JP3797974B2 - クローン化トランスジェニック有蹄動物における異種(ヒト)免疫グロブリンの発現 - Google Patents
クローン化トランスジェニック有蹄動物における異種(ヒト)免疫グロブリンの発現 Download PDFInfo
- Publication number
- JP3797974B2 JP3797974B2 JP2002570676A JP2002570676A JP3797974B2 JP 3797974 B2 JP3797974 B2 JP 3797974B2 JP 2002570676 A JP2002570676 A JP 2002570676A JP 2002570676 A JP2002570676 A JP 2002570676A JP 3797974 B2 JP3797974 B2 JP 3797974B2
- Authority
- JP
- Japan
- Prior art keywords
- nucleic acid
- ungulate
- cell
- human
- antibody
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 230000009261 transgenic effect Effects 0.000 title claims abstract description 103
- 108060003951 Immunoglobulin Proteins 0.000 title claims description 38
- 102000018358 immunoglobulin Human genes 0.000 title claims description 38
- 229940072221 immunoglobulins Drugs 0.000 title description 3
- 241000283690 Bos taurus Species 0.000 claims abstract description 174
- 238000004519 manufacturing process Methods 0.000 claims abstract description 25
- 210000004507 artificial chromosome Anatomy 0.000 claims abstract description 23
- 210000004027 cell Anatomy 0.000 claims description 368
- 102000039446 nucleic acids Human genes 0.000 claims description 213
- 108020004707 nucleic acids Proteins 0.000 claims description 213
- 150000007523 nucleic acids Chemical class 0.000 claims description 213
- 108090000623 proteins and genes Proteins 0.000 claims description 152
- 210000003754 fetus Anatomy 0.000 claims description 151
- 238000000034 method Methods 0.000 claims description 151
- 210000002950 fibroblast Anatomy 0.000 claims description 93
- 239000012634 fragment Substances 0.000 claims description 73
- 230000035772 mutation Effects 0.000 claims description 72
- 108700028369 Alleles Proteins 0.000 claims description 67
- 230000001605 fetal effect Effects 0.000 claims description 67
- 108700005091 Immunoglobulin Genes Proteins 0.000 claims description 58
- 210000000349 chromosome Anatomy 0.000 claims description 50
- 239000003550 marker Substances 0.000 claims description 48
- 230000008707 rearrangement Effects 0.000 claims description 45
- 210000000287 oocyte Anatomy 0.000 claims description 42
- 241001465754 Metazoa Species 0.000 claims description 39
- 239000000427 antigen Substances 0.000 claims description 36
- 108091007433 antigens Proteins 0.000 claims description 36
- 102000036639 antigens Human genes 0.000 claims description 36
- 210000001161 mammalian embryo Anatomy 0.000 claims description 32
- 210000003719 b-lymphocyte Anatomy 0.000 claims description 31
- 241001494479 Pecora Species 0.000 claims description 27
- 238000012217 deletion Methods 0.000 claims description 19
- 230000037430 deletion Effects 0.000 claims description 19
- 210000004080 milk Anatomy 0.000 claims description 19
- 239000008267 milk Substances 0.000 claims description 19
- 235000013336 milk Nutrition 0.000 claims description 19
- 239000002773 nucleotide Substances 0.000 claims description 19
- 125000003729 nucleotide group Chemical group 0.000 claims description 19
- 230000002759 chromosomal effect Effects 0.000 claims description 18
- 241000283707 Capra Species 0.000 claims description 17
- 230000003053 immunization Effects 0.000 claims description 16
- 210000002966 serum Anatomy 0.000 claims description 15
- 210000004940 nucleus Anatomy 0.000 claims description 12
- 108010077544 Chromatin Proteins 0.000 claims description 11
- 210000003483 chromatin Anatomy 0.000 claims description 11
- 210000004408 hybridoma Anatomy 0.000 claims description 11
- 230000004927 fusion Effects 0.000 claims description 10
- 210000004291 uterus Anatomy 0.000 claims description 10
- 101150008942 J gene Proteins 0.000 claims description 6
- 101150117115 V gene Proteins 0.000 claims description 6
- 101150097493 D gene Proteins 0.000 claims description 5
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 4
- 201000000050 myeloid neoplasm Diseases 0.000 claims description 4
- 108020004485 Nonsense Codon Proteins 0.000 claims description 2
- 230000037434 nonsense mutation Effects 0.000 claims description 2
- 230000002779 inactivation Effects 0.000 abstract description 11
- 238000012239 gene modification Methods 0.000 abstract description 2
- 230000005017 genetic modification Effects 0.000 abstract description 2
- 235000013617 genetically modified food Nutrition 0.000 abstract description 2
- 108020004414 DNA Proteins 0.000 description 103
- 230000008685 targeting Effects 0.000 description 85
- 239000013598 vector Substances 0.000 description 83
- 210000000688 human artificial chromosome Anatomy 0.000 description 70
- 238000003752 polymerase chain reaction Methods 0.000 description 62
- 238000012546 transfer Methods 0.000 description 62
- 210000001519 tissue Anatomy 0.000 description 48
- 101000840258 Homo sapiens Immunoglobulin J chain Proteins 0.000 description 42
- 102100029571 Immunoglobulin J chain Human genes 0.000 description 39
- 239000002609 medium Substances 0.000 description 36
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 36
- 239000000047 product Substances 0.000 description 32
- 239000011541 reaction mixture Substances 0.000 description 31
- 238000003757 reverse transcription PCR Methods 0.000 description 30
- 210000000952 spleen Anatomy 0.000 description 30
- 239000002299 complementary DNA Substances 0.000 description 27
- 239000000203 mixture Substances 0.000 description 27
- 238000002744 homologous recombination Methods 0.000 description 26
- 230000006801 homologous recombination Effects 0.000 description 26
- 229930193140 Neomycin Natural products 0.000 description 24
- 230000003321 amplification Effects 0.000 description 24
- 239000003814 drug Substances 0.000 description 24
- 229960004927 neomycin Drugs 0.000 description 24
- 238000003199 nucleic acid amplification method Methods 0.000 description 24
- 238000006243 chemical reaction Methods 0.000 description 22
- 108091000054 Prion Proteins 0.000 description 21
- 238000004458 analytical method Methods 0.000 description 21
- 230000003115 biocidal effect Effects 0.000 description 21
- 239000000872 buffer Substances 0.000 description 21
- 244000309466 calf Species 0.000 description 21
- 238000011534 incubation Methods 0.000 description 21
- 229940079593 drug Drugs 0.000 description 20
- 230000035935 pregnancy Effects 0.000 description 20
- 241000894007 species Species 0.000 description 19
- 108700024394 Exon Proteins 0.000 description 18
- 238000010363 gene targeting Methods 0.000 description 18
- 210000003917 human chromosome Anatomy 0.000 description 18
- 229950010131 puromycin Drugs 0.000 description 18
- 239000002953 phosphate buffered saline Substances 0.000 description 17
- 238000001890 transfection Methods 0.000 description 16
- 241000699670 Mus sp. Species 0.000 description 15
- 238000010222 PCR analysis Methods 0.000 description 15
- 102000029797 Prion Human genes 0.000 description 15
- 239000011543 agarose gel Substances 0.000 description 15
- 241000282412 Homo Species 0.000 description 14
- 210000002257 embryonic structure Anatomy 0.000 description 14
- 102100031181 Glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 13
- 230000000875 corresponding effect Effects 0.000 description 13
- 238000002474 experimental method Methods 0.000 description 13
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 13
- 238000002649 immunization Methods 0.000 description 13
- 230000010354 integration Effects 0.000 description 13
- 102000004169 proteins and genes Human genes 0.000 description 13
- 238000010561 standard procedure Methods 0.000 description 13
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 12
- 238000010240 RT-PCR analysis Methods 0.000 description 12
- 238000004520 electroporation Methods 0.000 description 12
- 238000003780 insertion Methods 0.000 description 12
- 230000037431 insertion Effects 0.000 description 12
- 210000004185 liver Anatomy 0.000 description 12
- 210000004556 brain Anatomy 0.000 description 11
- 238000010367 cloning Methods 0.000 description 11
- 210000001082 somatic cell Anatomy 0.000 description 11
- 239000013607 AAV vector Substances 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 description 10
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 description 10
- 238000004925 denaturation Methods 0.000 description 10
- 230000036425 denaturation Effects 0.000 description 10
- 238000010494 dissociation reaction Methods 0.000 description 10
- 230000005593 dissociations Effects 0.000 description 10
- 239000012528 membrane Substances 0.000 description 10
- 239000008188 pellet Substances 0.000 description 10
- 241000282898 Sus scrofa Species 0.000 description 9
- 239000011777 magnesium Substances 0.000 description 9
- 244000052769 pathogen Species 0.000 description 9
- 108091033319 polynucleotide Proteins 0.000 description 9
- 102000040430 polynucleotide Human genes 0.000 description 9
- 239000002157 polynucleotide Substances 0.000 description 9
- 239000000523 sample Substances 0.000 description 9
- 238000012216 screening Methods 0.000 description 9
- 238000012360 testing method Methods 0.000 description 9
- 238000000137 annealing Methods 0.000 description 8
- 210000002459 blastocyst Anatomy 0.000 description 8
- 239000011575 calcium Substances 0.000 description 8
- 210000004602 germ cell Anatomy 0.000 description 8
- 239000001963 growth medium Substances 0.000 description 8
- 230000004044 response Effects 0.000 description 8
- 230000002441 reversible effect Effects 0.000 description 8
- 239000000243 solution Substances 0.000 description 8
- 206010059866 Drug resistance Diseases 0.000 description 7
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 7
- 241000699666 Mus <mouse, genus> Species 0.000 description 7
- 241000282887 Suidae Species 0.000 description 7
- 241000700605 Viruses Species 0.000 description 7
- 239000003242 anti bacterial agent Substances 0.000 description 7
- 238000010276 construction Methods 0.000 description 7
- 230000013011 mating Effects 0.000 description 7
- 239000013641 positive control Substances 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 108090000631 Trypsin Proteins 0.000 description 6
- 102000004142 Trypsin Human genes 0.000 description 6
- 238000013459 approach Methods 0.000 description 6
- 230000005540 biological transmission Effects 0.000 description 6
- 239000006285 cell suspension Substances 0.000 description 6
- 239000013612 plasmid Substances 0.000 description 6
- 239000012588 trypsin Substances 0.000 description 6
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 6
- 241000702421 Dependoparvovirus Species 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 241000124008 Mammalia Species 0.000 description 5
- 238000012408 PCR amplification Methods 0.000 description 5
- 230000009824 affinity maturation Effects 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 239000012091 fetal bovine serum Substances 0.000 description 5
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 5
- 230000016784 immunoglobulin production Effects 0.000 description 5
- 108020004999 messenger RNA Proteins 0.000 description 5
- 239000002245 particle Substances 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 238000012163 sequencing technique Methods 0.000 description 5
- 239000006228 supernatant Substances 0.000 description 5
- 239000013603 viral vector Substances 0.000 description 5
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 4
- 241000282472 Canis lupus familiaris Species 0.000 description 4
- 102000004127 Cytokines Human genes 0.000 description 4
- 108090000695 Cytokines Proteins 0.000 description 4
- 238000002965 ELISA Methods 0.000 description 4
- 241000282326 Felis catus Species 0.000 description 4
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 4
- 108091028043 Nucleic acid sequence Proteins 0.000 description 4
- 150000001413 amino acids Chemical group 0.000 description 4
- 238000009395 breeding Methods 0.000 description 4
- 230000001488 breeding effect Effects 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 108010030074 endodeoxyribonuclease MluI Proteins 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 239000013642 negative control Substances 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 108091008146 restriction endonucleases Proteins 0.000 description 4
- 238000011282 treatment Methods 0.000 description 4
- 238000002604 ultrasonography Methods 0.000 description 4
- 238000011144 upstream manufacturing Methods 0.000 description 4
- PRDFBSVERLRRMY-UHFFFAOYSA-N 2'-(4-ethoxyphenyl)-5-(4-methylpiperazin-1-yl)-2,5'-bibenzimidazole Chemical compound C1=CC(OCC)=CC=C1C1=NC2=CC=C(C=3NC4=CC(=CC=C4N=3)N3CCN(C)CC3)C=C2N1 PRDFBSVERLRRMY-UHFFFAOYSA-N 0.000 description 3
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- 108010067770 Endopeptidase K Proteins 0.000 description 3
- 241000701959 Escherichia virus Lambda Species 0.000 description 3
- 108700039887 Essential Genes Proteins 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- 101150095194 NEO1 gene Proteins 0.000 description 3
- 238000009004 PCR Kit Methods 0.000 description 3
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 3
- 238000002105 Southern blotting Methods 0.000 description 3
- 239000007983 Tris buffer Substances 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 230000003497 anti-pneumococcal effect Effects 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 150000001720 carbohydrates Chemical class 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 239000013599 cloning vector Substances 0.000 description 3
- 238000012790 confirmation Methods 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 229930182830 galactose Natural products 0.000 description 3
- 125000002519 galactosyl group Chemical group C1([C@H](O)[C@@H](O)[C@@H](O)[C@H](O1)CO)* 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 210000005260 human cell Anatomy 0.000 description 3
- 230000000415 inactivating effect Effects 0.000 description 3
- 238000002955 isolation Methods 0.000 description 3
- 238000001638 lipofection Methods 0.000 description 3
- 210000004698 lymphocyte Anatomy 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 238000000386 microscopy Methods 0.000 description 3
- 210000004681 ovum Anatomy 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 239000006152 selective media Substances 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 229960005322 streptomycin Drugs 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 239000003104 tissue culture media Substances 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 3
- CNNSWSHYGANWBM-UHFFFAOYSA-N 6-chloro-2,3-dimethylquinoxaline Chemical compound C1=C(Cl)C=C2N=C(C)C(C)=NC2=C1 CNNSWSHYGANWBM-UHFFFAOYSA-N 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- 241000283705 Capra hircus Species 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 108091026890 Coding region Proteins 0.000 description 2
- 108010051219 Cre recombinase Proteins 0.000 description 2
- 238000007399 DNA isolation Methods 0.000 description 2
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 2
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- 241000287828 Gallus gallus Species 0.000 description 2
- 208000034951 Genetic Translocation Diseases 0.000 description 2
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 2
- 241000606768 Haemophilus influenzae Species 0.000 description 2
- 102100030500 Heparin cofactor 2 Human genes 0.000 description 2
- 101001082432 Homo sapiens Heparin cofactor 2 Proteins 0.000 description 2
- 102000012745 Immunoglobulin Subunits Human genes 0.000 description 2
- 108010079585 Immunoglobulin Subunits Proteins 0.000 description 2
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- 229920002274 Nalgene Polymers 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- 208000012902 Nervous system disease Diseases 0.000 description 2
- 208000025966 Neurological disease Diseases 0.000 description 2
- 239000004677 Nylon Substances 0.000 description 2
- 108010004729 Phycoerythrin Proteins 0.000 description 2
- 108020005067 RNA Splice Sites Proteins 0.000 description 2
- 239000013614 RNA sample Substances 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 241000725643 Respiratory syncytial virus Species 0.000 description 2
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- 208000018756 Variant Creutzfeldt-Jakob disease Diseases 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 210000004102 animal cell Anatomy 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- 208000005881 bovine spongiform encephalopathy Diseases 0.000 description 2
- 230000030833 cell death Effects 0.000 description 2
- 230000011712 cell development Effects 0.000 description 2
- 239000008004 cell lysis buffer Substances 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000013611 chromosomal DNA Substances 0.000 description 2
- 239000013601 cosmid vector Substances 0.000 description 2
- YPHMISFOHDHNIV-FSZOTQKASA-N cycloheximide Chemical compound C1[C@@H](C)C[C@H](C)C(=O)[C@@H]1[C@H](O)CC1CC(=O)NC(=O)C1 YPHMISFOHDHNIV-FSZOTQKASA-N 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 235000013601 eggs Nutrition 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 238000013401 experimental design Methods 0.000 description 2
- 239000013604 expression vector Substances 0.000 description 2
- 239000012894 fetal calf serum Substances 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 2
- 239000005090 green fluorescent protein Substances 0.000 description 2
- 229940047650 haemophilus influenzae Drugs 0.000 description 2
- 230000000521 hyperimmunizing effect Effects 0.000 description 2
- 230000028993 immune response Effects 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 238000009169 immunotherapy Methods 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 238000002372 labelling Methods 0.000 description 2
- 210000003563 lymphoid tissue Anatomy 0.000 description 2
- 238000013507 mapping Methods 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000001823 molecular biology technique Methods 0.000 description 2
- 238000007857 nested PCR Methods 0.000 description 2
- 229920001778 nylon Polymers 0.000 description 2
- 210000001986 peyer's patch Anatomy 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 239000011535 reaction buffer Substances 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- 230000003393 splenic effect Effects 0.000 description 2
- 230000001360 synchronised effect Effects 0.000 description 2
- 229940095064 tartrate Drugs 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 230000005030 transcription termination Effects 0.000 description 2
- 238000010361 transduction Methods 0.000 description 2
- 230000026683 transduction Effects 0.000 description 2
- 239000012096 transfection reagent Substances 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 241000701161 unidentified adenovirus Species 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- NNJPGOLRFBJNIW-HNNXBMFYSA-N (-)-demecolcine Chemical compound C1=C(OC)C(=O)C=C2[C@@H](NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-HNNXBMFYSA-N 0.000 description 1
- PXGPLTODNUVGFL-BRIYLRKRSA-N (E,Z)-(1R,2R,3R,5S)-7-(3,5-Dihydroxy-2-((3S)-(3-hydroxy-1-octenyl))cyclopentyl)-5-heptenoic acid Chemical compound CCCCC[C@H](O)C=C[C@H]1[C@H](O)C[C@H](O)[C@@H]1CC=CCCCC(O)=O PXGPLTODNUVGFL-BRIYLRKRSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 241000282979 Alces alces Species 0.000 description 1
- 235000002198 Annona diversifolia Nutrition 0.000 description 1
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 1
- 101100177160 Arabidopsis thaliana HAC2 gene Proteins 0.000 description 1
- 101100302211 Arabidopsis thaliana RNR2A gene Proteins 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 108010077805 Bacterial Proteins Proteins 0.000 description 1
- 241000588832 Bordetella pertussis Species 0.000 description 1
- 241000283725 Bos Species 0.000 description 1
- 241000282817 Bovidae Species 0.000 description 1
- 241000282832 Camelidae Species 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 241000282994 Cervidae Species 0.000 description 1
- 241000193403 Clostridium Species 0.000 description 1
- 241000193155 Clostridium botulinum Species 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 102000004594 DNA Polymerase I Human genes 0.000 description 1
- 108010017826 DNA Polymerase I Proteins 0.000 description 1
- 108020003215 DNA Probes Proteins 0.000 description 1
- 238000007400 DNA extraction Methods 0.000 description 1
- 239000003298 DNA probe Substances 0.000 description 1
- 241000450599 DNA viruses Species 0.000 description 1
- NNJPGOLRFBJNIW-UHFFFAOYSA-N Demecolcine Natural products C1=C(OC)C(=O)C=C2C(NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-UHFFFAOYSA-N 0.000 description 1
- 108010053187 Diphtheria Toxin Proteins 0.000 description 1
- 241001115402 Ebolavirus Species 0.000 description 1
- 241001635598 Enicostema Species 0.000 description 1
- 241000709661 Enterovirus Species 0.000 description 1
- 241000283074 Equus asinus Species 0.000 description 1
- 241001331845 Equus asinus x caballus Species 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 230000010190 G1 phase Effects 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 108700007698 Genetic Terminator Regions Proteins 0.000 description 1
- 108010044091 Globulins Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 241000606790 Haemophilus Species 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 1
- 101000664600 Homo sapiens Tripartite motif-containing protein 3 Proteins 0.000 description 1
- 101000639792 Homo sapiens U2 small nuclear ribonucleoprotein A' Proteins 0.000 description 1
- GRRNUXAQVGOGFE-UHFFFAOYSA-N Hygromycin-B Natural products OC1C(NC)CC(N)C(O)C1OC1C2OC3(C(C(O)C(O)C(C(N)CO)O3)O)OC2C(O)C(CO)O1 GRRNUXAQVGOGFE-UHFFFAOYSA-N 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 108010065825 Immunoglobulin Light Chains Proteins 0.000 description 1
- 102000013463 Immunoglobulin Light Chains Human genes 0.000 description 1
- 108010004020 Immunoglobulin lambda-Chains Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 108091092195 Intron Proteins 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- 241000282838 Lama Species 0.000 description 1
- 208000035752 Live birth Diseases 0.000 description 1
- 241000712079 Measles morbillivirus Species 0.000 description 1
- 101100070236 Mus musculus Hcn1 gene Proteins 0.000 description 1
- 241000588653 Neisseria Species 0.000 description 1
- 241000588650 Neisseria meningitidis Species 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 241001502414 Ovis canadensis Species 0.000 description 1
- 241000283089 Perissodactyla Species 0.000 description 1
- 229920002535 Polyethylene Glycol 1500 Polymers 0.000 description 1
- 241000283080 Proboscidea <mammal> Species 0.000 description 1
- 241000125945 Protoparvovirus Species 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 1
- 101150002896 RNR2 gene Proteins 0.000 description 1
- 241000711798 Rabies lyssavirus Species 0.000 description 1
- 206010057190 Respiratory tract infections Diseases 0.000 description 1
- 241000191940 Staphylococcus Species 0.000 description 1
- 241000194017 Streptococcus Species 0.000 description 1
- 241000193998 Streptococcus pneumoniae Species 0.000 description 1
- 241001493546 Suina Species 0.000 description 1
- 241000282890 Sus Species 0.000 description 1
- 101710120037 Toxin CcdB Proteins 0.000 description 1
- 102100038798 Tripartite motif-containing protein 3 Human genes 0.000 description 1
- 239000007984 Tris EDTA buffer Substances 0.000 description 1
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 102100034465 U2 small nuclear ribonucleoprotein A' Human genes 0.000 description 1
- GBOGMAARMMDZGR-UHFFFAOYSA-N UNPD149280 Natural products N1C(=O)C23OC(=O)C=CC(O)CCCC(C)CC=CC3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 GBOGMAARMMDZGR-UHFFFAOYSA-N 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- 241001416177 Vicugna pacos Species 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 238000001261 affinity purification Methods 0.000 description 1
- 238000007605 air drying Methods 0.000 description 1
- APKFDSVGJQXUKY-INPOYWNPSA-N amphotericin B Chemical compound O[C@H]1[C@@H](N)[C@H](O)[C@@H](C)O[C@H]1O[C@H]1/C=C/C=C/C=C/C=C/C=C/C=C/C=C/[C@H](C)[C@@H](O)[C@@H](C)[C@H](C)OC(=O)C[C@H](O)C[C@H](O)CC[C@@H](O)[C@H](O)C[C@H](O)C[C@](O)(C[C@H](O)[C@H]2C(O)=O)O[C@H]2C1 APKFDSVGJQXUKY-INPOYWNPSA-N 0.000 description 1
- 238000003975 animal breeding Methods 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 230000001857 anti-mycotic effect Effects 0.000 description 1
- 238000009175 antibody therapy Methods 0.000 description 1
- 239000002543 antimycotic Substances 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 244000052616 bacterial pathogen Species 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000003969 blast cell Anatomy 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 210000002798 bone marrow cell Anatomy 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 239000003710 calcium ionophore Substances 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 210000000234 capsid Anatomy 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 230000006037 cell lysis Effects 0.000 description 1
- 210000002230 centromere Anatomy 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 238000011281 clinical therapy Methods 0.000 description 1
- 230000005757 colony formation Effects 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 230000024203 complement activation Effects 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 238000009402 cross-breeding Methods 0.000 description 1
- 238000005138 cryopreservation Methods 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- JVHIPYJQMFNCEK-UHFFFAOYSA-N cytochalasin Natural products N1C(=O)C2(C(C=CC(C)CC(C)CC=C3)OC(C)=O)C3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 JVHIPYJQMFNCEK-UHFFFAOYSA-N 0.000 description 1
- GBOGMAARMMDZGR-JREHFAHYSA-N cytochalasin B Natural products C[C@H]1CCC[C@@H](O)C=CC(=O)O[C@@]23[C@H](C=CC1)[C@H](O)C(=C)[C@@H](C)[C@@H]2[C@H](Cc4ccccc4)NC3=O GBOGMAARMMDZGR-JREHFAHYSA-N 0.000 description 1
- GBOGMAARMMDZGR-TYHYBEHESA-N cytochalasin B Chemical compound C([C@H]1[C@@H]2[C@@H](C([C@@H](O)[C@@H]3/C=C/C[C@H](C)CCC[C@@H](O)/C=C/C(=O)O[C@@]23C(=O)N1)=C)C)C1=CC=CC=C1 GBOGMAARMMDZGR-TYHYBEHESA-N 0.000 description 1
- ZMAODHOXRBLOQO-UHFFFAOYSA-N cytochalasin-A Natural products N1C(=O)C23OC(=O)C=CC(=O)CCCC(C)CC=CC3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 ZMAODHOXRBLOQO-UHFFFAOYSA-N 0.000 description 1
- 235000013365 dairy product Nutrition 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 206010013023 diphtheria Diseases 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- 241001493065 dsRNA viruses Species 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 230000012173 estrus Effects 0.000 description 1
- CJAONIOAQZUHPN-KKLWWLSJSA-N ethyl 12-[[2-[(2r,3r)-3-[2-[(12-ethoxy-12-oxododecyl)-methylamino]-2-oxoethoxy]butan-2-yl]oxyacetyl]-methylamino]dodecanoate Chemical compound CCOC(=O)CCCCCCCCCCCN(C)C(=O)CO[C@H](C)[C@@H](C)OCC(=O)N(C)CCCCCCCCCCCC(=O)OCC CJAONIOAQZUHPN-KKLWWLSJSA-N 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 238000007667 floating Methods 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 description 1
- 229960002963 ganciclovir Drugs 0.000 description 1
- 238000003209 gene knockout Methods 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical class O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- GRRNUXAQVGOGFE-NZSRVPFOSA-N hygromycin B Chemical compound O[C@@H]1[C@@H](NC)C[C@@H](N)[C@H](O)[C@H]1O[C@H]1[C@H]2O[C@@]3([C@@H]([C@@H](O)[C@@H](O)[C@@H](C(N)CO)O3)O)O[C@H]2[C@@H](O)[C@@H](CO)O1 GRRNUXAQVGOGFE-NZSRVPFOSA-N 0.000 description 1
- 229940097277 hygromycin b Drugs 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 238000007901 in situ hybridization Methods 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000009399 inbreeding Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 229940090213 lutalyse Drugs 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 238000013160 medical therapy Methods 0.000 description 1
- 230000021121 meiosis Effects 0.000 description 1
- 244000000010 microbial pathogen Species 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 238000002559 palpation Methods 0.000 description 1
- 238000004091 panning Methods 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 102000054765 polymorphisms of proteins Human genes 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000001902 propagating effect Effects 0.000 description 1
- 230000013777 protein digestion Effects 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 238000003259 recombinant expression Methods 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 238000012827 research and development Methods 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 239000012487 rinsing solution Substances 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 239000013049 sediment Substances 0.000 description 1
- 238000010187 selection method Methods 0.000 description 1
- 210000000582 semen Anatomy 0.000 description 1
- 238000002864 sequence alignment Methods 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 238000010374 somatic cell nuclear transfer Methods 0.000 description 1
- 238000009987 spinning Methods 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 238000003153 stable transfection Methods 0.000 description 1
- 238000012289 standard assay Methods 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 229940031000 streptococcus pneumoniae Drugs 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 229960000814 tetanus toxoid Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000002463 transducing effect Effects 0.000 description 1
- 238000003151 transfection method Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 210000003954 umbilical cord Anatomy 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 108700026220 vif Genes Proteins 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 244000052613 viral pathogen Species 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 210000001835 viscera Anatomy 0.000 description 1
Images



































Classifications
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New or modified breeds of animals
- A01K67/027—New or modified breeds of vertebrates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/8509—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells for producing genetically modified animals, e.g. transgenic
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New or modified breeds of animals
- A01K67/027—New or modified breeds of vertebrates
- A01K67/0273—Cloned vertebrates
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New or modified breeds of animals
- A01K67/027—New or modified breeds of vertebrates
- A01K67/0275—Genetically modified vertebrates, e.g. transgenic
- A01K67/0276—Knock-out vertebrates
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K67/00—Rearing or breeding animals, not otherwise provided for; New or modified breeds of animals
- A01K67/027—New or modified breeds of vertebrates
- A01K67/0275—Genetically modified vertebrates, e.g. transgenic
- A01K67/0278—Knock-in vertebrates, e.g. humanised vertebrates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/1048—Glycosyltransferases (2.4)
- C12N9/1051—Hexosyltransferases (2.4.1)
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2207/00—Modified animals
- A01K2207/15—Humanized animals
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2217/00—Genetically modified animals
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2217/00—Genetically modified animals
- A01K2217/05—Animals comprising random inserted nucleic acids (transgenic)
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2217/00—Genetically modified animals
- A01K2217/07—Animals genetically altered by homologous recombination
- A01K2217/075—Animals genetically altered by homologous recombination inducing loss of function, i.e. knock out
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2227/00—Animals characterised by species
- A01K2227/10—Mammal
- A01K2227/101—Bovine
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01K—ANIMAL HUSBANDRY; AVICULTURE; APICULTURE; PISCICULTURE; FISHING; REARING OR BREEDING ANIMALS, NOT OTHERWISE PROVIDED FOR; NEW BREEDS OF ANIMALS
- A01K2267/00—Animals characterised by purpose
- A01K2267/01—Animal expressing industrially exogenous proteins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2800/00—Nucleic acids vectors
- C12N2800/30—Vector systems comprising sequences for excision in presence of a recombinase, e.g. loxP or FRT
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Zoology (AREA)
- Environmental Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Husbandry (AREA)
- Biodiversity & Conservation Biology (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Biomedical Technology (AREA)
- General Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Biophysics (AREA)
- Medicinal Chemistry (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Description
ないからである。また、補体活性化、サイトカイン放出の誘導および抗原除去を仲介するヒトFc受容体機能が、ウシの系において正常であるか否かも明確ではない。
、かつ抗体重鎖または軽鎖核酸と実質的な配列同一性を有する第1の核酸に作用可能に連結された第1のプロモーターを含む第1のカセットを有する核酸を前記細胞に挿入することにより細胞を調製する。この第1のカセットは抗体重鎖または軽鎖核酸の第1の内因性対立遺伝子に組み込まれて、第1のトランスジェニック細胞を生成する。
上述のように、本発明は、Homo H/L胎仔または仔ウシの作製に関する。本手法は図1に要約してある。図には3つのスキームの概要を示してある。第1のスキームでは再生胎仔細胞系における連続ノックアウトを利用する。この手法は技術的に最も難しく、リスクレベルが最も高いが、上記したように育種手法よりも結果が速く得られる可能性がある。他の2つのスキームでは動物の育種を利用する。第2のスキームでは、オスおよびメス細胞系においてそれぞれ、重鎖および軽鎖遺伝子の単一ノックアウトのみが必要である。このスキームは細胞系の再生を利用せず、技術的には最も簡単な手法であるが、終了するまでに最も時間がかかる。スキーム3はスキーム1と2の中間にある。全てのスキームにおいて、Homo H/Lノックアウト仔ウシの生存および維持は困難である可能性があるため、Homo H/L胎仔のみを作製する。必要であれば、受動免疫療法を使用してHomo H/Lノックアウト仔ウシの生存率を高めることもできる。
本発明では、ヘミ接合オス重鎖ノックアウト(M Hemi H)およびヘミ接合メス軽鎖ノックアウト(F Hemi L)を作製し、これらのターゲッティング欠失体から40日齢胎仔を作製することが好ましい。胚由来の細胞を回収し、M Hemi H細胞において軽鎖遺伝子座の一方の対立遺伝子をターゲッティングし、F Hemi L細胞において重鎖遺伝子座の一方の対立遺伝子をターゲッティングして、HおよびL遺伝子座の両方のヘミ接合欠失を有する細胞(Hemi H/L)を得る。これらの細胞を使用して40日齢胎仔を誘導し、これから繊維芽細胞を単離する。
胎仔繊維芽細胞を使用してゲノムライブラリーを構築する。ターゲッティング構築物がクローニングに使用する細胞と同遺伝子型であることが重要であることが報告されているが、本発明において必須ではない。例えば、同遺伝子型、実質的に同遺伝子型、または非同遺伝子型構築物を使用して、内因性免疫グロブリン遺伝子に突然変異を生成することができる。1つの可能性のある方法では、他のウシ品種と比べて近親交配が遺伝的に高レベルなホルスタイン牛を使用する。本発明者らは、異なる動物間で、免疫グロブリン遺伝子中の多形を全く検出していない。これは、配列相同性が高く、非同遺伝子型構築物でのターゲッティングが上手くいくことを示唆している。
重鎖および軽鎖遺伝子を単離したら、構築物を作製する。IgM構築物は、IgM定常領域膜ドメインを欠失することにより作製される。Rajewskyおよび共同研究者らによりマウスで示されているように、IgMの膜ドメインの欠失はB細胞発達をブロックする。なぜなら、表面IgMは継続的なB細胞発達のために必要なシグナルであるからである(Kitamuraら, Nature 350:423-6)。従って、ホモ接合IgMウシはB細胞を欠く。本方法においては、機能性Igを欠く動物の生仔出生(live birth)は必要ないため、問題にはならない。しかし、必要であれば、受動免疫療法を使用して、ヒトIg遺伝子座が導入される最後のステップまで動物の生存を改善してもよい。
ターゲッティング構築物を、例えばエレクトロポレーションにより、胚繊維芽細胞に導入する。適切な抗生物質を使用して、ターゲッティングベクターを取り込んだ細胞を選択する。使用した薬剤に耐性なクローンを選択して培養する。次いで、これらのクローンを、ガンシクロビルでのネガティブ選択に供し、適切に組み込まれたクローンを選択する。あるいはまた、薬剤選択で生き残ったクローンをPCRで選択する。適切にターゲッティングされたクローンを見つけるために、少なくとも500〜1000のクローンをスクリーニングする必要があると推定される。本発明者らの推測は、IgM重鎖定常領域の膜ドメインをターゲッティングすると、300のneo耐性クローンのうち約1つが正確にターゲッティングされるというKitamuraの知見(Kitamuraら, Nature 350:423-6, 1991)に基づいている。従って、クローンを、96ウェルプレート中で10クローンずつのグループにプールして、10クローンのプールを、目的とするターゲッティングクローンについてスクリーニングすることが考えられる。陽性が同定されたら、プールしたクローンから単離した単一のクローンをスクリーニングする。この方法により、ターゲッティングクローンの同定が可能になるはずである。
上述したように、例示のターゲッティング構築物は、cre/lox系を用いてマーカーを効率的に欠失し易くするために、loxP部位に挟まれた選択可能マーカーを含有している。ターゲッティングベクターを担持する胎仔繊維芽細胞を、エレクトロポレーションを介してCre含有プラスミドでトランスフェクトする。最近発表されたGFPcre融合遺伝子を含むCreプラスミド[Gagneten S.ら, Nucleic Acids Res 25:3326-31 (1997)]を使用してもよい。これにより、Creタンパク質を含む全てのクローンの迅速な選択が可能になる。これらの細胞は、FACSソーティング、または顕微鏡操作を介したグリーン蛍光発光細胞の手操作による回収のいずれかにより、選択される。緑色の細胞は、活発に転写されるCreリコンビナーゼを担持し、つまり、薬剤耐性マーカーを欠失していると期待される。Cre発現について選択した細胞をクローニングし、PCR分析を介して薬剤耐性マーカーの欠失についてクローンを分析する。切除されたと判断された細胞を小さいクローンにまで培養し、2つに分割し、1つのアリコートを選択培地中で試験して、薬剤耐性遺伝子が確実に欠失されたことを確認する。他のアリコートは、次のラウンドのターゲッティング欠失に使用する。

1 Southern PJ, Berg P. 1982. Transformation of mammalian cells to antibiotic resistance with a bacterial gene under control of the SV40 early region promoter. J Mol Appl Genet 1:327-41.
2 Santerre RF, Allen NE, Hobbs JN Jr, Rao RN, Schmidt RJ. 1984. Expression of prokaryotic genes for hygromycin B and G418 resistance as dominant-selection markers in mouse L cells. Gene 30:147-56.
3 Wirth M, Bode J, Zettlmeissl G, Hauser H. 1988. Isolation of overproducing recombinant mammalian cell lines by a fast and simple selection procedure. Gene 73:419-26.
4 Drews RE, Kolker MT, Sachar DS, Moran GP, Schnipper LE. 1996. Passage to nonselective media transiently alters growth of mycophenolic acid-resistant mammalian cells expressing the escherichia coli xanthine-guanine phosphoribosyltransferase gene: implications for sequential selection strategies. Anal Biochem 235:215-26.
5 Karreman C. 1998. New positive/negative selectable markers for mammalian cells on the basis of Blasticidin deaminase-thymidine kinase fusions. Nucleic Acids Res 26:2508-10.
6 Hartman SC, Mulligan RG. 1988. Two dominant-acting selectable markers for gene transfer studies in mammalian cells. Proc Natl Acad Sci USA 85:8047-51.
7 Yagi T, Nada S., Watanabe N, Tamemoto H, Kohmura N, Ikawa Y, Aizawa S. 1993. A novel negative selection for homologous recombinants using diphtheria toxin A fragment gene. Anal Biochem 214:77-86。
他の有蹄動物の免疫グロブリン遺伝子を改変するために、3つの主要領域を含むようにターゲッティングベクターを設計した。第1の領域は、ターゲッティングの遺伝子座に相同である。第2の領域は、そのターゲッティング遺伝子座の一部と特異的に置き換わる薬剤選択マーカーである。第3の領域は、第1の領域と同様にターゲッティング遺伝子座に相同であるが、野生型ゲノムにおいて第1の領域と隣接していない。ターゲッティングベクターと所望の野生型遺伝子座との相同組換えにより、ターゲッティングベクター中で相当する2つの相同領域間の遺伝子座配列が欠失され、該配列が薬剤耐性マーカーで置き換えられる。好適な実施形態では、2つの相同領域の合計の大きさが約6キロ塩基であり、ターゲッティング遺伝子座の一部と置き換わる第2の領域の大きさは約2キロ塩基である。このターゲッティング法は、原核生物細胞からヒト細胞まで幅広い種に対して概して有用である。使用する各ベクターの特異性は、遺伝子ターゲッティング手順のために選択される遺伝子座、およびこの方法で使用する配列にある。この手法は、全ての有蹄動物(ヤギ(Capra hircus)、ヒツジ(Ovis aries)、ブタ(Sus scrufa)、およびウシ(Bos taurus)が挙げられるがこれらに限定されない)に使用できる。
原則的に、ヒト人工染色体配列(#14fg.、#2fg.および#22fg.)を含むオスおよびメスウシ胎仔繊維芽細胞系を入手、選択および使用して、これらの系統からクローン化仔ウシを作製する。
HACの生殖細胞系伝達は、HACをIgノックアウト動物に導入するため、および動物を生成集団として増殖させるために有用であろう。生殖細胞系を介したHACの増殖で懸念されるのは、減数分裂の間の染色体物質の不完全な対合である。しかし、生殖細胞系伝達は、Tomizukaら(Proc. Natl. Acad. Sci. USA, 97:722, 2000)が示したように、マウスにおいて成功している。
細胞系の最初のスクリーニングから細胞を得る。これらは、ホルスタインであっても、または上で使用したものと異なった系統であってもよい。これにより、集団中で可能な限りの遺伝多様性を維持しながらの交雑が可能になる。次いで、HACを細胞系に導入し、陽性細胞系の選択を行う。選択された細胞系を核移植に使用して、仔ウシを作製する。12月齢から始めて、精液および卵子を回収し、受精させ、レシピエント動物に移す。DNAマーカー分析および核型分析のために細胞サンプルを取る。出生時から始めて、血液サンプルを取り、ヒトIgタンパク質の存在について分析する。
この実験のゴールは、オスHomo H細胞およびクローン化胎仔を作製し、ヒトIgHおよびヒトIgL遺伝子座を含む1つ以上のHAC(HAC#14fg.および#22fg.等)をHomo H細胞に挿入して仔ウシを作製し、免疫感作および親和性成熟に応答するヒトIgの発現を試験することである。これは、以下のように行う。
先に記載したように作製したHemi H細胞から、Homo H細胞を作製する。抗生物質選択または第2の挿入により二重ノックアウトを生成する。先に記載したようにHACをこれらの細胞に導入する。核移植により仔ウシを作製する。HACを保持する仔ウシの試験は、出生後直ぐに開始し、(1)ヒトIg発現、(2)免疫感作への応答、(3)親和性成熟、および(4)子孫へのHACの伝達、について評価することを含む。
ウシ(または他の有蹄動物)は、内因的に曝露されるあらゆる抗原または外因的に投与される抗原に対するトランスジェニック抗血清を産生する。例えば、抗原を有蹄動物に投与して、該抗原(病原体(例えば、細菌、ウイルス、原生動物、酵母または菌類)、腫瘍抗原、受容体、酵素、サイトカイン等の抗原が挙げられる)に反応する所望の抗体を産生させることができる。抗体産生のための病原体の例としては、肝炎ウイルス(例えばC型肝炎)、免疫不全ウイルス(例えばHIV)、ヘルペスウイルス、パルボウイルス、エンテロウイルス、エボラウイルス、狂犬病ウイルス、麻疹ウイルス、ワクシニアウイルス、ストレプトコッカス(例えば、肺炎球菌)、ヘモフィルス(Haemaphilus)(例えば、インフルエンザ菌)、ナイセリア(例えば、髄膜炎菌)、ジフテリア菌、ヘモフィルス(例えば、Haemophilus pertussis)、クロストリジウム(例えば、ボツリヌス菌)、スタフィロコッカス、シュードモナス(例えば、緑膿菌)、および呼吸器合胞体ウイルス(RSV)が挙げられるがこれらに限定されない。
以下の手順を用いて、免疫グロブリン重鎖(μ)遺伝子座の一方の対立遺伝子が相同組換えにより破壊されているウシ繊維芽細胞系を作製した。μ遺伝子座(IgM重鎖遺伝子に対応)のエキソン1〜4を除去し、ネオマイシン耐性遺伝子のコピーで置き換えることにより、IgMノックアウトをもたらすためのDNA構築物を作製した。この構築物を使用して、核移植法で上手く使用されるネオマイシン耐性細胞系を得て、これらの細胞系に由来する胚盤胞をレシピエントウシに移植した。さらに、これらの胚盤胞のいくつかをPCR法を用いて試験して、μ遺伝子座においてターゲッティング挿入が適切に生じたことを確認した。得られた細胞系の一部から核移植法により得た胚盤胞により、ヘテロ接合IgM-KO胎仔を妊娠していることが示された。さらに、単一のIgM重鎖(μ)ノックアウトを含むオスおよびメス細胞系の両方を作製した。これらの細胞系からクローニングされた動物を交配させることにより、μのコピーの両方が不活性化された子孫が得られることが予測される。以下、これらの手順をより詳細に述べる。
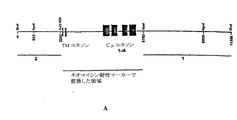
本明細書中に記載した全てのトランスフェクションに使用したDNAは以下のように作製した。4つの主要エキソン(膜貫通ドメインエキソンを除く)であるCH1〜4は、下流(CH4)端部のXhoI制限部位と、上流(CH1)端部のXbaI部位との間に挟まれている。トランスフェクション法に使用した構築物は、XhoI部位の下流にある1.5 kbのゲノム配列、およびXbaI部位の上流にある3.1 Kbのゲノム配列から構成した(図3Dおよび3E)。これらの配列を、マサチューセッツの酪農用ホルスタイン牛集団の1匹から、本明細書に記載するようにして単離した。ネオマイシン耐性マーカーを、3.5 Kb断片に載せてこれらの2つの断片の間に挿入し、元のゲノム配列に由来するCH1〜4を元々含む2.4 KbのDNAと置き換えた。ベクターの骨格はpBluescriptII SK+(Stratagene)であった。8.1 Kbのインサートを精製し、ウシ胎仔繊維芽細胞のトランスフェクションに使用した。この構築物を図3A〜3Cに示す。他の相同領域を含む、および/または別の抗生物質耐性遺伝子を含む他のμノックアウト構築物を、標準的方法を使用して構築し、内因性μ重鎖遺伝子を突然変異させるのに使用してもよい。
市販の試薬であるSuperfectトランスフェクション試薬(Qiagen, Valencia, CA, USA)カタログ番号301305を用いて、胎仔ウシをトランスフェクトした。
上述したように、DNA構築物を含むトランスフェクタントをスクリーニングするためのDNA供給源は、分析する一連の細胞の入った35 mm組織培養皿とした。DNAを以下のように調製し、Lairdらにより発表された手順を採用した(Lairdら, "Simplified mammalian DNA isolation procedure", Nucleic Acids Research, 19:4293)。簡単に言うと、DNAを以下のように調製した。以下の成分で、細胞溶解緩衝液を調製した。すなわち、100 mM Tris-HCl緩衝液(pH 8.5)、5 mM EDTA(pH 8.0)、0.2%ドデシル硫酸ナトリウム、200 mM NaCl、および100μg/mlプロテイナーゼKであった。
細胞系8-1C: 6/8
細胞系10-1C: 2/16
細胞系5-3C: 0/16
系統8-1C:2胎仔、PCRにより1胎仔がターゲッティング挿入について陽性
系統10-1C:1胎仔、PCRにより1胎仔がターゲッティング挿入について陽性
系統5-3C:1胎仔、PCRにより1胎仔がターゲッティング挿入について陰性
K/O細胞系(8-1-C(18))で核移植を行い、8つの胚を作製した。このバッチから得た合計6つの胚を、Trans Ova Genetics(「TOG」、Iowa)の疾患を患っていない3匹のレシピエントに移植した。
ノックアウト胎仔細胞由来のクローン化胚を移植された18匹のレシピエントの妊娠状態を、超音波検査により調べた。結果を以下にまとめる。
ノックアウト細胞由来のクローン化胚(8-1C)を移植した3匹のレシピエントの妊娠状態を調べた。1匹は妊娠しておらず、残りの2匹は1ヵ月後に再確認する必要があった。
40日目においてK/O胚で11匹の妊娠体を得た。60日目で、4匹の生存胎仔を取り出した。4匹全てから細胞系を樹立し、後に使用するために凍結保存した。また、胎仔由来の組織サンプルを回収および急速冷凍(snap freeze)し、PCR/サザンブロット分析のためにHematech分子生物学研究所に送付した。
追加の実験を行い、免疫グロブリン重鎖(μ)およびλ軽鎖が、単独または組み合わせられて、ウシ宿主により産生され得ることを実証した。さらに、これらの実験は、免疫グロブリン鎖が再配列し、ポリクローナル血清が得られたことを実証した。これらの手順において、免疫グロブリン発現遺伝子を、ヒト人工染色体を用いて、ウシ繊維芽細胞に導入した。次いで、繊維芽細胞を核移植のために利用し、胎仔を得て、抗体産生について分析した。これらの手順および結果は以下により詳細に記載する。
先に記載された染色体クローニング系(Kuroiwaら, Nature Biotech. 18:1086-1090, 2000)を用いてヒト人工染色体(HAC)を構築した。簡単に言うと、ΔHACを構築するために、loxP配列がHCF2遺伝子座に組み込まれた、先に報告したヒト第22染色体断片(hChr22)を、テロメア配列挿入による染色体トランケーション(Kuroiwaら, Nucleic Acid Res., 26: 3447-3448, 1998)によりAP000344遺伝子座でトランケートした。次に、AP000344遺伝子座でトランケートされた上記hChr22断片(hCF22)を含むDT40細胞クローンを、安定かつ生殖細胞系伝達性のヒトミニクロモソームSC20ベクターを含むDT40細胞クローン(「Rクローン」と称する)と融合させることにより、細胞ハイブリッドを形成した。SC20ベクターは、loxP配列を、S20断片のRNR2遺伝子座に挿入することにより作製された。SC20断片は、ヒトIg重鎖遺伝子の領域全体を含むヒト第14染色体から誘導される天然の断片である(Tomizukaら, Proc. Natl. Acad. Sci. USA 97:722, 2000)。得られたDT40細胞ハイブリッドは、両方のhChr断片を含んでいた。DT40ハイブリッドを、Creリコンビナーゼ発現ベクターでトランスフェクトして、hCF22とSC20ベクター間のCre/loxP仲介型染色体転座を誘導した。nested PCRを用いて安定トランスフェクタントを分析して、HCF2およびAP000344遺伝子座により規定される2.5メガ塩基のhChr22領域の、SC20ベクターのloxPクローニング部位へのクローニングを確認した。次いで、ΔHACを含むと予想されるPCR陽性細胞を、コードされるグリーン蛍光タンパク質の蛍光に基づくFACSソーティングにより単離した。ソートされた細胞を、蛍光in situハイブリダイゼーション(FISH)分析にもかけて、2.5メガ塩基のhChr22インサートを含むΔHACの存在を確認した。
ウシ胎仔繊維芽細胞を作製するために、45〜60日齢の胎仔を、オスおよびメス両親の系図が連続する3世代について記録された、Trans Ova(Iowa)で飼育された疾患試験済みホルスタイン牛またはジャージー牛から回収した。回収した胎仔は、一次胎仔繊維芽細胞の作製のために、HematechのWorcester分子生物学部門まで、保冷剤(wet ice)に載せて輸送した。到着後、胎仔を、組織培養フード内の非組織培養グレードの100 mmプラスチック製ペトリ皿に移した。滅菌ピンセットおよびハサミを用いて、胚外膜および臍帯を胎仔から取り除いた。胎仔を、新しいプラスチック製ペトリ皿に移した後、頭部、四肢および内臓を取り除いた。除臓した胎仔を、以下からなる約10 mlの胎仔濯ぎ溶液を含む第3のペトリ皿に移した。すなわち、Ca2+およびMg2+を含む125 ml 1×ダルベッコ-PBS(D-PBS)(Gibco-BRL、カタログ番号14040)、0.5 ml酒石酸チロシン(8 mg/ml、Sigma、カタログ番号T-3397)、2 mlペニシリン-ストレプトマイシン(Sigma、カタログ番号P-3539)、および1 mlのファンギゾン(Gibco-BRL、カタログ番号15295-017)(混合し、0.2μmナイロンフィルターユニット[Nalgene、カタログ番号150-0020]を通して濾過した)であった。
ミクロセル仲介型染色体移入(MMCT)(Kuroiwaら Nature Biotech. 18:1086-1090, 2000)を使用して、ΔHACおよびΔΔHACを、DT40細胞ハイブリッドからチャイニーズハムスター卵巣(CHO)細胞に移した。ΔHACを含むCHOクローン(「D15クローン」)を、10%FBS(Gibco)、1 mg/mlのG418および0.2 mg/mlのハイグロマイシンBを補足したF12(Gibco)培地中で、37℃および5%CO2にて、培養した。D15クローンを、12のT25フラスコにおいて増殖させた。集密度が80〜90%に到達したとき、コルセミド(Sigma)を、最終濃度を0.1μg/mlとして培地に添加した。3日後、培地を、10μg/mlのサイトカラシンB(Sigma)を補足したDMEM(Gibco)と交換した。フラスコを、60分間、8,000 rpmにて遠心分離にかけて、ミクロセルを回収した。ミクロセルを、8μm、5μmおよび3μmフィルター(Costar)に通して精製し、その後DMEM培地に再懸濁させた。これらのミクロセルを、以下に記載するように、ウシ繊維芽細胞との融合のために使用した。
核移植手順は、本質的に既に記載されているようにして実施した(Cibelliら, Science 1998: 280:1256-1258)。in vitro成熟卵母細胞を、成熟後18〜20時間(hpm)で除核し、紫外光(UV)下でのビスベンズイミド(Hoechst 33342, Sigma)標識化により、染色体の除去を確認した。これらの細胞質体-ドナー細胞対(couplet)を、2.4 kV/cmの単一の電気パルスを20μ秒間用いて(Electrocell manipulator 200, Genetronics, San Diego, CA)、融合させた。3〜4時間後、合計移植対(couplet)の25%をランダムなサブセットとして取り、移植された核のビスベンズイミド標識化により融合を確認した。30 hpmにて、再構築した卵母細胞および対照を、カルシウムイオノホア(5μM)で4分間(Cal Biochem, San Diego, CA)、そしてACM培養培地に入った10μgシクロへキシミドおよび2.5μgサイトカラシンD(Sigma)で6時間、先に記載されているように(Linら, Mol. Reprod. Dev. 1998:49:298-307;Presicceら, Mol. Reprod. Dev. 1994:38:380-385)活性化した。活性化の後、卵子を、HEPES緩衝化ハムスター胚培養培地(HECM-Hepes)中で5回洗浄し、0.2 mlの胚試験鉱油(Sigma)で覆われた、照射マウス胎仔繊維芽細胞および0.5 mlの胚培養培地を含む4ウェル組織培養プレートに入れて培養した。25〜50の胚を各ウェルに入れ、38.5℃にて、周囲雰囲気中5% CO2下で、インキュベートした。4日目に、10% FCSを、培養培地に添加した。
7および8日目に核移植胚盤胞を、6および7日目に同期化した未出産メスレシピエントにそれぞれ移植した。ルタリス(Lutalyse)(Pharmacia and Upjohn, Kalamazoo, MI)を1回注射した後、発情期を検出することで、レシピエント動物を同期化した。胚移植の30日後および60日後に、レシピエントを、超音波検査により受胎産物の存在について検査し、その後30日ごとに、直腸触診により270日目まで検査した。これらのウシ胎仔におけるHACの保持は、表3にまとめてあり、以下の節においてより詳細に記載する。
SC20断片、ヒト第14染色体断片(Ig重鎖遺伝子を含む「hchr.14fg」)を、上述したのと実質的に同じようにして、胎仔繊維芽細胞に導入した。任意の他の標準的な染色体移入方法を使用して、このHAC、またはヒトIg遺伝子を含む別のHACを、ドナー細胞に挿入してもよい。得られるドナー細胞を、上述したような技術等の標準的な核移植技術において用いて、HACを有するトランスジェニック有蹄動物を作製することができる。
クローン化ΔHAC-トランスジェニックウシ胎仔を、様々な妊娠日数において取り出し、ヒト免疫グロブリン遺伝子座の存在、再配列および発現について分析した。これらの胎仔の1匹の脾臓ならびに非リンパ系組織(肝臓および脳)からRT-PCRにより得られたゲノムDNAおよびcDNAの分析は、ΔHACの存在、再配列および発現を示していた。
ヒト重鎖および軽鎖がΔHAC胎仔に保持されているか否かを決定するため、肝臓DNAをΔHAC胎仔から単離し、PCRにより、ヒト重鎖および軽鎖をコードするゲノムDNAの存在について分析した。
Cμ3およびCμ4の部分を含むmRNA転写産物に特異的なプライマーを使用して、ΔHACが存在し、胎仔#5996のヒトμ遺伝子座の定常領域をコードする転写産物を発現しているか否かを決定した。
ΔHAC胎仔#5996を試験して、免疫グロブリン重鎖遺伝子座再配列に必要な組換え系の発現および活性化に必要な発達プロセスを経たか否かを決定した。この分析のために、標準RT-PCR分析を行い、μ-VH再配列体をコードするmRNA転写産物の存在を検出した。胎仔#5996の脾臓、肝臓および脳から単離したRNAを、プライマー「17L」(5'-ccctcctctttgtgctgtca-3'、配列番号9)および「P9」(5'-caccgtgctctcatcggatg-3'、配列番号10)を用いて、RT-PCRにより分析した。PCR反応混合液を、95℃にて3分間インキュベートし、その後、以下の条件を使用して、変性、アニーリングおよび増幅を35サイクル行った。すなわち、95℃にて1分間、58℃にて1分間および72℃にて2分間である。次いで、反応混合液を72℃にて10分間インキュベートした。
ヒト重鎖遺伝子座の再配列および発現は、CμおよびVH領域の部分を含むDNAのセグメントが増幅することにより実証された。Cμ(Cμ1)およびVH(VH3-30)の部分を含むRNA転写産物に特異的なプライマーを用いて、再配列ヒトCμ-VDJ配列を含むRNA転写産物が存在するか否かを決定した(図8)。
RT-PCR分析は、ΔHAC胎仔#5996における重鎖遺伝子座中でのVDJ再配列をさらに実証するためにも行った。第1の反応のためにプライマーCμ-1(5'-CAGGAGAAAGTGATGGAGTC-3'、配列番号15)、第2の反応のためにプライマーCμ-2(5'-AGGCAGCCAACGGCCACGCT-3'、配列番号16)、そして両方の反応のためにプライマーVH3-30.3(5'-CAGGTGCAGCTGGTGGAGTCTGG-3'、配列番号17)を用いて、nested RT-PCRを行った。RT-PCR反応混合液は、18.9μlの水、3μlの10×Ex Taq緩衝液、4.8μlのdNTP混合液、10 pmolのフォワードプライマー、10 pmolのリバースプライマー、1μlのcDNA、および0.3μlのEx Taqを含んでいた。RT-PCRは、第1の反応について以下の条件下で38サイクル行うことにより行った。すなわち、85℃にて3分間、94℃にて1分間、98℃にて10秒間、65℃にて30秒間、および72℃にて30秒間であった。第2の反応については、38サイクルを以下の条件下で行った。すなわち、プライマーVH3-30.3およびCμ-2(5'-AGGCAGCCAACGGCCACGCT-3'、配列番号16)を用いて、85℃にて3分間、94℃にて1分間、98℃にて10秒間、65℃にて30秒間、および72℃にて30秒間であった。
ΔHAC胎仔#5996の脾臓由来のRNAの逆転写により得たcDNAを、再配列ヒトμに特異的なプライマーで増幅し、アガロースゲル上を移動させた。Cμ1-VH3-30プライマー対での増幅により生成したバンドをゲルから切り出した。増幅したcDNAをバンドから回収し、クローニングした。再配列ヒトμについてPCR陽性である得られたクローンから得たDNAを、精製および配列決定した(図11A)。
ΔΔHACが染色体移入(transchromosomal)されたウシ胎仔繊維芽細胞から誘導されたクローン胎仔を、様々な妊娠日数にあるレシピエントウシから取り出した。HAC担持ヒト免疫グロブリン重鎖およびλ軽鎖遺伝子座の存在および再配列について、胎仔を分析した。これらの組織に由来するゲノムDNAの調査から、一部の胎仔におけるヒト免疫グロブリン重鎖および軽鎖の存在が示された。これらの胎仔の脾臓から誘導されたcDNAの調査から、これらの胎仔の一部における免疫グロブリン重鎖および軽鎖遺伝子座の再配列および発現が示された。FACS分析も、2匹の胎仔における脾臓リンパ球の表面上のヒトλ軽鎖タンパク質の発現を実証した。
ΔΔHAC胎仔が、ヒト重鎖および軽鎖遺伝子座を保持しているか否かを判断するために、58日齢胎仔#5580、57日齢胎仔#5848、ならびに91日齢胎仔#5442Aおよび5442Bの肝臓由来のゲノムDNAに対してPCR分析を行った。重鎖遺伝子座を検出するために使用したPCRプライマーは、VH3-F(5'-AGTGAGATAAGCAGTGGATG-3'、配列番号18)およびVH3-R(5'-CTTGTGCTACTCCCATCACT-3'、配列番号19)、そして軽鎖を検出するために使用したプライマーは、IgL-F(5'-GGAGACCACCAAACCCTCCAAA-3'、配列番号20)およびIgL-R(5'-GAGAGTTGCAGAAGGGGTYGACT-3'、配列番号21)であった。PCR反応混合液は、18.9μlの水、3μlの10×Ex Taq緩衝液、4.8μlのdNTP混合液、10 pmolのフォワードプライマー、10 pmolのリバースプライマー、1μlのゲノムDNA、および0.3μlのEx Taqを含んでいた。以下の通り38サイクルのPCRを行った。すなわち、85℃にて3分間、94℃にて1分間、98℃にて10秒間、56℃にて30秒間、および72℃にて30秒間であった(図13および図14)。
RT-PCRを使用して、ΔΔHAC胎仔#5542Aにおける再配列ヒト重鎖RNA転写産物の発現を検出した。使用したRT-PCRプライマーは、CH3-F3(5'-GGAGACCACCAAACCCTCCAAA-3'、配列番号22)およびCH4-R2(5'-GAGAGTTGCAGAAGGGGTGACT-3'、配列番号23)であった。RT-PCR反応混合液は、18.9μlの水、3μlの10×Ex Taq緩衝液、4.8μlのdNTP混合液、10 pmolのフォワードプライマー、10 pmolのリバースプライマー、1μlのcDNA、および0.3μlのEx Taqを含んでいた。以下の通り、40サイクルのRT-PCRサイクルを行った。すなわち、85℃にて3分間、94℃にて1分間、98℃にて10秒間、60℃にて30秒間、および72℃にて30秒間であった。
RT-PCRを行って、妊娠119日目のΔΔHAC胎仔(胎仔#5868A)の脾臓における再配列ヒト重鎖RNA転写産物の発現を検出した。この分析のために使用したプライマーは、VH30-3(5'-caggtgcagctggtggagtctgg-3'、配列番号24)およびCM-1(5'-caggagaaagtgatggagtc-3'、配列番号25)であった。さらに、プライマー「GAPDH up」(5'-gtcatcatctctgccccttctg-3'、配列番号26)および「GAPDH down」(5'-aacaacttcttgatgtcatcat-3'、配列番号27)を用いて、GAPDH対照転写産物を増幅した。このPCR分析のために、反応混合液を、95℃にて5分間インキュベートし、その後、95℃にて1分間、58℃にて1分間および72℃にて2分間インキュベートすることで変性、アニーリングおよび増幅を複数サイクル行った。次いで、混合液を72℃にて10分間インキュベートした。
ヒトλの部分を含む転写産物の増幅に特異的なプライマーを用いて、再配列ヒトλ軽鎖遺伝子座由来のRNA転写産物を検出した。
再配列ヒトλ軽鎖遺伝子座由来のRNA転写産物を、ΔΔHAC胎仔#5868Aにおいても検出した。この分析のために、ヒトλの部分を含む転写産物の増幅に特異的なプライマーを用いて、再配列ヒトλ遺伝子座の部分をコードする転写産物のΔΔHACコード発現を検出した。プライマーVL1 LEAI(5'-cccccaagcttRccKgStYYcctctcctc-3'、配列番号38)、ならびにプライマーCL1(5'-gggaattcgggtagaagtcactgatcag-3'、配列番号39)、CL2-3(5'-gggaattcgggtagaagtcacttatgag-3'、配列番号40)およびCL7(5'-gggaattcgggtagaagtcacttacgag-3'、配列番号41)の等モル混合液をこの分析のために使用した。このRT-PCR反応のために、反応混合液を、95℃にて5分間インキュベートし、次いで、95℃にて1分間、60℃にて1分間および72℃にて2分間インキュベートすることで変性、アニーリングおよび増幅を複数サイクル行った。次いで、混合液を72℃にて10分間インキュベートした。
胎仔#5442A由来の脾臓サンプルに対して、プライマーCλ1、Cλ2-3およびCλ7の等モル混合液をプライマーVλ1LEA1と共に、またはプライマーVλ3LEA1、Vλ3JLEADおよびVλBACK4の等モル混合液、ならびにプライマーCλ1、Cλ2-3およびCλ7の等モル混合液を用いて、RT-PCR分析を行った。PCR生成物を、CHROMA SPINカラム(CLONETECH)を用いて精製し、pCR2.1 TAクローニングベクター(Invitrogen)に製造元のプロトコールに従ってクローニングした。Dye Terminator配列決定反応(ABI Applied System)を、等モル混合液中のCλ1、Cλ2-3およびCλ7プライマーを用いて実施した。96℃にて1分間、96℃にて10秒間、55℃にて5秒間および60℃にて4分間を25サイクル行った。10μl反応混合液は、BigDye Terminator反応混合液(3μl)、鋳型プラスミド(200 ng)、ならびにCλ1、Cλ2-3およびCλ7プライマー(1.6 pmol)を含んでいた。反応混合液は、ABI 3700シーケンサーを用いて分析した。
ΔΔHAC胎仔#5442Aおよび5442B由来の脾臓リンパ球を、ヒトλ軽鎖およびウシ重鎖タンパク質の発現について分析した。これらの細胞を、フィコエリトリン標識化抗ヒトλ抗体と(図22Cおよび図22D)、FITC標識化抗ウシIgM抗体と(図22Dおよび22H)、または抗体無しで(図22A、図22B、図22Eおよび図22F)、4℃にて20分間反応させた。その後、細胞を、2% FCSを加えたPBSで2回洗浄し、FASCaliburセルソーター上で分析した。電子的にゲートを設定するために、非抗体対照を用いて、抗体と反応した細胞のパーセンテージを計算した。これらのパーセンテージは、各ヒストグラムの下に表示している。胎仔#5442A(図22A〜22D)および胎仔#5442B(図22E−22H)は、ヒトλ軽鎖タンパク質およびウシ重鎖タンパク質の両方を発現した。
異種抗体を発現し、かつ内因性抗体の発現のレベルが低くなったトランスジェニック有蹄動物も作製し得る。機能性内因性重鎖または軽鎖遺伝子の数に対する、機能性異種免疫グロブリン重鎖または軽鎖遺伝子の数を増やすことで、異種抗体を発現するB細胞のパーセンテージが高まるはずである。
上記したμ重鎖(図2A)、λ軽鎖、κ軽鎖、α-(1,3)-ガラクトシルトランスフェラーゼ、プリオンおよび/またはJ鎖ノックアウト構築物を使用してもよい。あるいはまた、以下に記載するピューロマイシン耐性μ重鎖構築物を使用してもよい(図3F)。このノックアウト構築物は、ウシμ重鎖遺伝子座の4つの主要コードエキソンを除去するが、膜貫通ドメインは無傷のまま残して、μ重鎖遺伝子座の不活性化を生じるように設計した。
エレクトロポレーションのために、限られた数の集団倍加を経た1×107ウシ胎仔繊維芽細胞(例えば、実施例2に記載のように、ΔHACまたはΔΔHACトランスジェニック胎仔から得た繊維芽細胞)の単一細胞懸濁液を、1200 rpmにて5分間遠心分離し、0.8 mlの無血清α-MEM培地に再懸濁させた。再懸濁細胞を、0.4 cmエレクトロポレーションキュベット(Invitrogen、カタログ番号P460-50)に移した。次に、制限酵素で直線化した遺伝子ターゲッティングベクターDNAを30μg加え、1 mlピペットを用いてキュベットの中身を混合し、その後室温にて2分間インキュベーションステップを行った。キュベットを、Gene Pulser IIエレクトロポレーションシステム(Biorad)のショックチャンバーに入れ、1000ボルトおよび50μFにてエレクトロポレーションした。キュベットを、組織培養フードに素早く移し、エレクトロポレーションした細胞を、約30 mlの完全繊維芽細胞培地にピペットで入れた。細胞を、30の100 mm組織培養皿(Corning、カタログ番号431079)に等しく分配し、緩やかに回して細胞を均一に行き渡らせ、38.5℃/5% CO2にて16〜24時間インキュベートした。吸引により培地を除去し、選択した選択薬剤を含む完全繊維芽細胞培地で置き換えた。培地は2日ごとに替え、合計7〜14日間の間続けた。薬剤選択プロセスの間、相当するプレートを目でモニターして、細胞死およびコロニー形成を調べた。遺伝子ターゲッティングベクターの不在下でエレクトロポレーションした繊維芽細胞を含み、薬剤選択プロセスの間にコロニーを生成しないはずの陰性対照プレートを用意した。
薬剤耐性コロニーは、薬剤選択ステップ(通常、7〜14日間)の終了後、肉眼で目に見え、すぐにも増殖用の48ウェル組織培養プレートに移せる状態である。この移すプロセスを助けるために、個々のコロニーについて、組織培養プレートの底に色ペン(Sharpie)で丸をつけた。コロニーを含む組織培養プレートを、1×D-PBS(Ca2+およびMg2+を含まない)で2回洗浄し、次いで、1プレートにつき細胞解離緩衝液の1:5希釈液を5 ml添加した。室温にて3〜5分間のインキュベーションステップの後、個々のコロニーが組織培養皿の底から剥がれ始める。コロニーが剥離する前に、P200ピペットメン(pipetmen)およびエアロゾルバリアーピペットチップ(200または250μl)を用いて、48ウェル組織培養プレートの単一のウェルに個々に移した。移した後、上下にピペッティングすることによりコロニーを完全に解離させ、1mlの完全繊維芽細胞培地を添加した。細胞が確実に薬剤耐性となるように、48ウェルステージにわたり薬剤選択を続けた。移したコロニーを、38.5℃/5% CO2にて培養し、倒立顕微鏡を用いて目でモニターした。2〜7日後、集密状態に達したウェルを、1×D-PBS(Ca2+およびMg2+を含まない)で2回洗浄し、0.2 mlの細胞解離緩衝液を添加することでウェルの底から剥がし、その後室温にて5分間インキュベーションステップを行った。剥離後、P1000ピペットメンおよびエアロゾルピペットチップ(1000μl)を用いて、上下にピペッティングすることにより細胞をさらに解離させる。解離した繊維芽細胞の約75%を、24ウェル組織培養プレートの個々のウェルに移し、後続するPCR分析のためにさらに増殖させ、残りの25%は、第2の24ウェルプレートの単一のウェルに移して増殖させ、最終的に体細胞核移植実験に使用する。元の細胞の75%を含むプレート中の細胞が集密状態近くまで増殖したら、そのクローンからDNAを単離して遺伝子分析する。
遺伝子分析のためにDNAを単離するのに使用する手順は、Lairdら, Nucleic Acids Research, 1991, Volume 19, No. 15を応用する。具体的には、24ウェルプレートの1つのウェルにおいて特定のクローンがほぼ集密状態になったら、そのウェルから培養培地を吸引し、付着細胞をPBSで2回洗浄する。PBSを吸引除去し、0.2 ml緩衝液と置き換えて、細胞を溶解し、単離しようとするDNA由来の余分なタンパク質を消化する。この緩衝液は、100 mM Tris-HCl(pH 8.5)、5 mM EDTA、0.2 % SDS、200 mM NaClおよび100μg/mlプロテイナーゼKからなる。24ウェルプレートを組織培養インキュベーターに、最低3時間戻して、DNAを放出させ、タンパク質を消化させる。この手順の粘性産物を、1.5 ml微量遠心管に移し、0.2 mlのイソプロパノールを添加して、DNAを沈降させる。遠心分離により沈降物を回収し、70%エタノールでDNAペレットを濯ぎ、通気乾燥後、10 mM Tris(pH 8)および1 mM EDTAを含む25〜50μlの緩衝液にペレットを再懸濁させる。このDNAを、クローンのPCR分析に使用する。
2つの異なる手法(両方ともポリメラーゼ連鎖反応(PCR)を採用する)を用いてクローンをスクリーニングする。本節において記載する全ての手法が、任意の他の遺伝子をターゲッティングするために応用でき、唯一の違いは遺伝子分析に使用するプライマーの配列である。
免疫グロブリン遺伝子が不活性化されている選択した繊維芽細胞を、実施例2に記載したように核移植に用いて、内因性免疫グロブリン遺伝子に突然変異を含み、異種免疫グロブリン遺伝子をコードするHACを含むトランスジェニック有蹄動物を作製することができる。あるいはまた、標準的な方法を用いて核移植を行い、選択したトランスジェニック繊維芽細胞から得た核またはクロマチン塊(すなわち、膜に覆われていない1つ以上の染色体)を、除核した卵母細胞(米国特許出願第60/258,151号;2000年12月22日出願)に挿入してもよい。これらの方法は、内因性α-(1,3)-ガラクトシルトランスフェラーゼ、プリオンおよび/またはJ鎖核酸が突然変異された細胞にも使用できる。
所望であれば、細胞(例えば、胎仔繊維芽細胞)を、第1ラウンドの核移植で生成したトランスジェニック有蹄動物から得てもよい。さらなるラウンドの遺伝子ターゲッティングを上述のように行って、第1のラウンドのターゲッティングで不活性化された遺伝子の第2の対立遺伝子を不活性化することができる。あるいはまた、別の免疫グロブリン(例えば、μ重鎖、λ軽鎖、κ軽鎖もしくはJ鎖)、α-(1,3)-ガラクトシルトランスフェラーゼ、またはプリオン遺伝子を、本ラウンドのターゲッティングで不活性化してもよい。この第2ラウンドのターゲッティングのためには、高濃度の抗生物質を使用するか、または異なる抗生物質耐性マーカーを有するノックアウト構築物を使用し得る。抗生物質耐性細胞は、上述したように選択できる。選択された細胞を上述したように第2ラウンドの核移植に使用して、例えば、内因性免疫グロブリン遺伝子に2つの突然変異を含み、そして異種免疫グロブリン遺伝子をコードするHACを含むトランスジェニック有蹄動物を作製することができる。あるいはまた、選択した抗生物質耐性細胞をまず処理して、以下に記載するようにG1期細胞を単離して、それを第2ラウンドの核移植に使用してもよい。
α-1,3-ガラクトシルトランスフェラーゼ遺伝子座の一方の対立遺伝子が突然変異したウシ繊維芽細胞系を、相同組換えにより作製した。α-ガラクトシルトランスフェラーゼノックアウト細胞を作製するためのDNA構築物を使用し、ピューロマイシン耐性遺伝子(puro、実施例3に記載)および転写終結カセット(STOP)を、触媒ドメインを含むエキソン9に挿入することにより、機能性完全長α-ガラクトシルトランスフェラーゼmRNAの転写を防いだ。従って、得られる未熟α-ガラクトシルトランスフェラーゼ転写産物は触媒ドメインを欠く。DNA構築物(すなわち、α-ガラクトシルトランスフェラーゼKOベクター)を、3つの独立したウシ繊維芽細胞系にエレクトロポレーションし、次いで、ピューロマイシン耐性コロニーを単離した。PCR分析によれば、いくつかのコロニーでエキソン9領域において相同組換えが生じた。こうして、α1,3-ガラクトシルトランスフェラーゼ遺伝子座の一方の対立遺伝子が突然変異されたウシ繊維芽細胞系が生成した。所望であれば、同じノックアウトベクターおよびより高濃度の抗生物質を(ホモ接合ノックアウト細胞を選択するために)用いるか、または別のノックアウトベクターを異なる抗生物質耐性遺伝子と共に用いて、第2の対立遺伝子を突然変異させてもよい。本方法を他の有蹄動物由来の細胞に適用し、本明細書に記載する核移植法に使用するトランスジェニック細胞を作製して、本発明のトランスジェニック有蹄動物を作製してもよい。
α-1,3-ガラクトシルトランスフェラーゼKOベクターを以下のように作製した(図23)。α-1,3-ガラクトシルトランスフェラーゼ遺伝子のエキソン9周辺のゲノムDNAを単離するために、以下のプライマー対5'-gatgatgtctccaggatgcc-3'(配列番号61)および5'-gacaagcttaatatccgcagg-3'(配列番号62)を用いたPCRにより、DNAプローブを増幅した。このプローブを用いて、ウシゲノムλファージライブラリーをスクリーニングし、7つの陽性λファージクローンを同定した。オスシャロレイ牛繊維芽細胞由来のDNAを含む1つのクローンを、制限マッピングによりさらに分析した。エキソン9を含むNotI−XhoIゲノム断片をpBluescriptII SK(-)にサブクローニングし、puroおよびSTOPカセットの両方を、触媒ドメインの5'側にあるNotI−XhoIゲノム断片のAviI部位に挿入した。ジフテリアトキシン遺伝子(DT-A, Gibco)もベクター構築物に付加して、ターゲッティングカセットが非相同的に組み込まれた細胞を殺した。
標準的なエレクトロポレーションプロトコルを用いて、以下のように3つの胎仔繊維芽細胞系(2つはオスジャージー牛由来のもの、1つはメスジャージー牛のもの)をトランスフェクトした。ウシ胎仔繊維芽細胞を培養するために使用した培地は、500 mlαMEM(Gibco, 12561-049)、50 mlウシ胎仔血清(Hy-Clone#ABL13080)、5 mlペニシリン-ストレプトマイシン(SIGMA)、および1 ml 2-メルカプトエタノール(Gibco/BRL #21985-023)を含んでいた。トランスフェクション前日、顕微鏡検査で判断する目標集密度を80〜100%として、細胞をT175組織培養フラスコに播種した。トランスフェクション当日、約107個のウシ繊維芽細胞をトリプシン処理し、αMEM培地で1回洗浄した。細胞を800μlのα-MEMに再懸濁した後、30μgのDNAを細胞懸濁液に添加し、ピペッティングによりよく混ぜた。細胞-DNA懸濁液を、エレクトロポレーションキュベットに移し、1,000Vおよび50μFにてエレクトロポレーションした。その後、エレクトロポレーションした細胞を、血清を補足したα-MEM培地と共に、20の24ウェルプレート上に植え付けた。48時間の培養後、培地を、1μg/mlのピューロマイシンを含む培地と置き換え、細胞を2〜3週間培養してピューロマイシン耐性細胞を選択した。選択後、100%に近い集密度に達した全てのコロニーを選び、ゲノムDNAをコロニーから抽出して、PCRにより所望の相同組換え事象についてスクリーニングした。
上述したように、PUREGENE DNA単離キット(Gentra SYSTEMS)を製造元のプロトコールに従って使用して、ゲノムDNAを各24ウェルから個別に抽出した。各ゲノムDNAサンプルを、20μlの10 mM Tris-Cl(pH 8.0)および1 mM EDTA(EDTA)に再懸濁させた。以下のプライマー対5'-aagaagagaaaggtagaagaccccaaggac-3'(配列番号63)および5'-cctgggtatagacaggtgggtattgtgc-3'(配列番号64)を使用して、PCRによるスクリーニングを行った。一方のプライマーの配列は、α-1,3-ガラクトシルトランスフェラーゼKOベクター中に位置し、他方のプライマーの配列はターゲッティング内因性遺伝子座中の組込まれたベクターのすぐ外側に位置する(図23)。従って、予想されるPCR生成物は、KOベクターが相同組換えにより標的遺伝子座に組み込まれた場合にのみ検出されるはずである。
細胞のゲノム中に存在する標的配列の特異的な置き換えのためにアデノ随伴ウイルス(AAV)を使用することができる(Inoueら, Mol. Ther. 3(4):526-530, 2001;Hirataら, J. Virol. 74(10):16536-42, 2000;Inoueら, J. Virol. 73(9):7376-80, 1999;およびRussellら, Nat. Genet. 18(4):325-30, 1998)。遺伝子ターゲッティング率は、より慣用的な遺伝子ターゲッティング手法と比べて非常に効率的である。AAVは、幅広い宿主範囲および組織特異性(ウシおよびヒト皮膚繊維芽細胞の両方に対する特異性を含む)を有する。従って、AAVを用いて、内因性免疫グロブリン(例えば、μ重鎖、λ軽鎖、κ軽鎖もしくはJ鎖)、α-(1,3)-ガラクトシルトランスフェラーゼまたはプリオン遺伝子中に1つ以上の突然変異を含むトランスジェニック有蹄動物細胞を作製することができる。次に、これらのトランスジェニック細胞を、本明細書に記載する核移植方法に用いて、本発明のトランスジェニック有蹄動物を作製することができる。
AAV構築物は、外来配列の単純な挿入、または内因性配列のAAVベクター中に存在する新しい配列による置き換えにより、遺伝子を破壊することができる。図24は、ウシ免疫グロブリン重鎖μ定常領域の4つのコードエキソンの全てが、2822塩基対のBamHI-XhoI断片上に存在するAAV構築物を示す。市販のベクターpMC1Neoに存在するネオマイシン耐性マーカーを含む1.16 Kb断片を、ホルスタイン牛由来のμ重鎖遺伝子座のエキソン4に存在するSacII部位に挿入した。この遺伝子座は、実施例1に記載したノックアウトベクターを作製するために単離されたファージクローンに含まれているものであった。AAVベクターを作製するために、μ重鎖遺伝子座中のSacII部位を充填して平滑末端とし、次いでこれを平滑末端SalIリンカー(New England Biolabs)にライゲートした。その後、ネオマイシン耐性遺伝子を含むpMC1NeoのXhoI断片を、SalIリンカーを介して、この遺伝子座に付加されたSalI部位にライゲートした。このライゲーションは、XhoIおよびSalI制限部位が互換性のある端部を有するために実施可能である。このノックアウトベクターにより、内因性μ重鎖遺伝子へのネオマイシン耐性遺伝子の破壊的挿入が生じ、μ重鎖遺伝子が不活性化される。この遺伝子不活性化は、内因性μ遺伝子座の領域を欠失させることなく生じる。
メスジャージー牛由来の繊維芽細胞を、48ウェル組織培養プレートの1つのウェルに、1ウェルにつき40,000細胞で播種し、完全培地中で38.5℃および5%CO2下にて、細胞がウェルの底表面に付着するまで培養した。細胞が付着したら、培地を除去し、図24に示すベクターの感染多重度(MOI)を500〜20,000粒子/細胞として、AAV粒子を含む0.2 mlの新しい培地と置き換えた。MOIは、得られるコロニー数および薬剤選択期間中のこのコロニーの間隔を判断した試験的な実験に基づき選択した。プレートを一晩インキュベートした。このインキュベーションの後、形質導入ウェルをカルシウムおよびマグネシウムを含まないPBSで濯ぎ、トリプシンまたは上記細胞解離緩衝液のいずれかを使用してウェルから剥離した。剥離した細胞をゆるやかにピペッティングすることで均一な細胞懸濁液を得、ウェル中の細胞を10の100 mm組織培養皿に再度分配した。皿を完全培地と共に一晩インキュベートした。
AAV粒子が形質導入された薬剤耐性クローン由来のDNAサンプルを、PCR分析を用いて、ベクターの適切なターゲッティングについてスクリーニングした。このスクリーニング法では、薬剤選択マーカーをコードするDNA内にアニールする一方のプライマー、および標的遺伝子座内にアニールするが、AAVターゲッティング粒子に存在する配列の外側にアニールする別のプライマーを使用する。PCR生成物は、AAVターゲッティングDNAが、内因性ゲノムの所望の位置に組み込まれた場合にのみ検出される。
以上の説明から、本明細書に記載した本発明を変更および改変して様々な用途および状況に応用できることが明らかであろう。このような実施形態も本発明の範囲内にある。
Claims (68)
- 再配列を経て1つ以上のヒト免疫グロブリン(Ig)分子を発現するヒト免疫グロブリン(Ig)遺伝子の全体、またはその一部、をコードする1つ以上の核酸を含み、かつウシ、ヒツジおよびヤギからなる群より選択されることを特徴とするトランスジェニック有蹄動物。
- 前記核酸がヒトのものである、請求項1に記載の有蹄動物。
- ヒト抗体をコードする核酸を含む、請求項1に記載の有蹄動物。
- 前記抗体がポリクローナル抗体である、請求項3に記載の有蹄動物。
- 前記Ig鎖または抗体が血清および/または乳汁中に発現される、請求項1に記載の有蹄動物。
- 前記核酸が染色体断片に含まれている、請求項1に記載の有蹄動物。
- 前記染色体断片が人工染色体ΔHAC(FERM BP-7582)である、請求項6に記載の有蹄動物。
- 前記染色体断片が人工染色体ΔΔHAC(FERM BP-7581)である、請求項6に記載の有蹄動物。
- 前記核酸が有蹄動物細胞中で宿主染色体とは独立に維持される、請求項6に記載の有蹄動物。
- 前記核酸が当該有蹄動物の染色体に組み込まれている、請求項1に記載の有蹄動物。
- 前記核酸が、V遺伝子セグメントをコードする全ての核酸セグメントが、1つ以上のヌクレオチドにより、J遺伝子セグメントをコードする全ての核酸セグメントから分離している未再配列抗体軽鎖核酸セグメントを含む、請求項3に記載の有蹄動物。
- 前記核酸が、(i)V遺伝子セグメントをコードする全ての核酸セグメントが、1つ以上のヌクレオチドにより、D遺伝子セグメントをコードする全ての核酸セグメントから分離しているか、かつ/または、(ii)D遺伝子セグメントをコードする全ての核酸セグメントが、1つ以上のヌクレオチドにより、J遺伝子セグメントをコードする全ての核酸セグメントから分離している、未再配列抗体重鎖核酸セグメントを含む、請求項3に記載の有蹄動物。
- ウシである、請求項1〜12のいずれか1項に記載の有蹄動物。
- 内因性Ig遺伝子の発現を低減する突然変異を該内因性Ig遺伝子上に含む、請求項1〜13のいずれか1項に記載の有蹄動物。
- 内因性抗体の発現を低減する突然変異を該内因性抗体をコードする遺伝子上に含む、請求項14に記載の有蹄動物。
- 前記突然変異が機能性IgM重鎖の発現を低減する、請求項14に記載の有蹄動物。
- 前記突然変異が機能性IgM重鎖の発現を喪失させる、請求項16に記載の有蹄動物。
- 前記突然変異が機能性Ig軽鎖の発現を低減する、請求項14に記載の有蹄動物。
- 前記突然変異が機能性Ig軽鎖の発現を喪失させる、請求項18に記載の有蹄動物。
- 前記突然変異が機能性IgM重鎖および機能性Ig軽鎖の発現を低減する、請求項14に記載の有蹄動物。
- 前記突然変異が機能性IgM重鎖および機能性Ig軽鎖の発現を喪失させる、請求項20に記載の有蹄動物。
- 請求項1〜21のいずれか1項に記載の有蹄動物に由来する細胞であって、再配列を経て1つ以上のヒトIg分子を発現することが可能なヒトIg遺伝子の全体、またはその一部、をコードする1つ以上の核酸を含む、前記細胞。
- 前記核酸がヒト抗体をコードする、請求項22に記載の細胞。
- 前記核酸が染色体断片に含まれている、請求項22に記載の細胞。
- 前記染色体断片が人工染色体ΔHAC(FERM BP-7582)またはΔΔHAC(FERM BP-7581)である、請求項24に記載の細胞。
- 前記核酸が有蹄動物細胞中で宿主染色体とは独立に維持される、請求項24に記載の細胞。
- 前記核酸が当該細胞の染色体に組み込まれている、請求項22に記載の細胞。
- 前記核酸がヒトのものである、請求項22に記載の細胞。
- 当該細胞が胎仔繊維芽細胞である、請求項22〜28のいずれか1項に記載の細胞。
- 当該細胞がB細胞である、請求項22〜28のいずれか1項に記載の細胞。
- 当該細胞がB細胞であって、再配列されたヒトIg分子を発現するものである、請求項22〜28のいずれか1項に記載の細胞。
- 前記有蹄動物がウシである、請求項22〜31のいずれか1項に記載の細胞。
- 内因性Ig重鎖および/もしくは軽鎖の発現を低減する突然変異を該内因性Ig重鎖および/もしくは軽鎖をコードする核酸上に含む、請求項22〜32のいずれか1項に記載の細胞。
- 前記突然変異がナンセンス変異または欠失変異である、請求項33に記載の細胞。
- 請求項31に記載の細胞と骨髄腫細胞との融合により形成されたハイブリドーマ。
- (a)1つ以上の目的の抗原を、ヒト抗体遺伝子座をコードする核酸を有する請求項1〜21のいずれか1項に記載の有蹄動物に投与するステップであって、該遺伝子座中の核酸セグメントが再配列を経て該抗原に特異的な抗体の産生をもたらす、ステップ;および
(b)該有蹄動物から該抗体を回収するステップ
を含む、抗体の製造方法。 - 前記核酸がヒトのものである、請求項36に記載の方法。
- 前記抗体の軽鎖がヒト核酸によりコードされている、請求項36に記載の方法。
- 前記抗体の重鎖がヒト核酸によりコードされている、請求項36に記載の方法。
- 前記有蹄動物がウシである、請求項36に記載の方法。
- 前記抗体がモノクローナルである、請求項36に記載の方法。
- 前記抗体がポリクローナルである、請求項36に記載の方法。
- 前記抗体を前記有蹄動物の血清または乳汁から回収する、請求項36〜42のいずれか1項に記載の方法。
- ヒト抗体遺伝子座をコードする核酸を有する請求項1〜21のいずれか1項に記載の有蹄動物から、ヒト抗体を回収することを含み、該遺伝子座中の核酸セグメントが再配列を経てヒト抗体の産生をもたらす、抗体の製造方法。
- 前記核酸がヒトのものである、請求項44に記載の方法。
- 前記抗体の軽鎖がヒト核酸によりコードされている、請求項45に記載の方法。
- 前記抗体の重鎖がヒト核酸によりコードされている、請求項45に記載の方法。
- 前記有蹄動物がウシである、請求項44に記載の方法。
- 前記抗体がモノクローナルである、請求項44に記載の方法。
- 前記抗体がポリクローナルである、請求項44に記載の方法。
- 前記抗体を前記有蹄動物の血清または乳汁から回収する、請求項44〜50のいずれか1項に記載の方法。
- (a)有蹄動物由来の細胞、該細胞由来のクロマチン塊、または該細胞由来の核を、卵母細胞に挿入するステップであって、該細胞が、再配列を経て1つ以上のヒトIg分子を発現するヒトIg遺伝子の全体、またはその一部、をコードする1つ以上の核酸を含み、かつ内因性抗体重鎖および/もしくは軽鎖の発現を低減する第1の突然変異を該内因性抗体重鎖および/もしくは軽鎖核酸上に含む、前記ステップ;ならびに
(b)該卵母細胞または該卵母細胞から形成された胚を、該卵母細胞または該胚から胎仔を発生させる条件下で宿主有蹄動物の子宮に移入するステップ
を含む、請求項1〜21のいずれか1項に記載のトランスジェニック有蹄動物の作製方法。 - (a)1つ以上のヒト核酸を有する有蹄動物由来の細胞、該細胞由来のクロマチン塊、または該細胞由来の核を卵母細胞に挿入するステップであって、該ヒト核酸はヒトIg遺伝子の全体または一部をコードし、該遺伝子はB細胞中で再配列を経て1つ以上のヒトIg分子を発現することができる、ステップ;および
(b)該卵母細胞または該卵母細胞から形成される胚を、該卵母細胞または該胚から胎仔が発生する条件下で宿主有蹄動物の子宮に導入するステップ
を含む、請求項1〜21のいずれか1項に記載のトランスジェニック有蹄動物の作製方法。 - 前記胎仔が生存能力のある仔に発育する、請求項52または53に記載の方法。
- 選択可能マーカーをコードする核酸に作用可能に連結され、かつ前記内因性抗体重鎖または軽鎖核酸と少なくとも98%の配列同一性を有する1つ以上の核酸に作用可能に連結されたプロモーターを有するカセットを含む核酸を該細胞に挿入することにより、該カセットが該抗体重鎖または軽鎖核酸の一方の内因性対立遺伝子に組み込まれる方法により、前記細胞を調製する、請求項52に記載の方法。
- (a)第1の選択可能マーカーをコードする核酸に作用可能に連結され、かつ前記抗体重鎖または軽鎖核酸と少なくとも98%の配列同一性を有する第1の核酸に作用可能に連結された第1のプロモーターを有する第1のカセットを含む核酸を該細胞に挿入することにより、該第1のカセットが該抗体重鎖または軽鎖核酸の第1の内因性対立遺伝子に組み込まれて、第1のトランスジェニック細胞を生成するステップ;および
(b)該第1の選択可能マーカーとは異なる第2の選択可能マーカーをコードする核酸に作用可能に連結され、かつ該抗体重鎖または軽鎖核酸と少なくとも98%の配列同一性を有する第2の核酸に作用可能に連結された第2のプロモーターを有する第2のカセットを含む核酸を該第1のトランスジェニック細胞に挿入することにより、該第2のカセットが該抗体重鎖または軽鎖核酸の第2の内因性対立遺伝子に組み込まれて、第2のトランスジェニック細胞を生成するステップ
を含む方法により、前記細胞を製造する、請求項52に記載の方法。 - さらに、
(c)前記胚、前記胎仔、または該胎仔から生成した仔から、細胞を単離するステップ;
(d)該細胞中の内因性抗体重鎖および/または軽鎖核酸に第2の突然変異を導入するステップ;
(e)該細胞、該細胞由来のクロマチン塊、または該細胞由来の核を、卵母細胞に挿入するステップ;ならびに
(f)該卵母細胞または該卵母細胞から形成された胚を、該卵母細胞または該胚から胎仔が発生する条件下で宿主有蹄動物の子宮に移入するステップ
を含む、請求項52に記載の方法。 - 前記核酸がヒト抗体をコードする、請求項52または53に記載の方法。
- 前記核酸が染色体断片に含まれている、請求項52または53に記載の方法。
- 前記染色体断片が人工染色体ΔHAC(FERM BP-7582)またはΔΔHAC(FERM BP-7581)である、請求項59に記載の方法。
- 前記核酸が有蹄動物細胞中で宿主染色体とは独立に維持される、請求項59に記載の方法。
- 前記核酸が前記細胞の染色体に組み込まれる、請求項52または53に記載の方法。
- 前記有蹄動物がウシである、請求項52または53に記載の方法。
- 前記抗体がポリクローナル抗体である、請求項58に記載の方法。
- 前記免疫グロブリン鎖または抗体が血清および/または乳汁中に発現される、請求項52または53に記載の方法。
- 請求項1〜21のいずれか1項に記載の有蹄動物を所望の抗原で免疫して、抗血清を採取することを含む、ポリクローナルヒト免疫グロブリン(Ig)を含む有蹄動物抗血清を製造する方法。
- 請求項1〜21のいずれか1項に記載の有蹄動物を所望の抗原で免疫して、乳汁を採取することを含む、ポリクローナルヒトIgを含む有蹄動物乳汁を製造する方法。
- 前記有蹄動物がウシである、請求項66または67に記載の方法。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US71418500A | 2000-11-17 | 2000-11-17 | |
| US25645800P | 2000-12-20 | 2000-12-20 | |
| US31162501P | 2001-08-09 | 2001-08-09 | |
| PCT/US2001/043128 WO2002070648A2 (en) | 2000-11-17 | 2001-11-16 | Expression of xenogenous (human) immunoglobulins in cloned, transgenic ungulates |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005136675A Division JP2005224250A (ja) | 2000-11-17 | 2005-05-09 | クローン化トランスジェニック有蹄動物における異種(ヒト)免疫グロブリンの発現 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2005504507A JP2005504507A (ja) | 2005-02-17 |
| JP2005504507A5 JP2005504507A5 (ja) | 2005-09-02 |
| JP3797974B2 true JP3797974B2 (ja) | 2006-07-19 |
Family
ID=27400960
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002570676A Expired - Fee Related JP3797974B2 (ja) | 2000-11-17 | 2001-11-16 | クローン化トランスジェニック有蹄動物における異種(ヒト)免疫グロブリンの発現 |
| JP2005136675A Pending JP2005224250A (ja) | 2000-11-17 | 2005-05-09 | クローン化トランスジェニック有蹄動物における異種(ヒト)免疫グロブリンの発現 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005136675A Pending JP2005224250A (ja) | 2000-11-17 | 2005-05-09 | クローン化トランスジェニック有蹄動物における異種(ヒト)免疫グロブリンの発現 |
Country Status (11)
| Country | Link |
|---|---|
| EP (2) | EP2042594B1 (ja) |
| JP (2) | JP3797974B2 (ja) |
| KR (2) | KR100882029B1 (ja) |
| CN (1) | CN1486365B (ja) |
| AT (2) | ATE503012T1 (ja) |
| AU (2) | AU2001297523B2 (ja) |
| CA (1) | CA2428053C (ja) |
| DE (2) | DE60138832D1 (ja) |
| MX (1) | MXPA03004313A (ja) |
| NZ (1) | NZ525664A (ja) |
| WO (1) | WO2002070648A2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018079857A1 (ja) | 2016-10-31 | 2018-05-03 | 国立大学法人鳥取大学 | ヒト抗体産生非ヒト動物及びそれを用いたヒト抗体作製法 |
Families Citing this family (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0110029D0 (en) | 2001-04-24 | 2001-06-13 | Grosveld Frank | Transgenic animal |
| IL165088A0 (en) * | 2002-05-17 | 2005-12-18 | Kirin Brewery | Transgenic ungulates capable of human antibody production |
| AU2003271095B2 (en) * | 2002-10-04 | 2009-06-11 | Kirin Beer Kabushiki Kaisha | Human artificial chromosome (HAC) vector |
| AU2003290689A1 (en) * | 2002-11-08 | 2004-06-03 | Kyowa Hakko Kirin Co., Ltd. | Transgenic ungulates having reduced prion protein activity and uses thereof |
| US7273610B2 (en) | 2003-08-14 | 2007-09-25 | Dyax Corp. | Endotheliase-2 ligands |
| MXPA06003553A (es) * | 2003-09-30 | 2007-02-02 | Sterrenbeld Biotechnologie North America Inc | Un proceso para producir proteina exogena en la leche de mamiferos transgenicos y un proceso para purificar proteinas de la leche. |
| US7420099B2 (en) | 2004-04-22 | 2008-09-02 | Kirin Holdings Kabushiki Kaisha | Transgenic animals and uses thereof |
| US20080026457A1 (en) | 2004-10-22 | 2008-01-31 | Kevin Wells | Ungulates with genetically modified immune systems |
| US20060130157A1 (en) * | 2004-10-22 | 2006-06-15 | Kevin Wells | Ungulates with genetically modified immune systems |
| US20090041783A1 (en) | 2005-04-28 | 2009-02-12 | Mochida Pharmaceutical Co., Ltd. | Anti-platelet membrane glycoprotein vi monoclonal antibody |
| ES2548377T3 (es) | 2008-10-27 | 2015-10-16 | Revivicor, Inc. | Ungulados inmunodeprimidos |
| RU2011129459A (ru) * | 2008-12-18 | 2013-01-27 | Эрасмус Юниверсити Медикал Сентр Роттердам | Трансгенные животные (не человек), экспрессирующие гуманизированные антитела, и их применение |
| CN102638971B (zh) | 2009-07-08 | 2015-10-07 | 科马布有限公司 | 动物模型及治疗分子 |
| US9445581B2 (en) | 2012-03-28 | 2016-09-20 | Kymab Limited | Animal models and therapeutic molecules |
| CA2781159A1 (en) | 2009-11-17 | 2011-05-26 | Kyowa Hakko Kirin Co., Ltd. | Human artificial chromosome vector |
| JP2014533930A (ja) | 2011-09-19 | 2014-12-18 | カイマブ・リミテッド | 免疫グロブリン遺伝子多様性の操作およびマルチ抗体治療薬 |
| JP6156144B2 (ja) | 2011-09-21 | 2017-07-05 | 富士レビオ株式会社 | 親和性複合体に対する抗体 |
| EP2761008A1 (en) | 2011-09-26 | 2014-08-06 | Kymab Limited | Chimaeric surrogate light chains (slc) comprising human vpreb |
| US9253965B2 (en) | 2012-03-28 | 2016-02-09 | Kymab Limited | Animal models and therapeutic molecules |
| EP3816284A1 (en) | 2011-12-22 | 2021-05-05 | F. Hoffmann-La Roche AG | Expression vector for antibody production in eukaryotic cells |
| US10251377B2 (en) | 2012-03-28 | 2019-04-09 | Kymab Limited | Transgenic non-human vertebrate for the expression of class-switched, fully human, antibodies |
| GB2502127A (en) | 2012-05-17 | 2013-11-20 | Kymab Ltd | Multivalent antibodies and in vivo methods for their production |
| EP2880055B1 (en) | 2012-08-03 | 2017-06-28 | Sab, Llc | Complex chromosome engineering for production of human antibodies in transgenic animals |
| US9788534B2 (en) | 2013-03-18 | 2017-10-17 | Kymab Limited | Animal models and therapeutic molecules |
| US9783618B2 (en) | 2013-05-01 | 2017-10-10 | Kymab Limited | Manipulation of immunoglobulin gene diversity and multi-antibody therapeutics |
| US9783593B2 (en) | 2013-05-02 | 2017-10-10 | Kymab Limited | Antibodies, variable domains and chains tailored for human use |
| US11707056B2 (en) | 2013-05-02 | 2023-07-25 | Kymab Limited | Animals, repertoires and methods |
| SG10201802295XA (en) | 2013-10-01 | 2018-04-27 | Kymab Ltd | Animal Models and Therapeutic Molecules |
| EP3229586A4 (en) | 2014-12-10 | 2018-10-24 | Regents of the University of Minnesota | Genetically modified cells, tissues, and organs for treating disease |
| EP3468356A4 (en) * | 2016-06-14 | 2020-02-26 | Regents Of The University Of Minnesota | GENETICALLY MODIFIED CELLS, TISSUES AND ORGANS FOR THE TREATMENT OF A DISEASE |
Family Cites Families (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE158021T1 (de) | 1990-08-29 | 1997-09-15 | Genpharm Int | Produktion und nützung nicht-menschliche transgentiere zur produktion heterologe antikörper |
| US5633425A (en) | 1990-08-29 | 1997-05-27 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5877397A (en) | 1990-08-29 | 1999-03-02 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US6300129B1 (en) | 1990-08-29 | 2001-10-09 | Genpharm International | Transgenic non-human animals for producing heterologous antibodies |
| US5545806A (en) | 1990-08-29 | 1996-08-13 | Genpharm International, Inc. | Ransgenic non-human animals for producing heterologous antibodies |
| US5661016A (en) | 1990-08-29 | 1997-08-26 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US5789650A (en) * | 1990-08-29 | 1998-08-04 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US5814318A (en) | 1990-08-29 | 1998-09-29 | Genpharm International Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US5625126A (en) * | 1990-08-29 | 1997-04-29 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US5874299A (en) | 1990-08-29 | 1999-02-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5770429A (en) * | 1990-08-29 | 1998-06-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| ES2301158T3 (es) * | 1992-07-24 | 2008-06-16 | Amgen Fremont Inc. | Produccion de anticuerpos xenogenicos. |
| EP1621627A3 (en) | 1993-03-16 | 2006-06-07 | The Austin Research Institute | Use of porcine gala (1,3) galactosyl transferase in xenograft therapies |
| WO1995016670A1 (en) | 1993-12-16 | 1995-06-22 | The Procter & Gamble Company | Process for making sulfonated fatty acid alkyl ester surfactant |
| US5827690A (en) | 1993-12-20 | 1998-10-27 | Genzyme Transgenics Corporatiion | Transgenic production of antibodies in milk |
| US5639940A (en) * | 1994-03-03 | 1997-06-17 | Pharmaceutical Proteins Ltd. | Production of fibrinogen in transgenic animals |
| DE69637481T2 (de) | 1995-04-27 | 2009-04-09 | Amgen Fremont Inc. | Aus immunisierten Xenomäusen stammende menschliche Antikörper gegen IL-8 |
| US5683993A (en) | 1995-06-22 | 1997-11-04 | Ciba Vision Corporation | Compositions and methods for stabilizing polymers |
| DE69636868T2 (de) | 1995-08-29 | 2007-10-25 | Kirin Beer Kabushiki Kaisha | Chimäres tier und methode zu dessen herstellung |
| JP3030092B2 (ja) | 1995-08-29 | 2000-04-10 | 麒麟麦酒株式会社 | キメラ動物およびその作製法 |
| KR20080059467A (ko) | 1996-12-03 | 2008-06-27 | 아브게닉스, 인크. | 복수의 vh 및 vk 부위를 함유하는 사람 면역글로불린유전자좌를 갖는 형질전환된 포유류 및 이로부터 생성된항체 |
| TWI255853B (en) | 1998-08-21 | 2006-06-01 | Kirin Brewery | Method for modifying chromosomes |
| WO2001035735A1 (en) * | 1999-11-19 | 2001-05-25 | Hematech, Llc | Production of ungulates, preferably bovines that produce human immunoglobulins |
| US6721750B1 (en) | 2001-07-03 | 2004-04-13 | Bellsouth Intellectual Property Corporation | System and method for broadband capacity tracking |
-
2001
- 2001-11-16 AT AT08016978T patent/ATE503012T1/de not_active IP Right Cessation
- 2001-11-16 KR KR1020037006659A patent/KR100882029B1/ko active IP Right Grant
- 2001-11-16 NZ NZ525664A patent/NZ525664A/en not_active IP Right Cessation
- 2001-11-16 EP EP08016978A patent/EP2042594B1/en not_active Expired - Lifetime
- 2001-11-16 MX MXPA03004313A patent/MXPA03004313A/es not_active Application Discontinuation
- 2001-11-16 DE DE60138832T patent/DE60138832D1/de not_active Expired - Lifetime
- 2001-11-16 EP EP01273248A patent/EP1343880B1/en not_active Expired - Lifetime
- 2001-11-16 CA CA2428053A patent/CA2428053C/en not_active Expired - Lifetime
- 2001-11-16 AU AU2001297523A patent/AU2001297523B2/en not_active Expired
- 2001-11-16 AT AT01273248T patent/ATE432343T1/de not_active IP Right Cessation
- 2001-11-16 CN CN018220762A patent/CN1486365B/zh not_active Expired - Lifetime
- 2001-11-16 WO PCT/US2001/043128 patent/WO2002070648A2/en active Application Filing
- 2001-11-16 JP JP2002570676A patent/JP3797974B2/ja not_active Expired - Fee Related
- 2001-11-16 DE DE60144305T patent/DE60144305D1/de not_active Expired - Lifetime
- 2001-11-16 KR KR1020077030804A patent/KR100882030B1/ko active IP Right Grant
-
2005
- 2005-05-09 JP JP2005136675A patent/JP2005224250A/ja active Pending
-
2007
- 2007-04-17 AU AU2007201700A patent/AU2007201700A1/en not_active Abandoned
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018079857A1 (ja) | 2016-10-31 | 2018-05-03 | 国立大学法人鳥取大学 | ヒト抗体産生非ヒト動物及びそれを用いたヒト抗体作製法 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20080005616A (ko) | 2008-01-14 |
| DE60144305D1 (de) | 2011-05-05 |
| MXPA03004313A (es) | 2007-02-27 |
| EP2042594A1 (en) | 2009-04-01 |
| DE60138832D1 (de) | 2009-07-09 |
| NZ525664A (en) | 2008-08-29 |
| KR20030059260A (ko) | 2003-07-07 |
| CA2428053A1 (en) | 2002-09-12 |
| EP1343880B1 (en) | 2009-05-27 |
| WO2002070648A3 (en) | 2003-02-13 |
| CN1486365B (zh) | 2010-05-12 |
| AU2007201700A1 (en) | 2007-05-10 |
| KR100882030B1 (ko) | 2009-02-09 |
| AU2001297523B2 (en) | 2007-01-18 |
| JP2005504507A (ja) | 2005-02-17 |
| JP2005224250A (ja) | 2005-08-25 |
| ATE503012T1 (de) | 2011-04-15 |
| EP1343880A2 (en) | 2003-09-17 |
| KR100882029B1 (ko) | 2009-02-09 |
| EP1343880A4 (en) | 2005-03-16 |
| CN1486365A (zh) | 2004-03-31 |
| WO2002070648A2 (en) | 2002-09-12 |
| EP2042594B1 (en) | 2011-03-23 |
| CA2428053C (en) | 2012-08-21 |
| ATE432343T1 (de) | 2009-06-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3797974B2 (ja) | クローン化トランスジェニック有蹄動物における異種(ヒト)免疫グロブリンの発現 | |
| US7491867B2 (en) | Expression of xenogenous (human) immunoglobulins in cloned, transgenic ungulates | |
| JP2005504507A5 (ja) | ||
| AU2001297523A1 (en) | Expression of xenogenous (human) immunoglobulins in cloned, transgenic ungulates | |
| JP3732407B2 (ja) | 染色体の改変方法 | |
| US7803981B2 (en) | Transgenic ungulates capable of human antibody production | |
| JPWO2002092812A1 (ja) | ヒト抗体λ軽鎖遺伝子を含むヒト人工染色体、および子孫伝達可能な該ヒト人工染色体を含む非ヒト動物 | |
| JP4082740B2 (ja) | 内因性遺伝子が破壊されている分化多能性保持細胞 | |
| JP2007533328A (ja) | トランスジェニック動物及びその用途 | |
| US7429690B2 (en) | Transgenic bovines having reduced prion protein production | |
| EP1513397B1 (en) | Transgenic ungulates capable of human antibody production | |
| WO2009111086A1 (en) | Transgenic non-human mammals with kappa light chain of xenogenous immunoglobulin | |
| AU2003245300B2 (en) | Transgenic ungulates capable of human antibody production | |
| JP2001231403A (ja) | 改変された外来染色体あるいはその断片を保持する非ヒト動物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A975 | Report on accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A971005 Effective date: 20041116 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20041124 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20050223 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20050307 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050509 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050810 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20050810 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20051206 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20060224 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20060404 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20060418 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 3797974 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090428 Year of fee payment: 3 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 Free format text: JAPANESE INTERMEDIATE CODE: R313111 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090428 Year of fee payment: 3 |
|
| R371 | Transfer withdrawn |
Free format text: JAPANESE INTERMEDIATE CODE: R371 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090428 Year of fee payment: 3 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 Free format text: JAPANESE INTERMEDIATE CODE: R313115 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090428 Year of fee payment: 3 |
|
| R360 | Written notification for declining of transfer of rights |
Free format text: JAPANESE INTERMEDIATE CODE: R360 |
|
| R360 | Written notification for declining of transfer of rights |
Free format text: JAPANESE INTERMEDIATE CODE: R360 |
|
| R371 | Transfer withdrawn |
Free format text: JAPANESE INTERMEDIATE CODE: R371 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090428 Year of fee payment: 3 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313117 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090428 Year of fee payment: 3 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090428 Year of fee payment: 3 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090428 Year of fee payment: 3 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090428 Year of fee payment: 3 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100428 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110428 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120428 Year of fee payment: 6 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130428 Year of fee payment: 7 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140428 Year of fee payment: 8 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140428 Year of fee payment: 8 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |