JP2004510705A - 方法 - Google Patents
方法 Download PDFInfo
- Publication number
- JP2004510705A JP2004510705A JP2002513457A JP2002513457A JP2004510705A JP 2004510705 A JP2004510705 A JP 2004510705A JP 2002513457 A JP2002513457 A JP 2002513457A JP 2002513457 A JP2002513457 A JP 2002513457A JP 2004510705 A JP2004510705 A JP 2004510705A
- Authority
- JP
- Japan
- Prior art keywords
- cox
- group
- inhibitor
- cells
- maids
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 CS(Nc1c(**)c(*CC=*)c(*)c(*)c1*)(=O)=O Chemical compound CS(Nc1c(**)c(*CC=*)c(*)c(*)c1*)(=O)=O 0.000 description 1
Images















Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/661—Phosphorus acids or esters thereof not having P—C bonds, e.g. fosfosal, dichlorvos, malathion or mevinphos
- A61K31/6615—Compounds having two or more esterified phosphorus acid groups, e.g. inositol triphosphate, phytic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/365—Lactones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
- A61K31/381—Heterocyclic compounds having sulfur as a ring hetero atom having five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
- A61K31/405—Indole-alkanecarboxylic acids; Derivatives thereof, e.g. tryptophan, indomethacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/5415—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame ortho- or peri-condensed with carbocyclic ring systems, e.g. phenothiazine, chlorpromazine, piroxicam
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Abstract
Description
本発明は、免疫不全およびウィルス感染の治療分野に属する。より詳しくは、本発明は、免疫不全およびウィルス性疾患、特にHIV感染およびAIDSおよび関連する状態の治療のための免疫調節における、シクロオキシゲナーゼ−2(COX−2)阻害剤またはその誘導体の使用に関する。
【0002】
プロスタグランジンは、炎症過程で重要な役割を担うため、プロスタグランジンの形成阻害は、抗炎症剤開発のための一般的な標的となっている。非ステロイド系抗炎症剤(NSAID類)は、アラキドン酸からのプロスタグランジン中間体の生合成に関与する酵素であるシクロオキシゲナーゼ(COX)を阻害する。臨床用途に使われるいくつかのNSAID類が存在し、たとえば、インドメタシン、ピロキシカム、テノキシカム、ジクロフェナク、メロキシカム、テニダップ(tenidap)、イソキシカム(isoxicam)、 アセチルサリチル酸、ジフルニサル、スリンダク、イブプロフェン、ナプロキセンおよび ケトプロフェンなどの薬剤が挙げられる。
【0003】
NSAID類は、今日、世界的に最も広く処方されている薬剤である。
これらNSAID類は、臨床的に有効な薬剤であり、解熱性、抗炎症性、および抗血栓性作用を有する。この種の薬剤の主な適応症は、関節炎であり、たとえば、骨関節炎、リューマチ性関節炎、筋骨格の疼痛状態および一般的な疼痛状態が挙げられる。しかしながら、これらの薬剤には深刻な副作用がある。最も頻発する副作用は、胃腸の潰瘍化および出血、血小板凝集の阻害および他の薬剤との相互作用である。
【0004】
1990年代初めに、前記酵素の第2のCOXアイソフォーム(COX isoform)がクローニングされた。この新しいCOXアイソフォームは、現在、COX−2として知られている(Vaneら、1998年、Ann. Rev. Pharmacol. Toxicol., 38, p97−120)。
現在、よく知られた2つのCOXのアイソフォームがある。COX−1とCOX−2である(最近、COX−3の存在も推定されている)。COX−1は、たいていの組織内に存在し、ハウスキーパー酵素としてみなすことができる。たとえば、COX−1酵素の活性は、消化管の内壁を保護する。これに対し、COX−2は、通常は存在せず、炎症の際に増加する。NSAID類のいくつかの副作用は、COX−1酵素の阻害に関係している。NSAID類は、COX−1およびCOX−2の双方を阻害する(表1−3参照)。
【0005】
表1: モルモットのマクロファージモデルにおける種々のNSAID類のIC50値およびCOX−2/COX−1比(Engelhartら、J. Inflammatory Res., 44, p422−43, 1995年からのIC50値)
【0006】
【表1】

表2: COX−1(ウシ内皮細胞)およびCOX−2(刺激されたマクロファージ)に関して無傷細胞におけるNSAID類のIC50値(Taketo, J. National Cancer Institute, 90, p1529−1536, 1998年 からのIC50値)
【0008】
【表2】
表3: 組換えヒトPGH合成の阻害(COX−1およびCOX−2)(Laneuvillら、J. Pharm. Exp. Ther., 271, p927−34,1994年からのIC50値)
【0010】
【表3】
この10年間に、いくつかの新しい選択的なCOX−2阻害剤、いわゆる「選択的」COX−2阻害剤が確認された。これらのCOX−2阻害剤のいくつかは開発され、そのうちのいくつかは最近、市販にこぎつけた。このような新しいCOX−2阻害剤のあるものは、臨床的な用量でCOX−1の阻害を示さない。これらのCOX−2阻害剤の利用に関する広範な臨床的研究および臨床での実施により、この新しいCOX−2阻害剤は、非選択的なNSAID類と比較して、安全性の点できわめて有利であることが示されている。COX−2阻害剤の概説については、たとえば、Goldenら,1999年, Osteoarthritis. 25, p359−379、 Mitchelら,1999年, Brit. J. Pharmacol., 128, p1121−1132、 Lipsky, 1999年, Am. J. Med., 106 (5B), p515−575、 Taketo, 1998年, J. National Cancer Inst., 90, p1529−1537、 Griswoldら 1996年, Med. Res. Rev., 16, p181−206 および Reitzら,1995年, Ann. Rep. Med. Chem., 30, p179−188を参照されたい。
【0012】
さらに、様々なCOX−2阻害剤に関する興味深い刊行物としては、たとえば、Lane, 1997年, J. Rheumatol., 24 (suppl. 49), p20−24、 Mehlishら,1998年, Clin. Pharmacol. Ther., 63, pl−8、 Zhaoら,1997年, Arthritis Rheum., 40 (suppl.), S88、 Ehrichら,1997年, Arthritis Rheum., 40 (suppl.), S93、 Maziaszら,1997年, Arthritis Rheum., 40 (suppl.), S195、 Mengle− Gaw ら,1997年, Arthritis Rheum., 40 (suppl.), S93、 Morrison, 1999年, Clin. Ther., 21, p943−953、Chanら,1995年, J. Pharmacol. Exp. Ther., 274, p1531−37、 Riendeauら,1997年, Br. J. Pharmacol., 121, p105−117、 Blackら,1999年, J. Med. Chem., 42, p1274−81、 Cuoら,1996年, J. Biol. Chem., 271, p19134−39、 Geiss, 1999年, Scand. J. Rheumatol., 109 (suppl.), p31−37、 Warnerら,1999年, PNAS USA, 96, p7563−68、 Bjarnsonら,1997年, Scand. J. Gastroenterol., 32, p126−130、 Danneberg ら,1999年, Semin. Oncol., 26, p499−504、 Mitchellら,1993年, PNAS USA, 90, p11693−97、 Futakiら,1994年、 Prostaglandins, 47, p55−9、 Futakiら,1993年, J. Pharm. Pharmacol., 45, p753−5、 Masferrerら,1994年, PNAS USA, 91, p3228−32、 Kleinら,1994年, Biochem. Pharmacol., 48, p1605−10、 Reitzら,1994年, J. Med. Chem., 37, p3878−81、 Seibertら,1994年, PNAS USA, 91, p12013−17、 Kleinら,1996年, Biochem. Pharmacol., 51, p285−90、 Nantalら,1998年,9th Intern. Conference Inflamm. Res. Assoc., Nov 1−5、 Pennig ら,1997年, J. Med. Chem., 40, p1347−65 および Puigら,2000年, J. Med. Chem., 43, p214−223が挙げられる。
【0013】
COX−2阻害剤は、化学構造の観点から見て比較的多様な化合物の集団である。選択的にCOX−2を阻害する化合物は、この10年間の多くの特許文献に記載されている。いくつかを挙げると、WO 94/26781, WO 94/20480, WO 94/13635, WO 95/00501, WO 94/27980, WO 94/15932, WO 95/21817, WO 95/15316, WO 96/06840, WO 96/03388, WO 96/03387, WO 96/03392, WO 96/25405, WO 96/24584, WO 96/03385, WO 96/16934, WO 98/41516, WO 98/43966, WO 99/12930, EPO 673 366, WO 98/41511, WO 98/47871, WO 99/20110, WO 99/23087, WO 99/14194, WO 99/14195, WO 99/15513 および WO 99/15503 および米国特許明細書5,380,738、5,344,991、5,393,790、5,434,178、5,474,995、5,475,018 および 5,510,368が挙げられる。
【0014】
2つの化合物、ロフェコキシブ、すなわち(4−(4−メチルスルホニル)フェニル)−3−フェニル−2(5H)−フラノン)(I)が「Vioxx(R)」として、ならびにセレコキシブ、すなわち(4−(5−(4−メチルフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル)−ベンゼンスルホンアミド)(II)が「Celebra(R)」として、現在市販されている。
【0015】
【化3】
ロフェコキシブ(rofecoxib)は、Merck Frosst Canada のWO 93/0500501、さらにMorrison, 1999年, Clin. Ther., 21, p943−953、Chanら,1995年, J. Pharmacol. Exp. Ther., 274, p1531−37 および Nantelら,1998年, 上掲に記載されている。
セレコキシブ(celecoxib)は、Geiss, 1999年, Scand. J. Rheumatol., 109 (suppl.), p31−37およびPenningら,1997年, J. Med. Chem., 40, p1347−65に記載されている。セレコキシブは、COX−1と比較して、COX−2に対して375倍選択的であると述べられている。
【0017】
他のいくつかのCOX−2阻害剤は、生物学的なシステムにおいて評価されており、それらのあるものとは、BF 389 (III)、 CGP 28232 (IV)、 DFP、 DFU (V)、 DuP 697 (VI)、 エトドラク(VII)、 FK 3311 (VIII)、 フロスリド(flosulide)(IX)、 L− 745,337 (X)、メロキシカム(「Mobic(R)」,US 4233299、4−ヒドロキシ−2−メチル−N−(5−メチル−2−チアゾリル)−1,1−ジオキシド−2H−1,2−ベンゾチアジン−3−カルボキサアミド)(XI)、 MF トリサイクリック (XII)、ニメスリド(XIII)、 NS−398 (XIV) および SC−58125 (XV)である。
【0018】
【化4】
【化5】
【化6】
さらに、COX−2阻害剤として記載された化合物として、たとえば、S−2474 (Shionogi, EP 595546、5(E)−(3,5−ジ−tert−ブチル−4−ヒドロキシ)ベンジリデン−2−エチル−1,2−イソチアゾリジン−1,1−ジオキシド)(XVI)、 JTE−522 または RWK−57504 (4−(4−シクロヘキシル−2−メチル−5−オキサゾリル)−2−フルオロベンゼンスルホンアミド)(XVII)、 メシル酸ダブフェロン(Darbufelone mesylate)(Pfizer, WO 94/03448、2−アミノ−5−((3,5−ビス(1,1−ジメチルエチル)−4−ヒドロキシフェニル)メチレン−4(5H)−チアゾロン)のモノメタンスルホン酸塩 )(XVIII)、 6089 ( Kotobuki Pharmaceuticalより)(XIX)、ヴァルデコキシブ(Valdecoxib)(Pharmacia、4−(5−メチル−3−フェニル−4−イソキサゾリル)−ベンゼンスルホンアミド)(XX)、パラコキシブナトリウム(Paracoxib sodium)(Pharmacia,N−((4−(5−メチル−3−フェニル−4−イソザゾリル)−フェニル)スルホニル−プロパンアミドのナトリウム塩 )(XXI)、4−(2−オキソ−3−フェニル−2,3−ジヒドロオキサゾル−4−イル)−ベンゼンスルホンアミド(Almirall− Prodespharma)(XXII)および エトリコキシブ(Etoricoxib)(MK−633, Merck and Co.)が挙げられる。
【0022】
【化7】
【化8】
上述の化合物は、後述する方法に用いるのに好ましいCOX−2阻害剤を形成する。
COX−2阻害剤の適応症は、古典的なNSAID類、たとえば、インドメタシン、ジクロフェナクおよびナプロキセンで治療されてきた、関節炎、筋骨格の疼痛状態、および一般的な疼痛である。最近、COX−2阻害剤をガン治療、さらにはおそらくガン予防にも使用することが提案されている。さらに、COX−2阻害剤を、アルツハイマー病や他の痴呆に関連する脳活動に関して使用できる可能性がある。
【0025】
COX−2阻害剤の臨床的有用性の可能性は、たとえば、Nature,367, p215−216 (1994年)、 Drug News and Perspectives, 7, P501−512 (1994年)、 Annual Reports in Medicinal Chem., 30, p179−188 (1995年) および Oncogene, 18, p7908−7916 (1999年)に述べられている。
抗ウィルス治療におけるCOX−2阻害剤の使用についての具体的な提案はなく、あるいはさらに具体的なHIV/AIDS治療についての提案はなく、COX−2阻害剤の抗HIV作用についても試験されていない。さらに、ウィルスおよび非ウィルス起源の免疫不全症の治療において、免疫賦活性剤としてCOX−2阻害剤(または非選択的なCOX阻害剤)を使用することについての提案はない。
【0026】
HIV感染およびAIDSは、世界中で3300万人を超える人々がウィルスに感染している主要な健康問題である。感染者の多くは、アフリカ(サハラ以南(sub−Sahara))およびアジアの一部に住んでいる。今日、臨床用途のために日常的に用いられる抗AIDS化合物には2つの種類がある。HIV逆転写酵素阻害剤およびHIVプロテアーゼ阻害剤である。HIV逆転写酵素阻害剤は、非ヌクレオシド系の逆転写酵素阻害剤(NNRTI)およびヌクレオシド系逆転写酵素阻害剤(NRTI)に分けられる。
【0027】
最も頻繁に使用されるNNRTIは、ネビラピン、デラビルジン、エファビレンツ、エミビリンおよびT180である。最も頻繁に使用されるNRTIとしては、たとえば、ジドブジン、ジダノシン、スタブジンおよびザルシタビンが挙げられる。臨床的に有用なHIVプロテアーゼ阻害剤として、インクリナビル(inclinavir)、パリナビル(palinavir)およびサクイラビル(saquiravir)が挙げられる。
【0028】
現代のHIV感染およびAIDSの治療法は、いくつかの薬剤の組合せ、いわゆる逆転写酵素阻害剤とプロテアーゼ阻害剤との混合剤に基づいている。これらの組合せは、HAART(高度に活性な抗レトロウィルス療法)と呼ばれ、極めて効果的であり、患者の血液中のウィルスを検出不能のレベルにまで減少させることができる。しかしながら、HAARTは患者を治癒しない。ウィルスは依然として免疫細胞内に存在しており、その疾患がいつでも再発可能だからである。治療を中断することにより、ウィルス血症が頂点に達し、またAIDSへの急速な進行が度々観察される。さらに、免疫不全およびHIVに特異的なT細胞機能不全が、HAARTの間、持続する。この治療法は、生涯にわたる治療を必要とし、しかも極めて高価である。薬剤の費用だけでも、しばしば15,000 USドルを超える。加えて、この治療法に関係する他の問題がいくつか存在する。すなわち患者の服薬遵守の難しさ(複雑な薬剤投与計画)、耐性ウィルスの発生、理想的ではない薬物動態ならびに副作用、たとえば、骨髄の抑制および長期代謝効果の抑制などが挙げられる。
【0029】
近年発行された、抗HIV治療の総説については、下記を参照されたい:たとえば、Hilgegroth, 1998年, Pharm. uns. Zeit., 1998年,27, p22−25、 Hilgegroth, 1998年, Pharm. uns Zeit., 7, p111−116、 Stellbrink, 1997年, Dk Arztebl., 94, p2497−2503、 Rettle ら,1998年, Int. J. STD AIDS, 9, p80−87、 De− Clercq, 1998年, Antiviral Res., 38, p153−179、 Gaitら,1995年, TIBTECH, 13, p430−438 および Redshawら,”Emerging Drugs: The Prospects of Improved Medicines“, Chapter 6, p127−154, 1997年が挙げられる。
【0030】
要するに、HAARTのような複数薬剤の組合せ併用は、HIV感染に苦しむ患者の予後を著しく改善したが、HIVの抗ウィルス治療おける新規化合物、とりわけ免疫系を刺激する薬剤に対する医療上の必要性が存在している。本発明は、この必要性に焦点をあてるものである。
COX−2の発現は、通常は脳/脳活動、関節炎の滑膜、および損傷組織部位に限定されている。COX−2は、通常のリンパ節またはリンパ球内には見出されない。しかしながら今や驚くべきことに、免疫不全病MAIDに感染したマウスのリンパ節細胞が、高レベルでCOX−2を発現していることが見出されている。さらに、MAIDSリンパ節からのB細胞ばかりでなく、正の選択をされたCD4+およびCD8+T細胞もまた、高レベルのCOX−2を含んでいた(実施例2を参照のこと)。このCOX−2が、免疫不全疾患の徴候を緩和すべく、標的にされているだろうということが見出された。具体的には、免疫刺激を作用させること、たとえば、抗原特異的な免疫応答を生起させることによって、T細胞機能不全を緩和するためである。
【0031】
一方、理論により拘束されることを望まないが、COX−2活性は、PGE2産生を増加させると信じられている。このことにより、cAMPのレベルを増加させ、次に、cAMP はPKAシグナル経路を活性化し、結果的にリンパ球機能を減退させる。MAIDSのマウスにインビボで実施した研究は、COX−2阻害剤がT細胞の免疫機能を改善することを説明している(実施例6参照)。
【0032】
本発明は、免疫不全の治療または予防、特にHIVおよびAIDSの治療または予防のための新規な方法を提供するものである。その方法は治療上有効な量のCOX−2阻害剤またはその誘導体あるいは製薬的に許容されるその塩により、患者を治療することを含むものである。
したがって、本発明の最初の面は、ヒトまたはヒト以外の動物において上昇したCOX−2活性によって特徴づけられる疾患、具体的には、(たとえば、COX−2発現の増加を通じて)免疫機能の減退により特徴づけられる疾患の治療方法または予防方法を提供することである。その方法は、前記動物に治療上有効な量のCOX−2阻害剤またはその誘導体またはその製薬的に許容される塩を投与することを特徴としている。
【0033】
本明細書において、増加したCOX−2活性とは、より多くのCOX−2分子の産生(たとえば、増大した発現)および/または、より活性な分子(たとえば、不活性型から活性型への転換あるいは活性型に対する阻害の除去)のいずれかを通して、上昇した活性のレベルを意味する。なるべくなら、前記疾患は、免疫機能の減退によって特徴づけられる。すなわち、免疫不全の状態、たとえば、リンパ球の機能不全を示す。なお、本明細書において、「免疫不全」とは、通常の免疫応答に関わる細胞、とりわけB細胞およびT細胞の機能障害を意味する。したがって、本明細書に記載された化合物は、免疫刺激効果を達成して免疫応答を増強するように使用してもよい。このように、COX−2阻害剤は、免疫調節作用を有すると考えられる。好ましくは治療の対象となる状態として、ウィルスによって引き起こされる免疫不全の疾患が挙げられる。
【0034】
かくして上記の方法は、これに限定されるものではないが、患者のHIVまたはAIDS関連疾患の治療に有用である。たとえば、一般の多様な免疫不全を有する患者の約50%は、HIV感染の場合と同様のT細胞機能障害を有しており、免疫刺激療法の恩恵を受け得るかもしれない。本発明によれば、いずれかのCOX−2阻害剤をHIV/AIDS治療を必要とする患者に投与することができる。したがって、本発明による治療に好適な状態としては、たとえば、レトロウィルスによる感染、具体的にはHIVによる感染(および他の動物における関連ウィルス、たとえばSIV、FIV、MAIDSの感染)ならびにその結果として生じるAIDS、さらには一般の様々な免疫不全および前記状態に関連する状態の治療が挙げられる。
【0035】
治療できる患者としては、好ましくは哺乳類、より好ましくはヒトおよびヒトに添う動物または農業用動物、たとえば、イヌ、ネコ、サル、ウマ、ヒツジ、ヤギ、ウシ、ウサギ、ラットおよびマウスが挙げられる。
換言的に述べるならば、本発明は、上述したように増加したCOX−2活性によって特徴づけられる疾患を治療または予防するためのCOX−2阻害剤またはその誘導体またはその製薬的に許容しうる塩を提供するか、あるいは上述したように増加したCOX−2活性によって特徴づけられる疾患を治療または予防するための医薬を調製する際において、COX−2阻害剤またはその誘導体またはその製薬的に許容しうる塩の使用の方法を与える。なお、本明細書において、「治療」とは、前記疾患、たとえば感染の1つ以上の徴候を、好ましくは通常のレベルまで、軽減または緩和、あるいは免疫機能障害の減少または緩和を意味する。「予防」とは、絶対的な予防、すなわち、検出可能な感染性因子(たとえば、ウィルス)が存在しないこと、および/または特定の徴候(たとえば、COX−2活性)について正常ベルの保持、またはそうした徴候発症の程度の減少または軽減もしくは時期調整(たとえば、遅延)を意味する。
【0036】
シクロオキシゲナーゼ2酵素は、HIV/AIDS治療の新しい標的である。「COX−2阻害剤」という用語は、ある特定の濃度で投与した場合に、シクロオキシゲナーゼ1を有意に阻害することなく、シクロオキシゲナーゼ2酵素を阻害することができる化合物を意味する。その化合物は、好ましくはシクロオキシゲナーゼ2の阻害について、シクロオキシゲナーゼ1阻害と比較した場合、少なくとも10、より好ましくは少なくとも50、さらに好ましくは少なくとも100の選択性(たとえば、WHAM試験によって、COX−1:COX−2 IC80比として測定される、下記参照)を有する化合物が挙げられる(一つの特定化合物に対する選択性の比率は、生物学的なアッセイならびにそれが表現される方式によって変化する(好ましくは、COX−1:COX−2 IC50またはIC80の比として表現される)、表1〜4を参照)。本明細書で述べる比は、1以上の適切な、公知のCOXアッセイ、好ましくは精製したヒト由来の酵素を使用して得られたデータを意味しており、たとえば、Engelhartら(1995年、上掲)によって測定されたIC50値の比を意味する。しかしながら、前記試験は、後述するようにWHAM試験が好ましい。
【0037】
COX−1およびCOX−2の力価比に関する多数の分析が、単離精製された酵素から様々な種からの無傷の(intact)細胞および細胞モデルまでわたる広範囲のアッセイシステムを用いて行われてきた。しかしながら、現在では、最も広く受け容れられているモデルは、ヒト全血アッセイ(WBA)ならびにWilliam Harvey変更ヒト全血アッセイ(WHMA)修正版であり、好ましいアッセイである。これらのアッセイは、ヒトに使用する前記化合物に対し好適である試験のために、容易に入手できるヒト細胞を利用している。また、NSAID類の血漿タンパク質への結合をも考慮に入れている。さらに、選択性の評価は、IC50よりもむしろIC80で行うことが好ましい。COX−2およびCOX−1の阻害に関する両濃度曲線が平行ではなく、また多くの化合物は、80%阻害に近い阻害となる定常状態血漿濃度を与える用量で使用されるからである(Warnerら, 1999年, PNAS USA, 96, p7563−7568)。
【0038】
WBAアッセイでは、COX−1分析のために、血液を試験試薬で処理し、60分後にカルシウムイオノフォアで処置し、30分間インキュベートした後、血漿を回収する。COX−2分析については、血液をアスピリンで処理してCOX−1を阻害し、6時間後にリポ多糖と試験試薬で処理し、18時間インキュベートした後、血漿を回収する。引き続き、血漿中のトロンボキサンB2含量を、COX活性の尺度として、ラジオイムノアッセイで評価する。
【0039】
WHMAアッセイでは、COX−1分析を上記のようにして実施する。COX−2分析については、インターロイキン1βに24時間曝したヒト気道上皮細胞(A549)の培養物から調節した培地で、血液を処理し、この培地と試験試薬で60分間インキュベートした後に、カルシウムイオノフォアを加えて、30分後にジクロフェナクを添加して、プロサノイド(prosanoid)産生を停止させる。その後、血漿を回収し、COX−2活性の尺度として、血漿中のプロスタグランジンE2含量をラジオイムノアッセイで分析する。COX−1およびCOX−2活性の評価に関して、インキュベートの回数は、この最後のアッセイと同様であるが、これにより活性をより比較できるものとし、またWHMAを好ましいアッセイとする。
【0040】
このアッセイを使用して、IC80でのCOX−2/WHMA−COX−1に基づく選択性を表4に示す。ここで、0.2および0.02は、それぞれCOX−2についての5倍および50倍の選択性を表している。
表4:(Warnerら(上掲)からのWHMA試験によるIC80でのCOX−2/COX−1比)
【0041】
【表4】
このため好ましい態様では、選択比(selectivity ratio)はWHMAアッセイによりIC80で決定され、COX−2:COX−1選択比が0.2未満、好ましくは0.05未満、たとえば0.02未満、好ましくは0.01未満、具体的には <0.005の選択比を有する化合物が本発明の方法での使用にとりわけ好ましい。別の表現をすれば、上述したように、好ましい化合物は、2より大、好ましくは5より大、特に好ましくは50もしくは100より大であるCOX−1:COX−2選択比(IC80でのWHMAアッセイによる)を有する。
【0043】
ここでいう「阻害」とは、測定可能なシクロオキシゲナーゼ−2活性の減少を意味する。これは、転写、翻訳、翻訳後の修飾またはCOX−2の活性に影響することにより達成されることができる。しかしながら好ましくは、阻害が酵素活性を阻害することにより達成され、すなわち既に存在する活性COX−2分子の活性部位への干渉である。
【0044】
望ましくは、免疫不全またはウィルス感染、とりわけHIV感染およびAIDSの治療のためのCOX−2阻害剤が、約0.5μmol/リットル未満、より好ましくは約0.2μmol/リットル未満のCOX−2 IC50を有する。
本発明で提供される方法は、免疫不全およびウィルス感染、とりわけHIVおよびAIDSを含むさまざまな症状の予防および治療における、COX−2阻害剤またはその誘導体の使用方法に関する。
【0045】
本発明のひとつの好ましい態様では、本発明に基づく治療のためのCOX−2阻害剤は、酸性スルホンアミド類(acidic sulfonamides)から選択される。
本発明のひとつの好ましい態様において、本発明で用いるCOX−2阻害剤は、メタンスルホンアミドエーテル類およびメタンスルホンアミドチオエーテル類を含む下記一般式Aで表される化合物、またはこれらの誘導体あるいはこれらの製薬的に許容しうる塩から選択される:
【0046】
【化9】
ここで、
Xは、酸素原子または硫黄原子またはアルキル基、好ましくは−CH2−基を表し、
R1は、必要に応じて1以上の基または原子、好ましくは1以上のフッ素などのハロゲン原子で置換されていてもよいシクロアルキル基あるいはアリール基を表し、
R2、R3、R4およびR5は、それぞれ独立に、水素原子、ニトロ基、アシル基、または必要に応じて1以上の基(たとえばアシル基)または原子で置換されていてもよいアルキル基、あるいはR2とR3、R3とR4、または、R4とR5とが互いに炭素原子を介在させてシクロペンタノン基を形成してもよい。
【0048】
好ましくは該化合物中において、Xは酸素原子である。またさらに好ましい化合物では、R1は、アリール基、または1以上のフッ素原子で置換されたアリール基、またはシクロアルキル基である。
より好ましい化合物中においては、R2およびR3が水素原子であり、R4が−NO2基または−COCH3基である。代わりの好ましい化合物は、そのR2が水素原子であり、R3とR4が一緒にシクロペンタノン基を形成するものを含む。
【0049】
特に好ましくは、本発明で用いる式Aの化合物は、本明細書に記載の名称の化合物であるフロスリド(flosulide)、NS−398 、ニメスリド、FK’ 3311およびL−745 337である。
本発明の別の好ましい態様では、本発明に用いるCOX−2阻害剤は、ジアリール複素環から選択される。
【0050】
本発明でCOX−2阻害剤として用いることのできるジアリール複素環の仲間の一例には、下記一般式Bで示される化合物、またはそれらの誘導体もしくはそれらの製薬的に許容しうる塩が含まれる。
【0051】
【化10】
ここで、
Yは、環状基、好ましくはオキサゾリル、イソキサゾリル、チエニル、ジヒドロフリル、フリル、ピロリル、ピラゾリル、チアゾリル、イミダゾリル、イソチアゾリル、シクロペンテニル、フェニルまたはピリジル基を表し、
nは、0〜3の整数であり、
mは、0〜4の整数であり、
R6は、環状ケト基(ketocyclyl)、シクロアルキル基またはアリール基を表し、これらの基は、必要に応じて1以上の基または原子、好ましくはフッ素といった1以上のハロゲン原子で置換されていてもよく、
R7は、それぞれ独立に任意の官能基であり得る置換基を表し、好ましくは、水素原子またはハロゲン原子、より好ましくはフッ素または臭素であり、あるいはアルキル基(好ましくは −CH3)であり、そのアルキル基は1以上の基または原子、好ましくは、たとえば −CF3などのように1以上のフッ素原子で置換されていてもよく
R8は、アルキル基、好ましくは −CH3またはNHR10、好ましくは −NH2を表し、
R9は、ハロゲン原子、好ましくはフッ素を表し、
R10は、水素原子、または必要に応じて1以上の基または原子で置換されたアルキル基、好ましくはアシル基で置換されたアルキル基を表す。
【0053】
この部類の化合物は、米国特許第6,025,353号における抗脈管形成剤として、請求されており、さらに、本発明による好ましい置換基および化合物の記載は、米国特許第6,025,353号明細書に記載のものと同じである。
このような化合物において、R8は −NH2または −CH3が望ましい。より好ましい化合物において、Yはピラゾリル基、フリル基またはチエニル基である。好ましくは、R6は必要に応じて1以上のフッ素原子で置換されたアリール基である。好ましくは、nは1または2である。R7は、臭素原子、アシル基または −CF3などの置換されたアルキル基であることが好ましくい。
【0054】
本発明で用いる、式Bの特に好ましい化合物は、本明細書に記載された、セレコキシブ、ロフェコキシブ、 DuP−697、 SC−58125、 DFP、 DFU、 CGP 28232 および三環MF(MF tricyclic)という化合物である。
本明細書で使用される用語「アルキル」は、特に他に言及のない限り、長鎖または短鎖、直鎖、分枝または環状の、必要に応じて水酸基、アルコキシ基、アシルオキシ基、ニトロ基、アルコキシカルボニルオキシ基、アミノ基、アリール基、オキソ基またはハロ基により、一置換または多置換されていてもよい脂肪族の飽和もしくは不飽和炭化水素基を含む。不飽和アルキル基は、一不飽和または多不飽和でもあってよく、アルケニル基およびアルキニル基の両方を含む。これらの基は40個まで、好ましくは1〜10個の炭素原子を含むことができる。
【0055】
本明細書で用いられる環式の環は、好ましくはC5−7であって、必要に応じて酸素、窒素および硫黄から選ばれる1以上のヘテロ原子を含有していてもよい。
本明細書で用いられる用語「アシル」は、カルボン酸基および炭酸基の両方を含み、よって、たとえば、アルキルカルボニルオキシアルキルなどのアシルオキシ置換アルキル基が挙げられる。そうした基において、いずれのアルキレン部分も、好ましくは上記でアルキル基に対し定義した炭素原子数を含む。好ましいアリール基として、フェニル基および単環の5〜7員複素環式芳香族、とりわけ必要に応じ置換されていてもよいフェニル基および単環5〜7員複素環式芳香族基が挙げられる。
【0056】
代表的な置換アルキル基R1として、アルコキシアルキル、ヒドロキシアルコキシアルキル、ポリヒドロキシアルキル、ヒドロキシポリアルキレンオキシアルキルなどの基、具体的には、アルコキシメチル、アルコキシエチルおよびアルコキシプロピル基、または、アシルオキシメチル、アシルオキシエチルおよびアシルオキシプロピル基、たとえばピバロイルオキシメチル(pivaloyloxyethyl)が挙げられる。
【0057】
本明細書で用いられる、「置換された基」としては、特に他に言及のない限り、水酸基、アルコキシ基、アシルオキシ基、ニトロ基、アルコキシカルボニルオキシ基、アミノ基、アリール基、オキソ基またはハロ基によって、一置換または多置換されたものであってもよい。
本発明の他の好ましい態様において、COX−2阻害剤は、古典的なNSAID類の修飾物、たとえばそれらのプロドラッグ類、エステル類または塩類から選択される。
【0058】
古典的なNSAID類の化学構造に基づいて、さらに新規な選択的COX−2阻害剤が調製されてきた。このような化合物は、オキセカム(よく知られたピロキシカムのCOX−2特異的アナログ)のひとつであるメロキシカム(meloxicam)、または、エトドラック(ジクロフェナックのCOX−2特異的アナログ)などの酢酸誘導体であってもよい。この部類におけるいくつかの最も好ましい化合物のあるものの例は、COX−2活性インドメタシン誘導体およびゾメピラックである。
【0059】
本発明に係る化合物のファミリーまたはサブファミリーに関するさらなる列挙は、COX−2阻害剤に関する特許および特許出願に見受けられ、たとえば、本明細書で先に掲げた特許文献中にある。これらの特許文献はまた、特定の化合物を例示および列挙しており、それらはまた本発明による最も好ましいCOX−2阻害剤でもある。
【0060】
特に好ましい化合物としては:
ジイソプロピルフルオロホスフェート、L−745337、ロフェコキシブ、NS 398、SC 58125、 エトドラク、メロキシカム、セレコキシブまたはニメスリドである。
本発明に基づいて使用するCOX−2阻害剤の製造方法は、当該技術分野でよく知られた方法であり、上記の文献中に具体的に記載されている。
【0061】
本明細書で記載する疾患、たとえば、免疫不全およびウィルス感染、特にHIV / AIDSの治療および予防に用いる、本発明によるCOX−2阻害剤は、1個以上の不斉中心および/または1個以上の二重結合を有していてもよい。すなわち、本発明は、本明細書が開示する化合物の異性体およびラセミ体の使用にも及ぶ。これらすべての可能な異性体は、本発明の範囲に含まれる。COX−2阻害剤は、化合物の異性体混合物の形態、より好ましくは精製した異性体またはその製薬的に許容される塩の形態であってもよい。
【0062】
本発明による症状の治療、たとえば免疫不全およびウィルス感染の治療のためのCOX−2阻害剤の医薬組成物は、製薬的に許容される塩として製剤されてもよく、さらに当該技術分野でよく知られ、製薬上許容される担体もまた含んでもよい。
このように、本発明は、COX−2阻害剤またはその誘導体あるいはその製薬的に許容される塩ならびに製薬的に許容される希釈剤、担体または賦型剤を含有する医薬組成物にも及ぶ。「製薬的に許容される」とは、成分が組成物中で他の原料成分と適合し、かつ、投与される者にとり生理的に許容しうるものであることを意味する。
【0063】
さらなる態様において本発明は、後述するように、このような組成物の使用、ならびにこのような組成物を用いた予防/治療の方法にもまた及ぶ。
COX−2阻害剤が塩基性である場合には、塩は、製薬的に許容される非毒性の無機酸および有機酸を含む酸とから調製することができる。特に好ましい塩は、塩酸塩、臭化水素酸塩、リン酸塩、硫酸塩、クエン酸塩、マレイン酸塩、クエン酸塩および酒石酸塩である。
【0064】
COX−2阻害剤が酸性である場合には、塩は、製薬的に許容される非毒性の無機塩基および有機塩基を含む塩基とから調製することができる。特に好ましい塩は、ナトリウム塩、カリウム塩およびマグネシウム塩である。
本明細書に記載の疾患、たとえばHIV/AIDSを含む免疫不全またはウィルス疾患などの、治療および予防のためには、COX−2阻害剤は、経口的に、経直腸的(坐薬)に、局所的に、舌下剤として、吸入、あるいは注射または注入の形態で非経口的(たとえば、筋肉内、皮下、腹腔内または静脈内)に投与することができる。好ましい投与形態は、経口、経直腸および注射または注入による投与であろう。最も好ましい投与形態は、経口投与に適したものである。
【0065】
すべての投与形態において、COX−2阻害剤は、通常、製薬的に許容される公知のキャリヤー、助剤および担体を含有する投薬単位の剤形で投与される。このようにして、活性成分は、錠剤、丸薬、散剤、トローチ剤、小袋剤、カシェ剤、エリキシル剤、懸濁剤、乳化剤、溶液剤、シロップ、煙霧剤(固体としてまたは液体媒体中で)、軟膏、軟質および硬質のゼラチンカプセル、坐剤、無菌注射剤、無菌包装粉剤およびその類似物を製造するために、必要に応じて配合調剤としての他の活性物質、1以上の通常の担体、希釈剤および/または賦形剤とともに一体化してもよい。生分解性ポリマー(ポリエステル、ポリ酸無水物(polyanhydrides)、ポリ乳酸またはポリグリコール酸など)もまた、固体インプラントに用いることができる。この組成物は、凍結乾燥、過冷法あるいはパーマザイム(Permazyme)を用いることにより安定化させることができる。
【0066】
適切な賦形剤、担体または希釈剤は、ラクトース、D型グルコース、ショ糖、ソルビトール、マンニトール、デンプン類、アラビアゴム、リン酸カルシウム、アグリネート、トラガカント、ゼラチン、ケイ酸カルシウム、微結晶セルロース、ポリビニルピロリドン、セルロース、水シロップ、水、水/エタノール、水/グリコール、水/ポリエチレン、グリコール、プロピレングリコール、メチルセルロース、メチルヒドロキシベンゾエート、プロピルヒドロキシベンゾエート、タルク、ステアリン酸マグネシウム、鉱油、または固い脂肪などの脂肪類、これらの適切な混合物である。この組成物は、必要に応じて、潤滑剤、湿潤剤、乳化剤、懸濁剤、保存剤、甘味剤、香料、たとえば点鼻用(胆汁酸塩、レシチン、界面活性剤、脂肪酸、キレート化剤)などの吸着促進剤などを含んでもよい。本発明の組成物は、当業界で周知の方法を用いることにより、患者への投与後、活性成分を即時の放出、持続的または遅延的な放出するように製剤することができる。
【0067】
投与する活性成分は、医薬組成物での利用のために、適切に修飾されてもよい。たとえば、活性成分は、塩類または非電解質、酢酸塩、SDS、EDTA、クエン酸塩または酢酸緩衝液、マンニトール、グリシン、HASまたはポリソルビン酸塩などの適切な添加剤を用いることなどにより安定化されてもよい。
結合物は、改善した親油性を与え、細胞輸送を増加させ、溶解性の向上またはターゲティングを可能とするように形成されてもよい。これらの結合は、切断可能であってもよく、このため該結合物が薬剤前駆体(プロドラッグ)のように作用する。安定性はまた、たとえばZn、CaまたはFeとの適切な金属錯体を用いることにより与えることができる。
【0068】
この活性成分は、送達のため、または特定の細胞、器官または組織をターゲティングするために、適切な媒体中で処方してもよい。このため、該医薬組成物では、活性物質が吸収、吸着、取り込みまたは結合されてもよい、ミクロエマルジョン、リポソーム、ニオソームまたはナノ粒子の形態をとることができ、これらに生成物を効果的に不溶性の形態に変換することができる。
【0069】
これらの粒子は、循環時間を向上するため適切な表面分子(たとえば、血清成分、界面活性剤、ポリオキサミン908、PEGなど)、または、たとえば特定細胞に担われている選択的レセプターに対するリガンドなど、部位特異的ターゲティングのための部分を持つことができる。ドラッグデリバリーのための、またターゲティングのための適切な技術は、当該技術分野で知られているが、たとえば、ドラッグターゲティングに関する、Kreuter, 1994年, Eur. J. Drug Metab. Pharmacokinet., 3,p253−256、Shen, 1997年, J. Drug Targeting, 5(1), p11−13、Mrsny, 1997年, J. Drug Targeting, 5(1), p5−9、Petit & Gomboz, 1998年, TIBTECH, 16, p343−349、およびDuncan, 1997年, J. Drug Targeting, 5(1), p1−4、および、ペプチドおよび核酸分子デリバリーに関する、Simari & Nabel, 1996年, Semin. Intervent. Cardiol., 1, p77−83、Torchilin, 1998年, J. Microencapsulation, 15(1), p1−19、Klyashchitsky & Owen, 1998年, J. Drug Targeting, 5(6), p443−458、Kreuter, 1996年, J. Anat., 189, p503−505、Fasano, 1998, TIBTECH, 16, p152−157、Kataokaら, 1993, 24, p119−132、Anderson, 1998年, Nature, 392(suppl), p25−30、Langer, 1998年, Nature, 392 (suppl), p5−10、Gregoriadis, 1995年, TIBTECH, 13, p527−536、Gregoriadis ら, 1997年, FEBS Lett., 402, p107−110、Rolland, 1998年, Critical Reviews in Therapeutic Drug Carrier Systems, 15 (2), pl43−198、Hope ら, 1998年, Molec. Memb. Biol., 15, p1−14、および Scherman ら, 1998年, Curr. Opinion Biotech., 9 (5), p480−485 が挙げられる。特定部位指向ターゲティングの例としては、たとえば、ナノ粒子がHIV感染マクロファージ内に蓄積される、Schaefer ら,1992年, Pharm. Res., 9, p541−546を参照。明らかにこれらの方法は、本明細書に記載の本発明の方法における具体的な適用例を明確に有する。
【0070】
これらの誘導体化または結合された活性成分は、本発明により用いられる阻害分子の定義の範囲に収まることを意味する。
このため経口的に使用される該医薬組成物は、たとえば活性成分の他に錠剤、カプセル剤、溶液剤、懸濁剤またはその他経口投与用として周知の剤形を形成するための生理学的に許容しうる適切な剤を含む。このような組成物は、経口医薬組成物の製造方法として知られたいずれの方法によっても調製することができる。このような組成物は、1以上の生物活性物質および保存剤、不活性希釈剤、増粘剤、着色剤、甘味剤、造粒剤、崩壊剤、結合剤、浸透性活性剤、湿潤剤、懸濁剤、遅延剤形の調製用物質、油類および水よりなる群から選ばれる1以上の剤を含むことができる。
【0071】
たとえば、直腸投与用の坐剤または注射もしくは注入用の溶液剤といった、経口使用以外の医薬組成物は、よく知られている方法およびそうした剤形のための添加剤を用いて調製できる。注射および注入用のすべての製剤は、無菌製剤であるべきである。
このような組成物中の活性成分は、製剤重量の約0.01%から約99%、好ましくは約0.1から約50%、たとえば10%含まれる。
【0072】
本発明によれば、たとえば免疫不全およびウィルス感染などの疾患の治療のために、COX−2阻害剤の一日あたりの用量水準は、0.005mgから約150mg/kg体重の範囲である。その投与量は、COX−2阻害剤化合物の選択、臨床的状態(ウィルスの種類、感染の進行状況および患者の状態)、患者の年齢および体重、投与の経路、および、患者の治療過程の長さを含め、薬剤の総使用量に大きく依存する。より好ましい投与量は、通常、一日あたり0.01mg〜50mg/kg体重の間、より好ましくは、一日あたり0.05mg〜20mg/kg体重である。よってたとえば、一日あたりロフェコキシブ25mgまたはセレコキシブ200mgが、経口投与でヒト成人1人に投与され得る。
【0073】
投与単位は、通常、活性成分1mgから500mgの間である。
本発明の一つの見地によれば、たとえば免疫不全またはウィルス感染などの本明細書に記載された疾患を治療するために、一つのCOX−2阻害剤に、さらに1以上のその他のCOX−2阻害剤を併用することができる。
本発明の別の見地によれば、免疫不全、HIV感染、またはAIDSなどの疾患の治療のために、COX−2阻害剤は1以上のさらなるCOX−2阻害剤、または異なる作用様式の1以上のさらなる薬剤と併用することができる。このような組み合わせの例としては、COX−2阻害剤と、1以上のNNRTIとの組み合わせ、または1以上のNRTI類との組み合わせ、または1以上のHIVプロテアーゼ阻害剤との組み合わせ、または、1以上のHAARTとCOX−2阻害剤との組み合わせが挙げられる。
【0074】
さらなる見地では本発明は、1以上のCOX−2阻害剤と、特に長期の治療の間に活性成分の耐性を向上させる化合物とを組み合わせる方法および/または組成物を提供する。その典型的な化合物として、抗ヒスタミンおよびプロトンポンプ阻害剤が挙げられる。
よって本発明は、前記のCOX−2阻害剤を、1以上の追加的なCOX−2阻害剤および/または1以上の追加的な活性成分とともに含む組成物にも及ぶ。本発明は、さらに前述のそのような組成物の使用ならびにそのような組成物を使用する方法にも及ぶ。本発明はまた上述した症状もしくは疾患の治療または予防において、同時または別個または逐次に使用される、混合製剤として上記の成分を含有する製品にも及ぶ。
【0075】
【実施例】
本発明は、以下の図を参照して以下の限定されない実施例でさらに詳述される。
【0076】
【実施例1】
げっ歯類後天性免疫不全症候群( MAIDS )のマウスは T 細胞機能障害を誘発する cAMP/PKA タイプ I を有する
MAIDS(げっ歯類の後天性免疫不全症候群)
多数の研究からMAIDS(Murine Acquired Immunodeficiency Syndrome)をヒトのHIVによる可能な感染モデルとして考えられている。この症候群は、変異したPr60gagポリタンパク質をコード化する複製−欠陥のレトロウィルスを用いる感染後に、進行する(Chattopadhyay ら, 1991年, J. Virol., 65, p4232−4241; Jolicoeur, 1991年, FASEB J., 5, p2398−2405)。本症候群は、脾臓およびリンパ節における進行性リンパ増殖および重症免疫不全と関係している。MAIDSの原因となる欠陥レトロウィルスは、主としてB細胞に感染する(Aziz, 1989年, Nature, 338, p505−508)が、CD4+ T細胞は、有糸分裂誘発因子のインビトロ刺激に対して、深在性の機能障害およびアネルギー(anergy)を示す。感染マウスのCD4+ T細胞(CD8+ T細胞ではなく)の大部分もまた、異常なThy−1のネガティブ表現型により特徴づけられる(Holmes ら, 1990年, Eur. J. Immunol., 20, p2783−2787; Moutschen ら, 1994年, Scand. J. Immunol., 39, p216−224 (MAIDS))。正常な感染していないマウスにおいては、CD4+ Thy−1− T細胞は、胚中心(該細胞はそこでは新しい抗原特異的な移出細胞に相当する)で選択的に認められる。
【0077】
変異したPr60gagタンパク質がT細胞異常を誘発する機構は知られていない。感染細胞によって分泌される「溶性因子」が、距離をおいてT細胞の機能に影響を及ぼすことが主張されているが(Simard, J. Virol., 68, p1903−1912)、そのような媒介物(mediator)の本性は決して解明されて来なかった。他の研究は、CD4+ T細胞と抗原提示細胞との間の直接的で同種の相互作用がT細胞欠陥の誘発に対して必要であることを提案している(Green, 2001, J. Virol., 70, p2569−2575; de Leval, 1998年, J. Virol., 72, p5285−5290)。
【0078】
アデニル酸シクラーゼ−cAMP−プロテインキナーゼA経路は、免疫応答の調節において重要な役割を果たす(Kammer, 1991年, Immunol. Today, 9, p222−229)。増加したcAMPの濃度は、抗CD3 mAbおよびインターロイキン−2といった種々の刺激に対するT細胞の増殖応答を阻害することが知られている。最近の報告は、JAK3チロシンキナーゼの下方制御が、cAMPによるT細胞増殖阻害の機構を表しているかもしれないということを提案している(Kolenko, 1999年, Blood, 93, p2308−2318)。ノルエピネフリンといった、cAMP誘導性の作用物質に暴露された、げっ歯類の胸腺細胞または胸腺腫細胞は、mRNAの不安定化を含むメカニズムによってThy−1発現を下方制御するため、環状AMPもまた膜タンパク質の下方制御を誘発できた(Wajeman−Chao, J. Immunol., 161, p4825−4833)。
【0079】
プロスタグランジンE2(PGE2)、すなわちcAMPの強力な誘導物質は、主に単球、マクロファージおよび活性化されたT細胞によって分泌される。PGE2は、IL−2を阻害することとIL−4産生を促進することによって、T−ヘルパー・タイプ1細胞からT−ヘルパー・タイプ2細胞の方へと釣合いを移動させる(Betz および Fox, 1991年, J. Immunol., 146, p108−113; Meyaard, 1997年, Blood, 89, p570−576)。それは、またB細胞の分化をIgE産生の方へと偏らせる(Fedyk および Phipps, 1996年, PNAS USA, 93, p1O978−10983)。プロスタグランジン合成は、シクロオキシゲナーゼ−1および−2(COX−1およびCOX−2)と特異的PGシンターゼとの逐次的な作用に由来する(Smith および DeWitt, 1996年, Adv. Immunol., 62, pl67−215)。COX−1発現は、主として構成的かつ普遍的であるのに対し、COX−2はある細胞タイプ(マクロファージ、繊維芽細胞、平滑筋細胞)において、IL−1およびTNF−αといった炎症性サイトカインおよびNOによって誘導されるのみである。
【0080】
MAIDSにおけるT細胞機能障害のもととなる機構は、まだ充分に理解されていない。CD4+ T細胞は優先的に関わるが、CD8+ T細胞の変化は、充分なCD4+ T細胞支援の欠如によるのみであるといくつかの報告は示唆している。反対に、B細胞応答の阻害は固有のものであり、欠陥のあるCD4+リンパ球によってだけでは説明できない。したがって、cAMPの選択的な増加がB細胞およびCD4+ T細胞中にあり、しかもCD8+ T細胞中にはないという本発明者の観察は、MAIDSに関係するアネルギープロセス(anergic process)へのcAMPの関与と矛盾しない。
【0081】
発明者が知る限り、これは疾患モデルにおける、サブセット選択的なcAMPの増加についての最初の実証である。プロスタグランジンE2などの溶性因子が本当にcAMP誘導を招いたとすると、何がその作用のサブセット選択性を説明できるであろうか。かつての研究は、CD4+およびCD8+ T細胞における様々なプロスタノイドレセプター(prostanoid receptor)の発現を比較して、両方のサブセットにおいて同じ発現パターンを結論づけた。通常のCD8+ T細胞は、cAMPが誘導するPGE2の効果に対して充分に感受性がある。可能な説明はポストレセプターレベル(post receptor level)でできた。記憶/活性化されたT細胞は、無垢な(naive)T細胞よりもPGE2に対して感受性がある。MAIDSにおいて、MHCクラスII−依存性プロセスが関係しているところでは、CD4+ T細胞は、活性化の特別の状態を獲得することができ、それにより該細胞は、任意の濃度のPGE2の効果をより受けやすくなった。プロスタノイド効果についてのポストレセプター変更は、対応する膜レセプターからプロテインGを分離するGレセプターキナーゼ(GRK)によって主に媒介される。リューマチ性関節炎などの炎症性状態は、GRKの下方制御により特徴づけられる。したがって、それは、カテコールアミンといった、cAMP誘発物質に対するリンパ球の増加した感受性によって特徴づけられる。感染マウスのCD4+およびCD8+ T細胞におけるGRK活性のレベルは知られていない。
実施例1および2で用いられた方法
マウスおよび細胞懸濁液
雄性C57BL/6マウスを発明者の施設で飼育した。4および5週齢において、マウスに0.25mlの無細胞ウィルス抽出液を2回腹腔内に注射した。週齢が一致した対照マウスには、0.25mlのリン酸緩衝化生理食塩水(PBS)を2回腹腔内に注射した。感染後の異なった時点で、マウスを炭酸ガス窒息により犠死させた。末梢リンパ節(鼠径部、腋窩および頸部)を、シリンジを用いて分離して単一細胞の懸濁液を得て、これをナイロンの細胞染色用具(cell stainer)に通し、RPMI1640完全培地で3回洗浄し、トリパンブルーの排除後にトーマ(Thoma)血球計算器上でカウントした。
【0082】
ウィルス
ウィルス抽出液を、前述したようにRadLV−Rを2ヶ月前に注射したマウスのリンパ節から調製した。リンパ節を集めてPBS中ですりつぶし、1.5×104gで30分間遠心分離した。上澄み液を再度30分間1.5×104gで回転させた。この無細胞性のウィルス抽出液を液体窒素中で保管した。XCプラーク(plaque)アッセイを、ウィルス粒子を定量するために用いた。ウィルス標品は、103粒子形成単位(PFU)/mlの狭宿主性(ecotropic)ウイルスを含んでいた。
【0083】
抗体
以下のポリクロナール抗体をウエスタンブロット実験に用いた。第一:ポリクロナールウサギ抗COX−1またはウサギ抗COX−2抗体(Santa Cruz Biotechnology)。第二段階:を共役させたセイヨウワサビ・ペルオキシダーゼ結合抗ウサギ抗体をTransduction Laboratories(Transduction Laboratories, 英国)から購入した。フローサイトメトリーのために、用いたmoAbは以下の通りである。PE−共役CD4/L3T4(YTS.191.1)、FITC−共役CD45R/B220(RA3−6B2)、FITC−共役CD11b/Mac−1(M1/70)、FITC−共役CD161/NK−1.1(PK136)、FITC−共役CD8a(Ly−2)およびCD16/CD32(FcγIII/IIレセプター)(2.4G2)(すべてPharmingenから入手:San Diego, カルフォルニア州, 米国)。CD3 moAb(145−2C11)を発明者の研究室で精製した。コンカナバリンA(ConA)をBoehringer Mannheim Biochemicaから、およびフィトヘマグルチニン−M(PHA)をDifcoから購入した。
【0084】
フローサイトメトリーおよびセルソーティング(細胞選別)
分析は、セルクエスト(Cellquest)ソフトウェア付きのFACStarプラス(FACStar−plus)フローセルソーター(Becton Dickinson)を使用することによって行った。前方及び側方散乱を、生存リンパ球をゲート(gate)するために用いた。FITC(緑)およびPE(橙)の二色分析のために、488nmでの青色励起光をアルゴンイオンレーザー(Air−to−Water cooled model Spinnaker 1161; Spectra Physics, Mountain View, カリフォルニア州)により供給した。セルソーティングのために、60×106個の細胞を、非特異的な相互作用を避けるために抗FcγRII(Fc Block)とともにインキュベートした後に、蛍光色素−結合抗体を用いて氷上で20分間標識した。CD4+ T細胞を、CD8+ B220+ CD11b+細胞を枯渇させることによってネガティブに選択した。同様に、CD8+ T細胞を、CD4+ B220+ CD11b+細胞を枯渇させることによって、およびB細胞を、CD8+ CD4+ CD11b+細胞を枯渇させることによってネガティブに選択した。各ソーティングについて、選別された画分を、フローサイトメトリーによって再分析し、純度を評価したところ、常に97%より高い純度であった。
【0085】
環状AMPの定量
リンパ節の単一細胞の懸濁液を上述したように調製し、RPMI1640を用いて2回洗浄し、1500×gで3分間遠心分離した。その後、細胞を超音波によって粉砕し、細胞内cAMPを抽出溶液(0.01N HCl、95%エタノール)中に容易に遊離させた。細胞溶解物を含む溶液を、13×104×gで15分間遠心分離し、上澄み液を新しい試験管に移した。抽出物を45℃でSpeed Vac濃縮装置中で蒸発濃縮し、ペレットを−20℃で保管した。使用直前に、ペレットをアッセイ用緩衝液中に再懸濁し、cAMPレベルを、125I−標識cAMPアッセイシステム(Amersham, 英国)を用いてラジオイムノアッセイ(RIA)により測定した。試験サンプル中のcAMPの濃度を、曲った直線形の検量線との比較によって求めた。正の対照および負の対照として、リンパ節細胞(1×106)を、1mMのdジブチリル−cAMPおよび0.5mMのDDA(アデニル酸シクラーゼ阻害剤)を用いて30分間37℃で、加湿した5%CO2空気の培養器中で、それぞれイキュベートし、cAMP濃度を測定した。
【0086】
細胞ホモジナイゼーションおよび免疫ブロット
細胞(50×106)を、250mMスクロース、1mMEDTA、0.1%トリトンX−100およびキモスタチン(chymostatin)、ロイペプチン、ペプスタチンAおよびアンチパイン(antipain)のプロテアーゼ阻害剤、各々10μg/ml、10mMリン酸カリウムを含む緩衝液(pH7.1)中、氷上で超音波(2×15秒)によってホモジナイズし(Taskenら, 1993年, J. Biol. Chem., 268, p21276−21283)、30分間(15,000×g)遠心分離して不溶性物質を除去した。タンパク質濃度をBradfordアッセイ法(BioRad)により求めた。免疫ブロットのために、40μgのタンパク質を10%SDS−PAGEにより分離してPVDF膜に移し、5%の非脂肪粉乳および0.1%BSA(Blotto)を含むTBS/Tween中の抗体とともにインキュベートした。一次抗体をHRP−結合二次抗体(Jackson Laboratories/Transduction Laboratories)およびECL(Amersham)により検出した。
【0087】
PKAのホスホトランスフェラーゼ活性
PKAの触媒活性を、[γ−32P]ATP(比活性0.25Ci/mMol、Amersham)を用いて、R. Roskoskiによって記載(Methods Enzymol., 1983年, 99, p36)されたアッセイ混合物中で、PKA−特異的基質(Leu−Arg−Arg−Ala−Ser−Leu−Gly)(Kemp ら, 1976年, PNAS USA, 73, p1038−1042)、Kemptide(Peninsula Laboratories INC.)をリン酸化することにより定量した。ホスホトランスフェラーゼ活性を、cAMP(5μM)およびPKI(1μM)の存在下および非存在下の両方について測定し、PKIによって阻害されない低レベルの活性を差し引くことにより、PKA−比活性を求めた。
【0088】
環状AMP結合の測定
可溶化されたPKA調節サブユニットの特異的[3H]cAMP結合の定量化を、CobbとCorbinによって記載(Methods in Enzymology, 159, p202−208, 1988年)されたように、[2,8−3H]cAMP(2.25μM;比活性5Ci/mMol;Du Pont−New England Nuclear)を含む混合物中で行った。Rサブユニットのモル比を各調節サブユニットモノマー上の2つのcAMP結合サイトに基づいて計算した。
【0089】
免疫細胞化学
対照および感染リンパ節のリンパ球を、冷アセトンで5分間固定し、それぞれ0.1%のサポニンPBS中で5分間2回洗浄した。内因性ペルオキシターゼを、0.1%サポニンPBS中の0.3%過酸化水素を用いて、15分間インキュベートすることによりブロッキングした。サポニン/PBS中でリンスした後、スライドグラスをブロッキング緩衝液(0.1%サポニン/PBS中、1.5%の通常のヤギ血清)を用いて30分間室温でインキュベートし、次いで一次抗体溶液を用いて加湿室中、室温で60分間インキュベートした。Caに対する抗体はサンタクルス(Santa Cruz)から入手し、0.1%のサポニンおよび0.5%の通常のヤギ血清を含むPBS中に1:1000で希釈した。その後、スライドグラスを前述の通り洗浄し、ビオチン化ヤギ抗ウサギ抗体を用いてインキュベートした。その後これをABC複合体(Novastain Super ABC Kit, Novocastra)により検出した。ペルオキシターゼを、H2O2の存在下で褐色の沈殿を与えるジアミノベンジジン(diaminobenzidine)(DAB)(Dako)を用いて、目に見えるようにした。スライドグラスを、ヘマトキシリン−エオシン(Sigma)を用いて対比染色した。特異性は、PKA−Cαサブユニットに対する特異的ペプチドとともにサイトスピン(cytospin)をインキュベートすることにより検証した。
【0090】
免疫組織化学
免疫組織化学を、固定された4%のパラホルムアルデヒド中で処理された2μm厚の組織切片および成形して埋め込まれた組織(JB4−JBポリサイエンス(JBPolyscience))について実施した。切片をトリプシン(0.24%)用いて1分間37℃で透過処理し、その後、Tween20(2%)を用いて30分間37℃で処理した。内因性ペルオキシターゼは、H2O2(1%)を用いて30分間、室温でインキュベートすることによりクエンチした。不特定部位(aspecific sites)を通常のヤギ血清(1.5%)を用いて37℃で1時間の間に飽和させた。その後、切片を、一晩4℃で一次ポリクロナールウサギ抗COX−1またはウサギ抗COX−2抗体(Santa Cruz Biotechnology)を用いてインキュベートした後、ビオチン化ヤギ抗ウサギ抗体を用いて2時間インキュベートした。この後のものをABC複合体(Novastain Super ABC Kit, Novocastra)により検出した。ペルオキシターゼを、H2O2の存在下で褐色沈殿を与えるジアミノベンジジン(diaminobenzidine)(DAB)(Dako)を用いて、目に見えるようにした。切片を、ヘマトキシリン−エオシン(Sigma)を用いて対比染色した。特異性を、一次抗体の代わりに通常のウサギ血清用いて切片をインキュベートすることにより分析した。
【0091】
MAIDSマウスについての増殖アッセイ
増殖アッセイを100μl容積の平坦底面96穴ミクロタイタープレート中で、1ml当たり0.1×106個のCD3+ T細胞のインキュベートにより行った。引き続き、活性化を、ヒツジ抗マウスIgG(Dynal, cat. no. 110.02)を細胞:ビーズ比として1:1でコートした単分散磁性ビーズを添加し、次いで、提示された実験について4μg/mlの最終希釈度で抗CD3(クローン2C11)を添加することにより達成した。抗体の最適濃度を初期設定の段階で慎重に滴定し、いくつかの異なった抗体希釈度での並行実験を常に行った。増殖は、72時間細胞をインキュベートすることにより分析したが、その間において、[3H]−チミジン(0.4μCi)を最後の4時間において含有させ、細胞回収器(cell harvester, Skatron, Sterling, ヴァージニア州, 米国)を用いてガラス繊維フィルター上に捕集した。
【0092】
取り込まれた前駆物質をシンチレーション分析器(Tri−Carb, Packard, Meriden,コネティカット、米国)でカウントした。cAMP類似化合物を使用した場合、その添加後30分してから、抗CD3抗体の添加により活性化した。8−CPT−cAMPはSigma(St. Louis, ミズーリ州)から入手し、SP−およびRP−8−Br−cAMPはBioLog Life Science Company(Bremen, ドイツ)から入手し、すべて4〜10mMの濃度までPBSに溶解し、濃度は、供給メーカーにより与えられる吸光係数を用いて計算した。インドメタシンを水に溶解し、50ng/mlの濃度で用いた。
【0093】
PGE2定量
対照および感染マウス由来のリンパ節細胞について、48時間−培養の上澄み液500μlをピペットで1.5mlのポリプロピレン製試験管に採取し、500μlの水:エタノール(1.4)および10μlの氷冷酢酸を加えた。試験管を穏やかに混合し、5分間室温で放置した。この後、2500×gで2分間遠心分離した。上澄み液を回収し、2カラム容積分の10%エタノールで詰めたAmprep C18ミニカラムに通した。その後、カラムを1容積分のH2Oおよび1カラム容積分のヘキサンで洗浄した。その後、PGE2を2×0.75mlの酢酸エチルで溶出した。溶出画分を集め、窒素下で乾燥するまで蒸発させた。最終的に、各画分を100μlのアッセイ緩衝液に再び戻して、PGE2を、メーカーによって推奨されているAmersham EIAキットを用いてアッセイした。
【0094】
統計解析
2群の個体の比較のために、Mann−WhitneyのU検定(両側の)を用いた。相関係数(r)をスピアマン(Spearman)の順位検定により計算した。統計的解析および曲線適合(curve−fit)解析を、それぞれスタティスティカ(Statistica)(Statsoft Inc., Tulsa, OK)およびシグマプロット(Sigma Plot)(Jandel Corporation, Erkrath, ドイツ)ソフトウェアパッケージを用いて行った。結果は中央値(median)および25位−75位の百分位数として与えられ、特にことわらない限り、p値は両側であり、0.05より小さい時には有意と見なされる。
実験
MAIDS感染は、CD4+ T細胞において上昇したcAMPを誘導する−
MAIDS発症の原因となる、RadLV−Rとして知られたレトロウィルスの混合物を接種したマウスを、感染後異なった時点で犠死せしめ、リンパ節細胞を、フローサイトメーター/セルソーター装置を用いて、ネガティブな選択により純粋なB細胞ならびにCD4+およびCD8+ T細胞に選別した。細胞内のcAMPレベルを感染後の様々な細胞集団群で評価した。図1に見られるように、cAMPレベルは、感染して数週間後のCD4+ T細胞中において顕著に増大した(20倍以上)。後の段階でもB細胞のcAMPレベルも増大したが、これに対しCD8+ T細胞では僅かな変化が観察されただけであった。さらに、CD4+ T細胞をポジティブなソーティングによりThy−1.2+およびThy−1.2−細胞に分離すると、Thy−1.2−細胞ではcAMPレベルの大きな変化があることが明らかになった(図2、6倍)。両細胞集団群を非感染マウスから集めて来ると、この正常には「低」が多い集団群も、Thy−1.2+のそれらに匹敵するよりも高いcAMP基底レベルを示した。
【0095】
界面活性剤で可溶化した抽出物の細胞核の後の(postnuclear)上澄み液におけるPKAホスホトランスフェラーゼ活性の分析により、cAMP依存キナーゼ活性の全レベルはMAIDSリンパ節細胞において減少したが、cAMPが存在しない場合には活性の僅かな変化が観察されただけであることが明らかとなった(図3A)。これは、慢性的な活性化、およびCサブユニットの分解またはCの転移のいずれかをもたらすPKAの分離と矛盾しない。cAMP結合の評価(図3B)は、PKA Rサブユニットの全レベルの変化はないことを明らかにした。MAIDSおよび対照マウスのリンパ節細胞の免疫細胞化学は、細胞核における免疫反応性PKA Cサブユニットレベルの上昇を明らかにした(図4)。これも、MAIDSにおけるcAMP−PKA経路の活性化と矛盾しない。
【0096】
PKAタイプIアンタゴニストはMAIDS T細胞のT細胞増殖を向上させる−
TCR/CD3−誘導T細胞増殖の阻害に与える、上昇したcAMPおよびPKAの活性化の効果を調べるために、PKAタイプIに対して充分なアンタゴニストとして作用する、硫黄置換したcAMP類似化合物(Rp−8B−Br−cAMPS)を使用した(Gjertsen, Mellgren, ら 1995年 1665/id)。図5Aは、MADIS感染マウス由来のT細胞において、TCR/CD3刺激による増殖が、非感染対照マウスT細胞(図5B)のそれの10%未満であることを示す。さらに、PKAタイプIアンタゴニストの効果をMAIDS T細胞で評価すると、より高い濃度では4倍を超えるというTCR/CD3誘導性の増殖について、濃度依存性の増加が観察された(図5A)。しかしながら、対照T細胞の処理では刺激は観察されなかった(図5B)。11匹のMAIDS感染マウスを見ると、対照と比較して、すべてT細胞増殖を著しく減少させており(p<0.001)、11匹のうち10匹のマウスで、PKAタイプIアンタゴニストはT細胞増殖を改善させた(p<0.01; 中央値 2.2倍, 表5)。cAMPアンタゴニストの刺激効果は使用した最も高濃度においても飽和していない(表5のすべてのマウスについて、図5Aおよび同様のデータ(未表示)を得た)。このことは、化合物の溶解性、細胞に対する親和力または利用可能性が、観察された効果に対する制限因子であるかもしれないということを示す。したがって、より透過性でしかも強力であるPKAタイプIアンタゴニストが入手できるならば、MAIDS T細胞のTCR/CD3誘導性増殖をさらに改善させるかもしれない。
【0097】
次に、TCR/CD3−誘導のT細胞増殖に与える、cAMPアゴニストの効果を、5匹のMAIDS感染マウスおよび4匹の対照で検討した。MAIDS感染マウス由来のT細胞は、外部から添加した8−CPT−cAMPにより、細胞増殖阻害に対する感受性において明らかな変化を示した(図5Cおよび表5)。さらに、MAIDS感染マウス由来のT細胞および対照T細胞の最大増殖速度を100%に正規化すると(図5Cおよびデータ未表示)、左方にシフトしたcAMP阻害曲線に加えて、それらの曲線の傾きが著しく異なった(正常のT細胞についてのHill係数2.2(1.9〜2.5)に対して、MAIDSマウス由来のT細胞についてのHill係数0.6(0.54〜1.52)、表5、p<0.05)。
cAMP類似化合物によって阻害への感受性が上昇したことは、外から加えたcAMP類似化合物に対するAサイト親和性が後に上昇することとともに、PKAタイプIのcAMP結合サイトBを初回刺激(prime)することにおいて、増加した内因性cAMPからの寄与を示唆する。
【0098】
曲線の傾きが、8−CPT−cAMPにより、協同的な2−リガンドサイト結合(2−ligandsite binding)の状況から、見かけの非協同的な阻害曲線へシフトすることも、又、増加した内因性cAMPによるBサイトの占有を示している。
【0099】
【表5】
【実施例2】
環状 AMP が誘発した MAIDS の T 細胞機能不全は、 COX ‐ 2 レベルの増加とともに CD11b− 陽性細胞による増大した PGE 2 産生による
MADISにおけるPGE2の増大した産生−
混合リンパ節細胞集団群をMAIDS感染マウスおよび対照マウスから分離し、インビトロで培養した。PGE2の分泌レベルを48時間培養後の培地上澄み液で評価すると、MAIDS感染細胞が対照細胞の7〜8倍も多いPGE2を分泌することが明らかとなった。
【0101】
MAIDSにおいてPGE2産生の阻害は、T細胞の増殖を回復させる−
次に、混合リンパ節細胞を抗CD3抗体によって活性化し、T細胞の増殖を誘導した。[3H]−チミジンの取り込みを72時間後に調べた。MAIDS感染マウス由来の細胞の増殖もまた非感染細胞のT細胞増殖の10〜20%だけであった。しかしながら、混合培養においてインドメタシンを培地に添加してPGE2の産生を阻害すると、これは、MAIDS感染マウス(5匹)由来の細胞の増殖を、対照マウスに匹敵するレベルまで強力に増大させた(図6)。10匹の追加したMAIDS感染マウスを見ると(表6)、混合リンパ球培養のT細胞の増殖に与えるインドメタシンの効果は、極めて有意であった(p<0.01)。対照的に、インドメタシンで対照培養を処置しても、増殖を変えなかった。
【0102】
COX−2がMAIDS感染マウスのリンパ節において高レベルで発現する−
構成的に発現されるCOX−1は、PGE2を産生するシクロオキシゲナーゼ活性の通常の源である。しかしながら、COX−1の増加が、PGE2の増大したレベルを説明し得るほどにMAIDSマウス中では見られなかった(データ未表示)。COX−2の発現は、通常、脳/脳プロセス、関節炎滑液および組織の損傷部分に限定される。COX−2は、たとえば、対照リンパ球について図8(上の図)に示されたように、リンパ節またはリンパ球では見出されない。驚くことに、MAIDS感染マウス由来の粗リンパ節細胞は、高レベルにCOX−2を発現することが分かった(図8、下の図)。さらに、MAIDSリンパ節から、B細胞と同様、ポジティブに選択されたCD4+およびCD8+ T細胞は、高いレベルのCOX−2を含有していた。対照的にネガティブに選択されたCD11b−細胞は、低レベルのCOX−2だけしか含有しなかった。
【0103】
MAIDS感染マウスおよび対照マウス由来のCD4+およびCD8+ T細胞ならびにB細胞(B220マーカー)をフローサイトメトリーにより調べると、CD11bマーカーは、通常TまたはB細胞上には発現されないことが明らかであった。しかしながら、MAIDS感染マウス由来のCD4+ T細胞とB細胞との両方の正確な分画は、CD11b明(bright)(標識R1を“ゲート(gate)”する)であり、CD8+ T細胞のみならずCD4+ T細胞とB細胞の追加プールは、CD11bの暗(dim)(標識R2を“ゲート”する)であった。このことは、これらの細胞が有意ではあるが低レベルのCD11b発現を有することを示している。したがって、MAIDS感染CD4+およびCD8+ T細胞の部分集団は、それぞれCD11b明、および暗(bright and dim)であるが、これに対しB細胞の大部分は陽性であった。CD11b+細胞はCOX−2を発現し、CD11b−細胞は発現しないという事実とを考え合わせると、これは、MAIDS感染マウスに由来するリンパ節のB細胞とT細胞との両方がCOX−2を発現するということを示す。
【0104】
MAIDS感染マウス由来の無処置のリンパ節を免疫組織化学により調べると、MAIDS(感染後19週)において、全体の構造は、対照マウスと比較すると胚中心の喪失でもって変更されていることが明らかである(図10、c対a)。COX−2について免疫染色したスライドグラスを高倍率で見れば、対照動物由来のリンパ節は、マクロファージに取り込まれた物質における、褐色HRP染色(偽りの陽性「薄染色できる実体」、図10b)を示すだけであるのに対し、MAIDSにおけるリンパ節細胞の大部分は、COX−2に対して陽性に染色することは明らかである(図10d)。
【0105】
【表6】
【実施例3】
HIV 患者は、インビボで非選択的な COX 阻害剤で治療されると、効果をかろうじて 示すだけである
方法
HIV患者由来の末梢血CD3+ T細胞のネガティブな選択
末梢血CD3+ T細胞を、健常者ドナーからのバフィーコート(buffycoats)からネガティブ選択により精製した(Ullevaal 大学病院血液センター、 オスロ、 ノルウェー)。随時、末梢血の単核細胞を密度勾配(Lymphoprep, NycoMed, オスロ, ノルウェー)遠心分離法により分離し、次いで、CD14およびCD19に対する抗体で直接コートした単分散の磁気ビーズと、CD56に対する抗体でコートしたラット抗マウスIgGビーズと磁石とを用いてネガティブ選択した。磁気ビーズはすべてDynal(オスロ, ノルウェー、それぞれcat. no. 111.12, 111.04, および110.11)から入手し、抗CD56抗体はPharmingen(San Diego, カルフォルニア州, cat. no. 31660.d)から入手した。すべての工程は4℃で実施した。細胞懸濁液をフローサイトメトリーにより分析し、90%を超える細胞がCD3+細胞からなることを示された。
【0107】
HIV患者のT細胞を用いた増殖アッセイ
増殖アッセイを100μl容積の平坦底面96穴ミクロタイタープレート中で0.75×106個/mlのCD3+ T細胞のインキュベーションにより実施した。引き続き、活性化は、ヒツジ抗マウスIgG(Dynal, cat. no. 110.02)を細胞:ビーズを1:1の比率でコートした単分散磁性ビーズを添加することにより達成され、次いで、抗CD3(クローンSpvT3b)を、示された実験について最終希釈度1:125000で添加した。抗体の最適濃度を初期設定の段階で慎重に滴定し、いくつかの異なった抗体希釈度での並行実験を常に実施した。増殖は、細胞を72時間、インキュベートすることにより分析したが、その間の最後の16時間[3H]−チミジンを含有させた。細胞を洗浄し、ガラスフィルター上に捕集し、次いでβ−シンチレーションカウンティングにより分析した。cAMP類似化合物を使用する場合には、抗CD3抗体の添加により活性化する30分前に加えた。8−CPT−cAMPはSigma(St. Louis, ミズーリ州)から入手した。
実験
進行中である第II相の臨床試験は、非選択的なCOX阻害剤(インドメタシン)を用いる短期治療の免疫賦活効果を、HIV感染患者由来のT細胞上の代理パラメーター(surrogate parameters)に関してテストしている。認可された手順によると、患者は50mgのインドメタシンを1日に3回(全投与量150mg/日)2週間、0日、14日、28日(中止後2週間目)にサンプリングしながら、受けることになっていた。しかしながら、上腹部痛および消化不良などの有害事象、ならびに初期患者間の研究の中断により、この投与量を25mgのインドメタシンを1日に3回にカットしなければならなかった(全投与量75mg/日)。図11は、研究をこれまで完了した3人の患者(患者1〜患者3)のT細胞免疫機能(活性化後の増殖として測定)を示す。
【0108】
上の図は開始時(0日)、インドメタシン治療の終了時(14日)、その2週間後(28日)におけるT細胞活性化後の増殖レベルを示す。見て分かるように、患者1および2は、インビボで投与された非選択的COXアンタゴニストによっては免疫機能を増加しなかった。しかしながら患者3におけるT細胞の応答は、約2.5倍に増加し、インドメタシン中止後2週間まで持続した。図11b、下図は、細胞培養においてPKA−I選択的cAMPアンタゴニスト、Rp−8−Br−cAMPとをインビトロでインキュベートした後のT細胞の増殖を示す。cAMP媒介によるT細胞機能不全の程度は、アンタゴニストによって得られた増殖の逆転から明らかである(上図と下図を比較。すべての時点で、増殖が入院患者1および3で約2倍増加、患者2では効果なし)。インドメタシンが納得できる効果を有しなかったことは図11から明らかであり、このことは有害事象による投与量の制限のみならず、COX−2選択性の欠如によるものかもしれない。
【0109】
【実施例4】
HIV 患者はインビボで非選択的 COX 阻害剤の投与した後、かろうじて効果を示す(実施例3の実験の続き)。
方法
使用した方法は実施例3で記載した方法であった。
実験
3剤の併用治療に加えて、インドメタシン25mgを1日に3回経口的に14日間受けた進行中の第II相臨床試験(実施例3の続き)における7人の患者の結果を図12に示す。患者1〜3は実施例3で述べた患者に相当する。インドメタシン投与に関わる問題は、上述(実施例3)したように、その投与量を1日3回25mgまでに制限される有害事象である。この許容される用量で、この非選択的COX阻害剤の効果は限界にある。治療の14日後、7人の患者のうち2人のみにおいて、T細胞免疫機能(T細胞活性化後の増殖として測定した)が明らかに上昇したが、1人の患者は免疫機能が減少し、4人の患者は僅かな変化であった。インドメタシンを中止して2週間後、7人の患者のうち5人は0日目と比べて免疫応答性が上昇した。しかしながら、2人の患者が2倍を超えるT細胞増殖の増加を示しただけであった。
【0110】
【実施例5】
COX−2 阻害剤はインビトロで MAIDS T 細胞の免疫機能を向上させる
方法
増殖アッセイで使用した方法は、実施例1に記載の方法であった。PGE2アッセイは実施例1に記載したとおりである。
実験
増殖アッセイ
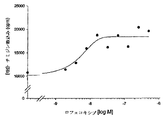
混合リンパ節細胞を感染後17週目のMAIDSマウスから分離した。細胞を抗CD3抗体により活性化してT細胞の増殖を誘導し、免疫機能の尺度として、[3H]−チミジンの取り込みを72時間後に試験した。MAIDS感染マウス由来の細胞の増殖もまた、非感染細胞のT細胞増殖の5〜20%だけであった(MAIDSの細胞において、2000〜12000cpmに対して、非感染マウス由来の細胞における平均55000cpm)。しかしながら、ロフェコキシブ(rofecoxib)(図13)またはセレコキシブ(celecoxib)(図14)を培養中に添加すると、これにより濃度依存的な様式で、MAIDS感染マウス由来の細胞増殖は2〜3倍に増加した。これと対照的に、ロフェコキシブまたはセレコキシブを用いる非感染マウス由来の対照培養の処置は、その増殖を増加させなかった(COX−2阻害剤の存在下で、0.8〜1.0倍の増加、すなわち増加しない。表示せず。)。MAIDSマウス由来のT細胞において、最大効果の半分(ED50)を示すロフェコキシブおよびセレコキシブの濃度は、ロフェコキシブについて約0.01μM、セレコキシブについて約0.03μMであった。マイクロモル濃度より下の濃度が効果的であるという事実は、明らかに、観察された免疫応答の増加がCOX−2の阻害を介しており、ロフェコキシブおよびセレコキシブのマイクロモル濃度においてのみ阻害されるCOX−1ではないことを示す(Warner らの値, 1999年, PNAS USA, 96, p7563−7568)。したがって、阻害されたT細胞免疫機能のロフェコキシブおよびセレコキシブによる反転は、混合培養における減少したPGE2産生となり、それによってCOX−2の阻害を介してT細胞cAMPレベルの低下をもたらす。
【0111】
PGE2産生
PGE2レベルに与えるCOX−2阻害剤ロフェコキシブおよびセレコキシブの効果も分析した。図15に見られるように、MAIDSマウス由来の粗リンパ節細胞は、健常マウス由来のリンパ節細胞よりも5〜6倍のPGE2を分泌した(図6も参照)。さらに、感染マウスではLPSに応答したPGE2レベルは、非感染マウスでの約2倍と対照的に8〜10倍に増加した。細胞をCOX−2阻害剤、ロフェコキシブおよびセレコキシブの存在下で培養すると、MAIDSリンパ節細胞のPGE2分泌は、非感染細胞と同様であった。インドメタシンの効果(図7の増殖との比較)を対照として含める。
【0112】
【実施例6】
COX−2 阻害剤はインビボで MAIDS T 細胞の免疫機能を向上させる
方法および実験
感染マウス(感染後17週目)を、経口的に(すなわち口から)1週間、ヒトへの使用に対して推奨される用量に相当する(ネズミでは7倍高いクリアランスを考慮して)ロフェコキシブの投与量で処置した。MAIDSマウスは通常、拡大されたリンパ節および脾臓とともに免疫増殖症候群を発生する。これによると、未治療の感染動物は、平均脾臓重量1.3gおよび集めたリンパ節の平均重量1.7gを有していた。対照的に、ロフェコキシブを7日間受けたMAIDSマウスは、平均脾臓重量0.8gおよび集めたリンパ節の平均重量0.3gを有していて、リンパ球増殖の反転を示した。
【0113】
結果を図16に示す。T細胞免疫機能を、感染した治療および未治療のマウスに由来する粗リンパ節細胞で評価した場合、未治療の感染動物は、抗CD3誘導性増殖を2000〜10000cpmの範囲(平均7300cpm)で有していたが、これに対して1週間ロフェコキシブを受けた感染マウスは、感染した未治療のマウスと比較して2.7〜5.6倍に増加した、抗CD3に対するT細胞応答を有することは明らかであった。さらに、感染した未治療のマウスは、Rp−8Br−cAMPSの存在下で、抗CD3誘導性のT細胞増殖を増加させたが、この2〜3倍の効果は、ロフェコキシブを用いて処置されたマウスでは失われた。このことは、インビボでロフェコキシブを用いる治療が、PGE2レベルを低下させ、T細胞機能のcAMP媒介による阻害を反転させたことを示した。
【0114】
【実施例7】
インビボでロフェコキシブまたはセレコキシブを用いた MAIDS マウスの治療は抗 CD3 に対する T 細胞応答および免疫応答を増加させる
方法および実験
感染マウスを、ヒトへの使用に対して推奨される用量に相当する投与量で(ネズミでは7倍高いクリアランスを考慮して、それぞれ3および20mg/kg/日)、ロフェコキシブおよびセレコキシブを用いて処置した。非経口的投与を、イントラリピッド中で処方されるCOX−2阻害剤を腹膜内に注射することにより実施した。結果を図17に示す。
【0115】
T細胞の免疫機能を、感染して18〜20日後の治療および未治療の感染マウスに由来する粗リンパ節細胞で評価すると、未治療の感染動物は抗CD3誘導性の増殖を10000cpmの範囲で有していたが、ロフェコキシブを18〜20日間受けた感染マウスは、感染した未治療マウスと比較して、約2倍に増加した抗CD3に対するT細胞応答を有することが明らかであった。同様にセレコキシブは、注射された多くのマウス群由来の細胞において、免疫応答を、未治療の非感染マウスの約3倍を超えるまで向上させた。
【0116】
【実施例8】
MAIDS マウスのメロキシカム( meloxicam )を用いたインビボ治療は T 細胞免疫機能を向上させる
方法および実験
感染および健康マウスを2.8mg/kg/日のメロキシカムを用いて治療した。このメロキシカムの投与量は、ネズミでは7倍高いクリアランスを考慮すると、ヒトへの使用に対して推奨される用量に相当する。非経口的投与は、水溶性メロキシカム注入化合物で満たされた浸透ポンプを皮下移植することにより実施された。T細胞機能を評価し、その結果を図18に示す。
【0117】
治療された感染マウスおよび対照の(PBS処置された)感染マウスに由来する粗リンパ節細胞において、処置して2週間後にT細胞免疫機能を評価すると、PBS処置された感染動物は、抗CD3誘導性の増殖を500cpmの範囲で有していたが、メロキシカムを14日間受けた感染マウスは、PBSのみを受けた感染マウスと比較して、抗CD3に対するT細胞の免疫応答が有意に増加していることが明らかであった(図18a、10倍を超える;p<0.05)。
【0118】
メロキシカムによるインビボ阻害からの解除およびそれによるCOX−2の再活性化を防止するために、3日のT細胞増殖インビトロアッセイ中、メロキシカムを細胞培養に再び添加したとき、メロキシカム治療群の免疫応答は、インビトロでメロキシカムを添加しないもの(p=0.005)よりも2倍高く、さらにPBSをインビボで受けたMAIDSマウスと比較しても、その効果は有意であった(図18b、p<0.05)。
【0119】
対照的に、PBSをインビボで受け、メロキシカム治療を受けなかったMAIDSマウスのみが、PKAタイプI選択性cAMPアンタゴニスト、Rp−8−Br−cAMPを、抗CD3刺激混合リンパ節培養にインビトロで添加した場合に、上昇した免疫応答を示した(図18c)。メロキシカム治療したMAIDSマウスにはcAMPアンタゴニストの効果がないという事実は、インビボのメロキシカム治療がMAIDSのcAMP誘導の免疫不全を減少させまたは除去し、免疫機能を復原することを示すものである。
【図面の簡単な説明】
【図1】図1は、MAIDS感染後においてCD8+ (A)、CD4+ (B)およびB(C)細胞におけるサイクリックAMPレベルを示す。単核細胞が、MAIDSで感染したマウスのリンパ節から種々の経過時間で分離され、FACS−セルソーターを用いるネガティブな選択(negative selection)により、CD4+、CD8+ およびB細胞に単離された。細胞内のcAMPレベルは、超音波処理およびラジオイムノアッセイにより測定された。バーは、平均値±SD(n=3、個々のマウス)を示す。
【図2】図2は、CD4+ 集団、Thy−1.2 陰性および陽性の集団におけるMAIDS cAMP レベルを示す。感染マウス3匹と週齢をマッチさせた対照マウス3匹とからのリンパ節細胞は、FACS−ソートされ、CD4+、Thy−1.2+(白のバー)およびCD4+、Thy−1.2−(濃いバー)の集団とし、細胞内cAMPレベルは図1と同様に測定した。バーは平均値±SD(n=3)を示す。
【図3】図3は、MAIDSマウス 対 野生型マウスについてプロテインキナーゼA活性のレベルを示す。(A)マウス脾臓から精製したリンパ節細胞の界面活性剤で可溶化した抽出物におけるキナーゼ活性は、基質として5μM cAMPの存在(総活性、ハッチバー)または非存在(フリー活性、濃いバー)のKemptideを用いて、分析された。PKA特異的プロテインキナーゼ阻害剤(PKI, 1μM)により阻害されないホスホトランスフェラーゼ活性は、PKA特異的活性のみを示すために差し引かれた。感染マウス(MAIDS、n=4)における活性は、野生型の同腹子のものとの比較で示される。(B)[3H−cAMP] 結合は、(A)と同じ抽出物中で測定し、Rモノマーのモル量を算出した。
【図4】図4は、MAIDSおよび野生型マウスの細胞におけるRKA C−サブユニットの免疫学的所在決定を示す。対照マウス(上パネル)およびMAIDS感染マウス(下の2個のパネル)からの単核細胞は、サイトスピン(400×g)によりガラススライドに付着させ、固定してから、非PKA−Cポリクローナル抗体およびHRP−結合二次抗体(茶色染色)で免疫染色した。対比染色はヘマトキシリン(クロマチン上で青色染色)による。
【図5】図5は、MAIDSおよび野生型マウスのT細胞機能への、PKAタイプIアンタゴニスト(抑制因子)、RP−8−ブロモ−cAMP−ホスホロチオエート(phosphorothioate)(Rp−8−Br−cAMPS)の効果を示す。TCR/CD刺激したT細胞増殖は、MAIDSマウス(A)および非感染の対照マウス(B)から分離したT細胞を用いて評価した。
MAIDSマウス(白丸、点線)および対照マウス(黒丸、実線)から分離したCD3+ T細胞のTCR/CD3刺激した増殖に対するcAMP作動薬(8−CTP−cAMP)の濃度増加効果は、同じ実験(C)で個別に試験した。3つの測定の平均値±SDを示す。表4に取りまとめたデータ(n=11)を示す。注:CではcAMP作動薬の非存在下で引き起こされたTCR/CD3増殖が、MAIDSおよび対照の両方のT細胞について100%正規化されたのに対して、AおよびBでは尺度が異なる。
【図6】図6は、インビトロでの、正常およびMAIDSリンパ節細胞によるPGE2の分泌を示す。感染後20週でのMAIDS感染マウス(濃いバー、n=9)および、週齢をマッチさせた対照マウス(陰影バー、n=4)からのソートしていないリンパ節細胞を、完全培地中で48時間培養し、その後ELISAにより上清中でのPGE2の分泌レベルを測定した。
【図7】図7は、正常およびMAIDS感染マウス中の、T細胞免疫機能に対する非選択的COX阻害剤の効果を示す。カラム1−対照マウス+抗CD3、カラム2−対照マウス+抗CD3+インドメタシン、カラム3−MAIDSマウス+抗CD3、カラム4−MAIDSマウス+抗CD3+インドメタシン。T細胞増殖応答は、ソートしていないリンパ節単核細胞の混合集団において、非選択的COX阻害剤であるインドメタシン(50ng/ml)の非存在および存在下で、[3H]−チミジン取り込みにより評価した。T細胞活性化は、抗CD3(mAb 2C11; 4mg)の交差連結(cross−ligation)により達成される。バーは、対照(n=3)およびMAIDS感染(n=5)マウスによる平均値±SDを示し、表5に付加的データを示す。細胞は、72時間培養し、その間最後の4時間[3H]−チミジンを含んだ。
【図8】図8は、正常(A)およびMAIDS感染(B)マウスにおける、リンパ節リンパ球の異なるサブセットによるCOX−2の発現を示す。CD4+ T、CD8+ TおよびB細胞は、それぞれCD4、CD8およびB220分子の発現に基づいて、ポジティブ選択(positive selection)によりFACSソートした。CD11b−細胞は、ネガティブ選択(CD11b不存在に基づく)によりソートした。MAIDS感染および正常マウスからの細胞は、次いで溶解させ、各サンプルからのタン白質10μgをCOX−2の発現の免疫ブロット分析に供した。対照としてブロットは、アクチンに対する抗体と同時に反応させた。
【図9】図9は、MAIDSおよび野生型リンパ節細胞中でのCD11bの発現を示す。MAIDS感染および対照マウスからの、リンパ節リンパ球(CD4+、CD8+ T細胞、および、B220+ B細胞)の異なるサブセットリンパ球によるCD11bの発現を示す(フローサイトメトリーによる)。R1:CD11b高、R2:CD11bはっきりしない、R3:CD11b−。
【図10】図10は、MAIDS感染マウスおよび野生型マウスのリンパ節におけるCOX−2の発現のレベルを示す。リンパ節は、凍結切片を作成し、COX−2免疫組織化学染色(茶色染色)に供した。(a)COX−2について染色された胚中心を有する正常な対照リンパ節。(b)高い倍率で拡大した正常なリンパ節。HRP−発色反応で陽性に染色される細胞は、取り込まれた物質を有する「染色できる実体」のマクロファージ(矢印)である。c. MAIDS感染マウス(感染後20週)からのリンパ節。注:変形した形態と構造 d. COX−2について染色されたMAIDSリンパ節の高倍率拡大。注:細胞質が茶色に染色した細胞多数および多数の有糸分裂像。
【図11】図11は、HIV感染患者のT細胞免疫機能への、非選択的COX阻害剤のインビボ投与の効果を示す。T細胞増殖応答は、第II相の臨床試験に参加しており3剤併用治療に加えて、インドメタシン25mgを1日3回、14日間経口的に摂取した患者3名(pat. 1から3)について、CD3+ T細胞中における[3H]−チミジン取り込みで評価した。上のパネルは、0日、14日(2週間の処置後)および28日(中止して2週間後)におけるT細胞免疫機能を、カラム1、2および3としてそれぞれ標識して示す。T細胞活性化は、抗CD3(mAb SpVT3b)の交差連結(cross−ligation)により達成される。A:T細胞活性化後の基底増殖; B:Rp−8−Br−cAMP(1mM)存在下での増殖; 注:cAMP仲介免疫欠損の程度は、上と下のパネルの比較から明らかである。バーは、3回の測定の平均値±SDを示す。細胞は72時間培養し、その間、最後の16時間は[3H]−チミジンを含ませた。
【図12】図12は、図11と同様であるが7人のHIV感染患者の、T細胞増殖への、非選択的COX阻害剤インドメタシンのインビボ投与の効果を示し、患者1から7はそれぞれ、黒丸、白丸、黒三角、白三角、黒正方形、白正方形、および黒菱形により表示する。3回測定の平均値がプロットされ、結合線は各患者の進展を示す。
【図13】図13は、COX−2特異的阻害剤であるロフェコキシブの、MAIDS感染マウスにおけるT細胞免疫機能への効果を示す。T細胞増殖応答は、ソートしていないリンパ節単核細胞の混合集団において、COX−2選択的阻害剤であるロフェコキシブの非存在下および濃度が増加する(1.9から500nM)存在の条件下で、[3H]−チミジン取り込みにより評価した。T細胞活性化は、抗CD3(mAb 2C11; 4μg/ml)の交差連結反応により達成される。3回測定の平均値は、S字型適合曲線とともに示される。細胞は、72時間培養し、その間、最後の4時間は[3H]−チミジンを含ませた。
【図14】図14は、ロフェコキシブに係る図13の記載のように、COX−2特異的阻害剤であるセレコキシブの、MAIDS感染マウス中でのT細胞免疫機能への効果を示す。
【図15】図15は、対照マウス(1)またはMAIDSマウス(2)について、生体外での(ex vivo)リンパ節(LN)細胞によるPGE2分泌に対するロフェコキシブおよびセレコキシブの効果(インドメタシンと比較した)を示す。ソートしていないLN細胞を、完全培地中で、PGE2誘導剤であるリポポリサッカライド(LPS;4μg/ml)、非選択的シクロオキシゲナーゼ阻害剤であるインドメタシン(50ng/ml)およびCOX−2特異的阻害剤であるロフェコキシブ(0.125μM)およびセレコキシブ(0.125μM)の存在下または非存在下で培養した。48時間後、上清におけるPGE2の濃度をEIAにより測定した。感染マウス(20週)3個体と、週齢をマッチさせた対照の3個体のプールとを分析した。平均±標準偏差を示す。
【図16】図16は、ロフェコキシブを用いたMAIDS感染マウスのインビボ処置の、T細胞免疫機能に対する効果を示す。MAIDSマウスは、非処置のままであるか(非処置1から3)、あるいはロフェコキシブを心室に挿入した管を経由して、経口で7日間投与して処置した(3mg/kg/日、1日1回投与、処置1および2)。続いてT細胞増殖応答を、処置および非処置動物からの、ソートしていないリンパ節単核細胞の混合集団について、Rp−8−Br−cAMPの非存在下(カラムA)および存在下(0.5または1.0mM、それぞれカラムBおよびC)で、[3H]−チミジン取り込みによりインビトロで評価した。T細胞活性化は、全サンプルにおいて、抗CD3(mAb 2C11; 4μg/ml)の交差連結(cross−ligation)により達成された。対照は、非感染マウスでのT細胞増殖を示す。3回測定からの平均値を示す。細胞は、72時間培養し、その間、最後の4時間は[3H]−チミジンを含ませた。
【図17】図17は、ロフェコキシブまたはセレコキシブを用いた、MAIDS感染マウスのインビボ処置のT細胞免疫機能に対する効果を示す。MAIDSマウスには、媒体(イントラリピッド)とともに注射した。すなわち、腹腔内注射(3mg/kg/日、1日1回投与、n=6)によりイントラリピッド中のロフェコキシブで、あるいは腹腔内注射(20mg/kg/日、1日1回投与、n=5)によりセレコキシブで、18から20日間処置した。続いて、T細胞増殖応答を、Rp−8−Br−cAMPを用いないこと除き、他は図16について記載されたようにしてインビトロで評価した。対照は、非感染マウス中のT細胞増殖を示す。25から75%パーセンタイル値(囲まれた領域)およびメジアン(囲み中の線)とともに、3回測定からの平均値を示す(黒丸)。バーはレンジを示す。
【図18】図18は、メロキシカムを用いた、MAIDS感染マウスのインビボ処置のT細胞免疫機能に対する効果を示す。メロキシカム(70μg/動物/日での放出速度)またはリン酸緩衝化生理的食塩水(PBS)を有する浸透ポンプ(Alzet、100μl)を、MAIDSマウス(感染後14週)および健康なマウスに14日間皮下に埋め込んだ。a)次いで、T細胞増殖応答は、図17に記載のようにインビトロで評価した。各グループにおける平均値±該平均の標準誤差(s.e.m.)を示す。MAIDSマウス(濃いバー)からの細胞の、抗CD3刺激した増殖へのメロキシカム処置の効果は、PBSを受けたMAIDSマウス(白バー)のものと比較すると有意性がある(p<0.05)。b)インビボでメロキシカムまたはPBSで処置したa)のマウスのグループからの混合リンパ節培養物に、インビトロでメロキシカム(2.5μg/ml)を細胞培養中に再び添加し、抗CD3誘導T細胞増殖をa)のように評価し、そしてインビトロで再度添加したメロキシカムの効果(白バー)をインビトロでの添加のない細胞の応答(濃いバー)と比較した(p=0.005)。c)a)においてメロキシカムまたはPBSでインビボ処置したマウスグループからの混合リンパ節細胞のインビトロ細胞培養物にRp−8−Br−cAMPS(0.5mM)を添加し、抗CD3誘導T細胞増殖をa)のように評価した。そして、インビトロでのRp−8−Br−cAMPSの効果(白バー)は、インビトロでの添加を受けていない細胞(濃いバー)を超える倍数(fold)誘導として表わされた。統計値は、2グループの動物の比較ではMann−Whitney U検定により、また2つの異なる処置をした同一グループの比較ではWilcoxon ペアマッチ検定により分析した。
Claims (30)
- 免疫不全で特徴づけられる疾患の治療用または予防用の医薬の調製において、COX−2阻害剤またはその誘導体あるいは製薬的に許容しうるその塩の使用方法。
- 前記疾患が、ウィルスによって引き起こされる免疫不全疾患であることを特徴とする請求項1に記載の使用方法。
- 前記疾患が、全般の多様な免疫不全、あるいはレトロウィルス、なるべくHIVまたは関連するウィルスによる感染に起因するもの、あるいはその結果として生じるAIDSまたは関連する状態であることを特徴とする請求項1または2に記載の使用方法。
- 前記医薬が、ヒトまたはヒトに添う動物または農業用動物に投与するためのものであることを特徴とする請求項1〜3のいずれかに記載の使用方法。
- 前記COX−2阻害剤が、IC80でのWHMA アッセイによるCOX−1:COX−2の選択性の比として >5 、好ましくは >50を有することを特徴とする請求項1〜4のいずれかに記載の使用方法。
- 前記COX−2阻害剤が、メタンスルホンアミドエーテル、メタンスルホンアミドチオエーテルまたはジアリール複素環であることを特徴とする請求項1〜5のいずれかに記載の使用方法。
- 前記COX−2阻害剤が、一般式Aの化合物またはその誘導体あるいは製薬的に許容しうるその塩であることを特徴とする請求項1〜6のいずれかに記載の使用方法;
Xは、酸素原子もしくは硫黄原子またはアルキル基、好ましくは−CH2−基を表し、
R1は、必要に応じて1以上の基または原子、好ましくは1以上のハロゲン原子で置換されていてもよいシクロアルキル基あるいはアリール基を表し、
R2、R3、R4およびR5は、それぞれ独立に水素原子、ニトロ基、アシル基、または必要に応じて1以上の基もしくは原子で置換されていてもよいアルキル基、あるいはR2とR3、R3とR4、またはR4とR5とが互いに炭素原子を介してシクロペンタノン基を形成してもよい。 - Xが酸素原子であることを特徴とする請求項7に記載の使用方法。
- R1がアリール基、または1以上のフッ素原子で置換されたアリール基、あるいはシクロアルキル基であることを特徴とする請求項7または8に記載の使用方法。
- R2およびR3が水素原子であり、R4が−NO2基または−COCH3基であることを特徴とする請求項7〜9のいずれかに記載の使用方法。
- R2が水素原子であり、R3とR4とがシクロペンタノン基を形成していることを特徴とする請求項7〜10のいずれかに記載の使用方法。
- 前記COX−2阻害剤が、フロスリド(flosulide)、NS−398 、ニメスリド、FK’ 3311、またはL−745 337であることを特徴とする請求項7〜11のいずれかに記載の使用方法。
- 前記COX−2阻害剤が、一般式Bの化合物またはその誘導体または製薬的に許容しうるその塩であることを特徴とする請求項1〜6のいずれかに記載の使用方法;
Yは、
環状基を、好ましくは、オキサゾリル、イソキサゾリル、チエニル、ジヒドロフリル、フリル、ピロリル、ピラゾリル、チアゾリル、イミダゾリル、イソチアゾリル、シクロペンテニル、フェニルまたはピリジル基を表し、
nは、0〜3の整数であり、
mは、0〜4の整数であり、
R6は、環状ケト基(ketocyclyl)、シクロアルキル基またはアリール基を表し、これらの基は、必要に応じて1以上の基または原子、好ましくは1以上のハロゲン原子で置換されていてもよく、
R7は、それぞれ独立に任意の官能基でもよい置換基を表し、好ましくは、水素原子またはハロゲン原子またはアルキル基であり、アルキル基は1以上の基または原子で置換されていてもよく、
R8は、アルキル基を表し、
R9は、ハロゲン原子を表し、
R10は、水素原子、または必要に応じて1以上の基または原子で置換されたアルキル基、好ましくはアシル基で置換されたアルキル基を表す。 - R8が−NH2または−CH3であることを特徴とする請求項13に記載の使用方法。
- Yがピラゾリル基、フリル基またはチエニル基であることを特徴とする請求項13または14に記載の使用方法。
- R6が必要に応じて1以上のフッ素原子で置換されたアリール基であることを特徴とする請求項13〜15のいずれかに記載の使用方法。
- nが1または2であることを特徴とする請求項13〜16のいずれかに記載の使用方法。
- R7が、臭素原子、アシル基、または置換されたアルキル基であることを特徴とする請求項13〜17のいずれかに記載の使用方法。
- 前記COX−2阻害剤が、セレコキシブ、ロフェコキシブ、 DuP−697、 SC−58125、 DFP、 DFU、 CGP 28232 または MF tricyclicであることを特徴とする請求項13〜18のいずれかに記載の使用方法。
- 前記COX−2阻害剤が、非ステロイド系抗炎症剤(NSAID)誘導体であることを特徴とする請求項1〜6のいずれかに記載の使用方法。
- 前記COX−2阻害剤が、ジイソプロピルフルオロホスフェート、L−745337、ロフェコキシブ、NS 398、SC 58125、 エトドラク、メロキシカム、セレコキシブまたはニメスリドであることを特徴とする請求項1〜20のいずれかに記載の使用方法。
- 前記COX−2阻害剤が、ロフェコキシブであることを特徴とする請求項21に記載の使用方法。
- 前記COX−2阻害剤が、セレコキシブであることを特徴とする請求項21に記載の使用方法。
- 前記COX−2阻害剤が、メロキシカムであることを特徴とする請求項21に記載の使用方法。
- 請求項1〜24のいずれかに記載のCOX−2阻害剤またはその誘導体または製薬的に許容しうるその塩、ならびに製薬的に許容しうる希釈剤、担体、または賦型剤を含有する医薬組成物。
- 1以上の追加のCOX−2阻害剤、その誘導体、または製薬的に許容しうるその塩、および/または、1以上の追加の活性成分をさらに含有する請求項25に記載の医薬組成物。
- 医薬として、好ましくは、請求項1〜4のいずれかに記載された疾患の治療または予防のために使用される請求項25または26に記載の医薬組成物。
- 請求項1〜24のいずれかに記載のCOX−2阻害剤またはその誘導体あるいは製薬的に許容しうるその塩、ならびに1以上の追加のCOX−2阻害剤、その誘導体、または製薬的に許容しうるその塩、および/または1以上の追加の活性成分を含有し、請求項1〜4のいずれかに記載された疾患の治療または予防において、同時または別個または逐次に使用される混合製剤である製品。
- 請求項1〜4のいずれかに記載された疾患の治療または予防のための医薬の調製における請求項25または26に記載の医薬組成物の使用方法。
- ヒトまたはヒト以外の動物に関し、前記動物に、請求項1〜24のいずれかに記載のCOX−2阻害剤またはその誘導体または製薬的に許容しうる塩を投与するか、請求項25または26に記載の医薬組成物を投与することを特徴とする請求項1〜4のいずれかに記載の疾患を治療する方法または予防の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0017908A GB0017908D0 (en) | 2000-07-20 | 2000-07-20 | Method |
| GB0109648A GB0109648D0 (en) | 2001-04-19 | 2001-04-19 | Method |
| PCT/GB2001/003284 WO2002007721A2 (en) | 2000-07-20 | 2001-07-20 | Use of cox-2 inhibitors for preventing immunodeficiency |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004510705A true JP2004510705A (ja) | 2004-04-08 |
| JP2004510705A5 JP2004510705A5 (ja) | 2005-02-17 |
Family
ID=26244695
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002513457A Pending JP2004510705A (ja) | 2000-07-20 | 2001-07-20 | 方法 |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US7790738B2 (ja) |
| EP (1) | EP1303265B1 (ja) |
| JP (1) | JP2004510705A (ja) |
| CN (1) | CN100488500C (ja) |
| AP (1) | AP1780A (ja) |
| AT (1) | ATE366569T1 (ja) |
| AU (1) | AU7090201A (ja) |
| CA (1) | CA2415577C (ja) |
| CY (1) | CY1107069T1 (ja) |
| CZ (1) | CZ302448B6 (ja) |
| DE (1) | DE60129330T2 (ja) |
| DK (1) | DK1303265T3 (ja) |
| ES (1) | ES2290157T3 (ja) |
| HK (1) | HK1055087A1 (ja) |
| HU (1) | HUP0302068A3 (ja) |
| NO (1) | NO330509B1 (ja) |
| NZ (1) | NZ524252A (ja) |
| OA (1) | OA12339A (ja) |
| PL (1) | PL359556A1 (ja) |
| PT (1) | PT1303265E (ja) |
| RU (1) | RU2303452C2 (ja) |
| WO (1) | WO2002007721A2 (ja) |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1284729A4 (en) * | 2000-04-13 | 2007-12-19 | Mayo Foundation | REDUCTION AGENTS OF A (BETA) 42 |
| PL359556A1 (en) | 2000-07-20 | 2004-08-23 | Lauras As | Method - use of cox-2 inhibitors for preventing immunodeficiency |
| GB0221443D0 (en) | 2002-09-16 | 2002-10-23 | Glaxo Group Ltd | Pyridine derivates |
| WO2004071431A2 (en) * | 2003-02-05 | 2004-08-26 | Myriad Genetics, Inc. | Method and composition for treating neurodegenerative disorders |
| US20050042284A1 (en) * | 2003-07-11 | 2005-02-24 | Myriad Genetics, Incorporated | Pharmaceutical methods, dosing regimes and dosage forms for the treatment of Alzheimer's disease |
| WO2005013902A2 (en) * | 2003-08-04 | 2005-02-17 | Mayo Foundation For Medical Education And Research | Methods and compositions for inhibiting the proliferation of prostate cancer cells |
| US20070293538A1 (en) * | 2004-04-13 | 2007-12-20 | Myriad Genetics, Incorporated | Pharmaceutical Composition And Methods For Treating Neurodegenerative Disorders |
| US20050252144A1 (en) * | 2004-04-29 | 2005-11-17 | Macdonald Robert A | Veneers for walls, retaining walls and the like |
| WO2006020850A2 (en) * | 2004-08-11 | 2006-02-23 | Myriad Genetics, Inc. | Pharmaceutical composition and method for treating neurodegenerative disorders |
| WO2006020853A2 (en) * | 2004-08-11 | 2006-02-23 | Myriad Genetics, Inc. | Pharmaceutical composition and method for treating neurodegenerative disorders |
| WO2006020852A2 (en) * | 2004-08-11 | 2006-02-23 | Myriad Genetics, Inc. | Pharmaceutical composition and method for treating neurodegenerative disorders |
| CA2615063A1 (en) * | 2005-07-22 | 2007-02-01 | Myriad Genetics, Inc. | High drug load formulations and dosage forms |
| US7713960B2 (en) * | 2005-07-22 | 2010-05-11 | University Of South Florida | Inhibition of the Raf/Mek/P-Erk pathway for treating cancer |
| US20070281927A1 (en) * | 2006-06-06 | 2007-12-06 | Shanthakumar Tyavanagimatt | Anti-inflammatory and analgesic compositions and related methods |
| EP2046119A2 (en) * | 2006-07-07 | 2009-04-15 | Myriad Genetics, Inc. | Treatment of psychiatric disorders |
| TWI466682B (zh) | 2006-12-15 | 2015-01-01 | Boehringer Ingelheim Vetmed | 以豬第二型環狀病毒(pcv2)抗原治療豬 |
| EP1941903A1 (en) | 2007-01-03 | 2008-07-09 | Boehringer Ingelheim Vetmedica Gmbh | Prophylaxis and treatment of PRDC |
| EP1958644A1 (en) | 2007-02-13 | 2008-08-20 | Boehringer Ingelheim Vetmedica Gmbh | Prevention and treatment of sub-clinical pcvd |
| EP3662927A3 (en) | 2013-02-05 | 2020-10-21 | Nitto Denko Corporation | Vaccine composition |
| CN108849912B (zh) * | 2018-07-26 | 2020-06-30 | 河南省农业科学院植物保护研究所 | 1,5-二芳基-3-三氟甲基吡唑类化合物在防治农业真菌病害中的应用 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996011002A1 (fr) * | 1994-10-05 | 1996-04-18 | Hisamitsu Pharmaceutical Co., Inc. | Agent anti-inflammatoire a usage externe |
Family Cites Families (138)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US1325832A (en) * | 1919-12-23 | Beak-axle mounting eor tractors | ||
| US2011239A (en) * | 1933-07-31 | 1935-08-13 | Packard Motor Car Co | Motor vehicle |
| US2163981A (en) * | 1938-12-06 | 1939-06-27 | Norman E Lawrence | Rear wheel suspension for motor vehicles |
| US2606036A (en) * | 1945-11-01 | 1952-08-05 | Six Wheels Inc | Mounting for wheel brake operating mechanism |
| US2635896A (en) * | 1949-07-26 | 1953-04-21 | Brown Trailers Inc | Vehicle wheel suspension |
| US2877010A (en) * | 1956-10-16 | 1959-03-10 | Gouirand Rene | Pneumatic suspension for motor vehicles |
| US2913252A (en) * | 1957-08-02 | 1959-11-17 | Pacific Car & Foundry Co | Vehicle suspension using air spring |
| NL232523A (ja) * | 1957-11-27 | |||
| DK105852A (ja) * | 1961-05-11 | |||
| US3434707A (en) * | 1966-06-02 | 1969-03-25 | John E Raidel | Suspensions |
| FR1573973A (ja) * | 1968-03-28 | 1969-07-11 | ||
| US3547215A (en) * | 1968-06-07 | 1970-12-15 | Neway Equipment Co | Automotive vehicle suspension structure |
| US3630541A (en) * | 1970-06-11 | 1971-12-28 | Int Harvester Co | Vehicle axle suspension |
| US3707298A (en) * | 1970-10-06 | 1972-12-26 | Lear Siegler Inc | Suspension structure for land vehicles |
| US3785673A (en) * | 1971-09-20 | 1974-01-15 | Western Unit Corp | Preloaded spring beam and method employed alone or in combination with air bellows spring |
| US3771812A (en) * | 1972-04-25 | 1973-11-13 | Lear Siegler Inc | Air suspension with improved axle lifting structure |
| US3961826A (en) * | 1975-03-06 | 1976-06-08 | American Carrier Equipment, Inc. | Suspension unit |
| DE2523121C2 (de) * | 1975-05-24 | 1984-06-20 | Volkswagenwerk Ag, 3180 Wolfsburg | Hinterachsaufhängung für Kraftfahrzeuge |
| US4027898A (en) * | 1976-04-07 | 1977-06-07 | Southwest Wheel And Manufacturing Company | Level load leaf spring drop axle |
| US4065153A (en) * | 1976-08-19 | 1977-12-27 | United States Steel Corporation | Vehicle wheel suspension assembly |
| US4174855A (en) * | 1977-01-13 | 1979-11-20 | Turner Quick-Lift Corporation | Wheeled vehicle axle suspension system |
| US4352509A (en) * | 1978-01-18 | 1982-10-05 | H. Neil Paton | Damped rubber tired vehicle suspension |
| US4293145A (en) * | 1978-02-28 | 1981-10-06 | Taylor Glenn E | Auxiliary lift axle wheel assembly |
| US4166640B1 (en) * | 1978-04-14 | 1994-03-22 | Boler Company Inc | Axle suspension for wheeled vehicles |
| US4371190A (en) * | 1980-01-28 | 1983-02-01 | Turner Quick-Lift Corporation | Axle suspension system |
| US4310171A (en) * | 1980-06-11 | 1982-01-12 | General Motors Corporation | Vehicle axle attachment |
| US4415179A (en) * | 1981-04-15 | 1983-11-15 | Marinelli Joseph A | Axle and air bag suspension |
| US4529224A (en) * | 1981-05-21 | 1985-07-16 | Raidel John E | Wide base air spring suspension assembly |
| US4537420A (en) * | 1981-07-28 | 1985-08-27 | Nissan Motor Company, Limited | Swing-arm-type suspension with a lateral rod for an automotive vehicle |
| US4427213A (en) * | 1982-01-21 | 1984-01-24 | Raidel Jr John E | Vehicle suspension with rigid torque beam |
| US4494771A (en) * | 1982-01-21 | 1985-01-22 | Raidel John E | Suspension system with U-joint mounted load carrying torque arm |
| US4504080A (en) * | 1983-05-16 | 1985-03-12 | Turner Quick-Lift Corporation | Leaf spring lift axle suspension system |
| US4541643A (en) * | 1983-06-27 | 1985-09-17 | Ivan Pavincic | Two wheel skating device |
| US4566719A (en) * | 1984-02-17 | 1986-01-28 | Turner Quick Lift Corporation | Spaced axle-to-beam connection for suspension of the rigid beam type |
| US4693486A (en) * | 1986-04-09 | 1987-09-15 | Lear Siegler, Inc. | Trailing arm suspension with wrapper compression axle mounting |
| US4691937A (en) * | 1986-05-19 | 1987-09-08 | Raidel John E | Vehicle suspension assembly |
| US5002305A (en) * | 1987-02-04 | 1991-03-26 | Raidel John E | Vehicle suspension system with standardized torque beam and special monopivot bushing assembly |
| US4902035A (en) * | 1987-02-04 | 1990-02-20 | Raidel John E | Suspension system with axle seat removable from universal torque beam |
| US4763923A (en) * | 1987-03-02 | 1988-08-16 | Raidel John E | Suspension assembly with air spring and self contained air lift spring |
| US4878691A (en) * | 1988-03-23 | 1989-11-07 | Dbx Corporation | Trailer suspension apparatus |
| GB8814057D0 (en) * | 1988-06-14 | 1988-07-20 | Lundbeck & Co As H | New enantiomers & their isolation |
| US4943081A (en) * | 1989-02-10 | 1990-07-24 | Rancho Industries, Inc. | Vehicle suspension assembly |
| DE8915893U1 (ja) * | 1989-04-26 | 1991-11-28 | Bergische Achsenfabrik Fr. Kotz & Soehne, 5276 Wiehl, De | |
| US4991868A (en) * | 1989-12-19 | 1991-02-12 | The Boler Company | Vehicle suspension beam pivot connection |
| US5504108A (en) | 1990-01-12 | 1996-04-02 | The Ohio State University Research Foundation | Optically pure 4-aryl-2-hydroxytetronic acids |
| US5058916A (en) * | 1990-06-18 | 1991-10-22 | Lear Siegler Truck Products Corp. | Apparatus for providing additional lift in a retractable suspension |
| US5037126A (en) * | 1990-09-07 | 1991-08-06 | The Boler Company | Lightweight beam suspension system |
| KR930703321A (ko) | 1990-12-13 | 1993-11-29 | 스튜어트 알. 슈터 | 신규한 사이토킨 억제성 소염 의약 |
| US5112078A (en) * | 1990-12-21 | 1992-05-12 | Neway Corp. | Axle mounting assembly |
| US5127668A (en) * | 1991-01-24 | 1992-07-07 | Raidel John E | Torque beam with clamped mono-pivot bushing and flexible axle seat |
| US5203585A (en) * | 1991-07-12 | 1993-04-20 | Neway Corp. | Split-beam suspension system |
| US5171036A (en) * | 1991-08-27 | 1992-12-15 | The Boler Company | Rebound strap |
| US5240929A (en) | 1992-08-03 | 1993-08-31 | Warner-Lambert Company | 2-heterocyclic-5-hydroxy-1,3-pyrimidines useful as antiinflammatory agents |
| AU666572B2 (en) * | 1992-10-19 | 1996-02-15 | Hendrickson International Corporation | Axle suspension systems |
| ATE135697T1 (de) | 1992-10-28 | 1996-04-15 | Shionogi & Co | Benzylidenderivate |
| JP3465277B2 (ja) * | 1992-11-26 | 2003-11-10 | トヨタ自動車株式会社 | アンダボデー構造 |
| US5604260A (en) | 1992-12-11 | 1997-02-18 | Merck Frosst Canada Inc. | 5-methanesulfonamido-1-indanones as an inhibitor of cyclooxygenase-2 |
| ATE160345T1 (de) | 1993-01-15 | 1997-12-15 | Searle & Co | 3,4-diarylthiophene und analoga davon, sowie deren verwendung als entzündungshemmende mittel |
| US5409944A (en) | 1993-03-12 | 1995-04-25 | Merck Frosst Canada, Inc. | Alkanesulfonamido-1-indanone derivatives as inhibitors of cyclooxygenase |
| EP0701570B1 (en) | 1993-05-18 | 2001-12-19 | Korea Institute Of Science And Technology | Thermoresistant alpha-1-antitrypsin mutein |
| US5380738A (en) | 1993-05-21 | 1995-01-10 | Monsanto Company | 2-substituted oxazoles further substituted by 4-fluorophenyl and 4-methylsulfonylphenyl as antiinflammatory agents |
| WO1994029724A1 (en) * | 1993-06-11 | 1994-12-22 | Coulter Corporation | Anti-cd3 antibody-aminodextran conjugates for induction of t-cell activation and proliferation |
| US5474995A (en) | 1993-06-24 | 1995-12-12 | Merck Frosst Canada, Inc. | Phenyl heterocycles as cox-2 inhibitors |
| US5375871A (en) * | 1993-07-02 | 1994-12-27 | Ridewell Corporation | Vehicle suspension system comprising a wide base beam and axle shell |
| US5593992A (en) | 1993-07-16 | 1997-01-14 | Smithkline Beecham Corporation | Compounds |
| US5344991A (en) | 1993-10-29 | 1994-09-06 | G.D. Searle & Co. | 1,2 diarylcyclopentenyl compounds for the treatment of inflammation |
| US5475018A (en) | 1993-11-30 | 1995-12-12 | G. D. Searle & Co. | 1,5-diphenyl pyrazole compounds for treatment of inflammation |
| RU2139281C1 (ru) | 1993-11-30 | 1999-10-10 | Джи Ди Сирл энд Компани | Пиразолилзамещенный бензолсульфонамид или его фармацевтически приемлемая соль, фармацевтическая композиция, способ лечения от воспаления или связанного с воспалением заболевания |
| US5434178A (en) | 1993-11-30 | 1995-07-18 | G.D. Searle & Co. | 1,3,5 trisubstituted pyrazole compounds for treatment of inflammation |
| US5403031A (en) * | 1993-12-08 | 1995-04-04 | The Boler Company | Parallelogram lift axle suspension system with a control for axle caster adjustment |
| US5393790A (en) | 1994-02-10 | 1995-02-28 | G.D. Searle & Co. | Substituted spiro compounds for the treatment of inflammation |
| DE69509223T2 (de) | 1994-02-10 | 1999-09-16 | Searle & Co | Substituierte spiroverbindungen zur behandlung von entzündungen |
| US5639110A (en) * | 1994-04-14 | 1997-06-17 | Nai Neway, Inc. | Trailing arm suspension |
| US5643960A (en) | 1994-04-15 | 1997-07-01 | Duke University | Method of delaying onset of alzheimer's disease symptoms |
| US5486534A (en) | 1994-07-21 | 1996-01-23 | G. D. Searle & Co. | 3,4-substituted pyrazoles for the treatment of inflammation |
| EP1125932A3 (en) | 1994-07-27 | 2001-08-29 | G.D. Searle & Co. | Substituted thiazoles for the treatment of inflammation |
| US5616601A (en) | 1994-07-28 | 1997-04-01 | Gd Searle & Co | 1,2-aryl and heteroaryl substituted imidazolyl compounds for the treatment of inflammation |
| US5620999A (en) | 1994-07-28 | 1997-04-15 | Weier; Richard M. | Benzenesulfonamide subtituted imidazolyl compounds for the treatment of inflammation |
| GB9415529D0 (en) | 1994-08-01 | 1994-09-21 | Wellcome Found | Phenyl xanthine derivatives |
| US5464245A (en) * | 1994-08-08 | 1995-11-07 | The Boler Company | Suspension for light duty trucks |
| US5521213A (en) | 1994-08-29 | 1996-05-28 | Merck Frosst Canada, Inc. | Diaryl bicyclic heterocycles as inhibitors of cyclooxygenase-2 |
| US5739166A (en) | 1994-11-29 | 1998-04-14 | G.D. Searle & Co. | Substituted terphenyl compounds for the treatment of inflammation |
| DE4442560A1 (de) | 1994-11-30 | 1996-06-05 | Basf Ag | Iminooxybenzylcrotonsäureester, Verfahren zu ihrer Herstellung und ihre Verwendung |
| RO119946B1 (ro) | 1995-02-13 | 2005-06-30 | G.D. Searle & Co. | Derivaţi de izoxazol, utilizaţi pentru tratamentul inflamaţiilor |
| US5603724A (en) | 1995-02-13 | 1997-02-18 | Tnco, Inc. | Suction punch |
| JPH11501049A (ja) | 1995-04-04 | 1999-01-26 | グラクソ、グループ、リミテッド | イミダゾ〔1,2−a〕ピリジン誘導体 |
| US5720489A (en) * | 1995-05-17 | 1998-02-24 | The Boler Company | Movable subframe for tractor-trailers |
| US5510368A (en) | 1995-05-22 | 1996-04-23 | Merck Frosst Canada, Inc. | N-benzyl-3-indoleacetic acids as antiinflammatory drugs |
| GB9514518D0 (en) | 1995-07-15 | 1995-09-13 | Sod Conseils Rech Applic | Guanidine salt inhibitors of NO synthase and cyclooxygenase |
| WO1998041516A1 (en) | 1997-03-14 | 1998-09-24 | Merck Frosst Canada & Co. | (methylsulfonyl)phenyl-2-(5h)-furanones with oxygen link as cox-2 inhibitors |
| US5785345A (en) * | 1995-10-16 | 1998-07-28 | The Boler Company | Means for and method of controlling frame rise in vehicle suspensions |
| ZA97175B (en) | 1996-01-11 | 1997-11-04 | Smithkline Beecham Corp | Novel substituted imidazole compounds. |
| CA2246336A1 (en) * | 1996-02-13 | 1997-08-21 | G.D. Searle & Co. | Combinations, having immunosuppressive effects, containing a cyclooxygenase-2 inhibitor and a leukotriene a4 hydrolase inhibitor |
| AP1009A (en) | 1996-04-12 | 2001-09-21 | Searle & Co | Substituted benzenesulfonamide derivatives as products of COX-2 inhibitors. |
| US5690353A (en) * | 1996-05-09 | 1997-11-25 | Suspensions Incorporated | Suspension system with improved beam |
| US5950971A (en) * | 1996-06-28 | 1999-09-14 | The Boler Company | Assembly for and method of mounting a suspension member to an axle housing |
| WO1998001443A1 (en) | 1996-07-09 | 1998-01-15 | Smithkline Beecham S.P.A. | Indole derivatives for the treatment of osteoporosis |
| JP2001506230A (ja) | 1996-08-09 | 2001-05-15 | スミスクライン・ビーチャム・コーポレイション | 新規ピペラジン含有化合物 |
| US6005000A (en) | 1996-08-22 | 1999-12-21 | Oxis International, Inc. | 5,5-Disubstituted-3, 4-dihydroxy-2(5H)-furanones and methods of use therefor |
| US5996981A (en) * | 1996-08-28 | 1999-12-07 | The Boler Company | Reduced size bushing for beam-type axle suspension system |
| DE29616257U1 (de) * | 1996-09-19 | 1996-11-07 | Sauer Achsenfab | Aufhängung für luftgefederte Fahrzeugradachse |
| ITMI962356A1 (it) | 1996-11-13 | 1998-05-13 | Uni Degli Studi Di Brescia D I | Uso di composti derivati da molecole ad attivita' antinfiammatoria di tipo non steroideo per la prevenzione e il trattamento di |
| US5921570A (en) * | 1996-11-21 | 1999-07-13 | The Boler Company | Weld-on axle bracket with U-bolt connection |
| JP4167733B2 (ja) | 1996-12-16 | 2008-10-22 | 花王株式会社 | NF−κB活性化抑制剤 |
| US5810377A (en) * | 1997-01-21 | 1998-09-22 | The Boler Company | Fabricated steer axle |
| ES2224366T3 (es) | 1997-03-14 | 2005-03-01 | MERCK FROSST CANADA & CO. | Piridazinonas como inhibidores de ciclooxigenasa-2. |
| EP0971910A1 (en) | 1997-04-02 | 2000-01-19 | Merck Frosst Canada & Co. | Alpha-methylene gamma lactones as selective cyclooxygenase-2 inhibitors |
| SE9701304D0 (sv) | 1997-04-09 | 1997-04-09 | Astra Pharma Prod | Compounds |
| TW492959B (en) | 1997-04-18 | 2002-07-01 | Merck & Co Inc | Process for making 2-aryl-3-aryl-5-halo pyridines useful as cox-2 inhibitors |
| US5944339A (en) * | 1997-05-06 | 1999-08-31 | Meritor Heavy Vehicle Systems, Llc | Integrated axle suspension anti-roll arrangement for push-pull suspension |
| CA2240791A1 (en) * | 1997-07-16 | 1999-01-16 | Neway Anchorlok International, Inc | Trailing arm suspension |
| US5988672A (en) * | 1997-08-11 | 1999-11-23 | Meritor Heavy Vehicle Suspension Systems, Inc. | Suspension system with integral box beam |
| US5887881A (en) * | 1997-08-11 | 1999-03-30 | The Boler Company. | Leaf spring attachment member |
| CN1155600C (zh) | 1997-09-05 | 2004-06-30 | 葛兰素集团有限公司 | 2,3-二芳基吡唑并[1,5-b]哒嗪衍生物,其制备方法和用作环氧酶2抑制剂 |
| AU741755B2 (en) | 1997-09-12 | 2001-12-06 | Merck Frosst Canada Ltd. | 2,3,5-trisubstituted pyridines as inhibitors of cyclooxygenase-2 |
| JP4368524B2 (ja) | 1997-09-12 | 2009-11-18 | メルク フロスト カナダ リミテツド | シクロオキシゲナーゼ−2阻害薬としての2−アミノピリジン類 |
| US6039336A (en) * | 1997-09-19 | 2000-03-21 | Otto Sauer Achsenfabrik Keilberg | Vehicle axle suspension assembly |
| EP1017687A4 (en) | 1997-09-24 | 2001-10-31 | Merck & Co Inc | METHOD FOR PRODUCING 3-ARYLOXY-4-ARYL-FURAN-2-ONE AS INHIBITORS OF COX-2 |
| AR015938A1 (es) | 1997-09-25 | 2001-05-30 | Merck Sharp & Dohme | Procedimiento para preparar diaril piridinas utiles como inhibidores cox-2 y compuesto intermediario |
| AU753657B2 (en) | 1997-10-22 | 2002-10-24 | Merck & Co., Inc. | Combination therapy for reducing the risks associated with cardio-and cerebrovascular disease |
| AU741790B2 (en) | 1997-10-30 | 2001-12-06 | Merck Frosst Canada & Co. | Diaryl-5-alkyl-5-methyl-2(5H)-furanones as selective cyclooxygenase-2 inhibitors |
| US6025353A (en) | 1997-11-19 | 2000-02-15 | G.D. Searle & Co. | Method of using cyclooxygenase-2 inhibitors as anti-angiogenic agents |
| US5938221A (en) * | 1997-12-08 | 1999-08-17 | The Boler Company | Tapered convolute leaf spring for truck suspensions |
| JP2002515891A (ja) | 1997-12-19 | 2002-05-28 | スミスクライン・ビーチャム・コーポレイション | 新規なピペリジン含有化合物 |
| EP1089889B1 (en) * | 1998-07-02 | 2003-02-26 | The Boler Company. | Trailing arm axle/suspension system |
| US6420403B1 (en) * | 1998-10-29 | 2002-07-16 | Edwin J. Iwanowicz | Inhibitors of IMPDH enzyme |
| US6209895B1 (en) * | 1998-12-03 | 2001-04-03 | Reyco Industries, Inc. | Axle suspension system for a wheeled vehicle |
| WO2000040243A1 (en) * | 1999-01-08 | 2000-07-13 | Smithkline Beecham Corporation | Novel compounds |
| US6123349A (en) * | 1999-04-09 | 2000-09-26 | Standen's Limited | Vehicle air suspension system |
| US20030138399A1 (en) * | 1999-05-14 | 2003-07-24 | Anton Peter A. | Anti-inflammatory therapy for inflammatory mediated infection |
| CN1353573A (zh) * | 1999-05-14 | 2002-06-12 | 加利福尼亚大学董事会 | 对炎症介导感染的抗炎症治疗 |
| HUP0200580A3 (en) | 1999-12-08 | 2002-12-28 | Pharmacia Corp Chicago | Polymorphic crystalline forms of celecoxib, process their preparation and pharmaceutical compositions containing them |
| US6264231B1 (en) * | 2000-03-20 | 2001-07-24 | The Boler Company | Axle suspension connection |
| ES2249426T3 (es) * | 2000-03-22 | 2006-04-01 | Hendrickson International Corporation | Dispositivo de suspension de chasis en aluminio para sistemas de eje/sispension. |
| US6306842B1 (en) * | 2000-06-02 | 2001-10-23 | Medinox, Inc. | Protected forms of a combination of pharmacologically active agents and uses therefor |
| PL359556A1 (en) | 2000-07-20 | 2004-08-23 | Lauras As | Method - use of cox-2 inhibitors for preventing immunodeficiency |
| GB0021716D0 (en) * | 2000-09-05 | 2000-10-18 | Meritor Heavy Vehicle Sys Ltd | Vehicle suspension axle wrap |
| US7007960B2 (en) * | 2002-02-01 | 2006-03-07 | Watson & Chalin Manufacturing, Inc. | Suspension system having reduced stress axle connection |
-
2001
- 2001-07-20 PL PL01359556A patent/PL359556A1/xx not_active Application Discontinuation
- 2001-07-20 AU AU7090201A patent/AU7090201A/xx not_active Withdrawn
- 2001-07-20 NZ NZ524252A patent/NZ524252A/en unknown
- 2001-07-20 PT PT01949787T patent/PT1303265E/pt unknown
- 2001-07-20 WO PCT/GB2001/003284 patent/WO2002007721A2/en active IP Right Grant
- 2001-07-20 AT AT01949787T patent/ATE366569T1/de not_active IP Right Cessation
- 2001-07-20 OA OA1200300011A patent/OA12339A/en unknown
- 2001-07-20 CN CNB018143199A patent/CN100488500C/zh not_active Expired - Fee Related
- 2001-07-20 JP JP2002513457A patent/JP2004510705A/ja active Pending
- 2001-07-20 CZ CZ20030177A patent/CZ302448B6/cs not_active IP Right Cessation
- 2001-07-20 US US10/333,657 patent/US7790738B2/en not_active Expired - Fee Related
- 2001-07-20 AP APAP/P/2003/002719A patent/AP1780A/en active
- 2001-07-20 HU HU0302068A patent/HUP0302068A3/hu unknown
- 2001-07-20 ES ES01949787T patent/ES2290157T3/es not_active Expired - Lifetime
- 2001-07-20 CA CA2415577A patent/CA2415577C/en not_active Expired - Fee Related
- 2001-07-20 RU RU2003104981/15A patent/RU2303452C2/ru not_active IP Right Cessation
- 2001-07-20 EP EP01949787A patent/EP1303265B1/en not_active Expired - Lifetime
- 2001-07-20 DK DK01949787T patent/DK1303265T3/da active
- 2001-07-20 DE DE60129330T patent/DE60129330T2/de not_active Expired - Lifetime
-
2003
- 2003-01-20 NO NO20030276A patent/NO330509B1/no not_active IP Right Cessation
- 2003-10-15 HK HK03107425A patent/HK1055087A1/xx not_active IP Right Cessation
-
2007
- 2007-10-08 CY CY20071101278T patent/CY1107069T1/el unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996011002A1 (fr) * | 1994-10-05 | 1996-04-18 | Hisamitsu Pharmaceutical Co., Inc. | Agent anti-inflammatoire a usage externe |
Non-Patent Citations (4)
| Title |
|---|
| AMBRUS,J.L. ET AL.: "Mechanism(s) of interferon inhibitory activity in blood from patients with AIDS and patients with lu", RES. COMMUN. MOL. PATHOL. PHARMACOL., vol. 96, no. 3, JPN6010063491, June 1997 (1997-06-01), pages 255 - 265, ISSN: 0001777095 * |
| EHRICH,E.W. ET AL.: "Characterization of rofecoxib as a cyclooxygenase-2 isoform inhibitor and demonstration of analgesia", CLIN. PHARMACOL. THER., vol. 65, no. 3, JPN6010063493, March 1999 (1999-03-01), pages 336 - 347, XP005168135, ISSN: 0001777096, DOI: 10.1016/S0009-9236(99)70113-X * |
| ENGELHARDT,G. ET AL.: "Meloxican: Influence on arachidonic acid metabolism", BIOCHEM. PHARMACOL., vol. 51, no. 1, JPN6010063496, 12 January 1996 (1996-01-12), pages 29 - 38, ISSN: 0001777098 * |
| WAKITANI,K. ET AL.: "Profile of JTE-522 as a human cyclooxyganase-2 inhibitor", JPN. J. PHARMACOL., vol. 78, no. 3, JPN6010063494, November 1998 (1998-11-01), JP, pages 365 - 371, ISSN: 0001777097 * |
Also Published As
| Publication number | Publication date |
|---|---|
| US20040082640A1 (en) | 2004-04-29 |
| HUP0302068A2 (hu) | 2003-11-28 |
| PT1303265E (pt) | 2007-10-09 |
| EP1303265A2 (en) | 2003-04-23 |
| OA12339A (en) | 2006-05-15 |
| ES2290157T3 (es) | 2008-02-16 |
| NO20030276L (no) | 2003-03-18 |
| HUP0302068A3 (en) | 2005-05-30 |
| AP1780A (en) | 2007-09-26 |
| WO2002007721A3 (en) | 2002-04-18 |
| RU2303452C2 (ru) | 2007-07-27 |
| CN100488500C (zh) | 2009-05-20 |
| ATE366569T1 (de) | 2007-08-15 |
| DE60129330T2 (de) | 2008-04-10 |
| AP2003002719A0 (en) | 2003-06-30 |
| NO20030276D0 (no) | 2003-01-20 |
| HK1055087A1 (en) | 2003-12-24 |
| WO2002007721A2 (en) | 2002-01-31 |
| CZ302448B6 (cs) | 2011-05-25 |
| DK1303265T3 (da) | 2007-11-12 |
| CY1107069T1 (el) | 2012-10-24 |
| NO330509B1 (no) | 2011-05-09 |
| CA2415577A1 (en) | 2002-01-31 |
| EP1303265B1 (en) | 2007-07-11 |
| PL359556A1 (en) | 2004-08-23 |
| CN1561204A (zh) | 2005-01-05 |
| AU7090201A (en) | 2002-02-05 |
| NZ524252A (en) | 2004-03-26 |
| CA2415577C (en) | 2010-10-19 |
| DE60129330D1 (de) | 2007-08-23 |
| US7790738B2 (en) | 2010-09-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2415577C (en) | Use of cox-2 inhibitors for preventing immunodeficiency | |
| Chen et al. | Predominance of cyclooxygenase 1 over cyclooxygenase 2 in the generation of proinflammatory prostaglandins in autoantibody‐driven K/BxN serum–transfer arthritis | |
| JP5460600B2 (ja) | 肥満症治療における肥満細胞安定化薬 | |
| KR20010089363A (ko) | 편두통 치료를 위한 5에이치티1 수용체 작용제 및메토클로프라미드 | |
| ES2371552T3 (es) | Uso de antagonistas del receptor cb1 para la fabricación de una composición útil para el tratamiento de enfermedades hepáticas. | |
| AU2002253181B2 (en) | Use of selective COX-2 inhibitors for the treatment of urinary incontinence | |
| JP2014522844A (ja) | 白血病を治療するための組成物、方法及びキット | |
| Mazaleuskaya et al. | Druggable prostanoid pathway | |
| KR20100032854A (ko) | 아스피린 민감성 및 다른 군의 심혈관 질환 치료를 위한 tp 조절자의 용도 | |
| JP2020516689A (ja) | 急性肺傷害を処置または予防する化合物、組成物および方法 | |
| WO2006049215A1 (ja) | 自己免疫疾患を治療するための併用薬 | |
| US20070072937A1 (en) | Pharmaceutical composition comprising alpha-lipoic acid for inflammatory diseases | |
| Rahmouni et al. | Cyclo-oxygenase type 2-dependent prostaglandin E2 secretion is involved in retrovirus-induced T-cell dysfunction in mice | |
| AU2001270902B2 (en) | Use of COX-2 inhibitors for preventing immunodeficiency | |
| US20040101902A1 (en) | Inhibition of Egr-1 expression by ppar-gamma agonists and related compositions and methods | |
| US8080579B2 (en) | Compositions and methods for treatment of inflammatory bowel disease | |
| ZA200300446B (en) | Use of cox-2 inhibitors for preventing immunodeficiency. | |
| Cahlin et al. | The effects of non-selective, preferential-selective and selective COX-inhibitors on the growth of experimental and human tumors in mice related to prostanoid receptors | |
| JP2001031568A (ja) | 片頭痛治療のための組み合わせ療法 | |
| JP2000344683A (ja) | 片頭痛の治療用薬剤組成物 | |
| CA2312631A1 (en) | 5ht1 receptor agonists, caffeine and either a cox-2 inhibitor or nsaid for the treatment of migraine | |
| CN111166756B (zh) | 20(S)-人参皂苷-Rg3在逆转胶质瘤细胞对化疗药物的耐药性中的用途 | |
| JP2007504164A (ja) | PPAR−γ経路およびHER−キナーゼ軸の調節に基づく癌および他の生理学的状態の処置のための組成物および方法 | |
| JP2021534141A (ja) | アルコール使用障害の治療剤 | |
| Potenza et al. | Endothelial cyclo-oxygenase-1 and-2 differentially affect reactivity of mesenteric vascular bed in portal hypertensive rats |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080205 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20101116 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20110203 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20110210 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20120124 |