KR20180049255A - 글루코실세라마이드 신타아제의 억제제로서 genz 112638 헤미타르타르산염의 무정형 및 결정질 형태 - Google Patents
글루코실세라마이드 신타아제의 억제제로서 genz 112638 헤미타르타르산염의 무정형 및 결정질 형태 Download PDFInfo
- Publication number
- KR20180049255A KR20180049255A KR1020187012588A KR20187012588A KR20180049255A KR 20180049255 A KR20180049255 A KR 20180049255A KR 1020187012588 A KR1020187012588 A KR 1020187012588A KR 20187012588 A KR20187012588 A KR 20187012588A KR 20180049255 A KR20180049255 A KR 20180049255A
- Authority
- KR
- South Korea
- Prior art keywords
- subject
- salt
- compound
- formula
- acid salt
- Prior art date
Links
- 102000044956 Ceramide glucosyltransferases Human genes 0.000 title claims abstract description 20
- 108091000114 ceramide glucosyltransferase Proteins 0.000 title claims abstract description 20
- 239000003112 inhibitor Substances 0.000 title claims description 18
- KUBARPMUNHKBIQ-VTHUDJRQSA-N eliglustat tartrate Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.C([C@@H](NC(=O)CCCCCCC)[C@H](O)C=1C=C2OCCOC2=CC=1)N1CCCC1.C([C@@H](NC(=O)CCCCCCC)[C@H](O)C=1C=C2OCCOC2=CC=1)N1CCCC1 KUBARPMUNHKBIQ-VTHUDJRQSA-N 0.000 title claims description 3
- 229960002856 eliglustat Drugs 0.000 title 1
- 150000003839 salts Chemical class 0.000 claims abstract description 327
- 239000002253 acid Substances 0.000 claims abstract description 193
- 150000001875 compounds Chemical class 0.000 claims abstract description 129
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 53
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 19
- -1 tartaric acid salt Chemical class 0.000 claims abstract description 17
- 201000010099 disease Diseases 0.000 claims abstract description 16
- 238000000034 method Methods 0.000 claims description 99
- 238000011282 treatment Methods 0.000 claims description 91
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 76
- 239000000945 filler Substances 0.000 claims description 66
- 239000000203 mixture Substances 0.000 claims description 63
- 239000003814 drug Substances 0.000 claims description 58
- 102000004190 Enzymes Human genes 0.000 claims description 50
- 108090000790 Enzymes Proteins 0.000 claims description 50
- 229940088598 enzyme Drugs 0.000 claims description 50
- 210000004185 liver Anatomy 0.000 claims description 48
- 239000007787 solid Substances 0.000 claims description 46
- 102100033342 Lysosomal acid glucosylceramidase Human genes 0.000 claims description 39
- 101150053185 P450 gene Proteins 0.000 claims description 36
- 239000011230 binding agent Substances 0.000 claims description 31
- 210000000952 spleen Anatomy 0.000 claims description 27
- 238000009472 formulation Methods 0.000 claims description 26
- 230000036470 plasma concentration Effects 0.000 claims description 26
- 229940124597 therapeutic agent Drugs 0.000 claims description 25
- 229920000168 Microcrystalline cellulose Polymers 0.000 claims description 24
- 239000008108 microcrystalline cellulose Substances 0.000 claims description 24
- 229940016286 microcrystalline cellulose Drugs 0.000 claims description 24
- 235000019813 microcrystalline cellulose Nutrition 0.000 claims description 24
- 208000015872 Gaucher disease Diseases 0.000 claims description 23
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 claims description 22
- 229960001021 lactose monohydrate Drugs 0.000 claims description 22
- 238000000634 powder X-ray diffraction Methods 0.000 claims description 20
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 claims description 17
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 claims description 17
- 208000024720 Fabry Disease Diseases 0.000 claims description 16
- 238000002360 preparation method Methods 0.000 claims description 16
- OKMWKBLSFKFYGZ-UHFFFAOYSA-N 1-behenoylglycerol Chemical compound CCCCCCCCCCCCCCCCCCCCCC(=O)OCC(O)CO OKMWKBLSFKFYGZ-UHFFFAOYSA-N 0.000 claims description 15
- 229940049654 glyceryl behenate Drugs 0.000 claims description 15
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 claims description 15
- 238000012360 testing method Methods 0.000 claims description 15
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical group O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 claims description 14
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 claims description 14
- 230000002401 inhibitory effect Effects 0.000 claims description 14
- 108010001237 Cytochrome P-450 CYP2D6 Proteins 0.000 claims description 13
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 claims description 13
- 229920002472 Starch Polymers 0.000 claims description 12
- 238000003556 assay Methods 0.000 claims description 12
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 claims description 12
- 239000008107 starch Substances 0.000 claims description 12
- 235000019698 starch Nutrition 0.000 claims description 12
- 108010081668 Cytochrome P-450 CYP3A Proteins 0.000 claims description 10
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 10
- AHOUBRCZNHFOSL-YOEHRIQHSA-N (+)-Casbol Chemical compound C1=CC(F)=CC=C1[C@H]1[C@H](COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-YOEHRIQHSA-N 0.000 claims description 9
- 102100021704 Cytochrome P450 2D6 Human genes 0.000 claims description 9
- AHOUBRCZNHFOSL-UHFFFAOYSA-N Paroxetine hydrochloride Natural products C1=CC(F)=CC=C1C1C(COC=2C=C3OCOC3=CC=2)CNCC1 AHOUBRCZNHFOSL-UHFFFAOYSA-N 0.000 claims description 9
- 229930006000 Sucrose Natural products 0.000 claims description 9
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 claims description 9
- 239000000314 lubricant Substances 0.000 claims description 9
- 229960002296 paroxetine Drugs 0.000 claims description 9
- 239000005720 sucrose Substances 0.000 claims description 9
- 108010056760 agalsidase beta Proteins 0.000 claims description 8
- LOUPRKONTZGTKE-LHHVKLHASA-N quinidine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@H]2[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-LHHVKLHASA-N 0.000 claims description 8
- 150000003892 tartrate salts Chemical class 0.000 claims description 8
- 230000001225 therapeutic effect Effects 0.000 claims description 7
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 claims description 6
- 229960004125 ketoconazole Drugs 0.000 claims description 6
- 239000011780 sodium chloride Substances 0.000 claims description 6
- 229920000881 Modified starch Polymers 0.000 claims description 5
- 238000003205 genotyping method Methods 0.000 claims description 5
- RTHCYVBBDHJXIQ-MRXNPFEDSA-N (R)-fluoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=C(C(F)(F)F)C=C1 RTHCYVBBDHJXIQ-MRXNPFEDSA-N 0.000 claims description 4
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 claims description 4
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 claims description 4
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 claims description 4
- 108010010803 Gelatin Proteins 0.000 claims description 4
- 102000001554 Hemoglobins Human genes 0.000 claims description 4
- 108010054147 Hemoglobins Proteins 0.000 claims description 4
- SQUHHTBVTRBESD-UHFFFAOYSA-N Hexa-Ac-myo-Inositol Natural products CC(=O)OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC(C)=O SQUHHTBVTRBESD-UHFFFAOYSA-N 0.000 claims description 4
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 claims description 4
- 229930195725 Mannitol Natural products 0.000 claims description 4
- 239000002202 Polyethylene glycol Substances 0.000 claims description 4
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 claims description 4
- 229960004977 anhydrous lactose Drugs 0.000 claims description 4
- 239000001506 calcium phosphate Substances 0.000 claims description 4
- 229910000389 calcium phosphate Inorganic materials 0.000 claims description 4
- 235000011010 calcium phosphates Nutrition 0.000 claims description 4
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 claims description 4
- 235000013539 calcium stearate Nutrition 0.000 claims description 4
- 239000008116 calcium stearate Substances 0.000 claims description 4
- 239000001768 carboxy methyl cellulose Substances 0.000 claims description 4
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 claims description 4
- 229960002464 fluoxetine Drugs 0.000 claims description 4
- 229920000159 gelatin Polymers 0.000 claims description 4
- 239000008273 gelatin Substances 0.000 claims description 4
- 235000019322 gelatine Nutrition 0.000 claims description 4
- 235000011852 gelatine desserts Nutrition 0.000 claims description 4
- 239000008172 hydrogenated vegetable oil Substances 0.000 claims description 4
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 claims description 4
- 239000001863 hydroxypropyl cellulose Substances 0.000 claims description 4
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 claims description 4
- 229960000367 inositol Drugs 0.000 claims description 4
- 239000000594 mannitol Substances 0.000 claims description 4
- 235000010355 mannitol Nutrition 0.000 claims description 4
- 229960001855 mannitol Drugs 0.000 claims description 4
- 238000012544 monitoring process Methods 0.000 claims description 4
- 229920001206 natural gum Polymers 0.000 claims description 4
- 229920001223 polyethylene glycol Polymers 0.000 claims description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 claims description 4
- 239000001267 polyvinylpyrrolidone Substances 0.000 claims description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 claims description 4
- 229960001404 quinidine Drugs 0.000 claims description 4
- CDAISMWEOUEBRE-UHFFFAOYSA-N scyllo-inosotol Natural products OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 claims description 4
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 claims description 4
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 claims description 4
- 229960002668 sodium chloride Drugs 0.000 claims description 4
- 239000000600 sorbitol Substances 0.000 claims description 4
- 239000006188 syrup Substances 0.000 claims description 4
- 235000020357 syrup Nutrition 0.000 claims description 4
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 claims description 4
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 claims description 3
- XVPZRKIQCKKYNE-UHFFFAOYSA-N Arborine Chemical compound N=1C(=O)C2=CC=CC=C2N(C)C=1CC1=CC=CC=C1 XVPZRKIQCKKYNE-UHFFFAOYSA-N 0.000 claims description 3
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 claims description 3
- 235000002017 Zea mays subsp mays Nutrition 0.000 claims description 3
- 235000005822 corn Nutrition 0.000 claims description 3
- 108010039650 imiglucerase Proteins 0.000 claims description 3
- 230000000977 initiatory effect Effects 0.000 claims description 3
- LXBIFEVIBLOUGU-DPYQTVNSSA-N migalastat Chemical group OC[C@H]1NC[C@H](O)[C@@H](O)[C@H]1O LXBIFEVIBLOUGU-DPYQTVNSSA-N 0.000 claims description 3
- UQRORFVVSGFNRO-UTINFBMNSA-N miglustat Chemical group CCCCN1C[C@H](O)[C@@H](O)[C@H](O)[C@H]1CO UQRORFVVSGFNRO-UTINFBMNSA-N 0.000 claims description 3
- 235000010413 sodium alginate Nutrition 0.000 claims description 3
- 239000000661 sodium alginate Substances 0.000 claims description 3
- 229940005550 sodium alginate Drugs 0.000 claims description 3
- 208000003311 Cytochrome P-450 Enzyme Inhibitors Diseases 0.000 claims description 2
- QPYJXFZUIJOGNX-HSUXUTPPSA-N afegostat Chemical group OC[C@H]1CNC[C@@H](O)[C@@H]1O QPYJXFZUIJOGNX-HSUXUTPPSA-N 0.000 claims description 2
- 229960004470 agalsidase beta Drugs 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 229960002127 imiglucerase Drugs 0.000 claims description 2
- 229950007469 migalastat Drugs 0.000 claims description 2
- 229960001512 miglustat Drugs 0.000 claims description 2
- 102000004328 Cytochrome P-450 CYP3A Human genes 0.000 claims 4
- 230000002503 metabolic effect Effects 0.000 claims 3
- 241000209149 Zea Species 0.000 claims 2
- 238000011260 co-administration Methods 0.000 claims 2
- 240000004307 Citrus medica Species 0.000 claims 1
- 239000003795 chemical substances by application Substances 0.000 claims 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 abstract description 14
- 235000002906 tartaric acid Nutrition 0.000 abstract description 9
- 239000011975 tartaric acid Substances 0.000 abstract description 9
- 238000000053 physical method Methods 0.000 abstract 1
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 159
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 126
- 241000699670 Mus sp. Species 0.000 description 95
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 66
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 51
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 45
- 210000004027 cell Anatomy 0.000 description 43
- 150000002305 glucosylceramides Chemical class 0.000 description 43
- 108010017544 Glucosylceramidase Proteins 0.000 description 37
- 239000002775 capsule Substances 0.000 description 32
- 239000002904 solvent Substances 0.000 description 32
- 210000001519 tissue Anatomy 0.000 description 32
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 30
- 230000000694 effects Effects 0.000 description 30
- 239000000758 substrate Substances 0.000 description 29
- 229940079593 drug Drugs 0.000 description 28
- 241001465754 Metazoa Species 0.000 description 24
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 24
- 235000013305 food Nutrition 0.000 description 24
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 22
- 238000009825 accumulation Methods 0.000 description 21
- 239000000243 solution Substances 0.000 description 20
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical class OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 19
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 18
- 210000004072 lung Anatomy 0.000 description 18
- 230000009467 reduction Effects 0.000 description 18
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 16
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 16
- 241000699666 Mus <mouse, genus> Species 0.000 description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- 239000013078 crystal Substances 0.000 description 14
- 230000005764 inhibitory process Effects 0.000 description 14
- 238000001556 precipitation Methods 0.000 description 14
- 238000004458 analytical method Methods 0.000 description 13
- 238000002641 enzyme replacement therapy Methods 0.000 description 13
- 239000012458 free base Substances 0.000 description 13
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 12
- 206010028980 Neoplasm Diseases 0.000 description 12
- 239000000523 sample Substances 0.000 description 12
- FEWJPZIEWOKRBE-LWMBPPNESA-N L-(+)-Tartaric acid Natural products OC(=O)[C@@H](O)[C@H](O)C(O)=O FEWJPZIEWOKRBE-LWMBPPNESA-N 0.000 description 11
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 11
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 10
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 10
- 239000008101 lactose Substances 0.000 description 10
- 229960001375 lactose Drugs 0.000 description 10
- 235000012054 meals Nutrition 0.000 description 10
- 239000002245 particle Substances 0.000 description 10
- 238000011002 quantification Methods 0.000 description 10
- 238000002560 therapeutic procedure Methods 0.000 description 10
- YDNKGFDKKRUKPY-JHOUSYSJSA-N C16 ceramide Natural products CCCCCCCCCCCCCCCC(=O)N[C@@H](CO)[C@H](O)C=CCCCCCCCCCCCCC YDNKGFDKKRUKPY-JHOUSYSJSA-N 0.000 description 9
- CRJGESKKUOMBCT-VQTJNVASSA-N N-acetylsphinganine Chemical compound CCCCCCCCCCCCCCC[C@@H](O)[C@H](CO)NC(C)=O CRJGESKKUOMBCT-VQTJNVASSA-N 0.000 description 9
- 108010030291 alpha-Galactosidase Proteins 0.000 description 9
- 102000005840 alpha-Galactosidase Human genes 0.000 description 9
- 229940106189 ceramide Drugs 0.000 description 9
- ZVEQCJWYRWKARO-UHFFFAOYSA-N ceramide Natural products CCCCCCCCCCCCCCC(O)C(=O)NC(CO)C(O)C=CCCC=C(C)CCCCCCCCC ZVEQCJWYRWKARO-UHFFFAOYSA-N 0.000 description 9
- 239000008187 granular material Substances 0.000 description 9
- VVGIYYKRAMHVLU-UHFFFAOYSA-N newbouldiamide Natural products CCCCCCCCCCCCCCCCCCCC(O)C(O)C(O)C(CO)NC(=O)CCCCCCCCCCCCCCCCC VVGIYYKRAMHVLU-UHFFFAOYSA-N 0.000 description 9
- RYHBNJHYFVUHQT-UHFFFAOYSA-N p-dioxane Substances C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 9
- 230000002829 reductive effect Effects 0.000 description 9
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 8
- 230000015572 biosynthetic process Effects 0.000 description 8
- 238000001816 cooling Methods 0.000 description 8
- 150000002632 lipids Chemical class 0.000 description 8
- 238000002844 melting Methods 0.000 description 8
- 230000008018 melting Effects 0.000 description 8
- 229910052757 nitrogen Inorganic materials 0.000 description 8
- 238000001953 recrystallisation Methods 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 229960001367 tartaric acid Drugs 0.000 description 8
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 7
- 101000934372 Homo sapiens Macrosialin Proteins 0.000 description 7
- 102100025136 Macrosialin Human genes 0.000 description 7
- 210000004369 blood Anatomy 0.000 description 7
- 239000008280 blood Substances 0.000 description 7
- 230000002132 lysosomal effect Effects 0.000 description 7
- 238000010186 staining Methods 0.000 description 7
- 102100039205 Cytochrome P450 3A4 Human genes 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- 239000008186 active pharmaceutical agent Substances 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 6
- 238000002648 combination therapy Methods 0.000 description 6
- 238000005538 encapsulation Methods 0.000 description 6
- 229940014516 fabrazyme Drugs 0.000 description 6
- 238000011049 filling Methods 0.000 description 6
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 238000010583 slow cooling Methods 0.000 description 6
- 108700028369 Alleles Proteins 0.000 description 5
- 108010029485 Protein Isoforms Proteins 0.000 description 5
- 102000001708 Protein Isoforms Human genes 0.000 description 5
- 238000010171 animal model Methods 0.000 description 5
- 238000013459 approach Methods 0.000 description 5
- 238000003304 gavage Methods 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 210000003734 kidney Anatomy 0.000 description 5
- 239000003960 organic solvent Substances 0.000 description 5
- 238000004806 packaging method and process Methods 0.000 description 5
- 210000002381 plasma Anatomy 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 239000011877 solvent mixture Substances 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 239000006228 supernatant Substances 0.000 description 5
- 208000024891 symptom Diseases 0.000 description 5
- 238000011870 unpaired t-test Methods 0.000 description 5
- 210000002700 urine Anatomy 0.000 description 5
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 4
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 4
- 206010018612 Gonorrhoea Diseases 0.000 description 4
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- 108010006519 Molecular Chaperones Proteins 0.000 description 4
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 241000700159 Rattus Species 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 230000009286 beneficial effect Effects 0.000 description 4
- 201000011510 cancer Diseases 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 208000001786 gonorrhea Diseases 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 150000002500 ions Chemical class 0.000 description 4
- 210000002540 macrophage Anatomy 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 238000010172 mouse model Methods 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- 239000008188 pellet Substances 0.000 description 4
- 238000011084 recovery Methods 0.000 description 4
- 238000012216 screening Methods 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 210000001835 viscera Anatomy 0.000 description 4
- 239000008096 xylene Substances 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- 208000007220 Cytochrome P-450 CYP2D6 Inhibitors Diseases 0.000 description 3
- 206010013710 Drug interaction Diseases 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- 239000001358 L(+)-tartaric acid Substances 0.000 description 3
- 235000011002 L(+)-tartaric acid Nutrition 0.000 description 3
- 102000005431 Molecular Chaperones Human genes 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 108091000080 Phosphotransferase Proteins 0.000 description 3
- HSCJRCZFDFQWRP-JZMIEXBBSA-N UDP-alpha-D-glucose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OP(O)(=O)OP(O)(=O)OC[C@@H]1[C@@H](O)[C@@H](O)[C@H](N2C(NC(=O)C=C2)=O)O1 HSCJRCZFDFQWRP-JZMIEXBBSA-N 0.000 description 3
- HSCJRCZFDFQWRP-UHFFFAOYSA-N Uridindiphosphoglukose Natural products OC1C(O)C(O)C(CO)OC1OP(O)(=O)OP(O)(=O)OCC1C(O)C(O)C(N2C(NC(=O)C=C2)=O)O1 HSCJRCZFDFQWRP-UHFFFAOYSA-N 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 238000007792 addition Methods 0.000 description 3
- 102000016679 alpha-Glucosidases Human genes 0.000 description 3
- 108010028144 alpha-Glucosidases Proteins 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000011278 co-treatment Methods 0.000 description 3
- 238000000354 decomposition reaction Methods 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- 229940000406 drug candidate Drugs 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 150000002339 glycosphingolipids Chemical class 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 238000000386 microscopy Methods 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 102000020233 phosphotransferase Human genes 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 235000021195 test diet Nutrition 0.000 description 3
- 239000003643 water by type Substances 0.000 description 3
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 102100037328 Chitotriosidase-1 Human genes 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 206010061818 Disease progression Diseases 0.000 description 2
- 102000005744 Glycoside Hydrolases Human genes 0.000 description 2
- 108010031186 Glycoside Hydrolases Proteins 0.000 description 2
- WZUVPPKBWHMQCE-UHFFFAOYSA-N Haematoxylin Chemical compound C12=CC(O)=C(O)C=C2CC2(O)C1C1=CC=C(O)C(O)=C1OC2 WZUVPPKBWHMQCE-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 208000015439 Lysosomal storage disease Diseases 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 206010027476 Metastases Diseases 0.000 description 2
- 229920003091 Methocel™ Polymers 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000001413 amino acids Chemical group 0.000 description 2
- 239000012296 anti-solvent Substances 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 230000003542 behavioural effect Effects 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- 108010057052 chitotriosidase Proteins 0.000 description 2
- 239000002131 composite material Substances 0.000 description 2
- 239000006059 cover glass Substances 0.000 description 2
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 2
- 230000001934 delay Effects 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- 238000004807 desolvation Methods 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- 230000005750 disease progression Effects 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 238000001493 electron microscopy Methods 0.000 description 2
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 239000010408 film Substances 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 210000002216 heart Anatomy 0.000 description 2
- 238000007489 histopathology method Methods 0.000 description 2
- 210000005260 human cell Anatomy 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 230000028993 immune response Effects 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 238000009434 installation Methods 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 230000001788 irregular Effects 0.000 description 2
- 238000011031 large-scale manufacturing process Methods 0.000 description 2
- 210000003712 lysosome Anatomy 0.000 description 2
- 230000001868 lysosomic effect Effects 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- 230000009401 metastasis Effects 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 210000001589 microsome Anatomy 0.000 description 2
- 239000012120 mounting media Substances 0.000 description 2
- KJONHKAYOJNZEC-UHFFFAOYSA-N nitrazepam Chemical compound C12=CC([N+](=O)[O-])=CC=C2NC(=O)CN=C1C1=CC=CC=C1 KJONHKAYOJNZEC-UHFFFAOYSA-N 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000012188 paraffin wax Substances 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- 208000033808 peripheral neuropathy Diseases 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 230000002685 pulmonary effect Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 238000009097 single-agent therapy Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 238000000527 sonication Methods 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- 239000010409 thin film Substances 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 238000001291 vacuum drying Methods 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 238000010792 warming Methods 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 239000008215 water for injection Substances 0.000 description 2
- 238000005550 wet granulation Methods 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- ULBPPCHRAVUQMC-MUMXBIPUSA-N (2r,3r)-2,3-dihydroxybutanedioic acid;(3r,4r,5r)-5-(hydroxymethyl)piperidine-3,4-diol Chemical compound OC[C@H]1CNC[C@@H](O)[C@@H]1O.OC(=O)[C@H](O)[C@@H](O)C(O)=O ULBPPCHRAVUQMC-MUMXBIPUSA-N 0.000 description 1
- AJIMMVOZOJXJKD-UHFFFAOYSA-N 2,3,4,5,6-pentafluoro-n-(3,3',6'-trihydroxyspiro[3h-2-benzofuran-1,9'-xanthene]-5-yl)benzamide Chemical compound C1=C2C(O)OC3(C4=CC=C(O)C=C4OC4=CC(O)=CC=C43)C2=CC=C1NC(=O)C1=C(F)C(F)=C(F)C(F)=C1F AJIMMVOZOJXJKD-UHFFFAOYSA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 1
- TVEXGJYMHHTVKP-UHFFFAOYSA-N 6-oxabicyclo[3.2.1]oct-3-en-7-one Chemical compound C1C2C(=O)OC1C=CC2 TVEXGJYMHHTVKP-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- CKLBXIYTBHXJEH-UHFFFAOYSA-J 75881-23-1 Chemical compound [Cl-].[Cl-].[Cl-].[Cl-].[Cu+2].[N-]1C(N=C2C3=CC=C(CSC(N(C)C)=[N+](C)C)C=C3C(N=C3C4=CC=C(CSC(N(C)C)=[N+](C)C)C=C4C(=N4)[N-]3)=N2)=C(C=C(CSC(N(C)C)=[N+](C)C)C=C2)C2=C1N=C1C2=CC(CSC(N(C)C)=[N+](C)C)=CC=C2C4=N1 CKLBXIYTBHXJEH-UHFFFAOYSA-J 0.000 description 1
- 230000035502 ADME Effects 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 241000605059 Bacteroidetes Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 102100022548 Beta-hexosaminidase subunit alpha Human genes 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 101150010738 CYP2D6 gene Proteins 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 102100024308 Ceramide synthase Human genes 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 208000029147 Collagen-vascular disease Diseases 0.000 description 1
- 235000014375 Curcuma Nutrition 0.000 description 1
- 244000164480 Curcuma aromatica Species 0.000 description 1
- 108010074922 Cytochrome P-450 CYP1A2 Proteins 0.000 description 1
- 108010020070 Cytochrome P-450 CYP2B6 Proteins 0.000 description 1
- 102000009666 Cytochrome P-450 CYP2B6 Human genes 0.000 description 1
- 108010000561 Cytochrome P-450 CYP2C8 Proteins 0.000 description 1
- 102000002263 Cytochrome P-450 CYP2C8 Human genes 0.000 description 1
- 108010001202 Cytochrome P-450 CYP2E1 Proteins 0.000 description 1
- 208000009011 Cytochrome P-450 CYP3A Inhibitors Diseases 0.000 description 1
- 102100026533 Cytochrome P450 1A2 Human genes 0.000 description 1
- 102100036194 Cytochrome P450 2A6 Human genes 0.000 description 1
- 102100024889 Cytochrome P450 2E1 Human genes 0.000 description 1
- 102000018832 Cytochromes Human genes 0.000 description 1
- 108010052832 Cytochromes Proteins 0.000 description 1
- FEWJPZIEWOKRBE-LWMBPPNESA-L D-tartrate(2-) Chemical compound [O-]C(=O)[C@@H](O)[C@H](O)C([O-])=O FEWJPZIEWOKRBE-LWMBPPNESA-L 0.000 description 1
- 241001539473 Euphoria Species 0.000 description 1
- 206010015535 Euphoric mood Diseases 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 1
- 102000002464 Galactosidases Human genes 0.000 description 1
- 108010093031 Galactosidases Proteins 0.000 description 1
- 241000295146 Gallionellaceae Species 0.000 description 1
- 208000020322 Gaucher disease type I Diseases 0.000 description 1
- 206010018364 Glomerulonephritis Diseases 0.000 description 1
- 102000000340 Glucosyltransferases Human genes 0.000 description 1
- 108010055629 Glucosyltransferases Proteins 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 229920002527 Glycogen Polymers 0.000 description 1
- 102000006263 Glycogen Debranching Enzyme System Human genes 0.000 description 1
- 108010058102 Glycogen Debranching Enzyme System Proteins 0.000 description 1
- 229930186217 Glycolipid Natural products 0.000 description 1
- 108060003393 Granulin Proteins 0.000 description 1
- 101000718525 Homo sapiens Alpha-galactosidase A Proteins 0.000 description 1
- 101000875170 Homo sapiens Cytochrome P450 2A6 Proteins 0.000 description 1
- 101000997662 Homo sapiens Lysosomal acid glucosylceramidase Proteins 0.000 description 1
- 101000859679 Homo sapiens Non-lysosomal glucosylceramidase Proteins 0.000 description 1
- 102000004867 Hydro-Lyases Human genes 0.000 description 1
- 108090001042 Hydro-Lyases Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 241000186660 Lactobacillus Species 0.000 description 1
- 208000033868 Lysosomal disease Diseases 0.000 description 1
- FEWJPZIEWOKRBE-XIXRPRMCSA-N Mesotartaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-XIXRPRMCSA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M Methanesulfonate Chemical compound CS([O-])(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 102100027814 Non-lysosomal glucosylceramidase Human genes 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 101710184309 Probable sucrose-6-phosphate hydrolase Proteins 0.000 description 1
- 102000001183 RAG-1 Human genes 0.000 description 1
- 108060006897 RAG1 Proteins 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 206010038468 Renal hypertrophy Diseases 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 102000002579 Sphingosine N-acyltransferase Human genes 0.000 description 1
- 108020004714 Sphingosine N-acyltransferase Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 102400000472 Sucrase Human genes 0.000 description 1
- 101710112652 Sucrose-6-phosphate hydrolase Proteins 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 208000022292 Tay-Sachs disease Diseases 0.000 description 1
- 206010057040 Temperature intolerance Diseases 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 230000001594 aberrant effect Effects 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- YTIVTFGABIZHHX-UHFFFAOYSA-L acetylenedicarboxylate(2-) Chemical compound [O-]C(=O)C#CC([O-])=O YTIVTFGABIZHHX-UHFFFAOYSA-L 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 108010049936 agalsidase alfa Proteins 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 210000001132 alveolar macrophage Anatomy 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- XORXDJBDZJBCOC-UHFFFAOYSA-N azanium;acetonitrile;acetate Chemical compound [NH4+].CC#N.CC([O-])=O XORXDJBDZJBCOC-UHFFFAOYSA-N 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 238000010876 biochemical test Methods 0.000 description 1
- 229940126587 biotherapeutics Drugs 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- GJPICJJJRGTNOD-UHFFFAOYSA-N bosentan Chemical compound COC1=CC=CC=C1OC(C(=NC(=N1)C=2N=CC=CN=2)OCCO)=C1NS(=O)(=O)C1=CC=C(C(C)(C)C)C=C1 GJPICJJJRGTNOD-UHFFFAOYSA-N 0.000 description 1
- 229960003065 bosentan Drugs 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000007963 capsule composition Substances 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 235000013339 cereals Nutrition 0.000 description 1
- 229940049197 cerezyme Drugs 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 238000009535 clinical urine test Methods 0.000 description 1
- 238000011284 combination treatment Methods 0.000 description 1
- NWFNSTOSIVLCJA-UHFFFAOYSA-L copper;diacetate;hydrate Chemical compound O.[Cu+2].CC([O-])=O.CC([O-])=O NWFNSTOSIVLCJA-UHFFFAOYSA-L 0.000 description 1
- 238000011262 co‐therapy Methods 0.000 description 1
- 229940109239 creatinine Drugs 0.000 description 1
- VFLDPWHFBUODDF-FCXRPNKRSA-N curcumin Chemical compound C1=C(O)C(OC)=CC(\C=C\C(=O)CC(=O)\C=C\C=2C=C(OC)C(O)=CC=2)=C1 VFLDPWHFBUODDF-FCXRPNKRSA-N 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000016396 cytokine production Effects 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 239000007857 degradation product Substances 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 1
- 108010061814 dihydroceramide desaturase Proteins 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 230000036267 drug metabolism Effects 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- YQGOJNYOYNNSMM-UHFFFAOYSA-N eosin Chemical compound [Na+].OC(=O)C1=CC=CC=C1C1=C2C=C(Br)C(=O)C(Br)=C2OC2=C(Br)C(O)=C(Br)C=C21 YQGOJNYOYNNSMM-UHFFFAOYSA-N 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 150000002118 epoxides Chemical class 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 210000003722 extracellular fluid Anatomy 0.000 description 1
- 239000004744 fabric Substances 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 238000001917 fluorescence detection Methods 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 108060003196 globin Proteins 0.000 description 1
- 102000018146 globin Human genes 0.000 description 1
- 206010061989 glomerulosclerosis Diseases 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 229940096919 glycogen Drugs 0.000 description 1
- 125000003147 glycosyl group Chemical group 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 230000008543 heat sensitivity Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- KKLGDUSGQMHBPB-UHFFFAOYSA-N hex-2-ynedioic acid Chemical compound OC(=O)CCC#CC(O)=O KKLGDUSGQMHBPB-UHFFFAOYSA-N 0.000 description 1
- 102000043404 human GLA Human genes 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 229960003943 hypromellose Drugs 0.000 description 1
- AUONNNVJUCSETH-UHFFFAOYSA-N icosanoyl icosanoate Chemical compound CCCCCCCCCCCCCCCCCCCC(=O)OC(=O)CCCCCCCCCCCCCCCCCCC AUONNNVJUCSETH-UHFFFAOYSA-N 0.000 description 1
- 230000007124 immune defense Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 235000011073 invertase Nutrition 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 201000008627 kidney hypertrophy Diseases 0.000 description 1
- 229940116298 l- malic acid Drugs 0.000 description 1
- 229940039696 lactobacillus Drugs 0.000 description 1
- 244000144972 livestock Species 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 238000011418 maintenance treatment Methods 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 201000008350 membranous glomerulonephritis Diseases 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 235000020938 metabolic status Nutrition 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- 238000006140 methanolysis reaction Methods 0.000 description 1
- IZYBEMGNIUSSAX-UHFFFAOYSA-N methyl benzenecarboperoxoate Chemical compound COOC(=O)C1=CC=CC=C1 IZYBEMGNIUSSAX-UHFFFAOYSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 238000002493 microarray Methods 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 230000003228 microsomal effect Effects 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- HZIRBXILQRLFIK-IEECYHMASA-N n-[(e,2s,3s)-1,3-dihydroxyoctadec-4-en-2-yl]-6-[(4-nitro-2,1,3-benzoxadiazol-7-yl)amino]hexanamide Chemical compound CCCCCCCCCCCCC\C=C\[C@H](O)[C@H](CO)NC(=O)CCCCCNC1=CC=C([N+]([O-])=O)C2=NON=C12 HZIRBXILQRLFIK-IEECYHMASA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 235000021590 normal diet Nutrition 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 238000003305 oral gavage Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 1
- 230000004526 pharmaceutical effect Effects 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 230000002974 pharmacogenomic effect Effects 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical compound CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 230000010287 polarization Effects 0.000 description 1
- 208000030761 polycystic kidney disease Diseases 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 230000001376 precipitating effect Effects 0.000 description 1
- 238000003672 processing method Methods 0.000 description 1
- 229940121649 protein inhibitor Drugs 0.000 description 1
- 239000012268 protein inhibitor Substances 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- 238000011555 rabbit model Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000009790 rate-determining step (RDS) Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 238000000935 solvent evaporation Methods 0.000 description 1
- 230000003393 splenic effect Effects 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 210000000352 storage cell Anatomy 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 238000004885 tandem mass spectrometry Methods 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- 238000002562 urinalysis Methods 0.000 description 1
- 230000002485 urinary effect Effects 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 239000002676 xenobiotic agent Substances 0.000 description 1
- 230000002034 xenobiotic effect Effects 0.000 description 1
- 229940099072 zavesca Drugs 0.000 description 1
Images














Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/357—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having two or more oxygen atoms in the same ring, e.g. crown ethers, guanadrel
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/4025—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil not condensed and containing further heterocyclic rings, e.g. cromakalim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7004—Monosaccharides having only carbon, hydrogen and oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D319/00—Heterocyclic compounds containing six-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D319/10—1,4-Dioxanes; Hydrogenated 1,4-dioxanes
- C07D319/14—1,4-Dioxanes; Hydrogenated 1,4-dioxanes condensed with carbocyclic rings or ring systems
- C07D319/16—1,4-Dioxanes; Hydrogenated 1,4-dioxanes condensed with carbocyclic rings or ring systems condensed with one six-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D319/00—Heterocyclic compounds containing six-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D319/10—1,4-Dioxanes; Hydrogenated 1,4-dioxanes
- C07D319/14—1,4-Dioxanes; Hydrogenated 1,4-dioxanes condensed with carbocyclic rings or ring systems
- C07D319/16—1,4-Dioxanes; Hydrogenated 1,4-dioxanes condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D319/18—Ethylenedioxybenzenes, not substituted on the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
- Heterocyclic Compounds That Contain Two Or More Ring Oxygen Atoms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Saccharide Compounds (AREA)
- Enzymes And Modification Thereof (AREA)
- Steroid Compounds (AREA)
Abstract
약학 용도에 사용될 수 있는 하기 구조식으로 표시되는 화합물의 헤미타르타르산염(화학식 I 헤미타르타르산염)이 기재된다. 특히, 화학식 (I) 헤미타르타르산염의 단일 결정질 형태는 다양한 특성과 물리적 측정값에 의하여 특징지워진다. 또한, 결정질 화학식 (I) 헤미타르타르산염을 생성하는 방법과, 다수의 질병을 치료하기 위한 대상에서 글루코실세라마이드 신타아제를 억제하거나 글리코스핀고리피드 농도를 저하하기 위하여 그것을 사용하는 것도 논의된다. 또한 약학 조성물이 기술된다.
Description
본 출원은 2009년 11월 27일에 제출된 미국 가출원 번호 제61/264,748호의 이익을 주장하며, 상기 가출원의 전체 내용은 본 명세서에 포함된다.
글리코스핀고리피드(GSL)은, 종양 세포들의 전이, 미생물들과 바이러스들의 세포들에의 결합, 세포들간 또는 세포들과 매트릭스 단백질들 간의 부착, 세포 분화 및 세포 성장을 촉진하는 능력을 비롯한, 다수의 생물학적 기능을 갖는 천연 발생 화합물의 한 부류이다. GSL은 효소 UDP-글루코오스: N-아실스핀고신 글루코실트랜스퍼라아제(GlcCer 신타아제)에 의하여 UDP-글루코오스 및 세라마이드로부터 생성되는 글루코실세라마이드(GlcCer)로부터 유래된다. 세라마이드의 구조는 하기에 도시된다:

세라마이드
GSL의 축적은 테이-삭스(Tay-Sachs) 병, 고셔(Gaucher) 병 및 패브리(Fabry) 병을 비롯한, 다수의 질병과 연관되어 왔다(예컨대, 미국 특허 번호 제6,051,598호 참고). GSL은 또한 특정 암과도 연관되어 왔다. 예컨대, 특정 GSL은 단지 종양에서만 또는 종양에서 비정상적으로 높은 농도로 발생하고; 배양 배지 중에서 종양 세포에 첨가되었을 때 종양 성장에 뚜렷한 자극 또는 억제 활성을 나타내며; 종양에 의하여 주변 세포 밖 유체로 스며들었을 때 신체의 정상적인 면역방어 시스템을 억제한다고 밝혀졌다. 종양이 점점 더 악성이 됨에 따라 종양의 GSL의 조성은 변화하고, 특정 GSL에 대한 항체가 종양의 성장을 억제한다.
GlcCer 신타아제를 억제하는 화합물은 GSL 농도를 낮출 수 있고, 전술한 질병 중 하나를 갖는 대상을 치료하는데 유용하다고 보고되었다. GlcCer의 많은 단백질 억제제 (이하, “아미노 세라마이드 유사 화합물”이라 함)은 미국 특허 제6,051,598호, 제5,952,370호, 제5,945,442호, 제5,916,911호 및 제6,030,995호에 기재되어 있다. 하기에 도시된 화학식 (I)의 화합물은 현재 고셔병의 치료를 위한 임상적 시도 중에 있는 GlcCer 신타아제 억제제이다.

결정질이고, 그렇지 않으면 대규모 제조를 할 수 있는 물리적 특성을 갖는 이 약물 후보자의 염 형태들의 요구가 있다. 또한, 이 화합물을 이용한 향상된 치료 방법 뿐만 아니라, 이 약물 후보자가 안정하고 환자에게 효과적으로 전달되는 약학 제형에 대한 요구가 있다.
화학식 (I) 화합물의 헤미타르타르산염(이하 “화학식 (I) 헤미타르타르산염”이라 함)은 특정한 비흡습성(non-hygroscopic) 결정질 형태를 제공하기 위하여 잘 한정된 조건하에서 결정질화될 수 있다는 것이 밝혀졌다. 화학식 (I) 헤미타르타르산염은 화학식 (I)의 다른 염들과 비교할 때 여러가지 이로운 특성들을 갖는다. 실시예 1에 추가로 기재된 바와 같이, 시트르산염, 말산염, 푸마르산, 메틸술폰산, 및 아세트산을 비롯한 많은 화학식 (I)의 염들은 고체 형태로 얻어질 수 없었다. 염화수소 및 1:1 화학식 (I)의 타르타르산염은 고체 형태로 얻어졌지만, 어느 것도 결정질이 아니었고 둘다 제형하기에 너무 흡습성이었다. 화학식 (I) 헤미타르타르산염은 자유 염기 및 다른 염들에 비하여 제형화하고 합성하기 쉽다. 화학식 (I) 헤미타르타르산염은 또한 대응하는 자유 염기(“이하 “화학식 (I) 자유 염기”라 함) 및 다른 염들 보다 더 잘 흐르고, 결정질, 비흡습성, 수용성이다. 따라서, 이들 유리한 특성들은 화학식 (I) 헤미타르타르산염이 약물 후보자로서 대규모 제작이 가능하도록 한다.
또한, 화학식 (I) 헤미타르타르산염의 캡슐 제형을 위한 안정한 과립이 수불용성 필러, 수용성 필러 및 화학식 (I) 헤미타르타르산염의 한정된 비율을 사용하여 제조될 수 있다는 것이 밝혀졌다. 이 발견을 기초로, 화학식 (I) 헤미타르타르산염의 안정한 약학 제형이 기재된다.
화학식 (I)의 화합물 또는 이의 약학적 허용염(화학식 (I) 헤미타르타르산염을 포함)은 간에 의하여, 주로 사이토크롬 P450 효소에 의하여 물질대사된다는 것이 밝혀졌다. 이 발견에 기초로, 약물/약물 상호작용에 대한 잠재성을 감소시키는 화학식 (I)의 화합물 또는 이의 약학적 허용염(화학식 (I) 헤미타르타르산염 포함)에 의한 치료 방법이 기재된다.
고셔병 마우스들은 재조합 글루코세레브로시다아제 및 이어서 화학식 (I) 헤미타르타르산염이 투여되었을 때, 글루코세레브로시다아제 단독 또는 화학식 (I) 헤미타르타르산염 단독으로 치료된 경우와 비교할 때, 내장 기관에서 GL1의 더 낮은 농도 및 간에서 고셔 세포의 더 감소된 수를 나타내었다. 이 발견에 기초하여, 화학식 (I) 화합물 또는 이의 약학적 허용염(화학식 (I) 헤미타르타르산염 포함)에 의한 병용 치료(combination therapy)도 기재된다.
본 출원의 하나의 실시상태는 화학식 (I)에 의하여 표시되는 화합물의 헤미타르타르산염이다. 전술한 바와 같이, 화학식 (I)에 의하여 표시되는 화합물의 헤미타르타르산염은 여기에서 “화학식 (I) 헤미타르타르산염”이라고 언급된다. 화학식 (I) 에 의하여 표시되는 화합물은 여기서 “화학식 (I) 자유 염기”라고 언급된다.
본 출원의 또 하나의 실시상태는 약학적 허용 담체 또는 희석제 및 화학식 (I) 헤미타르타르산염을 포함하는 약학 조성물을 제공한다.
또 하나의 실시상태는 화학식 (I) 헤미타르타르산염의 유효량을 대상에게 투여함으로써 그것이 필요한 대상 내의 글리코스핀고리피드 농도를 낮추거나 글루코실세라마이드 신타아제를 억제하는 방법을 제공한다.
또 하나의 실시상태는 그것을 필요로 하는 대상에서 글리코스핀고리피드 농도를 낮추거나 글루코실세라마이드 신타아제를 억제하기 위한 의약 제작을 위한 화학식 (I) 헤미타르타르산염의 용도를 제공한다.
또 하나의 실시상태는 그것을 필요로 하는 대상에서 글리코스핀고리피드 농도를 낮추거나 글루코실세라마이드 신타아제를 억제하기 위한 화학식 (I) 헤미타르타르산염의 용도를 제공한다.
또 하나의 실시상태는 고셔병을 갖는 대상을 치료하는 방법이다. 상기 방법은 제2 치료 제제의 유효량과 함께 제1 치료 제제의 유효량을 대상에게 투여하는 것을 포함한다. 상기 제1 치료 제제는 화학식 (I)로 표시되거나 이의 약학적 허용염이고; 상기 제2 치료제제는 고셔병의 치료에 효과적이다.
또 하나의 실시상태는 패브리병을 갖는 대상의 치료 방법이다. 상기 방법은 제2 치료 제제의 유효량과 함께 제1 치료 제제의 유효량을 대상에게 투여하는 것을 포함한다. 상기 제1 치료 제제는 화학식 (I)로 표시되거나 이의 약학적 허용염이고; 상기 제2 치료제제는 패브리병의 치료에 효과적이다.
또 하나의 실시상태는 화학식 (I)으로 표시되는 화합물의 헤미타르타르산염; 적어도 하나의 수용성 필러; 적어도 하나의 수불용성 필러; 적어도 하나의 바인더; 및 적어도 하나의 활택제를 포함하는 약학 조성물을 제공한다.
본 발명의 추가의 실시상태는 패브리병을 갖는 대상을 치료하는 방법이다. 상기 방법은
a) 화학식 (I)의 화합물 또는 이의 약학적 허용염의 유효량을 대상에게 투여하는 단계;
b) 상기 대상을 시험하여, 상기 대상이 좋지 못한, 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저(metabolizer)인지 여부를 결정하는 단계;
c) 상기 대상이 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인 경우, 상기 화합물의 조정된 유효량을 결정하는 단계; 및
d) 상기 대상이 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인 경우, 화학식 (I)의 화합물의 조정된 유효량을 대상에게 투여하고, 상기 대상이 좋지 못한 P450 메타볼라이저인 경우 상기 화학식 (I)의 화합물의 유효량을 상기 대상에게 투여하는 단계
를 포함한다.
본 발명의 또 하나의 실시상태는 고셔병을 갖는 대상을 치료하는 방법이다. 상기 방법은
a) 화학식 (I)의 화합물 또는 이의 약학적 허용염의 유효량을 대상에게 투여하는 단계;
b) 상기 대상을 시험하여 상기 대상이 좋지 못한, 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저(metabolizer)인지 여부를 결정하는 단계;
c) 상기 대상이 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인 경우, 상기 화합물의 조정된 유효량을 결정하는 단계; 및
d) 상기 대상이 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인 경우, 화학식 (I)의 화합물의 조정된 유효량을 대상에게 투여하고, 상기 대상이 좋지 못한 P450 메타볼라이저인 경우 상기 화학식 (I)의 화합물의 유효량을 상기 대상에게 투여하는 단계
를 포함한다.
본 발명의 또 하나의 실시상태는 패브리병을 갖는 대상을 치료하는 방법이다. 상기 방법은
a) 하기 구조식으로 표시되는 화합물 또는 이의 약학적 허용염의 유효량을 대상에게 투여하는 단계;

b) 상기 대상 중의 상기 화합물의 최저 혈장 농도를 평가하는 단계; 및
c) 상기 대상에게 투여하는 화합물의 양을 조정하여 상기 대상 중의 상기 화합물의 최저 혈장 농도가 적어도 5 ng/ml이 되도록 하는 단계를 포함한다.
대안으로서, 상기 대상 중의 화합물의 Cmax 및 최저 혈장 농도가 단계 b)에서 평가되고, 단계 c)에서 상기 대상에게 투여되는 화합물의 양이 조정되어 상기 대상 중의 화합물의 최저 혈장 농도가 적어도 5 ng/ml이 되고 상기 대상 중의 화합물의 Cmax가 100 ng/ml 미만이 되도록 한다.
본 발명의 또 하나의 실시상태는 고셔병을 갖는 대상을 치료하는 방법이다. 상기 방법은
a) 하기 구조식으로 표시되는 화합물 또는 이의 약학적 허용염의 유효량을 대상에게 투여하는 단계;

b) 상기 대상 중의 상기 화합물의 최저 혈장 농도를 평가하는 단계; 및
c) 상기 대상에게 투여하는 화합물의 양을 조정하여 상기 대상 중의 상기 화합물의 최저 혈장 농도가 적어도 5 ng/ml이 되도록 하는 단계를 포함한다.
대안으로서, 상기 대상 중의 화합물의 Cmax 및 최저 혈장 농도가 단계 b)에서 평가되고, 단계 c)에서 상기 대상에게 투여되는 화합물의 양이 조절되어 상기 대상 중의 화합물의 최저 혈장 농도가 적어도 5 ng/ml이 되고 상기 대상 중의 화합물의 Cmax가 100 ng/ml 미만이 되도록 한다.
본 출원은 화학식 (I) 헤미타르타르산염의 독특한 결정질 형태 및 본 명세서에 기재된 화학식 (I) 헤미타르타르산염의 결정질 형태를 포함하는 화학식 (I) 헤미타르타르산염의 신규한 약학 조성물을 제공한다. 본 출원은 또한 그것을 필요로 하는 대상에서 글리코스핀고리피드 농도를 낮추거나 글루코실세라마이드 신타아제를 억제하는 방법을 제공한다. 또한, 본 출원은 화학식 (I) 헤미타르타르산염의 특이적 결정질 형태를 제조하는 방법을 제공한다. 본 출원은 또한 약물/약물 상호작용의 위험을 최소화하는 화학식 (I) 헤미타르타르산염의 안정한 약학 제형, 화학식 (I)의 화합물 또는 이의 약학적 허용염(화학식 (I) 헤미타르타르사염 포함)과의 병용 요법 및 화학식 (I)의 화합물 또는 이의 약학적 허용염(화학식 (I) 헤미타르타르산염 포함)에 의한 치료 방법을 제공한다.
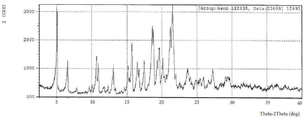
도 1은 화학식 (I) 헤미타르타르산염에 대한 실험적 XRPD 패턴(실온)을 나타낸다.
도 2는 고셔 마우스 간에서 글루코실세라마이드 수준을 낮추는 데 효소 및 기질 감소 치료의 효능 그래프이다. 간 GL1 수준은 미처리 생후 3개월의 고셔 마우스(A)에서 그리고 재조합 글루코세레브로시다아제에 의한 처리(B) 2주 후에 측정되었다. 재조합 글루코세레브로시다아제에 의하여 처리된 마우스는 추가의 처리 없이(C) 또는 화학식 (I) 헤미타르타르산염 150 mg/kg 공급에 의한 치료 후(D) 10주 후에 분석되었다.
전체 연구 기간동안 화학식 (I) 헤미타르타르산염 단독만이 투여된 마우스의 간(E)에서의, 그리고 미처리된 같은 연령의 대조군(F)에서의 GL1 수준도 나타내었다. 데이터는 평균 ± 그 평균 표준 오류(SEM) (n=5)로서 표시된다. 통계적인 의미는 언페어드 t 테스트(unpaired t test)를 이용하여 결정되었다.
도 3은 고셔 마우스의 비장에서의 글루코실세라마이드 수준을 낮추는 데 효소 및 기질 감소 치료의 효능 그래프이다. 비장 GL1 수준는 미처리 생후 3개월의 고셔 마우스(A)에서 그리고 재조합 글루코세레브로시다아제에 의한 처리(B) 2주 후에 측정되었다. 재조합 글루코세레브로시다아제에 의하여 처리된 마우스는 추가의 처리 없이(C) 또는 화학식 (I) 헤미타르타르산염 150 mg/kg 공급에 의한 치료 후(D) 10주 후에 분석되었다. 전체 연구 기간동안 화학식 (I) 헤미타르타르산염 단독만이 투여된 마우스의 비장(E)에서의, 그리고 미처리된 같은 연령의 대조군(F)에서의 GL1 수준도 나타내었다. 데이터는 평균 ± 그 평균 표준 오류(SEM) (n=5)로서 표시된다. 통계적인 의미는 언페어드 t 테스트(unpaired t test)를 이용하여 결정되었다.
도 4는 고셔 마우스의 폐에서의 글루코실세라마이드 수준를 낮추는 데 효소 및 기질 감소 치료의 효능 그래프이다. 폐 GL1 수준은 미처리 생후 3개월의 고셔 마우스(A)에서 그리고 재조합 글루코세레브로시다아제에 의한 처리(B) 2주 후에 측정되었다. 재조합 글루코세레브로시다아제에 의하여 처리된 마우스는 추가의 처리 없이(C) 또는 화학식 (I) 헤미타르타르산염에 의한 치료 후(D) 10주 후에 분석되었다. 전체 연구 기간동안 화학식 (I) 헤미타르타르산염 단독만이 투여된 마우스의 폐(E)에서의, 그리고 미처리된 같은 연령의 대조군(F)에서의 GL1 수준도 나타내었다. 데이터는 평균 ± 그 평균 표준 오류(SEM) (n=5)로서 표시된다. 통계적인 의미는 언페어드 t 테스트(unpaired t test)를 이용하여 결정되었다.
도 5는 간에서의 CD68 스테이닝(staining)의 정도의 정량을 나타내는 그래프이다. 간 섹션에서의 CD68-양성 스테이닝 정도는 MetaMorph 소프트웨어를 이용하여 정량되었다. 미처리 생후 3개월의 고셔 간(A)에서 또는 글루코세레브로시다아제에 의한 처리(B) 후에의 수준이 도시된다. 효소에 의하여 처리되고, 이어서 추가의 치료적 개입 없이(C) 또는 화학식 (I) 헤미타르타르산염에 의한 치료 후(D) 10주 후에 분석된 마우스도 역시 이용된다. 화학식 (I) 헤미타르타르산염 단독만이 투여된 고셔 마우스의 간(E)에서의, 그리고 미처리된 같은 연령의 대조군 마우스(F)에서의 스테이닝 정도도 나타내었다. 데이터는 각 마우스로부터의 섹션마다 십 400x 이미지들의 분석으로부터 수집되었다. 통계적인 의미는 언페어드 t 테스트(unpaired t test)를 이용하여 결정되었다.
도 6은 어린 D409V/null 마우스에서 화학식 (I) 헤미타르타르산염의 효능을 나타내는 그래프이다. 화학식 (I) 헤미타르타르산염이 10주간 75 또는 150 mg/kg의 투여량으로 구강 위관영양법에 의하여 매일 생후 10주 D409/null 마우스에 투여되었다. 간, 폐, 맥관구조 및 비장에서의 글루코실세라마이드 수준이 연구 말기에 HP-TLC 에 의하여 평가되었다. 데이터는 미처리된 동일 연령의 대조군 마우스에서의 GL-1의 백분율로서 제시된다. 대쉬 라인은 정상 야생형 마우스에서 관찰된 글루코실세라마이드 수준를 나타낸다. 미처리된 대조군에 상대적으로 *p<0.05; **p<0.01 (양측(two-tailed), 언페어드 t-테스트). 데이터는 평균 ± 그 평균 표준 오류(SEM) (75 mg/kg에 대하여는 n=5, 150 mg/kg에 대하여는 n=6)로서 표시된다.
도 7은 패브리 마우스 간, 심장, 신장, 비장, 뇌 및 혈액에서의 축적에 대한 화학식 (I) 헤미타르타르산염 치료의 효과를 나타낸다.
도 8은 패브리 마우스에서의 말초신경증의 시작 및 진전에 대한 화학식 (I) 헤미타르타르산염 치료의 효과 그래프를 나타낸다.
도 9는 화학식 (I) 헤미타르타르산염에 의하여 처리된 패브리 마우스에서의 신장 기능의 몇몇 마커의 측정 그래프를 나타낸다.
도 10은 상이한 약물 치료를 받은 마우스 군집의 ERT 및 SRT 연구의 타임라인을 나타낸다: A) 격월 패브라자임(fabrazyme), 화학식 (I) 헤미타르타르산염 없음; B) 격월 패브라자임, 음식 중의 화학식 (I) 헤미타르타르산염; C) 연구 시작시 및 연구 4개월째 패브라자임 투여, 음식 중의 화학식 (I) 헤미타르타르산염; D) 패브라자임 없음, 음식 중의 화학식 (I) 헤미타르타르산염; 및 E) 약물 치료 없음.
도 11은 마우스(A-E 패브리-래그; 및 F 야생형)의 6 군집(n=?)의 혈액의 ng/mL으로의 혈액 GL-3 수준의 그래프를 나타내며; 마우스 군집들은 다음의 치료를 받았다: A) 격월 패브라자임(fabrazyme), 화학식 (I) 헤미타르타르산염 없음; B) 격월 패브라자임, 음식 중의 화학식 (I) 헤미타르타르산염; C) 연구 시작시 및 연구 4개월째 패브라자임 투여, 음식 중의 화학식 (I) 헤미타르타르산염; D) 패브라자임 없음, 음식 중의 화학식 (I) 헤미타르타르산염; 및 E) 약물 치료 없음.
도 12는 패브리-래그 마우스들의 간 및 신장에서의 GL-3 수준의 그래프를 나타내며; 마우스 군집들(n=?)은 다음의 치료를 받았다: A) 격월 패브라자임(fabrazyme), 화학식 (I) 헤미타르타르산염 없음; B) 격월 패브라자임, 음식 중의 화학식 (I) 헤미타르타르산염; C) 연구 시작시 및 연구 4개월째 패브라자임 투여, 음식 중의 화학식 (I) 헤미타르타르산염; D) 패브라자임 없음, 음식 중의 화학식 (I) 헤미타르타르산염; 및 E) 약물 치료 없음.
도 13은 패브리-래그 마우스들에서의 소변 GL-3 수준의 그래프를 나타내며; 마우스 군집들(n=?)은 다음의 치료를 받았다: A) 격월 패브라자임(fabrazyme), 화학식 (I) 헤미타르타르산염 없음; B) 격월 패브라자임, 음식 중의 화학식 (I) 헤미타르타르산염; C) 연구 시작시 및 연구 4개월째 패브라자임 투여, 음식 중의 화학식 (I) 헤미타르타르산염; D) 패브라자임 없음, 음식 중의 화학식 (I) 헤미타르타르산염; 및 E) 약물 치료 없음.
도 14는 다음의 치료를 받고 있는 패브리-래그 마우스들의 열 민감성의 잠복기간(seconds)을 나타내는 그래프이다: 격월 패브라자임(fabrazyme), 화학식 (I) 헤미타르타르산염 없음; 격월 패브라자임, 음식 중의 화학식 (I) 헤미타르타르산염; 연구 시작시 및 연구 4개월째 패브라자임 투여, 음식 중의 화학식 (I) 헤미타르타르산염; 패브라자임 없음, 음식 중의 화학식 (I) 헤미타르타르산염; 약물 치료 없음; 야생형 마우스; 및 3개월에 미처리.
도 15는 3 일동안 85 ℃에 노출된 후, 화학식 (I) 헤미타르타르산염, 락토오스 일수화물 캡슐화 그레이드 및 아비셀(Avicel) PH 301(미정질 셀룰로오스)을 포함하는 다양한 블렌드의 HPLC 트레이스(trace)의 분해 영역(degradation area)의 총 양을 나타내는 그래프이다. HPLC 트레이스의 분해 영역은 화학식 (I) 헤미타르타르산염 및 분해 생성물에 대응하는 피크들의 총 영역에 대한 분해에 대응하는 피크들의 총 영역의 비이다.
도 2는 고셔 마우스 간에서 글루코실세라마이드 수준을 낮추는 데 효소 및 기질 감소 치료의 효능 그래프이다. 간 GL1 수준은 미처리 생후 3개월의 고셔 마우스(A)에서 그리고 재조합 글루코세레브로시다아제에 의한 처리(B) 2주 후에 측정되었다. 재조합 글루코세레브로시다아제에 의하여 처리된 마우스는 추가의 처리 없이(C) 또는 화학식 (I) 헤미타르타르산염 150 mg/kg 공급에 의한 치료 후(D) 10주 후에 분석되었다.
전체 연구 기간동안 화학식 (I) 헤미타르타르산염 단독만이 투여된 마우스의 간(E)에서의, 그리고 미처리된 같은 연령의 대조군(F)에서의 GL1 수준도 나타내었다. 데이터는 평균 ± 그 평균 표준 오류(SEM) (n=5)로서 표시된다. 통계적인 의미는 언페어드 t 테스트(unpaired t test)를 이용하여 결정되었다.
도 3은 고셔 마우스의 비장에서의 글루코실세라마이드 수준을 낮추는 데 효소 및 기질 감소 치료의 효능 그래프이다. 비장 GL1 수준는 미처리 생후 3개월의 고셔 마우스(A)에서 그리고 재조합 글루코세레브로시다아제에 의한 처리(B) 2주 후에 측정되었다. 재조합 글루코세레브로시다아제에 의하여 처리된 마우스는 추가의 처리 없이(C) 또는 화학식 (I) 헤미타르타르산염 150 mg/kg 공급에 의한 치료 후(D) 10주 후에 분석되었다. 전체 연구 기간동안 화학식 (I) 헤미타르타르산염 단독만이 투여된 마우스의 비장(E)에서의, 그리고 미처리된 같은 연령의 대조군(F)에서의 GL1 수준도 나타내었다. 데이터는 평균 ± 그 평균 표준 오류(SEM) (n=5)로서 표시된다. 통계적인 의미는 언페어드 t 테스트(unpaired t test)를 이용하여 결정되었다.
도 4는 고셔 마우스의 폐에서의 글루코실세라마이드 수준를 낮추는 데 효소 및 기질 감소 치료의 효능 그래프이다. 폐 GL1 수준은 미처리 생후 3개월의 고셔 마우스(A)에서 그리고 재조합 글루코세레브로시다아제에 의한 처리(B) 2주 후에 측정되었다. 재조합 글루코세레브로시다아제에 의하여 처리된 마우스는 추가의 처리 없이(C) 또는 화학식 (I) 헤미타르타르산염에 의한 치료 후(D) 10주 후에 분석되었다. 전체 연구 기간동안 화학식 (I) 헤미타르타르산염 단독만이 투여된 마우스의 폐(E)에서의, 그리고 미처리된 같은 연령의 대조군(F)에서의 GL1 수준도 나타내었다. 데이터는 평균 ± 그 평균 표준 오류(SEM) (n=5)로서 표시된다. 통계적인 의미는 언페어드 t 테스트(unpaired t test)를 이용하여 결정되었다.
도 5는 간에서의 CD68 스테이닝(staining)의 정도의 정량을 나타내는 그래프이다. 간 섹션에서의 CD68-양성 스테이닝 정도는 MetaMorph 소프트웨어를 이용하여 정량되었다. 미처리 생후 3개월의 고셔 간(A)에서 또는 글루코세레브로시다아제에 의한 처리(B) 후에의 수준이 도시된다. 효소에 의하여 처리되고, 이어서 추가의 치료적 개입 없이(C) 또는 화학식 (I) 헤미타르타르산염에 의한 치료 후(D) 10주 후에 분석된 마우스도 역시 이용된다. 화학식 (I) 헤미타르타르산염 단독만이 투여된 고셔 마우스의 간(E)에서의, 그리고 미처리된 같은 연령의 대조군 마우스(F)에서의 스테이닝 정도도 나타내었다. 데이터는 각 마우스로부터의 섹션마다 십 400x 이미지들의 분석으로부터 수집되었다. 통계적인 의미는 언페어드 t 테스트(unpaired t test)를 이용하여 결정되었다.
도 6은 어린 D409V/null 마우스에서 화학식 (I) 헤미타르타르산염의 효능을 나타내는 그래프이다. 화학식 (I) 헤미타르타르산염이 10주간 75 또는 150 mg/kg의 투여량으로 구강 위관영양법에 의하여 매일 생후 10주 D409/null 마우스에 투여되었다. 간, 폐, 맥관구조 및 비장에서의 글루코실세라마이드 수준이 연구 말기에 HP-TLC 에 의하여 평가되었다. 데이터는 미처리된 동일 연령의 대조군 마우스에서의 GL-1의 백분율로서 제시된다. 대쉬 라인은 정상 야생형 마우스에서 관찰된 글루코실세라마이드 수준를 나타낸다. 미처리된 대조군에 상대적으로 *p<0.05; **p<0.01 (양측(two-tailed), 언페어드 t-테스트). 데이터는 평균 ± 그 평균 표준 오류(SEM) (75 mg/kg에 대하여는 n=5, 150 mg/kg에 대하여는 n=6)로서 표시된다.
도 7은 패브리 마우스 간, 심장, 신장, 비장, 뇌 및 혈액에서의 축적에 대한 화학식 (I) 헤미타르타르산염 치료의 효과를 나타낸다.
도 8은 패브리 마우스에서의 말초신경증의 시작 및 진전에 대한 화학식 (I) 헤미타르타르산염 치료의 효과 그래프를 나타낸다.
도 9는 화학식 (I) 헤미타르타르산염에 의하여 처리된 패브리 마우스에서의 신장 기능의 몇몇 마커의 측정 그래프를 나타낸다.
도 10은 상이한 약물 치료를 받은 마우스 군집의 ERT 및 SRT 연구의 타임라인을 나타낸다: A) 격월 패브라자임(fabrazyme), 화학식 (I) 헤미타르타르산염 없음; B) 격월 패브라자임, 음식 중의 화학식 (I) 헤미타르타르산염; C) 연구 시작시 및 연구 4개월째 패브라자임 투여, 음식 중의 화학식 (I) 헤미타르타르산염; D) 패브라자임 없음, 음식 중의 화학식 (I) 헤미타르타르산염; 및 E) 약물 치료 없음.
도 11은 마우스(A-E 패브리-래그; 및 F 야생형)의 6 군집(n=?)의 혈액의 ng/mL으로의 혈액 GL-3 수준의 그래프를 나타내며; 마우스 군집들은 다음의 치료를 받았다: A) 격월 패브라자임(fabrazyme), 화학식 (I) 헤미타르타르산염 없음; B) 격월 패브라자임, 음식 중의 화학식 (I) 헤미타르타르산염; C) 연구 시작시 및 연구 4개월째 패브라자임 투여, 음식 중의 화학식 (I) 헤미타르타르산염; D) 패브라자임 없음, 음식 중의 화학식 (I) 헤미타르타르산염; 및 E) 약물 치료 없음.
도 12는 패브리-래그 마우스들의 간 및 신장에서의 GL-3 수준의 그래프를 나타내며; 마우스 군집들(n=?)은 다음의 치료를 받았다: A) 격월 패브라자임(fabrazyme), 화학식 (I) 헤미타르타르산염 없음; B) 격월 패브라자임, 음식 중의 화학식 (I) 헤미타르타르산염; C) 연구 시작시 및 연구 4개월째 패브라자임 투여, 음식 중의 화학식 (I) 헤미타르타르산염; D) 패브라자임 없음, 음식 중의 화학식 (I) 헤미타르타르산염; 및 E) 약물 치료 없음.
도 13은 패브리-래그 마우스들에서의 소변 GL-3 수준의 그래프를 나타내며; 마우스 군집들(n=?)은 다음의 치료를 받았다: A) 격월 패브라자임(fabrazyme), 화학식 (I) 헤미타르타르산염 없음; B) 격월 패브라자임, 음식 중의 화학식 (I) 헤미타르타르산염; C) 연구 시작시 및 연구 4개월째 패브라자임 투여, 음식 중의 화학식 (I) 헤미타르타르산염; D) 패브라자임 없음, 음식 중의 화학식 (I) 헤미타르타르산염; 및 E) 약물 치료 없음.
도 14는 다음의 치료를 받고 있는 패브리-래그 마우스들의 열 민감성의 잠복기간(seconds)을 나타내는 그래프이다: 격월 패브라자임(fabrazyme), 화학식 (I) 헤미타르타르산염 없음; 격월 패브라자임, 음식 중의 화학식 (I) 헤미타르타르산염; 연구 시작시 및 연구 4개월째 패브라자임 투여, 음식 중의 화학식 (I) 헤미타르타르산염; 패브라자임 없음, 음식 중의 화학식 (I) 헤미타르타르산염; 약물 치료 없음; 야생형 마우스; 및 3개월에 미처리.
도 15는 3 일동안 85 ℃에 노출된 후, 화학식 (I) 헤미타르타르산염, 락토오스 일수화물 캡슐화 그레이드 및 아비셀(Avicel) PH 301(미정질 셀룰로오스)을 포함하는 다양한 블렌드의 HPLC 트레이스(trace)의 분해 영역(degradation area)의 총 양을 나타내는 그래프이다. HPLC 트레이스의 분해 영역은 화학식 (I) 헤미타르타르산염 및 분해 생성물에 대응하는 피크들의 총 영역에 대한 분해에 대응하는 피크들의 총 영역의 비이다.
본 출원은 화학식 (I) 헤미타르타르산염의 독특한 결정질 형태 및 본 명세서에 기재된 화학식 (I) 헤미타르타르산염의 결정질 형태를 포함하는 화학식 (I) 헤미타르타르산염의 신규한 약학 조성물을 제공한다. 본 출원은 또한 그것을 필요로 하는 대상에서 글리코스핀고리피드 농도를 낮추거나 글루코실세라마이드 신타아제를 억제하는 방법을 제공한다. 또한, 본 출원은 화학식 (I) 헤미타르타르산염의 특이적 결정질 형태를 제조하는 방법을 제공한다. 본 출원은 또한 약물/약물 상호작용의 위험을 최소화하는 화학식 (I) 헤미타르타르산염의 안정한 약학 제형, 화학식 (I)의 화합물 또는 이의 약학적 허용염(화학식 (I) 헤미타르타르사염 포함)과의 병용 요법 및 화학식 (I)의 화합물 또는 이의 약학적 허용염(화학식 (I) 헤미타르타르산염 포함)에 의한 치료 방법을 제공한다.
화학식 (I) 헤미타르타르산염의 결정질 형태
특정 실시상태에 있어서, 화학식 (I) 헤미타르타르산염의 적어도 특정 중량 퍼센트가 결정질이다. 특정 중량 퍼센트는 70%, 72%, 75%, 77%, 80%, 82%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, 99.5%, 99.9%, 또는 70%와 100% 사이의 퍼센트를 포함한다.
또 하나의 특정 실시상태에 있어서, 화학식 (I) 헤미타르타르산염의 적어도 특정 중량 퍼센트가 화학식 (I) 헤미타르타르산염의 단일 결정질 형태이다. 특정 중량 퍼센트는 70%, 72%, 75%, 77%, 80%, 82%, 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, 99.5%, 99.9%, 또는 70%와 100% 사이의 퍼센트를 포함한다.
여기에 사용된 바와 같이, "결정질"은 개별 분자가 매우 동질의 규칙적인 고정된 화학적 배열을 갖는 결정 구조를 갖는 고체를 말한다. 결정질 화학식 (I) 헤미타르타르산염은 화학식 (I) 헤미타르타르산염의 단일 결정질 형태의 결정들 또는 상이한 단일 결정질 형태들의 혼합물일 수 있다. 단일 결정질 형태는 단일 결정 또는 각 결정이 동일한 결정 형태를 갖는 복수의 결정들로서 화학식 (I) 헤미타르타르산염를 의미한다.
화학식 (I) 헤미타르타르산염의 특정 중량 퍼센트가 단일 결정질 형태인 경우, 화학식 (I) 헤미타르타르산염의 나머지는 단일 결정질 형태를 제외한 화학식 (I) 헤미타르타르산염의 하나 이상의 다른 결정질 형태들, 및/또는 무정형 화학식 (I) 헤미타르타르산염의 몇몇 조합이다. 결정질 화학식 (I) 헤미타르타르산염이 화학식 (I) 헤미타르타르산염의 하나의 특정 결정질 형태의 특정된 퍼센트로서 한정되는 경우, 그 나머지는 특정된 하나 이상의 특정 형태들 이외의 결정질 형태들 및/또는 무정형 형태로 이루어진다. 단일 결정질 형태의 예들은 여기에 논의된 바와 같이 하나 이상의 특성에 의하여 특징지워지는 화학식 (I) 헤미타르타르산염의 형태 A를 포함한다.
타르타르산은 2개의 카복실산 기를 갖기 때문에, 타르타르산 염(타르타르산의 콘쥬게이트 염기)에 대한 화학식 (I)에 의하여 표시되는 화합물의 몰비가 상이한 염들을 형성할 수 있다. 예컨대, 화학식 (I)에 대한 타르타르산염의 약 일 대 일의 몰비가 있는 염은 화학식 (I) 타르타르산염 (1 타르타르산염 : 1 화학식 (I))이고; 화학식 (I)에 대한 타르타르산염의 약 일 대 이의 몰비가 있는 염은 화학식 (I) 헤미타르타르산염 (1 타르타르산염 : 2 화학식 (I))이다.
헤미타르타르산염은 다양한 스테레오이성질체 형태로 존재할 수 있다. 스테레오이성질체들은 단지 이들의 공간적 배치가 상이한 화합물들이다. 거울상이성질체는 거울 이미지들이 포개질 수 없는 스테레오 이성질체들의 쌍이며, 대부분은 통상 이들이 카이럴 중심으로서 작용하는 비대칭적으로 치환된 탄소 원자를 함유하기 때문이다. 부분입체이성질체는 거울 이미지들로서 관계되지 않는 스테레오이성질체이고, 대부분 통상 이들이 2 이상의 비대칭적으로 치환된 탄소 원자를 함유하기 때문이다.
입체 화학이 명명되거나(여기에서와 같이, 예컨대, L-(+)-타르타르산) 구조에 의하여 묘사되는 경우(여기에서와 같이, 예컨대 화학식 (I)), 명명되거나 묘사된 스테레오이성질체는 다른 스테레오이성질체에 비하여 순수 중량이 적어도 60%, 70%, 80%, 90%, 99% 또는 99.9%이다. 단일 거울상이성질체가 명명되거나(여기에서와 같이, 예컨대, L-(+)-타르타르산) 구조에 의하여 묘사되는 경우(여기에서와 같이, 예컨대 화학식 (I)), 묘사되거나 명명된 거울상이성질체는 광학적으로 순수한 중량이 적어도 80%, 90%, 99% 또는 99.9% 이다. 광학적 순도의 중량 퍼센트는 거울상이성질체의 중량 더하기 광학적 이성질체의 중량에 대하여 거울상 이성질체의 중량의 비이다.
"라세미체" 또는 "라세미 혼합물"은 2개의 거울상이성질체들의 같은 몰량의 화합물을 의미하고, 여기서 상기 혼합물은 광학적 활성을 나타내지 않는, 즉 이들은 편광된 광의 평면을 회전하지 않는다.
타르타르산은 3개의 스테레오이성질체를 갖는다: L-(+)-타르타르산 또는 덱스트로타르타르산 및 이의 거울상이성질체, 레보타르타르산 또는 D-(-)-타르타르산, 및 아카이럴(achiral) 형태, 메조타르타르산. L 또는 D 명칭은 편광된 광의 평면을 회전하는 산의 능력을 나타내지 않는다.
타르타르산의 임의의 스테레오이성질체들은 화학식 (I) 헤미타르타르산염을 제조하기 위하여 사용될 수 있다. 예컨대, 헤미타르타르산염은 그것의 스테레오이성질체들 중의 단지 하나 또는 이들의 조합으로부터 형성될 수 있다. 헤미타르타르산염은 D-헤미타르타르산염, L-헤미타르타르산염, 헤미메조타르타르산 또는 라세미 D,L-헤미타르타르산염으로부터 선택된다. 특정한 실시상태에 있어서, 헤미타르타르산염은 L-헤미타르타르산염이다. "L-헤미타르타르산염"은 헤미타르타르산염이 L-타르타르산으로부터 형성된 것을 의미한다. 라세미 D,L-헤미타르타르산염은 D-타르타르산염 및 L-타르타르산염이 모두 화학식 (I) 헤미타르타르산염의 제조에 사용되었다는 것을 의미한다. 라세미 D,L-헤미타르타르산염 중의 D-타르타르산염의 양은 존재하는 L-타르타르산염의 양 보다 많거나, 동등하거나, 적을 수 있다.
"좌측회전성(levorotatory)"은 편광된 광이 비대칭 화합물을 통과하는 경우 왼쪽으로 회전하는 것을 뜻한다. 좌측회전성을 표시하는 접두사는 "L"이다.
"우측회전성(dextrorotatory)"은 편광된 광이 비대칭 화합물을 통과하는 경우 오른쪽으로 회전하는 것을 뜻한다. 우측회전성을 표시하는 접두사는 "D"이다.
화학식 (I) 헤미타르타르산염의 제조
화학식 (I) 헤미타르타르산염은 적절한 용매 중에서 화학식 (I)을 L-타르타르산과 혼합함으로써 제조될 수 있다. 화학식 (I) 헤미타르타르산염의 침전은 시드(seed) 결정의 첨가에 의하여 도움이 될 수 있다. 사용될 수 있는 용매는 메탄올, 물, 에탄올, 아세톤, 에틸아세테이트 또는 이들의 조합이다.
화학식 (I) 헤미타르타르산염의 특정 고체 형태는 예컨대 느린 증발, 느린 냉각 및 역용매(antisolvent) 침전에 의하여 제조될 수 있다. 이들 방법에서 사용될 수 있는 용매는 물, 헵탄, 헥산, 톨루엔, 디클로로메탄, 에탄올, 이소프로필 알코올, 아세토니트릴, 에틸 아세테이트, 메탄올, 아세톤, 메틸 터셔리-부틸 에테르(이하 "TBME"라 함), p-디옥산 및 테트라히드로푸란(이하 "THF"라 함)을 포함한다.
화학식 (I) 헤미타르타르산염 고체 형태는 용매 또는 용매 혼합물 중의 화학식 (I) 헤미타르타르산염의 용액으로부터의 용매 증발에 의하여 제조될 수 있다. 적당한 용매 혼합물은 메탄올, 에탄올, 아세톤, 물, 에틸아세테이트 및 디클로로메탄을 포함한다. 바람직한 용매 혼합물은 에탄올, 메탄올, 물 및 아세톤을 포함한다.
화학식 (I) 헤미타르타르산염 고체 형태는 용매 중의 화학식 (I) 헤미타르타르산염의 가열된 용액의 느린 냉각을 통하여 제조될 수 있다. 적당한 용매는 에탄올, 메탄올, 물, 아세톤 및 에틸아세테이트를 포함한다.
화학식 (I) 헤미타르타르산염 고체 형태는 용액을 냉각 배스에 놓음으로써 용매 중의 화학식 (I) 헤미타르타르산염의 가열된 용액의 빠른 냉각을 통하여 제조될 수 있다. 적당한 용매는 에탄올, 메탄올, 아세톤, 물, 에틸아세테이트 또는 이들 용매의 혼합물을 포함할 수 있다.
화학식 (I) 헤미타르타르산염 고체 형태는 전술한 바와 같이 용매 중의 화학식 (I) 헤미타르타르산염의 용액을 주어진 온도에서 역용매에 첨가함으로써 제조될 수 있다. 좀더 구체적으로, 상기 역용매는 에틸아세테이트, 아세톤, 아세토니트릴, 톨루엔, THF, TBME, p-디옥산, 이소프로판올 또는 헵탄이다. 특정 용매/역용매 혼합물은 메탄올/에틸에세테이트, 메탄올/아세톤, 메탄올/헥산, 메탄올/헵탄, 메탄올/아세토니트릴, 메탄올/톨루엔, 메탄올/THF, 메탄올/TBME, 메탄올/p-디옥산, 에탄올/에틸아세테이트, 에탄올/헥산, 에탄올/헵탄, 에탄올, 아세톤, 에탄올/아세토니 트릴, 에탄올/톨루엔, 에탄올/TBME, 에탄올/THF, 물/THF, 물/이소프로판올, 물/아세토니트릴, 물/아세톤, 디클로로메탄/헵탄, 디클로로메탄/아세톤, 디클로로메탄/에틸아세테이트, 디클로로메탄/아세토니트릴, 디클로로메탄/톨루엔, 디클로로메탄/THF, 디클로로메탄/TBME, 디클로로메탄/p-디옥산, 및 디클로로메탄/이소프로판올을 포함한다.
바람직한 용매/역용매 혼합물은 메탄올/에틸아세테이트, 메탄올/아세톤, 메탄올/TBME, 및 물/아세톤을 포함한다.
여기에 사용된 바와 같이, "역용매"는 화학식 (I) 헤미타르타르산염이 낮은 용해도를 가지고 헤미타르타르산염이 미세 분말 또는 결정의 형태로 용액 밖으로 침전하도록 하는 용매를 말한다.
화학식 (I) 헤미타르타르산염의 고체 형태를 만들어내는 추가의 방법은 에틸아세테이트/아세톤으로부터 고체를 침전시키는 것 및 임의로 실온에서 형성된 고체를 건조시키는 것을 포함한다. 또 하나의 방법에 있어서, 상기 용매는 이어서 시드 결정을 첨가하거나 첨가하지 않고 아세톤으로부터 재결정화될 수 있다. 대안으로서, 화학식 (I) 헤미타르타르산염은 에틸아세테이트/아세톤 용매로부터 침전되고 에틸아세테이트로부터 재결정화될 수 있다. 대안으로서, 화학식 (I) 헤미타르타르산염은 이어서 이소프로판올으로부터 재결정화될 수 있다. 대안으로서, 화학식 (I) 헤미타르타르산염은 추가의 재결정화 없이 단지 아세톤을 사용함으로써 제조될 수 있다. 대안으로서, 화학식 (I) 헤미타르타르산염은 추가의 재결정화없이, 간단한 환류(reflux) 후에 아세톤으로부터 침전될 수 있다.
대안으로서, 화학식 (I) 헤미타르타르산염은 이어서 시드 결정을 첨가하거나 첨가하지 않고 메탄올/아세톤으로부터 재결정화될 수 있다. 대안으로서, 화학식 (I) 헤미타르타르산염는 이어서 시드 결정을 첨가하거나 첨가하지 않고 물/아세톤으로부터 재결정화될 수 있다.
화학식 (I) 헤미타르타르산염의 결정질 형태의 특징 설명
특정 실시상태에 있어서, 화학식 (I) 헤미타르타르산염의 결정질 형태, 결정 형태 A는 5.1°, 6.6°, 10.7°, 11.0°, 15.9° 및 21.7°의 2θ각에서의 적어도 하나, 두 개, 세 개, 네 개 또는 다섯 개의 주요 XRPD 피크에 의하여 특징지워진다. 더 특정한 실시상태에 있어서, 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 13.3°, 15.1°, 15.9°, 16.5°, 17.6°, 18.6°, 18.7°, 19.0°, 20.2°, 21.7° 및 23.5°의 2θ 각에서의 XRPD 피크에 의하여 특징지워진다. 특정한 2θ각은 특정한 값 ± 0.2°를 의미하는 것이 이해되어야 한다.
여기에 사용된 바와 같이 "주요한 XRPD 피크"는 상대 강도가 25%보다 큰 XRPD 피크를 말한다. 상대 강도는 가장 큰 피크의 피크 강도에 대하여 관심 피크의 피크 강도의 비로서 계산된다.
화학식 (I) 헤미타르타르산염을 이용한 치료 방법
여기에 사용된 바와 같이, 대상은 포유동물, 바람직하게 인간 환자이지만, 또한 반려동물(예컨대, 개, 고양이 등), 가축(예컨대, 소, 양, 돼지, 말 등) 또는 실험 동물(예컨대, 래트, 마우스, 기니 피그 등)과 같이 수의과적 치료가 필요한 동물일 수도 있다. 대상 및 환자는 교환가능하게 사용된다.
본 출원의 하나의 실시상태는 전술한 바와 같이 그것의 결정질 형태를 포함하는 화학식 (I) 헤미타르타르산염의 유효량을 대상에게 투여함으로써 그것을 필요로 하는 대상에서 글리코스핀고리피드 농도를 낮추거나 글루코실세라마이드 신타아제의 활성을 느리게, 예컨대 억제 또는 감소시키는 방법이다.
치료를 필요로 하는 대상은 세포, 특히 리소좀 또는 멤브레인 세포에서 글리코스핀고리피드 농도를 낮추거나 글루코실세라마이드 신타아제를 억제하는 것이 이로운 질병 또는 상태를 갖는 대상이다. 글루코실세라마이드 신타아제의 억제제가 테이-삭스, 고셔 또는 패브리병과 같은 리소좀 저장 병을 치료하는데 유용한 것으로 나타났다(예컨대 미국 특허 제6,569,889호; 제6,255,336호; 제5,916,911호; 제5,302,609호; 제6,660,749호; 제6,610,703호; 제5,472,969호; 제5,525,616호를 참고하라. 상기 미국 특허 문헌들의 전체 내용은 참고로서 본 명세서에 포함된다).
상태 또는 질병의 예는 다낭성 신장 병 및 막성 사구체병증(membranous glomerulopathy)(미국 특허 가출원 제61/130,401호 및 제61/102,541호를 참고하라, 이들 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.), 사구체 신염 및 사구체 경화증(미국 특허 가출원 제 61/137,214호를 참고하라), 낭창(PCT/US2009/001773를 참고하라, 이들 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.), 타입 2 당뇨병을 비롯한 당뇨병(WO 2006/053043를 참고하라, 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.); 당뇨병 환자의 신장 비대, 아테롬성 동맥 경화증, 교원질 혈관성 질환(collagen vascular disease), 암을 비롯한 세포 성장 및 분할(division)을 포함하는 장애 치료(미국 특허 제6,916,802호 및 제5,849,326호를 참고하라, 이들 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.); 동맥 상피 세포의 성장 억제(미국 특허 제6,916,802호 및 제5,849,326호를 참고하라.); 감염에 걸린 환자의 치료(문헌 [Svensson, M. et al., "Epithelial Glucosphingolipid Expression as a Determinant of Bacterial Adherence and Cytokine Production," Infect, and Immun., 62:4404-4410 (1994)]를 참고하라, 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.); 호스트, 즉 환자가 종양에 대하여 항체를 만들어내는 것의 방지(문헌 [Inokuchi, J. et al., "Antitumor Activity in Mice of an Inhibitor of Glycosphingolipid Biosynthesis," Cancer Lett., 38:23-30(1987)]를 참고하라, 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.); 및 종양의 치료(문헌 [Hakomori, S. "New Directions in Cancer Therapy Based on Aberrant Expression of Glycosphingolipids: Anti-adhesion and Ortho-Signaling Therapy," Cancer Cells 3:461-470 (1991)], 문헌 [Inokuchi, J. et al, "Inhibition of Experimental Metastasis of Murine Lewis Long Carcinoma by an Inhibitor of Glucosylceramide Synthase and its Possible Mechanism of Action," Cancer Res., 50:6731-6737 (1990)] 및 문헌 [Ziche, M. et al., "Angiogenesis Can Be Stimulated or Repressed in In Vivo by a Change in GM3 :GD3 Ganglioside Ratio," Lab. Invest., 67:711 -715 (1992)]을 참고하라, 이들 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.)를 포함한다.
화학식 (I) 헤미타르타르산염은 또한 암 백신-유사물 제조에 사용될 수 있다(예컨대, 미국 특허 제6,569,889호; 제6,255,336호; 제5,916,911호; 제5,302,609호; 제6,660,749호; 제6,610,703호; 제5,472,969호; 제5,525,616호를 참고하라).
화학식 (I)의 화합물 또는 이의 약학적 허용염(이의 헤미타르타르산염 포함)은 단일 치료, 즉 징후를 치료하기 위하여 투여되는 유일한 약학적 활성 성분으로서 기재된 방법에서 사용될 수 있다.
대안으로서, 화학식 (I)의 화합물 또는 이의 약학적 허용염(이의 헤미타르타르산염을 포함)은 원하는 병 또는 징후를 치료하기 위하여 당 기술분야에서 알려진 다른 치료적 활성 약물과 함께 병용 치료로서 기재된 방법에서 사용될 수 있다. "공-치료(Co-therapy)" 또는 "병용(combination)" 또는 "병용 치료(combination therapy)" 또는 "공-투여(co-administered)"는 여기에서 교환가능하게 사용되며, 화학식 (I)의 화합물 또는 이의 약학적 허용염(헤미타르타르산염을 포함)이 하나 이상의 다른 치료 제제의 전, 후 또는 함께 투여되는 것을 의미한다. 하나의 실시상태에 있어서, 병용 치료는 고셔병 또는 패브리병과 같은 리소좀 병을 치료하기 위하여 사용된다. 대안으로서, 화학식 (I)의 화합물 또는 이의 약학적 허용 염(헤미타르타르산염을 포함)은 별도의 제형으로서 또는 조인트 제형으로서 일제히(예컨대, 동시에) 공-투여된다. 대안으로서,
상기 제제들은 숙련된 임상의에 의하여 결정된, 적절한 시간 프레임 내에서(예컨대, 치료의 약학적 효과의 중첩을 허용하기에 충분한 시간) 별도의 조성으로서 순차적으로 투여될 수 있다. 화학식 (I)의 화합물 또는 이의 약학적 허용염(헤미타르타르산염을 포함)과 하나 이상의 다른 치료 제제는 원하는 치료 효과를 달성하기 위하어 적절한 스케줄에 따라 순서대로 단일 투여량 또는 복합 투여량으로 투여될 수 있다.
고셔병의 치료를 위하여 유효한 치료 제제는 글루코세레브로시다아제, 글루코세레브로시다아제의 유사체, 글루코실세레마이드 신타아제 억제제 및 글루코세레브로시다아제와 결합하고 그것의 정확한 배열을 회복하는 샤프론(chaperone)을 포함한다. 글루코세레브로시다아제 또는 이의 유사체는 인간 또는 포유동물 유래일 수 있다.
대안으로서, 글루코세레브로시다아제 및 이의 유사체는 재조합적으로 얻어질 수 있다. 글루코세레브로시다아제의 유사체는 생물학적 활성은 보유되고, 천연 효소의 천연 아미노 서열에 비하여 아미노산 치환체를 갖는 효소 및/또는 효소의 잘려진 형태를 포함한다.
글루코세레브로시다아제의 유사체의 예는 인간 β -글루코세레브로시다아제의 재조합 DNA-생성된 유사체인 벨라글루세라제 알파(Shire PLC에 의하여 개발), 탈리글루세라제 알파(상품명 Uplyso®으로 시판, Protalix Biotherapeutics, Inc.에 의하여 개발), 및 이미글루세라제 (Genzyme Corporation 에 의하여 상품명 Cerezyme®으로 판매)을 포함한다.
분자 샤프론의 예는 이소파고민(Amicus Therapeutics(Cranbury, NJ)에 의하여 상품명 PliceraTM으로 개발)을 포함한다. 이소파고민은 또한 아페고스타트 타르타르산염으로도 알려져 있고, 그것의 활성 성분으로서 이소파고민의 타르타르산염 형태를 함유한다. 글루코세레브로시다아제 억제제의 예는 미글루스타트(Actelion Pharmaceuticals Ltd.(Allschwil, Switzerland)에 의하여 상품명 ZavescaTM으로 개발)를 포함한다.
패브리병의 치료를 위하여 효과적인 치료 제제는 α 갈락토시다아제 A, α 갈락토시다아제 A의 유사체 및 α 갈락토시다아제 A에 결합하고 그것의 배열을 회복하는 분자 샤프론을 포함한다. α 갈락토시다아제 A 또는 이의 유사체는 인간 또는 포유동물 유래일 수 있다. 대안으로서, α 갈락토시다아제 A 및 이의 유도체는 재조합적으로 얻어질 수 있다. α 갈락토시다아제 A의 유사체는 생물학적 활성은 보유되고, 천연 효소의 천연 아미노 서열에 비하여 아미노산 치환체를 갖는 효소 및/또는 효소의 잘려진 형태를 포함한다. α 갈락토시다아제 A의 유사체의 예는 아갈시다아제 베타(Genzyme Corporation에 의하여 동결건조 의약품으로서 상품명 Fabrazyme®로 판매되는 재조합 인간 α-갈락토시다아제) 및 아갈시다아제 알파(Shire PLC에 의하여 상품명 Replagal®으로 판매되는 재조합 단백질)을 포함한다. 분자 샤프론의 예들은 미갈라스타트(활성 성분으로서 미갈라스타트 염화수소를 함유하는 약물로서 Amicus Therapeutics(Cranbury, NJ)에 의하여 상품명 AmigalTM으로 개발)을 포함한다.
하나의 실시상태에 있어서, 고셔 또는 패브리병의 치료를 위한 병용 치료는 2 스테이지로 수행된다. 첫번째 스테이지에 있어서, 고셔병 또는 패브리병의 치료에 효과적인 약물(전형적으로, 고셔병을 위해서는 글루코세레브로시다아제 또는 그 유사체 및 패브리병을 위해서는 갈락토시다제 A 또는 그 유사체)가 대상을 안정화하기 위하여 사용된다. 예컨대, 고셔병(또는 패브리병)에 있어서, 이들 약물 중 하나는 간, 비장, 폐 및/또는 신장과 같은 내장 기관들에서 GL-1 저장의 부담을 감소시키기 위하여 사용된다.
이것이 완료되면, 편리한 유지 치료로서 두번째 스테이지에 화학식 (I)의 화합물 또는 이의 약학적 허용염(헤미타르타르산염을 포함)이 사용된다. 상기 첫번째 스테이지는 전형적으로 1, 2, 3 또는 4주까지, 또는 1, 2, 3, 4, 6, 9 또는 12개월까지, 또는 대상의 혈소판 수가 100,000 mm3과 동등하거나 더 많아질 때까지; 헤모글로빈 농도가 11 g/dl (암컷) 또는 12 g/dl (수컷)과 동등하거나 더 많아질 때까지; 및/또는 대상의 비장 용적이 정상의 10 배 보다 작거나 동등하고 간 용적이 정상의 1.5배 보다 작거나 동등할 때까지 지속된다. 상기 첫번째 스테이지의 투여는 전형적으로 화학식 (I)의 화합물에 의한 치료가 개시될 때 종료한다.
여기에 사용된 바와 같이, "유효량"은
대상에서 허용할 수 없는 부작용을 최소화하면서 치료되는 대상의 존재하는 증상을 완화하기에 효과적인 양을 말한다. 정확한 제형, 투여 경로, 투여량은 환자의 상태를 고려하여 개별 의사에 의하여 선택된다. 투여량 및 간격은 원하는 치료 효과를 유지하기에 충분한 활성 화합물의 혈장 수준를 제공하기 위하여 개별적으로 조정될 수 있다. 환자의 상태 및 투여 모드 이외에, 투여되는 투여량은 환자의 증상의 심각성 및 환자의 나이와 체중에 의존할 것이다. 유효량은 전형적으로 상기 화합물의 혈장 최저 수준이 적어도 5 ng/ml이상이 되도록 할 것이다. 상기 화합물의 유효량의 투여 후에 혈장 최저 수준이 5 ng/ml 미만인 경우, 그 대상에 대하여 투여되는 투여량은 "조정된 유효량"으로 변화되어, 상기 화합물의 최저 수준이 적어도 5 ng/ml가 되도록 한다. 대안으로서, 상기 화합물의 유효량의 투여 후에 상기 화합물의 최저 혈장 수준이 5 ng/ml 미만이고/이거나 Cmax가 100 ng/ml 초과인 경우, 대상에게 투여되는 투여량은 "조정된 유효량"으로 변화되어, 상기 화합물의 최저 수준이 적어도 5 ng/ml이고 Cmax가 100 ng 미만이 되도록 한다. 유효량은 0.1 내지 500 mg/일의 범위일 수 있다. 대안으로서, 유효량은 50 - 300 mg/일의 범위이다. 또하나의 대안으로서 유효량은 100-300mg/day의 범위이다. 본 출원의 화합물은 특정 시간의 간격으로 또는 연속적으로 투여될 수 있다. 예컨대, 본 출원의 화합물은 예컨대 매일 또는 하루에 2회 제형과 같이, 하루에 1, 2, 3 또는 4회 투여될 수 있다. 상업적으로 시판가능한 분석법(assay)이 최적의 투여량 범위 및/또는 투여 스케즐을 결정하기 위하여 이용될 수 있다.
하나의 실시상태에 있어서, 화학식 (I)의 화합물 또는 이의 약학적 허용염(전술한 헤미타르타르산염을 포함)에 대한 유효량은 (단일 치료로서이든 또는 공-치료로서이든) 하루에 25, 50, 100, 200 또는 300 밀리그램과 같이 25 밀리그램 내지 300 밀리그램의 일일 투여량이다(대안적으로 25 밀리그램 내지 150 밀리그램; 또 하나의 대안으로서 50 밀리그램 내지 300 밀리그램; 및 또 하나의 대안으로서 100 밀리그램 내지 300 밀리그램). 특정한 실시상태에 있어서, 화학식 (I)의 화합물 또는 이의 약학적 허용염(화학식 (I) 헤미타르타르산염을 포함)의 유효량은 (단일 치료로서이든 또는 공-치료로서이든) 하루 2회 투여량 50 밀리그램 (하루 전체 100 밀리그램), 100 밀리그램(하루 전체 200 밀리그램) 또는 150 밀리그램(하루 전체 300 밀리그램)이다. 대안적인 실시상태에 있어서, 화학식 (I)의 화합물 또는 이의 약학적 허용염(화학식 (I) 헤미타르타르산염을 포함)에 대한 유효량은 (단일 치료로서이든 또는 공-치료로서이든) 하루 1회 투여량 100 밀리그램/일, 200 밀리그램/일 또는 300 밀리그램/일이다.
또 하나의 실시상태에 있어서, 유효량은 대상이 "좋지 못한 P450 메타볼라이저"라고 가정하고 이어서 최저 혈장 수준 및/또는 Cmax를 평가함으로써 결정된다. 이어서 후술하는 바와 같이, 최저 혈장수준이 5 ng/ml 미만이거나; 상기 화합물의 최저 수준이 5 ng/ml 미만이고/이거나 Cmax이 100 ng/ml를 초과하는 경우; 또는 대상이 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저라고 결정되는 경우, 대상에게 투여되는 양은 조정된 유효량으로 변화된다. 좋지 못한 P450 메타볼라이저에 대한 유효량은 (단일 치료로서이든 또는 공-치료로서이든) 하루 1회 투여량 또는 하루 2회 투여량으로서 통상 하루 100 - 200 밀리그램 사이, 예컨대 100 또는 200 밀리그램이다.
전형적으로, 본 출원의 약학 조성물은 식사 전 또는 후에 또는 식사와 함께 투여될 수 있다. 여기서 사용된 바와 같이, 식사 "전" 또는 "후"는 전형적으로 식사를 시작하거나 마칠 때 각각 2 시간 이내, 바람직하게는 1 시간 이내, 더 바람직하게는 30분 이내, 가장 바람직하게는 10분 이내이다.
화학식 (I)의 화합물 및 이의 약학적 허용염(화학식 (I) 헤미타르타르산염)이 간에 의하여, 주로 사이토크롬 P450 효소에 의하여 물질대사된다는 것이 밝혀졌다. 사이토크롬 P450("CYP")은 주요한 간의 생체 이물(xenobiotic) 물질대사 효소이다. 전형적인 인간 간에서 발현되는 11개의 생체 이물-물질대사 사이토크롬 P450(즉, CYP1A2, CYP2A6, CYP2B6, CYP2C8/9/18/19, CYP2D6, CYP2E1 및 CYP3A4/5)가 있다. 현재 또한 CYP2D6 및 CYP3A4가 화학식 (I)의 화합물 및 이의 약학적 허용염, 예컨대 화학식 (I) 헤미타르타르산염의 독성을 없애는 역할을 하는 주된 사이토크롬 P450 이소폼(isoform)이라는 것이 밝혀졌다. P450 효소의 활성수준은 개인에 따라 상이하다. 예컨대, 개인들은 좋지 못한, 중간의 및 광범위한/극도의 빠른 P450 메타볼라이저로 분류될 수 있다. 개인에서의 P450 활성의 더 낮은 수준은 약물/약물 상호작용("DDI")을 생기게 할 수 있기 때문에, 본 발명의 또 하나의 실시상태는 대상이 좋지 못한, 중간의 및 광범위한/극도의 빠른 P450 메타볼라이저인지 결정하는 것이다. 대상이 중간의 또는 광범위한/극도의 빠른 메타볼라이저인 경우, 이어서 2대상에게 투여되는 투여량은 "조정된 유효 투여량", 즉 상기 화합물의 최저 혈장 수준이 적어도 5 ng/ml이 되도록 하는 양; 또는 상기 화합물의 최저 농도가 적어도 5 ng/ml이고 화합물의 Cmax이 100 ng/ml 미만이 되도록 하는 양으로 높아져야 한다. 투여량이 점차 높아질 수 있고, 대상은 1회, 2회, 3회, 4회 또는 조정된 유효 투여량을 달성하기 위하여 필요한 횟수 만큼 재시험된다.
CYP2D6 유전자에 대하여 4가지 예상되는 표현형이 있다:
"좋지 못한 P450 메타볼라이저"는 효소 활성을 완전히 손실시키는 2개의 돌연변이 대립형질을 운반한다.
"중간의 P450 메타볼라이저"는 하나의 감소된 활성의 대립형질과 하나의 널(null) 대립형질을 갖는다.
"광범위한 P450 메타볼라이저"는 적어도 하나 및 2개 이하의 정상 기능 대립형질을 갖는다.
"극도의 빠른 P450 메타볼라이저"는 기능 대립형질의 다수의 복제물(3-13)을 운반하고 과도한 촉매 활성을 생성한다.
대상은 전형적으로 유전자형 분석을 통하여 또는 CYP2D6 또는 CYP3A4와 같은 P450 효소에 의하여 물질대사되는 약물의 최저 혈장 수준의 모니터링을 통하여 좋지 못한, 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인지 평가된다. 통상, 화학식 (I)의 화합물 또는 화학식 (I) 헤미타르타르산염을 비롯한 이의 약학적 허용염의 최저 혈장 수준 및/또는 Cmax가 상기 화합물에 의한 처리의 개시 이후 1주, 2주, 3주 또는 4주까지, 또는 1개월, 2개월, 3개월, 6개월, 9개월 또는 12개월 또는 그 이상까지 상기 대상에서 모니터링된다. 필요한 경우 전술한 한정 사항, 즉 최저 혈장 수준 5 ng/ml 또는 그 이상 내에서의 수준을 유지하기 위하여 투여량 조정이 이루어진다.
대상은 P450 효소 억제제인 특정 약물에 의한 처리의 결과로서 좋지 못한 P450 메타볼라이저가 될 수 있다. 상기 약물의 예들은 파록세틴(paroxetine), 플루옥세틴(fluoxetine), 퀴니딘(quinidine) 또는 케토코나졸(ketoconazole)을 포함한다. 대안으로서, 대상은 P450 효소의 낮은 발현의 결과로서 좋지 못한 P450 메타볼라이저이다. 상기 예에 있어서, 상기 낮은 발현은 대상에서 P450 효소 발현을 결정함으로써, 즉 대상을 P450 효소에 대한 유전자형 분석함으로써 평가될 수 있다. 예컨대, CYP2D6의 발현은 통상 PCR (문헌 [McElroy et.al. "CYP2D6 Genotyping as an Alternative to Phenotyping for Determination of Metabolic Status in a Clinical Trial Setting", AAPS Pharmsi (2000) 2(4):article 33 (http://www.pharmsci.org/)]) 또는 마이크로어레이 기반 약물유전체학 검사(microarray based pharmacogenomic testing) (Background Information, Roche Diagnostics "The CYP450 Gene Family and Drug Metabolism", Hoffmann La Roche Ltd.)에 의하여 평가되며, 상기 문헌들의 전체 내용은 본 명세서에 참고로서 포함된다. 상기와 같이, 대상은 편리하게 치료 시작 전에 P450 발현(예컨대, CYP2D6)에 대하여 유전자형 분석될수 있고 필요한 경우 조정된 유효량이 투여될 수 있다. 치료 시작 전에 유전자형 분석을 하는 경우에, 상기 화합물의 최저 혈장 수준 및 Cmax를 모니터링하고 필요한 경우 투여량을 조정하는 것이 여전히 권장된다.
미갈라스타트(migalastat), 아갈시다아제 베타(agalsidase β),이미글루세라아제(imiglucerase), 이소파고민(isofagomine) 및 미글루스타트(miglustat)에 대한 유효량은 약물 라벨상에 기재되어 있거나 각 약물의 임상적 시험에서 수행된 대로이다.
화학식 (I)의 화합물은 약학적 허용산과 반응하여 약학적 허용염을 형성할 수 있다. 약학적 허용산의 예들은 염화수소산, 브롬화수소산, 요오드화수소산, 황산, 인산 및 그 유사물과 같은 무기 산, 및 p-톨루엔설폰산, 메탄설폰산, 옥살산, p-브로모페닐-설폰산, 카본산, 숙신산, 시트르산, 벤조산, 아세트산, 및 그 유사물과 같은 유기산을 포함한다. 상기 염들의 예들은 설페이트, 피로설페이트, 바이설페이트, 설파이트, 바이설파이트, 포스페이트, 모노하이드로겐포스페이트, 다이하이드로겐포스페이트, 메타포스페이트, 피로포스페이트, 염화물, 브롬화물, 요오드화물, 아세테이트, 프로피오네이트, 데카노에이트, 카프릴레이트, 아크릴레이트, 포르메이트, 이소부티레이트, 카프로에이트, 헵타노에이트, 프로피올레이트, 옥살레이트, 말로네이트, 숙시네이트, 수베레이트, 세바케이트, 푸마레이트, 말레에이트, 부틴-1,4-디오에이트, 헥신-1,6-디오에이트, 벤조에이트, 클로로벤조에이트, 메틸벤조에이트, 디니트로벤조에이트, 히드록시벤조에이트, 메톡시벤조에이트, 프탈레이트, 설포네이트, 크실렌설포네이트, 페닐아세테이트, 페닐프로피오네이트, 페닐부티레이트, 시트레이트, 락테이트, 감마-히드록시부티레이트, 글리콜레이트, 타르타르산염, 메탄설포네이트, 프로판설포네이트, 나프탈렌-1-설포네이트, 나프탈렌-2-설포네이트, 만델레이트 및 그 유사물을 포함한다.
화학식 (I) 헤미타르타르산염을 포함하는 약학 조성물
화학식 (I)의 화합물 또는 이의 약학적 허용염(이의 헤미타르타르산염을 포함)에 대한 적절한 제형 및 투여 모드는 미국 특허 제7,253,185호에 기재된 것들을 포함하고, 상기 문헌의 전체 내용은 참고로서 본 명세서에 포함된다. 화학식 (I) 헤미타르타르산염에 대한 바람직한 제형은 후술하는 문단에 서술된다.
본 발명의 하나의 실시상태는 화학식 (I) 헤미타르타르산염, 적어도 하나의 수용성 필러, 적어도 하나의 수불용성 필러, 적어도 하나의 바인더 및 적어도 하나의 활택제를 포함하는 약학 조성물이다. 적절한 수용성 필러는 예컨대 무수 락토오스, 락토오스 일수화물, 만니톨, 염화나트륨, 분말화된 당(powdered sugar), 소르비톨, 수크로오스, 이노시톨 및 전호화분 전분(pregelatinized starch)을 포함할 수 있다. 적절한 수불용성 필러는 예컨대 미정질(microcrystalline) 셀룰로오스, 칼슘 포스페이트 및 전분을 포함할 수 있다. 적절한 바인더는 예컨대 전호화분 전분, 나트륨 카복시메틸 셀룰로오스, 히드록시프로필 셀룰로오스, 히드록시프로필 메틸 셀룰로오스, 폴리비닐 피롤리돈, 코폴리비돈, 젤라틴, 천연검(natural gum), 전분 페이스트, 수크로오스, 곡물 시럽(corn syrup), 폴리에틸렌 글리콜 및 나트륨 알긴산염을 포함할 수 있다. 적절한 활택제는 예컨대 수소화 식물성 오일(hydrogenated vegetable oil), 칼슘 스테아르산염 및 글리세릴 베헤네이트(grlceryl behenate)를 포함할 수 있다. 상기 약학 조성물의 하나의 실시상태에 있어서, 상기 수용성 필러는 무수 락토오스, 락토오스 일수화물, 만니톨, 염화나트륨, 분말화된 당, 소르비톨, 수크로오스, 이노시톨 및 전호화분 전분(pregelatinized starch)로 이루어진 군으로부터 선택되고; 상기 수불용성 필러는 미정질 셀룰로오스, 칼슘 포스페이트 및 전분으로 이루어진 군으로부터 선택되며; 상기 바인더는 전호화분 전분, 나트륨 카복시메틸 셀룰로오스, 히드록시프로필 셀룰로오스, 히드록시프로필 메틸 셀룰로오스, 폴리비닐 피롤리돈, 코폴리비돈, 젤라틴, 천연검, 전분 페이스트, 수크로오스, 곡물 시럽, 폴리에틸렌 글리콜 및 나트륨 알긴산염으로 이루어진 군으로부터 선택되고; 상기 활택제는 수소화 식물성 오일, 칼슘 스테아르산염 및 글리세릴 베헤네이트로 이루어진 군으로부터 선택된다.
약학 제형은 건조 고형분 기준으로 수불용성 필러 8 wt% 내지 32 wt% 사이, 8 wt% 내지 24 wt% 사이, 12 wt% 내지 20 wt% 사이 또는 14 wt% 내지 18 wt%사이를 포함한다.
약학 제형은 건조 고형분 기준으로 수용성 필러 26 wt% 내지 50 wt% 사이, 30 wt% 내지 46 wt% 사이, 34 wt% 내지 46 wt% 사이 또는 38 wt% 내지 44 wt% 사이를 포함한다.
약학 조성물은 건조 고형물 기준으로 화학식 (I) 헤미타르타르산염 30 wt%와 45 wt% 사이, 35 wt%와 40 wt % 사이 및 36 wt% 내지 39 wt%를 포함한다.
약학 제형은 전형적으로 건조 고형물 기준으로 바인더 2 wt%와 6 wt% 사이를 포함한다.
약학 제형은 전형적으로 건조 고형물 기준으로 바인더 0.1 wt%와 2 wt% 사이를 포함한다.
특정한 실시상태에 있어서, 약학 제형은, 모두 건조 고형물 기준으로, 8 wt% 내지 32 wt% 사이의 수불용성 필러, 26 wt% 내지 50 wt% 사이의 수용성 필러, 30 wt%와 45 wt% 사이의 화학식 (I) 헤미타르타르산염, 2 wt%과 6 wt% 사이의 바인더 및 0.1 wt%와 2 wt% 사이의 바인더를 포함한다. 더 구체적으로, 상기 수용성 필러는 락토오스 일수화물이고; 상기 수불용성 필러는 미정질 셀룰로오스이다. 좀더 구체적으로, 상기 수용성 필러는 락토오스 일수화물이고; 상기 수불용성 필러는 미정질 셀룰로오스이며; 상기 바인더는 히드록시프로필 메틸 셀룰로오스이고, 상기 활택제는 글리세릴 베헤네이트이다.
특정한 실시상태에 있어서, 약학 제형은, 모두 건조 고형물 기준으로, 8 wt% 내지 32 wt% 사이의 수불용성 필러, 26 wt% 내지 50 wt% 사이의 수용성 필러, 35 wt%와 40 wt% 사이의 화학식 (I) 헤미타르타르산염, 2 wt%와 6 wt% 사이의 바인더 및 0.1 wt%와 2 wt% 사이의 바인더를 포함한다. 더 구체적으로, 상기 수용성 필러는 락토오스 일수화물이고; 상기 수불용성 필러는 미정질 셀룰로오스이다. 좀더 구체적으로, 상기 수용성 필러는 락토오스 일수화물이고; 상기 수불용성 필러는 미정질 셀룰로오스이며; 상기 바인더는 히드록시프로필 메틸 셀룰로오스이고, 상기 활택제는 글리세릴 베헤네이트이다.
또 하나의 특정한 실시상태에 있어서, 상기 약학 제형은, 모두 건조 고형물 기준으로, 8 wt% 내지 24 wt% 사이의 수불용성 필러, 30 wt% 내지 46 wt% 사이의 수용성 필러, 35 wt%와 40 wt% 사이의 화학식 (I) 헤미타르타르산염, 2 wt%와 6 wt% 사이의 바인더 및 0.1 wt%와 2 wt% 사이의 바인더를 포함한다. 더 구체적으로, 상기 수용성 필러는 락토오스 일수화물이고; 상기 수불용성 필러는 미정질 셀룰로오스이다. 좀더 구체적으로, 상기 수용성 필러는 락토오스 일수화물이고; 상기 수불용성 필러는 미정질 셀룰로오스이며; 상기 바인더는 히드록시프로필 메틸 셀룰로오스이고, 상기 활택제는 글리세릴 베헤네이트이다.
또 하나의 특정한 실시상태에 있어서, 상기 약학 제형은, 모두 건조 고형물 기준으로, 12 wt% 내지 20 wt% 사이의 수불용성 필러, 34 wt% 내지 46 wt% 사이의 수용성 필러, 35 wt%와 40 wt% 사이의 화학식 (I) 헤미타르타르산염, 2 wt%와 6 wt% 사이의 바인더 및 0.1 wt%와 2 wt% 사이의 바인더를 포함한다. 더 구체적으로, 상기 수용성 필러는 락토오스 일수화물이고; 상기 수불용성 필러는 미정질 셀룰로오스이다. 좀더 구체적으로, 상기 수용성 필러는 락토오스 일수화물이고; 상기 수불용성 필러는 미정질 셀룰로오스이며; 상기 바인더는 히드록시프로필 메틸 셀룰로오스이고, 상기 활택제는 글리세릴 베헤네이트이다.
또 하나의 특정한 실시상태에 있어서, 상기 약학 제형은, 모두 건조 고형물 기준으로, 14 wt% 내지 18 wt% 사이의 수불용성 필러, 38 wt% 내지 44 wt% 사이의 수용성 필러, 35 wt%와 40 wt% 사이의 화학식 (I) 헤미타르타르산염, 2 wt%와 6 wt% 사이의 바인더 및 0.1 wt%와 2 wt% 사이의 바인더를 포함한다. 더 구체적으로, 상기 수용성 필러는 락토오스 일수화물이고; 상기 수불용성 필러는 미정질 셀룰로오스이다. 좀더 구체적으로, 상기 수용성 필러는 락토오스 일수화물이고; 상기 수불용성 필러는 미정질 셀룰로오스이며; 상기 바인더는 히드록시프로필 메틸 셀룰로오스이고, 상기 활택제는 글리세릴 베헤네이트이다.
또 하나의 특정한 실시상태에 있어서, 상기 약학 제형은, 모두 건조 고형물 기준으로, 14 wt% 내지 18 wt% 사이의 수불용성 필러, 38 wt% 내지 44 wt% 사이의 수용성 필러, 36 wt%와 39 wt% 사이의 화학식 (I) 헤미타르타르산염, 2 wt%와 6 wt% 사이의 바인더 및 0.1 wt%와 2 wt% 사이의 바인더를 포함한다. 더 구체적으로, 상기 수용성 필러는 락토오스 일수화물이고; 상기 수불용성 필러는 미정질 셀룰로오스이다. 좀더 구체적으로, 상기 수용성 필러는 락토오스 일수화물이고; 상기 수불용성 필러는 미정질 셀룰로오스이며; 상기 바인더는 히드록시프로필 메틸 셀룰로오스이고, 상기 활택제는 글리세릴 베헤네이트이다.
본 발명은 후술하는 실시예에 의하여 예시하며, 이하의 실시예는 어떠한 방식으로도 제한될 것을 의도한 것은 아니다.
실험
실시예 1: 화학식 (I)의 염의 제조
화학식 I의 헤미타르타르산염은 쉽게 결정화되고, 다른 염들에 비하여 많은 이로운 특성들을 나타낸다. 예컨대, 화학식 (I)에 의하여 표시되는 화합물의 염들의 제조에 하기의 산들이 사용되었다: 시트르산(1:1, 1:2, 및 1:3 (염:화학식 I) 비율로 염을 생성); L-말산(L-malic acid)(1:1 및 1:2); 메탄 설폰산(1:1); 푸마르산(1:1 및 1:2); 염화수소산(1:1); 아세트산(1:1) 및 타르타르산(1:1 및 1:2). 염화수소산(1:1); 타르타르산(1:1) 및 타르타르산(1:2)에 의하여 생성된 염들만이 고체 형태이었다. 이들 세 가지 염들 중 염화수소산(1:1) 및 타르타르산(1:1)이 흡습성이고 비결정질인 것이 밝혀졌고, 따라서 약학 생성물로의 용도로 허용될 수 없다. 화학식 I로 표시되는 화합물의 헤미타르타르산염(1 염: 2 화학식 I)은 결정질이고 비흡습성인 것으로 밝혀졌다.
화학식 (I) 헤미타르타르산염의 아세톤 제조
L-타르타르산(6.02 g, 40.11 mmol, 0.497 당량)을 아세톤(175 mL) 중에, 용액을 환류하고 이어서 실온으로 냉각함으로써, 용해시켰다. 화학식 (I) 자유 염기(32.67 g, 80.76 mmol)을 실온에서 아세톤(300 mL)에 용해시켰다. L-타르타르산 용액을 실온에서 15 분에 걸쳐 상기 화학식 (I) 자유 염기 용액에 첨가하였다. 상기 첨가를 통하여 흰색 침전물이 부분적으로(half way) 형성되었다. 상기 혼합물을 실온에서 0.5 시간 동안 교반하였고, 이어서 간단히 환류하고 실온으로 냉각하였다. 실온에서 0.5 시간 동안 교반한 후, 흰색 침전물을 여과하였다. 흰색 고체를 아세톤(2 x 130 mL)으로 2회 세척하였다. 상기 고체를 에어 건조시키고, 이어서 55-60 ℃에서 진공 건조시켰다. 그 수율은 36.66g(95 %)이었다.
화학식 (I) 헤미타르타르산염의 아세톤 중의 5% 메탄올 제조
화학식 (I) 자유 염기(10 g/24.7 mmol)을 5% 메탄올/아세톤 120 mL 또는 240 mL에 용해시켰다. L-타르타르산(1.85 g/12.3 mmol)을 40-45 ℃로 가온함으로써 5% 메탄올/아세톤 60 mL 또는 120 mL (N 또는 2N)에 용해시키고, 이 용액을 상기 첫번째 용액에 첨가하였다. 1 시간 후에 침전 없이 화학식 (I) 헤미타르타르산염 1 mg을 시드 결정(seed crystal)으로 첨가하였다. 5 분 후 침전이 일어났고, 30 분간 더 교반하여 반응을 지속하였다. 이어서, 상기 반응을 5 분간 환류에서 가열하였고(침전물은 완전히 가용성이었음), 이어서 수조 20-22 ℃ 내에서 실온까지 냉각하였다. 침전이 일어나고, 3 시간 교반하면서 반응을 계속하였다. 최종 생성물을 여과에 의하여 수집하고 아세톤 2 x 40 mL으로 세척하고, 이어서 55-60 ℃ 진공 오븐에서 16 시간 건조하였다. 생성물 중량은 8.72 g/74% 수율이었다.
화학식 (I) 헤미타르타르산염의 아세톤 중의 1% 물 제조
화학식 (I) 자유 염기(10 g/24.7 mmol)을 실온에서 1% 물/아세톤 120 mL 또는 240 mL에 용해시켰다. L-타르타르산(1.85 g/12.3 mmol)을 40-45 ℃로 가온함으로써 1% 물/아세톤 60 mL 또는 120 mL (N 또는 2N)에 용해시키고, 이 용액을 상기 첫번째 용액에 첨가하였다. 1 시간 후에 침전 없이 화학식 (I) 헤미타르타르산염 1 mg을 시드 결정(seed crystal)으로 첨가하였다. 5 분 후 침전이 일어났고, 30 분간 교반하여 반응을 지속하였다. 이어서, 상기 반응을 5 분간 환류에서 가열하였고(침전물은 완전히 가용성이 아니었음), 이어서 수조 20-22 ℃ 내에서 실온까지 냉각하였다. 침전이 일어나고, 3 시간 교반하면서 반응을 계속하였다. 최종 생성물을 여과에 의하여 수집하고 아세톤 2 x 40 mL으로 세척하고, 이어서 55-60 ℃ 진공 오븐에서 16 시간 건조하였다. 생성물 중량은 8.62 g 73% 수율이었다.
화학식 (I) 헤미타르타르산염의 아세톤 중의 5% 메탄올 재결정화
화학식 (I) 헤미타르타르산염(3.06 g)을 아세톤 중의 5% 메탄올 116 mL 중에 환류에서 용해시켰다. 상기 용액을 실온으로 냉각하고 실온에서 2 시간 동안 교반하였다. 흰색 침전물을 여과하고 아세톤 중의 5% 메탄올 10 mL로, 이어서 아세톤(15 mL)으로 세척하였다. 55-60 ℃에서 18 시간 진공 건조한 후에, 화학식 (I) 헤미타르타르산염 2.38 g을 얻었다(78 % 회복).
화학식 (I) 헤미타르타르산염의 아세톤 중의 1% H
2
0 재결정화
화학식 (I) 헤미타르타르산염(3.05 g)을 아세톤 중의 1% H20 125 mL 중에 환류에서 용해시켰다. 상기 용액을 실온으로 냉각하고 실온에서 2 시간 동안 교반하였다. 흰색 침전물을 여과하고 아세톤 중의 1% H20 10 mL로, 이어서 아세톤(15 mL)으로 세척하였다. 55-60 ℃에서 밤새 진공 건조한 후에, 화학식 (I) 헤미타르타르산염 2.35 g(77% 회복)을 얻었다.
실시예 2: 결정질 화학식 (I) 헤미타르타르산염의 제조
화학식 (I) 헤미타르타르산염을 여러가지 방법에 의하여 결정화하였다. 배치 1을 에틸 아세테이트/아세톤 용매를 이용하여 제조하고, 실온에서 건조하였다. 배치 3을 에틸 아세테이트/아세톤 용매를 이용하여 제조하고, 에틸 아세테이트로부터 재결정화하였다. 배치 4를 배치 1 물질을 이용하여 아세톤으로부터 재결정화하였다. 배치 5를 이소프로판올으로부터 재결정화하였다. 배치 7을 배치 1과 유사하지만 대규모로 에틸 아세테이트/아세톤 용매를 이용하여 제조하였고, 배치 8을 추가의 재결정화 없이 아세톤만을 이용하여 제조하였다. 배치 9를 간단한 환류로 아세톤만을 이용하여 제조하였고, 다시 추가의 재결정화는 하지 않았다.
| 배치 번호 | 프로세싱 방법 | DSC 융점(℃) 엔탈피(J/g) |
현미경 | TGA | |
| 1 | 아세톤/에틸 아세테이트 침전* | 162 | -81.4 | 결정 | 100 ℃에서 99.91% 175 ℃에서 98.73% |
| 2 | 아세톤/에틸 아세테이트 침전-실온에서 건조* | 164 | -95.6 | 결정 | N/A |
| 3 | 아세톤/에틸 아세테이트 침전-55-60℃ 건조 | 166 | -97.8 | 결정 | 100 ℃에서 100.0% 153 ℃에서 99.98% |
| 4 | 아세톤으로부터 재결정화 | 166 | -107.2 | 결정 | 100 ℃에서 100.2% 153 ℃에서 100.2% |
| 5 | 이소프로판올로부터 재결정화 | 166 | -102.6 | 결정 | 100 ℃에서 100.0% 153 ℃에서 100.0% |
| 7 | 아세톤/에틸 아세테이트 침전 | 166 | -99.4 | 결정** | 100 ℃에서100.1% 153 ℃에서 99.91% |
| 8 | 아세톤 침전 | 165 | -100.7 | 결정** | 100 ℃에서 100.0% 153 ℃에서 100.0% |
| 9 | 간단한 환류를 수반한 아세톤 침전 | 165 | -100.2 | 결정** | |
*: DSC 온도기록(thermogram)에 몇몇의 자유 염기를 함유
**: 이들 배치에서 막대형(rod), 평판형(plate-shaped)로부터 바늘형(needle), 막대형 및 불규칙한 형태로 변화되는 습관을 함유
화학식 (I) 헤미타르타르산염의 결정 형태도 역시 다양한 용매를 이용한 역용매 침전, 느린 증발, 느린 냉각, 빠른 냉각을 이용하여 제조되었다.
느린 증발 방법. 계량된 샘플(보통 20 mg)을 시험 용매의 부분 표본(aliquot)으로 처리하였다. 부분 표본은 전형적으로 100-200 μL이었다. 용매들의 첨가 사이에, 혼합물을 흔들거나 초음파 처리하였다(sonication). 시각 관찰에 의하여 판단시, 고체가 용해되었을 때, 핀홀로 구멍이 뚫어진 알루미늄 호일로 덮힌 열린 유리병(open vial) 내에서 주변 조건(ambient condition) 하에 용액이 증발되도록 하였다. 깨끗한 용액을 얻기 위하여 첨가된 전체 용매를 기초로 이들 실험들로부터 용해도를 추정하였다.
| 유기 용매 | 용해도 근사치(mg/mL) |
| 헵탄 | 얻을 수 없음 |
| 헥산 | 얻을 수 없음 |
| 톨루엔 | <5 |
| 디클로로메탄 | 100 |
| 에탄올 | 29 |
| 이소프로필 알코올 | <5 |
| 아세토니트릴 | <5 |
| 에틸 아세테이트 | <5 |
| 메탄올 | >200 |
| 아세톤 | <5 |
| 메틸 t-부틸 에테르(TBME) | <5 |
| p-디옥산 | <5 |
| 테트라히드로푸란(THF) | <5 |
| 유기 용매 | 느린 증발로부터 생성된 고체 형태 | DSC 융점(℃) 엔탈피(J/g) |
현미경 | TGA | |
| 메탄올 | 없음 | N/A | N/A | N/A | N/A |
| 에탄올 | 있음 | 165 | -95.0 | 결정** | 100 ℃에서 100.0% 150 ℃에서 100.0% |
**: 입자들이 평판형 및 막대형이었음.
느린/빠른 냉각 방법. 화학식 (I) 헤미타르타르산염을 50-60 ℃에서 시험 용매에 용해시켰다. 결과로 얻어진 용액을 이어서 주변 온도까지 냉각되도록 하였다(느린 냉각). 하루 후에 고체가 형성되지 않는다면, 유리병을 냉장고에 두었다. 빠른 냉각 실험을 위하여, 결과로 얻어진 용액을 이어서 냉장고에서 냉각되도록 하였다. 고체가 여과에 의하여 수집되고 에어 건조되었다.
| 유기 용매 | 느린 냉각으로부터 생성된 고체 형태 | DSC 융점(℃) 엔탈피(J/g) |
현미경 | TGA | |
| 에탄올 | 있음 | 167 | -106.2 | 결정** | 100 ℃에서 100.1% 150 ℃에서 100.1% |
**: 입자들이 평판형 및 막대형이었음.
| 유기 용매 | 빠른 냉각으로부터 생성된 고체 형태 | DSC 융점(℃) 엔탈피(J/g) |
현미경 | TGA | |
| 에탄올 | 있음 | 167 | -106.2 | 결정** | 100 ℃에서 100.0% 150 ℃에서 100.0% |
**: 입자들이 평판형 및 막대형이었음.
역용매 방법. 화학식 (I) 헤미타르타르산염을 용매에 용해시켰다. 역용매를 상기 용액에 첨가하였다. 형성된 고체를 여과에 의하여 수집하고 에어 건조하였다.
| 유기 용매 | 역용매 접근법으로부터 생성된 고체 형태 | DSC 융점(℃) 엔탈피(J/g) |
현미경 | TGA | |
| 메탄올/에틸 아세테이트 | 있음 | 167 | -99.5 | 결정* | 100 ℃에서 100.1% 150 ℃에서 100.1% |
| 메탄올/아세톤 | 있음 | 167 | -106.2 | 결정* | 100 ℃에서 100.3% 150 ℃에서 100.2% |
| 메탄올/아세토니트릴 | 없음 | N/A | N/A | N/A | N/A |
| 메탄올/톨루엔 | 없음 | N/A | N/A | N/A | N/A |
| 메탄올/THF | 없음 | N/A | N/A | N/A | N/A |
| 메탄올/ TBME | 있음 | 167 | -102.0 | 결정* | 100 ℃에서 100.2% 150 ℃에서 100.1% |
| 메탄올/p-디옥산 | 없음 | N/A | N/A | N/A | N/A |
| 물/THF | 없음 | N/A | N/A | N/A | N/A |
| 물/TMBE | 없음 | N/A | N/A | N/A | N/A |
| 물/이소프로판올 | 없음 | N/A | N/A | N/A | N/A |
| 물/아세토니트릴 | 없음 | N/A | N/A | N/A | N/A |
| 물/아세톤 | 없음 | N/A | N/A | N/A | N/A |
| 디클로로메탄/헵탄 | 있음 | 165 | -89.2 | 결정** | 100 ℃에서 100.0% 150 ℃에서 99.99% |
| 디클로로메탄/에틸 아세테이트 | 있음 | 167 | -97.8 | 결정* | 100 ℃에서 100.2% 150 ℃에서 100.1% |
| 디클로로메탄/톨루엔 | 있음 | 164 | -89.8 | 결정* | 100 ℃에서 99.95% 150 ℃에서 99.86% |
| 디클로로메탄/TBME | 있음 | 167 | -98.6 | 결정** | 100 ℃에서 100.0% 150 ℃에서 99.91% |
| 디클로로메탄/p-디옥산 | 있음(약간) | N/A | N/A | N/A | N/A |
| 디클로로메탄/이소프로판올 | 없음 | N/A | N/A | N/A | N/A |
*: 입자들이 평판형 및 막대형이었음.
**: 개별 입자들이 하나 이상의 복굴절 색상을 가졌음.
***: 입자들은 바늘형 및 막대형이었음.
실시예 3: 화학식 (I) 헤미타르타르산염의 물리적 특성
시차주사열량법(Differential Scanning Calorimater, DSC). DSC 데이터를 퍼지 가스(purge gas)로서 질소를 이용하는 TA Q100 설비 상에서 수집하였다. 약 2-5 mg의 샘플을 알루미늄 DSC 팬으로 정확하게 계량하였다. 상기 팬을 덮개로 덮고 겸자(forceps)를 이용하여 구멍을 뚫었다. 샘플 세포를 30 ℃에서 평형을 유지시키고 분당 10 ℃의 속도로 최종 온도 220 ℃까지 가열하였다.
핫 스테이지 현미경법(hot stage microscopy). 이미지 수집을 위하여 Sony DXC-970MD 3CCD 카메라가 구비된 Leica DM LP 현미경 상에 설치된 Linkam hot stage (model FTIR 600)을 이용하여 핫 스테이지 현미경법을 수행하였다. 40x 대물렌즈가 샘플을 보기 위한 편광과 함께 사용되었다. 각 샘플을 2개의 커버 슬라이드 사이에 놓았다. 스테이지가 가열되었을 때 각 샘플을 시각적으로 관찰하였다. Links version 2.27 (Linkam)를 이용하여 이미지를 캡쳐하였다. USP 융점 표준을 이용하여 핫 스테이지를 보정하였다(calibrate).
DSC 프로파일에서 관찰된 흡열성 이행(endothermic transition)이 핫 스테이지 현미경법에 의하여 160-163 ℃ 사이의 온도에서 용융 이행으로 확인되었다.
실시예 4: 화학식 (I) 헤미타르타르산염의 X-선 분말 회절법
모든 X-선 분말 회절법(XRPD) 분석들은 SSCI, Inc.(West Lafayette, IN 47906)에서 수행하였다. Cu K α 조사를 이용하는 Shimadzu XRD-6000 X-선 분말 회절계를 이용하여 XPRD 분석들을 수행하였다. 상기 설비는 미세 초점 X-선 튜브(fine focus X-ray tube)가 구비되어 있다. 상기 튜브 전압 및 암페어수(amperage)는 각각 40 kV 및 40 mA로 설정되었다. 발산성(divergence) 및 분산성(scattering) 슬릿(slit)을 1 °로 설정하였고, 수용(receiving) 슬릿을 0.15 mm로 설정하였다. 회절된 방사선은 NaI 신틸레이션 검출기(scintillation detector)에 의하여 검출되었다. 3 °/min (0.4 sec/0.02 °step)에서 2.5 내지 40 °2 θ까지 세타-2 세타 연속 스캔이 사용되었다. 실리콘 표준이 설비 배치(alignment)를 체크하기 위하여 분석되었다. XRD-6000 v 4.1을 이용하여 데이터를 수집하고 분석하였다.
실시예 5: 화학식 (I) 헤미타르타르산염과 화학식 (I) 자유 염기의 비교
자유 염기 및 헤미타르타르산염의 고체 특징이 표 7에 요약된다. 화학식 I 헤미타르타르산염은 화학식 I 자유 염기에 비하여 우월한 특성들을 갖는다. 예컨대, 화학식 I 헤미타르타르산염은 화학식 I 자유 염기에 비하여 더 높은 융점(>150 ℃), 더 높은 팩킹 에너지(더 큰 흡열성 엔탈피), 더 낮은 입경 변동, 더 높은 수 용해도(물 중의 300 mg/mL 이상), 적절한 결정 형태 및 더 높은 벌크 밀도를 갖는다.
| 물리적 특성 | 화학식 (I) 자유 염기 | 화학식 (I) 헤미타르타르산염 |
| 융점 (℃) | 86~88 | 163 |
| 흡열성 엔탈피(J/g) | 75-82 | 96-106 |
| 입자 사이즈(μm) | <10 내지 100 | ~3(평균) |
| 수 용해도(mg/mL) | 0.04 | >216 |
| 결정성 | 있음 | 있음 |
| 결정 형태 | 바늘형 | 평판형, 막대형, 약간 불규칙형 |
| 흡습성 (40 ℃/75% RH) | 없음 | 없음 |
| 벌크 밀도 | ~0.2 | 0.4~0.5 |
실시예
6: 체외 활성 및 특이성
체외에서 글리코스핀고리피드 합성을 억제하는, 화학식 (I) 헤미타르타르산염의 활성. 글루코실세라마이드 신타아제에 대한 화학식 (I) 헤미타르타르산염의 억제 활성을 정량화하기 위하여 2가지 분석법이 사용되었다. 글루코실세라마이드는 글리코스핀고리피드의 생합성에서 첫번째이고 속도 제한 단계이기 때문에, GM 1 및 GM3의 세포 표면 수준을 측정하는 유세포분석법(flow cytometry assay)이 사용되어 간접적으로 온전한 세포에서 억제제의 활성을 평가하였다. 화학식 (I) 헤미타르타르산염(0.6-1000 nM)의 양을 증가시키면서 K562 또는 B16/F10 세포들을 72 시간 동안 배양한 결과 GM1 및 GM3 둘 다의 세포 표면 수준이 투여량 의존 감소를 나타내었다. K562 세포에서의 GM1의 세포 표면 제공을 억제하기 위한 평균 IC50 값은 24 nM(14-34 nM 범위)(표 8)이었고, B16/F10 세포들에서의 GM3에 대한 것은 29 nM이었다(12-48 nM 범위). 가장 높은 투여량에서 시험된 경우에도, 명시적인 세포 독성은 어떠한 셀 라인에서도 나타나지 않았다.
활성에 대한 대안적인 분석법이 인간 세포 유레 마이크로좀에서의 클루코실세라마이드 신타아제의 억제를 측정하였다. 이 분석법에 있어서, 마이크로솜은 인간 흑색종(melanoma) A375 세포로부터 초음파 처리 및 원심 분리에 의하여 제조되었다. 마이크로솜 제조물을 형광성 세라마이드 기질(NBD-C6-세라마이드), UDP-글루코오스와 함께 화학식 (I) 헤미타르타르산염의 (0-1000 nM)양을 증가시키면서 1 시간 동안 실온에서 배양하였다. 배양 후에, 형광 표지된 글루코실세라마이드 및 미반응된 세라마이드를 분리하고, 역상 HPLC 및 형광성 검출에 의하여 정량화하였다. 이 분석법에 있어서, 클루코실세라마이드 합성을 억제하기 위한 IC50 값은 20 내지 40 nM 범위이었다. 이 값은 GM1 및 GM3에 대하여 상기에서 얻어진 것들과 유사하였으며, 이들 셀 표면 글리코리피드의 측정이 글루코실세라마이드 신타아제에 대한 화학식 (I) 헤미타르타르산염의 활성의 우수한 척도(surrogate)라는 것을 제안한다.
화학식 (I) 헤미타르타르산염의 기질 합성 억제의 특이성. 화학식 (I) 헤미타르타르산염의 특이성은 체외 세포 기반 및 무세포(cell-free) 분석법에서 연속적으로 평가되었다. 장 내 글리코시다제 효소는 래트 조직 균질 현탁액(homogenate)에서 분석되었고(참고 문헌 [U. Andersson, et a!., Biochem. Pharm. 59 (2000) 821 -829], 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다), 글리코겐 탈분지 효소는 전술한 무세포 분석법에서 분석되었다(참고 문헌 [U. Andersson, et al., Biochem. Pharm. 67 (2004) 697-705], 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다). 장내 글리코시다제(락토오스, 말타아제, 수크라아제), α-글루코시다제 I 및 II, 및 사이토졸 탈분지 효소(α-1,6-글루코시다아제)의 검출불가능한 억제가 2500 μM까지의 농도에서 밝혀졌다(표 8).
비리소좀(non-lysomal) 글루코실세라미다아제 및 리소좀 글루코세레브로시다아제는 기질로서 C6-NBD-글루코실세라마이드를 이용하여 온전한 인간 세포에서 분석되었다(참고 문헌 [H.S. Overkleeft, et al. J. Biol. Chem. 273 (1998) 26522-26527], 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.). 콘듀리톨 β 에폭사이드(conduritol β epoxide, 리소좀 글루코세레브로시다아제의 특이적 억제제)가 비리소좀 활성에 대한 리소좀 활성을 구별하는 데에 사용되었다. 글루코세레브로시다아제 활성은 또한 형광성-활성화된 세포 소팅(FACS)에 의하여도 측정되었다. K562 세포는 1 μM 5-(펜타플루오로벤조일아미노)-플루오레세인 디-β-D-글루코피라노사이드(PFB-FDGlu, 분자 프로브/ Invitrogen. Carlsbad, CA)의 존재하에 화학식 (I) 헤미타르타르산염의 양을 증가시키면서 30-60 분간 배양되었다. 세포는 즉시 얼음위에서 냉각되었고, 정량화된 형광성이 전술한 바와 같다. 비리소좀 글루코실세라미다아제는 IC50 1600 μM으로 약하게 억제되었다. 가장 높은 농도 2500 μM 까지, 고셔병에 결함이 있는 효소인 리소좀 글루코세레브로시다아제의 억제는 없었다(표 8). 따라서, 시험된 다른 효소 중 임의의 것과 비교될 때, 글루코실세라마이드 신타아제를 억제하기 위하여 농도에서 대략 40,000의 차이가 요구되었다.
| 기질 억제 잠재성 (체외 IC50): | ~0.024 μM |
| 효소 특이성, IC50: | |
| α-글루코시다제 I 및 II: | >2500 μM |
| 리소좀 글루코세레브로시다아제(GBA1): | >2500 μM μM |
| 비리소좀 글루코실세라미다제(GBA2): | 1600 μM |
| 글리코겐 탈분지 효소: | >2500 μM |
| 효소 특이성, Ki: | |
| 수크라아제 억제: | 10 μM 까지 억제 없음 |
| 말타아제 억제: | 10 μM 까지 억제 없음 |
실시예
7: 마우스 모델에서 리소좀
글루코실세라마이드
수준의 향상된
관리
A.
패브리병
효소 대체 치료(enzyme replacement therapy, ERT)와 기질 감소 치료(substrate reduction therapy, SRT) 모두의 병합된 사용이 효소 디벌킹(debulking)을 유지할 수 있거나 추가의 이점을 제공할 수 있는지 결정하기 위하여, 패브리병의 쥐과 동물 모델(패브리-래그, Fabry-Rag)에서 별도의 그리고 병합된 치료의 상대적인 효능이 비교되었다. 부모 패브리 마우스들은 문헌[ Wang, AM et al. Am. J. Hum. Genet. 59: A208 (1996)]에 기재되어 있다. 패브리-래그는 RAG-1 마우스와 교배되고 성숙한 림포사이트 또는 T-세포를 발달시키지 못한다(immune-comprimised).
동물 연구
단일 치료 연구를 위하여, 패브리 마우스들이 생후 1개월에 연구에 투입되었다(예방 모델). 치료군은 펠렛 음식 식사의 성분으로서 화학식 (I) 헤미타르타르산염(Genzyme Corp., Cambridge, MA)을 받았다. 약물은 표준 5053 마우스 먹이(TestDiet, Richmond, IN) 중에 0.15%(w/w)로 제형화되었고, 임의(자의)로 제공되었다. 이 제형은 하루에 화학식 (I) 헤미타르타르산염 300 mg/kg를 25 g 마우스에게 제공하였다.
병용 치료 연구를 위하여, 패브리-래그 마우스들이 생후 3개월에 연구에 투입되었다(치료 모델). 그룹 A의 마우스들은 재조합 인간 알파-갈락토시다아제 A(Genzyme Corp.)의 정맥 주사 주입을 2 개월 마다(즉, 생후 3, 5, 7 및 9 개월) 1 mg/kg의 투여량으로 받았다. 그룹 B는 동일한 정맥 주사 효소 투여량을 받은데 더하여 그것은 펠렛 음식 식사의 성분으로서 화학식 (I) 헤미타르타르산염(Genzyme Corp., Cambridge, MA)을 받았다. 약물은 표준 5053 마우스 먹이(TestDiet, Richmond, IN) 중에 0.15%(w/w)로 제형화되었고, 임의(자의)로 제공되었다. 이 제형은 하루에 화학식 (I) 헤미타르타르산염 300 mg/kg을 25 g 마우스에게 제공하였다. 그룹 C는 4 개월 마다 (즉, 생후 3 및 7개월) 효소 주입을 받았고, 그룹 B와 동일한 음식 식사 내 약물을 받았다. 그룹 D는 단지 음식 식사 내 약물(그룹 B 및 C와 동일)만을 받았다. 그룹 E는 미처리 패브리-래그 마우스들이었고, 그룹 F는 야생형 대조군이었다. 도 10을 참고하라.
조직 글로보트리아오실세라마이드(GL-3, Gb3) 수준의 정량화
GL-3의 정량화는 기본적으로는 GL-1을 위한 것인 Tandem Mass Spectrometry에 의하여 이루어졌다.
핫 플레이트 분석법이 전술한 바와 같이 수행되었다(Ziegler, RJ et al Molec. Ther. 15(3), 492-500 (2007)).
결과
화학식 (I) 헤미타르타르산염에 의한 패브리 마우스들의 단일 치료
α-갈락토시다제 A 활성의 결함에 의하여 야기된 패브리병의 마우스 모델에서 SRT가 평가되었다. 화학식 (I) 헤미타르타르산염에 의한 치료는 생후 1개월의 패브리 마우스들을 가지고 시작하였으며, 상기 마우스들이 생후 1년이 될 때까지 지속되었다. 동물들은 매일 그들의 식사에 300 mg/kg의 화학식 (I) 헤미타르타르산염이 투여되었다. 마우스들의 행동 테스트(즉, 핫-플레이트 분석법) 및 생화학적 테스트(즉, 소변 검사 및 조직/혈액/소변 중의 GL-3 농도 분석)이 2개월마다 수행되었다.
도 7에 도시된 바와 같이, 11 개월의 기간에 걸쳐 화학식 (I) 헤미타르타르산염을 패브리-래그 마우스들에게 투여한 것은 신체 기관(간, 신장, 심장 및 비장)에서 글로보트리아오슬리세라마이드(GL-3)의 리소좀 축적 속도를 약 50%까지 약화시켰다. 이것은 혐오적인 열자극에 대한 둔감성의 추후 제시에 의하여 입증된 바와 같이 병 진전의 지연 (도 8 참조) 및 소변 검사 요인, 예컨대 소변 용적, 크레아티닌 및 나트륨 수준의 악화 예방(도 9 참조)으로 해석된다. 따라서, 글리코스핀고리피드의 합성의 첫번째 단계를 촉매화하는 글루코실세라마이드 신타이제의 화학식 (I) 헤미타르타르산염-매개 억제는 고셔병의 동물 모델에서 이로울 뿐만 아니라, 다른 글리코스핀고리피도시스(glycosphingolipidoses)에서도 긍정적인 효과를 가질 수 있었다.
α갈락토시다제 A 및 화학식 (I) 헤미타르타르산염에 의한 패브리 마우스들의 병용 치료
ERT 단독의 효능 및 화학식 (I) 헤미타르타르산염을 이용한 SRT와의 병용의 효능이 패브리-래그 마우스들의 5개의 군집(n=12/그룹)에서 평가되었다. 도 10에 도시된 바와 같이, 3개월령에서 시작하여, 마우스들을 행동 테스트(즉, 핫-플레이트 분석법) 및 생화학적 테스트(즉, 조직/혈액/소변에서의 GL-3 수준 분석)의 스케줄에 적용시켰다. ERT에 적용된 마우스들에서, α -갈락토시다제 A의 1 mg/kg 투여량이 도 10에 도시된 바와 같은 스케줄에 투여되었다. SRT에 적용된 마우스들에서 화학식 (I) 헤미타르타르산염의 300 mg/kg 투여량이 마우스 식사에서 매일 투여되었다. 도 11에 도시된 바와 같이, ERT는 패브리-래그 마우스들에서 혈액 GL-3 수준을 감소시키는 반면, SRT는 그렇지 아니하였다. 도 12에 도시된 바와 같이, 조합 ERT/SRT는 패브리-래그 마우스들의 간 및 신장에서 GL-3 수준을 감소시키는데 가장 효과적이다. 도 13에 도시된 바와 같이, SRT는 패브리-래그 마우스들에서 소변 GL-3 수준을 감소시키는 반면 ERT는 그렇지 아니하였다. 도 14에 도시된 바와 같이 SRT는 패브리-래그 마우스들에서 열-둔감성의 시작을 지연시키지만, ERT는 그렇지 아니하였다.
요컨대, 패브라자임과 화학식 (I) 헤미타르타르산염을 병용하여 처리한 패브리-래그 마우스들은 치료 모델에서 ERT 또는 SRT 단독에 비하여 병 마커에서 다음의 방식으로 향상을 나타내었다: 병용 치료에 의하여 간 및 신장 GL-3 축적의 상당한 감소; SRT 그룹에서의 소변 GL-3 향상; ERT 그룹에서의 혈액 GL-3 향상; 및 SRT 그룹에서 말초신경증의 지연.
B. 고셔병. 효소 대체 치료(enzyme replacement therapy, ERT)와 기질 감소 치료(substrate reduction therapy, SRT) 모두의 순차적인 사용이 추가의 이점을 제공할 수 있는지 결정하기 위하여, 고셔병의 쥐과 동물 모델(D409V/null)에서 별도의 그리고 순차적인 치료의 상대적인 효능이 비교되었다.
방법
동물 연구. 동물을 수반한 절차는, AAALAC(Association for Assessment and Accreditation of Laboratory Animal Care)에 의하여 제안된 가이드라인을 따라서, IACUC(Institutional Animal Care and Use Committee)에 의하여 리뷰되고 승인되었다. 고셔 마우스(D409V/null)는 간, 비장 및 폐에서 글루코실세라마이드의 축적을 나타내지만 뼈 또는 뇌의 병리는 없는 타입 1 고셔병 모델이다(참고 문헌 [Y-H. Xu, et ai, Am. J. Pathol. 163, 2003, 2093-2101], 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.). 이전 실험에서 수컷과 암컷 사이에서 재조합 글루코세레브로시다아제 또는 화학식 (I) 헤미타르타르산염에 대한 응답에서 차이가 없다고 나타났기 때문에, 양성(both sexes)의 동물들을 3개월령에 연구에 투입하였다. 연구는 조직 글루코실세라마이드의 기준선 수준을 제공하기 위하여 2 주 후에 희생되는 그룹 A와 함께 6 그룹의 마우스들을 가졌다. 그룹 B, C 및 D는 모두 재조합 인간 글루코세레브로시다아제(Genzyme Corp., Cambridge, MA) (lOmg/kg)를 꼬리-정맥을 통하여 정맥 주사(100 μL)로 2일 마다 총 8회 주입을 받았다. 그룹 B는 이 요법의 종료시(그룹 A와 동일한 시간) 희생되어 조직 글루코실세라마이드의 효소-감소된 수준을 제공하였다. 그룹 D 및 E는 모두 펠렛 음식 식사의 성분으로서 화학식 (I) 헤미타르타르산염(Genzyme Corp., Cambridge, MA)을 공급받았다. 약물은 표준 5053 마우스 먹이(TestDiet, Richmond, IN) 중에 0.075%(w/w)로 제형화되었고, 임의(자의)로 제공되었다. 이 제형은 하루에 화학식 (I) 헤미타르타르산염 150 mg/kg을 25 g 마우스에게 제공하였다. 그룹 F는 치료를 받지 아니하였고 연구 시작 후 12주에 그룹 C, D 및 E를 따라 희생되었다. 음식 소비 및 마우스 체중은 1주일에 3회 모니터링되어, 약물 흡수 및 전체 건강에서 약물의 잠재적 충격을 결정하였다. 동물들을 이산화탄소 흡입에 의하여 죽이고, 즉시 조직을 얻었다. 각 조직의 반을 드라이아이스 상에서 급속냉동(snap freezing)시키고, 추가의 처리를 위하여 준비될 때까지 - 80 ℃에서 저장되었다. 나머지 반은 조직학적 분석을 위하여 처리되었다.
조직 글루코실세라마이드 수준의 정량화 글루코실세라마이드 수준은 전술한 바와 같이 질량 분광 분석법에 의하여 정량화되었다(참고 문헌 [McEachern, et ah, J. Gene. Med. 8 (2006) 719-729];[ T. Doering, J. Biol. Chem. 274 (1999) 11038- 11045], 이들 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.). 공지된 질량의 조직을 2:1 (v/v) 클로로포름:메탄올 중에서 균질화하고 37 ℃에서 15 분간 배양하였다. 샘플을 원심분리하고, 상청액을 4℃에서 밤새 물 0.2 용적으로 추출하였다. 상기 샘플을 원심분리하고, 수성 상을 폐기하였으며, 유기 상을 질소 하에서 필름으로 건조하였다. 전기분무 이온화 질량 분석법(ESI/MS) 분석을 위하여, 1 ml 클로로포름:메탄올 (2:1, v/v) 중에서 조직 샘플을 50 ng 최초(original) 조직 중량의 동등량으로 재구성하고, 5 분동안 와류(vortex)시켰다. 각 샘플의 부분 표본(40 μL)을 물 전체 회수 유리병으로 운반하고, 10 μg/mL d3-C16-GL-1 내부 표준(Matreya, Inc., Pleasant Gap, PA) 50 μL을 첨가하였다. 샘플들을 질소하에서 건조시키고 200 μL의 1:4(v/v) DMSO:메탄올로 재구성하였다. 상이한 탄소 체인 길이의 글루코실세라마이드의 ESI/MS 분석은 전기분무 이온 소스가 구비된 Micromass Quattro Micro system에 연결된(coupled) Waters alliance HPLC (Separation Module 2695)에서 수행되었다. 리피드 추출 샘플(20 μL)을 45 ℃에서 C8 컬럼(4 mL X 3 mm i.d; Phenomenex, Torrance, CA)으로 주입하였고, 0.5 mL/min 에서 50 내지 100%의 아세토니트릴 구배(gradient)로 (2 mM 암모늄 아세테이트, 0.1% 포름산)으로 용출하였다. 첫번째 0.5 분을 50% 유기에서 유지하였고, 이어서 최종 3.5분동안 대하여 신속하게 100%로 전환되었다. 소스 온도는 150 ℃에서 일정하게 유지되었고, 질소가 유속 670 L/h에서 탈용매(desolvation) 가스로 사용되었다. 콘 전압(cone voltage) 23V에서 모세관 전압이 3.80 KV로 유지되는 한편, 각 이온 종들에 대한 체류 시간(dwell time)이 100 ms이었다. MRM 모드에 의하여 스펙트럼이 얻어져서, 8개의 우세한 이소폼들(C16:0, C18:0, C20:0, C22:l, C22:0, C22:l-OH, C24:l, 및 C24:0) 을 모니터링 하였다. 글루코실세라마이드의 정량화는 내부 표준에 상대적으로 이들 8개의 이소폼들의 합에 기초하였고, 보정 커브(calibration curve)는 0.1 내지 10 μg/mL의 범위내이다.
조직학. 조직학적 분석을 위하여, 조직을 실온에서 24시간 동안 아연 포르말린에 고정하고, 이어서 추가의 처리에 대한 준비가 될 때까지 4 ℃에서 PBS 중에 보관하였다. 모든 샘플들은 에탄올 중에서 탈수소화처리되었고, 크실렌 중에서 세정되었으며(cleared), Surgipath R paraffin (Surgipath, Richmond, IL) 중에 침투시켜 끼워넣었다. 5개의 마이크론 섹션들은 회전 마이크로톰(rotatory microtome)을 이용하여 절단되었고, 스테이닝(staining)하기 전에 60℃오븐에서 건조하였다. 섹션들을 Hemo-De(Scientific Safety Solvents, Keller, TX)에서 탈파라핀화하고 에탄올의 디센딩 농도(descending concentration)에서 재수화한 후 PBS 세척하였다. 상기 섹션들을 헤마톡실린 및 에오신(H&E)에 의하여 스테이닝하고 래트 안티-마우스 CD68 모노클로날 항체(Serotec, Raleigh, NC)를 이용하여 라벨링하여 매크로파지(macrophage)를 식별하였다. PBS에서 5 분간 세척한 후에, 슬라이드를 에탄올에서 탈수시키고, SHUR/MountTM 커버글라스 설치 배지(TBS, Durham, NC)로의 설치 전에 Hemo-De에서 세정하였다. 간에서 CD68 면역양성의 퍼센트 영역을 조직 섹션 당 십 400X 이미지의 MetaMorph (MDS Analytical Technologies, Toronto, Canada) 분석을 이용하여 정량화하였다. 전문의 수의학 병리학자(board certified veterinary pathologist)는 그룹 지정되어 심사되는 모든 섹션들에 대하여 맹검하였다.
결과
생후 3개월 고셔 마우스들의 간, 비장 및 폐에서 축적된 GL1을 디벌킹하기 위한 글루코세레브로시다아제의 투여 요법. 효소 또는 기질의 감소(reduction) 치료에 의한 병용 또는 단일 치료의 상대적인 장점을 조사하기 위하여, 고셔 마우스의 내장 기관에서 GL1 수준을 최대로 대폭 경감시키는 효소요법을 먼저 결정하였다. 생후 3개월 고셔 마우스(D409V/null)에 10 mg/kg 재조합 인간 글루코세레브로시다아제의 2,4 또는 8 투여량을 정맥 주사로 투여하였다. 상기 효소의 2 또는 4 투여량으로 처리된 마우스들은 3일마다 약물 투입을 받았고, 8 투여량으로 처리된 마우스들은 2일마다 상기 효소를 받았다. 8 처리를 받은 동물들에서 투입들 간에 더 짧은 간격을 사용한 것은 투여되는 인간 효소에 임의의 면역 반응의 잠재적 충격을 최소화하기 위하여 설계되었다. 동물들은 마지막 효소 투입 후 7일째에 죽여졌고, 이들의 간, 비장 및 폐에 남아 있는 GL1의 양을 측정하였다.
글루코세레브로시다아제의 2 투여량에 의한 치료는 간에서 GL1의 수준을 50%까지 감소시켰다. 효소 투입의 수를 4 또는 8로 증가시키는 것은 기대한 바와 같이 간 GL1 수준을 더 큰 정도로(대략 75% 까지) 감소시켰다. 8 투여량에서도, GL1 수준을 완전히 낮추는 것 보다 적은 것은, 치료의 연장 기간 후에만 간비종대증이 감소한다는 것을 나타내는 고셔 대상에서의 경험과 일치한다(참고 문헌 [G.A. Grabowski, et ah, Ann. Int. Med. 122 (1995) 33-39], 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.). 고셔 마우스의 비장에서의 기질 수준은 효소 치료에 대하여 더 반응하지 않았다. 글루코세레브로시다아제의 2 투여량의 투여는 미치료된 대조군에서 나타난 것으로부터 GL1 수준을 별로 변화시키지 않았다. 효소 투입의 수를 4 또는 8로 증가시키는 것은 비장 GL1 수준을 약 50%까지 감소시켰다. 폐에서, 미처리된 대조군의 약 60%까지의 감소는 8 투여량 후에 관찰되었다. 폐에서 약간 더 낮은 정도의 기질 감소는 아마도 지질 함유 알비올라(alveolar) 매크로파지에 대하여 투입된 효소의 접근성이 더 좋지 못하기 때문이다. 비장 및 폐와 비교할 때 간에서 더 GL1 제거(clearance)가 관찰되는 것은 침투성(systemic) 투입 이후에 효소의 생체 분포를 반영하는 것 같다(참고 문헌 [S.M. Van Patten, et al. Glycobiology 17 (2007 ) 467-478], 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.). 이들 결과를 기초로, 2일 간격으로 투여된 10 mg/kg 글루코세레브로시다아제의 8회 연이은 투여로 이루어진 치료 요법은 그 이후의 연구를 위하여 사용되었다.
고셔 마우스의 간에서 GL1 수준을 낮추기 위한 효소 및 기질 감소 치료의 상대적인 활성. 생후 3개월 고셔 마우스의 집단들을 재조합 글루코세레브로시다아제 또는 화학식 (I) 헤미타르타르산염으로 별개로 또는 순차적으로 처리하였다. 그룹 B, C 및 D의 마우스들은 축적된 GL1을 제거하기 위하여 전술한 바와 같이 효소 8회 투여(2주 기간에 걸침)를 받았다. 이어서, 다른 그룹들에게는 추가의 10주 동안 규칙적인 먹이 또는 화학식 (I) 헤미타르타르산염(150 mg/kg/일)이 공급되었고, 그룹 F는 어떠한 치료도 받지 않고 순수한 대조군으로서 역할을 하였다. 먹이의 제형과 관계없이, 마우스들은 비슷한 양의 음식을 먹었고, 체중 증가에서 식별할 수 있는 차이가 없었다. 효소 단독 치료 2주 후에 간으로부터 저장된 GL1 수준의 대략 80%가 제거되었다. 이들 동물들이 10주간 추가 치료없이 진전되도록 허용되었을 때, 그들의 간 GL1 수준이 증가했고, 이는 그 사이의 기간 동안 기질의 재축적이 발생했다는 것을 나타낸다(도 2, 컬럼 C). 이들 수준은 미처리된 대조군의 것들과 별로 다르지 않았다(도 2, 컬럼 F). 그러나, 마우스들이 10주의 기간에 걸쳐 효소 및 이어서 그들의 음식 중에 화학식 (I) 헤미타르타르산염으로 처리되었다면, 그들의 간 GL1 농도은 미처리 대조군에 비하여 상당히 낮아졌다(도 2, 컬럼 D&F). 이 결과는 화학식 (I) 헤미타르타르산염에 의한 추가적인 처리가 기질의 재축적을 늦추었다는 것을 제안한다. 흥미롭게도, 전체 연구기간(12주) 동안 화학식 (I) 헤미타르타르산염만으로 치료된 고셔 마우스들도 역시 미처리, 동일 연령의 대조군(도 2, 컬럼 F)과 비교할 때, 그 차이가 크지 않지만, 더 낮은 GL-1 수준(도 2, 컬럼 E)을 나타내었다. 이 동물 모델에서의 GL1 수준을 감소시키는 SRT 단독의 활성은 우리의 이전 리포트와 일치하며(참고 문헌 [K.A. McEachem, et al., Mol. Genet. Metab. 91 (2007) 259-267], 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다), 고셔 마우스(D409V/null)가 잔여 효소적 활성을 보유한다는 사실을 반영하는 것 같다(참고 문헌 [Y-H. Xu, et ah, Am. J. Pathol. 163, 2003, 2093-2101], 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다).
고셔 마우스의 비장에서의 GL1 수준을 낮추기 위한 효소 및 기질 감소 치료의 상대적인 활성. 생후 3개월 고셔 마우스를 재조합 글루코세레브로시다아제만으로 2 주간 처리하는 것은 비장 GL1 수준을 대략 60%까지 감소시켰다(도 3, 컬럼 B). 이들 동물들이 추가의 간섭 없이 추가의 10 주간 시간이 지나도록 허용하는 경우, 기질 수준은 본 연구의 시작시에 관찰되었던 것으로 되돌아갔고(도 3, 컬럼 C), 미처리 대조군과 별로 다르지 않았다(도 3, 컬럼 F). 이것은 비장에서의 GL1의 재축적 속도가 간에서보다 더 높다는 것을 제안한다. 이 추정은 또한 간에서(~500 mg/g 조직; 도 3, 컬럼 A) 보다 비장에서(~1500 mg/g 조직; 도 2, 컬럼 A) 기질의 기초 수준가 더 높게 관찰되는 것에 의하여 뒷받침된다. 효소 및 이어서 화학식 (I) 헤미타르타르산염으로 그후 10 주간 처리된 동물들은 비장 GL1 수준에서 가장 많은 감소를 나타내었고(도 3, 컬럼 D), 이들은 미처리 대조군 비장에서보다 상당히 낮아졌다(도 3, 컬럼 F). 이것은 SRT의 전개(deployment)가 기질의 재축적을 지연시킬 뿐만 아니라, 이 기관에서의 축적의 부담을 추가로 감소시키는 역할을 한다는 것을 나타낸다. 적어도 이 예에 있어서, 잔류 내생 효소 및 기질 감소의 순수한 효과는 전체 기질 수준에서 추가의 감소를 초래했던 것 같다. 미처리 대조군에서(도 3, 컬럼 F) 보다 12 주간 화학식 (I) 헤미타르타르산염만으로 치료한 마우스들에서 더 낮은 비장 GL1 수준(도 3, 컬럼 E)이 관찰된 것은, 그 차이가 크지는 않지만, 이 생각과 일치한다. 따라서, 높은 잔류 효소 활성을 갖는 마일드 고셔 타입 1 환자들에서, SRT에 이어서 ERT에 의한 치료는 그 속도 및 아마도 문제가 되는 기질의 제거 정도 조차도 잠재적으로 가속화할 수 있었다.
고셔 마우스들의 폐에서의 GL1 농도를 낮추기 위한 효소 및 기질 감소 치료의 상대적인 활성. 앞에서 언급한 바와 같이, 폐 GL1 수준은 재조합 글루코세레브로시다아제의 정맥 주사 투여에 의하여 덜 효과적으로 제거되었다. 2주 동안 효소에 의한 생후 3개월 고셔 마우스의 치료는 폐에서 기질 농도의 30% 감소만을 초래하였다(도 4, 컬럼 B). 그 다음 10 주 동안 정상적인 먹이가 공급된 동물들의 집단은 예상대로 GL1의 재축적을 나타내었고, 미처리 농도과 별로 다르지 않았다(도 4, 컬럼 C&F). 반대로 동일한 간섭 기간에 걸쳐 화학식 (I) 헤미타르타르산염을 함유하는 먹이가 공급된 동물들은 효소만이 투여된 것 미만으로 기질 수준의 감소를 나타내었고(도 4, 컬럼 D), 미처리 대조군에서의 것보다 상당히 낮았다(도 4, 컬럼 F). 다시, 이것은, 비장에서와 같이, 폐에서 화학식 (I) 헤미타르타르산염의 순수한 효과(잔류 내생 효소 활성의 존재하)는 GL1의 재축적을 지연시킬 뿐만 아니라 시작 농도 미만으로 그것을 감소시키는 역할을 한다는 것을 제안한다. 다른 내장 기관과 같이, 화학식 (I) 헤미타르타르산염 단독에 의한 치료는, 미처리 대조군(도 4, 컬럼 F)과 비교할 때, 폐의 GL1수준을 낮추는데 효과적이었다(도 4, 컬럼 E).
효소 및 기질 감소 치료 후에 고셔 마우스들의 간의 조직병리학적 분석. 간에서의 상이한 치료 요법의 효과를 시각화하기 위하여, 조직 섹션들을 CD68 매크로파지 마커로 스테이닝하였다 미처리 생후 3개월 고셔 세포들로부터 얻은 간 섹션들의 분석은 12 주 후에 분석하였을 때 상당히 변화되지 않고 남아 있는 다수의 지질-울혈성(lipid-engorged) CD68-양성 고셔 마우스들의 존재를 나타내었다. 상기 생화학적 데이터와 일치하게, 재조합 글루코세레브로시다아제가 2주 기간동안 투여된 동물들의 간은 이들 비정상적인 매크로파지에서 지질의 실질적인 제거가 나타났다. 이들 동물들이 추가의 치료 없이 추가의 10주가 지나도록 허용된다면, 고셔 세포의 재출현에 의하여 나타난 바와 같이 GL1의 재축적의 증거가 있었다. 그러나, 고셔 세포에서의 이러한 증가는 만약 마우스들이 동일한 간섭 기간에 걸쳐 화학식 (I) 헤미타르타르산염에 의한 기질 감소 치료를 받았다면 효력이 없어졌다. 앞에서 언급한 바와 같이, 화학식 (I) 헤미타르타르산염만을 받은 고셔 마우스들도 역시, ERT 및 SRT의 조합을 받았던 것들과 동일한 정도는 아니지만, 기질의 축적 감소를 나타내었다. 다양한 섹션에서의 CD68-양성 스테이닝의 정도도 역시 MetaMorph 소프트웨어를 이용하여 정량화되었다(도 18). 이들 섹션에서의 스테이닝의 정도는 생화학적으로 결정된 간 GL1 수준의 양을 반영하였으며(도 15), 추가로 다른 치료 요법의 상대적인 장점에 대한 제안을 뒷받침한다.
실시예 8: 고셔병의 마우스 모델에서의 화학식 (I) 헤미타르타르산염의 효능
동물 연구. 동물을 수반한 절차는, AAALAC(Association for Assessment and Accreditation of Laboratory Animal Care)에 의하여 제안된 가이드라인을 따라서, IACUC(Institutional Animal Care and Use Committee)에 의하여 리뷰되고 승인되었다.
주 및 연방 가이드라인. 고셔 gbaD409V/null 마우스들(참고 문헌 [Y.-H. Xu. et ai , Am. J. Pathol. 163 (2003) 2093-2101], 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다)은 연구 필요조건에 따라 성숙하도록 허용되었다. 표현형이나 화학식 (I) 헤미타르타르산염에 대한 응답은 수컷과 암컷 사이에 차이가 발견되지 않았고, 따라서 양성(both sexes)이 연구에 사용되었다. 화학식 (I) 헤미타르타르산염의 전달은 10 mL/kg의 용적으로 매일 단일 위관영양법에 의하여 이루어졌다. 동물들은 치료 시작 전에 1주일간 동일한 용적의 물에 의한 위관영양법에 의하여 적응되어 있었다. 화학식 (I) 헤미타르타르산염은 주입용 물(WFI; VWR, West Chester, PA)에 용해되었고 9일 과정에 걸쳐 75 mg/kg/일 내지 150 mg/kg/일로서, 3일간 각 투여량을 유지하고 25 mg/kg/일을 증가시키는 투여량의 단계적 증가로 투여되었다. 전체 건강상 약물의 잠재적 충격을 모니터링하기 위하여, 마우스들은 3회 체중이 측정되었다. 동물들은 이산화탄소 흡입에 의하여 죽여졌고, 즉시 그들의 조직을 획득하였다. 각 조직의 반은 드라이아이스 상에서 급속냉동시키고 추가의 처리가 준비될 때까지 -80 ℃에서 저장되었다. 나머지 반은 조직학적 분석을 위하여 수집되었다.
고성능 박막 크로마토그래피에 의한 조직 글루코실세라마이드 수준의 정량화. 고성능 박막 크로마트그래피(HPTLC) 분석은 전술한 바와 같았다(참고 문헌[A. Abe, et al. , J. Clin. Inv. 105 (2000) 1563-1571]; [H. Zhao, et al. Diabetes 56 (2007) 1341 -1349]; 및 [S.P.F. Miller, et al. J. Lab. Clin. Med. 127 (1996) 353-358], 이들 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.). 간단히, 전체 지질 분획을, 차가운 PBS 중에서 조직을 균질화하고, 2:1(v/v) 클로로포름:메탄올로 추출하고, 수조 소니케이터(sonicator)에서 초음파처리함으로써 얻었다. 샘플들을 원심분리하여 상들을 나누고, 상청액을 회수하였다. 펠렛을 클로로포름:메탄올:염수(saline) 중에서 다시 초음파 처리하고, 원심분리하고, 생성된 두번째 상청액을 수집하여 첫번째 것과 혼합하였다. 1:1 (v/v) 클로로포름:염수 혼합물을 혼합한 상청액에 넣고, 와류시키고, 원심분리하였다. 상부 수성층을 제거한 후, 메탄올:염수를 첨가하고, 와류시키고, 다시 원심분리하였다. 유기상을 취하고, 질소하에서 건조시키고, 원래 중량 0.1 g 당 1 mL의 2:1(v/v) 클로로포름:메탄올 중에서 용해시키고 -20 ℃에서 저장하였다.
지질 추출물 부분을 전체 포스페이트(참고 문헌 [B.N. Ames, Methods Enzymol. 8 (1966) 115-118], 이 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.), 즉 내부 표준으로서 사용하기 위한 포스포리피드 함량을 측정하기 위하여 사용되었다. 나머지는 알칼리성 메탄올 분해를 시켜 HP-TLC 플레이트 상에서 글루코실세라마이드와 함께 이동하는 포스포리피드를 제거하였다. 전체 포스페이트의 동등한 양을 함유하는 추출물의 부분 표본을 공지된 글루코실세라마이드 표준(Matreya inc. Pleasant Gap, PA)과 함께 HP-TLC 플레이트 상에 떨어뜨렸다. 상기 지질을 용해하고, 3% 구리 아세테이트 일수화물(w/v), 15% 인산(v/v)을 가지고 시각화하였고, 이어서 150℃에서 10분간 베이킹 하였다. 지질 밴드가 농도계(GS-700, Bio-Rad, Hercules, CA) 상에 검출되고, Quantity One software (Bio-Rad)에 의하여 분석되었다.
질량 분광분석법에 의하여 조직 글루코실세라마이드 농도의 정량화. 글루코실세라마이드를 전술한 바와 같이 질량 분광분석법에 의하여 정량화하였다(참고 문헌 [K. McEachern, et al. J. Gene Med. 8 (2006) 719-729]; [T. Doering, et al, J. Biol. Chem. 274 (1999) 11038-11045], 이들 문헌의 전체 내용은 참고로서 본 명세서에 포함된다.). 조직을 2:1(v/v) 클로로포름:메탄올 중에서 균질화하고 37℃에서 배양하였다. 샘플을 원심분리하고 상청액을 밤새 물 0.2 용적을 가지고 추출하였다. 샘플을 다시 원심분리하고, 수성 상을 제거하고, 유기상을 질소하에서 필름으로 건조하였다.
전기분무 이온화 질량 분석법(ESI/MS) 분석을 위하여, 조직 샘플을 1 mL 클로로포름/메탄올(2:1, v/v) 중에서 50 ng 원래 조직 중량의 동등량으로 재구성하였고, 5분간 와류시켰다. 각 샘플의 부분 표본(40μM)을 물 전체 회수 유리병으로 운반하고, 10 μg/mL d3-C16-GL-1 내부 표준(Matreya, Inc., Pleasant Gap, PA) 50 μL을 첨가하였다. 샘플들을 질소하에서 건조시키고 200 μL의 1:4(v/v) DMSO:메탄올로 재구성하였다. 상이한 탄소 체인 길이의 글루코실세라마이드의 ESI/MS 분석은 전기분무 이온 소스가 구비된 Micromass Quattro Micro system에 연결된(coupled) Waters alliance HPLC (Separation Module 2695)에서 수행되었다. 20 마이크로리터 지질 추출물 샘플을 45 ℃에서 C8 컬럼(4 mL X 3 mm i.d; Phenomenex, Torrance, CA)에 주입하였고, 0.5 mL/min 에서 50 내지 100%의 구배(gradient)의 아세토니트릴(2 mM 암모늄 아세테이트, 0.1% 포름산)로 용출하였다. 첫번째 0.5분을 50% 유기에서 유지하였고, 이어서 최종 3.5분 동안 신속하게 100%로 전환되었다. 소스 온도는 150 ℃에서 일정하게 유지되었고, 질소가 유속 670 L/h에서 탈용매(desolvation) 가스로 사용되었다. 콘 전압(cone voltage) 23 V에서 모세관 전압이 3.80 KV로 유지되는 한편, 각 이온 종들에 대한 체류 시간(dwell time)이 100 ms이었다. MRM 모드에 의하여 스펙트럼이 얻어져서, 8개의 우세한 이소폼들(C16:0, C18:0, C20:0, C22:l, C22:0, C22:l-OH, C24:l, 및 C24:0) 을 모니터링 하였다. 글루코실세라마이드의 정량화는 내부 표준에 대하여 이들 8개의 이소폼들의 합에 기초하였고, 보정 커브(calibration curve)는 0.1 내지 10 μg/mL의 범위내이다.
조직학. 조직학적 분석을 위하여, 조직을 실온에서 24시간 동안 아연 포르말린(Electron Microscopy Sciences, Hatfield, PA)에 고정하고, 이어서 추가의 처리에 대한 준비가 될 때까지 4 ℃에서 PBS 중에 보관되었다. 모든 샘플들은 알코올의 디센딩 농도(descending concentration) 중에서 탈수소화처리되었고, 크실렌 중에서 세정되었으며(cleared), Surgipath R paraffin (Surgipath, Richmond, IL) 중에 침투시켜 끼워넣었다. 5개의 마이크론 섹션들은 회전 마이크로톰(rotatory microtome)을 이용하여 절단되었고, 스테이닝(staining)하기 전에 60℃에서 건조하였다. 섹션들을 크실렌 중에서 탈파라핀화하고 알코올의 디센딩 농도(descending concentration)에서 재수화하고, 물세척을 하였다. 3% 아세트산에서 1분간 헹군 후, 3% 아세트산 pH 2.0 중의 1% Alcian Blue 8GX(Electron Microscopy Sciences)에서 40 분간 스테이닝 하였다. 물에서 헹구고, 1% 주기적인 산(periodic acid) 중에서 1분간 산화한 후, 슬라이드를 Schiff's reagent (Surgipath)를 이용하여 12분간 스테이닝하였다 뜨거운 물로 5분간 세척한 후, 슬라이드를 알코올에서 탈수시키고, SHUR/MountTM 커버글라스 설치 배지(TBS, Durham, NC)로의 설치 전에 크실렌에서 세정하였다. 간에서 형태상으로 식별된 고셔 세포는 10 고 전력 필드(HPF, 400x) 당 매뉴얼 세포 카운트를 이용하여 정량화되었다.
결과
D409V/null 마우스들에 대한 화학식 (I) 헤미타르타르산염의 투여의 효과. D4009V/null 마우스들에게 화학식 (I) 헤미타르타르산염을 투여한 효과가 평가되었다. 대략 생후 7개월 마우스들에게 10 주간 위관영양법에 의하여 150 mg/kg/일dml 화학식 (I) 헤미타르타르산염(예비 연구에서 글루코실세라마이드 신타아제를 억제하기에 효과적인 것으로 보이는 투여량)이 투여되었다. 이 처리는 마우스들의 행복감 또는 급식(feeding) 습관에 주목할만한 효과를 갖지 않았다 연구 전체 기간 동안 그들의 체중의 측정은 미처리된 마우스들의 체중과 별로 편차를 나타내지 않았으며, 이는 화학식 (I) 헤미타르타르산염이 신타아제 억제에 효과적인 것으로 나타난 투여량에서 잘 수용되었다(tolerated)는 것을 제안한다.
어리고, 증상발현전(presymtomatic)인 고셔 마우스들의 치료에서의 화학식 (I) 헤미타르타르산염의 효능. 화학식 (I) 헤미타르타르산염은 어린(생후 10 주) D409V/null 마우스에서 고셔 세포의 출현 및 글루코실세라마이드의 리소좀 축적의 감소에 대하여 평가되었다. 이들 어린 고셔 마우스들은 감염된 조직에서 낮은 수준의 GL-1을 나타내었다. 생후 10주 동물들은 10주간 위관영양법에 의하여 75 또는 150 mg/kg/일의 화학식 (I) 헤미타르타르산염이 투여되었다. 글루코실세라마이드 수준의 측정은 동일 연령의 비히클(vehicle) 처리된 대조군과 비교할 때 투여량 의존적인 감소를 나타내었다. 150 mg/kg/일로 처리된 집단에서, 글르코실세라마이드 수준은 간, 폐 및 비장 각각에서 대조군의 것의 60, 40 및 75%이었다(도 6).
처리된 D409V/null 마우스들의 간 및 폐에서 통계학적으로 상당히 더 낮은 수준의 글루코실세라마이드가 관찰된 것은 화학식 (I) 헤미타르타르산염이 이들 조직에서 글리코스핀고리피드의 축적을 감소시키는데 효과적이었다는 것을 나타내었다.
연구 종료시의 미처리된 D409V/null 마우스들의 간의 조직병리학적 평가(20주령)는 간 전체에 고셔 세포의 존재를 나타내었다. 10 주간 화학식 (I) 헤미타르타르산염의 150 mg/kg/일로 처리된 마우스들은 크기가 모두 더 작은 고셔 세포들이 단지 간혹 출현하는 것을 나타내었다. 많은 상이한 섹션들에서의 이들 세포들의 정량화는 고셔 세포의 빈도가 화학식 (I) 헤미타르타르산염이 처리된 마우스들에서 상당히 더 낮았다는 것을 확인하였다. 함께, 이들 생화학적 및 조직학적 발견들은 증상발현전의 고셔 마우스들에게 화학식 (I) 헤미타르타르산염이 매일 구강 투여되었을 때 감염된 조직에서 글루코실세라마이드의 축적 및 그 결과로서 간에서의 고셔 세포의 생성을 감소시키는데 효과적이었다는 것을 제안하였다.
이전부터 존재하는 병리학적 면을 갖는 늙은 고셔 마우스들을 치료하는데 화학식 (I) 헤미타르타르산염의 효능. 증상을 보이는 늙은 고셔 마우스들에서 병의 진전을 막거나 역전시키는데 화학식 (I) 헤미타르타르산염의 효능도 역시 평가되었다.
생후 7개월 D409V/null 마우스들은 10주간 위관영양법에 의하여 150 mg/kg/일의 화학식 (I) 헤미타르타르산염이 투여되었다. 처리된 마우스들의 간, 폐 및 비장에서, 처리 후 5주 및 10 주에 글루코실세라마이드 수준의 분석은 그들이 연구 초기에 관찰된 것을 넘어서 증가하지 않았다는 것을 나타내었다. 처리 10주 후에 글루코실세라마이드 수준은 비히클 처리된 마우스들에 비하여 간에서 60% 더 낮아지고 폐에서 50% 더 낮아지고 비장에서 40% 더 낮아졌다.이들 결과는 화학식 (I) 헤미타르타르산염이 저장 병리학적 면의 존재하는 부담을 갖는 마우스들에서 글리코실세라마이드의 추가의 축적을 억제하는데 효과적이었다는 것을 나타내었다.
조직 섹션의 조직병리학적 분석은 미처리된 대조군과 비교할 때 처리된 D409V/null 마우스들의 간에서 고셔 세포의 감소된 수를 나타내었다. 고셔 세포들의 수의 정량화는 생화학적 발견들을 확증하였고; 처리된 D409V/null 마우스들은 5주 및 10주 시점 모두에서 처리 시작에서의 것과 크게 다르지 않은 고셔 세포 총 수를 나타내었다. 이들 시점 모두에서 고셔 세포 수는 미처리된 D409V/null 마우스들의 것에 비하여 상당히 낮아졌다. 함께, 이들 데이터는 이전부터 존재하는 병리학적 면을 갖는 동물에서 추가의 글루코실세라마이드 축적 및 고셔 세포의 성장을 효과적으로 억제하였다는 것을 나타낸다.
논의
화학식 (I) 헤미타르타르산염은 효소 글루코실세라마이드 신타아제에 대하여 고도의 특이성을 나타내었다 또한, 대다수가 잔류 글루코세레브로시다아제 활성을 보유한 고셔병 타입 1 환자들을 치료할 때 중요한 특징인, 유효 투여량에서 글루코세레브로시다아제 활성의 주목할 만한 억제가 없었다. 150 mg/kg/일의 유효 투여량에서, 처리된 그룹과 대조군의 미처리된 그룹 사이에 체중 차이가 없었고, 식별할 수 있는 위장 조직이 없었다. IC50(24-40 nM)에서 또는 그 이상에서의 혈청 농도는 최대 수용 농도 미만인 구강 투여량을 가지고 쉽게 달성할 수 있었다. 화학식 (I) 헤미타르타르산염은 또한 쉽게 물질대사되고 제거되었다: 래트 및 개에서 14C-방사능표지된 화합물에 의한 단일 및 반복 구강 투여 ADME 연구에서 나타난 바와 같이, 부모(parent) 화합물과 대사산물 모두가 24 시간 내에 효과적으로 제거되었다.
매일 단일 위장영양법의 비최적화 투여 요법을 사용하는 것은 어리고 증상발현전의 마우스들에서와 이미 저장 병리학적 면을 나타내는 늙은 고셔 마우스들 모두에서 글루코실세라마이드 축적을 성공적으로 예방하였다. 어린 생후 10주의 마우스들은, 야생형 대조군에 비하여 상대적으로 높은 글루코실세라마이드 수준이 잠복하고 있다고 하더라도, 고셔 세포라고 하는 특징적인 울혈성 조직 매크로파지를 아직 발달하지 못했다. 화학식 (I) 헤미타르타르산염의 150 mg/kg/일에 의한 처리는 모든 측정가능한 병 진전을 중단시키고, 고셔 세포의 발달을 억제하였다. 리소좀 글루코실세라마이드의 높은 수준 및 다수의 고셔 세포를 나타내는 늙은 마우스에 있어서, 처리 5 주 또는 10 주후에 저장 세포 수에서 또는 글리코스핀고리스드의 수준에서 추가의 증가는 없었다. 고셔 세포의 글루코실세라마이드의 주요원이 원래 세포 밖이라고 보고되어 있기 때문에, 이들 결과는 화학식 (I) 헤미타르타르산염의 글루코실세라마이드 신타아제의 억제가 침투성(systemic)이라는 것을 암시하였다.
화학식 (I) 헤미타르타르산염이 글루코실세라마이드의 추가의 축적을 예방하는 데 효과적이었다는 관찰은 고셔병의 치료를 더욱 향상시킬 수 있는 치료적 전략을 제안한다.
요컨대, 여기에 제시된 데이터는, 화학식 (I) 헤미타르타르산염이, 고셔병의 마우스 모델에서 명시적인 부작용을 나타내지 않으면서, 글루코실세라마이드 신타아제에 대하여 활성이 있고 특이적인 억제제라는 것을 나타내었다. 그것은 징후발현전과 늙은 병에 걸린 고셔 마우스 마두에서 글루코실세라마이드 축적 및 고셔 세포 형성을 억제함으로써 병의 진전을 성공적으로 예방하였다. 이들 발견은 화학식 (I) 헤미타르타르산염이 소아와 성인 모두의 고셔 타입 1 병 및 잠재적으로 다른 글리코스핀고리피드 저장 장애에 대한 또 하나의 치료적 옵션을 제시할 수 있다는 것을 제안한다.
실시예 9: 화학식 (I) 헤미타르타르산염의 상 2 임상적 시도
방법. 50 또는 100 mg 구강 처방된 화학식 (I) 헤미타르타르산염의 임상적 시도는 5 국가의 7 위치에 있는 고셔병 타입 1(GD1)에 걸린 26명 성인(16F:10M; 평균 나이 34세; 범위 18세 내지 60세; 모두 백인)을 처리하였다. 환자들은 비장 비대증(용적 10 정상)을 갖고 혈소판 감소증(혈소판 45,000-100,000/mm3) 또는 빈혈증(헤모글로빈 8-10 g/bl, 여성; 8-11 g/bl 남성)을 가졌다. 어느 누구도 12개월 전부터 효소 치환(replacement) 또는 기질 감소 치료를 받지 않았다. 합성물(composite)의 주요 효능의 종점은 치료 52주 후에 글로빈 농도(+0.5 g/dl) 또는 혈소판 총수(+15%)이다. 간 용적, 키토트리오시다제(chitotriosidase), 글루코실세라마이드도 역시 평가되었다. 환자들은 연속하여 치료되고 장기적으로 모니터링된다.
결과. 52 주 데이터를 20명의 환자로부터 이용가능하였고, 4 명은 조기에 중단하였고, 2 명은 계속 진행중이었다. 합성물의 주요 종점은 20 명의 환자 중 19 명까지 만족하였다. 기준선으로부터 52 주까지의 평균(1SD) 변화는 각각 헤모글로빈 +1.6 (11.35) g/dL; 혈소판 총수 +43.6% (137.59%); 비장 및 간 용적(정상의 배수) 40.2% (110.44%) 및 15.8% (110.39%); 및 키토트리오시다제 49.9% (120.75 %)이었다. 혈장 글루코실세라마이드 수준은 모든 환자에서 4주 후에 정상화되었고, 화학식 (I) 헤미타르타르산염은 허용가능한 안전성 프로파일로 잘 수용되었다. 6명의 환자에서 7개의 관련된 부정적 이벤트가 보고되었고, 모두 가볍고 성질상 일시적이었다.
실시예 10: 화학식 (I) 헤미타르타르산염 약학 조성물, 100mg 캡슐
100 mg 캡슐의 제조방법: 화학식 (I) 헤미타르타르산염, 미정질 셀룰로오스, 락토오스 일수화물 및 하이프로멜로오즈(hypromellose) E15를 20 메쉬 스크린에 별도로 통과시켰다. 표 9에 표시된 선별된 성분들의 양을 높은 전단 과립기(high-shear granulator)에서 9 내지 12분간 블렌딩하였다.
| 단위량 | |||
| 성분 | 단위량 100 mg 캡슐 | 단위 투여량 당 % (%w/w) |
작은(nominal) 배치 사이즈: 71,000 캡슐 전체량 19.2 kg |
| 화학식 (I) 헤미타르타르산염 | 100.0 | 37.0 | 7.1 |
| 미정질 셀룰로오스 | 45.0 | 16.7 | 3.2 |
| 락토오스 일수화물 | 111.5 | 41.3 | 7.9 |
| 히프로멜로오즈, E15 | 10.8 | 4.0 | 0.8 |
| 글리세릴 베헤네이트 | 2.7 | 1.0 | 0.2 |
| 충진 중량(mg) | 270 | 248-292 mg | |
| 전체 % 조성 | 100.0 | 19.2 kg | |
이어서, 정제된 물(2.2 kg; 11.7% 건조 성분중량 기준)을 시각적으로 확인하여 완료될 때까지 과립기 보울(bowl)에 첨가하여 성분들을 습식 과립화하였다. 습식 과립물을 보울로부터 제거하고 회전 임펠러(rotating impellor), 스크리닝 밀을 통과시켰다. 이어서, 습식 과립물을 직접 가열, 고정(static), 고체, 베드(bed), 트래이(tray) 드라이 오븐에서 50±5℃에서 공정 중 체크에 의하여 확인하여 수분 함량 3.5 % 이하까지 건조시켰다. 이어서, 건조 과립들을 스크리닝 밀을 통과시키고, 선별된 과립들을 V-블렌더로 이송하였다. 글리세릴 베헤네이트(0.2 kg)을 V-블렌더에 첨가하고, 최종 블랜드를 균일해질 때까지, 라인 내(in-line) 또는 라인 밖(off-line) 블렌드 균일성 테스트에 의하여 측정하면서, 전형적으로 10 내지 20분간, 혼합하였다. 이어서, 최종 블렌드를, 반 자동 캡슐 필러를 사용하여 적절한 충진 중량(평균 270 mg)으로 #2-사이즈 캡슐 내에 캡슐화하였고, 충진된 캡슐은 포장 전에 제진(dedust)되었다.
실시예 11A: 화학식 (I) 헤미타르타르산염 약학 조성물, 10 mg 캡슐
10 mg 캡슐 제조방법: 실시예 10의 과정을 캡슐화 단계까지 진행하였다. 10 mg 캡슐을 행성하기 위하여, 최종 블렌드를, 캡슐 충진 기계를 이용하여 적절한 충진 중량으로(평균 27 mg) #4 또는 #5-사이즈 캡슐 내에 캡슐화하였고, 충진된 캡슐은 포장 전에 제진하였다.
실시예 11B: 화학식 (I) 헤미타르타르산염 약학 조성물, 50 mg 캡슐
50 mg 캡슐 제조방법: 실시예 10의 과정을 캡슐화 단계까지 진행하였다. 50 mg 캡슐을 행성하기 위하여, 최종 블렌드를, 캡슐 충진 기계를 이용하여 적절한 충진 중량으로(평균 135 mg) #3-사이즈 캡슐 내에 캡슐화하였고, 충진된 캡슐은 포장 전에 제진하였다.
실시예 11C: 화학식 (I) 헤미타르타르산염 약학 조성물, 150 mg 캡슐
150 mg 캡슐 제조방법: 실시예 10의 과정을 캡슐화 단계까지 진행하였다. 150 mg 캡슐을 행성하기 위하여, 최종 블렌드를, 캡슐 충진 기계를 이용하여 적절한 충진 중량으로(평균 405 mg) #0-사이즈 캡슐 내에 캡슐화하였고, 충진된 캡슐은 포장 전에 제진하였다.
실시예 12: 화학식 (I) 헤미타르타르산염 약학 조성물, 25 mg 캡슐
25 mg 캡슐 제조방법: 실시예 10의 과정을 캡슐화 단계까지 진행하였다. 25 mg 캡슐을 행성하기 위하여, 최종 블렌드를, 캡슐 충진 기계를 이용하여 적절한 충진 중량으로(평균 67.5 mg) #4-사이즈 캡슐 내에 캡슐화하였고, 충진된 캡슐은 포장 전에 제진하였다.
실시예 13: 화학식 (I) 헤미타르타르산염 약물 상호작용 - CYP2D6 억제제
CYP2D6의 강한 억제제 파록세틴(30 mg 하루 1회)과 함께 또는 그것 없이 투여되는 화학식 (I) 헤미타르타르산염 (100 mg BID)의 다수의 구강 투여량의 약물 역학(phamacokinetics), 안전성 및 허용도(tolerability)를 평가하기 위하여 연구를 수행하였다. 이것은 36명의 건강한 대상(17명 남성 및 19명 여성)에서 개방 표지(open-label), 정해진 순서의 연구이었다. 2차 목적은 건강한 대상에서 화학식 (I) 헤미타르타르산염(100 mg BID)의 다수의 투여량과 함께 파록세틴의 PK를 평가하고, 추가로 단일 투여량의 화학식 (I) 헤미타르타르산염 투여와 비교할 때 다수 투여량에 따른 화학식 (I) 헤미타르타르산염 PK를 평가하는 것이었다.
화학식 (I) 헤미타르타르산염의 자유 염기의 평균 PK 파라미터는 그것이 혈장에 존재할 때 비선형이었고, 단일 투여량 투여와 비교할 때 반복된 투여(100 mg BID)로 Cmax 및 AUC에서 2배 축적을 나타내었다. 화학식 (I) 헤미타르타르산염과 파록세틴을 함께 투여하는 것은, 화학식 (I) 헤미타르타르산염 단독을 다수 투여량 투여하는 것과 비교할 때, Cmax의 7배 증가 및 AUC의 9배 증가를 초래하였다. 이들 결과는 파록세틴이 화학식 (I) 헤미타르타르산염의 대사를 억제할 수 있고, 약물의 혈액 혈장 농도를 증가시킨다는 것을 나타낸다. 유사한 효과가 다른 강한 CYP2D6 억제제(예컨대, 플루오세틴 및 퀴니딘)에도 기대될 수 있고, 화학식 (I) 헤미타르타르산염을 강한 CYP2D6 억제제로 알려진 약물과 공-투여하는 경우 약물 혈장 농도 및 잠재적 투여량 조절의 주의깊은 모니터링이 필요하다. 파록세틴 농도는 기대한 것 보다 약 1.5배 내지 2배 높았으며, 이는 화학식 (I) 헤미타르타르산염 또는 이의 대사산물 중 하나가 CYP2D6의 가벼운 억제제일 수 있다는 것을 제안한다.
실시예 14: 화학식 (I) 헤미타르타르산염 약물 상호작용 - CYP3A4 억제제 및 p-글리코프로테인(PGP) 억제제
건강한 남성 및 여성 대상에서 케토코나졸(400 mg 하루 1회)의 다수 투여와 함께 또는 그것 없이 화학식 (I) 헤미타르타르산염 (100 mg 하루 2회)의 다수의 구강 투여량의 약물 역학(phamacokinetics), 안전성 및 허용도(tolerability)를 평가하기 위하여 연구를 수행하였다. 이것은 36명의 건강한 대상(18명 남성 및 여성)에서 개방 표지(open-label), 정해진 순서의 연구이었으며, 이것은 화학식 (I) 헤미타르타르산염의 100-mg 단일 투여량 투여, 화학식 (I) 헤미타르타르산염 100 mg의 다수 투여량 투여, 및 화학식 (I) 헤미타르타르산염 100 mg(하루 2회)를 케토코나졸 400 mg(하루 1회)와 함께 투여하는 것을 포함하는 3개의 기간으로 이루어진다. 화학식 (I) 헤미타르타르산염과 사이토크롬 p450 3A4("CYP3A4") 및 p-글리코프로테인의 강한 억제제인 케토코나졸의 반복된 투여는 화학식 (I) 헤미타르타르산염의 자유 염기의 노출시 그것이 혈장에서 안정한 상태로 존재할 때 4배 증가를 초래한다. 따라서, 이미 화학식 (I) 헤미타르타르산염을 이미 받은 환자는 CYP3A4 또는 p-글리코프로테인의 강한 억제제와 함께 이루어진 치료에서 임시적인 투여량 감소가 필요할 수 있다.
실시예 15 - 화학식 (I) 헤미타르타르산염 제형의 안정성 연구
약 2 그램 스케일에서 신틸레이션 유리병 내에서 화학식 (I) 헤미타르타르산염 과 부형제(excipient)(락토즈 일수화물 캡슐화 그레이드, 아비셀 PH 301 (미정질 셀룰로오스) 및 Methocel El5 Prem LV (히드록시프로필메틸셀룰로오스))를 혼합함으로써 블렌드를 제조하였다. 15.6% 물을 블렌드에 첨가하고 혼합하여 습식 과립을 형성하였다. 습식 과립을 #10 체(sieve)(2000 마이크론의 개구)를 이용하여 선별하였다. 선별된 과립을 이어서 50℃에서 2시간 오븐에서 건조하였다. 건조된 과립을 #18 체(1000 마이크론의 개구)를 이용하여 선별하였다. 활택제인 글리세릴 베헤네이트를 블렌드에 첨가하고 혼합하여 최종 블렌드를 형성하였다. 제조된 블랜드를 하기 표에 나타내었다.
| Lot# | AP | 락토즈 일수화물 |
아비셀 PH 101 |
50 mg/100 mg 제형 |
코멘트 |
| 1 | 1 | 2.1 | 2.1 | 50 | 대조군 |
| 2 | 1 | 2.1 | 0 | 50 | 아비셀 없음 |
| 3 | 1 | 0 | 2.1 | 50 | 락토즈 없음 |
| 4 | 1 | 2.1 | 1.1 | 50 | 아비셀 적음 |
| 5 | 1 | 1.1 | 2.1 | 50 | 락토즈 적음 |
| 6 | 1 | 2.1 | 0.8 | 50 | 100 mg과 유사한 아비셀과 락토즈 비 |
| 7 | 1 | 1.1 | 0.4 | 100 | 대조군 |
Methocel(HPMC)가 2 내지 4% 범위로 사용되었다. Compritol ATO 88이 1 내지 1.6% 범위로 사용되었다.
상기에 열거된 상이한 API:락토즈:아비셀 비율을 갖는 7개 제형 블렌드는 각 제형의 분해 속도 및 안정성을 이해하기 위하여 고온 85℃에서 3일간(강제적인 분해 연구 조건) 노출되었다. 이 가속화된 조건은 24개월에 50 mg 약물 제품의 분해 생성물의 정도가 85℃에서 3일간 얻어진 것과 유사하였다는 연구 결과에 기초하여 선택되었다.
강제적인 분해 연구는 C18 컬럼(Waters T3, 3μm, 100 x 4.6 mm), 0.1 % 트리플루오로아세트산(TFA)를 갖는 물 및 아세토니트릴로 이루어진 유동상(mobile phase), 280 nm에서의 UV 검출, 40℃에서의 컬럼 온도 및 유석 2 mL/min을 사용한 역상 구배(gradient) HPLC 방법을 이용하여 수행하였다. 구배는 0.5분간 5% B(아세토니트릴 및 0.1% TFA)에서 유지를 시작하였고, 이어서 15분까지 분당 4.83% B에서 유기 성분을 증가시켰다.
각 제형 블렌드의 전체 분해가 계산되고, API:락토즈:아비셀의 비에 대하여 그래프로 그려졌으며, 그 결과를 도 15에 나타내었다. 이 연구는 API와 락토즈 비가 일정하게 유지될 때 아비셀의 양의 감소는 제형의 안정성을 향상시킨다. 아비셀이 제고되는 경우, 제형은 1:2.1:0의 API:락토즈:아비셀 비를 가지며, 이것이 가장 안정한 제형이다. 락토즈가 제거되는 경우, 제형은 1:0:2.1의 API:락토즈:아비셀 비를 가지며, 이 제형은 다른 비와 비교할 때 가장 불안정하지 않다. 조합된 정보는 락토즈가 제형을 안정화하는 한편, 아비셀은 제형을 불안정하게 한다는 것을 제안한다. 그러나, 부형제들이 모두 존재하는 경우, 이들은 서로 반응한다. 상기 비 대부분은 안정한 제형을 얻기 위하여 조정되어야 한다.
수용성인 화학식 (I) 헤미하이드레이트와 같은 활성 약학 성분을 위하여, 미정질 셀룰로오스는 수불용성이기 때문에 습식 과립화 도중 과립을 형성하는 것을 돕는다. 미정질 셀룰로오스가 사용되지 않는다면, 과립 단계에서 페이스트 형태로 예민한 변화가 발생한다. 페이스트 형태는 취급하기 어렵고, 건조 후 생성되는 입자가 적당한 기P적 강도 및 입자 사이즈 분포를 갖지 않는다. 건조 고형물 기준으로, 화학식 (I) 헤미타르타르산염 37 wt%, 수용성 필러 41.0 wt%, 불용성 필러 16.7 wt%, 바인더 2wt% 내지 약 6 wt% 및 활택제 약 0.1 wt% 내지 약 2 wt%를 갖는 약학 조성물이 형성되는 분해물의 양에 대하여 가장 안정성 있는 프로파일을 갖는다.
여기에 인용된 모든 특허, 공개된 출원 및 참고문헌은 그 전체가 참고로서 본 명세서에 포함된다.
이것은 특히 실시상태들을 예시하기 위하여 참고로 도시되고 기술되었지만, 당 기술분야의 숙련자들에게는 첨부된 청구범위에 포함되는 범위를 벗어남이 없이 여기서 형태 및 상세 내용에서의 다양한 변화가 이루어질 수 있다는 것이 이해될 것이다.
Claims (93)
- 하기 구조식으로 표시되는 화합물의 헤미타르타르산염:

여기서, 상기 염은 무정형 염이다. - 하기 구조식으로 표시되는 화합물의 헤미타르타르산염:

여기서, 상기 염의 적어도 70 중량%는 결정질이다. - 하기 구조식으로 표시되는 화합물의 헤미타르타르산염:

여기서, 상기 염의 적어도 70 중량%는 단일 결정질 형태이다. - 하기 구조식으로 표시되는 화합물의 헤미타르타르산염:

여기서, 상기 염의 적어도 99 중량%는 결정질이다. - 하기 구조식으로 표시되는 화합물의 헤미타르타르산염:

여기서, 상기 염의 적어도 99 중량%는 단일 결정질 형태이다. - 청구항 1 내지 5 중 어느 하나의 항에 있어서, 상기 헤미타르타르산염은 D-헤미타르타르산염, L-헤미타르타르산염, 헤미메조타르타르산 또는 라세미 D,L-헤미타르타르산염 중에서 선택되는 것인 헤미타르타르산염.
- 청구항 1 내지 5 중 어느 하나의 항에 있어서, 상기 헤미타르타르산염은 L-헤미타르타르산염인 것인 헤미타르타르산염.
- 청구항 3 또는 5에 있어서, 상기 염의 적어도 70 중량%는 단일 결정질 형태인 형태 A인 것인 헤미타르타르산염.
- 청구항 3 또는 5에 있어서, 상기 단일 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 15.9° 및 21.7°의 2θ각에서의 적어도 하나의 주요 x-레이 분말 회절 피크에 의하여 특징지워지는 것인 헤미타르타르산염.
- 청구항 3 또는 5에 있어서, 상기 단일 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 15.9° 및 21.7°의 2θ각에서의 적어도 두 개의 주요 x-레이 분말 회절 피크에 의하여 특징지워지는 것인 헤미타르타르산염.
- 청구항 3 또는 5에 있어서, 상기 단일 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 15.9° 및 21.7°의 2θ각에서의 적어도 세 개의 주요 x-레이 분말 회절 피크에 의하여 특징지워지는 것인 헤미타르타르산염.
- 청구항 3 또는 5에 있어서, 상기 단일 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 15.9° 및 21.7°의 2θ각에서의 적어도 네 개의 주요 x-레이 분말 회절 피크에 의하여 특징지워지는 것인 헤미타르타르산염.
- 청구항 3 또는 5에 있어서, 상기 단일 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 15.9° 및 21.7°의 2θ각에서의 주요 x-레이 분말 회절 피크에 의하여 특징지워지는 것인 헤미타르타르산염.
- 청구항 3 또는 5에 있어서, 상기 단일 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 13.3°, 15.1°, 15.9°, 16.5°, 17.6°, 18.6°, 18.7°, 19.0°, 20.2°, 21.7° 및 23.5°의 2θ각에서의 x-레이 분말 회절 피크에 의하여 특징지워지는 것인 헤미타르타르산염.
- 청구항 3 또는 5에 있어서, 상기 단일 결정질 형태는 도 1의 x-레이 분말 회절 패턴에 의하여 특정지워지는 것인 헤미타르타르산염.
- 하기 구조식에 의하여 표시되는 화합물의 헤미타르타르산염 및 약학적 허용 담체 또는 희석제를 포함하는 약학 조성물:

- 청구항 16에 있어서, 상기 염은 무정형 염인 것인 약학 조성물.
- 청구항 16에 있어서, 상기 염의 적어도 70 중량%는 결정성인 것인 약학 조성물.
- 청구항 16에 있어서, 상기 염의 적어도 70 중량%는 단일 결정질 형태인 것인 약학 조성물.
- 청구항 16에 있어서, 상기 염의 적어도 99 중량%는 결정성인 것인 약학 조성물.
- 청구항 16에 있어서, 상기 염의 적어도 99 중량%는 단일 결정질 형태인 것인 약학 조성물.
- 청구항 16에 있어서, 상기 헤미타르타르산염은 D-헤미타르타르산염, L-헤미타르타르산염, 헤미메조타르타르산 또는 라세미 D,L-헤미타르타르산염 중에서 선택되는 것인 약학 조성물.
- 청구항 16에 있어서, 상기 헤미타르타르산염은 L-헤미타르타르산염인 것인 약학 조성물.
- 청구항 19 또는 21에 있어서, 상기 염의 적어도 70 중량%는 단일 결정질 형태인 형태 A인 것인 약학 조성물.
- 청구항 19 또는 21에 있어서, 상기 단일 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 15.9° 및 21.7°의 2θ각에서의 적어도 하나의 주요 x-레이 분말 회절 피크에 의하여 특징지워지는 것인 약학 조성물.
- 청구항 19 또는 21에 있어서, 상기 단일 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 15.9° 및 21.7°의 2θ각에서의 적어도 두 개의 주요 x-레이 분말 회절 피크에 의하여 특징지워지는 것인 약학 조성물.
- 청구항 19 또는 21에 있어서, 상기 단일 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 15.9° 및 21.7°의 2θ각에서의 적어도 세 개의 주요 x-레이 분말 회절 피크에 의하여 특징지워지는 것인 약학 조성물.
- 청구항 19 또는 21에 있어서, 상기 단일 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 15.9° 및 21.7°의 2θ각에서의 적어도 네 개의 주요 x-레이 분말 회절 피크에 의하여 특징지워지는 것인 약학 조성물.
- 청구항 19 또는 21에 있어서, 상기 단일 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 15.9° 및 21.7°의 2θ각에서의 주요 x-레이 분말 회절 피크에 의하여 특징지워지는 것인 약학 조성물.
- 청구항 19 또는 21에 있어서, 상기 단일 결정질 형태는 5.1°, 6.6°, 10.7°, 11.0°, 13.3°, 15.1°, 15.9°, 16.5°, 17.6°, 18.6°, 18.7°, 19.0°, 20.2°, 21.7° 및 23.5°의 2θ각에서의 x-레이 분말 회절 피크에 의하여 특징지워지는 것인 약학 조성물.
- 청구항 19 또는 21에 있어서, 상기 단일 결정질 형태는 도 1의 x-레이 분말 회절 패턴에 의하여 특정지워지는 것인 약학 조성물.
- 청구항 1 내지 15 중 어느 하나의 항에 따른 헤미타르타르산염의 유효량을 대상(subject)에게 투여하는 것을 포함하는 고셔병(Gaucher disease)을 갖는 대상의 치료 방법.
- 청구항 1 내지 15 중 어느 하나의 항에 따른 헤미타르타르산염의 유효량을 대상(subject)에게 투여하는 것을 포함하는 패브리병(Fabry disease)을 갖는 대상의 치료 방법.
- 청구항 1 내지 15 중 어느 하나의 항에 따른 헤미타르타르산염의 유효량을 대상(subject)에게 투여함으로써 그것을 필요로 하는 대상에서 글리코스핀고리피드 농도를 낮추거나 글루코실세라마이드 신타아제를 억제하는 방법.
- 청구항 32, 33 또는 34에 있어서, 상기 헤미타르타르산염을 25 밀리그램 내지 200 밀리그램의 투여량으로 하루 2회 투여하는 것을 포함하는 것인 방법.
- 청구항 32, 33 또는 34에 있어서, 상기 헤미타르타르산염을 50 밀리그램의 투여량으로 하루 2회 투여하는 것을 포함하는 것인 방법.
- 하기 구조식으로 표시되는 제1 화합물 또는 이의 약학적 허용염의 유효량을 고셔병을 치료하기에 유효한 제2 치료 제제의 유효량과 함께 대상(subject)에게 투여하는 것을 포함하는 고셔병을 갖는 대상의 치료 방법.

- 청구항 37에 있어서, 상기 제2 치료 제제는 이미글루세라제(imiglucerase)인 것인 방법.
- 청구항 37에 있어서, 상기 제2 치료 제제는 이소파고민(isofagomine)인 것인 방법.
- 청구항 37에 있어서, 상기 제2 치료 제제는 미글루스타트(miglustat)인 것인 방법.
- 하기 구조식으로 표시되는 제1 치료 제제 또는 이의 약학적 허용염의 유효량을 패브리병을 치료하기에 유효한 제2 치료 제제의 유효량과 함께 대상에게 투여하는 것을 포함하는 패브리병을 갖는 대상의 치료 방법.

- 청구항 41에 있어서, 상기 제2 치료 제제는 미갈라스타트(migalastat)인 것인 방법.
- 청구항 41에 있어서, 제2 치료 제제는 아갈시다아제 베타(agalsidase β)인 것인 방법.
- 청구항 37 내지 43 중 어느 하나의 항에 있어서, 상기 제1 치료 제제는 청구항 1 내지 15 중 어느 하나의 항에 따른 헤미타르타르산염으로서 투여되는 것인 방법.
- 청구항 37 내지 43 중 어느 하나의 항에 있어서, 상기 제1 치료 제제에 의한 치료는 상기 제2 치료 제제에 의한 적어도 10주 기간에 걸친 치료 후에 시작되는 것인 방법.
- 청구항 37 내지 43 중 어느 하나의 항에 있어서, 상기 제1 치료 제제에 의한 치료는 상기 제2 치료 제제에 의한 치료 후에 시작되고, 상기 제1 치료 제제에 의한 치료는 상기 대상의 혈소판 수가 100,000 mm3과 동등하거나 더 많아지고/많아지거나; 헤모글로빈 농도가 11 g/dl (암컷) 또는 12 g/dl (수컷)과 동등하거나 더 많아지고/많아지거나; 상기 대상의 비장 용적이 정상의 10 배 보다 작거나 그와 동등하고 간 용적이 정상의 1.5 배 보다 작거나 그와 동등해진 후에 개시되는 것인 방법.
- 청구항 45 또는 46에 있어서, 상기 제2 치료 제제에 의한 치료는 상기 제1 치료 제제에 의한 치료의 개시 후에 종료되는 것인 방법.
- 청구항 37 내지 47 중 어느 하나의 항에 있어서, 상기 제1 치료 제제를 25 밀리그램 내지 200 밀리그램의 투여량으로 하루 2회 투여하는 것을 포함하는 것인 방법.
- 청구항 37 내지 47 중 어느 하나의 항에 있어서, 상기 제1 치료 제제를 50 밀리그램의 투여량으로 하루 2회 투여하는 것을 포함하는 것인 방법.
- 하기 화학식으로 표시되는 화합물의 헤미타르타르산염:

적어도 하나의 수용성 필러;
적어도 하나의 수불용성 필러;
적어도 하나의 바인더; 및
적어도 하나의 활택제를 포함하는 약학 조성물. - 청구항 50에 있어서, 상기 헤미타르타르산염은 청구항 1 내지 15 중 어느 하나의 항에 따른 헤미타르타르산염인 것인 약학 조성물.
- 청구항 50에 있어서, 상기 수용성 필러는 무수 락토오스, 락토오스 일수화물, 만니톨, 염화나트륨, 분말화된 당(powdered sugar), 소르비톨, 수크로오스, 이노시톨 및 전호화분 녹말(pregelatinized starch)로 이루어진 군으로부터 선택되는 것인 약학 조성물.
- 청구항 50에 있어서, 상기 수불용성 필러는 미정질(microcrystalline) 셀룰로오스, 칼슘 포스페이트 및 전분으로 이루어진 군으로부터 선택되는 것인 약학 조성물.
- 청구항 50에 있어서, 상기 바인더는 전호화분 전분, 나트륨 카복시메틸 셀룰로오스, 히드록시프로필 셀룰로오스, 히드록시프로필 메틸 셀룰로오스, 폴리비닐 피롤리돈, 코폴리비돈, 젤라틴, 천연검(natural gum), 전분 페이스트, 수크로오스, 곡물 시럽(corn syrup), 폴리에틸렌 글리콜 및 나트륨 알긴산염으로 이루어진 군으로부터 선택되는 것인 약학 조성물.
- 청구항 50에 있어서, 상기 활택제는 수소화 식물성 오일(hydrogenated vegetable oil), 칼슘 스테아르산염 및 글리세릴 베헤네이트(grlceryl behenate)로 이루어진 군으로부터 선택되는 것인 약학 조성물.
- 청구항 50에 있어서, 상기 수용성 필러는 무수 락토오스, 락토오스 일수화물, 만니톨, 염화나트륨, 분말화된 당(powdered sugar), 소르비톨, 수크로오스, 이노시톨 및 전호화분 녹말(pregelatinized starch)로 이루어진 군으로부터 선택되고; 상기 수불용성 필러는 미정질(microcrystalline) 셀룰로오스, 칼슘 포스페이트 및 전분으로 이루어진 군으로부터 선택되며; 상기 바인더는 전호화분 전분, 나트륨 카복시메틸 셀룰로오스, 히드록시프로필 셀룰로오스, 히드록시프로필 메틸 셀룰로오스, 폴리비닐 피롤리돈, 코폴리비돈, 젤라틴, 천연검(natural gum), 전분 페이스트, 수크로오스, 곡물 시럽(corn syrup), 폴리에틸렌 글리콜 및 나트륨 알긴산염으로 이루어진 군으로부터 선택되고; 상기 활택제는 수소화 식물성 오일(hydrogenated vegetable oil), 칼슘 스테아르산염 및 글리세릴 베헤네이트(grlceryl behenate)로 이루어진 군으로부터 선택되는 것인 약학 조성물.
- 청구항 56에 있어서, 상기 조성물은 건조 고형물 기준으로 상기 수용성 필러를 26 wt% 내지 50 wt% 포함하는 것인 약학 조성물.
- 청구항 56에 있어서, 상기 조성물은 건조 고형물 기준으로 상기 수불용성 필러를 8 wt% 내지 32 wt% 포함하는 것인 약학 조성물.
- 청구항 56에 있어서, 상기 조성물은 건조 고형물 기준으로 상기 수불용성 필러를 8 wt% 내지 24 wt% 포함하는 것인 약학 조성물.
- 청구항 56에 있어서, 상기 조성물은 건조 고형물 기준으로 상기 수불용성 필러를 12 wt% 내지 20 wt% 포함하는 것인 약학 조성물.
- 청구항 56에 있어서, 상기 조성물은 건조 고형물 기준으로 상기 수불용성 필러를 14 wt% 내지 18 wt% 포함하는 것인 약학 조성물.
- 청구항 56에 있어서, 상기 조성물은 건조 고형물 기준으로 상기 바인더를 2 wt% 내지 6 wt% 포함하는 것인 약학 조성물.
- 청구항 56에 있어서, 상기 조성물은 건조 고형물 기준으로 상기 활택제를 0.1 wt% 내지 2 wt% 포함하는 것인 약학 조성물.
- 청구항 56에 있어서, 상기 조성물은, 모두 건조 고형물 기준으로, 상기 헤미타르타르산염 35 wt% 내지 40 wt%, 상기 수용성 필러 26 wt% 내지 50 wt%; 상기 수불용성 필러 8 wt% 내지 32 wt%; 상기 바인더 2 wt% 내지 6 wt%; 및 상기 활택제 0.1 wt% 내지 2 wt%를 포함하는 것인 약학 조성물.
- 청구항 56에 있어서, 상기 수용성 필러는 락토오스 일수화물이고, 상기 수불용성 필러는 미정질 셀룰로오스이며, 상기 바인더는 히드록시프로필 메틸 셀룰로오스이고, 상기 활택제는 글리세릴 베헤네이트인 것인 약학 조성물.
- 청구항 65에 있어서, 상기 조성물은, 모두 건조 고형물 기준으로, 상기 헤미타르타르산염 35 wt% 내지 40 wt%, 상기 락토오스 일수화물 26 wt% 내지 50 wt%; 상기 미정질 셀룰로오스 8 wt% 내지 32 wt%; 상기 히드록시프로필 메틸 셀룰로오스 2 wt% 내지 6 wt%; 및 상기 글리세릴 베헤네이트 0.1 wt% 내지 2 wt%를 포함하는 것인 약학 조성물.
- 패브리병을 갖는 대상을 치료하는 방법으로서, 상기 방법은
a) 화학식 (I)의 화합물 또는 이의 약학적 허용염의 유효량을 상기 대상에게 투여하는 단계;

b) 상기 대상을 시험하여 상기 대상이 좋지 못한, 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저(metabolizer)인지 여부를 결정하는 단계;
c) 상기 대상이 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인 경우, 상기 화합물의 조정된 유효량을 결정하는 단계; 및
d) 상기 대상이 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인 경우, 화학식 (I)의 화합물의 조정된 유효량을 대상에게 투여하고, 상기 대상이 좋지 못한 P450 메타볼라이저인 경우 상기 화학식 (I)의 화합물의 유효량을 상기 대상에게 투여하는 단계
를 포함하는 것인 패브리병을 갖는 대상을 치료하는 방법. - 고셔병을 갖는 대상을 치료하는 방법으로서, 상기 방법은
a) 화학식 (I)의 화합물 또는 이의 약학적 허용염의 유효량을 대상에게 투여하는 단계;

b) 상기 대상을 시험하여 상기 대상이 좋지 못한, 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인지 여부를 결정하는 단계;
c) 상기 대상이 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인 경우, 상기 화합물의 조정된 유효량을 결정하는 단계; 및
d) 상기 대상이 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인 경우, 화학식 (I)의 화합물의 조정된 유효량을 대상에게 투여하고, 상기 대상이 좋지 못한 P450 메타볼라이저인 경우 상기 화학식 (I)의 화합물의 유효량을 상기 대상에게 투여하는 단계
를 포함하는 것인 고셔병을 갖는 대상을 치료하는 방법. - 청구항 67 또는 68에 있어서, 상기 화합물은 청구항 1 내지 15 중 어느 하나의 항에 따른 헤미타르타르산염인 것인 방법.
- 청구항 67 내지 69 중 어느 하나의 항에 있어서, 시험은 적어도 1 주일의 기간 동안 상기 대상 중의 상기 화합물의 최저 혈장 농도(trough plasma level)를 모니터링하는 것을 포함하는 것인 방법.
- 청구항 67 내지 69 중 어느 하나의 항에 있어서, 시험은 적어도 10 주일의 기간 동안 상기 대상 중의 상기 화합물의 최저 혈장 농도를 모니터링하는 것을 포함하는 것인 방법.
- 청구항 67 내지 71 중 어느 하나의 항에 있어서, 상기 대상 중의 상기 화합물의 혈장 최저 농도가 5 ng/ml 미만인 경우 상기 대상은 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저 효소이고, 상기 대상 중의 상기 화합물의 최저 농도가 적어도 5 ng/ml가 되도록 상기 화합물의 조정된 유효량이 선택되는 것인 방법.
- 청구항 67 내지 71 중 어느 하나의 항에 있어서, 상기 화합물의 유효량이 투여된 후에 상기 화합물의 혈장 최저 농도가 적어도 5 ng/ml인 경우, 상기 대상은 좋지 못한 P450 효소인 것인 방법.
- 청구항 67 또는 68에 있어서, 단계 b)의 시험은 치료 시작 전 또는 후에 발생하는 것인 방법.
- 청구항 67 내지 74 중 어느 하나의 항에 있어서, 상기 P450 효소는 CYP2D6 효소 및/또는 CYP3A4 효소인 것인 방법.
- 청구항 67 내지 75 중 어느 하나의 항에 있어서, 상기 대상은 CYP2D6 효소 또는 CYP3A4 효소의 억제제를 공-투여한 결과로서 좋지 못한 P450 메타볼라이저인 것인 방법.
- 청구항 76에 있어서, 상기 억제제는 파록세틴(paroxetine), 플루옥세틴(fluoxetine), 퀴니딘(quinidine) 및 케토코나졸(ketoconazole)로 이루어진 군으로부터 선택되는 것인 방법.
- 청구항 67 또는 69에 있어서, 단계 b)에서 상기 시험은 P450 효소에 대한 유전자형 분석을 하는 것인 방법.
- 청구항 78에 있어서, 상기 P450 효소는 CYP2D6인 것인 방법.
- 청구항 67 내지 79 중 어느 하나의 항에 있어서, 상기 화합물의 유효량은 25 밀리그램 내지 200 밀리그램의 투여량을 하루 2회 투여하는 것인 방법.
- 청구항 67 내지 79 중 어느 하나의 항에 있어서, 상기 화합물의 유효량은 50 밀리그램의 투여량을 하루 2회 투여하는 것인 방법.
- 패브리병을 갖는 대상을 치료하는 방법으로서, 상기 방법은
a) 하기 구조식으로 표시되는 화합물 또는 이의 약학적 허용염의 유효량을 대상에게 투여하는 단계;

b) 상기 대상 중의 상기 화합물의 최저 혈장 농도를 평가하는 단계;
c) 상기 대상에게 투여하는 화합물의 양을 조정하여 상기 대상 중의 상기 화합물의 최저 혈장 농도가 적어도 5 ng/ml이 되도록 하는 단계
를 포함하는 것인 패브리병을 갖는 대상을 치료하는 방법. - 고셔병을 갖는 대상을 치료하는 방법으로서, 상기 방법은
a) 하기 구조식으로 표시되는 화합물 또는 이의 약학적 허용염의 유효량을 대상에게 투여하는 단계;

b) 상기 대상 중의 상기 화합물의 최저 혈장 농도를 평가하는 단계;
c) 상기 대상에게 투여하는 화합물의 양을 조정하여 상기 대상 중의 상기 화합물의 최저 혈장 농도가 적어도 5 ng/ml이 되도록 하는 단계
를 포함하는 것인 고셔병을 갖는 대상을 치료하는 방법. - 청구항 82 또는 83에 있어서, 상기 화합물에 의한 치료를 시작하기 전에, 상기 대상이 좋지 못한, 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인지 여부를 평가하기 위하여 상기 대상을 시험하고, 상기 대상이 좋지 못한, 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인지 여부에 따라 상기 유효량이 결정되는 것인 방법.
- 청구항 82, 83 또는 84에 있어서, 상기 화합물은 청구항 1 내지 15 중 어느 하나의 항에 따른 헤미타르타르산염인 것인 방법.
- 청구항 82 또는 83에 있어서, 단계 b)에서 상기 대상 중의 화합물의 Cmax가 평가되고, 단계 c)에서 상기 대상에게 투여되는 화합물의 양이 조정되어, 상기 대상 중의 화합물의 최저 혈장 농도가 적어도 5 ng/ml이고 상기 대상 중의 화합물의 Cmax는 100 ng/ml 미만인 것인 방법.
- 청구항 82 내지 86 중 어느 하나의 항에 있어서, 상기 P450 효소는 CYP2D6 효소 및/또는 CYP3A4 효소인 것인 방법.
- 청구항 82 내지 86 중 어느 하나의 항에 있어서, P450 효소 억제제를 공-투여한 결과로서 상기 대상은 좋지 못한 P450 효소 메타볼라이저인 것인 방법.
- 청구항 88에 있어서, 상기 억제제는 파록세틴(paroxetine), 플루옥세틴(fluoxetine), 퀴니딘(quinidine) 및 케토코나졸(ketoconazole)로 이루어진 군으로부터 선택되는 것인 방법.
- 청구항 82 내지 86 중 어느 하나의 항에 있어서, 상기 대상이 좋지 못한, 중간의 또는 광범위한/극도의 빠른 P450 메타볼라이저인지 여부를 결정하기 위하여 상기 대상 중의 P450 효소 발현이 평가되는 것이 방법.
- 청구항 90에 있어서, 상기 P450 효소는 CYP2D6 효소 또는 CYP3A4 효소인 것인 방법.
- 청구항 82 내지 90 중 어느 하나의 항에 있어서, 상기 화합물의 유효량은 25 밀리그램 내지 200 밀리그램의 투여량을 하루 2회 투여하는 것인 방법.
- 청구항 82 내지 90 중 어느 하나의 항에 있어서, 상기 화합물의 유효량은 50 밀리그램의 투여량을 하루 2회 투여하는 것인 방법.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US26474809P | 2009-11-27 | 2009-11-27 | |
| US61/264,748 | 2009-11-27 | ||
| PCT/US2010/057952 WO2011066352A1 (en) | 2009-11-27 | 2010-11-24 | An amorphous and a crystalline form of genz 112638 hemitartrat as inhibitor of glucosylceramide synthase |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020157030851A Division KR102073207B1 (ko) | 2009-11-27 | 2010-11-24 | 글루코실세라마이드 신타아제의 억제제로서 genz 112638 헤미타르타르산염의 무정형 및 결정질 형태 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20180049255A true KR20180049255A (ko) | 2018-05-10 |
Family
ID=43431870
Family Applications (5)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020187012588A KR20180049255A (ko) | 2009-11-27 | 2010-11-24 | 글루코실세라마이드 신타아제의 억제제로서 genz 112638 헤미타르타르산염의 무정형 및 결정질 형태 |
| KR1020127016671A KR20120115972A (ko) | 2009-11-27 | 2010-11-24 | 글루코실세라마이드 신타아제의 억제제로서 genz 112638 헤미타르타르산염의 무정형 및 결정질 형태 |
| KR1020227040651A KR20220162824A (ko) | 2009-11-27 | 2010-11-24 | 글루코실세라마이드 신타아제의 억제제로서 genz 112638 헤미타르타르산염의 무정형 및 결정질 형태 |
| KR1020207002751A KR20200013105A (ko) | 2009-11-27 | 2010-11-24 | 글루코실세라마이드 신타아제의 억제제로서 genz 112638 헤미타르타르산염의 무정형 및 결정질 형태 |
| KR1020157030851A KR102073207B1 (ko) | 2009-11-27 | 2010-11-24 | 글루코실세라마이드 신타아제의 억제제로서 genz 112638 헤미타르타르산염의 무정형 및 결정질 형태 |
Family Applications After (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020127016671A KR20120115972A (ko) | 2009-11-27 | 2010-11-24 | 글루코실세라마이드 신타아제의 억제제로서 genz 112638 헤미타르타르산염의 무정형 및 결정질 형태 |
| KR1020227040651A KR20220162824A (ko) | 2009-11-27 | 2010-11-24 | 글루코실세라마이드 신타아제의 억제제로서 genz 112638 헤미타르타르산염의 무정형 및 결정질 형태 |
| KR1020207002751A KR20200013105A (ko) | 2009-11-27 | 2010-11-24 | 글루코실세라마이드 신타아제의 억제제로서 genz 112638 헤미타르타르산염의 무정형 및 결정질 형태 |
| KR1020157030851A KR102073207B1 (ko) | 2009-11-27 | 2010-11-24 | 글루코실세라마이드 신타아제의 억제제로서 genz 112638 헤미타르타르산염의 무정형 및 결정질 형태 |
Country Status (41)
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8889127B2 (en) | 2004-07-01 | 2014-11-18 | Icahn School Of Medicine At Mount Sinai | Targeted protein replacement for the treatment of lysosomal storage disorders |
| HUE029371T2 (en) | 2009-11-27 | 2017-02-28 | Genzyme Corp | GENZ 112638 For treatment of Gaucher or Fabry disease in combination therapy |
| CN103764824B (zh) * | 2011-06-20 | 2018-04-24 | 西奈山医学院 | 粘多糖贮积症和其它溶酶体病症的抗TNF-α疗法 |
| HUE051021T2 (hu) | 2011-09-07 | 2021-01-28 | Sinai School Medicine | Ceramidáz és sejtek differenciálódása |
| WO2013078413A1 (en) * | 2011-11-22 | 2013-05-30 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Modulators of lipid storage |
| DK2854910T3 (da) | 2012-06-01 | 2020-05-04 | Icahn School Med Mount Sinai | Ceramidniveauer i behandling og forebyggelse af infektioner |
| CA2905449A1 (en) | 2013-03-14 | 2014-10-02 | Icahn School Of Medicine At Mount Sinai | Therapeutic acid ceramidase compositions and methods of making and using them |
| AU2014321397C1 (en) * | 2013-09-20 | 2019-08-01 | Biomarin Pharmaceutical Inc. | Glucosylceramide synthase inhibitors for the treatment of diseases |
| CA2954030A1 (en) * | 2014-07-03 | 2016-01-07 | Dr. Reddy's Laboratories Limited | Amorphous form of eliglustat hemitartarate |
| WO2016166170A1 (en) | 2015-04-14 | 2016-10-20 | Sandoz Ag | Crystalline eliglustat hydrochloride |
| GB201508025D0 (en) | 2015-05-11 | 2015-06-24 | Ucl Business Plc | Fabry disease gene therapy |
| MX2018006250A (es) | 2015-11-18 | 2018-09-05 | Genzyme Corp | Biomarcador de enfermedad poliquistica renal y usos del mismo. |
| CN107445938B (zh) * | 2016-05-31 | 2020-04-03 | 北京启慧生物医药有限公司 | 依利格鲁司他半酒石酸盐的结晶形式、制备方法和含有所述结晶形式的药用组合物 |
| CN106349210A (zh) * | 2016-08-24 | 2017-01-25 | 北京阳光诺和药物研究有限公司 | 一种制备酒石酸艾力骨司坦的方法 |
| EP3318277A1 (en) | 2016-11-04 | 2018-05-09 | Institut du Cerveau et de la Moelle Epiniere-ICM | Inhibitors of glucosylceramide synthase for the treatment of motor neuron diseases |
| WO2018193090A2 (en) | 2017-04-21 | 2018-10-25 | Amneal Pharmaceuticals Company Gmbh | Process for preparation of eliglustat hemitartrate and intermediates thereof |
| WO2018225085A1 (en) | 2017-06-05 | 2018-12-13 | Cipla Limited | Stable solid dispersions of eliglustat hemitartrate |
| JP7045726B2 (ja) * | 2017-06-16 | 2022-04-01 | ベータ ファーマ,インコーポレイテッド | N-(2-(2-(ジメチルアミノ)エトキシ)-4-メトキシ-5-((4-(1-メチル-1h-インドール-3-イル)ピリミジン-2-イル)アミノ)フェニル)アクリルアミドおよびその塩の医薬品製剤 |
| US20200222310A1 (en) * | 2017-08-08 | 2020-07-16 | Kashiv Biosciences, Llc | Pharmaceutical composition comprising eliglustat |
| EP3664798A4 (en) * | 2017-08-08 | 2021-05-05 | Kashiv Biosciences, LLC | PHARMACEUTICAL COMPOSITION WITH ELIGLUSTAT |
| MX2020006272A (es) * | 2017-12-15 | 2020-09-14 | Genzyme Corp | Metodos para tratar la enfermedad de gaucher. |
| WO2019123476A1 (en) * | 2017-12-20 | 2019-06-27 | Sarudbhava Formulations Private Limited | Stable amorphous eliglustat premix and process for the preparation thereof |
| US11760741B2 (en) | 2018-05-02 | 2023-09-19 | Kashiv Biosciences, Llc | Pro-drugs of eliglustat |
| AU2019276882A1 (en) * | 2018-05-27 | 2021-01-14 | Bioasis Technolgies Inc. | Treatment of gaucher disease |
| CN110878079A (zh) * | 2018-12-31 | 2020-03-13 | 北京启慧生物医药有限公司 | 一种高纯度依利格鲁司他的制备方法 |
| CN116120274A (zh) * | 2021-11-12 | 2023-05-16 | 曙方(上海)医药科技有限公司 | 依利格鲁司他可药用盐及其晶型 |
Family Cites Families (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8311286D0 (en) | 1983-04-26 | 1983-06-02 | Searle & Co | Carboxyalkyl peptide derivatives |
| DE3522475A1 (de) | 1985-06-22 | 1987-01-02 | Kali Chemie Pharma Gmbh | Neue aromatische verbindungen, ihre herstellung und verwendung |
| US5041441A (en) | 1988-04-04 | 1991-08-20 | The Regents Of The University Of Michigan | Method of chemotherapy using 1-phenyl-2-decanoylamino-3-morpholino-1-propanol |
| ZA929008B (en) | 1991-12-13 | 1993-05-21 | Bristol Myers Squibb Co | Piperazinyl- and piperidinyl-cyclohexanols. |
| US5302609A (en) | 1992-12-16 | 1994-04-12 | The Regents Of The University Of Michigan | Treatment of diabetic nephropathy |
| US5399567A (en) | 1993-05-13 | 1995-03-21 | Monsanto Company | Method of treating cholera |
| WO1995005177A1 (fr) | 1993-08-13 | 1995-02-23 | Seikagaku Corporation | Remede contre les maladies nerveuses |
| US6309823B1 (en) | 1993-10-26 | 2001-10-30 | Affymetrix, Inc. | Arrays of nucleic acid probes for analyzing biotransformation genes and methods of using the same |
| PT742789E (pt) | 1994-02-02 | 2000-12-29 | Liposome Co Inc | Lipossomas e compostos farmaceuticamente activos e metodos para a sua utilizacao |
| CN1152907A (zh) | 1994-06-10 | 1997-06-25 | 生化学工业株式会社 | 2-酰胺基丙醇化合物及其药物组合物 |
| FR2734819B1 (fr) | 1995-05-31 | 1997-07-04 | Adir | Nouveaux composes de la piperazine, de la piperidine et de la 1,2,5,6-tetrahydropyridine, leur procede de preparation et les compositions pharmaceutiques les contenant |
| US5916911A (en) | 1995-09-20 | 1999-06-29 | The Regents Of The University Of Michigan | Amino ceramide--like compounds and therapeutic methods of use |
| US20030073680A1 (en) | 1995-09-20 | 2003-04-17 | The Regents Of The University Of Michigan | Amino ceramide-like compounds and therapeutic methods of use |
| US6255336B1 (en) | 1995-09-20 | 2001-07-03 | The Regents Of The University Of Michigan | Amino ceramide-like compounds and therapeutic methods of use |
| WO2001004108A1 (en) | 1999-07-09 | 2001-01-18 | Regents Of The University Of Michigan | Amino ceramide-like compounds and therapeutic methods of use |
| US6890949B1 (en) | 1999-07-09 | 2005-05-10 | The Regents Of The University Of Michigan | Amino ceramide-like compounds and therapeutic methods of use |
| NO965193L (no) | 1995-12-08 | 1997-06-09 | Seikagaku Kogyo Kk Seikagaku C | Aminalkoholderivat og fremgangsmåte for fremstilling derav |
| JP3993908B2 (ja) | 1995-12-08 | 2007-10-17 | 生化学工業株式会社 | アミノアルコール誘導体及び該誘導体の製造方法 |
| JP4140984B2 (ja) | 1995-12-20 | 2008-08-27 | 生化学工業株式会社 | 分化誘導作用を有する薬剤 |
| US5972928A (en) | 1997-05-21 | 1999-10-26 | Johns Hopkins University | Methods for treatment of conditions associated with lactosylceramide |
| JP4036500B2 (ja) | 1997-05-23 | 2008-01-23 | 生化学工業株式会社 | アミノアルコール誘導体及びそれを含有する医薬 |
| JP4176170B2 (ja) | 1997-06-06 | 2008-11-05 | 生化学工業株式会社 | アミノアルコール誘導体を含む医薬及び異常増殖性疾患治療薬 |
| US6465488B1 (en) | 1997-12-11 | 2002-10-15 | Chancellor, Masters & Scholars Of The University Of Oxford | Inhibition of glycolipid biosynthesis |
| AU756008B2 (en) | 1998-07-27 | 2003-01-02 | Johns Hopkins University, The | Methods for treating conditions modulated by lactosylceramide |
| US6610703B1 (en) | 1998-12-10 | 2003-08-26 | G.D. Searle & Co. | Method for treatment of glycolipid storage diseases |
| AU774960B2 (en) * | 1999-07-09 | 2004-07-15 | Regents Of The University Of Michigan, The | Amino ceramide-like compounds and therapeutic methods of use |
| KR20020071931A (ko) * | 2000-01-07 | 2002-09-13 | 트렌스폼 파마수티컬스 인코퍼레이티드 | 다양한 고체-형태들의 고도의 자료 처리 편성, 확인 및분석 |
| US6977723B2 (en) | 2000-01-07 | 2005-12-20 | Transform Pharmaceuticals, Inc. | Apparatus and method for high-throughput preparation and spectroscopic classification and characterization of compositions |
| ES2277301T3 (es) | 2000-04-24 | 2007-07-01 | Teva Pharmaceutical Industries Ltd. | Solvato de hemitartrato de zolpidem. |
| US6436987B1 (en) | 2000-06-08 | 2002-08-20 | Pfizer Inc. | Crystalline forms of (3S-trans)-2-[3,4-dihydro-4-hydroxy-3-(phenylmethyl)-2H-1-benzopyran-7-yl]-4-(trifluoromethyl)-benzoic acid |
| US7148251B2 (en) | 2001-01-10 | 2006-12-12 | The Regents Of The University Of Michigan | Amino ceramide-like compounds and therapeutic methods of use |
| US20020198240A1 (en) | 2001-01-10 | 2002-12-26 | Shayman James A. | Amino ceramide - like compounds and therapeutic methods of use |
| WO2002062777A2 (en) | 2001-01-10 | 2002-08-15 | The Regents Of The University Of Michigan | Amino ceramide-like compounds and therapeutic methods of use |
| US20040260099A1 (en) | 2001-01-10 | 2004-12-23 | The Regents Of The University Of Michigan | Amino ceramide-like compounds and therapeutic methods of use |
| EP1392826A2 (en) * | 2001-01-18 | 2004-03-03 | MERCK PATENT GmbH | Bifunctional fusion proteins with glucocerebrosidase activity |
| PT2266968E (pt) | 2001-07-16 | 2013-02-25 | Genzyme Corp | Síntese de udp-glucose: inibidores de n-acil esfingosinaglucosiltransferase |
| EP1281755A3 (en) | 2001-07-31 | 2003-06-18 | Pfizer Products Inc. | Variants of the human cyp2d6 gene |
| JP4316203B2 (ja) | 2001-08-08 | 2009-08-19 | トビラ セラピューティクス インク | 二環性化合物、その製造法および用途 |
| US7893101B2 (en) | 2002-03-20 | 2011-02-22 | Celgene Corporation | Solid forms comprising (+)-2-[1-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4-acetylaminoisoindoline-1,3-dione, compositions thereof, and uses thereof |
| US20060217560A1 (en) | 2002-04-29 | 2006-09-28 | Shayman James A | Amino ceramide-like compounds and therapeutic methods of use |
| US6916802B2 (en) | 2002-04-29 | 2005-07-12 | Genzyme Corporation | Amino ceramide-like compounds and therapeutic methods of use |
| WO2004004702A2 (en) | 2002-07-09 | 2004-01-15 | The Scripps Research Institute | Method to inhibit ischemia and reperfusion injury |
| US20050032070A1 (en) | 2003-08-05 | 2005-02-10 | Sebastian Raimundo | Polymorphisms in the human gene for CYP2D6 and their use in diagnostic and therapeutic applications |
| ATE504588T1 (de) * | 2004-01-27 | 2011-04-15 | Synthon Bv | Stabile salze von olanzapin |
| CA3113166A1 (en) | 2004-09-30 | 2006-04-13 | Vanda Pharmaceuticals Inc. | Methods for the administration of iloperidone |
| BRPI0516001A (pt) | 2004-10-13 | 2008-08-19 | Pharmacia & Upjohn Co Llc | formas cristalinas de 3-[5-cloro-4-[(2, 4-difluorobenzil) óxi]-6-oxopirimidin-1(6h)-il]-n-(2-hidroxietil)-4-metilben zamida |
| JP2008519840A (ja) | 2004-11-10 | 2008-06-12 | ジェンザイム・コーポレイション | 糖尿病の処置方法 |
| WO2007038676A2 (en) * | 2005-09-28 | 2007-04-05 | Teva Pharmaceutical Industries Ltd. | Polymorphic forms of ladostigil tartrate |
| AU2006297475A1 (en) * | 2005-10-03 | 2007-04-12 | Mallinckrodt Inc. | Process for preparing zolpidem hemitartrate and tartrate polymorphs |
| MX2007016179A (es) | 2006-04-17 | 2008-03-11 | Teva Pharma | Formas cristalinas de o-desmetilvenlafaxina. |
| PL2032134T3 (pl) | 2006-05-09 | 2015-11-30 | Genzyme Corp | Sposoby leczenia stłuszczeniowej choroby wątroby obejmujące hamowanie syntezy glikosfingolipidów |
| KR101271225B1 (ko) | 2006-10-31 | 2013-06-03 | 삼성디스플레이 주식회사 | 발광 다이오드 칩 및 발광 다이오드 광원 모듈의 제조 방법 |
| TWI314226B (en) | 2006-12-07 | 2009-09-01 | Ind Tech Res Inst | Piezoelectricity-driving optical lens module |
| EP1961765A1 (en) | 2006-12-08 | 2008-08-27 | Zealand Pharma A/S | Truncated PTH peptides with a cyclic conformation |
| EP2142197A4 (en) | 2007-03-30 | 2010-11-10 | Amicus Therapeutics Inc | PROCESS FOR THE TREATMENT OF FABRY DISEASE USING PHARMACOLOGICAL CHAPERONS |
| WO2008134628A2 (en) * | 2007-04-26 | 2008-11-06 | Amicus Therapeutics, Inc. | Dosing regimens for the treatment of lysosomal storage diseases using pharmacological chaperones |
| EP2594564B1 (en) * | 2007-05-31 | 2016-09-28 | Genzyme Corporation | 2-acylaminopropanol-type glucosylceramide synthase inhibitors |
| DE102007029581B4 (de) | 2007-06-26 | 2020-04-09 | GM Global Technology Operations LLC (n. d. Ges. d. Staates Delaware) | Kraftfahrzeugdach und Kraftfahrzeugkarosserie |
| US20090299645A1 (en) | 2008-03-19 | 2009-12-03 | Brandon Colby | Genetic analysis |
| WO2009117150A2 (en) | 2008-03-20 | 2009-09-24 | Genzyme Corporation | Method of treating lupus with ceramide derivatives |
| AU2010323068B2 (en) | 2009-11-27 | 2015-09-03 | Boehringer Ingelheim International Gmbh | Treatment of genotyped diabetic patients with DPP-IV inhibitors such as linagliptin |
| FR2953139B1 (fr) | 2009-11-27 | 2012-04-13 | Servier Lab | Composition pharmaceutique comprenant un sel de strontium, de la vitamine d et une cyclodextrine |
| TR201816146T4 (tr) | 2009-11-27 | 2018-11-21 | Adverio Pharma Gmbh | Meti̇l-{4,6-di̇ami̇no-2-[1-(2-florobenzi̇l)-1h-pi̇razolo[3,4-b]pi̇ri̇di̇n-3-i̇l]pi̇ri̇mi̇di̇n-5-i̇lmeti̇l}karbamatin farmasöti̇k etken madde olarak kullanima yöneli̇k olarak üreti̇lmesi̇ne yöneli̇k yöntem. |
| HUE029371T2 (en) | 2009-11-27 | 2017-02-28 | Genzyme Corp | GENZ 112638 For treatment of Gaucher or Fabry disease in combination therapy |
| EP2723733A1 (en) | 2011-06-21 | 2014-04-30 | Novartis AG | Polymorphs of (s)-pyrrolidine-1,2-dicarboxylic acid 2-amide 1-({4-methyl-5-[2-(2,2,2-trifluoro-1,1-dimethyl-ethyl)-pyridin-4-yl]-thiazol-2-yl}-amide |
| JO3316B1 (ar) | 2013-05-30 | 2019-03-13 | Lilly Co Eli | مركبات 3، 4-داي هيدرو أيزو كوينولين -2(1h)-يل |
| JP6728842B2 (ja) | 2016-03-24 | 2020-07-22 | オムロン株式会社 | 光学計測装置 |
| CN107445938B (zh) | 2016-05-31 | 2020-04-03 | 北京启慧生物医药有限公司 | 依利格鲁司他半酒石酸盐的结晶形式、制备方法和含有所述结晶形式的药用组合物 |
| WO2018193090A2 (en) | 2017-04-21 | 2018-10-25 | Amneal Pharmaceuticals Company Gmbh | Process for preparation of eliglustat hemitartrate and intermediates thereof |
-
2010
- 2010-11-24 HU HUE14164650A patent/HUE029371T2/en unknown
- 2010-11-24 LT LT16175117T patent/LT3133070T/lt unknown
- 2010-11-24 TW TW099140523A patent/TWI586663B/zh active
- 2010-11-24 IL IL310635A patent/IL310635A/en unknown
- 2010-11-24 PL PL19182732T patent/PL3599237T3/pl unknown
- 2010-11-24 CA CA3075788A patent/CA3075788C/en active Active
- 2010-11-24 RS RS20140446A patent/RS53503B2/sr unknown
- 2010-11-24 NZ NZ715108A patent/NZ715108A/en unknown
- 2010-11-24 PT PT191827328T patent/PT3599237T/pt unknown
- 2010-11-24 PL PL10785289T patent/PL2504332T5/pl unknown
- 2010-11-24 LT LTEP14164650.5T patent/LT2796457T/lt unknown
- 2010-11-24 SI SI201030715T patent/SI2504332T2/sl unknown
- 2010-11-24 PL PL14164650T patent/PL2796457T3/pl unknown
- 2010-11-24 EA EA201890254A patent/EA201890254A3/ru unknown
- 2010-11-24 EP EP19182732.8A patent/EP3599237B1/en active Active
- 2010-11-24 CN CN202010962663.7A patent/CN112521367B/zh active Active
- 2010-11-24 JP JP2012541183A patent/JP2013512252A/ja active Pending
- 2010-11-24 CN CN201610218210.7A patent/CN105753846B/zh active Active
- 2010-11-24 UA UAA201207602A patent/UA113491C2/uk unknown
- 2010-11-24 NZ NZ600155A patent/NZ600155A/en unknown
- 2010-11-24 CA CA3140959A patent/CA3140959A1/en active Pending
- 2010-11-24 PT PT107852899T patent/PT2504332E/pt unknown
- 2010-11-24 EA EA201270646A patent/EA023923B1/ru not_active IP Right Cessation
- 2010-11-24 SG SG10201800136QA patent/SG10201800136QA/en unknown
- 2010-11-24 PL PL16175117T patent/PL3133070T3/pl unknown
- 2010-11-24 US US13/511,768 patent/US11458119B2/en active Active
- 2010-11-24 BR BR112012012947A patent/BR112012012947B8/pt active IP Right Grant
- 2010-11-24 ES ES14164650.5T patent/ES2586947T3/es active Active
- 2010-11-24 SG SG10201407881WA patent/SG10201407881WA/en unknown
- 2010-11-24 CA CA2781676A patent/CA2781676C/en active Active
- 2010-11-24 CN CN202010962639.3A patent/CN112521366A/zh active Pending
- 2010-11-24 PE PE2016002590A patent/PE20171255A1/es unknown
- 2010-11-24 RS RS20160645A patent/RS54978B1/sr unknown
- 2010-11-24 KR KR1020187012588A patent/KR20180049255A/ko not_active IP Right Cessation
- 2010-11-24 HU HUE16175117A patent/HUE045784T2/hu unknown
- 2010-11-24 DK DK16175117T patent/DK3133070T3/da active
- 2010-11-24 MX MX2012006083A patent/MX358345B/es active IP Right Grant
- 2010-11-24 EP EP14164650.5A patent/EP2796457B1/en active Active
- 2010-11-24 ES ES19182732T patent/ES2875382T3/es active Active
- 2010-11-24 PT PT161751177T patent/PT3133070T/pt unknown
- 2010-11-24 MY MYPI2019004988A patent/MY192644A/en unknown
- 2010-11-24 TW TW105126084A patent/TWI606827B/zh active
- 2010-11-24 NZ NZ625712A patent/NZ625712A/en unknown
- 2010-11-24 CN CN201610219151.5A patent/CN105777707B/zh active Active
- 2010-11-24 TW TW106124135A patent/TWI656873B/zh active
- 2010-11-24 WO PCT/US2010/057952 patent/WO2011066352A1/en active Application Filing
- 2010-11-24 AR ARP100104348A patent/AR079152A1/es not_active Application Discontinuation
- 2010-11-24 MY MYPI2012002297A patent/MY160542A/en unknown
- 2010-11-24 EA EA201592195A patent/EA029990B1/ru not_active IP Right Cessation
- 2010-11-24 PE PE2012000717A patent/PE20121337A1/es not_active Application Discontinuation
- 2010-11-24 EP EP16175117.7A patent/EP3133070B1/en active Active
- 2010-11-24 ME MEP-2016-163A patent/ME02477B/me unknown
- 2010-11-24 KR KR1020127016671A patent/KR20120115972A/ko active Search and Examination
- 2010-11-24 DK DK14164650.5T patent/DK2796457T3/en active
- 2010-11-24 ES ES16175117T patent/ES2754398T3/es active Active
- 2010-11-24 DK DK10785289.9T patent/DK2504332T4/da active
- 2010-11-24 ES ES10785289T patent/ES2493940T5/es active Active
- 2010-11-24 EP EP10785289.9A patent/EP2504332B2/en active Active
- 2010-11-24 SI SI201031936T patent/SI3133070T1/sl unknown
- 2010-11-24 KR KR1020227040651A patent/KR20220162824A/ko not_active Application Discontinuation
- 2010-11-24 SI SI201031252A patent/SI2796457T1/sl unknown
- 2010-11-24 AU AU2010324810A patent/AU2010324810B2/en active Active
- 2010-11-24 EP EP21160074.7A patent/EP3896069A1/en active Pending
- 2010-11-24 KR KR1020207002751A patent/KR20200013105A/ko not_active IP Right Cessation
- 2010-11-24 PT PT141646505T patent/PT2796457T/pt unknown
- 2010-11-24 RS RS20191404A patent/RS59543B1/sr unknown
- 2010-11-24 CN CN201080061535.XA patent/CN102712629B/zh active Active
- 2010-11-24 KR KR1020157030851A patent/KR102073207B1/ko active IP Right Grant
-
2012
- 2012-05-18 TN TNP2012000237A patent/TN2012000237A1/en unknown
- 2012-05-20 IL IL219892A patent/IL219892A0/en unknown
- 2012-05-23 DO DO2012000141A patent/DOP2012000141A/es unknown
- 2012-05-24 EC ECSP12011926 patent/ECSP12011926A/es unknown
- 2012-05-24 CL CL2012001348A patent/CL2012001348A1/es unknown
- 2012-05-24 CR CR20120277A patent/CR20120277A/es unknown
- 2012-05-24 NI NI201200096A patent/NI201200096A/es unknown
- 2012-05-25 GT GT201200161A patent/GT201200161A/es unknown
- 2012-06-20 MA MA34989A patent/MA33838B1/fr unknown
- 2012-12-14 HK HK12112977.8A patent/HK1172031A1/xx unknown
- 2012-12-14 HK HK15103822.1A patent/HK1203485A1/zh unknown
-
2014
- 2014-08-18 HR HRP20140780TT patent/HRP20140780T4/hr unknown
- 2014-08-28 CY CY20141100690T patent/CY1115880T1/el unknown
-
2015
- 2015-07-06 JP JP2015134942A patent/JP6370264B2/ja active Active
- 2015-11-02 PH PH12015502514A patent/PH12015502514A1/en unknown
-
2016
- 2016-01-13 US US14/994,489 patent/US20160120842A1/en not_active Abandoned
- 2016-02-22 US US15/049,946 patent/US10888547B2/en active Active
- 2016-03-04 JP JP2016041843A patent/JP6452635B2/ja active Active
- 2016-04-22 AU AU2016202591A patent/AU2016202591B2/en active Active
- 2016-08-11 SM SM201600273T patent/SMT201600273B/it unknown
- 2016-08-17 CY CY20161100807T patent/CY1117996T1/el unknown
- 2016-08-17 HR HRP20161038TT patent/HRP20161038T1/hr unknown
- 2016-09-19 DO DO2016000250A patent/DOP2016000250A/es unknown
- 2016-10-12 CL CL2016002589A patent/CL2016002589A1/es unknown
-
2017
- 2017-11-27 AU AU2017265180A patent/AU2017265180B2/en active Active
-
2018
- 2018-06-27 IL IL260299A patent/IL260299A/en unknown
- 2018-08-23 EC ECSENADI201863798A patent/ECSP18063798A/es unknown
- 2018-09-07 JP JP2018167432A patent/JP7150528B2/ja active Active
-
2019
- 2019-09-12 HR HRP20191647 patent/HRP20191647T1/hr unknown
- 2019-11-13 CY CY20191101196T patent/CY1122698T1/el unknown
-
2020
- 2020-08-14 JP JP2020136889A patent/JP2020189873A/ja active Pending
-
2021
- 2021-01-07 US US17/143,821 patent/US20210369672A1/en active Pending
- 2021-03-18 AR ARP210100682A patent/AR121611A2/es unknown
- 2021-03-18 AR ARP210100683A patent/AR121612A2/es unknown
- 2021-06-13 IL IL283935A patent/IL283935A/en unknown
- 2021-09-03 US US17/465,994 patent/US20210393590A1/en not_active Abandoned
-
2022
- 2022-07-14 US US17/865,195 patent/US20230172903A1/en active Pending
-
2023
- 2023-06-21 JP JP2023101301A patent/JP2023116764A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20210369672A1 (en) | Inhibitors of glucosylceramide synthase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A107 | Divisional application of patent | ||
| AMND | Amendment | ||
| E902 | Notification of reason for refusal | ||
| AMND | Amendment | ||
| E601 | Decision to refuse application | ||
| AMND | Amendment | ||
| E902 | Notification of reason for refusal | ||
| AMND | Amendment | ||
| X601 | Decision of rejection after re-examination | ||
| J201 | Request for trial against refusal decision | ||
| J301 | Trial decision |
Free format text: TRIAL NUMBER: 2020101002036; TRIAL DECISION FOR APPEAL AGAINST DECISION TO DECLINE REFUSAL REQUESTED 20200814 Effective date: 20210809 |