JP2008538282A - 装置および循環腫瘍細胞および他の粒子の濃縮および変更のための方法 - Google Patents
装置および循環腫瘍細胞および他の粒子の濃縮および変更のための方法 Download PDFInfo
- Publication number
- JP2008538282A JP2008538282A JP2008505508A JP2008505508A JP2008538282A JP 2008538282 A JP2008538282 A JP 2008538282A JP 2008505508 A JP2008505508 A JP 2008505508A JP 2008505508 A JP2008505508 A JP 2008505508A JP 2008538282 A JP2008538282 A JP 2008538282A
- Authority
- JP
- Japan
- Prior art keywords
- cells
- cell
- cancer
- sample
- microns
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000000034 method Methods 0.000 title claims abstract description 475
- 239000002245 particle Substances 0.000 title abstract description 96
- 208000005443 Circulating Neoplastic Cells Diseases 0.000 title abstract description 91
- 230000004048 modification Effects 0.000 title description 7
- 238000012986 modification Methods 0.000 title description 7
- 210000004027 cell Anatomy 0.000 claims abstract description 1054
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 282
- 201000011510 cancer Diseases 0.000 claims abstract description 205
- 239000000523 sample Substances 0.000 claims description 568
- 108060006698 EGF receptor Proteins 0.000 claims description 162
- 102000001301 EGF receptor Human genes 0.000 claims description 154
- 239000003153 chemical reaction reagent Substances 0.000 claims description 126
- 230000035772 mutation Effects 0.000 claims description 112
- 210000002919 epithelial cell Anatomy 0.000 claims description 92
- 239000012530 fluid Substances 0.000 claims description 91
- 108020004414 DNA Proteins 0.000 claims description 88
- 150000007523 nucleic acids Chemical class 0.000 claims description 84
- 102000039446 nucleic acids Human genes 0.000 claims description 81
- 108020004707 nucleic acids Proteins 0.000 claims description 81
- 238000002372 labelling Methods 0.000 claims description 80
- 210000004369 blood Anatomy 0.000 claims description 68
- 239000008280 blood Substances 0.000 claims description 68
- 239000002773 nucleotide Substances 0.000 claims description 64
- 125000003729 nucleotide group Chemical group 0.000 claims description 61
- 238000001514 detection method Methods 0.000 claims description 57
- 108091000080 Phosphotransferase Proteins 0.000 claims description 55
- 238000004458 analytical method Methods 0.000 claims description 55
- 102000020233 phosphotransferase Human genes 0.000 claims description 55
- 108090000623 proteins and genes Proteins 0.000 claims description 44
- 230000000694 effects Effects 0.000 claims description 43
- 210000003743 erythrocyte Anatomy 0.000 claims description 42
- 238000003199 nucleic acid amplification method Methods 0.000 claims description 41
- 238000012545 processing Methods 0.000 claims description 41
- 230000003321 amplification Effects 0.000 claims description 40
- 239000000047 product Substances 0.000 claims description 39
- 230000027455 binding Effects 0.000 claims description 37
- 239000000872 buffer Substances 0.000 claims description 37
- 239000012634 fragment Substances 0.000 claims description 36
- 210000001519 tissue Anatomy 0.000 claims description 36
- 238000012217 deletion Methods 0.000 claims description 34
- 230000037430 deletion Effects 0.000 claims description 34
- 238000006467 substitution reaction Methods 0.000 claims description 33
- 238000012163 sequencing technique Methods 0.000 claims description 30
- 230000008685 targeting Effects 0.000 claims description 30
- 208000020816 lung neoplasm Diseases 0.000 claims description 27
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 26
- 108700021358 erbB-1 Genes Proteins 0.000 claims description 26
- 210000000265 leukocyte Anatomy 0.000 claims description 26
- 210000000130 stem cell Anatomy 0.000 claims description 26
- 238000003491 array Methods 0.000 claims description 24
- 201000010099 disease Diseases 0.000 claims description 24
- 239000003814 drug Substances 0.000 claims description 24
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 24
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 23
- 229920001184 polypeptide Polymers 0.000 claims description 22
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 20
- 210000000601 blood cell Anatomy 0.000 claims description 20
- 239000011324 bead Substances 0.000 claims description 19
- 201000005202 lung cancer Diseases 0.000 claims description 19
- 108020004999 messenger RNA Proteins 0.000 claims description 19
- 239000005411 L01XE02 - Gefitinib Substances 0.000 claims description 18
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 18
- 150000001413 amino acids Chemical group 0.000 claims description 18
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 claims description 18
- 230000001613 neoplastic effect Effects 0.000 claims description 18
- 230000008569 process Effects 0.000 claims description 18
- 208000000453 Skin Neoplasms Diseases 0.000 claims description 17
- 239000012472 biological sample Substances 0.000 claims description 17
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 17
- 201000000849 skin cancer Diseases 0.000 claims description 17
- 206010006187 Breast cancer Diseases 0.000 claims description 16
- 206010039491 Sarcoma Diseases 0.000 claims description 16
- 238000003745 diagnosis Methods 0.000 claims description 16
- 210000005168 endometrial cell Anatomy 0.000 claims description 16
- 230000001605 fetal effect Effects 0.000 claims description 16
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 claims description 16
- 241000894006 Bacteria Species 0.000 claims description 15
- 208000026310 Breast neoplasm Diseases 0.000 claims description 15
- 102000004190 Enzymes Human genes 0.000 claims description 15
- 108090000790 Enzymes Proteins 0.000 claims description 15
- 241000233866 Fungi Species 0.000 claims description 15
- 229960002584 gefitinib Drugs 0.000 claims description 15
- 210000002993 trophoblast Anatomy 0.000 claims description 15
- 239000005483 tyrosine kinase inhibitor Substances 0.000 claims description 15
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 14
- 101150039808 Egfr gene Proteins 0.000 claims description 14
- 206010060862 Prostate cancer Diseases 0.000 claims description 14
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 14
- 210000002798 bone marrow cell Anatomy 0.000 claims description 14
- 238000009826 distribution Methods 0.000 claims description 14
- 230000014509 gene expression Effects 0.000 claims description 14
- 208000015181 infectious disease Diseases 0.000 claims description 14
- 208000014674 injury Diseases 0.000 claims description 14
- 230000003211 malignant effect Effects 0.000 claims description 14
- 201000008968 osteosarcoma Diseases 0.000 claims description 14
- 102000004169 proteins and genes Human genes 0.000 claims description 14
- 230000009870 specific binding Effects 0.000 claims description 14
- 210000002700 urine Anatomy 0.000 claims description 14
- 201000009273 Endometriosis Diseases 0.000 claims description 13
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 claims description 13
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 claims description 13
- 210000001608 connective tissue cell Anatomy 0.000 claims description 13
- 238000009396 hybridization Methods 0.000 claims description 13
- 230000000302 ischemic effect Effects 0.000 claims description 13
- 238000005259 measurement Methods 0.000 claims description 13
- 230000008733 trauma Effects 0.000 claims description 13
- 210000001185 bone marrow Anatomy 0.000 claims description 12
- 210000000987 immune system Anatomy 0.000 claims description 12
- 230000004968 inflammatory condition Effects 0.000 claims description 12
- 108010008707 Mucin-1 Proteins 0.000 claims description 11
- 206010036790 Productive cough Diseases 0.000 claims description 11
- 239000012468 concentrated sample Substances 0.000 claims description 11
- 206010061289 metastatic neoplasm Diseases 0.000 claims description 11
- 210000005170 neoplastic cell Anatomy 0.000 claims description 11
- 210000003802 sputum Anatomy 0.000 claims description 11
- 208000024794 sputum Diseases 0.000 claims description 11
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 10
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 10
- 206010038389 Renal cancer Diseases 0.000 claims description 10
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 10
- 210000001175 cerebrospinal fluid Anatomy 0.000 claims description 10
- 238000002405 diagnostic procedure Methods 0.000 claims description 10
- 230000000968 intestinal effect Effects 0.000 claims description 10
- 201000010982 kidney cancer Diseases 0.000 claims description 10
- 201000007270 liver cancer Diseases 0.000 claims description 10
- 208000014018 liver neoplasm Diseases 0.000 claims description 10
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 10
- 201000002528 pancreatic cancer Diseases 0.000 claims description 10
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 10
- 201000002510 thyroid cancer Diseases 0.000 claims description 10
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 10
- 108091032973 (ribonucleotides)n+m Proteins 0.000 claims description 9
- 206010003445 Ascites Diseases 0.000 claims description 9
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 9
- 210000004381 amniotic fluid Anatomy 0.000 claims description 9
- 210000001772 blood platelet Anatomy 0.000 claims description 9
- 238000000701 chemical imaging Methods 0.000 claims description 9
- 229940079593 drug Drugs 0.000 claims description 9
- 230000002489 hematologic effect Effects 0.000 claims description 9
- 238000003384 imaging method Methods 0.000 claims description 9
- 238000003780 insertion Methods 0.000 claims description 9
- 230000037431 insertion Effects 0.000 claims description 9
- 210000002751 lymph Anatomy 0.000 claims description 9
- 230000001394 metastastic effect Effects 0.000 claims description 9
- 235000013336 milk Nutrition 0.000 claims description 9
- 210000004080 milk Anatomy 0.000 claims description 9
- 239000008267 milk Substances 0.000 claims description 9
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 9
- 210000003296 saliva Anatomy 0.000 claims description 9
- 230000028327 secretion Effects 0.000 claims description 9
- 210000000582 semen Anatomy 0.000 claims description 9
- 239000010703 silicon Substances 0.000 claims description 9
- 229910052710 silicon Inorganic materials 0.000 claims description 9
- 210000004243 sweat Anatomy 0.000 claims description 9
- 210000001138 tear Anatomy 0.000 claims description 9
- 102100031940 Epithelial cell adhesion molecule Human genes 0.000 claims description 8
- 101000920667 Homo sapiens Epithelial cell adhesion molecule Proteins 0.000 claims description 8
- 102000011782 Keratins Human genes 0.000 claims description 8
- 108010076876 Keratins Proteins 0.000 claims description 8
- 108091008611 Protein Kinase B Proteins 0.000 claims description 8
- 208000001647 Renal Insufficiency Diseases 0.000 claims description 8
- 210000004556 brain Anatomy 0.000 claims description 8
- -1 fluorophore Proteins 0.000 claims description 8
- 239000011521 glass Substances 0.000 claims description 8
- 201000006370 kidney failure Diseases 0.000 claims description 8
- 208000037841 lung tumor Diseases 0.000 claims description 8
- 238000002493 microarray Methods 0.000 claims description 8
- 229920000642 polymer Polymers 0.000 claims description 8
- 230000002062 proliferating effect Effects 0.000 claims description 8
- 239000002096 quantum dot Substances 0.000 claims description 8
- 150000004917 tyrosine kinase inhibitor derivatives Chemical group 0.000 claims description 8
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 7
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 7
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 7
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 claims description 7
- 208000003200 Adenoma Diseases 0.000 claims description 7
- 206010001233 Adenoma benign Diseases 0.000 claims description 7
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 7
- 206010004146 Basal cell carcinoma Diseases 0.000 claims description 7
- 206010005949 Bone cancer Diseases 0.000 claims description 7
- 208000018084 Bone neoplasm Diseases 0.000 claims description 7
- 206010008263 Cervical dysplasia Diseases 0.000 claims description 7
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims description 7
- 206010009944 Colon cancer Diseases 0.000 claims description 7
- 208000006168 Ewing Sarcoma Diseases 0.000 claims description 7
- 208000022072 Gallbladder Neoplasms Diseases 0.000 claims description 7
- 208000003098 Ganglion Cysts Diseases 0.000 claims description 7
- 208000037147 Hypercalcaemia Diseases 0.000 claims description 7
- 208000009164 Islet Cell Adenoma Diseases 0.000 claims description 7
- 206010023825 Laryngeal cancer Diseases 0.000 claims description 7
- 206010025323 Lymphomas Diseases 0.000 claims description 7
- 108700011259 MicroRNAs Proteins 0.000 claims description 7
- 201000003793 Myelodysplastic syndrome Diseases 0.000 claims description 7
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims description 7
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 7
- 208000005890 Neuroma Diseases 0.000 claims description 7
- 208000000821 Parathyroid Neoplasms Diseases 0.000 claims description 7
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 7
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 7
- 208000015634 Rectal Neoplasms Diseases 0.000 claims description 7
- 201000000582 Retinoblastoma Diseases 0.000 claims description 7
- 102000004495 STAT3 Transcription Factor Human genes 0.000 claims description 7
- 108010017324 STAT3 Transcription Factor Proteins 0.000 claims description 7
- 201000010208 Seminoma Diseases 0.000 claims description 7
- 208000021712 Soft tissue sarcoma Diseases 0.000 claims description 7
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 7
- 208000005400 Synovial Cyst Diseases 0.000 claims description 7
- 208000008383 Wilms tumor Diseases 0.000 claims description 7
- 230000001154 acute effect Effects 0.000 claims description 7
- 208000009956 adenocarcinoma Diseases 0.000 claims description 7
- 201000005188 adrenal gland cancer Diseases 0.000 claims description 7
- 238000001574 biopsy Methods 0.000 claims description 7
- 208000002458 carcinoid tumor Diseases 0.000 claims description 7
- 230000001684 chronic effect Effects 0.000 claims description 7
- 208000029742 colonic neoplasm Diseases 0.000 claims description 7
- 201000010175 gallbladder cancer Diseases 0.000 claims description 7
- 208000001130 gallstones Diseases 0.000 claims description 7
- 206010017758 gastric cancer Diseases 0.000 claims description 7
- 230000000148 hypercalcaemia Effects 0.000 claims description 7
- 208000030915 hypercalcemia disease Diseases 0.000 claims description 7
- 206010020718 hyperplasia Diseases 0.000 claims description 7
- 206010023841 laryngeal neoplasm Diseases 0.000 claims description 7
- 208000026037 malignant tumor of neck Diseases 0.000 claims description 7
- 208000026045 malignant tumor of parathyroid gland Diseases 0.000 claims description 7
- 201000001441 melanoma Diseases 0.000 claims description 7
- 239000002679 microRNA Substances 0.000 claims description 7
- 201000000050 myeloid neoplasm Diseases 0.000 claims description 7
- AHLKSOXEVRYIRD-UHFFFAOYSA-N n-phenylquinazolin-2-amine Chemical group N=1C=C2C=CC=CC2=NC=1NC1=CC=CC=C1 AHLKSOXEVRYIRD-UHFFFAOYSA-N 0.000 claims description 7
- 210000005036 nerve Anatomy 0.000 claims description 7
- 208000028591 pheochromocytoma Diseases 0.000 claims description 7
- 206010038038 rectal cancer Diseases 0.000 claims description 7
- 201000001275 rectum cancer Diseases 0.000 claims description 7
- 238000003757 reverse transcription PCR Methods 0.000 claims description 7
- 206010040882 skin lesion Diseases 0.000 claims description 7
- 231100000444 skin lesion Toxicity 0.000 claims description 7
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 7
- 201000011549 stomach cancer Diseases 0.000 claims description 7
- 208000033826 Promyelocytic Acute Leukemia Diseases 0.000 claims description 6
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 claims description 6
- 238000010195 expression analysis Methods 0.000 claims description 6
- 230000000527 lymphocytic effect Effects 0.000 claims description 6
- 239000011159 matrix material Substances 0.000 claims description 6
- 201000005962 mycosis fungoides Diseases 0.000 claims description 6
- 230000001717 pathogenic effect Effects 0.000 claims description 6
- 102000005962 receptors Human genes 0.000 claims description 6
- 108020003175 receptors Proteins 0.000 claims description 6
- 201000009410 rhabdomyosarcoma Diseases 0.000 claims description 6
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 6
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical group NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 claims description 5
- 108091023037 Aptamer Proteins 0.000 claims description 5
- 239000005551 L01XE03 - Erlotinib Substances 0.000 claims description 5
- 102000001712 STAT5 Transcription Factor Human genes 0.000 claims description 5
- 108010029477 STAT5 Transcription Factor Proteins 0.000 claims description 5
- 208000017733 acquired polycythemia vera Diseases 0.000 claims description 5
- 230000009089 cytolysis Effects 0.000 claims description 5
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 claims description 5
- 239000003446 ligand Substances 0.000 claims description 5
- 239000012528 membrane Substances 0.000 claims description 5
- 230000005012 migration Effects 0.000 claims description 5
- 238000013508 migration Methods 0.000 claims description 5
- 208000037244 polycythemia vera Diseases 0.000 claims description 5
- 239000003755 preservative agent Substances 0.000 claims description 5
- 230000002335 preservative effect Effects 0.000 claims description 5
- 230000000241 respiratory effect Effects 0.000 claims description 5
- 239000000243 solution Substances 0.000 claims description 5
- 239000000107 tumor biomarker Substances 0.000 claims description 5
- ARSRBNBHOADGJU-UHFFFAOYSA-N 7,12-dimethyltetraphene Chemical compound C1=CC2=CC=CC=C2C2=C1C(C)=C(C=CC=C1)C1=C2C ARSRBNBHOADGJU-UHFFFAOYSA-N 0.000 claims description 4
- 206010069754 Acquired gene mutation Diseases 0.000 claims description 4
- 208000005440 Basal Cell Neoplasms Diseases 0.000 claims description 4
- PLUBXMRUUVWRLT-UHFFFAOYSA-N Ethyl methanesulfonate Chemical compound CCOS(C)(=O)=O PLUBXMRUUVWRLT-UHFFFAOYSA-N 0.000 claims description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 4
- 208000007766 Kaposi sarcoma Diseases 0.000 claims description 4
- 108010070511 Keratin-8 Proteins 0.000 claims description 4
- 102000005712 Keratin-8 Human genes 0.000 claims description 4
- ZRKWMRDKSOPRRS-UHFFFAOYSA-N N-Methyl-N-nitrosourea Chemical compound O=NN(C)C(N)=O ZRKWMRDKSOPRRS-UHFFFAOYSA-N 0.000 claims description 4
- 239000004793 Polystyrene Substances 0.000 claims description 4
- 108010026552 Proteome Proteins 0.000 claims description 4
- 108010017842 Telomerase Proteins 0.000 claims description 4
- 230000017531 blood circulation Effects 0.000 claims description 4
- DENRZWYUOJLTMF-UHFFFAOYSA-N diethyl sulfate Chemical compound CCOS(=O)(=O)OCC DENRZWYUOJLTMF-UHFFFAOYSA-N 0.000 claims description 4
- 229960001433 erlotinib Drugs 0.000 claims description 4
- 239000000819 hypertonic solution Substances 0.000 claims description 4
- 229940021223 hypertonic solution Drugs 0.000 claims description 4
- 230000001900 immune effect Effects 0.000 claims description 4
- 239000012139 lysis buffer Substances 0.000 claims description 4
- 210000004379 membrane Anatomy 0.000 claims description 4
- MBABOKRGFJTBAE-UHFFFAOYSA-N methyl methanesulfonate Chemical compound COS(C)(=O)=O MBABOKRGFJTBAE-UHFFFAOYSA-N 0.000 claims description 4
- 210000000944 nerve tissue Anatomy 0.000 claims description 4
- 229920002223 polystyrene Polymers 0.000 claims description 4
- 230000037439 somatic mutation Effects 0.000 claims description 4
- 239000000725 suspension Substances 0.000 claims description 4
- 206010005003 Bladder cancer Diseases 0.000 claims description 3
- 108091026890 Coding region Proteins 0.000 claims description 3
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 claims description 3
- 230000007067 DNA methylation Effects 0.000 claims description 3
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 claims description 3
- 102000008394 Immunoglobulin Fragments Human genes 0.000 claims description 3
- 108010021625 Immunoglobulin Fragments Proteins 0.000 claims description 3
- 108020005196 Mitochondrial DNA Proteins 0.000 claims description 3
- VZUNGTLZRAYYDE-UHFFFAOYSA-N N-methyl-N'-nitro-N-nitrosoguanidine Chemical compound O=NN(C)C(=N)N[N+]([O-])=O VZUNGTLZRAYYDE-UHFFFAOYSA-N 0.000 claims description 3
- 102000008297 Nuclear Matrix-Associated Proteins Human genes 0.000 claims description 3
- 108010035916 Nuclear Matrix-Associated Proteins Proteins 0.000 claims description 3
- 108020004711 Nucleic Acid Probes Proteins 0.000 claims description 3
- 238000012408 PCR amplification Methods 0.000 claims description 3
- 208000016624 Retinal neoplasm Diseases 0.000 claims description 3
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 3
- 238000000149 argon plasma sintering Methods 0.000 claims description 3
- 230000005284 excitation Effects 0.000 claims description 3
- 230000013595 glycosylation Effects 0.000 claims description 3
- 201000010536 head and neck cancer Diseases 0.000 claims description 3
- 238000003364 immunohistochemistry Methods 0.000 claims description 3
- 238000002513 implantation Methods 0.000 claims description 3
- 238000007901 in situ hybridization Methods 0.000 claims description 3
- 230000031700 light absorption Effects 0.000 claims description 3
- 238000002595 magnetic resonance imaging Methods 0.000 claims description 3
- 201000011682 nervous system cancer Diseases 0.000 claims description 3
- 239000002853 nucleic acid probe Substances 0.000 claims description 3
- 230000009822 protein phosphorylation Effects 0.000 claims description 3
- 201000008933 retinal cancer Diseases 0.000 claims description 3
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 3
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 claims description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 2
- 230000009087 cell motility Effects 0.000 claims description 2
- 229960004630 chlorambucil Drugs 0.000 claims description 2
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 claims description 2
- 229960004397 cyclophosphamide Drugs 0.000 claims description 2
- 229940008406 diethyl sulfate Drugs 0.000 claims description 2
- 238000001943 fluorescence-activated cell sorting Methods 0.000 claims description 2
- 238000003119 immunoblot Methods 0.000 claims description 2
- 238000003365 immunocytochemistry Methods 0.000 claims description 2
- 238000001114 immunoprecipitation Methods 0.000 claims description 2
- 239000006249 magnetic particle Substances 0.000 claims description 2
- 229960004961 mechlorethamine Drugs 0.000 claims description 2
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 claims description 2
- 229960001924 melphalan Drugs 0.000 claims description 2
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 claims description 2
- 238000003127 radioimmunoassay Methods 0.000 claims description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 claims description 2
- 229960004528 vincristine Drugs 0.000 claims description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 claims description 2
- 238000001262 western blot Methods 0.000 claims description 2
- 210000002889 endothelial cell Anatomy 0.000 claims 18
- 230000004907 flux Effects 0.000 claims 9
- 238000003752 polymerase chain reaction Methods 0.000 claims 9
- 102000000905 Cadherin Human genes 0.000 claims 7
- 108050007957 Cadherin Proteins 0.000 claims 7
- 102100034256 Mucin-1 Human genes 0.000 claims 7
- 101150084967 EPCAM gene Proteins 0.000 claims 6
- 244000052769 pathogen Species 0.000 claims 6
- 230000007170 pathology Effects 0.000 claims 4
- 208000009458 Carcinoma in Situ Diseases 0.000 claims 3
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 claims 3
- 201000005179 adrenal carcinoma Diseases 0.000 claims 3
- 201000011143 bone giant cell tumor Diseases 0.000 claims 3
- 206010012818 diffuse large B-cell lymphoma Diseases 0.000 claims 3
- 208000005017 glioblastoma Diseases 0.000 claims 3
- 201000004933 in situ carcinoma Diseases 0.000 claims 3
- 201000002529 islet cell tumor Diseases 0.000 claims 3
- 210000004153 islets of langerhan Anatomy 0.000 claims 3
- 239000003550 marker Substances 0.000 claims 3
- 230000002175 menstrual effect Effects 0.000 claims 3
- 201000008026 nephroblastoma Diseases 0.000 claims 3
- 208000022102 pancreatic neuroendocrine neoplasm Diseases 0.000 claims 3
- 208000030266 primary brain neoplasm Diseases 0.000 claims 3
- 201000006845 reticulosarcoma Diseases 0.000 claims 3
- 208000029922 reticulum cell sarcoma Diseases 0.000 claims 3
- 208000010485 smooth muscle tumor Diseases 0.000 claims 3
- 208000011580 syndromic disease Diseases 0.000 claims 3
- 238000002965 ELISA Methods 0.000 claims 2
- 208000002966 Giant Cell Tumor of Bone Diseases 0.000 claims 2
- FUSGACRLAFQQRL-UHFFFAOYSA-N N-Ethyl-N-nitrosourea Chemical compound CCN(N=O)C(N)=O FUSGACRLAFQQRL-UHFFFAOYSA-N 0.000 claims 2
- 206010033128 Ovarian cancer Diseases 0.000 claims 2
- 238000002705 metabolomic analysis Methods 0.000 claims 2
- 239000003471 mutagenic agent Substances 0.000 claims 2
- 231100000707 mutagenic chemical Toxicity 0.000 claims 2
- UMFJAHHVKNCGLG-UHFFFAOYSA-N n-Nitrosodimethylamine Chemical compound CN(C)N=O UMFJAHHVKNCGLG-UHFFFAOYSA-N 0.000 claims 2
- 230000001850 reproductive effect Effects 0.000 claims 2
- 238000007789 sealing Methods 0.000 claims 2
- 238000003325 tomography Methods 0.000 claims 2
- 230000002485 urinary effect Effects 0.000 claims 2
- FSRFQOAAESLDSG-UHFFFAOYSA-N 1-nitro-2-nitrosoguanidine Chemical compound [O-][N+](=O)NC(=N)NN=O FSRFQOAAESLDSG-UHFFFAOYSA-N 0.000 claims 1
- DZBIMLCMMYGJFY-UHFFFAOYSA-N 6-(methylideneamino)-1,3,5-triazine-2,4-diamine Chemical compound NC1=NC(N)=NC(N=C)=N1 DZBIMLCMMYGJFY-UHFFFAOYSA-N 0.000 claims 1
- 206010061692 Benign muscle neoplasm Diseases 0.000 claims 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 claims 1
- 201000004458 Myoma Diseases 0.000 claims 1
- 201000000053 blastoma Diseases 0.000 claims 1
- 239000002458 cell surface marker Substances 0.000 claims 1
- 230000008878 coupling Effects 0.000 claims 1
- 238000010168 coupling process Methods 0.000 claims 1
- 238000005859 coupling reaction Methods 0.000 claims 1
- 230000000779 depleting effect Effects 0.000 claims 1
- 201000008184 embryoma Diseases 0.000 claims 1
- 239000005350 fused silica glass Substances 0.000 claims 1
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 claims 1
- 239000003112 inhibitor Substances 0.000 claims 1
- 230000007625 mitochondrial abnormality Effects 0.000 claims 1
- 230000003505 mutagenic effect Effects 0.000 claims 1
- 235000015097 nutrients Nutrition 0.000 claims 1
- 230000004936 stimulating effect Effects 0.000 claims 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 claims 1
- 230000004888 barrier function Effects 0.000 description 98
- 239000000306 component Substances 0.000 description 48
- 239000013615 primer Substances 0.000 description 45
- 229920002477 rna polymer Polymers 0.000 description 45
- 210000003705 ribosome Anatomy 0.000 description 35
- 238000006243 chemical reaction Methods 0.000 description 32
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 32
- 210000003556 vascular endothelial cell Anatomy 0.000 description 31
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical class CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 26
- 108700024394 Exon Proteins 0.000 description 21
- 238000000926 separation method Methods 0.000 description 18
- 238000012360 testing method Methods 0.000 description 18
- 238000013461 design Methods 0.000 description 17
- 108020004635 Complementary DNA Proteins 0.000 description 16
- 235000001014 amino acid Nutrition 0.000 description 15
- 238000010804 cDNA synthesis Methods 0.000 description 15
- 230000004087 circulation Effects 0.000 description 15
- 238000011156 evaluation Methods 0.000 description 15
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 14
- 239000002299 complementary DNA Substances 0.000 description 14
- 230000002829 reductive effect Effects 0.000 description 14
- 229940124597 therapeutic agent Drugs 0.000 description 14
- 239000012141 concentrate Substances 0.000 description 13
- 229940113082 thymine Drugs 0.000 description 13
- 108020004705 Codon Proteins 0.000 description 12
- 230000004913 activation Effects 0.000 description 12
- 210000001124 body fluid Anatomy 0.000 description 12
- 230000001413 cellular effect Effects 0.000 description 12
- 230000008859 change Effects 0.000 description 12
- 239000000463 material Substances 0.000 description 11
- 235000018102 proteins Nutrition 0.000 description 11
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 10
- 229940024606 amino acid Drugs 0.000 description 10
- 239000012491 analyte Substances 0.000 description 10
- 239000010839 body fluid Substances 0.000 description 10
- 241001515965 unidentified phage Species 0.000 description 10
- 108091093088 Amplicon Proteins 0.000 description 9
- 150000001875 compounds Chemical class 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 238000004519 manufacturing process Methods 0.000 description 9
- 244000000010 microbial pathogen Species 0.000 description 9
- 230000004044 response Effects 0.000 description 9
- 230000001225 therapeutic effect Effects 0.000 description 9
- 108700028369 Alleles Proteins 0.000 description 8
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 8
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 8
- 108091034117 Oligonucleotide Proteins 0.000 description 8
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 8
- 230000006037 cell lysis Effects 0.000 description 8
- 238000003776 cleavage reaction Methods 0.000 description 8
- 239000000284 extract Substances 0.000 description 8
- 230000003834 intracellular effect Effects 0.000 description 8
- 235000005772 leucine Nutrition 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 238000011002 quantification Methods 0.000 description 8
- 229930024421 Adenine Natural products 0.000 description 7
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 7
- 239000004471 Glycine Substances 0.000 description 7
- 102100034343 Integrase Human genes 0.000 description 7
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 7
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 7
- 229960000643 adenine Drugs 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 230000000875 corresponding effect Effects 0.000 description 7
- 238000002651 drug therapy Methods 0.000 description 7
- 230000036541 health Effects 0.000 description 7
- 210000004072 lung Anatomy 0.000 description 7
- 230000005291 magnetic effect Effects 0.000 description 7
- 229920003023 plastic Polymers 0.000 description 7
- 239000004033 plastic Substances 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 230000007017 scission Effects 0.000 description 7
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 6
- 241000700605 Viruses Species 0.000 description 6
- 230000001580 bacterial effect Effects 0.000 description 6
- 210000000481 breast Anatomy 0.000 description 6
- 230000006378 damage Effects 0.000 description 6
- 230000002068 genetic effect Effects 0.000 description 6
- 210000000936 intestine Anatomy 0.000 description 6
- 239000002609 medium Substances 0.000 description 6
- 239000000203 mixture Substances 0.000 description 6
- 210000004940 nucleus Anatomy 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 238000005728 strengthening Methods 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 5
- 238000011161 development Methods 0.000 description 5
- 230000018109 developmental process Effects 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 210000005259 peripheral blood Anatomy 0.000 description 5
- 239000011886 peripheral blood Substances 0.000 description 5
- 230000003334 potential effect Effects 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 230000002787 reinforcement Effects 0.000 description 5
- 241000894007 species Species 0.000 description 5
- 238000012546 transfer Methods 0.000 description 5
- 210000004881 tumor cell Anatomy 0.000 description 5
- 239000002699 waste material Substances 0.000 description 5
- 239000004475 Arginine Substances 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- 229920000089 Cyclic olefin copolymer Polymers 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- 208000007569 Giant Cell Tumors Diseases 0.000 description 4
- 108091027305 Heteroduplex Proteins 0.000 description 4
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 4
- 206010027476 Metastases Diseases 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- 102000007298 Mucin-1 Human genes 0.000 description 4
- 206010029260 Neuroblastoma Diseases 0.000 description 4
- 108091093037 Peptide nucleic acid Proteins 0.000 description 4
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 208000024447 adrenal gland neoplasm Diseases 0.000 description 4
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 4
- 235000009697 arginine Nutrition 0.000 description 4
- 238000006073 displacement reaction Methods 0.000 description 4
- 238000001502 gel electrophoresis Methods 0.000 description 4
- 210000003714 granulocyte Anatomy 0.000 description 4
- 210000002768 hair cell Anatomy 0.000 description 4
- 206010022498 insulinoma Diseases 0.000 description 4
- 210000004698 lymphocyte Anatomy 0.000 description 4
- 230000009401 metastasis Effects 0.000 description 4
- 238000007857 nested PCR Methods 0.000 description 4
- 210000003463 organelle Anatomy 0.000 description 4
- 208000021255 pancreatic insulinoma Diseases 0.000 description 4
- 238000003753 real-time PCR Methods 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 201000009030 Carcinoma Diseases 0.000 description 3
- 238000001712 DNA sequencing Methods 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 3
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 3
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 3
- 108010083644 Ribonucleases Proteins 0.000 description 3
- 102000006382 Ribonucleases Human genes 0.000 description 3
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 3
- 238000002835 absorbance Methods 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 230000001464 adherent effect Effects 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- 230000002457 bidirectional effect Effects 0.000 description 3
- 238000005251 capillar electrophoresis Methods 0.000 description 3
- 238000005266 casting Methods 0.000 description 3
- 238000004891 communication Methods 0.000 description 3
- 238000000748 compression moulding Methods 0.000 description 3
- 238000011109 contamination Methods 0.000 description 3
- 239000013068 control sample Substances 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000003935 denaturing gradient gel electrophoresis Methods 0.000 description 3
- 239000000975 dye Substances 0.000 description 3
- 229940121647 egfr inhibitor Drugs 0.000 description 3
- 238000004049 embossing Methods 0.000 description 3
- 238000001125 extrusion Methods 0.000 description 3
- 238000005194 fractionation Methods 0.000 description 3
- 102000054766 genetic haplotypes Human genes 0.000 description 3
- 229940084651 iressa Drugs 0.000 description 3
- 206010023332 keratitis Diseases 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- 230000033001 locomotion Effects 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 210000003470 mitochondria Anatomy 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 238000005191 phase separation Methods 0.000 description 3
- 238000000206 photolithography Methods 0.000 description 3
- 210000002381 plasma Anatomy 0.000 description 3
- 238000004393 prognosis Methods 0.000 description 3
- 210000002307 prostate Anatomy 0.000 description 3
- 230000029058 respiratory gaseous exchange Effects 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 230000035945 sensitivity Effects 0.000 description 3
- 238000004513 sizing Methods 0.000 description 3
- 210000002460 smooth muscle Anatomy 0.000 description 3
- 238000010183 spectrum analysis Methods 0.000 description 3
- 230000006641 stabilisation Effects 0.000 description 3
- 238000011105 stabilization Methods 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 230000009261 transgenic effect Effects 0.000 description 3
- 235000011178 triphosphate Nutrition 0.000 description 3
- 239000001226 triphosphate Substances 0.000 description 3
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical compound OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 description 3
- 210000001635 urinary tract Anatomy 0.000 description 3
- 239000004474 valine Substances 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 102000053642 Catalytic RNA Human genes 0.000 description 2
- 108090000994 Catalytic RNA Proteins 0.000 description 2
- 239000004713 Cyclic olefin copolymer Substances 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- 108020001019 DNA Primers Proteins 0.000 description 2
- 238000000018 DNA microarray Methods 0.000 description 2
- 239000003155 DNA primer Substances 0.000 description 2
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 2
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 2
- 206010061819 Disease recurrence Diseases 0.000 description 2
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 101710163270 Nuclease Proteins 0.000 description 2
- 229930040373 Paraformaldehyde Natural products 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 208000008601 Polycythemia Diseases 0.000 description 2
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 2
- 108020001027 Ribosomal DNA Proteins 0.000 description 2
- 238000012300 Sequence Analysis Methods 0.000 description 2
- 108010006785 Taq Polymerase Proteins 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 230000005856 abnormality Effects 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 238000001261 affinity purification Methods 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 230000004075 alteration Effects 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 239000000427 antigen Substances 0.000 description 2
- 108091007433 antigens Proteins 0.000 description 2
- 102000036639 antigens Human genes 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 2
- 229960002685 biotin Drugs 0.000 description 2
- 239000011616 biotin Substances 0.000 description 2
- 239000012503 blood component Substances 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- 235000014633 carbohydrates Nutrition 0.000 description 2
- 150000001720 carbohydrates Chemical class 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 230000020411 cell activation Effects 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 210000003850 cellular structure Anatomy 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- 238000011961 computed axial tomography Methods 0.000 description 2
- 230000008602 contraction Effects 0.000 description 2
- 230000002596 correlated effect Effects 0.000 description 2
- 230000001351 cycling effect Effects 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical class NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 230000003511 endothelial effect Effects 0.000 description 2
- 230000007613 environmental effect Effects 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 238000000799 fluorescence microscopy Methods 0.000 description 2
- 210000000497 foam cell Anatomy 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 229930195712 glutamate Natural products 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 239000010931 gold Substances 0.000 description 2
- 229910052737 gold Inorganic materials 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 230000003394 haemopoietic effect Effects 0.000 description 2
- 230000005661 hydrophobic surface Effects 0.000 description 2
- 210000002865 immune cell Anatomy 0.000 description 2
- 230000002055 immunohistochemical effect Effects 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 238000001746 injection moulding Methods 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- PHTQWCKDNZKARW-UHFFFAOYSA-N isoamylol Chemical compound CC(C)CCO PHTQWCKDNZKARW-UHFFFAOYSA-N 0.000 description 2
- 210000003292 kidney cell Anatomy 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 238000004020 luminiscence type Methods 0.000 description 2
- 230000002934 lysing effect Effects 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- 238000005459 micromachining Methods 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- 230000009871 nonspecific binding Effects 0.000 description 2
- 210000001672 ovary Anatomy 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- 229920001084 poly(chloroprene) Polymers 0.000 description 2
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 2
- 229920002401 polyacrylamide Polymers 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 239000004926 polymethyl methacrylate Substances 0.000 description 2
- 229920002689 polyvinyl acetate Polymers 0.000 description 2
- 239000011118 polyvinyl acetate Substances 0.000 description 2
- 239000004800 polyvinyl chloride Substances 0.000 description 2
- 229920000915 polyvinyl chloride Polymers 0.000 description 2
- 239000011148 porous material Substances 0.000 description 2
- 238000002600 positron emission tomography Methods 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000008707 rearrangement Effects 0.000 description 2
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 2
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 2
- 230000003014 reinforcing effect Effects 0.000 description 2
- 210000005000 reproductive tract Anatomy 0.000 description 2
- 108091008146 restriction endonucleases Proteins 0.000 description 2
- 108091092562 ribozyme Proteins 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 208000011581 secondary neoplasm Diseases 0.000 description 2
- 238000007873 sieving Methods 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 230000000392 somatic effect Effects 0.000 description 2
- 238000010186 staining Methods 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 230000005748 tumor development Effects 0.000 description 2
- 230000005740 tumor formation Effects 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- NERXPXBELDBEPZ-RMKNXTFCSA-N (e)-n-[4-[3-chloro-4-[(3-fluorophenyl)methoxy]anilino]-3-cyano-7-ethoxyquinolin-6-yl]-4-(dimethylamino)but-2-enamide Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC(C=C1Cl)=CC=C1OCC1=CC=CC(F)=C1 NERXPXBELDBEPZ-RMKNXTFCSA-N 0.000 description 1
- KKVYYGGCHJGEFJ-UHFFFAOYSA-N 1-n-(4-chlorophenyl)-6-methyl-5-n-[3-(7h-purin-6-yl)pyridin-2-yl]isoquinoline-1,5-diamine Chemical compound N=1C=CC2=C(NC=3C(=CC=CN=3)C=3C=4N=CNC=4N=CN=3)C(C)=CC=C2C=1NC1=CC=C(Cl)C=C1 KKVYYGGCHJGEFJ-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- AIQFOGOHLOICRK-UHFFFAOYSA-N 6-amino-4-(3-chloro-4-fluoroanilino)-7-ethoxyquinoline-3-carbonitrile Chemical compound C=12C=C(N)C(OCC)=CC2=NC=C(C#N)C=1NC1=CC=C(F)C(Cl)=C1 AIQFOGOHLOICRK-UHFFFAOYSA-N 0.000 description 1
- 102100024439 Adhesion G protein-coupled receptor A2 Human genes 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 102100031323 Anthrax toxin receptor 1 Human genes 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-N Betaine Natural products C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- 229920000742 Cotton Polymers 0.000 description 1
- 108020001738 DNA Glycosylase Proteins 0.000 description 1
- 102000012410 DNA Ligases Human genes 0.000 description 1
- 108010061982 DNA Ligases Proteins 0.000 description 1
- 102000028381 DNA glycosylase Human genes 0.000 description 1
- 230000009946 DNA mutation Effects 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 108010067770 Endopeptidase K Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 206010017533 Fungal infection Diseases 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000833358 Homo sapiens Adhesion G protein-coupled receptor A2 Proteins 0.000 description 1
- 101000796095 Homo sapiens Anthrax toxin receptor 1 Proteins 0.000 description 1
- 101000851181 Homo sapiens Epidermal growth factor receptor Proteins 0.000 description 1
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- 206010027202 Meningitis bacterial Diseases 0.000 description 1
- 206010027260 Meningitis viral Diseases 0.000 description 1
- 102000015728 Mucins Human genes 0.000 description 1
- 108010063954 Mucins Proteins 0.000 description 1
- 102000007474 Multiprotein Complexes Human genes 0.000 description 1
- 108010085220 Multiprotein Complexes Proteins 0.000 description 1
- 101100381978 Mus musculus Braf gene Proteins 0.000 description 1
- 101100118551 Mus musculus Egfr gene Proteins 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-O N,N,N-trimethylglycinium Chemical compound C[N+](C)(C)CC(O)=O KWIUHFFTVRNATP-UHFFFAOYSA-O 0.000 description 1
- 108091005461 Nucleic proteins Proteins 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 108020002230 Pancreatic Ribonuclease Proteins 0.000 description 1
- 102000005891 Pancreatic ribonuclease Human genes 0.000 description 1
- 206010033799 Paralysis Diseases 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 229920001328 Polyvinylidene chloride Polymers 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 102000009516 Protein Serine-Threonine Kinases Human genes 0.000 description 1
- 108010009341 Protein Serine-Threonine Kinases Proteins 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- 241001522306 Serinus serinus Species 0.000 description 1
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 206010066901 Treatment failure Diseases 0.000 description 1
- 239000007984 Tris EDTA buffer Substances 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 230000004520 agglutination Effects 0.000 description 1
- 208000036878 aneuploidy Diseases 0.000 description 1
- 231100001075 aneuploidy Toxicity 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 1
- 201000009904 bacterial meningitis Diseases 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 239000000090 biomarker Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000013626 chemical specie Substances 0.000 description 1
- 210000003763 chloroplast Anatomy 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 230000007797 corrosion Effects 0.000 description 1
- 238000005260 corrosion Methods 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 230000002380 cytological effect Effects 0.000 description 1
- 230000000120 cytopathologic effect Effects 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 231100000517 death Toxicity 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 239000003398 denaturant Substances 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000001066 destructive effect Effects 0.000 description 1
- 239000010432 diamond Substances 0.000 description 1
- 229910003460 diamond Inorganic materials 0.000 description 1
- 239000005546 dideoxynucleotide Substances 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 238000001312 dry etching Methods 0.000 description 1
- 238000013399 early diagnosis Methods 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- 230000005684 electric field Effects 0.000 description 1
- 238000005370 electroosmosis Methods 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 238000012869 ethanol precipitation Methods 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000004744 fabric Substances 0.000 description 1
- 238000009501 film coating Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229920002313 fluoropolymer Polymers 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 208000024386 fungal infectious disease Diseases 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 102000054767 gene variant Human genes 0.000 description 1
- 210000004392 genitalia Anatomy 0.000 description 1
- 230000001434 glomerular Effects 0.000 description 1
- 239000003292 glue Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- ZJYYHGLJYGJLLN-UHFFFAOYSA-N guanidinium thiocyanate Chemical compound SC#N.NC(N)=N ZJYYHGLJYGJLLN-UHFFFAOYSA-N 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000008241 heterogeneous mixture Substances 0.000 description 1
- 229960002885 histidine Drugs 0.000 description 1
- 230000001744 histochemical effect Effects 0.000 description 1
- 238000000265 homogenisation Methods 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 238000007849 hot-start PCR Methods 0.000 description 1
- 102000045108 human EGFR Human genes 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 239000000815 hypotonic solution Substances 0.000 description 1
- 239000003547 immunosorbent Substances 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000004941 influx Effects 0.000 description 1
- 239000003978 infusion fluid Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 238000005304 joining Methods 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 201000005296 lung carcinoma Diseases 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 239000000696 magnetic material Substances 0.000 description 1
- 239000006247 magnetic powder Substances 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 210000001161 mammalian embryo Anatomy 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 230000008774 maternal effect Effects 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229940056960 melamin Drugs 0.000 description 1
- JDSHMPZPIAZGSV-UHFFFAOYSA-N melamine Chemical compound NC1=NC(N)=NC(N)=N1 JDSHMPZPIAZGSV-UHFFFAOYSA-N 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000037353 metabolic pathway Effects 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 238000003801 milling Methods 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000002715 modification method Methods 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 101150029137 mutY gene Proteins 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- 208000018305 neoplasm of myocardium Diseases 0.000 description 1
- 229950008835 neratinib Drugs 0.000 description 1
- JWNPDZNEKVCWMY-VQHVLOKHSA-N neratinib Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 JWNPDZNEKVCWMY-VQHVLOKHSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 210000000633 nuclear envelope Anatomy 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000001282 organosilanes Chemical class 0.000 description 1
- 230000001151 other effect Effects 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 239000011860 particles by size Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- WVUNYSQLFKLYNI-AATRIKPKSA-N pelitinib Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC1=CC=C(F)C(Cl)=C1 WVUNYSQLFKLYNI-AATRIKPKSA-N 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 230000003836 peripheral circulation Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 238000001020 plasma etching Methods 0.000 description 1
- 238000007747 plating Methods 0.000 description 1
- 210000004910 pleural fluid Anatomy 0.000 description 1
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920002338 polyhydroxyethylmethacrylate Polymers 0.000 description 1
- 229920001195 polyisoprene Polymers 0.000 description 1
- 102000054765 polymorphisms of proteins Human genes 0.000 description 1
- 229920000098 polyolefin Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229940068984 polyvinyl alcohol Drugs 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 230000035935 pregnancy Effects 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 239000002987 primer (paints) Substances 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 238000012827 research and development Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 238000005096 rolling process Methods 0.000 description 1
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 229910000077 silane Inorganic materials 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 210000001082 somatic cell Anatomy 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 230000007480 spreading Effects 0.000 description 1
- 238000003892 spreading Methods 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 229940120982 tarceva Drugs 0.000 description 1
- 238000000123 temperature gradient gel electrophoresis Methods 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- BFKJFAAPBSQJPD-UHFFFAOYSA-N tetrafluoroethene Chemical group FC(F)=C(F)F BFKJFAAPBSQJPD-UHFFFAOYSA-N 0.000 description 1
- 238000005382 thermal cycling Methods 0.000 description 1
- 238000003856 thermoforming Methods 0.000 description 1
- 229920001169 thermoplastic Polymers 0.000 description 1
- 239000012815 thermoplastic material Substances 0.000 description 1
- 239000004416 thermosoftening plastic Substances 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 210000000115 thoracic cavity Anatomy 0.000 description 1
- 125000000341 threoninyl group Chemical group [H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 description 1
- 229950001353 tretamine Drugs 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 201000010044 viral meningitis Diseases 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 239000002351 wastewater Substances 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
Images











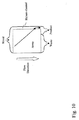

















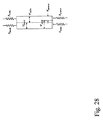





















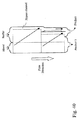










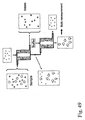




















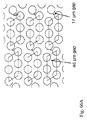



























Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/543—Immunoassay; Biospecific binding assay; Materials therefor with an insoluble carrier for immobilising immunochemicals
- G01N33/54366—Apparatus specially adapted for solid-phase testing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01L—CHEMICAL OR PHYSICAL LABORATORY APPARATUS FOR GENERAL USE
- B01L3/00—Containers or dishes for laboratory use, e.g. laboratory glassware; Droppers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01L—CHEMICAL OR PHYSICAL LABORATORY APPARATUS FOR GENERAL USE
- B01L3/00—Containers or dishes for laboratory use, e.g. laboratory glassware; Droppers
- B01L3/50—Containers for the purpose of retaining a material to be analysed, e.g. test tubes
- B01L3/502—Containers for the purpose of retaining a material to be analysed, e.g. test tubes with fluid transport, e.g. in multi-compartment structures
- B01L3/5027—Containers for the purpose of retaining a material to be analysed, e.g. test tubes with fluid transport, e.g. in multi-compartment structures by integrated microfluidic structures, i.e. dimensions of channels and chambers are such that surface tension forces are important, e.g. lab-on-a-chip
- B01L3/502753—Containers for the purpose of retaining a material to be analysed, e.g. test tubes with fluid transport, e.g. in multi-compartment structures by integrated microfluidic structures, i.e. dimensions of channels and chambers are such that surface tension forces are important, e.g. lab-on-a-chip characterised by bulk separation arrangements on lab-on-a-chip devices, e.g. for filtration or centrifugation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N15/00—Investigating characteristics of particles; Investigating permeability, pore-volume or surface-area of porous materials
- G01N15/10—Investigating individual particles
- G01N15/14—Optical investigation techniques, e.g. flow cytometry
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/483—Physical analysis of biological material
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/574—Immunoassay; Biospecific binding assay; Materials therefor for cancer
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01L—CHEMICAL OR PHYSICAL LABORATORY APPARATUS FOR GENERAL USE
- B01L2200/00—Solutions for specific problems relating to chemical or physical laboratory apparatus
- B01L2200/06—Fluid handling related problems
- B01L2200/0647—Handling flowable solids, e.g. microscopic beads, cells, particles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01L—CHEMICAL OR PHYSICAL LABORATORY APPARATUS FOR GENERAL USE
- B01L2300/00—Additional constructional details
- B01L2300/08—Geometry, shape and general structure
- B01L2300/0809—Geometry, shape and general structure rectangular shaped
- B01L2300/0816—Cards, e.g. flat sample carriers usually with flow in two horizontal directions
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01L—CHEMICAL OR PHYSICAL LABORATORY APPARATUS FOR GENERAL USE
- B01L2300/00—Additional constructional details
- B01L2300/08—Geometry, shape and general structure
- B01L2300/0861—Configuration of multiple channels and/or chambers in a single devices
- B01L2300/0864—Configuration of multiple channels and/or chambers in a single devices comprising only one inlet and multiple receiving wells, e.g. for separation, splitting
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01L—CHEMICAL OR PHYSICAL LABORATORY APPARATUS FOR GENERAL USE
- B01L2400/00—Moving or stopping fluids
- B01L2400/04—Moving fluids with specific forces or mechanical means
- B01L2400/0403—Moving fluids with specific forces or mechanical means specific forces
- B01L2400/0409—Moving fluids with specific forces or mechanical means specific forces centrifugal forces
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01L—CHEMICAL OR PHYSICAL LABORATORY APPARATUS FOR GENERAL USE
- B01L2400/00—Moving or stopping fluids
- B01L2400/04—Moving fluids with specific forces or mechanical means
- B01L2400/0475—Moving fluids with specific forces or mechanical means specific mechanical means and fluid pressure
- B01L2400/0487—Moving fluids with specific forces or mechanical means specific mechanical means and fluid pressure fluid pressure, pneumatics
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01L—CHEMICAL OR PHYSICAL LABORATORY APPARATUS FOR GENERAL USE
- B01L2400/00—Moving or stopping fluids
- B01L2400/08—Regulating or influencing the flow resistance
- B01L2400/084—Passive control of flow resistance
- B01L2400/086—Passive control of flow resistance using baffles or other fixed flow obstructions
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y10/00—Nanotechnology for information processing, storage or transmission, e.g. quantum computing or single electron logic
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y5/00—Nanobiotechnology or nanomedicine, e.g. protein engineering or drug delivery
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N15/00—Investigating characteristics of particles; Investigating permeability, pore-volume or surface-area of porous materials
- G01N15/10—Investigating individual particles
- G01N2015/1006—Investigating individual particles for cytology
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N15/00—Investigating characteristics of particles; Investigating permeability, pore-volume or surface-area of porous materials
- G01N15/10—Investigating individual particles
- G01N2015/1029—Particle size
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N21/00—Investigating or analysing materials by the use of optical means, i.e. using sub-millimetre waves, infrared, visible or ultraviolet light
- G01N21/01—Arrangements or apparatus for facilitating the optical investigation
- G01N21/03—Cuvette constructions
- G01N2021/0346—Capillary cells; Microcells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/52—Predicting or monitoring the response to treatment, e.g. for selection of therapy based on assay results in personalised medicine; Prognosis
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Analytical Chemistry (AREA)
- Urology & Nephrology (AREA)
- General Health & Medical Sciences (AREA)
- Pathology (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- General Physics & Mathematics (AREA)
- Microbiology (AREA)
- Food Science & Technology (AREA)
- Biotechnology (AREA)
- Medicinal Chemistry (AREA)
- Cell Biology (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Dispersion Chemistry (AREA)
- Hospice & Palliative Care (AREA)
- Oncology (AREA)
- Clinical Laboratory Science (AREA)
- Biophysics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Tropical Medicine & Parasitology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Apparatus Associated With Microorganisms And Enzymes (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
Description
癌は、異常な細胞の抑制されていない激増によって記録される疾患である。正常組織において、細胞は、分かれて、細胞を囲むことから、信号に応答して組織の範囲内で有機化する。癌細胞は同様にこれらの信号に反応しない。そして、それらに増殖して、多くの器官において、腫瘍を形成させる。腫瘍の成長が続けるように、遺伝子の変更は蓄積することができる。そして、癌細胞のより積極的な発展フェノタイプとして現れる。
無処置のままにされる場合、転移(リンパ系または血流としての本体の遠い領域に対する癌細胞の拡散)は起こることができる。転移は多重サイトで結果として二次腫瘍の形成になる。そして、健康な組織に損傷を与える。大部分の癌死は、この種の二次腫瘍によって生じる。
本発明は、以下によって細胞サンプルの癌細胞を検出する方法を特徴とする:
a)細胞サンプルを第一の方向に癌細胞に癌細胞において濃縮される第一出力端サンプルおよび第2の細胞において豊かにされる第2の出力サンプルを生じる第2の指示の一つ以上の第2の細胞を生産するように指示する構造を有するチャネルを含んでいる装置にもたらすこと;
そして、
b)第一出力端サンプルの癌細胞の有無を検出すること。
少なくとも一つの原種血管内皮細胞は、第一出力端サンプルにおいてある。
a)磁粉を使用することのない、または、抗体または断片単独を使用することのない細胞サンプルから癌細胞の一つ以上を豊かにすること、
そこにおいて、
豊かにすることは、細胞サイズ、形状または変形能に基づく;
そして、
b)豊かな癌細胞の数を決定すること。
i)細胞サンプルから一つ以上の血管内皮細胞を豊かにすること;
そして、
ii)例えば、血管内皮細胞に癌細胞の比率を決定するために豊かな血管内皮細胞の数を決定すること。
例えば、ステップa)およびb)は第2の細胞サンプルによって繰り返されることができる。そして、第1と同じ主題からとられる。
a)第一の方向に一つ以上の癌細胞に癌細胞において濃縮される第一出力端サンプルおよび第2の細胞において豊かにされる第2の出力サンプルを生じる第2の指示の一つ以上の第2の細胞を生産するように指示する構造を有するチャネルを含んでいる装置に主題から細胞サンプルを導くこと;
b)第一出力端サンプルの癌細胞の有無を検出すること;
そして、
c)ステップb)の結果に基づいて状態の有無を診断すること。
i)細胞サンプルの血管内皮細胞(例えば原種血管内皮細胞)の数を決定すること;
そして、
ii)血管内皮細胞に癌細胞の比率を決定すること、
そこにおいて、診断は、比率に更に基づく。
a) 第一の方向に一つ以上の第1の細胞に第1の細胞において濃縮される第一出力端サンプルおよび第2の細胞において豊かにされる第2の出力サンプルを生じる第2の指示の一つ以上の第2の細胞を生産するように指示する構造を有するチャネルを含んでいる装置に主題から細胞サンプルを導くこと;
b) 第一出力端サンプルを分析すること;
そして、
c) ステップb)の結果に基づいて状態の有無を診断すること。
いくつかの実施形態では、癌は、甲状腺癌でない。
構造は、以下を含む:すきまおよび障壁のネットワークを形成するずらりと並んだ障害物は、選択的に前記第1の細胞を捕獲することができる;そして、
障壁間のギャップは、15ミクロン以上、20ミクロン以上または60ミクロン未満である。
ステップb)は、第一出力端サンプルの粒度分布を分析することが必要である;そして、
ステップb)は、第1の細胞の数を決定することから成る。
a)複数の対照被検者および複数のケースの各々から細胞サンプルを得ることは、興味がある状態があることを従属させる;
b)各細胞サンプルから、サイズによって12ミクロンより大きい水力学的サイズを有する細胞を濃縮すること;
c)ステップb)において豊かにされる分析電池;
そして、
d)ステップc)において得られた結果を使用している関連研究を実行すること。
本発明も、主題の状態を診断する方法を特徴とする;
方法は、次のステップを含む:
a)状態と関連した細胞パターンを提供すること;
b)主題から細胞サンプルを得ること;
c)細胞サンプルから、サイズによって12ミクロンより大きい水力学的サイズを有する細胞を濃縮すること;
d)ステップc)において豊かにされる分析電池;
そして、
e)ステップd)の分析と共にステップa)の細胞パターンに基づいて主題の状態の有無を診断すること。
a)処理の前に主題から第1の細胞サンプルを得ること;
b)第1の細胞サンプルを第一の方向に一つ以上の第1の細胞に第1の細胞において濃縮される第一出力端サンプルおよび第2の細胞において豊かにされる第2の出力サンプルを生じる第2の指示の一つ以上の第2の細胞を生産するように指示する構造を有するチャネルを含んでいる装置にもたらすこと;
c)第一出力端サンプルを分析すること;
d)主題(と)同時にから第2の細胞サンプルを得ているか、薬物療法に続く;
e)第2の細胞サンプルのための繰り返す段階b)およびc);そして、
f)第1の細胞サンプルおよび第2の細胞サンプルのためのステップc)の結果を比較すること、そこにおいて、比較は、薬物療法の有効性を決定する。
リボゾームリボ核酸は、メッセンジャーRNAまたはマイクロ・リボゾームリボ核酸でありえる。
状態は、例えば、炎症性の状態、虚血性の状態、感染症、外傷、子宮内膜症または腫瘍性の状態(例えば肺、胸部または前立腺癌)である。体液は、例えば、血液、汗、裂け目、リンパ、耳フロー、痰、骨髄中止、尿、唾液、精液、膣の流れ、脳脊髄液、脳流体、腹水、牛乳、呼吸、腸の、および、尿生殖器の路の分泌物、羊水または本体組織エキスである。評価は、数および/または細胞のタイプを確認するかまたは細胞のDNAの総量を決定することを含むことができる。あるいは、評価は、血管内皮細胞の数を決定することからなるグループから選択される、血管内皮細胞に癌細胞の比率を決定すること、DNAまたはリボゾームリボ核酸の突然変異を検出して、SNPを分析して、細胞の形態を決定して、細胞(例えば、細胞を標識に特有である特定の結着剤と作用することによって)の標識を検出して、細胞の活性を検出すること。評価は、細胞のパターンを確認することを含むこともできる。方法は、複数の強化および評価ステップを更に含むことができる、そこにおいて、評価ステップの結果は、治療の有効性の徴候として比較される。例えば、毎日、毎週、隔週に、隔月で、毎月、年四回、二年に一度、または、毎年、複数の強化および評価ステップは、時間によって典型的に切り離される。方法は、少なくとも1の間、患者に対する治療に時間を提供することを更に含むことができる。強化ステップは、装置(例えば障壁の配列を含むミクロな液体の装置)で、好ましくは実行される。
方法は、ふるい分けにおいて確認される最も有効な治療薬患者を治療することを更に含むことができる。評価は、細胞の細胞またはパターンの数を確認することを含むことができる。望ましく、細胞のいくつかは、保存される。保存された細胞のいくつかは、それから治療薬によって接触することができて、治療薬の有効性の徴候として評価されることができる。状態は、例えば、炎症性の状態、虚血性の状態、感染症、外傷、子宮内膜症または腫瘍性の状態(例えば肺、胸部または前立腺癌)である。体液は、例えば、血液、汗、裂け目、リンパ、耳フロー、痰、骨髄中止、尿、唾液、精液、膣の流れ、脳脊髄液、脳流体、腹水、牛乳、呼吸、腸の、および、尿生殖器の路の分泌物、羊水または本体組織エキスである。
装置は、以下を含む:
a)第一の方向に一つ以上の第1の細胞に第1の細胞において濃縮される第一出力端サンプルおよび第2の細胞において豊かにされる第2の出力サンプルを生じる第2の指示の一つ以上の第2の細胞を生産するように指示する構造を含んでいるチャネル、
そこにおいて、
装置も、構成される:
i)第1の方向の12ミクロンより大きい水力学的サイズを有する細胞および第2の方向の12ミクロン以下の水力学的サイズを有する細胞を導くこと;または
ii)第1の方向の6ミクロンおよび12ミクロン以下以上の水力学的サイズを有する直接循環セルに対する、および6ミクロン未満の水力学的サイズまたは第2の方向の12ミクロンより大きい水力学的サイズを有する細胞を有する細胞;そして、
b)第一出力端サンプルまたは第2の出力サンプルを分析するための検出モジュール、そこにおいて、検出モジュールは、チャネルに流体工学的に連結する。
a)第一の方向に一つ以上の癌細胞に癌細胞において濃縮される第一出力端サンプルおよび第2の細胞において豊かにされる第2の出力サンプルを生じる第2の指示の一つ以上の第2の細胞を生産するように指示する構造を含んでいるチャネル;そして、
b)癌細胞または第2の細胞を捕獲するための捕獲モジュール、そこにおいて、捕獲モジュールはチャネルに流体工学的に連結する、そして、捕獲モジュールは選択的に癌細胞または第2の細胞を結合する一つ以上の結合部分を含む。
装置は、以下を含む:第一の方向に一つ以上の第1の細胞に第1の細胞において濃縮される第一出力端サンプルおよび第2の細胞において豊かにされる第2の出力サンプルを生じる第2の指示の一つ以上の第2の細胞を生産するように指示する構造を有するチャネル、そこにおいて、構造は、以下を含む:すきまのネットワークを形成する、そして、少なくとも障壁のいくつかが非常に選択的にモノクローン抗―EpCAM抗体またはその断片を含むずらりと並んだ障壁は、第1の細胞または第2の細胞を結合する。
装置は、以下を含む:
a)サイズに基づいて細胞サンプルの細胞を濃縮することができる強化モジュール;そして、
b)強化モジュールによって濃縮される細胞の数を決定するための細胞計数モジュール、そこにおいて、細胞計数モジュールは、強化モジュールに流体工学的に連結する。
第一の方向に一つ以上の第1の細胞に第1の細胞において濃縮される第一出力端サンプルおよび第2の細胞において豊かにされる第2の出力サンプルを生じる第2の指示の一つ以上の第2の細胞を生産するように指示する構造を含んでいるチャネル、
そこにおいて、
装置は、少なくとも20mLおよび時間当たりの好ましくは少なくとも50mLの流体を処理することができる。
第一出口、そして、第二出口、そこにおいて、細胞サンプルは第1の入口に適用される、第一出力端サンプルは第一出口から流出する、そして、第2の出力サンプルは第二出口から流出する。
検出器モジュールは、以下を含むことができる:システムおよびそれを撮像している顕微鏡、細胞カウンタ、磁石、バイオ空腔レーザー、マススペクトロメータ、PCR装置、RT―PCR装置、マトリックス、マイクロアレイまたはハイパー分光は、選択的に第1の細胞を結合するラベルを検出することができる。
方法は、次のステップを含む:
a)細胞サンプルを処理する装置に主題から細胞サンプルを導くこと。そして、それは、以下を含む:
第一の方向に一つ以上の第1の細胞に第1の細胞において濃縮される第一出力端サンプルおよび第2の細胞において豊かにされる第2の出力サンプルを生じる第2の指示の一つ以上の第2の細胞を生産するように指示する構造を有するチャネル、
そこにおいて、
装置は、時間当たりの少なくとも20mLの流体を処理することができる;
b)第一出力端サンプルを分析すること;
そして、
c)ステップb)の結果に基づいて状態の有無を診断すること。
ステップb)は、粘着力、遊走、締め具、形態学、分割、遺伝子発現レベルまたは体細胞突然変異の存在の特徴の一つ以上のための第一出力端サンプルの細胞を分析することが必要でありえる。
細胞は、あるサイトケラチン・メッセンジャーRNAのレベルを決定することによって分析されることもできる。
a.細胞サンプルを提供すること;
b.上皮細胞において豊かにされるサンプルを生じるために細胞サンプルを装置にもたらすこと;そして、
c.上皮細胞を定量化する。
a.通常の血液細胞より大きな細胞において豊かにされるサンプルを生じるために装置に患者から細胞サンプルを導くこと;そして、疾病状態を決定するサンプルに存在する通常の血液細胞より大きな細胞の数を決定しているb。
a.通常の血液細胞より大きな細胞において豊かにされるサンプルを生じるために装置に患者から細胞サンプルを導くこと;そして、疾病状態を決定するために豊かな検体に対してDNA鑑定、リボゾームリボ核酸分析、プロテオーム分析またはmetabolome分析を行っているb。
この方法は、次のステップを含むことができる:
i.豊かなサンプルから単離ゲノムDNA;そして、
ii。表皮成長因子受容体遺伝子の一つ以上のエキソン18、19、20、および21を増幅すること;この方法は、次のステップを更に含むことができる:
iii.周知の上皮細胞成長因子レセプタ突然変異に対応する増幅製品の亜区を増幅すること;(結果として更なる増幅製品になる)
iv.更なる増幅製品を配列決定すること;そして、
v.結果として生じるシーケンスを表皮成長因子受容体遺伝子および表皮成長因子受容体遺伝子の周知の突然変異のリストの野生型シーケンスと比較すること。
a.すきまのネットワークを形成している障壁の配列を有するチャネル、そこにおいて、
臨界サイズより大きな細胞の平均移動方向が流体流れの平均方向と平行でないように流体が等しくなく大きな流動および軽微な流動に分けられるように、流体はすきまの中を流れる;
b.放出口は、通常の血液細胞より大きな細胞を集めるために配置した;そして、
c。細胞を計数する細胞探知器。
天井(それは、透明でありえる)を含むチャネルは、上に、すきまのネットワークを形成する障壁の配列を配置した;装置は、臨界サイズの以下の直径を有する細胞がすきまのネットワークの中を流れて、天井および障壁との間に前記臨界サイズを超える直径を有する細胞を捕獲することができるように構成される。
モル浸透圧濃度を調整している試薬は、以下を含むことができる:
リリースされるための天井および障害との間に臨界サイズを超える直径を有する細胞が生じる高張液。
癌再発生の可能性の徴候として所定サイズより大きい細胞を評価すること。身体組織サンプルは、例えば、血液、骨髄中止、体液または本体組織エキスである。方法は複数の強化および評価ステップを含むことができる。そして、それは時間によって切り離されることができる。患者は、強化および評価ステップのうちの少なくとも1つの後の癌のための既治療でもよい。さらに、複数の評価ステップの上の細胞の変化は、癌再発生の可能性の徴候である。評価することは、細胞の細胞または活性の数を決定することを含むことができる。保健専門家にとって、方法は、例えば、評価する結果について、providing.a報告を含むこともできる。強化ステップは、例えば、障壁の配列から成る装置で実行される。
化学2005、48、1107−1131;そして、Tejparなど、J.Clin。
Oncol.ASCO年会会報vol 22、No.14S:3579 (2004)).本発明の一実施例では、EGFRは、癌を呈するための危険をもつ、または、の患者から、生体試料から得られる。EGFR(またはerbBl遺伝子)のキナーゼドメインの異型は、ATP結合ポケットの配座の構造に影響を及ぼす。好ましくは、EGFRのキナーゼドメインの異型は、1インチ・フレーム削除またはエキソン18、19、20、または21の置換である。
他の実施形態では、有無または少なくとも一つの異型の検出は、少なくとも一つの核酸プローブを有する異型サイトを含んでいるEGFR核酸を接触させることを提供する。プローブは、選択的なハイブリダイゼーション状況の下で異型サイトで異型サイトおよび含んでいる補完的なヌクレオチド塩基を含む核酸一次構造によって、優先して交雑する。ハイブリダイゼーションは、検出可能なラベルによって検出されることができる。
実施例において、異型は、ATP結合ポケットの構造変化を与えるerbBlのキナーゼドメインの突然変異である。
他の実施形態では、細胞は、トランスジェニック動物から得られる。実施例において、トランスジェニック動物は、マウスである。このマウスモデルにおいて、調査される細胞は、腫瘍生検から得られる。本発明によって選択されるerbBlキナーゼドメインの第2の突然変異を含んでいる細胞が、キナーゼドメインの第2の突然変異を有する異型EGFRのキナーゼ活性を阻害する化合物を選択するために、上記方法で用いられることができる。
抗体は、ポリクローン性でありえるか、より好ましくは、モノクローナルでありえる。
完全な抗体または断片単独(例えば、FabまたはF、(ab)2)が、使われることができる。プローブまたは抗体の直接の標識は、検出可能な物質をプローブまたは抗体(プローブの間接的な標識または直接標識である他の試薬を有する反応性による抗体と同様に)に連結する(すなわち、物理的に連結している)ことによって達成されることができる。間接的な標識の例としては、それが蛍光性に標識ストレプトアビジンによって検出されることができるように、蛍光性に標識二次抗体を使用している抗体およびDNAのend―標識がビオチンによって探索する第一の検出が挙げられる。
他の実施形態では、異型は、複数の異型である、それによって複数は、以上1、2、3遺伝子座から、異型を含むことができる。
処理が効果的であると述べることは、有益な治療効果の確率がerbBl遺伝子のキナーゼドメインの特定のキナーゼ活性を増加させている核酸異型の適当な存在を有する人の超過でないことを意味する。
しばしば、キットも、一つ以上の制御テンプレート(例えば正常78から分離される核酸)を提供する組織サンプルおよび/またはerbBl遺伝子のキナーゼドメインの異なる異型を表している一連のサンプル。
用語も、溶菌液を含む。
チャネルは、水性流体が限られるそれ以外は疎水表面上の毛細管、導管または1片の親水性パターンでもよい。
細胞構成要素は、オルガネラ(例えば核、核周囲の区画、核膜、ミトコンドリア、クロロプラストまたは細胞膜)ポリマーまたは分子複合体(例えば脂質、多糖、タンパク質(膜、トランス膜または細胞内可溶質)核酸(固有であるか、治療的であるか、病原性の)ウィルス粒子またはリボソーム)または他の分子(例えばホルモン類、イオン、コファクタまたは薬)でもよい。
本発明の好ましい実施例において、すきまのネットワークは、障壁の配列によって定義される。本実施例において、すきまは、隣接する障壁とのすきまである。好ましい実施例において、すきまのネットワークは、基質の表面上の障壁の配列によって造られる。
外部の細胞膜抗原の変調による細胞内の活性化は、レセプタ輸送の変化に導くこともできる。
表面に関する「ミクロ構造」によって、少なくとも一つの寸法の1mm未満を計量している一つ以上の個々の特徴を含む表面の顕微鏡建造物は、意味される。典型的なマイクロ特徴は、微障壁、マイクロ・ポスト、狭い溝の長時間レコード、マイクロ・フィンおよびマイクロ・コルゲーションである。
試薬を増大することは、体積を増加させるか、変形能を減らすか、または、粒子の形状を変えることによって行うことができる。
本発明は、循環腫瘍細胞(CTC)および他の粒子を検出して、濃縮して、分析する装置および方法を特徴とする。更なる本発明は、主題から細胞サンプルを分析することによって主題(例えば癌)の状態を診断する方法を特徴とする。いくつかの実施形態では、本発明の装置は、CTCまたは他の液成分の置換を許容する障壁の配列を含む。
固形腫瘍からはぎ落される循環腫瘍細胞(CTC)Epithelial細胞は高度な乳癌、コロン、肝臓、卵巣、前立腺および肺をもつ患者の循環の超低濃度で見つかった、そして、これらの細胞の存在または相対数は血液中全体の予後および治療への反応と相関していた。これらのCTCは、腫瘍展開の早期指標または臨床症状の体裁の前の転移でもよい。CTCは概してほぼ1日の短い半減期を有する、そして、それらの存在は増殖性腫瘍から一般に最近の流入を示す。従って、CTCは、患者の病および治療反応の現在の臨床状態を反映することができる動的過程の一部である。
一般に、装置はCTCの横変位および体液の他の構成要素を許容する障壁の一つ以上の配列を含む。それによって、この種の構成要素を豊かにするかまたはさもなければ処理する機構を提供する。それらと異なる先行技術装置本発明、しかし、本発明のそれらがこの目的のために障壁を使用するように、いずれが、例えば、HuangなどScience 304、987―990の(2004)およびU.S.公開番号20040144651に記載されているか。サイズによる析出粒子のための本発明の装置は、概してすきまのネットワークの配列を使用する、そこにおいて、すきまが寸法において同一でもよい場合であっても、すきまを通過している流体は等しくなく次のすきまに分けられる。方法は、すきまの配列で分離される細胞を担持する流れを使用する。流れは、配列の見通し線に関して、小角(流れ角度)で整列配置される。臨界サイズより大きい水力学的サイズを有する細胞は配列によって見通し線に沿って、すなわち、横に、移動するが、臨界サイズより小さい水力学的サイズを有するそれらは平均流れの向きに続く。装置の流れは、薄層状の流動状態の下で発生する。本発明の装置は、連続流通系装置として任意に構成される。
臨界サイズは、いくつかの設計パラメータの機能である。
パラメータΔλ/λ(「分岐比」ε)は、次の障壁の左に二またに分けられる流れの比率を決定する。図1A〜1Cにおいて、説明の便宜のために、εは、1/3である。一般に、2つの障壁間のギャップによる流動がφである場合、軽微な流動はεφである、そして、大きな流動は(l―ε)φ(図2)である。この例では、すきまによる流動は、基本的に三等品(図IB)に分けられる。すきまによる3つの束の各々が障壁の配列周辺で織る一方、各流動の平均方向は全体の流れの方向においてある。
図は、1C、配列による臨界サイズより上に大きさを設定される粒子の動きを例示する。
この種の粒子は大きな流動とともに移動する。そして、順次、各すきまを通過している大きな流動へ移される。
本発明は、加えて、細胞強化(例えばCTCの強化、その雇用試料動員)の装置を含む。装置の特徴(例えば障壁)と関連して、試料動員装置は、細胞の動きまたは流体サンプルの他の構成要素を引き起こす。例えば、本発明の1台の装置は、以下を含む:細胞サンプルを保持することができる容器、選択的に興味がある細胞を捕獲する一つ以上の捕捉残基を含んでいる職能化された蓋面を含む容器の範囲内で適合するように構成される着脱可能に、取付けられた蓋、そして、サンプルmobilizerは、容器か蓋に結合させた。任意には、容器は、選択的に第2の細胞型を捕獲する一つ以上の捕捉残基を含んでいる職能化された表面を有する。蓋面は、いかなる形状も、例えば、四角いか、矩形であるか、円を描くようにすることができる。装置は、公知技術のいかなる材料(例えばガラス、シリコンまたはプラスチック)も使用して製造されることができる。場合によっては、蓋面または容器面は、以下を含む:ミクロ構造(例えばマイクロ障壁、マイクロ・コルゲーション、狭い溝の長時間レコードまたはマイクロ・フィン)。捕捉残基は特に特定の細胞型と結合する一つ以上の抗体を含むことができる、そして、これらの抗体は配列において構成されることができる。本発明の他の装置と同様に、抗体は、特に多種多様な細胞(例えば白血球または上皮細胞)のいずれかと結合することができる。好ましくは、抗体は、特にCTCと結合することが可能である。さらにまた、抗体は、特に表1から選択される細胞表面癌標識(例えばEpCAM、ECadherin、Mucin―1、Cytokeratin 8、上皮細胞成長因子レセプタ(EGFR)および白血球関連のレセプタ(LAR))または標識を結合することができる。場合によっては、試料動員装置の蓋は、容器の壁に関して非直交性角度で容器に収まるように設計されていてもよい。容器は、サンプル(例えば10mLまたは50mL)のいかなる望ましい量も入るように設計されていてもよい。

本発明はCTCおよび他の粒子の濃縮の改良型の装置を特徴とする。そして、バクテリア、ウイルス、菌類、細胞、細胞構成要素、ウイルス、核酸、タンパク質およびタンパク質複合体を含む。そして、寸法通りに一致する。装置は、サンプルの粒子上のさまざまな操作を遂行するために用いることができる。この種の操作は、サイズ・ベースの分画を含んで、粒子または粒子自体の変更または粒子を担持している流体の強化または集中を含む。好ましくは、例えば、中止の液体を交換することによって、または、試薬を有する粒子を接触させることによって、装置は、不均質混合物からCTCまたは他の珍しい粒子を濃縮するかまたは珍しい粒子を変えるために使用される。この種の装置は、細胞(例えば細胞の還元した機械の細胞溶解または細胞内の活性化)に対する限られた応力を有する高度な強化を考慮に入れる。
一段式配列を配列する。実施例において、単段は、すきまのネットワークを形成して、障壁(例えば円筒状障壁(図ID))の配列を含む。ある種の実施形態では、配列は、血液サンプルの他の細胞からCTCを豊かにするときに、例えば、分離限界粒径より数回大きい最大パススルー・サイズを有する。この結果は、大きな間隙の大きさdおよび小さい分岐比εの組合せを使用して成し遂げられることができる。他の好ましい実施形態では、εは、多くても1/2(例えば多くても1/3、1/10、1/30、1/100、1/300または1/1000)である。このような実施例では、例えば、障壁形状はすきまの流れプロフィールに影響を及ぼすことができる。そうすると、すきまによる流体流れは障壁周辺に不規則に配布される;しかしながら、配列を短く(図IE)するために、障壁は、流れの向きにおいて圧縮されることができる。本願明細書において記載されているにつれて、単段配列はバイパスチャンネルを含むことができる。
このように、本発明の装置は、配列から出力を取り除くバイパスチャンネルを含むことができる。臨界サイズより上に粒子を除去する観点からここで記載されているにもかかわらず、バイパスチャンネルは配列のいかなる部分からも出力を取り除くために使用されることもできる。
障壁の複数の列、以前の列の期間の半分より少ないもので相殺されている各連続した列、この種のその少なくとも50%、60%、70%、80%、90%、95%または各々障壁間の99%のギャップさえ、第1の長さパラメータにほぼ等しい長さを有する、そして、多くても50%、40%、30%、20%、10%、5%または1%さえ、それぞれ、障壁間のギャップの中で、各々、第1の長さパラメータより短い第2の長さパラメータにほぼ等しい長さを有する。第2の長さパラメータにほぼ等しい長さを有するすきまは、一様に、または、非一様に、配列の全体にわたって配布されることができる。第2の長さパラメータは、細胞サンプルからの所定サイズより大きい関心の細胞を捕獲するために大きさを設定されることができる。第1の長さパラメータは、第2の長さパラメータより長く、例えば、1.1、1.5、2、3、5、10、20、50または100倍にさえある。第1の長さパラメータのための典型的な距離は30〜100ミクロンの範囲である、そして、第2の長さパラメータのための典型的な距離は10〜50ミクロンの範囲である。
各サブ配列は、いかなる数の障壁(例えば2〜200、3〜50または6〜20)を含むことができる。サブ配列障壁のための典型的な直径は、例えば、25〜200ミクロンの範囲である。一般に、本発明の配列における2つの障害間のギャップは、例えば、少なくとも20、40、60、80、または100ミクロンでもよい;上記の制限されたすきまの場合、このすきまは、例えば、最高でも100、80、60、40、または20ミクロンでもよい。他のギャップの長さも、可能である。
本発明の流れDevicesを定めて、安定させるためのデバイス設計On―チップ・フロー・レジスタは、配列の中で流れを定めて、安定させて、また、配列から集められる流れを定めるために、流体レジスタを使用することもできる。図26は、プレーナーデバイスの回路図を示す;サンプル(例えば血液を含んでいるCTC、取水路)緩衝取水路、荒廃した排水路、そして、製品排水路は、配列に各々接続している。入口および放出口は、流れレジスタとして働く。図26も、これらの異なる装置構成要素の対応する流体抵抗を示す。
装置が恒常的な深さを有して、所与の呼吸抵抗によって作動されるときに、図27および28は配列の中でサンプルおよび緩衝流れの流れおよび対応する幅を示す。流れは、抵抗によって分けられる呼吸抵抗で測定される。この特定の装置において、IbloodおよびIbufferは等価である、そして、これは血液の等価幅および配列の緩衝流れを決定する。
明確さが製品および荒廃した排水路の相対抵抗を制御して、1は、各分数に対する収集抵抗力を調整することができる。例えば、回路図のこの特定の一組で。そのとき、Rproductは、Rwasteより大きい>不用な断片にとっての潜在的に増加した損失および薄めることの代価のより集中製品分数意志結果。逆にいえば、RproductがRwaste未満のときに、より薄いおよびより高い収率製品分数は廃液流から潜在的汚染の代価で集められる。
本発明は、多重化配列を特徴とする。複数の配列を1台の装置に付けることは、CTCのサンプル処理スループットまたは興味がある他の細胞を増加させて、複数のサンプルの並列処理または異なる分数または操作のためのサンプルの部分を考慮に入れる。
多重は、調製用装置にとって更に望ましい。最も単純な複合装置は、連続(すなわちカスケード)において取り付けられる2台の装置を含む。例えば、1台の装置の主な束から出る出力は、第2の装置の入力に連結することができる。あるいは、1台の装置の軽微な束から出る出力は、第2の装置の入力に連結することができる。
例えば、複数の配列が同じ臨界サイズを有するときに、放出口は高いスループット・サンプル処理のために接続されることができる。他の例では、配列の全ては同じ臨界サイズを有することができない、または、配列の粒子の全ては同様に処理されることができない、そして、放出口は流体工学的に接続されることができない。
加えてすきまの配列に付加的な成分、本発明の装置は、CTCの中で隔離、強化、収集、操作または検出のために、例えば、添加元素またはモジュールを、例えば、含むことができる。この種の要素は、公知技術である。例えば、装置は、サンプルまたは緩衝入力のための一つ以上の入口およびサンプル出力の一つ以上の放出口を含むことができる。配列は他のタイプの強化または他の操作のための構成要素を有する装置に携わっていることもできる。そして、親和性(磁気、電気泳動的、遠心性の、および、ジ電気泳動的強化)を含む。本発明の装置は、装置(例えばウェルまたは平坦面の配列)から出る出力の二次元のイメージングのための構成要素によって使用されることもできる。
好ましくは、すきまの配列、本願明細書において記載されているにつれて、親和性と連動する就業者は、強化である。
本発明の二次加工法Devicesは、公知技術の技術を使用して製造されることができる。製作技術の選択は、装置および配列のサイズのために使用される材料次第である。
本発明の装置を組み立てることのための典型的な材料は、ガラス、シリコン、鋼、ニッケル、ポリマーを含む、ポリメタクリル酸メチルで例えばある)(PMMA)ポリカーボネート、ポリスチレン、ポリエチレン、ポリオレフィン、シリコーン(例えばポリ(ジメチルシロキサン))ポリプロピレン、cis―ポリイソプレン(ゴム)ポリ(塩化ビニル)(PVC)ポリ(酢酸ビニル)(PVAc)ポリクロロプレン(ネオプレン)四フッ化エチレン樹脂(テフロン(登録商標))ポリ(塩化ビニリデン)(SaranA)そして、環状オレフィン・ポリマー(COP)そして、環状オレフィン・コポリマー(COC)およびそれらの組み合わせ。他の材料は、従来技術において周知である。例えば、深いReactive Ion Etch(DRIE)は、小ギャップ、小さい障壁およびかなりの縦横比(横方向寸法に対する障壁高さの比率)を有するsilicon―ベースのデバイスを組み立てるために用いる。例えば、プラスチック器具の熱成形(射出成形して、型押しする)が使われることもできる。そのとき、最も小さい横方向の特徴は>20ミクロンである、そして、これらの特徴のアスペクトレシオは<10である。付加的な方法は、写真平版(例えばステレオリソグラフィまたはX線写真平版)応形機能、エンボシング、シリコン・マイクロマシニング、湿式であるか乾燥した化学腐食、ミリング、ダイヤモンド・カット、Lithographie GalvanoformungおよびAbformung(LIGA)および電気鍍金を含む。例えば、ガラスのために、湿気(KOH)またはドライエッチング(フッ素または他の反応性ガスを有する反応性イオンエッチング)が続く写真平版の従来のシリコン製作技術は、使用されることができる。レーザー・マイクロマシニングのような技術は、高い光子吸収効率を有する塑性物質のために採用されることができる。この技術は、方法の連続性質のため、下のスループット製作に適している。大量生産のプラスチック器具のために、射出成形していて、圧縮成形している熱可塑性物質は、適切でもよい。コンパクトディスク(それは、下位数ミクロンの特徴の忠実度を保存する)の大量加工のために使用する従来の熱可塑性押出鋳込は、本発明の装置を組み立てるために使用されることもできる。例えば、装置機能は、従来の写真平版によってガラス・マスターに複製される。ガラス・マスターは、堅い、熱耐衝撃(熱伝導、堅い型)を産生するために電鋳される。この型は、プラスチック器具に特徴を射出成形するかまたは圧縮成形することのマスター・テンプレートとして役立つ。装置および完成品の光学的特性およびスループット上の要件を作るために用いる塑性物質に応じて、圧縮成形または押出鋳込は、製造方法に選ばれることができる。圧縮成形(また、熱いエンボシングまたは救助刷込みと呼ばれる)は、高分子重合体と互換性を持つことの効果がある、それは、小さい構造のために優秀で、高いアスペクトレシオ構造を複製することができるより長いサイクルタイムを有する。押出鋳込は、低いアスペクトレシオ構造のためによくなって、低分子量のポリマーに最も適している。
装置が組み立てられる前に、又は、その後に、壁は職能化されることができる。
通信路の壁は、サンプル(例えば膜断片またはタンパク質)の材料を捕獲するために被覆されていることもできる。
本発明の装置は、臨界サイズの上下に不変化詞で豊かにされるサンプルの生産が要求されるいかなるアプリケーションでも使用されることができる。装置の好適な使用は、CTCまたは他の珍しい細胞において豊かにされるサンプルを生じることである。一旦強化試料が生じると、それは分析のために集められることができるかまたはさもなければ操作されることができる。
実施例において、本発明の装置は、所望の水力学的サイズのかけらにおいて豊かにされるサンプルを生じるために使用される。この種の強化の応用は、CTCまたは関心およびサイズfractionization(例えばサイズ・フィルタリング(特定のサイズ範囲のセレクティング・セル))の他の細胞に集中することを含む。装置は、細胞(例えば核)の構成要素を豊かにするために用いることもできる。望ましく、潜在的に一つ以上の望ましくない粒子と関連して少なくとも100、1,000、10,000、100,000、1,000,000、10,000,000または100,000,000倍にさえ所望の粒子を濃縮すると共に、本発明の方法は初期混合物と比較して所望の粒子の少なくとも50%、75%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%または99%さえ保持する。望ましく、装置が強化試料に加えて出力サンプルを生じる場合、この付加的な出力サンプルは初期混合物と比較して所望の粒子の50%未満、25%、20%、19%、18%、17%、16%、15%、14%、13%、12%、11%、10%、9%、8%、7%、6%、5%、4%、3%、2%または1%さえ含む。強化は原物のサンプルと比較して結果として強化粒子の希釈剤になることもできる。但し、サンプルの他の粒子と関連する強化粒子の濃度は増加した。好ましくは、希釈剤は、多くても90%(例えば多くても75%、50%、33%、25%、10%または1%)である。
胎児の赤血球は、性交を決定して、発育中の胎児の異数性または遺伝子の特徴(例えば突然変異)を確認するために、母性末しょう血液から、例えば、豊かにされることができる。CTC(それは上皮のタイプおよび起源である)は、診断の目的で末しょう血液から豊かにされることもできて、治療的な発展をモニタしていることもできる。
循環血管内皮細胞は、末しょう血液から同じように豊かにされることができる。
身体の流体または環境サンプルは大腸菌群のために病原微生物のために、例えば、映写されることもできる。そして、血液が疾患(例えば敗血症または細菌性であるかウィルス性髄膜炎)を抱かれる。珍しい細胞も、他の有機体(例えば移植臓器からの細胞)に存在する1つの有機体から、細胞を含む。
臨界サイズ(例えば上皮細胞)を越える細胞(CTC)は、特に偏って、流体流入口によって加えられる緩衝媒体に含まれる中心の放出口から現れる。装置の動作は、このように、臨界サイズの上下に細胞において豊かにされるサンプルを生じる。上皮細胞が血流において最も大きな細胞の一つであるので、装置のすきまのサイズおよびジオメトリは実質的に他の全ての細胞型を端放出口にあてるために選択されることができる、その一方で、サンプルを装置を通る単一の流路の後で上皮細胞において実質的に豊かにされる中心の放出口から作り出す。
他の実施態様において、強化に加えて、CTCまたは興味がある他の細胞は、粒子または中止の流体を化学的に、または、物理的に変えることができる変わっている試薬によって接触する。この種のアプリケーションは、浄化、緩衝交換、標識(例えば免疫組織化学的、磁気、および、組織化学的標識、細胞染色および流れその場蛍光ハイブリダイゼーション(FISH))細胞固定、細胞安定化、溶菌および細胞活性化を含む。
この装置において、血液粉末粒子は粒子と反応する摂政に血液から移動する、そして、反応された粒子はそれからバッファに持ち込まれる。それによって、未反応の試薬または反応副産物を取り除く。追加ステップは、加えられることができる。
本発明の装置は、遊離標識試薬をCTCまたは他のセル行きの標識試薬から分離するために使用されることができる。図45に示すように、標識試薬は、装置への導入の前に、細胞サンプルによって前暖められることができる。望ましく、標識試薬は、特に、または、優先して、興味がある菌体群(例えば上皮細胞(例えばCTC))を結合する。典型的な標識試薬には、抗体、量子ドット、バクテリオファージ、アプタマ、蛍光団含有分子、検出可能な化学反応を実施することができる酵素または職能化されたビードが含まれる。
通常、標識試薬は興味がある細胞より小さい、または、興味がある細胞はビードと結合した;このように、標識試薬と結合される細胞サンプルが装置にもたらされるときに、遊離標識試薬は非偏る装置によって移動して、端放出口から現れる。その一方で、縛られた標識試薬は上皮細胞とともに中心の放出口から現れる。都合のよいことに、この方法は、縛られた標識試薬から同時に遊離標識試薬のサイズ分離および分離を成し遂げる。加えて、後述するように、分離のこの方法は、解放ステップまたは分析の破壊的な方法の下流のサンプル分析を容易にする。
本発明の装置は、緩衝交換のために使用されることができる。
この結果を成し遂げるために、強化のために使用されるそれと類似のプロトコルは、従われる:細胞サンプルは装置の試料導入口によって加えられる、そして、所望の最終的な緩衝媒体は流体流入口によって加えられる。上述の通り、臨界サイズより上の細胞は、偏って、バッファに入る。
本発明の装置は、興味がある細胞サンプル(例えばCTCを含んでいるサンプル)に集中するために使用されることができる。図47に示すように、細胞サンプルは、装置の試料導入口にもたらされる。流体流入口にもたらされるバッファ量を減らすことによって、この体積が細胞サンプルの量より著しく小さいように、より少ない体積の的状赤血球の集中は起こる。この濃縮段階は、実行されるいかなる下流の分析の結果も改善することができる。
本発明の装置は、溶菌のために使用されることができる。これを達成するために、強化のために使用されるそれと類似のプロトコルは、従われる:細胞サンプルは装置(図48)の試料導入口によって加えられる、そして、溶解緩衝液は流体流入口によって加えられる。上述の通り、臨界サイズより上の細胞は偏って、溶解緩衝液に入る。そして、これらの細胞の細胞溶解に導く。その結果、中心の放出口から現れているサンプルはオルガネラを含んでいる溶解された細胞成分を含む。その一方で、非偏向する細胞全体は他の放出口から現れる。このように、装置は、方法を的状赤血球を選択的に溶解するために提供する。
本発明の装置は、強化および反応の複数の段階を提供するために結合されることができる。例えば、図43 Aは、「カスケード」構成を示す、 そこにおいて、1台の装置の放出口1は、第2の装置の試料導入口に取り付けられる。第2の装置にもたらされるサンプルが興味がある細胞のためにすでに豊かにされるように、これは第一装置を使用している最初の強化ステップを考慮に入れる。2台の装置は同一であるか異なる臨界サイズを有することができる。そして、意図されたアプリケーション次第である。
遊離標識試薬の的状赤血球および分離の更なる強化は成し遂げられる、そして、強化試料は更に分析されることができる。あるいは、標識試薬は、直接、第2の装置への導入の前に第一装置の中心の放出口から現れているサンプルに加えられることができる。
カスケード構成の使用は、図55の単一デバイス構成より少ない出費で、より少ない量の使用または標識試薬のより高い濃度を許すことができる;加えて、発生することができるいかなる非特異的結合も、第一装置を使用している最初の強化ステップの存在下で、著しく還元している。
多くの症候のための役立つステップが検定する下流の分析Aは、分析されるサンプルからの遊離標識試薬の除去である。上述の通り、本発明の装置は、遊離標識試薬をセル(例えばCTC)行きの標識試薬から分離することが可能である。遊離標識試薬から背景干渉の有意水準のない標識サンプルの大きさ測定を実行することは、それから可能である。
例えば、EpCAMのような特定の上皮細胞標識のために選択的な蛍光抗体が、使われることができる。蛍光部分は、Cy染料、Alexa染料または他の蛍光団含有分子を含むことができる。結果として生じる標識サンプルは、それから、蛍光光度計を使用している標識強化細胞の結果として生じる試料の蛍光を測定することによって分析される。
あるいは、発色団を含むラベルが、分光計(例えばUVまたは可視分光計)と連動して用いられることができる。得られた測定値は、的状赤血球の数またはサンプルのすべての細胞を定量化するために用いることができる。あるいは、サンプル(例えば血管内皮細胞に対する癌細胞の比率)の2セル・タイプの比率は、決定されることができる。この比率は各タイプの細胞の数の比率でもよい、または、あるいは、それは各タイプの細胞のいかなる測定された特徴の比率でもあってもよい。
例えば、装置は、サンプルのより小さい細胞から、12ミクロン、14ミクロン、16ミクロン、18ミクロンまたは20ミクロンよりさえ大きい水力学的サイズを有する細胞を濃縮する。あるいは、装置は、他の細胞から6ミクロンおよび12ミクロン以下(例えば8ミクロンおよび10ミクロン以下以上の水力学的サイズを有する細胞)以上の水力学的サイズを有する細胞を濃縮することができる。装置は、10ミクロンより大きい水力学的サイズを有する細胞から、5ミクロンおよび10ミクロン以下以上の水力学的サイズを有する細胞を濃縮することもできる;あるいは、それは、8ミクロンより大きい水力学的サイズを有する細胞から、4ミクロンおよび8ミクロン以下以上の水力学的サイズを有する細胞を濃縮することができる。細胞のこれらのサブセットの各々はそれから集められることができて、例えば、例えば、特定の細胞型の存在を検出することによって分析されることができる。そして、サンプルのうちの1つの珍しい細胞(例えば上皮細胞または原種血管内皮細胞)がこのように集められる。この方法が一般に成し遂げる強化のため、珍しい細胞の濃度は始まっている細胞サンプルにおいてより回復されたサンプルにおいて高くてもよい。そして、様々な手段によって珍しい細胞検出を考慮に入れる。実施例において、細胞サンプルは、装置の入口に適用される;例えば、第2の試薬(例えばバッファ)緩衝含んでいるBSA(細胞溶解試薬、核酸増幅試薬、モル浸透圧濃度を調整している試薬、標識試薬、防腐剤または固定試薬)は、第2の入口に任意に適用される;そして、2つの出力サンプルは、装置の2つの放出口から流出する。例えば、装置の入口に癌細胞を含んでいる細胞サンプルの塗布は、結果としてこの種の細胞において豊かにされる1つの出力サンプルになることができる、他のサンプルがこれらの細胞において減少する、または、それらの中でさえ完全に欠けている。上記の第2の試薬のいずれか、装置および本発明(例えば装置が第2の入口を含むそれら)の方法のいずれかで使用されることができる。
例えば、第2の細胞型は白血球を含むことができる、または、赤血球は、例えば、赤血球を摘出した。
細胞サイズ、形状または変形能に基づくいかなる方法も、興味がある細胞を濃縮するために用いられることができる;その後、細胞検出または他のいかなる下流のアプリケーション(例えば本願明細書において記載されているそれら)も、実行されることができる。
本発明の方法は、細胞の同一セットの強化、定量化および分子生物学分析を考慮に入れる。本発明(大きさ測定の記載されている方法に連結する)の装置の細胞の穏やかな処理は、更なる分析が必要に応じて実行されることができるように細胞の健全性を維持する。
例えば、細胞の健全性を破壊する技術は、大きさ測定に続いて実行されることができる;
この種の技術は、DNAまたはリボゾームリボ核酸分析、プロテオーム分析またはmetabolome分析を含む。例えば、DNAの総量またはサンプルのリボゾームリボ核酸は、決定されることができる;あるいは、例えば、特定のシーケンスの存在またはDNAまたはリボゾームリボ核酸の突然変異(例えば削除)は検出されることができる。そして、遺伝子の突然変異が表1にリストされるポリペチドをコード化する。
さらにまた、サンプルのミトコンドリアDNA、テロメラーゼまたは核マトリックス蛋白は分析されることができる(癌の糸粒体の突然変異のために、Parrellaなど(イチョウガニ科Re)参照。61:7623−7626 (2001)ジョーンズなど、癌財産。61:1299―1304の(2001)およびFlissなど(Science 287):2017―2019の(2000);テロメラーゼのために、Soriaなど(Clin)参照。癌財産。5:971−975 (1999)).例えば、あらゆる糸粒体の異常であるにせよ、サンプルは定量に分析されることができる(Carewその他を参照、例えば、MoI。癌1:9つの(2002)およびWallace(Science 283):1482―1488の(1999))または、核周囲の区画は、ある。DNAを分析する1つの役立つ方法は、PCRである、そこにおいて、細胞は溶解される、そして、特定のDNAの塩基配列のレベルは増幅される。分離される的状赤血球の数が非常に低いときに、この種の技術は特に役立つ。細胞のPCRは、使用されることができる;加えて、遺伝子形質発現分析(Giordanoその他を参照、例えば、Am。J..Pathol.159:1231−1238 (2001)そして、Buckhaultsなど(癌財産)。63:4144−4149 .(2003))または、蛍光その場ハイブリダイゼーションは、例えば、分析されている細胞の源の組織または組織を決定するために用いることができる。様々な細胞特徴は、上記の技術(例えばタンパク質リン酸化、タンパク質グリコシル化、DNAメチル化)のいずれかを使用して測定されることができる(Dasその他を参照、例えば、J.Clin。Oncol.
22:4632−4642(2004))マイクロ・リボゾームリボ核酸レベル(を参照、例えば、彼その他、自然435:828―833の(2005)Luなど、自然435:834―838の(2005)O´Donnellなど、自然435:839―843の(2005)およびCalinなど(N.Engl)。J.med.353:1793―1801の(2005))細胞形態または他の構造特徴(例えばpleomorphisrhs、粘着力、遊走、締め具、分割)は、遺伝子形質発現および体細胞突然変異の存在の中で水平になる。この分析はいかなる数の細胞に対して行われることができる。そして、興味がある単細胞(例えば癌細胞)を含む。加えて、細胞の粒度分布は、分析されることができる。望ましく、好ましくは同じ主題から、下流の分析(例えば検出)は、一つ以上の検体に対して行われる。
Cellsが血液中、さまざまなタイプの中であって、サイズの範囲にわたることを発見した細胞の定量化。本発明の方法を用いて、区別して、糊付けして、血液菌体群(例えばCTC)を計数することは、可能である。例えば、コールターカウンタが、使われることができる。図は、33A、通常の血液サンプルのための典型的粒度分布を示す。
若干の状況(例えば剥脱性腫瘍細胞である本体の腫瘍の存在)の下で、血液固有でない細胞は、末梢循環に現れることができる。隔てて、大細胞(または他の所望の細胞)が血液に非常に現れることができることを計数する能力は、強力な機会を疾病状態を診断するために提供する。
本発明の装置を使用している強化および変更方法は、他の懸濁物質試料操作技術と結合されることができる。特に、CTCまたは他の粒子の更なる濃縮または浄化は、望ましくてもよい。更なる強化はいかなる技術によっても発生することができる。そして、親和性強化を含む。好適な親和性強化技術には、障壁の壁または配列を向けるにちがいない親和性剤によって興味がある粒子を接触させることが含まれる。この種の親和性剤は、いかなる細胞型(例えば癌細胞)のためにも選択的でもよい。これは、的状赤血球に特有の抗体が装置の範囲内で固定される本発明の装置を使用することを含む。これは、装置の範囲内で締め具および的状赤血球の強化を考慮に入れる;その後、的状赤血球は、より高い移動比、競争しているリガンドまたは他の方法を使用して溶出させられる。
本願明細書において記載されているにつれて、固形腫瘍からはぎ落される上皮細胞は乳癌、コロン、肝臓、卵巣、前立腺および肺をもつ患者の循環で見つかった。一般に、治療の後のCTCの存在は、腫瘍発達および治療(疾患の再発)への広げられた、劣った反応と関係していておよび/または数年の期間にわたる生存を減少させた。従って、CTCの列挙は、最初の危険を予測する基本的な特徴および治療に対する応答に基づく次の危険のための患者を層化する手段を提供する。
役立つ血管内皮細胞表面マーカは、CD105、CD106、CD144およびCD146を含む;役立つ腫瘍血管内皮細胞表面マーカは、TEMl、TEM5およびTEM8を含む(Carson―Walterその他を参照、例えば、イチョウガニ科Re。61:
6649−6655 (2001));そして、役立つ間葉系細胞表面マーカは、CD 133を含む。
標識が得られることができるこれらまたはその他に対する、例えば抗体、Chemicon、真横および研究開発Systems。
既存の癌患者の場合、これは、疾患の進行の役立つ徴候を提供して、町医者が増加、減少または患者の血流の上皮細胞(例えばCTC)の変化の不足に基づいて適切な治療的な選択をするのを援助する。癌の危険にさらされたそれらのために、検出される細胞の数の急増は、患者が腫瘍を呈した初期警告を提供することができる。この早期診断(次の治療的な介入に連結する)は、診断情報の欠如と比較して結果として改良された寛容な結果になりそうである。
加えて、リボゾームリボ核酸分析、プロテオーム分析またはmetabolome分析は、患者に存在する癌の種類または種類を診断する手段として実行されることができる。
肺癌および他の癌のための1つの重要な診断インジケータは、EGFR(International出版物WO 2005/094357を参照、例えば)の特定の突然変異の有無である。
そして、細胞内のチロシンキナーゼ(TK)領域。EGFRの通常の生理学的役割はErbBリガンドを結合することである。そして、細胞増殖、生存、運動性および他の関連した動作に導いている下流の細胞内の信号のカスケードを起動させる細胞外の結合部で、上皮細胞成長因子(EGF)を含む。EGFR突然変異を有する多くの非小細胞肺腫瘍は、小分子EGFRインヒビター(例えばゲフィチニブ)に反応する(Iressa;AstraZeneca)しかし、しばしば結局、それらを薬剤抵抗性ようにする第2の突然変異を得る。本発明の装置および方法を用いて、1は、以下によってこの種の薬を飲んでいる患者をモニタすることができる:血液の頻繁なサンプルをすること、そして、時間の関数として、各サンプルの上皮細胞(例えばCTC)の数を決定すること。これは、疾患の過程に関しては、情報を提供する。例えば、減少の数の循環上皮細胞は、時間とともに疾患のひどさおよび腫瘍または腫瘍のサイズの減少を示唆する。すぐ上皮細胞の定量化の後に、これらの細胞は、どんな突然変異が上皮細胞において表されるEFGR遺伝子に存在してもよいかについて決定するために、PCRによって分析されることができる。特定の突然変異(例えばEGFR TK領域のATP結合ポケット周辺で集まっているそれら)は、癌細胞をゲフィチニブ抑制に影響されやすくすることは公知である。このように、これらの突然変異の存在は、処理を使用しているゲフィチニブに反応しそうである癌の診断法をサポートする。しかしながら、結局ゲフィチニブに応える多くの患者は第2の突然変異(しばしばTK領域のエキソン20の位置790のスレオニンに対するメチオニン置換)を開発する。そして、それによってそれらがゲフィチニブに対して耐性を示しなる。
本発明の装置および方法を用いて、1は同様にこの突然変異のために試験することができる。そして、疾患の過程およびそれがゲフィチニブまたは類似化合物に反応するという可能性について更なる診断情報を提供する。多くのEGFR突然変異が、すべてのEGFR突然変異を肺癌が感度またはゲフィチニブに対する耐性を与えることは公知であると現在まで報告したNSCに含んで、エキソン18〜21のコード領域の中で位置するので、EGFR遺伝子のこの領域は突然変異(実施例4―6を参照)の存在のための分析法の開発において強調されることができる。
方法は、野生型erbBl遺伝子と関連して前記患者のerbBl遺伝子のキナーゼドメインの少なくとも一つの核酸異型の有無を検出することを含む。少なくとも一つの異型の存在は、EGFRターゲッティング処理が効果的になりそうなことを示す。好ましくは、核酸異型は、EGFRのキナーゼ活性を増加させる。患者は、それからEGFRターゲッティング治療で治療されることができる。本発明の一実施例では、EGFRターゲッティング処理は、チロシンキナーゼ・インヒビターである。
診断検査は、以下を含むことができる:しばしば固体担体に、異型の中でパネル。そして、それは、一つ以上の異型の同時定量を可能にする。方法は、患者のEGFRのキナーゼドメインが少なくとも一つの核酸異型を含むかどうかについて決定することが必要である。この種の異型の存在は、EGFR目標とされた処理(例えばチロシンキナーゼ・インヒビターの管理)の効果を表す。
例えばのように、International公開番号WO 00/04194で、決定ハプロタイプの方法は当業者に知られている。好ましくは、キナーゼ活性を増加させている核酸異型の有無の決定は、方法(例えばPCR法(PCR))で異型サイトまたはサイトのシーケンスを測定することが必要である。あるいは、キナーゼ活性を増加させている核酸異型の有無の決定は、チェーンを終了しているDNA塩基配列決定またはミニ・シークエンシング(オリゴヌクレオチド・ハイブリダイゼーションまたはマススペクトル分析)を含むことができる。
例えば、erbBlのキナーゼドメインの少なくとも一つの異型の存在は、患者がEGFRターゲッティング化合物(例えばチロシンキナーゼ・インヒビター)を有する処理からたぶん利益を得ることを示す。
コントロール試料に対する試験生体試料のPCR製品の比較は、試験生体試料の異型を示す。変化は、試験生体試料の核酸異型の欠如か存在でありえる。
1173―1177)Qb Replicase(Lizardi、その他を参照、例えば、1988(BioTechnology 6:1197))または他のいかなる核酸増幅方法も、よく当業者に知られている技術を使用している増幅された分子の検出によって続いた。この種の分子が超低数に存在する場合、これらの検出方式は特に核酸分子の検出に役立つ。
例えば、レセプタ・チロシンキナーゼの相補DNAシーケンスは、GenBankから得られることができて、エキソン/イントロン境界線を確認するためにBLAT配列を使用しているヒトゲノム組立てと比較されることができる。外部の遺伝子に特有のプライマ対は、PrimerSプログラムを使用している両側上のイントロン・シーケンスまたは隣接するエキソン・シーケンスの側面に位置するエキソン・シーケンスおよび少なくとも250の塩基対を増幅するように設計されている。結果として生じる予測されたアンプリコンは、それから、エキソン(一般にエキソン/イントロン境界線から50の塩基対より大きい)の側面に位置していて、添付のM13前方または後方のプライマ尾部を含んでいる内部プライマを設計するために用いる。これらの入れ子にされたプライマ・セットは、コントロールDNAから適当なアンプリコン・サイズおよび高級のシーケンスを見つけるため検査される。47のチロシンキナーゼのレセプタ・チロシン・キナーゼ活性化ループをコード化しているエキソンを含んでいるアンプリコンは、増幅されて、測定用試料(例えば本発明のミクロな液体の装置を利用して濃縮される細胞)から順序付けた。加えて、完全長EGFRをカバーしているアンプリコンも、増幅される。本発明の方法に用いられる典型的なプライマおよび他のシーケンスは、表2に示される。
少なくとも一つの核酸異型の有無の検出は、レセプタをコード化している核酸の部分を増幅することによって決定されることができる。好ましくは、増幅される部分は、長さの1000のヌクレオチドである長さの500のヌクレオチドおよび長さまたはより少ないものの大部分の好ましくは100のヌクレオチド。増幅される部分は、複数の異型を含むことができる。他の実施形態では、有無または少なくとも一つの異型の検出は、少なくとも一つの核酸プローブを有する異型サイトを含んでいるEGFR核酸を接触させることを提供する。プローブは、選択的なハイブリダイゼーション状況の下で異型サイトで異型サイトおよび含んでいる補完的なヌクレオチド塩基を含む核酸一次構造によって、優先して交雑する。ハイブリダイゼーションは、検出可能なラベルによって検出されることができる。
代替実施形態では、試料セルからのEGFR遺伝子の突然変異は、限定酵素裂開パターンの変更によって確認されることができる。例えば、サンプルおよびコントロールDNAは、任意に増幅されて、一つ以上の限定エンドヌクレアーゼによって消化されて分離される、そして、断片長サイズはゲル電気泳動で測定されて、比較される。サンプルDNAのサンプルおよびコントロールDNA指示突然変異間の断片長サイズの違い。さらに、シーケンスに特有のリボザイム(米国特許番号5,493,531を参照、例えば)の使用は、現像またはリボザイム切断部位の損失によって特定の突然変異の存在のためにスコアするために用いることができる。
1242.一般に、「不適当な組合せ裂開」の技術は、潜在的に突然変異のリボゾームリボ核酸を有する野生型EGFRシーケンスを含んでいる(標識)リボゾームリボ核酸またはDNAの雑種を作ることによって形成されるヘテロデュプレックスまたは組織サンプルから得られるDNAを形成することから始める。二本鎖デュプレックスは、いずれが制御およびサンプル索の間にbasepair不適当な組合せのため存在するかというデュプレックスの一本鎖領域を切断する剤を有する既治療である。例えば、リボゾームリボ核酸/DNA二重螺旋はRNアーゼで処理されることができる、そして、酵素処理でにSlヌクレアーゼで処理されるDNA/DNAハイブリッドは不釣合いな地方を消化する。他の実施態様において、DNA/DNAかリボゾームリボ核酸/DNA二重螺旋は、不釣合いな地方を消化するために、ヒドロキシルアミンまたはオスミウム酸によって、そして、ピペリジンによって処理されることができる。不釣合いな地方の消化の後、結果として生じる材料は、それから、突然変異のサイトを決定するためにポリアクリルアミドゲルを変性することのサイズによって分離される。Cottonなど、1988、85:4397会報Natl. Acad. Sci.USAおよびSaleebaなど(1992)を参照手法Enzymol。2 17:286−295.実施例において、コントロールDNAまたはリボゾームリボ核酸は、検出のために標識でありえる。
さらに別の実施形態では、変性剤の勾配を含んでいるポリアクリルアミドゲルの変異体または野生型断片の動きは、変性勾配ゲル電気泳動(DGGE)を使用して検定される。
313:495にMyersなど、1985、Nature参照。DGGEが分析の方法として使われるときに、DNAはそれが、例えば、PCRによって高融点のGCの豊富なDNAのほぼ40の塩基対のGCクランプを加えることによって完全に変性しないことを確実にするために変更される。さらなる態様において、温度勾配が、制御およびサンプルDNAの可動性の違いを確認するために、変性勾配の代わりに使われる。RosenbaumおよびReissner、1987、Biophys参照。265:12753化学。
6230.オリゴヌクレオチドが交雑している膜に取り付けられて、標識目標DNAによって交雑するときに、この種のアレレに特有のオリゴヌクレオチドはPCR増幅された目標DNAまたは多くの異なる突然変異に交雑する。
酸財産。17:2437―24―48)または、適当な状況の下で、不適当な組合せがRNA依存性DNAポリメラーゼ拡張を防止することができるかまたは減らすことができる1つのプライマの極度の3―終点で(Prossnerを参照、例えば、1993、Tibteeli。11:238).加えて、裂開ベースの検出をつくるために突然変異の領域の新しい制限サイトを導くことは、望ましくてもよい。Gasparini、など、1992、MoI参照。細胞は、6:1に深く探る。特定の実施例で、増幅が増幅のためのTaq DNAリガーゼを使用して実行されることもできることが予期される。Barany 1991(会報Natl. Acad. Sci.USA 88:189)参照。そのような場合、完璧なマッチが3つ時にある場合だけ、ライゲーションは発生する―5´シーケンスの終点(増幅の有無を探すことによって特定のサイトで周知の突然変異の存在を検出することを可能にする)。
15分間の1サイクルの95度C、20秒間の35サイクルの95度C、30秒間の600Cおよび1分(3分間の720Cの1つのサイクルが続く)間の72度C。結果として生じるPCR製品は、汎用Ml 3下塗りを有する双方向性染料―ターミネータ蛍光シークエンシングが続く固相可逆固定化化学によって精製される。シークエンシング断片は、キャピラリー電気泳動法を使用しているABI Prism 3700 DNA Analyzer(印加Biosy茎、Foster City、CA)を介して検出される。PCRおよびシークエンシングは、Agencourt BioscienceCorporation(Beverly、MA)によって実行される。
ゲフィチニブへの著明な反応をもつ患者からのCTCサンプルは、本質的事項がこの種の患者に存在する腫瘍のこの成育因子の信号を送っている経路によって役割を演じたことを示しているEGFR1の突然変異を収容しているかもしれない。この種の突然変異を探すために、EGFRの細胞外の領域の中の再編成はテストされることができる。そして、膠腫に特徴的である。この種の再編成が検出されない場合、全てのコード領域は個々のエキソンのPCR―増幅を使用して配列決定されることができる。ヘテロの突然変異は、アミノ酸746―750(delE756―A750)747〜750(delL747―T751)および747〜752(delL747―P753)を取り除いているインフレーム削除を含むことができる。後の2つの削除は、削除限界点で新しいコードンの生成から生じているセリン残基の挿入と関係している。更なる突然変異は、エキソン21の中でアミノ酸置換を含むことができる:コードン858(L858R)のアルギニンに対するロイシンおよびコードン861(L861Q)のグルタミンに対するロイシン。L861Q突然変異は特に興味がある。−その理由は、次のことにある。マウスegfr遺伝子の同じアミノ酸変化はDark Skin(dsk5)特徴(変えられたEGFRシグナリングと関連した)の原因となる。キナーゼドメインのミスセンス突然変異は、エキソン18(G719C)の中で、コードン719でシステイン置換に結果としてグリシンになることができる。
(2253_2276 del)削除は、先に述べたエキソン19削除に重なる。
削除は、2つのグループのうちの1つに分類されることができる:最低限(アミノ酸配列LKE)のそれらの橋を架けているコードン747―749およびそれらの橋を架けているコードン752―759。19の削除が現在まで報告したすべてのエキソンの分析は、多種多様なアミノ酸がコードン747―759にわたっているTK領域から削除されることができることを示唆する。削除されるコードンは、必要な共有地であるために現れない。
例えば、ゲノムDNAのエキソン・シークエンシングでEGFRのミスセンスおよび欠失突然変異がみられる。そして、それは通常キナーゼドメインのエキソン18〜21の中で発生する。シーケンス変更は、腫瘍DNAの異種接合体でもよい;いずれの場合においても、同じ患者からの対正常はい組織は野生型シーケンスを有する。そして、突然変異が起源において身体であることを確認する。置換突然変異の若干の実施例は、G719SおよびL858Rである。「G719S」突然変異は、セリンに位置719でグリシン(G)を変える。これらの突然変異は、領域またはP環を結合していて、それぞれ、活性化ループの非常に節約されたDFGのモティーフと隣接しているヌクレオチド三燐酸のGXGXXGのモティーフに位置する。変異する残基はすべてのプロテインキナーゼにおいてほとんど不変量不変である、そして、B―Rafタンパク質セリンスレオニンキナーゼの類似した残基(G463およびL596)は結腸直腸ものにおいて変異する体細胞性、卵巣性および肺癌腫である。
例えば、強化試料(例えば12ミクロンより大きい水力学的サイズを有する細胞を含んでいるサンプル)の細胞の粒度分布を状態(例えば癌)に関連した粒度分布と比較することによって、診断は、この比較に基づくようにされることができる。比較のA細胞ひな型は、いかなる方法によっても発生することができる。例えば、関連研究は複数の対照被検者(例えば50)および興味がある状態を有する複数のケース主題(例えば50)からのどの細胞サンプルが処理されるかについて実行されることができる、例えば、12ミクロンより大きい水力学的サイズを有する細胞を濃縮することによって、結果サンプルは分析される、そして、分析の結果は比較される。この種の研究を実行するために、強化細胞のリボゾームリボ核酸レベル(例えばメッセンジャーRNAまたはマイクロ・リボゾームリボ核酸レベル)を分析することは、役立ってもよい。あるいは、いずれの場合においても濃縮される細胞の数を計数するか、または顕微鏡、細胞カウンタまたはマイクロアレイ装置を用いて細胞粒度分布を、例えば、決定することは、役立つ。特定の細胞型(例えば珍しい細胞)の存在は、確認されることもできる。
本発明の試料動員装置は、サンプルからCTCまたは他の細胞を濃縮するために用いることができる。実施例において、例えば、細胞サンプルは、試料動員装置に置かれる
以下を含む装置:容器、職能化された表面を有する蓋、そして、サンプルmobilizer。サンプルを含んでいる容器はそれから蓋でおおわれている、サンプルmobilizerはサンプルを起動させるために使用される、そして、蓋は取り外される。この種の装置は、関心のCTCまたは他の細胞を濃縮するために用いることができる。
付加的な力は、例えば、力を遠心力に印加されることもできる;さらに、力は繰り返し印加されることができる、そして、交替で、各々のアプリケーション間の任意の時間的間隔については、強制する。
一般の考慮すべき問題Samplesは、浄化(例えば特定の構成要素の安定化および除去)の有無にかかわらず本願明細書において記載されている方法で使用されることができる。若干のサンプルは、装置に希釈されることができるかまたははじめにの前に集中されることができる。
Samplesは、操作(例えば特定の構成要素の安定化および除去)の有無にかかわらず本願明細書において記載されている方法で使用されることができる。
実施例において、サンプルは、装置への本発明の導入の前に、関心のCTCまたは他の細胞において豊かにされる。菌体群を豊かにする方法は技術(例えば親和性機構、アグルチネーション)において公知である、そして、サイズ、形状および変形能は強化の基礎を形成した。興味がある細胞のサンプルを豊かにする典型的な方法は、米国特許番号5,837,115および5,641,628(International出版物数字WO 2004/029221およびWO 2004/113877)およびU.S.公開番号2004/0144651で見つかる。
本発明のミクロな液体の装置は、CAD(CAD)によって設計されて、写真平版によって微細作製された。二段法は、血液サンプルが小細胞の大集団を取り除くために非かさばって最初にいずれであるかについて開発された、そうすると、珍しい目標上皮細胞的状赤血球は、免疫親和性捕獲によって回復される。装置は、写真平版によって定義されて、CAD―生成の設計に基づいてシリコン基板にエッチングされた。細胞強化モジュール(それはほぼ標準顕微鏡スライドの寸法である)は、一般のサンプルおよび緩衝入口および製品に接続して、放出口を浪費するチャネルを扱っている14の平行したサンプル処理セクションおよび付随するサンプルを含む。各断面は、生成物流により大きな細胞の置換を経て水力学的サイズによって的状赤血球タイプを豊かにするために最適化される微細作製された障壁の配列を含む。この例では、マイクロチップは、赤血球(RBC)および血小板をより大きい白血球およびCTCから切り離すように設計されていた。的状赤血球の豊かな人口は、装置を通過する全血液から取り戻された。細胞強化マイクロチップのパフォーマンスは、RBCおよび血小板を通常の全血液(図52)の白血球(WBC)から切り離すことによって評価された。癌患者において、CTCは、より大きいWBC分数で見つかる。血液は最小限薄められた(30%)そして、6mlのサンプルは最高6ml/時間の流速で、処理された。製品および廃液流はCoulterモデル「Ac―T diff」臨床面血液アナライザで評価された。そして、それは自動的に区別して、糊付けして、異なる血液菌体群を計数する。強化チップは、WBCからRBCの分離を成し遂げた、 そこにおいて、WBC分数は、有核細胞の>99%保持を有した、>RBCの99%の減少および血小板の>97%減少。これらの細胞分画の代表的なヒストグラムは、図53に示される。ルーチン細胞学は、WBCおよびRBC分数(図54)の高い濃縮効果を確認した。
上皮細胞と白血球を区別するために試験において、フルオレセインのラベルが付いたCD45パン白血球単クローン抗体の原液の0.5mlは、捕獲モジュールに通過して、30分間の室温で暖められた。モジュールは解放された抗体を除去するためにバッファによって洗われた、そして、細胞は1%のパラホルムアルデヒドを有するチップに取り付けられて、蛍光顕微鏡法によって観察された。図55に示すように、上皮細胞は、捕獲モジュールの障壁および床行きだった。CD45パン白血球抗体を有する流通通路の背景染色は見える。そして、明らかに非特異的捕獲の低次のため、そのことはいくつかの着色された白血球である。
装置実施例本発明の好適な装置実施例のための計画は図57Aに示される、そして、この設計を伴う3つの好適な装置実施例に対応するパラメータは図57Bに示される。
これらの実施例は、特に血液から濃縮上皮細胞に役立つ。
非上皮細胞タイプUsingのためのカウントを決定する本発明の中で方法、1は、CTCまたは他の上皮細胞以外の細胞型を計数することに基づいて、診断をすることができる。癌の欠如、存在または進行の診断は、特定の区分のサイズより大きい細胞サンプルの細胞の数に基づいてもよい。例えば、14ミクロン以上の水力学的サイズを有する細胞は、選択されることができる。この区分のサイズは、大部分の白血球を除去する。これらの細胞の性質は、それから下流の分子であるか細胞学的分析で測定されることができる。
EGFR突然変異の検出のための方法
癌患者から血液サンプルは、結果として上皮細胞を含んでいるCTCの豊かなサンプルになって、装置および本発明(例えば実施例1のそれら)の方法を使用して処理されて、分析される。
このサンプルは、それから、潜在的EGFR突然変異を確認するために分析される。
方法は、新しい突然変異の発見と同様に周知の、臨床的に関連したEGFR突然変異の両方の識別ができるようにする。
このプロセスの概要は、図58に示される。
下記は、検出に対する戦略の概略およびEGFR突然変異の確認である:
1) 血液サンプルからシーケンスCTC EGFRメッセンジャーRNA a)精製CTC;
b) CTCから完全リボゾームリボ核酸を浄化する;
c) リボゾームリボ核酸を相補DNAを使用しているリバーストランスクリプターゼに変換する;
d) シークエンシング・テンプレートを生成するための第1および第2のPCR反応を実行するために、結果として生じる相補DNAを使用する;
そして、
e) 入れ子にされたPCRアンプリコンおよびシーケンスEGFRエキソン18―21に対するシークエンシング・テンプレートとしての用途を浄化する。
2) CTCゲノムDNA a)を使用しているリボゾームリボ核酸シーケンスが血液サンプルからCTCを浄化することを確認する;
b) CTCからゲノムDNA(gDNA)を浄化する;
c) エキソン18、19、20および/またはPCR反応を経た21を増幅する;
そして、
d) リボゾームリボ核酸シークエンシングを介して発見される突然変異の連続を確認するために、リアルタイム定量的アレレに特有のPCR反応の結果として生じるPCRアンプリコンを使用する。
a)精製CTC。
CTCは、本発明のサイズ・ベースの濃縮および/または親和性浄化装置のいずれかを使用して分離される。
完全リボゾームリボ核酸は、それから、例えば、Qiagen Micro RNeasyキットまたは他の製造業者からの類似の全体のリボゾームリボ核酸浄化プロトコルを使用している単離されたCTC人口から浄化される;あるいは、石炭酸/クロロホルム抽出およびエタノール沈殿が続くグアニジウム・イソチオシアネート・ホモジナイゼーションのような標準リボゾームリボ核酸浄化プロトコルが、使われることができる。そのような方法は、J.Sambrook(E.F)によって、「分子Cloning ― A Laboratory Manual(Secondエディション) ― 」(1989)に記載されている。FritchおよびT.Maniatis(7.24ページ)。
シークエンシングは、蛍光ジデオキシヌクレオチド混合物を使用しているサンガー・スタイルのシークエンシング反応を経て発生するABIオートメーション化した蛍光シークエンシング機械および蛍光のラベルが付いたDNA塩基配列決定はしごによって実行される。PCR製品は、他のベンダーから得られるQiagen QuickSpin柱、Agencourt AMPure PCR Purification SystemまたはPCR製品浄化キットを使用して精製される。PCRの後、製品は精製されると、集中および純度がそうであるヌクレオチドがNanodrop 7000分光光度計によって決定した、そして、PCR製品濃度は25ng/マイクロリットルの集中に至る。優良な抑制措置として、1.8より大きい紫外光吸光度比(A26o/A28o)を有するPCR製品だけが、シークエンシングのために用いられる。シークエンシング・プライマは、3.2pmol/マイクロリットルの集中に至る。
上のように、CTCは、本発明のサイズ・ベースの濃縮および/または親和性浄化装置のいずれかを使用して分離される。
ゲノムDNAは、the1 Qiagen DNeasy Miniキット、Invitrogen ChargeSwitch gDNAキットまたは他の市販キットを用いて、または、以下のプロトコルで浄化される:
1. 細胞ペレットは、新たに溶解されるかまたは記憶される―8O0C、そして、細胞溶解の直前に解凍する。
2. 500マイクロリットルの5OmMトリスpH 7.9/10OmM EDTA/0.5%SDS(TEバッファ)を加える。
3.=0.5mg/ml最終版[ProtK]を生成して、12.5マイクロリットルのProteinase K(IBI5406、20mg/ml)を加える。
4. 回転保育器の55度C前夜に孵化する。
5. 20マイクロリットルのRNアーゼ・カクテル(500u/mlのRNアーゼA+20,000u/milliliter RNアーゼTl、Ambion#2288)を加えて、370Cで4時間を暖める。
6.Phenol(コダック、pH 8が平衡させたトリス)によって抽出して、混合物に振動して、卓上の遠心機の5分を回転させる。
7. 水相を新しい管へ転送する。
8. 石炭酸/クロロホルム/イソアミルアルコールを有する抽出物(EMD、25:
24:1つの比率、pH 8が平衡させたトリス)混じって、卓上の遠心機の5分を回転させるために、振動する。
9. 50マイクロリットルの3M NaOAc pH=6を加える。
10. 500マイクロリットルのEtOHを加える。
11. 混じるために、振動する。
沈殿するDNAの列は、見えてもよい。
予想されたDNA濃度が非常に低い場合、キャリア・ヌクレオチド(通常イースト運搬RNA)を加える。
12. 卓上の遠心機の最大速度で、1分を回転させる。
13. 上澄を取り除く。
14. 500マイクロリットル70%のEtOH(室温(RT))を加える
15. 混じるために、振動する。
16.卓上の遠心機の最大速度で、1分を回転させる。
17. Teを加える前に、10―20分を空気乾燥する。
18. 400マイクロリットルのTeにおいて再懸濁する。
10分間の650Cで孵化する、そして、Nanodrop上の定量化の前に終夜RTで去る。
ホットスタート入れ子にされたPCR増幅はステップIdで先に述べた通りに行われる。但し、次の場合は除く−増幅の入れ子にされた回がない。
最初のPCRステップは、増幅の間、allele―特殊情報の可能な損失を最小化するために、対数期の間、停止されることができる。
EGFRエキソン18―21の展開のために使用するプライマ・セットは、表6(また、Paezその他を参照、Science 304:1497―1500(補助機材)(2004))にリストされる。
本当のTime PCRは、入力DNA濃度の8つのログの上の対立形質のシーケンスの定量化を提供する;このように、汚い人口のヘテロの突然変異さえ、この方法を使用して容易に検出される。
TaqManを用いて5´ヌクレアーゼが試験すること、野生型配列のために、または、周知のEGFR突然変異のために特有のアレレに特有の標識プローブは、開発される。
オリゴヌクレオチドは容易にマッチと不適当な組合せを区別する融解温度を有するように設計されている、そして、Real Time PCR状況は野生型および突然変異対立遺伝子を区別するために最適化される。すべてのReal Time PCR反応は、三通りにおいて実施される。
EGFRが背景白血球においてでなく目標癌細胞において表される場合、実施例4のプロトコルは最も役立つだろう。EGFRメッセンジャーRNAが白血球に存在するかどうか検査するために、いくつかのPCR実験は、実行された。4つのプライマーセット(表8に示される)は、4つの対応する遺伝子を増幅するように設計されていた:
1) BCKDK(分枝鎖a―ketoacidデヒドロゲナーゼ複合体キナーゼ)−全ての種類の細胞(白血球および腫瘍細胞のための正の制御)において表される「家事」遺伝子;
2) CD45−白血球(白血球のための正の制御および腫瘍細胞のための陰性対照)において、特に表される;
3) EpCaM−上皮細胞(白血球のための陰性対照および腫瘍細胞のための正の制御)において、特に表される;
そして、
4) EGFR−検討される目標メッセンジャーRNA。
実施例4に記載されている分析法の感度を決定するために、入力NSCLC細胞系合計リボゾームリボ核酸のさまざまな量はテストされた。そして、100pgから50ngにわたった。第1および第2のEGFR PCR反応(ステップId、実施例4)の結果は、図63に示される。第1のPCR反応は1ngの入力リボゾームリボ核酸を検出するために十分に感度が高いことを示した。その一方で、2回は感度を100pg以下の入力リボゾームリボ核酸に増やした。これは10セル、7―に対応する。そして、極めて希釈したサンプルさえこの分析を使用している検出可能な信号を生成することができることを証明する。
図64Aに示すように、PCR反応の第1のセットは、(増幅が全例で起こると共に)疑似バンドが最高汚染レベルで現れることを証明した。しかしながら、図64Bに示すように、PCR反応の第2の、入れ子にされた一組の後、所望の特定のアンプリコンが、最高汚染レベルでさえ疑似バンドなしでできた。従って、この実施例は、目標リボゾームリボ核酸がテストされているサンプルの完全リボゾームリボ核酸の小さい何分の1を占めるときでも、本願明細書において記載されているEGFR PCR分析が効果的なことを証明する。
これらのステップは、第2の血液サンプルおよび第2のチップで繰り返された。
H1650および白血球相補DNAとともにchiplおよびchip2リボゾームリボ核酸から合成される相補DNAは、EGFR_5(前方のプライマ、5.prime.―GTTCGGCACGGTGTATAAGG―S´)と同様に2つのプライマーセットを使用して増幅されるPCR、CD45_1およびCD45_2(表8)であった(配列番号である52) そして、EGFR_6(復帰プライマ、5´―CTGGCCATCACGTAGGCTTC―S´)(配列番号である53).138の塩基対野生型が増幅したEGFR_5およびEGFR_6生産物は分解する、そして、123の塩基対変異体は1650セル、Hの断片を増幅した。PCR製品は、2.5%のアガロースゲルに分離された。図65に示すように、EGFR分析が強くて、非常に精製されたサンプルを必要としないことを証明して、高い白血球背景にもかかわらず、EGFR野生型および変異体増幅された断片は、直ちに検出された。
健康な血液の検体が腫瘍細胞によってスパイクをつけた捕獲モジュールを含んでいる本発明の装置は、能力が腫瘍細胞を血液から豊かにして、とるためにテストされた。
選ばれた区分のサイズより大きな本発明の一実施例、CTCまたは他の細胞において、サブ配列の配列に配置される障壁を含む装置を使用して捕えられることができる。
サブ配列はわずかな食違いまたは平坦でない間隔を有する分野を通じて配列される。そして、まず最初に、フローラインにおける変化を導いて、障壁を有する細胞の相互作用を促すために設計される。図66 Aに示すように、この装置の1つの効果は、各サブ配列が流路が狭くなる領域を引き起こすということである。図に示される配列において、障壁間の規則的なギャップは46マイクロメートルである。その一方で、狭くなるすきまは17マイクロメートルである。配列およびサブ配列は、結果としていかなる望ましい間隙の大きさ(規則的なすきまに関する狭くなるすきまのいかなる所望の密度と同様にも)にもなるために変化することができる。
捕獲モジュール・チップ(図57C)は、Hl 650肺癌細胞の試料を処理するために用いた。捕獲モジュールのパラメータは、以下の通りである:チップ寸法は、66.0x24.9ミリメートルである;障壁分野寸法は、51.3x18.9ミリメートルである;障壁直径は、104マイクロメートルである;ポート寸法は、書類の表面上の2.83x2.83ミリメートルおよび裏面上の1.66×1.66mmである;基質は、シリコンである;そして、エッチング深は、100マイクロメートルである。Hl 650肺癌細胞は、バフィーコートに10,000セル/mlで釘で打ちつけられて、1.6ml/時間(66G図)に動いた。約12,700のHlは、650セル、装置を通過した。
装置は、放射面のほぼ7,230の捕獲場所を含んだ。実験後のH1650細胞の収率は16%であった。そして、利用できる捕獲場所の実質部分がH1650細胞によって占められたことを示した。
癌細胞(細胞系が装置(図67A)を計数しているベックマンCoulterモデルZ2で渡されたいくつかの癌)の粒度分布を決定する癌細胞In$orderの粒度分布。
この実験においてテストされた細胞系はH358、H1650、H1975、HT29およびMCF7細胞を含んだ。そして、それはコロン、肺および乳がん細胞を含む。
図67Aが示すように、これらの細胞系の各々は大部分の白血球より大きい細胞から成る。各癌細胞線の粒度分布は、各々と類似していて、示される白血球の配布から、よく切り離される。サイズ(図67C)の8、10または12マイクロメートル以下のさえ細胞の小さい少数だけについては、癌細胞(図67B)の粒度分布のクローズアップは、各ケースの細胞の一般にガウス配布を明らかにする。これらのデータは、サイズに基づいて他の血液細胞から強いサポートをCTCの強化の原則に対して提供する。
本発明は、様々な細胞キャプチャ装置および方法を含む。実施例において、本発明のキャプチャ装置は、図68Aに示すように職能化された表面(例えばガラス顕微鏡スライド)を利用する。スライドは、抗体または興味がある細胞型に特有の他の捕捉残基によって(例えば標準化学を用いてCTC)職能化されることができる。装置は、以下を含む:
サンプル流体室。そして、それは、例えば、室の一番下上の職能化されたスライドで、10ml以上の収容力を有することができる。いかなる流体(例えば血液または血液分数)も、処理のための室の中に配置されることができる。
流体室(それは空気で満たされることができる)は、あるいは満たされて、装置の主チャンバ内で流体運動を引き起こすために空にされる。気室は、それらを切り離している柔軟壁を有されて、満たされることができて、いかなる機構も使用して空にされる。装置は細胞サンプルまたは他の流体サンプルを起動させる。そして、沈殿させられた電池を転落して、職能化された表面上の捕捉部位の妨害物を防止させ続ける。
そして、もう一度それを高い遠心力(図69C)に従属させること。このステップは、非常に、職能化された表面に取り付けられるままである細胞を汚染することの数を減らす。
実施例において、白血球のような細胞を汚染することに特有の抗体は興味がある(図69D)細胞を捕獲するために用いる表面の反対側に職能化された表面に連結することができる。それによって、汚染している細胞を捕獲して、更に興味がある捕獲された細胞の汚染を最小化する。他のバリエーションにおいて、興味がある細胞を捕獲するために用いる職能化された表面はある角度に傾けられることができる。そして、結果として、遠心力(69E図)の垂直な成分に加えて、平坦面に沿って転がっている電池を動かす遠心性の力の成分になる。転がっている電池を動かす遠心力の成分は、細胞のクラスタを広げるのを助けて、細胞捕獲の効率を上昇させる。
キャプチャ装置
障壁(図7OAおよび記載されている上記)を含む本発明のパワー系統において、大細胞は一般に障壁を有する多数の相互作用を有する。その一方で、小細胞は障壁との最小の接触を有する装置の中を流れることが可能である。
配列された障壁の表面に取り付けられる抗体または他の結合部分を含むキャプチャ装置は、類似の原理を使用して設計されることができて、サイズおよび親和性選択性を結合する。
上記の仕様において記載のすべての刊行物、特許および特許出願は、本願明細書に引用したものとする。ここに記載した本発明の方法とシステムに本発明の範囲および精神から垂離することなく各種の改変および変更を加えることは当業者であれば自明のことであろう。本発明が特定実施例と関連して記載されていたにもかかわらず、それは、請求されてその本発明を理解されなければならないこの種の特定実施例に過度に制限してはならない。実際、当業者にとって明らかである本発明を実施するための記載されているモードのさまざまな変更態様は、本発明の範囲内のことを目的とする。
Claims (304)
- 細胞サンプルにおいて癌細胞を検出する方法であって、該方法は、
(a)該癌細胞を第1の方向に方向付けて該癌細胞について濃縮された第1の出力サンプルを産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて該第2の細胞について濃縮された第2の出力サンプルを産生する構造を備えるチャンネルを備えるデバイスに、該細胞サンプルを導入する工程;および
(b)該第1の出力サンプルにおいて該癌細胞の存在または非存在を検出する工程;
を包含する、方法。 - 前記構造物は、ギャップのネットワークを形成する障害物のアレイを備える、請求項1に記載の方法。
- 前記障害物は、前記癌細胞を選択的に捕捉し得る、請求項2に記載の方法。
- 前記細胞サンプルは、血液サンプルである、請求項1に記載の方法。
- 工程(b)は、前記第1の出力サンプルを前記癌細胞についてのマーカーに対する抗体と反応させる工程を包含する、請求項1に記載の方法。
- 前記マーカーは、表1から選択される、請求項5に記載の方法。
- 工程(b)は、前記第1の出力サンプルにおいて細胞の数を決定する工程を包含する、請求項1に記載の方法。
- 前記決定する工程は、前記第1の出力サンプルにおいてDNAの総量を決定する工程を包含する、請求項7に記載の方法。
- 工程(b)は、前記第1の出力サンプルにおいて前記癌細胞の数を決定する工程を包含する、請求項1に記載の方法。
- 請求項9に記載の方法であって、該方法は、前記細胞サンプルにおいて内皮細胞の数を決定する工程をさらに包含する、方法。
- 前記内皮細胞に対する前記癌細胞の比を決定する工程をさらに包含する、請求項10に記載の方法。
- 工程(b)は、前記第1の出力サンプルにおいてDNA中の変異またはRNA中の変異を検出する工程を包含する、請求項1に記載の方法。
- 前記変異は、表1に列挙されるポリペプチドをコードする遺伝子に存在する、請求項12に記載の方法。
- 工程(b)は、前記第1の出力サンプルにおいてタンパク質のリン酸化、タンパク質のグリコシル化、DNAのメチル化、マイクロRNAのレベル、または細胞の形態を分析する工程を包含する、請求項1に記載の方法。
- 工程(b)は、前記第1の出力サンプルにおいてミトコンドリアDNA、テロメラーゼ、または核マトリックスタンパク質を検出する工程を包含する、請求項1に記載の方法。
- 工程(b)は、前記第1の出力サンプルにおいて1種以上のミトコンドリア異常を検出する工程を包含する、請求項1に記載の方法。
- 工程(b)は、前記第1の出力サンプルの細胞において核周囲部の区画の存在または非存在を検出する工程を包含する、請求項1に記載の方法。
- 工程(b)は、前記第1の出力サンプルの遺伝子発現分析、インセルPCR、または蛍光インサイチュハイブリダイゼーションを行う工程を包含する、請求項1に記載の方法。
- 前記遺伝子発現分析は、前記癌細胞の起源の組織を決定するために使用される、請求項18に記載の方法。
- 前記遺伝子発現分析は、単一の癌細胞に対して行われる、請求項18に記載の方法。
- 前記細胞サンプルは、1種以上の前駆内皮細胞を含み、そして該前駆内皮細胞の少なくとも1種は、前記第1の出力サンプルに存在する、請求項1に記載の方法。
- 請求項1に記載の方法であって、前記デバイスは、第1の入口と、第1の出口と、第2の出口とを備える連続フローデバイスを備え、前記細胞サンプルは、該第1の入口に適用され、前記第1の出力サンプルは、該第1の出口から流出し、そして前記第2の出力サンプルは、該第2の出口から流出する、方法。
- 前記第2の細胞は、非癌細胞を含む、請求項22に記載の方法。
- 前記デバイスは、第2の入口を備え、そして第2の流体が、該第2の入口に適用される、請求項22に記載の方法。
- 前記第2の流体は、緩衝液、溶解試薬、核酸増幅試薬、容量オスモル濃度調節試薬、標識試薬、保存剤、または固定試薬を含む、請求項24に記載の方法。
- 細胞サンプルにおいて癌細胞を検出する方法であって、該方法は、
(a)磁性粒子を使用せずに該細胞サンプルから該癌細胞の1種以上を濃縮する工程であって、該濃縮する工程が、細胞のサイズ、細胞の形状、または細胞の変形能に基づく、工程;および
(b)該濃縮された癌細胞の数を決定する工程;
を包含する、方法。 - 細胞サンプルにおいて癌細胞を検出する方法であって、該方法は、
(a)抗体またはそのフラグメントを使用せずに該細胞サンプルから該癌細胞の1種以上を濃縮する工程であって、該濃縮する工程が、細胞のサイズ、細胞の形状、または細胞の変形能に基づく、工程;および
(b)該濃縮された癌細胞の数を決定する工程;
を包含する、方法。 - 請求項26または27に記載の方法であって、該方法は、
(i)前記細胞サンプルから1種以上の内皮細胞を濃縮する工程;および
(ii)該濃縮された内皮細胞の数を決定する工程;
を包含する、方法。 - 請求項28に記載の方法であって、該方法は、前記内皮細胞に対する前記癌細胞の比を決定する工程をさらに包含する、方法。
- 工程(b)は、前記濃縮された癌細胞を計測する工程を包含する、請求項26または27に記載の方法。
- 工程(b)は、前記濃縮された癌細胞においてDNAの総量を決定する工程を包含する、請求項26または27に記載の方法。
- 第2の細胞サンプルを用いて工程(a)および工程(b)を繰り返す工程をさらに包含する、請求項26または27に記載の方法。
- 前記細胞サンプルおよび前記第2の細胞サンプルは、単一の被験体から採取される、請求項32に記載の方法。
- 前記検出する工程は、前記第1の出力サンプルのハイパースペクトルイメージングを包含する、請求項1に記載の方法。
- 前記細胞サンプルは、工程(a)の前または工程(a)と同時に、前記癌細胞を優先的に標識する標識試薬に接触される、請求項1に記載の方法。
- 請求項35に記載の方法であって、前記標識試薬は、ビーズを含み、標識された癌細胞の流体力学的サイズは、該標識の非存在下での該癌細胞の流体力学的サイズよりも少なくとも10%大きい、方法。
- 被験体において病態を診断するための方法であって、該方法は、
(a)1種以上の癌細胞を第1の方向に方向付けて該癌細胞について濃縮された第1の出力サンプルを産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて該第2の細胞について濃縮された第2の出力サンプルを産生する構造を備えるチャンネルを備えるデバイスに、該被験体由来の細胞サンプルを導入する工程;
(b)該第1の出力サンプルにおいて該癌細胞の存在または非存在を検出する工程;および
(c)工程(b)の結果に基づいて該病態の存在または非存在を診断する工程;
を包含する、方法。 - 前記診断の前に前記被験体の一部を画像化する工程をさらに包含し、該診断が、さらに、該画像化する工程の結果に基づく、請求項37に記載の方法。
- 前記画像化する工程は、コンピュータ連動断層撮影、陽電子断層撮影法、または磁気共鳴画像法を包含する、請求項38に記載の方法。
- 請求項37に記載の方法であって、該方法は、
(i)前記細胞サンプルにおいて内皮細胞の数を決定する工程;および
(ii)該内皮細胞に対する前記癌細胞の比を決定する工程であって、前記診断が、さらに、該比に基づく、工程;
をさらに包含する、方法。 - 前記内皮細胞は、前駆内皮細胞を含む、請求項40に記載の方法。
- 前記病態は、血液学的な病態、炎症性の病態、虚血性の病態、腫瘍性の病態、感染、外傷、子宮内膜症、または腎不全である、請求項40に記載の方法。
- 前記チャンネルは、ギャップのネットワークを形成する障害物のアレイを備え、そして流体が、該流体が大きい流束および小さい流束へと不等に分けられるように該ギャップを通って流れる、請求項1に記載の方法。
- 被験体において病態を診断するための方法であって、
(a)1種以上の第1の細胞を第1の方向に方向付けて該第1の細胞について濃縮された第1の出力サンプルを産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて該第2の細胞について濃縮された第2の出力サンプルを産生する構造を備えるチャンネルを備えるデバイスに、該被験体由来の細胞サンプルを導入する工程;
(b)該第1の出力サンプルを分析する工程;および
(c)工程(b)の結果に基づいて該病態の存在または非存在を診断する工程;
を包含する、方法。 - 前記デバイスは、12ミクロンより大きい流体力学的サイズを有する細胞を前記第1の方向に方向付け、かつ12ミクロン以下の流体力学的サイズを有する細胞を前記第2の方向に方向付けるように構成される、請求項44に記載の方法。
- 請求項44に記載の方法。前記デバイスは、14ミクロンより大きい流体力学的サイズを有する細胞を前記第1の方向に方向付け、かつ14ミクロン以下の流体力学的サイズを有する細胞を前記第2の方向に方向付けるように構成される、請求項44に記載の方法。
- 前記デバイスは、5ミクロン以上かつ10ミクロン以下の流体力学的サイズを有する細胞を前記第1の方向に方向付け、かつ10ミクロンより大きい流体力学的サイズを有する細胞を前記第2の方向に方向付けるように構成される、請求項44に記載の方法。
- 前記デバイスは、4ミクロン以上かつ8ミクロン以下の流体力学的サイズを有する細胞を前記第1の方向に方向付け、かつ8ミクロンより大きい流体力学的サイズを有する細胞を前記第2の方向に方向付けるように構成される、請求項44に記載の方法。
- 前記第1の出力サンプルは、前記細胞サンプル中の前記第1の細胞の少なくとも90%を含む、請求項44に記載の方法。
- 工程(a)〜工程(c)を前記被験体由来のさらなる細胞サンプルを用いて1回以上繰り返す工程をさらに包含する、請求項44に記載の方法。
- 前記細胞サンプルは、一定間隔にて得られる、請求項50に記載の方法。
- 前記一定間隔は、1日間、2日間、3日間、1週間、2週間、1ヶ月間、2ヶ月間、3ヶ月間、6ヶ月間、または1年間である、請求項51に記載の方法。
- 前記第1の出力サンプルは、前記細胞サンプルと比較して前記第1の細胞について少なくとも1,000倍濃縮される、請求項44に記載の方法。
- 前記病態は、血液学的な病態、炎症性の病態、虚血性の病態、腫瘍性の病態、感染、外傷、子宮内膜症、または腎不全である、請求項44に記載の方法。
- 前記腫瘍性の病態は、急性リンパ芽球性白血病、急性または慢性のリンパ球性腫瘍または顆粒球性腫瘍、急性骨髄性白血病、急性前骨髄球性白血病、腺癌、腺腫、副腎癌、基底細胞癌、骨の癌、脳の癌、乳癌、気管支の癌、子宮頚部形成異常、慢性骨髄性白血病、結腸癌、類表皮癌、ユーイング肉腫、胆嚢癌、胆石性腫瘍、骨巨細胞腫、多形性神経膠芽腫、ヘアリーセル腫瘍、頭の癌、過形成、増殖性の角膜神経腫瘍、上皮内癌、腸の神経節細胞腫、ランゲルハンス島細胞腫瘍、カポジ肉腫、腎臓癌、喉頭癌、平滑筋の腫瘍、肝臓癌、肺癌、リンパ腫、悪性カルチノイド、悪性高カルシウム血症、悪性黒色腫、マルファン様体型の腫瘍、髄様癌、転移性皮膚癌、粘膜神経腫、菌状息肉腫、骨髄異形成症候群、骨髄腫、首の癌、神経組織の癌、神経芽腫、骨原性肉腫、骨肉腫、卵巣腫瘍、膵臓癌、副甲状腺癌、褐色細胞腫、真性赤血球増加症、原発性脳腫瘍、前立腺癌、直腸癌、腎細胞腫瘍、網膜芽腫、横紋筋肉腫、精上皮腫、皮膚癌、小細胞肺腫瘍、軟部組織肉腫、扁平上皮癌、胃癌、甲状腺癌、局所的な皮膚病変、細網肉腫、およびウィルムス腫瘍からなる群より選択される、請求項54に記載の方法。
- 前記細胞サンプルは、容量が50mL未満である、請求項44に記載の方法。
- 工程(b)は、前記第1の出力サンプルにおいて癌生物マーカーの存在または非存在を検出する工程を包含する、請求項44に記載の方法。
- 前記癌生物マーカーは、表1から選択されるポリペプチド、または該ポリペプチドをコードする核酸である、請求項57に記載の方法。
- 工程(b)は、癌に関連する核酸の存在または非存在を同定する工程を包含する、請求項44に記載の方法。
- 前記核酸は、ゲノムDNA、mRNA、またはマイクロRNAを含む、請求項59に記載の方法。
- 工程(b)は、癌に関連する核酸の発現パターンを分析する工程を包含する、請求項44に記載の方法。
- 前記核酸は、ゲノムDNA、mRNA、またはマイクロRNAを含む、請求項61に記載の方法。
- 前記核酸は、変異を含み、そして該核酸は、表1から選択されるポリペプチドをコードする、請求項59に記載の方法。
- 工程(b)は、癌に関連する細胞表面マーカーを有する細胞の存在または非存在を同定する工程を含む、請求項44に記載の方法。
- 前記細胞表面マーカーは、EpCAM、E−カドヘリン、ムチン−1、サイトケラチン8、EGFR、および白血球関連レセプター(LAR)からなる群より選択される、請求項64に記載の方法。
- 工程(b)は、前記第1の出力サンプルと、該第1の出力サンプル由来の1種以上の細胞を選択的に結合する1種以上の結合部分を有する表面を備えるデバイスとを接触させる工程をさらに包含する、請求項44に記載の方法。
- 前記第1の出力サンプル由来の前記細胞は、上皮細胞または腫瘍性細胞を含む、請求項66に記載の方法。
- 前記腫瘍性細胞の型は、急性リンパ芽球性白血病、急性または慢性のリンパ球性腫瘍または顆粒球性腫瘍、急性骨髄性白血病、前骨髄球性白血病、腺癌、腺腫、副腎癌、基底細胞癌、骨の癌、脳の癌、乳癌、気管支の癌、子宮頚部形成異常、慢性骨髄性白血病、結腸癌、類表皮癌、ユーイング肉腫、胆嚢癌、胆石性腫瘍、骨巨細胞腫、多形性神経膠芽腫、ヘアリーセル腫瘍、頭の癌、過形成、増殖性の角膜神経腫瘍、上皮内癌、腸の神経節細胞腫、ランゲルハンス島細胞腫瘍、カポジ肉腫、腎臓癌、喉頭癌、平滑筋の腫瘍、肝臓癌、肺癌、リンパ腫、悪性カルチノイド、悪性高カルシウム血症、悪性黒色腫、マルファン様体型の腫瘍、髄様癌、転移性皮膚癌、粘膜神経腫、菌状息肉腫、骨髄異形成症候群、骨髄腫、首の癌、神経組織の癌、神経芽腫、骨原性肉腫、骨肉腫、卵巣腫瘍、膵臓癌、副甲状腺癌、褐色細胞腫、真性赤血球増加症、原発性脳腫瘍、前立腺癌、直腸癌、腎細胞腫瘍、網膜芽腫、横紋筋肉腫、精上皮腫、皮膚癌、小細胞肺腫瘍、軟部組織肉腫、扁平上皮癌、胃癌、甲状腺癌、局所的な皮膚病変、細網肉腫、およびウィルムス腫瘍からなる群より選択される癌に関連する、請求項67に記載の方法。
- 前記癌は、甲状腺癌ではない、請求項68に記載の方法。
- 前記結合部分は、ポリペプチドを含む、請求項66に記載の方法。
- 前記ポリペプチドは、抗体またはそのフラグメントを含む、請求項70に記載の方法。
- 前記抗体またはそのフラグメントは、モノクローナル性である、請求項71に記載の方法。
- 前記モノクローナル抗体またはそのフラグメントは、EpCAMに結合する、請求項72に記載の方法。
- 前記構造は、ギャップのネットワークを形成する障害物のアレイを含む、請求項44に記載の方法。
- 前記障害物は、前記第1の細胞を選択的に捕捉し得る、請求項74に記載の方法。
- 前記障害物の間のギャップは、15ミクロンより大きいか、20ミクロンより大きいか、または60ミクロン未満である、請求項74に記載の方法。
- 、請求項44に記載の方法。前記細胞サンプルは、血液、汗、涙、耳の流出物、痰、リンパ、骨髄懸濁物、尿、唾液、精液、経血、脳脊髄液、脳の流体、腹水、乳汁、呼吸器の分泌物、腸もしくは尿生殖器の管、羊水、または水サンプル.
- 工程(b)は、前記第1の出力サンプルのサイズ分布を分析する工程を包含する、請求項44に記載の方法。
- 工程(b)は、前記第1の細胞の数を決定する工程を包含する、請求項44に記載の方法。
- 目的とする病態に関連する細胞パターンを同定するための方法であって、該方法は、
(a)複数のコントロール被験体の各々および該目的とする病態を有する複数の症例被験体の各々から細胞サンプルを得る工程;
(b)12ミクロンより大きい流体力学的サイズを有する細胞をそれぞれの該細胞サンプルからサイズによって濃縮する工程;
(c)工程(b)で濃縮された細胞を分析する工程;ならびに
(d)工程(c)で得られた結果を使用して関連研究を行なう工程;
を包含する、方法。 - 被験体において病態を診断するための方法であって、該方法は、
(a)該病態に関連する細胞パターンを提供する工程;
(b)該被験体から細胞サンプルを得る工程;
(c)12ミクロンより大きい流体力学的サイズを有する細胞をそれぞれの該細胞サンプルからサイズによって濃縮する工程;
(d)工程(c)で濃縮された細胞を分析する工程;および
(e)工程(a)の該細胞パターンと工程(d)の該分析とに基づいて該被験体における該病態の存在または非存在を診断する工程;
を包含する、方法。 - 工程(c)は、工程(b)で濃縮された前記細胞においてRNAレベルを検出する工程を包含する、請求項80に記載の方法。
- 前記RNAは、mRNAまたはマイクロRNAを含む、請求項82に記載の方法。
- 前記複数の症例被験体は、少なくとも50の症例被験体を含み、そして前記複数のコントロール被験体は、少なくとも50のコントロール被験体を含む、請求項80に記載の方法。
- 工程(c)は、工程(b)で濃縮された前記細胞の数を決定する工程を包含する、請求項80に記載の方法。
- 工程(c)は、インピーダンス、光吸収、光散乱、および静電容量からなる群より選択される細胞の特徴を利用する、請求項85に記載の方法。
- 工程(c)は、工程(b)で濃縮された前記細胞のサイズ分布を分析する工程を包含する、請求項80に記載の方法。
- 工程(c)は、前記サイズ分布を決定するために顕微鏡、細胞計数器、磁石、生体腔レーザー、質量分析器、PCRデバイス、RT−PCRデバイス、マトリックス、マイクロアレイ、またはハイパースペクトルイメージングシステムを使用する工程を包含する、請求項87に記載の方法。
- 前記細胞サンプルは、血液、汗、涙、耳の流出物、痰、リンパ、骨髄懸濁物、尿、唾液、精液、経血、脳脊髄液、脳の流体、腹水、乳汁、呼吸器の分泌物、腸もしくは尿生殖器の管、羊水、または水サンプルを含む、請求項80に記載の方法。
- 工程(c)は、工程(b)で濃縮された前記の起源の組織を決定する工程を包含する、請求項80に記載の方法。
- 工程(c)は、工程(b)で濃縮された前記細胞から、1種以上の上皮細胞、癌細胞、骨髄細胞、胎児細胞、前駆細胞、幹細胞、泡沫細胞、間葉細胞、免疫系細胞、内皮細胞、子宮内膜細胞、結合組織細胞、栄養膜、細菌、真菌、または病原体を同定する工程を包含する、請求項80に記載の方法。
- 工程(e)は、工程(b)で濃縮された前記細胞と、前記第1の細胞を選択的に結合する1種以上の結合部分とを接触させる工程を包含する、請求項80に記載の方法。
- 前記結合部分は、ポリペプチドを含む、請求項92に記載の方法。
- 前記ポリペプチドは、抗体またはそのフラグメントを含む、請求項93に記載の方法。
- 前記抗体またはそのフラグメントは、モノクローナル性である、請求項94に記載の方法。
- 前記モノクローナル抗体またはそのフラグメントは、EpCAMに結合する、請求項95に記載の方法。
- 前記抗体またはそのフラグメントは、抗Ber−Ep4、抗EpCAM、抗E−カドヘリン、抗ムチン−1、抗サイトケラチン8、および抗CD34+からなる群より選択される、請求項94に記載の方法。
- 前記デバイスは、12ミクロンより大きい流体力学的サイズを有する前記細胞を前記第1の方向に方向付け、かつ12ミクロン以下の流体力学的サイズを有する細胞を前記第2の方向に方向付けるように構成される、請求項80に記載の方法。
- 前記デバイスは、工程(b)で濃縮された前記細胞を選択的に捕捉する、請求項80に記載の方法。
- 被験体に適用された薬物処置の効力を決定するための方法であって、該方法は、
(a)該処置の前に該被験体から第1の細胞サンプルを得る工程;
(b)1種以上の第1の細胞を第1の方向に方向付けて該第1の細胞について濃縮された第1の出力サンプルを産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて該第2の細胞について濃縮された第2の出力サンプルを産生する構造を備えるチャンネルを備えるデバイスに、該第1の細胞サンプルを導入する工程;
(c)該第1の出力サンプルを分析する工程;
(d)該薬物処置と同時または該薬物処置の後に該被験体から第2の細胞サンプルを得る工程;
(e)該第2の細胞サンプルに対して工程(b)および工程(c)を繰り返す工程;ならびに
(f)該第1の細胞サンプルに対する工程(c)の結果と該第2の細胞サンプルに対する工程(c)の結果とを比較する工程であって、該比較する工程が、該薬物処置の効力を決定する、工程;
を包含する、方法。 - 前記デバイスは、12ミクロンより大きい流体力学的サイズを有する細胞を前記第1の方向に方向付け、かつ12ミクロン以下の流体力学的サイズを有する細胞を前記第2の方向に方向付けるように構成される、請求項100に記載の方法。
- 前記デバイスは、6ミクロン以上かつ12ミクロン以下の流体力学的サイズを有する細胞を前記第1の方向に方向付け、かつ6ミクロン未満の流体力学的サイズを有する細胞または12ミクロンより大きい流体力学的サイズを有する細胞を前記第2の方向に方向付けるように構成される、請求項100に記載の方法。
- 工程(c)は、前記第1の細胞サンプル由来の第1の細胞または前記第2の細胞サンプル由来の第1の細胞においてRNAレベルを検出する工程を包含する、請求項100に記載の方法。
- 前記RNAは、mRNAまたはマイクロRNAを含む、請求項103に記載の方法。
- 工程(c)は、前記第1の細胞サンプル由来の第1の細胞または前記第2の細胞サンプル由来の第1の細胞の数を決定する工程を包含する、請求項100に記載の方法。
- 工程(d)は、前記第1の細胞サンプル由来の第1の細胞または前記第2の細胞サンプル由来の第1の細胞から、1種以上の上皮細胞、癌細胞、骨髄細胞、胎児細胞、前駆細胞、幹細胞、泡沫細胞、間葉細胞、免疫系細胞、内皮細胞、子宮内膜細胞、結合組織細胞、栄養膜、細菌、真菌、または病原体を同定する工程を包含する、請求項100に記載の方法。
- 前記チャンネルは、ギャップのネットワークを形成する障害物のアレイを備え、そして流体が、該流体が大きい流束および小さい流束へと不等に分けられるように該ギャップを通って流れる、請求項44に記載の方法。
- 細胞サンプルを処理するためのデバイスであって、該デバイスは、
(a)1種以上の第1の細胞を第1の方向に方向付けて該第1の細胞について濃縮された第1の出力サンプルを産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて該第2の細胞について濃縮された第2の出力サンプルを産生する構造を備えるチャンネルであって、ここで該デバイスが、(i)12ミクロンより大きい流体力学的サイズを有する細胞を前記第1の方向に方向付け、かつ12ミクロン以下の流体力学的サイズを有する細胞を前記第2の方向に方向付けるか、あるいは(ii)6ミクロン以上かつ12ミクロン以下の流体力学的サイズを有する細胞を前記第1の方向に方向付け、かつ6ミクロン未満の流体力学的サイズを有する細胞または12ミクロンより大きい流体力学的サイズを有する細胞を前記第2の方向に方向付けるかのいずれかであるように構成される、チャネル;および
(b)該第1の出力サンプルまたは該第2の出力サンプルを分析するための検出モジュールであって、該検出モジュールが、流動工学的に該チャンネルに対して連結される、検出モジュール;
を備える、デバイス。 - 前記デバイスは、6ミクロン以上かつ12ミクロン以下の流体力学的サイズを有する細胞を前記第1の方向に方向付け、かつ6ミクロン未満の流体力学的サイズを有する細胞または12ミクロンより大きい流体力学的サイズを有する細胞を前記第2の方向に方向付けるように構成される、請求項108に記載のデバイス。
- 前記デバイスは、8ミクロン以上かつ10ミクロン以下の流体力学的サイズを有する細胞を前記第1の方向に方向付け、かつ8ミクロン未満の流体力学的サイズを有する細胞または10ミクロンより大きい流体力学的サイズを有する細胞を前記第2の方向に方向付けるように構成される、請求項108に記載のデバイス。
- 前記検出モジュールは、前記第1の細胞において癌に関連するマーカーを同定するように適合される、請求項108に記載のデバイス。
- 前記検出モジュールは、前記第1の細胞を特異的に結合する抗体を含む、請求項108に記載のデバイス。
- 前記抗体は、表1から選択される1種以上のマーカーを特異的に結合する、請求項112に記載のデバイス。
- 前記検出モジュールは、1種以上の上皮細胞、癌細胞、骨髄細胞、胎児細胞、前駆細胞、幹細胞、泡沫細胞、間葉細胞、免疫系細胞、内皮細胞、子宮内膜細胞、結合組織細胞、栄養膜、細菌、真菌、または病原体を検出するように構成される、請求項108に記載のデバイス。
- 前記検出モジュールは、顕微鏡、細胞計数器、磁石、生体腔レーザー、質量分析器、PCRデバイス、RT−PCRデバイス、マトリックス、マイクロアレイ、またはハイパースペクトルイメージングシステムを含む、請求項108に記載のデバイス。
- 細胞サンプルを処理するためのデバイスであって、該デバイスは、
(a)1種以上の第1の癌細胞を第1の方向に方向付けて該癌細胞について濃縮された第1の出力サンプルを産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて該第2の細胞について濃縮された第2の出力サンプルを産生する構造を備えるチャンネル;および
(b)該癌細胞または該第2の細胞を捕捉するための捕捉モジュールであって、該捕捉モジュールが、流動工学的に該チャンネルに対して連結され、そして該捕捉モジュールが、該癌細胞または該第2の細胞を選択的に結合する1種以上の結合部分を備える、捕捉モジュール;
を備える、デバイス。 - 前記構造は、ギャップのネットワークを形成する障害物のアレイを備える、請求項116に記載のデバイス。
- 前記1種以上の結合部分は、1種以上の上皮細胞、癌細胞、骨髄細胞、胎児細胞、前駆細胞、幹細胞、泡沫細胞、間葉細胞、免疫系細胞、内皮細胞、子宮内膜細胞、結合組織細胞、栄養膜、細菌、真菌、または病原体を特異的に結合する、請求項116に記載のデバイス。
- 前記障害物は、前記結合部分を含む、請求項117に記載のデバイス。
- 前記デバイスは、12ミクロンより大きい流体力学的サイズを有する細胞を前記第1の方向に方向付けるように構成される、請求項116に記載のデバイス。
- 前記デバイスは、14ミクロンより大きい流体力学的サイズを有する細胞を前記第1の方向に方向付けるように構成される、請求項116に記載のデバイス。
- 前記デバイスは、16ミクロンより大きい流体力学的サイズを有する細胞を前記第1の方向に方向付けるように構成される、請求項116に記載のデバイス。
- 流体工学的に前記捕捉モジュールに対して連結された細胞計数モジュールをさらに備える、請求項116に記載のデバイス。
- 前記1種以上の結合部分は、ポリペプチドを含む、請求項116に記載のデバイス。
- 前記ポリペプチドは、抗体またはそのフラグメントを含む、請求項124に記載のデバイス。
- 前記抗体またはそのフラグメントは、モノクローナル性である、請求項125に記載のデバイス。
- 前記モノクローナル抗体またはそのフラグメントは、EpCAMに結合する、請求項126に記載のデバイス。
- 細胞サンプルを処理するためのデバイスであって、該デバイスは、1種以上の第1の細胞を第1の方向に方向付けて該第1の細胞について濃縮された第1の出力サンプルを産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて該第2の細胞について濃縮された第2の出力サンプルを産生する構造を備えるチャンネルを備え、該構造は、ギャップのネットワークを形成する障害物のアレイを備え、そして該障害物のうちの少なくともいくつかは、該第1の細胞または該第2の細胞を選択的に結合するモノクローナル抗EpCAM抗体またはそのフラグメントを含む、デバイス。
- 細胞サンプルを処理するためのデバイス、該デバイスは、
(a)該細胞サンプルにおいて細胞をサイズに基づいて濃縮し得る濃縮モジュール;および
(b)該濃縮モジュールによって濃縮された細胞の数を決定するための細胞計数モジュールであって、該細胞計数モジュールが、流体工学的に該濃縮モジュールに対して連結される、濃縮モジュール;
を備える、デバイス - 前記濃縮モジュールは、1種以上の第1の細胞を第1の方向に方向付けて該第1の細胞について濃縮された第1の出力サンプルを産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて該第2の細胞について濃縮された第2の出力サンプルを産生する構造を備えるチャンネルを備える、請求項129に記載のデバイス。
- 前記デバイスは、12ミクロンより大きい流体力学的サイズを有する細胞を前記第1の方向に方向付け、かつ12ミクロン以下の流体力学的サイズを有する細胞を前記第2の方向に方向付けるように構成される、請求項130に記載のデバイス。
- 前記デバイスは、6ミクロン以上かつ12ミクロン以下の流体力学的サイズを有する細胞を前記第1の方向に方向付け、かつ6ミクロン未満の流体力学的サイズを有する細胞または12ミクロンより大きい流体力学的サイズを有する細胞を前記第2の方向に方向付けるように構成される、請求項130に記載のデバイス。
- 前記第1の細胞は、癌細胞を含む、請求項130に記載のデバイス。
- 前記構造は、ギャップのネットワークを形成する障害物のアレイを備える、請求項130に記載のデバイス。
- 前記細胞計数モジュールは、前記第1の出力サンプルまたは前記第2の出力サンプルにおいて細胞の数を決定するためにインピーダンス、光学、または静電容量を利用する、請求項129に記載のデバイス。
- 前記デバイスは、前記第1の出力サンプルまたは前記第2の出力サンプルを可視化するように適合された検出器をさらに備え、該検出器は、流体工学的に前記捕捉モジュールに対して連結される、請求項116、128、または129に記載のデバイス。
- 前記チャンネルは、ギャップのネットワークを形成する障害物のアレイを備え、そして流体が、該流体が大きい流束および小さい流束へと不等に分けられるように該ギャップを通って流れる、請求項108に記載のデバイス。
- 細胞サンプルを処理するためのデバイスであって、該デバイスは、1種以上の第1の細胞を第1の方向に方向付けて該第1の細胞について濃縮された第1の出力サンプルを産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて該第2の細胞について濃縮された第2の出力サンプルを産生する構造を備えるチャンネルを備え、該デバイスは、1時間あたり少なくとも20mLの流体を処理し得る、デバイス。
- 前記構造は、ギャップのネットワークを形成する障害物のアレイを備える、請求項138に記載のデバイス。
- 前記ギャップは、サイズが20ミクロンと100ミクロンとの間である、請求項139に記載のデバイス。
- 前記障害物のアレイは、障害物の互い違いに配置された二次元アレイを含む、請求項139に記載のデバイス。
- 前記障害物のアレイは、複数の列を含み、それぞれの連続した列は、先の列の半分未満の周期でずらされる、請求項139に記載のデバイス。
- 第1の障害物のアレイと直列または並列で障害物の1種以上のさらなるアレイをさらに備える、請求項139に記載のデバイス。
- 前記第1の細胞は、前記第2の細胞よりも大きい平均流体力学的サイズを有する、請求項138に記載のデバイス。
- 前記デバイスは、1時間あたり少なくとも50mLの流体を処理し得る、請求項138に記載のデバイス。
- 前記細胞サンプルは、血液またはその分画を含む、請求項138に記載のデバイス。
- 前記デバイスは、12ミクロンより大きい流体力学的サイズを有する細胞を前記第1の方向に方向付けるように構成される、請求項138に記載のデバイス。
- 前記デバイスは、14ミクロンより大きい流体力学的サイズを有する細胞を前記第1の方向に方向付けるように構成される、請求項138に記載のデバイス。
- 前記デバイスは、16ミクロンより大きい流体力学的サイズを有する細胞を前記第1の方向に方向付けるように構成される、請求項138に記載のデバイス。
- 請求項138に記載のデバイスであって、前記デバイスは、第1の入口と、第1の出口と、第2の出口とを備える連続フローデバイスを備え、前記細胞サンプルは、該第1の入口に適用され、前記第1の出力サンプルは、該第1の出口から流出し、そして前記第2の出力サンプルは、該第2の出口から流出する、デバイス。
- 前記デバイスは、前記第1の細胞について濃縮された第1の出力サンプルを産生し得、該第1の出力サンプルの容量は、前記細胞サンプルの容量よりも小さい、請求項138に記載のデバイス。
- 前記第1の出力サンプルは、前記細胞サンプル中の前記第1の細胞の少なくとも80%を含む、請求項138に記載のデバイス。
- 前記第2の出力サンプルは、前記細胞サンプル中の前記第1の細胞の20%未満を含む、請求項138に記載のデバイス。
- 前記デバイスは、第2の入口を備え、そして第2の流体が、該第2の入口に適用される、請求項150に記載のデバイス。
- 前記第1の細胞は、上皮細胞、癌細胞、骨髄細胞、胎児細胞、前駆細胞、幹細胞、泡沫細胞、間葉細胞、免疫系細胞、内皮細胞、子宮内膜細胞、結合組織細胞、栄養膜、細菌、真菌、または病原体を含む、請求項138に記載のデバイス。
- 請求項138に記載のデバイスであって、該デバイスは、流体工学的に前記チャンネルに対して連結された検出器モジュールをさらに備える、デバイス。
- 前記検出器モジュールは、顕微鏡、細胞計数器、磁石、生体腔レーザー、質量分析器、PCRデバイス、RT−PCRデバイス、マトリックス、マイクロアレイ、またはハイパースペクトルイメージングシステムを備える、請求項156に記載のデバイス。
- 前記検出器モジュールは、前記第1の細胞を選択的に結合する標識を検出する、請求項157に記載のデバイス。
- 前記デバイスは、被験体における移植に適合される、請求項138に記載のデバイス。
- 前記デバイスは、被験体の循環系または循環系の近傍における配置に適合される、請求項159に記載のデバイス。
- 流体工学的に被験体の循環系に対して連結され得るシステムであって、該システムは、細胞サンプルを処理するためのデバイスを備え、該デバイスは、1種以上の第1の細胞を第1の方向に方向付けて該第1の細胞について濃縮された第1の出力サンプルを産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて該第2の細胞について濃縮された第2の出力サンプルを産生する構造を備えるチャンネルを備える、システム。
- 前記システムは、チュービングまたは動静脈シャントによって流体工学的に前記循環系に対して連結される、請求項161に記載のシステム。
- 前記システムは、1種以上の被検体を前記循環系から取り出し得る、請求項161に記載のシステム。
- 前記システムは、前記デバイスを通る連続的な血流に適合される、請求項161に記載のシステム。
- 前記デバイスは、使い捨て可能である、請求項161に記載のシステム。
- 被検体を細胞サンプルから枯渇させるための方法であって、該方法は、細胞サンプルを処理するためのデバイスに該細胞サンプルを導入する工程を包含し、該デバイスは、1種以上の第1の細胞を第1の方向に方向付けて該第1の細胞について濃縮された第1の出力サンプルを産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて該第2の細胞について濃縮された第2の出力サンプルを産生する構造を備えるチャンネルを備え、該第1の出力サンプルまたは該第2の出力サンプルは、該細胞サンプルと比較して該被検体について枯渇している、方法。
- 前記細胞サンプルは、血液、汗、涙、耳の流出物、痰、リンパ、骨髄懸濁物、尿、唾液、精液、経血、脳脊髄液、脳の流体、腹水、乳汁、呼吸器の分泌物、腸もしくは尿生殖器の管、羊水、または水サンプルを含む、請求項166に記載の方法。
- 前記細胞サンプルは、血液学的な病態、炎症性の病態、虚血性の病態、腫瘍性の病態、感染、外傷、子宮内膜症、または腎不全を患う被験体から採取される、請求項166に記載の方法。
- 前記腫瘍性の病態は、急性リンパ芽球性白血病、急性または慢性のリンパ球性腫瘍または顆粒球性腫瘍、急性骨髄性白血病、急性前骨髄球性白血病、腺癌、腺腫、副腎癌、基底細胞癌、骨の癌、脳の癌、乳癌、気管支の癌、子宮頚部形成異常、慢性骨髄性白血病、結腸癌、類表皮癌、ユーイング肉腫、胆嚢癌、胆石性腫瘍、骨巨細胞腫、多形性神経膠芽腫、ヘアリーセル腫瘍、頭の癌、過形成、増殖性の角膜神経腫瘍、上皮内癌、腸の神経節細胞腫、ランゲルハンス島細胞腫瘍、カポジ肉腫、腎臓癌、喉頭癌、平滑筋の腫瘍、肝臓癌、肺癌、リンパ腫、悪性カルチノイド、悪性高カルシウム血症、悪性黒色腫、マルファン様体型の腫瘍、髄様癌、転移性皮膚癌、粘膜神経腫、菌状息肉腫、骨髄異形成症候群、骨髄腫、首の癌、神経組織の癌、神経芽腫、骨原性肉腫、骨肉腫、卵巣腫瘍、膵臓癌、副甲状腺癌、褐色細胞腫、真性赤血球増加症、原発性脳腫瘍、前立腺癌、直腸癌、腎細胞腫瘍、網膜芽腫、横紋筋肉腫、精上皮腫、皮膚癌、小細胞肺腫瘍、軟部組織肉腫、扁平上皮癌、胃癌、甲状腺癌、局所的な皮膚病変、細網肉腫、およびウィルムス腫瘍からなる群より選択される、請求項166に記載の方法。
- 被験体において病態を診断するための方法であって、該方法は、
(a)細胞サンプルを処理するためのデバイスに該被験体由来の細胞サンプルを導入する工程であって、該デバイスは、1種以上の第1の細胞を第1の方向に方向付けて該第1の細胞について濃縮された第1の出力サンプルを産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて該第2の細胞について濃縮された第2の出力サンプルを産生する構造を備えるチャンネルを備え、該デバイスは、1時間あたり少なくとも20mLの流体を処理し得る、工程;
(b)該第1の出力サンプルを分析する工程;および
(c)工程(b)の結果に基づいて該病態の存在または非存在を診断する工程;
を包含する、方法。 - 工程(b)は、前記第1の出力サンプルの細胞を、接着、移動、結合、形態、分裂、遺伝子発現のレベル、および体細胞変異の存在からなる群より選択される1種以上の特徴について分析する工程を包含する、請求項170に記載の方法。
- 工程(b)は、表1から選択される1種以上のマーカーの存在または非存在を検出する工程、表1から選択される1種以上のマーカーをコードする核酸において変異の存在または非存在を検出する工程、表1から選択される1種以上のマーカーをコードする核酸において欠失の存在または非存在を検出する工程、表1から選択される1種以上のマーカーの発現のレベルを検出する工程、または前記第1の出力サンプルにおいてマイクロRNAのレベルを検出する工程を包含する、請求項170に記載の方法。
- 前記病態は、血液学的な病態、炎症性の病態、虚血性の病態、腫瘍性の病態、感染、外傷、子宮内膜症、または腎不全である、請求項170に記載の方法。
- 工程(b)は、前記第1の出力サンプルにおいて前記第1の細胞の数を決定する工程を包含する、請求項170に記載の方法。
- 前記チャンネルは、ギャップのネットワークを形成する障害物のアレイを備え、そして流体が、該流体が大きい流束および小さい流束へと不等に分けられるように該ギャップを通って流れる、請求項138に記載のデバイス。
- 細胞サンプルを処理するためのデバイスであって、該デバイスは、ギャップのネットワークを形成する障害物のアレイを備えるチャンネルを備え、流体が、該流体が大きい流束および小さい流束へと不等に分けられるように該ギャップを通って流れ、そして該アレイは、上皮細胞を、該アレイにおける流れの平均した方向に平行ではない方向に方向付けるように構成される、デバイス。
- 前記デバイスは、2個の入口をさらに備える、請求項176に記載のデバイス。
- 前記デバイスは、2個の出口をさらに備える、請求項176に記載のデバイス。
- 請求項176に記載の第2のデバイスをさらに備え、該デバイスは、流体工学的に連結される、請求項176に記載のデバイス。
- 前記ギャップは、上皮細胞を前記出口のうちの1個へと方向付けるようにサイジングされる、請求項178に記載のデバイス。
- 前記ギャップは、上皮細胞を、1つの前記入口に流入する流体から、別の前記入口に流入する流体へと方向付けるようにサイジングされる、請求項177に記載のデバイス。
- 前記障害物は、ポリマー、ケイ素、ガラス、または融合したシリカを含む、請求項176に記載のデバイス。
- 細胞サンプルを処理するためのデバイスであって、該デバイスは、
a.ギャップのネットワークを形成する障害物のアレイを備えるチャンネルであって、ここで流体が、該流体が大きい流束および小さい流束へと不等に分けられるように該ギャップを通って流れ、その結果、臨界サイズよりも大きい細胞の動きの平均した方向が、流体の流れの平均した方向に平行ではない、チャンネル;
b.正常血球よりも大きい細胞を収集するように配置された出口;および
c.細胞を計測する細胞検出器;
を備える、デバイス。 - 上皮細胞の濃縮されたサンプルを産生する方法であって、該方法は、請求項176に記載のデバイスに細胞サンプルを導入する工程を包含し、ここで、該細胞サンプルが、該デバイスを通って流れる場合、該細胞サンプル中に存在する上皮細胞は、該細胞サンプルの平均した流れの方向に平行ではない方向に方向付けられ、そして該細胞サンプルの別の成分は、該平均した流れの方向に沿って方向付けられ、それによって該他の成分と比較して上皮細胞について濃縮されたサンプルが、産生される、方法。
- 前記他の成分は、赤血球、血小板、白血球、または内皮細胞を含む、請求項184に記載の方法。
- 前記上皮細胞は、直径が8μm〜30μmの範囲である、請求項184に記載の方法。
- 前記細胞は、DNA分析、RNA分析、タンパク質分析、またはメタボローム分析を受け得る、請求項184に記載の方法。
- 前記細胞は、インセルPCRを使用して分析される、請求項187に記載の方法。
- 前記細胞は、存在するサイトケラチンmRNAのレベルを決定することによって分析される、請求項187に記載の方法。
- 前記上皮細胞は、癌細胞である、請求項184に記載の方法。
- 前記癌細胞は、癌幹細胞である、請求項190に記載の方法。
- 前記濃縮されたサンプルの容量が、前記細胞サンプルの容量よりも実質的に小さく、前記上皮細胞の濃縮を生じる、請求項184に記載の方法。
- 溶解緩衝液が、前記デバイスに同時導入され、そして前記濃縮されたサンプルは、上皮細胞の成分を含む、請求項184に記載の方法。
- 交換緩衝液は、前記デバイスに同時導入され、そして前記濃縮されたサンプルは、交換緩衝液を含む、請求項184に記載の方法。
- 前記濃縮されたサンプルと、上皮細胞を優先的に標識する標識試薬とを接触させる工程をさらに包含する、請求項184に記載の方法。
- 前記濃縮されたサンプルにおいて細胞の数を定量する工程をさらに包含する、請求項184に記載の方法。
- 前記デバイスは、前記濃縮されたサンプルにおいて500fLよりも大きい細胞容量の細胞の数を決定する、請求項196に記載の方法。
- 前記デバイスは、前記濃縮されたサンプルにおいて14μmよりも大きい直径の細胞の数を決定する、請求項196に記載の方法。
- 前記デバイス内部の前記細胞の各々に対するずれ応力は、全ての時間において10ダイン/cm2未満である、請求項184に記載の方法。
- 前記細胞サンプルは、上皮細胞を優先的に標識する標識試薬に接触され、ここで該標識試薬は、最初に、前記デバイスに対する該細胞サンプルの導入前または該デバイスに対する該細胞サンプルの導入後のいずれかにおいて該細胞サンプルと合わせられる、請求項184に記載の方法。
- 前記標識試薬は、粒状標識試薬である、請求項200に記載の方法。
- 請求項200に記載の方法であって、該方法は、前記標識された細胞を定量する工程をさらに包含する、方法。
- 前記標識試薬は、量子ドットを含み、そして前記標識された細胞は、2−光子励起を使用して分析される、請求項202に記載の方法。
- 請求項203に記載の方法であって、該方法は、前記濃縮されたサンプルの容積測定を行なう工程をさらに包含する、方法。
- 前記細胞に結合された標識試薬は、該細胞に結合されていないままの標識試薬から分離される、請求項200に記載の方法。
- 前記標識試薬は、前記デバイスに同時導入され、該デバイス内において前記細胞の標識化をもたらす、請求項200に記載の方法。
- 前記標識試薬は、量子ドット、抗体、ファージ、アプタマー、フルオロフォア、酵素、またはビーズを含む、請求項200に記載の方法。
- 前記ビーズは、アフィニティー試薬を含む、請求項207に記載の方法。
- 前記ビーズは、ポリスチレンを含む、請求項207に記載の方法。
- 前記ビーズは、天然に浮遊する、請求項207に記載の方法。
- 前記ビーズは、抗体を含む、請求項207に記載の方法。
- 前記標識試薬は、前記細胞のサイズを増加させる、請求項200に記載の方法。
- 前記サイズが、少なくとも10%増加する、請求項212に記載の方法。
- 前記サイズが、少なくとも100%増加する、請求項212に記載の方法。
- 前記サイズが、少なくとも1000%増加する、請求項212に記載の方法。
- 上皮細胞を定量する方法であって、該方法は、
a.細胞サンプルを提供する工程;
b.請求項176に記載のデバイスに該細胞サンプルを導入して、上皮細胞について濃縮されたサンプルを産生する工程;および
c.該上皮細胞を定量する工程;
を包含する、方法。 - a.請求項176に記載のデバイスに患者由来の細胞サンプルを導入して、正常血球よりも大きい細胞について濃縮されたサンプルを産生する工程;および
b.該サンプルに存在する正常血球よりも大きい細胞の数を決定して、疾患状態を決定する工程;
を包含する、診断方法。 - a.請求項176に記載のデバイスに患者由来の細胞サンプルを導入して、正常血球よりも大きい細胞について濃縮されたサンプルを産生する工程;および
b.疾患状態を決定するために、DNA分析、RNA分析、プロテオーム分析、またはメタボローム分析を該濃縮されたサンプルに対して行なう工程;
を包含する、診断方法。 - 前記DNA分析は、上皮増殖因子レセプター遺伝子において1種以上の変異の存在、および同一性、すなわち非存在を決定する工程を包含する、請求項218に記載の方法。
- 前記DNA分析は、
i.前記濃縮されたサンプルからゲノムDNAを単離する工程;ならびに
ii.前記上皮増殖因子レセプター遺伝子のエキソン18、エキソン19、エキソン20、およびエキソン21のうちの1種以上を増幅する工程;
を包含する、請求項219に記載の方法。 - iii.公知の上皮増殖因子レセプター変異に対応する前記増幅産物の下位領域を増幅し、さらなる増幅産物を生じる工程;
iv.該さらなる増幅産物を配列決定する工程;および
v.該生じた配列を、該上皮増殖因子レセプター遺伝子の野生型配列および該上皮増殖因子レセプター遺伝子の公知の変異の一覧と比較する工程;
をさらに包含する、請求項220に記載の方法。 - 前記正常血球よりも大きい細胞は、上皮細胞を含む、請求項217または218に記載の方法。
- 前記疾患状態は、癌を含む、請求項217または218に記載の方法。
- 前記正常血球よりも大きい細胞は、上皮細胞を含む、請求項183に記載のデバイス。
- 細胞サンプルを処理するためのデバイスであって、前記デバイスは、ギャップのネットワークを形成する障害物のアレイ上に位置決めされたシーリングを備えるチャンネルを備え、該デバイスは、臨界サイズ以下の直径を有する細胞を該ギャップのネットワークを通して流させ、かつ該シーリングと該障害物との間の該臨界サイズよりも大きい直径を有する細胞を捕捉するように構成される、デバイス。
- 前記臨界サイズは、5ミクロンと20ミクロンとの間である、請求項225に記載のデバイス。
- 前記臨界サイズは、12ミクロンである、請求項225に記載のデバイス。
- 前記臨界サイズは、14ミクロンである、請求項225に記載のデバイス。
- 前記臨界サイズよりも大きい直径を有する細胞は、1種以上の稀な細胞を含む、請求項225に記載のデバイス。
- 前記稀な細胞は、1種以上の上皮細胞、癌細胞、骨髄細胞、胎児細胞、前駆細胞、幹細胞、泡沫細胞、間葉細胞、免疫系細胞、内皮細胞、子宮内膜細胞、結合組織細胞、栄養膜、細菌、真菌、または病原体を含む、請求項229に記載のデバイス。
- 前記シーリングは、透明である、請求項225に記載のデバイス。
- 細胞サンプルにおいて臨界サイズよりも大きい直径を有する細胞を処理する方法であって、該方法は、ギャップのネットワークを形成する障害物のアレイ上に位置決めされたシーリングを備えるチャンネルを備えるデバイスに、該細胞サンプルを導入する工程を包含し、該デバイスは、該臨界サイズ以下の直径を有する細胞を該ギャップのネットワークを通して流し得、かつ該シーリングと該障害物との間の該臨界サイズよりも大きい直径を有する細胞を捕捉し得るように構成される、方法。
- 前記シーリングと前記障害物との間の前記臨界サイズよりも大きい直径を有する前記細胞の存在または非存在を検出する工程をさらに包含する、請求項232に記載の方法。
- 前記シーリングは、透明であり、そして前記検出する工程は、顕微鏡を使用する工程を包含する、請求項233に記載の方法。
- 前記デバイスに、緩衝液、溶解試薬、核酸増幅試薬、容量オスモル濃度調節試薬、標識試薬、保存剤、または固定試薬を導入する工程を、前記細胞サンプルを導入する工程の後にさらに包含する、請求項232に記載の方法。
- 前記容量オスモル濃度調節試薬は、高浸透圧溶液を含む、請求項235に記載の方法。
- 前記高浸透圧溶液は、前記シーリングと前記障害物との間の前記臨界サイズよりも大きい直径を有する前記細胞を放出させる、請求項236に記載の方法。
- 癌に罹患したかまたは癌を発症する危険性があるヒト患者における上皮増殖因子レセプター(EGFR)を標的する処置の効力の可能性を決定するための方法であって、該方法は、
1つの流体力学的サイズの第1の細胞を1つの方向に方向付けて第1の細胞について濃縮された第1の出力を産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて第2の出力を産生する構造を備えるチャンネルを備えるデバイスに、該患者由来の細胞サンプルを適用する工程;および
該第1の出力の細胞または該第2の出力の細胞において、EGFR遺伝子における少なくとも1種の所定の核酸改変体の存在または非存在を検出する工程であって、該少なくとも1種の核酸改変体の存在は、EGFRを標的する処置が有効である可能性が高いことを示す、工程;
を包含する、方法。 - 前記改変体は、前記第1の出力の細胞のerbB1遺伝子のキナーゼドメインに位置し、該改変体は、野生型erbB1遺伝子である、請求項238に記載の方法。
- 前記核酸改変体は、キナーゼ活性を上昇させる、請求項239に記載の方法。
- 前記第1の細胞は、癌細胞である、請求項238に記載の方法。
- 前記癌は、胃腸癌、前立腺癌、卵巣癌、乳癌、頭および首の癌、肺癌、非小細胞肺癌、神経系の癌、腎臓癌、網膜の癌、皮膚癌、肝臓癌、膵癌、生殖器−泌尿器の癌、および膀胱癌からなる群より選択される、請求項240に記載の方法。
- 前記癌は、非小細胞肺癌である、請求項241に記載の方法。
- 前記erbB1遺伝子のキナーゼドメインにおける改変体は、ATP結合ポケットの立体配座構造に影響を及ぼす、請求項239に記載の方法。
- erbB 1の前記キナーゼドメインにおける前記改変体は、エキソン18、エキソン19、エキソン20、およびエキソン21からなる群より選択される前記erbB1遺伝子のエキソンに存在する、請求項239に記載の方法。
- 前記改変体は、エキソン18、エキソン19またはエキソン21に存在する、請求項245に記載の方法。
- 前記erbB1遺伝子のキナーゼドメインにおける改変体は、インフレームの欠失、置換、または挿入である、請求項239に記載の方法。
- 前記少なくとも1種の改変体の存在または非存在の検出は、核酸のセグメントを増幅する工程を包含する、請求項238に記載の方法。
- 前記少なくとも1種の改変体の存在または非存在の検出は、前記erbB1核酸と少なくとも1種の核酸プローブとを接触させる工程を包含し、該少なくとも1種のプローブは、該改変体を含む核酸配列と、選択的ハイブリダイゼーション条件下で優先的にハイブリダイズする、請求項239に記載の方法。
- 少なくとも1種の改変体の存在または非存在の検出は、ポリメラーゼ連鎖反応(PCR)を行って前記erbB1コード配列を含む核酸を増幅する工程、および該増幅された核酸のヌクレオチド配列を決定する工程を包含する、請求項239に記載の方法。
- 前記構造は、前記細胞を分離するギャップのネットワークを形成する障害物の二次元アレイを含む、請求項238に記載の方法。
- 前記第1の出力と、上皮細胞または腫瘍性細胞を選択的に結合する特異的結合部分を備えるデバイスとを接触させる工程をさらに包含する、請求項251に記載の方法。
- 前記特異的結合部分は、抗体またはそのフラグメントである、請求項252に記載の方法。
- 前記抗体は、Ber−Ep4、EpCam、E−カドヘリン、ムチン−1、サイトケラチンまたはCD−34を特異的に結合する、請求項253に記載の方法。
- 患者におけるEGFRを標的する処置の効力の可能性を決定するための方法であって、該方法は、
1つの流体力学的サイズの第1の細胞を1つの方向に方向付けて第1の細胞について濃縮された第1の出力を産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて第2の出力を産生する構造を備えるチャンネルを備えるデバイスに、該患者由来の細胞サンプルを適用する工程;
該細胞をEGFRリガンドによって刺激する工程;および
該第1の細胞においてerbB1遺伝子にコードされるキナーゼのキナーゼ活性を決定する工程であって、コントロールと比較したキナーゼ活性の上昇は、EGFRを標的する処置が有効である可能性が高いことを示す、工程;
を包含する、方法。 - 前記第1の細胞は、癌細胞である、請求項255に記載の方法。
- 前記癌は、胃腸癌、前立腺癌、卵巣癌、乳癌、頭および首の癌、肺癌、非小細胞肺癌、神経系の癌、腎臓癌、網膜の癌、皮膚癌、肝臓癌、膵癌、生殖器−泌尿器の癌、および膀胱癌からなる群より選択される、請求項256に記載の方法。
- 前記癌は、非小細胞肺癌である、請求項256に記載の方法。
- 前記構造は、前記細胞を分離するギャップのネットワークを形成する障害物の二次元アレイを含む、請求項255に記載の方法。
- 前記第1の出力と、上皮細胞または腫瘍性細胞を選択的に結合する特異的結合部分を備える前記デバイスとを接触させる工程をさらに包含する、請求項259に記載の方法。
- 前記特異的結合部分は、抗体またはそのフラグメントである、請求項260に記載の方法。
- 前記抗体は、Ber−Ep4、EpCam、E−カドヘリン、ムチン−1、サイトケラチンまたはCD−34を特異的に結合する、請求項261に記載の方法。
- 前記EGFRを標的する処置は、チロシンキナーゼインヒビターである、請求項255に記載の方法。
- 前記チロシンキナーゼインヒビターは、アニリノキナゾリンである、請求項263に記載の方法。
- 前記アニリノキナゾリンは、合成アニリノキナゾリンである、請求項264に記載の方法。
- 前記合成アニリノキナゾリンは、ゲフィチニブおよびエルロチニブからなる群より選択される、請求項265に記載の方法。
- 癌に罹患したかまたは癌を発症する危険性があるヒト患者における上皮増殖因子レセプター(EGFR)を標的する処置の効力の可能性を決定するための方法であって、該方法は、
1つの流体力学的サイズの第1の細胞を1つの方向に方向付けて第1の細胞について濃縮された第1の出力を産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて第2の出力を産生する構造を備えるチャンネルを備えるデバイスに、該患者由来の細胞サンプルを適用する工程;
エキソン18、エキソン19、エキソン20、またはエキソン21の一部を増幅するポリメラーゼ連鎖反応(PCR)を行うことによってエキソン18、エキソン19、エキソン20、またはエキソン21において少なくとも1種の核酸改変体の存在または非存在を検出する工程;および
該増幅されたエキソン18、エキソン19、エキソン20、またはエキソン21の少なくとも一部を配列決定することによって、該増幅された核酸のヌクレオチド配列を決定する工程であって、野生型erbB1コントロールと比較してエキソン18、エキソン19、エキソン20、またはエキソン21における少なくとも1種のヌクレオチド改変体の存在は、EGFRを標的する処置が有効である可能性が高いことを示す、工程;
を包含する、方法。 - 前記構造は、前記細胞を分離するギャップのネットワークを形成する障害物の二次元アレイを含む、請求項255に記載の方法。
- 前記第1の出力と、上皮細胞または腫瘍性細胞を選択的に結合する特異的結合部分を備えるデバイスとを接触させる工程をさらに包含する、請求項268に記載の方法。
- 前記特異的結合部分は、抗体またはそのフラグメントである、請求項269に記載の方法。
- 前記抗体は、Ber−Ep4、EpCam、E−カドヘリン、ムチン−1、サイトケラチンまたはCD−34を特異的に結合する、請求項270に記載の方法。
- 1つの流体力学的サイズの第1の細胞を1つの方向に方向付けて第1の細胞について濃縮された第1の出力を産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて第2の出力を産生する構造を備えるチャンネルを備えるデバイス;および
EGFR遺伝子において少なくとも1種の核酸変異の存在または非存在を検出するための試薬;
を備える、キット。 - 前記試薬は、核酸および生成物にアニーリングするように設計された少なくとも1つの縮重プライマー対ならびにPCR増幅を行うのに必要とされる試薬を含む、請求項272に記載のキット。
- 前記構造は、前記細胞を分離するギャップのネットワークを形成する障害物の二次元アレイを含む、請求項272に記載のキット。
- 前記第1の出力を受容するように適合されたデバイスをさらに備える請求項274に記載のキットであって、該デバイスは、上皮細胞または腫瘍性細胞を選択的に結合する特異的結合部分を備える、キット。
- 前記特異的結合部分は、抗体またはそのフラグメントである、請求項275に記載のキット。
- 前記抗体は、Ber−Ep4、EpCam、E−カドヘリン、ムチン−1、サイトケラチンまたはCD−34を特異的に結合する、請求項276に記載のキット。
- 前記試薬は、前記EGFRキナーゼドメインタンパク質のATP結合ポケットに結合し得る少なくとも1種のプローブを含む、請求項272に記載のキット。
- 前記試薬は、抗体、抗体フラグメントまたはキメラ抗体を含む、請求項272または278記載のキット。
- 前記試薬は、検出可能な標識をさらに含む、請求項279に記載のキット。
- 患者由来の癌細胞のEGFR遺伝子における二次変異の獲得を予測するための方法であって、該方法は、
該患者由来の細胞サンプルと、1つの流体力学的サイズの第1の細胞を1つの方向に方向付けて第1の細胞について濃縮された第1の出力を産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて第2の出力を産生する構造を備えるチャンネルを備えるデバイスとを接触させる工程;
該第1の細胞と、致死量以下の用量のチロシンキナーゼインヒビターとを接触させる工程;
該チロシンインヒビターの効果に抵抗性である細胞を選択する工程;および
該抵抗性の細胞由来の核酸を二次変異の存在について分析する工程;
を包含する、方法。 - 前記第1の細胞は、erbB1遺伝子の改変体を有する、請求項281に記載の方法。
- 前記構造は、前記細胞を分離するギャップのネットワークを形成する障害物の二次元アレイを含む、請求項281に記載の方法。
- 前記第1の出力と、上皮細胞または腫瘍性細胞を選択的に結合する特異的結合部分を備えるデバイスとを接触させる工程をさらに包含する、請求項273に記載の方法。
- 前記特異的結合部分は、抗体またはそのフラグメントである、請求項284に記載の方法。
- 前記抗体は、Ber−Ep4、EpCam、E−カドヘリン、ムチン−1、サイトケラチンまたはCD−34を特異的に結合する、請求項285に記載の方法。
- 前記細胞は、腫瘍生検から得られる、請求項281に記載の方法。
- 最初に前記第1の細胞と有効量の変異誘発因子とを接触させる工程をさらに包含する、請求項281に記載の方法。
- 前記変異誘発因子は、エチルメタンスルホネート(EMS)、N−エチル−N−ニトロソ尿素(ENU)、N−メチル−N−ニトロソ尿素(MNU)、塩酸ホカルバキシン(Prc)、メチルメタンスルホネート(MeMS)、クロラムブシル(Chi)、メルファラン、塩酸ポルカルバジン、シクロホスファミド(Cp)、ジエチル硫酸 (Et2SO4)、アクリルアミドモノマー(AA)、メチレンメラミン(TEM)、ナイトロジェンマスタード、ビンクリスチン、ジメチルニトロソアミン、N−メチル−N’−ニトロ−ニトロソグアニジン(MNNG)、7,12 ジメチルベンズアントラセン(DMBA)、酸化エチレン、ヘキサメチルホスホラミド、ビスルファン、およびエチルメタンスルホレート(EtMs)からなる群より選択される、請求項288に記載の方法。
- 癌に罹患したかまたは癌を発症する危険性がある患者における上皮増殖因子レセプター(EGFR)を標的する処置の効力の可能性を決定するための方法であって、該方法は、
患者由来の生物学的サンプルと、1つの流体力学的サイズの第1の細胞を1つの方向に方向付けて第1の細胞について濃縮された第1の出力を産生し、かつ1種以上の第2の細胞を第2の方向に方向付けて第2の出力を産生する構造を備えるチャンネルを備えるデバイスとを接触させる工程であって、該第1の細胞は、上皮細胞または腫瘍性細胞である、工程;および
Akt、STAT5、またはSTAT3が該第1の出力由来の該第1の細胞において活性化されているか否かを決定する工程であって、活性化されたAkt、STATS、またはSTATSは、該EGFRを標的する処置が有効である可能性が高いことを示す、工程 - 前記生物学的サンプルは、生検または吸引物である、請求項290に記載の方法。
- 活性化されたAkt、STAT3、またはSTATSは、リン酸化されている、請求項290に記載の方法。
- 前記活性化されたAkt、STAT3、またはSTATSは、免疫学的に決定される、請求項290に記載の方法。
- 前記免疫学的検出方法は、免疫組織化学、免疫細胞学、FACSスキャニング、免疫ブロッティング、ラジオイムノアッセイ、ウェスタンブロッティング、免疫沈降、または酵素結合イムノソルベントアッセイ(ELISA)からなる群より選択される、請求項293に記載の方法。
- 前記免疫学的検出方法は、抗ホスホAkt抗体、抗ホスホSTATS抗体または抗ホスホSTATS抗体を使用した免疫組織化学または免疫細胞化学である、請求項294に記載の方法。
- 前記構造は、前記細胞を分離するギャップのネットワークを形成する障害物の二次元アレイを含む、請求項290に記載の方法。
- 前記第1の出力と、上皮細胞または腫瘍性細胞を選択的に結合する特異的結合部分を備えるデバイスとを接触させる工程をさらに包含する、請求項296に記載の方法。
- 前記特異的結合部分は、抗体またはそのフラグメントである、請求項297に記載の方法。
- 前記抗体は、Ber−Ep4、EpCam、E−カドヘリン、ムチン−1、サイトケラチン、またはCD−34を特異的に結合する、請求項298に記載の方法。
- 前記EGFR遺伝子は、配列番号511の野生型配列を含む、請求項238に記載の方法。
- 前記改変体は、1個以上のヌクレオチド位置において配列番号511と異なる、請求項238に記載の方法。
- 前記EGFR遺伝子によってコードされるポリペプチドは、配列番号512の野生型配列を含む、請求項238に記載の方法。
- 前記改変体によってコードされるポリペプチドは、1個以上のアミノ酸位置において配列番号512と異なる、請求項238に記載の方法。
- 前記改変体は、表3から選択される、請求項238に記載の方法。
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US66841505P | 2005-04-05 | 2005-04-05 | |
| US70383305P | 2005-07-29 | 2005-07-29 | |
| US11/324,041 US20070026418A1 (en) | 2005-07-29 | 2005-12-29 | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US11/323,945 US20070026414A1 (en) | 2005-07-29 | 2005-12-29 | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US11/323,946 US20070026415A1 (en) | 2005-07-29 | 2005-12-29 | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US11/322,790 US20070026413A1 (en) | 2005-07-29 | 2005-12-29 | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US11/323,962 US20070026417A1 (en) | 2005-07-29 | 2005-12-29 | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| PCT/US2006/012778 WO2006108087A2 (en) | 2005-04-05 | 2006-04-05 | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2008538282A true JP2008538282A (ja) | 2008-10-23 |
| JP2008538282A5 JP2008538282A5 (ja) | 2010-10-14 |
Family
ID=37074090
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008505508A Pending JP2008538282A (ja) | 2005-04-05 | 2006-04-05 | 装置および循環腫瘍細胞および他の粒子の濃縮および変更のための方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US20070099207A1 (ja) |
| EP (2) | EP2594631A1 (ja) |
| JP (1) | JP2008538282A (ja) |
| GB (1) | GB2427468B (ja) |
| HK (1) | HK1154422A1 (ja) |
| WO (1) | WO2006108087A2 (ja) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012002447A1 (ja) * | 2010-06-30 | 2012-01-05 | 武田薬品工業株式会社 | 変異ポリヌクレオチドの検出方法 |
| JP2012504956A (ja) * | 2008-10-10 | 2012-03-01 | セントレ ナショナル デ ラ レシェルシェ サイエンティフィーク−ディーエーイー | 細胞ソート・デバイス |
| JP2012529983A (ja) * | 2009-06-19 | 2012-11-29 | コミサリア ア レネルジィ アトミーク エ オ ゼネ ルジイ アルテアナティーフ | 液相間で成分を移動させるマイクロ流体システム及び対応する方法、並びに上記成分を抽出するための上記システムの使用 |
| JP2013505429A (ja) * | 2009-09-21 | 2013-02-14 | マウント シナイ ホスピタル | 甲状腺癌を診断および処置するための方法および組成物 |
| WO2012151501A3 (en) * | 2011-05-05 | 2013-04-25 | Anpac Bio-Medical Science Co., Ltd. | Apparatus for detecting tumor cells |
| JP2017000921A (ja) * | 2015-06-05 | 2017-01-05 | 国立大学法人 東京大学 | 曲げ弾性係数が小さい粒子の分離捕捉装置 |
| JP2021510516A (ja) * | 2018-01-19 | 2021-04-30 | インターナショナル・ビジネス・マシーンズ・コーポレーションInternational Business Machines Corporation | 粒子の精製および分画のための方法およびマイクロ流体装置 |
| US11085923B2 (en) | 2011-03-24 | 2021-08-10 | Anpac Bio-Medical Science Co., Ltd | Micro-devices for disease detection |
| US11458474B2 (en) | 2018-01-19 | 2022-10-04 | International Business Machines Corporation | Microfluidic chips with one or more vias |
| US11566982B2 (en) | 2018-01-19 | 2023-01-31 | International Business Machines Corporation | Microscale and mesoscale condenser devices |
| WO2023189095A1 (ja) * | 2022-03-28 | 2023-10-05 | 国立大学法人茨城大学 | 白血球捕捉デバイス |
| JP7455339B2 (ja) | 2020-09-29 | 2024-03-26 | Nok株式会社 | 白血球捕捉デバイス |
Families Citing this family (78)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6913697B2 (en) | 2001-02-14 | 2005-07-05 | Science & Technology Corporation @ Unm | Nanostructured separation and analysis devices for biological membranes |
| WO2004029221A2 (en) | 2002-09-27 | 2004-04-08 | The General Hospital Corporation | Microfluidic device for cell separation and uses thereof |
| EP1838442B1 (en) | 2005-01-18 | 2013-09-11 | Biocept, Inc. | Cell separation using microchannel having patterned posts |
| US20090136982A1 (en) * | 2005-01-18 | 2009-05-28 | Biocept, Inc. | Cell separation using microchannel having patterned posts |
| US8158410B2 (en) | 2005-01-18 | 2012-04-17 | Biocept, Inc. | Recovery of rare cells using a microchannel apparatus with patterned posts |
| US20060252087A1 (en) * | 2005-01-18 | 2006-11-09 | Biocept, Inc. | Recovery of rare cells using a microchannel apparatus with patterned posts |
| US20070196820A1 (en) | 2005-04-05 | 2007-08-23 | Ravi Kapur | Devices and methods for enrichment and alteration of cells and other particles |
| US8921102B2 (en) | 2005-07-29 | 2014-12-30 | Gpb Scientific, Llc | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US20070059716A1 (en) * | 2005-09-15 | 2007-03-15 | Ulysses Balis | Methods for detecting fetal abnormality |
| US20070059718A1 (en) * | 2005-09-15 | 2007-03-15 | Mehmet Toner | Systems and methods for enrichment of analytes |
| US20110052570A1 (en) * | 2005-10-26 | 2011-03-03 | Children's Medical Center Corporation | Method to prognose response to anti-egfr therapeutics |
| US7695956B2 (en) | 2006-01-12 | 2010-04-13 | Biocept, Inc. | Device for cell separation and analysis and method of using |
| US20080070792A1 (en) | 2006-06-14 | 2008-03-20 | Roland Stoughton | Use of highly parallel snp genotyping for fetal diagnosis |
| US20080050739A1 (en) | 2006-06-14 | 2008-02-28 | Roland Stoughton | Diagnosis of fetal abnormalities using polymorphisms including short tandem repeats |
| WO2007147018A1 (en) * | 2006-06-14 | 2007-12-21 | Cellpoint Diagnostics, Inc. | Analysis of rare cell-enriched samples |
| EP2589668A1 (en) | 2006-06-14 | 2013-05-08 | Verinata Health, Inc | Rare cell analysis using sample splitting and DNA tags |
| US8137912B2 (en) | 2006-06-14 | 2012-03-20 | The General Hospital Corporation | Methods for the diagnosis of fetal abnormalities |
| US20080007838A1 (en) * | 2006-07-07 | 2008-01-10 | Omnitech Partners, Inc. | Field-of-view indicator, and optical system and associated method employing the same |
| US8014577B2 (en) * | 2007-01-29 | 2011-09-06 | Institut National D'optique | Micro-array analysis system and method thereof |
| US8999634B2 (en) | 2007-04-27 | 2015-04-07 | Quest Diagnostics Investments Incorporated | Nucleic acid detection combining amplification with fragmentation |
| US20100167948A1 (en) * | 2007-05-22 | 2010-07-01 | The Brigham And Women's Hospital, Inc. | MicroRNA Expression Profiling of Cerebrospinal Fluid |
| JP2010536565A (ja) * | 2007-08-23 | 2010-12-02 | シンベニオ・バイオシステムズ・インコーポレーテッド | 目標種用のトラップ用磁気選別システム |
| US8008032B2 (en) | 2008-02-25 | 2011-08-30 | Cellective Dx Corporation | Tagged ligands for enrichment of rare analytes from a mixed sample |
| EP2291509B1 (en) * | 2008-05-16 | 2023-11-15 | Board of Supervisors of Louisiana State University and Agricultural and Mechanical College | Microfluidic isolation of tumor cells or other rare cells from whole blood or other liquids |
| JPWO2010016533A1 (ja) * | 2008-08-08 | 2012-01-26 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Neph3(65B13)とE−カドヘリンを用いたプルキンエ前駆細胞の取得方法 |
| EP2334812B1 (en) | 2008-09-20 | 2016-12-21 | The Board of Trustees of The Leland Stanford Junior University | Noninvasive diagnosis of fetal aneuploidy by sequencing |
| US20100304978A1 (en) * | 2009-01-26 | 2010-12-02 | David Xingfei Deng | Methods and compositions for identifying a fetal cell |
| US20120270209A1 (en) * | 2009-05-15 | 2012-10-25 | Massachusetts Institute Of Technology | Systems, devices, and methods for specific capture and release of biological sample components |
| WO2011063416A2 (en) | 2009-11-23 | 2011-05-26 | The General Hospital Corporation | Microfluidic devices for the capture of biological sample components |
| CN102140518B (zh) * | 2011-01-18 | 2013-02-06 | 杭州迪安医学检验中心有限公司 | 肺癌相关表皮生长因子受体egfr外显子突变定量检测试剂盒及方法 |
| JP5934921B2 (ja) * | 2011-04-08 | 2016-06-15 | パナソニックIpマネジメント株式会社 | 診断キット及びその使用方法 |
| WO2013003624A2 (en) | 2011-06-29 | 2013-01-03 | Academia Sinica | The capture, purification and release of biological substance using a surface coating |
| WO2013116523A1 (en) * | 2012-01-31 | 2013-08-08 | The Regents Of The University Of Michigan | Circulating tumor cell capturing techniques and devices |
| ES2635549T3 (es) * | 2012-02-16 | 2017-10-04 | National Research Council Of Canada | Aparato y procedimiento de mezclado microfluídico centrífugo |
| TWI618932B (zh) * | 2012-03-08 | 2018-03-21 | 上海新申派科技有限公司 | 用於改進疾病檢測的微器件 |
| JP2016512197A (ja) | 2013-03-05 | 2016-04-25 | ボード・オブ・リージエンツ,ザ・ユニバーシテイ・オブ・テキサス・システム | 間葉及び上皮間葉形質転換循環腫瘍細胞のための特異的検出ツール |
| US20150064153A1 (en) | 2013-03-15 | 2015-03-05 | The Trustees Of Princeton University | High efficiency microfluidic purification of stem cells to improve transplants |
| CN113512522A (zh) | 2013-03-15 | 2021-10-19 | 普林斯顿大学理事会 | 用于高通量纯化的方法和设备 |
| CN110186835B (zh) | 2013-03-15 | 2022-05-31 | Gpb科学有限公司 | 颗粒的片上微流体处理 |
| US11262361B2 (en) | 2013-07-18 | 2022-03-01 | The General Hospital Corporation | Selective capture and release of rare mammalian cells using photodegradable hydrogels in a microfluidic platform |
| KR102113310B1 (ko) * | 2013-09-03 | 2020-05-20 | 아주대학교 산학협력단 | 암 세포 분리용 조성물, 키트 및 이를 이용한 분리 방법 |
| JP2016536303A (ja) | 2013-10-21 | 2016-11-24 | ザ ジェネラル ホスピタル コーポレイション | 末梢循環腫瘍細胞クラスターおよび癌処置に関する方法 |
| JP2014158468A (ja) * | 2014-03-05 | 2014-09-04 | Clinical Genomics Pty Ltd | 検出方法 |
| WO2015153816A2 (en) | 2014-04-01 | 2015-10-08 | Academia Sinica | Methods and systems for cancer diagnosis and prognosis |
| ES2755406T3 (es) * | 2014-04-17 | 2020-04-22 | Siemens Healthcare Diagnostics Inc | Método para evitar positivos falsos en un ensayo de células tumorales circulantes |
| US10502674B2 (en) * | 2014-06-27 | 2019-12-10 | The Regents Of The University Of California | Apparatus and method for label-free analysis of rare cells from bodily fluids |
| JP6339432B2 (ja) * | 2014-07-16 | 2018-06-06 | オリンパス株式会社 | 細胞観察情報処理システム、細胞観察情報処理方法、細胞観察情報処理プログラム |
| US10112198B2 (en) | 2014-08-26 | 2018-10-30 | Academia Sinica | Collector architecture layout design |
| US10656157B2 (en) * | 2014-09-24 | 2020-05-19 | Purdue Research Foundation | Rare event detection using mass tags |
| WO2016049658A1 (en) * | 2014-09-26 | 2016-03-31 | The Regents Of The University Of California | Method of assessing disease condition of cancer |
| JP6744302B2 (ja) | 2014-11-03 | 2020-08-19 | ザ ジェネラル ホスピタル コーポレイション | ミクロ流体装置における粒子の分類 |
| US10304188B1 (en) | 2015-03-27 | 2019-05-28 | Caleb J. Kumar | Apparatus and method for automated cell analysis |
| US10976232B2 (en) | 2015-08-24 | 2021-04-13 | Gpb Scientific, Inc. | Methods and devices for multi-step cell purification and concentration |
| US12083512B2 (en) * | 2016-01-14 | 2024-09-10 | European Molecular Biology Laboratory | Microfluidic analysis of ligand induced cell expression |
| US10371610B2 (en) | 2016-02-23 | 2019-08-06 | Noul Co., Ltd. | Contact-type patch, staining method using the same, and manufacturing method thereof |
| KR20170099787A (ko) * | 2016-02-23 | 2017-09-01 | 노을 주식회사 | 표지 물질을 저장하는 패치, 이를 이용하는 조직 진단 방법 및 장치 |
| KR102406596B1 (ko) * | 2016-02-23 | 2022-06-10 | 노을 주식회사 | 패치를 이용하는 ctc 진단 방법 및 장치 |
| KR20170099737A (ko) | 2016-02-23 | 2017-09-01 | 노을 주식회사 | 접촉식 염색 패치 및 이를 이용하는 염색 방법 |
| KR102478639B1 (ko) | 2016-02-23 | 2022-12-19 | 노을 주식회사 | 표지 물질을 저장하는 패치, 이를 이용하는 조직 진단 방법 및 장치 |
| US10107726B2 (en) | 2016-03-16 | 2018-10-23 | Cellmax, Ltd. | Collection of suspended cells using a transferable membrane |
| US10010883B2 (en) | 2016-09-20 | 2018-07-03 | International Business Machines Corporation | Deterministic lateral displacement arrays |
| JP6779483B2 (ja) | 2016-09-29 | 2020-11-04 | 住友ゴム工業株式会社 | 医療用検査装置及び細胞検査方法 |
| JP6518985B2 (ja) * | 2016-09-29 | 2019-05-29 | 住友ゴム工業株式会社 | がん細胞捕捉方法 |
| CA3041522A1 (en) * | 2016-10-24 | 2018-05-03 | Gpb Scientific, Llc | Deterministic lateral displacement in the preparation of cells and compositions for therapeutic uses |
| CN107041361A (zh) * | 2017-04-20 | 2017-08-15 | 北京奥康华医学检验所有限公司 | 一种肿瘤组织的保存方法及保存试剂 |
| CN111065399B (zh) | 2017-09-01 | 2023-11-28 | Gpb科学有限公司 | 使用微流体制备治疗活性细胞的方法 |
| EP3691536A4 (en) | 2017-10-03 | 2021-03-17 | The Regents of the University of California | APPARATUS AND METHOD FOR DETERMINING THE SPATIAL PROBABILITY OF PROSTATE CANCER |
| JP7109719B2 (ja) | 2018-02-14 | 2022-08-01 | 住友ゴム工業株式会社 | 特定細胞捕捉方法 |
| JP7158671B2 (ja) * | 2018-02-14 | 2022-10-24 | 住友ゴム工業株式会社 | 特定細胞捕捉方法 |
| JP7170254B2 (ja) * | 2018-02-14 | 2022-11-14 | 住友ゴム工業株式会社 | 特定細胞捕捉方法 |
| WO2020056162A1 (en) * | 2018-09-12 | 2020-03-19 | Oregon Health & Science University | Detecting and/or subtyping circulating hybrid cells that correlate with stage and survival |
| US20220074932A1 (en) * | 2018-12-27 | 2022-03-10 | The General Hospital Corporation | Size-based particle separation and concentration using particle size amplification |
| US11614440B2 (en) | 2019-01-24 | 2023-03-28 | Sumitomo Rubber Industries, Ltd. | Specific cell fractionating and capturing methods |
| US20220267726A1 (en) | 2019-07-18 | 2022-08-25 | Gpb Scientific, Inc. | Ordered processing of blood products to produce therapeutically active cells |
| CA3166192A1 (en) | 2019-12-28 | 2021-07-01 | Gpb Scientific, Inc. | Microfluidic cartridges for processing particles and cells |
| CN111537422B (zh) * | 2020-06-10 | 2022-09-23 | 兰州大学 | 表征及调控包晶合金定向凝固时糊状区内渗透率的方法 |
| CN111676289A (zh) * | 2020-06-19 | 2020-09-18 | 安徽微分基因科技有限公司 | 一种egfr基因19号外显子e746-a750突变基因的检测方法 |
| EP4172315A1 (en) * | 2020-06-25 | 2023-05-03 | Arrive Pte Ltd. | System and methods for characterizing cells and microenvironments |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003085379A2 (en) * | 2002-04-01 | 2003-10-16 | Fluidigm Corporation | Microfluidic particle-analysis systems |
| JP2004045358A (ja) * | 2001-08-03 | 2004-02-12 | Nec Corp | 分離装置および分離装置の製造方法 |
Family Cites Families (114)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3906929A (en) * | 1973-11-23 | 1975-09-23 | Lynn Lawrence Augspurger | Processes for reproduction of cellular bodies |
| US4508625A (en) * | 1982-10-18 | 1985-04-02 | Graham Marshall D | Magnetic separation using chelated magnetic ions |
| US4675286A (en) * | 1985-01-28 | 1987-06-23 | Aspen Diagnostics, Inc. | Fetal cell separation and testing |
| US4999283A (en) * | 1986-01-10 | 1991-03-12 | University Of Kentucky Research Foundation | Method for x and y spermatozoa separation |
| US5459039A (en) | 1989-05-12 | 1995-10-17 | Duke University | Methods for mapping genetic mutations |
| US5641628A (en) * | 1989-11-13 | 1997-06-24 | Children's Medical Center Corporation | Non-invasive method for isolation and detection of fetal DNA |
| US5153117A (en) * | 1990-03-27 | 1992-10-06 | Genetype A.G. | Fetal cell recovery method |
| US5296375A (en) * | 1992-05-01 | 1994-03-22 | Trustees Of The University Of Pennsylvania | Mesoscale sperm handling devices |
| US5304487A (en) * | 1992-05-01 | 1994-04-19 | Trustees Of The University Of Pennsylvania | Fluid handling in mesoscale analytical devices |
| US5726026A (en) * | 1992-05-01 | 1998-03-10 | Trustees Of The University Of Pennsylvania | Mesoscale sample preparation device and systems for determination and processing of analytes |
| US5637469A (en) * | 1992-05-01 | 1997-06-10 | Trustees Of The University Of Pennsylvania | Methods and apparatus for the detection of an analyte utilizing mesoscale flow systems |
| US5629147A (en) * | 1992-07-17 | 1997-05-13 | Aprogenex, Inc. | Enriching and identifying fetal cells in maternal blood for in situ hybridization |
| US5457024A (en) * | 1993-01-22 | 1995-10-10 | Aprogenex, Inc. | Isolation of fetal erythrocytes |
| US5427663A (en) | 1993-06-08 | 1995-06-27 | British Technology Group Usa Inc. | Microlithographic array for macromolecule and cell fractionation |
| EP0657811B1 (en) | 1993-12-09 | 1998-09-02 | STMicroelectronics S.r.l. | Integrated circuitry for checking the utilization rate of redundancy memory elements in a semiconductor memory device |
| US5432054A (en) * | 1994-01-31 | 1995-07-11 | Applied Imaging | Method for separating rare cells from a population of cells |
| US20020042079A1 (en) * | 1994-02-01 | 2002-04-11 | Sanford M. Simon | Methods and agents for measuring and controlling multidrug resistance |
| US5750339A (en) * | 1994-11-30 | 1998-05-12 | Thomas Jefferson University | Methods for identifying fetal cells |
| US5648220A (en) * | 1995-02-14 | 1997-07-15 | New England Medical Center Hospitals, Inc. | Methods for labeling intracytoplasmic molecules |
| US5715946A (en) * | 1995-06-07 | 1998-02-10 | Reichenbach; Steven H. | Method and apparatus for sorting particles suspended in a fluid |
| US6582904B2 (en) * | 1995-11-16 | 2003-06-24 | Michael W. Dahm | Method of quantifying tumour cells in a body fluid and a suitable test kit |
| US5972721A (en) * | 1996-03-14 | 1999-10-26 | The United States Of America As Represented By The Secretary Of The Air Force | Immunomagnetic assay system for clinical diagnosis and other purposes |
| US5891651A (en) * | 1996-03-29 | 1999-04-06 | Mayo Foundation For Medical Education And Research | Methods of recovering colorectal epithelial cells or fragments thereof from stool |
| US6074827A (en) * | 1996-07-30 | 2000-06-13 | Aclara Biosciences, Inc. | Microfluidic method for nucleic acid purification and processing |
| AU4164597A (en) * | 1996-08-26 | 1998-03-19 | Princeton University | Reversibly sealable microstructure sorting devices |
| WO1998010267A1 (en) * | 1996-09-04 | 1998-03-12 | Technical University Of Denmark | A micro flow system for particle separation and analysis |
| GB9704876D0 (en) * | 1997-03-08 | 1997-04-23 | Univ Dundee | Diagnostic methods |
| US6066449A (en) * | 1997-04-15 | 2000-05-23 | The Trustees Of Columbia University In The City Of New York | Method of detecting metastatic thyroid cancer |
| US6156273A (en) * | 1997-05-27 | 2000-12-05 | Purdue Research Corporation | Separation columns and methods for manufacturing the improved separation columns |
| US6368871B1 (en) * | 1997-08-13 | 2002-04-09 | Cepheid | Non-planar microstructures for manipulation of fluid samples |
| US6197523B1 (en) * | 1997-11-24 | 2001-03-06 | Robert A. Levine | Method for the detection, identification, enumeration and confirmation of circulating cancer and/or hematologic progenitor cells in whole blood |
| SE521415C2 (sv) * | 1998-02-17 | 2003-10-28 | Hans Goeran Evald Martin | Metod för att framställa en gassensortillhörig detektor, samt en detektor framställd enligt metoden |
| US6537505B1 (en) * | 1998-02-20 | 2003-03-25 | Bio Dot, Inc. | Reagent dispensing valve |
| US6036857A (en) * | 1998-02-20 | 2000-03-14 | Florida State University Research Foundation, Inc. | Apparatus for continuous magnetic separation of components from a mixture |
| US6027623A (en) * | 1998-04-22 | 2000-02-22 | Toyo Technologies, Inc. | Device and method for electrophoretic fraction |
| US6200765B1 (en) * | 1998-05-04 | 2001-03-13 | Pacific Northwest Cancer Foundation | Non-invasive methods to detect prostate cancer |
| JP2002528699A (ja) * | 1998-05-22 | 2002-09-03 | カリフォルニア インスティチュート オブ テクノロジー | 微細製作細胞分類器 |
| US6296752B1 (en) * | 1998-06-05 | 2001-10-02 | Sarnoff Corporation | Apparatus for separating molecules |
| US6576478B1 (en) * | 1998-07-14 | 2003-06-10 | Zyomyx, Inc. | Microdevices for high-throughput screening of biomolecules |
| EP1100964A1 (en) | 1998-07-20 | 2001-05-23 | Variagenics, Inc. | Gene sequence variances with utility in determining the treatment of disease |
| US6337472B1 (en) * | 1998-10-19 | 2002-01-08 | The University Of Texas System Board Of Regents | Light imaging microscope having spatially resolved images |
| US6632655B1 (en) * | 1999-02-23 | 2003-10-14 | Caliper Technologies Corp. | Manipulation of microparticles in microfluidic systems |
| EP1173744A4 (en) * | 1999-03-02 | 2002-10-16 | Qualigen Inc | METHODS AND APPARATUS FOR SEPARATING BIOLOGICAL FLUIDS |
| US6942978B1 (en) * | 1999-03-03 | 2005-09-13 | The Board Of Trustees Of The University Of Arkansas | Transmembrane serine protease overexpressed in ovarian carcinoma and uses thereof |
| US6858439B1 (en) * | 1999-03-15 | 2005-02-22 | Aviva Biosciences | Compositions and methods for separation of moieties on chips |
| US6368562B1 (en) * | 1999-04-16 | 2002-04-09 | Orchid Biosciences, Inc. | Liquid transportation system for microfluidic device |
| US6589791B1 (en) * | 1999-05-20 | 2003-07-08 | Cartesian Technologies, Inc. | State-variable control system |
| US6395232B1 (en) * | 1999-07-09 | 2002-05-28 | Orchid Biosciences, Inc. | Fluid delivery system for a microfluidic device using a pressure pulse |
| US6623945B1 (en) * | 1999-09-16 | 2003-09-23 | Motorola, Inc. | System and method for microwave cell lysing of small samples |
| US6692952B1 (en) | 1999-11-10 | 2004-02-17 | Massachusetts Institute Of Technology | Cell analysis and sorting apparatus for manipulation of cells |
| US6361958B1 (en) * | 1999-11-12 | 2002-03-26 | Motorola, Inc. | Biochannel assay for hybridization with biomaterial |
| US20030170631A1 (en) * | 2000-04-03 | 2003-09-11 | Corixa Corporation | Methods, compositions and kits for the detection and monitoring of breast cancer |
| SE0001790D0 (sv) * | 2000-05-12 | 2000-05-12 | Aamic Ab | Hydrophobic barrier |
| DE60142691D1 (de) * | 2000-09-09 | 2010-09-09 | Univ New York State Res Found | Verfahren und zusammensetzungen zur isolierung von metastatischen krebszellen und anwendung zur messung des metastatischen potentials eines krebses |
| US6689615B1 (en) * | 2000-10-04 | 2004-02-10 | James Murto | Methods and devices for processing blood samples |
| CA2424941A1 (en) * | 2000-10-10 | 2002-04-18 | Aviva Biosciences Corporation | An integrated biochip system for sample preparation and analysis |
| US6521188B1 (en) * | 2000-11-22 | 2003-02-18 | Industrial Technology Research Institute | Microfluidic actuator |
| US6778724B2 (en) * | 2000-11-28 | 2004-08-17 | The Regents Of The University Of California | Optical switching and sorting of biological samples and microparticles transported in a micro-fluidic device, including integrated bio-chip devices |
| FR2817967B1 (fr) * | 2000-12-08 | 2003-02-28 | Diagast | Procede de magnetisation de marqueurs chimiques ou biologiques |
| US6453928B1 (en) * | 2001-01-08 | 2002-09-24 | Nanolab Ltd. | Apparatus, and method for propelling fluids |
| US6939670B2 (en) * | 2001-03-12 | 2005-09-06 | Monogen, Inc. | Cell-based detection and differentiation of lung cancer |
| US7323140B2 (en) * | 2001-03-28 | 2008-01-29 | Handylab, Inc. | Moving microdroplets in a microfluidic device |
| EP1377821A2 (en) * | 2001-04-03 | 2004-01-07 | Micronics, Inc. | Pneumatic valve interface for use in microfluidic structures |
| FR2824144B1 (fr) * | 2001-04-30 | 2004-09-17 | Metagenex S A R L | Methode de diagnostic prenatal sur cellule foetale isolee du sang maternel |
| US6743636B2 (en) * | 2001-05-24 | 2004-06-01 | Industrial Technology Research Institute | Microfluid driving device |
| US20050026167A1 (en) * | 2001-06-11 | 2005-02-03 | Mark Birch-Machin | Complete mitochondrial genome sequences as a diagnostic tool for the health sciences |
| KR20040032884A (ko) * | 2001-08-03 | 2004-04-17 | 닛본 덴끼 가부시끼가이샤 | 분리 장치 및 분리 장치의 제조 방법 |
| US7863012B2 (en) * | 2004-02-17 | 2011-01-04 | Veridex, Llc | Analysis of circulating tumor cells, fragments, and debris |
| CA2458704A1 (en) * | 2001-09-04 | 2003-03-13 | Iq Corporation B.V. | Determination and quantification of red blood cell populations in samples |
| ATE309393T1 (de) * | 2001-09-06 | 2005-11-15 | Adnagen Ag | Verfahren zum qualitativen und/oder quantitativen nachweis von zellen |
| DE10143776A1 (de) * | 2001-09-06 | 2003-04-03 | Adnagen Ag | Verfahren und Kit zur Diagnostik oder Behandlungskontrolle von Brustkrebs |
| WO2003044224A1 (de) * | 2001-11-22 | 2003-05-30 | Adnagen Ag | Diagnose-kit, dns-chip sowie verfahren zur diagnostik oder behandlungskontrolle bei hodenkrebs |
| US6783647B2 (en) * | 2001-10-19 | 2004-08-31 | Ut-Battelle, Llc | Microfluidic systems and methods of transport and lysis of cells and analysis of cell lysate |
| AU2002347468A1 (en) * | 2001-11-30 | 2003-06-10 | Pfizer Products Inc. | Methods for detecting cells with numerical chromosomal abnormalities |
| EP1495106A2 (en) * | 2002-03-20 | 2005-01-12 | Advanced Sensor Technologies, Inc. | Personal monitor to detect exposure to toxic agents |
| SE0201738D0 (sv) * | 2002-06-07 | 2002-06-07 | Aamic Ab | Micro-fluid structures |
| US20040101444A1 (en) * | 2002-07-15 | 2004-05-27 | Xeotron Corporation | Apparatus and method for fluid delivery to a hybridization station |
| US20060008807A1 (en) * | 2002-08-23 | 2006-01-12 | O'hara Shawn M | Multiparameter analysis of comprehensive nucleic acids and morphological features on the same sample |
| US7455770B2 (en) * | 2002-09-09 | 2008-11-25 | Cytonome, Inc. | Implementation of microfluidic components in a microfluidic system |
| WO2004029221A2 (en) * | 2002-09-27 | 2004-04-08 | The General Hospital Corporation | Microfluidic device for cell separation and uses thereof |
| AU2003299553A1 (en) | 2002-10-23 | 2004-05-13 | The Trustees Of Princeton University | Method for continuous particle separation using obstacle arrays asymmetrically aligned to fields |
| US20070160503A1 (en) | 2003-06-13 | 2007-07-12 | Palaniappan Sethu | Microfluidic systems for size based removal of red blood cells and platelets from blood |
| MXPA06001110A (es) | 2003-08-01 | 2006-04-11 | Wyeth Corp | Uso de una combinacion de un inhibidor de la cinasa del receptor del factor de crecimiento epidermico y agentes citotoxicos para el tratamiento e inhibicion del cancer. |
| US20050069528A1 (en) * | 2003-08-21 | 2005-03-31 | Genzyme Corporation | Mammalian endothelial cell model systems |
| WO2005022147A1 (en) * | 2003-08-28 | 2005-03-10 | Celula, Inc. | Methods and apparatus for sorting cells using an optical switch in a microfluidic channel network |
| EP1678475A4 (en) * | 2003-10-29 | 2009-11-11 | Mec Dynamics Corp | MICROMECHANICAL METHODS AND SYSTEMS FOR PERFORMING TESTS |
| US7939249B2 (en) * | 2003-12-24 | 2011-05-10 | 3M Innovative Properties Company | Methods for nucleic acid isolation and kits using a microfluidic device and concentration step |
| US20050181353A1 (en) * | 2004-02-17 | 2005-08-18 | Rao Galla C. | Stabilization of cells and biological specimens for analysis |
| US20060121624A1 (en) * | 2004-03-03 | 2006-06-08 | Huang Lotien R | Methods and systems for fluid delivery |
| WO2005084380A2 (en) | 2004-03-03 | 2005-09-15 | The General Hospital Corporation | System for delivering a diluted solution |
| US20050266433A1 (en) | 2004-03-03 | 2005-12-01 | Ravi Kapur | Magnetic device for isolation of cells and biomolecules in a microfluidic environment |
| US7390388B2 (en) * | 2004-03-25 | 2008-06-24 | Hewlett-Packard Development Company, L.P. | Method of sorting cells on a biodevice |
| PT1733056E (pt) | 2004-03-31 | 2013-08-29 | Gen Hospital Corp | Método para determinar a responsividade do cancro a tratamentos visando o receptor do factor de crescimento epidérmico |
| WO2005108963A1 (en) * | 2004-05-06 | 2005-11-17 | Nanyang Technological University | Microfluidic cell sorter system |
| DE102004047953A1 (de) * | 2004-10-01 | 2006-04-20 | Rudolf Rigler | Selektion von Partikeln im laminaren Fluss |
| JP2008526255A (ja) * | 2005-01-13 | 2008-07-24 | マイクロニクス, インコーポレイテッド | 微量流体希薄細胞検出デバイス |
| US8158410B2 (en) * | 2005-01-18 | 2012-04-17 | Biocept, Inc. | Recovery of rare cells using a microchannel apparatus with patterned posts |
| US20070026418A1 (en) * | 2005-07-29 | 2007-02-01 | Martin Fuchs | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US20070026413A1 (en) * | 2005-07-29 | 2007-02-01 | Mehmet Toner | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US20070026415A1 (en) * | 2005-07-29 | 2007-02-01 | Martin Fuchs | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US20070196820A1 (en) * | 2005-04-05 | 2007-08-23 | Ravi Kapur | Devices and methods for enrichment and alteration of cells and other particles |
| US20070026414A1 (en) * | 2005-07-29 | 2007-02-01 | Martin Fuchs | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US20070026417A1 (en) * | 2005-07-29 | 2007-02-01 | Martin Fuchs | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US20070059680A1 (en) * | 2005-09-15 | 2007-03-15 | Ravi Kapur | System for cell enrichment |
| US8921102B2 (en) * | 2005-07-29 | 2014-12-30 | Gpb Scientific, Llc | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US20070026416A1 (en) * | 2005-07-29 | 2007-02-01 | Martin Fuchs | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US20070026419A1 (en) * | 2005-07-29 | 2007-02-01 | Martin Fuchs | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| US20070059781A1 (en) * | 2005-09-15 | 2007-03-15 | Ravi Kapur | System for size based separation and analysis |
| US20070059716A1 (en) * | 2005-09-15 | 2007-03-15 | Ulysses Balis | Methods for detecting fetal abnormality |
| JP2009509143A (ja) * | 2005-09-15 | 2009-03-05 | アルテミス ヘルス,インク. | 分析改善システムおよび方法 |
| US20070059718A1 (en) * | 2005-09-15 | 2007-03-15 | Mehmet Toner | Systems and methods for enrichment of analytes |
| US20070059719A1 (en) * | 2005-09-15 | 2007-03-15 | Michael Grisham | Business methods for prenatal Diagnosis |
| US20070059683A1 (en) * | 2005-09-15 | 2007-03-15 | Tom Barber | Veterinary diagnostic system |
| US20070059774A1 (en) * | 2005-09-15 | 2007-03-15 | Michael Grisham | Kits for Prenatal Testing |
-
2006
- 2006-04-05 WO PCT/US2006/012778 patent/WO2006108087A2/en active Application Filing
- 2006-04-05 EP EP12180052.8A patent/EP2594631A1/en not_active Withdrawn
- 2006-04-05 GB GB0612649A patent/GB2427468B/en active Active
- 2006-04-05 EP EP06749394A patent/EP1874920A4/en not_active Withdrawn
- 2006-04-05 JP JP2008505508A patent/JP2008538282A/ja active Pending
- 2006-06-08 US US11/449,161 patent/US20070099207A1/en not_active Abandoned
-
2009
- 2009-07-09 US US12/500,541 patent/US20110306043A1/en not_active Abandoned
-
2011
- 2011-08-15 HK HK11108515.6A patent/HK1154422A1/xx unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004045358A (ja) * | 2001-08-03 | 2004-02-12 | Nec Corp | 分離装置および分離装置の製造方法 |
| WO2003085379A2 (en) * | 2002-04-01 | 2003-10-16 | Fluidigm Corporation | Microfluidic particle-analysis systems |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012504956A (ja) * | 2008-10-10 | 2012-03-01 | セントレ ナショナル デ ラ レシェルシェ サイエンティフィーク−ディーエーイー | 細胞ソート・デバイス |
| JP2012529983A (ja) * | 2009-06-19 | 2012-11-29 | コミサリア ア レネルジィ アトミーク エ オ ゼネ ルジイ アルテアナティーフ | 液相間で成分を移動させるマイクロ流体システム及び対応する方法、並びに上記成分を抽出するための上記システムの使用 |
| JP2013505429A (ja) * | 2009-09-21 | 2013-02-14 | マウント シナイ ホスピタル | 甲状腺癌を診断および処置するための方法および組成物 |
| WO2012002447A1 (ja) * | 2010-06-30 | 2012-01-05 | 武田薬品工業株式会社 | 変異ポリヌクレオチドの検出方法 |
| US11085923B2 (en) | 2011-03-24 | 2021-08-10 | Anpac Bio-Medical Science Co., Ltd | Micro-devices for disease detection |
| CN106929401B (zh) * | 2011-05-05 | 2020-06-09 | 安派科生物医学科技有限公司 | 肿瘤细胞检测仪 |
| WO2012151501A3 (en) * | 2011-05-05 | 2013-04-25 | Anpac Bio-Medical Science Co., Ltd. | Apparatus for detecting tumor cells |
| TWI547691B (zh) * | 2011-05-05 | 2016-09-01 | 上海新申派科技有限公司 | 腫瘤細胞檢測儀 |
| CN106929401A (zh) * | 2011-05-05 | 2017-07-07 | 安派科生物医学科技有限公司 | 肿瘤细胞检测仪 |
| JP2014512839A (ja) * | 2011-05-05 | 2014-05-29 | アンパック バイオ−メディカル サイエンス カンパニー リミテッド | 腫瘍細胞を検出するための装置 |
| US10895573B2 (en) | 2011-05-05 | 2021-01-19 | Anpac Bio-Medical Science Co., Ltd. | Apparatus for detecting tumor cells |
| US9408565B2 (en) | 2011-05-05 | 2016-08-09 | Shanghai Xinshenpai Technology Co., Ltd. | Apparatus for detecting tumor cells |
| JP2017000921A (ja) * | 2015-06-05 | 2017-01-05 | 国立大学法人 東京大学 | 曲げ弾性係数が小さい粒子の分離捕捉装置 |
| JP2021510516A (ja) * | 2018-01-19 | 2021-04-30 | インターナショナル・ビジネス・マシーンズ・コーポレーションInternational Business Machines Corporation | 粒子の精製および分画のための方法およびマイクロ流体装置 |
| US11458474B2 (en) | 2018-01-19 | 2022-10-04 | International Business Machines Corporation | Microfluidic chips with one or more vias |
| US11566982B2 (en) | 2018-01-19 | 2023-01-31 | International Business Machines Corporation | Microscale and mesoscale condenser devices |
| US11754476B2 (en) | 2018-01-19 | 2023-09-12 | International Business Machines Corporation | Microscale and mesoscale condenser devices |
| US11872560B2 (en) | 2018-01-19 | 2024-01-16 | International Business Machines Corporation | Microfluidic chips for particle purification and fractionation |
| JP7455339B2 (ja) | 2020-09-29 | 2024-03-26 | Nok株式会社 | 白血球捕捉デバイス |
| WO2023189095A1 (ja) * | 2022-03-28 | 2023-10-05 | 国立大学法人茨城大学 | 白血球捕捉デバイス |
Also Published As
| Publication number | Publication date |
|---|---|
| GB2427468A (en) | 2006-12-27 |
| GB0612649D0 (en) | 2006-08-16 |
| GB2427468B (en) | 2011-03-02 |
| EP1874920A2 (en) | 2008-01-09 |
| EP2594631A1 (en) | 2013-05-22 |
| EP1874920A4 (en) | 2009-11-04 |
| US20070099207A1 (en) | 2007-05-03 |
| HK1154422A1 (en) | 2012-04-20 |
| WO2006108087A2 (en) | 2006-10-12 |
| US20110306043A1 (en) | 2011-12-15 |
| WO2006108087A3 (en) | 2009-06-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2008538282A (ja) | 装置および循環腫瘍細胞および他の粒子の濃縮および変更のための方法 | |
| JP2008538282A5 (ja) | ||
| US8921102B2 (en) | Devices and methods for enrichment and alteration of circulating tumor cells and other particles | |
| Vaidyanathan et al. | Cancer diagnosis: from tumor to liquid biopsy and beyond | |
| Poudineh et al. | Profiling circulating tumour cells and other biomarkers of invasive cancers | |
| US20070026418A1 (en) | Devices and methods for enrichment and alteration of circulating tumor cells and other particles | |
| US20070026413A1 (en) | Devices and methods for enrichment and alteration of circulating tumor cells and other particles | |
| US20070026415A1 (en) | Devices and methods for enrichment and alteration of circulating tumor cells and other particles | |
| US20070026414A1 (en) | Devices and methods for enrichment and alteration of circulating tumor cells and other particles | |
| US20130302796A1 (en) | Devices And Methods For Enrichment And Alteration Of Circulating Tumor Cells And Other Particles | |
| US20070026416A1 (en) | Devices and methods for enrichment and alteration of circulating tumor cells and other particles | |
| US20070026419A1 (en) | Devices and methods for enrichment and alteration of circulating tumor cells and other particles | |
| US10613015B2 (en) | Methods for classification and sorting of cancer cells | |
| WO2007079229A2 (en) | Devices and methods for enrichment and alteration of circulating tumor cells and other particles | |
| US20080090239A1 (en) | Rare cell analysis using sample splitting and dna tags | |
| US20170232439A1 (en) | Separation of low-abundance cells from fluid using surface acoustic waves | |
| US20150232936A1 (en) | Rare Cell Analysis Using Sample Splitting And DNA Tags | |
| WO2011063416A2 (en) | Microfluidic devices for the capture of biological sample components | |
| GB2472927A (en) | Microfluidic cell separation device comprising micro-corrugations | |
| KR101794218B1 (ko) | 비침습적 산전 진단을 위한 산모 혈액 내 유핵 적혈구의 농축 분리 방법 | |
| Costa et al. | Methodology for the isolation and analysis of CTCs | |
| Johnson et al. | Isolating rare cells and circulating tumor cells with high purity by sequential eDAR | |
| Yee-de León et al. | Characterization of a novel automated microfiltration device for the efficient isolation and analysis of circulating tumor cells from clinical blood samples | |
| Khoo et al. | Advancing techniques and insights in circulating tumor cell (ctc) research | |
| Teixeira et al. | Advances in Microfluidics for the Implementation of Liquid Biopsy in Clinical Routine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090403 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090403 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20100810 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100819 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110902 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20111202 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20111202 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20111209 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20111209 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20111209 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20120123 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120302 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120326 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120625 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120702 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20121213 |