JP4157600B2 - 化合物、製剤用組成物及びこれらを含む診断装置とこれらの利用 - Google Patents
化合物、製剤用組成物及びこれらを含む診断装置とこれらの利用 Download PDFInfo
- Publication number
- JP4157600B2 JP4157600B2 JP50766296A JP50766296A JP4157600B2 JP 4157600 B2 JP4157600 B2 JP 4157600B2 JP 50766296 A JP50766296 A JP 50766296A JP 50766296 A JP50766296 A JP 50766296A JP 4157600 B2 JP4157600 B2 JP 4157600B2
- Authority
- JP
- Japan
- Prior art keywords
- leu
- ala
- dnr
- cells
- ligand
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 150000001875 compounds Chemical class 0.000 title claims description 56
- 239000000203 mixture Substances 0.000 title claims description 12
- 239000003814 drug Substances 0.000 claims abstract description 51
- 239000003446 ligand Substances 0.000 claims abstract description 45
- 229940124597 therapeutic agent Drugs 0.000 claims abstract description 39
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 claims description 85
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 claims description 74
- 229960000975 daunorubicin Drugs 0.000 claims description 74
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 claims description 73
- 229960004679 doxorubicin Drugs 0.000 claims description 56
- 206010028980 Neoplasm Diseases 0.000 claims description 29
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 28
- 229940045799 anthracyclines and related substance Drugs 0.000 claims description 27
- 239000003550 marker Substances 0.000 claims description 23
- -1 vinca alkaloid Chemical compound 0.000 claims description 20
- 150000001413 amino acids Chemical class 0.000 claims description 12
- 239000008194 pharmaceutical composition Substances 0.000 claims description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 8
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 8
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 8
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 claims description 7
- 206010061218 Inflammation Diseases 0.000 claims description 7
- 230000004054 inflammatory process Effects 0.000 claims description 7
- FSBIGDSBMBYOPN-VKHMYHEASA-N L-canavanine Chemical compound OC(=O)[C@@H](N)CCONC(N)=N FSBIGDSBMBYOPN-VKHMYHEASA-N 0.000 claims description 6
- 208000025747 Rheumatic disease Diseases 0.000 claims description 6
- 229940122803 Vinca alkaloid Drugs 0.000 claims description 6
- HXCHCVDVKSCDHU-LULTVBGHSA-N calicheamicin Chemical compound C1[C@H](OC)[C@@H](NCC)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@](C/3=C/CSSSC)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HXCHCVDVKSCDHU-LULTVBGHSA-N 0.000 claims description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 6
- OIRDTQYFTABQOQ-KQYNXXCUSA-N Adenosine Natural products C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 claims description 5
- 229930195731 calicheamicin Natural products 0.000 claims description 5
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 claims description 5
- 229960001156 mitoxantrone Drugs 0.000 claims description 5
- 239000002126 C01EB10 - Adenosine Substances 0.000 claims description 4
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 claims description 4
- 229960005305 adenosine Drugs 0.000 claims description 4
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 claims description 4
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 claims description 4
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 claims description 4
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 claims description 4
- GOLORTLGFDVFDW-UHFFFAOYSA-N 3-(1h-benzimidazol-2-yl)-7-(diethylamino)chromen-2-one Chemical compound C1=CC=C2NC(C3=CC4=CC=C(C=C4OC3=O)N(CC)CC)=NC2=C1 GOLORTLGFDVFDW-UHFFFAOYSA-N 0.000 claims description 3
- 108010006654 Bleomycin Proteins 0.000 claims description 3
- 229930192392 Mitomycin Natural products 0.000 claims description 3
- FSBIGDSBMBYOPN-UHFFFAOYSA-N O-guanidino-DL-homoserine Natural products OC(=O)C(N)CCON=C(N)N FSBIGDSBMBYOPN-UHFFFAOYSA-N 0.000 claims description 3
- 239000002671 adjuvant Substances 0.000 claims description 3
- 229960001561 bleomycin Drugs 0.000 claims description 3
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 claims description 3
- 229960005304 fludarabine phosphate Drugs 0.000 claims description 3
- 229960001924 melphalan Drugs 0.000 claims description 3
- 229960004857 mitomycin Drugs 0.000 claims description 3
- JVXXKQIRGQDWOJ-UHFFFAOYSA-N naphthalene-2-carboxamide Chemical compound C1=CC=CC2=CC(C(=O)N)=CC=C21 JVXXKQIRGQDWOJ-UHFFFAOYSA-N 0.000 claims description 3
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 3
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 claims description 2
- 229940127093 camptothecin Drugs 0.000 claims description 2
- 125000004432 carbon atom Chemical group C* 0.000 claims description 2
- BFMYDTVEBKDAKJ-UHFFFAOYSA-L disodium;(2',7'-dibromo-3',6'-dioxido-3-oxospiro[2-benzofuran-1,9'-xanthene]-4'-yl)mercury;hydrate Chemical compound O.[Na+].[Na+].O1C(=O)C2=CC=CC=C2C21C1=CC(Br)=C([O-])C([Hg])=C1OC1=C2C=C(Br)C([O-])=C1 BFMYDTVEBKDAKJ-UHFFFAOYSA-L 0.000 claims description 2
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 claims description 2
- TUFFYSFVSYUHPA-UHFFFAOYSA-M rhodamine 123 Chemical compound [Cl-].COC(=O)C1=CC=CC=C1C1=C(C=CC(N)=C2)C2=[O+]C2=C1C=CC(N)=C2 TUFFYSFVSYUHPA-UHFFFAOYSA-M 0.000 claims description 2
- 229960000303 topotecan Drugs 0.000 claims description 2
- UCMIRNVEIXFBKS-UHFFFAOYSA-N beta-alanine Chemical compound NCCC(O)=O UCMIRNVEIXFBKS-UHFFFAOYSA-N 0.000 claims 2
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 claims 2
- 239000000825 pharmaceutical preparation Substances 0.000 claims 2
- SFKZPTYRENGBTJ-UHFFFAOYSA-N 4-methoxynaphthalen-2-amine Chemical compound C1=CC=C2C(OC)=CC(N)=CC2=C1 SFKZPTYRENGBTJ-UHFFFAOYSA-N 0.000 claims 1
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 claims 1
- 229940000635 beta-alanine Drugs 0.000 claims 1
- 229960000304 folic acid Drugs 0.000 claims 1
- 235000019152 folic acid Nutrition 0.000 claims 1
- 239000011724 folic acid Substances 0.000 claims 1
- 229940002612 prodrug Drugs 0.000 abstract description 15
- 239000000651 prodrug Substances 0.000 abstract description 15
- 210000004369 blood Anatomy 0.000 abstract description 12
- 239000008280 blood Substances 0.000 abstract description 12
- 230000003834 intracellular effect Effects 0.000 abstract description 10
- 210000002966 serum Anatomy 0.000 abstract description 7
- 230000035515 penetration Effects 0.000 abstract 1
- 210000004027 cell Anatomy 0.000 description 125
- 241000699670 Mus sp. Species 0.000 description 31
- 230000037396 body weight Effects 0.000 description 24
- 210000004881 tumor cell Anatomy 0.000 description 21
- 230000001988 toxicity Effects 0.000 description 19
- 231100000419 toxicity Toxicity 0.000 description 19
- 102000035195 Peptidases Human genes 0.000 description 18
- 108091005804 Peptidases Proteins 0.000 description 18
- 235000018102 proteins Nutrition 0.000 description 18
- 102000004169 proteins and genes Human genes 0.000 description 18
- 108090000623 proteins and genes Proteins 0.000 description 18
- 230000003013 cytotoxicity Effects 0.000 description 17
- 231100000135 cytotoxicity Toxicity 0.000 description 17
- 210000002950 fibroblast Anatomy 0.000 description 17
- 229960004528 vincristine Drugs 0.000 description 17
- 239000003636 conditioned culture medium Substances 0.000 description 16
- 238000000034 method Methods 0.000 description 16
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 16
- 239000002207 metabolite Substances 0.000 description 15
- 208000026310 Breast neoplasm Diseases 0.000 description 14
- 125000003277 amino group Chemical group 0.000 description 14
- 206010006187 Breast cancer Diseases 0.000 description 13
- 230000035508 accumulation Effects 0.000 description 13
- 238000009825 accumulation Methods 0.000 description 13
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 13
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 11
- 239000004472 Lysine Substances 0.000 description 11
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 11
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- 201000011510 cancer Diseases 0.000 description 11
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 10
- 102000004190 Enzymes Human genes 0.000 description 10
- 108090000790 Enzymes Proteins 0.000 description 10
- 238000011785 NMRI mouse Methods 0.000 description 10
- 230000007059 acute toxicity Effects 0.000 description 10
- 231100000403 acute toxicity Toxicity 0.000 description 10
- 229940024606 amino acid Drugs 0.000 description 10
- 235000001014 amino acid Nutrition 0.000 description 10
- 229940079593 drug Drugs 0.000 description 10
- 238000002347 injection Methods 0.000 description 10
- 239000007924 injection Substances 0.000 description 10
- 235000019833 protease Nutrition 0.000 description 10
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 9
- 230000008859 change Effects 0.000 description 9
- 238000002474 experimental method Methods 0.000 description 9
- DZINNPYMYFDDHL-RCQRPICHSA-N n-[6-[[(1s,3s)-3-acetyl-3,5,12-trihydroxy-10-methoxy-6,11-dioxo-2,4-dihydro-1h-tetracen-1-yl]oxy]-3-hydroxy-2-methyloxan-4-yl]-2-amino-4-methylpentanamide Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)C1CC(NC(=O)C(N)CC(C)C)C(O)C(C)O1 DZINNPYMYFDDHL-RCQRPICHSA-N 0.000 description 9
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- 241001465754 Metazoa Species 0.000 description 8
- 239000004365 Protease Substances 0.000 description 8
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 7
- 241000699666 Mus <mouse, genus> Species 0.000 description 7
- 230000000259 anti-tumor effect Effects 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 230000015556 catabolic process Effects 0.000 description 6
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N coumarin Chemical compound C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 6
- 238000006731 degradation reaction Methods 0.000 description 6
- 230000007062 hydrolysis Effects 0.000 description 6
- 238000006460 hydrolysis reaction Methods 0.000 description 6
- 229960000485 methotrexate Drugs 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 5
- 229930012538 Paclitaxel Natural products 0.000 description 5
- 230000001413 cellular effect Effects 0.000 description 5
- 238000002512 chemotherapy Methods 0.000 description 5
- 238000001990 intravenous administration Methods 0.000 description 5
- 210000004882 non-tumor cell Anatomy 0.000 description 5
- 229960001592 paclitaxel Drugs 0.000 description 5
- 235000019419 proteases Nutrition 0.000 description 5
- 239000000377 silicon dioxide Substances 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- 230000004580 weight loss Effects 0.000 description 5
- 230000009471 action Effects 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 230000037041 intracellular level Effects 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 206010009944 Colon cancer Diseases 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 3
- 238000004364 calculation method Methods 0.000 description 3
- 125000003636 chemical group Chemical group 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 208000029742 colonic neoplasm Diseases 0.000 description 3
- 229960000956 coumarin Drugs 0.000 description 3
- 235000001671 coumarin Nutrition 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000003745 diagnosis Methods 0.000 description 3
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 3
- 239000003480 eluent Substances 0.000 description 3
- 150000002224 folic acids Chemical class 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 238000007912 intraperitoneal administration Methods 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 230000010534 mechanism of action Effects 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000010532 solid phase synthesis reaction Methods 0.000 description 3
- 229960003048 vinblastine Drugs 0.000 description 3
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 3
- CILBMBUYJCWATM-PYGJLNRPSA-N vinorelbine ditartrate Chemical class OC(=O)[C@H](O)[C@@H](O)C(O)=O.OC(=O)[C@H](O)[C@@H](O)C(O)=O.C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC CILBMBUYJCWATM-PYGJLNRPSA-N 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 238000009010 Bradford assay Methods 0.000 description 2
- 102000004157 Hydrolases Human genes 0.000 description 2
- 108090000604 Hydrolases Proteins 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- 231100000111 LD50 Toxicity 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 206010061309 Neoplasm progression Diseases 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 150000003797 alkaloid derivatives Chemical class 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 210000001185 bone marrow Anatomy 0.000 description 2
- 230000007665 chronic toxicity Effects 0.000 description 2
- 231100000160 chronic toxicity Toxicity 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 210000003743 erythrocyte Anatomy 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 238000010253 intravenous injection Methods 0.000 description 2
- 229960003136 leucine Drugs 0.000 description 2
- 208000032839 leukemia Diseases 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 210000001616 monocyte Anatomy 0.000 description 2
- 210000000653 nervous system Anatomy 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 231100000161 signs of toxicity Toxicity 0.000 description 2
- 231100000027 toxicology Toxicity 0.000 description 2
- 230000005751 tumor progression Effects 0.000 description 2
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 2
- 229960004355 vindesine Drugs 0.000 description 2
- HJEZFVLKJYFNQW-PRFXOSGESA-N (13S)-13-dihydrodaunorubicin Chemical group O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)[C@H](C)O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 HJEZFVLKJYFNQW-PRFXOSGESA-N 0.000 description 1
- NAALWFYYHHJEFQ-ZASNTINBSA-N (2s,5r,6r)-6-[[(2r)-2-[[6-[4-[bis(2-hydroxyethyl)sulfamoyl]phenyl]-2-oxo-1h-pyridine-3-carbonyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid Chemical compound N([C@@H](C(=O)N[C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C=1C=CC(O)=CC=1)C(=O)C(C(N1)=O)=CC=C1C1=CC=C(S(=O)(=O)N(CCO)CCO)C=C1 NAALWFYYHHJEFQ-ZASNTINBSA-N 0.000 description 1
- XVXGYZFARCOVHS-BINOZUKVSA-N (7s,9s)-7-[(2r,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-8,10-dihydro-7h-tetracene-5,12-dione Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(=O)CO)C1 XVXGYZFARCOVHS-BINOZUKVSA-N 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- FUXVKZWTXQUGMW-FQEVSTJZSA-N 9-Aminocamptothecin Chemical compound C1=CC(N)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 FUXVKZWTXQUGMW-FQEVSTJZSA-N 0.000 description 1
- 208000031648 Body Weight Changes Diseases 0.000 description 1
- 102000005600 Cathepsins Human genes 0.000 description 1
- 108010084457 Cathepsins Proteins 0.000 description 1
- 241000557626 Corvus corax Species 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- ZZZCUOFIHGPKAK-UHFFFAOYSA-N D-erythro-ascorbic acid Natural products OCC1OC(=O)C(O)=C1O ZZZCUOFIHGPKAK-UHFFFAOYSA-N 0.000 description 1
- HJEZFVLKJYFNQW-UHFFFAOYSA-N Daunorubicinol Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)O)CC1OC1CC(N)C(O)C(C)O1 HJEZFVLKJYFNQW-UHFFFAOYSA-N 0.000 description 1
- OWCHPBVMSHIYCQ-UHFFFAOYSA-N Dihydro-dauno-mycinon Natural products C1C(O)(C(C)O)CC(O)C2=C1C(O)=C1C(=O)C(C=CC=C3OC)=C3C(=O)C1=C2O OWCHPBVMSHIYCQ-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 102000005593 Endopeptidases Human genes 0.000 description 1
- 108010059378 Endopeptidases Proteins 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 206010022998 Irritability Diseases 0.000 description 1
- 241000764238 Isis Species 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- 239000004395 L-leucine Substances 0.000 description 1
- 235000019454 L-leucine Nutrition 0.000 description 1
- 108010057150 Peplomycin Proteins 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229930003268 Vitamin C Natural products 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 229960003767 alanine Drugs 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000007234 antiinflammatory process Effects 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 239000003899 bactericide agent Substances 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 238000005415 bioluminescence Methods 0.000 description 1
- 230000029918 bioluminescence Effects 0.000 description 1
- 230000004579 body weight change Effects 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- XREUEWVEMYWFFA-CSKJXFQVSA-N carminomycin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=C(O)C=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XREUEWVEMYWFFA-CSKJXFQVSA-N 0.000 description 1
- 229930188550 carminomycin Natural products 0.000 description 1
- XREUEWVEMYWFFA-UHFFFAOYSA-N carminomycin I Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=C(O)C=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XREUEWVEMYWFFA-UHFFFAOYSA-N 0.000 description 1
- 229950001725 carubicin Drugs 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229910001429 cobalt ion Inorganic materials 0.000 description 1
- XLJKHNWPARRRJB-UHFFFAOYSA-N cobalt(2+) Chemical compound [Co+2] XLJKHNWPARRRJB-UHFFFAOYSA-N 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 229960003109 daunorubicin hydrochloride Drugs 0.000 description 1
- 229950000950 daunorubicinol Drugs 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 150000004985 diamines Chemical class 0.000 description 1
- 238000006193 diazotization reaction Methods 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000012202 endocytosis Effects 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 230000007071 enzymatic hydrolysis Effects 0.000 description 1
- 238000006047 enzymatic hydrolysis reaction Methods 0.000 description 1
- ITSGNOIFAJAQHJ-BMFNZSJVSA-N esorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)C[C@H](C)O1 ITSGNOIFAJAQHJ-BMFNZSJVSA-N 0.000 description 1
- 229950002017 esorubicin Drugs 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 231100000086 high toxicity Toxicity 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000007154 intracellular accumulation Effects 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 230000001678 irradiating effect Effects 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 150000002576 ketones Chemical group 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 231100000636 lethal dose Toxicity 0.000 description 1
- 231100000225 lethality Toxicity 0.000 description 1
- 108700020781 liblomycin Proteins 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- YIQATJBOCJPFCF-XQLDGQACSA-N methyl (1R,9R,10S,11R,12R,19R)-12-ethyl-4-[(13S,15R,17S)-17-ethyl-17-hydroxy-13-methoxycarbonyl-1,11-diazatetracyclo[13.3.1.04,12.05,10]nonadeca-4(12),5,7,9-tetraen-13-yl]-8-formyl-10,11-dihydroxy-5-methoxy-8,16-diazapentacyclo[10.6.1.01,9.02,7.016,19]nonadeca-2,4,6,13-tetraene-10-carboxylate Chemical compound CC[C@]1(O)C[C@@H]2CN(C1)CCc1c([nH]c3ccccc13)[C@@](C2)(C(=O)OC)c1cc2c(cc1OC)N(C=O)[C@@H]1[C@]22CCN3CC=C[C@](CC)([C@@H]23)[C@@H](O)[C@]1(O)C(=O)OC YIQATJBOCJPFCF-XQLDGQACSA-N 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- 229940086322 navelbine Drugs 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- QIMGFXOHTOXMQP-GFAGFCTOSA-N peplomycin Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCCN[C@@H](C)C=1C=CC=CC=1)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C QIMGFXOHTOXMQP-GFAGFCTOSA-N 0.000 description 1
- 229950003180 peplomycin Drugs 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000004451 qualitative analysis Methods 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 239000012925 reference material Substances 0.000 description 1
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- GBABOYUKABKIAF-IELIFDKJSA-N vinorelbine Chemical class C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-IELIFDKJSA-N 0.000 description 1
- 235000019154 vitamin C Nutrition 0.000 description 1
- 239000011718 vitamin C Substances 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Images





























Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K9/00—Peptides having up to 20 amino acids, containing saccharide radicals and having a fully defined sequence; Derivatives thereof
- C07K9/001—Peptides having up to 20 amino acids, containing saccharide radicals and having a fully defined sequence; Derivatives thereof the peptide sequence having less than 12 amino acids and not being part of a ring structure
- C07K9/005—Peptides having up to 20 amino acids, containing saccharide radicals and having a fully defined sequence; Derivatives thereof the peptide sequence having less than 12 amino acids and not being part of a ring structure containing within the molecule the substructure with m, n > 0 and m+n > 0, A, B, D, E being heteroatoms; X being a bond or a chain, e.g. muramylpeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/65—Peptidic linkers, binders or spacers, e.g. peptidic enzyme-labile linkers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/0019—Fluorescence in vivo characterised by the fluorescent group, e.g. oligomeric, polymeric or dendritic molecules
- A61K49/0021—Fluorescence in vivo characterised by the fluorescent group, e.g. oligomeric, polymeric or dendritic molecules the fluorescent group being a small organic molecule
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/0019—Fluorescence in vivo characterised by the fluorescent group, e.g. oligomeric, polymeric or dendritic molecules
- A61K49/0021—Fluorescence in vivo characterised by the fluorescent group, e.g. oligomeric, polymeric or dendritic molecules the fluorescent group being a small organic molecule
- A61K49/0041—Xanthene dyes, used in vivo, e.g. administered to a mice, e.g. rhodamines, rose Bengal
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/005—Fluorescence in vivo characterised by the carrier molecule carrying the fluorescent agent
- A61K49/0052—Small organic molecules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0013—Luminescence
- A61K49/0017—Fluorescence in vivo
- A61K49/005—Fluorescence in vivo characterised by the carrier molecule carrying the fluorescent agent
- A61K49/0056—Peptides, proteins, polyamino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Biomedical Technology (AREA)
- Pharmacology & Pharmacy (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Crystallography & Structural Chemistry (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Pulmonology (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Immunology (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicinal Preparation (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Peptides Or Proteins (AREA)
- Hydrogenated Pyridines (AREA)
- Multicomponent Fibers (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Description
本発明は、新規の化合物、製剤用組成物、及びこれらを含む診断装置、並びに癌性腫瘍及び/または炎症反応を診断及び/または治療するための薬剤の製造を目的とするこれらの利用に係わる。
公知技術及び本発明の技術的背景
数年来、アントラサイクリンやビンカアルカロイドのような種々の腫瘍治療剤が開発され、癌の治療に特に効果を上げている。しかし、これらの分子は、生体内で急性毒性、特に骨髄及び粘膜に対する毒性を示すだけでなく、アントラサイクリンの場合には心臓に対して慢性毒性を、ビンカアルカロイドの場合には神経系に対して慢性毒性を示すことが多い。
そこで、腫瘍細胞に対してもっと有効な特異性の高い抗腫瘍剤を開発してこの種の製品の副作用(毒性、非腫瘍細胞の破壊など)を軽減する努力がなされている。
米国特許第4,296,105号は、随意に置換可能なアミノ酸と結合しているドキソルビシン誘導体を開示しており、この誘導体はドキソルビシンよりも抗腫瘍作用が強く、毒性が低い。
しかし、この誘導体は細胞内活性部位を有し、依然として腫瘍細胞と正常細胞とに進入し易い。
そこで、数年前からプロドラッグの形で新規の治療剤が提案されている。
プロドラッグは、その構造が化学的作用または酵素作用で変化することによってドラッグ(活性治療化合物)に変換できる分子である。
しかし、このプロドラッグにも、この分子を不活性化する酵素を含んでいる血液及び血清中では安定度が低いという特徴がある。
文献 ケミカルアブストラクト(Chemical Abstract)97:150635,ジャーナル オブ メディカル ケミストリー(Journal of Medicinal Chemistry),Vol.23,pp.1171−1174(1980);ドラッグス Exp.Clin.(Drugs Exp.Clin.),Vol.9,pp.303−311(1983);ジャーナル オブ メディカル ケミストリー(Journal of Medical Chemistry),Vol.23,pp.1166−1170(1980)及び特許出願BE−882.541号は、ペプチドアームを介してドラッグと結合するキャリアを含むプロドラッグを記述している。好ましくは、アームは4個のアミノ酸から成り、遊離カルボキシル官能基を介して例えばダウノルビシンのようなアントラサイクリン誘導体の遊離アミン官能基と結合している。
また、このようなプロドラッグのアームは、ターゲット細胞の選択的なエンドサイトーシスを可能にする巨大分子(BSA、イムノグロブリンなどのような蛋白質)から成るキャリアと、遊離アミン官能基を介して結合している。
リウマチ疾患のような炎症反応の治療にメトトレキサートを利用できることも公知であるが、その高い毒性がその利用範囲を狭めている。
発明の目的
本発明の目的は、抗腫瘍性治療剤またはマーカを含む新規の化合物、特に抗腫瘍性治療剤を含み、公知製品よりもすぐれた治療性能を示し、特に癌性腫瘍の治療及び/またはリウマチ疾患のような炎症反応の治療においてすぐれた治療性能を示すプロドラッグを提供することにある。
本発明の具体的な目的は、作用、低い毒性、及び血清と血液中での高い安定性という高度の特異性を示すプロドラッグを得ることにある。
本発明の他の目的は、腫瘍を特徴づけること(腫瘍の診断、進行、腫瘍細胞から分泌される因子の検査など)を可能にするマーカを含む化合物を得ることにある。
本発明の特徴
本発明は、末端基(W)とそれに結合するアーム(Z)とを含むリガンド(W−Z)、及びマーカと治療剤とからなる群から選択されて前記リガンド(W−Z)と結合する成分(M)を有する化合物(W−Z−M)において、リガンド(W−Z)のアーム(Z)と成分(M)との間の結合が化合物(W−Z−M)の細胞内進入を妨げ及び/またはマーカ(M)の発現を抑止し、ターゲット細胞から分泌される因子によって前記結合を選択的に開裂することにより、マーカ(M)の発現または前記ターゲット細胞への治療剤(M)の進入を可能にすることができ、末端基(W)とアーム(Z)との間の結合及びアーム(Z)と成分(M)との間の結合が、化合物(W−Z−M)の性質に影響を及ぼさない共有結合であり、末端基(W)が、哺乳動物に存在しないアミノ酸であることを特徴とする化合物(W−Z−M)に係わる。
ターゲット細胞、特に腫瘍細胞及び炎症反応を伴なっている細胞(マクロファージ、単球など)から分泌される因子は、前記細胞から特に細胞外媒質中へ分泌されるプロテアーゼまたはペプチダーゼのような酵素を意味すると考えられる。
従って、これらの酵素は成分(M)とリガンド(W−Z)のアーム(Z)との間に存在する結合を選択的に開裂することによって、(好ましくは前記細胞の領域における)マーカの発現及び/または前記細胞への治療剤の優先的な進入を可能にして前記細胞の破壊及び/またはその増殖の阻止を達成することができる。
末端基(W)とアーム(Z)との間の結合も、アーム(Z)と成分(M)との間の結合も、本発明の化合物(W−Z−M)の性質に影響を及ぼさない共有結合ならどのような形でもよい。
マーカ(M)の発現を抑止するリカンド(W−Z)のアーム(Z)とマーカ(M)との間の結合は、マーカ(M)の検出及び/または定量を妨げる共有結合を意味すると考えられる。
遊離マーカ(M)の発現は、当業者に周知の方法または装置、例えば、着色、蛍光、生体発光、化学発光などのほか、場合によっては1種類または2種類以上の中間試薬を併用して検出することができる。
好ましくは、前記マーカをクマリン、7−アミド−4−(トリフルオロメチル)−クマリン、(遊離の形では、ジアゾ化反応後に比色計によって検出できる)パラ−ニトロアニリド、β−ナフチルアミド及び4−メトキシ−β−ナフチルアミドから成るグールプから選択され、リガンド(W−Z)と結合していなければ蛍光によって検出できる。
血清及び循環血液中において本発明の化合物を安定させる機能を果す末端基(W)は、血清及び循環血液中における本発明の化合物の開裂を減少または抑止する基、特に血清及び/または循環血液中に存在するプロテイナーゼ及びペプチダーゼによる本発明の化合物の加水分解を減少または抑止する基、特に赤血球と関連するペプチダーゼ及びプロテイナーゼによる加水分解を減少または抑止する基を意味すると考えられる。
具体的には、37℃のヒトの血液中に2時間以上滞留している間、化合物の20%以下、好ましくは2%以下が前記酵素によって開裂されても、末端基(W)は本発明の化合物の安定性を維持する。
末端基(W)は、哺乳動物には存在していないアミノ酸(すなわち、哺乳動物によっては遺伝的にコード化され得ないアミノ酸)から選ばれる。
本発明の好ましい実施例では、基(W)はカルボキシ官能基を介してアーム(Z)と結合する式:
NH2−CH2−CH2−COOH
で表わされるβ−アラニンである。
アーム(Z)は、成分(M)との結合がターゲット細胞から分泌される因子によって選択的に開裂されることで前記細胞の領域におけるマーカ(M)の発現及び/または前記細胞への治療剤(M)の優先的な進入を許すような化学構造(多糖類、ペプチドなど)であればよい。
本発明では、成分(M)とリガンド(W−Z)のアーム(Z)との間の結合はペプチド結合から成る。好ましくは、アーム(Z)は少なくとも2つの随意に置換可能なアミノ酸から成るペプチドである。
リガンド(W−Z)のアーム(Z)は、好ましくは下記のアミノ酸の配列から成る:即ち、カルボキシ官能基を介して成分(M)と結合するL−ロイシル−L−アラニル−L−ロイシル、L−ロイシル−L−アラニルまたはL−アラニル−L−ロイシル−L−フェニルアラニルまたはL−アラニル−L−ロイシルから成る。
本発明では、治療剤(M)として好ましくはブルース エイ.チャブナ(Bruce A.Chabner)及びジェリ エム.コリンズ(Jerry M.Collins)(カンサケモテラピー,リッピンコット Ed.ISBN(Cancer Chemotherapy,Lippincott Ed.,ISBN)0−397−50900−6(1990))によって記述されている治療剤から選択された癌の化学療法に使用されている治療剤か、またはメトトレキサートのような抗炎症剤であり、リガンド(W−Z)と結合可能な、好ましくはペプチド結合を介してリガンド(W−Z)の基(Z)と結合可能なものである。
具体的には、治療剤を、アントラサイクリン、葉酸誘導体、ビンカアルカロイド、ミトザントロン、カリケアマイシン、シトシン アラビノシド(ARA−C)またはアデノシン アラビノシド(ARA−A)、リン酸フルダラビン、メルファラン、ブレオマイシン、ミトマイシン、L−カナバニン、トキソイド、カンプトテシン及びこれらの誘導体、特にTOPOTECAN(商品名、9−ジメチル−アミノメチル−10−ヒドロキシカンプトテシンヒドロクロリド)、ローダミン123のようなフルオロクロム誘導体、特にローダミンイソチオシアネート、及び随意に置換または非置換アミノ酸と結合しているそれらの誘導体から選択する。アミノ酸における置換は、本発明の化合物(W−Z−M)の性質に影響を及ぼさない置換であればよい。
これらの分子の誘導体は、元の分子の治療作用に影響を及ぼさない共有結合を介してリガンド(W−Z)のアーム(Z)と結合することを可能にする化学基で変性させられた前記分子を意味する。
本発明の化合物においては、成分(M)を随意に置換または非置換アミノ酸と結合しているアントラサイクリン、特にドキソルビシン(DOX)及びダウノルビシン(DNR)から成るグループから選択するのが好ましい。
成分(M)は、好ましくは次の一般式に対応するものである:

−R1は、水素原子またはOH基、
−R2は、水素原子または次式のラジカル:
−R3は、水素原子または随意に置換されているアルキルラジカルを表わし、
−R4は、水素原子、またはR3と共に3または4個の炭素原子を有するアルキレンラジカルを形成する。
R2は、好ましくは次式のラジカルであるか、またはその同位体の1つである:
本発明は、本発明の化合物、及び場合によっては製剤上許容できる補薬または賦形剤を含む製剤用組成物にも係わる。
これらの組成物は、例えば、非経口または静脈内投与することができる。非経口投与用の本発明組成物は、特に滅菌溶液、水性または非水性の懸濁液または乳化液の形を取ることができる。製剤上許容できる溶剤または賦形剤としては、プロピレングリコール、ポリエチレングリコール、注射可能な有機エステル、例えば、オレイン酸エチル、またはシクロデクストリンを使用できる。これらの組成物は、湿潤剤、乳化剤及び/または分散剤をも含むことができる。
滅菌の方法としては、例えば細菌フィルタを使用したり、組成物に殺菌剤を入れたり、照射したりする種々の方法がある。使用時に滅菌水やその他の滅菌注射液に溶かすことができる滅菌固形組成物の形に製造することもできる。
本発明は、癌性腫瘍の治療効果を高め、持続させるため本発明の化合物と併用できる(ビタミンC、酸化防止剤などのような)当業者に公知の補薬をも含むことができる。
患者への本発明化合物の最少投与量は、特にBruce A.Chabner and Jerry N Collins(Cancer Chemotherapy,Lippincott Ed.,ISBN 0−397−50900−6(1990))によって記述されている上記抗腫瘍治療剤の投与量またはそれ以上の投与量である。
投与量は、本発明の化合物の調製に使用される治療剤に応じて異なる。
本発明は、癌性腫瘍の治療薬の製造を目的とする本発明の製剤用組成物の利用にも係わり、また、本発明の製剤用組成物を患者に対して特に非経口または静脈内投与することから成る癌性腫瘍の治療方法にも係わる。
本発明はまた、炎症反応、特にリウマチ疾患の治療薬の製造を目的とする本発明の製剤用組成物の利用にも係わり、また、本発明の製剤用組成物、特に治療剤(M)がメトトレキサートまたはメトトレキサート誘導体である本発明の化合物を含む製剤用組成物を患者に対して特に非経口または静脈内投与することから成る炎症反応、特にリウマチ疾患の治療方法にも係わる。
本発明はまた、本発明の化合物、特にクマリンをマーカとする化合物を含む診断及び/または検査装置にも係わる。
【図面の簡単な説明】
図1は、本発明の化合物の、ターゲット細胞(T.C.)における活性部位(A.S.)における作用メカニズムを示す。
図2は、本発明の化合物の、正常細胞(N.C.)における作用メカニズムを示す。
図3は、MCF7/6ヒト乳癌細胞から得たならし培地中でのβ−Ala−Leu−Ala−Leu−ダウノルビシンの加水分解を示す((a)は0時点、(b)は2時間の培養後におけるクロマトグラム)。
図4は、MCF7/ADRヒト乳癌細胞から得たならし培地中でのβ−Ala−Leu−Ala−Leu−ダウノルビシンの加水分解を示す((a)は0時点、(b)は1時間の培養後におけるクロマトグラム)。
図5は、HT29結腸癌細胞から得たならし培地中でのβ−Ala−Leu−Ala−Leu−ダウノルビシンの加水分解を示す((a)は0時点、(b)は2時間の培養後におけるクロマトグラム)。
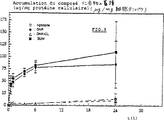
図6は、融合性MCF7/6細胞による濃度10μg/mlでのダウノルビシン(DNR)の蓄積量を示す。
図7は、融合性MCF7/6細胞による濃度10μg当量DNR/mlでのN−L−ロイシルダウノルビシン(Leu−DNR)の蓄積量を示す。
図8は、融合性MCF7/6細胞による濃度10μg当量DNR/mlでのβ−Ala−Leu−Ala−Leu−ダウノルビシンの蓄積量を示す。
図9は、融合性MRC5線維芽細胞による濃度10μg/mlでのダウノルビシン(DNR)の蓄積量を示す。
図10は、融合性MRC5線維芽細胞による濃度10μg当量DNR/mlでのN−L−ロイシルダウノルビシン(Leu−DNR)の蓄積量を示す。
図11は、融合性MRC5線維芽細胞による濃度10μg当量DNR/mlでのβ−Ala−Leu−Ala−Leu−ダウノルビシンの蓄積量を示す。
図12は、アントラサイクリンの存在において72時間培養されたMCF7/6細胞に対するダウノルビン(DNR)、L−Leu−ダウノルビシン及びβ−Ala−Leu−Ala−Leu−ダウノルビシンの細胞毒性を示す。
図13は、アントラサイクリンの存在において72時間培養されたMRC5線維芽細胞に対するダウノルビシン(DNR)、L−Leu−ダウノルビシン及びβ−Ala−Leu−A1a−Leu−ダウノルビシンの細胞毒性を示す。
図14は、連続5日間にわたり総量2.0〜3.5mg/kgのダウノルビシン(DNR)を腹腔内投与したのち、雌NMRIマウスに現われた死亡率を示す。
図15は、連続5日間にわたり総量2.0〜3.5mg/kgのダウノルビシンを腹腔内投与された雌NMRIマウスの平均体重の変化を示す。体重は、グループごとの平均初期体重の%で表わしてある。
図16は、連続5日間にわたり総量10〜60mg/kgのβ−Ala−Leu−Ala−Leu−ダウノルビシンを腹腔内投与された雌NMRIマウスの死亡率を示す。
図17は、連続5日間にわたり総量10〜60mg/kgのβ−Ala−Leu−Ala−Leu−ダウノルビシンを1回投与された雌NMRIマウスの平均体重の変化を示す。体重は、グループごとの平均初期体重の%で表わしてある。
図18は、10〜35mg/kgの投与量でダウノルビシン(DNR)を1回静脈内投与された雌NMRIマウスの死亡率を示す。
図19は、10〜35mg/kgのダウノルビシンを静脈内投与された雌NMRIマウスの平均体重の変化を示す。体重は、グループごとの平均初期体重の%で表わしてある。
図20は、30〜60mg/kgのβ−Ala−Leu−Ala−Leu−ダウノルビシンを静脈内投与された雌NMRIマウスの平均体重の変化を示す。体重はグループごとの平均初期体重の%で表わしてある。
図21は、30mg/kgのβ−Ala−Leu−Ala−Leu−ダウノルビシンを1回静脈投与するか、2日間にわたり30mg/kgずつを2回投与した場合に、雌NMRIマウスの平均体重に現われた変化を示す。体重は、グループごとの平均初期体重を%で表わしてある。
図22は、MCF7/6細胞から得たならし培地中で起こるβ−Ala−Leu−Ala−Leu−ドキソルビシンからL−Ala−L−Leu−ドキソルビシン及びL−Leu−ドキソルビシンへの酵素による加水分解の速度を示す。
図23は、融合性MCF7/6細胞による濃度10μg/mlでのドキソルビシン(DOX)の蓄積量を示す。
図24は、融合性MCF7/6細胞による濃度10μg/mlでのL−Leu−ドキソルビシンの蓄積量を示す。
図25は、融合性MCF7/6細胞による濃度10μg/mlでのβ−Ala−Leu−Ala−Leu−ドキソルビシンの蓄積量を示す。
図26は、融合性MRC5線維芽細胞による濃度10μg/mlでのドキソルビシン(DOX)の蓄積量を示す。
図27は、融合性MRC5線維芽細胞による濃度10μg/mlでのL−Leu−ドキソルビシンの蓄積量を示す。
図28は、融合性MRC5線維芽細胞による濃度10μg/mlでのβ−Ala−Leu−Ala−Leu−ドキソルビシンの蓄積量を示す。
図29は、前記アントラサイクリン誘導体の存在において72時間培養されたMCF7/6細胞に対するドキソルビシン(DOX)、L−Leu−ドキソルビシン及びβ−Ala−Leu−Ala−Leu−ドキソルビシンの細胞毒性を示す。
図30は前記アントラサイクリン誘導体の存在において72時間培養されたMRC5線維芽細胞に対するドキソルビシンの(DOX)、L−Leu−ドキソルビシン及びβ−Ala−Leu−Ala−Leu−ドキソルビシンの細胞毒性を示す。
図31は、T−21の日に無感覚状態のマウスに移植したMCF7/6ヒト乳腫瘍の平均腫瘍質量において、ドキソルビシン(DOX)及びβ−Ala−Leu−Ala−Leu−ドキソルビシン(スーパーDOX)の投与に応じて現われた変化を示す。
図32は、ドキソルビシン(DOX)及びβ−Ala−Leu−Ala−Leu−ドキソルビシン(スーパーDOX)の投与によって処置されたマウスの平均体重(g)の変化を示す。
図33は、本発明の化合物が、腫瘍細胞のホモジネート、形質転換細胞のホモジネート及び正常細胞のホモジネートと共に含まれる試験管内における遊離クマリンマーカの発現を示す(細胞タンパク質1mgにつき遊離したクマリンのモル数)。
発明の好ましい実施態様の説明
本発明は、マーカの発現を抑止するか、または正常細胞(N.C.)及びターゲット細胞(T.C.)への治療剤(M)の細胞内進入を阻止するリガンド(W−Z)との結合によって不活性化されているマーカまたは治療剤(M)、特に抗腫瘍及び/または抗炎症治療剤を製造できるという予想外の発見に基づく。
本発明では、前記ターゲット細胞が腫瘍細胞または抗炎症反応に関与する細胞、特にマクロファージ、単球などのようなリウマチ疾患と関連する細胞である。
予想外の所見として、ターゲット細胞(T.C.)はリガンド(W−Z)のアーム(Z)とマーカ(M)または治療剤(M)との間の共有結合を選択的に加水分解することによってマーカ(M)の発現または前記ターゲット細胞(T.C.)への治療剤(M)の進入を可能にするプロテアーゼやペプチダーゼのような酵素を細胞外媒質中に放出する。ターゲット細胞において、治療剤(M)は特定の細胞内活性部位(A.S.)に直接作用するか、または細胞内プロテアーゼの作用下に変性したのち、別の治療剤(M*)となり、ターゲット細胞(T.C.)を殺すか、またはその増殖を阻止する(図1)。
正常細胞は、生体内に前記酵素を殆ど、または全く放出しないから、本発明の化合物は不活性のままであり、正常細胞に進入しない(図2)。
具体的には、(例えば中間的な発光による)マーカの発現は癌細胞の特徴づけを可能にし、これにより癌診断、腫瘍の進行状況検査、腫瘍細胞からの分泌因子の同定などを改善する。
本願に記載されている化合物は、効果的に投与されるのに必要な条件を満たすから、このような作用メカニズムと合致する。
1.治療剤(M):−細胞内活性部位を有し、
−高度の特異作用を有し、
−リガンド(W−Z)との共有結合を可能にする化学基を有し、
−所要の化学基が治療剤(M)の作用にとって重要でなければ、前記共有結合が酵素によって加水分解されたのち、元の完全な状態に回復しなくてもよい。
2.リガンド(W−Z)との結合は、正常細胞だけでなくターゲット細胞へも治療剤(M)が細胞内進入するのを阻止する。
3.本発明の化合物は、血清及び血液中で安定性を維持し、赤血球と共に循環するプロテイナーゼ及びペプチダーゼに対して反応しない。
4.治療剤(M)とリガンド(W−Z)との間に存在する共有結合は、ターゲット細胞から分泌される酵素によって部分的にまたは完全に減成する。
5.マーカまたは治療剤(M)に対するリガンド(W−Z)及びその結合の性質は、ターゲット細胞から分泌される酵素に応じて決定される。
6.化合物は、出発の治療剤よりも生体内での毒性が低い。この毒性低下は、特に骨髄及び粘膜毒性、並びに心臓または神経系毒性のような急性効果に当てはまる。
アントラサイクリン誘導体
本発明の治療剤(M)としては、アントラサイクリン誘導体、特にドキソルビシン、ダウノルビシン、4−エピドキソルビシン、4−デメトキシドキソルビシン(イダルビシン)、4′−テトラヒドロピラニルドキソルビシン(ピラルビシン)、カルミノマイシン、エソルビシン及び4′−ヨードドキソルビシンを使用できる。
この場合、リガンド(W−Z)は、そのカルボキシル官能基を介して治療剤のα−アミノ末端と結合する。
葉酸誘導体
抗腫瘍治療剤として、葉酸誘導体、特にフィッツパトリック等(Fitzpatrick et al.)(アンチ−カンサー ドラッグ デザイン(Anti−cancer Drug Design))10,pp.1−9及びpp.11−24(1995))によって述べられているようなメトトレキサート(MTX)またはその誘導体、例えば、リジン(ε−γ)−MTXまたはリジン(ε−α)−MTX、またはアミノプテリン、特にリジン(ε−γ)−AMPTやリジン(ε−α)−AMPTも利用できる。
この場合、リガンド(W−Z)はそのカルボキシル官能基を介して治療剤のα−アミノ末端と結合する。
ビンカアルカロイド誘導体
本発明のビンカアルカロイド誘導体は、具体的にはビンブラスチン、ビンクリスチン、ビンデシン、及びナベルビンの誘導体である。
A.位置3における結合
1.リジン(ε)−dAcVCRを得るためε−アミノ基にリジンを加えることによって形成されるデアセチルビンクリスチン酸。この場合、リガンド(W−Z)は、そのカルボキシル末端を介してリジン(ε)−dAcVCRのα−アミノ基と結合する。
2.デアセチルビンクリスチン酸は、ビンクリスチンに脂肪族ジアミン(NH 2 −アルキル−NH 2 )を加えることによってNH2−アルキル−dAcVCRを得、リガンド(W−Z)をそのカルボキシル末端を介してリジン(ε)−dAcVCRのα−アミノ基と結合させて形成することもできる。
ビンブラスチン(VBL)及びナベルビン(5′−ノルアンヒドロ−ビンブラスチン)誘導体は、ビンクリスチンに関連して上述したのと同じ態様でリガンド(W−Z)を結合することができる。
B.位置4における結合
1.V4−ヘミアスパテート−ビンクリスチンは、ビンクリスチン(V4)の位置4におけるヒドロキシル基にアスパラギン酸をそのγ−カルボキシル基を介して結合させることによってビンクリスチンから形成される。この場合、リガンド(W−Z)は、そのカルボキシル末端を介してV4−ヘミアスパテート−ビンクリスチンのα−アミノ基と結合する。
2.V4−リシルビンクリスチンは、そのα−カルボキシル基を介してリジンをビンクリスチン(V4)の位置4におけるヒドロキシル基と結合させることによってビンクリスチンから形成される。この場合、リガンド(W−Z)は、そのカルボキシル末端を介してV4−リシルビンクリスチンのα−アミノ基と結合する。
3.V4−リシルビンクリスチンは、リジンをそのα−カルボキシル基を介してビンクリスチン(V4)の位置4におけるヒドロキシル基と結合させることによってビンクリスチンから形成される。この場合、リガンド(W−Z)は、そのカルボキシル末端を介してV4−リシルビンクリスチンのリジンのε−アミノ基と結合する。リガンド(W−Z)をV4−リシルビンクリスチンのα−及びε−アミノ基の双方と結合させてもよい。
4.V4−β−アラニルビンクリスチンは、β−アラニルをそのカルボキシル基を介してビンクリスチン(V4)の位置4におけるヒドロキシル基と結合させることによってビンクリスチンから形成される。この場合、リガンド(W−Z)は、そのカルボキシル末端を介してV4−β−アラニルビンクリスチンのβ−アラニルのアミノ基と結合する。
ビンクリスチン、ビンデシン及びナベルビン誘導体はビンクリスチンに関して上述したのと同じ態様でリガンド(W−Z)と結合させることができる。
カリケアマイシン誘導体
カリケアマイシンから誘導されるN−アセチルジメチルヒドラジドは、チオールヒドラジドをカリケアマイシンと反応させることによって得られる。
この場合、リガンド(W−Z)はそのカルボキシル末端を介してカリケアマイシンから誘導されたN−アセチルジメチルヒドラジドと結合する。
ミトキサントロン誘導体
β−アラニルのカルボキシル官能基をミトキサントロンのヒドロキシル側鎖と結合することによって、ミトキサントロンから誘導されたβ−アラニルを製造する。
この場合、1置換または2置換が可能である。リガンド(W−Z)は、そのカルボキシル末端を介してβ−アラニルミトキサントロンのアミノ基と結合する。
シトシンアラビノシド(ARA−C)誘導体
リガンド(W−Z)は、そのカルボキシル末端を介してシトシンアラビノシドのアミノ基と結合する。
アデノシンアラビノシド(Ara−A)誘導体
リガンド(W−Z)は、そのカルボキシル末端を介してアデノシンアラビノシドのアミノ基と結合する。
リン酸フルダラビン誘導体
リガンド(W−Z)は、そのカルボキシル末端を介してリン酸フルダラビンのアミノ基と結合する。
メルファラン誘導体
リガンド(W−Z)は、そのカルボキシル末端を介してメルファランのアミノ基と結合する。
ブレオマイシン誘導体
リガンド(W−Z)は、そのカルボキシル末端を介してブレオマイシン、ペプロマイシンまたはリブロマイシンのアミノ基と結合する。
ミトマイシン誘導体
リガンド(W−Z)は、そのカルボキシル末端を介してミトマイシンの位置7におけるアミノ基と結合する。
L−カナバニン誘導体
リガンド(W−Z)は、そのカルボキシル末端を介してL−カナバニンのα−アミノ基と結合する。
タキソイド誘導体
タキソールのβ−アラニル誘導体は、β−アラニルのカルボキシル官能基をタキソールの位置7におけるヒドロキシル側鎖と結合させることによって製造される。
タキソールのヒドロキシル基は、反応性であるがタキソールの抗腫瘍作用にとって不可欠ではない(ニカラウ ケイ.シー.等,ケミストリ アンド バイオロジー オブ タキソール(Nicalaou K.C.et al.,Chemistry and Biology of Taxol),Angew.Chem.Int.Ed.Engl.(1994),33,pp.15−44)。
リガンド(W−Z)は、そのカルボキシル末端を介してβ−アラニルタキソールのアミノ基と結合する。
リガンド(W−Z)は誘導体β−アラニルタキソテーレとも結合できる。
カンプトテシン誘導体
リガンド(W−Z)は、そのカルボキシル末端を介して9−アミノカンプトテシンまたは7−アミノメチルカンプトテシンのα−アミノ基と結合する。
本発明を実施例に従って以下に詳しく説明するが、これらの実施例は本発明の範囲を制限するものではない。
例1
N−L−ロイシルダウノルビシン(Leu−DNR)の合成
FMOC(フルオレニルメトキシカルボニル,fluorenylmethoxycarbonyl)基によってアミン官能基を保護されているL−ロイシンと塩基の形のダウノルビシン(DNR)とを反応させてカルボキシル基をIBCF(クロロホルム酸イソブチル,isobutylchloroformate)で活性化し、次いでアミン基に対する保護を除くことによってL−ロイシルダウノルビシンを合成する。
塩基型のDNRは、500μlのDMF(ジメチルホルマミド)に200mgのダウノルビシンヒドロクロリド(R.Bellon)を溶解させ、これに1.2当量のN−メチルモルホリンを添加することによって調製する。
1.2当量のFMOC−N−ロイシン(NOVA BIOCHEM)を500μlのDMF(ジメチルホルマミド)に溶解させ、マイナス20℃において1.2当量のN−メチルモルホリン及び1.2当量のIBCFを添加する。15分後、この溶液をDNR塩基の溶液に添加し、この混合物を暗所で16時間撹拌する。
150mlのエーテル/石油エーテルの1:1混合物(40−60℃)中にFMOC−N−L−ロイシルダウノルビシンを沈殿させ、次いでNo.4グラスフィルターで濾過する。クロロホルムを溶離液としてシリカ(70−230メッシュのシリカ、E.MERCK社のSi−60)をカラムとするクロマトグラフィで生成物を精製する。残留DNRを10%メタノールで溶離する。FMOC−N−L−ロイシル−DNRを含有する画分を回転蒸発器で濃縮し、生成物を500μlのDMF中に取り出す。マイナス20℃で5当量のジエチルアミン(LABSCAN社製)を添加することによって、保護基を分離する。
1時間後、エーテル/石油エーテルの1:1混合物(40−60℃)でN−L−ロイシル−DNRを沈殿させ、No.4グラスフィルターで濾過する。生成物をクロロホルム/メタノールの4:1容積比混合物中に取り出し、1当量のHClでジエチルアミンを中和する。
次いで、シリカ(70−230メッシュのシリカ、E.MERCK社のSi−60)をカラムとするクロマトグラフィでクロロホルムを溶離液とし、さらに15%のメタノールを含有するクロロホルムを溶離液として生成物を精製する。N−L−ロイシル−DNRを含有する画分を回転蒸発器で濃縮し、生成物を蒸留水中に取り出し、1NのHClでpHを7に調節することによってヒドロクロリドを形成する。次いで生成物を変成シリカゲル(WATERS社のSeppak C18)で濾過し、メタノールでN−L−ロイシルダウノルビシンヒドロクロリドを溶離して、回転蒸発器で濃縮する。平均的な収率は、70〜80%である。
生成物の特性
メリフィールド法による固相合成でβ−L−アラニル−L−ロイシル−L−アラニンを製造する(ザ ケミストリー オブ ポリペプチド,(ピー.ジー.カツオヤニス Ed.),プレナム プレス,ニューヨーク,pp.336−36(1973),(The Chemistry of Polypeptides,(P.G.Katsoyannis Ed.),Plenum Press,New York,pp.336−361(1973)))。
フルオロクロム誘導体
リガンド(W−Z)は、カルボキシル末端を介してローダミン123のアミノ基と結合している。
β−Ala−Leu−Ala−Leu−DNRの合成
固相合成によって得られたFMOC−β−L−アラニル−L−ロイシル−L−アラニンをN−L−ロイシルダウノルビシンにグラフトすることによって、β−Ala−Leu−Ala−Leu−DNRを合成する。
DMFに溶解させたFMOC−β−L−アラニル−L−ロイシル−L−アラニン(1.5当量)に1.5当量(eq.)のN−メチルモルホリン及び1.5当量のクロロホルム酸イソブチルを加える。マイナス20℃に10分間放置したのち、1.5当量のHOBTを加える。さらに5分後、DMFに溶解させた1当量のN−L−ロイシルダウノルビシン塩基を添加する。反応後、FMOC−β−Ala−Leu−Ala−Leu−DNRをエーテルで沈殿させ、ジエチルアミンで保護基を分離する。シリカをカラムとするクロマトグラフィでβ−Ala−Leu−Ala−Leu−DNRを精製し、1N HClを添加してヒドロクロリドを形成する。
β−Ala−Leu−Ala−Leu−DOXの合成
β−Ala−Leu−Ala−Leu−DNRの合成に関して上述したのと同様に、固相合成で得たβ−L−アラニル−L−ロイシル−L−アラニンをN−L−ロイシルドキソルビシンにグラフトすることによってβ−Ala−Leu−Ala−Leu−DOXを合成する。
この合成においては、β−Ala−Leu−Ala−Leu−DOXを形成する場合の本発明化合物の収量をさらに高めるため結合剤(クロロホルム酸イソブチル)の使用を省いてもよい。
例2
MCF7/6乳癌細胞から得たならし培地中におけるβ−Ala−Leu−Ala−Leu−DNRの分解
MCF7/6細胞から得たならし培地中に2時間にわたり37℃でβ−Ala−Leu−Ala−Leu−DNRを培養し、20〜40倍に濃縮し、この化合物を消化生成物と共にpH9で抽出し、HPLCで分析した(図3)。
例3
MCF7/6乳癌細胞から得たならし培地中におけるβ−Ala−Leu−Ala−Leu−DOXの分解
MCF7/6細胞から得たならし培地中で2時間、β−Ala−Leu−Ala−Leu−DOXを37℃で培養し、20倍に濃縮すると、代謝産物としてL−Leu−DOX及びDOXだけが得られる。
例4
ヒト血液中におけるβ−Ala−Leu−Ala−Leu−DNR及びβ−Ala−Leu−Ala−Leu−DOXの分解
β−Ala−Leu−Ala−Leu−DNRと同様に、β−Ala−Leu−Ala−Leu−DOXもヒト血液中に37℃で培養される場合、安定である。即ち、2時間後に加水分解でL−Leu−DOXとなるのはプロドラッグの2%以下であり、これに反して、合成された他の誘導体はヒト血液の存在において急速に加水分解する(表1下方)。
MCF7/ADR乳癌細胞から得たならし培地中におけるβ−Ala−Leu−Ala−Leu−DNRの分解
β−Ala−Leu−Ala−Leu−DNRをアントラサイクリン耐性MCF7細胞(MCF7/ADRライン)から得たならし培地中で1時間にわたり37℃で培養すると、L−Leu−DNRだけが代謝産物として検出される(図4)。
アントラサイクリン感性MCF7/6細胞も、アントラサイクリン耐性MCF7/6細胞も、本発明化合物を加水分解できるプロテアーゼを分泌する。
例6
HepG2肝癌細胞から得たならし培地中におけるβ−Ala−Leu−Ala−Leu−DNRの分解
HepG2肝癌細胞から得たならし培地中で2時間にわたり、β−Ala−Leu−Ala−Leu−DNRを37℃で培養すると、主な代謝産物としてL−Leu−DNR(全蛍光の27%)とL−Ala−L−Leu−DNR(8%)とが検出される。ならし培地を構成する細胞数もならし培地の濃度も異なるから、MCF7乳癌細胞とHepG2肝癌細胞との加水分解の%を比較することはできない。しかし、定性分析の結果に照らして、ペプチダーゼはMCF7細胞に特異なものではない。
例7
HT29結腸癌細胞から得たならし培地中におけるβ−Ala−Leu−Ala−Leu−DNRの分解
HT29結腸癌細胞から得た40倍濃縮ならし培地中でβ−Ala−Leu−Ala−Leu−DNRを2時間にわたり37℃で培養すると、主要な代謝物としてL−Leu−DNR(全蛍光の82%)及びL−Leu−L−Ala−L−Leu−DNR及び/またはDNR(3%)が検出される(図5)。
例8
MCF7/6腫瘍細胞中でのDNR,L−Leu−DNR及びβ−Ala−L−Leu−L−Ala−L−Leu−DNRの蓄積
10μg当量DNR/mlの濃度のβ−Ala−Leu−Ala−Leu−DNRの存在においてMCF7/6ヒト乳癌細胞を培養した。種々の経過時間におけるプロドラッグ及びその蛍光性代謝産物の蓄積を、実験室で開発された方法に従って、塩基性pHの生成物として抽出したのち、HPLCによって測定した。細胞タンパクのμg/mgで表わされる蓄積量をDNR,DOX,L−Leu−DNR及びL−Leu−DOXの蓄積量と比較した。実験室で調製された基準生成物の保持時間を測定することにより代謝産物を識別した。
DNRは、MCF7/6細胞中でほぼ変化しない形で急速に蓄積し、6時間の培養後、最大蓄積量(±15μg/mgの細胞タンパク)に達した。DNR蓄積量は、24時間後まで減少する。主要な細胞内代謝産物は、位置C13におけるDNRのケトン官能基が細胞内レダクターゼによって還元されたことに起因するダウノルビシノールである(図6)。
L−Leu−DNRもMCF7/6細胞によって極めて迅速に蓄積されるが、そのレベルは比較的低く、24時間の培養後に細胞タンパクで±14μg/mgの限界値に達する。DNRは経時的に細胞内に形成され、24時間にわたる培養後には細胞内の全蛍光の14%に達する(図7)。
MCF7/6細胞の存在において24時間培養されたβ−Ala−Leu−Ala−Leu−DNRは、変化しない形でDNRよりもL−Leu−DNRよりもはるかに少ない量が蓄積し、蓄積レベルは細胞タンパクの±1μg/mgに過ぎない(図8)。細胞外で形成されるL−Leu−DNR及びDNRは、主として24時間の培養後に細胞内で検出されるものである。細胞内に検出されるDNRは、24時間の培養後に全蛍光のほぼ40%に相当する。
例9.
MRC5正常細胞におけるDNR,L−Leu−DNR及び−Ala−Leu−Ala−Leu−DNRの蓄積
10μg当量DNR/mlの濃度のβ−Ala−Leu−Ala−Leu−DNRの存在において、MRC5線維芽細胞を培養した。プロドラッグ及びその蛍光性代謝産物の蓄積量を、実験室で開発された方法に従って塩基性pHの生成物を抽出したのち、HPLCによって種々の経過時間で測定した。細胞タンパクのμg/mgで表わされる蓄積量をDNR及びL−Leu−DNRの蓄積量と比較した。実験室で合成された基準生成物の保持時間を測定することによって、代謝産物を同定した。
DNRは、主としてMRC5細胞中に変化しない形で蓄積し、蓄積量は6時間後に細胞タンパク±76μg/mgの最大値に達し、主要な代謝産物はダウノルビシノールである(図9)。
L−Leu−DNRは、MRC5細胞によっても極めて迅速に蓄積されるが、そのレベルは比較的低く、蓄積量は24時間の培養後に±40μg/mgの細胞タンパクに相当する最大値に達する。主要な代謝産物は、DNR及びL−Leu−DNR−OLである(図−0)。
MRC5細胞の存在において24時間培養されたβ−Ala−Leu−Ala−Leu−DNRは、DNRよりもL−Leu−DNRよりもはるかに蓄積量が少なく、そのレベルは細胞タンパク±3.3μg/mgに過ぎない(図11)。細胞外で形成されたL−Leu−DNRは、主として細胞内で検出される物質である。その蓄積量は、培養時間が24時間に達するまで線形的に増大する。
これらの結果は、MCF7/6細胞で得られた結果を裏づける。即ち、化合物β−Ala−Leu−Ala−Leu−DNRはほとんど細胞内に進入せず(6時間後はDNR 1/300)、細胞外形成L−Leu−DNRは主としてMRC5細胞内に蓄積する(表3)。
Leu−DNR及びDNRの存在において細胞を培養すると、細胞内DNRレベルの比はそれぞれ0.37及び0.19に過ぎない。
例10
MCF7/6腫瘍細胞及びMRC5正常細胞に対するβ−Ala−Leu−Ala−Leu−DNRの細胞毒性
DNRプロドラック、Leu−DNR及びDNRの細胞毒性を種々の化合物の存在において72時間増殖させたMCF7/6及びMRC5細胞において比較した。
β−Ala−Leu−Ala−Leu−DNR,L−Leu−DNR及びDNRの細胞毒性を、濃度の異なる種々の化合物
の存在において培養され、96−ウエルディシュで増殖するMCF7/6細胞で測定した。72時間後、アントラサイクリンが存在しない状態で細胞を48時間培養し、ブラッドフォード法によって細胞タンパクを測定することによって細胞毒性を測定する。700μg/mlから0.0035μg/mlまで9つの濃度を使用し、各測定値が6個の値の平均及び標準偏差を表わす。実験点は、細胞の半分が生存する投与量(IC50)に対応する変曲点の計算を可能にするS字曲線を画くように調整する。
図12は、DNRのIC50が0.090±0.004μg/mlであることを示す。L−Leu−DNRの存在において72時間培養された細胞では、IC50は1.30±0.56μg/mlである。β−Ala−Leu−Ala−Leu−DNRの場合、IC50は22.00±7.31μg/mlである。
従って、Leu−DNR及びβ−Ala−Leu−Ala−Leu−DNRは,アントラサイクリンの存在において72時間増殖したMCF7/6ヒト乳癌細胞に対するDNRに比較して、細胞毒性はそれぞれ1/14及び1/244である。
β−Ala−Leu−Ala−Leu−DNR,L−Leu−DNR及びDNRの細胞毒性を、次第に濃度が高くなる種々の化合物の存在において培養され、96−ウエルディシュで増殖したMRC5細胞を対象に測定した。72時間後、アントラサイクリンが存在しない状態で48時間培養し、プラッドフォード法で細胞タンパクを測定することによって細胞毒性を測定する。700μg/mlから0.0035μg/mlまで9つの濃度を採用し、6つの値の平均及び標準偏差をそれぞれの測定値とする。細胞の半数が生き残る投与量(IC50)に対応する変曲点の算出を可能にするS字曲線を画くように実験点を調整する。
図13は、DNRのIC50が0.010±0.006μg/mlであることを示す。L−Leu−DNRの存在において72時間培養された細胞に対しては、IC50は1.78±0.23μg/mlである。β−Ala−Leu−Ala−Leu−DNRの場合、IC50は23.14±4.81μg/mlである。
従って、アントラサイクリンの存在において72時間増殖したMRC5線維芽細胞に対するLeu−DNR及びβ−Ala−Leu−Ala−Leu−DNRの細胞毒性は、それぞれDNRの1/172及び1/2230である。
腫瘍細胞に対しても線維芽細胞に対しても、DNR先駆物質の毒性は親化合物よりもはるかに低い。試験管中で観察されるこの毒性低下が生体内でも観察されるかどうかを確認しなければならない。
例11
生体内急性毒性 β−Ala−Leu−Ala−Leu−DNRの毒性をマウスを対象にダウノルビシンの毒性と比較した。医薬品の急性毒性を生体内で検査するには、50%致死量(動物の50%に対する致死量、LD50)が重要な要素となる。生物学的定数を表わすものではないが、注入薬の急性毒性に関する情報を得る手がかりとなる。LD50は、試験薬の量を増やしながら連続5日間にわたり1日1回ずつの注射で腹腔内(i.p.)投与すると共に1回だけの注射で静脈内(i.v.)投与する簡単な試験である。経時的に動物の死亡率をモニターする。通常は、30日間に及ぶ観察期間の終りに、投与量の対数に応じて(プロビット目盛りで)%死亡率を線形回帰することによって得られる(オー.ケー.チャン アンド エイ.ダブリュ ヘイ,「プリンシプルズ アンド メソッズ フォア アキュート トキシシティ アンド アイ イリタンシィ」,イン プリンシプルズ アンド メソッズ オブ トキソコロジィ,セコンドエディション.Ed.バイ エイ.ダブリュ.ヘイズ,ラブン プレス,ニューヨーク,ユエスヱイ(1989),pp.169−220;ディ.デプレッツ−デ カンペニール アンド エイ.トラウト.「DNA−アントラサイクリン コンプレックシズ.I.トキシシティ イン マイス アンド ケモテラプリィック アクティビティ アゲインスト L1210 ルーカミア オブ ダウノルビシン−DNA アンド アドリアマイシン−DNA」,Enr.ジェイ.カンサー,16(1980),pp.981−986;エイチ.イー.スキッパー,エル.エイチ.シュミット,ヱイ.ゴールディン アンド ジェイ.エム.ベンディティ.「ア マニュアル オン コンティティブ ドラッグ エバリュエイション イン エクスペリメンタル テュモア システム」,カンサー Chemather,Rep,17(1962),pp.1−178,(O.K.Chanand A.W.Haye,“Principlecs and methods for acute toxicityand eye irritancy”,in Principles and methods of toxicology,Second edition.Ed.by A.W.Hayes,Raven Press,New York,USA(1989),pp.169−220;D.Deprez−De Campeneere and A.Trouet.“DNA−Anthracycline Complexes.I.Toxicity in mice and chemotherapeutic activity against L1210 Leukaemia of Daunorubicin−DNA and Adriamycin−DNA”,Eur.J.Cancer,16(1980),pp.981−986;H.E Skipper,L.H.Schmidt,A.Goldin and J.M.Venditti.“A manual on quantitative drug evaluation in experimental tumor system”,Cancer Chemother.Rep.17(1962),pp.1−178))。
LD50に達しない場合、マウスの毎回の体重変化も投与薬の急性毒性に関する情報を与えてくれる。
1.材料及び方法
単一種のマウスに対してi.v.投与及びi.p.投与することによって、これら2種類の薬剤の急性毒性を観察した。(ベルギーのIFFA−CREDOから供給された生後約5週間、平均体重14.6gの非血縁SPF−Hanである)雌のNMRIマウスを1週間隔離した。注射の前日、マウスと注射による投与量ごとに5匹(β−Ala−Leu−Ala−Leu−DNR)と7匹(DNR)のグループに分け、あらためて体重を記録した(平均約22g)。
注射は朝規則的に行われ(静脈内投与では尾の静脈に1回、腹腔内投与では5日間連続で腹腔に注射)、1.0ml注射筒と無菌30G(i.v.)及び27G(i.p.)注射針を使用した。動物の取り扱いは必らず手袋を着用して行われ、動物の保守が毎週規則的に行われた。
β−Ala−Leu−Ala−Leu−DNRは、30,60,及び120mg/kgの投与量でi.v.注射し、10,15,20,25,30,45及び60mg/kgの総投与量でi.p.注射した。別の2グループに対しては、1回では30mg/kg、2回では60mg/kgの総投与量でβ−Ala−Leu−Ala−Leu−DNRを再注射し、2回に分けて注射する場合には連続して30mg/kgずつ再注射した。
DNRは、10,15,20,25,30及び35mg/kgの投与量でi.v.注射し、2.0,2.5,3.0及び3.5mg/kgの投与量でi.p.注射した。
注射量がマウスの体重に対して0.1ml/10gとなるように、生理的食塩水でβ−Ala−Leu−Ala−Leu−DNR及びDNRの溶液を調製した。溶液の濃度を分光測定によって確認した。
注射の日(DO)からマウスを臨床的にモニターし、死亡したマウスを毎日記録した。マウスの体重をほぼ毎日測定した。遅れて現われる毒性の徴候を容易にスポット分析できるように、観察期間を1カ月以上に延ばした。実験の終了時に生き残ったマウスを、動物実験に関する規定(ディ.ビー.マクレガ,「エシックス イン エクスペリメンツ オン アニマルズ」,イン エクスペリメンツ イン トキシコロジィ,ファーストエディション.Ed.バイ ディ.アンダーソン アンド ディ.エム.コニング.ザ ロイヤル ササイアティ オブ ケミストリ アンド ザ ユニバシティズ プレス.ベルファスト,アイルランド(1988),pp.512−522(D.B.McGregor,“Ethics in experiments on animals”,in Experiments in toxicology,First edition.Ed.by D.Anderson and D.M.Conning.The Royal Society of Chemistry and The Universities Press.Belfast,Ireland(1988),pp.512−522))に従って犠牲にした。
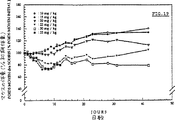
2.結果腹腔内投与の場合の毒性
図14に示すように、DNRの場合、濃度が高くなるに従って強い毒性が観察され、毒性はマウスの死亡率に反映し、投与量が3.5mg/kgの場合には7日目(7匹のうち1匹のマウスが死亡)から、投与量が3.0mg/kgの場合には9日目(7匹のうち2匹のマウスが死亡)から、投与量が2.5及び2.0mg/kgの場合には11日目(7匹のうち2匹及び1匹のマウスがそれぞれ死亡)からマウスの死亡となって毒性が顕在化する。
12日目(投与量3.5mg/kgの場合)、19日目(投与量3.0mg/kgの場合)、29日目(投与量2.0mg/kgの場合)には生き残るマウスは皆無である。2.5mg/kgを注射されたマウスのうち1匹だけが43日目まで生き残った。この実験結果は、これまでに報告された結果を裏づけた(D.Deprez−De Campeneere and A.Trouet.“DNA−Anthracycline Complexes.I.Toxicity in mice and chemotherapeutic activity against L1210 Leukaemia of Daunorubicin−DNA and Adriamycin−DNA,Eur.J.Cancer,16(1980),pp.981−986)。
図15から明らかなように、投与量に関係なく、マウスには初期体重の30%に達する体重減が現われる。この体重減は、30日目に初期体重を回復した2.5mg/kg投与の1匹を除いて回復不能である。
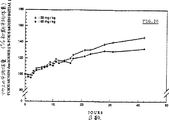
5日間連続して総量で10〜45mg/kgのβ−Ala−Leu−Ala−Leu−DNRを腹腔内投与した場合は、死亡率は0であり、投与量60mg/kgの場合、2匹のマウスが22日目に死亡し、35日目に3匹目のマウスが死亡した(図16)。
総投与量が10〜45mg/kgの場合、体重減は観察されず、むしろ40日後で±40%の体重増が観察された(図17)。体重曲線を分析した結果、著しい変化は認められない。他方、投与量が45mg/kgの場合、マウスの平均体重は最初の7日間は安定であり、以後30日間で20%だけ体重が増える。総投与量60mg/kgの場合、マウスの平均体重は15日後に±15%低下する。生き残った2匹のマウスは36日目に初期体重を回復する。
これらのデータから明らかなように、β−Ala−Leu−Ala−Leu−DNRの投与量60mg/kgは、LD50に近く、従って、5日間連続して腹腔内投与したあとの急性毒性に関してβ−Ala−Leu−Ala−Leu−DNRは、DNRの少なくとも1/30である。
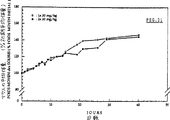
静脈内投与に伴なう毒性
図18に示すように、雌NMRIマウスにDNRを1回だけ静脈投与する場合、投与量が低ければ(10,15及び20mg/kg)死亡率は0である。25mg/kgを投与した場合、6匹のマウスが12,17,20,28,34及び42日目にそれぞれ死亡する。DNRの濃度がさらに高くなると、(30mg/kg投与の場合には7匹のうちの1匹が)7日目に、(35mg/kg投与の場合には7匹のうち2匹が)6日目にそれぞれ死亡する。この実験結果はすでに報告されている実験結果(D.Deprez−De Campaneere and A.Trouet.“DNA−Anthracycline Complexes.I.Toxicity in mice and chemotherapeutic activity against L1210 Leukaemia of Daunorubicin−DNA and Adriamycin−DNA,Eur.J.Cancer,16(1980),pp.981−986)と一致する。
図19に示す実験結果から明らかなように、平均体重減は7日目に最大となり、この体重減は投与量と共に増大する。30及び35mg/kgの投与で生き残ったマウスは、DNRを静脈内注射されてから30日経過しても初期体重を回復しない。
30mg/kgのβ−Ala−Leu−Ala−Leu−DNRを静脈内投与すると、死亡率は0であり、マウスの平均体重は40日後に±40%増える(図20)。実験終了時のマウスの平均体重は、β−Ala−Leu−Ala−Leu−DNRを同じ量だけ腹腔内投与した場合に観察される平均体重とほぼ同じである。しかし、投与量が60mg/kgの場合には、注射から5分後に2匹のマウスが死亡する。この死亡は、注射後の血液量増大があまりに急激に起こるためと考えられる。2日目までやや体重減を示したのち、生き残った3匹のマウスは初期体重を回復し、さらに体重増を示す(40日後に±30%)。投与量120mg/kgの場合、投与から7分以内に5匹のマウスが死亡し、臨床的な毒性徴候を示す。
総投与量60mg/kgのβ−Ala−Leu−Ala−Leu−DNRを2回の静脈内30mg/kg注射に分けて2日連続して投与した場合、死亡率は0である。マウスの平均体重変化は、β−Ala−Leu−Ala−Leu−DNRを30mg/kgだけ1回投与したマウスと2回投与したマウスとの間に相違はない(図21)。
3.結論
得られた実験結果に照らして、静脈内投与でも腹腔内投与でも、致死率という点でβ−Ala−Leu−Ala−Leu−DNRの急性毒性は、DNRの急性毒性よりもはるかに低い。これらの実験結果は、誘導体β−Ala−Leu−Ala−Leu−DNRの毒性の低下を立証する。
上述した実験と同じ実験をドキソルビシン誘導体で実施した。
例12
MCF7/6腫瘍細胞及びMRC5非腫瘍細胞中でのDOX,L−Leu−DOX及び−Ala−Leu−Ala−Leu−DOXの蓄積
濃度10μg当量DOX/mlのβ−Ala−Leu−Ala−Leu−DOXの存在においてMCF7/6ヒト乳癌細胞及びMRC5ヒト線維芽細胞系を培養した。実験室で開発された方法で塩基性pHの生成物を抽出したのち、HPLCによりプロドラッグ及びその蛍光性代謝物質の蓄積量を種々の経過時間で測定した。細胞タンパクのμg/mgで表わされる蓄積量をDOX及びL−Leu−DOXの蓄積量と比較した。代謝物質は、実験室で合成された基準物質の保持時間を測定することによって同定した。
DOXは、ほとんど変化しない形で主としてMCF7/6細胞中に蓄積され、6時間後には細胞内レベルが細胞タンパク6.9μg/mgに達する(図23)。
10μg当量DOX/mlの濃度のアントラサイクリンの存在において6時間培養されたMRC5線維芽細胞において、DOXはほとんど変化しない形で蓄積し、6時間後に蓄積量は±11.2μg/mg細胞タンパクのレベルに達する(図26)。
L−Leu−DOXは、比較的低いレベルで蓄積し、蓄積量は1.4μg/mg細胞タンパクのレベルに達する。主要な代謝産物は、DOX(0.3μg/mg細胞タンパク)である(図27)。
6時間後におけるβ−Ala−Leu−Ala−Leu−DOXの蓄積量は、DOXの蓄積量の1/112である。β−Ala−Leu−Ala−Leu−DOXの存在において細胞を培養したDOX蓄積レベルは、1/1100に低下する(図28)。
これらの結果に照らして、β−Ala−Leu−Ala−Leu−DOXは、ほとんど細胞内に進入せず、細胞内に進入する前に外部媒質中であらかじめ加水分解されてLeu−DOXの形となる必要があり、これが細胞内に進入してDOXを生成させることになる(図22)。
これらの結果は、活性治療剤の細胞内レベルは、DOXの場合には腫瘍細胞中のレベルに比較して正常細胞中では2倍になることも示している。他方、β−Ala−Leu−Ala−Leu−DOXの場合、DOXの細胞内レベルはMRC5非腫瘍細胞と比較してMCF7腫瘍細胞では22倍となる。
例13
MCF7/6腫瘍細胞及びMRC5非腫瘍細胞に対するインビトロ細胞毒性
濃度の異なる種々の化合物の存在において培養し、96−ウエルディッシュで増殖したMCF7/6及びMRC5細胞に対するβ−Ala−Leu−Ala−Leu−DOX、L−Leu−DOX及びDOXの細胞毒性を測定した。72時間後、アントラサイクリンが存在しない状態で48時間にわたり細胞を培養し、ブラッドフォード法で細胞タンパクを測定することによって細胞毒性を測定した。700μg/mlから0.0035μg/mlまで9つの濃度を採用し、それぞれの測定値を6個の値の平均及び標準偏差で表わした。細胞の半数が生き残る投与量(IC50)に対応する変曲点の算出を可能にするS字曲線を画くように実験点を調整した。
例12の表は、MCF7/6ヒト乳癌細胞に対するDOX、L−Leu−DOX及びβ−Ala−Leu−Ala−Leu−DOXのそれぞれのIC50値、即ち、0.0025,0.020及び3.0μg/mlを記録したものである。MRC5ヒト線維芽細胞系に対しては、この値はそれぞれ0.018,0.30及び120μg/mlである。
これらの結果から明らかなように、アントラサイクリンの存在において72時間増殖した線維芽MCF7/6ヒト乳癌細胞に対するLeu−DOX及びβ−Ala−Leu−Ala−Leu−DOXの細胞毒性は、DOXの細胞毒性のそれぞれ1/8及び1/1000である。アントラサイクリンの存在において72時間増殖したMRC5細胞に対しては、Leu−DOX及びβ−Ala−Leu−Ala−Leu−DOXの細胞毒性はDOXのそれぞれ1/17及び1/6,700である。
MCF7腫瘍細胞に対してもMRC5非腫瘍細胞に対しても、β−Ala−Leu−Ala−Leu−DOXの毒性は40倍になる。この毒性は、β−Ala−Leu−Ala−Leu−DOXの存在において培養されたMCF7細胞において観察されたDOXの高い細胞内レベルと同じレベルである(図29及び30)。
本発明の化合物の特徴は、腫瘍細胞を静脈内注射したあとに得られる“白血病”型のモデルに対してよりも、(例えば腫瘍細胞を皮下注射して得られる)固相腫瘍型モデルに対して顕著に作用することである。
腫瘍細胞を皮下注射すると、注射部位に固相腫瘍が形成され、この細胞から分泌される加水分解酵素の局所濃度は高い状態を維持する。腫瘍細胞を静脈内注射すると、この細胞から分泌される加水分解酵素はたちまち血流中に希釈される。
例14
インビボ急性毒性
MCF7/6ヒト乳癌腫瘍を移植された無感覚状態のマウスにβ−Ala−Leu−Ala−Leu−DOXを投与すると、マウスの平均体重に著しく影響を及ぼすことなく癌性腫瘍の進行(図31)が抑制される(図32)。
これらの実験は、例11で述べたプロトコルに従って行われた。
発明者らは、ヒト乳癌細胞の細胞外媒質中へ分泌されてβ−Ala−Leu−Ala−Leu−DOXを加水分解することができるプロテアーゼをも検出した。このプロテアーゼは、すでに述べたどのプロテアーゼとも一致しないことが確認された。
即ち、仮に“COUM”と呼称するこの酵素は、例えばEDTAのような金属キレーターによって抑制することができ、作用するにはコバルトイオンを必要とする金属プロテアーゼである。その最適pHは7.5〜8.0であり、カテプシンである可能性は除外される。
高性能クロマトグラフィ及び硫酸ラウリルの存在における電気泳動により、70kDを超えるいくつかの分子量帯が観察される。
図33は、腫瘍細胞(肺、乳房、卵巣などの癌)、変換細胞(不朽化された、ただし非癌性の正常細胞)、及び正常細胞(線維芽及び筋肉細胞)のホモジネート中に本発明の化合物を含む試験管内でノクマリンの発現測定結果を示す。
従って、本発明の化合物を利用すれば、マーカの発現によって癌性腫瘍を診断することができるから、癌の診断、腫瘍進行の検査、腫瘍細胞から分泌される因子の同定などの精度を高めることができる。
本発明の化合物は、当業者に公知の種々の試薬から成る診断及び/または検査装置に組み込むことができる。
本発明の診断装置は、患者から組織、細胞または生理的流体サンプルを採取し、遊離マーカ(場合によっては1種類または2種類以上の中間試薬を含む)の発現を可能にする条件下で前記サンプルを本発明の化合物と接触させ、遊離したマーカを検出及び/または定量するステップから成る組織学的または生化学的診断及び/または検査方法に利用することができる。
Claims (11)
- 末端基(W)とそれに結合するアーム(Z)とを含むリガンド(W−Z)、及びマーカと治療剤とからなる群から選択されて前記リガンド(W−Z)と結合する成分(M)を有する化合物(W−Z−M)において、
リガンド(W−Z)のアーム(Z)と成分(M)との間の結合が化合物(W−Z−M)の細胞内進入を妨げ及び/またはマーカ(M)の発現を抑止し、ターゲット細胞から分泌される因子によって前記結合を選択的に開裂することにより、マーカ(M)の発現または前記ターゲット細胞への治療剤(M)の進入を可能にすることができ、
前記結合がペプチド結合であり、リガンド(W−Z)のアーム(Z)が少なくとも2つの随意に置換されるアミノ酸から成り、末端基(W)が、式:
NH2−CH2−CH2−COOH
で表わされるβ−アラニンであり、
前記成分(M)がアントラサイクリン,葉酸,ビンカアルカロイド,カリケアマイシン,ミトザントロン,シトシンアラビノシド(ARA−C),アデノシンアラビノシド(ARA−A),リン酸フルダラビン,メルファラン,ブレオマイシン,ミトマイシン,L−カナバニン,トキソイド,カンプトテシン,フルオロクロム,TOPOTECAN(商品名,9−ジメチルアミノメチル−10−ヒドロキシカンプトテシンヒドロクロリド),ローダミン123,クマリン、7−アミド−4−(トリフルオロメチル)クマリン、パラ−ニトロアニリド、β−ナフチルアミド及び4−メトキシ−β−ナフチルアミドであって、それら各々が随意に置換または非置換アミノ酸と結合しているものからなるグループから選択したことを特徴とする前記化合物(W−Z−M)。 - リガンド(W−Z)のアーム(Z)を次のペプチド配列:L−ロイシル−L−アラニル−L−ロイシル−,L−ロイシル−L−アラニル−,L−アラニル−L−ロイシル−L−フェニルアラニル−,L−アラニル−L−ロイシル−から成るグループから選択したことを特徴とする請求項1に記載の化合物。
- 前記アントラサイクリンが、随意に置換または非置換アミノ酸と結合しているドキソルビシンまたはダウノルビシンである請求項1または請求項2に記載の化合物。
- 成分(M)が次の一般式に対応することを特徴とする請求項3に記載の化合物:
−R1は水素原子またはOH基、
−R2は水素原子または次式のラジカル:
−R3は水素原子または随意に置換されているアルキルラジカル、
−R4は水素原子またはR3と共に3または4個の炭素原子を有するアルキレンラジカル、である。 - R2が次式で表わされるラジカルまたはその同位体の1つであることを特徴とする請求項4に記載の化合物:
- 請求項1項から請求項5までのいずれか1項に記載の化合物のほかに製剤上許容できる補薬または賦形剤をも適宜含む製剤用組成物。
- 癌性腫瘍の治療薬の製造を目的とする請求項6に記載の製剤用組成物の利用。
- 炎症反応治療薬の製造を目的とする請求項6に記載の製剤用組成物の利用。
- リウマチ疾患治療薬の製造を目的とする請求項6に記載の製剤用組成物の利用。
- 前記成分(M)をクマリン、7−アミド−4−(トリフルオロメチル)クマリン、パラ−ニトロアニリド、β−ナフチルアミド及び4−メトキシ−β−ナフチルアミドから成るグループから選択したことを特徴とする請求項1または請求項2に記載の化合物。
- 請求項1または請求項10に記載の化合物を含む診断及び/または検査装置。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BE9400752A BE1008581A3 (fr) | 1994-08-19 | 1994-08-19 | Prodrogues, composition pharmaceutique les comprenant et leur utilisation. |
| BE9400751A BE1008580A3 (fr) | 1994-08-19 | 1994-08-19 | Prodrogues, composition pharmaceutiques les comprenant et leur utilisation. |
| BE9400752 | 1994-08-19 | ||
| BE9400751 | 1994-08-19 | ||
| PCT/BE1995/000076 WO1996005863A1 (fr) | 1994-08-19 | 1995-08-21 | Composes, composition pharmaceutique et dispositif de diagnostic les comprenant et leur utilisation |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007301893A Division JP2008069175A (ja) | 1994-08-19 | 2007-11-21 | 化合物、製剤用組成物及びこれらを含む診断装置とこれらの利用 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH10508291A JPH10508291A (ja) | 1998-08-18 |
| JP4157600B2 true JP4157600B2 (ja) | 2008-10-01 |
Family
ID=25662908
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50766296A Expired - Lifetime JP4157600B2 (ja) | 1994-08-19 | 1995-08-21 | 化合物、製剤用組成物及びこれらを含む診断装置とこれらの利用 |
| JP2007301893A Pending JP2008069175A (ja) | 1994-08-19 | 2007-11-21 | 化合物、製剤用組成物及びこれらを含む診断装置とこれらの利用 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007301893A Pending JP2008069175A (ja) | 1994-08-19 | 2007-11-21 | 化合物、製剤用組成物及びこれらを含む診断装置とこれらの利用 |
Country Status (14)
| Country | Link |
|---|---|
| US (5) | US5962216A (ja) |
| EP (2) | EP0769967B1 (ja) |
| JP (2) | JP4157600B2 (ja) |
| AT (1) | ATE380559T1 (ja) |
| AU (1) | AU694546C (ja) |
| CA (1) | CA2203622C (ja) |
| DE (1) | DE69535665T2 (ja) |
| DK (1) | DK0769967T3 (ja) |
| ES (1) | ES2297832T3 (ja) |
| NO (1) | NO324045B1 (ja) |
| NZ (1) | NZ291368A (ja) |
| PT (1) | PT769967E (ja) |
| SI (1) | SI0769967T1 (ja) |
| WO (1) | WO1996005863A1 (ja) |
Families Citing this family (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6517824B1 (en) * | 1990-05-14 | 2003-02-11 | University Of Medicine & Denistry Of New Jersey | Polymer compositions comprising antifibrotic agents, and methods of treatment, pharmaceutical compositions, and methods of preparation therefor |
| US5866679A (en) * | 1994-06-28 | 1999-02-02 | Merck & Co., Inc. | Peptides |
| US6143864A (en) * | 1994-06-28 | 2000-11-07 | Merck & Co., Inc. | Peptides |
| AU694546C (en) * | 1994-08-19 | 2001-09-06 | La Region Wallonne | Compounds, pharmaceutical composition and diagnostic device comprising same and their use |
| EP0923603A1 (en) * | 1996-07-16 | 1999-06-23 | Novo Nordisk A/S | Oligopeptide transporters |
| US5998362A (en) * | 1996-09-12 | 1999-12-07 | Merck & Co., Inc. | Conjugates useful in the treatment of prostate cancer |
| US5948750A (en) * | 1996-10-30 | 1999-09-07 | Merck & Co., Inc. | Conjugates useful in the treatment of prostate cancer |
| US6127333A (en) * | 1997-07-10 | 2000-10-03 | Merck & Co., Inc. | Conjugates useful in the treatment of prostate cancer |
| US6391305B1 (en) | 1997-09-10 | 2002-05-21 | Merck & Co., Inc. | Conjugates useful in the treatment of prostate cancer |
| US6174858B1 (en) * | 1998-11-17 | 2001-01-16 | Merck & Co., Inc. | Conjugates useful in the treatment of prostate cancer |
| US7425541B2 (en) | 1998-12-11 | 2008-09-16 | Medarex, Inc. | Enzyme-cleavable prodrug compounds |
| PT1144011E (pt) * | 1998-12-11 | 2010-06-16 | Coulter Pharm Inc | Compostos pró-fármacos e processo para a sua preparação |
| CA2385528C (en) | 1999-10-01 | 2013-12-10 | Immunogen, Inc. | Compositions and methods for treating cancer using immunoconjugates and chemotherapeutic agents |
| GB9924759D0 (en) | 1999-10-19 | 1999-12-22 | Merck Sharp & Dohme | Process for preparing peptide intermediates |
| AU781531B2 (en) | 1999-10-27 | 2005-05-26 | Merck & Co., Inc. | Salt form of a conjugate useful in the treatment of prostate cancer |
| AU4583601A (en) * | 2000-03-15 | 2001-09-24 | Du Pont Pharm Co | Peptidase-cleavable, targeted antineoplastic drugs and their therapeutic use |
| US20040014652A1 (en) * | 2000-06-01 | 2004-01-22 | Andre Trouet | Tumor activated prodrug compounds and methods of making and using the same |
| EP1286700A2 (en) * | 2000-06-01 | 2003-03-05 | Universite Catholique De Louvain | Tumor activated prodrug compounds |
| WO2001095945A2 (en) * | 2000-06-14 | 2001-12-20 | Medarex, Inc. | Prodrug compounds cleavable by thimet oligopeptidase |
| AU2001266853B2 (en) * | 2000-06-14 | 2005-02-17 | Medarex, Inc. | Prodrug compounds with an oligopeptide having an isoleucine residue |
| CA2411545A1 (en) * | 2000-06-14 | 2002-01-03 | Medarex, Inc. | Tripeptide prodrug compounds |
| AU2001286727A1 (en) * | 2000-08-24 | 2002-03-04 | Coulter Pharmaceutical, Inc. | Prodrugs activated by plasmin and their use in cancer chemotherapy |
| IL155696A0 (en) | 2000-11-09 | 2003-11-23 | Neopharm Inc | Sn-38 lipid complexes and methods of use |
| JP2006516948A (ja) * | 2000-11-14 | 2006-07-13 | ニュー リバー ファーマシューティカルズ インコーポレイテッド | 硫酸アバカビルを含有する新規な薬剤化合物および同化合物の製造ならびに使用方法 |
| EP1355675A1 (en) * | 2001-01-30 | 2003-10-29 | Universite Catholique De Louvain | Anti-tumor compounds |
| GB0105929D0 (en) * | 2001-03-09 | 2001-04-25 | Btg Int Ltd | Physiologically activated prodrugs |
| WO2003030864A1 (en) | 2001-05-29 | 2003-04-17 | Neopharm, Inc. | Liposomal formulation of irinotecan |
| MXPA03011094A (es) * | 2001-05-31 | 2004-12-06 | Medarex Inc | Citotoxinas, profarmacos, ligadores, y estabilizadores utiles para ello. |
| CA2450316A1 (en) * | 2001-06-11 | 2002-12-19 | Medarex, Inc. | Cd10-activated prodrug compounds |
| US20050069549A1 (en) | 2002-01-14 | 2005-03-31 | William Herman | Targeted ligands |
| GB0310593D0 (en) * | 2003-05-08 | 2003-06-11 | Leuven K U Res & Dev | Peptidic prodrugs |
| FR2858936A1 (fr) * | 2003-08-22 | 2005-02-25 | Diatos | Potentialisation de l'activation de prodrogues de haut poids moleculaire |
| EP3075247B1 (en) * | 2004-02-02 | 2022-10-12 | BioSight Ltd. | Conjugates for cancer therapy and diagnosis |
| WO2005086951A2 (en) * | 2004-03-10 | 2005-09-22 | Threshold Pharmaceuticals, Inc. | Hypoxia-activated anti-cancer agents |
| JP4658507B2 (ja) * | 2004-03-31 | 2011-03-23 | 竹本容器株式会社 | 自開式キャップ機構 |
| RU2402548C2 (ru) | 2004-05-19 | 2010-10-27 | Медарекс, Инк. | Химические линкеры и их конъюгаты |
| EP1747021B1 (en) * | 2004-05-19 | 2015-09-09 | E. R. Squibb & Sons, L.L.C. | Self-immolative linkers and drug conjugates |
| JP4433918B2 (ja) * | 2004-07-15 | 2010-03-17 | コニカミノルタエムジー株式会社 | 画像形成方法 |
| KR100651728B1 (ko) * | 2004-11-10 | 2006-12-06 | 한국전자통신연구원 | 정착기를 갖는 전자 소자용 화합물 및 이를 포함하는 전자소자와 이들의 제조 방법 |
| MX2007009878A (es) * | 2005-02-18 | 2007-10-03 | Medarex Inc | Anticuerpos monoclonales para antigeno de membrana especifica para prostata (amep). |
| US7714016B2 (en) * | 2005-04-08 | 2010-05-11 | Medarex, Inc. | Cytotoxic compounds and conjugates with cleavable substrates |
| US20070060534A1 (en) * | 2005-06-30 | 2007-03-15 | Threshold Pharmaceuticals, Inc. | Anthracycline analogs |
| BRPI0617546A2 (pt) * | 2005-09-26 | 2011-07-26 | Medarex Inc | conjugado de fÁrmaco-anticorpo, formulaÇço farmacÊutica, mÉtodo para matar uma cÉlula de tumor, mÉtodo para retardar ou interromper o crescimento de um tumor em um sujeito mamÍfero e composto |
| BRPI0619331A2 (pt) | 2005-10-26 | 2011-09-27 | Medarex Inc | método para fazer um composto para preparar um análogo de cbi cc-1065, método para preparar um análogo de cbi cc-1065 e composto |
| WO2007059404A2 (en) | 2005-11-10 | 2007-05-24 | Medarex, Inc. | Duocarmycin derivatives as novel cytotoxic compounds and conjugates |
| CA2645347A1 (en) * | 2006-03-10 | 2007-09-20 | Diatos | Anticancer drugs conjugated to antibody via an enzyme cleavable linker |
| WO2008005942A2 (en) * | 2006-06-30 | 2008-01-10 | The Govt. Of The Usa As Represented By The Secretary Of The Dept. Of Health And Human Services. | Activatable probes and methods of use |
| US7718044B2 (en) * | 2006-09-11 | 2010-05-18 | Seagate Technology Llc | Method for controlling shaft coating taper |
| TWI412367B (zh) | 2006-12-28 | 2013-10-21 | Medarex Llc | 化學鏈接劑與可裂解基質以及其之綴合物 |
| JP2010519310A (ja) | 2007-02-21 | 2010-06-03 | メダレックス インコーポレイテッド | 単一のアミノ酸を有する化学リンカーおよびその複合体 |
| EP2190861A4 (en) | 2007-08-22 | 2011-03-30 | Univ California | ACTIVE BINDING POLYPEPTIDES AND METHOD FOR THEIR IDENTIFICATION AND USE |
| CN106995495A (zh) | 2009-01-12 | 2017-08-01 | 希托马克斯医疗有限责任公司 | 修饰抗体组合物及其制备和使用方法 |
| JP5861223B2 (ja) | 2009-02-23 | 2016-02-16 | サイトムエックス セラピューティクス, インク.CytomX Therapeutics, Inc. | プロタンパク質およびその使用方法 |
| FR2960153B1 (fr) * | 2010-05-20 | 2012-08-17 | Centre Nat Rech Scient | Nouveaux bras autoreactifs et prodrogues les comprenant |
| CA2827170A1 (en) | 2011-02-11 | 2012-08-16 | David M. Hilbert | Monovalent and multivalent multispecific complexes and uses thereof |
| CA2837169C (en) | 2011-05-24 | 2021-11-09 | Zyngenia, Inc. | Multispecific complexes comprising angiopoietin-2-binding peptide and their uses |
| WO2014080251A1 (en) | 2012-11-24 | 2014-05-30 | Hangzhou Dac Biotech Co., Ltd. | Hydrophilic linkers and their uses for conjugation of drugs to cell binding molecules |
| EP2781919A1 (en) | 2013-03-19 | 2014-09-24 | Roche Diagniostics GmbH | Method / device for generating a corrected value of an analyte concentration in a sample of a body fluid |
| DK3122757T3 (da) | 2014-02-28 | 2023-10-09 | Hangzhou Dac Biotech Co Ltd | Ladede linkere og anvendelse deraf til konjugering |
| JP6759326B2 (ja) | 2015-07-12 | 2020-09-23 | ハンジョウ ディーエーシー バイオテック シーオー.,エルティディ.Hangzhou Dac Biotech Co.,Ltd. | 細胞結合分子の共役のための架橋連結体 |
| US9839687B2 (en) | 2015-07-15 | 2017-12-12 | Suzhou M-Conj Biotech Co., Ltd. | Acetylenedicarboxyl linkers and their uses in specific conjugation of a cell-binding molecule |
| US12064445B2 (en) | 2015-12-03 | 2024-08-20 | Biosight Ltd. | Cytarabine conjugates for cancer therapy |
| US12071450B2 (en) | 2015-12-03 | 2024-08-27 | Biosight Ltd. | Salts of conjugates for cancer therapy |
| US11058701B2 (en) | 2015-12-03 | 2021-07-13 | Biosight Ltd. | Cytarabine conjugates for cancer therapy |
| RU2747528C2 (ru) | 2015-12-03 | 2021-05-06 | Биосайт Лтд. | Соли конъюгатов для лечения рака |
| CN106074556B (zh) * | 2016-06-14 | 2018-11-16 | 河南师范大学 | L型胞嘧啶丙氨酸在制备抗癌药物中的应用 |
| AU2016429272A1 (en) | 2016-11-14 | 2019-05-02 | Hangzhou Dac Biotech Co., Ltd. | Conjugation linkers, cell binding molecule-drug conjugates containing the likers, methods of making and uses such conjugates with the linkers |
| WO2022043256A1 (en) | 2020-08-23 | 2022-03-03 | Cobiores Nv | Synergistic combinations of anticancer drugs linked to a tetrapeptidic moiety and immunotherapeutic agents |
| CA3203072A1 (en) | 2020-12-22 | 2022-06-30 | Andrea CASAZZA | Compounds comprising a tetrapeptidic moiety |
| WO2022167664A1 (en) | 2021-02-07 | 2022-08-11 | Cobiores Nv | Compounds comprising a tetrapeptidic moiety |
Family Cites Families (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE869485A (fr) * | 1978-08-03 | 1978-12-01 | Inst Internat De Pathologie Ce | Nouveaux derives de la doxorubicine, leur preparation et les compositions qui les contiennent |
| US4296105A (en) * | 1978-08-03 | 1981-10-20 | Institut International De Pathologie Cellulaire Et Moleculaire | Derivatives of doxorubicine, their preparation and use |
| US4277466A (en) | 1978-08-29 | 1981-07-07 | Institut International De Pathologie Cellulaire Et Moleculaire | Complexes of DNA and esters derived from daunorubicine, their preparation and use |
| EP0009944B1 (en) * | 1978-10-02 | 1983-05-18 | Merck & Co. Inc. | Lysosomotropic fluorinated amine therapeutic agents and compositions containing them |
| US4719312A (en) * | 1978-10-02 | 1988-01-12 | Merck & Co., Inc. | Lysosometropic detergent therapeutic agents |
| WO1981001145A1 (en) | 1979-10-18 | 1981-04-30 | Univ Illinois | Hydrolytic enzyme-activatible pro-drugs |
| DE3167679D1 (en) * | 1980-03-31 | 1985-01-24 | Inst Int Pathologie Cellulaire | Pharmaceutical forms, their preparation and compositions containing the same |
| US4376765A (en) * | 1980-03-31 | 1983-03-15 | Institut International De Pathologie Cellulaire Et Moleculaire | Medicaments, their preparation and compositions containing same |
| BE882541A (fr) * | 1980-03-31 | 1980-07-16 | Inst Internat De Pathologie Ce | Nouvelles formes pharmaceutiques leur preparation et les compositions qui les contiennent |
| OA06421A (fr) * | 1980-06-10 | 1981-09-30 | Omnium Chimique Sa | Procédé de préparation de dérivés N-(vinblastinoyl-23) d'acides aminés et de peptides. |
| US4639456A (en) * | 1980-06-10 | 1987-01-27 | Omnichem S.A. | Vinblastin-23-oyl amino acid derivatives |
| US4671958A (en) * | 1982-03-09 | 1987-06-09 | Cytogen Corporation | Antibody conjugates for the delivery of compounds to target sites |
| GR81790B (ja) | 1983-04-29 | 1984-12-12 | Omnichem Sa | |
| FR2546163B1 (fr) * | 1983-05-16 | 1987-10-09 | Centre Nat Rech Scient | Nouveaux derives acyles hydrosolubles de peptides ou d'amino-acides, leur preparation et leur application |
| EP0126344A2 (en) | 1983-05-20 | 1984-11-28 | Abbott Laboratories | Tripeptide esters of therapeutic agents |
| JPS60233642A (ja) * | 1984-05-07 | 1985-11-20 | Fuji Photo Film Co Ltd | 写真画像の形成方法 |
| FR2584293B1 (fr) * | 1985-07-04 | 1989-03-17 | Ire Celltarg Sa | Anticorps utiles comme agents de pilotage et conjugues les incorporant |
| AU618029B2 (en) | 1987-11-02 | 1991-12-12 | Imperial Chemical Industries Plc | Polypeptide compounds |
| FR2626882B1 (fr) | 1988-02-08 | 1991-11-08 | Ire Celltarg Sa | Conjugues de derives de vinca comportant une chaine detergente en position c-3 |
| DE3841764A1 (de) | 1988-12-12 | 1990-06-13 | Basf Ag | Neue tnf-peptide |
| US5220001A (en) | 1989-10-25 | 1993-06-15 | Zaidan Hojim Biseibutsu Dong-A Pharm Co. | Anthracycline glycoside derivatives |
| FI91777C (fi) * | 1990-04-02 | 1994-08-10 | Elomit Oy | Menetelmä plasmiiniaktiivisuuden määrittämiseksi |
| US5618790A (en) | 1990-10-05 | 1997-04-08 | Queen's University At Kingston | Protease mediated drug delivery system |
| WO1993002703A1 (en) | 1991-08-05 | 1993-02-18 | Igen, Inc. | Prodrugs activated by targeted catalytic proteins |
| WO1994007518A1 (en) | 1992-09-25 | 1994-04-14 | Abbott Laboratories | Anaphylatoxin receptor ligands containing lipophilic residues |
| EP0679153A4 (en) | 1993-01-14 | 1996-05-15 | Magainin Pharma | FINALLY MODIFIED AMINO ACIDS AND PEPTIDES. |
| US5599686A (en) | 1994-06-28 | 1997-02-04 | Merck & Co., Inc. | Peptides |
| US6143864A (en) | 1994-06-28 | 2000-11-07 | Merck & Co., Inc. | Peptides |
| AU694546C (en) * | 1994-08-19 | 2001-09-06 | La Region Wallonne | Compounds, pharmaceutical composition and diagnostic device comprising same and their use |
| US5659061A (en) * | 1995-04-20 | 1997-08-19 | Drug Innovation & Design, Inc. | Tumor protease activated prodrugs of phosphoramide mustard analogs with toxification and detoxification functionalities |
| EP1181055A2 (en) | 1999-05-14 | 2002-02-27 | Boehringer Ingelheim Pharmaceuticals Inc. | Enzyme-activated anti-tumor prodrug compounds |
| CA2388700A1 (en) | 1999-10-19 | 2001-04-26 | Merck & Co., Inc. | Conjugates useful in the treatment of prostate cancer |
| AU781531B2 (en) | 1999-10-27 | 2005-05-26 | Merck & Co., Inc. | Salt form of a conjugate useful in the treatment of prostate cancer |
-
1995
- 1995-08-21 AU AU32486/95A patent/AU694546C/en not_active Expired
- 1995-08-21 US US08/793,910 patent/US5962216A/en not_active Expired - Lifetime
- 1995-08-21 EP EP95928905A patent/EP0769967B1/fr not_active Expired - Lifetime
- 1995-08-21 DK DK95928905T patent/DK0769967T3/da active
- 1995-08-21 CA CA2203622A patent/CA2203622C/en not_active Expired - Lifetime
- 1995-08-21 SI SI9530732T patent/SI0769967T1/sl unknown
- 1995-08-21 PT PT95928905T patent/PT769967E/pt unknown
- 1995-08-21 JP JP50766296A patent/JP4157600B2/ja not_active Expired - Lifetime
- 1995-08-21 EP EP07119790A patent/EP1880737A1/fr not_active Withdrawn
- 1995-08-21 DE DE69535665T patent/DE69535665T2/de not_active Expired - Lifetime
- 1995-08-21 NZ NZ291368A patent/NZ291368A/en not_active IP Right Cessation
- 1995-08-21 AT AT95928905T patent/ATE380559T1/de active
- 1995-08-21 WO PCT/BE1995/000076 patent/WO1996005863A1/fr active IP Right Grant
- 1995-08-21 ES ES95928905T patent/ES2297832T3/es not_active Expired - Lifetime
-
1997
- 1997-02-18 NO NO19970748A patent/NO324045B1/no not_active IP Right Cessation
-
1999
- 1999-04-23 US US09/298,330 patent/US6342480B1/en not_active Expired - Lifetime
-
2001
- 2001-11-09 US US10/012,576 patent/US7037898B2/en not_active Expired - Fee Related
-
2006
- 2006-03-08 US US11/370,290 patent/US7390629B2/en not_active Expired - Fee Related
-
2007
- 2007-11-21 JP JP2007301893A patent/JP2008069175A/ja active Pending
-
2008
- 2008-06-10 US US12/157,363 patent/US7951772B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| MX9701283A (es) | 1997-09-30 |
| AU694546C (en) | 2001-09-06 |
| NO324045B1 (no) | 2007-08-06 |
| US7037898B2 (en) | 2006-05-02 |
| NZ291368A (en) | 1999-04-29 |
| US20060148682A1 (en) | 2006-07-06 |
| DE69535665T2 (de) | 2009-04-02 |
| ES2297832T3 (es) | 2008-05-01 |
| US6342480B1 (en) | 2002-01-29 |
| NO970748D0 (no) | 1997-02-18 |
| JP2008069175A (ja) | 2008-03-27 |
| CA2203622A1 (en) | 1996-02-29 |
| US5962216A (en) | 1999-10-05 |
| DE69535665D1 (de) | 2008-01-24 |
| US20020160943A1 (en) | 2002-10-31 |
| US7390629B2 (en) | 2008-06-24 |
| SI0769967T1 (sl) | 2008-06-30 |
| AU694546B2 (en) | 1998-07-23 |
| JPH10508291A (ja) | 1998-08-18 |
| EP1880737A1 (fr) | 2008-01-23 |
| EP0769967A1 (fr) | 1997-05-02 |
| US7951772B2 (en) | 2011-05-31 |
| NO970748L (no) | 1997-04-10 |
| CA2203622C (en) | 2011-11-01 |
| US20090137494A1 (en) | 2009-05-28 |
| AU3248695A (en) | 1996-03-14 |
| WO1996005863A1 (fr) | 1996-02-29 |
| PT769967E (pt) | 2008-02-18 |
| DK0769967T3 (da) | 2008-03-31 |
| ATE380559T1 (de) | 2007-12-15 |
| EP0769967B1 (fr) | 2007-12-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4157600B2 (ja) | 化合物、製剤用組成物及びこれらを含む診断装置とこれらの利用 | |
| EP2347770B1 (de) | Verfahren zur Herstellung einer injizierbaren Arzneimittelzubereitung | |
| EP1198254B1 (de) | Träger-pharmaka-konjugate | |
| JP4675028B2 (ja) | トリメチルロック型テトラパルテートプロドラッグ | |
| US6265540B1 (en) | Tissue specific prodrug | |
| US8822406B2 (en) | Tumor activated prodrugs | |
| US20080161245A1 (en) | Protein-Binding Anthracycline Peptide Derivatives and Drugs Containing Them | |
| US20190375837A1 (en) | Immunocytokine combination therapy | |
| DE10310082A1 (de) | Protein-bindende Doxorubicin-Peptid-Derivate | |
| US6504014B1 (en) | Tissue specific prodrug | |
| MXPA97001283A (en) | Compounds, pharmaceutical composition and diagnostic device containing them and its |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20060711 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20061011 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20061127 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20070110 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20070227 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20070528 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20070718 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20070809 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20070821 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20071121 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080122 |
|
| RD13 | Notification of appointment of power of sub attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7433 Effective date: 20080415 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080422 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20080415 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20080617 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20080714 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110718 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110718 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120718 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120718 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130718 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |