JP2010013458A - クラブラン酸カリウムの製造方法 - Google Patents
クラブラン酸カリウムの製造方法 Download PDFInfo
- Publication number
- JP2010013458A JP2010013458A JP2009197755A JP2009197755A JP2010013458A JP 2010013458 A JP2010013458 A JP 2010013458A JP 2009197755 A JP2009197755 A JP 2009197755A JP 2009197755 A JP2009197755 A JP 2009197755A JP 2010013458 A JP2010013458 A JP 2010013458A
- Authority
- JP
- Japan
- Prior art keywords
- clavulanic acid
- solution
- potassium
- organic solvent
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
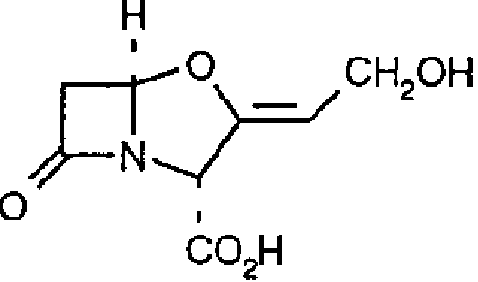
- ABVRVIZBZKUTMK-JSYANWSFSA-M potassium clavulanate Chemical compound [K+].[O-]C(=O)[C@H]1C(=C/CO)/O[C@@H]2CC(=O)N21 ABVRVIZBZKUTMK-JSYANWSFSA-M 0.000 title claims abstract description 31
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 8
- HZZVJAQRINQKSD-PBFISZAISA-N clavulanic acid Chemical compound OC(=O)[C@H]1C(=C/CO)/O[C@@H]2CC(=O)N21 HZZVJAQRINQKSD-PBFISZAISA-N 0.000 claims abstract description 90
- HZZVJAQRINQKSD-UHFFFAOYSA-N Clavulanic acid Natural products OC(=O)C1C(=CCO)OC2CC(=O)N21 HZZVJAQRINQKSD-UHFFFAOYSA-N 0.000 claims abstract description 89
- 229960003324 clavulanic acid Drugs 0.000 claims abstract description 78
- 239000000243 solution Substances 0.000 claims abstract description 41
- 239000003960 organic solvent Substances 0.000 claims abstract description 32
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 claims abstract description 14
- -1 amine salt Chemical class 0.000 claims abstract description 11
- 239000007864 aqueous solution Substances 0.000 claims abstract description 11
- 239000012736 aqueous medium Substances 0.000 claims abstract description 8
- 238000000855 fermentation Methods 0.000 claims abstract description 8
- 230000004151 fermentation Effects 0.000 claims abstract description 8
- 238000001556 precipitation Methods 0.000 claims abstract description 6
- 229910001414 potassium ion Inorganic materials 0.000 claims abstract description 3
- 238000000034 method Methods 0.000 claims description 45
- 239000002904 solvent Substances 0.000 claims description 22
- 239000002253 acid Substances 0.000 claims description 17
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 12
- 238000001914 filtration Methods 0.000 claims description 9
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical group CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 8
- 238000000108 ultra-filtration Methods 0.000 claims description 4
- 150000007524 organic acids Chemical class 0.000 claims description 3
- 230000001376 precipitating effect Effects 0.000 claims description 3
- 238000004255 ion exchange chromatography Methods 0.000 claims description 2
- ZUFQCVZBBNZMKD-UHFFFAOYSA-M potassium 2-ethylhexanoate Chemical group [K+].CCCCC(CC)C([O-])=O ZUFQCVZBBNZMKD-UHFFFAOYSA-M 0.000 claims description 2
- 238000001223 reverse osmosis Methods 0.000 claims description 2
- 238000006243 chemical reaction Methods 0.000 claims 1
- 238000003828 vacuum filtration Methods 0.000 claims 1
- 238000000605 extraction Methods 0.000 abstract description 8
- NPYPAHLBTDXSSS-UHFFFAOYSA-N Potassium ion Chemical compound [K+] NPYPAHLBTDXSSS-UHFFFAOYSA-N 0.000 abstract 1
- 230000001737 promoting effect Effects 0.000 abstract 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 28
- 150000003839 salts Chemical class 0.000 description 22
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 21
- 239000002243 precursor Substances 0.000 description 16
- 150000001875 compounds Chemical class 0.000 description 12
- 239000000203 mixture Substances 0.000 description 12
- 229940090805 clavulanate Drugs 0.000 description 11
- 150000001412 amines Chemical class 0.000 description 10
- 238000009472 formulation Methods 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- 239000000284 extract Substances 0.000 description 8
- 238000000746 purification Methods 0.000 description 8
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 7
- 239000012535 impurity Substances 0.000 description 7
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- 239000008346 aqueous phase Substances 0.000 description 6
- 239000002609 medium Substances 0.000 description 6
- 125000000217 alkyl group Chemical group 0.000 description 5
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- 229910052783 alkali metal Inorganic materials 0.000 description 4
- DKPFZGUDAPQIHT-UHFFFAOYSA-N butyl acetate Chemical compound CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 4
- 229910052799 carbon Inorganic materials 0.000 description 4
- 238000002955 isolation Methods 0.000 description 4
- 239000008194 pharmaceutical composition Substances 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 238000000638 solvent extraction Methods 0.000 description 4
- OBETXYAYXDNJHR-UHFFFAOYSA-N 2-Ethylhexanoic acid Chemical compound CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 3
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 3
- 150000001450 anions Chemical class 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 229940088710 antibiotic agent Drugs 0.000 description 3
- 230000003115 biocidal effect Effects 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 229960003975 potassium Drugs 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 description 3
- LAIUFBWHERIJIH-UHFFFAOYSA-N 3-Methylheptane Chemical compound CCCCC(C)CC LAIUFBWHERIJIH-UHFFFAOYSA-N 0.000 description 2
- 208000035143 Bacterial infection Diseases 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 241000187433 Streptomyces clavuligerus Species 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 239000006286 aqueous extract Substances 0.000 description 2
- 208000022362 bacterial infectious disease Diseases 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000006184 cosolvent Substances 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 2
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 2
- 235000019797 dipotassium phosphate Nutrition 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- MFIGJRRHGZYPDD-UHFFFAOYSA-N n,n'-di(propan-2-yl)ethane-1,2-diamine Chemical compound CC(C)NCCNC(C)C MFIGJRRHGZYPDD-UHFFFAOYSA-N 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 238000005191 phase separation Methods 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 235000012239 silicon dioxide Nutrition 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- OBETXYAYXDNJHR-SSDOTTSWSA-M (2r)-2-ethylhexanoate Chemical compound CCCC[C@@H](CC)C([O-])=O OBETXYAYXDNJHR-SSDOTTSWSA-M 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- LIEYAZJYCTUZBM-UHFFFAOYSA-N C(CCCCC)(=O)O.CC[K] Chemical compound C(CCCCC)(=O)O.CC[K] LIEYAZJYCTUZBM-UHFFFAOYSA-N 0.000 description 1
- 229930186147 Cephalosporin Natural products 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- JLTDJTHDQAWBAV-UHFFFAOYSA-N N,N-dimethylaniline Chemical compound CN(C)C1=CC=CC=C1 JLTDJTHDQAWBAV-UHFFFAOYSA-N 0.000 description 1
- AFBPFSWMIHJQDM-UHFFFAOYSA-N N-methylaniline Chemical compound CNC1=CC=CC=C1 AFBPFSWMIHJQDM-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000274 adsorptive effect Effects 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- LSQZJLSUYDQPKJ-NJBDSQKTSA-N amoxicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=C(O)C=C1 LSQZJLSUYDQPKJ-NJBDSQKTSA-N 0.000 description 1
- 229960003022 amoxicillin Drugs 0.000 description 1
- 239000003957 anion exchange resin Substances 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 239000003782 beta lactam antibiotic agent Substances 0.000 description 1
- 239000003781 beta lactamase inhibitor Substances 0.000 description 1
- 229940126813 beta-lactamase inhibitor Drugs 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- HFNQLYDPNAZRCH-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O.OC(O)=O HFNQLYDPNAZRCH-UHFFFAOYSA-N 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 229940124587 cephalosporin Drugs 0.000 description 1
- 150000001780 cephalosporins Chemical class 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 239000002274 desiccant Substances 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000004088 foaming agent Substances 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- LSQZJLSUYDQPKJ-UHFFFAOYSA-N p-Hydroxyampicillin Natural products O=C1N2C(C(O)=O)C(C)(C)SC2C1NC(=O)C(N)C1=CC=C(O)C=C1 LSQZJLSUYDQPKJ-UHFFFAOYSA-N 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 229940086066 potassium hydrogencarbonate Drugs 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 238000005185 salting out Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229940083542 sodium Drugs 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M sodium bicarbonate Substances [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- OHKOGUYZJXTSFX-KZFFXBSXSA-N ticarcillin Chemical compound C=1([C@@H](C(O)=O)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)C=CSC=1 OHKOGUYZJXTSFX-KZFFXBSXSA-N 0.000 description 1
- 229960004659 ticarcillin Drugs 0.000 description 1
- 150000004684 trihydrates Chemical group 0.000 description 1
- 239000002132 β-lactam antibiotic Substances 0.000 description 1
- 229940124586 β-lactam antibiotics Drugs 0.000 description 1
- 229940126085 β‑Lactamase Inhibitor Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D503/00—Heterocyclic compounds containing 4-oxa-1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. oxapenicillins, clavulanic acid derivatives; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Compounds Of Unknown Constitution (AREA)
- Saccharide Compounds (AREA)
- Glass Compositions (AREA)
- Iron Core Of Rotating Electric Machines (AREA)
Abstract
【解決手段】発酵プロセスから生じるクラブラン酸の遊離酸が有機溶媒中に抽出されることにより有機溶媒中でクラブラン酸の溶液を形成させ、カリウム塩を有機溶媒中で溶液中のクラブラン酸に添加し、次いで、かかる形成されたクラブラン酸カリウムを、有機溶媒から水性媒体に逆抽出することにより有機溶媒に対する分離相としての水性媒体中にクラブラン酸カリウムの溶液を形成させ、次いで、クラブラン酸カリウムの水溶液への有機沈殿溶媒の添加によりクラブラン酸カリウムの生成物を水溶液から沈殿させる方法。
【選択図】なし
Description
a)ブロスのRVF濾過、ついで、発酵ブロスのイオン交換クロマトグラフィー
b)ブロスの限外濾過、ついで、発酵ブロスのRO濃縮
生成物重量=2.03g
生成物純度=純粋な遊離酸として83.0%
豊富溶媒からの収率=78%
生成物重量=21.0g
結晶化段階での収率=86.3%
生成物純度=純粋な遊離酸として83.2%
生成物水分含量=0.3%
Claims (14)
- 中間体アミン塩を形成することによって前精製されていないクラブラン酸をカリウム塩として直接沈殿させることによる、そして、溶液中のクラブラン酸と溶液中のカリウムイオンとの反応による、医薬上許容される品質のクラブラン酸カリウムの製造方法であって、発酵プロセスから生じるクラブラン酸の遊離酸が有機溶媒中に抽出されることにより有機溶媒中でクラブラン酸の溶液を形成させる方法であって、
カリウム塩を有機溶媒中で溶液中のクラブラン酸に添加し、
次いで、かかる形成されたクラブラン酸カリウムを、有機溶媒から水性媒体に逆抽出することにより有機溶媒に対する分離相としての水性媒体中にクラブラン酸カリウムの溶液を形成させ、
次いで、クラブラン酸カリウムの水溶液への有機沈殿溶媒の添加によりクラブラン酸カリウムの生成物を水溶液から沈殿させること、
を特徴とする方法。 - 医薬上許容される品質のクラブラン酸カリウムが、純粋な遊離酸として少なくとも80%の純度を有することを特徴とする、請求項1記載の方法。
- 前記カリウム塩が、有機酸のカリウム塩であることを特徴とする、請求項1または2記載の方法。
- 前記カリウム塩が、2−エチルヘキサン酸カリウムであることを特徴とする、請求項3記載の方法。
- 水性媒体中のクラブラン酸の溶液が濃縮されることを特徴とする、請求項1〜3のいずれか1項記載の方法。
- 沈殿溶媒が、水不混和性溶媒と混合されてもよい水混和性溶媒であることを特徴とする、請求項1〜5のいずれか1項記載の方法。
- 沈殿溶媒が、アセトンまたはイソプロパノールであることを特徴とする、請求項6記載の方法。
- クラブラン酸の力価が、1ミリリットルあたり100,000ないし200,000マイクログラムの範囲であることを特徴とする、請求項1〜7のいずれか1項記載の方法。
- クラブラン酸が、全ブロス発酵物より得られることを特徴とする、請求項1〜8のいずれか1項記載の方法。
- 全ブロス発酵物が濾過されることを特徴とする、請求項9記載の方法。
- 濾過が、限外濾過によることを特徴とする、請求項10記載の方法。
- 濾過が、逆浸透の前に行われることを特徴とする、請求項11記載の方法。
- 濾過が、RVF(回転真空濾過)によるものである、請求項11記載の方法。
- 濾過が、イオン交換クロマトグラフィーの前に行われることを特徴とする、請求項13記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9515809.3 | 1995-08-02 | ||
| GBGB9515809.3A GB9515809D0 (en) | 1995-08-02 | 1995-08-02 | Process |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50725297A Division JP2002515022A (ja) | 1995-08-02 | 1996-07-30 | クラブラン酸カリウムの製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010013458A true JP2010013458A (ja) | 2010-01-21 |
| JP5184465B2 JP5184465B2 (ja) | 2013-04-17 |
Family
ID=10778623
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50725297A Withdrawn JP2002515022A (ja) | 1995-08-02 | 1996-07-30 | クラブラン酸カリウムの製造方法 |
| JP2009197755A Expired - Lifetime JP5184465B2 (ja) | 1995-08-02 | 2009-08-28 | クラブラン酸カリウムの製造方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50725297A Withdrawn JP2002515022A (ja) | 1995-08-02 | 1996-07-30 | クラブラン酸カリウムの製造方法 |
Country Status (10)
| Country | Link |
|---|---|
| EP (2) | EP1088826A1 (ja) |
| JP (2) | JP2002515022A (ja) |
| AT (1) | ATE205498T1 (ja) |
| AU (1) | AU6740296A (ja) |
| DE (1) | DE69615204T2 (ja) |
| DK (1) | DK0843680T3 (ja) |
| ES (1) | ES2163038T3 (ja) |
| GB (1) | GB9515809D0 (ja) |
| PT (1) | PT843680E (ja) |
| WO (1) | WO1997005142A1 (ja) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AT400033B (de) | 1992-03-10 | 1995-09-25 | Biochemie Gmbh | Neues verfahren zur isolierung und reinigung von clavulansäure und zur herstellung von pharmakologisch verträglichen salzen derselben |
| SI9400107A (en) | 1994-03-02 | 1995-10-31 | Lek Tovarna Farmacevtskih | New process of the isolation of clavulanic acid and its pharmaceutical salts from fermented broth of streptomyces sp.p 6621 ferm p 2804. |
| SI9500074A (en) * | 1995-03-10 | 1996-10-31 | Lek Tovarna Farmacevtskih | Process for preparation of alkani salts of clavulanic acid. |
| AT403375B (de) * | 1995-11-15 | 1998-01-26 | Biochemie Gmbh | Verfahren zur fällung von alkalisalzen der clavulansäure |
| ZA975198B (en) | 1996-06-13 | 1997-12-15 | Smithkline Beecham Corp | Improved process for preparing potassium clavulanate. |
| WO1998042858A1 (en) * | 1997-03-24 | 1998-10-01 | Cipan-Companhia Industrial Produtora De Antibióticos, S.A. | Process for the isolation of a pharmaceutically acceptable alkali metal salt of clavulanic acid |
| DE69700903T2 (de) | 1997-03-24 | 2000-07-06 | Cipan-Companhia Industrial Produtora De Antibioticos, S.A. | Verfahren zur Isolierung eines pharmazeutisch verträglichen Alkalimetallsalzes der Clavulansäure |
| DZ2456A1 (fr) | 1997-04-04 | 2003-01-18 | Smithkline Beecham Plc | Procédé de préparation de sels de l'acide clavulanique. |
| KR100827898B1 (ko) | 2000-05-13 | 2008-05-07 | 스미스클라인비이참피이엘시이 | 클라불란산의 염의 정제 방법 |
| CN103833774A (zh) * | 2014-03-21 | 2014-06-04 | 山东新时代药业有限公司 | 一种从克拉维酸钾混粉中回收有效成分的方法 |
| CN117534683B (zh) * | 2023-11-13 | 2024-08-16 | 山西双雁药业有限公司 | 克拉维酸钾的制备方法 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS50142591A (ja) * | 1974-04-20 | 1975-11-17 | ||
| JPS5448787A (en) * | 1977-09-01 | 1979-04-17 | Beecham Group Ltd | Manufacture of potassium clavulanate |
| JPS5634685A (en) * | 1979-08-24 | 1981-04-06 | Beecham Group Ltd | Salt of clavulanic acid* its manufacture and use |
| JPS61119195A (ja) * | 1984-10-27 | 1986-06-06 | ビーチャム・グループ・パブリック・リミテッド・カンパニー | クラブラン酸の製法 |
| JPS6259281A (ja) * | 1985-08-29 | 1987-03-14 | ビ−チヤム・グル−プ・ピ−エルシ− | β−ラクタム化合物およびその製造方法 |
| JPH01101879A (ja) * | 1987-09-15 | 1989-04-19 | Henkel Kgaa | 生物工学的に生産された有用物質の培養液からの分離方法 |
| JPH05294982A (ja) * | 1991-12-04 | 1993-11-09 | Ministero Dell Univ E Della Ric Scient & Tecnol | 抗生物質ab−041およびその製造法 |
| JPH06197782A (ja) * | 1992-10-21 | 1994-07-19 | Pharma Dev Ltd | 粗クラブラン酸の精製方法 |
| JP4074336B2 (ja) * | 1994-02-02 | 2008-04-09 | スミスクライン・ビーチャム・パブリック・リミテッド・カンパニー | クラブラン酸の塩の製法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB2003863B (en) * | 1977-09-01 | 1982-03-31 | Beecham Group Ltd | Chemical process |
| GB2200355B (en) * | 1987-01-29 | 1990-08-01 | Beecham Group Plc | Potassium clavulanate |
| SI9300296B (sl) * | 1992-06-11 | 1998-06-30 | Smithkline Beecham P.L.C. | Postopek in intermedianti za pripravo klavulanske kisline |
| AT400846B (de) * | 1994-02-25 | 1996-03-25 | Fermic S A De C V | Verfahren zur gewinnung und reinigung von alkalimetallsalzen der clavulansäure |
| SI9400107A (en) * | 1994-03-02 | 1995-10-31 | Lek Tovarna Farmacevtskih | New process of the isolation of clavulanic acid and its pharmaceutical salts from fermented broth of streptomyces sp.p 6621 ferm p 2804. |
| SI9500074A (en) * | 1995-03-10 | 1996-10-31 | Lek Tovarna Farmacevtskih | Process for preparation of alkani salts of clavulanic acid. |
| KR100200242B1 (ko) * | 1995-05-16 | 1999-06-15 | 김충환 | 클라불란산염의 제조 방법 |
-
1995
- 1995-08-02 GB GBGB9515809.3A patent/GB9515809D0/en active Pending
-
1996
- 1996-07-30 ES ES96927643T patent/ES2163038T3/es not_active Expired - Lifetime
- 1996-07-30 AU AU67402/96A patent/AU6740296A/en not_active Abandoned
- 1996-07-30 EP EP00127571A patent/EP1088826A1/en not_active Ceased
- 1996-07-30 EP EP96927643A patent/EP0843680B1/en not_active Expired - Lifetime
- 1996-07-30 JP JP50725297A patent/JP2002515022A/ja not_active Withdrawn
- 1996-07-30 DE DE69615204T patent/DE69615204T2/de not_active Expired - Lifetime
- 1996-07-30 DK DK96927643T patent/DK0843680T3/da active
- 1996-07-30 WO PCT/EP1996/003375 patent/WO1997005142A1/en not_active Ceased
- 1996-07-30 PT PT96927643T patent/PT843680E/pt unknown
- 1996-07-30 AT AT96927643T patent/ATE205498T1/de active
-
2009
- 2009-08-28 JP JP2009197755A patent/JP5184465B2/ja not_active Expired - Lifetime
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS50142591A (ja) * | 1974-04-20 | 1975-11-17 | ||
| JPS5448787A (en) * | 1977-09-01 | 1979-04-17 | Beecham Group Ltd | Manufacture of potassium clavulanate |
| JPS5634685A (en) * | 1979-08-24 | 1981-04-06 | Beecham Group Ltd | Salt of clavulanic acid* its manufacture and use |
| JPS61119195A (ja) * | 1984-10-27 | 1986-06-06 | ビーチャム・グループ・パブリック・リミテッド・カンパニー | クラブラン酸の製法 |
| JPS6259281A (ja) * | 1985-08-29 | 1987-03-14 | ビ−チヤム・グル−プ・ピ−エルシ− | β−ラクタム化合物およびその製造方法 |
| JPH01101879A (ja) * | 1987-09-15 | 1989-04-19 | Henkel Kgaa | 生物工学的に生産された有用物質の培養液からの分離方法 |
| JPH05294982A (ja) * | 1991-12-04 | 1993-11-09 | Ministero Dell Univ E Della Ric Scient & Tecnol | 抗生物質ab−041およびその製造法 |
| JPH06197782A (ja) * | 1992-10-21 | 1994-07-19 | Pharma Dev Ltd | 粗クラブラン酸の精製方法 |
| JP4074336B2 (ja) * | 1994-02-02 | 2008-04-09 | スミスクライン・ビーチャム・パブリック・リミテッド・カンパニー | クラブラン酸の塩の製法 |
Also Published As
| Publication number | Publication date |
|---|---|
| DE69615204T2 (de) | 2002-06-13 |
| JP2002515022A (ja) | 2002-05-21 |
| PT843680E (pt) | 2002-01-30 |
| EP0843680A1 (en) | 1998-05-27 |
| EP1088826A1 (en) | 2001-04-04 |
| DE69615204D1 (de) | 2001-10-18 |
| WO1997005142A1 (en) | 1997-02-13 |
| ATE205498T1 (de) | 2001-09-15 |
| JP5184465B2 (ja) | 2013-04-17 |
| EP0843680B1 (en) | 2001-09-12 |
| AU6740296A (en) | 1997-02-26 |
| ES2163038T3 (es) | 2002-01-16 |
| DK0843680T3 (da) | 2002-01-21 |
| GB9515809D0 (en) | 1995-10-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5184465B2 (ja) | クラブラン酸カリウムの製造方法 | |
| RU2206613C2 (ru) | Способ выделения клавулановой кислоты | |
| JP2873143B2 (ja) | 新規なアルキレンジアンモニウム ジクラブラネート誘導体、その製造方法並びにその使用 | |
| JP4074336B2 (ja) | クラブラン酸の塩の製法 | |
| EP0813536B1 (en) | Process for the preparation of pharmaceutically acceptable salts of clavulanic acid | |
| JPH0641143A (ja) | クラブラン酸塩の製造方法 | |
| SI9500134A1 (en) | Preparation procedure of pure alkali salts of clavulanic acid | |
| AU776184B2 (en) | Process for the purification of a salt of clavulanic acid | |
| KR100537732B1 (ko) | 클라불란산염의제조 | |
| EP0941229B1 (en) | Purification of fermented clavulanic acid | |
| US5801241A (en) | Cyclohexanone extraction of 3-hydroxymethylcephalosporins | |
| JP2001521393A (ja) | クラブラン酸の薬学的に許容されるアルカリ金属塩の単離方法 | |
| JP2002509933A (ja) | セファロスポリンの発酵製造の新規な方法 | |
| CA2337072A1 (en) | Improved process for the preparation of salts and esters of clavulanic acid | |
| HK1036621A (en) | Process for the preparation of potassium clavulanate | |
| US20010029038A1 (en) | Purification of fermented clavulanic acid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20120309 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A132 Effective date: 20120703 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120911 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20121002 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20121206 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121225 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130116 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20160125 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |