JP4835159B2 - ポリオレフィン系樹脂の結晶化速度制御のための組成物及び方法、樹脂組成物及び樹脂成形体 - Google Patents
ポリオレフィン系樹脂の結晶化速度制御のための組成物及び方法、樹脂組成物及び樹脂成形体 Download PDFInfo
- Publication number
- JP4835159B2 JP4835159B2 JP2005516714A JP2005516714A JP4835159B2 JP 4835159 B2 JP4835159 B2 JP 4835159B2 JP 2005516714 A JP2005516714 A JP 2005516714A JP 2005516714 A JP2005516714 A JP 2005516714A JP 4835159 B2 JP4835159 B2 JP 4835159B2
- Authority
- JP
- Japan
- Prior art keywords
- temperature
- molding
- resin
- polyolefin
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- UAEPNZWRGJTJPN-UHFFFAOYSA-N CC1CCCCC1 Chemical compound CC1CCCCC1 UAEPNZWRGJTJPN-UHFFFAOYSA-N 0.000 description 1
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/09—Carboxylic acids; Metal salts thereof; Anhydrides thereof
- C08K5/098—Metal salts of carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/18—Oxygen-containing compounds, e.g. metal carbonyls
- C08K3/20—Oxides; Hydroxides
- C08K3/22—Oxides; Hydroxides of metals
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/0008—Organic ingredients according to more than one of the "one dot" groups of C08K5/01 - C08K5/59
- C08K5/0083—Nucleating agents promoting the crystallisation of the polymer matrix
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/20—Carboxylic acid amides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Manufacture Of Macromolecular Shaped Articles (AREA)
- Injection Moulding Of Plastics Or The Like (AREA)
- Extrusion Moulding Of Plastics Or The Like (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
Description
これらの問題点を改善するために、アミド系化合物等を造核剤として活用する技術が提案されている(特開平6−192496号、特開平7−242610号、特開平7−278374号、特開平8−100088号)。これらアミド系化合物を含有するポリオレフィン系樹脂組成物を成形した場合、透明性、機械強度に優れた成形体を得ることができる。
しかしながら、商業的な生産においてそのような優れた特性を有する成形体を得るためには、成形方法に合わせて、成形加工条件を最適化する必要があった。
成形方法には、射出成形、押出成形、シート成形、フィルム成形、ブロー成形などの種々の成形方法がある。成形体製造メーカーは、独自に、成形方法に合わせて成形機の成形加工条件(射出または押出速度、射出圧力、樹脂温度、金型及びチルロールの温度、金型形状など)を設定し、成形加工条件の最適化を図らなければならない。
しかしながら、その成形加工条件の最適化は、設定項目が多い上に、その設定作業が繁雑であり、時間を要するものであった。しかも、その設定を誤ると、成形体中に造核剤の未分散物に基づく白点が生じる、成形体の透明性が劣る、成形体の機械的強度が劣る等の問題が生じることもあった。
また、或る特定の核剤を使用すると、ポリオレフィン系樹脂の結晶化温度がある程度高くなり、成形サイクルタイムが短縮できることも知られている。しかし、かかる核剤の使用は、その核剤の使用に特有の結晶化温度がもたらされるだけであって、結晶化温度が制御できるわけではないため、成形加工条件の設定の簡便化、迅速化、成形加工条件の柔軟化、多様化等には大きく寄与していないのが現状である。
本発明者らは、かかる現状に鑑み、前記課題を解決すべく鋭意検討した。特に、本発明者らは、成形条件の設定について検討した結果、その条件設定の煩雑さが、かなりの程度、ポリオレフィン系樹脂の結晶化速度に起因しており、成形機による成形条件の設定のみに依存することなく、ポリオレフィン系樹脂の結晶化速度を制御できれば、成形条件設定が簡便かつ迅速となり、また、成形加工条件を柔軟化、多様化できることを見いだした。
より詳しくは、成形方法や成形すべき樹脂によっては、その結晶化速度が速い故に成形加工が困難になるという逆効果になることがある。例えば、ポリオレフィン系樹脂の結晶化速度が適切な範囲内になければ、フィルム又はシート成形、ブロー成形、大型製品の射出成形などでは均質な成形体を得られにくくなる。しかし、ポリオレフィン系樹脂の結晶化速度を低減させて上記適切な範囲内にするためには、成形機での成形条件(例えば、樹脂の排出速度、排出量、射出成形時の背圧、チルロール回転速度、金型又はチルロール温度、金型形状、冷却時間、冷却速度等)の煩雑な設定作業に依存しなければならない。一方では、小型製品の射出成形などでは、生産コストをできるだけ低減させる為に、ポリオレフィン系樹脂の結晶化速度をさらに高めて成形サイクルタイムを短縮すること(高速成形)ができれば、工業上きわめて有利である。しかし、ポリオレフィン系樹脂の成形サイクルタイムを短縮しようとしても、従来は、ある種の核剤が結晶化温度を高めることが知られているだけであり、結晶化速度を高めるには、やはり成形機での成形条件(例えば、冷却時間、冷却速度等)の煩雑な設定作業に依存せざるを得ない。
かかる状況に鑑み、本発明者らは、ポリオレフィン系樹脂の結晶化速度が制御された樹脂組成物が提供できれば、煩雑な成形機での成形条件設定にのみに依存せずに、広範囲の成形加工に対応することが可能となると考えた。
本発明者らは、更に検討を重ねた結果、ポリオレフィン系樹脂に(A)特定のアミド系化合物と(B)特定の脂肪酸金属塩とを特定の重量比で配合することにより、該ポリオレフィン系樹脂の結晶化速度(結晶化終了時間)を制御できることを見出した。より詳しくは、次の知見を得た。
(a) 特定のアミド系化合物(A)が、溶解型の核剤である。
(b) ポリオレフィン系樹脂中に該アミド系化合物(A)を溶解させて得られる溶融ポリオレフィン系樹脂組成物の温度変化に対する貯蔵弾性率の変化を測定すると、昇温時及び降温時のいずれの曲線においても転移点があり、貯蔵弾性率の温度依存曲線の微分曲線が極値を示す。
(c) 上記(b)の知見は、特定の温度範囲において、溶融ポリオレフィン系樹脂中に上記アミド系化合物の繊維状粒子により形成される網目構造が存在することを示す。このことは、従来全く知られておらず、本発明者らにより初めて見いだされた知見である。
(d) 成形加工時の樹脂温度(成形温度)Tを、昇温時の貯蔵弾性率の転移温度Tsh以下の温度とするか、又はTshよりも高い温度に設定し、且つ、アミド系化合物(A)と特定の脂肪酸金属塩(B)とを特定割合で併用すると、該ポリオレフィン系樹脂の結晶化速度(結晶化終了時間)を制御できる。
(e) より詳しくは、図8に示すように、成形加工時の樹脂温度(成形温度)Tを、ポリオレフィン系樹脂の融解温度Tm以上であって、且つ、昇温時の貯蔵弾性率の転移温度Tsh以下の温度に設定する場合(図8の成形方法(I))、該特定の脂肪酸金属塩(B)の配合比率を増加させると(領域(IA)→領域(IAB))、ポリオレフィン系樹脂の結晶化速度を速くする(結晶化終了時間を短縮させる)ことができる。
(f) また、成形加工時の樹脂温度(成形温度)Tを、昇温時の貯蔵弾性率の転移温度Tshを超える温度に設定する場合(図8の成形方法(II))、該特定の脂肪酸金属塩(B)の配合比率を増加させると(領域(IIA)→領域(IIAB))、ポリオレフィン系樹脂の結晶化速度を遅くする(結晶化終了時間を延長させる)ことができる。
(g) 即ち、昇温時の貯蔵弾性率の転移温度Tsh以下の温度領域と、Tshを超える温度領域とからなる上記成形温度範囲の全体を通して見れば、また、Tm〜Tshの樹脂温度範囲、又は、Tshよりも高い樹脂温度範囲のそれぞれにおいても、特定の脂肪酸金属塩の配合比の調整により、前記(f)に記載の成形方法(II)において特定のアミド系化合物のみを含み脂肪酸金属塩を含まない結晶化速度制御組成物により達成される結晶化速度に比べて、ポリオレフィン系樹脂の結晶化速度を、制御、即ち、低減又は増加することができることとなる。
(h) 上記(e)の成形方法(I)は、結晶化速度を速くすることができ、そのため、核剤の元来の目的である成形サイクル時間の短縮が更に可能となり、主に小型製品の高速度での射出成形に適している。この場合、昇温時の貯蔵弾性率の転移温度Tsh以下の樹脂温度で成形するので、図7の(I)に示すように、上記(c)の網目構造が存在した状態で成形され、該網目構造を構成する繊維状粒子が配向し、そのため、得られる成形体においてポリオレフィン系樹脂の結晶ラメラを配向させることができ、その結果、特に剛性に優れた成形体を製造することができる。そして、脂肪酸金属塩(B)の割合を増減することにより、ポリオレフィン系樹脂の結晶化速度を制御できる。
(i) 一方、上記(f)の成形方法(II)は、結晶化速度を遅くすることができ、均質な成形体が得られやすくなるので、フィルム成形、シート成形、大型製品の射出成形において有利である。この場合、昇温時の貯蔵弾性率の転移温度Tshを超える温度で成形するので、図7の(II)に示すように、上記(c)の網目構造が溶解して消失した状態で成形が行われる。そのため、その溶融したポリオレフィン系樹脂組成物を冷却すると、アミド系化合物の纎維状粒子が形成され、網目構造が再構成される。それを起点として、ポリオレフィン系樹脂の微少な結晶(球晶)が形成され、その結果、特に透明性に優れた成形体を製造できる。そして、脂肪酸金属塩(B)の割合を増減することにより、ポリオレフィン系樹脂の結晶化速度を制御できる。
(j)このように、本発明によると、ポリオレフィン系樹脂の結晶化速度を制御する(変化させる)ことができるが、前記アミド系化合物の他の核剤機能は実質的に変化させない。従って、本発明によれば、成形装置の条件設定による結晶化速度制御に加えて、本発明の結晶化速度制御組成物によっても結晶化速度が制御できるため、成形工程での条件設定の自由度が大きく拡大され、その結果、優れた物性を有する成形体を容易に製造することができる。
本発明は、かかる知見に基づき、更に検討を重ねて完成されたものであって、以下のポリオレフィン系樹脂の結晶化速度制御組成物、該結晶化速度制御組成物を含有してなる樹脂組成物、該樹脂組成物を成形して得られる成形体及びその製造方法、並びにポリオレフィン系樹脂の結晶化速度を制御する方法及び該制御方法を利用したポリオレフィン系樹脂成形体の製造方法を提供するものである。
項1 (A)一般式(1)

で表される少なくとも一種のアミド系化合物、及び
(B)一般式(2)
で表される少なくとも一種の脂肪酸金属塩を含有し、成分(A):成分(B)の重量比が、100:0〜30:70である(即ち、成分(A)及び成分(B)の合計量に対して、成分(A)を100〜30重量%、成分(B)を0〜70重量%含有している)ポリオレフィン系樹脂の結晶化速度制御組成物。
項2 成分(A):成分(B)の重量比が、95:5〜30:70(即ち、成分(A)及び成分(B)の合計量に対して、成分(A)を95〜30重量%、成分(B)を5〜70重量%含有している)である上記項1に記載の組成物。
項3 一般式(1)における3個又は4個のR2が、同一又は異なって、シクロヘキシル基、又は1個の炭素数1〜4のアルキル基で置換されたシクロヘキシル基である上記項1又は項2に記載の組成物。
項4 一般式(1)における3個又は4個のR2が、同一又は異なって、シクロヘキシル基又は2−メチル基、3−メチル基若しくは4−メチル基で置換されたシクロヘキシル基である上記項1に記載の組成物。
項5 一般式(1)におけるR1が1,2,3−プロパントリカルボン酸から全てのカルボキシル基を除いて得られる残基であり、kが3である上記項1〜4のいずれかに記載の組成物。
項6 一般式(2)におけるMが、アルカリ金属、アルカリ土類金属及び亜鉛からなる群より選ばれる少なくとも1種の金属である上記項1〜5のいずれかに記載の組成物。
項7 一般式(2)におけるR3が、分子内に1個以上の水酸基を有していてもよい炭素数10〜18の飽和若しくは不飽和の脂肪族モノカルボン酸からカルボキシル基を除いて得られる残基である上記項1〜6のいずれかに記載の組成物。
項8 脂肪族モノカルボン酸が、ラウリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、オレイン酸及び12−ヒドロキシステアリン酸からなる群より選ばれる少なくとも1種である上記項7に記載の組成物。
項9 ポリオレフィン系樹脂の成形時のポリオレフィン系樹脂の結晶化速度を制御する方法であって、
(A)一般式(1)
で表される少なくとも一種のアミド系化合物、及び
(B)一般式(2)
で表される少なくとも一種の脂肪酸金属塩を含有し、成分(A):成分(B)の重量比が100:0〜30:70である(即ち、成分(A)及び成分(B)の合計量に対して、成分(A)を100〜30重量%、成分(B)を0〜70重量%含有している)ポリオレフィン系樹脂結晶化速度制御組成物を該ポリオレフィン系樹脂に配合するか、又は、上記成分(A)と成分(B)とを同時に又は別々に、成分(A):成分(B)の重量比が100:0〜30:70となるように(即ち、成分(A)及び成分(B)の合計量に対して、成分(A)が100〜30重量%、成分(B)が0〜70重量%となるように)、該ポリオレフィン系樹脂に配合することにより、ポリオレフィン系樹脂組成物を得ること、及び
該樹脂組成物を成形すること
を含む方法。
項10 成分(A):成分(B)の重量比が、95:5〜30:70である(即ち、成分(A)及び成分(B)の合計量に対して、成分(A)が95〜30重量%使用され、成分(B)が5〜70重量%使用される)上記項9に記載の方法。
項11 樹脂組成物を、昇温時の貯蔵弾性率の転移温度を超える樹脂温度で成形する上記項9又は項10に記載の方法。
項12 樹脂組成物を、ポリオレフィン系樹脂の融解温度以上であって、且つ、昇温時の貯蔵弾性率の転移温度以下の樹脂温度で、成形(特に、射出工程又は押し出し工程を含む成形法で成形)する上記項9又は項10に記載の方法。
項13 (A)一般式(1)
kは3又4の整数を表す。3個又は4個のR2は互いに同一又は異なって、シクロヘキシル基又は1個の炭素数1〜10の直鎖状若しくは分岐鎖状のアルキル基で置換されたシクロヘキシル基を表す。]
で表される少なくとも一種のアミド系化合物、及び
(B)一般式(2)
で表される少なくとも一種の脂肪酸金属塩を、成分(A):成分(B)の重量比が100:0〜30:70の割合で含有する(即ち、成分(A)及び成分(B)の合計量に対して、成分(A)を100〜30重量%、成分(B)を0〜70重量%含有している)組成物の、ポリオレフィン系樹脂の成形時のポリオレフィン系樹脂の結晶化速度を制御するための使用。
項14 成分(A):成分(B)の重量比が、95:5〜30:70である(即ち、成分(A)及び成分(B)の合計量に対して、成分(A)が95〜30重量%の量で存在し、成分(B)が5〜70重量%の量で存在している)上記項13に記載の使用。
項15 (A)一般式(1)
で表される少なくとも一種のアミド系化合物、及び
(B)一般式(2)
で表される少なくとも一種の脂肪酸金属塩を含有し、成分(A):成分(B)の重量比が100:0〜30:70である(即ち、成分(A)及び成分(B)の合計量に対して、成分(A)を100〜30重量%、成分(B)を0〜70重量%含有している)ポリオレフィン系樹脂の結晶化速度制御組成物をポリオレフィン系樹脂に配合するか、又は、上記成分(A)と成分(B)とを、同時にまたは別々に、成分(A):成分(B)の重量比が100:0〜30:70となるように(即ち、成分(A)及び成分(B)の合計量に対して、成分(A)が100〜30重量%、成分(B)が0〜70重量%となるように)、ポリオレフィン系樹脂に配合することにより、ポリオレフィン系樹脂組成物を得ること、及び
該樹脂組成物を成形すること
を包含するポリオレフィン系樹脂成形体の製造方法。
項16 成分(A):成分(B)の重量比が、95:5〜30:70である(即ち、成分(A)及び成分(B)の合計量に対して、成分(A)が95〜30重量%使用され、成分(B)が5〜70重量%使用される)上記項15に記載の製造方法。
項17 樹脂組成物を、昇温時の貯蔵弾性率の転移温度を超える樹脂温度で成形する項15又は項16に記載の製造方法。
項18 樹脂組成物を、ポリオレフィン系樹脂の融解温度以上であって、且つ、昇温時の貯蔵弾性率の転移温度以下の樹脂温度で成形する項15又は16に記載の製造方法。
項19 一般式(1−p)
で表される少なくとも1種のアミド系化合物の繊維状粒子により形成された網目構造を含む溶融ポリオレフィン系樹脂組成物を、該網目構造を構成する繊維状粒子が溶解又は溶融しない温度条件で、成形する工程を備えているポリオレフィン系樹脂成形体の製造法(又は項18に記載の製造法)。
項20 (a) 一般式(1−p)で表される少なくとも1種のアミド系化合物を溶融ポリオレフィン系樹脂に溶解して溶融混合物を得る工程、
(b) 該溶融混合物を、降温時の貯蔵弾性率の転移温度以下に冷却して、一般式(1−p)で表される少なくとも1種のアミド系化合物の繊維状粒子により形成される網目構造を含むポリオレフィン系樹脂組成物を得る工程、及び
(c) 該ポリオレフィン系樹脂組成物を、ポリオレフィン系樹脂の溶融温度以上であって、且つ、昇温時の貯蔵弾性率の転移温度以下の樹脂温度で成形する工程を備えている項19に記載の製造法。
項21 上記ポリオレフィン系樹脂組成物が、ペレットの形態にある項20に記載の製造法。
項22 上記ポリオレフィン系樹脂組成物が、更に、一般式(2)
で表される少なくとも1種の脂肪酸金属塩を含有する項19〜21のいずれかに記載の製造法。
項23 上記繊維状粒子により形成される網目構造を含むポリオレフィン系樹脂組成物を、射出工程又は押し出し工程を含む成形方法により成形する項19〜22のいずれかに記載の製造法。
項24 前記射出工程又は押し出し工程を含む成形方法が、射出成形、押し出し成形、射出ブロー成形、射出押出ブロー成形、射出圧縮成形、押し出しブロー成形、押し出しサーモフォーム成形又は溶融紡糸である項23に記載の製造法。
項25 上記ポリオレフィン系樹脂が、プロピレンホモポリマー及びプロピレンコポリマーから選ばれる少なくとも1種である項15〜24のいずれかに記載の製造法。
項26 上記項18又は項19に記載の製造法により得られ、広角X線回折により求められる(040)反射強度の(110)反射強度に対する比で表される配向度が2以上であるポリオレフィン系樹脂成形体。
項27 ポリオレフィン系樹脂、
(A)一般式(1)
で表される少なくとも一種のアミド系化合物、及び
(B)一般式(2)
で表される少なくとも一種の脂肪酸金属塩を、成分(A):成分(B)の重量比が、100:0〜30:70の比率の範囲で含有し、(即ち、成分(A)及び成分(B)の合計量に対して、成分(A)が100〜30重量%の量で存在し、成分(B)が0〜70重量%の量で存在しており、)広角X線回折により求められる(040)反射強度の(110)反射強度に対する比で表される配向度が2以上であるポリオレフィン系樹脂成形体。
項28 ポリオレフィン系樹脂、及び
(a) 一般式(1−p)
(b) 上記一般式(1−p)で表される少なくとも1種のアミド系化合物及び一般式(2)
で表される少なくとも1種の脂肪酸金属塩
を含有し、
広角X線回折により求められる(040)反射強度の(110)反射強度に対する比で表される配向度が2以上であるポリオレフィン系樹脂成形体。
項29 ポリオレフィン系樹脂及び上記項1〜8のいずれかに記載の結晶化速度制御組成物を含有するポリオレフィン系樹脂組成物。
項30 ポリオレフィン系樹脂100重量部に対し、結晶化速度制御組成物を0.01〜10重量部含有する上記項29に記載のポリオレフィン系樹脂組成物。
項31 上記項29又は項30に記載のポリオレフィン系樹脂組成物を成形することにより得ることができる(又は得られた)ポリオレフィン系樹脂成形体。
(I) 成形加工時の樹脂温度(成形温度)Tが、ポリオレフィン系樹脂の融解温度Tm以上であって、且つ、昇温時の貯蔵弾性率の転移温度Tsh以下の温度の場合、成分(A):成分(B)=100:0〜30:70(重量比)の範囲において、成分(B)の比率を高めることにより、そのポリオレフィン系樹脂の結晶化速度を速くする(結晶化終了時間を短縮させる)ことができる。
(II) 成形加工時の樹脂温度(成形温度)Tが、昇温時の貯蔵弾性率の転移温度Tshを超える温度の場合、成分(A):成分(B)=100:0〜30:70(重量比)の範囲において、成分(B)の比率を高めることにより、ポリオレフィン系樹脂の結晶化速度を低下させる(結晶化終了時間を延長させる)ことができる。
従って、成形温度範囲の全体を通して見ても、また、Tm〜Tshの樹脂温度範囲、又は、Tshよりも高い樹脂温度範囲のそれぞれにおいても、成分(A):成分(B)の比を、重量比100:0〜30:70の範囲で調整することにより、前記(I)の方法における成分(A):成分(B)=100:0の場合の結晶化速度に比べて、そのポリオレフィン系樹脂の結晶化速度を制御、即ち、増大又は低減できる。
そのため、該結晶化速度制御組成物をポリオレフィン系樹脂に配合することにより、ポリオレフィン系樹脂の成形時の結晶化速度が制御できるので、所望の成形体を得るための成形条件設定のための更なる選択肢が提供される。従って、成形条件設定を、成形装置のみに依存することなく、容易にすることできる。換言すると、本発明は、射出成形、押し出し成形、シート成形、フィルム成形、ブロー成形等の種々の成形方法に好適に且つ柔軟に用いることができるポリオレフィン系樹脂組成物を提供する。
また、こうして得られるポリオレフィン系樹脂組成物を、上記(I)又は(II)の方法で成形することにより、優れた特性の成形品を得ることができる。具体的には、上記(I)の成形加工では、ポリオレフィン系樹脂成形体の剛性の向上に大きく寄与し、特にホモポリプロピレン樹脂、ブロックポリプロピレン樹脂、高密度ポリエチレン樹脂で顕著である。また、上記(II)の成形加工では、ポリオレフィン系樹脂成形体の透明性の向上に大きく寄与し、特にホモポリプロピレン樹脂やランダムポリプロピレン樹脂において顕著である。
図2は、ポリオレフィン系樹脂及び一般式(1)で表されるアミド系化合物を含む樹脂組成物の昇温時(実線)及び降温時(破線)の貯蔵弾性率の温度依存曲線を模式的に示すグラフである。
図3は、図2の温度依存曲線の微分曲線を模式的に示すグラフである。
図4は、実施例及び比較例で使用した試験片のTHROUGH方向、END方向及びEDGE方向を示す斜視図である。
図5は、実施例I−2で得られたポリオレフィン系樹脂組成物について測定された昇温時及び降温時の貯蔵弾性率の温度依存曲線を示すグラフである。白丸(○)は昇温時の貯蔵弾性率の温度依存曲線であり、黒丸(●)は降温時の貯蔵弾性率の温度依存曲線である。
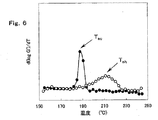
図6は、図5の温度依存曲線の微分曲線である。白丸(○)は昇温時の貯蔵弾性率の温度依存曲線の微分曲線であり、黒丸(●)は降温時の貯蔵弾性率の温度依存曲線の微分曲線である。
図7は、成形体の製造法の各段階におけるポリオレフィン系樹脂組成物の状態を示す概念図である。図7において、(I)は本発明の成形方法(I)の条件を採用した製造法を示す概念図であり、(II)は本発明の成形方法(II)の条件を採用した製造法の概念図である。
図8は、本発明のポリオレフィン系樹脂の結晶化速度制御に関する、本発明の結晶化速度制御組成物の組成と成形加工時の樹脂温度と結晶化速度との関係を示す概念図である。
図面における符号は次の意味を有する。
(a) 延長線
(b) 接線
(c) 交点
1 射出成形機
2 ホッパー
3 金型
4 ノズル
発明の詳細な記載
ポリオレフィン系樹脂の結晶化速度制御組成物
本発明のポリオレフィン系樹脂の結晶化速度制御組成物は、(A)一般式(1)で表される少なくとも一種のアミド系化合物、又は該成分(A)及び(B)一般式(2)で表される少なくとも一種の脂肪酸金属塩を必須成分として含有する。
成分(A):成分(B)の重量比は、広い範囲から適宜選択することできるが、一般には、成分(A):成分(B)の重量比が100:0〜30:70であり、好ましくは95:5〜30:70、より好ましくは90:10〜60:40、特に好ましくは90:10〜70:30の範囲である。
換言すると、本発明のポリオレフィン系樹脂の結晶化速度制御組成物は、成分(A)と成分(B)との合計量に対して、成分(A)を100〜30重量%(特に100重量%未満〜30重量%)、好ましくは95〜30重量%、より好ましくは90〜60重量%、特に好ましくは90〜70重量%、成分(B)を0〜70重量%(特に0重量%超〜70重量%以下)、好ましくは5〜70重量%、より好ましくは10〜40重量%、特に好ましくは10〜30重量%含有する。
成分(A)と成分(B)との合計量に対して、成分(A)の割合が30重量%より小さいと、得られる樹脂成形体の透明性或いは剛性の有意な改善効果が認められにくくなる。
成分(A):アミド系化合物
本発明に係る一般式(1)で表されるアミド系化合物(A)は、脂肪族ポリカルボン酸成分と置換又は非置換シクロヘキシルアミンとを、従来公知の方法、例えば特開平7−242610号に記載の方法に従ってアミド化することにより容易に調製することができる。
上記ポリカルボン酸成分としては、ポリカルボン酸又はその反応性誘導体(典型的には、その酸塩化物、該ポリカルボン酸と炭素数1〜4の低級アルコールとのエステル等の誘導体)を用いることができ、その製造方法には特に限定がなく、いずれの方法で製造されたものであってもよい。
脂肪族ポリカルボン酸としては、1,2,3−プロパントリカルボン酸、1,2,3,4−ブタンテトラカルボン酸であり、特に1,2,3−プロパントリカルボン酸が推奨される。
置換又は非置換シクロヘキシルアミンとしては、炭素数1〜10の直鎖状又は分岐鎖状のアルキル基で置換されていてもよいシクロヘキシルアミンが挙げられ、具体的には、シクロヘキシルアミン、2−メチルシクロヘキシルアミン、2−エチルシクロヘキシルアミン、2−n−プロピルシクロヘキシルアミン、2−iso−プロピルシクロヘキシルアミン、2−n−ブチルシクロヘキシルアミン、2−iso−ブチルシクロヘキシルアミン、2−sec−ブチルシクロヘキシルアミン、2−tert−ブチルシクロヘキシルアミン、2−n−ペンチルシクロヘキシルアミン、2−n−ヘキシルシクロヘキシルアミン、2−n−ヘプチルシクロヘキシルアミン、2−n−オクチルシクロヘキシルアミン、2−(2−エチルヘキシル)シクロヘキシルアミン、2−n−ノニルシクロヘキシルアミン、2−n−デシルシクロヘキシルアミン、3−メチルシクロヘキシルアミン、3−エチルシクロヘキシルアミン、3−n−プロピルシクロヘキシルアミン、3−iso−プロピルシクロヘキシルアミン、3−n−ブチルシクロヘキシルアミン、3−iso−ブチルシクロヘキシルアミン、3−sec−ブチルシクロヘキシルアミン、3−tert−ブチルシクロヘキシルアミン、3−n−ペンチルシクロヘキシルアミン、3−n−ヘキシルシクロヘキシルアミン、3−n−ヘプチルシクロヘキシルアミン、3−n−オクチルシクロヘキシルアミン、3−(2−エチルヘキシル)シクロヘキシルアミン、3−n−ノニルシクロヘキシルアミン、3−n−デシルシクロヘキシルアミン、4−メチルシクロヘキシルアミン、4−エチルシクロヘキシルアミン、4−n−プロピルシクロヘキシルアミン、4−iso−プロピルシクロヘキシルアミン、4−n−ブチルシクロヘキシルアミン、4−iso−ブチルシクロヘキシルアミン、4−sec−ブチルシクロヘキシルアミン、4−tert−ブチルシクロヘキシルアミン、4−n−ペンチルシクロヘキシルアミン、4−n−ヘキシルシクロヘキシルアミン、4−n−ヘプチルシクロヘキシルアミン、4−n−オクチルシクロヘキシルアミン、4−(2−エチルヘキシル)シクロヘキシルアミン、4−n−ノニルシクロヘキシルアミン、4−n−デシルシクロヘキシルアミン等が例示される。
これらの中でも、シクロヘキシルアミンと、置換基として炭素数1〜4の直鎖状若しくは分岐鎖状のアルキル基、特にメチル基を有するシクロヘキシルアミンが好ましい。
また、置換基がメチル基である場合、その置換位置は2位、3位及び4位のいずれでもよいが、特に2位が好ましい。
また、置換基が炭素数2〜4の直鎖状若しくは分岐鎖状のアルキル基の場合、その置換位置は2位が好ましい。
これらの置換基を有する好ましいシクロヘキシルアミンの具体例としては、2−メチルシクロヘキシルアミン、3−メチルシクロヘキシルアミン、4−メチルシクロヘキシルアミン、2−エチルシクロヘキシルアミン、2−n−プロピルシクロヘキシルアミン、2−iso−プロピルシクロヘキシルアミン、2−n−ブチルシクロヘキシルアミン、2−iso−ブチルシクロヘキシルアミン、2−sec−ブチルシクロヘキシルアミン、2−tert−ブチルシクロヘキシルアミン等の2−アルキル(炭素数1〜4)シクロヘキシルアミンが挙げられる。
上記アルキルシクロヘキシルアミンは、シス体、トランス体及びこれら立体異性体の混合物のいずれでもよい。この立体異性体の混合物のシス体−トランス体の比率は、GLC(ガスクロマトグラフィー)で測定できる。
これら置換又は非置換シクロヘキシルアミンは単独で又は2種以上を混合してアミド化に供することができる。
好ましいアミド系化合物
本発明に係るアミド系化合物の中でも、一般式(1)におけるR2が、シクロヘキシル基又は炭素数1〜4の直鎖状若しくは分岐鎖状のアルキル基で置換されたシクロヘキシル基であるアミド系化合物は、造核作用が高く好ましい。
上記の好ましいアミド系化合物の中で、R1が1,2,3−プロパントリカルボン酸残基であるアミド系化合物の具体例としては、
1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(2−エチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(3−エチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(4−エチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(2−n−プロピルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(3−n−プロピルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(4−n−プロピルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(2−iso−プロピルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(3−iso−プロピルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(4−iso−プロピルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(2−n−ブチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(3−n−ブチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(4−n−ブチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(2−iso−ブチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(3−iso−ブチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(4−iso−ブチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(2−sec−ブチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(3−sec−ブチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(4−sec−ブチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(2−tert−ブチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(3−tert−ブチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(4−tert−ブチルシクロヘキシルアミド)等が挙げられる。
また、R1が1,2,3,4−ブタンテトラカルボン酸残基である好ましいアミド系化合物の具体例としては、
1,2,3,4−ブタンテトラカルボン酸テトラシクロヘキシルアミド、
1,2,3,4−ブタンテトラカルボン酸テトラキス(2−メチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(3−メチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(4−メチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(2−エチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(3−エチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(4−エチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(2−n−プロピルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(3−n−プロピルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(4−n−プロピルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(2−iso−プロピルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(3−iso−プロピルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(4−iso−プロピルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(2−n−ブチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(3−n−ブチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(4−n−ブチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(2−iso−ブチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(3−iso−ブチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(4−iso−ブチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(2−sec−ブチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(3−sec−ブチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(4−sec−ブチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(2−tert−ブチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(3−tert−ブチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(4−tert−ブチルシクロヘキシルアミド)等が挙げられる。
これら好ましいアミド系化合物の中でも、一般式(1)におけるR2が、シクロヘキシル基又はメチル基で置換されたシクロヘキシル基であるアミド系化合物は、造核作用が特に高く、また、原料入手が容易な点から好ましい。
具体的には、1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラシクロヘキシルアミド、
1,2,3,4−ブタンテトラカルボン酸テトラキス(2−メチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(3−メチルシクロヘキシルアミド)、
1,2,3,4−ブタンテトラカルボン酸テトラキス(4−メチルシクロヘキシルアミド)が例示される。
中でも、特に1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)が好ましい。
本発明に係るアミド系化合物の結晶系は、本発明の効果が得られる限り特に限定されず、六方晶、単斜晶、立方晶等の任意の結晶系が使用できる。これらの結晶も公知であるか又は公知の方法に従い製造できる。
本発明に係るアミド系化合物は、若干不純物を含むものであってもよい。一般式(1)で表されるアミド系化合物の純度が90重量%以上、好ましくは95重量%以上、特に好ましくは97重量%以上が推奨される。不純物としては、反応中間体又は未反応体由来のモノアミドジカルボン酸若しくはそのエステル、ジアミドモノカルボン酸若しくはそのエステル、モノアミドトリカルボン酸若しくはそのエステル、ジアミドジカルボン酸若しくはそのエステル、トリアミドカルボン酸若しくはそのエステル、アミド−イミド構造やビスイミド構造等のイミド骨格を有するものなどが例示される。
本発明に係るアミド系化合物の粒径は、本発明の効果が得られる限り特に限定されないが、溶融したポリオレフィン系樹脂に対する溶解速度又は分散性の点からできる限り粒径が小さいものが好ましく、通常、レーザー回折光散乱法で測定した最大粒径が200μm以下、好ましくは100μm以下、さらに好ましくは50μm以下、特に好ましくは10μm以下である。
最大粒子径を上記範囲内に調製する方法としては、この分野で公知の慣用装置を用いて微粉砕し、これを分級する方法等が挙げられる。具体的には、流動層式カウンタージェットミル100AFG(商品名、ホソカワミクロン社製)、超音速ジェットミルPJM−200(商品名、日本ニューマチック社製)等を用いて微粉砕並びに分級する方法が例示される。
本発明に係る一般式(1)で表されるアミド系化合物においては、該アミド系化合物を構成するアルキルシクロヘキシルアミン残基(特に、2−アルキルシクロヘキシルアミン残基)の立体異性構造におけるシス配置部分とトランス配置部分とが混合していてもよい。また、該アミド系化合物は、トランス配置部分とシス配置部分との比が異なる2種以上のアミド化合物の混合物であってもよい。
上記少なくとも1種のアミド系化合物中のトランス配置部分の総和とシス配置部分の総和の比率は、FT−IR法を用いて測定されたトランス構造部分のN−H伸縮振動ピークの吸光度のシス構造部分のN−H伸縮振動ピークの吸光度に対する比により確認することができる。
本明細書において、一般式(1)で表されるアミド系化合物を構成するアルキルシクロヘキシルアミン残基(即ち、アルキルシクロヘキシルアミンからアミノ基を除いて得られる残基、特に2−アルキルシクロヘキシルアミンからアミノ基を除いて得られる残基)の立体異性構造におけるトランス配置部分とは、一般式(X)
本明細書において、上記一般式(X)又は(X1)で示される構造は、トランス構造のアルキルシクロヘキシルアミンからアミノ基を除いて得られる基であるので、「トランス−アルキルシクロヘキシルアミン残基」という。
また、一般式(1)で表されるアミド系化合物を構成するアルキルシクロヘキシルアミン残基(特に2−アルキルシクロヘキシルアミン残基)の立体異性構造におけるシス配置部分とは、一般式(Y)
本明細書において、上記一般式(Y)又は(Y1)で示される構造は、シス構造のアルキルシクロヘキシルアミンからアミノ基を除いて得られる基であるので、「シス−アルキルシクロヘキシルアミン残基」という。
換言すると、上記好ましいアミド系化合物は、下記一般式(1Z)
で表されるアミド系化合物又は該アミド系化合物の少なくとも2種の混合物であって、
上記一般式(1Z)のR1が1,2,3−プロパントリカルボン酸から全てのカルボキシル基を除いて得られる残基(上記一般式(1Z)のkは3である。)である場合、上記一般式(X)で表されるトランス−アルキルシクロヘキシルアミン残基の割合が好ましくは50〜100%、より好ましくは65〜100%である化合物又は混合物であるか、又は、
上記一般式(1Z)のR1が1,2,3,4−ブタンテトラカルボン酸から全てのカルボキシル基を除いて得られる残基(上記一般式(1Z)のkは4である。)である場合、上記一般式(X)で表されるトランス−アルキルシクロヘキシルアミン残基の割合が好ましくは10〜80%、より好ましくは25〜60%である化合物又は混合物である。
なお、本発明に係るアミド系化合物(R2がアルキルシクロヘキシル基であるアミド系化合物)の混合物中のトランス配置部分とシス配置部分との割合(トランス:シス)は、原料のアルキルシクロヘキシルアミンのトランス体:シス体の比(GLCにより求めたモル比。以下、「GLC組成比」という。)と実質上同一割合となっていることが明らかとなった。これは、(a)原料アルキルシクロヘキシルアミン中のトランス体:シス体の比と、当該原料をアミド化反応に供した後に残留した未反応アルキルシクロヘキシルアミン中のトランス体:シス体の比とが一致すること、及び、(b)本発明で得られた生成物アミドを、アミド化反応条件と実質上同様の温度条件(室温〜280℃)で処理しても、FT−IRスペクトル及び融点が処理前のそれらと完全に一致し、アミド化反応により立体配置が変更されないことが確認されることによる。従って、アミド系化合物の混合物中のトランス配置部分とシス配置部分との割合(トランス:シス)は、原料アミンのトランス体:シス体の比によりコントロールできる。
成分(B):脂肪酸金属塩
本発明に係る脂肪酸金属塩(B)は、分子内に1個以上(特に1個又は2個)の水酸基を有していてもよい炭素数8〜32、好ましくは炭素数10〜18の飽和若しくは不飽和脂肪族モノカルボン酸と、一価若しくは二価の金属とから得られる脂肪酸金属塩である。該脂肪酸金属塩は、通常市販品を用いることができる他、例えば、脂肪族モノカルボン酸と、金属又はその塩化物、酸化物若しくは水酸化物とを用いて、ポリオレフィン系樹脂中で、当該脂肪酸金属塩が生成し得る形態で用いてもよい。
上記脂肪族モノカルボン酸としては、具体的には、カプリル酸、ノナン酸、カプリン酸、ウンデカン酸、ラウリン酸、トリデカン酸、ミリスチン酸、ペンタデカン酸、パルミチン酸、ヘプタデカン酸、ステアリン酸、ノナデカン酸、イコサン酸、ヘンイコサン酸、ドコサン酸、トリコサン酸、テトラコサン酸、ペンタコサン酸、ヘキサコサン酸、ヘプタコサン酸、オクタコサン酸、ノナコサン酸、トリアコンタン酸、ヘントリアコンタン酸、ドトリアコンタン酸等の飽和モノカルボン酸;
オクテン酸、ノネン酸、デセン酸、ウンデセン酸、ドデセン酸、トリデセン酸、テトラデセン酸、ペンタデセン酸、ヘキサデセン酸、オレイン酸、リノール酸、リノレン酸、ノナデセン酸、イコセン酸、ヘンイコセン酸、ドコセン酸、トリコセン酸、テトラコセン酸、ペンタコセン酸、ヘキサコセン酸、ヘプタコセン酸、オクタコセン酸、ノナコセン酸等の不飽和モノカルボン酸;
12−ヒドロキシステアリン酸、リシノール酸等の水酸基を有する脂肪族モノカルボン酸が例示される。中でも、ラウリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、オレイン酸、12−ヒドロキシステアリン酸が推奨される。
これらの脂肪族モノカルボン酸は、1種又は2種以上の混合物であってもよい。
一般式(2)における金属としては、アルカリ金属、アルカリ土類金属及び周期律表第12族(化学と工業,日本化学会編,第57巻第4号(2004)参照)の金属等の一価又は二価の金属が例示され、その中でも、アルカリ金属、アルカリ土類金属及び亜鉛が好ましい。特に、ナトリウム、カリウム、マグネシウム、カルシウム、亜鉛が推奨される。
これらの金属は1種又は2種以上の混合物であってもよい。
好ましい脂肪酸金属塩
好ましい脂肪酸金属塩としては、ポリオレフィン系樹脂に比較的高い結晶化温度を与える点から、上記脂肪族モノカルボン酸と一価金属、特に、ナトリウム、カリウムとの脂肪酸一価金属塩が好ましい。また、アミド系化合物のポリオレフィン系樹脂に対する溶解性・分散性を向上させる効果に優れる点から、脂肪族モノカルボン酸と二価金属、特にカルシウム、マグネシウム、亜鉛との脂肪酸二価金属塩が好ましい。
また、耐熱性及び入手容易性の点から、脂肪族モノカルボン酸としては、ラウリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、オレイン酸及び12−ヒドロキシステアリン酸からなる群より選ばれる少なくとも1種の脂肪族モノカルボン酸が好ましい。
脂肪酸一価金属塩の好ましい具体例としては、ラウリン酸ナトリウム、ミリスチン酸ナトリウム、パルミチン酸ナトリウム、ステアリン酸ナトリウム、12−ヒドロキシステアリン酸ナトリウム、オレイン酸ナトリウム、ラウリン酸カリウム、ミリスチン酸カリウム、パルミチン酸カリウム、ステアリン酸カリウム、12−ヒドロキシステアリン酸カリウム、オレイン酸カリウム等が挙げられる。
脂肪酸二価金属塩の好ましい具体例としては、ラウリン酸カルシウム、ミリスチン酸カルシウム、パルミチン酸カルシウム、ステアリン酸カルシウム、12−ヒドロキシステアリン酸カルシウム、オレイン酸カルシウム、ラウリン酸マグネシウム、ミリスチン酸マグネシウム、パルミチン酸マグネシウム、ステアリン酸マグネシウム、12−ヒドロキシステアリン酸マグネシウム、オレイン酸マグネシウム、ラウリン酸亜鉛、ミリスチン酸亜鉛、パルミチン酸亜鉛、ステアリン酸亜鉛、12−ヒドロキシステアリン酸亜鉛及びオレイン酸亜鉛等が挙げられる。
これらの脂肪酸金属塩は、それぞれ単独で又は2種以上を適宜組み合わせて用いることができる。
好ましい結晶化速度制御組成物
好ましい結晶化速度制御組成物としては、上記の好ましい成分(A)と好ましい成分(B)夫々から選ばれる組み合わせが挙げられる。その中でも、成分(A)が、1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド、1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)、1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)並びに1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)からなる群より選ばれる少なくとも1種、及び成分(B)が、ステアリン酸ナトリウム、ステアリン酸カリウム、ステアリン酸カルシウム、ステアリン酸マグネシウム、ステアリン酸亜鉛、12−ヒドロキシステアリン酸ナトリウム、12−ヒドロキシステアリン酸カリウム、12−ヒドロキシステアリン酸カルシウム、12−ヒドロキシステアリン酸マグネシウム及び12−ヒドロキシステアリン酸亜鉛から選ばれる少なくとも1種である組み合わせが特に推奨される。
上記の特に好ましい組み合わせとして、成分(B)が脂肪酸一価金属塩である好ましい組み合わせの具体例としては、
1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド+ステアリン酸ナトリウム、
1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド+ステアリン酸カリウム、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)+ステアリン酸ナトリウム、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)+ステアリン酸カリウム、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)+ステアリン酸ナトリウム、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)+ステアリン酸カリウム、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)+ステアリン酸ナトリウム、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)+ステアリン酸カリウム、
1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド+12−ヒドロキシステアリン酸ナトリウム、
1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド+12−ヒドロキシステアリン酸カリウム、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸ナトリウム、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸カリウム、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸ナトリウム、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸カリウム、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸ナトリウム、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸カリウム
等が挙げられる。
成分(B)が脂肪酸二価金属塩である好ましい組み合わせの具体例としては、
1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド+ステアリン酸カルシウム、
1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド+ステアリン酸マグネシウム、
1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド+ステアリン酸亜鉛、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)+ステアリン酸カルシウム、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)+ステアリン酸マグネシウム、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)+ステアリン酸亜鉛、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)+ステアリン酸カルシウム、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)+ステアリン酸マグネシウム、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)+ステアリン酸亜鉛、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)+ステアリン酸カルシウム、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)+ステアリン酸マグネシウム、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)+ステアリン酸亜鉛1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド+12−ヒドロキシステアリン酸カルシウム、
1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド+12−ヒドロキシステアリン酸マグネシウム、
1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド+12−ヒドロキシステアリン酸亜鉛、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸カルシウム、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸マグネシウム、
1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸亜鉛、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸カルシウム、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸マグネシウム、
1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸亜鉛、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸カルシウム、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸マグネシウム、
1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)+12−ヒドロキシステアリン酸亜鉛等が挙げられる。
さらに、次の組み合わせも好ましい。
・1,2,3,4−ブタンテトラカルボン酸テトラシクロヘキシルアミドと、ステアリン酸ナトリウム、ステアリン酸カリウム、ステアリン酸カルシウム、ステアリン酸マグネシウム、ステアリン酸亜鉛、12−ヒドロキシステアリン酸ナトリウム、12−ヒドロキシステアリン酸カリウム、12−ヒドロキシステアリン酸カルシウム、12−ヒドロキシステアリン酸マグネシウム及び12−ヒドロキシステアリン酸亜鉛からなる群から選ばれる少なくとも1種との組み合わせ、・1,2,3,4−ブタンテトラカルボン酸テトラキス(2−メチルシクロヘキシルアミド)と、ステアリン酸ナトリウム、ステアリン酸カリウム、ステアリン酸カルシウム、ステアリン酸マグネシウム、ステアリン酸亜鉛、12−ヒドロキシステアリン酸ナトリウム、12−ヒドロキシステアリン酸カリウム、12−ヒドロキシステアリン酸カルシウム、12−ヒドロキシステアリン酸マグネシウム及び12−ヒドロキシステアリン酸亜鉛からなる群から選ばれる少なくとも1種との組み合わせ。
本発明の結晶化速度制御組成物は、ポリオレフィン系樹脂の結晶化速度を制御するだけでなく、成分(B)が配合されることにより、アミド系化合物のポリオレフィン系樹脂への分散性・溶解性がより向上する。
特に、アミド系化合物と脂肪酸金属塩を予め混合した状態で使用することにより、それらの効果が顕著になる傾向がある。その混合方法として、特に制限がなく、粉体のまま混合する方法、脂肪酸金属塩の融点以上で混合する方法、アミド系化合物と脂肪酸金属塩を、溶媒中で溶解又分散した状態で混合し、溶媒を留去乾燥した後、必要に応じて造粒、粉砕、解砕する方法などが例示される。
また、その形態も特に制限はなく、一般的な粉末、顆粒、タブレット、ペレットなどの形態を任意に選択することができる。
粉末若しくは粒状の場合、その粒径は、本発明の効果が得られる限り特に限定されないが、溶融ポリオレフィン系樹脂に対する溶解速度又は分散性の点からできる限り粒径が小さいものが好ましく、通常、レーザー回折光散乱法で測定した最大粒径が、200μm以下、好ましくは100μm以下、さらに好ましくは50μm以下、特に好ましくは10μm以下である。
顆粒、タブレット、ペレットなどの造粒物の場合には、任意の形状、粒径を選択することができ、これらは、公知の造粒機、粉砕機・解砕機、分級機などを用いて製造することができる。それらの造粒物とすることで、粉体流動性の改善や粉塵発生の抑制(粉塵爆発の発生の低減)等に寄与する。
造粒機としては、乾式又は湿式押出造粒機、混合撹拌造粒機、タブレットマシーン、乾式圧縮ロール造粒機、マルメライザー(商品名、(株)ダルトン社製)が、粉砕機・解砕機としては、ピンミル、ジェットミル、パルベライザー、カッターミル、ハンマーミル、プレーナークラッシャー、ニブラーが、分級機としては、振動篩分機、風力分級機などが例示される。
また、アミド系化合物及び脂肪酸金属塩以外の他の成分として、必要に応じてポリオレフィン用改質剤等を、本発明の効果を損なわない範囲で配合してもよい。
上記ポリオレフィン用改質剤としては、例えば、ポリオレフィン等衛生協議会編「ポジティブリストの添加剤要覧」(2002年1月)に記載されている各種添加剤が挙げられ、より具体的には、安定剤(金属化合物、エポキシ化合物、窒素化合物、燐化合物、硫黄化合物等)、紫外線吸収剤(ベンゾフェノン系化合物、ベンゾトリアゾール系化合物等)、酸化防止剤(フェノール系化合物、亜リン酸エステル系化合物、イオウ系化合物等)、界面活性剤(モノグリセリンステアレート等のグリセリン脂肪酸エステル等)、滑剤(パラフィン、ワックス(ポリプロピレンワックス、ポリエチレンワックス等)等の脂肪族炭化水素、炭素数8〜22の高級脂肪酸、炭素数8〜22の高級脂肪族アルコール(ステアリルアルコール等)、ポリグリコール、炭素数4〜22の高級脂肪酸と炭素数4〜18の脂肪族1価アルコールとのエステル、炭素数8〜22の高級脂肪酸アマイド、シリコーン油、ロジン誘導体等)、発泡剤、発泡助剤、ポリマー添加剤の他、可塑剤(ジアルキルフタレート、ジアルキルヘキサヒドロフタレート等)、架橋剤、架橋促進剤、帯電防止剤、難燃剤、分散剤、有機無機の顔料、加工助剤、フィラー、他の核剤等の各種添加剤が例示される。その添加量は、本発明の所定の効果に悪影響を及ぼさない限り、特に限定されない。
前記フィラーとしては、タルク、クレー、マイカ、ハイドロタルサイト、アスベスト、ゼオライト、ガラス繊維、ガラスフレーク、ガラスビーズ、パーライト、ケイ酸カルシウム、炭酸カルシウム、モンモリロナイト、ベントナイト、グラファィト、アルミニウム粉末、アルミナ、シリカ、ケイ藻土、酸化チタン、酸化マグネシウム、軽石粉末、軽石バルーン、水酸化アルミニウム、水酸化マグネシウム、塩基性炭酸マグネシウム、ドロマイト、硫酸カルシウム、チタン酸カリウム、硫酸バリウム、亜硫酸カルシウム、硫化モリブデン等が挙げられる。これらのうちでも、タルク、ハイドロタルサイト、マイカ、ゼオライト、パーライト、珪藻土、炭酸カルシウム等が好ましい。これらフィラーは、1種単独で又は2種以上を適宜組み合わせて使用できる。
これらの他の成分を配合する場合は、特に限定されないが、例えば、アミド系化合物、脂肪酸金属塩及び他の成分をドライブンレンドして均一混合物とする等の方法が好ましい。
ポリオレフィン系樹脂組成物
本発明のポリオレフィン系樹脂組成物は、ポリオレフィン系樹脂、上記本発明に係る成分(A)及び成分(B)、並びに必要に応じてポリオレフィン用改質剤等を含有するものであり、例えば、上記本発明の結晶化速度制御組成物を、ポリオレフィン系樹脂に常法に従って配合することにより得られる。
本発明のポリオレフィン系樹脂組成物の製造方法は、所望の該樹脂組成物が得られれば特に限定されることなく、常法を用いることができる。例えば、ポリオレフィン系樹脂(粉末、顆粒又はペレット)と上述の本発明の結晶化速度制御組成物とを(或いは、ポリオレフィン系樹脂(粉末、顆粒又はペレット)と本発明に係る成分(A)及び成分(B)並びに必要に応じて上記のポリオレフィン用改質剤とを)、慣用の混合機、例えば、ヘンシェルミキサー、リボンブレンダー、Vブレンダー等を用いて混合したブレンドタイプのポリオレフィン系樹脂組成物を得る方法、又は、このブレンドタイプのポリオレフィン系樹脂組成物を、慣用の混練機、例えば、一軸又は二軸の押し出し機等を用いて、通常160〜300℃、好ましくは180〜280℃、特に好ましくは200℃〜260℃の温度で溶融混練し、押し出されたストランドを、冷却し、得られたストランドをカッティングすることでペレットタイプのポリオレフィン系樹脂組成物とする方法などが例示される。
本発明に係るポリオレフィン系樹脂に対する本発明の結晶化速度制御組成物の添加方法としては、前記の慣用されている装置、例えば、1軸又は2軸の押出機等を用いる一段添加法が好ましいが、2〜15重量%程度の高濃度マスターバッチの形態による2段添加法を採用しても何ら差し支えない。
また、本発明に係るアミド系化合物(A)と脂肪酸金属塩(B)は、結晶化速度制御組成物の形態で添加してもよいが、成分(A):成分(B)の重量比が、100:0〜30:70、好ましくは95:5〜30:70の範囲となるように、ポリオレフィン系樹脂に対し、同時にまたは別々に添加してもよい。
例えば、本発明に係る成分(B)を、予めポリオレフィン系樹脂に溶解等させて樹脂組成物(粉末・ペレット・顆粒等の形態)とした後、その樹脂組成物に本発明に係る成分(A)及び必要に応じて上記のポリオレフィン用改質剤等を配合して、本発明のポリオレフィン系樹脂組成物としてもよい。
本発明のポリオレフィン系樹脂組成物中の結晶化速度制御組成物の含有量は、所定の効果が得られる限り特に限定されるものではなく、広い範囲から適宜選択することができる。通常、ポリオレフィン系樹脂100重量部に対し、本発明の結晶化速度制御組成物を0.01〜10重量部、好ましくは0.05〜5重量部、特に好ましくは0.05〜2重量部含有することが推奨される。これらの範囲内で配合することにより十分に本発明の効果を得ることができる。
成分(A)及び成分(B)を同時に又は別々にポリオレフィン系樹脂に配合することもでき、その場合、成分(A)及び成分(B)の合計量が、ポリオレフィン系樹脂100重量部に対し、0.01〜10重量部、好ましくは0.05〜5重量部、特に好ましくは0.05〜2重量部含有することが推奨される。
また、核剤効果の観点から、一般式(1)で表される少なくとも1種のアミド系化合物(成分(A))の使用量は、ポリオレフィン系樹脂100重量部に対し、好ましくは0.01〜5重量部、より好ましくは0.05〜2重量部が推奨される。この範囲の量で使用することにより、透明性又は剛性の有意な向上が認められる。
上記ポリオレフィン系樹脂組成物は、一般式(1)で表されるアミド系化合物を単独で使用しても(即ち、成分(B)を使用することなく、成分(A)のみを使用しても)、透明性又は剛性に優れた成形体を与えることができる。しかしながら、得られる樹脂成形体の透明性又は剛性を向上させる上で、また、ポリオレフィン系樹脂の結晶化速度を制御するために、脂肪酸金属塩(成分(B))を含有させることが好ましい。
該脂肪酸金属塩(成分(B))の添加量は、ポリオレフィン系樹脂100重量部に対し、通常、0〜5重量部、好ましくは0〜1重量部、より好ましくは0.005〜0.5重量部、特に好ましくは0.01〜0.3重量部であるのが有利である。そして、成分(A):成分(B)=100:0〜30:70の範囲において、成分(B)の比率を高めることにより、そのポリオレフィン系樹脂組成物の結晶化速度を制御する(結晶化終了時間を制御する)ことができる。
本発明に係るポリオレフィン系樹脂としては、ポリエチレン系樹脂、ポリプロピレン系樹脂、ポリブテン系樹脂、ポリメチルペンテン系樹脂、ポリブタジエン系樹脂等が例示され、より具体的には、高密度ポリエチレン、中密度ポリエチレン、直鎖状ポリエチレン、エチレン含量50重量%以上、好ましくは70重量%以上のエチレンコポリマー、プロピレンホモポリマー、プロピレン50重量%以上、好ましくは70重量%以上のプロピレンコポリマー、ブテンホモポリマー、ブテン含量50重量%以上、好ましくは70重量%以上のブテンコポリマー、メチルペンテンホモポリマー、メチルペンテン含量50重量%以上、好ましくは70重量%以上のメチルペンテンコポリマー、ポリブタジエン等が例示される。
上記コポリマーはランダムコポリマーであってもよく、ブロックコポリマーであってもよい。これらの樹脂の立体規則性がある場合は、アイソタクチックでもシンジオタクチックでもよい。
上記コポリマーを構成し得るコモノマーとして、具体的にはエチレン、プロピレン、ブテン、ペンテン、ヘキセン、ヘプテン、オクテン、ノネン、デセン、ウンデセン、ドデセン等の炭素数2〜12のα−オレフィン、1,4−エンドメチレンシクロヘキセン等のビシクロ型モノマー、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル等の(メタ)アクリル酸エステル、酢酸ビニル等が例示できる。
かかる重合体を製造するために適用される触媒としては、一般に使用されているチーグラー・ナッタ型触媒はもちろん、遷移金属化合物(例えば、三塩化チタン、四塩化チタン等のチタンのハロゲン化物)を塩化マグネシウム等のハロゲン化マグネシウムを主成分とする担体に担持してなる触媒と、アルキルアルミニウム化合物(テトラエチルアルミニウム、ジエチルアルミニウムクロリド等)とを組み合わせてなる触媒系やメタロセン触媒も使用できる。
本発明に係るポリオレフィン系樹脂の推奨されるメルトフローレート(以下、「MFR」と略記する。JIS K 7210−1995)は、その適用する成形方法により適宜選択されるが、通常、0.01〜200g/10分、好ましくは0.05〜100g/10分である。
本発明に係るポリオレフィン系樹脂組成物には、使用目的やその用途に応じて適宜、従来公知の前記ポリオレフィン用改質剤を本発明の効果を損なわない範囲で添加することができる。
かくして得られる本発明のポリオレフィン系樹脂組成物は、成形加工時の樹脂温度条件及び成分(A)と成分(B)との比率を調節することにより、結晶化速度の制御ができる。より詳しくは、次の通りである。
(I)成形加工時の樹脂温度(成形温度)Tが、ポリオレフィン系樹脂の融解温度Tm以上であって、且つ、昇温時の貯蔵弾性率の転移温度Tsh以下の温度の場合、成分(A):成分(B)=100:0〜30:70の範囲において、成分(B)の比率を高めることにより、そのポリオレフィン系樹脂の結晶化速度を速くする(結晶化終了時間を短縮させる)ことができる。また、この成形方法(I)により、特に剛性に優れた成形体を製造することができる。
(II)成形加工時の樹脂温度(成形温度)Tが、昇温時の貯蔵弾性率の転移温度Tshを超える温度の場合、成分(A):成分(B)=100:0〜30:70の範囲において、成分(B)の比率を高めることにより、ポリオレフィン系樹脂の結晶化速度を低下させる(結晶化終了時間を延長させる)ことができる。この成形方法(II)により、特に未分散核剤が少なく、透明性に優れた成形体を製造することができる。
即ち、Tm〜Tshの樹脂温度範囲とTshよりも高い樹脂温度範囲との樹脂温度範囲の全体を通して見れば、また、Tm〜Tshの樹脂温度範囲、又は、Tshよりも高い樹脂温度範囲のそれぞれにおいても、成分(B)の比率を、成分(A):成分(B)=100:0〜30:70の範囲で調整することにより、前記(II)の方法における成分(A):成分(B)=100:0の場合の結晶化速度に比べて、そのポリオレフィン系樹脂の結晶化速度を制御、即ち、増加又は低減できる。
ポリオレフィン系樹脂の結晶化速度の制御方法
上記のように、本発明のポリオレフィン系樹脂組成物は、成形時の樹脂温度条件及び成分(A)と成分(B)との比率を調節することにより、成形加工時の結晶化速度を制御できる。即ち、本発明は、ポリオレフィン系樹脂に、成分(A)及び成分(B)を、成分(A):成分(B)の重量比が100:0〜30:70となるよう配合することを含む、成形時のポリオレフィン系樹脂の結晶化速度(結晶化終了時間)を制御する方法を提供するものでもある。
本発明の制御方法において、ポリオレフィン系樹脂の結晶化速度制御組成物の使用量は、所定の効果が得られる限り特に限定されるものではなく、広い範囲から適宜選択することができる。通常、ポリオレフィン系樹脂100重量部に対し、本発明の結晶化速度制御組成物を0.01〜10重量部、好ましくは0.05〜5重量部、特に好ましくは0.05〜2重量部使用することが推奨される。
成分(A)及び成分(B)を同時に又は別々にポリオレフィン系樹脂に配合する場合も、成分(A)及び成分(B)の合計量が、ポリオレフィン系樹脂100重量部に対し、0.01〜10重量部、好ましくは0.05〜5重量部、特に好ましくは0.05〜2重量部であることが推奨される。
本明細書及び請求の範囲において、「結晶化終了時間」という用語は、下記に記載するように、また、後述する実施例の項に記載するように、示差走査熱量計(商品名「DSC7」、パーキンエルマー社製)を用いて測定した結果から求められる値である。
具体的には、ポリオレフィン系樹脂組成物の温度を成形加工する際の樹脂温度とした後、結晶化終了時間の測定温度(該組成物の結晶化温度(実施例の項に記載の方法により求められる。)よりも0〜20℃高温側の範囲)まで、100〜200℃/分の冷却速度で急冷し、その測定温度でポリオレフィン系樹脂を等温結晶化させる。
得られた示差走査熱量法で得られたチャート(図1参照)において、長時間側のベースラインを短時間側に延長した延長線(図1の(a)で示される)を描き、また、発熱ピークの長時間側の曲線(d)上に勾配が最大になる点で接線(図1の(b)で示される)を引き、上記延長線と上記接線との交点(図1の(c)で示される)を求める。その交点の時間を、その“ポリオレフィン系樹脂組成物の「結晶化終了時間」”とする。
その結晶化終了時間が長い程、成形可能な時間が長いことを示す。尚、結晶化終了時間の測定における測定開始時間は、急冷したポリオレフィン系樹脂組成物の温度が測定温度に達した時間とした。
成形方法や成形すべき樹脂によっては、その結晶化速度が速い故に成形加工が困難になるという逆効果になることがある。例えば、ポリオレフィン系樹脂の結晶化速度が適切な範囲内になければ、フィルム・シート成形、ブロー成形、大型製品の射出成形などでは均質な成形体を得られにくくなる。
一方では、小型製品の射出成形などでは、生産コストをできるだけ低減させる為に、ポリオレフィン系樹脂の結晶化速度をさらに高めて結晶化時間を短縮することができれば、工業上有利である。
即ち、ポリオレフィン系樹脂の結晶化速度を制御する方法、或いは結晶化速度が制御された樹脂組成物があれば、煩雑な成形機の機械的設定のみに依存することなく、広範囲の成形加工に対応することが可能となり、上記本発明の課題が解決される。本発明は、ポリオレフィン系樹脂の結晶化速度の制御方法、種々の成形方法に適応したポリオレフィン系樹脂組成物を提供するものでもある。
本発明のポリオレフィン系樹脂の結晶化速度の制御方法は、ポリオレフィン系樹脂の結晶化速度制御組成物をポリオレフィン系樹脂に配合して、ポリオレフィン系樹脂組成物を得、該ポリオレフィン系樹脂組成物を成形することを包含する制御方法である。
又は、本発明に係る成分(B)を、予めポリオレフィン系樹脂に溶解などさせて樹脂組成物(粉末・ペレット・顆粒等の形態)とした後、その樹脂組成物に本発明に係る成分(A)を配合して、本発明のポリオレフィン系樹脂組成物とした後、該ポリオレフィン系樹脂組成物を成形することを包含する制御方法であってもよい。
より具体的には、本発明の結晶化速度制御方法は、
(1)ポリオレフィン系樹脂の結晶化速度制御組成物を構成する成分(A)及び成分(B)を、同時又は別々に、ポリオレフィン系樹脂に配合して本発明のポリオレフィン系樹脂組成物を製造する工程、及び
(2)ポリオレフィン系樹脂組成物を成形する工程
を包含している。
該工程(1)は、前記「ポリオレフィン系樹脂組成物」の項で上述した通りである。尚、本発明に係るアミド系化合物(A)は、溶解型の核剤であるため、前記工程(2)を行う前に、本発明の結晶化速度制御組成物をポリオレフィン系樹脂に十分に溶解させておくことは、結晶化速度制御組成物の性能を最大限発揮する上で重要であり、また本発明の効果を得る上でも重要なことである。
上記工程(2)について、以下に詳述する。
なお、本明細書及び請求の範囲において、降温時の貯蔵弾性率の転移温度(「Tsc」という)及び昇温時の貯蔵弾性率転移温度(「Tsh」という。)は、次のように定義される。
本発明で使用する一般式(1)で表されるアミド系化合物は、ポリオレフィン系樹脂中で繊維状粒子により形成される熱可逆性の網目構造を形成する。本発明に係る貯蔵造弾性率は、このアミド系化合物の繊維状粒子により形成される網目構造の形成に起因している。該アミド系化合物を含む溶融ポリオレフィン系樹脂の貯蔵弾性率(G’)は温度に対して、非連続的に変化する。その一例を図2に模式的に示す。
図2の破線で示すように、降温時に繊維状粒子により形成される網目構造の形成に由来して該アミド系化合物を含む溶融ポリオレフィン系樹脂の貯蔵弾性率(G’)が非連続的に変化(上昇)するが、その変化率が最大となる温度をTscと定義する。また、図2の実線で示すように、昇温時に該アミド系化合物の繊維状粒子の溶解による網目構造の消失に起因してG’が変化(低下)するが、その変化率が最大となる温度をTshと定義する。
このTsc及びTshは、レオメーターを用いて動的粘弾性を測定することにより得られる。測定法の詳細は、後述の実施例の項に記載している通りである。これにより、図2に示すような昇温時のカーブ(実線)及び降温時のカーブ(破線)が得られ、これらカーブを微分型に変換すると、図3に示すようにピークが現れるので、これらピークを示す温度をそれぞれTsh及びTscとする。
Tsh及びTscは、使用する一般式(1)で表されるアミド系化合物の種類及び使用量等により変わり得る。例えば、Tsh及びTscは、該アミド系化合物の含有量が高いほど、又は、該アミド系化合物を構成するアルキルシクロヘキシルアミン残基の立体異性体であるトランス配置部分の比率が高いほど、高温側へシフトする。
従って、本発明のポリオレフィン系樹脂成形体を製造する前に、当該結晶化速度制御組成物を溶解させたポリオレフィン系樹脂組成物を試験的に調製し、そして、予めその樹脂組成物についてTsh及びTscを測定しておく。こうして測定されたTsh及びTscに従って、実際のポリオレフィン系樹脂成形体の製造時の温度条件を調整すればよい。
一般に、R1が1,2,3−プロパントリカルボン酸から全てのカルボキシル基を除いて得られる残基を示す一般式(1)のアミド系化合物の少なくとも1種を使用する場合、Tshは、170〜250℃、特に180〜240℃である。また、R1が1,2,3,4−ブタンテトラカルボン酸から全てのカルボキシル基を除いて得られる残基を示す一般式(1)のアミド系化合物の少なくとも1種を使用する場合、Tshは、一般には180〜280℃、特に190〜270℃である。
ポリオレフィン系樹脂の結晶化速度を制御するに際し、工程(2)における樹脂温度(成形温度)Tは、特に重要な条件の一つである。以下、本発明の結晶化速度制御方法を、樹脂温度TがTsh以下(且つ、ポリオレフィン系樹脂の融解温度Tm以上)である場合(成形方法(I))について説明し、次いで、樹脂温度TがTshよりも高い場合(成形方法(II))について説明する。
<成形方法(I)>
前記樹脂温度Tがポリオレフィン系樹脂の融解温度(以下「Tm」という)以上であって、且つTsh以下の樹脂温度の範囲(即ち、Tm≦T≦Tsh)の場合、成分(A):成分(B)=100:0〜30:70の重量比の範囲において、成分(B)の比率を高めることにより、そのポリオレフィン系樹脂組成物の結晶化速度を速くする(結晶化終了時間を短縮させる)ことができる。
より詳しくは、図8に示すように、成形加工時の樹脂温度(成形温度)Tを、ポリオレフィン系樹脂の融解温度Tm以上であって、且つ、昇温時の貯蔵弾性率の転移温度Tsh以下の温度に設定する場合(図7の成形方法(I))、該特定の脂肪酸金属塩(B)の配合比率を増加させると(領域(IA)→領域(IAB))、ポリオレフィン系樹脂の結晶化速度を速くする(結晶化終了時間を短縮させる)ことができる。その結晶化速度の上昇の度合いは、成分(A)及び成分(B)の組み合わせと、成分(A)に対する成分(B)の重量比に主に依存する。
この成形方法(I)では、ポリオレフィン系樹脂成形体の剛性の向上に大きく寄与し、特にホモポリプロピレン樹脂やブロックポリプロピレン樹脂、高密度ポリエチレン樹脂で顕著である。
この樹脂温度の領域では、上述の網目構造を残した状態で(即ち、該網目構造を構成する繊維状粒子が溶解又は溶融しない温度)成形加工を行うので、該網目構造を構成する繊維状粒子が配向し、ポリオレフィン系樹脂を配向結晶化させている。
より詳しくは、図7の(I)に示すように、昇温時の貯蔵弾性率の転移温度Tsh以下の温度で成形するために上記網目構造が存在した状態で成形されるので、該網目構造を構成する繊維状粒子が配向し、そのため、得られる成形体においてポリオレフィン系樹脂の結晶ラメラを配向させることができ、その結果、特に剛性に優れた成形体を製造することができる。
工程(2)におけるポリオレフィン系樹脂組成物の成形方法は、射出工程又は押し出し工程を含む全ての成形方法に広く適応できる。具体的には、射出成形、押し出し成形、射出ブロー成形、射出押出ブロー成形、射出圧縮成形、押し出しブロー成形、押し出しサーモフォーム成形又は溶融紡糸による繊維の製造などが挙げられる。
工程(2)において、本発明のポリオレフィン系樹脂の結晶化速度制御組成物の使用に加え、一般式(1)で表されるアミド系化合物がポリオレフィン系樹脂中で網目構造を形成することを利用することにより、より広い領域でポリオレフィン系樹脂の結晶化速度を制御することが可能となった。
以下、上記成形方法(I)について、詳述する。成形方法(I)は、成分(B)の使用により結晶化速度を制御(増大)することを可能とした成形体製造法でもある。ポリオレフィン系樹脂組成物(ペレットの製造)の製造方法及び本発明のポリオレフィン系樹脂成形体の製造方法(成形方法)の概念図である図7の(I)を参照して説明すると、次の通りである。
<ポリオレフィン系樹脂組成物(ペレット)の製造方法>
上述した通り、本発明のポリオレフィン系樹脂組成物は、典型的には、次の方法で製造される。
例えば、ポリオレフィン系樹脂(粉末、顆粒又はペレット)と上述の本発明の結晶化速度制御組成物とを(或いは、ポリオレフィン系樹脂(粉末、顆粒又はペレット)と本発明に係る成分(A)及び成分(B)並びに必要に応じて上記のポリオレフィン用改質剤とを)、慣用の混合機、例えば、ヘンシェルミキサー、リボンブレンダー、Vブレンダー等を用いてドライブレンドする。得られるドライブレンド物を、慣用の混練機、例えば、一軸又は二軸の押し出し機等を用いて、本発明で使用する一般式(1)で表されるアミド系化合物が溶解する温度以上で溶融混練して溶融混合物を得る(図7の(a1)参照)。
この溶融混合物を押し出し、得られたストランドの樹脂温度Tを、降温時の貯蔵弾性率の転移温度Tsc以下の温度に冷却することにより、該アミド系化合物の繊維状粒子により形成される網目構造を含むポリオレフィン系樹脂組成物が得られる(図7の(a2)参照)。図7の(a2)に示されている細線は、繊維状粒子を模式的に表している(図7の他の図においても同じ)。
得られるポリオレフィン系樹脂組成物の樹脂温度Tは、Tsc以下の温度にまで冷却されていれば足り、ポリオレフィン系樹脂自体が固体状態である必要はなく、溶融状態であってもよい。
しかし、必要であれば、得られたストランドの樹脂温度Tをポリオレフィン系樹脂組成物の結晶化温度(以下、「Tc」という。)以下の温度、例えば室温以下とし、ストランドをカッティングすることにより、該ポリオレフィン系樹脂組成物のペレットを得ることができる(図7の(a3)参照)。
こうして得られるペレットにおいては、ポリオレフィン系樹脂の結晶ラメラが一般式(1)で表されるアミド系化合物の繊維状粒子を起点として成長している。図7の(a3)に示されている波線は、ポリオレフィン系樹脂の結晶ラメラを模式的に示している(図7の他の図においても同じ)。
上記のペレットの製造方法は、下記の成形方法(II)におけるペレットの製造方法と同様である。
<ポリオレフィン系樹脂成形体の製造方法>
本発明に係るポリオレフィン系樹脂成形体の製造方法(成形方法(I))の好ましい実施形態としては、例えば、次の[1]〜[5]の工程を採用するものが例示できる。以下、これらの工程について、図7の(I)を参照して説明する。なお、図7では、射出工程を含む成形方法について記載しているが、以下の記載では押出工程を含む成形法についても併せて説明する。
[1]まず、図7の(a1)に示すように、溶融ポリオレフィン系樹脂に、本発明の結晶化速度制御組成物(或いは、本発明に係る成分(A)及び成分(B)、並びに必要に応じて、ポリオレフィン用改質剤等)を、可能な限り均一に溶解する。この時の樹脂温度Tは、例えば、ポリプロピレン系樹脂の場合には160〜300℃が挙げられる。
[2]次に、図7の(a2)に示すように、当該ポリオレフィン系樹脂組成物を、降温時の貯蔵弾性率の転移温度Tsc以下の樹脂温度まで冷却すると、該アミド系化合物の繊維状粒子により形成される網目構造が形成される。
Tscは、該アミド系化合物の含有量が高いほど、又は該アミド系化合物を構成するアルキルシクロヘキシルアミン残基の立体異性体であるトランス配置部分の比率が高いほど、高温側へシフトする。
必要であれば、図7の(a3)に示すように、更に樹脂温度Tをポリオレフィン系樹脂の結晶化温度Tc以下の温度に下げて、該溶融ポリオレフィン系樹脂組成物中のポリオレフィン系樹脂を結晶化させ、ペレットとすることもできる。この時、ペレット中には、一般式(1)で表されるアミド系化合物の繊維状粒子により形成される網目構造が保持されている。
[3]本発明において、射出工程を含む成形方法を採用する場合、該アミド系化合物の繊維状粒子により形成される網目構造を含むポリオレフィン系樹脂組成物(図7の(a2)の状態のもの)を、一旦ペレット(図7の(a3)の状態のもの)とし、得られたペレットを射出成形機1のホッパー2から導入する。次いで、図7の(a4)に示すように、特定の温度条件下(即ち、Tm≦T≦Tsh)、上記網目構造を保ったままの状態で、射出成形を行う。これにより、図7の(a5)に示すように、射出の際に生じる剪断力、特に射出成形機1のノズル4内での流れ及び金型3内での流れにより生じる剪断力により、一般式(1)で表されるアミド系化合物の網目構造を構成する微小な繊維状粒子が配向される。
他方、押出工程を含む成形方法を採用する場合、一般式(1)で表されるアミド系化合物の繊維状粒子により形成される網目構造を含むポリオレフィン系樹脂組成物(図7の(a2)の状態のもの)を、ペレットにすることなくそのまま、特定の温度条件下、上記網目構造を保ったままの状態で、押出工程を含む成形方法に供する。或いは、該網目構造を含むポリオレフィン系樹脂組成物(図7の(a2)の状態のもの)を、図7の(a3)に示すように一旦ペレットにし、該ペレットを、特定の温度条件下、上記網目構造を保ったままの状態で、押出工程を含む成形方法に供する。これにより、押出の際に生じる剪断力により、一般式(1)で表されるアミド系化合物の網目構造を構成する微小な繊維状粒子が配向される。
これら成形方法において採用する上記特定の温度条件は、樹脂温度Tを、ポリオレフィン系樹脂の溶融温度Tm以上であって、且つ、当該ポリオレフィン系樹脂組成物の昇温時の貯蔵弾性率の転移温度Tsh以下の樹脂温度の範囲に調整するものである(即ち、Tm≦T≦Tsh)。なお、Tshも、該アミド系化合物の含有量が高いほど、又は該アミド系化合物を構成するアルキルシクロヘキシルアミン残基の立体異性体であるトランス配置部分の比率が高いほど、高温側へシフトする。
上記特定の温度条件としては、例えば、ポリプロピレン樹脂組成物を射出工程又は押し出し工程を含む成形方法により成形する場合、R1が1,2,3−プロパントリカルボン酸から全てのカルボキシル基を除いて得られる残基を示す一般式(1)のアミド系化合物の少なくとも1種を使用する場合、樹脂温度Tは、一般には170〜250℃、特に180〜240℃である。また、R1が1,2,3,4−ブタンテトラカルボン酸から全てのカルボキシル基を除いて得られる残基を示す一般式(1)のアミド系化合物の少なくとも1種を使用する場合、樹脂温度Tは、一般には180〜280℃、特に190〜270℃である。
[4]こうして射出工程を含む成形法で金型に到達した溶融ポリオレフィン系樹脂組成物は、金型により冷却され、一般式(1)で表されるアミド系化合物の微小な繊維状粒子が、樹脂の流れ方向に配向された状態に保持される(図7の(a6)参照)。
同様に、押出工程を含む成形法でチルロールに到達した溶融ポリオレフィン樹脂組成物は、チルロールにより冷却され、一般式(1)で表されるアミド系化合物の微小な繊維状粒子が、樹脂の流れ方向に配向された状態に保持される。
[5]次いで、図7の(a6)に示す一般式(1)で表されるアミド系化合物の微小な繊維状粒子が配向された状態にある樹脂組成物を、更にポリオレフィン系樹脂の結晶化温度(Tc)以下に下げることにより、ポリオレフィン系樹脂の結晶化が起こる。その際に、繊維状粒子が配向しているので、それら繊維状粒子を起点として成長したポリオレフィン系樹脂の結晶ラメラも配向した状態となる(図7の(a7)参照)。押出工程を含む成形法の場合も同様である。
この時、設定される金型温度又はチルロール温度としては、ポリオレフィン系樹脂の結晶化温度Tc以下の温度であることが必要であり、例えば、10〜80℃が好ましい。
その結果、本発明の製造方法により得られるポリオレフィン系樹脂成形体は、ポリオレフィン系樹脂の結晶ラメラが配向しているという特徴を有する。
このように、本発明に係る成形方法(I)は、一般式(1)で表されるアミド系化合物が、上記特定温度条件で網目構造を形成することを利用し、該網目構造を残した状態で(即ち、該網目構造を構成する繊維状粒子が溶解又は溶融しない温度で)、成形を行うことにより、該繊維状粒子を配向させ、ポリオレフィン系樹脂を配向結晶化させるものである。
その結果、本発明の成形方法(I)により、機械的強度、特に剛性(曲げ弾性率)の高い成形体を得ることができる。
従って、本発明は、前記項18に記載のように、ポリオレフィン系樹脂の融解温度以上であって、且つ、昇温時の貯蔵弾性率の転移温度以下の樹脂温度で、本発明の樹脂組成物を成形する工程を備えたポリオレフィン系樹脂成形体の製造方法を提供するものでもある。
この製造方法においては、アミド系化合物は、一般式(1)において、R1が1,2,3−プロパントリカルボン酸からすべてのカルボキシル基を除いて得られる残基であるのが好ましく、また、3個のR2が同一又は異なってシクロヘキシル基を示すか又は1個の炭素数1〜4の直鎖状又は分岐鎖状のアルキル基で置換されたシクロヘキシル基を示すことが好ましい(即ち、前記項19に記載の製造法)。
また、上記製造法(成形方法(I))においては、一般式(1)で表される少なくとも1種のアミド系化合物(成分(A))の使用量は、ポリオレフィン系樹脂100重量部に対し、好ましくは0.01〜5重量部、より好ましくは0.05〜2重量部が推奨される。この範囲の量で使用することにより、剛性の有意な向上が認められる。なお、5重量部を越える含有量で使用することも可能である。
上記製造方法では、一般式(1)で表されるアミド系化合物を単独で使用しても(即ち、成分(B)を使用することなく、成分(A)のみを使用しても)、剛性に優れた成形体を得ることができる。しかしながら、得られる樹脂成形体の剛性を向上させる上で、また、ポリオレフィン系樹脂の結晶化速度を制御するために、脂肪酸金属塩(成分(B))を含有させることが好ましい。
該脂肪酸金属塩(成分(B))の添加量は、ポリオレフィン系樹脂100重量部に対し、通常、0〜5重量部、好ましくは0〜1重量部、より好ましくは0.005〜0.5重量部、特に好ましくは0.01〜0.3重量部であるのが有利である。そして、成分(A):成分(B)=100:0〜30:70の範囲において、成分(B)の比率を高めることにより、そのポリオレフィン系樹脂組成物の結晶化速度を速くする(結晶化終了時間を短縮させる)ことができる。
<成形方法(II)>
前記樹脂温度TがTshを超える温度範囲(即ち、Tsh<T)の場合、成分(A):成分(B)=100:0〜30:70の範囲において、成分(B)の比率を高めることにより、そのポリオレフィン系樹脂の結晶化速度を低下させる(結晶化終了時間を延長させる)ことができる。
より詳しくは、図8に示すように、成形加工時の樹脂温度(成形温度)Tを、昇温時の貯蔵弾性率の転移温度Tshを超える温度に設定する場合(図7の成形方法(II))、該脂肪酸金属塩(B)の配合比率を増加させると(領域(IIA)→領域(IIAB))、ポリオレフィン系樹脂の結晶化速度を遅くする(結晶化終了時間を延長させる)ことができる。
また、成分(B)の比率を高めることにより、ポリオレフィン系樹脂の結晶化温度Tcを低下させることもできる。
その結晶化速度の低下及び結晶化温度の低下の度合いは、成分(A)及び成分(B)の組み合わせと成分(A)に対する成分(B)の比率に主に依存する。
この成形方法(II)は、ポリオレフィン系樹脂成形体の透明性の向上に大きく寄与し、特にホモポリプロピレン樹脂やランダムポリプロピレン樹脂において顕著である。
なお、この成形方法(II)において、樹脂温度Tがあまりに高いと樹脂が劣化することもあるので、樹脂温度Tは、300℃以下、特に280℃以下であるのが好ましい。
上記樹脂温度の領域では、上述のアミド系化合物の繊維状粒子が溶融又は溶解し、網目構造が消失した状態にある。
より詳しくは、成形方法(II)によると、図7の(II)に示すように、昇温時の貯蔵弾性率の転移温度Tshを超える温度で成形するために網目構造が溶解して消失した状態で成形され、そのため、その溶融したポリオレフィン系樹脂組成物を冷却すると、アミド系化合物の繊維状粒子が形成され、網目構造が再構成される。それを起点として、ポリオレフィン系樹脂の微少な結晶(球晶)が形成され、その結果、特に透明性に優れた成形体を製造できる。
<ポリオレフィン系樹脂成形体の製造方法>
以下、本発明に係るポリオレフィン系樹脂組成物の成形方法(II)について、詳述する。成形方法(II)は、成分(B)の使用により結晶化速度を制御(低減)することを可能とした成形体製造法でもある。本発明のポリオレフィン系樹脂成形体の製造方法(成形方法)の概念図である図7の(II)を参照して説明すると、次の通りである。
尚、ポリオレフィン系樹脂組成物(ペレットの製造)の製造方法(図7の(II)の(b1)〜(b3)は、上記成形方法(I)における図7の(I)の(a1)〜(a3)と同じである。
図7の(b4)及び(b5)に示すように、樹脂温度TをTshよりも高い温度に設定して成形すると、繊維状粒子が溶融又は溶解し、網目構造が消失した状態で成形することとなる。
その結果、射出工程又は押し出し工程後に、図7の(b6)及び(b7)に示すように、溶融ポリオレフィン系樹脂組成物は網目構造が消失した状態で金型又はチルロールに到達し、そこで、溶融ポリオレフィン系樹脂組成物を降温時の貯蔵弾性率の転移温度Tsc以下に冷却することによって再び網目構造が形成されるが、その際に射出又は押出工程を経るので、網目構造は若干配向するが、その程度は低い。
このように、繊維状粒子の配向の程度が低いので、図7の(b8)に示すように、冷却温度をポリオレフィン系樹脂組成物の結晶化温度Tc以下に下げて成形体を得ても、得られる成形体においてはポリオレフィン系樹脂の結晶ラメラの配向度は低い。
上記成形方法(II)によると、透明性が高い未分散核剤の少ない成形体を得ることができる。従って、本発明は、前記項17に記載のように、昇温時の貯蔵弾性率の転移温度を超える樹脂温度で、本発明のポリオレフィン系樹脂組成物を成形することを特徴とするポリオレフィン系樹脂成形体の製造方法を提供するものでもある。
上記成形方法(II)において、成形法としては、上記射出工程又は押し出し工程を含む成形法が挙げられる。具体的には、射出成形、押し出し成形、射出ブロー成形、射出押出ブロー成形、射出圧縮成形、押し出しブロー成形、押し出しサーモフォーム成形又は溶融紡糸による繊維の製造などが挙げられる。
上記成形方法(II)により達成される本発明の効果(透明性が高く、未分散核剤の少ない成形体が得られる効果)は、特に、成分(A):成分(B)の重量比が、好ましくは100:0〜30:70の範囲、より好ましくは95:5〜30:70の範囲、より好ましくは90:10〜60:40、特に好ましくは90:10〜70:30の範囲となるように配合することにより、得られる。また、この範囲で成分(B)の割合を変化させることにより、ポリオレフィン系樹脂の結晶化速度を小さくすることができる。
また、上記製造法(成形方法(II))においても、一般式(1)で表される少なくとも1種のアミド系化合物(成分(A))の使用量は、ポリオレフィン系樹脂100重量部に対し、好ましくは0.01〜5重量部、より好ましくは0.05〜2重量部が推奨される。この範囲の量で使用することにより、透明性の有意な向上が認められる。なお、5重量部を越える含有量で使用することも可能である。
上記製造方法では、一般式(1)で表されるアミド系化合物を単独で使用しても(即ち、成分(B)を使用することなく、成分(A)のみを使用しても)、透明性に優れた成形体を得ることができる。しかしながら、得られる樹脂成形体の透明性を向上させる上で、また、ポリオレフィン系樹脂の結晶化速度を制御するために、脂肪酸金属塩(成分(B))を含有させることが好ましい。
該脂肪酸金属塩(成分(B))の添加量は、ポリオレフィン系樹脂100重量部に対し、通常、0〜5重量部、好ましくは0〜1重量部、より好ましくは0.005〜0.5重量部、特に好ましくは0.01〜0.3重量部であるのが有利である。そして、成分(A):成分(B)=100:0〜30:70の範囲において、成分(B)の比率を高めることにより、そのポリオレフィン系樹脂組成物の結晶化速度を低減させる(結晶化終了時間を延長させる)ことができる。
上記成形方法(I)及び成形方法(II)の記載から判るように、本発明では、特定割合の成分(A)及び成分(B)を使用することにより、ポリオレフィン系樹脂組成物の成形時のポリオレフィン系樹脂の結晶化速度を制御することができる。従って、本発明は、成分(A):成分(B)の重量比が100:0〜30:70の割合で含有する組成物の、ポリオレフィン系樹脂成形時のポリオレフィン系樹脂の結晶化速度を制御するための使用を提供するものでもある。
ポリオレフィン系樹脂成形体
本発明の成形体は、上記本発明のポリオレフィン系樹脂組成物を、上記成形方法(I)及び(II)に要求される条件を用いる限り、慣用されている成形法に従って成形することにより得られる。本発明のポリオレフィン系樹脂組成物は、結晶化速度が制御できるので、射出成形、押出成形、ブロー成形、圧空成形、回転成形、シート成形、フィルム成形等の従来公知の成形方法のいずれをも採用でき、成形条件としては、従来採用されている条件が広い範囲から適宜選択できる。
また、上記の成形方法(II)を採用した場合、透明性に優れた成形体が得られ、また成形方法(I)を採用した場合剛性に優れた成形体が得られる。
また、本発明の成形方法(I)により得られるポリオレフィン系樹脂成形体、特に、汎用されているエチレン−プロピレンランダム共重合体(特にエチレン含量2〜4重量%程度であって、残部がプロピレンであるもの)、プロピレンホモポリマー又はエチレン−プロピレンブロック共重合体(特にエチレン含量が5〜15重量%程度であって、残部がプロピレンであるもの)、と一般式(1)で表されるアミド系化合物の少なくとも1種を用いて得られる成形体は、広角X線回折により求められる(040)反射強度の(110)反射強度に対する比で表される配向度が2以上(特に2〜10)であり、有意に高い剛性を有する。上記の配向度の測定法は、次の通りである。
通常の対称反射X線回折法により、射出成形体試料表面に対して入射するX線の入射角θと回折線検出角2θがθ−2θの関係を保って回転する集中光学系でX線回折強度を測定する。次いで、得られたX線回折強度曲線を非晶質ハローと各結晶質ピークとにピーク分離を行ない、ポリプロピレン結晶からの(110)面反射(2θ=約13.9度)と(040)面反射(2θ=約16.6度)のピーク強度の比より下記(F)式で配向度が求められる。
配向度=I(040)/I(110) (F)
上記式(F)において、I(110)は(110)面反射のピーク強度(cps)を示し、I(040)は(040)面反射のピーク強度(cps)を示す。
本発明のポリオレフィン系樹脂組成物の成形方法(I)を適用することにより、ポリオレフィン系樹脂成形体に対して優れた剛性を付与することができる。この特質は、成形品の肉厚を薄くして軽量化を達成することにも役立つ。
かくして得られた本発明のポリオレフィン系樹脂成形体は、従来、リン酸金属塩類、芳香族カルボン酸金属塩類、ベンジリデンソルビトール類等を核剤として配合してなるポリオレフィン系樹脂組成物が用いられてきたと同様の分野において適用され、具体的には、熱や放射線等により滅菌されるディスポーザブル注射器、輸液・輸血セット、採血器具等の医療用器具類;放射線等により滅菌される食品・植物等の包装物;衣料ケースや衣料保存用コンテナ等の各種ケース類;食品を熱充填するためのカップ、レトルト食品の包装容器;電子レンジ用容器;ジュース、茶等の飲料用、化粧品用、医薬品用、シャンプー用等の缶、ビン等の容器;味噌、醤油等の調味料用容器及びキャップ;水、米、パン、漬物等の食品用ケース及び容器;冷蔵庫用ケース等の雑貨;文具;電気・機械部品;自動車用部品等の素材として好適である。
本発明に係るポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度(℃)、本発明のポリオレフィン系樹脂組成物より得られる成形体の結晶化温度(℃)、結晶化終了時間(分)、ヘイズ値(%)、分散性(白点数)、曲げ弾性率(MPa)、X線回折測定及び配向度を以下の方法により測定し、評価した。
(1)結晶化温度Tc(℃)
示差走査熱量法(パーキンエルマー社製、商品名「DSC7」)を用いて、測定サンプル(サンプル重量=10mg、各実施例及び比較例で得られた成形体(試験片)から切り出して作成)をポリオレフィン系樹脂組成物の成形時の樹脂温度まで昇温させ、その温度の到達後3分間保持した以外は、JIS K 7121に準じて、発熱ピークのピーク温度を測定した。
結晶化温度(Tc)が高い程、成形サイクルを短くすることができる。
(2)結晶化終了時間(分)(以下「Te」という。)
示差走査熱量計(パーキンエルマー社製、商品名「DSC7」)を用いて、測定サンプル(サンプル重量=10mg、各実施例及び比較例で得られた成形体(試験片)から切り出して作成)を昇温速度200℃/分でポリオレフィン系樹脂組成物の成形時の樹脂温度まで昇温させ、その温度の到達後3分間保持した。次に、冷却速度100℃/分で「結晶化終了時間」の測定温度(アイソタクチックエチレン−プロピレンランダム共重合樹脂;120℃、アイソタクチックホモポリプロピレン樹脂及びエチレン−プロピレンブロック共重合樹脂;130℃)まで急冷し、ポリオレフィン系樹脂を等温結晶化させた。尚、急冷して測定温度に達した時間を「結晶化終了時間」の測定開始時間とした。
得られた示差走査熱量法(DSC)のチャートから、長時間側のベースラインを短時間側に延長した延長線(図1の(a))と、発熱ピークの長時間側の曲線(図1の(d))に勾配が最大になる点で引いた接線(図1の(b))の交点(図1の(c))に対応する時間を「結晶化終了時間」(分)とした。「結晶化終了時間」が長い程、成形可能な時間が長いことを示す。
(3)ヘイズ値(%)
各実施例及び比較例で得られた5cmx5cmx1mmのサイズの試験片について、東洋精機製作所製のヘイズメーターを用いて、JIS K 7136(2000)に準じて測定した。得られた数値が小さい程、透明性に優れていることを示す。
(4)分散性(白点数)
各実施例及び比較例で得られた5cmx5cmx1mmのサイズの試験片10枚中の未分散又は未溶解のアミド系化合物による白点の数を目視で測定し1枚あたりの平均値を求めて、下記判定を行った。
判定:平均白点数
S:0.5個/枚以下
A:1個/枚以下
B:1個/枚より多く、2個/枚以下
C:2個/枚より多く、10個/枚以下
D:10個/枚より多い
(5)貯蔵弾性率(G’)の転移温度(℃)
動的粘弾性測定装置((株)レオロジ社製、商品名「MR−500ソリキッドメータ」)を用いて、下記の測定条件下で、各実施例で得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の温度依存曲線を作成した。Tscは降温時の温度依存曲線の微分曲線のピークトップの温度に対応し、Tshは昇温時の温度依存曲線の微分曲線のピークトップの温度に対応する。
〈動的粘弾性率の測定条件〉
測定治具:パラレルプレート(直径20mm)
周波数:0.5Hz
歪角:0.5度
温度条件:
(i)降温時の貯蔵弾性率の転移温度(Tsc):ポリオレフィン系樹脂組成物(ペレット)を250℃で(但し、BTC−2MeCHAを用いた実施例については280℃で、PTC−2MeCHA[100]又はPTC−2MeCHA(100)を用いた実施例については260℃で)1分間保持した後、降温速度を5℃/分で、150℃まで降温させて測定した。
(ii)昇温時の貯蔵弾性率の転移温度(Tsh):降温時の貯蔵弾性率の測定終了後、上記ポリオレフィン系樹脂組成物を、昇温速度5℃/分で250℃まで(但し、BTC−2MeCHAを用いた実施例については280℃まで、PTC−2MeCHA[100]又はPTC−2MeCHA(100)を用いた実施例については260℃まで)再昇温させて測定した。
(6)曲げ弾性率(MPa)
インストロン万能試験機を用いて、JIS K 7203(1982)に準じて、ポリオレフィン系樹脂成形体の曲げ弾性率(MPa)を測定した。尚、試験温度は25℃、試験速度は10mm/分とした。曲げ弾性率の値が大きいほど剛性に優れている。
なお、曲げ弾性率を測定している実施例及び比較例において、曲げ弾性率測定用試験片は、特に断らない限り、長さ90mm、幅10mm、高さ4mmである。
(7)X線回折測定
X線回折装置(理学電機社製、商品名「RINT2000」)を用いて、長さ90mm、幅10mm、高さ4mmの試験片を用いてX線回折を測定した。尚、X線の入射方向は、試験片中央部をThrough方向(図4参照)から行った。
<X線回折測定条件>
X線ビーム:CuKα
X線管球:Cu
管電圧:40kV,管電流:100mA
X線入射角(θ):θ−2θ連動
測定角度(2θ)範囲:5〜30度
スキャンスピード:4度/分
(8)配向度
上記(7)に記載の方法で得られたX線回折強度曲線を非晶質ハローと各結晶質ピークとにピーク分離を行ない、ポリプロピレン結晶からの(110)面反射(2θ=約13.9度)と(040)面反射(2θ=約16.6度)のピーク強度の比から下記の式(F)を用いて配向度を求めた。
配向度=I(040)/I(110) (F)
[式中、I(110)は(110)面反射のピーク強度(cps)を表し、I(040)は(040)面反射のピーク強度(cps)を表す。]
配向度の値が大きいほど、ポリオレフィンの結晶の配向の度合いが大きい。
製造例1
(1)攪拌機、温度計、冷却管及びガス導入口を備えた500mlの4ツ口フラスコに1,2,3−プロパントリカルボン酸(以下「PTC」と略記する。)9.7g(0.055モル)とN−メチル−2−ピロリドン100gを秤取り、窒素雰囲気下、室温にて攪拌しながらPTCを完全溶解させた。続いて、2−メチルシクロヘキシルアミン(トランス体:シス体=74.3:25.7、GLC組成比)20.5g(0.1815モル)、亜リン酸トリフェニル56.3g(0.1815モル)、ピリジン14.4g(0.1815モル)及びN−メチル−2−ピロリドン50gを加え、窒素雰囲気下、攪拌しながら100℃で4時間反応を行った。冷却後、反応溶液をイソプロピルアルコール500mlと水500mlの混合溶液中にゆっくり注ぎ込み、約40℃で1時間攪拌後、析出した白色沈殿物を濾別した。更に、得られた白色固体を約40℃のイソプロピルアルコール500mlで2回洗浄した後、100℃、133Paにて6時間乾燥した。
得られた乾燥物を乳鉢で粉砕し、目開き106μmの標準篩い(JIS Z 8801規格)に通して、1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)(以下、「PTC−2MeCHA」と略記する。)18.8g(収率74%)を得た。
(2)また、アミド化反応後に回収した未反応2−メチルシクロヘキシルアミンをGLC分析に供したところ、該未反応アミンのトランス体:シス体の比は74.3:25.7であり、原料として使用した2−メチルシクロヘキシルアミンのトランス体:シス体の比(74.3:25.7、GLC組成比)と一致した。
更に、上記で得られた生成物であるアミド化合物を、100℃で熱処理しても、FT−IRスペクトル、融点が処理前のそれらと一致することから、上記アミド化反応により、該アミド系化合物を構成する2−メチルシクロヘキシルアミン残基の立体配置が変化していないことを確認した。
その結果、本製造例1の生成物であるアミド化合物において、トランス配置の2−メチルシクロヘキシルアミン残基とシス配置の2−メチルシクロヘキシルアミン残基との比は、原料2−メチルシクロヘキシルアミンのトランス体とシス体との比と一致することを確認した。
以下、製造例2,3,5及び7においても、上記と同様に、生成物のアミド化合物において、トランス配置のアルキルシクロヘキシルアミン残基とシス配置のアルキルシクロヘキシルアミン残基との比は、原料アルキルシクロヘキシルアミンのトランス体とシス体との比と一致することを確認した。
製造例2
2−メチルシクロヘキシルアミンに代えて、3−メチルシクロヘキシルアミン(トランス体:シス体=75.4:24.6、GLC組成比)を用いた以外は製造例1と同様に行い、1,2,3−プロパントリカルボン酸トリス(3−メチルシクロヘキシルアミド)(以下、「PTC−3MeCHA」と略記する。)15.5g(収率61%)を得た。
製造例3
2−メチルシクロヘキシルアミンに代えて、4−メチルシクロヘキシルアミン(トランス体:シス体=58.4:41.6、GLC組成比)を用いた以外は製造例1と同様に行い、1,2,3−プロパントリカルボン酸トリス(4−メチルシクロヘキシルアミド)(以下、「PTC−4MeCHA」と略記する。)9.7g(収率38%)を得た。
製造例4
2−メチルシクロヘキシルアミンに代えて、シクロヘキシルアミンを用いた以外は製造例1と同様に行い、1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド(以下、「PTC−CHA」と略記する。)17.3g(収率75%)を得た。
製造例5
1,2,3−プロパントリカルボン酸に代えて、1,2,3,4−ブタンテトラカルボン酸12.9g(0.055モル)と2−メチルシクロヘキシルアミン(トランス体:シス体=74.3:25.7、GLC組成比)27.4g(0.242モル)、亜リン酸トリフェニル75.1g(0.242モル)及びピリジン19.1g(0.242モル)を用いた以外は、製造例1と同様にして行い、1,2,3,4−ブタンテトラカルボン酸テトラキス(2−メチルシクロヘキシルアミド)(以下、「BTC−2MeCHA」と略記する。)21.3g(収率63%)を得た。
製造例6
2−メチルシクロヘキシルアミン(トランス体:シス体=74.3:25.7、GLC組成比)に代えて、2−メチルシクロヘキシルアミン(トランス体:シス体=100:0、GLC組成比)を用いた以外は製造例1と同様に行い、1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)(以下、「PTC−2MeCHA[100]」と略記する。)20.3g(収率80%)を得た。
製造例7
2−メチルシクロヘキシルアミン(トランス体:シス体=74.3:25.7、GLC組成比)に代えて、2−メチルシクロヘキシルアミン(トランス体:シス体=50:50、GLC組成比)を用いた以外は製造例1と同様に行い、1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)(以下、「PTC−2MeCHA[50]」と略記する。)18.0g(収率71%)を得た。
以下に本発明の実施例を挙げる。実施例1〜51は、前記成形方法(II)(T>Tsh)についての実施例であり、実施例1〜33は樹脂としてエチレン−プロピレンランダム共重合樹脂を使用した例であり、実施例34〜41は樹脂としてホモポリプロピレン樹脂を使用した例であり、実施例42〜51は樹脂としてエチレン−プロピレンブロック共重合樹脂を使用した例である。
[実施例1]
エチレン含有量3.0重量%のアイソタクチックエチレン−プロピレンランダム共重合樹脂(MFR=20g/10分、融解温度=153℃、以下「r−PP」と略記する。)100重量部に対して、アミド系化合物として製造例1で調製したPTC−2MeCHA0.2重量部と、脂肪酸金属塩として、ステアリン酸カルシウム0.05重量部を添加し、更に、テトラキス[メチレン−3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオネート]メタン(チバスペシャルティーケミカルズ社製、商品名「IRGANOX1010」)0.05重量部及びテトラキス(2,4−ジ−t−ブチルフェニル)ホスファイト(チバスペシャルティーケミカルズ社製、商品名「IRGAFOS168」)0.05重量部を配合し、ヘンシェルミキサーで1000rpm、5分間ドライブレンドした。
次に、樹脂温度240℃で直径20mmの一軸押出機を用いて溶融混練して、押し出されたストランドを水冷し、次に得られたストランドを切断してペレット状ポリオレフィン系樹脂組成物を得た。
得られたペレットを樹脂温度(成形温度)240℃、金型温度40℃の条件下で射出成形し、ポリオレフィン系樹脂成形体(試験片:サイズ=5cmx5cmx1mm)を調製した。
得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
[実施例2及び実施例3]
ステアリン酸カルシウムの添加量を表1記載の添加量に変えた以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
[実施例4〜8]
脂肪酸金属塩を表1記載の脂肪酸金属塩に変更した以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
[実施例9]
PTC−2MeCHAに代えて製造例2で調製したPTC−3MeCHAを用いた以外は、実施例1と同様にして行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その評価結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
[実施例10]
PTC−2MeCHAに代えて製造例3で調製したPTC−4MeCHAを用いた以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
[実施例11]
PTC−2MeCHAに代えて製造例4で調製したPTC−CHAを用いた以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
を表1に示した。
[実施例12]
PTC−2MeCHAに代えて製造例6で調製したPTC−2MeCHA[100]を用い、溶融混練時の樹脂温度を260℃、射出成形時の樹脂温度を260℃とした以外は、実施例1と同様にして行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
[実施例13]
PTC−2MeCHAに代えて製造例7で調製したPTC−2MeCHA[50]を用いた以外は、実施例1と同様にして行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
[実施例14〜16]
脂肪酸金属塩を表1記載の脂肪酸金属塩に変更した以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
[実施例17]
攪拌機、温度計、冷却管及びガス導入口を備えた200mLの4ツ口フラスコに、メタノール5重量部、アミド系化合物として製造例1で調製したPTC−2MeCHA0.2重量部及び脂肪酸金属塩としてステアリン酸カルシウム0.05重量部を入れて、室温で0.5時間攪拌した。次に、メタノールを減圧留去した後、室温で133Paにて6時間乾燥した。得られた乾燥物を乳鉢で粉砕し、目開き106μmの標準篩い(JIS Z 8801規格)に通して、本発明の結晶化速度制御組成物を得た。
r−PP100重量部に対して、前記結晶化速度制御組成物0.25重量部、テトラキス[メチレン−3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオネート]メタン(チバスペシャルティーケミカルズ社製、商品名「IRGANOX1010」)0.05重量部及びテトラキス(2,4−ジ−t−ブチルフェニル)ホスファイト(チバスペシャルティーケミカルズ社製、商品名「IRGAFOS168」)0.05重量部を配合し、ヘンシェルミキサーで1000rpm、5分間ドライブレンドした。
次に、樹脂温度240℃で直径20mmの一軸押出機を用いて溶融混練して、押し出されたストランドを水冷し、次に得られたストランドを切断してペレット状ポリオレフィン系樹脂組成物を得た。
得られたペレットを樹脂温度(成形温度)240℃、金型温度40℃の条件下で射出成形し、ポリオレフィン系樹脂成形体(試験片、サイズ:5cmx5cmx1mm)を調製した。
得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
[実施例18]
PTC−2MeCHAに代えて製造例5で調製したBTC−2MeCHA0.15重量部を用い、溶融混練時の樹脂温度を280℃、射出成形時の樹脂温度を280℃とした以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
[実施例19]
PTC−2MeCHAに代えて製造例5で調製したBTC−2MeCHA0.1重量部を用い、溶融混練時の樹脂温度を260℃とした以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
[実施例20]
ステアリン酸カルシウムに代えて、ステアリン酸カリウムを用いた以外は、実施例19と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表1に示した。
[実施例21]
脂肪酸金属塩を用いない以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表2に示した。
[実施例22及び実施例23]
脂肪酸金属塩を用いず、アミド系化合物を表2記載の添加量に変更した以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表2に示した。
[実施例24〜26]
脂肪酸金属塩に代えて、表2記載の添加剤を用いた以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表2に示した。
[実施例27]
脂肪酸金属塩を用いない以外は、実施例9と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表2に示した。
[実施例28]
脂肪酸金属塩を用いない以外は、実施例10と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表2に示した。
[実施例29]
脂肪酸金属塩を用いない以外は、実施例11と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表2に示した。
[実施例30]
脂肪酸金属塩を用いない以外は、実施例12と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表2に示した。
[実施例31]
脂肪酸金属塩を用いない以外は、実施例13と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表2に示した。
[実施例32]
脂肪酸金属塩を用いない以外は、実施例18と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表2に示した。
[実施例33]
脂肪酸金属塩を用いない以外は、実施例19と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表2に示した。
(1)結晶化終了時間
成形加工時の樹脂温度(成形温度)に昇温時の貯蔵弾性率の転移温度を越える温度を選択した場合(即ちT>Tsh)、成分(A)と成分(B)の比率を制御すること、特に成分(B)の比率を高める制御により、ポリオレフィン系樹脂の結晶化速度を低減させること(結晶化終了時間を延長させること)ができる(実施例1〜3、実施例17、実施例21〜23参照)。
また、ステアリン酸カルシウム以外の本発明に係る脂肪酸金属塩によっても、同様にポリオレフィン系樹脂の結晶化速度を低減させること(結晶化終了時間を延長させること)ができる(実施例4〜8、実施例14〜16参照)。
更に、PTC−2MeCHA以外の本発明に係るアミド系化合物によっても、同様にポリオレフィン系樹脂の結晶化速度を低減させること(結晶化終了時間を延長させること)ができる(実施例9〜13、実施例18〜20、実施例27〜33参照)。
本発明に係るアミド系化合物に、ポリエチレンワックス、ステアリルアルコール又はグリセリンステアレートを併用しても、本発明に係る脂肪酸金属塩と比較して、ポリオレフィン系樹脂の結晶化速度を制御すること(結晶化終了時間を制御すること)への貢献度は非常に少ない(実施例24〜26参照)。
(2)結晶化温度
上記効果に加え、成形加工時の樹脂温度(成形温度)に昇温時の貯蔵弾性率の転移温度を越える温度を選択した場合(即ちT>Tsh)、本発明の結晶化速度制御組成物は、成分(A)と成分(B)との比率或いはその種類の組み合わせにより、そのポリオレフィン系樹脂の結晶化温度を低下させることもできる(実施例1〜5、実施例9〜10、実施例14〜17参照)。
(3)ヘイズ値及び分散性
本発明に係るアミド系化合物と脂肪酸金属塩(特に脂肪酸二価金属塩)との組み合わせを含む結晶化速度制御組成物を用いることにより、特にポリオレフィン系樹脂に対する溶解性・分散性が向上し、その成形体には未分散物が少ない(実施例1〜5、実施例8〜17、実施例19参照)。
また、本発明のポリオレフィン系樹脂組成物を成形加工して得られた成形体は、後述の比較例1及び比較例2と比較して透明性が格段に向上し、1,2,3−プロパントリカルボン酸と2−メチルシクロヘキシルアミンとの組み合わせで構成されるアミド系化合物を含有する結晶化速度制御組成物を用いた場合、特にその透明性が優れる(実施例1〜8、実施例12〜17、比較例1及び2参照)。
[実施例34]
r−PPをアイソタクチックホモポリプロピレン樹脂(MFR=10g/10分、融解温度=163℃、以下「h−PP」と略記する。)に代えた以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表3に示した。
[実施例35及び実施例36]
脂肪酸金属塩を表3記載の脂肪酸金属塩に変更した以外は、実施例34と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表3に示した。
[実施例37]
PTC−2MeCHAに代えて製造例4で調製したPTC−CHAを用いた以外は、実施例34と同様にして行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表3に示した。
[実施例38]
PTC−2MeCHAに代えて製造例6で調製したPTC−2MeCHA[100]を用い、溶融混練時の樹脂温度を260℃、射出成形時の樹脂温度を260℃とした以外は、実施例34と同様にして行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表3に示した。
[実施例39]
脂肪酸金属塩を用いない以外は、実施例34と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表3に示した。
[実施例40]
脂肪酸金属塩を用いない以外は、実施例37と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表3に示した。
[実施例41]
脂肪酸金属塩を用いない以外は、実施例38と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、ヘイズ値及び分散性を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表3に示した。
(1)結晶化終了時間
上記r−PPと同様にh−PPにおいても、成形加工時の樹脂温度(成形温度)に昇温時の貯蔵弾性率の転移温度を越える温度を選択した場合、成分(A)と成分(B)の比率を制御すること、特に成分(B)の比率を高めることにより、ポリオレフィン系樹脂の結晶化速度を低減させること(結晶化終了時間を延長させること)ができる(実施例34〜38、実施例39〜41参照)。
(2)結晶化温度
上記r−PPと同様にh−PPにおいても、成形加工時の樹脂温度(成形温度)に昇温時の貯蔵弾性率の転移温度を越える温度を選択した場合、本発明の結晶化速度制御組成物は、成分(A)と成分(B)との比率或いはその種類の組み合わせにより、そのポリオレフィン系樹脂の結晶化温度を低下させることもできる(実施例34〜38参照)。
(3)ヘイズ値及び分散性
上記r−PPと同様にh−PPにおいても、本発明に係るアミド系化合物と脂肪酸金属塩との組み合わせの結晶化速度制御組成物を用いることにより、特にポリオレフィン系樹脂に対する溶解性・分散性が向上し、その成形体は未分散物が少ない(実施例34〜38参照)。
また、本発明のポリオレフィン系樹脂組成物を成形加工して得られた成形体は、後述の比較例3及び4に比較して透明性に優れる(実施例34〜38、実施例39〜41、比較例3及び4参照)。
[実施例42]
r−PPをエチレン含有量9.5重量%のエチレン−プロピレンブロック共重合樹脂(MFR=26g/10分、融解温度=164℃、以下「b−PP」と略記する。)に代え、且つ、5cm×5cm×1mmの試験片に加えて90mm×10mm×4mmのサイズの試験片を同様にして得た以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間及び曲げ弾性率を測定した。なお、結晶化温度及び結晶化終了時間の測定には、5cm×5cm×1mmのサイズの試験片を使用し、曲げ弾性率の測定には、90mm×10mm×4mmのサイズの試験片を使用した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表4に示した。
[実施例43及び実施例44]
脂肪酸金属塩を表4に脂肪酸金属塩に変更した以外は、実施例42と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間及び曲げ弾性率を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表4に示した。
[実施例45]
PTC−2MeCHAに代えて製造例4で調製したPTC−CHAを用いた以外は、実施例42と同様にして行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間及び曲げ弾性率を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表4に示した。
[実施例46]
PTC−2MeCHAに代えて製造例6で調製したPTC−2MeCHA[100]を用い、溶融混練時の樹脂温度を260℃、射出成形時の樹脂温度を260℃とした以外は、実施例42と同様にして行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間及び曲げ弾性率を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表4に示した。
[実施例47]
PTC−2MeCHAに代えて製造例7で調製したPTC−2MeCHA[50]を用いた以外は、実施例42と同様にして行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間及び曲げ弾性率を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表4に示した。
[実施例48]
脂肪酸金属塩を用いない以外は、実施例42と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間及び曲げ弾性率を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表4に示した。
[実施例49]
脂肪酸金属塩を用いない以外は、実施例45と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間及び曲げ弾性率を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表4に示した。
[実施例50]
脂肪酸金属塩を用いない以外は、実施例46と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間及び曲げ弾性率を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表4に示した。
[実施例51]
脂肪酸金属塩を用いない以外は、実施例47と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間及び曲げ弾性率を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表4に示した。
(1)結晶化終了時間
上記r−PPと同様にb−PPにおいても、成形加工時の樹脂温度(成形温度)に昇温時の貯蔵弾性率の転移温度を越える温度を選択した場合、成分(A)と成分(B)の比率を制御すること、特に成分(B)の比率を高めることにより、ポリオレフィン系樹脂の結晶化速度を低減させること(結晶化終了時間を延長させること)ができる(実施例42〜47,48〜51参照)。
(2)結晶化温度
上記r−PPと同様にb−PPにおいても、成形加工時の樹脂温度(成形温度)に昇温時の貯蔵弾性率の転移温度を越える温度を場合、本発明の結晶化速度制御組成物は、成分(A)と成分(B)との比率或いはその種類の組み合わせにより、そのポリオレフィン系樹脂組成物の結晶化温度を低下させることもできる。(実施例42〜47参照)。
(3)曲げ弾性率
成形加工時の樹脂温度(成形温度)に昇温時の貯蔵弾性率の転移温度を越える温度を選択した場合、本発明のポリオレフィン系樹脂組成物を成形して得られた成形体は、後述の比較例5及び6に比較して曲げ弾性率が改善されている(実施例42〜51、比較例5及び6参照)。
以下の実施例52〜69は、成形方法(I)(Tm≦T≦Tsh)についての実施例である。
[実施例52]
(射出)成形工程の樹脂温度を200℃に変え、且つ、5cm×5cm×1mmのサイズの試験片に加えて90mm×10mm×4mmのサイズの試験片を同様にして得た以外は、実施例1と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。なお、結晶化温度及び結晶化終了時間の測定には、5cm×5cm×1mmの試験片を使用し、曲げ弾性率の測定には、90mm×10mm×4mmのサイズの試験片を使用した。(以下の実施例53〜69及び比較例1〜6においても同じ)。
[実施例53及び実施例54]
ステアリン酸カルシウムの添加量を表5記載の添加量に変えた以外は、実施例52と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例55]
(射出)成形工程の樹脂温度を200℃に変えた以外は、実施例4と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例56]
(射出)成形工程の樹脂温度を200℃に変えた以外は、実施例5と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例57]
(射出)成形工程の樹脂温度を200℃に変えた以外は、実施例34と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例58]
(射出)成形工程の樹脂温度を200℃に変えた以外は、実施例35と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例59]
(射出)成形工程の樹脂温度を200℃に変えた以外は、実施例36と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例60]
(射出)成形工程の樹脂温度を200℃に変えた以外は、実施例42と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例61]
(射出)成形工程の樹脂温度を200℃に変えた以外は、実施例43と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例62]
(射出)成形工程の樹脂温度を200℃に変えた以外は、実施例44と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例63]
(射出)成形工程の樹脂温度を240℃に変えた以外は、実施例18と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例64]
(射出)成形工程の樹脂温度を200℃に変えた以外は、実施例21と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例65]
(射出)成形工程の樹脂温度を200℃に変えた以外は、実施例22と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例66]
(射出)成形工程の樹脂温度を180℃に変えた以外は、実施例23と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例67]
(射出)成形工程の樹脂温度を200℃に変えた以外は、実施例39と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例68]
(射出)成形工程の樹脂温度を200℃に変えた以外は、実施例48と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
[実施例69]
(射出)成形工程の樹脂温度を240℃に変えた以外は、実施例32と同様に行い、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに得られた試験片の結晶化温度、結晶化終了時間、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表5に示した。
(1)結晶化終了時間
成形加工時の樹脂温度(成形温度)に、ポリオレフィン系樹脂の融解温度以上、且つ昇温時の貯蔵弾性率の転移温度以下の温度を選択した場合、成分(A)と成分(B)の比率を制御すること、特に成分(B)の比率を高めることにより、ポリオレフィン系樹脂の結晶化速度を高めること(結晶化終了時間を短縮させること)ができる(実施例52〜54、実施例64〜66参照)。
また、ステアリン酸カルシウム以外の本発明に係る脂肪酸金属塩によっても、同様にポリオレフィン系樹脂の結晶化速度を高めること(結晶化終了時間を短縮させること)ができる(実施例55、実施例56参照)。
更に、r−PP以外の本発明に係るポリオレフィン系樹脂を使用する場合も、同様に結晶化速度を高めること(結晶化終了時間を短縮させること)ができる(実施例57〜62、実施例67及び68参照)。
(2)結晶化温度
上記効果に加え、成形加工時の樹脂温度(成形温度)に、ポリオレフィン系樹脂の融解温度以上、且つ昇温時の貯蔵弾性率の転移温度以下の温度を選択した場合、本発明の結晶化速度制御組成物を含有するポリオレフィン系樹脂組成物の結晶化温度は、成形方法(II)において、成分(A):成分(B)=100:0である本発明の結晶化速度制御組成物を含有するポリオレフィン系樹脂組成物により達成される結晶化温度と比較して、殆ど低下が認められない。即ち、高い結晶化速度を保持していることを示している。(例えば、r−PPについては、実施例52〜56、実施例64〜66,実施例21〜23参照)
(3)曲げ弾性率
成形加工時の樹脂温度(成形温度)に、ポリオレフィン系樹脂の融解温度以上、且つ昇温時の貯蔵弾性率の転移温度以下の温度を選択した場合、本発明のポリオレフィン系樹脂組成物を成形加工して得られた成形体は、剛性に優れている。上記の成形方法(II)で得られた成形体と比較しても顕著である(実施例52〜69、実施例42〜51、比較例1〜6参照)。
PTC−2MeCHAの他の本発明に係るアミド系化合物においても、同様に当該成形体の剛性が向上する(実施例63、実施例69参照)。
(4)配向度
成形加工時の樹脂温度(成形温度)が、ポリオレフィン系樹脂の融解温度以上、昇温時の貯蔵弾性率の転移温度以下の範囲の場合、本発明のポリオレフィン系樹脂組成物を成形加工して得られた成形体は、配向度が何れも2以上を有していた。一方、上記の成形方法(II)で得られた成形体は2未満であった(実施例52〜69、実施例42〜51、比較例1〜6参照)。
比較例1
アミド系化合物及び脂肪酸金属塩を用いない他は実施例1と同様に行い、結晶化温度、結晶化終了時間、ヘイズ値、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表6に示した。
比較例2
ドライブレンド時に、ステアリン酸カルシウム0.05重量部を用いた以外は、比較例1と同様に行い、結晶化温度、結晶化終了時間、ヘイズ値、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表6に示した。
比較例3
アミド系化合物及び脂肪酸金属塩を用いない他は実施例34と同様に行い、結晶化温度、結晶化終了時間、ヘイズ値、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表6に示した。
比較例4
ドライブレンド時に、ステアリン酸カルシウム0.05重量部を用いた以外は、比較例3と同様に行い、結晶化温度、結晶化終了時間、ヘイズ値、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表6に示した。
比較例5
アミド系化合物及び脂肪酸金属塩を用いない他は実施例42と同様に行い、結晶化温度、結晶化終了時間、ヘイズ値、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表6に示した。
比較例6
ドライブレンド時に、ステアリン酸カルシウム0.05重量部を用いた以外は、比較例5と同様に行い、結晶化温度、結晶化終了時間、ヘイズ値、曲げ弾性率及び配向度を測定した。その測定結果、並びに混練工程の樹脂温度及び(射出)成形工程の樹脂温度を表6に示した。
以下の記載において、実施例I−1のように、「I−」を伴って記載されている実施例は成形方法(I)に関する実施例であり、実施例II−1のように、「II−」を伴って記載されている実施例は成形方法(II)に関する実施例である。
尚、得られたペレット状ポリオレフィン系樹脂組成物の貯蔵弾性率の転移温度、並びに本発明のポリオレフィン系樹脂組成物より得られる成形体の曲げ弾性率、X線回折測定及び配向度を上述の方法と同様に測定し、評価した。また、当該成形体の耐衝撃性(デュポン法衝撃強度)を下記の方法により測定し、評価した。
(9)耐衝撃性(デュポン法衝撃強度)
落錘衝撃試験方法JIS K 7211に準拠して、23℃における厚さ2mmのシートの50%破壊エネルギーを求めた。数値が大きい程、耐衝撃性に優れる。
製造例I−1
(1)攪拌機、温度計、冷却管及びガス導入口を備えた500mlの4ツ口フラスコに、1,2,3−プロパントリカルボン酸(PTC)9.7g(0.055モル)とN−メチル−2−ピロリドン100gを秤取り、窒素雰囲気下、室温にて攪拌しながらPTCを完全溶解させた。次に、2−メチルシクロヘキシルアミン(トランス体:シス体=100:0、GLC組成比)20.5g(0.18モル)、亜リン酸トリフェニル56.3g(0.18モル)、ピリジン14.4g(0.18モル)及びN−メチル−2−ピロリドン50gを加え、窒素雰囲気下、攪拌しながら100℃で4時間反応を行った。冷却後、その反応溶液をイソプロピルアルコール500mlと水500mlとの混合液中にゆっくり注ぎ込み、約40℃で1時間攪拌後、析出した白色沈殿物を濾別した。更に、得られた白色固体を約40℃のイソプロピルアルコール500mlで2回洗浄した後、100℃、133Paにて6時間減圧乾燥した。
得られた乾燥物を乳鉢で粉砕し、目開き106μmの標準篩い(JIS Z 8801規格)に通して、1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)(以下、「PTC−2MeCHA(100)」という。)20.3g(収率80%)を得た。
製造例I−2
2−メチルシクロヘキシルアミン(トランス体:シス体=100:0、GLC組成比)に代えて2−メチルシクロヘキシルアミン(トランス体:シス体=74:26、GLC組成比)を用いた以外は製造例I−1と同様に行い、1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)(以下、「PTC−2MeCHA(74)」という。)18.8g(収率74%)を得た。
尚、上記のアミド化反応後に回収した未反応2−メチルシクロヘキシルアミンをGLC分析に供したところ、該未反応アミンのトランス体:シス体の比は74:26であり、原料として使用した2−メチルシクロヘキシルアミンのトランス体:シス体の比(74:26、GLC組成比)と一致した。
更に、上記で得られた生成物であるアミド化合物を、100℃で熱処理しても、FT−IRスペクトル、融点が処理前のそれらと一致することから、上記アミド化反応により、該アミド系化合物を構成する2−メチルシクロヘキシルアミン残基の立体配置が変化していないことを確認した。
その結果、本製造例1の生成物であるアミド化合物において、トランス配置の2−メチルシクロヘキシルアミン残基とシス配置の2−メチルシクロヘキシルアミン残基との比は、原料2−メチルシクロヘキシルアミンのトランス体とシス体との比と一致することを確認した。
製造例I−3
2−メチルシクロヘキシルアミン(トランス体:シス体=100:0、GLC組成比)に代えて2−メチルシクロヘキシルアミン(トランス体:シス体=50:50、GLC組成比)を用いた以外は製造例I−1と同様に行い、1,2,3−プロパントリカルボン酸トリス(2−メチルシクロヘキシルアミド)(以下、「PTC−2MeCHA(50)」という。)18.0g(収率71%)を得た。
また、製造例I−2と同様に前記アミド化合物の立体配置を確認したところ、アミド化合物のトランス配置部分とシス配置部分との比が、原料アミンのトランス体とシス体との比と一致することを確認した。
製造例I−4
2−メチルシクロヘキシルアミンに代えてシクロヘキシルアミンを用いた以外は製造例I−1と同様に行い、1,2,3−プロパントリカルボン酸トリシクロヘキシルアミド(以下(以下、「PTC−CHA」と略記する。)17.3g(収率75%)を得た。
実施例I−1
r−PP100重量部に対して、PTC−2MeCHA(100)0.2重量部、テトラキスメチレン−3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオネート]メタン(チバ・スペシャルティ・ケミカルズ社製、商品名「IRGANOX1010」)0.05重量部及びテトラキス(2,4−ジ−t−ブチルフェニル)ホスファイト(チバ・スペシャルティ・ケミカルズ社製、商品名「IRGAFOS168」)0.05重量部を秤取り、r−PP100重量部と共にヘンシェルミキサーで1000rpm、5分間ドライブレンドした。
次に、前記ドライブレンド物を混練温度260℃(樹脂温度)で一軸押出機(L/D=440mm/20mm)を用いて溶融混練し、押し出されたストランドを水冷し、そのストランドを切断してペレット状ポリオレフィン系樹脂組成物を得た。
このペレットを用いて、成形温度220℃(樹脂温度)、金型温度40℃で射出成形して試験片(長さ90mm、幅10mm、高さ4mm)を作製した。
こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表7に示した。
実施例I−2
PTC−2MeCHA(100)に代えてPTC−2MeCHA(74)を用いて、混練温度240℃及び成形温度200℃に変えた以外は実施例I−1と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表7に示した。
また、実施例I−2で得られたポリオレフィン系樹脂組成物の昇温時及び降温時の貯蔵弾性率の温度依存曲線を図5に示し、その微分曲線を図6に示す。図5において、白丸(○)は昇温時の貯蔵弾性率の温度依存曲線であり、黒丸(●)は降温時の貯蔵弾性率の温度依存曲線である。同様に、図6において、白丸(○)は昇温時の貯蔵弾性率の温度依存曲線の微分曲線であり、黒丸(●)は降温時の貯蔵弾性率の温度依存曲線の微分曲線である。
実施例I−3
PTC−2MeCHA(74)0.2重量部を0.1重量部及び成形温度を180℃に変えた以外は実施例I−2と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表7に示した。
実施例I−4
PTC−2MeCHA(100)に代えてPTC−2MeCHA(50)を用いて、混練温度240℃及び成形温度180℃に変えた以外は実施例I−1と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表7に示した。
実施例I−5
PTC−2MeCHA(100)に代えてPTC−CHAを用いて、混練温度240℃及び成形温度180℃に変えた以外は実施例I−1と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表7に示した。
実施例I−6
ドライブレンド時に、さらにステアリン酸カルシウム0.05重量部を用いた以外は実施例I−1と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表7に示した。
実施例I−7
ドライブレンド時に、さらにステアリン酸カルシウム0.05重量部を用いた以外は実施例I−2と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表7に示した。
実施例I−8
ドライブレンド時に、さらにステアリン酸亜鉛0.05重量部を用いた以外は実施例I−2と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表7に示した。
実施例I−9
ドライブレンド時に、さらにステアリン酸マグネシウム0.05重量部を用いた以外は実施例I−2と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表7に示した。
実施例I−10
ドライブレンド時に、さらにステアリン酸カルシウム0.1重量部を用いた以外は実施例I−2と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表7に示した。
実施例I−11
ドライブレンド時に、さらにステアリン酸カルシウム0.05重量部を用いた以外は実施例I−5と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表7に示した。
実施例II−1
実施例I−1で調製したペレットを用いて、成形温度260℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表8に示した。
実施例II−2
実施例I−2で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表8に示した。
実施例II−3
実施例I−3で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表8に示した。
実施例II−4
実施例I−4で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表8に示した。
実施例II−5
実施例I−5で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表8に示した。
実施例II−6
実施例I−6で調製したペレットを用いて、成形温度260℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表8に示した。
実施例II−7
実施例I−7で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表8に示した。
実施例II−8
実施例I−8で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表8に示した。
実施例II−9
実施例I−9で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表8に示した。
実施例II−10
実施例I−10で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表8に示した。
実施例II−11
実施例I−11で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表8に示した。
このことから、本発明に係る成形方法(I)に従い得られ、ポリプロピレン樹脂の結晶ラメラが配向している成形体は、有意に高い剛性を有することが明らかである。
実施例I−12
h−PP100重量部に対して、PTC−2MeCHA(100)0.2重量部、テトラキスメチレン−3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオネート]メタン(チバ・スペシャルティ・ケミカルズ社製:IRGANOX1010(商品名))0.05重量部及びテトラキス(2,4−ジ−t−ブチルフェニル)ホスファイト(チバ・スペシャルティ・ケミカルズ社製:IRGAFOS168(商品名))0.05重量部を秤取り、h−PP100重量部と共にヘンシェルミキサーで1000rpm、5分間ドライブレンドした。
次に、前記ドライブレンド物を混練温度260℃(樹脂温度)で一軸押出機(L/D=440mm/20mm)を用いて溶融混練し、押し出されたストランドを水冷し、そのストランドを切断してペレット状ポリオレフィン樹脂組成物を得た。
このペレットを用いて、成形温度220℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表9に示した。
実施例I−13
PTC−2MeCHA(100)に代えてPTC−2MeCHA(74)を用いて、混練温度240℃及び成形温度200℃に変えた以外は実施例I−12と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表9に示した。
実施例I−14
PTC−2MeCHA(74)0.2重量部を0.1重量部、成形温度を180℃に変えた以外は実施例I−13と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表9に示した。
実施例I−15
PTC−2MeCHA(100)に代えてPTC−2MeCHA(50)を用いて、混練温度240℃及び成形温度180℃に変えた以外は実施例I−12と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表9に示した。
実施例I−16
PTC−2MeCHA(100)に代えてPTC−CHAを用いて、混練温度240℃及び成形温度180℃に変えた以外は実施例I−12と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表9に示した。
実施例I−17
ドライブレンド時に、さらにステアリン酸カルシウム0.05量部を用いた以外は実施例I−12と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表9に示した。
実施例I−18
ドライブレンド時に、さらにステアリン酸カルシウム0.05量部を用いた以外は実施例I−13と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表9に示した。
実施例I−19
ドライブレンド時に、さらにステアリン酸マグネシウム0.05量部を用いた以外は実施例I−13と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表9に示した。
実施例I−20
ドライブレンド時に、さらにステアリン酸亜鉛0.05量部を用いた以外は実施例I−13と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表9に示した。
実施例I−21
ドライブレンド時に、さらにステアリン酸カルシウム0.05量部を用いた以外は実施例I−16と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表9に示した。
実施例II−12
実施例I−12で調製したペレットを用いて、成形温度260℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表10に示した。
実施例II−13
実施例I−13で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表10に示した。
実施例II−14
実施例I−14で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表10に示した。
実施例II−15
実施例I−15で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表10に示した。
実施例II−16
実施例I−16で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表10に示した。
実施例II−17
実施例I−17で調製したペレットを用いて、成形温度260℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表10に示した。
実施例II−18
実施例I−18で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表10に示した。
実施例II−19
実施例I−19で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表10に示した。
実施例II−20
実施例I−20で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表10に示した。
実施例II−21
実施例I−21で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表10に示した。
このことから、本発明に係る成形方法(I)に従い得られ、ポリプロピレン樹脂の結晶ラメラが配向している成形体は、有意に高い剛性を有することが明らかである。
実施例I−22
b−PP100重量部に対して、PTC−2MeCHA(100)0.2重量部、テトラキスメチレン−3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオネート]メタン(チバ・スペシャルティ・ケミカルズ社製:IRGANOX1010(商品名))0.05重量部及びテトラキス(2,4−ジ−t−ブチルフェニル)ホスファイト(チバ・スペシャルティ・ケミカルズ社製:IRGAFOS168(商品名))0.05重量部を秤取り、b−PP100重量部と共にヘンシェルミキサーで1000rpm、5分間ドライブレンドした。
次に、前記ドライブレンド物を混練温度260℃(樹脂温度)で一軸押出機(L/D=440mm/20mm)を用いて溶融混練し、押し出されたストランドを水冷し、そのストランドを切断してペレット状ポリオレフィン樹脂組成物を得た。
このペレットを用いて、成形温度220℃(樹脂温度)、金型温度40℃で射出成形して試験片(長さ90mm、幅10mm、高さ4mm)を作製した。得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表11に示した。
実施例I−23
PTC−2MeCHA(100)に代えてPTC−2MeCHA(74)を用いて、混練温度240℃及び成形温度200℃に変えた以外は実施例I−22と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表11に示した。
実施例I−24
PTC−2MeCHA(74)0.2重量部を0.1重量部、成形温度を180℃に変えた以外は実施例I−23と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表11に示した。
実施例I−25
PTC−2MeCHA(100)に代えてPTC−2MeCHA(50)を用いて、混練温度240℃及び成形温度180℃に変えた以外は実施例I−22と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表11に示した。
実施例I−26
PTC−2MeCHA(100)に代えてPTC−CHAを用いて、混練温度240℃及び成形温度180℃に変えた以外は実施例I−22と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表11に示した。
実施例I−27
ドライブレンド時に、さらにステアリン酸カルシウム0.05量部を用いた以外は実施例I−22と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表11に示した。
実施例I−28
ドライブレンド時に、さらにステアリン酸カルシウム0.05量部を用いた以外は実施例I−23と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表11に示した。
実施例I−29
ドライブレンド時に、さらにステアリン酸マグネシウム0.05量部を用いた以外は実施例I−23と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表11に示した。
実施例I−30
ドライブレンド時に、さらにステアリン酸亜鉛0.05量部を用いた以外は実施例I−23と同様にして試験片を作製した。こうして得られた樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表11に示した。
実施例I−31
ドライブレンド時に、さらにステアリン酸カルシウム0.05量部を用いた以外は実施例I−26と同様にして試験片を作製した。こうして得られたポリオレフィン系樹脂組成物のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表11に示した。
実施例II−22
実施例I−22で調製したペレットを用いて、成形温度260℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表12に示した。
実施例II−23
実施例I−23で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表12に示した。
実施例II−24
実施例I−24で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表12に示した。
実施例II−25
実施例I−25で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表12に示した。
実施例II−26
実施例I−26で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表12に示した。
実施例II−27
実施例I−27で調製したペレットを用いて、成形温度260℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表12に示した。
実施例II−28
実施例I−28で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表12に示した。
実施例II−29
実施例I−29で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表12に示した。
実施例II−30
実施例I−30で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表12に示した。
実施例II−31
実施例I−31で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して試験片を作製した。上記ペレット(樹脂組成物)のTsc(℃)及びTsh(℃)並びに試験片の曲げ弾性率(MPa)及び配向度を表12に示した。
このことから、本発明に係る成形方法(I)に従い得られ、ポリプロピレン樹脂の結晶ラメラが配向している成形体は、有意に高い剛性を有することが明らかである。
実施例I−32
実施例I−28で調製したペレットを用いて、成形温度200℃(樹脂温度)、金型温度40℃で射出成形して耐衝撃性試験用の試験片(厚さ2mmのシート)を作製した。耐衝撃性試験の評価結果を表13に示した。
実施例II−32
実施例I−28で調製したペレットを用いて、成形温度240℃(樹脂温度)、金型温度40℃で射出成形して耐衝撃性試験用の試験片を作製した。耐衝撃性試験の評価結果を表13に示した。
比較例7
比較例6で調製したペレットを用いて、成形温度200℃(樹脂温度)、金型温度40℃で射出成形して耐衝撃性試験用の試験片を作製した。耐衝撃性試験の評価結果を表13に示した。
Claims (27)
- (A)一般式(1)
で表される少なくとも一種のアミド系化合物、及び
(B)一般式(2)
nは、1又は2の整数を表し、n=2の場合、2個のR3は同一又は異なっていてもよい。Mは1価又は2価の金属を表す。]
で表される少なくとも一種の脂肪酸金属塩を含有し、
成分(A):成分(B)の重量比が、95:5〜30:70であるポリオレフィン系樹脂の結晶化速度制御組成物。 - 一般式(1)における3個又は4個のR2が、同一又は異なって、シクロヘキシル基、又は1個の炭素数1〜4のアルキル基で置換されたシクロヘキシル基である請求項1に記載の組成物。
- 一般式(1)における3個又は4個のR2が、同一又は異なって、シクロヘキシル基又は2−メチル基、3−メチル基若しくは4−メチル基で置換されたシクロヘキシル基である請求項1に記載の組成物。
- 一般式(1)におけるR1が1,2,3−プロパントリカルボン酸から全てのカルボキシル基を除いて得られる残基であり、kが3である請求項1に記載の組成物。
- 一般式(2)におけるMが、アルカリ金属、アルカリ土類金属及び亜鉛からなる群より選ばれる少なくとも1種の金属である請求項1に記載の組成物。
- 一般式(2)におけるR3が、分子内に1個以上の水酸基を有していてもよい炭素数10〜18の飽和若しくは不飽和の脂肪族モノカルボン酸からカルボキシル基を除いて得られる残基である請求項1に記載の組成物。
- 脂肪族モノカルボン酸が、ラウリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、オレイン酸及び12−ヒドロキシステアリン酸からなる群より選ばれる少なくとも1種である請求項6に記載の組成物。
- ポリオレフィン系樹脂の成形時のポリオレフィン系樹脂の結晶化速度を制御する方法であって、
(A)一般式(1)
で表される少なくとも一種のアミド系化合物、及び
(B)一般式(2)
nは、1又は2の整数を表し、n=2の場合、2個のR3は同一又は異なっていてもよい。Mは1価又は2価の金属を表す。]
で表される少なくとも一種の脂肪酸金属塩を含有し、成分(A):成分(B)の重量比が95:5〜30:70であるポリオレフィン系樹脂結晶化速度制御組成物を該ポリオレフィン系樹脂に配合するか、又は、
上記成分(A)と成分(B)とを、同時に又は別々に、成分(A):成分(B)の重量比が95:5〜30:70となるように、該ポリオレフィン系樹脂に配合することにより、ポリオレフィン系樹脂組成物を得ること、及び
該樹脂組成物を成形することを含む方法。 - 樹脂組成物を、昇温時の貯蔵弾性率の転移温度を超える樹脂温度で成形する請求項8に記載の方法。
- 樹脂組成物を、ポリオレフィン系樹脂の融解温度以上であって、且つ、昇温時の貯蔵弾性率の転移温度以下の樹脂温度で成形する請求項8に記載の方法。
- (A)一般式(1)
で表される少なくとも一種のアミド系化合物、及び
(B)一般式(2)
nは、1又は2の整数を表し、n=2の場合、2個のR3は同一又は異なっていてもよい。Mは1価又は2価の金属を表す。]
で表される少なくとも一種の脂肪酸金属塩を、成分(A):成分(B)の重量比が95:5〜30:70の割合で含有する組成物の、ポリオレフィン系樹脂の成形時のポリオレフィン系樹脂の結晶化速度を制御するための使用。 - (A)一般式(1)
で表される少なくとも一種のアミド系化合物、及び
(B)一般式(2)
nは、1又は2の整数を表し、n=2の場合、2個のR3は同一又は異なっていてもよい。Mは1価又は2価の金属を表す。]
で表される少なくとも一種の脂肪酸金属塩を含有し、成分(A):成分(B)の重量比が95:5〜30:70であるポリオレフィン系樹脂結晶化速度制御組成物をポリオレフィン系樹脂に配合するか、又は、
上記成分(A)と成分(B)とを、同時にまたは別々に、成分(A):成分(B)の重量比が95:5〜30:70となるように、ポリオレフィン系樹脂に配合することにより、ポリオレフィン系樹脂組成物を得ること、及び
該樹脂組成物を成形することを含むポリオレフィン系樹脂成形体の製造方法。 - 樹脂組成物を、昇温時の貯蔵弾性率の転移温度を超える樹脂温度で成形する請求項12に記載の製造方法。
- 樹脂組成物を、ポリオレフィン系樹脂の融解温度以上であって、且つ、昇温時の貯蔵弾性率の転移温度以下の樹脂温度で成形することを特徴とする請求項12に記載の製造方法。
- 一般式(1−p)
で表される少なくとも1種のアミド系化合物の繊維状粒子により形成された網目構造を含む溶融ポリオレフィン系樹脂組成物を、該網目構造を構成する繊維状粒子が溶解又は溶融しない温度条件で、成形する請求項14に記載の製造法。 - (a)一般式(1−p)で表される少なくとも1種のアミド系化合物及び成分(B)を含むポリオレフィン系樹脂結晶化速度制御組成物をポリオレフィン系樹脂に配合するか、又は、
一般式(1−p)で表される少なくとも1種のアミド系化合物及び成分(B)とを、同時に又は別々に、ポリオレフィン系樹脂に配合し、
該ポリオレフィン系樹脂を溶融させ、
一般式(1−p)で表される少なくとも1種のアミド系化合物を溶融ポリオレフィン系樹脂に溶解させて溶融混合物を得る工程、
(b)該溶融混合物を、降温時の貯蔵弾性率の転移温度以下に冷却して、一般式(1−p)で表される少なくとも1種のアミド系化合物の繊維状粒子により形成される網目構造を含むポリオレフィン系樹脂組成物を得る工程、及び
(c)該ポリオレフィン系樹脂組成物を、ポリオレフィン系樹脂の溶融温度以上であって、且つ、昇温時の貯蔵弾性率の転移温度以下の樹脂温度で成形する工程
を備えている請求項15に記載の製造法。 - 上記ポリオレフィン系樹脂組成物が、ペレットの形態にある請求項16に記載の製造法。
- 一般式(2)
- 上記繊維状粒子により形成される網目構造を含むポリオレフィン系樹脂組成物を、射出工程又は押し出し工程を含む成形方法により成形する請求項15に記載の製造法。
- 前記射出工程又は押し出し工程を含む成形方法が、射出成形、押し出し成形、射出ブロー成形、射出押出ブロー成形、射出圧縮成形、押し出しブロー成形、押し出しサーモフォーム成形又は溶融紡糸である請求項19に記載の製造法。
- 上記ポリオレフィン系樹脂が、プロピレンホモポリマー及びプロピレンコポリマーから選ばれる少なくとも1種である請求項12に記載の製造法。
- 請求項14に記載のポリオレフィン系樹脂成形体の製造法により得られ、広角X線回折により求められる(040)反射強度の(110)反射強度に対する比で表される配向度が2以上であるポリオレフィン系樹脂成形体。
- ポリオレフィン系樹脂、
(A)一般式(1)
で表される少なくとも一種のアミド系化合物、及び
(B)一般式(2)
nは、1又は2の整数を表し、n=2の場合、2個のR3は同一又は異なっていてもよい。Mは1価又は2価の金属を表す。]
で表される少なくとも一種の脂肪酸金属塩を、成分(A):成分(B)の重量比が、95:5〜30:70の比率の範囲で含有し、広角X線回折により求められる(040)反射強度の(110)反射強度に対する比で表される配向度が2以上であるポリオレフィン系樹脂成形体。 - ポリオレフィン系樹脂、
(A)一般式(1−p)
(B)一般式(2)
nは、1又は2の整数を表し、n=2の場合、2個のR3は同一又は相異なっていてもよい。Mは1価又は2価の金属を表す。]
で表される少なくとも1種の脂肪酸金属塩を、成分(A):成分(B)の重量比が、95:5〜30:70の比率の範囲で含有し、広角X線回折により求められる(040)反射強度の(110)反射強度に対する比で表される配向度が2以上であるポリオレフィン系樹脂成形体。 - ポリオレフィン系樹脂及び請求項1に記載の結晶化速度制御組成物を含有するポリオレフィン系樹脂組成物。
- ポリオレフィン系樹脂100重量部に対し、結晶化速度制御組成物を0.01〜10重量部含有する請求項25に記載のポリオレフィン系樹脂組成物。
- 請求項25に記載のポリオレフィン系樹脂組成物を成形して得ることができるポリオレフィン系樹脂成形体。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005516714A JP4835159B2 (ja) | 2003-12-26 | 2004-12-22 | ポリオレフィン系樹脂の結晶化速度制御のための組成物及び方法、樹脂組成物及び樹脂成形体 |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003432138 | 2003-12-26 | ||
| JP2003432138 | 2003-12-26 | ||
| JP2004169266 | 2004-06-07 | ||
| JP2004169266 | 2004-06-07 | ||
| PCT/JP2004/019701 WO2005063874A1 (ja) | 2003-12-26 | 2004-12-22 | ポリオレフィン系樹脂の結晶化速度制御のための組成物及び方法、樹脂組成物及び樹脂成形体 |
| JP2005516714A JP4835159B2 (ja) | 2003-12-26 | 2004-12-22 | ポリオレフィン系樹脂の結晶化速度制御のための組成物及び方法、樹脂組成物及び樹脂成形体 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2005063874A1 JPWO2005063874A1 (ja) | 2007-07-19 |
| JP4835159B2 true JP4835159B2 (ja) | 2011-12-14 |
Family
ID=34742132
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005516714A Expired - Fee Related JP4835159B2 (ja) | 2003-12-26 | 2004-12-22 | ポリオレフィン系樹脂の結晶化速度制御のための組成物及び方法、樹脂組成物及び樹脂成形体 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US7723413B2 (ja) |
| EP (2) | EP2083045A1 (ja) |
| JP (1) | JP4835159B2 (ja) |
| KR (1) | KR101154564B1 (ja) |
| CN (1) | CN1898317B (ja) |
| AT (1) | ATE460457T1 (ja) |
| BR (1) | BRPI0418175A (ja) |
| DE (1) | DE602004025980D1 (ja) |
| SA (1) | SA04250433B1 (ja) |
| TW (1) | TW200535185A (ja) |
| WO (1) | WO2005063874A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012246377A (ja) * | 2011-05-26 | 2012-12-13 | Canon Inc | 顔料分散体、イエロートナー |
| JP2012251135A (ja) * | 2011-05-10 | 2012-12-20 | Canon Inc | 顔料分散体及び該顔料分散体を用いたインク組成物、カラーフィルター用黄色レジスト組成物 |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4792973B2 (ja) | 2003-10-17 | 2011-10-12 | 新日本理化株式会社 | アミド系化合物、ポリオレフィン樹脂組成物及び成形体 |
| JP4940601B2 (ja) * | 2005-09-01 | 2012-05-30 | 新日本理化株式会社 | 新規なポリプロピレン樹脂組成物、該組成物を用いたシート又はフィルム成形体及びその製造方法 |
| JP2007092020A (ja) * | 2005-09-01 | 2007-04-12 | New Japan Chem Co Ltd | ブロー成形用ポリプロピレン樹脂組成物、該組成物を用いたブロー成形品及びその製造方法 |
| JP2007231134A (ja) * | 2006-02-28 | 2007-09-13 | New Japan Chem Co Ltd | 改良されたポリプロピレン樹脂組成物及びその成形体 |
| WO2007107579A1 (en) * | 2006-03-21 | 2007-09-27 | Basell Poliolefine Italia S.R.L. | A controlled nucleated polymer composition |
| US20120028006A1 (en) * | 2009-01-31 | 2012-02-02 | New Japan Chemical Co., Ltd. | Polypropylene resin molded article |
| JP5722002B2 (ja) * | 2010-11-10 | 2015-05-20 | 株式会社Adeka | ポリプロピレン系樹脂組成物 |
| US9200142B2 (en) | 2013-09-23 | 2015-12-01 | Milliken & Company | Thermoplastic polymer composition |
| US9580575B2 (en) | 2013-09-23 | 2017-02-28 | Milliken & Company | Polyethylene articles |
| US9193845B2 (en) | 2013-09-23 | 2015-11-24 | Milliken & Company | Thermoplastic polymer composition |
| US9200144B2 (en) | 2013-09-23 | 2015-12-01 | Milliken & Company | Thermoplastic polymer composition |
| US9120914B2 (en) | 2013-09-23 | 2015-09-01 | Milliken & Company | Thermoplastic polymer composition |
| JP6875051B2 (ja) | 2014-10-15 | 2021-05-19 | 株式会社ブリヂストン | 改善されたゴム組成物及びその使用 |
| CN107001707B (zh) | 2014-10-15 | 2019-02-22 | 株式会社普利司通 | 改善的橡胶组合物以及其应用 |
| KR101674609B1 (ko) * | 2015-05-07 | 2016-11-09 | 롯데케미칼 주식회사 | 폴리올레핀 수지 단일칩 제조방법 및 폴리올레핀 수지 단일칩 |
| WO2019082930A1 (ja) * | 2017-10-25 | 2019-05-02 | 株式会社Adeka | 結晶化遅延剤、これを含有する組成物、オレフィン系樹脂組成物、その成形品、およびオレフィン樹脂の結晶化の遅延方法 |
| US11535733B2 (en) * | 2019-02-28 | 2022-12-27 | Fina Technology, Inc. | Clarified polypropylene for long term color performance |
| US11667603B2 (en) | 2019-12-12 | 2023-06-06 | Milliken & Company | Trisamide compounds and compositions comprising the same |
| KR20220114022A (ko) | 2019-12-12 | 2022-08-17 | 밀리켄 앤드 캄파니 | 트리스아미드 화합물 및 이를 포함하는 조성물 |
| CN116583543A (zh) | 2020-12-14 | 2023-08-11 | 美利肯公司 | 三酰胺化合物和包含所述三酰胺化合物的组合物 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06192496A (ja) * | 1992-10-22 | 1994-07-12 | New Japan Chem Co Ltd | ポリプロピレン系樹脂組成物 |
| JPH06234890A (ja) * | 1993-02-08 | 1994-08-23 | New Japan Chem Co Ltd | ポリエチレン系樹脂組成物 |
| JPH07242610A (ja) * | 1994-03-07 | 1995-09-19 | New Japan Chem Co Ltd | 新規なアミド系化合物 |
| JPH07278374A (ja) * | 1994-04-06 | 1995-10-24 | New Japan Chem Co Ltd | ポリプロピレン系樹脂組成物 |
| JPH10279739A (ja) * | 1997-04-08 | 1998-10-20 | New Japan Chem Co Ltd | ポリオレフィン樹脂用核剤組成物並びにポリオレフィン樹脂組成物 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2983314B2 (ja) * | 1991-02-14 | 1999-11-29 | 旭電化工業株式会社 | 透明性の改善されたポリオレフィン系樹脂組成物 |
| JP3480064B2 (ja) | 1994-09-29 | 2003-12-15 | 新日本理化株式会社 | ポリプロピレン系樹脂組成物 |
| JP3991085B2 (ja) * | 1994-10-04 | 2007-10-17 | 新日本理化株式会社 | 結晶性合成樹脂組成物 |
| JPH10273569A (ja) * | 1997-03-28 | 1998-10-13 | Grand Polymer:Kk | ポリプロピレン系樹脂組成物 |
| US5969021A (en) * | 1997-08-07 | 1999-10-19 | Fina Technology, Inc. | Method of improving crystallization rate and temperature of syndiotactic polypropylene and compositions and products made therefrom |
| EP1431335A4 (en) * | 2001-09-12 | 2004-11-17 | New Japan Chem Co Ltd | POLYPROPYLENE RESIN MOLDED BODIES AND PRODUCTION METHOD THEREFOR |
| JP4261447B2 (ja) | 2004-09-10 | 2009-04-30 | 株式会社日立製作所 | 通信システム |
-
2004
- 2004-12-22 EP EP09157014A patent/EP2083045A1/en not_active Withdrawn
- 2004-12-22 EP EP04808052A patent/EP1715000B1/en not_active Expired - Lifetime
- 2004-12-22 WO PCT/JP2004/019701 patent/WO2005063874A1/ja active Application Filing
- 2004-12-22 DE DE602004025980T patent/DE602004025980D1/de not_active Expired - Lifetime
- 2004-12-22 BR BRPI0418175-1A patent/BRPI0418175A/pt not_active IP Right Cessation
- 2004-12-22 AT AT04808052T patent/ATE460457T1/de not_active IP Right Cessation
- 2004-12-22 US US10/583,000 patent/US7723413B2/en not_active Expired - Fee Related
- 2004-12-22 CN CN2004800389531A patent/CN1898317B/zh not_active Expired - Fee Related
- 2004-12-22 KR KR1020067014976A patent/KR101154564B1/ko not_active Expired - Lifetime
- 2004-12-22 JP JP2005516714A patent/JP4835159B2/ja not_active Expired - Fee Related
- 2004-12-23 TW TW093140251A patent/TW200535185A/zh not_active IP Right Cessation
- 2004-12-25 SA SA04250433A patent/SA04250433B1/ar unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06192496A (ja) * | 1992-10-22 | 1994-07-12 | New Japan Chem Co Ltd | ポリプロピレン系樹脂組成物 |
| JPH06234890A (ja) * | 1993-02-08 | 1994-08-23 | New Japan Chem Co Ltd | ポリエチレン系樹脂組成物 |
| JPH07242610A (ja) * | 1994-03-07 | 1995-09-19 | New Japan Chem Co Ltd | 新規なアミド系化合物 |
| JPH07278374A (ja) * | 1994-04-06 | 1995-10-24 | New Japan Chem Co Ltd | ポリプロピレン系樹脂組成物 |
| JPH10279739A (ja) * | 1997-04-08 | 1998-10-20 | New Japan Chem Co Ltd | ポリオレフィン樹脂用核剤組成物並びにポリオレフィン樹脂組成物 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012251135A (ja) * | 2011-05-10 | 2012-12-20 | Canon Inc | 顔料分散体及び該顔料分散体を用いたインク組成物、カラーフィルター用黄色レジスト組成物 |
| JP2012246377A (ja) * | 2011-05-26 | 2012-12-13 | Canon Inc | 顔料分散体、イエロートナー |
Also Published As
| Publication number | Publication date |
|---|---|
| DE602004025980D1 (de) | 2010-04-22 |
| EP1715000A1 (en) | 2006-10-25 |
| JPWO2005063874A1 (ja) | 2007-07-19 |
| ATE460457T1 (de) | 2010-03-15 |
| EP1715000B1 (en) | 2010-03-10 |
| TWI359842B (ja) | 2012-03-11 |
| US20070142514A1 (en) | 2007-06-21 |
| CN1898317A (zh) | 2007-01-17 |
| KR20070007057A (ko) | 2007-01-12 |
| WO2005063874A1 (ja) | 2005-07-14 |
| SA04250433B1 (ar) | 2010-01-20 |
| CN1898317B (zh) | 2010-11-24 |
| EP2083045A1 (en) | 2009-07-29 |
| TW200535185A (en) | 2005-11-01 |
| US7723413B2 (en) | 2010-05-25 |
| KR101154564B1 (ko) | 2012-06-08 |
| EP1715000A4 (en) | 2007-01-24 |
| BRPI0418175A (pt) | 2007-04-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4835159B2 (ja) | ポリオレフィン系樹脂の結晶化速度制御のための組成物及び方法、樹脂組成物及び樹脂成形体 | |
| JP3826187B2 (ja) | ポリオレフィン用改質剤 | |
| JP5239869B2 (ja) | 新規なポリオレフィン系樹脂組成物及びその樹脂成形体 | |
| JP2001507395A (ja) | ポリオレフィン用改質剤 | |
| JP3358659B2 (ja) | ジアセタール組成物、該組成物を含むポリオレフィン用核剤、該ジアセタール組成物を含有するポリオレフィン樹脂組成物、該樹脂組成物の製造法、及び成形体 | |
| WO1999018108A1 (fr) | Composition de diacetal, procede de preparation, agent de nucleation pour polyolefines contenant une telle composition, compositions de resines de polyolefines, et moulages | |
| EP0861877B1 (en) | Rosin-based molecular crystal, nucleating agent for polyolefin resin, polyolefin resin composition, and moldings thereof | |
| JP4792973B2 (ja) | アミド系化合物、ポリオレフィン樹脂組成物及び成形体 | |
| JPH08239386A (ja) | ジアセタールの六方晶結晶、該六方晶結晶を含む核剤、該六方晶結晶を含むポリオレフィン系樹脂組成物及び成形物、並びに該組成物の成形方法 | |
| JP5794145B2 (ja) | アミド化合物の結晶成長速度を抑制する方法及びポリオレフィン系樹脂成形体の製造方法 | |
| JP3921750B2 (ja) | ロジン系分子結晶及びポリオレフィン樹脂用核剤並びにポリオレフィン樹脂組成物とその成形体 | |
| JPH11130964A (ja) | ロジン系ガラス化物及びポリオレフィン樹脂用核剤並びにポリオレフィン樹脂組成物とその成形体 | |
| JP5525830B2 (ja) | ポリ乳酸樹脂組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070523 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20101124 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110117 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110322 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110330 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20110525 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110614 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110803 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20110830 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20110912 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20141007 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20141007 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20141007 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |