JP6992084B2 - 高リン血症治療剤 - Google Patents
高リン血症治療剤 Download PDFInfo
- Publication number
- JP6992084B2 JP6992084B2 JP2019549288A JP2019549288A JP6992084B2 JP 6992084 B2 JP6992084 B2 JP 6992084B2 JP 2019549288 A JP2019549288 A JP 2019549288A JP 2019549288 A JP2019549288 A JP 2019549288A JP 6992084 B2 JP6992084 B2 JP 6992084B2
- Authority
- JP
- Japan
- Prior art keywords
- particles
- mass
- hyperphosphatemia
- therapeutic agent
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images


















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/74—Synthetic polymeric materials
- A61K31/785—Polymers containing nitrogen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Description
CKD患者および透析患者における高リン血症治療のリン吸着薬としては、炭酸カルシウム、炭酸ランタンなどの金属系のリン吸着薬、また、セベラマー塩酸塩、ビキサロマーなどのポリマー系のリン吸着薬が使用されている。これらの治療薬は、いずれも摂取した食物中のリンを消化管内で吸着することにより血清リン濃度低下作用を示す。
セベラマー塩酸塩は、例えば、媒体中に乳化した粒子の形態として調製する場合がある。特許文献4~5では、乳化したポリアリルアミン塩酸塩に架橋剤を添加して、架橋ポリアリルアミンポリマーを製造している。架橋剤としては、エピクロロヒドリンを使用している。
セベラマー塩酸塩の添付文書における投与量は3~9g/日、服薬錠数は12~36錠/日である。セベラマー塩酸塩は、透析患者にとって規定の用量における服薬量の負荷が大きい薬剤である。
ビキサロマーの添付文書における投与量は1.5~7.5g/日、服薬カプセル数は6~30カプセル/日である。ビキサロマーは、セベラマー塩酸塩と同様に透析患者では服薬量の負荷が大きい薬剤である。
このような現状から、医療現場においては,少ない服薬量で血清リン濃度の管理が可能な高リン血症治療のためのポリマー系のリン吸着薬が必要とされている。
本発明は、血清リン濃度を十分に低下でき、服薬量の少ない高リン血症治療剤を提供することを解決すべき課題とする。
<1> 下記式(1-1)または(1-2)で表わされる繰り返し単位Aと、
下記式(2-1)または(2-2)で表わされる繰り返し単位Bと、
を少なくとも有する架橋ポリマーを含む粒子を、有効成分として含有する高リン血症治療剤;

R6、R7およびR8は、各々独立に、水素原子、炭素数1~20のアルキル基、炭素数1~20のアミノアルキル基またはその塩、炭素数2~20のアルキルアミノアルキル基またはその塩、炭素数3~20のジアルキルアミノアルキル基またはその塩、炭素数4~20のトリアルキルアンモニウムアルキル基、炭素数1~20のアルキルカルボニル基、炭素数1~20のカルボキシアルキル基または炭素数1~20のヒドロキシアルキル基を表し、
X-は、負に荷電した対イオンであり、
nは、5~7の整数を表し、
*は、繰り返し単位Aの側鎖の窒素原子との結合手を意味し、この場合、R6、R7およびR8のうちの少なくとも1つは結合手となる。
<2> nが6である、<1>に記載の高リン血症治療剤。
<3> R1、R2、R3、R4およびR5は、各々独立に、水素原子または炭素数1~20のアルキル基を表し、R6、R7およびR8は、各々独立に、水素原子または炭素数1~20のアルキル基を表す、<1>または<2>に記載の高リン血症治療剤。
<4> R1、R2、R3、R4およびR5は、水素原子を表し、R6、R7およびR8は、水素原子を表す、<1>から<3>の何れか一に記載の高リン血症治療剤。
<5> 粒子は、平均粒径が20~150μmである、<1>から<4>の何れか一に記載の高リン血症治療剤;平均粒径は、光学顕微鏡写真の1000個以上の水分散状態の粒子画像の面積から直径に換算し、その直径を用いて体積平均粒径として算出する。
<6> 粒子は、膨潤率が8~20mL/gmL/gである、<1>から<5>の何れか一に記載の高リン血症治療剤;
膨潤率は、20℃、2-モルホリノエタンスルホン酸ナトリウム2.2質量%および塩化ナトリウム0.5質量%でありpH6.3の水溶液中で、振盪および1時間以上の静置を20回以上繰り返した膨潤後の粒子体積を、膨潤前の粒子質量で除することにより算出する。
<7> 粒子が球状粒子である、<1>から<6>の何れか一に記載の高リン血症治療剤。
<8> 全架橋ポリマー中、
繰り返し単位Aの含有量が90~99モル%であり、
繰り返し単位Bの含有量が1~10モル%である、
<1>から<7>の何れか一に記載の高リン血症治療剤。
<9> 上記粒子は、パルスNMRで得られる自由誘導減衰信号を最小二乗法によってスピン-スピン緩和時間T2の長い成分から順に差し引き、波形分離することにより、スピン-スピン緩和時間の長い方から順に非拘束部、半拘束部、拘束部の3成分に分けた場合において、半拘束部の割合が25~70%である、<1>から<8>の何れか一に記載の高リン血症治療剤。
<10> 上記粒子は、パルスNMRで得られる自由誘導減衰信号を最小二乗法によってスピン-スピン緩和時間T2の長い成分から順に差し引き、波形分離することにより、スピン-スピン緩和時間の長い方から順に非拘束部、半拘束部、拘束部の3成分に分けた場合において、拘束部の割合が30~70%である、<1>から<9>の何れか一に記載の高リン血症治療剤。
<11> リン酸吸着能が6.0~10.0mmol/gである、<1>から<10>の何れか一に記載の高リン血症治療剤;
但し、リン酸吸着能は、2.2質量%モルホリノエタンスルホン酸ナトリウム、0.47質量%塩化ナトリウム、および0.24質量%リン酸でありpH=6.4の水溶液20mL中において、粒子30mgを37℃で1時間混合撹拌した際の、混合前後の上澄み中のリン酸濃度をICP発光分光分析法により定量し、その減少量を粒子質量で除し、乾燥減量値を用いて補正することにより算出したものである。
<12> アミン価が11.0~17.5mmol/gである、<1>から<11>の何れか一に記載の高リン血症治療剤;
但し、アミン価は、超純水に分散した粒子を5規定の塩酸で処理し、0.1規定の水酸化ナトリウム水溶液による中和滴定を行うことによりアミノ基の量を定量し、それを粒子質量で除し、乾燥減量値を用いて補正することにより算出したものである。
本発明は、さらに以下を提供する。
<A> 上記の架橋ポリマーを含む粒子を、対象(ヒトを含む哺乳動物、好ましくはヒト)に投与する工程を含む、高リン血症の治療方法。
<B> 高リン血症の治療に使用するための、上記の架橋ポリマーを含む粒子。
<C> 高リン血症治療剤の製造のための、上記の架橋ポリマーを含む粒子の使用。
本発明において、特に断らない限り、各用語は、次の意味を有する。
本発明において、特に断らない限り、「~」を用いて示された数値範囲は、「~」の前後に記載される数値をそれぞれ最小値および最大値として含む範囲を意味する。
炭素数1~20のアルキル基(C1-20アルキル基)とは、メチル、エチル、プロピル、イソプロピル、ブチル、sec-ブチル、イソブチル、tert-ブチル、ペンチル、イソペンチル、2-メチルブチル、2-ペンチル、3-ペンチル、ヘキシル基などの直鎖状または分枝鎖状のC1-20アルキル基を意味する。アルキル基の炭素数は、1~10が好ましく、1~6がより好ましく、1~3がさらに好ましい。
炭素数1~20のアルキルアミノ基(C1-20アルキルアミノ基)とは、メチルアミノ、エチルアミノ、プロピルアミノ、イソプロピルアミノ、シクロプロピルアミノ、ブチルアミノ、sec-ブチルアミノ、tert-ブチルアミノ、シクロブチルアミノ、ペンチルアミノ、シクロペンチルアミノ、ヘキシルアミノ、シクロヘキシルアミノ基などの直鎖状または分枝鎖状のC1-20アルキルアミノ基を意味する。好ましい炭素数は、1~10であり、1~6がより好ましく、1~3がさらに好ましい。
炭素数2~20のジアルキルアミノ基(ジ(C1-20アルキル)アミノ基)とは、ジメチルアミノ、ジエチルアミノ、ジプロピルアミノ、ジイソプロピルアミノ、ジブチルアミノ、ジ(tert-ブチル)アミノ、ジペンチルアミノ、ジヘキシルアミノ、(エチル)(メチル)アミノ、(メチル)(プロピル)アミノ、(シクロプロピル)(メチル)アミノ、(シクロブチル)(メチル)アミノ、(シクロヘキシル)(メチル)アミノ基などの直鎖状または分枝鎖状のジ(C1-20アルキル)アミノ基を意味する。好ましい炭素数は、2~10であり、2~6がより好ましい。それらアルキル基は同一でも異なっていてもよい。
炭素数2~20のアルキルアミノアルキル基とは、アミノアルキル基におけるアミノ基の水素原子がアルキルで置換されたものであり、2つのアルキルの炭素数の合計が2~20の範囲内のものである。好ましい炭素数は、2~10であり、2~6がより好ましい。
炭素数3~20のジアルキルアミノアルキル基とは、アミノアルキル基におけるアミノ基の2つの水素原子がそれぞれアルキルで置換されたものであり、3つのアルキルの炭素数の合計が3~20の範囲内のものである。好ましい炭素数は、3~10であり、3~6がより好ましい。それらアルキルは同一でも異なっていてもよい。
炭素数1~20のアミノアルキル基の塩、炭素数2~20のアルキルアミノアルキル基の塩、および炭素数3~20のジアルキルアミノアルキル基の塩とは、アミノアルキル基、アルキルアミノアルキル基およびジアルキルアミノアルキル基における窒素原子がアンモニウム塩を形成している場合を意味する。アンモニウム塩としては有機酸または無機酸との塩が挙げられ、有機酸としてはギ酸、酢酸、シュウ酸、コハク酸、またはクエン酸などが挙げられ、無機酸としては塩酸、炭酸、硫酸、硝酸、またはリン酸などが挙げられる。
炭素数4~20のトリアルキルアンモニウムアルキル基とは、上記炭素数1~20のアルキル基のうちの炭素数1~16のアルキル基(好ましい炭素数は1~10であり、より好ましくは1~6である)の少なくとも1つの水素原子がトリアルキルアンモニウム基で置換されたものであり、アルキル基の末端の炭素原子上の水素原子が置換されたものが好ましい。トリアルキルアンモニウム基のアルキル基は、炭素数1~8のアルキル基(好ましい炭素数は1~6であり、より好ましくは1~3である)である。それらアルキルは同一でも異なっていてもよい。
炭素数1~20のカルボキシアルキル基とは、具体的には、-(CH2)n-COOHであり、式中nは1~20の整数を示す。nは好ましくは、1~10であり、1~6がより好ましい。
炭素数1~20のヒドロキシアルキル基とは、具体的には、-(CH2)n-OHであり、式中nは1~20の整数を示す。nは好ましくは、1~10であり、1~6がより好ましい。
装置:東ソー社製HLC-8320GPC
カラム:東ソー社製TSK-GEL G5000PWXL
カラム温度:40℃
流速:1.0mL/min
検量線:TOSOH TSKstandard POLY(ETHYLENE OXIDE)
溶離液:硝酸ナトリウム42.5gを水/アセトニトリル(9/1)の混合物にて5000gに希釈した溶液。

R6、R7およびR8は、各々独立に、水素原子、炭素数1~20のアルキル基、炭素数1~20のアミノアルキル基またはその塩、炭素数2~20のアルキルアミノアルキル基またはその塩、炭素数3~20のジアルキルアミノアルキル基またはその塩、炭素数4~20のトリアルキルアンモニウムアルキル基、炭素数1~20のアルキルカルボニル基、炭素数1~20のカルボキシアルキル基または炭素数1~20のヒドロキシアルキル基を表し、
X-は、負に荷電した対イオンであり、
nは、5~7の整数を表し、
*は、繰り返し単位Aの側鎖の窒素原子との結合手を意味する。
nは、6~7の整数がより好ましく、6が特に好ましい。
R1、R2、R3、R4およびR5は、各々独立に、水素原子または炭素数1~20のアルキル基であることが好ましく、水素原子であることが特に好ましい。
R6、R7およびR8は、各々独立に、水素原子または炭素数1~20のアルキル基であることが好ましく、水素原子であることが特に好ましい。
全架橋ポリマー中、繰り返し単位Aの含有量が90~99モル%であり、繰り返し単位Bの含有量が1~10モル%であることが好ましい。
本発明に係る架橋ポリマー粒子は、膨潤率の上限値が20mL/gであることが好ましく、16mL/gであることがより好ましく、14mL/gであることがさらに好ましい。また膨潤率の下限値は8mL/gであることが好ましく、9mL/gであることがより好ましく、10mL/gであることがさらに好ましい。膨潤率は8~20mL/gであることが好ましく、9~16mL/gであることがより好ましく、10~14mL/gであることがさらに好ましい。この数値範囲を満たすことで、より高い血清リン濃度の低下作用を発現する傾向がある。
本発明に係る架橋ポリマー粒子は、真円度の上限値は1である。また真円度の下限値は0.80であることが好ましく、0.90であることがより好ましい。この数値範囲を満たすことで、より高い血清リン濃度の低下作用を発現する傾向がある。なお、真円度は、光学顕微鏡写真の50個以上の水分散状態の粒子画像からの平均値として算出できる。光学顕微鏡での確認結果から、個々の粒子については真円度が1に近いものほど真球状に近いと判断した。また50個以上の水分散状態の粒子画像からの平均値が1に近いほど球状ではない架橋ポリマー粒子の含有率が低く、球状の架橋ポリマー粒子の含有率が高いと判断できる。
なお、上記平均粒径、膨潤率、真円度などの物性の測定は、実施例に記載の方法と同様の方法で測定できる。
架橋ポリマーの疎密構造は、膨潤させた粒子を凍結乾燥させ、その断面の走査電子顕微鏡像により評価できる。走査電子顕微鏡像において、本発明に係る粒子は2層構造を示す。外殻部には孔が存在しないため黒色に見え、内部には多数の孔が存在するため白色に見える。孔が存在しない領域は架橋ポリマーの存在量が多い領域であり、多数の孔が存在する領域は架橋ポリマーの存在量が少ない領域である。また、孔が存在しない領域は架橋度の高い領域であり、多数の孔が存在する領域は架橋度が低い領域である。
孔が存在しない領域は、架橋度が高いため膨潤しにくく、膨潤させた粒子でも架橋ポリマーの存在量が多いと推定される。他方、多数の孔が存在する領域は、架橋度が低いため膨潤し易く、膨潤させた粒子を凍結乾燥させるとその膨潤領域に多数の孔が生じると推定され、架橋ポリマーの存在量が少なくなると推定される。
パルスNMRの測定では、まず、測定試料を準備する。例えば、粒子100mgに重水(CIL(Cambridge Isotope Laboratories, Inc.)社製)を5mL加え、1分間振り混ぜて均一に分散させたのち、遠心沈降し上澄みをデカンテーションすることで、重水により膨潤した粒子を得る。得られた粒子に対し、上記と同様に重水を混合およびデカンテーションする操作を3回繰り返し、測定用の重水膨潤粒子(測定試料)を得る。
本発明に係る粒子は、パルスNMRより求められる非拘束部の割合が、拘束部と半拘束部の合計の残りの部分となる。本発明に係る粒子は、パルスNMRより求められる非拘束部の割合の上限値が25%であることが好ましく、20%であることがより好ましく、15%であることがさらに好ましく、10%であることが特に好ましい。非拘束部の割合は0~25%であることが好ましく、0~20%であることがより好ましく、0~15%であることがより好ましく、0~10%であることがさらに好ましい。この数値範囲を満たすことで、より高い血清リン濃度の低下作用を発現する傾向がある。
(a)パルスNMRで得られる自由誘導減衰信号を最小二乗法によってスピン-スピン緩和時間T2の長い成分から順に差し引き、波形分離することにより、スピン-スピン緩和時間の長い方から順に非拘束部、半拘束部、拘束部の3成分に分けた場合において、半拘束部の割合が25~70%であること;
(b)パルスNMRで得られる自由誘導減衰信号を最小二乗法によってスピン-スピン緩和時間T2の長い成分から順に差し引き、波形分離することにより、スピン-スピン緩和時間の長い方から順に非拘束部、半拘束部、拘束部の3成分に分けた場合において、拘束部の割合が30~70%であること;
(c)リン酸吸着能が6.0~10.0mmol/gであること;並びに
(d)アミン価が11.0~17.5mmol/gであること、
の何れか1以上の特徴を有することが好ましい。
具体的には、特徴(a)~(d)の何れか1以上の特徴としては、特徴(a)、特徴(b)、特徴(c)、特徴(d)、特徴(a)と特徴(b)の組み合わせ、特徴(a)と特徴(c)の組み合わせ、特徴(a)と特徴(d)の組み合わせ、特徴(b)と特徴(c)の組み合わせ、特徴(b)と特徴(d)の組み合わせ、特徴(c)と特徴(d)の組み合わせ、特徴(a)と特徴(b)と特徴(c)との組み合わせ、特徴(a)と特徴(b)と特徴(d)との組み合わせ、特徴(a)と特徴(c)と特徴(d)との組み合わせ、特徴(b)と特徴(c)と特徴(d)との組み合わせ、特徴(a)と特徴(b)と特徴(c)と特徴(d)との組み合わせなどが挙げられる。
本発明に係る架橋ポリマー粒子(好ましくは、架橋ポリアリルアミン粒子)は、ポリマーの乳化液を調製する乳化液調製工程と、ポリマーの乳化液に架橋剤を添加して架橋反応を行う架橋工程を経て製造されることが好ましい。乳化液調製工程と架橋工程は連続して行ってもよいし、所定時間をおいて独立した工程として行ってもよい。
乳化液調製工程は、ポリマーと親水性溶媒とを含有し粘度が10~2000mPa・sである第一溶液、および、疎水性溶媒を含有し、粘度が1~100mPa・sである第二溶液を混合して攪拌することによって、ポリマーの乳化液を得ることが好ましい。ここで、第一溶液の粘度と第二溶液の粘度との比は、0.1:1~300:1の範囲内であることが好ましい。
乳化液調製工程においては、上記式(1-1)または(1-2)で表わされる繰り返し単位Aを有するポリマーと親水性溶媒とを含有する第一溶液を使用する。
一般式(1-1)または一般式(1-2)で表される繰り返し単位を有するポリマー(以下、ポリマーAともいう)は、一般式(1-1)で表される繰り返し単位と一般式(1-2)で表される繰り返し単位の両方を含んでいてもよい。
一般式(1-1)および(1-2)において、R1~R8は、その好ましい範囲は上記と同様であり、水素原子であることが原料の入手性の観点から好ましい。
ポリマーAは、一般式(1-1)および一般式(1-2)で表される繰り返し単位以外に、さらに他の繰り返し単位を共重合成分として含んでいてもよい。
ポリマーAの塩においては、ポリマー中の全アミノ基の0%を超え50%以下が中和されていることが好ましい。
ポリマーAまたはその塩としては、ポリマーAまたは炭酸塩が好ましい。
第一溶液の粘度の測定は、25℃で行う。粘度の測定は、公知の手法によって測定できる。例えば、東輝産業社製R215型粘度計(RE-215L)により行うことができる。100mPa・sを超える場合は高粘度用コーンローター(3°×R9.7)を用いてサンプル量0.6mLで測定する。100mPa・s以下の場合は低粘度用コーンローター(0.8°×R24)を用いて、サンプル量0.2mLで測定する。指度値(TQ)が50~100%の範囲で安定するように、回転速度を設定し、粘度を読み取る。
乳化液調製工程においては、疎水性溶媒を含有し、粘度が1~100mPa・sである第二溶液を使用する。
疎水性溶媒としては特に限定はされないが、例えば、芳香族炭化水素系溶媒(例えば、ベンゼン、トルエン、キシレン、メシチレン、エチルベンゼン、ジエチルベンゼン、プロピルベンゼン、クロロベンゼン、o-ジクロロベンゼンまたはt-ブチルベンゼン等)、エステル系溶媒(例えば、酢酸エチル、酢酸ブチル、プロピレングリコールモノメチルエーテルアセテート等)、ケトン系溶媒(例えば、シクロヘキサノン等)、ハロゲン系溶媒(例えば、塩化メチレン、クロロホルム、ブロモホルムまたは四塩化炭素等)、飽和炭化水素系溶媒(例えば、流動パラフィン、ヘキサン、ヘプタン、シクロヘキサン等)、鉱油、オリーブオイルなどが挙げられる。これらは単独で用いてもよいし、2種以上を混合して用いてもよい。疎水性溶媒は、好ましくは、芳香族炭化水素系溶媒、エステル系溶媒、またはオリーブオイルであり、より好ましくは、芳香族炭化水素系溶媒であり、特に好ましくはトルエンまたはキシレンである。
第二溶液は、疎水性溶媒に加えて、疎水性溶媒以外の溶媒を含有していてもよい。疎水性溶媒以外の溶媒としては、アルコール(例えば、メタノール、エタノール、2-プロパノール、ヘキサノール、エチレングルコールモノプロピルエーテル、ポリエチレングリコールなど)、エーテル(ビス[2-メトキシエトキシエチル]、ジブチルエーテルなど)、テトラヒドロフラン、アセトニトリル等の親水性溶媒を使用してもよい。親水性溶媒としては、好ましくはアルコール、エーテルであり、さらに好ましくはアルコールであり、最も好ましくはエタノールである。
第二溶液が疎水性溶媒以外の溶媒を含有する場合、疎水性溶媒以外の溶媒の含有量は、疎水性溶媒の含有量に対して、質量比で50%以下であり、好ましくは30%以下であり、より好ましくは20%以下であり、さらに好ましくは15%以下である。含有量の下限値は、0.1%である。
親水性溶媒を含有する場合、第二溶液の粘度としては、好ましくは1~50mPa・sであり、より好ましくは1~30mPa・sであり、さらに好ましくは1~20mPa・sである。
第二溶液の粘度の測定は、第一溶液の粘度の測定と同様の方法で行うことができる。
ポリスチレン、ポリヒドロキシスチレン、ポリスチレンスルホン酸、ビニルフェノール-(メタ)アクリル酸エステル共重合体、スチレン-(メタ)アクリル酸エステル共重合体、またはスチレン-ビニルフェノール-(メタ)アクリル酸エステル共重合体等のポリスチレン誘導体;
ポリ(メタ)アクリル酸エステル共重合体、ポリ(メタ)アクリル酸メチル、ポリ(メタ)アクリルアミド、ポリアクリロニトリル、ポリエチル(メタ)アクリレート、またはポリブチル(メタ)アクリレート等のポリ(メタ)アクリル酸誘導体;
ポリメチルビニルエーテル、ポリエチルビニルエーテル、ポリブチルビニルエーテル、またはポリイソブチルビニルエーテル等のポリビニルアルキルエーテル誘導体;
ポリプロピレングリコール等のポリアルキレングリコール誘導体;
セルロース、エチルセルロース、セルロースプロピオネート、セルロースアセテートプロピオネート、セルロースアセテート、セルロースブチレート、セルロースアセテートブチレート、セルロースフタレートまたは硝酸セルロース等のセルロース誘導体(糖類);
ポリビニルブチラール、ポリビニルホルマール、ポリ酢酸ビニル等のポリ酢酸ビニル誘導体;
ポリビニルピリジン、ポリビニルピロリドン、またはポリ-2-メチル-2-オキサゾリン等の含窒素ポリマー誘導体;
ポリ塩化ビニル、またはポリ塩化ビニリデン等のポリハロゲン化ビニル誘導体;
ポリジメチルシロキサン等のポリシロキサン誘導体、
カルボジイミド樹脂、エポキシ樹脂、フェノール樹脂、メラミン樹脂、ユリア樹脂、ウレタン樹脂、ポリエチレン、ポリプロピレン、ポリアミド、ポリイミド、ポリカーボネート、液晶ポリマー、ポリエチレンテレフタレート、またはポリブチレンテレフタレート等の各種乳化剤。
第二溶液における乳化剤の含有量の上限値は、30質量%が好ましく、20質量%がより好ましく、10質量%がさらに好ましく、7質量%がよりさらに好ましい。第二溶液における乳化剤の含有量の下限値は、0.1質量%が好ましく、0.2質量%がより好ましく、0.3質量%がさらに好ましく、0.5質量%がよりさらに好ましい。
第二溶液における乳化剤の含有量は、好ましくは0.1~20質量%であり、より好ましくは0.1~10質量%であり、さらに好ましくは0.2~7質量%であり、よりさらに好ましくは0.3~5質量%であり、特に好ましくは0.4~3質量%である。
乳化液調製工程においては、上記の第一溶液および上記の第二溶液を混合し、ポリマーAの乳化液を得る。混合溶液は20~500回転/分で攪拌することが好ましい。乳化液調製工程においては、上記で規定した第一溶液および第二溶液を用いることにより、このような低速の回転でも、乳化安定性が高く、乳化粒径の分散度が小さいポリマーAの乳化液が製造できる。
攪拌を行う容器の容量は、特に限定されないが、一般的には、100mL~100,000Lの範囲内である。
攪拌を行う際の温度は、特に限定されないが、一般的には2℃~98℃であり、5℃~80℃が好ましく、10℃~70℃がより好ましい。
攪拌速度は、好ましくは20~500回転/分であり、より好ましくは30~400回転/分であり、さらに好ましくは40~300回転/分であり、特に好ましくは50~300回転/分である。
容器の最大内部径と攪拌羽根の長さの比は、容器の最大内部径(円筒形の容器の場合は直径)に対し、攪拌羽根の長さが3/10以上最大内部径未満であることが好ましく、5/10以上9/10以下がより好ましい。
容器の容量が変わった場合でも、回転数によって攪拌条件を調整できる。また攪拌羽根の大きさまたは形状と回転数を調整することで攪拌条件を最適化することが好ましい。例えば、攪拌羽根が大きければ回転数は小さめに設定し、攪拌羽根が小さい場合には回転数は大きめに設定するなど、攪拌羽根の大きさおよび形状によって回転数を調整することが好ましい。
平均乳化粒径の測定は、公知の手法によって測定でき、例えば、以下の方法で行うことができる。攪拌により得られたポリマーの乳化液を1時間静置した後、-78℃のドライアイスメタノール中に滴下し、ポリマーの粒子を凝固させる。ランダムに選んだ1000個以上の凍結粒子の光学顕微鏡写真を撮影して電子データとして保存し、アメリカ国立衛生研究所製のソフトウェアImageJを用いて凍結粒子の平均粒径を算出する。
架橋工程は、(1)ポリマーAの乳化液に架橋剤を添加して架橋反応を行うか、(2)予め第二溶液に架橋剤を混合したのちに第一溶液と第二溶液を混合して乳化し、架橋反応を行う。
架橋工程の反応時間は、1~36時間が好ましく、3~24時間がさらに好ましく、6~20時間が特に好ましい。
架橋工程は、高反応率化の観点から、第一溶液中の水を除去してから架橋反応を進行することが望ましい。そのため、ディーンスターク管等を使用し、95℃以上の温度で架橋反応を実施することが好ましい。
すなわち、水の留去が完了した後に、1~24時間反応させることが好ましい。反応時間は、2~20時間がさらに好ましく、3~16時間が特に好ましい。
架橋剤としては、炭素数5~7のアルキレン型架橋剤が好ましい。より具体的には、1,5-ジクロロペンタン、1,5-ジブロモペンタン、1,6-ジクロロヘキサン、1,6-ジブロモヘキサン、1,6-ビス(パラトルエンスルホニル)ヘキサン、1,7-ジクロロヘプタン、1,7-ジブロモヘプタンが挙げられる。これらの中でも、ジハロヘキサンが好ましく、1,6-ジブロモヘキサンまたは1,6-ジクロロヘキサンがより好ましく、1,6-ジクロロヘキサンが特に好ましい。このような疎水性の架橋剤を使用することで、より高い血清リン濃度の低下作用を発現する傾向がある。架橋剤の使用量としては、繰り返し単位Bの含有率が1~10モル%となる量であることが好ましく、1.25モル%~8モル%となる量であることがより好ましく、1.5モル%~6モル%となる量であることがさらに好ましい。
その後、架橋ポリマー粒子を所定の溶液で洗浄してろ過し、得られた粒子を乾燥させることで、本発明に係る架橋ポリマー粒子が得られる。得られた粒子は、上記式(1-1)または(1-2)で表わされる繰り返し単位Aと、上記式(2-1)または(2-2)で表わされる繰り返し単位Bと、を少なくとも有する架橋ポリマーAを含む。
なお、ビキサロマーは球状であるが、コアシェル構造を発現しない。
本発明に係る医薬製剤は、活性成分として上記架橋ポリマー粒子を単独で、または任意の他の治療のための有効成分との混合物として含有することができる。また、それら医薬製剤は、活性成分を薬学的に許容される一種またはそれ以上の担体(希釈剤、溶剤、賦形剤など)と一緒に混合し、製剤学の技術分野においてよく知られている任意の方法により製造される。
投与形態としては、錠剤などがあげられる。
経口投与に適当な、錠剤などは、乳糖などの賦形剤、澱粉などの崩壊剤、ステアリン酸マグネシウムなどの滑沢剤、ヒドロキシプロピルセルロースなどの結合剤などを用いて製造できる。
本発明に用いられる架橋ポリマー粒子の投与量および投与回数は、投与形態、患者の年齢、体重、治療すべき症状の性質もしくは重篤度などにより異なるが、通常経口の場合、成人一人あたり、0.01g~30gの範囲で、1日1回ないし数回投与する。しかしながら、これら投与量および投与回数に関しては、前述の種々の条件により変動する。
本発明の別の態様によれば、本発明に係る粒子または医薬製剤を、対象(該対象とは、人を含む哺乳動物であって、好ましくは人であり、より好ましくは高リン血症を有するCKD患者および透析患者である)に投与する工程を含む治療方法が提供される。本発明の治療方法は、高リン血症の治療のために用いられる。
本発明の別の態様によれば、高リン血症の治療に使用するための、本発明に係る粒子が提供される。
本発明の別の態様によれば、高リン血症治療剤の製造のための、本発明に係る粒子の使用が提供される。
膨潤率は、20℃、2-モルホリノエタンスルホン酸ナトリウム2.2質量%および塩化ナトリウム0.5質量%でありpH6.3の水溶液中で、振盪および1時間以上の静置を20回以上繰り返した膨潤後の粒子体積を、膨潤前の粒子質量で除することにより算出する。
振盪および1時間以上の静置を繰り返す回数は、膨潤粒子体積の変化が無くなるまで行えばよい。
より具体的には、1Lメスフラスコ中に2-モルホリノエタンスルホン酸ナトリウム(アルドリッチ社製)21.7g、塩化ナトリウム(和光純薬製)4.7gを量りとり、水を加えて1Lとした。完全に溶解した後、30質量%塩酸をpHが6.3になるまで加えて、バッファーを調整した。
各実施例および各比較例で得られた粒子を10mLメスシリンダーに0.30g秤量し、10mLのバッファーを混合し、スパチュラを用いて1分間撹拌することで粒子を一様に懸濁させた後に静置した。24時間後に沈降した膨潤粒子の体積をメスシリンダーの目盛から読み取った後、弱い振盪を1分間与えて更に24時間静置した。上記の振盪・静置を膨潤粒子体積の変化が無くなるまで繰り返し行った。変化が無くなった際の膨潤粒子体積を粒子質量(0.30g)で除することにより、膨潤率(mL/g)を算出した。
粒子の形状は、光学顕微鏡写真から判定した。より具体的には、各実施例および各比較例で得られた粒子を水に分散させたのち、ランダムに選んだ500個以上の粒子の光学顕微鏡(Nikon社製 ECLIPSE E600POL)写真を撮影した。その写真における全粒子の投影面積のうち、略円形の粒子の投影面積が60%以上である場合、それらの粒子は球状であると判定した。略円形の粒子の投影面積は、80%以上が好ましく、90%以上がより好ましく、95%以上がさらに好ましい。略円形の粒子の投影面積が高いほど好ましい。
なお、水への分散は、乾燥後の粒子をサンプル瓶に0.1g秤量し、純水を10mL加え、ふり混ぜたのち、25℃で10分間静置することで、水分散液を調整した。
平均粒径は、光学顕微鏡写真の1000個以上の水分散状態の粒子画像の面積から直径に換算し、その直径を用いて体積平均粒径として算出する。
より具体的には、各実施例および各比較例で得られた粒子を水に分散させたのち、ランダムに選んだ1000個以上の粒子の光学顕微鏡(Nikon社製 ECLIPSE E600POL)写真を撮影して電子データとして保存し、アメリカ国立衛生研究所製のソフトウェアImageJを用いて粒子の平均粒径を算出した。
光学顕微鏡での撮影は倍率50倍(接眼レンズ10倍、対物レンズ5倍)にて反射光を観察した。一枚あたりの粒子数が1000個に満たない場合は、複数枚の写真を解析し、合算した。
ImageJでの粒子解析においては、
(a)光学顕微鏡で撮影した写真をImageJにて読み込む。
(b)スムージング処理、8bit化処理、白黒2色化、穴埋め処理、および結合粒子の分割処理を実施する。
(c)ノイズを除去するため、解析範囲として粒子径10μm以上かつ真円度0.5以上に限定して解析処理を実行した。
真円度は、光学顕微鏡写真の50個以上の粒子画像の真円度:4π×(面積)/(周長の2乗)の平均値である。真円度が1のとき、正円であることを示す。
より具体的には、各実施例及び各比較例で得られた粒子を水に分散させたのち、ランダムに選んだ50個以上の粒子の光学顕微鏡(Nikon社製 ECLIPSE E600POL)写真を撮影して電子データとして保存し、アメリカ国立衛生研究所製のソフトウェアImageJを用いて粒子の真円度を算出した。
光学顕微鏡での撮影は倍率50倍(接眼レンズ10倍、対物レンズ5倍)にて反射光を観察した。一枚あたりの粒子数が50個に満たない場合は、複数枚の写真を解析し、合算した。ImageJでの粒子解析においては、
(a)光学顕微鏡で撮影した写真をImageJにて読み込む。
(b)スムージング処理、8bit化処理、白黒2色化、および穴埋め処理を実施する。
(c)粒子同士が重なっているもの、および写真の淵で切れている粒子については、真円度の算出に影響を与えるため、手動で除外した。
(d)ノイズを除去するため、解析範囲として粒子径10μm以上に限定して解析処理を実行した。
東輝産業社製R215型粘度計(RE-215L)により、25℃での粘度を測定した。100mPa・sを超える場合は高粘度用コーンローター(3°×R9.7)を用いて、サンプル量0.6mLで測定した。100mPa・s以下の場合は低粘度用コーンローター(0.8°×R24)を用いて、サンプル量0.2mLで測定した。いずれの場合も指度値(TQ)が50~100%の範囲で安定するように、回転速度を設定し、粘度を読み取った。
膨潤状態の粒子構造観察には、凍結乾燥粒子を用いた。凍結乾燥工程では、実施例や比較例で作製した粒子0.2gに超純水20mLを混合し、振り混ぜたのち1時間放置することで水分散液を調整した。次に、3000Gにて10分間遠心分離し、デカンテーションにより上澄みを除いた後、エタノール20mLを加える溶媒置換工程を3回繰り返し、エタノール分散粒子を得た。続いて、エタノールを遠心分離により除いた後、t-ブタノール20mLで溶媒置換する工程を3回繰り返し、t-ブタノール分散粒子を得た。そのt-ブタノール分散粒子を、-18℃以下にて凍結し、常法により凍結乾燥を行なった。なお、この工程は、水分散時とt-ブタノール分散時における粒径がほぼ同じになるように操作した。
実施例および比較例で作製した粒子100mgに重水(CIL社製)を5mL加え、1分間振り混ぜて均一に分散させたのち、遠心沈降し上澄みをデカンテーションすることで、重水により膨潤した粒子を得た。得られた粒子に対し、上記と同様に重水を混合およびデカンテーションする操作を3回繰り返し、測定用の重水膨潤粒子(測定試料)を得た。次に、Bruker Biospin社製MQ-20を用いて測定試料を測定した。測定はsolidecho法にて、90°パルス間隔は1μ秒、取り込み時間は5m秒、データ点数は2000点で測定した。また、核種は1H、積算回数は64回 、繰り返し時間は2秒とし、24℃で測定した。得られた自由誘導減衰信号を、式(1)に示す3成分の和として最小二乗法によりフィッティングすることにより、各成分のスピン―スピン緩和時間T2,1、T2,2、T2,3と強度A1、A2、A3を算出した。
分子運動性が低く緩和時間が短い成分を拘束部、分子運動性が高く緩和時間が長い成分を非拘束部とし、それらの中間の緩和時間をもつ成分を半拘束部とし、それぞれの成分比および緩和時間を評価した。拘束部の割合、半拘束部の割合、非拘束部の割合とは、全体に占める各領域の割合を表し、A1~A3の各々の値をA1~A3の合計値で除することにより算出した。
アミン価およびリン酸吸着能の測定においては、試料の保管中の経時による水分等の増加の影響を除外するため、別途、乾燥減量値を測定し、補正することで水分等の影響を除外した。乾燥減量の測定としてはじめに、秤量瓶を120℃で30分間乾燥し、シリカゲル入りのデシケーター中で室温まで冷却し、その重量(W1)を正確に測定した。実施例および比較例で作製した粒子を約100mg上記の秤量瓶にとり、秤量瓶の重量(W2)を測定した。次に、水酸化カリウム約20gが入った50mLバイアル瓶を減圧乾燥機中に配置し、上記の粒子入りの秤量瓶を同一の減圧乾燥機中で、120℃にて5時間乾燥処理を行い、その後シリカゲル入りのデシケーターにて室温まで冷却し、秤量瓶の重量(w3)を測定した。乾燥減量D(%)の値は式(2)により算出した。
実施例および比較例で作製した粒子100mgを4mLバイアル瓶に秤量し(正確な秤量値をWgとする)、秤量瓶を120℃で30分間乾燥し、シリカゲル入りのデシケーター中で室温まで冷却した。乾燥した粒子入りバイアル瓶に水1mLを添加し膨潤させた後、5規定の塩酸を500μL添加し、さらに2mLの水を添加した後、マグネチックスターラーにより30分間撹拌した。その後、4mLバイアル瓶の内容物を200mLビーカーに移し、超純水90mLを添加し、京都電子製の電位差自動滴定装置MCU-610およびAT-610を用い、0.1規定の水酸化ナトリウム水溶液による中和滴定を行い、中和点を与える滴定量(VmL)を求めた。アミン価は、式(3)により算出した。fHCl、fNaOHはファクター値を表す。
[(5×0.5×fHCl-0.1×V×fNaOH)/W]×{100/(100-D)} (3)
1Lメスシリンダーにモルホリノエタンスルホン酸ナトリウム(Aldrich製)21.72g、塩化ナトリウム(和光純薬製)4.67g、85%リン酸(和光純薬製)2.88gを秤量し、超純水で1Lに調整し、pH6.4のリン酸含有バッファーとした。実施例および比較例で作製した粒子30mg(正確な秤量値をW’mgとする)を20mLのポリエチレンテレフタレート製容器に秤量し、上記のリン酸含有バッファー20mLを混合し、37℃の恒温槽にて、マグネチックスターラーにより1時間撹拌した。粒子と混合する前の水溶液および粒子混合撹拌後の水溶液をシリンジフィルターにて濾過し、ICP発光分光分析法により濾液中のリン元素を定量した。リン酸吸着能は式(4)により算出した。ここで、混合前のリン濃度をP0 ppmとし、混合撹拌後のリン濃度をP ppmとした。
[(P-P0)×20/(30.97×W’)]×{100/(100-D)} (4)
15.0質量%ポリアリルアミン水溶液(ニットーボーメディカル株式会社製PAA-15C、アミン価17.5mmol/g)400gを、減圧下で水を留去することにより、40.0質量%ポリアリルアミン水溶液150g(第一溶液)を調製した。
エチルセルロース(和光純薬株式会社製エチルセルロース(約49%エトキシ)45、重量平均分子量は125,000)15.0gをトルエン303gに溶解することにより、第二溶液318gを調製した。
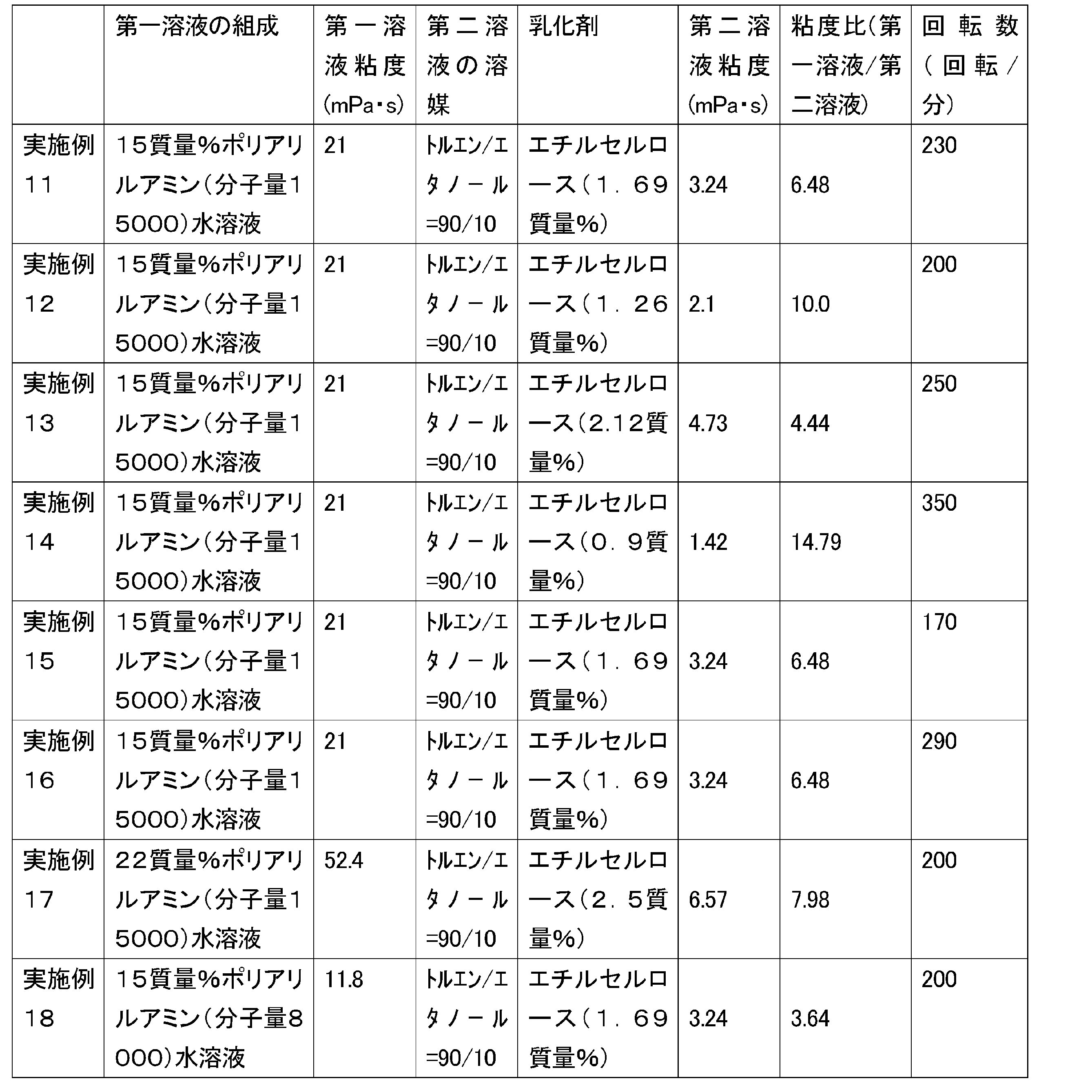
上記の第一溶液と上記の第二溶液とをディーン・スターク装置を備えた500mLセパラブルフラスコ中で混合することによって混合物を得た。ステンレス製平型攪拌羽根(IKA社製R1375、羽根径70mm)および新東科学株式会社製スリーワンモーター(BL600)を用いて、上記混合物を60℃で120回転/分で60分間攪拌することによって、ポリアリルアミン乳化液を得た。得られた乳化液の物性を、表1に記載した。以下、各実施例および比較例の乳化液の物性も、表1に記載した。表中の分子量は重量平均分子量である。
得られた乳化液に対し、1,6-ジクロロヘキサン(東京化成工業株式会社製)4.08gをトルエン10mLで希釈した溶液を5分間かけて滴下した。滴下終了後、浴温度を120℃に昇温し4時間還流することで、74mLの水を除去した。フラスコ温度を室温まで冷却し、デカンテーションにより上澄み液を取り除いた。得られた粒子をエタノール(500mL、3回)、1mol/LのNaOH水溶液:水(60mL:440mL,1回)、水(500mL,2回)、エタノール(500mL,1回)によって、それぞれリスラリーとろ過を繰り返すことで精製した。得られた粒子を送風乾燥機で50℃下48時間、減圧乾燥機で70℃下12時間乾燥させ、架橋ポリアリルアミン球状粒子を得た。反応式は下記参照。

各実施例および各比較例におけるパルスNMRにより求めた分子領域の比率、アミン価およびリン酸吸着能を表3に示す。



15.0質量%ポリアリルアミン水溶液(ニットーボーメディカル株式会社製PAA-15C、アミン価17.5mmol/g)480gを、減圧下で水を留去することにより、40.0質量%ポリアリルアミン水溶液180g(第一溶液)を調製した。
エチルセルロース(和光純薬株式会社製エチルセルロース(約49%エトキシ)45、重量平均分子量は125,000)18.0gをトルエン364gに溶解することにより、第二溶液382gを調製した。
上記第一溶液と上記第二溶液とをディーン・スターク装置を備えた500mLセパラブルフラスコ中で混合することによって混合物を得た。ステンレス製平型攪拌羽根(IKA社製R1375、羽根径70mm)および新東科学株式会社製スリーワンモーター(BL600)を用いて、上記混合物を50℃で120回転/分で60分間攪拌することによって、ポリアリルアミン乳化液を得た。
得られた乳化液に対し、1,6-ジクロロヘキサン(東京化成工業株式会社製)4.90gをトルエン12mLで希釈した溶液を10分間かけて滴下した。滴下終了後、2.5時間撹拌し、浴温度を120℃に昇温し4時間還流することで、88mLの水を除去した。以降、実施例1-1と同様にして架橋ポリアリルアミン球状粒子を得た。
攪拌時の温度を50℃から80℃に変更したこと以外は、実施例2と同様にして架橋ポリアリルアミン球状粒子を得た。
[実施例4]
攪拌時の温度を50℃から60℃に変更し、1,6-ジクロロヘキサンの重量を4.90gから9.79gに変更し、還流時間を4時間から5.5時間に変更したこと以外は、実施例2と同様にして架橋ポリアリルアミン球状粒子を得た。
実施例1-1の架橋ポリアリルアミン球状粒子248gに対し水5Lを加え、室温で100回転/分で30分間撹拌した。得られた懸濁液に対して、30質量%塩酸(和光純薬株式会社製)173mLを加え、室温で100回転/分で1時間撹拌した。反応液をろ過し、水(5L,2回)によってリスラリーとろ過を繰り返すことで精製した。得られた粒子を送風乾燥機で50℃下48時間、減圧乾燥機で70℃下12時間乾燥させ、架橋ポリアリルアミン球状粒子を得た。
実施例1-1の架橋ポリアリルアミン球状粒子150gに対し水3Lを加え、室温で100回転/分で30分間撹拌した。得られた懸濁液に対して、30質量%塩酸(和光純薬株式会社製)105mLを加え、室温で100回転/分で1時間撹拌した。反応液をろ過し、水(3L,2回)によって、リスラリーとろ過を繰り返すことで精製した。
得られた粒子に対し水3Lと炭酸ナトリウム(和光純薬株式会社製)215gを加え、室温で100回転/分で2時間撹拌した。反応液をろ過し、水(3L,4回)によってリスラリーとろ過を繰り返すことで精製した。送風乾燥機で50℃下48時間、減圧乾燥機で70℃下12時間乾燥させ、架橋ポリアリルアミン球状粒子を得た。
常法により、次の組成からなる錠剤を調製する。実施例1-1で得た架橋ポリアリルアミン球状粒子(40g)、乳糖(286.8g)及び馬鈴薯澱粉(60g)を混合し、これにヒドロキシプロピルセルロースの10%水溶液(120g)を加える。この混合物を常法により練合し、造粒して乾燥させた後、整粒し打錠用顆粒とする。これにステアリン酸マグネシウム(1.2g)を加えて混合し、径8mmの杵をもった打錠機(菊水社製RT-15型)で打錠を行って、錠剤(1錠あたり活性成分20mgを含有する)を得る。
処方 実施例1-1で得た架橋ポリアリルアミン球状粒子 20mg
乳糖 143.4mg
馬鈴薯澱粉 30mg
ヒドロキシプロピルセルロース 6mg
ステアリン酸マグネシウム 0.6mg
200mg
セベラマー塩酸塩としては、試験例1および試験例2においては、協和発酵キリン社製フォスブロック(登録商標)の錠剤を粉砕したものを用いた。各試験例で対応する実施例と等量投与したものを比較例1とし、実施例の2倍量投与したものを比較例2とした。走査電子顕微鏡、光学顕微鏡の撮影および膨潤率の測定においては、協和発酵キリン社製フォスブロック(登録商標)の錠剤1gあたり超純水50gと混合し、1時間以上振とうした後、濾過、乾燥することで、原薬に相当する架橋ポリマーを取り出し、評価に用いた。
図3にセベラマー塩酸塩の走査電子顕微鏡像を、図4にセベラマー塩酸塩の光学顕微鏡写真を示した。
ビキサロマーとしては、アステラス製薬社製キックリン(登録商標)カプセルを準備し、カプセルから原薬を取り出したものを用いた。各試験例で対応する実施例と等量投与したものを比較例3とし、実施例の2倍量投与したものを比較例4とした。図5にビキサロマーの走査電子顕微鏡像を、図6にビキサロマーの光学顕微鏡写真を示した。
特許文献7の実施例2に基づき合成した。
すなわち、40.0質量%ポリアリルアミン水溶液35.6gと水24.3mLと30質量%塩酸8.50mLと塩化ナトリウム10.0gを25℃から35℃で混合してポリアリルアミン塩酸塩の22.4重量%水溶液を得た。溶液を5℃から15℃に冷却し、200mLのトルエンと1gのスパン-85(トリオレイン酸ソルビタン)を加えた。次に、反応混合物の温度を20℃から25℃に上げ、250回転/分で15分間維持した。反応混合物を25℃から35℃でろ過して異物をすべて除去した。
ステンレス製平型攪拌羽根(IKA社製R1375、羽根径70mm)および新東科学株式会社製スリーワンモーター(BL600)を備えた500mLセパラブルフラスコへろ液を移液し、温度を55℃から60℃に上げ、250回転/分で15分間維持した。55℃から60℃の一定の温度で反応混合物に2.25gのエピクロロヒドリンを加え、55℃から60℃で3時間維持した。反応混合物を25℃から35℃に冷却し、生成物をろ別によって単離した。含液ケーキをさらに水(3×375mL)で25℃から50℃でほぐして45分間洗浄し、ろ過し、送風乾燥機中50℃で乾燥させ、架橋ポリアリルアミン球状粒子を得た。図7に比較例5の走査電子顕微鏡像を、図8に比較例5の光学顕微鏡写真を示した。図8からわかるように、比較例5の粒子は真円から外れて歪んだ形状の粒子が存在することから真円度が低くなる。
特許文献6の実施例2に基づき合成した。
すなわち、ステンレス製平型攪拌羽根(IKA社製R1375、羽根径70mm)および新東科学株式会社製スリーワンモーター(BL600)を備えた500mLセパラブルフラスコ中、40.0質量%ポリアリルアミン水溶液22.8gと30質量%塩酸5.79mLと塩化ナトリウム6.10gを混合してポリアリルアミン塩酸塩の32.3重量%水溶液を得た。得られた混合物を5℃まで冷却した。120mLのトルエンと0.57gのスパン-85(トリオレイン酸ソルビタン)を加えた。1.8mLのエピクロロヒドリンを全て一度に、部分的に中和されたポリアリルアミン塩酸塩溶液に加えた。この溶液を直ちに1200回転/分で撹拌してトルエン中に分散させた。混合物を60℃まで加熱して3時間撹拌した。トルエンをデカンテーションした。形成された架橋ポリアリルアミン塩酸塩を、150mLの脱イオン水中に45分間撹拌することによって懸濁させて3回洗浄し、続いてろ過した。架橋された固体を200mLのイソプロパノール中に45分間撹拌して懸濁させることによって1回すすぎ、続いてろ過した。固体を8時間真空乾燥させ、架橋ポリアリルアミン球状粒子を得た。図9に比較例6の走査電子顕微鏡像を、図10に比較例6の光学顕微鏡写真を示した。図10からわかるように、比較例6の粒子は歪な形状の粒子が存在するため真円度が小さい。
Sprague Dawley(登録商標)ラット(雄性、6週齢、日本チャールス・リバー社供給)を購入し、室温19~25℃、湿度30~70%、1日12時間照明(午前7時~午後7時)の飼育室にて2~4匹ずつ収容し、FR-2固形飼料(フナバシファーム社製)および水を自由に摂取させて飼育した。1週間の検疫・馴化飼育後、外見上異常が認められない個体を実験に使用し、FR-2固形飼料からFR-2粉末飼料(フナバシファーム社製)に切り替えて、さらに2~3日間粉末飼料の馴化を行った。
その後、体重測定および尾静脈から採血し、血液を400μL血清分離チューブに回収した。3000rpm、4℃および20分の条件で遠心分離機(CF15RX1、HITACHI社製)にて遠心分離し、上清を血清サンプルとし、フォスファCテストワコー(和光純薬工業株式会社)にて血清リン濃度を測定した。血清リン濃度および体重を指標に各群(N=6)が均等になるように群分けし、以下の薬剤を各群それぞれFR-2粉末飼料に混餌投与した。
実施例1-1投与群;実施例1-1で得た架橋ポリアリルアミン球状粒子を1質量%混餌投与した。
実施例1-2投与群;実施例1-2で得た架橋ポリアリルアミン球状粒子を1質量%混餌投与した。
実施例1-3投与群;実施例1-3で得た架橋ポリアリルアミン球状粒子を1質量%混餌投与した。
比較例5投与群;比較例5で得た架橋ポリアリルアミン球状粒子を1質量%混餌投与した。
比較例6投与群;比較例6で得た架橋ポリアリルアミン球状粒子を1質量%混餌投与した。
セベラマー塩酸塩投与群;比較例1のセベラマー塩酸塩を1質量%混餌投与した。
2倍量セベラマー塩酸塩投与群;比較例2のセベラマー塩酸塩を2質量%混餌投与した。
2倍量ビキサロマー投与群;比較例4のビキサロマーを2質量%混餌投与した。
コントロール群;薬剤を含まないFR-2粉末飼料を与えた。
混餌投与開始から3日後に、尾静脈より採血し、血清リン濃度を測定することで薬剤の血清リン濃度低下作用を評価した。血清リン濃度は各群の個体値の平均値±標準誤差を算出し、表4に記載した。

また、実施例1-1投与群、実施例1-2投与群および実施例1-3投与群は、いずれも比較例5投与群、比較例6投与群またはセベラマー塩酸塩投与群と比較して血清リン濃度が低かった。すなわち、実施例1-1、実施例1-2および実施例1-3で得られた架橋ポリアリルアミン球状粒子は、いずれも比較例5で得られた架橋ポリアリルアミン球状粒子、比較例6で得られた架橋ポリアリルアミン球状粒子またはセベラマー塩酸塩と比較して、より強力なリン吸着作用を有することが示唆された。
さらに、実施例1-1投与群、実施例1-2投与群および実施例1-3投与群は、いずれも2倍量のセベラマー塩酸塩投与群または2倍量のビキサロマー投与群と比較して同等の血清リン濃度であった。すなわち、実施例1-1、実施例1-2および実施例1-3で得られた架橋ポリアリルアミン球状粒子は、いずれも2倍量のセベラマー塩酸塩または2倍量のビキサロマーと同等のリン吸着作用を有することが示唆された。
Sprague Dawley(登録商標)ラット(雄性、6週齢、日本チャールス・リバー社供給)を購入し、室温19~25℃、湿度30~70%、1日12時間照明(午前7時~午後7時)の飼育室にて2~4匹ずつ収容し、FR-2固形飼料(フナバシファーム社製)および水を自由に摂取させて飼育した。1週間の検疫・馴化飼育後、外見上異常が認められない個体を実験に使用し、体重測定および個体番号を付し、2~3匹/ケージに割り振った。同時にFR-2固形飼料を取り除き、高リン高アデニン含有FR-2粉末飼料(オリエンタル酵母工業株式会社)をアルミ製給餌器に入れ、2週間摂餌させた。正常対照群(N=6)にはFR-2粉末飼料(フナバシファーム社製)を摂餌させた。その間、週に1~2回の頻度で適宜、飼料を補充した。ケージ交換は週2~3回の頻度で実施した。
2週間摂餌させた翌日に体重測定し、尾静脈より採血針にて採血を行い、血液を400μL血清分離チューブに回収した。回収した血液を、3000rpm、4℃および20分の条件にて遠心分離機(CF15RX1、HITACHI)を用いて遠心分離し、上清を血清サンプルとし、フォスファCテストワコー(和光純薬工業株式会社)にて血清リン濃度を測定した。さらに同血清サンプルを用いて日立7010型自動分析装置にて血中尿素窒素(BUN)および血清クレアチニン(CRNN)濃度を測定した。高リン高アデニン含有FR-2粉末飼料を摂餌させたラット(以下、病態ラットと呼ぶ)の血清リン濃度、BUNおよび血清CRNN濃度が正常対照群に比較し上昇しているのを確認した後、血清リン濃度、BUN、血清CRNN濃度および体重を指標に、病態ラットのそれぞれの値がコントロール群および各薬剤投与群で均等になるように群分けした(各群N=8~9)。採血および測定の翌日から2週間、各群それぞれ以下の薬剤を高リン高アデニン含有FR-2粉末飼料に混餌投与した。コントロール群にはセルロース(ナカライテスク社製)を高リン高アデニン含有FR-2粉末飼料に混餌投与した。正常対照群にはセルロースをFR-2粉末飼料に混餌投与した。その間、週1~2回の頻度で飼料を補充した。ケージ交換は週2~3回の頻度で実施した。
実施例2投与群;病態ラットに、実施例2で得た架橋ポリアリルアミン球状粒子を2質量%混餌投与した。さらに、セルロースを2質量%混餌することで薬剤の全量で4質量%混餌投与した。
実施例3投与群;病態ラットに、実施例3で得た架橋ポリアリルアミン球状粒子を2質量%混餌投与した。さらに、セルロースを2質量%混餌することで薬剤の全量で4質量%混餌投与した。
実施例4投与群;病態ラットに、実施例4で得た架橋ポリアリルアミン球状粒子を2質量%混餌投与した。さらに、セルロースを2質量%混餌することで薬剤の全量で4質量%混餌投与した。
ビキサロマー投与群;病態ラットに、比較例3のビキサロマーを2質量%混餌投与した。さらに、セルロースを2質量%混餌することで薬剤の全量で4質量%混餌投与した。
2倍量ビキサロマー投与群;病態ラットに、比較例4のビキサロマーを4質量%混餌投与した。
コントロール群;病態ラットに、セルロースを4質量%混餌投与した。
正常対照群;正常ラットに、セルロースを4質量%混餌投与した。
混餌投与開始から13日後に尾静脈より採血し、血清リン濃度を測定することで薬剤の血清リン濃度低下作用を評価した。血清リン濃度は各群の個体値の平均値±標準誤差を算出し、表5に記載した。

実施例1-1投与群、実施例2投与群、実施例3投与群および実施例4投与群は、いずれもコントロール群と比較して血清リン濃度が低かった。すなわち、実施例1-1、実施例2、実施例3および実施例4で得られた架橋ポリアリルアミン球状粒子は、いずれもリン吸着作用を有することが示唆された。
また、実施例1-1投与群、実施例2投与群、実施例3投与群および実施例4投与群は、ビキサロマー投与群と比較して血清リン濃度が低かった。すなわち、実施例1-1、実施例2、実施例3および実施例4で得られた架橋ポリアリルアミン球状粒子は、いずれもビキサロマーと比較して、より強力なリン吸着作用を有することが示唆された。
さらに、実施例1-1投与群、実施例2投与群、実施例3投与群および実施例4投与群は、2倍量のビキサロマー投与群と比較して同等の血清リン濃度であった。すなわち、実施例1-1、実施例2、実施例3および実施例4で得られた架橋ポリアリルアミン球状粒子は、いずれも2倍量のビキサロマーと同等のリン吸着作用を有することが示唆された。
実施例1-1投与群;病態ラットに、実施例1-1で得た架橋ポリアリルアミン球状粒子を2質量%混餌投与した。さらに、セルロースを2質量%混餌することで薬剤の全量で4質量%混餌投与した。
2倍量セベラマー塩酸塩投与群;病態ラットに、比較例2のセベラマー塩酸塩を4質量%混餌投与した。
ビキサロマー投与群;病態ラットに、比較例3のビキサロマーを2質量%混餌投与した。さらに、セルロースを2質量%混餌することで薬剤の全量で4質量%混餌投与した。
2倍量ビキサロマー投与群;病態ラットに、比較例4のビキサロマーを4質量%混餌投与した。
コントロール群;病態ラットに、セルロースを4質量%混餌投与した。
正常対照群;正常ラットに、セルロースを4質量%混餌投与した。
混餌投与開始から13日後に尾静脈より採血し、血清リン濃度を測定することで薬剤の血清リン濃度低下作用を評価した。血清リン濃度は各群の個体値の平均値±標準誤差を算出し、表6に記載した。

実施例1-1投与群は、ビキサロマー投与群と比較して血清リン濃度が低かった。すなわち、実施例1-1で得られた架橋ポリアリルアミン球状粒子は、ビキサロマーと比較して、より強力なリン吸着作用を有することが示唆された。
さらに、実施例1-1投与群は、2倍量のセベラマー塩酸塩投与群および2倍量のビキサロマー投与群と比較して同等の血清リン濃度であった。すなわち、実施例1-1で得られた架橋ポリアリルアミン球状粒子は、2倍量のセベラマー塩酸塩または2倍量のビキサロマーと同等のリン吸着作用を有することが示唆された。
本発明によれば、セベラマー塩酸塩およびビキサロマーではそれぞれ多量の処方が必要であった高リン血症患者に対して、その半分以下の処方量とすることができる。この結果、服用量の多さに起因する服薬コンプライアンスを改善し、良好な血清リン濃度のコントロールと服薬ストレス軽減が期待される。
ディーン・スターク装置を備え、撹拌翼としてPTFEオール被覆撹拌棒(ツイスタータイプ、フロンケミカル社製、羽根径80mm)および新東科学株式会社製スリーワンモーター(BL600)を備えた1Lセパラブルフラスコ(筒型、内径120mm、品番6-741-10、アズワン社製)に、エチルセルロース(和光純薬株式会社製エチルセルロース45(約49%エトキシ)、重量平均分子量は125,000)8.00g、1、6―ジクロロヘキサン(東京化成工業株式会社製)1.24g、トルエン425.9g、エタノール47.3gを加え、40℃、230回転/分で1時間撹拌し、エチルセルロースを完溶させた。その後、15.0質量%ポリアリルアミン水溶液(ニットーボーメディカル株式会社製PAA-15C、アミン価17.5mmol/g)162gを、1時間かけて滴下した。上記混合物を40℃で200回転/分で60分間攪拌することによって、ポリアリルアミン乳化液を得た。その後、浴温度を120℃に昇温し20時間還流することで、180mLの水を除去した。
フラスコ温度を室温まで冷却し、濾過後、エタノールで洗浄後得られた粒子をビーカーに入れ、水300ml、2N-NaOH水溶液3mlで1時間撹拌し、その後水300mlで洗浄を5回行った後、エタノール(300mL,1回)で洗浄し、得られる粒子を減圧乾燥機で70℃下20時間乾燥させ架橋ポリマー球状粒子を得た。
ディーン・スターク装置を備え、撹拌翼としてステンレス製平型攪拌羽根(IKA社製R1375、羽根径70mm)および新東科学株式会社製スリーワンモーター(BL600)を備えた500mlセパラブルフラスコ(SIBATA製円筒形平底タイプ、品番005820-500)に、エチルセルロース(和光純薬株式会社製エチルセルロース45(約49%エトキシ)、重量平均分子量は125,000)3.32g、1、6-ジクロロヘキサン(東京化成工業株式会社製)0.92g、トルエン237g、エタノール26.3gを加え、40℃、200回転/分で1時間撹拌し、エチルセルロースを完溶させた。その後、15.0質量%ポリアリルアミン水溶液(ニットーボーメディカル株式会社製PAA-15C、アミン価17.5mmol/g)90gを、1時間かけて滴下した。上記混合物を40℃で200回転/分で60分間攪拌することによって、ポリアリルアミン乳化液を得た。その後、浴温度を120℃に昇温し20時間還流することで、88mLの水を除去した。 フラスコ温度を室温まで冷却し、濾過後、エタノールで洗浄後得られた粒子をビーカーに入れ、水200ml、2N-NaOH水溶液2mlで1時間撹拌し、その後水200mlで洗浄を5回行った後、エタノール(200mL,1回)で洗浄し、得られる粒子を減圧乾燥機で70℃下20時間乾燥させ架橋ポリマー球状粒子を得た。
撹拌数を200回転/分から250回転/分に変更し、エチルセルロースの質量を3.32gから5.59gに変更したこと以外は、実施例12と同様にして架橋ポリアリルアミン球状粒子を得た。
乳化温度を40℃から22℃に、撹拌数を200回転/分から350回転/分に変更し、エチルセルロースの質量を3.32gから5.59gに変更したこと以外は、実施例11と同様にして架橋ポリアリルアミン球状粒子を得た。
撹拌数を230回転/分から170回転/分に変更したこと以外は、実施例11と同様にして架橋ポリアリルアミン球状粒子を得た。
撹拌数を230回転/分から290回転/分に変更したこと以外は、実施例11と同様にして架橋ポリアリルアミン球状粒子を得た。
15.0質量%ポリアリルアミン水溶液90gを、22.0質量%ポリアリルアミン水溶液(ニットーボーメディカル株式会社製PAA-15C、アミン価17.5mmol/g、15wt%を濃縮したもの)90gに、ジクロロヘキサンの質量を1.01gに、エチルセルロースの質量を3.32gから6.57gに変更したこと以外は、実施例12と同様にして架橋ポリアリルアミン球状粒子を得た。
15.0質量%ポリアリルアミン水溶液(ニットーボーメディカル株式会社製PAA-15C、平均分子量15000)90gを、 15.0質量%ポリアリルアミン水溶液(ニットーボーメディカル株式会社製PAA-8、平均分子量8000)90gに、ジクロロヘキサンの質量を0.92gから1.00gに、エチルセルロースの質量を3.32gから4.45gに変更したこと以外は、実施例12と同様にして架橋ポリアリルアミン球状粒子を得た。
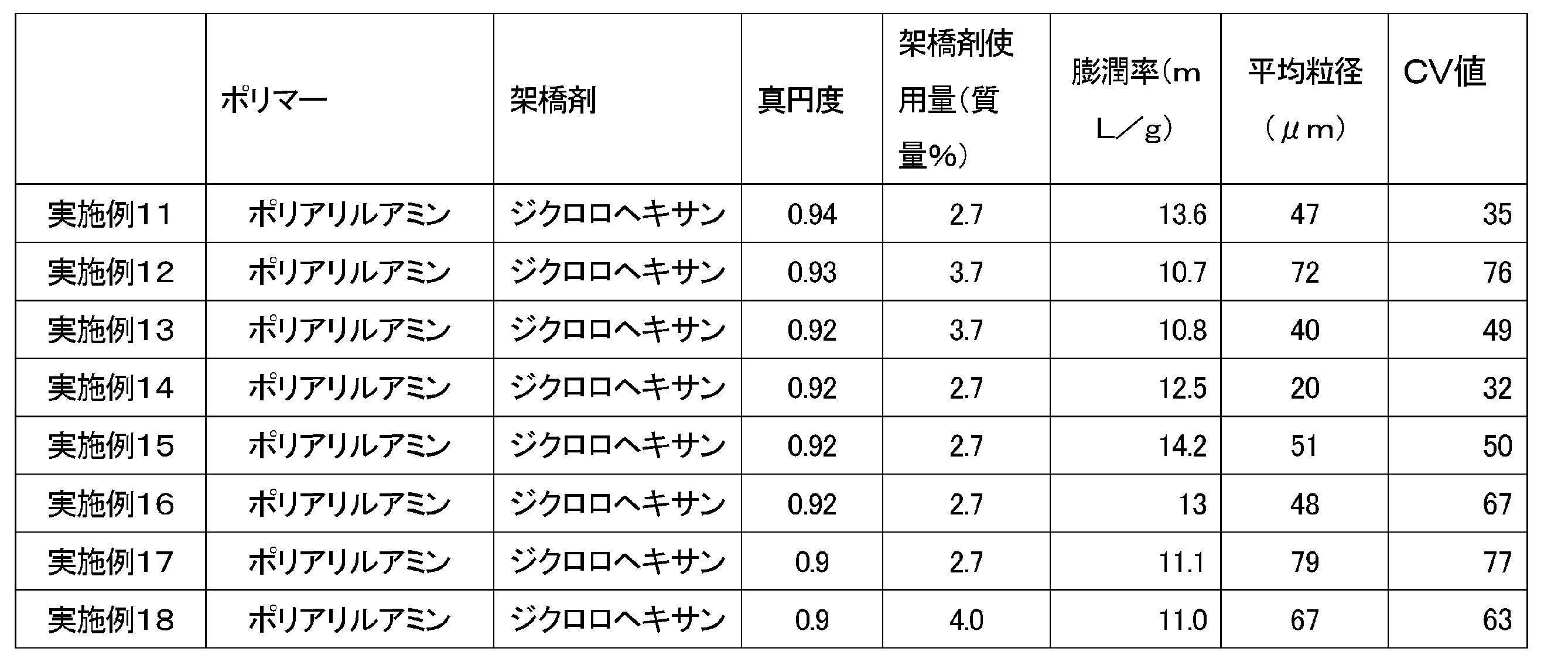
エチルセルロースの質量を8.00gから3.79gに、ジクロロヘキサンの質量を1.24gから1.65gに、乳化時の撹拌数を230回転/分から350回転/分に変更したこと以外は、実施例11と同様にして架橋ポリアリルアミン球状粒子を得た。
エチルセルロースの質量を8.00gから3.79gに、ジクロロヘキサンの質量を1.24gから1.06gに、乳化時の撹拌数を230回転/分から350回転/分に変更したこと以外は、実施例11と同様にして架橋ポリアリルアミン球状粒子を得た。
ジクロロヘキサンの質量を1.24gから1.65gに、乳化時の撹拌数を230回転/分から500回転/分に変更したこと以外は、実施例11と同様にして架橋ポリアリルアミン球状粒子を得た。
ジクロロヘキサンの質量を1.24gから1.06gに、乳化時の撹拌数を230回転/分から500回転/分に変更したこと以外は、実施例11と同様にして架橋ポリアリルアミン球状粒子を得た。


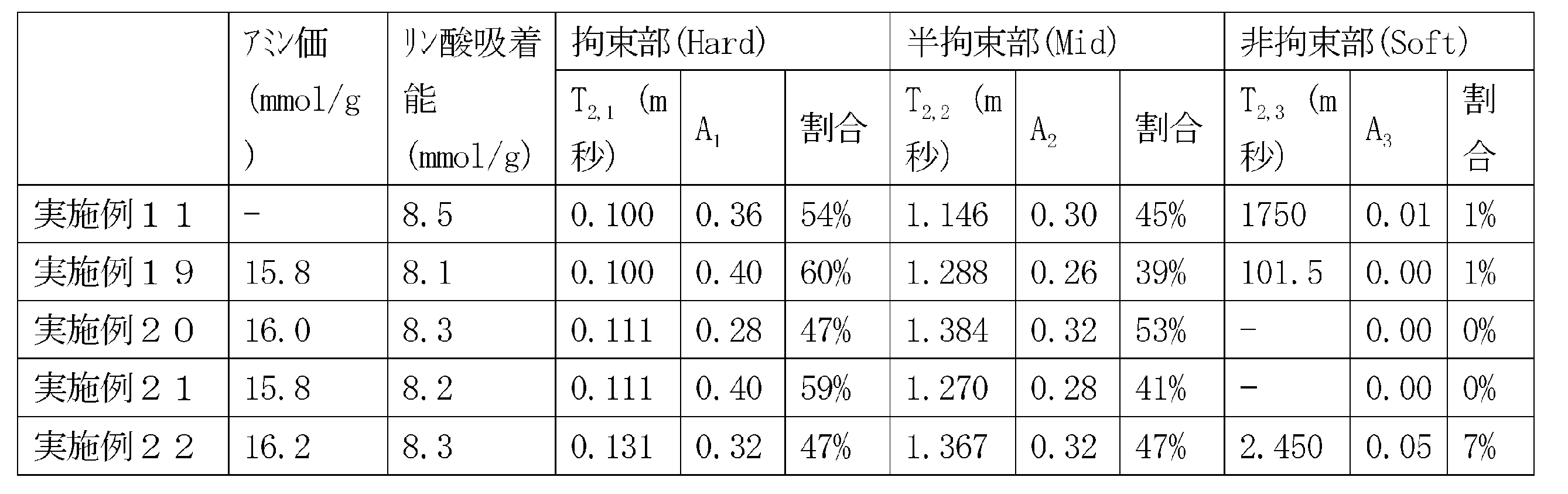
Claims (9)
- 下記式(1-1)または(1-2)で表わされる繰り返し単位Aと、
下記式(2-1)または(2-2)で表わされる繰り返し単位Bと、
を少なくとも有する架橋ポリマーを含む粒子を、有効成分として含有する高リン血症治療剤;

R6、R7およびR8は、各々独立に、水素原子または結合手を表し、
X-は、負に荷電した対イオンであり、
nは、6を表し、
*は、繰り返し単位Aの側鎖の窒素原子との結合手を意味し、この場合、R6、R7およびR8のうちの少なくとも1つは結合手となる。 - 粒子は、平均粒径が20~150μmである、請求項1に記載の高リン血症治療剤;
平均粒径は、光学顕微鏡写真の1000個以上の水分散状態の粒子画像の面積から直径に換算し、その直径を用いて体積平均粒径として算出する。 - 粒子は、膨潤率が8~20mL/gである、請求項1又は2に記載の高リン血症治療剤;
膨潤率は、20℃、2-モルホリノエタンスルホン酸ナトリウム2.2質量%および塩化ナトリウム0.5質量%でありpH6.3の水溶液中で、振盪および1時間以上の静置を20回以上繰り返した膨潤後の粒子体積を、膨潤前の粒子質量で除することにより算出する。 - 粒子が球状粒子である、請求項1から3の何れか一項に記載の高リン血症治療剤。
- 全架橋ポリマー中、
繰り返し単位Aの含有量が90~99モル%であり、
繰り返し単位Bの含有量が1~10モル%である、
請求項1から4の何れか一項に記載の高リン血症治療剤。 - 前記粒子は、パルスNMRで得られる自由誘導減衰信号を最小二乗法によってスピン-スピン緩和時間T2の長い成分から順に差し引き、波形分離することにより、スピン-スピン緩和時間の長い方から順に非拘束部、半拘束部、拘束部の3成分に分けた場合において、半拘束部の割合が25~70%である、請求項1から5の何れか一項に記載の高リン血症治療剤。
- 前記粒子は、パルスNMRで得られる自由誘導減衰信号を最小二乗法によってスピン-スピン緩和時間T2の長い成分から順に差し引き、波形分離することにより、スピン-スピン緩和時間の長い方から順に非拘束部、半拘束部、拘束部の3成分に分けた場合において、拘束部の割合が30~70%である、請求項1から6の何れか一項に記載の高リン血症治療剤。
- リン酸吸着能が6.0~10.0mmol/gである、請求項1から7の何れか一項に記載の高リン血症治療剤;
但し、リン酸吸着能は、2.2質量%モルホリノエタンスルホン酸ナトリウム、0.47質量%塩化ナトリウム、および0.24質量%リン酸でありpH=6.4の水溶液20mL中において、粒子30mgを37℃で1時間混合撹拌した際の、混合前後の上澄み中のリン酸濃度をICP発光分光分析法により定量し、その減少量を粒子質量で除し、乾燥減量値を用いて補正することにより算出したものである。 - アミン価が11.0~17.5mmol/gである、請求項1から8の何れか一項に記載の高リン血症治療剤;
但し、アミン価は、超純水に分散した粒子を5規定の塩酸で処理し、0.1規定の水酸化ナトリウム水溶液による中和滴定を行うことによりアミノ基の量を定量し、それを粒子質量で除し、乾燥減量値を用いて補正することにより算出したものである。
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017200016 | 2017-10-16 | ||
| JP2017200016 | 2017-10-16 | ||
| JP2017252895 | 2017-12-28 | ||
| JP2017252895 | 2017-12-28 | ||
| JP2018121071 | 2018-06-26 | ||
| JP2018121071 | 2018-06-26 | ||
| PCT/JP2018/038465 WO2019078197A1 (ja) | 2017-10-16 | 2018-10-16 | 高リン血症治療剤 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2019078197A1 JPWO2019078197A1 (ja) | 2020-11-05 |
| JP6992084B2 true JP6992084B2 (ja) | 2022-01-13 |
Family
ID=66174122
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019549288A Expired - Fee Related JP6992084B2 (ja) | 2017-10-16 | 2018-10-16 | 高リン血症治療剤 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US11147833B2 (ja) |
| EP (1) | EP3698799B1 (ja) |
| JP (1) | JP6992084B2 (ja) |
| CN (1) | CN111225674A (ja) |
| TW (1) | TW201922267A (ja) |
| WO (1) | WO2019078197A1 (ja) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6792640B2 (ja) | 2016-12-28 | 2020-11-25 | 富士フイルム株式会社 | 窒素原子含有ポリマー又はその塩の乳化液、その製造方法、及び粒子の製造方法 |
Family Cites Families (72)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6090243A (ja) * | 1983-10-25 | 1985-05-21 | Nitto Boseki Co Ltd | 小球状モノアリルアミン橋かけ重合体の製造方法 |
| JPS6151006A (ja) | 1984-08-20 | 1986-03-13 | Mitsubishi Chem Ind Ltd | ポリビニルアミン類の架橋造球方法 |
| US4799962A (en) | 1987-12-24 | 1989-01-24 | Aqualon Company | Water-soluble polymer dispersion |
| JPH048710A (ja) | 1990-04-27 | 1992-01-13 | Nippon Zeon Co Ltd | アミノ基含有重合体粒子の製造方法 |
| US5496545A (en) * | 1993-08-11 | 1996-03-05 | Geltex Pharmaceuticals, Inc. | Phosphate-binding polymers for oral administration |
| US5496845A (en) | 1994-05-25 | 1996-03-05 | American Cyanamid Co. | Suspension concentrate compositions of arylpyrrole insecticidal and acaricidal agents |
| CN1252829A (zh) * | 1997-02-19 | 2000-05-10 | 普罗克特和甘保尔公司 | 混合床离子交换可形成水凝胶的聚合物组合物以及含相对高浓度这些组合物的吸收性部件 |
| TW592727B (en) * | 1997-04-04 | 2004-06-21 | Chugai Pharmaceutical Co Ltd | Phosphate-binding polymer preparations |
| JP3389493B2 (ja) | 1997-04-04 | 2003-03-24 | 中外製薬株式会社 | リン酸結合性ポリマー製剤 |
| JP3952223B2 (ja) | 1997-05-28 | 2007-08-01 | 日東紡績株式会社 | アリルアミン重合体 |
| US6005035A (en) | 1997-09-18 | 1999-12-21 | Eastman Chemical Company | Stable waterborne polymer compositions containing poly(alkylenimines) |
| AU3005599A (en) | 1998-03-19 | 1999-10-11 | Geltex Pharmaceuticals, Inc. | Continuous crosslinking of polymer gels |
| DE19846413A1 (de) | 1998-10-08 | 2000-04-13 | Basf Ag | Verfahren zur Herstellung von hydrophilen wasserquellbaren Polymeren sowie deren Verwendung |
| TW568788B (en) | 1998-10-12 | 2004-01-01 | Chugai Pharmaceutical Co Ltd | Polymer combining with phosphoric acid and preparation containing the same |
| US6482872B2 (en) | 1999-04-01 | 2002-11-19 | Programmable Materials, Inc. | Process for manufacturing a biodegradable polymeric composition |
| CA2362410A1 (en) | 1999-04-16 | 2000-10-26 | Abbott Laboratories | Process for producing cross-linked polyallylamine hydrochloride |
| US6180754B1 (en) | 1999-09-03 | 2001-01-30 | The Dow Chemical Company | Process for producing cross-linked polyallylamine polymer |
| US6362266B1 (en) | 1999-09-03 | 2002-03-26 | The Dow Chemical Company | Process for reducing cohesiveness of polyallylamine polymer gels during drying |
| US6733780B1 (en) | 1999-10-19 | 2004-05-11 | Genzyme Corporation | Direct compression polymer tablet core |
| US20040059065A1 (en) * | 2000-03-09 | 2004-03-25 | Takeshi Goto | Crosslinked anion-exchange resin or salt thereof |
| AT409629B (de) | 2000-09-14 | 2002-09-25 | Dsm Fine Chem Austria Gmbh | Waschverfahren zur reinigung von n-bzw. amino- oder ammoniumgruppen haltigen polymeren |
| WO2002066543A1 (en) | 2001-02-16 | 2002-08-29 | Genzyme Corporation | Method of drying a material having a cohesive phase |
| BR0209133A (pt) | 2001-04-18 | 2004-06-15 | Genzyme Corp | Método para tratar gota e para reduzir os nìveis de ácido úrico n0 sangue |
| EP1923064B1 (en) | 2001-04-18 | 2017-06-28 | Genzyme Corporation | Use of amine polymer for lowering serum glucose |
| WO2002085383A1 (en) | 2001-04-18 | 2002-10-31 | Genzyme Corporation | Method for reducing copper levels and treating copper toxicosis |
| WO2002085379A1 (en) | 2001-04-18 | 2002-10-31 | Geltex Pharmaceuticals, Inc. | Method for improving vascular access in patients with vascular shunts |
| ES2304437T3 (es) | 2001-04-18 | 2008-10-16 | Genzyme Corporation | Uso de colesevelam o de hidrocloruro de sevelamer para reducir la glucosa serica. |
| WO2002085380A1 (en) | 2001-04-18 | 2002-10-31 | Geltex Pharmaceuticals, Inc. | Method for treating gout and reducing serum uric acid |
| KR20040018357A (ko) | 2001-04-18 | 2004-03-03 | 젠자임 코포레이션 | 저염 형태의 폴리알릴아민 |
| MXPA03009562A (es) | 2001-04-18 | 2004-02-12 | Genzyme Corp | METODO PARA TRATAR EL SiNDROME X CON POLIAMINAS ALIFATICAS. |
| DE60225710T2 (de) | 2001-10-09 | 2009-04-02 | Genzyme Corp., Cambridge | Verfahren zum reinigen und trocknen von polymerhydrogelen |
| JP2004059747A (ja) | 2002-07-30 | 2004-02-26 | Hymo Corp | 水溶性高分子エマルジョン及びその使用方法 |
| US7449605B2 (en) | 2003-11-03 | 2008-11-11 | Ilypsa, Inc. | Crosslinked amine polymers |
| US7608674B2 (en) * | 2003-11-03 | 2009-10-27 | Ilypsa, Inc. | Pharmaceutical compositions comprising cross-linked small molecule amine polymers |
| US7459502B2 (en) * | 2003-11-03 | 2008-12-02 | Ilypsa, Inc. | Pharmaceutical compositions comprising crosslinked polyamine polymers |
| WO2006043984A2 (en) * | 2004-10-13 | 2006-04-27 | Ilypsa, Inc. | Crosslinked amine polymers |
| US7275928B2 (en) | 2004-11-23 | 2007-10-02 | Rohm And Haas Electronic Materials Cmp Holdings, Inc. | Apparatus for forming a striation reduced chemical mechanical polishing pad |
| JP4547620B2 (ja) | 2004-12-02 | 2010-09-22 | 日東紡績株式会社 | 架橋アリルアミン類重合体の製造方法 |
| JP2006169292A (ja) | 2004-12-13 | 2006-06-29 | Yokohama Rubber Co Ltd:The | 天然ゴム/無機フィラーマスターバッチの製造 |
| JP2008533272A (ja) | 2005-03-16 | 2008-08-21 | ユーエスヴィー リミテッド | 架橋ポリアリルアミンポリマーの調製のための改良された方法 |
| DE102005023107A1 (de) * | 2005-05-13 | 2006-11-16 | Basf Ag | Modifizierte Polyamine |
| DE102005037777A1 (de) | 2005-08-10 | 2007-02-15 | Construction Research & Technology Gmbh | Additiv für bauchemische Anwendung |
| US20090162314A1 (en) * | 2005-11-08 | 2009-06-25 | Huval Chad C | Magnesium-Containing Polymers for the Treatment of Hyperphosphatemia |
| CA2661987C (en) | 2006-09-01 | 2012-11-06 | Usv Limited | Process for the preparation of sevelamer hydrochloride and formulation thereof |
| US7964182B2 (en) | 2006-09-01 | 2011-06-21 | USV, Ltd | Pharmaceutical compositions comprising phosphate-binding polymer |
| KR20100045965A (ko) * | 2007-07-11 | 2010-05-04 | 도레이 카부시키가이샤 | 가교 폴리알릴아민 또는 그 산부가염 및 그 의약 용도 |
| EP2016947A1 (en) | 2007-07-17 | 2009-01-21 | Chemo Ibérica, S.A. | Novel one step process for preparing cross-linked poly(allylamine) polymers |
| JP5332520B2 (ja) * | 2007-11-07 | 2013-11-06 | アステラス製薬株式会社 | 医薬用錠剤 |
| PA8807201A1 (es) * | 2007-12-14 | 2009-07-23 | Genzyme Corp | Composiciones farmaceuticas |
| US7943597B2 (en) | 2008-04-08 | 2011-05-17 | Cypress Pharmaceutical, Inc. | Phosphate-binding chitosan and uses thereof |
| WO2009128085A1 (en) | 2008-04-15 | 2009-10-22 | Lupin Limited | Process for the preparation of cross-linked polyallylamine polymer |
| EP2628795A1 (en) | 2008-06-06 | 2013-08-21 | Danisco US Inc. | Compositions and methods comprising cellulase variants with reduced affinity to non-cellulosic materials |
| US20100330175A1 (en) | 2009-06-24 | 2010-12-30 | Jobdevairakkam Christopher N | Cross-linked polyallylamine tablet core |
| JP2011094128A (ja) | 2009-09-30 | 2011-05-12 | Toray Ind Inc | ポリアミド微粒子の製造方法 |
| JP5865847B2 (ja) | 2010-02-24 | 2016-02-17 | レリプサ, インコーポレイテッド | 胆汁酸捕捉剤として使用するための架橋されたポリビニルアミン、ポリアリルアミンおよびポリエチレンイミン |
| BR112012021448A2 (pt) | 2010-02-24 | 2016-05-31 | Relypsa Inc | polímero de amina, método para tratar doença, método de remoção de sais biliares de um indivíduo animal, método para melhorar o controle glicêmico em um indivíduo, e, processo para preparar o polímero de amina. |
| WO2012042542A1 (en) | 2010-10-01 | 2012-04-05 | Usv Limited | Process for preparation of crosslinked polymer |
| JP5609606B2 (ja) | 2010-12-08 | 2014-10-22 | 三菱レイヨン株式会社 | 水溶性ポリマーの製造方法 |
| JP5669308B2 (ja) | 2011-01-24 | 2015-02-12 | ハイモ株式会社 | ポリビニルアミンの安定な油中水型エマルジョンの製造方法 |
| IT1404163B1 (it) | 2011-02-01 | 2013-11-15 | Chemi Spa | Processo per la preparazione di poliallilamine reticolate o loro sali farmaceuticamente accettabili |
| EP2719730A4 (en) | 2011-06-13 | 2015-05-27 | Nat Inst For Materials Science | SOLUTION FROM NANOPARTICULAR FIBERS, METHOD FOR THEIR PRODUCTION AND FILTERS FROM NANOPARTICULAR FIBERS |
| US20130123433A1 (en) | 2011-11-14 | 2013-05-16 | Formosa Laboratories, Inc. | Method for preparing poly(allylamine) hydrochloride and derivatives therefrom |
| EP2791183A1 (en) | 2011-12-13 | 2014-10-22 | Synthon BV | Preparation of sevelamer with reduced content of allylamine |
| JP2013209617A (ja) | 2012-02-29 | 2013-10-10 | Toray Ind Inc | ポリマー微粒子の製造方法 |
| CN102942646B (zh) | 2012-10-26 | 2016-06-29 | 青岛正大海尔制药有限公司 | 一种高分子聚合物的合成与分离纯化方法 |
| CN103111247A (zh) | 2013-02-01 | 2013-05-22 | 江南大学 | 一种聚胺基微球的制备方法 |
| KR102633597B1 (ko) | 2013-06-05 | 2024-02-06 | 트리시다, 인크. | 경구 투여를 위한 양성자-결합 중합체 |
| CN103724518A (zh) | 2013-11-08 | 2014-04-16 | 绍兴鼎翔纺织品贸易有限公司 | 一种固色剂乳液及其制备方法 |
| KR20160127052A (ko) | 2014-04-08 | 2016-11-02 | 미쯔비시 레이온 가부시끼가이샤 | 비닐아민 단위 함유 중합체의 제조 방법 및 중합 생성물 |
| KR20240008418A (ko) | 2014-12-10 | 2024-01-18 | 트리시다, 인크. | 경구 투여용 양성자-결합 폴리머 |
| WO2016135065A1 (en) | 2015-02-23 | 2016-09-01 | Amneal Pharmaceuticals Company Gmbh | Process for granulating sevelamer carbonate |
| TW201922268A (zh) * | 2017-10-16 | 2019-06-16 | 日商富士軟片股份有限公司 | 高磷血症治療劑及粒子 |
-
2018
- 2018-10-16 TW TW107136310A patent/TW201922267A/zh unknown
- 2018-10-16 CN CN201880067531.9A patent/CN111225674A/zh active Pending
- 2018-10-16 EP EP18869094.5A patent/EP3698799B1/en not_active Not-in-force
- 2018-10-16 WO PCT/JP2018/038465 patent/WO2019078197A1/ja not_active Ceased
- 2018-10-16 JP JP2019549288A patent/JP6992084B2/ja not_active Expired - Fee Related
-
2019
- 2019-06-28 US US16/456,822 patent/US11147833B2/en not_active Expired - Fee Related
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6792640B2 (ja) | 2016-12-28 | 2020-11-25 | 富士フイルム株式会社 | 窒素原子含有ポリマー又はその塩の乳化液、その製造方法、及び粒子の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3698799A1 (en) | 2020-08-26 |
| TW201922267A (zh) | 2019-06-16 |
| EP3698799A4 (en) | 2020-11-25 |
| EP3698799B1 (en) | 2022-01-26 |
| US11147833B2 (en) | 2021-10-19 |
| CN111225674A (zh) | 2020-06-02 |
| JPWO2019078197A1 (ja) | 2020-11-05 |
| US20190321392A1 (en) | 2019-10-24 |
| WO2019078197A1 (ja) | 2019-04-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7018451B2 (ja) | 高リン血症治療剤および粒子 | |
| US20250009785A1 (en) | Hcl-binding compositions for and methods of treating acid-base disorders | |
| US20190240252A1 (en) | Pharmaceutical compositions | |
| JP6423506B2 (ja) | 架橋型カチオン結合性ポリマーを含む組成物 | |
| EP2897594B1 (en) | Pharmaceutical composition | |
| DE112006002617T5 (de) | Verfahren zur Herstellung von Core-Shell-Kompositen bzw. Kern-Hüllen-Kompositen mit vernetzten Hüllen und daraus entstehende Core-Shell-Komposite | |
| JP6992084B2 (ja) | 高リン血症治療剤 | |
| JP2011506449A (ja) | コーティング医薬組成物 | |
| HK40030228A (en) | Hyperphosphatemia treatment agent | |
| HK40030230A (en) | Hyperphosphatemia treatment agent, and particles | |
| JPH07309766A (ja) | コレステロール低下剤 | |
| HK40055721A (en) | Compositions for treating acid-base disorders | |
| JP2003321515A (ja) | 胆汁酸吸着性ポリマー及びコレステロール低下剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20200410 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20201118 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210420 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210617 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20210907 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20211101 |
|
| C60 | Trial request (containing other claim documents, opposition documents) |
Free format text: JAPANESE INTERMEDIATE CODE: C60 Effective date: 20211101 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20211111 |
|
| C21 | Notice of transfer of a case for reconsideration by examiners before appeal proceedings |
Free format text: JAPANESE INTERMEDIATE CODE: C21 Effective date: 20211116 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20211130 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20211208 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6992084 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |