JP4316794B2 - イソキノリン誘導体及び医薬 - Google Patents
イソキノリン誘導体及び医薬 Download PDFInfo
- Publication number
- JP4316794B2 JP4316794B2 JP2000516962A JP2000516962A JP4316794B2 JP 4316794 B2 JP4316794 B2 JP 4316794B2 JP 2000516962 A JP2000516962 A JP 2000516962A JP 2000516962 A JP2000516962 A JP 2000516962A JP 4316794 B2 JP4316794 B2 JP 4316794B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- pharmaceutical composition
- composition according
- cerebral
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 239000003814 drug Substances 0.000 title claims description 28
- 125000002183 isoquinolinyl group Chemical class C1(=NC=CC2=CC=CC=C12)* 0.000 title description 5
- 150000001875 compounds Chemical class 0.000 claims description 96
- 208000026106 cerebrovascular disease Diseases 0.000 claims description 21
- 239000008194 pharmaceutical composition Substances 0.000 claims description 16
- 208000001286 intracranial vasospasm Diseases 0.000 claims description 14
- 229940124597 therapeutic agent Drugs 0.000 claims description 14
- 206010008118 cerebral infarction Diseases 0.000 claims description 13
- 150000003839 salts Chemical class 0.000 claims description 9
- 230000000069 prophylactic effect Effects 0.000 claims description 8
- 230000000302 ischemic effect Effects 0.000 claims description 7
- 239000003795 chemical substances by application Substances 0.000 claims description 6
- 206010002383 Angina Pectoris Diseases 0.000 claims description 5
- 206010008111 Cerebral haemorrhage Diseases 0.000 claims description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 5
- 206010019196 Head injury Diseases 0.000 claims description 4
- 239000004480 active ingredient Substances 0.000 claims description 4
- 201000010099 disease Diseases 0.000 claims description 4
- 230000007654 ischemic lesion Effects 0.000 claims description 4
- 210000002569 neuron Anatomy 0.000 claims description 4
- 239000003112 inhibitor Substances 0.000 claims description 3
- 208000010125 myocardial infarction Diseases 0.000 claims description 3
- 208000031225 myocardial ischemia Diseases 0.000 claims description 2
- 206010028851 Necrosis Diseases 0.000 claims 1
- 208000032109 Transient ischaemic attack Diseases 0.000 claims 1
- 230000017074 necrotic cell death Effects 0.000 claims 1
- 239000003223 protective agent Substances 0.000 claims 1
- 201000010875 transient cerebral ischemia Diseases 0.000 claims 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 48
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 46
- 239000000243 solution Substances 0.000 description 45
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 32
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 30
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 27
- 238000012360 testing method Methods 0.000 description 25
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 24
- 230000017531 blood circulation Effects 0.000 description 23
- 238000006243 chemical reaction Methods 0.000 description 23
- 239000000203 mixture Substances 0.000 description 22
- 229960002435 fasudil Drugs 0.000 description 21
- LFVPBERIVUNMGV-UHFFFAOYSA-N fasudil hydrochloride Chemical compound Cl.C=1C=CC2=CN=CC=C2C=1S(=O)(=O)N1CCCNCC1 LFVPBERIVUNMGV-UHFFFAOYSA-N 0.000 description 21
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 19
- 229910001868 water Inorganic materials 0.000 description 19
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 18
- 238000000034 method Methods 0.000 description 18
- 239000002904 solvent Substances 0.000 description 17
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 16
- 235000019341 magnesium sulphate Nutrition 0.000 description 16
- -1 lipid peroxide Chemical class 0.000 description 15
- 230000000694 effects Effects 0.000 description 14
- UFMZFWOWTOGZIK-QMMMGPOBSA-N 4-fluoro-n-[(2s)-1-hydroxypropan-2-yl]isoquinoline-5-sulfonamide Chemical compound N1=CC(F)=C2C(S(=O)(=O)N[C@H](CO)C)=CC=CC2=C1 UFMZFWOWTOGZIK-QMMMGPOBSA-N 0.000 description 13
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- 241000700159 Rattus Species 0.000 description 13
- 210000003657 middle cerebral artery Anatomy 0.000 description 13
- 229940079593 drug Drugs 0.000 description 12
- 230000001965 increasing effect Effects 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 11
- COBSKSLDBJBGEK-MERQFXBCSA-N 4-fluoro-5-[[(2s)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline;hydrochloride Chemical compound [Cl-].C[C@H]1C[NH2+]CCCN1S(=O)(=O)C1=CC=CC2=CN=CC(F)=C12 COBSKSLDBJBGEK-MERQFXBCSA-N 0.000 description 10
- 230000009471 action Effects 0.000 description 10
- 230000002490 cerebral effect Effects 0.000 description 10
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 239000002585 base Substances 0.000 description 9
- 238000001816 cooling Methods 0.000 description 9
- 238000010898 silica gel chromatography Methods 0.000 description 9
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 9
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 8
- 210000004204 blood vessel Anatomy 0.000 description 8
- 210000003710 cerebral cortex Anatomy 0.000 description 8
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 8
- 150000001412 amines Chemical class 0.000 description 7
- 238000002474 experimental method Methods 0.000 description 7
- 238000009472 formulation Methods 0.000 description 7
- 238000004519 manufacturing process Methods 0.000 description 7
- 239000012044 organic layer Substances 0.000 description 7
- 125000006239 protecting group Chemical group 0.000 description 7
- 206010002091 Anaesthesia Diseases 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 206010008089 Cerebral artery occlusion Diseases 0.000 description 6
- 229940126062 Compound A Drugs 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 6
- 206010061216 Infarction Diseases 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 6
- 230000037005 anaesthesia Effects 0.000 description 6
- 239000008280 blood Substances 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 6
- 230000000052 comparative effect Effects 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 230000007574 infarction Effects 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 201000007309 middle cerebral artery infarction Diseases 0.000 description 6
- 210000001577 neostriatum Anatomy 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 239000007789 gas Substances 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- WEXRUCMBJFQVBZ-UHFFFAOYSA-N pentobarbital Chemical compound CCCC(C)C1(CC)C(=O)NC(=O)NC1=O WEXRUCMBJFQVBZ-UHFFFAOYSA-N 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 4
- 208000032843 Hemorrhage Diseases 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 208000032851 Subarachnoid Hemorrhage Diseases 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- 206010047163 Vasospasm Diseases 0.000 description 4
- 230000036772 blood pressure Effects 0.000 description 4
- 210000004556 brain Anatomy 0.000 description 4
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 4
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 235000019359 magnesium stearate Nutrition 0.000 description 4
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 4
- 235000015097 nutrients Nutrition 0.000 description 4
- 239000002504 physiological saline solution Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 230000002265 prevention Effects 0.000 description 4
- 239000002994 raw material Substances 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 4
- 230000001052 transient effect Effects 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- UZUCKWMDQGESLP-UHFFFAOYSA-N 4-fluoroisoquinoline-5-sulfonyl chloride Chemical compound C1=CC(S(Cl)(=O)=O)=C2C(F)=CN=CC2=C1 UZUCKWMDQGESLP-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 3
- 241000282472 Canis lupus familiaris Species 0.000 description 3
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 3
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 3
- 239000004372 Polyvinyl alcohol Substances 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- WUGQZFFCHPXWKQ-UHFFFAOYSA-N Propanolamine Chemical compound NCCCO WUGQZFFCHPXWKQ-UHFFFAOYSA-N 0.000 description 3
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 3
- 208000007536 Thrombosis Diseases 0.000 description 3
- 235000011114 ammonium hydroxide Nutrition 0.000 description 3
- 210000001841 basilar artery Anatomy 0.000 description 3
- 230000000740 bleeding effect Effects 0.000 description 3
- 238000010531 catalytic reduction reaction Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000008602 contraction Effects 0.000 description 3
- 238000000921 elemental analysis Methods 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 3
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 3
- 208000028867 ischemia Diseases 0.000 description 3
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 229960001412 pentobarbital Drugs 0.000 description 3
- 229920002451 polyvinyl alcohol Polymers 0.000 description 3
- 230000003449 preventive effect Effects 0.000 description 3
- 230000002633 protecting effect Effects 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 210000003462 vein Anatomy 0.000 description 3
- 239000008215 water for injection Substances 0.000 description 3
- 239000008096 xylene Substances 0.000 description 3
- BKMMTJMQCTUHRP-VKHMYHEASA-N (S)-2-aminopropan-1-ol Chemical compound C[C@H](N)CO BKMMTJMQCTUHRP-VKHMYHEASA-N 0.000 description 2
- UEHCZLGUARZVFU-UHFFFAOYSA-N 4-fluoro-5-nitroisoquinoline Chemical compound N1=CC(F)=C2C([N+](=O)[O-])=CC=CC2=C1 UEHCZLGUARZVFU-UHFFFAOYSA-N 0.000 description 2
- WSUBMVBAMAKLFM-UHFFFAOYSA-N 4-fluoroisoquinolin-5-amine Chemical compound N1=CC(F)=C2C(N)=CC=CC2=C1 WSUBMVBAMAKLFM-UHFFFAOYSA-N 0.000 description 2
- VFFQGPWQVYUFLV-UHFFFAOYSA-N 4-fluoroisoquinoline Chemical compound C1=CC=C2C(F)=CN=CC2=C1 VFFQGPWQVYUFLV-UHFFFAOYSA-N 0.000 description 2
- HIYAVKIYRIFSCZ-CYEMHPAKSA-N 5-(methylamino)-2-[[(2S,3R,5R,6S,8R,9R)-3,5,9-trimethyl-2-[(2S)-1-oxo-1-(1H-pyrrol-2-yl)propan-2-yl]-1,7-dioxaspiro[5.5]undecan-8-yl]methyl]-1,3-benzoxazole-4-carboxylic acid Chemical compound O=C([C@@H](C)[C@H]1O[C@@]2([C@@H](C[C@H]1C)C)O[C@@H]([C@@H](CC2)C)CC=1OC2=CC=C(C(=C2N=1)C(O)=O)NC)C1=CC=CN1 HIYAVKIYRIFSCZ-CYEMHPAKSA-N 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 238000001061 Dunnett's test Methods 0.000 description 2
- 239000004129 EU approved improving agent Substances 0.000 description 2
- 0 FC1=CNCc2cccc(*(I)I)c12 Chemical compound FC1=CNCc2cccc(*(I)I)c12 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- 239000004677 Nylon Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical class OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 2
- 206010057469 Vascular stenosis Diseases 0.000 description 2
- TXFACSSUKSDROL-JTQLQIEISA-N [(2s)-2-(phenylmethoxycarbonylamino)propyl] methanesulfonate Chemical compound CS(=O)(=O)OC[C@H](C)NC(=O)OCC1=CC=CC=C1 TXFACSSUKSDROL-JTQLQIEISA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 238000010306 acid treatment Methods 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 230000007059 acute toxicity Effects 0.000 description 2
- 231100000403 acute toxicity Toxicity 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 238000002583 angiography Methods 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- JRDLCUCACRHAQS-LBPRGKRZSA-N benzyl n-[(2s)-1-(3-hydroxypropylamino)propan-2-yl]carbamate Chemical compound OCCCNC[C@H](C)NC(=O)OCC1=CC=CC=C1 JRDLCUCACRHAQS-LBPRGKRZSA-N 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 230000003925 brain function Effects 0.000 description 2
- 239000003710 calcium ionophore Substances 0.000 description 2
- HIYAVKIYRIFSCZ-UHFFFAOYSA-N calcium ionophore A23187 Natural products N=1C2=C(C(O)=O)C(NC)=CC=C2OC=1CC(C(CC1)C)OC1(C(CC1C)C)OC1C(C)C(=O)C1=CC=CN1 HIYAVKIYRIFSCZ-UHFFFAOYSA-N 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 230000004087 circulation Effects 0.000 description 2
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 2
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 125000002576 diazepinyl group Chemical group N1N=C(C=CC=C1)* 0.000 description 2
- 239000012954 diazonium Substances 0.000 description 2
- 150000001989 diazonium salts Chemical class 0.000 description 2
- 238000001647 drug administration Methods 0.000 description 2
- CJAONIOAQZUHPN-KKLWWLSJSA-N ethyl 12-[[2-[(2r,3r)-3-[2-[(12-ethoxy-12-oxododecyl)-methylamino]-2-oxoethoxy]butan-2-yl]oxyacetyl]-methylamino]dodecanoate Chemical compound CCOC(=O)CCCCCCCCCCCN(C)C(=O)CO[C@H](C)[C@@H](C)OCC(=O)N(C)CCCCCCCCCCCC(=O)OCC CJAONIOAQZUHPN-KKLWWLSJSA-N 0.000 description 2
- 210000003191 femoral vein Anatomy 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- BCQZXOMGPXTTIC-UHFFFAOYSA-N halothane Chemical compound FC(F)(F)C(Cl)Br BCQZXOMGPXTTIC-UHFFFAOYSA-N 0.000 description 2
- 229960003132 halothane Drugs 0.000 description 2
- 239000007902 hard capsule Substances 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- ISIUXVGHQFJYHM-UHFFFAOYSA-N isoquinolin-4-amine Chemical compound C1=CC=C2C(N)=CN=CC2=C1 ISIUXVGHQFJYHM-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 229940031703 low substituted hydroxypropyl cellulose Drugs 0.000 description 2
- 235000011090 malic acid Nutrition 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 229920001778 nylon Polymers 0.000 description 2
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- FGIUAXJPYTZDNR-UHFFFAOYSA-N potassium nitrate Chemical compound [K+].[O-][N+]([O-])=O FGIUAXJPYTZDNR-UHFFFAOYSA-N 0.000 description 2
- 238000004393 prognosis Methods 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 230000029058 respiratory gaseous exchange Effects 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 210000003625 skull Anatomy 0.000 description 2
- 235000010288 sodium nitrite Nutrition 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 238000010186 staining Methods 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 150000003460 sulfonic acids Chemical class 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- FWPIDFUJEMBDLS-UHFFFAOYSA-L tin(II) chloride dihydrate Chemical compound O.O.Cl[Sn]Cl FWPIDFUJEMBDLS-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 208000019553 vascular disease Diseases 0.000 description 2
- 229940124549 vasodilator Drugs 0.000 description 2
- 239000003071 vasodilator agent Substances 0.000 description 2
- BJEPYKJPYRNKOW-UWTATZPHSA-N (R)-malic acid Chemical compound OC(=O)[C@H](O)CC(O)=O BJEPYKJPYRNKOW-UWTATZPHSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- USVVENVKYJZFMW-ONEGZZNKSA-N (e)-carboxyiminocarbamic acid Chemical compound OC(=O)\N=N\C(O)=O USVVENVKYJZFMW-ONEGZZNKSA-N 0.000 description 1
- LRANPJDWHYRCER-UHFFFAOYSA-N 1,2-diazepine Chemical compound N1C=CC=CC=N1 LRANPJDWHYRCER-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- BKMMTJMQCTUHRP-UHFFFAOYSA-N 2-aminopropan-1-ol Chemical compound CC(N)CO BKMMTJMQCTUHRP-UHFFFAOYSA-N 0.000 description 1
- URWZLMUTDCFNFV-UHFFFAOYSA-N 2-methyl-1h-1,4-diazepine Chemical compound CC1=CN=CC=CN1 URWZLMUTDCFNFV-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- SCRBSGZBTHKAHU-UHFFFAOYSA-N 4-bromoisoquinoline Chemical compound C1=CC=C2C(Br)=CN=CC2=C1 SCRBSGZBTHKAHU-UHFFFAOYSA-N 0.000 description 1
- QSKQVZWVLOIIEV-UHFFFAOYSA-N 4-fluoro-5-[(2-methyl-1,4-diazepan-1-yl)sulfonyl]isoquinoline Chemical compound CC1CNCCCN1S(=O)(=O)C1=CC=CC2=CN=CC(F)=C12 QSKQVZWVLOIIEV-UHFFFAOYSA-N 0.000 description 1
- QSKQVZWVLOIIEV-LLVKDONJSA-N 4-fluoro-5-[[(2R)-2-methyl-1,4-diazepan-1-yl]sulfonyl]isoquinoline Chemical compound C[C@@H]1CNCCCN1S(=O)(=O)c1cccc2cncc(F)c12 QSKQVZWVLOIIEV-LLVKDONJSA-N 0.000 description 1
- HYOFAZNITIMWJS-NSHDSACASA-N 4-fluoro-n-[(2s)-1-(3-hydroxypropylamino)propan-2-yl]isoquinoline-5-sulfonamide Chemical compound N1=CC(F)=C2C(S(=O)(=O)N[C@H](CNCCCO)C)=CC=CC2=C1 HYOFAZNITIMWJS-NSHDSACASA-N 0.000 description 1
- CVICEEPAFUYBJG-UHFFFAOYSA-N 5-chloro-2,2-difluoro-1,3-benzodioxole Chemical group C1=C(Cl)C=C2OC(F)(F)OC2=C1 CVICEEPAFUYBJG-UHFFFAOYSA-N 0.000 description 1
- 206010005746 Blood pressure fluctuation Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- AWDORCFLUJZUQS-UHFFFAOYSA-N CC(CNCCC1)N1S(c1c(c(C)cnc2)c2ccc1)(=O)=O Chemical compound CC(CNCCC1)N1S(c1c(c(C)cnc2)c2ccc1)(=O)=O AWDORCFLUJZUQS-UHFFFAOYSA-N 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- ZDQWESQEGGJUCH-UHFFFAOYSA-N Diisopropyl adipate Chemical compound CC(C)OC(=O)CCCCC(=O)OC(C)C ZDQWESQEGGJUCH-UHFFFAOYSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 208000000386 Hypertensive Intracranial Hemorrhage Diseases 0.000 description 1
- 208000004044 Hypesthesia Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- FEWJPZIEWOKRBE-XIXRPRMCSA-N Mesotartaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-XIXRPRMCSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 208000016285 Movement disease Diseases 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 206010033799 Paralysis Diseases 0.000 description 1
- 208000031481 Pathologic Constriction Diseases 0.000 description 1
- QGMRQYFBGABWDR-UHFFFAOYSA-M Pentobarbital sodium Chemical compound [Na+].CCCC(C)C1(CC)C(=O)NC(=O)[N-]C1=O QGMRQYFBGABWDR-UHFFFAOYSA-M 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102000003923 Protein Kinase C Human genes 0.000 description 1
- 108090000315 Protein Kinase C Proteins 0.000 description 1
- 206010039330 Ruptured cerebral aneurysm Diseases 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 208000005392 Spasm Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 206010067347 Thrombotic cerebral infarction Diseases 0.000 description 1
- 208000026062 Tissue disease Diseases 0.000 description 1
- 235000011054 acetic acid Nutrition 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 238000005273 aeration Methods 0.000 description 1
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 1
- 150000008041 alkali metal carbonates Chemical class 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 230000003276 anti-hypertensive effect Effects 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229960004676 antithrombotic agent Drugs 0.000 description 1
- 210000000709 aorta Anatomy 0.000 description 1
- 210000002376 aorta thoracic Anatomy 0.000 description 1
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical class CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 125000005098 aryl alkoxy carbonyl group Chemical group 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 239000012752 auxiliary agent Substances 0.000 description 1
- 125000005604 azodicarboxylate group Chemical group 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 1
- AFPHMHSLDRPUSM-VIFPVBQESA-N benzyl n-[(2s)-1-hydroxypropan-2-yl]carbamate Chemical compound OC[C@H](C)NC(=O)OCC1=CC=CC=C1 AFPHMHSLDRPUSM-VIFPVBQESA-N 0.000 description 1
- 210000000133 brain stem Anatomy 0.000 description 1
- 210000005013 brain tissue Anatomy 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- QYKRDDCDQPHRTK-UHFFFAOYSA-N carbon dioxide;ethoxyethane Chemical compound O=C=O.CCOCC QYKRDDCDQPHRTK-UHFFFAOYSA-N 0.000 description 1
- 238000003763 carbonization Methods 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 210000001168 carotid artery common Anatomy 0.000 description 1
- 210000004004 carotid artery internal Anatomy 0.000 description 1
- 150000003943 catecholamines Chemical class 0.000 description 1
- 230000005779 cell damage Effects 0.000 description 1
- 208000037887 cell injury Diseases 0.000 description 1
- 238000002585 cerebral angiography Methods 0.000 description 1
- 230000003727 cerebral blood flow Effects 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 210000000038 chest Anatomy 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 210000003703 cisterna magna Anatomy 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910000365 copper sulfate Inorganic materials 0.000 description 1
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 210000004351 coronary vessel Anatomy 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 229960003280 cupric chloride Drugs 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 230000000916 dilatatory effect Effects 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- ODCCJTMPMUFERV-UHFFFAOYSA-N ditert-butyl carbonate Chemical compound CC(C)(C)OC(=O)OC(C)(C)C ODCCJTMPMUFERV-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 238000009297 electrocoagulation Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- LBAQSKZHMLAFHH-UHFFFAOYSA-N ethoxyethane;hydron;chloride Chemical compound Cl.CCOCC LBAQSKZHMLAFHH-UHFFFAOYSA-N 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 238000004508 fractional distillation Methods 0.000 description 1
- 238000010575 fractional recrystallization Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 208000034783 hypoesthesia Diseases 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 208000023589 ischemic disease Diseases 0.000 description 1
- 150000002537 isoquinolines Chemical class 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229940041476 lactose 100 mg Drugs 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L magnesium chloride Substances [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000005948 methanesulfonyloxy group Chemical group 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 208000022084 motor paralysis Diseases 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 230000003961 neuronal insult Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 231100000862 numbness Toxicity 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 229960002275 pentobarbital sodium Drugs 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 239000004323 potassium nitrate Substances 0.000 description 1
- 235000010333 potassium nitrate Nutrition 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 229960003857 proglumide Drugs 0.000 description 1
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000002040 relaxant effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 230000036262 stenosis Effects 0.000 description 1
- 208000037804 stenosis Diseases 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 210000003582 temporal bone Anatomy 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 125000005424 tosyloxy group Chemical group S(=O)(=O)(C1=CC=C(C)C=C1)O* 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 230000002455 vasospastic effect Effects 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 238000005550 wet granulation Methods 0.000 description 1
- 210000000216 zygoma Anatomy 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Description
本発明は、脳血管攣縮抑制作用や血流増加作用を有し、医薬として有用なイソキノリン誘導体に関する。
背 景 技 術
脳血管障害や心臓病は、がんと並んで、先進国における主要な死因となっている。
脳血管障害は出血群と虚血群に大別される。出血群においては脳動脈瘤破裂によるクモ膜下出血と高血圧性脳出血、頭部外傷が代表的である。クモ膜下出血が発生すると、脳の主幹動脈において出血後4〜5日から約2週間継続する血管内腔の狭小化が起こる。これは脳血管攣縮と称されているものである。その発現機序についてはまだ不明な点が多く、さまざまな生理活性物質(例、カテコールアミン、ヘモグロビン、過酸化脂質、アラキドン酸代謝物、ペプタイド、プロテインキナーゼC)が関与しており、これら種々の生理活性物質の相互作用によるものとされている。血管狭窄に伴って虚血となって神経学的障害が出現した場合、機能予後のみならず、時には生命予後をも左右する。虚血群においては、脳梗塞、一過性脳虚血発作(TIA)が代表的である。虚血又は出血に伴う血管障害や神経細胞損傷により急性期から慢性期にかけてしびれや四肢の運動麻痺等の運動障害や神経・精神障害を生じ、重症になると意識障害から死に至ることもある。
これら脳血管障害の治療には、抗血栓剤や脳循環・代謝改善剤が使用されている。しかし、致死性の脳血管攣縮や、痴呆に進展する神経細胞の障害を抑制する薬剤はほとんどなく、治療剤が渇望されている。クモ膜下出血後の脳血管攣縮の治療剤としては、塩酸ファスジル[ヘキサヒドロ−1−(5−イソキノリニルスルホニル)−1H−1,4−ジアゼピン]が唯一臨床的に用いられている医薬品である(薬理と治療,1992,20(Suppl 6),1515等参照)が効果や副作用の点からみて十分満足できる薬剤とは言えない)。
一方、虚血性心疾患としては、狭心症が代表的である。狭心症においては、血管狭窄に伴って虚血となり、心筋梗塞を起こし、死に至ることもある。薬物治療としては、主として血管拡張剤が使用されるが、その多くは降圧作用も併有し、また、血管攣縮まで治療する薬剤は殆どない。
イソキノリン骨格の5位が環状アミノスルホニルで置換されている化合物は、循環器官用剤(血管拡張剤、脳循環改善剤、狭心症治療薬、脳心血管系の血栓症の予防および治療薬、高血圧症の予防治療薬、脳機能改善剤)として有用であることが知られている(特開昭57−156463号公報、特開昭57−200366号公報、特開昭58−121278号公報、特開昭58−121279号公報、特開昭59−93054号公報、特開昭60−81168号公報、特開昭61−152658号公報、特開昭61−227581号公報参照)。さらに脳機能改善剤(脳組織の機能、状態(代謝能を含む)の障害およびそれに伴う症状、後遺症の予防、改善、もしくは当該障害の進行を緩やかにする薬剤)として有用であることが知られている(特開平2−256617号公報参照)。
本発明者らは、イソキノリン骨格の5位が環状アミノスルホニルで置換されていて、且つ、4位に置換基を有する化合物がこれらの既知の化合物より優れた脳血管障害予防剤又は治療剤であることを見出し、先に特許出願した(国際公開WO97/28130号公報)。この中には、イソキノリン骨格の4位にフッ素を有し、5位がヘキサヒドロ−4H−1,4−ジアゼピン−1−イルスルホニルで置換されていて、且つ、このヘキサヒドロ−4H−1,4−ジアゼピンの2位がメチルで置換されている本発明化合物は、具体的に開示されていない。
発 明 の 開 示
本発明の目的は、安全性が高く、脳血管障害予防・治療剤として、特に脳血管攣縮抑制剤として、また、虚血性病変を伴う疾患の予防・治療剤として、既存の薬物より優れた化合物を提供することにあった。
本発明者らは、上記目的を達成するために、新規な構造を有する種々の化合物を合成し、検討する過程において下記の式〔I〕で表される化合物が、非常に低い用量で、脳血管攣縮抑制作用を有し、加えて全身血圧を低下させることなく、冠血管や脳血管を拡張して優れた血流増加作用を有することを見いだし、本発明を完成した。
従って、本発明は、次の式〔I〕
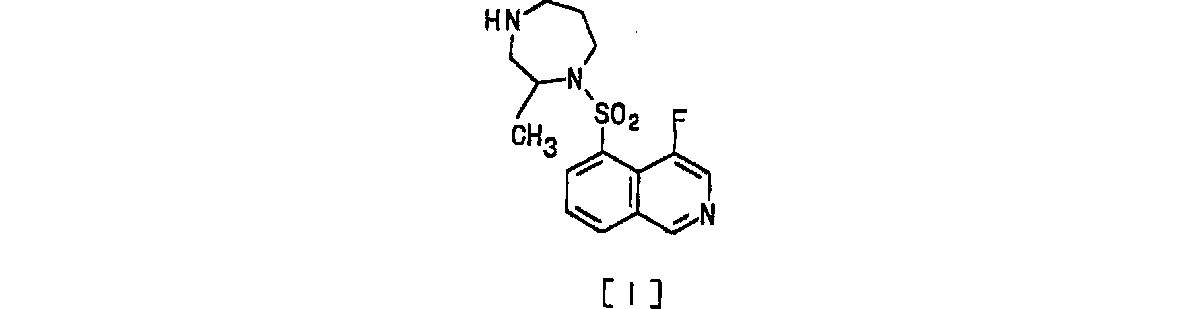
本発明化合物の化学構造上の特徴は、イソキノリン骨格の4位がフッ素で置換されていて、且つ、5位のヘキサヒドロ−4H−1,4ジアゼピン−1−イルスルホニルのヘキサヒドロ−4H−1,4−ジアゼピンの2位がメチルで置換されている点にある。
本発明化合物〔I〕は、不斉炭素を有し、光学異性体が存在する。これらの各異性体及びこれらの混合物のいずれも本発明に包含される。なかでも(S)体のものが好ましい。
「医薬上許容される塩」としては、例えば、塩酸、硫酸、硝酸、リン酸、フッ化水素酸、臭化水素酸等の無機酸の塩又は酢酸、酒石酸、乳酸、クエン酸、フマール酸、マレイン酸、コハク酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、トルエンスルホン酸、ナフタレンスルホン酸、カンファースルホン酸等の有機酸の塩を挙げることができる。
本発明化合物〔I〕は、例えば、国際公開WO97/28130号公報に記載された方法又は以下の方法によって製造することができる。
製造法1
脱離基としてのL1は、後記するスルホン酸の反応性誘導体の残基を挙げることができる。R31として表される保護基としては、例えば、ホルミル、アセチル、ペンゾイル等のアシル、ベンジルオキシカルボニル等のアラルキルオキシカルボニル、tert−ブチルオキシカルボニル等のアルコキシカルボニル、ベンジル等のアラルキルを挙げることができる。
式〔III〕で表されるアミンを適当な溶媒中、式〔II〕で表されるスルホン酸の反応性誘導体と反応させ、保護基を除去して化合物〔I〕を製造する。反応溶媒としては、反応に支障のないものであればよく、例えば、テトラヒドロフラン(THF)、ジオキサン、ジエチルエーテル等のエーテル類、ベンゼン、トルエン等の炭化水素類、塩化メチレン、クロロホルム等のハロゲン化炭化水素類、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド等の非プロトン性極性溶媒、ピリジン、アセトニトリル、又はこれらの混合物を用いることができる。スルホン酸の反応性誘導体としては、スルホン酸ハライド(例、スルホン酸クロライド、スルホン酸プロマイド)、スルホン酸無水物、N−スルホニルイミダゾリド等が用いられる。特にスルホン酸ハライドが好ましい。
本反応は、適当な塩基存在下に行うのが好ましい。かかる塩基としては、アルカリ金属炭酸水素塩(例、炭酸水素ナトリウム)、アルカリ金属炭酸塩(例、炭酸カリウム)、アルカリ金属水酸化物(例、水酸化ナトリウム、水酸化カリウム)のようなアルカリ、トリエチルアミン、トリエチレンジアミン等の有機第3級アミンを用いることができる。溶媒としてピリジンのような塩基性溶媒を使用すれば、かかる塩基は不要であり、好ましい。
本反応は、通常、室温で進行する場合が多いが、必要に応じて冷却又は加熱して、−78〜150℃、好ましくは、0〜120℃で行うことができる。塩基を使用する場合、アミン〔III〕の使用量は、反応性誘導体〔II〕に対して1〜5倍モルの範囲が好ましく、より好ましくは、1〜2倍モルである。塩基の使用量は、アミン〔III〕に対して1〜10倍モルの範囲が好ましく、より好ましくは、1〜8倍モルである。塩基を使用しない場合は、反応性誘導体〔II〕の使用量は、アミン〔III)に対して等モル以下、好ましくは、0.5〜0.1倍モルの範囲である。反応時間は、使用する原料、溶媒、反応温度等によって異なるが、通常、5分〜70時間である。次いでそれ自体公知の方法で保護基を除去する。酸処理、アルカリ処理、接触還元等のそれ自体公知の方法で保護基を除去することができる。
出発原料〔II〕は、後述する参考例1と同様にして製造することができる。
出発原料〔III〕は、参考例2と同様にして製造することができる。
製造法2
(第1工程) 2−アミノ−1−プロパノールと化合物〔II〕を製造法1と同様に反応させることにより、化合物〔IV〕を製造する。
(第2工程) 化合物〔IV〕のヒドロキシ基をそれ自体公知の方法で、ハロゲン(例、塩素、臭素)、スルホニルオキシ(例、メタンスルホニルオキシ)、又はアシルオキシ(例、トシルオキシ、アセチルオキシ)に変換した後、3−アミノ−1−プロパノールを適当な溶媒中、塩基の存在又は不存在下で、製造法1と同様に反応させることにより、化合物〔V〕を製造する。
(第3工程) 化合物〔V〕の2級アミノの窒素原子をそれ自体公知の方法で保護した後、適当な溶媒中、塩基の存在下、閉環、脱保護することにより、化合物〔I〕を得ることができる。
また、化合物〔V〕をトリフェニルホスフィンとアゾジカルボン酸ジアルキルで処理して分子内脱水縮合反応を行うことによっても化合物〔I〕を製造することができる。
上記の製造法において、アミノ基は、必要により、通常用いられる保護基で保護し、上記反応に付した後、酸処理、アルカリ処理、接触還元等のそれ自体公知の方法で保護基を除去することができる。アミノ基の保護基としては、例えば、ベンジル、ベンジルオキシカルボニル、トリフルオロアセチルを用いることができる。
本発明化合物〔I〕は、通常、ラセミ体で得られる。これらのラセミ体はそのままでも薬理活性を有するが、所望によりそれぞれの異性体に分割することができる。例えば、異性体混合物を公知の光学分割法、例えば、光学活性なカルボン酸(例、(+)−又は(−)−酒石酸、(+)−又は(−)−リンゴ酸)又は光学活性なスルホン酸(例、(+)−カンファースルホン酸)との塩を生成させ、分別再結晶する方法、光学活性カラムを用いる方法によって分離することができる。また、光学異性体は、原料化合物〔III〕に対応する光学活性な原料化合物(S配置又はR配置)を用いて各反応を行うことにより得ることができる。
本発明化合物〔I〕は、公知の方法により、前記した塩を形成させることができる。例えば、本発明化合物〔I〕の塩酸塩は、本発明化合物〔I〕を塩化水素のアルコール溶液又はエチルエーテル溶液に溶解し、溶液を濃縮し、析出結晶をろ取することにより得ることができる。
このようにして製造される本発明化合物は、それ自体公知の手段により、遊離塩基の形又は酸付加塩の形で、例えば、濃縮、液性変換、転溶、溶媒抽出、結晶化、分留、クロマトグラフィー、再結晶により単離精製することができる。
本発明化合物は、脳血管攣縮抑制作用を有し、且つ安全性が高いので、脳血管障害、特に脳出血後の脳血管攣縮による脳組織障害の予防・治療に有用である。また、血流増加作用を有するので虚血性疾患に伴う病変の予防剤又は治療剤としても有用である。
本発明化合物を医薬として投与する場合、本発明化合物はそのまま又は医薬的に許容される無毒性かつ不活性の担体中に、例えば、0.1%〜99.5%、好ましくは0.5%〜90%含有する医薬組成物として、人を含む哺乳動物に投与される。
担体としては、固形、半固形、又は液状の希釈剤、充填剤、及びその他の処方用の助剤一種以上が用いられる。医薬組成物は、投与単位形態で投与することが望ましい。本発明医薬組成物は、経口投与、組織内投与、局所投与(経皮投与等)又は経直腸的に投与することができる。これらの投与方法に適した剤型で投与されるのはもちろんである。中でも、静脈内投与又は経口投与が好ましい。
医薬としての用量は、年齢、体重等の患者の状態、投与経路、病気の性質と程度等を考慮した上で調整することが望ましいが、通常は、脳血管攣縮予防・治療剤としては、成人に対して本発明の有効成分量として、静脈内投与の場合、1日あたり、0.1〜100mg/ヒトの範囲、好ましくは、1〜30mg/ヒトの範囲である。経口投与の場合、1日あたり、1〜1,000mg/ヒトの範囲、好ましくは、10〜30mg/ヒトの範囲である。虚血性病変を伴う疾患の予防・治療剤としては、成人に対して本発明の有効成分量として、静脈内投与の場合、1日あたり、0.1〜100mg/ヒトの範囲、好ましくは、1〜30mg/ヒトの範囲である。経口投与の場合、1日あたり、1〜1,000mg/ヒトの範囲、好ましくは、10〜30mg/ヒトの範囲である。場合によっては、これ以下でも足りるし、また逆にこれ以上の用量を必要とすることもある。また1日2〜3回に分割して投与することもできる。
発明を実施するための最良の形態
以下に、代表的な原料の製造を参考例を以て、本発明化合物の製造を実施例を以て及び代表的化合物の製剤例、試験例を掲げて、本発明を更に詳しく説明するが、本発明はこれらに限定されるものではない。なお、比旋光度は20℃で測定した。
参考例1
5−クロロスルホニル−4−フルオロイソキノリンの合成
(第1工程)
300mlの封管に29%アンモニア水160ml、硫酸銅CuSO4・5H2O)3g、4−ブロモイソキノリン49.92gを入れ、170℃で16時間撹拌した。冷後、反応液に2N水酸化ナトリウム40mlを加え、ベンゼン400mlで5回抽出した。有機層を硫酸マグネシウムで乾燥し、活性炭処理後、溶媒を留去して、4−アミノイソキノリン29.5gを得た。
(第2工程)
42%ほうフッ化水素酸100mlに4−アミノイソキノリン28.8gを加え、溶解した。この混合物をドライアイス−エーテルで冷却し、固化し始めたところで亜硝酸ナトリウム13.8gを1時間かけて加えた。反応液を濾過し、得られた固化物(ジアゾニウム塩)を低温でエーテルを用いて素早く洗浄し、可及的に水分を除いた。このジアゾニウム塩をキシレン中に加え、撹拌下に熱風(ドライヤー)で加熱し、ガスが発生し始めた後、2時間室温で攪拌した。得られたタール状物からキシレンをデカントして、キシレン溶液を希塩酸100mlで3回抽出した。塩酸層とタール状物を混合し、この混合物に水100mlを加え、炭酸カリウムでアルカリ性とし、エーテル400mlで3回抽出した。抽出液を硫酸マグネシウムで乾燥し、濾過した後、活性炭を加え、セライト濾過した。溶媒を留去し、残留物をシリカゲルカラムクロマトグラフィー(クロロホルム)で精製した。溶媒を留去し4−フルオロイソキノリン14.5gを得た。
(第3工程)
4−フルオロイソキノリン22.05gを硫酸90mlに加えた。これに氷冷下、硝酸カリウム18.18gの硫酸(90ml)溶液を滴下した。室温で2時間撹拌した後、反応液を氷に注入し、冷却下にアンモニア水を加え、塩基性とした。析出した結晶を濾取、水洗、乾燥して4−フルオロ−5−ニトロイソキノリン22.16gを得た。
(第4工程)
4−フルオロ−5−ニトロイソキノリン23.04gをエタノール60mlに懸濁した。これに濃塩酸144mlを加え、氷冷攪拌下、塩化第一スズ・二水和物90.24gのエタノール(144ml)溶液を滴下し、次いで室温で3時間撹拌した。反応液を濃縮し、氷水200mlを加え、つづいて2N水酸化ナトリウムを加えてアルカリ性とし、クロロホルム500mlで3回抽出した。抽出液は硫酸マグネシウムで乾燥した後、濃縮した。残留物をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール=100:1)で精製し、5−アミノ−4−フルオロイソキノリン15.05gを得た。
(第5工程)
5−アミノ−4−フルオロイソキノリン8.1gを濃塩酸100mlに懸濁した。これに亜硝酸ナトリウム5.18gを水20mlに溶かして−5℃以下で滴下して加え、次いで室温で30分撹拌した(A溶液の調製)。別に、亜硫酸ガス飽和酢酸溶液62.5mlに塩化第二銅・二水和物2.88gの水(12.5ml)溶液を加えた溶液を調製し、これにA溶液を滴下した。反応液を次いで室温で1時間撹拌した後、さらに30℃の水浴上で温め、未反応の亜硫酸ガスを除いた。反応液をクロロホルム400mlで3回抽出した。抽出液を炭酸水素ナトリウム水溶液で中和洗浄し、乾燥した後、溶媒を留去し5−クロロスルホニル−4−フルオロイソキノリン9.6gを得た。
参考例2
(S)−ヘキサヒドロ−4−(tert−プトキシカルボニル)−2−メチル−1H−1,4−ジアゼピンの合成
(第1工程)
(S)−(+)−2−アミノ−1−プロパノール15gをクロロホルム150mlに溶解し、トリエチルアミン20.21gを加えた。これにベンジルオキシカルボニルクロライド(ZCl)34.07gをクロロホルム50mlに溶解した溶液を氷冷攪拌下に徐々に滴下し、混合物を室温にて一夜撹拌した。反応液に水200mlを加え、クロロホルム200mlで3回抽出した。有機層を飽和食塩水200mlで2回洗浄し、硫酸マグネシウムで乾燥し、溶媒を留去した。得られた結晶性の残留物にn−ヘキサン200mlを加え、残留物を破砕した後、濾取し、n−ヘキサンで洗浄し、乾燥して、(S)−2−ベンジルオキシカルボニルアミノ−1−プロパノール37.04gを得た。
(第2工程)
(S)−2−ベンジルオキシカルボニルアミノ−1−プロパノール36.52gを塩化メチレン140mlに溶解し、続いてトリエチルアミン18.67gを加えた。これにメシルクロライド21.40gを塩化メチレン60mlに溶かした溶液を、氷冷攪拌下、徐々に加え、20分間撹拌した。反応液に水200mlを加え、クロロホルム200mlで3回抽出した。有機層を飽和食塩水200mlで洗浄して、硫酸マグネシウムで乾燥した後、溶媒を留去して(S)−2−ベンジルオキシカルボニルアミノ−1−メタンスルホニルオキシプロパン49.53gを得た。
(第3工程)
3−アミノ−1−プロパノール70.0gをテトラヒドロフラン(THF)60mlに溶解し、これに室温で(S)−2−ベンジルオキシカルボニルアミノ−1−メタンスルホニルオキシプロパン48.97gをTHF140mlに懸濁した懸濁液を加えた。この混合物を攪拌下に80℃にて一夜加熱還流した。冷後、反応液に水200mlを加え、クロロホルム200mlで3回抽出した。有機層を飽和食塩水200mlで2回洗浄し、硫酸マグネシウムで乾燥した後、溶媒を留去し、(S)−6−ベンジルオキシカルボニルアミノ−4−アザヘプタン−1−オールを淡黄色油状物として37.61g得た。本油状物は暫時放置すると結晶化した。
(第4工程)
(S)−6−ベンジルオキシカルボニルアミノ−4−アザヘプタン−1−オール37.1gをTHF150mlに溶解し、これに氷冷撹拌下、ジ−tert−ブチルカーボネート34.7gをTHF50mlに溶かした溶液を滴下し、次いで室温で一夜撹拌した。反応液に水200mlを加え、クロロホルム200mlで3回抽出した。有機層を飽和食塩水200mlで2回洗浄した後、硫酸マグネシウムで乾燥し、溶媒を留去した。得られた残留物をシリカゲルカラムクロマトグラフィーを用いて精製(ワコーゲルC−200/登録商標1kg、クロロホルム;メタノール=100:1→100:5で溶出)し、(S)−6−ベンジルオキシカルボニルアミノ−4−(tert−ブトキシカルボニル)−4−アザヘプタン−1−オールを淡黄色油状物として44.42g得た。
(第5工程)
(S)−6−ベンジルオキシカルボニルアミノ−4−(tert−プトキシカルボニル)−4−アザヘプタン−1−オール10.0gを塩化メチレン50mlに溶解し、トリエチルアミン3.0gを加え、氷冷撹拌した。これにメシルクロライド8.32gを塩化メチレン25mlに溶かした溶液を加え、室温で10分間撹拌した。反応液に水200mlを加え、クロロホルム100mlで3回抽出した。有機層を飽和食塩水200mlで洗浄した後、硫酸マグネシウムで乾燥し、溶媒を留去し、(S)−6−ベンジルオキシカルボニルアミノ−4−(tert−プトキシカルボニル)−4−アザヘプチル メタンスルホネートを淡黄色油状物として11.74g得た。
(第6工程)
(S)−6−ベンジルオキシカルボニルアミノ−4−(tert−プトキシカルボニル)−4−アザヘプチル メタンスルホネート11.74gをジメチルスルホキシド(DMSO)に溶解し、60%水素化ナトリウム2.2gを加え、室温で30分撹拌した。反応液に水及び酢酸エチルを加え、分液し、有機層を水洗した後、飽和食塩水200mlで5回洗浄し、硫酸マグネシウムで乾燥し、溶媒を留去した。得られた残留物をシリカゲルカラムクロマトグラフィーにて精製(ワコーゲルC−200/登録商標、200g使用、クロロホルムで溶出)して、(S)−ヘキサヒドロ−1−ベンジルオキシカルボニル−4−(tert−プトキシカルボニル)−2−メチル−1H−1,4−ジアゼピンを淡黄色油状物として8.58g得た。
(第7工程)
(S)−ヘキサヒドロ−1−ベンジルオキシカルボニル−4−(tert−プトキシカルボニル)−2−メチル−1H−1,4−ジアゼピン8.53gを500mlの常圧還元装置を用い、メタノール80mlに溶解した後、5%パラジウム/カーボン4.0gを加え、室温にて常圧で接触還元した。6時間後、セライト濾過し、溶媒を留去して、(S)−ヘキサヒドロ−4−(tert−プトキシカルボニル)−2−メチル−1H−1,4−ジアゼピンを淡黄色油状物として3.97g得た。
同様にして(R)−ヘキサヒドロ−4−(tert−プトキシカルボニル)−2−メチル−1H−1,4−ジアゼピン及びヘキサヒドロ−4−(tert−プトキシカルボニル)−2−メチル−1H−1,4−ジアゼピンを合成した。
実施例1
ヘキサヒドロ−1−(4−フルオロ−5−イソキノリンスルホニル)−2−メチル−1H−1,4−ジアゼピン
(1)ヘキサヒドロ−4−(tert−プトキシカルボニル)−2−メチル−1H−1,4−ジアゼピン0.51gとトリエチルアミン0.30gを塩化メチレン10mlに溶かした溶液に5−クロロスルホニル−4−フルオロイソキノリン0.49gを加え、12時間室温攪拌した。反応液を濃縮し、残渣を塩化メチレンに溶解した。塩化メチレン溶液を水洗し、硫酸マグネシウムで乾燥した後、濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィ(クロロホルム/メタノール=100/1)にて精製し、ヘキサヒドロ−4−(tert−プトキシカルボニル)−1−(4−フルオロ−5−イソキノリンスルホニル)−2−メチル−1H−1,4−ジアゼピン0.51gを得た。
(2)(1)で得た化合物0.99gを塩化メチレン10mlに溶解し、トリフルオロ酢酸10mlを滴下し、2時間室温攪拌した。反応液を飽和炭酸水素ナトリウム水溶液にて中和し、クロロホルムで抽出した。クロロホルム層を硫酸マグネシウムで乾燥し、濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィ(クロロホルム/メタノール=10/1)にて精製し、目的化合物0.24gを得た。
NMRスペクトル(CDCl3):δ(ppm)0.91(3H,d),1.69−1.88(3H,m),2.47−2.77(2H,m),3.13−3.36(3H,m),3.92−4.10(2H,m),7.73(1H,t),8.22(1H,dd),8.57(1H,d),8.84(1H,d),9.15(1H,s)
実施例2
(S)−(−)−ヘキサヒドロ−1−(4−フルオロ−5−イソキノリンスルホニル)−2−メチル−1H−1,4−ジアゼピン塩酸塩
(1)(S)−ヘキサヒドロ−4−(tert−プトキシカルボニル)−2−メチル−1H−1,4−ジアゼピン0.54gとトリエチルアミン0.38gを塩化メチレン10mlに溶かした溶液に5−クロロスルホニル−4−フルオロイソキノリン0.62gを加え12時間室温攪拌した。反応液を濃縮し、残渣を塩化メチレンに溶解した。塩化メチレン溶液を水洗し、硫酸マグネシウムで乾燥した後、濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィ(クロロホルム/メタノール=100/1)にて精製し、(S)−ヘキサヒドロ−4−(tert−プトキシカルボニル)−1−(4−フルオロ−5−イソキノリンスルホニル)−2−メチル−1H−1,4−ジアゼピン0.99gを得た。
(2)(1)で得た化合物0.99gを塩化メチレン10mlに溶解し、トリフルオロ酢酸を滴下し、2時間室温攪拌した。反応液を飽和炭酸水素ナトリウム水溶液にて中和し、クロロホルムで抽出した。クロロホルム層を硫酸マグネシウムで乾燥し、濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィ(クロロホルム/メタノール=10/1)にて精製し(S)−ヘキサヒドロ−1−(4−フルオロ−5−イソキノリンスルホニル)−2−メチル−1H−1,4−ジアゼピン0.58gを得た。
NMRスペクトル(CDCl3):δ(ppm)0.91(3H,d),1.69−1.88(3H,m),2.47−2.77(2H,m),3.13−3.36(3H,m),3.92−4.10(2H,m),7.73(1H,t),8.22(1H,dd),8.57(1H,d),8.84(1H,d),9.15(1H,s)
(3)(2)で得た化合物0.58gをメタノール10mlに溶解し、1N塩酸水溶液1.80mlを加え、濃縮して目的化合物0.50gを得た。
元素分析値(C15H18FN3O2S・HCl)として
計算値(%)C:50.01 H:5.32 N:11.68
実測値(%)C:50.26 H:5.60 N:11.44
[α]D=−9.02°(c=1.086,H2O)
実施例3
(R)−(+)−ヘキサヒドロ−1−(4−フルオロ−5−イソキノリンスルホニル)−2−メチル−1H−1,4−ジアゼピン塩酸塩
(1)(R)−ヘキサヒドロ−4−(tert−プトキシカルボニル)−2−メチル−1H−1,4−ジアゼピン0.54gを用いて実施例2(1)及び(2)と同様に操作して、(R)−(+)−ヘキサヒドロ−1−(4−フルオロ−5−イソキノリンスルホニル)−2−メチル−1H−1,4−ジアゼピンを得た。
NMRスペクトル(CDCl3);δ(ppm)0.91(3H,d),1.69−1.88(3H,m),2.47−2.77(2H,m),3.13−3.36(3H,m),3.92−4.10(2H,m),7.73(1H,t),8.22(1H,dd),8.57(1H,d),8.84(1H,d),9.15(1H,s)
(2)(1)で得た化合物を実施例2(3)と同様に操作して目的化合物0.45gを得た。
元素分析値(C15H18FN3O2S・HCl)
計算値(%)C:50.01 H:5.32 N:11.68
実測値(%)C:50.27 H:5.05 N:11.82
[α]D=7.70°(c=1.116,H2O)
実施例4
(S)−(−)−ヘキサヒドロ−1−(4−フルオロ−5−イソキノリンスルホニル)−2−メチル−1H−1,4−ジアゼピン塩酸塩(別法)
(1)(S)−(+)−2−アミノ−1−プロパノール3.00gとトリエチルアミン6.06gを塩化メチレン100mlに溶解し、5−クロロスルホニル−4−フルオロイソキノリン9.82gを加え、12時間室温撹拌した。反応液を水洗、硫酸マグネシウムで乾燥後、濃縮した。析出結晶をイソプロピルエーテルで洗浄し、(S)−4−フルオロ−5−[N−(1−ヒドロシプロパン−2−イル)アミノスルホニル]イソキノリン7.96gを得た。
(2)(1)で得た化合物1.42gとトリエチルアミン0.76を塩化メチレン30mlに溶解し、氷冷下、メタンスルホニルクロライド0.63gを滴下し1時間室温撹拌した。反応液を水洗、硫酸マグネシウムで乾燥後、濃縮した。残渣にTHF20mlを加え、3−アミノプロパノール1.88gを滴下し、12時間室温撹拌した。反応液を濃縮し、残渣をシリカゲルカラムクロマトグラフィー(塩化メチレン/メタノーノル/29%アンモニア水=90/10/1)にて精製し、(S)−4−フルオロ−5−[N−(1−ヒドロキシ−4−アザヘプタン−6−イル)アミノスルホニル]イソキノリン1.56gを得た。
(3)(2)で得た化合物1.54gとトリフェニルホスフィン1.77gをTHF15mlに溶解し、アゾジカルボン酸ジイソプロピルエステル40%トルエン溶液3.41gを滴下し、2時間室温撹拌した。反応液を濃縮し、残渣に酢酸エチルを加え、1N塩酸にて抽出した。水層を炭酸水素ナトリウムで弱アルカリ性とし、生じた油状物をクロロホルムで抽出した。抽出液を水洗、硫酸マグネシウムで乾燥後、濃縮した。残渣をエタノールに溶解し、1N塩化水素エーテル溶液を加え析出結晶を濾取した。結晶をエタノールで洗浄し、目的物0.60gを得た。
元素分析値(C15H18FN3O2S・HCl)として
計算値(%)C:50.01 H:5.32 N:11.68
実測値(%)C:49.82 H:5.21 N:11.43
融点 258〜260℃
製剤例1
処方(1ml中)
実施例2の化合物 3mg
塩化ナトリウム 9mg
注射用水 適量
1ml
調製法
実施例2の化合物及び塩化ナトリウムを上記の比率で注射用水に溶解後、メンプランフィルター(0.22μm)を用いて濾過を行い、アンプルに充填後、滅菌を行い水性注射剤とする。
製剤例2
処方(1バイアルあたり)
実施例2の化合物 3mg
マンニトール 50mg
調製法
実施例2の化合物及びマンニトールを上記の比率で注射用水に溶解後、メンプランフィルター(0.22μm)を用いて無菌濾過を行い、バイアルに充填後、常法により凍結乾燥を行い、用時溶解型注射剤とする。
製剤例3
処方(1錠180mg中)
実施例2の化合物 10mg
乳糖 100mg
トウモロコシ澱粉 55mg
低置換度ヒドロキシプロピルセルロース 9mg
ポリビニルアルコール(部分ケン化物) 5mg
ステアリン酸マグネシウム 1mg
調製法
上記の比率で、ポリビニルアルコール及びステアリン酸マグネシウムを除く上記成分を均一に混合した後、ポリビニルアルコール水溶液を結合剤として湿式造粒法にて打錠用顆粒を製造する。これにステアリン酸マグネシウムを混合した後に、打錠機を用いて1錠重量180mgに成形し内服錠とする。
製剤例4
処方(1カプセル220mg中)
実施例2の化合物 10mg
乳糖 187mg
微結晶セルロース 20mg
ステアリン酸マグネシウム 3mg
220mg
調製法
上記の比率で成分を均一に混合した後、カプセル充填機で硬カプセルに上記の220mgを充填し、硬カプセル剤とする。
製剤例5
処方(顆粒1g中)
実施例2の化合物 10mg
乳糖 880mg
低置換度ヒドロキシプロピルセルロース 70mg
ヒドロキシプロピルセルロース 40mg
1000mg
調製法
上記の比率で、ヒドロキシプロピルセルロースを除く上記成分を均一に混合した後、ヒドロキシプロピルセルロース水溶液を結合剤として練合した後、造粒機にて造粒し、顆粒剤とする。
次に本発明化合物の代表的化合物の試験例を掲げる。被験化合物としては実施例2の化合物、比較対照化合物としては、先行技術のWO97/28130号公報に具体的に開示されている化合物で本発明化合物と構造的に最も近似しているヘキサヒドロ−1−(4−フルオロ−5−イソキノリンスルホニル)−1H−1,4−ジアゼピン塩酸塩(以下化合物Aと称する)と市販品の塩酸ファスジルを用いた。
試験例1
ラット大動脈のカルシウムイオノフォア収縮に対する作用
エーテル麻酔したラット(SD、雄、10−14週齢)を放血致死させ、胸部大動脈(約3cm)を摘出した。脂肪および結合組織を除去し、約3mm幅の輪状標本とした後、内腔を擦り、血管内皮細胞を除去した。標本を栄養液を満たしたマグヌス槽の等尺性張力トランスデューサーに装着し、1gの静止張力を負荷した。マグヌス槽の栄養液は混合ガス(95%O2+5%CO2)通気下、37℃に保ち、約20分毎に栄養液を交換しながら、標本を約1時間安定化させた。標本にカルシウムイオノフォアA23187を最終濃度1μMとなるよう投与し、収縮が安定した後、被験化合物を累積的に投与した。この間の収縮弛緩反応を記録し、被験化合物の、A23187血管収縮に対する50%阻害濃度(IC50(μM))を求めた。その結果、実施例2の化合物のIC50は、1.7であった。一方、比較対照化合物の化合物A及び塩酸ファスジルのIC50は、それぞれ10.6及び7.4であった。なお、実験に用いた栄養液の組成は以下のごとくである。NaCl 115.9mM(以下同じ);KC15.9;CaCl22.5;MgCl21.2;NaH2PO41.2;NaHCO825.0:グルコース11.5。これらを蒸留脱イオン水に溶解した。混合ガス飽和時のpHは、7.4であった。
本発明化合物は、カルシウムイオノフォアにより収縮した血管を弛緩させる作用を有し、その作用強度は、比較対照化合物の化合物A及び塩酸ファスジルよりもはるかに大きかった。
試験例2
ラット中大脳動脈血流増加作用
ウレタン麻酔したラット(SD,雄性,11−12週齢)の頭部を固定し、頬筋を剥離した後、頬骨を露出した。歯科用電気ドリルを用いで中大脳動脈(MCA)直上の頭蓋骨に、直径約5mmの孔をあけ、MCAを直視できるようにした。レーザードップラー血流計のプロープ(直径:1.0mm)をMCAと近接させ、MCA血流の変化を測定した。被験化合物は、生理食塩水に溶解、希釈し、大腿静脈からカニューレを介して5分間かけて3mg/kgを投与した。各化合物の作用は、薬物投与前に対する血流増加率で示した。その結果、実施例2の化合物の血流増加率は、18.8%であった。一方、比較対照化合物の化合物A及び塩酸ファスジルの血流増加率は、それぞれ12.4%及び5.2%であった。また、被験化合物を30分間かけて投与して、同様にして血流増加率を測定した。その結果、実施例2の化合物は、1mg/kg及び3mg/kg投与により血流量をそれぞれ5.2%、9.6%と有意に増加させたが、塩酸ファスジルは3mg/kgで増加が認められず、10mg/kgによって5.4%の有意な増加がみられた。
本発明化合物は、ラット中大脳動脈血流を増加させる作用を有し、その作用は、比較対照化合物の化合物A及び塩酸ファスジルよりもはるかに大きかった。
試験例3
イヌのクモ膜下出血モデルにおける脳血管攣縮緩解作用
Varsosらの方法[J.Neurosurgery58,11−17(1983)]に準じて自家血大槽内二回注入モデル犬を作製した。実験初日に、ペントバルビタール麻酔下、血管造影により化合物投与前の脳底動脈血管を撮影した。その後、大槽内より4mlの脳脊髄液を除去し、同量の自家血を2ml/分の速度で注入した。血液注入後、80分間、頭部を下に30度傾斜させ、脳底動脈からウィリス環に均一に血液を分布させた。実験3日目に、再度4mlの自家血の大槽内注入を行った。実験7日目に、ペントバルビタール麻酔下に脳血管造影を行い、遅延性脳血管攣縮が誘発されていることを確認した後、薬物の評価を行った。被験化合物は、左大腿静脈から1分間かけて投与した。経時的に血管造影を行った。各化合物の作用は、脳底動脈最狭窄部の直径の最大値を、実験初日の同一部位の直径を100%として示した。
その結果、実施例2の化合物は、血管攣縮に対して、0.3mg/kg以上の投与で有意な緩解作用を示し、3mg/kgで攣縮を完全に緩解させた。血圧は、0.3mg/kgでは有意な変化が認められなかったが、1m/kg以上で低下した。一方、塩酸ファスジルは、10mg/kgで持続注入直後にのみ有意な緩解作用を示し、血圧は3mg/kg以上で低下した。
すなわち、実施例2の化合物投与群3mg/kgを試験例A(使用動物数=5)、比較対照化合物の塩酸ファスジル3mg/kg投与群を試験例B(使用動物数=4)、同10mg/kg投与群を試験例C(使用動物数=4)とすると、7日目における薬物投与前の血管径は、試験例Aでは58%であった。一方、試験例B及びCでは、それぞれ58%、57%であった。実施例2の化合物は、静脈内投与により107%と攣縮血管を血液注入以前の内径にまで拡張させた。一方、塩酸ファスジルは、3mg/kgの静脈内投与では明確な緩解作用を示さず、10mg/kgの静脈内投与でも82%まで緩解させたにすぎなかった。
本発明化合物は、0.3mg/kgという非常に低用量で、かつ、血圧変化を現さない用量で有意に血管攣縮を緩解させた。
試験例4
ラット一過性中大脳動脈閉塞による脳梗塞に対する作用
SD系雄性ラット(210−240g、7週令、1群12匹)の総頸動脈をハロセン麻酔下切開し、同部より内頸動脈を経て中大脳動脈起始部に達するまでナイロン栓子を挿入した。中大脳動脈血流遮断後、麻酔を止め、2時間後、ナイロン栓子を抜き去ることにより血流の再開通を行なった。血流再開6時間後、脳を摘出し、TTC(2,3,5−tri−phenyltetrazolium chloride)染色により梗塞領域を判定し、梗塞体積を大脳皮質と皮質以外の上位脳幹部に分けて測定し、Dunnett’s testで検定した。実施例2の化合物0.3,1mg/kgおよび塩酸ファスジル3mg/kgを中大脳動脈閉塞−再還流直後より全量を80分間かけて静脈内に持続投与した。その結果、実施例2の化合物は、1mg/kgの投与で一過性中大脳動脈閉塞による脳梗塞に対して大脳皮質において脳梗塞を298.7mm3から181.7mm3に有意に抑制した(P〈0.05)。一方、塩酸ファスジルは、8mg/kgの投与で脳梗塞に対して抑制傾向を示しただけであった。
試験例5
ラット永久中大脳動脈閉塞による脳梗塞に対する作用
SD系雄性ラット(210−240g、7週令、1群10−11匹)の側頭骨底部に、ハロセン麻酔下、小孔をあけ、左中大脳動脈本幹を電気凝固により焼灼切断した。中大脳動脈閉塞48時間後、脳を摘出し、TTC染色により梗塞領域を判定し、梗塞体積を大脳皮質と線条体に分けて測定し、Dunnett’s testで検定した。実施例2の化合物0.03、0.1、0.3又は1mg/kgおよび塩酸ファスジル1又は3mg/kgを中大脳動脈閉塞直後から全量を30分間かけて、静脈内に持続投与した。その結果、大脳皮質および線条体で、実施例2の化合物は、0.3mg/kgの投与でそれぞれ脳梗塞体積を341.0mm3から198.0mm3、141.8mm3から103.8mm3に有意に抑制したが(P〈0.01)、塩酸ファスジルは3mg/kgの投与で脳梗塞体積を大脳皮質では341.0mm3から224.7mm3、線条体では141.8mm3から103.7mm3に有意に抑制したにすぎない(P〈0.01)。すなわち、実施例2の化合物は、塩酸ファスジルに比較して10倍以上の強い作用を有する。
試験例6
ラットの光化学誘発血栓による脳梗塞に対する作用
SD系雄性ラット(7週令、1群8匹)の中大脳動脈直上の頭蓋骨に孔をあけ、中大脳動脈を直視できるようにした。ローズベンガルを静脈内投与し、血栓モデル作製用光源を中大脳動脈と近接させ、緑色光を照射した。薬物は緑色光照射終了直後に静脈内に投与した。24時間後に脳を摘出し、連続冠状断切片を作製しTTC溶液にて染色した。その結果、実施例2の化合物は、大脳皮質及び線条体のいずれにおいても脳梗塞抑制作用を示し、大脳皮質においてその作用は明確であった。すなわち、0.3および1mg/kgの投与で大脳皮質において脳梗塞体積を86.3mm3から64.5mm3及び55.6mm3に有意に抑制し(それぞれP〈0.05、P〈0.01)、1mg/kgの投与で線条体における梗塞体積を58.2mm3から37.3mm3に有意に抑制した(P〈0.05)。一方、塩酸ファスジルは、1及び3mg/kgの投与で線条体における梗塞体積を59.9mm3からそれぞれ43.5m3及び44.5mm3に有意に抑制したたけであった(P〈0.01)。
実施例2の化合物は、脳血栓型の脳梗塞に対し、塩酸ファスジルよりもはるかに優れた改善効果を示した。
試験例7
麻酔開胸イヌにおける実施例2の化合物の冠状動脈血流量に対する作用
実験方法
体重12.8〜13.4kgのビーグル犬(1群4匹)をペントバルビタールナトリウム(30mg/kg i.v.)により麻酔した後、背位に固定した。以後、実験終了まで麻酔を維持するために右側橈側皮静脈内に留置したカニューレよりペントバルビタールナトリウム(3〜5mg/kg/hr)を持続注入した。気管カニューレを挿入し、人工呼吸器による人工呼吸下(1回換気量15ml/kg,呼吸頻度20breaths/min)に左第4およぴ5肋骨間で開胸し、次いで心嚢膜を切開し、ハンモック状に心臓を吊るして固定した。左冠状動脈前下行枝に電磁血流計用プローブを装着して電磁血流計および生体電気用プリアンプを用いて冠状動脈血流量を測定し、レクチコーダ上に記録した。実施例2の化合物及び塩酸ファスジルは動物1匹あたりの投与容量が30mlとなるように、生理食塩液に用時溶解し、予め左側橈側皮静脈内に留置したカニューレより30分かけて持続注入した。持続注入開始前値に対する%変化率を算出した。測定は投与中を含め60分間行った。その結果、生理食塩液投与では、冠状動脈血流量にはほとんど変化が見られなかった。被験薬静脈内投与開始後の冠状動脈血流量の経時変化を観察した。投与開始後5〜10分に実施例2の化合物は、0.1、0.3、1mg/kgでそれぞれ29.7%、69.3%及び86.7%の有意な増加を示した。一方、対照化合物の塩酸ファスジルは、3mg/kgで増加傾向、10mg/kgで53.1%の有意な増加を示したにすぎない。
試験例1〜7から明らかなように、本発明化合物は、本発明化合物に類似した公知化合物と比較しても格段に優れた脳血管攣縮緩解作用、血流増加作用等を示した。
試験例8
急性毒性
ラットを用いてワイル氏法に従って実施した。6週齢の雄性slcSD系ラット(1群5例)に被験薬物を尾静脈より60秒間かけて投与し、以後24時間の死亡の有無を観察した。被験薬物は、生理食塩水に溶解、希釈して用いた。その結果、実施例2の化合物の急性毒性は低かった。
産業上の利用可能性
以上のように、本発明化合物に類似した対照化合物の化合物Aや塩酸ファスジルに比較して、本発明化合物は、はるかに低い用量で、かつ、血圧変化を示さない用量で強い脳血管攣縮緩解作用を示した。しかも、本発明化合物は、攣縮血管を血液注入以前の内径にまで完全に回復させた。この事実から、本発明化合物は、脳血管障害、特に脳出血後の脳血管攣縮による脳組織障害の予防・治療に有用である。また、本発明化合物は、上記の脳血管攣縮緩解作用に加えて、対照化合物の塩酸ファスジルに比較して、はるかに低い用量で、臓器選択性に優れた脳血管や心臓血管の拡張作用、血流増加作用、及び虚血性神経細胞保護作用をあわせもつので、虚血性病変を伴う疾患の予防剤又は治療剤として有用である。即ち、本発明化合物は脳血管を拡張し、脳血流増加作用及び、虚血性神経細胞保護作用を有するので、脳出血、脳梗塞、一過性脳虚血発作、頭部外傷等に伴う後遺症(例、運動麻痺)の予防又は治療剤として有用である。さらに、冠血管を拡張し、優れた冠血流増加作用を示すことから、心筋梗塞や狭心症の予防・治療に有用である。
Claims (14)
- 次の式〔I〕
で表される化合物又はその医薬上許容される塩。 - 絶対配置がS配置である請求項1記載の化合物又はその医薬上許容される塩。
- 請求項1記載の化合物又はその医薬上許容される塩を有効成分とする医薬組成物。
- 医薬組成物が脳血管障害予防剤又はその治療剤である請求項3記載の医薬組成物。
- 医薬組成物が虚血性病変を伴う疾患の予防剤又はその治療剤である請求項3記載の医薬組成物。
- 医薬組成物が脳血管攣縮抑制剤である請求項3記載の医薬組成物。
- 医薬組成物が虚血性神経細胞壊死保護剤である請求項3記載の医薬組成物。
- 脳血管障害の病型が脳梗塞であることを特徴とする請求項3記載の医薬組成物。
- 脳血管障害の病型が一過性脳虚血発作であることを特徴とする請求項3記載の医薬組成物。
- 脳血管障害の病型が脳出血及び頭部外傷であることを特徴とする請求項3記載の医薬組成物。
- 医薬組成物が、脳出血及び頭部外傷に伴う後遺症の予防剤又はその治療剤である請求項3記載の医薬組成物。
- 医薬組成物が虚血性心疾患予防又は治療剤である請求項3記載の医薬組成物。
- 医薬組成物が心筋梗塞抑制剤である請求項3記載の医薬組成物。
- 医薬組成物が狭心症治療剤である請求項3記載の医薬組成物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP28968897 | 1997-10-22 | ||
| PCT/JP1998/004781 WO1999020620A1 (fr) | 1997-10-22 | 1998-10-22 | Derive d'isoquinoleine et medicament |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP4316794B2 true JP4316794B2 (ja) | 2009-08-19 |
Family
ID=17746466
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000516962A Expired - Lifetime JP4316794B2 (ja) | 1997-10-22 | 1998-10-22 | イソキノリン誘導体及び医薬 |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP4316794B2 (ja) |
| AU (1) | AU9646198A (ja) |
| WO (1) | WO1999020620A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103951624A (zh) * | 2010-08-26 | 2014-07-30 | 兴和株式会社 | 1,4-二氮杂环庚烷衍生物或其盐的制造方法 |
Families Citing this family (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7094789B2 (en) | 2002-07-22 | 2006-08-22 | Asahi Kasei Pharma Corporation | 5-substituted isoquinoline derivatives |
| US7160894B2 (en) | 2003-06-06 | 2007-01-09 | Asahi Kasei Pharma Corporation | Tricyclic compound |
| MY139797A (en) * | 2004-11-29 | 2009-10-30 | Kowa Co | (s)-(-)-1-(4-fluoroisoquinolin-5-yl)sulfonyl-2-methyl-1, 4-homopiperazine hydrochloride dihydrate |
| EP1852421B1 (en) * | 2005-02-25 | 2011-05-04 | Kowa Company, Ltd. | Process for production of 4-fluoroisoquinoline-5-sulfonyl halide or salt thereof |
| JP4257383B2 (ja) * | 2005-04-25 | 2009-04-22 | 株式会社デ・ウエスタン・セラピテクス研究所 | 高選択的Rhoキナーゼ阻害剤 |
| WO2006115245A1 (ja) * | 2005-04-25 | 2006-11-02 | D. Western Therapeutics Institute, Inc. | 4-エチニルイソキノリン誘導体及びこれを含有する医薬 |
| WO2006115244A1 (ja) * | 2005-04-25 | 2006-11-02 | D. Western Therapeutics Institute, Inc. | 4-ブロモイソキノリン誘導体及びこれを含有する医薬 |
| JP4972551B2 (ja) * | 2005-06-21 | 2012-07-11 | 興和株式会社 | 緑内障の予防又は治療剤 |
| WO2007007737A1 (ja) * | 2005-07-12 | 2007-01-18 | Kowa Co., Ltd. | 緑内障を予防又は治療する薬剤 |
| CN101253166B (zh) | 2005-08-30 | 2013-07-10 | 旭化成制药株式会社 | 磺酰胺化合物 |
| JP4800720B2 (ja) * | 2005-09-21 | 2011-10-26 | 興和株式会社 | 点眼用組成物 |
| WO2008105058A1 (ja) | 2007-02-27 | 2008-09-04 | Asahi Kasei Pharma Corporation | スルホンアミド化合物 |
| AU2008220104B2 (en) | 2007-02-28 | 2012-09-27 | Asahi Kasei Pharma Corporation | Sulfonamide derivative |
| US8232292B2 (en) | 2007-07-02 | 2012-07-31 | Asahi Kasei Pharma Corporation | Sulfonamide compound and crystal thereof |
| CA2697895C (en) | 2007-08-29 | 2017-10-31 | Senju Pharmaceutical Co., Ltd. | Agent for promoting corneal endothelial cell adhesion |
| TW201729813A (zh) | 2011-02-04 | 2017-09-01 | Kowa Co Ltd | 青光眼預防或治療之藥物療法 |
| ES2716200T3 (es) | 2011-12-06 | 2019-06-11 | Astellas Inst For Regenerative Medicine | Método de diferenciación dirigida que produce células endoteliales corneales |
| ES2897740T3 (es) | 2011-12-28 | 2022-03-02 | Kyoto Prefectural Public Univ Corp | Normalización del cultivo de células endoteliales de la córnea |
| CN105050600B (zh) | 2013-04-24 | 2018-09-28 | 国立大学法人九州大学 | 眼底疾病治疗剂 |
| WO2015016371A1 (ja) | 2013-07-30 | 2015-02-05 | 京都府公立大学法人 | 角膜内皮細胞マーカー |
| US11382904B2 (en) | 2013-10-31 | 2022-07-12 | Kyoto Prefectural Public University Corporation | Therapeutic drug for diseases related to endoplasmic reticulum cell death in corneal endothelium |
| JP6704721B2 (ja) * | 2014-12-12 | 2020-06-03 | 興和株式会社 | 水性の組成物 |
| JP7008337B2 (ja) | 2016-02-15 | 2022-02-10 | 京都府公立大学法人 | ヒト機能性角膜内皮細胞およびその応用 |
| CN107216311B (zh) * | 2016-03-21 | 2019-09-03 | 山东诚创医药技术开发有限公司 | (s)-4-[(4-氟代异喹啉-5-基)磺酰基]-3-甲基-1,4-二氮杂环庚烷盐酸盐的精制方法 |
| CN106496189A (zh) * | 2016-10-10 | 2017-03-15 | 江苏礼华生物技术有限公司 | 盐酸Ripasudil晶型 |
| CA3082643A1 (en) | 2017-11-14 | 2019-05-23 | The Schepens Eye Research Institute, Inc. | Runx1 inhibition for treatment of proliferative vitreoretinopathy and conditions associated with epithelial to mesenchymal transition |
| EP3843845A4 (en) | 2018-08-29 | 2022-05-11 | University Of Massachusetts | INHIBITION OF PROTEIN KINASE FOR THE TREATMENT OF FRIEDREICH'S ATAXIA |
| JPWO2020045642A1 (ja) | 2018-08-31 | 2021-08-12 | 学校法人同志社 | 眼細胞を保存または培養するための組成物および方法 |
| US11445723B2 (en) | 2018-10-02 | 2022-09-20 | The Doshisha | Method and container for preserving corneal endothelial cells |
| CN111116555B (zh) * | 2018-10-30 | 2023-06-02 | 北京盈科瑞创新药物研究有限公司 | 一种Rho激酶抑制剂及其制备方法和应用 |
| US20230257700A1 (en) | 2020-02-27 | 2023-08-17 | Kyoto Prefectural Public University Corporation | Functional human corneal endothelial cells and application thereof |
| CN111909088B (zh) * | 2020-08-04 | 2022-03-01 | 浙江工业大学 | 利用BTC/Ph3PO氯代体系制备异喹啉类盐酸盐中间体及Rho激酶抑制剂的方法 |
| AU2022270611A1 (en) | 2021-05-03 | 2023-10-12 | Astellas Institute For Regenerative Medicine | Methods of generating mature corneal endothelial cells |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1210521A (zh) * | 1996-02-02 | 1999-03-10 | 日本新药株式会社 | 异喹啉衍生物及医药 |
-
1998
- 1998-10-22 WO PCT/JP1998/004781 patent/WO1999020620A1/ja not_active Application Discontinuation
- 1998-10-22 AU AU96461/98A patent/AU9646198A/en not_active Abandoned
- 1998-10-22 JP JP2000516962A patent/JP4316794B2/ja not_active Expired - Lifetime
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103951624A (zh) * | 2010-08-26 | 2014-07-30 | 兴和株式会社 | 1,4-二氮杂环庚烷衍生物或其盐的制造方法 |
| CN103951624B (zh) * | 2010-08-26 | 2019-04-16 | 兴和株式会社 | 1,4-二氮杂环庚烷衍生物或其盐的制造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU9646198A (en) | 1999-05-10 |
| WO1999020620A1 (fr) | 1999-04-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4316794B2 (ja) | イソキノリン誘導体及び医薬 | |
| JP3834663B2 (ja) | イソキノリン誘導体及び医薬 | |
| JP6896113B2 (ja) | ジアザ二環式置換イミダゾピリミジンおよび呼吸障害を治療するためのその使用 | |
| KR102143257B1 (ko) | 인자 XIa 억제제의 결정질 형태 | |
| JP5620340B2 (ja) | 光学活性ピリジル−4h−1,2,4−オキサジアジン誘導体およびその血管疾患の治療における使用 | |
| EP1415990B1 (en) | Diazepan derivatives useful as factor X inhibitor | |
| KR20180088462A (ko) | 수면-관련 호흡기 장애의 치료를 위한 TASK-1 및 TASK-2 채널의 차단제로서의 2-페닐-3-(피페라지노메틸)이미다조[1,2-a]피리딘 유도체 | |
| JP2022031367A (ja) | 置換ジアザヘテロ-二環式化合物およびそれらの使用 | |
| PT1877390E (pt) | Compostos benzisoxazole-piridina e métodos para a sua utilização | |
| BR112012007828B1 (pt) | compostos inibidores da xantina oxidase, processo para preparar os compostos, e, composição farmacêutica para a inibição da xantina oxidase | |
| JP2011522843A (ja) | 新規なカリウムチャンネルブロッカー及びその使用 | |
| KR100264807B1 (ko) | 혈관내막비후억제제 | |
| JP2011201916A5 (ja) | ||
| EP2138482A1 (en) | Bicyclic heterocyclic compound | |
| WO2012174856A1 (zh) | 一类可用于口服的凝血酶抑制剂及其制法以及医药用途 | |
| PT86082B (pt) | Processo para a preparacao de agentes di-hidropiridinicos anti-alergicos e anti-inflamatorios | |
| WO2007055183A1 (ja) | ベンゼン誘導体又はその塩 | |
| WO2016206576A1 (zh) | 一种氘代噻吩并哌啶衍生物、制备方法及其应用 | |
| JP5796872B2 (ja) | 第Xa因子阻害剤の結晶性塩 | |
| JP3748935B2 (ja) | オキシインドール誘導体 | |
| CN114671878B (zh) | 取代的含氮双环化合物及其用途 | |
| CZ448999A3 (cs) | 5-substituované 1,2,4-thiadiazolylové deriváty, způsob jejich výroby a farmaceutické kompozice | |
| JP2006316064A (ja) | 3(2h)−ピリダジノン誘導体及びこれら化合物の使用 | |
| JP2837318B2 (ja) | アンジオテンシンii拮抗性ピリジン誘導体 | |
| WO2000068231A1 (fr) | Dihydrate derive de purine, medicaments le contenant comme principe actif et intermediaire utilise dans sa preparation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050606 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090519 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090521 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 4316794 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120529 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120529 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130529 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140529 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R153 | Grant of patent term extension |
Free format text: JAPANESE INTERMEDIATE CODE: R153 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S802 | Written request for registration of partial abandonment of right |
Free format text: JAPANESE INTERMEDIATE CODE: R311802 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |