JP7050747B2 - 低分子量芳香族リグニン由来化合物を製造するための方法 - Google Patents
低分子量芳香族リグニン由来化合物を製造するための方法 Download PDFInfo
- Publication number
- JP7050747B2 JP7050747B2 JP2019503620A JP2019503620A JP7050747B2 JP 7050747 B2 JP7050747 B2 JP 7050747B2 JP 2019503620 A JP2019503620 A JP 2019503620A JP 2019503620 A JP2019503620 A JP 2019503620A JP 7050747 B2 JP7050747 B2 JP 7050747B2
- Authority
- JP
- Japan
- Prior art keywords
- branched
- linear
- arbitrarily substituted
- lignin
- derived
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims description 598
- 150000001875 compounds Chemical class 0.000 title claims description 212
- 125000003118 aryl group Chemical group 0.000 title claims description 131
- 229920005610 lignin Polymers 0.000 claims description 484
- 230000008569 process Effects 0.000 claims description 352
- 238000004537 pulping Methods 0.000 claims description 112
- -1 hydroxy, carboxy Chemical group 0.000 claims description 100
- 238000005336 cracking Methods 0.000 claims description 79
- 238000007254 oxidation reaction Methods 0.000 claims description 78
- 230000003647 oxidation Effects 0.000 claims description 72
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 claims description 70
- 125000003545 alkoxy group Chemical group 0.000 claims description 69
- 239000012978 lignocellulosic material Substances 0.000 claims description 58
- 239000000047 product Substances 0.000 claims description 58
- 125000000217 alkyl group Chemical group 0.000 claims description 54
- 229910052739 hydrogen Inorganic materials 0.000 claims description 54
- 238000004519 manufacturing process Methods 0.000 claims description 53
- 239000001257 hydrogen Substances 0.000 claims description 52
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 51
- AZQWKYJCGOJGHM-UHFFFAOYSA-N para-benzoquinone Natural products O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 claims description 50
- 239000002023 wood Substances 0.000 claims description 48
- 150000001299 aldehydes Chemical class 0.000 claims description 45
- 238000002955 isolation Methods 0.000 claims description 45
- 241000196324 Embryophyta Species 0.000 claims description 40
- 238000001914 filtration Methods 0.000 claims description 40
- 238000006722 reduction reaction Methods 0.000 claims description 40
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N 1,4-Benzenediol Natural products OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 claims description 38
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 37
- 230000009467 reduction Effects 0.000 claims description 37
- 229920002678 cellulose Polymers 0.000 claims description 36
- 239000001913 cellulose Substances 0.000 claims description 36
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical group C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 claims description 34
- 238000000354 decomposition reaction Methods 0.000 claims description 33
- 125000004043 oxo group Chemical group O=* 0.000 claims description 33
- 238000000926 separation method Methods 0.000 claims description 33
- 229920002488 Hemicellulose Polymers 0.000 claims description 32
- 150000002148 esters Chemical class 0.000 claims description 30
- 238000000605 extraction Methods 0.000 claims description 30
- 150000003839 salts Chemical class 0.000 claims description 30
- 238000000108 ultra-filtration Methods 0.000 claims description 29
- 125000004103 aminoalkyl group Chemical group 0.000 claims description 28
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 28
- 125000004181 carboxyalkyl group Chemical group 0.000 claims description 28
- 230000001590 oxidative effect Effects 0.000 claims description 28
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 27
- 238000001728 nano-filtration Methods 0.000 claims description 27
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical group [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 26
- 239000007864 aqueous solution Substances 0.000 claims description 26
- 239000002815 homogeneous catalyst Substances 0.000 claims description 26
- 238000007363 ring formation reaction Methods 0.000 claims description 26
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 25
- 125000000524 functional group Chemical group 0.000 claims description 24
- 229910052751 metal Inorganic materials 0.000 claims description 24
- 239000002184 metal Substances 0.000 claims description 24
- 239000012634 fragment Substances 0.000 claims description 23
- 239000002638 heterogeneous catalyst Substances 0.000 claims description 23
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 claims description 21
- 229910052752 metalloid Inorganic materials 0.000 claims description 21
- 235000010290 biphenyl Nutrition 0.000 claims description 20
- 238000001556 precipitation Methods 0.000 claims description 20
- 150000001298 alcohols Chemical class 0.000 claims description 19
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 18
- 239000004305 biphenyl Substances 0.000 claims description 18
- 239000003795 chemical substances by application Substances 0.000 claims description 18
- 150000002431 hydrogen Chemical group 0.000 claims description 18
- 150000002738 metalloids Chemical class 0.000 claims description 18
- 238000000746 purification Methods 0.000 claims description 18
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 17
- 238000010411 cooking Methods 0.000 claims description 17
- 229910052736 halogen Inorganic materials 0.000 claims description 17
- 150000002367 halogens Chemical class 0.000 claims description 17
- 230000002378 acidificating effect Effects 0.000 claims description 16
- 239000007800 oxidant agent Substances 0.000 claims description 16
- 125000005499 phosphonyl group Chemical group 0.000 claims description 16
- 229910021645 metal ion Inorganic materials 0.000 claims description 15
- 238000006467 substitution reaction Methods 0.000 claims description 15
- UUEVFMOUBSLVJW-UHFFFAOYSA-N oxo-[[1-[2-[2-[2-[4-(oxoazaniumylmethylidene)pyridin-1-yl]ethoxy]ethoxy]ethyl]pyridin-4-ylidene]methyl]azanium;dibromide Chemical compound [Br-].[Br-].C1=CC(=C[NH+]=O)C=CN1CCOCCOCCN1C=CC(=C[NH+]=O)C=C1 UUEVFMOUBSLVJW-UHFFFAOYSA-N 0.000 claims description 14
- 238000001212 derivatisation Methods 0.000 claims description 13
- 241000218657 Picea Species 0.000 claims description 12
- 238000002144 chemical decomposition reaction Methods 0.000 claims description 12
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 11
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 11
- 239000003929 acidic solution Substances 0.000 claims description 11
- 150000001768 cations Chemical class 0.000 claims description 11
- KLIDCXVFHGNTTM-UHFFFAOYSA-N 2,6-dimethoxyphenol Chemical compound COC1=CC=CC(OC)=C1O KLIDCXVFHGNTTM-UHFFFAOYSA-N 0.000 claims description 10
- 150000001412 amines Chemical class 0.000 claims description 10
- PYKYMHQGRFAEBM-UHFFFAOYSA-N anthraquinone Natural products CCC(=O)c1c(O)c2C(=O)C3C(C=CC=C3O)C(=O)c2cc1CC(=O)OC PYKYMHQGRFAEBM-UHFFFAOYSA-N 0.000 claims description 10
- 239000012071 phase Substances 0.000 claims description 10
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 10
- 238000005863 Friedel-Crafts acylation reaction Methods 0.000 claims description 9
- 239000012530 fluid Substances 0.000 claims description 9
- 229910052759 nickel Inorganic materials 0.000 claims description 9
- 238000005406 washing Methods 0.000 claims description 9
- 239000011575 calcium Substances 0.000 claims description 8
- 239000003638 chemical reducing agent Substances 0.000 claims description 8
- LHGVFZTZFXWLCP-UHFFFAOYSA-N guaiacol Chemical compound COC1=CC=CC=C1O LHGVFZTZFXWLCP-UHFFFAOYSA-N 0.000 claims description 8
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 7
- 229910052791 calcium Inorganic materials 0.000 claims description 7
- 239000007795 chemical reaction product Substances 0.000 claims description 7
- 125000005647 linker group Chemical group 0.000 claims description 7
- MWOOGOJBHIARFG-UHFFFAOYSA-N vanillin Chemical compound COC1=CC(C=O)=CC=C1O MWOOGOJBHIARFG-UHFFFAOYSA-N 0.000 claims description 7
- RGHHSNMVTDWUBI-UHFFFAOYSA-N 4-hydroxybenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1 RGHHSNMVTDWUBI-UHFFFAOYSA-N 0.000 claims description 6
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 claims description 6
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 claims description 6
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 claims description 6
- RRAFCDWBNXTKKO-UHFFFAOYSA-N eugenol Chemical compound COC1=CC(CC=C)=CC=C1O RRAFCDWBNXTKKO-UHFFFAOYSA-N 0.000 claims description 6
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 claims description 6
- 229910052707 ruthenium Inorganic materials 0.000 claims description 6
- 239000011734 sodium Substances 0.000 claims description 6
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 6
- 235000018185 Betula X alpestris Nutrition 0.000 claims description 5
- 235000018212 Betula X uliginosa Nutrition 0.000 claims description 5
- 240000000731 Fagus sylvatica Species 0.000 claims description 5
- 235000010099 Fagus sylvatica Nutrition 0.000 claims description 5
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 claims description 5
- 238000007664 blowing Methods 0.000 claims description 5
- 239000011777 magnesium Substances 0.000 claims description 5
- 229910052763 palladium Inorganic materials 0.000 claims description 5
- 150000002989 phenols Chemical class 0.000 claims description 5
- 229910052697 platinum Inorganic materials 0.000 claims description 5
- 238000007873 sieving Methods 0.000 claims description 5
- 239000000377 silicon dioxide Substances 0.000 claims description 5
- 235000012141 vanillin Nutrition 0.000 claims description 5
- FGQOOHJZONJGDT-UHFFFAOYSA-N vanillin Natural products COC1=CC(O)=CC(C=O)=C1 FGQOOHJZONJGDT-UHFFFAOYSA-N 0.000 claims description 5
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 claims description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 claims description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 4
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 claims description 4
- 229960001867 guaiacol Drugs 0.000 claims description 4
- 229910052749 magnesium Inorganic materials 0.000 claims description 4
- BVJSUAQZOZWCKN-UHFFFAOYSA-N p-hydroxybenzyl alcohol Chemical class OCC1=CC=C(O)C=C1 BVJSUAQZOZWCKN-UHFFFAOYSA-N 0.000 claims description 4
- 239000011347 resin Substances 0.000 claims description 4
- 229920005989 resin Polymers 0.000 claims description 4
- 229910052708 sodium Inorganic materials 0.000 claims description 4
- 239000005711 Benzoic acid Substances 0.000 claims description 3
- NPBVQXIMTZKSBA-UHFFFAOYSA-N Chavibetol Natural products COC1=CC=C(CC=C)C=C1O NPBVQXIMTZKSBA-UHFFFAOYSA-N 0.000 claims description 3
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 claims description 3
- 239000005770 Eugenol Substances 0.000 claims description 3
- UVMRYBDEERADNV-UHFFFAOYSA-N Pseudoeugenol Natural products COC1=CC(C(C)=C)=CC=C1O UVMRYBDEERADNV-UHFFFAOYSA-N 0.000 claims description 3
- 235000010233 benzoic acid Nutrition 0.000 claims description 3
- 239000012876 carrier material Substances 0.000 claims description 3
- 238000004821 distillation Methods 0.000 claims description 3
- 238000007336 electrophilic substitution reaction Methods 0.000 claims description 3
- 229960002217 eugenol Drugs 0.000 claims description 3
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 claims description 3
- 229910052737 gold Inorganic materials 0.000 claims description 3
- 239000010931 gold Substances 0.000 claims description 3
- 238000005342 ion exchange Methods 0.000 claims description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 3
- 238000002414 normal-phase solid-phase extraction Methods 0.000 claims description 3
- 238000005935 nucleophilic addition reaction Methods 0.000 claims description 3
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 claims description 3
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 claims description 3
- 238000011045 prefiltration Methods 0.000 claims description 3
- KCDXJAYRVLXPFO-UHFFFAOYSA-N syringaldehyde Chemical compound COC1=CC(C=O)=CC(OC)=C1O KCDXJAYRVLXPFO-UHFFFAOYSA-N 0.000 claims description 3
- COBXDAOIDYGHGK-UHFFFAOYSA-N syringaldehyde Natural products COC1=CC=C(C=O)C(OC)=C1O COBXDAOIDYGHGK-UHFFFAOYSA-N 0.000 claims description 3
- 229940117960 vanillin Drugs 0.000 claims description 3
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 claims description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 claims description 2
- 235000008331 Pinus X rigitaeda Nutrition 0.000 claims description 2
- 241000018646 Pinus brutia Species 0.000 claims description 2
- 235000011613 Pinus brutia Nutrition 0.000 claims description 2
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 claims description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 claims description 2
- 159000000007 calcium salts Chemical class 0.000 claims description 2
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 claims description 2
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 claims description 2
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 claims description 2
- 125000005017 substituted alkenyl group Chemical group 0.000 claims 13
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims 1
- 244000166124 Eucalyptus globulus Species 0.000 claims 1
- 239000003054 catalyst Substances 0.000 description 99
- 238000006243 chemical reaction Methods 0.000 description 75
- 239000000463 material Substances 0.000 description 64
- 239000000243 solution Substances 0.000 description 64
- 229920001732 Lignosulfonate Polymers 0.000 description 55
- 239000000123 paper Substances 0.000 description 48
- 239000007858 starting material Substances 0.000 description 42
- 239000000203 mixture Substances 0.000 description 38
- 239000000178 monomer Substances 0.000 description 38
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 37
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 36
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 35
- 235000002639 sodium chloride Nutrition 0.000 description 32
- 229920000642 polymer Polymers 0.000 description 31
- 239000000126 substance Substances 0.000 description 31
- 229920005611 kraft lignin Polymers 0.000 description 28
- 239000000539 dimer Substances 0.000 description 26
- 239000000376 reactant Substances 0.000 description 24
- 239000002904 solvent Substances 0.000 description 23
- 239000002655 kraft paper Substances 0.000 description 22
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- 210000004027 cell Anatomy 0.000 description 20
- 239000007787 solid Substances 0.000 description 20
- 239000012528 membrane Substances 0.000 description 19
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 18
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 18
- 239000007788 liquid Substances 0.000 description 18
- 125000003342 alkenyl group Chemical group 0.000 description 17
- 239000006227 byproduct Substances 0.000 description 16
- 238000006731 degradation reaction Methods 0.000 description 15
- 238000006056 electrooxidation reaction Methods 0.000 description 15
- 230000015556 catabolic process Effects 0.000 description 14
- 239000010949 copper Substances 0.000 description 14
- 125000004122 cyclic group Chemical group 0.000 description 14
- 229910052760 oxygen Inorganic materials 0.000 description 14
- 229910052799 carbon Inorganic materials 0.000 description 13
- 239000003960 organic solvent Substances 0.000 description 13
- 239000001301 oxygen Substances 0.000 description 13
- 238000011084 recovery Methods 0.000 description 13
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 12
- 238000002485 combustion reaction Methods 0.000 description 12
- 239000002270 dispersing agent Substances 0.000 description 12
- 239000000543 intermediate Substances 0.000 description 12
- 150000002500 ions Chemical class 0.000 description 12
- 239000011572 manganese Substances 0.000 description 11
- 238000000197 pyrolysis Methods 0.000 description 11
- 229910052717 sulfur Inorganic materials 0.000 description 11
- 235000001508 sulfur Nutrition 0.000 description 11
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 10
- 150000004056 anthraquinones Chemical class 0.000 description 10
- 239000000835 fiber Substances 0.000 description 10
- 238000005470 impregnation Methods 0.000 description 10
- 239000000344 soap Substances 0.000 description 10
- 239000011593 sulfur Substances 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 229920003043 Cellulose fiber Polymers 0.000 description 9
- VTLYFUHAOXGGBS-UHFFFAOYSA-N Fe3+ Chemical compound [Fe+3] VTLYFUHAOXGGBS-UHFFFAOYSA-N 0.000 description 9
- 239000002608 ionic liquid Substances 0.000 description 9
- LAQYHRQFABOIFD-UHFFFAOYSA-N methoxyhydroquinone Natural products COC1=CC(O)=CC=C1O LAQYHRQFABOIFD-UHFFFAOYSA-N 0.000 description 9
- 239000002243 precursor Substances 0.000 description 9
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- 125000004429 atom Chemical group 0.000 description 8
- 238000006555 catalytic reaction Methods 0.000 description 8
- 239000000460 chlorine Substances 0.000 description 8
- 239000003792 electrolyte Substances 0.000 description 8
- 239000000446 fuel Substances 0.000 description 8
- 239000007789 gas Substances 0.000 description 8
- 238000012545 processing Methods 0.000 description 8
- 150000004763 sulfides Chemical class 0.000 description 8
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 7
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 7
- 238000005516 engineering process Methods 0.000 description 7
- 238000004880 explosion Methods 0.000 description 7
- 239000002657 fibrous material Substances 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 235000019357 lignosulphonate Nutrition 0.000 description 7
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 7
- 150000004053 quinones Chemical class 0.000 description 7
- 125000001424 substituent group Chemical group 0.000 description 7
- 238000006277 sulfonation reaction Methods 0.000 description 7
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 7
- 239000010457 zeolite Substances 0.000 description 7
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 6
- 238000005698 Diels-Alder reaction Methods 0.000 description 6
- 239000012670 alkaline solution Substances 0.000 description 6
- 239000012298 atmosphere Substances 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 230000003197 catalytic effect Effects 0.000 description 6
- 230000004048 modification Effects 0.000 description 6
- 238000012986 modification Methods 0.000 description 6
- 239000002245 particle Substances 0.000 description 6
- 239000005077 polysulfide Substances 0.000 description 6
- 229920001021 polysulfide Polymers 0.000 description 6
- 150000008117 polysulfides Polymers 0.000 description 6
- 150000003384 small molecules Chemical class 0.000 description 6
- AKEJUJNQAAGONA-UHFFFAOYSA-N sulfur trioxide Chemical compound O=S(=O)=O AKEJUJNQAAGONA-UHFFFAOYSA-N 0.000 description 6
- 125000003396 thiol group Chemical group [H]S* 0.000 description 6
- JSHLOPGSDZTEGQ-UHFFFAOYSA-N 3-methoxy-4-phenylmethoxybenzaldehyde Chemical compound COC1=CC(C=O)=CC=C1OCC1=CC=CC=C1 JSHLOPGSDZTEGQ-UHFFFAOYSA-N 0.000 description 5
- JGMBQAGNZLBZCE-UHFFFAOYSA-N 3-methoxy-4-phenylmethoxybenzoic acid Chemical compound COC1=CC(C(O)=O)=CC=C1OCC1=CC=CC=C1 JGMBQAGNZLBZCE-UHFFFAOYSA-N 0.000 description 5
- 239000002028 Biomass Substances 0.000 description 5
- 239000004117 Lignosulphonate Substances 0.000 description 5
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 5
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 5
- 125000001931 aliphatic group Chemical group 0.000 description 5
- 125000002619 bicyclic group Chemical group 0.000 description 5
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 5
- 239000000920 calcium hydroxide Substances 0.000 description 5
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 5
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical compound C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 description 5
- 239000007857 degradation product Substances 0.000 description 5
- 238000005868 electrolysis reaction Methods 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 5
- 229910052742 iron Inorganic materials 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- 150000002894 organic compounds Chemical class 0.000 description 5
- PXIKRTCSSLJURC-UHFFFAOYSA-N p-propylguaiacol Natural products CCCC1=CC=C(O)C(OC)=C1 PXIKRTCSSLJURC-UHFFFAOYSA-N 0.000 description 5
- 239000011148 porous material Substances 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 230000000717 retained effect Effects 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- GXAVBFNRWXCOPY-UHFFFAOYSA-N 1,4-dihydroxy-2,6-dimethoxybenzene Chemical compound COC1=CC(O)=CC(OC)=C1O GXAVBFNRWXCOPY-UHFFFAOYSA-N 0.000 description 4
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 4
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 4
- RAHZWNYVWXNFOC-UHFFFAOYSA-N Sulphur dioxide Chemical compound O=S=O RAHZWNYVWXNFOC-UHFFFAOYSA-N 0.000 description 4
- 229910010413 TiO 2 Inorganic materials 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 238000005917 acylation reaction Methods 0.000 description 4
- 150000001336 alkenes Chemical class 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical compound C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 4
- 150000001491 aromatic compounds Chemical class 0.000 description 4
- 229910052796 boron Inorganic materials 0.000 description 4
- 238000004517 catalytic hydrocracking Methods 0.000 description 4
- 210000002421 cell wall Anatomy 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 238000004140 cleaning Methods 0.000 description 4
- 238000003776 cleavage reaction Methods 0.000 description 4
- 238000009833 condensation Methods 0.000 description 4
- 230000005494 condensation Effects 0.000 description 4
- 150000004696 coordination complex Chemical class 0.000 description 4
- 229910052802 copper Inorganic materials 0.000 description 4
- 238000009826 distribution Methods 0.000 description 4
- 230000005611 electricity Effects 0.000 description 4
- 238000005265 energy consumption Methods 0.000 description 4
- 239000012467 final product Substances 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 239000011121 hardwood Substances 0.000 description 4
- 239000000833 heterodimer Substances 0.000 description 4
- 125000002950 monocyclic group Chemical group 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 150000004032 porphyrins Chemical class 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 239000010948 rhodium Substances 0.000 description 4
- 230000007017 scission Effects 0.000 description 4
- 229910052938 sodium sulfate Inorganic materials 0.000 description 4
- 235000011152 sodium sulphate Nutrition 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 description 3
- 150000005208 1,4-dihydroxybenzenes Chemical class 0.000 description 3
- VEUMANXWQDHAJV-UHFFFAOYSA-N 2-[2-[(2-hydroxyphenyl)methylideneamino]ethyliminomethyl]phenol Chemical compound OC1=CC=CC=C1C=NCCN=CC1=CC=CC=C1O VEUMANXWQDHAJV-UHFFFAOYSA-N 0.000 description 3
- VRJLWFYHYPQLLO-UHFFFAOYSA-N 3-methoxy-4-phenylmethoxybenzoyl chloride Chemical compound COC1=CC(C(Cl)=O)=CC=C1OCC1=CC=CC=C1 VRJLWFYHYPQLLO-UHFFFAOYSA-N 0.000 description 3
- YHEWWEXPVKCVFY-UHFFFAOYSA-N 4-propylsyringol Natural products CCCC1=CC(OC)=C(O)C(OC)=C1 YHEWWEXPVKCVFY-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 3
- 235000008733 Citrus aurantifolia Nutrition 0.000 description 3
- 241000218631 Coniferophyta Species 0.000 description 3
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- YNPNZTXNASCQKK-UHFFFAOYSA-N Phenanthrene Natural products C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 229920001131 Pulp (paper) Polymers 0.000 description 3
- 235000011941 Tilia x europaea Nutrition 0.000 description 3
- 229920002522 Wood fibre Polymers 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 150000001263 acyl chlorides Chemical class 0.000 description 3
- 230000010933 acylation Effects 0.000 description 3
- 238000007259 addition reaction Methods 0.000 description 3
- 239000003513 alkali Substances 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 150000001450 anions Chemical class 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cis-cyclohexene Natural products C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 3
- VZHHNBNSMNNUAD-UHFFFAOYSA-N cobalt 2-[2-[(2-hydroxyphenyl)methylideneamino]ethyliminomethyl]phenol Chemical compound [Co].OC1=CC=CC=C1C=NCCN=CC1=CC=CC=C1O VZHHNBNSMNNUAD-UHFFFAOYSA-N 0.000 description 3
- 239000008139 complexing agent Substances 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 239000012847 fine chemical Substances 0.000 description 3
- 238000005194 fractionation Methods 0.000 description 3
- 239000000710 homodimer Substances 0.000 description 3
- 239000004571 lime Substances 0.000 description 3
- 238000006011 modification reaction Methods 0.000 description 3
- 229910052750 molybdenum Inorganic materials 0.000 description 3
- 150000002791 naphthoquinones Chemical class 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- 150000002825 nitriles Chemical class 0.000 description 3
- 230000000269 nucleophilic effect Effects 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 125000003367 polycyclic group Chemical group 0.000 description 3
- 238000006116 polymerization reaction Methods 0.000 description 3
- 229920001282 polysaccharide Polymers 0.000 description 3
- 239000005017 polysaccharide Substances 0.000 description 3
- 150000004804 polysaccharides Chemical class 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 229910052703 rhodium Inorganic materials 0.000 description 3
- 239000011122 softwood Substances 0.000 description 3
- 238000003797 solvolysis reaction Methods 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 230000019635 sulfation Effects 0.000 description 3
- 238000005670 sulfation reaction Methods 0.000 description 3
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 3
- 239000004753 textile Substances 0.000 description 3
- 238000005979 thermal decomposition reaction Methods 0.000 description 3
- 229910052723 transition metal Inorganic materials 0.000 description 3
- 239000002699 waste material Substances 0.000 description 3
- 239000002025 wood fiber Substances 0.000 description 3
- TXBDGLVJCSOBLF-UHFFFAOYSA-N 1-(2,5-dihydroxy-4-methoxyphenyl)ethanone Chemical compound COC1=CC(O)=C(C(C)=O)C=C1O TXBDGLVJCSOBLF-UHFFFAOYSA-N 0.000 description 2
- OLBNOBQOQZRLMP-UHFFFAOYSA-N 2,6-dimethoxy-p-benzoquinone Chemical compound COC1=CC(=O)C=C(OC)C1=O OLBNOBQOQZRLMP-UHFFFAOYSA-N 0.000 description 2
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 2
- CHWNEIVBYREQRF-UHFFFAOYSA-N 4-Ethyl-2-methoxyphenol Chemical compound CCC1=CC=C(O)C(OC)=C1 CHWNEIVBYREQRF-UHFFFAOYSA-N 0.000 description 2
- HXDOZKJGKXYMEW-UHFFFAOYSA-N 4-ethylphenol Chemical compound CCC1=CC=C(O)C=C1 HXDOZKJGKXYMEW-UHFFFAOYSA-N 0.000 description 2
- PCFMUWBCZZUMRX-UHFFFAOYSA-N 9,10-Dihydroxyanthracene Chemical compound C1=CC=C2C(O)=C(C=CC=C3)C3=C(O)C2=C1 PCFMUWBCZZUMRX-UHFFFAOYSA-N 0.000 description 2
- RSWGJHLUYNHPMX-UHFFFAOYSA-N Abietic-Saeure Natural products C12CCC(C(C)C)=CC2=CCC2C1(C)CCCC2(C)C(O)=O RSWGJHLUYNHPMX-UHFFFAOYSA-N 0.000 description 2
- 241000609240 Ambelania acida Species 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- 229930192627 Naphthoquinone Natural products 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- KHPCPRHQVVSZAH-HUOMCSJISA-N Rosin Natural products O(C/C=C/c1ccccc1)[C@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 KHPCPRHQVVSZAH-HUOMCSJISA-N 0.000 description 2
- 239000002262 Schiff base Substances 0.000 description 2
- 150000004753 Schiff bases Chemical class 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 241000222645 Trametes cinnabarina Species 0.000 description 2
- 230000021736 acetylation Effects 0.000 description 2
- 238000006640 acetylation reaction Methods 0.000 description 2
- 239000011149 active material Substances 0.000 description 2
- 150000001266 acyl halides Chemical class 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 239000012736 aqueous medium Substances 0.000 description 2
- 239000003125 aqueous solvent Substances 0.000 description 2
- 239000010905 bagasse Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 230000027455 binding Effects 0.000 description 2
- 238000009739 binding Methods 0.000 description 2
- WTEOIRVLGSZEPR-UHFFFAOYSA-N boron trifluoride Chemical compound FB(F)F WTEOIRVLGSZEPR-UHFFFAOYSA-N 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- RYAGRZNBULDMBW-UHFFFAOYSA-L calcium;3-(2-hydroxy-3-methoxyphenyl)-2-[2-methoxy-4-(3-sulfonatopropyl)phenoxy]propane-1-sulfonate Chemical compound [Ca+2].COC1=CC=CC(CC(CS([O-])(=O)=O)OC=2C(=CC(CCCS([O-])(=O)=O)=CC=2)OC)=C1O RYAGRZNBULDMBW-UHFFFAOYSA-L 0.000 description 2
- 125000002837 carbocyclic group Chemical group 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000001569 carbon dioxide Substances 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000013626 chemical specie Substances 0.000 description 2
- 239000003245 coal Substances 0.000 description 2
- 239000004567 concrete Substances 0.000 description 2
- JMFRWRFFLBVWSI-NSCUHMNNSA-N coniferol Chemical group COC1=CC(\C=C\CO)=CC=C1O JMFRWRFFLBVWSI-NSCUHMNNSA-N 0.000 description 2
- 125000000753 cycloalkyl group Chemical group 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- 230000001066 destructive effect Effects 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 150000001993 dienes Chemical class 0.000 description 2
- 238000010494 dissociation reaction Methods 0.000 description 2
- 230000005593 dissociations Effects 0.000 description 2
- 239000012153 distilled water Substances 0.000 description 2
- 230000007515 enzymatic degradation Effects 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 230000005484 gravity Effects 0.000 description 2
- 238000000227 grinding Methods 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 238000007172 homogeneous catalysis Methods 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 150000002466 imines Chemical class 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 239000011261 inert gas Substances 0.000 description 2
- 229910010272 inorganic material Inorganic materials 0.000 description 2
- 239000011147 inorganic material Substances 0.000 description 2
- 230000010354 integration Effects 0.000 description 2
- TWBYWOBDOCUKOW-UHFFFAOYSA-N isonicotinic acid Chemical compound OC(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-N 0.000 description 2
- 239000011968 lewis acid catalyst Substances 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 229920002521 macromolecule Polymers 0.000 description 2
- 229910052748 manganese Inorganic materials 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- 150000002772 monosaccharides Chemical class 0.000 description 2
- PCILLCXFKWDRMK-UHFFFAOYSA-N naphthalene-1,4-diol Chemical compound C1=CC=C2C(O)=CC=C(O)C2=C1 PCILLCXFKWDRMK-UHFFFAOYSA-N 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 239000011368 organic material Substances 0.000 description 2
- 239000003348 petrochemical agent Substances 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 238000007539 photo-oxidation reaction Methods 0.000 description 2
- 239000003495 polar organic solvent Substances 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 238000010248 power generation Methods 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 230000008707 rearrangement Effects 0.000 description 2
- 230000002468 redox effect Effects 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 2
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 235000017550 sodium carbonate Nutrition 0.000 description 2
- FQENQNTWSFEDLI-UHFFFAOYSA-J sodium diphosphate Chemical compound [Na+].[Na+].[Na+].[Na+].[O-]P([O-])(=O)OP([O-])([O-])=O FQENQNTWSFEDLI-UHFFFAOYSA-J 0.000 description 2
- CSMWJXBSXGUPGY-UHFFFAOYSA-L sodium dithionate Chemical compound [Na+].[Na+].[O-]S(=O)(=O)S([O-])(=O)=O CSMWJXBSXGUPGY-UHFFFAOYSA-L 0.000 description 2
- 229940075931 sodium dithionate Drugs 0.000 description 2
- 229910052979 sodium sulfide Inorganic materials 0.000 description 2
- GRVFOGOEDUUMBP-UHFFFAOYSA-N sodium sulfide (anhydrous) Chemical compound [Na+].[Na+].[S-2] GRVFOGOEDUUMBP-UHFFFAOYSA-N 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-L sulfite Chemical class [O-]S([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-L 0.000 description 2
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 2
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 description 2
- 239000003784 tall oil Substances 0.000 description 2
- 235000019818 tetrasodium diphosphate Nutrition 0.000 description 2
- KHPCPRHQVVSZAH-UHFFFAOYSA-N trans-cinnamyl beta-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OCC=CC1=CC=CC=C1 KHPCPRHQVVSZAH-UHFFFAOYSA-N 0.000 description 2
- LZFOPEXOUVTGJS-ONEGZZNKSA-N trans-sinapyl alcohol Chemical compound COC1=CC(\C=C\CO)=CC(OC)=C1O LZFOPEXOUVTGJS-ONEGZZNKSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 150000003624 transition metals Chemical class 0.000 description 2
- 230000007306 turnover Effects 0.000 description 2
- 239000003039 volatile agent Substances 0.000 description 2
- LUEWUZLMQUOBSB-FSKGGBMCSA-N (2s,3s,4s,5s,6r)-2-[(2r,3s,4r,5r,6s)-6-[(2r,3s,4r,5s,6s)-4,5-dihydroxy-2-(hydroxymethyl)-6-[(2r,4r,5s,6r)-4,5,6-trihydroxy-2-(hydroxymethyl)oxan-3-yl]oxyoxan-3-yl]oxy-4,5-dihydroxy-2-(hydroxymethyl)oxan-3-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound O[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@@H](O[C@@H]2[C@H](O[C@@H](OC3[C@H](O[C@@H](O)[C@@H](O)[C@H]3O)CO)[C@@H](O)[C@H]2O)CO)[C@H](O)[C@H]1O LUEWUZLMQUOBSB-FSKGGBMCSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- 150000004055 1,2-benzoquinones Chemical class 0.000 description 1
- WLDGDTPNAKWAIR-UHFFFAOYSA-N 1,4,7-trimethyl-1,4,7-triazonane Chemical compound CN1CCN(C)CCN(C)CC1 WLDGDTPNAKWAIR-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- 238000007115 1,4-cycloaddition reaction Methods 0.000 description 1
- PRBHEGAFLDMLAL-UHFFFAOYSA-N 1,5-Hexadiene Natural products CC=CCC=C PRBHEGAFLDMLAL-UHFFFAOYSA-N 0.000 description 1
- DSJWOERHCVJUFY-UHFFFAOYSA-N 1-[2-(1,4,7-triazonan-1-yl)ethyl]-1,4,7-triazonane Chemical compound C1CNCCNCCN1CCN1CCNCCNCC1 DSJWOERHCVJUFY-UHFFFAOYSA-N 0.000 description 1
- ZPTRYWVRCNOTAS-UHFFFAOYSA-M 1-ethyl-3-methylimidazol-3-ium;trifluoromethanesulfonate Chemical compound CC[N+]=1C=CN(C)C=1.[O-]S(=O)(=O)C(F)(F)F ZPTRYWVRCNOTAS-UHFFFAOYSA-M 0.000 description 1
- NADHCXOXVRHBHC-UHFFFAOYSA-N 2,3-dimethoxycyclohexa-2,5-diene-1,4-dione Chemical compound COC1=C(OC)C(=O)C=CC1=O NADHCXOXVRHBHC-UHFFFAOYSA-N 0.000 description 1
- ZJKWJHONFFKJHG-UHFFFAOYSA-N 2-Methoxy-1,4-benzoquinone Chemical compound COC1=CC(=O)C=CC1=O ZJKWJHONFFKJHG-UHFFFAOYSA-N 0.000 description 1
- TZUORCZPIKYDQG-UHFFFAOYSA-N 2-methoxy-5-propylphenol Chemical compound CCCC1=CC=C(OC)C(O)=C1 TZUORCZPIKYDQG-UHFFFAOYSA-N 0.000 description 1
- ABDVJABGTGHCPI-UHFFFAOYSA-N 3-ethyl-1h-imidazol-3-ium;chloride Chemical compound Cl.CCN1C=CN=C1 ABDVJABGTGHCPI-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- RZVHIXYEVGDQDX-UHFFFAOYSA-N 9,10-anthraquinone Chemical compound C1=CC=C2C(=O)C3=CC=CC=C3C(=O)C2=C1 RZVHIXYEVGDQDX-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 229910015900 BF3 Inorganic materials 0.000 description 1
- 235000017166 Bambusa arundinacea Nutrition 0.000 description 1
- 235000017491 Bambusa tulda Nutrition 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- 229920002101 Chitin Polymers 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 229910020632 Co Mn Inorganic materials 0.000 description 1
- 229910020678 Co—Mn Inorganic materials 0.000 description 1
- 229910017566 Cu-Mn Inorganic materials 0.000 description 1
- 229910002482 Cu–Ni Inorganic materials 0.000 description 1
- 229910017871 Cu—Mn Inorganic materials 0.000 description 1
- 125000002353 D-glucosyl group Chemical group C1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 229920000875 Dissolving pulp Polymers 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- 244000004281 Eucalyptus maculata Species 0.000 description 1
- 229910002551 Fe-Mn Inorganic materials 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 229920002581 Glucomannan Polymers 0.000 description 1
- 229920001706 Glucuronoxylan Polymers 0.000 description 1
- 240000000797 Hibiscus cannabinus Species 0.000 description 1
- 102000004157 Hydrolases Human genes 0.000 description 1
- 108090000604 Hydrolases Proteins 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 1
- 229910000990 Ni alloy Inorganic materials 0.000 description 1
- 229910018054 Ni-Cu Inorganic materials 0.000 description 1
- 229910003296 Ni-Mo Inorganic materials 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- JCXJVPUVTGWSNB-UHFFFAOYSA-N Nitrogen dioxide Chemical compound O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 1
- 229910018481 Ni—Cu Inorganic materials 0.000 description 1
- FBVSKRAYVSAFBU-UHFFFAOYSA-N OC1=C(C=2C(C3=CC(=C(C=C3C(C=2C=C1)=O)O)OC)=O)OC Chemical compound OC1=C(C=2C(C3=CC(=C(C=C3C(C=2C=C1)=O)O)OC)=O)OC FBVSKRAYVSAFBU-UHFFFAOYSA-N 0.000 description 1
- YGLUCVSOUOEMJC-UHFFFAOYSA-N OC1=CC=2C(C3=CC(=C(C=C3C(C=2C=C1OC)=O)O)OC)=O Chemical compound OC1=CC=2C(C3=CC(=C(C=C3C(C=2C=C1OC)=O)O)OC)=O YGLUCVSOUOEMJC-UHFFFAOYSA-N 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108700020962 Peroxidase Proteins 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- 244000082204 Phyllostachys viridis Species 0.000 description 1
- 235000015334 Phyllostachys viridis Nutrition 0.000 description 1
- 235000005018 Pinus echinata Nutrition 0.000 description 1
- 241001236219 Pinus echinata Species 0.000 description 1
- 235000017339 Pinus palustris Nutrition 0.000 description 1
- 241000219000 Populus Species 0.000 description 1
- NPYPAHLBTDXSSS-UHFFFAOYSA-N Potassium ion Chemical compound [K+] NPYPAHLBTDXSSS-UHFFFAOYSA-N 0.000 description 1
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 1
- 240000000111 Saccharum officinarum Species 0.000 description 1
- 235000007201 Saccharum officinarum Nutrition 0.000 description 1
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- 241000779819 Syncarpia glomulifera Species 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- 229920002000 Xyloglucan Polymers 0.000 description 1
- 229910021536 Zeolite Inorganic materials 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 1
- UGXQOOQUZRUVSS-ZZXKWVIFSA-N [5-[3,5-dihydroxy-2-(1,3,4-trihydroxy-5-oxopentan-2-yl)oxyoxan-4-yl]oxy-3,4-dihydroxyoxolan-2-yl]methyl (e)-3-(4-hydroxyphenyl)prop-2-enoate Chemical compound OC1C(OC(CO)C(O)C(O)C=O)OCC(O)C1OC1C(O)C(O)C(COC(=O)\C=C\C=2C=CC(O)=CC=2)O1 UGXQOOQUZRUVSS-ZZXKWVIFSA-N 0.000 description 1
- SROAVWOKUOROGU-UHFFFAOYSA-N [Cl-].C(=O)(O)C=1C=[N+](C=CC=1)C1=C(C=CC(=C1)O)O Chemical compound [Cl-].C(=O)(O)C=1C=[N+](C=CC=1)C1=C(C=CC(=C1)O)O SROAVWOKUOROGU-UHFFFAOYSA-N 0.000 description 1
- LJSAJMXWXGSVNA-UHFFFAOYSA-N a805044 Chemical compound OC1=CC=C(O)C=C1.OC1=CC=C(O)C=C1 LJSAJMXWXGSVNA-UHFFFAOYSA-N 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 239000003377 acid catalyst Substances 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 238000005273 aeration Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 239000003570 air Substances 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 125000002723 alicyclic group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 150000001454 anthracenes Chemical class 0.000 description 1
- 239000002518 antifoaming agent Substances 0.000 description 1
- 229910052787 antimony Inorganic materials 0.000 description 1
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 229920000617 arabinoxylan Polymers 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 229910052785 arsenic Inorganic materials 0.000 description 1
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical compound [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 description 1
- 239000002956 ash Substances 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 239000011425 bamboo Substances 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 239000003637 basic solution Substances 0.000 description 1
- WVHBHPATSLQXGC-UHFFFAOYSA-N benzene;ethanol Chemical compound CCO.C1=CC=CC=C1 WVHBHPATSLQXGC-UHFFFAOYSA-N 0.000 description 1
- 150000001555 benzenes Chemical class 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 150000004054 benzoquinones Chemical class 0.000 description 1
- KCXMKQUNVWSEMD-UHFFFAOYSA-N benzyl chloride Chemical compound ClCC1=CC=CC=C1 KCXMKQUNVWSEMD-UHFFFAOYSA-N 0.000 description 1
- 229940073608 benzyl chloride Drugs 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 229920001222 biopolymer Polymers 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- 238000004061 bleaching Methods 0.000 description 1
- 238000010504 bond cleavage reaction Methods 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000000337 buffer salt Substances 0.000 description 1
- 239000004566 building material Substances 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 229920005551 calcium lignosulfonate Polymers 0.000 description 1
- GBAOBIBJACZTNA-UHFFFAOYSA-L calcium sulfite Chemical group [Ca+2].[O-]S([O-])=O GBAOBIBJACZTNA-UHFFFAOYSA-L 0.000 description 1
- 235000010261 calcium sulphite Nutrition 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 150000001721 carbon Chemical class 0.000 description 1
- 239000006229 carbon black Substances 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 150000001793 charged compounds Polymers 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- LZFOPEXOUVTGJS-UHFFFAOYSA-N cis-sinapyl alcohol Natural products COC1=CC(C=CCO)=CC(OC)=C1O LZFOPEXOUVTGJS-UHFFFAOYSA-N 0.000 description 1
- 239000004020 conductor Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 230000007797 corrosion Effects 0.000 description 1
- 238000005260 corrosion Methods 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 150000001923 cyclic compounds Chemical class 0.000 description 1
- 150000001935 cyclohexenes Chemical class 0.000 description 1
- 239000002274 desiccant Substances 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- HQWOEDCLDNFWEV-UHFFFAOYSA-M diethyl phosphate;1-ethyl-3-methylimidazol-3-ium Chemical compound CC[N+]=1C=CN(C)C=1.CCOP([O-])(=O)OCC HQWOEDCLDNFWEV-UHFFFAOYSA-M 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- HTXDPTMKBJXEOW-UHFFFAOYSA-N dioxoiridium Chemical compound O=[Ir]=O HTXDPTMKBJXEOW-UHFFFAOYSA-N 0.000 description 1
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000011143 downstream manufacturing Methods 0.000 description 1
- 238000005553 drilling Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000005684 electric field Effects 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 150000002118 epoxides Chemical class 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000003546 flue gas Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 239000010881 fly ash Substances 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 238000004508 fractional distillation Methods 0.000 description 1
- 235000011389 fruit/vegetable juice Nutrition 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 238000002309 gasification Methods 0.000 description 1
- 238000012239 gene modification Methods 0.000 description 1
- 230000005017 genetic modification Effects 0.000 description 1
- 235000013617 genetically modified food Nutrition 0.000 description 1
- 229910052732 germanium Inorganic materials 0.000 description 1
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 description 1
- 229910021397 glassy carbon Inorganic materials 0.000 description 1
- 229940046240 glucomannan Drugs 0.000 description 1
- 238000004442 gravimetric analysis Methods 0.000 description 1
- 159000000011 group IA salts Chemical class 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 229920000140 heteropolymer Polymers 0.000 description 1
- PYGSKMBEVAICCR-UHFFFAOYSA-N hexa-1,5-diene Chemical compound C=CCCC=C PYGSKMBEVAICCR-UHFFFAOYSA-N 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 125000000687 hydroquinonyl group Chemical group C1(O)=C(C=C(O)C=C1)* 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 230000009474 immediate action Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052741 iridium Inorganic materials 0.000 description 1
- 229910000457 iridium oxide Inorganic materials 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 238000010667 large scale reaction Methods 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000010985 leather Substances 0.000 description 1
- 239000002029 lignocellulosic biomass Substances 0.000 description 1
- 239000007791 liquid phase Substances 0.000 description 1
- 238000000622 liquid--liquid extraction Methods 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 229910001510 metal chloride Inorganic materials 0.000 description 1
- 239000003863 metallic catalyst Substances 0.000 description 1
- 238000001465 metallisation Methods 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- GDOPTJXRTPNYNR-UHFFFAOYSA-N methyl-cyclopentane Natural products CC1CCCC1 GDOPTJXRTPNYNR-UHFFFAOYSA-N 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 238000000874 microwave-assisted extraction Methods 0.000 description 1
- 238000003801 milling Methods 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 238000005065 mining Methods 0.000 description 1
- DDTIGTPWGISMKL-UHFFFAOYSA-N molybdenum nickel Chemical compound [Ni].[Mo] DDTIGTPWGISMKL-UHFFFAOYSA-N 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 229920005690 natural copolymer Polymers 0.000 description 1
- 239000003345 natural gas Substances 0.000 description 1
- 239000005445 natural material Substances 0.000 description 1
- 229920005615 natural polymer Polymers 0.000 description 1
- AOPCKOPZYFFEDA-UHFFFAOYSA-N nickel(2+);dinitrate;hexahydrate Chemical compound O.O.O.O.O.O.[Ni+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O AOPCKOPZYFFEDA-UHFFFAOYSA-N 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 230000009972 noncorrosive effect Effects 0.000 description 1
- 229910052755 nonmetal Inorganic materials 0.000 description 1
- 235000019645 odor Nutrition 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 238000006053 organic reaction Methods 0.000 description 1
- 238000012261 overproduction Methods 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 229930015763 p-coumaryl alcohol Natural products 0.000 description 1
- 238000010979 pH adjustment Methods 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000001739 pinus spp. Substances 0.000 description 1
- 229920000867 polyelectrolyte Polymers 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- ODLMAHJVESYWTB-UHFFFAOYSA-N propylbenzene Chemical group CCCC1=CC=CC=C1 ODLMAHJVESYWTB-UHFFFAOYSA-N 0.000 description 1
- 238000005086 pumping Methods 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 239000002296 pyrolytic carbon Substances 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 239000002265 redox agent Substances 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 229910052702 rhenium Inorganic materials 0.000 description 1
- WUAPFZMCVAUBPE-UHFFFAOYSA-N rhenium atom Chemical compound [Re] WUAPFZMCVAUBPE-UHFFFAOYSA-N 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 238000009938 salting Methods 0.000 description 1
- 238000005185 salting out Methods 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 239000004576 sand Substances 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 230000001932 seasonal effect Effects 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- JVBXVOWTABLYPX-UHFFFAOYSA-L sodium dithionite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])=O JVBXVOWTABLYPX-UHFFFAOYSA-L 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000002689 soil Substances 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 235000011150 stannous chloride Nutrition 0.000 description 1
- 150000001629 stilbenes Chemical class 0.000 description 1
- 235000021286 stilbenes Nutrition 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 238000000859 sublimation Methods 0.000 description 1
- 230000008022 sublimation Effects 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- XTHPWXDJESJLNJ-UHFFFAOYSA-N sulfurochloridic acid Chemical class OS(Cl)(=O)=O XTHPWXDJESJLNJ-UHFFFAOYSA-N 0.000 description 1
- 238000003815 supercritical carbon dioxide extraction Methods 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000011885 synergistic combination Substances 0.000 description 1
- 229910052714 tellurium Inorganic materials 0.000 description 1
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical compound [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- IFLREYGFSNHWGE-UHFFFAOYSA-N tetracene Chemical compound C1=CC=CC2=CC3=CC4=CC=CC=C4C=C3C=C21 IFLREYGFSNHWGE-UHFFFAOYSA-N 0.000 description 1
- 238000002411 thermogravimetry Methods 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- AXZWODMDQAVCJE-UHFFFAOYSA-L tin(II) chloride (anhydrous) Chemical compound [Cl-].[Cl-].[Sn+2] AXZWODMDQAVCJE-UHFFFAOYSA-L 0.000 description 1
- PTNLHDGQWUGONS-UHFFFAOYSA-N trans-p-coumaric alcohol Natural products OCC=CC1=CC=C(O)C=C1 PTNLHDGQWUGONS-UHFFFAOYSA-N 0.000 description 1
- PTNLHDGQWUGONS-OWOJBTEDSA-N trans-p-coumaryl alcohol Chemical group OC\C=C\C1=CC=C(O)C=C1 PTNLHDGQWUGONS-OWOJBTEDSA-N 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 229910001428 transition metal ion Inorganic materials 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 229940036248 turpentine Drugs 0.000 description 1
- 238000002137 ultrasound extraction Methods 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 238000009834 vaporization Methods 0.000 description 1
- 230000008016 vaporization Effects 0.000 description 1
- 229920001221 xylan Polymers 0.000 description 1
- 150000004823 xylans Chemical class 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- D—TEXTILES; PAPER
- D21—PAPER-MAKING; PRODUCTION OF CELLULOSE
- D21C—PRODUCTION OF CELLULOSE BY REMOVING NON-CELLULOSE SUBSTANCES FROM CELLULOSE-CONTAINING MATERIALS; REGENERATION OF PULPING LIQUORS; APPARATUS THEREFOR
- D21C11/00—Regeneration of pulp liquors or effluent waste waters
- D21C11/0007—Recovery of by-products, i.e. compounds other than those necessary for pulping, for multiple uses or not otherwise provided for
-
- E—FIXED CONSTRUCTIONS
- E05—LOCKS; KEYS; WINDOW OR DOOR FITTINGS; SAFES
- E05C—BOLTS OR FASTENING DEVICES FOR WINGS, SPECIALLY FOR DOORS OR WINDOWS
- E05C17/00—Devices for holding wings open; Devices for limiting opening of wings or for holding wings open by a movable member extending between frame and wing; Braking devices, stops or buffers, combined therewith
- E05C17/02—Devices for holding wings open; Devices for limiting opening of wings or for holding wings open by a movable member extending between frame and wing; Braking devices, stops or buffers, combined therewith by mechanical means
- E05C17/04—Devices for holding wings open; Devices for limiting opening of wings or for holding wings open by a movable member extending between frame and wing; Braking devices, stops or buffers, combined therewith by mechanical means with a movable bar or equivalent member extending between frame and wing
- E05C17/12—Devices for holding wings open; Devices for limiting opening of wings or for holding wings open by a movable member extending between frame and wing; Braking devices, stops or buffers, combined therewith by mechanical means with a movable bar or equivalent member extending between frame and wing consisting of a single rod
- E05C17/24—Devices for holding wings open; Devices for limiting opening of wings or for holding wings open by a movable member extending between frame and wing; Braking devices, stops or buffers, combined therewith by mechanical means with a movable bar or equivalent member extending between frame and wing consisting of a single rod pivoted at one end, and with the other end running along a guide member
- E05C17/28—Devices for holding wings open; Devices for limiting opening of wings or for holding wings open by a movable member extending between frame and wing; Braking devices, stops or buffers, combined therewith by mechanical means with a movable bar or equivalent member extending between frame and wing consisting of a single rod pivoted at one end, and with the other end running along a guide member with braking, clamping or securing means at the connection to the guide member
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07G—COMPOUNDS OF UNKNOWN CONSTITUTION
- C07G1/00—Lignin; Lignin derivatives
-
- E—FIXED CONSTRUCTIONS
- E05—LOCKS; KEYS; WINDOW OR DOOR FITTINGS; SAFES
- E05F—DEVICES FOR MOVING WINGS INTO OPEN OR CLOSED POSITION; CHECKS FOR WINGS; WING FITTINGS NOT OTHERWISE PROVIDED FOR, CONCERNED WITH THE FUNCTIONING OF THE WING
- E05F5/00—Braking devices, e.g. checks; Stops; Buffers
- E05F5/12—Braking devices, e.g. checks; Stops; Buffers specially for preventing the closing of a wing before another wing has been closed
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/02—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of sulfonic acids or halides thereof
- C07C303/04—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of sulfonic acids or halides thereof by substitution of hydrogen atoms by sulfo or halosulfonyl groups
- C07C303/06—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides of sulfonic acids or halides thereof by substitution of hydrogen atoms by sulfo or halosulfonyl groups by reaction with sulfuric acid or sulfur trioxide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/28—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/41—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing singly-bound oxygen atoms bound to the carbon skeleton
- C07C309/43—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing singly-bound oxygen atoms bound to the carbon skeleton having at least one of the sulfo groups bound to a carbon atom of a six-membered aromatic ring being part of a condensed ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/01—Sulfonic acids
- C07C309/28—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C309/45—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing nitrogen atoms, not being part of nitro or nitroso groups, bound to the carbon skeleton
- C07C309/46—Sulfonic acids having sulfo groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing nitrogen atoms, not being part of nitro or nitroso groups, bound to the carbon skeleton having the sulfo groups bound to carbon atoms of non-condensed six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08H—DERIVATIVES OF NATURAL MACROMOLECULAR COMPOUNDS
- C08H8/00—Macromolecular compounds derived from lignocellulosic materials
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G1/00—Production of liquid hydrocarbon mixtures from oil-shale, oil-sand, or non-melting solid carbonaceous or similar materials, e.g. wood, coal
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G1/00—Production of liquid hydrocarbon mixtures from oil-shale, oil-sand, or non-melting solid carbonaceous or similar materials, e.g. wood, coal
- C10G1/08—Production of liquid hydrocarbon mixtures from oil-shale, oil-sand, or non-melting solid carbonaceous or similar materials, e.g. wood, coal with moving catalysts
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G15/00—Cracking of hydrocarbon oils by electric means, electromagnetic or mechanical vibrations, by particle radiation or with gases superheated in electric arcs
- C10G15/08—Cracking of hydrocarbon oils by electric means, electromagnetic or mechanical vibrations, by particle radiation or with gases superheated in electric arcs by electric means or by electromagnetic or mechanical vibrations
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G27/00—Refining of hydrocarbon oils in the absence of hydrogen, by oxidation
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G29/00—Refining of hydrocarbon oils, in the absence of hydrogen, with other chemicals
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G3/00—Production of liquid hydrocarbon mixtures from oxygen-containing organic materials, e.g. fatty oils, fatty acids
- C10G3/42—Catalytic treatment
-
- D—TEXTILES; PAPER
- D21—PAPER-MAKING; PRODUCTION OF CELLULOSE
- D21C—PRODUCTION OF CELLULOSE BY REMOVING NON-CELLULOSE SUBSTANCES FROM CELLULOSE-CONTAINING MATERIALS; REGENERATION OF PULPING LIQUORS; APPARATUS THEREFOR
- D21C11/00—Regeneration of pulp liquors or effluent waste waters
- D21C11/0042—Fractionating or concentration of spent liquors by special methods
-
- D—TEXTILES; PAPER
- D21—PAPER-MAKING; PRODUCTION OF CELLULOSE
- D21C—PRODUCTION OF CELLULOSE BY REMOVING NON-CELLULOSE SUBSTANCES FROM CELLULOSE-CONTAINING MATERIALS; REGENERATION OF PULPING LIQUORS; APPARATUS THEREFOR
- D21C11/00—Regeneration of pulp liquors or effluent waste waters
- D21C11/0057—Oxidation of liquors, e.g. in order to reduce the losses of sulfur compounds, followed by evaporation or combustion if the liquor in question is a black liquor
-
- D—TEXTILES; PAPER
- D21—PAPER-MAKING; PRODUCTION OF CELLULOSE
- D21H—PULP COMPOSITIONS; PREPARATION THEREOF NOT COVERED BY SUBCLASSES D21C OR D21D; IMPREGNATING OR COATING OF PAPER; TREATMENT OF FINISHED PAPER NOT COVERED BY CLASS B31 OR SUBCLASS D21G; PAPER NOT OTHERWISE PROVIDED FOR
- D21H11/00—Pulp or paper, comprising cellulose or lignocellulose fibres of natural origin only
- D21H11/02—Chemical or chemomechanical or chemothermomechanical pulp
- D21H11/04—Kraft or sulfate pulp
-
- D—TEXTILES; PAPER
- D21—PAPER-MAKING; PRODUCTION OF CELLULOSE
- D21H—PULP COMPOSITIONS; PREPARATION THEREOF NOT COVERED BY SUBCLASSES D21C OR D21D; IMPREGNATING OR COATING OF PAPER; TREATMENT OF FINISHED PAPER NOT COVERED BY CLASS B31 OR SUBCLASS D21G; PAPER NOT OTHERWISE PROVIDED FOR
- D21H11/00—Pulp or paper, comprising cellulose or lignocellulose fibres of natural origin only
- D21H11/02—Chemical or chemomechanical or chemothermomechanical pulp
- D21H11/06—Sulfite or bisulfite pulp
-
- D—TEXTILES; PAPER
- D21—PAPER-MAKING; PRODUCTION OF CELLULOSE
- D21H—PULP COMPOSITIONS; PREPARATION THEREOF NOT COVERED BY SUBCLASSES D21C OR D21D; IMPREGNATING OR COATING OF PAPER; TREATMENT OF FINISHED PAPER NOT COVERED BY CLASS B31 OR SUBCLASS D21G; PAPER NOT OTHERWISE PROVIDED FOR
- D21H17/00—Non-fibrous material added to the pulp, characterised by its constitution; Paper-impregnating material characterised by its constitution
- D21H17/03—Non-macromolecular organic compounds
- D21H17/05—Non-macromolecular organic compounds containing elements other than carbon and hydrogen only
- D21H17/12—Organo-metallic compounds
-
- D—TEXTILES; PAPER
- D21—PAPER-MAKING; PRODUCTION OF CELLULOSE
- D21H—PULP COMPOSITIONS; PREPARATION THEREOF NOT COVERED BY SUBCLASSES D21C OR D21D; IMPREGNATING OR COATING OF PAPER; TREATMENT OF FINISHED PAPER NOT COVERED BY CLASS B31 OR SUBCLASS D21G; PAPER NOT OTHERWISE PROVIDED FOR
- D21H17/00—Non-fibrous material added to the pulp, characterised by its constitution; Paper-impregnating material characterised by its constitution
- D21H17/71—Mixtures of material ; Pulp or paper comprising several different materials not incorporated by special processes
- D21H17/74—Mixtures of material ; Pulp or paper comprising several different materials not incorporated by special processes of organic and inorganic material
-
- E—FIXED CONSTRUCTIONS
- E05—LOCKS; KEYS; WINDOW OR DOOR FITTINGS; SAFES
- E05F—DEVICES FOR MOVING WINGS INTO OPEN OR CLOSED POSITION; CHECKS FOR WINGS; WING FITTINGS NOT OTHERWISE PROVIDED FOR, CONCERNED WITH THE FUNCTIONING OF THE WING
- E05F3/00—Closers or openers with braking devices, e.g. checks; Construction of pneumatic or liquid braking devices
- E05F3/22—Additional arrangements for closers, e.g. for holding the wing in opened or other position
- E05F3/223—Hydraulic power-locks, e.g. with electrically operated hydraulic valves
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M8/00—Fuel cells; Manufacture thereof
- H01M8/18—Regenerative fuel cells, e.g. redox flow batteries or secondary fuel cells
- H01M8/184—Regeneration by electrochemical means
- H01M8/188—Regeneration by electrochemical means by recharging of redox couples containing fluids; Redox flow type batteries
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D2257/00—Components to be removed
- B01D2257/70—Organic compounds not provided for in groups B01D2257/00 - B01D2257/602
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D61/00—Processes of separation using semi-permeable membranes, e.g. dialysis, osmosis or ultrafiltration; Apparatus, accessories or auxiliary operations specially adapted therefor
- B01D61/02—Reverse osmosis; Hyperfiltration ; Nanofiltration
- B01D61/027—Nanofiltration
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D61/00—Processes of separation using semi-permeable membranes, e.g. dialysis, osmosis or ultrafiltration; Apparatus, accessories or auxiliary operations specially adapted therefor
- B01D61/14—Ultrafiltration; Microfiltration
- B01D61/145—Ultrafiltration
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/04—One of the condensed rings being a six-membered aromatic ring
- C07C2602/10—One of the condensed rings being a six-membered aromatic ring the other ring being six-membered, e.g. tetraline
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/02—Ortho- or ortho- and peri-condensed systems
- C07C2603/04—Ortho- or ortho- and peri-condensed systems containing three rings
- C07C2603/22—Ortho- or ortho- and peri-condensed systems containing three rings containing only six-membered rings
- C07C2603/24—Anthracenes; Hydrogenated anthracenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/02—Ortho- or ortho- and peri-condensed systems
- C07C2603/04—Ortho- or ortho- and peri-condensed systems containing three rings
- C07C2603/22—Ortho- or ortho- and peri-condensed systems containing three rings containing only six-membered rings
- C07C2603/26—Phenanthrenes; Hydrogenated phenanthrenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C303/00—Preparation of esters or amides of sulfuric acids; Preparation of sulfonic acids or of their esters, halides, anhydrides or amides
- C07C303/42—Separation; Purification; Stabilisation; Use of additives
- C07C303/44—Separation; Purification
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/10—Feedstock materials
- C10G2300/1011—Biomass
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2300/00—Aspects relating to hydrocarbon processing covered by groups C10G1/00 - C10G99/00
- C10G2300/10—Feedstock materials
- C10G2300/1011—Biomass
- C10G2300/1014—Biomass of vegetal origin
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2400/00—Products obtained by processes covered by groups C10G9/00 - C10G69/14
- C10G2400/30—Aromatics
-
- D—TEXTILES; PAPER
- D21—PAPER-MAKING; PRODUCTION OF CELLULOSE
- D21D—TREATMENT OF THE MATERIALS BEFORE PASSING TO THE PAPER-MAKING MACHINE
- D21D5/00—Purification of the pulp suspension by mechanical means; Apparatus therefor
- D21D5/18—Purification of the pulp suspension by mechanical means; Apparatus therefor with the aid of centrifugal force
-
- E—FIXED CONSTRUCTIONS
- E05—LOCKS; KEYS; WINDOW OR DOOR FITTINGS; SAFES
- E05F—DEVICES FOR MOVING WINGS INTO OPEN OR CLOSED POSITION; CHECKS FOR WINGS; WING FITTINGS NOT OTHERWISE PROVIDED FOR, CONCERNED WITH THE FUNCTIONING OF THE WING
- E05F3/00—Closers or openers with braking devices, e.g. checks; Construction of pneumatic or liquid braking devices
- E05F3/22—Additional arrangements for closers, e.g. for holding the wing in opened or other position
- E05F2003/228—Arrangements where the end of the closer arm is sliding in a track
-
- E—FIXED CONSTRUCTIONS
- E05—LOCKS; KEYS; WINDOW OR DOOR FITTINGS; SAFES
- E05Y—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES E05D AND E05F, RELATING TO CONSTRUCTION ELEMENTS, ELECTRIC CONTROL, POWER SUPPLY, POWER SIGNAL OR TRANSMISSION, USER INTERFACES, MOUNTING OR COUPLING, DETAILS, ACCESSORIES, AUXILIARY OPERATIONS NOT OTHERWISE PROVIDED FOR, APPLICATION THEREOF
- E05Y2201/00—Constructional elements; Accessories therefor
- E05Y2201/60—Suspension or transmission members; Accessories therefor
- E05Y2201/622—Suspension or transmission members elements
- E05Y2201/638—Cams; Ramps
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2300/00—Electrolytes
- H01M2300/0002—Aqueous electrolytes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2300/00—Electrolytes
- H01M2300/0002—Aqueous electrolytes
- H01M2300/0005—Acid electrolytes
- H01M2300/0011—Sulfuric acid-based
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/30—Hydrogen technology
- Y02E60/50—Fuel cells
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P30/00—Technologies relating to oil refining and petrochemical industry
- Y02P30/20—Technologies relating to oil refining and petrochemical industry using bio-feedstock
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P80/00—Climate change mitigation technologies for sector-wide applications
- Y02P80/20—Climate change mitigation technologies for sector-wide applications using renewable energy
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Physics & Mathematics (AREA)
- Electromagnetism (AREA)
- Polymers & Plastics (AREA)
- Medicinal Chemistry (AREA)
- Biochemistry (AREA)
- Materials Engineering (AREA)
- Health & Medical Sciences (AREA)
- Mechanical Engineering (AREA)
- Sustainable Energy (AREA)
- Electrochemistry (AREA)
- Manufacturing & Machinery (AREA)
- Sustainable Development (AREA)
- Inorganic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Fuel Cell (AREA)
- Compounds Of Unknown Constitution (AREA)
- Paper (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
Description


R1、R3、又はR5の少なくとも1つは、好ましくはヒドロキシ、又は直鎖又は分枝の任意に置換されたC1-6アルコキシであり、
R6は、水素、ヒドロキシ、直鎖又は分枝の任意に置換されたC1-6カルボキシル、直鎖又は分枝の任意に置換されたC1-6アルデヒド、及び直鎖又は分枝の任意に置換されたC1-6アルコールからなる群から選択される。

R10は、水素、ヒドロキシ、直鎖又は分枝の任意に置換されたC1-6カルボキシル、直鎖又は分枝の任意に置換されたC1-6アルデヒド、及び直鎖又は分枝の任意に置換されたC1-6アルコールからなる群から選択される。

R6は、水素、ヒドロキシ、直鎖又は分枝のC1-6カルボキシ、直鎖又は分枝のC1-6アルデヒド、及び直鎖又は分枝のC1-6アルコールからなる群から選択される。)

R10は、水素、ヒドロキシ、直鎖又は分枝のC1-6カルボキシル、直鎖又は分枝のC1-6アルデヒド、及び直鎖又は分枝のC1-6アルコールからなる群から選択される。)によって特徴付けることができる。



式(II)のR1及びR4は、水素、ヒドロキシ、直鎖又は分枝の任意に置換されたC1-6カルボキシル、直鎖又は分枝の任意に置換されたC1-6アルデヒド、及び直鎖又は分枝の任意に置換されたC1-6アルコールからなる群から選択され、
式(III)のR1~R10は、それぞれ独立して、水素、ヒドロキシ、カルボキシ、直鎖又は分枝の任意に置換されたC1-6アルキル、直鎖又は分枝の任意に置換されたC1-6アルケニル、直鎖又は分枝の任意に置換されたC1-6アルコール、直鎖又は分枝の任意に置換されたC1-6アミノアルキル、直鎖又は分枝の任意に置換されたC1-6カルボキシアルキル、直鎖又は分枝の任意に置換されたC1-6アルコキシ、直鎖又は分枝の任意に置換されたC1-6アルデヒド、エステル、オキソ、又はカルボニルから選択され、
好ましくは、R2、R5、R6、及びR8の少なくとも1つは、ヒドロキシ又はC1-3アルコキシであり、
好ましくは、式(III)のR1、R4、R9、及びR10は、水素、ヒドロキシ、直鎖又は分枝の任意に置換されたC1-6カルボキシル、直鎖又は分枝の任意に置換されたC1-6アルデヒド、及び直鎖又は分枝の任意に置換されたC1-6アルコールからなる群から選択され、
式(IV)のR2、R3、及びR7~R10は、それぞれ独立して、水素、ヒドロキシ、カルボキシ、直鎖又は分枝の任意に置換されたC1-6アルキル、直鎖又は分枝の任意に置換されたC1-6アルケニル、直鎖又は分枝の任意に置換されたC1-6アルコール、直鎖又は分枝の任意に置換されたC1-6アミノアルキル、直鎖又は分枝の任意に置換されたC1-6カルボキシアルキル、直鎖又は分枝の任意に置換されたC1-6アルコキシ、直鎖又は分枝の任意に置換されたC1-6アルデヒド、エステル、オキソ、又はカルボニルから選択され、
好ましくは、R2、R3、及びR7~R10の少なくとも1つは、ヒドロキシ又はC1-3アルコキシであり、
式(IV)のR1、R4、R5、及びR6は、水素、ヒドロキシ、直鎖又は分枝の任意に置換されたC1-6カルボキシル、直鎖又は分枝の任意に置換されたC1-6アルデヒド、及び直鎖又は分枝の任意に置換されたC1-6アルコールからなる群から選択される。
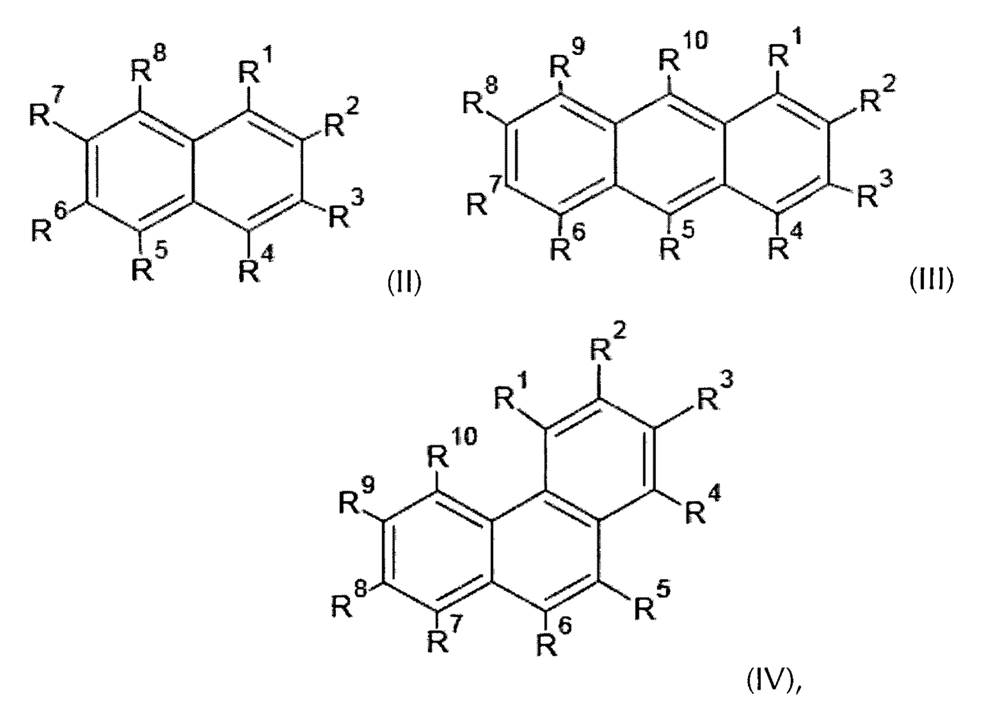
式(II)のR1及び/又はR8は、水素、ヒドロキシ、直鎖又は分枝のC1-6カルボキシル、直鎖又は分枝のC1-6アルデヒド、及び直鎖又は分枝のC1-6アルコールからなる群から選択され、
式(III)のR2~R8は、それぞれ独立して、H、任意に置換されたC1-6アルキル、任意に置換されたC1-6アルケニル、ハロゲン、任意に置換されたC1-6アルコキシ、アミノ、ニトロ、ホスホリル、ホスホニルから選択され、R2、R4、R5、R6、及びR8の少なくとも1つは、ヒドロキシ又はC1-3アルコキシであり、
式(III)のR1、R9、及び/又はR10は、水素、ヒドロキシ、直鎖又は分枝のC1-6カルボキシル、直鎖又は分枝のC1-6アルデヒド、及び直鎖又は分枝のC1-6アルコールからなる群から選択され、
式(IV)のR2~R9は、それぞれ独立して、H、任意に置換されたC1-6アルキル、任意に置換されたC1-6アルケニル、ハロゲン、任意に置換されたC1-6アルコキシ、アミノ、ニトロ、ホスホリル、ホスホニルから選択され、
R2、R4、R7、及びR9の少なくとも1つは、ヒドロキシ又はC1-3アルコキシであり、
式(IV)のR1及び/又はR10は、水素、ヒドロキシ、直鎖又は分枝のC1-6カルボキシル、直鎖又は分枝のC1-6アルデヒド、及び直鎖又は分枝のC1-6アルコールからなる群から選択される。

又は一般式(Vb):

によって特徴付けられる少なくとも1つのヒドロキノン化合物(工程(H.1)を生成する。




によって特徴付けられる少なくとも1つキノン化合物を提供する(工程H.2)。

又は一般式(Vb):

によって特徴付けられる少なくとも1つのヒドロキノン化合物を生成することができる(工程H.1)。







式(VII)のR8及びR5又はR1及びR4の少なくとも1つは、ヒドロキシ又はオキソであり、又は式(VIII)のR9及びR6、R10及びR5、又はR1及びR4の少なくとも1つは、ヒドロキシ又はオキソであり、又は式(IX)のR10及びR7又はR1及びR4の少なくとも1つは、ヒドロキシ又はオキソである。

式(VII)のR8及びR5又はR1及びR4の少なくとも1つは、ヒドロキシ又はオキソであり、又は式(VIII)のR9及びR6、R10及びR5、又はR1及びR4の少なくとも1つは、ヒドロキシ又はオキソであり、又は式(IX)のR10及びR7又はR1及びR4の少なくとも1つは、ヒドロキシ又はオキソである。



式(VII)のR8及びR5又はR1及びR4の少なくとも1つがヒドロキシ又はオキソである、又は式(VIII)のR9及びR6、R10及びR5、又はR1及びR4の少なくとも1つがヒドロキシ又はオキソである、又は式(IX)のR10及びR7又はR1及びR4の少なくとも1つがヒドロキシ又はオキソである、又はR7及びR10、R5及びR6、又はR6及びR2の少なくとも1つがヒドロキシル又はオキソである。)

本発明方法の工程(E.2)による修飾リグニン由来成分の還元クラッキング(クラッキング及び還元)は、例えば、ニッケルを含む触媒(活性炭に担持されたもの(Ni/C)など)によって行うことができる。触媒は、典型的には、溶液滴下含浸法(incipient-wetness impregnation method)によって調製され、更に、当技術分野で知られた炭素熱還元法(carbothermal reduction method)によって処理される。
リグノスルホナートは、本発明に係る工程(D)によって与えられる。1%(W/W)のリグノスルホナートを含む1M NaOH水溶液を調製する。前記溶液を、工程(E.3)にしたがう電気酸化に付す。そこでは、この溶液はアノライトとして使用される。1M水溶液はカタライト(katalyte)として使用される。250ml/分間の流速を有するフローセルが使用される。電気分解は、8時間定電流で1mA/cm2の電流を印加して行うことができる。通常得られる電圧は1.4Vである。電圧曲線は典型的には漸近的であり、溶液は好ましくは褐色から暗褐色に変色する。
低分子量芳香族リグニン由来化合物としてのバニリンは、本発明に係る工程(F)によって提供される。前記化合物は、更に、工程(G)にしたがって環化され、本発明に係る工程(H)にしたがって以下のように5工程で酸化される。






実施例4.1 ジメトキシベンゾキノンの還元


2-メトキシベンゼン-1,4-ジオール8.24g(0.059モル、1.0当量)を、還流冷却器を備えた250mL反応フラスコに秤量した。60mLのジクロロエタン及び15mL(0.159モル、2.7当量)の無水酢酸を添加した。次いで、三フッ化ホウ素エーテル溶液12mL(0.096モル、1.63当量)を攪拌しながら室温でゆっくり添加した。反応混合物を90℃に20時間加熱した。混合物を60℃に冷却し、30mLのH2Oを加え、続いて10mLのHCl(6M)を加えた。得られた混合物を100℃に30分間加熱し、冷却し、酢酸エチル(150mL×3)で抽出した。合わせた抽出物をH2O(100mL)、飽和重炭酸ナトリウム(100mL)、及びH2O(100mL)で順次洗浄し、次いで無水硫酸ナトリウムで乾燥させた。溶媒を真空下で除去して褐色固体残渣を得、これをメタノールで洗浄して、ベージュの固体として1-(2,5-ジヒドロキシ-4-メトキシフェニル)エタン-1-オンを7.49g(0.041モル、70%収率)得た。
p-ベンゾキノン2.16g(0.02モル、1.0当量)を酢酸6.4mLに懸濁させた。2.46g(0.02mol、1.0当量)のニコチン酸を加え、混合物を室温で2時間撹拌した。得られた暗色混合物を3mLの水で希釈し、6.6mLのHCl(6M)で処理した。冷却すると、固体が析出し、これを濾別し、60℃で一晩乾燥して、黄色の固体として3-カルボキシ-1-(2,5-ジヒドロキシフェニル)ピリジン-1-イウムクロライド3.13g(0.012モル、59%収率)を得た。




Claims (70)
- 少なくとも1つの低分子量芳香族リグニン由来化合物を製造するための方法であって、
(A)リグノセルロース材料を提供する工程と、
(B)前記リグノセルロース材料をパルプ化プロセスに付す工程と、
(C)パルプ分離工程において工程(B)で得られたセルロースを工程(B)から得られるプロセス流から分離して、実質的にセルロースを含まないプロセス流を提供し、前記プロセス流が、修飾リグニン由来成分、ヘミセルロース、及び/又はそのフラグメントを含み、前記プロセス流が、単一のプロセス流又は少なくとも2つの部分プロセス流として提供される工程と、
(D)(D.1)工程(C)のプロセス流に含まれる、又は
(D.2)工程(C)における少なくとも2つの部分プロセス流のうちの少なくとも1つに含まれる、
修飾リグニン由来成分のフラクションを、前記プロセス流のいずれかから単離する工程と、
(E)工程(D)の修飾リグニン由来成分のフラクションを化学分解工程に付し、前記化学分解工程が、
(E.2)金属イオン又は半金属成分を含む不均一系又は均一系触媒の存在下での前記修飾リグニン成分の還元クラッキング(クラッキング及び還元)を含む工程と、
(F)工程(E)で得られた前記修飾リグニン由来生成物を、単離及び任意に精製工程に付し、低分子量芳香族リグニン由来化合物をより高分子量の芳香族リグニン由来成分及び/又は他の非リグニン由来残留成分から単離し、任意に精製する工程と
を含み、
前記工程(B)が、以下の工程:
(a)任意に、前記リグノセルロース材料をプレスチーミングし、前記リグノセルロース材料を蒸気で湿潤し、予熱する工程と、
(b)前記リグノセルロース材料を、亜硫酸塩又は重亜硫酸塩剤を含む水性の溶液に添加する工程と、及び
(c)前記リグノセルロース材料を前記水性の溶液中で蒸解する工程と
を含む(B.2)亜硫酸塩プロセスであることを特徴とする方法。 - 前記リグノセルロース材料が、細断されたリグノセルロース材料である請求項1に記載の方法。
- 前記工程(B)における前記水性の溶液が、酸性の溶液である請求項1から2のいずれかに記載の方法。
- 前記リグノセルロース材料が、低シリカ及び樹脂含量の木材に由来することができる請求項1から3のいずれかに記載の方法。
- 前記リグノセルロース材料が、北方木材に由来することができる請求項4に記載の方法。
- 前記リグノセルロース材料が、ブナ、マツ、バーチ、ユーカリ、及びスプルースからなる群から選択される少なくともいずれかに由来することができる請求項5に記載の方法。
- 前記リグノセルロース材料が、ブナに由来することができる請求項6に記載の方法。
- 前記リグノセルロース材料は、木材チップの形態で提供される請求項1から7のいずれかに記載の方法。
- 工程(B.2)のサブ工程(b)における前記酸性水溶液のpHが1~5であり、及び/又は工程(B.2)のサブ工程(b)における前記水性の溶液の温度が100℃未満である請求項1から8のいずれかに記載の方法。
- 工程(B.2)のサブ工程(b)にしたがって添加される亜硫酸塩又は重亜硫酸塩剤が、ナトリウム、カルシウム、マグネシウム、及びアンモニウムからなる群から選択されるカウンターカチオンとの塩である請求項1から9のいずれかに記載の方法。
- 工程(B.2)のサブ工程(c)における蒸解が、加圧容器内で、少なくとも3時間、少なくとも120℃の温度で行われる請求項1から10のいずれかに記載の方法。
- 亜硫酸塩プロセス(B.2)のサブ工程(c)が、4~24時間行われる請求項1から11のいずれかに記載の方法。
- 亜硫酸塩プロセス(B.2)のサブ工程(c)が、4~6時間行われる請求項12に記載の方法。
- 工程(B.2)の亜硫酸塩プロセスサブ工程(c)が、120~170℃の温度で行われる請求項1から13のいずれかに記載の方法。
- 工程(B.2)の亜硫酸塩プロセスサブ工程(c)が、130~160℃の温度で行われる請求項14に記載の方法。
- 工程(B.2)のサブ工程(c)が、加圧容器内で少なくとも4バールの圧力で行われる請求項1から15のいずれかに記載の方法。
- 工程(B.2)のサブ工程(c)が、加圧容器内で5~10バールの圧力で行われる請求項16に記載の方法。
- (B.2)のサブ工程(c)が、バッチモード又は連続モードで行われる請求項1から17のいずれかに記載の方法。
- (B.2)のサブ工程(c)が、連続モードで行われる請求項18に記載の方法。
- 工程(C)の分離が、吹き込み、篩い分け、遠心分離、濾過、及び/又は洗浄、又はそれらの任意の組合せによって行われる請求項1から19のいずれかに記載の方法。
- 工程(D)の単離が、抽出、向流、ストリッピング、イオン交換、二価又は多価カチオンによる沈殿、酸性溶液中CO 2 による沈殿、濾過、又はそれらの任意の組合せによって行われる請求項1から20のいずれかに記載の方法。
- 前記二価又は多価カチオンによる沈殿が、カルシウム塩による沈殿である請求項21に記載の方法。
- 前記濾過が、限外濾過及び/又はナノ濾過である請求項21に記載の方法。
- 工程(E.2)において、前記修飾リグニン由来成分の還元クラッキング(クラッキング及び還元)が、還元剤及び不均一触媒の存在下で行われ、前記不均一触媒が、ニッケル、白金、パラジウム、ルテニウム、レニウム、及び金から選択される金属を含む請求項1から23のいずれかに記載の方法。
- 前記還元剤が、水素又は水素供与性アルコールである請求項24に記載の方法。
- 前記不均一触媒が、担体材料の表面上に設けられている請求項24から25のいずれかに記載の方法。
- 前記担体材料が、活性炭、シリカ、酸化チタン、及び酸化アルミニウムからなる群から選択される請求項26に記載の方法。
- 単離工程(D)及び/又は単離工程(F)が、濾過及び/又は抽出を含む請求項1から27のいずれかに記載の方法。
- 単離工程(D)及び/又は単離工程(F)が、限外濾過及び/又はナノ濾過セルによる限外濾過及び/又はナノ濾過を含む請求項28に記載の方法。
- 前記限外濾過及び/又はナノ濾過セルが、前濾過セクションを有する請求項29に記載の方法。
- 濾過が、少なくとも1つの分子量カットオフユニットを含む限外濾過及び/又はナノ濾過セルで行われる請求項1から30のいずれかに記載の方法。
- 前記限外濾過及び/又はナノ濾過セルが、少なくとも2つの分子量カットオフユニットを含む請求項31に記載の方法。
- 前記少なくとも1つの分子量カットオフユニットが、工程(D)では0.5kDa~2kDaのカットオフレベルを有する請求項31から32のいずれかに記載の方法。
- 前記少なくとも1つの分子量カットオフユニットが、工程(F)では1kDa~1.5kDaのカットオフレベルを有する請求項31から33のいずれかに記載の方法。
- 前記少なくとも1つの低分子量芳香族リグニン由来化合物が、1つ又は2つの芳香族環を含む請求項1から34のいずれかに記載の方法。
- 前記少なくとも1つの低分子量芳香族リグニン由来化合物が、2つの非環化芳香族環を含む請求項35に記載の方法。
- 前記少なくとも1つの低分子量芳香族リグニン由来化合物が、2つの芳香族環を含み、前記2つの芳香族環が、リンカー部分によって、又は結合によって連結される請求項35から36のいずれかに記載の方法。
- 前記リンカー部分が、脂肪族リンカーである請求項37に記載の方法。
- 前記少なくとも1つの低分子量芳香族リグニン由来化合物が、ビフェニル部分を形成する2つの芳香族環を含む請求項35から38のいずれかに記載の方法。
- 前記1つ又は2つの芳香族環が炭素環である請求項35から39のいずれかに記載の方法。
- 前記芳香族環が少なくとも1つの位置で官能基で置換されている請求項35から40のいずれかに記載の方法。
- 前記芳香族環が2つの位置で官能基で置換されている請求項41に記載の方法。
- 前記官能基の少なくとも1つがアルコキシ又はヒドロキシルである請求項41から42のいずれかに記載の方法。
- 前記少なくとも1つの低分子量芳香族リグニン由来化合物が、式(Ia):

R 6 は、水素、ヒドロキシ、直鎖又は分枝のC 1-6 カルボキシル、直鎖又は分枝の任意に置換されたC 1-6 アルデヒド、及び直鎖又は分枝の任意に置換されたC 1-6 アルコールからなる群から選択される。)又は
式(Ib):

R 10 は、水素、ヒドロキシ、直鎖又は分枝の任意に置換されたC 1-6 カルボキシル、直鎖又は分枝の任意に置換されたC 1-6 アルデヒド、及び直鎖又は分枝の任意に置換されたC 1-6 アルコールからなる群から選択される。)によって特徴付けられる請求項1から43のいずれかに記載の方法。 - 前記少なくとも1つの低分子量芳香族リグニン由来化合物が、ビフェニル、ベンジルアルコール、ベンズアルデヒド、及び安息香酸のフェノール誘導体、及び/又は前記のいずれかの誘導体、及び/又は前記の組合せからなる群から選択される請求項1から44のいずれかに記載の方法。
- 前記少なくとも1つの低分子量芳香族リグニン由来化合物が、p-ヒドロキシベンジルアルコール、p-ヒドロキシベンズアルデヒド、及びp-ヒドロキシ安息香酸の誘導体、及び/又は前記のいずれかの誘導体、及び/又は前記の組合せからなる群から選択される請求項45に記載の方法。
- 前記少なくとも1つの低分子量芳香族リグニン由来化合物が、バニリン、グアイアコール、オイゲノール、シリンゴール、フェノール、シリンガアルデヒド、及び/又は前記のいずれかの誘導体、及び/又は前記の組合せからなる群から選択される請求項45に記載の方法。
- 工程(F)によって提供される前記少なくとも1つの低分子量芳香族リグニン由来化合物が1つの芳香族環を含み、工程(G)において更に処理され、1つの芳香族環を含む前記低分子量芳香族リグニン由来化合物が環化反応に付され、環化反応生成物は、低分子量芳香族二環式又は三環式環化芳香族リグニン由来化合物であり、前記化合物が、以下の式(II)、(III)、又は(IV)によって特徴付けられる請求項1から38及び40から47のいずれかに記載の方法。

式(II)のR 1 及びR 4 は、水素、ヒドロキシ、直鎖又は分枝の任意に置換されたC 1-6 カルボキシル、直鎖又は分枝の任意に置換されたC 1-6 アルデヒド、及び直鎖又は分枝の任意に置換されたC 1-6 アルコールからなる群から選択され、
式(III)のR 1 ~R 10 は、それぞれ独立して、水素、ヒドロキシ、カルボキシ、直鎖又は分枝の任意に置換されたC 1-6 アルキル、直鎖又は分枝の任意に置換されたC 1-6 アルケニル、直鎖又は分枝の任意に置換されたC 1-6 アルコール、直鎖又は分枝の任意に置換されたC 1-6 アミノアルキル、直鎖又は分枝の任意に置換されたC 1-6 カルボキシアルキル、直鎖又は分枝の任意に置換されたC 1-6 アルコキシ、直鎖又は分枝の任意に置換されたC 1-6 アルデヒド、エステル、オキソ、又はカルボニルから選択され、
式(IV)のR 2 、R 3 、及びR 7 ~R 10 は、それぞれ独立して、水素、ヒドロキシ、カルボキシ、直鎖又は分枝の任意に置換されたC 1-6 アルキル、直鎖又は分枝の任意に置換されたC 1-6 アルケニル、直鎖又は分枝の任意に置換されたC 1-6 アルコール、直鎖又は分枝の任意に置換されたC 1-6 アミノアルキル、直鎖又は分枝の任意に置換されたC 1-6 カルボキシアルキル、直鎖又は分枝の任意に置換されたC 1-6 アルコキシ、直鎖又は分枝の任意に置換されたC 1-6 アルデヒド、エステル、オキソ、又はカルボニルから選択され、
式(IV)のR 1 、R 4 、R 5 、及びR 6 は、水素、ヒドロキシ、直鎖又は分枝の任意に置換されたC 1-6 カルボキシル、直鎖又は分枝の任意に置換されたC 1-6 アルデヒド、及び直鎖又は分枝の任意に置換されたC 1-6 アルコールからなる群から選択される。) - 前記環化反応が、フリーデル-クラフツアシル化である請求項48に記載の方法。
- 前記式(II)のR 2 、R 3 、及びR 5 ~R 8 の少なくとも1つは、ヒドロキシ又はC 1-3 アルコキシである請求項48から49のいずれかに記載の方法。
- 前記式(III)のR 2 、R 4 、R 5 、R 6 、及びR 8 の少なくとも1つは、ヒドロキシ又はC 1-3 アルコキシである請求項48から50のいずれかに記載の方法。
- 前記式(IV)のR 2 、R 3 、及びR 7 ~R 10 の少なくとも1つは、ヒドロキシ又はC 1-3 アルコキシである請求項48から51のいずれかに記載の方法。
- 工程(F)によって提供される前記少なくとも1つの低分子量芳香族リグニン由来化合物が、
(i.)H 2 O 2 、O 2 、及び空気からなる群から選択される酸化剤、及び
(ii.)均一系又は不均一系触媒の存在下で、前記少なくとも1つの低分子量芳香族リグニン由来化合物を酸化することにより工程(H)で更に修飾される請求項1から52のいずれかに記載の方法。 - 前記均一系又は不均一系触媒が、金属イオン又は半金属成分を含む請求項53に記載の方法。
- 工程(H)が、少なくとも1つのヒドロキノン化合物を提供し(工程H.1)、前記化合物が、式(Va):

式(Vb):

- 工程(H)が、(工程H.1)よりも過酷な酸化条件下で、少なくとも1つキノン化合物を提供し(工程H.2)、前記化合物が、式(VIa)~(VIb):




- 工程(H.1)によって提供される前記少なくとも1つのヒドロキノン化合物が、工程(I)において更に酸化され、請求項56に記載の式(VIa)~(VId)によって特徴付けられるキノン化合物を生成する請求項55に記載の方法。
- 前記更なる酸化が、電池のセルスタック中又は不均一系触媒によって行われる請求項57に記載の方法。
- 工程(G)によって提供される前記低分子量芳香族二環式又は三環式環化化合物が、
(i.)H 2 O 2 、O 2 、及び空気からなる群から選択される酸化剤、及び
(ii.)不均一系又は均一系触媒の存在下で、前記少なくとも1つの低分子量芳香族二環式又は三環式環化化合物を酸化することにより工程(H)で更に修飾され、少なくとも1つのキノン及び/又はヒドロキノン化合物を提供し、前記化合物が、以下の式(VII)、(VIII)、及び/又は(IX)によって特徴付けられる請求項48から52のいずれかに記載の方法。

- 前記均一系又は不均一系触媒が、金属イオン又は半金属成分を含む請求項59に記載の方法。
- 工程(H)、(H.1)、(H.2)又は(H.1)及び(I)によって提供される前記少なくとも1つのキノン及び/又はヒドロキノン化合物を精製工程(J)に付し、抽出又は蒸留によって、前記少なくとも1つのキノン及び/又はヒドロキノン化合物を残留化合物から分離する請求項53から60のいずれかに記載の方法。
- 前記抽出が、固相抽出又は流体-流体相抽出である請求項61に記載の方法。
- 前記少なくとも1つのキノン及び/又はヒドロキノン化合物が、誘導体化工程(K)に付されて更に修飾され、1つ以上の水素、ヒドロキシ、カルボキシ、直鎖又は分岐の任意に置換されたC 1-6 アルキル、直鎖又は分枝の任意に置換されたC 1-6 アルケニル、直鎖又は分枝の任意に置換されたC 1-6 アルコール、直鎖又は分枝の任意に置換されたC 1-6 アミノアルキル、直鎖又は分枝の任意に置換されたC 1-6 カルボキシアルキル、直鎖又は分枝の任意に置換されたC 1-6 アルコキシ、直鎖又は分枝の任意に置換されたC 1-6 アルデヒド、エステル、ハロゲン、アミン、アミノ、アミド、ニトロ、オキソ、カルボニル、ホスホリル、ホスホニル、シアニド、又はスルホニルの基が、式(I)から(IX)のいずれかで表される化合物に、オキソ又はヒドロキシル基によって特徴付けられる以外のアリール構造の位置に導入され、
前記基が、前記アリール構造に直接結合している、又はアルキルリンカーを介して前記アリール構造に結合している請求項53から62のいずれかに記載の方法。 - 前記アルキルリンカーが、メチルリンカーである請求項63に記載の方法。
- 前記誘導体化工程(K)が、少なくとも1つの還元反応、酸化反応、置換反応、求核付加及び/又は求電子置換反応を含む請求項63から64のいずれかに記載の方法。
- 前記少なくとも1つのキノン及び/又はヒドロキノン化合物がアントラキノン化合物である請求項59から65のいずれかに記載の方法。
- 前記アントラキノン化合物が、式(X)によって特徴付けられる請求項66に記載の方法。

- (i)任意に、流セパレータ、(ii)任意に、単離ユニット、(iii)分解ユニット、及び(iv)分離ユニットを含む請求項1から47のいずれかに記載の工程(C)~(F)を行うためのアセンブリ。
- (v)任意に、環化ユニット、(vi)酸化ユニット、(vii)誘導体化ユニット、及び任意に(viii)精製ユニットを更に含む請求項1から68のいずれかに記載の工程(C)~(K)を行うためのアセンブリ。
- 前記パルプ化プロセスを用いるパルプ及び/又は紙製造プロセスをプラントによって適用するための方法であって、前記プラントが請求項68から69のいずれかに記載のアセンブリを備えていることを特徴とする方法。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/EP2016/000575 WO2017174098A1 (en) | 2016-04-07 | 2016-04-07 | Method for producing low molecular weight aromatic lignin-derived compounds |
| EPPCT/EP2016/000575 | 2016-04-07 | ||
| EP2017000198 | 2017-02-13 | ||
| EPPCT/EP2017/000198 | 2017-02-13 | ||
| PCT/EP2017/000462 WO2017174207A1 (en) | 2016-04-07 | 2017-04-07 | Method for producing low molecular weight aromatic lignin-derived compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2019513831A JP2019513831A (ja) | 2019-05-30 |
| JP7050747B2 true JP7050747B2 (ja) | 2022-04-08 |
Family
ID=58536933
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019503620A Active JP7050747B2 (ja) | 2016-04-07 | 2017-04-07 | 低分子量芳香族リグニン由来化合物を製造するための方法 |
| JP2019503619A Pending JP2019516781A (ja) | 2016-04-07 | 2017-04-07 | スルホン化芳香族化合物 |
| JP2021142062A Pending JP2022000431A (ja) | 2016-04-07 | 2021-09-01 | スルホン化芳香族化合物 |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019503619A Pending JP2019516781A (ja) | 2016-04-07 | 2017-04-07 | スルホン化芳香族化合物 |
| JP2021142062A Pending JP2022000431A (ja) | 2016-04-07 | 2021-09-01 | スルホン化芳香族化合物 |
Country Status (11)
| Country | Link |
|---|---|
| US (5) | US11225756B2 (ja) |
| EP (4) | EP4180504A3 (ja) |
| JP (3) | JP7050747B2 (ja) |
| KR (2) | KR20180134369A (ja) |
| CN (2) | CN109072089B (ja) |
| AU (3) | AU2017246493A1 (ja) |
| BR (2) | BR112018069518A8 (ja) |
| CA (2) | CA3017991A1 (ja) |
| MY (1) | MY191139A (ja) |
| WO (2) | WO2017174206A1 (ja) |
| ZA (2) | ZA201806216B (ja) |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109072089B (zh) | 2016-04-07 | 2021-06-29 | Cmblu企划股份公司 | 经磺化的芳香族化合物 |
| WO2019072386A1 (en) * | 2017-10-11 | 2019-04-18 | Cmblu Projekt Ag | NOVEL PROCESSES FOR TREATING LIGNOCELLULOSIC MATERIAL |
| WO2018146344A1 (en) | 2017-02-13 | 2018-08-16 | Cmblu Projekt Ag | Process for the production of sulphonated low molecular weight derivatives from lignin |
| US11788228B2 (en) | 2017-02-13 | 2023-10-17 | Cmblu Energy Ag | Methods for processing lignocellulosic material |
| WO2019072385A1 (en) * | 2017-10-11 | 2019-04-18 | Cmblu Projekt Ag | REDOX BATTERY ELECTROLYTES |
| US11014951B2 (en) * | 2017-04-27 | 2021-05-25 | National Technology & Engineering Solutions Of Sandia, Llc | Compositions and methods for depolymerizing lignin using a chelator-mediated Fenton reaction |
| WO2019068918A1 (en) * | 2017-10-05 | 2019-04-11 | Cmblu Projekt Ag | PROCESSES FOR TREATING LIGNOCELLULOSIC MATERIAL |
| AU2019220364A1 (en) * | 2018-02-13 | 2020-07-30 | Cmblu Energy Ag | Aminated lignin-derived compounds and uses thereof |
| US11831017B2 (en) | 2018-02-13 | 2023-11-28 | Cmblu Energy Ag | Redox flow battery electrolytes |
| CN109456496A (zh) * | 2018-11-26 | 2019-03-12 | 广州楹鼎生物科技有限公司 | 一种木质素的纯化方法 |
| FI129846B (en) * | 2019-02-01 | 2022-09-30 | Andritz Oy | Method for producing oxidized lignin in kraft pulp mills |
| EP3693508A1 (en) * | 2019-02-08 | 2020-08-12 | Scitech-service OY | Method for lignin production |
| CN109824497B (zh) * | 2019-03-06 | 2020-10-27 | 华南理工大学 | 一种微波协同金属卟啉催化氧化降解碱木质素制备单苯环化合物的方法 |
| WO2020219021A1 (en) * | 2019-04-23 | 2020-10-29 | United States Of America As Represented By The Secretary Of Agriculture | Self-expanding lignofoam compositions and lignofoams made therefrom |
| WO2021058483A1 (en) | 2019-09-23 | 2021-04-01 | Université Catholique de Louvain | Process for producing lignin components from lignocellulosic biomass |
| KR102194179B1 (ko) * | 2020-03-26 | 2020-12-22 | 서울과학기술대학교 산학협력단 | 활물질 및 이의 전구체의 혼합물을 포함하는 전해액 |
| EP4139374A1 (en) * | 2020-04-23 | 2023-03-01 | Woodchem S.A. | Process of catalytic cracking of solid waste from pine derivatives industry |
| CN111821861B (zh) * | 2020-08-21 | 2022-01-07 | 烟台大学 | 一种星型分子化合物制备高通量有机溶剂纳滤膜的方法 |
| CN112176754A (zh) * | 2020-09-30 | 2021-01-05 | 青岛科技大学 | 一种实现木质纤维素分离的装置及工艺 |
| CN112871146B (zh) * | 2021-01-14 | 2023-05-23 | 常州大学 | 一种双功能金属-有机框架材料改性复合膜及其制备方法和应用 |
| CA3110357A1 (en) * | 2021-02-25 | 2022-08-25 | Sixring Inc. | Modified sulfuric acid and uses thereof |
| WO2023008548A1 (ja) * | 2021-07-29 | 2023-02-02 | 三菱ケミカル株式会社 | 水系電解液及びそれに用いる塩、並びに該水系電解液を用いたレドックスフロー電池 |
| CN113598195B (zh) * | 2021-08-06 | 2022-06-03 | 南京林业大学 | 小分子木质素作为植物生长调节剂的应用 |
| WO2023044364A1 (en) | 2021-09-15 | 2023-03-23 | Enko Chem, Inc. | Protoporphyrinogen oxidase inhibitors |
| EP4413094A1 (en) * | 2021-10-07 | 2024-08-14 | Sund Group S.R.O. | A process for production of fossil free hydrocarbons from lignocellulosic material |
| JP2023092820A (ja) * | 2021-12-22 | 2023-07-04 | 三菱重工業株式会社 | アントラキノン類活物質 |
| JPWO2023120445A1 (ja) * | 2021-12-22 | 2023-06-29 | ||
| CN114307362A (zh) * | 2021-12-30 | 2022-04-12 | 无锡东恒新能源科技有限公司 | 一种碳纳米管浆料过滤装置 |
| CN114989449B (zh) * | 2022-06-21 | 2023-04-18 | 南京林业大学 | 一种木质素-蒽醌电解质材料及其制备方法和应用 |
| CN115821619A (zh) * | 2022-12-27 | 2023-03-21 | 上海昶法新材料有限公司 | 一种利用硫酸盐制浆废液提取木质素的方法 |
| CN117691160B (zh) * | 2023-12-07 | 2024-05-07 | 温州锌时代能源有限公司 | 液流电池电解液及其应用 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011057636A (ja) | 2009-09-11 | 2011-03-24 | Tosoh Corp | アントラキノン誘導体の製造方法 |
| US20110268652A1 (en) | 2008-09-08 | 2011-11-03 | Basf Se | Method for the integrated production of cellulose and low-molecular-weight reusable materials |
| US20130232852A1 (en) | 2012-03-09 | 2013-09-12 | Thesis Chemistry, Llc | Method for tiered production of biobased chemicals and biofuels from lignin |
| JP2015534708A (ja) | 2012-09-26 | 2015-12-03 | プレジデント アンド フェローズ オブ ハーバード カレッジ | 低分子有機化合物ベースのフロー電池 |
| JP2019516781A (ja) | 2016-04-07 | 2019-06-20 | ツェーエムブルー プロイェクト アーゲー | スルホン化芳香族化合物 |
Family Cites Families (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1916627A (en) * | 1930-02-20 | 1933-07-04 | Gen Aniline Works Inc | Process of preparing 1.7-dihydroxynaphthalene |
| US1963383A (en) * | 1930-03-20 | 1934-06-19 | Nat Aniline & Chem Co Inc | Process of sulphonating anthraquinone and its derivatives |
| CH489565A (de) | 1968-04-08 | 1970-04-30 | Sandoz Ag | Verfahren zur Herstellung von Anthrachinonverbindungen |
| GB1502275A (en) | 1975-05-01 | 1978-03-01 | Ici Ltd | Naphthoquinone derivatives |
| JPS51138666A (en) | 1975-05-26 | 1976-11-30 | Kawasaki Kasei Chem Ltd | Purification of anthraquinone |
| JPS51100064A (ja) | 1975-06-05 | 1976-09-03 | Kogyo Gijutsuin | Antorakinonnosurupponkahoho |
| JPS52144662A (en) | 1976-05-26 | 1977-12-02 | Nippon Jiyouriyuu Kougiyou Kk | Production of alphaa anthraquinone sulphonic acid |
| US4124606A (en) * | 1976-12-08 | 1978-11-07 | Buffalo Color Corporation | Sulfonation of anthraquinone in sulfur dioxide solvent |
| RO76126A2 (ro) | 1978-07-13 | 1981-05-30 | Combinatul De Produse Organice,Ro | Procedeu pentru prepararea acizilor antrachinon 2,6-si-2,7-disulfonici |
| US4420644A (en) | 1981-08-24 | 1983-12-13 | Hydrocarbon Research, Inc. | Lignin hydrocracking process to produce phenol and benzene |
| SU1129204A1 (ru) | 1981-10-23 | 1984-12-15 | Предприятие П/Я В-8611 | Способ получени сернокислотной пасты 1,5-диокси-2,6-дисульфоантрахинона |
| JPS6094401A (ja) | 1983-10-31 | 1985-05-27 | Asahi Chem Ind Co Ltd | 吸液特性のすぐれたセルロース誘導体およびその製造方法 |
| JPH07117695B2 (ja) | 1988-04-15 | 1995-12-18 | 富士写真フイルム株式会社 | 輻射線感応性組成物 |
| US5002634A (en) | 1988-08-23 | 1991-03-26 | Institute Of Paper Science And Technology, Inc. | Method for the delignification of wood pulp utilizing fused ring quinone compounds prepared from lignin or lignin derived substances |
| US5932752A (en) * | 1995-12-29 | 1999-08-03 | Council Of Scientific And Industrial Research | Process for the C-C bond forming reaction using solid acid catalysts |
| JPH09227499A (ja) | 1996-02-23 | 1997-09-02 | Konika Chem:Kk | フェノ−ル類のスルホン化方法 |
| US5944953A (en) | 1996-03-12 | 1999-08-31 | Le Centre Specialise En Pates Et Papiers (Cspp) Du College D'enseignement General Et Professionnel De Trois-Riveres | Process for simultaneous mechanical and chemical defibration of corn stalks and straw materials |
| KR0182834B1 (ko) | 1996-05-16 | 1999-05-15 | 김흥기 | 안트라퀴논류의 제조방법 |
| GB9620357D0 (en) | 1996-09-27 | 1996-11-13 | Yorkshire Chemicals Plc | Fluxing agents for the reflowing of electro-deposited tinplate |
| JP3813864B2 (ja) | 2001-11-29 | 2006-08-23 | 株式会社日立製作所 | 抵抗体素子 |
| WO2004106624A1 (en) | 2003-06-03 | 2004-12-09 | Pacific Pulp Resources Inc. | Method for producing pulp and lignin |
| US8652705B2 (en) | 2005-09-26 | 2014-02-18 | W.L. Gore & Associates, Inc. | Solid polymer electrolyte and process for making same |
| US7365220B2 (en) | 2005-09-29 | 2008-04-29 | Momentive Performance Materials Inc. | Process for the recovery of alkoxysilanes obtained from the direct reaction of silicon with alkanols |
| WO2009083940A2 (en) | 2008-01-03 | 2009-07-09 | Action Medicines S.L. | Processes for the preparation of 2,5-dihydroxybenzenesulfonic acid salts |
| CN101475758B (zh) | 2009-01-16 | 2012-01-18 | 山西永东化工股份有限公司 | 黄腐植酸降解改性物接枝普通炭黑制备色素炭黑的方法 |
| DE102009032294A1 (de) | 2009-07-09 | 2011-01-13 | Aktiebolaget Skf | Lageranordnung |
| CN102040483B (zh) | 2009-10-21 | 2013-08-28 | 中国石油化工股份有限公司 | 利用木质素生产芳基化合物的方法 |
| GB201006488D0 (en) * | 2010-04-19 | 2010-06-02 | Univ Belfast | Battery |
| CN103000924B (zh) | 2011-09-16 | 2015-02-18 | 清华大学 | 一种有机相双液流电池 |
| US20130079566A1 (en) | 2011-09-27 | 2013-03-28 | Nevada, | Catalytic process for conversion of biomass into hydrocarbon fuels |
| US20130116424A1 (en) | 2011-11-09 | 2013-05-09 | Thesis Chemistry, Llc | Method for producing biobased chemicals from plant biomass |
| US20130232853A1 (en) | 2012-03-09 | 2013-09-12 | Thesis Chemistry, Llc | Method for selective production of biobased chemicals and biofuels from plant lignin |
| JP2013254685A (ja) | 2012-06-08 | 2013-12-19 | National Institute Of Advanced Industrial & Technology | レドックスフロー電池及びそれを用いた燃料電池システム |
| KR101532306B1 (ko) | 2012-11-23 | 2015-06-30 | 한국화학연구원 | 2개 이상의 술폰화 방향족기로 치환된 페닐 펜던트를 포함하는 고분자 이온전도체 |
| PE20160101A1 (es) | 2013-03-08 | 2016-03-09 | Univ Central Florida Res Found | Catalizadores para la despolimerizacion oxidativa mecanocatalitica de materiales que contienen un polimero y metodos para fabricar productos de reaccion oxidados utilizandolos |
| JP3203665U (ja) | 2013-03-15 | 2016-04-14 | グラフテック インターナショナル ホールディングス インコーポレーテッドGrafTech International Holdings Inc. | フローバッテリー用の改良された電極 |
| SE1550117A1 (sv) * | 2013-05-29 | 2015-02-04 | Kiram Ab | A method for the treatment of spent pulping liquor for the removal and production of a lignin containing product |
| EP3011627B1 (en) | 2013-06-17 | 2022-05-04 | University of Southern California | Inexpensive metal-free organic redox flow battery (orbat) for grid-scale storage |
| JP6643983B2 (ja) | 2013-09-26 | 2020-02-12 | プレジデント アンド フェローズ オブ ハーバード カレッジ | キノン及びヒドロキノン系フロー電池 |
| WO2015148357A1 (en) | 2014-03-24 | 2015-10-01 | Cornell University | Symmetric redox flow battery containing organic redox active molecule |
| US10458066B2 (en) * | 2014-08-01 | 2019-10-29 | Organic Chemical Llc | Process for the isolation of lignin from black liquor and modification of the lignin for plastic applications |
| FR3030561B1 (fr) | 2014-12-18 | 2017-01-27 | Ifp Energies Now | Procede de transformation de biomasse lignocellulosique en molecules oxygenees et en hydrocarbures sans etape de pretraitement |
| US9825323B2 (en) | 2015-01-06 | 2017-11-21 | Toyota Motor Engineering & Manufacturing North America, Inc. | Quinone-based high energy density liquid active material for flow battery |
| US20180048011A1 (en) | 2015-03-06 | 2018-02-15 | President And Fellows Of Harvard College | HIGH pH ORGANIC FLOW BATTERY |
| JP6759233B2 (ja) * | 2015-04-01 | 2020-09-23 | フンダシオン セントロ デ インベスティガシオン コオペラティバ デ エネルヒアス アルテルナティバス セイセ エネルヒグネ フンダツィオアFundacion Centro De Investigacion Cooperativa De Energias Alternativas Cic Energigune Fundazioa | レドックスフロー電池用の有機電解質化合物 |
| EP3392784B1 (en) | 2016-01-14 | 2021-12-15 | Huawei Technologies Co., Ltd. | Method and system for managing resource objects |
| WO2017174098A1 (en) | 2016-04-07 | 2017-10-12 | Cmblu Projekt Ag | Method for producing low molecular weight aromatic lignin-derived compounds |
| US10833345B2 (en) | 2016-09-30 | 2020-11-10 | University Of Southern California | Materials for high-performance aqueous organic redox flow batteries |
| US20180099917A1 (en) | 2016-10-11 | 2018-04-12 | University Of Kentucky Research Foundation | Reversibly reducible materials and use thereof |
| WO2018145720A1 (en) | 2017-02-10 | 2018-08-16 | Cmblu Projekt Ag | Flow-by electrode unit and use thereof, redox flow battery system and use thereof, method of manufacturing a flow-by electrode unit, method of operating a redox flow battery system |
| US11788228B2 (en) | 2017-02-13 | 2023-10-17 | Cmblu Energy Ag | Methods for processing lignocellulosic material |
| WO2018146344A1 (en) | 2017-02-13 | 2018-08-16 | Cmblu Projekt Ag | Process for the production of sulphonated low molecular weight derivatives from lignin |
| EP4102617A1 (en) | 2017-02-13 | 2022-12-14 | CMBlu Energy AG | Compositions comprising combinations of group substituted benzene , naphtalene and anthracene sulfonated compounds |
| AU2019220364A1 (en) | 2018-02-13 | 2020-07-30 | Cmblu Energy Ag | Aminated lignin-derived compounds and uses thereof |
| US11831017B2 (en) | 2018-02-13 | 2023-11-28 | Cmblu Energy Ag | Redox flow battery electrolytes |
-
2017
- 2017-04-07 CN CN201780022496.4A patent/CN109072089B/zh active Active
- 2017-04-07 JP JP2019503620A patent/JP7050747B2/ja active Active
- 2017-04-07 BR BR112018069518A patent/BR112018069518A8/pt not_active Application Discontinuation
- 2017-04-07 EP EP22203648.5A patent/EP4180504A3/en active Pending
- 2017-04-07 CA CA3017991A patent/CA3017991A1/en active Pending
- 2017-04-07 AU AU2017246493A patent/AU2017246493A1/en not_active Abandoned
- 2017-04-07 KR KR1020187032231A patent/KR20180134369A/ko not_active Application Discontinuation
- 2017-04-07 CN CN201780022306.9A patent/CN109072088B/zh active Active
- 2017-04-07 WO PCT/EP2017/000461 patent/WO2017174206A1/en active Application Filing
- 2017-04-07 EP EP22203539.6A patent/EP4180503A1/en active Pending
- 2017-04-07 US US16/091,437 patent/US11225756B2/en active Active
- 2017-04-07 EP EP17716793.9A patent/EP3440158A1/en not_active Withdrawn
- 2017-04-07 US US16/091,436 patent/US11008284B2/en active Active
- 2017-04-07 KR KR1020187032226A patent/KR20180133888A/ko not_active Application Discontinuation
- 2017-04-07 WO PCT/EP2017/000462 patent/WO2017174207A1/en active Application Filing
- 2017-04-07 BR BR112018069113-5A patent/BR112018069113B1/pt active IP Right Grant
- 2017-04-07 MY MYPI2018703302A patent/MY191139A/en unknown
- 2017-04-07 EP EP17716792.1A patent/EP3440157A1/en not_active Withdrawn
- 2017-04-07 CA CA3017989A patent/CA3017989A1/en active Pending
- 2017-04-07 JP JP2019503619A patent/JP2019516781A/ja active Pending
- 2017-04-07 AU AU2017246494A patent/AU2017246494B2/en active Active
-
2018
- 2018-09-14 ZA ZA2018/06216A patent/ZA201806216B/en unknown
- 2018-09-14 ZA ZA2018/06214A patent/ZA201806214B/en unknown
-
2021
- 2021-02-17 US US17/177,567 patent/US11773537B2/en active Active
- 2021-09-01 JP JP2021142062A patent/JP2022000431A/ja active Pending
-
2023
- 2023-05-05 AU AU2023202814A patent/AU2023202814B2/en active Active
- 2023-05-15 US US18/197,415 patent/US20230304221A1/en not_active Abandoned
-
2024
- 2024-05-08 US US18/657,949 patent/US20240318382A1/en active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20110268652A1 (en) | 2008-09-08 | 2011-11-03 | Basf Se | Method for the integrated production of cellulose and low-molecular-weight reusable materials |
| JP2011057636A (ja) | 2009-09-11 | 2011-03-24 | Tosoh Corp | アントラキノン誘導体の製造方法 |
| US20130232852A1 (en) | 2012-03-09 | 2013-09-12 | Thesis Chemistry, Llc | Method for tiered production of biobased chemicals and biofuels from lignin |
| JP2015534708A (ja) | 2012-09-26 | 2015-12-03 | プレジデント アンド フェローズ オブ ハーバード カレッジ | 低分子有機化合物ベースのフロー電池 |
| JP2019516781A (ja) | 2016-04-07 | 2019-06-20 | ツェーエムブルー プロイェクト アーゲー | スルホン化芳香族化合物 |
Non-Patent Citations (2)
| Title |
|---|
| B.Moodley et al ,The electro-oxidation of lignin in sappi saiccor dissolving pulp mill effluent ,Water SA,2011年,volume 37, number 1,p33-40 |
| Joseph Zakzeski et al.,The Catalytic Valorization of Lignin for the Production of Renewable Chemicals,Chemical Reviews,2010年,volume 110, number 6,p3552-3599 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7050747B2 (ja) | 低分子量芳香族リグニン由来化合物を製造するための方法 | |
| US11788228B2 (en) | Methods for processing lignocellulosic material | |
| EP3580304A1 (en) | Process for the production of sulphonated low molecular weight derivatives from lignin | |
| WO2017174098A1 (en) | Method for producing low molecular weight aromatic lignin-derived compounds | |
| AU2024204002A1 (en) | Aminated lignin-derived compounds and uses thereof | |
| EP4102617A1 (en) | Compositions comprising combinations of group substituted benzene , naphtalene and anthracene sulfonated compounds | |
| EA040994B1 (ru) | Способ получения обладающих низкой молекулярной массой ароматических образованных из лигнина соединений | |
| EA038804B1 (ru) | Сульфированные образованные из лигнина соединения и их применение |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20190207 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20200324 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20201118 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210112 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210511 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20210810 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210901 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20220301 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20220329 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7050747 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |