JP5016926B2 - 架橋したヒアルロナンおよび/またはハイランに由来する粘着性ゲル、その調製および使用法 - Google Patents
架橋したヒアルロナンおよび/またはハイランに由来する粘着性ゲル、その調製および使用法 Download PDFInfo
- Publication number
- JP5016926B2 JP5016926B2 JP2006547530A JP2006547530A JP5016926B2 JP 5016926 B2 JP5016926 B2 JP 5016926B2 JP 2006547530 A JP2006547530 A JP 2006547530A JP 2006547530 A JP2006547530 A JP 2006547530A JP 5016926 B2 JP5016926 B2 JP 5016926B2
- Authority
- JP
- Japan
- Prior art keywords
- gel
- solution
- dvs
- polymer
- ipc
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CO[C@](C(C1O)O)C(C([*+])=O)O[C@]1O[C@@](C(*)[C@](OC)OC1CO)C1O Chemical compound CO[C@](C(C1O)O)C(C([*+])=O)O[C@]1O[C@@](C(*)[C@](OC)OC1CO)C1O 0.000 description 1
Images



Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0024—Solid, semi-solid or solidifying implants, which are implanted or injected in body tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0014—Skin, i.e. galenical aspects of topical compositions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/006—Heteroglycans, i.e. polysaccharides having more than one sugar residue in the main chain in either alternating or less regular sequence; Gellans; Succinoglycans; Arabinogalactans; Tragacanth or gum tragacanth or traganth from Astragalus; Gum Karaya from Sterculia urens; Gum Ghatti from Anogeissus latifolia; Derivatives thereof
- C08B37/0063—Glycosaminoglycans or mucopolysaccharides, e.g. keratan sulfate; Derivatives thereof, e.g. fucoidan
- C08B37/0072—Hyaluronic acid, i.e. HA or hyaluronan; Derivatives thereof, e.g. crosslinked hyaluronic acid (hylan) or hyaluronates
Description
本発明は、天然ポリマーおよび合成ポリマーをジビニルスルホン(DVS)で修飾することによって作られた材料(例えば、ゲル、流体、および固体)の生成、ならびにこれらの材料が有する独特の化学的特性、物理化学的特性、および機械的特性に関する。本発明はまた、多くの応用分野におけるこれらの材料の使用(例えば、医療分野および外科分野における、注射可能な装置および/または移植可能な装置としての使用;高分子薬物および低分子薬物ならびに他の治療剤のための薬物送達システムとしての使用;化粧品用途および局所用途のための使用)に関する。なお、本願は、2003年12月30日に出願された米国特許出願第60/533,429号に対して優先権を主張する。米国特許出願第60/533,429号はその全体が参照により本発明に組み入れられる。
並外れて優れた生体適合性を有するヒドロゲルが開発されている。これらのゲルは、ヒアルロナン(ヒアルロン酸およびその塩)、ならびに/またはハイラン(hylan)およびその塩をベースとしている。これらのゲルはまた、DVSと架橋したヒアルロナンもしくはハイラン(図1および図2,米国特許第4,605,691号および同第6,521,223号も参照)、ならびに/またはヒアルロナンと他のポリマーまたは低分子量物質との架橋混合物(米国特許第4,582,865号)もベースとしている。ハイランAは、少量のアルデヒド(一般的に、ホルムアルデヒド)との共有結合架橋によって化学修飾された水溶性ヒアルロナン調製物であるのに対して、ハイランBは、ハイランAがDVSによってさらに架橋されたものである(米国特許第4,713,448号参照)。ヒアルロナン、化学修飾ヒアルロナン、およびハイランから調製されたゲルスラリーも説明されている(米国特許第5,143,724号)。このようなヒドロゲルは、薬物送達(米国特許第4,636,524)および医療分野での他の目的に使用することができる。しかしながら、これらのゲルおよびゲルスラリーには、弾力性、粘着性、および接着性がない。
本発明は、その1つの局面において、架橋したヒアルロナンおよび/もしくはハイラン、またはヒアルロナンもしくはハイランと、DVSと架橋することができる他の親水性ポリマー(グリコサミノグリカン(例えば、コンドロイチン4-硫酸およびコンドロイチン6-硫酸、キトサン);ポリアニオン多糖(例えば、セルロースのアルキルカルボキシエーテル誘導体およびその塩、アルギン酸およびその塩、ならびにポリヘキスロン酸(例えば、ペクチン、ポリグルクロン酸、およびポリマンヌロン酸);他の非荷電多糖(例えば、デンプン、グルコマンナン、ガラクトマンナン、プルラン、カードラン、イヌリン、ならびにセルロースおよびそのヒドロキシアルキルエーテル誘導体);多糖ゴム(例えば、キサンタンガム、アラビアゴム(Arabic)、アラビアゴム(Acacia)、およびガーゴム(Guar)、ならびにこれらのアルキルカルボキシエーテル誘導体およびヒドロキシアルキルエーテル誘導体);ならびに合成ポリマー(例えば、ポリビニルアルコールおよびポリエチレンイミン)などがあるが、これに限定されない)との混合ゲルからなる粘着性、接着性、および弾力性の高いゲルを提供する。これらのゲルは、選択されたポリマー初濃度(IPC)と特定のDVS濃度またはDVS:ヒアルロナンおよび/もしくはハイラン比、ならびに/あるいは水性酸性媒体において行われる洗浄手順から調製される。本発明の様々な態様において、用語「ポリマー初濃度」(以下「IPC」)は、出発反応条件下での反応物の重量による濃度を意味する。2種類以上の出発ポリマーが反応に用いられる場合、ポリマー初濃度は、出発反応条件下での出発ポリマーの重量による総濃度を意味する。用語「ポリマー終濃度」(以下「FPC」)は、全ての処理が完了した後の修飾ポリマーの重量による濃度を意味する。2種類以上の出発ポリマーが反応に用いられる場合、ポリマー終濃度は、全ての処理が完了した後の修飾ポリマーの重量による総濃度を意味する。
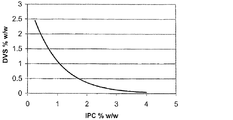
本発明は、IPC、出発試薬の比などのプロセスパラメータを制御しながら、修飾試薬としてDVSを用いて、ヒアルロナンおよび/またはハイランを、単独で、および/または他のポリマー(天然および合成)と混合して修飾し、さらに、処理された材料を洗浄することによって、独特かつ有用な特性を有する材料を形成することができるという発見に基づいている。本発明は、DVSとの架橋反応によって形成された、均一な、単一の、かつ弾力性のある構造を有するゲルを提供する。これらのゲルは、もろいまたは壊れやすいゲルの場合のように小さな粒子に容易に分解されない。これらの生成物から「パテ」様の材料または弾力性のある材料が得られ、細かく切られた後に、粒子は粘着性が高いために合体する傾向がある。これらのタイプの粘着性のある壊れにくいゲルは、架橋反応条件、洗浄、IPC、およびDVS量のある特定の組み合わせを使用することによって得られることが観察されている。これらのゲルの機械的特性はこれらの条件の全てに左右されるが、最も重要なことには、使用されるDVS:ポリマー比(DVS:Pol)および酸洗浄に左右される。DVS:Pol比は、図3に示したようにIPCに反比例する。特定の機序によって限定されることを意図しないが、ポリマー濃度が低いほど、ゲル形成に必要とされるDVSの量は多くなると考えられる。これは、DVS濃度がIPCと正比例する、以前に述べられたゲルとは異なる。
a.0.25重量%〜50重量%のポリマー初濃度(IPC)で、ヒアルロナン、ハイラン、またはその混合物を含む少なくとも1種類の出発ポリマー(Pol)の溶液を作成する工程;
b.少なくとも1種類の出発ポリマーとジビニルスルホン(DVS)を反応させる工程;および
c.工程bにおいて形成されたゲルをpH<4の水溶液で洗浄する工程
を含む、粘着性ゲルを調製する方法を提供する。
a.8重量%より大(例えば、8.1重量%、8.5重量%、9重量%、9.5重量%、10重量%、11重量%、12重量%、13重量%、15重量%、および20重量%)〜25重量%、30重量%、40重量%、または50重量%のポリマー初濃度(IPC)で、ヒアルロナン、ハイラン、またはその混合物を含む少なくとも1種類の出発ポリマー(Pol)の溶液を作成する工程;および
b.出発ポリマーとジビニルスルホン(DVS)を反応させて、ゲルを形成する工程であって、DVS:Pol(w:w)比は、約0.0025〜約0.033、約0.05〜約0.033、または約0.01〜約0.033から選択することができる工程
を含む、粘着性ゲルを調製する方法を提供する。任意に、この方法は、工程bにおいて形成されたゲルをpH<4の水溶液で洗浄する工程をさらに含んでもよい。
a.0.25重量%〜50重量%(例えば、3重量%〜8重量%、0.25重量%〜8重量%、3重量%〜6重量%、3重量%〜10重量%、3重量%〜15重量%、8重量%〜15重量%、10重量%〜20重量%、9重量%〜20重量%、8%〜30%、10重量%〜12重量%、約12重量%、12重量%〜30%、12重量%〜50重量%、および20重量%〜50重量%のポリマー初濃度(IPC)で、ヒアルロナン、ハイラン、またはその混合物を含む少なくとも1種類の出発ポリマー(Pol)の溶液を作成する工程;および
b.約0.025〜0.05、約0.0025〜約0.033、約0.05〜約0.033、または約0.01〜約0.033から選択されるジビニルスルホン:ポリマー比(DVS:Pol)(w:w)で、出発ポリマーとジビニルスルホン(DVS)を反応させて、ゲルを形成する工程
を含む、粘着性ゲルを調製する方法を提供する。任意に、この方法は、工程bにおいて形成されたゲルをpH<4の水溶液で洗浄する工程をさらに含んでもよい。
a.0.25重量%〜0.9重量%(例えば、0.25重量%〜0.5重量%、0.3重量%〜0.8重量%、0.5重量%〜0.8%、約0.5重量%)のポリマー初濃度(IPC)で、ヒアルロナン、ハイラン、またはその混合物を含む少なくとも1種類の出発ポリマー(Pol)の溶液を作成する工程;
b.出発ポリマーとジビニルスルホン(DVS)を反応させる工程
を含む、粘着性ゲルを調製する方法を提供する。ある態様において、DVS:Pol(w:w)比は、約1.4〜約17.7、約2〜15、約5〜約15、約2〜約10から選択される。任意に、この方法は、工程bにおいて形成されたゲルをpH<4の水溶液で洗浄する工程をさらに含んでもよい。
本実施例は、IPC 0.75%およびDVS:Pol比2:1でのゲルの調製を説明する。
本実施例は、IPC 0.5%およびDVS:Pol比4:1でのゲルの調製を説明する。
本実施例は、IPC 0.38%およびDVS:Pol比6:1でのゲルの調製を説明する。
本実施例は、IPC 0.25%およびDVS:Pol比8:1でのゲルの調製を説明する。
本実施例は、IPC 0.15%およびDVS:Pol比17.7:1での材料の調製を説明する。
本実施例は、IPC 4%およびDVS:Pol比1:17でのゲルの調製を説明する。
本実施例は、IPC 4.8%およびDVS:Pol比1:48でのゲルの調製を説明する。
本実施例は、IPC 4.8%およびDVS:Pol比1:96でのゲルの調製を説明する。
本実施例は、IPC 5.6%およびDVS:Pol比1:48でのゲルの調製を説明する。
本実施例は、IPC 5.6%およびDVS:Pol比1:96でのゲルの調製を説明する。
本実施例は、IPC 6%およびDVS:Pol比1:48でのゲルの調製を説明する。
本実施例は、IPC 6%およびDVS:Pol比1:96でのゲルの調製を説明する。
本実施例は、IPC 8%およびDVS:Pol比1:100でのゲルの調製を説明する。
本実施例は、IPC 3%およびDVS:Pol比4:17でのゲルの調製を説明する。
本実施例は、IPC 3%およびDVS:Pol比4:17でのゲルの調製を説明する。
本実施例は、IPC 1%およびDVS:Pol比5:1でのゲルの調製を説明する。
本実施例は、IPC 0.9%およびDVS:Pol比5:1でのゲルの調製を説明する。
本実施例は、IPC 1%およびDVS:Pol比1.4:1でのゲルの調製を説明する。
本実施例は、IPC 0.9%およびDVS:Pol比1.4:1でのゲルの調製を説明する。
本実施例は、IPC 4%およびDVS:Pol比約1:15でのゲルの調製を説明する。
本実施例は、IPC 8%およびDVS:Pol比2:35でのゲルの調製を説明する。溶解するまで攪拌しながら、0.2M NaOH溶液(90.63g)にNaCl(5.85g)を添加した。急速に機械的に攪拌しながら、中MW細菌発酵ヒアルロン酸ナトリウム(8.40g)を添加した。溶解するまで攪拌を120分間続けて、IPC 8.0%のポリマー溶液を得た。IPA(0.610mL)に溶解したDVS(0.390mL)の溶液を、ポリマー溶液に、1分間にわたってピペットで添加した(5×〜0.2mL)。反応混合物を2〜3分間、高速攪拌し、室温で4時間保存して、ゲルを得た。次いで、2M HCl溶液(21.00mL)が添加されている中性食塩水(3.0L)が入っているガラス容器に、ゲルを移し、オービタルシェーカーで室温で約25時間攪拌した。ふるいを用いて洗浄液を流し、ゲルのpHを記録し(2.2)、さらにゲルをオービタルシェーカーで中性食塩水(4.0L)で17時間洗浄した。1M NaOH溶液(15.00mL)を洗浄液に7日間にわたって添加することで、ゲルのpHを5.0〜6.5にゆっくりと調節した。次いで、ゲルを0.01M PBS溶液(2.0L)で約24時間洗浄し、次いで、さらに0.01M PBS溶液4.0Lで7日間洗浄した(この時、ゲルのpHは7.4であった)。ゲルの最終収量は416.3gであり、FPCは1.73%であった。材料の一部の濃度を0.01M PBS溶液で1.5%に調節した。両方の材料の一部を126℃で10分間オートクレーブした。表2に示したレオロジーデータおよび観察から、ゲルは非常に硬く、弾力性および粘着性がないが、相分離することなく、わずかに希釈できることが分かった。
本実施例は、IPC 8%およびDVS:Pol比1:15でのゲルの調製を説明する。
本実施例は、ヒアルロン酸ナトリウムおよびコンドロイチン6硫酸のIPCがそれぞれ0.75%であり、DVS:ヒアルロン酸ナトリウム比が2:1であるゲルをどのように調製できるかを説明する。
本実施例は、ヒアルロン酸ナトリウムおよびポリビニルアルコールのIPCがそれぞれ0.75%であり、DVS:ヒアルロン酸ナトリウム比が2:1であるゲルをどのように調製できるかを説明する。
本実施例は、ヒアルロン酸ナトリウムおよびカルボキシメチルセルロースのIPCがそれぞれ0.75%であり、DVS:ヒアルロン酸ナトリウム比が1.4:1であるゲルをどのように調製できるかを説明する。
本実施例は、IPC 4.0%およびDVS:Pol比1:24でゲルを調製できることを説明する。
無菌処理条件下(層流フード)にある実施例9のゲル(50.00g,0.75%FPC)に、非ステロイド性抗炎症剤(例えば、ジクロフェナク(16.80mL,9mg/mL));局所麻酔薬(例えば、塩酸ブピバカイン(35.00mL,5mg/mL));および抗新生物薬(例えば、メトトレキセート(5.00mL,25mg/mL))または抗不整脈薬(例えば、塩酸プロプラノロール(5.00mL,25mg/mL))の0.01M PBS溶液を添加することができる。薬物を含むゲルを、Turbula T2Fミキサーで23min-1の速度で室温で約24時間混合する。次いで、混合物を凍らし、凍結乾燥させて、乾燥した発泡体様の材料にする。滅菌0.01M PBSまたは滅菌0.15M食塩水(49.63g)を添加し、Turbula T2Fミキサーで約24時間混合することによって、ゲルをFPC 0.75%まで再構成する。所望であれば、最後に、材料をオートクレーブで滅菌してもよい。
双極性非プロトン溶媒(例えば、ジメチルスルホキシド)に溶解した、ステロイド性抗炎症剤(例えば、デキサメタゾン(5.00mL,25mg/mL));および抗新生物薬(例えば、パクリタキセル(25.00mL,5mg/mL))または抗不整脈薬(例えば、アミオダロン(5.00mL,5mg/mL))の溶液を、無菌処理条件下(層流フード)にある実施例9のゲル(50.00g,0.75%FPC)に添加することができる。ゲルおよび薬物溶液をTurbula T2Fミキサーで毎分23min-1の速度で、室温で約24時間混合してもよい。次いで、混合物を滅菌透析チューブに入れ、無菌条件下で2.0L部の中性食塩水で大規模に透析するか、または過剰量のエタノールもしくは薬物が溶けない他の水混和性溶媒から沈殿させ、真空下で乾燥させる。滅菌0.01M PBSまたは滅菌0.15M食塩水(49.63g)を添加し、Turbula T2Fミキサーで約24時間混合することによって、ゲルをFPC 0.75%まで再構成する。所望であれば、最後に、材料をオートクレーブで滅菌してもよい。
ラット盲腸擦過(cecal-abration)モデルを用いて、実施例6および7の癒着軽減能力の評価を行った。この研究は、Guide for the Care and Use of Laboratory Animals, National Academy Press, 1996に記載のNIHガイドラインに従って行った。盲腸の腹面および背面を機械的擦過装置で4回擦過し(これにより、作業者に左右されずに、ある決まった部位を制御して擦過することができる)、盲腸を腹腔内の解剖学的位置に戻した。動物を3つの群(1つは手術対照群、2つは処置群、それぞれ10匹の動物からなる)に割り当てた。2つの処置群の盲腸の両側に、実施例6および7に従って調製された1.5cm3のゲルを投与し、合計3cm3の盲腸表面に均一に塗布した。手術対照群には擦過後、何も処置をしなかった。手術後7日目に、動物の癒着形成を等級で評価した。

*p<0.05 対対照群(カイ二乗分析)
ハートレイ系モルモットの膝において、試験物品(実施例31に従って調製されたゲルを含む)の関節内注射の安全性を評価した。これは、関節内での安全性を評価するのに適したモデルである。この研究は、Guide for the Care and Use of Laboratory Animals, National Academy Press, 1996に記載のNIHガイドラインに従って行った。モルモットを、それぞれ6匹の動物からなる群(1つは対照群)に分けた。群には、後左の大腿膝蓋関節に、週1回の間隔で、3回注射を行った。それぞれの注射について注射前および注射の翌日に、ノギスを用いて、関節(脛骨プラトー)を基準にして両後肢の関節幅を評価した。動物の歩行異常も大まかに評価した。関節軟部組織の炎症を確かめるために組織学的分析も行った。評価した変数(運動の範囲、死体解剖日の膝の幅)について、対照と試験物品との間には違いがなかった。組織学的分析から、ゲルをモルモットの膝に注射したことに関連する炎症性または変形性の有意な変化は認められなかった。
本実施例は、IPC 5.25%およびDVS:Pol比1:96でのゲルの調製を説明する。
本実施例は、滅菌1.0〜1.25%ヒアルロン酸ナトリウム溶液の調製を説明する。
本実施例はハイラスタンSGL-80の調製を説明する。
実施例34および35は、皮膚充填材として使用するのに適したゲルの調製を説明する。一般的に、このようなゲルは、前記の実施例で説明したように、DVSと架橋したヒアルロナン(例えば、細菌発酵HA)から調製される。一般的に、皮膚充填材ゲルは、以下の特徴:(a)IPC 8〜12%(好ましくは、10〜12%);(b)HA MW 500〜2500KDa(好ましくは、500〜600KDa);(c)DVS:Pol比1:200〜1:15(好ましくは、1:100〜1:15、例えば、1:50および1:60);(d)FPC約1%〜2.5%を有する。ゲルは平衡状態まで洗浄されてもよく、その他の状態になるまで洗浄されてもよい。好ましくは、ゲルは酸洗浄されてもよく、または、中性食塩水で洗浄されてもよい。
本実施例は、IPC 約12%およびDVS:Pol比1:50でのゲルの調製を説明する。
様々な超低MW HA(500KDa未満)の溶液を、前記の実施例で述べたように0.2N NaOHに溶解して調製した。粘度は、Bohlin C-VORレオメーターを用いて、1sec-1の剪断速度で測定した。図4に示したように、ある決まった粘度値でのHA MWと1/IPCの関係は正比例関数である。従って、望ましい粘度、弾力性、および軟らかさを得るために、超低MWから調製されるゲルのDVS:Pol比の範囲は、ポリマーのIPCおよびMWに応じて、約0.0025〜約20、例えば、約0.05〜約20、0.01〜約20であることが予想される。
Claims (32)
- 粘着性ゲルを調製する方法であって、以下の工程を含む方法:
a.0.25重量%〜50重量%のポリマー初濃度(IPC)で、ヒアルロナン、ハイラン、またはその混合物を含む少なくとも1種類の出発ポリマー(Pol)の溶液を作成する工程;
b.出発ポリマーとジビニルスルホン(DVS)を反応させて、ゲルを形成する工程;および
c.工程bにおいて形成されたゲルをpH<4の水溶液で洗浄し、ゲルが4以下のpHを示すようにする、工程。 - IPCが0.25重量%〜8重量%である、請求項1記載の方法。
- IPCが3重量%〜10重量%である、請求項1記載の方法。
- IPCが3重量%〜15重量%である、請求項1記載の方法。
- 工程cにおいて、ゲルが洗浄されて、pH 2.0〜3.0になる、請求項1記載の方法。
- ゲルのpHを4以下から6.9〜7.5に調節する工程をさらに含む、請求項1記載の方法。
- ゲルのpHを4以下から4.5〜6.5に調節する工程をさらに含む、請求項1記載の方法。
- 工程bがpH>9で行われる、請求項1記載の方法。
- 工程bが24時間以下続けられる、請求項1記載の方法。
- 出発ポリマーの平均分子量(MW)が0.5MDa〜4MDaから選択される、請求項1記載の方法。
- 出発ポリマーの1つがヒアルロナンである、請求項10記載の方法。
- 出発ポリマーの平均分子量(MW)が30KDa〜500KDaから選択される、請求項1記載の方法。
- ジビニルスルホン:ポリマー比(DVS:Pol)(w:w)が0.0025:1〜17.7:1から選択される、請求項1記載の方法。
- ジビニルスルホン:ポリマー比(DVS:Pol)(w:w)が0.01:1〜17.7:1から選択される、請求項1記載の方法。
- 出発ポリマーの平均分子量(MW)が0.5MDa〜4MDaから選択され、DVS:Pol(w:w)比が0.01:1〜17.7:1から選択される、請求項1記載の方法。
- 粘着性ゲルを調製する方法であって、以下の工程を含む方法:
a.8重量%より大きく50重量%までのポリマー初濃度(IPC)で、ヒアルロナン、ハイラン、またはその混合物を含む少なくとも1種類の出発ポリマー(Pol)の溶液を作成する工程;
b.出発ポリマーとジビニルスルホン(DVS)を反応させて、ゲルを形成する工程;および
c.工程bにおいて形成されたゲルをpH<4の水溶液で洗浄し、ゲルが4以下のpHを示すようにする、工程。 - 工程cにおいて、ゲルが洗浄されて、pHが2.0〜3.0になる、請求項16記載の方法。
- ゲルのpHを4以下から6.9〜7.5に調節する工程をさらに含む、請求項16記載の方法。
- ゲルのpHを4以下から4.5〜6.5に調節する工程をさらに含む、請求項16記載の方法。
- 工程bがpH>9で行われる、請求項16記載の方法。
- 工程bが24時間以下続けられる、請求項16記載の方法。
- 出発ポリマーの平均分子量(MW)が0.5MDa〜4MDaから選択される、請求項16記載の方法。
- 出発ポリマーの1つがヒアルロナンである、請求項22記載の方法。
- 出発ポリマーの平均分子量(MW)が30KDa〜500KDaから選択される、請求項16記載の方法。
- ジビニルスルホン:ポリマー比(DVS:Pol)(w:w)が0.0025:1〜0.033:1から選択される、請求項16記載の方法。
- ジビニルスルホン:ポリマー比(DVS:Pol)(w:w)が0.01:1〜0.033:1から選択される、請求項16記載の方法。
- 粘着性ゲルを調製する方法であって、以下の工程を含む方法:
a.0.25重量%〜50重量%のポリマー初濃度(IPC)で、ヒアルロナン、ハイラン、またはその混合物を含む少なくとも1種類の出発ポリマー(Pol)の溶液を作成する工程;
b. 出発ポリマーとジビニルスルホン(DVS)を、0.0025:1〜0.05:1から選択されるジビニルスルホン:ポリマー比(DVS:Pol)(w:w)で反応させて、ゲルを形成する工程;および
c.工程bにおいて形成されたゲルをpH<4の水溶液で洗浄し、ゲルが4以下のpHを示すようにする、工程。 - 工程bがpH>9で行われる、請求項27記載の方法。
- ジビニルスルホン:ポリマー比(DVS:Pol)(w:w)が0.0025:1〜0.033:1から選択される、請求項27記載の方法。
- 粘着性ゲルを調製する方法であって、以下の工程を含む方法:
a.0.025重量%〜0.9重量%のポリマー初濃度(IPC)で、ヒアルロナン、ハイラン、またはその混合物を含む少なくとも1種類の出発ポリマー(Pol)の溶液を作成する工程;
b.出発ポリマーとジビニルスルホン(DVS)を反応させて、ゲルを形成する工程;および
c.工程bにおいて形成されたゲルをpH<4の水溶液で洗浄し、ゲルが4以下のpHを示すようにする、工程。 - 出発ポリマーの平均分子量(MW)が3MDa〜10MDaから選択される、請求項30記載の方法。
- ジビニルスルホン:ポリマー比(DVS:Pol)(w:w)が1.4:1〜17.7:1から選択される、請求項30記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US53342903P | 2003-12-30 | 2003-12-30 | |
| US60/533,429 | 2003-12-30 | ||
| PCT/US2004/043811 WO2005066215A1 (en) | 2003-12-30 | 2004-12-30 | Cohesive gels form cross-linked hyaluronan and/or hylan, their preparation and use |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011169777A Division JP5680501B2 (ja) | 2003-12-30 | 2011-08-03 | 架橋したヒアルロナンおよび/またはハイランに由来する粘着性ゲル、その調製および使用法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2007519779A JP2007519779A (ja) | 2007-07-19 |
| JP5016926B2 true JP5016926B2 (ja) | 2012-09-05 |
Family
ID=34748900
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006547530A Active JP5016926B2 (ja) | 2003-12-30 | 2004-12-30 | 架橋したヒアルロナンおよび/またはハイランに由来する粘着性ゲル、その調製および使用法 |
| JP2011169777A Active JP5680501B2 (ja) | 2003-12-30 | 2011-08-03 | 架橋したヒアルロナンおよび/またはハイランに由来する粘着性ゲル、その調製および使用法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011169777A Active JP5680501B2 (ja) | 2003-12-30 | 2011-08-03 | 架橋したヒアルロナンおよび/またはハイランに由来する粘着性ゲル、その調製および使用法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20050142152A1 (ja) |
| EP (1) | EP1701981B1 (ja) |
| JP (2) | JP5016926B2 (ja) |
| CN (1) | CN100537606C (ja) |
| AU (1) | AU2004312532B2 (ja) |
| BR (1) | BRPI0418309B8 (ja) |
| CA (1) | CA2550718C (ja) |
| IL (1) | IL176465A (ja) |
| MX (1) | MXPA06007556A (ja) |
| WO (1) | WO2005066215A1 (ja) |
Families Citing this family (81)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7338433B2 (en) | 2002-08-13 | 2008-03-04 | Allergan, Inc. | Remotely adjustable gastric banding method |
| EP2181655B1 (en) | 2002-08-28 | 2016-12-07 | Apollo Endosurgery, Inc. | Fatigue-restistant gastric banding device |
| FR2861734B1 (fr) | 2003-04-10 | 2006-04-14 | Corneal Ind | Reticulation de polysaccharides de faible et forte masse moleculaire; preparation d'hydrogels monophasiques injectables; polysaccharides et hydrogels obtenus |
| EP2399528B1 (en) | 2004-01-23 | 2013-01-09 | Allergan, Inc. | Releasably-securable one-piece adjustable gastric band |
| EP1725193B1 (en) | 2004-03-08 | 2009-09-30 | Allergan Medical S.A. | Closure system for tubular organs |
| ES2368149T3 (es) | 2004-03-18 | 2011-11-14 | Allergan, Inc. | Aparato para el ajuste del volumen de globos intragástricos. |
| DK1817347T3 (en) * | 2004-11-24 | 2017-08-14 | Albumedix As | Process for Crosslinking Hyaluronic Acid with Divinyl Sulfone |
| CN101094680A (zh) * | 2004-12-30 | 2007-12-26 | 建新公司 | 用于关节内粘弹性补充的方案 |
| US8251888B2 (en) | 2005-04-13 | 2012-08-28 | Mitchell Steven Roslin | Artificial gastric valve |
| US20070067045A1 (en) * | 2005-09-19 | 2007-03-22 | Ar2 Partners, Inc. | Systems and methods for skin wrinkle removal |
| US8043206B2 (en) | 2006-01-04 | 2011-10-25 | Allergan, Inc. | Self-regulating gastric band with pressure data processing |
| US7798954B2 (en) | 2006-01-04 | 2010-09-21 | Allergan, Inc. | Hydraulic gastric band with collapsible reservoir |
| US8071757B2 (en) | 2006-03-02 | 2011-12-06 | Novozymes Biopharma Dk A/S | Aryl/alkyl vinyl sulfone hyaluronic acid derivatives |
| FR2909560B1 (fr) | 2006-12-06 | 2012-12-28 | Fabre Pierre Dermo Cosmetique | Gel d'acide hyaluronique pour injection intradermique |
| WO2008147867A2 (en) * | 2007-05-23 | 2008-12-04 | Allergan, Inc. | Cross-linked collagen and uses thereof |
| US8658148B2 (en) | 2007-06-22 | 2014-02-25 | Genzyme Corporation | Chemically modified dendrimers |
| US8318695B2 (en) * | 2007-07-30 | 2012-11-27 | Allergan, Inc. | Tunably crosslinked polysaccharide compositions |
| US8697044B2 (en) | 2007-10-09 | 2014-04-15 | Allergan, Inc. | Crossed-linked hyaluronic acid and collagen and uses thereof |
| WO2009065116A1 (en) | 2007-11-16 | 2009-05-22 | Aspect Pharmaceuticals Llc | Compositions and methods for treating purpura |
| US8394782B2 (en) | 2007-11-30 | 2013-03-12 | Allergan, Inc. | Polysaccharide gel formulation having increased longevity |
| US8394784B2 (en) | 2007-11-30 | 2013-03-12 | Allergan, Inc. | Polysaccharide gel formulation having multi-stage bioactive agent delivery |
| US20090143348A1 (en) * | 2007-11-30 | 2009-06-04 | Ahmet Tezel | Polysaccharide gel compositions and methods for sustained delivery of drugs |
| AU2015252122A1 (en) * | 2008-08-04 | 2015-11-26 | Allergan Industrie Sas | Hyaluronic acid-based gels including anesthetic agents |
| US8357795B2 (en) * | 2008-08-04 | 2013-01-22 | Allergan, Inc. | Hyaluronic acid-based gels including lidocaine |
| ES2829971T3 (es) | 2008-09-02 | 2021-06-02 | Tautona Group Lp | Hilos de ácido hialurónico y/o derivados de los mismos, métodos para fabricar los mismos y usos de los mismos |
| EP2341953B1 (en) | 2008-09-04 | 2018-11-21 | The General Hospital Corporation | Hydrogels for vocal cord and soft tissue augmentation and repair |
| EP2362762A1 (en) | 2008-10-06 | 2011-09-07 | Allergan Medical Sàrl | Mechanical gastric band with cushions |
| US20100305397A1 (en) * | 2008-10-06 | 2010-12-02 | Allergan Medical Sarl | Hydraulic-mechanical gastric band |
| US20100185049A1 (en) | 2008-10-22 | 2010-07-22 | Allergan, Inc. | Dome and screw valves for remotely adjustable gastric banding systems |
| FR2938187B1 (fr) * | 2008-11-07 | 2012-08-17 | Anteis Sa | Composition injectable a base d'acide hyaluronique ou l'un de ses sels, de polyols et de lidocaine, sterilisee a la chaleur |
| US8390326B2 (en) * | 2009-05-05 | 2013-03-05 | William Marsh Rice University | Method for fabrication of a semiconductor element and structure thereof |
| FR2945293B1 (fr) * | 2009-05-11 | 2011-06-17 | Teoxane | Procede de preparation d'un gel reticule. |
| KR101551681B1 (ko) | 2009-07-30 | 2015-09-09 | 카르빌란 테라퓨틱스, 인코포레이티드 | 변형된 히알루론산 폴리머 조성물 및 관련방법 |
| US8273725B2 (en) | 2009-09-10 | 2012-09-25 | Genzyme Corporation | Stable hyaluronan/steroid formulation |
| US20110171311A1 (en) * | 2010-01-13 | 2011-07-14 | Allergan Industrie, Sas | Stable hydrogel compositions including additives |
| US9114188B2 (en) | 2010-01-13 | 2015-08-25 | Allergan, Industrie, S.A.S. | Stable hydrogel compositions including additives |
| US20110171286A1 (en) * | 2010-01-13 | 2011-07-14 | Allergan, Inc. | Hyaluronic acid compositions for dermatological use |
| US20110172180A1 (en) | 2010-01-13 | 2011-07-14 | Allergan Industrie. Sas | Heat stable hyaluronic acid compositions for dermatological use |
| US8758221B2 (en) | 2010-02-24 | 2014-06-24 | Apollo Endosurgery, Inc. | Source reservoir with potential energy for remotely adjustable gastric banding system |
| US8840541B2 (en) * | 2010-02-25 | 2014-09-23 | Apollo Endosurgery, Inc. | Pressure sensing gastric banding system |
| WO2011109730A2 (en) | 2010-03-04 | 2011-09-09 | The General Hospital Corporation | Methods and systems of matching voice deficits with a tunable mucosal implant to restore and enhance individualized human sound and voice production |
| CA2792729C (en) | 2010-03-12 | 2016-06-28 | Allergan Industrie, Sas | Fluid compositions for improving skin conditions |
| EP3078388B1 (en) | 2010-03-22 | 2019-02-20 | Allergan, Inc. | Cross-linked hydrogels for soft tissue augmentation |
| US9028394B2 (en) | 2010-04-29 | 2015-05-12 | Apollo Endosurgery, Inc. | Self-adjusting mechanical gastric band |
| US20110270024A1 (en) | 2010-04-29 | 2011-11-03 | Allergan, Inc. | Self-adjusting gastric band having various compliant components |
| US9044298B2 (en) | 2010-04-29 | 2015-06-02 | Apollo Endosurgery, Inc. | Self-adjusting gastric band |
| US20110270025A1 (en) | 2010-04-30 | 2011-11-03 | Allergan, Inc. | Remotely powered remotely adjustable gastric band system |
| EP2575909B1 (en) * | 2010-06-03 | 2020-01-15 | Technology Innovation Momentum Fund (Israel) Limited Partnership | Malleable hydrogel hybrids made of self-assembled peptides and biocompatible polymers and uses thereof |
| US8517915B2 (en) | 2010-06-10 | 2013-08-27 | Allergan, Inc. | Remotely adjustable gastric banding system |
| US8716204B2 (en) | 2010-07-27 | 2014-05-06 | Zimmer, Inc. | Synthetic synovial fluid compositions and methods for making the same |
| US8697057B2 (en) | 2010-08-19 | 2014-04-15 | Allergan, Inc. | Compositions and soft tissue replacement methods |
| US9005605B2 (en) | 2010-08-19 | 2015-04-14 | Allergan, Inc. | Compositions and soft tissue replacement methods |
| US8889123B2 (en) | 2010-08-19 | 2014-11-18 | Allergan, Inc. | Compositions and soft tissue replacement methods |
| US8883139B2 (en) | 2010-08-19 | 2014-11-11 | Allergan Inc. | Compositions and soft tissue replacement methods |
| US20120059216A1 (en) | 2010-09-07 | 2012-03-08 | Allergan, Inc. | Remotely adjustable gastric banding system |
| NZ708247A (en) | 2010-11-08 | 2017-01-27 | Allergan Ind Sas | Hyaluronic acid based formulations |
| US8961393B2 (en) | 2010-11-15 | 2015-02-24 | Apollo Endosurgery, Inc. | Gastric band devices and drive systems |
| KR102238406B1 (ko) | 2011-06-03 | 2021-04-08 | 알러간 인더스트리 에스에이에스 | 항산화제를 포함하는 피부 충전제 조성물 |
| US20130096081A1 (en) * | 2011-06-03 | 2013-04-18 | Allergan, Inc. | Dermal filler compositions |
| US9408797B2 (en) | 2011-06-03 | 2016-08-09 | Allergan, Inc. | Dermal filler compositions for fine line treatment |
| US9393263B2 (en) | 2011-06-03 | 2016-07-19 | Allergan, Inc. | Dermal filler compositions including antioxidants |
| RU2613887C2 (ru) | 2011-08-10 | 2017-03-21 | Гликорес 2000 С.Р.Л. | Устойчивый к расщеплению сшитый низкомолекулярный гиалуронат |
| US20130244943A1 (en) | 2011-09-06 | 2013-09-19 | Allergan, Inc. | Hyaluronic acid-collagen matrices for dermal filling and volumizing applications |
| US9662422B2 (en) | 2011-09-06 | 2017-05-30 | Allergan, Inc. | Crosslinked hyaluronic acid-collagen gels for improving tissue graft viability and soft tissue augmentation |
| KR20220013588A (ko) * | 2011-09-14 | 2022-02-04 | 알러간, 인코포레이티드 | 잔주름 치료를 위한 진피 필러 조성물 |
| CN102558600A (zh) * | 2011-12-01 | 2012-07-11 | 上海白衣缘生物工程有限公司 | 交联透明质酸海绵及其制备方法 |
| US8876694B2 (en) | 2011-12-07 | 2014-11-04 | Apollo Endosurgery, Inc. | Tube connector with a guiding tip |
| US8961394B2 (en) | 2011-12-20 | 2015-02-24 | Apollo Endosurgery, Inc. | Self-sealing fluid joint for use with a gastric band |
| CA2876070C (en) * | 2012-06-15 | 2021-01-26 | Merz Pharma Gmbh & Co. Kgaa | Method of preparing a composition based on hyaluronic acid |
| US9579388B2 (en) | 2012-11-29 | 2017-02-28 | Rene Gauthier | System and method for alleviating the appearance of scars and/or scar tissue |
| US9421198B2 (en) | 2013-07-30 | 2016-08-23 | Teoxane | Composition comprising hyaluronic acid and mepivacaine |
| SG11201609593RA (en) * | 2014-05-16 | 2016-12-29 | Ultrast Inc | Phase-shifting formulations |
| US10722444B2 (en) | 2014-09-30 | 2020-07-28 | Allergan Industrie, Sas | Stable hydrogel compositions including additives |
| WO2016128783A1 (en) | 2015-02-09 | 2016-08-18 | Allergan Industrie Sas | Compositions and methods for improving skin appearance |
| AR105319A1 (es) | 2015-06-05 | 2017-09-27 | Sanofi Sa | Profármacos que comprenden un conjugado agonista dual de glp-1 / glucagón conector ácido hialurónico |
| WO2017119198A1 (ja) * | 2016-01-05 | 2017-07-13 | デンカ株式会社 | 半月板変性治療用組成物 |
| AR110299A1 (es) * | 2016-12-02 | 2019-03-13 | Sanofi Sa | Conjugados que comprenden un agonista dual de glp-1 / glucagón, un conector y ácido hialurónico |
| CN108250457A (zh) * | 2017-05-08 | 2018-07-06 | 上海利康瑞生物工程有限公司 | 一种剪切粘度可控的双相交联透明质酸钠凝胶及其制备方法和制剂 |
| US20190240335A1 (en) * | 2018-02-07 | 2019-08-08 | Promedon S.A. | Biocompatible Hydrogel Compositions |
| WO2022051060A1 (en) * | 2020-09-01 | 2022-03-10 | Orthogenrx, Inc. | Crosslinking of non-animal-derived hyaluronic acid with divinyl sulfone |
| WO2023135135A1 (en) | 2022-01-11 | 2023-07-20 | Gpq S.R.L. | New hyaluronic acid derivatives as innovative fillers |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE442820B (sv) * | 1984-06-08 | 1986-02-03 | Pharmacia Ab | Gel av tverbunden hyaluronsyra for anvendning som glaskroppssubstitut |
| US4582865A (en) * | 1984-12-06 | 1986-04-15 | Biomatrix, Inc. | Cross-linked gels of hyaluronic acid and products containing such gels |
| US4713448A (en) * | 1985-03-12 | 1987-12-15 | Biomatrix, Inc. | Chemically modified hyaluronic acid preparation and method of recovery thereof from animal tissues |
| EP0224987B1 (en) * | 1985-11-29 | 1992-04-15 | Biomatrix, Inc. | Drug delivery systems based on hyaluronan, derivatives thereof and their salts and method of producing same |
| US4795741A (en) * | 1987-05-06 | 1989-01-03 | Biomatrix, Inc. | Compositions for therapeutic percutaneous embolization and the use thereof |
| AU619760B2 (en) * | 1987-12-10 | 1992-02-06 | Genzyme Biosurgery Corporation | Hylan preparation and method of recovery thereof from animal tissues |
| SE8900422D0 (sv) * | 1989-02-08 | 1989-02-08 | Pharmacia Ab | Tvaerbundna hyaluronatgeler samt foerfarande foer framstaellning av dessa |
| US5143724A (en) * | 1990-07-09 | 1992-09-01 | Biomatrix, Inc. | Biocompatible viscoelastic gel slurries, their preparation and use |
| US5246698A (en) * | 1990-07-09 | 1993-09-21 | Biomatrix, Inc. | Biocompatible viscoelastic gel slurries, their preparation and use |
| US6635267B1 (en) * | 1998-11-10 | 2003-10-21 | Denki Kagaku Kogyo Kabushiki Kaisha | Hyaluronic acid gel, process for the preparation thereof and medical materials containing the same |
| US6521223B1 (en) * | 2000-02-14 | 2003-02-18 | Genzyme Corporation | Single phase gels for the prevention of adhesions |
| EP1265630B1 (en) * | 2000-03-24 | 2006-06-07 | Genentech, Inc. | Use of insulin for the treatment of cartilagenous disorders |
-
2004
- 2004-12-30 BR BRPI0418309A patent/BRPI0418309B8/pt active IP Right Grant
- 2004-12-30 MX MXPA06007556A patent/MXPA06007556A/es active IP Right Grant
- 2004-12-30 WO PCT/US2004/043811 patent/WO2005066215A1/en active Application Filing
- 2004-12-30 EP EP04815811.7A patent/EP1701981B1/en active Active
- 2004-12-30 AU AU2004312532A patent/AU2004312532B2/en active Active
- 2004-12-30 CA CA2550718A patent/CA2550718C/en active Active
- 2004-12-30 CN CNB2004800391832A patent/CN100537606C/zh active Active
- 2004-12-30 US US11/026,388 patent/US20050142152A1/en not_active Abandoned
- 2004-12-30 JP JP2006547530A patent/JP5016926B2/ja active Active
-
2006
- 2006-06-21 IL IL176465A patent/IL176465A/en active IP Right Grant
-
2011
- 2011-08-03 JP JP2011169777A patent/JP5680501B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CA2550718A1 (en) | 2005-07-21 |
| EP1701981B1 (en) | 2017-06-07 |
| BRPI0418309B1 (pt) | 2016-08-02 |
| MXPA06007556A (es) | 2006-08-31 |
| WO2005066215A1 (en) | 2005-07-21 |
| BRPI0418309B8 (pt) | 2021-05-25 |
| JP2012025955A (ja) | 2012-02-09 |
| AU2004312532A1 (en) | 2005-07-21 |
| AU2004312532B2 (en) | 2010-05-20 |
| BRPI0418309A (pt) | 2007-05-02 |
| CA2550718C (en) | 2013-11-05 |
| US20050142152A1 (en) | 2005-06-30 |
| JP5680501B2 (ja) | 2015-03-04 |
| CN100537606C (zh) | 2009-09-09 |
| CN1902232A (zh) | 2007-01-24 |
| EP1701981A1 (en) | 2006-09-20 |
| IL176465A (en) | 2013-07-31 |
| IL176465A0 (en) | 2006-10-05 |
| JP2007519779A (ja) | 2007-07-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5016926B2 (ja) | 架橋したヒアルロナンおよび/またはハイランに由来する粘着性ゲル、その調製および使用法 | |
| US8524213B2 (en) | Polymeric materials, their preparation and use | |
| JP6063867B2 (ja) | ジスルフィド結合架橋生体適合性高分子ヒドロゲルおよびこれを含む製剤 | |
| EP1753787B1 (en) | Method of covalently linking hyaluronan and chitosan | |
| US8222399B2 (en) | Photoreactive polysaccharide, photocrosslinked polysaccharide product and method of making same and medical materials made from the crosslinked polysaccharide | |
| CN107281549B (zh) | 与多糖交联的蛋白质的制备和/或制剂 | |
| ES2248817T3 (es) | Procedimiento de preparacion de particulas reticuladas de polimeros hidrosolubles, las particulas obtenidas y su utilizacion. | |
| PT2572702E (pt) | Hidrogel coesivo monofásico biodegradável | |
| US20150080333A1 (en) | Hyaluronic acid particles and their use in biomedical applications | |
| CN117511002A (zh) | 一种改性透明质酸水凝胶及其制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070913 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110207 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20110506 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20110513 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110803 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120523 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120611 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150615 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5016926 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |