JP2008519069A - プロトンポンプ阻害剤用の新規な改変された放出のペレット製剤 - Google Patents
プロトンポンプ阻害剤用の新規な改変された放出のペレット製剤 Download PDFInfo
- Publication number
- JP2008519069A JP2008519069A JP2007540283A JP2007540283A JP2008519069A JP 2008519069 A JP2008519069 A JP 2008519069A JP 2007540283 A JP2007540283 A JP 2007540283A JP 2007540283 A JP2007540283 A JP 2007540283A JP 2008519069 A JP2008519069 A JP 2008519069A
- Authority
- JP
- Japan
- Prior art keywords
- layer
- dosage form
- ppi
- coating
- pellet
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- OFPUAQDJLPMNKL-UHFFFAOYSA-N Cc(cnc(CS(c1nc(cc(cc2)C3=NCCO3)c2[nH]1)=O)c1C)c1OC Chemical compound Cc(cnc(CS(c1nc(cc(cc2)C3=NCCO3)c2[nH]1)=O)c1C)c1OC OFPUAQDJLPMNKL-UHFFFAOYSA-N 0.000 description 1
- ZBFDAUIVDSSISP-UHFFFAOYSA-N Cc(cnc(CS(c1nc(nc(cc2)OC)c2[nH]1)=O)c1C)c1OC Chemical compound Cc(cnc(CS(c1nc(nc(cc2)OC)c2[nH]1)=O)c1C)c1OC ZBFDAUIVDSSISP-UHFFFAOYSA-N 0.000 description 1
Images







Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2072—Pills, tablets, discs, rods characterised by shape, structure or size; Tablets with holes, special break lines or identification marks; Partially coated tablets; Disintegrating flat shaped forms
- A61K9/2086—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat
- A61K9/209—Layered tablets, e.g. bilayer tablets; Tablets of the type inert core-active coat containing drug in at least two layers or in the core and in at least one outer layer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5026—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone, poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5036—Polysaccharides, e.g. gums, alginate; Cyclodextrin
- A61K9/5042—Cellulose; Cellulose derivatives, e.g. phthalate or acetate succinate esters of hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5073—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals having two or more different coatings optionally including drug-containing subcoatings
- A61K9/5078—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals having two or more different coatings optionally including drug-containing subcoatings with drug-free core
Abstract
Description

Het1は
ベンゾイミダゾール部分におけるNは、R6〜R9で置換された環炭素原子のうちの1つが、置換基を有さない窒素原子と場合により交換されてもよいことを意味し;
R1、R2及びR3は、同じであるかまたは異なり、そして水素、アルキル、場合によりフッ素で置換されるアルコキシ、アルキルチオ、アルコキシアルコキシ、ジアルキルアミノ、ピペリジノ、モルホリノ、ハロゲン、フェニル及びフェニルアルコキシより選択され;
R4及びR5は、同じか又は異なり、そして水素、アルキル及びアリールアルキルより選択され;
R6’は、水素、ハロゲン、トリフルオロメチル、アルキル又はアルコキシであり;
R6〜R9は、同じか若しくは異なり、そして水素、アルキル、アルコキシ、ハロゲン、ハロ−アルコキシ、アルキルカルボニル、アルコキシカルボニル、オキサゾリニル、及びトリフルオロアルキルより選択されるか、又は隣接する基R6〜R9が、さらに置換され得る環構造を形成し;
R10は、水素であるか又はR3と共にアルキレン鎖を形成し、そして
R11及びR12は、同じか又は異なり、そして水素、ハロゲン又はアルキルより選択される]
の化合物、そのアルカリ性塩、その単一の鏡像異性体のうちの1つ又は鏡像異性体のうちの1つのアルカリ性塩である。
本発明の使用される二重パルス実施態様において使用されると予測される用量は、等用量(例えば60mg+60mg)で適切に組み合わされた、酸感受性PPIの1つの即時放出部分と1つの遅延放出部分に分割された2〜500mgの範囲である。
酸感受性PPI含有コアは、場合により薬学的に許容しうる賦形剤と共に従来の方法によるペレットの形態のコア材料中に活性薬物を入れて配合されている。
本発明による酸性化合物は、10%w/wの濃度で(室温すなわち約20℃にて)精製水に溶解または懸濁され、そしてガラス電極またはISFET電極を備えたpHメーターで測定した場合に5またはそれ以下のpHを示す化合物である。
コア材料上に塗布され、そして遅滞時間制御層をPPI含有コアと分離している遅延放出改変層は、水溶性ポリマーベースの層に疎水性化剤及びタルクを組み込むことにより疎水性にされる。
遅滞時間制御層は、例えばヒドロキシプロピルメチルセルロース 4000のような高粘性水溶性ポリマーを必須成分として含む。用語「水溶性ポリマー」は、本明細書で使用される場合、水溶性ポリマー、水溶性コポリマー、又はこのようなポリマーの混合物を意味する。本発明における「高粘性」は、1つ目の選択肢として欧州薬局方及び2つ目の選択肢として米国薬局方に従って試験されて100mPas(cps)から約150000mPas(cps)までのみかけ粘度とみなされる。試験が両方の薬局方に記載される場合、欧州薬局方における方法が普及している。
以前に記載した遅滞時間制御層を有するペレットは、本発明の1つの代替の実施態様として、水溶性結合剤及び場合により界面活性剤とともに、PPIの分散液/溶液/懸濁液で、例えばスプレーされて、コーティングされる。このコーティングは、適切なコーティング装置で行われて、遅滞時間制御層の上面上に堆積された第2PPI含有層を有するペレットコアを得、これが最終製剤を投与した場合に即時放出パルスを生じる。
層を形成されたペレット上に腸溶性コーティング層を塗布する前に、それらを、例えばpH−緩衝化化合物のようなアルカリ性化合物を場合により含む薬学的賦形剤を含む1つまたはそれ以上の水溶性かまたは水中で急速に崩壊するサブコーティング層で場合により被覆し得る。このサブコーティング層は、層を形成されたペレットの組成物を外部腸溶性コーティング層と分離する。
本発明の最終製剤は、第1の代替の実施態様についての以下の原則的な方法に従って製造される;
I) 唯一の活性薬物として酸感受性プロトンポンプ阻害剤(PPI)を含むペレットの形態でコア材料を調製する工程;
II) 工程I)で得られたペレットコアを、遅延放出改変層でコーティングする工程;
III) 工程II)から得られた遅延放出改変層を形成されたペレットコアを、必須成分として高粘性水溶性ポリマーを含む遅滞時間制御層でコーティングする工程;
IV) 工程III)から得られた遅滞時間制御層を形成されたペレットを、外部腸溶性コーティングでコーティングする工程であって、任意のサブコーティング層が、腸溶性コーティング層が塗布される前に塗布される、工程;
V) 工程IV)で得られたペレット生成物を、外部腸溶性コーティング及び任意のサブコーティング層を有しPPIの即時放出を生じる他のペレットと共に、カプセル剤、サシェ剤又は複数単位ペレット系錠剤に組み込む工程。
I) 唯一の活性薬物として酸感受性プロトンポンプ阻害剤(PPI)を含むペレットの形態でコア材料を調製する工程;
II) 工程I)から得られたペレットコアを、遅延放出改変層でコーティングする工程;
III) 工程II)から得られた遅延放出改変層を形成されたペレットコアを、必須成分として高粘性水溶性ポリマーを含む遅滞時間制御層でコーティングする工程;
IV) 工程III)から得られた遅滞時間制御層を形成されたペレットを、第2のPPI部分を含む層でコーティングする工程;
V) 工程IV)から得られたペレットを、任意のサブコーティング層で場合によりコーティングする工程;並びに
VI) 工程V)から得られたペレット生成物を、外部腸溶性コーティングでコーティングする工程;
VII) 工程VI)から得られた腸溶性コーティングを施されたペレットを、カプセル剤、サシェ剤、又は複数単位ペレット系錠剤に製剤化する工程。
a)高粘性水溶性ポリマーを非溶媒中に分散すること;及び
b)水性液体又は水を加えて分散したポリマー粒子の水和形態を形成すること;
により製造された高粘性水溶性ポリマーの分散液を利用することが特に有益である。
このような分散系が、最初にポリマーを含水液体に溶解し、次いでその系を沈降させることでは得ることができないことは理解されるべきである。
実施態様は、遅延(第2)パルスについての遅滞時間が1〜10時間、好ましくは1〜8時間又は最も好ましくは1〜6時間の範囲であるように設計される。
患者への提示前の本発明の剤形を、カプセル剤、サシェ剤、又は複数単位ペレット系錠剤の形態になるように仕上げることが企図されている。完成した剤形は、それぞれ遅延放出パルス、即時放出パルスを生じる、ペレット、他の種類のペレット及び錠剤の選択的組み合わせを含み得る。遅延放出パルスは、本発明によればペレットを起源とする。以下の組み合わせが企図されている;
遅滞時間/遅延時間:ペレット/錠剤の形態の腸溶性コーティングを施されたコアが露出された後でさえも、pH1.2を有する第1の溶出媒体については2時間、次いでpH6.8を有する第2の溶出媒体中で、インビトロでのPPIの溶出が遅延されることを本発明については意味する。
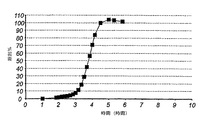
遅滞時間は、(第2の)溶出媒体中において、(遅延パルスにおける用量の)薬物の10%が放出されるまでに要する時間として定義される。例証のため、図1を参照のこと。
勾配は、溶出した量(80%)を(遅延用量の)10−90%の間の溶出に必要な時間(分)で割ったものとして定義される。これは、1分あたりの%(10-90)で表わされるので、この期間の平均速度としての勾配を与える。例証のために、図1を参照のこと。
遅延パルス放出ペレット
組成物において示される全ての量は、供給した量であり、収率に対して補正していない。
遅延パルス放出ペレットの製造のための概要の方式は、以下の順序でシードに層をコーティングすることによる;
活性薬物(PPI)含有層→遅延放出改変層→遅滞時間制御層→腸溶性コーティング層。
賦形剤 量(g)
エソメプラゾール−Mg三水和物 250
ポリソルベート80 5.0
ヒドロキシプロピルメチルセルロース6cps 37.5
精製水 1170
活性薬物層形成のためのシード
糖シード(ノンパレイユ)1.0−1.18mm 250
ヒドロキシプロピルメチルセルロース(以下ではHPMCとも呼ぶ)及びポリソルベート80を水に溶解し、その後エソメプラゾール−Mg三水和物をその中に懸濁させた。この懸濁液をアジテーターミル(Dyno−MillTM)で湿式微粉化工程にかけた。
調製した層形成懸濁液を、直径0.8mmの開口を有する液体ノズルを備えたWurster方式に従う流動床装置で糖シード上にスプレーコートした。
入口空気温度は80℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.5bar、噴霧装置空気流量は2.5Nm3/時、スプレー速度は12−19g/分であり、結果として約40℃の出口空気温度となった。
遅延放出改変層溶液/懸濁液
賦形剤 量(g)
タルク粉末 112.5
ヒドロキシプロピルセルロース(75−150cps) 30
Mg−ステアレート 7.5
精製水 1050
前の工程と同じコーティング装置でコーティングを行った。
入口空気温度は75℃であり、流動空気流量は40m3/時、噴霧装置空気圧2.8bar、噴霧装置空気流量2.8Nm3/時、スプレー速度は6−11g/分であり、結果として約45℃の出口空気温度となった。
遅滞時間制御層のための溶液/懸濁液
賦形剤 量(g)
HPMC 4000cps* 80
HPMC 6cps 11
EtOH 99.5% 1350
精製水 172
* 欧州薬局方に従って試験したpHは7.5であった。
前の工程と同じコーティング装置でコーティングを行った。
入口空気温度は40℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.5bar、噴霧装置空気流量は2.5Nm3/時、スプレー速度は14−16g/分であり、結果として約20℃の出口空気温度となった。
腸溶性コーティング懸濁液
賦形剤 量(g)
メタクリル酸コポリマーC型、30%分散液 100
タルク 6
クエン酸トリエチル 3
精製水 126
前の工程と同じコーティング装置でコーティングを行った。
入口空気温度は65℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.8bar、噴霧装置空気流量は2.8Nm3/時、スプレー速度は6−10g/分であり、結果として約38℃の出口空気温度となった。
遅延パルス放出ペレット
組成物において示される全ての量は、供給した量であり、収率に対して補正していない。
遅延パルス放出ペレットの製造のための概要の方式は、以下の順序でシードに層をコーティングすることによる;活性薬物(PPI)含有層→遅延放出改変層→遅滞時間制御層→腸溶性コーティング層。
遅延放出改変層を形成されたコアを実施例1に従って得た。
遅滞時間制御層のための溶液/懸濁液
賦形剤 量(g)
HPMC 4000cps 120
HPMC 6cps 16.5
EtOH 99.5% 2025
精製水 258
直径0.8mmの開口を有する液体ノズルを備えたWurster方式に従う流動床装置でコーティングを行った。
腸溶性コーティング懸濁液
賦形剤 量(g)
メタクリル酸コポリマーC型、30%分散液 100
タルク 6
クエン酸トリエチル 3
精製水 126
前の工程と同じコーティング装置でコーティングを行った。
得られた生成物のサンプルをインビトロ溶出について試験した。得られた溶出プロフィールを図2に示す。
溶出を実施例1のように試験した。
評価された遅滞時間は約2.5時間であった。勾配は約1.0〜1.1%/分(10-90)であった。
遅延パルス放出ペレット
組成物において示される全ての量は、供給した量であり、収率に対して補正していない。
遅延パルス放出ペレットの製造のための概要の方式は、以下の順序でシードに層をコーティングすることによる;活性薬物(PPI)含有層→遅延放出改変層→遅滞時間制御層→腸溶性コーティング層。
遅延放出改変層を形成されたコアを実施例1に従って得た。
遅滞時間制御層のための溶液/懸濁液
賦形剤 量(g)
HPMC 4000cps* 240
HPMC 6cps 33
EtOH 99.5% 4050
精製水 516
* 欧州薬局方に従って試験したpHは7.5であった。
直径0.8mmの開口を有する液体ノズルを備えたWurster方式に従う流動床装置でコーティングを行った。
腸溶性コーティング懸濁液
賦形剤 量(g)
メタクリル酸コポリマー C型、30%分散液 100
タルク 6
クエン酸トリエチル 3
精製水 126
前の工程と同じコーティング装置でコーティングを行った。
得られた生成物のサンプルをインビトロ溶出について試験した。得られた溶出プロフィールを図4に示す。
溶出試験を実施例1のように行った。
評価された遅滞時間は約4.5時間であった。勾配は約0.6〜0.7%/分(10-90)であった。
エソメプラゾールマグネシウム(40mg+40mg)の即時放出パルス及び遅延放出パルスを示すカプセル剤
二相性パルス放出カプセル剤の製造のための概略的方式は、即時放出のペレット及び遅延放出ペレットを混合し(すなわち、本発明による遅延放出改変層及び遅滞時間制御層の組み合わせを有するペレット)、そしてそれらをカプセルに充てんする。すなわち以下の順序に従った;
遅延放出ペレット(本発明による遅滞時間ペレット)を調製すること→従来技術に従って調製された即時放出ペレットと混合すること→カプセルに充てんすること。
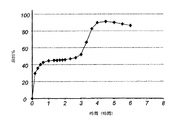
得られた生成物のサンプルをインビトロ溶出について試験した。得られた溶出プロフィールを図5に示す。
溶出を実施例1のように試験した。
遅延放出部分について評価した遅滞時間は約2.5時間であり、そして勾配は約1.3%/分(10-90)であった。
時間 分解生成物の量 *
0 0.2%
1年 0.2%
* オメプラゾールピークの面積に対する分解生成物のピークの面積の合計としてHPLCにより測定した。
遅延パルス放出ペレット
組成物において示される全ての量は、供給した量であり、収率に対して補正していない。
遅延パルス放出ペレットの製造のための概要の方式は、以下の順序でシードを層でコーティングすることによる;
活性薬物(PPI)含有層→遅延放出改変層→遅滞時間制御層→サブコーティング層→腸溶性コーティング層。
賦形剤 量(g)
エソメプラゾール−Mg三水和物 300
ポリソルベート80 6.0
ヒドロキシプロピルメチルセルロース6cps 45
精製水 1404
活性薬物層形成のためのシード
糖シード(ノンパレイユ)1.0−1.18mm 300
ヒドロキシプロピルメチルセルロース及びポリソルベート80を水に溶解し、その後エソメプラゾール−Mg三水和物をその中に懸濁させた。この懸濁液をアジテーターミル(Dyno−MillTM)で湿式微粉化工程にかけた。
調製した層形成懸濁液を直径0.8mmの開口を有する液体ノズルを備えたWurster方式に従う流動床装置で糖シード上にスプレーコートした。
遅延放出改変層溶液/懸濁液
賦形剤 量(g)
タルク粉末 45
ヒドロキシプロピルセルロース(75−150cps) 12
Mg−ステアレート 3.0
精製水 420
前の工程と同じコーティング装置でコーティングを行った。
遅滞時間制御層を2つの操作で上記の前の工程からの出発材料上に塗布し、結果として131gの出発材料が240gのHPMC 4000cps*(この工程において唯一のポリマーとして)でコーティングされ、その他の点では以前の実施例と同様であった(例えば同じ溶媒の組み合わせを使用した)。
(* 欧州薬局方に従って試験されたpHは7.5であった)。
前の工程と同じコーティング装置でコーティングを行った。
サブコーティング懸濁液
賦形剤 量(g)
タルク粉末 25
ヒドロキシプロピルセルロース(75−150cps) 6.7
Mg−ステアレート 1.7
精製水 234
前の工程と同じコーティング装置でコーティングを行った。
腸溶性コーティング懸濁液
賦形剤 量(g)
メタクリル酸コポリマーC型、30%分散液 100
タルク 6
クエン酸トリエチル 3
精製水 126
前の工程と同じコーティング装置でコーティングを行った。
得られた生成物のサンプルをインビトロ溶出について(実施例1のように)試験した。得られた溶出プロフィールを図6に示す。
評価された遅滞時間は約4時間であった。勾配は約0.7%/分(10-90)であった。
時間 分解生成物の量 *
0 0.2−0.3%
1か月 0.2−0.3%
2か月 0.67%
* オメプラゾールピークの面積に対する分解生成物のピークの面積の合計としてHPLCにより測定した。
時間 分解生成物の量 *
0 0.2−0.3%
1年 0.2−0.3%
2年 0.2−0.3%
* オメプラゾールピークの面積に対する分解生成物のピークの面積の合計としてHPLCにより測定した。
遅延パルス放出ペレット
組成物において示される全ての量は、供給した量であり、収率に対して補正していない。
遅延パルス放出ペレットの製造のための概要の方式は、以下の順序でシードに層をコーティングすることによる;
活性薬物(PPI)含有層→遅延放出改変層→遅滞時間制御層→腸溶性コーティング層
賦形剤 量(g)
エソメプラゾール−Mg三水和物 250
ポリソルベート80 5.0
ヒドロキシプロピルメチルセルロース6cps 37.5
精製水 1170
活性薬物層形成のためのシード
糖シード(ノンパレイユ)1.0−1.18mm 250
ヒドロキシプロピルメチルセルロース及びポリソルベート80を水に溶解し、その後エソメプラゾール−Mg三水和物をその中に懸濁させた。懸濁液をアジテーターミル(Dyno−MillTM)で湿式微粉化工程にかけた。
調製した層形成懸濁液を直径0.8mmの開口を有する液体ノズルを備えたWurster方式に従う流動床装置で糖シード上にスプレーコートした。
入口空気温度は80℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.5bar、噴霧装置空気流量は2.5Nm3/時、スプレー速度は12−19g/分であり、結果として約40℃の出口空気温度となった。
遅延放出改変層溶液/懸濁液
賦形剤 量(g)
タルク粉末 20.0
ヒドロキシプロピルセルロース(75−150cps) 9.0
ステアリルフマル酸ナトリウム(Pruv(登録商標)) 2.3
精製水 250
入口空気温度は75℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.8bar、噴霧装置空気流量は2.8Nm3/時、スプレー速度は6−11g/分であり、結果として約45℃の出口空気温度となった。
遅滞時間制御層のための溶液/懸濁液
賦形剤 量(g)
HPMC 4000cps 115.5
HPMC 6cps 15.9
EtOH 99.5% 1950
精製水 248
前の工程と同じコーティング装置でコーティングを行った。
入口空気温度は40℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.5bar、噴霧装置空気流量は2.5Nm3/時、スプレー速度は14−16g/分であり、結果として約20℃の出口空気温度となった。
腸溶性コーティング懸濁液
賦形剤 量(g)
メタクリル酸コポリマーC型、30%分散液 100
タルク 6
クエン酸トリエチル 3
精製水 126
前の工程と同じコーティング装置でコーティングを行った。
入口空気温度は65℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.8bar、噴霧装置空気流量は2.8Nm3/時、スプレー速度は6−10g/分であり、結果として約38℃の出口空気温度となった。
得られた生成物のサンプルをインビトロ溶出について試験した。得られた溶出プロフィールを図7に示す。
溶出試験を実施例1に記載されるように行った。
評価された遅滞時間は約2.5時間であり、そして勾配は約1.0−1.1%/分(10-90)であった。
時間が離れている2つのパルスを有する遅延パルス放出ペレット
組成物において示される全ての量は、供給した量であり、収率に対して補正していない。
遅延パルス放出ペレットの製造のための概要の方式は、以下の順序でシードに層をコーティングすることによる;
活性薬物(PPI)含有(第1)層→遅延放出改変層→遅滞時間制御層→活性薬物(PPI)含有(第2)層→サブコーティング層→腸溶性コーティング層。
賦形剤 量(g)
エソメプラゾール−Mg三水和物 250
ポリソルベート80 5.0
ヒドロキシプロピルメチルセルロース6cps 37.5
精製水 1170
活性薬物層形成のためのシード
糖シード(ノンパレイユ)1.0−1.18mm 250
ヒドロキシプロピルメチルセルロース(以下ではHPMCとも呼ぶ)及びポリソルベート80を水に溶解し、その後エソメプラゾール−Mg三水和物をその中に懸濁させた。この懸濁液をアジテーターミル(Dyno−MillTM)で湿式微粉化工程にかけた。
調製した層形成懸濁液を直径0.8mmの開口を有する液体ノズルを備えたWurster方式に従う流動床装置で糖シード上にスプレーコートした。
入口空気温度は80℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.5bar、噴霧装置空気流量は2.5Nm3/時、スプレー速度は12−19g/分であり、結果として約40℃の出口空気温度となった。
遅延放出改変層溶液/懸濁液
賦形剤 量(g)
タルク粉末 112.5
ヒドロキシプロピルセルロース(75−150cps) 30
Mg−ステアレート 7.5
精製水 1050
前の工程と同じコーティング装置でコーティングを行った。
入口空気温度は75℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.8bar、噴霧装置空気流量は2.8Nm3/時、スプレー速度は6−11g/分であり、結果として約45℃の出口空気温度となった。
遅滞時間制御層のための溶液/懸濁液
賦形剤 量(g)
HPMC 4000cps 120
HPMC 6cps 16.5
EtOH 99.5% 2025
精製水 258
第2の活性薬物(PPI)層のための層形成懸濁液
賦形剤 量(g)
微粉化したオメプラゾール粉末 40
ポリソルベート80 0.8
ヒドロキシプロピルメチルセルロース6cps 6
精製水 187
ヒドロキシプロピルメチルセルロース及びポリソルベート80を水に溶解し、その後オメプラゾール粉末をその中に懸濁させた。
調製した層形成懸濁液を、上記に従い先に得られたペレット上に同じ流動床装置でスプレーコートした。
入口空気温度は80℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.5bar、噴霧装置空気流量は2.5Nm3/時、スプレー速度は10−13g/分であり、結果として約40℃の出口空気温度となった。
サブコーティング層懸濁液
賦形剤 量(g)
タルク粉末 37.5
ヒドロキシプロピルセルロース(75−150cps) 10
ステアリン酸マグネシウム 2.5
精製水 350
前の工程と同じコーティング装置でコーティングを行った。
入口空気温度は75℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.8bar、噴霧装置空気流量は2.8Nm3/時、スプレー速度は6−11g/分であり、結果として約45℃の出口空気温度となった。
腸溶性コーティング懸濁液
賦形剤 量(g)
メタクリル酸コポリマーC型、30%分散液 100
タルク 6
クエン酸トリエチル 3
精製水 126
前の工程と同じコーティング装置でコーティングを行った。
得られた生成物のサンプルをインビトロ溶出について試験した。得られた溶出プロフィールを図8に示す。
溶出を実施例1のように試験した。
評価された第2パルスについての遅滞時間は約3時間であった。勾配は約1.4%/分(10-90)であった。
遅延パルス放出ペレット
組成物において示される全ての量は、供給した量であり、収率に対して補正していない。
遅延パルス放出ペレットの製造のための概要の方式は、以下の順序でシードに層をコーティングすることによる;
活性薬物(PPI)含有層→遅延放出改変層→遅滞時間制御層→腸溶性コーティング層。
賦形剤 量(g)
エソメプラゾール−Mg三水和物 250
ポリソルベート80 5.0
ヒドロキシプロピルメチルセルロース6cps 37.5
精製水 1170
活性薬物層形成のためのシード
糖シード(ノンパレイユ)1.0−1.18mm 250
ヒドロキシプロピルメチルセルロース及びポリソルベート80を水に溶解し、その後エソメプラゾール−Mg三水和物をその中に懸濁させた。懸濁液をアジテーターミル(Dyno−MillTM)で湿式微粉化工程にかけた。
調製した層形成懸濁液を直径0.8mmの開口を有する液体ノズルを備えたWurster方式に従う流動床装置で糖シード上にスプレーコートした。
入口空気温度は80℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.5bar、噴霧装置空気流量は2.5Nm3/時、スプレー速度は12−19g/分であり、結果として約40℃の出口空気温度となった。
遅延放出改変層溶液/懸濁液
賦形剤 量(g)
タルク粉末 112.5
ヒドロキシプロピルセルロース(75−150cps) 30
Mg−ステアレート 7.5
精製水 1050
前の工程と同じコーティング装置でコーティングを行った。
入口空気温度は75℃であり、流動空気流量は40m3/時、噴霧装置空気圧は2.8bar、噴霧装置空気流量は2.8Nm3/時、スプレー速度は6−11g/分であり、結果として約45℃の出口空気温度となった。
遅滞時間制御層のための溶液/懸濁液
賦形剤 量(g)
ヒドロキシエチルセルロース(Natrosol 250 HHX(登録商標))、
篩分け<100μm 90
HPMC 6cps 13.5
EtOH 99.5% 900
精製水 212
直径0.8mmの開口を有する液体ノズルを備えたWurster方式に従う流動床装置でコーティングを行った。
腸溶性コーティング懸濁液
賦形剤 量(g)
メタクリル酸コポリマーC型、30%分散液 100
タルク 6
クエン酸トリエチル 3
精製水 126
前の工程と同じコーティング装置でコーティングを行った。
得られた生成物のサンプルをインビトロ溶出について試験した。溶出プロフィールを図9に示す。
溶出を実施例1のように試験した。
評価された第2パルスについての遅滞時間は約2時間であった。勾配は約1.2%/分(10-90)であった。
遅延パルス放出ランソプラゾールペレット
組成物において示される全ての量は、供給した量であり、収率に対して補正していない。
遅延パルス放出ペレットの製造のための概要の方式は、以下の順序でシードに層をコーティングすることによる;
活性薬物(PPI)含有層→遅延放出改変層→遅滞時間制御層→腸溶性コーティング層。
賦形剤 量(g)
ランソプラゾール 250
ポリソルベート80 5.0
ヒドロキシプロピルメチルセルロース6cps 37.5
精製水 1170
活性薬物層形成のためのシード
糖シード(ノンパレイユ)1.0−1.18mm 250
ヒドロキシプロピルメチルセルロース及びポリソルベート80を水に溶解し、その後ランソプラゾールをその中に懸濁させた。この懸濁液をアジテーターミル(Dyno−MillTM)で湿式微粉化工程にかけた。
調製した層形成懸濁液を、直径0.8mmの開口を有する液体ノズルを備えたWurster方式に従う流動床装置で糖シード上にスプレーコートした。
入口空気温度を80℃に設定し、流動空気流量を40m3/時、噴霧装置空気圧を2.5bar、噴霧装置空気流量を2.5Nm3/時、スプレー速度を12−19g/分に設定した。
遅延放出改変層溶液/懸濁液
賦形剤 量(g)
タルク粉末 112.5
ヒドロキシプロピルセルロース(75−150cps) 30
Mg−ステアレート 7.5
精製水 1050
前の工程と同じコーティング装置でコーティングを行った。
入口空気温度を75℃に設定し、流動空気流量を40m3/時、噴霧装置空気圧を2.8bar、噴霧装置空気流量を2.8Nm3/時、スプレー速度を6−11g/分に設定した。
遅滞時間制御層のための溶液/懸濁液
賦形剤 量(g)
HPMC 4000cps 80
HPMC 6cps 11
EtOH 99.5% 1350
精製水 172
前の工程と同じ装置でコーティングを行った。
入口空気温度を40℃に設定し、流動空気流量を40m3/時、噴霧装置空気圧を2.5bar、噴霧装置空気流量を2.5Nm3/時、スプレー速度を14−16g/分に設定した。
腸溶性コーティング懸濁液
賦形剤 量(g)
メタクリル酸コポリマーC型、30%分散液 100
タルク 6
クエン酸トリエチル 3
精製水 126
前の工程と同じコーティング装置でコーティングを行った。
入口空気温度を65℃に設定し、流動空気流量を40m3/時、噴霧装置空気圧を2.8bar、噴霧装置空気流量を2.8Nm3/時、スプレー速度を6−10g/分に設定した。
Claims (23)
- 活性薬物として酸感受性プロトンポンプ阻害剤(PPI)を含む経口用固形医薬剤形であって、該剤形は、2つのPPI放出部分、遅延放出パルスでPPIを放出するペレット、及び即時放出パルスでPPIを放出するペレットを含み、PPIは、ペレットの形態でコア材料中に配合されており、そして遅延放出パルスを生じるペレットが、所定の順序でコア材料上に以下の層;
−水溶性ポリマー、タルク及び疎水性化剤を含む遅延放出改変層;
−必須成分として高粘性水溶性ポリマーを含む遅滞時間制御層;
−任意のサブコーティング層;及び
−外部腸溶性コーティング層;
を有し、
そして即時放出パルスを生じるペレットが、コア材料上に以下の層;
−任意のサブコーティング層;及び
−外部腸溶性コーティング層
を有することを特徴とする、上記剤形。 - 活性薬物として酸感受性プロトンポンプ阻害剤(PPI)を含む経口用固形医薬剤形であって、該剤形は、2つのPPI放出部分と共にペレットの一集団を含み、各ペレットは、遅延放出パルス及び即時放出パルスを生じ、PPIがペレットの形態でコア材料中に配合されており、そしてペレットが所定の順序でコア材料上に以下の層;
−水溶性ポリマー、タルク及び疎水性化剤を含む遅延放出改変層;
−必須成分として高粘性水溶性ポリマーを含む遅滞時間制御層;
−PPIの第2の部分を含む層;
−任意のサブコーティング層;及び
−外部腸溶性コーティング層
を有することを特徴とする、上記剤形。 - 最終剤形がカプセル剤である、請求項1又は2に記載の経口用医薬剤形。
- 最終剤形がサシェ剤である、請求項1又は2に記載の経口用医薬剤形。
- 即時放出パルスを生じるペレットが1つ又はそれ以上の錠剤の形態であり、そして最終剤形が遅延放出パルスのペレット及び即時放出パルスの錠剤を含む、請求項1、3又は4のいずれか1項に記載の経口用医薬剤形。
- 酸感受性プロトンポンプ阻害剤が、エソメプラゾールのアルカリ塩である、請求項1〜5のいずれか1項に記載の経口用医薬剤形。
- 酸感受性プロトンポンプ阻害剤が、エソメプラゾールマグネシウムである、請求項1〜5のいずれか1項に記載の経口用医薬剤形。
- 酸感受性プロトンポンプ阻害剤が、オメプラゾールマグネシウムである、請求項1〜5のいずれか1項に記載の経口用医薬剤形。
- 1〜10時間の範囲の、遅延(第2)パルスについての遅滞時間を有する、請求項1〜8のいずれか1項に記載の経口用医薬剤形。
- 2〜8時間の範囲の遅滞時間を有する、請求項9に記載の経口用医薬剤形。
- 遅滞時間制御層が、コーティング過程からのあらゆる残留物以外の唯一の成分として高粘性水溶性ポリマーを含む、請求項1〜10のいずれか1項に記載の経口用医薬剤形。
- 遅滞時間制御層における必須成分が、高粘性ヒドロキシプロピルメチルセルロース又は高粘性ヒドロキシエチルセルロースである、請求項1〜11のいずれか1項に記載の経口用医薬剤形。
- 高粘性ヒドロキシプロピルメチルセルロース又はヒドロキシエチルセルロースが、欧州薬局方に従って測定した場合に7.0〜9.0の間のpHを示す、請求項12に記載の経口用医薬剤形。
- 遅延放出改変層が、水溶性ポリマー、タルク並びにMg−ステアレート、ベヘン酸グリセリル及びステアリルフマル酸ナトリウムからなる群より選択される疎水性化剤を含む、請求項1〜13のいずれか1項に記載の経口用医薬剤形。
- 遅延放出改変層が、50〜90%の範囲のヒドロキシプロピル含量及び180cps以下の粘度を有するヒドロキシプロピルセルロース、タルク、並びにMg−ステアレートのみから構成される、請求項1〜14のいずれか1項に記載の経口用医薬剤形。
- 請求項1に記載の経口用医薬剤形を製造するための方法であって、以下の工程;
I) 活性薬物として酸感受性プロトンポンプ阻害剤(PPI)を含むペレットの形態でコア材料を調製する工程;
II) 工程I)で得られたペレットコアを、遅延放出改変層でコーティングする工程;
III) 工程II)からの遅延放出改変層を形成されたペレットコアを、必須成分として高粘性水溶性ポリマーを含む遅滞時間制御層でコーティングする工程;
IV) 工程III)からの遅滞時間制御層を形成されたペレットを、外部腸溶性コーティングでコーティングする工程であって、任意のサブコーティング層が、腸溶性コーティング層が塗布される前に塗布される工程;並びに
V) 工程IV)で得られたペレット生成物を、外部腸溶性コーティング及び任意のサブコーティング層を有しPPIの即時放出を生じる他のペレットと共に、カプセル剤、サシェ剤又は複数単位ペレット系錠剤に組み込む工程、
を含む、上記方法。 - 請求項2に記載の経口用医薬剤形を製造するための方法であって、以下の工程;
I) 活性薬物として酸感受性プロトンポンプ阻害剤(PPI)を含むペレットの形態でコア材料を調製する工程;
II) 工程I)からのペレットコアを、遅延放出改変層でコーティングする工程;
III) 工程II)からの遅延放出改変層を形成されたペレットコアを、必須成分として高粘性水溶性ポリマーを含む遅滞時間制御層でコーティングする工程;
IV) 工程III)からの遅滞時間制御層を形成されたペレットを、第2のPPI部分を含む層でコーティングする工程;
V) 工程IV)から得られたペレットを、任意のサブコーティング層で場合によりコーティングする工程;並びに
VI) 工程V)から得られたペレット生成物を、外部腸溶性コーティングでコーティングする工程;
VII) 工程VI)から得られた腸溶性コーティングを施されたペレットを、カプセル剤、サシェ剤、又は複数単位ペレット系錠剤に製剤化する工程、
を含む、上記方法。 - 工程V)における即時放出パルスを生じるペレットが、1つ又はそれ以上の錠剤の形態であり、そして剤形が遅延放出パルスのペレット及び即時放出パルスの錠剤を含む、請求項16に記載の方法。
- 工程II)からの遅延放出改変層を形成されたペレットコアを遅滞時間制御層でコーティングする工程III)が、
a) 高粘性水溶性ポリマーを非溶媒中に分散させること;及び
b) 水性液体又は水を加えて分散したポリマー粒子の水和形態を形成すること、
により製造される高粘性水溶性ポリマーの分散液を利用することにより行われる、請求項16又は17に記載の方法。 - 工程IIにより得られる遅延放出改変層が、コーティング過程からの溶媒/分散媒体/懸濁媒体のあらゆる残留物以外はヒドロキシプロピルセルロース、タルク及びMg−ステアレートの成分のみから構成される、請求項16〜19のいずれか1項に記載の方法。
- 得られた生成物が、1〜10時間の範囲の遅滞時間を有する、請求項16〜20のいずれか1項に記載の方法。
- 胃酸分泌の抑制を改善することを必要とする患者に、請求項1〜15のいずれか1項に規定される経口用医薬剤形を投与することからなる、胃酸分泌の抑制を改善するための方法。
- 胃腸疾患の処置における、請求項1〜15のいずれか1項に記載の医薬剤形の使用。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US62562104P | 2004-11-04 | 2004-11-04 | |
| PCT/SE2005/001642 WO2006049564A1 (en) | 2004-11-04 | 2005-11-02 | New modified release pellet formulations for proton pump inhibitors |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2008519069A true JP2008519069A (ja) | 2008-06-05 |
| JP2008519069A5 JP2008519069A5 (ja) | 2008-12-11 |
Family
ID=36319457
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007540283A Pending JP2008519069A (ja) | 2004-11-04 | 2005-11-02 | プロトンポンプ阻害剤用の新規な改変された放出のペレット製剤 |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US20080095853A1 (ja) |
| EP (1) | EP1809263A4 (ja) |
| JP (1) | JP2008519069A (ja) |
| KR (1) | KR20070073867A (ja) |
| CN (1) | CN101094660A (ja) |
| AR (1) | AR051654A1 (ja) |
| AU (1) | AU2005301368A1 (ja) |
| BR (1) | BRPI0517933A (ja) |
| CA (1) | CA2584417A1 (ja) |
| IL (1) | IL182696A0 (ja) |
| MX (1) | MX2007004986A (ja) |
| NO (1) | NO20072254L (ja) |
| RU (1) | RU2007115537A (ja) |
| TW (1) | TW200624127A (ja) |
| UY (1) | UY29192A1 (ja) |
| WO (1) | WO2006049564A1 (ja) |
| ZA (1) | ZA200703112B (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014088356A (ja) * | 2012-10-31 | 2014-05-15 | Shin Etsu Chem Co Ltd | 高粘度ヒプロメロースを分散したコーティング液並びに固形製剤及びその製造方法 |
| JP2019507158A (ja) * | 2016-02-29 | 2019-03-14 | 株式会社柳英製薬Yoo Young Pharm Co., Ltd. | エソメプラゾールを含有する製剤 |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102406628A (zh) * | 2010-09-26 | 2012-04-11 | 上海复星普适医药科技有限公司 | 一种稳定的埃索美拉唑肠溶小丸的制备方法 |
| DE102010052847A1 (de) * | 2010-11-29 | 2012-05-31 | Temmler Werke Gmbh | Verfahren zur Herstellung einer PPI-haltigen pharmazeutischen Zubereitung |
| TWI483749B (zh) * | 2010-12-03 | 2015-05-11 | Nippon Soda Co | 羥烷基纖維素 |
| UA112855C2 (uk) * | 2010-12-29 | 2016-11-10 | Др. Редді'С Лабораторіс Лтд. | Композиції бензімідазолів з модифікованим вивільненням |
| CN103565770A (zh) * | 2012-07-31 | 2014-02-12 | 北京阜康仁生物制药科技有限公司 | 一种右兰索拉唑肠溶缓控释微丸片 |
| CN104586809A (zh) * | 2015-01-08 | 2015-05-06 | 浙江亚太药业股份有限公司 | 一种埃索美拉唑镁微丸肠溶片及制备方法 |
| EP3288556A4 (en) | 2015-04-29 | 2018-09-19 | Dexcel Pharma Technologies Ltd. | Orally disintegrating compositions |
| US10076494B2 (en) | 2016-06-16 | 2018-09-18 | Dexcel Pharma Technologies Ltd. | Stable orally disintegrating pharmaceutical compositions |
| CN111991367A (zh) * | 2020-09-21 | 2020-11-27 | 青岛吉达巴尔国际贸易有限公司 | 一种埃索美拉唑镁脉冲微丸胶囊剂及制备方法 |
| WO2022154687A1 (ru) * | 2021-01-14 | 2022-07-21 | Общество C Ограниченной Ответственностью "Новамедика" | Фармацевтическая композиция, включающая эзомепразол |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001526213A (ja) * | 1997-12-22 | 2001-12-18 | アストラゼネカ・アクチエボラーグ | 経口医薬パルス放出剤形 |
| JP2003171277A (ja) * | 2001-12-07 | 2003-06-17 | Wyeth Lederle Japan Ltd | 薬物放出時間制御型固形製剤 |
Family Cites Families (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL75400A (en) * | 1984-06-16 | 1988-10-31 | Byk Gulden Lomberg Chem Fab | Dialkoxypyridine methyl(sulfinyl or sulfonyl)benzimidazoles,processes for the preparation thereof and pharmaceutical compositions containing the same |
| IT1230576B (it) * | 1988-10-20 | 1991-10-28 | Angeli Inst Spa | Formulazioni farmaceutiche per via orale a liberazione selettiva nel colon |
| JPH07223970A (ja) * | 1994-02-10 | 1995-08-22 | Tanabe Seiyaku Co Ltd | 消化管内適所放出製剤 |
| US5945124A (en) * | 1995-07-05 | 1999-08-31 | Byk Gulden Chemische Fabrik Gmbh | Oral pharmaceutical composition with delayed release of active ingredient for pantoprazole |
| US20050054682A1 (en) * | 1996-01-04 | 2005-03-10 | Phillips Jeffrey O. | Pharmaceutical compositions comprising substituted benzimidazoles and methods of using same |
| SE9600072D0 (sv) * | 1996-01-08 | 1996-01-08 | Astra Ab | New oral formulation of two active ingredients II |
| CA2173818A1 (fr) * | 1996-04-10 | 1997-10-11 | Francois Chouinard | Comprime pharmaceutique a liberation controlee contenant un support a base d'amylose reticule et d'hydroxypropylmethylcellulose |
| AU5179898A (en) * | 1996-11-06 | 1998-05-29 | Sharmatek, Inc. | Delayed delivery system for acid-sensitive drugs |
| US5885616A (en) * | 1997-08-18 | 1999-03-23 | Impax Pharmaceuticals, Inc. | Sustained release drug delivery system suitable for oral administration |
| US6645503B1 (en) * | 1998-03-10 | 2003-11-11 | Wyeth Holdings Corporation | Antigenic conjugates of conserved lipopolysaccharides of gram negative bacteria |
| PT1126826E (pt) * | 1998-11-02 | 2008-11-25 | Elan Pharma Int Ltd | Composição de metilfenidato de libertação modificada multiparticulada |
| US6632451B2 (en) * | 1999-06-04 | 2003-10-14 | Dexcel Pharma Technologies Ltd. | Delayed total release two pulse gastrointestinal drug delivery system |
| EP1064938A1 (en) * | 1999-06-28 | 2001-01-03 | Sanofi-Synthelabo | Pharmaceutical dosage forms for controlled release producing at least a timed pulse |
| EP1074249A1 (en) * | 1999-07-27 | 2001-02-07 | Jagotec Ag | A pharmaceutical tablet system for releasing at least one active substance during a release period subsequent to a no-release period |
| ES2168043B1 (es) * | 1999-09-13 | 2003-04-01 | Esteve Labor Dr | Forma farmaceutica solida oral de liberacion modificada que contiene un compuesto de bencimidazol labil en medio acido. |
| GB9923436D0 (en) * | 1999-10-04 | 1999-12-08 | American Home Prod | Pharmaceutical compositions |
| US20030180352A1 (en) * | 1999-11-23 | 2003-09-25 | Patel Mahesh V. | Solid carriers for improved delivery of active ingredients in pharmaceutical compositions |
| US6627223B2 (en) * | 2000-02-11 | 2003-09-30 | Eurand Pharmaceuticals Ltd. | Timed pulsatile drug delivery systems |
| US6749867B2 (en) * | 2000-11-29 | 2004-06-15 | Joseph R. Robinson | Delivery system for omeprazole and its salts |
| CN100408029C (zh) * | 2001-09-28 | 2008-08-06 | 麦克内尔-Ppc股份有限公司 | 有镶嵌部分的组合剂型 |
| CA2496044A1 (en) * | 2002-08-16 | 2004-02-26 | Themis Laboratories Private Limited | A process for manufacture of stable oral multiple units pharamceutical composition containing benzimidazoles |
| US8487002B2 (en) * | 2002-10-25 | 2013-07-16 | Paladin Labs Inc. | Controlled-release compositions |
| US20040110781A1 (en) * | 2002-12-05 | 2004-06-10 | Harmon Troy M. | Pharmaceutical compositions containing indistinguishable drug components |
| WO2004071374A2 (en) * | 2003-02-11 | 2004-08-26 | Torrent Pharmaceuticals Limited | Once a day orally administered pharmaceutical compositions |
| US7670624B2 (en) * | 2004-01-29 | 2010-03-02 | Astella Pharma Inc. | Gastrointestinal-specific multiple drug release system |
| US20050239845A1 (en) * | 2004-04-16 | 2005-10-27 | Santarus, Inc. | Combination of proton pump inhibitor, buffering agent, and prokinetic agent |
| AR052225A1 (es) * | 2004-11-04 | 2007-03-07 | Astrazeneca Ab | Formulaciones de tabletas de liberacion modificada par inhibidores de la bomba de protones |
| US20060165797A1 (en) * | 2005-01-12 | 2006-07-27 | Pozen Inc. | Dosage form for treating gastrointestinal disorders |
-
2005
- 2005-10-31 AR ARP050104548A patent/AR051654A1/es not_active Application Discontinuation
- 2005-11-02 KR KR1020077010116A patent/KR20070073867A/ko not_active Application Discontinuation
- 2005-11-02 BR BRPI0517933-5A patent/BRPI0517933A/pt not_active Application Discontinuation
- 2005-11-02 MX MX2007004986A patent/MX2007004986A/es not_active Application Discontinuation
- 2005-11-02 WO PCT/SE2005/001642 patent/WO2006049564A1/en active Application Filing
- 2005-11-02 JP JP2007540283A patent/JP2008519069A/ja active Pending
- 2005-11-02 RU RU2007115537/15A patent/RU2007115537A/ru unknown
- 2005-11-02 US US11/718,583 patent/US20080095853A1/en not_active Abandoned
- 2005-11-02 CA CA002584417A patent/CA2584417A1/en not_active Abandoned
- 2005-11-02 AU AU2005301368A patent/AU2005301368A1/en not_active Abandoned
- 2005-11-02 EP EP05801799A patent/EP1809263A4/en not_active Withdrawn
- 2005-11-02 CN CNA2005800457444A patent/CN101094660A/zh active Pending
- 2005-11-04 TW TW094138679A patent/TW200624127A/zh unknown
- 2005-11-04 UY UY29192A patent/UY29192A1/es not_active Application Discontinuation
-
2007
- 2007-04-16 ZA ZA200703112A patent/ZA200703112B/xx unknown
- 2007-04-19 IL IL182696A patent/IL182696A0/en unknown
- 2007-05-02 NO NO20072254A patent/NO20072254L/no not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001526213A (ja) * | 1997-12-22 | 2001-12-18 | アストラゼネカ・アクチエボラーグ | 経口医薬パルス放出剤形 |
| JP2003171277A (ja) * | 2001-12-07 | 2003-06-17 | Wyeth Lederle Japan Ltd | 薬物放出時間制御型固形製剤 |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014088356A (ja) * | 2012-10-31 | 2014-05-15 | Shin Etsu Chem Co Ltd | 高粘度ヒプロメロースを分散したコーティング液並びに固形製剤及びその製造方法 |
| JP2019507158A (ja) * | 2016-02-29 | 2019-03-14 | 株式会社柳英製薬Yoo Young Pharm Co., Ltd. | エソメプラゾールを含有する製剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2006049564A1 (en) | 2006-05-11 |
| ZA200703112B (en) | 2008-10-29 |
| CA2584417A1 (en) | 2006-05-11 |
| UY29192A1 (es) | 2006-06-30 |
| US20080095853A1 (en) | 2008-04-24 |
| RU2007115537A (ru) | 2008-12-10 |
| NO20072254L (no) | 2007-07-30 |
| MX2007004986A (es) | 2007-06-14 |
| AU2005301368A1 (en) | 2006-05-11 |
| EP1809263A4 (en) | 2012-09-26 |
| AR051654A1 (es) | 2007-01-31 |
| IL182696A0 (en) | 2007-09-20 |
| CN101094660A (zh) | 2007-12-26 |
| KR20070073867A (ko) | 2007-07-10 |
| EP1809263A1 (en) | 2007-07-25 |
| TW200624127A (en) | 2006-07-16 |
| BRPI0517933A (pt) | 2008-10-21 |
| WO2006049564A8 (en) | 2007-06-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2008519070A (ja) | プロトンポンプ阻害剤用の新規な改変された放出の錠剤製剤 | |
| JP2008519069A (ja) | プロトンポンプ阻害剤用の新規な改変された放出のペレット製剤 | |
| KR101752014B1 (ko) | 고용량 및 저용량 약물들의 조합을 포함하는 구강붕해정 조성물 | |
| RU2428176C2 (ru) | Системы доставки лекарственного средства, содержащие слабоосновные лекарственные средства и органические кислоты | |
| JP4885347B2 (ja) | 酸に不安定ベンズイミダゾールを含有する修飾された放出特性の経口用固形製剤 | |
| JP2020059755A (ja) | 時限パルス放出システム | |
| TWI440480B (zh) | 包含弱鹼性藥物及有機酸之藥物遞送系統 | |
| JP2016014048A (ja) | 弱塩基性選択性セロトニン5−ht3遮断剤および有機酸を含む薬物送達系 | |
| JP2002532425A (ja) | 新規な医薬製剤 | |
| JP5925318B2 (ja) | 有核錠 | |
| SG190905A1 (en) | Orally disintegrating tablet | |
| CN108348475B (zh) | 处于液体剂型的多层药学活性化合物释放微粒 | |
| EP2773348B1 (en) | Pharmaceutical composition of omeprazole | |
| EP4007568A1 (en) | Pharmaceutical composition comprising dabigatran etexilate | |
| JP2009542669A (ja) | ピペリジノアルカノールと充血除去剤の組み合わせを含んで成る医薬組成物 | |
| EP2535045A1 (en) | Pharmaceutical composition of lansoprazole | |
| WO2010018593A2 (en) | Gastric acid resistant benzimidazole multiple unit tablet composition | |
| EP3380084B1 (en) | Omeprazole formulations | |
| AU2013204408B2 (en) | Drug delivery systems comprising weakly basic selective serotonin 5-HT3 blocking agent and organic acids | |
| MX2014008975A (es) | Composicion de nitazoxanida mejorada y proceso para prepararla. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20081022 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20081022 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20111121 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20111129 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20120515 |