JP2013010761A - 新規分子機能の進化 - Google Patents
新規分子機能の進化 Download PDFInfo
- Publication number
- JP2013010761A JP2013010761A JP2012165977A JP2012165977A JP2013010761A JP 2013010761 A JP2013010761 A JP 2013010761A JP 2012165977 A JP2012165977 A JP 2012165977A JP 2012165977 A JP2012165977 A JP 2012165977A JP 2013010761 A JP2013010761 A JP 2013010761A
- Authority
- JP
- Japan
- Prior art keywords
- dna
- template
- reaction
- library
- synthesis
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 230000004879 molecular function Effects 0.000 title description 6
- 238000006243 chemical reaction Methods 0.000 abstract description 221
- 150000007523 nucleic acids Chemical class 0.000 abstract description 175
- 102000039446 nucleic acids Human genes 0.000 abstract description 162
- 108020004707 nucleic acids Proteins 0.000 abstract description 161
- 238000000034 method Methods 0.000 abstract description 151
- 150000001875 compounds Chemical class 0.000 abstract description 60
- 108020004566 Transfer RNA Proteins 0.000 abstract description 59
- 108020005098 Anticodon Proteins 0.000 abstract description 49
- 238000012546 transfer Methods 0.000 abstract description 38
- 238000009396 hybridization Methods 0.000 abstract description 20
- 239000000203 mixture Substances 0.000 abstract description 16
- 230000002194 synthesizing effect Effects 0.000 abstract description 12
- 108020004414 DNA Proteins 0.000 description 333
- 239000003153 chemical reaction reagent Substances 0.000 description 127
- 230000015572 biosynthetic process Effects 0.000 description 118
- 150000003384 small molecules Chemical class 0.000 description 118
- 238000003786 synthesis reaction Methods 0.000 description 110
- 239000000047 product Substances 0.000 description 106
- 239000003054 catalyst Substances 0.000 description 86
- 239000002585 base Substances 0.000 description 85
- 239000000178 monomer Substances 0.000 description 85
- 108020004705 Codon Proteins 0.000 description 76
- 125000005647 linker group Chemical group 0.000 description 72
- 238000005287 template synthesis Methods 0.000 description 65
- 230000027455 binding Effects 0.000 description 64
- 102000004169 proteins and genes Human genes 0.000 description 61
- 108090000623 proteins and genes Proteins 0.000 description 60
- 239000000126 substance Substances 0.000 description 58
- 108091034117 Oligonucleotide Proteins 0.000 description 56
- 125000003729 nucleotide group Chemical group 0.000 description 56
- 238000003752 polymerase chain reaction Methods 0.000 description 55
- 239000002773 nucleotide Substances 0.000 description 53
- 238000013459 approach Methods 0.000 description 49
- 229910052751 metal Inorganic materials 0.000 description 47
- 239000002184 metal Substances 0.000 description 47
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 46
- 238000006116 polymerization reaction Methods 0.000 description 45
- 239000003446 ligand Substances 0.000 description 44
- 229920005615 natural polymer Polymers 0.000 description 43
- 239000000376 reactant Substances 0.000 description 42
- 229920002521 macromolecule Polymers 0.000 description 41
- 238000005859 coupling reaction Methods 0.000 description 40
- 230000008878 coupling Effects 0.000 description 37
- 238000010168 coupling process Methods 0.000 description 37
- 229920000642 polymer Polymers 0.000 description 37
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 36
- 239000000758 substrate Substances 0.000 description 35
- 102000006495 integrins Human genes 0.000 description 33
- 108010044426 integrins Proteins 0.000 description 33
- -1 2′-deoxyribose Chemical compound 0.000 description 32
- 239000011324 bead Substances 0.000 description 32
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 30
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 30
- 230000000694 effects Effects 0.000 description 30
- 238000003776 cleavage reaction Methods 0.000 description 29
- 108090000765 processed proteins & peptides Proteins 0.000 description 29
- 230000007017 scission Effects 0.000 description 29
- 150000001413 amino acids Chemical class 0.000 description 28
- 230000003321 amplification Effects 0.000 description 26
- 238000000338 in vitro Methods 0.000 description 26
- 238000003199 nucleic acid amplification method Methods 0.000 description 26
- 238000007792 addition Methods 0.000 description 25
- 238000006664 bond formation reaction Methods 0.000 description 25
- 239000001226 triphosphate Substances 0.000 description 25
- 150000001412 amines Chemical class 0.000 description 24
- 229960002685 biotin Drugs 0.000 description 24
- 239000011616 biotin Substances 0.000 description 24
- 235000011178 triphosphate Nutrition 0.000 description 24
- 108091093037 Peptide nucleic acid Proteins 0.000 description 23
- 235000020958 biotin Nutrition 0.000 description 23
- 238000005755 formation reaction Methods 0.000 description 23
- 125000000524 functional group Chemical group 0.000 description 23
- 239000000872 buffer Substances 0.000 description 22
- 230000014616 translation Effects 0.000 description 22
- 108010090804 Streptavidin Proteins 0.000 description 21
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 21
- 230000000295 complement effect Effects 0.000 description 21
- 229920001222 biopolymer Polymers 0.000 description 20
- 239000013615 primer Substances 0.000 description 20
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 19
- 238000013519 translation Methods 0.000 description 19
- 239000011780 sodium chloride Substances 0.000 description 18
- 150000001336 alkenes Chemical class 0.000 description 17
- 239000007795 chemical reaction product Substances 0.000 description 17
- 230000003197 catalytic effect Effects 0.000 description 16
- 238000000746 purification Methods 0.000 description 16
- 239000011347 resin Substances 0.000 description 16
- 229920005989 resin Polymers 0.000 description 16
- 108010074860 Factor Xa Proteins 0.000 description 15
- 230000006870 function Effects 0.000 description 15
- 239000003960 organic solvent Substances 0.000 description 15
- 108091028043 Nucleic acid sequence Proteins 0.000 description 14
- 238000010976 amide bond formation reaction Methods 0.000 description 14
- 238000003556 assay Methods 0.000 description 14
- 230000002068 genetic effect Effects 0.000 description 14
- 230000001404 mediated effect Effects 0.000 description 14
- 229920002635 polyurethane Polymers 0.000 description 14
- 102000004196 processed proteins & peptides Human genes 0.000 description 14
- 239000000243 solution Substances 0.000 description 14
- 125000002827 triflate group Chemical group FC(S(=O)(=O)O*)(F)F 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 13
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 13
- 150000002148 esters Chemical class 0.000 description 13
- 230000001965 increasing effect Effects 0.000 description 13
- 230000003993 interaction Effects 0.000 description 13
- 230000035772 mutation Effects 0.000 description 13
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 13
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 13
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 12
- 102000053602 DNA Human genes 0.000 description 12
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 12
- 238000005580 one pot reaction Methods 0.000 description 12
- 150000003141 primary amines Chemical class 0.000 description 12
- 125000006239 protecting group Chemical group 0.000 description 12
- 150000003573 thiols Chemical class 0.000 description 12
- 238000001712 DNA sequencing Methods 0.000 description 11
- 238000000137 annealing Methods 0.000 description 11
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 11
- 238000012216 screening Methods 0.000 description 11
- 238000005406 washing Methods 0.000 description 11
- 238000012408 PCR amplification Methods 0.000 description 10
- 108010067902 Peptide Library Proteins 0.000 description 10
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 10
- 150000001299 aldehydes Chemical class 0.000 description 10
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 10
- 238000007306 functionalization reaction Methods 0.000 description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 10
- 238000004949 mass spectrometry Methods 0.000 description 10
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 10
- 239000002253 acid Substances 0.000 description 9
- 238000006668 aldol addition reaction Methods 0.000 description 9
- 239000003814 drug Substances 0.000 description 9
- 230000000269 nucleophilic effect Effects 0.000 description 9
- 102000005962 receptors Human genes 0.000 description 9
- 108020003175 receptors Proteins 0.000 description 9
- HDZZVAMISRMYHH-KCGFPETGSA-N tubercidin Chemical compound C1=CC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O HDZZVAMISRMYHH-KCGFPETGSA-N 0.000 description 9
- HSJKGGMUJITCBW-UHFFFAOYSA-N 3-hydroxybutanal Chemical compound CC(O)CC=O HSJKGGMUJITCBW-UHFFFAOYSA-N 0.000 description 8
- 108090001008 Avidin Proteins 0.000 description 8
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 8
- 239000007864 aqueous solution Substances 0.000 description 8
- 210000004027 cell Anatomy 0.000 description 8
- 238000006352 cycloaddition reaction Methods 0.000 description 8
- 239000012634 fragment Substances 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- 230000007246 mechanism Effects 0.000 description 8
- 238000002703 mutagenesis Methods 0.000 description 8
- 231100000350 mutagenesis Toxicity 0.000 description 8
- 239000012038 nucleophile Substances 0.000 description 8
- 102000040430 polynucleotide Human genes 0.000 description 8
- 108091033319 polynucleotide Proteins 0.000 description 8
- 239000002157 polynucleotide Substances 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 239000002904 solvent Substances 0.000 description 8
- 239000007858 starting material Substances 0.000 description 8
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical compound OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 description 8
- 230000007306 turnover Effects 0.000 description 8
- BMTZEAOGFDXDAD-UHFFFAOYSA-M 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholin-4-ium;chloride Chemical compound [Cl-].COC1=NC(OC)=NC([N+]2(C)CCOCC2)=N1 BMTZEAOGFDXDAD-UHFFFAOYSA-M 0.000 description 7
- 238000010640 amide synthesis reaction Methods 0.000 description 7
- 125000002619 bicyclic group Chemical group 0.000 description 7
- 238000010504 bond cleavage reaction Methods 0.000 description 7
- 230000007423 decrease Effects 0.000 description 7
- 239000005547 deoxyribonucleotide Substances 0.000 description 7
- 238000011161 development Methods 0.000 description 7
- 238000006077 hetero Diels-Alder cycloaddition reaction Methods 0.000 description 7
- 238000010348 incorporation Methods 0.000 description 7
- 238000004519 manufacturing process Methods 0.000 description 7
- 238000007040 multi-step synthesis reaction Methods 0.000 description 7
- 230000006798 recombination Effects 0.000 description 7
- 238000005215 recombination Methods 0.000 description 7
- 238000006268 reductive amination reaction Methods 0.000 description 7
- 108091008146 restriction endonucleases Proteins 0.000 description 7
- 238000012163 sequencing technique Methods 0.000 description 7
- RPENMORRBUTCPR-UHFFFAOYSA-M sodium;1-hydroxy-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].ON1C(=O)CC(S([O-])(=O)=O)C1=O RPENMORRBUTCPR-UHFFFAOYSA-M 0.000 description 7
- 125000006850 spacer group Chemical group 0.000 description 7
- 238000006467 substitution reaction Methods 0.000 description 7
- 239000003774 sulfhydryl reagent Substances 0.000 description 7
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 6
- 239000007993 MOPS buffer Substances 0.000 description 6
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 6
- 101150003085 Pdcl gene Proteins 0.000 description 6
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 6
- 108020004682 Single-Stranded DNA Proteins 0.000 description 6
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 6
- 238000007295 Wittig olefination reaction Methods 0.000 description 6
- 125000003277 amino group Chemical group 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 238000006555 catalytic reaction Methods 0.000 description 6
- 125000003636 chemical group Chemical group 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 230000000977 initiatory effect Effects 0.000 description 6
- 238000000816 matrix-assisted laser desorption--ionisation Methods 0.000 description 6
- 230000004048 modification Effects 0.000 description 6
- 238000012986 modification Methods 0.000 description 6
- 239000002777 nucleoside Substances 0.000 description 6
- XYFCBTPGUUZFHI-UHFFFAOYSA-N phosphine group Chemical group P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 6
- 229920003023 plastic Polymers 0.000 description 6
- 239000004033 plastic Substances 0.000 description 6
- 229920001184 polypeptide Polymers 0.000 description 6
- 238000011160 research Methods 0.000 description 6
- 238000007152 ring opening metathesis polymerisation reaction Methods 0.000 description 6
- 238000010186 staining Methods 0.000 description 6
- 230000007704 transition Effects 0.000 description 6
- 108020004635 Complementary DNA Proteins 0.000 description 5
- 239000007987 MES buffer Substances 0.000 description 5
- 241000872931 Myoporum sandwicense Species 0.000 description 5
- 238000006445 Nitro-Michael reaction Methods 0.000 description 5
- 108090000190 Thrombin Proteins 0.000 description 5
- 208000034953 Twin anemia-polycythemia sequence Diseases 0.000 description 5
- 239000012190 activator Substances 0.000 description 5
- 150000003838 adenosines Chemical class 0.000 description 5
- 150000001408 amides Chemical class 0.000 description 5
- 239000012148 binding buffer Substances 0.000 description 5
- 238000010804 cDNA synthesis Methods 0.000 description 5
- 238000012512 characterization method Methods 0.000 description 5
- 239000002299 complementary DNA Substances 0.000 description 5
- 125000004122 cyclic group Chemical group 0.000 description 5
- AFOSIXZFDONLBT-UHFFFAOYSA-N divinyl sulfone Chemical compound C=CS(=O)(=O)C=C AFOSIXZFDONLBT-UHFFFAOYSA-N 0.000 description 5
- 230000037433 frameshift Effects 0.000 description 5
- 239000000543 intermediate Substances 0.000 description 5
- 150000002576 ketones Chemical class 0.000 description 5
- 150000003833 nucleoside derivatives Chemical class 0.000 description 5
- 238000007254 oxidation reaction Methods 0.000 description 5
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 5
- 230000009467 reduction Effects 0.000 description 5
- 238000006722 reduction reaction Methods 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 231100000241 scar Toxicity 0.000 description 5
- 229960004072 thrombin Drugs 0.000 description 5
- 229940045145 uridine Drugs 0.000 description 5
- 239000013598 vector Substances 0.000 description 5
- BQWBEDSJTMWJAE-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-[(2-iodoacetyl)amino]benzoate Chemical compound C1=CC(NC(=O)CI)=CC=C1C(=O)ON1C(=O)CCC1=O BQWBEDSJTMWJAE-UHFFFAOYSA-N 0.000 description 4
- MXHRCPNRJAMMIM-SHYZEUOFSA-N 2'-deoxyuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 MXHRCPNRJAMMIM-SHYZEUOFSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 4
- 238000010485 C−C bond formation reaction Methods 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 238000007341 Heck reaction Methods 0.000 description 4
- 238000006842 Henry reaction Methods 0.000 description 4
- 238000006736 Huisgen cycloaddition reaction Methods 0.000 description 4
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- 108010006785 Taq Polymerase Proteins 0.000 description 4
- 239000012445 acidic reagent Substances 0.000 description 4
- 230000002378 acidificating effect Effects 0.000 description 4
- 239000012736 aqueous medium Substances 0.000 description 4
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 4
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 4
- 150000007942 carboxylates Chemical class 0.000 description 4
- 150000001735 carboxylic acids Chemical class 0.000 description 4
- 150000001780 cephalosporins Chemical class 0.000 description 4
- 230000021615 conjugation Effects 0.000 description 4
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 4
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 4
- 238000010511 deprotection reaction Methods 0.000 description 4
- 238000013461 design Methods 0.000 description 4
- 238000003379 elimination reaction Methods 0.000 description 4
- 229940088598 enzyme Drugs 0.000 description 4
- 238000001502 gel electrophoresis Methods 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 230000002209 hydrophobic effect Effects 0.000 description 4
- 230000001976 improved effect Effects 0.000 description 4
- 238000002955 isolation Methods 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 150000002739 metals Chemical class 0.000 description 4
- 125000003835 nucleoside group Chemical group 0.000 description 4
- 238000006053 organic reaction Methods 0.000 description 4
- 230000003647 oxidation Effects 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 235000021317 phosphate Nutrition 0.000 description 4
- 230000003252 repetitive effect Effects 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 239000007790 solid phase Substances 0.000 description 4
- 230000008685 targeting Effects 0.000 description 4
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 4
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 4
- SWHAGWLVMRLFKO-UHFFFAOYSA-N (2-nitrophenyl)methyl carbamate Chemical compound NC(=O)OCC1=CC=CC=C1[N+]([O-])=O SWHAGWLVMRLFKO-UHFFFAOYSA-N 0.000 description 3
- NJBIVXMQFIQOGE-KVQBGUIXSA-N (2r,3s,5r)-5-(6-amino-8-bromopurin-9-yl)-2-(hydroxymethyl)oxolan-3-ol Chemical compound BrC1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 NJBIVXMQFIQOGE-KVQBGUIXSA-N 0.000 description 3
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 3
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 3
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 3
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical group NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 3
- 108091033380 Coding strand Proteins 0.000 description 3
- 108010069514 Cyclic Peptides Proteins 0.000 description 3
- 102000001189 Cyclic Peptides Human genes 0.000 description 3
- 108010017826 DNA Polymerase I Proteins 0.000 description 3
- 102000004594 DNA Polymerase I Human genes 0.000 description 3
- 102100031780 Endonuclease Human genes 0.000 description 3
- 108010049003 Fibrinogen Proteins 0.000 description 3
- 102000008946 Fibrinogen Human genes 0.000 description 3
- 108060003951 Immunoglobulin Proteins 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 3
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 238000007239 Wittig reaction Methods 0.000 description 3
- 239000003377 acid catalyst Substances 0.000 description 3
- 238000007259 addition reaction Methods 0.000 description 3
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 3
- 125000000539 amino acid group Chemical group 0.000 description 3
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000005557 antagonist Substances 0.000 description 3
- 150000001500 aryl chlorides Chemical class 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000006287 biotinylation Effects 0.000 description 3
- 238000007413 biotinylation Methods 0.000 description 3
- 230000023555 blood coagulation Effects 0.000 description 3
- 210000004899 c-terminal region Anatomy 0.000 description 3
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 3
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 3
- 150000001768 cations Chemical class 0.000 description 3
- 239000002738 chelating agent Substances 0.000 description 3
- 238000006757 chemical reactions by type Methods 0.000 description 3
- 238000010276 construction Methods 0.000 description 3
- 238000006114 decarboxylation reaction Methods 0.000 description 3
- 230000002950 deficient Effects 0.000 description 3
- 238000004925 denaturation Methods 0.000 description 3
- 230000036425 denaturation Effects 0.000 description 3
- 238000000326 densiometry Methods 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 230000005595 deprotonation Effects 0.000 description 3
- 238000010537 deprotonation reaction Methods 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- 239000012039 electrophile Substances 0.000 description 3
- 230000008030 elimination Effects 0.000 description 3
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 3
- 229960005542 ethidium bromide Drugs 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 229940012952 fibrinogen Drugs 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 238000002523 gelfiltration Methods 0.000 description 3
- 238000007429 general method Methods 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 238000002744 homologous recombination Methods 0.000 description 3
- 230000006801 homologous recombination Effects 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 102000018358 immunoglobulin Human genes 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 108020004999 messenger RNA Proteins 0.000 description 3
- 230000003505 mutagenic effect Effects 0.000 description 3
- 229910052759 nickel Inorganic materials 0.000 description 3
- 238000006772 olefination reaction Methods 0.000 description 3
- 125000002524 organometallic group Chemical group 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 150000008300 phosphoramidites Chemical class 0.000 description 3
- 239000002685 polymerization catalyst Substances 0.000 description 3
- 235000013406 prebiotics Nutrition 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 238000004007 reversed phase HPLC Methods 0.000 description 3
- 210000003705 ribosome Anatomy 0.000 description 3
- JXOHGGNKMLTUBP-HSUXUTPPSA-N shikimic acid Chemical compound O[C@@H]1CC(C(O)=O)=C[C@@H](O)[C@H]1O JXOHGGNKMLTUBP-HSUXUTPPSA-N 0.000 description 3
- JXOHGGNKMLTUBP-JKUQZMGJSA-N shikimic acid Natural products O[C@@H]1CC(C(O)=O)=C[C@H](O)[C@@H]1O JXOHGGNKMLTUBP-JKUQZMGJSA-N 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 238000010532 solid phase synthesis reaction Methods 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 150000007970 thio esters Chemical class 0.000 description 3
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 3
- 125000002264 triphosphate group Chemical class [H]OP(=O)(O[H])OP(=O)(O[H])OP(=O)(O[H])O* 0.000 description 3
- XUDGDVPXDYGCTG-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-[2-(2,5-dioxopyrrolidin-1-yl)oxycarbonyloxyethylsulfonyl]ethyl carbonate Chemical compound O=C1CCC(=O)N1OC(=O)OCCS(=O)(=O)CCOC(=O)ON1C(=O)CCC1=O XUDGDVPXDYGCTG-UHFFFAOYSA-N 0.000 description 2
- JKHVDAUOODACDU-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(2,5-dioxopyrrol-1-yl)propanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCN1C(=O)C=CC1=O JKHVDAUOODACDU-UHFFFAOYSA-N 0.000 description 2
- PVGATNRYUYNBHO-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-(2,5-dioxopyrrol-1-yl)butanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCCN1C(=O)C=CC1=O PVGATNRYUYNBHO-UHFFFAOYSA-N 0.000 description 2
- XXNHOXKGISCJIY-WOUKDFQISA-N (2r,3r,4s,5r)-2-(6-aminopurin-9-yl)-2-ethenyl-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@]1(C=C)O[C@H](CO)[C@@H](O)[C@H]1O XXNHOXKGISCJIY-WOUKDFQISA-N 0.000 description 2
- ZHWHMUIGIKPSJN-WOUKDFQISA-N (2r,3r,4s,5r)-2-(6-aminopurin-9-yl)-2-ethyl-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C1=NC2=C(N)N=CN=C2N1[C@]1(CC)O[C@H](CO)[C@@H](O)[C@H]1O ZHWHMUIGIKPSJN-WOUKDFQISA-N 0.000 description 2
- HDBGFRPOJPTJFA-FCKMSMMTSA-N (2s,3r,4s,5r)-2-(6-aminopurin-9-yl)-5-(hydroxymethyl)-2-(methylamino)oxolane-3,4-diol Chemical compound C1=NC2=C(N)N=CN=C2N1[C@]1(NC)O[C@H](CO)[C@@H](O)[C@H]1O HDBGFRPOJPTJFA-FCKMSMMTSA-N 0.000 description 2
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 2
- BSSNZUFKXJJCBG-OWOJBTEDSA-N (e)-but-2-enediamide Chemical compound NC(=O)\C=C\C(N)=O BSSNZUFKXJJCBG-OWOJBTEDSA-N 0.000 description 2
- 0 *C(O[C@]1C2)O[C@@]1[C@@](*)C=C2C(O)=O Chemical compound *C(O[C@]1C2)O[C@@]1[C@@](*)C=C2C(O)=O 0.000 description 2
- 150000000180 1,2-diols Chemical class 0.000 description 2
- LWTIGYSPAXKMDG-UHFFFAOYSA-N 2,3-dihydro-1h-imidazole Chemical compound C1NC=CN1 LWTIGYSPAXKMDG-UHFFFAOYSA-N 0.000 description 2
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- LILXDMFJXYAKMK-UHFFFAOYSA-N 2-bromo-1,1-diethoxyethane Chemical compound CCOC(CBr)OCC LILXDMFJXYAKMK-UHFFFAOYSA-N 0.000 description 2
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 2
- QCPFFGGFHNZBEP-UHFFFAOYSA-N 4,5,6,7-tetrachloro-3',6'-dihydroxyspiro[2-benzofuran-3,9'-xanthene]-1-one Chemical group O1C(=O)C(C(=C(Cl)C(Cl)=C2Cl)Cl)=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 QCPFFGGFHNZBEP-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- HDZZVAMISRMYHH-UHFFFAOYSA-N 9beta-Ribofuranosyl-7-deazaadenin Natural products C1=CC=2C(N)=NC=NC=2N1C1OC(CO)C(O)C1O HDZZVAMISRMYHH-UHFFFAOYSA-N 0.000 description 2
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 2
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 2
- JPRWMFLCIVUTMH-UHFFFAOYSA-N C(C)OC(COC(CC#N)=O)OCC Chemical compound C(C)OC(COC(CC#N)=O)OCC JPRWMFLCIVUTMH-UHFFFAOYSA-N 0.000 description 2
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 2
- 239000008001 CAPS buffer Substances 0.000 description 2
- 108090000994 Catalytic RNA Proteins 0.000 description 2
- 102000053642 Catalytic RNA Human genes 0.000 description 2
- 229930186147 Cephalosporin Natural products 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 108020004394 Complementary RNA Proteins 0.000 description 2
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 2
- 108020001019 DNA Primers Proteins 0.000 description 2
- 239000003155 DNA primer Substances 0.000 description 2
- 230000006820 DNA synthesis Effects 0.000 description 2
- 108010016626 Dipeptides Proteins 0.000 description 2
- 229940123583 Factor Xa inhibitor Drugs 0.000 description 2
- 102000009123 Fibrin Human genes 0.000 description 2
- 108010073385 Fibrin Proteins 0.000 description 2
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 2
- 101000945318 Homo sapiens Calponin-1 Proteins 0.000 description 2
- 101000652736 Homo sapiens Transgelin Proteins 0.000 description 2
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 2
- 239000002841 Lewis acid Substances 0.000 description 2
- 102000003960 Ligases Human genes 0.000 description 2
- 108090000364 Ligases Proteins 0.000 description 2
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 229920002396 Polyurea Polymers 0.000 description 2
- 101000781681 Protobothrops flavoviridis Disintegrin triflavin Proteins 0.000 description 2
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 2
- 108091028664 Ribonucleotide Proteins 0.000 description 2
- 229920005654 Sephadex Polymers 0.000 description 2
- 239000012507 Sephadex™ Substances 0.000 description 2
- 108010022999 Serine Proteases Proteins 0.000 description 2
- 102000012479 Serine Proteases Human genes 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- 238000003477 Sonogashira cross-coupling reaction Methods 0.000 description 2
- 108091081024 Start codon Proteins 0.000 description 2
- 102100031013 Transgelin Human genes 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- POIOOCHMXHKUHV-UHFFFAOYSA-N [nitro-[nitro(phenyl)methoxy]methyl]benzene Chemical compound C=1C=CC=CC=1C([N+](=O)[O-])OC([N+]([O-])=O)C1=CC=CC=C1 POIOOCHMXHKUHV-UHFFFAOYSA-N 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 238000005917 acylation reaction Methods 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 229960005305 adenosine Drugs 0.000 description 2
- 125000003172 aldehyde group Chemical group 0.000 description 2
- 238000005575 aldol reaction Methods 0.000 description 2
- 238000005865 alkene metathesis reaction Methods 0.000 description 2
- 125000003275 alpha amino acid group Chemical group 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 239000003125 aqueous solvent Substances 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- 150000001503 aryl iodides Chemical class 0.000 description 2
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 2
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 150000001718 carbodiimides Chemical class 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 125000002843 carboxylic acid group Chemical group 0.000 description 2
- 229940124587 cephalosporin Drugs 0.000 description 2
- RCTYPNKXASFOBE-UHFFFAOYSA-M chloromercury Chemical compound [Hg]Cl RCTYPNKXASFOBE-UHFFFAOYSA-M 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- APEJMQOBVMLION-UHFFFAOYSA-N cinnamamide Chemical compound NC(=O)C=CC1=CC=CC=C1 APEJMQOBVMLION-UHFFFAOYSA-N 0.000 description 2
- 239000003184 complementary RNA Substances 0.000 description 2
- 229940104302 cytosine Drugs 0.000 description 2
- SUYVUBYJARFZHO-RRKCRQDMSA-N dATP Chemical class C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-RRKCRQDMSA-N 0.000 description 2
- RGWHQCVHVJXOKC-SHYZEUOFSA-J dCTP(4-) Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)C1 RGWHQCVHVJXOKC-SHYZEUOFSA-J 0.000 description 2
- HAAZLUGHYHWQIW-KVQBGUIXSA-N dGTP Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 HAAZLUGHYHWQIW-KVQBGUIXSA-N 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000005828 desilylation reaction Methods 0.000 description 2
- QQJDHWMADUVRDL-UHFFFAOYSA-N didodecyl(dimethyl)azanium Chemical compound CCCCCCCCCCCC[N+](C)(C)CCCCCCCCCCCC QQJDHWMADUVRDL-UHFFFAOYSA-N 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- GNGACRATGGDKBX-UHFFFAOYSA-N dihydroxyacetone phosphate Chemical compound OCC(=O)COP(O)(O)=O GNGACRATGGDKBX-UHFFFAOYSA-N 0.000 description 2
- 238000009510 drug design Methods 0.000 description 2
- 238000007876 drug discovery Methods 0.000 description 2
- 238000006735 epoxidation reaction Methods 0.000 description 2
- 238000012869 ethanol precipitation Methods 0.000 description 2
- ZIUSEGSNTOUIPT-UHFFFAOYSA-N ethyl 2-cyanoacetate Chemical compound CCOC(=O)CC#N ZIUSEGSNTOUIPT-UHFFFAOYSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 229950003499 fibrin Drugs 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 238000012248 genetic selection Methods 0.000 description 2
- 108010064365 glycyl- arginyl-glycyl-aspartyl-seryl-prolyl-lysine Proteins 0.000 description 2
- LEQAOMBKQFMDFZ-UHFFFAOYSA-N glyoxal Chemical compound O=CC=O LEQAOMBKQFMDFZ-UHFFFAOYSA-N 0.000 description 2
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 150000002466 imines Chemical class 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 239000003999 initiator Substances 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- PGLTVOMIXTUURA-UHFFFAOYSA-N iodoacetamide Chemical group NC(=O)CI PGLTVOMIXTUURA-UHFFFAOYSA-N 0.000 description 2
- 150000007517 lewis acids Chemical class 0.000 description 2
- 239000007791 liquid phase Substances 0.000 description 2
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 2
- 238000005710 macrocyclization reaction Methods 0.000 description 2
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 2
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 2
- 231100000243 mutagenic effect Toxicity 0.000 description 2
- SQDFHQJTAWCFIB-UHFFFAOYSA-N n-methylidenehydroxylamine Chemical compound ON=C SQDFHQJTAWCFIB-UHFFFAOYSA-N 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 125000004971 nitroalkyl group Chemical group 0.000 description 2
- 210000002997 osteoclast Anatomy 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- RLOWWWKZYUNIDI-UHFFFAOYSA-N phosphinic chloride Chemical compound ClP=O RLOWWWKZYUNIDI-UHFFFAOYSA-N 0.000 description 2
- GWLJTAJEHRYMCA-UHFFFAOYSA-N phospholane Chemical compound C1CCPC1 GWLJTAJEHRYMCA-UHFFFAOYSA-N 0.000 description 2
- 238000011907 photodimerization Methods 0.000 description 2
- 239000004417 polycarbonate Substances 0.000 description 2
- 229920000515 polycarbonate Polymers 0.000 description 2
- 229920000728 polyester Polymers 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 239000012041 precatalyst Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 230000037452 priming Effects 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 238000000159 protein binding assay Methods 0.000 description 2
- 238000002818 protein evolution Methods 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 239000002464 receptor antagonist Substances 0.000 description 2
- 229940044551 receptor antagonist Drugs 0.000 description 2
- 239000002336 ribonucleotide Substances 0.000 description 2
- 125000002652 ribonucleotide group Chemical group 0.000 description 2
- 108091092562 ribozyme Proteins 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 239000012064 sodium phosphate buffer Substances 0.000 description 2
- 230000002269 spontaneous effect Effects 0.000 description 2
- 230000000707 stereoselective effect Effects 0.000 description 2
- 238000005556 structure-activity relationship Methods 0.000 description 2
- JJAHTWIKCUJRDK-UHFFFAOYSA-N succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate Chemical compound C1CC(CN2C(C=CC2=O)=O)CCC1C(=O)ON1C(=O)CCC1=O JJAHTWIKCUJRDK-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- 150000003568 thioethers Chemical class 0.000 description 2
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 2
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 2
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 2
- PVSXPSHNTNUYHR-PPSWKLDYSA-N (2S,3R,4S,5R)-2-(6-aminopurin-9-yl)-2-(ethylamino)-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C(C)N[C@@]1([C@H](O)[C@H](O)[C@@H](CO)O1)N1C=NC=2C(N)=NC=NC12 PVSXPSHNTNUYHR-PPSWKLDYSA-N 0.000 description 1
- MXYRZDAGKTVQIL-IOSLPCCCSA-N (2r,3r,4s,5r)-2-(6-aminopurin-9-yl)-5-(hydroxymethyl)-2-methyloxolane-3,4-diol Chemical compound C1=NC2=C(N)N=CN=C2N1[C@]1(C)O[C@H](CO)[C@@H](O)[C@H]1O MXYRZDAGKTVQIL-IOSLPCCCSA-N 0.000 description 1
- RIFDKYBNWNPCQK-IOSLPCCCSA-N (2r,3s,4r,5r)-2-(hydroxymethyl)-5-(6-imino-3-methylpurin-9-yl)oxolane-3,4-diol Chemical compound C1=2N(C)C=NC(=N)C=2N=CN1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O RIFDKYBNWNPCQK-IOSLPCCCSA-N 0.000 description 1
- ZRVZOBGMZWVJOS-VMXHOPILSA-N (2s)-6-amino-2-[[(2s)-1-[(2s)-2-[[(2s)-2-[[2-[[(2s)-2-[(2-aminoacetyl)amino]-5-(diaminomethylideneamino)pentanoyl]amino]acetyl]amino]-3-carboxypropanoyl]amino]-3-hydroxypropanoyl]pyrrolidine-2-carbonyl]amino]hexanoic acid Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@@H]1CCCN1C(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CCCN=C(N)N)NC(=O)CN ZRVZOBGMZWVJOS-VMXHOPILSA-N 0.000 description 1
- LXJXRIRHZLFYRP-VKHMYHEASA-L (R)-2-Hydroxy-3-(phosphonooxy)-propanal Natural products O=C[C@H](O)COP([O-])([O-])=O LXJXRIRHZLFYRP-VKHMYHEASA-L 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- ODIGIKRIUKFKHP-UHFFFAOYSA-N (n-propan-2-yloxycarbonylanilino) acetate Chemical compound CC(C)OC(=O)N(OC(C)=O)C1=CC=CC=C1 ODIGIKRIUKFKHP-UHFFFAOYSA-N 0.000 description 1
- XMKLTEGSALONPH-UHFFFAOYSA-N 1,2,4,5-tetrazinane-3,6-dione Chemical compound O=C1NNC(=O)NN1 XMKLTEGSALONPH-UHFFFAOYSA-N 0.000 description 1
- GJFNRSDCSTVPCJ-UHFFFAOYSA-N 1,8-bis(dimethylamino)naphthalene Chemical compound C1=CC(N(C)C)=C2C(N(C)C)=CC=CC2=C1 GJFNRSDCSTVPCJ-UHFFFAOYSA-N 0.000 description 1
- RKSLVDIXBGWPIS-UAKXSSHOSA-N 1-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-iodopyrimidine-2,4-dione Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 RKSLVDIXBGWPIS-UAKXSSHOSA-N 0.000 description 1
- PISWNSOQFZRVJK-XLPZGREQSA-N 1-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-methyl-2-sulfanylidenepyrimidin-4-one Chemical compound S=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 PISWNSOQFZRVJK-XLPZGREQSA-N 0.000 description 1
- HRSBDXXXHGAUJF-QJPTWQEYSA-N 1-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-pent-1-ynylpyrimidine-2,4-dione Chemical compound O=C1NC(=O)C(C#CCCC)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 HRSBDXXXHGAUJF-QJPTWQEYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 1
- YKBGVTZYEHREMT-KVQBGUIXSA-N 2'-deoxyguanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 YKBGVTZYEHREMT-KVQBGUIXSA-N 0.000 description 1
- NIJSNUNKSPLDTO-DJLDLDEBSA-N 2'-deoxytubercidin Chemical compound C1=CC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 NIJSNUNKSPLDTO-DJLDLDEBSA-N 0.000 description 1
- CKTSBUTUHBMZGZ-SHYZEUOFSA-N 2'‐deoxycytidine Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 CKTSBUTUHBMZGZ-SHYZEUOFSA-N 0.000 description 1
- YQAMRGPPAKDUIK-UHFFFAOYSA-N 2,2,2-trifluoro-n-prop-1-ynylacetamide Chemical compound CC#CNC(=O)C(F)(F)F YQAMRGPPAKDUIK-UHFFFAOYSA-N 0.000 description 1
- NRKYWOKHZRQRJR-UHFFFAOYSA-N 2,2,2-trifluoroacetamide Chemical group NC(=O)C(F)(F)F NRKYWOKHZRQRJR-UHFFFAOYSA-N 0.000 description 1
- XBNGYFFABRKICK-UHFFFAOYSA-N 2,3,4,5,6-pentafluorophenol Chemical compound OC1=C(F)C(F)=C(F)C(F)=C1F XBNGYFFABRKICK-UHFFFAOYSA-N 0.000 description 1
- GVJXGCIPWAVXJP-UHFFFAOYSA-N 2,5-dioxo-1-oxoniopyrrolidine-3-sulfonate Chemical compound ON1C(=O)CC(S(O)(=O)=O)C1=O GVJXGCIPWAVXJP-UHFFFAOYSA-N 0.000 description 1
- ZDTFMPXQUSBYRL-UUOKFMHZSA-N 2-Aminoadenosine Chemical compound C12=NC(N)=NC(N)=C2N=CN1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O ZDTFMPXQUSBYRL-UUOKFMHZSA-N 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- SLTGLTLBIVDQKE-UHFFFAOYSA-N 2-amino-3-(2-aminoethoxy)propanoic acid Chemical compound NCCOCC(N)C(O)=O SLTGLTLBIVDQKE-UHFFFAOYSA-N 0.000 description 1
- JRYMOPZHXMVHTA-DAGMQNCNSA-N 2-amino-7-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1h-pyrrolo[2,3-d]pyrimidin-4-one Chemical compound C1=CC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O JRYMOPZHXMVHTA-DAGMQNCNSA-N 0.000 description 1
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 1
- ASJSAQIRZKANQN-CRCLSJGQSA-N 2-deoxy-D-ribose Chemical compound OC[C@@H](O)[C@@H](O)CC=O ASJSAQIRZKANQN-CRCLSJGQSA-N 0.000 description 1
- LXBGSDVWAMZHDD-UHFFFAOYSA-N 2-methyl-1h-imidazole Chemical compound CC1=NC=CN1 LXBGSDVWAMZHDD-UHFFFAOYSA-N 0.000 description 1
- MYHNUQVKTSTVDI-UHFFFAOYSA-N 2-methyl-2-[(2-methylpropan-2-yl)oxy]propane;potassium Chemical compound [K].CC(C)(C)OC(C)(C)C MYHNUQVKTSTVDI-UHFFFAOYSA-N 0.000 description 1
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 1
- RHFUOMFWUGWKKO-XVFCMESISA-N 2-thiocytidine Chemical compound S=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 RHFUOMFWUGWKKO-XVFCMESISA-N 0.000 description 1
- OBJOREYUKFGGKX-UHFFFAOYSA-N 3-[2-[3-(4-methylpiperazin-1-yl)anilino]pyrrolo[2,3-d]pyrimidin-7-yl]benzamide Chemical compound C1CN(C)CCN1C1=CC=CC(NC=2N=C3N(C=4C=C(C=CC=4)C(N)=O)C=CC3=CN=2)=C1 OBJOREYUKFGGKX-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- ASYBZHICIMVQII-UHFFFAOYSA-N 4-hydroxylysine Chemical compound NCCC(O)CC(N)C(O)=O ASYBZHICIMVQII-UHFFFAOYSA-N 0.000 description 1
- ZAYHVCMSTBRABG-UHFFFAOYSA-N 5-Methylcytidine Natural products O=C1N=C(N)C(C)=CN1C1C(O)C(O)C(CO)O1 ZAYHVCMSTBRABG-UHFFFAOYSA-N 0.000 description 1
- AGFIRQJZCNVMCW-UAKXSSHOSA-N 5-bromouridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(Br)=C1 AGFIRQJZCNVMCW-UAKXSSHOSA-N 0.000 description 1
- FHIDNBAQOFJWCA-UAKXSSHOSA-N 5-fluorouridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 FHIDNBAQOFJWCA-UAKXSSHOSA-N 0.000 description 1
- ZAYHVCMSTBRABG-JXOAFFINSA-N 5-methylcytidine Chemical compound O=C1N=C(N)C(C)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 ZAYHVCMSTBRABG-JXOAFFINSA-N 0.000 description 1
- KDOPAZIWBAHVJB-UHFFFAOYSA-N 5h-pyrrolo[3,2-d]pyrimidine Chemical compound C1=NC=C2NC=CC2=N1 KDOPAZIWBAHVJB-UHFFFAOYSA-N 0.000 description 1
- BXJHWYVXLGLDMZ-UHFFFAOYSA-N 6-O-methylguanine Chemical compound COC1=NC(N)=NC2=C1NC=N2 BXJHWYVXLGLDMZ-UHFFFAOYSA-N 0.000 description 1
- UEHOMUNTZPIBIL-UUOKFMHZSA-N 6-amino-9-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-7h-purin-8-one Chemical compound O=C1NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O UEHOMUNTZPIBIL-UUOKFMHZSA-N 0.000 description 1
- HSHGZXNAXBPPDL-HZGVNTEJSA-N 7beta-aminocephalosporanic acid Chemical compound S1CC(COC(=O)C)=C(C([O-])=O)N2C(=O)[C@@H]([NH3+])[C@@H]12 HSHGZXNAXBPPDL-HZGVNTEJSA-N 0.000 description 1
- HCAJQHYUCKICQH-VPENINKCSA-N 8-Oxo-7,8-dihydro-2'-deoxyguanosine Chemical compound C1=2NC(N)=NC(=O)C=2NC(=O)N1[C@H]1C[C@H](O)[C@@H](CO)O1 HCAJQHYUCKICQH-VPENINKCSA-N 0.000 description 1
- 208000035657 Abasia Diseases 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 101710134784 Agnoprotein Proteins 0.000 description 1
- OSDWBNJEKMUWAV-UHFFFAOYSA-N Allyl chloride Chemical compound ClCC=C OSDWBNJEKMUWAV-UHFFFAOYSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 108010031480 Artificial Receptors Proteins 0.000 description 1
- RENMDAKOXSCIGH-UHFFFAOYSA-N Chloroacetonitrile Chemical compound ClCC#N RENMDAKOXSCIGH-UHFFFAOYSA-N 0.000 description 1
- 208000032544 Cicatrix Diseases 0.000 description 1
- RGJOEKWQDUBAIZ-IBOSZNHHSA-N CoASH Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCS)O[C@H]1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-IBOSZNHHSA-N 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- 206010010904 Convulsion Diseases 0.000 description 1
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 1
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 description 1
- 150000008574 D-amino acids Chemical class 0.000 description 1
- LXJXRIRHZLFYRP-VKHMYHEASA-N D-glyceraldehyde 3-phosphate Chemical compound O=C[C@H](O)COP(O)(O)=O LXJXRIRHZLFYRP-VKHMYHEASA-N 0.000 description 1
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- 102000012410 DNA Ligases Human genes 0.000 description 1
- 108010061982 DNA Ligases Proteins 0.000 description 1
- 108010069941 DNA receptor Proteins 0.000 description 1
- 238000012270 DNA recombination Methods 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- CKTSBUTUHBMZGZ-UHFFFAOYSA-N Deoxycytidine Natural products O=C1N=C(N)C=CN1C1OC(CO)C(O)C1 CKTSBUTUHBMZGZ-UHFFFAOYSA-N 0.000 description 1
- 108091027757 Deoxyribozyme Proteins 0.000 description 1
- 238000006117 Diels-Alder cycloaddition reaction Methods 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 1
- 208000005189 Embolism Diseases 0.000 description 1
- 108010042407 Endonucleases Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 108060002716 Exonuclease Proteins 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 229910003771 Gold(I) chloride Inorganic materials 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 208000005331 Hepatitis D Diseases 0.000 description 1
- 208000037262 Hepatitis delta Diseases 0.000 description 1
- 108010007267 Hirudins Proteins 0.000 description 1
- 102000007625 Hirudins Human genes 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- MBYJMFIRGBGDHP-KQYNXXCUSA-N I[N+]1=CN([C@H]2[C@H](O)[C@H](O)[C@@H](CO)O2)C=2N=CN=C(C1=2)N Chemical compound I[N+]1=CN([C@H]2[C@H](O)[C@H](O)[C@@H](CO)O2)C=2N=CN=C(C1=2)N MBYJMFIRGBGDHP-KQYNXXCUSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 229910021617 Indium monochloride Inorganic materials 0.000 description 1
- 229930010555 Inosine Natural products 0.000 description 1
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 1
- 229940127449 Integrin Receptor Antagonists Drugs 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- GHSJKUNUIHUPDF-BYPYZUCNSA-N L-thialysine Chemical compound NCCSC[C@H](N)C(O)=O GHSJKUNUIHUPDF-BYPYZUCNSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 238000006683 Mannich reaction Methods 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 238000006845 Michael addition reaction Methods 0.000 description 1
- 125000003047 N-acetyl group Chemical group 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- GDIHDSKOVXEJQQ-UHFFFAOYSA-N NC(O)=O.NC(O)=O.NC(O)=O.N Chemical compound NC(O)=O.NC(O)=O.NC(O)=O.N GDIHDSKOVXEJQQ-UHFFFAOYSA-N 0.000 description 1
- 108091005461 Nucleic proteins Proteins 0.000 description 1
- JXOHGGNKMLTUBP-HCWXCVPCSA-N O[C@@H](CC(C(O)=O)=C[C@@H]1O)[C@@H]1O Chemical compound O[C@@H](CC(C(O)=O)=C[C@@H]1O)[C@@H]1O JXOHGGNKMLTUBP-HCWXCVPCSA-N 0.000 description 1
- 108010038807 Oligopeptides Proteins 0.000 description 1
- 102000015636 Oligopeptides Human genes 0.000 description 1
- 108700026244 Open Reading Frames Proteins 0.000 description 1
- 239000012807 PCR reagent Substances 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 108090000279 Peptidyltransferases Proteins 0.000 description 1
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical class ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 1
- 102000006335 Phosphate-Binding Proteins Human genes 0.000 description 1
- 108010058514 Phosphate-Binding Proteins Proteins 0.000 description 1
- 206010034960 Photophobia Diseases 0.000 description 1
- 206010034972 Photosensitivity reaction Diseases 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 108010021757 Polynucleotide 5'-Hydroxyl-Kinase Proteins 0.000 description 1
- 102000008422 Polynucleotide 5'-hydroxyl-kinase Human genes 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 108010094028 Prothrombin Proteins 0.000 description 1
- 102100027378 Prothrombin Human genes 0.000 description 1
- 230000007022 RNA scission Effects 0.000 description 1
- 239000007868 Raney catalyst Substances 0.000 description 1
- 229910000564 Raney nickel Inorganic materials 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- AUNGANRZJHBGPY-SCRDCRAPSA-N Riboflavin Chemical compound OC[C@@H](O)[C@@H](O)[C@@H](O)CN1C=2C=C(C)C(C)=CC=2N=C2C1=NC(=O)NC2=O AUNGANRZJHBGPY-SCRDCRAPSA-N 0.000 description 1
- 102000002278 Ribosomal Proteins Human genes 0.000 description 1
- 108010000605 Ribosomal Proteins Proteins 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 229910021607 Silver chloride Inorganic materials 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 238000006619 Stille reaction Methods 0.000 description 1
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 1
- 101000865057 Thermococcus litoralis DNA polymerase Proteins 0.000 description 1
- 208000001435 Thromboembolism Diseases 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- QYKIQEUNHZKYBP-UHFFFAOYSA-N Vinyl ether Chemical compound C=COC=C QYKIQEUNHZKYBP-UHFFFAOYSA-N 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- DILVWDXOASDRPN-MROZADKFSA-N [(2r,3r,4r)-3,4,5-trihydroxy-1-oxopentan-2-yl] dihydrogen phosphate Chemical group OC[C@@H](O)[C@@H](O)[C@H](C=O)OP(O)(O)=O DILVWDXOASDRPN-MROZADKFSA-N 0.000 description 1
- WYBZFSGKJOYRQH-UHFFFAOYSA-N [nitro(phenyl)methyl] carbamate Chemical compound NC(=O)OC([N+]([O-])=O)C1=CC=CC=C1 WYBZFSGKJOYRQH-UHFFFAOYSA-N 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- CIHUOJZDKZENCP-UHFFFAOYSA-N acetonitrile;triethylazanium;acetate Chemical compound CC#N.CC(O)=O.CCN(CC)CC CIHUOJZDKZENCP-UHFFFAOYSA-N 0.000 description 1
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 125000005262 alkoxyamine group Chemical group 0.000 description 1
- 125000000746 allylic group Chemical group 0.000 description 1
- RNBGYGVWRKECFJ-ZXXMMSQZSA-N alpha-D-fructofuranose 1,6-bisphosphate Chemical compound O[C@H]1[C@H](O)[C@](O)(COP(O)(O)=O)O[C@@H]1COP(O)(O)=O RNBGYGVWRKECFJ-ZXXMMSQZSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 230000006229 amino acid addition Effects 0.000 description 1
- IVHKZGYFKJRXBD-UHFFFAOYSA-N amino carbamate Chemical group NOC(N)=O IVHKZGYFKJRXBD-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 230000002491 angiogenic effect Effects 0.000 description 1
- 238000010719 annulation reaction Methods 0.000 description 1
- 230000001745 anti-biotin effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- PYMYPHUHKUWMLA-WDCZJNDASA-N arabinose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)C=O PYMYPHUHKUWMLA-WDCZJNDASA-N 0.000 description 1
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 238000005899 aromatization reaction Methods 0.000 description 1
- 150000001502 aryl halides Chemical class 0.000 description 1
- 125000003288 aryl iodide group Chemical group 0.000 description 1
- 238000000376 autoradiography Methods 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 1
- 230000002457 bidirectional effect Effects 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 230000001851 biosynthetic effect Effects 0.000 description 1
- OIRCOABEOLEUMC-GEJPAHFPSA-N bivalirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)CNC(=O)CNC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 OIRCOABEOLEUMC-GEJPAHFPSA-N 0.000 description 1
- 108010055460 bivalirudin Proteins 0.000 description 1
- 229960001500 bivalirudin Drugs 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000002805 bone matrix Anatomy 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- 235000013877 carbamide Nutrition 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 239000007809 chemical reaction catalyst Substances 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- XOYLJNJLGBYDTH-UHFFFAOYSA-M chlorogallium Chemical compound [Ga]Cl XOYLJNJLGBYDTH-UHFFFAOYSA-M 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 239000003593 chromogenic compound Substances 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- RGJOEKWQDUBAIZ-UHFFFAOYSA-N coenzime A Natural products OC1C(OP(O)(O)=O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-UHFFFAOYSA-N 0.000 description 1
- 239000005516 coenzyme A Substances 0.000 description 1
- 229940093530 coenzyme a Drugs 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 239000005289 controlled pore glass Substances 0.000 description 1
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 1
- 239000007819 coupling partner Substances 0.000 description 1
- 230000037029 cross reaction Effects 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- MBVZEDIBUZMQOR-UHFFFAOYSA-N cyclopenten-1-yl acetate Chemical compound CC(=O)OC1=CCCC1 MBVZEDIBUZMQOR-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 1
- SUYVUBYJARFZHO-UHFFFAOYSA-N dATP Natural products C1=NC=2C(N)=NC=NC=2N1C1CC(O)C(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-UHFFFAOYSA-N 0.000 description 1
- NHVNXKFIZYSCEB-XLPZGREQSA-N dTTP Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)C1 NHVNXKFIZYSCEB-XLPZGREQSA-N 0.000 description 1
- 125000001295 dansyl group Chemical group [H]C1=C([H])C(N(C([H])([H])[H])C([H])([H])[H])=C2C([H])=C([H])C([H])=C(C2=C1[H])S(*)(=O)=O 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- KDTSHFARGAKYJN-UHFFFAOYSA-N dephosphocoenzyme A Natural products OC1C(O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 KDTSHFARGAKYJN-UHFFFAOYSA-N 0.000 description 1
- 230000027832 depurination Effects 0.000 description 1
- 238000009795 derivation Methods 0.000 description 1
- MXHRCPNRJAMMIM-UHFFFAOYSA-N desoxyuridine Natural products C1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 MXHRCPNRJAMMIM-UHFFFAOYSA-N 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 150000001993 dienes Chemical class 0.000 description 1
- 238000005906 dihydroxylation reaction Methods 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- JGUQDUKBUKFFRO-CIIODKQPSA-N dimethylglyoxime Chemical compound O/N=C(/C)\C(\C)=N\O JGUQDUKBUKFFRO-CIIODKQPSA-N 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- KAKKHKRHCKCAGH-UHFFFAOYSA-L disodium;(4-nitrophenyl) phosphate;hexahydrate Chemical compound O.O.O.O.O.O.[Na+].[Na+].[O-][N+](=O)C1=CC=C(OP([O-])([O-])=O)C=C1 KAKKHKRHCKCAGH-UHFFFAOYSA-L 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- OAEGRYMCJYIXQT-UHFFFAOYSA-N dithiooxamide Chemical compound NC(=S)C(N)=S OAEGRYMCJYIXQT-UHFFFAOYSA-N 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 230000009881 electrostatic interaction Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- FXPHJTKVWZVEGA-UHFFFAOYSA-N ethenyl hydrogen carbonate Chemical group OC(=O)OC=C FXPHJTKVWZVEGA-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 102000013165 exonuclease Human genes 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 125000004030 farnesyl group Chemical group [H]C([*])([H])C([H])=C(C([H])([H])[H])C([H])([H])C([H])([H])C([H])=C(C([H])([H])[H])C([H])([H])C([H])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005313 fatty acid group Chemical group 0.000 description 1
- IRXSLJNXXZKURP-UHFFFAOYSA-N fluorenylmethyloxycarbonyl chloride Chemical compound C1=CC=C2C(COC(=O)Cl)C3=CC=CC=C3C2=C1 IRXSLJNXXZKURP-UHFFFAOYSA-N 0.000 description 1
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 1
- 229940025237 fructose 1,6-diphosphate Drugs 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 230000034659 glycolysis Effects 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- YMAWOPBAYDPSLA-UHFFFAOYSA-N glycylglycine Chemical compound [NH3+]CC(=O)NCC([O-])=O YMAWOPBAYDPSLA-UHFFFAOYSA-N 0.000 description 1
- 229940015043 glyoxal Drugs 0.000 description 1
- FDWREHZXQUYJFJ-UHFFFAOYSA-M gold monochloride Chemical compound [Cl-].[Au+] FDWREHZXQUYJFJ-UHFFFAOYSA-M 0.000 description 1
- 230000005283 ground state Effects 0.000 description 1
- 229960004198 guanidine Drugs 0.000 description 1
- 229940029575 guanosine Drugs 0.000 description 1
- 230000023597 hemostasis Effects 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 208000029570 hepatitis D virus infection Diseases 0.000 description 1
- 150000002402 hexoses Chemical class 0.000 description 1
- 229920006158 high molecular weight polymer Polymers 0.000 description 1
- 229940006607 hirudin Drugs 0.000 description 1
- WQPDUTSPKFMPDP-OUMQNGNKSA-N hirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(OS(O)(=O)=O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H]1NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H]2CSSC[C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@H](C(NCC(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N2)=O)CSSC1)C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)C(C)C)[C@@H](C)O)CSSC1)C(C)C)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 WQPDUTSPKFMPDP-OUMQNGNKSA-N 0.000 description 1
- 238000004845 hydriding Methods 0.000 description 1
- 150000001261 hydroxy acids Chemical class 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- APHGZSBLRQFRCA-UHFFFAOYSA-M indium(1+);chloride Chemical compound [In]Cl APHGZSBLRQFRCA-UHFFFAOYSA-M 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 229960003786 inosine Drugs 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 239000013067 intermediate product Substances 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000003402 intramolecular cyclocondensation reaction Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 239000011967 lanthanide triflate Substances 0.000 description 1
- 238000002898 library design Methods 0.000 description 1
- 208000013469 light sensitivity Diseases 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 150000002668 lysine derivatives Chemical class 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 238000006263 metalation reaction Methods 0.000 description 1
- 238000001465 metallisation Methods 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 238000005649 metathesis reaction Methods 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- WAERZCCNJRHZFS-QXUBKYEPSA-N mismatched pna Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCCN)NC(=O)[C@@H]1CCCN1C(=O)CN(C(=O)CN1C2=C(C(NC(N)=N2)=O)N=C1)CCNC(=O)CN(C(=O)CN1C2=C(C(NC(N)=N2)=O)N=C1)CCNC(=O)CN(C(=O)CN1C2=NC=NC(N)=C2N=C1)CCNC(=O)CN(C(=O)CN1C2=NC=NC(N)=C2N=C1)CCNC(=O)CN(C(=O)CN1C(NC(=O)C(C)=C1)=O)CCNC(=O)CN(C(=O)CN1C2=NC=NC(N)=C2N=C1)CCNC(=O)CN(C(=O)CN1C2=C(C(NC(N)=N2)=O)N=C1)CCNC(=O)CN(C(=O)CN1C(N=C(N)C=C1)=O)CCNC(=O)CN(C(=O)CN1C2=NC=NC(N)=C2N=C1)CCNC(=O)CN(C(=O)CN1C2=NC=NC(N)=C2N=C1)CCNC(=O)CN(C(=O)CN1C2=C(C(NC(N)=N2)=O)N=C1)CCNC(=O)CN(C(=O)CN1C(NC(=O)C(C)=C1)=O)CCNC(=O)CN(C(=O)CN1C(NC(=O)C(C)=C1)=O)CCNC(=O)CN(C(=O)CN1C(NC(=O)C(C)=C1)=O)CCNC(=O)CN(CCN)C(=O)CN1C(=O)NC(=O)C(C)=C1 WAERZCCNJRHZFS-QXUBKYEPSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 230000004001 molecular interaction Effects 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 229960005181 morphine Drugs 0.000 description 1
- 230000008450 motivation Effects 0.000 description 1
- 231100000219 mutagenic Toxicity 0.000 description 1
- HQMBYXUPHJBVMF-UHFFFAOYSA-N n-chlorophosphanyl-n-propan-2-ylpropan-2-amine Chemical compound CC(C)N(PCl)C(C)C HQMBYXUPHJBVMF-UHFFFAOYSA-N 0.000 description 1
- UOXMJKDGJMTPSI-UHFFFAOYSA-N n-ethenyl-2,2,2-trifluoroacetamide Chemical compound FC(F)(F)C(=O)NC=C UOXMJKDGJMTPSI-UHFFFAOYSA-N 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 230000010807 negative regulation of binding Effects 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 125000006502 nitrobenzyl group Chemical group 0.000 description 1
- 238000006384 oligomerization reaction Methods 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 150000002902 organometallic compounds Chemical class 0.000 description 1
- 238000010653 organometallic reaction Methods 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 1
- IZUPBVBPLAPZRR-UHFFFAOYSA-N pentachloro-phenol Natural products OC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl IZUPBVBPLAPZRR-UHFFFAOYSA-N 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 150000004713 phosphodiesters Chemical class 0.000 description 1
- 125000005541 phosphonamide group Chemical group 0.000 description 1
- SWFDTFBWXCNRGN-UHFFFAOYSA-N phosphonato phosphate;tributylazanium Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O.CCCC[NH+](CCCC)CCCC.CCCC[NH+](CCCC)CCCC.CCCC[NH+](CCCC)CCCC.CCCC[NH+](CCCC)CCCC SWFDTFBWXCNRGN-UHFFFAOYSA-N 0.000 description 1
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical group 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 230000003711 photoprotective effect Effects 0.000 description 1
- 230000036211 photosensitivity Effects 0.000 description 1
- 230000015843 photosynthesis, light reaction Effects 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 238000013492 plasmid preparation Methods 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229920001281 polyalkylene Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229930001119 polyketide Natural products 0.000 description 1
- 125000000830 polyketide group Chemical group 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 150000004032 porphyrins Chemical class 0.000 description 1
- ZNNZYHKDIALBAK-UHFFFAOYSA-M potassium thiocyanate Chemical compound [K+].[S-]C#N ZNNZYHKDIALBAK-UHFFFAOYSA-M 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 239000002987 primer (paints) Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- JKANAVGODYYCQF-UHFFFAOYSA-N prop-2-yn-1-amine Chemical compound NCC#C JKANAVGODYYCQF-UHFFFAOYSA-N 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 229940039716 prothrombin Drugs 0.000 description 1
- 238000002708 random mutagenesis Methods 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 238000006479 redox reaction Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229920002477 rna polymer Polymers 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- RHFUOMFWUGWKKO-UHFFFAOYSA-N s2C Natural products S=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 RHFUOMFWUGWKKO-UHFFFAOYSA-N 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 229910052706 scandium Inorganic materials 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- 230000037387 scars Effects 0.000 description 1
- 150000003333 secondary alcohols Chemical class 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- 238000010187 selection method Methods 0.000 description 1
- JRPHGDYSKGJTKZ-UHFFFAOYSA-N selenophosphoric acid Chemical compound OP(O)([SeH])=O JRPHGDYSKGJTKZ-UHFFFAOYSA-N 0.000 description 1
- HKZLPVFGJNLROG-UHFFFAOYSA-M silver monochloride Chemical compound [Cl-].[Ag+] HKZLPVFGJNLROG-UHFFFAOYSA-M 0.000 description 1
- KZJPVUDYAMEDRM-UHFFFAOYSA-M silver;2,2,2-trifluoroacetate Chemical compound [Ag+].[O-]C(=O)C(F)(F)F KZJPVUDYAMEDRM-UHFFFAOYSA-M 0.000 description 1
- 238000006884 silylation reaction Methods 0.000 description 1
- 230000015590 smooth muscle cell migration Effects 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 150000003505 terpenes Chemical class 0.000 description 1
- 235000007586 terpenes Nutrition 0.000 description 1
- FGTJJHCZWOVVNH-UHFFFAOYSA-N tert-butyl-[tert-butyl(dimethyl)silyl]oxy-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)O[Si](C)(C)C(C)(C)C FGTJJHCZWOVVNH-UHFFFAOYSA-N 0.000 description 1
- 125000005207 tetraalkylammonium group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 125000005413 thiopyridyl group Chemical group 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- 238000007070 tosylation reaction Methods 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000006276 transfer reaction Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- XQKBFQXWZCFNFF-UHFFFAOYSA-K triiodosamarium Chemical compound I[Sm](I)I XQKBFQXWZCFNFF-UHFFFAOYSA-K 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- WVLBCYQITXONBZ-UHFFFAOYSA-N trimethyl phosphate Chemical compound COP(=O)(OC)OC WVLBCYQITXONBZ-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- 210000005167 vascular cell Anatomy 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Images














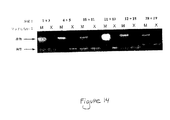









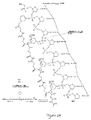

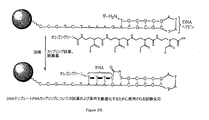

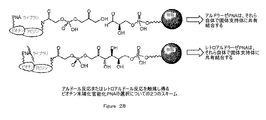

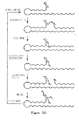




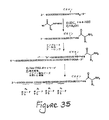











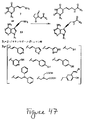










Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1068—Template (nucleic acid) mediated chemical library synthesis, e.g. chemical and enzymatical DNA-templated organic molecule synthesis, libraries prepared by non ribosomal polypeptide synthesis [NRPS], DNA/RNA-polymerase mediated polypeptide synthesis
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P1/00—Preparation of compounds or compositions, not provided for in groups C12P3/00 - C12P39/00, by using microorganisms or enzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/26—Preparation of nitrogen-containing carbohydrates
- C12P19/28—N-glycosides
- C12P19/30—Nucleotides
- C12P19/34—Polynucleotides, e.g. nucleic acids, oligoribonucleotides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
Abstract
【解決手段】1つ以上の化学物質を合成する方法のための組成物であって、該方法は、以下の工程:
1つ以上のテンプレートを提供する工程であって、該1つ以上のテンプレートは、該テンプレートに会合する反応性単位を必要に応じて有する、工程;
1つ以上のアンチコドンの該テンプレートへのハイブリダイゼーションおよび反応性単位の反応を可能にする条件下で、アンチコドンを有する1つ以上の移動単位および反応性単位を、該1つ以上のテンプレートに接触させる工程、
を包含する。
【選択図】なし
Description
新たな機能を有する分子を作製するための古典的「化学アプローチ」は、薬物の発見から合成方法論まで、そして材料科学までの範囲にわたる適用において、前世紀をとおして広範に用いられている。このアプローチ(図1、黒色)では、研究者は、候補分子を合成または単離し、これらの候補を所望の特性についてアッセイし、未知の場合は活性化合物の構造を決定し、このアッセイおよび構造データに基づいて構造活性相関をまとめ、次いで改善された特性を保有するように設計された新世代の分子を合成する。コンビナトリアルケミストリーの方法(例えば、非特許文献1;非特許文献2;非特許文献3を参照のこと)はこのアプローチのスループットを増大させてきたが、その基本的限界は変化していないままである。いくつかの要因は、分子機能を生じる化学的アプローチの有効性を制限している。第一に、新たな機能をもたらすのを構造変化を正確に予測する能力は、しばしば、分子のわずかなコンホメーション再配置、不測の溶媒相互作用または結合事象もしくは反応事象の未知の立体化学的必要条件に起因して不充分である。構造活性相関より得られる複雑さは、頻繁に、合理的リガンド設計もしく触媒設計の成功(ハイスループット様式で行われる試みを含む)を制限する。第二に、候補のコレクションの各メンバーを、選択するのではなく、アッセイまたはスクリーニングする必要性は、各実験において探索され得る分子数を制限する。最終的に、合成分子を増幅する方法がないことは、特徴付け、スクリーニングおよび構造解明のために産生されなければならない物質の最少量を必要条件とする。その結果、ほぼ106より多くの異なる合成化合物のライブラリーを作製することは困難であり得る。
本願は、米国仮特許出願第60/277,081号(2001年3月19日出願;発明の名称「Nucleic Acids Directed Synthesis of Chemical Compounds」);同第60/277,094号(2001年3月19日出願、発明の名称「Approaches to Generating New Molecular Function」);および同第60/306,691号(2001年7月20日;発明の名称「Approaches to Generating New Molecular Function」)(これらの出願の各々の全内容は、本明細書中に参考として援用される)に対する米国特許法119(e)条のもとでの優先権を主張する。
核酸およびタンパク質以外のクラスの分子を増幅および進化させ得る必要性の認識は、本発明をもたらし、テンプレートによって指向される、分子の合成、増幅および進化のための方法および組成物を提供した。一般に、これらの方法は、進化可能なテンプレートを用いて、化合物または化合物のライブラリーの合成を導く(すなわち、テンプレートは実際に、化合物の合成をコードする)。核酸のようなテンプレートを用いてコードまたは合成されるライブラリーに基づいて、ライブラリーを増幅、進化およびスクリーニングするための方法が提供される。特に目的の特定の実施形態では、化合物は、核酸でもそのアナログでもなく、核酸にもそのアナログにも類似していない化合物である。特定の実施形態では、テンプレートにコードされるこれらのコンビナトリアルライブラリーの化合物は高分子であり、より好ましくは非天然の高分子(すなわち、天然のペプチド、タンパク質およびポリヌクレオチドを除く)である。他の実施形態では、これらの化合物は、低分子である。
本発明は、例えば、以下を提供する:
(項目1)
1つ以上の化学物質を合成する方法であって、該方法は、以下の工程:
1つ以上のテンプレートを提供する工程であって、該1つ以上のテンプレートは、該テンプレートに会合する反応性単位を必要に応じて有する、工程;
1つ以上のアンチコドンの該テンプレートへのハイブリダイゼーションおよび反応性単位の反応を可能にする条件下で、アンチコドンを有する1つ以上の移動単位および反応性単位を、該1つ以上のテンプレートに接触させる工程、
を包含する、方法。
(項目2)
項目1に記載の方法であって、前記1つ以上の化学物質の一部分が、1つ以上の核酸テンプレートにハイブリダイズするヌクレオチド配列を含むアンチコドンを含有する、方法。
(項目3)
1つより多い化合物のライブラリーが合成される、項目1に記載の方法であって、該方法は、以下の工程:
所望の特性を提示する前記反応性単位の反応産物を含むライブラリーメンバーを検出するためにライブラリーを検索する工程;
該反応産物についての構造情報を検出する工程;および
反応産物の新規の産生を生じるために、1つ以上のさらなる種を用いて該方法を繰り返す工程であって、該反応産物の少なくともいくつかが該所望の特性を提示する、工程、
をさらに包含する、方法。
(項目3)
合成された前記化学物質が、核酸または核酸アナログ以外の化合物である、項目1に記載の方法。
(項目4)
前記化学物質が非天然の高分子である、項目1に記載の方法。
(項目5)
前記化学物質が低分子である、項目1に記載の方法。
(項目6)
前記化学物質は低分子であり、そして接触させる工程が、一連の様式で2つ以上の移動単位を接触させる工程を包含する、項目1に記載の方法。
(項目7)
前記テンプレートが核酸テンプレートである、項目1に記載の方法。
(項目8)
前記テンプレートがDNAまたはRNAを含む、項目1に記載の方法。
(項目9)
前記テンプレートがDNAを含む、項目1に記載の方法。
(項目10)
1つ以上の化学物質を含むライブラリーであって、該化学物質の各々は、増幅可能なテンプレートに結合され、該テンプレートのヌクレオチド配列は化学物質の構造の情報を与える、ライブラリー。
(項目11)
前記化学物質のライブラリーが、低分子のライブラリーを含む、項目10に記載のライブラリー。
(項目12)
化学物質のライブラリーの合成方法であって、該方法は、以下の工程:
1つ以上の反応性単位と、必要に応じて会合される1つ以上のテンプレートを提供する工程;
該1つ以上のテンプレートと1つ以上の移動単位とを、同時にかまたは連続して接触させる工程であって、該移動単位は、複数の異なるライブラリーメンバーを産生するために、該アンチコドンの該テンプレートへのハイブリダイゼーションおよび反応性単位の反応に適切な条件下で、1つ以上の反応単位と会合するアンチコドンを含む、工程、
を包含する、方法。
(項目13)
前記化合物の一部分が、1つ以上の核酸テンプレートとハイブリダイズするヌクレオチド配列を含むアンチコドンを含有する、項目12に記載の方法。
(項目14)
項目12の方法であって、該方法は、以下の工程:
所望の特性を提示する反応性単位の反応産物を含むライブラリーメンバーを検出するためのライブラリーを探索する工程;
該反応産物についての構造的情報を検出する工程;および
反応産物の新規の産生を生じるために、1つ以上のさらなる種を用いて該方法を繰り返す工程であって、該反応産物の少なくともいくつかが該所望の特性を提示する、工程、
をさらに包含する、方法。
(項目15)
前記所望の特性が、標的タンパク質に結合することを含む、項目14に記載の方法。
(項目16)
前記所望の特性が、化学反応を触媒することを含む、項目14に記載の方法。
(項目17)
1つ以上の化学物質を単離する工程をさらに包含する、項目14に記載の方法。
(項目18)
前記合成された化学物質が、低分子である、項目14に記載の方法。
(項目19)
1つ以上の非天然の高分子の合成のための方法であって、該方法は、以下の工程:
1つ以上の核酸テンプレートを提供する工程;
ハイブリダイゼーションおよび該テンプレートに沿って並ぶ隣接するモノマー単位の間に結合を形成するための反応を可能にする条件下で、1つ以上の移動単位と、1つ以上の核酸テンプレートとを接触させる工程、
を包含する、方法。
(項目20)
非天然の高分子を単離する工程をさらに包含する、項目19に記載の方法。
(項目21)
前記非天然の高分子の一部分が、1つ以上の核酸テンプレートにハイブリダイズするヌクレオチド配列を含むアンチコドンを含有する、項目19に記載の方法。
(項目22)
項目19に記載の方法であって、該方法は、以下の工程:
所望の特性を提示する反応性単位の反応産物を含むライブラリーメンバーを検出するためにライブラリーを探索する工程;
反応産物についての構造情報を検出する工程;
反応産物の新規の産生を生じるために、1つ以上のさらなる種を用いて該方法を繰り返す工程であって、該反応産物の少なくともいくつかが該所望の特性を提示する、工程、
をさらに包含する、方法。
(項目24)
前記所望の特性が化学反応を触媒することを含む、項目23に記載の方法。
(項目25)
前記核酸テンプレートがDNAである、項目12または19に記載の方法。
(項目26)
前記核酸テンプレートが一本鎖である、項目12または19に記載の方法。
(項目27)
前記核酸テンプレートが二本鎖である、項目12または19に記載の方法。
(項目28)
前記核酸テンプレートがDNA、RNA、DNAとRNAのハイブリッド、DNA誘導体、およびRNA誘導体からなる群より選択される、項目12または19に記載の方法。
(項目29)
前記アンチセンスコドンが、2、3、4、5、6、7、8、9または10塩基を含む、項目12または19に記載の方法。
(項目30)
前記アンチコドンがフレームシフトし得ない、項目12または19に記載の方法。
(項目31)
前記アンチコドンがDNA、RNA、DNAとRNAのハイブリッド、DNA誘導体、およびRNA誘導体からなる群より選択される、項目12または19に記載の方法。
(項目32)
前記アンチコドンが共有結合を介して、モノマー単位と会合される、項目12または19に記載の方法。
(項目33)
化合物のライブラリーを進化させる方法であって、該方法は、以下の工程:
所望の活性を有する化合物を単離する工程であって、該化合物が、該化合物の合成がコードされる核酸テンプレートに結合されている、工程;
核酸テンプレートを変異および増幅させる工程;ならびに
変異および増幅された核酸テンプレートを用いて、関連する化合物の新規のライブラリーを合成する工程、
を包含する、方法。
(項目34)
項目33に記載の方法であって、該方法は、以下の工程:
所望の活性について化合物をアッセイする工程;および
所望の活性を有する高分子のテンプレートに基づいて、項目33に記載の工程を繰り返す工程、
をさらに包含する、方法。
(項目35)
前記所望の活性が、触媒活性および結合活性からなる群より選択される、項目34に記載の方法。
(項目36)
前記変異および増幅の工程が、ポリメラーゼ連鎖反応を用いて実行される、項目34に記載の方法。
(項目37)
前記変異および増幅の工程が、エラープローンPCRを用いて実行される、項目34に記載の方法。
(項目38)
前記変異および増幅の工程が、インビトロ相同的組み換え(DNAシャフリング)を用いて実行される、項目34に記載の方法。
(項目39)
1つ以上の化学物質を含む、化学物質のライブラリーであって、ここで、該化学物質の各々は増幅可能なテンプレートに結合され、該テンプレートのヌクレオチド配列は化学物質の構造の情報を与え、そして該ライブラリーは項目1、12または19に従って合成される、ライブラリー。
(項目40)
前記ライブラリーが、低分子または非天然の高分子のライブラリーである、項目39に記載のライブラリー。
(項目41)
1位以上の核酸テンプレートおよび1つ以上の移動単位を備える、キット。
(項目42)
緩衝液、熱安定性DNAポリメラーゼ、ヌクレオチド、および制限エンドヌクレアーゼをさらに備える、項目41に記載のキット。
(項目43)
前記核酸テンプレートが、低分子の骨格と会合される、項目41に記載のキット。
(項目44)
複数の核酸テンプレートを備える、項目41に記載のキット。
(項目45)
前記移動単位が、モノマー単位を含む、項目41に記載のキット。
(項目46)
前記移動単位が、低分子の骨格を改変するために使用される反応物を備える、項目41に記載のキット。
用語抗体とは、天然であるか、または完全もしくは部分的に合成によって生成されたかによらず、免疫グロブリンをいう。特異的結合能力を保持するその全ての誘導体もまた、この用語に含まれる。この用語はまた、免疫グロブリン結合ドメインに相同であるかまたは大部分相同である結合ドメインを有する任意のタンパク質を包含する。これらのタンパク質は、天然の供給源から誘導され得るか、または部分的もしくは完全に合成によって生成され得る。抗体は、モノクローナルまたはポリクローナルであり得る。抗体は、ヒトの以下のクラスのいずれかを含め、任意の免疫グロブリンクラスのメンバーであり得る:IgG、IgM、IgA、IgD、およびIgE。しかし、IgGクラスの誘導体は、本発明において好ましい。
上記に議論されるように、化学物質(限定されないが低分子および高分子を含む)が、生体高分子(例えば、ポリヌクレオチドおよびタンパク質)増幅し、そして進化し得るのと同じ方法で、成長および増幅し得ることが所望される。DNAテンプレート合成は、DNA配列における情報を合成的低分子へと翻訳する可能な手段を提供することが証明された。一般的に、1つの反応物に結合されるDNAテンプレートは、産物を得るために、相補的なDNA分子に結合される第2の反応基を補充され得る。DNAハイブリダイゼーションは、配列特異的であるので、DNAテンプレート反応の結果は、対応する反応産物への特定のDNA配列の翻訳である。図2に示されるように、相補的なオリゴヌクレオチド(T.Inoueら J.Am.Chem.Soc.1981,103,7666;L.Inouら J.Mol.Biol.1984,178,669−76)の配列特異的オリゴマー形成を触媒するような一本鎖核酸テンプレートの能力が、示された。この発見のすぐ後に、DNAテンプレートまたはRNAテンプレートが相補的なDNAまたはRNAの、モノヌクレオチド、ジヌクレオチド、トリヌクレオチドまたはオリゴヌクレオチドの重合を触媒し得ることが見出された(T.Inoueら J.Am.Chem.Soc.1981,103,7666;L.E.Orgelら Acc.Chem.Res.1995,28,109−118;H.Remboldら J.Mol.Evol.1994,38,205;L.Rodriguezら J.Mol.Evol.1991,33,
477;C.B.Chenら J.Mol.Biol.1985,181,271)。DNAテンプレートまたはRNAテンプレートは、種々の非天然核酸アナログ(ペプチド核酸(C.Bohlerら Nature 1995,376,578)、ホスホロチオネート含有核酸(M.K.Herrleinら J.Am.Chem.Soc.1995,117,10151−10152)、ホスホロセレネート含有核酸(Y.Xuら J.Am.Chem.Soc.2000,122,9040−9041;Y.Xuら Nat.Biotechnol.2001,19,148−152)、およびホスホロアミデート含有核酸(A.Lutherら Nature 1998,396,245−8)核酸、非リボース核酸(M.Bolliら Chem.Biol.1997,4,309−20)およびリン酸結合がアミノエチル基(Y.Gatら Biopolymers 1998,48,19−28)で置き変えられたDNAアナログを含む)の形成を加速することを示した。核酸テンプレートはまた、ヌクレオチドアナログの間のアミンのアシル化を触媒し得る(R.K.Bruickら Chem.Biol.1996,3,49−56)。
上記に議論されるように、1つ以上のテンプレートが、本発明の方法において使用され、そして移動単位にハイブリダイズして化学物質の合成を指示する。当業者に理解されるように、任意のテンプレートは、本発明の方法および組成物に使用され得る。変異されそしてそれによって成長したテンプレートを使用して、本明細書中に記載されるような別の化学物質または化学物質のライブラリーの合成を導き得る。本明細書中により詳細に記載されるように、成長可能なテンプレートは、化学物質の合成をコードし、そして化学物質の合成履歴を後にデコードするために、化学物質を間接的に増幅するために、そして/または化学物質を進化する(すなわち、多様化、選択および増幅)ために使用され得る。特定の実施形態において、成長可能なテンプレートは、核酸である。本発明の特定の実施形態において、テンプレートは、核酸に基づく。
上記のように、本発明の方法において、アンチコドンおよび反応性単位を含む移動単位はまた提供される。本発明において使用されるアンチコドンは、核酸テンプレート内に存在するコドンに相補的であるように設計され、そしてその中で使用される核酸テンプレートおよびコドンを考慮して設計されるべきであることが理解される。例えば、テンプレートに使用される配列ならびにコドンの長さは、アンチコドンの設計において考慮する必要がある。テンプレートに使用されるコドンに相補的である任意の分子が、本発明の方法において使用され得る(例えば、ヌクレオチドまたは非天然ヌクレオチド)。特定の他の実施形態において、コドンは、自然界で見出される1つ以上の塩基(すなわち、チミジン、ウラシル、グアニジン、シトシン、アデニン)を含む。特定の他の実施形態において、アンチコドンは、塩基、糖および任意のリン酸基を有する通常自然界で見出される1つ以上のヌクレオチドを含む。なお他の実施形態において、塩基は、通常自然界で見出される糖−リン酸骨格(例えば、非天然ヌクレオチド)でない骨格に沿って延長される。
種々の化合物および/またはライブラリーが、本発明の方法を用いて調製され得ることが理解される。上記のように、特別な目的の特定の実施形態において、核酸でもそのアナログでもなく、それらに似てもいない化合物が、本発明の方法に従って合成される。
の実施例2および4に記載されるような水性媒体および有機媒体中の反応を参照のこと。移動単位は、ヒドリド化(hydridizing)条件下でテンプレートを接触させ、そして接着させられた反応物または構築ブロックは、低分子骨格の部位と反応させられる。特定の実施形態において、低分子テンプレート上の保護基は、機能付与される部位から1つずつ除去され、移動単位の反応物は、骨格上の所望の位置のみで反応する。当業者によって理解されるように、アンチコドンは、リンカー部分を介して反応物と結合され得る(実施例3を参照のこと)。リンカーは、反応物と低分子骨格との接触を容易にし、そして特定の実施形態において、所望の反応に依存して、脱離基としてDNAを位置づける(「オートクレーブ可能な」ストラテジー)、または「瘢痕なしの」リンカーストラテジー(これは、さらなる化学機能を残すことなく産物を産出する)、もしくは「有用な瘢痕」ストラテジー(ここで、リンカーが残され、そしてリンカーの切断後の実質的な工程で機能付与され得る)を介してテンプレートに反応基を連結し得る。反応条件、リンカー、反応物、および機能付与される部位は、分子間反応を避け、そして分子内反応を促進するように選択される。本発明の方法が、合成される特定の化合物に依存して、テンプレートと移動単位とを連続的に接触および同時に接触させることの両方を企図することがまた、理解される。特別な目的の特定の実施形態において、化学物質の多工程合成が提供され、ここで、テンプレートは、2つ以上の移動単位と連続的に接触されて複雑な化学物質の多工程合成を容易にする。
本発明の方法において、上述のような核酸テンプレートは、非天然の高分子、低分子、または任意の他の型の目的の分子の合成を指向するように提供される。一般に、複数の核酸テンプレートが提供され、ここで、提供される異なる配列の数の範囲は、2〜1015である。本発明の1つの実施形態において、複数の核酸テンプレートが提供され、好ましくは少なくとも100個の異なる核酸テンプレート、より好ましくは少なくとも10000個の異なる核酸テンプレート、そして最も好ましくは少なくとも1000000個の異なる核酸テンプレートが提供される。提供される各テンプレートは、特定の非天然の高分子または低分子の合成をコードするために使用される特有の核酸配列を含む。上述のように、テンプレートはまた、重合化が開始される第1級アミンのような機能性を有し得るか、または低分子骨格であり得る。特定の実施形態において、核酸テンプレートは、1つの「ポット(pot)」に提供される。特定の実施形態において、テンプレートは、水性媒体中に提供され、そしてこれに続く反応が、水性媒体中で実行される。
本発明の方法および組成物は、所望の特性を有する分子を生成するための新しい方法を提供する。このアプローチは、非常に強力な遺伝的方法と一緒になり、分子生物学者は、有機化学の柔軟性および力を数十年間利用してきた。遺伝的選択による非天然の高分子の調製、増幅および展開の能力は、活性、バイオアベイラビリティ、安定性、蛍光、感光、またはタンパク質および核酸中に見出される制限された1組の構築ブロックを用いて達成することが困難であるか、もしくは不可能である他の特性を保有する、新しいクラスの触媒を導き得る。同様に、変異および選択の反復する周期によって低分子を調製、増幅、および展開するために開発された新しいシステムは、より遅い従来の薬物送達方法によって単離されたものよりも優れた特性を有する新規リガンドまたは薬物の単離を導き得る(実施例7を参照のこと)。
本発明はまた、本発明の方法における使用のためのキットおよび組成物を提供する。このキットは、本発明の実施において有用な、任意の物品または組成物を備え得る。このキットは、テンプレート、アンチコドン、移動単位、モノマー単位、構築ブロック、反応物、低分子骨格、緩衝液、溶媒、酵素(例えば、熱安定ポリメラーゼ、逆転写酵素、リガーゼ、制限エンドヌクレアーゼ、エキソヌクレアーゼ、クレノウフラグメント、ポリメラーゼ、アルカリホスファターゼ、ポリヌクレオチドキナーゼ)、リンカー、保護基、ポリヌクレオチド、ヌクレオシド、ヌクレオチド、塩、酸、塩基、固体支持体、または任意のこれらの組み合わせを備え得るが、これらに限定されない。
以下に示される代表的な実施例は、発明の例示を助けることを意図し、本発明の範囲を制限することを意図せず、またそのように解釈すべきでない。確かに、本明細書中に示され、そして記載されるものに加えて、本発明の種々の改変および多くのさらなるその実施形態は、この文書の全部の内容から当業者に理解され、この文書は、本明細書中で引用される科学文献および特許文献に従い、そしてこれらを参照する例を含む。引用された参照の内容が、当該分野の状態の例示を助けるために、本明細書中で参考として援用されることが、さらに理解される。
明瞭に、上述の低分子展開アプローチを実行するために、DNAテンプレート化合成の普遍性の確立が必要とされる。第1に、本発明は、このアプローチに対する普遍性を確立し、従ってDNAテンプレート化合成を用いた種々の化学的化合物の合成を可能にする。図6aに示されるように、液相DNAテンプレート化合成を支持する2つのDNA構成の能力を確立した。求電子マレイミド基を有するヘアピン(H)テンプレートおよびエンドオブヘリックス(E)テンプレートは、相補的DNAオリゴヌクレオチドに連結した1当量のチオール試薬と効果的に反応して、−25℃でチオエーテル生成物を生じた。DNAテンプレート化反応速度(κapp=約105M−1s−1)は、HおよびEの構造に類似していた一方で、その反応基の相対的な方向とは有意に異なっていた。反対に、配列不適合を含む試薬を用いた場合、または過剰のβ−メルカプトエタノールで予めクエンチされたテンプレートを用いた場合、生成物は観察されなかった(図6a)。故に、両方のテンプレートは、たとえ生じた産物の構造が、天然のDNA骨格の構造と著しく異なっていたとしても、マレイミドへのチオールの配列特異的DNAテンプレート化添加を支持する。反応条件(pH7.5、25℃、250mM NaCl、60nM テンプレートおよび試薬)下でテンプレートの分子間反応物は、ほとんどまたは全く観察されない。
(DNA合成) DNAオリゴヌクレオチドを、標準的なプロトコルを用いてPerSeptive Biosystems Expedite 8909 DNA合成機で合成し、そして逆相HPLCによって精製した。オリゴヌクレオチドを、分光光度的および変性ポリアクリルアミドゲル電気泳動(PAGE)に続くエチジウムブロマイドまたはSYBR Green(Molecular Probes)を用いる染色およびStratagene Eagle Eye II濃度計を用いる定量化によって定量化した。ホスホルアミダイト(5’−NH2−dT基、5’テトラクロロフルオレセイン基、無塩基の骨格スペーサー基、C3骨格スペーサー基、9−結合ポリエチレングリコールスペーサー基、12−結合飽和炭化水素スペーサー基、および5’ビオチン基の合成を可能にする)は、Glen Researchから購入された。チオール連結オリゴヌクレオチド試薬を、C3ジスルフィドコントロールドポアガラス(Glen Research)上で合成した。
SIAB反応およびSBAP反応において適合した試薬:5’−CCCGAGTCGAAGTCGTACC−SH;図6b SIAB反応およびSBAP反応において不適合な試薬:5’−GGGCTCAGCTTCCCCATAA−SH;図6b、図6c、図6d、および図8aにおける他の反応についての不適合な試薬;5’−FAAATCTTCCC−SH(F=テトラクロロフルオレセイン);1つのミスマッチを含む図6cおよび図6dにおける試薬:5’−FAATTCTTACC−SH;図6a、図6b、および図8a SMCC反応、GMBS反応、BMPS反応、およびSVSB反応におけるEテンプレート:5’−(NH2dT)−CGCGAGCGTACGCTCGCGATGGTACGAATTCGACTCGGGAATACCACCTTCGACTCGAGG;図6b SIAB反応、SBAP反応、およびSIA反応におけるHテンプレート:5’−(NH2dT)−CGCGAGCGTACGCTCGCGATGGTACGAATTC;図8bにおけるクランプオリゴヌクレオチド:5’−ATTCGTACCA。
上記されるように、DNAテンプレート合成化学の一般性を、試験した(Liuら、J.Am.Chem.Soc.2001,123,6961を参照のこと)。詳細には、DNA様コンフォメーションへの反応性基の正確な整列化を必要とせずに、化学反応の適度な収集に関するDNAテンプレート合成の能力を実証した。実際に、DNAテンプレート合成の距離非依存性および配列の正確さは、1,000を越えるテンプレートのモデルライブラリを、対応するチオエステル生成物へと同時にワンポット変換させ、これらの1つを、タンパク質ストレプトアビジンへの結合についてインビトロでの選択によって濃縮し、そしてPCRによって増幅した。
官能化したテンプレートおよび試薬を、代表的には、5’−NH2末端化オリゴヌクレオチド(テンプレート1について)、5’−NH2−(CH2O)2末端化オリゴヌクレオチド(他の全てのテンプレートについて)または3’−OPO3−CH2CH(CH2OH)(CH2)4NH2末端化ヌクレオチド(全ての試薬について)を、0.2Mリン酸ナトリウム緩衝液(pH7.2)中の適切なNHSエステル(DMF中20mg/mL溶液の0.1容量)と25℃で1時間、反応させることによって調製して、図13および図15に示される構造のテンプレートおよび試薬を提供した。アミノ酸連結試薬6〜9について、0.2Mリン酸ナトリウム緩衝液(pH7.2)中の3’−OPO3CH2CH(CH2OH)(CH2)4NH2末端化オリゴヌクレオチドを、DMF中100mMビス[2−(スクシンイミジルオキシカルボニルオキシ)エチル]スルホン(BSOCOES,Pierce)溶液の0.1容量と25℃で10分間、続いて300mM NaOH中の300mMアミノ酸の0.3容量と25℃で30分間反応させた。
(例示的なリンカーの開発)
当業者に理解されるように、ゲル電気泳動による分析を容易にすることは、反応の進行の間、生成物に結合する試薬のDNA部分を除去するためにしばしば有用である。しかし、インビトロ選択に適当な合成小分子の対応するライブラリーへ、DNAのライブラリーを翻訳するためにDNAテンプレート合成を使用することは、試薬の反応性基をそれらの解読DNAオリゴヌクレオチドと結合する切断可能なリンカーの開発を必要とする。以下および本明細書中に記載されるように、3つの例示的な型のリンカーが、開発されてきた(図18を参照のこと)。1つの反応性基を有する試薬について、反応部分へ脱離基としてのDNAを位置決めすることが望ましい。この「自己切断可能な」リンカーの戦略下で、DNA反応性基の結合が、反応の自然な結果として切断される。しかし、このアプローチの1つの例として、蛍光Wittigホスホラン試薬(14、図19を参照しながら)を合成し、ここで解読DNAオリゴヌクレオチドは、アリールホスフィン基の1つに結合される(図19左を参照のこと)。アルデヒド結合テンプレートを用いたDNAテンプレートWittig反応は、蛍光基の、Wittig試薬からテンプレートへのほとんど定量的な移動および試薬のDNA部分からのアルケン生成物の同時の遊離が生じる。さらに、1つを超える反応性基を有する試薬は、2つのさらなるリンカー戦略の1つを介して、それらの解読DNAオリゴヌクレオチドに結合され得る。「スカーレス(scarless)」リンカー戦略において、1つの反応性基のDNAテンプレート反応の後、第2の反応性基を介して結合されるリンカーの切断が生じ、あとにさらなる化学官能基を残さずに生成物を生じる。例えば、解読DNAオリゴヌクレオチドにカルバモイルエチルスルホンリンカーを介して結合された一連のアミノ酸試薬を、合成した(図19、中央)。これらのアミノ酸試薬を使用するDNAテンプレートアミド結合形成の生成物を、水性アルカリ緩衝液で処理し、カルバモイル基の定量的な脱離および自発的な脱炭酸を行った。従って、このスカーレスリンカーの脱離の生成物は、完全に移行されたアミノ酸部分である。本発明のなお他の実施形態において、第3のリンカー戦略(「有用なスカー(scar)」)は、有用な化学基を誘導することが、リンカー切断の結果として有利であり得る戦略において使用され得る。特に、「有用なスカー」は、以後の工程において、官能化され得、リンカー切断後に残される。例えば、1,2−ジオールを介して解読DNAオリゴヌクレオチドへと連結されるアミノ酸試薬を、作製した。アミド結合生成に続いて、このリンカーを、NaIO4を用いた酸化によって定量的に切断し、有用なアルデヒド基を保持する生成物を得た(図19、右を参照のこと)。すぐ上に記載されるリンカーに加えて、種々のリンカーを使用し得る。例えば、図20に示されるように、チオエステルリンカーを、チオール末端化DNAとカルボン酸含有試薬とのカルボジイミド媒介性カップリングによって、生成し得、水性塩基で切断し得る。カルボキシレート基が、上記に記載されるDNAテンプレートアミド結合形成反応への入り口を提供するために、このリンカーは、切断されたとき、「有用なスカー」を遊離する(図20を参照のこと)。あるいは、チオエステルリンカーを、Ag(I)カチオンの存在下で、アミンアシル化反応の間、自己切断可能なリンカーとして使用し得る(Zhangら、J.Am.Chem.Soc.1999,121,3311−3320を参照のこと)。なぜならこの試薬のチオールDNA部分が、反応の自然の結果として遊離されるからである。pH11で酸化され除去され、ビニルスルホンを遊離し得るチオエステルリンカーが、「有用なスカー」リンカーとして使用され得ることは、理解される。本明細書中に示されるように、このビニルスルホン基は、続いてのDNAテンプレート反応の多くにおいて基質として作用する。
(有機溶媒中の例示的な反応:)
本明細書中に示されるように、種々のDNAテンプレート反応は、水性溶媒中で生じ得る。以下に考察されるように、DNAテンプレート反応が、有機溶媒中で生じ得、従って、DNAテンプレート合成の範囲を大いに拡大することもまた示されてきた。具体的には、DNAテンプレートおよびDNA試薬は、長鎖のテトラアルキルアンモニウムカチオンと複合体を形成し得(Jostら、Nucleic Acids Res.1989,17,2143;Mel’nikovら、Langmuir,1999,15,1923−1928を参照のこと)、CH2Cl2、CHCl3、DMFおよびMeOHを含む無水有機溶媒中の反応成分の定量的な溶解を可能にする。驚いたことに、DNAテンプレート合成が、実は、高い配列選択性を有する無水有機溶媒において生じ得ることが見出された。DNAテンプレートアミド形成反応(reacation)が、図2に示され、ここで、試薬およびテンプレートが、別々の容器中または前もってアニーリングされた後、水中のいずれかにおいて、ジメチルジドデシルアンモニウムカチオンと複合体を形成し、乾燥状態まで凍結乾燥され、CH2Cl2に溶解され、そして一緒に混合される。マッチした反応は、反応物を水溶液中で前もってアニーリングした場合およびこれらの反応物をCH2Cl2中で最初に混合した場合に、生成物を提供したが、ミスマッチの反応は、生成物を提供しなかった(図21を参照のこと)。無水DMF中でのDNAテンプレートアミド形成およびPd媒介性Heckカップリングもまた、配列特異的に進行した。明らかに、有機溶媒中での配列特異的DNAテンプレート合成のこれらの観察は、有機媒体中のテトラアルキルアンモニウム複合体化DNA中の少なくともいくつかの二次構造の存在を意味し、DNAレセプターおよびDNA触媒を、有機溶媒中での立体選択的結合特性および立体選択的触媒特性の方へ進化させることを可能にするはずである。具体的には、共役付加、付加環化、置換反応、およびPd媒介性カップリングを含む、水性媒体中で生じることが公知であるDNAテンプレート反応は、有機溶媒中でもまた実施され得る。特定の他の実施形態において、水中で実施することが非効率または不可能な有機溶媒中の反応が使用され得る。例えば、水中でのRu触媒オレフィン置換が、Grubbsおよびその共同研究者らによって報告されているのに対して(Lynnら、J.Am.Chem.Soc.1998,120,1627−1628;Lynnら、J.Am.Chem.Soc.2000,122,6601−6609;Mohrら、Organometallics 1996,15,4317−4325を参照のこと)、水性置換系は、極端に官能基感受性である。しかし、有機溶媒におけるRu触媒オレフィン置換の耐性官能基は、有意に、より強靭である。有機溶媒中で使用するためのいくつかの例示的な反応としては、基底状態の出発物質よりも極性ではない遷移状態を通って進行し得る、ニトロンとオレフィンとの間の1,3−双極子環状付加が挙げられるがこれに限定されない。
(例示的な化合物および化合物のライブラリーの合成:)
(A)ポリカルバメートライブラリーの合成:)
上記に記載される戦略の1つの実施形態は、増幅可能なポリカルバメートライブラリーの作製である。ライブラリーをコードするために使用される16の可能なジヌクレオチドのうち、1つは、開始コドンの機能を割り当てられ、そして1つは、終始コドンとして作用するよう割り当てられる。次いで、異なるモノマーに対して14までの残された各ジヌクレオチドを割り当てる、人工的な遺伝コードを作製する。幾何学的な理由のために、1つのモノマーは、実際に2つの側鎖を含むジカルバメートを含む。各モノマー中で、ジカルバメートは、フッ素を用いた処理の際に、ネイティブなDNAおよび遊離カルバメートを遊離するシリルエノールエーテルリンカーを介して、対応するジヌクレオチド(tRNAアンチコドンに類似)へと結合される。このジヌクレオチド部分は、活性化された5’−2−メチルイミダゾールホスフェートとして存在することが示され(Inoueら、J.Mol.Biol.162:201,1982;Remboldら、J.Mol.Evol.38:205,1994;Chenら、J.Mol.Biol.181:271,1985;Acevedoら、J.Mol.Biol.197:187,1987;Inoueら、J.Am.Chem.Soc.103:7666,1981;各々は、参考として本明細書中に援用される)、ヌクレオチドのテンプレート指向性オリゴマー化のための優れた脱離基として作用するが、この脱離基は、中性水性条件下または塩基性水性条件下で、比較的安定である(Schwartzら、Science 228:585,1985;参考として本明細書中に援用される)。ジカルバメート部分は、ビニルオキシカーボネート(vinyloxycarbonate)リンカーを介して結合された環状形態で存在する。ビニルカーボネート基は、中性水性条件下または塩基性水性条件下で、安定であることが示され(Olofsonら、Tetrahedron Lett.18:1563,1977;Olofsonら、Tetrahedron Lett.18:1567,1977;Olofsonら、Tetrahedron Lett.18:1571,1977;各々は、参考として本明細書中に援用される)、さらに、アミンの添加の際に、非常に高収率でカルバメートを提供することが示された(Olofsonら、Tetrahedron Lett.18:1563,1977;参考として本明細書中に援用される)。

新生ポリカルバメート鎖からのアミンによって攻撃される場合、m−クレゾールの芳香族化によって推進されるビニルカルボネートリンカーは、遊離アミンを遊離する。この遊離アミンは、続いてのビニルオキシカーボネートを攻撃するための求核試薬として作用し、成長するカルバメート鎖の重合を増大させる。このような戦略は、ただ1つの求核試薬が、重合の間の任意の時点で存在することを保証することによって、交差反応および二方向性重合の可能性を最小化する。
モノマーを合成した後、DNA骨格(scaffold)上のモノマーのアセンブリおよび重合が、自発的に生じるはずである。市販入手可能で、生合成され(Davis Adv.Enzymol.16:287,1955;参考として本明細書中に援用される)、またはD−マンノースから簡単に合成することによる(Fleetら、J.Chem.Soc.,Perkins Trans.I 905,1984;Harveyら、Tetrahedron Lett.32:4111,1991;各々は、参考として本明細書中に援用される)、シキミ酸1は、モノマー合成のための便利な開始点として作用する。synヒドロキシル基を、p−メトキシベンジリデンとして保護し、残りのヒドロキシル基を、tert−ブチルジメチルシリルエーテルとして保護して、2を得る。次いで、保護されたシキミ酸のカルボキシレート部分は、LAH還元、得られるアルコールのトシル化、そしてさらにLAHを用いた還元によって完全に還元され、3を提供する。
市販の合成可能なN−保護アミノ酸は、各モノマーのジカルバメート部分に対する出発物質として作用する。反応性側鎖を、光分解性エーテル、エステル、アセタール、カルバメート、またはチオエーテルとして保護する。以前に開発された化学(Choら、Science 261:1303、1993;参考として本明細書中に援用される)に従って、所望のアミノ酸4を、イソブチルクロロホルメートを用いる混合アルデヒド形成、続く水素化ホウ素ナトリウムによる還元によって、対応するアミノアルコール5に転換する。次いで、このアミノアルコールを、p−ニトロフェニルクロロホルメートでの処理によって活性化カーボネートに転換し、6を生成し、次いで、この6を、第2のアミノアルコール7とカップリングし、ヒドロキシル基のシリル化およびFMOC脱保護の後に、カルバメート8を得る。
シキミ酸誘導性リンカーへのカルバメート8のカップリングは、以下のように進行する。3のアリルヒドロキシル基を、TBAFで脱保護し、トリフラート(triflic)無水物で処理し、第2のトリフラートを形成し、次いでアミノカルバメート8で置換し、9を得る。3中のビニルメチル基の存在は、SN2’付加から生じる所望されない生成物の量を最小化することを補助するはずである(Magid Tetrahedron 36:1901,1980;参考として本明細書中に援用される)。α,β−不飽和エステルへの脱プロトン化されたカルバメートのMichael付加は、十分に示されてきた(Colladoら、Tetrahedron Lett.35:8037,1994;Hiramaら、J.Am.Chem.Soc.107:1797,1985;Nagasakaら、Heterocycles 29:155,1989;Shishidoら、J.Chem.Soc.Perkins Trans.I 993,1987;Hiramaら、Heterocycles 28:1229,1989;各々は、参考として本明細書中に援用される)。類推によって、二級アミンを、o−ニトロベンジルカルバメート(NBOC)として保護し、得られた化合物を、カルバメート窒素上で脱プロトン化する。この脱プロトン化を、代表的には、水素化ナトリウムまたはカリウムtert−ブチルオキシドのいずれかを用いて実施し得るが(Colladoら、Tetrahedron Lett.35:8037,1994;Hiramaら、J.Am.Chem.Soc.107:1797,1985;Nagasakaら、Heterocycles 29:155,1989;Shishidoら、J.Chem.Soc.Perkins Trans.I 993,1987;Hiramaら、Heterocycles 28:1229,1989;各々は、参考として本明細書中に援用される)、他の塩基を、ニトロベンジルプロトンの脱プロトン化を最小にするために使用し得る。脱プロトン化カルバメートを、α,β−不飽和ケトン10に付加し、続いて得られたエノラートをTBSCIでトラップすることによって、シリルエノールエーテル11を生じるはずである。以前に見出された、5−置換エノン(例えば、10)への共役付加の立体選択性(Houseら、J.Org.Chem.33:949,1968;Stillら、Tetrahedron 37:3981,1981;各々は、参考として本明細書中に援用される)は、そのジアステレオマー上の11の優先的な形成を示唆する。ケトン10(フッ素切断カルバメートホスフェートリンカーに対する前駆体)を、ワンポットでの脱炭酸(Bartonら、Tetrahedron 41:3901,1985;参考として本明細書中に援用される)、続いて、TBAFで処理し、得られたアルコールをSwern酸化し、12を得、DDQで脱プロトン化し、低障害性アルコールを選択的ニトロベンジルエーテル形成し、そしてヨウ化サマリウムでα−ヒドロキシル基の還元することによって得る(Molander In Organic Reactions,Paquette,編.46:211,1994;参考として本明細書中に援用される)。
11のp−メトキシベンジリデン基を、シアノ水素化ホウ素ナトリウムおよびTMSクロリドを使用して、α−ヒドロキシPMBエーテルに変換し(Johanssonら、J.Chem.Soc.Perkin Trans.I 2371,1984;参考として本明細書中に援用される)、TES基を、2%のHFを用いて脱プロトン化し(TBSエーテルに影響しないはずの条件(Boschelliら、Tetrahedron Lett.26:5239,1985;参考として本明細書中に援用される))、13を得る。先例に従って(Johanssonら、J.Chem.Soc.Perkin Trans.I 2371,1984;Sutherlinら、Tetrahedron Lett.34:4897,1993;各々は、参考として本明細書中に援用される)、PMB基は、より障害性の二級アルコール上に残るはずである。2つの遊離ヒドロキシル基を、クロロ蟻酸p−ニトロフェノール(または、別のホスゲンアナログ)の溶液への非常にゆっくりとした13の添加により、大環状化し得、14を得る。PMBエーテルを、脱プロトン化し、得られたアルコールを、トリフラートに転換し、立体障害性の塩基を用いて反応動力学的条件下で、除去して、ビニルオキシカルボネート15を得る。ニトロベンジルエーテルおよびニトロベンジルカルバメートの光脱プロトン化は、アルコール16を生じる。
モノマー合成を、3つの成分の連続的なカップリングによって完了する。クロロジイソプロピルアミノホスフィン17を、PCl3とジイソプロピルアミンとの反応によって合成する(Kingら、J.Org.Chem.49:1784,1984;参考として本明細書中に援用される)。樹脂結合ヌクレオシド(または、3’−o−ニトロベンジルエーテル保護ヌクレオシド)18を、17とカップリングして、ホスホルアミダイト19を得る。19とヌクレオシド20との続いてのカップリング(Inoueら、J.Am.Chem.Soc.103:7666,1981;参考として本明細書中に援用される)は、21を提供する。次いで、アルコール16を、21と反応させ、MCPBAまたはI2を使用した慎重な酸化、続いての樹脂からの切断(または光脱プロトン化)の後に、完成したモノマー22を生じる。17とアルコールとの連続的なカップリングのこの戦略を、連続的に使用し、良好な収率で3つの異なるアルコキシ置換基を保有するホスフェートを生成する(Bannwarthら、Helv.Chim.Acta 70:175,1987;参考として本明細書中に援用される)。
別の実施形態において、増幅可能なペプチド核酸ライブラリーを作製する。Orgelおよびその共同研究者らは、ペプチド核酸(PNA)オリゴマーを、相補的DNAテンプレートまたは相補的RNAテンプレート上で効率的に重合し得ることを示した(Boehlerら、Nature 376:578,1995;Schmidtら、Nucl.Acids Res.25:4792,1997;各々は、参考として本明細書中に援用される)。この知見ならびにアミノ酸側鎖を保有するキラルなペプチド核酸の最近の合成および特徴付け(Haaimaら、Angew.Chem.Int.Ed.Engl.35:1939−1942,1996;Puschlら、Tetrahedron Lett.39:4707,1998;各々は、参考として本明細書中に援用される)は、高分子骨格および成長する核酸鎖を単一の構造に結合することを可能にする。この例において、各テンプレートは、5’アミノ基で終了するDNAヘアピンからなり;このような分子の固相合成および溶液合成は、以前に記載されている(Uhlmannら、Angew.Chem.Int.Ed.Engl.35:2632,1996;参考として本明細書中に援用される)。各伸長モノマーは、目的の官能基を含有する側鎖を有するPNAトリマー(またはもっと長い)からなる。人工的な遺伝コードは、各トリヌクレオチドを異なるセットの側鎖へと割り当てるために書かれる。カルボキシ末端からアミノ末端の方向での相補的DNAテンプレートに沿った、PNAダイマーのアセンブリ、活性化(例えば、カルボジイミドおよび適切な脱離基を用いる)、および重合は、非天然の高分子与える(図20)。ビオチン化されたN末端を有する「停止」モノマーを選択することは、伸長終結および全長高分子の精製の都合の良い方法を提供する。得られた高分子(これらをコードするDNAに共有結合された)は、選択、配列決定、または変異の準備ができている。
本発明のなお別の実施形態において、合成薬物の骨格を含む、増幅可能かつ進化可能な非天然非高分子性分子の調製において、革新的な方法を使用する。求核性基または求電子基のそれぞれは、単結合の連結によって接着される低分子骨格上か、またはコードするオリゴヌクレオチドテンプレートに対する通常の固体支持体を介して、マスクが外される。短い核酸配列に連結された求電子試薬または求核試薬は、対応するテンプレートにハイブリダイズされる。適切な試薬との配列特異的な反応が、近接触媒によって起こる(図29)。
分子進化は、進化する分子の構造中に増幅可能な情報担体の配列特異的な翻訳を必要とする。この要件により、直接的に進化する分子型が、2種(つまり、タンパク質および核酸)に限定される。なぜなら、これらのクラスの分子のみが核酸配列から翻訳され得るからである。一般的に上記のように、タンパク質および核酸以外に分子進化に対する有望なアプローチは、DNA配列を合成低分子に翻訳する方法として、DNAテンプレートの合成を使用する。DNAテンプレートの合成は、高い配列特異性を有し、かつDNA骨格の構造的模倣物を必要とはしない、広範な種々の強力な化学反応へと方向付け得る。しかし、このアプローチの、利用的に複雑なものの合成分子への適用は、DNAテンプレート反応の生成物がその後DNAテンプレートの変換を受けることを可能とする一般的な方法の開発を必要とする。第1のDNAテンプレート多段階低分子合成は、本明細書中で詳細に記載される。DNAテンプレート合成の反応範囲における最近の伸展を併せると、これらの知見は、合成低分子ライブラリーのインビトロ進化のための段階を設置する。
1980,124,913)。この試薬およびアミン末端のテンプレート(5)を使用するDNAテンプレートアミド結合の形成(本明細書中に記載される)の生成物(8)は、塩基性水溶液で処理されてリンカーの量的排除および自発性の脱カルボキシル化をもたらし、上手く転換されるアミノ酸基を含む生成物9を提供する(図33)。このスルホンリンカーは、pH7.5以下の緩衝液中で25℃で24時間より長く安定であるが、pH11.8の緩衝液に37℃で2時間曝される場合に量的切断を受ける。
各々の反応工程、精製工程およびスルホンリンカー切断工程の進行の後に、変性ポリアクリルアミドゲル電気泳動を続けた。テンプレート(16)に連結した最終トリペプチドを、制限エンドヌクレアーゼEcoRIで消化し、そしてトリペプチドを含む消化フラグメントをMALDI質量分析によって特徴付けた。2nmol(約20μg)の出発材料より開始し、十分なトリペプチド生成物を生成して、106より多くのインビトロ選択およびPCR反応に対するテンプレートとして扱った(Kramerら、Current Protocols in Molecular Biology、第3巻(編者:F.M.Ausubel)、Wiely、1999、15.1頁)(推定で1/10,000の分子が残る選択)。出発材料テンプレートを無水酢酸でキャップした場合も、配列ミスマッチを含むコントロール試薬を相補的な試薬の代わりに使用した場合も、有効な生成物を全く生成しなかった(図39)。
本発明のなお別の実施形態において、核酸(例えば、DNA、RNA、これらの誘導体)は、重合化触媒に結合される。核酸が、複合体構造に折りたたまれ得るために、その核酸は成長する高分子鎖の重合化を指向するおよび/またはその重合化に影響を与えるために使用され得る。例えば、核酸は、重合化されるモノマー単位の選択、およびどのような重合化反応がおこるか(例えば、立体化学、立体規則性、活性)に影響し得る。この合成高分子は、分子量、密度、疎水性、立体規則性、立体選択性などのような特定の特性について選択され得、そしてその合成を指向する触媒の不可欠な部分を形成する核酸は、増幅され、そして進化され得る(図41)。リガンドの多様化、選択、および増幅の繰り返しサイクルは、所望の特性への、触媒および高分子の真の進化を可能にする。
上記の非天然ペプチドおよび二環式のライブラリは、種々の段階で特徴付けられる。各候補試薬は、その翻訳DNAオリゴヌクレオチドに結合体化され、次いでマッチテンプレートおよびミスマッチテンプレートでのモデル反応に供される。これらの反応物からの生成物を、変性ポリアクリルアミドゲル電気泳動によって分析し、反応効率を評価し、そして質量分析によって予測された生成物構造を裏付けた。一旦強固(robust)な試薬の完全なセットが同定されると、大スケールでの代表的な単一ライブラリの完全多段階DNAテンプレート合成が実施され、そして最終生成物が質量分析によって特徴付けられる。
1994,79,1157−64)(新たな血管の生成)を含む)を媒介する。腫瘍誘発性血管新生の間、侵襲性内皮細胞は、それらのαVβ3インテグリンレセプターによって、細胞外マトリクス成分へと結合する。いくつかの研究(Brooksら、Cell 1994,79,1157−64;Brooksら、Cell 1998,92,391−400;Friedlanderら、Science 1995,270,1500−2;Varnerら、Cell Adhes Commun 1995,3,367−74;Brooksら、J.Clin Invest 1995,96,1815−22)は、抗体または低分子合成ペプチドを用いてのこのインテグリン結合事象の阻害が、増殖性脈管形成性血管細胞のアポトーシスを誘導し、そして腫瘍転移を阻害し得ることを実証した。
1999、18、377−91;J.A.Lathamら、Nucleic Acids Res.1994、22、2817−22;T.Gourlainら、Nucleic Acids Res.2001、29、1898−1905;S.E.Leeら、Nucleic Acids Res.2001、29、1565−73;K.Sakthivelら、Angew.Chem.Int.Ed.Engl.1998、37、2872−2875)。この収束性アプローチは、幅広い種類の金属結合配位子を、いずれかのヌクレオチドアナログに迅速に取り組むことを可能にする。以前に報告された(K.Sakthivelら、Angew.Chem.Int.Ed.Engl.1998、37、2872−2875)経路(図48、Phillips、Chorba、Liu、未公開結果)に従い、7の合成が完成されている。N−アリルトリフルオロアセトアミドとの市販の5−ヨード−2’−デオキシウリジン(22)のHeckカップリングは23を生成した。トリメチルホスフェート、POCl3、およびプロトンスポンジ(proton sponge)(1,8−ビス(ジメチルアミノ)−ナフタレン)、その後のトリ−n−ブチルアンモニウムピロホスフェートでの23の処理により、5’−三リン酸基が導入され、次いでトリフルオロアセトアミド基が水性アンモニアで除去され、7を生成した。
DNAポリメラーゼによる7の受容に関するこれまでの知見は、報告された非受容(K.Sakthivelら、Angew.Chem.Int.Ed.Engl.1998、37、2872−2875)および受容(S.E.Leeら、Nucleic Acids Res.2001、29、1565−73)の両方で矛盾がある。
それぞれの選択ラウンドの後に、活性なライブラリメンバーは、非天然ヌクレオチドを用いるPCRによって増幅され、所望の触媒についてライブラリを富化するために、さらなる選択ラウンドに供される。これらのライブラリは、それぞれの選択ラウンド前に多様化工程を導入することにより、真に進化される。ライブラリは、エラープローンPCRを使用する、ランダム変異誘発による(Caldwellら、PCR Methods A
pplic.1992、2、28−33)か、もしくは小さな非相同核酸フラグメントを組み換える、改変DNAシャッフリング方法を使用する組換えにより多様化される。エラープローンPCRは、本質的に通常のPCRよりも効率が低いので、エラープローンPCR多様化は、天然のdATP、dTTP、dCTPおよびdGTPのみと共に、そして化学的ハンドルまたはビオチン基を欠くプライマーを使用して行なわれる。次に、得られる変異化産物は、非天然ヌクレオチド、ビオチン化プライマー、およびアミノ終端プライマーまたはチオール終端プライマーを含む、標準PCR反応物を使用して、非天然核酸高分子へのPCR翻訳に供される。
数回の進化に供されたライブラリーは、混合された配列のプールまたは個々のライブラリーメンバーの両方として、目的の反応を触媒する能力について特徴付けられる。個々のメンバーを、DNAベクター中にPCR増幅した配列を連結すること、コンピテントな細菌細胞内に連結したベクターの希釈溶液を形質転換すること、および形質転換体の単一コロニーを選択することによって、進化したプールから取り出す。プールまたは個々の配列に関するアッセイを、単一の代謝回転形態と本質的に複数の代謝回転触媒性形態の両方において実行する。単一代謝回転アッセイについて、遊離のビオチン化基質の存在下で、基質に連結した結合形成触媒がそれ自体のビオチン化をもたらす速度を測定するか、またはビオチン化結合破壊触媒がそのビオチン基の欠損をもたらす速度を測定する。複数代謝回転アッセイを、低分子型の基質と共に進化した触媒をインキュベートし、そしてtlc、NMR、質量分析、HPLCまたは分光分析によって産物形成速度を分析することによって実施する。
Claims (1)
- 本明細書および図面に記載される発明。
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US27708101P | 2001-03-19 | 2001-03-19 | |
| US27709401P | 2001-03-19 | 2001-03-19 | |
| US60/277,094 | 2001-03-19 | ||
| US60/277,081 | 2001-03-19 | ||
| US30669101P | 2001-07-20 | 2001-07-20 | |
| US60/306,691 | 2001-07-20 | ||
| US10/101,030 | 2002-03-19 | ||
| US10/101,030 US7070928B2 (en) | 2001-03-19 | 2002-03-19 | Evolving new molecular function |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009169425A Division JP2009232869A (ja) | 2001-03-19 | 2009-07-17 | 新規分子機能の進化 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2013010761A true JP2013010761A (ja) | 2013-01-17 |
| JP5509270B2 JP5509270B2 (ja) | 2014-06-04 |
Family
ID=27493169
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002574322A Expired - Fee Related JP4128453B2 (ja) | 2001-03-19 | 2002-03-19 | 新規分子機能の進化 |
| JP2008042250A Expired - Fee Related JP4384229B2 (ja) | 2001-03-19 | 2008-02-22 | 新規分子機能の進化 |
| JP2009169425A Withdrawn JP2009232869A (ja) | 2001-03-19 | 2009-07-17 | 新規分子機能の進化 |
| JP2012165977A Expired - Fee Related JP5509270B2 (ja) | 2001-03-19 | 2012-07-26 | 新規分子機能の進化 |
Family Applications Before (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002574322A Expired - Fee Related JP4128453B2 (ja) | 2001-03-19 | 2002-03-19 | 新規分子機能の進化 |
| JP2008042250A Expired - Fee Related JP4384229B2 (ja) | 2001-03-19 | 2008-02-22 | 新規分子機能の進化 |
| JP2009169425A Withdrawn JP2009232869A (ja) | 2001-03-19 | 2009-07-17 | 新規分子機能の進化 |
Country Status (12)
| Country | Link |
|---|---|
| US (7) | US7070928B2 (ja) |
| EP (3) | EP1832567A3 (ja) |
| JP (4) | JP4128453B2 (ja) |
| AT (1) | ATE335754T1 (ja) |
| AU (2) | AU2002306777C1 (ja) |
| CA (1) | CA2441820C (ja) |
| DE (1) | DE60213826T3 (ja) |
| DK (1) | DK1423400T4 (ja) |
| ES (1) | ES2271306T5 (ja) |
| IL (4) | IL158000A0 (ja) |
| PT (1) | PT1423400E (ja) |
| WO (1) | WO2002074929A2 (ja) |
Families Citing this family (118)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040121313A1 (en) | 2002-12-06 | 2004-06-24 | Ecker David J. | Methods for rapid detection and identification of bioagents in organs for transplantation |
| US7226739B2 (en) | 2001-03-02 | 2007-06-05 | Isis Pharmaceuticals, Inc | Methods for rapid detection and identification of bioagents in epidemiological and forensic investigations |
| US7666588B2 (en) | 2001-03-02 | 2010-02-23 | Ibis Biosciences, Inc. | Methods for rapid forensic analysis of mitochondrial DNA and characterization of mitochondrial DNA heteroplasmy |
| US20030027135A1 (en) | 2001-03-02 | 2003-02-06 | Ecker David J. | Method for rapid detection and identification of bioagents |
| IL158000A0 (en) | 2001-03-19 | 2004-03-28 | Harvard College | Evolving new molecular function |
| WO2002102820A1 (en) | 2001-06-20 | 2002-12-27 | Nuevolution A/S | Nucleoside derivatives for library preparation |
| DK1402024T3 (da) * | 2001-06-20 | 2007-12-27 | Nuevolution As | Fremgangsmåde til syntese af et template-molekyle |
| US8073627B2 (en) * | 2001-06-26 | 2011-12-06 | Ibis Biosciences, Inc. | System for indentification of pathogens |
| US7217510B2 (en) | 2001-06-26 | 2007-05-15 | Isis Pharmaceuticals, Inc. | Methods for providing bacterial bioagent characterizing information |
| DE10147074A1 (de) * | 2001-09-25 | 2003-05-08 | Beru Ag | Verfahren zum Betreiben einer aus mehreren Heizelementen bestehenden mehrstufigen elektrischen Heizung |
| DE20200926U1 (de) * | 2002-01-23 | 2002-04-18 | Hegenscheidt Mfd Gmbh & Co Kg | Festwalzgerät einer Festwalzmaschine für Kurbelwellen |
| DE60231436D1 (de) | 2002-03-08 | 2009-04-16 | Eidgenoess Tech Hochschule | Kodierte, selbst-assemblierende chemische bibliotheken (esachel) |
| EP1487849A2 (en) * | 2002-03-15 | 2004-12-22 | Nuevolution A/S | A building block capable of transferring a functional entity |
| US20050221318A1 (en) * | 2002-03-15 | 2005-10-06 | Gouliaev Alex H | Building block forming a c-c bond upon reaction |
| DE60324778D1 (de) * | 2002-03-15 | 2009-01-02 | Nuevolution As | Eine verbesserte methode zur synthese von matritzenabhängigen molekülen |
| WO2003078446A2 (en) * | 2002-03-15 | 2003-09-25 | Nuevolution A/S | A building block forming a c-c or a c-hetero atom bond upon reaction |
| US10730906B2 (en) | 2002-08-01 | 2020-08-04 | Nuevolutions A/S | Multi-step synthesis of templated molecules |
| JP4657919B2 (ja) * | 2002-08-19 | 2011-03-23 | プレジデント アンド フェローズ オブ ハーバード カレッジ | 進化する新しい分子機能 |
| EP1539953A2 (en) * | 2002-09-12 | 2005-06-15 | Nuevolution A/S | Proximity-aided synthesis of templated molecules |
| EP1545762A2 (en) | 2002-09-27 | 2005-06-29 | Carlsberg A/S | Spatially encoded polymer matrix |
| ATE513055T1 (de) | 2002-10-30 | 2011-07-15 | Nuevolution As | Enzymatisches codieren |
| AU2011226815B2 (en) * | 2002-10-30 | 2014-09-25 | Nuevolution A/S | Enzymatic encoding |
| JP2006516193A (ja) * | 2002-12-06 | 2006-06-29 | アイシス・ファーマシューティカルス・インコーポレーテッド | ヒトおよび動物における病原体の迅速な同定方法 |
| US9121110B2 (en) | 2002-12-19 | 2015-09-01 | Nuevolution A/S | Quasirandom structure and function guided synthesis methods |
| EP1597395A2 (en) * | 2003-02-21 | 2005-11-23 | Nuevolution A/S | Method for producing second-generation library |
| US20060269920A1 (en) * | 2003-02-21 | 2006-11-30 | Nuevolution A/S | Method for obtaining structural information about an encoded molecule |
| WO2004083427A2 (en) * | 2003-03-20 | 2004-09-30 | Nuevolution A/S | Ligational encoding of small molecules |
| US8017323B2 (en) | 2003-03-26 | 2011-09-13 | President And Fellows Of Harvard College | Free reactant use in nucleic acid-templated synthesis |
| US8046171B2 (en) | 2003-04-18 | 2011-10-25 | Ibis Biosciences, Inc. | Methods and apparatus for genetic evaluation |
| US8057993B2 (en) | 2003-04-26 | 2011-11-15 | Ibis Biosciences, Inc. | Methods for identification of coronaviruses |
| WO2005016851A1 (en) * | 2003-05-08 | 2005-02-24 | Valent Technologies, Llc | Recombined molecules and preparation thereof |
| US8158354B2 (en) | 2003-05-13 | 2012-04-17 | Ibis Biosciences, Inc. | Methods for rapid purification of nucleic acids for subsequent analysis by mass spectrometry by solution capture |
| US7964343B2 (en) | 2003-05-13 | 2011-06-21 | Ibis Biosciences, Inc. | Method for rapid purification of nucleic acids for subsequent analysis by mass spectrometry by solution capture |
| WO2004110964A2 (en) * | 2003-06-16 | 2004-12-23 | Nuevolution A/S | Encoded molecules by translation (emt) |
| WO2005003778A2 (en) * | 2003-07-02 | 2005-01-13 | Nuevolution A/S | A method for identifying a synthetic molecule having affinity towards a target |
| US7956175B2 (en) | 2003-09-11 | 2011-06-07 | Ibis Biosciences, Inc. | Compositions for use in identification of bacteria |
| US8546082B2 (en) | 2003-09-11 | 2013-10-01 | Ibis Biosciences, Inc. | Methods for identification of sepsis-causing bacteria |
| US8097416B2 (en) | 2003-09-11 | 2012-01-17 | Ibis Biosciences, Inc. | Methods for identification of sepsis-causing bacteria |
| EP1670939B1 (en) * | 2003-09-18 | 2009-11-04 | Nuevolution A/S | A method for obtaining structural information concerning an encoded molecule and method for selecting compounds |
| CN1898257B (zh) | 2003-12-17 | 2010-06-23 | 普雷西斯药品公司 | 合成编码文库的方法 |
| US7972994B2 (en) | 2003-12-17 | 2011-07-05 | Glaxosmithkline Llc | Methods for synthesis of encoded libraries |
| WO2005078122A2 (en) | 2004-02-17 | 2005-08-25 | Nuevolution A/S | Method for enrichment involving elimination by mismatch hybridisation |
| US7666592B2 (en) | 2004-02-18 | 2010-02-23 | Ibis Biosciences, Inc. | Methods for concurrent identification and quantification of an unknown bioagent |
| US8119336B2 (en) | 2004-03-03 | 2012-02-21 | Ibis Biosciences, Inc. | Compositions for use in identification of alphaviruses |
| EP1730277B1 (en) * | 2004-03-22 | 2009-10-28 | Nuevolution A/S | Ligational encoding using building block oligonucleotides |
| CA2567839C (en) | 2004-05-24 | 2011-06-28 | Isis Pharmaceuticals, Inc. | Mass spectrometry with selective ion filtration by digital thresholding |
| US20050266411A1 (en) | 2004-05-25 | 2005-12-01 | Hofstadler Steven A | Methods for rapid forensic analysis of mitochondrial DNA |
| EP1774033A4 (en) * | 2004-06-10 | 2008-04-23 | Perkinelmer Las Inc | MULTIPLEX ASSAYS FOR DETECTION OF ANALYTES |
| US7811753B2 (en) | 2004-07-14 | 2010-10-12 | Ibis Biosciences, Inc. | Methods for repairing degraded DNA |
| WO2006135400A2 (en) | 2004-08-24 | 2006-12-21 | Isis Pharmaceuticals, Inc. | Methods for rapid identification of recombinant organisms |
| ES2320361T3 (es) | 2004-11-08 | 2009-05-21 | Vipergen Aps | Sintesis quimica guiada por el acido nucleico estructural. |
| EP1828381B1 (en) | 2004-11-22 | 2009-01-07 | Peter Birk Rasmussen | Template directed split and mix systhesis of small molecule libraries |
| US8182992B2 (en) | 2005-03-03 | 2012-05-22 | Ibis Biosciences, Inc. | Compositions for use in identification of adventitious viruses |
| US8084207B2 (en) | 2005-03-03 | 2011-12-27 | Ibis Bioscience, Inc. | Compositions for use in identification of papillomavirus |
| DE602006017365D1 (de) * | 2005-04-13 | 2010-11-18 | Ibis Biosciences Inc | Zusammensetzungen für die identifizierung von adenoviren |
| US7968289B2 (en) * | 2005-05-03 | 2011-06-28 | Ensemble Therapeutics Corporation | Turn over probes and use thereof for nucleic acid detection |
| EP1885891A2 (en) * | 2005-05-26 | 2008-02-13 | Ensemble Discovery Corporation | Biodetection by nucleic acid-templated chemistry |
| WO2006135654A2 (en) * | 2005-06-07 | 2006-12-21 | President And Fellows Of Harvard College | Polymer evolution via templated synthesis related applications |
| ES2401485T3 (es) | 2005-06-09 | 2013-04-22 | Glaxosmithkline Llc | Método de identificación de compuestos que se unen a una molécula diana biológica |
| WO2006138560A2 (en) * | 2005-06-17 | 2006-12-28 | President And Fellows Of Harvard College | Nucleic acid-templated chemistry in organic solvents |
| DE602006013134D1 (de) * | 2005-06-17 | 2010-05-06 | Harvard College | Iterierte verzweigende reaktionswege über nukleinsäure-vermittelte chemie |
| WO2007011722A2 (en) | 2005-07-15 | 2007-01-25 | President And Fellows Of Harvard College | Reaction discovery system |
| AU2006272776B2 (en) | 2005-07-21 | 2012-01-19 | Ibis Biosciences, Inc. | Methods for rapid identification and quantitation of nucleic acid variants |
| DE602005003374T2 (de) * | 2005-10-17 | 2008-09-11 | Pls-Design Gmbh | Verfahren zur Bestimmung einer unbekannten PNS Seqenz und seine Verwendungen |
| WO2007053358A2 (en) | 2005-10-28 | 2007-05-10 | Praecis Pharmaceuticals, Inc. | Methods for identifying compounds of interest using encoded libraries |
| ES2901551T3 (es) | 2005-12-01 | 2022-03-22 | Nuevolution As | Métodos de codificación enzimática para la síntesis eficiente de bibliotecas grandes |
| WO2007124758A1 (en) | 2006-05-03 | 2007-11-08 | Vipergen Aps | A method for preparing compounds by nucleic acid directed synthesis |
| EP2064332B1 (en) | 2006-09-14 | 2012-07-18 | Ibis Biosciences, Inc. | Targeted whole genome amplification method for identification of pathogens |
| US20100159455A1 (en) * | 2006-09-18 | 2010-06-24 | Ensemble Discovery | Receptor family profiling |
| CA2664649A1 (en) | 2006-09-28 | 2008-05-08 | Ensemble Discovery Corporation | Compositions and methods for biodetection by nucleic acid-templated chemistry |
| WO2008104002A2 (en) | 2007-02-23 | 2008-08-28 | Ibis Biosciences, Inc. | Methods for rapid forensic dna analysis |
| US9598724B2 (en) | 2007-06-01 | 2017-03-21 | Ibis Biosciences, Inc. | Methods and compositions for multiple displacement amplification of nucleic acids |
| JP2010534836A (ja) * | 2007-07-27 | 2010-11-11 | アンサンブル ディスカバリー コーポレイション | 検出アッセイとそれらの使用 |
| WO2009134470A2 (en) * | 2008-01-18 | 2009-11-05 | The Johns Hopkins University | Methods for identifying eubacteria |
| WO2010022249A2 (en) * | 2008-08-20 | 2010-02-25 | Ensemble Discovery Corporation | Macrocyclic compounds for inhibition of tumor necrosis factor alpha |
| JP2012502911A (ja) * | 2008-09-14 | 2012-02-02 | インサイター インコーポレイテッド | ナノスケール分子合成 |
| EP2349549B1 (en) | 2008-09-16 | 2012-07-18 | Ibis Biosciences, Inc. | Mixing cartridges, mixing stations, and related kits, and system |
| EP2344893B1 (en) | 2008-09-16 | 2014-10-15 | Ibis Biosciences, Inc. | Microplate handling systems and methods |
| EP2347254A2 (en) | 2008-09-16 | 2011-07-27 | Ibis Biosciences, Inc. | Sample processing units, systems, and related methods |
| EP2396803A4 (en) | 2009-02-12 | 2016-10-26 | Ibis Biosciences Inc | IONIZATION PROBE ASSEMBLIES |
| DK2396459T3 (en) | 2009-02-13 | 2017-08-28 | X-Chem Inc | Method for preparing and screening DNA-encoded libraries |
| WO2010150103A2 (en) | 2009-06-22 | 2010-12-29 | Universite De Strasbourg | Method of preparing an adduct |
| WO2011008972A1 (en) | 2009-07-17 | 2011-01-20 | Ibis Biosciences, Inc. | Systems for bioagent identification |
| US8950604B2 (en) | 2009-07-17 | 2015-02-10 | Ibis Biosciences, Inc. | Lift and mount apparatus |
| EP3225695A1 (en) | 2009-10-15 | 2017-10-04 | Ibis Biosciences, Inc. | Multiple displacement amplification |
| WO2011127933A1 (en) | 2010-04-16 | 2011-10-20 | Nuevolution A/S | Bi-functional complexes and methods for making and using such complexes |
| DK3399035T3 (da) | 2010-09-27 | 2021-05-17 | Vipergen Aps | Fremgangsmåde til fremstilling af et beriget bibliotek |
| WO2012159089A1 (en) * | 2011-05-19 | 2012-11-22 | Sequenom, Inc. | Products and processes for multiplex nucleic acid identification |
| KR102124613B1 (ko) | 2011-09-07 | 2020-06-19 | 엑스-켐, 인크. | Dna-코딩된 라이브러리에 태그부착하는 방법 |
| NZ703766A (en) | 2012-07-13 | 2018-03-23 | X Chem Inc | Dna-encoded libraries having encoding oligonucleotide linkages not readable by polymerases |
| US10983118B2 (en) * | 2013-03-15 | 2021-04-20 | Arizona Board Of Regents On Behalf Of Arizona State University | Biosensor microarray compositions and methods |
| US9163284B2 (en) | 2013-08-09 | 2015-10-20 | President And Fellows Of Harvard College | Methods for identifying a target site of a Cas9 nuclease |
| US9340799B2 (en) | 2013-09-06 | 2016-05-17 | President And Fellows Of Harvard College | MRNA-sensing switchable gRNAs |
| GB201322692D0 (en) | 2013-12-20 | 2014-02-05 | Philochem Ag | Production of encoded chemical libraries |
| AU2015298571B2 (en) | 2014-07-30 | 2020-09-03 | President And Fellows Of Harvard College | Cas9 proteins including ligand-dependent inteins |
| US10040048B1 (en) | 2014-09-25 | 2018-08-07 | Synthego Corporation | Automated modular system and method for production of biopolymers |
| US20180340174A1 (en) | 2014-11-11 | 2018-11-29 | Nanocore Aps | Method for identification of molecules with desired characteristics |
| CN107532162A (zh) * | 2014-12-12 | 2018-01-02 | 托德·M·伍尔夫 | 用于利用寡核苷酸编辑细胞中核酸的组合物和方法 |
| US10233489B2 (en) | 2015-04-24 | 2019-03-19 | Agena Bioscience, Inc. | Multiplexed method for the identification and quantitation of minor alleles and polymorphisms |
| CA3024875A1 (en) | 2016-06-16 | 2017-12-21 | Richard Edward Watts | Oligonucleotide directed and recorded combinatorial synthesis of encoded probe molecules |
| CA3033327A1 (en) | 2016-08-09 | 2018-02-15 | President And Fellows Of Harvard College | Programmable cas9-recombinase fusion proteins and uses thereof |
| WO2018039438A1 (en) | 2016-08-24 | 2018-03-01 | President And Fellows Of Harvard College | Incorporation of unnatural amino acids into proteins using base editing |
| WO2018081534A1 (en) | 2016-10-28 | 2018-05-03 | President And Fellows Of Harvard College | Assay for exo-site binding molecules |
| US10745677B2 (en) | 2016-12-23 | 2020-08-18 | President And Fellows Of Harvard College | Editing of CCR5 receptor gene to protect against HIV infection |
| US11898179B2 (en) | 2017-03-09 | 2024-02-13 | President And Fellows Of Harvard College | Suppression of pain by gene editing |
| CN110914310A (zh) | 2017-03-10 | 2020-03-24 | 哈佛大学的校长及成员们 | 胞嘧啶至鸟嘌呤碱基编辑器 |
| EP3619340A4 (en) | 2017-05-02 | 2021-01-20 | Haystack Sciences Corporation | MOLECULES FOR THE VERIFICATION OF OLIGONUCLEOTIDE DIRECTED COMBINATIONAL SYNTHESIS AND METHOD OF MANUFACTURING AND USING THEREOF |
| US11560566B2 (en) | 2017-05-12 | 2023-01-24 | President And Fellows Of Harvard College | Aptazyme-embedded guide RNAs for use with CRISPR-Cas9 in genome editing and transcriptional activation |
| WO2019023680A1 (en) | 2017-07-28 | 2019-01-31 | President And Fellows Of Harvard College | METHODS AND COMPOSITIONS FOR EVOLUTION OF BASIC EDITORS USING PHAGE-ASSISTED CONTINUOUS EVOLUTION (PACE) |
| WO2019139645A2 (en) | 2017-08-30 | 2019-07-18 | President And Fellows Of Harvard College | High efficiency base editors comprising gam |
| EP3681637A4 (en) * | 2017-09-11 | 2021-06-02 | Synthego Corporation | SYSTEM AND PROCEDURE FOR BIOPOLYMER SYNTHESIS |
| WO2019079347A1 (en) | 2017-10-16 | 2019-04-25 | The Broad Institute, Inc. | USES OF BASIC EDITORS ADENOSINE |
| US11674136B2 (en) | 2018-02-09 | 2023-06-13 | President And Fellows Of Harvard College | DNA-templated macrocycle library |
| CA3120895A1 (en) | 2018-12-20 | 2020-06-25 | Sergey N. Krylov | Binder selection using capillary electrophoresis |
| JP2022518242A (ja) | 2019-01-22 | 2022-03-14 | ビペルゲン エーピーエス | 細胞内のインビトロディスプレイライブラリのスクリーニング方法 |
| WO2020191239A1 (en) | 2019-03-19 | 2020-09-24 | The Broad Institute, Inc. | Methods and compositions for editing nucleotide sequences |
| DE112021002672T5 (de) | 2020-05-08 | 2023-04-13 | President And Fellows Of Harvard College | Vefahren und zusammensetzungen zum gleichzeitigen editieren beider stränge einer doppelsträngigen nukleotid-zielsequenz |
| WO2023164224A1 (en) * | 2022-02-28 | 2023-08-31 | The Board Of Trustees Of The Leland Stanford Junior University | Functionalized nanoclusters and their use in treating bacterial infections |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001509828A (ja) * | 1997-01-08 | 2001-07-24 | プロリゴ・エルエルシー | 高分子のバイオコンジュゲーション |
| JP2003529328A (ja) * | 1999-08-12 | 2003-10-07 | マキシジェン, インコーポレイテッド | コンビナトリアル化学および医化学のための酵素の進化および使用 |
| JP2004536162A (ja) * | 2001-03-19 | 2004-12-02 | プレジデント アンド フェロウズ オブ ハーバード カレッジ | 新規分子機能の進化 |
Family Cites Families (86)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US277081A (en) | 1883-05-08 | Scalding napped hats and sizing or felting hat and other fabrics | ||
| US4194550A (en) * | 1976-01-26 | 1980-03-25 | Insulating Shade (Limited Partnership) | Apparatus for insulating against conductive, convective and radiant heat transmission |
| US4863857A (en) | 1985-03-01 | 1989-09-05 | Board Of Regents, The University Of Texas System | Polypeptide complementary to peptides or proteins having an amino acid sequence or nucleotide coding sequence at least partially known |
| US5506337A (en) | 1985-03-15 | 1996-04-09 | Antivirals Inc. | Morpholino-subunit combinatorial library and method |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4703034A (en) * | 1986-04-28 | 1987-10-27 | Merck & Co., Inc. | Novel cyclic tetrapeptide |
| US5449602A (en) * | 1988-01-13 | 1995-09-12 | Amoco Corporation | Template-directed photoligation |
| US5162218A (en) | 1988-11-18 | 1992-11-10 | The Regents Of The University Of California | Conjugated polypeptides and methods for their preparation |
| US6291160B1 (en) | 1989-05-16 | 2001-09-18 | Scripps Research Institute | Method for producing polymers having a preselected activity |
| US6291161B1 (en) | 1989-05-16 | 2001-09-18 | Scripps Research Institute | Method for tapping the immunological repertiore |
| US6680192B1 (en) | 1989-05-16 | 2004-01-20 | Scripps Research Institute | Method for producing polymers having a preselected activity |
| ATE168416T1 (de) | 1989-10-05 | 1998-08-15 | Optein Inc | Zellfreie synthese und isolierung von genen und polypeptiden |
| US5660985A (en) | 1990-06-11 | 1997-08-26 | Nexstar Pharmaceuticals, Inc. | High affinity nucleic acid ligands containing modified nucleotides |
| US5637682A (en) * | 1990-06-11 | 1997-06-10 | Nexstar Pharmaceuticals, Inc. | High-affinity oligonucleotide ligands to the tachykinin substance P |
| US5723289A (en) | 1990-06-11 | 1998-03-03 | Nexstar Pharmaceuticals, Inc. | Parallel selex |
| US20020038000A1 (en) | 1990-08-02 | 2002-03-28 | Larry Gold | Systematic polypeptide evolution by reverse translation |
| US5843701A (en) | 1990-08-02 | 1998-12-01 | Nexstar Pharmaceticals, Inc. | Systematic polypeptide evolution by reverse translation |
| WO1992002536A1 (en) | 1990-08-02 | 1992-02-20 | The Regents Of The University Of Colorado | Systematic polypeptide evolution by reverse translation |
| US6140496A (en) | 1990-10-09 | 2000-10-31 | Benner; Steven Albert | Precursors for deoxyribonucleotides containing non-standard nucleosides |
| US6037120A (en) | 1995-10-12 | 2000-03-14 | Benner; Steven Albert | Recognition of oligonucleotides containing non-standard base pairs |
| US5641625A (en) | 1992-05-22 | 1997-06-24 | Isis Pharmaceuticals, Inc. | Cleaving double-stranded DNA with peptide nucleic acids |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US5719262A (en) | 1993-11-22 | 1998-02-17 | Buchardt, Deceased; Ole | Peptide nucleic acids having amino acid side chains |
| US5574141A (en) | 1991-08-28 | 1996-11-12 | Boehringer Mannheim Gmbh | Functionalized carrier materials for the simultaneous synthesis and direct labeling of oligonucleotides as primers for template-dependent enzymatic nucleic acid syntheses |
| US5639603A (en) | 1991-09-18 | 1997-06-17 | Affymax Technologies N.V. | Synthesizing and screening molecular diversity |
| DK0604552T3 (da) | 1991-09-18 | 1997-08-04 | Affymax Tech Nv | Fremgangsmåde til syntese af forskellige samlinger af oligomerer |
| US5270170A (en) | 1991-10-16 | 1993-12-14 | Affymax Technologies N.V. | Peptide library and screening method |
| EP0916396B1 (en) | 1991-11-22 | 2005-04-13 | Affymetrix, Inc. (a Delaware Corporation) | Combinatorial strategies for polymer synthesis |
| US5573905A (en) | 1992-03-30 | 1996-11-12 | The Scripps Research Institute | Encoded combinatorial chemical libraries |
| US5721099A (en) | 1992-10-01 | 1998-02-24 | Trustees Of Columbia University In The City Of New York | Complex combinatorial chemical libraries encoded with tags |
| US5565324A (en) * | 1992-10-01 | 1996-10-15 | The Trustees Of Columbia University In The City Of New York | Complex combinatorial chemical libraries encoded with tags |
| US6436635B1 (en) | 1992-11-06 | 2002-08-20 | Boston University | Solid phase sequencing of double-stranded nucleic acids |
| EP0679196B1 (en) | 1993-01-07 | 2004-05-26 | Sequenom, Inc. | Dna sequencing by mass spectrometry |
| US5605798A (en) * | 1993-01-07 | 1997-02-25 | Sequenom, Inc. | DNA diagnostic based on mass spectrometry |
| US6194144B1 (en) | 1993-01-07 | 2001-02-27 | Sequenom, Inc. | DNA sequencing by mass spectrometry |
| US6074823A (en) | 1993-03-19 | 2000-06-13 | Sequenom, Inc. | DNA sequencing by mass spectrometry via exonuclease degradation |
| DE69430909T2 (de) | 1993-03-19 | 2003-02-27 | Sequenom Inc | Dns-sequenzbestimmung durch massenspektrometrie auf dem weg des abbaus mit exonuklease |
| US5840485A (en) | 1993-05-27 | 1998-11-24 | Selectide Corporation | Topologically segregated, encoded solid phase libraries |
| GB9315847D0 (en) * | 1993-07-30 | 1993-09-15 | Isis Innovation | Tag reagent and assay method |
| US5922545A (en) | 1993-10-29 | 1999-07-13 | Affymax Technologies N.V. | In vitro peptide and antibody display libraries |
| US5925517A (en) * | 1993-11-12 | 1999-07-20 | The Public Health Research Institute Of The City Of New York, Inc. | Detectably labeled dual conformation oligonucleotide probes, assays and kits |
| WO1995014106A2 (en) * | 1993-11-17 | 1995-05-26 | Id Biomedical Corporation | Cycling probe cleavage detection of nucleic acid sequences |
| US6127154A (en) | 1994-02-10 | 2000-10-03 | Mosbach; Klaus | Methods for direct synthesis of compounds having complementary structure to a desired molecular entity and use thereof |
| US5605793A (en) | 1994-02-17 | 1997-02-25 | Affymax Technologies N.V. | Methods for in vitro recombination |
| US6048698A (en) | 1994-09-20 | 2000-04-11 | Nexstar Pharmaceuticals, Inc. | Parallel SELEX™ |
| US20030099945A1 (en) | 1994-09-20 | 2003-05-29 | Invenux, Inc. | Parallel selex |
| US5597697A (en) | 1994-09-30 | 1997-01-28 | Diamond; Paul | Screening assay for inhibitors and activators of RNA and DNA-dependent nucleic acid polymerases |
| GB9524630D0 (en) * | 1994-12-24 | 1996-01-31 | Zeneca Ltd | Chemical compounds |
| US5559000A (en) | 1995-01-18 | 1996-09-24 | The Scripps Research Institute | Encoded reaction cassette |
| DE19646372C1 (de) | 1995-11-11 | 1997-06-19 | Evotec Biosystems Gmbh | Genotyp und Phänotyp koppelnde Verbindung |
| US5846839A (en) | 1995-12-22 | 1998-12-08 | Glaxo Group Limited | Methods for hard-tagging an encoded synthetic library |
| US5888971A (en) * | 1996-02-20 | 1999-03-30 | Ortho Pharmaceutical Corporation, Inc. | Macrocyclic peptides useful in the treatment of thrombin related disorders |
| CA2252048C (en) * | 1996-04-12 | 2008-03-11 | The Public Health Research Institute Of The City Of New York, Inc. | Detection probes, kits and assays |
| US5998140A (en) | 1996-07-31 | 1999-12-07 | The Scripps Research Institute | Complex formation between dsDNA and oligomer of cyclic heterocycles |
| US6140053A (en) | 1996-11-06 | 2000-10-31 | Sequenom, Inc. | DNA sequencing by mass spectrometry via exonuclease degradation |
| US5958703A (en) | 1996-12-03 | 1999-09-28 | Glaxo Group Limited | Use of modified tethers in screening compound libraries |
| US6080826A (en) | 1997-01-06 | 2000-06-27 | California Institute Of Technology | Template-directed ring-closing metathesis and ring-opening metathesis polymerization of functionalized dienes |
| AU738328B2 (en) | 1997-01-21 | 2001-09-13 | General Hospital Corporation, The | Selection of proteins using RNA-protein fusions |
| US6261804B1 (en) | 1997-01-21 | 2001-07-17 | The General Hospital Corporation | Selection of proteins using RNA-protein fusions |
| US6969584B2 (en) | 1997-06-12 | 2005-11-29 | Rigel Pharmaceuticals, Inc. | Combinatorial enzymatic complexes |
| US6607878B2 (en) | 1997-10-06 | 2003-08-19 | Stratagene | Collections of uniquely tagged molecules |
| AU752220B2 (en) * | 1998-01-27 | 2002-09-12 | British Biocell International Limited | Modified nucleic acid probes and uses thereof |
| US5945325A (en) * | 1998-04-20 | 1999-08-31 | California Institute Of Technology | Thermally stable para-nitrobenzyl esterases |
| US6287765B1 (en) | 1998-05-20 | 2001-09-11 | Molecular Machines, Inc. | Methods for detecting and identifying single molecules |
| US6175001B1 (en) | 1998-10-16 | 2001-01-16 | The Scripps Research Institute | Functionalized pyrimidine nucleosides and nucleotides and DNA's incorporating same |
| CA2346989A1 (en) * | 1998-10-19 | 2000-04-27 | The Board Of Trustees Of The Leland Stanford Junior University | Dna-templated combinatorial library chemistry |
| CA2403209A1 (en) | 1999-04-08 | 2000-10-19 | Pavel V. Sergeev | Synthesis of biologically active compounds in cells |
| WO2001016352A1 (en) * | 1999-08-27 | 2001-03-08 | Phylos, Inc. | Methods for encoding and sorting in vitro translated proteins |
| US7306904B2 (en) * | 2000-02-18 | 2007-12-11 | Olink Ab | Methods and kits for proximity probing |
| US6511809B2 (en) * | 2000-06-13 | 2003-01-28 | E. I. Du Pont De Nemours And Company | Method for the detection of an analyte by means of a nucleic acid reporter |
| WO2004001042A2 (en) | 2002-06-20 | 2003-12-31 | Nuevolution A/S | Microarrays displaying encoded molecules |
| DK1402024T3 (da) | 2001-06-20 | 2007-12-27 | Nuevolution As | Fremgangsmåde til syntese af et template-molekyle |
| WO2002102820A1 (en) | 2001-06-20 | 2002-12-27 | Nuevolution A/S | Nucleoside derivatives for library preparation |
| US20030143561A1 (en) | 2001-06-20 | 2003-07-31 | Nuevolution A/S | Nucleoside derivatives for library preparation |
| DE60231436D1 (de) | 2002-03-08 | 2009-04-16 | Eidgenoess Tech Hochschule | Kodierte, selbst-assemblierende chemische bibliotheken (esachel) |
| WO2003078446A2 (en) | 2002-03-15 | 2003-09-25 | Nuevolution A/S | A building block forming a c-c or a c-hetero atom bond upon reaction |
| US20050221318A1 (en) | 2002-03-15 | 2005-10-06 | Gouliaev Alex H | Building block forming a c-c bond upon reaction |
| DE60324778D1 (de) | 2002-03-15 | 2009-01-02 | Nuevolution As | Eine verbesserte methode zur synthese von matritzenabhängigen molekülen |
| EP1487849A2 (en) | 2002-03-15 | 2004-12-22 | Nuevolution A/S | A building block capable of transferring a functional entity |
| AU2003214032A1 (en) | 2002-03-15 | 2003-09-29 | Nuevolution A/S | A building block forming a c=c double bond upon reaction |
| WO2003082901A2 (en) | 2002-03-22 | 2003-10-09 | Emory University | Template-driven processes for synthesizing polymers and components related to such processes |
| JP4657919B2 (ja) | 2002-08-19 | 2011-03-23 | プレジデント アンド フェローズ オブ ハーバード カレッジ | 進化する新しい分子機能 |
| US8017323B2 (en) * | 2003-03-26 | 2011-09-13 | President And Fellows Of Harvard College | Free reactant use in nucleic acid-templated synthesis |
| EP1888746B1 (en) * | 2005-06-07 | 2010-03-17 | President And Fellows Of Harvard College | Ordered multi-step synthesis by nucleic acid-mediated chemistry |
| DE602006013134D1 (de) | 2005-06-17 | 2010-05-06 | Harvard College | Iterierte verzweigende reaktionswege über nukleinsäure-vermittelte chemie |
| WO2006138560A2 (en) * | 2005-06-17 | 2006-12-28 | President And Fellows Of Harvard College | Nucleic acid-templated chemistry in organic solvents |
-
2002
- 2002-03-19 IL IL15800002A patent/IL158000A0/xx unknown
- 2002-03-19 EP EP06016511A patent/EP1832567A3/en not_active Withdrawn
- 2002-03-19 JP JP2002574322A patent/JP4128453B2/ja not_active Expired - Fee Related
- 2002-03-19 DK DK02753671.3T patent/DK1423400T4/da active
- 2002-03-19 US US10/101,030 patent/US7070928B2/en not_active Expired - Lifetime
- 2002-03-19 AT AT02753671T patent/ATE335754T1/de active
- 2002-03-19 EP EP02753671.3A patent/EP1423400B2/en not_active Expired - Lifetime
- 2002-03-19 ES ES02753671T patent/ES2271306T5/es not_active Expired - Lifetime
- 2002-03-19 DE DE60213826T patent/DE60213826T3/de not_active Expired - Lifetime
- 2002-03-19 EP EP10182548A patent/EP2332896A3/en not_active Withdrawn
- 2002-03-19 WO PCT/US2002/008546 patent/WO2002074929A2/en active IP Right Grant
- 2002-03-19 AU AU2002306777A patent/AU2002306777C1/en not_active Ceased
- 2002-03-19 PT PT02753671T patent/PT1423400E/pt unknown
- 2002-03-19 CA CA2441820A patent/CA2441820C/en not_active Expired - Fee Related
-
2003
- 2003-09-18 IL IL158000A patent/IL158000A/en not_active IP Right Cessation
- 2003-12-23 US US10/744,605 patent/US7557068B2/en not_active Expired - Lifetime
-
2004
- 2004-09-24 US US10/949,163 patent/US7223545B2/en not_active Expired - Lifetime
- 2004-09-24 US US10/949,162 patent/US20050042669A1/en not_active Abandoned
-
2005
- 2005-05-31 US US11/141,542 patent/US7442160B2/en not_active Expired - Lifetime
- 2005-05-31 US US11/141,164 patent/US7998904B2/en not_active Expired - Fee Related
-
2008
- 2008-01-03 AU AU2008200027A patent/AU2008200027B2/en not_active Ceased
- 2008-02-22 JP JP2008042250A patent/JP4384229B2/ja not_active Expired - Fee Related
-
2009
- 2009-07-17 JP JP2009169425A patent/JP2009232869A/ja not_active Withdrawn
-
2010
- 2010-06-23 IL IL206585A patent/IL206585A/en not_active IP Right Cessation
- 2010-06-23 IL IL206586A patent/IL206586A/en not_active IP Right Cessation
-
2011
- 2011-06-30 US US13/173,593 patent/US20120165228A1/en not_active Abandoned
-
2012
- 2012-07-26 JP JP2012165977A patent/JP5509270B2/ja not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001509828A (ja) * | 1997-01-08 | 2001-07-24 | プロリゴ・エルエルシー | 高分子のバイオコンジュゲーション |
| JP2003529328A (ja) * | 1999-08-12 | 2003-10-07 | マキシジェン, インコーポレイテッド | コンビナトリアル化学および医化学のための酵素の進化および使用 |
| JP2004536162A (ja) * | 2001-03-19 | 2004-12-02 | プレジデント アンド フェロウズ オブ ハーバード カレッジ | 新規分子機能の進化 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5509270B2 (ja) | 新規分子機能の進化 | |
| JP5254934B2 (ja) | 進化する新しい分子機能 | |
| AU2002306777A1 (en) | Evolving new molecular function | |
| JP2005536234A5 (ja) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20131023 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140117 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140224 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140324 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5509270 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |