JP2004513070A - β−L−2’−デオキシヌクレオシドを用いたデルタ型肝炎ウイルス感染の処置方法 - Google Patents
β−L−2’−デオキシヌクレオシドを用いたデルタ型肝炎ウイルス感染の処置方法 Download PDFInfo
- Publication number
- JP2004513070A JP2004513070A JP2001587753A JP2001587753A JP2004513070A JP 2004513070 A JP2004513070 A JP 2004513070A JP 2001587753 A JP2001587753 A JP 2001587753A JP 2001587753 A JP2001587753 A JP 2001587753A JP 2004513070 A JP2004513070 A JP 2004513070A
- Authority
- JP
- Japan
- Prior art keywords
- alkyl
- deoxy
- acyl
- erythro
- use according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 238000000034 method Methods 0.000 title claims abstract description 65
- 241000724709 Hepatitis delta virus Species 0.000 title claims description 65
- 230000009385 viral infection Effects 0.000 title description 9
- 150000003839 salts Chemical class 0.000 claims abstract description 81
- 239000000651 prodrug Substances 0.000 claims abstract description 71
- 229940002612 prodrug Drugs 0.000 claims abstract description 71
- 208000037262 Hepatitis delta Diseases 0.000 claims description 65
- 239000001226 triphosphate Substances 0.000 claims description 61
- 235000011178 triphosphate Nutrition 0.000 claims description 61
- UNXRWKVEANCORM-UHFFFAOYSA-N triphosphoric acid Chemical compound OP(O)(=O)OP(O)(=O)OP(O)(O)=O UNXRWKVEANCORM-UHFFFAOYSA-N 0.000 claims description 57
- -1 lobukavir Chemical compound 0.000 claims description 52
- 125000002252 acyl group Chemical group 0.000 claims description 51
- 150000003013 phosphoric acid derivatives Chemical class 0.000 claims description 41
- 125000000217 alkyl group Chemical group 0.000 claims description 35
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 26
- 125000000539 amino acid group Chemical group 0.000 claims description 26
- 125000005140 aralkylsulfonyl group Chemical group 0.000 claims description 26
- 125000004391 aryl sulfonyl group Chemical group 0.000 claims description 26
- 150000001413 amino acids Chemical class 0.000 claims description 24
- 239000003814 drug Substances 0.000 claims description 22
- 239000001177 diphosphate Substances 0.000 claims description 21
- 235000011180 diphosphates Nutrition 0.000 claims description 21
- CKTSBUTUHBMZGZ-CHKWXVPMSA-N 4-amino-1-[(2s,4r,5s)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)[C@H](O)C1 CKTSBUTUHBMZGZ-CHKWXVPMSA-N 0.000 claims description 20
- 239000003795 chemical substances by application Substances 0.000 claims description 20
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 claims description 20
- 150000004712 monophosphates Chemical class 0.000 claims description 18
- KDCGOANMDULRCW-UHFFFAOYSA-N Purine Natural products N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 claims description 13
- 208000002672 hepatitis B Diseases 0.000 claims description 13
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 claims description 11
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 claims description 11
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 claims description 11
- 229910052736 halogen Inorganic materials 0.000 claims description 11
- 150000002367 halogens Chemical class 0.000 claims description 11
- 239000004474 valine Substances 0.000 claims description 11
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 10
- 229910052739 hydrogen Inorganic materials 0.000 claims description 10
- IQFYYKKMVGJFEH-CSMHCCOUSA-N telbivudine Chemical compound O=C1NC(=O)C(C)=CN1[C@H]1O[C@@H](CO)[C@H](O)C1 IQFYYKKMVGJFEH-CSMHCCOUSA-N 0.000 claims description 9
- 239000002253 acid Substances 0.000 claims description 7
- 238000004519 manufacturing process Methods 0.000 claims description 7
- IGFXRKMLLMBKSA-UHFFFAOYSA-N purine Chemical compound N1=C[N]C2=NC=NC2=C1 IGFXRKMLLMBKSA-UHFFFAOYSA-N 0.000 claims description 7
- 229960005311 telbivudine Drugs 0.000 claims description 7
- 125000002264 triphosphate group Chemical class [H]OP(=O)(O[H])OP(=O)(O[H])OP(=O)(O[H])O* 0.000 claims description 7
- WOZSCQDILHKSGG-UHFFFAOYSA-N adefovir depivoxil Chemical compound N1=CN=C2N(CCOCP(=O)(OCOC(=O)C(C)(C)C)OCOC(=O)C(C)(C)C)C=NC2=C1N WOZSCQDILHKSGG-UHFFFAOYSA-N 0.000 claims description 6
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 claims description 4
- GBBJCSTXCAQSSJ-XQXXSGGOSA-N clevudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1[C@H](F)[C@@H](O)[C@H](CO)O1 GBBJCSTXCAQSSJ-XQXXSGGOSA-N 0.000 claims description 4
- 229960000329 ribavirin Drugs 0.000 claims description 4
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 claims description 4
- 229960004396 famciclovir Drugs 0.000 claims description 3
- GGXKWVWZWMLJEH-UHFFFAOYSA-N famcyclovir Chemical compound N1=C(N)N=C2N(CCC(COC(=O)C)COC(C)=O)C=NC2=C1 GGXKWVWZWMLJEH-UHFFFAOYSA-N 0.000 claims description 3
- LLYJISDUHFXOHK-GOCONZMPSA-N ferroptocide Chemical compound C[C@@H]1CC[C@@]23C[C@@H](C(=O)[C@]2([C@@]1([C@@H](C[C@H]([C@@H]3C)C4=CCN5C(=O)N(C(=O)N5C4)C6=CC=CC=C6)OC(=O)CCl)C)O)O LLYJISDUHFXOHK-GOCONZMPSA-N 0.000 claims description 3
- JNTOCHDNEULJHD-UHFFFAOYSA-N Penciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(CCC(CO)CO)C=N2 JNTOCHDNEULJHD-UHFFFAOYSA-N 0.000 claims description 2
- RLAHNGKRJJEIJL-RFZPGFLSSA-N [(2r,4r)-4-(2,6-diaminopurin-9-yl)-1,3-dioxolan-2-yl]methanol Chemical compound C12=NC(N)=NC(N)=C2N=CN1[C@H]1CO[C@@H](CO)O1 RLAHNGKRJJEIJL-RFZPGFLSSA-N 0.000 claims description 2
- 229960001997 adefovir Drugs 0.000 claims description 2
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 claims description 2
- 229960002963 ganciclovir Drugs 0.000 claims description 2
- 229960001179 penciclovir Drugs 0.000 claims description 2
- AFINAILKDBCXMX-PBHICJAKSA-N (2s,3r)-2-amino-3-hydroxy-n-(4-octylphenyl)butanamide Chemical compound CCCCCCCCC1=CC=C(NC(=O)[C@@H](N)[C@@H](C)O)C=C1 AFINAILKDBCXMX-PBHICJAKSA-N 0.000 claims 18
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims 9
- 239000001257 hydrogen Substances 0.000 claims 9
- 239000000203 mixture Substances 0.000 abstract description 58
- 208000005331 Hepatitis D Diseases 0.000 abstract description 26
- 238000011282 treatment Methods 0.000 abstract description 25
- 150000001875 compounds Chemical class 0.000 description 111
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 86
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 81
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 68
- 239000002777 nucleoside Substances 0.000 description 58
- 239000000243 solution Substances 0.000 description 53
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 51
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 51
- 229910001868 water Inorganic materials 0.000 description 47
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 42
- 208000015181 infectious disease Diseases 0.000 description 42
- 241000700721 Hepatitis B virus Species 0.000 description 41
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 36
- 230000015572 biosynthetic process Effects 0.000 description 36
- 230000000694 effects Effects 0.000 description 34
- 230000000840 anti-viral effect Effects 0.000 description 33
- 238000003786 synthesis reaction Methods 0.000 description 33
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N DMSO-d6 Substances [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 31
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 30
- 238000005160 1H NMR spectroscopy Methods 0.000 description 27
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 26
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 24
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 22
- 125000003729 nucleotide group Chemical group 0.000 description 22
- 210000004027 cell Anatomy 0.000 description 20
- 229940079593 drug Drugs 0.000 description 20
- 150000003833 nucleoside derivatives Chemical class 0.000 description 20
- 125000003835 nucleoside group Chemical group 0.000 description 20
- 239000002773 nucleotide Substances 0.000 description 20
- 230000002829 reductive effect Effects 0.000 description 20
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 17
- 208000006454 hepatitis Diseases 0.000 description 16
- 150000002632 lipids Chemical class 0.000 description 16
- 239000011159 matrix material Substances 0.000 description 16
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 15
- JTEGQNOMFQHVDC-NKWVEPMBSA-N lamivudine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)SC1 JTEGQNOMFQHVDC-NKWVEPMBSA-N 0.000 description 15
- 238000010898 silica gel chromatography Methods 0.000 description 15
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 14
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 14
- 102000036639 antigens Human genes 0.000 description 14
- 108091007433 antigens Proteins 0.000 description 14
- 125000001424 substituent group Chemical group 0.000 description 14
- 241000700605 Viruses Species 0.000 description 13
- 241001492404 Woodchuck hepatitis virus Species 0.000 description 13
- 239000000427 antigen Substances 0.000 description 13
- 238000009472 formulation Methods 0.000 description 13
- 229960001627 lamivudine Drugs 0.000 description 13
- 229930024421 Adenine Natural products 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical group O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 12
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 12
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 11
- 229960000643 adenine Drugs 0.000 description 11
- 201000010099 disease Diseases 0.000 description 11
- 239000006260 foam Substances 0.000 description 11
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 11
- 239000011541 reaction mixture Substances 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- 239000002904 solvent Substances 0.000 description 11
- 230000003612 virological effect Effects 0.000 description 11
- 229960002555 zidovudine Drugs 0.000 description 11
- DRTQHJPVMGBUCF-PSQAKQOGSA-N 1-[(2s,3s,4r,5s)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidine-2,4-dione Chemical compound O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 10
- 241000283923 Marmota monax Species 0.000 description 10
- 241001465754 Metazoa Species 0.000 description 10
- 210000004185 liver Anatomy 0.000 description 10
- 238000002360 preparation method Methods 0.000 description 10
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 9
- 230000001684 chronic effect Effects 0.000 description 9
- 229940104302 cytosine Drugs 0.000 description 9
- 238000000921 elemental analysis Methods 0.000 description 9
- 239000003480 eluent Substances 0.000 description 9
- 238000011156 evaluation Methods 0.000 description 9
- 108020004707 nucleic acids Proteins 0.000 description 9
- 102000039446 nucleic acids Human genes 0.000 description 9
- 239000012074 organic phase Substances 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- 229910052938 sodium sulfate Inorganic materials 0.000 description 9
- 239000007858 starting material Substances 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 229940035893 uracil Drugs 0.000 description 9
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 8
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- 229910002091 carbon monoxide Inorganic materials 0.000 description 8
- 239000000969 carrier Substances 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 208000029570 hepatitis D virus infection Diseases 0.000 description 8
- 230000000670 limiting effect Effects 0.000 description 8
- 150000007523 nucleic acids Chemical class 0.000 description 8
- UIIMBOGNXHQVGW-UHFFFAOYSA-M sodium bicarbonate Substances [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- 238000002560 therapeutic procedure Methods 0.000 description 8
- 206010008909 Chronic Hepatitis Diseases 0.000 description 7
- 241000282412 Homo Species 0.000 description 7
- 229910019142 PO4 Inorganic materials 0.000 description 7
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 7
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 7
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 7
- PYMYPHUHKUWMLA-MROZADKFSA-N aldehydo-L-ribose Chemical compound OC[C@H](O)[C@H](O)[C@H](O)C=O PYMYPHUHKUWMLA-MROZADKFSA-N 0.000 description 7
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 7
- 229910052786 argon Inorganic materials 0.000 description 7
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 7
- 239000003937 drug carrier Substances 0.000 description 7
- 238000012986 modification Methods 0.000 description 7
- 235000021317 phosphate Nutrition 0.000 description 7
- 230000010076 replication Effects 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- 235000011152 sodium sulphate Nutrition 0.000 description 7
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 7
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 6
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 6
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 6
- SRBFZHDQGSBBOR-HWQSCIPKSA-N L-arabinopyranose Chemical compound O[C@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-HWQSCIPKSA-N 0.000 description 6
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 6
- 229960000583 acetic acid Drugs 0.000 description 6
- 239000004480 active ingredient Substances 0.000 description 6
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 6
- 239000003443 antiviral agent Substances 0.000 description 6
- 239000002552 dosage form Substances 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 238000004949 mass spectrometry Methods 0.000 description 6
- 230000004048 modification Effects 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 239000008194 pharmaceutical composition Substances 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 6
- 238000000967 suction filtration Methods 0.000 description 6
- 235000000346 sugar Nutrition 0.000 description 6
- 230000002194 synthesizing effect Effects 0.000 description 6
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 6
- OLXZPDWKRNYJJZ-VQVTYTSYSA-N (2s,3r,5s)-5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-ol Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1C[C@@H](O)[C@H](CO)O1 OLXZPDWKRNYJJZ-VQVTYTSYSA-N 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- 241000725303 Human immunodeficiency virus Species 0.000 description 5
- 102000006601 Thymidine Kinase Human genes 0.000 description 5
- 108020004440 Thymidine kinase Proteins 0.000 description 5
- 235000011054 acetic acid Nutrition 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 125000003118 aryl group Chemical group 0.000 description 5
- 230000004071 biological effect Effects 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 238000002648 combination therapy Methods 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 230000001965 increasing effect Effects 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 239000002245 particle Substances 0.000 description 5
- 239000010452 phosphate Substances 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- 210000002966 serum Anatomy 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- 0 *c1nc2c(*)nc(*)nc2[n]1[C@](CC12)C3[C@]1(CO)C23O Chemical compound *c1nc2c(*)nc(*)nc2[n]1[C@](CC12)C3[C@]1(CO)C23O 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 206010016654 Fibrosis Diseases 0.000 description 4
- 241000700588 Human alphaherpesvirus 1 Species 0.000 description 4
- 208000001940 Massive Hepatic Necrosis Diseases 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- GCZABPLTDYVJMP-TVQWTUMOSA-N [(2s,3s,4s,5r)-5-acetyloxy-3,4-dibenzoyloxyoxolan-2-yl]methyl benzoate Chemical compound C([C@@H]1O[C@@H]([C@H]([C@H]1OC(=O)C=1C=CC=CC=1)OC(=O)C=1C=CC=CC=1)OC(=O)C)OC(=O)C1=CC=CC=C1 GCZABPLTDYVJMP-TVQWTUMOSA-N 0.000 description 4
- 230000009102 absorption Effects 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 230000036436 anti-hiv Effects 0.000 description 4
- 230000000259 anti-tumor effect Effects 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- 238000012925 biological evaluation Methods 0.000 description 4
- 230000007882 cirrhosis Effects 0.000 description 4
- 208000019425 cirrhosis of liver Diseases 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 230000008030 elimination Effects 0.000 description 4
- 238000003379 elimination reaction Methods 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 239000000796 flavoring agent Substances 0.000 description 4
- 231100000283 hepatitis Toxicity 0.000 description 4
- 230000002779 inactivation Effects 0.000 description 4
- 230000002458 infectious effect Effects 0.000 description 4
- 239000004615 ingredient Substances 0.000 description 4
- 239000003112 inhibitor Substances 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 239000002502 liposome Substances 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 230000004060 metabolic process Effects 0.000 description 4
- 239000006186 oral dosage form Substances 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 239000002342 ribonucleoside Substances 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 239000000829 suppository Substances 0.000 description 4
- 239000006188 syrup Substances 0.000 description 4
- 235000020357 syrup Nutrition 0.000 description 4
- 231100000419 toxicity Toxicity 0.000 description 4
- 230000001988 toxicity Effects 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 3
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 3
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 description 3
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 3
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 3
- 208000010334 End Stage Liver Disease Diseases 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- 206010019759 Hepatitis chronic persistent Diseases 0.000 description 3
- 206010019799 Hepatitis viral Diseases 0.000 description 3
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 3
- 102100034349 Integrase Human genes 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Natural products O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- IVOMOUWHDPKRLL-UHFFFAOYSA-N UNPD107823 Natural products O1C2COP(O)(=O)OC2C(O)C1N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-UHFFFAOYSA-N 0.000 description 3
- 208000036142 Viral infection Diseases 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 231100000354 acute hepatitis Toxicity 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 229960005305 adenosine Drugs 0.000 description 3
- PYMYPHUHKUWMLA-WISUUJSJSA-N aldehydo-L-xylose Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)C=O PYMYPHUHKUWMLA-WISUUJSJSA-N 0.000 description 3
- 229930195726 aldehydo-L-xylose Natural products 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 230000003602 anti-herpes Effects 0.000 description 3
- 230000000798 anti-retroviral effect Effects 0.000 description 3
- 239000004599 antimicrobial Substances 0.000 description 3
- 239000002246 antineoplastic agent Substances 0.000 description 3
- 125000003710 aryl alkyl group Chemical group 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 208000011444 chronic liver failure Diseases 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000002299 complementary DNA Substances 0.000 description 3
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 206010016256 fatigue Diseases 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 235000013355 food flavoring agent Nutrition 0.000 description 3
- 230000008570 general process Effects 0.000 description 3
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 3
- 125000000623 heterocyclic group Chemical group 0.000 description 3
- 230000002163 immunogen Effects 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 208000018191 liver inflammation Diseases 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 229920001184 polypeptide Polymers 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 230000000707 stereoselective effect Effects 0.000 description 3
- 229940104230 thymidine Drugs 0.000 description 3
- 241001430294 unidentified retrovirus Species 0.000 description 3
- 229960005486 vaccine Drugs 0.000 description 3
- 230000029812 viral genome replication Effects 0.000 description 3
- 210000002845 virion Anatomy 0.000 description 3
- OIRDTQYFTABQOQ-DEGSGYPDSA-N (2S,3S,4R,5S)-2-(6-aminopurin-9-yl)-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound [C@H]1([C@@H](O)[C@@H](O)[C@@H](O1)CO)N1C2=NC=NC(=C2N=C1)N OIRDTQYFTABQOQ-DEGSGYPDSA-N 0.000 description 2
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 2
- IVWWFWFVSWOTLP-YVZVNANGSA-N (3'as,4r,7'as)-2,2,2',2'-tetramethylspiro[1,3-dioxolane-4,6'-4,7a-dihydro-3ah-[1,3]dioxolo[4,5-c]pyran]-7'-one Chemical compound C([C@@H]1OC(O[C@@H]1C1=O)(C)C)O[C@]21COC(C)(C)O2 IVWWFWFVSWOTLP-YVZVNANGSA-N 0.000 description 2
- IVFVSTOFYHUJRU-KLVWXMOXSA-N (3as,5s,6s,6ar)-2-amino-5-(hydroxymethyl)-3a,5,6,6a-tetrahydrofuro[2,3-d][1,3]oxazol-6-ol Chemical compound O1[C@@H](CO)[C@H](O)[C@H]2OC(N)=N[C@H]21 IVFVSTOFYHUJRU-KLVWXMOXSA-N 0.000 description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- OBOHMJWDFPBPKD-UHFFFAOYSA-N 1-[chloro(diphenyl)methyl]-4-methoxybenzene Chemical compound C1=CC(OC)=CC=C1C(Cl)(C=1C=CC=CC=1)C1=CC=CC=C1 OBOHMJWDFPBPKD-UHFFFAOYSA-N 0.000 description 2
- OLXZPDWKRNYJJZ-RRKCRQDMSA-N 2'-deoxyadenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 OLXZPDWKRNYJJZ-RRKCRQDMSA-N 0.000 description 2
- ZOOGRGPOEVQQDX-UUOKFMHZSA-N 3',5'-cyclic GMP Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-UUOKFMHZSA-N 0.000 description 2
- CWNPOQFCIIFQDM-UHFFFAOYSA-N 3-nitrobenzyl alcohol Chemical compound OCC1=CC=CC([N+]([O-])=O)=C1 CWNPOQFCIIFQDM-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- DDFHBQSCUXNBSA-UHFFFAOYSA-N 5-(5-carboxythiophen-2-yl)thiophene-2-carboxylic acid Chemical compound S1C(C(=O)O)=CC=C1C1=CC=C(C(O)=O)S1 DDFHBQSCUXNBSA-UHFFFAOYSA-N 0.000 description 2
- PNWOYKVCNDZOLS-UHFFFAOYSA-N 6-amino-5-chloro-1h-pyrimidin-2-one Chemical compound NC=1NC(=O)N=CC=1Cl PNWOYKVCNDZOLS-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- 101000621943 Acholeplasma phage L2 Probable integrase/recombinase Proteins 0.000 description 2
- 101000618348 Allochromatium vinosum (strain ATCC 17899 / DSM 180 / NBRC 103801 / NCIMB 10441 / D) Uncharacterized protein Alvin_0065 Proteins 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 101000781117 Autographa californica nuclear polyhedrosis virus Uncharacterized 12.4 kDa protein in CTL-LEF2 intergenic region Proteins 0.000 description 2
- 101000708323 Azospirillum brasilense Uncharacterized 28.8 kDa protein in nifR3-like 5'region Proteins 0.000 description 2
- 101000770311 Azotobacter chroococcum mcd 1 Uncharacterized 19.8 kDa protein in nifW 5'region Proteins 0.000 description 2
- 101000748761 Bacillus subtilis (strain 168) Uncharacterized MFS-type transporter YcxA Proteins 0.000 description 2
- 101000765620 Bacillus subtilis (strain 168) Uncharacterized protein YlxP Proteins 0.000 description 2
- 101000916134 Bacillus subtilis (strain 168) Uncharacterized protein YqxJ Proteins 0.000 description 2
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 2
- 101000754349 Bordetella pertussis (strain Tohama I / ATCC BAA-589 / NCTC 13251) UPF0065 protein BP0148 Proteins 0.000 description 2
- BGGALFIXXQOTPY-NRFANRHFSA-N C1(=C(C2=C(C=C1)N(C(C#N)=C2)C[C@@H](N1CCN(CC1)S(=O)(=O)C)C)C)CN1CCC(CC1)NC1=NC(=NC2=C1C=C(S2)CC(F)(F)F)NC Chemical compound C1(=C(C2=C(C=C1)N(C(C#N)=C2)C[C@@H](N1CCN(CC1)S(=O)(=O)C)C)C)CN1CCC(CC1)NC1=NC(=NC2=C1C=C(S2)CC(F)(F)F)NC BGGALFIXXQOTPY-NRFANRHFSA-N 0.000 description 2
- 101000827633 Caldicellulosiruptor sp. (strain Rt8B.4) Uncharacterized 23.9 kDa protein in xynA 3'region Proteins 0.000 description 2
- 241001227713 Chiron Species 0.000 description 2
- 206010057573 Chronic hepatic failure Diseases 0.000 description 2
- 101000947628 Claviceps purpurea Uncharacterized 11.8 kDa protein Proteins 0.000 description 2
- 101000686796 Clostridium perfringens Replication protein Proteins 0.000 description 2
- 108010033174 Deoxycytidine kinase Proteins 0.000 description 2
- 102100029588 Deoxycytidine kinase Human genes 0.000 description 2
- 206010059866 Drug resistance Diseases 0.000 description 2
- 101710091045 Envelope protein Proteins 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- 101000788129 Escherichia coli Uncharacterized protein in sul1 3'region Proteins 0.000 description 2
- 101000788370 Escherichia phage P2 Uncharacterized 12.9 kDa protein in GpA 3'region Proteins 0.000 description 2
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 2
- 101000787096 Geobacillus stearothermophilus Uncharacterized protein in gldA 3'region Proteins 0.000 description 2
- 101000976889 Haemophilus phage HP1 (strain HP1c1) Uncharacterized 19.2 kDa protein in cox-rep intergenic region Proteins 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 101000827627 Klebsiella pneumoniae Putative low molecular weight protein-tyrosine-phosphatase Proteins 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 241000701076 Macacine alphaherpesvirus 1 Species 0.000 description 2
- 101001130841 Middle East respiratory syndrome-related coronavirus (isolate United Kingdom/H123990006/2012) Non-structural protein ORF5 Proteins 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 101100170604 Mus musculus Dmap1 gene Proteins 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- 239000007832 Na2SO4 Substances 0.000 description 2
- 206010028980 Neoplasm Diseases 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 241000282579 Pan Species 0.000 description 2
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 2
- 108091000080 Phosphotransferase Proteins 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 102000001253 Protein Kinase Human genes 0.000 description 2
- 101710188315 Protein X Proteins 0.000 description 2
- 101000974028 Rhizobium leguminosarum bv. viciae (strain 3841) Putative cystathionine beta-lyase Proteins 0.000 description 2
- 101000756519 Rhodobacter capsulatus (strain ATCC BAA-309 / NBRC 16581 / SB1003) Uncharacterized protein RCAP_rcc00048 Proteins 0.000 description 2
- 101000948219 Rhodococcus erythropolis Uncharacterized 11.5 kDa protein in thcD 3'region Proteins 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 101000936711 Streptococcus gordonii Accessory secretory protein Asp4 Proteins 0.000 description 2
- 101000929863 Streptomyces cinnamonensis Monensin polyketide synthase putative ketoacyl reductase Proteins 0.000 description 2
- 101000788468 Streptomyces coelicolor Uncharacterized protein in mprR 3'region Proteins 0.000 description 2
- 101000845085 Streptomyces violaceoruber Granaticin polyketide synthase putative ketoacyl reductase 1 Proteins 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 241000701093 Suid alphaherpesvirus 1 Species 0.000 description 2
- 101000711771 Thiocystis violacea Uncharacterized 76.5 kDa protein in phbC 3'region Proteins 0.000 description 2
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 2
- 101000711318 Vibrio alginolyticus Uncharacterized 11.6 kDa protein in scrR 3'region Proteins 0.000 description 2
- 108010067390 Viral Proteins Proteins 0.000 description 2
- HBHJTJKGFOGKMG-LADGJGSJSA-N [(2r)-3-[[(2s,3s,5r)-3-azido-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-2-tetradecanoyloxypropyl] tetradecanoate Chemical compound C1[C@H](N=[N+]=[N-])[C@@H](COP(O)(=O)OC[C@@H](COC(=O)CCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCC)O[C@H]1N1C(=O)NC(=O)C(C)=C1 HBHJTJKGFOGKMG-LADGJGSJSA-N 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 125000002877 alkyl aryl group Chemical group 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 230000000118 anti-neoplastic effect Effects 0.000 description 2
- 229940034982 antineoplastic agent Drugs 0.000 description 2
- OIRDTQYFTABQOQ-UHFFFAOYSA-N ara-adenosine Natural products Nc1ncnc2n(cnc12)C1OC(CO)C(O)C1O OIRDTQYFTABQOQ-UHFFFAOYSA-N 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 244000309464 bull Species 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- ZOOGRGPOEVQQDX-UHFFFAOYSA-N cyclic GMP Natural products O1C2COP(O)(=O)OC2C(O)C1N1C=NC2=C1NC(N)=NC2=O ZOOGRGPOEVQQDX-UHFFFAOYSA-N 0.000 description 2
- 229940095074 cyclic amp Drugs 0.000 description 2
- 238000006392 deoxygenation reaction Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 230000009266 disease activity Effects 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 229910001851 flerovium Inorganic materials 0.000 description 2
- XRECTZIEBJDKEO-UHFFFAOYSA-N flucytosine Chemical compound NC1=NC(=O)NC=C1F XRECTZIEBJDKEO-UHFFFAOYSA-N 0.000 description 2
- 229960004413 flucytosine Drugs 0.000 description 2
- 229960002949 fluorouracil Drugs 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 229910001867 inorganic solvent Inorganic materials 0.000 description 2
- 239000003049 inorganic solvent Substances 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 238000010907 mechanical stirring Methods 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 2
- KOSYAAIZOGNATQ-UHFFFAOYSA-N o-phenyl chloromethanethioate Chemical compound ClC(=S)OC1=CC=CC=C1 KOSYAAIZOGNATQ-UHFFFAOYSA-N 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 235000006408 oxalic acid Nutrition 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- 150000008298 phosphoramidates Chemical class 0.000 description 2
- 102000020233 phosphotransferase Human genes 0.000 description 2
- 239000002504 physiological saline solution Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 230000000069 prophylactic effect Effects 0.000 description 2
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- 229910001220 stainless steel Inorganic materials 0.000 description 2
- 239000010935 stainless steel Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000013589 supplement Substances 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- 229940113082 thymine Drugs 0.000 description 2
- 230000002110 toxicologic effect Effects 0.000 description 2
- 238000002054 transplantation Methods 0.000 description 2
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 201000001862 viral hepatitis Diseases 0.000 description 2
- TXUICONDJPYNPY-UHFFFAOYSA-N (1,10,13-trimethyl-3-oxo-4,5,6,7,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-17-yl) heptanoate Chemical compound C1CC2CC(=O)C=C(C)C2(C)C2C1C1CCC(OC(=O)CCCCCC)C1(C)CC2 TXUICONDJPYNPY-UHFFFAOYSA-N 0.000 description 1
- NALRCAPFICWVAQ-JDJSBBGDSA-N (2r,3s,4r)-2-(hydroxymethyl)-5-methoxyoxolane-3,4-diol Chemical compound COC1O[C@H](CO)[C@@H](O)[C@H]1O NALRCAPFICWVAQ-JDJSBBGDSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- FYADHXFMURLYQI-UHFFFAOYSA-N 1,2,4-triazine Chemical compound C1=CN=NC=N1 FYADHXFMURLYQI-UHFFFAOYSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- YNGDWRXWKFWCJY-UHFFFAOYSA-N 1,4-Dihydropyridine Chemical compound C1C=CNC=C1 YNGDWRXWKFWCJY-UHFFFAOYSA-N 0.000 description 1
- TUSDEZXZIZRFGC-UHFFFAOYSA-N 1-O-galloyl-3,6-(R)-HHDP-beta-D-glucose Natural products OC1C(O2)COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC1C(O)C2OC(=O)C1=CC(O)=C(O)C(O)=C1 TUSDEZXZIZRFGC-UHFFFAOYSA-N 0.000 description 1
- BBYWOYAFBUOUFP-JOCHJYFZSA-N 1-stearoyl-sn-glycero-3-phosphoethanolamine zwitterion Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](O)COP(O)(=O)OCCN BBYWOYAFBUOUFP-JOCHJYFZSA-N 0.000 description 1
- FUFLCEKSBBHCMO-UHFFFAOYSA-N 11-dehydrocorticosterone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 FUFLCEKSBBHCMO-UHFFFAOYSA-N 0.000 description 1
- HKJAWHYHRVVDHK-UHFFFAOYSA-N 15,16,17-trihydroxyhentriacontane-14,18-dione Chemical compound CCCCCCCCCCCCCC(=O)C(O)C(O)C(O)C(=O)CCCCCCCCCCCCC HKJAWHYHRVVDHK-UHFFFAOYSA-N 0.000 description 1
- MGAXHFMCFLLMNG-UHFFFAOYSA-N 1h-pyrimidine-6-thione Chemical compound SC1=CC=NC=N1 MGAXHFMCFLLMNG-UHFFFAOYSA-N 0.000 description 1
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 1
- BTOTXLJHDSNXMW-POYBYMJQSA-N 2,3-dideoxyuridine Chemical compound O1[C@H](CO)CC[C@@H]1N1C(=O)NC(=O)C=C1 BTOTXLJHDSNXMW-POYBYMJQSA-N 0.000 description 1
- YPFDNCJJFMAZPJ-UHFFFAOYSA-N 2,6-dibromo-7h-purine Chemical compound BrC1=NC(Br)=C2NC=NC2=N1 YPFDNCJJFMAZPJ-UHFFFAOYSA-N 0.000 description 1
- RMFWVOLULURGJI-UHFFFAOYSA-N 2,6-dichloro-7h-purine Chemical compound ClC1=NC(Cl)=C2NC=NC2=N1 RMFWVOLULURGJI-UHFFFAOYSA-N 0.000 description 1
- HNCFPTJMAVGCEI-UHFFFAOYSA-N 2,6-diiodo-7h-purine Chemical compound IC1=NC(I)=C2NC=NC2=N1 HNCFPTJMAVGCEI-UHFFFAOYSA-N 0.000 description 1
- HRVFEVBDZIXIGY-UHFFFAOYSA-N 2-(6-aminopurin-9-yl)oxyethoxymethylphosphonic acid Chemical compound NC1=NC=NC2=C1N=CN2OCCOCP(O)(O)=O HRVFEVBDZIXIGY-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- LULAYUGMBFYYEX-UHFFFAOYSA-M 3-chlorobenzoate Chemical compound [O-]C(=O)C1=CC=CC(Cl)=C1 LULAYUGMBFYYEX-UHFFFAOYSA-M 0.000 description 1
- UHDGCWIWMRVCDJ-YDKYIBAVSA-N 4-amino-1-[(2r,3s,4r,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-YDKYIBAVSA-N 0.000 description 1
- NQUVCRCCRXRJCK-UHFFFAOYSA-N 4-methylbenzoyl chloride Chemical compound CC1=CC=C(C(Cl)=O)C=C1 NQUVCRCCRXRJCK-UHFFFAOYSA-N 0.000 description 1
- 102100022464 5'-nucleotidase Human genes 0.000 description 1
- OOXNYFKPOPJIOT-UHFFFAOYSA-N 5-(3-bromophenyl)-7-(6-morpholin-4-ylpyridin-3-yl)pyrido[2,3-d]pyrimidin-4-amine;dihydrochloride Chemical compound Cl.Cl.C=12C(N)=NC=NC2=NC(C=2C=NC(=CC=2)N2CCOCC2)=CC=1C1=CC=CC(Br)=C1 OOXNYFKPOPJIOT-UHFFFAOYSA-N 0.000 description 1
- LMNPKIOZMGYQIU-UHFFFAOYSA-N 5-(trifluoromethyl)-1h-pyrimidine-2,4-dione Chemical compound FC(F)(F)C1=CNC(=O)NC1=O LMNPKIOZMGYQIU-UHFFFAOYSA-N 0.000 description 1
- BLXGZIDBSXVMLU-OWOJBTEDSA-N 5-[(e)-2-bromoethenyl]-1h-pyrimidine-2,4-dione Chemical compound Br\C=C\C1=CNC(=O)NC1=O BLXGZIDBSXVMLU-OWOJBTEDSA-N 0.000 description 1
- SVXNJCYYMRMXNM-UHFFFAOYSA-N 5-amino-2h-1,2,4-triazin-3-one Chemical compound NC=1C=NNC(=O)N=1 SVXNJCYYMRMXNM-UHFFFAOYSA-N 0.000 description 1
- NPYPQKXJJZZSAX-UHFFFAOYSA-N 5-benzylpyrimidine Chemical compound C=1N=CN=CC=1CC1=CC=CC=C1 NPYPQKXJJZZSAX-UHFFFAOYSA-N 0.000 description 1
- NOEOKMYYFIKGCV-UHFFFAOYSA-N 5-bromo-6-ethenyl-1h-pyrimidine-2,4-dione Chemical compound OC1=NC(O)=C(Br)C(C=C)=N1 NOEOKMYYFIKGCV-UHFFFAOYSA-N 0.000 description 1
- WOVKYSAHUYNSMH-RRKCRQDMSA-N 5-bromodeoxyuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-RRKCRQDMSA-N 0.000 description 1
- LQLQRFGHAALLLE-UHFFFAOYSA-N 5-bromouracil Chemical compound BrC1=CNC(=O)NC1=O LQLQRFGHAALLLE-UHFFFAOYSA-N 0.000 description 1
- HXXVIKZQIFTJOQ-UHFFFAOYSA-N 5-ethenylpyrimidine Chemical compound C=CC1=CN=CN=C1 HXXVIKZQIFTJOQ-UHFFFAOYSA-N 0.000 description 1
- FHIDNBAQOFJWCA-UAKXSSHOSA-N 5-fluorouridine Chemical class O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 FHIDNBAQOFJWCA-UAKXSSHOSA-N 0.000 description 1
- KSNXJLQDQOIRIP-UHFFFAOYSA-N 5-iodouracil Chemical compound IC1=CNC(=O)NC1=O KSNXJLQDQOIRIP-UHFFFAOYSA-N 0.000 description 1
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 1
- NOYDQGFVFOQSAJ-UHFFFAOYSA-N 5-nitropyrimidine Chemical compound [O-][N+](=O)C1=CN=CN=C1 NOYDQGFVFOQSAJ-UHFFFAOYSA-N 0.000 description 1
- ZKBQDFAWXLTYKS-UHFFFAOYSA-N 6-Chloro-1H-purine Chemical compound ClC1=NC=NC2=C1NC=N2 ZKBQDFAWXLTYKS-UHFFFAOYSA-N 0.000 description 1
- OHILKUISCGPRMQ-UHFFFAOYSA-N 6-amino-5-(trifluoromethyl)-1h-pyrimidin-2-one Chemical compound NC1=NC(=O)NC=C1C(F)(F)F OHILKUISCGPRMQ-UHFFFAOYSA-N 0.000 description 1
- QFVKLKDEXOWFSL-UHFFFAOYSA-N 6-amino-5-bromo-1h-pyrimidin-2-one Chemical compound NC=1NC(=O)N=CC=1Br QFVKLKDEXOWFSL-UHFFFAOYSA-N 0.000 description 1
- UFVWJVAMULFOMC-UHFFFAOYSA-N 6-amino-5-iodo-1h-pyrimidin-2-one Chemical compound NC=1NC(=O)N=CC=1I UFVWJVAMULFOMC-UHFFFAOYSA-N 0.000 description 1
- PVRBGBGMDLPYKG-UHFFFAOYSA-N 6-benzyl-7h-purine Chemical compound N=1C=NC=2N=CNC=2C=1CC1=CC=CC=C1 PVRBGBGMDLPYKG-UHFFFAOYSA-N 0.000 description 1
- CTGFGRDVWBZYNB-UHFFFAOYSA-N 6-bromo-7h-purine Chemical compound BrC1=NC=NC2=C1NC=N2 CTGFGRDVWBZYNB-UHFFFAOYSA-N 0.000 description 1
- NIBFSSALYRLHPY-UHFFFAOYSA-N 6-iodo-7h-purine Chemical compound IC1=NC=NC2=C1NC=N2 NIBFSSALYRLHPY-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- OYXZMSRRJOYLLO-UHFFFAOYSA-N 7alpha-Hydroxycholesterol Natural products OC1C=C2CC(O)CCC2(C)C2C1C1CCC(C(C)CCCC(C)C)C1(C)CC2 OYXZMSRRJOYLLO-UHFFFAOYSA-N 0.000 description 1
- OYXZMSRRJOYLLO-KGZHIOMZSA-N 7beta-hydroxycholesterol Chemical compound C([C@@H]1O)=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 OYXZMSRRJOYLLO-KGZHIOMZSA-N 0.000 description 1
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical compound NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 description 1
- 208000007788 Acute Liver Failure Diseases 0.000 description 1
- 206010000804 Acute hepatic failure Diseases 0.000 description 1
- 108010076278 Adenosine kinase Proteins 0.000 description 1
- 102100032534 Adenosine kinase Human genes 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- WOVKYSAHUYNSMH-UHFFFAOYSA-N BROMODEOXYURIDINE Natural products C1C(O)C(CO)OC1N1C(=O)NC(=O)C(Br)=C1 WOVKYSAHUYNSMH-UHFFFAOYSA-N 0.000 description 1
- FARWHCSIWKBXQJ-OWJIYDKWSA-N CC(C)[Si](C(C)C)(OC12)O[Si+](C(C)C)(C(C)C)OC[C@@H]1OC([n]1c3ncnc(N)c3nc1)=C2OC(Oc1ccccc1)=S Chemical compound CC(C)[Si](C(C)C)(OC12)O[Si+](C(C)C)(C(C)C)OC[C@@H]1OC([n]1c3ncnc(N)c3nc1)=C2OC(Oc1ccccc1)=S FARWHCSIWKBXQJ-OWJIYDKWSA-N 0.000 description 1
- DWXUERRMXZCVHC-NRBRWOBXSA-N CC(C)[Si](C(C)C)(OC[C@@H]1O[C@@H]2[n]3c4ncnc(N)c4nc3)O[Si+](C(C)C)(C(C)C)OC1C2N=O Chemical compound CC(C)[Si](C(C)C)(OC[C@@H]1O[C@@H]2[n]3c4ncnc(N)c4nc3)O[Si+](C(C)C)(C(C)C)OC1C2N=O DWXUERRMXZCVHC-NRBRWOBXSA-N 0.000 description 1
- FPGZYYSCYNFYHT-UHFFFAOYSA-N CC(C1)OC(CO)C1[O]=C Chemical compound CC(C1)OC(CO)C1[O]=C FPGZYYSCYNFYHT-UHFFFAOYSA-N 0.000 description 1
- UGDCZCKMDBQTFN-RXUZKVILSA-N CC(CC1)[C@@H]2C1(C)C[C@]1(C)[C@@H]2C1 Chemical compound CC(CC1)[C@@H]2C1(C)C[C@]1(C)[C@@H]2C1 UGDCZCKMDBQTFN-RXUZKVILSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 241000589518 Comamonas testosteroni Species 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- MFYSYFVPBJMHGN-ZPOLXVRWSA-N Cortisone Chemical compound O=C1CC[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 MFYSYFVPBJMHGN-ZPOLXVRWSA-N 0.000 description 1
- MFYSYFVPBJMHGN-UHFFFAOYSA-N Cortisone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)(O)C(=O)CO)C4C3CCC2=C1 MFYSYFVPBJMHGN-UHFFFAOYSA-N 0.000 description 1
- XZMCDFZZKTWFGF-UHFFFAOYSA-N Cyanamide Chemical compound NC#N XZMCDFZZKTWFGF-UHFFFAOYSA-N 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 108010058222 Deoxyguanosine kinase Proteins 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical group [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 239000001263 FEMA 3042 Substances 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 241000700739 Hepadnaviridae Species 0.000 description 1
- 206010019663 Hepatic failure Diseases 0.000 description 1
- 208000037319 Hepatitis infectious Diseases 0.000 description 1
- 101000800463 Homo sapiens Transketolase Proteins 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 1
- 235000003332 Ilex aquifolium Nutrition 0.000 description 1
- 235000002296 Ilex sandwicensis Nutrition 0.000 description 1
- 235000002294 Ilex volkensiana Nutrition 0.000 description 1
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 1
- 229930010555 Inosine Natural products 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 244000246386 Mentha pulegium Species 0.000 description 1
- 235000016257 Mentha pulegium Nutrition 0.000 description 1
- 235000004357 Mentha x piperita Nutrition 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- RJUFJBKOKNCXHH-UHFFFAOYSA-N Methyl propionate Chemical compound CCC(=O)OC RJUFJBKOKNCXHH-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241000714177 Murine leukemia virus Species 0.000 description 1
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 1
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 1
- BYEIJZFKOAXBBV-ATZCPNFKSA-N N-[(5S)-5-amino-5-carboxypentanoyl]-L-cysteinyl-D-valine Chemical compound CC(C)[C@H](C(O)=O)NC(=O)[C@H](CS)NC(=O)CCC[C@H](N)C(O)=O BYEIJZFKOAXBBV-ATZCPNFKSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 229920002230 Pectic acid Polymers 0.000 description 1
- LRBQNJMCXXYXIU-PPKXGCFTSA-N Penta-digallate-beta-D-glucose Natural products OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@@H]2[C@H]([C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-PPKXGCFTSA-N 0.000 description 1
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 108010020346 Polyglutamic Acid Proteins 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 229940123066 Polymerase inhibitor Drugs 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 229940121659 Prenyltransferase inhibitor Drugs 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 102000004389 Ribonucleoproteins Human genes 0.000 description 1
- 108010081734 Ribonucleoproteins Proteins 0.000 description 1
- 102000044437 S1 domains Human genes 0.000 description 1
- 108700036684 S1 domains Proteins 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 102000007501 Thymosin Human genes 0.000 description 1
- 108010046075 Thymosin Proteins 0.000 description 1
- 229910021627 Tin(IV) chloride Inorganic materials 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 206010058874 Viraemia Diseases 0.000 description 1
- 108010003533 Viral Envelope Proteins Proteins 0.000 description 1
- WREGKURFCTUGRC-POYBYMJQSA-N Zalcitabine Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)CC1 WREGKURFCTUGRC-POYBYMJQSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- GCZABPLTDYVJMP-CBUXHAPBSA-N [(2r,3r,4r,5s)-5-acetyloxy-3,4-dibenzoyloxyoxolan-2-yl]methyl benzoate Chemical compound C([C@H]1O[C@H]([C@@H]([C@@H]1OC(=O)C=1C=CC=CC=1)OC(=O)C=1C=CC=CC=1)OC(=O)C)OC(=O)C1=CC=CC=C1 GCZABPLTDYVJMP-CBUXHAPBSA-N 0.000 description 1
- UDMBCSSLTHHNCD-UHTZMRCNSA-N [(2r,3s,4s,5r)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl dihydrogen phosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O UDMBCSSLTHHNCD-UHTZMRCNSA-N 0.000 description 1
- PLGZGEBVJIRKLN-UHFFFAOYSA-N [2,2-dimethylpropanoyloxymethoxy-[[2-[hydroxy(methyl)amino]-2-oxoethoxy]methyl]phosphoryl]oxymethyl 2,2-dimethylpropanoate Chemical compound CN(O)C(=O)COCP(=O)(OCOC(=O)C(C)(C)C)OCOC(=O)C(C)(C)C PLGZGEBVJIRKLN-UHFFFAOYSA-N 0.000 description 1
- 238000000862 absorption spectrum Methods 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 229960004150 aciclovir Drugs 0.000 description 1
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 description 1
- 238000001994 activation Methods 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 150000003862 amino acid derivatives Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000002259 anti human immunodeficiency virus agent Substances 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000000719 anti-leukaemic effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 125000001769 aryl amino group Chemical group 0.000 description 1
- 125000005160 aryl oxy alkyl group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 229910052788 barium Inorganic materials 0.000 description 1
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- AQHMSOCWYFWQRO-UHFFFAOYSA-N benzene;methylsulfinylmethane Chemical compound CS(C)=O.C1=CC=CC=C1 AQHMSOCWYFWQRO-UHFFFAOYSA-N 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- HMFHBZSHGGEWLO-FCAWWPLPSA-N beta-L-ribose Chemical compound OC[C@@H]1O[C@H](O)[C@@H](O)[C@H]1O HMFHBZSHGGEWLO-FCAWWPLPSA-N 0.000 description 1
- 229920000249 biocompatible polymer Polymers 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 229910052797 bismuth Inorganic materials 0.000 description 1
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 229950004398 broxuridine Drugs 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- DQXBYHZEEUGOBF-UHFFFAOYSA-N but-3-enoic acid;ethene Chemical compound C=C.OC(=O)CC=C DQXBYHZEEUGOBF-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052793 cadmium Inorganic materials 0.000 description 1
- BDOSMKKIYDKNTQ-UHFFFAOYSA-N cadmium atom Chemical compound [Cd] BDOSMKKIYDKNTQ-UHFFFAOYSA-N 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 238000003763 carbonization Methods 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000006652 catabolic pathway Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- XMPZTFVPEKAKFH-UHFFFAOYSA-P ceric ammonium nitrate Chemical compound [NH4+].[NH4+].[Ce+4].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O XMPZTFVPEKAKFH-UHFFFAOYSA-P 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 229940112822 chewing gum Drugs 0.000 description 1
- 235000015218 chewing gum Nutrition 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 238000005100 correlation spectroscopy Methods 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 229960004544 cortisone Drugs 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 230000009615 deamination Effects 0.000 description 1
- 238000006481 deamination reaction Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 230000000368 destabilizing effect Effects 0.000 description 1
- 230000001066 destructive effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 229960005215 dichloroacetic acid Drugs 0.000 description 1
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 230000000311 effect on hepatitis Effects 0.000 description 1
- QDGZDCVAUDNJFG-FXQIFTODSA-N entecavir (anhydrous) Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@H]1C[C@H](O)[C@@H](CO)C1=C QDGZDCVAUDNJFG-FXQIFTODSA-N 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 239000005038 ethylene vinyl acetate Substances 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- LRBQNJMCXXYXIU-QWKBTXIPSA-N gallotannic acid Chemical compound OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@H]2[C@@H]([C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-QWKBTXIPSA-N 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 238000005469 granulation Methods 0.000 description 1
- 230000003179 granulation Effects 0.000 description 1
- 229940029575 guanosine Drugs 0.000 description 1
- RQFCJASXJCIDSX-UUOKFMHZSA-N guanosine 5'-monophosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O RQFCJASXJCIDSX-UUOKFMHZSA-N 0.000 description 1
- 108010036383 guanosine kinase Proteins 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 208000005252 hepatitis A Diseases 0.000 description 1
- 210000003494 hepatocyte Anatomy 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 231100000086 high toxicity Toxicity 0.000 description 1
- 235000001050 hortel pimenta Nutrition 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- CBOIHMRHGLHBPB-UHFFFAOYSA-N hydroxymethyl Chemical compound O[CH2] CBOIHMRHGLHBPB-UHFFFAOYSA-N 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229960003786 inosine Drugs 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 208000007903 liver failure Diseases 0.000 description 1
- 231100000835 liver failure Toxicity 0.000 description 1
- 210000005228 liver tissue Anatomy 0.000 description 1
- 238000011866 long-term treatment Methods 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 210000003563 lymphoid tissue Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229940099690 malic acid Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 210000004779 membrane envelope Anatomy 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000037323 metabolic rate Effects 0.000 description 1
- LULAYUGMBFYYEX-UHFFFAOYSA-N metachloroperbenzoic acid Natural products OC(=O)C1=CC=CC(Cl)=C1 LULAYUGMBFYYEX-UHFFFAOYSA-N 0.000 description 1
- HRDXJKGNWSUIBT-UHFFFAOYSA-N methoxybenzene Chemical group [CH2]OC1=CC=CC=C1 HRDXJKGNWSUIBT-UHFFFAOYSA-N 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229940017219 methyl propionate Drugs 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 231100000324 minimal toxicity Toxicity 0.000 description 1
- PJUIMOJAAPLTRJ-UHFFFAOYSA-N monothioglycerol Chemical compound OCC(O)CS PJUIMOJAAPLTRJ-UHFFFAOYSA-N 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 210000004165 myocardium Anatomy 0.000 description 1
- ACTNHJDHMQSOGL-UHFFFAOYSA-N n',n'-dibenzylethane-1,2-diamine Chemical compound C=1C=CC=CC=1CN(CCN)CC1=CC=CC=C1 ACTNHJDHMQSOGL-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical compound C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- 230000001338 necrotic effect Effects 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- LBQAJLBSGOBDQF-UHFFFAOYSA-N nitro azanylidynemethanesulfonate Chemical compound [O-][N+](=O)OS(=O)(=O)C#N LBQAJLBSGOBDQF-UHFFFAOYSA-N 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000000346 nonvolatile oil Substances 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid group Chemical group C(CCCCCCC\C=C/CCCCCCCC)(=O)O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 238000003305 oral gavage Methods 0.000 description 1
- 239000007935 oral tablet Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002892 organic cations Chemical class 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-N palmitic acid group Chemical group C(CCCCCCCCCCCCCCC)(=O)O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 239000003961 penetration enhancing agent Substances 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 description 1
- 150000003014 phosphoric acid esters Chemical class 0.000 description 1
- 150000008299 phosphorodiamidates Chemical class 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000010318 polygalacturonic acid Substances 0.000 description 1
- 229920002643 polyglutamic acid Polymers 0.000 description 1
- 239000004633 polyglycolic acid Substances 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- QMLAXRXNTUYJMM-UHFFFAOYSA-N propane-1,2,3-triol;3-sulfanylpropane-1,2-diol Chemical compound OCC(O)CO.OCC(O)CS QMLAXRXNTUYJMM-UHFFFAOYSA-N 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 108010043671 prostatic acid phosphatase Proteins 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 235000011962 puddings Nutrition 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- FVLAYJRLBLHIPV-UHFFFAOYSA-N pyrimidin-5-amine Chemical compound NC1=CN=CN=C1 FVLAYJRLBLHIPV-UHFFFAOYSA-N 0.000 description 1
- 239000002718 pyrimidine nucleoside Substances 0.000 description 1
- HBCQSNAFLVXVAY-UHFFFAOYSA-N pyrimidine-2-thiol Chemical compound SC1=NC=CC=N1 HBCQSNAFLVXVAY-UHFFFAOYSA-N 0.000 description 1
- XVIAPHVAGFEFFN-UHFFFAOYSA-N pyrimidine-5-carbonitrile Chemical compound N#CC1=CN=CN=C1 XVIAPHVAGFEFFN-UHFFFAOYSA-N 0.000 description 1
- 238000003753 real-time PCR Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000003362 replicative effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003333 secondary alcohols Chemical class 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- XXFQGNXPWZSRRK-UHFFFAOYSA-N sodium;n-chlorobenzenesulfonamide Chemical compound [Na+].ClNS(=O)(=O)C1=CC=CC=C1 XXFQGNXPWZSRRK-UHFFFAOYSA-N 0.000 description 1
- 238000000956 solid--liquid extraction Methods 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000001119 stannous chloride Substances 0.000 description 1
- 235000011150 stannous chloride Nutrition 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-M succinate(1-) Chemical compound OC(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-M 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 125000000446 sulfanediyl group Chemical class *S* 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- FIAFUQMPZJWCLV-UHFFFAOYSA-N suramin Chemical compound OS(=O)(=O)C1=CC(S(O)(=O)=O)=C2C(NC(=O)C3=CC=C(C(=C3)NC(=O)C=3C=C(NC(=O)NC=4C=C(C=CC=4)C(=O)NC=4C(=CC=C(C=4)C(=O)NC=4C5=C(C=C(C=C5C(=CC=4)S(O)(=O)=O)S(O)(=O)=O)S(O)(=O)=O)C)C=CC=3)C)=CC=C(S(O)(=O)=O)C2=C1 FIAFUQMPZJWCLV-UHFFFAOYSA-N 0.000 description 1
- 229960005314 suramin Drugs 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 229940033123 tannic acid Drugs 0.000 description 1
- 229920002258 tannic acid Polymers 0.000 description 1
- 235000015523 tannic acid Nutrition 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000001973 tert-pentyl group Chemical group [H]C([H])([H])C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- VXKWYPOMXBVZSJ-UHFFFAOYSA-N tetramethyltin Chemical compound C[Sn](C)(C)C VXKWYPOMXBVZSJ-UHFFFAOYSA-N 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- 229940035024 thioglycerol Drugs 0.000 description 1
- LCJVIYPJPCBWKS-NXPQJCNCSA-N thymosin Chemical compound SC[C@@H](N)C(=O)N[C@H](CO)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CO)C(=O)N[C@H](CO)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@H]([C@H](C)O)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](C(C)C)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@H](CCC(O)=O)C(O)=O LCJVIYPJPCBWKS-NXPQJCNCSA-N 0.000 description 1
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 239000003104 tissue culture media Substances 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 231100000759 toxicological effect Toxicity 0.000 description 1
- 231100000723 toxicological property Toxicity 0.000 description 1
- 230000014616 translation Effects 0.000 description 1
- 230000032895 transmembrane transport Effects 0.000 description 1
- DBGVGMSCBYYSLD-UHFFFAOYSA-N tributylstannane Chemical compound CCCC[SnH](CCCC)CCCC DBGVGMSCBYYSLD-UHFFFAOYSA-N 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- FIQMHBFVRAXMOP-UHFFFAOYSA-N triphenylphosphane oxide Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=O)C1=CC=CC=C1 FIQMHBFVRAXMOP-UHFFFAOYSA-N 0.000 description 1
- SCHZCUMIENIQMY-UHFFFAOYSA-N tris(trimethylsilyl)silicon Chemical compound C[Si](C)(C)[Si]([Si](C)(C)C)[Si](C)(C)C SCHZCUMIENIQMY-UHFFFAOYSA-N 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000017613 viral reproduction Effects 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
- 229940075420 xanthine Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- MJIBOYFUEIDNPI-HBNMXAOGSA-L zinc 5-[2,3-dihydroxy-5-[(2R,3R,4S,5R,6S)-4,5,6-tris[[3,4-dihydroxy-5-(3,4,5-trihydroxybenzoyl)oxybenzoyl]oxy]-2-[[3,4-dihydroxy-5-(3,4,5-trihydroxybenzoyl)oxybenzoyl]oxymethyl]oxan-3-yl]oxycarbonylphenoxy]carbonyl-3-hydroxybenzene-1,2-diolate Chemical class [Zn++].Oc1cc(cc(O)c1O)C(=O)Oc1cc(cc(O)c1O)C(=O)OC[C@H]1O[C@@H](OC(=O)c2cc(O)c(O)c(OC(=O)c3cc(O)c(O)c(O)c3)c2)[C@H](OC(=O)c2cc(O)c(O)c(OC(=O)c3cc(O)c(O)c(O)c3)c2)[C@@H](OC(=O)c2cc(O)c(O)c(OC(=O)c3cc(O)c(O)c(O)c3)c2)[C@@H]1OC(=O)c1cc(O)c(O)c(OC(=O)c2cc(O)c([O-])c([O-])c2)c1 MJIBOYFUEIDNPI-HBNMXAOGSA-L 0.000 description 1
Images

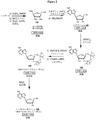




Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
- A61K31/7072—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid having two oxo groups directly attached to the pyrimidine ring, e.g. uridine, uridylic acid, thymidine, zidovudine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
- A61K31/708—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid having oxo groups directly attached to the purine ring system, e.g. guanosine, guanylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Biotechnology (AREA)
- Engineering & Computer Science (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description
本出願は、米国仮特許第60/207,538号(2000年5月26日出願)の優先権を主張する。
【0002】
発明の分野
本発明は、効果的な量の規定されたβ−L−2’−デオキシヌクレオシドまたはその医薬として許容される塩もしくはプロドラッグを投与することを含む、デルタ型肝炎ウイルス(これはまた「HDV」と呼ばれる)に感染した宿主の処置を行うための方法および組成物の領域である。
【0003】
発明の背景
D型肝炎は、ウイルス性肝炎の最も重篤な形態であり、B型肝炎ウイルス(HBV)のサブウイルスサテライトであるD(デルタ)型肝炎ウイルス(HDV)による感染によって引き起こされる(Smedile,A.他、Prog Liver Dis、1994、12、157〜75)。ウイルス性肝炎の他の病原体と比較して、急性HDV感染は、劇症肝炎、急速な進行、多くの場合には肝臓の大部分が破壊される致死的な疾患形態を伴うことが非常に多い。慢性型のD型肝炎は、慢性的なHBV感染と類似した壊死性炎症病巣を典型的には特徴としているが、それよりも重症であり、肝硬変および肝不全に急速に進行することが多く、慢性HDV感染が末期肝疾患を不相応に伴う原因となっている(Smedile,A.他、Prog Liver Dis、1994、12、157〜75;Rizzetto,M.他、Ann Intern Med、1983、98、437〜41)。HDV感染には、HBV単独よりも少ない人が罹患しているが、生じる急性肝不全または慢性肝不全は、欧州ならびに北アメリカにおける肝臓移植に対する一般的な症例である(Smedile,A.およびRizzetto,M.、Int J Clin Lab Res、1992、22、211〜215;Wright,T.L.およびPereira,B.、Liver Transplant Surgery、1995、1、30〜42)。慢性的な疾患には世界中で1500万人が罹患し、そのうち約70,000人が合衆国に存在する。疾病対策センターは、合衆国では年間1,000人がHDV感染により死亡していると推定している(Alter,M.J.およびHadler,S.C.、Prog Clin Biol Res、1993、382、243〜50;Alter,M.J.およびMast,E.E.、Gastroenterol Clin North Am、1994、23、437〜55)。
【0004】
現在、D型肝炎に対して一般的に受け入れられている効果的な治療法はなく、肝臓移植が、付随した終末期の肝疾患に対する唯一の選択肢である。インターフェロンαが、一部のD型肝炎患者の処置においてほどよく成功しているが、より良好な処置選択肢の必要性が、非常に大きい用量を必要とすること、応答が一定していないこと、処置を中断した後の再発が多いこと、そして薬物投与の困難さによって示されている(Thomas,H.C.他、Prog Clin Biol Res、1987、234、277〜90;Hoofnagle,J.他、Prog Clin Biol Res、1987、234、291〜8;Rosina,F.他、Prog Clin Biol Res、1987、234、299〜303;Rosina,F.他、Hepatology、1991、13、1052〜6;Farci,P.他、N Engl J Med、1994、330、88〜94;Hadziyannis,S.J.、J Hepatol、1991、13(増刊1)、S21〜6;Di Marco,V.他、J Viral Hepat、1996、3、123〜8;Porres,J.C.他、J Hepatol、1989、9、338〜44)。
【0005】
HDVビリオンはリボヌクレオタンパク質コアおよびエンベロープからなる。コアは、HDV−RNAと、このウイルスによってコードされる唯一のタンパク質であるデルタ型肝炎抗原(HDAg)とを含む(Wang,K.S.他、Nature、1986、323、508〜14)。エンベロープは、ヘルパーウイルスであるB型肝炎の表面抗原タンパク質(B型肝炎表面抗原またはHBsAg)により形成される(Bonino,F.、Infect Immun、1984、43、1000〜5;Bonino,F.他、Hepatology、1981、1、127〜31;Bonino,F.他、J Virol、1986、58、945〜50)。エンベロープは、HBVによってもたらされる唯一のヘルパー機能である。HDVは、HBVが存在しない細胞内でそのRNAを複製することができる(Kuo,M.Y.他、J Virol、1989、63、1945〜50)が、HBsAgを、HDVビリオンのパッケージングおよび放出のために要求し(Wu、J.C.他、J Virol、1991、65、1099〜104;Ryu,W.S.他、J Virol、1992、66、2310〜2315)、そしてまた感染性のために要求する(Sureau,C.他、J Virol、1992、66、1241〜5)。HDVがHBVに依存している結果、HDVは、HBVを伴う場合にのみ個体に感染する。
【0006】
ラミブジン(β−L−2’,3’−ジデオキシ−3’−チアシチジン、3TC)は、HIV感染およびHBV感染を処置することにおいて有効であることが示されている合成ヌクレオシドである。米国特許第5,539,116号(Liotta他)を参照のこと。ラミブジンは、処置期間中のHBVの複製を持続的に抑制することが知られている(Nevens,F.他、Gastroenterology、1997、113、1258〜1263)。しかしながら、ラミブジンは、慢性的なデルタ型肝炎患者において、疾患活動の改善をもたらさず、またはHDV−RNAのレベルを低下させない(Lau,D.T.他、Hepatology、1999、30、546〜9)。ラミブジンは、最近、慢性HBV感染の処置に対して合衆国および他の国々で承認された。慢性HBV保有者のラミブジンによる長期間の処置で、血清中のHBVレベルが低下し、肝臓組織が改善される(Lai,C.L.他、N Engl J Med、1998、339、61〜8;Tyrrell,D.他、Hepatology、1993、18、112A;Nevens,F.他、Gastroenterology、1997、113、1258〜63;Dienstag,J.L.他、N Engl J Med、1995、333、1657〜61)。HBVに対する劇的な効果にもかかわらず、HBVおよびHDVの両方に慢性的に感染している患者のラミブジン処置は、HDVの循環レベルに対する効果をほとんど有していない。より重要なことは、HBVレベルが抑制されたとしても、疾患活動の改善が全く見られない(Honkoop,P.他、Hepatology、1997、24(増刊)、1219(要約);Lau,D.T.他、Hepatology、1999、30、546〜9)。
【0007】
さらに様々な処置形態が試みられている。例えば、スラミンは、肝細胞へのビリオンの進入をインビトロで阻止するが、毒性が大きいために、ヒトにおける長期の使用には許容され得ない(Smedile,A.他、Prog Liver Dis、1994、12、157〜75)。アシクロビルはHDVの複製をインビトロで高める(Smedile,A.他、Prog Liver Dis、1994、12、157〜75)。リバビリンは、ウイルス学的または生化学的なパラメーターを有意に変化させず、重篤な副作用を有していた(Smedile,A.他、Prog Liver Dis、1994、12、157〜75)。チモシンの合成アナログもまたHDV感染の処置において有効ではなかった(Smedile,A.他、Prog Liver Dis、1994、12、157〜75)。
【0008】
HDV感染に対する記載された処置はどれも、有効であると一般には受け入れられていない。
【0009】
ウッドチャック肝炎ウイルス(WHV)は、HBVに対して近縁(約85%の核酸相同性)であるので、その自然界の宿主であるイースタンウッドチャック(M.monax)におけるHBV感染および疾患に対するモデルとして広く使用されている(Gerin,J.L.、Gastroenterol Jpn、1990、25増刊、38〜42;Tennant,B.C.他、Viral Hepatitis and Liver Disease、1988、462〜464)。実験的に感染させたウッドチャックもまた、抗HBV治療薬の分析および開発のために広範囲に使用されている(Zahm,F.E.他、Ital J Gastroenterol Hepatol、1998、30、510〜6;Tennant,B.C.他、Hepatology、1998、28、179〜91;Mason,W.S.他、Virology、1998、245、18〜32;Korba,B.E.他、Hepatology、1996、23、958〜63;Hurwitz,S.他、Antimicrob Agents Chemother、1998、42、2804〜2809;Block,T.M.他、Nat Med、1998、4、610〜4;Cullen,J.M.他、Antimicrob Agents Chemother、1997、41、2076〜82;Fourel,G.他、Nature、1990、347、294〜8;Gangemi,J.他、Antivir Therap、1997、1、64〜70;Genovesi,E.V.他、Antimicrob Agents Chemother、1998、42、3209〜17;Korba,B.E.他、Antiviral Res、2000、45、19〜32;Cote,P.J.他、Hepatology、2000、31、190〜200;Korba,B.E.他、Antiviral Therapy、2000、5(2)、95〜104;Korba,B.E.他、Antimicrobial Agents and Chemotherapy、2000、44(6)、1757〜60;Korba,B.E.他、Antimicrobial Agents and Chemotherapy、2000、44(7)、1964〜1969)。ウッドチャックにおける慢性WHV感染を実験的に処置するために使用されたいくつかの抗HBV剤(araAMP、リバビリン、AZT、ACV、3TC、ファムシクロビル、FTC)の効力は、臨床試験途中で処置されているHBV患者に投与されたこれらの薬剤の効力と正確に一致していた。抗HBV剤で処置されたWHV感染ウッドチャックおよびHBV感染者において認められるこの類似した効力は、ウッドチャック動物モデルがヒトにおける抗HBV治療法の予測のために使用できることを明らかにしている(Zahm,F.E.他、Ital J Gastroenterol Hepatol、1998、30、510〜6;Tennant,B.C.他、Hepatology、1998、28、179〜91;Mason,W.S.他、Virology、1998、245、18〜32;Hurwitz,S.J.他、Antimicrob Agents Chemother、1998、42(11)、2804〜2809;Fourel,G.他、Nature、1990、347、294〜8;Gangemi,J.他、Antivir Therap、1997、1、64〜70;Genovesi,E.V.他、Antimicrob Agents Chemother、1998、42、3209〜17;Korba,B.E.他、Antiviral Res、2000、45(1)、19〜32;Korba,B.E.他、Hepatology、2000、32(4Pt1)、807〜817;Korba,B.E.他、Hepatology、2000、31(5)、1165〜1175;Korba,B.E.他、Antiviral Therapy、2000、5(2)、95〜104)。WHVは、HDVの粒子形成および感染をHBVと同様に支えることができ、したがってイースタンウッドチャックは、HDV感染に対する有用なモデルである(Negro,F.他、J Virol、1989、63、1612〜8;Parana,R.、Gerard,F.、Lesbordes,J.L.、Pichoud,C.、Vitvitski,L.、Lyra,L.G.&Trepo,C.、J Hepatol、1995、22、468〜73;Ciccaglione,A.R.他、Arch Virol、1993、増刊8、15〜21;Bergmann,K.F.他、J Immunol、1989、143、3714〜21;Ponzetto,A.他、Proc Natl Acad Sci USA、1984、81、2208〜12;Ponzetto,A.他、Prog Clin Biol Res、1987、234、37〜46)。
【0010】
HDVがそのヘルパーウイルスのHBVに依存していることにより、HDV感染の好成績な処置は、支援するHBV感染の好成績な処置から生じることが示唆され得るが、薬物のラミブジン(Glaxo−Wellcome,Inc.)を用いて得られた最近の結果によって例示されるように、このことは当てはまらないようである(Honkoop,P.他、Hepatology、1997、24(増刊)、1219(要約);Lau,D.T.他、Hepatology、1999、30、546〜9)。HBV−HDV感染患者における疾患に対するラミブジンの効果がないことにより、そのような患者の疾患重篤度におけるHDVの直接的な役割が強調される。ラミブジンはHBVおよびWHVの複製を阻害するが、ウイルス表面抗原の産生には影響を及ぼさない(Lau,D.T.他、Hepatology、1999、30、546〜9;Doong,S.L.他、Proc Natl Acad Sci USA、1991、88、8495〜9;Korba,B.E.他、Hepatology、2000、32(4Pt1)、807〜817;Korba,B.E.他、Hepatology、31(5)、1165〜1175)。HBVおよびこのウイルスファミリーの他の代表物(例えば、WHV)の生活環は、ウイルスのゲノムコピーを複製するプロセスとウイルスタンパク質(例えば、HBVまたはWHVの表面抗原)の産生とが異なって調節されているとい点で特異である(Ganem,D.、Hepadnaviridae、「Fields Virology」、Fields BN、Knipe DM、Howley P.編、Lippincott−Raven、1996、Philadelphia、2703〜2737)。したがって、ウイルスのポリメラーゼを標的とする合成ヌクレオシド(例えば、ラミブジン)などの抗ウイルス剤は、(例えば、ウイルス血症の低下によって測定されるような)HBVの複製を著しく阻害することができるが、(例えば、血漿中または血清中のHBV表面抗原のレベルによって測定されるような)ウイルスmRNAまたはウイルスタンパク質の産生のレベルに影響を及ぼすことができない。表面抗原タンパク質によるウイルスエンベロープの形成は、HDVに対する重要な、HBVおよびWHVの唯一の機能であるので、HBsAg産生が阻害されないことが、ラミブジンがHDV複製および疾患に影響を及ぼさないことにおいて一定の役割を果たしていると考えられる。
【0011】
米国特許第5,747,044号は、ワクチンとして有用な組換え産生された免疫原性のHDVポリペプチドを開示する。
【0012】
米国特許第5,932,219号(Chiron)は、D型肝炎ウイルスの全ゲノム、すなわち、HDVゲノム全体のcDNA複製体の集団を開示し、そしてこれらのcDNA配列の様々な部分が、臨床サンプルにおけるウイルスの存在を診断するためのプローブとして有用であることを教示している。この特許には、ワクチン製造において有用な、cDNAによってコードされるタンパク質もまた開示される。具体的には、‘219号特許は、p24およびp27のウイルスポリペプチドを含むD型肝炎ワクチンを開示している。米国特許第5,750,350号(Chiron)では、HDVゲノムのORF5によってコードされるペプチドを含む、D型肝炎ウイルスの分析において有用なキットが特許請求されている。米国特許第5,747,044号では、HDVヌクレオチド配列のORF5またはその相補体にコードされている免疫原性ポリペプチドを含む、HDVに対する抗体を惹起させる組換え産生された免疫原性粒子が特許請求されている。
【0013】
米国特許第6,020,167号(Medeva Holdings B.V.に譲渡)は、HBsAgを含有する組成物を投与することを含む慢性肝炎(特にB型肝炎)の処置方法を開示する。
【0014】
米国特許第5,770,584号は、アルキル脂質またはアルキル脂質誘導体を投与することによる肝炎ウイルス感染の処置方法を開示する。
【0015】
米国特許第4,619,896号は、動物の血液中のデルタ抗原を暴露させる方法を開示する。この方法は、血清を界面活性剤および場合によっては抗体−抗原解離剤で処理することを含む。血液由来のデルタ抗原が、D型肝炎ウイルスに対する抗体の種々のクラスを検出および決定する際の診断剤として使用される。
【0016】
合衆国法定発明登録H1,345号は、タンパク質−プレニルトランスフェラーゼ阻害剤を投与することによって肝炎ウイルスを防止または処置する方法を開示する。
【0017】
Sureau他、「感染性デルタ型肝炎ウイルスのインビトロでの産生およびB型肝炎ウイルスのプレS抗原に対する抗体による中和」(Journal of Virology、1992、1241〜1245)は、インビトロで産生されたHDV粒子が感染性であること、そして(i)感染性粒子は、プレS1領域およびプレS2領域を含有するHBVエンベロープタンパク質で覆われていること、および(ii)HBVエンベロープタンパク質のプレS1ドメインおよびプレS2ドメインのエピトープがHDV粒子の表面に露出していること、および(iii)そのようなエピトープに対する抗体はHDVに対する中和活性を有することを開示している。
【0018】
最近、L−FMAUが慢性的な感染動物におけるHDVの強力な阻害剤であることが報告されている(Casey,J.L.他、Antiviral Therapy、2000、5(増刊1)、32、要約057)。
【0019】
合成ヌクレオシドのβ−L−2’−デオキシシチジン(β−L−2’−dC)、β−L−2’−デオキシチミジン(β−L−dT)およびβ−L−2’−デオキシアデノシン(β−L−2’−dA)もまたこの分野では知られている。Antonin Holyにより、β−L−dCおよびβ−L−dTが1972年に初めて開示された(「核酸化合物およびそのアナログ.CLIII.ピリミジン系列の2’−デオキシ−L−リボヌクレオシドの調製」、Collect Czech Chem Commun、1972、37(12)、4072〜87)。Morris S.Zedeck他により、β−L−dAが、シュードモナステストステロニにおける誘導型酵素の合成を阻害するために初めて開示された(Mol Phys、1967、3(4)、386〜95)。
【0020】
ある種の2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが抗新生物活性および選択された抗ウイルス活性を有することが知られている。Verri他により、抗新生物剤および抗ヘルペス剤としての2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドの使用が開示されている(Mol Pharmacol、1997、51(1)、132〜138;Biochem J、1997、328(1)、317〜20)。Saneyoshi他により、レトロウイルスの抑制およびAIDSの処置に対する逆転写酵素(I)阻害剤としての2’−デオキシ−L−リボヌクレオシドの使用が明らかにされている(日本公開特許公報JP06293645(1994))。
【0021】
Giovanni他は部分的に偽狂犬病ウイルス(PRV)に対して2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドを調べた(Biochem J、1993、294(2)、381〜5)。
【0022】
2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドの化学療法的使用がTyrsted他(Biochem Biophys Acta、1968、155(2)、619〜22)およびBloch他(J Med Chem、1967、10(5)、908〜12)によって研究された。
【0023】
β−L−2’−デオキシチミジン(β−L−dT)は1型ヘルペス単純ウイルス(HSV−1)のチミジンキナーゼ(TK)を阻害することがこの分野では知られている。Iotti他の国際特許出願公開WO92/08727には、β−L−dTが、ヒトのTKではなく、HSV−1のTKによるD−チミジンのリン酸化を選択的に阻害することが教示されている。Spaldari他は、L−チミジンが1型ヘルペス単純ウイルスのチミジンキナーゼによってリン酸化され、ウイルス増殖を阻害することを報告した(J Med Chem、1992、35(22)、4214〜20)。
【0024】
合成ヌクレオシドのβ−L−2’−デオキシシチジン(β−L−2’−dC)、β−L−2’−デオキシチミジン(β−L−dT)、β−L−2’−デオキシイノシン(β−L−dI)およびβ−L−2’−デオキシアデノシン(β−L−2’−dA)が、最近、B型肝炎ウイルスを処置するためにこの分野で開示された。Gilles Gosselin他により、β−L−dT、β−L−dA、β−L−dCおよびβ−L−dIならびにそれらの医薬として許容される塩およびプロドラッグをB型肝炎ウイルスの処置のために使用することが国際特許出願公開WO00/09531(PCT/US99/18149)に開示された。
【0025】
PCT/US01/09987(ジョージタウン大学、コーネル大学およびジョージア大学研究財団によって出願)には、宿主におけるB型肝炎表面抗原(明細書中ではHBsAgとして示される)のレベルを実質的に低下させるヌクレオシドまたはヌクレオシドアナログの投与がその宿主におけるデルタ型肝炎ウイルス感染の処置において有用であることが記載されている。1つの実施形態において、PCT/US01/09987は、2’−フルオロ−5−メチル−β−L−アラビノフラノシルウリジン(L−FMAU)がB型肝炎表面抗原のレベルを著しく低下させ、したがってデルタ型肝炎感染症の処置において有用であることを記載している。
【0026】
デルタ型肝炎ウイルスの感染者数が非常に多く、そして個体に対するデルタ型肝炎ウイルス感染の作用は破壊的であり、そして効果的な処置がないために、デルタ型肝炎ウイルス感染を処置するための新しい効果的な方法が非常に求められている。
【0027】
したがって、デルタ型肝炎ウイルスに感染した宿主(ヒトを含む)を処置するための方法を提供することは本発明の目的である。
【0028】
発明の概要
ヒトおよび他の宿主におけるデルタ型肝炎の感染を処置するための方法が開示される。この方法は、場合によっては医薬として許容されるキャリアにおいて、効果的な量の生物学的に活性な2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド(これは代わりに本明細書中ではβ−L−d−ヌクレオシドまたはβ−L−2’−d−ヌクレオシドとして示される)またはその医薬として許容される塩もしくはプロドラッグを投与することを含む。この場合、これらは単独または組合わせもしくは交互のいずれでも投与される。本明細書中で使用される用語の2’−デオキシは、2’位に置換基を有しないヌクレオシドを示す。
【0029】
開示される2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドまたは医薬として許容されるプロドラッグもしくは塩、あるいはこれらの化合物を含有する医薬として許容される配合物は、D型肝炎感染症および他の関連する状態(HDVによって引き起こされる慢性的な肝臓炎症、肝硬変、急性肝炎、劇症肝炎、慢性の持続性肝炎、および疲労など)の防止および処置において有用である。これらの化合物または配合物はまた、HDVに感染している個体、またはHDVにさらされている個体における臨床的疾患の進行を防止または遅延させるために予防的に使用することができる。
【0030】
本発明の一実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0031】
【化29】

R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;かつ
BASEは、場合によっては置換され得るプリン塩基またはピリミジン塩基である。)
の化合物である。
【0032】
本発明の別の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0033】
【化30】
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;
YはOR3、NR3R4またはSR3であり;かつ
X1およびX2は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、ハロゲン、OR5、NR5NR6またはSR5からなる群から独立して選択され;そして
R3、R4、R5およびR6は独立して、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体である。)
のβ−L−2’−デオキシプリンまたはその医薬として許容される塩もしくはプロドラッグである。
【0034】
特定の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0035】
【化31】
のβ−L−2’−デオキシアデノシンまたはその医薬として許容される塩もしくはプロドラッグである。
【0036】
好ましい実施形態では、R1はHである。
【0037】
別の特定の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0038】
【化32】
のβ−L−2’−デオキシグアノシンまたはその医薬として許容される塩もしくはプロドラッグである。
【0039】
別の特定の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0040】
【化33】
のβ−L−2’−デオキシイノシンまたはその医薬として許容される塩もしくはプロドラッグである。
【0041】
本発明の別の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0042】
【化34】
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;
YはOR3、NR3R4またはSR3であり;かつ
X1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、ハロゲン、OR5、NR5NR6またはSR5からなる群から選択され;そして
R3、R4、R5およびR6は独立して、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体である。)
のβ−L−2’−デオキシピリミジンまたはその医薬として許容される塩もしくはプロドラッグである。
【0043】
特定の一実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0044】
【化35】
のβ−L−2’−デオキシシチジンまたはその医薬として許容される塩もしくはプロドラッグである。
【0045】
好ましい実施形態では、R1はHである。
【0046】
別の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0047】
【化36】
のβ−L−2’−デオキシウリジンまたはその医薬として許容される塩もしくはプロドラッグである。
【0048】
別の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0049】
【化37】
のβ−L−チミジンまたはその医薬として許容される塩もしくはプロドラッグである。
【0050】
好ましい実施形態において、R1はHである。
【0051】
別の実施形態において、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド、その医薬として許容される塩またはプロドラッグは、1つ以上の他の2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド、その医薬として許容される塩もしくはプロドラッグ、またはD型肝炎ウイルスに対する活性を示す1つ以上の他の化合物と交互に、またはそれらと組み合わせて投与される。一般に、交互療法のときには、それぞれの薬剤の効果的な投薬量が儒順次投与され、これに対して、混合療法では、2つ以上の薬剤の効果的な投薬量が一緒に投与される。投薬量は、薬物の吸収、不活性化および排出速度、ならびに当業者に知られている他の要因に依存する。投薬量の値はまた、緩和すべき状態の重篤度により変化することに留意しなければならない。任意の特定の対象体について、具体的な投薬法およびスケジュールは、個々の必要性、および組成物を投与する医師または組成物の投与を監督する医師の専門的判断に従って時間とともに調節すべきであることをさらに理解しなければならない。
【0052】
別の実施形態において、本発明は、HDVに感染しているヒトを処置するための方法を包含する。この方法は、開示される2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体のプロドラッグのHDV処置量を投与することを含む。本明細書中で使用されるプロドラッグは、インビボ投与したときにそのヌクレオシドに変換される化合物を示す。非限定的な例には、医薬として許容される塩(これは代わりに「生理学的に受容可能な塩」として示される)、活性な化合物の5’位および/またはN4位(シチジン)および/またはN6位(アデノシン)のアシル化誘導体またはアルキル化誘導体、活性な化合物の5’−リン脂質および/または5’−エーテル脂質が挙げられる。
【0053】
好ましい実施形態において、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドは、5’−ヒドロキシルがアミノ酸でアシル化されているという点で、医薬として許容されるプロドラッグの形態である。さらにより好ましい実施形態において、アミノ酸はバリンである。
【0054】
(図面の簡単な説明)
図1は出発物質としてL−リボースまたはL−キシロースを使用してβ−L−エリスロ−ペンタフラノヌクレオシド(β−L−dN)を得るための一般的なプロセスを例示する図である。
図2は出発物質としてL−リボースを使用してL−デオキシアデノシンを合成する非限定的な例を例示する図である。
図3aは出発物質としてL−アラビノースを使用してβ−L−dCを合成する非限定的な例を例示する図である。
図3bは出発物質としてL−アラビノースを使用してβ−L−dTを合成する非限定的な例を例示する図である。
図4は蓄積および減少に関してヒトHepG2細胞におけるL−dA、L−dCおよびL−dTの代謝を例示するグラフである。細胞は10μMの化合物とともにインキュベーションされた。
図5はウッドチャック慢性肝炎モデルにおけるβ−L−dA、β−L−dTおよびβ−L−dCの抗ウイルス作用を例示するグラフである。
【0055】
発明の詳細な説明
ヒトおよび他の宿主におけるデルタ型肝炎の感染を処置するための方法が開示される。この方法は、場合によって医薬として許容されるキャリアにおいて、効果的な量の生物学的に活性な2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド(これは代わりに本明細書中ではβ−L−d−ヌクレオシドまたはβ−L−2’−d−ヌクレオシドとして示される)またはその医薬として許容される塩もしくはプロドラッグを投与することを含む。この場合、これらは単独または組合わせのいずれでも投与される。本明細書中で使用される用語の2’−デオキシは、2’位に置換基を有しないヌクレオシドを示す。
【0056】
開示される2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドまたは医薬として許容されるプロドラッグもしくは塩、あるいはこれらの化合物を含有する医薬として許容される配合物は、D型肝炎感染症および他の関連する状態(HDVによって引き起こされる慢性的な肝臓炎症、肝硬変、急性肝炎、劇症肝炎、慢性の持続性肝炎、および疲労など)の防止および処置において有用である。これらの化合物または配合物はまた、HDVに感染している個体、またはHDVにさらされている個体における臨床的疾患の進行を防止または遅延させるために予防的に使用することができる。
【0057】
本発明の一実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0058】
【化38】
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;かつ
BASEは、場合によっては置換され得るプリン塩基またはピリミジン塩基である。)
の化合物である。
【0059】
本発明の別の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0060】
【化39】
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;
YはOR3、NR3R4またはSR3であり;かつ
X1およびX2は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、ハロゲン、OR5、NR5NR6またはSR5からなる群から独立して選択され;そして
R3、R4、R5およびR6は独立して、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体である。)
のβ−L−2’−デオキシプリンまたはその医薬として許容される塩もしくはプロドラッグである。
【0061】
特定の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0062】
【化40】
のβ−L−2’−デオキシアデノシンまたはその医薬として許容される塩もしくはプロドラッグである。
【0063】
好ましい実施形態では、R1はHである。
【0064】
別の特定の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0065】
【化41】
のβ−L−2’−デオキシグアノシンまたはその医薬として許容される塩もしくはプロドラッグである。
【0066】
別の特定の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0067】
【化42】
のβ−L−2’−デオキシイノシンまたはその医薬として許容される塩もしくはプロドラッグである。
【0068】
本発明の別の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0069】
【化43】
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;
YはOR3、NR3R4またはSR3であり;かつ
X1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、ハロゲン、OR5、NR5NR6またはSR5からなる群から選択され;そして
R3、R4、R5およびR6は独立して、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体である。)
のβ−L−2’−デオキシピリミジンまたはその医薬として許容される塩もしくはプロドラッグである。
【0070】
特定の一実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0071】
【化44】
のβ−L−2’−デオキシシチジンまたはその医薬として許容される塩もしくはプロドラッグである。
【0072】
好ましい実施形態では、R1はHである。
【0073】
別の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0074】
【化45】
のβ−L−2’−デオキシウリジンまたはその医薬として許容される塩もしくはプロドラッグである。
【0075】
別の実施形態では、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体は、下記式:
【0076】
【化46】
のβ−L−チミジンまたはその医薬として許容される塩もしくはプロドラッグである。
【0077】
好ましい実施形態において、R1はHである。
【0078】
別の実施形態において、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド、その医薬として許容される塩またはプロドラッグは、1つ以上の他の2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド、その医薬として許容される塩もしくはプロドラッグ、またはD型肝炎ウイルスに対する活性を示す1つ以上の他の化合物と交互に、またはそれらと組み合わせて投与される。一般に、交互療法のときには、それぞれの薬剤の効果的な投薬量が順次投与され、これに対して、混合療法では、2つ以上の薬剤の効果的な投薬量が一緒に投与される。投薬量は、薬物の吸収、不活性化および排出速度、ならびに当業者に知られている他の要因に依存する。投薬量の値はまた、緩和すべき状態の重篤度により変化することに留意しなければならない。任意の特定の対象体について、具体的な投薬法およびスケジュールは、個々の必要性、および組成物を投与する医師または組成物の投与を監督する医師の専門的判断に従って時間とともに調節すべきであることをさらに理解しなければならない。
【0079】
別の実施形態において、本発明は、HDVに感染しているヒトを処置するための方法を包含する。この方法は、開示される2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体のプロドラッグのHDV処置量を投与することを含む。本明細書中で使用されるプロドラッグは、インビボ投与したときにそのヌクレオシドに変換される化合物を示す。非限定的な例には、医薬として許容される塩(これは代わりに「生理学的に受容可能な塩」として示される)、活性な化合物の5’位および/またはN4位(シチジン)および/またはN6位(アデノシン)のアシル化誘導体またはアルキル化誘導体、活性な化合物の5’−リン脂質および/または5’−エーテル脂質が挙げられる。
【0080】
好ましい実施形態において、本発明の2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドは、5’−ヒドロキシルがアミノ酸でアシル化されているという点で、医薬として許容されるプロドラッグの形態である。さらにより好ましい実施形態において、アミノ酸はバリンである。
【0081】
立体化学
下記に示されるように、ヌクレオシドは少なくとも2つの重要なキラル炭素原子(*)を含有する。一般に、ヌクレオシドのキラル炭素における置換基[指定されたプリン塩基またはピリミジン塩基(これは糖環の中間番号付けを使用したときにはC1置換基として示される)およびCH2OH(これはC4置換基として示される)]は、糖の環系に関してcis(同じ側)またはtrans(反対側)のいずれかであり得る。cisおよびtransの両方のラセミ体は1対の光学異性体からなる。従って、各化合物は4つの異なる立体異性体を有する。4つの立体異性体は、(−O−成分が後側に位置するように糖成分を水平面で配向させたときの)下記の立体配置によって表される:(1)両方の基が「上側」にあるcis、これはβ−Dと呼ばれる;(2)その鏡像体、すなわち、両方の基が「下側」にあるcis、これは鏡像体であり、β−Lと呼ばれる;(3)C4置換基が「上側」にあり、C1置換基が「下側」にあるtrans(これはα−Dと呼ばれる);および(4)C4置換基が「下側」にあり、C1置換基が「上側」にあるtrans(これはα−Lと呼ばれる)。2つのcisエナンチオマーは一緒にしてβ−エナンチオマーのラセミ混合物と呼ばれ、2つのtransエナンチオマーはα−エナンチオマーのラセミ混合物と呼ばれる。
【0082】
【化47】
【0083】
【化48】
【化49】
定義
本明細書中で使用される用語「実質的に単一異性体の形態で」または用語「単離された形態で」は、指定された立体配置が少なくとも約95%であり、好ましくは少なくとも98%または99%である2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドを示す。好ましい実施形態において、活性な化合物は、少なくともこのレベルの純度で、治療が必要な宿主に投与される。
【0086】
本明細書中で使用される用語のD型肝炎および関連する状態は、D型肝炎の感染、HDVに付随する慢性的な肝臓炎症、肝硬変、急性肝炎、劇症肝炎、慢性の持続性肝炎および疲労を示す。本発明の方法は、HBVに感染している個体、またはHBVにさらされている個体における臨床的疾患の進行を防止または遅延させるために予防的に、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体、その医薬として許容される塩またはそのプロドラッグを使用することを含む。
【0087】
本明細書中で使用される用語のアルキルは、別途示されない限り、典型的にはC1〜C18(好ましくはC1〜C6)の飽和した直鎖状または分枝状または環状の一級または二級または三級の炭化水素を示し、具体的には、メチル、トリフルオロメチル、エチル、プロピル、ブチル、ペンチル、ヘキシル、イソプロピル、イソブチル、sec−ブチル、t−ブチル、イソペンチル、アミル、t−ペンチル、シクロペンチルおよびシクロヘキシルを含むが、これらに限定されない。アルキル基は、当業者に知られているように、例えば、Greene他、「Protective Groups in Organic Synthesis」(John Wiley and Sons、第2版、1991;これはここに参照により組み込まれる)に教示されているように、ヒドロキシル、アミノ、アルキルアミノ、アリールアミノ、アルコキシ、アリールオキシ、ニトロ、シアノ、スルホン酸、スルファート、ホスホン酸、リン酸またはホスホナート(これらは必要な場合には非保護または保護のいずれであってもよい)からなる群から選択される1つ以上の成分で場合によっては置換することができる。
【0088】
本明細書中で使用される用語のアシルは、式−C(O)R’の成分を示す(式中、R’は、アルキル、アリール、アルカリール、アラルキル、複素芳香族、アルコキシアルキル(メトキシメチルを含む)、アリールアルキル(ベンジルを含む)、アリールオキシアルキル(フェノキシメチルなど)またはアリール(フェニルを含む)であり、これらは、ハロゲン、C1〜C4アルキルもしくはC1〜C4アルコキシ、またはアミノ酸の残基で場合によっては置換される)。用語アシルは、具体的には、アセチル、プロピオニル、ブチリル、ペンタノイル、3−メチルブチリル、ハイドロジェンスクシナート、3−クロロベンゾアート、ベンゾイル、アセチル、ピバロイル、メシラート、プロピオニル、バレリル、カプロイック、カプリリック、カプリック、ラウリック、ミリスチック、パルミチック、ステアリックおよびオレイック(これらに限定されない)を含み、そしてまたアミノ酸の残基であり得る。
【0089】
本明細書中で使用される用語のプリン塩基またはピリミジン塩基には、アデニン、N6−アルキルプリン、N6−アシルプリン(この場合、アシルはC(O)(アルキル、アリール、アルキルアリールまたはアリールアルキル)である)、N6−ベンジルプリン、N6−ハロプリン、N6−ビニルプリン、N6−アセチレン性プリン、N6−アシルプリン、N6−ヒドロキシアルキルプリン、N6−チオアルキルプリン、N2−アルキルプリン、N2−アルキル−6−チオプリン、チミン、シトシン、5−フルオロシトシン、5−メチルシトシン、6−アザピリミジン(6−アザシトシンを含む)、2−メルカプトピリミジンおよび/または4−メルカプトピリミジン、ウラシル、5−ハロウラシル(5−フルオロウラシルを含む)、C5−アルキルピリミジン、C5−ベンジルピリミジン、C5−ハロピリミジン、C5−ビニルピリミジン、C5−アセチレン性ピリミジン、C5−アシルピリミジン、C5−ヒドロキシアルキルプリン、C5−アミドピリミジン、C5−シアノピリミジン、C5−ニトロピリミジン、C5−アミノピリミジン、N2−アルキルプリン、N2−アルキル−6−チオプリン、5−アザシチジニル、5−アザウラシリル、トリアゾロピリジニル、イミダゾロピリジニル、ピロロピリミジニルおよびピラゾロピリミジニルが含まれるが、これらに限定されない。
【0090】
塩基の例には、シトシン、5−フルオロシトシン、5−ブロモシトシン、5−ヨードシトシン、5−クロロシトシン、ウラシル、5−フルオロウラシル、5−ブロモウラシル、5−ヨードウラシル、5−メチルウラシル、チミン、アデニン、グアニン、イノシン、キサンチン、2,6−ジアミノプリン、6−アミノプリン、6−クロロプリンおよび2,6−ジクロロプリン、6−ブロモプリン、2,6−ジブロモプリン、6−ヨードプリン、2,6−ジヨードプリン、ヒポキサンチン、アミノ基またはカルボニル基を含む置換基を6位に場合により有する2−(Br、Fl、ClまたはI)プリン、ならびにアミノ基またはカルボニル基を含む置換基を2位に場合により有する6−(Br、Fl、ClまたはI)プリン、5−ブロモビニルシトシン、5−ブロモビニルウラシル、5−ブロモエテニルシトシン、5−ブロモエテニルウラシル、5−トリフルオロメチルシトシン、ならびに5−トリフルオロメチルウラシルが挙げられる。
【0091】
本明細書中で使用される用語のプロドラッグは、インビボ投与したときにそのヌクレオシドに変換される化合物を示す。非限定的な例には、医薬として許容される塩(これは代わりに「生理学的に受容可能な塩」として示される)、活性な化合物の5’位および/またはN4位(シチジン)またはN6位(アデノシン)のアシル化誘導体またはアルキル化誘導体、ならびに活性な化合物の5’−リン脂質誘導体および5’−エーテル脂質誘導体が挙げられる。
【0092】
本明細書中で使用される用語の宿主は、細胞株および動物(好ましくはヒト)を含む、ウイルスが複製し得る単細胞生物または多細胞生物を示す。あるいは、宿主は、本発明の化合物によってその複製または機能が変化し得るデルタ型肝炎ウイルスのゲノムの一部分を保有し得る。このような用語の宿主は、具体的には、感染した細胞、HDVゲノムのすべてまたは一部でトランスフェクションされた細胞、および動物(特に、霊長類(チンパンジーを含む)およびヒト)を示す。本発明の大部分の動物適用において、宿主はヒト患者である。しかし、ある種の適用症における獣医学的適用が本発明によって明らかに予想される(チンパンジーなど)。
【0093】
医薬として許容される塩およびプロドラッグ
本明細書中で使用される用語の医薬として許容される塩または複合体は、元の化合物の所望される生物学的活性を保持し、かつ存在する場合でも、最小限の望ましくない毒物学的作用を示す、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドの塩または複合体をいう。そのような塩の非限定的な例として、(a)無機酸(例えば、塩酸、臭化水素酸、硫酸、リン酸、硝酸など)によって形成される酸付加塩、ならびに酢酸、シュウ酸、酒石酸、コハク酸、リンゴ酸、アスコルビン酸、安息香酸、タンニン酸、パモ酸、アルギン酸、ポリグルタミン酸、ナフタレンスルホン酸、ナフタレンジスルホン酸およびポリガラクツロン酸などの有機酸との形成される塩;(b)ナトリウム、カリウム、亜鉛、カルシウム、ビスマス、バリウム、マグネシウム、アルミニウム、銅、コバルト、ニッケル、カドミウム、ナトリウム、カリウムなどのカチオンとの形成される塩基付加塩、またはN,N−ジベンジルエチレンジアミン、アンモニウムもしくはエチレンジアミンから形成される有機カチオンとの形成される塩基付加塩;あるいは(c)(a)および(b)の組合せ、例えば、タンニン酸亜鉛塩などが挙げられる。
【0094】
2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドは、下記の参考文献に開示されるように、5’−リン脂質または5’−エーテル脂質として提供され得る:Kucera,L.S.、Lyer,N.、Leake,E.、Raben,A.、Modest,E.J.、D.L.W.およびPiantadosi,C.、「感染性HIV−1の産生を阻害し、かつ欠陥ウイルスの形成を誘導する新規な膜相互作用性エーテル脂質アナログ」、AIDS Res Hum Retroviruses、1990、6、491〜501;Piantadosi,C.、J.Marasco C.J.、S.L.Morris−Natschke、K.L.Meyer、F.Gumus、J.R.Surles、K.S.Ishaq、L.S.Kucera、N.Lyer、C.A.Wallen、S.PiantadosiおよびE.J.Modest、「抗HIV活性に必要な新規なエーテル脂質ヌクレオシド結合体の合成および評価」、J Med Chem、1991、34、1408〜1414;Hostetler,K.Y.、Richman,D.D.、Carson,D.A.、Stuhmiller,L.M.、van Wijk,G.M.T.およびvan den Bosch,H.、「31−デオキシチミジン二リン酸ジミリストイルグリセロール(31−デオキシチミジンの脂質プロドラッグ)によるCEM細胞およびHT4−6C細胞における1型ヒト免疫不全症ウイルス複製の非常に強化された阻害」、Antimicrob Agents Chemother、1992、36、2025〜2029;Hostetler,K.Y.、Stuhmiller,L.M.、Lenting,H.B.、van den Bosch,H.およびRichman,D.D.、「アジドチミジンおよび他の抗ウイルス性ヌクレオシドのリン脂質アナログの合成および抗レトロウイルス活性」、J Biol Chem、1990、265、6112〜7。
【0095】
2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドは、適切なエステル化剤(例えば、酸ハロゲン化物または酸無水物)との反応によって医薬として許容されるエステルに変換され得る。ヌクレオシドまたはその医薬として許容されるプロドラッグは、従来の様式で、例えば、適切な塩基または酸との処理によってその医薬として許容される塩に変換され得る。エステルまたは塩は、例えば、加水分解によって元のヌクレオシドに変換され得る。
【0096】
活性な化合物の修飾、特にN4位、N6位および5’−O位における修飾は、活性な化学種の生物利用性および代謝速度に影響を及ぼすことがあり、したがって、活性な化学種の送達を制御することができる。
【0097】
本発明の好ましい実施形態は、ヒトまたは他の宿主動物におけるHDV感染症を処置するための方法である。この方法は、β−L−2’−デオキシアデノシン、β−L−2’−デオキシシチジン、β−L−2’−デオキシウリジン、β−L−2’−グアノシン、β−L−2’−デオキシイノシンおよびβ−L−2’−デオキシチミジン、あるいはその医薬として許容されるプロドラッグ(リン酸、5’位および/またはN4位またはN6位のアルキル化誘導体またはアシル化誘導体を含む)、あるいはその生理学的に受容可能な塩からなる群から選択される2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体の1つまたは複数の効果的な量を、場合によっては医薬として許容されるキャリアにおいて投与することを含む。本発明の化合物は、直接的な抗HDV活性を有するか、または抗HDV活性を示す化合物(1つもしくは複数)に代謝されるか、そのいずれでもある。好ましい実施形態において、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドは、実質的には単一異性体の形態で、すなわち、指定された立体配置が少なくとも約95%で投与される。
【0098】
本明細書中に記載されるヌクレオシドはどれも、活性、生物利用性、安定性、またはヌクレオシドを変化させる他の性質を増大させるために安定化されたプロドラッグとして投与することができる。多数のヌクレオチドプロドラッグリガンドが知られている。一般に、ヌクレオシドの一リン酸または二リン酸または三リン酸のアルキル化、アシル化または他の親油性修飾はヌクレオチドの安定性を増大させる。リン酸基における1つ以上の水素を置換し得る置換基の例には、アルキル、アリール、ステロイド、炭水化物(糖を含む)、1,2−ジアシルグルセロールおよびアルコールが挙げられる。多くが、R.JonesおよびN.Bischofberger、Antiviral Research、1995、27、1〜17に記載される。これらはいずれも、所望する効果を達成するために、開示されたヌクレオシドと組み合わせて使用することができる。
【0099】
1つの実施形態において、2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドは5’−ヒドロキシル親油性プロドラッグとして提供される。ヌクレオシドまたは親油性調製物の好ましくは5’−OH位においてヌクレオシドに共有結合的に取り込まれ得る好適な親油性置換基を開示する米国特許の非限定的な例には、米国特許第5,149,794号(1992年9月22日、Yatvin他)、同第5,194,654号(1993年3月16日、Hostetler他)、同第5,223,263号(1993年6月29日、Hostetler他)、同第5,256,641号(1993年10月26日、Yatvin他)、同第5,411,947号(1995年5月2日、Hostetler他)、同第5,463,092号(1995年10月31日、Hostetler他)、同第5,543,389号(1996年8月6日、Yatvin他)、同第5,543,390号(1996年8月6日、Yatvin他)、同第5,543,391号(1996年8月6日、Yatvin他)および同第5,554,728号(1996年9月10日、Basava他)がある。
【0100】
本発明の2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体または親油性調製物に結合させることができる親油性置換基を開示する外国特許出願明細書には、国際特許出願公開WO89/02733、同WO90/00555、同WO91/16920、同WO91/18914、同WO93/00910、同WO94/26273、同WO96/15132、欧州特許EP0350287、同EP93917054.4および国際特許出願公開WO91/19721が挙げられる。
【0101】
2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドのさらなる非限定的な例には、下記の刊行物に記載されるような置換基を含むヌクレオシドがある。これらの誘導体化された2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドは、本明細書中に記載される適応症に対して使用することができ、またはそれ以外の場合には、抗HBV剤としての使用を含む、抗ウイルス剤としての使用が可能である(Ho,D.H.W.、「ヒトおよびマウスの組織における1β−D−アラビノフラノシルシトシンのキナーゼおよびデアミナーゼの分布」、Cancer Res、1973、33、2816〜2820;Holy,A.、「等極性リン修飾ヌクレオチドアナログ」、Adavances in Antiviral Drug Design、De Clercq(編)、JAI Press:1993、第I巻、179〜231;Hong,C.I.、Nechaev,A.およびWest,C.R.、「コルチゾールおよびコルチゾンの1β−D−アラビノフラノシルシトシン結合体の合成および抗腫瘍活性」、Biochem Biophys Rs Commun、1979a、88、1223〜1229;Hong,C.I.、Nechaev,A.、Kirisits,A.J.、Buchheit,D.J.およびWest,C.R.、「潜在的な抗腫瘍剤としてのヌクレオシド結合体.3.コルチコステロイドおよび選択された親油性アルコールの1−(β−D−アラビノフラノシル)シトシン結合体の合成および抗腫瘍活性」、J Med Chem、1980、28、171〜177;Hostetler,K.Y.、Stuhmiller,L.M.、Lenting,H.B.M.、van den Bosch,H.およびRichman,D.D.、「アジドチミジンおよび他の抗ウイルス性ヌクレオシドのリン脂質アナログの合成および抗レトロウイルス活性」、J Biol Chem、1990、265、6112〜6117;Hostetler,K.Y.、Carson,D.A.およびRichman,D.D.、「ホスファチジルアジドチミジン:CEM細胞における抗レトロウイルス作用の機構」、J Biol Chem、1991、266、11714〜11717;Hostetler,K.Y.、Korba,B.、Sridhar,C.、Gardener,M.、「B型肝炎感染細胞におけるホスファチジルジデオキシシチジンの抗ウイルス活性およびマウスにおける増強された幹細胞取り込み」、Antiviral Res、1994a、24、59〜67;Hostetler,K.Y.、Richman,D.D.、Sridhar,C.N.、Felgner,P.L.、Felgner,J.、Ricci,J.、Gardener,M.F.、Selleseth,D.W.およびEllis,M.N.、「ホスファチジルアジドチミジンおよびホスファチジル−ddC:マウスリンパ組織における取り込みならびにヒト免疫不全症ウイルス感染細胞およびラウシャー白血病ウイルス感染マウスにおける抗ウイルス活性の評価」、Antimicrobial Agents Chemother、1994b、38、2792〜2797;Hunston,R.N.、Jones,A.A.、McGuigan,C.、Walker,R.T.、Balzarini,J.およびDe Clercq,E.、「2’−デオキシ−5−フルオロウリジンに由来するいくつかの環状ホスホトリエステルの合成および生物学的性質」、J Med Chem、1984、27、440〜444;Ji,Y.H.、Moog,C.、Schmitt,G.、Bischoff,P.およびLuu,B.、「潜在的な抗腫瘍剤としての7β−ヒドロキシコレステロールおよびピリミジンヌクレオシドのモノリン酸ジエステル:合成および抗腫瘍活性の予備的評価」、J Med Chem、1990、33、2264〜2270;Jones,A.S.、McGuigan,C.、Walker,R.T.、Balzarini,J.およびDe Clercq,E.、「いくつかのヌクレオシド環状ホスホルアミダートの合成、性質および生物学的活性」、J Chem Soc Perkin Trans I、1984、1471〜1474;Juodka,B.A.およびSmart,J.、「ジリボヌクレオシドa(P→N)アミノ酸誘導体の合成」、Coll Czech Chem Comm、1974、39、363〜968;Kataoka,S.、Imai,J.、Yamaji,N.、Kato,M.、Saito,M.、Kawada,T.およびImai,S.、「アルキル化されたcAMP誘導体:選択的合成および生物学的活性」、Nucleic Acids Res Sym Ser、1989、21、1〜2;Kataoka,S.、Uchida,R.およびYamaji,N.、「アデノシン3’,5’環状リン酸(cAMP)のベンジルトリエステルおよびメチルトリエステルの簡便な合成」、Heterocycles、1991、32、1351〜1356;Kinchington,D.、Harvey,J.J.、O’Connor,T.J.、Jones,B.C.N.M.、Devine,K.G.、Taylor−Robinson,D.、Jeffries,D.J.およびMcGuigan,C.、「インビトロでのHIVおよびMuLVに対するジドブジンのホスホルアミダート誘導体およびホスホロジアミダート誘導体の抗ウイルス作用の比較」、Antiviral Chem Chemother、1992、3、107〜112;Kodama,K.、Morozumi,M.、Saitoh,K.I.、Kuninaka,H.、Yoshino,H.およびSaneyoshi,M.、「1−β−D−アラビノフラノシルシトシン−5’−ステアリルリン酸(1−β−D−アラビノフラノシルシトシンの経口活性な誘導体)の抗腫瘍活性および薬理学」、Jpn J Cancer Res、1989、80、679〜685;Korty,M.およびEngels,J.、「モルモット心室心筋に対するアデノシン−3’,5’−リン酸およびグアノシン−3’,5’−リン酸およびベンジルエステルの影響」、Naunyn−Schmiedeberg’s Arch Pharmacol、1979、310、103〜111;Kumar,A.、Goe,P.L.、Jones,A.S.、Walker,R.T.、Balzarini,J.およびDe Clercq,E.、「いくつかの環状ホスホルアミダートヌクレオシド誘導体の合成および生物学的評価」、J Med Chem、1990、33、2368〜2375;LeBec,C.およびHuynh−Dinh,T.、「抗ガン性プロドラッグとしての5−フルオロウリジンおよびアラビノシチジンの親油性リン酸トリエステル誘導体の合成」、Tetrahedron Lett、1991、32、6553〜6556;Lichtenstein,J.、Barner,H.D.およびCohen,S.S.、「外部から供給されたヌクレオチドの大腸菌による代謝」、J Biol Chem、1960、235、457〜465;Lucthy,J.、Von Daenilken,A.、Friederich,J.、Manthey,B.、Zweifel,J.、Schlatter,C.およびBenn,M.H.、「3つの天然に存在するシアノエピチオアルカンの合成および毒物学的性質」、Mitt Geg Lebensmittelunters Hyg、1981、72、131〜133(Chem Abstr、95、127093);McGuigan,C.、Tollerfield,S.M.およびRiley,P.A.、「抗ウイルス性薬物Araのいくつかのリン酸トリエステル誘導体の合成および生物学的評価」、Nucleic Acids Res、1989、17、6065〜6075;McGuigan,C.、Devine,K.G.、O’Connor,T.J.、Galpin,S.A.、Jeffries,D.J.およびKinchington,D.、「抗HIV化合物としての3’−アジド−3’−デオキシチミジン(AZT)のいくつかの新規なホスホルアミダート誘導体の合成および評価」、Antiviral Chem Chemother、1990a、1、107〜113;McGuigan,C.、O’Connor,T.J.、Nicholls,S.R.、Nickson,C.およびKinchington,D.、「AZTおよびddCydのいくつかの新規な置換ジアルキルリン酸誘導体の合成および抗HIV活性」、Antiviral Chem Chemother、1990b、1、355〜360;McGuigan,C.、Nicholls,S.R.、O’Connor,T.J.およびKinchington,D.、「潜在的な抗AIDS薬としての3’修飾ヌクレオシドのいくつかの新規なジアルキルリン酸誘導体の合成」、Antiviral Chem Chemother、1990c、1、25〜33;McGuigan,C.、Devine,K.G.、O’Connor,T.J.およびKinchington,D.、「3’−アジド−3’−デオキシチミジン(AZT)のいくつかのハロアルキルホスホルアミダート誘導体の合成および抗HIV活性;トリクロロエチルメトキシアラニニル化合物の強力な活性」、Antiviral Res、1991、15、255〜263;McGuigan,C.、Pathirana,R.N.、Mahmood,N.、Devine,K.G.およびHay,A.J.、「AZTのアリールリン酸誘導体は、AZTの作用に対して抵抗性の細胞株においてHIV−1に対する活性を保持する」、Antiviral Res、1992、17、311〜321;McGuigan,C.、Pathirana,R.N.、Choi,S.M.、Kinchington,D.およびO’Connor,T.J.、「HIV阻害剤としてのAZTのホスホルアミダート誘導体:カルボキシル末端に対する研究」、Antiviral Chem Chemother、1993a、4、97〜101;McGuigan,C.、Pathirana,R.N.、Balzarini,J.およびDe Clercq,E.、「AZTのアリールリン酸誘導体による生物活性なAZTヌクレオチドの細胞内送達」、J Med Chem、1993b、36、1048〜1052。
【0102】
ヌクレオシド環状3’,5’−一リン酸のリン酸環に対するチェア−ツイスト平衡の問題が、チミジンフェニル環状3’,5’−一リン酸のジアステレオマーの1HNMRおよびX線結晶学による研究によって分析されている(J Am Chem Soc、109、4058〜4064;Nerbonne,J.M.、Richard,S.、Nargeot,J.およびLester,H.A.、「新しい光活性化可能な環状ヌクレオチドは環状AMPおよび環状GMPの濃度の細胞内上昇をもたらす」、Nature、1984、301、74〜76;Neumann,J.M.、Herve,M.、Debouzy,J.C.、Guerra,F.I.、Gouyette,C.、Dupraz,B.およびHuynh−Dinh,T.、「チミジンのグルコシルリン脂質の合成およびNMRによる膜貫通輸送研究」、J Am Chem Soc、1989、111、4270〜4277;Ohno,R.、Tatsumi,N.、Hirano,M.、Imai,K.、Mizoguchi,H.、Nakamura,T.、Kosaka,M.、Takatuski,K.、Yamaya,T.、Toyama,K.、Yoshida,T.、Masaoka,T.、Hashimoto,S.、Ohshima,T.、Kimura,I.、Yamada,K.およびKimura,J.、「経口投与された1−β−D−アラビノフラノシルシトシン−5’−ステアリルリン酸を用いた骨髄異形成症候群の処置」、Oncology、1991、48、451〜455)。
【0103】
Palomino,E.、Kessle,D.およびHorwitz,J.P.、「2’,3’−ジデオキシヌクレオシドを脳に持続的に送達するためのジヒドロピリジンキャリアシステム」、J Med Chem、1989、32、622〜625;Perkins,R.M.、Barney,S.、Wittrock,R.、Clark,P.H.、Levin,R.、Lambert,D.M.、Petteway,S.R.、Serafinowska,H.T.、Bailey,S.M.、Jackson,S.、Harnden,M.R.、Ashton,R.、Sutton,D.、Harvey,J.J.およびBrown,A.G.、「マウスにおけるラウシャーネズミ白血病ウイルスに対するBRL47923およびその経口プロドラッグSB203657Aの活性」、Antiviral Res、1993、20(増刊I)、84;Piantadosi,C.、Marasco,C.J.,Jr.、Morris−Natschke,S.L.、Meyer,K.L.、Gumus,F.、Surles,J.R.、Ishaq,K.S.、Kucera,L.S.、Iyer,N.、Wallen,C.A.、Piantadosi,S.およびModest,E.J.、「抗HIV−1活性に対する新規なエーテル脂質ヌクレオシド結合体の合成および評価」、J Med Chem、1991、34、1408〜1414;Pompon,A.、Lefebvre,I.、Imbach,J.L.、Kahn,S.およびFarquhar,D.、「細胞抽出物および組織培養培地におけるアジドチミジン−5’−一リン酸のモノ(ピバロイルオキシメチル)エステルおよびビス(ピバロイルオキシメチル)エステルの分解経路;‘オンラインISRPクリーニング’HPLC技術の適用」、Antiviral Chem Chemother、1994、5、91〜98;Postemark,T.、「環状AMPおよび環状GMP」、Annu Rev Pharmacol、1974、14、23〜33;Prisbe,E.J.、Martin,J.C.M.、McGee,D.P.C.、Barker,M.F.、Smee,D.F.、Duke,A.E.、Matthews,T.R.およびVerheyden,J.P.J.、「9−[(1,3−ジヒドロキシ−2−プロポキシ)メチル]グアニンのリン酸誘導体およびホスホナート誘導体の合成および抗ヘルペスウイルス活性」、J Med Chem、1986、29、671〜675;Puech,F.、Gosselin,G.、Lefebvre,I.、Pompon,A.、Aubertin,A.M.、Dirn,A.およびImbach,J.L.、「レダクターゼにより媒介される活性化プロセスによるヌクレオシド一リン酸の細胞内送達」、Antiviral Res、1993、22、155〜174;Pugaeva,V.P.、Klochkeva,S.L,、Mashbits,F.D.およびEizengart,R.S.(1969)Robins,R.K.、「レトロウイルスおよび腫瘍の阻害剤としてのヌクレオチドアナログの可能性」、Pharm Res、1984、11〜18;Rosowsky,A.、Kim,S.H.、Ross,J.およびWick,M.M.、「潜在的なプロドラッグとしての1−β−D−アラビノフラノシルシトシンならびにそのN4−アシル誘導体および2,2’−アンヒドロ−3’−O−アシル誘導体の親油性5’−(アルキルリン酸)エステル」、J Med Chem、1982、25、171〜178;Ross,W.、「グルコース前処理後の塩基性側鎖を有する芳香族ナイトロジェンマスタードに対するウォーカーターンアウトの増大した感受性」、Biochem Pharm、1961、8、235〜240;Ryu,E.K.、Ross,R.J.、Matsushita,T.、MacCoss,M.、Hong,C.I.およびWest,C.R.、「リン脂質ヌクレオシド結合体.3.1−β−D−アラビノフラノシルシトシン5’二リン酸[−],2−ジアシルグリセロールの合成および予備的な生物学的評価」、J Med Chem、1982、25、1322〜1329;Saffhill,R.およびHume,W.J.、「種々の供給源の血清による5−ヨードデオキシウリジンおよび5−ブロモデオキシウリジンの分解ならびにDNA内に取り込ませるためのこれらの化合物の使用に対するその結果」、Chem Biol Interact、1986、57、347〜355;Saneyoshi,M.、Morozumi,M.、Kodama,K.、Machida,J.、Kuninaka,A.およびYoshino,H.、「合成ヌクレオシドおよび合成ヌクレオチド.XVI.一連の1−β−D−アラビノフラノシルシトシンの5’−アルキルリン酸または5’−アリールリン酸の合成および生物学的評価」、Chem Pharm Bull、1980、28、2915〜2923;Sastry,J.K.、Nehete,P.N.、Khan,S.、Nowak,B.J.、Plunkett,W.、Arlinghaus,R.B.およびFarquhar,D.、「膜透過性ジデオキシウリジン5’−一リン酸アナログはヒト免疫不全症ウイルスの感染を阻害する」、Mol Pharmacol、1992、41、441〜445;Shaw,J.P.、Jones,R.J.、Arimilli,M.N.、Louie,M.S.、Lee,W.A.およびCundy,K.C.、「オスSprague−DawleyラットにおけるPMEAプロドラッグに由来するPMEAの経口による生物利用性」、9th Annual AAPS Meeting、San Diego、CA、1994(要約)。Shuto,S.、Ueda,S.、Imamura,S.、Fukukawa,K.、Matsuda,A.およびUeda,T.、「酵素二相反応による5’−ホスファチジルヌクレオシドの容易な一段階合成」、Tetrahedron Lett、1987、28、199〜202;Shuto,S.、Itoh,H.、Ueda,S.、Imamura,S.、Kukukawa,K.、Tsujino,M.、Matsuda,A.およびUeda,T.、「5’−(3−sn−ホスファチジル)ヌクレオシドの容易な酵素的合成およびその抗白血病活性」、Chem Pharm Bull、1988、36、209〜217。1つの好ましいリン酸プロドラッグ基はS−アシル−2−チオエチル基であり、これはまた「SATE」としても示される。
【0104】
薬学的組成物
本発明の2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体に基づく薬学的組成物で、D型肝炎の感染を処置するために治療効果的な量の上記化合物またはその塩もしくはプロドラッグを、場合によっては医薬として許容される添加剤、キャリアまたは賦形剤との組合せで含む薬学的組成物を調製することができる。治療効果的な量は、処置される感染または状態、その重篤度、用いられる処置法、使用される薬剤の薬物動態学、ならびに処置される患者により変化し得る。
【0105】
本発明による1つの態様において、本発明による化合物は、医薬として許容されるキャリアとの混合物で好ましくは配合される。一般には、薬学的組成物を経口投与可能な形態で投与することが好ましいが、配合物は、非経口的、静脈内、筋肉内、経皮的、口内、皮下、坐薬または他の経路によって投与することができる。静脈内および筋肉内に投与される配合物は、好ましくは、無菌の生理学的食塩水において投与される。当業者は、本発明の組成物を不安定にすることなく、またはその治療活性を損なうことなく、特定の投与経路に対する多数の配合物を提供するために本明細書の教示の範囲に含まれる配合物を改変することができる。具体的には、例えば、水または他のビヒクルにおける安定性をより大きくするために、所望する化合物の修飾を、日常的な修飾(塩の形成、エステル化など)によって容易に達成することができる。
【0106】
いくつかの薬学的投薬形態において、化合物のプロドラッグ形態が好ましく、これには、特に、本発明の化合物のアシル化誘導体(アセチル化誘導体など)およびエーテル誘導体、リン酸エステルおよび様々な塩形態が含まれる。当業者は、宿主生物または患者における標的化部位への活性な化合物の送達を促進するために、本発明の化合物をプロドラッグ形態に容易に修飾する方法を理解する。当業者はまた、適用可能な場合には、D型肝炎感染の処置において化合物の意図された作用を最大限にするために、所望する化合物を宿主生物または患者における標的化部位に送達する際にプロドラッグ形態の有利な薬物動態学的パラメーターを利用する。
【0107】
治療活性な配合物に含まれる化合物の量は、本発明によれば、感染または状態を処置するために効果的な量であり、好ましい実施形態ではD型肝炎の感染を処置するために効果的な量である。一般に、薬学投薬形態における本発明の化合物の治療効果的な量は、使用される化合物、処置される状態または感染、および投与経路に依存して、通常的には約0.1mg/kg〜約100mg/kg以上の範囲である。本発明のためには、組成物の予防的または防止的に効果的な量は、本発明によれば、治療効果的な量について上記に示されたのと同じ濃度範囲に含まれ、通常的には治療効果的な量と同じである。
【0108】
活性な化合物の投与は、連続的(静脈内点滴)から1日あたり数回(例えば、1日4回、1日2回など)の経口投与まで変化させることができ、そして他の投与経路の中でも、経口投与、局所投与、非経口投与、筋肉内投与、静脈内投与、皮下投与、経皮的投与(浸透強化剤を含む場合がある)、口内投与および坐薬投与を含み得る。腸溶性コーティングが施された経口錠剤もまた、経口投与経路からの化合物の生物利用性および安定性を高めるために使用することができる。最も効果的な投薬形態は、患者における疾患の重篤度だけでなく、選ばれた特定の薬剤の薬物動態学に依存する。投与が容易であり、そして患者が指示に従いやすいことが期待されるので、経口投薬形態が特に好ましい。
【0109】
本発明による薬学的組成物を調製するために、治療効果的な量の1つまたは複数の本発明による化合物が好ましくは、服用量を製造するための従来の製薬配合技術に従って、医薬として許容されるキャリアと混合される。キャリアは、投与(例えば、経口または非経口)のために所望される調製物の形態に依存して広範囲の形態を取ることができる。経口投薬形態の薬学的組成物を調製する際、通常の薬学的媒体はいずれも使用することができる。例えば、懸濁物、エリキシル剤および溶液剤などの液体の経口調製物の場合、水、グリコール、オイル、アルコール、矯味矯臭剤、保存剤、着色剤などを含む好適なキャリアおよび添加剤を使用することができる。粉末剤、錠剤、カプセルなどの固体の経口調製物の場合、そして坐薬などの固体の調製物の場合、デンプン、糖キャリア(デキストロース、マンニトール、ラクトースおよび関連するキャリアなど)、希釈剤、造粒化剤、滑剤、結合剤、崩壊剤などを含む好適なキャリアおよび添加剤を使用することができる。所望する場合には、錠剤またはカプセルは、標準的な技術によって、持続した放出のために腸溶性コーティングすることができる。これらの投薬形態の使用は患者における化合物の生物利用性に著しい影響を与え得る。
【0110】
非経口配合物の場合、キャリアは、通常、滅菌された水または塩化ナトリウム水溶液を含むが、分散補助成分などの他の成分もまた使用することができる。滅菌水が使用され、無菌として維持される場合、組成物およびキャリアもまた滅菌されなければならない。注射可能な懸濁物もまた調製することができ、そのような懸濁物が調製される場合、適切な液体のキャリア、懸濁剤などを用いることができる。
【0111】
リポソーム懸濁物(ウイルス抗原に対して標的化されたリポソームを含む)もまた、医薬として許容されるキャリアを製造するための従来の方法によって調製することができる。これは、本発明によるヌクレオシド化合物の遊離ヌクレオシド、アシルヌクレオシドまたはリン酸エステルプロドラッグ形態を送達するためには適切であり得る。
【0112】
本発明による特に好ましい実施形態において、本発明の化合物および組成物は、D型肝炎感染症を処置し、またはその発症を防止し、またはその発症を遅延させるために使用される。好ましくは、D型肝炎感染症を処置し、またはその発症を防止し、またはその発症を遅延させるために、組成物は、約250マイクログラムから約1グラム以上までの範囲の量で、1日に少なくとも1回、または好ましくは1日に4回までで、経口投薬形態で投与される。本発明の化合物は、好ましくは経口投与されるが、非経口的に、局所的に、または坐薬形態で投与することができる。
【0113】
本発明による化合物は、いくつかの場合には宿主細胞に対するその毒性が低いため、D型肝炎の感染を防止するために、またはウイルス感染に付随する臨床的症状の発生を防止するために好都合には予防的に用いることができる。したがって、本発明はまた、ウイルス感染(特にD型肝炎の感染)を予防的に処置するための方法を包含する。この態様において、本発明によれば、本発明の組成物が、D型肝炎の感染の発症を防止または遅延させるために使用される。この予防方法は、そのような処置を必要とする患者に、またはHDV疾患を発症する危険性を有する患者に、ウイルス感染の発症を緩和または防止または遅延させるために効果的な量の本発明による化合物を投与することを含む。本発明による予防的処置では、利用される抗ウイルス性化合物は患者に対して毒性が低く、好ましくは毒性がないことが好ましい。本発明のこの態様では、使用される化合物が、ウイルスに対して最大限に効果的であり、かつ患者に対する最小限の毒性を示すことが特に好ましい。このような疾患状態を処置するために使用され得る本発明による化合物は、D型肝炎の感染の増殖を防止するか、あるいは臨床的症状を発現するD型肝炎感染の発症を引き延ばすための予防剤として、治療的処置の場合と同じ範囲内で投与することができる(すなわち、経口投薬形態については約250マイクログラムから約1グラム以上、1日あたり1回から4回)。
【0114】
さらに、本発明による化合物は、本発明の他の化合物を含んで、1つまたは複数の抗ウイルス剤、抗HBV剤、抗HCV剤、抗HDV剤もしくは抗ヘルペス剤またはインターフェロン、抗ガン剤もしくは抗菌剤と組み合わせて、またはそれらと交互に投与することができる。本発明によるいくつかの化合物は、他の化合物の代謝、異化作用または不活性化を低下させることによって本発明によるいくつかの薬剤の生物学的活性を高めることに対して効果的であることがあり、そのため、この意図された作用のために同時に投与される。
【0115】
組合せ療法または交互療法
肝炎ウイルスの薬物耐性変化体が抗ウイルス剤による長期間の処置の後に出現し得ることが認識されている。D型肝炎ウイルスの生活環におけるB型肝炎ウイルスの役割は不可欠であるため、抗B型肝炎ウイルス活性を有する化合物を、開示されたβ−2’−L−デオキシヌクレオシド、その医薬として許容される塩またはプロドラッグと組み合わせて、またはそれらと交互に投与することができる。薬物耐性は、最も典型的には、ウイルス生活環において使用される酵素をコードする遺伝子が変異することによって生じ、HBVの場合には、最も典型的にはDNAポリメラーゼの変異によって生じる。近年、HBV感染に対する薬物の効力が、主成分の薬剤によって引き起こされる変異とは異なる変異を誘導する第2の抗ウイルス化合物および場合によっては第3の抗ウイルス化合物と組み合わせて、またはそれらと交互に化合物を投与することによって持続または増強または回復し得ることが明らかにされている。あるいは、薬物の薬物動態学、生物分布または他のパラメーターをそのような組合せ療法または交互療法によって変化させることができる。一般に、組合せ療法は典型的には、ウイルスに対して多重の同時ストレスが誘導されるために交互療法よりも好ましい。
【0116】
β−L−2’−dA、β−L−2’−dC、β−L−2’−dU、β−L−2’−dG、β−L−2’−dT、β−L−dI、または本明細書中に示される他のβ−L−2’−ヌクレオシド、またはこれらの化合物のプロドラッグ、リン酸もしくは塩の抗D型肝炎ウイルス活性は、2つ以上のこれらのヌクレオシドを組合せまたは交互で投与することによって高めることができる。あるいは、例えば、1つまたは複数のβ−L−2’−dA、β−L−2’−dC、β−L−2’−dU、β−L−2’−dG、β−L−2’−dT、β−L−dI、または本明細書中に示される他のβ−L−2’−ヌクレオシドを、3TC、FTC、L−FMAU、DAPD、ファムシクロビル、ペンシクロビル、BMS−200475、ビスポムPMEA(アデホビル、ジピボキシル)、ロブカビル、ガンシクロビルまたはリバビリンと組み合わせて、またはそれらと交互に投与することができる。
【0117】
本明細書中に記載される実施形態のいずれにおいても、本発明のβ−L−2’−ヌクレオシドは、活性な形態にリン酸化される第2のヌクレオシド系ポリメラーゼ阻害剤または非ヌクレオシド系ポリメラーゼ阻害剤と組み合わせて、またはそれらと交互に投与されるならば、選択された本発明のβ−L−2’−ヌクレオシドをインビボでリン酸化する酵素とは異なる酵素によって第2の化合物がリン酸化されることが好ましい。キナーゼ酵素の例には、チミジンキナーゼ、シトシンキナーゼ、グアノシンキナーゼ、アデノシンキナーゼ、デオキシシチジンキナーゼ、5’−ヌクレオチダーゼおよびデオキシグアノシンキナーゼがある。
【0118】
活性な化合物の調製
本発明の2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド誘導体はこの分野では知られており、Holy(Collect Czech Chem Commun、1972、37(12)、4072〜87;およびMol Phys、1967、3(4)、386〜95)によって開示される方法に従って調製することができる。
【0119】
β−L−エリスロ−ペントフラノヌクレオシド(β−L−dN)を得るための一般的なプロセスが、出発物質としてL−リボースまたはL−キシロースを使用して、図1に示される。
【0120】
活性なヌクレオシドの一リン酸誘導体、二リン酸誘導体および三リン酸誘導体を、発表された方法に従って記載される通りにして調製することができる。一リン酸は、Imai他(J Org Chem、1969、34(6)、1547〜1550)の手順に従って調製することができる。二リン酸は、Davisson他(J Org Chem、1987、52(9)、1794〜1801)の手順に従って調製することができる。三リン酸は、Hoard他(J Am Chem Soc、1965、87(8)、1785〜1788)の手順に従って調製することができる。
【0121】
実施例
実験プロトコル
融点は、封じていない毛細管でGallenkamp MFB−595−010M装置において測定されたが、未補正値である。UV吸収スペクトルはUvikon931(KONTRON)分光光度計でエタノール中で測定された。1H−NMRスペクトルは、Bruker AC250分光計または400分光計を用いてDMSO−d6中で室温において測定された。ケミカルシフトはppm単位で示され、DMSO−d5が基準として2.49ppmに設定されている。重水素交換、デカップリング実験または2D−COSYを行い、プロトンの帰属を確認した。シグナル多重度は、s(一重)、d(二重)、dd(二重の二重)、t(三重)、q(四重)、br(広い)、m(多重)によって表される。すべてのJ値はHz単位である。FAB質量スペクトルはJEOL DX300質量分析計において陽イオンモード(FAB>0)または負イオンモード(FAB<0)で記録された。マトリックスは3−ニトロベンジルアルコール(NBA)、またはグリセロールとチオグリセロール(GT)との混合物(50:50、v/v)であった。比旋光度はPerkin−Elmer241分光旋光計(光路長:1cm)で測定され、10−1 deg cm2 g−1の単位で示される。元素分析は「Service de Microanalyses du CNRS,Division de Vernaison」(フランス)によって行われた。元素記号または官能基によって示される分析は理論値の±0.4%以内であった。薄層クロマトグラフィーはシリカゲル60F254(Merck、製品番号5554)のプレコーティングされたアルミニウムシートで行われ、生成物の可視化が、10%のエタノール性硫酸および加熱による炭化後のUV吸収性によって行われた。カラムクロマトグラフィーは、大気圧下、シリカゲル60(Merck、製品番号9385)で行われた。
【0122】
実施例1 9−(3,5−ジ−O−ベンゾイル−β−L−キシロフラノシル)アデニン(3)
9−(2−O−アセチル−3,5−ジ−O−ベンゾイル−β−L−キシロフラノシル)アデニン2[参考文献:Gosselin,G.、Bergogne,M.−C.、Imbach,J.−L.、「5個の天然に存在する核酸塩基のβ−L−キシロフラノシルヌクレオシドの合成および抗ウイルス評価」、Journal of Heterocyclic Chemistry、1993、30、1229〜1233](8.30g、16.05mmol)および98%ヒドラジン水和物(234mL、48.5mmol)をピリジン/氷酢酸の混合物(4/1、v/v、170mL)に含む溶液を室温で22時間撹拌した。反応を、アセトン(40mL)を加えることによって停止させ、撹拌をさらに1時間続けた。反応混合物を、その容量を半分にして、水(250mL)で希釈し、クロロホルムで抽出した(150mLで2回)。有機層をNaHCO3飽和水溶液(100mLで3回)および水(100mLで3回)で順に洗浄し、乾燥し、ろ過して、濃縮し、そしてトルエンおよびメタノールと同時に蒸発させた。残渣をシリカゲルカラムクロマトグラフィー(0〜3%MeOH/ジクロロメタン)によって精製して、3を、ジイソプロピルエーテルから析出させて得た(5.2g、68%):1H NMR(DMSO−d6):δ4.5〜4.9(m,4H,H−2’,H−4’,H−5’およびH−5”)、5.64(t,1H,H−3’,J2’,3’=J3’,4’=3.5Hz)、6.3(br s,1H,OH−2’)、6.45(d,1H,H−1’,J1’,2’=4.6Hz)、7.3(br s,2H,NH2−6)、7.4〜7.9(m,10H,2 ベンゾイル)、8.07および8.34(2s,2H,H−2およびH−8);ms:マトリックス G/T、(FAB+)m/z 476[M+H]+、136[BH2]+、(FAB−)m/z 474[M−H]−、134[B]−;UV(95% エタノール):λmax 257nm(ε 16400)、230nm(ε 29300)、λmin 246nm(ε 14800);[α]D 20=−64(c 1.07,CHCl3)。元素分析C24H21N5O4(M=475.45)の計算値:C,60.43;H,4.45;N,14.73。実測値:C,60.41;H,4.68;N,14.27。
【0123】
実施例2 9−(3,5−ジ−O−ベンゾイル−2−デオキシ−β−L−threo−ペントフラノシル)アデニン(4)
化合物3(1.00g、2.11mmol)を含む乾燥アセトニトリル(65mL)の溶液に4−(ジメチルアミノ)ピリジン(0.77g、6.32mmol)およびフェノキシチオカルボニル塩化物(0.44mL、3.16mmol)を加えた。混合物を室温で2時間撹拌した。濃縮後、残渣をジクロロメタン(50mL)に溶解して、水(30mLで2回)および0.5N塩酸水溶液(30mL)および水(30mLで3回)で順に洗浄した。有機層を乾燥し、ろ過して、濃縮乾固した。粗製のチオカルボニル化中間体を、乾燥ジオキサン(17mL)中で2時間還流しながら、トリス(トリメチルシリル)シラン水素化物(0.78mL、5.23mmol)およびα,α’−アゾイソブチロニトリル(AIBN、0.112g、0.69mmol)で直接処理した。溶媒を減圧下で除き、残渣をシリカゲルカラムクロマトグラフィー(0〜5%MeOH/ジクロロメタン)によって精製して、純粋な4を泡状物として得た(0.93g、96%):1H NMR(DMSO−d6):δ2.9〜3.1(m,2H,H−2’およびH−2”)、4.6〜4.7(m,3H,H−4’,H−5’およびH−5”)、5.8(br s,1H,H−3’)、6.43(dd,1H,H−1’,J1’,2’=3.1Hz,J1’,2”=7.6Hz)、7.3(br s,2H,NH2−6)、7.4〜7.9(m,10H,2 ベンゾイル)、8.05および8.33(2s,2H,H−2およびH−8);ms:マトリックス G/T、(FAB+)m/z 460[M+H]+、325[S]+、136[BH2]+、(FAB−)m/z 458[M−H]−、134[B]−;UV(95% エタノール):λmax 261nm(ε 14400)、231nm(ε 26300)、λmin 249nm(ε 12000);[α]D 20=−38(c 1.04,DMSO)。
【0124】
実施例3 6−N−(4−モノメトキシトリチル)−9−(3,5−ジ−O−ベンゾイル−2−デオキシ−β−L−threo−ペントフラノシル)アデニン(5)
化合物4(0.88g、1.92mmol)を含む乾燥ピリジン(40mL)の溶液に4−モノメトキシトリチル塩化物(1.18g、3.84mmol)を加えた。混合物を60℃で24時間撹拌した。メタノール(5mL)を添加した後、溶液を濃縮乾固して、残渣をジクロロメタン(50mL)に溶解し、水(30mL)およびNaHCO3飽和水溶液(30mL)および水(30mL)で順に洗浄した。有機層を乾燥し、ろ過し、濃縮して、トルエンと同時に蒸発させ、純粋な5を泡状物として得た(1.01g、72%):1H NMR(CDCl3):δ2.9〜3.0(m,2H,H−2’およびH−2”)、3.62(s,3H,OCH3)、4.6〜4.8(m,3H,H−4’,H−5’およびH−5”)、5.85(pt,1H,H−3’)、6.44(dd,1H,H−1’,J1’,2’=3.1Hz,J1’,2”=7.3Hz)、6.9(br s,1H,NH−6)、6.7〜6.8および7.2〜7.4(2m,24H,2 ベンゾイルおよびMMTr)、7.97および8.13(2s,2H,H−2およびH−8);ms:マトリックス G/T、(FAB+)m/z 732[M+H]+、(FAB−)m/z 730[M−H]−;UV(95% エタノール):λmax 274nm(ε 12100)、225nm(ε 24200)、λmin 250nm(ε 5900);[α]D 20=−16(c 1.12,DMSO)。
【0125】
実施例4 6−N−(4−モノメトキシトリチル)−9−(2−デオキシ−β−L−threo−ペントフラノシル)アデニン(6)
化合物5(0.95g、1.30mmol)を(−10℃で飽和させた)メタノール性アンモニアの溶液(40mL)で室温で一晩処理した。濃縮後、残渣をジクロロメタン(60mL)に溶解し、水(30mL)で洗浄した。水層をジクロロメタン(10mL)で2回抽出した。有機層を一緒にして、乾燥し、ろ過して、濃縮した。残渣をシリカゲルカラムクロマトグラフィー(0〜5%MeOH/ジクロロメタン)によって精製して、純粋な6を泡状物として得た(0.67g、98%):1H NMR(CDCl3):δ2.6〜2.9(m,2H,H−2’およびH−2”)、3.5(br s,1H,OH−5’)、3.55(s,3H,OCH3)、3.9〜4.0(m,3H,H−4’,H−5’およびH−5”)、4.5〜4.6(m,1H,H−3’)、6.03(dd,1H,H−1’,J1’,2’=4.0Hz,J1’,2”=8.8Hz)、7.0(br s,1H,NH−6)、6.7〜6.8および7.1〜7.4(2m,14H,MMTr)、7.40(d,1H,OH−3’,JH,OH=10.6Hz)、7.80および7.99(2s,2H,H−2およびH−8);ms:マトリックス G/T、(FAB+)m/z 524[M+H]+、408[BH2]+、(FAB−)m/z 1045[2M−H]−、522[M−H]−、406[B]−;UV(95% エタノール):λmax 275nm(ε 12300)、λmin 247nm(ε 3600);[α]D 20=+28(c 0.94,DMSO)。
【0126】
実施例5 6−N−(4−モノメトキシトリチル)−9−(2−デオキシ−5−O−(4−モノメトキシトリチル)−β−L−threo−ペントフラノシル)アデニン(7)
化合物6(0.62g、1.24mmol)を、乾燥ピリジン(25mL)中で、4−モノメトキシトリチル塩化物(0.46g、1.49mmol)で室温で16時間処理した。メタノール(5mL)を添加した後、混合物を濃縮乾固した。残渣をジクロロメタン(60mL)に溶解し、水(40mL)およびNaHCO3飽和水溶液(40mL)および水(40mLで3回)で順に洗浄した。有機層を乾燥し、ろ過し、濃縮して、トルエンおよびメタノールと同時に蒸発させた。残渣をシリカゲルカラムクロマトグラフィー(0〜10%MeOH/ジクロロメタン)によって精製し、純粋な7を泡状物として得た(0.71g、72%):1H NMR(DMSO−d6):δ2.21(d,1H,H−2’J2’,2”=14.3Hz)、2.6〜2.7(m,1H,H−2”)、3.1〜3.3(2m,2H,H−5’およびH−5”)、3.64および3.65(2s,6H,2×OCH3)、4.1〜4.2(m,1H,H−4’)、4.2〜4.3(m,1H,H−3’)、5.68(d,1H,OH−3’,JH,OH=5.2Hz)、6.24(d,1H,H−1’,J1’,2”=7.0Hz)、6.7〜6.8および7.1〜7.3(2m,29H,2 MMTrおよびNH−6)、7.83および8.21(2s,2H,H−2およびH−8);ms:マトリックス G/T、(FAB+)m/z 796[M+H]+、408[BH2]+、(FAB−)m/z 794[M−H]−、406[B]−;UV(95% エタノール):λmax 275nm(ε 30900)、λmin 246nm(ε 12800);[α]D 20=+14(c 1.03,DMSO)。
【0127】
実施例6 6−N−(4−モノメトキシトリチル)−9−(3−O−ベンゾイル−2−デオキシ−5−O−(4−モノメトキシトリチル)−β−L−エリスロ−ペントフラノシル)アデニン(8)
ジエチルアゾジカルボキシラート(0.38mL、2.49mmol)を含む乾燥テトラヒドロフラン(20mL)の溶液を、ヌクレオシド7(0.66g、0.83mmol)、トリフェニルホスフィン(0.66g、2.49mmol)および安息香酸(0.30g、2.49mmol)を含む乾燥THF(20mL)の冷却した溶液(0℃)に滴下して加えた。混合物を室温で18時間撹拌して、メタノール(1mL)を加えた。溶媒を減圧下で除き、粗製物をシリカゲルカラムクロマトグラフィー(0〜5%酢酸エチル/ジクロロメタン)によって精製して、トリフェニルホスフィンオキシドをわずかに含有する化合物8を得た。
【0128】
実施例7 6−N−(4−モノメトキシトリチル)−9−(2−デオキシ−5−O−(4−モノメトキシトリチル)−β−L−エリスロ−ペントフラノシル)アデニン(9)
化合物8を、(−10℃で飽和させた)メタノール性アンモニアの溶液(20mL)によって室温で24時間処理し、その後、反応混合物を濃縮乾固した。残渣をジクロロメタン(30mL)に溶解し、水(20mL)で洗浄した。水層をジクロロメタン(20mLで2回)で抽出し、一緒にした有機相を乾燥し、ろ過し、濃縮した。純粋な化合物9(0.50g、7から76%)を、シリカゲルカラムクロマトグラフィー(0〜2%MeOH/ジクロロメタン)による精製の後に泡状物として得た:1H NMR(DMSO−d6):δ2.2〜2.3(m,1H,H−2’)、2.8〜2.9(m,1H,H−2”)、3.1〜3.2(m,2H,H−5’およびH−5”)、3.64および3.65(2s,6H,2×OCH3)、3.97(pq,1H,H−4’)、4.4〜4.5(m,1H,H−3’)、5.36(d,1H,OH−3’,JH,OH=4.5Hz)、6.34(t,1H,H−1’,J1’,2’=J1’,2”=6.4Hz)、6.8〜6.9および7.1〜7.4(2m,29H,2 MMTrおよびNH−6)、7.81および8.32(2s,2H,H−2およびH−8);ms:マトリックス G/T、(FAB+)m/z 796[M+H]+、408[BH2]+、(FAB−)m/z 794[MH]−、406[B]−;UV(95% エタノール):λmax 276nm(ε 42600)、λmin 248nm(ε 23300);[α]D 20=+29(c 1.05,DMSO)。
【0129】
実施例8 2’−デオキシ−β−L−アデノシン(β−L−dA)
化合物9(0.44g、0.56mmol)を80%酢酸水溶液(17mL)で室温において5時間処理した。混合物を濃縮乾固して、残渣を水(20mL)に溶解し、ジエチルエーテルで洗浄した(15mLで2回)。水層を濃縮して、トルエンおよびメタノールと同時に蒸発させた。所望する2’−デオキシ−β−L−アデノシン(β−L−dA)(0.12g、83%)を、シリカゲルカラムクロマトグラフィー(0〜12%MeOH/ジクロロメタン)による精製およびMillex HV−4ユニット(0.45μ、Millipore)によるのろ過の後に得た:mp193〜194℃(水からの結晶化物)(文献値:184〜185℃(L−エナンチオマー)[参考文献:Robins,M.J.、Khwaja,T.A.、Robins,R.K.、J Org Chem、1970、35、636〜639]および187〜189℃(D−エナンチオマー)[参考文献:Ness,R.K.、Synthetic Procedures in Nucleic Acid Chemistry;Zorbach,W.W.、Tipson,R.S.編、J.Wiley and sons、New York、1968、第1巻、183〜187];1H NMR(DMSO−d6):δ2.2〜2.3および2.6〜2.7(2m,2H,H−2’およびH−2”)、3.4〜3.6(2m,2H,H−5’およびH−5”)、3.86(pq,1H,H−4’)、4.3〜4.4(m,1H,H−3’)、5.24(t,1H,OH−5’,JH,OH=5.8Hz)、5.30(d,1H,OH−3’,JH,OH=4.0Hz)、6.32(dd,1H,H−1’,J1’,2’=6.2Hz,J1’,2”=7.8Hz)、7.3(br s,2H,NH2−6)、8.11および8.32(2s,2H,H−2およびH−8);ms:マトリックス G/T、(FAB+)m/z 252[M+H]+、136[BH2]+、(FAB−)m/z 250[M−H]−、134[B]−;UV(95% エタノール):λmax 258nm(ε 14300)、λmin 226nm(ε 2100);[α]D 20=+25(c 1.03,H2O)、(Lit.[α]D 20=+23(c 1.0,H2O)(文献値:[α]D 20=+23(c 1.0、H2O)(L−エナンチオマー)[参考文献:Robins,M.J.、Khwaja,T.A.、Robins,R.K.、J Org Chem、1970、35、636〜639]および[α]D 20=−25(c 0.47、H2O)(D−エナンチオマー)[参考文献:Ness,R.K.、Synthetic Procedures in Nucleic Acid Chemistry;Zorbach,W.W.、Tipson,R.S.編、J.Wiley and sons、New York、1968、第1巻、183〜187]。元素分析、計算値:C10H13N5O3+1.5H2O(M=278.28):C、43.16;H、5.80;N、25.17。実測値:C、43.63;H、5.45;N、25.33。
【0130】
実施例9 1−O−アセチル−2,3,5−トリ−O−ベンゾイル−β−L−リボフラノース(143)
スキーム1に示されるように、L−リボース140(150g、1mol;Cultor Science Food、CAS[24259−59−4]、バッチRIB9711013)を含むメタノール(2リットル、P.A.Prolabo;参照番号20847.295)の溶液を95%〜97%の硫酸(12mL;Merck;参照番号1.00731.1000)で処理して、+4℃で12時間放置し、その後、ピリジン(180mL;99%Acros;参照番号131780025)で中和した。留去により、メチルリボフラノシド141のα、βの混合物をシロップとして得た。このアノマー混合物を含むピリジン(1.3リットル)の溶液を、冷却および機械的撹拌を行いながら塩化ベンゾイル(580mL、5mol;Fluka;参照番号12930)で処理した。溶液を室温で12時間放置し、その後、絶えず撹拌しながら氷(約10リットル)に注いだ。混合物(水中のオイル)をセライト床でろ過した。セライト床上の得られたオイルを水(3リットルで3回)で洗浄し、その後、酢酸エチル(3リットル)を用いて溶解させた。有機相を5%NaHCO3溶液(2リットル)および水(2リットル)で洗浄し、硫酸ナトリウム(Prolabo;参照番号28111.365)で乾燥し、ろ過し、蒸発させて、1−O−メチル−2,3,5−トリ−O−ベンゾイル−α/β−L−リボフラノース142を濃厚なシロップとして得た。このオイルを無水酢酸(560mL;Fluka;参照番号45830)および酢酸(240mL;P.A.carlo erba;参照番号20104298)に溶解した。溶液を、濃硫酸(80mL)に滴下して加えた後、機械的に撹拌しながら低温(+4℃)で10時間保った。その後、溶液を連続的な撹拌下で氷(約10リットル)に注いだ。混合物(水中の油状化合物)をセライト床上でろ過した。セライト床上の得られたガム状固体を、水(3リットルで3回)で洗浄した後、ジクロロメタン(2.5リットル;P.A.Merck;参照番号1.06050.6025)に溶解した。有機相を5%NaHCO3溶液(1リットル)および水(2リットルで2回)で洗浄し、硫酸ナトリウムで乾燥し、ろ過し、蒸発させて、ガム状固体143を得た。これをエタノール95(Prolabo;参照番号20823.293)から結晶化して、225gの生成物(44%)を得た:mp129〜130℃(EtOH95)(Recondo,E.F.およびRinderknecht,H.(「1−O−アセチル−2,3,5−トリ−O−β−D−リボフラノシドの新しい容易な合成」、Helv.Chim.Acta、1959、1171〜1173)によって報告される文献参考値は130〜131℃のmpを示している);1H NMR(200MHz,CDCl3):δ8.09〜7.87(m,6H,HArom)、7.62〜7.31(m,9H,HArom)6.43(s,1H,H1)、5.91(dd,1H,H3,J3,4 6.7Hz;J3,2 4.9Hz)、5.79(pd,1H,H2,J2,3 4,9Hz;J1,2<1)、4,78(m,2H,H4およびH5)、4,51(dd,1H,H5,J5,5’ 13,1Hz,J5’,4 5,5Hz)、2,00(s,3H,CH3CO);(市販の1−O−アセチル−2,3,5−トリ−O−ベンゾイル−β−D−リボフラノースと同一)、質量分析(FAB+、GT)m/z445(M−OAc)+、元素分析C28H24O9、計算値C66.66、H4.79;実測値C、H。
【0131】
【化50】
実施例10 β−L−アデノシン(145)
スキーム2に示されるように、アデニン(19.6g、144mmol;Pharma−Waldhof;参照番号400134.001、ロット45276800)をアセトニトリル(400mL;Riedel−de Hean;参照番号33019;CaH2上で蒸留)に1−O−アセチル−2,3,5−トリ−O−ベンゾイル−β−L−リボフラノース143(60g、119mmol)とともに懸濁した。この懸濁物に発煙塩化第二スズ(22mL、187mmol;Fluka;参照番号96558)を加えた。12時間後、反応物を小容量(約100mL)に濃縮して、重炭酸ナトリウム(110g)および水(120mL)を加えた。得られた白色固体(スズ塩)を熱クロロホルムで抽出した(200mLで5回;Acros;参照番号22706463)。抽出液を一緒にしてセライト床でろ過した。有機相を5%NaHCO3溶液および水で洗浄し、硫酸ナトリウム(Prolabo;参照番号28111.365)で乾燥し、ろ過し、蒸発させて、化合物144(60g、無色の泡状物)を得た。泡状物を、撹拌下、室温で、密閉容器においてアンモニア飽和メタノール(220mL)で4日間処理した。溶媒を減圧下で蒸発させ、得られた粉末を酢酸エチル(400mL;Carlo erba;参照番号528299)に1時間還流しながら懸濁した。ろ過後、粉末を水(220mL)から再結晶化して、L−アデノシン145(24g、結晶、75%)を得た:mp233〜234℃(Saneyoshi,M.およびSatoh,E.(「合成ヌクレオシドおよび合成ヌクレオチド.XIII.いくつかの6−置換プリンの塩化第二スズ触媒によるリボシル化」、Chem Pharm Bull、1979、27、2518〜2521);Nakayama,C.およびSaneyoshi,M.(「合成ヌクレオシドおよび合成ヌクレオチド.XX.様々な1−β−キシロフラノシル−5−アルキルウラシルおよび関連ヌクレオシドの合成」、Nucleosides Nucleotides、1982、1、139〜146)は235℃〜238℃のmpを報告している);1H NMR(200MHz,DMSO−D6):δ8.34および8.12(2s,2H,H2およびH8)、7.37(ls,2H,NH2)、5.86(d,1H,H1’,J1’,2’ 6.2Hz)、5.43(m,2H,OH2’およびOH5’)、5.19(d,1H,OH3’,J 3.7Hz)、4,60(m,H2’)、4.13(m,1H,H3’)、3.94(m,1H,H4’)、3.69〜3.49(m,2H,H5’aおよびH5’b)、(市販のD−アデノシンと同一);質量分析(FAB+,GT)m/z 268(M+H)+、136(BH2)+。
【0133】
【化51】
実施例11 3’,5’−O−(1,1,3,3−テトライソプロピル−1,3−ジシロキサニル)−β−L−アデノシン(146)
スキーム3に示されるように、L−アデノシン145(47.2g、177mmol)をピリジン(320mL;99%、Acrosから入手;参照番号131780025)に懸濁して、1,3−ジクロロ−1,1,3,3−テトライソプロピルジシロキサン(63mL、201mmol;Fluka;参照番号36520)を加え、混合物を室温で12時間撹拌した。ピリジンを蒸発させ、残渣を、酢酸エチル(1L;Carlo erba;参照番号528299)および5%NaHCO3溶液(600mL)を用いて分配した。有機相を0.5NのHCl溶液(500mLで2回)および水(500mL)で洗浄し、硫酸ナトリウム(Prolabo;参照番号28111.365)で乾燥し、ろ過し、濃縮乾固した。得られた固体をアセトニトリル(Riedel−de Haen;参照番号33019)から結晶化して、化合物146(81g、90%)を得た:mp97〜98℃(Robins,M.J.他(「核酸関連化合物.42.二級アルコールを効率的に脱酸素化する一般的手法.リボヌクレオシドの2’−デオキシヌクレオシドへの位置特異的および立体選択的な変換」、J Am Chem Soc、1983、105、4059〜4065)はD−エナンチオマーについて98℃のmpを報告している);1H NMR(200MHz,CDCl3):δ8.28および7.95(2s,2H,H2およびH8)、5.96(d,1H,J1’,2’ 1,1Hz)、5.63(s,2H,NH2)、5.10(dd,1H,H3’,J3’,4’ 7.6Hz,J3’,2’ 5.5Hz)、4.57(dd,1H,H2’,J2’,1’ 1.2Hz;J2’,3’ 7.6Hz)、4.15〜3.99(m,3H,H4’,H5’aおよびH5’b)3.31(s1,1H,OH2’)、1.06(m,28H,イソプロピルのプロトン);質量分析(FAB−,GT)m/z 508(M−H)−、134(B)−;(FAB+,GT)m/z 510(M+H)+、136(BH2)+。
【0135】
【化52】
実施例12 3’,5’−O−(1,1,3,3−テトライソプロピル−1,3−ジシロキサニル)−2’−デオキシ−β−L−アデノシン(148)
スキーム4に示されるように、化合物146(34g、67mmol)にアセトニトリル(280mL;Riedel−de Haen;参照番号33019)、DMAP(16.5g、135mmol;99%、Acrosから入手;参照番号1482702050)およびフェニルクロロチオノカーボナート(10.2mL、73mmol;99%、Acrosから入手;参照番号215490050)を加えた。溶液を室温で12時間撹拌した。溶媒を蒸発させ、残渣を、酢酸エチル(400mL;Carlo Erba;参照番号528299)および0.5NのHCl溶液(400mL)を用いて分配した。有機相を0.5NのHCl溶液(400mL)および水(400mLで2回)で洗浄し、硫酸ナトリウム(Prolabo;参照番号28111.365)で乾燥し、ろ過し、濃縮乾固して、中間体を淡黄色の固体として得た。この粗製物147をジオキサン(Merck;参照番号1.09671.1000)に溶解して、AIBN(3.3g、20mmol;α,α’−アゾイソブチロニトリル、Flukaから入手、参照番号11630)およびTTMSS(33mL、107mmol;トリス(トリメチルシリル)シラン、Flukaから入手;参照番号93411)を加えた。溶液を徐々に加熱して還流させ、2時間撹拌した。反応物を濃縮して黄色のオイルにし、これをクロマトグラフィー処理(溶出液、ジクロロメタン(Merck;参照番号1.06050.6025):メタノール(Carlo Erba;参照番号309002)の95:5)して、化合物148(23g、無色の泡状物、70%)を得た。一部をエタノール/石油エーテルから結晶化した:mp110〜111℃(Robins,M.J.、Wilson,J.S.およびHansske,F.、「核酸関連化合物.42.二級アルコールを効率的に脱酸素化する一般的手法.リボヌクレオシドの2’−デオキシヌクレオシドへの位置特異的および立体選択的な変換」、J Am Chem Soc、1983、105、4059〜4065)は113℃〜114℃のmpを報告している);1H NMR(200MHz,CDCl3):δ8.33および8.03(2s,2H,H2およびH8)、6.30(dd,1H,H1’,J 2.85Hz,J 7.06Hz)、5.63(s1,2H,NH2)、4.96(m,1H,H3’)、4.50(m,2H,H5’aおよびH5’b)、2,68(m,2H,H2’aおよびH2’b)、1.08(m,28H,イソプロピルのプロトン);質量分析(FAB+,GT)m/z 494(M+H)+、136(BH2)+。
【0137】
【化53】
実施例13 2’−デオキシ−β−L−アデノシン(149)
Zhang,W.およびRobins,M.J.(「メタノール中のフッ化アンモニウムによるヒドロキシル官能基からのシリル保護基の除去」、Tetrahedron Lett、1992、33、1177〜1180)により教示されるように、3’,5’−O−(1,1,3,3−テトライソプロピル−1,3−ジシロキサニル)−2’−デオキシ−L−アデノシン148(32g、65mmol)およびフッ化アンモニウム(32g、mmol;Fluka;参照番号09742)を含むメタノール(Prolabo;参照番号20847.295)の溶液を還流下で2時間撹拌した(スキーム5)。シリカゲル(Merck;参照番号1.07734.2500)を加えて、混合物を慎重に蒸発させ、白色の粉末を得た。この粉末をシリカカラムの上部に加え、シリカカラムをジクロロメタン(Merck;参照番号1.06050.6025)/メタノールの9/1で溶出した。適切な画分を一緒にして蒸発させ、白色の粉末を得た。これをエタノール95(Prolabo;参照番号20823.293)から結晶化し、12.1gの生成物(75%)を得た:mp189〜190℃(EtOH95)(市販の2’−デオキシ−D−アデノシンと同一);1H NMR(200MHz,DMSO−D6):δ8.35および8.14(2s,2H,H2およびH8)、7.34(s1,2H,NH2)、6.35(dd,1H,H1’,J 6.1Hz,J 7.85Hz)、5.33(d,1H,OH2’,J 4.0Hz)、5.28(dd,1H,H3’,J 4.95Hz;J 6.6Hz)、4.42(m,1H,OH5’)、3.88(m,1H,H4’)、3.63〜3.52(m,2H,H5’aおよびH5’b)、2,71(m,1H,H2’a)、2.28(m,1H,H2’b)。(市販の2’−デオキシ−D−アデノシンと同一);αD +26°(c 0.5 水)(市販の2’−デオキシ−D−アデノシン−25°(c 0.5 水));UV λmax 260nm(ε 14100)(H2O);質量分析(FAB+,GT)m/z 252(M+H)+、136(BH2)+。
【0139】
【化54】
実施例14 1−(3,5−ジ−O−ベンゾイル−β−L−キシロフラノシル)ウラシル(11)
ヒドラジン水和物(1.4mL、28.7mmol)を、1−(2−O−アセチル−3,5−ジ−O−ベンゾイル−β−L−キシロフラノシル)ウラシル10[Gosselin,G.、Bergogne,M.−C.およびImbach,J.−L.、「5つの天然に存在する核酸塩基のβ−L−キシロフラノシルヌクレオシドの合成および抗ウイルス評価」、Journal of Heterocyclic Chemistry、1993、30、1229〜1233](4.79g、9.68mmol)を含むピリジン(60mL)と酢酸(15mL)との溶液に加えた。この溶液を室温で一晩撹拌した。アセトン(35mL)を加えて、混合物を30分間撹拌した。反応混合物を減圧下で蒸発させた。得られた残渣をシリカゲルカラムクロマトグラフィー[溶出液、ジクロロメタンにおけるメタノール(0〜4%)の段階的勾配]によって精製して、11(3.0g、68%)を得た。これをシクロヘキサン/ジクロロメタンから結晶化した:mp=111〜114℃;1H−NMR(DMSO−d6):δ11.35(br s,1H,NH)、7.9〜7.4(m,11H,2 C6H5CO,H−6)、6.38(d,1H,OH−2’,JOH−2’=4.2Hz)、5.77(d,1H,H−1’,J1’−2’=1.9Hz)、5.55(d,1H,H−5,J5−6=8Hz)、5.54(dd,1H,H−3’,J3’−2’=3.9HzおよびJ3’−4’=1.8Hz)、4.8(m,1H,H−4’)、4.7(m,2H,H−5’およびH−5”)、4.3(m,1H,H−2’);MS:FAB>0(マトリックス GT)m/z 453(M+H)+、105(C6H5CO)+;FAB<0(マトリックス GT)m/z 451(M−H)−、121(C6H5CO2)−、111(B)−;元素分析C23H20N2O8−H2Oの計算値:C,58.09;H,4.76;N,5.96。実測値:C,57.71;H,4.42;N,5.70。
【0141】
実施例15 1−(3,5−ジ−O−ベンゾイル−β−L−アラビノフラノシル)ウラシル(12)
1−(3,5−ジ−O−ベンゾイル−β−L−キシロフラノシル)ウラシル11(8g、17.7mL)を含む無水ベンゼン−DMSO混合物(265mL、6:4、v/v)の溶液に、無水ピリジン(1.4mL)、ジシクロヘキシルカルボジイミド(10.9g、53mmol)およびジクロロ酢酸(0.75mL)を加えた。得られた混合物を室温で4時間撹拌し、その後、酢酸エチル(400mL)で希釈して、シュウ酸(4.8g、53mmol)を含むメタノール(14mL)の溶液を加えた。1時間撹拌した後、溶液をろ過した。ろ液を飽和NaCl溶液(500mLで2回)、3%NaHCO3溶液(500mLで2回)および水(500mLで2回)で洗浄した。有機相をNa2SO4で乾燥して、減圧下で蒸発させた。その後、得られた残渣を無水EtOH/ベンゼンの混合物(140mL、2:1、v/v)に溶解させた。この溶液に0℃でNaBH4(0.96g、26.5mmol)を加えた。1時間撹拌した後、溶液を酢酸エチル(400mL)で希釈し、ろ過した。ろ液を飽和NaCl溶液(400mL)および水(400mL)で洗浄した。有機相をNa2SO4で乾燥して、減圧下で蒸発させた。得られた粗製物をシリカゲルカラムクロマトグラフィー[溶出液、ジクロロメタンにおけるメタノール(0〜3%)の段階的勾配]によって精製して、12(5.3g、66%)を得た。これをアセトニトリルから結晶化した:mp=182〜183℃;1H−NMR(DMSO−d6):δ11.35(br s,1H,NH)、8.0〜7.5(m,11H,2 C6H5CO,H−6)、6.23(br s,1H,OH−2’)、6.15(d,1H,H−1’,J1’−2’=4Hz)、5.54(d,1H,H−5,J5−6=8.1Hz)、5.37(t,1H,H−3’,J3’−2’=J3’−4’=2.6Hz)、4.7〜4.6(m,2H,H−5’およびH−5”)、4.5(m,1H,H−4’)、4.4(m,1H,H−2’);MS:FAB>0(マトリックス GT)m/z 453(M+H)+、341(S)+、113(BH2)+、105(C6H5CO)+;FAB<0(マトリックス GT)m/z 451(M−H)−、121(C6H5CO2)−、111(B)−;元素分析C23H20N2O8の計算値:C,61.06;H,4.46;N,6.19。実測値:C,60.83;H,4.34;N,6.25。
【0142】
実施例16 1−(3,5−ジ−O−ベンゾイル−2−デオキシ−β−L−エリスロ−ペントフラノシル)ウラシル(13)
1−(3,5−ジ−O−ベンゾイル−β−L−アラビノフラノシル)ウラシル12(5.2g、11.4mmol)を含む無水1,2−ジクロロエタン(120mL)の溶液に、フェノキシチオカルボニル塩化物(4.7mL、34.3mmol)および4−(ジメチルアミノ)ピリジン(DMAP、12.5g、102.6mmol)を加えた。得られた溶液をアルゴン雰囲気下において室温で1時間撹拌し、その後、減圧下で蒸発させた。残渣をジクロロメタン(300mL)に溶解して、有機溶液を、氷冷した0.2N塩酸(200mLで3回)および水(200mLで2回)で洗浄し、Na2SO4で乾燥し、その後、減圧下で蒸発させた。粗製物を、数回、無水ジオキサンと同時に蒸発させて、この溶媒(110mL)に溶解した。得られた溶液にアルゴン下においてトリス(トリメチルシリル)シラン水素化物(4.2mL、13.7mmol)およびα,α’−アゾイソブチロニトリル(AIBN、0.6g、3.76mmol)を加えた。反応混合物を加熱し、アルゴン下において100℃で1時間撹拌し、その後、室温に冷却し、減圧下で蒸発させた。残渣をシリカゲルカラムクロマトグラフィー[溶出液、メタノール(0〜5%)の段階的勾配]によって精製して、13(2.78g、56%)を得た。これをEtOHから結晶化した:mp=223〜225℃;H−NMR(DMSO−d6):δ11.4(br s,1H,NH)、8.0〜7.5(m,11H,2 C6H5CO,H−6)、6.28(t,1H,H−1’,J=7Hz)、5.5(m,2H,H−1’およびH−5)、4.6〜4.4(m,3H,H−4’,H−5’およびH−5”)、2.6(m,2H,H−2’およびH−2”);MS:FAB>0(マトリックス GT)m/z 437(M+H)+、3325(S)+;FAB<0(マトリックス GT)m/z 435(M−H)−、111(B)−;元素分析C23H20N2O7の計算値:C,63.30;H,4.62;N,6.42。実測値:C,62.98;H,4.79;N,6.40。
【0143】
実施例17 2’−デオキシ−β−L−シチジン(β−L−dC)
Lawesson試薬(1.72g、4.26mmol)を、1−(3,5−ジ−O−ベンゾイル−2−デオキシ−β−L−エリスロ−ペントフラノシル)ウラシル13(2.66g、6.1mmol)を含む無水1,2−ジクロロエタン(120mL)の溶液にアルゴン下で加えて、反応混合物を還流下で2時間撹拌した。その後、溶媒を減圧下で蒸発させて、残渣をシリカゲルカラムクロマトグラフィー[溶出液、ジクロロメタンにおける酢酸エチル(0〜8%)の段階的勾配]によって精製して、4−チオ中間体を黄色の泡状物として得た。このチオ中間体(1.5g、3.31mmol)を含むメタノール性アンモニア(事前に−10℃で飽和させ、きつく栓をしたもの)(50mL)の溶液をステンレススチール製ボンベにおいて100℃で3時間加熱し、その後、0℃に冷却した。溶液を減圧下で蒸発させた。得られた粗製物をシリカゲルカラムクロマトグラフィー[溶出液、ジクロロメタンにおけるメタノール(0〜20%)の段階的勾配]によって精製した。最後に、適切な画分をまとめて、Millex HV−4ユニット(0.45μm、Millipore)でろ過し、減圧下で蒸発させて、所望する2’−デオキシ−β−L−シチジン(β−L−dC)を泡状物として得た(0.6g、80%)。これを無水EtOTから結晶化した:mp=198〜199℃;1H−NMR(DMSO−d6):δ7.77(d,1H,H−6,J6−5=7.4Hz)、7.10(br d,2H,NH−2)、6.13(t,1H,H−1’,J=6.7Hz)、5.69(d,1H,H−5,J5−6=7.4Hz)、5.19(d,1H,OH−3’,JOH−3’=4.1Hz)、4.96(t,1H,OH−5’,JOH−5’=JOH−5”=5.2Hz)、4.1(m,1H,H−3’)、3.75(m,1H,H−4’)、3.5(m,2H,H−5’およびH−5”)、2.0(m,1H,H−2’)、1.9(m,1H,H−2”);MS:FAB>0(マトリックス GT)m/z 228(M+H)+、112(BH2)+;FAB<0(マトリックス GT)m/z 226(M−H)−;[α]20 D=−69(c 0.52,DMSO)[市販のD−エナンチオマーの塩酸塩についての[α]20 D=+76(c 0.55,DMSO)]。元素分析C9H13N3O4の計算値:C,47.57;H,5.77;N,18.49。実測値:C,47.35;H,5.68;N,18.29。
【0144】
実施例18 2−アミノ−β−L−アラビノフラノ[1’,2’:4,5]オキサゾリン(151)
L−アラビノース(170g、1.13mol;Fluka、>99.5%、参照番号10839)、シアナミド(100g、2.38mol;Fluka、>98%、参照番号28330)、メタノール(300mL)および6M−NH4OH(50mL)の混合物を室温で3日間撹拌し、その後、−10℃で一晩保った。生成物を吸引ろ過で集め、メタノールおよびエーテルで順に洗浄し、真空乾燥した。収量:130g(66.0%)の分析的に純粋な化合物151、m.p.170〜172℃;1H NMR(DMSO−d6)δppm 6.35(br s,2H,NH2)、5.15(d,1H,H−1,J=5.6Hz)、5.45(br s,1H,OH−3)、4.70(br s,1H,OH−5)、4.55(d,1H,H−2,J=5.6Hz)、4.00(br s,1H,H−3)、3.65(m,1H,H−4)、3.25(m,2H,H−5,H−5’)。
【0145】
実施例19 O2,2’−アンヒドロ−β−L−ウリジン(152)
化合物151(98.8g、0.57mol)およびプロピオン酸メチル(98mL、Fluka、>97%、参照番号81863)を含む50%水性エタノール(740mL)の溶液を5時間還流して、冷却し、そして最初の容量の1/2に減圧下で濃縮した。アセトン(600mL)で沈殿させた後、生成物を吸引ろ過で集め、エタノールおよびエーテルで洗浄して、乾燥した。母液を部分的に濃縮して、濃縮物をアセトン(1000mL)で沈殿させ、固体を吸引ろ過で集め、アセトンおよびエーテルで洗浄して、生成物の二番晶を得た。全収量:80g(62%)の化合物152、m.p.236〜240℃;1H NMR(DMSO−d6)δppm 7.87(d,1H,H−6,J=7.4Hz)、6.35(d,1H,H−1’,J=5.7Hz)、5.95(d,1H,H−5,J=7.4Hz)、5.90(d,1H,OH−3’)、5.20(d,1H,H−2’,J=5.7Hz)、5.00(m,1H,OH−3’)、4.44(br s,1H,H−3’)、4.05(m,1H,H−4’)、3.25(m,2H,H−5,H−5’)。
【0146】
実施例20 3’,5’−ジ−O−ベンゾイル−O2,2’−アンヒドロ−β−L−ウリジン(153)
化合物152(71.1g、0.31mol)を含む無水ピリジン(1200mL)の溶液に、アルゴン下、0℃で、塩化ベンゾイル(80.4mL;Fluka,p.a.、参照番号12930)を加えた。反応混合物を、雰囲気中の水分を除きながら室温で5時間撹拌し、そしてエタノールの添加によって停止させた。溶媒を減圧下で蒸発させ、得られた残渣をトルエンおよび無水エタノールと同時に蒸発させた。その後、粗製の混合物をエタノールで希釈して、沈殿を吸引ろ過で集め、エタノールおよびエーテルで順に洗浄し、乾燥した。収量:129g(95.8%)の化合物153、m.p.254℃:1H NMR(DMSO−d6)δppm 8.1〜7.4(m,11H,C6H5CO,H−6)、6.50(d,1H,H−l’,J=5.7Hz)、5.90(d,1H,H−5,J=7.5Hz)、5.80(d,1H,H−2’,J=5.8Hz)、5.70(d,1H,H−3’)4.90(m,1H,H−4’)、4.35(m,2H,H−5,H−5’)。
【0147】
実施例21 3’,5’−ジ−O−ベンゾイル−2’−クロロ−2’−デオキシ−β,L−ウリジン(154)
化合物153(60.3g、0.139mol)を含むジメチルホルムアミド(460mL)の溶液に、0℃で、3.2N−HCl/DMF溶液(208mL、47.2mLの塩化アセチル(Fluka,p.a.、参照番号00990)を0℃で27.3mLのメタノールと133.5mLのジメチルホルムアミドとの溶液に加えることによってその場で調製)を加えた。反応混合物を、雰囲気中の水分を除きながら100℃で1時間撹拌して、冷却し、そして水(4000mL)に注いだ。化合物154の沈殿を吸引ろ過で集め、水で洗浄して、エタノールから再結晶した。結晶を集め、冷エタノールおよびエーテルで洗浄して、減圧下で乾燥した。収量:60.6g(92.6%)の化合物154、m.p.164〜165℃;1H NMR(DMSO−d6)δppm 8.7(br s,1H,NH)、8.1〜7.3(m,11H,C6H5CO,H−6)、6.15(d,1H,H−1’,J=4.8Hz)、5.5(m,2H,H−5,H−2’)、4.65(m,4H,H−3’,H−4’,H−5’,H−5”)。
【0148】
実施例22 3’,5’−ジ−O−ベンゾイル−2’−デオキシ−β,L−ウリジン(155)
化合物154(60.28g、0.128mol)、トリ−n−ブチルスズ水素化物(95mL;Fluka、>98%、参照番号90915)およびアザビスイソブチロニトリル(0.568g;Fluka、>98%、参照番号11630)を含む乾燥トルエン(720mL)の混合物を撹拌下で5時間還流して、冷却した。固体を吸引ろ過で集め、冷トルエンおよび石油エーテルで洗浄した。ろ液を減圧下で濃縮し、石油エーテルで希釈して、化合物155の二番晶を析出させた。収量:54.28g(97.2%)の化合物155、m.p.220〜221℃:1H NMR(CDCl3)δppm 8.91(br s,1H,NH)、8.1〜7.5(m,11H,C6H5COおよびH−6)、6.43(q,1H,H−1’,J1’,2’=5.7HzおよびJ1’,2”=8.3Hz)、5.7〜5.6(m,2H,H−3’およびH−5)、4.8〜4.6(m,3H,H−5’,H−5”およびH−4’)、2.8〜2.7(m,1H,H−2’)、2.4〜2.3(m,1H,H−2”)。
【0149】
実施例23 3’,5’−ジ−O−ベンゾイル−2’−デオキシ−β−L−4−チオウリジン(156)
化合物155(69g、0.158mol)およびLawesson試薬(74g;Fluka、>98%、参照番号61750)を含む無水塩化メチレン(3900mL)の溶液をアルゴン下で一晩還流した。溶媒を蒸発させた後、粗製の残渣をシリカゲルカラムクロマトグラフィー[溶出液、塩化メチレンにおけるメタノール(0〜2%)の勾配]によって精製して、純粋な化合物156(73g)を定量的収率で得た;1H NMR(CDCl3)δppm 9.5(br s,1H,NH)、8.1〜7.4(m,10H,C6H5CO)、7.32(d,1H,H−6,J=7.7Hz)、6.30(dd,1H,H−1’,J=5.6HzおよびJ=8.2Hz)、6.22(d,1H,H−5,J=7.7Hz)、5.6(m,1H,H−3’)、4.7(m,2H,H−5’,H−5”)、4.5(m,1H,H−4’)、2.8(m,1H,H−2’)、2.3(m,1H,H−2”)。
【0150】
実施例24 2’−デオキシ−β−L−シトシン
化合物156(7.3g、0.016mol)を含むアンモニア飽和メタノール(73mL)の溶液をステンレススチール製ボンベにおいて100℃で3時間加熱した。注意深く冷却した後、溶媒を減圧下で蒸発させた。残渣の水溶液を酢酸エチルで洗浄し、蒸発乾固した。そのような手順を化合物156の9個の他のサンプル(それぞれ7.3g)について行った(2’−デオキシ−β−L−シトシンの収量:73g)。10個の残渣を一緒にして、無水エタノールで希釈し、冷却して、2’−デオキシ−β−L−シトシンを結晶として得た。微量のベンズアミドを、(酢酸エチル中で1時間の還流での)固液抽出法によって2’−デオキシ−β−L−シトシンの結晶から除いた。収量:28.75g(78.6%)の化合物2’−デオキシ−β−L−シトシン;m.p.141〜145℃;1H NMR(DMSO)δppm 8.22および8.00(2 br s,2H,NH2)、7.98(d,1H,H−6,J=7.59Hz)、6.12(t,1H,H−1’,J=6.5HzおよびJ=7.6Hz)、5.89(d,1H,H−5,J=7.59Hz)、5.3(br s,1H,OH−3’)、5.1(br s,1H,OH−5’)、4.2(m,1H,H−3’)、3.80(q,1H,H−4’,J=3.6HzおよびJ=6.9Hz)、3.6〜3.5(m,2H,H−5’,H−5”)、2.2〜2.0(m,2H,H−2’,H−2”);FAB<0(GT)m/z 226(M−H)−、110(B)−;FAB>0(GT)228(M+H)+、112(B+2H)+;[α]D 20−56.48(DMSO中のc=1.08);UV(pH 7)λmax=270nm(ε=10000)。
【0151】
実施例25 3’,5’−ジ−O−ベンゾイル−2’−デオキシ−5−ヨード−β−L−ウリジン(157)
化合物155(105.8g、0.242mol)、ヨウ素(76.8g;Fluka、99.8%、参照番号57650)、CAN(66.4g;硝酸セリウムアンモニウム、Aldrichから入手、>98.5%、参照番号21,547−3)およびアセトニトリル(2550mL)の混合物を80℃で3時間撹拌し、その後、反応混合物を室温に冷却して、化合物157を結晶化させた(86.6g、63.5%);m.p.192〜194℃;1H NMR(DMSO)δppm:8.34(s,1H,NH)、8.2〜7.2(m,11H,2 C6H5CO,H−6)、6.31(q,1H,H−1’,J=5.5HzおよびJ=8.7Hz)、5.5(m,1H,H−3’)、4.7(m,2H,H−5’,H−5”)、4.5(m,1H,H−4’)、2.7(m,1H,H−2’)、2.3(m,1H,H−2”);FAB<0、(GT)m/e 561(M−H)−、237(B)−;FAB>0(GT)563(M+H)+;[α]D 20+39.05(DMSO中のc=1.05);UV(EtOH 95)υmax=281nm(ε=9000)、υmin=254nm(ε=4000)、υmax=229nm(ε=31000);元素分析C23H19IN2O7の計算値:C,49.13 H,3.41 N,4.98 I,22.57。実測値:C,49.31 H,3.53 N,5.05 I,22.36。
【0152】
実施例26 3’,5’−ジ−O−ベンゾイル−2’−デオキシ−3−N−トルオイル−β−L−チミジン(159)
N−エチルジイソプロピルアミン(53.6mL;Aldrich、>99.5%、参照番号38,764−9)を含有する無水ピリジン(1530mL)における化合物157(86.6g、0.154mol)の溶液に、0℃で、p−トルオイル塩化物(40.6mL、Aldrich、98%、参照番号10,663−1)を少量ずつ加えた。反応混合物を室温で2時間撹拌し、その後、水を加えて反応を停止させ、反応混合物を塩化メチレンで抽出した。有機相を水で洗浄し、硫酸ナトリウムで乾燥し、蒸発乾固して、粗製の3’,5’−ジ−O−ベンゾイル−2’−デオキシ−3−N−トルオイル−5’−ヨード−β−L−ウリジン(158)を得た。これを、さらに精製することなく次の段階で使用した。
【0153】
粗混合物158、酢酸パラジウム(3.44g、Aldrich、>99.98%、参照番号37,987−5)、トリフェニルホスフィン(8.0g;Fluka、>97%、参照番号93092)をトリエチルアミン(4.3mL)とともに含むN−メチルピロリジノン(1375mL;Aldrich、>99%、参照番号44,377−8)の溶液を室温で45分間撹拌した。その後、テトラメチルスズ(42.4mL;Aldrich、>99%、参照番号14,647−1)をアルゴン下において0℃で滴下して加えた。100℃〜110℃で一晩撹拌した後、反応混合物を水に注ぎ、ジエチルエーテルで抽出した。有機溶液を硫酸ナトリウムで乾燥して、減圧下で濃縮した。残渣をシリカゲルカラムクロマトグラフィー[溶出液、トルエンにおける酢酸エチル(0〜10%)の段階的勾配]によって精製して、化合物159を泡状物として得た(2段階について、42.3g、48.3%)。1H NMR(DMSO)δppm.8.3〜7.2(m,15H,2 C6H5CO,1 CH3C6H4CO,H−6)、6.29(t,1H,H−1’,J=7.0Hz)、5.7(m,1H,H−3’)、4.7〜4.5(m,3H,H−5’,H−5”,H−4’)、2.7〜2.6(m,2H,H−2’,H−2”);FAB<0、(GT)m/e 567(M−H)−、449(M−CH3C6H4CO)−、243(B)−、121(C6H5COO)−;FAB>0(GT)1137(2M+H)+、569(M+H)+、325(M−B)−、245(B+2H)+、119(CH3C6H5CO)+。
【0154】
実施例27 2’−デオキシ−β−L−チミジン
化合物159(42.3g、0.074mol)を含むアンモニア飽和メタノール(1850mL)の溶液を室温で2日間撹拌した。溶媒を蒸発させた後、残渣を水で希釈して、酢酸エチルで数回洗浄した。水層を分離して減圧下で蒸発させ、残渣をシリカゲルカラムクロマトグラフィー[溶出液、塩化メチレンにおけるメタノール(0〜10%)の段階的勾配]によって精製して、純粋な2’−デオキシ−β−L−チミジン(11.62g、64.8%)を得た。これをエタノールから結晶化した;m.p.185〜188℃;1H NMR(DMSO)δppm 11.3(s,1H,NH)、7.70(s,1H,H−6)、6.2(pt,1H,H−1’)、5.24(d,1H,OH−3’,J=4.2Hz)、5.08(t,1H,OH−5’,J=5.1Hz)、4.2(m,1H,H−3’)、3.7(m,1H,H−4’)、3.5〜3.6(m,2H,H−5’,H−5”)、2.1〜2.0(m,2H,H−2’,H−2”);FAB<0、(GT)m/e 483(2M−H)−、349(M+T−H)−、241(M−H)−、125(B)−;FAB>0(GT)243(M+H)+、127(B+2H)+;)+;[α]D 20−13.0(DMSO中のc=1.0);UV(pH 1)υmax=267nm(ε=9700)、υmin=234nm(ε=2000)。
【0155】
実施例28 2’−デオキシ−β−L−イノシン(β−L−dI)の立体選択的合成
β−L−dIを、9−D−グルコピラノシル系列において以前に記載された手順に従った2’−デオキシ−β−L−アデノシン(β−L−dA)の脱アミノ化によって合成した(参考文献:I.Iwai、T.NishimuraおよびB.Shimizu、「Synthetic Procedures in Nucleic Acid Chemistry」(W.W.AorbachおよびR.S.Tipson編)、John Wiley&Sons,Inc.、New York、1968、1、135〜138)。
【0156】
【化55】
例えば、酢酸(0.61mL)と水(19mL)との混合物におけるβ−L−dA(200mg)の溶液を亜硝酸ナトリウム(495mg)と一緒に加熱し、混合物を室温で一晩撹拌した。その後、溶液を減圧下で蒸発乾固した。残渣の水溶液をIR−120(H+)イオン交換樹脂カラムに加え、カラムを水で溶出した。適切な画分を集め、蒸発乾固して、純粋なβ−L−dIを得た。これをメタノールから結晶化した(106mg、53%、収率は最適化されていない):m.p.=209〜211℃;UV(H2O)、λmax=247nm;1H−NMR(DMSO−d6)=8.32および8.07(2s,各1H,H−2およびH−8)、6.32(pt,1H,H−1;J=6.7Hz)、4.4(m,1H,H−3’)、3.9(m,1H,H−4’)、3.7〜3.4(m,2H HODで部分的に不明確,H−5’,5”)、2.6および2.3(2m,各1H,H−2’およびH−2”);質量分析(マチュアー,グリセロール−チオグリセロール,1:1,v/v)、FAB>0:253(M+H)+、137(塩基+2H)+;FAB<0:251(m−H)−、135(塩基)−;[α]D 20=+19.3(−c 0.88,H2O)。
【0158】
実施例29 化合物の毒性
毒性分析を、何らかの認められる抗ウイルス作用が細胞生存性に対する一般的な作用のためであるかどうかを評価するために行った。使用された方法は、ラムビジン(Lamuvidine)と比較して、ヒト骨髄クロロゲンアッセイにおける細胞増殖に対する、β−L−dA、β−L−dCおよびβ−L−dTの作用の測定である。結果を表1に示す。
【0159】
【表1】
実施例30 リン酸化された化合物の生物学的活性
β−L−dA、β−L−dC、β−L−dU、β−L−2’−dG、β−L−dIおよびβ−L−dTの各三リン酸誘導体のB型肝炎阻害能を調べた。表2には、ウッドチャック肝炎ウイルス(WHV)DNAポリメラーゼ、ヒトDNAポリメラーゼのα、βおよびγに対する、β−L−dTの三リン酸(β−L−dT−TP)、β−L−dCの三リン酸(β−L−dC−TP)、β−L−dUの三リン酸(β−L−dU−TP)およびβ−L−dAの三リン酸(β−L−dA−TP)の比較される阻害活性が記載される。
【0161】
【表2】
実施例31 化合物の抗ウイルス活性
β−L−dA、β−L−dC、β−L−dU、β−L−2’−dGおよびβ−L−dTの抗B型肝炎ウイルス活性を、トランスフェクションされたHepG−2(2.2.15)細胞で調べた。表3には、トランスフェクションされたHepG−2(2.2.15)細胞におけるB型肝炎ウイルスの複製に対するβ−L−dA、β−L−dC、β−L−dUおよびβ−L−dTの作用が例示される。
【0163】
【表3】
実施例32 化合物の組合せ療法
β−L−dA、β−L−dCおよびβ−L−dTを組み合わせたときのB型肝炎増殖に対する作用を2.2.15細胞で測定した。結果を表4に示す。
【0165】
【表4】
それぞれの組合せは相乗的な抗HBV活性をもたらした。さらに、L−dA+L−dC+L−dTの組合せもまたこのモデルでは相乗的であった。
【0167】
実施例33
β−L−dAおよびβ−L−dCの単独および組合せによる2.2.15細胞におけるB型肝炎の複製阻害を測定した。結果を表5に示す。
【0168】
【表5】
実施例35 化合物の効力
ウッドチャック肝炎ウイルス(WHV)に慢性的に感染しているウッドチャック(Marmota monax)におけるヘパドナウイルス感染に対するL−dA、L−dTおよびL−dCの効力を測定した。HBV感染のこの動物モデルは広く受け入れられており、HBVに対する抗ウイルス剤の評価に有用であることが証明されている。
【0170】
薬物群あたり3匹の動物およびコントロールあたり4匹の動物が使用された。群1において、動物にはビヒクルコントロールが投与された;群2にはラミブジン(3TC)が投与された(10mg/kg/日);群3〜6にはL−dAが投与された(それぞれ、0.01、0.1、1.0、10mg/kg/日);群7〜10にはL−dTが投与された(それぞれ、0.01、0.1、1.0、10mg/kg/日);群11〜14にはL−dCが投与された(それぞれ、0.01、0.1、1.0、10mg/kg/日)。
【0171】
薬物は、1日に1回、経口胃管によって投与され、血液サンプルが、0日目、1日目、3日目、7日目、14日目、21日目、28日目に、そして処置後の+1日目、+3日目、+7日目、+14日目、+28日目および+56日目に採取された。活性および毒性の評価は血清中のWHVのDNAの減少に基づいた:ドットブロット、定量的PCR。
【0172】
結果が図5および表6に例示される。
【0173】
【表6】
データは、L−dA、L−dTおよびL−dCがこのインビボモデルにおいて非常に活性であることを示している。第1に、ウイルス量が、検出できないレベルにまで低下している(L−dT)か、または検出限界に近いレベルにまで低下している(L−dA、L−dC)。第2に、L−dA、L−dTおよびL−dCは、このモデルでは3TC(ラミブジン)よりも活性であることを示している。第3に、ウイルスの反跳が、L−dTの中断後少なくとも2週間にわたって検出されない。第4に、用量応答曲線は、L−dAおよびL−dCの用量をある程度増大することにより、L−dTに類似する抗ウイルス活性が示されることを示唆している。第5に、薬物が投与された動物はすべて、体重が増大し、薬物に関連した毒性は全く検出されなかった。
【0175】
実施例34 薬学的組成物の調製
D型肝炎に感染しているヒトまたは他の宿主は、効果的な量のβ−2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド(例えば、β−L−2’−デオキシアデノシン、β−L−2’−デオキシシチジン、β−L−2’−デオキシウリジン、β−L−2’−デオキシグアノシンまたはβ−L−2’−デオキシチミジン)またはその医薬として許容されるプロドラッグもしくは塩を、医薬として許容されるキャリアまたは希釈剤の存在下で投与することによって処置することができる。活性な物質は、液体形態または固体形態において、任意の適切な経路によって、例えば、経口的、非経口的、静脈内、皮内、皮下または局所的に投与することができる。
【0176】
活性な化合物は、処置される患者において重大な毒性作用を引き起こすことなくウイルスの複製をインビボで阻害するために治療効果的な量の化合物を患者に送達するために十分な量で医薬として許容されるキャリアまたは希釈剤に含められる。「阻害的な量」により、例えば、本明細書中に記載されるアッセイなどのアッセイによって測定されるような阻害作用を示すために十分な有効成分の量が意味される。
【0177】
上記に記載された状態のすべてに対して好ましい用量は、1日あたり約1mg/kg体重〜50mg/kg体重の範囲であり、好ましくは1mg/kg体重〜20mg/kg体重の範囲であり、より一般的には1日あたり受容者の体重1kgについて0.1mg〜約100mgである。医薬として許容されるプロドラッグの効果的な投薬量範囲は、送達される親ヌクレオシドの重量に基づいて計算することができる。プロドラッグがそれ自体、活性を示す場合、効果的な投薬量は、プロドラッグの重量を使用して上記のように推定することができ、または当業者に知られている他の手段によって推定することができる。
【0178】
本発明の化合物はユニット型の任意の好適な投薬形態で都合よく投与される。そのような形態には、ユニット投薬形態物あたり7mg〜3000mg(好ましくは、70mg〜1400mg)の有効成分を含有する形態が含まれるが、これに限定されない。50mg〜1000mgの経口投薬量が通常の場合には好ましい。
【0179】
理想的には、有効成分は、約0.2μM〜70μM(好ましくは、約1.0μM〜10μM)の活性な化合物の最大血漿濃度が達成されるように投与されるべきである。これは、例えば、有効成分の0.1%〜5%の溶液を場合によっては生理的食塩水において静脈内注射することによって達成することができ、または有効成分のボーラス剤として投与することができる。
【0180】
薬物組成物における活性な化合物の濃度は、薬物の吸収速度、不活性化速度および排出速度、ならびに当業者に知られている他の要因に依存する。投薬量の値はまた、緩和すべき状態の重篤度により変化することには留意しなければならない。任意の特定の対象者については、具体的な投薬法を、個々の必要性、および組成物を投与する医師または組成物の投与を監督する医師の専門的判断に従って時間とともに調節すべきであること、そして本明細書中に示されている濃度範囲は例示にすぎず、請求項に記載される組成物の範囲または実施を制限するものではないことをさらに理解しなければならない。有効成分は一度に投与することができ、または様々な時間間隔で投与される多数の小量用量物に分割することができる。
【0181】
活性な化合物の好ましい投与経路は経口である。経口組成物は、不活性な希釈剤または食用のキャリアを一般に含む。それらはゼラチンカプセルに包むことができ、または錠剤に圧縮成形することができる。経口による治療的投与の場合、活性な化合物は賦形剤と配合され、そして錠剤、トローチ剤またはカプセル剤の形態で使用することができる。薬学的に適合し得る結合剤および/またはアジュバント物質を組成物の一部として含めることができる。
【0182】
錠剤、ピル剤、カプセル剤およびトローチ剤などは、下記の成分または性質が類似する化合物のいずれをも含有することができる:微結晶性セルロース、トラガカントゴムまたはゼラチンなどの結合剤;デンプンまたはラクトースなどの賦形剤;アルギン酸、プリモゲル(Primogel)またはトウモロコシデンプンなどの崩壊剤;ステアリン酸マグネシウムまたはステロート(Sterote)などの滑剤;コロイド状二酸化ケイ素などのグリダント;スクロースまたはサッカリンなどの甘味剤;またはペパーミント、サリチル酸メチルもしくはオレンジ風味剤などの矯味矯臭剤。投薬ユニット形態物がカプセルである場合、カプセルは、上記タイプの物質に加えて、油脂などの液体キャリアを含有することができる。さらに、投薬ユニット形態物は、投薬ユニットの物理的形態物を修飾する様々な他の物質を含有することができ、例えば、糖、セラックまたは他の腸溶剤のコーティングを含有することができる。
【0183】
本発明の化合物は、エリキシル剤、懸濁剤、シロップ剤、カシェ剤またはチューインガムなどの成分として投与することができる。シロップ剤は、活性な化合物に加えて、甘味剤としてのスクロース、ならびにある種の保存剤、色素および着色剤および香味剤を含有することができる。
【0184】
本発明の化合物またはその医薬として許容される誘導体もしくは塩はまた、所望する作用を損なわない他の活性な物質と、または所望する作用を補う物質と混合することができ、例えば、抗生物質、抗真菌剤、抗炎症剤、プロテアーゼ阻害剤、または他のヌクレオシド抗ウイルス剤もしくは非ヌクレオシド抗ウイルス剤と混合することができる。非経口適用、皮内適用、皮下適用または局所適用のために使用される溶液剤または懸濁剤は下記の成分を含むことができる:注射用水、生理的食塩水、不揮発性油、ポリエチレングリコール、グリセリン、プロピレングリコールまたは他の合成溶媒などの無菌希釈剤;ベンジルアルコールまたはメチルパラベンなどの抗菌剤;アスコルビン酸または重亜硫酸ナトリウムなどの抗酸化剤;エチレンジアミン四酢酸などのキレート化剤;酢酸塩、クエン酸塩またはリン酸塩などの緩衝剤、および塩化ナトリウムまたはデキストロースなどの張性調節剤。非経口用の調製物は、ガラス製またはプラスチック製のアンプル、ディスポーザブルシリンジまたは多回用量バイアルに入れることができる。
【0185】
静脈内投与される場合、好ましいキャリアは生理学的食塩水またはリン酸塩緩衝化生理的食塩水(PBS)である。
【0186】
好ましい実施形態において、活性な化合物は、身体から迅速に除去されないようにように化合物を保護するキャリアを用いて、例えば、インプラントおよびマイクロカプセル化送達システムを含む制御放出配合物などを用いて調製される。生分解性の生体適合性ポリマーを使用することができ、例えば、エチレンビニルアセタート、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステルおよびポリ乳酸を使用することができる。そのような配合物の調製方法は当業者には明らかである。これらの物質もまた、Alza Corporationから市販品を得ることができる。
【0187】
リポソーム懸濁物(ウイルス抗原に対するモノクローナル抗体で感染細胞に対して標的化されたリポソームを含む)もまた医薬として許容されるキャリアとして好ましい。これらは、例えば米国特許第4,522,811号に記載されるように、当業者に知られている方法に従って調製することができる。例えば、リポソーム配合物を、適切な脂質(1つまたは複数)(ステアロイルホスファチジルエタノールアミン、ステアロイルホスファチジルコリン、アラカドイルホスファチジルコリンおよびコレステロール)を無機溶媒に溶解し、その後、無機溶媒を蒸発させて、乾燥した脂質の薄い膜を容器表面に残すことによって調製することができる。その後、活性な化合物あるいはその一リン酸誘導体および/または二リン酸誘導体および/または三リン酸誘導体の水溶液が容器内に導入される。その後、容器を手で回して、容器の側面から脂質物質を遊離させ、脂質凝集体を分散させ、それにより、リポソーム懸濁物が形成される。
【0188】
本発明がその好ましい実施形態を参照して記載されている。本発明の様々な変化および改変が、本発明の前記の詳細な説明から当業者には明らかである。これらの変化および改変はすべて本発明の範囲内に含まれるものとする。
【図面の簡単な説明】
【図1】
出発物質としてL−リボースまたはL−キシロースを使用してβ−L−エリスロ−ペンタフラノヌクレオシド(β−L−dN)を得るための一般的なプロセスを例示する図である。
【図2】
出発物質としてL−リボースを使用してL−デオキシアデノシンを合成する非限定的な例を例示する図である。
【図3a】
出発物質としてL−アラビノースを使用してβ−L−dCを合成する非限定的な例を例示する図である。
【図3b】
出発物質としてL−アラビノースを使用してβ−L−dTを合成する非限定的な例を例示する図である。
【図4】
蓄積および減少に関してヒトHepG2細胞におけるL−dA、L−dCおよびL−dTの代謝を例示するグラフである。細胞は10μMの化合物とともにインキュベーションされた。
【図5】
ウッドチャック慢性肝炎モデルにおけるβ−L−dA、β−L−dTおよびβ−L−dCの抗ウイルス作用を例示するグラフである。
Claims (65)
- 下記式:
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;かつ
BASEは、場合によって置換され得るプリン塩基またはピリミジン塩基である。)
の2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドまたはその医薬として許容される塩の処置有効量を投与することを含むD型肝炎ウイルスに感染した宿主を処置するための方法。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;
YはOR3、NR3R4またはSR3であり;かつ
X1およびX2は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、ハロゲン、OR5、NR5NR6またはSR5からなる群から独立して選択され;そして
R3、R4、R5およびR6は独立して、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体である。)
のβ−L−2’−デオキシプリンまたはその医薬として許容される塩である請求項1に記載の方法。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシアデノシンまたはその医薬として許容される塩である請求項2に記載の方法。 - R1が水素である請求項3に記載の方法。
- R1がアシルである請求項3に記載の方法。
- アシルがアミノ酸に由来する請求項5に記載の方法。
- アミノ酸がバリンである請求項6に記載の方法。
- 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシグアノシンまたはその医薬として許容される塩である請求項2に記載の方法。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシイノシンまたはその医薬として許容される塩である請求項2に記載の方法。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;
YはOR3、NR3R4またはSR3であり;かつ
X1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、ハロゲン、OR5、NR5NR6またはSR5からなる群から選択され;そして
R3、R4、R5およびR6は独立して、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体である。)
のβ−L−2’−デオキシピリミジンまたはその医薬として許容される塩もしくはプロドラッグである請求項1に記載の方法。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシシチジンまたはその医薬として許容される塩である請求項10に記載の方法。 - R1が水素である請求項11に記載の方法。
- R1がアシルである請求項11に記載の方法。
- アシルがアミノ酸に由来する請求項13に記載の方法。
- アミノ酸がバリンである請求項14に記載の方法。
- 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシウリジンまたはその医薬として許容される塩である請求項10に記載の方法。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−チミジンまたはその医薬として許容される塩である請求項10に記載の方法。 - R1が水素である請求項17に記載の方法。
- R1がアシルである請求項17に記載の方法。
- アシルがアミノ酸に由来する請求項19に記載の方法。
- アミノ酸がバリンである請求項20に記載の方法。
- D型肝炎処置有効量の少なくとも2つの2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドを組み合わせてまたは交互に投与することを含み、それぞれの2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが独立して、下記の式:
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;かつ
BASEは、場合によって置換され得るプリン塩基またはピリミジン塩基である。)
またはその医薬として許容される塩であるD型肝炎ウイルスに感染した宿主を処置するための方法。 - D型肝炎処置有効量の生物学的に活性な2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドまたはその医薬として許容される塩を抗B型肝炎剤と組み合わせてまたは抗B型肝炎剤と交互に投与することを含み、さらなる抗B型肝炎剤が、FTC、L−FMAU、DAPD、ファムシクロビル、ペンシクロビル、BMS−200475、ビスポムPMEA(アデホビル、ジピボキシル)、ロブカビル、ガンシクロビルまたはリバビリンからなる群から選択されるD型肝炎ウイルスに感染した宿主を処置するための方法。
- 下記式の2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシド:
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;かつ
BASEは、必要によっては置換され得るプリン塩基またはピリミジン塩基である。)
またはその医薬として許容される塩の、D型肝炎ウイルスに感染した宿主を処置または予防するための使用。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;
YはOR3、NR3R4またはSR3であり;かつ
X1およびX2は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、ハロゲン、OR5、NR5NR6またはSR5からなる群から独立して選択され;そして
R3、R4、R5およびR6は独立して、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体である。)
のβ−L−2’−デオキシプリンまたはその医薬として許容される塩である請求項24に記載の使用。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシアデノシンまたはその医薬として許容される塩である請求項25に記載の使用。 - R1が水素である請求項26に記載の使用。
- R1がアシルである請求項26に記載の使用。
- アシルがアミノ酸に由来する請求項28に記載の使用。
- アミノ酸がバリンである請求項29に記載の使用。
- 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシグアノシンまたはその医薬として許容される塩である請求項25に記載の使用。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシイノシンまたはその医薬として許容される塩である請求項25に記載の使用。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;
YはOR3、NR3R4またはSR3であり、かつ
X1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、ハロゲン、OR5、NR5NR6またはSR5からなる群から選択され;そして
R3、R4、R5およびR6は独立して、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体である。)
のβ−L−2’−デオキシピリミジンまたはその医薬として許容される塩である請求項24に記載の使用。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシシチジンまたはその医薬として許容される塩である請求項33に記載の使用。 - R1が水素である請求項34に記載の使用。
- R1がアシルである請求項34に記載の使用。
- アシルがアミノ酸に由来する請求項36に記載の使用。
- アミノ酸がバリンである請求項37に記載の使用。
- 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシウリジンまたはその医薬として許容される塩である請求項33に記載の使用。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−チミジンまたはその医薬として許容される塩である請求項33に記載の使用。 - R1が水素である請求項40に記載の使用。
- R1がアシルである請求項40に記載の使用。
- アシルがアミノ酸に由来する請求項42に記載の使用。
- アミノ酸がバリンである請求項43に記載の使用。
- 下記式:
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;かつ
BASEは、必要によっては置換され得るプリン塩基またはピリミジン塩基である。)
の2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドまたはその医薬として許容される塩の、D型肝炎ウイルスに感染した宿主を処置または予防するための医薬品を製造することにおける使用。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;
YはOR3、NR3R4またはSR3であり;かつ
X1およびX2は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、ハロゲン、OR5、NR5NR6またはSR5からなる群から独立して選択され;そして
R3、R4、R5およびR6は独立して、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体である。)
のβ−L−2’−デオキシプリンまたはその医薬として許容される塩である請求項45に記載の使用。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシアデノシンまたはその医薬として許容される塩である請求項46に記載の使用。 - R1が水素である請求項47に記載の使用。
- R1がアシルである請求項47に記載の使用。
- アシルがアミノ酸に由来する請求項49に記載の使用。
- アミノ酸がバリンである請求項50に記載の使用。
- 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシグアノシンまたはその医薬として許容される塩である請求項46に記載の使用。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシイノシンまたはその医薬として許容される塩である請求項46に記載の使用。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
R1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体からなる群から選択され;
YはOR3、NR3R4またはSR3であり;かつ
X1は、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、ハロゲン、OR5、NR5NR6またはSR5からなる群から選択され;そして
R3、R4、R5およびR6は独立して、H、直鎖状もしくは分枝状もしくは環状のアルキル、CO−アルキル、CO−アリール、CO−アルコキシアルキル、CO−アリールオキシアルキル、CO−置換アリール、アルキルスルホニル、アリールスルホニル、アラルキルスルホニル、アミノ酸残基、一リン酸もしくは二リン酸もしくは三リン酸、またはリン酸誘導体である。)
のβ−L−2’−デオキシピリミジンまたはその医薬として許容される塩である請求項45に記載の使用。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシシチジンまたはその医薬として許容される塩である請求項54に記載の使用。 - R1が水素である請求項55に記載の使用。
- R1がアシルである請求項55に記載の使用。
- アシルがアミノ酸に由来する請求項57に記載の使用。
- アミノ酸がバリンである請求項58に記載の使用。
- 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−2’−デオキシウリジンまたはその医薬として許容される塩である請求項54に記載の使用。 - 2’−デオキシ−β−L−エリスロ−ペントフラノヌクレオシドが、下記式:
のβ−L−チミジンまたはその医薬として許容される塩である請求項54に記載の使用。 - R1が水素である請求項61に記載の使用。
- R1がアシルである請求項61に記載の使用。
- アシルがアミノ酸に由来する請求項63に記載の使用。
- アミノ酸がバリンである請求項64に記載の使用。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US20753800P | 2000-05-26 | 2000-05-26 | |
| PCT/US2001/017301 WO2001091737A2 (en) | 2000-05-26 | 2001-05-29 | Methods for treating hepatitis delta virus infection with beta-l-2' deoxy-nucleosides |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004513070A true JP2004513070A (ja) | 2004-04-30 |
| JP2004513070A5 JP2004513070A5 (ja) | 2008-08-14 |
Family
ID=22771005
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2001587753A Pending JP2004513070A (ja) | 2000-05-26 | 2001-05-29 | β−L−2’−デオキシヌクレオシドを用いたデルタ型肝炎ウイルス感染の処置方法 |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US6596700B2 (ja) |
| EP (1) | EP1292313B1 (ja) |
| JP (1) | JP2004513070A (ja) |
| KR (1) | KR100854398B1 (ja) |
| CN (1) | CN100490818C (ja) |
| AT (1) | ATE275406T1 (ja) |
| AU (3) | AU6348401A (ja) |
| BR (1) | BR0111195A (ja) |
| CA (1) | CA2410488C (ja) |
| DE (1) | DE60105424T2 (ja) |
| EA (1) | EA005890B1 (ja) |
| ES (1) | ES2227203T3 (ja) |
| HK (1) | HK1050999A1 (ja) |
| IL (1) | IL153078A0 (ja) |
| MX (1) | MXPA02011641A (ja) |
| TR (1) | TR200402565T4 (ja) |
| WO (1) | WO2001091737A2 (ja) |
| ZA (2) | ZA200210100B (ja) |
Families Citing this family (63)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA986614B (en) * | 1997-07-25 | 1999-01-27 | Gilead Sciences | Nucleotide analog composition |
| EP2415776B1 (en) * | 1998-08-10 | 2016-05-25 | Novartis AG | Beta-L-2'-Deoxy-Nucleosides for the Treatment of Hepatitis B |
| US6444652B1 (en) | 1998-08-10 | 2002-09-03 | Novirio Pharmaceuticals Limited | β-L-2'-deoxy-nucleosides for the treatment of hepatitis B |
| US20020056123A1 (en) * | 2000-03-09 | 2002-05-09 | Gad Liwerant | Sharing a streaming video |
| JP2003532643A (ja) | 2000-04-13 | 2003-11-05 | フアーマセツト・リミテツド | 肝炎ウイルス感染症を治療するための3’−または2’−ヒドロキシメチル置換ヌクレオシド誘導体 |
| MY164523A (en) | 2000-05-23 | 2017-12-29 | Univ Degli Studi Cagliari | Methods and compositions for treating hepatitis c virus |
| NZ547204A (en) | 2000-05-26 | 2008-01-31 | Idenix Cayman Ltd | Methods and compositions for treating flaviviruses and pestiviruses |
| US6787526B1 (en) * | 2000-05-26 | 2004-09-07 | Idenix Pharmaceuticals, Inc. | Methods of treating hepatitis delta virus infection with β-L-2′-deoxy-nucleosides |
| KR20090089922A (ko) * | 2000-10-18 | 2009-08-24 | 파마셋 인코포레이티드 | 바이러스 감염 및 비정상적인 세포 증식의 치료를 위한 변형된 뉴클레오시드 |
| CN101172993A (zh) | 2002-06-28 | 2008-05-07 | 埃迪尼克斯(开曼)有限公司 | 用于治疗黄病毒感染的2′-c-甲基-3′-o-l-缬氨酸酯核糖呋喃基胞苷 |
| US7456155B2 (en) * | 2002-06-28 | 2008-11-25 | Idenix Pharmaceuticals, Inc. | 2′-C-methyl-3′-O-L-valine ester ribofuranosyl cytidine for treatment of flaviviridae infections |
| PT1523489E (pt) * | 2002-06-28 | 2014-06-24 | Centre Nat Rech Scient | Profármacos de nucleósido modificado em 2' e 3' para tratamento de infecções por flaviridae |
| PL374792A1 (en) * | 2002-06-28 | 2005-10-31 | Idenix (Cayman) Limited | 2' and 3'-nucleoside prodrugs for treating flaviviridae infections |
| US7608600B2 (en) * | 2002-06-28 | 2009-10-27 | Idenix Pharmaceuticals, Inc. | Modified 2′ and 3′-nucleoside prodrugs for treating Flaviviridae infections |
| WO2004024095A2 (en) * | 2002-09-13 | 2004-03-25 | Idenix (Cayman) Limited | ß-L-2'-DEOXYNUCLEOSIDES FOR THE TREATMENT OF RESISTANT HBV STRAINS AND COMBINATION THERAPIES |
| US7148349B2 (en) * | 2002-10-31 | 2006-12-12 | Metabasis Therapeutics, Inc. | Cyclic phosphate diesters of 1,3-propane-1-aryl diols and their use in preparing prodrugs |
| HUE033832T2 (en) | 2002-11-15 | 2018-01-29 | Idenix Pharmaceuticals Llc | 2'-methyl nucleosides in combination with interferon and Flaviviridae mutation |
| CN1303089C (zh) * | 2002-11-19 | 2007-03-07 | 天津药物研究院 | 阿德福韦酯结晶形态及其制备方法 |
| CN1330373C (zh) * | 2002-12-02 | 2007-08-08 | 美德(江西)生物科技有限公司 | 含alfa-干扰素,阿地福韦和二脱氧氟硫代胞嘧啶的药物组合物 |
| PL377287A1 (pl) | 2002-12-12 | 2006-01-23 | Idenix (Cayman) Limited | Sposób wytwarzania 2'-rozgałęzionych nukleozydów |
| PL3521297T3 (pl) | 2003-05-30 | 2022-04-04 | Gilead Pharmasset Llc | Zmodyfikowane fluorowane analogi nukleozydów |
| WO2006031725A2 (en) | 2004-09-14 | 2006-03-23 | Pharmasset, Inc. | Preparation of 2'fluoro-2'- alkyl- substituted or other optionally substituted ribofuranosyl pyrimidines and purines and their derivatives |
| RU2427372C2 (ru) | 2005-06-17 | 2011-08-27 | Новартис Аг | Применение санглиферина для лечения вируса гепатита с |
| CA2634749C (en) | 2005-12-23 | 2014-08-19 | Idenix Pharmaceuticals, Inc. | Process for preparing a synthetic intermediate for preparation of branched nucleosides |
| GB0623493D0 (en) * | 2006-11-24 | 2007-01-03 | Univ Cardiff | Chemical compounds |
| US7964580B2 (en) | 2007-03-30 | 2011-06-21 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| EP2376514A2 (en) | 2008-12-23 | 2011-10-19 | Pharmasset, Inc. | Nucleoside analogs |
| NZ593648A (en) | 2008-12-23 | 2013-09-27 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| CL2009002206A1 (es) | 2008-12-23 | 2011-08-26 | Gilead Pharmasset Llc | Compuestos derivados de pirrolo -(2-3-d]-pirimidin-7(6h)-tetrahidrofuran-2-il fosfonamidato, composicion farmaceutica; y su uso en el tratamiento de enfermedades virales. |
| US8618076B2 (en) | 2009-05-20 | 2013-12-31 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| TWI583692B (zh) | 2009-05-20 | 2017-05-21 | 基利法瑪席特有限責任公司 | 核苷磷醯胺 |
| KR101715981B1 (ko) | 2010-03-31 | 2017-03-13 | 길리애드 파마셋 엘엘씨 | 뉴클레오사이드 포스포르아미데이트 |
| EA025341B1 (ru) | 2010-09-22 | 2016-12-30 | Алиос Биофарма, Инк. | Замещенные аналоги нуклеотидов |
| US8841275B2 (en) | 2010-11-30 | 2014-09-23 | Gilead Pharmasset Llc | 2′-spiro-nucleosides and derivatives thereof useful for treating hepatitis C virus and dengue virus infections |
| MX2013006127A (es) * | 2010-12-10 | 2013-09-26 | Sigmapharm Lab Llc | Composiciones sumamente estables de analogos nucleotidicoas oralmente activos o profarmacos analogos nucleotidicos oralmente activos. |
| DE202012013074U1 (de) | 2011-09-16 | 2014-10-29 | Gilead Pharmasset Lcc | Zusammensetzungen zur Behandlung von HCV |
| US8889159B2 (en) | 2011-11-29 | 2014-11-18 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| CA2860234A1 (en) | 2011-12-22 | 2013-06-27 | Alios Biopharma, Inc. | Substituted phosphorothioate nucleotide analogs |
| WO2013142124A1 (en) | 2012-03-21 | 2013-09-26 | Vertex Pharmaceuticals Incorporated | Solid forms of a thiophosphoramidate nucleotide prodrug |
| NZ630805A (en) | 2012-03-22 | 2016-01-29 | Alios Biopharma Inc | Pharmaceutical combinations comprising a thionucleotide analog |
| SI2950786T1 (sl) | 2013-01-31 | 2020-03-31 | Gilead Pharmasset Llc | Formulacija kombinacije dveh protivirusnih spojin |
| RS56211B1 (sr) * | 2013-07-31 | 2017-11-30 | Merck Patent Gmbh | Oksohinazolinil-butanamid derivati |
| ES2900570T3 (es) | 2013-08-27 | 2022-03-17 | Gilead Pharmasset Llc | Formulación de combinación de dos compuestos antivirales |
| EP3623364A1 (en) | 2014-02-13 | 2020-03-18 | Ligand Pharmaceuticals, Inc. | Prodrug compounds and their uses |
| CN106687118A (zh) | 2014-07-02 | 2017-05-17 | 配体药物公司 | 前药化合物及其用途 |
| KR20220018623A (ko) | 2015-06-17 | 2022-02-15 | 더 트러스티스 오브 컬럼비아 유니버시티 인 더 시티 오브 뉴욕 | 미토콘드리아 dna 고갈 증후군을 포함하는 불균형 뉴클레오타이드 풀에 의해 야기된 질환에 대한 데옥시뉴클레오타이드 요법 |
| EP3344642A1 (en) | 2015-09-02 | 2018-07-11 | AbbVie Inc. | Anti-viral tetrahydrofurane derivatives |
| CN108264524A (zh) * | 2016-12-31 | 2018-07-10 | 正大天晴药业集团股份有限公司 | 恩曲他滨膦酸酯化合物 |
| RU2644156C1 (ru) | 2017-02-28 | 2018-02-08 | Александр Васильевич Иващенко | Пролекарство ингибитора NS5B HCV полимеразы, способ его получения и применения |
| ES2938859T3 (es) | 2017-05-01 | 2023-04-17 | Gilead Sciences Inc | Una forma cristalina de (S)-2-etilbutilo 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopirrolo[2,1-f][1,2,4]triazin-7-il)-5-ciano-3,4-dihidroxitetrahidrofuran-2-il)metoxi)(fenoxi)fosforil)amino)propanoato |
| RU2662160C9 (ru) * | 2017-07-03 | 2018-10-22 | Александрович Иващенко Андрей | Комбинированный лекарственный препарат для терапии вирусных инфекций |
| CN111093627B (zh) | 2017-07-11 | 2024-03-08 | 吉利德科学公司 | 用于治疗病毒感染的包含rna聚合酶抑制剂和环糊精的组合物 |
| RU2662161C1 (ru) * | 2017-08-11 | 2018-07-24 | Васильевич Иващенко Александр | Ингибитор входа вируса гепатита и фармацевтическая композиция для лечения гепатита |
| JP2021509907A (ja) | 2018-01-09 | 2021-04-08 | リガンド・ファーマシューティカルズ・インコーポレイテッド | アセタール化合物およびその治療的使用 |
| CN118766947A (zh) | 2020-01-27 | 2024-10-15 | 吉利德科学公司 | 用于治疗SARS CoV-2感染的方法 |
| WO2021183750A2 (en) | 2020-03-12 | 2021-09-16 | Gilead Sciences, Inc. | Methods of preparing 1'-cyano nucleosides |
| KR20220164784A (ko) | 2020-04-06 | 2022-12-13 | 길리애드 사이언시즈, 인코포레이티드 | 1'-시아노 치환된 카르바뉴클레오시드 유사체의 흡입 제형 |
| CN115666570A (zh) | 2020-05-29 | 2023-01-31 | 吉利德科学公司 | 瑞德西韦治疗方法 |
| EP4172160A2 (en) | 2020-06-24 | 2023-05-03 | Gilead Sciences, Inc. | 1'-cyano nucleoside analogs and uses thereof |
| MX2023002233A (es) | 2020-08-24 | 2023-03-15 | Gilead Sciences Inc | Compuestos fosfolipidos y usos de estos. |
| CA3190702A1 (en) | 2020-08-27 | 2022-03-03 | Elaine Bunyan | Compounds and methods for treatment of viral infections |
| TWI811812B (zh) | 2020-10-16 | 2023-08-11 | 美商基利科學股份有限公司 | 磷脂化合物及其用途 |
| TW202400185A (zh) | 2022-03-02 | 2024-01-01 | 美商基利科學股份有限公司 | 用於治療病毒感染的化合物及方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06293645A (ja) * | 1993-04-09 | 1994-10-21 | Minero Saneyoshi | 逆転写酵素阻害剤 |
| WO2000009531A2 (en) * | 1998-08-10 | 2000-02-24 | Novirio Pharmaceuticals Limited | β-L-2'-DEOXY-NUCLEOSIDES FOR THE TREATMENT OF HEPATITIS B |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE8802687D0 (sv) | 1988-07-20 | 1988-07-20 | Astra Ab | Nucleoside derivatives |
| WO1996011204A1 (de) | 1994-10-07 | 1996-04-18 | Max-Delbrück-Centrum für Molekulare Medizin | NEUE β-L-NUCLEOSIDE UND IHRE VERWENDUNG |
-
2001
- 2001-05-29 DE DE60105424T patent/DE60105424T2/de not_active Expired - Fee Related
- 2001-05-29 CN CNB018134297A patent/CN100490818C/zh not_active Expired - Fee Related
- 2001-05-29 EP EP01937785A patent/EP1292313B1/en not_active Expired - Lifetime
- 2001-05-29 AU AU6348401A patent/AU6348401A/xx active Pending
- 2001-05-29 IL IL15307801A patent/IL153078A0/xx not_active IP Right Cessation
- 2001-05-29 US US09/867,110 patent/US6596700B2/en not_active Expired - Fee Related
- 2001-05-29 JP JP2001587753A patent/JP2004513070A/ja active Pending
- 2001-05-29 KR KR1020027015985A patent/KR100854398B1/ko not_active IP Right Cessation
- 2001-05-29 AT AT01937785T patent/ATE275406T1/de not_active IP Right Cessation
- 2001-05-29 BR BR0111195-7A patent/BR0111195A/pt not_active Application Discontinuation
- 2001-05-29 ES ES01937785T patent/ES2227203T3/es not_active Expired - Lifetime
- 2001-05-29 EA EA200201263A patent/EA005890B1/ru not_active IP Right Cessation
- 2001-05-29 WO PCT/US2001/017301 patent/WO2001091737A2/en active IP Right Grant
- 2001-05-29 TR TR2004/02565T patent/TR200402565T4/xx unknown
- 2001-05-29 MX MXPA02011641A patent/MXPA02011641A/es active IP Right Grant
- 2001-05-29 AU AU2001263484A patent/AU2001263484B2/en not_active Ceased
- 2001-05-29 CA CA002410488A patent/CA2410488C/en not_active Expired - Fee Related
-
2002
- 2002-12-12 ZA ZA200210100A patent/ZA200210100B/en unknown
-
2003
- 2003-04-22 HK HK03102836A patent/HK1050999A1/xx not_active IP Right Cessation
-
2004
- 2004-06-01 ZA ZA200404304A patent/ZA200404304B/xx unknown
-
2006
- 2006-12-21 AU AU2006252214A patent/AU2006252214A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06293645A (ja) * | 1993-04-09 | 1994-10-21 | Minero Saneyoshi | 逆転写酵素阻害剤 |
| WO2000009531A2 (en) * | 1998-08-10 | 2000-02-24 | Novirio Pharmaceuticals Limited | β-L-2'-DEOXY-NUCLEOSIDES FOR THE TREATMENT OF HEPATITIS B |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2410488A1 (en) | 2001-12-06 |
| AU2001263484B2 (en) | 2006-12-14 |
| AU2006252214A1 (en) | 2007-01-18 |
| US6596700B2 (en) | 2003-07-22 |
| KR20030031487A (ko) | 2003-04-21 |
| IL153078A0 (en) | 2003-06-24 |
| ATE275406T1 (de) | 2004-09-15 |
| ES2227203T3 (es) | 2005-04-01 |
| WO2001091737A2 (en) | 2001-12-06 |
| DE60105424T2 (de) | 2005-09-22 |
| AU6348401A (en) | 2001-12-11 |
| EA200201263A1 (ru) | 2003-06-26 |
| KR100854398B1 (ko) | 2008-08-26 |
| CN1444483A (zh) | 2003-09-24 |
| ZA200404304B (en) | 2005-03-30 |
| EP1292313B1 (en) | 2004-09-08 |
| DE60105424D1 (de) | 2004-10-14 |
| WO2001091737A3 (en) | 2002-05-30 |
| MXPA02011641A (es) | 2004-07-30 |
| BR0111195A (pt) | 2004-07-20 |
| EA005890B1 (ru) | 2005-06-30 |
| CN100490818C (zh) | 2009-05-27 |
| HK1050999A1 (en) | 2003-07-18 |
| EP1292313A2 (en) | 2003-03-19 |
| US20020035085A1 (en) | 2002-03-21 |
| TR200402565T4 (tr) | 2004-12-21 |
| CA2410488C (en) | 2009-10-27 |
| ZA200210100B (en) | 2004-06-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6596700B2 (en) | Methods of treating hepatitis delta virus infection with β-L-2'-deoxy-nucleosides | |
| US6787526B1 (en) | Methods of treating hepatitis delta virus infection with β-L-2′-deoxy-nucleosides | |
| US6395716B1 (en) | β-L-2′-deoxy-nucleosides for the treatment of hepatitis B | |
| US9290533B2 (en) | β-L-2′-deoxy-nucleosides for the treatment of hepatitis B | |
| AU2001263484A1 (en) | Methods of treating hepatitis delta virus infection with beta-l-2'- deoxy-nucleosides | |
| EP1431304B1 (en) | Beta - L-2'-Deoxy-Nucleosides for the treatment of Hepatitis B |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080528 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080528 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20080527 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110830 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20120207 |