JP4965745B2 - 含窒素脂環式骨格を有する一本鎖核酸分子 - Google Patents
含窒素脂環式骨格を有する一本鎖核酸分子 Download PDFInfo
- Publication number
- JP4965745B2 JP4965745B2 JP2011537085A JP2011537085A JP4965745B2 JP 4965745 B2 JP4965745 B2 JP 4965745B2 JP 2011537085 A JP2011537085 A JP 2011537085A JP 2011537085 A JP2011537085 A JP 2011537085A JP 4965745 B2 JP4965745 B2 JP 4965745B2
- Authority
- JP
- Japan
- Prior art keywords
- region
- bases
- nucleic acid
- acid molecule
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images



















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/08—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by hetero atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/11—Antisense
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/14—Type of nucleic acid interfering N.A.
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/35—Nature of the modification
- C12N2310/351—Conjugate
- C12N2310/3519—Fusion with another nucleic acid
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/50—Physical structure
- C12N2310/53—Physical structure partially self-complementary or closed
- C12N2310/531—Stem-loop; Hairpin
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2320/00—Applications; Uses
- C12N2320/50—Methods for regulating/modulating their activity
- C12N2320/51—Methods for regulating/modulating their activity modulating the chemical stability, e.g. nuclease-resistance
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2330/00—Production
- C12N2330/30—Production chemically synthesised
Description
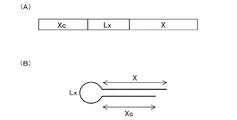
本発明の一本鎖核酸分子は、前述のように、標的遺伝子の発現を抑制する発現抑制配列を含む一本鎖核酸分子であって、領域(X)、リンカー領域(Lx)および領域(Xc)を含み、前記領域(X)と前記領域(Xc)との間に、前記リンカー領域(Lx)が連結され、前記領域(X)および前記領域(Xc)の少なくとも一方が、前記発現抑制配列を含み、前記リンカー領域(Lx)が、ピロリジン骨格およびピペリジン骨格の少なくとも一方を含む非ヌクレオチド構造を有することを特徴とする。
5’−GUUGUCAUACUUCUCAUGG−3’ (配列番号4)
5’−AAAGUCAAUGUACAGCUGCUU−3’ (配列番号18)
5’−AUUGUAACGAGACAAACAC−3’ (配列番号6)
5’−UUGCGCUUUUUGGUGACGC−3’ (配列番号30)
X1およびX2は、それぞれ独立して、H2、O、SまたはNHであり;
Y1およびY2は、それぞれ独立して、単結合、CH2、NH、OまたはSであり;
R3は、環A上のC−3、C−4、C−5またはC−6に結合する水素原子または置換基であり、
L1は、n個の原子からなるアルキレン鎖であり、ここで、アルキレン炭素原子上の水素原子は、OH、ORa、NH2、NHRa、NRaRb、SH、もしくはSRaで置換されても置換されていなくてもよく、または、
L1は、前記アルキレン鎖の一つ以上の炭素原子が、酸素原子で置換されたポリエーテル鎖であり、
ただし、Y1が、NH、OまたはSの場合、Y1に結合するL1の原子は炭素であり、OR1に結合するL1の原子は炭素であり、酸素原子同士は隣接せず;
L2は、m個の原子からなるアルキレン鎖であり、ここで、アルキレン炭素原子上の水素原子は、OH、ORc、NH2、NHRc、NRcRd、SHもしくはSRcで置換されても置換れていなくてもよく、または、
L2は、前記アルキレン鎖の一つ以上の炭素原子が、酸素原子で置換されたポリエーテル鎖であり、
ただし、Y2が、NH、OまたはSの場合、Y2に結合するL2の原子は炭素であり、OR2に結合するL2の原子は炭素であり、酸素原子同士は隣接せず;
Ra、Rb、RcおよびRdは、それぞれ独立して、置換基または保護基であり;
lは、1または2であり;
mは、0〜30の範囲の整数であり;
nは、0〜30の範囲の整数であり;
環Aは、前記環A上のC−2以外の1個の炭素原子が、窒素、酸素、硫黄で置換されてもよく、
前記環A内に、炭素−炭素二重結合または炭素−窒素二重結合を含んでもよく、
前記領域(Yc)および前記領域(Y)は、それぞれ、−OR1−または−OR2−を介して、前記リンカー領域(Ly)に結合し、
ここで、R1およびR2は、存在しても存在しなくてもよく、存在する場合、R1およびR2は、それぞれ独立して、ヌクレオチド残基または前記構造(I)である。
条件(1)
前記領域(Xc)は、−OR2−を介して、前記領域(X)は、−OR1−を介して、前記式(I)の構造と結合する。
条件(2)
前記領域(Xc)は、−OR1−を介して、前記領域(X)は、−OR2−を介して、前記式(I)の構造と結合する。
前記第1のssPN分子は、例えば、前記領域(X)、前記領域(Xc)および前記リンカー領域(Lx)からなる分子である。
X>Xc ・・・(3)
X−Xc=1〜10、好ましくは1、2または3、
より好ましくは1または2 ・・・(11)
X=Xc ・・・(5)
前記第2のssPN分子は、例えば、前記領域(X)、前記リンカー領域(Lx)および前記領域(Xc)の他に、さらに、領域(Y)および前記領域(Y)に相補的な領域(Yc)を有する分子である。前記第2のssPN分子において、前記領域(X)と前記領域(Y)とが連結して、内部領域(Z)を形成している。なお、特に示さない限り、前記第2のssPN分子は、前記第1のssPN分子の記載を援用できる。
条件(1)
前記領域(Xc)は、−OR2−を介して、前記領域(X)は、−OR1−を介して、前記式(I)の構造と結合し、
前記領域(Yc)は、−OR1−を介して、前記領域(Y)は、−OR2−を介して、前記式(I)の構造と結合する。
条件(2)
前記領域(Xc)は、−OR2−を介して、前記領域(X)は、−OR1−を介して、前記式(I)の構造と結合し、
前記領域(Yc)は、−OR2−を介して、前記領域(Y)は、−OR1−を介して、前記式(I)の構造と結合する。
条件(3)
前記領域(Xc)は、−OR1−を介して、前記領域(X)は、−OR2−を介して、前記式(I)の構造と結合し、
前記領域(Yc)は、−OR1−を介して、前記領域(Y)は、−OR2−を介して、前記式(I)の構造と結合する。
条件(4)
前記領域(Xc)は、−OR1−を介して、前記領域(X)は、−OR2−を介して、前記式(I)の構造と結合し、
前記領域(Yc)は、−OR2−を介して、前記領域(Y)は、−OR1−を介して、前記式(I)の構造と結合する。
Z=X+Y ・・・(1)
Z≧Xc+Yc ・・・(2)
X=Y ・・・(19)
X<Y ・・・(20)
X>Y ・・・(21)
(a)下記式(3)および(4)の条件を満たす。
X>Xc ・・・(3)
Y=Yc ・・・(4)
(b)下記式(5)および(6)の条件を満たす。
X=Xc ・・・(5)
Y>Yc ・・・(6)
(c)下記式(7)および(8)の条件を満たす。
X>Xc ・・・(7)
Y>Yc ・・・(8)
(d)下記式(9)および(10)の条件を満たす。
X=Xc ・・・(9)
Y=Yc ・・・(10)
(a)下記式(11)および(12)の条件を満たす。
X−Xc=1〜10、好ましくは1、2、3または4、
より好ましくは1、2または3 ・・・(11)
Y−Yc=0 ・・・(12)
(b)下記式(13)および(14)の条件を満たす。
X−Xc=0 ・・・(13)
Y−Yc=1〜10、好ましくは1、2、3または4、
より好ましくは1、2または3 ・・・(14)
(c)下記式(15)および(16)の条件を満たす。
X−Xc=1〜10、好ましくは、1、2または3、
より好ましくは1または2 ・・・(15)
Y−Yc=1〜10、好ましくは、1、2または3、
より好ましくは1または2 ・・・(16)
(d)下記式(17)および(18)の条件を満たす。
X−Xc=0 ・・・(17)
Y−Yc=0 ・・・(18)
(1)非修飾ヌクレオチド残基
(2)修飾ヌクレオチド残基
(3)非修飾ヌクレオチド残基および修飾ヌクレオチド残基
(4)非ヌクレオチド残基
(5)非ヌクレオチド残基および非修飾ヌクレオチド残基
(6)非ヌクレオチド残基および修飾ヌクレオチド残基
(7)非ヌクレオチド残基、非修飾ヌクレオチド残基および修飾ヌクレオチド残基
(1)非修飾ヌクレオチド残基
(2)修飾ヌクレオチド残基
(3)非修飾ヌクレオチド残基および修飾ヌクレオチド残基
(4)非ヌクレオチド残基
(5)非ヌクレオチド残基および非修飾ヌクレオチド残基
(6)非ヌクレオチド残基および修飾ヌクレオチド残基
(7)非ヌクレオチド残基、非修飾ヌクレオチド残基および修飾ヌクレオチド残基
前記ヌクレオチド残基は、例えば、構成要素として、糖、塩基およびリン酸を含む。前記ヌクレオチド残基は、前述のように、例えば、リボヌクレオチド残基およびデオキシリボヌクレオチド残基等があげられる。前記リボヌクレオチド残基は、例えば、糖としてリボース残基を有し、塩基として、アデニン(A)、グアニン(G)、シトシン(C)およびU(ウラシル)を有し、前記デオキシリボース残基は、例えば、糖としてデオキシリボース残基を有し、塩基として、アデニン(A)、グアニン(G)、シトシン(C)およびチミン(T)を有る。
本発明のssPN分子の合成方法は、特に制限されず、従来公知の方法が採用できる。前記合成方法は、例えば、遺伝子工学的手法による合成法、化学合成法等があげられる。遺伝子工学的手法は、例えば、インビトロ転写合成法、ベクターを用いる方法、PCRカセットによる方法等があげられる。前記ベクターは、特に制限されず、プラスミド等の非ウイルスベクター、ウイルスベクター等があげられる。これに限定されない。前記化学合成法は、特に制限されず、例えば、ホスホロアミダイト法およびH−ホスホネート法等があげられる。前記化学合成法は、例えば、市販の自動核酸合成機を使用可能である。前記化学合成法は、一般に、アミダイトが使用される。前記アミダイトは、特に制限されず、市販のアミダイトとして、例えば、RNA Phosphoramidites(2’−O−TBDMSi、商品名、三千里製薬)、ACEアミダイト、TOMアミダイト、CEEアミダイト、CEMアミダイト、TEMアミダイト等があげられる。本発明のssPN分子については、例えば、前記式(I)で表わされるリンカー領域の合成の際、後述する本発明のモノマーを使用することが好ましい。
本発明の発現抑制用組成物は、前述のように、標的遺伝子の発現を抑制するための組成物であり、前記本発明のssPN分子を含むことを特徴とする。本発明の組成物は、前記本発明のssPN分子を含むことが特徴であり、その他の構成は、何ら制限されない。本発明の発現抑制用組成物は、例えば、発現抑制用試薬ということもできる。
本発明の発現抑制方法は、前述のように、標的遺伝子の発現を抑制する方法であって、前記本発明のssPN分子を使用することを特徴とする。本発明の発現抑制方法は、前記本発明のssPN分子を使用することが特徴であって、その他の工程および条件は、何ら制限されない。
本発明の疾患の治療方法は、前述のように、前記本発明のssPN分子を、患者に投与する工程を含み、前記ssPN分子が、前記発現抑制配列として、前記疾患の原因となる遺伝子の発現を抑制する配列を有することを特徴とする。本発明の治療方法は、前記本発明のssPN分子を使用することが特徴であって、その他の工程および条件は、何ら制限されない。本発明の治療方法は、例えば、前記本発明の発現抑制方法を援用できる。
本発明の使用は、前記標的遺伝子の発現抑制のための、前記本発明のssPN分子の使用である。また、本発明の使用は、RNA干渉の誘導のための、前記本発明のssPN分子の使用である。
本発明のモノマーは、核酸合成用のモノマーであって、下記式(II)の構造を有することを特徴とする。本発明のモノマーは、特に示さない限り、前記本発明のssPN分子の説明を援用できる。
X1およびX2は、それぞれ独立して、H2、O、SまたはNHであり;
Y1およびY2は、それぞれ独立して、単結合、CH2、NH、OまたはSであり;
R1およびR2は、それぞれ独立して、H、保護基またはリン酸保護基であり;
R3は、環A上のC−3、C−4、C−5またはC−6に結合する水素原子または置換基であり;
L1は、n個の原子からなるアルキレン鎖であり、ここで、アルキレン炭素原子上の水素原子は、OH、ORa、NH2、NHRa、NRaRb、SHもしくはSRaで置換されても置換されていなくてもよく、または、
L1は、前記アルキレン鎖の一つ以上の炭素原子が、酸素原子で置換されたポリエーテル鎖であり、
ただし、Y1が、NH、OまたはSの場合、Y1に結合するL1の原子は炭素であり、OR1に結合するL1の原子は炭素であり、酸素原子同士は隣接せず;
L2は、m個の原子からなるアルキレン鎖であり、ここで、アルキレン炭素原子上の水素原子は、OH、ORc、NH2、NHRc、NRcRd、SHもしくはSRcで置換されても置換されていなくてもよく、または、
L2は、前記アルキレン鎖の一つ以上の炭素原子が、酸素原子で置換されたポリエーテル鎖であり、
ただし、Y2が、NH、OまたはSの場合、Y2に結合するL2の原子は炭素であり、OR2に結合するL2の原子は炭素であり、酸素原子同士は隣接せず;
Ra、Rb、RcおよびRdは、それぞれ独立して、置換基または保護基であり;
lは、1または2であり;
mは、0〜30の範囲の整数であり;
nは、0〜30の範囲の整数であり;
環Aは、前記環A上のC−2以外の1個の炭素原子が、窒素、酸素または硫黄で置換されてもよく、
前記環A内に、炭素−炭素二重結合または炭素−窒素二重結合を含んでもよい。
−P(OR6)(NR7R8)
前記式において、R6は、水素原子または任意の置換基である。置換基R6は、例えば、炭化水素基が好ましく、前記炭化水素基は、電子吸引基で置換されていてもよいし、置換されていなくてもよい。置換基R6は、例えば、ハロゲン、ハロアルキル、ヘテロアリール、ヒドロキシアルキル、アルコキシアルキル、アミノアルキル、シリル、シリルオキシアルキル、ヘテロシクリルアルケニル、ヘテロシクリルアルキル、ヘテロアリールアルキル、および、アルキル、アルケニル、アルキニル、アリール、アリールアルキル、シクロアルキル、シクロアルケニル、シクロアルキルアルキル、シクリルアルキル等の炭化水素等があげられ、さらに、電子吸引基で置換されていてもよいし、置換されていなくてもよい。置換基R6は、具体的には、例えば、β−シアノエチル基、ニトロフェニルエチル基、メチル基等があげられる。
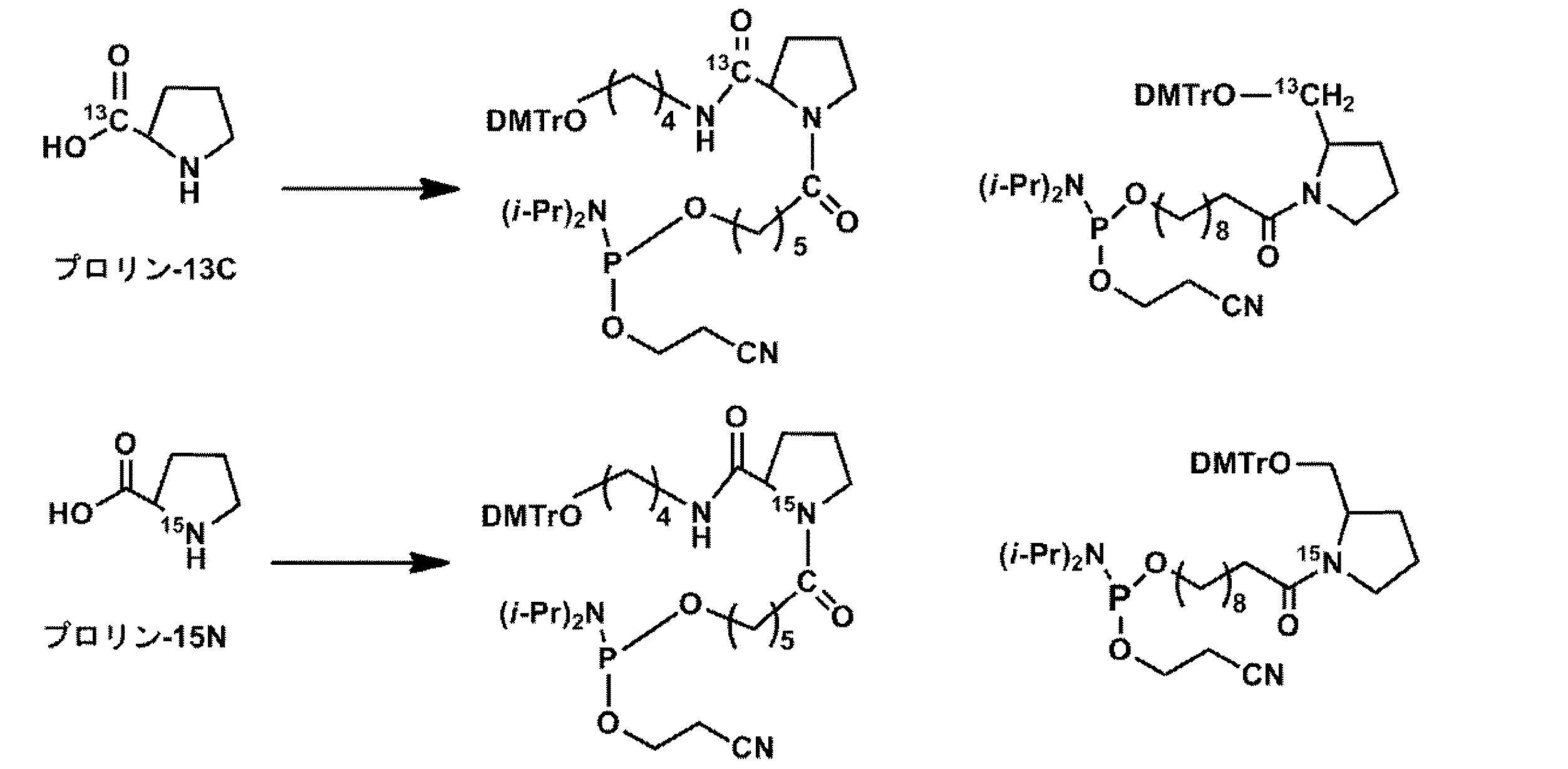
1.プロリノールの合成
下記式に示すスキーム1に従い、ジメトキシトリチル基で保護されたプロリノールを合成した。
L−プロリノール(化合物1)(0.61g、6.0mmol)を、純水70mLに溶解し、L−プロリノール水溶液を調製した。N−(9−フルオレニルメトキシカルボニロキシ)スクシンイミド(Fmoc−OSu)(2.0g、6.0mmol)を、THF10mLに溶解した。このTHF溶液を、前記L−プロリノール水溶液に加え、1時間撹拌して、両者を反応させた。この反応液を、液体画分と沈殿画分とに分離し、それぞれの画分を酢酸エチルで抽出し、それぞれ有機層を回収した。そして、それぞれの有機層を合わせた後、無水硫酸ナトリウムを添加して、水分を吸収させた(以下、乾燥という)。前記有機層をろ過して、ろ液を回収し、前記ろ液を減圧濃縮した。得られた残渣を、シリカゲルカラムクロマトグラフィー(展開溶媒 ヘキサン:酢酸エチル=1:1)により精製し、化合物2を得た(1.4g、収率74%)。以下に、化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.77(2H,d,J=7.7Hz,Ar-H),7.60(2H,d,J=7.3Hz,Ar−H),7.40(2H,t,J=7.5Hz,Ar−H),7.31(2H,t,J=7.6Hz,Ar−H),4.40−4.50(2H,m,COOCH2),4.22(1H,t,J=6.5Hz,Ar−CH),3.20−3.80(5H,m,H−5,H−6),1.75(3H,m,H−3,H−4),1.40(1H,m,H−3).
前記Fmoc−L−プロリノール(化合物2)(1.4g、4.3mmol)を、ピリジン20mLに溶解して、3回共沸した。得られた残留物を、ピリジン20mLに溶解した。この溶液を、アルゴン下、氷浴中で、撹拌しながら、4,4’−ジメトキシトリチルクロリド(DMTr−Cl)(1.8g、5.3mmol)を添加した。この反応液について、クロロホルム/メタノールのTLCにより反応を追跡し、Fmoc−L−プロリノールのスポットが消えるまで、4時間反応させた。そして、過剰のDMTr−Clをクエンチするために、前記反応液に、メタノール3mLを加えて10分撹拌した。前記反応液に、さらに、クロロホルムを加えた後、有機層を回収した。回収した前記有機層に、飽和食塩水による洗浄、5%炭酸水素ナトリウム水溶液による洗浄を行い、もう一度、飽和食塩水による洗浄を行った。洗浄後の有機層を、無水硫酸ナトリウムで乾燥させた。前記有機層をろ過して、得られたろ液を減圧濃縮した。得られた残渣を、シリカゲルカラムクロマトグラフィー(展開溶媒 クロロホルム、1%ピリジン)により精製し、化合物3を得た(2.0g、収率74%)。以下に、化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.77(2H,d,J=7.7Hz,Ar−H),7.60(2H,d,J=7.3Hz,Ar−H),7.40−7.18(13H,m,Ar−H),6.89(4H,d,J=8.6Hz,Ar−H),4.20−4.40(2H,m,COOCH2),4.02(1H,t,J=6.5Hz,Ar−CH),3.80−3.10(5H,m,H−5,H−6),3.73(s,6H,OCH3),1.84(3H,m,H−3,H−4),1.58(1H,m,H−3).
前記Fmoc−DMTr−L−プロリノール(化合物3)(2.0g、3.2mmol)を、20%ピペリジンを含むDMF溶液25mLに溶解し、12時間撹拌した。この溶液を減圧濃縮し、得られた残渣を、シリカゲルカラムクロマトグラフィー(クロロホルム:メタノール=85:15、1%ピリジン含有)で精製し、化合物4を得た(1.0g、収率78%)。以下に、化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H),6.82(4H,d,J=8.6Hz,Ar−H),3.78(6H,s,OCH3),3.31(1H,m,H−6),3.07(2H,m,H−2,H−6),2.90(2H,m,H−5),1.84(3H,m,H−3,H−4),1.40(1H,m,H−3).
つぎに、下記式に示すスキーム2に従い、プロリノールを有するアミダイト誘導体を合成した。以下、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩を、「EDC」、N,N−ジメチルアミノピリジン(4−ジメチルアミノピリジン)を「DMAP」という。
前記DMTr−L−プロリノール(化合物4)(0.80g、2.0mmol)、EDC(0.46g、2.4mmol)およびDMAP(0.29g、2.4mmol)を、ジクロロメタン20mLに溶解して撹拌した。この溶液に、10−ヒドロキシデカン酸(0.45g、2.4mmol)を添加し、撹拌した。この反応液について、酢酸エチルのTLCにより反応を追跡し、DMTr−L−プロリノールのスポットが消えるまで、20時間反応させた。そして、前記反応液に、ジクロロメタンを加えた後、有機層を回収した。回収した前記有機層を、飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥させた。前記有機層をろ過して、得られたろ液を減圧濃縮し、その残渣を、シリカゲルカラムクロマトグラフィー(酢酸エチル、1%ピリジン含有)により精製し、化合物5を得た(0.71g,収率62%)。以下に、化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H), 6.82(4H,d,J=8.6Hz,Ar−H),3.78(6H,s,OCH3),3.68−2.93(7H,m,H−2,H−5,H−6),2.27−1.72(6H,m,アルキル,H−3,H−4),1.58(4H,s,アルキル),1.30(10H,s,アルキル).
前記DMTr−L−プロリノール(化合物4)(0.80g、2.0mmol)を、メタノール15mLに溶解し、5−ヒドロキシペンタナール(0.31g、3.0mmol)を加えて撹拌した。この溶液に、シアノ水素化ホウ素ナトリウム(0.25g、4.0mmol)を加え、さらに撹拌した。この反応液について、酢酸エチル/ヘキサンのTLCにより反応を追跡し、DMTr−L−プロリノールのスポットが消えるまで、24時間反応させた。そして、前記反応液に、酢酸エチルを加え、有機層を回収した。回収した前記有機層を、飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥させた。前記有機層をろ過して、得られたろ液を減圧濃縮し、その残渣を、シリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1、1%ピリジン含有)により精製し、化合物6を得た(0.62g、収率63%)。以下に、化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H),6.82(4H,d,J=8.6Hz,Ar−H),3.78(6H,s,OCH3),3.70−2.86(4H,m,CH2OH,H−6),2.06−1.79(5H,m,アルキル,H−2,H−5),1.74−1.49(6H,m,アルキル,H−3,H−4),1.45−1.27(4H,m,アルキル).
1,4−ブタンジオール(0.90g、10mmol)を、ジクロロメタン30mLに溶解し、さらに、カルボニルジイミダゾール(1.4g、8.6mmol)を加え、3時間撹拌した。この反応液の有機層を、飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥させた。前記有機層をろ過して、得られたろ液を減圧濃縮し、その残渣を、シリカゲルカラムクロマトグラフィー(クロロホルム:メタノール=9:1)により精製した。これによって、1,4−ブタンジオールの一方の末端がカルボニルジイミダゾールで活性化された化合物を得た(0.25g,1.5mmol)。この化合物をジクロロメタン15mLに溶解し、前記DMTr−L−プロリノール(化合物4)(0.6g、1.5mmol)を添加し、24時間撹拌した。この混合液に、さらに、酢酸エチルを加え、有機層を回収した。回収した前記有機層を、飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥させた。前記有機層をろ過して、得られたろ液を減圧濃縮し、その残渣を、シリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1、1%ピリジン含有)により精製し、化合物7を得た(0.61g、収率77%)。以下に、化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H),6.82(4H,d,J=8.6Hz,Ar−H),4.24−3.94(2H,m,COOCH2),3.78(s,6H,OCH3),3.72−2.96(7H,m,アルキル,H−2,H−5,H−6),2.10−1.30(8H,m,アルキル,H−3,H−4).
前記DMTr−L−プロリノール(化合物4)(0.50g、1.2mmol)およびトリホスゲン(0.12g、0.40mmol)を、ジクロロメタン8mLに溶解し、アルゴン下、氷浴中で、撹拌した。そして、前記溶液に、N,N−ジイソプロピルエチルアミン(0.31g、2.4mmol)を添加し、1時間撹拌した。さらに、前記溶液に、8−アミノ−1−オクタノール(0.17g、1.2mmol)を添加し、同様にして氷浴中で30分撹拌した後、室温で20時間撹拌した。前記溶液に、ジクロロメタンを加え、有機層を回収した。回収した前記有機層を、飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥させた。前記有機層をろ過して、得られたろ液を減圧濃縮し、その残渣を、シリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=4:1、1%トリエチルアミン含有)により精製し、化合物8を得た(0.44g、収率62%)。以下に、化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H),6.82(4H,m,Ar−H),3.78(s,6H,OCH3),3.68−3.25(9H,m,CH2NH,CH2OH,H−2,H−5,H−6),1.74−1.18(16H,m,アルキル,H−3,H−4).
前記修飾プロリノール(化合物5〜8)を、それぞれ原料として、以下に示す方法により、化合物9〜12を合成した。前記修飾プロリノールおよび5−ベンジルチオ−1H−テトラゾールを、アセトニトリル3mLに溶解した。前記修飾プロリノールの使用量は、化合物5の場合、0.69g(1.2mmol)、化合物6の場合、0.60g(1.2mmol)、化合物7の場合、0.60g(1.2mmol)、化合物8の場合、0.25g(0.43mmol)とした。また、5−ベンジルチオ−1H−テトラゾールの使用量は、化合物5〜7に対しては、0.15g(0.78mmol)、化合物8に対しては、54mg(0.15mmol)とした。前記溶液に、アルゴン下、2−シアノエチルN,N,N’,N’−テトライソプロピルホスホロジアミダイトを添加し、2時間撹拌した。前記2−シアノエチルN,N,N’,N’−テトライソプロピルホスホロジアミダイトの添加量は、前記化合物5〜7を使用した系では、0.54g(1.8mmol)とし、前記化合物8を使用した系では、0.19g(0.64mmol)とした。そして、前記溶液に、飽和炭酸水素ナトリウム水溶液を添加し、さらに、ジクロロメタンで抽出し、有機層を回収した。回収した前記有機層を、無水硫酸ナトリウムで乾燥させた。前記有機層をろ過して、得られたろ液を減圧濃縮し、その残渣を、シリカゲルカラムクロマトグラフィー(ヘキサン:酢酸エチル=1:1、1%トリエチルアミン含有)により精製し、化合物9〜12を得た。以下に、各化合物のNMRの結果を示す。
1H−NMR(CDCl3)δ7.40−7.14(9H,m,Ar−H),6.82(4H,d,J=8.6Hz,Ar−H),3.78(6H,s,OCH3),3.68−2.93(11H,m,CH2O,POCH2,CHCH3,H−2,H−5,H−6),2.58(2H,m,CH2CN),2.27−1.72(6H,m,アルキル,H−3,H−4),1.58(4H,s,アルキル),1.30(22H,s,アルキル,CHCH3).
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H),6.82(4H,d,J=8.6Hz,Ar−H),3.78(6H,s,OCH3),3.70−2.86(8H,m,CH2O,POCH2,CHCH3,H−6),2.58(2H,m,CH2CN),2.06−1.79(5H,m,アルキル,H−2,H−5),1.74−1.49(6H,m,アルキル,H−3,H−4),1.37−1.10(16H,m,アルキル,CHCH3).
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H),6.82(4H,d,J=8.6Hz,Ar−H),4.24−3.94(2H,m,COOCH2),3.78(s,6H,OCH3),3.72−2.96(11H,m,CH2O,POCH2,CHCH3,H−2,H−5,H−6),2.58(2H,m,CH2CN),2.10−1.46(8H,m,アルキル,H−3,H−4),1.34−1.10(12H,m,CHCH3).
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H),6.82(4H,m,Ar−H),3.78(s,6H,OCH3),3.65−3.25(13H,m,CH2O,POCH2,CHCH3,H−2,CH2NH,CH2OH,H−2,H−5,H−6),2.73(2H,m,CH2CN),2.10−1.48(16H,m,アルキル,H−3,H−4),1.35−1.10(12H,m,CHCH3).
つぎに、下記式に示すスキーム3に従い、L−プロリンを有するアミダイト誘導体を合成した。
DMTr−アミド−L−プロリン(化合物6)(1.00g、2.05mmol)および5−ヒドロキシペンタナール(0.33g、3.07mmol)を含むエタノール溶液(7mL)に、氷冷下、酢酸緩衝液(7mL)を加えた。この混合液を、氷冷下、20分撹拌した後、シアノ水素化ホウ素ナトリウム(0.77g、12.28mmol)を加え、さらに、室温下、7時間撹拌した。前記混合液をジクロロメタンで希釈し、水で洗浄した後、さらに飽和食塩水で洗浄した。そして、前記有機層を回収し、硫酸ナトリウムで乾燥させた。前記有機層をろ過し、ろ液について、減圧下で溶媒を留去した。得られた残渣を、シリカゲルカラムクロマトグラフィー(展開溶媒 CH2Cl2:CH3OH=98:2、0.05%ピリジン含有)に供した。ついで、得られた生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒 CH2Cl2:CH3OH=98:2、0.05%ピリジン含有)に供し、さらに、得られた生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒 ジクロロメタン:アセトン=7:3、0.05%ピリジン含有)に供した。これによって、無色シロップ状の化合物11を得た(0.49g、収率41%)。
Ms(FAB+): m/z 575(M+)、303(DMTr+)
得られた前記DMTr−ヒドロキシアミドアミノ−L−プロリン(化合物11)(0.50g、0.87mmol)を無水アセトニトリルと混合し、室温で共沸乾燥した。得られた残留物に、ジイソプロピルアンモニウムテトラゾリド(178mg、1.04mmol)を加え、減圧下で脱気し、アルゴンガスを充填した。前記混合物に対し、無水アセトニトリル(1mL)を加え、さらに、2−シアノエトキシ−N,N,N’,N’−テトライソプロピルホスホロジアミダイト(313mg、1.04mmol)の無水アセトニトリル溶液(1mL)を加えた。この混合物を、アルゴン雰囲気下、室温で4時間撹拌した。そして、前記混合物をジクロロメタンで希釈し、飽和重曹水および飽和食塩水で、順次洗浄した。有機層を回収し、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られた前記ろ液について、減圧下で溶媒を留去した。得られた残渣を、充填剤としてアミノシリカを用いたカラムクロマトグラフィー(展開溶媒 ヘキサン:アセトン=7:3、0.05%ピリジン含有)に供し、無色シロップ状の化合物12(0.57g、純度93%、収率79%)を得た。前記純度は、HPLCにより測定した(以下、同様)。以下に、化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.41−7.43(m,2H,Ar−H)、7.28−7.32(m,4H,Ar−H)、7.25−7.27(m,2H,Ar−H)、7.18−7.21(m,1H,Ar−H)、6.80−6.84(m,4H,Ar−H)、3.73−3.84(m,1H)、3.79(s,6H,OCH3)、3.47−3.64(m,3H)、3.12−3.26(m,2H)、3.05(t,J=6.4Hz,2H,CH2)、2.98−2.02(m,2H)、2.61(t,J=5.8Hz,2H,CH2)、2.55−2.63(m,2H)、2.27−2.42(m,1H,CH)、2.31(t,7.8Hz,2H,CH2)、2.03−2.19(m,1H,CH)、1.40−1.90(m,8H)、1.23−1.33(m,5H)、1.14−1.20(m,12H,CH3);
P−NMR(CDCl3): δ146.91;
Ms(FAB+): m/z 774(M+)、303(DMTr+),201(C8H19N2OP+).
DMTr−アミド−L−プロリン(化合物6)(1.00g、2.05mmol)を溶解した無水アセトニトリル溶液(10mL)に、1−イミダゾリルカルボニルオキシ−8−ヒドロキシオクタン(1.12g,4.92mmol)を溶解した無水アセトニトリル溶液(20mL)を、アルゴン雰囲気下、室温で加えた。この混合液を、40〜50℃で2日間加熱した後、5日間室温で放置した。前記混合液について、減圧下で溶媒を留去した。得られた残渣を、シリカゲルカラムクロマトグラフィー(展開溶媒 ジクロロメタン:アセトン=4:1、0.05%ピリジン含有)に供した。これによって、無色シロップ状の化合物13を得た(0.68g、収率50%)。以下に、化合物のNMRの結果を示す。
1H−NMR (CDCl3): δ7.40−7.42(m,2H,Ar−H)、7.27−7.31 (m,6H,Ar−H)、7.17−7.21(m,1H,Ar−H)、6.79−6.82(m,4H, Ar−H)、4.23−4.30(m,1H)、4.05−4.10(m,2H)、3.79(s,6H,OCH3)、3.60−3.65(m,2H)、3.32−3.55(m,2H)、3.16−3.29(m,2H),3.01−3.07(m,2H)、2.38−2.40(m,1H,CH)、1.83−1.90(m,2H)、1.57−1.69(m,8H)、1.26−1.36(m,2H);
Ms(FAB+): m/z 602(M+)、303(DMTr+).
得られた前記DMTr−ヒドロキシアミドカルバモイル−L−プロリン(化合物13)(0.63g、1.00mmol)を無水ピリジンと混合し、室温で共沸乾燥した。得られた残留物に、ジイソプロピルアンモニウムテトラゾリド(206mg、1.20mmol)を加え、減圧下に脱気し、アルゴンガスを充填した。前記混合物に対し、無水アセトニトリル(1mL)を加え、さらに、2−シアノエトキシ−N,N,N’,N’−テトライソプロピルホスホロジアミダイト(282mg、1.12mmol)の無水アセトニトリル溶液(1mL)を加えた。この混合物を、アルゴン雰囲気下、室温で4時間撹拌した。そして、前記混合物をジクロロメタンで希釈し、飽和重曹水および飽和食塩水で順次洗浄した。有機層を回収し、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒を留去した。得られた残渣を、充填剤としてアミノシリカを用いたカラムクロマト(展開溶媒 ヘキサン:アセトン=7:3、0.5%ピリジン含有)に供し、無色シロップ状の化合物14(0.74g、純度100%、収率87%)を得た。以下に、化合物のNMRの結果を示す。
P−NMR(CDCl3): δ147.19;
Ms(FAB+): m/z 860 (M+)、303(DMTr+),201(C8H19N2OP+).
トリホスゲン(1.22g、4.10mmol)に、アルゴン雰囲気および氷冷下、無水テトラヒドロフラン溶液(10mL)を加えた。この混合液に、アルゴン雰囲気および氷冷下、DMTr−アミド−L−プロリン(化合物6)(1.00g、2.05mmol)およびDIEA(9.80g、75.8mmol)を溶解した無水テトラヒドロフラン溶液(10mL)を、30分間で滴下し、その後、室温で1時間撹拌した。前記混合液に、アルゴン雰囲気および氷冷下、10−アミノ−1−t−ブチルジメチルシロキシデカン(2.66g、10.25mmol)およびDIEA(3.20g、24.76mmol)を溶解した無水テトラヒドロフラン溶液(20mL)を、45分間で滴下した。そして、前記混合物を、アルゴン雰囲気下、室温で一晩撹拌した。この混合液を酢酸エチル(200mL)で希釈し、有機層を回収した。前記有機層を、飽和重曹水で洗浄した後、さらに、飽和食塩水で洗浄した。そして、有機層を回収し、硫酸ナトリウムで乾燥させた。前記有機層をろ過し、ろ液について、減圧下で溶媒を留去した。得られた残渣を、シリカゲルカラムクロマトグラフィー(展開溶媒 ジクロロメタン:アセトン=4:1、0.05%ピリジン含有)に供した。これによって、無色シロップ状の化合物15を得た(0.87g、収率55%)。
得られた前記DMTr−t−ブチルジメチルシロキシアミドウレイド−L−プロリン(15)(0.87g、1.12mmol)に、アルゴン雰囲気下、無水テトラヒドロフランジクロロメタン溶液(10mL)を室温で加えた。前記混合液に、アルゴン雰囲気下、1mol/Lテトラブチルアンモニウムフルオリド含有テトラヒドロフラン溶液(4.69mL、東京化成)を加え、室温で3日間撹拌した。前記混合液をジクロロメタン(150mL)で希釈し、水で洗浄した後、さらに飽和食塩水で洗浄した。有機層を回収し、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒を留去した。得られた残渣を、シリカゲルカラムクロマトグラフィー(展開溶媒 ジクロロメタン:アセトン=1:1、0.05%ピリジン含有)に供し、無色シロップ状の化合物16を得た(0.68g、収率92%)。以下に、化合物のNMRの結果を示す。
1H−NMR (CDCl3): δ7.41−7.43 (m,2H,Ar−H)、7.27−7.31(m,4H,Ar−H)、7.19−7.26(m,2H,Ar−H)、7.19−7.21(m,1H,Ar−H)、6.80−6.83(m,4H,Ar−H)、4.34(t,2H,CH2)、3.79(s,6H,OCH3)、3.63(d,1H,J=6.4 Hz,CH2)、3.61(d,1H,J=6.4Hz,CH2)、3.34−3.37(m,1H,CH)、3.16−3.27(m,5H),3.04(t,J=5.9Hz,2H,CH2)、2.38−2.45(m,1H,CH)、1.83−2.05(m,3H)、1.45−1.64(m,8H)、1.25−1.38(m,7H).
得られた前記DMTr−ヒドロキシアミドウレイド−L−プロリン(化合物16)(0.62g、0.94mmol)を無水アセトニトリルと混合し、室温で共沸乾燥した。得られた残留物に、ジイソプロピルアンモニウムテトラゾリド(192mg、1.12mmol)を加え、減圧下で脱気し、アルゴンガスを充填した。前記混合液に対し、無水アセトニトリル(1mL)を加え、さらに、2−シアノエトキシ−N,N,N’,N’−テトライソプロピルホスホロジアミダイト(282mg、1.12mmol)の無水アセトニトリル溶液(1mL)を加えた。この混合物を、アルゴン雰囲気下、室温で4時間撹拌した。そして、前記混合物をジクロロメタンで希釈し、飽和重曹水および飽和食塩水で、順次洗浄した。有機層を回収し、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られた前記ろ液について、減圧下で溶媒を留去した。得られた残渣を、充填剤としてアミノシリカを用いたカラムクロマト(展開溶媒 ヘキサン:アセトン=1:1、0.05%ピリジン含有)に供し、無色シロップ状の化合物17を得た(0.77g、純度88%、収率84%)。以下に、化合物のNMRの結果を示す。
P−NMR (CDCl3): δ147.27;
Ms(FAB+): m/z 860(M++1)、303(DMTr+),201(C8H19N2OP+).
プロリン骨格を有するリンカーを含む本発明の核酸分子を生成するため、前記スキーム3により、L−プロリンジアミドアミダイトおよびD−プロリンジアミドアミダイトを合成した。
(1)Fmoc−ヒドロキシアミド−L−プロリン(化合物4)
前記スキーム3の化合物2(Fmoc−L−プロリン)を開始原料とした。前記化合物2(10.00g、29.64mmol)、4−アミノ−1−ブタノール(3.18g、35.56mmol)および1−ヒドロキシベンゾトリアゾール(10.90g、70.72mmol)を混合し、前記混合物に対し、減圧下で脱気し、アルゴンガスを充填した。前記混合物に、無水アセトニトリル(140mL)を室温で加え、さらに、ジシクロヘキシルカルボジイミド(7.34g、35.56mmol)の無水アセトニトリル溶液(70mL)を添加した後、アルゴン雰囲気下、室温で15時間撹拌した。反応終了後、生成した沈殿をろ別し、回収したろ液について、減圧下で溶媒を留去した。得られた残渣にジクロロメタン(200mL)を加え、飽和重曹水(200mL)で洗浄した。そして、有機層を回収し、硫酸マグネシウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒を留去し、その残渣にジエチルエーテル(200mL)を加え、粉末化した。生じた粉末を濾取することにより、無色粉末状の化合物4(10.34g、収率84%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.76−7.83(m,2H,Ar−H)、7.50−7.63(m,2H,Ar−H)、7.38−7.43 (m,2H,Ar−H)、7.28−7.33(m,2H,Ar−H),4.40−4.46(m,1H,CH),4.15−4.31(m,2H,CH2),3.67−3.73(m,2H,CH2)、3.35−3.52(m,2H,CH2)、3.18−3.30(m,2H,CH2)、2.20−2.50(m,4H)、1.81−2.03(m,3H)、1.47−1.54(m,2H);
Ms(FAB+): m/z409(M+H+).
Fmoc−ヒドロキシアミド−L−プロリン(化合物4)(7.80g、19.09mmol)を無水ピリジン(5mL)と混合し、室温で2回共沸乾燥した。得られた残留物に、4,4’−ジメトキシトリチルクロリド(8.20g、24.20mmol)、DMAP(23mg、0.19mmol)および無水ピリジン(39mL)を加えた。この混合物を、室温で1時間撹拌した後、メタノール(7.8mL)を加え、室温で30分撹拌した。この混合物を、ジクロロメタン(100mL)で希釈し、飽和重曹水(150mL)で洗浄後、有機層を分離した。前記有機層を、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒を留去した。得られた未精製の残渣に、無水ジメチルホルムアミド(39mL)およびピペリジン(18.7mL、189mmol)を加え、室温で1時間撹拌した。反応終了後、前記混合液について、減圧下、室温で、溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(商品名Wakogel C−300、展開溶媒 CH2Cl2:CH3OH=9:1、0.05%ピリジン含有に供し、淡黄色油状の化合物6(9.11g、収率98%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR (CDCl3): δ7.39−7.43(m,2H,Ar−H)、7.30(d,J=8.8Hz,4H,Ar−H)、7,21(tt,1H,4.9,1.3Hz,Ar−H)、6.81(d,J=8.8Hz,4H,Ar−H)、3.78(s,6H,OCH3)、3.71(dd,H,J=6.3Hz,5.4Hz,CH)、3.21(2H,12.9,6.3Hz,2H,CH2)、3.05(t,J=6.3Hz,2H,CH2)、2.85−2.91(m,2H,CH2)、2.08−2.17(m,1H,CH)、1.85−2.00(m,3H)、1.55−1.65(m,5H);
Ms(FAB+); m/z 489(M+H+)、303(DMTr+).
得られた前記DMTr−アミド−L−プロリン(化合物6)(6.01g、12.28mmol)、EDC(2.83g、14.74mmol)、1−ヒドロキシベンゾトリアゾール(3.98g、29.47mmol)およびトリエチルアミン(4.47g、44.21mmol)の無水ジクロロメタン溶液(120mL)を混合した。この混合液に、さらに、アルゴン雰囲気下、室温で、6−ヒドロキシヘキサン酸(1.95g、14.47mmol)を加え、その後、アルゴン雰囲気下、室温で、1時間撹拌した。前記混合液をジクロロメタン(600mL)で希釈し、飽和食塩水(800mL)で3回洗浄した。有機層を回収し、前記有機層を、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒を留去した。これにより、淡黄色泡状の前記化合物8(6.29g、収率85%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR (CDCl3): δ7.41−7.43(m,2H,Ar−H)、7.27−7.31(m,4H,Ar−H)、7.19−7.26(m,2H,Ar−H)、7.17−7.21(m,1H,Ar−H)、6.79−6.82(m,4H,Ar−H)、4.51−4.53(m,1H,CH)、3.79(s,6H,OCH3)、3.61(t,2H,J=6.4Hz,CH2)、3.50−3.55(m,1H,CH)、3.36−3.43(m,1H,CH),3.15−3.24(m,2H,CH2),3.04(t,J=6.3Hz,2H,CH2)、2.38−2.45(m,1H,CH)、2.31(t,6.8Hz,2H,CH2)、2.05−2.20(m,1H,CH)、1.92−2.00(m,1H,CH)、1.75−1.83(m,1H,CH)、1.48−1.71(m,8H)、1.35−1.44(m,2H,CH2);
Ms(FAB+): m/z 602(M+)、303(DMTr+).
得られた前記DMTr−ヒドロキシジアミド−L−プロリン(化合物8)(8.55g、14.18mmol)を無水アセトニトリルと混合し、室温で3回共沸乾燥した。得られた残留物に、ジイソプロピルアンモニウムテトラゾリド(2.91g、17.02mmol)を加え、減圧下で脱気し、アルゴンガスを充填した。前記混合物に対し、無水アセトニトリル(10mL)を加え、さらに、2−シアノエトキシ−N,N,N’,N’−テトライソプロピルホスホロジアミダイト(5.13g、17.02mmol)の無水アセトニトリル溶液(7mL)を加えた。この混合物を、アルゴン雰囲気下、室温で2時間撹拌した。そして、前記混合物をジクロロメタンで希釈し、飽和重曹水(200mL)で3回洗浄した後、飽和食塩水(200mL)で洗浄した。有機層を回収し、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られた前記ろ液について、減圧下に溶媒を留去した。得られた残渣を、充填剤としてアミノシリカゲルを用いたカラムクロマトグラフィー(展開溶媒 ヘキサン:酢酸エチル=1:3、0.05%ピリジン含有)に供し、無色シロップ状の化合物10(10.25g、純度92%、収率83%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.40−7.42(m,2H,Ar−H)、7.29−7.31(m,4H,Ar−H)、7.25−7.27(m,2H, Ar−H)、7.17−7.21(m,1H,Ar−H)、6.80−6.82(m,4H,Ar−H)、4.51−4.53(m,1H,CH)、3.75−3.93(m,4H)、3.79(s,6H,OCH3)、3.45−3.60(m,4H)、3.35−3.45(m,1H,CH)、3.20−3.29(m,1H)、3.04(t,J=6.4Hz,2H,CH2)、2.62(t,J=5.8Hz,2H,CH2)、2.40−2.44(m,1H,CH)、2.31(t,7.8Hz,2H,CH2)、2.03−2.19(m,1H,CH)、1.92−2.02(m,1H,CH)、1.70−1.83(m,1H,CH)、1.51−1.71(m,8H)、1.35−1.44(m,2H,CH2)、1.18(d,J=6.8Hz,6H,CH3)、1.16(d,J=6.8Hz,6H,CH3);
P−NMR(CDCl3): Msδ147.17;
Ms(FAB+): m/z 802(M+)、303(DMTr+),201(C8H19N2OP+).
(1)Fmoc−ヒドロキシアミド−D−プロリン(化合物3)
前記スキーム3の化合物1(Fmoc−D−プロリン)を開始原料とした。前記化合物1(1.5g、4.45mmol)、ジシクロヘキシルカルボジイミド(1.1g、5.34mmol)および1−ヒドロキシベンゾトリアゾール(1.5g、10.69mmol)の混合物に対し、減圧下で脱気し、アルゴンガスを充填した。前記混合物に、無水アセトニトリル(24mL)を室温で加え、さらに、4−アミノ−1−ブタノール(0.48g、5.34mmol)の無水アセトニトリル溶液(6mL)添加した後、アルゴン雰囲気下、室温で15時間撹拌した。反応終了後、生成した沈殿をろ別し、回収したろ液について、減圧下で溶媒を留去した。得られた残渣にジクロロメタンを加え、酢酸緩衝液(pH4.0)で3回、飽和重曹水で3回洗浄した。そして、有機層を回収し、硫酸マグネシウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒留去し、その残渣にジエチルエーテル(50mL)を加え、粉末化した。生じた粉末を濾取することにより、白色粉末状の化合物3を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR (400MHz,CDCl3): δ7.77(d,J=7.3Hz,2H);7.58(br,2H);7.41(t,J=7.3Hz,2H);7.32(t,J=7.3Hz,2H);4.25−4.43(m,4H);3.25−3.61(m,6H);1.57−1.92(m,8H).
MS(FAB+): m/z 409(M+H+).
Fmoc−ヒドロキシアミド−D−プロリン(化合物3)(1.0g、2.45mmol)を無水ピリジン(5mL)と混合し、室温で2回共沸乾燥した。得られた残留物に、4,4’−ジメトキシトリチルクロリド(1.05g、3.10mmol)、DMAP(3mg、0.024mmol)および無水ピリジン(5mL)を加えた。この混合物を、室温で1時間撹拌した後、メタノール(1mL)を加え、室温で30分撹拌した。この混合物を、ジクロロメタンで希釈し、飽和重曹水で洗浄後、有機層を分離した。前記有機層を、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒を留去した。得られた未精製の残渣に、無水ジメチルホルムアミド(5mL)およびピペリジン(2.4mL、24mmol)を加え、室温で1時間撹拌した。反応終了後、前記混合液について、減圧下、室温で、溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(商品名Wakogel C−300、展開溶媒 CH2Cl2:CH3OH=9:1、0.05%ピリジン含有)に供し、淡黄色油状の化合物5(1.26g、収率96%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(400MHz, CDCl3): δ7.62(br,1H);7.41−7.44(m,2H);7.26−7.33(m,6H);7.17−7.22(m,1H);6.80−6.84(m,4H);3.78(s,6H);3.71(dd,J=8.8,5.4Hz,1H); 3.22(q,6.5Hz,2H);3.07(t,J=6.1Hz,2H);2.97−3.03(m,1H);2.85−2.91(m,1H);1.85−2.15(m,3H);1.55−1.73(m,6H);
MS (FAB+): m/z 489(M+H+),303(DMTr+).
得られた前記DMTr−アミド−D−プロリン(化合物5)(1.2g、2.45mmol)、EDC(566mg、2.95mmol)、1−ヒドロキシベンゾトリアゾール(796mg、5.89mmol)、およびトリエチルアミン(1.2mL、8.84mmol)の無水ジクロロメタン溶液(24mL)を混合した。この混合液に、さらに、アルゴン雰囲気下、室温で、6−ヒドロキシヘキサン酸(390mg、2.95mmol)を加え、その後、アルゴン雰囲気下、室温で1時間撹拌した。前記混合液をジクロロメタンで希釈し、飽和重曹水で3回洗浄した。有機層を回収し、前記有機層を、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒を留去した。これにより、淡黄色油状の化合物7(1.4g、収率95%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(400MHz,CDCl3): δ7.40−7.43(m,2H);7.25−7.32 (m,6H);7.17−7.22(m,1H);6.79−6.83(m,4H);3.79(s,6H);3.58−3.63(m,2H);3.49−3.55(m,1H);3.15−3.26(m,2H);3.02−3.07(m,2H);2.30−2.33(m,2H);2.11−2.20(m,1H);1.50−1.99(m,13H);1.36−1.43(m,2H);
MS (FAB+): m/z 602(M+),303(DMTr+).
得られた前記DMTr−ヒドロキシジアミド−D−プロリン(化合物7)(1.2g、1.99mmol)を無水アセトニトリルと混合し、室温で3回共沸乾燥した。得られた残留物に、ジイソプロピルアンモニウムテトラゾリド(410mg、2.40mmol)を加え、減圧下で脱気し、アルゴンガスを充填した。前記混合物に対し、無水アセトニトリル(2.4mL)を加え、さらに、2−シアノエトキシ−N,N,N’,N’−テトライソプロピルホスホロジアミダイト(722mg、2.40mmol)を加えた。この混合物を、アルゴン雰囲気下、室温で2時間撹拌した。そして、前記混合物をジクロロメタンで希釈し、飽和重曹水で3回洗浄後、飽和食塩水で洗浄した。有機層を回収し、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒を留去した。得られた残渣を、充填剤としてアミノシリカゲルを用いたカラムクロマトグラフィー(展開溶媒 ヘキサン:酢酸エチル=1:3)に供し、無色油状の化合物9(1.4g、純度95%、収率83%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(400MHz,CDCl3): δ7.40−7.43(m,2H);7.25−7.32(m,6H);7.14−7.21(m,1H);6.80−6.83(m,4H);3.80−3.85(m,2H);3.79(s,6H);3.49−3.65(m,5H);3.02−3.06(m,2H);2.60−2.63(m,2H);2.29−2.33 (m, 2H);1.77−1.82(m,2H);1.56−1.68(m,8H);1.38−1.43(m,2H);1.15−1.29(m,18H);
31P−NMR(162MHz,CDCl3): δ146.94;
MS(FAB+): m/z 802(M+), 303(DMTr+),201(C8H19N2OP+).
プロリン骨格を有するリンカーを含む本発明の核酸分子を生成するため、下記スキーム4により、L−プロリンジアミドアミダイトタイプBを合成した。
Fmoc−ヒドロキシアミド−L−プロリン(化合物4)(2.00g、30mmol)、t−ブチル−ジメチルシリルクロリド(1.11g、35mmol)およびイミダゾール(10.90g、71mmol)を混合した。前記混合物に対し、減圧下に脱気し、アルゴンガスを充填した。前記混合物に、無水アセトニトリル(20mL)を室温で加え、アルゴン雰囲気下、室温で終夜撹拌した。反応終了後、前記混合物にジクロロメタン(150mL)を加え、水で3回洗浄し、飽和食塩水で洗浄した。有機層を回収し、硫酸マグネシウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒を留去し、その残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 CH2Cl2:CH3OH=95:5)に供し、無色シロップ状の化合物18(2.35g、収率92%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.76−7.78(m,2H,Ar−H)、7.50−7.63(m,2H,Ar−H)、7.38−7.42(m,2H,Ar−H)、7.29−7.34(m,2H,Ar−H),4.10−4.46(m,4H,CH2),3.47−3.59(m,4H,CH2)、3.20−3.26(m,2H,CH)、1.85−1.95(m,2H)、1.42−1.55(m,6H)、0.96(s,9H,t−Bu)、0.02(s,6H,SiCH3);
Ms(FAB+): m/z 523(M+H+).
得られた前記Fmoc−t−ブチル−ジメチルシロキシアミド−L−プロリン(化合物18)(1.18g、2.5mmol)に対し、無水アセトニトリル(5mL)およびピペリジン(2.4mL)を加え、室温で1時間撹拌した。反応終了後、前記混合物にアセトニトリル(50mL)を加え、不溶物をろ別した。得られたろ液について、減圧下で溶媒を留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 CH2Cl2:CH3OH=9:1)に供し、無色シロップ状の化合物19(0.61g、収率90%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ3.71(dd,1H,J=9.0Hz,5.2Hz,CH)、3.61−3.64(m,2H,CH2)、3.22−3.28(m,2H,CH2)、2.98−3.04(m,1H,CH)、2.86−2.91(m,1H,CH)、2.08−2.17(m,1H,CH)、1.86−1.93(m,1H,CH)、1.66−1.75(m,2H,CH2)、1.52−1.57(m,4H)、0.89(s,9H,t−Bu)、0.05(s,6H,SiCH3);
Ms(FAB+); m/z 301(M+H+).
得られた前記t−ブチル−ジメチルシロキシアミド−L−プロリン(化合物19)(550mg、1.8mmol)、6−ヒドロキシヘキサン酸(300mg、2.3mmol)、EDC(434mg、2.3mmol)、および1−ヒドロキシベンゾトリアゾール(695mg、4.5mmol)の無水ジクロロメタン溶液(20mL)を混合した。前記混合物に、アルゴン雰囲気下、室温で、トリエチルアミン(689mg、6.8mmol)を加え、その後、アルゴン雰囲気下、室温で、終夜撹拌した。前記混合液を飽和食塩水で洗浄した。有機層を回収し、前記を硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 CH2Cl2:CH3OH=9:1)に供し、無色シロップ状の化合物20(696mg、収率92%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3):δ4.54(d,1H,CH)、3.58−3.67(m,5H)、3.52−3.56(m,1H,CH),3.32−3.39(m,1H),3.20−3.25(m,2H)、2.40−2.43(m,1H,CH)、2.33(t,J=7.3Hz,2H,CH2)、2.05−2.25(m,2H)、1.93−2.03(m,1H,CH)、1.75−1.85(m,1H,CH)、1.50−1.73(m,8H)、1.37−1.46(m,2H,CH2)、0.87(s,9H,t−Bu)、0.04(s,6H,SiCH3);
Ms(FAB+): m/z 415(M++1).
得られた前記t−ブチル−ジメチルシロキシアミドヒドロキシアミド−L−プロリン(化合物20)(640mg、1.54mmol)を無水ピリジン(1mL)と混合し、室温で共沸乾燥した。得られた残留物に、4,4’−ジメトキシトリチルクロリド(657mg、1.85mmol)、DMAP(2mg)および無水ピリジン(5mL)を加え、室温で4時間撹拌した後、メタノール(1mL)を加え、30分室温で撹拌した。前記混合物をジクロロメタンで希釈し、飽和重曹水で洗浄した。有機層を回収し、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒を留去した。得られた残渣に、無水アセトニトリル(5mL)および1mol/Lテトラブチルアンモニウムフルオリド含有テトラヒドロフラン溶液(1.42mL、テトラブチルアンモニウムフルオリド1.42mmol)を加え、室温で終夜撹拌した。反応終了後、前記混合物に酢酸エチル(100mL)を加え、水で洗浄した後、飽和食塩水で洗浄した。有機層を回収し、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下に溶媒を留去した。得られた残渣を、シリカゲルカラムクロマトグラフィー(展開溶媒 CH2Cl2:CH3OH=95:5、0.05%ピリジン含有)に供し、無色シロップ状の化合物21(680mg、収率73%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.41−7.44(m,2H,Ar−H)、7.26−7.33(m,4H,Ar−H)、7.18−7.21(m,2H,Ar−H)、7.17−7.21(m,1H,Ar−H)、6.80−6.84(m,4H,Ar−H)、4.51−4.53(d,6.8Hz,1H,CH)、3.79(s,6H,OCH3)、3.61(dd,2H,J=11Hz,5.4Hz,CH2)、3.50−3.54(m,1H,CH)、3.36−3.43(m,1H,CH),3.20−3.26(m,2H,CH2),3.05(t,J=6.4Hz,2H,CH2)、2.38−2.45(m,1H,CH)、2.30(t,J=7.8Hz,2H,CH2)、2.05−2.25(m,1H,CH)、1.92−2.00(m,1H,CH)、1.75−1.83(m,1H,CH)、1.52−1.67(m,8H)、1.35−1.45(m,2H,CH2);
Ms (FAB+): m/z 602(M+)、303(DMTr+).
得られた前記DMTr−ヒドロキシジアミド−L−プロリン タイプB(化合物21)(637mg、1.06mmol)を無水アセトニトリルと混合し、室温で共沸乾燥した。得られた残留物に、ジイソプロピルアンモニウムテトラゾリド(201mg、1.16mmol)を加え、減圧下で脱気し、アルゴンガスを充填した。前記混合物に対し、無水アセトニトリル(1mL)を加え、さらに、2−シアノエトキシ−N,N,N’,N’−テトライソプロピルホスホロジアミダイト(350mg、1.16mmol)の無水アセトニトリル溶液(1mL)を加えた。この混合物を、アルゴン雰囲気下、室温で4時間撹拌した。前記混合物をジクロロメタンで希釈し、飽和重曹水および飽和食塩水で洗浄した。有機層を回収し、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られたろ液について、減圧下で溶媒を留去した。得られた残渣を、充填剤としてアミノシリカゲルを用いたカラムクロマトグラフィー(展開溶媒 ヘキサン:アセトン=7:3)に供し、無色シロップ状の化合物22(680mg、純度95%、収率76%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.41−7.43(m,2H,Ar−H)、7.25−7.32(m,4H,Ar−H)、7.17−7.22(m,2H,Ar−H)、6.80−6.83(m,4H,Ar−H)、4.53(d,J=7.8Hz,1H,CH)、3.75−3.93(m,3H)、3.79(s,6H,OCH3)、3.46−3.68(m,5H)、3.34−3.41(m,1H,CH)、3.10−3.31(m,1H,CH)、3.05(t,J=6.3Hz,2H,CH2)、2.62(t,J=6.3Hz,2H,CH2)、2.39−2.46(m,1H,CH)、2.29(t,7.3Hz,2H,CH2)、2.03−2.19(m,1H,CH)、1.90−2.00(m,1H,CH)、1.70−1.83(m,1H,CH)、1.51−1.71(m,8H)、1.35−1.45(m,2H,CH2)、1.18(d,J=6.4 Hz,6H, CH3)、1.16(d,J=6.4Hz,6H,CH3);
P−NMR (CH3CN) δ146.90;
Ms (FAB+): m/z 803(M++1)、303(DMTr+).
プロリン骨格を有するリンカーを含む本発明の核酸分子を生成するため、下記スキーム5により、DMTr−アミドエチレンオキシエチルアミノ−L−プロリンアミダイト(以下、PEGスペーサータイプという)を合成した。
DMTr−アミド−L−プロリン(化合物6)(1.00g、2.05mmol)、4−トルエンスルホン酸2−(2−ヒドロキシエトキシ)エチルエステル(3.10g、12.30mmol)、および炭酸カリウム(0.85g、6.15mmol)の無水ジメチルホルムアミド溶液(10mL)を混合し、アルゴン雰囲気下、室温で4日間撹拌した。前記混合物について、減圧化、室温で溶媒を留去した後、ジクロロメタン(20mL)を加え、ろ過した。ろ液を濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィーに供した。前記シリカゲルカラムクロマトグラフィーの展開溶媒は、まず、0.05%ピリジンを含む酢酸エチルを使用した後、0.05%ピリジンを含むCH2Cl2とCH3OHの混合液(CH2Cl2:CH3OH=9:1)を使用した。その結果、無色シロップ状の化合物23(1.15g、収率97%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.41−7.45(m,2H,Ar−H)、7.27−7.31(m,6H,Ar−H)、7.17−7.21(m,1H,Ar−H)、6.79−6.82(m,4H,Ar−H)、3.79(s,6H,OCH3)、3.60−3.70(m,2H)、3.39−3.57(m,4H),3.13−3.27(m,3H),3.07−3.08(m,2H)、2.71−2.84(m,1H)、2.38−2.46(m,1H)、2.14−2.19(m,1H)、1.84−1.87(m,1H)、1.57−1.76(m,8H).
得られた前記DMTr−アミドヒドロキシエトキシエチルアミノ−L−プロリン(化合物23)(0.63g、1.00mmol)を無水ピリジンと混合し、室温で共沸乾燥した。得られた残留物に、ジイソプロピルアンモニウムテトラゾリド(206mg、1.20mmol)を加え、減圧下で脱気し、アルゴンガスを充填した。前記混合物に対し、無水アセトニトリル(1mL)を加え、さらに、2−シアノエトキシ−N,N,N’,N’−テトライソプロピルホスホロジアミダイト(282mg、1.12mmol)の無水アセトニトリル溶液(1mL)を加えた。この混合物を、アルゴン雰囲気下、室温で4時間撹拌した。そして、前記混合物をジクロロメタンで希釈し、飽和重曹水および飽和食塩水で洗浄した。有機層を回収し、硫酸ナトリウムで乾燥した後、前記有機層をろ過した。得られた前記ろ液について、減圧下で溶媒を留去した。得られた残渣を、充填剤としてアミノシリカゲルを用いたカラムクロマトグラフィー(展開溶媒 ヘキサン:アセトン=7:3、0.05%ピリジン含有)に供し、無色シロップ状の化合物24(0.74g、純度100%、収率87%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CD3CN): δ7.41−7.43(m,2H,Ar−H)、7.28−7.31(m,6H,Ar−H)、7.18−7.22(m,1H,Ar−H)、6.84−6.86(m,4H,Ar−H)、3.73−3.84(m,2H,CH2)、3.79(s,6H,OCH3)、3.47−3.64(m,7H)、3.15−3.23(m,1H)、3.11(t,J=6.4Hz,2H,CH2)、3.01(t,J=5.9Hz,2H,CH2)、2.95−2.99(m,1H)、2.58−2.63(m,2H)、2.31−2.35(m,1H,CH)、2.03−2.19(m,1H,CH)、1.48−1.78(m,10H)、1.12−1.57(m,12H,CH3);
P−NMR(CD3CN): δ148.00;
Ms(FAB+): m/z 776(M+)、303(DMTr+) 201(C8H19N2OP+).
1.保護プロリノールの合成
以下に示すスキーム6に従い、ジメトキシトリチル基で保護されたプロリノール(化合物3)を合成した。
L−プロリノール(2.0g、20mmol)をTHF20mLに溶解した。他方、トリフルオロ酢酸エチル(3.0g、21mmol)をTHF20mLに溶解した。そして、後者のTHF溶液を、前者のL−プロリノール含有THF溶液に滴下し、12時間撹拌した。この反応液を減圧濃縮し、化合物1を得た(3.7g、収率97%)。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ4.28−4,23(1.0H,m,OH),3.90−3.41(5H,H−2,H−5,H−6,m),2.27−1.77(4H,H−3,H−4,m).
得られた前記トリフルオロアセチル−L−プロリノール(化合物1)(3.7g、19mmol)をピリジンに溶解して、3回、室温で共沸乾燥した。得られた残留物をピリジン15mLに溶かし、アルゴン下、氷浴中で撹拌しながら、4,4’−ジメトキシトリチルクロリド(DMTr−Cl)(8.1g、24mmol)を加え、さらに、室温で4時間反応させた。そして、過剰のDMTr−Clをクエンチするために、前記反応液に、さらに、メタノール10mLを加え10分撹拌した。その後、前記反応液に、ジクロロメタンを加え飽和炭酸水素ナトリウム水溶液と飽和食塩水で洗浄した。洗浄後の回収した有機層を、硫酸ナトリウムで乾燥させた。前記有機層をろ過して、得られたろ液を減圧濃縮し、その残渣を、シリカゲルカラムクロマトグラフィー(展開溶媒CH2Cl2:CH3OH=95:5、0.1%ピリジン含有)に供し、精製した化合物2を得た(8.5g、収率89%)。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.39−7.18(9H,m,Ar−H),6.82(4H,d,J=8.6Hz,Ar−H),3.78(6H,s,OCH3),3.70−3.41(5H,H−2,H−5,H−6,m),2.19−1.85(4H,H−3,H−4,m).
得られた前記トリフルオロアセチル−DMTr−L−プロリノール(化合物2)(5g、10mmol)をTHF100mLに溶解した。このTHF溶液に5%水酸化ナトリウム水溶液100mLを加え、撹拌した。この溶液に、1M フッ化テトラ−n−ブチルアンモニウム(TBAF)溶液5mL加え、室温で12時間撹拌した。この反応液を、飽和炭酸水素ナトリウム水溶液と飽和食塩水で洗浄した。洗浄後の回収した有機層を、硫酸ナトリウムで乾燥させた。前記有機層をろ過して、得られたろ液を減圧濃縮し、化合物3を得た(3.6g、収率90%)。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H),6.82(4H,d,J=8.6Hz,Ar−H),3.78(6H,s,OCH3),3.31(1H,m,H−6),3.07(2H,m,H−2,H−6),2.90(2H,m,H−5),1.84(3H,m,H−3,H−4),1.40(1H,m,H−3).
前記「1.」で合成した保護プロリノール(化合物3)を用いて、下記スキーム7により、結合形式が異なる、プロリノールを有するアミダイト誘導体を合成した。
1,8−オクタンジオール(9.0g、62mmol)をTHF90mLに溶解し、アルゴン下に置いた。他方、カルボニルジイミダゾール(2.0g、12mmol)をTHF10mLに溶解した。後者のTHF溶液を、前者のTHF溶液に加え、室温で1時間撹拌した。この反応液を、1,8−オクタンジオールのTLCスポットが消えるまで、水で洗浄した。さらに、洗浄後に回収した有機層を、飽和食塩水で洗浄し、回収した有機層を、無水硫酸ナトリウムで乾燥させた。前記有機層をろ過し、得られたろ液を減圧濃縮した。その残渣を、シリカゲルカラムクロマトグラフィー(展開溶媒 CH2Cl2:CH3OH=95:5)に供し、精製した化合物を得た。この化合物は、1,8−オクタンジオールの片末端がカルボニルジイミダゾールで活性化された化合物であった(2.3g、収率77%)。
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H),6.82(4H,d,J=8.6Hz,Ar−H),4.24−3.94(2H,m,COOCH2),3.78(s,6H,OCH3),3.72−2.96(7H,m,alkyl,H−2,H−5,H−6),2.10−1.30(16H,m,alkyl,H−3,H−4);
FAB−MS: 576[M+H]+.
アルゴン下、トリホスゲン(2.0g、6.7mmol)をTHF10mLに溶解し、0℃で撹拌した。他方、DMTr−L−プロリノール(化合物3)(1.3g、3.2mmol)およびN,N−ジイソプロピルエチルアミン(16g、124mmol)をTHF10mLに溶解し、前記トリホスゲンのTHF溶液に滴下した。この反応液を、0℃で1時間、続いて、室温で2時間撹拌した。そして、8−アミノ−1−オクタノール(2.3g、16mmol)およびN,N−ジイソプロピルエチルアミン(5.0g、38mmol)をTHF30mLに溶解した。このTHF溶液に、前記撹拌後の反応液を滴下し、0℃で1時間、続いて、室温で48時間撹拌した。この反応液を減圧濃縮し、その残渣をジクロロメタンに溶解した。この溶液を、飽和炭酸水素ナトリウム水溶液と飽和食塩水で洗浄し、回収した有機層を、無水硫酸ナトリウムで乾燥させた。前記有機層をろ過して、得られたろ液を減圧濃縮し、その残渣を、逆相シリカゲルカラムクロマトグラフィーに供して、精製した。この際、展開溶媒は、0.1%ピリジンを含有するアセトンと水との混合溶媒を使用し、前記アセトンと水との混合割合は、ステップワイズとし、具体的には、アセトン:水のモル比を、2:8、3:7、4:6および5:5の順に変化させた。目的の化合物5を含むフラクションを、ジクロロメタンで抽出し、この有機層を無水硫酸ナトリウムで乾燥させた。前記有機層をろ過し、得られたろ液を減圧濃縮し、化合物5(プロリノールウレイドアミダイト)を得た(0.9g、収率49%)。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H),6.82(4H,m,Ar−H),3.78(s,6H,OCH3),3.68−3.25(9H,m,CH2NH,CH2OH,H−2,H−5,H−6),1.74−1.18(16H,m,alkyl,H−3,H−4);
FAB−MS :575[M+H]+.
修飾プロリノールとして、得られた前記化合物4(0.80g、1.4mmol)をアセトニトリルに溶解し、室温で3回共沸乾燥した。得られた残留物をアセトニトリル1mLに溶解し、アルゴン下においた。このアセトニトリル溶液に、ジイソプロピルアンモニウムテトラゾリド(0.24g,1.4mmol)を添加し、反応液とした。他方、2−シアノエチルN,N,N’,N’−テトライソプロピルホスホロジアミダイト(0.50g、1.7mmol)をアセトニトリル1mLに溶解した。これを、前記反応液に添加し、室温で4時間撹拌した。前記反応液に、ジクロロメタンを加え、飽和炭酸水素ナトリウム水溶液と飽和食塩水で洗浄した。洗浄後の回収した有機層を無水硫酸ナトリウムで乾燥させ、前記有機層をろ過して、得られたろ液を減圧濃縮した。その残渣を、アミノシリカゲルカラムクロマトグラフィー(展開溶媒ヘキサン:アセトン=10:1、0.1%ピリジン含有)に供し、精製した化合物6(DMTr−ウレタン−L−プロリノールアミダイト)(0.90g、収率83%)を得た。以下に、前記化合物のNMRの結果を示す。
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H),6.82(4H,d,J=8.6Hz,Ar−H),4.24−3.94(2H,m,COOCH2),3.78(s,6H,OCH3),3.72−2.96(11H,m,CH2O,POCH2,CHCH3,H−2,H−5,H−6),2.58(2H,m,CH2CN),2.10−1.46(16H,m,alkyl,H−3,H−4),1.34−1.10(12H,m,CHCH3);
31P−NMR(CD3CN): δ146.82;
FAB−MS: 776[M+H]+.
1H−NMR(CDCl3): δ7.40−7.14(9H,m,Ar−H),6.82(4H,m,Ar−H),3.78(s,6H,OCH3),3.65−3.25(13H,m,CH2O,POCH2,CHCH3,H−2,CH2NH,CH2OH,H−2,H−5,H−6),2.73(2H,m,CH2CN),2.10−1.48(16H,m,alkyl,H−3,H−4),1.35−1.10(12H,m,CHCH3);
31P−NMR(CD3CN): δ 146.83;
FAB−MS:775 [M+H]+.
本発明のリンカーを有するRNAを合成した。RNAは、ホスホロアミダイト法に基づき、核酸合成機(商品名ABI Expedite(登録商標) 8909 Nucleic Acid Synthesis System、アプライドバイオシステムズ)により、3’側から5’側に向かって合成した。前記合成には、RNAアミダイトとして、RNA Phosphoramidites(2’−O−TBDMSi、商品名、三千里製薬)を用いた(以下、同様)。前記アミダイトの脱保護は、定法に従い、合成したRNAは、HPLCにより精製した。以下の実施例において、RNAの合成は、特に示さない限り、同様に行った。
5’-GGCUGUUGUCAUACUUCUCAUGGUU-3’(配列番号1)
5’-CCAUGAGAAGUAUGACAACAGCC-3’(配列番号2)
Ex:PH−0001(配列番号3)
5’-CCAUGAGAAGUAUGACAACAGCC-Lx-GGCUGUUGUCAUACUUCUCAUGGUU-3’
Pc:NH−0001(配列番号5)
5’-CCAUGAGAAGUAUGACAACAGCCccacaccGGCUGUUGUCAUACUUCUCAUGGUU-3’
本発明のRNAを用いて、in vitroにおけるGAPDH遺伝子の発現抑制を確認した。
実施例のRNA(Ex)として、前記実施例B1のssRNA(PH−0001)を使用した。前記RNAを、所望の濃度(1μmol/L、5μmol/L、25μmol/L)となるように、注射用蒸留水(大塚製薬、以下同様)に溶解し、RNA溶液を調製した。
GAPDH遺伝子用PCRプライマーセット
5’-GGAGAAGGCTGGGGCTCATTTGC-3’(配列番号7)
5’-TGGCCAGGGGTGCTAAGCAGTTG-3’(配列番号8)
β−アクチン遺伝子用プライマーセット
5’-GCCACGGCTGCTTCCAGCTCCTC-3’(配列番号9)
5’-AGGTCTTTGCGGATGTCCACGTCAC-3’(配列番号10)
これらの結果を、図4に示す。図4は、GAPDH遺伝子発現量の相対値を示すグラフであり、縦軸は、相対遺伝子発現量である。図4に示すように、前記実施例B1のPH−0001は、発現阻害活性を損なうことがなかった。また、前記PH−0001は、本発明のリンカーである前記化合物12によって安定化されていると考えられる。
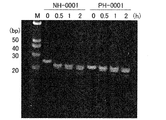
本発明のRNAについて、ヒト血清中での安定性を確認した。
実施例のRNA(Ex)として、前記実施例B1のssRNA(PH−0001)を使用した。比較例のRNAとして、前記実施例B1に示す、RNAiポジティブコントロール(Pc)の前記shRNA(NH−0001)を使用した。
この結果を図5に示す。図5は、安定性を示す電気泳動写真である。図5において、レーン「M」は、分子量マーカーであり、(h)は、インキュベート時間を示す。
プロリンを含む下記式のリンカーを有するssRNAを合成し、GAPDH遺伝子の発現抑制効果を確認した。
(1.1)ssRNAの固相合成
前記RNAは、前記実施例B1と同様にして、ホスホロアミダイト法に基づき合成した。
凍結保存した前記RNAを、20μmol/Lとなるように、注射用蒸留水(大塚製薬)に溶解し、RNA溶液を調製した。
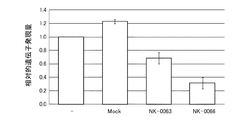
これらの結果を、図6に示す。図6は、GAPDH遺伝子発現量の相対値を示すグラフである。図6に示すように、本発明のリンカーを有する実施例のPK−0004は、強い遺伝子発現抑制活性を示し、その活性は投与量依存的であった。他方、ネガティブコントロールのPK−0003は、抑制効果が観察されなかった。
プロリンを有するリンカーで置換したssRNAに対する、組換えヒトダイサータンパク質の反応性を確認した。
実施例のRNA(Ex)として、前記実施例B4のssRNA(PK−0004)を使用した。比較例のRNAとして、前記実施例B4に示す、RNAiネガティブコントロール(Nc)のssRNA(PK−0003)、以下に示すRNAiポジティブコントロール(Pc)のssRNA(NK−0016)を使用した。前記NK−0016は、前記5’側領域(Xc)および前記内部5’側領域(X)、前記3’側領域(Yc)および前記内部3’側領域(Y)が、前記PK−0004と同じ配列であり、前記XcとXとの間、YcとYとの間に、前記PK−0004の前記式(化22)のリンカー(Lx、Ly)に代えて、ポリヌクレオチドをリンカーとして有する構造とした。
これらの結果を図7に示す。図7は、ssRNAに対するダイサータンパク質の反応性を示す電気泳動の結果である。図7において、レーン「M」は、分子量マーカー(20bp、30bp、40bpおよび50bp)であり、(h)は、前記インキュベートの時間を示す。
プロリンを有するリンカーで置換したssRNAを用いて、in vitroにおけるGAPDH遺伝子の発現抑制を確認した。
実施例のRNA(Ex)として、前記実施例B4のssRNA(PK−0004)を使用した。比較例のRNAとして、前記実施例B4に示す、RNAiネガティブコントロール(Nc)のssRNA(PK−0003)を使用した。前記RNAを、20μmol/Lとなるように、注射用蒸留水(大塚製薬)に溶解し、RNA溶液を調製した。
これらの結果を、図8および図9に示す。図8は、A549細胞の結果であり、図9は、293細胞の結果である。図8および図9は、GAPDH遺伝子発現量の相対値を示すグラフである。図8および図9に示すように、実施例のPK−0004は、強い遺伝子発現抑制活性を示し、濃度依存的に効果を示すことがわかった。他方、ネガティブコントロールのPK−0003は、抑制効果が観察されなかった。
プロリンまたはプロリノールを有するリンカーで置換したssRNAを用いて、HCT116細胞でのGAPDH発現抑制効果を確認した。
(1.1)ssRNAの固相合成
実施例のRNA(Ex ssRNA)として、前記実施例B4と同じEx ssRNAを合成した。前記RNAの合成は、特に示さない限り、前記実施例B4に準じた。
前記RNAを使用した以外は、前記実施例B4と同様にして、HCT116細胞へのトランスフェクション、培養、RNA回収、cDNA合成およびPCRを行い、GAPDH遺伝子の相対的発現量を測定した。
これらの結果を、図10に示す。図10は、HCT116細胞のGAPDH遺伝子発現量の相対値を示すグラフである。図10に示すように、プロリンまたはプロリノールを含むssRNA(PK−0004、PK−0006、PK−0010、PK−0012、PK−0016)は、強い遺伝子発現抑制活性を示し、濃度依存的に効果を示すことがわかった。
プロリンを有するリンカーで置換したssRNAを用いて、HCT116細胞でのGAPDH発現抑制効果を確認した。
(1.1)ssRNAの固相合成
実施例のRNA(Ex ssRNA)として、前記実施例B4と同じEx ssRNAを合成した。前記RNAの合成は、特に示さない限り、前記実施例B4に準じた。
前記RNAを使用した以外は、前記実施例B4と同様にして、HCT116細胞へのトランスフェクション、培養、RNA回収、cDNA合成およびPCRを行い、GAPDH遺伝子の相対的発現量を測定した。
これらの結果を、図11に示す。図11は、HCT116細胞のGAPDH遺伝子発現量の相対値を示すグラフである。図11に示すように、プロリンを含むssRNA(PK−0004、PK−0034、PK−0036)は、強い遺伝子発現抑制活性を示し、濃度依存的に効果を示すことがわかった。
プロリンを有するリンカーで置換したssRNAを用いて、Hepa1−6細胞でのTGF発現抑制効果を確認した。
(1.1)ssRNAの固相合成
実施例のRNAとして、以下に示すPK−0007、PK−0026、PK−0027、PK−0028を合成した。前記RNAの合成は、特に示さない限り、前記実施例B4に準じた。リンカー用アミダイトとして、前記実施例A3−1で合成したL−プロリンジアミドアミダイト(スキーム3の化合物10)を用いた。各RNAは、TGF−β1遺伝子の発現を抑制する21塩基長の下記配列を有している。この配列は、Chengらが用いたsiRNA(Mol.Pharm.,2009,6,772−779)に基づいて、設計した。下記配列において、「*」は、フリー塩基を示す。
TGF−β1遺伝子発現抑制配列(配列番号18)
5’−AAAGUCAAUGUACAGCUGCUU−3’
凍結保存した前記RNAを、20μmol/Lとなるように、注射用蒸留水(大塚製薬)に溶解し、RNA溶液を調製した。
TGF−β1遺伝子用PCRプライマーセット
5’-CCATTGCTGTCCCGTGCAGAGCTG-3’(配列番号19)
5’-ATGGTAGCCCTTGGGCTCGTGGATC-3’(配列番号20)
β−アクチン遺伝子用プライマーセット
5’-GTCGTACCACAGGCATTGTGATGG-3’(配列番号21)
5’-GCAATGCCTGGGTACATGGTGG-3’(配列番号22)
これらの結果を、図12に示す。図12は、TGF−β1遺伝子発現量の相対値を示すグラフである。図12に示すように、プロリンを含むssRNAは、全て、強い遺伝子発現抑制活性を示した。
プロリンを有するリンカーで置換したssRNAを用いて、in vivoでの遺伝子発現抑制および急性肺傷害抑制の効果を確認した。前記効果の確認は、Takagiら(J.Thromb Hemost 2009;7:2053−2063)に記載の方法に従って行った。
プロリンを有するリンカーで置換したssRNAを用いて、in vivoでのTGF−β1遺伝子の発現抑制効果を確認した。
(1.1)急性肺傷害マウスへのRNAの投与
実施例のRNA(Ex)は、前記実施例B9におけるssRNA(PK−0007)を使用した。比較例のRNAは、以下に示す、ネガティブコントロール(Nc)のssRNA(PK−0008)、ポジティブコントロール(Pc)のssRNA(NK−0033)およびそのネガティブコントロール(Nc)のssRNA(NK−0035)、ポジティブコントロール(Pc)のdsRNA(NI−0030)およびそのネガティブコントロール(Nc)のdsRNA(NI−0031)を使用した。
・投与群1
滅菌生理食塩水75μlの投与から5分後、滅菌生理食塩水50μlを投与
・投与群2
滅菌生理食塩水75μlの投与から5分後、前記LPS溶液50μlを投与
・投与群3
RNA溶液(PK−0007)75μlの投与から5分後、前記LPS溶液50μlを投与
・投与群4
RNA溶液(PK−0008)75μlの投与から5分後、前記LPS溶液50μlを投与
・投与群5
RNA溶液(NK−0033)75μLの投与から5分後、前記LPS溶液50μlを投与
・投与群6
RNA溶液(NK−0035)75μlの投与から5分後、前記LPS溶液50μlを投与
・投与群7
RNA溶液(NI−0030)75μlの投与から5分後、前記LPS溶液50μlを投与
・投与群8
RNA溶液(NI−0031)50μlの投与から5分後に、前記LPS溶液50μlを投与
前記LPS溶液または滅菌生理食塩水(LPSに対するネガティブコントロール)を滴下してから24時間後、前記マウスの腹腔に、過剰量のペントバルビタールを投与して安楽死させた。そして、肺を採取し、サンプルとした。
その結果を、図13に示す。図13は、各投与群における単位重量の肺あたりのTGF−β1遺伝子発現量を示すグラフであり、横軸は、TGF−β1タンパク質の発現量を示す。LPS(+)/PK−0007(+)の投与群3は、LPS(+)/ssRNA(−)の投与群2と比較して、TGF−β1タンパク質の発現量が著しく抑制された。この抑制効果は、LPS(+)/ポジティブコントロールNK−0033(+)の投与群5およびLPS(+)/ポジティブコントロールNI−0030の投与群7よりも、強いことが明らかとなった。なお、ネガティブコントロールPK−0008(+)の投与群4、ネガティブコントロールNK−0035(+)の投与群6、ネガティブコントロールNI−0031(+)の投与群8では、抑制効果は確認されなかった。
プロリンを有するリンカーで置換したssRNAを用いて、in vivoでのオフターゲット効果を確認し、副作用を評価した。
・投与群1
滅菌生理食塩水75μlを投与
・投与群2
ssRNA溶液(PK−0007)75μlを投与
・投与群3
ssRNA溶液(PK−0008)75μlを投与
本発明のssRNAについて、リボヌクレアーゼ耐性を確認した。
実施例のRNA(Ex)として、前記実施例B9のssRNA(PK−0007)を使用した。比較例のRNAとして、前記実施例B10−1に示す、ポジティブコントロール(Pc)のdsRNA(NI−0030)を使用した。
この結果を図15に示す。図15は、リボヌクレアーゼ耐性を示す電気泳動写真である。図15において、レーン「M」は、分子量マーカーであり、(min)は、インキュベート時間を示す。
本発明のssRNAについて、ヌクレアーゼ耐性を確認した。
実施例のRNA(Ex)として、前記実施例B9のssRNA(PK−0007)を使用した。比較例のRNAとして、前記実施例B10−1に示す、RNAiポジティブコントロール(Pc)のdsRNA(NI−0030)を使用した。
この結果を図16に示す。図16は、S7ヌクレアーゼ耐性を示す電気泳動写真である。図16において、レーン「M」は、分子量マーカーであり、(h)は、インキュベート時間を示す。
フリー塩基の位置が異なるssRNAを使用して、in vitroにおけるGAPDH遺伝子の発現抑制を確認した。
RNAとして、図17に示すssRNAを使用した。図17において、右端の番号は、配列番号を示す。図17において、5’側から、小文字下線の領域は、前記領域(Xc)、大文字下線の領域は、前記内部領域(Z)、小文字下線の領域は、前記領域(Yc)を示す。前記Xcと前記Zとの間が、リンカー領域(Lx)であり、前記Zと前記Ycとの間が、リンカー領域(Ly)である。また、「Xc/Yc」は、前記領域(Xc)の塩基長(Xc)と、前記領域(Yc)の塩基長(Yc)との比を示す。図17において、「*」は、フリー塩基を示す。
これらの結果を、図18に示す。図18は、終濃度10nmol/LのRNAを使用した場合におけるGAPDH遺伝子発現量の相対値を示すグラフである。図18に示すように、前記5’側領域(Xc)および前記3’側領域(Yc)の長さを変化させたいずれのssRNAについても、GAPDH遺伝子の発現抑制が確認できた。
フリー塩基の位置が異なるssRNAを使用して、in vitroにおけるTGF−β1遺伝子の発現抑制効果を確認した。
RNAとして、以下に示すssRNAを使用した。下記配列において、「*」は、フリー塩基を示す。
凍結保存した前記RNAを、20μmol/Lとなるように、注射用蒸留水に溶解し、RNA溶液を調製した。そして、前記RNA溶液を使用した以外は、前記実施例B9と同様にして、Hepal−6細胞への前記ssRNAのトランスフェクション、RNA回収、cDNA合成およびPCRを行い。TGF−β1遺伝子の相対的発現量を測定した。トランスフェクション時のRNA濃度は、1nmol/Lとした。
これらの結果を、図19に示す。図19は、TGF−β1遺伝子発現量の相対値を示すグラフである。図19に示すように、いずれのssRNAも、遺伝子発現抑制活性を示した。また、フリー塩基の位置を、前記内部領域(Z)における3’末端から2番目および3番目としたNK−0055およびNK−0062は、フリー塩基の位置を、前記内部領域(Z)における3’末端から4番目および5番目としたNK−0033およびNK−0061よりも、高い発現抑制活性を示した。この結果は、異なる遺伝子をターゲットとする前記参考例1と同様の挙動であった。
フリー塩基の位置が異なるssRNAを使用して、in vitroにおけるLAMA1遺伝子の発現抑制を確認した。
RNAとして、以下に示すssRNAを使用した。下記配列において、「*」は、フリー塩基を示す。
LAMA1遺伝子用プライマーセット
5’-AAAGCTGCCAATGCCCCTCGACC-3’(配列番号55)
5’-TAGGTGGGTGGCCCTCGTCTTG-3’(配列番号56)
これらの結果を、図20に示す。図20は、293細胞におけるLAMA1遺伝子の発現量の相対値を示すグラフである。図20に示すように、いずれのssRNAも、遺伝子発現抑制活性を示した。また、フリー塩基の位置を、前記内部領域(Z)における3’末端から2番目としたNK−0064は、フリー塩基の位置を、前記内部領域(Z)における3’末端から4番目としたNK−0043よりも、高い発現抑制活性を示した。この結果は、異なる遺伝子をターゲットとする前記参考例1および参考例2と同様の挙動であった。
フリー塩基の位置が異なるssRNAを用いて、in vitroにおけるLMNA遺伝子の発現抑制を確認した。
RNAとして、以下に示すssRNAを使用した。下記配列において、「*」は、フリー塩基を示す。
LMNA遺伝子用プライマーセット
5’-CTGGACATCAAGCTGGCCCTGGAC-3’(配列番号59)
5’-CACCAGCTTGCGCATGGCCACTTC-3’(配列番号60)
これらの結果を、図21に示す。図21は、A549細胞におけるLMNA遺伝子の発現量の相対値を示すグラフである。図21に示すように、いずれのssRNAも、遺伝子発現抑制活性を示した。また、フリー塩基の位置を、前記内部領域(Z)における3’末端から2番目としたNK−0066は、フリー塩基の位置を、前記内部領域(Z)における3’末端から4番目としたNK−0063よりも、高い発現抑制活性を示した。この結果は、異なる遺伝子をターゲットとする前記参考例1〜参考例3と同様の挙動であった。
前記内部5’側領域(X)、前記5’側領域(Xc)、前記内部3’側領域(Y)および前記3’側領域(Yc)の各長さを変化させたssRNAを用いて、in vitroにおけるGAPDH遺伝子の発現抑制を確認した。
RNAとして、図22に示すssRNAを使用した。図22において、右端の番号は、配列番号を示す。図22において、5’側から、小文字下線の領域は、前記領域(Xc)、大文字下線の領域は、前記内部領域(Z)、小文字下線の領域は、前記領域(Yc)を示す。また、「Xc+Yc/X+Y」は、前記領域(Xc)と前記領域(Yc)の塩基長の合計と、前記領域(X)と前記領域(Y)の塩基長の合計との比を示す。図22において、「*」は、フリー塩基を示す。
これらの結果を、図23に示す。図23は、終濃度1nmol/LのRNAを使用した場合におけるGAPDH遺伝子発現量の相対値を示すグラフである。図23に示すように、前記領域(X)、前記領域(Xc)、前記領域(Y)および前記領域(Yc)の長さを変化させたいずれのssRNAについても、GAPDH遺伝子の発現抑制が確認できた。
Claims (38)
- 標的遺伝子の発現を抑制する発現抑制配列を含む一本鎖RNA核酸分子であって、
領域(Xc)、リンカー領域(Lx)、領域(X)、領域(Y)および領域(Yc)を、この順序で含み、
前記領域(X)と前記領域(Y)とが連結して、内部領域(Z)を形成し、
前記領域(Xc)が、前記領域(X)と相補的であり、
前記領域(Yc)が、前記領域(Y)と相補的であり、
前記内部領域(Z)が、前記発現抑制配列を含み、
前記内部領域(Z)の長さ(Z)が、19〜30塩基であり、
前記内部領域(Z)の長さ(Z)と、前記領域(Xc)および領域(Yc)の合計長さ(Xc+Yc)との差(Z−(Xc+Yc))が、0〜2塩基であり、
5’末端と3’末端とが未結合であって、
前記リンカー領域(Lx)が、ピロリジン骨格およびピペリジン骨格の少なくとも一方を含む非ヌクレオチド構造を有することを特徴とする、標的遺伝子の発現を抑制する一本鎖RNA核酸分子。 - 前記リンカー領域(Lx)が、下記式(I)で表わされる、請求項1記載の一本鎖RNA核酸分子。
C=X1およびC=X2は、それぞれ独立して、CH2、CO、CSまたはCNHであり;
Y1およびY2は、それぞれ独立して、単結合、CH2、NH、OまたはSであり;
R3は、環A上のC−3、C−4、C−5またはC−6に結合する水素原子または置換基であり;
L1は、n個の原子からなるアルキレン鎖であり、ここで、アルキレン炭素原子上の水素原子は、OH、ORa、NH2、NHRa、NRaRb、SH、もしくはSRaで置換されても置換されていなくてもよく、または、
L1は、前記アルキレン鎖の一つ以上の炭素原子が、酸素原子で置換されたポリエーテル鎖であり、
ただし、Y1が、NH、OまたはSの場合、Y1に結合するL1の原子は炭素であり、OR1に結合するL1の原子は炭素であり、酸素原子同士は隣接せず;
L2は、m個の原子からなるアルキレン鎖であり、ここで、アルキレン炭素原子上の水素原子は、OH、ORc、NH2、NHRc、NRcRd、SHもしくはSRcで置換されても置換されていなくてもよく、または、
L2は、前記アルキレン鎖の一つ以上の炭素原子が、酸素原子で置換されたポリエーテル鎖であり、
ただし、Y2が、NH、OまたはSの場合、Y2に結合するL2の原子は炭素であり、OR2に結合するL2の原子は炭素であり、酸素原子同士は隣接せず;
Ra、Rb、RcおよびRdは、それぞれ独立して、置換基または保護基であり;
lは、1または2であり;
mは、0〜30の範囲の整数であり;
nは、0〜30の範囲の整数であり;
環Aは、前記環A上のC−2以外の1個の炭素原子が、窒素、酸素または硫黄で置換されてもよく、
前記環A内に、炭素−炭素二重結合または炭素−窒素二重結合を含んでもよく、
前記領域(Xc)および前記領域(X)は、それぞれ、−OR1−または−OR2−を介して、前記リンカー領域(Lx)に結合し、
ここで、R1およびR2は、存在しても存在しなくてもよく、存在する場合、R1およびR2は、それぞれ独立して、ヌクレオチド残基または前記構造(I)である。 - 前記領域(X)の塩基数(X)および前記領域(Xc)の塩基数(Xc)が、下記式(3)または式(5)の条件を満たす、請求項1または2記載の一本鎖RNA核酸分子。
X>Xc ・・・(3)
X=Xc ・・・(5) - 前記領域(X)の塩基数(X)および前記領域(Xc)の塩基数(Xc)が、下記式(11)の条件を満たす、請求項3記載の一本鎖RNA核酸分子。
X−Xc=1、2または3 ・・・(11) - さらに、リンカー領域(Ly)を有し、
前記領域(Y)と前記領域(Yc)との間に、前記リンカー領域(Ly)が連結している、請求項1から4のいずれか一項に記載の一本鎖RNA核酸分子。 - 前記リンカー領域(Ly)が、ピロリジン骨格またはピペリジン骨格を含む非ヌクレオチド構造を有する、請求項5記載の一本鎖RNA核酸分子。
- 前記リンカー領域(Ly)が、下記式(I)で表わされる、請求項6記載の一本鎖RNA核酸分子。
C=X 1 およびC=X 2 は、それぞれ独立して、CH 2 、CO、CSまたはCNHであり;
Y 1 およびY 2 は、それぞれ独立して、単結合、CH 2 、NH、OまたはSであり;
R 3 は、環A上のC−3、C−4、C−5またはC−6に結合する水素原子または置換基であり;
L 1 は、n個の原子からなるアルキレン鎖であり、ここで、アルキレン炭素原子上の水素原子は、OH、OR a 、NH 2 、NHR a 、NR a R b 、SH、もしくはSR a で置換されても置換されていなくてもよく、または、
L 1 は、前記アルキレン鎖の一つ以上の炭素原子が、酸素原子で置換されたポリエーテル鎖であり、
ただし、Y 1 が、NH、OまたはSの場合、Y 1 に結合するL 1 の原子は炭素であり、OR 1 に結合するL 1 の原子は炭素であり、酸素原子同士は隣接せず;
L 2 は、m個の原子からなるアルキレン鎖であり、ここで、アルキレン炭素原子上の水素原子は、OH、OR c 、NH 2 、NHR c 、NR c R d 、SHもしくはSR c で置換されても置換れていなくてもよく、または、
L 2 は、前記アルキレン鎖の一つ以上の炭素原子が、酸素原子で置換されたポリエーテル鎖であり、
ただし、Y 2 が、NH、OまたはSの場合、Y 2 に結合するL 2 の原子は炭素であり、OR 2 に結合するL 2 の原子は炭素であり、酸素原子同士は隣接せず;
R a 、R b 、R c およびR d は、それぞれ独立して、置換基または保護基であり;
lは、1または2であり;
mは、0〜30の範囲の整数であり;
nは、0〜30の範囲の整数であり;
環Aは、前記環A上のC−2以外の1個の炭素原子が、窒素、酸素、硫黄で置換されてもよく、
前記環A内に、炭素−炭素二重結合または炭素−窒素二重結合を含んでもよく、
前記領域(Yc)および前記領域(Y)は、それぞれ、−OR 1 −または−OR 2 −を介して、前記リンカー領域(Ly)に結合し、
ここで、R 1 およびR 2 は、存在しても存在しなくてもよく、存在する場合、R 1 およびR 2 は、それぞれ独立して、ヌクレオチド残基または前記構造(I)である。 - 前記領域(Xc)および前記領域(X)と、前記リンカー領域(Lx)の前記式(I)の構造との結合、ならびに、
前記領域(Yc)および前記領域(Y)と、前記リンカー領域(Ly)の前記式(I)の構造との結合が、それぞれ、下記(1)〜(4)のいずれか一つの条件を満たす、請求項7記載の一本鎖RNA核酸分子。
条件(1)
前記領域(Xc)は、−OR 2 −を介して、前記領域(X)は、−OR 1 −を介して、前記式(I)の構造と結合し、
前記領域(Yc)は、−OR 1 −を介して、前記領域(Y)は、−OR 2 −を介して、前記式(I)の構造と結合する。
条件(2)
前記領域(Xc)は、−OR 2 −を介して、前記領域(X)は、−OR 1 −を介して、前記式(I)の構造と結合し、
前記領域(Yc)は、−OR 2 −を介して、前記領域(Y)は、−OR 1 −を介して、前記式(I)の構造と結合する。
条件(3)
前記領域(Xc)は、−OR 1 −を介して、前記領域(X)は、−OR 2 −を介して、前記式(I)の構造と結合し、
前記領域(Yc)は、−OR 1 −を介して、前記領域(Y)は、−OR 2 −を介して、前記式(I)の構造と結合する。
条件(4)
前記領域(Xc)は、−OR 1 −を介して、前記領域(X)は、−OR 2 −を介して、前記式(I)の構造と結合し、
前記領域(Yc)は、−OR 2 −を介して、前記領域(Y)は、−OR 1 −を介して、前記式(I)の構造と結合する。 - 前記式(I)において、L 1 は、前記ポリエーテル鎖であり、前記ポリエーテル鎖が、ポリエチレングリコールである、請求項2から8のいずれか一項に記載の一本鎖RNA核酸分子。
- 前記式(I)において、L 1 の原子個数(n)とL 2 の原子個数(m)との合計(m+n)が、0〜30の範囲である、請求項2から9のいずれか一項に記載の一本鎖RNA核酸分子。
- 前記式(I)の構造が、下記式(I−1)〜式(I−9)のいずれか一つであり、下記式において、nは、0〜30の整数、mは、0〜30の整数、qは、0〜10の整数である、請求項2から10のいずれか一項に記載の一本鎖RNA核酸分子。
- 前記式(I−1)において、n=8、前記(I−2)において、n=3、前記式(I−3)において、n=4または8、前記(I−4)において、n=7または8、前記式(I−5)において、n=3およびm=4、前記(I−6)において、n=8およびm=4、前記式(I−7)において、n=8およびm=4、前記(I−8)において、n=5およびm=4、前記式(I−9)において、q=1およびm=4である、請求項11記載の一本鎖RNA核酸分子。
- 前記式(I−4)が、下記式(I−4a)であり、前記式(I−8)が、下記式(I−8a)である、請求項12記載の一本鎖RNA核酸分子。
- 前記領域(X)の塩基数(X)、前記領域(Y)の塩基数(Y)、前記領域(Xc)の塩基数(Xc)および前記領域(Yc)の塩基数(Yc)が、下記式(2)の条件を満たす、請求項1から13のいずれか一項に記載の一本鎖RNA核酸分子。
Z≧Xc+Yc ・・・(2) - 前記領域(X)の塩基数(X)、前記(Xc)の塩基数(Xc)、前記領域(Y)の塩基数(Y)および前記領域(Yc)の塩基数(Yc)が、下記(a)〜(d)のいずれかの条件を満たす、請求項1から14のいずれか一項に記載の一本鎖RNA核酸分子。
(a)下記式(3)および(4)の条件を満たす。
X>Xc ・・・(3)
Y=Yc ・・・(4)
(b)下記式(5)および(6)の条件を満たす。
X=Xc ・・・(5)
Y>Yc ・・・(6)
(c)下記式(7)および(8)の条件を満たす。
X>Xc ・・・(7)
Y>Yc ・・・(8)
(d)下記式(9)および(10)の条件を満たす。
X=Xc ・・・(9)
Y=Yc ・・・(10) - 前記(a)〜(d)において、前記領域(X)の塩基数(X)と前記領域(Xc)の塩基数(Xc)の差、前記領域(Y)の塩基数(Y)と前記領域(Yc)の塩基数(Yc)の差が、下記条件を満たす、請求項15記載の一本鎖RNA核酸分子。
(a)下記式(11)および(12)の条件を満たす。
X−Xc=1、2または3 ・・・(11)
Y−Yc=0 ・・・(12)
(b)下記式(13)および(14)の条件を満たす。
X−Xc=0 ・・・(13)
Y−Yc=1、2または3 ・・・(14)
(c)下記式(15)および(16)の条件を満たす。
X−Xc=1、2または3 ・・・(15)
Y−Yc=1、2または3 ・・・(16)
(d)下記式(17)および(18)の条件を満たす。
X−Xc=0 ・・・(17)
Y−Yc=0 ・・・(18) - 前記領域(Xc)の塩基数(Xc)が、1〜11塩基である、請求項1から16のいずれか一項に記載の一本鎖RNA核酸分子。
- 前記領域(Xc)の塩基数(Xc)が、1〜7塩基である、請求項17記載の一本鎖RNA核酸分子。
- 前記領域(Xc)の塩基数(Xc)が、1〜3塩基である、請求項17記載の一本鎖RNA核酸分子。
- 前記領域(Yc)の塩基数(Yc)が、1〜11塩基である、請求項1から19のいずれか一項に記載の一本鎖RNA核酸分子。
- 前記領域(Yc)の塩基数(Yc)が、1〜7塩基である、請求項20記載の一本鎖RNA核酸分子。
- 前記領域(Yc)の塩基数(Yc)が、1〜3塩基である、請求項20記載の一本鎖RNA核酸分子。
- 少なくとも1つの修飾された残基を含む、請求項1から22のいずれか一項に記載の一本鎖RNA核酸分子。
- 標識物質を含む、請求項1から23のいずれか一項に記載の一本鎖RNA核酸分子。
- 安定同位体を含む、請求項1から24のいずれか一項に記載の一本鎖RNA核酸分子。
- 前記一本鎖RNA核酸分子において、塩基数の合計が、50塩基以上である、請求項1から25のいずれか一項に記載の一本鎖RNA核酸分子。
- 前記遺伝子の発現抑制が、RNA干渉による発現抑制である、請求項1から26のいずれか一項に記載の一本鎖RNA核酸分子。
- 標的遺伝子の発現を抑制するための組成物であって、
請求項1から27のいずれか一項に記載の一本鎖RNA核酸分子を含むことを特徴とする、発現抑制用組成物。 - 請求項1から27のいずれか一項に記載の一本鎖RNA核酸分子を含むことを特徴とする、薬学的組成物。
- 炎症治療用である、請求項29記載の薬学組成物。
- 非ヒト動物において、標的遺伝子の発現を抑制する方法であって、
請求項1から27のいずれか一項に記載の一本鎖RNA核酸分子を使用することを特徴とする発現抑制方法。 - 前記一本鎖RNA核酸分子を、細胞、組織または器官に投与する工程を含む、請求項31記載の発現抑制方法。
- 前記一本鎖RNA核酸分子を、in vivoまたはin vitroで投与する、請求項32記載の発現抑制方法。
- 前記遺伝子の発現抑制が、RNA干渉による発現抑制である、請求項31から33のいずれか一項に記載の発現抑制方法。
- in vitroにおいて、標的遺伝子の発現を抑制する方法であって、
請求項1から27のいずれか一項に記載の一本鎖RNA核酸分子を使用することを特徴とする発現抑制方法。 - 非ヒト動物において、標的遺伝子の発現を抑制するRNA干渉を誘導する方法であって、
請求項1から27のいずれか一項に記載の一本鎖RNA核酸分子を使用することを特徴とする発現誘導方法。 - in vitroにおいて、標的遺伝子の発現を抑制するRNA干渉を誘導する方法であって、
請求項1から27のいずれか一項に記載の一本鎖RNA核酸分子を使用することを特徴とする発現誘導方法。 - 非ヒト動物の疾患の治療方法であって、
請求項1から27のいずれか一項に記載の一本鎖RNA核酸分子を、非ヒト動物に投与する工程を含み、
前記一本鎖RNA核酸分子が、前記発現抑制配列として、前記疾患の原因となる遺伝子の発現を抑制する配列を有することを特徴とする治療方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011537085A JP4965745B2 (ja) | 2010-08-03 | 2011-07-28 | 含窒素脂環式骨格を有する一本鎖核酸分子 |
Applications Claiming Priority (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010174915 | 2010-08-03 | ||
| JP2010174915 | 2010-08-03 | ||
| JP2010230806 | 2010-10-13 | ||
| JP2010230806 | 2010-10-13 | ||
| JP2010269823 | 2010-12-02 | ||
| JP2010269823 | 2010-12-02 | ||
| JP2011152381 | 2011-07-08 | ||
| JP2011152381 | 2011-07-08 | ||
| PCT/JP2011/067292 WO2012017919A1 (ja) | 2010-08-03 | 2011-07-28 | 含窒素脂環式骨格を有する一本鎖核酸分子 |
| JP2011537085A JP4965745B2 (ja) | 2010-08-03 | 2011-07-28 | 含窒素脂環式骨格を有する一本鎖核酸分子 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012026878A Division JP5261677B2 (ja) | 2010-08-03 | 2012-02-10 | 含窒素脂環式骨格を有する一本鎖核酸分子 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP4965745B2 true JP4965745B2 (ja) | 2012-07-04 |
| JPWO2012017919A1 JPWO2012017919A1 (ja) | 2013-10-03 |
Family
ID=45559420
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011537085A Active JP4965745B2 (ja) | 2010-08-03 | 2011-07-28 | 含窒素脂環式骨格を有する一本鎖核酸分子 |
| JP2012026878A Active JP5261677B2 (ja) | 2010-08-03 | 2012-02-10 | 含窒素脂環式骨格を有する一本鎖核酸分子 |
| JP2013048777A Active JP5555346B2 (ja) | 2010-08-03 | 2013-03-12 | 含窒素脂環式骨格を有する一本鎖核酸分子 |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012026878A Active JP5261677B2 (ja) | 2010-08-03 | 2012-02-10 | 含窒素脂環式骨格を有する一本鎖核酸分子 |
| JP2013048777A Active JP5555346B2 (ja) | 2010-08-03 | 2013-03-12 | 含窒素脂環式骨格を有する一本鎖核酸分子 |
Country Status (10)
| Country | Link |
|---|---|
| EP (4) | EP2436767B1 (ja) |
| JP (3) | JP4965745B2 (ja) |
| KR (3) | KR101849801B1 (ja) |
| CN (3) | CN103052711B (ja) |
| DK (1) | DK2674494T3 (ja) |
| ES (4) | ES2533129T3 (ja) |
| HK (2) | HK1189910A1 (ja) |
| PT (1) | PT2674494E (ja) |
| TW (2) | TWI572716B (ja) |
| WO (1) | WO2012017919A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018182008A1 (ja) | 2017-03-31 | 2018-10-04 | 株式会社ボナック | 遺伝子発現制御機能を有する環状型核酸分子 |
| WO2021070494A1 (ja) | 2019-10-11 | 2021-04-15 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021153770A1 (en) | 2020-01-29 | 2021-08-05 | Sumitomo Chemical Company, Limited | Process of preparing nucleic acid oligomer |
| WO2021153047A1 (ja) | 2020-01-29 | 2021-08-05 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021193954A1 (ja) | 2020-03-27 | 2021-09-30 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2022009959A1 (ja) | 2020-07-09 | 2022-01-13 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2022064908A1 (ja) | 2020-09-24 | 2022-03-31 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
Families Citing this family (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8691782B2 (en) | 2010-08-03 | 2014-04-08 | Bonac Corporation | Single-stranded nucleic acid molecule having nitrogen-containing alicyclic skeleton |
| WO2013077446A1 (ja) * | 2011-11-26 | 2013-05-30 | 株式会社ボナック | 遺伝子発現制御のための一本鎖核酸分子 |
| CN108715594B (zh) * | 2012-03-04 | 2020-12-04 | 株式会社博纳克 | 微小rna抑制剂 |
| WO2013133393A1 (ja) * | 2012-03-07 | 2013-09-12 | 学校法人東京医科大学 | Vegf遺伝子の発現抑制用一本鎖核酸分子 |
| EP2832860A4 (en) * | 2012-03-29 | 2016-01-27 | Univ Kyushu Nat Univ Corp | NUCLEIC ACID MOLECULE CAPABLE OF INHIBITING EXPRESSION OF PERIOSTINE GENE, METHOD FOR INHIBITING PERIOSTINE EXPRESSION AND USE OF SAID NUCLEIC ACID MOLECULE |
| US9663784B2 (en) | 2012-05-26 | 2017-05-30 | Bonac Corporation | Single-stranded nucleic acid molecule for regulating expression of gene having delivering function |
| WO2015046451A1 (ja) * | 2013-09-27 | 2015-04-02 | 株式会社アクアセラピューティクス | 眼科疾患を除くペリオスチン発現に起因する疾患用医薬、およびその用途 |
| WO2015093495A1 (ja) * | 2013-12-16 | 2015-06-25 | 株式会社ボナック | TGF-β1遺伝子発現制御のための一本鎖核酸分子 |
| WO2015099122A1 (ja) | 2013-12-26 | 2015-07-02 | 学校法人東京医科大学 | 遺伝子発現制御のための人工ミミックmiRNAおよびその用途 |
| RU2697094C2 (ru) | 2013-12-27 | 2019-08-12 | Бонак Корпорейшн | ИСКУССТВЕННАЯ мкРНК С СООТВЕТСТВИЕМ ДЛЯ КОНТРОЛЯ ЭКСПРЕССИИ ГЕНОВ И ЕЕ ПРИМЕНЕНИЕ |
| JP6492014B2 (ja) * | 2013-12-27 | 2019-03-27 | 株式会社ボナック | 遺伝子発現制御のための人工マッチ型miRNAおよびその用途 |
| US10337009B2 (en) * | 2014-12-15 | 2019-07-02 | Bonac Corporation | Single-stranded nucleic acid molecule for inhibiting TGF-β1 expression |
| KR20170098914A (ko) * | 2014-12-27 | 2017-08-30 | 가부시키가이샤 보낙 | 유전자 발현 제어를 위한 천연형 miRNA 및 그의 용도 |
| WO2016158809A1 (ja) | 2015-03-27 | 2016-10-06 | 株式会社ボナック | デリバリー機能と遺伝子発現制御能を有する一本鎖核酸分子 |
| WO2016167366A1 (ja) * | 2015-04-17 | 2016-10-20 | 国立大学法人 東京大学 | 眼疾患治療剤 |
| US10751426B2 (en) * | 2015-10-30 | 2020-08-25 | Bonac Corporation | Composition stably containing single-stranded nucleic acid molecule that suppresses expression of TGF-β1 gene |
| WO2017115872A1 (ja) * | 2015-12-29 | 2017-07-06 | 国立大学法人北海道大学 | プロレニン遺伝子またはプロレニン受容体遺伝子の発現を抑制する一本鎖核酸分子およびその用途 |
| BR112018015164A2 (pt) | 2016-01-26 | 2018-12-26 | Nissan Chemical Corp | oligonucleotídeo de fita simples |
| JP6800171B2 (ja) * | 2016-01-30 | 2020-12-16 | 株式会社ボナック | 人工単一ガイドrna及びその用途 |
| CN109071579B (zh) * | 2016-04-26 | 2021-05-14 | 住友化学株式会社 | 单链核酸分子用单体的制造方法 |
| MX2019009305A (es) * | 2017-02-06 | 2019-09-19 | Nissan Chemical Corp | Oligonucleotido de cadena simple. |
| WO2018199338A1 (ja) * | 2017-04-27 | 2018-11-01 | 国立大学法人広島大学 | B型肝炎治療用核酸分子 |
| WO2019022257A1 (ja) * | 2017-07-28 | 2019-01-31 | 杏林製薬株式会社 | 線維症治療剤 |
| US20210188895A1 (en) * | 2017-10-13 | 2021-06-24 | Bonac Corporation | Single-stranded nucleic acid molecule, and production method therefor |
| US20210040525A1 (en) | 2018-02-09 | 2021-02-11 | Sumitomo Chemical Company, Limited | Manufacturing method for nucleic acid molecule |
| BR112020018902A2 (pt) | 2018-03-20 | 2021-01-26 | Tokyo Institute Of Technology | oligonucleotídeo antissenso com toxicidade reduzida |
| EP3778886A4 (en) | 2018-03-30 | 2022-11-02 | Toray Industries, Inc. | METHOD FOR PRODUCING A HAIRPIN SINGLE-STRANDED RNA MOLECULE |
| EP3778914A4 (en) * | 2018-03-30 | 2022-01-19 | Sumitomo Chemical Company, Ltd. | PROCEDURE FOR DELIVERING SINGLE STRAND RNA |
| US11891602B2 (en) | 2018-10-02 | 2024-02-06 | Toray Industries, Inc. | Method of producing hairpin single-stranded RNA molecules |
| JP7241098B2 (ja) | 2019-01-25 | 2023-03-16 | 杏林製薬株式会社 | 線維症治療剤 |
| CN113631264A (zh) | 2019-03-29 | 2021-11-09 | 住友化学株式会社 | 无机多孔质担载体及使用其的核酸的制造方法 |
| US20220177319A1 (en) | 2019-03-29 | 2022-06-09 | Sumitomo Chemical Company, Limited | Inorganic porous support and method for producing nucleic acid using same |
| JPWO2020202949A1 (ja) | 2019-03-29 | 2020-10-08 | ||
| WO2020202952A1 (ja) | 2019-03-29 | 2020-10-08 | 住友化学株式会社 | 無機多孔質担体、及びこれを用いた核酸の製造方法 |
| US20220106347A1 (en) | 2019-03-29 | 2022-04-07 | Sumitomo Chemical Company, Limited | Porous inorganic carrier and method for producing nucleic acid using same |
| WO2021230293A1 (ja) | 2020-05-13 | 2021-11-18 | 住友化学株式会社 | 無機多孔質基材、無機多孔質担体、および核酸の製造方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004015075A2 (en) * | 2002-08-08 | 2004-02-19 | Dharmacon, Inc. | Short interfering rnas having a hairpin structure containing a non-nucleotide loop |
| WO2004090108A2 (en) * | 2003-04-03 | 2004-10-21 | Alnylam Pharmaceuticals | Irna conjugates |
| WO2005019453A2 (en) * | 2001-05-18 | 2005-03-03 | Sirna Therapeutics, Inc. | RNA INTERFERENCE MEDIATED INHIBITION OF GENE EXPRESSION USING CHEMICALLY MODIFIED SHORT INTERFERING NUCLEIC ACID (siNA) |
| WO2008116094A2 (en) * | 2007-03-21 | 2008-09-25 | Brookhaven Science Associates, Llc | Combined hairpin-antisense compositions and methods for modulating expression |
| WO2009073809A2 (en) * | 2007-12-04 | 2009-06-11 | Alnylam Pharmaceuticals, Inc. | Carbohydrate conjugates as delivery agents for oligonucleotides |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| GB9621367D0 (en) * | 1996-10-14 | 1996-12-04 | Isis Innovation | Chiral peptide nucleic acids |
| US6509323B1 (en) | 1998-07-01 | 2003-01-21 | California Institute Of Technology | Linear cyclodextrin copolymers |
| US6962906B2 (en) * | 2000-03-14 | 2005-11-08 | Active Motif | Oligonucleotide analogues, methods of synthesis and methods of use |
| TWI321054B (en) | 2000-12-19 | 2010-03-01 | California Inst Of Techn | Compositions containing inclusion complexes |
| US20060111312A1 (en) * | 2002-02-22 | 2006-05-25 | The John Hopkins University | Antigene locks and therapeutic uses thereof |
| NL1022811C2 (nl) * | 2003-02-28 | 2004-08-31 | Fountain Patents B V | Systeem en werkwijze voor opslag en transport van zeecontainers. |
| WO2005030960A1 (ja) * | 2003-09-30 | 2005-04-07 | Anges Mg, Inc. | ステイプル型オリゴヌクレオチドおよびそれからなる医薬 |
| EP1789553B1 (en) * | 2004-06-30 | 2014-03-26 | Alnylam Pharmaceuticals Inc. | Oligonucleotides comprising a non-phosphate backbone linkage |
| EP1838875A4 (en) * | 2004-12-30 | 2010-08-25 | Todd M Hauser | COMPOSITIONS AND METHODS FOR MODULATING GENE EXPRESSION USING SELF-PROTECTED OLIGONUCLEOTIDES |
| WO2009074076A1 (fr) * | 2007-11-29 | 2009-06-18 | Suzhou Ribo Life Science Co., Ltd | Molécule complexe interférant avec l'expression de gènes cibles, et ses méthodes de préparation |
| US20110159586A1 (en) * | 2007-12-07 | 2011-06-30 | Halo-Bio Rnai Therapeutics, Inc. | Compositions and methods for modulating gene expression using asymmetrically-active precursor polynucleotides |
| JP2010174915A (ja) | 2009-01-27 | 2010-08-12 | Toyota Motor Corp | 高圧ガスタンク |
| JP5449815B2 (ja) | 2009-03-26 | 2014-03-19 | 住友化学株式会社 | 偏光板の製造方法 |
| JP2010269823A (ja) | 2009-05-21 | 2010-12-02 | British American Tobacco Japan Kk | 喫煙品用容器 |
| JP2011152381A (ja) | 2010-01-25 | 2011-08-11 | Ucom:Kk | 箸置き |
-
2011
- 2011-07-28 WO PCT/JP2011/067292 patent/WO2012017919A1/ja active Application Filing
- 2011-07-28 KR KR1020137004646A patent/KR101849801B1/ko active IP Right Grant
- 2011-07-28 PT PT131841785T patent/PT2674494E/pt unknown
- 2011-07-28 EP EP11748250.5A patent/EP2436767B1/en active Active
- 2011-07-28 EP EP13167541.5A patent/EP2639307B1/en active Active
- 2011-07-28 EP EP13184178.5A patent/EP2674494B1/en active Active
- 2011-07-28 KR KR1020187010010A patent/KR101894701B1/ko active IP Right Grant
- 2011-07-28 CN CN201180037592.9A patent/CN103052711B/zh active Active
- 2011-07-28 ES ES13167541.5T patent/ES2533129T3/es active Active
- 2011-07-28 ES ES13167536.5T patent/ES2528285T3/es active Active
- 2011-07-28 CN CN201410217071.7A patent/CN104004018B/zh active Active
- 2011-07-28 JP JP2011537085A patent/JP4965745B2/ja active Active
- 2011-07-28 ES ES13184178.5T patent/ES2527660T3/es active Active
- 2011-07-28 ES ES11748250.5T patent/ES2443346T3/es active Active
- 2011-07-28 EP EP13167536.5A patent/EP2628801B1/en active Active
- 2011-07-28 CN CN201410217512.3A patent/CN104059911B/zh active Active
- 2011-07-28 KR KR1020187010011A patent/KR101894702B1/ko active IP Right Grant
- 2011-07-28 DK DK13184178.5T patent/DK2674494T3/en active
- 2011-08-02 TW TW104132132A patent/TWI572716B/zh active
- 2011-08-02 TW TW100127384A patent/TWI515294B/zh active
-
2012
- 2012-02-10 JP JP2012026878A patent/JP5261677B2/ja active Active
-
2013
- 2013-03-12 JP JP2013048777A patent/JP5555346B2/ja active Active
-
2014
- 2014-03-26 HK HK14102959.9A patent/HK1189910A1/xx not_active IP Right Cessation
-
2015
- 2015-01-28 HK HK15100974.3A patent/HK1200460A1/xx unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005019453A2 (en) * | 2001-05-18 | 2005-03-03 | Sirna Therapeutics, Inc. | RNA INTERFERENCE MEDIATED INHIBITION OF GENE EXPRESSION USING CHEMICALLY MODIFIED SHORT INTERFERING NUCLEIC ACID (siNA) |
| WO2004015075A2 (en) * | 2002-08-08 | 2004-02-19 | Dharmacon, Inc. | Short interfering rnas having a hairpin structure containing a non-nucleotide loop |
| WO2004090108A2 (en) * | 2003-04-03 | 2004-10-21 | Alnylam Pharmaceuticals | Irna conjugates |
| WO2008116094A2 (en) * | 2007-03-21 | 2008-09-25 | Brookhaven Science Associates, Llc | Combined hairpin-antisense compositions and methods for modulating expression |
| WO2009073809A2 (en) * | 2007-12-04 | 2009-06-11 | Alnylam Pharmaceuticals, Inc. | Carbohydrate conjugates as delivery agents for oligonucleotides |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018182008A1 (ja) | 2017-03-31 | 2018-10-04 | 株式会社ボナック | 遺伝子発現制御機能を有する環状型核酸分子 |
| WO2021070494A1 (ja) | 2019-10-11 | 2021-04-15 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021153770A1 (en) | 2020-01-29 | 2021-08-05 | Sumitomo Chemical Company, Limited | Process of preparing nucleic acid oligomer |
| WO2021153047A1 (ja) | 2020-01-29 | 2021-08-05 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2021193954A1 (ja) | 2020-03-27 | 2021-09-30 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2022009959A1 (ja) | 2020-07-09 | 2022-01-13 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
| WO2022064908A1 (ja) | 2020-09-24 | 2022-03-31 | 住友化学株式会社 | 核酸オリゴマーの製造方法 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5555346B2 (ja) | 含窒素脂環式骨格を有する一本鎖核酸分子 | |
| JP6162182B2 (ja) | 遺伝子発現制御のための一本鎖核酸分子 | |
| JP5876890B2 (ja) | アミノ酸骨格を有する一本鎖核酸分子 | |
| US9206422B2 (en) | Single-stranded nucleic acid molecule having nitrogen-containing alicyclic skeleton | |
| US8785121B2 (en) | Single-stranded nucleic acid molecule for controlling gene expression | |
| WO2013077446A1 (ja) | 遺伝子発現制御のための一本鎖核酸分子 | |
| JP2013153736A (ja) | ペプチド骨格を有する一本鎖核酸分子 | |
| JP2013055913A (ja) | 遺伝子発現制御のための一本鎖rna分子 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120321 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120329 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4965745 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150406 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |