,6a-hexahydrofuro[3,2-b]furan Chemical compound CO[C@H]1CO[C@@H]2[C@H](OC)CO[C@@H]21 MEJYDZQQVZJMPP-ULAWRXDQSA-N 0.000 description 2
- ZGMJYTYLTJFNCS-VQYXCCSOSA-N (e)-but-2-enedioic acid;1-[4-(2-hydroxy-3-quinolin-5-yloxypropyl)piperazin-1-yl]-2,2-diphenylethanone Chemical compound OC(=O)\C=C\C(O)=O.OC(=O)\C=C\C(O)=O.OC(=O)\C=C\C(O)=O.C=1C=CC2=NC=CC=C2C=1OCC(O)CN(CC1)CCN1C(=O)C(C=1C=CC=CC=1)C1=CC=CC=C1.C=1C=CC2=NC=CC=C2C=1OCC(O)CN(CC1)CCN1C(=O)C(C=1C=CC=CC=1)C1=CC=CC=C1 ZGMJYTYLTJFNCS-VQYXCCSOSA-N 0.000 description 2
- LVYLCBNXHHHPSB-UHFFFAOYSA-N 2-hydroxyethyl salicylate Chemical compound OCCOC(=O)C1=CC=CC=C1O LVYLCBNXHHHPSB-UHFFFAOYSA-N 0.000 description 2
- DODQJNMQWMSYGS-QPLCGJKRSA-N 4-[(z)-1-[4-[2-(dimethylamino)ethoxy]phenyl]-1-phenylbut-1-en-2-yl]phenol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 DODQJNMQWMSYGS-QPLCGJKRSA-N 0.000 description 2
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 2
- 102000043966 ABC-type transporter activity proteins Human genes 0.000 description 2
- 108010006533 ATP-Binding Cassette Transporters Proteins 0.000 description 2
- 241000321096 Adenoides Species 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 238000012935 Averaging Methods 0.000 description 2
- 206010006298 Breast pain Diseases 0.000 description 2
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 2
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 2
- 108010036949 Cyclosporine Proteins 0.000 description 2
- 108010000543 Cytochrome P-450 CYP2C9 Proteins 0.000 description 2
- 229920003143 Eudragit® FS 30 D Polymers 0.000 description 2
- 229920003139 Eudragit® L 100 Polymers 0.000 description 2
- 229920003135 Eudragit® L 100-55 Polymers 0.000 description 2
- 229920003138 Eudragit® L 30 D-55 Polymers 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 208000006662 Mastodynia Diseases 0.000 description 2
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 2
- 244000246386 Mentha pulegium Species 0.000 description 2
- 235000016257 Mentha pulegium Nutrition 0.000 description 2
- 235000004357 Mentha x piperita Nutrition 0.000 description 2
- RTHCYVBBDHJXIQ-UHFFFAOYSA-N N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]propan-1-amine Chemical compound C=1C=CC=CC=1C(CCNC)OC1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- BPQQTUXANYXVAA-UHFFFAOYSA-N Orthosilicate Chemical compound [O-][Si]([O-])([O-])[O-] BPQQTUXANYXVAA-UHFFFAOYSA-N 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- 235000019483 Peanut oil Nutrition 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 239000004372 Polyvinyl alcohol Substances 0.000 description 2
- 208000001647 Renal Insufficiency Diseases 0.000 description 2
- 208000034189 Sclerosis Diseases 0.000 description 2
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 2
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 229920002494 Zein Polymers 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- IYKJEILNJZQJPU-UHFFFAOYSA-N acetic acid;butanedioic acid Chemical compound CC(O)=O.OC(=O)CCC(O)=O IYKJEILNJZQJPU-UHFFFAOYSA-N 0.000 description 2
- ZUAAPNNKRHMPKG-UHFFFAOYSA-N acetic acid;butanedioic acid;methanol;propane-1,2-diol Chemical compound OC.CC(O)=O.CC(O)CO.OC(=O)CCC(O)=O ZUAAPNNKRHMPKG-UHFFFAOYSA-N 0.000 description 2
- 238000007792 addition Methods 0.000 description 2
- 210000002534 adenoid Anatomy 0.000 description 2
- 201000008395 adenosquamous carcinoma Diseases 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 229940023476 agar Drugs 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 229940072056 alginate Drugs 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 235000013871 bee wax Nutrition 0.000 description 2
- 239000012166 beeswax Substances 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 210000001124 body fluid Anatomy 0.000 description 2
- 239000010839 body fluid Substances 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 239000001506 calcium phosphate Substances 0.000 description 2
- 229910000389 calcium phosphate Inorganic materials 0.000 description 2
- 235000011010 calcium phosphates Nutrition 0.000 description 2
- 239000004203 carnauba wax Substances 0.000 description 2
- 235000013869 carnauba wax Nutrition 0.000 description 2
- 125000002091 cationic group Chemical group 0.000 description 2
- 229920006217 cellulose acetate butyrate Polymers 0.000 description 2
- 229960000541 cetyl alcohol Drugs 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 235000015218 chewing gum Nutrition 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 229960001265 ciclosporin Drugs 0.000 description 2
- 150000001860 citric acid derivatives Chemical class 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 229960000935 dehydrated alcohol Drugs 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- DOIRQSBPFJWKBE-UHFFFAOYSA-N dibutyl phthalate Chemical compound CCCCOC(=O)C1=CC=CC=C1C(=O)OCCCC DOIRQSBPFJWKBE-UHFFFAOYSA-N 0.000 description 2
- XXJWXESWEXIICW-UHFFFAOYSA-N diethylene glycol monoethyl ether Chemical compound CCOCCOCCO XXJWXESWEXIICW-UHFFFAOYSA-N 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 238000012377 drug delivery Methods 0.000 description 2
- 230000002357 endometrial effect Effects 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- 201000003585 eunuchism Diseases 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 125000003976 glyceryl group Chemical group [H]C([*])([H])C(O[H])([H])C(O[H])([H])[H] 0.000 description 2
- 238000001631 haemodialysis Methods 0.000 description 2
- 230000000322 hemodialysis Effects 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 235000001050 hortel pimenta Nutrition 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 description 2
- 238000003364 immunohistochemistry Methods 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 201000006370 kidney failure Diseases 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 239000007937 lozenge Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 208000037106 male hypogonadism Diseases 0.000 description 2
- 230000035800 maturation Effects 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 229960001047 methyl salicylate Drugs 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 239000004200 microcrystalline wax Substances 0.000 description 2
- 235000019808 microcrystalline wax Nutrition 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- GOQYKNQRPGWPLP-UHFFFAOYSA-N n-heptadecyl alcohol Natural products CCCCCCCCCCCCCCCCCO GOQYKNQRPGWPLP-UHFFFAOYSA-N 0.000 description 2
- ZCYXXKJEDCHMGH-UHFFFAOYSA-N nonane Chemical compound CCCC[CH]CCCC ZCYXXKJEDCHMGH-UHFFFAOYSA-N 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 235000008390 olive oil Nutrition 0.000 description 2
- 238000011275 oncology therapy Methods 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- 235000019809 paraffin wax Nutrition 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 239000000312 peanut oil Substances 0.000 description 2
- 235000019271 petrolatum Nutrition 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 238000003752 polymerase chain reaction Methods 0.000 description 2
- 208000001061 polyostotic fibrous dysplasia Diseases 0.000 description 2
- 229920002744 polyvinyl acetate phthalate Polymers 0.000 description 2
- 229920002451 polyvinyl alcohol Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 230000002028 premature Effects 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 229940035613 prozac Drugs 0.000 description 2
- LOUPRKONTZGTKE-LHHVKLHASA-N quinidine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@H]2[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-LHHVKLHASA-N 0.000 description 2
- 238000003753 real-time PCR Methods 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- URAYPUMNDPQOKB-UHFFFAOYSA-N triacetin Chemical compound CC(=O)OCC(OC(C)=O)COC(C)=O URAYPUMNDPQOKB-UHFFFAOYSA-N 0.000 description 2
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 2
- 125000005591 trimellitate group Chemical group 0.000 description 2
- 235000015112 vegetable and seed oil Nutrition 0.000 description 2
- 239000008158 vegetable oil Substances 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 235000012431 wafers Nutrition 0.000 description 2
- 229940093612 zein Drugs 0.000 description 2
- 239000005019 zein Substances 0.000 description 2
- KTGRHKOEFSJQNS-BDQAORGHSA-N (1s)-1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-3h-2-benzofuran-5-carbonitrile;oxalic acid Chemical compound OC(=O)C(O)=O.C1([C@]2(C3=CC=C(C=C3CO2)C#N)CCCN(C)C)=CC=C(F)C=C1 KTGRHKOEFSJQNS-BDQAORGHSA-N 0.000 description 1
- DNXIKVLOVZVMQF-UHFFFAOYSA-N (3beta,16beta,17alpha,18beta,20alpha)-17-hydroxy-11-methoxy-18-[(3,4,5-trimethoxybenzoyl)oxy]-yohimban-16-carboxylic acid, methyl ester Natural products C1C2CN3CCC(C4=CC=C(OC)C=C4N4)=C4C3CC2C(C(=O)OC)C(O)C1OC(=O)C1=CC(OC)=C(OC)C(OC)=C1 DNXIKVLOVZVMQF-UHFFFAOYSA-N 0.000 description 1
- SVJMLYUFVDMUHP-MGBGTMOVSA-N (4R)-2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylic acid O5-[3-(4,4-diphenyl-1-piperidinyl)propyl] ester O3-methyl ester Chemical compound C1([C@H]2C(=C(C)NC(C)=C2C(=O)OC)C(=O)OCCCN2CCC(CC2)(C=2C=CC=CC=2)C=2C=CC=CC=2)=CC=CC([N+]([O-])=O)=C1 SVJMLYUFVDMUHP-MGBGTMOVSA-N 0.000 description 1
- YYGNTYWPHWGJRM-UHFFFAOYSA-N (6E,10E,14E,18E)-2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene Chemical compound CC(C)=CCCC(C)=CCCC(C)=CCCC=C(C)CCC=C(C)CCC=C(C)C YYGNTYWPHWGJRM-UHFFFAOYSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- WSEQXVZVJXJVFP-UHFFFAOYSA-N 1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydro-2-benzofuran-5-carbonitrile Chemical compound O1CC2=CC(C#N)=CC=C2C1(CCCN(C)C)C1=CC=C(F)C=C1 WSEQXVZVJXJVFP-UHFFFAOYSA-N 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- CTPDSKVQLSDPLC-UHFFFAOYSA-N 2-(oxolan-2-ylmethoxy)ethanol Chemical compound OCCOCC1CCCO1 CTPDSKVQLSDPLC-UHFFFAOYSA-N 0.000 description 1
- MUZDXNQOSGWMJJ-UHFFFAOYSA-N 2-methylprop-2-enoic acid;prop-2-enoic acid Chemical compound OC(=O)C=C.CC(=C)C(O)=O MUZDXNQOSGWMJJ-UHFFFAOYSA-N 0.000 description 1
- RSJCLODJSVZNQA-BQYQJAHWSA-N 4-[2-[4-[(e)-3-ethoxyprop-1-enyl]phenyl]-4-[4-(propan-2-ylamino)phenyl]-1h-imidazol-5-yl]-n-propan-2-ylaniline Chemical compound C1=CC(/C=C/COCC)=CC=C1C1=NC(C=2C=CC(NC(C)C)=CC=2)=C(C=2C=CC(NC(C)C)=CC=2)N1 RSJCLODJSVZNQA-BQYQJAHWSA-N 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- WBZFUFAFFUEMEI-UHFFFAOYSA-M Acesulfame k Chemical compound [K+].CC1=CC(=O)[N-]S(=O)(=O)O1 WBZFUFAFFUEMEI-UHFFFAOYSA-M 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- 229910002016 Aerosil® 200 Inorganic materials 0.000 description 1
- 208000028060 Albright disease Diseases 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 244000099147 Ananas comosus Species 0.000 description 1
- 235000007119 Ananas comosus Nutrition 0.000 description 1
- 206010059245 Angiopathy Diseases 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 206010061692 Benign muscle neoplasm Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- 101150051438 CYP gene Proteins 0.000 description 1
- 101100460513 Caenorhabditis elegans nlt-1 gene Proteins 0.000 description 1
- 102100033620 Calponin-1 Human genes 0.000 description 1
- 241000222122 Candida albicans Species 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 241001112696 Clostridia Species 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- UDIPTWFVPPPURJ-UHFFFAOYSA-M Cyclamate Chemical compound [Na+].[O-]S(=O)(=O)NC1CCCCC1 UDIPTWFVPPPURJ-UHFFFAOYSA-M 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- 102000002269 Cytochrome P-450 CYP2C9 Human genes 0.000 description 1
- PHOQVHQSTUBQQK-SQOUGZDYSA-N D-glucono-1,5-lactone Chemical compound OC[C@H]1OC(=O)[C@H](O)[C@@H](O)[C@@H]1O PHOQVHQSTUBQQK-SQOUGZDYSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- MQIUGAXCHLFZKX-UHFFFAOYSA-N Di-n-octyl phthalate Natural products CCCCCCCCOC(=O)C1=CC=CC=C1C(=O)OCCCCCCCC MQIUGAXCHLFZKX-UHFFFAOYSA-N 0.000 description 1
- 206010013710 Drug interaction Diseases 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 206010058314 Dysplasia Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 201000009273 Endometriosis Diseases 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 229920003149 Eudragit® E 100 Polymers 0.000 description 1
- 229920003150 Eudragit® E 12,5 Polymers 0.000 description 1
- 229920003136 Eudragit® L polymer Polymers 0.000 description 1
- 229920003141 Eudragit® S 100 Polymers 0.000 description 1
- 229920003142 Eudragit® S 12,5 Polymers 0.000 description 1
- 229920003137 Eudragit® S polymer Polymers 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 235000016623 Fragaria vesca Nutrition 0.000 description 1
- 240000009088 Fragaria x ananassa Species 0.000 description 1
- 235000011363 Fragaria x ananassa Nutrition 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- 101150050733 Gnas gene Proteins 0.000 description 1
- 229920002907 Guar gum Polymers 0.000 description 1
- CMBYOWLFQAFZCP-UHFFFAOYSA-N Hexyl dodecanoate Chemical compound CCCCCCCCCCCC(=O)OCCCCCC CMBYOWLFQAFZCP-UHFFFAOYSA-N 0.000 description 1
- 101000945318 Homo sapiens Calponin-1 Proteins 0.000 description 1
- 101000652736 Homo sapiens Transgelin Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 108010076876 Keratins Proteins 0.000 description 1
- 102000011782 Keratins Human genes 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 208000004059 Male Breast Neoplasms Diseases 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 201000001853 McCune-Albright syndrome Diseases 0.000 description 1
- 238000006519 Mcmurry reaction Methods 0.000 description 1
- 208000007054 Medullary Carcinoma Diseases 0.000 description 1
- 235000014749 Mentha crispa Nutrition 0.000 description 1
- 244000078639 Mentha spicata Species 0.000 description 1
- 229920003091 Methocel™ Polymers 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 201000004458 Myoma Diseases 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- 239000008118 PEG 6000 Substances 0.000 description 1
- 241000282320 Panthera leo Species 0.000 description 1
- IQPSEEYGBUAQFF-UHFFFAOYSA-N Pantoprazole Chemical compound COC1=CC=NC(CS(=O)C=2NC3=CC=C(OC(F)F)C=C3N=2)=C1OC IQPSEEYGBUAQFF-UHFFFAOYSA-N 0.000 description 1
- 208000004605 Persistent Truncus Arteriosus Diseases 0.000 description 1
- 229920002556 Polyethylene Glycol 300 Polymers 0.000 description 1
- 229920002584 Polyethylene Glycol 6000 Polymers 0.000 description 1
- HLCFGWHYROZGBI-JJKGCWMISA-M Potassium gluconate Chemical compound [K+].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O HLCFGWHYROZGBI-JJKGCWMISA-M 0.000 description 1
- 102100025803 Progesterone receptor Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 208000003251 Pruritus Diseases 0.000 description 1
- 206010037660 Pyrexia Diseases 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 229920000297 Rayon Polymers 0.000 description 1
- LCQMZZCPPSWADO-UHFFFAOYSA-N Reserpilin Natural products COC(=O)C1COCC2CN3CCc4c([nH]c5cc(OC)c(OC)cc45)C3CC12 LCQMZZCPPSWADO-UHFFFAOYSA-N 0.000 description 1
- QEVHRUUCFGRFIF-SFWBKIHZSA-N Reserpine Natural products O=C(OC)[C@@H]1[C@H](OC)[C@H](OC(=O)c2cc(OC)c(OC)c(OC)c2)C[C@H]2[C@@H]1C[C@H]1N(C2)CCc2c3c([nH]c12)cc(OC)cc3 QEVHRUUCFGRFIF-SFWBKIHZSA-N 0.000 description 1
- 235000011552 Rhamnus crocea Nutrition 0.000 description 1
- 235000004443 Ricinus communis Nutrition 0.000 description 1
- 240000007651 Rubus glaucus Species 0.000 description 1
- 235000011034 Rubus glaucus Nutrition 0.000 description 1
- 235000009122 Rubus idaeus Nutrition 0.000 description 1
- YJDYDFNKCBANTM-QCWCSKBGSA-N SDZ PSC 833 Chemical compound C\C=C\C[C@@H](C)C(=O)[C@@H]1N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C(=O)[C@H](C(C)C)NC1=O YJDYDFNKCBANTM-QCWCSKBGSA-N 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 239000004376 Sucralose Substances 0.000 description 1
- 235000019486 Sunflower oil Nutrition 0.000 description 1
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 1
- LGGHDPFKSSRQNS-UHFFFAOYSA-N Tariquidar Chemical compound C1=CC=CC2=CC(C(=O)NC3=CC(OC)=C(OC)C=C3C(=O)NC3=CC=C(C=C3)CCN3CCC=4C=C(C(=CC=4C3)OC)OC)=CN=C21 LGGHDPFKSSRQNS-UHFFFAOYSA-N 0.000 description 1
- BHEOSNUKNHRBNM-UHFFFAOYSA-N Tetramethylsqualene Natural products CC(=C)C(C)CCC(=C)C(C)CCC(C)=CCCC=C(C)CCC(C)C(=C)CCC(C)C(C)=C BHEOSNUKNHRBNM-UHFFFAOYSA-N 0.000 description 1
- 241000863032 Trieres Species 0.000 description 1
- DOOTYTYQINUNNV-UHFFFAOYSA-N Triethyl citrate Chemical compound CCOC(=O)CC(O)(C(=O)OCC)CC(=O)OCC DOOTYTYQINUNNV-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 208000037258 Truncus arteriosus Diseases 0.000 description 1
- 206010046798 Uterine leiomyoma Diseases 0.000 description 1
- 241000759263 Ventia crocea Species 0.000 description 1
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 1
- BLGXFZZNTVWLAY-CCZXDCJGSA-N Yohimbine Natural products C1=CC=C2C(CCN3C[C@@H]4CC[C@@H](O)[C@H]([C@H]4C[C@H]33)C(=O)OC)=C3NC2=C1 BLGXFZZNTVWLAY-CCZXDCJGSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 239000000619 acesulfame-K Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 208000009956 adenocarcinoma Diseases 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000010775 animal oil Substances 0.000 description 1
- 230000002280 anti-androgenic effect Effects 0.000 description 1
- 239000000051 antiandrogen Substances 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 239000000164 antipsychotic agent Substances 0.000 description 1
- 229940005529 antipsychotics Drugs 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- 238000003339 best practice Methods 0.000 description 1
- BLGXFZZNTVWLAY-UHFFFAOYSA-N beta-Yohimbin Natural products C1=CC=C2C(CCN3CC4CCC(O)C(C4CC33)C(=O)OC)=C3NC2=C1 BLGXFZZNTVWLAY-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- BJQHLKABXJIVAM-UHFFFAOYSA-N bis(2-ethylhexyl) phthalate Chemical compound CCCCC(CC)COC(=O)C1=CC=CC=C1C(=O)OCC(CC)CCCC BJQHLKABXJIVAM-UHFFFAOYSA-N 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 201000008275 breast carcinoma Diseases 0.000 description 1
- 201000010983 breast ductal carcinoma Diseases 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 239000004067 bulking agent Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229960003563 calcium carbonate Drugs 0.000 description 1
- 239000004227 calcium gluconate Substances 0.000 description 1
- 235000013927 calcium gluconate Nutrition 0.000 description 1
- 229960004494 calcium gluconate Drugs 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- NEEHYRZPVYRGPP-UHFFFAOYSA-L calcium;2,3,4,5,6-pentahydroxyhexanoate Chemical compound [Ca+2].OCC(O)C(O)C(O)C(O)C([O-])=O.OCC(O)C(O)C(O)C(O)C([O-])=O NEEHYRZPVYRGPP-UHFFFAOYSA-L 0.000 description 1
- 239000010495 camellia oil Substances 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 229940084030 carboxymethylcellulose calcium Drugs 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 229940047493 celexa Drugs 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 229940112822 chewing gum Drugs 0.000 description 1
- 150000003841 chloride salts Chemical class 0.000 description 1
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 1
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 238000011220 combination immunotherapy Methods 0.000 description 1
- 235000009508 confectionery Nutrition 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 229960000913 crospovidone Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 229940109275 cyclamate Drugs 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 229950002422 dexniguldipine Drugs 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 229940031569 diisopropyl sebacate Drugs 0.000 description 1
- XFKBBSZEQRFVSL-UHFFFAOYSA-N dipropan-2-yl decanedioate Chemical compound CC(C)OC(=O)CCCCCCCCC(=O)OC(C)C XFKBBSZEQRFVSL-UHFFFAOYSA-N 0.000 description 1
- SZXQTJUDPRGNJN-UHFFFAOYSA-N dipropylene glycol Chemical compound OCCCOCCCO SZXQTJUDPRGNJN-UHFFFAOYSA-N 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- 238000007922 dissolution test Methods 0.000 description 1
- OJLOUXPPKZRTHK-UHFFFAOYSA-N dodecan-1-ol;sodium Chemical compound [Na].CCCCCCCCCCCCO OJLOUXPPKZRTHK-UHFFFAOYSA-N 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 229950005476 elacridar Drugs 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- AHJUHHDDCJQACA-UHFFFAOYSA-N encequidar Chemical compound C1=CC=C2OC(C(=O)NC3=CC(OC)=C(OC)C=C3C=3N=NN(N=3)C3=CC=C(C=C3)CCN3CCC=4C=C(C(=CC=4C3)OC)OC)=CC(=O)C2=C1 AHJUHHDDCJQACA-UHFFFAOYSA-N 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 229960003649 eribulin Drugs 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 230000005713 exacerbation Effects 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 150000002191 fatty alcohols Chemical class 0.000 description 1
- 230000003176 fibrotic effect Effects 0.000 description 1
- 235000021323 fish oil Nutrition 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 229960003681 gluconolactone Drugs 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 239000001087 glyceryl triacetate Substances 0.000 description 1
- 235000013773 glyceryl triacetate Nutrition 0.000 description 1
- 229960002389 glycol salicylate Drugs 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000000665 guar gum Substances 0.000 description 1
- 235000010417 guar gum Nutrition 0.000 description 1
- 229960002154 guar gum Drugs 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000010224 hepatic metabolism Effects 0.000 description 1
- 229940100463 hexyl laurate Drugs 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 230000008105 immune reaction Effects 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 201000004933 in situ carcinoma Diseases 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229910001867 inorganic solvent Inorganic materials 0.000 description 1
- 239000003049 inorganic solvent Substances 0.000 description 1
- 210000002570 interstitial cell Anatomy 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 230000001788 irregular Effects 0.000 description 1
- 229920003049 isoprene rubber Polymers 0.000 description 1
- 229940111707 ixempra Drugs 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229940054157 lexapro Drugs 0.000 description 1
- 229940059904 light mineral oil Drugs 0.000 description 1
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 1
- 229940071260 lithium gluconate Drugs 0.000 description 1
- ZOTSUVWAEYHZRI-JJKGCWMISA-M lithium;(2r,3s,4r,5r)-2,3,4,5,6-pentahydroxyhexanoate Chemical compound [Li+].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O ZOTSUVWAEYHZRI-JJKGCWMISA-M 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 235000015250 liver sausages Nutrition 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 210000001365 lymphatic vessel Anatomy 0.000 description 1
- 239000001755 magnesium gluconate Substances 0.000 description 1
- 235000015778 magnesium gluconate Nutrition 0.000 description 1
- 229960003035 magnesium gluconate Drugs 0.000 description 1
- 229940057948 magnesium stearate Drugs 0.000 description 1
- IAKLPCRFBAZVRW-XRDLMGPZSA-L magnesium;(2r,3s,4r,5r)-2,3,4,5,6-pentahydroxyhexanoate;hydrate Chemical compound O.[Mg+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O IAKLPCRFBAZVRW-XRDLMGPZSA-L 0.000 description 1
- 201000003175 male breast cancer Diseases 0.000 description 1
- 208000010907 male breast carcinoma Diseases 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 235000010449 maltitol Nutrition 0.000 description 1
- 239000000845 maltitol Substances 0.000 description 1
- VQHSOMBJVWLPSR-WUJBLJFYSA-N maltitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-WUJBLJFYSA-N 0.000 description 1
- 229940035436 maltitol Drugs 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 208000023356 medullary thyroid gland carcinoma Diseases 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000037353 metabolic pathway Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229940117841 methacrylic acid copolymer Drugs 0.000 description 1
- IWVKTOUOPHGZRX-UHFFFAOYSA-N methyl 2-methylprop-2-enoate;2-methylprop-2-enoic acid Chemical compound CC(=C)C(O)=O.COC(=O)C(C)=C IWVKTOUOPHGZRX-UHFFFAOYSA-N 0.000 description 1
- IQSHMXAZFHORGY-UHFFFAOYSA-N methyl prop-2-enoate;2-methylprop-2-enoic acid Chemical compound COC(=O)C=C.CC(=C)C(O)=O IQSHMXAZFHORGY-UHFFFAOYSA-N 0.000 description 1
- 230000002906 microbiologic effect Effects 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- JXTPJDDICSTXJX-UHFFFAOYSA-N n-Triacontane Natural products CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC JXTPJDDICSTXJX-UHFFFAOYSA-N 0.000 description 1
- 238000009099 neoadjuvant therapy Methods 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000003956 nonsteroidal anti androgen Substances 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- KSCKTBJJRVPGKM-UHFFFAOYSA-N octan-1-olate;titanium(4+) Chemical compound [Ti+4].CCCCCCCC[O-].CCCCCCCC[O-].CCCCCCCC[O-].CCCCCCCC[O-] KSCKTBJJRVPGKM-UHFFFAOYSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- SBQLYHNEIUGQKH-UHFFFAOYSA-N omeprazole Chemical compound N1=C2[CH]C(OC)=CC=C2N=C1S(=O)CC1=NC=C(C)C(OC)=C1C SBQLYHNEIUGQKH-UHFFFAOYSA-N 0.000 description 1
- 229960000381 omeprazole Drugs 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 239000005022 packaging material Substances 0.000 description 1
- 229960005019 pantoprazole Drugs 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 229960000292 pectin Drugs 0.000 description 1
- 229940046159 pegylated liposomal doxorubicin Drugs 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920001083 polybutene Polymers 0.000 description 1
- 239000008389 polyethoxylated castor oil Substances 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 235000013809 polyvinylpolypyrrolidone Nutrition 0.000 description 1
- 229920000523 polyvinylpolypyrrolidone Polymers 0.000 description 1
- 235000007686 potassium Nutrition 0.000 description 1
- 235000010408 potassium alginate Nutrition 0.000 description 1
- 239000000737 potassium alginate Substances 0.000 description 1
- MZYRDLHIWXQJCQ-YZOKENDUSA-L potassium alginate Chemical compound [K+].[K+].O1[C@@H](C([O-])=O)[C@@H](OC)[C@H](O)[C@H](O)[C@@H]1O[C@@H]1[C@@H](C([O-])=O)O[C@@H](O)[C@@H](O)[C@H]1O MZYRDLHIWXQJCQ-YZOKENDUSA-L 0.000 description 1
- 239000004224 potassium gluconate Substances 0.000 description 1
- 235000013926 potassium gluconate Nutrition 0.000 description 1
- 229960003189 potassium gluconate Drugs 0.000 description 1
- 229940114930 potassium stearate Drugs 0.000 description 1
- ANBFRLKBEIFNQU-UHFFFAOYSA-M potassium;octadecanoate Chemical compound [K+].CCCCCCCCCCCCCCCCCC([O-])=O ANBFRLKBEIFNQU-UHFFFAOYSA-M 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 235000019814 powdered cellulose Nutrition 0.000 description 1
- 229920003124 powdered cellulose Polymers 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000004886 process control Methods 0.000 description 1
- 108090000468 progesterone receptors Proteins 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- FLALGSYYVIWTFQ-UHFFFAOYSA-K propan-2-olate;titanium(4+);trichloride Chemical compound [Cl-].[Cl-].[Cl-].CC(C)O[Ti+3] FLALGSYYVIWTFQ-UHFFFAOYSA-K 0.000 description 1
- 229940121649 protein inhibitor Drugs 0.000 description 1
- 239000012268 protein inhibitor Substances 0.000 description 1
- 238000010298 pulverizing process Methods 0.000 description 1
- 230000001698 pyrogenic effect Effects 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 229960001404 quinidine Drugs 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 229960003147 reserpine Drugs 0.000 description 1
- BJOIZNZVOZKDIG-MDEJGZGSSA-N reserpine Chemical compound O([C@H]1[C@@H]([C@H]([C@H]2C[C@@H]3C4=C([C]5C=CC(OC)=CC5=N4)CCN3C[C@H]2C1)C(=O)OC)OC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 BJOIZNZVOZKDIG-MDEJGZGSSA-N 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- MDMGHDFNKNZPAU-UHFFFAOYSA-N roserpine Natural products C1C2CN3CCC(C4=CC=C(OC)C=C4N4)=C4C3CC2C(OC(C)=O)C(OC)C1OC(=O)C1=CC(OC)=C(OC)C(OC)=C1 MDMGHDFNKNZPAU-UHFFFAOYSA-N 0.000 description 1
- 239000005060 rubber Substances 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229960001852 saquinavir Drugs 0.000 description 1
- QWAXKHKRTORLEM-UGJKXSETSA-N saquinavir Chemical compound C([C@@H]([C@H](O)CN1C[C@H]2CCCC[C@H]2C[C@H]1C(=O)NC(C)(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)C=1N=C2C=CC=CC2=CC=1)C1=CC=CC=C1 QWAXKHKRTORLEM-UGJKXSETSA-N 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 229940095743 selective estrogen receptor modulator Drugs 0.000 description 1
- 239000000333 selective estrogen receptor modulator Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 229920002545 silicone oil Polymers 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 235000011008 sodium phosphates Nutrition 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 229960002920 sorbitol Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 229940032094 squalane Drugs 0.000 description 1
- 229940031439 squalene Drugs 0.000 description 1
- TUHBEKDERLKLEC-UHFFFAOYSA-N squalene Natural products CC(=CCCC(=CCCC(=CCCC=C(/C)CCC=C(/C)CC=C(C)C)C)C)C TUHBEKDERLKLEC-UHFFFAOYSA-N 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- JJAHTWIKCUJRDK-UHFFFAOYSA-N succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate Chemical compound C1CC(CN2C(C=CC2=O)=O)CCC1C(=O)ON1C(=O)CCC1=O JJAHTWIKCUJRDK-UHFFFAOYSA-N 0.000 description 1
- 235000019408 sucralose Nutrition 0.000 description 1
- BAQAVOSOZGMPRM-QBMZZYIRSA-N sucralose Chemical compound O[C@@H]1[C@@H](O)[C@@H](Cl)[C@@H](CO)O[C@@H]1O[C@@]1(CCl)[C@@H](O)[C@H](O)[C@@H](CCl)O1 BAQAVOSOZGMPRM-QBMZZYIRSA-N 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 235000021092 sugar substitutes Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000002600 sunflower oil Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 238000009121 systemic therapy Methods 0.000 description 1
- 229960001967 tacrolimus Drugs 0.000 description 1
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 1
- 239000003784 tall oil Substances 0.000 description 1
- UWHCKJMYHZGTIT-UHFFFAOYSA-N tetraethylene glycol Chemical compound OCCOCCOCCOCCO UWHCKJMYHZGTIT-UHFFFAOYSA-N 0.000 description 1
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 1
- 229960005026 toremifene Drugs 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 229960002622 triacetin Drugs 0.000 description 1
- 239000001069 triethyl citrate Substances 0.000 description 1
- 235000013769 triethyl citrate Nutrition 0.000 description 1
- VMYFZRTXGLUXMZ-UHFFFAOYSA-N triethyl citrate Natural products CCOC(=O)C(O)(C(=O)OCC)C(=O)OCC VMYFZRTXGLUXMZ-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 201000007423 tubular adenocarcinoma Diseases 0.000 description 1
- 230000005748 tumor development Effects 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- 229950010938 valspodar Drugs 0.000 description 1
- 108010082372 valspodar Proteins 0.000 description 1
- 208000019553 vascular disease Diseases 0.000 description 1
- 230000001457 vasomotor Effects 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 239000000341 volatile oil Substances 0.000 description 1
- 239000000811 xylitol Substances 0.000 description 1
- 235000010447 xylitol Nutrition 0.000 description 1
- 229960002675 xylitol Drugs 0.000 description 1
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 1
- 229960000317 yohimbine Drugs 0.000 description 1
- GQDDNDAYOVNZPG-SCYLSFHTSA-N yohimbine Chemical compound C1=CC=C[C]2C(CCN3C[C@@H]4CC[C@H](O)[C@@H]([C@H]4C[C@H]33)C(=O)OC)=C3N=C21 GQDDNDAYOVNZPG-SCYLSFHTSA-N 0.000 description 1
- AADVZSXPNRLYLV-UHFFFAOYSA-N yohimbine carboxylic acid Natural products C1=CC=C2C(CCN3CC4CCC(C(C4CC33)C(O)=O)O)=C3NC2=C1 AADVZSXPNRLYLV-UHFFFAOYSA-N 0.000 description 1
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical compound [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 1
- 229940057977 zinc stearate Drugs 0.000 description 1
- 229940020965 zoloft Drugs 0.000 description 1
Images
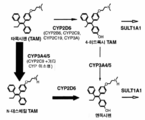












Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/138—Aryloxyalkylamines, e.g. propranolol, tamoxifen, phenoxybenzamine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/02—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C217/04—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C217/06—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having only one etherified hydroxy group and one amino group bound to the carbon skeleton, which is not further substituted
- C07C217/14—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having only one etherified hydroxy group and one amino group bound to the carbon skeleton, which is not further substituted the oxygen atom of the etherified hydroxy group being further bound to a carbon atom of a six-membered aromatic ring
- C07C217/18—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having only one etherified hydroxy group and one amino group bound to the carbon skeleton, which is not further substituted the oxygen atom of the etherified hydroxy group being further bound to a carbon atom of a six-membered aromatic ring the six-membered aromatic ring or condensed ring system containing that ring being further substituted
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/02—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions involving the formation of amino groups from compounds containing hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/08—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions not involving the formation of amino groups, hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/10—Separation; Purification; Stabilisation; Use of additives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/09—Geometrical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Abstract
본 개시는 (Z)-엔독시펜 또는 그의 염을 제조하는 산업적으로 확장 가능한 방법, 엔독시펜의 결정질 형태, 및 그들을 포함하는 조성물을 제공한다. 본 개시는 또한 호르몬 의존적 유방 및 호르몬 의존적 생식관 장애를 치료하는 방법을 제공한다.
Description
상호 참조
본 출원은 2017년 9월 11일에 출원된 미국 가출원 번호 62/556,799; 2017년 9월 11일에 출원된 미국 가출원 번호 62/556,884; 2018년 1월 31일에 출원된 미국 가출원 번호 62/624,787; 및 2018년 7월 3일에 출원된 미국 가출원 번호 62/693,885(각각 전부 참조로 본원에 포함됨)의 이익을 주장한다.
발명의 배경
유방암은 여성에게 단연코 가장 흔한 형태의 암이며, 인간에서 암 사망의 두 번째 주요 원인이다. 유방암 진단 및 치료의 발전에도 불구하고, 이 질환의 유병률은 1940년 이후 매년 약 1%의 비율로 꾸준히 증가하고 있다. 오늘날, 북미에 사는 여성이 그녀의 일생 동안 유방암에 걸릴 가능성은 8분의 1이다. 유방암 이외에 많은 여성들에게 영향을 끼치는 다른 유방 장애는 양성이지만 종종 전암성인 병변, 예컨대 유관(ductal) 증식증, 소엽 증식증, 비정형 유관 증식증, 및 비정형 소엽 증식증을 포함한다.
현재 유방암 치료를 위한 최선의 실시는 유방 촬영으로 유방암을 진단하고 이어서 환자를 수술, 방사선 요법 및 화학요법으로 치료하는 것이다. 현재 널리 사용되는 유방촬영은 유방암의 검출을 개선하였다. 유방암은 여성보다 남성에서 약 100배 덜 일반적이지만 남성이 평생 유방암에 걸릴 위험은 1,000명 중 1명이다. 침습성 유방암의 약 2,470개의 새로운 사례가 남성에서 진단될 것이며 약 460명의 남성이 유방암으로 사망할 것이다. 미국 암학회(American Cancer Society)는 2017년에 유방암의 255,180개의 새로운 사례가 남녀 모두에서 진단될 것이며 41,070명의 대상체가 유방암으로 사망할 것으로 추정한다. 그럼에도 불구하고 유방암으로 인한 사망률은 여성 10만명당 약 21명 및 남성 10만명당 0.4명으로 상대적으로 변동이 없다. 너무 자주, 유방암은 치료 옵션 및 생존율이 심각하게 제한되는 너무 진행된 단계에서 발견된다.
유방암은 유방 세포의 임의의 악성 종양을 포함한다. 유방암에는 여러 유형이 있다. 예시적인 유방암은 관 제자리암종, 소엽 제자리암종, 침습적 (또는 침윤) 관 암종, 침습적 (또는 침윤) 소엽 암종, 염증성 유방암, 삼중 음성 유방암, ER+ 유방암, HER2+ 유방암, 선양 낭성 (또는 샘낭성) 암종, 낮은 등급 선편평상피 암종, 수질성 암종, 점액 (또는 콜로이드) 암종, 유두 암종, 관상(tubular) 암종, 화생 암종, 및 미세유두 암종을 포함하지만 이에 제한되지 않는다. 단일 유방 종양은 이러한 유형의 조합이거나 침습적 암과 제자리 암의 혼합일 수 있다.
타목시펜은 내분비 반응성 유방암, 즉 호르몬 의존성 또는 호르몬 민감성 유방암을 갖는 여성의 치료에 사용되는 선택적인 에스트로겐 수용체 조정제이다. 타목시펜의 경구 전달을 통한 보조 요법은 혈관운동 증상, 예컨대 열감 및 생식관(부인과) 암과 같은 심각한 부작용을 갖는 것으로 공지되어 있다. 환자 순응도는 타목시펜 요법에서 여전히 문제가 된다. 또한, 보조 타목시펜 요법을 받는 개체의 대다수는 약물에 반응하지 않으며 환자의 30-50%가 이후에 그들의 질병으로 사망한다.
몇몇 시토크롬 P450(CYP) 돌연변이가 타목시펜의 그의 활성 대사산물인 엔독시펜으로의 전환을 감소시키고 타목시펜 효능을 감소시키며 약물에 대한 내성을 증가시키는 것으로 제안되었다(Dickschen et al. Front Pharmacol. 2012; 3: 92). PMCID: PMC3357105). 지금까지, CYP2D6의 140개 초과의 대립유전자 변이체가 기술되었으며, 이들 중 상당 부분은 암호화된 효소의 활성이 감소되거나 부재하는 것과 연관된다. 포함된 대립유전자의 조합에 기초하여, 각각의 개별 대상체는 4가지의 표현형 그룹 중 하나로 분류될 수 있다: 혈청 엔독시펜 수준 차이를 반영하여, 30 nM 미만의 혈장 엔도시펜 수준을 갖는 저조한 대사자(PM), 중간 대사자(IM), 광범위 대사자(EM) 및 초고속 대사자(UM). 그러나, CYP 유전형의 변화는 일부 대상체에서 관찰되는 타목시펜 내성 및 감소된 엔독시펜 수준을 충분히 설명하지 못한다.
따라서, 타목시펜의 몇몇 대안이 유방암 치료를 위해 개발되고 있으며, 이는 저용량 타목시펜(Lazzeroni M et al. Breast Cancer Res. 2012 Oct 29;14(5):214) 및 타목시펜의 활성 대사산물인 아피목시펜(미국 특허 번호 7,485,623; 7,507,769; 7,704,516; 7,786,172; 7,968,532; 및 8,048,927; Mansel R et al. Breast Cancer Res Treat. 2007 Dec;106(3):389-97. PubMed PMID: 17351746; Rouanet P et al. J ClinOncol. 2005 May 1;23(13):2980-7. PubMed PMID: 15860853), 엔독시펜(US9333190; 미국 특허 번호 9,220,680; 9,090,640; 및 9,200,045; 미국 공개 번호 2009/0291134 및 US20100112041), 및 그들의 유도체(미국 특허 번호 8,063,249; 미국 공개 번호 2015/0080339 및 2014/0193334)를 포함한다. (Z)-엔독시펜은 타목시펜의 임상 효능을 담당하는 주요 활성 대사산물인 것으로 널리 인정되고 있다.
엔독시펜의 히드로클로라이드 및 시트레이트 염(예컨대, 문헌(Fauq et al., Bioorganic & Medicinal Chemistry Letters. 20 (2010) 3036 - 3038; Stearns et al., J. Natl. Cancer Inst. Vol 95, No. 23, 2003; US Publication Nos. 2009/0291134 and 2010/0112041; Clinical Trials Gov. Identifier Nos. NCT01273168 and NCT02311933; Goetz et al., 2015, San Antonio Cancer Symposium; Ahmad et al., Clinical Pharmacology & Therapeutics. 88(6) 814-817, 2010; and J Clin. Oncol. 30, 2012 (suppl; abstr 3089); Ahmad et al. Breast Cancer Research and Treatment 2010, 122, 579-58) 참조)은 당해 분야에 공지되어 있고 현재 전이성 암에 대한 평가를 받고 있지만, 호르몬 의존적 유방 및 생식관(부인과) 장애의 치료 및/또는 예방을 위한 새로운 조성물 및 방법에 대한 의학적 요구가 여전히 충족되지 않고 있다.
본 개시는 호르몬 의존적 유방 및 생식관(부인과) 장애의 치료 및/또는 예방을 위한 조성물 및 방법을 제공함으로써 이러한 요구를 해결한다. 특정 측면에서, 본 개시는 Z-엔독시펜 또는 그의 염을 제조하는 새로운 산업적으로 확장 가능한 방법, 엔독시펜의 결정질 형태, 및 그들을 포함하는 조성물을 제공한다. 특정 측면에서, 본 개시는 다른 결정질 또는 무정형 형태에 비해 개선된 생체이용률 및 안정성을 포함하는 이점을 제공할 수 있는 엔독시펜의 새로운 결정질 형태를 제공한다.
특정 측면에서, 본 개시는 화학식 (III)의 화합물의 결정질 형태를 포함하는 조성물을 제공한다:

화학식 (III).
일부 실시양태에서, 조성물 중의 화학식 (III)의 화합물의 적어도 적어도 90중량%는 (Z)-이성질체이다. 일부 실시양태에서, 결정질 형태는 화학식 (III)의 화합물의 형태 I이다. 결정질 형태 I은 16.8 ± 0.3°, 17.1 ± 0.3° 및 21.8 ± 0.3°2 쎄타에서 주요 피크를 포함하고, 임의적으로 16.0 ± 0.3°, 18.8 ± 0.3° 및 26.5 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 추가로 포함하는 x-선 분말 회절 패턴으로 특징지어질 수 있다. 일부 실시양태에서, x-선 분말 회절 패턴은 12.3 ± 0.3°, 28.0 ± 0.3° 및 29.0 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 추가로 포함한다. x-선 분말 회절 패턴은 12.3 ± 0.3°, 16.0 ± 0.3°, 18.8 ± 0.3°, 26.5 ± 0.3°, 28.0 ± 0.3° 및 29.0 ± 0.3°2 쎄타에서 피크를 추가로 포함할 수 있다. 일부 실시양태에서, 결정질 형태 I은 실질적으로 도 9 또는 도 10에 제시된 바와 같은 x-선 분말 회절 패턴으로 특징지어진다. 조성물 중의 화학식 (III)의 화합물의 90중량%, 95중량% 또는 99중량% 초과는 결정질 형태 I일 수 있다. 일부 실시양태에서, 조성물은 0.01 mg 내지 200 mg의 결정질 형태 I, 예컨대 약 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 결정질 형태 I을 포함할 수 있다.
일부 실시양태에서, 조성물은 화학식 (III)의 화합물의 (E)-이성질체 및 (Z)-이성질체를 0.9 내지 1.3, 예컨대 약 1.1의 E/Z 비율로 포함한다. 일부 실시양태에서, 결정질 형태는 화학식 (III)의 화합물의 형태 II이다. 결정질 형태 II는 7.0 ± 0.3°, 11.9 ± 0.3°, 14.0 ± 0.3° 및 18.4 ± 0.3°2 쎄타에서 주요 피크를 포함하고, 임의적으로 22.0 ± 0.3°2 쎄타에서 피크를 추가로 포함하는 x-선 분말 회절 패턴으로 특징지어질 수 있다. 일부 실시양태에서, x-선 분말 회절 패턴은 6.6 ± 0.3°, 13.3 ± 0.3° 및 20.0 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 추가로 포함한다. x-선 분말 회절 패턴은 6.6 ± 0.3°, 13.3 ± 0.3°, 20.0 ± 0.3° 및 22.0 ± 0.3°2 쎄타에서 피크를 추가로 포함할 수 있다. 일부 실시양태에서, 결정질 형태 II는 실질적으로 도 11 또는 도 12에 제시된 바와 같은 x-선 분말 회절 패턴으로 특징지어진다. 조성물 중의 화학식 (III)의 화합물의 90중량%, 95중량% 또는 99중량% 초과는 결정질 형태 II이다. 일부 실시양태에서, 조성물은 0.01 mg 내지 200 mg의 결정질 형태 II, 예컨대 약 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 결정질 형태 II를 포함한다.
일부 실시양태에서, 조성물은 화학식 (III)의 화합물의 (E)-이성질체 및 (Z)-이성질체를 0.9 내지 1.3, 예컨대 약 1.1의 E/Z 비율로 포함한다. 일부 실시양태에서, 결정질 형태는 화학식 (III)의 화합물의 형태 III이다. 결정질 형태 III은 11.9± 0.3°, 13.9 ± 0.3°, 17.1 ± 0.3° 및 17.7 ± 0.3°2 쎄타에서 주요 피크를 포함하고, 임의적으로 25.3 ± 0.3°2 쎄타에서 피크를 추가로 포함하는 x-선 분말 회절 패턴으로 특징지어질 수 있다. 일부 실시양태에서, x-선 분말 회절 패턴은 18.2 ± 0.3°, 22.5 ± 0.3° 및 26.8 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 추가로 포함한다. x-선 분말 회절 패턴은 18.2 ± 0.3°, 22.5 ± 0.3°, 25.3 ± 0.3° 및 26.8 ± 0.3°2 쎄타에서 피크를 추가로 포함할 수 있다. 일부 실시양태에서, 결정질 형태 III은 실질적으로 도 13에 제시된 바와 같은 x-선 분말 회절 패턴으로 특징지어진다. 조성물 중의 화학식 (III)의 화합물의 90중량%, 95중량% 또는 99중량% 초과는 결정질 형태 III일 수 있다. 일부 실시양태에서, 조성물은 0.01 mg 내지 200 mg의 결정질 형태 III, 예컨대 약 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 결정질 형태 III을 포함한다.
특정 측면에서, 본 개시는 약제학적으로 허용되는 담체 또는 희석제 및 화학식 (III)의 화합물의 결정질 형태를 포함하는 조성물을 포함하는 약제학적 조성물을 제공한다. 본 개시의 조성물은 경구, 비경구, 국소, 또는 관내 전달을 위해 제형화될 수 있다. 일부 실시양태에서, 조성물은 경구 전달을 위해 정제, 캐플릿, 캡슐, 또는 환제로 제형화된다. 일부 실시양태에서, 조성물로 치료되는 대상체에서 엔독시펜의 평균 반감기는 30시간 내지 60시간이다. 화학식 (III)의 화합물의 결정질 형태를 포함하는 조성물은 경구 전달을 위해 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿 또는 지연 방출 캡슐로 제형화될 수 있다. 일부 실시양태에서, 조성물은 대상체에서 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다의 치료 또는 예방을 위해 대상체에게 투여된다.
특정 측면에서, 본 개시는 단위 용량당 1 mg 내지 200 mg의 화학식 (III)의 화합물의 결정질 형태를 포함하는 조성물을 포함하고 이를 필요로 하는 대상체에게 투여하기 위한 경구 조성물로서, 여기서 경구 조성물의 일일 투여로 대상체에서
7 내지 21일 이내에 엔독시펜의 항정 상태 혈장 수준;
25 nM 내지 300 nM 범위의 엔독시펜의 항정 상태 혈장 수준;
30 nM 초과의 엔독시펜의 항정 상태 혈장 수준;
투여 후 2 내지 10시간 이내에 엔독시펜의 최대 혈장 수준; 또는
그의 임의의 조합
을 달성하는 것인 경구 조성물을 제공한다.
일부 실시양태에서, 화학식 (III)의 화합물의 결정질 형태를 포함하는 조성물로 치료되는 대상체에서 엔독시펜의 평균 반감기는 40시간 내지 55시간이다. 일부 실시양태에서, 조성물은 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿 또는 지연 방출 캡슐로 제형화된다. 일부 실시양태에서, 조성물 중의 엔독시펜의 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 또는 적어도 90%는 장에서 방출된다. 일부 실시양태에서, 화학식 (III)의 화합물의 결정질 형태를 포함하는 조성물은 200 hr*ng/mL 내지 10000 hr*ng/mL, 300 hr*ng/mL 내지 8000 hr*ng/mL, 400 hr*ng/mL 내지 6000 hr*ng/mL 또는 700 hr*ng/mL 내지 6000 hr*ng/mL의 엔독시펜의 무한대 시간으로 외삽된 곡선 아래 평균 면적(AUC0-inf)을 나타낸다.
특정 측면에서, 본 개시는 화학식 (III)의 화합물의 결정질 형태를 포함하는 조성물을 대상체에게 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법을 제공한다. 일부 실시양태에서, 대상체는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있다. 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애는 양성 유방 장애, 증식증, 비정형(atypia), 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군(McCune-Albright Syndrome), 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 또는 외음부암일 수 있다. 일부 실시양태에서, 대상체는 전립선암을 갖고 여성형 유방증을 갖거나 가질 위험이 있다. 대상체는 타목시펜 불응성 또는 타목시펜 내성 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 가질 수 있다. 일부 실시양태에서, 대상체는 시탈로프람, 에스시탈로프람, 플루옥세틴, 파록세틴, 세르트랄린, 및 빌라조돈으로 이루어진 군으로부터 선택되는 SSRI 약물로 치료되거나 치료될 것이다.
본 방법 중 임의의 것을 실시함에 있어서, 조성물은 0.01 mg 내지 200 mg의 (Z)-엔독시펜을 포함할 수 있다. 일부 실시양태에서, 대상체에게 약 1 mg, 2 mg, 4 mg, 또는 20 mg의 (Z)-엔독시펜이 매일 투여된다. 일부 실시양태에서, 대상체에서 엔독시펜의 항정 상태 혈장 수준은 30 nM 초과이다. 엔독시펜의 항정 상태 혈장 수준은 조성물의 제1 투여의 7 내지 21일 이내에 달성될 수 있다. 일부 실시양태에서, 엔독시펜의 최대 혈장 수준까지의 시간은 조성물 투여 후 2시간 내지 10시간 또는 4시간 내지 8시간의 범위이다.
특정 측면에서, 본 개시는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 대상체를 치료하는 방법으로서, 방법은 화학식 (III)의 화합물의 결정질 형태를 포함하는 조성물을 투여하는 단계를 포함하고, 여기서 조성물의 투여는
대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기;
투여 후 4시간 내지 8시간 범위의 엔독시펜의 최대 혈장 수준까지의 시간; 및
30 nM 초과의 엔독시펜의 항정 상태 혈장 수준
을 달성하는 것인 방법을 제공한다.
일부 실시양태에서, 호르몬 의존적 유방 장애 및 호르몬 의존적 생식관 장애는 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 및 외음부암로 이루어진 군으로부터 선택된다.
본 방법 중 임의의 것을 실시함에 있어서, 조성물은 0.01 mg 내지 200 mg의 (Z)-엔독시펜을 포함할 수 있다. 일부 실시양태에서, 대상체에게 약 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 (Z)-엔독시펜이 투여된다. 엔독시펜의 무한대 시간으로 외삽된 곡선 아래 평균 면적(AUC0-inf)은 200 hr*ng/mL 내지 10000 hr*ng/mL, 300 hr*ng/mL 내지 8000 hr*ng/mL, 400 hr*ng/mL 내지 6000 hr*ng/mL 또는 700 hr*ng/mL 내지 6000 hr*ng/mL일 수 있다. 일부 실시양태에서, 조성물은 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿 또는 지연 방출 캡슐로 제형화된다. 일부 실시양태에서, 조성물 중의 엔독시펜의 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 또는 적어도 90%가 장에서 방출된다. 조성물은 1일 1회, 1일 2회, 1일 3회, 1일 4회, 격일, 1주 2회, 매주, 격주, 1개월 2회, 매월, 분기별, 6개월 1회, 또는 매년 투여될 수 있다.
일 측면에서, 본 개시는 (a) 하기로 표시되는 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 제1 용매로부터 분별 결정화하여 제1 결정질 고체 및 제1 결정질 모액을 형성하는 단계로서, 여기서 제1 모액은 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물의 E/Z 비율과 비교하여 적어도 50% 더 높은 (Z) 엔독시펜의 E/Z 비율을 갖는 것인 단계:

(b) 제1 모액을 농축시키거나 제1 모액으로부터의 제1 용매를 제2 용매와 1회 이상 교환함으로써 제1 모액을 제2 용매로부터 재결정화하여 제2 결정질 고체 및 제2 모액을 형성하는 단계로서, 여기서 제2 결정질 고체는 ≥ 90% (Z)-엔독시펜인 단계; 및 (c) 임의적으로, 제2 결정질 고체를 제3 용매 또는 크로마토그래피 처리로부터 1회 이상 재결정화하여 제3 결정질 고체를 형성하는 단계를 포함하는, (Z)-엔독시펜 또는 그의 염을 제조하는 산업적으로 확장 가능한 방법을 제공한다. 일부 실시양태에서, 제1 용매, 제2 용매, 및 제3 용매 중 임의의 하나 이상은 40℃ 내지 80℃ 범위의 온도로 예열된다.
일부 측면에서, 본 개시는 (a) 하기로 표시되는 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 제1 용매로부터 분별 결정화하여 제1 결정질 고체 및 제1 모액을 형성하는 단계로서, 여기서 제1 모액은 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물의 E/Z 비율과 비교하여 적어도 50% 더 높은 (Z) 엔독시펜의 E/Z 비율을 갖는 것인 단계:

(b) 제1 모액을 농축시키거나 제1 모액으로부터의 제1 용매를 제2 용매와 1회 이상 교환함으로써 제1 모액을 제2 용매로부터 재결정화하여 제2 결정질 고체 및 제2 모액을 형성하는 단계로서, 여기서 제2 결정질 고체는 ≥ 90% (Z)-엔독시펜인 단계; 및 (c) 제2 결정질 고체를 제3 용매 또는 크로마토그래피 처리로부터 1회 이상 재결정화하여 제3 결정질 고체를 형성하는 단계로서, 여기서 제3 결정질 고체는 화학식 (III)의 화합물의 결정질 형태 I인 단계를 포함하는, 화학식 (III)의 화합물의 결정질 형태 I을 제조하는 산업적으로 확장 가능한 방법을 제공한다.
특정 측면에서, 본 개시는 본원에 기재된 방법에 따라 생성되는 화학식 (III)의 화합물의 결정질 형태를 제공한다. 일부 실시양태에서, 결정질 형태는 화학식 (III)의 화합물의 형태 I이다. 일부 실시양태에서, 결정질 형태는 화학식 (III)의 화합물의 형태 II이다. 일부 실시양태에서, 결정질 형태는 화학식 (III)의 화합물의 형태 III이다.
또 다른 측면에서, 본 개시는 단계 (a)에서 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물이 6N HCl로 전처리되고 8N NaOH로 중화되는 것을 제공한다.
또 다른 측면에서, 산업적으로 확장 가능한 방법은, 불활성 유기 용매 중의 티타늄 염 및 환원제를 통해 맥머리 반응(McMurry reaction)에 의해 매개되는 프로피오페논에 하기로 표시되는 구조를 갖는 화학식 (II)의 화합물인 (4-히드록시페닐)(4-(2-(메틸아미노)에톡시)페닐) 메탄온을 커플링시켜 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성함으로써 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 제조하는 단계를 추가로 포함한다:

또 다른 측면에서, 산업적으로 확장 가능한 방법은 하기 구조를 갖는 화학식 (I)의 화합물인 [4-[2-(디메틸아미노)에톡시]페닐](4-히드록시페닐)메탄온을 불활성 유기 용매 중의 탈메틸화제 및 양성자 억셉터로 탈메틸화시켜 화학식 (II)의 화합물을 형성함으로써 화학식 (II)의 화합물을 제조하는 단계를 추가로 포함한다:

본 개시는 또한 (a) 하기로 표시되는 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 에틸 아세테이트로부터 분별 결정화하여 제1 결정질 고체 및 제1 결정질 모액을 형성하는 단계로서, 여기서 제1 모액은 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물의 E/Z 비율과 비교하여 적어도 30% 더 높은 (Z) 엔독시펜의 E/Z 비율을 갖는 것인 단계:

(b) 제1 모액을 농축시키거나 제1 모액으로부터의 에틸 아세테이트를 IPA 또는 IPA/PPW와 1회 이상 교환함으로써 제1 모액을 IPA 또는 IPA/PPW(1:1 v/v)로부터 재결정화하여 제2 결정질 고체 및 제2 모액을 형성하는 단계로서, 여기서 제2 결정질 고체는 ≥ 90% (Z)-엔독시펜인 단계; 및 (c) 임의적으로, 제2 결정질 고체를 에탄올 또는 크로마토그래피 처리로부터 1회 이상 재결정화하여 제3 결정질 고체를 형성하는 단계를 포함하는, (Z)-엔독시펜 및 그의 염을 제조하는 산업적으로 확장 가능한 방법을 제공한다.
일부 실시양태에서, 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물은 화학식 (II)의 화합물인-히드록시페닐)(4-(2-(메틸아미노)에톡시)페닐) 메탄온을 맥머리 반응에 의해 매개되는 프로피오페논에 커플링시킴으로써 제조되며, (a) 화학식 (II)의 화합물을 불활성 유기 용매(1:1 내지 1: 20 wt/wt) 중의 프로피오페논(1:0.01 내지 1.5 wt/wt)과 반응시키는 단계; (b) 불활성 유기 용매(1:1 내지 1: 20 wt/wt) 중의 티타늄 염(1:0.1 내지 1:12 wt/wt) 및 환원제 (1:0.01 내지 1:10 wt/wt)을 제조하는 단계; 및 (c) 단계 (a)의 화학식 (II)의 화합물을 단계 (b)의 불활성 유기 용매 중의 티타늄 염 및 환원제와 반응시켜 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성하는 단계를 포함하고, 여기서 wt/wt는 화학식 (II)의 화합물에 대한 것이다.
다른 실시양태에서, 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물은 화학식 (II)의 화합물인 (4-히드록시페닐)(4-(2-(메틸아미노)에톡시)페닐) 메탄온을 맥머리 반응에 의해 매개되는 프로피오페논에 커플링시킴으로써 제조되며, (a) 화학식 (II)의 화합물을 THF(1:1 내지 1:20 wt/wt) 중의 프로피오페논(1:0.01 내지 1:5 wt/wt)과 반응시키는 단계; (b) THF(1:1 내지 1:20 wt/wt) 중의 TiCl4(0.1 내지 4 wt/wt) 및 Zn(0.01 내지 1:2 wt./wt)을 제조하는 단계; 및 (c) 단계 (a)의 화학식 (II)의 화합물을 단계 (b)의 THF 중의 TiCl4 및 Zn과 반응시켜 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성하는 단계를 포함하고, 여기서 wt/wt는 화학식 (II)의 화합물에 대한 것이다.
일부 실시양태에서, 화학식 (II)의 화합물은 화학식 (I)의 화합물을 불활성 유기 용매(1:1 내지 1: 20 wt/wt) 중의 탈메틸화제 (1:0.5 내지 1:10 wt/wt) 및 양성자 억셉터(1:0.5 내지 1:10 wt/wt)로 탈메틸화시켜 화학식 (II)의 화합물을 형성함으로써 생성되며, 여기서 wt/wt는 화학식 (I)의 화합물에 대한 것이다.
일부 실시양태에서, 화학식 (II)의 화합물은 화학식 (I)의 화합물을 THF 중의 1-클로로에틸 클로로포르메이트 및 DIPEA로 탈메틸화시켜 화학식 (II)의 화합물을 형성함으로써 제조되며, (a) 화학식 (I)의 화합물을 THF(1:20 wt/wt) 중의 DIPEA(1:0.5 내지 1:10 wt/wt)와 반응시키는 단계; (b) 1-클로로에틸 클로로포르메이트(1:0.5 내지 1:10 wt./wt)를 첨가하는 단계; (c) 메탄올(1:1 내지 1:10 wt./wt)로 1회 이상 증류시키는 단계; (d) 메탄올(1:1 내지 1:5 wt/wt)/HCl(1:1 내지 1:10 wt/wt)과 반응시키는 단계; 및 (e) NaOH(1:1 내지 1:10 wt/wt)로 중화시키는 단계를 포함하고, 여기서 wt/wt는 화학식 (I)의 화합물에 대한 것이다.
또 다른 측면에서, 본 개시는 (Z)-엔독시펜을 D-글루코네이트 또는 L-글루코네이트와 반응시켜 (Z)-엔독시펜 L-글루코네이트 또는 (Z)-엔독시펜 D-글루코네이트를 형성함으로써 (Z)-엔독시펜 글루코네이트 염을 제조하는 방법을 제공한다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜, (E)-엔독시펜, 화학식 (III)의 화합물, 화학식 (II)의 화합물, 및 그의 염이 안정하다는 것을 제공한다. 특정 측면에서, (Z)-엔독시펜 유리 염기는 적어도 9개월 동안 주위 온도에서 안정하다.
특정 측면에서, 본 개시는 (a) 99:1 내지 60:40 범위의 E/Z-비율을 갖는 (E)-엔독시펜과 (Z)-엔독시펜의 출발 혼합물을 에틸 아세테이트(1:1 내지 1:20 wt/wt) 중의 6N HCL(1:1 내지 1:5 wt/wt)에 반응시키는 단계; (b) 8N NaOH(1:1 내지 1:20 wt/wt)로 중화시키는 단계; (c) 에틸 아세테이트로 1회 이상 세척하는 단계; (d) 에틸 아세테이트와 n-헵탄의 혼합물로 1회 이상 세척하는 단계; 및 (e) 화학식 (III)의 화합물의 결정질 형태 II 또는 III을 회수하는 단계를 포함하고; 여기서 wt/wt는 (E)-엔독시펜과 (Z)-엔독시펜의 출발 혼합물에 대한 것인, 화학식 (III)의 화합물의 결정질 형태 II 또는 III을 제조하는 산업적으로 확장 가능한 방법을 제공한다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 유리 염기 또는 그의 염을 포함하는 조성물을 제공한다. 조성물은 경구, 비경구, 국소, 및 관내 전달을 위해 제형화된다. 조성물은 경구 전달을 위해 정제, 캐플릿, 캡슐, 또는 환제로 제형화된다. 일부 실시양태에서, 조성물은 경구 전달 정제를 위해 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿, 및 지연 방출 캡슐로 제형화된다.
특정 측면에서, 본 개시는 특정한 약동학 프로파일을 갖는 조성물을 제공한다. 일 측면에서, 본 개시는 조성물 중의 엔독시펜의 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 적어도 90%가 장에서 방출되는 것을 제공한다. 또 다른 측면에서, 본 개시는 조성물 중의 엔독시펜이 대상체에서 투여 후 30시간 내지 60시간 범위의 평균 반감기를 갖는 것을 제공한다. 일부 실시양태에서, 대상체에서 엔독시펜의 평균 반감기는 투여 후 40시간 내지 55시간의 범위이다. 또 다른 측면에서, 본 개시는 200 hr*ng/mL 내지 10000 hr*ng/mL, 300 hr*ng/mL 내지 8000 hr*ng/mL, 400 hr*ng/mL 내지 6000 hr*ng/mL 또는 700 hr*ng/mL 내지 6000 hr*ng/mL의 무한대 시간으로 외삽된 곡선 아래 평균 면적(AUC0-inf)을 제공한다.
또 다른 측면에서, 본 개시는 단위 용량당 1 mg 내지 200 mg의 (Z)-엔독시펜 유리 염기 또는 그의 염을 포함하는 이를 필요로 하는 대상체에게 투여하기 위한 경구 조성물을 제공하며, 여기서 경구 조성물의 일일 투여는 대상체에서 (i) 7 내지 21일 이내에 엔독시펜의 항정 상태 혈장 수준; 또는 (ii) 25 nM 내지 300 nM 범위의 엔독시펜의 항정 상태 혈장 수준; 또는 (iii) 30 nM 초과의 엔독시펜의 항정 상태 혈장 수준; 또는 (iv) 투여 후 2 내지 10시간 이내에 엔독시펜의 최대 혈장 수준; 또는 (v) 그의 임의의 조합을 달성한다. 본 개시는 또한 조성물이 대상체에서 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 치료 또는 예방하기 위해 이를 필요로 하는 대상체에게 투여되는 것을 제공한다.
특정 측면에서, 본 개시는 본원에 개시된 경구 조성물을 대상체에게 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법을 제공한다. 일부 실시양태에서, 대상체는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있다. 다양한 실시양태에서, 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애는 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 또는 외음부암이다.
일부 실시양태에서, 대상체는 전립선암을 갖고 대상체는 여성형 유방증을 갖거나 가질 위험이 있다.
특정 측면에서, 본 개시는 이러한 치료 방법으로부터 이익을 얻을 수 있는 특정한 환자 집단을 제공한다. 일부 실시양태에서, 환자 집단은 타목시펜 불응성 또는 타목시펜 내성 집단이다. 특정 실시양태에서, 대상체는 타목시펜 불응성 또는 타목시펜 내성 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖는다. 다른 실시양태에서, 환자 집단은 시탈로프람, 에스시탈로프람, 플루옥세틴, 파록세틴, 세르트랄린, 및 빌라조돈으로 이루어진 군으로부터 선택되는 SSRI 약물로 치료되거나 이로 치료될 대상체를 포함한다.
일 측면에서, 본 개시는 이를 필요로 하는 대상체에게 투여될 특정 용량의 (Z)-엔독시펜을 제공한다. 다양한 실시양태에서, 대상체에게 0.01 mg 내지 200 mg의 (Z)-엔독시펜이 투여된다. 특정 실시양태에서, 대상체에게 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 (Z)-엔독시펜이 매일 투여된다.
또 다른 측면에서, 본 개시는 대상체에서 엔독시펜의 항정 상태 혈장 수준이 조성물 투여 시 30 nM 초과인 대상체의 치료 방법을 제공한다. 일부 실시양태에서, 엔독시펜의 이러한 항정 상태 혈장 수준은 조성물의 제1 투여의 7 내지 21일 이내에 달성된다. 일부 실시양태에서, 엔독시펜의 최대 혈장 수준까지의 시간은 조성물 투여 후 2시간 내지 10시간 또는 4시간 내지 8시간의 범위이다.
또 다른 측면에서, 본 개시는 (Z)-엔독시펜 또는 그의 염을 포함하는 경구 조성물을 투여하는 단계를 포함하는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 대상체를 치료하는 방법을 제공하며, 여기서 조성물의 투여는 (i) 대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기; (ii) 투여 후 4시간 내지 8시간 범위의 엔독시펜의 최대 혈장 수준까지의 시간; 및 (iii) 30 nM 초과의 엔독시펜의 항정 상태 혈장 수준을 달성한다. 특정 실시양태에서, 대상체에게 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 (Z)-엔독시펜이 투여된다. 다른 실시양태에서, 무한대 시간으로 외삽된 곡선 아래 평균 면적(AUC0-inf)은 200 hr*ng/mL 내지 10000 hr*ng/mL, 300 hr*ng/mL 내지 8000 hr*ng/mL, 400 hr*ng/mL 내지 6000 hr*ng/mL 또는 700 hr*ng/mL 내지 6000 hr*ng/mL이다. 일부 실시양태에서, 조성물은 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿 또는 지연 방출 캡슐로 제형화된다. 일부 실시양태에서, 엔독시펜의 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 적어도 90%가 장에서 방출된다. 일부 실시양태에서, 조성물은 1일 1회, 1일 2회, 1일 3회, 1일 4회, 격일, 1주 2회, 매주, 격주, 1개월 2회, 매월, 분기별, 6개월 1회, 또는 매년 투여된다. 일부 실시양태에서, 호르몬 의존적 유방 장애 및 호르몬 의존적 생식관 장애는 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 및 외음부암로 이루어진 군으로부터 선택된다.
참조에 의한 삽입
본 명세서에서 언급된 모든 공개문, 특허 및 특허 출원은 각각의 개별 공개문, 특허 또는 특허 출원이 구체적이고 개별적으로 참조로 포함되는 것으로 지시되는 것과 동일한 정도로 본원에 참조로 포함된다.
본 발명의 새로운 특징은 첨부된 청구범위에 구체적으로 제시되어 있다. 본 발명의 특징 및 이점에 대한 더 나은 이해는 본 발명의 원리가 이용되는 예시적인 실시양태를 제시하는 하기의 상세한 설명 및 첨부 도면을 참조함으로써 달성될 것이다.
도 1은 타목시펜의 엔독시펜 및 다른 대사산물로의 대사 경로를 도시하는 도해이다. 4-히드록시타목시펜의 혈장 농도는 N-데스메틸 타목시펜에 비해 낮으며, 이는 모 약물의 대사에 대한 1차 경로가 시토크롬 P450 3A4(CYP3A4)에 의한 N-데스메틸 타목시펜으로의 N-탈메틸화에 이어 시토크롬 P450 2D6(CYP2D6)에 의해 엔독시펜으로 히드록실화된다는 것을 나타낸다.
도 2는 본원에 개시된 방법을 이용하여 제조되는 (Z)-엔독시펜 유리 염기의 대표적인 적외선 스펙트럼이다.
도 3은 위약 또는 1 mg, 2 mg 또는 4 mg 엔독시펜 캡슐로 처리한 후 엔독시펜의 최대 혈청 수준(nM)까지의 시간을 도시한다.
도 4는 제1일 및 제21일에 위약 또는 1 mg, 2 mg 또는 4 mg 엔독시펜 캡슐으로 처리한 후 엔독시펜의 최대 혈청 수준까지의 시간을 도시한다. 투여 후 시간(시간)은 X-축에 플롯팅되고 평균 혈청 농도(ng/mL)는 Y-축에 플로팅된다.
도 5는 제0일(제0일은 X-축 상의 제8일을 지칭함)에서 제21일까지 위약 또는 1 mg, 2 mg 또는 4 mg 엔독시펜 캡슐로 처리한 후 엔독시펜의 항정 상태 혈청 수준을 달성하는 시간을 도시한다. X-축은 일(제8일 내지 제21일)의 시간이고 Y-축은 ng/mL의 평균 혈청 농도이다. 혈액을 제0일에서 임상 시험 기간 끝까지 각각의 일에 채취하였다. 대상체에게 제1일에 투여하고 혈장 엔독시펜 수준을 표 17에 나타낸 바와 같이 측정하였다. 이어서, 대상체에게 제8일에 시작하여(도 5에서 제8일로 표시됨) 연구 기간 끝까지 매일 투여하였다.
도 6은 제1일 및 제21일에 위약 또는 1 mg, 2 mg 또는 4 mg 엔독시펜 캡슐로 처리한 후 시간에 따른 평균 혈청 농도(ng/mL)를 도시한다(선형).
도 7은 제1일 및 제21일에 위약 또는 1 mg, 2 mg 또는 4 mg 엔독시펜 캡슐로 처리한 후 시간에 따른 평균 혈청 농도(ng/mL)를 도시한다(세미-로그).
도 8은 제1일 및 제21일에 위약 또는 1 mg, 2 mg 또는 4 mg 엔독시펜 캡슐로 처리한 후 시간에 따른 평균 혈청 농도(nM)를 도시한다(선형).
도 9는 화학식 (III)의 화합물의 형태 I의 샘플로부터 수득된 XRPD 패턴이다.
도 10은 화학식 (III)의 화합물의 형태 I의 샘플로부터 수득된 XRPD 패턴이다.
도 11은 화학식 (III)의 화합물의 형태 II의 샘플로부터 수득된 XRPD 패턴이다.
도 12는 화학식 (III)의 화합물의 형태 II의 샘플로부터 수득된 XRPD 패턴이다.
도 13은 화학식 (III)의 화합물의 형태 III의 샘플로부터 수득된 XRPD 패턴이다.
도 1은 타목시펜의 엔독시펜 및 다른 대사산물로의 대사 경로를 도시하는 도해이다. 4-히드록시타목시펜의 혈장 농도는 N-데스메틸 타목시펜에 비해 낮으며, 이는 모 약물의 대사에 대한 1차 경로가 시토크롬 P450 3A4(CYP3A4)에 의한 N-데스메틸 타목시펜으로의 N-탈메틸화에 이어 시토크롬 P450 2D6(CYP2D6)에 의해 엔독시펜으로 히드록실화된다는 것을 나타낸다.
도 2는 본원에 개시된 방법을 이용하여 제조되는 (Z)-엔독시펜 유리 염기의 대표적인 적외선 스펙트럼이다.
도 3은 위약 또는 1 mg, 2 mg 또는 4 mg 엔독시펜 캡슐로 처리한 후 엔독시펜의 최대 혈청 수준(nM)까지의 시간을 도시한다.
도 4는 제1일 및 제21일에 위약 또는 1 mg, 2 mg 또는 4 mg 엔독시펜 캡슐으로 처리한 후 엔독시펜의 최대 혈청 수준까지의 시간을 도시한다. 투여 후 시간(시간)은 X-축에 플롯팅되고 평균 혈청 농도(ng/mL)는 Y-축에 플로팅된다.
도 5는 제0일(제0일은 X-축 상의 제8일을 지칭함)에서 제21일까지 위약 또는 1 mg, 2 mg 또는 4 mg 엔독시펜 캡슐로 처리한 후 엔독시펜의 항정 상태 혈청 수준을 달성하는 시간을 도시한다. X-축은 일(제8일 내지 제21일)의 시간이고 Y-축은 ng/mL의 평균 혈청 농도이다. 혈액을 제0일에서 임상 시험 기간 끝까지 각각의 일에 채취하였다. 대상체에게 제1일에 투여하고 혈장 엔독시펜 수준을 표 17에 나타낸 바와 같이 측정하였다. 이어서, 대상체에게 제8일에 시작하여(도 5에서 제8일로 표시됨) 연구 기간 끝까지 매일 투여하였다.
도 6은 제1일 및 제21일에 위약 또는 1 mg, 2 mg 또는 4 mg 엔독시펜 캡슐로 처리한 후 시간에 따른 평균 혈청 농도(ng/mL)를 도시한다(선형).
도 7은 제1일 및 제21일에 위약 또는 1 mg, 2 mg 또는 4 mg 엔독시펜 캡슐로 처리한 후 시간에 따른 평균 혈청 농도(ng/mL)를 도시한다(세미-로그).
도 8은 제1일 및 제21일에 위약 또는 1 mg, 2 mg 또는 4 mg 엔독시펜 캡슐로 처리한 후 시간에 따른 평균 혈청 농도(nM)를 도시한다(선형).
도 9는 화학식 (III)의 화합물의 형태 I의 샘플로부터 수득된 XRPD 패턴이다.
도 10은 화학식 (III)의 화합물의 형태 I의 샘플로부터 수득된 XRPD 패턴이다.
도 11은 화학식 (III)의 화합물의 형태 II의 샘플로부터 수득된 XRPD 패턴이다.
도 12는 화학식 (III)의 화합물의 형태 II의 샘플로부터 수득된 XRPD 패턴이다.
도 13은 화학식 (III)의 화합물의 형태 III의 샘플로부터 수득된 XRPD 패턴이다.
화합물은 표준 명명법을 사용하여 기재된다. 달리 정의되지 않는 한, 본원에 사용된 모든 기술 및 과학 용어는 본 발명이 속하는 기술 분야의 숙련가에 의해 일반적으로 이해되는 것과 동일한 의미를 갖는다.
본원에 사용된 단수 형태의 용어는 문맥이 달리 지시하지 않는 한 복수 참조를 포함한다.
본원에 사용된 용어 "활성 약제학적 성분", "활성 성분", "API", "약물", "활성물", "활성물들" 또는 "치료제"는 약제학적 조성물 중의 약제학적으로 활성인 화합물(들)을 지칭하기 위해 상호교환적으로 사용될 수 있다. 이는 실질적으로 또는 완전히 약제학적으로 불활성인 조성물 중의 다른 성분, 예컨대 부형제와 대조적이다. 본 개시에 따른 적합한 API는 특정 질환, 병태, 또는 장애를 치료하기 위한 환자 순응 문제가 있거나 있을 수 있는 것이다. 본원에 사용된 치료제는 활성 화합물 및 그의 염, 전구약물 및 대사산물을 포함한다. 본원에 사용된 용어 "약물"은 인간 또는 다른 동물에서 질환의 진단, 치유, 완화, 치료 및/또는 예방에 사용하기 위한 화합물을 의미한다.
본원에 사용된 "보조 요법"은 1차 요법에 잇따르고 재발 위험이 있는 대상체에게 투여되는 요법을 지칭한다. 유방암 또는 생식관 암의 경우, 예컨대 타목시펜으로의 보조 전신 요법은 일반적으로 재발을 지연시키거나 생존을 연장시키거나 대상체를 치유하기 위해 1차 요법 직후에 시작된다.
본원에 사용된 용어 "타목시펜"은 (Z)-2-[4-(1,2-디페닐-1-부테닐)페녹시]-N,N-디메틸에탄아민을 지칭한다. 타목시펜은 또한 E-이성질체 또는 E-이성질체와 Z-이성질체의 조합을 지칭할 수 있다.
본원에서 사용된 용어 "4-히드록시타목시펜", "아피목시펜", 및 "4-OHT"는 상호교환적으로 사용되며 4-1-[4-[2-(디메틸아미노)에톡시]페닐]-2-페닐부트-1-에닐]페놀을 지칭하고, 타목시펜의 활성 대사산물을 구성한다. 4-OHT는 Z-이성질체, E-이성질체 또는 이들의 조합을 지칭할 수 있다.
본원에 사용된 용어 "엔독시펜"은 4-히드록시-N-데스메틸-타목시펜을 지칭한다. 이는 타목시펜의 2차 활성 대사산물이다.
명세서 전반에서 "화합물"에 참조 사항이 달린 구체물, 예컨대 화학식 (I), 화학식 (II), 화학식 (III) 및 화학식 (IV)의 화합물은 본원에 개시된 화학식 및/또는 화합물의 다형체, 염, 유리 염기, 공동-결정, 및 용매화물 형태를 포함한다. 따라서, 어구 "화합물", "화학식 (I)의 화합물", "화학식 (II)의 화합물", "화학식 (III)의 화합물" 및 "화학식 (IV)의 화합물"의 출현은 화학식 (IV)의 화합물의 형태 I, 화학식 (III)의 화합물의 형태 II-III, 화학식 (IV)의 화합물의 유리 염기, 화학식 (III)의 화합물의 유리 염기, 및/또는 본원에 기재된 글루코네이트 염을 포함한다.
용어 "결정질 형태", "다형체" 및 "형태"는 본원에서 상호교환적으로 사용될 수 있으며, 특정 결정질 또는 무정형 형태가 언급되지 않는 한, 예컨대 다형체, 슈도다형체, 염, 용매화물, 수화물, 용매화되지 않은 다형체(무수물 포함), 입체구조적 다형체 및 무정형 형태, 및 그의 혼합물을 포함하는 화합물의 모든 결정질 및 무정형 형태를 포함하는 것을 의미한다. 본 개시의 화합물은, 예컨대 화합물의 다형체, 슈도다형체, 염, 용매화물, 수화물, 용매화되지 않은 다형체(무수물 포함), 입체구조적 다형체, 및 무정형 형태, 및 그의 혼합물을 포함하는 그들 화합물의 결정질 및 무정형 형태를 포함한다.
본원에 개시된 모든 화합물은 또한 화합물에서 나타나는 원자의 모든 가능한 동위 원소를 포함하는 것으로 이해된다. 동위 원소는 원자 번호는 같지만 질량수는 상이한 원자를 포함한다. 예로서 및 제한 없이, 수소의 동위 원소는 삼중수소 및 중수소를 포함하고 탄소의 동위 원소는 11C, 13C 및 14C를 포함한다.
본원 및 청구범위에 사용된 용어 "포함하는" 및 "함유하는"은 포괄적인 개방형이며 추가의 언급되지 않은 요소, 조성 성분 또는 방법 단계를 배제하지 않는다. 따라서, 용어 "포함하는"은 보다 제한적인 용어 "이루어진" 및 "본질적으로 이루어진"을 포괄한다.
본원에 사용된 용어 "조합 요법"은 하나 이상의 추가 치료와 조합하여 본원에 기재된 조성물을 사용하는 것을 지칭한다. 조합 요법에서의 치료는 임의의 예방제, 치료제(예컨대, 화학요법), 방사선 요법, 수술 등과 같은 임의의 치료일 수 있다. 조합물은 본원에 개시된 조성물과 동일한 조성물에(예컨대, 동일한 캡슐, 정제, 연고 등에) 또는 별도의 조성물에(예컨대, 2개의 별도의 캡슐에)에 치료제 또는 예방제를 포함시키는 것을 지칭할 수 있다. 별도의 조성물은 상이한 투여 형태일 수 있다. 용어 "조합 요법" 및 "과 조합하여"의 사용은 본원에 기재된 조성물 및 예방제 및/또는 치료제 및/또는 치료가 이를 필요로 하는 대상체에게 투여되는 순서를 제한하지 않는다. 본 개시의 조성물은 이를 필요로 하는 대상체에게 하나 이상의 예방제 및/또는 치료제 및/또는 치료의 투여 전(예컨대, 1분(min), 5 min, 15 min, 30 min, 45 min, 1시간(h), 2 h, 4 h, 6 h, 8 h, 10 h, 12 h, 24 h, 36 h, 48 h, 72 h, 96 h, 1주(wk), 2 wk, 3 wk, 4 wk, 5 wk, 6 wk, 8 wk, 12 wk, 6개월(m), 9 m, 또는 1년 전), 투여와 동시에, 또는 투여 후(예컨대, 1분(min), 5 min, 15 min, 30 min, 45 min, 1시간(h), 2 h, 4 h, 6 h, 8 h, 10 h, 12 h, 24 h, 36 h, 48 h, 72 h, 96 h, 1주(wk), 2 wk, 3 wk, 4 wk, 5 wk, 6 wk, 8 wk, 12 wk, 6개월(m), 9 m, 또는 1년 후)에 투여될 수 있다. 본원에 사용된 조합 요법은 또한 단일 질환 또는 다수 질환, 예컨대 남성에서 전립선암 및 여성형 유방증을 갖는 대상체의 치료를 지칭할 수 있다.
본원에 사용된 용어 "시험 샘플"은 대상체로부터 수득되는 혈액 샘플을 의미한다. 혈액 샘플이 대상체로부터 수득되는 경우, 대상체의 혈액이 대상체의 엔독시펜 수준 및/또는 측정되거나 시험될 수 있는 다른 바이오마커를 결정하기 위해 사용된다는 것을 이해해야 한다. 본원에 사용된 "혈장 엔독시펜"은 시험이 전혈, 혈장 또는 혈청에 대해 수행되는지에 관계없이 대상체의 시험 샘플 중의 엔독시펜 수준을 지칭하기 위해 사용된다.
본원에 사용된 용어 "투여 형태"는 본 개시의 화합물 또는 조성물이 환자에게 전달되는 형태를 의미한다.
본원에 사용된 용어 "약제학적으로 허용되는" 또는 "약리학적으로 허용되는"은 제형의 다른 성분과 함께 사용할 수 있고 대상체에게 투여될 때 유해 반응, 예컨대 독성, 알레르기, 또는 면역학적 반응을 실질적으로 생성하지 않는 물질, 조성물, 또는 비히클을 의미한다. 그들은, 예컨대 미국 연방 또는 주정부의 규제 기관에 의해 승인되거나 미국 약전 또는 동물, 및 특히 인간에서 사용하기 위해 일반적으로 인정되는 다른 약전에 열거될 수 있다.
본원에 사용된 용어 "약제학적으로 허용되는 담체" 또는 "담체"는, 하나의 조직, 기관 또는 신체의 일부로부터 또는 피부를 가로질러 본 개시의 화합물 중 하나 이상을 운반 또는 수송하는 데 관여되는, 약제학적으로 허용되는 물질, 조성물 또는 비히클, 예컨대 액체 또는 고체 충전제, 희석제, 부형제, 용매 또는 캡슐화 물질을 의미한다.
본원에 사용된 용어 "약제학적으로 허용되는 염"은 대상체(예컨대, 포유동물, 및/또는 생체내, 생체외, 시험관내 세포, 조직 또는 기관)에서 생리적으로 허용되는 본 개시의 화합물의 임의의 염(예컨대, 산 또는 염기와의 반응에 의해 수득됨)을 지칭한다. 본 개시의 화합물의 "염"은 무기 또는 유기 산 및 염기로부터 유래될 수 있다. 적합한 음이온 염은 아레콜린, 베실레이트, 비카르보네이트, 비타르타레이트, 부틸브로마이드, 시트레이트, 카미실레이트, 글루코네이트, 글루타메이트, 글리콜릴라르사닐레이트, 헥시레소르시네이트, 히드라바민, 히드로브로마이드, 히드로클로라이드, 히드록시나프타노에이트, 이세티오네이트, 말레이트, 만델레이트, 메실레이트, 메틸브로마이드, 메틸브로마이드, 메틸니트레이트, 메틸술페이트, 무케이트, 납실레이트, 니트레이트, 파마오에이트(엠보네이트), 판토테네이트, 포스페이트/디포스페이트, 폴리갈라쿠로네이트, 살리실레이트, 스테아레이트, 술페이트, 탄네이트, 테오클레이트, 지방산 음이온, 및 트리에티오다이드를 포함한다.
적합한 양이온은 벤자틴, 클레미졸, 클로로프로카인, 콜린, 디에틸아민, 디에탄올아민, 에틸렌디아민, 메글루민, 피페라진, 프로카인, 알루미늄, 바륨, 비스무트, 리튬, 마그네슘, 칼륨, 및 아연을 포함한다.
본 출원의 목적 상, 본 개시의 화합물의 염은 치료적 이용을 위해 약제학적으로 허용되는 것으로 고려된다. 그러나, 약제학적으로 허용되지 않는 산 및 염기의 염이 또한, 예컨대 약제학적으로 허용되는 화합물의 제조 또는 정제에 유용할 수 있다.
본원에 사용된 용어 "약제학적 조성물"은 활성제(예컨대, 활성 약제학적 화합물 또는 성분, API)와, 조성물을 시험관내, 생체내 또는 생체외에서 진단 또는 치료적 이용에 특별히 적합하게 하는, 불활성 또는 활성(예컨대, 인지질)인 담체의 조합을 의미 한다.
본원에 사용된 "1차 요법"은 대상체에서 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다의 초기 진단 시 치료의 제1 라인을 지칭한다. 예시적인 1차 요법은 수술, 광범위한 화학요법, 및 방사선 요법을 포함할 수 있다.
본원에 사용된 용어 "대상체", "환자", "참가자" 및 "개체"는 본원에서 상호교환적으로 사용될 수 있고 포유동물, 예컨대 인간을 지칭할 수 있다. 포유동물은 또한 애완동물, 예컨대 개, 고양이, 실험실 동물, 예컨대 래트, 마우스, 및 농장 동물, 예컨대 소 및 말을 포함한다. 달리 명시되지 않는 한, 포유동물은 임의의 성별 또는 성일 수 있다.
본원에 사용된 용어 "타목시펜 불응성"은 적어도 2일 동안 타목시펜으로 매일 투여되었고 30 nM 미만(예컨대, 20 nM 미만, 25 nM 미만 또는 30 nM 미만)의 혈장 엔독시펜 수준을 갖는 대상체를 지칭한다. 본원에 사용된 용어 "타목시펜 내성"은 2가지 부류의 내성을 지칭한다: (a) 신생 내성, 즉 치료 시작부터 타목시펜 요법에 대한 비반응성, 또는 (b) 획득 내성, 즉 에스트로겐 수용체를 계속 발현하면서 초기 반응성 또는 타목시펜 의존적 성장/자극 성장 후 타목시펜 요법에 대한 비반응성(Minsun Chang. Biomol. Ther. 20(3), 256-267 (2012)). 타목시펜에 대한 획득 내성은 빠르면 3 m에서 1년 내지 늦으면 5-10년에 발생할 수 있다. 본원에 사용된 용어 "참조 혈장 엔독시펜 수준"은 30 nM의 값을 지칭한다.
본원에 사용된 용어 "단위 투여 형태"는 대상체에 대한 단위 용량에 적합한 물리적으로 분리된 단위를 지칭하며, 각각의 단위는 적합한 약제학적 부형제와 관련하여 원하는 치료 효과를 생성하도록 계산된 미리 결정된 양의 활성 물질을 함유한다 .
본원에 언급된 임의의 수치는 하한값에서 상한값까지의 모든 값, 즉 열거된 최저값과 최고값 사이의 모든 가능한 수치 조합이 본원에서 명시적으로 언급되는 것으로 간주되고 모든 범위의 종점이 범위 내에 포함되고 독립적으로 조합될 수 있는 것으로 구체적으로 이해된다. 예컨대, 농도 범위 또는 유익한 범위가 1% 내지 50%로 언급되는 경우, 2% 내지 40%, 10% 내지 30%, 또는 1% 내지 3% 등과 같은 값이 본 명세서에 명시적으로 열거되도록 의도된다. 또한, 농도 또는 용량이 1 mg 또는 10 mg과 같은 특정 값으로 언급되는 경우, 10% 변동을 포함하는 것으로 의도된다는 것을 이해해야 한다. 또 다른 예로서, 20%의 언급된 농도는 값 ± 10%의 값을 포함하는 것으로 의도된다. 또 다른 예에서, 1:10 내지 10:1의 비율이 언급되는 경우, 이는 1:9 내지 9:1, 1:8 내지 8:1, 1:7 내지 7:1, 1:6 내지 6:1, 1:5 내지 5:1, 1:4 내지 4:1, 1:3 내지 3:1, 1:2 내지 2:1, 1:1 내지 2:1 또는 2:5 내지 3:5 등과 같은 비율이 구체적으로 의도된다. 구체적으로 의도된 것에 대한 몇 가지 예만 있다. 달리 명시되지 않는 한, 조성물의 구성요소 또는 성분의 값은 성분 내의 각각의 구성성분의 중량%로 표현된다.
본원에 기재된 모든 방법은 달리 지시되거나 문맥에 의해 달리 명백하게 모순되지 않는 한 적합한 순서로 수행될 수 있다. 임의의 및 모든 예 또는 예시적인 언어(예컨대, "과 같은" 및 "등")의 사용은 단지 본 발명을 설명하기 위한 것이며 달리 청구되지 않는 한 본 발명의 범위를 제한하지 않는다. 본 명세서의 어떠한 언어도 임의의 비청구된 요소를 나타내는 임의의 것이 본원에 사용된 본 발명을 실시하는 데 필수적인 것으로 해석되어서는 안된다.
본원에 사용된 용어 "호르몬 의존적 유방 장애", "호르몬 의존적 생식관 장애", "호르몬 의존적 유방 및 생식관 장애"는 각각 및 일괄하여, 제한 없이, 감소가 필요한 높은 에스트로겐 또는 정상적인 에스트로겐 수준과 관련되거나 이에 민감한 임의의 유방 또는 생식관(부인과) 장애, 에스트로겐 수용체 양성(ER+)을 갖는 장애 및/또는 프로게스테론 수용체 양성(PR+) 장애, 예컨대 유방 장애, 자궁내막증, 자궁 근종(평활근종으로도 불림) 등을 포함한다. 생식관 장애는 자궁내막암, 난소암, 자궁경부암, 자궁암, 질암, 및 외음부암을 포함한다. 용어 "에스트로겐 관련된 장애" 및 "에스트로겐 수용체 관련된 장애"는 앞서의 호르몬 의존적 장애를 지칭하기 위해 상호교환적으로 사용될 수 있다. 장애는 기저 질환, 예컨대 전립선암 또는 다른 장애, 예컨대 간 질환에 대해 일차적으로 또는 이차적으로 나타날 수 있다. 호르몬 의존적 유방 및 생식관 장애는, 예컨대 뼈, 피부 및 몇몇 호르몬 생성(내분비) 조직에 영향을 끼치는 GNAS 유전자의 돌연변이에 의해 야기되는 장애인 맥쿤-알브라이트 증후군을 포함하며, 종종 이러한 돌연변이를 갖는 개체에서 그들의 뼈에 비정상적인 흉터 유사 (섬유) 조직, 다골성 섬유 형성이상으로 불리는 병태, 갑상선기능항진증, 및 소녀에서 성조숙증을 초래한다.
본원에 사용된 "유방 장애"는 유방의 임의의 이상 또는 한 무리의 이상을 의미한다. 이러한 이상은 증식, 비증식, 양성 또는 악성일 수 있다. 유방 장애는 유방의 양성 병변(예컨대, 증식증), 증가된 유방 밀도, 여성형 유방증, 유방통, 및 유방암을 포함한다. 양성 유방 병변은 증식증, 비정형, 관 증식증, 소엽 증식증, 비정형 유관 증식증(ADH), 및 비정형 소엽 증식증(ALH)을 포함하지만 이에 제한되지 않는다. ADH와 ALH는 암은 아니지만 유방암에 대한 소인을 나타낼 수 있다.
유방 밀도는 시각 기술, 예컨대 유방촬영에 의해 확인되는 유방 장애이며, 유방 내의 증가된 섬유선 조직, 즉 유방에서 간질 세포 및 상피 세포의 과도성장을 반영한다. 유방 밀도는 밀도의 심각도에 기초하여 4가지 부류(부류 A, B, C 및 D)로 분류된다. 이것은 유방암에 대한 독립적인 위험 인자이다. 미국의 적어도 23개 주는 대상체가 조밀한 유방(들)을 갖는다는 경우 의사가 대상체에게 이를 알릴 것을 규정한다. 대상체들이 건강한 생활방식을 선택하고 유방 변화를 모니터링하기 위해 정규적인 유방촬영상을 받도록 상기되지만, 현재 조밀한 유방에 대한 치료법은 없다.
여성형 유방증은 상피 증식증을 포함하여 유방 조직의 증가된 증식증을 반영하는 흔한 남성 유방 병태이며, 무증상 여성형 유방증의 유병률은 신생아에서 60% 내지 90%, 청소년에서 50% 내지 60%, 및 50세에서 69세의 남성에서 최대 70%이다(Therapeutics and Clinical Risk Management 2011:7, 145 - 148). 신생아 여성형 유방증은 보통 출생 후 4주 이내에 스스로 해결되며, 남성 청소년의 적어도 절반은 13 내지 14세에 전형적인 발병을 갖는 여성형 유방증을 경험한다(타너(Tanner) 단계 3 또는 4). 여성형 유방증은 남성 유방암에 대한 위험 인자로 제안되었다.
또한, 여성형 유방증은 종종 그 자체를 기저 장애, 예컨대 전립선암, 경화증 및 간 질환, 남성 성선기능저하증, 갑상선기능항진증, 신부전에 대해 및 혈액투석, 제I형 당뇨병 등을 겪는 환자에서 이차적으로 나타난다. 또한, 약물, 예컨대 항안드로겐 약물 또는 특정 항정신병약 자체가 여성형 유방증 사례의 최대 25%를 유발하는 것으로 보고되었으며, 그들의 호르몬 유사 작용에 의해 분류될 수 있다. 예컨대, 전립선암 치료에 사용되는 비스테로이드성 항안드로겐인 비칼루타미드에 기인하는 가장 흔한 부작용은 여성형 유방증 및 유방 통증이다.
본원에 사용된 "유방암"은 유방 세포의 임의의 악성 종양을 의미한다. 유방암은 전암, 초기 단계 암, 비-전이성 암, 전-전이성 암, 국소 진행 암, 및 전이성 암의 단계를 포함하는 임의의 단계의 유방암일 수 있다. 유방암에는 여러 유형이 있다. 예시적인 유방암은 관 제자리암종(DCIS), 소엽 제자리암종(LCIS), 침습적 (또는 침윤) 소엽 암종(ILC), 침습적 (또는 침윤) 관 암종(IDC), 미세침습적 유방 암종(MIC), 염증성 유방암, ER-양성(ER+) 유방암, ER-음성(ER-) 유방암, HER2+ 유방암, 삼중 음성 유방암(TNBC), 선양 낭성(샘낭성) 암종, 낮은 등급 선편평상피 암종, 수질성 암종, 점액 (또는 콜로이드) 암종, 유두 암종, 관상 암종, 화생 암종, 또는 미세유두 암종을 포함하지만 이에 제한되지 않는다. 단일 유방암 종양은 이들 유형의 조합이거나 침습적 암과 제자리 암의 혼합일 수 있다.
DCIS는 가장 흔한 비침습적 유방암이다. 이는 유방관의 세포 내층을 관여시킨다. DCIS에서, 세포는 관의 벽을 넘어 주변 유방 조직으로 퍼지지 않는다. 5개의 새로운 유방암 사례 중 약 1개가 DCIS이다. LCIS는 전암성 신생물이다. 이는 침습적 암에 대한 소인을 나타낼 수 있다. LCIS는 제자리(관 또는 소엽) 유방암의 약 15%만을 차지한다.
IDC는 가장 침습적인 유방암이다. 명칭이 적용되듯이 유방관에서 시작하여 주변 지방 조직을 침범하는 암종이다. 약 8 내지 10개의 침습적 유방암이 침윤성 관 암종이다. IDC는 종종 암 조직을 잘라내기 위한 수술 및 방사선 요법으로 치료된다. 또한, 면역요법과 조합된 화학요법(예컨대, 타목시펜 및 트라투주맙)이 종종 IDC를 치료하는 데 사용된다. 종양이 4 cm보다 큰 경우, 근치 유방절제가 수행될 수 있다.
ILC는 유방의 소엽에서 발병하고 주변 조직을 침범하는 암이다. 10개의 침습적 유방암 중 약 1개가 ILC이다. ILC는 암성 조직을 잘라내기 위한 수술 및 방사선 요법에 의해 치료된다. 또한, 화학요법 및 면역요법 조합(예컨대, 타목시펜 및 트라투주맙)은 종종 ILC를 치료하기 위한 보조 요법으로서 이용된다.
염증성 유방암은 모든 유방암의 약 1% 내지 3%를 차지한다. 염증성 유방암에서 암 세포는 피부의 림프관을 차단하여 유방이 붉게 변하고 따뜻함을 느끼게 한다. 이환된 유방은 더 커지거나 더 단단해지거나, 부드러워지거나, 가려워질 수 있다. 염증성 유방암은 화학요법, 면역요법, 방사선 요법 및 일부의 경우 수술로 치료된다.
에스트로겐 수용체 양성(ER+) 유방암은 암성 세포의 표면에 에스트로겐 수용체의 존재로 특징지어진다. ER+ 암 세포의 성장은 에스트로겐(호르몬 의존적 또는 호르몬 민감성 유방암)의 이용 가능성과 연관된다. 모든 유방암의 대략 80%가 ER+ 유방암이다. ER+ 유방암의 치료 옵션은 에스트로겐을 차단하는 화학치료제(예컨대, 타목시펜)을 포함한다.
본 개시는 (Z)-엔독시펜 유리 염기, (E)-엔독시펜과 (Z)-엔독시펜의 혼합물(E/Z-혼합물), 및 그의 염의 안정한 제제를 제조하는 방법, 및 그의 용도에 관한 것이다. 본 개시는 또한 (Z)-엔독시펜 또는 그의 염을 포함하는 경구 조성물 및 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이다. 본 개시는 또한 호르몬 의존적 유방 장애, 호르몬 의존적 생식관(부인과) 장애, 또는 둘 다를 갖거나 가질 위험이 있는 대상체에게 (Z)-엔독시펜, E/Z-혼합물, 또는 그의 염을 포함하는 경구 조성물을 투여함으로써 상기 대상체를 치료하는 방법을 제공한다.
한 측면에서, 본 개시는 엔독시펜 또는 그의 염을 포함하는 경구 조성물에 관한 것이다. 본 개시의 조성물에 포함된 엔독시펜은 (Z)-엔독시펜, (E)-엔독시펜 또는 그의 조합일 수 있다. 일 측면에서, 본 개시는 안정한 (Z)-엔독시펜 유리 염기를 포함하는 조성물에 관한 것이다.
일 측면에서, 본 개시는 (Z)-엔독시펜 유리 염기, (E)-엔독시펜과 (Z)-엔독시펜의 혼합물(E/Z-혼합물), 및 그의 염을 제조하는 산업적으로 확장 가능한 방법을 제공한다. 일 측면에서, 산업적으로 확장 가능한 방법은 안정한 (Z)-엔독시펜 유리 염기, E/Z-혼합물, 및 그의 염을 제조하는 합성 방법이다. 또 다른 측면에서, 본 개시는 안정한 (Z)-엔독시펜 유리 염기, E/Z-혼합물, 및 그의 염을 포함하는 조성물의 제조 방법을 제공한다.
특정 측면에서, 본 개시는 (Z)-엔독시펜 유리 염기의 결정질 형태 및 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물의 결정질 형태, 및 본원에 기재된 결정질 형태를 포함하는 엔독시펜의 약제학적 조성물을 제공한다.
(Z)-엔독시펜 유리 염기의 합성
엔독시펜의 합성 제조를 위한 몇몇 방법은 당해 분야에 공지되어 있다. 예컨대, 엔독시펜 및 그들의 전구약물 및 염의 합성 제조 방법이 미국 특허 번호 9,333,190(Ahmad, Jina Pharmaceuticals); WO 2008/070463(Ahmad, Jina Pharmaceuticals), 미국 공개 번호 2010/0112041(Ahmad, Jina Pharmaceuticals), WO 2012/050263(Ahmad, Jina Pharmaceuticals), WO 2014/141292(Desai, Intas Pharmaceuticals), WO 2017/070651(USA/Alchem Lab. Corp.); WO 2009/120999A2(Kushner), 미국 특허 번호 8,063,249(Kushner, Olema Pharmaceuticals), 미국 특허 번호 7,531,578 및 8,119,695(Forman and Yu), 및 WO 2012/050263(Song, CJ Cheiljedang Corp)에 기재되어 있다.
엔독시펜의 합성 제조 방법은 또한 연구 문헌에 공개되어 있다(Gauthier et al., J. Org. Chem, 61, 3890 - 3893 (1996), Fauq et al., Bioorg Med Chem Lett. 2010 May 15;20(10):3036-3038); Stearns et al., J. Natl. Cancer Inst. Vol 95, No. 23, 2003; Johnson et al., Breast Cancer Research and Treatment. 85:151-159, 2004; Ogawa, et al. Chem. Pharm. Bull. 39, 911 - 916, 1991). 그러나, 대규모의 산업적으로 확장 가능한 제조에 대한 요구가 충족되지 않고 있다. 안정한 (Z)-엔독시펜 유리 염기는 하기에서 및 실시예 1 내지 9에서 추가로 기재되는 바와 같이 도식 1에 따라 제조될 수 있다.
도식 1.
(Z)-엔독시펜 유리 염기의 합성

탈메틸화
일 측면에서, 본 개시는 화학식 (I)의 화합물인 [4-[2-(디메틸아미노)에톡시]페닐](4-히드록시페닐)메탄온)(AstaTech Pharmaceuticals, Inc.(중국)으로부터 입수 가능)을 탈메틸화하여 화학식 (II)의 화합물을 형성하는 단계를 포함하여, 실질적으로 정제된 (Z)-엔독시펜 유리 염기, E/Z-혼합물, 또는 그의 염을 제조하는 산업적으로 확장 가능한 방법에 관한 것이다. 따라서, 일부 실시양태에서, 산업적 방법은 [4-[2-(디메틸아미노)에톡시]페닐](4-히드록시페닐)메탄온)을 불활성 유기 용매 중의 탈메틸화제 및 양성자 억셉터로 탈메틸화하여 화학식 (II)의 화합물을 형성함으로써 화학식 (II)의 화합물을 제조하는 단계를 포함한다. 화학식 (II)의 화합물, (Z)-엔독시펜 유리 염기, E/Z-혼합물, 또는 그의 염을 제조하는 산업적으로 확장 가능한 방법은 화학식 (I)의 화합물을 반응당 1 mg 내지 1000 kg 범위의 양으로 탈메틸화시키는 단계를 포함한다.
일부 실시양태에서, 화학식 (I)의 화합물은 화학식 (I)의 화합물을 탈메틸화제와 반응시킴으로써 탈메틸화될 수 있다. 탈메틸화제는 목적에 적합한 임의의 작용제이다. 적합한 탈메틸화제의 예는 N-요오도숙신아미드(NIS), 에틸 클로로포르메이트(예컨대, 1-클로로에틸 클로로포르메이트, 디클로로에틸 클로로포르메이트, 트리클로로에틸 클로로포르메이트, α-클로로에틸클로로포르메이트(ACE-Cl)), 비닐 클로로포르메이트(VO-Cl), 브롬화시아노겐(BrCN: 폰 브라운 반응(von Braun's reaction)), 디에틸 아조디카르복실레이트, 염화피리디늄 등을 포함한다.
일부 실시양태에서, 탈메틸화제는 1-클로로에틸 클로로포르메이트, 디클로로에틸 클로로포르메이트, 트리클로로에틸 클로로포르메이트, α-클로로에틸클로로포르메이트(ACE-Cl), 및 비닐 클로로포르메이트(VO-Cl)로 이루어진 군으로부터 선택되는 클로로포르메이트이다. 다른 실시양태에서, 탈메틸화제는 에틸 클로로포르메이트, 예컨대 1-클로로에틸 클로로포르메이트, 디클로로에틸 클로로포르메이트, 트리클로로에틸 클로로포르메이트, 및 α-클로로에틸클로로포르메이트(ACE-Cl)로 이루어진 군으로부터 선택되는 에틸 클로로포르메이트이다. 적어도 하나의 실시양태에서, 탈메틸화제는 1-클로로에틸 클로로포르메이트이다.
일부 실시양태에서, 탈메틸화제는 1:0.5 내지 1:10 범위의 화학식 (I)의 화합물 대 탈메틸화제의 wt/wt 비율로 반응 혼합물에 첨가된다. 다른 실시양태에서, 탈메틸화제는 1:2: 내지 1:5 범위의 화학식 (I)의 화합물 대 탈메틸화제의 비율(wt/wt)로 존재한다. 적어도 하나의 실시양태에서, 탈메틸화제는 1:2의 화학식 (I)의 화합물 대 탈메틸화제의 비율(wt/wt)로 존재한다. 적어도 하나의 실시양태에서, 탈메틸화제는 1:3.3의 화학식 (I)의 화합물 대 탈메틸화제의 비율(wt/wt)로 존재한다. 다른 실시양태에서, 탈메틸화제 1-클로로에틸 클로로포르메이트는 1:0.5 내지 1:10 범위의 화학식 (I)의 화합물 대 탈메틸화제의 비율(wt/wt)로 존재한다.
탈메틸화 반응은 양성자 억셉터의 존재 하에서 탈메틸화 반응에 적합한 불활성 유기 용매 중에서 수행될 수 있다. 이러한 무기 용매는 디클로로메탄, 디클로로에탄, 클로로포름, 사염화탄소, 클로로벤젠, 디에틸 에테르, 1,4-디옥산, 3차-부틸 메틸 에테르(TBME), 테트라히드로푸란(THF), N,N-디메틸포름아미드(DMF), N-메틸피롤리돈(NMP), 디글림, 니트로메탄, 1,2-디메톡시에탄(DME), 피리딘, 아세톤, 아세토니트릴, 벤젠, o-자일렌, m-자일렌, p-자일렌, 자일렌, 헥산, 시클로헥산, 헵탄, 옥탄, 노난, 및 데칸, 또는 그의 조합을 포함한다. 일 측면에서, 산업적으로 확장 가능한 방법은 1:1 내지 1:50 범위의 화학식 (I)의 화합물 대 불활성 유기 용매의 (wt/wt) 비율로 탈메틸화를 위한 하나 이상의 불활성 유기 용매를 포함한다. 일부 실시양태에서, 탈메틸화 용매는 1:1 내지 1:50 범위의 화학식 (I)의 화합물 대 THF의 비율(wt/wt)의 THF이다. 다른 실시양태에서, THF는 1:1 내지 1:20 범위의 화학식 (I)의 화합물 대 THF의 비율(wt/wt)로 존재한다.
본 개시의 목적에 적합한 양성자 억셉터는 탄산염, 예컨대 탄산나트륨 및 탄산칼륨, 및 중탄산염, 예컨대 중탄산나트륨 및 중탄산칼륨, 양성자 스폰지, 및 에틸디이소프로필아민, (N',N-디이소프로필레아민, "DIPEA")을 포함하지만 이에 제한되지 않는다. 일 측면에서, 산업적으로 확장 가능한 방법은 1:0.5 내지 1:10 범위의 화학식 (I)의 화합물 대 양성자 억셉터의 wt/wt 비율로 반응 혼합물에 첨가되는 양성자 억셉터를 포함한다. 일부 실시양태에서, 양성자 억셉터는 1:1:8의 화학식 (I)의 화합물 대 양성자 억셉터의 비율(wt/wt)로 존재한다.
당업자는 본 개시의 탈메틸화 반응에 적합한 당해 분야에 공지된 다른 용매 및 양성자 억셉터를 쉽게 결정할 수 있을 것이다.
탈메틸화제, 탈메틸화 반응을 위한 용매, 및 화학식 (I)의 화합물은 임의의 순서로 첨가될 수 있다. 각각의 시약은 단일 볼러스(bolus)로 또는 다수 볼러스로 적합한 반응기에 첨가되고 교반될 수 있다.
일부 실시양태에서, 화학식 (I)의 화합물은 적합한 반응기에 충전되고 여기에 불활성 유기 용매 THF 및 양성자 억셉터 DIPEA가 탈메틸화 반응을 위해 첨가되고, 0℃ 내지 20℃로 냉각되고, 이어서 하나 이상의 탈메틸화제가 첨가된다. THF는 1:1 내지 1:20의 wt/wt 비율로 첨가되고, 양성자 억셉터 DIPEA는 1:0.5 내지 1:10의 wt/wt 비율로 탈메틸화 반응 혼합물에 첨가되고, 여기서 wt/wt는 화학식 (I)의 화합물에 대한 것이다. 일부 실시양태에서, 반응 혼합물은 15℃ 이하(NMT)로 냉각되고, 이어서 하나 이상의 탈메틸화제가 서서히 첨가된다. 반응은 불활성 조건, 예컨대 질소 또는 아리곤 하에서 수행될 수 있다.
하나 이상의 불활성 유기 용매 중의 화학식 (I)의 화합물, 하나 이상의 탈메틸화제(예컨대, 1-클로로에틸 클로로포르메이트)를 포함하는 반응 혼합물은 20℃ 내지 250℃, 예컨대 40℃ 내지 80℃, 50℃ 내지 230℃, 50℃ 내지 120℃, 및 150℃ 내지 200℃ 범위의 온도에서 가열될 수 있다. 일부 실시양태에서, 반응 혼합물은 환류 하에 가열된다. 다른 실시양태에서, 화학식 (I)의 화합물은 5시간 이상(NLT), NLT 8시간, NLT 12시간, NLT 24시간, NLT 36시간, NLT 48시간 및 NLT 72시간 동안 탈메틸화제 및 양성자 억셉터와 반응된다. 적어도 하나의 실시양태에서, 반응은 환류 하에 12시간 NLT 동안 가열된다. 반응은 추가의 사용시까지 유지되고 보관될 수 있다. 일부 실시양태에서, 반응 혼합물은 환류 조건 하에 교반하면서 24시간 이하(NMT)로 유지된다.
혼합물은 감압 하에서 하나 이상의 라운드의 증류에 적용될 수 있다. 증류는 증류에 적합한 용매, 예컨대 에틸 아세테이트, 저급 알콜(비제한적 예는 메탄올, 에탄올, n-프로판올, 및 이소프로판올을 포함), 벤젠, 아세톤, 아세토니트릴, 톨루엔, 디클로로메탄, 1,2-디클로로에탄, 및 클로로포름를 사용하여 NMT 100℃, NMT 95℃, NMT 90℃, NMT 85℃, NMT 80℃, 또는 NMT 70℃에서 수행될 수 있다. 당업자는 목적에 적합한 추가의 적합한 용매를 쉽게 결정할 수 있을 것이다.
적어도 하나의 실시양태에서, 증류에 사용되는 용매는 메탄올이다. 비제한적 예로서, 메탄올이 감압 하에서 2 내지 5 라운드의 증류를 위한 용매 교환에 사용될 수 있다.
용매, 예컨대 메탄올은 증류 라운드당 1:1 내지 1:10 범위의 화학식 (I)의 화합물 대 용매의 (wt/wt) 비율로 증류에 사용될 수 있다.
이어서, 혼합물은 교반 및 환류 하에 가열하면서 용매에 산을 첨가함으로써 (1:1 내지 1:10 범위의 화학식 (I)의 화합물 대 산의 wt/wt 비율) 용매/산 혼합물과 반응될 수 있다. 산은 임의의 적합한 산일 수 있다. HCl이 본 개시의 목적에 적합한 산의 예이다. 적합한 용매/산 혼합물의 비제한적 예는 메탄올/HCl, 에탄올/HCl, 프로판올/HCl, 이소프로판올/HCl, 메탄올/황산, 메탄올/인산, 에탄올/황산, 에탄올/인산, 프로판올/황산, 프로판올/인산, 이소프로판올/황산, 이소프로판올/인산, 메탄올/아세트산, 에탄올/아세트산, 프로판올/아세트산, 이소프로판올/아세트산, 메탄올/포름산, 에탄올/포름산, 프로판올/포름산, 및 이소프로판올/포름산을 포함한다. 적어도 하나의 실시양태에서, 용매/산 혼합물은 메탄올/6N HCl이다. 비제한적 예로서, 메탄올/HCl 혼합물 중의 메탄올은 1:3.2의 화학식 (I)의 화합물 대 용매/산 혼합물 중의 메탄올의 wt/wt 비율로 첨가될 수 있고, 메탄올/HCl 혼합물 중의 HCl은 1:4의 화학식 (I)의 화합물 대 용매/산 혼합물 중의 HCl의 wt/wt 비율일 수 있다. 적어도 하나의 실시양태에서, 메탄올/6N HCl 혼합물은 1:1 내지 1:10, 예컨대 1:1 내지 1:5 범위의 화학식 (I)의 화합물 대 메탄올/6N HCl의 wt/wt 비율로 첨가된다.
증류는 감압 하에서 NLT 5시간, NLT 8시간, NLT 10시간, NLT 12시간, NLT 14시간 동안 수행될 수 있다. 증류는 NMT 70℃, NMT 75℃, NMT 80℃, NMT 85℃, NMT 90℃ 및 NMT 90℃의 온도에서 수행될 수 있다.
반응 혼합물은 추가의 사용시까지 유지되고 보관될 수 있다. 일부 실시양태에서, 반응 혼합물은 환류 조건 하에 교반하면서 NMT 24시간 동안 유지될 수 있다.
반응 혼합물은 중화제, 예컨대 수산화나트륨(NaOH), 수산화암모늄, 아미노메틸프로판올 등으로 중화되고, 여과될 수 있다. 생성된 습윤 케이크는 물 및 유기 용매, 예컨대 에틸 아세테이트(EtOAc)로 세척되어 화학식 (II)의 화합물인 (4-히드록시페닐)(4-(2-(메틸아미노)에톡시)페닐)메탄온을 수득할 수 있다.
중화제는 1:1 내지 1:10 범위의 화학식 (I)의 화합물 대 중화제의 wt/wt 비율일 수 있다. 일부 실시양태에서, 중화제는 1:1 내지 1:10 범위의 비율의 8N 수산화나트륨이다. 다른 실시양태에서, 중화제는 1:2 내지 1:8 범위의 비율의 8N 수산화나트륨이다.
또 다른 측면에서, (Z)-엔독시펜 유리 염기, E/Z-혼합물 또는 그의 염을 제조하는 산업적으로 확장 가능한 방법은 여과된 생성물(화학식 (II)의 화합물)을 정제수(1:1 내지 1:5 wt/wt) 및 유기 용매, 예컨대 에틸 아세테이트(EtOAc)(1:0.5 내지 1:10 wt/wt)로 세척하는 하나 이상의 단계를 포함하며, 여기서 wt/wt는 화학식 (I)의 화합물에 대한 것이다. 습윤 케이크는 감압/진공 하에서 건조된다. 온도는 25℃ 내지 60℃의 범위일 수 있다. 일부 실시양태에서, 건조는 NMT 50℃의 온도에서 수행된다.
맥머리 반응
또 다른 측면에서, 본 개시는 화학식 (II)의 화합물을 맥머리 반응에 적용하여 화학식 (III)의 화합물인 E/Z-혼합물(즉, (E)-엔독시펜과 (Z)-엔독시펜 유리 염기의 혼합물)을 제공하는 단계를 포함하는 (Z)-엔독시펜 유리 염기, E/Z-혼합물, 및 그의 염을 제조하는 산업적으로 확장 가능한 방법에 관한 것이다.
맥머리 반응은 타목시펜(유럽 특허 출원 번호 168175)을 제조하는 데 이용되었다. 본 개시는 화학식 (II)의 화합물인 (4-히드록시페닐)(4-(2-(메틸아미노)에톡시)페닐) 메탄온이 불활성 유기 용매 중의 티타늄 염, 예컨대 티타늄의 염화물 염(예컨대, 삼염화티타늄 및 사염화티타늄(TiCl4)) 및 환원제를 통해 맥머리 반응에 의해 매개되는 프로피오페논에 커플링되어 화학식 (III)의 화합물의 E/Z 혼합물을 형성하는 산업적으로 확장 가능한 방법에 관한 것이다.
본 개시에 유용한 티타늄의 염은 티타늄 할로겐화물(예컨대, 삼염화티타늄(TiCl3), 사염화티타늄(TiCl4), 요오드화티타늄, 브롬화티타늄, 및 불화티타늄), 삼염화티타늄(IV) 이소프로폭사이드, 및 티타늄 이소프로폭사이드를 포함한다. 일부 실시양태에서, 티타늄 염은 TiCl4이다. 티타늄 염, 예컨대 TiCl4는 1:0.1 내지 1:12 범위의 화학식 (II)의 화합물 대 티타늄 염의 wt/wt 비율로 첨가된다.
환원제는 아연, 지르코늄, 바나듐, 니오븀, 몰리브덴, 텅스텐, 알루미늄, 마그네슘, 칼륨, 아연-구리 커플, 알칼리 및 알칼리 토 금속, 부틸륨, 리튬, 및 리튬 알루미늄 수소화물을 포함한다. 적어도 하나의 실시양태에서, 환원제는 아연이다. 맥머리 합성은 편리하게는 1:0.1 내지 1:10 범위의 화학식 (II)의 화합물 대 환원제의 wt/wt 비율로 환원제, 예컨대 아연을 사용하여 수행된다. 일부 실시양태에서, 환원제의 비율은 티타늄 염과 비교하여 과량이다.
맥머리 합성은 하나 이상의 불활성 유기 용매 중에서 수행될 수 있다. 맥머리 반응에 유용한 불활성 유기 용매는 디클로로메탄, 디클로로에탄, 클로로포름, 사염화탄소, 클로로벤젠, 디에틸 에테르, 1,4-디옥산, 3차-부틸 메틸 에테르(TBME), 테트라히드로푸란(THF), N,N-디메틸포름아미드(DMF), N-메틸피롤리돈(NMP), 디글림, 니트로메탄, 1,2-디메톡시에탄(DME), 피리딘, 아세톤, 아세토니트릴, 벤젠, o-자일렌, m-자일렌, p-자일렌, 자일렌, 헥산, 시클로헥산, 헵탄, 옥탄, 노난, 및 데칸, 또는 그의 조합을 포함한다. 일부 실시양태에서, 불활성 유기 용매는 1:1 내지 1:50 범위의 화학식 (II)의 화합물 대 용매의 wt/wt 비율이다. 다른 실시양태에서, 불활성 유기 용매는 1:1 내지 1:20 범위의 화학식 (II)의 화합물 대 용매의 wt/wt 비율이다. 일부 실시양태에서, 맥머리 반응에 사용되는 불활성 유기 용매는 THF이다. 일부 실시양태에서, THF는 1:1 내지 1:20 범위의 화학식 (II)의 화합물 대 THF의 wt/wt 비율이다.
불활성 유기 용매 중의 티타늄 염 및 환원제를 배합하여 예비-혼합물을 생성하는 것이 유리하다. 티타늄 염은 NMT 75℃, 예컨대 NMT 65℃, NMT 55℃, NMT 45℃, NMT 40℃, NMT 35℃, NMT 30℃, NMT 25℃, NMT 20℃, 및 NMT 15℃의 내부 온도를 유지하기 위해 이러한 비율로 환원제 및 불활성 유기 용매에 첨가된다. 따라서, 불활성 유기 용매 중의 티타늄 염 및 환원제는 배합되어 예비-혼합물을 생성한다. 일부 실시양태에서, Zn, TiCl4 및 THF는 배합되어 Zn/TiCl4/THF 혼합물을 생성한다. 적어도 하나의 실시양태에서, TiCl4는 Zn 및 THF에 첨가되고, NLT 10분 동안 내부 온도를 NMT 20℃로 유지하면서 혼합된다.
불활성 유기 용매 중의 티타늄 염 및 환원제의 제조는 불활성 유기 용매 중의 티타늄 염 및 환원제를 20℃ 내지 250℃, 예컨대 40℃ 내지 80℃, 50℃ 내지 230℃, 50℃ 내지 120℃, 및 150℃ 내지 200℃ 범위의 온도로 가열하는 단계를 추가로 포함할 수 있다. 일부 실시양태에서, 불활성 유기 용매 중에 존재하는 티타늄 염 및 환원제는 NLT 60℃에서 가열된다. 일부 실시양태에서, 불활성 유기 용매 중의 티타늄 염 및 환원제의 제조는 불활성 유기 용매 중의 티타늄 염 및 환원제를 환류 하에 가열하는 단계를 추가로 포함한다. 불활성 유기 용매 중의 티타늄 염 및 환원제는 환류 하에 NLT 30분, 예컨대 NLT 1시간, NLT 2시간, NLT 4시간, NLT 6시간 및 NLT 8시간 동안 불활성 조건 하에서, 예컨대 N2 또는 아르곤 하에서 가열된다.
또한, 화학식 (II)의 화합물을 불활성 유기 용매, 예컨대 THF 및 프로피오페논과 예비-혼합하고, 이어서 화학식 (II)의 화합물을 예비-혼합된 환원제/티타늄 염/용매 혼합물, 예컨대 Zn/TiCl4/THF 혼합물에 반응시켜 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성하는 것이 유리하다. 피로피오페논은 1:0.01 내지 1:5 범위의 화학식 (II)의 화합물 대 프로피오페논의 wt/wt 비율로 첨가될 수 있다. 불활성 유기 용매는 1:1 내지 1:20 범위의 화학식 (II)의 화합물 대 용매의 wt/wt 비율일 수 있다. 단계(a)의 화학식 (II)의 화합물은 유기 용매 중의 티타늄 염 및 환원제와 환류 하에 NLT 0.5시간, NLT 1시간, NLT 2시간, NLT 4시간, NLT 6시간, NLT 8시간, NLT 12시간, NLT 24시간, 및 NLT 48시간 동안 반응된다. 적어도 하나의 실시양태에서, 화학식 (II)의 화합물은 THF 및 프로피오페논과 혼합되고, Zn, TiCl4, THF의 예비-혼합물에 반응되고, 환류 하에 NLT 2시간 동안 가열된다. 또 다른 실시양태에서, 화학식 (II)의 화합물은 THF 및 프로피오페논과 혼합되고, 상술된 바와 같이 Zn/THF/TiCL4 혼합물에 반응되고, 환류 하에 NLT 8시간 동안 가열된다. 이어서, 반응 혼합물은 0℃ 내지 35℃로 냉각될 수 있다.
이어서, 반응 혼합물 중의 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물은 추출 정제, 증류 및 결정화되어 (E)-엔독시펜과 (Z)-엔독시펜의 정제된 혼합물을 제공할 수 있다. 반응 혼합물 중의 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물은 불활성 유기 용매, 예컨대 THF 및 MeTHF로 추출함으로써, 또는 염, 예컨대 탄산칼륨, 염화암모늄, 염화나트륨, 수산화나트륨을 반응 혼합물에 첨가하고 불활성 유기 용매, 예컨대 THF 및 MeTHF로 추출함으로써 추출 정제될 수 있다.
일부 실시양태에서, 반응 혼합물은 염화암모늄, 예컨대 25% 염화암모늄(1:1 내지 1:30 wt/wt); 실리카(Celite®) 층(1:0.01 내지 1:5 wt/wt); 및/또는 용매, 예컨대 THF(1:1 내지 1:10 wt/wt)로 1회 이상 추출된다. 다른 실시양태에서, 반응 혼합물은 탄산칼륨(K2CO3), 예컨대 40% K2CO3(1:1 내지 1:10 wt/wt) 및 MeTHF(1:1 내지 1:10 wt/wt)로 1회 이상 추출된다. 일부 실시양태에서, E/Z 혼합물은 NaOH, 예컨대 1N NaOH(1:1 내지 1:20 wt/wt)로 추가로 추출될 수 있다. 적어도 하나의 실시양태에서, NaCl(1:0.1 내지 1:0.5 wt/wt)은 추출 단계를 위해 1N NaOH에 첨가될 수 있다.
일부 실시양태에서, 반응 혼합물은 THF 또는 MeTHF로 1회 이상 추가로 추출될 수 있다. 적어도 하나의 실시양태에서, 반응 혼합물은 MeTHF로 3회 이상 추출된다. 본 출원인은 MeTHF가 놀랍게도 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물의 추출 단계에 적합하여 보다 높은 수율의 (Z)-엔독시펜와 (E)-엔독시펜의 정제된 혼합물을 제공한다는 것을 밝혀내었다. 적어도 하나의 실시양태에서, 혼합물은 20% 염화나트륨(1:1 내지 1:10 wt/wt)으로 추가로 추출될 수 있다.
이어서, 반응 혼합물은 적합한 용매, 예컨대 에틸 아세테이트, IPA, 및 IPA/PPW(화학식 (III)의 화합물에 대해 1:1 내지 1:10 wt/wt)로 2 내지 5 라운드의 용매 교환 및 증류에 적용될 수 있다. 증류는 감압/진공 하에 30℃ 내지 90℃ 범위의 온도에서 수행될 수 있다. 일부 실시양태에서, 증류는 NMT 30℃, NMT 35℃, NMT 40℃, NMT 45℃, NMT 50℃, NMT 55℃, NMT 60℃, NMT 65℃, NMT 70℃, NMT 75℃, NMT 80℃ 및 NMT 90℃의 온도에서 수행되고 여과될 수 있다. 여과 생성물은 용매, 예컨대 EtOAc, IPA, IPA/PPW 또는 n-헵탄으로 세척되고, 이어서 결정화 시스템, 예컨대 EtOAc/n-헵탄(1:2 v/v) 또는 IPA/n-헵탄(1:2.7 v/v)으로 1:1 내지 1:20 범위의 화학식 (III)의 화합물 대 EtOAc/n-헵탄 또는 IPA/n-헵탄의 wt/wt 비율로 결정화되고, 예컨대 NMT 60℃에서 건조되어 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜 유리 염기의 결정질 고체 혼합물을 제공할 수 있다.
또 다른 측면에서, 본 개시는 대략 1:1(45:55 내지 55:45)의 E/Z 비율을 갖는 E/Z-혼합물을 제조 또는 재평형화하는 산업적으로 확장 가능한 방법에 관한 것이다. 적합한 반응기는 상술된 바와 같이 맥머리 반응으로 제조된 불활성 유기 용매, 예컨대 에틸 아세테이트 중에 용해된 화학식 (III)의 화합물로 충전될 수 있다. 화학식 (III)의 화합물은 99:1 내지 60:40의 E/Z 비율을 가질 수 있다. 혼합물은 40℃ 내지 85℃ 범위의 온도에서 농축될 수 있다. 일부 실시양태에서, 혼합물은 부피가 5 vol.에 도달할 때까지 NMT 75℃의 온도에서 농축된다. 혼합물은 환류로 가열되고, 이어서 40℃ 내지 60℃ 범위의 온도로 냉각된다. 일부 실시양태에서, 혼합물의 온도는 50±5℃로 냉각된다. 1:1 내지 1:20 범위의 화학식 (III)의 화합물 대 n-헵탄의 비율로 n-헵탄이 혼합물에 서서히 첨가될 수 있고, 이어서 혼합물은 0±5℃로 냉각될 수 있다. 혼합물은 NLT 0.5시간, 예컨대 NLT 1시간, NLT 2시간, NLT 4시간, NLT 8시간, NLT 12시간 또는 NLT 24시간 동안 0±5℃에서 교반될 수 있다. 혼합물은 여과되고, 1:1 내지 1:10 범위의 화학식 (III)의 화합물 대 에틸 아세테이트/n-헵탄의 wt/wt 비율로 에틸 아세테이트/n-헵탄(1:2 v/v)으로 세척될 수 있다. 습윤 케이크는 감압 하에서 건조되어 대략 1:1의 E/Z-비율을 갖는 E/Z-엔독시펜 혼합물을 제공할 수 있다. 건조는 30℃ 내지 70℃ 범위의 온도에서 수행될 수 있다. 일부 실시양태에서, 습윤 케이트는 NMT 60℃에서 감압 또는 진공 하에서 건조되어 대략 1:1(45:55 내지 55:45)의 E/Z-비율을 갖는 E/Z-엔독시펜 혼합물을 제공할 수 있다.
또 다른 측면에서, 화학식 (III)의 화합물은 하기에 기재되는 바와 같이 실질적으로 순수한 (Z)-엔독시펜을 수득하기 위해 추가로 정제, 농축 또는 재평형화될 수 있다.
(Z)-엔독시펜 유리 염기의 농축 정제
또 다른 측면에서, 본 개시는 농축 및 정제에 의해 (Z)-엔독시펜 유리 염기을 제조하는 산업적으로 확장 가능한 방법에 관한 것이다. (Z)-엔독시펜의 산업적으로 확장 가능한 농축 및 정제는 예시적인 도식 1의 단계 3의 방법을 이용하여 본원에 기재되는 바와 같이 및 실시예 1, 2, 4 및 9에 추가로 기재되는 바와 같이 수행될 수 있다. 분별 결정화에 사용되는 (E)-엔독시펜과 (Z)-엔독시펜의 출발 혼합물은 임의의 E/Z 비율, 예컨대 99:1 내지 1:10 범위의 E/Z 비율을 가질 수 있다. 일부 실시양태에서, 출발 (E)/(Z)-엔독시펜 혼합물의 E/Z-비율은 30:70 내지 70:30의 범위이다. 일부 실시양태에서, 출발 (E)/(Z)-엔독시펜 혼합물의 E/Z-비율은 99:1 내지 1:99의 범위이다. 일부 실시양태에서, 출발 (E)/(Z)-엔독시펜 혼합물의 E/Z-비율은 51:1, 1:1.8 또는 1:5.6이다.
E/Z-혼합물은 상술된 바와 같이 수득되는 화학식 (III)의 화합물일 수 있거나 (예컨대, 시그마 알드리치(Sigma Aldrich)로부터) 상업적으로 공급될 수 있다. (E)-엔독시펜과 (Z)-엔독시펜의 혼합물(E/Z-혼합물)은 분별 결정화되어 (Z)-엔독시펜 유리 염기가 농축된 제1 결정질 고체 및 제1 모액을 수득한다(실시예 1). 분별 결정화는 (Z)-엔독시펜이 여과액에 잔류되도록 엔독시펜 및 그의 유도체를 분쇄할 수 있는 제1 용매를 사용하여 수행된다. 적합한 제1 용매는 엔독시펜 이성질체를 차별적으로 가용화시키는 것이며, 제한 없이, 에틸 아세테이트, 이소프로판올, 이소프로판올/PPW, 아세토니트릴, 아세토니트릴/PPW, 및 디클로로메탄을 포함한다. 일부 실시양태에서, 제1 용매는 에틸 아세테이트이다. 제1 용매, 예컨대 에틸 아세테이트는 1:1 내지 1:20 범위의 화학식 (III)의 화합물 대 제1 용매의 wt/wt 비율로 첨가된다. 일부 실시양태에서, 화학식 (III)의 화합물은 제1 용매에 용해되고, 50℃ 내지 80℃ 범위의 온도로 가열되고, NMT 35℃로 냉각된다.
혼합물의 산성화가 (E)-엔독시펜의 (Z)-엔독시펜으로의 전환을 향상시킨다는 것은 놀라운 발견이었다. 따라서, 일부 실시양태에서, 화학식 (III)의 화합물은 산으로 전처리되고, 이어서 염기로 중화된다.
비제한적 예로서, 적합한 반응기는 화학식 (III)의 화합물로 충전되고 이에 제1 용매가 첨가되고, 0℃ - 5℃로 냉각된다. 이어서, 산, 예컨대 HCl 또는 TFA가 서서히 첨가될 수 있다. 일부 실시양태에서, 산은 1:1 내지 1:5 범위의 화학식 (III)의 화합물 대 산의 wt/wt 비율로 E/Z-엔독시펜 혼합물에 첨가된다. 이어서, 반응 혼합물은 교반하면서 50℃ 내지 70℃ 범위의 온도로 가열될 수 있다. 일부 실시양태에서, 반응을 환류 하에 수행된다. 반응은 NLT 4시간, 예컨대 NLT 6시간, NLT 12시간, NLT 24시간, 및 NLT 48시간 동안 수행될 수 있다. 반응 혼합물은 0℃ - 5℃로 냉각되고, 중화제로 중화된다.
일부 실시양태에서, 중화제는 1:1 내지 1:5 범위의 화학식 (III)의 화합물 대 중화제의 wt/wt 비율로 반응 혼합물에 첨가된다. 적합한 중화제는 수산화나트륨, 수산화칼륨, 수산화암모늄, 아미노메틸프로판올 등을 포함한다. 일부 실시양태에서, 중화제는 8N 수산화나트륨이다. 반응 혼합물의 pH는 바람직하게는 알칼리이다. 일부 실시양태에서, pH는 ≥ 10, 예컨대 ≥ 11 또는 ≥ 12이다.
일부 실시양태에서, (Z)-엔독시펜은 유기층으로 추출되고, 수집되며, 수상은 제1 용매, 예컨대 에틸 아세테이트(1:1 내지 1:10 범위의 화학식 (III)의 화합물 대 제1 용매의 wt/wt 비율로 첨가됨)로 1회 이상 세척된다. 유기층을 모으고, 염수(20% NaCl; 1:1 내지 1:10 범위의 화학식 (III)의 화합물 대 NaCl의 wt/wt 비율로 첨가됨)로 1회 이상 세척한다. 유기층은 활성탄으로 처리되고, 이산화규소(Celite®) 층(1.01 내지 1:0.1 범위의 화학식 (III)의 화합물 대 실리카의 wt/wt 비율로 첨가됨) 상에서 여과된다. 생성물은 제1 용매(1:1 내지 1:10 범위의 화학식 (III)의 화합물 대 제1 용매의 wt/wt 비율로 첨가됨)로 1회 이상 다시 세척되고, 증류된다. 증류는 50℃ 내지 80℃ 범위의 온도에서 수행될 수 있다. 일부 실시양태에서, 온도는 NMT 75℃일 수 있다.
제1 모액은 실시예 1 및 표 2 및 3에서 보여지는 바와 같이 (Z)-엔독시펜이 농축된다. 일부 실시양태에서, 제1 모액은 E-엔독시펜과 Z-엔독시펜의 혼합물의 E/Z-비율과 비교하여 적어도 50% 농축된다. 다른 실시양태에서, 제1 모액은 E-엔독시펜과 Z-엔독시펜의 혼합물의 E/Z-비율과 비교하여 (Z)-엔독시펜이 적어도 70% 농축된다.
제1 모액은 제1 모액을 농축시키거나, 제1 모액으로부터의 제1 용매를 1회 이상 교환함으로써 제2 결정질 고체 및 제2 모액을 수득함으로써 재결정화될 수 있다(표 5). 재결정화는 교환을 위한 제2 용매를 사용하여 수행된다. 제2 용매는 1:1 내지 1:10 범위의 화학식 (III)의 화합물 대 제2 용매의 wt/wt 비율로 첨가되어 슬러리를 생성한다. 적합한 제2 용매는 IPA, IPA/PPW, 아세톤, 아세톤/MTBE, 에탄올, EtOAc, EtOAc/n-헵탄을 포함한다. 일부 실시양태에서, 제2 용매는 IPA이다. 놀랍게도, IPA는 (Z)-엔독시펜을 EtOAc보다 높은 수준으로 고체 분획으로 분쇄한다는 것이 밝혀졌다. 따라서, 일부 실시양태에서, 제1 용매가 EtOAc인 경우, 제2 용매는 IPA 또는 IPA/PPW이다. 이는 (Z)-엔독시펜을 먼저 EtOAc로부터 여과액(제1 모액)으로 유도하고 이어서 IPA 또는 IPA/PPW로부터 고체 분획으로 유도하는 데 유용하다. 적어도 하나의 실시양태에서, 제2 용매는 IPA/PPW이다(표 7). 또 다른 실시양태에서, 제2 용매는 아세톤/MTBE이다. 제2 결정질 고체는 화학식 (IV)의 화합물인 (Z)-엔독시펜, (Z)-4-(1-(4-(2-(메틸아미노)에톡시)페닐)-2-페닐부트-1-에닐)페놀이다(분자량 373.49; 분자식: C25H27NO2; 융점 139℃ - 143℃). 제2 결정질 고체는 ≥ 70% (Z)-엔독시펜, 예컨대 ≥ 75% (Z)-엔독시펜, ≥ 80% (Z)-엔독시펜 또는 ≥ 90% (Z)-엔독시펜이다. 일부 실시양태에서, 제2 결정질 고체는 ≥ 90% (Z)-엔독시펜이다.
제2 결정질 고체는 임의적으로 재결정화되어 제3 결정질 고체인 (Z)-엔독시펜을 수득할 수 있다(표 5). 제3 결정질 고체는 ≥90% (Z)-엔독시펜, 예컨대 ≥91% (Z)-엔독시펜, ≥92% (Z)-엔독시펜, ≥93% (Z)-엔독시펜, ≥94% (Z)-엔독시펜, ≥95% (Z)-엔독시펜, ≥96% (Z)-엔독시펜, ≥97% (Z)-엔독시펜, ≥98% (Z)-엔독시펜, 또는 ≥99% (Z)-엔독시펜일 수 있다. 일부 실시양태에서, 제3 결정질 고체는 ≥90% (Z)-엔독시펜이다. 일부 실시양태에서, 제3 결정질 고체는 ≥95% (Z)-엔독시펜이다. 이 임의적인 재결정화는 제3 용매를 사용하여 수행된다. 제3 용매는 에탄올, 메탄올, 에틸 아세테이트, IPA, IPA/PPW, 아세톤, 아세톤/MTBE 및 EtOAc/n-헵탄으로 이루어진 군으로부터 선택된다.
일 측면에서, 본 개시는 제1 용매, 제2 용매, 및 제3 용매를 사용하기 전에 예열하는 것에 관한 것이다(표 2). 일부 실시양태에서, 제1 용매, 제2 용매, 및 제3 용매 중 하나 이상은 각각 40℃ 내지 80℃ 범위의 온도로 독립적으로 예열될 수 있다. 분별 결정화 및 재결정화 단계는 또한 60℃ 내지 80℃에서의 증류 및/또는 생성된 용액의 0℃ 내지 35℃ 범위의 온도로의 냉각 단계를 포함할 수 있다.
일부 실시양태에서, 상술된 바와 같이 수득되는 제2 모액 뿐만 아니라 제1, 제2 및 제3 결정질 고체는 정제된 (Z)-엔독시펜을 수득하기 위해 1회 이상 상술된 바와 같이 분별 결정화 및 재결정화에 추가도 적용될 수 있음을 이해해야 한다. 또한, 수득되는 제1, 제2 및/또는 제3 결정질 고체는 더 많은 (Z)-엔독시펜을 수득하기 위해 컬럼 크로마토그래피 기술을 이용하여 임의적으로 재처리될 수 있음을 이해해야 한다.
특정 실시양태에서, 본원에 기재된 산업적으로 확장 가능한 방법은 독립적으로 본원의 실시예에서 구체화되는 바와 같이 (예컨대, 반응 부산물을 제거하거나, 반응 생성물을 후처리하거나 단리하거나 정제하기 위해) 추가의 단계 또는 절차를 포함한다. 일부 실시양태에서, (Z)-엔독시펜 유리 염기는 <2%, <1%, 및 <0.5% 불순물을 갖는다. 다른 실시양태에서, 화학식 (III)의 화합물은 <2%, <1%, 및 <0.5% 불순물을 갖는다.
당업자는 바람직한 결과를 얻기 위해 변경될 수 있는 전술한 공정의 여러 파라미터를 인식할 것이다. 이들 파라미터는, 예컨대 반응 성분 및 용매의 정제 방법 및 수단, 상기 반응 성분 및 용매를 반응 혼합물에 첨가하는 순서, 상기 반응 성분 및 용매의 반응 지속기간; 및 반응 동안 반응 성분 및 용매의 교반, 혼합 또는 뒤섞기의 온도 및 속도를 포함한다.
본원의 방법(또한 특정 공정 단계를 포함)에 의해 구현되는 공정은 하기 기준 중 하나 이상을 충족한다는 것이 밝혀졌다: 화학식 (II), (III) 및 (IV)의 화합물을 제조하는 공지된 공정과 비교하여 보다 우수한 안정성, 보다 안전함, 보다 단순함, 보다 높은 수율 및 보다 경제적임. 하기에 개시된 공정에 의해 제조되는 (Z)-엔독시펜은 5℃ 및 25℃에서 60% 상대 습도(25℃/60% RH)에서 적어도 9개월 동안 및 40℃/75% RH에서 적어도 3개월 동안 안정하다(실시예 9). 승온에서의 안정성은 장기가 안정성을 나타낸다. 따라서, 본 개시의 방법에 의해 제조되는 (Z)-엔독시펜 유리 염기는 적어도 6개월 동안, 예컨대 적어도 9개월, 적어도 12개월, 또는 적어도 18개월 동안 안정하다. 또한, 본원에 기재된 방법은 수 킬로그램 작업에서 확장 가능한 것으로 간주되어 상업적 생산에 적합하다.
엔독시펜 염
또 다른 측면에서, 본 출원은 엔독시펜 염 및 엔독시펜 염을 제조하는 방법을 제공한다. 당해 분야에 공지된 엔독시펜 염은 엔독시펜의 히드로클로라이드(Fauq et al., Bioorg Med Chem Lett. 2010 May 15;20(10):3036-3038) 및 시트레이트 염(미국 특허 번호 9333190; 미국 공개 번호 2010/0112041)을 포함하며, 대상체에 대한 경구 투여에 대해 평가되고 있다.
특정 실시양태에서, 본 개시는 아레콜린, 베실레이트, 비카르보네이트, 비타르타레이트, 부틸브로마이드, 시트레이트, 카미실레이트, 글루코네이트, 글루타메이트, 글리콜릴라르사닐레이트, 헥시레소르시네이트, 히드라바민, 히드로브로마이드, 히드로클로라이드, 히드록시나프타노에이트, 이세티오네이트, 말레이트, 만델레이트, 메실레이트, 메틸브로마이드, 메틸브로마이드, 메틸니트레이트, 메틸술페이트, 무케이트, 납실레이트, 니트레이트, 파마오에이트(엠보네이트), 판토테네이트, 포스페이트/디포스페이트, 폴리갈라쿠로네이트, 살리실레이트, 스테아레이트, 술페이트, 탄네이트, 테오클레이트, 및 트리에티오다이드로 이루어진 군으로부터 선택되는 엔독시펜의 음이온 염을 제공한다.
다른 실시양태에서, 본 개시는 벤자틴, 클레미졸, 클로로프로카인, 콜린, 디에틸아민, 디에탄올아민, 에틸렌디아민, 메글루민, 피페라진, 프로카인, 알루미늄, 바륨, 비스무트, 리튬, 마그네슘, 칼륨, 및 아연으로 이루어진 군으로부터 선택되는 엔독시펜의 양이온 염을 제공한다.
본 개시의 엔독시펜 염은 상술된 바와 같이 제조되는 화학식 (I), (II), (III) 및 (IV)의 임의의 화합물을 사용하여 제조될 수 있다.
엔독시펜 글루코네이트
일부 실시양태에서, 본 개시는 엔독시펜의 글루코네이트 염, 또는 그의 화학적 등가물을 제공한다. 따라서, 적어도 하나의 실시양태에서, 본 개시의 조성물 중에 포함되는 엔독시펜은 엔독시펜 글루코네이트 또는 그의 화학적 등가물이다. 달리 명시되지 않는 한, 본원에서 엔독시펜 글루코네이트를 언급할 때, 그의 화학적 등가물이 또한 포괄될 것으로 이해될 것이다.
본 개시의 목적 상, 엔독시펜 글루코네이트의 화학적 등가물은 엔독시펜 분자와 글루코네이트 모이어티 사이의 모든 음이온성, 양이온성, 및 비이온성 반응 복합체를 포함한다. 이러한 복합체는 전형적으로 엔독시펜 분자의 히드록실기와 반응한다. 글루코네이트 모이어티는 D-글루콘산, 글루콘산, 글리코겐산, 글리칸-Δ-락톤 등을 포함한다. 일부 실시양태에서, 글루코네이트 모이어티는 약제학적으로 허용되는 글루코네이트 염이다. 이러한 염은 칼슘 글루코네이트, 나트륨 글루코네이트, 및 알칼리 금속 및 알칼리 토 금속의 염, 예컨대 칼륨 글루코네이트, 마그네슘 글루코네이트, 리튬 글루코네이트 등을 포함한다.
글루코네이트의 입체이성질체 D-형 및 L-형 모두가 본 개시에 포괄된다. 일부 실시양태에서, 본 개시의 조성물 중에 포함되는 엔독시펜 글루코네이트는 (Z)-엔독시펜 D-글루코네이트, (Z)-엔독시펜 L-글루코네이트, (E)-엔독시펜 D-글루코네이트, (E)-엔독시펜 L-글루코네이트 및 그의 조합으로 이루어진 군으로부터 선택된다. 일부 실시양태에서, 약제학적 조성물은 (Z)-엔독시펜 L-글루코네이트이다. 특정 실시양태에서, 약제학적 조성물은 (Z)-엔독시펜 D-글루코네이트를 포함한다. 달리 지시되지 않는 한, 조성물은 라세미 혼합물, 순수 입체이성질체(예컨대, 에난티오머 및 디아스테레오머), 입체-농축된 혼합물 등으로 존재할 수 있다. 특정 입체이성질체가 본원에 표시되거나 명명될 때, 조성물 전체의 유용성이 다른 이성질체의 존재에 의해 제거되지 않는다면, 달리 지시되지 않는 한, 소량의 다른 입체이성질체가 존재할 수 있다는 것이 당업자에 의해 이해될 것이다. 개별 이성질체는 적합한 키랄 정지상 또는 지지체를 사용하는 키랄 크로마토그래피를 포함하는 당해 분야에 널리 공지된 다수의 방법에 의해, 또는 그들을 디아스테레오머로 화학적으로 전환시키고 통상의 수단, 예컨대 크로마토그래피 또는 재결정화에 의해 디아스테레오머를 분리시킨 후 최초 입체이성질체를 재생함으로써 수득될 수 있다.
본 개시는 또한 엔독시펜 글루코네이트 또는 그의 화학적 등가물을 제조하는 방법을 포함하며, 이는 엔독시펜을 글루코네이트 모이어티와 혼합하여 엔독시펜 글루코네이트를 수득하는 단계를 포함한다. 치료제의 글루코네이트 염을 제조하는 방법은 당해 분야에 공지되어 있다(예컨대, 미국 공개 번호 2002/0127665).
일부 실시양태에서, 엔독시펜 D-글루코네이트(예컨대, (Z)-엔독시펜 D-글루코네이트) 염은 유리 염기로서의 엔독시펜(예컨대, 상술된 방법에 의해 제조되는 (Z)-엔독시펜)의 에탄올성 슬러리를 물 중의 D-글루코놀락톤의 20% w/v 용액을 15 내지 30분 동안 70℃에서 가열함으로써 가수분해시켜 수득된 D-글루콘산의 수용액과 혼합함으로써 수득될 수 있다. 일부 실시양태에서, 최소 부피의 에탄올이 사용되며, 엔독시펜 유리 염기 1 g당 5 ml의 수성 D-글루콘산 용액이 첨가된다. 이어서, 투명 용액이 수득될 때까지 교반을 계속한다. 이어서, 이 용액의 필요한 부피를 하나 이상의 다른 부형제에 첨가하여 "활성 성분"이 (Z)-엔독시펜 D-글루코네이트 또는 (Z)-엔독시펜 L-글루코네이트 염인 제형을 생성한다. 염은 당해 분야 공지되고 본원에 개시된 임의의 방법에 의해 정제될 수 있다.
정제된 (Z)-엔독시펜 유리 염기 또는 (E)/(Z)-엔독시펜의 혼합물은 모두 엔독시펜 글루코네이트 염의 제조 목적에 유용하다. (Z)-엔독시펜 D-글루코네이트 또는 (Z)-엔독시펜 L-글루코네이트의 용액은 또한 분별 결정화 또는 재결정화에 의해 본원에 기재되는 바와 같이 (E)/(Z)-엔독시펜 글루코네이트 염의 혼합물로부터 정제되어(실시예 1-3) 고체의 정제된 (Z)-엔독시펜 염을 수득할 수 있다. 고체 염은 추가의 사용 시까지 N2 하에 -5℃에서 저장될 수 있다.
다른 엔독시펜 염이 또한 글루코네이트 모이어티 또는 글루코네이트 염과 함께 출발 물질로서 사용되어 엔독시펜 글루코네이트 또는 그의 화학적 등가물을 수득할 수 있다는 것이 당업자에 의해 이해될 것이다.
엔독시펜 글루코네이트 또는 그의 화학적 등가물은 상술된 바와 같이 제조되는 (Z)-엔독시펜을 사용하여 또는 쉽게 이용 가능한 출발 반응물로 제조될 수 있다. 예컨대, 조 엔독시펜 HCl(≥ 98%) 및 나트륨 글루코네이트는 시그마 알드리치(미국)로부터 쉽게 입수 가능하다.
일부 실시양태에서, 엔독시펜 HCl 및 나트륨 글루코네이트는 적합한 용매 중에 용해되어 엔독시펜 글루코네이트 또는 그의 화학적 등가물을 수득한다. 적어도 하나의 실시양태에서, 엔독시펜 글루코네이트를 포함하는 조성물은 또한 엔독시펜 HCl을 포함한다. 다른 실시양태에서, (E)/(Z)-엔독시펜 혼합물(화학식 (III)의 화합물) 및 나트륨 글루코네이트는 적합한 용매 중에 용해되어 엔독시펜 글루코네이트 또는 그의 화학적 등가물을 제공한다.
엔독시펜 글루코네이트 또는 그의 화학적 등가물을 제조하기에 적합한 용매는 유기 용매, 예컨대 알콜, 아세톤, DMSO, 폴리에틸렌 글리콜, 지방산 및 지방 알콜 및 그들의 유도체, 히드록실산, 피롤리돈, 우레아, 식물성유, 동물유, 예컨대 어유, 정유 등 또는 그의 혼합물, 및 물-혼화성 용매, 예컨대 물 혼화성 알콜, 디메틸술폭사이드, 디메틸포름아미드, 물-혼화성 에테르, 예컨대 테트라히드로푸란, 물-혼화성 니트릴, 예컨대 아크릴로니트릴, 물 혼화성 케톤, 예컨대 아세톤 또는 메틸 에틸 케톤, 아미드, 예컨대 디메틸아세타미드, 프로필렌 글리콜, 글리세린, 폴리에틸렌 글리콜 400, 글리코푸롤, 테트라글리콜 등, 또는 그의 혼합물을 포함하지만 이에 제한되지 않는다.
엔독시펜 글루코네이트의 제조에 유용한 수-혼화성 용매는 글리세린, 에탄올, 프로판올, 이소프로판올, 프로필렌 글리콜, 폴리에틸렌 글리콜, 또는 그의 혼합물이다. 유용한 추가의 용매는 디글리콜 모노에틸 에테르(트란스쿠톨); 알켈렌 글리콜, 예컨대 디프로필렌 글리콜, 프로필렌 글리콜, 폴리에틸렌 글리콜, 예컨대 PEG 300, 400, 3395, 4450 등; 디메틸 이소소르바이드; 및 탈수 알콜을 포함한다. 일부 실시양태에서, 용매는 탈수 알콜, 예컨대 무수 알콜이다. 특정 실시양태에서, 용매의 양은 엔독시펜(유리 염기, 염) 및 글루코네이트 염을 용해시키기에 충분하다. 필요한 경우, 용매의 농도가 또한 조정될 수 있다. 반응은 실온에서 및 대기압에서 수행될 수 있다.
엔독시펜 글루코네이트 또는 그의 화학적 등가물을 제조하는 데 사용될 수 있는 엔독시펜 유리 염기 또는 엔독시펜 염(예컨대, 엔독시펜 HCl 등) 및 글루코네이트 염(예컨대, 및 나트륨 글루코네이트 등)의 양은 사용되는 반응물의 양에 따라 변화할 수 있다. 생성된 엔독시펜 글루코네이트는 1:1 비율로 엔독시펜:글루코네이트 모이어티를 가질 것이다.
일부 실시양태에서, 엔독시펜 글루코네이트를 제조하는 데 사용되는 엔독시펜 또는 엔독시펜 염의 양은 총 조성물(wt/wt)의 0.01중량% 내지 40중량%(예컨대, 1중량% 내지 10중량%, 또는 3중량% 내지 5중량%)이다. 일부 실시양태에서, 엔독시펜 글루코네이트을 제조하는 데 사용되는 글루코네이트 염은 0.01% 내지 40%(wt/wt)(예컨대, 1% 내지 10%(wt/wt), 또는 3% 내지 5%(wt/wt))이다. 당업자는, 제한 없이, 목적하는 수율을 달성하기에 효과적인 반응물의 양을 포함하는 당해 분야 및 본 개시의 기술 및 지식에 의해 안내될 것이다.
결정질 형태
특정 측면에서, 본 개시는 (Z)-엔독시펜 유리 염기의 결정질 형태 및 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물의 결정질 형태를 포함하는 엔독시펜의 결정질 형태를 제공한다. 본 개시는 또한 본원에 기재된 결정질 형태를 포함하는 엔독시펜의 약제학적 조성물을 제공한다. 엔독시펜의 결정질 형태는 약제학적 조성물 중의 활성 성분으로서 사용하기에 적합한 생체이용률 및 안정성의 이점을 제공할 수 있다. 약제학적 약물 물질 또는 활성 성분의 결정 구조의 변화는 약제학적 약물 제품 또는 활성 성분의 용해 속도(생체이용률 등에 영향을 끼칠 수 있음), 제조가능성(예컨대, 취급 용이성, 공지된 강도의 용량을 일정하게 제조하는 능력) 및 안정성(예컨대, 열적 안정성, 저장 수명 등)에 영향을 끼칠 수 있다. 이러한 변화는 상이한 투여 또는 전달 형태, 예컨대 정제 및 캡슐을 포함하는 고체 경구 투여 형태의 약제학적 조성물의 제제 또는 제형에 영향을 끼칠 수 있다. 다른 형태, 예컨대 비결정질 또는 무정형 형태와 비교하여, 결정질 형태는 목적하는 또는 적합한 흡습성, 입자 크기 제어, 용해 속도, 용해도, 순도, 물리적 및 화학적 안정성, 제조가능성, 수율, 및/또는 공정 제어를 제공할 수 있다. 따라서, 엔독시펜의 결정질 형태는 하기와 같은 이점을 제공할 수 있다: 활성제의 제조 공정 또는 화합물 또는 활성 성분의 약물 제품 형태의 안정성 또는 저장성을 개선함, 및/또는 적합한 생체이용률 및/또는 활성제로서 안정성을 가짐.
특정 용매 및 분별 결정화 방법의 이용은 상술된 하나 이상의 유리한 특징을 나타낼 수 있는 다형체 형태 I, II 및 III 중 임의의 하나 이상을 포함하는 엔독시펜의 상이한 다형체 형태를 생성하는 것으로 밝혀졌다. 본원에 기재된 다형체의 제조 방법, 및 이들 다형체의 특징규명이 하기에서 보다 상세히 기재된다.
화학식 III, 형태 I
특정 측면에서, 본 개시는 화학식 (III)의 화합물의 다형체 형태 I을 제공하며, 여기서 조성물 중의 화학식 (III)의 화합물의 적어도 90중량%는 (Z)-이성질체(즉, (Z)-엔독시펜)이다. 일부 실시양태에서, 다형체 형태 I은 실질적으로 도 9 또는 도 10에 제시된 바와 같은 x-선 분말 회절(XRPD) 패턴을 나타낸다. 일부 실시양태에서, 다형체 형태 I은 실질적으로 도 9 또는 도 10에 제시된 바와 같은 XRPD 패턴으로 적어도 2개, 적어도 3개, 적어도 4개, 적어도 5개, 또는 적어도 6개의 주요 피크를 포함하는 XRPD 패턴을 갖는다.
예컨대, XRPD 패턴을 언급할 때 용어 "실질적으로 ~에 제시된 바와 같은"은 본원에 도시된 것과 반드시 동일하지는 않지만 당업자에 의해 고려될 때 실험 오차 또는 편차의 한계 내에 속하는 패턴을 포함한다. XRPD 피크의 상대적 강도는 입자 크기, 샘플 제조 기술, 샘플 장착 절차 및 사용된 특정 기기에 따라 변화할 수 있다. 또한, 기기 변동 및 다른 인자가 2 쎄타(2θ) 값에 영향을 끼칠 수 있다.
따라서, 지정된 2 쌔타 각이 제공될 때, 지정된 2 쎄타 각은 지정된 지정된 값 ± 0.5°, 예컨대 ± 0.4°, ± 0.3°, ± 0.2°, 또는 ± 0.1°로 변화할 수 있다는 것을 이해해야 한다. 본원에 사용된 "주요 피크"는 30% 초과, 예컨대 35% 초과의 상대적 강도를 갖는 XRPD 피크를 지칭한다. 상대적 강도는 XRPD 패턴에서 가장 큰 피크의 피크 강도에 대한 관심 피크의 피크 강도의 비율로 계산된다.
특정 실시양태에서, 다형체 형태 I은 16.8 ± 0.3°, 17.1 ± 0.3° 및 21.8 ± 0.3°2 쎄타에서 주요 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 I은 16.0 ± 0.3°, 18.8 ± 0.3° 및 26.5 ± 0.3°2 쎄타에서 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 I은 16.8 ± 0.3°, 17.1 ± 0.3° 및 21.8 ± 0.3°2 쎄타에서 주요 피크 및 16.0 ± 0.3°, 18.8 ± 0.3° 및 26.5 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 I은 12.3 ± 0.3°, 28.0 ± 0.3° 및 29.0 ± 0.3°2 쎄타에서 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 I은 16.8 ± 0.3°, 17.1 ± 0.3° 및 21.8 ± 0.3°2 쎄타에서 주요 피크 및 12.3 ± 0.3°, 28.0 ± 0.3° 및 29.0 ± 0.3°2 쎄타로 부터 선택되는 적어도 하나의 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 I은 16.8 ± 0.3°, 17.1 ± 0.3° 및 21.8 ± 0.3°2 쎄타에서 주요 피크 및 12.3 ± 0.3°, 16.0 ± 0.3°, 18.8 ± 0.3°, 26.5 ± 0.3°, 28.0 ± 0.3° 및 29.0 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 I은 16.8 ± 0.3°, 17.1 ± 0.3° 및 21.8 ± 0.3°2 쎄타에서 주요 피크 및 12.3 ± 0.3°, 16.0 ± 0.3°, 18.8 ± 0.3°, 26.5 ± 0.3°, 28.0 ± 0.3° 및 29.0 ± 0.3°2 쎄타에서 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다.
특정 실시양태에서, 본 개시는 다형체 형태 I을 포함하는 조성물을 제공한다. 조성물 중의 화학식 (III)의 화합물의 90중량%, 95중량% 또는 99중량% 초과는 다형체 형태 I일 수 있다. 일부 실시양태에서, 조성물은 0.01 mg 내지 200 mg의 다형체 형태 I을 포함한다. 일부 실시양태에서, 조성물은 약 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 다형체 형태 I을 포함한다.
화학식 III, 형태 II
특정 측면에서, 본 개시는 화학식 (III)의 화합물의 다형체 형태 II를 제공하며, 여기서 조성물은 0.9 내지 1.3, 예컨대 약 1.1의 E/Z 비율의 화학식 (III)의 화합물의 (E)-이성질체 및 (Z)-이성질체(즉, (E)-엔독시펜 및 (Z)-엔독시펜)을 포함한다. 일부 실시양태에서, 다형체 형태 II는 실질적으로 도 11 또는 도 12에 제시된 바와 같은 x-선 분말 회절(XRPD) 패턴을 나타낸다. 일부 실시양태에서, 다형체 형태 II는 실질적으로 도 11 또는 도 12에 제시된 바와 같은 XRPD 패턴으로 적어도 2개, 적어도 3개, 적어도 4개, 적어도 5개, 또는 적어도 6개의 주요 피크를 포함하는 XRPD 패턴을 갖는다.
특정 실시양태에서, 다형체 형태 II는 7.0 ± 0.3°, 11.9 ± 0.3°, 14.0 ± 0.3° 및 18.4 ± 0.3°2 쎄타에서 주요 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 II는 22.0 ± 0.3°2 쎄타에서 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 II는 7.0 ± 0.3°, 11.9 ± 0.3°, 14.0 ± 0.3° 및 18.4 ± 0.3°2 쎄타에서 주요 피크 및 22.0 ± 0.3°2 쎄타에서 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 II는 6.6 ± 0.3°, 13.3 ± 0.3° 및 20.0 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 II는 7.0 ± 0.3°, 11.9 ± 0.3°, 14.0 ± 0.3° 및 18.4 ± 0.3°2 쎄타에서 주요 피크 및 6.6 ± 0.3°, 13.3 ± 0.3° 및 20.0 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 II는 7.0 ± 0.3°, 11.9 ± 0.3°, 14.0 ± 0.3° 및 18.4 ± 0.3°2 쎄타에서 주요 피크 및 6.6 ± 0.3°, 13.3 ± 0.3°, 20.0 ± 0.3° 및 22.0 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 II는 7.0 ± 0.3°, 11.9 ± 0.3°, 14.0 ± 0.3° 및 18.4 ± 0.3°2 쎄타에서 주요 피크 및 6.6 ± 0.3°, 13.3 ± 0.3°, 20.0 ± 0.3° 및 22.0 ± 0.3°2 쎄타에서 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다.
특정 실시양태에서, 본 개시는 다형체 형태 II를 포함하는 조성물을 제공한다. 조성물 중의 화학식 (III)의 화합물의 90중량%, 95중량% 또는 99중량% 초과는 다형체 형태 II일 수 있다. 일부 실시양태에서, 조성물은 0.01 mg 내지 200 mg의 다형체 형태 II를 포함한다. 일부 실시양태에서, 조성물은 약 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 다형체 형태 II를 포함한다.
화학식 III, 형태 III
특정 측면에서, 본 개시는 화학식 (III)의 화합물의 다형체 형태 III을 제공하며, 여기서 조성물은 0.9 내지 1.3, 예컨대 약 1.1의 E/Z 비율의 화학식 (III)의 화합물의 (E)-이성질체 및 (Z)-이성질체(즉, (E)-엔독시펜 및 (Z)-엔독시펜)을 포함한다. 일부 실시양태에서, 다형체 형태 III은 실질적으로 도 13에 제시된 바와 같은 x-선 분말 회절(XRPD) 패턴을 나타낸다. 일부 실시양태에서, 다형체 형태 III은 실질적으로 도 13에 제시된 바와 같은 XRPD 패턴으로 적어도 2개, 적어도 3개, 적어도 4개, 적어도 5개, 또는 적어도 6개의 주요 피크를 포함하는 XRPD 패턴을 갖는다.
특정 실시양태에서, 다형체 형태 III은 11.9 ± 0.3°, 13.9 ± 0.3°, 17.1 ± 0.3° 및 17.7 ± 0.3°2 쎄타에서 주요 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 III은 25.3 ± 0.3°2 쎄타에서 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 III은 11.9 ± 0.3°, 13.9 ± 0.3°, 17.1 ± 0.3° 및 17.7 ± 0.3°2 쎄타에서 주요 피크 및 25.3 ± 0.3°2 쎄타에서 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 III은 18.2 ± 0.3°, 22.5 ± 0.3° 및 26.8 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 III은 11.9 ± 0.3°, 13.9 ± 0.3°, 17.1 ± 0.3° 및 17.7 ± 0.3°2 쎄타에서 주요 피크 및 18.2 ± 0.3°, 22.5 ± 0.3° 및 26.8 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 III은 11.9 ± 0.3°, 13.9 ± 0.3°, 17.1 ± 0.3° 및 17.7 ± 0.3°2 쎄타에서 주요 피크 및 18.2 ± 0.3°, 22.5 ± 0.3°, 25.3 ± 0.3° 및 26.8 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다. 일부 실시양태에서, 다형체 형태 III은 11.9 ± 0.3°, 13.9 ± 0.3°, 17.1 ± 0.3° 및 17.7 ± 0.3°2 쎄타에서 주요 피크 및 18.2 ± 0.3°, 22.5 ± 0.3°, 25.3 ± 0.3° 및 26.8 ± 0.3°2 쎄타에서 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어진다.
특정 실시양태에서, 본 개시는 다형체 형태 III을 포함하는 조성물을 제공한다. 조성물 중의 화학식 (III)의 화합물의 90중량%, 95중량% 또는 99중량% 초과는 다형체 형태 III일 수 있다. 일부 실시양태에서, 조성물은 0.01 mg 내지 200 mg의 다형체 형태 III을 포함한다. 일부 실시양태에서, 조성물은 약 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 다형체 형태 III을 포함한다.
엔독시펜 유리 염기 조성물
일 측면에서, 본 개시는 안정한 (Z)-엔독시펜 유리 염기 또는 그의 염, 및 (Z)-엔독시펜 유리 염기 또는 그의 염을 포함하는 조성물을 제공한다. 일부 실시양태에서, 약제학적 조성물은 주로 (Z)-엔독시펜 유리 염기로서 엔독시펜을 포함한다.
특정 실시양태에서, 조성물은 조성물 중 총 엔독시펜의 적어도 0.1%, 적어도 0.2%, 적어도 0.3%, 적어도 0.4%, 적어도 0.5%, 적어도 1%, 적어도 5%, 적어도 10%, 적어도 20%, 적어도 25%, 적어도 30%, 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 적어도 90%, 적어도 91%, 적어도 92%, 적어도 93%, 적어도 94%, 적어도 95%, 적어도 96%, 적어도 97%, 적어도 98%, 적어도 99%, 적어도 99.5%, 적어도 99.99%, 또는 100%의 (Z)-엔독시펜 유리 염기 wt/wt로서 엔독시펜을 포함할 수 있다. 적어도 하나의 조성물에서, 조성물은 조성물 중 총 엔독시펜의 ≥ 90%의 (Z)-엔독시펜 유리 염기 wt/wt를 포함한다. 또 다른 실시양태에서, 조성물은 조성물 중 총 엔독시펜의 ≥ 95%의 (Z)-엔독시펜 유리 염기 wt/wt를 포함한다. 또 다른 실시양태에서, 조성물은 조성물 중 총 엔독시펜의 ≥ 96%, ≥ 97%, ≥ 98%, ≥ 99%, 또는 ≥ 99.5%의 (Z)-엔독시펜 유리 염기 wt/wt를 포함한다.
다른 실시양태에서, 엔독시펜을 포함하는 조성물은 조성물의 0.01% 내지 20%, 0.05% 내지 15%, 또는 0.1% 내지 10%의 (Z)-엔독시펜 wt/wt 또는 w/v을 포함한다. 적어도 하나의 실시양태에서, 엔독시펜을 포함하는 조성물은 조성물의 0.01% 내지 20%의 (Z)-엔독시펜 wt/wt 또는 w/v을 포함한다. 다양한 다른 실시양태에서, 엔독시펜을 포함하는 조성물은 조성물의 0.01%, 0.02%, 0.03%, 0.04%, 0.05%, 0.06%, 0.07%, 0.08%, 0,09%, 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1%, 2%, 3%, 4%, 5%, 10%, 또는 20%의 (Z)-엔독시펜 wt/wt을 포함한다.
일 측면에서, (Z)-엔독시펜을 포함하는 조성물은 (E)-엔독시펜을 추가로 포함한다. 일부 실시양태에서, 조성물 중의 엔독시펜은 1:99; 5:95; 10:90, 15:85; 20:80, 25:75; 30:70; 40:70, 45:55; 50:50; 55:45; 60:40; 65:45; 및 70:30의 (E)-엔독시펜 대 (Z)-엔독시펜(E/Z-비율)의 비율을 갖는다. 다른 실시양태에서, 조성물은 10:90 내지 70:30 범위의 E/Z-비율을 갖는 엔독시펜을 포함한다. 다른 실시양태에서, 조성물은 45:55 내지 55:45 범위의 E/Z-비율을 갖는 엔독시펜을 포함한다.
접두사 (Z), (E) 또는 (E/Z)에 의해 구체적으로 언급되지 않는 한, 접두사 없이 일반적으로 사용되는 엔독시펜은 본원에서 임의의 또는 모든 엔독시펜 이소형을 포함하도록 사용된다.
엔독시펜 염 조성물
일부 실시양태에서, 본 개시는 엔독시펜의 염을 포함하는 조성물을 제공한다. 일부 실시양태에서, 본 개시는 엔독시펜의 약제학적으로 허용되는 염을 포함하는 조성물을 제공한다. 본원에는 특정 실시양태에서 1%, 5%, 10%, 20%, 25%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 96%, 97%, 98%, 99%, 99.5%, 99.99% 또는 100%의 엔독시펜 염을 포함하는 조성물이 제공된다.
일부 실시양태에서, 염은 아레콜린, 베실레이트, 비카르보네이트, 비타르타레이트, 부틸브로마이드, 시트레이트, 카미실레이트, 글루코네이트, 글루타메이트, 글리콜릴라르사닐레이트, 헥시레소르시네이트, 히드라바민, 히드로브로마이드, 히드로클로라이드, 히드록시나프타노에이트, 이세티오네이트, 말레이트, 만델레이트, 메실레이트, 메틸브로마이드, 메틸브로마이드, 메틸니트레이트, 메틸술페이트, 무케이트, 납실레이트, 니트레이트, 파마오에이트(엠보네이트), 판토테네이트, 포스페이트/디포스페이트, 폴리갈라쿠로네이트, 살리실레이트, 스테아레이트, 술페이트, 탄네이트, 테오클레이트, 트리에티오다이드, 벤자틴, 클레미졸, 클로로프로카인, 콜린, 디에틸아민, 디에탄올아민, 에틸렌디아민, 메글루민, 피페라진, 프로카인, 알루미늄, 바륨, 비스무트, 리튬, 마그네슘, 칼륨, 및 아연으로 이루어진 군으로부터 선택된다. 일부 실시양태에서, 염은 엔독시펜 글루코네이트이다. 엔독시펜 글루코네이트는 (Z)-엔독시펜 D-글루코네이트, (E)-엔독시펜 D-글루코네이트, (Z)-엔독시펜 L-글루코네이트, (E)-엔독시펜 L-글루코네이트 또는 그의 조합으로 이루어진 군으로부터 선택될 수 있다.
일부 실시양태에서, 엔독시펜 글루코네이트를 포함하는 조성물은 조성물 중 총 엔독시펜 글루코네이트의 wt/wt 기준으로 10% 내지 100%의 (Z)-엔독시펜 D-글루코네이트를 포함한다. 일부 실시양태에서, 엔독시펜 글루코네이트를 포함하는 조성물은 조성물 중 총 엔독시펜의 wt/wt 기준으로 10% 내지 100%의 (Z)-엔독시펜 L-글루코네이트를 포함한다.
다른 실시양태에서, 엔독시펜 글루코네이트를 포함하는 조성물은 총 엔독시펜 글루코네이트에 대해 10%, 20%, 30%, 40%, 50%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, 99.5%, 99.75%, 99.99%, 또는 100%의 (Z)-엔독시펜 D-글루코네이트 또는 (Z)-엔독시펜 L-글루코네이트를 포함한다. 일부 실시양태에서, 조성물은 적어도 80%, 적어도 85%, 적어도 90%, 적어도 91%, 적어도 92%, 적어도 93%, 적어도 94%, 적어도 95%, 적어도 96%, 적어도 97%, 적어도 98%, 적어도 99%, 적어도 99.5%, 또는 적어도 99.99%의 (Z)-엔독시펜 D-글루코네이트, (Z)-엔독시펜 L-글루코네이트 또는 그의 조합을 포함한다.
본원에는 일부 실시양태에서 (Z)-엔독시펜 D-글루코네이트 및 (E)-엔독시펜 D-글루코네이트를 포함하는 조성물이 제공된다. (Z)-엔독시펜 D-글루코네이트 및 (E)-엔독시펜 D-글루코네이트는 각각 10:90 내지 99:1 wt/wt 또는 v/v 범위의 비율로 조성물 중에 존재할 수 있다. 일부 실시양태에서, (Z)-엔독시펜 D-글루코네이트 대 (E)-엔독시펜 D-글루코네이트의 비율(wt/wt 또는 v/v)은 각각 10:90 내지 99:1(예컨대, 45:55, 50:50, 60:40, 70:30, 80: 20, 90:10; 91:9; 92:8; 93:7; 94:8; 95:5, 96:4, 97:3, 98:2, 99:1, 99.5:0.5, 또는 99.99:0.01)이다. 특정 실시양태에서, (Z)-엔독시펜 D-글루코네이트 대 (E)-엔독시펜 D-글루코네이트의 비율(wt/wt 또는 v/v)은 90:10; 91:9; 92:8; 93:7; 94:8; 95:5, 96:4, 97:3, 98:2, 99:1, 99.5:0.5, 또는 99.99:0.01이다. 당업자는 엔독시펜 글루코네이트 이성질체의 다른 조합이 본 개시에 포괄된다는 것을 인식할 것이다.
일부 실시양태에서, 엔독시펜 글루코네이트를 포함하는 조성물은 조성물의 0.01%, 0.05%, 0.01%, 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19% 또는 20%의 엔독시펜 글루코네이트 (wt/wt) 또는 (w/v)를 포함한다. 일부 실시양태에서, 엔독시펜 글루코네이트를 포함하는 조성물은 조성물의 0.01%, 0.05%, 0.01%, 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 1%, 2%, 3%, 4%, 5%, 6%, 7%, 8%, 9%, 10%, 11%, 12%, 13%, 14%, 15%, 16%, 17%, 18%, 19% 또는 20%의 (Z)-엔독시펜 글루코네이트 (wt/wt) 또는 (w/v)를 포함한다.
본 개시의 화합물 및 조성물은 경구, 비경구, 국소, 및 관내 전달을 포함하지만 이에 제한되지 않는 당해 분야에 공지된 임의의 경로에 의해 이를 필요로 하는 대상체에게 투여될 수 있다. 따라서, 본원에 개시된 조성물은 의도된 투여 경로에 적합하게 제형화된다.
일부 실시양태에서, 엔독시펜을 포함하는 조성물은 부형제를 추가로 포함한다. 이러한 부형제는 의도된 투여 경로와 적합할 수 있다.
엔독시펜 다형체 조성물
일 측면에서, 본 개시는 엔독시펜의 하나 이상의 다형체 형태, 예컨대 본원에 기재된 형태 I, 형태 II, 또는 형태 II를 포함하는 조성물을 제공한다. 일부 실시양태에서, 약제학적 조성물은 주로 다형체 형태 I로서 엔독시펜을 포함한다. 일부 실시양태에서, 약제학적 조성물은 주로 다형체 형태 II로서 엔독시펜을 포함한다. 일부 실시양태에서, 약제학적 조성물은 주로 다형체 형태 III으로서 엔독시펜을 포함한다.
특정 실시양태에서, 조성물은 조성물 중의 총 엔독시펜의 적어도 0.1%, 적어도 0.2%, 적어도 0.3%, 적어도 0.4%, 적어도 0.5%, 적어도 1%, 적어도 5%, 적어도 10%, 적어도 20%, 적어도 25%, 적어도 30%, 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 적어도 90%, 적어도 91%, 적어도 92%, 적어도 93%, 적어도 94%, 적어도 95%, 적어도 96%, 적어도 97%, 적어도 98%, 적어도 99%, 적어도 99.5%, 적어도 99.99%, 또는 100%의 엔독시펜의 단일 다형체 형태, 예컨대 형태 I, 형태 II, 또는 형태 III wt/wt를 포함한다. 적어도 하나의 조성물에서, 조성물은 조성물 중의 총 엔독시펜의 ≥ 90%의 엔독시펜의 단일 다형체 형태, 예컨대 형태 I, 형태 II, 또는 형태 III wt/wt를 포함한다. 또 다른 실시양태에서, 조성물은 조성물 중의 총 엔독시펜의 ≥ 95%의 엔독시펜의 단일 다형체 형태, 예컨대 형태 I, 형태 II, 또는 형태 III wt/wt를 포함한다. 또 다른 실시양태에서, 조성물은 조성물 중의 총 엔독시펜의 ≥96%, ≥97%, ≥98%, ≥99%, 또는 ≥99.5%의 엔독시펜의 단일 다형체 형태, 예컨대 형태 I, 형태 II, 또는 형태 III wt/wt를 포함한다. 엔독시펜의 특정 중량%가 단일 다형체 형태일 때, 조성물 중의 엔독시펜의 나머지는 무정형 엔독시펜 및/또는 단일 다형체 형태를 제외한 하나 이상의 다형체 형태의 엔독시펜의 일부 조합이다. 다형체 엔독시펜이 하나의 특정 형태의 엔독시펜으로 정의될 때, 나머지는 무정형 엔독시펜 및/또는 지정된 특정 형태 이외의 하나 이상의 다형체 형태로 구성된다. 단일 다형체 형태의 예는 엔독시펜의 형태 I, II 및 III 뿐만 아니라 본원에 기재된 바와 같은 하나 이상의 특성으로 특징지어지는 단일 다형체 형태의 성상을 포함한다.
다른 실시양태에서, 엔독시펜을 포함하는 조성물은 조성물의 0.01% 내지 20%, 0.05% 내지 15%, 또는 0.1% 내지 10%의 엔독시펜의 단일 다형체 형태, 예컨대 형태 I, 형태 II, 또는 형태 III wt/wt 또는 w/v를 포함한다. 적어도 하나의 실시양태에서, 엔독시펜을 포함하는 조성물은 조성물의 0.01% 내지 20%의 엔독시펜의 단일 다형체 형태, 예컨대 형태 I, 형태 II, 또는 형태 III wt/wt 또는 w/v를 포함한다. 다양한 다른 실시양태에서, 엔독시펜을 포함하는 조성물은 조성물의 0.01%, 0.02%, 0.03%, 0.04%, 0.05%, 0.06%, 0.07%, 0.08%, 0,09%, 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.6%, 0.7%, 0.8%, 0.9%, 1%, 2%, 3%, 4%, 5%, 10%, 또는 20%의 엔독시펜의 단일 다형체 형태, 예컨대 형태 I, 형태 II, 또는 형태 III wt/wt를 포함한다. 일 측면에서, 엔독시펜의 단일 다형체 형태, 예컨대 형태 I, 형태 II, 또는 형태 III을 포함하는 조성물은 엔독시펜의 제2의 다형체 형태를 추가로 포함한다.
경구 제형
일부 실시양태에서, 본 개시의 약제학적 조성물은 경구 전달을 위해 제형화된다. 경구 사용이 의도되는 조성물은 고체 또는 유체 단위 투여 형태로 제조될 수 있다. 적어도 일부 실시양태에서, 조성물은 경구 전달을 위해 정제, 캐플릿, 캡슐, 환제, 분말, 트로키, 엘릭시르, 현탁액, 시럽, 웨이퍼, 츄잉 검, 당의정, 로젠지 등으로 제형화된다.
일부 실시양태에서, 경구 투여 형태는 고체 경구 투여 형태, 예컨대 정제, 캐플릿, 및 캡슐이다. 일부 실시양태에서, 캡슐은 경질 캡슐 또는 연질 캡슐이다. 다른 실시양태에서, 캡슐은 젤라틴 캡슐, 젤라틴-비함유 캡슐, "캡-인-캡(cap-in-cap)" 캡슐, 알기네이트 캡슐, 히드록시프로필메틸 셀룰로스(HPMC) 캡슐, 폴리비닐 알콜(PVA) 캡슐, 히프로멜로스 캡슐, 또는 전분 캡슐이다.
부형제
일부 실시양태에서, (Z)-엔독시펜 또는 그의 염을 포함하는 경구 조성물은 하나 이상의 부형제를 추가로 포함한다. 일부 실시양태에서, 엔독시펜 또는 그의 다형체를 포함하는 경구 조성물은 하나 이상의 부형제를 추가로 포함한다. 따라서, 경구 투여용으로 설계된 조성물은 본원에 개시되는 바와 같이 불활성 또는 활성 부형제로 또는 식용 담체로 제조될 수 있다.
다양한 실시양태에서, 본원에 제공된 조성물은 약 1중량% 내지 약 99.99중량%, 약 5중량% 내지 약 95중량%, 약 5중량% 내지 약 90중량%, 약 10중량% 내지 약 80중량%, 약 15중량% 내지 약 70중량%, 약 20중량% 내지 약 60중량%, 약 30중량% 내지 약 95중량%, 약 50중량% 내지 약 90중량%, 약 60중량% 내지 약 90중량%, 약 60중량% 내지 약 80중량%, 또는 약 70중량% 내지 약 80중량%의 하나 이상의 부형제를 포함한다. 특정 실시양태에서, 본원에 제공된 조성물은 약 99.99중량%, 약 95중량%, 약 90중량%, 약 85중량%, 약 80중량%, 약 75중량%, 약 70중량%, 약 65중량%, 약 60중량%, 약 55중량%, 또는 약 50중량%의 하나 이상의 부형제를 포함한다. 특정 실시양태에서, 본원에 제공된 조성물은 약 99.99중량%, 약 99중량%, 약 98중량%, 약 97중량%, 약 96중량%, 약 95중량%, 약 94중량%, 약 93중량%, 약 92중량%, 약 91중량%, 약 90중량%, 약 89중량%, 약 88중량%, 약 87중량%, 약 86중량%, 또는 약 85중량%의 하나 이상의 부형제를 포함한다. 특정 실시양태에서, 본원에 제공된 조성물은 약 85중량%, 약 84중량%, 약 83중량%, 약 82중량%, 약 80중량%, 약 79중량%, 약 78중량%, 약 77중량%, 약 76중량%, 약 75중량%, 약 74중량%, 약 73중량%, 약 72중량%, 약 71중량%, 약 70중량%, 약 69중량%, 약 68중량%, 약 67중량%, 약 66중량%, 또는 약 65중량%의 하나 이상의 부형제를 포함한다. 특정 실시양태에서, 본원에 제공된 조성물은 약 55중량%, 약 54중량%, 약 53중량%, 약 52중량%, 약 51중량%, 약 50중량%, 약 49중량%, 약 48중량%, 약 47중량%, 약 46중량%, 또는 약 45중량%의 하나 이상의 부형제를 포함한다. 특정 실시양태에서, 본원에 제공된 조성물은 약 30중량%, 약 29중량%, 약 28중량%, 약 27중량%, 약 26중량%, 약 25중량%, 약 24중량%, 약 23중량%, 약 22중량%, 약 21중량%, 또는 약 20중량%의 하나 이상의 부형제를 포함한다.
경구 투여를 위해 제형화되는 조성물에 사용될 수 있는 부형제의 예가 본원에 제공되며, 벌킹제, 결합제, 충전제, 붕해제, 윤활제, 활주제, 방출 조절제, 장용 코팅제, 필름 형성제, 가소제, 착색제, 스위트너, 향미제 등, 또는 그의 임의의 조합 중 하나 이상을 포함하지만 이에 제한되지 않을 수 있다.
본원에서 제공되는 약제학적 조성물에 사용하기에 적합한 결합제는 수크로스, 전분, 예컨대 옥수수 전분, 감자 전분, 또는 전분, 예컨대 전분 페이스트, 전호화 전분, 및 전분 1500, PEG 6000, 메토셀, 왈로셀 HM, 루비텍, 루비카파롤락탐, 아비셀, SMCC, UNIPURE, 젤라틴, 천연 및 합성 검, 예컨대 아카시아, 나트륨 알기네이트, 알긴산, 다른 알기네이트, 트라가카트, 구아 검, 셀룰로스 및 그의 유도체(예컨대, 에틸 셀룰로스, 셀룰로스 아세테이트, 카르복시메틸 셀룰로스 칼슘, 나트륨 카르복시메틸 셀룰로스), 폴리비닐 피롤리돈 메틸 셀룰로스, 폴리비닐 피롤리돈 히드록시프로필 메틸 셀룰로스(예컨대, Nos. 2208, 2906, 2910), 미세결정질 셀룰로스, 및 그의 혼합물을 포함하지만 이에 제한되지 않는다. 미세결정질 셀룰로스의 적합한 형태는 아비셀 PH 101, 아비셀 PH 103 아비셀 RC 581, 아비셀 PH 105(입수처: FMC Corporation, American Viscose Division, Avicl Sales, Marcus Hook, Pa.)로 시판되는 물질, 및 그의 혼합물을 포함하지만 이에 제한되지 않는다. 일부 실시양태에서, 결합제는 미세결정질 셀룰로스와 나트륨 카르복시메틸 셀룰로스의 혼합물이다. 적합한 무수 또는 낮은 수분 부형제 또는 첨가제는 아비셀 PH 103 및 전분 1500 LM을 포함한다.
본원에서 제공되는 약제학적 조성물에 사용하기에 적합한 충전제의 예는 활석, 탄산칼슘(예컨대, 과립 또는 분말), 슈가, 예컨대 덱스트로스, 수크로스, 락토스, 염, 예컨대 탄산칼슘, 인산칼슘, 탄산나트륨, 인산나트륨, 전분, 미세결정질 셀룰로스, 분말화된 셀룰로스, 셀룰로스 베이스, 예컨대 메틸 셀룰로스, 카르복시메틸 셀룰로스 덱스트레이트, 카올린, 만니톨, 규산, 소르비톨, 전분, 전호화 전분, 및 그의 혼합물을 포함하지만 이에 제한되지 않는다.
조성물 중의 하나 이상의 결합제 또는 충전제는 전형적으로 조성물 또는 투여 형태의 약 10% 내지 약 99%(wt/wt)로 존재한다. 일부 실시양태에서, 조성물 중의 결합제 및/또는 충전제는 조성물의 약 15% 내지 99%, 약 20% 내지 60%, 약 25% 내지 55%, 약 30% 내지 50%, 약 35% 내지 60%, 약 50% 내지 99%(wt/wt)를 포함한다.
붕해제는 수성 환경에 노출될 때 붕해하는 정제를 제공하기 위해 조성물 중에 사용될 수 있다. 너무 많은 붕해제를 함유하는 정제는 저장 시 붕해할 수 있고, 너무 적게 함유하는 것은 원하는 속도로 또는 원하는 조건에서 붕해하지 않을 수 있다. 따라서, 활성 성분의 방출을 불리하게 변경하기에는 너무 많지도 너무 적지도 않은 충분한 양의 붕해제를 사용하여 고체 경구 투여 형태를 형성해야 한다. 일부 실시양태에서, 붕해제는 경구 고체 투여 형태에서 깊어서 붕해를 지연시킨다. 사용되는 붕해제의 양은 제형의 유형에 기초하여 다양하며, 당업자에게 쉽게 구별될 수 있다.
전형적 조성물은 0.5% 내지 15%(wt/wt)의 붕해제를 포함한다. 일부 실시양태에서, 조성물은 조성물 중에 1% 내지 5%(wt/wt)의 붕해제를 포함한다. 또 다른 실시양태에서, 붕해제는 조성물의 1% 내지 25%, 2% 내지 20%, 5% 내지 15%, 8% 내지 12%, 또는 약 10%(wt/wt)이다.
본원에서 제공되는 약제학적 조성물에 사용될 수 있는 붕해제는 한천, 알긴산, 탄산칼슘, 미세결정질 셀룰로스, 크로스카르멜로스 나트륨, 크로스포비돈, 폴락릴린 칼륨, 나트륨 전분 글리콜레이트, 감자 또는 타피오카 전분, 전호화 전분, 다른 전분, 점토, 다른 알긴, 다른 셀룰로스, 검, 및 그의 혼합물을 포함하지만 이에 제한되지 않는다.
본원에서 제공되는 약제학적 조성물에 사용될 수 있는 윤활제는 칼슘 스테아레이트, 마그네슘 스테아레이트, 무기질유, 경유(light mineral oil), 글리세린, 소르비톨, 만니톨, 폴리에틸렌 글리콜, 다른 글리콜, 스테아르산, 나트륨 라우릴 술페이트, 활석, 수소첨가 식물성유(예컨대, 낙화생유, 면실유, 해바라기유, 참기름, 올리브유, 옥수수유, 및 대두유), 아연 스테아레이트, 마그네슘 스테아레이트 또는 칼륨 스테아레이트, 에틸 올레에이트, 에틸 라우레에이트, 한천, 및 그의 혼합물을 포함하지만 이에 제한되지 않는다. 추가의 윤활제는, 예컨대 실로이드 실리카 겔(AEROSIL 200, 제조사: W.R. Grace Co., 메릴랜드주 볼티모어), 합성 실리카의 컨주게이션된 에어로졸(시판사: Degussa Co., 텍사스주 플라노), CAB O SIL(발열성 이산화규소 제품, 시판사: Cabot Co., 메사추세츠주 보스톤), Q7-9120(Dow Corning), 및 그의 혼합물을 포함한다. 조금이라도 사용되는 경우, 윤활제는 전형적으로 그들이 혼입되는 조성물 또는 투여 형태의 1%(wt/wt) 미만의 양으로 사용된다. 또 다른 실시양태에서, 윤활제는 조성물의 0.1% 내지 3%, 예컨대 0.5% 내지 1%(wt/wt)이다.
경구 투여 형태, 예컨대 캡슐, 캐플릿 또는 정제의 껍질의 부드러움 또는 유연성을 제어하기 위해 가소제가 첨가될 수 있으며, 따라서 경구 투여 형태 상의 코팅물의 pH 민감성 물질의 기계적 특성을 개선할 수 있다. 적합한 가소제는 석유 오일(예컨대, 파라핀 공정 오일, 나프탈렌 공정 오일, 및 방향족 공정 오일), 스쿠알렌, 스쿠알란, 식물유(예컨대, 올리브유, 카멜리아 오일, 피마자유, 톨유, 및 낙화생유), 실리콘 오일, 이염기산 에스테르(예컨대, 디부틸 프탈레이트, 및 디옥틸 프탈레이트), 액체 고무(예컨대, 폴리부텐 및 액체 이소프렌 고무), 액체 지방산 에스테르(예컨대, 이소프로필 미리스테이트 ISM), 헥실 라우레이트, 디에틸 세바케이트, 및 디이소프로필 세바케이트, 트리에틸 시트레이트, 트리아세틴, 디에틸렌 글리콜, 폴리에틸렌 글리콜, 폴리프로필렌 글리콜, 프탈레이트, 소르비톨, 글리콜 살리실레이트, 크로타민톤, 및 글리세린 또는 그의 혼합물을 포함하지만 이에 제한되지 않는다. 가소제의 양은 약제학적 제제의 화학적 조성에 따라 다양할 수 있다. 일 실시양태에서, 적어도 하나의 가소제는 소르비톨, 디메틸 이소소르바이드, 또는 글리세롤이다. 또 다른 실시양태에서, 가소제는 조성물의 1% 내지 10%, 예컨대 3% 내지 5%(wt/wt)이다.
활주제의 예는 콜로이드성 이산화규소, 셀룰로스, 칼슘 포스페이트, 이 또는 삼-염기물 등을 포함하지만 이에 제한되지 않는다.
스위트너 또는 감미제의 예로서 수크로스, 사카린, 덱스트로스, 말토스, 슈가 대체물, 아스파탐, 자일리톨, 만니톨, 시클라메이트, 수크랄로스, 말티톨, 소르비톨, 아세술팜 K 등이 포함된다.
향미제의 예는 페퍼민트, 메틸 살리실레이트, 페퍼민트, 스피어민트, 메틸 살리실레이트, 라즈베리, 레드 베리, 스트로베리, 파인애플, 오렌지, 체리 등을 포함한다.
본원에 개시되는 바와 같은 경구 전달을 위해 제형화되는 조성물, 예컨대 정제, 캐플릿, 및 캡슐은 위장관에서 엔독시펜 또는 그의 염을 포함하는 조성물의 붕해 및 흡수를 제어 또는 지연시키기 위해 하나 이상의 장용 코팅제, 방출 조절제 또는 필름 형성제로 코팅될 수 있으며, 따라서 보다 긴 기간에 걸처 지속적인 작용을 제공한다. 따라서, 일부 실시양태에서, 정제는 장용 정제일 수 있거나, 캐플릿은 장용 캐플릿일 수 있거나, 캡슐은 장용 캡슐일 수 있다. 본 개시의 장용 정제, 장용 캐플릿, 또는 장용 캡슐은 당해 분야에 공지된 기술에 의해 제조될 수 있다.
본원에 개시된 약제학적 제제는 방출 조절제를 포함할 수 있다. 사용하기에 적합한 방출 조절제의 예는 pH-의존적 중합체, 산-불용성 중합체, 메틸 아크릴레이트-메타크릴산 공중합체, 셀룰로스 아세테이트 프탈레이트(CAP), 셀룰로스 아세테이트 숙시네이트, 히드록시프로필 메틸 셀룰로스 프탈레이트, 히드록시프로필 메틸 셀룰로스 아세테이트 숙시네이트(히프로멜로스 아세테이트 숙시네이트), 폴리비닐 아세테이트 프탈레이트(PVAP), 메틸 메타크릴레이트-메타크릴산 공중합체, 셸락, 셀룰로스 아세테이트 트리멜리테이트, 나트륨 알기네이트, 제인, 왁스(합성 왁스를 포함), 미세결정질 왁스, 파라핀 왁스, 카나우바 왁스, 및 밀랍; 폴리에톡실화된 피마자유 유도체, 수소첨가 오일, 글리세릴 모노-, 디- 트리베네이트, 글리세릴 모노스테아레이트, 글리세릴 디스테아레이트, 장쇄 알콜, 예컨대 스테아릴 알콜, 세틸 알콜, 및 폴리에틸렌 글리콜; 및 그의 혼합물을 포함하지만 이에 제한되지 않는다. 일부 실시양태에서, 시간 지연 물질, 예컨대 글리세릴 모노스테아레이트 또는 글리세릴 디스테아레이트가 사용될 수 있다. 다른 실시양태에서, 방출 조절 시약은 소화 가능한 왁스 물질, 예컨대 경질 파라핀 왁스이다.
일부 실시양태에서, 조성물은 pH-의존적 중합체, 예컨대 산 불용성 중합체 중 하나 이상을 포함할 수 있다. pH-의존적 중합체는 pH 5.0 초과에서는 점진적으로 투과성이지만 pH 5.0 미만에서는 불투과성인 반면, 산 불용성 중합체는 중성 내지 약알칼리성 조건에서 가용성이 된다. 이러한 방출 조절 중합체는 상부 소장 및 결장을 표적으로 한다. 산 불용성 중합체의 비제한적 예는 셀룰로스 아세테이트 프탈레이트, 셀룰로스 아세테이트 부티레이트, 히드록시프로필 메틸 셀룰로스 프탈레이트, 알젠산 염, 예컨대 나트륨 또는 칼륨 알기네이트, 셸락, 펙틴, 아크릴산-메틸아크릴산 공중합체(분말 또는 30% 수성 분산액으로 Rohm America Inc.(뉴저지주 피스카타웨이)로부터 상품명 EUDRAGIT® L 및 EUDRAGIT® S 하에 상업적으로 입수 가능; 또는 30% 분산액으로 Eastman Chemical Co.(테네시주 킹스포트)로부터 상품명 EASTACRYL® 하에 상업적으로 입수 가능)를 포함한다. 추가의 예는 EUDRAGIT® L100-55, EUDRAGIT® L30D-55, EUDRAGIT® L100, EUDRAGIT® L100 12,5, EUDRAGIT® S100, EUDRAGIT® S12,5, EUDRAGIT® FS 30D, EUDRAGIT® E100, EUDRAGIT® E 12,5, 및 EUDRAGIT® PO를 포함한다. 적어도 하나의 실시양태에서, 조성물은 EUDRAGIT® L100-55를 포함한다. L100-55. EUDRAGIT® RS 및 RL 및 EUDRAGIT®NE 및 NM도 또한 본 개시의 목적 상 유용한 중합체이다. 일부 실시양태에서, 조성물은 EUDRAGIT ® L30D 55를 포함한다. 또 다른 실시양태에서, 제제는 EUDRAGIT® FS 30D를 포함한다. 당업자는 본원에 열거된 적어도 일부의 산 불용성 중합체가 또한 생분해가능할 것이라는 것을 인식할 것이다.
경구 투여 형태의 시간 지연 또는 지연 방출 약제학적 제제를 위해, 글리세릴 모노스테아레이트, 글리세릴 디스테아레이트, 및 산-불용성 중합체, 예컨대 폴리메타크릴레이트 pH-민감성 중합체-기반 코팅물이 (예컨대, 캡슐, 캐플릿, 및 정제의 장용 코팅을 위한 코팅 물질, 즉 장용 코팅제로서) 사용될 수 있다. 지연 방출 경구 투여 형태의 상업적 공급원, 예컨대 캅수겔(Capsugel, USA)로부터의 히프로멜로스(HPMC)로 제조된 DRCaps가 이용 가능하다. 이러한 지연 방출 경구 투여 형태는 내산성이며, 적어도 30분, 예컨대 적어도 1시간, 적어도 1.5시간, 또는 적어도 2시간 동안 위에 보여지는 바와 같이 산도에 저항할 수 있다. 이러한 지연 방출 경구 투여 형태는 장(소장, 대장/결장 등)에서 엔독시펜 또는 그의 염의 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80% 또는 적어도 90%를 방출할 수 있다.
본 개시의 일 측면에서, 장용 정제, 장용 캐플릿, 및 장용 캡슐은 코팅되지 않을 수 있다. 본질적으로 장 캡슐 기술(예컨대, 캅수겔로부터 입수 가능한 EnTrinsic Drug Delivery)을 이용하는 장 능력을 갖는 경질의 비코팅된 캡슐이 본 개시의 목적에 적합하다.
다양한 실시양태에서, 장용 정제는 (Z)-엔독시펜 또는 그의 염의 자유 유동 분말로 제조된 경질 정제이다. 다양한 실시양태에서, 장용 캡슐은 (Z)-엔독시펜 또는 그의 염의 자유 유동 분말로 제조되는 캡슐이다. 다양한 실시양태에서, 장용 정제는 엔독시펜 또는 그의 다형체의 자유 유동 분말로 제조되는 경질 정제이다. 다양한 실시양태에서, 장용 캡슐은 엔독시펜 또는 그의 다형체의 자유 유동 분말로 제조되는 캡슐이다.
일부 실시양태에서, 장용 캡슐은 비-동물 기반 캡슐, 예컨대 히프로멜로스 캡슐이다(예컨대, 상업적으로 입수 가능한 자가-겔화 Vcaps, VCaps Plus, VCaps 엔테릭, 캅수겔로부터의 엑스셀로도스(Xcellodose), ENCODE 결장 전달 기술, 및 EnTrinsicTM 약물 전달 기술을 이용하여 제조되는 다른 장용 캡슐). 경구 고체 투여 형태의 장용 형태를 제형화하기 위한 당해 분야에 공지되고 상업적으로 입수 가능한(예컨대, Qualicaps(USA), Nutrascience(USA) 등) 다른 기술이 또한 이용될 수 있다. 적어도 하나의 실시양태에서, 캡슐은 (Z)-엔독시펜 유리 염기 또는 그의 염이 캡슐로 순수하게(neat) 충전됨을 의미하는 API-인-캡슐(API-in-capsule)이다. 이러한 API-인-캡슐 경구 투여 형태에서, 활성 성분인 (Z)-엔독시펜 또는 그의 염은 자유 유동 분말 또는 미분화된 분말일 수 있다. 투여 형태가 캡슐인 경우, 적어도 하나의 실시양태에서, 캡슐은 이음이 없는 캡슐 또는 띠가 있는 캡슐일 수 있다.
암 성장 속도를 더욱 감소시킬 수 있는 항암 치료제, 예컨대 엔독시펜의 빠른 흡수 및 생체이용률이 매우 바람직하다. 일 측면에서, 본 개시는 조성물이 특정 약동학(PK) 특성을 위해 제형화되는 것을 제공한다.
일 측면에서, 엔독시펜의 최대 및 항정 상태 혈장 수준의 빠른 달성이 본 개시의 특정 측면이다. 본 개시는 조성물 투여 후 2 내지 30시간 이내, 3 내지 20시간 이내, 2 내지 10시간 이내 또는 4 내지 8시간 이내 범위에서 엔독시펜의 최대 혈장 수준을 달성하는 조성물을 제공한다. 따라서, 일부 실시양태에서, 엔독시펜의 최대(피크) 혈장 수준까지의 시간은 조성물 투여 후 2 내지 10시간의 범위이다. 일부 실시양태에서, 엔독시펜의 최대 혈장 수준까지의 시간은 본원에 개시된 조성물 투여 후 4 내지 8시간의 범위이다.
엔독시펜의 항정 상태 혈장 수준의 빠른 달성이 또한 매우 바람직하며, 본 개시의 조성물은 빠르게 항정 상태를 달성하는 엔독시펜의 다형체 형태, (Z)-엔독시펜 또는 그의 염를 포함하는 조성물이 투여되는 대상체에서 엔독시펜의 혈장 수준을 제공할 수 있다. 항정 상태 혈장 수준은 제7일 내지 제21일에 달성될 수 있다. 일부 실시양태에서, 항정 상태 혈장 수준은 본원에 개시된 조성물의 일일 투여 시 제7일(도 5)에 달성될 수 있다.
일 측면에서, 본 개시는 본원에 개시된 조성물로부터 방출되는 순환 엔독시펜이 타목시펜보다 빠르게 제거될 수 있다는 것을 제공한다. 타목시펜의 터미널(terminal) 제거 반감기는 5-7일인 것으로 알려져 있으며(Jordan C. Steroids. 2007 Nov; 72(13): 829-842), 타목시펜의 피크 농도 시간은 투여 후 대략 5시간이다. 본원에 개시된 조성물로부터 방출되는 엔독시펜은 타목시펜보다 상당히 더 낮은 30 내지 60시간 범위의 터미널 제거 반감기를 가질 수 있다. 일부 실시양태에서, 평균 반감기는 40 내지 53시간의 범위이다. AUC24hr(제21일)/AUC0-inf(제1일)의 평균 비율은 전형적으로 1 mg 내지 4 mg의 (Z)-엔독시펜, 또는 그의 염을 포함하는 조성물에 대해 0.7 내지 1.2의 범위이다. 따라서, 본원에 개시된 조성물로부터 방출되는 엔독시펜의 축적은 지속된 치료 동안 크게 변화되지 않는다.
또 다른 측면에서, 본원에 개시된 조성물은 치료적으로 유효한 엔독시펜의 흡수를 달성한다.
곡선 아래 면적 AUC(0-24hr)("AUC24hr")는 투여 시간(0 hr)으로부터 24시간 동안 약물에 대한 대상체의 총 노출을 기술한다. (Z)-엔독시펜 또는 그의 염을 포함하는 조성물은 전형적으로 1 mg 내지 4 mg의 (Z)-엔독시펜을 포함하는 조성물의 초기(제1) 용량의 제1일에 150 hr*ng/mL 내지 600 hr*ng/mL의 평균(AUC24hr)을 달성한다. (Z)-엔독시펜 또는 그의 염을 포함하는 조성물은 전형적으로 1 mg 내지 4 mg의 (Z)-엔독시펜을 포함하는 조성물의 초기(제1) 용량의 제21일에 400 hr*ng/mL 내지 2500 hr*ng/mL이 평균 AUC24hr를 달성한다.
분석되는 체액(일반적으로 혈장, 혈액 또는 혈청)에서 순환하는 약물의 시간-평균 농도인 AUC0-inf("AUC0-inf")는 약물에 대한 대상체의 총 노출을 기술한다. 본 개시는 엔독시펜에 대한 대상체의 노출(AUC0-inf)이 용량에 비례할 수 있다는 것을 제공한다. 일부 실시양태에서, AUC0-inf는 200 hr*ng/mL 내지 10000 hr*ng/mL의 범위이다. 다른 실시양태에서, AUC0-inf는 300 hr*ng/mL 내지 8000 hr*ng/mL의 범위이다. 특정 실시양태에서, 1 mg 내지 4 mg의 (Z)-엔독시펜의 투여 범위에 걸처 AUC0-inf는 400 hr*ng/mL 내지 6000 hr*ng/mL의 범위이다(예컨대, 표 17 참조).
본원에 개시된 경구 투여 형태의 용해는 USP 711의 현재 방법에 따른 용해 시험에 의해 시험된다. 일부 실시양태에서, 본원에 개시되는 경구 투여 형태는 위의 산성 환경으로부터 보호되며, 적어도 2시간, 적어도 3시간, 적어도 4시간, 적어도 5시간, 6시간, 적어도 7시간 또는 적어도 8시간 동안 용해되지 않는다. 적어도 하나의 실시양태에서, 경구 투여 형태는 적어도 6시간 동안 엔독시펜을 방출하지 않는다. 또 다른 실시양태에서, 경구 투여 형태는 적어도 2시간 동안 엔독시펜 또는 그의 염을 방출하지 않는다.
다른 실시양태에서, USP 711의 방법으로 시험 시, 본원에 개시된 엔독시펜 또는 그의 염을 포함하는 조성물 중의 (Z)-엔독시펜의 10% 미만이 투여 2시간 후 위에서 방출되거나; (Z)-엔독시펜의 40% 미만이 투여 4시간 후 위에서 방출되거나; (Z)-엔독시펜의 50% 미만이 투여 6시간 후 위에서 방출된다.
또 다른 실시양태에서, USP 711의 방법으로 시험 시, 본원에 개시된 조성물은 투여 후 2시간에 (Z)-엔독시펜의 10% 미만; 투여 후 4시간에 (Z)-엔독시펜의 40% 미만; 및 투여 후 6시간에 (Z)-엔독시펜의 50% 미만을 위에 방출한다.
또 다른 실시양태에서, 조성물은 소장에서 방출하기 위해 제형화되어 엔독시펜의 적어도 10%가 투여 후 4시간 후 방출되거나; 엔독시펜의 적어도 30%가 투여 후 6시간 후 방출되거나; 엔독시펜의 적어도 40%가 투여 후 7시간 후 방출되거나; 엔독시펜의 적어도 50%가 투여 후 8시간 후 방출된다.
추가의 실시양태에서, USP 711의 방법으로 결정 시, 조성물은 투여 후 8시간 후 엔독시펜의 적어도 50%를 결장에 방출하도록 제형화된다.
추가의 실시양태에서, 조성물은 투여 후 4시간 후 엔독시펜의 적어도 20%; 투여 후 6시간 후 엔독시펜의 적어도 40%; 투여 후 7시간 후 엔독시펜의 적어도 60%; 투여 후 8시간 후 엔독시펜의 적어도 80%를 결장에 방출하도록 제형화된다.
경구 투여 형태는 경구 투여에 적합한 임의의 형태, 예컨대 구형(0.05 - 5 mL), 난형(0.05 - 7 mL), 타원형, 배형(0.3 - 5 mL), 원통형, 입방체형, 규칙적인 형 및/또는 불규칙적인 형일 수 있다. 경구 투여 형태는 경구 투여에 적합한 임의의 크기, 예컨대 크기 0, 크기 2 등일 수 있다.
당업자는 또한 본원에 개시된 조성물이 당해 분야에 공지되어 있고 본원에 개시된 하나 이상의 부형제를 원하는 제형 또는 제제에 적합한 임의의 조합으로 포함할 수 있다는 것을 인식할 것이다. 추가의 부형제는 일반적으로 문헌(Remington's The Science and Practice of Pharmacy, Meade Publishing Co., United States Pharmacopeia/National Formulary)에서 찾을 수 있다. 당업자는 당해 분야의 기술 및 지식 및 본원에 기재된 개시 내용에 기초하여 제형의 제조에 필요한 적합한 부형제 및 투여 경로에 적합한 적절한 투여 형태를 선택할 수 있을 것이다. 모든 경우에 있어서, 최종 투여 형태는 제조 및 저장 조건 하에서 멸균되고 안정적이어야한다.
본원에 개시된 고체 투여 조성물의 제형의 경우, 수분 활성도(Aw)가 0.75 미만이므로, 일반적으로 총 호기성 플레이트 카운트(TAC) 및 USP 지표 유기체를 시험할 필요가 없다. 공개문("Microbial Bioburden on Oral Solid Dosage Form," by Jose E. Martinez, Pharmaceutical Technology, February 2002, pages 58 to 70)이 모두 참조로 본원에 포함된다.
또한, 본원에 개시된 조성물의 제형은 또한 0.75 미만의 수분 활성도를 가지므로, 일반적으로 상세한 미생물 시험은 필요하지 않다. TAC는 원료, 공정 중 물질 또는 완제품의 샘플에 존재하는 총 생존 호기성 미생물의 추정치이다. 샘플은 가장 최신 USP 39 <61>, "비멸균 제품의 미생물 검사: 미생물 계수 시험"에 따라 분석된다.
경구 고체 투여 제형(OSDF)에 대해 허용되는 TAC가 경계 및 작용 수준의 관점에서 본 발명의 조성물의 제형에 대해 확립되며, 이는 각각 1000 cfu g/mL, 및 10,000 cfu g/mL일 수 있다. 20,000 cfu g/mL인 TAC는 허용되지 않는 것으로 간주될 수 있다.
다른 제형, 예컨대 0.75 미만의 수분 활성도를 갖는 액체 또는 유체 제형의 경우, USP 지침 62장에 따르는, 지정된 미생물(에스. 아우레우스(S. aureus), 피에스. 아에루기노사(Ps. aeruginosa), 살모넬라(Salmonella), 씨. 알비칸스(C. albicans), 클로스트리디아(Clostridia), 이. 콜라이(E. coli) 및 담즙 내성 그램 음성 박테리아)에 대한 시험을 수행할 필요가 없을 수 있다.
이용 방법
화학식 (I), (II), (III), 및 (IV)의 화합물, 본원에 개시된 엔독시펜 염, 본원에 개시된 엔독시펜의 다형체 형태, 그들을 포함하는 조성물은 이를 필요로 하는 대상체, 예컨대 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 대상체의 치료에 사용하기 위한 약제의 제조에 사용될 수 있다.
본 개시의 조성물은, 의도가 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 대상체를 개선하거나 치유하는 것인 경우, 1차 요법으로, (1차 요법에 대한) 선행보조(neo-adjuvant) 요법의 일부로서, 또는 보조 요법의 일부로서 사용될 수 있다.
특정 실시양태에서, 장애는 호르몬 의존적 유방 장애이다. 다른 실시양태에서, 장애는 호르몬 의존적 생식관 장애이다. 다른 실시양태에서, 대상체는 호르몬 의존적 유방 장애 및 호르몬 의존적 생식관 장애 둘 다를 갖는다. 일부 실시양태에서, 호르몬 의존적 장애는 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암 또는 외음부암이다.
일부 실시양태에서, 유방 장애는 증가된 유방 밀도이다. 예컨대, 유방 장애는 부류 B(이전에는 부류 II), 부류 C(이전에는 부류 III) 또는 부류 D(이전에는 부류 IV) 유방 밀도이다.
일부 실시양태에서, 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애는 성조숙증이다. 다른 실시양태에서, 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애는 맥쿤-알부라이트 증후군이다.
일부 실시양태에서, 유방 장애는 여성형 유방증이다. 일부 실시양태에서, 여성형 유방증은 기저 질환에 이차적으로 나타난다. 따라서, 일부 실시양태에서, 대상체는 또한 전립선암, 경화증 및 간 질환, 남성 성선기능저하증, 갑상선기능항진증, 신부전 및 혈액투석을 받는 환자, 또는 제I형 당뇨병으로 이루어진 군으로부터 선택되는 기저 질환을 갖는다. 특정 실시양태에서, 대상체는 기저 질환으로 전립선암을 가지며 여기서 대상체는 여성형 유방증을 갖거나 가질 위험이 있다.
특정 실시양태에서, 유방암은 DCIS, LCIS, ILC, IDC, MIC, 염증성 유방암, ER-양성(ER+) 유방암, HER2+ 유방암, 선양 낭성(샘낭성) 암종, 낮은 등급 선편평상피 암종, 수질성 암종, 점액 (또는 콜로이드) 암종, 유두 암종, 관상 암종, 화생 암종, 또는 미세유두 암종이다. 적어도 하나의 실시양태에서, 단일 유방암 종양은 상기의 조합이거나 침습적 암과 제자리 암의 혼합일 수 있다.
본 개시는 수술 또는 방사선요법과 같은 치료의 국소적 양식에 의해 치료할 수 없는 대상체에서의 진행성 및/또는 공격성 신생물, 즉 현성 질환, 전이성 질환, 또는 국소 진행 질환을 포함하여 종양 발달 및 진행의 다양한 단계에서 본원에 개시된 화합물 및 조성물의 사용을 고려한다. 따라서, 일부 실시양태에서, 유방암은 전암, 초기 단계 암, 비-전이성 암, 전-전이성 암, 또는 국소 진행 암이다. 적어도 하나의 실시양태에서, 유방 장애는 전이성 암이다. 일부 실시양태에서, 대상체는 추가로 전립선암을 갖는다.
이러한 장애의 치료를 위한 현재의 선택은 심각한 유해 효과, 저조한 환자 순응도 및 대상체에서 보여지는 낮은 혈장 엔독시펜 수준에 기인한 약물에 대한 내성에도 불구하고 타목시펜으로 남아 있다. 이러한 대상체는, 예컨대 CYP2D6, CYP3A4, 또는 CYP2C9에서 CYP 유전자 돌연변이를 갖거나(이는 타목시펜을 그의 활성 대사산물인 엔독시펜으로 대사할 수 없게 함) 충분한 타목시펜 흡수를 막는 (또는 감소시키는) 낮은 또는 기능장애의 에스트로겐 수용체와 같은 다수의 이유로, 아직까지 확인되지 않은 다른 이유로 타목시펜으로 투여 시 낮은 엔독시펜 수준을 가질 수 있다. 20 mg의 경구 타목시펜으로 투여된 대상체에서 혈장 타목시펜의 보고된 치료 수준은 ≥30 nM이다(Lyon et al. Genet Med. 2012 Dec;14(12):990-1000). 대상체에서 낮은 혈장 엔독시펜의 기초가 되는 메카니즘에도 불구하고, 본 개시의 조성물은 대상체가 낮은 엔독시펜을 갖거나 대상체가 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖거나 가질 위험이 있는 임의의 상태에서 유용하다. 따라서, 본 개시의 조성물은 타목시펜 내성, 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애의 치료에 특히 중요할 수 있다.
본원에는 특정 실시양태에서 약제학적 조성물이 특히 유용한 환자 집단이 제공된다. 본 개시의 조성물은 또한 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖는 타목시펜 불응성 환자의 치료에 특히 중요하다. 따라서, 일부 실시양태에서, 본원에 개시된 조성물은 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 타목시펜 불응성 또는 타목시펜 내성 대상체의 치료에 유용하다. 일부 실시양태에서, 본원에 개시된 용량으로 이러한 대상체에게 투여되는 엔독시펜 염, 예컨대 엔독시펜 글루코네이트을 포함하는 조성물이 유리할 것이다.
또한, 도네용(Donneyong) 등은 티목시펜과 프로작(Prozac) 및 팍실(Paxil)(파록세틴)과 같은 선택적 세로토닌 재흡수 억제제(SSRI) 약물 사이에 약물 상호작용이 존재하고 유방암 대상체에게 유해하다는 것을 입증하였다(Donneyong et al. BMJ 2016;354:i5014). SSRI 약물은 SSRI 약물에 대한 대상체에서 타목시펜의 엔독시펜으로의 간 대사작용을 감소시키거나 정지시킨다. 따라서, 본원에는 특정 실시양태에서 본 개시의 조성물로의 처리로 이익을 얻을 수 있는 SSRI 약물로 치료되고 있거나 이로 치료될 환자 집단이 제공된다.
경구로 투여되는 본원에 개시된 조성물은 대상체의 혈장 엔독시펜을 30 nM 초과의 항정 상태 수준, 예컨대 30 nM 내지 80 nM 범위의 수준 또는 30 nM 내지 300 nM 범위의 수준으로 유지한다. 일부 실시양태에서, 혈장 항정 상태 엔독시펜 수준은 >40 nM로 유지된다. 혈장 엔독시펜을 30 nM 초과의 항정 상태 수준으로 유지하는 것은 30 nM 미만의 혈장 엔독시펜 수준에서 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애, 특히 유방암의 재발(악화) 가능성을 감소시킨다는 점에서 유리하다. (16 nM 미만의 혈장 엔독시펜 수준을 갖는) 타목시펜의 저조한 대사자, (27 nM 미만의 혈장 엔독시펜 수준을 갖는) 타목시펜의 중간 대사자인 대상체에 대해 본원에 개시된 조성물을 투여하는 것이 특히 유리하다. 또한, 항우울 약물, 예컨대 SSRI 약물, 예컨대 시탈로프람(셀렉사), 에스시탈로프람(렉사프로), 플루옥세틴(프로작), 파록세틴(팍실, 펙세바), 세르트랄린(졸로프트), 빌라조돈(비이브리드) 등으로 치료되고 있거나 이로 치료될 대상체, 예컨대 우울증을 갖거나 가질 수 있는 대상체에 대해 유리하다.
대상체가 타목시펜 불응성인지의 여부는 대상체에게 초기 용량의 타목시펜을 투여하고 대상체의 혈장 엔독시펜 항정 상태 수준을 결정함으로써 결정될 수 있다. 타목시펜이 투여되는 대상체에서 혈장 엔독시펜 항정 상태 수준은 타목시펜 불응성 대상체에 대한 바이오마커로서 역할을 한다. 혈장 엔독시펜 수준(급성 및/또는 항정 상태)은 대상체에게 타목시펜을 투여한 후 대상체로부터 수집되는 혈액 샘플일 수 있는 시험 샘플을 대상체로부터 수득함으로써 결정될 수 있다. 혈장 또는 혈청이 바이오마커 엔독시펜 수준을 시험하기 위해 혈액 샘플로부터 수득될 수 있다. 초기 용량은 적어도 1일, 2일, 3일, 15일, 1주, 2주, 4주, 1개월, 2개월, 3개월, 4개월, 5개월, 또는 6개월 동안 매일 타목시펜을 투여하는 단계를 포함할 수 있다. 또한, 대상체에게 적어도 1일, 2일, 3일, 15일, 1주, 2주, 4주, 1개월, 2개월, 3개월, 4개월, 5개월, 6개월, 1년, 2년, 3년, 4년, 5년 또는 10년 동안 매일 타목시펜을 포함하는 제1 조성물이 투여될 수 있다.
대상체의 혈장 엔독시펜 항정 상태 수준은 시험 샘플에서 엔독시펜을 측정함으로써 결정될 수 있다. 대상체의 혈장 엔독시펜 항정 상태 수준은 참조 혈장 엔독시펜 수준과 비교된다. 본 개시의 목적 상, 참조 혈장 수준은 30 nM이다. 대상체의 혈장 엔독시펜 수준이 30 nM 미만인 것으로 결정되는 경우, 대상체는 타목시펜 불응성으로 정의된다. 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖거나 가질 위험이 있을 수 있는 이러한 타목시펜 불응성 대상체는 대상체에게 본원에 개시된 (Z)-엔독시펜 또는 그의 염, 또는 본원에 개시된 엔독시펜의 다형체 형태를 포함하는 경구 조성물을 투여함으로써 치료된다. 일부 실시양태에서, 이러한 대상체에게 투여되는 조성물은 (Z)-엔독시펜 유리 염기를 포함한다. 일부 실시양태에서, 이러한 대상체에게 투여되는 조성물물은 엔독시펜의 다형체 형태, 예컨대 형태 I, 형태 II 또는 형태 III을 포함한다. 다른 실시양태에서, 이러한 대상체에게 투여되는 조성물은 (Z)-엔독시펜 D-글루코네이트, (Z)-엔독시펜 L-글루코네이트, (E)-엔독시펜 D-글루코네이트, (E)-엔독시펜 L-글루코네이트, 또는 그의 조합으로 이루어진 군으로부터 선택되는 엔독시펜 글루코네이트를 포함한다. 다른 실시양태에서, 엔독시펜을 포함하는 조성물은 엔독시펜 HCl 또는 엔독시펜 시트레이트이다. 또한, 본 개시는 대상체의 혈장 엔독시펜 수준을 주기적으로 또는 필요한 경우 추적하거나 모니터링하는 것을 고려한다. 필요한 경우, 초기 용량의 타목시펜이 투여된 대상체는 시험 결과에 기초하여 지속적으로 엔독시펜을 포함하는 조성물을 투여함으로써 그의 또는 그녀의 혈장 엔독시펜 항정 상태 수준이 조정될 수 있다.
일부 실시양태에서, 대상체의 타목시펜 불응성 상태는 대조군 또는 정상적인 대상체에서 보여지는 참조 타목시펜-대사산물 프로파일과 비교되는 대상체의 타목시펜-대사산*타목시펜 대사산물 프로파일을 결정함으로써 결정될 수 있다. 참조 타목시펜-대사산물 프로파일과 비교하여 대상체의 타목시펜-대사산물 프로파일에서 낮은 혈장 엔독시펜 수준을 갖는 대상체에게 엔독시펜 또는 그의 염을 포함하는 경구 조성물이 투여된다. 이러한 조성물은 합성적으로 제조되는 엔독시펜을 포함할 수 있다.
혈장 엔독시펜은 당해 분야에 공지된 임의의 방법으로 측정될 수 있다. 시험 샘플 중의 혈장 엔독시펜의 수준은 대상체의 유전자, DNA, RNA, 단백질, 타목시펜-대사산*타목시펜 대사산물 프로파일 또는 그의 조합에 기초하여 결정될 수 있다. 타목시펜-대사산물 프로파일은 적어도 타목시펜, 4-OHT, N-데스메틸타목시펜, 및/또는 엔독시펜을 포함할 수 있다. 일부 실시양태에서, 시험 샘플 중의 혈장 엔독시펜의 수준 및/또는 타목시펜-대사산물 프로파일은 고성능 액체 크로마토그래피(HPLC), 가스 크로마토그래피 질량 분광측정(GC-MS), 액체 크로마토그래피 질량 분광측정(LC-MS), 액체 크로마토그래피 탠덤 질량 분광측정(LC-MS/MS), 면역조직화학(IHC), 폴리머라제 연쇄 반응(PCR), 정량적 PCR(qPCR) 등에 의해 측정된다. 일부 실시양태에서, 타목시펜-대사산*타목시펜 대사산물 프로파일은 대상체의 유전자 조성에 기초하여 예측된다. 일부 실시양태에서, 대상체의 CYP 유전형은 CYP2D6, CYP3A4, CYP2C9 유전자의 분석을 포함하지만 이에 제한되지 않는다. 일부 실시양태에서, 대상체의 에스트로겐 수용체 수준이 분석될 수 있다. 다른 실시양태에서, 혈장 엔독시펜의 결정은 제3자 실험실에서 수행될 수 있다.
따라서, 본원에는 대상체에게 엔독시펜 또는 그의 염을 포함하는 조성물을 투여함으로써 이를 필요로 하는 대상체에서 혈장 엔독시펜을 30 nM 초과의 수준으로 유지하는 방법이 제공된다. 일부 실시양태에서, 대상체의 혈장 엔독시펜 수준은 30 nM 초과의 항정 상태 수준으로 유지된다. 일부 실시양태에서, 대상체의 혈장 엔독시펜 수준은 30 nM 내지 300 nM(예컨대, 30 nM 내지 200 nM, 또는 30 nm 내지 80 nM) 범위의 항정 상태 수준으로 유지된다. 일부 실시양태에서, 대상체의 혈장 엔독시펜 수준은 >40 nM의 항정 상태 수준으로 유지된다.
또 다른 측면에서, 대상체는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 나타내거나 모니터링할 수 있는 그들의 바이오마커 프로파일에 대해 시험되는 그들의 시험 샘플을 가질 수 있다. 이러한 바이오마커는 당해 분야에 공지되어 있고, 비제한적 예로서 바이오마커, 예컨대 CYP2D6, BRCA-1, BRCA-2, ER, PR, Her2, uPA, PAI, Tf, p53, Ki67, 시토케라틴, 암 종양 항원, 및 Mammaprint, OncotypeDx, PAM50, EndoxPredict, MammoStrat, 및 다른 진단 및 예후 시험에 의해 측정되는 바이오마커를 포함한다. 대상체가 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있다는 것을 나타내는 바이오마커 프로파일을 갖는 대상체에게 본원에 개시된 조성물이 투여될 수 있다. 일 측면에서, 본 개시는 대상체의 타목시펜 불응성 또는 타목시펜 내성 상태를 결정하는 단계 및 대상체에게 본원에 기재된 조성물을 투여하는 단계를 포함하여, 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 대상체를 치료하는 방법을 제공한다.
일부 측면에서, 본원에는 대상체에게 엔독시펜, 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여, 타목시펜 불응성 또는 타목시펜 내성 대상체를 치료하는 방법이 제공된다.
일부 실시양태에서, 본원에는 대상체에게 엔독시펜, 또는 그의 염을 포함하는 경구 조성물을 투여하는 단계를 포함하여, 호르몬 민감성 유방 장애, 호르몬 민감성 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 타목시펜 불응성 대상체를 치료하는 방법이 제공되며, 여기서 대상체는 30 nM 미만, 25 nM 미만, 20 nM 미만, 15 nM 미만, 10 nM 미만, 5 nM 미만 또는 1 nM 미만의 혈장 엔독시펜 수준을 갖는다. 특정 실시양태에서, 엔독시펜 염을 포함하는 조성물은 엔독시펜 글루코네이트, 엔독시펜 HCl, 또는 엔독시펜 시트레이트이다. 다른 실시양태에서, 적어도 90%의 (Z)-엔독시펜 또는 그의 염을 포함하는 경구 고체 투여 형태가 투여된다. 다른 실시양태에서, 적어도 90%의 엔독시펜의 다형체 형태 I, 형태 II, 또는 형태 III을 포함하는 경구 고체 투여 형태가 투여된다.
또한, 본원에는 (a) 대상체로부터 수득되는 시험 샘플 중의 혈장 엔독시펜 수준을 결정하는 또는 결정한 단계:(b) 시험 샘플 중의 혈장 엔독시펜의 수준을 참조 혈장 엔독시펜 수준과 비교하는 또는 비교 또는 결정한 단계; (c) 참조 혈장 엔독시펜 수준과 비교하여 시험 샘플 중의 혈장 엔독시펜의 감소된 수준을 결정하는 또는 결정한 단계; 및 (d) 엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 대상체에게 투여하는 단계를 포함하여, 타목시펜 불응성 대상체를 치료하는 방법이 제공된다. 엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물의 투여는 대상체에서 혈장 엔독시펜의 수준을 30 nM 초과의 항정 상태 수준으로 유지한다. 일부 실시양태에서, 대상체에서 혈장 엔독시펜의 수준은 30 nM 내지 80 nM 범위의 항정 상태 수준으로 유지된다.
본원에는 (a) 대상체에게 타목시펜을 포함하는 제1 조성물을 투여하는 단계; (b) 대상체로부터 수득되는 시험 샘플 중의 혈장 엔독시펜의 수준을 결정하는 또는 결정한 단계; (c) 혈장 엔독시펜의 참조 수준과 비교하여 시험 샘플 중의 혈장 엔독시펜의 감소된 수준을 결정하는 또는 결정한 단계; 및 (d) 대상체에게 본원에 개시된 경구 조성물을 투여하는 단계를 포함하여, 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖거나 가질 위험이 있는 대상체를 치료하는 방법이 제공된다. 대상체에게 타목시펜을 포함하는 제1 조성물이 적어도 1일, 2일, 3일, 15일, 1주, 2주, 4주, 1개월, 2개월, 3개월, 4개월, 5개월, 6개월, 1년, 2년, 3년, 4년, 5년 또는 10년 동안 매일 투여될 수 있다. 일부 실시양태에서, 엔독시펜 또는 그의 염 또는 다형체을 포함하는 경구 조성물의 투여는 대상체의 혈장 엔독시펜을 30 nM 초과의 수준으로 유지한다. 다른 실시양태에서, 엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물의 투여는 대상체의 혈장 엔독시펜을 30 nM 내지 300 nM(예컨대, 30 nM 내지 200 nM, 또는 30 nM 내지 80 nM) 범위의 수준으로 유지한다. 일부 실시양태에서, 대상체에게 (Z)-엔독시펜 D-글루코네이트, (Z)-엔독시펜 L-글루코네이트, (E)-엔독시펜 D-글루코네이트, (E)-엔독시펜 L-글루코네이트, 또는 그의 조합을 포함하는 경구 조성물이 투여된다. 다른 실시양태에서, 엔독시펜 염을 포함하는 경구 조성물은 엔독시펜 HCl 또는 엔독시펜 시트레이트이다. 일부 실시양태에서, 대상체에게 본원에 개시된 엔독시펜의 다형체 형태 I, 형태 II 또는 형태 III을 포함하는 경구 조성물이 투여된다.
본원에는 (a) 대상체에게 타목시펜을 포함하는 제1 조성물을 투여하는 단계; (b) 대상체로부터 수득되는 시험 샘플에서 대상체의 타목시펜-대사산물 프로파일을 결정하는 또는 결정한 단계; (c) 참조 타목시펜-대사산물 프로파일에서의 참조 혈장 엔독시펜의 수준과 비교하기 위해 대상체의 타목시펜-대사산물 프로파일에 기초하여 대상체의 혈장 엔독시펜의 감소된 수준을 결정하는 단계; 및 (d) 대상체에게 엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물을 투여하는 단계를 포함하여, 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖는 대상체를 치료하는 방법이 제공된다. 특정 실시양태에서, 엔독시펜을 포함하는 조성물은 엔독시펜 글루코네이트, 엔독시펜 HCl, 또는 엔독시펜 시트레이트이다. 일부 실시양태에서, 조성물은 엔독시펜의 다형체 형태 I, 형태 II, 또는 형태 III을 포함한다.
본원에는 (a) 타목시펜의 초기 용량 후 대상체의 혈장 엔독시펜 수준을 측정하는 단계; (b) 대상체의 혈장 엔독시펜 수준을 참조 혈장 엔독시펜 수준과 비교하는 단계; (c) 대상체에게 엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물을 투여하여 대상체의 혈장 엔독시펜 수준을 30 nM 초과의 수준으로 유지하는 단계를 포함하여, 하나 이상의 CYP2D6 또는 CYP3A4 돌연변이를 갖거나 이전에 초기 용량의 타목시펜이 투여되었고, 참조 혈장 엔독시펜 수준보다 낮은 혈장 엔독시펜 수준을 갖는, 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애가 치료되고 있는 대상체에서 혈장 엔독시펜 수준을 조정하는 방법이 제공된다. 일부 실시양태에서, 엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물의 투여는 대상체의 혈장 엔독시펜을 30 nM 내지 300 nM(예컨대, 30 nM 내지 200 nM, 또는 30 nM 내지 80 nM) 범위의 수준으로 유지한다. 일부 실시양태에서, 대상체의 혈장 엔독시펜 수준은 항정 상태 수준으로 유지된다. 대상체에게 초기 용량의 타목시펜이 적어도 1일, 2일, 3일, 15일, 1주, 2주, 4주, 1개월, 2개월, 3개월, 4개월, 5개월, 또는 6개월 동안 매일 투여될 수 있다.
본원에는 (a) 초기 용량의 타목시펜 후 대상체의 혈장 타목시펜-대사산물 엔독시펜의 수준을 측정하는 단계; (b) 타목시펜-대사산물 엔독시펜의 혈장 수준을 정상적인 타목시펜-대사산물 엔독시펜 수준에 대한 참조 수준과 비교하는 단계; (c) 합성적으로 제조된 엔독시펜을 포함하는 조성물의 조정 용량을 투여하는 단계로서, 여기서 합성적으로 제조된 엔독시펜의 용량은 대상체의 혈장 엔독시펜을 30 nM 초과의 수준으로 유지하기에 충분한 것인 단계를 포함하여, 이전에 초기 용량의 타목시펜이 투여되었고, 30 nM 미만의 혈장 엔독시펜 수준을 갖는, 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애가 치료되고 있는 대상체에서 혈장 엔독시펜의 수준을 조정하는 방법이 제공된다. 일부 실시양태에서, 합성적으로 제조된 엔독시펜을 포함하는 제2 조성물의 투여는 대상체의 혈장 엔독시펜을 30 nM 내지 300 nM(예컨대, 30 nM 내지 200 nM, 30 nM 내지 80 nM) 범위의 수준으로 유지한다. 일부 실시양태에서, 단계 (a) 내지 (c)는 대상체가 엔독시펜의 원하는 혈장 수준을 나타낼 때까지 반복될 수 있다.
일 측면에서, 본 개시는 대상체의 유방 조직을 절제하거나 대상체에게 방사선요법을 투여하는 단계 및 본원에 개시된 엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물을 투여하는 단계를 포함하여, 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 대상체를 치료하는 방법을 고려한다. 또 다른 측면에서, 본 개시는 대상체의 유방 조직을 절제하거나 대상체에게 방사선요법을 투여하기 전에 본원에 개시된 경구 조성물을 투여하는 단계를 포함하여, 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 대상체를 치료하는 방법을 고려한다.
대상체에게 투여될 용량은 일반적으로 단위 투여 형태일 것이다. 각각의 투여 단위 형태에서 엔독시펜에 대한 범위의 예는 0.01 mg 내지 200 mg이다. 용량은 일반적으로 그의 원하는 약리학적 및 생리학적 효과를 달성하기 위해 활성 유리 약물의 대사적 방출 시 투여 제형에 의해 생성되는 약리학적 활성 (Z)-유리 형태의, 몰량 기준으로, 유효량 및 등가물이어야 한다. 일부 실시양태에서, 엔독시펜 또는 엔독시펜 염 또는 다형체를 포함하는 조성물은 대상체에게 0.01 mg 내지 200.0 mg의 용량으로 투여된다. 다른 실시양태에서, 엔독시펜 또는 엔독시펜 염 또는 다형체를 포함하는 경구 조성물은 대상체에게 1 mg 내지 200.0 mg의 용량으로 투여된다. 일부 실시양태에서, 엔독시펜 또는 엔독시펜 염 또는 다형체를 포함하는 경구 조성물은 대상체에게 단위 용량당 0.01 mg, 0.05 mg, 0.1 mg, 0.25 mg, 0.5 mg, 0.75 mg, 1.0 mg, 1.5 mg, 2.0 mg, 4.0 mg, 6 mg, 8 mg, 10 mg, 20 mg, 40 mg, 50 mg, 100 mg 또는 200 mg의 용량으로 투여된다. 특정 실시양태에서, 적어도 90% (Z)-엔독시펜(wt/wt)의 엔독시펜을 포함하는 경구 조성물은 단위 용량당 1 mg, 2.0 mg, 4.0 mg 6 mg, 8 mg, 10 mg, 20 mg, 40 mg, 50, 100 mg 또는 200 mg의 용량으로 투여된다. 일부 실시양태에서, 엔독시펜 글루코네이트를 포함하는 조성물은 0.01 내지 20 mg 범위의 용량으로 투여된다. 일부 실시양태에서, (Z)-엔독시펜 D-글루코네이트를 포함하는 조성물은 단위 용량당 0.5 mg, 1 mg, 2 mg 4.0 mg, 6 mg, 8 mg, 10 mg, 20 mg, 40 mg, 50 mg, 100 mg, 및 200 mg으로 투여된다. 일부 실시양태에서, 1 mg의 (Z)-엔독시펜 D-글루코네이트를 포함하는 조성물이 투여된다. 다른 실시양태에서, 1 mg의 (Z)-엔독시펜 L-글루코네이트를 포함하는 조성물이 투여된다. 또 다른 실시양태에서, 2 mg의 (Z)-엔독시펜 D-글루코네이트 및 (E)-엔독시펜 D-글루코네이트를 포함하는 조성물이 투여된다. 특정 실시양태에서, 적어도 90%의 엔독시펜의 다형체, 예컨대 다형체 형태 I, 형태 II 또는 형태 III(wt/wt)을 포함하는 경구 조성물이 단위 용량당 1 mg, 2.0 mg, 4.0 mg 6 mg, 8 mg, 10 mg, 20 mg, 40 mg, 50, 100 mg 또는 200 mg의 용량으로 투여된다. 일부 실시양태에서, 엔독시펜의 다형체 형태, 예컨대 형태 I, 형태 II 또는 형태 III을 포함하는 조성물이 0.01 내지 20 mg 범위의 용량으로 투여된다.
유방암 성장 속도 연구는 유방암을 갖는 대상체의 유방촬영 스크리닝을 이용하여 50 내지 59세 여성의 25번째 백분위수에서 유방암 성장 속도가 치료제에 대한 대상체의 빠른 노출에 대한 충족되지 않은 필요성을 나타낸다는 것을 입증하였다(Weeden-Fekjaer et al. Breast Cancer Research200810:R41). 암 성장 속도를 더욱 감소시킬 수 있는 항암 치료제, 예컨대 엔독시펜의 빠른 흡수 및 생체이용률이 매우 바람직하다.
일 측면에서, 엔독시펜의 최대 및 항정 상태 혈장 수준의 빠른 달성이 본 개시의 특정 측면이다. 본 개시는 대상체에 대한 본원에 개시된 조성물의 투여가 조성물 투여 후 2 내지 30시간 이내, 3 내지 20시간 이내, 2 내지 10시간 이내 또는 4 내지 8시간 이내의 범위에서 엔독시펜의 최대 혈장 수준을 달성한다는 것을 제공한다. 따라서, 일부 실시양태에서, 엔독시펜의 최대(피크) 혈장 수준까지의 시간은 조성물 투여 후 2 내지 10시간의 범위이다. 일부 실시양태에서, 엔독시펜의 최대 혈장 수준까지의 시간은 본원에 개시된 조성물 투여 후 4 내지 8시간의 범위이다.
엔독시펜의 항정 상태 혈장 수준의 빠른 달성이 또한 매우 바람직하다. (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 본원에 개시된 조성물로 투여되는 대상체에서 엔독시펜의 혈장 수준이 빠르게 항정 상태를 달성한다. 항정 상태 혈장 수준은 제7일 내지 제21일에 달성될 수 있다. 일부 실시양태에서, 항정 상태 혈장 수준은 본원에 개시된 조성물의 일일 투여 후 제7일에 달성될 수 있다(도 5).
일 측면에서, 본 개시는 본원에 개시된 조성물로부터 방출되는 순환 엔독시펜이 타목시펜보다 빠르게 제거될 수 있다는 것을 제공한다. 타목시펜의 터미널 제거 반감기는 5-7일인 것으로 알려져 있으며(Jordan C. Steroids. 2007 Nov; 72(13): 829-842), 타목시펜의 피크 농도 시간은 투여 후 대략 5시간이다. 본원에 개시된 조성물로부터 방출되는 엔독시펜은 타목시펜보다 상당히 더 낮은 30 내지 60시간 범위의 터미널 제거 반감기를 가질 수 있다. 일부 실시양태에서, 평균 반감기는 40 내지 53시간의 범위이다. AUC24hr(제21일)/AUC0-inf(제1일)의 평균 비율은 전형적으로 1 mg 내지 4 mg의 (Z)-엔독시펜, 또는 그의 염을 포함하는 조성물에 대해 0.7 내지 1.2의 범위이다. 따라서, 본원에 개시된 조성물로부터 방출되는 엔독시펜의 축적은 지속된 치료 동안 크게 변화되지 않는다.
또 다른 측면에서, 본 개시는 본원에 개시된 조성물의 투여가 치료적으로 유효한 엔독시펜의 흡수를 달성한다는 것을 제시한다.
곡선 아래 면적 AUC(0-24hr)("AUC24hr")는 투여 시간(0 hr)으로부터 24시간 동안 약물에 대한 대상체의 총 노출을 기술한다. (Z)-엔독시펜 또는 그의 염을 포함하는 조성물은 전형적으로 1 mg 내지 4 mg의 (Z)-엔독시펜을 포함하는 조성물의 초기(제1) 용량의 제1일에 150 hr*ng/mL 내지 600 hr*ng/mL의 평균(AUC24hr)을 달성한다. (Z)-엔독시펜 또는 그의 염을 포함하는 조성물은 전형적으로 1 mg 내지 4 mg의 (Z)-엔독시펜을 포함하는 조성물의 초기(제1) 용량의 제21일에 평균 AUC24hr 400 hr*ng/mL 내지 2500 hr*ng/mL를 달성한다.
분석되는 체액(일반적으로 혈장, 혈액 또는 혈청)에서 순환하는 약물의 시간-평균 농도인 AUC(0-inf)("AUC0-inf")는 약물에 대한 대상체의 총 노출을 기술한다. 본 개시는 엔독시펜에 대한 대상체의 노출(AUC0-inf)이 용량에 비례할 수 있다는 것을 제공한다. 일부 실시양태에서, AUC0-inf는 200 hr*ng/mL 내지 10000 hr*ng/mL의 범위이다. 다른 실시양태에서, AUC0-inf는 300 hr*ng/mL 내지 8000 hr*ng/mL의 범위이다. 특정 실시양태에서, 1 mg 내지 4 mg의 (Z)-엔독시펜의 투여 범위에 걸처 AUC0-inf는 400 hr*ng/mL 내지 6000 hr*ng/mL의 범위이다(예컨대, 표 17 참조).
건강관리 전문가, 예컨대 주치의는 대상체에서 조성물의 약동학 프로파일에 기초하여 투여 요법을 조정할 수 있다.
일 측면에서, 본 개시의 조성물은 단독으로 또는 조합 요법으로 사용될 수 있다. 예컨대, 본원에 개시된 조성물은 1차 요법, 선행보조 요법, 또는 보조 요법의 일부로 하나 이상의 치료제와 조합하여 사용될 수 있다. 본 개시의 조성물이 다른 요법, 예컨대 수술 및 방사선과 조합하여 선행보조 또는 보조 요법으로 사용될 수 있다는 것이 본 개시의 일 측면이다. 조성물의 조합은 치료제의 효능을 개선하도록 작용할 수 있으므로, 표준 암 요법을 개선하는 데 사용될 수 있다. 예컨대, 대상체가 전립선암을 갖고 전립선암의 치료를 위해 비칼루타미드 또는 엔잘루타미드 요법을 받고 있는 경우, 대상체는 요법의 결과로 여성형 유방증이 발병할 가능성이 있다. 본원에 개시된 조성물은 여성형 유방증을 예방 및/또는 치료하기 위해 전립선암을 갖는 대상체에게 조합 요법으로 투여될 수 있다. 또 다른 예로서, ER+/Her2+ 양성 유방암을 갖는 대상체는 트라스투주맙 또는 다른 종양학 약물, 예컨대 항신생물제와의 조합 요법 또는 면역요법을 받고 있을 것이며, 본원에 개시된 조성물이 ER+/Her2+ 양성 유방암을 갖는 이러한 환자를 치료하는 데 사용될 수 있다. 따라서, 일부 실시양태에서, 조성물은 또한 비칼루타미드, 엔잘루타미드 또는 항암 약물, 예컨대 트라스투주맙, 항신생물제, 예컨대 카페시타빈(젤로다), 카르보플라틴(파라플라틴), 시스플라틴(플라티놀), 시클로포스파미드(네오사르), 도세탁셀(도세프레즈, 탁소테레), 독소루비신(아드리아마이신), 페길화된 리포솜 독소루비신(독실), 에피루비신(엘렌스), 플루오로우라실(5-FU, 아드루실), 겜시타빈(젬저), 메토트렉세이트(다수의 브랜드명), 파클리탁셀(탁솔), 단백질 결합된 파클리탁셀(아브락산), 비노렐빈(나벨빈), 에리불린(할라벤), 익사베필론(익셈프라), 및 ATP-카세트 결합 단백질 억제제를 포함한다.
또 다른 측면에서, 본원에 개시된 조성물은 대상체에서 엔독시펜의 생체이용률을 증가시키는 치료제를 포함할 수 있다. P-당단백질(P-gp, ABCB1)은 뇌, 간, 및 소장 뿐만 아니라 암 세포에서도 발현되는 매우 효율적인 약물 유출 펌프이며, 약동학에 영향을 끼치고 많은 항암 약물에 대한 요법 내성을 부여한다. 따라서, 일부 실시양태에서, 조성물은 또한 ATP-결합 카세트(ABC 계열) 수송체의 억제제, 예컨대 유방암 내성 단백질(BCRP 단백질) 및 P-gp의 억제제를 포함한다. BCRP 단백질 및 P-Gp의 몇몇 억제제가 당해 분야에 공지되어 있다. 예컨대, BCRP 단백질의 억제제는 시클로스포린, 오메프라졸, 판토프라졸, 사퀴나비르, 및 타크롤리무스를 포함한다.
P-gp 억제제의 비제한적 예는 1세대 억제제, 예컨대 베라파밀, 시클로스포린 A, 레세르핀, 퀴니딘, 요힘빈, 타목시펜 및 토레미펜, 2세대 억제제, 예컨대 덱스베라파밀, 덱스니굴디핀, 발스포다르(PSC 833), 및 도페퀴다르 푸마레이트(MS-209), 3세대 P-gp 억제제, 예컨대 시클로프로필디벤조수베란 조수퀴다르(LY335979), 라니퀴다르(R101933), 미토탄(NSC-38721), 비리코다르(VX-710), 엘라크리다르(GF120918/GG918), ONT-093, 타리퀴다르(XR9576), 및 HM30181 및 항-P-gp 모노클로날 항체, 예컨대 MRK-16)를 포함한다.
또한, 본 개시는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 대상체의 치료에 사용하기 위한 조성물 중 하나 이상을 함유하는 치료 키트를 제공한다. 본 개시의 키트는 본원에 개시된 경구 조성물, 조성물을 수용하기 위한 밀폐된 용기, 및 경구로 투여되는 조성물의 사용 설명서를 포함할 수 있다. 일 측면에서, 본 개시의 키트는 제2 치료제를 포함할 수 있다. 이러한 제2 치료제는 비칼루타미드, 엔잘루타미드 또는 항암 약물, 예컨대 트라스투주맙, 항신생물제, 예컨대 카페시타빈(젤로다), 카르보플라틴(파라플라틴), 시스플라틴(플라티놀), 시클로포스파미드(네오사르), 도세탁셀(도세프레즈, 탁소테레), 독소루비신(아드리아마이신), 페길화된 리포솜 독소루비신(독실), 에피루비신(엘렌스), 플루오로우라실(5-FU, 아드루실), 겜시타빈(젬저), 메토트렉세이트(다수의 브랜드명), 파클리탁셀(탁솔), 단백질 결합된 파클리탁셀(아브락산), 비노렐빈(나벨빈), 에리불린(할라벤), 익사베필론(익셈프라), 및 ATP-결합 카세트(ABC 수송체) 억제제, 예컨대 P-gp 억제제일 수 있다.
예시적인 조성물
예시적인 비제한적인 조성물이 하기에 제공된다. 상술된 바와 같이, 퍼센트(%)는 조성물의 총 중량에 기반한 중량에 의한 양(wt/wt)을 지칭한다. 조성물의 상이한 성분의 합은 총 조성물의 100%(wt/wt)까지 첨가된다. 적어도 90%(≥ 90%)의 (Z)-엔독시펜 유리 염기는 임의의 조성물 중의 엔톡시펜의 총 중량과 비교하여 (Z)-엔독시펜 이성질체의 중량 퍼센트를 지칭한다.
일 측면에서, 본 개시는 (Z)-엔독시펜, 화학식 (III)의 화합물, 화학식 (II)의 화합물, 및 그의 염 및 다형체를 제조하는 산업적으로 확장 가능한 방법에 관한 것이다.
일 측면에서, 본 개시는 (a) 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 EtOAc(1:1 내지 1:20 wt/wt) 중의 6N HCl(1:1 내지 1:5 wt/wt)과 반응시키는 단계, (b) 8N NaOH(1:1 내지 1:20 wt/wt)로 중화시키는 단계; (c) EtOAc(1:1 내지 1:10 wt/wt)로 1회 이상 세척하는 단계; (d) 20% NaCl(1:1 내지 1:5 wt/wt)로 추출하는 단계; (e) 활성탄(1:0.01 내지 1:1)과 반응시키는 단계; (f) IPA(1:1 내지 1:10 wt/wt)로 1회 이상 세척하고 MeOH/PPW(1:1 내지 1:10 wt/wt)로 추출하는 단계를 포함하고; 여기서 wt/wt는 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물, 예컨대 화학식 (III)의 화합물에 대한 것인, (Z)-엔독시펜을 제조하는 산업적으로 확장 가능한 방법에 관한 것이다.
일 측면에서, 본 개시는 (a) 화학식 (II)의 화합물을 THF(4.4 wt/wt) 중의 프로피오페논과 반응시키는 단계; (b) THF(8.9 wt/wt) 중의 TiCl4(1.4 wt/wt) 및 Zn(0.9 wt/wt)의 용액을 제조하는 단계; 및 (c) 단계 (a)의 화학식 (II)의 화합물을 단계 (b)로부터의 THF 중의 TiCl4 및 Zn과 반응시켜 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성하는 단계를 포함하고; 여기서 wt/wt는 화학식 (II)의 화합물에 대한 것인, (E)-엔독시펜과 (Z)-엔독시펜의 혼합물(화학식 (III)의 화합물)을 제조하는 산업적으로 확장 가능한 방법에 관한 것이다.
일 측면에서, 본 개시는 (a) 25% 염화암모늄(1:20 wt/wt) 및 실리카(Celite)(1:1 wt/wt)로 추출; (b) THF(1:1 내지 1:5 wt/wt)로 1회 이상 세척; (c) 20% 염화나트륨(1:3 wt/wt)으로 1회 이상 세척; (c) EtOAc(1:4.5 wt/wt)로 증류; 및 (d) (1:2 v/v) EtOAc/n-헵탄(1:3.8 wt/wt)으로 결정화 중 하나 이상의 단계를 포함하고, 여기서 wt/wt는 화학식 (II)의 화합물에 대한 것인, 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 제조하는 산업적으로 확장 가능한 방법에 관한 것이다.
일 측면에서, 본 개시는 (a) 40% K2CO3(1:2 wt/wt)로 1회 이상 추출; (b) 1N NaOH(1:10 wt/wt) 및 MeTHF(1:1 내지 1:10 wt/wt)로 추출; (c) MeTHF(1:1 내지 1:20 wt/wt)로 2회 이상 추출; (d) 20% 염화나트륨(1:5 wt/wt)으로 추출; (e) IPA(1:4.5 wt/wt)로 증류; 및 (f) (1:2.7 v/v) IPA/n-헵탄(1:3.4 wt/wt)으로 결정화 중 하나 이상의 단계를 추가로 포함하고; 여기서 wt/wt는 화학식 (II)의 화합물에 대한 것인, 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 제조하는 산업적으로 확장 가능한 방법에 관한 것이다.
일 측면에서, 본 개시는 (a) 화학식 (I)의 화합물(1 equiv.)을 THF(4.9 wt/wt) 중의 DIPEA(3 wt/wt)와 반응시키는 단계; (b) 1-클로로에틸 클로로포르메이트(3.3 wt/wt)를 첨가하는 단계; (c) 메탄올(4.0 wt/wt)로 1 내지 4회 증류시키는 단계; (d) 메탄올(3.2 wt/wt)로 증류시키는 단계; (e) 메탄올(3.2 wt/wt)/6N HCl(4 wt/wt)과 반응시키는 단계; 및 (f) 8N NaOH(5 wt/wt)로 중화시키는 단계를 포함하고; 여기서 wt/wt는 화학식 (I)의 화합물에 대한 것인, 화학식 (II)의 화합물을 제조하는 산업적으로 확장 가능한 방법에 관한 것이다.
일 측면에서, 본 개시는 본원에 개시된 방법에 따라 제조되는 (Z)-엔독시펜, (E)-엔독시펜, 화학식 (III)의 화합물, 및 화학식 (II)의 화합물, 및 그의 염에 관한 것이다. 일 측면에서, 본 개시는 본원에 개시된 방법에 따라 제조되는 (Z)-엔독시펜에 관한 것이며, 여기서 (Z)-엔독시펜은 적어도 6개월, 적어도 9개월, 적어도 12개월, 또는 적어도 18개월 동안 주위 온도에서 안정하다.
또 다른 측면에서, 본원에 개시된 방법에 따라 제조되는 (Z)-엔독시펜 유리 염기는 <1% 불순물을 갖는다. 또 다른 측면에서, 본원에 개시된 방법에 따라 제조되는 (E)/(Z)-엔독시펜 유리 염기는 <1% 불순물을 갖는다.
일 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 유리 염기 또는 그의 염을 포함하는 조성물에 관한 것이다.
또 다른 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 유리 염기 또는 그의 염을 포함하는 조성물에 관한 것이고, 여기서 (Z)-엔독시펜은 조성물 중의 총 엔독시펜의 적어도 90%의 (Z)-엔독시펜 유리 염기 wt/wt이다.
또 다른 측면에서, 조성물은 또한 (E)-엔독시펜을 포함하며, 여기서 (E)-엔독시펜 대 (Z)-엔독시펜(E/Z-비율)은 1:99; 5:95; 10:90, 15:85; 20:80, 25:75; 30:70; 40:70, 45:55; 50:50; 55:45; 60:40; 65:45; 또는 70:30이다.
추가의 측면에서, 조성물은 또한 (E)-엔독시펜을 포함하고, 여기서 E/Z-비율은 45:55 내지 55:45의 범위이거나 대략 1:1이다.
일 측면에서, 조성물은 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체을 포함하고, 여기서 (Z)-엔독시펜은 적어도 6개월, 적어도 9개월, 적어도 12개월 또는 적어도 18개월 동안 주위 온도에서 안정하다.
또 다른 측면에서, 조성물은 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하고,
a. 여기서 조성물은 20,000 g/mL 이하의 호기성 박테리아 플레이트 카운트를 가지거나;
b. 여기서 조성물의 물 함량은 USP 921의 방법 Ic로 시험 시 1.0% 이하이거나;
c. 여기서 조성물의 수분 활성도(Aw)는 0.9 미만이거나,
d. 여기서 점화 잔류물은 USP 281의 방법으로 시험 시 0.1% 이하이거나;
e. 여기서 중금속은 USP 231의 방법 II로 시험 시 20 ppm 이하이거나;
f. 여기서 검증된 HPLC 방법으로 시험 시 메탄올은 NMT 3000 ppm이고, 테트라히드로푸란은 NMT 720 ppm이고, 이소프로판올은 NMT 5000 ppm이고, 에틸 아세테이트는 NMT 5000 ppm이고; n-헵탄은 NMT 5000 ppm이고, 에탄올은 NMT 5000 ppm이다.
또 다른 측면에서, 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물은 단위 용량당 0.01 mg 내지 200 mg의 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함한다.
또 다른 측면에서, 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물은 단위 용량당 1 mg 내지 20 mg의 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함한다.
추가의 측면에서, 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물은 0.01% 내지 20%(wt/wt)의 엔독시펜 또는 그의 염 또는 다형체를 포함한다.
추가의 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이고, 여기서 조성물은 0.1% 내지 10%(wt/wt)의 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함한다.
또 다른 측면에서, 본 개시는 하나 이상의 부형제를 추가로 포함하는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이다.
또 다른 측면에서, 본 개시는 하나 이상의 부형제를 추가로 포함하는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 부형제는 결합제, 충전제, 붕해제, 윤활제, 활주제, 방출 조*방출 제어제, 장용 코팅제, 필름 형성제, 가소제, 감미제, 향미제, 또는 그의 조합이다.
또 다른 측면에서, 본 개시는 하나 이상의 부형제를 추가로 포함하는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 부형제는 조성물의 약 0.1% 내지 약 99% wt/wt이다.
추가의 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이고, 여기서 조성물은 산-불용성 중합체, 메틸 아크릴레이트-메타크릴산 공중합체, 셀룰로스 아세테이트 프탈레이트(CAP), 셀룰로스 아세테이트 숙시네이트, 히드록시프로필 메틸 셀룰로스 프탈레이트, 히드록시프로필 메틸 셀룰로스 아세테이트 숙시네이트(히프로멜로스 아세테이트 숙시네이트), 폴리비닐 아세테이트 프탈레이트(PVAP), 메틸 메타크릴레이트-메타크릴산 공중합체, 셸락, 셀룰로스 아세테이트 트리멜리테이트, 나트륨 알기네이트, 제인, 왁스(합성 왁스 포함), 미세결정질 왁스, 파라핀 왁스, 카나우바 왁스, 및 밀랍, 폴리에톡실화된 피마자유 유도체, 수소첨가 오일, 글리세릴 모노-, 디- 트리베네이트, 글리세릴 모노스테아레이트, 글리세릴 디스테아레이트, 장쇄 알콜, 예컨대 스테아릴 알콜, 세틸 알콜, 폴리에틸렌 글리콜, 및 그의 혼합물로 이루어진 군으로부터 선택되는 하나 이상의 방출 조절제를 포함한다.
일 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 조성물은 경구, 비경구, 국소, 및 관내 전달을 위해 제형화된다.
또 다른 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 경구 전달을 위해 제형화되는 조성물은 정제, 캐플릿, 캡슐, 환제, 분말, 트로키, 엘릭시르, 현탁액, 시럽, 웨이퍼, 츄잉 검, 당의정, 및 로젠지이다.
또 다른 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 경구 전달을 위해 제형화되는 조성물은 장용 정제로 제형화되는 정제, 장용 캐플릿으로 제형화되는 캐플릿, 및 장용 캡슐로 제형화되는 캡슐이다.
또 다른 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 경구 전달을 위해 제형화되는 조성물은 지연 방출 정제로 제형화되는 정제, 지연 방출 캐플릿으로 제형화되는 캐플릿, 또는 지연 방출 캡슐로 제형화되는 캡슐이다.
또 다른 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 조성물은 경구 전달을 위해 제형화되며, USP 711 또는 701의 방법으로 시험 시, 조성물은 위에서
a. 투여 후 2시간에 10% 미만의 엔톡시펜;
b. 투여 후 2시간에 20% 미만의 엔톡시펜;
c. 투여 후 2시간에 30% 미만의 엔톡시펜;
d. 투여 후 4시간에 40% 미만의 엔톡시펜; 또는
e. 투여 후 6시간에 50% 미만의 엔톡시펜
을 방출한다.
또 다른 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 USP 711 또는 701의 방법으로 시험 시, 조성물은 위에서
a. 투여 후 2시간에 10% 미만의 (Z) 엔독시펜;
b. 투여 후 2시간에 20% 미만의 (Z) 엔독시펜;
c. 투여 후 4시간에 40% 미만의 (Z) 엔독시펜; 또는
d. 투여 후 6시간에 50% 미만의 (Z) 엔독시펜
을 방출한다.
또 다른 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 조성물은 경구 전달을 위해 제형화되며, USP 711 또는 701의 방법으로 시험 시, 조성물은 장에서 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 또는 적어도 90%의 엔독시펜을 방출한다.
또 다른 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 조성물은 경구 전달을 위해 제형화되며, USP 711 또는 701으로 시험 시, 조성물은 장에서 투여 후 2시간에 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 또는 적어도 90%의 엔독시펜을 방출한다.
또 다른 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 조성물은 경구 전달을 위해 제형화되며, USP 711 또는 701으로 시험 시, 조성물은 장에서 투여 후 3시간에 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 또는 적어도 90%의 엔독시펜을 방출한다.
또 다른 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 조성물은 경구 전달을 위해 제형화되며, USP 711 또는 701으로 시험 시, 조성물은 장에서 투여 후 4시간에 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 또는 적어도 90%의 엔독시펜을 방출한다.
또 다른 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 조성물은 경구 전달을 위해 제형화되며, USP 711 또는 701의 방법으로 시험 시, 조성물은 소장에서
a. 투여 후 4시간 후 적어도 10%의 엔독시펜;
b. 투여 후 6시간 후 적어도 30%의 엔독시펜;
c. 투여 후 7시간 후 적어도 40%의 엔독시펜;
d. 투여 후 8시간 후 적어도 50%의 엔독시펜;
e. 투여 후 2시간 후 적어도 50%의 엔독시펜;
f. 투여 후 2시간 후 적어도 60%의 엔독시펜;
g. 투여 후 2시간 후 적어도 70%의 엔독시펜;
h. 투여 후 2시간 후 적어도 80%의 엔독시펜; 또는
i. 투여 후 2시간 후 적어도 90%의 엔독시펜
을 방출한다.
또 다른 측면에서, 본 개시는 본원에 개시된 임의의 방법에 따라 제조되는 (Z)-엔독시펜 또는 그의 염을 포함하는 조성물에 관한 것이며, 여기서 조성물은 경구 전달을 위해 제형화되고, USP 711의 방법으로 시험 시, 조성물은 결장에서 투여 후 8시간 후 적어도 50%의 엔독시펜을 방출하도록 제형화된다.
또 다른 측면에서, 본 개시는 단위 용량당 1 mg 내지 200 mg의 엔독시펜을 포함하는 경구 투여를 위해 제형화되는 조성물에 관한 것이며; 여기서 조성물은 적어도 6개월 동안 안정하다..
또 다른 측면에서, 본 개시는 단위 용량당 1 mg 내지 200 mg의 엔독시펜을 포함하는 경구 투여를 위해 제형화되는 조성물에 관한 것이며;
여기서 엔독시펜은 적어도 90%의 (Z)-엔독시펜 유리 염기이고;
여기서 조성물은 20,000 g/ml 이하의 호기성 박테리아 플레이트 카운트를 갖거나;
(Z)-엔독시펜의 물 함량은 USP 921의 방법 Ic로 결정 시 1% 이하이거나;
(Z)-엔독시펜의 점화 잔류물은 USP 281의 방법으로 시험 시 0.10% 이하이거나;
중금속은 USP 231의 방법 II로 시험 시 20 ppm 이하이다.
또 다른 측면에서, 본 개시는 단위 용량당 1 mg 내지 200 mg의 엔독시펜을 포함하는 경구 투여를 위해 제형화되는 조성물에 관한 것이며,
여기서 조성물은 장용 정제, 장용 캐플릿, 및 장용 캡슐로 제형화되거나;
여기서 USP 711의 방법으로 결정 시, 조성물은 소장에서
투여 후 4시간 후 적어도 25%의 엔독시펜;
투여 후 6시간 후 적어도 30%의 엔독시펜;
투여 후 7시간 후 적어도 40%의 엔독시펜; 또는
투여 후 8시간 후 적어도 50%의 엔독시펜
을 방출하도록 제형화되거나;
여기서 USP 711의 방법으로 결정 시, 조성물은 결장에서 적어도 50%의 엔독시펜을 방출하도록 제형화된다.
또 다른 측면에서, 본 개시는 단위 용량당 1 mg 내지 200 mg의 엔독시펜을 포함하는 경구 고체 투여 형태 조성물에 관한 것이며;
여기서 엔독시펜은 적어도 90%의 (Z)-엔독시펜 유리 염기이고;
여기서 조성물은 20,000 g/ml 이하의 호기성 박테리아 플레이트 카운트를 갖거나;
(Z)-엔독시펜의 물 함량은 USP 921의 방법 Ic로 결정 시 1% 이하이거나;
(Z)-엔독시펜의 점화 잔류물은 USP 281의 방법으로 시험 시 0.10% 이하이거나;
중금속은 USP 231의 방법 II로 시험 시 20 ppm 이하이고;
여기서 조성물은 장용 정제, 장용 캐플릿 및 장용 캡슐로 제형화되고;
여기서 엔독시펜은 순수하거나;
여기서 조성물은 결합제, 충전제, 붕해제, 윤활제, 활주제, 방출 조절제, 장용 코팅제, 필름 형성제, 가소제, 착색제, 감미제, 및 향미제, 또는 그의 조합으로 이루어진 군으로부터 선택되는 부형제를 추가로 포함한다.
또 다른 측면에서, 본 개시는 본원에 기재된 방법에 의해 제조되는 (Z)-엔독시펜 유리 염기를 포함하는 장용 캡슐에 관한 것이다.
또 다른 측면에서, 본 개시는 단위 용량당 1 mg 내지 200 mg의 순수 엔독시펜을 포함하는 장용 캡슐에 관한 것이며;
여기서 엔독시펜은 적어도 90%의 (Z)-엔독시펜 유리 염기이고;
여기서 조성물은 20,000 g/ml 이하의 호기성 박테리아 플레이트 카운트를 갖고;
(Z)-엔독시펜의 물 함량은 USP 921의 방법 Ic로 결정 시 1% 이하이고;
(Z)-엔독시펜의 점화 잔류물은 USP 281의 방법으로 시험 시 0.10% 이하이고;
중금속은 USP 231의 방법 II로 시험 시 20 ppm 이하이다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 엔독시펜 염은 아레콜린, 베실레이트, 비카르보네이트, 비타르타레이트, 부틸브로마이드, 시트레이트, 카미실레이트, 글루코네이트, 글루타메이트, 글리콜릴라르사닐레이트, 헥시레소르시네이트, 히드라바민, 히드로브로마이드, 히드로클로라이드, 히드록시나프타노에이트, 이세티오네이트, 말레이트, 만델레이트, 메실레이트, 메틸브로마이드, 메틸브로마이드, 메틸니트레이트, 메틸술페이트, 무케이트, 납실레이트, 니트레이트, 파마오에이트(엠보네이트), 판토테네이트, 포스페이트/디포스페이트, 폴리갈라쿠로네이트, 살리실레이트, 스테아레이트, 술페이트, 탄네이트, 테오클레이트, 트리에티오다이드, 벤자틴, 클레미졸, 클로로프로카인, 콜린, 디에틸아민, 디에탄올아민, 에틸렌디아민, 메글루민, 피페라진, 프로카인, 알루미늄, 바륨, 비스무트, 리튬, 마그네슘, 칼륨, 및 아연으로 이루어진 군으로부터 선택된다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 엔독시펜 염은 엔독시펜 글루코네이트이다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 엔독시펜 염은 (Z)-엔독시펜 D-글루코네이트, (E)-엔독시펜 D-글루코네이트, (Z)-엔독시펜 L-글루코네이트, 및 (E)-엔독시펜 L-글루코네이트, 또는 그의 조합으로 이루어진 군으로부터 선택되는 엔독시펜 글루코네이트이다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 엔독시펜 염은 엔독시펜 글루코네이트이고, 여기서 엔독시펜 글루코네이트는 1%, 5%, 10%, 20%, 25%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 96%, 97%, 98%, 99%, 99.5%, 99.99% 또는 100%(wt/wt)의 (Z)-엔독시펜 D-글루코네이트 또는 (Z)-엔독시펜 L-글루코네이트를 포함한다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 엔독시펜 염은 엔독시펜 글루코네이트이고, 여기서 엔독시펜 글루코네이트는 (Z)-엔독시펜 D-글루코네이트 및 (E)-엔독시펜 D-글루코네이트(wt/wt)를 10:90, 20:80, 30:70; 40:60; 50:50, 60:40, 70:30, 80:20, 90:10; 91:9; 92:8; 93:7; 94:8; 95:5, 96:4, 97:3, 98:2, 99:1; 99.5:0.5; 또는 99.99:0.01의 Z/E-비율로 포함한다.
또 다른 측면에서, 본 개시는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다의 치료 및 예방을 위한 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이다.
또 다른 측면에서, 본 개시는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다의 치료 및 예방을 위한 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애는 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 또는 외음부암이다.
또 다른 측면에서, 본 개시는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다의 치료 및 예방을 위한 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 호르몬 의존적 유방 장애는 유방암이다.
또 다른 측면에서, 본 개시는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다의 치료 및 예방을 위한 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 호르몬 의존적 유방 장애는 유방암이고, 여기서 유방암은 전암, 초기 단계 암, 비-전이성 암, 전-전이성 암, 국소 진행 암, 또는 전이성 암이다.
또 다른 측면에서, 본 개시는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다의 치료 및 예방을 위한 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애는 타목시펜 불응성 또는 타목시펜 내성이다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 이를 필요로 하는 대상체에게 투여된다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 이를 필요로 하는 대상체에게 투여되며, 여기서 대상체는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 이를 필요로 하는 대상체에게 투여되며, 여기서 대상체의 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애는 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 또는 외음부암이다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 이를 필요로 하는 대상체에게 투여되며, 여기서 대상체는 유방암을 가지며, 여기서 유방암은 전암, 초기 단계 암, 비-전이성 암, 전-전이성 암, 국소 진행 암, 또는 전이성 암이다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며,여기서 조성물은 이를 필요로 하는 대상체에게 투여되며, 여기서 대상체는 전립선암을 가지며, 여기서 대상체는 전립선암 요법을 갖거나 이를 시작하려고 한다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 이를 필요로 하는 대상체에게 투여되며, 여기서 대상체는 전립선암을 가지며, 여기서 대상체는 여성형 유방증을 갖거나 가질 위험이 있다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 대상체에게 투여되며, 여기서 대상체의 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애는 타목시펜 불응성 또는 타목시펜 내성이다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 대상체에게 0.01 mg, 0.05 mg, 0.1 mg, 0.25 mg, 0.5 mg, 0.75 mg, 1.0 mg, 1.5 mg, 2.0 mg, 4 mg, 5 mg, 10 mg, 20 mg, 25 mg, 50 mg 또는 200 mg의 (Z)-엔독시펜의 단위 용량으로 투여된다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 대상체에게 0.01 mg, 0.05 mg, 0.1 mg, 0.25 mg, 0.5 mg, 0.75 mg, 1.0 mg, 1.5 mg, 2.0 mg, 4 mg, 5 mg, 10 mg, 20 mg, 25 mg, 50 mg 또는 200 mg의 (Z)-엔독시펜의 용량으로 투여된다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 대상체에게 1일 1회, 1일 2회, 1일 3회, 1일 4회, 격일, 1주 2회, 매주, 격주, 1개월 2회, 매월, 분기별, 6개월 1회, 또는 매년 투여된다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 경구로 조성물의 투여는 대상체의 혈장 엔독시펜을 30 nM 초과의 수준으로 유지한다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 대상체에게 조합 요법으로 투여된다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 1차 요법, 선행보조 요법, 또는 보조 요법으로 투여된다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 대상체에게 단독으로 또는 제2 치료제와 조합하여 투여된다.
또 다른 측면에서, 본 개시는 본원에 기재된 임의의 방법에 의해 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물에 관한 것이며, 여기서 조성물은 대상체에게 단독으로 또는 제2 치료제와 조합하여 투여되며, 여기서 제2 치료제는 비칼루타미드, 엔잘루타미드, 또는 항암 약물, 예컨대 트라스투주맙, 항신생물제, 예컨대 카페시타빈(젤로다), 카르보플라틴(파라플라틴), 시스플라틴(플라티놀), 시클로포스파미드(네오사르), 도세탁셀(도세프레즈, 탁소테레), 독소루비신(아드리아마이신), 페길화된 리포솜 독소루비신(독실), 에피루비신(엘렌스), 플루오로우라실(5-FU, 아드루실), 겜시타빈(젬저), 메토트렉세이트(다수의 브랜드명), 파클리탁셀(탁솔), 단백질 결합된 파클리탁셀(아브락산), 비노렐빈(나벨빈), 에리불린(할라벤), 익사베필론(익셈프라), 또는 ATP-결합 카세트 수송체의 억제제이다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 대상체는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 대상체는 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있으며, 여기서 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애는 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 또는 외음부암이다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 대상체는 유방암을 갖는다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 대상체는 유방암을 가지며, 여기서 유방암은 전암, 초기 단계 암, 비-전이성 암, 전-전이성 암, 국소 진행 암, 또는 전이성 암이다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 대상체는 전립선암을 가지며, 여기서 대상체는 여성형 유방증을 갖거나 가질 위험이 있다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 대상체는 타목시펜 불응성 또는 타목시펜 내성 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖는다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 조성물은 대상체에게 0.01 mg, 0.05 mg, 0.1 mg, 0.25 mg, 0.5 mg, 0.75 mg, 1.0 mg, 1.5 mg, 2.0 mg, 4 mg, 5 mg, 6 mg, 8 mg, 10 mg, 20 mg, 25 mg, 50 mg, 100 mg 또는 200 mg의 단위 용량으로 투여된다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 조성물은 1일 1회, 1일 2회, 1일 3회, 1일 4회, 격일, 1주 2회, 매주, 격주, 1개월 2회, 매월, 분기별, 6개월 1회, 또는 매년 투여된다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 경구로 조성물의 투여는 대상체의 혈장 엔독시펜을 30 nM 초과의 항정 상태 수준으로 유지한다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 경구로 조성물의 투여는 대상체의 혈장 엔독시펜을 제1 용량 투여(제1일) 후 제14일까지 30 nM 초과의 항정 상태 수준으로 달성한다.
또 다른 측면에서, 본 개시는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 경구로 조성물의 투여는 대상체의 혈장 엔독시펜을 제1 용량 투여(제1일) 후 제14일까지 30 nM 초과의 항정 상태 수준으로 달성한다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 경구로 조성물의 투여는 대상체에서 엔독시펜의 최대 혈장 수준까지의 시간을 투여 후(post-dose) 2 내지 10시간에 달성한다.
또 다른 측면에서, 본 개시는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 경구로 조성물의 투여는 대상체에서 엔독시펜의 최대 혈장 수준까지의 시간을 투여 후(post-dose) 2 내지 10시간에 달성한다.
또 다른 측면에서, 본 개시는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 대상체에서 엔독시펜의 평균 터미널 제거 반감기는 투여 후 30 내지 60시간의 범위이다.
또 다른 측면에서, 본 개시는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 대상체에서 엔독시펜의 평균 터미널 제거 반감기는 투여 후 40 내지 55시간의 범위이다.
또 다른 측면에서, 본 개시는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물을 투여하는 단계를 포함하여 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 대상체를 치료하는 방법에 관한 것이며, 여기서 조성물의 투여는
a. 대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기;
b. 투여 후 2시간 내지 10시간 범위의 엔독시펜의 최대 혈장 수준까지의 시간; 및
c. 30 nM 초과의 엔독시펜의 항정 상태 혈장 수준
을 달성한다.
또 다른 측면에서, 본 개시는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물을 투여하는 단계를 포함하여 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 및 외음부암으로 이루어진 군으로부터 선택되는 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖거나 가질 위험이 있는 대상체를 치료하는 방법에 관한 거이며, 여기서 조성물의 투여는
a. 대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기;
b. 투여 후 2시간 내지 10시간 범위의 엔독시펜의 최대 혈장 수준까지의 시간; 및
c. 30 nM 초과의 엔독시펜의 항정 상태 혈장 수준
을 달성한다..
또 다른 측면에서, 본 개시는 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿, 또는 지연 방출 캡슐로 제형화된 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물을 투여하는 단계를 포함하여 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암 및 외음부암으로 이루어진 군으로부터 선택되는 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖거나 가질 위험이 있는 대상체를 치료하는 방법에 관한 것이며, 여기서 조성물의 투여는
a. 대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기;
b. 투여 후 2시간 내지 10시간 범위의 엔독시펜의 최대 혈장 수준까지의 시간; 및
c. 30 nM 초과의 엔독시펜의 항정 상태 혈장 수준
을 달성한다.
또 다른 측면에서, 본 개시는 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿, 또는 지연 방출 캡슐로 제형화된 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물을 투여하는 단계를 포함하여 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 및 외음부암으로 이루어진 군으로부터 선택되는 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖거나 가질 위험이 있는 대상체를 치료하는 방법에 관한 것이며, 여기서 조성물의 투여는
a. 대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기;
b. 투여 후 2시간 내지 10시간 범위의 엔독시펜의 최대 혈장 수준까지의 시간; 및
c. 30 nM 초과의 엔독시펜의 항정 상태 혈장 수준
을 달성하며;
여기서 엔독시펜의 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80% 또는 적어도 90%는 장에서 방출된다.
또 다른 측면에서, 본 개시는 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿, 또는 지연 방출 캡슐로 제형화된 0.01 mg 내지 200 mg의 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물을 투여하는 단계를 포함하여 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 및 외음부암으로 이루어진 군으로부터 선택되는 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖거나 가질 위험이 있는 대상체를 치료하는 방법에 관한 것이며, 여기서 조성물의 투여는
a. 대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기;
b. 투여 후 2시간 내지 10시간 범위의 엔독시펜의 최대 혈장 수준까지의 시간; 및
c. 30 nM 초과의 엔독시펜의 항정 상태 혈장 수준
을 달성하며;
여기서 엔독시펜의 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80% 또는 적어도 90%는 장에서 방출된다.
또 다른 측면에서, 본 개시는 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿, 또는 지연 방출 캡슐로 제형화된 0.01 mg 내지 200 mg의 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물을 매일 투여하는 단계를 포함하여 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 및 외음부암으로 이루어진 군으로부터 선택되는 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖거나 가질 위험이 있는 대상체를 치료하는 방법이 제공되며, 여기서 조성물의 투여는
a. 대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기;
b. 투여 후 2시간 내지 10시간 범위의 엔독시펜의 최대 혈장 수준까지의 시간; 및
c. 30 nM 초과의 엔독시펜의 항정 상태 혈장 수준
을 달성한다.
또 다른 측면에서, 본 개시는 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿, 또는 지연 방출 캡슐로 제형화된 0.01 mg 내지 200 mg의 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물을 매일 투여하는 단계를 포함하여 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 및 외음부암으로 이루어진 군으로부터 선택되는 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖거나 가질 위험이 있는 대상체를 치료하는 방법에 관한 것이며, 여기서 조성물의 투여는
a. 대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기;
b. 투여 후 2시간 내지 10시간 범위의 엔독시펜의 최대 혈장 수준까지의 시간; 및
c. 30 nM 초과의 엔독시펜의 항정 상태 혈장 수준
을 달성하며;
여기서 엔독시펜의 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80% 또는 적어도 90%는 장에서 방출된다.
또 다른 측면에서, 본 개시는 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿, 또는 지연 방출 캡슐로 제형화된 0.01 mg 내지 200 mg의 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 경구 조성물을 매일 투여하는 단계를 포함하여 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 및 외음부암으로 이루어진 군으로부터 선택되는 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖거나 가질 위험이 있는 대상체를 치료하는 방법에 관한 것이며, 여기서 조성물의 투여는
a. 대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기;
b. 투여 후 2시간 내지 10시간 범위의 엔독시펜의 최대 혈장 수준까지의 시간; 및
c. 30 nM 초과의 엔독시펜의 항정 상태 혈장 수준
을 달성하며;
여기서 무한대 시간으로 외삽된 곡선 아래 평균 면적(AUC0-inf)은 200 hr*ng/mL 내지 10000 hr*ng/mL, 300 hr*ng/mL 내지 8000 hr*ng/mL, 400 hr*ng/mL 내지 6000 hr*ng/mL 또는 700 hr*ng/mL 내지 6000 hr*ng/mL이고;
여기서 엔독시펜의 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80% 또는 적어도 90%는 장에서 방출된다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서방법은 1차 요법, 선행보조 요법, 또는 보조 요법이다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 조성물은 대상체에게 조합 요법으로 투여된다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 조성물은 대상체에게 단독으로 또는 제2 치료제와의 조합으로 투여된다.
또 다른 측면에서, 본 개시는 본원에 개시된 방법 중 어느 하나 하나에 따라 제조되는 (Z)-엔독시펜 또는 그의 염 또는 다형체를 포함하는 조성물을 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법에 관한 것이며, 여기서 조성물은 대상체에게 비칼루타미드, 엔잘루타미드, 또는 항암 약물, 예컨대 트라스투주맙, 항신생물제, 예컨대 카페시타빈(젤로다), 카르보플라틴(파라플라틴), 시스플라틴(플라티놀), 시클로포스파미드(네오사르), 도세탁셀(도세프레즈, 탁소테레), 독소루비신(아드리아마이신), 페길화된 리포솜 독소루비신(독실), 에피루비신(엘렌스), 플루오로우라실(5-FU, 아드루실), 겜시타빈(젬저), 메토트렉세이트(다수의 브랜드명), 파클리탁셀(탁솔), 단백질 결합된 파클리탁셀(아브락산), 비노렐빈(나벨빈), 에리불린(할라벤), 익사베필론(익셈프라), 및 ATP-카세트 결합 단백질 수송 억제제로 이루어진 군으로부터 선택되는 제2 치료제와 함께 투여된다.
또 다른 측면에서, 본 개시는 대상체에서 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다의 치료 및 예방을 위한 용도에 관한 것이며, 용도는
대상체에게 타목시펜을 포함하는 제1 조성물을 투여하는 단계;
대상체로부터 수득되는 시험 샘플에서 대상체의 타목시펜-대사산물 프로파일을 결정하는 단계;
참조 타목시펜-대사산물 프로파일에서 혈장 엔독시펜의 수준과 비교하여 대상체의 타목시펜-대사산물 프로파일에 기초하여 대상체의 혈장 엔독시펜의 감소된 수준을 결정하는 단계; 및
대상체에게 본 개시의 조성물을 투여하는 단계
를 포함한다.
또 다른 측면에서, 본 개시는 대상체에서 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다의 치료 및 예방을 위한 용도에 관한 것이며, 용도는
대상체에게 타목시펜을 포함하는 제1 조성물을 투여하는 단계;
대상체로부터 수득되는 시험 샘플에서 대상체의 타목시펜-대사산물 프로파일을 결정하는 단계;
참조 타목시펜-대사산물 프로파일에서 혈장 엔독시펜의 수준과 비교하여 대상체의 타목시펜-대사산물 프로파일에 기초하여 대상체의 혈장 엔독시펜의 감소된 수준을 결정하는 단계; 및
대상체에게 본 개시의 조성물을 투여하는 단계
를 포함하며;
여기서 조성물은 대상체의 혈장 엔독시펜을 30 nM 초과의 항정 상태 수준으로 유지하기에 충분한 용량으로 경구 투여되거나;
여기서 타목시펜-대사산물 프로파일은 적어도 타목시펜, 4-히드록시타목시펜, N-데스메틸타목시펜, 또는 엔독시펜의 패널을 포함하거나;
여기서 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애는 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 또는 외음부암이거나;
여기서 호르몬 의존적 유방 장애는 전암, 초기 단계 암, 비-전이성 암, 전-전이성 암, 국소 진행 암, 또는 전이성 암이거나;
여기서 호르몬 의존적 유방 장애는 비-전이성 암이거나;
여기서 대상체는 전립선암을 갖고, 여기서 대상체는 또한 여성형 유방증을 갖거나 가질 위험이 있거나; 또는
여기서 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애는 타목시펜 불응성 또는 타목시펜 내성이다.
또 다른 측면에서, 본 개시는 (a) 본 개시의 조성물; 및 (b) 조성물을 수용하기 위한 밀폐된 용기; 및 c) 경구로 투여되는 조성물의 사용 설명서를 포함하는 이를 필요로 하는 대상체에게 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖거나 가질 위험이 있는 대상체를 치료하기 위한 키트에 관한 것이다.
또 다른 측면에서, (a) 본 개시의 조성물; 및 (b) 조성물을 수용하기 위한 밀폐된 용기; 및 c) 경구로 투여되는 조성물의 사용 설명서를 포함하는 이를 필요로 하는 대상체에게 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖거나 가질 위험이 있는 대상체를 치료하기 위한 키트에 관한 것이며, 여기서 키트는 비칼루타미드, 엔잘루타미드 및 항암 약물, 예컨대 트라스투주맙, 항신생물제, 예컨대 카페시타빈(젤로다), 카르보플라틴(파라플라틴), 시스플라틴(플라티놀), 시클로포스파미드(네오사르), 도세탁셀(도세프레즈, 탁소테레), 독소루비신(아드리아마이신), 페길화된 리포솜 독소루비신(독실), 에피루비신(엘렌스), 플루오로우라실(5-FU, 아드루실), 겜시타빈(젬저), 메토트렉세이트(다수의 브랜드명), 파클리탁셀(탁솔), 단백질 결합된 파클리탁셀(아브락산), 비노렐빈(나벨빈), 에리불린(할라벤), 익사베필론(익셈프라), 및 ATP-카세트 결합 단백질 수송 억제제로 이루어진 군으로부터 선택되는 제2 치료제를 포함한다.
또 다른 측면에서, 본 개시는 조성물을 포함하는 키트에 포함된 사용 설명서에 따라 이를 필요로 하는 대상체에게 본원에 기재된 임의의 방법에 따라 제조되는 조성물을 투여하는 방법에 관한 것이다.
실시예
본원에 사용된 약어
ACN/PPW = 아세토니트릴/공정 정제수
NaHCO3 = 탄산수소나트륨
HCl = 염산
THF = 테트라히드로푸란
MeTHF = 2-메틸테트라히드로푸란
CH2Cl2 = 디클로로메탄
EtOH = 에탄올
MeOH = 메탄올
EtOAc = 에틸 아세테이트
IPA = 이소프로필 알콜; 이소프로판올
TFA = 트리플루오로아세트산
TCA = 트리클로로아세트산
PPW = 공정 정제수
IPA/PPW = 이소프로필 알콜/공정 정제수
실시예 1:
(Z)-엔독시펜 유리 염기의 농축 및 제조
A. 분별 결정화
(Z)-엔독시펜 유리 염기(약물 물질)은 본원에 기재된 분별 결정화로 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물로부터 제조될 수 있다. 간략히, 엔독시펜 (E)-엔독시펜과 (Z)-엔독시펜 이소형의 상업적으로 입수 가능한 조(crude) 혼합물(Astatech Ltd. China)을 출발 물질로 사용하여 유리 염기로서의 (Z)-엔독시펜을 제조하였다.
i. E/Z-엔독시펜 용액은 각각 51/1, 1/1.8, 및 1/5.8의 비율(E/Z 비율)로 (E)-엔독시펜 및 (Z)-엔독시펜을 갖는 1g의 엔독시펜을 10x 부피의 용매인 이소프로판올(IPA)에 현탁시킴으로써 제조하였다. 제2 세트의 용액은 51/1, 1/1.8, 및 1/5.8의 E/Z 비율의 각각 1 g의 엔독시펜을 10x 부피의 용매 EtOAc에 현탁시킴으로써 제조하였다. 용액을 50℃의 제1 온도로 가열하여 고온(hot) 슬러리를 생성하고, 50℃에서 밤새 교반하였다. 고온 슬러리 샘플을 셀룰로스 필터 공극 크기 0.5 μm를 사용하여 여과시켰다. 이어서, 혼합물을 12시간 동안 교반하면서 제2 온도인 실온 23℃로 냉각시켜 결정화를 수행하였다. 냉각된 샘플의 분취량을 셀룰로스 필터 공극 크기 0.5 μm를 사용하여 여과시켰다. 온도 50℃ 및 23℃ 각각에서의 고체 및 여과액의 분취량을 취하고 HPLC-UV로 모니터링하여 그들의 E/Z-엔독시펜 비율을 결정하였다. (Z)-엔독시펜 농축된 여과액 및 E-엔독시펜 농축된 여과액을 그들의 각각의 모액에 다시 첨가하였다.
샘플의 HPLC-UV 분석은 루나(Luna) 페닐-헥실 컬럼(4.6 mm x 150 mm x 3 μm의 입자 크기)을 사용하여 수행하였다. 컬럼 온도는 40℃였으며, 샘플 구획 온도는 주위 온도로 유지되었다. 샘플은 샘플 버퍼로서 메탄올 중에 0.2 mg/ml의 샘플로 고체 및 여과액을 용해시킴으로써 제조하였다. 이어서, 샘플을 20 μl의 주입 부피로 컬럼에 주입하였다. 마찬가지로, 구배 용출로 이동상에 약산성(pH 4.3) 암모늄 포르메이트 버퍼/메탄올을 사용하였다. 이동상 시스템은 하기로 이루어졌다: 이동상 A(MPA) 버퍼 물 중 0.03% HCOOH 버퍼를 갖는 10 mM HCOONH4이고, 이동상 B(MBP) 버퍼는 사용된 구배 프로그램을 갖는 메탄올 중의 10 mM HCOONH4였다.

작업 시간은 30 m이었고, 유속은 1.0 mL/m이었으며, 지연 시간은 5 m이었다. 샘플을 243 nm에서 UV 검출로 검출하였다. 보유 시간은 전형적으로 (E)-엔독시펜에 대해 12.95 내지 13.20 m이고, (Z)-엔독시펜에 대해 14.0 내지 14.33 m이었다. (E)-엔독시펜에 대한 상대적 보유 시간(RRT)은 0.4 내지 0.9이고 (Z)-엔독시펜에 대해서는 1.0이었다.

결과(표 2)는 Z-엔독시펜이 상업적 조 E/Z-엔독시펜 출발 물질을 사용하여 IPA 및 EtOAc로 제조된 엔독시펜 혼합물의 고온 슬러리로부터 농축될 수 있다는 것을 나타낸다. Z-엔독시펜은 1:1의 E/Z를 갖는 E/Z-엔독시펜 혼합물로부터 및 혼합물 중 더 높은 Z-엔독시펜 수준, 예컨대 1:8, 및 1:5.8로 정제될 수 있다. 실질적인 Z-엔독시펜 농축은 분결 결정화 방법을 이용하여, 예컨대 본원의 실시예 1A에 기재된 1:90의 E/Z 비율로 단일 단계로 달성되었다. (Z)-엔독시펜은 EtOAc 중에서 (E)-엔독시펜보다 더 가용성인 것으로 나타났다. (Z)-엔독시펜은 또한 IPA와 비교하여 EtOAc에서 더 다량으로 여과액에 남아있는 경향이 있었다. 고체 및 여과액 모두 (Z)-엔독시펜 유리 염기의 제조에 유용하다. 여과액을 제1 여과액(제1 모액)에 첨가함으로써 Z-엔독시펜의 연속 농축이 또한 수행될 수 있다.
유사하게, E-엔독시펜은 E/Z 혼합물에 더 높은 E-엔독시펜 수준을 갖는 E/Z-엔독시펜 혼합물로부터 정제될 수 있다.
ii. E/Z-엔독시펜 혼합물을 IPA/PPW(1:1 v/v)의 혼합물로 제조된 고온 슬러리에 현탁시키고 추출하고 미리냉각된 IPA/PPW(1:1)로 세척하여 농축된 (Z)-엔독시펜을 갖는 결정질 고체 및 여과액을 형성한 실험에서 유사한 결과가 얻어졌다.
B. 고체 분획으로부터의 재결정화
(Z)-엔독시펜의 연속 농축은 51:1, 1:1.8 및 1:5.6의 E/Z-엔독시펜 비율을 갖는 엔독시펜 혼합물 및 분별 결정화로부터 수득된 고체 및 모액을 하나 이상의 라운드의 재결정화에 적용함으로써 수행될 수 있다.
(Z)-엔독시펜은 조 E/Z-엔독시펜 혼합물, (Z)-농축된 결정질 고체 및 (Z)-농축된 모액으로부터 본원에 기재된 바와 같은 조건 하에서 용매, 예컨대 에탄올, 에틸 아세테이트/n-헵탄, 아세톤/MTBE, IPA, IPA/PPW로 재결정화함으로써 실질적으로 정제된 형태로 농축될 수 있다.
에탄올. 상기의 E/Z-엔독시펜의 고온 IPA 슬러리 혼합물로부터 수득된 결정질 고체를 사용하여 더욱 정제된 Z-엔독시펜을 수득하였다. 23℃로 냉각시킨 후, 상기의 표 2의 샘플 2(80 g) 및 샘플 3(79 g)의 고체 분획을 합하고, 1000 mL(1L) EtOH를 사용하여 재결정화하였다. 에탄올 혼합물을 환류로 가열하였다. 용해점은 75℃였다. 가열된 용액을 셀룰로스 필터(공극 크기 0.5 μm)를 사용하여 여과시켜 이질 불순물, 예컨대 폴리싱 여과에 의한 면섬유를 제거하였다. 이어서, 여과액 용액을 30분 동안 70℃에서 초기에 교반하였다. 이어서, 용액을 0℃ - 5℃로 냉각시키고, 5 h 동안 0℃ - 5℃에서 연속적으로 교반하였다. 용액을 다시 여과시켰다. 필터 상의 결정을 200 mL의 EtOH로 세척하였다.
결과는, 엔독시펜의 고체 결정은 ≥1/100(0.22%/99.48%)의 E/Z 비율을 가졌으며 여과액 중의 엔독시펜은 EtOH로 재결정화될 때 1/5.9(13.93%/82.48%)의 E/Z 비율을 가졌다는 것을 나타낸다. 이 방법으로 수행된 적어도 3개의 상이한 실험의 HPLC 분석은, (Z) 엔독시펜 보유 시간은 14.00 m이고 E-엔독시펜 보유 시간은 13.14 m이라는 것을 나타내었다. (Z)-엔독시펜 수준은 97% 내지 99.48%의 범위였고, (E)-엔독시펜 수준은 0.22% 내지 2.14%의 범위였다. 다른 불순물은 <1%였다.
에틸 아세테이트/n-헵탄. EtOAc/n-헵탄으로의 재결정화의 가능성이 하기에 기재되는 바와 같이 연구되었다.
5 g의 엔독시펜(E/Z 비율 1/5.8)을 EtOAc/n-헵탄(140 mL, 1/1 v/v)에 첨가하고, 밤새 교반하면서 환류(~82℃)로 가열하여 투명한 용액을 수득하였다. 혼합물의 흐림점은 30℃였다. 이어서, 혼합물을 실온으로 냉각시켜 생성물을 침전시키고, 여과시켰다. 상술된 바와 같이 단리시킨 후, 고체 생성물의 엔독시펜은 HPLC에 의해 66%(3.3 g)의 생성물 수율로 1/8.1(10.9%:88.60%)의 E/Z 비율을 갖는 것으로 결정되었다. 여과액 중의 (Z)-엔독시펜을 1:3(23.94%:74.47%)의 E/Z 비율로 농축시켰다. EtOAc/n-헵탄을 사용한 재결정화는 또한 표 3에 나타난 바와 같이 고온 IPA 슬러리와 유사한 비율로 정제된 Z-엔독시펜을 제공할 수 있다.

아세톤/MTBE. 0.5 g의 엔독시펜(E/Z 비율 1/1.8)을 아세톤(10 v; 5 mL) 용매에 첨가하고, 반용매 MTBE(9 mL)를 용액에 첨가하고 밤새 교반하면서 환류(~82℃)로 가열하여 투명한 용액을 수득하였다. 혼합물의 흐림점은 ~22℃였다. 이어서, 혼합물을 실온으로 냉각시켜 생성물을 침전시키고 여과시켰다. 고체 생성물 중의 엔독시펜은 HPLC에 의해 39/1의 E/Z 비율을 갖는 것으로 결정되었으며, 여과액은 1:2.6의 E/Z 비율을 갖는 (Z)-엔독시펜으로 농축되었다. 아세톤/MTBE 용매 시스템은 여과액 중의 (Z)-엔독시펜으로부터 E-엔독시펜을 고체로 분리하는 데 유용하다
C. 모액으로부터 재결정화
정제된 (Z)-엔독시펜은 하기 표 4 및 5에 나타난 바와 같이 (Z)-엔독시펜으로 농축된 모액으로부터 회수될 수 있다.
i. 조 E/Z-엔독시펜 혼합물의 출발 물질은 표 4에 나타난 바와 같이 제1 용매인 에틸 아세테이트 중에 용해되어 제1 모액을 형성할 수 있으며, 이로부터 제2 용매, 예컨대 n-헵탄 또는 에탄올의 제2 단계 첨가에 의해 (Z)-엔독시펜이 회수될 수 있다.

ii. (Z)-엔독시펜은 조 E/Z-엔독시펜 혼합물을 제1 용매인 에틸 아세테이트 중에 용해시켜 (Z)-엔독시펜을 제1 모액에 농축시키는 제1 단계; 제2 용매인 IPA를 첨가하여 결정질 (Z)-엔독시펜 및 제2 모액을 형성하는 제2 단계; 및 제3 용매인 에탄올을 첨가하여 제2 모액으로부터 (Z)-엔독시펜을 재결정화 및 회수하는 단계를 포함하는 공정에 의해 제조되었다.
간략히, 엔독시펜(E/Z = 1/1.8, 30 g) 및 (E/Z = 1.2/1, 50 g 비-GMP)을 각각 EtOAc 300 mL 및 500 mL 중에 용해시키고, 용해되게 가열하였다(~70℃). 이어서, 혼합물을 침전을 위해 밤새 교반하면서 25℃로 냉각시켰다. 현탁액을 여과시키고, 고체 및 여과액의 E/Z 비율을 HPLC로 결정하였다.
이어서, 여과액을 건조되게 농축시키고, 각각 IPA 230 mL 및 900 mL를 첨가하여 교반하면서 고온 슬러리(50℃)를 형성하였다. 고온 슬러리를 50℃에서 밤새 계속해서 교반하였다. 이어서, 슬러리를 각각 0.5 h 및 1 h 동안 25℃로 냉각시키고 여과시켰다. 고체 및 여과액 중의 Z-엔독시펜을 다시 결정하였다.
각각의 샘플의 고체(14.8 g 및 21.6 g)를 NMT 70℃에서 용해되게 가열하고 각각 0.5 h 및 1 h 동안 25℃로 냉각시킴으로써 148 mL 및 220 mL EtOH로부터 추가로 재결정화하였다. 샘플을 1 h 동안 0℃ - 5℃로 추가로 냉각시키고 여과시켰다. 고체 및 여과액의 E/Z 비율을 HPLC로 다시 결정하였다. 각각의 샘플의 E/Z 비율 및순도가 하기 표 5에 제공된다.

결과는 90% 초과의 정제된 결정질 (Z)-엔독시펜이 E/Z-엔독시펜 혼합물을 제1 용매인 에틸 아세테이트로 분별 결정화하여 결정질 고체 및 제1 모액을 형성하고, 제1 모액을 제2 용매, 예컨대 고온 IPA로 결정화하여 제2 결정질 고체 및 제2 모액을 형성한 후, 고체를 제3 용매, 예컨대 에탄올로부터 재결정화함으로써 수득될 수 있다는 것을 나타낸다. 95% 초과의 (Z)-엔독시펜의 수율도 또한 가능하다. (E)/(Z)-엔독시펜의 비율은 ~1.2:1 및 1:1.8에서 1:51 내지 1:118 이상으로 개선될 수 있다. (Z)-엔독시펜의 불순물 프로파일을 상술된 바와 같이 HPLC 방법으로 분석하였으며, <2% 미만인 것으로 밝혀졌다. (Z)-엔독시펜 및 (E)-엔독시의 보유 시간은 각각 14.19 m 및 13.2 m이었다.
실시예 2:
(E)-엔독시펜-풍부 E/Z-혼합물의 재평형화
(E)-엔독시펜-풍부 E/Z-엔독시펜 혼합물은 또한 (정제된 (E)-엔독시펜의 제조에 유용할 뿐만 아니라) Z-엔독시펜 유리 염기 제조에도 유용하다. (E)-엔독시펜-풍부 E/Z-엔독시펜 혼합물은 ≥ 1.5:1의 E/Z 비율(예컨대, 51:1의 E/Z 비율)에서 약 ~1:1 혼합물로 재평형화된다. 본 개시는 재평형화가 임의의 E/Z 비율, 예컨대 99:1 내지 1:99을 갖는 출발 E/Z-엔독시펜 혼합물로 수행될 수 있다는 아이디어를 포함한다. 이러한 재평형화는 상기 실시예 1에서 기재되는 바와 같이 안정한 (E)/(Z)-엔독시펜 ~1:1 혼합물의 제조 뿐만 아니라 Z-엔독시펜 유리 염기의 추가의 농축 및 정제에도 유용하다.
간략히, 조 E/Z-엔독시펜을 산, 예컨대 염산, 트리클로로아세트산 또는 트리플루오로아세트산의 존재 하에서 불활성 용매, 예컨대 아세토니트릴/물(8:1) 또는 디클로로메탄 중에 용해시킴으로써 (E)-엔독시펜을 (Z)-엔독시펜으로 전환시킨다. ACN/PPW와의 반응 혼합물을 1시간 동안 60℃에서 교반하고, 디클로로메탄 및 TFA과의 반응 혼합물을 1시간 동안 RT에서 교반하였다. 표 6에 기재되는 바와 같이 후처리를 수행한 후, 생성물의 E/Z 비율은 90% 초과의 순도를 갖는 E/Z-엔독시펜(비율 약 1:1)인 것으로 결정되었다.

실시예 3:
(E)-엔독시펜의 정제
반응 혼합물을 NMT 70℃로 가열하여 투명한 용액을 수득하고 0℃ - 5℃로 냉각시켜 생성물을 침전시킴으로써 EtOH(500 mL)에서 재결정화하여 (E)-엔독시펜을 유사하게 정제하였다. 본원에 기재된 방법으로 정제한 후, 38.1 g의 (E)-엔독시펜을 97.6% 순도로 76% 수율로 수득하였다.
실시예 4: 스케일 업
본원에 개시된 합성 및 정제 방법은 또한 대상체에서 임상 사용을 위한 산업적 약물 제품 투여 형태, 예컨대 경구, 경피/국소, 비강, 관내 비경구 투여 형태를 제조하기 위해 (Z)-엔독시펜의 제조를 확장하는 데 유용하다. 1/5.8의 E/Z 비율의 5 g, 100 g, 및 110 g의 화학식 (III)의 화합물인 E/Z-엔독시펜 혼합물을 사용하는 결정화의 가능성 연구는 E/Z-엔독시펜 혼합물을 10x 부피의 IPA에 현탁시키고 50℃로 가열하고 4 h 동안 50℃에서 교반함으로써 수행되었다. 샘플을 셀룰로스 필터 공극 크기 0.5 μm를 사용하여 여과시키고, E/Z 비율을 고체에서 결정하였다. 샘플을 23℃로 냉각시키고, 4 h 동안 23℃에서 계속해서 교반하였다. 샘플을 다시 여과시켰다. 고체 및 여과액을 상술된 바와 같이 HPLC에 의해 (E)-엔독시펜 및 (Z)-엔독시펜 수준에 대해 모니터링하였으며, 결과가 하기 표 7에 제공된다.

결과는 Z-엔독시펜이 결정질 고체로서 ≥ 90% 초과 뿐만 아니라 여과액 중 ≥ 65% (Z)-엔독시펜 유리 염기의 농도로 IPA 용매로부터 E/Z-엔독시펜 혼합물로부터 수득될 수 있다는 것을 나타낸다. 따라서, IPA는 제1 용매, 또는 제2 용매 또는 둘 다로서 유용하다.
실시예 5:
[4-[2-(메틸아미노)에톡시]페닐](4-히드록시페닐)메탄온의 산업적으로 확장 가능한 제조

적합한 10L 반응기를 N2 분위기 하에서 화학식 (I)의 화합물인 출발 물질 [4-[2-(디메틸아미노)에톡시]페닐](4-히드록시페닐)메탄온(0.5 kg, 1.0 equiv.), DIPEA(1.5 kg, 6.65 equiv., 3.0 wt./wt.) 및 테트라히드로푸란(5 L, 10 vol./wt., 8.9 wt./wt.)으로 충전하였다. 내부 온도를 NMT 20℃로 유지하면서 1-클로로에틸 클로로포르메이트(1.7 kg, 6.65 equiv., 3.3 wt./wt.)를 서서히 첨가하였다. 혼합물을 환류로 가열하고, NLT 12 hr 동안 환류에서 교반하였다. 혼합물을 부피가 교반 가능한 최저 부피에 도달할 때까지 NMT 75℃에서 감압 하에 증발시켰다. 메탄올(2.5 L, 5 vol./wt., 4.0 wt./wt.)을 서서히 첨가하고, 혼합물을 부피가 교반 가능한 최저 부피에 도달할 때까지 NMT 75℃에서 감압 하에 증류시켰다. 메탄올(2.5 L, 5 vol./wt., 4.0 wt./wt.)을 첨가하고, 혼합물을 부피가 교반 가능한 최저 부피에 도달할 때까지 NMT 75℃에서 감압 하에 증류시켰다. 메탄올(2.5 L, 5 vol./wt., 4.0 wt./wt.)을 다시 한번 첨가하고, 혼합물을 부피가 다시 한번 교반 가능한 최저 부피에 도달할 때까지 NMT 75℃에서 감압 하에 추가로 증류시켰다. 메탄올(2 L, 4 vol./wt., 3.2 wt./wt.) 및 6N HCl(2 L, 4 vol./wt., 4.0 wt./wt.)을 혼합물에 첨가하고, 혼합물을 환류로 가열하였다. 혼합물을 NLT 12 hr 동안 환류에서 교반하였다. 반응 완료 후, 혼합물을 대부분의 MeOH가 제거될 때까지 NMT 75℃에서 감압 하에 증발시켰다. 혼합물을 25±5℃로 냉각시키고, 8N NaOH(2.5 L, 5 vol./wt., 5.0 wt./wt.)를 혼합물의 pH가 11-12일 때까지 서서히 첨가하였다. 혼합물을 NLT 2 hr 동안 0-5℃에서 교반하였다. 혼합물을 여과시키고, H2O(1 L, 2 vol./wt.) 및 에틸 아세테이트(1 L, 2 vol./wt., 1.8 wt./wt.)로 세척하였다. 습윤 케이크를 감압 하에 NMT 50℃에서 건조시켜 화학식 (II)의 화합물인 (4-히드록시페닐)(4-(2-(메틸아미노)에톡시)페닐)메탄온을 수득하였다. 수율: 297 gm, 63%; 순도: 100%( 예상 수율 - 60 - 90%)
실시예 6:
[4-[2-(메틸아미노)에톡시]페닐](4-히드록시페닐)메탄온의 산업적으로 확장 가능한 제조

적합한 2 L 반응기를 N2 분위기 하에서 화학식 (I)의 화합물인 출발 물질 [4-[2-(디메틸아미노)에톡시]페닐](4-히드록시페닐)메탄온(70 gm, 1.0 equiv.), N-에틸디이소프로필아민(126 gm, 4.0 equiv., 1.8 wt.) 및 테트라히드로푸란(700 mL, 10.0 vol., 8.9 wt.)으로 충전하였다. 혼합물을 NLT 60℃로 가열하고, 1-클로로에틸 클로로포르메이트(140 gm, 4.0 equiv., 2.0 wt.)를 첨가하였다. 혼합물을 환류로 가열하고, NLT 12 hr 동안 교반하였다. 혼합물을 부피가 3 vol.에 도달할 때까지 농축시켰다. 메탄올(350 mL, 5.0 vol., 4.0 wt.)을 서서히 첨가하고, 혼합물을 부피가 3 vol.(210 mL)에 도달할 때까지 농축시켰다. 이어서, 메탄올(350 mL, 5.0 vol., 4.0 wt.)을 첨가하고, 혼합물을 부피가 4 vol.(280 mL)에 도달할 때까지 농축시켰다. 6N HCl(280 mL, 4.0 vol., 4.0 wt.)을 첨가하고, 혼합물을 환류로 가열하였다. 혼합물을 NLT 12 hr 동안 환류에서 교반하였다. 반응 완료 후, 혼합물을 부피가 ~4 vol.(280 mL)에 도달할 때까지 농축시켰다. 혼합물을 주위 온도로 냉각시키고, 8N NaOH(NLT 350 mL, 5.0 vol., 5.0 wt.)를 혼합물의 pH가 NLT 13일 때까지 서서히 첨가하였다. 에틸 아세테이트(280 mL, 4.0 vol., 3.6 wt.)를 추출을 위해 혼합물에 첨가하였다. 상 분리 후, 수층에 pH가 8-10일 때까지 6N HCl(NLT 42 mL, 0.6 vol., 0.6 wt.)을 첨가하였다. 혼합물을 0±5℃로 냉각시키고, NLT 2 hr 동안 교반하였다. 혼합물을 여과시키고, 정제수(NLT 140 mL, 2.0 vol., 2.0 wt.) 및 에틸 아세테이트(NLT 140 mL, 2 vol., 1.8 wt.)로 세척하였다. 습윤 케이크를 감압 하에 NMT 60℃에서 건조시켜 화학식 (II)의 화합물인 (4-히드록시페닐)(4-(2-(메틸아미노)에톡시)페닐)메탄온을 수득하였다. 수율: 55.2 gm, 83%; 순도: 100%( 예상 수율 - NLT 70%) .
실시예 7:
맥머리 반응으로 E/Z-엔독시펜 혼합물의 산업적으로 확장 가능한 제조

적합한 10 L 반응기를 N2 분위기 하에서 Zn 분말(0.27 kg, 4.0 equiv., 0.9 wt./wt.) 및 테트라히드로푸란(1.5 L, 5 vol./wt., 4.4 wt./wt.)으로 충전하였다. 내부 온도를 NMT 15℃로 유지하면서 TiCl4(0.42 kg, 2.0 equiv., 1.4 wt./wt.)를 서서히 첨가하였다. 반응물을 환류로 가열하고, NLT 2 hr 동안 환류에서 교반하였다. 테트라히드로푸란(3.0 L, 10 vol./wt., 8.9 wt./wt.) 중의 단계 1로부터 수득된 화학식 (II)의 화합물인 (4-히드록시페닐)(4-(2-(메틸아미노)에톡시)페닐)메탄온(0.297 kg, 1.0 equiv.) 및 프로피오페논(0.21 kg, 1.5 equiv., 0.7 wt./wt.)의 현탁액을 첨가하고, NLT 8 hr 동안 환류를 계속하였다. 혼합물을 20-30℃로 냉각시키고, 25% 염화암모늄(5.9 L, 20 vol./wt., 20 wt./wt.)/시그마 알드리치로부터 입수 가능한 실리카(규조토/이산화규소; 상품명 Celite® S)(0.3 kg, 1.0 wt./wt.) 혼합물로 첨가하였다. 혼합물을 여과시키고, 테트라히드로푸란(0.9 L, 3 vol./wt., 2.7 wt./wt.)으로 세척하였다.
혼합물을 상 분리를 위해 정치시켰다. 유기층을 수집하고, 수층을 테트라히드로푸란(0.9 L, 3 vol./wt., 2.7 wt./wt.)으로 세척하였다. 유기층을 다시 수집하고, 제1 유기층과 합하였다. 합한 유기층을 40% K2CO3(1.2 L, 4 vol./wt., 5.6 wt./wt.)로 세척하였다. 유기층을 부피가 교반 가능한 최저 부피에 도달할 때까지 감압 하에 NMT 75℃에서 농축시켰다. 에틸 아세테이트(1.5 L, 5 vol./wt., 4.5 wt./wt.)를 첨가하고, 혼합물을 부피가 교반 가능한 최저 부피에 도달할 때까지 NMT 75℃에서 감압 하에 증류시켰다. 에틸 아세테이트(1.5 L, 5 vol./wt., 4.5 wt./wt.)를 첨가하고, 혼합물을 부피가 5 vol.(1.5 L)에 도달할 때까지 NMT 75℃에서 감압 하에 증류시켰다. 혼합물을 용해되게 가열하고, n-헵탄(3.0 L, 10 vol./wt., 6.8 wt./wt.)을 첨가하였다. 혼합물을 0±5℃로 냉각시키고, NLT 2 hr 동안 0±5℃에서 교반하였다. 혼합물을 여과시키고, 잔사를 에틸 아세테이트/n-헵탄=1/2(v/v, 1.5 L, 5 vol./wt., 3.7 wt./wt.)으로 세척하였다. 잔류 습윤 케이크를 NMT 60℃에서 감압 하에 건조시켜 화학식 (III)의 화합물인 (Z)-엔독시펜과 (E)-엔독시펜의 혼합물, (E/Z)-4-[1-[4-[2-(메틸아미노)에톡시]페닐]-2-페닐-1-부텐-1-일]-페놀을 수득하였다. 수율: 176 g, 42%; 순도: 81.96%. E/Z 비율: 3.1:1(예상 수율 - 40 - 60%).
적합한 10 L 반응기를 상기 (E/Z)-엔독시펜(0.250 kg, 1.0 wt.), 이소프로판올(1.25 L, 5 vol./wt., 3.9 wt./wt.) 및 정제수(1.25 L, 5 vol./wt., 5.0 wt./wt.)로 충전하였다. 혼합물을 70±5℃로 가열하고, NLT 2 hr 동안 70±5℃에서 교반하였다. 혼합물을 0±5℃로 냉각시키고, NLT 2 hr 동안 0-5℃에서 교반하였다. 혼합물을 여과시키고, 미리냉각된 이소프로판올/정제수=1/1(0.5 L, 2 vol./wt., 3.6 wt./wt.)로 세척하였다. 잔류 습윤 케이크를 NMT 60℃에서 건조시켜 화학식 (III)의 화합물인 (Z)-엔독시펜과 (E)-엔독시펜의 혼합물, (E/Z)-4-[1-[4-[2-(메틸아미노)에톡시]페닐]-2-페닐-1-부텐-1-일]-페놀을 수득하였다. 수율: 105 g, 41%; 순도: 96.79%. E/Z 비율: 48.6:1 (예상 수율 - 40 - 60%).
실시예 8:
맥머리 반응으로 E/Z-엔독시펜 혼합물의 산업적으로 확장 가능한 제조

적합한 반응기에 N2 분위기 하에서 아연 분말(10 gm, 4.0 equiv., 1.0 wt.) 및 테트라히드로푸란(50 mL, 5.0 vol., 4.3 wt.)을 충전하였다. 내부 온도를 NMT 45℃로 유지하면서 염화티타늄(IV)(14 gm, 2.0 equiv., 1.4 wt.)을 첨가하였다. 반응물을 65±5℃로 가열하고, NLT 2 hr 동안 교반하였다. 테트라히드로푸란(100 mL, 10.0 vol., 8.5 wt.) 중의 실시예 5로부터 수득된 화학식 (II)의 화합물인 (4-히드록시페닐)(4-(2-(메틸아미노)에톡시)페닐)메탄온(10 gm, 1.0 equiv.) 및 프로피오페논(5 gm, 1.0 equiv., 0.5 wt.)의 현탁액을 상기 혼합물에 첨가하고, NLT 8 hr 동안 65±5℃에서 교반하였다. 혼합물을 NMT 30℃로 냉각시키고, 40% K2CO3(20 mL, 2.0 vol., 2.0 wt.)를 첨가한다. 혼합물을 NLT 1 hr 동안 교반하였다. 혼합물을 여과시키고, Me-THF(40 mL, 4.0 vol., 3.5 wt.)로 세척하였다. 여과액을 40% K2CO3(30 mL, 3.0 vol., 3.0 wt.)에 첨가하고, NLT 30분 동안 교반하였다. 혼합물을 여과시키고, Me-THF(20 mL, 2.0 vol., 1.7 wt.)로 세척하였다. 여과액을 상 분리를 위해 정치시키고, 유기층을 부피가 3 vol.(30 mL)에 도달할 때까지 농축시켰다. 혼합물을 1N NaOH(100 mL, 10.0 vol., 10.0 wt.)로 추출하고, 유기층을 버렸다. 수층에 염화나트륨(1 gm, 0.10 wt.)을 첨가하고, 이어서 Me-THF(50 mL, 5.0 vol., 4.3 wt.)로 4회 추출하였다. 4개의 유기층을 합하고, 부피가 5 vol.가 될 때까지 농축시켰다. 혼합물을 20% NaCl(50 mL, 5.0 vol., 5.0 wt.)로 추출하였다. 유기층에 이소프로판올(50 mL, 5.0 vol., 3.9 wt.)을 첨가하고, 혼합물을 부피가 3 vol.에 도달할 때까지 농축시켰다. 이소프로판올(50 mL, 5.0 vol., 4.5 wt.)을 첨가하고, 혼합물을 부피가 3 vol.(30 mL)에 도달할 때까지 농축시켰다. 혼합물을 65±5℃로 가열하고, n-헵탄(120 mL, 12.0 vol., 8.2 wt.)을 첨가하였다. 혼합물을 NMT 5℃로 냉각시키고, NLT 2 hr 동안 NMT 5℃에서 교반하였다. 혼합물을 여과시키고, n-헵탄(50 mL, 5 vol., 3.4 wt.)으로 세척하였다. 습윤 케이크를 감압 하에 NMT 70℃에서 건조시켜 화학식 (III)의 화합물인 (Z)-엔독시펜과 (E)-엔독시펜의 혼합물, (E/Z)-4-[1-[4-[2-(메틸아미노)에톡시]페닐]-2-페닐-1-부텐-1-일]-페놀을 수득하였다. 수율: 6.7 g, 45%; 순도: 96.79%. E/Z 비율: 48.6:1 (예상 수율 - NLT 30%).
실시예 9:
Z-엔독시펜의 산업적으로 확장 가능한 농축

적합한 반응기를 화학식 III의 화합물인 (Z)-엔독시펜과 (E)-엔독시펜의 혼합물 및 제1 용매인 에틸 아세테이트(1.1 L, 10 vol./wt., 9.0 wt./wt.)로 충전하였다. 혼합물을 0-5℃로 냉각시키고, 6N HCl(0.3 L, 3 vol./wt., 3.0 wt./wt.)을 혼합물에 서서히 첨가하였다. 혼합물을 60±5℃로 가열하고, NLT 6 hr 동안 60±5℃에서 교반하였다. 혼합물을 0±5℃로 냉각시키고, 8N NaOH(0.3 L, 3.0 vol./wt., 3.0 wt./wt.)를 혼합물의 pH가 ≥ 12일 때까지 서서히 첨가하였다.
혼합물을 상 분리를 위해 정치시키고, 유기층을 수집하고, 수층을 제1 용매인 에틸 아세테이트(0.5 L, 5 vol./wt., 4.5 wt./wt.)로 세척하였다. 유기층을 수집하였다. 합한 유기층을 20% NaCl(0.3 L, 3 vol./wt., 3.0 wt./wt.)로 세척하였다. 유기층을 활성탄(숯)(0.005 kg, 0.05 wt.)으로 처리하고, NLT 1 hr 동안 50±5℃에서 교반하였다. 혼합물을 규조토/실리카(Celite® S)의 층을 통해 여과시키고, 에틸 아세테이트(0.5 L, 5 vol./wt., 4.5 wt./wt.)로 세척하였다. 여과액을 부피가 10 vol.(1.1 L)에 도달할 때까지 NMT 75℃에서 농축시켰다. 혼합물을 0-5℃로 냉각시키고, NLT 2 hr 동안 0-5℃에서 교반하였다. 혼합물을 여과시키고, 여과액을 수집하였다. 여과액을 부피가 교반 가능한 최저 부피에 도달할 때까지 NMT 75℃에서 농축시켰다.
제2 용매인 이소프로판올(1.1 L, 10 vol./wt., 7.9 wt./wt.)을 첨가하고, 혼합물을 부피가 교반 가능한 최저 부피에 도달할 때까지 NMT 75℃에서 농축시켰다. 이소프로판올(1.1 L, 10 vol./wt., 7.9 wt./wt.)을 다시 첨가하고, 혼합물을 부피가 교반 가능한 최저 부피에 도달할 때까지 NMT 75℃에서 농축시켰다. 이소프로판올(1.1 L, 10 vol./wt., 7.9 wt./wt.)을 첨가하고, 혼합물을 부피가 5 vol.(0.5 L)에 도달할 때까지 NMT 75℃에서 농축시켰다. 현탁액을 NLT 6 hr 동안 50±5℃에서 교반하였다. 혼합물을 20±5℃로 냉각시키고, NLT 4 hr 동안 20±5℃에서 교반하였다. 혼합물을 여과시키고, 이소프로판올(0.2 L, 2 vol./wt., 1.6 wt./wt.)로 세척하였다. 습윤 케이크를 NMT 60℃에서 건조시켜 화학식 (IV)의 정제된 (Z-엔독시펜) 고체 화합물을 회백색 고체로서 수득하였다. 수율: 39.5 g, 36%, (E)/(Z) 비율: 1/12.2; 순도(Z): 89.59% by HPLC, 예상 수율 (20-40%).
적합한 1 L 반응기를 상기 화학식 (IV)의 Z-엔독시펜(39 g) 고체 화합물 및 제3 용매인 이소프로판올(390 mL, 10 vol./wt.)로 충전하였다. 현탁액을 NLT 6 hr 동안 50±5℃에서 교반하였다. 혼합물을 20±5℃로 냉각시키고, NLT 4 hr 동안 20±5℃에서 교반하였다. 혼합물을 여과시키고, 이소프로판올(78 mL, 2 vol./wt.)로 세척하였다. 습윤 케이크를 NMT 60℃에서 건조시켜 화학식 (IV)의 정제된 (Z-엔독시펜) 고체 화합물을 회백색 고체로서 수득하였다. 수율: 25 g, 61%, (E)/(Z) 비율: 1/55.1; 순도(Z): 95.81%, 예상 수율 (50-70%).
이런 방법으로, 엔독시펜의 다형체 형태 I을 제조하였다. 생성된 다형체의 2개의 배치에 대한 XRPD 패턴이 도 9 및 10.에 제공된다. 도 9 및 10로부터의 XRPD 피크가 표 8에 제공된다.

실시예 10:
~1:1 E/엔독시펜/Z-엔독시펜 혼합물의 산업적으로 확장 가능한 농축
적합한 반응기를 화학식 III의 화합물인 (Z)-엔독시펜과 (E)-엔독시펜의 혼합물(0.420 kg, 1.0 wt.) 및 에틸 아세테이트(4.2 L, 10 vol./wt., 9.0 wt./wt.)로 충전하였다. 혼합물을 0-5℃로 냉각시키고, 6N HCl(1.3 L, 3 vol./wt., 3.0 wt./wt.)을 서서히 첨가하였다. 혼함물을 60±5℃로 가열하고, NLT 6 hr 동안 60±5℃에서 교반하였다. 혼합물을 0±5℃로 냉각시키고, 8N NaOH(1.3 L, 3.0 vol./wt., 3.0 wt./wt.)를 혼합물의 pH가 ≥ 12일 때까지 서서히 첨가하였다.
혼합물을 상 분리를 위해 정치시키고, 유기층을 수집하고, 수층을 에틸 아세테이트(2.0 L, 5 vol./wt., 4.5 wt./wt.)로 세척하였다. 유기층을 수집하였다. 합한 유기층을 20% NaCl(1.3 L, 3 vol./wt., 3.0 wt./wt.)로 세척하였다. 유기층을 활성탄(숯)(0.02 kg, 0.05 wt.)으로 처리하고, NLT 1 hr 동안 50±5℃에서 교반하였다. 혼합물을 규조토/실리카(Celite® S) 층을 통해 여과시키고, 에틸 아세테이트(2.0 L, 5 vol./wt., 4.5 wt./wt.)로 세척하였다. 여과액 부피가 5 vol.(2.0 L)에 도달할 때까지 NMT 75℃에서 농축시켰다. 혼합물을 환류로 가열하고, 이어서 50±5℃로 냉각시켰다. n-헵탄을 50±5℃에서 서서히 첨가하였다. 혼합물을 0±5℃로 냉각시키고, NLT 2 hr 동안 0±5℃에서 교반하였다. 혼합물을 여과시키고, EtOAc/n-헵탄=1/2(v/v)로 세처하였다. 습윤 케이크를 감압 하에 NMT 60℃에서 건조시켜 실질적으로 순수한 ~1:1 E/Z-엔독시펜을 수득하였다. [수율: 106 g, 25%; (E)/(Z) 비율: 1.1/1; 순도(E/Z): 97.94%, 예상 수율 (20-40%)].
이런 방법으로, 엔독시펜의 다형체 형태 II 및 III이 제조되었다. 다형체 형태 II 및 III의 제조를 위한 상세한 파라미터가 표 9에 제공되며, XRPD 패턴이 도 11-13에 제공된다. 도 11-13으로부터의 XRPD 피크가 표 10에 제공된다.


실시예 11:
Z-엔독시펜 유리 염기의 안정성
상업적으로, 엔독시펜은 E/Z 이성질체 유리 염기 혼합물 뿐만 아니라 > 98% (Z)-엔독시펜 HCl 및 (Z)-엔독시펜 시트레이트 염으로서 입수 가능하다. 수성 및 고체 형태의 Z-엔독시펜 HCl 염의 안정성은 이미 공개되어 있다(Elkins et al. J Pharm Biomed Anal. 2014 January; 88: 174-179). 엘킨(Elkins) 등은 25℃/60% RH에서 53개월 및 40℃/75% RH에서 4.3개월의 고체 상태에서의 (Z)-엔독시펜 HCl의 (E)-엔독시펜으로의 10% 전환에 대한 예측된 t90 값을 제공한다. 전환은 용액 환경에 더 빠를 것으로 예상된다. 수성 매질에서, 엘킨 등은 실온에서 149일 및 45℃에서 9일의 (Z)-엔독시펜 HCl t90 값을 제공한다. 예컨대, 주위 온도 뿐만 아니라 보다 높은 온도 및 습도에서 대상체에게 투여하기에 적합한 약제학적 조성물을 제조하기에 충분히 안정한 (Z)-엔독시펜 유리 염기 제조가 필요하다.
본원에는 실질적으로 순수하고 (적어도 90% (Z)-엔독시펜 유리 염기) 및 주위 및 높은 온도 및 습도의 조건 하에서 안정한 엔독시펜 유리 염기의 제조가 제공된다. 본 개시의 목적 상 안정성은 적어도 6개월 동안 조성물 중 적어도 90% (Z)-엔독시펜의 지속적인 존재로 정의되고, 합성일로부터 시작하여 (E)-엔독시펜으로의 (Z)-엔독시펜 전환에 의해 측정할 수 있다.
A. (Z)-엔독시펜 유리 염기의 가속된 10일 안정성 연구
표 11의 (Z)-엔독시펜 유리 염기 샘플 1의 안정성을 다양한 온도에서 고체 형태로 및 에탄올(EtOH) 용액 중에 저장하는 경우에 대해 연구하였다. 무수 N2 하에서 180 mL HDPE 병에 1 g 분취량의 정제된 (Z)-엔독시펜 유리 염기를 넣고 이어서 이중 LDPE 백에 넣은 후 (E)-엔독시펜 및 (Z)-엔독시펜 수준을 제2일, 제7일 및 제10일에 측정하였다. 각각의 샘플을 함유하는 이중 백 HDPE 병을 개별적으로 알루미늄 호일 백에 포장하고, 샘플의 안정성을 시험하기 위해 개방할 때까지 열로 밀봉하였다. 엔독시펜의 (E)- 및 (Z)-이성질체를 상술된 바와 같이 HPLC로 모니터링하였다. 고체 형태의 (Z)-엔독시펜의 순도는 10일까지 40℃, 60℃, 및 80℃에서 약 97%로 유지되었다. 가속된 10일 연구의 결과는 고체 형태의 (Z)-엔독시펜이 10일까지 시험된 온도에서 (E)-엔독시펜으로 상호전환되지 않는다는 것을 나타낸다.


결과는 (Z)-엔독시펜 유리 염기가 놀랍게도 10일에 걸쳐 더 높은 농도에서 심지어 상승된 온도(40℃)에서도 알콜(예컨대, 에탄올 및 이소프로판올(데이터 제시되지 않음)) 용액에서 안정하다는 것을 나타낸다. 더 높은 온도에서 가속된 안정성은 일반적으로 주위 온도에서 장기간(적어도 18 m) 안정성을 예측하는 것으로 간주된다.
B. 벌크 약물 안정성 연구
본원에 개시된 방법에 의해 제조되는 본 개시의 (Z)-엔독시펜 유리 염기는 놀랍게도 벌크 형태로 안정하다. 벌크 안정성 연구를 위해, ~ 1 g 분취량의 (Z)-엔독시펜 유리 염기를 나일론 케이블 타이로 묶인 이중 LDPE 백에 개별적으로 넣었다. 이어서, LDPE 백을 알루미늄 호일 백에 넣고 열로 밀봉하였다. 안정성을 위한 샘플은 12개월 동안 5℃ 및 25℃/60% RH에서 및 3개월 동안 40℃/75% RH에서 불활성 조건(무수 질소) 하에 단단히 캡핑된 병에 넣었다. 샘플을 제0일, 10일, 1 m 및 3 m의 시점에서 (E)-엔독시펜 및 (Z)-엔독시펜 농도 및 불순물, 칼 피셔(Karl Fischer) 적정에 의한 수분 또는 물 함량, 호기성 박테리아 콜로니 형성 단위 및 외관에 대해 시험하였다. 하기 표 13은 다양한 저장 조건에서 엔독시펜 유리 염기에 대한 9 m에서의 벌크 약물 안정성 데이터를 제공한다.

결과는 고체 Z-엔독시펜 유리 염기가 5℃ 및 25℃/60% RH에서 적어도 9개월 동안 안정하다는 것을 나타낸다. 높은 온도에서의 가속된 안정성은 주위 온도에서 장기간(적어도 18 m) 안정성을 예측하는 것으로 간주된다. (Z)-대-(E) 엔독시펜 상호전환은 연구된 저장 조건에서 최소이고, 물 함량은 1% 이하이고, 호기성 박테리아 플레이트 카운트 TAC(cfu)는 20,000 cfu g/mL 미만이다. 최대 개별 불순물은 각각의 시점에서 측정되었으며, 0.10% 내지 0.11%의 범위였다.
실시예 12:
정제된 고체 Z-엔독시펜 유리 염기의 특징
본원에 개시된 방법에 의해 생성된 (Z)-엔독시펜 유리 염기는 백색 내지 회백색 분말이었다. 이러한 분말의 물 함량은 USP 921의 방법 1c(칼 피셔 적정)를 이용하여 결정 시 1% 이하였다. 잔류 용매를 측정하였다. 메탄올은 3000 ppm 이하(NMT)이고, 테트라히드로푸란은 NMT 720 ppm이고, 이소프로판올, 에틸 아세테이트, n-헵탄, 및 에탄올은 각각 NMT 5000 ppm이었다.
(Z)-엔독시펜의 점화 잔류물은 USP 281의 방법 II로 결정 시 0.1% 이하였다. (Z)-엔독시펜의 점화 잔류물은 0.02% 내지 0.099%의 범위였다. 일부 실시양태에서, 점화 시 잔류물은 0.02%, 0.03%, 0.04%, 0.05%, 0.06%, 0.07%, 0.08%, 0.09%, 및 0.1% 이하이다. 또 다른 측면에서, 중금속은 USP 231의 방법 II로 시험 시 NMT 20 ppm이었다.
고체 (Z)-엔독시펜 유리 염기 중의 원소 수준은 USP 232 및 USP 40의 시험 방법을 이용하여 시험된다.
따라서, 본원에 기재된 공정 및 이들 공정을 이용하여 제조되는 엔독시펜은 임상 및 상업적 등급의 (Z)-엔독시펜을 제조하는 데 적합하다.
Z-엔독시펜의 FTIR 스펙트럼
(Z)-엔독시펜을 표준 광원 및 TGS 검출기와 함께 푸리에(Fourier) 변환 적외선 분광법(기기 유형: FT/IR4600Type A; Jasco: S/N 비율 25,000:1, 최대 해상도 0.7 cm-1)을 이용하여 분석하였다. 적외선 스펙트럼은 트리글리신 술페이트(TGS) 검출기 및 KBr 빔 분리기가 장착된 FTIR 분광기(모델 FT/IR 4600 Type A; 제조자 Jasco)에 AgBr 윈도우를 사용하여 기록되었다. 약 10 mg의 (Z)-엔독시펜 유리 염기를 ATR 결정에 놓고, 압력을 팁으로 적용하고, 샘플 스펙트럼을 수집하였다. 스펙트럼을 수화된 필름을 사용하여 2 mm/sec로의 스캔 속도로 수집하였다. 인터페로그램을 공칭 해상도 0.4 cm-1 및 32 스캔으로 스펙트럼 범위 4000-650 cm-1에 걸쳐 축적하였다. 도 2는 (Z)-엔독시펜 유리 염기의 대표적인 IR 스펙트럼을 나타낸다.
도 2는 정제된 (Z)-엔독시펜이 702.0, 730.9, 794.5, 825.4, 907.3, 1027.9, 1167.7, 1238.1, 1278.6, 1508.1, 1575.6, 1607.4, 3310.2 파장 cm-1 +/- 0.4 cm-1에서 피크를 갖는다는 것을 나타낸다. 유리 엔독시펜의 이전에 공개된 FT/IR 스펙트럼은 706, 831, 1053, 1171, 1240, 1465, 1507 및 1604 파장 cm-1이다(Agudelo et al. PLoS ONE 8(3): e60250. doi:10.1371/journal.pone.0060250).

실시예 13:
Z-엔독시펜의 경구 조성물
API-인-캡슐. > 90% Z-엔독시펜 유리 염기를 상술된 바와 같이 제조하였다. 무수 백색 내지 회백색 분말을 안정한 자유 유동 분말로 제형화하고, 약물-인-캡슐(DIC, 또한 API-인-캡슐(AIC)로도 불림) 경구 고체 투여 형태로 캡슐로 순수하게 충전하였다. 적어도 90%의 (Z)-엔독시펜인 1 mg, 2 mg, 및 4 mg의 엔독시펜을 엑스셀로도스 기술(Xcellodose technolgy)(Capsugel)을 이용하여 VCaps® Plus 장용 캡슐에 순수하게 충전하였다. AIC는 크기가 0이고 스웨덴 오렌지색이었다.
VCaps® Plus 장용 캡슐(Capsugel)은 글루텐이 함유되지 않은 비-동물성 자가-겔화 제품인 히프로멜로스(셀룰로스의 메틸 및 히드록시프로필 혼합된 에테르)로 제조되며 수분 함량이 낮아 수분-민감성 성분, 예컨대 (Z)-엔독시펜에 적합하다. 캡슐은 문헌(Cole et al., Int. Journal of Pharmaceutics Vol. 231 83-95, 2002)의 방법에 의해 장(상부 GI 및 결장) 표적화를 달성하도록 설계된 장용 코팅물로로 코팅된다. Eudragit FS D30은 결장 표적 방출을 위한 장용 코팅물로서 사용되고, Eudragit L30 D55는 상부 위장 표적 방출을 위한 장용 코팅물로서 사용된다.
AIC 중의 (Z)-엔독시펜 유리 염기는 주로 장(상부 GI 및 결장)에서 방출되며, 적어도 6시간 동안 위의 산성 환경으로부터 보호된다. 캡슐의 장용 코팅물은 USP 711의 방법으로 시험 시 적어도 6시간 동안 위에서 (Z)-엔독시펜의 방출을 방지한다.
본원에 개시된 장용 AIC는 적어도 6개월 동안 안정하다.
본 개시의 경구 고체 투여 형태의 미생물 오염에 대한 표준 및 시험. 본원에 개시된 고체 투여 조성물의 제형에 대해 물 함량이 NMT 1% 이고 수분 활성도(Aw)가 0.75 미만이므로, TAC 및 USP 지표 유기체를 시험할 필요가 없다. 공개문("Microbial Bioburden on Oral Solid Dosage Form," Jose E. Martinez, Pharmaceutical Technology, February 2002, pages 58 to 70)이 전부 참조로 본원에 포함된다.
또한, 본원에 개시된 조성물의 제형은 또한 수분 활성도가 0.75 미만이므로, 이에 대한 상세한 미생물 시험을 수행할 필요가 없다. 총 호기성 플레이트 카운트(TAC)는 원료 물질, 공정 중 물질, 또는 완제품의 샘플에 존재하는 총 생존 호기성 박테리아의 추정치이다. 샘플은 가장 최근의 USP 지침 61장(미생물 한계 시험)에 따라 분석된다.
경구 고체 투여 형태(OSDF)에 대한 허용되는 TAC가 경계 및 작용 수준의 관점에서 본 발명의 조성물의 제형에 대해 확립되며, 이는 각각 1000 cfu g/ml, 및 10,000 cfu g/mL일 수 있다. 20,000 cfu g/mL인 TAC는 허용되지 않는 것으로 간주된다.
실시예 14:
(Z)-엔독시펜 염의 제조
(Z)-엔독시펜 D-글루코네이트 염은 유리 염기로서의 (Z)-엔독시펜의 에탄올 슬러리를 D-글루콘산의 수용액과 혼합한 후 물 중의 D-글루코놀락톤의 20% w/v 용액을 가수분해하고 15 내지 30분 동안 70℃에서 가열함으로써 제조된다. 최소 부피의 에탄올이 사용되고, 5 ml의 수성 D-글로콘산 용액이 1 g의 엔독시펜 유리 염기에 첨가된다. 이어서, 투명한 용액이 수득될 때까지 교반을 계속한다. (Z)-엔독시펜 D-글루코네이트는 실시예 1 내지 4에서 상기 개시된 방법을 이용하여 결정화되어 (Z)-엔독시펜 D-글루코네이트 염을 제공한다.
1 mg, 2 mg, 5 mg, 10 mg, 20 mg 및 40 mg AIC 장용 캡슐은 상술된 바와 같이 제조될 것이다. 유사하게, 소장 및 결장을 표적하기 위해 장용 코팅물 EUDRAGIT ® FS D30 및 EUDRAGIT ® L30D 55와 함께 (Z)-엔독시펜 D-글루코네이트 염을 사용하여 1 mg 내지 50 mg 정제가 제조될 것이다.

실시예 15:
경구 (Z)-엔독시펜의 위약 대조, 용량 상승 안전성 및 약동학 연구
연구의 목적은 건강한 여성 지원자에게 유방에 대해 국소 적용으로 또는 캡슐로 경구 투여될 때 경구 (Z)-엔독시펜의 안전성 및 내약성을 평가하는 것이었다. 제2 목적은 대상체에서 다중 용량의 경구 (Z)-엔독시펜의 약동학을 평가하는 것이었다.
일반적으로, ≥ 18세 및 ≤ 65세의 건강한 여성 지원자를 등록하였다. 24명의 참가자를 3개의 코호트에 등록하였으며, 연구 약물을 경구로 투여할 것이다. 경구로 투여되는 (Z)-엔독시펜의 3개의 투여 수준을 3개의 코호트에서 조사하였다. 각각의 코호트에서 참가자는 맹검 방식으로 경구 (Z)-엔독시펜 또는 위약을 받도록 무작위 처리되었다. 본 연구에 사용된 장 내성 캡슐은 위 조건 하에서 적어도 30분 동안, 전형적으로 약 2시간 동안 파열에 저항한 후 장액에서 완전히 개방되도록 지연 방출 캡슐로 설계되었다(DRCaps; Capsugel, Lonza Company, USA). GMP 규정에 따라 순수하게 1, 2 또는 4 mg의 (Z)-엔독시펜으로 수동으로 충전된 장 내성(스웨덴 오렌지색, 크기 제로(0); Capsugel, USA) DRCaps 캡슐을 사용하였다. 위약 캡슐은 불활성의 일반적인 캡슐 및 정제 부형제인 미세결정질 셀룰로스를 함유하였다.
각각의 코호트는 8명의 참가자를 등록하였고, 6명의 참가자는 1 mg(코호트 1), 2 mg(코호트 2) 또는 4 mg(코호트 3) 용량의 (Z)-엔독시펜 1 mg 캡슐을 경구로 받고, 2명의 참가자는 대응 위약 캡슐을 받았다. 코호트 1(용량 수준 = 1 mg)에서 2명의 참가자(파수)는 나머지 참가자가 참가하기 24시간 전에 투여되었다. 1명의 파수에는 (Z)-엔독시펜이 투여되고 다른 파수에는 위약이 투여되었다. 파수 참가자에서 안전성 문제가 확인되지 않았으므로 코호트 1의 나머지 6명의 참가자에게 투여되었다. 코호트 1의 비-파수 참가자는 파수보다 적어도 1일 후에 임상 연구 시설에 들어왔다. 코호트 2 및 3에는 파수가 없었다.
이 연구에서 참여자 참여 기간은 56일이었다. 이들은 28일의 스크리닝 기간 및 약동학(PK) 샘플링을 위한 치료 기간 후 7일을 포함하였다. 2가지의 억류(confinement) 기간이 있었으며, 하나는 시작시이고, 하나는 투여 기간 말이었다. 참여자는 제-1일부터 제2일까지 및 제21일 및 제22일에 임상 시설에 억류되었다. 참여자가 연구 동안 임의의 유의한 유해 사례를 경험하는 경우, 연구 책임자(PI)의 재량으로 추가의 관찰을 위해 임상 시설에 남아 있어야 하였다.
건강한 여성 지원자는 투여 개시 전 28일 이내에 스크리닝되었다. 참자가는 제-1일에 최대 3일 동안 임상 시설에 들어왔다. (Z)-엔독시펜 또는 위약 캡슐의 제1 용량을 제1일에 투여하였다. PK 분석을 위한 모든 안전성 평가 및 샘플링 후, 참가자는 제2일에 임상 시설로부터 내보내졌다. 제1일의 제1 용량 후 6일의 처리가 없는 기간(제2일-제7일)이 이어졌다. 각각의 참가자는 처리가 없는 기간 동안 연구 제4일 및 제6일에 PK 채혈 및 안전성 평가를 위해 임상 시설로 돌아왔다. 제8일에 참가자는 연속 14일(제8일-제21일) 동안 (Z)-엔독시펜 캡슐 또는 위약 캡슐의 매일 투여를 시작하였다. 참가자에게 연구 약물 캡슐이 제공되었으며, 매일 자가 관리가 요청되었다. 참가자는 PK 채혈 및 안전성 평가를 위해 용량 투여 전 제11일, 제14일 및 제17일에 임상 시설을 방문하였다. 제21일 아침에, 각각의 참가자는 용량 투여 전 임상 시설로 돌아왔고, PK 채혈 및 안전성 평가의 수집이 가능하도록 제22일까지 시설에 억류되었다. 제21일 투여는 연구 약물의 마지막 투여였다. 참가자는 제22일에 임상 시설로부터 내보내졌으며, 연구 제24일, 제26일 및 제28일에 PK 채혈 및 안전성 평가를 위해 복귀되었다.
연구 평가는 임의의 연구 유해 사례 및 수반 약물 사용의 평가; 키 및 체중; 신체 검사; 주기적 활력 징후(체온, 심박수, 호흡률, 혈압); 주기적 12-리드 ECG를 포함하여 대상체의 의료 병력을 수집하는 것을 포함하였다. 실험실 시험은 혈액학, 응고, 소변분석, 혈청 화학 및 바이오마커 분석(예컨대, CYP2D6, BRCA1/2, Ki67, 타목시펜 대사산물 등)을 포함하였다. 치료 안전성을 평가하기 위한 구체적인 평가는 유해 사례의 빈도 및 유형, 임상 실험실 시험, 12-리드 ECG 및 활력 징후를 포함하였다. 변형된 FACT-ES® 채점 설문지를 사용하여 증상을 평가하였다.
PK 분석을 위한 채혈을 투여 전(10분 이내) 및 제1일 및 제21일에 연구 약물을 투여한 후 0.5, 1, 2, 3, 4, 6, 8, 10, 12 및 24시간에 수집하였다. 모든 다른 연구일에 대한 PK 샘플을 용량 투여 전 10분 이내에 수집하였다. PK 샘플을 처리가 없는 기간 동안 제4일 및 제6일에 수득하였다. 추가의 PK 샘플을 매일 투여 기간 동안 제8일, 제11일, 제14일 및 제17일에 (투여 전 10분 이내) 수집하였다. 처리 완료 후 PK 샘플을 제24일, 제26일 및 제28일에 수집할 것이다. AUC(inf)를 제1일 채혈 샘플에서 측정하였다. 이 결정을 위해 제8일(제1일로 사용됨)부터 시작하여 항정 상태 혈장 수준을 결정하였다.
안전성 및 내약성
안전성 종점은 용량 코호트에 의해 요약되었고, 위약은 코호트 전체에 걸쳐 수집되었다. 처리-응급 AE는 말 그대로의 용어로 분류된 SOC(System Organ Class) 및 선호 용어(Preferred Term)에 의해 최신 버전의 MedDRA를 사용하여 코드화되었다. AE, 및 SAE의 발생률 및 빈도는 SOC 및 선호 용어에 따라 코호트에 의해 및 중증도 및 관계에 의해 요약되었다. AE의 지속기간이 결정되었고, 취해진 조치 및 결과와 함께 목록에 포함되었다. 활력 징후, ECG 및 안전성 실험실 파라미터는 기술 통계를 사용하여 각각의 예약된 시점에 요약되었다. 투여 후 평가는 기선 측정치와 비교되었다. 실험실 이상 발생이 요약되었다. 신체 검사 발견이 목록에 제시되었다.
연구 약물과 (아마도 또는 가능하게) 관련되는 것으로 간주되는 처리-응급 AE는 (Z)-엔독시펜을 받은 18명의 대상체 중 15명(83%, 41 AE) 및 위약을 받은 6명의 대상체 중 4명(67%, 20 AE)에서 보고되었다. 대부분의 처리 관련된 AE는 중증도가 경증이었으며, 중등도의 처리 관련된 AE는 (Z)-엔독시펜을 받은 18명의 대상체 중 5명(28%, 6 AE) 및 위약을 받은 6명의 대상체 중 2명(33%, 2 AE)이었다.
모든 AE는 중증도가 경증(84 AE 중 75) 또는 중등도(84 AE 중 9)로 분류되었으며, 중증도로 분류된 AE는 없었다. 긴장성 두통, 두통, 복통, 구역, 월경통 및 피로의 일반적 유해 사례가 경구 (Z)-엔독시펜를 받은 대상체 및 위약을 받은 대상체 모두에서 보고되었다. 상부 호흡 감염, 홍조, 복부 팽창, 입안 건조, 및 월경 지연이 (Z)-엔독시펜을 받은 대상체에서만 보고되었다.
위약을 받은 대상체와 비교하여 국소 또는 경구 (Z)-엔독시펜을 받은 대상체에서 임상 실험실 시험, 활력 징후, ECG 평가, FACT-ES 반응으로 평가된 안전성의 차이는 뚜렷하지 않았으며, (Z)-엔독시펜을 받은 대상체에서 이들 안전성 평가에 용량 관련된 경향은 없었다.
약동학
평균 및 개별 (Z)-엔독시펜 혈청 농도-시간 곡선을 각각의 용량 코호트에 대해 표로 만들고, 선형 및 로그 척도로 표시된 농도를 그래픽으로 제시하였다. 약동학 파라미터를 각각의 참가자에 대해 결정하고 기술 통계(산술 평균, 표준 편차, 분산 계수, 샘플 크기, 최소, 최대 및 중앙치)를 사용하여 코호트로 요약하였다. 또한 AUC 및 Cmax에 대한 기하 평균을 계산하였다. 선형 모델을 사용한 분석을 수행하여 용량 비례성(단일 용량 및 다중 용량 후 모두), 시간 의존성 및 축적(경구 다중 용량) 및 항정 상태의 달성(다중 용량)을 평가하였다.
제1 및 최종 용량(제1일 및 제21일)에 대해 결정된 파라미터는 최대 농도까지의 시간(Tmax), 최대 농도(Cmax), 약물 투여 후 0시간에서 24시간까지의 농도-시간 곡선 아래 면적(AUC0-24h), 터미널 제거 속도 상수(kel), 터미널 반감기(t1/2), 터미널 제거(CL/F) 및 분포 부피(Vd/F)를 포함하였다. 0시간에서 무한대까지의 농도-시간 곡선 아래 면적(AUC0-inf)이 또한 제1일에 제1 용량에 대해 결정되었다.
약동학 파라미터는 비구획적 방법(들)을 이용하여 결정되었다. 약동학 파라미터의 기술 통계는 평균, 표준 편차(SD), 및 분산 계수(CV), 최소(min) 및 최대(max)를 포함하였다. 약동학 파라미터에서 용량 관련된 경향을 평가하였다.
각각의 용량 코호트에 대한 평균 및 개별 엔독시펜 혈청 농도-시간 곡선을 표로 만들었다. 약동학 파라미터를 각각의 참가자에 대해 결정하고, 기술 통계(산술 평균, SD, CV, 샘플 크기[N], 최소, 최대 및 중앙치)를 사용하여 코호트로 요약하였다. 또한, AUC 및 Cmax에 대한 기하 평균을 계산하였다. 선형 모델을 사용한 분석을 수행하여 용량 비례성(단일 용량(경구) 및 다중 용량(국소 및 경구) 후 모두)을 평가하였다. SAS v9.3 및 Phoenix WinNonLin 버전 7.0 이상을 사용하여 약동학 파라미터에 대한 통계 분석을 수행하였다.
사용된 약어:
CV
분산 계수
Tmax
최대 농도까지의 시간
Cmax
최대 농도
AUC0-24h
("AUC24hr") 약물 투여 후 0시간에서 24시간까지의 농도-시간 곡선 아래 면적
kel
터미널 제거 속도 상수, 및 부피
t1/2
터미널 반감기
CL/F
터미널 제거
Vd/F
분포
AUC0-inf
("AUC0-inf") 0시간에서 무한대까지의 농도-시간 곡선 아래 면적은 또한 제1 용량에 대해 제1일에 결정되었다.
(Z)-엔독시펜의 터미널 반감기, 터미널 제거 반감기, Tmax, Cmax, 및 AUC를 갖는 혈청 약동학 파라미터의 요약이 하기 표 16에 제공된다.

지연 방출(내산성) (Z)-엔독시펜 캡슐의 1일 1회 경구 투여의 경우, 연구에 사용된 3가지 용량 수준에 걸쳐 엔독시펜의 Cmax 및 AUC24hr에서 용량 비례성(선형 용량 반응)이 있었다. Cmax는 제1일에 1 mg 내지 4 mg 용량에 대해 9.6 ng/mL에서 32.5 ng/mL로 증가하였고, 제21일에 24.6 ng/mL에서 115.0 ng/mL로 증가하였다. 제1일 및 제21일에 용량 수준에 의한 중앙치 Tmax는 4 내지 8시간의 범위였다. 용량 수준에 의한 겉보기 터미널 반감기(t1/2)는 42 내지 53시간의 범위였으며, 대략 7일 후에 항정 상태에 도달하는 것으로 나타났다(도 5 - 제8일에 투여 및 제14일에 Css). 최대 혈액 수준에 도달하는 시간은 4 내지 8시간의 범위였다(도 4).

[표 18a]

[표 18b]

[표 18c]

[표 19a]

[표 19b]

[표 19c]

[표 19d]

[표 19e]

[표 19f]


평균 축적 비율은 제1일과 비교한 제21일에서의 파라미터의 비율로서 Cmax에 대해 2.74 내지 3.56 및 AUC24hr에 대해 3.04 내지 3.47의 범위였다. 제1일 및 제21일에 제거(Cl/F) 및 분포 부피(Vd/F)에서 용량 수준에 의한 뚜렷한 경향은 없었다.
최대 혈청 엔독시펜 수준까지의 시간은 약 10시간까지 달성되며, 혈청 엔독시펜 수준에 용량 관련된 증가가 있다(도 3 및도 4). 혈청 엔독시펜 수준은 약 25 nM 내지 약 80 nM의 범위였다.
제8일에서 제21일까지의 투여 전 최저 농도(trough concentration)의 약동학 평가(도 4)는 제14일 후 직선의 달성을 나타내었다. 혈청 엔독시펜의 항정 상태 수준은 일일 투여와 함께 제7일까지 달성되는 것으로 보인다. 투여 전 혈청 농도는 4mg의 (Z)-엔독시펜에 대해 17일 후 거의 축적되지 않음을 나타낸다.


공개된 문헌
1. Wu X et al. Cancer Res. 2009 Mar 1;69(5):1722-7. PubMed PMID: 19244106
2. Ahmad A et al. Breast Cancer Res Treat. 2010 Jul;122(2):579-84. PubMed PMID:20052538
3. Ahmad A et al. J Clin Oncol 30, 2012 (suppl; abstr 3089)
4. Goetz MP et al. San Antonio Breast Conference 2013 [PD3-4]
5. Goetz MP et al. San Antonio Breast Conference 2015. [PD203]
6. Ahmad A et al. Clin Transl Sci. 2016 Jun 27. doi:10.1111/cts.12407. PubMed PMID: 27346789
7. Lee O et al. Cancer Chemother Pharmacol. 2015 Dec;76(6):1235-46
따라서, 이 연구로부터의 데이터는 경구 엔독시펜의 안전성 및 내약성이 일반적으로 경구 타목시펜과 비교될 수 있고 (Z)-엔독시펜 조성물로 투여된 대상체는 유방암을 갖는 여성의 보조 세팅에서 치료 효과를 갖는 것으로 입증된 범위 내에 있는 혈장 엔독시펜 수준을 갖는다는 것을 제시한다. (Z)-엔독시펜은 빠르게 흡수되고 전신적으로 이용 가능하며, 용량 범위 1 mg - 4.0 mg에 걸쳐 혈청 중 피크 약물 농도(C(max)) 및 0에서 무한대로 외삽된 농도-시간 곡선 아래 면적(AUC0-inf)에서 용량 비례성을 나타낸다.
실시예 16:
Z-엔독시펜 유리 염기의 경구 투여에 의한 타목시펜 불응성 유방암의 치료적 처치
이 연구의 목적은 타목시펜을 (Z)-엔독시펜으로 보충함으로써 타목시펜 불응성 환자에서 안정하고 치료적인 (Z)-엔독시펜 수준이 달성될 수 있음을 입증하는 것이다. 혈장 엔독시펜 수준은 이 집단에서 임상적 이익 뿐만 아니라 재발률을 예측하고 타목시펜과 비교하여 (Z)-엔독시펜 투여의 안전성 및 내약성을 확인하기 위한 대용 종점으로서 사용될 것이다.
타목시펜의 보조 요법으로 적어도 30일 동안 치료 중인 초기 단계의 에스트로겐 수용체 양성 유방암을 갖는 건강한 대상체가 일반적으로 6개월까지 연구에 등록될 것이다. 평가 가능한 집단이 적어도 25명의 타목시펜 및 25명의 (Z)-엔독시펜 대상체로 구성되도록 하기 위해 적어도 75명의 타목시펜 불응성 유방암 대상체가 연구에 등록될 것이다. 원하는 타목시펜 불응성 대상체 집단을 달성하기 위해 추가의 대상체가 등록될 수 있다. 이들 대상체는 유방암 진단을 받고 유방절제술 또는 종괴절제술을 받게 될 것이다.
경구 타목시펜의 적어도 30일 후, 엔독시펜 혈장 수준이 측정될 것이다. 엔독시펜 수준이 30 또는 40 nM 미만인 경우, 경구 타목시펜 보조 요법에 1 mg의 경구 (Z)-엔독시펜이 첨가될 것이다. 40 nM 이상 80 nM 이하의 안정한 엔독시펜이 달성될 때까지 추가의 (Z)-엔독시펜 용량이 첨가될 것이다. 종점은 적어도 6개월 동안 타목시펜+엔독시펜 그룹에서 치료적으로 안정한 엔독시펜 수준을 확립하는 것이다.
실시예 17:
Z-엔독시펜-D-글루코네이트의 경구 투여에 의한 에스트로겐 수용체 양성 유방암의 수술 전 처리
목적은 Z-엔독시펜-D-글루코네이트가 수술 전 에스트로겐 수용체 양성 유방암 환자에서 종양 활성을 감소시키는지의 여부를 결정하는 것이다. ER+ 유방암의 초기 진단시, 8명의 환자가 3개의 그룹 중 하나에 배정될 것이며, 여기서 각각의 환자는 수술 전 21일 동안 1, 2 또는 4 mg의 (Z)-엔독시펜-D-글루코네이트를 받을 것이다.
종양의 바이오 마커 KI-67 수준은 초기 생검의 시간으로부터 비교될 것이고, 수술 샘플은 3개의 용량 중 하나가 종양 활성의 감소를 초래하는지를 결정하기 위해 비교될 것이다.
실시예 18:
(Z)-엔독시펜 D-글루코네이트의 경구 투여에 의한 유방암의 치료적 처치
본 연구의 목적은 보조 유방암 요법을 개시하는 타목시펜 불응성 환자에서 대상체 혈장 중에 치료 (Z)-엔독시펜 수준이 달성될 수 있음을 입증하는 것이다. 혈장 엔독시펜 수준은 이 집단에서 임상 이익 뿐만 아니라 재발률을 예측하고, 타목시펜과 비교하여 경구 (Z)-엔독시펜 D-글루코네이트 투여의 안전성 및 내약성을 확인하기 위한 대용 종점으로서 사용될 것이다.
타목시펜이 완전한 치료 요법의 일부로 표시되는 에스트로겐 수용체 양성 유방암을 갖는 건강한 대상체가 일반적으로 6개월까지 연구에 등록될 것이다. 평가 가능한 집단이 적어도 25명의 타목시펜 및 25명의 (Z)-엔독시펜 대상체로 구성되도록 하기 위해 적어도 75명의 타목시펜 불응성 유방암 대상체가 연구에 등록될 것이다. 원하는 타목시펜 불응성 대상체 집단을 달성하기 위해 추가의 대상체가 등록될 수 있다. 이들 대상체는 유방암 진단을 받고 유방절제술 또는 종괴절제술을 받게 될 것이다.
유방절제술 또는 종괴절제술 직후, 25명의 대상체가 타목시펜 그룹에 배정되고 3개월의 기간 동안 매일 20mg의 타목시펜이 투여될 것이다. 추가로 25명의 대상체가 (Z)-엔독시펜 그룹에 배정되고 3개월의 기간 동안 (Z)-엔독시펜 D-글루코네이트의 경구 10mg 정제가 매일 투여될 것이다. 제0일(기선) 및 제7일, 제14일, 제21일, 제28일, 제60일 및 제90일에 각각의 대상체로부터 혈액을 채취할 것이다. 혈장 (Z)-엔독시펜 수준, 혈액학, 화학 및 응고 파라미터는 두 처리 세트 모두에 대해 제0일(기선 측정) 뿐만 아니라 제7일, 제14일, 제21일, 제28일, 제60일 및 제90일에 취해질 것이다.
2개의 처리 세트의 혈장 (Z)-엔독시펜 수준을 서로 비교할 것이다. 타목시펜 처리된 대상체의 혈장 엔독시펜 수준이 30 nM 미만인 경우, 이들 대상체는 (Z)-엔독시펜 요법 그룹으로 옮겨지고 추가의 6개월 동안 처리가 계속될 것이다. 타목시펜 처리된 대상체의 혈장 엔독시펜 수준이 30 nM을 초과하면, 타목시펜으로 계속 치료될 것이다. 혈액 샘플은 대상체의 혈장 (Z)-엔독시펜 수준, 혈액학, 화학 및 응고 파라미터를 모니터링하기 위해 연구가 끝날 때까지 매주 계속 채취될 것이다. 대상체에서 암의 재발이 모니터링되고 처리 그룹 사이에서 비교될 것이다. 안전성 및 효능은 연구가 끝날 때까지 3개월마다 평가될 것이다.
본 발명의 바람직한 실시양태를 본원에 제시하고 설명하였지만, 이러한 실시양태는 단지 예로서 제공된다는 것이 당업자에게 명백할 것이다. 다수의 변형, 변경 및 치환이 본 발명을 벗어나지 않고 당업자에게 일어날 것이다. 본원에 기재된 본 발명의 실시양태에 대한 다양한 대안이 본 발명을 실시하는 데 사용될 수 있음을 이해해야 한다. 하기의 청구범위는 본 발명의 범위를 정의하고 이들 청구범위의 범위 내의 방법 및 구조물 및 그들의 등가물이 이에 의해 포함되도록 의도된다.
Claims (157)
- 화학식 (III)의 화합물의 결정질 형태를 포함하는 조성물:

- 제1항에 있어서, 조성물 중의 화학식 (III)의 화합물의 적어도 90 중량%가 (Z)-이성질체인 조성물.
- 제2항에 있어서, 결정질 형태가 화학식 (III)의 화합물의 형태 I인 조성물.
- 제3항에 있어서, 결정질 형태가 16.8 ± 0.3°, 17.1 ± 0.3° 및 21.8 ± 0.3°2 쎄타에서 주요 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어지는 것인 조성물.
- 제4항에 있어서, x-선 분말 회절 패턴이 16.0 ± 0.3°, 18.8 ± 0.3° 및 26.5 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 추가로 포함하는 것인 조성물.
- 제4항 또는 제5항에 있어서, x-선 분말 회절 패턴이 12.3 ± 0.3°, 28.0 ± 0.3° 및 29.0 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 추가로 포함하는 것인 조성물.
- 제4항에 있어서, x-선 분말 회절 패턴이 12.3 ± 0.3°, 16.0 ± 0.3°, 18.8 ± 0.3°, 26.5 ± 0.3°, 28.0 ± 0.3° 및 29.0 ± 0.3°2 쎄타에서 피크를 추가로 포함하는 것인 조성물.
- 제3항 내지 제7항 중 어느 한 항에 있어서, 결정질 형태가 실질적으로 도 9 또는 도 10에 제시된 바와 같은 x-선 분말 회절 패턴으로 특징지어지는 것인 조성물.
- 제3항 내지 제8항 중 어느 한 항에 있어서, 조성물 중의 화학식 (III)의 화합물의 90 중량%, 95 중량% 또는 99 중량% 초과가 결정질 형태 I인 조성물.
- 제3항 내지 제9항 중 어느 한 항에 있어서, 조성물이 0.01 mg 내지 200 mg의 결정질 형태 I를 포함하는 것인 조성물.
- 제10항에 있어서, 조성물이 약 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 결정질 형태 I를 포함하는 것인 조성물.
- 제1항에 있어서, 조성물이 화학식 (III)의 화합물의 (E)-이성질체 및 (Z)-이성질체를 0.9 내지 1.3의 E/Z 비율로 포함하는 것인 조성물.
- 제12항에 있어서, E/Z 비율이 약 1.1인 조성물.
- 제12항 또는 제13항에 있어서, 결정질 형태가 화학식 (III)의 화합물의 형태 II인 조성물.
- 제14항에 있어서, 결정질 형태가 7.0 ± 0.3°, 11.9 ± 0.3°, 14.0 ± 0.3° 및 18.4 ± 0.3°2 쎄타에서 주요 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어지는 것인 조성물.
- 제15항에 있어서, x-선 분말 회절 패턴이 22.0 ± 0.3°2 쎄타에서 피크를 추가로 포함하는 것인 조성물.
- 제15항 또는 제16항에 있어서, x-선 분말 회절 패턴이 6.6 ± 0.3°, 13.3 ± 0.3° 및 20.0 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 추가로 포함하는 것인 조성물.
- 제15항에 있어서, x-선 분말 회절 패턴이 6.6 ± 0.3°, 13.3 ± 0.3°, 20.0 ± 0.3° 및 22.0 ± 0.3°2 쎄타에서 피크를 추가로 포함하는 것인 조성물.
- 제14항 내지 제18항 중 어느 한 항에 있어서, 결정질 형태가 실질적으로 도 11 또는 도 12에 제시된 바와 같은 x-선 분말 회절 패턴으로 특징지어지는 것인 조성물.
- 제14항 내지 제19항 중 어느 한 항에 있어서, 조성물 중의 화학식 (III)의 화합물의 90 중량%, 95 중량% 또는 99 중량% 초과가 결정질 형태 II인 조성물.
- 제14항 내지 제20항 중 어느 한 항에 있어서, 조성물이 0.01 mg 내지 200 mg의 결정질 형태 II를 포함하는 것인 조성물.
- 제21항에 있어서, 조성물이 약 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 결정질 형태 II를 포함하는 것인 조성물.
- 제12항 또는 제13항에 있어서, 결정질 형태가 화학식 (III)의 화합물의 형태 III인 조성물.
- 제23항에 있어서, 결정질 형태가 11.9 ± 0.3°, 13.9 ± 0.3°, 17.1 ± 0.3° 및 17.7 ± 0.3°2 쎄타에서 주요 피크를 포함하는 x-선 분말 회절 패턴으로 특징지어지는 것인 조성물.
- 제24항에 있어서, x-선 분말 회절 패턴이 25.3 ± 0.3°2 쎄타에서 피크를 추가로 포함하는 것인 조성물.
- 제24항 또는 제25항에 있어서, x-선 분말 회절 패턴이 18.2 ± 0.3°, 22.5 ± 0.3° 및 26.8 ± 0.3°2 쎄타로부터 선택되는 적어도 하나의 피크를 추가로 포함하는 것인 조성물.
- 제24항에 있어서, x-선 분말 회절 패턴이 18.2 ± 0.3°, 22.5 ± 0.3°, 25.3 ± 0.3° 및 26.8 ± 0.3°2 쎄타에서 피크를 추가로 포함하는 것인 조성물.
- 제23항 내지 제27항 중 어느 한 항에 있어서, 결정질 형태가 실질적으로 도 13에 제시된 바와 같은 x-선 분말 회절 패턴으로 특징지어지는 것인 조성물.
- 제23항 내지 제28항 중 어느 한 항에 있어서, 조성물 중의 화학식 (III)의 화합물의 90 중량%, 95 중량% 또는 99 중량% 초과가 결정질 형태 III인 조성물.
- 제23항 내지 제29항 중 어느 한 항에 있어서, 조성물이 0.01 mg 내지 200 mg의 결정질 형태 III를 포함하는 것인 조성물.
- 제30항에 있어서, 조성물이 약 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 결정질 형태 III를 포함하는 것인 조성물.
- 약제학적으로 허용되는 담체 또는 희석제와 제1항 내지 제31항 중 어느 한 항의 조성물을 포함하는 약제학적 조성물.
- 제1항 내지 제32항 중 어느 한 항에 있어서, 조성물이 경구, 비경구, 국소, 또는 관내 전달을 위해 제형화되는 것인 조성물.
- 제1항 내지 제33항 중 어느 한 항에 있어서, 조성물이 경구 전달을 위해 정제, 캐플릿, 캡슐, 또는 환제로 제형화되는 것인 조성물.
- 제1항 내지 제34항 중 어느 한 항에 있어서, 조성물로 치료되는 대상체에서 엔독시펜의 평균 반감기가 30시간 내지 60시간인 조성물.
- 제1항 내지 제35항 중 어느 한 항에 있어서, 조성물이 경구 전달을 위해 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿 또는 지연 방출 캡슐로 제형화되는 것인 조성물.
- 제1항 내지 제36항 중 어느 한 항에 있어서, 조성물이 대상체에서 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다의 치료 또는 예방을 위해 대상체에게 투여되는 것인 조성물.
- 단위 용량당 1 mg 내지 200 mg의 제1항 내지 제37항 중 어느 한 항의 조성물을 포함하고 이를 필요로 하는 대상체에게 투여하기 위한 경구 조성물로서, 경구 조성물의 일일 투여로 대상체에서
7 내지 21일 이내에 엔독시펜의 항정 상태 혈장 수준;
25 nM 내지 300 nM 범위의 엔독시펜의 항정 상태 혈장 수준;
30 nM 초과의 엔독시펜의 항정 상태 혈장 수준;
투여 후 2 내지 10시간 이내에 엔독시펜의 최대 혈장 수준; 또는
그의 임의의 조합
이 달성되는 것인 경구 조성물. - 제38항에 있어서, 조성물로 치료되는 대상체에서 엔독시펜의 평균 반감기가 40시간 내지 55시간인 경구 조성물.
- 제38항 또는 제39항에 있어서, 조성물이 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿 또는 지연 방출 캡슐로 제형화되는 것인 경구 조성물.
- 제1항 내지 제40항 중 어느 한 항에 있어서, 조성물 중의 엔독시펜의 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 또는 적어도 90%가 장에서 방출되는 것인 조성물.
- 제1항 내지 제41항 중 어느 한 항에 있어서, 200 hr*ng/mL 내지 10000 hr*ng/mL, 300 hr*ng/mL 내지 8000 hr*ng/mL, 400 hr*ng/mL 내지 6000 hr*ng/mL 또는 700 hr*ng/mL 내지 6000 hr*ng/mL의 엔독시펜의 무한대 시간으로 외삽된 곡선 아래 평균 면적(AUC0-inf)을 갖는 것인 조성물.
- 제1항 내지 제42항 중 어느 한 항의 조성물을 대상체에게 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법.
- 제43항에 있어서, 대상체가 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 것인 방법.
- 제44항에 있어서, 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애가 양성 유방 장애, 증식증, 비정형(atypia), 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 또는 외음부암인 방법.
- 제43항 내지 제45항 중 어느 한 항에 있어서, 대상체가 전립선암을 갖고 대상체가 추가로 여성형 유방증을 갖거나 가질 위험이 있는 것인 방법.
- 제43항 내지 제46항 중 어느 한 항에 있어서, 대상체가 타목시펜 불응성 또는 타목시펜 내성 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖는 것인 방법.
- 제43항 내지 제47항 중 어느 한 항에 있어서, 대상체가 시탈로프람, 에스시탈로프람, 플루옥세틴, 파록세틴, 세르트랄린, 및 빌라조돈으로 이루어진 군으로부터 선택되는 SSRI 약물로 치료되거나 치료될 것인 방법.
- 제43항 내지 제48항 중 어느 한 항에 있어서, 조성물이 0.01 mg 내지 200 mg의 (Z)-엔독시펜을 포함하는 것인 방법.
- 제43항 내지 제49항 중 어느 한 항에 있어서, 대상체에게 약 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 (Z)-엔독시펜이 매일 투여되는 것인 방법.
- 제43항 내지 제50항 중 어느 한 항에 있어서, 대상체에서 엔독시펜의 항정 상태 혈장 수준이 30 nM 초과인 방법.
- 제43항 내지 제51항 중 어느 한 항에 있어서, 엔독시펜의 항정 상태 혈장 수준이 조성물의 제1 투여의 7 내지 21일 이내에 달성되는 것인 방법.
- 제43항 내지 제52항 중 어느 한 항에 있어서, 엔독시펜의 최대 혈장 수준까지의 시간이 조성물 투여 후 2시간 내지 10시간 또는 4시간 내지 8시간의 범위인 방법.
- 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 대상체를 치료하는 방법으로서, 제1항 내지 제42항 중 어느 한 항의 조성물을 투여하는 단계를 포함하고, 여기서 조성물의 투여는
대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기;
투여 후 4시간 내지 8시간 범위의 엔독시펜의 최대 혈장 수준까지의 시간; 및
30 nM 초과의 엔독시펜의 항정 상태 혈장 수준
을 달성하는 것인 방법. - 제54항에 있어서, 호르몬 의존적 유방 장애 및 호르몬 의존적 생식관 장애가 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 및 외음부암으로 이루어진 군으로부터 선택되는 것인 방법.
- 제54항 또는 제55항에 있어서, 조성물이 0.01 mg 내지 200 mg의 (Z)-엔독시펜을 포함하는 것인 방법.
- 제56항에 있어서, 대상체에게 약 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 (Z)-엔독시펜이 투여되는 것인 방법.
- 제43항 내지 제57항 중 어느 한 항에 있어서, 엔독시펜의 무한대 시간으로 외삽된 곡선 아래 평균 면적(AUC0-inf)이 200 hr*ng/mL 내지 10000 hr*ng/mL, 300 hr*ng/mL 내지 8000 hr*ng/mL, 400 hr*ng/mL 내지 6000 hr*ng/mL 또는 700 hr*ng/mL 내지 6000 hr*ng/mL인 방법.
- 제43항 내지 제58항 중 어느 한 항에 있어서, 조성물이 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿 또는 지연 방출 캡슐로 제형화되는 것인 방법.
- 제43항 내지 제59항 중 어느 한 항에 있어서, 조성물 중의 엔독시펜의 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 또는 적어도 90%가 장에서 방출되는 것인 방법.
- 제43항 내지 제60항 중 어느 한 항에 있어서, 조성물이 1일 1회, 1일 2회, 1일 3회, 1일 4회, 격일, 1주 2회, 매주, 격주, 1개월 2회, 매월, 분기별, 6개월 1회, 또는 매년 투여되는 것인 방법.
- 하기 단계를 포함하는, (Z)-엔독시펜을 제조하는 산업적으로 확장 가능한 방법:
(a)
로 표시되는 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 제1 용매로부터 분별 결정화하여 제1 결정질 고체 및 제1 모액을 형성하는 단계로서, 여기서 제1 모액은 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물의 E/Z 비율과 비교하여 적어도 50% 더 높은 (Z) 엔독시펜의 E/Z 비율을 갖는 것인 단계;
(b) 제1 모액을 농축시키거나 제1 모액으로부터 제1 용매를 제2 용매와 1회 이상 교환함으로써 제1 모액을 제2 용매로부터 재결정화하여 제2 결정질 고체 및 제2 모액을 형성하는 단계로서, 여기서 제2 결정질 고체는 ≥ 90% (Z)-엔독시펜인 단계; 및
(c) 임의적으로, 제2 결정질 고체를 제3 용매 또는 크로마토그래피 처리로부터 1회 이상 재결정화하여 제3 결정질 고체를 형성하는 단계. - 하기 단계를 포함하는, 제1항 내지 제11항 중 어느 한 항의 결정질 형태를 제조하는 산업적으로 확장 가능한 방법:
(a)
로 표시되는 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 제1 용매로부터 분별 결정화하여 제1 결정질 고체 및 제1 모액을 형성하는 단계로서, 여기서 제1 모액은 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물의 E/Z 비율과 비교하여 적어도 50% 더 높은 (Z) 엔독시펜의 E/Z 비율을 갖는 것인 단계;
(b) 제1 모액을 농축시키거나 제1 모액으로부터 제1 용매를 제2 용매와 1회 이상 교환함으로써 제1 모액을 제2 용매로부터 재결정화하여 제2 결정질 고체 및 제2 모액을 형성하는 단계로서, 여기서 제2 결정질 고체는 ≥ 90% (Z)-엔독시펜인 단계; 및
(c) 제2 결정질 고체를 제3 용매 또는 크로마토그래피 처리로부터 1회 이상 재결정화하여 제3 결정질 고체를 형성하는 단계로서, 여기서 제3 결정질 고체는 제1항 내지 제11항 중 어느 한 항의 결정질 형태인 단계. - 제62항 또는 제63항에 있어서, (E)-엔독시펜과 (Z)-엔독시펜의 혼합물은, 불활성 유기 용매 중의 티타늄 염 및 환원제를 통해 맥머리 반응에 의해 매개되는 프로피오페논에 화학식 (II)의 화합물인 (4-히드록시페닐)(4-(2-(메틸아미노)에톡시)페닐) 메탄온을 커플링시켜 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성함으로써 제조되고, 화학식 (II)의 화합물은

로 표시되는 구조를 갖는 것인 방법. - 제64항에 있어서, 화학식 (I)의 화합물인 [4-[2-(디메틸아미노)에톡시]페닐](4-히드록시페닐)메탄온을 불활성 유기 용매 중의 탈메틸화제 및 양성자 억셉터로 탈메틸화시켜 화학식 (II)의 화합물을 형성함으로써 화학식 (II)의 화합물을 제조하는 단계를 추가로 포함하며, 화학식 (I)의 화합물은

를 갖는 것인 방법. - 제62항 내지 제65항 중 어느 한 항에 있어서, 제1 용매가 에틸 아세테이트, IPA, IPA/PPW, ACN, ACN/PPW 또는 아세톤인 방법.
- 제62항 내지 제66항 중 어느 한 항에 있어서, 제2 용매가 IPA, IPA/PPW, 아세톤, 에탄올, 에틸 아세테이트, 또는 아세톤/MTBE인 방법.
- 제62항 내지 제67항 중 어느 한 항에 있어서, 제3 용매가 에탄올, 메탄올, 에틸 아세테이트, IPA, IPA/PPW, n-헵탄, 또는 아세톤인 방법.
- 제62항 내지 제68항 중 어느 한 항에 있어서, 제1 용매, 제2 용매 및 제3 용매 중 임의의 하나 이상을 40℃ 내지 80℃ 범위의 온도로 예열하는 단계를 추가로 포함하는 것인 방법.
- 제62항 내지 제69항 중 어느 한 항에 있어서, 각각의 분별 결정화 및 재결정화 단계가 독립적으로 50℃ 내지 80℃에서의 증류 단계; 및 용액를 0℃ 내지 NMT 35℃ 범위의 온도로 냉각시키는 단계를 포함하는 것인 방법.
- 제64항 내지 제70항 중 어느 한 항에 있어서, 하기 단계를 추가로 포함하는 것인 방법:
(a) 화학식 (II)의 화합물을 불활성 유기 용매 중의 프로피오페논과 반응시키는 단계;
(b) 불활성 유기 용매 중의 티타늄 염 및 환원제를 제조하는 단계; 및
(c) 단계 (a)의 화학식 (II)의 화합물을 단계 (b)의 불활성 유기 용매 중의 티타늄 염 및 환원제와 반응시켜 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성하는 단계. - 제64항 내지 제71항 중 어느 한 항에 있어서,
(a) 화학식 (II)의 화합물을 불활성 유기 용매(1:1 내지 1:20 wt/wt) 중의 프로피오페논(1:0.01 내지 1:5 wt/wt)과 반응시키는 단계;
(b) 불활성 유기 용매(1:1 내지 1:20 wt/wt) 중의 티타늄 염(1:0.1 내지 1:12 wt/wt) 및 환원제(1:0.01 내지 1:10 wt/wt)를 제조하는 단계; 및
(c) 단계 (a)의 화학식 (II)의 화합물을 단계 (b)의 불활성 유기 용매 중의 티타늄 염 및 환원제와 반응시켜 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성하는 단계
를 추가로 포함하고;
여기서 wt/wt는 화학식 (II)의 화합물에 대한 것인 방법. - 제64항 내지 제72항 중 어느 한 항에 있어서, 티타늄 염이 티타늄 할로겐화물(예컨대, 삼염화티타늄(TiCl3), 사염화티타늄(TiCl4), 요오드화티타늄, 브롬화티타늄, 및 불화티타늄), 삼염화티타늄(IV) 이소프로폭사이드, 및 티타늄 이소프로폭사이드로 이루어진 군으로부터 선택되는 것인 방법.
- 제64항 내지 제73항 중 어느 한 항에 있어서, 환원제가 아연, 지르코늄, 바나듐, 니오븀, 몰리브덴, 텅스텐, 알루미늄, 마그네슘, 칼륨, 아연-구리 커플, 알칼리 및 알칼리 토 금속, 부틸륨, 리튬, 및 리튬 알루미늄 수소화물로 이루어진 군으로부터 선택되는 것인 방법.
- 제64항 내지 제74항 중 어느 한 항에 있어서, 불활성 유기 용매가 디클로로메탄, 디클로로에탄, 클로로포름, 사염화탄소, 클로로벤젠, 디에틸 에테르, 1,4-디옥산, 3차-부틸 메틸 에테르, 테트라히드로푸란, N,N-디메틸포름아미드, N-메틸피롤리돈, 디글림, 니트로메탄, 1,2-디메톡시에탄, 피리딘, 아세톤, 아세토니트릴, 벤젠, o-자일렌, m-자일렌, p-자일렌, 자일렌, 헥산, 시클로헥산, 헵탄, 옥탄, 노난, 및 데칸, 또는 그의 조합으로 이루어진 군으로부터 선택되는 것인 방법.
- 제64항 내지 제75항 중 어느 한 항에 있어서, 단계 (b)에서 불활성 유기 용매 중의 티타늄 염 및 환원제의 제조가, 티타늄 염이 환원제 및 불활성 유기 용매에 첨가되는 경우, 반응 온도를 NMT 75℃, NMT 65℃, NMT 55℃, NMT 50℃, NMT 45℃, NMT 40℃, NMT 35℃, NMT 30℃, NMT 25℃, NMT 20℃, 또는 NMT 15℃의 온도로 유지하는 단계를 추가로 포함하는 것인 방법.
- 제64항 내지 제76항 중 어느 한 항에 있어서, 불활성 유기 용매 중의 티타늄 염 및 환원제의 제조가, 티타늄 염이 환원제 및 불활성 유기 용매에 첨가되는 경우, 반응 온도를 NMT 75℃, NMT 70℃, NMT 65℃, NMT 60℃, NMT 55℃, NMT 50℃, 또는 NMT 45℃의 온도로 유지하는 단계를 추가로 포함하는 것인 방법.
- 제64항 내지 제77항 중 어느 한 항에 있어서, 단계 (b)에서 불활성 유기 용매 중의 티타늄 염 및 환원제의 제조가, 불활성 유기 용매 중의 티타늄 염 및 환원제를 20℃ 내지 250℃, 40℃ 내지 70℃, 50℃ 내지 230℃, 50℃ 내지 120℃, 또는 150℃ 내지 200℃ 범위의 온도로 가열하는 단계를 추가로 포함하는 것인 방법.
- 제64항 내지 제78항 중 어느 한 항에 있어서, 단계 (b)에서 불활성 유기 용매 중의 티타늄 염 및 환원제의 제조가, 불활성 유기 용매 중의 티타늄 염 및 환원제를 N2 또는 아르곤 하에서 NLT 30분, NLT 1시간, NLT 2시간, NLT 4시간, NLT 6시간, 또는 NLT 8시간 동안 환류 하에 가열하는 단계를 추가로 포함하는 것인 방법.
- 제64항 내지 제79항 중 어느 한 항에 있어서, 단계 (a)의 화학식 (II)의 화합물이 환류 하에 단계 (b)의 불활성 유기 용매 중의 티타늄 염 및 환원제과 반응하여 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성하는 것인 방법.
- 제64항 내지 제80항 중 어느 한 항에 있어서, 단계 (a)의 화학식 (II)의 화합물이 40℃ 내지 80℃ 범위의 온도에서 단계 (b)의 불활성 유기 용매 중의 티타늄 염 및 환원제와 반응하여 NLT 4시간, NLT 6시간, NLT 8시간, NLT 12시간, NLT 24시간, 또는 NLT 48시간 동안 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성하는 것인 방법.
- 제64항 내지 제81항 중 어느 한 항에 있어서, (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 0℃ 내지 30℃ 범위의 온도로 냉각시키는 단계를 추가로 포함하는 것인 방법.
- 제64항 내지 제82항 중 어느 한 항에 있어서, 하기 중 하나 이상의 단계를 추가로 포함하는 것인 방법:
(a) (E)-엔독시펜과 (Z)-엔독시펜의 혼합물의 추출;
(b) (E)-엔독시펜과 (Z)-엔독시펜의 혼합물의 세척;
(c) (E)-엔독시펜과 (Z)-엔독시펜의 혼합물의 증류; 및
(d) 결정화하여 (E)-엔독시펜과 (Z)-엔독시펜의 결정질 고체 혼합물을 수득. - 제83항에 있어서, 추출이 MeTHF 또는 THF 중에서 1회 이상 수행되는 것인 단계.
- 제65항 내지 제84항 중 어느 한 항에 있어서, 화학식 (I)의 화합물을 불활성 유기 용매(1:1 내지 1:20 wt/wt) 중의 탈메틸화제(1:0.5 내지 1:10 wt/wt) 및 양성자 억셉터(1:0.5 내지 1:10 wt/wt)로 탈메틸화시켜 화학식 (II)의 화합물을 형성함으로써 화학식 (II)의 화합물을 생성하는 단계를 추가로 포함하고, 여기서 wt/wt 비율은 화학식 (I)의 화합물에 대한 것인 방법.
- 제65항 내지 제85항 중 어느 한 항에 있어서, 양성자 억셉터가 탄산염, 예컨대 탄산나트륨 및 탄산칼륨, 및 중탄산염, 예컨대 중탄산나트륨 및 중탄산칼륨, 양성자 스폰지, 및 DIPEA로 이루어진 군으로부터 선택되는 것인 방법.
- 제65항 내지 제86항 중 어느 한 항에 있어서, 탈메틸화제가 N-요오도숙신아미드, 에틸 클로로포르메이트(예컨대, 1-클로로에틸 클로로포르메이트, 디클로로에틸 클로로포르메이트, 트리클로로에틸 클로로포르메이트, α-클로로에틸 클로로포르메이트), 비닐 클로로포르메이트, 브롬화시아노겐, 디에틸 아조디카르복실레이트, 및 염화피리디늄으로 이루어진 군으로부터 선택되는 것인 방법.
- 제65항 내지 제87항 중 어느 한 항에 있어서, 화학식 (I)의 화합물이 20℃ 내지 250℃, 40℃ 내지 80℃, 50℃ 내지 230℃, 50℃ 내지 120℃, 및 150℃ 내지 200℃ 범위의 온도에서 탈메틸화제 및 양성자 억셉터와 반응하는 것인 방법.
- 제65항 내지 제88항 중 어느 한 항에 있어서, 화학식 (I)의 화합물이 환류 하에 탈메틸화제 및 양성자 억셉터와 반응하는 것인 방법.
- 제65항 내지 제89항 중 어느 한 항에 있어서, 화학식 (I)의 화합물이 NLT 5시간, NLT 8시간, NLT 12시간, NLT 24시간, NLT 36시간, NLT 48시간 또는 NLT 72시간 동안 탈메틸화제 및 양성자 억셉터와 반응하는 것인 방법.
- 제3항 및 제85항 내지 제90항 중 어느 한 항에 있어서, 하기 중 하나 이상의 단계를 추가로 포함하는 것인 방법:
(a) 증류;
(b) 용매/산 혼합물과의 반응;
(c) 중화제로 중화; 및
(d) 감압 하에 건조. - 제91항에 있어서, 증류 단계가 에틸 아세테이트, 알콜, 예컨대 메탄올, 에탄올, n-프로판올, 및 이소프로판올, 벤젠, 아세톤, 아세토니트릴, 톨루엔, 디클로로메탄, 1,2-디클로로에탄, 및 클로로포름으로 이루어진 군으로부터 선택되는 유기 증류 용매와의 하나 이상 용매 교환을 포함하는 것인 방법.
- 제91항 또는 제92항에 있어서, 용매/산 혼합물이 메탄올/HCl, 에탄올/HCl, 프로판올/HCl, 이소프로판올/HCl, 메탄올/황산, 메탄올/인산, 에탄올/황산, 에탄올/인산, 프로판올/황산, 프로판올/인산, 이소프로판올/황산, 이소프로판올/인산, 메탄올/아세트산, 에탄올/아세트산, 프로판올/아세트산, 이소프로판올/아세트산, 메탄올/포름산, 에탄올/포름산, 프로판올/포름산, 및 이소프로판올/포름산으로 이루어진 군으로부터 선택되는 것인 방법.
- 제91항 내지 제93항 중 어느 한 항에 있어서, 중화제가 수산화나트륨, 수산화암모늄, 수산화칼륨, 또는 아미노메틸프로판올인 방법.
- 제62항 내지 제94항 중 어느 한 항에 있어서, (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 용매(1:1 내지 1:20 wt/wt) 중의 산(1:1 내지 1:5 wt/wt)에 반응시킴으로써 (E)-엔독시펜을 (Z)-엔독시펜으로 전환시키는 단계를 추가로 포함하고, 여기서 wt/wt 비율은 화학식 (III)의 화합물에 대한 것인 방법.
- 제95항에 있어서, 산이 HCl, TCA, 또는 TFA인 방법.
- 제95항 또는 제96항에 있어서, 용매가 아세토니트릴, 아세토니트릴/PPW, IPA, IPA/PPW, 디클로로메탄, 또는 에틸 아세테이트인 방법.
- 제95항 내지 제97항 중 어느 한 항에 있어서, (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 환류 하에 용매 중의 산과 함께 가열하고 NLT 4시간, NLT 6시간, NLT 12시간, NLT 24시간, 또는 NLT 48시간 동안 교반하는 것인 방법.
- 제95항 내지 제98항 중 어느 한 항에 있어서, 하기 중 하나 이상의 단계를 추가로 포함하는 것인 방법:
(a) 중화제로 중화;
(b) 추출;
(c) 1회 이상 세척; 및
(d) 활성탄으로 처리. - 하기 단계를 포함하는, (Z)-엔독시펜을 제조하는 산업적으로 확장 가능한 방법:
(a)
로 표시되는 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 에틸 아세테이트로부터 분별 결정화하여 제1 결정질 고체 및 제1 결정질 모액을 형성하는 단계로서, 여기서 제1 모액은 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물의 E/Z 비율과 비교하여 적어도 50% 더 높은 (Z) 엔독시펜의 E/Z 비율을 갖는 것인 단계;
(b) 제1 모액을 농축시키거나 제1 모액으로부터 에틸 아세테이트를 IPA 또는 IPA/PPW와 1회 이상 교환함으로써 제1 모액을 IPA 또는 IPA/PPW(1:1 v/v)로부터 재결정화하여 제2 결정질 고체 및 제2 모액을 형성하는 단계로서, 여기서 제2 결정질 고체는 ≥ 90% (Z)-엔독시펜인 단계; 및
(c) 임의적으로, 제2 결정질 고체를 에탄올 또는 컬럼 크로마토그래피 처리로부터 1회 이상 재결정화하여 제3 결정질 고체를 형성하는 단계. - 제99항 또는 제100항에 있어서, 단계 (a)에서 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물이 6N HCl로 전처리되고 8N NaOH로 중화되는 것인 방법.
- 제99항 내지 제101항 중 어느 한 항에 있어서, 화학식 (III)의 화합물인 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물은, THF 중의 TiCl4 및 Zn을 통해 맥머리 반응으로 촉매되는 프로피오페논에 화학식 (II)의 화합물인 (4-히드록시페닐)(4-(2-(메틸아미노)에톡시)페닐)메탄온을 커플링시켜 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성함으로써 제조되고, 화학식 (III)의 화합물은

로 표시되는 것인 방법. - 제102항에 있어서, 하기의 단계를 추가로 포함하는 것인 방법:
(a) 화학식 (II)의 화합물을 THF 중의 프로피오페논과 반응시키는 단계;
(b) THF 중의 TiCl4 및 Zn을 제조하는 단계; 및
(c) 단계 (a)의 화학식 (II)의 화합물을 단계 (b)의 THF 중의 TiCl4 및 Zn과 반응시켜 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성하는 단계. - 제102항 또는 제103항에 있어서,
(a) 화학식 (II)의 화합물을 THF(1:1 내지 1:20 wt/wt) 중의 프로피오페논과 반응시키는 단계;
(b) THF(1:1 내지 1:20 wt/wt) 중의 TiCl4(0.1 내지 12 wt/wt) 및 Zn(0.01 내지 1:10 wt/wt)을 제조하는 단계; 및
(c) 단계 (a)의 화학식 (II)의 화합물을 단계 (b)의 THF 중의 TiCl4 및 Zn과 반응시켜 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 형성하는 단계
를 포함하고,
여기서 wt/wt는 화학식 (II)의 화합물에 대한 것인 방법. - 제102항 내지 제104항 중 어느 한 항에 있어서, THF 중의 TiCl4 및 Zn의 제조가 N2 하에서 NLT 2시간 동안 환류 하에 가열하는 단계를 추가로 포함하는 것인 방법.
- 제102항 내지 제105항 중 어느 한 항에 있어서, 추출 정제, 증류, 및 결정화의 단계를 추가로 포함하는 것인 방법.
- 제102항 내지 제106항 중 어느 한 항에 있어서, (E)-엔독시펜과 (Z)-엔독시펜의 혼합물에 대해 하기의 단계를 포함하는 추출 정제를 실시하는 것인 방법:
(a) 염화암모늄, 이산화규소, 40% K2CO3, 및 THF로 1회 이상 추출;
(b) K2CO3 및 MeTHF로 1회 이상 추출;
(c) NaOH, NaCl, 및 MeTHF로 1회 이상의 추출;
(d) MeTHF 또는 THF로 1회 이상의 추출;
(e) 20% NaCl로 1회 이상의 추출; 또는
(f) 그의 조합. - 제102항 내지 제107항 중 어느 한 항에 있어서, (E)-엔독시펜과 (Z)-엔독시펜의 혼합물에 대해
(a) 40% K2CO3(1:1 내지 1:10 wt/wt) 및 MeTHF(1:1 내지 1:10 wt/wt)로 1회 이상 추출;
(b) 1N NaOH(1:1 내지 1:20 wt/wt), NaCl(1:0.01 내지 1:0.5 wt/wt) 및 MeTHF(1:1 내지 1:10 wt/wt)로 1회 이상 추출;
(c) MeTHF(1:1 내지 1:5 wt/wt)로 1회 이상 추출; 및
(d) 20% NaCl(1:1 내지 1:10 wt/wt)로 추출
의 단계를 포함하는 추출 정제를 실시하고;
여기서 wt/wt는 화학식 (II)의 화합물에 대한 것인 방법. - 제102항 내지 제108항 중 어느 한 항에 있어서, (E)-엔독시펜과 (Z)-엔독시펜의 혼합물에 대해
(a) 25% 염화암모늄(1:10 내지 1:30 wt/wt), 이산화규소(1:0.01 내지 1:5 wt/wt) 및 THF(1:1 내지 1:5 wt/wt)로 추출;
(b) THF(1:1 내지 1:5 wt/wt)로 1회 이상 세척; 및
(c) 40% K2CO3(1:1 내지 1:10 wt/wt)로 1회 이상 세척
의 단계를 포함하는 추출 정제를 실시하고;
여기서 wt/wt는 화학식 (II)의 화합물에 대한 것인 방법. - 제108항 또는 제109항에 있어서, 증류 단계가 EtOAc(1:1 내지 1:10 wt/wt) 또는 IPA(1:1 내지 1:10 wt/wt)로 1 내지 5회 수행되는 것인 방법.
- 제106항 내지 제110항 중 어느 한 항에 있어서, 증류 단계가 30℃ 내지 90℃ 범위의 온도에서 수행되는 것인 방법.
- 제106항 내지 제111항 중 어느 한 항에 있어서, 증류가 NMT 75℃에서 수행되는 것인 방법.
- 제102항 내지 제112항 중 어느 한 항에 있어서, 화학식 (II)의 화합물이, 화학식 (I)의 화합물인 [4-[2-(디메틸아미노)에톡시]페닐](4-히드록시페닐)메탄온을 THF 중의 1-클로로에틸 클로로포르메이트 및 DIPEA로 탈메틸화시켜 화학식 (II)의 화합물을 형성함으로써 제조되고, 상기 화학식 (I)의 화합물은

의 구조를 갖는 것인 방법. - 제113항에 있어서,
(a) 화학식 (I)의 화합물을 테트라히드로푸란 중의 DIPEA와 반응시키는 단계;
(b) 1-클로로에틸 클로로포르메이트를 첨가하는 단계;
(c) 메탄올로 증류시키는 단계;
(d) 메탄올/6N HCl과 반응시키는 단계; 및
(e) 8N NaOH로 중화시키는 단계
를 포함하는 것인 방법. - 제113항 또는 제114항에 있어서,
(a) 화학식 (I)의 화합물을 THF(1:20 wt/wt) 중의 DIPEA(1:1 내지 1:10 wt/wt)와 반응시키는 단계;
(b) 1-클로로에틸 클로로포르메이트(1:1 내지 1:10 wt./wt)를 첨가하는 단계;
(c) 메탄올(1:1 내지 1:10 wt./wt)로 1회 이상 증류시키는 단계;
(d) 메탄올(1:1 내지 1:5 wt/wt)/6N HCl(1:1 내지 1:10 wt/wt)과 반응시키는 단계; 및
(e) 8N NaOH(1:1 내지 1:10 wt/wt)로 중화시키는 단계
를 포함하고;
여기서 wt/wt는 화학식 (I)의 화합물에 대한 것인 방법. - 제113항 내지 제115항 중 어느 한 항에 있어서,
(a) 화학식 (II)의 화합물을 정제수(1:1 내지 1:5 vol/wt)로 세척하는 단계;
(b) 화학식 (II)의 화합물을 에틸 아세테이트(1:1 내지 1:5 wt/wt)로 세척하는 단계; 및
(c) 화학식 (II)의 화합물을 감압 하에 NMT 50℃에서 건조시키는 단계
중 하나 이상의 단계를 추가로 포함하고;
여기서 wt/wt는 화학식 (I)의 화합물에 대한 것인 방법. - 제62항 내지 제116항 중 어느 한 항에 있어서, (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 EtOAc(1:1 내지 1:20 wt/wt) 중의 6N HCl(1:1 내지 1:5 wt/wt)에 반응시킴으로써 (E)-엔독시펜을 (Z)-엔독시펜으로 전환시키는 단계를 추가로 포함하고, 여기서 wt/wt는 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물에 대한 것인 방법.
- 제117항에 있어서,
(a) 8N NaOH(1:1 내지 1:20 wt/wt)로 중화시키는 단계;
(b) 에틸 아세테이트(1:1 내지 1:10 wt/wt)로 추출하는 단계;
(c) 20% NaCl(1:1 내지 1:10 wt/wt)로 1회 이상 세척하는 단계; 및
(d) 활성탄(1:0.01 내지 1:0.1 wt/wt)으로 처리하는 단계
를 추가로 포함하고;
여기서 wt/wt는 화학식 (III)의 화합물에 대한 것인 방법. - 제62항 내지 제118항 중 어느 한 항의 방법에 따라 생성된 화학식 (III)의 화합물의 결정질 형태.
- 제119항에 있어서, 결정질 형태가 화학식 (III)의 화합물의 형태 I인 결정질 형태.
- 45:55 내지 55:45 범위의 E/Z 비율을 갖는 (E)-엔독시펜과 (Z)-엔독시펜의 혼합물을 재평형화시키는 산업적으로 확장 가능한 방법으로서,
(a) 99:1 내지 60:40 범위의 E/Z-비율을 갖는 (E)-엔독시펜과 (Z)-엔독시펜의 출발 혼합물을 에틸 아세테이트(1:1 내지 1:20 wt/wt) 중의 6N HCL(1:1 내지 1:5 wt/wt)에 반응시키는 단계;
(b) 8N NaOH(1:1 내지 1:20 wt/wt)로 중화시키는 단계;
(c) 에틸 아세테이트로 1회 이상 세척하는 단계; 및
(d) IPA로 1회 이상 세척하는 단계
를 포함하고;
여기서 wt/wt는 (E)-엔독시펜과 (Z)-엔독시펜의 출발 혼합물에 대한 것인 방법. - 제12항 내지 제31항 중 어느 한 항의 결정질 형태를 제조하는 산업적으로 확장 가능한 방법으로서,
(a) 99:1 내지 40:60 범위의 E/Z-비율을 갖는 (E)-엔독시펜과 (Z)-엔독시펜의 출발 혼합물을 에틸 아세테이트(1:1 내지 1:20 wt/wt) 중의 6N HCL(1:1 내지 1:5 wt/wt)에 반응시키는 단계;
(b) 8N NaOH(1:1 내지 1:20 wt/wt)로 중화시키는 단계;
(c) 에틸 아세테이트로 1회 이상 세척하는 단계;
(d) 에틸 아세테이트와 n-헵탄의 혼합물로 1회 이상 세척하는 단계; 및
(e) 제12항 내지 제31항 중 어느 한 항의 결정질 형태를 회수하는 단계
를 포함하고;
여기서 wt/wt는 (E)-엔독시펜과 (Z)-엔독시펜의 출발 혼합물에 대한 것인 방법. - 제121항 또는 제122항의 방법에 따라 생성된 화학식 (III)의 화합물의 결정질 형태.
- 제123항에 있어서, 결정질 형태가 화학식 (III)의 화합물의 형태 II 또는 형태 III인 결정질 형태.
- 제62항 내지 제122항 중 어느 한 항에 있어서, (Z)-엔독시펜이 D-글루코네이트 또는 L-글루코네이트와 반응하여 (Z)-엔독시펜 L-글루코네이트 또는 (Z)-엔독시펜 D-글루코네이트를 형성하는 것인 방법.
- 제62항 내지 제125항 중 어느 한 항에 있어서, (Z)-엔독시펜 유리 염기가 < 1% 불순물을 갖는 것인 방법.
- 제62항 내지 제126항 중 어느 한 항에 있어서, (Z)-엔독시펜 유리 염기가 적어도 9개월 동안 주위 온도에서 안정한 것인 방법.
- 제62항 내지 제127항 중 어느 한 항의 방법에 의해 제조된, (Z)-엔독시펜, (E)-엔독시펜, 화학식 (III)의 화합물, 화학식 (II)의 화합물, 또는 그의 염.
- 제62항 내지 제127항 중 어느 한 항의 방법에 의해 제조된 (Z)-엔독시펜 유리 염기 또는 그의 염을 포함하는 조성물.
- 제129항에 있어서, 조성물이 경구, 비경구, 국소, 또는 관내 전달을 위해 제형화되는 것인 조성물.
- 제129항 또는 제130항에 있어서, 조성물이 경구 전달을 위해 정제, 캐플릿, 캡슐, 또는 환제로 제형화되는 것인 조성물.
- 제131항에 있어서, 대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기를 갖는 것인 조성물.
- 제129항 내지 제132항 중 어느 한 항에 있어서, 조성물이 경구 전달을 위해 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿 또는 지연 방출 캡슐로 제형화되는 것인 조성물.
- 제129항 내지 제133항 중 어느 한 항에 있어서, 조성물이 대상체에서 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다의 치료 또는 예방을 위해 대상체에게 투여되는 것인 조성물.
- 단위 용량당 1 mg 내지 200 mg의 (Z)-엔독시펜 유리 염기 또는 그의 염을 포함하고 이를 필요로 하는 대상체에게 투여하기 위한 경구 조성물로서, 여기서 경구 조성물의 일일 투여는 대상체에서
(a) 7 내지 21일 이내에 엔독시펜의 항정 상태 혈장 수준;
(b) 25 nM 내지 300 nM 범위의 엔독시펜의 항정 상태 혈장 수준;
(c) 30 nM 초과의 엔독시펜의 항정 상태 혈장 수준;
(d) 투여 후 2 내지 10시간 이내에 엔독시펜의 최대 혈장 수준; 또는
(e) 그의 임의의 조합
을 달성하는 것인 경구 조성물. - 제135항에 있어서, 대상체에서 투여 후 40시간 내지 55시간 범위의 엔독시펜의 평균 반감기를 갖는 것인 경구 조성물.
- 제135항 또는 제136항에 있어서, 조성물이 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿 또는 지연 방출 캡슐로 제형화되는 것인 경구 조성물.
- 제133항 또는 제137항에 있어서, 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 또는 적어도 90%의 엔독시펜이 장에서 방출되는 것인 경구 조성물.
- 제129항 내지 제138항 중 어느 한 항에 있어서, 200 hr*ng/mL 내지 10000 hr*ng/mL, 300 hr*ng/mL 내지 8000 hr*ng/mL, 400 hr*ng/mL 내지 6000 hr*ng/mL 또는 700 hr*ng/mL 내지 6000 hr*ng/mL의 무한대 시간으로 외삽된 곡선 아래 평균 면적(AUC0-inf)을 갖는 것인 조성물 또는 경구 조성물.
- 제129항 내지 제139항 중 어느 한 항의 경구 조성물을 대상체에게 투여하는 단계를 포함하여 이를 필요로 하는 대상체를 치료하는 방법.
- 제140항에 있어서, 대상체가 호르몬 의존적 유방 장애, 호르몬 의존적 생식관 장애, 또는 둘 다를 갖거나 가질 위험이 있는 것인 방법.
- 제141항에 있어서, 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애가 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 또는 외음부암인 방법.
- 제141항 또는 제142항에 있어서, 대상체가 전립선암을 갖고 대상체가 추가로 여성형 유방증을 갖거나 가질 위험이 있는 것인 방법.
- 제141항 내지 제143항 중 어느 한 항에 있어서, 대상체가 타목시펜 불응성 또는 타목시펜 내성 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애를 갖는 것인 방법.
- 제141항 내지 제144항 중 어느 한 항에 있어서, 대상체가 시탈로프람, 에스시탈로프람, 플루옥세틴, 파록세틴, 세르트랄린, 및 빌라조돈으로 이루어진 군으로부터 선택되는 SSRI 약물로 치료되거나 치료될 것인 방법.
- 제141항 내지 제145항 중 어느 한 항에 있어서, 대상체에게 0.01 mg 내지 200 mg의 (Z)-엔독시펜이 투여되는 것인 방법.
- 제141항 내지 제146항 중 어느 한 항에 있어서, 대상체에게 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 (Z)-엔독시펜이 매일 투여되는 것인 방법.
- 제141항 내지 제147항 중 어느 한 항에 있어서, 대상체에서 엔독시펜의 항정 상태 혈장 수준이 30 nM 초과인 방법.
- 제141항 내지 제148항 중 어느 한 항에 있어서, 엔독시펜의 항정 상태 혈장 수준이 조성물의 제1 투여 후 7 내지 21일 이내에 달성되는 것인 방법.
- 제141항 내지 제149항 중 어느 한 항에 있어서, 엔독시펜의 최대 혈장 수준까지의 시간이 조성물을 투여한 후 2시간 내지 10시간 또는 4시간 내지 8시간의 범위인 방법.
- 호르몬 의존적 유방 장애 또는 호르몬 의존적 생식관 장애 또는 둘 다를 갖거나 가질 위험이 있는 대상체를 치료하는 방법으로서, (Z)-엔독시펜 또는 그의 염을 포함하는 경구 조성물을 투여하는 단계를 포함하고, 여기서 조성물의 투여는
(a) 대상체에서 투여 후 30시간 내지 60시간 범위의 엔독시펜의 평균 반감기;
(b) 투여 후 4시간 내지 8시간 범위의 엔독시펜의 최대 혈장 수준까지의 시간; 및
(c) 30 nM 초과의 엔독시펜의 항정 상태 혈장 수준
을 달성하는 것인 방법. - 제151항에 있어서, 대상체에게 1 mg, 2 mg, 4 mg, 6 mg, 10 mg 또는 20 mg의 (Z)-엔독시펜이 투여되는 것인 방법.
- 제151항 또는 제152항에 있어서, 무한대 시간으로 외삽된 곡선 아래 평균 면적(AUC0-inf)이 200 hr*ng/mL 내지 10000 hr*ng/mL, 300 hr*ng/mL 내지 8000 hr*ng/mL, 400 hr*ng/mL 내지 6000 hr*ng/mL 또는 700 hr*ng/mL 내지 6000 hr*ng/mL인 방법.
- 제151항 내지 제153항 중 어느 한 항에 있어서, 조성물이 장용 정제, 장용 캐플릿, 장용 캡슐, 지연 방출 정제, 지연 방출 캐플릿 또는 지연 방출 캡슐로 제형화되는 것인 방법.
- 제151항 내지 제154항 중 어느 한 항에 있어서, 엔독시펜의 적어도 40%, 적어도 50%, 적어도 60%, 적어도 70%, 적어도 80%, 또는 적어도 90%가 장에서 방출되는 것인 방법.
- 제151항 또는 제155항에 있어서, 조성물이 1일 1회, 1일 2회, 1일 3회, 1일 4회, 격일, 1주 2회, 매주, 격주, 1개월 2회, 매월, 분기별, 6개월 1회, 또는 매년 투여되는 것인 방법.
- 제151항 내지 제156항 중 어느 한 항에 있어서, 호르몬 의존적 유방 장애 및 호르몬 의존적 생식관 장애가 양성 유방 장애, 증식증, 비정형, 비정형 유관 증식증, 비정형 소엽 증식증, 증가된 유방 밀도, 여성형 유방증, DCIS, LCIS, 유방암, 성조숙증, 맥쿤-알부라이트 증후군, 자궁내막암, 난소암, 자궁암, 자궁경부암, 질암, 및 외음부암으로 이루어진 군으로부터 선택되는 것인 방법.
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762556884P | 2017-09-11 | 2017-09-11 | |
| US201762556799P | 2017-09-11 | 2017-09-11 | |
| US62/556,884 | 2017-09-11 | ||
| US62/556,799 | 2017-09-11 | ||
| US201862624787P | 2018-01-31 | 2018-01-31 | |
| US62/624,787 | 2018-01-31 | ||
| US201862693885P | 2018-07-03 | 2018-07-03 | |
| US62/693,885 | 2018-07-03 | ||
| PCT/US2018/050272 WO2019051416A1 (en) | 2017-09-11 | 2018-09-10 | METHODS FOR THE MANUFACTURE AND USE OF ENDOXIFEN |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20200052349A true KR20200052349A (ko) | 2020-05-14 |
Family
ID=65635248
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020207010196A KR20200052349A (ko) | 2017-09-11 | 2018-09-10 | 엔독시펜을 제조 및 사용하는 방법 |
Country Status (12)
| Country | Link |
|---|---|
| US (4) | US11261151B2 (ko) |
| EP (1) | EP3681491A4 (ko) |
| JP (2) | JP2020533283A (ko) |
| KR (1) | KR20200052349A (ko) |
| CN (2) | CN111328280A (ko) |
| AU (2) | AU2018329202B2 (ko) |
| CA (1) | CA3073836A1 (ko) |
| IL (2) | IL272924B2 (ko) |
| MX (1) | MX2020002649A (ko) |
| SG (1) | SG11202002105WA (ko) |
| TW (1) | TWI793165B (ko) |
| WO (1) | WO2019051416A1 (ko) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2018329202B2 (en) | 2017-09-11 | 2022-10-27 | Atossa Therapeutics, Inc. | Methods for making and using endoxifen |
| CN114302717A (zh) * | 2019-07-03 | 2022-04-08 | 阿托萨治疗学公司 | 内昔芬的持续释放组合物 |
| CN110564962B (zh) * | 2019-10-14 | 2020-12-22 | 中南大学 | 一种黑白钨混合矿的冶炼方法 |
| CN113975279A (zh) * | 2021-10-13 | 2022-01-28 | 复旦大学附属肿瘤医院 | 维拉佐酮及其衍生物在制备抗肿瘤药物中的应用 |
| TW202340135A (zh) * | 2022-01-12 | 2023-10-16 | 美商阿托薩醫療公司 | (z)—因多昔芬之組合物及其富集方法 |
| WO2023211939A1 (en) * | 2022-04-26 | 2023-11-02 | Atossa Therapeutics, Inc. | High dose endoxifen formulations and methods of use |
Family Cites Families (79)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IT951631B (it) | 1971-03-18 | 1973-07-10 | Richardson Merrell Spa | Composti utili per la separazione di isomeri ottici geometrici e strutturali e relativo procedimen to di sintesi |
| GB8604528D0 (en) | 1986-02-24 | 1986-04-03 | Ici Plc | Therapeutic agents |
| US6881548B2 (en) | 1997-05-23 | 2005-04-19 | A&G Pharmaceutical, Inc. | Methods and kits for diagnosing tumorigenicity and determining resistance to the antineoplastic effects of antiestrogen therapy |
| US6638727B1 (en) | 1999-01-26 | 2003-10-28 | Cytyc Health Corporation | Methods for identifying treating or monitoring asymptomatic patients for risk reduction or therapeutic treatment of breast cancer |
| US6398765B1 (en) | 1999-03-01 | 2002-06-04 | Pro Duct Health, Inc. | Apparatus, methods and kits for simultaneous delivery of a substance to multiple breast milk ducts |
| US6245352B1 (en) | 1999-04-27 | 2001-06-12 | Eli Lilly And Company | Pharmaceutical formulation |
| MXPA02000053A (es) | 1999-07-05 | 2003-07-21 | Idea Ag | Un metodo para mejorar el tratamiento a traves de barreras adaptables semipermeables. |
| GB0000313D0 (en) | 2000-01-10 | 2000-03-01 | Astrazeneca Uk Ltd | Formulation |
| GB0008172D0 (en) | 2000-04-05 | 2000-05-24 | Astrazeneca Ab | Therapy |
| DE60327363D1 (de) | 2002-12-18 | 2009-06-04 | Besins Int Lab | Verringerung der dichte des brustgewebes durch 4-hydroxy tamoxifen |
| CA2509660C (en) | 2002-12-18 | 2011-08-30 | Bruno De Lignieres (Deceased) | Treatment of mastalgia with 4-hydroxy tamoxifen |
| EP1952810B1 (en) | 2003-04-01 | 2014-01-08 | Besins Healthcare Luxembourg SARL | Treatment of breast cancer with 4-hydroxytamoxifen |
| EP1663142A2 (en) | 2003-06-23 | 2006-06-07 | MacroChem Corporation | Compositons and methods for topical administration |
| US7531578B2 (en) | 2003-09-18 | 2009-05-12 | City Of Hope | Compounds and methods for treating breast cancer and other diseases |
| US7968532B2 (en) | 2003-12-15 | 2011-06-28 | Besins Healthcare Luxembourg | Treatment of gynecomastia with 4-hydroxy tamoxifen |
| WO2005070951A1 (en) | 2004-01-16 | 2005-08-04 | Cedarburg Pharmaceuticals, Inc. | Exemestane and its intermediates and methods of making the same |
| US8436029B2 (en) | 2004-03-19 | 2013-05-07 | Transform Pharmaceuticals, Inc. | Pharmaceutical forms, and methods of making and using the same |
| US7507769B2 (en) | 2004-03-22 | 2009-03-24 | Laboratoires Besins International | Treatment and prevention of benign breast disease with 4-hydroxy tamoxifen |
| JP2008537942A (ja) | 2005-03-31 | 2008-10-02 | マイトジェン, インコーポレイテッド | 心疾患のための処置 |
| EP1871750A1 (en) | 2005-07-06 | 2008-01-02 | Sicor, Inc. | Improved process for the preparation of letrozole |
| US20080319092A1 (en) | 2005-08-05 | 2008-12-25 | Nuvo Research Inc. | Transdermal Drug Delivery Formulation |
| WO2007043580A1 (ja) | 2005-10-12 | 2007-04-19 | Kowa Co., Ltd. | 乳癌及び/又は乳腺炎治療用イオントフォレシス製剤 |
| WO2007067773A2 (en) | 2005-12-09 | 2007-06-14 | Mayo Foundation For Medical Education And Research | Assessing outcomes for breast cancer patients by determining cyp2d6 genotype |
| US8450351B2 (en) | 2005-12-20 | 2013-05-28 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| US9308181B2 (en) | 2006-03-06 | 2016-04-12 | Nuvo Research Inc. | Topical formulations, systems and methods |
| WO2007141799A1 (en) | 2006-06-05 | 2007-12-13 | Cadila Healthcare Limited | A process for preparing pure anastrozole |
| CL2007002851A1 (es) | 2006-10-05 | 2008-01-18 | M S Panacea Biotec Ltd | Composicion inyectable que comprende micro o nano -particulas biodegradables que incluyen un agente activo, un polimero biodegardable, un agente para intensificar la viscosidad y excipiente farmaceuticamente aceptables; y procedimientos de preparacion |
| US20090291134A1 (en) | 2006-11-21 | 2009-11-26 | Jina Pharmaceuticals, Inc. | Endoxifen methods and compositions in the treatment of psychiatric and neurodegenerative diseases |
| EP2101731B1 (en) | 2006-11-21 | 2018-01-31 | Jina Pharmaceuticals Inc. | Endoxifen for use in the treatment of cancer |
| US20120164075A1 (en) * | 2006-11-21 | 2012-06-28 | Jina Pharmaceuticals, Inc. | Endoxifen methods and compositions in the treatment of mammalian diseases |
| EP2097085A4 (en) | 2006-11-27 | 2010-02-10 | Ariad Pharma Inc | THERAPEUTIC MATERIALS AND METHOD |
| WO2008067991A2 (en) | 2006-12-08 | 2008-06-12 | Antares Pharma Ipl Ag | Skin-friendly drug complexes for transdermal administration |
| WO2008113177A1 (en) | 2007-03-20 | 2008-09-25 | Centre De Recherche Sur Les Biotechnologies Marines | Compositions comprising polyunsaturated fatty acid monoglycerides or derivatives thereof and uses thereof |
| CA2680825C (en) | 2007-03-22 | 2013-10-29 | Cytotech Labs, Llc | Topical formulations having enhanced bioavailability |
| WO2009032699A1 (en) | 2007-08-29 | 2009-03-12 | Drugtech Corporation | Anti-proliferative combinations |
| AR068409A1 (es) | 2007-09-14 | 2009-11-18 | Drugtech Corp | Composiciones farmaceuticas, transdermicas sin alcohol |
| ATE547409T1 (de) | 2007-11-28 | 2012-03-15 | Fresenius Kabi Oncology Ltd | Verbessertes verfahren zur herstellung von letrozol und zwischenprodukten davon |
| WO2009120999A2 (en) | 2008-03-28 | 2009-10-01 | Olema Pharmaceuticals, Inc. | Use of an endoxifen prodrug for treatment of breast cancer |
| US20100069781A1 (en) | 2008-04-15 | 2010-03-18 | Johansen Jerald A | Device and method for accessing and treating ducts of mammary glands |
| US8063249B1 (en) | 2008-04-25 | 2011-11-22 | Olema Pharmaceuticals, Inc. | Substituted triphenyl butenes |
| KR101783004B1 (ko) | 2008-07-08 | 2017-09-28 | 인사이트 홀딩스 코포레이션 | 인돌아민 2,3-디옥시게나아제의 억제제로서의 1,2,5-옥사디아졸 |
| WO2010009335A1 (en) | 2008-07-17 | 2010-01-21 | Micell Technologies, Inc. | Drug delivery medical device |
| WO2010043404A1 (en) | 2008-10-15 | 2010-04-22 | Synthon B.V. | Processes and intermediates for the production of fulvestrant |
| US20100098659A1 (en) | 2008-10-22 | 2010-04-22 | Revalesio Corporation | Compositions and methods for treating matrix metalloproteinase 9 (mmp9)-mediated conditions |
| DK2373305T3 (en) | 2008-12-11 | 2017-05-15 | Besins Healthcare Lu Sarl | TRANSDERMAL PHARMACEUTICAL COMPOSITIONS INCLUDING A SERM |
| CN103896786B (zh) * | 2009-05-21 | 2015-11-25 | 扬子江药业集团有限公司 | 枸橼酸他莫昔芬的晶型及晶型a的制备方法 |
| US9132084B2 (en) * | 2009-05-27 | 2015-09-15 | Neothetics, Inc. | Methods for administration and formulations for the treatment of regional adipose tissue |
| US20100317635A1 (en) | 2009-06-16 | 2010-12-16 | Endorecherche, Inc. | Treatment of hot flushes, vasomotor symptoms, and night sweats with sex steroid precursors in combination with selective estrogen receptor modulators |
| WO2011031561A2 (en) | 2009-08-27 | 2011-03-17 | Ensisheim Partners Llc | Nucleic acid molecules and uses thereof |
| US8637569B2 (en) | 2009-10-22 | 2014-01-28 | Api Genesis, Llc | Methods of increasing solubility of poorly soluble compounds and methods of making and using formulations of such compounds |
| WO2011072244A1 (en) | 2009-12-10 | 2011-06-16 | Mount Sinai School Of Medicine Of New York University | Method of treatment of breast cancer with tamoxifen |
| EP2425833A1 (de) | 2010-09-03 | 2012-03-07 | Fresenius Kabi Deutschland GmbH | Die intravenöse Applikation von Fischöl/DHA +EPA vor bzw. mit Beginn der Chemotherapie |
| KR101308258B1 (ko) | 2010-10-15 | 2013-09-13 | 씨제이제일제당 (주) | 엔독시펜의 신규한 제조 방법 |
| US9200045B2 (en) | 2011-03-11 | 2015-12-01 | President And Fellows Of Harvard College | Small molecule-dependent inteins and uses thereof |
| US20120301541A1 (en) | 2011-05-24 | 2012-11-29 | Haronsky Elina | Compressed core for pharmaceutical composition |
| WO2013033430A1 (en) | 2011-09-02 | 2013-03-07 | Wake Forest School Of Medicine | Targeted delivery and prodrug designs for platinum-acridine anti-cancer compounds and methods thereof |
| FR2980972B1 (fr) | 2011-10-05 | 2014-02-28 | Commissariat Energie Atomique | Formulations pour le diagnostic et le traitement de cancers hormonodependants et de cancers des organes de synthese d'hormones steroidiennes. |
| EP2822955B1 (en) | 2012-03-05 | 2017-07-26 | Xavier University | Boron-based 4-hydroxytamoxifen and endoxifen prodrugs as treatment for breast cancer |
| US9220680B2 (en) | 2012-07-13 | 2015-12-29 | South Dakota State University | Compositions and methods for localized drug delivery through mammary papillae |
| US20150321983A1 (en) | 2012-10-19 | 2015-11-12 | Fermion Oy | A process for the preparation of ospemifene |
| CA2891412A1 (en) | 2012-11-20 | 2014-05-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of indoleamine 2,3-dioxygenase |
| KR102363191B1 (ko) | 2013-02-26 | 2022-02-17 | 메모리얼 슬로안 케터링 캔서 센터 | 면역치료용 조성물 및 방법 |
| IN2013MU00646A (ko) * | 2013-03-04 | 2015-06-26 | Intas Pharmaceuticals Ltd | |
| TW201536278A (zh) * | 2013-05-31 | 2015-10-01 | Del Mar Pharmaceuticals | 雙脫水半乳糖醇及其類似物與衍生物用於治療復發性惡性神經膠瘤或進行性繼發腦瘤之用途 |
| AU2014334531B2 (en) | 2013-10-18 | 2019-05-16 | The Regents Of The University Of Michigan | Systems and methods for determining a treatment course of action |
| HUP1300646A2 (en) | 2013-11-12 | 2015-05-28 | Druggability Technologies Ip Holdco Jersey Ltd | Complexes of fulvestrant and its derivatives, process for the preparation thereof and pharmaceutical compositions containing them |
| US20160375234A1 (en) | 2014-01-10 | 2016-12-29 | Atossa Genetic Inc. | Transpapillary methods and compositions for diagnosing and treating breast conditions |
| US9744177B2 (en) | 2014-03-10 | 2017-08-29 | Endorecherche, Inc. | Treatment of male androgen deficiency symptoms or diseases with sex steroid precursor combined with SERM |
| CA2942301A1 (en) | 2014-03-11 | 2015-09-17 | Repros Therapeutics Inc. | Clomiphene synthesis using a single solvent |
| US20170145515A1 (en) | 2014-06-04 | 2017-05-25 | Atossa Genetics Inc. | Molecular mammography |
| CN104230723B (zh) | 2014-08-21 | 2016-08-24 | 凯莱英医药集团(天津)股份有限公司 | 托瑞米芬的合成方法 |
| US20180049999A1 (en) | 2015-04-14 | 2018-02-22 | Atossa Genetics Inc. | Compositions and methods of treatment of breast disorders and estrogen-related disorders |
| US20180200206A1 (en) | 2015-07-14 | 2018-07-19 | Atossa Genetics Inc. | Transpapillary methods and compositions for treating breast disorders |
| WO2017070651A1 (en) * | 2015-10-22 | 2017-04-27 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Process for preparing (z)-endoxifen of high purity |
| DE102015222031A1 (de) * | 2015-11-10 | 2017-05-11 | Robert Bosch Gesellschaft Für Medizinische Forschung Mbh | Synthese von (Z)-Endoxifenhydrochlorid |
| US20170304232A1 (en) | 2016-04-20 | 2017-10-26 | Northwestern University | Endoxifen For Local Transdermal Therapy To The Breast |
| AU2018329202B2 (en) | 2017-09-11 | 2022-10-27 | Atossa Therapeutics, Inc. | Methods for making and using endoxifen |
| EP3681478A4 (en) | 2017-09-11 | 2021-07-14 | Atossa Therapeutics, Inc. | TOPICAL COMPOSITIONS AND METHODS OF TREATMENT |
| KR20200051690A (ko) | 2017-09-11 | 2020-05-13 | 아토사 테라퓨틱스, 인크. | 국소 조성물 |
-
2018
- 2018-09-10 AU AU2018329202A patent/AU2018329202B2/en active Active
- 2018-09-10 CN CN201880072905.6A patent/CN111328280A/zh active Pending
- 2018-09-10 MX MX2020002649A patent/MX2020002649A/es unknown
- 2018-09-10 CN CN202310795504.6A patent/CN116832020A/zh active Pending
- 2018-09-10 CA CA3073836A patent/CA3073836A1/en active Pending
- 2018-09-10 IL IL272924A patent/IL272924B2/en unknown
- 2018-09-10 JP JP2020511956A patent/JP2020533283A/ja active Pending
- 2018-09-10 TW TW107131790A patent/TWI793165B/zh active
- 2018-09-10 US US16/641,985 patent/US11261151B2/en active Active
- 2018-09-10 EP EP18853361.6A patent/EP3681491A4/en active Pending
- 2018-09-10 SG SG11202002105WA patent/SG11202002105WA/en unknown
- 2018-09-10 KR KR1020207010196A patent/KR20200052349A/ko not_active Application Discontinuation
- 2018-09-10 IL IL304863A patent/IL304863A/en unknown
- 2018-09-10 WO PCT/US2018/050272 patent/WO2019051416A1/en unknown
-
2022
- 2022-01-20 US US17/580,428 patent/US11572334B2/en active Active
- 2022-12-29 US US18/090,757 patent/US11680036B1/en active Active
-
2023
- 2023-01-03 AU AU2023200013A patent/AU2023200013A1/en active Pending
- 2023-03-30 US US18/128,536 patent/US20230365490A1/en active Pending
- 2023-07-18 JP JP2023116961A patent/JP2023126540A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| AU2023200013A1 (en) | 2023-02-09 |
| IL304863A (en) | 2023-10-01 |
| MX2020002649A (es) | 2020-09-25 |
| TWI793165B (zh) | 2023-02-21 |
| CN111328280A (zh) | 2020-06-23 |
| WO2019051416A1 (en) | 2019-03-14 |
| EP3681491A4 (en) | 2021-06-30 |
| EP3681491A1 (en) | 2020-07-22 |
| CA3073836A1 (en) | 2019-03-14 |
| US11572334B2 (en) | 2023-02-07 |
| IL272924A (en) | 2020-04-30 |
| AU2018329202B2 (en) | 2022-10-27 |
| TW201919596A (zh) | 2019-06-01 |
| IL272924B2 (en) | 2024-01-01 |
| US20220242816A1 (en) | 2022-08-04 |
| US20230365490A1 (en) | 2023-11-16 |
| US11261151B2 (en) | 2022-03-01 |
| AU2018329202A1 (en) | 2020-03-19 |
| JP2020533283A (ja) | 2020-11-19 |
| US20230183166A1 (en) | 2023-06-15 |
| CN116832020A (zh) | 2023-10-03 |
| US20200207704A1 (en) | 2020-07-02 |
| JP2023126540A (ja) | 2023-09-07 |
| US11680036B1 (en) | 2023-06-20 |
| SG11202002105WA (en) | 2020-04-29 |
| IL272924B1 (en) | 2023-09-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11572334B2 (en) | Methods for making and using endoxifen | |
| TWI761476B (zh) | 尼拉帕尼(niraparib)組合物 | |
| EP3227273B1 (en) | Compositions comprising 2-((1-(2(4-fluorophenyl)-2-oxoethyl)piperidin-4-yl)methyl)isoindolin-1-one for treating schizophrenia | |
| JP2018525414A (ja) | 癌を処置するための方法 | |
| WO2019196957A1 (zh) | 亲水性小檗碱型衍生物及其制备药物的应用 | |
| TW202233606A (zh) | Cdk2抑制劑之固體形式 | |
| KR20140129164A (ko) | 히스톤 데아세틸라아제 억제제 및 파조파닙의 조합물 및 이의 용도 | |
| KR20150123248A (ko) | 유기 화합물의 제제 | |
| US20230026232A1 (en) | Ahr inhibitors and uses thereof | |
| WO2011085606A1 (zh) | 用于抗感染的普卢利沙星的光学活性化合物和制备方法 | |
| TW202340135A (zh) | (z)—因多昔芬之組合物及其富集方法 | |
| EP3241562A1 (en) | Zonisamide for use in the treatment of breast cancer | |
| TWI814468B (zh) | 藥用組合物、其製備方法及用途 | |
| WO2017000270A1 (zh) | 枸橼酸舍曲林的2种晶型及其制备方法 | |
| JP2019529502A (ja) | アルツハイマー病及びパーキンソン病を処置するための組成物及び方法 | |
| TW202404585A (zh) | 含有匹密特匹(Pimitespib)之醫藥組合物 | |
| AU2022378576A1 (en) | Endoxifen for treatment of cancers | |
| WO2023158715A1 (en) | Oxphos inhibitors for use in treating cancer | |
| KR20240054333A (ko) | 공결정 | |
| KR20230026415A (ko) | 경구 제제 및 이의 용도 | |
| CN117157073A (zh) | 用墨蝶呤治疗胶质母细胞瘤的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| E902 | Notification of reason for refusal |