JP5367521B2 - リチウム二次電池の負極用炭素材料及びその製造方法 - Google Patents
リチウム二次電池の負極用炭素材料及びその製造方法Info
- Publication number
- JP5367521B2 JP5367521B2 JP2009217842A JP2009217842A JP5367521B2 JP 5367521 B2 JP5367521 B2 JP 5367521B2 JP 2009217842 A JP2009217842 A JP 2009217842A JP 2009217842 A JP2009217842 A JP 2009217842A JP 5367521 B2 JP5367521 B2 JP 5367521B2
- Authority
- JP
- Japan
- Prior art keywords
- negative electrode
- graphite
- secondary battery
- lithium secondary
- lithium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images
Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
- H01M4/583—Carbonaceous material, e.g. graphite-intercalation compounds or CFx
- H01M4/587—Carbonaceous material, e.g. graphite-intercalation compounds or CFx for inserting or intercalating light metals
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/20—Graphite
- C01B32/205—Preparation
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/70—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data
- C01P2002/77—Crystal-structural characteristics defined by measured X-ray, neutron or electron diffraction data by unit-cell parameters, atom positions or structure diagrams
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/80—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70
- C01P2002/82—Crystal-structural characteristics defined by measured data other than those specified in group C01P2002/70 by IR- or Raman-data
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02T—CLIMATE CHANGE MITIGATION TECHNOLOGIES RELATED TO TRANSPORTATION
- Y02T10/00—Road transport of goods or passengers
- Y02T10/60—Other road transportation technologies with climate change mitigation effect
- Y02T10/70—Energy storage systems for electromobility, e.g. batteries
Landscapes
- Chemical & Material Sciences (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Manufacturing & Machinery (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Life Sciences & Earth Sciences (AREA)
- Geology (AREA)
- Materials Engineering (AREA)
- Battery Electrode And Active Subsutance (AREA)
Description
とを備えたリチウム二次電池である。
また、シート状、ペレット状等の形状に成形された負極材スラリーと集電体との一体化は、例えば、ロール、プレス、もしくはこれらの組み合わせ等、公知の方法により行うことができる。
正極に用いる活物質としては、特に制限はなく、例えば、リチウムイオンをドーピング又はインターカレーション可能な金属化合物、金属酸化物、金属硫化物、又は導電性高分子材料を用いればよく、例示するのであれば、コバルト酸リチウム(LiCoO2)、ニッケル酸リチウム(LiNiO2)、マンガン酸リチウム(LiMn2O4)、及び複酸化物(LiCoXNiYMnZO2、X+Y+Z=1)、リチウムバナジウム化合物、V2O5、V6O13、VO2、MnO2、TiO2、MoV2O8、TiS2、V2S5、VS2、MoS2、MoS3、Cr3O8、Cr2O5、オリビン型LiMPO4(M:Co、Ni、Mn、Fe)、ポリアセチレン、ポリアニリン、ポリピロール、ポリチオフェン、ポリアセン等の導電性ポリマー、多孔質炭素等及びこれらの混合物を挙げることができる。
セパレータとしては、例えば、ポリエチレン、ポリプロピレン等のポリオレフィンを主成分とした不織布、クロス、微多孔性フィルム又はそれらを組み合わせたものを使用することができる。なお、作製するリチウムイオン二次電池の正極と負極が直接接触しない構造にした場合は、セパレータを使用する必要はない。
<物性の測定>
(1)黒鉛粉末の結晶子の大きさL(112)の算出
黒鉛粉末に、内部標準としてSi標準試料を10wt%混合し、ガラス製回転試料ホルダー(25mmφ×0.2mmt)に詰め、JIS R7651(2007)に基づき、広角X線回折法で測定を行い、黒鉛粉末の結晶子の大きさL(112)を算出した。X線回折装置は、Bruker−AXS社製 D8 ADVANCE(封入管型)、X線源はCuKα線(KβフィルターNiを使用)、X線管球への印可電圧及び電流は40kV及び40mAとした。
得られた回折図形についても、JIS R7651(2007)に準拠した方法で解析を行った。具体的には、測定データにスムージング処理、バックグラウンド除去の後、吸収補正、偏光補正、Lorentz 補正を施し、Si標準試料の(422)回折線のピーク位置、及び値幅を用いて、黒鉛粉末の(112)回折線に対して補正を行い、結晶子サイズを算出した。なお、結晶子サイズは、補正ピークの半値幅から以下のScherrerの式を用いて計算した。測定・解析は3 回ずつ実施し、その平均値をL(112)とした。
L=K×λ/(β0×cosθB)- - - - - -Scherrerの式
ここで、L :結晶サイズ(nm)
K:形状因子定数(=1.0)
λ :X線の波長(=0.15406nm)
θB:ブラッグ角
β0:半値幅(補正値)
実施例及び比較例に記載された黒鉛粉末のL(112)が測定された結果は、表1に示された通りである。
光源をAr+レーザー(励起波長514.5 nm)としたラマン分光分析を行った。測定はマクロモードで、レーザーのスポット径は約100μmであり、レーザー照射範囲全体からの平均的な情報が得られるように設定した。測定装置はRamanor T−64000 (Jobin Yvon/愛宕物産)、測定配置は60°、レーザーパワーは10mWである。
得られたラマンスペクトル図において、1580cm−1±100cm−1の波長領域に存在するピークの半価幅ΔνGを、最小二乗法による直接読み取りにより算出した。なお測定・解析は3回ずつ実施し、その平均値をΔνGとした。
実施例及び比較例に記載された黒鉛粉末のΔνGが測定された結果は、表1に示された通りである。
原料油組成物のノルマルパラフィンの含有率は、キャピラリーカラムが装着されたガスクロマトグラフによって測定した。具体的には、ノルマルパラフィンの標準物質によって検定した後、溶出クロマトグラフィー法によって分離された非芳香族成分の試料をキャピラリーカラムに通して測定した。この測定値から原料油組成物の全質量を基準とした含有率を算出した。
また芳香族炭素分率(fa)は、Knight法により算出した。すなわち、重質油組成物の13C−NMR法による芳香族炭素のスペクトルとして3つの成分(A1,A2,A3)に分割する。ここで、A1は芳香族環内部炭素数、置換されている芳香族炭素と置換していない芳香族炭素の半分(13C−NMRの約40〜60ppmのピークに相当)、A2は置換していない残りの半分の芳香族炭素(13C−NMRの約60〜80ppmのピークに相当)、A3は脂肪族炭素数(13C−NMRの約130〜190ppmのピークに相当)であり、これらからfaは、fa=(A1+A2)/(A1+A2+A3)により算出した。比較例8以外の全ての実施例及び比較例に記載された黒鉛粉末の原料油のノルマルパラフィン含有率、及び芳香族指数faは、表1に示された通りである。
(1)負極材料評価用セルの作製方法
負極材料として、下記実施例又は比較例で得られた黒鉛粉末と結着剤のポリフッ化ビニリデン(クレハ社製KF#9310)、アセチレンブラック(デンカ社製のデンカブラック)を重量比で90:2:8に混合し、N−メチル−2−ピロリジノンを加えて混練した後、ペースト状にして、厚さ18μmの銅箔の片面に塗布し、乾燥及び圧延した。得られたシート状の電極を直径φ15mmに打ち抜き作用極とした。この作用極及びその他の必要部材を十分に乾燥させ、露点−100℃のアルゴンガスが満たされたグローブボックス内に導入し、負極材料評価用セルを組み立てた。乾燥条件は、作用極が減圧状態の下150℃で12時間以上、その他部材が減圧状態の下70℃で12時間以上である。
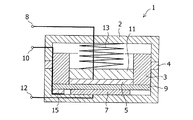
図1に負極材料評価用セル1の断面図を示す。評価用セル1は、四弗化エチレン製パッキング4により内部の気密が保持可能な中空金属体2を容器としている。当該中空金属体2にはまず、参照極15と上記工程により得られた作用極7とを離間して配置した。次に、これらの電極上に直径φ24mmのポリプロピレン製のマイクロポーラスフィルム(セルガード社製#2400)からなるセパレータ9と、厚さ0.7mm、直径φ17mmの円盤状リチウム金属箔からなる対極5とを順に積層した。なおリチウム金属箔と作用極との積層位置関係は、リチウム金属箔を作用極側に投影したときにその外周部が作用極7の外周を包囲するように押さえ治具3によって保持した。さらに、対極5、作用極7および参照極15から各々金属枠2の外部に延びる端子8、10、12を設けた。
次いで、前記中空金属体3に電解液6を注入すると共に、この積層体が、厚さ1mm、直径φ20mmのステンレス(SUS304)製円盤11を介してステンレス製のバネ13で加圧され、帯状のニッケル製リード板(厚さ50μm,幅3mm)にリチウム金属が巻きつけられた参照極15が作用極7近傍で固定されるように前記中空金属体3を封止し、負極材料評価用セル1を作製した。使用した電解液6は、エチレンカーボネートとエチルエチルメチルカーボネートとを体積比で3:7に混合した溶媒にヘキサフルオロリン酸リチウム(LiPF6)を1mol/Lの濃度となるように溶解したものである。
負極材料評価用セルを25℃の恒温室内に設置し、以下に示す充放電試験を行った。先ず作用極の面積を基準とし、電流密度が0.1mA/cm2となるような電流値で対極及び作用極の間を通電(放電)し、参照極に対する作用極の電位が0.01Vになるまで作用極にリチウムをドープした。10分間の休止の後、同じ電流値で参照極に対する作用極の電位が1.2Vになるまで通電(充電)し、作用極に吸蔵されたリチウムを脱ドープした。得られたリチウムドープ容量(mAh/g)とリチウム脱ドープ容量(mAh/g)を確認し、これらの値から初期充放電サイクルの充放電効率(%)を以下の式から算出した。
実施例及び比較例に記載された黒鉛粉末のリチウム脱ドープ容量、及び充放電効率は、表1に示された通りである。
(1)電池の作製方法
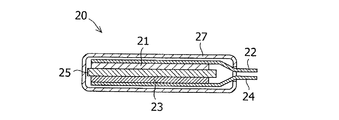
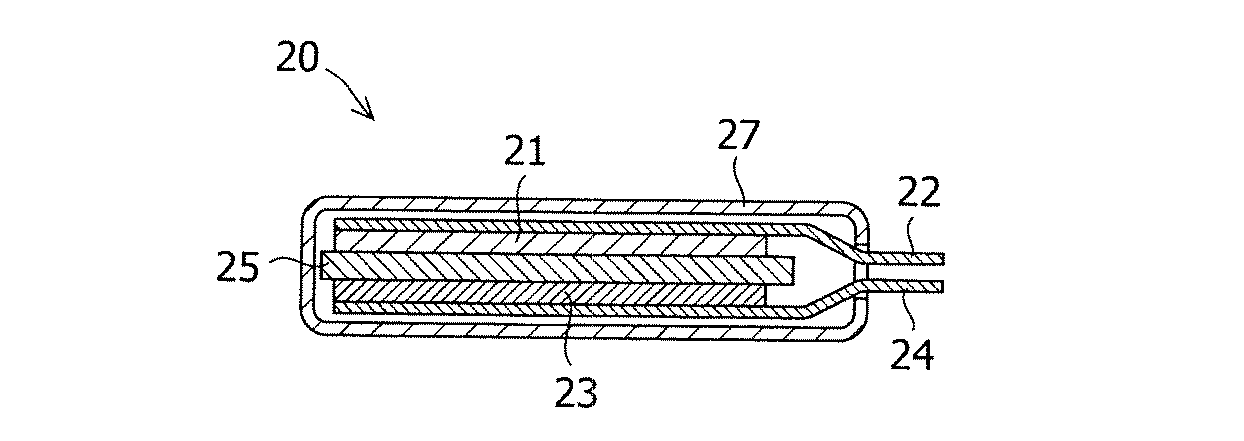
図2に作製した電池20の断面図を示す。正極21は、正極材料である平均粒子径6μmのニッケル酸リチウム(戸田工業社製LiNi0.8Co0.15Al0.05)と結着剤のポリフッ化ビニリデン(クレハ社製KF#1320)、アセチレンブラック(デンカ社製のデンカブラック)を重量比で89:6:5に混合し、N−メチル−2−ピロリジノンを加えて混練した後、ペースト状にして、厚さ30μmのアルミニウム箔の片面に塗布し、乾燥及び圧延操作を行い、塗布部のサイズが、幅30mm、長さ50mmとなるように切断されたシート電極である。このシート電極の一部はシートの長手方向に対して垂直に正極合剤が掻き取られ、その露出したアルミニウム箔が塗布部の集電体22(アルミニウム箔)と一体化して繋がっており、正極リード板としての役割を担っている。
負極23は、負極材料である下記実施例又は比較例で得られた黒鉛粉末と結着剤のポリフッ化ビニリデン(クレハ社製KF#9310)と、アセチレンブラック(デンカ社製のデンカブラック)とを重量比で90:2:8に混合し、N−メチル−2−ピロリジノンを加えて混練した後、ペースト状にして、厚さ18μmの銅箔の片面に塗布し、乾燥及び圧延操作を行い、塗布部のサイズが、幅32mm、長さ52mmとなるように切断されたシート電極である。このシート電極の一部はシートの長手方向に対して垂直に負極合剤が掻き取られ、その露出した銅箔が塗布部の集電体24(銅箔)と一体化して繋がっており、負極リード板としての役割を担っている。
電池20の作製は、正極21、負極23、セパレータ25、外装27及びその他部品を十分に乾燥させ、露点−100℃のアルゴンガスが満たされたグローブボックス内に導入して組み立てた。乾燥条件は、正極21及び負極23が減圧状態の下150℃で12時間以上、セパレータ25及びその他部材が減圧状態の下70℃で12時間以上である。
このようにして乾燥された正極21及び負極23を、正極の塗布部と負極の塗布部とが、ポリポロピレン製のマイクロポーラスフィルム(セルガード社製#2400)を介して対向させる状態で積層し、ポリイミドテープで固定した。なお、正極及び負極の積層位置関係は、負極の塗布部に投影される正極塗布部の周縁部が、負極塗布部の周縁部の内側で囲まれるように対向させた。得られた単層電極体を、アルミラミネートフィルムで包埋させ、電解液を注入し、前述の正・負極リード板がはみ出した状態で、ラミネートフィルムを熱融着することにより、密閉型の単層ラミネート電池を作製した。使用した電解液は、エチレンカーボネートとエチルメチルカーボネートが体積比で3:7に混合された溶媒にヘキサフルオロリン酸リチウム(LiPF6)が1mol/Lの濃度となるように溶解されたものである。
得られた電池を25℃の恒温室内に設置し、以下に示す充放電試験を行った。先ず1.5mAの電流で、電池電圧が4.2Vとなるまで定電流で充電した。10分間休止の後、同じ電流で電池電圧が3.0Vとなるまで定電流で放電する充放電サイクルを10回繰り返した。この充放電サイクルは、電池の異常を検地するためのものであるため、充放電サイクル試験のサイクル数には含まなかった。本実施例で作製された電池は、全て異常がないことを確認した。
次の充放電サイクルを第1サイクル(初期サイクル)とする。75mAの電流で、電池電圧が4.2Vとなるまで定電流で充電し、1分間休止の後、同じ電流で電池電圧が3.0Vとなるまで定電流で放電する充放電サイクルを設定し、このサイクルを1000回繰り返した。充放電サイクルの容量維持率として、初期放電容量に対する1000サイクル目の放電容量の割合(%)を算出した。実施例及び比較例で作製した黒鉛粉末を負極として使用した電池の充放電サイクルの容量維持率を表1中に示す。
(比較例1)
硫黄分3.1質量%の常圧蒸留残油を、触媒存在下、水素化分解率が25%以下となるように水素化脱硫し、水素化脱硫油を得た。水素化脱硫条件は、全圧180MPa、水素分圧160MPa、温度380℃である。また、常圧蒸留残渣油を減圧蒸留し、更に水素化脱硫したもの(硫黄分380質量ppm、15℃における密度0.83g/cm3)を、反応温度530℃、全圧2.3kgf/cm2、触媒/油比13、接触時間7秒で流動接触分解し、流動接触分解残油を得た。触媒としては、シリカ・アルミナ触媒に白金が担持されたものを使用した。次に、前述の水素化脱硫油と流動接触分解残油及びエチレンボトム油とを、質量比1:3:1で混合し、コークスの原料となる重質油組成物(以下、特に原料油組成物と略記)を得た。この原料油組成物のノルマルパラフィン含有率、及び芳香族指数faを表1に示す。
この原料油組成物をディレードコーカー装置に導入して、不活性ガス雰囲気下、550℃でコーキング処理し、原料炭組成物を得た。次いで、当該原料炭組成物をロータリーキルンに導入して1400℃でか焼し、炭素材料を得た。得られた炭素材料を機械式粉砕機(スーパーローターミル/日清エンジニアリング製)で粉砕し、精密空気分級機(ターボクラシファイヤー/日清エンジニアリング製)で分級することにより、平均粒子径12μmの炭素微粒子材料を得た。この炭素微粒子材料をるつぼに投入し、電気炉に設置して、80L/分の窒素ガス気流中、最高到達温度2100℃で熱処理した。このとき昇温速度は200℃/時間、最高到達温度の保持時間は16時間、降温速度は1000℃までが100℃/時間とし、その後窒素気流を保持させた状態で室温まで放冷させた。炭素微粒子材料を得た後の熱処理を、以下「黒鉛化」と呼称する。得られた黒鉛粉末を、黒鉛Aとする。
比較例1と同様にして得た炭素微粒子材料に対し、最高到達温度を2200℃とした以外は比較例1と同様な方法で黒鉛化した。得られた黒鉛粉末を、黒鉛Bとする。
比較例1と同様にして得た炭素微粒子材料に対し、最高到達温度を2400℃とした以外は比較例1と同様な方法で黒鉛化した。得られた黒鉛粉末を、黒鉛Cとする。
比較例1と同様にして得た炭素微粒子材料に対し、最高到達温度を2600℃とした以外は比較例1と同様な方法で黒鉛化した。得られた黒鉛粉末を、黒鉛Dとする。
比較例1と同様にして得た炭素微粒子材料に対し、最高到達温度を2700℃とした以外は比較例1と同様な方法で黒鉛化した。得られた黒鉛粉末を、黒鉛Eとする。
比較例1と同様にして得た原料炭組成物をるつぼに投入し、最高到達温度を2400℃とした以外は比較例1と同様な方法で黒鉛化した。この黒鉛を機械式粉砕機(スーパーローターミル/日清エンジニアリング製)で粉砕し、精密空気分級機(ターボクラシファイヤー/日清エンジニアリング製)で分級することにより、平均粒子径12μmの炭素微粒子材料を得た。得られた黒鉛粉末を黒鉛Fとする。
比較例1記載の流動接触分解残油のノルマルパラフィン含有率、及び芳香族指数faを表1に示す。この流動接触分解残油を原料油組成物とし、比較例1に記載された方法と同様にして原料炭組成物及び炭素微粒子を得た。この炭素微粒子材料に対し、最高到達温度を2400℃とした以外は比較例1と同様な方法で黒鉛化した。得られた黒鉛粉末を、黒鉛Gとする。
比較例1記載の流動接触分解残油を、同体積のn−ヘプタンを加え混合した後、ジメチルホルムアミドで選択抽出し、芳香族分と飽和分に分離させた。このうちの芳香族分と比較例1記載の水素化脱硫油とを質量比4:1で混合し、コークスの原料油組成物を得た。この原料油組成物のノルマルパラフィン含有率、及び芳香族指数faを表1に示す。この原料油を、比較例1に記載された方法と同様に処理し、原料炭組成物及び炭素微粒子を得た。この炭素微粒子材料に対し、最高到達温度を2400℃とした以外は比較例1と同様な方法で黒鉛化した。得られた黒鉛粉末を、黒鉛Hとする。
比較例1記載の水素化脱硫油と流動接触分解残油とを質量比3:1で混合し、コークスの原料油組成物を得た。この原料油組成物のノルマルパラフィン含有率、及び芳香族指数faを表1に示す。この原料油組成物を、比較例1に記載された方法と同様に処理し、原料炭組成物及び炭素微粒子を得た。この炭素微粒子材料に対し、最高到達温度を2400℃とした以外は比較例1と同様な方法で黒鉛化した。得られた黒鉛粉末を、黒鉛Iとする。
比較例1記載の流動接触分解残油に、同体積のn−ヘプタンを加え混合した後、ジメチルホルムアミドで選択抽出し、芳香族分と飽和分に分離させた。このうちの飽和分と比較例1記載の流動接触分解残油とを質量比1:1で混合し、コークスの原料油組成物を得た。この原料油組成物のノルマルパラフィン含有率、及び芳香族指数faを表1に示す。この原料油組成物を、比較例1に記載された方法と同様に処理し、原料炭組成物及び炭素微粒子を得た。この炭素微粒子材料に対し、最高到達温度を2400℃とした以外は比較例1と同様な方法で黒鉛化した。得られた黒鉛粉末を、黒鉛Jとする。
比較例1記載の水素化脱硫油と流動接触分解残油とを質量比4:1で混合し、コークスの原料油組成物を得た。この原料油組成物のノルマルパラフィン含有率、及び芳香族指数faを表1に示す。この原料油組成物を、比較例1に記載された方法と同様に処理し、原料炭組成物及び炭素微粒子を得た。この炭素微粒子材料に対し、最高到達温度を2400℃とした以外は比較例1と同様な方法で黒鉛化した。得られた黒鉛粉末を、黒鉛Kとする。
比較例1記載の流動接触分解残油を、ジメチルホルムアミドで選択抽出し、芳香族分と飽和分に分離させた。このようにして得られた芳香族分と飽和分を質量比4:1で混合し、コークスの原料油組成物を得た。この原料油組成物のノルマルパラフィン含有率、及び芳香族指数faを表1に示す。この原料油を、比較例1に記載された方法と同様に処理し、原料炭組成物及び炭素微粒子を得た。この炭素微粒子材料に対し、最高到達温度を2400℃とした以外は比較例1と同様な方法で黒鉛化した。得られた黒鉛粉末を、黒鉛Lとする。
高純度化処理された市販のブラジル産鱗片状天然黒鉛(固定炭素=99.6%,灰分=0.1%,揮発分=0.3%)を機械式粉砕機(スーパーローターミル/日清エンジニアリング製)で粉砕し、精密空気分級機(ターボクラシファイヤー/日清エンジニアリング製)で分級することにより、平均粒子径12μmの炭素微粒子材料を得た。得られた黒鉛粉末を黒鉛Mとする。
比較例1、実施例1〜3及び比較例2は同一の原料油組成物から得られた黒鉛粉末で、黒鉛化温度のみが異なっている。比較例1の黒鉛Aは、L(112)が1.5nm、ΔνGが22.3cm−1で、本出願に係る第一の態様の範囲であるL(112)が2〜4.2nm、ΔνGが15〜19cm−1の2つの範囲から共に外れている。
本発明の技術的範囲を以下の考察が何ら制約するものではないが、L(112)が2nm以下と相対的に小さくなった要因は、黒鉛化温度が2100℃と低かったからであると考えられる。このため負極としての容量(負極材料評価用セルのリチウム脱ドープ容量)が低くなった。
2 中空金属体
3 押さえ治具
4 パッキン
5、21 対極(正極)
6 電解液
7、23 作用極(負極)
8、10,12 端子
9、25 セパレータ
11 対極押さえ板
13 ばね
15 参照極
20 電池
22 正極集電体
24 負極集電体
27 外装
Claims (5)
- X線広角回折法によって得られた(112)回折線から算出されるc軸方向の結晶子の大きさL(112)が2.0〜4.2nmであって、且つ、波長5145オングストロームのアルゴンイオンレーザー光を用いたラマンスペクトル分析において、1580cm−1±100cm−1の波長領域に存在するピークの半価幅ΔνGが15〜19cm−1であるリチウム二次電池負極用人造黒鉛。
- 重質油組成物をディレードコーキングプロセスによってコーキング処理した後、熱処理する工程を経て得られたリチウム二次電池負極用人造黒鉛であって、前記重質油組成物は、ノルマルパラフィン含有率が5〜20wt%、Knight法により求められた芳香族指数faが0.3〜0.65である請求項1に記載のリチウム二次電池負極用人造黒鉛。
- 前記熱処理する工程が、1500℃以下で炭素化し粉砕することにより炭素微粒子材料を得るステップと、該炭素微粒子材料を不活性ガス雰囲気下最高到達温度が2200から2600℃になるまで加熱して前記人造黒鉛を得るステップとを含む請求項2に記載のリチウム二次電池負極用人造黒鉛。
- 重質油組成物をディレードコーキングプロセスによってコーキング処理した後、熱処理する工程を含む、請求項1〜3のいずれか1項に記載のリチウム二次電池負極用人造黒鉛の製造方法であって、前記重質油組成物は、ノルマルパラフィン含有率が5〜20wt%、Knight法により求められた芳香族指数faが0.3〜0.65であるリチウム二次電池負極用人造黒鉛の製造方法。
- リチウムの可逆的なインターカレーションが可能なリチウムを含んだ正極と、請求項1〜3のいずれか1項に記載のリチウム二次電池負極用人造黒鉛を含む負極と、非水電解質とを備えたリチウム二次電池。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009217842A JP5367521B2 (ja) | 2009-09-18 | 2009-09-18 | リチウム二次電池の負極用炭素材料及びその製造方法 |
| KR1020127008412A KR101820071B1 (ko) | 2009-09-18 | 2010-09-16 | 리튬 이차 전지의 음극용 탄소 재료 및 그 제조 방법 |
| PCT/JP2010/066096 WO2011034152A1 (ja) | 2009-09-18 | 2010-09-16 | リチウム二次電池の負極用炭素材料及びその製造方法 |
| EP10817265.1A EP2479823A4 (en) | 2009-09-18 | 2010-09-16 | NEGATIVE ELECTRODE CARBON MATERIAL FOR A LITHIUM ION SECONDARY BATTERY AND METHOD OF MANUFACTURING THEREOF |
| CN201080041837.0A CN102511096B (zh) | 2009-09-18 | 2010-09-16 | 锂二次电池的负极用碳材料及其制造方法 |
| US13/422,513 US8617508B2 (en) | 2009-09-18 | 2012-03-16 | Carbon material for negative electrode of lithium secondary battery and method for producing the same |
| US14/089,392 US20140079622A1 (en) | 2009-09-18 | 2013-11-25 | Carbon material for negative electrode of lithium secondary battery and method for producing the same |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009217842A JP5367521B2 (ja) | 2009-09-18 | 2009-09-18 | リチウム二次電池の負極用炭素材料及びその製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2011065961A JP2011065961A (ja) | 2011-03-31 |
| JP5367521B2 true JP5367521B2 (ja) | 2013-12-11 |
Family
ID=43758752
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009217842A Active JP5367521B2 (ja) | 2009-09-18 | 2009-09-18 | リチウム二次電池の負極用炭素材料及びその製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US8617508B2 (ja) |
| EP (1) | EP2479823A4 (ja) |
| JP (1) | JP5367521B2 (ja) |
| KR (1) | KR101820071B1 (ja) |
| CN (1) | CN102511096B (ja) |
| WO (1) | WO2011034152A1 (ja) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102947986B (zh) * | 2010-05-31 | 2015-12-09 | 吉坤日矿日石能源株式会社 | 锂离子二次电池负极材料用原料炭组合物 |
| JP5528923B2 (ja) * | 2010-06-25 | 2014-06-25 | Jx日鉱日石エネルギー株式会社 | リチウムイオン二次電池負極材料用の原料炭組成物 |
| JP5490636B2 (ja) * | 2010-06-30 | 2014-05-14 | Jx日鉱日石エネルギー株式会社 | リチウムイオン二次電池負極炭素材料用の原料油組成物 |
| JP5498279B2 (ja) * | 2010-06-30 | 2014-05-21 | Jx日鉱日石エネルギー株式会社 | リチウムイオン二次電池負極炭素材料用の原料油組成物 |
| JP5623262B2 (ja) * | 2010-12-13 | 2014-11-12 | Jx日鉱日石エネルギー株式会社 | リチウムイオン二次電池負極用黒鉛材料およびその製造方法、リチウムイオン二次電池 |
| JP5914460B2 (ja) * | 2011-03-30 | 2016-05-11 | Jxエネルギー株式会社 | リチウムイオン二次電池の負極用炭素材料の製造方法 |
| KR102008534B1 (ko) * | 2011-03-30 | 2019-08-07 | 제이엑스티지 에네루기 가부시키가이샤 | 리튬이온 이차전지의 음극용 탄소재료의 원료탄 조성물 및 그 제조방법 |
| JP5269231B1 (ja) * | 2012-06-29 | 2013-08-21 | エム・ティー・カーボン株式会社 | リチウムイオン二次電池負極用の黒鉛材料、それを用いたリチウムイオン二次電池及びリチウムイオン二次電池用の黒鉛材料の製造方法 |
| JP6242716B2 (ja) | 2014-03-04 | 2017-12-06 | Jxtgエネルギー株式会社 | リチウムイオン二次電池負極用人造黒鉛材料およびその製造方法 |
| JP7178271B2 (ja) | 2019-01-15 | 2022-11-25 | Eneos株式会社 | 人造黒鉛材料、人造黒鉛材料の製造方法、リチウムイオン二次電池用負極およびリチウムイオン二次電池 |
| JP7178269B2 (ja) * | 2019-01-15 | 2022-11-25 | Eneos株式会社 | 人造黒鉛材料、人造黒鉛材料の製造方法、リチウムイオン二次電池用負極およびリチウムイオン二次電池 |
| JP7178270B2 (ja) * | 2019-01-15 | 2022-11-25 | Eneos株式会社 | 人造黒鉛材料、人造黒鉛材料の製造方法、リチウムイオン二次電池用負極およびリチウムイオン二次電池 |
| CN111211315B (zh) * | 2020-02-26 | 2023-02-28 | 中国科学院山西煤炭化学研究所 | 一种沥青基片层碳材料及其制备方法和应用 |
| US20230216052A1 (en) * | 2020-06-18 | 2023-07-06 | Eneos Corporation | Artificial graphite material for lithium ion secondary battery negative electrode, and production method therefor |
| TWI756928B (zh) * | 2020-11-19 | 2022-03-01 | 台灣中油股份有限公司 | 人工石墨的製備方法 |
| JP7623229B2 (ja) * | 2021-06-18 | 2025-01-28 | Eneos株式会社 | リチウムイオン二次電池負極用人造黒鉛材料の製造方法、リチウムイオン二次電池負極用人造黒鉛材料、リチウムイオン二次電池用負極、及び、リチウムイオン二次電池 |
| CN113644257B (zh) * | 2021-07-27 | 2024-07-16 | 江苏正力新能电池技术有限公司 | 一种负极材料及其制备方法、负极片以及电化学装置 |
| CN118908199B (zh) * | 2024-08-16 | 2025-10-17 | 山东亿维新材料有限责任公司 | 一种长循环石墨负极材料及其制备方法和应用 |
Family Cites Families (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0955209A (ja) * | 1995-08-10 | 1997-02-25 | Fuji Elelctrochem Co Ltd | リチウム二次電池用負極炭素質材料およびその製造方法 |
| EP0917223A4 (en) | 1997-02-04 | 2006-10-04 | Mitsubishi Chem Corp | SECONDARY ACCUMULATOR WITH LITHIUM IONS |
| JPH10284081A (ja) * | 1997-02-04 | 1998-10-23 | Mitsubishi Chem Corp | リチウムイオン二次電池 |
| JP3886285B2 (ja) * | 1999-02-23 | 2007-02-28 | 大阪瓦斯株式会社 | リチウム二次電池用負極材料およびその製造方法、リチウム二次電池用負極並びにリチウム二次電池 |
| JP3654790B2 (ja) * | 1999-04-22 | 2005-06-02 | 三菱化学株式会社 | 電極用黒鉛材料およびそれを用いたリチウムイオン二次電池 |
| JP2000348727A (ja) * | 1999-06-01 | 2000-12-15 | Fuji Elelctrochem Co Ltd | 非水電解液2次電池 |
| JP2001313032A (ja) * | 2000-04-27 | 2001-11-09 | Mitsubishi Chemicals Corp | 非水系二次電池 |
| CA2364651A1 (en) * | 2001-12-07 | 2003-06-07 | Hydro-Quebec | Carbon-carbon composite as negative electrode for li-ion batteries |
| JP4014151B2 (ja) * | 2002-09-30 | 2007-11-28 | 日立マクセル株式会社 | リチウム二次電池 |
| AU2003272947A1 (en) | 2002-10-11 | 2004-05-04 | Fdk Corporation | Nonaqueous electrolyte secondary battery and process for producing positive electrode for use in nonaqueous electrolyte secondary battery |
| JP4215633B2 (ja) * | 2002-12-19 | 2009-01-28 | Jfeケミカル株式会社 | 複合黒鉛粒子の製造方法 |
| WO2004075319A1 (ja) * | 2003-02-20 | 2004-09-02 | Mitsubishi Chemical Corporation | リチウム二次電池用負極活物質、リチウム二次電池負極及びリチウム二次電池 |
| JP4765253B2 (ja) * | 2003-02-20 | 2011-09-07 | 三菱化学株式会社 | リチウム二次電池用負極活物質、リチウム二次電池負極及びリチウム二次電池 |
| JP4729716B2 (ja) | 2003-02-20 | 2011-07-20 | 三菱化学株式会社 | リチウム二次電池負極及びリチウム二次電池 |
| WO2006109497A1 (ja) * | 2005-03-30 | 2006-10-19 | Osaka Gas Co., Ltd. | メソカーボンマイクロビーズの製造方法 |
| JP5916268B2 (ja) * | 2005-12-07 | 2016-05-11 | 三菱化学株式会社 | 二次電池用非水系電解液及びそれを用いた非水系電解液二次電池 |
| KR20090094098A (ko) * | 2006-12-22 | 2009-09-03 | 도요탄소 가부시키가이샤 | 흑연 재료 및 그의 제조 방법 |
| JP5415684B2 (ja) | 2007-10-02 | 2014-02-12 | Jx日鉱日石エネルギー株式会社 | リチウムイオン二次電池負極用人造黒鉛及びその製造方法 |
| JP5270906B2 (ja) * | 2007-11-08 | 2013-08-21 | Jx日鉱日石エネルギー株式会社 | リチウムイオン二次電池負極材料用原料炭組成物及びその製造方法 |
| EP2352192B1 (en) * | 2008-10-31 | 2016-06-01 | Mitsubishi Chemical Corporation | Negative electrode material for nonaqueous secondary battery |
| JP5400064B2 (ja) * | 2008-12-26 | 2014-01-29 | Jx日鉱日石エネルギー株式会社 | リチウムイオン二次電池負極材料用の原料油組成物 |
-
2009
- 2009-09-18 JP JP2009217842A patent/JP5367521B2/ja active Active
-
2010
- 2010-09-16 WO PCT/JP2010/066096 patent/WO2011034152A1/ja not_active Ceased
- 2010-09-16 CN CN201080041837.0A patent/CN102511096B/zh active Active
- 2010-09-16 KR KR1020127008412A patent/KR101820071B1/ko active Active
- 2010-09-16 EP EP10817265.1A patent/EP2479823A4/en not_active Withdrawn
-
2012
- 2012-03-16 US US13/422,513 patent/US8617508B2/en active Active
-
2013
- 2013-11-25 US US14/089,392 patent/US20140079622A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| CN102511096A (zh) | 2012-06-20 |
| KR101820071B1 (ko) | 2018-01-18 |
| US20140079622A1 (en) | 2014-03-20 |
| KR20120081114A (ko) | 2012-07-18 |
| WO2011034152A1 (ja) | 2011-03-24 |
| CN102511096B (zh) | 2014-11-26 |
| JP2011065961A (ja) | 2011-03-31 |
| US8617508B2 (en) | 2013-12-31 |
| EP2479823A1 (en) | 2012-07-25 |
| EP2479823A4 (en) | 2016-03-02 |
| US20120171572A1 (en) | 2012-07-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5367521B2 (ja) | リチウム二次電池の負極用炭素材料及びその製造方法 | |
| JP5612428B2 (ja) | 格子歪を有するリチウムイオン二次電池負極用黒鉛材料及びリチウムイオン二次電池 | |
| CN101842924B (zh) | 锂离子二次电池负极材料用原料炭组合物及其制造方法 | |
| US10035707B2 (en) | Artificial graphite material for negative electrode of lithium ion secondary battery, and method for producing same | |
| JP5931727B2 (ja) | リチウム二次電池負極用黒鉛材料およびその製造方法、およびそれを用いたリチウム二次電池 | |
| JP5728475B2 (ja) | リチウムイオン二次電池負極材料用原料炭組成物 | |
| US20180019472A1 (en) | Method for manufacturing graphite powder for negative-electrode material for lithium-ion secondary battery, negative electrode for lithium-ion secondary battery, and lithium-ion secondary battery | |
| WO2012081439A1 (ja) | リチウムイオン二次電池負極用黒鉛材料およびその製造方法、リチウムイオン二次電池 | |
| JP5615673B2 (ja) | リチウムイオン二次電池負極用非晶質系炭素材料の製造方法及びリチウムイオン二次電池 | |
| JP5657348B2 (ja) | リチウムイオン二次電池負極用炭素材料およびそれを使用した非水系二次電池 | |
| JP5914460B2 (ja) | リチウムイオン二次電池の負極用炭素材料の製造方法 | |
| JP6030958B2 (ja) | リチウムイオン二次電池負極炭素材料用石油生コークスの製造方法及び同炭素材料の製造方法 | |
| JP2020111490A (ja) | 人造黒鉛材料、人造黒鉛材料の製造方法、リチウムイオン二次電池用負極およびリチウムイオン二次電池 | |
| JP2012012488A (ja) | 石油生コークス及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20111116 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130618 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130808 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130903 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130911 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5367521 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |