JP6482459B2 - 細胞サンプル中の生存細胞を検出する方法 - Google Patents
細胞サンプル中の生存細胞を検出する方法 Download PDFInfo
- Publication number
- JP6482459B2 JP6482459B2 JP2015510465A JP2015510465A JP6482459B2 JP 6482459 B2 JP6482459 B2 JP 6482459B2 JP 2015510465 A JP2015510465 A JP 2015510465A JP 2015510465 A JP2015510465 A JP 2015510465A JP 6482459 B2 JP6482459 B2 JP 6482459B2
- Authority
- JP
- Japan
- Prior art keywords
- membrane
- cells
- viable cells
- porous membrane
- viable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims description 82
- 239000012528 membrane Substances 0.000 claims description 348
- 239000007850 fluorescent dye Substances 0.000 claims description 176
- 238000001514 detection method Methods 0.000 claims description 96
- 239000002245 particle Substances 0.000 claims description 54
- 239000007788 liquid Substances 0.000 claims description 42
- 239000012530 fluid Substances 0.000 claims description 39
- 238000010186 staining Methods 0.000 claims description 33
- 244000005700 microbiome Species 0.000 claims description 26
- 230000005284 excitation Effects 0.000 claims description 20
- 230000034994 death Effects 0.000 claims description 15
- 230000000717 retained effect Effects 0.000 claims description 14
- 239000011148 porous material Substances 0.000 claims description 11
- 238000002372 labelling Methods 0.000 claims description 8
- 230000010261 cell growth Effects 0.000 claims description 4
- 241000894007 species Species 0.000 claims description 4
- 238000012258 culturing Methods 0.000 claims description 3
- 230000004663 cell proliferation Effects 0.000 claims description 2
- 230000005855 radiation Effects 0.000 claims description 2
- 210000004027 cell Anatomy 0.000 description 465
- 239000000523 sample Substances 0.000 description 75
- 239000000975 dye Substances 0.000 description 62
- 108020004707 nucleic acids Proteins 0.000 description 46
- 102000039446 nucleic acids Human genes 0.000 description 46
- 150000007523 nucleic acids Chemical class 0.000 description 46
- 241000588724 Escherichia coli Species 0.000 description 35
- 238000000295 emission spectrum Methods 0.000 description 34
- 239000000243 solution Substances 0.000 description 33
- 238000013459 approach Methods 0.000 description 31
- 239000007787 solid Substances 0.000 description 19
- 238000002073 fluorescence micrograph Methods 0.000 description 16
- 238000010791 quenching Methods 0.000 description 15
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 14
- 230000000171 quenching effect Effects 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- 108090000371 Esterases Proteins 0.000 description 11
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 11
- 239000011521 glass Substances 0.000 description 11
- 239000001963 growth medium Substances 0.000 description 11
- 239000002953 phosphate buffered saline Substances 0.000 description 11
- -1 support member Substances 0.000 description 11
- 239000006285 cell suspension Substances 0.000 description 9
- 230000006870 function Effects 0.000 description 9
- 230000000813 microbial effect Effects 0.000 description 9
- 239000000758 substrate Substances 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 238000011109 contamination Methods 0.000 description 8
- 239000011780 sodium chloride Substances 0.000 description 8
- 241000894006 Bacteria Species 0.000 description 7
- 241000222122 Candida albicans Species 0.000 description 7
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 7
- 230000003834 intracellular effect Effects 0.000 description 7
- 229920000515 polycarbonate Polymers 0.000 description 7
- 239000004417 polycarbonate Substances 0.000 description 7
- 239000013641 positive control Substances 0.000 description 7
- 230000035899 viability Effects 0.000 description 7
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 6
- 210000000170 cell membrane Anatomy 0.000 description 6
- 238000003570 cell viability assay Methods 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 239000004926 polymethyl methacrylate Substances 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 235000010378 sodium ascorbate Nutrition 0.000 description 6
- PPASLZSBLFJQEF-RKJRWTFHSA-M sodium ascorbate Substances [Na+].OC[C@@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RKJRWTFHSA-M 0.000 description 6
- 229960005055 sodium ascorbate Drugs 0.000 description 6
- PPASLZSBLFJQEF-RXSVEWSESA-M sodium-L-ascorbate Chemical compound [Na+].OC[C@H](O)[C@H]1OC(=O)C(O)=C1[O-] PPASLZSBLFJQEF-RXSVEWSESA-M 0.000 description 6
- 238000000862 absorption spectrum Methods 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 235000013305 food Nutrition 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- GHTWDWCFRFTBRB-UHFFFAOYSA-M oxazine-170 Chemical compound [O-]Cl(=O)(=O)=O.N1=C2C3=CC=CC=C3C(NCC)=CC2=[O+]C2=C1C=C(C)C(N(C)CC)=C2 GHTWDWCFRFTBRB-UHFFFAOYSA-M 0.000 description 5
- 229920003023 plastic Polymers 0.000 description 5
- 239000004033 plastic Substances 0.000 description 5
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- 230000035945 sensitivity Effects 0.000 description 5
- 238000001228 spectrum Methods 0.000 description 5
- 238000007447 staining method Methods 0.000 description 5
- 238000005406 washing Methods 0.000 description 5
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 241000233866 Fungi Species 0.000 description 4
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 235000013361 beverage Nutrition 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 229940095731 candida albicans Drugs 0.000 description 4
- 238000003384 imaging method Methods 0.000 description 4
- 230000014759 maintenance of location Effects 0.000 description 4
- 230000013011 mating Effects 0.000 description 4
- 231100000252 nontoxic Toxicity 0.000 description 4
- 230000003000 nontoxic effect Effects 0.000 description 4
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 4
- 239000004810 polytetrafluoroethylene Substances 0.000 description 4
- 150000003839 salts Chemical class 0.000 description 4
- 230000003595 spectral effect Effects 0.000 description 4
- 230000001954 sterilising effect Effects 0.000 description 4
- 230000004083 survival effect Effects 0.000 description 4
- 238000003260 vortexing Methods 0.000 description 4
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- 150000003973 alkyl amines Chemical class 0.000 description 3
- BQRGNLJZBFXNCZ-UHFFFAOYSA-N calcein am Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC(CN(CC(=O)OCOC(C)=O)CC(=O)OCOC(C)=O)=C(OC(C)=O)C=C1OC1=C2C=C(CN(CC(=O)OCOC(C)=O)CC(=O)OCOC(=O)C)C(OC(C)=O)=C1 BQRGNLJZBFXNCZ-UHFFFAOYSA-N 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- 238000009792 diffusion process Methods 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 210000003527 eukaryotic cell Anatomy 0.000 description 3
- 230000002209 hydrophobic effect Effects 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 230000036512 infertility Effects 0.000 description 3
- 239000004816 latex Substances 0.000 description 3
- 229920000126 latex Polymers 0.000 description 3
- 238000004020 luminiscence type Methods 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- VUQUOGPMUUJORT-UHFFFAOYSA-N methyl 4-methylbenzenesulfonate Chemical compound COS(=O)(=O)C1=CC=C(C)C=C1 VUQUOGPMUUJORT-UHFFFAOYSA-N 0.000 description 3
- 230000002906 microbiologic effect Effects 0.000 description 3
- 235000013336 milk Nutrition 0.000 description 3
- 239000008267 milk Substances 0.000 description 3
- 210000004080 milk Anatomy 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 210000001236 prokaryotic cell Anatomy 0.000 description 3
- 230000027756 respiratory electron transport chain Effects 0.000 description 3
- 239000012192 staining solution Substances 0.000 description 3
- 238000004659 sterilization and disinfection Methods 0.000 description 3
- 239000008399 tap water Substances 0.000 description 3
- 235000020679 tap water Nutrition 0.000 description 3
- 230000037303 wrinkles Effects 0.000 description 3
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 2
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 2
- RQFCJASXJCIDSX-UHFFFAOYSA-N 14C-Guanosin-5'-monophosphat Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(COP(O)(O)=O)C(O)C1O RQFCJASXJCIDSX-UHFFFAOYSA-N 0.000 description 2
- RJPSHDMGSVVHFA-UHFFFAOYSA-N 2-[carboxymethyl-[(7-hydroxy-4-methyl-2-oxochromen-8-yl)methyl]amino]acetic acid Chemical compound OC(=O)CN(CC(O)=O)CC1=C(O)C=CC2=C1OC(=O)C=C2C RJPSHDMGSVVHFA-UHFFFAOYSA-N 0.000 description 2
- BZTDTCNHAFUJOG-UHFFFAOYSA-N 6-carboxyfluorescein Chemical compound C12=CC=C(O)C=C2OC2=CC(O)=CC=C2C11OC(=O)C2=CC=C(C(=O)O)C=C21 BZTDTCNHAFUJOG-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 229920000742 Cotton Polymers 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 102000003886 Glycoproteins Human genes 0.000 description 2
- 108090000288 Glycoproteins Proteins 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 239000004677 Nylon Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 239000004698 Polyethylene Substances 0.000 description 2
- 229920002125 Sokalan® Polymers 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 2
- 239000000370 acceptor Substances 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 230000000712 assembly Effects 0.000 description 2
- 238000000429 assembly Methods 0.000 description 2
- 235000013405 beer Nutrition 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000001124 body fluid Anatomy 0.000 description 2
- 239000010839 body fluid Substances 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- MMYYTPYDNCIFJU-UHFFFAOYSA-N calix[6]arene Chemical compound C1C(C=2)=CC=CC=2CC(C=2)=CC=CC=2CC(C=2)=CC=CC=2CC(C=2)=CC=CC=2CC(C=2)=CC=CC=2CC2=CC=CC1=C2 MMYYTPYDNCIFJU-UHFFFAOYSA-N 0.000 description 2
- 235000014633 carbohydrates Nutrition 0.000 description 2
- 150000001720 carbohydrates Chemical class 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000003599 detergent Substances 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 230000035622 drinking Effects 0.000 description 2
- 235000020188 drinking water Nutrition 0.000 description 2
- 239000003651 drinking water Substances 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 2
- 229960005542 ethidium bromide Drugs 0.000 description 2
- GTSMOYLSFUBTMV-UHFFFAOYSA-N ethidium homodimer Chemical compound [H+].[H+].[Cl-].[Cl-].[Cl-].[Cl-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2C(C)=[N+]1CCCNCCNCCC[N+](C1=CC(N)=CC=C1C1=CC=C(N)C=C11)=C1C1=CC=CC=C1 GTSMOYLSFUBTMV-UHFFFAOYSA-N 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 239000010931 gold Substances 0.000 description 2
- 229910052737 gold Inorganic materials 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- RQFCJASXJCIDSX-UUOKFMHZSA-N guanosine 5'-monophosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O RQFCJASXJCIDSX-UUOKFMHZSA-N 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 229920001903 high density polyethylene Polymers 0.000 description 2
- 239000004700 high-density polyethylene Substances 0.000 description 2
- 230000005865 ionizing radiation Effects 0.000 description 2
- 238000007834 ligase chain reaction Methods 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 210000002751 lymph Anatomy 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 235000015097 nutrients Nutrition 0.000 description 2
- 229920001778 nylon Polymers 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 210000003463 organelle Anatomy 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 230000035515 penetration Effects 0.000 description 2
- 229920002492 poly(sulfone) Polymers 0.000 description 2
- 239000004584 polyacrylic acid Substances 0.000 description 2
- 229920000728 polyester Polymers 0.000 description 2
- 229920000573 polyethylene Polymers 0.000 description 2
- 238000003752 polymerase chain reaction Methods 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 235000008476 powdered milk Nutrition 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 230000010076 replication Effects 0.000 description 2
- 238000005096 rolling process Methods 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 238000012163 sequencing technique Methods 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 210000002700 urine Anatomy 0.000 description 2
- 238000012800 visualization Methods 0.000 description 2
- 235000014101 wine Nutrition 0.000 description 2
- CHADEQDQBURGHL-UHFFFAOYSA-N (6'-acetyloxy-3-oxospiro[2-benzofuran-1,9'-xanthene]-3'-yl) acetate Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(OC(C)=O)C=C1OC1=CC(OC(=O)C)=CC=C21 CHADEQDQBURGHL-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- CDAWCLOXVUBKRW-UHFFFAOYSA-N 2-aminophenol Chemical compound NC1=CC=CC=C1O CDAWCLOXVUBKRW-UHFFFAOYSA-N 0.000 description 1
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 1
- 229940093475 2-ethoxyethanol Drugs 0.000 description 1
- BCHZICNRHXRCHY-UHFFFAOYSA-N 2h-oxazine Chemical compound N1OC=CC=C1 BCHZICNRHXRCHY-UHFFFAOYSA-N 0.000 description 1
- WPUZGNPQMIWOHE-UHFFFAOYSA-N 3',6'-diacetyloxy-3-oxospiro[2-benzofuran-1,9'-xanthene]-5-carboxylic acid Chemical compound O1C(=O)C2=CC(C(O)=O)=CC=C2C21C1=CC=C(OC(C)=O)C=C1OC1=CC(OC(=O)C)=CC=C21 WPUZGNPQMIWOHE-UHFFFAOYSA-N 0.000 description 1
- DNUJVGBNIXGTHC-UHFFFAOYSA-N 3,6-dihydro-2h-oxazine Chemical compound C1NOCC=C1 DNUJVGBNIXGTHC-UHFFFAOYSA-N 0.000 description 1
- QJZYHAIUNVAGQP-UHFFFAOYSA-N 3-nitrobicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic acid Chemical compound C1C2C=CC1C(C(=O)O)C2(C(O)=O)[N+]([O-])=O QJZYHAIUNVAGQP-UHFFFAOYSA-N 0.000 description 1
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 1
- DDGMDTGNGDOUPX-UHFFFAOYSA-N 7-methyliminophenothiazin-3-amine;hydrochloride Chemical compound [Cl-].C1=C(N)C=C2SC3=CC(=[NH+]C)C=CC3=NC2=C1 DDGMDTGNGDOUPX-UHFFFAOYSA-N 0.000 description 1
- IVRMZWNICZWHMI-UHFFFAOYSA-N Azide Chemical compound [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 description 1
- ROFVEXUMMXZLPA-UHFFFAOYSA-N Bipyridyl Chemical compound N1=CC=CC=C1C1=CC=CC=N1 ROFVEXUMMXZLPA-UHFFFAOYSA-N 0.000 description 1
- TYBKADJAOBUHAD-UHFFFAOYSA-J BoBo-1 Chemical compound [I-].[I-].[I-].[I-].S1C2=CC=CC=C2[N+](C)=C1C=C1C=CN(CCC[N+](C)(C)CCC[N+](C)(C)CCCN2C=CC(=CC3=[N+](C4=CC=CC=C4S3)C)C=C2)C=C1 TYBKADJAOBUHAD-UHFFFAOYSA-J 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- QTANTQQOYSUMLC-UHFFFAOYSA-O Ethidium cation Chemical compound C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 QTANTQQOYSUMLC-UHFFFAOYSA-O 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- 241000192125 Firmicutes Species 0.000 description 1
- 229930186217 Glycolipid Natural products 0.000 description 1
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229920005439 Perspex® Polymers 0.000 description 1
- QBKMWMZYHZILHF-UHFFFAOYSA-L Po-Pro-1 Chemical compound [I-].[I-].O1C2=CC=CC=C2[N+](C)=C1C=C1C=CN(CCC[N+](C)(C)C)C=C1 QBKMWMZYHZILHF-UHFFFAOYSA-L 0.000 description 1
- CZQJZBNARVNSLQ-UHFFFAOYSA-L Po-Pro-3 Chemical compound [I-].[I-].O1C2=CC=CC=C2[N+](C)=C1C=CC=C1C=CN(CCC[N+](C)(C)C)C=C1 CZQJZBNARVNSLQ-UHFFFAOYSA-L 0.000 description 1
- BOLJGYHEBJNGBV-UHFFFAOYSA-J PoPo-1 Chemical compound [I-].[I-].[I-].[I-].O1C2=CC=CC=C2[N+](C)=C1C=C1C=CN(CCC[N+](C)(C)CCC[N+](C)(C)CCCN2C=CC(=CC3=[N+](C4=CC=CC=C4O3)C)C=C2)C=C1 BOLJGYHEBJNGBV-UHFFFAOYSA-J 0.000 description 1
- GYPIAQJSRPTNTI-UHFFFAOYSA-J PoPo-3 Chemical compound [I-].[I-].[I-].[I-].O1C2=CC=CC=C2[N+](C)=C1C=CC=C1C=CN(CCC[N+](C)(C)CCC[N+](C)(C)CCCN2C=CC(=CC=CC3=[N+](C4=CC=CC=C4O3)C)C=C2)C=C1 GYPIAQJSRPTNTI-UHFFFAOYSA-J 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920002367 Polyisobutene Polymers 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- DPXHITFUCHFTKR-UHFFFAOYSA-L To-Pro-1 Chemical compound [I-].[I-].S1C2=CC=CC=C2[N+](C)=C1C=C1C2=CC=CC=C2N(CCC[N+](C)(C)C)C=C1 DPXHITFUCHFTKR-UHFFFAOYSA-L 0.000 description 1
- QHNORJFCVHUPNH-UHFFFAOYSA-L To-Pro-3 Chemical compound [I-].[I-].S1C2=CC=CC=C2[N+](C)=C1C=CC=C1C2=CC=CC=C2N(CCC[N+](C)(C)C)C=C1 QHNORJFCVHUPNH-UHFFFAOYSA-L 0.000 description 1
- ULHRKLSNHXXJLO-UHFFFAOYSA-L Yo-Pro-1 Chemical compound [I-].[I-].C1=CC=C2C(C=C3N(C4=CC=CC=C4O3)C)=CC=[N+](CCC[N+](C)(C)C)C2=C1 ULHRKLSNHXXJLO-UHFFFAOYSA-L 0.000 description 1
- ZVUUXEGAYWQURQ-UHFFFAOYSA-L Yo-Pro-3 Chemical compound [I-].[I-].O1C2=CC=CC=C2[N+](C)=C1C=CC=C1C2=CC=CC=C2N(CCC[N+](C)(C)C)C=C1 ZVUUXEGAYWQURQ-UHFFFAOYSA-L 0.000 description 1
- GRRMZXFOOGQMFA-UHFFFAOYSA-J YoYo-1 Chemical compound [I-].[I-].[I-].[I-].C12=CC=CC=C2C(C=C2N(C3=CC=CC=C3O2)C)=CC=[N+]1CCC[N+](C)(C)CCC[N+](C)(C)CCC[N+](C1=CC=CC=C11)=CC=C1C=C1N(C)C2=CC=CC=C2O1 GRRMZXFOOGQMFA-UHFFFAOYSA-J 0.000 description 1
- JSBNEYNPYQFYNM-UHFFFAOYSA-J YoYo-3 Chemical compound [I-].[I-].[I-].[I-].C12=CC=CC=C2C(C=CC=C2N(C3=CC=CC=C3O2)C)=CC=[N+]1CCC(=[N+](C)C)CCCC(=[N+](C)C)CC[N+](C1=CC=CC=C11)=CC=C1C=CC=C1N(C)C2=CC=CC=C2O1 JSBNEYNPYQFYNM-UHFFFAOYSA-J 0.000 description 1
- ZHAFUINZIZIXFC-UHFFFAOYSA-N [9-(dimethylamino)-10-methylbenzo[a]phenoxazin-5-ylidene]azanium;chloride Chemical compound [Cl-].O1C2=CC(=[NH2+])C3=CC=CC=C3C2=NC2=C1C=C(N(C)C)C(C)=C2 ZHAFUINZIZIXFC-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 229920006243 acrylic copolymer Polymers 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000012790 adhesive layer Substances 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- PYKYMHQGRFAEBM-UHFFFAOYSA-N anthraquinone Natural products CCC(=O)c1c(O)c2C(=O)C3C(C=CC=C3O)C(=O)c2cc1CC(=O)OC PYKYMHQGRFAEBM-UHFFFAOYSA-N 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- YCOXTKKNXUZSKD-UHFFFAOYSA-N as-o-xylenol Natural products CC1=CC=C(O)C=C1C YCOXTKKNXUZSKD-UHFFFAOYSA-N 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- PGWTYMLATMNCCZ-UHFFFAOYSA-M azure A Chemical compound [Cl-].C1=CC(N)=CC2=[S+]C3=CC(N(C)C)=CC=C3N=C21 PGWTYMLATMNCCZ-UHFFFAOYSA-M 0.000 description 1
- KFZNPGQYVZZSNV-UHFFFAOYSA-M azure B Chemical compound [Cl-].C1=CC(N(C)C)=CC2=[S+]C3=CC(NC)=CC=C3N=C21 KFZNPGQYVZZSNV-UHFFFAOYSA-M 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical group C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 229920005549 butyl rubber Polymers 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000006229 carbon black Substances 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 210000003850 cellular structure Anatomy 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 229910000365 copper sulfate Inorganic materials 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- IUNMPGNGSSIWFP-UHFFFAOYSA-N dimethylaminopropylamine Chemical compound CN(C)CCCN IUNMPGNGSSIWFP-UHFFFAOYSA-N 0.000 description 1
- BVQAWSJMUYMNQN-UHFFFAOYSA-N dipyridophenazine Chemical compound C1=CC=C2C3=NC4=CC=CC=C4N=C3C3=CC=CN=C3C2=N1 BVQAWSJMUYMNQN-UHFFFAOYSA-N 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000004043 dyeing Methods 0.000 description 1
- 230000005670 electromagnetic radiation Effects 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000007071 enzymatic hydrolysis Effects 0.000 description 1
- 238000006047 enzymatic hydrolysis reaction Methods 0.000 description 1
- SEACYXSIPDVVMV-UHFFFAOYSA-L eosin Y Chemical compound [Na+].[Na+].[O-]C(=O)C1=CC=CC=C1C1=C2C=C(Br)C(=O)C(Br)=C2OC2=C(Br)C([O-])=C(Br)C=C21 SEACYXSIPDVVMV-UHFFFAOYSA-L 0.000 description 1
- 210000003013 erythroid precursor cell Anatomy 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000002189 fluorescence spectrum Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000005283 ground state Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 239000004021 humic acid Substances 0.000 description 1
- 238000005286 illumination Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- SNHMUERNLJLMHN-UHFFFAOYSA-N iodobenzene Chemical compound IC1=CC=CC=C1 SNHMUERNLJLMHN-UHFFFAOYSA-N 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 230000031852 maintenance of location in cell Effects 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- CXKWCBBOMKCUKX-UHFFFAOYSA-M methylene blue Chemical compound [Cl-].C1=CC(N(C)C)=CC2=[S+]C3=CC(N(C)C)=CC=C3N=C21 CXKWCBBOMKCUKX-UHFFFAOYSA-M 0.000 description 1
- 229960000907 methylthioninium chloride Drugs 0.000 description 1
- SORARJZLMNRBAQ-UHFFFAOYSA-N n,n',n'-trimethylpropane-1,3-diamine Chemical compound CNCCCN(C)C SORARJZLMNRBAQ-UHFFFAOYSA-N 0.000 description 1
- SHXOKQKTZJXHHR-UHFFFAOYSA-N n,n-diethyl-5-iminobenzo[a]phenoxazin-9-amine;hydrochloride Chemical compound [Cl-].C1=CC=C2C3=NC4=CC=C(N(CC)CC)C=C4OC3=CC(=[NH2+])C2=C1 SHXOKQKTZJXHHR-UHFFFAOYSA-N 0.000 description 1
- 210000002445 nipple Anatomy 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 230000009635 nitrosylation Effects 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 1
- FIKAKWIAUPDISJ-UHFFFAOYSA-L paraquat dichloride Chemical compound [Cl-].[Cl-].C1=C[N+](C)=CC=C1C1=CC=[N+](C)C=C1 FIKAKWIAUPDISJ-UHFFFAOYSA-L 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 229920001083 polybutene Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 150000004032 porphyrins Chemical class 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 1
- 238000006862 quantum yield reaction Methods 0.000 description 1
- 238000005956 quaternization reaction Methods 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- TUIHPLOAPJDCGN-UHFFFAOYSA-M rhodamine 800 Chemical compound [Cl-].C1CCN2CCCC3=C2C1=C1OC2=C(CCC4)C5=[N+]4CCCC5=CC2=C(C#N)C1=C3 TUIHPLOAPJDCGN-UHFFFAOYSA-M 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 229920002379 silicone rubber Polymers 0.000 description 1
- 239000004945 silicone rubber Substances 0.000 description 1
- 238000005245 sintering Methods 0.000 description 1
- IFGCUJZIWBUILZ-UHFFFAOYSA-N sodium 2-[[2-[[hydroxy-(3,4,5-trihydroxy-6-methyloxan-2-yl)oxyphosphoryl]amino]-4-methylpentanoyl]amino]-3-(1H-indol-3-yl)propanoic acid Chemical compound [Na+].C=1NC2=CC=CC=C2C=1CC(C(O)=O)NC(=O)C(CC(C)C)NP(O)(=O)OC1OC(C)C(O)C(O)C1O IFGCUJZIWBUILZ-UHFFFAOYSA-N 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 230000004763 spore germination Effects 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 229920006132 styrene block copolymer Polymers 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- AWDBHOZBRXWRKS-UHFFFAOYSA-N tetrapotassium;iron(6+);hexacyanide Chemical compound [K+].[K+].[K+].[K+].[Fe+6].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-] AWDBHOZBRXWRKS-UHFFFAOYSA-N 0.000 description 1
- ANRHNWWPFJCPAZ-UHFFFAOYSA-M thionine Chemical compound [Cl-].C1=CC(N)=CC2=[S+]C3=CC(N)=CC=C3N=C21 ANRHNWWPFJCPAZ-UHFFFAOYSA-M 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- 239000011135 tin Substances 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- XJCQPMRCZSJDPA-UHFFFAOYSA-L trimethyl-[3-[4-[(e)-(3-methyl-1,3-benzothiazol-2-ylidene)methyl]pyridin-1-ium-1-yl]propyl]azanium;diiodide Chemical compound [I-].[I-].S1C2=CC=CC=C2N(C)\C1=C\C1=CC=[N+](CCC[N+](C)(C)C)C=C1 XJCQPMRCZSJDPA-UHFFFAOYSA-L 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
Images










































Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/536—Immunoassay; Biospecific binding assay; Materials therefor with immune complex formed in liquid phase
- G01N33/542—Immunoassay; Biospecific binding assay; Materials therefor with immune complex formed in liquid phase with steric inhibition or signal modification, e.g. fluorescent quenching
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/58—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving labelled substances
- G01N33/582—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving labelled substances with fluorescent label
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N21/00—Investigating or analysing materials by the use of optical means, i.e. using sub-millimetre waves, infrared, visible or ultraviolet light
- G01N21/62—Systems in which the material investigated is excited whereby it emits light or causes a change in wavelength of the incident light
- G01N21/63—Systems in which the material investigated is excited whereby it emits light or causes a change in wavelength of the incident light optically excited
- G01N21/64—Fluorescence; Phosphorescence
- G01N21/6486—Measuring fluorescence of biological material, e.g. DNA, RNA, cells
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Biomedical Technology (AREA)
- Molecular Biology (AREA)
- Chemical & Material Sciences (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Analytical Chemistry (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Physics & Mathematics (AREA)
- Cell Biology (AREA)
- Medicinal Chemistry (AREA)
- Food Science & Technology (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Virology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Apparatus Associated With Microorganisms And Enzymes (AREA)
- Investigating, Analyzing Materials By Fluorescence Or Luminescence (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
Description
本出願は、それぞれ参照により全開示内容が本明細書に組み込まれる、2012年5月2日出願の同時係属の米国仮特許出願第61/641,809号明細書および2013年3月14日出願の同時係属の米国仮特許出願第61/784,789号の優先権および恩典を請求するものである。
本明細書で説明される細胞収集システムは、生存細胞の存在を検出する光検出システムと共に使用することができる。この結果を使用して、目的の特定のサンプルのバイオバーデン(例えば、サンプル中の生存細胞の数および/またはパーセンテージおよび/または画分)を測定することができる。検出システムの例が、例えば、2010年11月3日出願の国際出願PCT/IB2010/054965号明細書、2011年2月24日出願の米国特許出願第13/034,402号明細書、2010年11月3日出願の国際出願PCT/IB2010/054966号明細書、2011年2月24日出願の米国特許出願第13/034,380号明細書、2010年11月3日出願の国際出願PCT/IB2010/054967号明細書、および2011年2月24日出願の米国特許出願第13/034,515号明細書に記載されている。図1Aに概略的に示されている例示的なシステム100の一実施形態は、(i)細胞が配置される多孔質膜が回転軸140を中心に回転する回転プラットフォーム130および(ii)光源180(例えば、白色光源またはレーザー光源(例えば、近赤外線レーザー))を備える検出システム170に対して線形に(経路160を参照)に移動する可動プラットフォーム150含むサンプルアセンブリ120、ならびに少なくとも1つの検出器190、例えば、蛍光検出器を備えている。光源180からの光ビーム(励起光)が、回転プラットフォーム130およびその上に配置された平面膜に衝当し、放射光が検出器190によって検出される。光源180および検出器190は、光ビームが衝突し、実質的に同じ角度でプラットフォーム130から離れるように、プラットフォーム130に対して同様の角度で配置することができる。特定の状況では、検出システムは、1つの波長範囲(図12Bを参照)または複数の波長範囲を検出する1つの検出器からなる。あるいは、検出システムは、それぞれが異なる波長範囲を検出する複数の検出器からなる。
図10Aは、例示的なカップ/ベースアセンブリ550の構成要素を示している。多孔質支持部材558および膜202が、ベース554の中心に配設される。次いで、カップ552が、膜202の上部に取り付けられ、これにより、この膜202が平らな位置に容易に維持される。カップ552の内部が汚染されないようにするためにカップ552の上部に蓋556を設けることができる。図10Bは、構成要素の取り付けを示している(膜202および支持部材558は示されていない。
細胞が透過性膜に収集されたら、細胞を、生死判別染色剤または生死判別染色システムを用いて染色して、生存細胞を検出する、または生存細胞と非生存細胞を区別することができる。特定の染色プロトコルは、様々な因子、例えば、検出される細胞、利用される染色剤または染色システム、ならびに細胞が迅速に染色されて検出されるか否か、または細胞が、一定期間、例えば、30分〜数時間培養されて細胞が増殖し、これにより1つの細胞ではなく複数の細胞が特定の位置で検出されるか否かによって決まる。例示的な染色、および必要な場合は培養プロトコルも、以下のセクションで説明される。

細胞収集システムを使用して、流体サンプル中に元々存在した細胞を収集したら、膜または膜アセンブリを、適切な検出システムの中に挿入するために膜ホルダー(例えば、ホルダー802)の中に挿入することができる。例示的な検出システムは、例えば、2010年11月3日出願の国際出願PCT/IB2010/054965号明細書、2011年2月24日出願の米国特許出願第13/034,402号明細書、2010年11月3日出願の国際出願PCT/IB2010/054966号明細書、2011年2月24日出願の米国特許出願第13/034,380号明細書、2010年11月3日出願の国際出願PCT/IB2010/054967号明細書、および2011年2月24日出願の米国特許出願第13/034,515号明細書に記載されている。上述の検出システムでは、励起光ビームが膜の表面に照射されるときに、膜が回転させられる。放射された光は、少なくとも1つの光検出器で検出される。
この実施例は、蛍光プローブおよび1回の消光剤洗浄を用いて固体支持体上の生菌(大腸菌(E.coli))を選択的に染色して画像化することが可能であることを実証する。
この実施例は、蛍光プローブおよび2つのPET消光剤を用いて溶液中の生菌(大腸菌(E.coli))を選択的に染色して画像化することが可能であることを実証する。
この実施例は、蛍光プローブおよびPET消光剤とFRET消光剤との組み合わせを用いて溶液中の生菌(大腸菌(E.coli))を選択的に染色して画像化することが可能であることを実証する。
この実施例は、図1Aに概略的に示されている検出システムを用いて、蛍光プローブおよび消光剤洗浄により生存微生物を選択的に染色して画像化することが可能であることを実証する。
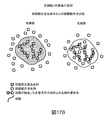
この実施例は、核酸結合膜不透過性消光剤と組み合わせられた核酸結合膜透過性蛍光染料を用いて生存微生物(大腸菌(E.coli)、黄色ブドウ球菌(S.aureus)、カンジダアルビカンス(C.albicans))を選択的に染色して画像化することが可能であることを実証する。
この実施例は、核酸結合膜不透過性消光剤と組み合わせて核酸結合膜透過性蛍光染料を用いて生存微生物(大腸菌(E.coli)および黄色ブドウ球菌(S.aureus))を選択的に染色して画像化し、次いで、生存細胞が回転膜上で検出される本明細書で説明された検出システムまたは落射蛍光顕微鏡のいずれかを用いて生存細胞を画像化することが可能であることを実証する。
本明細書で言及した特許文献および科学論文のそれぞれの全開示は、あらゆる目的のために参照により組み込まれる。米国仮特許出願第61/641,805号明細書;同第61/641,809号明細書;同第61/641,812号明細書;同第61/784,759号明細書;同第61/784,789号明細書;および同第61/784,807号明細書の全ての開示内容は、あらゆる目的のために参照により本明細書に組み込まれる。
本発明は、本発明の概念または本質的な特徴から逸脱することなく、他の特定の形態で実施することができる。従って、上述の実施形態は、本明細書で説明される本発明を限定するのではなく、全ての点で例示であると見なされるべきである。様々な実施形態の様々な構造要素および開示される様々な方法のステップは、様々な組み合わせおよび順で利用することができ、このような全ての変更形態が、本発明の形態と見なされる。従って、本発明の範囲は、上述の説明ではなく添付の特許請求の範囲によって示され、請求項と均等の意味および範囲に含まれる全ての変更が、本発明に包含されるものとする。
Claims (21)
- 液体サンプル中の生存細胞の存在および/または量を検出する方法であって、前記方法が、
(a)液体サンプルを、実質的に平面の多孔質膜の少なくとも一部分を通過させ、その結果、生存細胞が存在する場合、生存細胞が多孔質膜の一部分に保持されるステップ、ここで、前記多孔質膜は、最大でも100μmの平面度公差を有し、最大でも100μmの平面度公差を有する膜接触表面を含む流体透過性支持部材の上に維持される;
(b)前記液体サンプルが多孔質膜を通過した後に、多孔質膜の一部分に保持された生存細胞が存在する場合は、生存細胞を蛍光標識で標識するステップ;
(c)前記多孔質膜を検出システムに対して回転させることによって、最大でも100μmの平面度公差を有する前記多孔質膜の一部分をスキャンするステップ、ここで、前記検出システムが、
(i)前記蛍光標識を励起して発光事象を生じさせるのに適した波長の光ビームを放射する光源、および
(ii)前記発光事象を検出することができる少なくとも1つの検出器
を含み、前記スキャンにより、前記平面の多孔質膜の複数の領域を調べ、全ての生存細胞に関連した蛍光標識の励起によって生じる発光事象を検出する;ならびに
(d)ステップ(c)で検出された発光事象に基づいて、前記膜によって収集された生存細胞の存在および/または量を決定するステップ、
を含み、前記生存細胞が微生物である、方法。 - ステップ(b)で、前記細胞が、生死判別染色剤および生死判別染色システムの少なくとも1つを用いて標識される、請求項1に記載の方法。
- 光源のビームが、350nm〜1000nmの範囲の波長を有する光を放射する、請求項1または2に記載の方法。
- 前記波長が、350nm〜600nmおよび600nm〜750nmの少なくとも1つの範囲である、請求項3に記載の方法。
- 前記検出器が、350nm〜1000nmの範囲の放射光を検出する、請求項1〜4のいずれか1項に記載の方法。
- 前記光検出器が、350nm〜450nm、450nm〜550nm、550nm〜650nm、650nm〜750nm、750nm〜850nm、および850nm〜950nmから選択される少なくとも1つの範囲の放射光を検出する、請求項5に記載の方法。
- 前記多孔質膜がディスクを含む、請求項1〜6のいずれか1項に記載の方法。
- 前記多孔質膜が、350nm〜1000nmの範囲の波長を有する光に曝露されたときに実質的に非自家蛍光性である、請求項1〜7のいずれか1項に記載の方法。
- 前記多孔質膜が、前記多孔質膜を流体は通過するが細胞は前記多孔質膜に保持されるように1μm未満の平均直径を有する複数の孔を画定している、請求項1〜8のいずれか1項に記載の方法。
- 前記多孔質膜が、1μm〜3,000μm;10μm〜2,000μm;および100μm〜1,000μmからなる群から選択される範囲の厚さを有する、請求項1〜9のいずれか1項に記載の方法。
- 前記流体透過性支持部材が、0.1mm〜10mm;0.5mm〜5mm;および1mm〜3mmからなる群から選択される範囲の厚さを有する、請求項1〜10のいずれか1項に記載の方法。
- 前記光源からの光によって活性化されると蛍光事象を生成する複数の蛍光粒子を前記多孔質膜上に収集するステップをさらに含む、請求項1〜11のいずれか1項に記載の方法。
- 前記液体サンプルの少なくとも一部における生存細胞の量を決定するステップをさらに含む、請求項1〜12のいずれか1項に記載の方法。
- 前記生存細胞の前記多孔質膜における位置を決定するステップをさらに含む、請求項1〜13のいずれか1項に記載の方法。
- ステップ(d)の後に、前記多孔質膜によって収集された前記生存細胞が成長および/または増殖できる条件下で前記多孔質膜を培養するステップをさらに含む、請求項1〜14のいずれか1項に記載の方法。
- 前記生存細胞の属および/または種を特定するステップをさらに含む、請求項1〜15のいずれか1項に記載の方法。
- 前記スキャンするステップ(c)が、前記多孔質膜上の入れ子状円形パターンおよび螺旋パターンの少なくとも1つを光ビームでトレースするステップを含む、請求項1〜16のいずれか1項に記載の方法。
- ステップ(b)の前、ステップ(b)の最中、またはステップ(b)の前および最中に、細胞増殖できる条件下で前記生存細胞が培養される、請求項1〜17のいずれか1項に記載の方法。
- 前記多孔質膜上に配置された前記生存細胞が、細胞増殖ができる条件下で培養される、請求項18に記載の方法。
- ステップ(b)の後であるが、ステップ(c)の前に、細胞増殖ができる条件下で前記生存細胞が培養される、請求項1〜19のいずれか1項に記載の方法。
- ステップ(c)において、前記多孔質膜を、30rpm〜1000rpmの速度で回転する、請求項1記載の方法。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261641809P | 2012-05-02 | 2012-05-02 | |
| US61/641,809 | 2012-05-02 | ||
| US201361784789P | 2013-03-14 | 2013-03-14 | |
| US61/784,789 | 2013-03-14 | ||
| PCT/US2013/039349 WO2013166337A1 (en) | 2012-05-02 | 2013-05-02 | Method of detecting viable cells in a cell sample |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2015517656A JP2015517656A (ja) | 2015-06-22 |
| JP2015517656A5 JP2015517656A5 (ja) | 2016-06-23 |
| JP6482459B2 true JP6482459B2 (ja) | 2019-03-13 |
Family
ID=48670053
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015510465A Active JP6482459B2 (ja) | 2012-05-02 | 2013-05-02 | 細胞サンプル中の生存細胞を検出する方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US9709500B2 (ja) |
| EP (1) | EP2780707B1 (ja) |
| JP (1) | JP6482459B2 (ja) |
| CN (1) | CN104755931B (ja) |
| WO (1) | WO2013166337A1 (ja) |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2769204B1 (en) | 2012-05-02 | 2016-02-17 | Charles River Laboratories, Inc. | Cell capture system and use thereof |
| US9709500B2 (en) | 2012-05-02 | 2017-07-18 | Charles River Laboratories, Inc. | Optical method for detecting viable microorganisms in a cell sample |
| CN104662425B (zh) | 2012-05-02 | 2017-10-10 | 查尔斯河实验室公司 | 活性染色方法 |
| US9898820B2 (en) * | 2013-03-18 | 2018-02-20 | Life Technologies Corporation | Methods and systems for analyzing biological reaction systems |
| USD724755S1 (en) | 2013-11-04 | 2015-03-17 | Charles River Laboratories, Inc. | Cup |
| JP6556155B2 (ja) | 2013-11-04 | 2019-08-07 | チャールズ リバー ラボラトリーズ, インコーポレイテッド | ろ過システム及びその用途 |
| WO2015066727A1 (en) * | 2013-11-04 | 2015-05-07 | Charles River Laboratories, Inc. | Method for detecting viable cells in a cell sample |
| US20150218612A1 (en) * | 2014-02-06 | 2015-08-06 | Tacount Exact Ltd. | Apparatus, system and method for live bacteria microscopy |
| US9436866B2 (en) | 2014-08-01 | 2016-09-06 | General Electric Company | High sensitivity flat panel microbiology detection and enumeration system |
| USD776295S1 (en) | 2014-11-04 | 2017-01-10 | Charles River Laboratories, Inc. | Base |
| USD776296S1 (en) | 2014-11-04 | 2017-01-10 | Charles River Laboratories, Inc. | Adapter |
| USD782694S1 (en) | 2014-11-04 | 2017-03-28 | Charles River Laboratories, Inc. | Filtration device |
| USD759836S1 (en) | 2014-11-04 | 2016-06-21 | Charles River Laboratories, Inc. | Cup |
| CA3211036A1 (en) | 2015-04-23 | 2016-10-27 | Bd Kiestra B.V. | A method and system for automated microbial colony counting from streaked sample on plated media |
| USD817509S1 (en) * | 2016-10-14 | 2018-05-08 | Spartan Bioscience Inc. | Diagnostic device |
| JP2018183088A (ja) * | 2017-04-26 | 2018-11-22 | アズビル株式会社 | 細胞生存率判定装置 |
| US10266870B2 (en) * | 2017-09-07 | 2019-04-23 | Eagle Analytical Services, Inc. | Process and system for flow cytometry fluorescent detection of reactive materials in viscous non-filterable materials |
| GB201716883D0 (en) * | 2017-10-13 | 2017-11-29 | Q-Linea Ab | Method for determining the concentration of intact microorganisms in a sample |
| US10564100B1 (en) * | 2018-10-12 | 2020-02-18 | SageMedic Corporation | Analysis of viable and nonviable cells |
| EP3884329A4 (en) | 2018-11-21 | 2022-11-09 | Cornell University | CLEARING AND AUTOFLUORESCENT OPTICAL SOLUTIONS AND METHODS FOR USE IN ENHANCED MICROSCOPIC IMAGING OF BIOLOGICAL TISSUE |
| KR102205473B1 (ko) * | 2018-12-21 | 2021-01-21 | 칸트사이언스 주식회사 | 세포 내 pH 측정방법 |
| WO2020127563A2 (en) * | 2018-12-21 | 2020-06-25 | Global Life Sciences Solutions Usa Llc | In-process device and method for cell culture monitoring |
| WO2021226113A1 (en) * | 2020-05-04 | 2021-11-11 | The Regents Of The University Of California | Electrochemical cellular circuits |
Family Cites Families (126)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1256360A (en) | 1984-02-10 | 1989-06-27 | John W. Bornhoeft | Apparatus and method for determining the effectiveness of sterlization |
| US4561553A (en) | 1985-01-22 | 1985-12-31 | Northern Engineering And Plastics Corp. | Snap on twist off tamper-proof closure for containers |
| WO1986005206A1 (en) | 1985-02-27 | 1986-09-12 | University Of Cincinnati | Viable microorganism detection by induced fluorescence |
| US4725891A (en) | 1986-09-15 | 1988-02-16 | Matrix Instruments Inc. | Film image digitizer |
| US4739416A (en) | 1986-09-15 | 1988-04-19 | Matrix Instruments Inc. | Digital image reproduction |
| US4783401A (en) | 1986-10-31 | 1988-11-08 | Smithkline Beckman Corporation | Viable cell labelling |
| US5037656A (en) | 1986-12-04 | 1991-08-06 | Millipore Corporation | Porous membrane having hydrophilic and cell growth promotions surface and process |
| JPH0813930B2 (ja) | 1987-12-28 | 1996-02-14 | 三井東圧化学株式会社 | 近赤外線吸収フィルター用高純度アントラキノン系色素 |
| US4930893A (en) | 1988-03-25 | 1990-06-05 | Molecular Dynamics | Electrophoresis imaging system |
| US4838632A (en) | 1988-05-06 | 1989-06-13 | Lumisys Inc. | Two-dimensional beam scanner |
| US5073488A (en) | 1988-11-29 | 1991-12-17 | Minnesota Mining And Manufacturing Company | Rapid method for determining efficacy of a sterilization cycle and rapid read-out biological indicator |
| WO1992002632A1 (en) | 1990-07-30 | 1992-02-20 | Sierra Cytometry | Fluorescent dyes for identification and enumeration of viable cells in milk |
| US5185450A (en) | 1991-02-19 | 1993-02-09 | University Of South Florida | Tetrazolium compounds for cell viability assays |
| US5296341A (en) | 1991-03-05 | 1994-03-22 | Lumisys, Inc. | Green light laser imaging method |
| US5172419A (en) | 1991-03-05 | 1992-12-15 | Lumisys, Inc. | Medical image processing system |
| US5314805A (en) | 1991-10-28 | 1994-05-24 | Molecular Probes, Inc. | Dual-fluorescence cell viability assay using ethidium homodimer and calcein AM |
| US5234585A (en) | 1991-12-19 | 1993-08-10 | Zuk, Incorporated | Vacuum filtration device |
| US5137609A (en) | 1992-01-31 | 1992-08-11 | Biometric Imaging Inc. | Differential separation assay |
| US5221454A (en) | 1992-01-31 | 1993-06-22 | Biometric Imaging Inc. | Differential separation assay |
| US5843680A (en) | 1992-01-31 | 1998-12-01 | Biometric Imaging, Inc. | Differential separation assay methods and test kits |
| DE69311944T2 (de) | 1992-04-01 | 1997-11-20 | Nihon Millipore Kogyo K K | Verfahren zur Bestimmung der Anzahl lebender Mikroorganismen |
| JP3228812B2 (ja) | 1993-02-10 | 2001-11-12 | 日本マイクロリス株式会社 | 生菌数を測定する方法 |
| US5547849A (en) | 1993-02-17 | 1996-08-20 | Biometric Imaging, Inc. | Apparatus and method for volumetric capillary cytometry |
| US5585246A (en) | 1993-02-17 | 1996-12-17 | Biometric Imaging, Inc. | Method for preparing a sample in a scan capillary for immunofluorescent interrogation |
| US5545535A (en) | 1993-04-13 | 1996-08-13 | Molecular Probes, Inc. | Fluorescent assay for bacterial gram reaction |
| US5658751A (en) | 1993-04-13 | 1997-08-19 | Molecular Probes, Inc. | Substituted unsymmetrical cyanine dyes with selected permeability |
| US5436134A (en) | 1993-04-13 | 1995-07-25 | Molecular Probes, Inc. | Cyclic-substituted unsymmetrical cyanine dyes |
| US5534416A (en) | 1993-04-13 | 1996-07-09 | Molecular Probes, Inc. | Fluorescent viability assay using cyclic-substituted unsymmetrical cyanine dyes |
| US5445946A (en) | 1993-04-13 | 1995-08-29 | Molecular Probes, Inc. | Intravacuolar stains for yeast and other fungi |
| US5437980A (en) | 1993-05-17 | 1995-08-01 | Molecular Probes, Inc. | Phenanthridium dye staining of nucleic acids in living cells |
| FR2707665A1 (fr) | 1993-06-28 | 1995-01-20 | Chemunex | Procédé de mesure et d'évaluation de la vitalité des microorganismes et ses applications. |
| JP3011036B2 (ja) | 1993-12-20 | 2000-02-21 | 三菱化学株式会社 | 光ディスク基板の複屈折の測定法 |
| IES940182A2 (en) * | 1994-03-01 | 1995-11-29 | Teagasc Agric Food Dev Authori | "Rapid detection of bacteria in liquid cultures" |
| JP3137160B2 (ja) | 1994-04-01 | 2001-02-19 | 日立電子エンジニアリング株式会社 | ウエハの位置ズレ補正方法 |
| US5550032A (en) | 1994-05-27 | 1996-08-27 | George Mason University | Biological assay for microbial contamination |
| JP3593161B2 (ja) | 1994-11-15 | 2004-11-24 | 株式会社トプコン | 回転体上異物位置測定装置 |
| DE69417900T2 (de) | 1994-11-17 | 1999-11-11 | Chemunex, Maisons-Alfort | Vorrichtung und Verfahren zum schnellen und hochempfindlichen Erkennen und Zählen von Mikroorganismen mittels Fluoreszenz |
| US5603900A (en) | 1995-05-19 | 1997-02-18 | Millipore Investment Holdings Limited | Vacuum filter device |
| US5565678A (en) | 1995-06-06 | 1996-10-15 | Lumisys, Inc. | Radiographic image quality assessment utilizing a stepped calibration target |
| WO1997012226A1 (en) | 1995-09-25 | 1997-04-03 | Tencor Instruments | Improved system for surface inspection |
| WO1997017463A1 (en) | 1995-11-08 | 1997-05-15 | Pioneer Hi-Bred International, Inc. | Methods of assessing viability of microbial cultures |
| WO1997024616A1 (fr) | 1995-12-29 | 1997-07-10 | Ishihara Sangyo Kaisha, Ltd. | Procede de determination du taux de viabilite de cellules |
| US6459805B1 (en) | 1996-03-26 | 2002-10-01 | Childrens Hospital Los Angeles | Fluorescence digital imaging microscopy system |
| DE19621312A1 (de) | 1996-05-28 | 1997-12-04 | Bayer Ag | Maskierung der Hintergrundfluoreszenz und Signalverstärkung bei der optischen Analyse biologisch medizinischer Assays |
| US5956146A (en) | 1997-01-29 | 1999-09-21 | Victor Company Of Japan, Ltd. | Birefringence measuring apparatus for optical disc substrate |
| US6221621B1 (en) | 1997-03-06 | 2001-04-24 | Bard Diagnostic Sciences, Inc. | Methods of screening for colorectal cancers in which a complement Factor I or related protein is associated |
| FR2764305B1 (fr) | 1997-06-04 | 2000-10-06 | Chemunex | Procede de detection et de numeration de cellules viables dans un echantillon biologique et kit pour sa mise en oeuvre |
| US6221612B1 (en) | 1997-08-01 | 2001-04-24 | Aurora Biosciences Corporation | Photon reducing agents for use in fluorescence assays |
| US6200762B1 (en) | 1997-08-01 | 2001-03-13 | Aurora Biosciences Corporation | Photon reducing agents and compositions for fluorescence assays |
| US6214563B1 (en) | 1997-08-01 | 2001-04-10 | Aurora Biosciences Corporation | Photon reducing agents for reducing undesired light emission in assays |
| US6043506A (en) | 1997-08-13 | 2000-03-28 | Bio-Rad Laboratories, Inc. | Multi parameter scanner |
| US5922617A (en) | 1997-11-12 | 1999-07-13 | Functional Genetics, Inc. | Rapid screening assay methods and devices |
| BR9814726A (pt) | 1997-12-30 | 2000-10-17 | Jose Remacle | Método compreendendo molécula de captura fixada em superficìe de disco |
| US6080868A (en) | 1998-01-23 | 2000-06-27 | The Perkin-Elmer Corporation | Nitro-substituted non-fluorescent asymmetric cyanine dye compounds |
| WO2000007007A1 (en) | 1998-07-28 | 2000-02-10 | Biometric Imaging, Inc. | Device and method for cell motility assay |
| US6130745A (en) | 1999-01-07 | 2000-10-10 | Biometric Imaging, Inc. | Optical autofocus for use with microtiter plates |
| JP2000232897A (ja) * | 1999-02-15 | 2000-08-29 | Nippon Mizushori Giken:Kk | 菌類の即時判別方法 |
| CA2362117C (en) | 1999-02-26 | 2004-11-30 | Cellomics, Inc. | A system for cell-based screening |
| JP2000304688A (ja) | 1999-04-16 | 2000-11-02 | Canon Inc | 基板測定方法および装置 |
| US6181413B1 (en) | 1999-09-03 | 2001-01-30 | Biometric Imaging, Inc. | Displacing volume in field of view |
| CA2427106C (en) | 1999-10-25 | 2014-09-16 | Genprime, Inc. | Method and apparatus for prokaryotic and eukaryotic cell quantitation |
| JP2001228088A (ja) | 2000-02-18 | 2001-08-24 | Nippon Laser & Electronics Lab | 生体試料光学的走査装置 |
| CA2337987A1 (en) | 2000-02-25 | 2001-08-25 | Elmex Limited | Membrane filtration system |
| JP2001242082A (ja) | 2000-02-29 | 2001-09-07 | Nippon Laser & Electronics Lab | 生体試料光学的走査装置 |
| GB0008787D0 (en) | 2000-04-10 | 2000-05-31 | Norsk Naeringsmiddelforskning | A method of cell detection |
| US6323337B1 (en) | 2000-05-12 | 2001-11-27 | Molecular Probes, Inc. | Quenching oligonucleotides |
| US7118878B1 (en) | 2000-06-09 | 2006-10-10 | Promega Corporation | Method for increasing luminescence assay sensitivity |
| IL139593A (en) | 2000-11-09 | 2010-12-30 | Biogem Optical Ltd | Method for the detection of viable microorganisms |
| US7546925B1 (en) | 2000-12-04 | 2009-06-16 | Roush Life Sciences, Llc | Disposable vacuum filtration apparatus capable of detecting microorganisms and particulates in liquid samples |
| US6633368B2 (en) | 2001-01-02 | 2003-10-14 | Becton, Dickinson And Company | Method for determining the volume of single red blood cells |
| US6714287B2 (en) | 2001-01-02 | 2004-03-30 | Becton, Dickinson And Company | Apparatus for determining the volume of single red blood cells |
| US20020098531A1 (en) | 2001-01-25 | 2002-07-25 | Thacker James D. | Rapid methods for microbial typing and enumeration |
| JP2002310886A (ja) | 2001-04-11 | 2002-10-23 | Canon Inc | ディスクサイトメトリーによる分析方法及び装置 |
| US7016087B2 (en) | 2001-08-08 | 2006-03-21 | Becton Dickinson And Company | Photon efficient scanner |
| US6750457B2 (en) | 2001-08-29 | 2004-06-15 | Becton Dickinson And Company | System for high throughput analysis |
| ES2329986T3 (es) * | 2001-09-06 | 2009-12-03 | Rapid Micro Biosystems Inc | Deteccion rapida de celulas en replicacion. |
| US7205100B2 (en) | 2002-10-10 | 2007-04-17 | Advanced Analytical Technologies, Inc. | Methods for reducing background fluorescence |
| JP4231920B2 (ja) * | 2003-08-01 | 2009-03-04 | 独立行政法人産業技術総合研究所 | ディスク状バイオチップ及びその読取り装置 |
| US7435576B2 (en) | 2003-09-30 | 2008-10-14 | Gen-Probe Incorporated | Filter snapper |
| ES2239886B1 (es) | 2003-11-05 | 2006-12-16 | Universidad De Barcelona | Metodo y aparato para la determinacion de la viabilidad celular. |
| EP1699351B1 (en) | 2003-12-22 | 2011-05-04 | Lightouch Medical, Inc. | Process for determination of cell viability |
| EP1728069A4 (en) | 2004-03-17 | 2009-08-19 | Ca Nat Research Council | METHOD AND DEVICE FOR DETECTING MICROORGANISMS |
| DE102004019685A1 (de) | 2004-04-20 | 2005-11-17 | DRäGER AEROSPACE GMBH | Passagiersauerstoffmaske |
| WO2006098752A2 (en) | 2004-07-29 | 2006-09-21 | Kim Laboratories | Ultrasensitive sensor and rapid detection of analytes |
| CN101432739A (zh) | 2004-07-29 | 2009-05-13 | 金实验室公司 | 超灵敏传感器和分析物的快速检测 |
| DE602005010515D1 (de) | 2004-08-06 | 2008-12-04 | Fuji Electric Holdings | Nachweisverfahren für lebenfähige Zellen |
| JP4779433B2 (ja) | 2004-08-26 | 2011-09-28 | パナソニック株式会社 | ろ過フィルタデバイス |
| US20060121443A1 (en) | 2004-12-03 | 2006-06-08 | Orion Biosolutions, Inc. | Determination of cell viability |
| US7018804B1 (en) | 2004-12-03 | 2006-03-28 | Orion Biosolutions, Inc. | Determination of cell viability and phenotype |
| WO2006091630A2 (en) | 2005-02-22 | 2006-08-31 | University Of Cincinnati | Determination of viable microorganisms using coated paramagnetic beads |
| JP2007033170A (ja) * | 2005-07-26 | 2007-02-08 | Fujifilm Corp | 蛍光検出方法および蛍光検出システム |
| WO2007028157A1 (en) | 2005-09-02 | 2007-03-08 | Zuk Peter Jr | Systems, apparatus and methods for vacuum filtration |
| US8268567B2 (en) | 2005-09-16 | 2012-09-18 | The Charles Stark Draper Laboratory | Reaction sensing in living cells |
| JP5268175B2 (ja) | 2006-01-12 | 2013-08-21 | バイオセンス テクノロジーズ インク. | 迅速に細胞の生存テストを実行するための方法及び構成物 |
| DE102006022877B4 (de) | 2006-05-15 | 2009-01-29 | Sartorius Stedim Biotech Gmbh | Verfahren zur Bestimmung der Viabilität von Zellen in Zellkulturen |
| GB0615302D0 (en) | 2006-08-01 | 2006-09-13 | Biotrace Internat Plc | Assay Method For The Detection Of Viable Microbial Cells In A Sample |
| US20080153125A1 (en) | 2006-11-10 | 2008-06-26 | Advanced Analytical Technologies, Inc. | Rapid detection of microorganisms in fluids |
| CN101201312A (zh) | 2006-12-15 | 2008-06-18 | 北京林业大学 | 一种中性红染色法检测藻细胞活性的方法 |
| AU2008239654A1 (en) | 2007-04-13 | 2008-10-23 | Promega Corporation | Luminescent live and dead cell assay |
| US20080305514A1 (en) * | 2007-06-06 | 2008-12-11 | Alcon Research, Ltd. | Method for detecting microbes |
| WO2009029039A1 (en) | 2007-08-28 | 2009-03-05 | Ridgeview Instruments Ab | Method for measurement of cell viability |
| US20090099525A1 (en) | 2007-10-11 | 2009-04-16 | Animas Corporation | Drug Delivery System with Breakaway Plunger Extractor |
| JP2011503579A (ja) | 2007-11-09 | 2011-01-27 | ジェンザイム・コーポレーション | 対照細胞を使用しない細胞生存率の測定方法 |
| EP2232243B1 (en) | 2007-12-20 | 2015-12-16 | Nederlandse Organisatie voor toegepast- natuurwetenschappelijk onderzoek TNO | Real-time method for the detection of viable micro-organisms |
| JP2011519959A (ja) | 2008-05-13 | 2011-07-14 | ウォルター アンド エリザ ホール インスティテュート オブ メディカル リサーチ | 破壊された細胞膜を有する細胞、病原体に感染した細胞、死にかけている細胞または死細胞を検出する方法 |
| US9128097B2 (en) | 2008-07-14 | 2015-09-08 | Chemometec A/S | Method and kit for assessing viable cells |
| CN101344476B (zh) | 2008-08-20 | 2011-03-16 | 吉林农业大学 | 细菌活的非可培养状态荧光显微镜观察与计数方法 |
| ES2540544T3 (es) | 2009-05-04 | 2015-07-10 | Novartis Ag | Microensayo rápido de esterilidad |
| FR2945457B1 (fr) | 2009-05-12 | 2011-07-15 | Aes Chemunex | Ensemble forme d'une membrane de filtration et d'une plaque de support, ainsi que dispositif de test microbiologique en faisant usage. |
| US8148515B1 (en) | 2009-06-02 | 2012-04-03 | Biotium, Inc. | Detection using a dye and a dye modifier |
| WO2010151131A2 (en) | 2009-06-26 | 2010-12-29 | Nederlandse Organisatie Voor Toegepast- Natuurwetenschappelijk Onderzoek Tno | Apparatus for detecting viable microorganisms or spores in a sample and use thereof. |
| JP4825313B2 (ja) | 2009-07-24 | 2011-11-30 | 森永乳業株式会社 | 微生物検出法及び微生物検出キット |
| US20120244571A1 (en) | 2010-03-26 | 2012-09-27 | Morinaga Milk Industry Co. Ltd | Measurement method for viable cell count, and culture medium |
| GB201005939D0 (en) | 2010-04-09 | 2010-05-26 | Biostatus Ltd | Method of analysing a cell or other biological material containing nucleic acid |
| US20110294206A1 (en) * | 2010-05-28 | 2011-12-01 | California Institute Of Technology | Methods and design of membrane filters |
| CN102971433A (zh) | 2010-06-03 | 2013-03-13 | 巴斯夫欧洲公司 | 微生物的检测和计数 |
| WO2012051437A1 (en) | 2010-10-13 | 2012-04-19 | Life Technologies Corporation | Compositions and assays for determining cell viability |
| WO2012059785A1 (en) | 2010-11-03 | 2012-05-10 | Reametrix Inc. | Methods for calibrating a raman spectrometer, and spectrometer |
| WO2012059784A1 (en) | 2010-11-03 | 2012-05-10 | Reametrix Inc | Method and device for fluorescent measurement of samples |
| EP2635895A1 (en) | 2010-11-03 | 2013-09-11 | Reametrix Inc. | Measurement system for fluorescent detection, and method therefor |
| EP2637787A1 (en) | 2010-11-08 | 2013-09-18 | Reactrix Systems, Inc. | Sample assembly for a measurement device |
| US9446411B2 (en) | 2011-02-24 | 2016-09-20 | Reametrix, Inc. | Sample assembly for a measurement device |
| US20130068310A1 (en) | 2011-09-15 | 2013-03-21 | University Of Washington Through Its Center Of Commercialization | Method and Apparatus for a Microfluidic Device |
| EP2769204B1 (en) | 2012-05-02 | 2016-02-17 | Charles River Laboratories, Inc. | Cell capture system and use thereof |
| CN104662425B (zh) | 2012-05-02 | 2017-10-10 | 查尔斯河实验室公司 | 活性染色方法 |
| US9709500B2 (en) | 2012-05-02 | 2017-07-18 | Charles River Laboratories, Inc. | Optical method for detecting viable microorganisms in a cell sample |
-
2013
- 2013-05-02 US US13/886,004 patent/US9709500B2/en active Active
- 2013-05-02 EP EP13730384.8A patent/EP2780707B1/en active Active
- 2013-05-02 JP JP2015510465A patent/JP6482459B2/ja active Active
- 2013-05-02 WO PCT/US2013/039349 patent/WO2013166337A1/en active Application Filing
- 2013-05-02 CN CN201380035732.8A patent/CN104755931B/zh not_active Expired - Fee Related
-
2017
- 2017-06-02 US US15/612,320 patent/US20180120231A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| US9709500B2 (en) | 2017-07-18 |
| EP2780707B1 (en) | 2016-02-24 |
| JP2015517656A (ja) | 2015-06-22 |
| CN104755931A (zh) | 2015-07-01 |
| CN104755931B (zh) | 2016-08-31 |
| EP2780707A1 (en) | 2014-09-24 |
| US20180120231A1 (en) | 2018-05-03 |
| US20130323745A1 (en) | 2013-12-05 |
| WO2013166337A1 (en) | 2013-11-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6285915B2 (ja) | 生死判別染色法 | |
| JP6482459B2 (ja) | 細胞サンプル中の生存細胞を検出する方法 | |
| JP6240162B2 (ja) | 細胞収集システムおよびその使用 | |
| Zarei | Portable biosensing devices for point-of-care diagnostics: Recent developments and applications | |
| AU2007207450B2 (en) | Method for rapid detection and evaluation of cultured cell growth | |
| KR20070061802A (ko) | 초음파 감응 센서 및 분석 대상물의 고속 검출 방법 | |
| US20160298162A1 (en) | Method for detecting viable cells in a cell sample | |
| Leader et al. | DEVELOPING AN OXGYEN DETECTION METHOD FOR A MICROFLUIDIC-BASED HYPOXIA CHAMBER |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160426 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20160426 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20170329 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170407 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20170707 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20170907 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171006 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180223 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20180523 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20180720 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180817 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190129 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190212 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6482459 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |