ES2864150T3 - Conjugados de fármacos con anticuerpos anti-cMet y métodos para su uso - Google Patents
Conjugados de fármacos con anticuerpos anti-cMet y métodos para su uso Download PDFInfo
- Publication number
- ES2864150T3 ES2864150T3 ES19197371T ES19197371T ES2864150T3 ES 2864150 T3 ES2864150 T3 ES 2864150T3 ES 19197371 T ES19197371 T ES 19197371T ES 19197371 T ES19197371 T ES 19197371T ES 2864150 T3 ES2864150 T3 ES 2864150T3
- Authority
- ES
- Spain
- Prior art keywords
- cmet
- antibody
- adc
- antibodies
- days
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229940049595 antibody-drug conjugate Drugs 0.000 title description 416
- 239000000611 antibody drug conjugate Substances 0.000 title description 410
- 238000000034 method Methods 0.000 title description 74
- 239000003814 drug Substances 0.000 claims abstract description 75
- 229940079593 drug Drugs 0.000 claims abstract description 64
- IEDXPSOJFSVCKU-HOKPPMCLSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[6-(2,5-dioxopyrrolidin-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl N-[(2S)-1-[[(2S)-1-[[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-[[(1S,2R)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]-N-methylcarbamate Chemical group CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c1ccccc1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCc1ccc(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CCC2=O)C(C)C)cc1)C(C)C IEDXPSOJFSVCKU-HOKPPMCLSA-N 0.000 claims abstract description 20
- 108010093470 monomethyl auristatin E Proteins 0.000 claims abstract description 18
- 239000008194 pharmaceutical composition Substances 0.000 claims description 12
- 125000005647 linker group Chemical group 0.000 description 168
- 206010028980 Neoplasm Diseases 0.000 description 100
- 210000004027 cell Anatomy 0.000 description 95
- 238000001802 infusion Methods 0.000 description 73
- 239000000824 cytostatic agent Substances 0.000 description 71
- 239000012634 fragment Substances 0.000 description 71
- 239000002254 cytotoxic agent Substances 0.000 description 69
- 231100000433 cytotoxic Toxicity 0.000 description 66
- 230000001472 cytotoxic effect Effects 0.000 description 66
- 108090000623 proteins and genes Proteins 0.000 description 62
- 239000000427 antigen Substances 0.000 description 60
- 102000036639 antigens Human genes 0.000 description 60
- 108091007433 antigens Proteins 0.000 description 60
- 238000001990 intravenous administration Methods 0.000 description 60
- 239000000203 mixture Substances 0.000 description 55
- -1 and the like Chemical class 0.000 description 51
- 238000002560 therapeutic procedure Methods 0.000 description 49
- 206010061818 Disease progression Diseases 0.000 description 45
- 239000003795 chemical substances by application Substances 0.000 description 45
- 230000005750 disease progression Effects 0.000 description 45
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 42
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 41
- 230000000295 complement effect Effects 0.000 description 40
- 239000003112 inhibitor Substances 0.000 description 40
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 37
- 108020004414 DNA Proteins 0.000 description 36
- 201000011510 cancer Diseases 0.000 description 36
- 230000008901 benefit Effects 0.000 description 34
- 230000002055 immunohistochemical effect Effects 0.000 description 34
- 108090000765 processed proteins & peptides Proteins 0.000 description 32
- 102100024952 Protein CBFA2T1 Human genes 0.000 description 31
- 230000001225 therapeutic effect Effects 0.000 description 31
- 125000003275 alpha amino acid group Chemical group 0.000 description 29
- 230000000694 effects Effects 0.000 description 29
- 229920002857 polybutadiene Polymers 0.000 description 29
- 238000010186 staining Methods 0.000 description 29
- 230000004044 response Effects 0.000 description 26
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 25
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 24
- 102000004169 proteins and genes Human genes 0.000 description 24
- 238000011282 treatment Methods 0.000 description 24
- 108060003951 Immunoglobulin Proteins 0.000 description 23
- 230000014509 gene expression Effects 0.000 description 23
- 102000018358 immunoglobulin Human genes 0.000 description 23
- 238000002360 preparation method Methods 0.000 description 23
- 235000018102 proteins Nutrition 0.000 description 23
- 229960001433 erlotinib Drugs 0.000 description 21
- 102000004196 processed proteins & peptides Human genes 0.000 description 20
- 238000012360 testing method Methods 0.000 description 20
- 238000003556 assay Methods 0.000 description 19
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 19
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 18
- 229960003301 nivolumab Drugs 0.000 description 18
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 17
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 17
- 235000001014 amino acid Nutrition 0.000 description 17
- 229960005277 gemcitabine Drugs 0.000 description 17
- 229920001184 polypeptide Polymers 0.000 description 17
- 239000000126 substance Substances 0.000 description 17
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 16
- 229940024606 amino acid Drugs 0.000 description 15
- 150000001413 amino acids Chemical class 0.000 description 15
- 238000003776 cleavage reaction Methods 0.000 description 15
- 239000013604 expression vector Substances 0.000 description 15
- 125000000524 functional group Chemical group 0.000 description 15
- 230000007017 scission Effects 0.000 description 15
- 239000013598 vector Substances 0.000 description 15
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 14
- 229960001592 paclitaxel Drugs 0.000 description 14
- 229930012538 Paclitaxel Natural products 0.000 description 13
- 230000001085 cytostatic effect Effects 0.000 description 13
- 229940127089 cytotoxic agent Drugs 0.000 description 13
- 229960002621 pembrolizumab Drugs 0.000 description 13
- 150000003839 salts Chemical class 0.000 description 13
- 238000009097 single-agent therapy Methods 0.000 description 13
- 102000004190 Enzymes Human genes 0.000 description 12
- 108090000790 Enzymes Proteins 0.000 description 12
- 125000000539 amino acid group Chemical group 0.000 description 12
- 231100000599 cytotoxic agent Toxicity 0.000 description 12
- 229940088598 enzyme Drugs 0.000 description 12
- 229940043355 kinase inhibitor Drugs 0.000 description 12
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 12
- 125000003396 thiol group Chemical group [H]S* 0.000 description 12
- 206010006187 Breast cancer Diseases 0.000 description 11
- 208000026310 Breast neoplasm Diseases 0.000 description 11
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 11
- 230000002776 aggregation Effects 0.000 description 11
- 238000004220 aggregation Methods 0.000 description 11
- 229960000397 bevacizumab Drugs 0.000 description 11
- 239000000562 conjugate Substances 0.000 description 11
- 238000005516 engineering process Methods 0.000 description 11
- 229960002949 fluorouracil Drugs 0.000 description 11
- 230000035772 mutation Effects 0.000 description 11
- 230000002829 reductive effect Effects 0.000 description 11
- 102100021866 Hepatocyte growth factor Human genes 0.000 description 10
- 201000010099 disease Diseases 0.000 description 10
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 10
- 239000012528 membrane Substances 0.000 description 10
- 230000002018 overexpression Effects 0.000 description 10
- 230000008569 process Effects 0.000 description 10
- 230000004083 survival effect Effects 0.000 description 10
- 210000001519 tissue Anatomy 0.000 description 10
- 229910052720 vanadium Inorganic materials 0.000 description 10
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 9
- 102000014914 Carrier Proteins Human genes 0.000 description 9
- 108010016626 Dipeptides Proteins 0.000 description 9
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 9
- 108091008324 binding proteins Proteins 0.000 description 9
- 150000001875 compounds Chemical class 0.000 description 9
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 9
- KLEAIHJJLUAXIQ-JDRGBKBRSA-N irinotecan hydrochloride hydrate Chemical compound O.O.O.Cl.C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 KLEAIHJJLUAXIQ-JDRGBKBRSA-N 0.000 description 9
- 208000020816 lung neoplasm Diseases 0.000 description 9
- 230000002132 lysosomal effect Effects 0.000 description 9
- 239000003550 marker Substances 0.000 description 9
- 230000004048 modification Effects 0.000 description 9
- 238000012986 modification Methods 0.000 description 9
- 229940048191 onivyde Drugs 0.000 description 9
- 238000003259 recombinant expression Methods 0.000 description 9
- 230000001105 regulatory effect Effects 0.000 description 9
- 206010009944 Colon cancer Diseases 0.000 description 8
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 8
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 8
- 230000000970 DNA cross-linking effect Effects 0.000 description 8
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 8
- 101000936922 Homo sapiens Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 Proteins 0.000 description 8
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 8
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 8
- 102100027732 Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 Human genes 0.000 description 8
- 230000002378 acidificating effect Effects 0.000 description 8
- 208000009956 adenocarcinoma Diseases 0.000 description 8
- 239000003080 antimitotic agent Substances 0.000 description 8
- 239000000872 buffer Substances 0.000 description 8
- 229960004316 cisplatin Drugs 0.000 description 8
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 8
- 230000021615 conjugation Effects 0.000 description 8
- 239000003431 cross linking reagent Substances 0.000 description 8
- 238000009472 formulation Methods 0.000 description 8
- 230000006870 function Effects 0.000 description 8
- 229940020967 gemzar Drugs 0.000 description 8
- 150000007857 hydrazones Chemical class 0.000 description 8
- 230000001965 increasing effect Effects 0.000 description 8
- GURKHSYORGJETM-WAQYZQTGSA-N irinotecan hydrochloride (anhydrous) Chemical compound Cl.C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 GURKHSYORGJETM-WAQYZQTGSA-N 0.000 description 8
- 201000005202 lung cancer Diseases 0.000 description 8
- 210000004379 membrane Anatomy 0.000 description 8
- 229920000642 polymer Polymers 0.000 description 8
- KVJXBPDAXMEYOA-CXANFOAXSA-N trilostane Chemical compound OC1=C(C#N)C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@@]32O[C@@H]31 KVJXBPDAXMEYOA-CXANFOAXSA-N 0.000 description 8
- 210000004881 tumor cell Anatomy 0.000 description 8
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 7
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 7
- 102000001301 EGF receptor Human genes 0.000 description 7
- 108060006698 EGF receptor Proteins 0.000 description 7
- 108010050904 Interferons Proteins 0.000 description 7
- 102000014150 Interferons Human genes 0.000 description 7
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 7
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 7
- 239000002202 Polyethylene glycol Substances 0.000 description 7
- 108010076504 Protein Sorting Signals Proteins 0.000 description 7
- 229960001686 afatinib Drugs 0.000 description 7
- ULXXDDBFHOBEHA-CWDCEQMOSA-N afatinib Chemical compound N1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-CWDCEQMOSA-N 0.000 description 7
- 230000000259 anti-tumor effect Effects 0.000 description 7
- 230000006907 apoptotic process Effects 0.000 description 7
- RITAVMQDGBJQJZ-FMIVXFBMSA-N axitinib Chemical compound CNC(=O)C1=CC=CC=C1SC1=CC=C(C(\C=C\C=2N=CC=CC=2)=NN2)C2=C1 RITAVMQDGBJQJZ-FMIVXFBMSA-N 0.000 description 7
- 239000011230 binding agent Substances 0.000 description 7
- 229960004562 carboplatin Drugs 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- 235000018417 cysteine Nutrition 0.000 description 7
- 238000000684 flow cytometry Methods 0.000 description 7
- 229960003180 glutathione Drugs 0.000 description 7
- 230000002209 hydrophobic effect Effects 0.000 description 7
- 229940127121 immunoconjugate Drugs 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 229960005386 ipilimumab Drugs 0.000 description 7
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 7
- 229920001223 polyethylene glycol Polymers 0.000 description 7
- 102000005962 receptors Human genes 0.000 description 7
- 108020003175 receptors Proteins 0.000 description 7
- 230000009467 reduction Effects 0.000 description 7
- 238000003860 storage Methods 0.000 description 7
- 229960000575 trastuzumab Drugs 0.000 description 7
- 229960001670 trilostane Drugs 0.000 description 7
- 229960005486 vaccine Drugs 0.000 description 7
- YCOYDOIWSSHVCK-UHFFFAOYSA-N vatalanib Chemical compound C1=CC(Cl)=CC=C1NC(C1=CC=CC=C11)=NN=C1CC1=CC=NC=C1 YCOYDOIWSSHVCK-UHFFFAOYSA-N 0.000 description 7
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 6
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 6
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 6
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 6
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 6
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 6
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 6
- 239000002146 L01XE16 - Crizotinib Substances 0.000 description 6
- 239000004472 Lysine Substances 0.000 description 6
- 241001529936 Murinae Species 0.000 description 6
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 6
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 6
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 6
- 125000000217 alkyl group Chemical group 0.000 description 6
- 229940100198 alkylating agent Drugs 0.000 description 6
- 239000002168 alkylating agent Substances 0.000 description 6
- 230000000340 anti-metabolite Effects 0.000 description 6
- 239000002256 antimetabolite Substances 0.000 description 6
- 229940100197 antimetabolite Drugs 0.000 description 6
- 239000002246 antineoplastic agent Substances 0.000 description 6
- 238000013459 approach Methods 0.000 description 6
- 108010044540 auristatin Proteins 0.000 description 6
- KVUAALJSMIVURS-ZEDZUCNESA-L calcium folinate Chemical compound [Ca+2].C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC([O-])=O)C([O-])=O)C=C1 KVUAALJSMIVURS-ZEDZUCNESA-L 0.000 description 6
- 210000000170 cell membrane Anatomy 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 230000000875 corresponding effect Effects 0.000 description 6
- KTEIFNKAUNYNJU-GFCCVEGCSA-N crizotinib Chemical compound O([C@H](C)C=1C(=C(F)C=CC=1Cl)Cl)C(C(=NC=1)N)=CC=1C(=C1)C=NN1C1CCNCC1 KTEIFNKAUNYNJU-GFCCVEGCSA-N 0.000 description 6
- 229960000684 cytarabine Drugs 0.000 description 6
- 230000007423 decrease Effects 0.000 description 6
- 150000002019 disulfides Chemical class 0.000 description 6
- 229960004679 doxorubicin Drugs 0.000 description 6
- 235000008191 folinic acid Nutrition 0.000 description 6
- 239000011672 folinic acid Substances 0.000 description 6
- 229960002584 gefitinib Drugs 0.000 description 6
- 230000012010 growth Effects 0.000 description 6
- 229960002885 histidine Drugs 0.000 description 6
- 235000014304 histidine Nutrition 0.000 description 6
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 6
- 210000004408 hybridoma Anatomy 0.000 description 6
- 230000001900 immune effect Effects 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 229960004768 irinotecan Drugs 0.000 description 6
- 229960001691 leucovorin Drugs 0.000 description 6
- 229960003646 lysine Drugs 0.000 description 6
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 6
- 229960000435 oblimersen Drugs 0.000 description 6
- MIMNFCVQODTQDP-NDLVEFNKSA-N oblimersen Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP(S)(=O)O[C@@H]2[C@H](O[C@H](C2)N2C3=NC=NC(N)=C3N=C2)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=NC=NC(N)=C3N=C2)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP(O)(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(NC(=O)C(C)=C2)=O)CO)[C@@H](O)C1 MIMNFCVQODTQDP-NDLVEFNKSA-N 0.000 description 6
- FDLYAMZZIXQODN-UHFFFAOYSA-N olaparib Chemical compound FC1=CC=C(CC=2C3=CC=CC=C3C(=O)NN=2)C=C1C(=O)N(CC1)CCN1C(=O)C1CC1 FDLYAMZZIXQODN-UHFFFAOYSA-N 0.000 description 6
- 201000002528 pancreatic cancer Diseases 0.000 description 6
- 208000008443 pancreatic carcinoma Diseases 0.000 description 6
- BTIHMVBBUGXLCJ-OAHLLOKOSA-N seliciclib Chemical compound C=12N=CN(C(C)C)C2=NC(N[C@@H](CO)CC)=NC=1NCC1=CC=CC=C1 BTIHMVBBUGXLCJ-OAHLLOKOSA-N 0.000 description 6
- 229960003787 sorafenib Drugs 0.000 description 6
- 239000003381 stabilizer Substances 0.000 description 6
- 231100000402 unacceptable toxicity Toxicity 0.000 description 6
- 229960004295 valine Drugs 0.000 description 6
- 239000004474 valine Substances 0.000 description 6
- 229960000241 vandetanib Drugs 0.000 description 6
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 6
- 229950011257 veliparib Drugs 0.000 description 6
- JNAHVYVRKWKWKQ-CYBMUJFWSA-N veliparib Chemical compound N=1C2=CC=CC(C(N)=O)=C2NC=1[C@@]1(C)CCCN1 JNAHVYVRKWKWKQ-CYBMUJFWSA-N 0.000 description 6
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 5
- ULXXDDBFHOBEHA-ONEGZZNKSA-N Afatinib Chemical compound N1=CN=C2C=C(OC3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-ONEGZZNKSA-N 0.000 description 5
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 5
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 5
- OFDNQWIFNXBECV-UHFFFAOYSA-N Dolastatin 10 Natural products CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)CC)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 OFDNQWIFNXBECV-UHFFFAOYSA-N 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 5
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 5
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 5
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 5
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 5
- 239000005536 L01XE08 - Nilotinib Substances 0.000 description 5
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 5
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 5
- 206010061535 Ovarian neoplasm Diseases 0.000 description 5
- 102000035195 Peptidases Human genes 0.000 description 5
- 108091005804 Peptidases Proteins 0.000 description 5
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 5
- 229940028652 abraxane Drugs 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 229940120638 avastin Drugs 0.000 description 5
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 5
- 229960004117 capecitabine Drugs 0.000 description 5
- 229960005395 cetuximab Drugs 0.000 description 5
- 230000008859 change Effects 0.000 description 5
- 229960002433 cysteine Drugs 0.000 description 5
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 5
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 5
- 210000000805 cytoplasm Anatomy 0.000 description 5
- 230000001086 cytosolic effect Effects 0.000 description 5
- OFDNQWIFNXBECV-VFSYNPLYSA-N dolastatin 10 Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C=1SC=CN=1)CC1=CC=CC=C1 OFDNQWIFNXBECV-VFSYNPLYSA-N 0.000 description 5
- 108010045524 dolastatin 10 Proteins 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 201000010536 head and neck cancer Diseases 0.000 description 5
- 208000014829 head and neck neoplasm Diseases 0.000 description 5
- 230000036541 health Effects 0.000 description 5
- 229940072221 immunoglobulins Drugs 0.000 description 5
- 229940079322 interferon Drugs 0.000 description 5
- 229960004891 lapatinib Drugs 0.000 description 5
- 210000003712 lysosome Anatomy 0.000 description 5
- 230000001868 lysosomic effect Effects 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 description 5
- 125000003729 nucleotide group Chemical group 0.000 description 5
- 229950000846 onartuzumab Drugs 0.000 description 5
- 238000011275 oncology therapy Methods 0.000 description 5
- CUIHSIWYWATEQL-UHFFFAOYSA-N pazopanib Chemical compound C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 CUIHSIWYWATEQL-UHFFFAOYSA-N 0.000 description 5
- WBXPDJSOTKVWSJ-ZDUSSCGKSA-N pemetrexed Chemical compound C=1NC=2NC(N)=NC(=O)C=2C=1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 WBXPDJSOTKVWSJ-ZDUSSCGKSA-N 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 5
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 5
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 5
- 238000001959 radiotherapy Methods 0.000 description 5
- 230000028327 secretion Effects 0.000 description 5
- 239000000243 solution Substances 0.000 description 5
- 229940124597 therapeutic agent Drugs 0.000 description 5
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 5
- 230000001988 toxicity Effects 0.000 description 5
- 231100000419 toxicity Toxicity 0.000 description 5
- 229950000578 vatalanib Drugs 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 4
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 4
- 239000004475 Arginine Substances 0.000 description 4
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 4
- 102000053602 DNA Human genes 0.000 description 4
- 102000009058 Death Domain Receptors Human genes 0.000 description 4
- 108010049207 Death Domain Receptors Proteins 0.000 description 4
- 238000002965 ELISA Methods 0.000 description 4
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 4
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 4
- 108700012941 GNRH1 Proteins 0.000 description 4
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 4
- 108010024636 Glutathione Proteins 0.000 description 4
- 239000004471 Glycine Substances 0.000 description 4
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 102100026120 IgG receptor FcRn large subunit p51 Human genes 0.000 description 4
- 102000009786 Immunoglobulin Constant Regions Human genes 0.000 description 4
- 108010009817 Immunoglobulin Constant Regions Proteins 0.000 description 4
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 4
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 4
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 4
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 4
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 4
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 4
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 4
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 4
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 4
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 4
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 4
- 241000699666 Mus <mouse, genus> Species 0.000 description 4
- 108091034117 Oligonucleotide Proteins 0.000 description 4
- 206010033128 Ovarian cancer Diseases 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- 108091007960 PI3Ks Proteins 0.000 description 4
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 4
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 4
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 4
- 239000004365 Protease Substances 0.000 description 4
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 4
- 206010041067 Small cell lung cancer Diseases 0.000 description 4
- 210000001744 T-lymphocyte Anatomy 0.000 description 4
- NAVMQTYZDKMPEU-UHFFFAOYSA-N Targretin Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C(=C)C1=CC=C(C(O)=O)C=C1 NAVMQTYZDKMPEU-UHFFFAOYSA-N 0.000 description 4
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 4
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 4
- 239000004473 Threonine Substances 0.000 description 4
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 4
- 229960003767 alanine Drugs 0.000 description 4
- 235000004279 alanine Nutrition 0.000 description 4
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 4
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 4
- 229960003121 arginine Drugs 0.000 description 4
- 229960001230 asparagine Drugs 0.000 description 4
- 235000009582 asparagine Nutrition 0.000 description 4
- 239000003719 aurora kinase inhibitor Substances 0.000 description 4
- 229960003005 axitinib Drugs 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- 238000007385 chemical modification Methods 0.000 description 4
- 230000004087 circulation Effects 0.000 description 4
- 229960005061 crizotinib Drugs 0.000 description 4
- 229960003901 dacarbazine Drugs 0.000 description 4
- 230000002354 daily effect Effects 0.000 description 4
- 231100000517 death Toxicity 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 238000010586 diagram Methods 0.000 description 4
- 239000000539 dimer Substances 0.000 description 4
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 4
- 229960003668 docetaxel Drugs 0.000 description 4
- 239000003623 enhancer Substances 0.000 description 4
- MNJVRJDLRVPLFE-UHFFFAOYSA-N etoricoxib Chemical compound C1=NC(C)=CC=C1C1=NC=C(Cl)C=C1C1=CC=C(S(C)(=O)=O)C=C1 MNJVRJDLRVPLFE-UHFFFAOYSA-N 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 238000002710 external beam radiation therapy Methods 0.000 description 4
- PJZDLZXMGBOJRF-CXOZILEQSA-L folfirinox Chemical compound [Pt+4].[O-]C(=O)C([O-])=O.[NH-][C@H]1CCCC[C@@H]1[NH-].FC1=CNC(=O)NC1=O.C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1.C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 PJZDLZXMGBOJRF-CXOZILEQSA-L 0.000 description 4
- 229940087158 gilotrif Drugs 0.000 description 4
- 229940097043 glucuronic acid Drugs 0.000 description 4
- 229960002989 glutamic acid Drugs 0.000 description 4
- 235000013922 glutamic acid Nutrition 0.000 description 4
- 239000004220 glutamic acid Substances 0.000 description 4
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 4
- 235000004554 glutamine Nutrition 0.000 description 4
- 229960002743 glutamine Drugs 0.000 description 4
- 229960002449 glycine Drugs 0.000 description 4
- HHXHVIJIIXKSOE-QILQGKCVSA-N histrelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC(N=C1)=CN1CC1=CC=CC=C1 HHXHVIJIIXKSOE-QILQGKCVSA-N 0.000 description 4
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 238000000111 isothermal titration calorimetry Methods 0.000 description 4
- 230000003902 lesion Effects 0.000 description 4
- 229960003136 leucine Drugs 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 239000008176 lyophilized powder Substances 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 4
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 4
- 230000001394 metastastic effect Effects 0.000 description 4
- 206010061289 metastatic neoplasm Diseases 0.000 description 4
- 229930182817 methionine Natural products 0.000 description 4
- 229960004452 methionine Drugs 0.000 description 4
- 235000006109 methionine Nutrition 0.000 description 4
- 229960000485 methotrexate Drugs 0.000 description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 4
- 229960004857 mitomycin Drugs 0.000 description 4
- 239000002105 nanoparticle Substances 0.000 description 4
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 239000002773 nucleotide Substances 0.000 description 4
- 229960003278 osimertinib Drugs 0.000 description 4
- DUYJMQONPNNFPI-UHFFFAOYSA-N osimertinib Chemical compound COC1=CC(N(C)CCN(C)C)=C(NC(=O)C=C)C=C1NC1=NC=CC(C=2C3=CC=CC=C3N(C)C=2)=N1 DUYJMQONPNNFPI-UHFFFAOYSA-N 0.000 description 4
- 229960001756 oxaliplatin Drugs 0.000 description 4
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 4
- 229960001972 panitumumab Drugs 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- 102000040430 polynucleotide Human genes 0.000 description 4
- 108091033319 polynucleotide Proteins 0.000 description 4
- 239000002157 polynucleotide Substances 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- 229960002429 proline Drugs 0.000 description 4
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 4
- YUOCYTRGANSSRY-UHFFFAOYSA-N pyrrolo[2,3-i][1,2]benzodiazepine Chemical class C1=CN=NC2=C3C=CN=C3C=CC2=C1 YUOCYTRGANSSRY-UHFFFAOYSA-N 0.000 description 4
- 230000010076 replication Effects 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- 238000001542 size-exclusion chromatography Methods 0.000 description 4
- 125000006850 spacer group Chemical group 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 150000005846 sugar alcohols Chemical class 0.000 description 4
- AYUNIORJHRXIBJ-TXHRRWQRSA-N tanespimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(NCC=C)C(=O)C=C1C2=O AYUNIORJHRXIBJ-TXHRRWQRSA-N 0.000 description 4
- 229940120982 tarceva Drugs 0.000 description 4
- 230000008685 targeting Effects 0.000 description 4
- 229960004964 temozolomide Drugs 0.000 description 4
- 229960000235 temsirolimus Drugs 0.000 description 4
- 229960002898 threonine Drugs 0.000 description 4
- 229960004528 vincristine Drugs 0.000 description 4
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 4
- 230000003442 weekly effect Effects 0.000 description 4
- BMKDZUISNHGIBY-ZETCQYMHSA-N (+)-dexrazoxane Chemical compound C([C@H](C)N1CC(=O)NC(=O)C1)N1CC(=O)NC(=O)C1 BMKDZUISNHGIBY-ZETCQYMHSA-N 0.000 description 3
- AGGWFDNPHKLBBV-YUMQZZPRSA-N (2s)-2-[[(2s)-2-amino-3-methylbutanoyl]amino]-5-(carbamoylamino)pentanoic acid Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCCNC(N)=O AGGWFDNPHKLBBV-YUMQZZPRSA-N 0.000 description 3
- CVCLJVVBHYOXDC-IAZSKANUSA-N (2z)-2-[(5z)-5-[(3,5-dimethyl-1h-pyrrol-2-yl)methylidene]-4-methoxypyrrol-2-ylidene]indole Chemical compound COC1=C\C(=C/2N=C3C=CC=CC3=C\2)N\C1=C/C=1NC(C)=CC=1C CVCLJVVBHYOXDC-IAZSKANUSA-N 0.000 description 3
- MYBLAOJMRYYKMS-RTRLPJTCSA-N 1-(2-chloroethyl)-1-nitroso-3-[(3r,4r,5s,6r)-2,4,5-trihydroxy-6-(hydroxymethyl)oxan-3-yl]urea Chemical compound OC[C@H]1OC(O)[C@H](NC(=O)N(CCCl)N=O)[C@@H](O)[C@@H]1O MYBLAOJMRYYKMS-RTRLPJTCSA-N 0.000 description 3
- PVCULFYROUOVGJ-UHFFFAOYSA-N 1-[2-chloroethyl(methylsulfonyl)amino]-3-methyl-1-methylsulfonylurea Chemical compound CNC(=O)N(S(C)(=O)=O)N(S(C)(=O)=O)CCCl PVCULFYROUOVGJ-UHFFFAOYSA-N 0.000 description 3
- 150000003923 2,5-pyrrolediones Chemical class 0.000 description 3
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 3
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 3
- 108010006654 Bleomycin Proteins 0.000 description 3
- 108010037003 Buserelin Proteins 0.000 description 3
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 3
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 3
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 3
- 101150013553 CD40 gene Proteins 0.000 description 3
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 3
- 102000000844 Cell Surface Receptors Human genes 0.000 description 3
- 108010001857 Cell Surface Receptors Proteins 0.000 description 3
- 108020004705 Codon Proteins 0.000 description 3
- 102100021906 Cyclin-O Human genes 0.000 description 3
- 241000701022 Cytomegalovirus Species 0.000 description 3
- SHZGCJCMOBCMKK-UHFFFAOYSA-N D-mannomethylose Natural products CC1OC(O)C(O)C(O)C1O SHZGCJCMOBCMKK-UHFFFAOYSA-N 0.000 description 3
- 229940123780 DNA topoisomerase I inhibitor Drugs 0.000 description 3
- 229940124087 DNA topoisomerase II inhibitor Drugs 0.000 description 3
- 108010092160 Dactinomycin Proteins 0.000 description 3
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- PNNNRSAQSRJVSB-SLPGGIOYSA-N Fucose Natural products C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C=O PNNNRSAQSRJVSB-SLPGGIOYSA-N 0.000 description 3
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 3
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 3
- 108010069236 Goserelin Proteins 0.000 description 3
- 101000897441 Homo sapiens Cyclin-O Proteins 0.000 description 3
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 3
- 101710177940 IgG receptor FcRn large subunit p51 Proteins 0.000 description 3
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 3
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 3
- SHZGCJCMOBCMKK-DHVFOXMCSA-N L-fucopyranose Chemical compound C[C@@H]1OC(O)[C@@H](O)[C@H](O)[C@@H]1O SHZGCJCMOBCMKK-DHVFOXMCSA-N 0.000 description 3
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 3
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 3
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 3
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 3
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 3
- 239000003798 L01XE11 - Pazopanib Substances 0.000 description 3
- 239000002118 L01XE12 - Vandetanib Substances 0.000 description 3
- 239000002145 L01XE14 - Bosutinib Substances 0.000 description 3
- 239000002144 L01XE18 - Ruxolitinib Substances 0.000 description 3
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 3
- 241000829100 Macaca mulatta polyomavirus 1 Species 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 3
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 3
- 108091008606 PDGF receptors Proteins 0.000 description 3
- 206010035226 Plasma cell myeloma Diseases 0.000 description 3
- 102000011653 Platelet-Derived Growth Factor Receptors Human genes 0.000 description 3
- 229940079156 Proteasome inhibitor Drugs 0.000 description 3
- 229940123573 Protein synthesis inhibitor Drugs 0.000 description 3
- 108020004511 Recombinant DNA Proteins 0.000 description 3
- 102000009203 Sema domains Human genes 0.000 description 3
- 108050000099 Sema domains Proteins 0.000 description 3
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 3
- 108020004459 Small interfering RNA Proteins 0.000 description 3
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 3
- 108010022394 Threonine synthase Proteins 0.000 description 3
- 239000000365 Topoisomerase I Inhibitor Substances 0.000 description 3
- 239000000317 Topoisomerase II Inhibitor Substances 0.000 description 3
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 3
- 102100040247 Tumor necrosis factor Human genes 0.000 description 3
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 3
- 108091008605 VEGF receptors Proteins 0.000 description 3
- HSRXSKHRSXRCFC-WDSKDSINSA-N Val-Ala Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](C)C(O)=O HSRXSKHRSXRCFC-WDSKDSINSA-N 0.000 description 3
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 3
- 108010023617 abarelix Proteins 0.000 description 3
- AIWRTTMUVOZGPW-HSPKUQOVSA-N abarelix Chemical compound C([C@@H](C(=O)N[C@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCNC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)N(C)C(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(O)C=C1 AIWRTTMUVOZGPW-HSPKUQOVSA-N 0.000 description 3
- 229960002184 abarelix Drugs 0.000 description 3
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 229930013930 alkaloid Natural products 0.000 description 3
- 125000002947 alkylene group Chemical group 0.000 description 3
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 3
- 229960000473 altretamine Drugs 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 230000003321 amplification Effects 0.000 description 3
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 3
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 3
- 239000004037 angiogenesis inhibitor Substances 0.000 description 3
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 3
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 3
- 229960005261 aspartic acid Drugs 0.000 description 3
- 235000003704 aspartic acid Nutrition 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 3
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 3
- ACWZRVQXLIRSDF-UHFFFAOYSA-N binimetinib Chemical compound OCCONC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1F ACWZRVQXLIRSDF-UHFFFAOYSA-N 0.000 description 3
- 238000001574 biopsy Methods 0.000 description 3
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 3
- UBPYILGKFZZVDX-UHFFFAOYSA-N bosutinib Chemical compound C1=C(Cl)C(OC)=CC(NC=2C3=CC(OC)=C(OCCCN4CCN(C)CC4)C=C3N=CC=2C#N)=C1Cl UBPYILGKFZZVDX-UHFFFAOYSA-N 0.000 description 3
- 210000000481 breast Anatomy 0.000 description 3
- 229960000455 brentuximab vedotin Drugs 0.000 description 3
- 239000006172 buffering agent Substances 0.000 description 3
- CUWODFFVMXJOKD-UVLQAERKSA-N buserelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](COC(C)(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 CUWODFFVMXJOKD-UVLQAERKSA-N 0.000 description 3
- 229940127093 camptothecin Drugs 0.000 description 3
- BLMPQMFVWMYDKT-NZTKNTHTSA-N carfilzomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)[C@]1(C)OC1)NC(=O)CN1CCOCC1)CC1=CC=CC=C1 BLMPQMFVWMYDKT-NZTKNTHTSA-N 0.000 description 3
- 229960002438 carfilzomib Drugs 0.000 description 3
- 229940076006 cell cycle modulator Drugs 0.000 description 3
- 230000030833 cell death Effects 0.000 description 3
- 108700008462 cetrorelix Proteins 0.000 description 3
- SBNPWPIBESPSIF-MHWMIDJBSA-N cetrorelix Chemical compound C([C@@H](C(=O)N[C@H](CCCNC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(O)C=C1 SBNPWPIBESPSIF-MHWMIDJBSA-N 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 229960002173 citrulline Drugs 0.000 description 3
- 235000013477 citrulline Nutrition 0.000 description 3
- 230000001268 conjugating effect Effects 0.000 description 3
- 210000000172 cytosol Anatomy 0.000 description 3
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 3
- 229960000640 dactinomycin Drugs 0.000 description 3
- 229960002448 dasatinib Drugs 0.000 description 3
- 230000034994 death Effects 0.000 description 3
- 238000002405 diagnostic procedure Methods 0.000 description 3
- 102000004419 dihydrofolate reductase Human genes 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 230000009977 dual effect Effects 0.000 description 3
- 239000012636 effector Substances 0.000 description 3
- 229950004255 emibetuzumab Drugs 0.000 description 3
- 229960004945 etoricoxib Drugs 0.000 description 3
- DBEPLOCGEIEOCV-WSBQPABSSA-N finasteride Chemical compound N([C@@H]1CC2)C(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H](C(=O)NC(C)(C)C)[C@@]2(C)CC1 DBEPLOCGEIEOCV-WSBQPABSSA-N 0.000 description 3
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 3
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 3
- 206010017758 gastric cancer Diseases 0.000 description 3
- 210000004602 germ cell Anatomy 0.000 description 3
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 3
- 229960002193 histrelin Drugs 0.000 description 3
- 108700020746 histrelin Proteins 0.000 description 3
- 238000001794 hormone therapy Methods 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 230000036039 immunity Effects 0.000 description 3
- CGIGDMFJXJATDK-UHFFFAOYSA-N indomethacin Chemical compound CC1=C(CC(O)=O)C2=CC(OC)=CC=C2N1C(=O)C1=CC=C(Cl)C=C1 CGIGDMFJXJATDK-UHFFFAOYSA-N 0.000 description 3
- 229940005319 inlyta Drugs 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 239000000138 intercalating agent Substances 0.000 description 3
- 229940084651 iressa Drugs 0.000 description 3
- 229960000310 isoleucine Drugs 0.000 description 3
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 3
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 3
- 229950002884 lexatumumab Drugs 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 238000011068 loading method Methods 0.000 description 3
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 3
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 3
- 229960001924 melphalan Drugs 0.000 description 3
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 3
- 239000002207 metabolite Substances 0.000 description 3
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 3
- 229960005225 mifamurtide Drugs 0.000 description 3
- JMUHBNWAORSSBD-WKYWBUFDSA-N mifamurtide Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCCCCCCCCCC)COP(O)(=O)OCCNC(=O)[C@H](C)NC(=O)CC[C@H](C(N)=O)NC(=O)[C@H](C)NC(=O)[C@@H](C)O[C@H]1[C@H](O)[C@@H](CO)OC(O)[C@@H]1NC(C)=O JMUHBNWAORSSBD-WKYWBUFDSA-N 0.000 description 3
- 229960003248 mifepristone Drugs 0.000 description 3
- VKHAHZOOUSRJNA-GCNJZUOMSA-N mifepristone Chemical compound C1([C@@H]2C3=C4CCC(=O)C=C4CC[C@H]3[C@@H]3CC[C@@]([C@]3(C2)C)(O)C#CC)=CC=C(N(C)C)C=C1 VKHAHZOOUSRJNA-GCNJZUOMSA-N 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 210000003470 mitochondria Anatomy 0.000 description 3
- 229950003063 mitumomab Drugs 0.000 description 3
- VOWOEBADKMXUBU-UHFFFAOYSA-J molecular oxygen;tetrachlorite;hydrate Chemical compound O.O=O.[O-]Cl=O.[O-]Cl=O.[O-]Cl=O.[O-]Cl=O VOWOEBADKMXUBU-UHFFFAOYSA-J 0.000 description 3
- 201000000050 myeloid neoplasm Diseases 0.000 description 3
- LWGJTAZLEJHCPA-UHFFFAOYSA-N n-(2-chloroethyl)-n-nitrosomorpholine-4-carboxamide Chemical compound ClCCN(N=O)C(=O)N1CCOCC1 LWGJTAZLEJHCPA-UHFFFAOYSA-N 0.000 description 3
- LBWFXVZLPYTWQI-IPOVEDGCSA-N n-[2-(diethylamino)ethyl]-5-[(z)-(5-fluoro-2-oxo-1h-indol-3-ylidene)methyl]-2,4-dimethyl-1h-pyrrole-3-carboxamide;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C LBWFXVZLPYTWQI-IPOVEDGCSA-N 0.000 description 3
- 229960001346 nilotinib Drugs 0.000 description 3
- XWXYUMMDTVBTOU-UHFFFAOYSA-N nilutamide Chemical compound O=C1C(C)(C)NC(=O)N1C1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 XWXYUMMDTVBTOU-UHFFFAOYSA-N 0.000 description 3
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 3
- 239000002736 nonionic surfactant Substances 0.000 description 3
- 230000030147 nuclear export Effects 0.000 description 3
- 238000003199 nucleic acid amplification method Methods 0.000 description 3
- 102000039446 nucleic acids Human genes 0.000 description 3
- 108020004707 nucleic acids Proteins 0.000 description 3
- 150000007523 nucleic acids Chemical class 0.000 description 3
- 229950006584 obatoclax Drugs 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 238000005192 partition Methods 0.000 description 3
- 229960005079 pemetrexed Drugs 0.000 description 3
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 3
- 230000035699 permeability Effects 0.000 description 3
- 229960002087 pertuzumab Drugs 0.000 description 3
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 3
- 229960005190 phenylalanine Drugs 0.000 description 3
- 235000008729 phenylalanine Nutrition 0.000 description 3
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 description 3
- 229910052697 platinum Inorganic materials 0.000 description 3
- 238000003752 polymerase chain reaction Methods 0.000 description 3
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 3
- 229960004618 prednisone Drugs 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 3
- 229960000624 procarbazine Drugs 0.000 description 3
- 239000003207 proteasome inhibitor Substances 0.000 description 3
- 239000000007 protein synthesis inhibitor Substances 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 3
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 3
- 239000002336 ribonucleotide Substances 0.000 description 3
- 229950009213 rubitecan Drugs 0.000 description 3
- VHXNKPBCCMUMSW-FQEVSTJZSA-N rubitecan Chemical compound C1=CC([N+]([O-])=O)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VHXNKPBCCMUMSW-FQEVSTJZSA-N 0.000 description 3
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 description 3
- 229960000215 ruxolitinib Drugs 0.000 description 3
- 229960001153 serine Drugs 0.000 description 3
- 229960002930 sirolimus Drugs 0.000 description 3
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 3
- 208000000587 small cell lung carcinoma Diseases 0.000 description 3
- 206010041823 squamous cell carcinoma Diseases 0.000 description 3
- 238000010561 standard procedure Methods 0.000 description 3
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 3
- 229960001796 sunitinib Drugs 0.000 description 3
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 3
- FQZYTYWMLGAPFJ-OQKDUQJOSA-N tamoxifen citrate Chemical compound [H+].[H+].[H+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O.C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 FQZYTYWMLGAPFJ-OQKDUQJOSA-N 0.000 description 3
- 150000003573 thiols Chemical class 0.000 description 3
- 229960001196 thiotepa Drugs 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- 229960004799 tryptophan Drugs 0.000 description 3
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 3
- 229960004441 tyrosine Drugs 0.000 description 3
- 229940124676 vascular endothelial growth factor receptor Drugs 0.000 description 3
- 229960003048 vinblastine Drugs 0.000 description 3
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 3
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- 229960000237 vorinostat Drugs 0.000 description 3
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 3
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 2
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- WVTKBKWTSCPRNU-KYJUHHDHSA-N (+)-Tetrandrine Chemical compound C([C@H]1C=2C=C(C(=CC=2CCN1C)OC)O1)C(C=C2)=CC=C2OC(=C2)C(OC)=CC=C2C[C@@H]2N(C)CCC3=CC(OC)=C(OC)C1=C23 WVTKBKWTSCPRNU-KYJUHHDHSA-N 0.000 description 2
- LVLLALCJVJNGQQ-SEODYNFXSA-N (1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-1-[(2r,3e,5e)-7-ethyl-7-hydroxynona-3,5-dien-2-yl]-7a-methyl-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)/C=C/C=C/C(O)(CC)CC)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C LVLLALCJVJNGQQ-SEODYNFXSA-N 0.000 description 2
- WZRFLSDVFPIXOV-LRQRDZAKSA-N (2s)-1-[(2s)-2-cyclohexyl-2-[[(2s)-2-(methylamino)propanoyl]amino]acetyl]-n-(4-phenylthiadiazol-5-yl)pyrrolidine-2-carboxamide Chemical compound C1([C@H](NC(=O)[C@H](C)NC)C(=O)N2[C@@H](CCC2)C(=O)NC2=C(N=NS2)C=2C=CC=CC=2)CCCCC1 WZRFLSDVFPIXOV-LRQRDZAKSA-N 0.000 description 2
- NECZZOFFLFZNHL-XVGZVFJZSA-N (2s)-2-amino-5-[[(2r)-3-[2-[bis[bis(2-chloroethyl)amino]-oxidophosphaniumyl]oxyethylsulfonyl]-1-[[(r)-carboxy(phenyl)methyl]amino]-1-oxopropan-2-yl]amino]-5-oxopentanoic acid;hydron;chloride Chemical compound Cl.ClCCN(CCCl)P(=O)(N(CCCl)CCCl)OCCS(=O)(=O)C[C@H](NC(=O)CC[C@H](N)C(O)=O)C(=O)N[C@@H](C(O)=O)C1=CC=CC=C1 NECZZOFFLFZNHL-XVGZVFJZSA-N 0.000 description 2
- MHFUWOIXNMZFIW-WNQIDUERSA-N (2s)-2-hydroxypropanoic acid;n-[4-[4-(4-methylpiperazin-1-yl)-6-[(5-methyl-1h-pyrazol-3-yl)amino]pyrimidin-2-yl]sulfanylphenyl]cyclopropanecarboxamide Chemical compound C[C@H](O)C(O)=O.C1CN(C)CCN1C1=CC(NC2=NNC(C)=C2)=NC(SC=2C=CC(NC(=O)C3CC3)=CC=2)=N1 MHFUWOIXNMZFIW-WNQIDUERSA-N 0.000 description 2
- UFPFGVNKHCLJJO-SSKFGXFMSA-N (2s)-n-[(1s)-1-cyclohexyl-2-[(2s)-2-[4-(4-fluorobenzoyl)-1,3-thiazol-2-yl]pyrrolidin-1-yl]-2-oxoethyl]-2-(methylamino)propanamide Chemical compound C1([C@H](NC(=O)[C@H](C)NC)C(=O)N2[C@@H](CCC2)C=2SC=C(N=2)C(=O)C=2C=CC(F)=CC=2)CCCCC1 UFPFGVNKHCLJJO-SSKFGXFMSA-N 0.000 description 2
- HCSMRSHIIKPNAK-LSAVBLLPSA-N (2s)-n-[(1s)-2-[(3ar,7as)-6-(2-phenylethyl)-3,3a,4,5,7,7a-hexahydro-2h-pyrrolo[2,3-c]pyridin-1-yl]-1-cyclohexyl-2-oxoethyl]-2-(methylamino)propanamide Chemical compound C1([C@H](NC(=O)[C@H](C)NC)C(=O)N2[C@@H]3CN(CCC=4C=CC=CC=4)CC[C@@H]3CC2)CCCCC1 HCSMRSHIIKPNAK-LSAVBLLPSA-N 0.000 description 2
- OMRPLUKQNWNZAV-CONSDPRKSA-N (6as)-3-[3-[[(6as)-2-methoxy-8-(4-methoxyphenyl)-11-oxo-6a,7-dihydropyrrolo[2,1-c][1,4]benzodiazepin-3-yl]oxy]propoxy]-8-(4-aminophenyl)-2-methoxy-6a,7-dihydropyrrolo[2,1-c][1,4]benzodiazepin-11-one Chemical compound C1=CC(OC)=CC=C1C1=CN2C(=O)C3=CC(OC)=C(OCCCOC=4C(=CC=5C(=O)N6C=C(C[C@H]6C=NC=5C=4)C=4C=CC(N)=CC=4)OC)C=C3N=C[C@@H]2C1 OMRPLUKQNWNZAV-CONSDPRKSA-N 0.000 description 2
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 2
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 2
- OPAKEJZFFCECPN-XQRVVYSFSA-N (z)-3-[3-[3,5-bis(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]-n'-pyridin-2-ylprop-2-enehydrazide Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(C2=NN(\C=C/C(=O)NNC=3N=CC=CC=3)C=N2)=C1 OPAKEJZFFCECPN-XQRVVYSFSA-N 0.000 description 2
- KHWIRCOLWPNBJP-UHFFFAOYSA-N 1-(2-chloroethyl)-3-(2,6-dioxopiperidin-3-yl)-1-nitrosourea Chemical compound ClCCN(N=O)C(=O)NC1CCC(=O)NC1=O KHWIRCOLWPNBJP-UHFFFAOYSA-N 0.000 description 2
- WPHKIQPVPYJNAX-UHFFFAOYSA-N 1-[4-[4-amino-7-[1-(2-hydroxyethyl)pyrazol-4-yl]thieno[3,2-c]pyridin-3-yl]phenyl]-3-(3-fluorophenyl)urea Chemical compound C1=2SC=C(C=3C=CC(NC(=O)NC=4C=C(F)C=CC=4)=CC=3)C=2C(N)=NC=C1C=1C=NN(CCO)C=1 WPHKIQPVPYJNAX-UHFFFAOYSA-N 0.000 description 2
- CZDWSKBKCZWXFI-UHFFFAOYSA-N 1-morpholin-4-yl-3-[4-oxo-3-[4-[2-oxo-2-(4-propan-2-ylpiperazin-1-yl)ethoxy]phenyl]-1h-indeno[1,2-c]pyrazol-5-yl]urea Chemical compound C1CN(C(C)C)CCN1C(=O)COC1=CC=C(C=2C=3C(=O)C4=C(NC(=O)NN5CCOCC5)C=CC=C4C=3NN=2)C=C1 CZDWSKBKCZWXFI-UHFFFAOYSA-N 0.000 description 2
- HAWSQZCWOQZXHI-FQEVSTJZSA-N 10-Hydroxycamptothecin Chemical compound C1=C(O)C=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 HAWSQZCWOQZXHI-FQEVSTJZSA-N 0.000 description 2
- QFWCYNPOPKQOKV-UHFFFAOYSA-N 2-(2-amino-3-methoxyphenyl)chromen-4-one Chemical compound COC1=CC=CC(C=2OC3=CC=CC=C3C(=O)C=2)=C1N QFWCYNPOPKQOKV-UHFFFAOYSA-N 0.000 description 2
- JNAHVYVRKWKWKQ-UHFFFAOYSA-N 2-(2-methyl-2-pyrrolidinyl)-1H-benzimidazole-4-carboxamide Chemical compound N=1C2=C(C(N)=O)C=CC=C2NC=1C1(C)CCCN1 JNAHVYVRKWKWKQ-UHFFFAOYSA-N 0.000 description 2
- QINPEPAQOBZPOF-UHFFFAOYSA-N 2-amino-n-[3-[[3-(2-chloro-5-methoxyanilino)quinoxalin-2-yl]sulfamoyl]phenyl]-2-methylpropanamide Chemical compound COC1=CC=C(Cl)C(NC=2C(=NC3=CC=CC=C3N=2)NS(=O)(=O)C=2C=C(NC(=O)C(C)(C)N)C=CC=2)=C1 QINPEPAQOBZPOF-UHFFFAOYSA-N 0.000 description 2
- BJVRNXSHJLDZJR-UHFFFAOYSA-N 3-(1-methyl-4-morpholin-4-ylpyrazolo[3,4-d]pyrimidin-6-yl)phenol Chemical compound N1=C2N(C)N=CC2=C(N2CCOCC2)N=C1C1=CC=CC(O)=C1 BJVRNXSHJLDZJR-UHFFFAOYSA-N 0.000 description 2
- HXHAJRMTJXHJJZ-UHFFFAOYSA-N 3-[(4-bromo-2,6-difluorophenyl)methoxy]-5-(4-pyrrolidin-1-ylbutylcarbamoylamino)-1,2-thiazole-4-carboxamide Chemical compound S1N=C(OCC=2C(=CC(Br)=CC=2F)F)C(C(=O)N)=C1NC(=O)NCCCCN1CCCC1 HXHAJRMTJXHJJZ-UHFFFAOYSA-N 0.000 description 2
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 2
- PRDJGNVQBVXXEO-UHFFFAOYSA-N 3-cyanopropyl carbamimidothioate Chemical compound NC(=N)SCCCC#N PRDJGNVQBVXXEO-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 2
- WJRRGYBTGDJBFX-UHFFFAOYSA-N 4-(2-methyl-3-propan-2-yl-4-imidazolyl)-N-(4-methylsulfonylphenyl)-2-pyrimidinamine Chemical compound CC(C)N1C(C)=NC=C1C1=CC=NC(NC=2C=CC(=CC=2)S(C)(=O)=O)=N1 WJRRGYBTGDJBFX-UHFFFAOYSA-N 0.000 description 2
- CLPFFLWZZBQMAO-UHFFFAOYSA-N 4-(5,6,7,8-tetrahydroimidazo[1,5-a]pyridin-5-yl)benzonitrile Chemical compound C1=CC(C#N)=CC=C1C1N2C=NC=C2CCC1 CLPFFLWZZBQMAO-UHFFFAOYSA-N 0.000 description 2
- XXJWYDDUDKYVKI-UHFFFAOYSA-N 4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-(1-pyrrolidinyl)propoxy]quinazoline Chemical compound COC1=CC2=C(OC=3C(=C4C=C(C)NC4=CC=3)F)N=CN=C2C=C1OCCCN1CCCC1 XXJWYDDUDKYVKI-UHFFFAOYSA-N 0.000 description 2
- HHFBDROWDBDFBR-UHFFFAOYSA-N 4-[[9-chloro-7-(2,6-difluorophenyl)-5H-pyrimido[5,4-d][2]benzazepin-2-yl]amino]benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1NC1=NC=C(CN=C(C=2C3=CC=C(Cl)C=2)C=2C(=CC=CC=2F)F)C3=N1 HHFBDROWDBDFBR-UHFFFAOYSA-N 0.000 description 2
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical class C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 2
- MDOJTZQKHMAPBK-UHFFFAOYSA-N 4-iodo-3-nitrobenzamide Chemical compound NC(=O)C1=CC=C(I)C([N+]([O-])=O)=C1 MDOJTZQKHMAPBK-UHFFFAOYSA-N 0.000 description 2
- XTWYTFMLZFPYCI-KQYNXXCUSA-N 5'-adenylphosphoric acid Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XTWYTFMLZFPYCI-KQYNXXCUSA-N 0.000 description 2
- SRSGVKWWVXWSJT-ATVHPVEESA-N 5-[(z)-(5-fluoro-2-oxo-1h-indol-3-ylidene)methyl]-2,4-dimethyl-n-(2-pyrrolidin-1-ylethyl)-1h-pyrrole-3-carboxamide Chemical compound CC=1NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C(C)C=1C(=O)NCCN1CCCC1 SRSGVKWWVXWSJT-ATVHPVEESA-N 0.000 description 2
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 2
- UPALIKSFLSVKIS-UHFFFAOYSA-N 5-amino-2-[2-(dimethylamino)ethyl]benzo[de]isoquinoline-1,3-dione Chemical compound NC1=CC(C(N(CCN(C)C)C2=O)=O)=C3C2=CC=CC3=C1 UPALIKSFLSVKIS-UHFFFAOYSA-N 0.000 description 2
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 2
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 2
- DUUGKQCEGZLZNO-UHFFFAOYSA-N 5-hydroxyindoleacetic acid Chemical compound C1=C(O)C=C2C(CC(=O)O)=CNC2=C1 DUUGKQCEGZLZNO-UHFFFAOYSA-N 0.000 description 2
- DQOGWKZQQBYYMW-LQGIGNHCSA-N 5-methyl-6-[(3,4,5-trimethoxyanilino)methyl]quinazoline-2,4-diamine;(2s,3s,4s,5r,6s)-3,4,5,6-tetrahydroxyoxane-2-carboxylic acid Chemical compound O[C@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O.COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 DQOGWKZQQBYYMW-LQGIGNHCSA-N 0.000 description 2
- DOCINCLJNAXZQF-LBPRGKRZSA-N 6-fluoro-3-phenyl-2-[(1s)-1-(7h-purin-6-ylamino)ethyl]quinazolin-4-one Chemical compound C1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)=NC2=CC=C(F)C=C2C(=O)N1C1=CC=CC=C1 DOCINCLJNAXZQF-LBPRGKRZSA-N 0.000 description 2
- 102100026802 72 kDa type IV collagenase Human genes 0.000 description 2
- FUXVKZWTXQUGMW-FQEVSTJZSA-N 9-Aminocamptothecin Chemical compound C1=CC(N)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 FUXVKZWTXQUGMW-FQEVSTJZSA-N 0.000 description 2
- 101150054149 ANGPTL4 gene Proteins 0.000 description 2
- BUROJSBIWGDYCN-GAUTUEMISA-N AP 23573 Chemical compound C1C[C@@H](OP(C)(C)=O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 BUROJSBIWGDYCN-GAUTUEMISA-N 0.000 description 2
- GBJVVSCPOBPEIT-UHFFFAOYSA-N AZT-1152 Chemical compound N=1C=NC2=CC(OCCCN(CC)CCOP(O)(O)=O)=CC=C2C=1NC(=NN1)C=C1CC(=O)NC1=CC=CC(F)=C1 GBJVVSCPOBPEIT-UHFFFAOYSA-N 0.000 description 2
- XTWYTFMLZFPYCI-UHFFFAOYSA-N Adenosine diphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(O)=O)C(O)C1O XTWYTFMLZFPYCI-UHFFFAOYSA-N 0.000 description 2
- 102100026439 Adhesion G protein-coupled receptor E1 Human genes 0.000 description 2
- LIWMQSWFLXEGMA-WDSKDSINSA-N Ala-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@H](C)N LIWMQSWFLXEGMA-WDSKDSINSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 206010073478 Anaplastic large-cell lymphoma Diseases 0.000 description 2
- 102000045205 Angiopoietin-Like Protein 4 Human genes 0.000 description 2
- 108700042530 Angiopoietin-Like Protein 4 Proteins 0.000 description 2
- 108010032595 Antibody Binding Sites Proteins 0.000 description 2
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 2
- 101100330725 Arabidopsis thaliana DAR4 gene Proteins 0.000 description 2
- GOLCXWYRSKYTSP-UHFFFAOYSA-N Arsenious Acid Chemical compound O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 102100028820 Aspartate-tRNA ligase, cytoplasmic Human genes 0.000 description 2
- 241000271566 Aves Species 0.000 description 2
- YUXMAKUNSXIEKN-BTJKTKAUSA-N BGT226 Chemical compound OC(=O)\C=C/C(O)=O.C1=NC(OC)=CC=C1C1=CC=C(N=CC2=C3N(C=4C=C(C(N5CCNCC5)=CC=4)C(F)(F)F)C(=O)N2C)C3=C1 YUXMAKUNSXIEKN-BTJKTKAUSA-N 0.000 description 2
- 102000051485 Bcl-2 family Human genes 0.000 description 2
- 108700038897 Bcl-2 family Proteins 0.000 description 2
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 2
- KHBQMWCZKVMBLN-UHFFFAOYSA-N Benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1 KHBQMWCZKVMBLN-UHFFFAOYSA-N 0.000 description 2
- VGGGPCQERPFHOB-MCIONIFRSA-N Bestatin Chemical compound CC(C)C[C@H](C(O)=O)NC(=O)[C@@H](O)[C@H](N)CC1=CC=CC=C1 VGGGPCQERPFHOB-MCIONIFRSA-N 0.000 description 2
- 206010065553 Bone marrow failure Diseases 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- 102100032367 C-C motif chemokine 5 Human genes 0.000 description 2
- 102100025248 C-X-C motif chemokine 10 Human genes 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- SHHKQEUPHAENFK-UHFFFAOYSA-N Carboquone Chemical compound O=C1C(C)=C(N2CC2)C(=O)C(C(COC(N)=O)OC)=C1N1CC1 SHHKQEUPHAENFK-UHFFFAOYSA-N 0.000 description 2
- 102000011727 Caspases Human genes 0.000 description 2
- 108010076667 Caspases Proteins 0.000 description 2
- 108090000994 Catalytic RNA Proteins 0.000 description 2
- 102000053642 Catalytic RNA Human genes 0.000 description 2
- 108010072135 Cell Adhesion Molecule-1 Proteins 0.000 description 2
- 102100024649 Cell adhesion molecule 1 Human genes 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 2
- 101710112752 Cytotoxin Proteins 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 2
- GJKXGJCSJWBJEZ-XRSSZCMZSA-N Deslorelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CNC2=CC=CC=C12 GJKXGJCSJWBJEZ-XRSSZCMZSA-N 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- LQKSHSFQQRCAFW-UHFFFAOYSA-N Dolastatin 15 Natural products COC1=CC(=O)N(C(=O)C(OC(=O)C2N(CCC2)C(=O)C2N(CCC2)C(=O)C(C(C)C)N(C)C(=O)C(NC(=O)C(C(C)C)N(C)C)C(C)C)C(C)C)C1CC1=CC=CC=C1 LQKSHSFQQRCAFW-UHFFFAOYSA-N 0.000 description 2
- XXPXYPLPSDPERN-UHFFFAOYSA-N Ecteinascidin 743 Natural products COc1cc2C(NCCc2cc1O)C(=O)OCC3N4C(O)C5Cc6cc(C)c(OC)c(O)c6C(C4C(S)c7c(OC(=O)C)c(C)c8OCOc8c37)N5C XXPXYPLPSDPERN-UHFFFAOYSA-N 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- ULGZDMOVFRHVEP-RWJQBGPGSA-N Erythromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 ULGZDMOVFRHVEP-RWJQBGPGSA-N 0.000 description 2
- 102100038595 Estrogen receptor Human genes 0.000 description 2
- 108010029961 Filgrastim Proteins 0.000 description 2
- 108700000266 GSK923295 Proteins 0.000 description 2
- 241000287828 Gallus gallus Species 0.000 description 2
- 206010062878 Gastrooesophageal cancer Diseases 0.000 description 2
- SITLTJHOQZFJGG-XPUUQOCRSA-N Glu-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@@H](N)CCC(O)=O SITLTJHOQZFJGG-XPUUQOCRSA-N 0.000 description 2
- 102000003886 Glycoproteins Human genes 0.000 description 2
- 108090000288 Glycoproteins Proteins 0.000 description 2
- 102100039619 Granulocyte colony-stimulating factor Human genes 0.000 description 2
- 102000006354 HLA-DR Antigens Human genes 0.000 description 2
- 108010058597 HLA-DR Antigens Proteins 0.000 description 2
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 2
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 2
- 102100028006 Heme oxygenase 1 Human genes 0.000 description 2
- 102000003745 Hepatocyte Growth Factor Human genes 0.000 description 2
- 108090000100 Hepatocyte Growth Factor Proteins 0.000 description 2
- 241001272567 Hominoidea Species 0.000 description 2
- 101000718225 Homo sapiens Adhesion G protein-coupled receptor E1 Proteins 0.000 description 2
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 2
- 101000696909 Homo sapiens Aspartate-tRNA ligase, cytoplasmic Proteins 0.000 description 2
- 101000797762 Homo sapiens C-C motif chemokine 5 Proteins 0.000 description 2
- 101000858088 Homo sapiens C-X-C motif chemokine 10 Proteins 0.000 description 2
- 101001079623 Homo sapiens Heme oxygenase 1 Proteins 0.000 description 2
- 101000994375 Homo sapiens Integrin alpha-4 Proteins 0.000 description 2
- 101000916644 Homo sapiens Macrophage colony-stimulating factor 1 receptor Proteins 0.000 description 2
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 2
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 2
- 101000666896 Homo sapiens V-type immunoglobulin domain-containing suppressor of T-cell activation Proteins 0.000 description 2
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 2
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 2
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 2
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 2
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 2
- 102100032818 Integrin alpha-4 Human genes 0.000 description 2
- 108010047761 Interferon-alpha Proteins 0.000 description 2
- 102000006992 Interferon-alpha Human genes 0.000 description 2
- 229940121730 Janus kinase 2 inhibitor Drugs 0.000 description 2
- 102000010638 Kinesin Human genes 0.000 description 2
- 108010063296 Kinesin Proteins 0.000 description 2
- FADYJNXDPBKVCA-UHFFFAOYSA-N L-Phenylalanyl-L-lysin Natural products NCCCCC(C(O)=O)NC(=O)C(N)CC1=CC=CC=C1 FADYJNXDPBKVCA-UHFFFAOYSA-N 0.000 description 2
- DEFJQIDDEAULHB-IMJSIDKUSA-N L-alanyl-L-alanine Chemical compound C[C@H](N)C(=O)N[C@@H](C)C(O)=O DEFJQIDDEAULHB-IMJSIDKUSA-N 0.000 description 2
- 239000002137 L01XE24 - Ponatinib Substances 0.000 description 2
- UIARLYUEJFELEN-LROUJFHJSA-N LSM-1231 Chemical compound C12=C3N4C5=CC=CC=C5C3=C3C(=O)NCC3=C2C2=CC=CC=C2N1[C@]1(C)[C@](CO)(O)C[C@H]4O1 UIARLYUEJFELEN-LROUJFHJSA-N 0.000 description 2
- CZQHHVNHHHRRDU-UHFFFAOYSA-N LY294002 Chemical compound C1=CC=C2C(=O)C=C(N3CCOCC3)OC2=C1C1=CC=CC=C1 CZQHHVNHHHRRDU-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 208000032004 Large-Cell Anaplastic Lymphoma Diseases 0.000 description 2
- 229920001491 Lentinan Polymers 0.000 description 2
- 108010000817 Leuprolide Proteins 0.000 description 2
- 102100028198 Macrophage colony-stimulating factor 1 receptor Human genes 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 108010016165 Matrix Metalloproteinase 2 Proteins 0.000 description 2
- 102000001776 Matrix metalloproteinase-9 Human genes 0.000 description 2
- 108010015302 Matrix metalloproteinase-9 Proteins 0.000 description 2
- 102000008135 Mechanistic Target of Rapamycin Complex 1 Human genes 0.000 description 2
- 108010035196 Mechanistic Target of Rapamycin Complex 1 Proteins 0.000 description 2
- 102000009308 Mechanistic Target of Rapamycin Complex 2 Human genes 0.000 description 2
- 108010034057 Mechanistic Target of Rapamycin Complex 2 Proteins 0.000 description 2
- 102000018697 Membrane Proteins Human genes 0.000 description 2
- 108010052285 Membrane Proteins Proteins 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-M Methanesulfonate Chemical compound CS([O-])(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 2
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 description 2
- 229930192392 Mitomycin Natural products 0.000 description 2
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 2
- SITLTJHOQZFJGG-UHFFFAOYSA-N N-L-alpha-glutamyl-L-valine Natural products CC(C)C(C(O)=O)NC(=O)C(N)CCC(O)=O SITLTJHOQZFJGG-UHFFFAOYSA-N 0.000 description 2
- MVTQIFVKRXBCHS-SMMNFGSLSA-N N-[(3S,6S,12R,15S,16R,19S,22S)-3-benzyl-12-ethyl-4,16-dimethyl-2,5,11,14,18,21,24-heptaoxo-19-phenyl-17-oxa-1,4,10,13,20-pentazatricyclo[20.4.0.06,10]hexacosan-15-yl]-3-hydroxypyridine-2-carboxamide (10R,11R,12E,17E,19E,21S)-21-hydroxy-11,19-dimethyl-10-propan-2-yl-9,26-dioxa-3,15,28-triazatricyclo[23.2.1.03,7]octacosa-1(27),6,12,17,19,25(28)-hexaene-2,8,14,23-tetrone Chemical compound CC(C)[C@H]1OC(=O)C2=CCCN2C(=O)c2coc(CC(=O)C[C@H](O)\C=C(/C)\C=C\CNC(=O)\C=C\[C@H]1C)n2.CC[C@H]1NC(=O)[C@@H](NC(=O)c2ncccc2O)[C@@H](C)OC(=O)[C@@H](NC(=O)[C@@H]2CC(=O)CCN2C(=O)[C@H](Cc2ccccc2)N(C)C(=O)[C@@H]2CCCN2C1=O)c1ccccc1 MVTQIFVKRXBCHS-SMMNFGSLSA-N 0.000 description 2
- QGZYDVAGYRLSKP-UHFFFAOYSA-N N-[7-(hydroxyamino)-7-oxoheptyl]-2-(N-phenylanilino)-5-pyrimidinecarboxamide Chemical compound N1=CC(C(=O)NCCCCCCC(=O)NO)=CN=C1N(C=1C=CC=CC=1)C1=CC=CC=C1 QGZYDVAGYRLSKP-UHFFFAOYSA-N 0.000 description 2
- BLXXJMDCKKHMKV-UHFFFAOYSA-N Nabumetone Chemical compound C1=C(CCC(C)=O)C=CC2=CC(OC)=CC=C21 BLXXJMDCKKHMKV-UHFFFAOYSA-N 0.000 description 2
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- SUDAHWBOROXANE-SECBINFHSA-N PD 0325901 Chemical compound OC[C@@H](O)CONC(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F SUDAHWBOROXANE-SECBINFHSA-N 0.000 description 2
- QIUASFSNWYMDFS-NILGECQDSA-N PX-866 Chemical compound CC(=O)O[C@@H]1C[C@]2(C)C(=O)CC[C@H]2C2=C1[C@@]1(C)[C@@H](COC)OC(=O)\C(=C\N(CC=C)CC=C)C1=C(O)C2=O QIUASFSNWYMDFS-NILGECQDSA-N 0.000 description 2
- SHGAZHPCJJPHSC-UHFFFAOYSA-N Panrexin Chemical compound OC(=O)C=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-UHFFFAOYSA-N 0.000 description 2
- UOZODPSAJZTQNH-UHFFFAOYSA-N Paromomycin II Natural products NC1C(O)C(O)C(CN)OC1OC1C(O)C(OC2C(C(N)CC(N)C2O)OC2C(C(O)C(O)C(CO)O2)N)OC1CO UOZODPSAJZTQNH-UHFFFAOYSA-N 0.000 description 2
- FADYJNXDPBKVCA-STQMWFEESA-N Phe-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@@H](N)CC1=CC=CC=C1 FADYJNXDPBKVCA-STQMWFEESA-N 0.000 description 2
- 102000012288 Phosphopyruvate Hydratase Human genes 0.000 description 2
- 108010022181 Phosphopyruvate Hydratase Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 108091000080 Phosphotransferase Proteins 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- HRHKSTOGXBBQCB-UHFFFAOYSA-N Porfiromycine Chemical compound O=C1C(N)=C(C)C(=O)C2=C1C(COC(N)=O)C1(OC)C3N(C)C3CN12 HRHKSTOGXBBQCB-UHFFFAOYSA-N 0.000 description 2
- 102100033237 Pro-epidermal growth factor Human genes 0.000 description 2
- 101100528525 Prochlorococcus marinus (strain SARG / CCMP1375 / SS120) rnc gene Proteins 0.000 description 2
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 2
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 2
- 239000005464 Radotinib Substances 0.000 description 2
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 description 2
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 2
- 208000006265 Renal cell carcinoma Diseases 0.000 description 2
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 2
- 108091028664 Ribonucleotide Proteins 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 102100024834 T-cell immunoreceptor with Ig and ITIM domains Human genes 0.000 description 2
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 2
- 108700011582 TER 286 Proteins 0.000 description 2
- 108010000449 TNF-Related Apoptosis-Inducing Ligand Receptors Proteins 0.000 description 2
- 102000002259 TNF-Related Apoptosis-Inducing Ligand Receptors Human genes 0.000 description 2
- WFWLQNSHRPWKFK-UHFFFAOYSA-N Tegafur Chemical compound O=C1NC(=O)C(F)=CN1C1OCCC1 WFWLQNSHRPWKFK-UHFFFAOYSA-N 0.000 description 2
- 239000004098 Tetracycline Substances 0.000 description 2
- 102000002938 Thrombospondin Human genes 0.000 description 2
- 108010078233 Thymalfasin Proteins 0.000 description 2
- 239000003819 Toceranib Substances 0.000 description 2
- IVTVGDXNLFLDRM-HNNXBMFYSA-N Tomudex Chemical compound C=1C=C2NC(C)=NC(=O)C2=CC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)S1 IVTVGDXNLFLDRM-HNNXBMFYSA-N 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- 108091023040 Transcription factor Proteins 0.000 description 2
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- 102000006275 Ubiquitin-Protein Ligases Human genes 0.000 description 2
- 108010083111 Ubiquitin-Protein Ligases Proteins 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 102100038282 V-type immunoglobulin domain-containing suppressor of T-cell activation Human genes 0.000 description 2
- UPJONISHZRADBH-XPUUQOCRSA-N Val-Glu Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCC(O)=O UPJONISHZRADBH-XPUUQOCRSA-N 0.000 description 2
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 2
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 2
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 2
- OGQICQVSFDPSEI-UHFFFAOYSA-N Zorac Chemical compound N1=CC(C(=O)OCC)=CC=C1C#CC1=CC=C(SCCC2(C)C)C2=C1 OGQICQVSFDPSEI-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- 108010052004 acetyl-2-naphthylalanyl-3-chlorophenylalanyl-1-oxohexadecyl-seryl-4-aminophenylalanyl(hydroorotyl)-4-aminophenylalanyl(carbamoyl)-leucyl-ILys-prolyl-alaninamide Proteins 0.000 description 2
- 230000021736 acetylation Effects 0.000 description 2
- 238000006640 acetylation reaction Methods 0.000 description 2
- USZYSDMBJDPRIF-SVEJIMAYSA-N aclacinomycin A Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@H]1C)O[C@H]1[C@H](C[C@@H](O[C@H]1C)O[C@H]1C[C@]([C@@H](C2=CC=3C(=O)C4=CC=CC(O)=C4C(=O)C=3C(O)=C21)C(=O)OC)(O)CC)N(C)C)[C@H]1CCC(=O)[C@H](C)O1 USZYSDMBJDPRIF-SVEJIMAYSA-N 0.000 description 2
- 229960004176 aclarubicin Drugs 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 229950009084 adecatumumab Drugs 0.000 description 2
- 201000008395 adenosquamous carcinoma Diseases 0.000 description 2
- 229940009456 adriamycin Drugs 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 108010056243 alanylalanine Proteins 0.000 description 2
- 125000003158 alcohol group Chemical group 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- KDGFLJKFZUIJMX-UHFFFAOYSA-N alectinib Chemical compound CCC1=CC=2C(=O)C(C3=CC=C(C=C3N3)C#N)=C3C(C)(C)C=2C=C1N(CC1)CCC1N1CCOCC1 KDGFLJKFZUIJMX-UHFFFAOYSA-N 0.000 description 2
- IHUNBGSDBOWDMA-AQFIFDHZSA-N all-trans-acitretin Chemical compound COC1=CC(C)=C(\C=C\C(\C)=C\C=C\C(\C)=C\C(O)=O)C(C)=C1C IHUNBGSDBOWDMA-AQFIFDHZSA-N 0.000 description 2
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 2
- 229960004701 amonafide Drugs 0.000 description 2
- 229960002932 anastrozole Drugs 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 229940124650 anti-cancer therapies Drugs 0.000 description 2
- 230000002927 anti-mitotic effect Effects 0.000 description 2
- 238000011319 anticancer therapy Methods 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 239000000074 antisense oligonucleotide Substances 0.000 description 2
- 238000012230 antisense oligonucleotides Methods 0.000 description 2
- 239000003443 antiviral agent Substances 0.000 description 2
- 229940121357 antivirals Drugs 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- MOTJMGVDPWRKOC-QPVYNBJUSA-N atrasentan Chemical compound C1([C@H]2[C@@H]([C@H](CN2CC(=O)N(CCCC)CCCC)C=2C=C3OCOC3=CC=2)C(O)=O)=CC=C(OC)C=C1 MOTJMGVDPWRKOC-QPVYNBJUSA-N 0.000 description 2
- 239000005441 aurora Substances 0.000 description 2
- KLNFSAOEKUDMFA-UHFFFAOYSA-N azanide;2-hydroxyacetic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OCC(O)=O KLNFSAOEKUDMFA-UHFFFAOYSA-N 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- PYXFVCFISTUSOO-UHFFFAOYSA-N betulafolienetriol Natural products C1CC(O)C(C)(C)C2CCC3(C)C4(C)CCC(C(C)(O)CCC=C(C)C)C4C(O)CC3C21C PYXFVCFISTUSOO-UHFFFAOYSA-N 0.000 description 2
- 229960002938 bexarotene Drugs 0.000 description 2
- 229960000997 bicalutamide Drugs 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 229960001561 bleomycin Drugs 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 229960001467 bortezomib Drugs 0.000 description 2
- 229960003736 bosutinib Drugs 0.000 description 2
- 239000004067 bulking agent Substances 0.000 description 2
- 229960002719 buserelin Drugs 0.000 description 2
- 229960002092 busulfan Drugs 0.000 description 2
- 239000012830 cancer therapeutic Substances 0.000 description 2
- 229940022399 cancer vaccine Drugs 0.000 description 2
- 238000009566 cancer vaccine Methods 0.000 description 2
- 229960002115 carboquone Drugs 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 108010021331 carfilzomib Proteins 0.000 description 2
- 229960005243 carmustine Drugs 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 2
- 229960002412 cediranib Drugs 0.000 description 2
- KQJSQWZMSAGSHN-JJWQIEBTSA-N celastrol Chemical compound C([C@H]1[C@]2(C)CC[C@@]34C)[C@](C)(C(O)=O)CC[C@]1(C)CC[C@]2(C)C4=CC=C1C3=CC(=O)C(O)=C1C KQJSQWZMSAGSHN-JJWQIEBTSA-N 0.000 description 2
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 2
- VERWOWGGCGHDQE-UHFFFAOYSA-N ceritinib Chemical compound CC=1C=C(NC=2N=C(NC=3C(=CC=CC=3)S(=O)(=O)C(C)C)C(Cl)=CN=2)C(OC(C)C)=CC=1C1CCNCC1 VERWOWGGCGHDQE-UHFFFAOYSA-N 0.000 description 2
- 229960003230 cetrorelix Drugs 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 235000013330 chicken meat Nutrition 0.000 description 2
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 2
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 2
- 229960004630 chlorambucil Drugs 0.000 description 2
- 229960001480 chlorozotocin Drugs 0.000 description 2
- 229960001338 colchicine Drugs 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 230000000139 costimulatory effect Effects 0.000 description 2
- 229940043378 cyclin-dependent kinase inhibitor Drugs 0.000 description 2
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 2
- GLNWREBYRLDPQP-MHZLTWQESA-N cyclopentyl (2s)-2-[[4-[[8-(hydroxyamino)-8-oxooctanoyl]amino]phenyl]methylamino]-2-phenylacetate Chemical compound C1=CC(NC(=O)CCCCCCC(=O)NO)=CC=C1CN[C@@H](C=1C=CC=CC=1)C(=O)OC1CCCC1 GLNWREBYRLDPQP-MHZLTWQESA-N 0.000 description 2
- 150000001945 cysteines Chemical class 0.000 description 2
- 239000002619 cytotoxin Substances 0.000 description 2
- 229950006418 dactolisib Drugs 0.000 description 2
- JOGKUKXHTYWRGZ-UHFFFAOYSA-N dactolisib Chemical compound O=C1N(C)C2=CN=C3C=CC(C=4C=C5C=CC=CC5=NC=4)=CC3=C2N1C1=CC=C(C(C)(C)C#N)C=C1 JOGKUKXHTYWRGZ-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- 229960002272 degarelix Drugs 0.000 description 2
- MEUCPCLKGZSHTA-XYAYPHGZSA-N degarelix Chemical compound C([C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCNC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)NC(=O)[C@H](CC=1C=CC(NC(=O)[C@H]2NC(=O)NC(=O)C2)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(NC(N)=O)C=C1 MEUCPCLKGZSHTA-XYAYPHGZSA-N 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 238000004925 denaturation Methods 0.000 description 2
- 230000036425 denaturation Effects 0.000 description 2
- WAZQAZKAZLXFMK-UHFFFAOYSA-N deracoxib Chemical compound C1=C(F)C(OC)=CC=C1C1=CC(C(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 WAZQAZKAZLXFMK-UHFFFAOYSA-N 0.000 description 2
- 229960005408 deslorelin Drugs 0.000 description 2
- 108700025485 deslorelin Proteins 0.000 description 2
- 239000003599 detergent Substances 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 229960000605 dexrazoxane Drugs 0.000 description 2
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 2
- WVYXNIXAMZOZFK-UHFFFAOYSA-N diaziquone Chemical compound O=C1C(NC(=O)OCC)=C(N2CC2)C(=O)C(NC(=O)OCC)=C1N1CC1 WVYXNIXAMZOZFK-UHFFFAOYSA-N 0.000 description 2
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 2
- 230000009699 differential effect Effects 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- HUPFGZXOMWLGNK-UHFFFAOYSA-N diflunisal Chemical compound C1=C(O)C(C(=O)O)=CC(C=2C(=CC(F)=CC=2)F)=C1 HUPFGZXOMWLGNK-UHFFFAOYSA-N 0.000 description 2
- GDLPAGOVHZLZEK-JBUFHSOLSA-L disodium;(4s)-4-amino-5-[[(1s)-1-carboxylato-2-(1h-indol-3-yl)ethyl]amino]-5-oxopentanoate Chemical compound [Na+].[Na+].C1=CC=C2C(C[C@H](NC(=O)[C@H](CCC([O-])=O)N)C([O-])=O)=CNC2=C1 GDLPAGOVHZLZEK-JBUFHSOLSA-L 0.000 description 2
- 125000002228 disulfide group Chemical group 0.000 description 2
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- HKXBNHCUPKIYDM-CGMHZMFXSA-N doxercalciferol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)/C=C/[C@H](C)C(C)C)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C HKXBNHCUPKIYDM-CGMHZMFXSA-N 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 238000012377 drug delivery Methods 0.000 description 2
- 229950009791 durvalumab Drugs 0.000 description 2
- BNFRJXLZYUTIII-UHFFFAOYSA-N efaproxiral Chemical compound CC1=CC(C)=CC(NC(=O)CC=2C=CC(OC(C)(C)C(O)=O)=CC=2)=C1 BNFRJXLZYUTIII-UHFFFAOYSA-N 0.000 description 2
- CTSPAMFJBXKSOY-UHFFFAOYSA-N ellipticine Chemical compound N1=CC=C2C(C)=C(NC=3C4=CC=CC=3)C4=C(C)C2=C1 CTSPAMFJBXKSOY-UHFFFAOYSA-N 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- INVTYAOGFAGBOE-UHFFFAOYSA-N entinostat Chemical compound NC1=CC=CC=C1NC(=O)C(C=C1)=CC=C1CNC(=O)OCC1=CC=CN=C1 INVTYAOGFAGBOE-UHFFFAOYSA-N 0.000 description 2
- WXCXUHSOUPDCQV-UHFFFAOYSA-N enzalutamide Chemical compound C1=C(F)C(C(=O)NC)=CC=C1N1C(C)(C)C(=O)N(C=2C=C(C(C#N)=CC=2)C(F)(F)F)C1=S WXCXUHSOUPDCQV-UHFFFAOYSA-N 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- QXRSDHAAWVKZLJ-PVYNADRNSA-N epothilone B Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 QXRSDHAAWVKZLJ-PVYNADRNSA-N 0.000 description 2
- XOZIUKBZLSUILX-GIQCAXHBSA-N epothilone D Chemical compound O1C(=O)C[C@H](O)C(C)(C)C(=O)[C@H](C)[C@@H](O)[C@@H](C)CCC\C(C)=C/C[C@H]1C(\C)=C\C1=CSC(C)=N1 XOZIUKBZLSUILX-GIQCAXHBSA-N 0.000 description 2
- 108010038795 estrogen receptors Proteins 0.000 description 2
- NNYBQONXHNTVIJ-UHFFFAOYSA-N etodolac Chemical compound C1COC(CC)(CC(O)=O)C2=C1C(C=CC=C1CC)=C1N2 NNYBQONXHNTVIJ-UHFFFAOYSA-N 0.000 description 2
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 2
- 229960005167 everolimus Drugs 0.000 description 2
- 229960000255 exemestane Drugs 0.000 description 2
- 229950000484 exisulind Drugs 0.000 description 2
- 229950011548 fadrozole Drugs 0.000 description 2
- 229950000133 filanesib Drugs 0.000 description 2
- LLXISKGBWFTGEI-FQEVSTJZSA-N filanesib Chemical compound C1([C@]2(CCCN)SC(=NN2C(=O)N(C)OC)C=2C(=CC=C(F)C=2)F)=CC=CC=C1 LLXISKGBWFTGEI-FQEVSTJZSA-N 0.000 description 2
- 229960004039 finasteride Drugs 0.000 description 2
- 229960000390 fludarabine Drugs 0.000 description 2
- 229960002074 flutamide Drugs 0.000 description 2
- JYEFSHLLTQIXIO-SMNQTINBSA-N folfiri regimen Chemical compound FC1=CNC(=O)NC1=O.C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1.C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 JYEFSHLLTQIXIO-SMNQTINBSA-N 0.000 description 2
- 229960004421 formestane Drugs 0.000 description 2
- OSVMTWJCGUFAOD-KZQROQTASA-N formestane Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1O OSVMTWJCGUFAOD-KZQROQTASA-N 0.000 description 2
- YAKWPXVTIGTRJH-UHFFFAOYSA-N fotemustine Chemical compound CCOP(=O)(OCC)C(C)NC(=O)N(CCCl)N=O YAKWPXVTIGTRJH-UHFFFAOYSA-N 0.000 description 2
- 229960004783 fotemustine Drugs 0.000 description 2
- 229960002258 fulvestrant Drugs 0.000 description 2
- 208000010749 gastric carcinoma Diseases 0.000 description 2
- 201000006974 gastroesophageal cancer Diseases 0.000 description 2
- 229960003297 gemtuzumab ozogamicin Drugs 0.000 description 2
- 229940080856 gleevec Drugs 0.000 description 2
- 239000003862 glucocorticoid Substances 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 229960002913 goserelin Drugs 0.000 description 2
- QBKSWRVVCFFDOT-UHFFFAOYSA-N gossypol Chemical compound CC(C)C1=C(O)C(O)=C(C=O)C2=C(O)C(C=3C(O)=C4C(C=O)=C(O)C(O)=C(C4=CC=3C)C(C)C)=C(C)C=C21 QBKSWRVVCFFDOT-UHFFFAOYSA-N 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- 229940022353 herceptin Drugs 0.000 description 2
- 239000008241 heterogeneous mixture Substances 0.000 description 2
- PPZMYIBUHIPZOS-UHFFFAOYSA-N histamine dihydrochloride Chemical compound Cl.Cl.NCCC1=CN=CN1 PPZMYIBUHIPZOS-UHFFFAOYSA-N 0.000 description 2
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 2
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 2
- 229940124866 human papillomavirus vaccine Drugs 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 238000004191 hydrophobic interaction chromatography Methods 0.000 description 2
- 229960001330 hydroxycarbamide Drugs 0.000 description 2
- 229960001680 ibuprofen Drugs 0.000 description 2
- 229960002411 imatinib Drugs 0.000 description 2
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 2
- 239000000367 immunologic factor Substances 0.000 description 2
- 239000002955 immunomodulating agent Substances 0.000 description 2
- DBIGHPPNXATHOF-UHFFFAOYSA-N improsulfan Chemical compound CS(=O)(=O)OCCCNCCCOS(C)(=O)=O DBIGHPPNXATHOF-UHFFFAOYSA-N 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 230000005917 in vivo anti-tumor Effects 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 2
- 229960000367 inositol Drugs 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 239000012444 intercalating antibiotic Substances 0.000 description 2
- 229940047124 interferons Drugs 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000007913 intrathecal administration Methods 0.000 description 2
- 230000002601 intratumoral effect Effects 0.000 description 2
- DKYWVDODHFEZIM-UHFFFAOYSA-N ketoprofen Chemical compound OC(=O)C(C)C1=CC=CC(C(=O)C=2C=CC=CC=2)=C1 DKYWVDODHFEZIM-UHFFFAOYSA-N 0.000 description 2
- 229940001447 lactate Drugs 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 108010021336 lanreotide Proteins 0.000 description 2
- 201000003445 large cell neuroendocrine carcinoma Diseases 0.000 description 2
- DXOJIXGRFSHVKA-BZVZGCBYSA-N larotaxel Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@@]23[C@H]1[C@@]1(CO[C@@H]1C[C@@H]2C3)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 DXOJIXGRFSHVKA-BZVZGCBYSA-N 0.000 description 2
- 229960002367 lasofoxifene Drugs 0.000 description 2
- GXESHMAMLJKROZ-IAPPQJPRSA-N lasofoxifene Chemical compound C1([C@@H]2[C@@H](C3=CC=C(C=C3CC2)O)C=2C=CC(OCCN3CCCC3)=CC=2)=CC=CC=C1 GXESHMAMLJKROZ-IAPPQJPRSA-N 0.000 description 2
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 2
- 229940115286 lentinan Drugs 0.000 description 2
- 229960003881 letrozole Drugs 0.000 description 2
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 2
- 229960004338 leuprorelin Drugs 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- MPVGZUGXCQEXTM-UHFFFAOYSA-N linifanib Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C(N)=NNC=3C=CC=2)=C1 MPVGZUGXCQEXTM-UHFFFAOYSA-N 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 239000012669 liquid formulation Substances 0.000 description 2
- 229960002247 lomustine Drugs 0.000 description 2
- KHPKQFYUPIUARC-UHFFFAOYSA-N lumiracoxib Chemical compound OC(=O)CC1=CC(C)=CC=C1NC1=C(F)C=CC=C1Cl KHPKQFYUPIUARC-UHFFFAOYSA-N 0.000 description 2
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 229950002736 marizomib Drugs 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 229960001428 mercaptopurine Drugs 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 2
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 2
- 229960002216 methylparaben Drugs 0.000 description 2
- 238000002493 microarray Methods 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 108700007621 mifamurtide Proteins 0.000 description 2
- 229960005485 mitobronitol Drugs 0.000 description 2
- 229960001156 mitoxantrone Drugs 0.000 description 2
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 2
- QXYYYPFGTSJXNS-UHFFFAOYSA-N mitozolomide Chemical compound N1=NN(CCCl)C(=O)N2C1=C(C(=O)N)N=C2 QXYYYPFGTSJXNS-UHFFFAOYSA-N 0.000 description 2
- PJUIMOJAAPLTRJ-UHFFFAOYSA-N monothioglycerol Chemical compound OCC(O)CS PJUIMOJAAPLTRJ-UHFFFAOYSA-N 0.000 description 2
- ZTFBIUXIQYRUNT-MDWZMJQESA-N mubritinib Chemical compound C1=CC(C(F)(F)F)=CC=C1\C=C\C1=NC(COC=2C=CC(CCCCN3N=NC=C3)=CC=2)=CO1 ZTFBIUXIQYRUNT-MDWZMJQESA-N 0.000 description 2
- ISGGVCWFTPTHIX-UHFFFAOYSA-N n'-(2-hydroxy-3-piperidin-1-ylpropoxy)pyridine-3-carboximidamide;dihydrochloride Chemical compound Cl.Cl.C1CCCCN1CC(O)CONC(=N)C1=CC=CN=C1 ISGGVCWFTPTHIX-UHFFFAOYSA-N 0.000 description 2
- SMFXSYMLJDHGIE-UHFFFAOYSA-N n-(3-aminopropyl)-n-[1-(5-benzyl-3-methyl-4-oxo-[1,2]thiazolo[5,4-d]pyrimidin-6-yl)-2-methylpropyl]-4-methylbenzamide Chemical compound N=1C=2SN=C(C)C=2C(=O)N(CC=2C=CC=CC=2)C=1C(C(C)C)N(CCCN)C(=O)C1=CC=C(C)C=C1 SMFXSYMLJDHGIE-UHFFFAOYSA-N 0.000 description 2
- 229960002009 naproxen Drugs 0.000 description 2
- 229950007221 nedaplatin Drugs 0.000 description 2
- QZGIWPZCWHMVQL-UIYAJPBUSA-N neocarzinostatin chromophore Chemical compound O1[C@H](C)[C@H](O)[C@H](O)[C@@H](NC)[C@H]1O[C@@H]1C/2=C/C#C[C@H]3O[C@@]3([C@@H]3OC(=O)OC3)C#CC\2=C[C@H]1OC(=O)C1=C(O)C=CC2=C(C)C=C(OC)C=C12 QZGIWPZCWHMVQL-UIYAJPBUSA-N 0.000 description 2
- 229960002653 nilutamide Drugs 0.000 description 2
- 229960001420 nimustine Drugs 0.000 description 2
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 2
- 229960000572 olaparib Drugs 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- OFPXSFXSNFPTHF-UHFFFAOYSA-N oxaprozin Chemical compound O1C(CCC(=O)O)=NC(C=2C=CC=CC=2)=C1C1=CC=CC=C1 OFPXSFXSNFPTHF-UHFFFAOYSA-N 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 2
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 description 2
- 210000000496 pancreas Anatomy 0.000 description 2
- UOZODPSAJZTQNH-LSWIJEOBSA-N paromomycin Chemical compound N[C@@H]1[C@@H](O)[C@H](O)[C@H](CN)O[C@@H]1O[C@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](N)C[C@@H](N)[C@@H]2O)O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)N)O[C@@H]1CO UOZODPSAJZTQNH-LSWIJEOBSA-N 0.000 description 2
- 229960001914 paromomycin Drugs 0.000 description 2
- 229960000639 pazopanib Drugs 0.000 description 2
- AQIXEPGDORPWBJ-UHFFFAOYSA-N pentan-3-ol Chemical compound CCC(O)CC AQIXEPGDORPWBJ-UHFFFAOYSA-N 0.000 description 2
- 229960002340 pentostatin Drugs 0.000 description 2
- 238000002823 phage display Methods 0.000 description 2
- ZJAOAACCNHFJAH-UHFFFAOYSA-N phosphonoformic acid Chemical compound OC(=O)P(O)(O)=O ZJAOAACCNHFJAH-UHFFFAOYSA-N 0.000 description 2
- 102000020233 phosphotransferase Human genes 0.000 description 2
- IIMIOEBMYPRQGU-UHFFFAOYSA-L picoplatin Chemical compound N.[Cl-].[Cl-].[Pt+2].CC1=CC=CC=N1 IIMIOEBMYPRQGU-UHFFFAOYSA-L 0.000 description 2
- LHNIIDJUOCFXAP-UHFFFAOYSA-N pictrelisib Chemical compound C1CN(S(=O)(=O)C)CCN1CC1=CC2=NC(C=3C=4C=NNC=4C=CC=3)=NC(N3CCOCC3)=C2S1 LHNIIDJUOCFXAP-UHFFFAOYSA-N 0.000 description 2
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 2
- 229960000952 pipobroman Drugs 0.000 description 2
- QYSPLQLAKJAUJT-UHFFFAOYSA-N piroxicam Chemical compound OC=1C2=CC=CC=C2S(=O)(=O)N(C)C=1C(=O)NC1=CC=CC=N1 QYSPLQLAKJAUJT-UHFFFAOYSA-N 0.000 description 2
- 230000010287 polarization Effects 0.000 description 2
- 229920001983 poloxamer Polymers 0.000 description 2
- 229920001515 polyalkylene glycol Polymers 0.000 description 2
- PHXJVRSECIGDHY-UHFFFAOYSA-N ponatinib Chemical compound C1CN(C)CCN1CC(C(=C1)C(F)(F)F)=CC=C1NC(=O)C1=CC=C(C)C(C#CC=2N3N=CC=CC3=NC=2)=C1 PHXJVRSECIGDHY-UHFFFAOYSA-N 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 108010090388 progesterone receptor A Proteins 0.000 description 2
- NLNGWFLRRRYNIL-PLNGDYQASA-N propan-2-yl (z)-3-[3-[3-methoxy-5-(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]prop-2-enoate Chemical compound FC(F)(F)C1=CC(OC)=CC(C2=NN(\C=C/C(=O)OC(C)C)C=N2)=C1 NLNGWFLRRRYNIL-PLNGDYQASA-N 0.000 description 2
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 2
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 2
- 229960003415 propylparaben Drugs 0.000 description 2
- 210000002307 prostate Anatomy 0.000 description 2
- 229940121649 protein inhibitor Drugs 0.000 description 2
- 239000012268 protein inhibitor Substances 0.000 description 2
- 238000001742 protein purification Methods 0.000 description 2
- 150000003212 purines Chemical class 0.000 description 2
- 150000003230 pyrimidines Chemical class 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 229950004043 radotinib Drugs 0.000 description 2
- DUPWHXBITIZIKZ-UHFFFAOYSA-N radotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3N=CC=NC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 DUPWHXBITIZIKZ-UHFFFAOYSA-N 0.000 description 2
- 229960004622 raloxifene Drugs 0.000 description 2
- 229960004432 raltitrexed Drugs 0.000 description 2
- 229960002185 ranimustine Drugs 0.000 description 2
- FNHKPVJBJVTLMP-UHFFFAOYSA-N regorafenib Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=C(F)C(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 FNHKPVJBJVTLMP-UHFFFAOYSA-N 0.000 description 2
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 229960000329 ribavirin Drugs 0.000 description 2
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 2
- 125000002652 ribonucleotide group Chemical group 0.000 description 2
- 108091092562 ribozyme Proteins 0.000 description 2
- 229960004641 rituximab Drugs 0.000 description 2
- RZJQGNCSTQAWON-UHFFFAOYSA-N rofecoxib Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C(=O)OC1 RZJQGNCSTQAWON-UHFFFAOYSA-N 0.000 description 2
- OHRURASPPZQGQM-GCCNXGTGSA-N romidepsin Chemical compound O1C(=O)[C@H](C(C)C)NC(=O)C(=C/C)/NC(=O)[C@H]2CSSCC\C=C\[C@@H]1CC(=O)N[C@H](C(C)C)C(=O)N2 OHRURASPPZQGQM-GCCNXGTGSA-N 0.000 description 2
- NGWSFRIPKNWYAO-SHTIJGAHSA-N salinosporamide A Chemical compound C([C@@H]1[C@H](O)[C@]23C(=O)O[C@]2([C@H](C(=O)N3)CCCl)C)CCC=C1 NGWSFRIPKNWYAO-SHTIJGAHSA-N 0.000 description 2
- WVYADZUPLLSGPU-UHFFFAOYSA-N salsalate Chemical compound OC(=O)C1=CC=CC=C1OC(=O)C1=CC=CC=C1O WVYADZUPLLSGPU-UHFFFAOYSA-N 0.000 description 2
- 229960005399 satraplatin Drugs 0.000 description 2
- 190014017285 satraplatin Chemical compound 0.000 description 2
- CDAISMWEOUEBRE-UHFFFAOYSA-N scyllo-inosotol Natural products OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 2
- 229950000055 seliciclib Drugs 0.000 description 2
- CYOHGALHFOKKQC-UHFFFAOYSA-N selumetinib Chemical compound OCCONC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1Cl CYOHGALHFOKKQC-UHFFFAOYSA-N 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- UPMFZISCCZSDND-JJKGCWMISA-M sodium gluconate Chemical compound [Na+].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O UPMFZISCCZSDND-JJKGCWMISA-M 0.000 description 2
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000012409 standard PCR amplification Methods 0.000 description 2
- 239000008227 sterile water for injection Substances 0.000 description 2
- 201000000498 stomach carcinoma Diseases 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 229960001052 streptozocin Drugs 0.000 description 2
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 2
- SUVMJBTUFCVSAD-UHFFFAOYSA-N sulforaphane Chemical compound CS(=O)CCCCN=C=S SUVMJBTUFCVSAD-UHFFFAOYSA-N 0.000 description 2
- 229960000894 sulindac Drugs 0.000 description 2
- MLKXDPUZXIRXEP-MFOYZWKCSA-N sulindac Chemical compound CC1=C(CC(O)=O)C2=CC(F)=CC=C2\C1=C/C1=CC=C(S(C)=O)C=C1 MLKXDPUZXIRXEP-MFOYZWKCSA-N 0.000 description 2
- MVGSNCBCUWPVDA-MFOYZWKCSA-N sulindac sulfone Chemical compound CC1=C(CC(O)=O)C2=CC(F)=CC=C2\C1=C/C1=CC=C(S(C)(=O)=O)C=C1 MVGSNCBCUWPVDA-MFOYZWKCSA-N 0.000 description 2
- 239000013589 supplement Substances 0.000 description 2
- 230000000153 supplemental effect Effects 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 229940037128 systemic glucocorticoids Drugs 0.000 description 2
- 229960003454 tamoxifen citrate Drugs 0.000 description 2
- 229940099419 targretin Drugs 0.000 description 2
- 229940069905 tasigna Drugs 0.000 description 2
- QFJCIRLUMZQUOT-UHFFFAOYSA-N temsirolimus Natural products C1CC(O)C(OC)CC1CC(C)C1OC(=O)C2CCCCN2C(=O)C(=O)C(O)(O2)C(C)CCC2CC(OC)C(C)=CC=CC=CC(C)CC(C)C(=O)C(OC)C(O)C(C)=CC(C)C(=O)C1 QFJCIRLUMZQUOT-UHFFFAOYSA-N 0.000 description 2
- 229960002180 tetracycline Drugs 0.000 description 2
- 229930101283 tetracycline Natural products 0.000 description 2
- 235000019364 tetracycline Nutrition 0.000 description 2
- 150000003522 tetracyclines Chemical class 0.000 description 2
- NZVYCXVTEHPMHE-ZSUJOUNUSA-N thymalfasin Chemical compound CC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O NZVYCXVTEHPMHE-ZSUJOUNUSA-N 0.000 description 2
- 229960004231 thymalfasin Drugs 0.000 description 2
- 229960004089 tigecycline Drugs 0.000 description 2
- FPZLLRFZJZRHSY-HJYUBDRYSA-N tigecycline Chemical class C([C@H]1C2)C3=C(N(C)C)C=C(NC(=O)CNC(C)(C)C)C(O)=C3C(=O)C1=C(O)[C@@]1(O)[C@@H]2[C@H](N(C)C)C(O)=C(C(N)=O)C1=O FPZLLRFZJZRHSY-HJYUBDRYSA-N 0.000 description 2
- 229960003087 tioguanine Drugs 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 2
- 229960005026 toremifene Drugs 0.000 description 2
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 2
- AKCRNFFTGXBONI-UHFFFAOYSA-N torin 1 Chemical compound C1CN(C(=O)CC)CCN1C1=CC=C(N2C(C=CC3=C2C2=CC(=CC=C2N=C3)C=2C=C3C=CC=CC3=NC=2)=O)C=C1C(F)(F)F AKCRNFFTGXBONI-UHFFFAOYSA-N 0.000 description 2
- 229960005267 tositumomab Drugs 0.000 description 2
- PKVRCIRHQMSYJX-AIFWHQITSA-N trabectedin Chemical compound C([C@@]1(C(OC2)=O)NCCC3=C1C=C(C(=C3)O)OC)S[C@@H]1C3=C(OC(C)=O)C(C)=C4OCOC4=C3[C@H]2N2[C@@H](O)[C@H](CC=3C4=C(O)C(OC)=C(C)C=3)N(C)[C@H]4[C@@H]21 PKVRCIRHQMSYJX-AIFWHQITSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000002103 transcriptional effect Effects 0.000 description 2
- 238000001890 transfection Methods 0.000 description 2
- 239000003558 transferase inhibitor Substances 0.000 description 2
- 230000014616 translation Effects 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- 229960001727 tretinoin Drugs 0.000 description 2
- 229960001099 trimetrexate Drugs 0.000 description 2
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 2
- 190014017283 triplatin tetranitrate Chemical compound 0.000 description 2
- 229950002860 triplatin tetranitrate Drugs 0.000 description 2
- XPFJYKARVSSRHE-UHFFFAOYSA-K trisodium;2-hydroxypropane-1,2,3-tricarboxylate;2-hydroxypropane-1,2,3-tricarboxylic acid Chemical compound [Na+].[Na+].[Na+].OC(=O)CC(O)(C(O)=O)CC(O)=O.[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O XPFJYKARVSSRHE-UHFFFAOYSA-K 0.000 description 2
- 229950010147 troxacitabine Drugs 0.000 description 2
- RXRGZNYSEHTMHC-BQBZGAKWSA-N troxacitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)OC1 RXRGZNYSEHTMHC-BQBZGAKWSA-N 0.000 description 2
- 229940094060 tykerb Drugs 0.000 description 2
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 2
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 2
- 229950009811 ubenimex Drugs 0.000 description 2
- 241000701161 unidentified adenovirus Species 0.000 description 2
- 229960001055 uracil mustard Drugs 0.000 description 2
- LNPDTQAFDNKSHK-UHFFFAOYSA-N valdecoxib Chemical compound CC=1ON=C(C=2C=CC=CC=2)C=1C1=CC=C(S(N)(=O)=O)C=C1 LNPDTQAFDNKSHK-UHFFFAOYSA-N 0.000 description 2
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 2
- 229960000604 valproic acid Drugs 0.000 description 2
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 2
- 229960003862 vemurafenib Drugs 0.000 description 2
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 2
- LQBVNQSMGBZMKD-UHFFFAOYSA-N venetoclax Chemical compound C=1C=C(Cl)C=CC=1C=1CC(C)(C)CCC=1CN(CC1)CCN1C(C=C1OC=2C=C3C=CNC3=NC=2)=CC=C1C(=O)NS(=O)(=O)C(C=C1[N+]([O-])=O)=CC=C1NCC1CCOCC1 LQBVNQSMGBZMKD-UHFFFAOYSA-N 0.000 description 2
- 229960001183 venetoclax Drugs 0.000 description 2
- AQTQHPDCURKLKT-JKDPCDLQSA-N vincristine sulfate Chemical compound OS(O)(=O)=O.C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C=O)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 AQTQHPDCURKLKT-JKDPCDLQSA-N 0.000 description 2
- 229940049068 xalkori Drugs 0.000 description 2
- 229940053867 xeloda Drugs 0.000 description 2
- 239000000811 xylitol Substances 0.000 description 2
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 2
- 235000010447 xylitol Nutrition 0.000 description 2
- 229960002675 xylitol Drugs 0.000 description 2
- 229940055760 yervoy Drugs 0.000 description 2
- 229950009268 zinostatin Drugs 0.000 description 2
- 229960000641 zorubicin Drugs 0.000 description 2
- FBTUMDXHSRTGRV-ALTNURHMSA-N zorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(\C)=N\NC(=O)C=1C=CC=CC=1)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 FBTUMDXHSRTGRV-ALTNURHMSA-N 0.000 description 2
- AADVCYNFEREWOS-UHFFFAOYSA-N (+)-DDM Natural products C=CC=CC(C)C(OC(N)=O)C(C)C(O)C(C)CC(C)=CC(C)C(O)C(C)C=CC(O)CC1OC(=O)C(C)C(O)C1C AADVCYNFEREWOS-UHFFFAOYSA-N 0.000 description 1
- AYIRNRDRBQJXIF-NXEZZACHSA-N (-)-Florfenicol Chemical compound CS(=O)(=O)C1=CC=C([C@@H](O)[C@@H](CF)NC(=O)C(Cl)Cl)C=C1 AYIRNRDRBQJXIF-NXEZZACHSA-N 0.000 description 1
- NNJPGOLRFBJNIW-HNNXBMFYSA-N (-)-demecolcine Chemical compound C1=C(OC)C(=O)C=C2[C@@H](NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-HNNXBMFYSA-N 0.000 description 1
- DOKWDOLZCMETIJ-ZAQUEYBZSA-N (12as)-9-[[3-[[(12as)-8-methoxy-6-oxo-11,12,12a,13-tetrahydroindolo[2,1-c][1,4]benzodiazepin-9-yl]oxymethyl]-5-[2-[2-(2-methoxyethoxy)ethoxy]ethyl-(2-methyl-2-sulfanylpropyl)amino]phenyl]methoxy]-8-methoxy-12a,13-dihydroindolo[2,1-c][1,4]benzodiazepin-6-o Chemical compound N1=C[C@@H]2CC3=CC=CC=C3N2C(=O)C(C=C2OC)=C1C=C2OCC1=CC(N(CC(C)(C)S)CCOCCOCCOC)=CC(COC=2C(=CC=3C(=O)N4C5=CC=CC=C5C[C@H]4CNC=3C=2)OC)=C1 DOKWDOLZCMETIJ-ZAQUEYBZSA-N 0.000 description 1
- IXXFZUPTQVDPPK-ZAWHAJPISA-N (1r,2r,4r,6r,7r,8r,10s,13r,14s)-17-[4-[4-(3-aminophenyl)triazol-1-yl]butyl]-7-[(2s,3r,4s,6r)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy-13-ethyl-10-fluoro-6-methoxy-2,4,6,8,10,14-hexamethyl-12,15-dioxa-17-azabicyclo[12.3.0]heptadecane-3,9,11,16-tet Chemical compound O([C@@H]1[C@@H](C)C(=O)[C@](C)(F)C(=O)O[C@@H]([C@]2(OC(=O)N(CCCCN3N=NC(=C3)C=3C=C(N)C=CC=3)[C@@H]2[C@@H](C)C(=O)[C@H](C)C[C@@]1(C)OC)C)CC)[C@@H]1O[C@H](C)C[C@H](N(C)C)[C@H]1O IXXFZUPTQVDPPK-ZAWHAJPISA-N 0.000 description 1
- AAFJXZWCNVJTMK-GUCUJZIJSA-N (1s,2r)-1-[(2s)-oxiran-2-yl]-2-[(2r)-oxiran-2-yl]ethane-1,2-diol Chemical compound C([C@@H]1[C@H](O)[C@H](O)[C@H]2OC2)O1 AAFJXZWCNVJTMK-GUCUJZIJSA-N 0.000 description 1
- PYXFVCFISTUSOO-HKUCOEKDSA-N (20S)-protopanaxadiol Chemical compound C1C[C@H](O)C(C)(C)[C@@H]2CC[C@@]3(C)[C@]4(C)CC[C@H]([C@@](C)(O)CCC=C(C)C)[C@H]4[C@H](O)C[C@@H]3[C@]21C PYXFVCFISTUSOO-HKUCOEKDSA-N 0.000 description 1
- DLMYFMLKORXJPO-FQEVSTJZSA-N (2R)-2-amino-3-[(triphenylmethyl)thio]propanoic acid Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(SC[C@H](N)C(O)=O)C1=CC=CC=C1 DLMYFMLKORXJPO-FQEVSTJZSA-N 0.000 description 1
- KZMHNEBMQDBQND-LBNZKSCFSA-N (2e,5s,6r,7s,9s,10e,12e,15r,16z,18e)-17-ethyl-6-hydroxy-9-(hydroxymethyl)-3,5,7,11,15-pentamethyl-19-[(2s,3s)-3-methyl-6-oxo-2,3-dihydropyran-2-yl]-8-oxononadeca-2,10,12,16,18-pentaenoic acid Chemical compound OC(=O)/C=C(C)/C[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@H](CO)/C=C(\C)/C=C/C[C@@H](C)/C=C(/CC)\C=C\[C@@H]1OC(=O)C=C[C@@H]1C KZMHNEBMQDBQND-LBNZKSCFSA-N 0.000 description 1
- XUSXOPRDIDWMFO-CTMSJIKGSA-N (2r,3r,4r,5r)-2-[(1s,2s,3r,4s,6r)-4,6-diamino-3-[[(2s,3r)-3-amino-6-[(1s)-1-aminoethyl]-3,4-dihydro-2h-pyran-2-yl]oxy]-2-hydroxycyclohexyl]oxy-5-methyl-4-(methylamino)oxane-3,5-diol Chemical compound O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H](CC=C(O2)[C@H](C)N)N)[C@@H](N)C[C@H]1N XUSXOPRDIDWMFO-CTMSJIKGSA-N 0.000 description 1
- WDQLRUYAYXDIFW-RWKIJVEZSA-N (2r,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-4-[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-[[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxy-6-(hydroxymethyl)oxane-2,3,5-triol Chemical compound O[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@@H](CO[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)O1 WDQLRUYAYXDIFW-RWKIJVEZSA-N 0.000 description 1
- RIWLPSIAFBLILR-WVNGMBSFSA-N (2s)-1-[(2s)-2-[[(2s,3s)-2-[[(2s)-2-[[(2s,3r)-2-[[(2r,3s)-2-[[(2s)-2-[[2-[[2-[acetyl(methyl)amino]acetyl]amino]acetyl]amino]-3-methylbutanoyl]amino]-3-methylpentanoyl]amino]-3-hydroxybutanoyl]amino]pentanoyl]amino]-3-methylpentanoyl]amino]-5-(diaminomethy Chemical compound CC(=O)N(C)CC(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1CCC[C@H]1C(=O)NCC RIWLPSIAFBLILR-WVNGMBSFSA-N 0.000 description 1
- ZZKNRXZVGOYGJT-VKHMYHEASA-N (2s)-2-[(2-phosphonoacetyl)amino]butanedioic acid Chemical compound OC(=O)C[C@@H](C(O)=O)NC(=O)CP(O)(O)=O ZZKNRXZVGOYGJT-VKHMYHEASA-N 0.000 description 1
- BXTJCSYMGFJEID-XMTADJHZSA-N (2s)-2-[[(2r,3r)-3-[(2s)-1-[(3r,4s,5s)-4-[[(2s)-2-[[(2s)-2-[6-[3-[(2r)-2-amino-2-carboxyethyl]sulfanyl-2,5-dioxopyrrolidin-1-yl]hexanoyl-methylamino]-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methoxy-5-methylheptanoyl]pyrrolidin-2-yl]-3-met Chemical compound C([C@H](NC(=O)[C@H](C)[C@@H](OC)[C@@H]1CCCN1C(=O)C[C@H]([C@H]([C@@H](C)CC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCCCN1C(C(SC[C@H](N)C(O)=O)CC1=O)=O)C(C)C)OC)C(O)=O)C1=CC=CC=C1 BXTJCSYMGFJEID-XMTADJHZSA-N 0.000 description 1
- KQRHTCDQWJLLME-XUXIUFHCSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-aminopropanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-4-methylpentanoic acid Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)N KQRHTCDQWJLLME-XUXIUFHCSA-N 0.000 description 1
- MALRUKQRDHTJPO-INIZCTEOSA-N (2s)-2-[[4-[(2,4-diamino-5-ethylquinazolin-6-yl)methylamino]benzoyl]amino]butanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C(CC)=C1CNC1=CC=C(C(=O)N[C@@H](CC(O)=O)C(O)=O)C=C1 MALRUKQRDHTJPO-INIZCTEOSA-N 0.000 description 1
- XNZUKUJONMYGIN-INIZCTEOSA-N (2s)-2-[[4-[(2,4-diaminopteridin-6-yl)methyl-methylamino]naphthalene-1-carbonyl]amino]pentanedioic acid Chemical compound C1=CC=C2C(N(CC=3N=C4C(N)=NC(N)=NC4=NC=3)C)=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C2=C1 XNZUKUJONMYGIN-INIZCTEOSA-N 0.000 description 1
- YXTKHLHCVFUPPT-YYFJYKOTSA-N (2s)-2-[[4-[(2-amino-5-formyl-4-oxo-1,6,7,8-tetrahydropteridin-6-yl)methylamino]benzoyl]amino]pentanedioic acid;(1r,2r)-1,2-dimethanidylcyclohexane;5-fluoro-1h-pyrimidine-2,4-dione;oxalic acid;platinum(2+) Chemical compound [Pt+2].OC(=O)C(O)=O.[CH2-][C@@H]1CCCC[C@H]1[CH2-].FC1=CNC(=O)NC1=O.C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 YXTKHLHCVFUPPT-YYFJYKOTSA-N 0.000 description 1
- QXOPTIPQEVJERB-JQWIXIFHSA-N (2s)-2-[[5-[2-[(6s)-2-amino-4-oxo-5,6,7,8-tetrahydro-1h-pyrido[2,3-d]pyrimidin-6-yl]ethyl]-4-methylthiophene-2-carbonyl]amino]pentanedioic acid Chemical compound C1=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)SC(CC[C@H]2CC=3C(=O)N=C(N)NC=3NC2)=C1C QXOPTIPQEVJERB-JQWIXIFHSA-N 0.000 description 1
- ZFKBWSREWJOSSJ-VIFPVBQESA-N (2s)-6,8-dichloro-2-(trifluoromethyl)-2h-chromene-3-carboxylic acid Chemical compound ClC1=CC(Cl)=C2O[C@H](C(F)(F)F)C(C(=O)O)=CC2=C1 ZFKBWSREWJOSSJ-VIFPVBQESA-N 0.000 description 1
- ZBVJFYPGLGEMIN-OYLNGHKZSA-N (2s)-n-[(2s)-1-[[(2s)-1-[[(2s)-1-[[(2s)-1-[[(2r)-1-[[(2s)-1-[[(2s)-1-[(2s)-2-[(2-amino-2-oxoethyl)carbamoyl]pyrrolidin-1-yl]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1h-indol-3-yl)-1-oxopropan-2-yl]amino]-3-( Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1.C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 ZBVJFYPGLGEMIN-OYLNGHKZSA-N 0.000 description 1
- GTXSRFUZSLTDFX-HRCADAONSA-N (2s)-n-[(2s)-3,3-dimethyl-1-(methylamino)-1-oxobutan-2-yl]-4-methyl-2-[[(2s)-2-sulfanyl-4-(3,4,4-trimethyl-2,5-dioxoimidazolidin-1-yl)butanoyl]amino]pentanamide Chemical compound CNC(=O)[C@H](C(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](S)CCN1C(=O)N(C)C(C)(C)C1=O GTXSRFUZSLTDFX-HRCADAONSA-N 0.000 description 1
- XSAKVDNHFRWJKS-IIZANFQQSA-N (2s)-n-benzyl-1-[(2s)-1-[(2s)-2-[[(2s)-2-[[(2s)-2-(dimethylamino)-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methylbutanoyl]pyrrolidine-2-carbonyl]pyrrolidine-2-carboxamide Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)N1[C@H](C(=O)NCC=2C=CC=CC=2)CCC1 XSAKVDNHFRWJKS-IIZANFQQSA-N 0.000 description 1
- PSVUJBVBCOISSP-SPFKKGSWSA-N (2s,3r,4s,5s,6r)-2-bis(2-chloroethylamino)phosphoryloxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound OC[C@H]1O[C@@H](OP(=O)(NCCCl)NCCCl)[C@H](O)[C@@H](O)[C@@H]1O PSVUJBVBCOISSP-SPFKKGSWSA-N 0.000 description 1
- HBJOXQRURQPDEX-MHXMMLMNSA-N (2s,4r)-n-[(1s,2s)-2-chloro-1-[(2r,3r,4s,5r,6r)-3,4,5-trihydroxy-6-methylsulfanyloxan-2-yl]propyl]-4-ethylpiperidine-2-carboxamide Chemical compound C1[C@H](CC)CCN[C@@H]1C(=O)N[C@H]([C@H](C)Cl)[C@@H]1[C@H](O)[C@H](O)[C@@H](O)[C@@H](SC)O1 HBJOXQRURQPDEX-MHXMMLMNSA-N 0.000 description 1
- PUDHBTGHUJUUFI-SCTWWAJVSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-n-[(2s,3r)-1-amino-3-hydroxy-1-oxobutan-2-yl]-19-[[(2r)-2-amino-3-naphthalen-2-ylpropanoyl]amino]-16-[(4-hydroxyphenyl)methyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-7-propan-2-yl-1,2-dithia-5,8,11,14,17-p Chemical compound C([C@H]1C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](N)CC=1C=C2C=CC=CC2=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(N)=O)=O)C(C)C)C1=CC=C(O)C=C1 PUDHBTGHUJUUFI-SCTWWAJVSA-N 0.000 description 1
- XIYOPDCBBDCGOE-IWVLMIASSA-N (4s,4ar,5s,5ar,12ar)-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methylidene-3,12-dioxo-4,4a,5,5a-tetrahydrotetracene-2-carboxamide Chemical compound C=C1C2=CC=CC(O)=C2C(O)=C2[C@@H]1[C@H](O)[C@H]1[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]1(O)C2=O XIYOPDCBBDCGOE-IWVLMIASSA-N 0.000 description 1
- RNIADBXQDMCFEN-IWVLMIASSA-N (4s,4ar,5s,5ar,12ar)-7-chloro-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methylidene-3,12-dioxo-4,4a,5,5a-tetrahydrotetracene-2-carboxamide Chemical compound C=C1C2=C(Cl)C=CC(O)=C2C(O)=C2[C@@H]1[C@H](O)[C@H]1[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]1(O)C2=O RNIADBXQDMCFEN-IWVLMIASSA-N 0.000 description 1
- SGKRLCUYIXIAHR-AKNGSSGZSA-N (4s,4ar,5s,5ar,6r,12ar)-4-(dimethylamino)-1,5,10,11,12a-pentahydroxy-6-methyl-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1=CC=C2[C@H](C)[C@@H]([C@H](O)[C@@H]3[C@](C(O)=C(C(N)=O)C(=O)[C@H]3N(C)C)(O)C3=O)C3=C(O)C2=C1O SGKRLCUYIXIAHR-AKNGSSGZSA-N 0.000 description 1
- FFTVPQUHLQBXQZ-KVUCHLLUSA-N (4s,4as,5ar,12ar)-4,7-bis(dimethylamino)-1,10,11,12a-tetrahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1C2=C(N(C)C)C=CC(O)=C2C(O)=C2[C@@H]1C[C@H]1[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]1(O)C2=O FFTVPQUHLQBXQZ-KVUCHLLUSA-N 0.000 description 1
- GUXHBMASAHGULD-SEYHBJAFSA-N (4s,4as,5as,6s,12ar)-7-chloro-4-(dimethylamino)-1,6,10,11,12a-pentahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1([C@H]2O)=C(Cl)C=CC(O)=C1C(O)=C1[C@@H]2C[C@H]2[C@H](N(C)C)C(=O)C(C(N)=O)=C(O)[C@@]2(O)C1=O GUXHBMASAHGULD-SEYHBJAFSA-N 0.000 description 1
- HBUJYEUPIIJJOS-PBHICJAKSA-N (5r)-3-[4-[1-[(2s)-2,3-dihydroxypropanoyl]-3,6-dihydro-2h-pyridin-4-yl]-3,5-difluorophenyl]-5-(1,2-oxazol-3-yloxymethyl)-1,3-oxazolidin-2-one Chemical compound C1N(C(=O)[C@@H](O)CO)CCC(C=2C(=CC(=CC=2F)N2C(O[C@@H](COC3=NOC=C3)C2)=O)F)=C1 HBUJYEUPIIJJOS-PBHICJAKSA-N 0.000 description 1
- QYNUQALWYRSVHF-OLZOCXBDSA-N (6R)-5,10-methylenetetrahydrofolic acid Chemical compound C([C@H]1CNC=2N=C(NC(=O)C=2N1C1)N)N1C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QYNUQALWYRSVHF-OLZOCXBDSA-N 0.000 description 1
- RWZVMMQNDHPRQD-SFTDATJTSA-N (6as)-3-[3-[[(6as)-2-methoxy-8-methylidene-11-oxo-7,9-dihydro-6ah-pyrrolo[2,1-c][1,4]benzodiazepin-3-yl]oxy]propoxy]-2-methoxy-8-methylidene-7,9-dihydro-6ah-pyrrolo[2,1-c][1,4]benzodiazepin-11-one Chemical compound N1=C[C@@H]2CC(=C)CN2C(=O)C(C=C2OC)=C1C=C2OCCCOC1=CC(N=C[C@H]2N(CC(=C)C2)C2=O)=C2C=C1OC RWZVMMQNDHPRQD-SFTDATJTSA-N 0.000 description 1
- FDZKMDGYTVIWIB-LCWDIZFYSA-N (6r,6as,8s)-3,8-dihydroxy-2,6-dimethoxy-5,6,6a,7,8,9-hexahydropyrrolo[2,1-c][1,4]benzodiazepin-11-one Chemical compound CO[C@H]1NC2=CC(O)=C(OC)C=C2C(=O)N2C[C@@H](O)C[C@@H]12 FDZKMDGYTVIWIB-LCWDIZFYSA-N 0.000 description 1
- RCFNNLSZHVHCEK-IMHLAKCZSA-N (7s,9s)-7-(4-amino-6-methyloxan-2-yl)oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;hydrochloride Chemical compound [Cl-].O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)C1CC([NH3+])CC(C)O1 RCFNNLSZHVHCEK-IMHLAKCZSA-N 0.000 description 1
- MWWSFMDVAYGXBV-FGBSZODSSA-N (7s,9s)-7-[(2r,4s,5r,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;hydron;chloride Chemical compound Cl.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-FGBSZODSSA-N 0.000 description 1
- HEQRYQONNHFDHG-TZSSRYMLSA-N (7s,9s)-7-[(2r,4s,5s,6s)-4,5-dihydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](O)[C@H](O)[C@H](C)O1 HEQRYQONNHFDHG-TZSSRYMLSA-N 0.000 description 1
- KQJSQWZMSAGSHN-UHFFFAOYSA-N (9beta,13alpha,14beta,20alpha)-3-hydroxy-9,13-dimethyl-2-oxo-24,25,26-trinoroleana-1(10),3,5,7-tetraen-29-oic acid Natural products CC12CCC3(C)C4CC(C)(C(O)=O)CCC4(C)CCC3(C)C2=CC=C2C1=CC(=O)C(O)=C2C KQJSQWZMSAGSHN-UHFFFAOYSA-N 0.000 description 1
- RXZBMPWDPOLZGW-XMRMVWPWSA-N (E)-roxithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=N/OCOCCOC)/[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 RXZBMPWDPOLZGW-XMRMVWPWSA-N 0.000 description 1
- AGBQKNBQESQNJD-SSDOTTSWSA-N (R)-lipoic acid Chemical compound OC(=O)CCCC[C@@H]1CCSS1 AGBQKNBQESQNJD-SSDOTTSWSA-N 0.000 description 1
- ZGNLFUXWZJGETL-YUSKDDKASA-N (Z)-[(2S)-2-amino-2-carboxyethyl]-hydroxyimino-oxidoazanium Chemical compound N[C@@H](C\[N+]([O-])=N\O)C(O)=O ZGNLFUXWZJGETL-YUSKDDKASA-N 0.000 description 1
- VYEWZWBILJHHCU-OMQUDAQFSA-N (e)-n-[(2s,3r,4r,5r,6r)-2-[(2r,3r,4s,5s,6s)-3-acetamido-5-amino-4-hydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-[2-[(2r,3s,4r,5r)-5-(2,4-dioxopyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]-2-hydroxyethyl]-4,5-dihydroxyoxan-3-yl]-5-methylhex-2-enamide Chemical compound N1([C@@H]2O[C@@H]([C@H]([C@H]2O)O)C(O)C[C@@H]2[C@H](O)[C@H](O)[C@H]([C@@H](O2)O[C@@H]2[C@@H]([C@@H](O)[C@H](N)[C@@H](CO)O2)NC(C)=O)NC(=O)/C=C/CC(C)C)C=CC(=O)NC1=O VYEWZWBILJHHCU-OMQUDAQFSA-N 0.000 description 1
- BWDQBBCUWLSASG-MDZDMXLPSA-N (e)-n-hydroxy-3-[4-[[2-hydroxyethyl-[2-(1h-indol-3-yl)ethyl]amino]methyl]phenyl]prop-2-enamide Chemical compound C=1NC2=CC=CC=C2C=1CCN(CCO)CC1=CC=C(\C=C\C(=O)NO)C=C1 BWDQBBCUWLSASG-MDZDMXLPSA-N 0.000 description 1
- AAFJXZWCNVJTMK-UHFFFAOYSA-N 1,2-bis(oxiran-2-yl)ethane-1,2-diol Chemical compound C1OC1C(O)C(O)C1CO1 AAFJXZWCNVJTMK-UHFFFAOYSA-N 0.000 description 1
- OUPZKGBUJRBPGC-UHFFFAOYSA-N 1,3,5-tris(oxiran-2-ylmethyl)-1,3,5-triazinane-2,4,6-trione Chemical compound O=C1N(CC2OC2)C(=O)N(CC2OC2)C(=O)N1CC1CO1 OUPZKGBUJRBPGC-UHFFFAOYSA-N 0.000 description 1
- HJTAZXHBEBIQQX-UHFFFAOYSA-N 1,5-bis(chloromethyl)naphthalene Chemical compound C1=CC=C2C(CCl)=CC=CC2=C1CCl HJTAZXHBEBIQQX-UHFFFAOYSA-N 0.000 description 1
- QTWDFPFYTVIXMP-UHFFFAOYSA-N 1-(2-chloroethyl)-4-(3-chloropropyl)piperazine;dihydrochloride Chemical compound Cl.Cl.ClCCCN1CCN(CCCl)CC1 QTWDFPFYTVIXMP-UHFFFAOYSA-N 0.000 description 1
- SWQQELWGJDXCFT-PNHWDRBUSA-N 1-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-ethynylimidazole-4-carboxamide Chemical compound C#CC1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 SWQQELWGJDXCFT-PNHWDRBUSA-N 0.000 description 1
- QBPISWMTLKTKMK-UHFFFAOYSA-N 1-[(4-aminophenyl)methyl]pyridin-2-one Chemical compound C1=CC(N)=CC=C1CN1C(=O)C=CC=C1 QBPISWMTLKTKMK-UHFFFAOYSA-N 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- WLKSPGHQGFFKGE-UHFFFAOYSA-N 1-chloropropan-2-yl n-(3-chlorophenyl)carbamate Chemical compound ClCC(C)OC(=O)NC1=CC=CC(Cl)=C1 WLKSPGHQGFFKGE-UHFFFAOYSA-N 0.000 description 1
- GZCWLCBFPRFLKL-UHFFFAOYSA-N 1-prop-2-ynoxypropan-2-ol Chemical compound CC(O)COCC#C GZCWLCBFPRFLKL-UHFFFAOYSA-N 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- AMDHQGZVCWWZCS-UHFFFAOYSA-N 109466-93-5 Chemical compound O=CC1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 AMDHQGZVCWWZCS-UHFFFAOYSA-N 0.000 description 1
- 108010058566 130-nm albumin-bound paclitaxel Proteins 0.000 description 1
- GVEZIHKRYBHEFX-MNOVXSKESA-N 13C-Cerulenin Natural products CC=CCC=CCCC(=O)[C@H]1O[C@@H]1C(N)=O GVEZIHKRYBHEFX-MNOVXSKESA-N 0.000 description 1
- KUFRQPKVAWMTJO-QSTRRNJOSA-N 17-dmag Chemical compound N1C(=O)\C(C)=C/C=C\[C@@H](OC)[C@H](OC(N)=O)\C(C)=C/[C@@H](C)[C@@H](O)[C@H](OC)C[C@@H](C)CC2=C(NCCN(C)C)C(=O)C=C1C2=O KUFRQPKVAWMTJO-QSTRRNJOSA-N 0.000 description 1
- ROZCIVXTLACYNY-UHFFFAOYSA-N 2,3,4,5,6-pentafluoro-n-(3-fluoro-4-methoxyphenyl)benzenesulfonamide Chemical compound C1=C(F)C(OC)=CC=C1NS(=O)(=O)C1=C(F)C(F)=C(F)C(F)=C1F ROZCIVXTLACYNY-UHFFFAOYSA-N 0.000 description 1
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 1
- XNTLXAUHLBBEKP-UHFFFAOYSA-N 2-(3,4-difluorophenyl)-4-(3-hydroxy-3-methylbutoxy)-5-(4-methylsulfonylphenyl)pyridazin-3-one Chemical compound O=C1C(OCCC(C)(O)C)=C(C=2C=CC(=CC=2)S(C)(=O)=O)C=NN1C1=CC=C(F)C(F)=C1 XNTLXAUHLBBEKP-UHFFFAOYSA-N 0.000 description 1
- PADGNZFOVSZIKZ-UHFFFAOYSA-N 2-(chloromethyl)oxirane;hydrogen carbonate;prop-2-enylazanium Chemical compound NCC=C.OC(O)=O.ClCC1CO1 PADGNZFOVSZIKZ-UHFFFAOYSA-N 0.000 description 1
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical compound O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 description 1
- ACTOXUHEUCPTEW-BWHGAVFKSA-N 2-[(4r,5s,6s,7r,9r,10r,11e,13e,16r)-6-[(2s,3r,4r,5s,6r)-5-[(2s,4r,5s,6s)-4,5-dihydroxy-4,6-dimethyloxan-2-yl]oxy-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy-10-[(2s,5s,6r)-5-(dimethylamino)-6-methyloxan-2-yl]oxy-4-hydroxy-5-methoxy-9,16-dimethyl-2-o Chemical compound O([C@H]1/C=C/C=C/C[C@@H](C)OC(=O)C[C@@H](O)[C@@H]([C@H]([C@@H](CC=O)C[C@H]1C)O[C@H]1[C@@H]([C@H]([C@H](O[C@@H]2O[C@@H](C)[C@H](O)[C@](C)(O)C2)[C@@H](C)O1)N(C)C)O)OC)[C@@H]1CC[C@H](N(C)C)[C@@H](C)O1 ACTOXUHEUCPTEW-BWHGAVFKSA-N 0.000 description 1
- AWVHFDOHKFRHKQ-UHFFFAOYSA-N 2-[10-(3-aminopropylimino)-6,8-dihydroxy-3-oxo-14,15-diazatetracyclo[7.6.1.02,7.013,16]hexadeca-1,4,6,8,11,13(16)-hexaen-14-yl]ethyl-(2-hydroxyethyl)azanium chloride Chemical compound C1=CC(=NCCCN)C2=C(C3=C(C=CC(=O)C3=C4C2=C1N(N4)CC[NH2+]CCO)O)O.[Cl-] AWVHFDOHKFRHKQ-UHFFFAOYSA-N 0.000 description 1
- OTLLEIBWKHEHGU-UHFFFAOYSA-N 2-[5-[[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy]-3,4-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-3,5-dihydroxy-4-phosphonooxyhexanedioic acid Chemical compound C1=NC=2C(N)=NC=NC=2N1C(C(C1O)O)OC1COC1C(CO)OC(OC(C(O)C(OP(O)(O)=O)C(O)C(O)=O)C(O)=O)C(O)C1O OTLLEIBWKHEHGU-UHFFFAOYSA-N 0.000 description 1
- WEZDRVHTDXTVLT-GJZGRUSLSA-N 2-[[(2s)-2-[[(2s)-2-[(2-aminoacetyl)amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]acetic acid Chemical compound OC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)CN)CC1=CC=CC=C1 WEZDRVHTDXTVLT-GJZGRUSLSA-N 0.000 description 1
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 1
- SGUMXALIXOMJSH-UHFFFAOYSA-N 2-[[2-chloro-5-[(2,4-diamino-5-methylquinazolin-6-yl)methylamino]benzoyl]amino]butanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C(C)=C1CNC1=CC=C(Cl)C(C(=O)NC(CC(O)=O)C(O)=O)=C1 SGUMXALIXOMJSH-UHFFFAOYSA-N 0.000 description 1
- RGHYDLZMTYDBDT-UHFFFAOYSA-N 2-amino-8-ethyl-4-methyl-6-(1H-pyrazol-5-yl)-7-pyrido[2,3-d]pyrimidinone Chemical compound O=C1N(CC)C2=NC(N)=NC(C)=C2C=C1C=1C=CNN=1 RGHYDLZMTYDBDT-UHFFFAOYSA-N 0.000 description 1
- SCVJRXQHFJXZFZ-JKUQZMGJSA-N 2-amino-9-[(2s,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-3h-purine-6-thione Chemical compound C1=2NC(N)=NC(=S)C=2N=CN1[C@@H]1C[C@H](O)[C@@H](CO)O1 SCVJRXQHFJXZFZ-JKUQZMGJSA-N 0.000 description 1
- HTCSFFGLRQDZDE-UHFFFAOYSA-N 2-azaniumyl-2-phenylpropanoate Chemical compound OC(=O)C(N)(C)C1=CC=CC=C1 HTCSFFGLRQDZDE-UHFFFAOYSA-N 0.000 description 1
- SEHSPJCWCBQHPF-UHFFFAOYSA-N 2-chloroethyl methylsulfonylmethanesulfonate Chemical compound CS(=O)(=O)CS(=O)(=O)OCCCl SEHSPJCWCBQHPF-UHFFFAOYSA-N 0.000 description 1
- WVCHIGAIXREVNS-UHFFFAOYSA-N 2-hydroxy-1,4-naphthoquinone Chemical compound C1=CC=C2C(O)=CC(=O)C(=O)C2=C1 WVCHIGAIXREVNS-UHFFFAOYSA-N 0.000 description 1
- GHWSMQHJFMAATF-DSHMRAQASA-N 20(S)-protopanaxadiol Natural products CC(=CCC[C@@](O)(CO)[C@H]1CC[C@]2(C)[C@@H]1CC[C@H]3[C@@]2(C)CC[C@H]4C(C)(C)[C@@H](O)CC[C@]34CO)C GHWSMQHJFMAATF-DSHMRAQASA-N 0.000 description 1
- YIMDLWDNDGKDTJ-QLKYHASDSA-N 3'-deamino-3'-(3-cyanomorpholin-4-yl)doxorubicin Chemical compound N1([C@H]2C[C@@H](O[C@@H](C)[C@H]2O)O[C@H]2C[C@@](O)(CC=3C(O)=C4C(=O)C=5C=CC=C(C=5C(=O)C4=C(O)C=32)OC)C(=O)CO)CCOCC1C#N YIMDLWDNDGKDTJ-QLKYHASDSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- HDBQZGJWHMCXIL-UHFFFAOYSA-N 3,7-dihydropurine-2-thione Chemical compound SC1=NC=C2NC=NC2=N1 HDBQZGJWHMCXIL-UHFFFAOYSA-N 0.000 description 1
- AXRCEOKUDYDWLF-UHFFFAOYSA-N 3-(1-methyl-3-indolyl)-4-[1-[1-(2-pyridinylmethyl)-4-piperidinyl]-3-indolyl]pyrrole-2,5-dione Chemical compound C12=CC=CC=C2N(C)C=C1C(C(NC1=O)=O)=C1C(C1=CC=CC=C11)=CN1C(CC1)CCN1CC1=CC=CC=N1 AXRCEOKUDYDWLF-UHFFFAOYSA-N 0.000 description 1
- FOFCCSJJVAOPFC-PILSHRGASA-N 3-[(3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]imidazole-4-carboxamide Chemical compound C1([C@H](O)[C@H](O)[C@H](O1)CO)N1C=NC=C1C(=O)N FOFCCSJJVAOPFC-PILSHRGASA-N 0.000 description 1
- MAUCONCHVWBMHK-UHFFFAOYSA-N 3-[(dimethylamino)methyl]-N-[2-[4-[(hydroxyamino)-oxomethyl]phenoxy]ethyl]-2-benzofurancarboxamide Chemical compound O1C2=CC=CC=C2C(CN(C)C)=C1C(=O)NCCOC1=CC=C(C(=O)NO)C=C1 MAUCONCHVWBMHK-UHFFFAOYSA-N 0.000 description 1
- QNKJFXARIMSDBR-UHFFFAOYSA-N 3-[2-[bis(2-chloroethyl)amino]ethyl]-1,3-diazaspiro[4.5]decane-2,4-dione Chemical compound O=C1N(CCN(CCCl)CCCl)C(=O)NC11CCCCC1 QNKJFXARIMSDBR-UHFFFAOYSA-N 0.000 description 1
- WUIABRMSWOKTOF-OYALTWQYSA-N 3-[[2-[2-[2-[[(2s,3r)-2-[[(2s,3s,4r)-4-[[(2s,3r)-2-[[6-amino-2-[(1s)-3-amino-1-[[(2s)-2,3-diamino-3-oxopropyl]amino]-3-oxopropyl]-5-methylpyrimidine-4-carbonyl]amino]-3-[(2r,3s,4s,5s,6s)-3-[(2r,3s,4s,5r,6r)-4-carbamoyloxy-3,5-dihydroxy-6-(hydroxymethyl)ox Chemical compound OS([O-])(=O)=O.N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C WUIABRMSWOKTOF-OYALTWQYSA-N 0.000 description 1
- HKPVIFTWECXNPY-UHFFFAOYSA-N 3-[[2-chloro-4-(4,6-diamino-2,2-dimethyl-1,3,5-triazin-1-yl)phenoxy]methyl]-n,n-dimethylbenzamide;ethanesulfonic acid Chemical compound CCS(O)(=O)=O.CN(C)C(=O)C1=CC=CC(COC=2C(=CC(=CC=2)N2C(N=C(N)N=C2N)(C)C)Cl)=C1 HKPVIFTWECXNPY-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- INZOTETZQBPBCE-NYLDSJSYSA-N 3-sialyl lewis Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@H]([C@H](O)CO)[C@@H]([C@@H](NC(C)=O)C=O)O[C@H]1[C@H](O)[C@@H](O[C@]2(O[C@H]([C@H](NC(C)=O)[C@@H](O)C2)[C@H](O)[C@H](O)CO)C(O)=O)[C@@H](O)[C@@H](CO)O1 INZOTETZQBPBCE-NYLDSJSYSA-N 0.000 description 1
- AKJHMTWEGVYYSE-AIRMAKDCSA-N 4-HPR Chemical compound C=1C=C(O)C=CC=1NC(=O)/C=C(\C)/C=C/C=C(C)C=CC1=C(C)CCCC1(C)C AKJHMTWEGVYYSE-AIRMAKDCSA-N 0.000 description 1
- SUVMJBTUFCVSAD-JTQLQIEISA-N 4-Methylsulfinylbutyl isothiocyanate Natural products C[S@](=O)CCCCN=C=S SUVMJBTUFCVSAD-JTQLQIEISA-N 0.000 description 1
- HEUVRFNVTLGKMZ-SANMLTNESA-N 4-[(2s)-2-(4-cyanophenyl)-2-hydroxy-2-(3-methylimidazol-4-yl)ethoxy]-3-[4-(trifluoromethoxy)phenyl]benzonitrile Chemical compound CN1C=NC=C1[C@@](O)(C=1C=CC(=CC=1)C#N)COC1=CC=C(C#N)C=C1C1=CC=C(OC(F)(F)F)C=C1 HEUVRFNVTLGKMZ-SANMLTNESA-N 0.000 description 1
- BQMGBEHGBILFLB-YUPPHXPYSA-N 4-[2-[4-[[2-(4-chlorophenyl)phenyl]methyl]piperazin-1-yl]benzoyl]-4-[[(2R)-4-(dimethylamino)-1-phenylsulfanylbutan-2-yl]amino]-3-nitrocyclohexa-1,5-diene-1-sulfonamide Chemical compound ClC1=CC=C(C=C1)C1=C(C=CC=C1)CN1CCN(CC1)C1=C(C(=O)C2(C(C=C(C=C2)S(=O)(=O)N)[N+](=O)[O-])N[C@H](CCN(C)C)CSC2=CC=CC=C2)C=CC=C1 BQMGBEHGBILFLB-YUPPHXPYSA-N 0.000 description 1
- NKABOCWFLUROCN-PNHWDRBUSA-N 4-amino-1-[(2r,3r,4s,5r)-2-ethyl-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound C1=CC(N)=NC(=O)N1[C@]1(CC)O[C@H](CO)[C@@H](O)[C@H]1O NKABOCWFLUROCN-PNHWDRBUSA-N 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- HEROWGICZZJWKP-UHFFFAOYSA-N 4-pyrrol-1-ylbenzenesulfonamide Chemical compound C1=CC(S(=O)(=O)N)=CC=C1N1C=CC=C1 HEROWGICZZJWKP-UHFFFAOYSA-N 0.000 description 1
- WUUGFSXJNOTRMR-IOSLPCCCSA-N 5'-S-methyl-5'-thioadenosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CSC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 WUUGFSXJNOTRMR-IOSLPCCCSA-N 0.000 description 1
- 102100022464 5'-nucleotidase Human genes 0.000 description 1
- HUNAOTXNHVALTN-UHFFFAOYSA-N 5-(5-chloro-2,4-dihydroxyphenyl)-N-ethyl-4-(4-methoxyphenyl)pyrazole-3-carboxamide Chemical compound CCNC(=O)C1=NNC(C=2C(=CC(O)=C(Cl)C=2)O)=C1C1=CC=C(OC)C=C1 HUNAOTXNHVALTN-UHFFFAOYSA-N 0.000 description 1
- NMUSYJAQQFHJEW-UHFFFAOYSA-N 5-Azacytidine Natural products O=C1N=C(N)N=CN1C1C(O)C(O)C(CO)O1 NMUSYJAQQFHJEW-UHFFFAOYSA-N 0.000 description 1
- ODACNRQBNVVGAI-UHFFFAOYSA-N 5-[2-chloroethyl(2-fluoroethyl)amino]-6-methyl-1h-pyrimidine-2,4-dione Chemical compound CC=1NC(=O)NC(=O)C=1N(CCF)CCCl ODACNRQBNVVGAI-UHFFFAOYSA-N 0.000 description 1
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical group OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 1
- LJIRBXZDQGQUOO-KVTDHHQDSA-N 6-amino-3-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,4-dihydro-1,3,5-triazin-2-one Chemical compound C1NC(N)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 LJIRBXZDQGQUOO-KVTDHHQDSA-N 0.000 description 1
- PAXWQORCRCBOCU-LURJTMIESA-N 6-fluoro-L-dopa Chemical compound OC(=O)[C@@H](N)CC1=CC(O)=C(O)C=C1F PAXWQORCRCBOCU-LURJTMIESA-N 0.000 description 1
- WLCZTRVUXYALDD-IBGZPJMESA-N 7-[[(2s)-2,6-bis(2-methoxyethoxycarbonylamino)hexanoyl]amino]heptoxy-methylphosphinic acid Chemical compound COCCOC(=O)NCCCC[C@H](NC(=O)OCCOC)C(=O)NCCCCCCCOP(C)(O)=O WLCZTRVUXYALDD-IBGZPJMESA-N 0.000 description 1
- KUAKYFCHUDSMNU-UHFFFAOYSA-N 7-chlorocamptothecin Chemical compound C1=CC=C2C(Cl)=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 KUAKYFCHUDSMNU-UHFFFAOYSA-N 0.000 description 1
- GOJJWDOZNKBUSR-UHFFFAOYSA-N 7-sulfamoyloxyheptyl sulfamate Chemical compound NS(=O)(=O)OCCCCCCCOS(N)(=O)=O GOJJWDOZNKBUSR-UHFFFAOYSA-N 0.000 description 1
- HFDKKNHCYWNNNQ-YOGANYHLSA-N 75976-10-2 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@@H](NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)N)C(C)C)[C@@H](C)O)C1=CC=C(O)C=C1 HFDKKNHCYWNNNQ-YOGANYHLSA-N 0.000 description 1
- NKGPJODWTZCHGF-UHFFFAOYSA-N 9-[3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-3h-purine-6-thione Chemical compound OC1C(O)C(CO)OC1N1C(NC=NC2=S)=C2N=C1 NKGPJODWTZCHGF-UHFFFAOYSA-N 0.000 description 1
- FUXVKZWTXQUGMW-UHFFFAOYSA-N 9-amino-20-(r,s)-camptothecin Chemical compound C1=CC(N)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 FUXVKZWTXQUGMW-UHFFFAOYSA-N 0.000 description 1
- TUOSCZDRWRYPRS-UHFFFAOYSA-N 9-butyl-8-(3,4,5-trimethoxybenzyl)-9h-purin-6-amine Chemical compound N=1C2=C(N)N=CN=C2N(CCCC)C=1CC1=CC(OC)=C(OC)C(OC)=C1 TUOSCZDRWRYPRS-UHFFFAOYSA-N 0.000 description 1
- XVMZDZFTCKLZTF-NRFANRHFSA-N 9-methoxycamptothecin Chemical compound C1=CC(OC)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 XVMZDZFTCKLZTF-NRFANRHFSA-N 0.000 description 1
- 229940127124 90Y-ibritumomab tiuxetan Drugs 0.000 description 1
- OONFNUWBHFSNBT-HXUWFJFHSA-N AEE788 Chemical compound C1CN(CC)CCN1CC1=CC=C(C=2NC3=NC=NC(N[C@H](C)C=4C=CC=CC=4)=C3C=2)C=C1 OONFNUWBHFSNBT-HXUWFJFHSA-N 0.000 description 1
- 102000013563 Acid Phosphatase Human genes 0.000 description 1
- 108010051457 Acid Phosphatase Proteins 0.000 description 1
- 102100036732 Actin, aortic smooth muscle Human genes 0.000 description 1
- 206010001150 Adenocarcinoma gastric Diseases 0.000 description 1
- TWCMVXMQHSVIOJ-UHFFFAOYSA-N Aglycone of yadanzioside D Natural products COC(=O)C12OCC34C(CC5C(=CC(O)C(O)C5(C)C3C(O)C1O)C)OC(=O)C(OC(=O)C)C24 TWCMVXMQHSVIOJ-UHFFFAOYSA-N 0.000 description 1
- ZGCSNRKSJLVANE-UHFFFAOYSA-N Aglycone-Rebeccamycin Natural products N1C2=C3NC4=C(Cl)C=CC=C4C3=C(C(=O)NC3=O)C3=C2C2=C1C(Cl)=CC=C2 ZGCSNRKSJLVANE-UHFFFAOYSA-N 0.000 description 1
- QXRNAOYBCYVZCD-BQBZGAKWSA-N Ala-Lys Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCCCN QXRNAOYBCYVZCD-BQBZGAKWSA-N 0.000 description 1
- 108010012934 Albumin-Bound Paclitaxel Proteins 0.000 description 1
- 108010003133 Aldo-Keto Reductase Family 1 Member C2 Proteins 0.000 description 1
- 102100026446 Aldo-keto reductase family 1 member C1 Human genes 0.000 description 1
- 102100024089 Aldo-keto reductase family 1 member C2 Human genes 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 102100040121 Allograft inflammatory factor 1 Human genes 0.000 description 1
- 102100021266 Alpha-(1,6)-fucosyltransferase Human genes 0.000 description 1
- 102100023635 Alpha-fetoprotein Human genes 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 108090000644 Angiozyme Proteins 0.000 description 1
- MXPOCMVWFLDDLZ-NSCUHMNNSA-N Apaziquone Chemical compound CN1C(\C=C\CO)=C(CO)C(C2=O)=C1C(=O)C=C2N1CC1 MXPOCMVWFLDDLZ-NSCUHMNNSA-N 0.000 description 1
- 102000013918 Apolipoproteins E Human genes 0.000 description 1
- 108010025628 Apolipoproteins E Proteins 0.000 description 1
- 101100330723 Arabidopsis thaliana DAR2 gene Proteins 0.000 description 1
- QJMCHPGWFZZRID-BQBZGAKWSA-N Asn-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@@H](N)CC(N)=O QJMCHPGWFZZRID-BQBZGAKWSA-N 0.000 description 1
- CPMKYMGGYUFOHS-FSPLSTOPSA-N Asp-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@@H](N)CC(O)=O CPMKYMGGYUFOHS-FSPLSTOPSA-N 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- PLMKQQMDOMTZGG-UHFFFAOYSA-N Astrantiagenin E-methylester Natural products CC12CCC(O)C(C)(CO)C1CCC1(C)C2CC=C2C3CC(C)(C)CCC3(C(=O)OC)CCC21C PLMKQQMDOMTZGG-UHFFFAOYSA-N 0.000 description 1
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 1
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 1
- 108010074708 B7-H1 Antigen Proteins 0.000 description 1
- 102100035656 BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 Human genes 0.000 description 1
- QULDDKSCVCJTPV-UHFFFAOYSA-N BIIB021 Chemical compound COC1=C(C)C=NC(CN2C3=NC(N)=NC(Cl)=C3N=C2)=C1C QULDDKSCVCJTPV-UHFFFAOYSA-N 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 244000056139 Brassica cretica Species 0.000 description 1
- 235000003351 Brassica cretica Nutrition 0.000 description 1
- 235000003343 Brassica rupestris Nutrition 0.000 description 1
- 102100035875 C-C chemokine receptor type 5 Human genes 0.000 description 1
- 101710149870 C-C chemokine receptor type 5 Proteins 0.000 description 1
- 102100025279 C-X-C motif chemokine 11 Human genes 0.000 description 1
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 1
- 102100027207 CD27 antigen Human genes 0.000 description 1
- 108010045374 CD36 Antigens Proteins 0.000 description 1
- 108010029697 CD40 Ligand Proteins 0.000 description 1
- 102100032937 CD40 ligand Human genes 0.000 description 1
- 102100025221 CD70 antigen Human genes 0.000 description 1
- HAWSQZCWOQZXHI-UHFFFAOYSA-N CPT-OH Natural products C1=C(O)C=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 HAWSQZCWOQZXHI-UHFFFAOYSA-N 0.000 description 1
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 1
- 229940045513 CTLA4 antagonist Drugs 0.000 description 1
- 102100035350 CUB domain-containing protein 1 Human genes 0.000 description 1
- FVLVBPDQNARYJU-XAHDHGMMSA-N C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O Chemical compound C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O FVLVBPDQNARYJU-XAHDHGMMSA-N 0.000 description 1
- 101100228196 Caenorhabditis elegans gly-4 gene Proteins 0.000 description 1
- 101100454807 Caenorhabditis elegans lgg-1 gene Proteins 0.000 description 1
- 101100454808 Caenorhabditis elegans lgg-2 gene Proteins 0.000 description 1
- 101100463133 Caenorhabditis elegans pdl-1 gene Proteins 0.000 description 1
- 102400000113 Calcitonin Human genes 0.000 description 1
- 108060001064 Calcitonin Proteins 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical group NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- 102000004225 Cathepsin B Human genes 0.000 description 1
- 108090000712 Cathepsin B Proteins 0.000 description 1
- 102000003908 Cathepsin D Human genes 0.000 description 1
- 102100032219 Cathepsin D Human genes 0.000 description 1
- 108090000258 Cathepsin D Proteins 0.000 description 1
- 102100035654 Cathepsin S Human genes 0.000 description 1
- 102000005600 Cathepsins Human genes 0.000 description 1
- 108010084457 Cathepsins Proteins 0.000 description 1
- AQKDBFWJOPNOKZ-UHFFFAOYSA-N Celastrol Natural products CC12CCC3(C)C4CC(C)(C(O)=O)CCC4(C)CCC3(C)C2=CC=C2C1=CC(=O)C(=O)C2C AQKDBFWJOPNOKZ-UHFFFAOYSA-N 0.000 description 1
- 229940123587 Cell cycle inhibitor Drugs 0.000 description 1
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 1
- 102100025832 Centromere-associated protein E Human genes 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 229940124957 Cervarix Drugs 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- 241001233914 Chelidonium majus Species 0.000 description 1
- FDZKMDGYTVIWIB-UHFFFAOYSA-N Chicamycin A Natural products COC1NC2=CC(O)=C(OC)C=C2C(=O)N2CC(O)CC12 FDZKMDGYTVIWIB-UHFFFAOYSA-N 0.000 description 1
- 241001227713 Chiron Species 0.000 description 1
- 239000004099 Chlortetracycline Substances 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- VWFCHDSQECPREK-LURJTMIESA-N Cidofovir Chemical compound NC=1C=CN(C[C@@H](CO)OCP(O)(O)=O)C(=O)N=1 VWFCHDSQECPREK-LURJTMIESA-N 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- 102100024335 Collagen alpha-1(VII) chain Human genes 0.000 description 1
- 108010060123 Conjugate Vaccines Proteins 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 101800000414 Corticotropin Proteins 0.000 description 1
- 239000000055 Corticotropin-Releasing Hormone Substances 0.000 description 1
- 229930188224 Cryptophycin Natural products 0.000 description 1
- IELOKBJPULMYRW-NJQVLOCASA-N D-alpha-Tocopheryl Acid Succinate Chemical compound OC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C IELOKBJPULMYRW-NJQVLOCASA-N 0.000 description 1
- HEBKCHPVOIAQTA-QWWZWVQMSA-N D-arabinitol Chemical compound OC[C@@H](O)C(O)[C@H](O)CO HEBKCHPVOIAQTA-QWWZWVQMSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- 102100026139 DNA damage-inducible transcript 4 protein Human genes 0.000 description 1
- 230000004543 DNA replication Effects 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 101100481408 Danio rerio tie2 gene Proteins 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- 102000010170 Death domains Human genes 0.000 description 1
- 108050001718 Death domains Proteins 0.000 description 1
- 102100033553 Delta-like protein 4 Human genes 0.000 description 1
- NNJPGOLRFBJNIW-UHFFFAOYSA-N Demecolcine Natural products C1=C(OC)C(=O)C=C2C(NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-UHFFFAOYSA-N 0.000 description 1
- FMTDIUIBLCQGJB-UHFFFAOYSA-N Demethylchlortetracyclin Natural products C1C2C(O)C3=C(Cl)C=CC(O)=C3C(=O)C2=C(O)C2(O)C1C(N(C)C)C(O)=C(C(N)=O)C2=O FMTDIUIBLCQGJB-UHFFFAOYSA-N 0.000 description 1
- 108010002156 Depsipeptides Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- ASXBYYWOLISCLQ-UHFFFAOYSA-N Dihydrostreptomycin Natural products O1C(CO)C(O)C(O)C(NC)C1OC1C(CO)(O)C(C)OC1OC1C(N=C(N)N)C(O)C(N=C(N)N)C(O)C1O ASXBYYWOLISCLQ-UHFFFAOYSA-N 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 102000016607 Diphtheria Toxin Human genes 0.000 description 1
- 108010053187 Diphtheria Toxin Proteins 0.000 description 1
- AADVCYNFEREWOS-OBRABYBLSA-N Discodermolide Chemical compound C=C\C=C/[C@H](C)[C@H](OC(N)=O)[C@@H](C)[C@H](O)[C@@H](C)C\C(C)=C/[C@H](C)[C@@H](O)[C@@H](C)\C=C/[C@@H](O)C[C@@H]1OC(=O)[C@H](C)[C@@H](O)[C@H]1C AADVCYNFEREWOS-OBRABYBLSA-N 0.000 description 1
- 230000010777 Disulfide Reduction Effects 0.000 description 1
- OJIYIVCMRYCWSE-UHFFFAOYSA-M Domiphen bromide Chemical compound [Br-].CCCCCCCCCCCC[N+](C)(C)CCOC1=CC=CC=C1 OJIYIVCMRYCWSE-UHFFFAOYSA-M 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 102100029722 Ectonucleoside triphosphate diphosphohydrolase 1 Human genes 0.000 description 1
- 102100038132 Endogenous retrovirus group K member 6 Pro protein Human genes 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- 101000877447 Enterobacteria phage T4 Endonuclease V Proteins 0.000 description 1
- 101710091045 Envelope protein Proteins 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 206010073064 Epithelioid mesothelioma Diseases 0.000 description 1
- XOZIUKBZLSUILX-SDMHVBBESA-N Epothilone D Natural products O=C1[C@H](C)[C@@H](O)[C@@H](C)CCC/C(/C)=C/C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C XOZIUKBZLSUILX-SDMHVBBESA-N 0.000 description 1
- 239000004386 Erythritol Substances 0.000 description 1
- UNXHWFMMPAWVPI-UHFFFAOYSA-N Erythritol Natural products OCC(O)C(O)CO UNXHWFMMPAWVPI-UHFFFAOYSA-N 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical group OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 108091029865 Exogenous DNA Proteins 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- UKCVAQGKEOJTSR-UHFFFAOYSA-N Fadrozole hydrochloride Chemical compound Cl.C1=CC(C#N)=CC=C1C1N2C=NC=C2CCC1 UKCVAQGKEOJTSR-UHFFFAOYSA-N 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 102000008857 Ferritin Human genes 0.000 description 1
- 108050000784 Ferritin Proteins 0.000 description 1
- 238000008416 Ferritin Methods 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 102100022629 Fructose-2,6-bisphosphatase Human genes 0.000 description 1
- 229940032072 GVAX vaccine Drugs 0.000 description 1
- 229910052688 Gadolinium Inorganic materials 0.000 description 1
- RVAQIUULWULRNW-UHFFFAOYSA-N Ganetespib Chemical compound C1=C(O)C(C(C)C)=CC(C=2N(C(O)=NN=2)C=2C=C3C=CN(C)C3=CC=2)=C1O RVAQIUULWULRNW-UHFFFAOYSA-N 0.000 description 1
- 229940124897 Gardasil Drugs 0.000 description 1
- 102400000921 Gastrin Human genes 0.000 description 1
- 108010052343 Gastrins Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- JRZJKWGQFNTSRN-UHFFFAOYSA-N Geldanamycin Natural products C1C(C)CC(OC)C(O)C(C)C=C(C)C(OC(N)=O)C(OC)CCC=C(C)C(=O)NC2=CC(=O)C(OC)=C1C2=O JRZJKWGQFNTSRN-UHFFFAOYSA-N 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 108010053070 Glutathione Disulfide Proteins 0.000 description 1
- 108010009504 Gly-Phe-Leu-Gly Proteins 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 102000001398 Granzyme Human genes 0.000 description 1
- 108060005986 Granzyme Proteins 0.000 description 1
- 102100030385 Granzyme B Human genes 0.000 description 1
- 244000041633 Grewia tenax Species 0.000 description 1
- 235000005612 Grewia tenax Nutrition 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 102100030595 HLA class II histocompatibility antigen gamma chain Human genes 0.000 description 1
- 102100032510 Heat shock protein HSP 90-beta Human genes 0.000 description 1
- 102100034458 Hepatitis A virus cellular receptor 2 Human genes 0.000 description 1
- 208000027927 Hereditary papillary renal cell carcinoma Diseases 0.000 description 1
- SQUHHTBVTRBESD-UHFFFAOYSA-N Hexa-Ac-myo-Inositol Natural products CC(=O)OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC(C)=O SQUHHTBVTRBESD-UHFFFAOYSA-N 0.000 description 1
- VLDVBZICYBVQHB-IUCAKERBSA-N His-Val Chemical compound CC(C)[C@@H](C([O-])=O)NC(=O)[C@@H]([NH3+])CC1=CN=CN1 VLDVBZICYBVQHB-IUCAKERBSA-N 0.000 description 1
- 108010033040 Histones Proteins 0.000 description 1
- 101000600756 Homo sapiens 3-phosphoinositide-dependent protein kinase 1 Proteins 0.000 description 1
- 101000678236 Homo sapiens 5'-nucleotidase Proteins 0.000 description 1
- 101000929319 Homo sapiens Actin, aortic smooth muscle Proteins 0.000 description 1
- 101000718028 Homo sapiens Aldo-keto reductase family 1 member C1 Proteins 0.000 description 1
- 101000890626 Homo sapiens Allograft inflammatory factor 1 Proteins 0.000 description 1
- 101000819490 Homo sapiens Alpha-(1,6)-fucosyltransferase Proteins 0.000 description 1
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 1
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 1
- 101000803294 Homo sapiens BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 Proteins 0.000 description 1
- 101000858060 Homo sapiens C-X-C motif chemokine 11 Proteins 0.000 description 1
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 1
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 description 1
- 101000737742 Homo sapiens CUB domain-containing protein 1 Proteins 0.000 description 1
- 101000914324 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 5 Proteins 0.000 description 1
- 101000914321 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 7 Proteins 0.000 description 1
- 101000869010 Homo sapiens Cathepsin D Proteins 0.000 description 1
- 101000947086 Homo sapiens Cathepsin S Proteins 0.000 description 1
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 1
- 101000914247 Homo sapiens Centromere-associated protein E Proteins 0.000 description 1
- 101000909498 Homo sapiens Collagen alpha-1(VII) chain Proteins 0.000 description 1
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 description 1
- 101000912753 Homo sapiens DNA damage-inducible transcript 4 protein Proteins 0.000 description 1
- 101000872077 Homo sapiens Delta-like protein 4 Proteins 0.000 description 1
- 101001012447 Homo sapiens Ectonucleoside triphosphate diphosphohydrolase 1 Proteins 0.000 description 1
- 101000823463 Homo sapiens Fructose-2,6-bisphosphatase Proteins 0.000 description 1
- 101001009603 Homo sapiens Granzyme B Proteins 0.000 description 1
- 101001082627 Homo sapiens HLA class II histocompatibility antigen gamma chain Proteins 0.000 description 1
- 101001016856 Homo sapiens Heat shock protein HSP 90-beta Proteins 0.000 description 1
- 101001068133 Homo sapiens Hepatitis A virus cellular receptor 2 Proteins 0.000 description 1
- 101001034652 Homo sapiens Insulin-like growth factor 1 receptor Proteins 0.000 description 1
- 101001044927 Homo sapiens Insulin-like growth factor-binding protein 3 Proteins 0.000 description 1
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 1
- 101001082070 Homo sapiens Interferon alpha-inducible protein 6 Proteins 0.000 description 1
- 101000599940 Homo sapiens Interferon gamma Proteins 0.000 description 1
- 101001039113 Homo sapiens Leucine-rich repeat-containing protein 15 Proteins 0.000 description 1
- 101000868279 Homo sapiens Leukocyte surface antigen CD47 Proteins 0.000 description 1
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 description 1
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 description 1
- 101001065609 Homo sapiens Lumican Proteins 0.000 description 1
- 101000934372 Homo sapiens Macrosialin Proteins 0.000 description 1
- 101001011906 Homo sapiens Matrix metalloproteinase-14 Proteins 0.000 description 1
- 101000956317 Homo sapiens Membrane-spanning 4-domains subfamily A member 4A Proteins 0.000 description 1
- 101001014567 Homo sapiens Membrane-spanning 4-domains subfamily A member 7 Proteins 0.000 description 1
- 101000623901 Homo sapiens Mucin-16 Proteins 0.000 description 1
- 101000604123 Homo sapiens Noggin Proteins 0.000 description 1
- 101000595746 Homo sapiens Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit delta isoform Proteins 0.000 description 1
- 101000579123 Homo sapiens Phosphoglycerate kinase 1 Proteins 0.000 description 1
- 101000935642 Homo sapiens Phosphoinositide 3-kinase adapter protein 1 Proteins 0.000 description 1
- 101000595923 Homo sapiens Placenta growth factor Proteins 0.000 description 1
- 101001126417 Homo sapiens Platelet-derived growth factor receptor alpha Proteins 0.000 description 1
- 101000617725 Homo sapiens Pregnancy-specific beta-1-glycoprotein 2 Proteins 0.000 description 1
- 101000595907 Homo sapiens Procollagen-lysine,2-oxoglutarate 5-dioxygenase 2 Proteins 0.000 description 1
- 101001117317 Homo sapiens Programmed cell death 1 ligand 1 Proteins 0.000 description 1
- 101000881678 Homo sapiens Prolyl hydroxylase EGLN3 Proteins 0.000 description 1
- 101000804792 Homo sapiens Protein Wnt-5a Proteins 0.000 description 1
- 101000654674 Homo sapiens Semaphorin-6A Proteins 0.000 description 1
- 101000984753 Homo sapiens Serine/threonine-protein kinase B-raf Proteins 0.000 description 1
- 101000701446 Homo sapiens Stanniocalcin-2 Proteins 0.000 description 1
- 101000577874 Homo sapiens Stromelysin-2 Proteins 0.000 description 1
- 101000831007 Homo sapiens T-cell immunoreceptor with Ig and ITIM domains Proteins 0.000 description 1
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 1
- 101000976959 Homo sapiens Transcription factor 4 Proteins 0.000 description 1
- 101000596771 Homo sapiens Transcription factor 7-like 2 Proteins 0.000 description 1
- 101000635938 Homo sapiens Transforming growth factor beta-1 proprotein Proteins 0.000 description 1
- 101000635958 Homo sapiens Transforming growth factor beta-2 proprotein Proteins 0.000 description 1
- 101000801234 Homo sapiens Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 description 1
- 101000851370 Homo sapiens Tumor necrosis factor receptor superfamily member 9 Proteins 0.000 description 1
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 1
- 101001117146 Homo sapiens [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 1, mitochondrial Proteins 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- GRRNUXAQVGOGFE-UHFFFAOYSA-N Hygromycin-B Natural products OC1C(NC)CC(N)C(O)C1OC1C2OC3(C(C(O)C(O)C(C(N)CO)O3)O)OC2C(O)C(CO)O1 GRRNUXAQVGOGFE-UHFFFAOYSA-N 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- 108010023610 IL13-PE38 Proteins 0.000 description 1
- MPBVHIBUJCELCL-UHFFFAOYSA-N Ibandronate Chemical compound CCCCCN(C)CCC(O)(P(O)(O)=O)P(O)(O)=O MPBVHIBUJCELCL-UHFFFAOYSA-N 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- BBIXOODYWPFNDT-CIUDSAMLSA-N Ile-Pro Chemical compound CC[C@H](C)[C@H](N)C(=O)N1CCC[C@H]1C(O)=O BBIXOODYWPFNDT-CIUDSAMLSA-N 0.000 description 1
- BCXBIONYYJCSDF-CIUDSAMLSA-N Ile-Val Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](C(C)C)C(O)=O BCXBIONYYJCSDF-CIUDSAMLSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 1
- 229930010555 Inosine Natural products 0.000 description 1
- 102100023915 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 1
- 102100022708 Insulin-like growth factor-binding protein 3 Human genes 0.000 description 1
- 102100022338 Integrin alpha-M Human genes 0.000 description 1
- 102100022297 Integrin alpha-X Human genes 0.000 description 1
- 102100040018 Interferon alpha-2 Human genes 0.000 description 1
- 102100027354 Interferon alpha-inducible protein 6 Human genes 0.000 description 1
- 102100037850 Interferon gamma Human genes 0.000 description 1
- 108010079944 Interferon-alpha2b Proteins 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- 102000003996 Interferon-beta Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- 102000004889 Interleukin-6 Human genes 0.000 description 1
- 102000002698 KIR Receptors Human genes 0.000 description 1
- 108010043610 KIR Receptors Proteins 0.000 description 1
- 101150069255 KLRC1 gene Proteins 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- 102000011743 Kisspeptin-1 Receptors Human genes 0.000 description 1
- 108010076800 Kisspeptin-1 Receptors Proteins 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- MLFKVJCWGUZWNV-UHFFFAOYSA-N L-alanosine Natural products OC(=O)C(N)CN(O)N=O MLFKVJCWGUZWNV-UHFFFAOYSA-N 0.000 description 1
- 101710159002 L-lactate oxidase Proteins 0.000 description 1
- 239000004395 L-leucine Substances 0.000 description 1
- 235000019454 L-leucine Nutrition 0.000 description 1
- 239000002138 L01XE21 - Regorafenib Substances 0.000 description 1
- 239000002177 L01XE27 - Ibrutinib Substances 0.000 description 1
- 108090001090 Lectins Proteins 0.000 description 1
- 102000004856 Lectins Human genes 0.000 description 1
- 101000988090 Leishmania donovani Heat shock protein 83 Proteins 0.000 description 1
- 108010062867 Lenograstim Proteins 0.000 description 1
- LMVRPBWWHMVLPC-KBPJCXPTSA-N Leptolstatin Natural products CC(CC=CC(=CC(C)C(=O)C(C)C(O)C(C)CC(=CCO)C)C)C=C(C)/C=C/C1CC=CC(=O)O1 LMVRPBWWHMVLPC-KBPJCXPTSA-N 0.000 description 1
- YACHGFWEQXFSBS-UHFFFAOYSA-N Leptomycin B Natural products OC(=O)C=C(C)CC(C)C(O)C(C)C(=O)C(C)C=C(C)C=CCC(C)C=C(CC)C=CC1OC(=O)C=CC1C YACHGFWEQXFSBS-UHFFFAOYSA-N 0.000 description 1
- 102100040645 Leucine-rich repeat-containing protein 15 Human genes 0.000 description 1
- 102100032913 Leukocyte surface antigen CD47 Human genes 0.000 description 1
- OJMMVQQUTAEWLP-UHFFFAOYSA-N Lincomycin Natural products CN1CC(CCC)CC1C(=O)NC(C(C)O)C1C(O)C(O)C(O)C(SC)O1 OJMMVQQUTAEWLP-UHFFFAOYSA-N 0.000 description 1
- 239000000232 Lipid Bilayer Substances 0.000 description 1
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 description 1
- 102100032114 Lumican Human genes 0.000 description 1
- 239000012819 MDM2-Inhibitor Substances 0.000 description 1
- 101100404845 Macaca mulatta NKG2A gene Proteins 0.000 description 1
- SVSFCSOFEPJFSF-UHFFFAOYSA-N Macbecin II Natural products N1C(=O)C(C)=CC=CC(C)C(OC(N)=O)C(C)=CC(C)C(OC)C(OC)CC(C)C(OC)C2=CC(O)=CC1=C2O SVSFCSOFEPJFSF-UHFFFAOYSA-N 0.000 description 1
- 102100025136 Macrosialin Human genes 0.000 description 1
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 102100030216 Matrix metalloproteinase-14 Human genes 0.000 description 1
- 229930126263 Maytansine Natural products 0.000 description 1
- 101150031009 Mcpt8 gene Proteins 0.000 description 1
- 208000000172 Medulloblastoma Diseases 0.000 description 1
- 102100032512 Membrane-spanning 4-domains subfamily A member 7 Human genes 0.000 description 1
- IMTUWVJPCQPJEE-IUCAKERBSA-N Met-Lys Chemical compound CSCC[C@H](N)C(=O)N[C@H](C(O)=O)CCCCN IMTUWVJPCQPJEE-IUCAKERBSA-N 0.000 description 1
- 206010027458 Metastases to lung Diseases 0.000 description 1
- 108700011259 MicroRNAs Proteins 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- DMUAPQTXSSNEDD-QALJCMCCSA-N Midecamycin Chemical compound C1[C@](O)(C)[C@@H](OC(=O)CC)[C@H](C)O[C@H]1O[C@H]1[C@H](N(C)C)[C@@H](O)[C@H](O[C@@H]2[C@H]([C@H](OC(=O)CC)CC(=O)O[C@H](C)C/C=C/C=C/[C@H](O)[C@H](C)C[C@@H]2CC=O)OC)O[C@@H]1C DMUAPQTXSSNEDD-QALJCMCCSA-N 0.000 description 1
- 239000005462 Mubritinib Substances 0.000 description 1
- 102100023123 Mucin-16 Human genes 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 101100481410 Mus musculus Tek gene Proteins 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 1
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 1
- 241001467552 Mycobacterium bovis BCG Species 0.000 description 1
- 206010073137 Myxoid liposarcoma Diseases 0.000 description 1
- NFIXBCVWIPOYCD-UHFFFAOYSA-N N,N-diethyl-2-[4-(phenylmethyl)phenoxy]ethanamine Chemical compound C1=CC(OCCN(CC)CC)=CC=C1CC1=CC=CC=C1 NFIXBCVWIPOYCD-UHFFFAOYSA-N 0.000 description 1
- HRNLUBSXIHFDHP-UHFFFAOYSA-N N-(2-aminophenyl)-4-[[[4-(3-pyridinyl)-2-pyrimidinyl]amino]methyl]benzamide Chemical compound NC1=CC=CC=C1NC(=O)C(C=C1)=CC=C1CNC1=NC=CC(C=2C=NC=CC=2)=N1 HRNLUBSXIHFDHP-UHFFFAOYSA-N 0.000 description 1
- URCVCIZFVQDVPM-UHFFFAOYSA-N N-[2-(4-hydroxyanilino)-3-pyridinyl]-4-methoxybenzenesulfonamide Chemical compound C1=CC(OC)=CC=C1S(=O)(=O)NC1=CC=CN=C1NC1=CC=C(O)C=C1 URCVCIZFVQDVPM-UHFFFAOYSA-N 0.000 description 1
- YALNUENQHAQXEA-UHFFFAOYSA-N N-[4-[(hydroxyamino)-oxomethyl]phenyl]carbamic acid [6-(diethylaminomethyl)-2-naphthalenyl]methyl ester Chemical compound C1=CC2=CC(CN(CC)CC)=CC=C2C=C1COC(=O)NC1=CC=C(C(=O)NO)C=C1 YALNUENQHAQXEA-UHFFFAOYSA-N 0.000 description 1
- LKJPYSCBVHEWIU-UHFFFAOYSA-N N-[4-cyano-3-(trifluoromethyl)phenyl]-3-[(4-fluorophenyl)sulfonyl]-2-hydroxy-2-methylpropanamide Chemical compound C=1C=C(C#N)C(C(F)(F)F)=CC=1NC(=O)C(O)(C)CS(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-UHFFFAOYSA-N 0.000 description 1
- UBQYURCVBFRUQT-UHFFFAOYSA-N N-benzoyl-Ferrioxamine B Chemical compound CC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCN UBQYURCVBFRUQT-UHFFFAOYSA-N 0.000 description 1
- TZYWCYJVHRLUCT-VABKMULXSA-N N-benzyloxycarbonyl-L-leucyl-L-leucyl-L-leucinal Chemical compound CC(C)C[C@@H](C=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)OCC1=CC=CC=C1 TZYWCYJVHRLUCT-VABKMULXSA-N 0.000 description 1
- PAWIYAYFNXQGAP-UHFFFAOYSA-N N-hydroxy-2-[4-[[(1-methyl-3-indolyl)methylamino]methyl]-1-piperidinyl]-5-pyrimidinecarboxamide Chemical compound C12=CC=CC=C2N(C)C=C1CNCC(CC1)CCN1C1=NC=C(C(=O)NO)C=N1 PAWIYAYFNXQGAP-UHFFFAOYSA-N 0.000 description 1
- 108010072915 NAc-Sar-Gly-Val-(d-allo-Ile)-Thr-Nva-Ile-Arg-ProNEt Proteins 0.000 description 1
- 102100031911 NEDD8 Human genes 0.000 description 1
- 108700004934 NEDD8 Proteins 0.000 description 1
- 101150107958 NEDD8 gene Proteins 0.000 description 1
- 108010057466 NF-kappa B Proteins 0.000 description 1
- 102000003945 NF-kappa B Human genes 0.000 description 1
- 102100022682 NKG2-A/NKG2-B type II integral membrane protein Human genes 0.000 description 1
- 101710204212 Neocarzinostatin Proteins 0.000 description 1
- 241001045988 Neogene Species 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 206010061309 Neoplasm progression Diseases 0.000 description 1
- KYRVNWMVYQXFEU-UHFFFAOYSA-N Nocodazole Chemical compound C1=C2NC(NC(=O)OC)=NC2=CC=C1C(=O)C1=CC=CS1 KYRVNWMVYQXFEU-UHFFFAOYSA-N 0.000 description 1
- 102100038454 Noggin Human genes 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 102000004473 OX40 Ligand Human genes 0.000 description 1
- 108010042215 OX40 Ligand Proteins 0.000 description 1
- 239000004104 Oleandomycin Substances 0.000 description 1
- RZPAKFUAFGMUPI-UHFFFAOYSA-N Oleandomycin Natural products O1C(C)C(O)C(OC)CC1OC1C(C)C(=O)OC(C)C(C)C(O)C(C)C(=O)C2(OC2)CC(C)C(OC2C(C(CC(C)O2)N(C)C)O)C1C RZPAKFUAFGMUPI-UHFFFAOYSA-N 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 101100532088 Oryza sativa subsp. japonica RUB2 gene Proteins 0.000 description 1
- 101100532090 Oryza sativa subsp. japonica RUB3 gene Proteins 0.000 description 1
- 239000004100 Oxytetracycline Substances 0.000 description 1
- 229940029536 PANVAC Drugs 0.000 description 1
- 239000012661 PARP inhibitor Substances 0.000 description 1
- KJWZYMMLVHIVSU-IYCNHOCDSA-N PGK1 Chemical compound CCCCC[C@H](O)\C=C\[C@@H]1[C@@H](CCCCCCC(O)=O)C(=O)CC1=O KJWZYMMLVHIVSU-IYCNHOCDSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108010025700 PR-171 Proteins 0.000 description 1
- 244000131316 Panax pseudoginseng Species 0.000 description 1
- 235000005035 Panax pseudoginseng ssp. pseudoginseng Nutrition 0.000 description 1
- 235000003140 Panax quinquefolius Nutrition 0.000 description 1
- VREZDOWOLGNDPW-ALTGWBOUSA-N Pancratistatin Chemical compound C1=C2[C@H]3[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)[C@@H]3NC(=O)C2=C(O)C2=C1OCO2 VREZDOWOLGNDPW-ALTGWBOUSA-N 0.000 description 1
- VREZDOWOLGNDPW-MYVCAWNPSA-N Pancratistatin Natural products O=C1N[C@H]2[C@H](O)[C@H](O)[C@H](O)[C@H](O)[C@@H]2c2c1c(O)c1OCOc1c2 VREZDOWOLGNDPW-MYVCAWNPSA-N 0.000 description 1
- 102000018886 Pancreatic Polypeptide Human genes 0.000 description 1
- 108010057150 Peplomycin Proteins 0.000 description 1
- 102000057297 Pepsin A Human genes 0.000 description 1
- 108090000284 Pepsin A Proteins 0.000 description 1
- 108090000279 Peptidyltransferases Proteins 0.000 description 1
- KHGNFPUMBJSZSM-UHFFFAOYSA-N Perforine Natural products COC1=C2CCC(O)C(CCC(C)(C)O)(OC)C2=NC2=C1C=CO2 KHGNFPUMBJSZSM-UHFFFAOYSA-N 0.000 description 1
- 206010057249 Phagocytosis Diseases 0.000 description 1
- OZILORBBPKKGRI-RYUDHWBXSA-N Phe-Arg Chemical compound NC(N)=NCCC[C@@H](C(O)=O)NC(=O)[C@@H](N)CC1=CC=CC=C1 OZILORBBPKKGRI-RYUDHWBXSA-N 0.000 description 1
- 102100036056 Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit delta isoform Human genes 0.000 description 1
- 102100028251 Phosphoglycerate kinase 1 Human genes 0.000 description 1
- 102100028238 Phosphoinositide 3-kinase adapter protein 1 Human genes 0.000 description 1
- 102100035194 Placenta growth factor Human genes 0.000 description 1
- 108010022233 Plasminogen Activator Inhibitor 1 Proteins 0.000 description 1
- 102100039418 Plasminogen activator inhibitor 1 Human genes 0.000 description 1
- 102100030485 Platelet-derived growth factor receptor alpha Human genes 0.000 description 1
- 229940121906 Poly ADP ribose polymerase inhibitor Drugs 0.000 description 1
- 229930182556 Polyacetal Natural products 0.000 description 1
- 102100025067 Potassium voltage-gated channel subfamily H member 4 Human genes 0.000 description 1
- 101710163352 Potassium voltage-gated channel subfamily H member 4 Proteins 0.000 description 1
- 102100022019 Pregnancy-specific beta-1-glycoprotein 2 Human genes 0.000 description 1
- 108010079780 Pristinamycin Proteins 0.000 description 1
- RLNUPSVMIYRZSM-UHFFFAOYSA-N Pristinamycin Natural products CC1OC(=O)C(C=2C=CC=CC=2)NC(=O)C2CC(=O)CCN2C(=O)C(CC=2C=CC(=CC=2)N(C)C)CCN(C)C(=O)C2CCCN2C(=O)C(CC)NC(=O)C1NC(=O)C1=NC=CC=C1O RLNUPSVMIYRZSM-UHFFFAOYSA-N 0.000 description 1
- 102100027467 Pro-opiomelanocortin Human genes 0.000 description 1
- 102100035198 Procollagen-lysine,2-oxoglutarate 5-dioxygenase 2 Human genes 0.000 description 1
- 108010076181 Proinsulin Proteins 0.000 description 1
- 102100037247 Prolyl hydroxylase EGLN3 Human genes 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 102000006010 Protein Disulfide-Isomerase Human genes 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 101710188315 Protein X Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102100026858 Protein-lysine 6-oxidase Human genes 0.000 description 1
- 102000052575 Proto-Oncogene Human genes 0.000 description 1
- 108700020978 Proto-Oncogene Proteins 0.000 description 1
- 108010090931 Proto-Oncogene Proteins c-bcl-2 Proteins 0.000 description 1
- 102000013535 Proto-Oncogene Proteins c-bcl-2 Human genes 0.000 description 1
- 238000003559 RNA-seq method Methods 0.000 description 1
- MUPFEKGTMRGPLJ-RMMQSMQOSA-N Raffinose Natural products O(C[C@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O[C@@]2(CO)[C@H](O)[C@@H](O)[C@@H](CO)O2)O1)[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 MUPFEKGTMRGPLJ-RMMQSMQOSA-N 0.000 description 1
- CAYGMWMWJUFODP-UWQYKGISSA-N Ratjadone Natural products CC=CC1OC(CC(O)C1C)C(O)C=CC=C(/C)CC(C)C=C(C)/C=C/C2CC=CC(=O)O2 CAYGMWMWJUFODP-UWQYKGISSA-N 0.000 description 1
- QEHOIJJIZXRMAN-UHFFFAOYSA-N Rebeccamycin Natural products OC1C(O)C(OC)C(CO)OC1N1C2=C3NC4=C(Cl)C=CC=C4C3=C3C(=O)NC(=O)C3=C2C2=CC=CC(Cl)=C21 QEHOIJJIZXRMAN-UHFFFAOYSA-N 0.000 description 1
- 102000004278 Receptor Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000873 Receptor Protein-Tyrosine Kinases Proteins 0.000 description 1
- 229940127361 Receptor Tyrosine Kinase Inhibitors Drugs 0.000 description 1
- 101710100968 Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- JVWLUVNSQYXYBE-UHFFFAOYSA-N Ribitol Natural products OCC(C)C(O)C(O)CO JVWLUVNSQYXYBE-UHFFFAOYSA-N 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- URWAJWIAIPFPJE-UHFFFAOYSA-N Rickamicin Natural products O1CC(O)(C)C(NC)C(O)C1OC1C(O)C(OC2C(CC=C(CN)O2)N)C(N)CC1N URWAJWIAIPFPJE-UHFFFAOYSA-N 0.000 description 1
- NCDNCNXCDXHOMX-UHFFFAOYSA-N Ritonavir Natural products C=1C=CC=CC=1CC(NC(=O)OCC=1SC=NC=1)C(O)CC(CC=1C=CC=CC=1)NC(=O)C(C(C)C)NC(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-UHFFFAOYSA-N 0.000 description 1
- VYWWNRMSAPEJLS-MDWYKHENSA-N Rokitamycin Chemical compound C1[C@](OC(=O)CC)(C)[C@@H](OC(=O)CCC)[C@H](C)O[C@H]1O[C@H]1[C@H](N(C)C)[C@@H](O)[C@H](O[C@@H]2[C@H]([C@H](O)CC(=O)O[C@H](C)C/C=C/C=C/[C@H](O)[C@H](C)C[C@@H]2CC=O)OC)O[C@@H]1C VYWWNRMSAPEJLS-MDWYKHENSA-N 0.000 description 1
- 108010044012 STAT1 Transcription Factor Proteins 0.000 description 1
- LOGJQOUIVKBFGH-UHFFFAOYSA-N SU6656 Chemical compound C1CCCC(N2)=C1C=C2C=C1C(=O)NC2=CC=C(S(=O)(=O)N(C)C)C=C21 LOGJQOUIVKBFGH-UHFFFAOYSA-N 0.000 description 1
- BFZKMNSQCNVFGM-UCEYFQQTSA-N Sagopilone Chemical compound O1C(=O)C[C@H](O)C(C)(C)C(=O)[C@H](CC=C)[C@@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@H]1C1=CC=C(SC(C)=N2)C2=C1 BFZKMNSQCNVFGM-UCEYFQQTSA-N 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 102100032795 Semaphorin-6A Human genes 0.000 description 1
- 102100027103 Serine/threonine-protein kinase B-raf Human genes 0.000 description 1
- 102100029904 Signal transducer and activator of transcription 1-alpha/beta Human genes 0.000 description 1
- 229920000519 Sizofiran Polymers 0.000 description 1
- 239000004187 Spiramycin Substances 0.000 description 1
- UQZIYBXSHAGNOE-USOSMYMVSA-N Stachyose Natural products O(C[C@H]1[C@@H](O)[C@H](O)[C@H](O)[C@@H](O[C@@]2(CO)[C@H](O)[C@@H](O)[C@@H](CO)O2)O1)[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@H](CO[C@@H]2[C@@H](O)[C@@H](O)[C@@H](O)[C@H](CO)O2)O1 UQZIYBXSHAGNOE-USOSMYMVSA-N 0.000 description 1
- 102100030510 Stanniocalcin-2 Human genes 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 108010034396 Streptogramins Proteins 0.000 description 1
- 241000187747 Streptomyces Species 0.000 description 1
- ZSJLQEPLLKMAKR-UHFFFAOYSA-N Streptozotocin Natural products O=NN(C)C(=O)NC1C(O)OC(CO)C(O)C1O ZSJLQEPLLKMAKR-UHFFFAOYSA-N 0.000 description 1
- 102100028848 Stromelysin-2 Human genes 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 239000012505 Superdex™ Substances 0.000 description 1
- 101100215487 Sus scrofa ADRA2A gene Proteins 0.000 description 1
- 101000983124 Sus scrofa Pancreatic prohormone precursor Proteins 0.000 description 1
- 101710090983 T-cell immunoreceptor with Ig and ITIM domains Proteins 0.000 description 1
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 1
- 102100033456 TGF-beta receptor type-1 Human genes 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical group OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- 108060008245 Thrombospondin Proteins 0.000 description 1
- 108010034949 Thyroglobulin Proteins 0.000 description 1
- 102100033504 Thyroglobulin Human genes 0.000 description 1
- IWEQQRMGNVVKQW-OQKDUQJOSA-N Toremifene citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 IWEQQRMGNVVKQW-OQKDUQJOSA-N 0.000 description 1
- 102100023489 Transcription factor 4 Human genes 0.000 description 1
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 1
- 229940123468 Transferase inhibitor Drugs 0.000 description 1
- 108010011702 Transforming Growth Factor-beta Type I Receptor Proteins 0.000 description 1
- 102000009618 Transforming Growth Factors Human genes 0.000 description 1
- 108010009583 Transforming Growth Factors Proteins 0.000 description 1
- 102100030742 Transforming growth factor beta-1 proprotein Human genes 0.000 description 1
- 102100030737 Transforming growth factor beta-2 proprotein Human genes 0.000 description 1
- 229930189037 Trapoxin Natural products 0.000 description 1
- YCPOZVAOBBQLRI-WDSKDSINSA-N Treosulfan Chemical compound CS(=O)(=O)OC[C@H](O)[C@@H](O)COS(C)(=O)=O YCPOZVAOBBQLRI-WDSKDSINSA-N 0.000 description 1
- 108010050144 Triptorelin Pamoate Proteins 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 102100033728 Tumor necrosis factor receptor superfamily member 18 Human genes 0.000 description 1
- 102100022153 Tumor necrosis factor receptor superfamily member 4 Human genes 0.000 description 1
- 101710165473 Tumor necrosis factor receptor superfamily member 4 Proteins 0.000 description 1
- 102100036856 Tumor necrosis factor receptor superfamily member 9 Human genes 0.000 description 1
- 206010054094 Tumour necrosis Diseases 0.000 description 1
- YJQCOFNZVFGCAF-UHFFFAOYSA-N Tunicamycin II Natural products O1C(CC(O)C2C(C(O)C(O2)N2C(NC(=O)C=C2)=O)O)C(O)C(O)C(NC(=O)C=CCCCCCCCCC(C)C)C1OC1OC(CO)C(O)C(O)C1NC(C)=O YJQCOFNZVFGCAF-UHFFFAOYSA-N 0.000 description 1
- 239000004182 Tylosin Substances 0.000 description 1
- 229930194936 Tylosin Natural products 0.000 description 1
- MUPFEKGTMRGPLJ-UHFFFAOYSA-N UNPD196149 Natural products OC1C(O)C(CO)OC1(CO)OC1C(O)C(O)C(O)C(COC2C(C(O)C(O)C(CO)O2)O)O1 MUPFEKGTMRGPLJ-UHFFFAOYSA-N 0.000 description 1
- VGQOVCHZGQWAOI-UHFFFAOYSA-N UNPD55612 Natural products N1C(O)C2CC(C=CC(N)=O)=CN2C(=O)C2=CC=C(C)C(O)=C12 VGQOVCHZGQWAOI-UHFFFAOYSA-N 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 229940091171 VEGFR-2 tyrosine kinase inhibitor Drugs 0.000 description 1
- JKHXYJKMNSSFFL-IUCAKERBSA-N Val-Lys Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCCCN JKHXYJKMNSSFFL-IUCAKERBSA-N 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 108010080702 Virginiamycin Proteins 0.000 description 1
- 239000004188 Virginiamycin Substances 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- 102000043366 Wnt-5a Human genes 0.000 description 1
- VUPBDWQPEOWRQP-RTUCOMKBSA-N [(2R,3S,4S,5R,6R)-2-[(2R,3S,4S,5S,6S)-2-[(1S,2S)-3-[[(2R,3S)-5-[[(2S,3R)-1-[[2-[4-[4-[[4-amino-6-[3-(4-aminobutylamino)propylamino]-6-oxohexyl]carbamoyl]-1,3-thiazol-2-yl]-1,3-thiazol-2-yl]-1-[(2S,3R,4R,5S,6S)-5-amino-3,4-dihydroxy-6-methyloxan-2-yl]oxy-2-hydroxyethyl]amino]-3-hydroxy-1-oxobutan-2-yl]amino]-3-hydroxy-5-oxopentan-2-yl]amino]-2-[[6-amino-2-[(1S)-3-amino-1-[[(2S)-2,3-diamino-3-oxopropyl]amino]-3-oxopropyl]-5-methylpyrimidine-4-carbonyl]amino]-1-(1H-imidazol-5-yl)-3-oxopropoxy]-4,5-dihydroxy-6-(hydroxymethyl)oxan-3-yl]oxy-3,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl] carbamate Chemical compound C[C@@H](O)[C@H](NC(=O)C[C@H](O)[C@@H](C)NC(=O)[C@@H](NC(=O)c1nc(nc(N)c1C)[C@H](CC(N)=O)NC[C@H](N)C(N)=O)[C@H](O[C@@H]1O[C@@H](CO)[C@@H](O)[C@H](O)[C@@H]1O[C@H]1O[C@H](CO)[C@@H](O)[C@H](OC(N)=O)[C@@H]1O)c1cnc[nH]1)C(=O)NC(O[C@@H]1O[C@@H](C)[C@@H](N)[C@@H](O)[C@H]1O)C(O)c1nc(cs1)-c1nc(cs1)C(=O)NCCCC(N)CC(=O)NCCCNCCCCN VUPBDWQPEOWRQP-RTUCOMKBSA-N 0.000 description 1
- SVSFCSOFEPJFSF-OEPVMNMSSA-N [(2r,3s,5r,6s,7r,8e,11s,12z,14e)-20,22-dihydroxy-2,5,6-trimethoxy-3,7,9,11,15-pentamethyl-16-oxo-17-azabicyclo[16.3.1]docosa-1(21),8,12,14,18(22),19-hexaen-10-yl] carbamate Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](C)C(OC(N)=O)\C(C)=C\[C@@H](C)[C@H](OC)[C@H](OC)C[C@H](C)[C@@H](OC)C2=CC(O)=CC1=C2O SVSFCSOFEPJFSF-OEPVMNMSSA-N 0.000 description 1
- LQKSHSFQQRCAFW-CCVNJFHASA-N [(2s)-1-[(2s)-2-benzyl-3-methoxy-5-oxo-2h-pyrrol-1-yl]-3-methyl-1-oxobutan-2-yl] (2s)-1-[(2s)-1-[(2s)-2-[[(2s)-2-[[(2s)-2-(dimethylamino)-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methylbutanoyl]pyrrolidine-2-carbonyl]pyrrolidine-2-carboxyl Chemical compound C([C@@H]1N(C(=O)C=C1OC)C(=O)[C@@H](OC(=O)[C@H]1N(CCC1)C(=O)[C@H]1N(CCC1)C(=O)[C@H](C(C)C)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C)C(C)C)C(C)C)C1=CC=CC=C1 LQKSHSFQQRCAFW-CCVNJFHASA-N 0.000 description 1
- JMNXSNUXDHHTKQ-QVMSTPCGSA-N [(3r,6r)-6-[(3s,5r,7r,8r,9s,10s,13r,14s,17r)-3-[3-(4-aminobutylamino)propylamino]-7-hydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylheptan-3-yl] hydrogen sulfate;(2s)-2-hydroxypropanoic ac Chemical compound C[C@H](O)C(O)=O.C([C@@H]1C[C@H]2O)[C@@H](NCCCNCCCCN)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@H](C)CC[C@H](C(C)C)OS(O)(=O)=O)[C@@]2(C)CC1 JMNXSNUXDHHTKQ-QVMSTPCGSA-N 0.000 description 1
- XJXKGUZINMNEDK-GPJOBVNKSA-L [(4r,5r)-5-(aminomethyl)-2-propan-2-yl-1,3-dioxolan-4-yl]methanamine;platinum(2+);propanedioate Chemical compound [Pt+2].[O-]C(=O)CC([O-])=O.CC(C)C1O[C@H](CN)[C@@H](CN)O1 XJXKGUZINMNEDK-GPJOBVNKSA-L 0.000 description 1
- XSMVECZRZBFTIZ-UHFFFAOYSA-M [2-(aminomethyl)cyclobutyl]methanamine;2-oxidopropanoate;platinum(4+) Chemical compound [Pt+4].CC([O-])C([O-])=O.NCC1CCC1CN XSMVECZRZBFTIZ-UHFFFAOYSA-M 0.000 description 1
- GZLGNNHEHXBCBI-UHFFFAOYSA-L [Na+].[Na+].OC(=O)C(O)C(O)C(O)=O.[O-]C(=O)C(O)C(O)C([O-])=O Chemical compound [Na+].[Na+].OC(=O)C(O)C(O)C(O)=O.[O-]C(=O)C(O)C(O)C([O-])=O GZLGNNHEHXBCBI-UHFFFAOYSA-L 0.000 description 1
- IREFLPJJRVTUAO-UHFFFAOYSA-L [Pt+2].C(=O)(O)C1=C(C(C(=O)[O-])=CC=C1)C(=O)[O-] Chemical compound [Pt+2].C(=O)(O)C1=C(C(C(=O)[O-])=CC=C1)C(=O)[O-] IREFLPJJRVTUAO-UHFFFAOYSA-L 0.000 description 1
- 102100024148 [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 1, mitochondrial Human genes 0.000 description 1
- 229950008805 abexinostat Drugs 0.000 description 1
- GZOSMCIZMLWJML-VJLLXTKPSA-N abiraterone Chemical compound C([C@H]1[C@H]2[C@@H]([C@]3(CC[C@H](O)CC3=CC2)C)CC[C@@]11C)C=C1C1=CC=CN=C1 GZOSMCIZMLWJML-VJLLXTKPSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- PENDGIOBPJLVBT-HMMOOPTJSA-N abt-773 Chemical compound O([C@@H]1[C@@H](C)C(=O)[C@@H](C)C(=O)O[C@@H]([C@]2(OC(=O)N[C@@H]2[C@@H](C)C(=O)[C@H](C)C[C@]1(C)OC\C=C\C=1C=C2C=CC=CC2=NC=1)C)CC)[C@@H]1O[C@H](C)C[C@H](N(C)C)[C@H]1O PENDGIOBPJLVBT-HMMOOPTJSA-N 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Chemical group CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 239000008351 acetate buffer Substances 0.000 description 1
- DEXPIBGCLCPUHE-UISHROKMSA-N acetic acid;(4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-n-[(2s,3r)-1-amino-3-hydroxy-1-oxobutan-2-yl]-19-[[(2r)-2-amino-3-naphthalen-2-ylpropanoyl]amino]-16-[(4-hydroxyphenyl)methyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-7-propan-2-yl-1,2-dithia-5, Chemical compound CC(O)=O.C([C@H]1C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](N)CC=1C=C2C=CC=CC2=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(N)=O)=O)C(C)C)C1=CC=C(O)C=C1 DEXPIBGCLCPUHE-UISHROKMSA-N 0.000 description 1
- 108010014659 acetyl-glycyl-valyl-allo-isoleucyl-seryl-glutaminyl-isoleucyl-arginyl-prolyl-cysteinamide Proteins 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 229960004150 aciclovir Drugs 0.000 description 1
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 229960005339 acitretin Drugs 0.000 description 1
- QAWIHIJWNYOLBE-OKKQSCSOSA-N acivicin Chemical compound OC(=O)[C@@H](N)[C@@H]1CC(Cl)=NO1 QAWIHIJWNYOLBE-OKKQSCSOSA-N 0.000 description 1
- 229950008427 acivicin Drugs 0.000 description 1
- YBCVMFKXIKNREZ-UHFFFAOYSA-N acoh acetic acid Chemical compound CC(O)=O.CC(O)=O YBCVMFKXIKNREZ-UHFFFAOYSA-N 0.000 description 1
- 229940099550 actimmune Drugs 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 208000024447 adrenal gland neoplasm Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 108010054982 alanyl-leucyl-alanyl-leucine Proteins 0.000 description 1
- 108700025316 aldesleukin Proteins 0.000 description 1
- 229940083773 alecensa Drugs 0.000 description 1
- 229960001611 alectinib Drugs 0.000 description 1
- 229960000548 alemtuzumab Drugs 0.000 description 1
- 229940060515 aleve Drugs 0.000 description 1
- 229940110282 alimta Drugs 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910001854 alkali hydroxide Inorganic materials 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 229940098174 alkeran Drugs 0.000 description 1
- 239000002295 alkylating antineoplastic agent Substances 0.000 description 1
- 108010026331 alpha-Fetoproteins Proteins 0.000 description 1
- AGBQKNBQESQNJD-UHFFFAOYSA-N alpha-Lipoic acid Natural products OC(=O)CCCCC1CCSS1 AGBQKNBQESQNJD-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229950010817 alvocidib Drugs 0.000 description 1
- BIIVYFLTOXDAOV-YVEFUNNKSA-N alvocidib Chemical compound O[C@@H]1CN(C)CC[C@@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O BIIVYFLTOXDAOV-YVEFUNNKSA-N 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 229960004821 amikacin Drugs 0.000 description 1
- LKCWBDHBTVXHDL-RMDFUYIESA-N amikacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)[C@@H](O)[C@H](O)[C@H]1O LKCWBDHBTVXHDL-RMDFUYIESA-N 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229960002550 amrubicin Drugs 0.000 description 1
- VJZITPJGSQKZMX-XDPRQOKASA-N amrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC=C4C(=O)C=3C(O)=C21)(N)C(=O)C)[C@H]1C[C@H](O)[C@H](O)CO1 VJZITPJGSQKZMX-XDPRQOKASA-N 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 229950000242 ancitabine Drugs 0.000 description 1
- KZOWNALBTMILAP-JBMRGDGGSA-N ancitabine hydrochloride Chemical compound Cl.N=C1C=CN2[C@@H]3O[C@H](CO)[C@@H](O)[C@@H]3OC2=N1 KZOWNALBTMILAP-JBMRGDGGSA-N 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- CIDNKDMVSINJCG-GKXONYSUSA-N annamycin Chemical compound I[C@@H]1[C@H](O)[C@@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(=O)CO)C1 CIDNKDMVSINJCG-GKXONYSUSA-N 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- VGQOVCHZGQWAOI-HYUHUPJXSA-N anthramycin Chemical compound N1[C@@H](O)[C@@H]2CC(\C=C\C(N)=O)=CN2C(=O)C2=CC=C(C)C(O)=C12 VGQOVCHZGQWAOI-HYUHUPJXSA-N 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000003432 anti-folate effect Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 238000009175 antibody therapy Methods 0.000 description 1
- 238000011394 anticancer treatment Methods 0.000 description 1
- 229940127074 antifolate Drugs 0.000 description 1
- 229950002465 apaziquone Drugs 0.000 description 1
- IOASYARYEYRREA-LQAJYKIKSA-N aphidicolin glycinate Chemical compound C1[C@]23[C@]4(C)CC[C@H](O)[C@](C)(CO)[C@H]4CC[C@@H]3C[C@@H]1[C@@](COC(=O)CN)(O)CC2 IOASYARYEYRREA-LQAJYKIKSA-N 0.000 description 1
- 201000007580 appendix carcinoma Diseases 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- MKKYBZZTJQGVCD-XTCKQBCOSA-N arbekacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)CC[C@H]1N MKKYBZZTJQGVCD-XTCKQBCOSA-N 0.000 description 1
- 229940078010 arimidex Drugs 0.000 description 1
- 229940087620 aromasin Drugs 0.000 description 1
- 210000004436 artificial bacterial chromosome Anatomy 0.000 description 1
- MCGDSOGUHLTADD-UHFFFAOYSA-N arzoxifene Chemical compound C1=CC(OC)=CC=C1C1=C(OC=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 MCGDSOGUHLTADD-UHFFFAOYSA-N 0.000 description 1
- 229950005529 arzoxifene Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- BIDUPMYXGFNAEJ-APGVDKLISA-N astromicin Chemical compound O[C@@H]1[C@H](N(C)C(=O)CN)[C@@H](OC)[C@@H](O)[C@H](N)[C@H]1O[C@@H]1[C@H](N)CC[C@@H]([C@H](C)N)O1 BIDUPMYXGFNAEJ-APGVDKLISA-N 0.000 description 1
- 229950004810 atamestane Drugs 0.000 description 1
- PEPMWUSGRKINHX-TXTPUJOMSA-N atamestane Chemical compound C1C[C@@H]2[C@@]3(C)C(C)=CC(=O)C=C3CC[C@H]2[C@@H]2CCC(=O)[C@]21C PEPMWUSGRKINHX-TXTPUJOMSA-N 0.000 description 1
- 229960003852 atezolizumab Drugs 0.000 description 1
- 229950010993 atrasentan Drugs 0.000 description 1
- 229940054720 avage Drugs 0.000 description 1
- 229950002916 avelumab Drugs 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- 229960002278 azidamfenicol Drugs 0.000 description 1
- SGRUZFCHLOFYHZ-MWLCHTKSSA-N azidamfenicol Chemical compound [N-]=[N+]=NCC(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 SGRUZFCHLOFYHZ-MWLCHTKSSA-N 0.000 description 1
- 229960004099 azithromycin Drugs 0.000 description 1
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 1
- 229960000190 bacillus calmette–guérin vaccine Drugs 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 229950001429 batabulin Drugs 0.000 description 1
- 229950008356 becatecarin Drugs 0.000 description 1
- 229960001192 bekanamycin Drugs 0.000 description 1
- 229960003094 belinostat Drugs 0.000 description 1
- NCNRHFGMJRPRSK-MDZDMXLPSA-N belinostat Chemical compound ONC(=O)\C=C\C1=CC=CC(S(=O)(=O)NC=2C=CC=CC=2)=C1 NCNRHFGMJRPRSK-MDZDMXLPSA-N 0.000 description 1
- LNHWXBUNXOXMRL-VWLOTQADSA-N belotecan Chemical compound C1=CC=C2C(CCNC(C)C)=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 LNHWXBUNXOXMRL-VWLOTQADSA-N 0.000 description 1
- 229950011276 belotecan Drugs 0.000 description 1
- 229960002707 bendamustine Drugs 0.000 description 1
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 1
- 229960001716 benzalkonium Drugs 0.000 description 1
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- YBHILYKTIRIUTE-UHFFFAOYSA-N berberine Chemical compound C1=C2CC[N+]3=CC4=C(OC)C(OC)=CC=C4C=C3C2=CC2=C1OCO2 YBHILYKTIRIUTE-UHFFFAOYSA-N 0.000 description 1
- 229940093265 berberine Drugs 0.000 description 1
- QISXPYZVZJBNDM-UHFFFAOYSA-N berberine Natural products COc1ccc2C=C3N(Cc2c1OC)C=Cc4cc5OCOc5cc34 QISXPYZVZJBNDM-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 1
- 229940110331 bextra Drugs 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 229950003054 binimetinib Drugs 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- HUTDDBSSHVOYJR-UHFFFAOYSA-H bis[(2-oxo-1,3,2$l^{5},4$l^{2}-dioxaphosphaplumbetan-2-yl)oxy]lead Chemical compound [Pb+2].[Pb+2].[Pb+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O HUTDDBSSHVOYJR-UHFFFAOYSA-H 0.000 description 1
- 229960003008 blinatumomab Drugs 0.000 description 1
- 229940101815 blincyto Drugs 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- JSKFWUPVIZYJMR-UDOAKELVSA-N bmy-27557 Chemical compound O=C1N(CCN(CC)CC)C(=O)C(C2=C3[CH]C=CC(Cl)=C3NC2=C23)=C1C2=C1C=CC=C(Cl)[C]1N3[C@@H]1O[C@H](CO)[C@@H](OC)[C@H](O)[C@H]1O JSKFWUPVIZYJMR-UDOAKELVSA-N 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 238000010504 bond cleavage reaction Methods 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- GJPICJJJRGTNOD-UHFFFAOYSA-N bosentan Chemical compound COC1=CC=CC=C1OC(C(=NC(=N1)C=2N=CC=CN=2)OCCO)=C1NS(=O)(=O)C1=CC=C(C(C)(C)C)C=C1 GJPICJJJRGTNOD-UHFFFAOYSA-N 0.000 description 1
- 229940083476 bosulif Drugs 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 238000002725 brachytherapy Methods 0.000 description 1
- PHEZJEYUWHETKO-UHFFFAOYSA-N brequinar Chemical compound N1=C2C=CC(F)=CC2=C(C(O)=O)C(C)=C1C(C=C1)=CC=C1C1=CC=CC=C1F PHEZJEYUWHETKO-UHFFFAOYSA-N 0.000 description 1
- 229950010231 brequinar Drugs 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 230000000981 bystander Effects 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- PPKJUHVNTMYXOD-PZGPJMECSA-N c49ws9n75l Chemical compound O=C([C@@H]1N(C2=O)CC[C@H]1S(=O)(=O)CCN(CC)CC)O[C@H](C(C)C)[C@H](C)\C=C\C(=O)NC\C=C\C(\C)=C\[C@@H](O)CC(=O)CC1=NC2=CO1.N([C@@H]1C(=O)N[C@@H](C(N2CCC[C@H]2C(=O)N(C)[C@@H](CC=2C=CC(=CC=2)N(C)C)C(=O)N2C[C@@H](CS[C@H]3C4CCN(CC4)C3)C(=O)C[C@H]2C(=O)N[C@H](C(=O)O[C@@H]1C)C=1C=CC=CC=1)=O)CC)C(=O)C1=NC=CC=C1O PPKJUHVNTMYXOD-PZGPJMECSA-N 0.000 description 1
- BMQGVNUXMIRLCK-OAGWZNDDSA-N cabazitaxel Chemical compound O([C@H]1[C@@H]2[C@]3(OC(C)=O)CO[C@@H]3C[C@@H]([C@]2(C(=O)[C@H](OC)C2=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=3C=CC=CC=3)C[C@]1(O)C2(C)C)C)OC)C(=O)C1=CC=CC=C1 BMQGVNUXMIRLCK-OAGWZNDDSA-N 0.000 description 1
- 229960001573 cabazitaxel Drugs 0.000 description 1
- GVEZIHKRYBHEFX-UHFFFAOYSA-N caerulein A Natural products CC=CCC=CCCC(=O)C1OC1C(N)=O GVEZIHKRYBHEFX-UHFFFAOYSA-N 0.000 description 1
- BBBFJLBPOGFECG-VJVYQDLKSA-N calcitonin Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(N)=O)C(C)C)C(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1 BBBFJLBPOGFECG-VJVYQDLKSA-N 0.000 description 1
- 229960004015 calcitonin Drugs 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 229940112129 campath Drugs 0.000 description 1
- 229940088954 camptosar Drugs 0.000 description 1
- OMZCMEYTWSXEPZ-UHFFFAOYSA-N canertinib Chemical compound C1=C(Cl)C(F)=CC=C1NC1=NC=NC2=CC(OCCCN3CCOCC3)=C(NC(=O)C=C)C=C12 OMZCMEYTWSXEPZ-UHFFFAOYSA-N 0.000 description 1
- 229950000772 canfosfamide Drugs 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 125000005587 carbonate group Chemical group 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 229940097647 casodex Drugs 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 150000003943 catecholamines Chemical class 0.000 description 1
- 229960000419 catumaxomab Drugs 0.000 description 1
- 229940047495 celebrex Drugs 0.000 description 1
- 229960000590 celecoxib Drugs 0.000 description 1
- 230000007910 cell fusion Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000005889 cellular cytotoxicity Effects 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 108010046713 cemadotin Proteins 0.000 description 1
- 229950009017 cemadotin Drugs 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229960001602 ceritinib Drugs 0.000 description 1
- GVEZIHKRYBHEFX-NQQPLRFYSA-N cerulenin Chemical compound C\C=C\C\C=C\CCC(=O)[C@H]1O[C@H]1C(N)=O GVEZIHKRYBHEFX-NQQPLRFYSA-N 0.000 description 1
- 229950005984 cerulenin Drugs 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 229950010329 cethromycin Drugs 0.000 description 1
- 229940112106 cetrotide Drugs 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- HZCWPKGYTCJSEB-UHFFFAOYSA-N chembl118841 Chemical compound C12=CC(OC)=CC=C2NC2=C([N+]([O-])=O)C=CC3=C2C1=NN3CCCN(C)C HZCWPKGYTCJSEB-UHFFFAOYSA-N 0.000 description 1
- OKVKOKPUEQTYKI-UHFFFAOYSA-N chembl141769 Chemical compound CS(O)(=O)=O.C12=CC(OC)=CC=C2NC2=C([N+]([O-])=O)C=CC3=C2C1=NN3CCCN(C)C OKVKOKPUEQTYKI-UHFFFAOYSA-N 0.000 description 1
- IQCIQDNWBGEGRL-UHFFFAOYSA-N chembl1614651 Chemical compound O=C1C2=C(O)C=CC(O)=C2N2N=C(CNCCO)C3=CC=C(NCCCN)C1=C32 IQCIQDNWBGEGRL-UHFFFAOYSA-N 0.000 description 1
- KPMVHELZNRNSMN-UHFFFAOYSA-N chembl1985849 Chemical compound N1=CC=C2NCCN21 KPMVHELZNRNSMN-UHFFFAOYSA-N 0.000 description 1
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 1
- ZFVRYNYOPQZKDG-MQMHXKEQSA-N chembl560895 Chemical compound O=C1CC(C)(C)CC2=C1C(C(F)(F)F)=NN2C(C=1)=CC=C(C(N)=O)C=1N[C@H]1CC[C@H](O)CC1 ZFVRYNYOPQZKDG-MQMHXKEQSA-N 0.000 description 1
- 238000012412 chemical coupling Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000012829 chemotherapy agent Substances 0.000 description 1
- 229950009221 chidamide Drugs 0.000 description 1
- 229960005091 chloramphenicol Drugs 0.000 description 1
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical compound ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 1
- CYDMQBQPVICBEU-UHFFFAOYSA-N chlorotetracycline Natural products C1=CC(Cl)=C2C(O)(C)C3CC4C(N(C)C)C(O)=C(C(N)=O)C(=O)C4(O)C(O)=C3C(=O)C2=C1O CYDMQBQPVICBEU-UHFFFAOYSA-N 0.000 description 1
- 229960004475 chlortetracycline Drugs 0.000 description 1
- 235000019365 chlortetracycline Nutrition 0.000 description 1
- CYDMQBQPVICBEU-XRNKAMNCSA-N chlortetracycline Chemical compound C1=CC(Cl)=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(O)=C(C(N)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O CYDMQBQPVICBEU-XRNKAMNCSA-N 0.000 description 1
- 229960000724 cidofovir Drugs 0.000 description 1
- 239000007979 citrate buffer Substances 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 229960002626 clarithromycin Drugs 0.000 description 1
- AGOYDEPGAOXOCK-KCBOHYOISA-N clarithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)OC)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 AGOYDEPGAOXOCK-KCBOHYOISA-N 0.000 description 1
- 229960002227 clindamycin Drugs 0.000 description 1
- KDLRVYVGXIQJDK-AWPVFWJPSA-N clindamycin Chemical compound CN1C[C@H](CCC)C[C@H]1C(=O)N[C@H]([C@H](C)Cl)[C@@H]1[C@H](O)[C@H](O)[C@@H](O)[C@@H](SC)O1 KDLRVYVGXIQJDK-AWPVFWJPSA-N 0.000 description 1
- 229960000928 clofarabine Drugs 0.000 description 1
- WDDPHFBMKLOVOX-AYQXTPAHSA-N clofarabine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1F WDDPHFBMKLOVOX-AYQXTPAHSA-N 0.000 description 1
- 229960004094 clomocycline Drugs 0.000 description 1
- BXVOHUQQUBSHLD-XCTBDMBQSA-N clomocycline Chemical compound C1=CC(Cl)=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(=O)C(=C(/O)NCO)/C(=O)[C@@]4(O)C(=O)C3=C(O)C2=C1O BXVOHUQQUBSHLD-XCTBDMBQSA-N 0.000 description 1
- 101150045189 cmo gene Proteins 0.000 description 1
- 229960002271 cobimetinib Drugs 0.000 description 1
- RESIMIUSNACMNW-BXRWSSRYSA-N cobimetinib fumarate Chemical compound OC(=O)\C=C\C(O)=O.C1C(O)([C@H]2NCCCC2)CN1C(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F.C1C(O)([C@H]2NCCCC2)CN1C(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F RESIMIUSNACMNW-BXRWSSRYSA-N 0.000 description 1
- XQRJFEVDQXEIAX-JFYQDRLCSA-M cobinamide Chemical compound [Co]N([C@@H]1[C@H](CC(N)=O)[C@@]2(C)CCC(=O)NC[C@H](O)C)\C2=C(C)/C([C@H](C\2(C)C)CCC(N)=O)=N/C/2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O XQRJFEVDQXEIAX-JFYQDRLCSA-M 0.000 description 1
- 239000000084 colloidal system Substances 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 229950007276 conatumumab Drugs 0.000 description 1
- 229940031670 conjugate vaccine Drugs 0.000 description 1
- 239000013068 control sample Substances 0.000 description 1
- 239000000599 controlled substance Substances 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 description 1
- 229960000258 corticotropin Drugs 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- CZZLLDDFQKSALH-UHFFFAOYSA-N cpg 8954 Chemical compound O=C1NC(=O)C(C)=CN1C1OC(COP(O)(=O)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=O)OC2C(OC(C2)N2C(N=C(N)C=C2)=O)COP(O)(=O)OC2C(OC(C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(=O)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=O)OC2C(OC(C2)N2C(N=C(N)C=C2)=O)COP(O)(=O)OC2C(OC(C2)N2C3=NC=NC(N)=C3N=C2)COP(O)(=O)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=O)OC2C(OC(C2)N2C(N=C(N)C=C2)=O)COP(O)(=O)OC2C(OC(C2)N2C3=NC=NC(N)=C3N=C2)COP(O)(=O)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=O)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=O)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=O)OC2C(OC(C2)N2C3=C(C(NC(N)=N3)=O)N=C2)CO)C(OP(O)(=O)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=O)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=O)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=O)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=O)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=O)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=O)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)O)C1 CZZLLDDFQKSALH-UHFFFAOYSA-N 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 101150054175 cro gene Proteins 0.000 description 1
- 108010006226 cryptophycin Proteins 0.000 description 1
- PSNOPSMXOBPNNV-VVCTWANISA-N cryptophycin 1 Chemical compound C1=C(Cl)C(OC)=CC=C1C[C@@H]1C(=O)NC[C@@H](C)C(=O)O[C@@H](CC(C)C)C(=O)O[C@H]([C@H](C)[C@@H]2[C@H](O2)C=2C=CC=CC=2)C/C=C/C(=O)N1 PSNOPSMXOBPNNV-VVCTWANISA-N 0.000 description 1
- PSNOPSMXOBPNNV-UHFFFAOYSA-N cryptophycin-327 Natural products C1=C(Cl)C(OC)=CC=C1CC1C(=O)NCC(C)C(=O)OC(CC(C)C)C(=O)OC(C(C)C2C(O2)C=2C=CC=CC=2)CC=CC(=O)N1 PSNOPSMXOBPNNV-UHFFFAOYSA-N 0.000 description 1
- DMSZORWOGDLWGN-UHFFFAOYSA-N ctk1a3526 Chemical compound NP(N)(N)=O DMSZORWOGDLWGN-UHFFFAOYSA-N 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 239000002875 cyclin dependent kinase inhibitor Substances 0.000 description 1
- 150000003999 cyclitols Chemical class 0.000 description 1
- CCQPAEQGAVNNIA-UHFFFAOYSA-N cyclobutane-1,1-dicarboxylic acid Chemical compound OC(=O)C1(C(O)=O)CCC1 CCQPAEQGAVNNIA-UHFFFAOYSA-N 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- UKJLNMAFNRKWGR-UHFFFAOYSA-N cyclohexatrienamine Chemical group NC1=CC=C=C[CH]1 UKJLNMAFNRKWGR-UHFFFAOYSA-N 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229960000978 cyproterone acetate Drugs 0.000 description 1
- UWFYSQMTEOIJJG-FDTZYFLXSA-N cyproterone acetate Chemical compound C1=C(Cl)C2=CC(=O)[C@@H]3C[C@@H]3[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 UWFYSQMTEOIJJG-FDTZYFLXSA-N 0.000 description 1
- 230000001461 cytolytic effect Effects 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 229940099418 d- alpha-tocopherol succinate Drugs 0.000 description 1
- 229960002465 dabrafenib Drugs 0.000 description 1
- BFSMGDJOXZAERB-UHFFFAOYSA-N dabrafenib Chemical compound S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 BFSMGDJOXZAERB-UHFFFAOYSA-N 0.000 description 1
- 229960002806 daclizumab Drugs 0.000 description 1
- 229940070230 daypro Drugs 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000008260 defense mechanism Effects 0.000 description 1
- 229960000958 deferoxamine Drugs 0.000 description 1
- 229960002398 demeclocycline Drugs 0.000 description 1
- 229960001251 denosumab Drugs 0.000 description 1
- 229960003314 deracoxib Drugs 0.000 description 1
- 229940051427 deramaxx Drugs 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- XOZIUKBZLSUILX-UHFFFAOYSA-N desoxyepothilone B Natural products O1C(=O)CC(O)C(C)(C)C(=O)C(C)C(O)C(C)CCCC(C)=CCC1C(C)=CC1=CSC(C)=N1 XOZIUKBZLSUILX-UHFFFAOYSA-N 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 229910003460 diamond Inorganic materials 0.000 description 1
- 239000010432 diamond Substances 0.000 description 1
- 229950000758 dianhydrogalactitol Drugs 0.000 description 1
- JJCQSGDBDPYCEO-XVZSLQNASA-N dibekacin Chemical compound O1[C@H](CN)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N JJCQSGDBDPYCEO-XVZSLQNASA-N 0.000 description 1
- 229960003807 dibekacin Drugs 0.000 description 1
- 229960001259 diclofenac Drugs 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- LFQCJSBXBZRMTN-OAQYLSRUSA-N diflomotecan Chemical compound CC[C@@]1(O)CC(=O)OCC(C2=O)=C1C=C1N2CC2=CC3=CC(F)=C(F)C=C3N=C21 LFQCJSBXBZRMTN-OAQYLSRUSA-N 0.000 description 1
- 229960000616 diflunisal Drugs 0.000 description 1
- 229960002222 dihydrostreptomycin Drugs 0.000 description 1
- ASXBYYWOLISCLQ-HZYVHMACSA-N dihydrostreptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](CO)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O ASXBYYWOLISCLQ-HZYVHMACSA-N 0.000 description 1
- CITHEXJVPOWHKC-UHFFFAOYSA-N dimyristoyl phosphatidylcholine Chemical compound CCCCCCCCCCCCCC(=O)OCC(COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCC CITHEXJVPOWHKC-UHFFFAOYSA-N 0.000 description 1
- 229960003724 dimyristoylphosphatidylcholine Drugs 0.000 description 1
- 206010013023 diphtheria Diseases 0.000 description 1
- KNKDZWFHOIKECV-UHFFFAOYSA-L dipotassium 2,3,4-trihydroxy-4-oxobutanoate Chemical compound [K+].[K+].OC(=O)C(O)C(O)C(O)=O.[O-]C(=O)C(O)C(O)C([O-])=O KNKDZWFHOIKECV-UHFFFAOYSA-L 0.000 description 1
- OQOQSRMIBLJVHE-UHFFFAOYSA-L dipotassium 2-hydroxy-2-oxoacetate Chemical compound [K+].[K+].OC(=O)C(O)=O.[O-]C(=O)C([O-])=O OQOQSRMIBLJVHE-UHFFFAOYSA-L 0.000 description 1
- 229960004100 dirithromycin Drugs 0.000 description 1
- WLOHNSSYAXHWNR-NXPDYKKBSA-N dirithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H]2O[C@H](COCCOC)N[C@H]([C@@H]2C)[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 WLOHNSSYAXHWNR-NXPDYKKBSA-N 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- WGFMTHGYKYEDHF-UHFFFAOYSA-L disodium 2-hydroxy-2-oxoacetate Chemical compound [Na+].[Na+].OC(=O)C(O)=O.[O-]C(=O)C([O-])=O WGFMTHGYKYEDHF-UHFFFAOYSA-L 0.000 description 1
- SILCDLWESNHZKB-UHFFFAOYSA-L disodium 4-hydroxy-4-oxobutanoate Chemical compound [Na+].[Na+].OC(=O)CCC([O-])=O.OC(=O)CCC([O-])=O SILCDLWESNHZKB-UHFFFAOYSA-L 0.000 description 1
- MSJMDZAOKORVFC-SEPHDYHBSA-L disodium fumarate Chemical compound [Na+].[Na+].[O-]C(=O)\C=C\C([O-])=O MSJMDZAOKORVFC-SEPHDYHBSA-L 0.000 description 1
- NYDXNILOWQXUOF-UHFFFAOYSA-L disodium;2-[[4-[2-(2-amino-4-oxo-1,7-dihydropyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]amino]pentanedioate Chemical compound [Na+].[Na+].C=1NC=2NC(N)=NC(=O)C=2C=1CCC1=CC=C(C(=O)NC(CCC([O-])=O)C([O-])=O)C=C1 NYDXNILOWQXUOF-UHFFFAOYSA-L 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 108010045552 dolastatin 15 Proteins 0.000 description 1
- 229940072701 dolobid Drugs 0.000 description 1
- 229960000413 doxercalciferol Drugs 0.000 description 1
- ZWAOHEXOSAUJHY-ZIYNGMLESA-N doxifluridine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ZWAOHEXOSAUJHY-ZIYNGMLESA-N 0.000 description 1
- 229950005454 doxifluridine Drugs 0.000 description 1
- 229960003722 doxycycline Drugs 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 229960005501 duocarmycin Drugs 0.000 description 1
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 description 1
- 229930184221 duocarmycin Natural products 0.000 description 1
- GVGYEFKIHJTNQZ-RFQIPJPRSA-N ecgonine benzoate Chemical compound O([C@@H]1[C@@H]([C@H]2CC[C@@H](C1)N2C)C(O)=O)C(=O)C1=CC=CC=C1 GVGYEFKIHJTNQZ-RFQIPJPRSA-N 0.000 description 1
- 229960001776 edrecolomab Drugs 0.000 description 1
- 229960000925 efaproxiral Drugs 0.000 description 1
- 229940121647 egfr inhibitor Drugs 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 229940087477 ellence Drugs 0.000 description 1
- 229940120655 eloxatin Drugs 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- 230000012202 endocytosis Effects 0.000 description 1
- 210000001163 endosome Anatomy 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- JOZGNYDSEBIJDH-UHFFFAOYSA-N eniluracil Chemical compound O=C1NC=C(C#C)C(=O)N1 JOZGNYDSEBIJDH-UHFFFAOYSA-N 0.000 description 1
- 229950010213 eniluracil Drugs 0.000 description 1
- HKSZLNNOFSGOKW-UHFFFAOYSA-N ent-staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 HKSZLNNOFSGOKW-UHFFFAOYSA-N 0.000 description 1
- 229950005837 entinostat Drugs 0.000 description 1
- 229960004671 enzalutamide Drugs 0.000 description 1
- 229950002189 enzastaurin Drugs 0.000 description 1
- 229950008631 eperezolid Drugs 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 229930013356 epothilone Natural products 0.000 description 1
- HESCAJZNRMSMJG-HGYUPSKWSA-N epothilone A Natural products O=C1[C@H](C)[C@H](O)[C@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C HESCAJZNRMSMJG-HGYUPSKWSA-N 0.000 description 1
- 150000003883 epothilone derivatives Chemical class 0.000 description 1
- 108700002672 epoxomicin Proteins 0.000 description 1
- DOGIDQKFVLKMLQ-JTHVHQAWSA-N epoxomicin Chemical compound CC[C@H](C)[C@H](N(C)C(C)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(=O)[C@@]1(C)CO1 DOGIDQKFVLKMLQ-JTHVHQAWSA-N 0.000 description 1
- 229950009760 epratuzumab Drugs 0.000 description 1
- 229950006835 eptaplatin Drugs 0.000 description 1
- 229940082789 erbitux Drugs 0.000 description 1
- UNXHWFMMPAWVPI-ZXZARUISSA-N erythritol Chemical compound OC[C@H](O)[C@H](O)CO UNXHWFMMPAWVPI-ZXZARUISSA-N 0.000 description 1
- 229940009714 erythritol Drugs 0.000 description 1
- 235000019414 erythritol Nutrition 0.000 description 1
- 229960003276 erythromycin Drugs 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 229960001842 estramustine Drugs 0.000 description 1
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 1
- GBPZYMBDOBODNK-SFTDATJTSA-N ethyl (2s)-2-[[(2s)-2-acetamido-3-[4-[bis(2-chloroethyl)amino]phenyl]propanoyl]amino]-4-methylpentanoate Chemical compound CCOC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(C)=O)CC1=CC=C(N(CCCl)CCCl)C=C1 GBPZYMBDOBODNK-SFTDATJTSA-N 0.000 description 1
- GBPZYMBDOBODNK-LBAQZLPGSA-N ethyl (2s)-2-[[2-acetamido-3-[4-[bis(2-chloroethyl)amino]phenyl]propanoyl]amino]-4-methylpentanoate Chemical compound CCOC(=O)[C@H](CC(C)C)NC(=O)C(NC(C)=O)CC1=CC=C(N(CCCl)CCCl)C=C1 GBPZYMBDOBODNK-LBAQZLPGSA-N 0.000 description 1
- 229960005293 etodolac Drugs 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 229940085363 evista Drugs 0.000 description 1
- ZVYVPGLRVWUPMP-FYSMJZIKSA-N exatecan Chemical compound C1C[C@H](N)C2=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC3=CC(F)=C(C)C1=C32 ZVYVPGLRVWUPMP-FYSMJZIKSA-N 0.000 description 1
- 229950009429 exatecan Drugs 0.000 description 1
- 230000028023 exocytosis Effects 0.000 description 1
- 231100000776 exotoxin Toxicity 0.000 description 1
- 239000002095 exotoxin Substances 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 229940043168 fareston Drugs 0.000 description 1
- 229940087861 faslodex Drugs 0.000 description 1
- 229940065410 feldene Drugs 0.000 description 1
- 229940087476 femara Drugs 0.000 description 1
- 229950003662 fenretinide Drugs 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 229960004177 filgrastim Drugs 0.000 description 1
- 229960003760 florfenicol Drugs 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 229960005304 fludarabine phosphate Drugs 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229960001398 flurithromycin Drugs 0.000 description 1
- XOEUHCONYHZURQ-HNUBZJOYSA-N flurithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@@](C)(F)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 XOEUHCONYHZURQ-HNUBZJOYSA-N 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- 230000022244 formylation Effects 0.000 description 1
- 238000006170 formylation reaction Methods 0.000 description 1
- 229960005102 foscarnet Drugs 0.000 description 1
- 229950005309 fostamatinib Drugs 0.000 description 1
- GKDRMWXFWHEQQT-UHFFFAOYSA-N fostamatinib Chemical compound COC1=C(OC)C(OC)=CC(NC=2N=C(NC=3N=C4N(COP(O)(O)=O)C(=O)C(C)(C)OC4=CC=3)C(F)=CN=2)=C1 GKDRMWXFWHEQQT-UHFFFAOYSA-N 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- 229960003704 framycetin Drugs 0.000 description 1
- PGBHMTALBVVCIT-VCIWKGPPSA-N framycetin Chemical compound N[C@@H]1[C@@H](O)[C@H](O)[C@H](CN)O[C@@H]1O[C@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](N)C[C@@H](N)[C@@H]2O)O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CN)O2)N)O[C@@H]1CO PGBHMTALBVVCIT-VCIWKGPPSA-N 0.000 description 1
- 229950004003 fresolimumab Drugs 0.000 description 1
- 230000033581 fucosylation Effects 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- UIWYJDYFSGRHKR-UHFFFAOYSA-N gadolinium atom Chemical compound [Gd] UIWYJDYFSGRHKR-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-GUCUJZIJSA-N galactitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-GUCUJZIJSA-N 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 229960002963 ganciclovir Drugs 0.000 description 1
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 description 1
- 150000002270 gangliosides Chemical class 0.000 description 1
- 108010063604 gastrin immunogen Proteins 0.000 description 1
- 238000001641 gel filtration chromatography Methods 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- QTQAWLPCGQOSGP-GBTDJJJQSA-N geldanamycin Chemical compound N1C(=O)\C(C)=C/C=C\[C@@H](OC)[C@H](OC(N)=O)\C(C)=C/[C@@H](C)[C@@H](O)[C@H](OC)C[C@@H](C)CC2=C(OC)C(=O)C=C1C2=O QTQAWLPCGQOSGP-GBTDJJJQSA-N 0.000 description 1
- 230000030279 gene silencing Effects 0.000 description 1
- 238000012226 gene silencing method Methods 0.000 description 1
- 229960002518 gentamicin Drugs 0.000 description 1
- UIVFUQKYVFCEKJ-OPTOVBNMSA-N gimatecan Chemical compound C1=CC=C2C(\C=N\OC(C)(C)C)=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UIVFUQKYVFCEKJ-OPTOVBNMSA-N 0.000 description 1
- 229950009073 gimatecan Drugs 0.000 description 1
- 235000008434 ginseng Nutrition 0.000 description 1
- 229950010415 givinostat Drugs 0.000 description 1
- 229950000918 glembatumumab Drugs 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229950011595 glufosfamide Drugs 0.000 description 1
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 1
- YPZRWBKMTBYPTK-BJDJZHNGSA-N glutathione disulfide Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@H](C(=O)NCC(O)=O)CSSC[C@@H](C(=O)NCC(O)=O)NC(=O)CC[C@H](N)C(O)=O YPZRWBKMTBYPTK-BJDJZHNGSA-N 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 208000027124 goblet cell carcinoma Diseases 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 239000003630 growth substance Substances 0.000 description 1
- PKWIYNIDEDLDCJ-UHFFFAOYSA-N guanazole Chemical compound NC1=NNC(N)=N1 PKWIYNIDEDLDCJ-UHFFFAOYSA-N 0.000 description 1
- 125000005179 haloacetyl group Chemical group 0.000 description 1
- 229950010152 halofuginone Drugs 0.000 description 1
- LVASCWIMLIKXLA-LSDHHAIUSA-N halofuginone Chemical compound O[C@@H]1CCCN[C@H]1CC(=O)CN1C(=O)C2=CC(Cl)=C(Br)C=C2N=C1 LVASCWIMLIKXLA-LSDHHAIUSA-N 0.000 description 1
- 239000003481 heat shock protein 90 inhibitor Substances 0.000 description 1
- 229940062743 hectorol Drugs 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 229960004931 histamine dihydrochloride Drugs 0.000 description 1
- 108010040030 histidinoalanine Proteins 0.000 description 1
- PFOARMALXZGCHY-UHFFFAOYSA-N homoegonol Natural products C1=C(OC)C(OC)=CC=C1C1=CC2=CC(CCCO)=CC(OC)=C2O1 PFOARMALXZGCHY-UHFFFAOYSA-N 0.000 description 1
- MFZWMTSUNYWVBU-UHFFFAOYSA-N hycanthone Chemical compound S1C2=CC=CC=C2C(=O)C2=C1C(CO)=CC=C2NCCN(CC)CC MFZWMTSUNYWVBU-UHFFFAOYSA-N 0.000 description 1
- 229950000216 hycanthone Drugs 0.000 description 1
- 125000005597 hydrazone group Chemical group 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Chemical class O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 229960004171 hydroxychloroquine Drugs 0.000 description 1
- XXSMGPRMXLTPCZ-UHFFFAOYSA-N hydroxychloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CCO)CC)=CC=NC2=C1 XXSMGPRMXLTPCZ-UHFFFAOYSA-N 0.000 description 1
- CSFWPUWCSPOLJW-UHFFFAOYSA-N hydroxynaphthoquinone Natural products C1=CC=C2C(=O)C(O)=CC(=O)C2=C1 CSFWPUWCSPOLJW-UHFFFAOYSA-N 0.000 description 1
- GRRNUXAQVGOGFE-NZSRVPFOSA-N hygromycin B Chemical compound O[C@@H]1[C@@H](NC)C[C@@H](N)[C@H](O)[C@H]1O[C@H]1[C@H]2O[C@@]3([C@@H]([C@@H](O)[C@@H](O)[C@@H](C(N)CO)O3)O)O[C@H]2[C@@H](O)[C@@H](CO)O1 GRRNUXAQVGOGFE-NZSRVPFOSA-N 0.000 description 1
- 229940097277 hygromycin b Drugs 0.000 description 1
- 230000001146 hypoxic effect Effects 0.000 description 1
- 229960005236 ibandronic acid Drugs 0.000 description 1
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 1
- 229960001507 ibrutinib Drugs 0.000 description 1
- XYFPWWZEPKGCCK-GOSISDBHSA-N ibrutinib Chemical compound C1=2C(N)=NC=NC=2N([C@H]2CN(CCC2)C(=O)C=C)N=C1C(C=C1)=CC=C1OC1=CC=CC=C1 XYFPWWZEPKGCCK-GOSISDBHSA-N 0.000 description 1
- 229940049235 iclusig Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960002751 imiquimod Drugs 0.000 description 1
- DOUYETYNHWVLEO-UHFFFAOYSA-N imiquimod Chemical compound C1=CC=CC2=C3N(CC(C)C)C=NC3=C(N)N=C21 DOUYETYNHWVLEO-UHFFFAOYSA-N 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 238000012151 immunohistochemical method Methods 0.000 description 1
- 238000003364 immunohistochemistry Methods 0.000 description 1
- 230000001024 immunotherapeutic effect Effects 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 229940089536 indocin Drugs 0.000 description 1
- 229960000905 indomethacin Drugs 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 229960003786 inosine Drugs 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 108010042414 interferon gamma-1b Proteins 0.000 description 1
- 229960001388 interferon-beta Drugs 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 206010073095 invasive ductal breast carcinoma Diseases 0.000 description 1
- 201000010985 invasive ductal carcinoma Diseases 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229950010897 iproplatin Drugs 0.000 description 1
- 229960000779 irinotecan hydrochloride Drugs 0.000 description 1
- 230000001788 irregular Effects 0.000 description 1
- 108010078274 isoleucylvaline Proteins 0.000 description 1
- 150000002540 isothiocyanates Chemical class 0.000 description 1
- FABUFPQFXZVHFB-PVYNADRNSA-N ixabepilone Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)N1)O)C)=C\C1=CSC(C)=N1 FABUFPQFXZVHFB-PVYNADRNSA-N 0.000 description 1
- 229960003648 ixazomib Drugs 0.000 description 1
- MXAYKZJJDUDWDS-LBPRGKRZSA-N ixazomib Chemical compound CC(C)C[C@@H](B(O)O)NC(=O)CNC(=O)C1=CC(Cl)=CC=C1Cl MXAYKZJJDUDWDS-LBPRGKRZSA-N 0.000 description 1
- 229960004144 josamycin Drugs 0.000 description 1
- XJSFLOJWULLJQS-NGVXBBESSA-N josamycin Chemical compound CO[C@H]1[C@H](OC(C)=O)CC(=O)O[C@H](C)C\C=C\C=C\[C@H](O)[C@H](C)C[C@H](CC=O)[C@@H]1O[C@H]1[C@H](O)[C@@H](N(C)C)[C@H](O[C@@H]2O[C@@H](C)[C@H](OC(=O)CC(C)C)[C@](C)(O)C2)[C@@H](C)O1 XJSFLOJWULLJQS-NGVXBBESSA-N 0.000 description 1
- 229960000318 kanamycin Drugs 0.000 description 1
- 229930027917 kanamycin Natural products 0.000 description 1
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 1
- 229930182823 kanamycin A Natural products 0.000 description 1
- SKKLOUVUUNMCJE-FQSMHNGLSA-N kanamycin B Chemical compound N[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SKKLOUVUUNMCJE-FQSMHNGLSA-N 0.000 description 1
- 229930182824 kanamycin B Natural products 0.000 description 1
- SKKLOUVUUNMCJE-UHFFFAOYSA-N kanendomycin Natural products NC1C(O)C(O)C(CN)OC1OC1C(O)C(OC2C(C(N)C(O)C(CO)O2)O)C(N)CC1N SKKLOUVUUNMCJE-UHFFFAOYSA-N 0.000 description 1
- 239000003835 ketolide antibiotic agent Substances 0.000 description 1
- 229960000991 ketoprofen Drugs 0.000 description 1
- 229960004752 ketorolac Drugs 0.000 description 1
- OZWKMVRBQXNZKK-UHFFFAOYSA-N ketorolac Chemical compound OC(=O)C1CCN2C1=CC=C2C(=O)C1=CC=CC=C1 OZWKMVRBQXNZKK-UHFFFAOYSA-N 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 229940000764 kyprolis Drugs 0.000 description 1
- 238000011005 laboratory method Methods 0.000 description 1
- 229960002437 lanreotide Drugs 0.000 description 1
- 208000003849 large cell carcinoma Diseases 0.000 description 1
- 229950000340 laromustine Drugs 0.000 description 1
- 229950005692 larotaxel Drugs 0.000 description 1
- 239000002523 lectin Substances 0.000 description 1
- 229960004942 lenalidomide Drugs 0.000 description 1
- 229960002618 lenograstim Drugs 0.000 description 1
- 229960003784 lenvatinib Drugs 0.000 description 1
- WOSKHXYHFSIKNG-UHFFFAOYSA-N lenvatinib Chemical compound C=12C=C(C(N)=O)C(OC)=CC2=NC=CC=1OC(C=C1Cl)=CC=C1NC(=O)NC1CC1 WOSKHXYHFSIKNG-UHFFFAOYSA-N 0.000 description 1
- YACHGFWEQXFSBS-XYERBDPFSA-N leptomycin B Chemical compound OC(=O)/C=C(C)/C[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@H](C)/C=C(\C)/C=C/C[C@@H](C)/C=C(/CC)\C=C\[C@@H]1OC(=O)C=C[C@@H]1C YACHGFWEQXFSBS-XYERBDPFSA-N 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- OJMMVQQUTAEWLP-KIDUDLJLSA-N lincomycin Chemical compound CN1C[C@H](CCC)C[C@H]1C(=O)N[C@H]([C@@H](C)O)[C@@H]1[C@H](O)[C@H](O)[C@@H](O)[C@@H](SC)O1 OJMMVQQUTAEWLP-KIDUDLJLSA-N 0.000 description 1
- 229960005287 lincomycin Drugs 0.000 description 1
- 229960003907 linezolid Drugs 0.000 description 1
- TYZROVQLWOKYKF-ZDUSSCGKSA-N linezolid Chemical compound O=C1O[C@@H](CNC(=O)C)CN1C(C=C1F)=CC=C1N1CCOCC1 TYZROVQLWOKYKF-ZDUSSCGKSA-N 0.000 description 1
- 229950002950 lintuzumab Drugs 0.000 description 1
- 238000001638 lipofection Methods 0.000 description 1
- 235000019136 lipoic acid Nutrition 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 229950011263 lirilumab Drugs 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 229950008991 lobaplatin Drugs 0.000 description 1
- 229940063718 lodine Drugs 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 229960000994 lumiracoxib Drugs 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 201000008443 lung non-squamous non-small cell carcinoma Diseases 0.000 description 1
- RVFGKBWWUQOIOU-NDEPHWFRSA-N lurtotecan Chemical compound O=C([C@]1(O)CC)OCC(C(N2CC3=4)=O)=C1C=C2C3=NC1=CC=2OCCOC=2C=C1C=4CN1CCN(C)CC1 RVFGKBWWUQOIOU-NDEPHWFRSA-N 0.000 description 1
- 229950002654 lurtotecan Drugs 0.000 description 1
- 229960004196 lymecycline Drugs 0.000 description 1
- AHEVKYYGXVEWNO-UEPZRUIBSA-N lymecycline Chemical compound C1=CC=C2[C@](O)(C)[C@H]3C[C@H]4[C@H](N(C)C)C(O)=C(C(=O)NCNCCCC[C@H](N)C(O)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O AHEVKYYGXVEWNO-UEPZRUIBSA-N 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 1
- 229940124302 mTOR inhibitor Drugs 0.000 description 1
- 229940092110 macugen Drugs 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 229950001474 maitansine Drugs 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 208000014699 malignant epithelioid mesothelioma Diseases 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 1
- 229960000733 mannosulfan Drugs 0.000 description 1
- UUVIQYKKKBJYJT-ZYUZMQFOSA-N mannosulfan Chemical compound CS(=O)(=O)OC[C@@H](OS(C)(=O)=O)[C@@H](O)[C@H](O)[C@H](OS(C)(=O)=O)COS(C)(=O)=O UUVIQYKKKBJYJT-ZYUZMQFOSA-N 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- WKPWGQKGSOKKOO-RSFHAFMBSA-N maytansine Chemical compound CO[C@@H]([C@@]1(O)C[C@](OC(=O)N1)([C@H]([C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(C)=O)CC(=O)N1C)C)[H])\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 WKPWGQKGSOKKOO-RSFHAFMBSA-N 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical class ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 229960000826 meclocycline Drugs 0.000 description 1
- 229940090004 megace Drugs 0.000 description 1
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 229940115256 melanoma vaccine Drugs 0.000 description 1
- LWYJUZBXGAFFLP-OCNCTQISSA-N menogaril Chemical compound O1[C@@]2(C)[C@H](O)[C@@H](N(C)C)[C@H](O)[C@@H]1OC1=C3C(=O)C(C=C4C[C@@](C)(O)C[C@H](C4=C4O)OC)=C4C(=O)C3=C(O)C=C12 LWYJUZBXGAFFLP-OCNCTQISSA-N 0.000 description 1
- 229950002676 menogaril Drugs 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229940100630 metacresol Drugs 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- 229940042016 methacycline Drugs 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- HRHKSTOGXBBQCB-VFWICMBZSA-N methylmitomycin Chemical compound O=C1C(N)=C(C)C(=O)C2=C1[C@@H](COC(N)=O)[C@@]1(OC)[C@H]3N(C)[C@H]3CN12 HRHKSTOGXBBQCB-VFWICMBZSA-N 0.000 description 1
- 229940099246 mevacor Drugs 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 229960002757 midecamycin Drugs 0.000 description 1
- UQRORFVVSGFNRO-UTINFBMNSA-N miglustat Chemical compound CCCCN1C[C@H](O)[C@@H](O)[C@H](O)[C@H]1CO UQRORFVVSGFNRO-UTINFBMNSA-N 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- PQLXHQMOHUQAKB-UHFFFAOYSA-N miltefosine Chemical compound CCCCCCCCCCCCCCCCOP([O-])(=O)OCC[N+](C)(C)C PQLXHQMOHUQAKB-UHFFFAOYSA-N 0.000 description 1
- 229960004023 minocycline Drugs 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 239000003226 mitogen Substances 0.000 description 1
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 1
- VFKZTMPDYBFSTM-GUCUJZIJSA-N mitolactol Chemical compound BrC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-GUCUJZIJSA-N 0.000 description 1
- 229950010913 mitolactol Drugs 0.000 description 1
- 229950005967 mitozolomide Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229950007812 mocetinostat Drugs 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 229960003063 molgramostim Drugs 0.000 description 1
- 108010032806 molgramostim Proteins 0.000 description 1
- DASWEROEPLKSEI-UIJRFTGLSA-N monomethyl auristatin e Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)C1=CC=CC=C1 DASWEROEPLKSEI-UIJRFTGLSA-N 0.000 description 1
- MFRNYXJJRJQHNW-NARUGQRUSA-N monomethyl auristatin f Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)C([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MFRNYXJJRJQHNW-NARUGQRUSA-N 0.000 description 1
- VYGYNVZNSSTDLJ-HKCOAVLJSA-N monorden Natural products CC1CC2OC2C=C/C=C/C(=O)CC3C(C(=CC(=C3Cl)O)O)C(=O)O1 VYGYNVZNSSTDLJ-HKCOAVLJSA-N 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 239000002524 monosodium citrate Substances 0.000 description 1
- 235000018342 monosodium citrate Nutrition 0.000 description 1
- JFOHFDSMPQIOES-UHFFFAOYSA-N motexafin Chemical compound C1=NC2=CC(OCCOCCOCCOC)=C(OCCOCCOCCOC)C=C2N=CC(C(=C2CCCO)C)=NC2=CC(C(CC)=C2CC)=NC2=CC2=C(CCCO)C(C)=C1N2 JFOHFDSMPQIOES-UHFFFAOYSA-N 0.000 description 1
- 229950011637 motexafin Drugs 0.000 description 1
- AARXZCZYLAFQQU-UHFFFAOYSA-N motexafin gadolinium Chemical compound [Gd].CC(O)=O.CC(O)=O.C1=C([N-]2)C(CC)=C(CC)C2=CC(C(=C2C)CCCO)=NC2=CN=C2C=C(OCCOCCOCCOC)C(OCCOCCOCCOC)=CC2=NC=C2C(C)=C(CCCO)C1=N2 AARXZCZYLAFQQU-UHFFFAOYSA-N 0.000 description 1
- 229940072709 motrin Drugs 0.000 description 1
- 229940126619 mouse monoclonal antibody Drugs 0.000 description 1
- 229950002212 mubritinib Drugs 0.000 description 1
- 201000010879 mucinous adenocarcinoma Diseases 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 235000010460 mustard Nutrition 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 1
- WXHHICFWKXDFOW-BJMVGYQFSA-N n-(2-amino-5-fluorophenyl)-4-[[[(e)-3-pyridin-3-ylprop-2-enoyl]amino]methyl]benzamide Chemical compound NC1=CC=C(F)C=C1NC(=O)C(C=C1)=CC=C1CNC(=O)\C=C\C1=CC=CN=C1 WXHHICFWKXDFOW-BJMVGYQFSA-N 0.000 description 1
- CMEGANPVAXDBPL-INIZCTEOSA-N n-[(7s)-1,2,3-trimethoxy-10-methylsulfanyl-9-oxo-6,7-dihydro-5h-benzo[a]heptalen-7-yl]acetamide Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(SC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC CMEGANPVAXDBPL-INIZCTEOSA-N 0.000 description 1
- SIMWTRCFFSTNMG-AWEZNQCLSA-N n-[[(5s)-3-[3-fluoro-4-[4-(2-hydroxyacetyl)piperazin-1-yl]phenyl]-2-oxo-1,3-oxazolidin-5-yl]methyl]acetamide Chemical compound O=C1O[C@@H](CNC(=O)C)CN1C(C=C1F)=CC=C1N1CCN(C(=O)CO)CC1 SIMWTRCFFSTNMG-AWEZNQCLSA-N 0.000 description 1
- 229960004270 nabumetone Drugs 0.000 description 1
- QRGHOAATPOLDPF-VQFNDLOPSA-N nanatinostat Chemical compound N1=CC(C(=O)NO)=CN=C1N1C[C@@H]([C@@H]2NCC=3N=C4C=CC(F)=CC4=CC=3)[C@@H]2C1 QRGHOAATPOLDPF-VQFNDLOPSA-N 0.000 description 1
- 229940090008 naprosyn Drugs 0.000 description 1
- 229960000513 necitumumab Drugs 0.000 description 1
- IXOXBSCIXZEQEQ-UHTZMRCNSA-N nelarabine Chemical compound C1=NC=2C(OC)=NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O IXOXBSCIXZEQEQ-UHTZMRCNSA-N 0.000 description 1
- 229960000801 nelarabine Drugs 0.000 description 1
- CTMCWCONSULRHO-UHQPFXKFSA-N nemorubicin Chemical compound C1CO[C@H](OC)CN1[C@@H]1[C@H](O)[C@H](C)O[C@@H](O[C@@H]2C3=C(O)C=4C(=O)C5=C(OC)C=CC=C5C(=O)C=4C(O)=C3C[C@](O)(C2)C(=O)CO)C1 CTMCWCONSULRHO-UHQPFXKFSA-N 0.000 description 1
- 229950010159 nemorubicin Drugs 0.000 description 1
- 101150091879 neo gene Proteins 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 108010068617 neonatal Fc receptor Proteins 0.000 description 1
- ZBGPYVZLYBDXKO-HILBYHGXSA-N netilmycin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@]([C@H](NC)[C@@H](O)CO1)(C)O)NCC)[C@H]1OC(CN)=CC[C@H]1N ZBGPYVZLYBDXKO-HILBYHGXSA-N 0.000 description 1
- 229940029345 neupogen Drugs 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229940063708 neutrexin Drugs 0.000 description 1
- 229940080607 nexavar Drugs 0.000 description 1
- 229940099637 nilandron Drugs 0.000 description 1
- 229940109551 nipent Drugs 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 229950006344 nocodazole Drugs 0.000 description 1
- XHWRWCSCBDLOLM-UHFFFAOYSA-N nolatrexed Chemical compound CC1=CC=C2NC(N)=NC(=O)C2=C1SC1=CC=NC=C1 XHWRWCSCBDLOLM-UHFFFAOYSA-N 0.000 description 1
- 229950000891 nolatrexed Drugs 0.000 description 1
- 229940085033 nolvadex Drugs 0.000 description 1
- 230000000683 nonmetastatic effect Effects 0.000 description 1
- 229960002351 oleandomycin Drugs 0.000 description 1
- 235000019367 oleandomycin Nutrition 0.000 description 1
- RZPAKFUAFGMUPI-KGIGTXTPSA-N oleandomycin Chemical compound O1[C@@H](C)[C@H](O)[C@@H](OC)C[C@@H]1O[C@@H]1[C@@H](C)C(=O)O[C@H](C)[C@H](C)[C@H](O)[C@@H](C)C(=O)[C@]2(OC2)C[C@H](C)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C RZPAKFUAFGMUPI-KGIGTXTPSA-N 0.000 description 1
- 229920001542 oligosaccharide Polymers 0.000 description 1
- 150000002482 oligosaccharides Chemical class 0.000 description 1
- 229950003600 ombrabulin Drugs 0.000 description 1
- IXWNTLSTOZFSCM-YVACAVLKSA-N ombrabulin Chemical compound C1=C(NC(=O)[C@@H](N)CO)C(OC)=CC=C1\C=C/C1=CC(OC)=C(OC)C(OC)=C1 IXWNTLSTOZFSCM-YVACAVLKSA-N 0.000 description 1
- 102000027450 oncoproteins Human genes 0.000 description 1
- 108091008819 oncoproteins Proteins 0.000 description 1
- 230000014207 opsonization Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000033667 organ regeneration Effects 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 150000002905 orthoesters Chemical class 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 229940039748 oxalate Drugs 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229960002739 oxaprozin Drugs 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- YPZRWBKMTBYPTK-UHFFFAOYSA-N oxidized gamma-L-glutamyl-L-cysteinylglycine Natural products OC(=O)C(N)CCC(=O)NC(C(=O)NCC(O)=O)CSSCC(C(=O)NCC(O)=O)NC(=O)CCC(N)C(O)=O YPZRWBKMTBYPTK-UHFFFAOYSA-N 0.000 description 1
- 229960000625 oxytetracycline Drugs 0.000 description 1
- IWVCMVBTMGNXQD-PXOLEDIWSA-N oxytetracycline Chemical compound C1=CC=C2[C@](O)(C)[C@H]3[C@H](O)[C@H]4[C@H](N(C)C)C(O)=C(C(N)=O)C(=O)[C@@]4(O)C(O)=C3C(=O)C2=C1O IWVCMVBTMGNXQD-PXOLEDIWSA-N 0.000 description 1
- 235000019366 oxytetracycline Nutrition 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 108700027936 paclitaxel poliglumex Proteins 0.000 description 1
- 229940090244 palladia Drugs 0.000 description 1
- 229960003978 pamidronic acid Drugs 0.000 description 1
- VREZDOWOLGNDPW-UHFFFAOYSA-N pancratistatine Natural products C1=C2C3C(O)C(O)C(O)C(O)C3NC(=O)C2=C(O)C2=C1OCO2 VREZDOWOLGNDPW-UHFFFAOYSA-N 0.000 description 1
- 229960005184 panobinostat Drugs 0.000 description 1
- FPOHNWQLNRZRFC-ZHACJKMWSA-N panobinostat Chemical compound CC=1NC2=CC=CC=C2C=1CCNCC1=CC=C(\C=C\C(=O)NO)C=C1 FPOHNWQLNRZRFC-ZHACJKMWSA-N 0.000 description 1
- 229940096763 panretin Drugs 0.000 description 1
- 201000010279 papillary renal cell carcinoma Diseases 0.000 description 1
- 210000002990 parathyroid gland Anatomy 0.000 description 1
- 229960004662 parecoxib Drugs 0.000 description 1
- TZRHLKRLEZJVIJ-UHFFFAOYSA-N parecoxib Chemical compound C1=CC(S(=O)(=O)NC(=O)CC)=CC=C1C1=C(C)ON=C1C1=CC=CC=C1 TZRHLKRLEZJVIJ-UHFFFAOYSA-N 0.000 description 1
- 229950007460 patupilone Drugs 0.000 description 1
- 230000006320 pegylation Effects 0.000 description 1
- WVUNYSQLFKLYNI-AATRIKPKSA-N pelitinib Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC1=CC=C(F)C(Cl)=C1 WVUNYSQLFKLYNI-AATRIKPKSA-N 0.000 description 1
- 229950003819 pelitrexol Drugs 0.000 description 1
- 229960003349 pemetrexed disodium Drugs 0.000 description 1
- QIMGFXOHTOXMQP-GFAGFCTOSA-N peplomycin Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCCN[C@@H](C)C=1C=CC=CC=1)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C QIMGFXOHTOXMQP-GFAGFCTOSA-N 0.000 description 1
- 229950003180 peplomycin Drugs 0.000 description 1
- 229940111202 pepsin Drugs 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 238000003359 percent control normalization Methods 0.000 description 1
- 229930192851 perforin Natural products 0.000 description 1
- 230000008782 phagocytosis Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 108010018625 phenylalanylarginine Proteins 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- SXADIBFZNXBEGI-UHFFFAOYSA-N phosphoramidous acid Chemical group NP(O)O SXADIBFZNXBEGI-UHFFFAOYSA-N 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229950005566 picoplatin Drugs 0.000 description 1
- 229950010773 pidilizumab Drugs 0.000 description 1
- JTHRRMFZHSDGNJ-UHFFFAOYSA-N piperazine-2,3-dione Chemical compound O=C1NCCNC1=O JTHRRMFZHSDGNJ-UHFFFAOYSA-N 0.000 description 1
- 229960001635 pirlimycin Drugs 0.000 description 1
- 229960002702 piroxicam Drugs 0.000 description 1
- 229960004403 pixantrone Drugs 0.000 description 1
- PEZPMAYDXJQYRV-UHFFFAOYSA-N pixantrone Chemical compound O=C1C2=CN=CC=C2C(=O)C2=C1C(NCCN)=CC=C2NCCN PEZPMAYDXJQYRV-UHFFFAOYSA-N 0.000 description 1
- BLFWHYXWBKKRHI-JYBILGDPSA-N plap Chemical compound N([C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(O)=O)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(=O)[C@@H]1CCCN1C(=O)[C@H](CO)NC(=O)[C@@H](N)CCC(O)=O BLFWHYXWBKKRHI-JYBILGDPSA-N 0.000 description 1
- 229940012957 plasmin Drugs 0.000 description 1
- 108050009312 plexin Proteins 0.000 description 1
- 102000002022 plexin Human genes 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920006324 polyoxymethylene Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 108010001062 polysaccharide-K Proteins 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229940068968 polysorbate 80 Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229940068965 polysorbates Drugs 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229960001131 ponatinib Drugs 0.000 description 1
- 229950004447 posizolid Drugs 0.000 description 1
- 231100000683 possible toxicity Toxicity 0.000 description 1
- 239000004224 potassium gluconate Substances 0.000 description 1
- 229960003189 potassium gluconate Drugs 0.000 description 1
- LCPMNMXCIHBTEX-UHFFFAOYSA-M potassium;2-hydroxypropanoate;2-hydroxypropanoic acid Chemical compound [K+].CC(O)C(O)=O.CC(O)C([O-])=O LCPMNMXCIHBTEX-UHFFFAOYSA-M 0.000 description 1
- JHDKZFFAIZKUCU-ZRDIBKRKSA-N pracinostat Chemical compound ONC(=O)/C=C/C1=CC=C2N(CCN(CC)CC)C(CCCC)=NC2=C1 JHDKZFFAIZKUCU-ZRDIBKRKSA-N 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 229940071643 prefilled syringe Drugs 0.000 description 1
- 229960003961 pristinamycin Drugs 0.000 description 1
- DAIKHDNSXMZDCU-OUDXUNEISA-N pristinamycin-IIA Natural products CC(C)[C@H]1OC(=O)C2=CCCN2C(=O)c3coc(CC(=O)C[C@H](O)C=C(C)C=CCNC(=O)C=C[C@@H]1C)n3 DAIKHDNSXMZDCU-OUDXUNEISA-N 0.000 description 1
- JOOMGSFOCRDAHL-XKCHLWDXSA-N pristinamycin-IIB Natural products CC(C)[C@@H]1OC(=O)[C@H]2CCCN2C(=O)c3coc(CC(=O)C[C@@H](O)C=C(C)C=CCNC(=O)C=C[C@H]1C)n3 JOOMGSFOCRDAHL-XKCHLWDXSA-N 0.000 description 1
- 229940087463 proleukin Drugs 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 229940117382 propecia Drugs 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 229940030749 prostate cancer vaccine Drugs 0.000 description 1
- 235000019419 proteases Nutrition 0.000 description 1
- 108020003519 protein disulfide isomerase Proteins 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 230000006337 proteolytic cleavage Effects 0.000 description 1
- 229940024999 proteolytic enzymes for treatment of wounds and ulcers Drugs 0.000 description 1
- SHCBCKBYTHZQGZ-CJPZEJHVSA-N protopanaxatriol Chemical compound C1C[C@H](O)C(C)(C)[C@@H]2[C@@H](O)C[C@@]3(C)[C@]4(C)CC[C@H]([C@@](C)(O)CCC=C(C)C)[C@H]4[C@H](O)C[C@@H]3[C@]21C SHCBCKBYTHZQGZ-CJPZEJHVSA-N 0.000 description 1
- BBEUDPAEKGPXDG-UHFFFAOYSA-N protopanaxatriol Natural products CC(CCC=C(C)C)C1CCC2(C)C1C(O)CC3C4(C)CCC(O)C(C)(C)C4C(O)CC23C BBEUDPAEKGPXDG-UHFFFAOYSA-N 0.000 description 1
- 229940034080 provenge Drugs 0.000 description 1
- 229940117820 purinethol Drugs 0.000 description 1
- NGVDGCNFYWLIFO-UHFFFAOYSA-N pyridoxal 5'-phosphate Chemical compound CC1=NC=C(COP(O)(O)=O)C(C=O)=C1O NGVDGCNFYWLIFO-UHFFFAOYSA-N 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 229940052337 quinupristin/dalfopristin Drugs 0.000 description 1
- 229950010654 quisinostat Drugs 0.000 description 1
- WVTKBKWTSCPRNU-UHFFFAOYSA-N rac-Tetrandrin Natural products O1C(C(=CC=2CCN3C)OC)=CC=2C3CC(C=C2)=CC=C2OC(=C2)C(OC)=CC=C2CC2N(C)CCC3=CC(OC)=C(OC)C1=C23 WVTKBKWTSCPRNU-UHFFFAOYSA-N 0.000 description 1
- BTTNOGHPGJANSW-IBGZPJMESA-N radezolid Chemical compound O=C1O[C@@H](CNC(=O)C)CN1C1=CC=C(C=2C=CC(CNCC=3NN=NC=3)=CC=2)C(F)=C1 BTTNOGHPGJANSW-IBGZPJMESA-N 0.000 description 1
- 229950009965 radezolid Drugs 0.000 description 1
- AECPBJMOGBFQDN-YMYQVXQQSA-N radicicol Chemical compound C1CCCC(=O)C[C@H]2[C@H](Cl)C(=O)CC(=O)[C@H]2C(=O)O[C@H](C)C[C@H]2O[C@@H]21 AECPBJMOGBFQDN-YMYQVXQQSA-N 0.000 description 1
- 229930192524 radicicol Natural products 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 239000012857 radioactive material Substances 0.000 description 1
- MUPFEKGTMRGPLJ-ZQSKZDJDSA-N raffinose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)O)O1 MUPFEKGTMRGPLJ-ZQSKZDJDSA-N 0.000 description 1
- 229960002633 ramucirumab Drugs 0.000 description 1
- PWHNTOQANLCTHN-KRWDZBQOSA-N ranbezolid Chemical compound O=C1O[C@@H](CNC(=O)C)CN1C(C=C1F)=CC=C1N1CCN(CC=2OC(=CC=2)[N+]([O-])=O)CC1 PWHNTOQANLCTHN-KRWDZBQOSA-N 0.000 description 1
- 108010061338 ranpirnase Proteins 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 229960005567 rebeccamycin Drugs 0.000 description 1
- INSACQSBHKIWNS-QZQSLCQPSA-N rebeccamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](OC)[C@@H](CO)O[C@H]1N1C2=C3N=C4[C](Cl)C=CC=C4C3=C3C(=O)NC(=O)C3=C2C2=CC=CC(Cl)=C21 INSACQSBHKIWNS-QZQSLCQPSA-N 0.000 description 1
- 229950005950 rebimastat Drugs 0.000 description 1
- 108091006082 receptor inhibitors Proteins 0.000 description 1
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 1
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 1
- 229960004836 regorafenib Drugs 0.000 description 1
- 229940087462 relafen Drugs 0.000 description 1
- 208000015347 renal cell adenocarcinoma Diseases 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- FECGNJPYVFEKOD-VMPITWQZSA-N resminostat Chemical compound C1=CC(CN(C)C)=CC=C1S(=O)(=O)N1C=C(\C=C\C(=O)NO)C=C1 FECGNJPYVFEKOD-VMPITWQZSA-N 0.000 description 1
- 229950002821 resminostat Drugs 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 229960002771 retapamulin Drugs 0.000 description 1
- STZYTFJPGGDRJD-NHUWBDDWSA-N retapamulin Chemical compound C([C@H]([C@@]1(C)[C@@H](C[C@@](C)(C=C)[C@@H](O)[C@@H]2C)OC(=O)CS[C@@H]3C[C@H]4CC[C@H](N4C)C3)C)C[C@]32[C@H]1C(=O)CC3 STZYTFJPGGDRJD-NHUWBDDWSA-N 0.000 description 1
- OAKGNIRUXAZDQF-TXHRRWQRSA-N retaspimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(O)C1=CC(O)=C2NCC=C OAKGNIRUXAZDQF-TXHRRWQRSA-N 0.000 description 1
- 229940002683 retin-a Drugs 0.000 description 1
- 150000004492 retinoid derivatives Chemical class 0.000 description 1
- 238000011268 retreatment Methods 0.000 description 1
- 229940120975 revlimid Drugs 0.000 description 1
- MYFATKRONKHHQL-UHFFFAOYSA-N rhodamine 123 Chemical compound [Cl-].COC(=O)C1=CC=CC=C1C1=C2C=CC(=[NH2+])C=C2OC2=CC(N)=CC=C21 MYFATKRONKHHQL-UHFFFAOYSA-N 0.000 description 1
- HEBKCHPVOIAQTA-ZXFHETKHSA-N ribitol Chemical compound OC[C@H](O)[C@H](O)[C@H](O)CO HEBKCHPVOIAQTA-ZXFHETKHSA-N 0.000 description 1
- 229960003485 ribostamycin Drugs 0.000 description 1
- NSKGQURZWSPSBC-NLZFXWNVSA-N ribostamycin Chemical compound N[C@H]1[C@H](O)[C@@H](O)[C@H](CN)O[C@@H]1O[C@@H]1[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@H](CO)O2)O)[C@H](O)[C@@H](N)C[C@H]1N NSKGQURZWSPSBC-NLZFXWNVSA-N 0.000 description 1
- 229930190553 ribostamycin Natural products 0.000 description 1
- NSKGQURZWSPSBC-UHFFFAOYSA-N ribostamycin A Natural products NC1C(O)C(O)C(CN)OC1OC1C(OC2C(C(O)C(CO)O2)O)C(O)C(N)CC1N NSKGQURZWSPSBC-UHFFFAOYSA-N 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229960000311 ritonavir Drugs 0.000 description 1
- NCDNCNXCDXHOMX-XGKFQTDJSA-N ritonavir Chemical compound N([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1SC=NC=1)CC=1C=CC=CC=1)C(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-XGKFQTDJSA-N 0.000 description 1
- 229920002477 rna polymer Polymers 0.000 description 1
- 229960000371 rofecoxib Drugs 0.000 description 1
- 229960001170 rokitamycin Drugs 0.000 description 1
- 229960005009 rolitetracycline Drugs 0.000 description 1
- HMEYVGGHISAPJR-IAHYZSEUSA-N rolitetracycline Chemical compound O=C([C@@]1(O)C(O)=C2[C@@H]([C@](C3=CC=CC(O)=C3C2=O)(C)O)C[C@H]1[C@@H](C=1O)N(C)C)C=1C(=O)NCN1CCCC1 HMEYVGGHISAPJR-IAHYZSEUSA-N 0.000 description 1
- 229960003452 romidepsin Drugs 0.000 description 1
- 108010091666 romidepsin Proteins 0.000 description 1
- OHRURASPPZQGQM-UHFFFAOYSA-N romidepsin Natural products O1C(=O)C(C(C)C)NC(=O)C(=CC)NC(=O)C2CSSCCC=CC1CC(=O)NC(C(C)C)C(=O)N2 OHRURASPPZQGQM-UHFFFAOYSA-N 0.000 description 1
- 229960005224 roxithromycin Drugs 0.000 description 1
- 101150024074 rub1 gene Proteins 0.000 description 1
- HMABYWSNWIZPAG-UHFFFAOYSA-N rucaparib Chemical compound C1=CC(CNC)=CC=C1C(N1)=C2CCNC(=O)C3=C2C1=CC(F)=C3 HMABYWSNWIZPAG-UHFFFAOYSA-N 0.000 description 1
- FCCGJTKEKXUBFZ-UHFFFAOYSA-N rucaparib phosphate Chemical compound OP(O)(O)=O.C1=CC(CNC)=CC=C1C(N1)=C2CCNC(=O)C3=C2C1=CC(F)=C3 FCCGJTKEKXUBFZ-UHFFFAOYSA-N 0.000 description 1
- NGWSFRIPKNWYAO-UHFFFAOYSA-N salinosporamide A Natural products N1C(=O)C(CCCl)C2(C)OC(=O)C21C(O)C1CCCC=C1 NGWSFRIPKNWYAO-UHFFFAOYSA-N 0.000 description 1
- 229960000953 salsalate Drugs 0.000 description 1
- 238000003118 sandwich ELISA Methods 0.000 description 1
- 229930182490 saponin Natural products 0.000 description 1
- 235000017709 saponins Nutrition 0.000 description 1
- 150000007949 saponins Chemical class 0.000 description 1
- 208000014212 sarcomatoid carcinoma Diseases 0.000 description 1
- FVLVBPDQNARYJU-UHFFFAOYSA-N semustine Chemical compound CC1CCC(NC(=O)N(CCCl)N=O)CC1 FVLVBPDQNARYJU-UHFFFAOYSA-N 0.000 description 1
- 229960003440 semustine Drugs 0.000 description 1
- 229950009921 seocalcitol Drugs 0.000 description 1
- 208000019694 serous adenocarcinoma Diseases 0.000 description 1
- 208000004548 serous cystadenocarcinoma Diseases 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 229960003693 sevelamer Drugs 0.000 description 1
- ZNSIZMQNQCNRBW-UHFFFAOYSA-N sevelamer Chemical compound NCC=C.ClCC1CO1 ZNSIZMQNQCNRBW-UHFFFAOYSA-N 0.000 description 1
- 229960005441 sevelamer carbonate Drugs 0.000 description 1
- RAGFPHFDFVNLCG-INYQBOQCSA-N sibiromycin Chemical compound O[C@@H]1[C@@](O)(C)[C@@H](NC)[C@H](C)O[C@H]1OC(C(=C1O)C)=CC(C2=O)=C1N[C@H](O)[C@H]1N2C=C(\C=C\C)C1 RAGFPHFDFVNLCG-INYQBOQCSA-N 0.000 description 1
- RAGFPHFDFVNLCG-UHFFFAOYSA-N sibiromycin Natural products OC1C(O)(C)C(NC)C(C)OC1OC(C(=C1O)C)=CC(C2=O)=C1NC(O)C1N2C=C(C=CC)C1 RAGFPHFDFVNLCG-UHFFFAOYSA-N 0.000 description 1
- 229960000714 sipuleucel-t Drugs 0.000 description 1
- URWAJWIAIPFPJE-YFMIWBNJSA-N sisomycin Chemical compound O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H](CC=C(CN)O2)N)[C@@H](N)C[C@H]1N URWAJWIAIPFPJE-YFMIWBNJSA-N 0.000 description 1
- 238000004513 sizing Methods 0.000 description 1
- 229950001403 sizofiran Drugs 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- KYOYLUVYCHVYGC-BUOKYLHBSA-M sodium (E)-but-2-enedioic acid (E)-4-hydroxy-4-oxobut-2-enoate Chemical compound [Na+].OC(=O)\C=C\C(O)=O.OC(=O)\C=C\C([O-])=O KYOYLUVYCHVYGC-BUOKYLHBSA-M 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000019294 sodium fumarate Nutrition 0.000 description 1
- 239000000176 sodium gluconate Substances 0.000 description 1
- 229940005574 sodium gluconate Drugs 0.000 description 1
- GNBVPFITFYNRCN-UHFFFAOYSA-M sodium thioglycolate Chemical compound [Na+].[O-]C(=O)CS GNBVPFITFYNRCN-UHFFFAOYSA-M 0.000 description 1
- 229940046307 sodium thioglycolate Drugs 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- LLVQEXSQFBTIRD-UHFFFAOYSA-M sodium;2,3,4-trihydroxy-4-oxobutanoate;hydrate Chemical compound O.[Na+].OC(=O)C(O)C(O)C([O-])=O LLVQEXSQFBTIRD-UHFFFAOYSA-M 0.000 description 1
- KMPHTYSTEHXSTL-UHFFFAOYSA-M sodium;2-hydroxypropanoate;2-hydroxypropanoic acid Chemical compound [Na+].CC(O)C(O)=O.CC(O)C([O-])=O KMPHTYSTEHXSTL-UHFFFAOYSA-M 0.000 description 1
- VDZDAHYKYRVHJR-UHFFFAOYSA-M sodium;2-hydroxypropanoate;hydrate Chemical compound [OH-].[Na+].CC(O)C(O)=O VDZDAHYKYRVHJR-UHFFFAOYSA-M 0.000 description 1
- DGPIGKCOQYBCJH-UHFFFAOYSA-M sodium;acetic acid;hydroxide Chemical compound O.[Na+].CC([O-])=O DGPIGKCOQYBCJH-UHFFFAOYSA-M 0.000 description 1
- VBGUQBPWJMPQBI-UHFFFAOYSA-M sodium;butanedioic acid;4-hydroxy-4-oxobutanoate Chemical compound [Na+].OC(=O)CCC(O)=O.OC(=O)CCC([O-])=O VBGUQBPWJMPQBI-UHFFFAOYSA-M 0.000 description 1
- JISIBLCXFLGVJX-UHFFFAOYSA-M sodium;butanedioic acid;hydroxide Chemical compound [OH-].[Na+].OC(=O)CCC(O)=O JISIBLCXFLGVJX-UHFFFAOYSA-M 0.000 description 1
- KIJIBEBWNNLSKE-UHFFFAOYSA-M sodium;oxalic acid;hydroxide Chemical compound [OH-].[Na+].OC(=O)C(O)=O KIJIBEBWNNLSKE-UHFFFAOYSA-M 0.000 description 1
- 229950008588 solithromycin Drugs 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 229940078986 somatuline Drugs 0.000 description 1
- 229940072291 soriatane Drugs 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 229960000268 spectinomycin Drugs 0.000 description 1
- UNFWWIHTNXNPBV-WXKVUWSESA-N spectinomycin Chemical compound O([C@@H]1[C@@H](NC)[C@@H](O)[C@H]([C@@H]([C@H]1O1)O)NC)[C@]2(O)[C@H]1O[C@H](C)CC2=O UNFWWIHTNXNPBV-WXKVUWSESA-N 0.000 description 1
- 229960001294 spiramycin Drugs 0.000 description 1
- 235000019372 spiramycin Nutrition 0.000 description 1
- 229930191512 spiramycin Natural products 0.000 description 1
- 210000004988 splenocyte Anatomy 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- UQZIYBXSHAGNOE-XNSRJBNMSA-N stachyose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@@H]3[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O3)O)O2)O)O1 UQZIYBXSHAGNOE-XNSRJBNMSA-N 0.000 description 1
- 238000011301 standard therapy Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical compound C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 1
- CGPUWJWCVCFERF-UHFFFAOYSA-N staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(OC)O1 CGPUWJWCVCFERF-UHFFFAOYSA-N 0.000 description 1
- SFVFIFLLYFPGHH-UHFFFAOYSA-M stearalkonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCC[N+](C)(C)CC1=CC=CC=C1 SFVFIFLLYFPGHH-UHFFFAOYSA-M 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 229940090374 stivarga Drugs 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 229940041030 streptogramins Drugs 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000008362 succinate buffer Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- MPUQHZXIXSTTDU-QXGSTGNESA-N sulfamic acid [(1S,2S,4R)-4-[4-[[(1S)-2,3-dihydro-1H-inden-1-yl]amino]-7-pyrrolo[2,3-d]pyrimidinyl]-2-hydroxycyclopentyl]methyl ester Chemical compound C1[C@H](O)[C@H](COS(=O)(=O)N)C[C@H]1N1C2=NC=NC(N[C@@H]3C4=CC=CC=C4CC3)=C2C=C1 MPUQHZXIXSTTDU-QXGSTGNESA-N 0.000 description 1
- 229960005559 sulforaphane Drugs 0.000 description 1
- 235000015487 sulforaphane Nutrition 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 229940034785 sutent Drugs 0.000 description 1
- FNDDDNOJWPQCBZ-ZDUSSCGKSA-N sutezolid Chemical compound O=C1O[C@@H](CNC(=O)C)CN1C(C=C1F)=CC=C1N1CCSCC1 FNDDDNOJWPQCBZ-ZDUSSCGKSA-N 0.000 description 1
- 229950000448 sutezolid Drugs 0.000 description 1
- 230000001839 systemic circulation Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 229930194539 taccalonolide Natural products 0.000 description 1
- 108700003774 talisomycin Proteins 0.000 description 1
- 229950002687 talisomycin Drugs 0.000 description 1
- 229950007866 tanespimycin Drugs 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 1
- RCINICONZNJXQF-XAZOAEDWSA-N taxol® Chemical compound O([C@@H]1[C@@]2(CC(C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3(C21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-XAZOAEDWSA-N 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 229960000565 tazarotene Drugs 0.000 description 1
- 229960003879 tedizolid Drugs 0.000 description 1
- XFALPSLJIHVRKE-GFCCVEGCSA-N tedizolid Chemical compound CN1N=NC(C=2N=CC(=CC=2)C=2C(=CC(=CC=2)N2C(O[C@@H](CO)C2)=O)F)=N1 XFALPSLJIHVRKE-GFCCVEGCSA-N 0.000 description 1
- 229950009112 tefinostat Drugs 0.000 description 1
- 229960003250 telithromycin Drugs 0.000 description 1
- LJVAJPDWBABPEJ-PNUFFHFMSA-N telithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)[C@@H](C)C(=O)O[C@@H]([C@]2(OC(=O)N(CCCCN3C=C(N=C3)C=3C=NC=CC=3)[C@@H]2[C@@H](C)C(=O)[C@H](C)C[C@@]1(C)OC)C)CC)[C@@H]1O[C@H](C)C[C@H](N(C)C)[C@H]1O LJVAJPDWBABPEJ-PNUFFHFMSA-N 0.000 description 1
- 229940061353 temodar Drugs 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- IWVCMVBTMGNXQD-UHFFFAOYSA-N terramycin dehydrate Natural products C1=CC=C2C(O)(C)C3C(O)C4C(N(C)C)C(O)=C(C(N)=O)C(=O)C4(O)C(O)=C3C(=O)C2=C1O IWVCMVBTMGNXQD-UHFFFAOYSA-N 0.000 description 1
- 229950007967 tesmilifene Drugs 0.000 description 1
- 229960003433 thalidomide Drugs 0.000 description 1
- 229940110675 theracys Drugs 0.000 description 1
- OTVAEFIXJLOWRX-NXEZZACHSA-N thiamphenicol Chemical compound CS(=O)(=O)C1=CC=C([C@@H](O)[C@@H](CO)NC(=O)C(Cl)Cl)C=C1 OTVAEFIXJLOWRX-NXEZZACHSA-N 0.000 description 1
- 229960003053 thiamphenicol Drugs 0.000 description 1
- 229960002663 thioctic acid Drugs 0.000 description 1
- 229940035024 thioglycerol Drugs 0.000 description 1
- 229960002175 thyroglobulin Drugs 0.000 description 1
- UURAUHCOJAIIRQ-QGLSALSOSA-N tiamulin Chemical compound CCN(CC)CCSCC(=O)O[C@@H]1C[C@@](C)(C=C)[C@@H](O)[C@H](C)[C@@]23CC[C@@H](C)[C@]1(C)[C@@H]2C(=O)CC3 UURAUHCOJAIIRQ-QGLSALSOSA-N 0.000 description 1
- 229960004885 tiamulin Drugs 0.000 description 1
- MIMJSJSRRDZIPW-UHFFFAOYSA-N tilmacoxib Chemical compound C=1C=C(S(N)(=O)=O)C(F)=CC=1C=1OC(C)=NC=1C1CCCCC1 MIMJSJSRRDZIPW-UHFFFAOYSA-N 0.000 description 1
- 230000025934 tissue morphogenesis Effects 0.000 description 1
- 230000007838 tissue remodeling Effects 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 229960000707 tobramycin Drugs 0.000 description 1
- NLVFBUXFDBBNBW-PBSUHMDJSA-N tobramycin Chemical compound N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N NLVFBUXFDBBNBW-PBSUHMDJSA-N 0.000 description 1
- 229960005048 toceranib Drugs 0.000 description 1
- 229960001017 tolmetin Drugs 0.000 description 1
- UPSPUYADGBWSHF-UHFFFAOYSA-N tolmetin Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC=C(CC(O)=O)N1C UPSPUYADGBWSHF-UHFFFAOYSA-N 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 229960000977 trabectedin Drugs 0.000 description 1
- 229940118436 tracleer Drugs 0.000 description 1
- 229960004066 trametinib Drugs 0.000 description 1
- LIRYPHYGHXZJBZ-UHFFFAOYSA-N trametinib Chemical compound CC(=O)NC1=CC=CC(N2C(N(C3CC3)C(=O)C3=C(NC=4C(=CC(I)=CC=4)F)N(C)C(=O)C(C)=C32)=O)=C1 LIRYPHYGHXZJBZ-UHFFFAOYSA-N 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 230000005945 translocation Effects 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 108010060597 trapoxin A Proteins 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 229940032510 trelstar Drugs 0.000 description 1
- 229950007217 tremelimumab Drugs 0.000 description 1
- 229960003181 treosulfan Drugs 0.000 description 1
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 description 1
- 229950001353 tretamine Drugs 0.000 description 1
- 229960004560 triaziquone Drugs 0.000 description 1
- PXSOHRWMIRDKMP-UHFFFAOYSA-N triaziquone Chemical compound O=C1C(N2CC2)=C(N2CC2)C(=O)C=C1N1CC1 PXSOHRWMIRDKMP-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical class CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 1
- 229960000538 trimetrexate glucuronate Drugs 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 150000004043 trisaccharides Chemical class 0.000 description 1
- 229940086984 trisenox Drugs 0.000 description 1
- 229960005041 troleandomycin Drugs 0.000 description 1
- LQCLVBQBTUVCEQ-QTFUVMRISA-N troleandomycin Chemical compound O1[C@@H](C)[C@H](OC(C)=O)[C@@H](OC)C[C@@H]1O[C@@H]1[C@@H](C)C(=O)O[C@H](C)[C@H](C)[C@H](OC(C)=O)[C@@H](C)C(=O)[C@@]2(OC2)C[C@H](C)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)OC(C)=O)[C@H]1C LQCLVBQBTUVCEQ-QTFUVMRISA-N 0.000 description 1
- 230000004565 tumor cell growth Effects 0.000 description 1
- 208000025444 tumor of salivary gland Diseases 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- MEYZYGMYMLNUHJ-UHFFFAOYSA-N tunicamycin Natural products CC(C)CCCCCCCCCC=CC(=O)NC1C(O)C(O)C(CC(O)C2OC(C(O)C2O)N3C=CC(=O)NC3=O)OC1OC4OC(CO)C(O)C(O)C4NC(=O)C MEYZYGMYMLNUHJ-UHFFFAOYSA-N 0.000 description 1
- 229960004059 tylosin Drugs 0.000 description 1
- WBPYTXDJUQJLPQ-VMXQISHHSA-N tylosin Chemical compound O([C@@H]1[C@@H](C)O[C@H]([C@@H]([C@H]1N(C)C)O)O[C@@H]1[C@@H](C)[C@H](O)CC(=O)O[C@@H]([C@H](/C=C(\C)/C=C/C(=O)[C@H](C)C[C@@H]1CC=O)CO[C@H]1[C@@H]([C@H](OC)[C@H](O)[C@@H](C)O1)OC)CC)[C@H]1C[C@@](C)(O)[C@@H](O)[C@H](C)O1 WBPYTXDJUQJLPQ-VMXQISHHSA-N 0.000 description 1
- 235000019375 tylosin Nutrition 0.000 description 1
- 210000001364 upper extremity Anatomy 0.000 description 1
- 229940045136 urea Drugs 0.000 description 1
- 229950005972 urelumab Drugs 0.000 description 1
- XGOYIMQSIKSOBS-UHFFFAOYSA-N vadimezan Chemical compound C1=CC=C2C(=O)C3=CC=C(C)C(C)=C3OC2=C1CC(O)=O XGOYIMQSIKSOBS-UHFFFAOYSA-N 0.000 description 1
- 229960002004 valdecoxib Drugs 0.000 description 1
- LLYYNOVSVPBRGV-MVNKZKPCSA-N valnemulin Chemical compound CC(C)[C@@H](N)C(=O)NCC(C)(C)SCC(=O)O[C@@H]1C[C@@](C)(C=C)[C@@H](O)[C@H](C)[C@@]23CC[C@@H](C)[C@]1(C)[C@@H]2C(=O)CC3 LLYYNOVSVPBRGV-MVNKZKPCSA-N 0.000 description 1
- 229950008166 valnemulin Drugs 0.000 description 1
- 229960000653 valrubicin Drugs 0.000 description 1
- 229940054937 valstar Drugs 0.000 description 1
- 108010073969 valyllysine Proteins 0.000 description 1
- 229940097704 vantas Drugs 0.000 description 1
- 229940099039 velcade Drugs 0.000 description 1
- 229950001544 verdinexor Drugs 0.000 description 1
- 229940094450 vetoryl Drugs 0.000 description 1
- 229960004982 vinblastine sulfate Drugs 0.000 description 1
- KDQAABAKXDWYSZ-PNYVAJAMSA-N vinblastine sulfate Chemical compound OS(O)(=O)=O.C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 KDQAABAKXDWYSZ-PNYVAJAMSA-N 0.000 description 1
- 229960002110 vincristine sulfate Drugs 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960000922 vinflunine Drugs 0.000 description 1
- NMDYYWFGPIMTKO-HBVLKOHWSA-N vinflunine Chemical compound C([C@@](C1=C(C2=CC=CC=C2N1)C1)(C2=C(OC)C=C3N(C)[C@@H]4[C@@]5(C3=C2)CCN2CC=C[C@]([C@@H]52)([C@H]([C@]4(O)C(=O)OC)OC(C)=O)CC)C(=O)OC)[C@H]2C[C@@H](C(C)(F)F)CN1C2 NMDYYWFGPIMTKO-HBVLKOHWSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 229940087652 vioxx Drugs 0.000 description 1
- 229960003842 virginiamycin Drugs 0.000 description 1
- 235000019373 virginiamycin Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 108010069784 vitespin Proteins 0.000 description 1
- 229940063674 voltaren Drugs 0.000 description 1
- 229940069559 votrient Drugs 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- QDLHCMPXEPAAMD-QAIWCSMKSA-N wortmannin Chemical compound C1([C@]2(C)C3=C(C4=O)OC=C3C(=O)O[C@@H]2COC)=C4[C@@H]2CCC(=O)[C@@]2(C)C[C@H]1OC(C)=O QDLHCMPXEPAAMD-QAIWCSMKSA-N 0.000 description 1
- QDLHCMPXEPAAMD-UHFFFAOYSA-N wortmannin Natural products COCC1OC(=O)C2=COC(C3=O)=C2C1(C)C1=C3C2CCC(=O)C2(C)CC1OC(C)=O QDLHCMPXEPAAMD-UHFFFAOYSA-N 0.000 description 1
- 229940085728 xtandi Drugs 0.000 description 1
- ZPUHVPYXSITYDI-HEUWMMRCSA-N xyotax Chemical compound OC(=O)[C@@H](N)CCC(O)=O.O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 ZPUHVPYXSITYDI-HEUWMMRCSA-N 0.000 description 1
- 229940004212 yondelis Drugs 0.000 description 1
- 229950009002 zanolimumab Drugs 0.000 description 1
- 229940099072 zavesca Drugs 0.000 description 1
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 1
- 229960002555 zidovudine Drugs 0.000 description 1
- 229940106067 zinbryta Drugs 0.000 description 1
- 229940033942 zoladex Drugs 0.000 description 1
- XRASPMIURGNCCH-UHFFFAOYSA-N zoledronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CN1C=CN=C1 XRASPMIURGNCCH-UHFFFAOYSA-N 0.000 description 1
- 229940002005 zometa Drugs 0.000 description 1
- 229940052129 zykadia Drugs 0.000 description 1
- 229940051084 zytiga Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6871—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting an enzyme
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68031—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being an auristatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68035—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a pyrrolobenzodiazepine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6811—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a protein or peptide, e.g. transferrin or bleomycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6857—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from lung cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2863—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for growth factors, growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
Abstract
-cMet, en donde el conjugado de fármaco es monometil auristatina E ("MMAE") y el ADC tiene la siguiente estructura: **(Ver fórmula)** en donde Ab es un anticuerpo anti-cMet que comprende una cadena VH que comprende tres CDR, denominadas VH CDR 1 (SEQ ID NO: 112), VH CDR 2 (SEQ ID NO: 113) y VH CDR 3 (SEQ ID NO: 114); una cadena VL que comprende tres CDR, denominadas VL CDR 1 (SEQ ID NO: 115), VL CDR 2 (SEQ ID NO: 116) y VL CDR 3 (SEQ ID NO: 117); y en donde n tiene un valor que varía de 2 a 4.
Description
DESCRIPCIÓN
Conjugados de fármacos con anticuerpos anti-cMet y métodos para su uso
1. Campo
Esta solicitud se refiere, entre otras cosas, a conjugados de fármacos con anticuerpos ("ADC") anti-cMet, composiciones que incluyen los ADC, métodos para fabricar los ADC, métodos para seleccionar poblaciones específicas de pacientes para el tratamiento del cáncer con un ADC anti-cMet y métodos de usar los ADC para tratar el cáncer.
2. Antecedentes
Las proteínas quinasas oncogénicas tal como cMet representan una clase de dianas biológicamente importantes para la intervención contra el cáncer. El cMet, un receptor de tirosina quinasa bien caracterizado codificado por el protooncogén MET, es el receptor de la superficie celular para el factor de crecimiento de hepatocitos (HGF; Gherardi E, Birchmeier W, Birchmeier C y otros. Targeting MET in cancer: rationale and progress. Nat Rev Can.
2012; 12:89-103). La sobreexpresión de cMet ocurre en aproximadamente 30 % - 50 % de los tumores sólidos, que incluyen el cáncer de pulmón de células no pequeñas (NSCLC), el cáncer colorrectal (CRC) y el cáncer gastroesofágico avanzado (AGEC) (Spigel DR, Ervin TJ, Ramlau RA, y otros. Randomized Phase II trial of onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol.
2013;31(32):41054114; Resnick MB, Routhier J, Konkin T y otros. Epidermal growth factor receptor, cMET, B-catenin, and p53 expression as prognostic indicators in stage II colon cancer: a tissue microarray study. Clin Can Res. 2004; 10:3069-3075; Lee HE, Kim MA, Lee HS, y otros. MET in gastric carcinomas: comparison between protein express and gene copy number and impact on outcome. Br J Can. 2012; 107(2):325-333).
La sobreexpresión de cMet se ha asociado con un mal resultado del paciente. Por lo tanto, aún existe la necesidad de terapéuticas del cáncer que se dirijan a los cánceres de tumores sólidos caracterizados por la sobreexpresión de cMet.
3. Resumen
La invención proporciona un conjugado de fármaco con anticuerpo (“ADC”) anti-cMet, en donde el conjugado de fármaco es monometil auristatina E ("MMAE"), y el ADC tiene la siguiente estructura:

en donde Ab es un anticuerpo anti-cMet que comprende una cadena VH que comprende tres CDR, denominadas VH CDR #1 (SEQ ID NO: 112), VH CDR #2 (SEQ ID NO: 113) y VH CDR #3 (SEQ ID NO: 114); una cadena VL que comprende tres CDR, denominadas VL CDR #1 (SEQ ID NO: 115), VL CDR #2 (SEQ ID NO: 116) y VL CDR #3 (SEQ ID NO: 117); y en donde n tiene un valor que varía de 2 a 4.
Las terapias descritas en la presente descripción se dirigen a cánceres de tumores sólidos en los que cMet se sobreexpresa en al menos el 10 % de la población de pacientes que tienen el cáncer. El cMet (factor de transición epitelio-mesenquimal celular) es un receptor de tirosina quinasa de la superficie celular que transduce señales de la matriz extracelular al citoplasma al unirse al ligando del factor de crecimiento de hepatocitos/HGF. Este receptor de la superficie celular se expresa en las células epiteliales de muchos órganos, que incluyen el hígado, el páncreas, la próstata, los riñones, los músculos y la médula ósea, tanto durante la embriogénesis como en la edad adulta. El cMet regula muchos procesos fisiológicos, que incluyen la proliferación y supervivencia celular, la migración y la dispersión (repulsión célula-célula), la morfogénesis de los tejidos, la regeneración de los órganos y la remodelación de los tejidos. En el cáncer y otros procesos patológicos, el cMet a menudo se activa de manera aberrante mediante mutación, amplificación o sobreexpresión de proteínas.
Los cánceres de tumores sólidos en los que cMet se sobreexpresa en al menos 10 % de la población de pacientes incluyen cáncer de pulmón, cáncer colorrectal, cáncer de cabeza y cuello, cáncer de páncreas, cáncer gástrico, glioblastoma, cáncer de ovario, mama, próstata, cuello uterino y esófago. Los datos presentados en la presente descripción demuestran, por primera vez, que los conjugados de fármacos con anticuerpos ("ADC") que se dirigen
específicamente a la sobreexpresión de cMet han demostrado actividad antitumoral en pacientes diagnosticados con cáncer de pulmón de células no pequeñas. Los datos que demuestran la eficacia antitumoral in vivo de los ADC anti -cMet administrados como monoterapia o combinación se proporcionan en los ejemplos 10-14 y 16, y las figuras 8-12 y 14-18.
La sobreexpresión de cMet puede definirse mediante una puntuación H de inmunohistoquímica (IHC) mayor o igual a 150 cuando se mide de acuerdo con el ensayo del ejemplo 17. Brevemente, el protocolo de tinción iHc para la sobreexpresión de cMet se ha desarrollado mediante el uso del estuche Ventana cMet CONFIRM (SP44). Las muestras de tejido se tiñen con el anticuerpo Ventana y después se evalúan mediante determinación de los porcentajes de tinción de células de tejidos diana a diversos niveles de intensidad de bajo a alto. La figura 20 representa puntuaciones H representativas mediante el uso del ensayo descrito en el ejemplo 17.
Alternativamente, el tejido tumoral que sobreexpresa cMet mediante el uso de una puntuación IHC de 0 a 3+ como se describe en el ejemplo 17. Las figuras 19 y 21 representan puntuaciones representativas de IHC mediante el uso del ensayo descrito en el ejemplo 17.
Los ADC anti-cMet pueden administrarse como agentes terapéuticos únicos (monoterapia) o de forma complementaria con, o, a otros tratamientos anticancerígenos y/o agentes terapéuticos, típicamente pero no necesariamente, aquellos usados para tratar el tipo de cáncer del que se trata. De hecho, los datos presentados en la presente descripción demuestran que los tumores que muestran resistencia a otras quimioterapias dirigidas o no dirigidas retienen la sensibilidad a los ADC anti-cMet (véase, por ejemplo, el ejemplo 14 y las figuras 12A-C). Por consiguiente, los ADC anti-cMet descritos en la presente descripción proporcionan beneficios significativos sobre los enfoques actuales dirigidos y no dirigidos hacia el tratamiento de cánceres de tumores sólidos que sobreexpresan cMet. Las terapias complementarias y/o los agentes terapéuticos se usarán, típicamente, a su dosis, vía de administración y frecuencia de administración aprobadas, pero pueden usarse a dosis más bajas y/o con menos frecuencia. Cuando se administra en monoterapia, el ADC anti-cMet típicamente se administrará en un programa que proporciona un beneficio terapéutico. Se contempla que los ADC anti-cMet administrados una vez por semana, una vez cada dos semanas, una vez cada tres semanas, una vez cada cuatro semanas, una vez cada cinco semanas, una vez cada seis semanas, una vez cada siete semanas o una vez cada ocho semanas proporcionarán beneficio terapéutico, aunque la administración más o menos frecuente puede ser beneficiosa. Cuando se administra de forma complementaria a o con otra terapia y/o agente, el ADC anti-cMet puede administrarse antes, después o simultáneamente con la otra terapia o agente.
Los ADC anti-cMet pueden administrarse a través de una variedad de vías o modos de administración, que incluyen, pero no se limitan a, infusión y/o inyección intravenosa e inyección subcutánea. La cantidad administrada dependerá de la vía de administración, el programa de dosificación, el tipo de cáncer que se trata, la etapa del cáncer que se trata y otros parámetros tales como la edad y el peso del paciente, así como es bien conocido en la técnica. Los programas de dosificación ilustrativos específicos que se espera proporcionen un beneficio terapéutico se proporcionan en la Descripción Detallada. Generalmente, se espera que una cantidad de ADC anti-cMet en el intervalo de aproximadamente 0,005 a 15 mg/kg cuando se administra por vía intravenosa semanalmente desde una vez a la semana hasta e incluido, una vez cada ocho semanas, proporcione un beneficio terapéutico.
Por consiguiente, en un aspecto, la presente divulgación proporciona los ADC que se unen específicamente a cMet ("ADC anti-cMet"). Los ADC anti-cMet comprenden agentes citotóxicos y/o citostáticos unidos mediante conectores a una porción de unión al antígeno que se une específicamente a cMet. Como se describe en la presente descripción, la porción de unión al antígeno es un anticuerpo y/o un fragmento de unión al antígeno.
Los anticuerpos y/o fragmentos de unión de los ADC anti-cMet generalmente comprenden una cadena pesada que comprende una región variable (V h) que tiene tres regiones determinantes de complementariedad ("CDR") referidas en la presente descripción (en orden N—>C) como V h CDR#1, V h CDR#2 y V h CDR#3 y una cadena ligera que comprende una región variable (V l) que tiene tres regiones determinantes de complementariedad referidas en la presente descripción (en orden N—>C) como V l CDR#1, V l CDR#2 y V l CDR#3. En la presente descripción se proporcionan las secuencias de aminoácidos de las CDR ilustrativas, así como también, la secuencia de aminoácidos de las regiones Vh y Vl de las cadenas pesadas y ligeras de anticuerpos anti-cMet y/o fragmentos de unión ilustrativos que pueden componer los ADC anti-cMet. Los ADC anti-cMet específicos incluyen, pero no se limitan a, ABT-700 y sTi-0602.
Para usos terapéuticos, puede ser conveniente utilizar ADC anti-cMet que se unan a cMet con una afinidad de al menos 100 nM. Por consiguiente, en algunas modalidades, los ADC anti-cMet comprenden un anti-cMet y/o fragmento de unión anti-cMet que se une a cMet con una afinidad de al menos aproximadamente 100 nM, o incluso mayor, por ejemplo, al menos aproximadamente 90 nM, 80 nM, 70 nM, 60 nM, 50 nM, 40 nM, 30 nM, 25 nM, 20 nM, 15 nM, 10 nM, 7 nM, 6 nM, 5 nM, 4 nM, 3 nM, 2 nM, 1 nM, 0,1 nM, 0,01 nM o mayor. La afinidad de los anticuerpos anti-cMet y/o los fragmentos de unión pueden determinarse mediante el uso de técnicas bien conocidas en la técnica o descritas en la presente descripción, tales como, por ejemplo, ELISA, calorimetría de titulación isotérmica (ITC), resonancia de plasmón superficial, citometría de flujo o ensayo de polarización fluorescente. En algunas modalidades, la afinidad se refiere valores de EC50 de afinidad aparente, medida de acuerdo con el ejemplo 5. En
una modalidad, el anticuerpo tiene un valor de EC50 de afinidad aparente inferior a aproximadamente 10 nanomoles/L, preferentemente de aproximadamente 1 picomol/L a 10 nanomol/L, preferentemente de aproximadamente 0,3 nanomol/L, tal como se determina de acuerdo con el ejemplo 5.
Los anticuerpos pueden estar en forma de anticuerpos de longitud completa, anticuerpos biespecíficos, anticuerpos de dominio variable dual, anticuerpos de cadena múltiple o cadena simple, surrobodies (que incluye constructo de cadena ligera sustituta), anticuerpos de dominio único, anticuerpos camelizados, anticuerpos scFv-Fc y similares. Pueden ser de cualquier isotipo, o derivarse de él, que incluyen, por ejemplo, IgA (por ejemplo, IgA1 o IgA2), IgD, IgE, IgG (por ejemplo, IgG1, IgG2 , IgG3 o IgG4), IgM o IgY. En algunas modalidades, el anticuerpo anti-cMet es una IgG (por ejemplo, IgG1, IgG2 , IgG3 o IgG4). Los anticuerpos pueden ser de origen humano o no humano. Los ejemplos de origen no humano incluyen, pero no se limitan a, origen de mamífero (por ejemplo, simios, roedores, cabras y conejos) o de origen aviar (por ejemplo, pollos). En modalidades específicas, los anticuerpos de los ADC anti-cMet son adecuados para la administración a humanos, tales como, por ejemplo, anticuerpos humanizados y/o anticuerpos completamente humanos.
Los fragmentos de unión al antígeno de los ADC anti-cMet pueden incluir cualquier fragmento de un anticuerpo capaz de unirse específicamente a cMet. Los ejemplos específicos de fragmentos de unión de anticuerpos que pueden incluirse en los ADC anti-cMet incluyen, pero no se limitan a, Fab, Fab’, (Fab’)2 , Fv y scFv.
Los anticuerpos y/o fragmentos de unión de los ADC anti-cMet pueden incluir modificaciones y/o mutaciones que alteren las propiedades de los anticuerpos y/o fragmentos, tales como aquellas que aumentan la vida media, aumentan o disminuyen la ADCC, etc., como se conoce en la técnica.
Los agentes citotóxicos y/o citostáticos de los ADC anti-cMet pueden ser cualquier agente conocido por inhibir el crecimiento y/o la replicación y/o matar células. En la literatura se conocen numerosos agentes que tienen propiedades citotóxicas y/o citostáticas. Los ejemplos no limitantes de clases de agentes citotóxicos y/o citostáticos incluyen, a modo de ejemplo y sin limitación, moduladores del ciclo celular, reguladores de apoptosis, inhibidores de quinasas, inhibidores de síntesis de proteínas, agentes alquilantes, agentes de reticulación de ADN, agentes intercalantes, inhibidores de mitocondrias, inhibidores de exportación nuclear, inhibidores de topoisomerasa I, inhibidores de topoisomerasa II, antimetabolitos de ARN/ADN y agentes antimitóticos.
En un aspecto específico, un agente citotóxico y/o citostático que compone un ADC anti-cMet es un agente antimitótico que permea las células, tal como, por ejemplo, una auristatina. Los ejemplos específicos de auristatinas que penetran en las células incluyen, pero no se limitan a, dolastatina-10 y monometil auristatina E ("MMAE"). En otro aspecto específico, un agente citotóxico y/o citostático del ADC anti-cMet es un agente de reticulación de ADN que penetra en las células, tal como un agente de reticulación de ADN de unión a surcos menores que penetran en las células. Los ejemplos específicos de agentes de unión a surcos menores de ADN que penetran en las células incluyen, pero no se limitan a, pirrolobenzodiazepinas ("PBD") y dímeros de PBD.
Los conectores que unen los agentes citotóxicos y/o citostáticos a la porción de unión al antígeno de un ADC anticMet pueden ser de naturaleza larga, corta, flexible, rígida, hidrófila o hidrófoba, o pueden comprender segmentos que tienen características diferentes, tales como segmentos de flexibilidad, segmentos de rigidez, etc. El conector puede ser químicamente estable en entornos extracelulares, por ejemplo, químicamente estable en el torrente sanguíneo, o puede incluir enlaces que no son estables y liberan los agentes citotóxicos y/o citostáticos en el medio extracelular. En algunas modalidades, los conectores incluyen enlaces que se diseñan para liberar los agentes citotóxicos y/o citostáticos tras la internalización del ADC anti-cMet dentro de la célula. En algunas modalidades específicas, los conectores incluyen enlaces diseñados para escindirse y/o inmolarse o de cualquier otra manera descomponerse específicamente o no específicamente dentro de las células. En la técnica se conoce una amplia variedad de conectores útiles para unir fármacos a porciones de unión al antígeno tales como anticuerpos en el contexto de los ADC. Puede usarse cualquiera de estos conectores, así como también otros conectores, para unir los agentes citotóxicos y/o citostáticos a la porción de unión al antígeno de los ADC anti-cMet descritos en la presente descripción.
El número de agentes citotóxicos y/o citostáticos unidos a la porción de unión al antígeno de un ADC anti-cMet puede variar (denominado "relación fármaco-a-anticuerpo" o "DAR"), y estará limitado solo por el número de sitios de unión disponibles en la porción de unión al antígeno y el número de agentes unidos a un único conector. Típicamente, un conector unirá un único agente citotóxico y/o citostático a la porción de unión al antígeno de un ADC anti-cMet. Cuando un ADC anti-cMet incluye más de un agente citotóxico y/o citostático, cada agente puede ser igual o diferente. Mientras el ADC anti-cMet no exhiba niveles inaceptables de agregación en las condiciones de uso y/o almacenamiento, se contemplan los ADC anti-cMet con DAR de veinte, o incluso superior. En algunas modalidades, los ADC anti-cMet descritos en la presente descripción pueden tener un DAR en el intervalo de aproximadamente 1-10, 1-8, 1-6 o 1-4. En ciertas modalidades específicas, los ADC anti-cMet pueden tener un DAR de 2, 3 o 4. En otras modalidades específicas, los ADC anti-cMet pueden tener un DAR promedio de 3.1.
4. Breve descripción de las figuras
El archivo de patente o solicitud contiene al menos un dibujo realizado a color. La Oficina proporcionará copias de esta patente o publicación de solicitud de patente con dibujo(s) a color, previa solicitud y pago de la tarifa necesaria. Las figuras 1 A-E muestran las secuencias de aminoácidos de varios anticuerpos a cMet.
Las figuras 2 A-B: ilustran el Proceso 1 de ABBV-399.
Las figuras 3 A-B ilustran el Proceso 2 de ABBV-399.
Las figuras 4 A-B representan la citotoxicidad de ABBV-399 en líneas celulares que expresan cMet.
La figura 5 proporciona resultados de inhibición de la proliferación con ABBV-399 y ABT-700 PBD.
Las figuras 6 A-B muestran la actividad in vitro de ABT-700 PBD en líneas celulares de cáncer colorrectal.
La figura 7 muestra la actividad in vitro de ABT-700 PBD en líneas celulares de cáncer de cerebro.
La figura 8 muestra la actividad ABT-700 PBD en xenoinjertos SW48.
Las figuras 9 A-C muestran la actividad de ABT-700 PBD y ABBV-399 en xenoinjertos de pacientes con NSCLC. Las figuras 10 A-B muestran la actividad de ABBV-399 en xenoinjertos de pacientes con NSCLC mediante el uso de diagramas de Kaplan-Meier.
Las figuras 11 A-B comparan la actividad de ABT-700 frente a ABBV-399 en xenoinjertos de tumor humano; la figura 11 C muestra la actividad de ABBV-339 solo o en combinación con FOLFIRI.
Las figuras 12 A-C representan la actividad de ABBV-399 en modelos de xenoinjerto humano refractarios a ABT-700.
La figura 13 proporciona el esquema de escalado de la dosis de ABBV-399 para el ensayo de fase I de monoterapia. La figura 14 proporciona un diagrama en cascada que muestra el mejor porcentaje de cambio en las lesiones diana. La figura 15 proporciona un diagrama en cascada que muestra el mejor porcentaje de cambio en las lesiones diana/niveles de cMet con la monoterapia con ABBV-399.
La figura 16 muestra el número de semanas antes de la progresión clínica en 16 pacientes tratados con ABBV-399. La figura 17 es un diagrama en cascada que muestra el mejor porcentaje de cambio en las lesiones diana de ABBV-399 en combinación con erlotinib.
La figura 18 muestra el número de semanas antes de la progresión clínica en 6 pacientes tratados con ABBV-399 y erlotinib.
La figura 19 ilustra la guía de puntuación SP44 de Ventana.
La figura 20 ilustra la selección de pacientes en base a la sobreexpresión de cMet.
La figura 21 proporciona calificaciones ilustrativas de IHC mediante el uso del método del ejemplo 17.
5. Descripción detallada
5.1 Abreviaturas
Los anticuerpos, fragmentos de unión, los ADC y polinucleótidos descritos en la presente descripción se describen mediante sus respectivas secuencias de polipéptidos o polinucleótidos. A menos que se indique de cualquier otra manera, las secuencias de polipéptidos se proporcionan en orientación N—>C; las secuencias de polinucleótidos en orientación 5’—>3’. Para secuencias de polipéptidos, pueden usarse las abreviaturas convencionales de tres o una letra para los aminoácidos codificados genéticamente, como se indica en la TABLA 1, más abajo.

Ciertas secuencias se definen mediante fórmulas estructurales que especifican residuos de aminoácidos que pertenecen a ciertas clases (por ejemplo, alifáticos, hidrófobos, etc.). Las diversas clases a las que pertenecen los aminoácidos codificados genéticamente como se usan en la presente descripción se indican en la TABLA 2, más abajo. Algunos aminoácidos pueden pertenecer a más de una clase. La cisteína, que contiene un grupo sulfhidrilo, y la prolina, que está restringida conformacionalmente, no son clases asignadas.

5.2 Definiciones
A menos que se defina de cualquier otra manera en la presente descripción, los términos científicos y técnicos usados en relación con la presente divulgación tendrán los significados que comúnmente entienden los expertos en la técnica.
5.3 Conjugados de fármacos con anticuerpos que se unen a cMet y ensayo de sobreexpresión de cMet La presente divulgación se refiere a conjugados de fármacos con anticuerpos que se unen específicamente a cMet humano, composiciones que comprenden los ADC, anticuerpos anti-cMet y/o fragmentos de unión que pueden comprender los ADC, polinucleótidos que codifican anticuerpos anti-cMet y/o fragmentos de unión que comprenden los ADC, células huésped capaces de producir los anticuerpos y/o fragmentos de unión, métodos y composiciones útiles para fabricar los anticuerpos, fragmentos de unión y los ADC, y diversos métodos para usar los ADC en el tratamiento del cáncer.
Los datos proporcionados en la presente descripción demuestran, por primera vez, que los conjugados de fármacos con anticuerpos ("ADC") dirigidos específicamente a cMet exhiben potentes efectos antitumorales, tanto solos como en combinación con otras terapias antitumorales dirigidas y no dirigidas, contra tumores sólidos en los que se sobreexpresa cMet, particularmente aquellos con una puntuación IHC de 2+ y 3+ cuando se mide por inmunohistoquímica con el anticuerpo SP44. En los ejemplos se proporcionan datos que demuestran la eficacia antitumoral in vivo de ABBV-399 administrado como monoterapia.
Para los fines de esta solicitud, que incluye las reivindicaciones, el ensayo particular usado en el estudio En la presente descripción se describe se denomina "protocolo de tinción cMet a BbV-ADC." Este protocolo se describe en detalle en el ejemplo 17 y los resultados se expresan en términos de puntuación H y pueden expresarse además en términos de puntuación IHC u otro sistema de puntuación bien conocido en la técnica.
El enfoque de la puntuación H proporciona una resolución óptima de los datos para determinar la variación en la intensidad y el porcentaje tumoral de tinción dentro y entre los tipos de tumor. Se proporciona además una buena herramienta para determinar los umbrales para la tinción positiva. En este método, se proporciona el porcentaje de células (0-100) dentro de un tumor con intensidades de tinción que varían de 0-3+. Este protocolo da como resultado la tinción de la proteína cMet tanto en el citoplasma como en la superficie/membrana celular. Se determina la intensidad de tinción para cada célula en un campo fijo (típicamente, 100 células) de la biopsia tumoral procesada, y se atribuye un valor individual a cada célula de la siguiente manera, en dependencia de la tinción de la superficie/membrana celular:
0 = sin tinción
1+ = tinción débil
2+ = tinción moderada
3+ = tinción fuerte
Para obtener una puntuación H, el porcentaje de células tumorales se multiplica por cada intensidad y se suman. La puntuación H máxima es 300 si el 100 % de las células tumorales se marcan con una intensidad de 3+. La puntuación H se calcula de la siguiente manera:
puntuación H = [1 x (% de células 1+) 2 x (% de células 2+) 3 x (% de células 3+)] Este protocolo da como resultado tanto la tinción de cMet citoplasmática como de membrana. Para los cálculos de la puntuación H mencionados en la presente descripción, se usó la tinción de membrana. La puntuación de la puntuación H final del tumor (0-300) da más peso relativo a la tinción de membrana de mayor intensidad (células 3+ > células 2+ > células 1+). La figura 20 muestra resultados de tinción ilustrativos para diversas puntuaciones H de tumor (15, 90, 180 y 290) obtenidas con el "protocolo de tinción cMet ABBV-ADC."
Cada tumor puede recibir además una puntuación IHC de IHC 0, IHC 1+, IHC 2+ o IHC 3+. Si bien las puntuaciones IHC y H implican valores 0, 1+, 2+ y 3+, no deben confundirse. Para la puntuación H, los valores 0, 1+, 2+ y 3+ se refieren a la intensidad de la tinción de una célula individual. Para la puntuación IHC, los valores 0, 1+, 2+ y 3+ se refieren a la tinción general de un área particular de la muestra tumoral. La figura 21 muestra resultados de tinción ilustrativos para diversas puntuaciones IHC0/1+/2+/3+ de tumor obtenidas con el "protocolo de tinción cMet ABBV-ADC."
Para los propósitos de esta divulgación, y al seguir el protocolo descrito en la presente descripción, si ninguna de las células en un campo fijo se tiñe, el valor atribuido al tumor es IHC 0. Si el nivel general de tinción en un campo fijo es bajo, el valor atribuido es IHC 1+. Si la mayoría de las células en un campo fijo exhiben una tinción moderada, el valor atribuido es IHC 2+. Si la mayoría de las células en un campo fijo exhiben una fuerte tinción, el valor atribuido es IHC 3+.
Para los propósitos de esta divulgación, y al seguir el protocolo descrito en la presente descripción, si ninguna de las células en un campo fijo se tiñe, el valor atribuido al tumor es IHC 0. Si el nivel general de tinción en un campo fijo es bajo, el valor atribuido es IHC 1+. Si al menos el 15 % de las células en un campo fijo exhiben una tinción moderada, el valor atribuido es IHC 2+. Si al menos 15 % de las células en un campo fijo exhiben una tinción fuerte, el valor atribuido es IHC 3+.
Para los propósitos de esta divulgación, una puntuación H entre 150 y 224 es equivalente a una puntuación IHC de 2+ y una puntuación H de 225 y superior es equivalente a una puntuación IHC de 3+.
Por consiguiente, en un aspecto, la presente divulgación proporciona los ADC que se unen específicamente a cMet ("ADC anti-cMet"). Los ADC anti-cMet comprenden agentes citotóxicos y/o citostáticos unidos mediante conectores a una porción de unión al antígeno que se une específicamente a cMet. En el caso de ABBV-399, la porción de unión
al antígeno (ABT-700) se une a cMet en el dominio 1 de IPT de cMet humana. En otros ADC anti-cMet, la porción de unión al antígeno puede ser cualquier porción capaz de unirse específicamente a cMet. En algunos aspectos, la porción de unión al antígeno es un anticuerpo y/o un fragmento de unión al anticuerpo.
En un aspecto específico, un agente citotóxico y/o citostático de un ADC anti-cMet es un agente antimitótico que penetra en las células, tal como, por ejemplo, una auristatina. Los ejemplos específicos de auristatinas que penetran en las células incluyen, pero no se limitan a, dolastatina-10 y monometil auristatina E ("MMAE"). En otro aspecto, un agente citotóxico y/o citostático del ADC anti-cMet es un agente de reticulación de ADN que penetra en las células, tal como un agente de reticulación de ADN de unión a surcos menores que penetran en las células. Los ejemplos específicos de agentes de unión a surcos menores de ADN que penetran en las células incluyen, pero no se limitan a, pirrolobenzodiazepinas ("PBD") y dímeros de PBD.
Como apreciarán los expertos en la técnica, los anticuerpos y/o fragmentos de unión son de naturaleza "modular". A lo largo de la divulgación, se describen "módulos" de los anticuerpos y/o fragmentos de unión. Se describen las CDR de Vh, las cadenas Vh, las CDR de Vl y las cadenas Vl, ilustrativas. Se pretende que todos los aspectos específicos puedan combinarse entre sí como si cada combinación específica se describiera explícitamente de forma individual. Los ADC divulgados en la presente descripción además son de naturaleza "modular". A lo largo de la divulgación, se describen los "módulos" de los ADC. Como ejemplos no limitantes, se describen modalidades específicas de anticuerpos, conectores y agentes citotóxicos y/o citostáticos que pueden componer los ADC. Se pretende que todos los aspectos específicos descritos puedan combinarse entre sí como si cada combinación específica se describiera explícitamente de forma individual.
Los expertos en la técnica apreciarán además que los diversos ADC descritos en la presente descripción pueden estar en forma de sales, tales como sales farmacéuticamente aceptables. Los ADC de la divulgación que poseen grupos funcionales suficientemente ácidos, suficientemente básicos o ambos, pueden reaccionar con cualquiera de una serie de bases inorgánicas y ácidos inorgánicos y orgánicos, para formar una sal. Alternativamente, los compuestos que están inherentemente cargados, tal como aquellos que tienen un nitrógeno cuaternario, pueden formar una sal con un contraión apropiado, por ejemplo, un haluro tal como un bromuro, cloruro o fluoruro.
Los ácidos comúnmente empleados para formar sales de adición ácida son ácidos inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido yodhídrico, ácido sulfúrico, ácido fosfórico y similares, y ácidos orgánicos tales como ácido p-toluenosulfónico, ácido metanosulfónico, ácido oxálico, ácido p-bromofenilsulfónico, ácido carbónico, ácido succínico, ácido cítrico, etc. Las sales de adición de bases incluyen aquellas derivadas de bases inorgánicas, tales como amonio e hidróxidos de metales alcalinos o alcalinotérreos, carbonatos, bicarbonatos y similares.
5.4 Anticuerpos contra cMet
En la presente descripción se describe, que la porción de unión al antígeno es un anticuerpo o un fragmento de unión al antígeno.
Como se usa en la presente descripción, el término "anticuerpo" (Ab) se refiere a una molécula de inmunoglobulina que se une específicamente a, o es inmunológicamente reactiva con, un antígeno particular, aquí, cMet. Los anticuerpos comprenden regiones determinantes de complementariedad (CDR), también conocidas como regiones hipervariables, en los dominios variables tanto de la cadena ligera como de la cadena pesada. Las porciones más altamente conservadas de los dominios variables se denominan el marco (FR). Como se sabe en la técnica, la posición/límite de aminoácidos que delimita una región hipervariable de un anticuerpo puede variar, en dependencia del contexto y las diversas definiciones conocidas en la técnica. Algunas posiciones dentro de un dominio variable pueden verse como posiciones híbridas hipervariables en que estas posiciones pueden considerarse estar dentro de una región hipervariable bajo un conjunto de criterios, mientras que se consideran fuera de una región hipervariable bajo un conjunto diferente de criterios. Una o más de estas posiciones pueden encontrarse además en regiones hipervariables extendidas. Los dominios variables de las cadenas pesadas y ligeras nativas comprenden cada uno cuatro regiones FR, en gran parte mediante la adopción de una configuración de lámina (3, conectada por tres CDR, que forman lazos que se conectan y, en algunos casos, forman parte de la estructura de lámina (3. Las CDR en cada cadena se mantienen juntas en estrecha proximidad por las regiones FR y, con las CDR de la otra cadena, contribuyen a la formación del sitio de unión al antígeno de los anticuerpos. Véase, Kabat y otros, Sequences of Proteins of Immunological Interest (National Institute of Health, Bethesda, Md. 1987). Como se usa en la presente descripción, la numeración de residuos de aminoácidos de inmunoglobulinas se realiza de acuerdo con el sistema de numeración de residuos de aminoácidos de inmunoglobulinas de Kabat y otros a menos que se indique de cualquier otra manera.
Los anticuerpos y/o fragmentos de unión de los ADC anti-cMet generalmente comprenden una cadena pesada que comprende una región variable (Vh) que tiene tres regiones determinantes de complementariedad ("CDR") referidas en la presente descripción (en orden N—>C) como Vh CDR#1, Vh CDR#2 y Vh CDR#3 y una cadena ligera que comprende una región variable (Vl) que tiene tres regiones determinantes de complementariedad referidas en la presente descripción (en orden N—>C) como Vl CDR#1, Vl CDR#2 y Vl CDR#3. En la presente descripción, se
divulgan las secuencias de aminoácidos ilustrativas de las CDR, así como también, la secuencia de aminoácidos de las regiones Vh y Vl de las cadenas pesadas y ligeras de anticuerpos y/o fragmentos de unión anti-cMet ilustrativos que pueden incluirse en las porciones de unión al antígeno de los ADC anti-cMet. Los ADC anti-cMet ilustrativos incluyen, pero no se limitan a, aquellos que comprenden anticuerpos y/o fragmentos de unión que incluyen estas CDR y/o secuencias Vh y/o Vl ilustrativas, así como también, anticuerpos y/o fragmentos de unión que compiten por la unión de cMet con tales anticuerpos y/o fragmentos de unión.
Los anticuerpos pueden estar en forma de anticuerpos de longitud completa, anticuerpos biespecíficos, anticuerpos de dominio variable dual, anticuerpos de cadena múltiple o cadena simple, surrobodies (que incluye constructo de cadena ligera sustituta), anticuerpos de dominio único, anticuerpos camelizados, anticuerpos scFv-Fc y similares. Pueden ser de cualquier isotipo, o derivarse de él, que incluyen, por ejemplo, IgA (por ejemplo, IgA1 o IgA2), IgD, IgE, IgG (por ejemplo, IgG1, IgG2 , IgG3 o IgG4), IgM o IgY. En algunas modalidades, el anticuerpo anti-cMet es una IgG (por ejemplo, IgG1, IgG2 , IgG3 o IgG4). Los anticuerpos pueden ser de origen humano o no humano. Los ejemplos de origen no humano incluyen, pero no se limitan a, origen de mamífero (por ejemplo, simios, roedores, cabras y conejos) o de origen aviar (por ejemplo, pollos). En modalidades específicas, los anticuerpos de los ADC anti-cMet son adecuados para la administración a humanos, tales como, por ejemplo, anticuerpos humanizados y/o anticuerpos completamente humanos.
Los anticuerpos de los ADC anti-cMet pueden ser por naturaleza policlonales, monoclonales, genéticamente modificados y/o modificados de cualquier otra manera, que incluyen, pero no se limitan a, anticuerpos quiméricos, anticuerpos humanizados, anticuerpos humanos, anticuerpos primatizados, anticuerpos monocatenarios, anticuerpos biespecíficos, anticuerpos de dominio doble variable, etc. En diversas modalidades, los anticuerpos comprenden todo o una porción de una región constante de un anticuerpo. En algunas modalidades, la región constante es un isotipo seleccionado de: IgA (por ejemplo, IgA1 o IgA2), IgD, IgE, IgG (por ejemplo, IgG1, IgG2 , IgG3 o IgG4), IgM e IgY. En modalidades específicas, los anticuerpos del ADC anti-c-Met comprenden un isotipo de región constante de IgG1.
El término "anticuerpo monoclonal" como se usa en la presente descripción no se limita a anticuerpos producidos mediante la tecnología de hibridomas. Un anticuerpo monoclonal se deriva de un solo clon, que incluye cualquier clon de eucariota, de procariota o de fago, por cualquier medio disponible o conocido en la técnica. Los anticuerpos monoclonales útiles con la presente divulgación pueden prepararse mediante el uso de una amplia variedad de técnicas conocidas en la técnica, que incluyen el uso de tecnologías de hibridomas, recombinantes y de presentación en fagos, o una combinación de las mismas. En muchos usos de la presente divulgación, que incluyen el uso in vivo de los ADC que incluyen anticuerpos anti-cMet en humanos, pueden usarse adecuadamente anticuerpos quiméricos, primatizados, humanizados o humanos.
El término anticuerpo "quimérico", como se usa en la presente descripción se refiere a un anticuerpo que tiene secuencias variables derivadas de una inmunoglobulina no humana, tal como un anticuerpo de rata o de ratón, y regiones constantes de inmunoglobulina humana, típicamente elegidas de un molde de inmunoglobulina humana. Los métodos para producir anticuerpos quiméricos se conocen en la técnica. Véase, por ejemplo, Morrison, 1985, Science 229(4719):1202-7; Oi y otros, 1986, BioTechniques 4:214-221; Gillies y otros, 1985, J. Immunol. Methods 125:191-202; las patentes de Estados Unidos núms. 5, 807,715; 4, 816,567; y 4, 816,397.
Las formas "humanizadas" de anticuerpos no humanos (por ejemplo, murinos) son inmunoglobulinas quiméricas que contienen secuencias mínimas derivadas de inmunoglobulina no humana. En general, un anticuerpo humanizado comprenderá sustancialmente todos, o al menos uno, y típicamente dos dominios variables, en los que todas o sustancialmente todas las regiones CDR corresponden a aquellas de una inmunoglobulina no humana y todas o sustancialmente todas las regiones FR son aquellas de una secuencia de inmunoglobulina humana. El anticuerpo humanizado puede comprender además al menos una porción de una región constante de inmunoglobulina (Fc), típicamente la de una secuencia consenso de inmunoglobulina humana. Los métodos de humanización de anticuerpos se conocen en la técnica. Véase, por ejemplo, Riechmann y otros, 1988, Nature 332: 323-7; Patentes de Estados Unidos núm. 5,530,101; 5,585,089; 5,693,761; 5,693,762; y 6,180,370 de Queen y otros; EP239400; Publicación de PCT WO 91/09967; patente de Estados Unidos núm. 5,225,539; EP592106; EP519596; Padlan, 1991, Mol. Immunol., 28: 489-498; Studnicka y otros, 1994, Prot. Eng. 7:805-814; Roguska y otros, 1994, Proc. Natl. Acad. Sci. 91: 969-973; y patente de Estados Unidos núm. 5, 565,332.
Los "anticuerpos humanos" son anticuerpos que tienen la secuencia de aminoácidos de una inmunoglobulina humana e incluyen anticuerpos aislados de bibliotecas de inmunoglobulinas humanas o de animales transgénicos para una o más inmunoglobulinas humanas y que no expresan inmunoglobulinas endógenas. Los anticuerpos humanos pueden prepararse mediante una variedad de métodos conocidos en la técnica, que incluyen los métodos de presentación en fagos que usan bibliotecas de anticuerpos derivadas de secuencias de inmunoglobulinas humanas. Véase las patentes de Estados Unidos núms. 4, 444,887 y 4, 716,111; y publicaciones PCT WO 98/46645; WO 98/50433; WO 98/24893; WO 98/16654; WO 96/34096; WO 96/33735; y WO 91/10741. Los anticuerpos humanos pueden producirse además mediante el uso de ratones transgénicos que son incapaces de expresar inmunoglobulinas endógenas funcionales pero que pueden expresar genes de inmunoglobulinas humanas. Véase, por ejemplo, las publicaciones de PCT WO 98/24893; WO 92/01047; WO 96/34096; WO 96/33735; las
patentes de Estados Unidos núms. 5,413,923; 5,625,126; 5,633,425; 5,569,825; 5,661,016; 5,545,806; 5,814,318; 5,885,793; 5,916,771; y 5,939,598. Además, empresas tales como Medarex (Princeton, NJ), Astellas Pharma (Deerfield, IL), Amgen (Thousand Oaks, CA) y Regeneron (Tarrytown, NY) pueden participar para proporcionar anticuerpos humanos dirigidos contra un antígeno seleccionado mediante el uso de similar tecnología a la descrita anteriormente. Pueden generarse anticuerpos completamente humanos que reconocen un epítopo seleccionado mediante el uso de una técnica denominada "selección guiada." En este enfoque, se usa un anticuerpo monoclonal no humano seleccionado, por ejemplo, un anticuerpo de ratón, para guiar la selección de un anticuerpo completamente humano que reconozca el mismo epítopo (véase, Jespers y otros, 1988, Biotechnology 12: 899-903). Los "anticuerpos primatizados" comprenden regiones variables de mono y regiones constantes humanas. Los métodos para producir anticuerpos primatizados se conocen en la técnica. Véase, por ejemplo, las patentes de Estados Unidos núms. 5, 658,570; 5, 681,722 y 5, 693,780.
Los ADC anti-cMet pueden comprender moléculas de anticuerpos de longitud completa (intactas), así como también, fragmentos de unión al antígeno que son capaces de unirse específicamente a cMet. Los ejemplos de fragmentos de unión de anticuerpos incluyen, a modo de ejemplo y sin limitación, Fab, Fab’, F (ab’)2, fragmentos Fv, fragmentos Fv de cadena simple y fragmentos de dominio único.
Un fragmento Fab contiene el dominio constante de la cadena ligera y el primer dominio constante (CH2) de la cadena pesada. Los fragmentos Fab’ se diferencian de los fragmentos Fab por la adición de unos pocos residuos en el extremo carboxilo del dominio CH2 de la cadena pesada que incluyen una o más cisternas de la región bisagra del anticuerpo. Los fragmentos F (ab’j se producen por escisión del enlace disulfuro en las cisternas bisagra del producto de digestión de pepsina F (ab’)2. Los expertos en la técnica conocen los acoplamientos químicos adicionales de fragmentos de anticuerpos. Los fragmentos Fab y F (ab’ )2 carecen del fragmento Fe del anticuerpo intacto, se eliminan más rápidamente de la circulación de los animales y pueden tener menos unión inespecífica de tejido que un anticuerpo intacto (véase, por ejemplo, Wahl y otros, 1983, J. Nucl. Med. 24:316).
Un fragmento "Fv" es el fragmento mínimo de un anticuerpo que contiene un sitio de reconocimiento y unión a la diana completo. Esta región consiste en un dímero de un dominio variable de una cadena pesada y una ligera en una asociación estrecha y no covalente (dímero Vh-Vl). Es en esta configuración que las tres CDR de cada dominio variable interactúan para definir un sitio de unión al antígeno en la superficie del dímero Vh-Vl. A menudo, las seis CDR confieren especificidad de unión al antígeno sobre el anticuerpo. Sin embargo, en algunos casos, incluso un dominio variable único (o la mitad de un Fv que comprende solo tres CDR específicas para una diana) puede tener la capacidad de reconocer y unirse al antígeno, aunque con una afinidad menor que el sitio de unión completo. Los fragmentos de unión de anticuerpos "Fv monocatenario" o "scFv" comprenden los dominios Vh y Vl de un anticuerpo, donde estos dominios están presentes en una sola cadena polipeptídica. Generalmente, el polipéptido Fv comprende además un conector polipeptídico entre los dominios Vh y Vl que permite a scFv formar la estructura deseada para la unión al antígeno.
Los anticuerpos y/o fragmentos de unión de los ADC anti-cMet pueden incluir modificaciones y/o mutaciones que alteran las propiedades de los anticuerpos y/o fragmentos, tales como aquellos que aumentan la vida media, aumentan o disminuyen la ADCC, etc., como se conoce en la técnica.
Los "anticuerpos de dominio único" se componen de dominios únicos Vh o Vl que exhiben suficiente afinidad por cMet. En un aspecto específico, el anticuerpo de dominio único es un anticuerpo camelizado (véase, por ejemplo, Riechmann, 1999, Journal of Immunological Methods 231:25-38).
Los anticuerpos de los ADC anti-cMet además pueden ser anticuerpos biespecíficos. Anticuerpos biespecíficos compuestos de anticuerpos monoclonales, a menudo humanos o humanizados, que tienen especificidades de unión para dos epítopos diferentes en el mismo o diferentes antígenos. En la presente divulgación, una de las especificidades de unión puede dirigirse hacia cMet, la otra puede ser para cualquier otro antígeno, por ejemplo, para una proteína de la superficie celular, receptor, subunidad del receptor, antígeno específico de tejido, proteína derivada de virus, proteína de la envoltura codificada por virus, proteína derivada de bacterias o proteína de superficie bacteriana, etc.
Los anticuerpos de los ADC anti-cMet pueden derivatizarse. Los anticuerpos derivatizados se modifican típicamente mediante glicosilación, acetilación, pegilación, fosforilación, amidación, derivatización por grupos protectores/bloqueadores conocidos, escisión proteolítica, enlace a un ligando celular u otra proteína. Cualquiera de las numerosas modificaciones químicas puede llevarse a cabo mediante técnicas conocidas, que incluyen, pero no se limitan a, escisión química específica, acetilación, formilación, síntesis metabólica de tunicamicina, etc. Adicionalmente, el derivado puede contener uno o más aminoácidos no naturales, por ejemplo, mediante el uso de la tecnología ambrx. Véase, por ejemplo, Wolfson, 2006, Chem. Biol. 13(10):1011 -2.
Los anticuerpos o fragmentos de unión de los ADC anti-cMet pueden ser anticuerpos o fragmentos cuyas secuencias se han modificado para alterar al menos una función efectora biológica mediada por la región constante.
Por ejemplo, en algunas modalidades, un anticuerpo anti-cMet puede modificarse para reducir al menos una función efectora biológica mediada por la región constante con respecto al anticuerpo no modificado, por ejemplo, unión reducida al receptor Fe (FcyR). La unión de FcyR puede reducirse al mutar el segmento de región constante de inmunoglobulina del anticuerpo en regiones particulares necesarias para las interacciones FcyR (véase, por ejemplo, Canfield y Morrison, 1991, J. Exp. Med. 173: 1483-1491; y Lund y otros, 1991, J. Immunol. 147:2657-2662). Al reducir la unión de FcyR también puede reducirse otras funciones efectoras que dependen de las interacciones de FcyR, tales como opsonización, fagocitosis y citotoxicidad celular dependiente de antígeno ("ADCC").
Los anticuerpos incluidos en los ADC anti-cMet pueden tener niveles bajos, o carecer, de fucosa. Los anticuerpos que carecen de fucosa se han correlacionado con una mayor actividad de ADCC, especialmente a bajas dosis de anticuerpo. Véase Shields y otros, 2002, J. Biol. Chem 277:26733-26740; Shinkawa y otros, 2003, J. Biol. Chem 278:3466-73. Los métodos para preparar anticuerpos con menos fucosa incluyen el crecimiento en células YB2/0 de mieloma de rata (ATCC CRL 1662). Las células YB2/0 expresan bajos niveles de ARNm de FUT8, que codifica la a-1,6-fucosiltransferasa, una enzima necesaria para la fucosilación de polipéptidos.
Los anticuerpos o fragmentos de unión de los ADC anti-cMet pueden incluir modificaciones que aumentan o disminuyen sus afinidades de unión al receptor Fc neonatal, FcRn, por ejemplo, al mutar el segmento de la región constante de inmunoglobulina en regiones particulares involucradas en interacciones de FcRn (véase, por ejemplo, el documento WO 2005/123780). En modalidades particulares, un anticuerpo anti-cMet de la clase IgG está mutado de tal manera que al menos uno de los residuos de aminoácidos 250, 314 y 428 de la región constante de la cadena pesada se sustituye solo, o en cualquier combinación de los mismos, tal como en las posiciones 250 y 428, o en las posiciones 250 y 314, o en las posiciones 314 y 428, o en las posiciones 250, 314 y 428, con la sustitución en las posiciones 250 y 428 que es una combinación específica. Para la posición 250, el residuo de aminoácido sustituyente puede ser cualquier residuo de aminoácido distinto de treonina, que incluye, pero no se limitan a, alanina, cisteína, ácido aspártico, ácido glutámico, fenilalanina, glicina, histidina, isoleucina, lisina, leucina, metionina, asparagina, prolina, glutamina, arginina, serina, valina, triptófano o tirosina. Para la posición 314, el residuo de aminoácido sustituyente puede ser cualquier residuo de aminoácido distinto de leucina, que incluye, pero no se limitan a, alanina, cisteína, ácido aspártico, ácido glutámico, fenilalanina, glicina, histidina, isoleucina, lisina, metionina, asparagina, prolina, glutamina, arginina, serina, treonina, valina, triptófano o tirosina. Para la posición 428, los residuos de aminoácidos sustituyentes pueden ser cualquier residuo de aminoácido distinto de metionina, que incluye, pero no se limitan a, alanina, cisteína, ácido aspártico, ácido glutámico, fenilalanina, glicina, histidina, isoleucina, lisina, leucina, asparagina, prolina, glutamina, arginina, serina, treonina, valina, triptófano o tirosina. Las combinaciones específicas de sustituciones de aminoácidos adecuadas se identifican en la TABLA 1 de la patente de Estados Unidos núm. 7, 217,797. Tales mutaciones aumentan la unión a FcRn, lo cual protege al anticuerpo de la degradación y aumenta su vida media.
Un anticuerpo y/o fragmento de unión anti-c-Met puede tener uno o más aminoácidos insertados en una o más de sus regiones hipervariables, por ejemplo, como se describe en Jung y Pluckthun, 1997, Protein Engineering 10:9, 959-966; Yazaki y otros, 2004, Protein Eng. Des Sel. 17(5):481-9; y la solicitud de patente de Estados Unidos núm.
2007/0280931.
Los anticuerpos anti-cMet y/o fragmentos de unión con alta afinidad por cMet pueden ser convenientes para usos terapéuticos. Por consiguiente, la presente divulgación contempla los ADC que comprenden anticuerpos anti-cMet y/o fragmentos de unión que tienen una alta afinidad de unión a cMet. En modalidades específicas, los anticuerpos y/o fragmentos de unión se unen a cMet con una afinidad de al menos aproximadamente 100 nM, pero pueden exhibir mayor afinidad, por ejemplo, al menos aproximadamente 90 nM, 80 nM, 70 nM, 60 nM, 50 nM, 40 nM, 30 nM, 25 nM, 20 nM, 15 nM, 10 nM, 7 nM, 6 nM, 5 nM, 4 nM, 3 nM, 2 nM, 1 nM, 0,1 nM, 0,01 nM, o incluso más. En algunas modalidades, los anticuerpos se unen a cMet con una afinidad en el intervalo de aproximadamente 1 pM a aproximadamente 100 nM, o una afinidad que varía entre cualquiera de los valores anteriores.
La afinidad de anticuerpos y/o fragmentos de unión para cMet puede determinarse mediante el uso de técnicas bien conocidas en la técnica o descritas en la presente descripción, como, por ejemplo, pero no a modo de limitación, ELISA, calorimetría de titulación isotérmica (ITC), resonancia de plasmón superficial, citometría de flujo o ensayos de polarización fluorescente. En algunas modalidades, la afinidad se refiere valores de EC50 de afinidad aparente medida de acuerdo con el ejemplo 5.
En el contexto de esta divulgación, los anticuerpos anti-cMet pueden servir al menos para dos propósitos diferentes. Los anticuerpos anti-cMet se usan con fines de diagnóstico, al ayudar y guiar la selección de pacientes. Por ejemplo, estos anticuerpos anti-cMet pueden usarse para ensayos de inmunohistoquímica de biopsias tumorales obtenidas de los pacientes a tratar o bajo tratamiento. Un experto en la técnica está familiarizado con las técnicas para seleccionar un anticuerpo particular con fines de diagnóstico para analizar los niveles de expresión de la proteína cMet en biopsias tumorales. Típicamente, las muestras se califican bajo una o más guías de puntuación, lo que incluye las puntuaciones IHC de 0/1+/2+/3+ o las puntuaciones H. La divulgación detalla un ejemplo de dicho ensayo de diagnóstico que está disponible comercialmente de Ventana. El anticuerpo Ventana SP44 y los anticuerpos con propiedades similares pueden fabricarse o adquirirse de otros proveedores y el protocolo puede ajustarse para que el método tenga el mismo o mejor poder diagnóstico que el ensayo Ventana. Además, los anticuerpos anti-cMet
distintos de SP44 pueden usarse además para este propósito. Un experto en la técnica sabría cómo ajustar adecuadamente el protocolo a un nuevo anticuerpo para obtener una prueba de diagnóstico para los niveles de expresión de cMet. Existen diagnósticos complementarios para una variedad de otros tratamientos contra el cáncer aprobados por la FDA y están dentro del nivel de habilidad ordinaria. La FDA mantiene una lista de pruebas de diagnóstico complementarias aprobadas por la FDA en, por ejemplo, www.fda.gov/.
Los ejemplos de anticuerpos anti-cMet que pueden usarse incluyen, por ejemplo, los anticuerpos de diagnóstico divulgados en la patente de Estados Unidos núm. 8, 673,302 (224D10 y 221C9) y la patente de Estados Unidos núm. 9, 120,852 (227D3 y 205A5). En un aspecto de la divulgación, el anticuerpo es 227D3.
El 227D3 se secreta por el hibridoma depositado en el CNCM el 18 de noviembre de 2009, con el número I-4247.

En otros aspectos, los anticuerpos anti-cMet se administran para fines de tratamiento, ya sea como componentes de conjugados de fármacos con anticuerpos (ADC) o antes/después/simultáneamente con la administración de los ADC.
5.6.1 ABT-700 y anticuerpos relacionados para fines de tratamiento
Para los fines de los anticuerpos de esta sección, las CDR se han identificado de acuerdo con el sistema de numeración de IMGT.
El ABBV-399 es un ADC compuesto por el anticuerpo dirigido a cMet ABT-700 (PR-1266688, h224G11) conjugado con la potente citotoxina MMAE a través de un conector de valina citrulina (vc). El ADC se une a cMet en la superficie de las células tumorales, se internaliza y después libera MMAE que conduce a la inhibición de la función de los microtúbulos y a la interrupción de los procesos celulares críticos y la muerte. El ABBV-399 es potente citotóxico para las células cancerosas con sobreexpresión de cMet o MET amplificado y demuestra actividad antitumoral en xenoinjertos tumorales humanos. Se ha demostrado además la actividad de ABBV-399 contra tumores refractarios a ABT-700 (véase, por ejemplo, el ejemplo 14).
ABT-700
ABT-700 es una versión humanizada del anticuerpo monoclonal de ratón 224G11, que se divulgó por primera vez en la patente de Estados Unidos núm. 8, 329,173. ABT-700 es una IgGlK recombinante "humanizada" (divulgada como 224G11 [TH7 Hz3] en la patente de Estados Unidos núm. 8,741,290) que se dirige a un epítopo único de cMet ubicado dentro del dominio 1 de homología del factor de transcripción inmunoglobulina-plexina (IPT), que da como resultado el bloqueo de la señalización de cMet tanto dependiente de HGF como independiente de HGF. ABT-700 compite por la unión a cMet con anticuerpos dirigidos contra SEMA blade 5 (y viceversa), pero no con anticuerpos dirigidos contra blades 1-3 o IPT 2-3. En contraste, 5D5 (el progenitor bivalente de un onartuzumab armado, discutido más abajo) se une a blade 5 del dominio SEMA.
Los cMet-ADC de esta divulgación abarcan cualquier anticuerpo que comprende una cadena pesada que comprende CDR-H1, CDR-H2 y CDR-H3 que comprenden, respectivamente, la secuencia de aminoácidos SEQ ID Nos. 1, 2 y 3; y una cadena ligera que comprende CDR-L1, CDR-L2 y CDR-L3 que comprenden, respectivamente, la secuencia de aminoácidos SEQ ID Nos. 5, 6 y 7, de acuerdo con la patente de Estados Unidos núm. 8, 741,290. Estas son las CDR del anticuerpo murino 224G11 original, según se define en base al sistema de numeración de IMGT.
Como se define en la nomenclatura IMGT, las secuencias CDR de ABT-700 comprenden las siguientes secuencias:
CDR-H1: GYIFTAYT (SEQ ID NO: 72)
CDR-H2: IKPNNGLA (SEQ ID NO: 73)
CDR-H3: ARSEITTEFDY (SEQ ID NO: 74)
CDR-L1: ESVDSYANSF (SEQ ID NO: 75)
CDR-L2: RAS (SEQ ID NO: 76)
CDR-L3: QQSKEDPLT (SEQ ID NO: 77)
En una modalidad, la región variable de la cadena pesada de 224G11 [TH7 Hz3] comprende la SEQ ID NO: 4 de la patente de Estados Unidos núm. 8, 741,290:
QVQLVQSGAEVKKPGASVKVSCKASGYIFTAYTMHWVRQAPGQGLEWMG WIKPNNGLANYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARSEITTEFDYWGQ GTLVTVSS (SEQ ID NO: 78):
y la región variable de la cadena ligera comprende la SEQ ID No. 10 de la patente de Estados Unidos núm.
8,741,290:
DIVMTQSPDSLAVSLGERATINCKSSESVDSYANSFLHWYQQKPGQPPKLLIYRASTRESGVPDRFSGSGSGTDFTL T ISSLQAEDVAVYYCQQSKED PLTFGGGTKVEIKR (SEQ ID NO: 79)
En otra modalidad, la región variable de la cadena pesada de 224G11[TH7 Hz3] comprende:
QVQLVQSGAEVKKPGASVKVSCKASGYIFTAYTMHWVRQAPGQGLEWMG WIKPNNGLANYAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCAR SEITTEFDYWGQGTLVTVSS (SEQ ID NO: 80),
y la región variable de la cadena ligera comprende:
DIVMTQSPDSLAVSLGERATINCKSSESVDSYANSFLHWYQQKPGQPPK
LLIYRASTRESGVPDRFSGSGSGTDFTLTISSLQAEDVAVYYCQQSKED PLTFGGGTKVEIK
(SEQ ID NO: 81)
En otra modalidad, el anticuerpo [224G11] [TH7 Hz3] comprende una cadena pesada completa que comprende la secuencia de aminoácidos SEQ ID No. 37 de la patente de Estados Unidos núm. 8,741,290 y una cadena ligera completa que comprende la secuencia de aminoácidos SEQ ID No. 40 de la patente de Estados Unidos núm.
8,741,290. La región de bisagra modificada tiene la secuencia de SEQ ID NO: 170.
En algunas modalidades, el anticuerpo anti-cMet comprende una región variable de cadena pesada que comprende la SEQ ID No. 4 de la patente de Estados Unidos núm. 8, 741,290:
QVQLVQSGAEVKKPGASVKVSCKASGYIFTAYTMHWVRQAPGQGLEWMGWIKPNNGLAN-YAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARSEITTEFDYWGQGTLVTVSS (SEQ ID NO: 78) unida a cualquier región constante de la cadena pesada;
y una región variable de la cadena ligera que comprende la SEQ ID No. 10 de la patente de Estados Unidos núm. 8, 741,290:
DIVMTQSPDSLAVSLGERATINCKSSESVDSYANSFLHWYQQKPGQPPKLLIYRASTRESGVPDRFSGSGSGTD
FTLTISSLQAEDVAVYYCQQSKED PLTFGGGTKVEIKR (SEQ ID NO: 79) unida a cualquier región constante de cadena ligera. Más abajo se proporcionan ejemplos de regiones constantes de cadena pesada y ligera adecuadas.
En algunas modalidades, el anticuerpo anti-cMet comprende una región variable de cadena pesada que comprende la SEQ ID No. 4 de la patente de Estados Unidos núm. 8, 741,290:
QVQLVQSGAEVKKPGASVKVSCKASGYIFTAYTMHWVRQAPGQGLEWMGWIKPNNGLAN-YAQKFQGRVTMTRDTSISTAYMELSRLRSDDTAVYYCARSEITTEFDYWGQGTLVTVSS (SEQ ID NO: 80
constante de cadena) unida a región constante de la cadena pesada;
y una región variable de cadena ligera que comprende:
DIVMTQSPDSLAVSLGERATINCKSSESVDSYANSFLHWYQQQKPGQPPKLLIYRASTRESGVPDRFSGSGSGTD FTLTISSLQAEDVAVYYCQQSKED PLTFGGGTKVEIK (SEQ ID NO: 81) unida a cualquier región constante de la cadena ligera. Más abajo se proporcionan ejemplos de regiones constantes de cadena pesada y ligera adecuadas.
En algunas modalidades, un anticuerpo y/o fragmento de unión anti-c-Met de un ADC anti-c-Met es una IgGi
En algunas modalidades, un anticuerpo anti-cMet de un ADC anti-cMet comprende una cadena pesada que tiene una región constante que comprende o que consiste en:
ASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTF
PAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDCHCPPCPAPELL
GGPSVF
LFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYR
VVSVLTVLHQ DWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKN
QVSLTCLVKG FYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGN
VFSCSVMHEALHNHYTQKSLSLSPG (SEQ ID NO: 82)
En algunas modalidades, un anticuerpo anti-cMet de un ADC anti-cMet comprende una cadena ligera que tiene una región constante que comprende o que consiste en:
RTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNS QESVTEQ DSKDSTYSLS STLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO: 83)
En algunas modalidades, un anticuerpo anti-cMet de un ADC anti-cMet comprende una cadena pesada que tiene una región constante que comprende o que consiste en:
A STKGPS VFPL AP SSK STSGGTAALGCLV KDFPEPVTV SWN SGALTSGVHTFP AVLQSSGL
YSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDCHCPPCPAPELLGGPSVF
LFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRV
V S VLTVLHQDWLNGKEYKCKV SNKALP APIEKTISKAKGQPREPQVYTLPPS REEMTKN QV S
LTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCS
VMHEALHNHYTQKSLSLSPGK (SEQ ID NO: 84)
y una cadena ligera que tiene una región constante que comprende o que consiste en:
TVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQ ESVTEQ DSKDSTYSLS STLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO: 85)
En algunas modalidades, la cadena pesada de un anticuerpo anti-cMet (ABT-700) de un ADC anti-cMet comprende o consiste en (las regiones constantes están en negrita; las CDR están subrayadas (las secuencias de CDR numeradas por Kabat divulgadas como SEQ ID NOS 112-114, respectivamente, en orden de aparición)):
QVQLVQSGAE VKKPGASVKV SCKASGYIFT AYTMHWVRQA PGQGLEWMGW 050
IKPNNGLANY AQKFQGRVTM TRDTSISTAY MELSRLRSDD TAVYYCARSE 100
ITTEFDYWGQ GTLVTVSSAS TKGPSVFPLA PSSKSTSGGT AALGCLVKDY 150
FPE PVTVSWN SGALTSGVHT FPAVLQSSGL Y S L S S W T V P SSSLG TQ TYI 200
CNVNHKPSNT KVDKRVEPKS CDCHCPPCPA PELLGGPSVF LFPPKPKDTL 25 0
MISRTPEVTC VW DVSHEDP EVKFNWYVDG VEVHNAKTKP REEQYNSTYR 30 0
W S V L TVLHQ DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL 3 50
PPSREEMTKN QVSLTCLVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD 400
GSFFLYSKLT VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPG 4 45
[0129] (secuencia de longitud completa divulgada como SEQ TD NO: 86)
y la cadena ligera comprende o consta de (las secuencias de CDR divulgadas como SEQ ID NOS 115-117, respectivamente, en orden de aparición):
DIVMTQSPDS LAVSLGERAT INCKSSESVD SYANSFLHWY QQKPGQPPKL 050
L IY RASTRES GVPDRFSGSG SGTDFTLTIS SLQAEDVAVY YCQQSKEDPL 100
TFGGGTKVEI KRTVAAPSVF  LK SG TASW CLL NNFYPREAKV 150
LK SG TASW CLL NNFYPREAKV 150
QWKVDNALQS GNSQESVTEQ DSKDSTYSLS STLTLSKADY EKHKVYACEV 200
THQGLSSFVT KSFNRGEC 218
[0131] (secuencia de longitud completa divulgada como SEQ ID NO: 87)
En algunas modalidades, la cadena pesada de un anticuerpo anti-cMet de un ADC anti-cMet comprende o consta de una región variable (aminoácidos 1-118 de SEQ ID NO: 88), una región constante (mostrada en negrita) y CDR (subrayadas: las secuencias de CDR divulgadas como SEQ ID NOS 118-120, respectivamente, en orden de aparición):
QVQLVQSGAE VKKPGASVKV SCKASGYIFT AYTMHWVRQA PGQGLEWMGW 050
IKPNNGLAHY AQKFQGRVTM TRDTSISTAY MELSRLRSDD TAVYYCARSE 100
ITTEFDYWGQ GTLVTVSSAS TKGPSVFPLA PSSKSTSGGT AALGCLVKDY 15 0
FPE PVTVSWN SGALTSGVHT FPAVLQSSGL Y S L S S W T V P SSSLG TQ TYI 200
CNVNHKPSNT KVDKRVEPKS CDCHCPPCPA PELLGGPSVF LFPPKPKDTL 2 50
MISRTPEVTC VW DVSH EDP EVKFNWYVDG VEVHNAKTKP REEQYNSTYR 30 0
W SVLTVLH Q DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL 35 0
PPSREEMTKN QVSLTCLVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD 4 00
GSFFLYSKLT VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPGK 44 6
[0133] (secuencia de cadena pesada de longitud completa divulgada como SEQ ID NO: 88)
y la cadena ligera comprende o consiste de una región variable (aminoácidos 1-110 en la SEQ ID NO: 89), una región constante (mostrada en negrita) y secuencias de las CDR (subrayadas y divulgadas como SEQ ID NOS 121 123, respectivamente, en orden de aparición):
DIVMTQSPDS LAVSLGERAT INCKSSESVD SYANSFLHWY QQKPGQPPKL 050
L IY RASTRES GVPDRFSGSG SGTDFTLTIS SLQAEDVAVY YCQQSKEDPL 100
TFGGGTKVEI KRTVAAPSVF IFPPSDEQ LK SG TASW CLL NNFYPREAKV 1 50 QWKVDNALQS GNSQESVTEQ DSKDSTYSLS STLTLSKADY EKHKVYACEV 200
THQGLSSFVT KSFNRGEC 218
(secuencia de cadena ligera de longitud completa divulgada como SEQ ID NO: 89)
En una modalidad, el anticuerpo es ABT-700 y la cadena pesada está codificada por la siguiente secuencia de nucleótidos (secuencia de longitud completa divulgada como SEQ ID NO: 90):
ATGGGATGGTCTTGGATCTTTCTGCTGTTTCTGTCTGGTACTGCTGGTGTGCTGAGC caggtccagctggtgcaatccggcgcagaggtgaagaagccaggcgcttccgtgaaggtgagctgtaaggcctctggctacatcttcacagcata caccatgcactgggtgaggcaagctcctgggcagggactggagtggatgggatggattaaacccaacaatgggctggccaactacgcccagaa attccagggtagggtcactatgac aagggataccagc atcagcacc ge atatatggagctgagc aggctgaggtctgacgacactgctgtctattat tgcgccaggagcgaaattacaacagaattcgattactgggggcagggcaccctggtgaccgtgtcctctgccagcaccaagggcccaagcgtg ttccccctggcccccagcagcaagagcaccagcggcggcacagccgccctgggctgcctggtgaaggactacttccccgagcccgtgacc gtgtcctggaacagcggagccctcacttctggagttcataccttcccagcagtattgcagagcagtggcctgtattcactgtcttccgtcgta acagttccatcctccagcctcgggacacagacttacatttgtaacgtgaatcacaagcctagcaacaccaaggtcgacaagagagttgaa ccaaagagttgtgattgccactgtcctccctgcccagctcctgagctgcttggcggtcccagtgtcttcttgtttccccctaaacccaaagaca ccctgatgatctcaaggactcccgaggtgacatgcgtggtggtggatgtgtctcatgaggacccagaggtgaagttcaactggtacgtgga cggcgtggaggtgcacaacgccaagaccaagcccagagaggagcagtacaacagcacctacagggtggtgtccgtgctgaccgtgctg caccaggactggctgaacggcaaggagtacaagtgtaaggtgtccaacaaggccctgccagccccaatcgaaaagaccatcagcaagg ccaagggccagccaagagagccccaggtgtacaccctgccacccagcagggaggagatgaccaagaaccaggtgtccctgacctgtct ggtgaagggcttctacccaagcgacatcgccgtggagtgggagagcaacggccagcccgagaacaactacaagaccacccccccagtg ctggacagcgacggcagcttcttcctgtacagcaagctgaccgtggacaagagcagatggcagcagggcaacgtgttcagctgctccgtg atgcacgaggccctgcacaaccactacacccagaagagcctgagcctgtccccaggctga
Péptido señal de secreción en letras negrita mayúsculas.
Incluye codón de parada final (TGA)
La región constante es negrita
Las CDR están subrayadas (las secuencias de las CDR divulgadas como SEQ ID NOS 124-126, respectivamente, en orden de aparición)
En una modalidad, el anticuerpo es ABT-700 y la cadena ligera está codificada por la siguiente secuencia de nucleótidos (secuencia de longitud completa divulgada como SEQ ID NO: 91):
ATGGAAACTGATACACTGCTGCTGTGGGTCCTGCTGCTGTGGGTCCC TGGAAGCACAGGGgacattgtgatgacccagtctcccgatagcctggccgtgtccctgggcgagagggctaccatcaactgtaaaa gctcc gaatctgtggactcttac ge aaacagctttctgc actggtatcagcaaaagccaggcc aacctccaaagctgctgatttac agg gcttctacc agggagagcggcgtgcccgataggttcagcggatctggcagcggcaccgactttacactgaccatctccagcctgcaggccgaagatgtggcag tctattactgccagcagtccaaggaggaccccctgactttcgggggtggtactaaagtggagatcaagcgtacggtggccgctcccagcgtgttc atcttccccccaagcgacgagcagctgaagagcggcaccgccagcgtggtgtgtctgctgaacaacttctaccccagggaggccaaggtg cagtggaaggtggacaacgccctgcagagcggcaacagccaggagagcgtcaccgagcaggacagcaaggactccacctacagcctg agcagcaccctgaccctgagcaaggccgactacgagaagcacaaggtgtacgcctgtgaggtgacccaccagggcctgtccagccccgt gaccaagagcttcaacaggggcgagtgctga
Péptido señal de secreción en negrita letras mayúsculas.
Incluye codón de parada final (tga)
La región constante es negrita
Las CDR están subrayadas (las secuencias de las CDR divulgadas como SEQ ID NOS 127-129, respectivamente, en orden de aparición)
En una modalidad, denominada en la presente descripción como ABBV399, la secuencia de la cadena pesada del anticuerpo está representada por la SEQ ID NO: 88, la secuencia de la cadena ligera está representada por la SEQ ID NO: 89 conjugada con monometil auristatina E (MMAE) a través de un conector de valina citrulina (vc).
La secuencia de ABT-700 PBD, que comprende la secuencia de ABT-700 que lleva una mutación S238C (se denomina además en la presente descripción como ABT-700 (S238C)-PBD) de acuerdo con la numeración de Kabat, es la siguiente (las CDR están subrayadas; el sistema de numeración es Kabat; y la mutación S238C está representada por C (negrita, cursiva y subrayada):
Secuencia de aminoácidos (10 AA por grupo, 5 grupos por línea)
Cadena pesada (SEQ ID NO: 171) (las secuencias de las CDR subrayadas divulgadas como las SEQ ID NOS 173 175, respectivamente, en orden de aparición):
QVQLVQSGAE VKKPGASVKV SCKASGYIFT AYTMHWVRQA PGQGLEWMGW 50
IKPNNGLANY AQKFQGRVTM TRDTSISTAY MELSRLRSDD TAVYYCARSE 100
ITTEFDYWGQ GTLVTVSSAS TKGPSVFPLA PSSKST3GGT AALGCLVKDY 150
FPEPVTVSWN SGALTSGVHT FPAVLQSSGL Y S L SSW T V P SSSLGTQTYI 200
CNVNHKPSNT KVDKRVEPKS CDCHCPPCPA PELLGGPCYF LFPPKPKDTL 250
MISRTPEVTC VWDVSHEDP EVKFNWYVDG VEVHNAKTKP REEQYNSTYR 300
VVSVLTVLHQ UWLJMGKEYKC KVSJMKALPAP lEK'i'ISKAKG QPREPQVYTL 350
PPSREEMTKN QVSLTCLVKC- FYPSDIAVEW ESNGQPENNY KTTPPVLDSD 400
GSFFLYSKLT VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPG 445
Cadena ligera (SEQ ID NO: 172) (las secuencias de las CDR subrayadas divulgadas como SEQ ID NOS 176-178, respectivamente, en orden de aparición):
DIVMTQSPDS LAVSLGERAT INCKSSESVD SYANSFLHWY QQKPGQPPKL 50
LIY RASTRES GVPDRFSGSG SGTDFTLTIS SLQAEDVAVY YCQQSKEDPL 100
TFGGGTKVEI KRTVAAPSVF IFPPSDEQLK SGTASWCLL NNFYPREAKV 150
QWKVDNALQS GNSQESVTEQ DSKDSTYSLS STLTLSKADY EKHKVYACEV 200
THQGLSSPVT KSFNRGEC 218
Por consiguiente, el anticuerpo ABT-700 PBD comprende dos moléculas de fármaco PBD-conector conjugadas a un mAb ABT-700 diseñado genéticamente con cys (S238C), y tiene una cadena pesada de SEQ ID NO: 171 y una cadena ligera de SEQ ID NO: 172.
En una modalidad, el aminoácido lisina C-terminal en la cadena pesada de 224G11[TH7 Hz3] se diseñó genéticamente para eliminar la heterogeneidad en el extremo C debido a la escisión incompleta de la lisina. En ABT -700, la cadena pesada se modifica postraduccionalmente mediante la adición de glicanos unidos a N a la asparagina-296. Los glicanos principales son oligosacáridos bicatenarios fucosilados que contienen cero, uno o dos residuos de galactosa. Además, en el extremo N de la cadena pesada hay un residuo de glutamina, que puede experimentar una ciclación espontánea para formar un residuo de piroglutamato.
El anticuerpo murino original 224G11 se ha quimerizado y humanizado adicionalmente. Los procesos de quimerización y humanización se describen en detalle en la patente de Estados Unidos núm. 8, 741,290. Durante el proceso de humanización del anticuerpo murino 224G11, la forma quimérica del Mab 224G11 (224G11chim/IgG1), que significa dominio variable (VH+VL) de m224G11 combinado con dominio constante de IgG1/kappa humano produjo fuerte (17 % del efecto máximo de HGF) actividad agonista asociada con una eficacia antagonista reducida (54 % de inhibición del efecto máximo de HGF en comparación con el m224G11 que produce 75 % de inhibición del efecto máximo de HGF). Tres formas humanizadas del Mab 224G11, [224G11] Hz1/IgG1, [224G11] Hz2/IgG1 y [224G11] Hz3/IgG1, construidas además en un esqueleto de IgG1/kappa humana, produjeron también una eficacia del antagonista disminuida y una actividad agonista significativa (11 a 24 % del nivel máximo de HGF) en comparación con 224G11 de ratón.
Las bisagras de algunas de las formas humanizadas del anticuerpo 224G11 se modificaron, como se describe en
detalle en la patente de Estados Unidos núm. 8, 741,290. Los anticuerpos resultantes, cuyos ADC también están dentro del alcance de esta divulgación, incluyeron 224G11[TH7Hz3].
El anticuerpo h224G11/ABT-700 se refiere a la forma humanizada de 224G11[TH7 Hz3]. Este anticuerpo representa el anticuerpo ABT-700 que forma parte del ABBV-399 de esta divulgación. Las actividades biológicas del anticuerpo ABT-700, o h224G11, se caracterizaron ampliamente en la patente de Estados Unidos núm. 8, 741,290.
Las versiones ilustrativas de otras versiones quimerizadas y humanizadas de conjugados de fármacos con anticuerpos 224G11 que están dentro del alcance de esta divulgación son las divulgadas en la patente de Estados Unidos núm. 8,741,290 como los anticuerpos [224G11][IgG2Hz1], [224G11][IgG2Hz2]; [224G11][IgG2Hz3];
[224G11][TH7Hz1]; [224G11][TH7z2]; [224G11][TH7Hz3]; [224G11][IgG2chim]; [224G11][TH7chim]; [224G11][C1];
[224G11][C2]; [224G11][C3]; [224G11][C5]; [224G11][C6]; [224G11][C7]; [224G11][C8]; y [224G11][C9].
Otros ejemplos incluyen los anticuerpos [224G11][A1-3]; [224G11][C7A6]; [224G11][C6A9]; [224G11][C2A5-7];
[224G11][C5A2-6]; [224G11][C9A2-7]; [224G11] AS-6-7-8]; [224G11 ][l gG1/lgG2]; [224G11 ][l gG2Hz1 ]; [224G11][lgG2Hz2];[224G11][lgG2Hz3];[224G11][TH7Hz1];[224G11][TH 7Hz2];[224G11][TH7Hz3]; [224G11][TH7chim]; [224G11][MHchim]; [224G11][MUP9Hchim] y [224G11][MMCHchim]. En ambas series de anticuerpos, el primer corchete se refiere al nombre del anticuerpo que se modifica (es decir, 224G11) y el segundo corchete identifica la modificación específica del anticuerpo, la mayoría de los cuales corresponden a cambios en la región de la bisagra, de acuerdo con la numeración única IMGT para dominios C. El símbolo A significa eliminación. Los detalles específicos de cada modificación pueden encontrarse en la patente de Estados Unidos núm. 8, 741,290.
En la presente descripción se describen ADC anti-cMet, un anticuerpo y/o fragmento de unión anti-c-Met que comprende un ADC anti-cMet es adecuado para la administración a seres humanos. En una modalidad específica, el anticuerpo anti-cMet se humaniza.
En algunas modalidades, los anticuerpos anti-cMet y/o fragmentos de unión que comprenden un ADC anti-anti-cMet compiten por la unión de cMet en células que expresan cMet, o la homología del factor de transcripción inmunoglobulina-plexina (IPT) de cMet humano, o Met-Fc o cMet diseñado genéticamente/recombinante en fase sólida, en ensayos in vitro con un anticuerpo de referencia. El anticuerpo de referencia puede ser cualquier anticuerpo que se una específicamente a la homología del factor de transcripción inmunoglobulina-plexina (IPT) del cMet humano. En una modalidad específica, el anticuerpo de referencia es 224G11 de ratón. En otra modalidad específica, el anticuerpo de referencia es ABT-700.
Los ensayos para la competencia incluyen, pero no se limitan a, un inmunoensayo marcado con material radioactivo (RIA), un ensayo por inmunoadsorción ligado a enzimas (ELISA), un ELISA sándwich, ensayos de citometría de flujo y ensayos de resonancia de plasmón superficial. Un método preferido es el descrito en Basilico C, Hultberg A, Blanchetot C, de Jonge N, Festjens E, Hanssens V, Osepa SI, De Boeck G, Mira A, Cazzanti M, Morello V, Dreier T, Saunders M, de Haard H, Michieli P. Four individually druggable MET hotspots mediate HGF-driven tumor progression. J Clin Invest. Julio de 2014; 124(7):3172-86. doi: 10.1172/JCI72316. Epub 27 de mayo de 2014.
En un ensayo de competencia de anticuerpos entre un anticuerpo de referencia y un anticuerpo de prueba (independientemente de la especie o el isotipo), uno primero puede marcar la referencia con un marcador detectable, tal como un fluoróforo, biotina o un marcador enzimático o radiactivo para permitir la detección posterior. En este caso, las células que expresan cMet o el dominio extracelular de cMet (o una subparte del mismo) se incuban con el anticuerpo de prueba no marcado, se añade el anticuerpo de referencia marcado y se mide la intensidad del marcador unido. Si el anticuerpo de prueba compite con el anticuerpo de referencia marcado por la unión al mismo epítopo proximal o superpuesto, la intensidad de la señal de detección disminuirá en relación con una reacción de control llevada a cabo sin el anticuerpo de prueba.
En un ensayo de competencia de anticuerpos, la concentración del anticuerpo de referencia marcado que produce el 80 % de unión máxima ("conc80%") en las condiciones del ensayo (por ejemplo, una densidad específica de células o una concentración específica de cMet/dominio extracelular de cMet o subparte del mismo) se determina en primer lugar, y un ensayo de competencia se lleva a cabo con una concentración80% 10X del anticuerpo de prueba no marcado y conc80% del anticuerpo de referencia marcado.
En otra modalidad ilustrativa de llevar a cabo un ensayo de competencia de citometría de flujo, las células que expresan cMet se incuban con una serie de titulación de anticuerpos que comprende concentraciones crecientes del anticuerpo de prueba no marcado frente a anticuerpo de referencia anti-cMet marcado con fluorescencia. El anticuerpo anti-cMet de referencia marcado se usa a una concentración fija X (por ejemplo, X = 1 pg/mL) y el anticuerpo de prueba no marcado se usa en un rango de concentraciones (por ejemplo, de 10_4X a 100X). Las células o cMet/dominio extracelular de cMet o subparte del mismo se incuban con el anticuerpo de prueba no marcado y con el anticuerpo de referencia marcado simultáneamente. Los datos de citometría de flujo se normalizan en relación con el anticuerpo de referencia marcado con fluorescencia solo, donde la intensidad de fluorescencia de
una muestra llevada a cabo sin anticuerpo de prueba no marcado se asigna al 100 % de unión. Si un anticuerpo de prueba compite por la unión a cMet con el anticuerpo de referencia marcado, un ensayo llevado a cabo con la misma concentración de cada uno (por ejemplo, 1 mg/mL de anticuerpo de prueba no marcado y 1 mg/mL de anticuerpo de referencia marcado) producirá aproximadamente 50 % de reducción en la intensidad de la fluorescencia en comparación con el control al 100 %, lo que indica aproximadamente un 50 % de unión. El uso de un anticuerpo de referencia marcado a una concentración de X y un anticuerpo de prueba no marcado que compita por la unión a cMet a una concentración de 10X produciría una reducción de aproximadamente el 90 % en la unión en comparación con el control del 100 %, lo que indica aproximadamente un 10 % de unión.
La inhibición puede expresarse como una constante de inhibición, o Ki, que se calcula de acuerdo con la siguiente fórmula:
Ki = IC50/ (1+ [concentración del Ab de referencia]/Kd),
donde IC50 es la concentración del anticuerpo de prueba que produce una reducción del 50 % en la unión del anticuerpo de referencia y Kd es la constante de disociación del anticuerpo de referencia, una medida de su afinidad por cMet. Los anticuerpos que compiten con los anticuerpos a c-Met de referencia puede tener una Ki de 10 pM a 100 nM en las condiciones de ensayo descritas en la presente descripción.
En varios aspectos, se considera que un anticuerpo de prueba compite con un anticuerpo de referencia si disminuye la unión del anticuerpo de referencia a las células que expresan cMet o cMet/dominio extracelular de cMet o subparte del mismo en al menos aproximadamente 20 % o más, por ejemplo, en al menos aproximadamente 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 %, 95 % o incluso más, o en un porcentaje que varía entre cualquiera de los valores anteriores, a una concentración de anticuerpo de referencia que es 80 % de la unión máxima en las condiciones de ensayo específicas usadas, y una concentración de anticuerpo de prueba que es 10 veces mayor que la concentración de anticuerpo de referencia.
En varios aspectos de un ensayo de competencia de citometría de flujo, se considera que un anticuerpo de prueba compite con un anticuerpo de referencia si disminuye la unión del anticuerpo de referencia a las células que expresan cMet en al menos aproximadamente 20 % o más, por ejemplo, en al menos aproximadamente 20 %, 30 %, 40%, 50 %, 60 %, 70 %, 80 %, 90 %, 95 % o incluso más, o en un porcentaje que varía entre cualquiera de los valores anteriores, a una concentración de anticuerpo de prueba que es 10x mayor que la del anticuerpo de referencia.
La detección de la expresión de cMet generalmente implica poner en contacto una muestra biológica (células, tejidos o fluido corporal de un individuo) con uno o más anticuerpos anti-cMet (opcionalmente conjugado con una porción detectable) y detectar si la muestra es positiva o no para la expresión de cMet, o si la muestra ha alterado (por ejemplo, reducido o aumentado) la expresión en comparación con una muestra control. Los expertos en la técnica conocen bien los métodos para hacerlo, los que incluyen los descritos en los ejemplos.
5.6.2 Algunos otros anticuerpos a cMet ilustrativas
Otro anticuerpo anti-cMet que puede usarse de acuerdo con esta divulgación se ha denominado 227H1, comprende una cadena pesada que comprende CDR-H1, CDR-H2 y CDR-H3 que comprenden, respectivamente, la secuencia de aminoácidos SEQ ID Nos. 4, 5 y 6; y una cadena ligera que comprende CDR-L1, CDR-L2 y CDR-L3 que comprenden, respectivamente, las secuencia de aminoácidos SEQ ID Nos. 13, 11 y 14 de la patente de Estados Unidos núm. 8,329,173 (SEQ ID NOS 4, 5, 6, 13, 11 y 14, respectivamente, de esta solicitud). Estos anticuerpos se han descrito en detalle en la patente de Estados Unidos núm. 8, 329,173. El Listado de Secuencias presentado simultáneamente con esta solicitud incluye las SEQ ID NOS 1-71 de la patente de Estados Unidos núm. 8, 329,173 como las SEQ ID NOS 1-71.
El anticuerpo 227H1 comprende una cadena pesada que comprende la secuencia de aminoácidos SEQ ID No. 19 y una cadena ligera que comprende la secuencia de aminoácidos SEQ ID No. 22 de la patente de Estados Unidos núm. 8,329,173 (SEQ ID NOS 19 y 20, respectivamente, de esta solicitud). Estos anticuerpos se han descrito en detalle en la patente de Estados Unidos núm. 8, 329,173.
Otro anticuerpo anti-cMet que puede usarse de acuerdo con esta divulgación se ha denominado 223C4, comprende una cadena pesada que comprende CDR-H1, CDR-H2 y CDR-H3 que comprenden, respectivamente, la secuencia de aminoácidos SEQ ID Nos. 7, 8 y 9; y una cadena ligera que comprende CDR-L1, CDR-L2 y CDR-L3 que comprenden, respectivamente, la secuencia de aminoácidos SEQ ID Nos. 15, 16 y 17 de la patente de Estados Unidos núm. 8,329,173 (SEQ ID NOS 7, 8, 9, 15, 16 y 17, respectivamente, de esta solicitud). Estos anticuerpos se han descrito en detalle en la patente de Estados Unidos núm. 8, 329,173 y sus descripciones se incorporan en la presente descripción como referencia en su totalidad.
El anticuerpo 223C4 comprende una cadena pesada que comprende la secuencia de aminoácidos SEQ ID No. 20 y una cadena ligera que comprende la secuencia de aminoácidos SEQ ID No. 23 de la patente de Estados Unidos
núm. 8,329,173 (SEQ ID NOS 20 y 23, respectivamente). Estos anticuerpos se han descrito en detalle en la patente de Estados Unidos núm. 8, 329,173.
Otro anticuerpo anti-cMet que puede usarse de acuerdo con esta divulgación se ha denominado 11E1, comprende una cadena pesada que comprende CDR-H1, CDR-H2 y CDR-H3 que comprenden, respectivamente, la secuencia de aminoácidos SEQ ID Nos. 56, 57 y 58; y una cadena ligera que comprende CDR-L1, CDR-L2 y CDR-L3 que comprenden, respectivamente, la secuencia de aminoácidos SEQ ID Nos. 59, 60 y 61 de la patente de Estados Unidos núm. 8,329,173 (SEQ ID NOS 56, 57, 58, 59, 60 y 61, respectivamente). Estos anticuerpos se han descrito en detalle en la patente de Estados Unidos núm. 8, 329,173.
En una modalidad, el anticuerpo 11E1 comprende una cadena pesada que comprende la secuencia de aminoácidos SEQ ID No. 62 y una cadena ligera que comprende la secuencia de aminoácidos SEQ ID No. 63 de la patente de Estados Unidos núm. 8,329,173 (SEQ ID NOS 62 y 63, respectivamente). Estos anticuerpos se han descrito en detalle en la patente de Estados Unidos núm. 8, 329,173.
Estos primeros anticuerpos monoclonales descritos anteriormente, o uno de sus fragmentos funcionales o derivados, se caracterizan porque dichos anticuerpos son secretados por el hibridoma depositado en la Colección Nacional de Cultivos de Microorganismos (CNCM, Colección Nacional de Cultivos de Microorganismos) (Instituto Pasteur, París, Francia) el 14/03/2007 con los números CNCM 1-3724 (correspondiente a 11E1), 1-3731 (correspondiente a 224G11), 1-3732 (correspondiente a 227H1) y el 07/06/2007 con el número 1-3786 (correspondiente a 223C4). Estos hibridomas consisten en hibridomas murinos que resultan de la fusión celular de esplenocitos de ratón inmunizado con una línea celular de mieloma (Sp20 Ag14).
Estos primeros anticuerpos, todos los cuales se divulgaron originalmente en la patente de Estados Unidos núm. 8, 329,173 y que están cubiertos por varias patentes, se resumen de la siguiente manera (las SEQ ID NO son las mismas en la patente "173 y en esta solicitud):
224G11 I-3731 227H1 I-3732 223C4 I-3786 11E1 I-3724 Prot. SEQ ID NO: Nucl. SEQ Prot; SEQ Nucl. SEQ Prot. SEQ Nucl. SEQ Prot. SEQ Nucl. SEQ ID NO: ID NO: ID NO: ID NO: ID NO: ID NO: ID NO:
CDR- 1 24 4 27 7 30 56 64
H1
CDR- 2 25 5 28 8 31 57 65
H2
CDR- 3 26 6 29 9 32 58 66
H3
Cadena 18 41 19 42 20 43 62 70

Cadena 21 44 22 45 23 46 63 71 L.
Los anticuerpos 224G11, 227H1 y 223C4 no se unen al dominio SEMA del receptor cMet. 11E1 es capaz de unirse al dominio SEMA.
En la presente descripción se describe, que el anticuerpo anti-cMet que comprende las CDR del anticuerpo STI-D0602 o STI-0602 (Sorrento Therapeutics). En la presente descripción se describe, que el anticuerpo anti-cMet es STI-D0602 o STI-0602, como se describe en Lingna Li, Cathrine Fells, Julia Guo, Pia Muyot, Edwige Gros, Yanliang Zhang, Yingqing Sun, Hong, Zhang, Yanwen Fu, Tong Zhu, Jian Cao, Gunnar Kaufmann, Gang Chen, Zhenwei Miao, A novel cMet targeting antibody drug conjugate for NSCLC, Resumen No. 3897, Reunión Anual de AACR, abril 16-20, Nueva Orleans, Estados Unidos.
En la presente descripción se describe, que el anticuerpo anti-cMet comprende las CDR del anticuerpo 5D5 (Genentech) o el derivado de un brazo (monovalente) onartuzumab. En la presente descripción se describe, que el anticuerpo anti-cMet es el anticuerpo 5D5 (Genentech) o el derivado de un brazo (monovalente) onartuzumab (figura 1B). La información adicional para onartuzumab es la siguiente:
Cadena pesada (SEQ ID NO: 92):
EVQLVESGGGLVQPGGSLRLSCAASGYTFTSYWLHWVRQAPGKGL
EWVGMIDPSNSDTRFNPNFKDRFTISADTSKNTAYLQMNSLRAED
TAVYYCATYRSYVTPLDYWGQGTLVTVS S A ST K G P 3 V F P L A P SS K
STSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSG
L Y SL S5W T V P5SSL G T Q T Y IC N V N H K P SN T K V D K K V E P K 5C D K T
H T C PPC PA PE L L G G PSV FL FPPK PK D T L M ISR T PE V T C V W D V SH
E D P EVKFNWYVDGVEVHNAKTKPREEQYNSTYRWS VLTVLHQ DW
LNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEM
TKHQVSLSCAVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGS
FFLVSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
Cadena ligera (SEQ ID NO: 93):
DIQMTQSPSSLSASVGDRVTITCKSSQSLLYTSSQKNYLAW YQQK
PG K A P K L L IY W A ST R E SG V P SR F SG SG SG T D FT L T ISSL Q P E D F A
TYYCQQYYAYPWTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGT
ASWCLLNNFYPREAKVQW KVDNALQSGNSQESVTEQDSKDSTYS
LSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFMRGEC
Bisagra-CH2-CH3 (SEQ ID NO: 94):
D K T H T CPPC PA PE L L G G PSVFL FPPK PK DT L M ISR T PE V T CV W D
VSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRWSVLTVLH
QDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSR
E EMT KNQV S LWC LVKG FY P S DIAVEW ES N GQPENNYKTTP PVL DS
DGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSP
GK
En la presente descripción se describe, que el anticuerpo anti-cMet comprende las CDR del anticuerpo emibetuzumab/LY2875358. En la presente descripción se describe, que el anticuerpo anti-cMet es emibetuzumab/ LY2875358 (Eli Lilly and Company, número CAS 1365287-97-3) (figura 1A). La información adicional para emibetuzumab es la siguiente:
Cadena pesada (SEQ ID NO: 95):
QVQLVQSGAEVKKPGASVKVSCKASGYTFTDYYMHWVRQAPGQGL
EWMGRVNPNRRGTTYNQKFEGRVTMTTDTSTSTAYMELRSLRSDD
TAVYYCARANWLDYWGQGTTVTVSSASTKGPSVFPLAPCSRSTSE
STAALGCLVKDYFPEPVTVSW NSGALTSGVHTFPAVLQSSGLYSL
SSW TVPSSSLG TK TYTCN VDH K PSNTK VDK RV ESK YG PPCPPCP
AP EAAGG P SV F L FP P K P K DT LMIS RT P EVT C V W D V S Q ED P EVQ F
NWYVDGVEVHNAKTKPREEQFNSTYRWSVLTVLHQDWLNGKEYK
CKVSMKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVSL
TCLV KG FYPSDIAVEW ESNGQPENNYKTTPPVLDSDGSFFLYSRL
TVDKSRWQEGNVFSCSVMHEALHNHYTQKSLSLSLG
Cadena ligera (SEQ ID NO: 96)
D IQ M T Q SP SSL SASVG DRV T IT C SVSSSVSSIYL H W YQ Q K PG K A P
K L L IY S T SN L A SG V P 3R F SG S G S G T D F T L T ISS L Q P E D F A T Y Y C Q
VY SG YPLTFG G GTK VE IK RTVA APSVFIFPPSDEQ LKSGT ASV VC
LLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTL
TLSKADYEKHKVYACEVTHQGLSSPVTKSFMRGEC
En la presente descripción se describe, que el anticuerpo anti-cMet comprende las CDR del anticuerpo AbF46 o SAIT301 (Samsung Electronics). En la presente descripción se describe, que el anticuerpo es AbF46 (figura 1C). En otra modalidad, el anticuerpo anti-cMet es SAIT301 (figura 1E).
En la presente descripción se describe, que el anticuerpo anti-cMet comprende las CDR del anticuerpo ARGX-111 (36C4) (arGEN-X BV). En la presente descripción se describe, que el anticuerpo anti-cMet es ARGX-111 (figura ID). En la presente descripción se describe, que el anticuerpo anti-cMet comprende las CDR de uno de los anticuerpos en Sym015 (Hu9006, Hu9338) (Symphogen A/S). En la presente descripción se describe, que el anticuerpo anticMet es Hu9006. En la presente descripción se describe, que el anticuerpo anti-cMet es Hu9338. Las secuencias de aminoácidos de estos anticuerpos, que incluyen sus CDR, se divulgan en el documento WO2016042412.
5.5. Sistemas de expresión y métodos para fabricar los anticuerpos
Los anticuerpos anti-cMet pueden prepararse mediante expresión recombinante de genes de las cadenas de inmunoglobulina ligera y pesada en una célula huésped a través de métodos bien conocidos por los expertos en la técnica. Para expresar un anticuerpo de forma recombinante, una célula huésped se transfecta con uno o más vectores de expresión recombinantes que portan fragmentos de ADN que codifican las cadenas de inmunoglobulina ligera y pesada del anticuerpo, de manera que las cadenas ligera y pesada se expresan en la célula huésped y, opcionalmente, se secretan en el medio en el que se cultivan las células huésped, a partir del cual pueden recuperarse los anticuerpos. Las metodologías estándar de ADN recombinante se usan para obtener genes de las cadenas pesada y ligera de anticuerpos, incorporar estos genes en vectores de expresión recombinantes e introducir los vectores en células huésped, tales como las descritas en Molecular Cloning; A Laboratory Manual, Segunda Edición (Sambrook, Fritsch y Maniatis (eds), Cold Spring Harbor, N.Y., 1989), Current Protocols in Molecular Biology (Ausubel, FM y otros, Eds., Greene Publishing Associates, 1989) y en la patente de Estados Unidos núm. 4,816,397. Para generar ácidos nucleicos que codifican tales anticuerpos anti-cMet, primero se obtienen fragmentos de ADN que codifican las regiones variables de las cadenas ligera y pesada. Estos ADN pueden obtenerse mediante amplificación y modificación de ADN de línea germinal o ADNc que codifican secuencias variables de las cadenas ligera y pesada, por ejemplo, mediante el uso de la reacción en cadena de la polimerasa (PCR). Las secuencias de ADN de línea germinal para los genes humanos de la región variable de las cadenas pesada y ligera se conocen en
la técnica (véase, por ejemplo, la base de datos de la secuencia de la línea germinal humana "VBASE"; véase además Kabat y otros, 1991, Sequences of Proteins of Immunological Interest, Quinta edición, Departamento de Salud y Servicios Humanos de Estados Unidos, Publicación NIH núm. 91-3242; Tomlinson y otros, 1992, J. Mol. Biol. 22t :116-198; y Cox y otros, 1994, Eur. J. Immunol. 24: 827-836. Los nucleótidos que codifican los anticuerpos 224G11, 227H1, 223C4 y 11E11 se han descrito en detalle en la patente de Estados Unidos núm. 8, 329,173.
Una vez que se obtienen los fragmentos de ADN que codifican los segmentos Vh y Vl relacionados con el anticuerpo anti-cMet, estos fragmentos de ADN pueden manipularse adicionalmente mediante técnicas estándar de ADN recombinante, por ejemplo, para convertir los genes de la región variable en genes de las cadenas de anticuerpos de longitud completa, en genes de fragmentos Fab o en un gen de scFv. En estas manipulaciones, un fragmento de ADN que codifica Vl o Vh se une operativamente a otro fragmento de ADN que codifica otra proteína, tal como una región constante de anticuerpo o un conector flexible. El término "unido operativamente," como se usa en este contexto, pretende significar que los dos fragmentos de ADN están unidos de tal manera que las secuencias de aminoácidos codificadas por los dos fragmentos de ADN permanecen dentro del marco.
El ADN aislado que codifica la región Vh puede convertirse en un gen de cadena pesada de longitud completa al unir operativamente la molécula de ADN que codifica la Vh a otra de ADN que codifica las regiones constantes de la cadena pesada (CH1, CH2 , CH3 y, opcionalmente, CH4). Las secuencias de los genes humanos de la región constante de la cadena pesada se conocen en la técnica (véase, por ejemplo, Kabat y otros, 1991, Sequences of Proteins of Immunological Interest, Quinta edición, Departamento de Salud y Servicios Humanos de Estados Unidos, Publicación NIH Núm. 91-3242) y los fragmentos de ADN que abarcan estas regiones pueden obtenerse mediante amplificación por PCR estándar. La región constante de la cadena pesada puede ser una región constante de IgG 1, IgG2 , IgG3, IgG4, IgA, IgE, IgM o IgD, pero en ciertas modalidades es una región constante de IgG1 o IgG4. Para un gen de la cadena pesada del fragmento Fab, el ADN que codifica la Vh puede unirse operativamente a otra molécula de ADN que codifica solo la región constante CH1 de la cadena pesada.
El ADN aislado que codifica la región Vl puede convertirse en un gen de cadena ligera de longitud completa (así como también, un gen de Fab de la cadena ligera) al unir operativamente el ADN que codifica la Vl a otra molécula de ADN que codifica la región constante de la cadena ligera, CL. Las secuencias de los genes humanos de la región constante de la cadena ligera se conocen en la técnica (véase, por ejemplo, Kabat y otros, 1991, Sequences of Proteins of Immunological Interest, Quinta edición, Departamento de Salud y Servicios Humanos de Estados Unidos, Publicación NIH Núm. 91-3242) y los fragmentos de ADN que abarcan estas regiones pueden obtenerse mediante amplificación por PCR estándar. La región constante de la cadena ligera puede ser una región constante kappa o lambda, pero en ciertas modalidades es una región constante kappa. Para crear un gen de scFv, los fragmentos de ADN que codifican las Vh y Vl se unen operativamente a otro fragmento que codifica un conector flexible, por ejemplo, que codifica la secuencia de aminoácidos (Gly4~Ser)3 (SEQ ID NO: 97), de modo que las secuencias Vh y Vl pueden expresarse como una proteína monocatenaria contigua, con las regiones Vl y Vh unidas por el conector flexible (véase, por ejemplo, Bird y otros, 1988, Science 242:423-426; Huston y otros, 1988, Proc. Natl. Acad. Sci. EE.UU. 85:5879-5883; McCafferty y otros, 1990, Nature 348:552-554).
Para expresar los anticuerpos anti-cMet, los ADN que codifican las cadenas ligera y pesada de longitud parcial o completa, obtenidas como se describió anteriormente, se insertan en vectores de expresión de manera que los genes se unen operativamente a secuencias de control transcripcional y traduccional. En este contexto, el término "unido operativamente" pretende significar que un gen de anticuerpo está ligado a un vector de tal manera que las secuencias de control transcripcional y traduccional dentro del vector cumplen su función prevista de regular la transcripción y la traducción del gen del anticuerpo. El vector de expresión y las secuencias de control de expresión se eligen para que sean compatibles con la célula huésped de expresión usada. El gen de la cadena ligera del anticuerpo y el gen de la cadena pesada del anticuerpo pueden insertarse en vectores separados o, más típicamente, ambos genes se insertan en el mismo vector de expresión.
Los genes del anticuerpo se insertan en el vector de expresión mediante métodos estándar (por ejemplo, ligadura de sitios de restricción complementarios en el fragmento del gen del anticuerpo y el vector, o ligadura de extremos romos si no hay sitios de restricción presentes). Antes de la inserción de las secuencias de las cadenas ligera o pesada, relacionadas con el anticuerpo anti-cMet, el vector de expresión ya puede portar secuencias de región constante del anticuerpo. Por ejemplo, un enfoque para convertir las secuencias Vh y Vl relacionadas con anticuerpos monoclonales anti-cMet en genes de anticuerpos de longitud completa es insertarlas dentro de vectores de expresión que ya codifican regiones constantes de cadena pesada y constante de cadena ligera, respectivamente, de modo que el segmento Vh está unido operativamente a segmento(s) CH dentro del vector y el segmento Vl está unido operativamente al segmento CL dentro del vector. Adicionalmente o alternativamente, el vector de expresión recombinante puede codificar un péptido señal que facilita la secreción de la cadena de anticuerpo a partir de una célula huésped. El gen de la cadena de anticuerpo puede clonarse en el vector de modo que el péptido señal esté unido en el marco al extremo amino del gen de la cadena de anticuerpos. El péptido señal puede ser un péptido señal de inmunoglobulina o un péptido señal heterólogo (es decir, un péptido señal de una proteína que no sea inmunoglobulina).
Además de los genes de las cadenas de anticuerpos, los vectores de expresión recombinantes portan secuencias
reguladoras que controlan la expresión de los genes de las cadenas de anticuerpos en una célula huésped. El término "secuencia reguladora" pretende incluir promotores, potenciadores y otros elementos de control de la expresión (por ejemplo, señales de poliadenilación) que controlan la transcripción o traducción de los genes de las cadenas de anticuerpos. Tales secuencias reguladoras se describen, por ejemplo, en Goeddel, Gene Expression Technology: Methods in Enzymology 185, Academic Press, San Diego, Ca , 1990. Los expertos en la técnica apreciarán que el diseño del vector de expresión, que incluye la selección de secuencias reguladoras, puede depender de factores tales como la elección de la célula huésped a transformar, el nivel de expresión de la proteína deseada, etc. Las secuencias reguladoras adecuadas para la expresión en células huésped de mamíferos incluyen elementos virales que dirigen altos niveles de expresión de proteínas en células de mamíferos, tales como promotores y/o potenciadores derivados del citomegalovirus (CMV) (tal como el promotor/potenciador de CMV), virus simio 40 (SV40) (tal como el promotor/potenciador de SV40), adenovirus (por ejemplo, el promotor tardío principal de adenovirus (AdMLP)) y el polioma. Para una descripción más detallada de los elementos reguladores virales y sus secuencias, véase, por ejemplo, la patente de Estados Unidos núm. 5, 168,062 de Stinski, la patente de Estados Unidos núm. 4, 510,245 de Bell y otros, y la patente de Estados Unidos núm. 4, 968,615 de Schaffner y otros.
Los vectores de expresión recombinantes de la divulgación pueden portar secuencias además de los genes de las cadenas de anticuerpos y las secuencias reguladoras, tales como secuencias que regulan la replicación del vector en las células huésped (por ejemplo, orígenes de replicación) y genes marcadores seleccionables. Los genes marcadores seleccionables facilitan la selección de células huésped en las que se ha introducido el vector (véase, por ejemplo, las patentes de Estados Unidos núms. 4, 399,216, 4, 634,665 y 5, 179,017, todas de Axel y otros). Por ejemplo, típicamente un gen marcador seleccionable confiere resistencia a fármacos, tales como G418, higromicina o metotrexato, en una célula huésped en la que se ha introducido el vector. Los genes marcadores seleccionables adecuados incluyen el gen de dihidrofolato reductasa (DHFR) (para usar en células huésped DHFR- con selección/amplificación por metotrexato) y el gen neo (para la selección por G418). Para la expresión de las cadenas ligera y pesada, los vectores de expresión que codifican las cadenas pesada y ligera se transfectan en una célula huésped mediante técnicas estándar. Las diversas formas del término "transfección" pretenden abarcar una amplia variedad de técnicas comúnmente usadas para la introducción de ADN exógeno en una célula huésped procariota o eucariota, por ejemplo, electroporación, lipofección, precipitación con fosfato de calcio, transfección con DEAE-dextrano y similares.
Es posible expresar anticuerpos anti-cMet de los ADC anti-cMet en células huésped procariotas o eucariotas. En la presente descripción se describe, que la expresión de anticuerpos se realiza en células eucariotas, por ejemplo, células huésped de mamífero, de secreción óptima de un anticuerpo plegado apropiadamente e inmunológicamente activo. Las células huésped de mamífero ilustrativas para expresar los anticuerpos recombinantes de la divulgación incluyen células de ovario de hámster chino (células CHO) (que incluyen células CHO DHFR-, descritas en Urlaub y Chasin, 1980, Proc. Natl. Acad. Sci. USA 77: 4216-4220, usado con un marcador seleccionable DHFR, por ejemplo, como se describe en Kaufman y Sharp, 1982, Mol. Biol. 159: 601-621), células de mieloma NS0, células COS y células SP2. Cuando los vectores de expresión recombinantes que codifican los genes de anticuerpos se introducen en células huésped de mamífero, los anticuerpos se producen mediante cultivo de las células huésped durante un período de tiempo suficiente para permitir la expresión del anticuerpo en las células huésped o la secreción del anticuerpo hacia el medio de cultivo en el que crecen las células huésped. Los anticuerpos pueden recuperarse del medio de cultivo mediante el uso de métodos estándar de purificación de proteínas. Las células huésped pueden usarse además para producir porciones de anticuerpos intactos, tales como moléculas de fragmentos Fab o scFv. Se entenderá que las variaciones en el procedimiento anterior están dentro del alcance de la presente divulgación. Por ejemplo, puede ser conveniente transfectar una célula huésped con ADN que codifica la cadena ligera o la cadena pesada (pero no ambas) de un anticuerpo anti-cMet.
La tecnología de ADN recombinante puede usarse además para eliminar parte o la totalidad del ADN que codifica cualquiera o ambas de las cadenas ligera y pesada que no sea necesaria para la unión a cMet. Las moléculas expresadas a partir de tales moléculas de ADN truncadas también se abarcan por los anticuerpos de la divulgación.
Para la expresión recombinante de un anticuerpo anti-cMet, la célula huésped puede cotransfectarse con dos vectores de expresión, el primer vector que codifica un polipéptido derivado de la cadena pesada y el segundo vector que codifica un polipéptido derivado de la cadena ligera. Los dos vectores pueden contener marcadores seleccionables idénticos, o cada uno puede contener un marcador seleccionable separado. Alternativamente, puede usarse un solo vector que codifica polipéptidos de las cadenas pesada y ligera.
Una vez que se obtiene un ácido nucleico que codifica una o más porciones de un anticuerpo anti-cMet, pueden introducirse más alteraciones o mutaciones en la secuencia de codificación, por ejemplo, para generar ácidos nucleicos que codifican los anticuerpos con diferentes secuencias de CDR, anticuerpos con afinidad reducida al receptor Fc o anticuerpos de diferentes subclases.
Los anticuerpos y/o fragmentos de unión de los ADC anti-cMet también pueden producirse mediante síntesis química (por ejemplo, mediante los métodos descritos en Solid Phase Peptide Synthesis, 2da ed., 1984 The Pierce Chemical Co., Rockford, Ill.). Los anticuerpos variantes pueden generarse además mediante el uso de una
plataforma libre de células, véase, por ejemplo, Chu y otros, Biochemia Núm. 2, 2001 (Roche Molecular Biologicals) y Murray y otros, 2013, Current Opinion in Chemical Biology, 17:420-426.
Una vez que se ha producido un anticuerpo y/o fragmento de unión anti-c-Met mediante expresión recombinante, puede purificarse mediante cualquier método conocido en la técnica para la purificación de una molécula de inmunoglobulina, por ejemplo, mediante cromatografía (por ejemplo, intercambio iónico, afinidad y cromatografía en columna de encolado), centrifugación, solubilidad diferencial o por cualquier otra técnica estándar para la purificación de proteínas. Además, los anticuerpos y/o los fragmentos de unión anti-cMet pueden fusionarse con secuencias de polipéptidos heterólogos descritos en la presente descripción o de cualquier otra manera conocida en la técnica para facilitar la purificación.
Una vez aislado, el anticuerpo y/o fragmento de unión anti-c-Met puede, si se desea, purificarse adicionalmente, por ejemplo, mediante cromatografía en columna. (véase, por ejemplo, Fisher, Laboratory Techniques In Biochemistry And Molecular Biology, Work y Burdon, eds., Elsevier, 1980), o por cromatografía de filtración en gel en una columna Superdex™ 75 (Pharmacia Biotech AB, Uppsala, Suecia).
5.6. Conjugados específicos de fármacos con anticuerpos anti-cMet
Como se mencionó, los ADC anti-cMet generalmente comprenden una porción de unión al antígeno anti-cMet, tal como un anticuerpo y/o fragmento de unión anti-cMet, que tiene uno o más agentes citotóxicos y/o citostáticos, que pueden ser iguales o diferentes, unidos a estos a través de uno o más conectores, que además pueden ser iguales o diferentes. Pueden unirse múltiples agentes citotóxicos/citostáticos diferentes a cada Ab para formar un ADC. Estos agentes pueden dirigirse a dos o más vías para matar o detener el crecimiento de células tumorales, atacar múltiples nodos de la misma vía o duplicarse en la misma diana (es decir, inhibir el crecimiento y/o matar células a través de dos o más mecanismos diferentes).
Los ADC anti-cMet son compuestos de acuerdo con la fórmula estructural (I):
(I) [D-L-XY]„-Ab
o sales de los mismos, donde cada "D" representa, independientemente de los demás, un agente citotóxico y/o citostático ("fármaco"); cada "L" representa, independientemente de los demás, un conector; "Ab" representa una porción de unión al antígeno anti-cMet, tal como un anticuerpo o fragmento de unión anti-cMet; cada "XY" representa un enlace formado entre un grupo funcional Rx en el conector y un grupo funcional "complementario" Ry en la porción de unión al antígeno; y n representa el número de fármacos ligados a Ab del ADC.
Las modalidades específicas de diversos anticuerpos o fragmentos de unión (Ab) que pueden constituir el ADC de acuerdo con la fórmula estructural (I) incluyen las diversas modalidades de anticuerpos y/o fragmentos de unión anticMet descritos anteriormente.
En ADC específicos o sales de fórmula estructural (I), cada D es el mismo y/o cada L es el mismo.
Los agentes citotóxicos y/o citostáticos (D) y los conectores (L) que pueden constituir los ADC anti-cMet, así como también, el número de agentes citotóxicos y/o citostáticos unidos a los ADC anti-cMet, se describen con más detalle más abajo.
En la presente descripción se describe, que todos los ADC anti-cMet son compuestos de acuerdo con la fórmula estructural (I) en la que cada "D" es el mismo y es una auristatina que penetra en las células (por ejemplo, dolastatina-10 o MMAE) o un agente de reticulación de ADN de unión a surcos menores que penetran en las células (por ejemplo, una PBD o dímero de PBD); cada "L" es el mismo y es un conector escindible por una enzima lisosomal; cada "XY" es un enlace formado entre una maleimida y un grupo sulfhidrilo; "Ab" es un anticuerpo que comprende seis CDR correspondientes a las seis CDR del anticuerpo ABT-700 (224G11), o un anticuerpo que compite por la unión de cMet con dicho anticuerpo; y n es 2, 3 o 4. En unos ADC anti-cMet ilustrativos de fórmula estructural (I), "Ab" es un anticuerpo humanizado, por ejemplo, un anticuerpo humanizado que comprende cadenas Vh y Vl correspondientes a las cadenas Vh y Vl del anticuerpo 5D5. En unos ADC anti-cMet ilustrativos de fórmula estructural (I), el Ab es el anticuerpo STI-D0602 (Sorrento).
Se proporciona un compuesto de acuerdo con la fórmula estructural (I) con la estructura de fórmula (IIa):

En una modalidad, el Ab en el compuesto de fórmula (IIa) es ABT-700.
Se proporciona un compuesto de fórmula estructural (I) con la siguiente estructura:

o una sal farmacéuticamente aceptable del mismo, en donde n tiene un valor promedio que varía de 2-4, y el Ab es un anticuerpo anti-cMet de longitud completa.
En una modalidad específica, el Ab en el compuesto de esta fórmula particular es ABT-700.
En una modalidad específica, n tiene un valor promedio que varía de 2-4 y Ab es un anticuerpo anti-cMet de longitud completa.
5.6.1 Agentes citotóxicos y/o citostáticos
Los agentes citotóxicos y/o citostáticos pueden ser cualquiera de los agentes conocidos por inhibir el crecimiento y/o la replicación y/o matar células, y en particular células cancerosas y/o tumorales. En la literatura se conocen numerosos agentes que tienen propiedades citotóxicas y/o citostáticas. Los ejemplos no limitantes de clases de agentes citotóxicos y/o citostáticos incluyen, a modo de ejemplo y sin limitación, radionúclidos, agentes alquilantes, agentes de reticulación de ADN, agentes intercalantes de ADN (por ejemplo, agentes de unión a surcos tales como aglutinantes de surcos menores), moduladores del ciclo celular, reguladores de apoptosis, inhibidores de quinasas, inhibidores de síntesis de proteínas, inhibidores de mitocondrias, inhibidores de exportación nuclear, inhibidores de topoisomerasa I, inhibidores de topoisomerasa II, antimetabolitos de ARN/ADN y agentes antimitóticos.
Más abajo se describen ejemplos específicos no limitantes de agentes dentro de ciertas de estas diversas clases. Agentes alquilantes: asaley (L-leucina, N-[N-acetil-4-[bis-(2-cloroetil)amino]-DL-fenilalanil]-, éster etílico); AZQ (ácido 1,4-ciclohexadieno-1,4-dicarbámico, 2, 5-bis(1-aziridinil)-3,6-dioxo-, éster dietílico); BCNU (N,N'-Bis(2-cloroetil)-N-nitrosourea); busulfano (dimetanosulfonato de 1,4-butanodiol); (carboxiftalato)platino; CBDCA (cis-(1,1-c¡clobutanod¡carbox¡lato)diam¡noplat¡no (II))); CCNU (N-(2-cloroetil)-N’-c¡clohex¡l-N-n¡trosourea); CHIP (iproplatino; NSC 256927); clorambucilo; clorozotocina (2-[[[(2-cloroetil)nitrosoamino]carbonil]amino]-2-desoxi-D-glucopiranosa); c/s-platino (cisplatino); clomesona; cianomorfolinodoxorrubicina; ciclodisona; dianhidrogalactitol (5,6-diepoxidulcitol); fluorodopa ((5-[(2-cloroetil)-(2-fluoroetil)amino]-6-metil-uracilo); hepsulfam; hicantona; dímero de indolinobenzodiazepina DGN462; melfalán; metil CCNU ((1-(2-cloroetil)-3-(trans-4-metilciclohexano)-1-nitrosourea); mitomicina C; mitozolamida; mostaza nitrogenada ((clorhidrato de bis(2-cloroetil)metilamina); PCNU ((1-(2-cloroetil)-3-(2,6-dioxo-3-piperidil)-1-nitrosourea)); alquilante de piperazina (diclorhidrato de (1 -(2-cloroetil)-4-(3-cloropropil)-piperazina)); piperazinadiona; pipobromán (N,N'-bis(3-bromopropionil)piperazina); porfiromicina (N-metilmitomicina C); mostaza de espirohidantoína; teroxirona (triglicidilisocianurato); tetraplatino; tiotepa (N,N’,N"-tri-1,2-etanod¡¡lt¡o fosforamida); trietilenomelamina, mostaza nitrogenada de uracilo (desmetildopan); Yoshi-864 ((clorhidrato de bis(3-mesiloxi propil)amina).
Agentes similares a alquilantes de ADN: cisplatino; carboplatino; nedaplatino; oxaliplatino; satraplatino;
tetranitrato de triplatino; procarbazina; altretamina; dacarbazina; mitozolomida; temozolomida.
Agentes antineoplásicos alquilantes: carbocuona; carmustina; clomafazina; clorozotocina; duocarmicina; evofosfamida; fotemustina; glufosfamida; lomustina; manosulfán; nimustina; fenantriplatino; pipobroman; ranimustina; semustina; estreptozotocina; tiotepa; treosulfán; triazicuona; trietilenmelamina; tetranitrato de triplatino.
Inhibidores de la replicación y reparación del ADN: altretamina; bleomicina; dacarbazina; dactinomicina; mitobronitol; mitomicina; pingiangmicina; plicamicina; procarbazina; temozolomida; ABT-888 (veliparib); olaparib; KU-59436; AZD-2281; AG-014699; BSI-201; BGP-15; INO-1001; ONO-2231.
Moduladores del Ciclo Celular: paclitaxel; nab-paclitaxel; docetaxel; vincristina; vinblastina; ABT-348; AZD-1152; MLN-8054; VX-680; inhibidores de quinasa específicos de Aurora A; inhibidores de quinasa específicos de Aurora B e inhibidores de quinasa pan-Aurora; AZD-5438; BMI-1040; BMS-032; BMS-387; CVT-2584; flavopiridol; GPC-286199; MCS-5A; PD0332991; PHA-690509; seliciclib (CYC-202, R-roscovitina); ZK-304709; AZD4877, ARRY-520; GSK923295A.
Reguladores de la apoptosis: AT-101 ((-) gosipol); G3139 u oblimersen (oligonucleótido antisentido dirigido a Bcl-2); IPI-194; I PI-565; N-(4-(4-((4’-cloro(1,1’-bifenil)-2-il)metil)piperazin-1-ilbenzoil)-4-(((1R)-3-(dimetilamino)-1-((fenilsulfanil)metil)propil)amino)-3-nitrobencenosulfonamida); N-(4-(4-((2-(4-clorofenil)-5,5-dimetil-1-ciclohex-1-en-1-il)metil)piperazin-1-il)benzoil)-4-(((1R)-3-(morfolin-4-il)-1-((fenilsulfanil)metil)propil)amino)-3-((trifluorometil)sulfonil)bencenosulfonamida; GX-070 (Obatoclax®; 1H-indol, 2-(2-((3,5-dimetil-1H-pirrol-2-il)metilen)-3-metoxi-2H-pirrol-5-il)-)); HGS1029; GDC-0145; GDC-0152; LCL-161; LBW-242; venetoclax; agentes que se dirigen a TRAIL o receptores de muerte (por ejemplo, DR4 y DR5) tales como ETR2-ST01, GDC0145, HGS-1029, LBY-135, PRO-1762; fármacos que se dirigen a caspasas, reguladores de caspasas, miembros de la familia BCL-2, proteínas del dominio de muerte, miembros de la familia TNF, miembros de la familia Toll y/o proteínas NF-kappa-B.
Inhibidores de la angiogénesis: ABT-869; AEE-788; axitinib (AG-13736); AZD-2171; CP-547,632; IM-862; pegaptamib; sorafenib; BAY43-9006; pazopanib (GW-786034); vatalanib (PTK-787, ZK-222584); sunitinib; SU-11248; trampa de VEGF; vandetanib; ABT-165; ZD-6474; inhibidores de DLL4.
Inhibidores del proteasoma: bortezomib; carfilzomib; epoxomicina; ixazomib; salinosporamida A.
Inhibidores de quinasa: afatinib; axitinib; bosutinib; crizotinib; dasatinib; erlotinib; fostamatinib; gefitinib; ibrutinib; imatinib; lapatinib; lenvatinib; mubritinib; nilotinib; pazopanib; pegaptanib; sorafenib; sunitinib; SU6656; vandetanib; vemurafenib; CEP-701 (lesaurtinib); XL019; INCB018424 (ruxolitinib); ARRY-142886 (selemetinib); ARRY-438162 (binimetinib); PD-325901; PD-98059; AP-23573; CCI-779; everolimus; RAD-001; rapamicina; temsirolimus; inhibidores de TORC1/TORC2 competitivos con ATP que incluyen PI-103, PP242, PP30, Torin 1; LY294002; XL-147; CAL-120; ONC-21; AEZS-127; ETP-45658; PX-866; GDC-0941; BGT226; BEZ235; XL765. Inhibidores de la síntesis de proteínas: estreptomicina; dihidrostreptomicina; neomicina; framicetina; paromomicina; ribostamicina; kanamicina; amikacina; arbekacina; bekanamicina; dibekacina; tobramicina; espectinomicina; higromicina B; paromomicina; gentamicina; netilmicina; sisomicina; isepamicina, verdamicina; astromicina; tetraciclina; doxiciclina; clortetraciclina; clomociclina; demeclociclina; limeciclina; meclociclina; metaciclina; minociclina; oxitetraciclina; penimepiciclina; rolitetraciclina; tetraciclina; glicilciclinas, tigeciclina; oxazolidinona; eperezolid; linezolid; posizolid; radezolid; ranbezolid; sutezolid; tedizolid; inhibidores de peptidil transferasa; cloranfenicol; azidamfenicol; tiamfenicol; florfenicol; pleuromutilinas; retapamulina; tiamulina; valnemulina; azitromicina; claritromicina; diritromicina; eritromicina; fluritromicina; josamicina; midecamicina; miocamicina; oleandomicina; rokitamicina; roxitromicina; espiramicina; troleandomicina; tilosina; cetólidos; telitromicina; cetromicina; solitromicina; clindamicina; lincomicina; pirlimicina; estreptograminas; pristinamicina; quinupristina/dalfopristina; virginiamicina.
Inhibidores de la histona deacetilasa: vorinostat; romidepsina; quidamida; panobinostat; ácido valproico; belinostat; mocetinostat; abexinostat; entinostat; SB939 (pracinostat); resminostat; givinostat; quisinostat; tioureidobutironitrilo (Kevetrin™); CUDC-10; CHR-2845 (tefinostato); CHR-3996; 4SC-202; CG200745; ACY-1215 (rocilinostat); ME-344; sulforafano.
Inhibidores de la topoisomerasa I: camptotecina; diversos derivados y análogos de camptotecina (por ejemplo, NSC 100880, NSC 603071, NSC 107124, NSC 643833, NSC 629971, NSC 295500, NSC 249910, NSC 606985, NSC 74028, NSC 176323, NSC 295501, NSC 606172, NSC 606173, NSC 610458, NSC 618939, NSC 610457, NSC 610459, NSC 606499, NSC 610456, NSC 364830 y NSC 606497); morfolinisoxorrubicina; SN-38.
Inhibidores de la topoisomerasa II: doxorrubicina; amonafida (benzisoquinolinindiona); m-AMSA (4’-(9-acridinilamino)-3’-metoximetanosulfonanilida); derivado de antrapirazol ((NSC 355644); etopósido (VP-16); pirazoloacridina ((pirazolo[3,4,5-kl]acridina-2(6H)-propanamina, 9-metoxi-N, N-dimetil-5-nitro-,
monometanosulfonato); clorhidrato de bisantreno; daunorrubicina; desoxidoxorrubicina; mitoxantrona; menogaril; N, N-dibencil daunomicina; oxantrazol; rubidazona; tenipósido.
Agentes intercalantes de ADN: antramicina; chicamicina A; tomaimicina; DC-81; sibiromicina; derivado de pirrolobenzodiazepina; SGD-1882 ((S)-2-(4-aminofenil)-7-metoxi-8-(3-(((S)-7-metoxi-2-(4-metoxifenil)-5-oxo-5,11 a-dihidro-1 H-benzo[e]pirrolo[1,2-a][1,4]diazepin-8-il)oxi)propoxi)-1 H-benzo[e]pirrolo[1,2-a][1,4] diazepin-5(11 aH)-ona); SG2000 (SJG-136; (11 aS, 11 a’S)-8,8’-(propano-1,3-diilbis(oxi))bis(7-metoxi-2-metMeno-2,3-dihidro-1 H-benzo[e]pirrolo[1,2-a][1,4]diazepin-5(11 aH)-ona)).
Antimetabolitos de ARN/ADN: L-alanosina; 5-azacitidina; 5-fluorouracilo; acivicina; derivado de aminopterina ácido N-[2-cloro-5-[[(2, 4-diamino-5-metil-6-quinazolinil)metil]amino]benzoil] L-aspártico (NSC 132483); derivado de aminopterina ácido N-[4-[[(2, 4-diamino-5-etil-6-quinazolinil)metil]amino]benzoil] L-aspártico; derivado de aminopterina ácido N-[2-cloro-4-[[(2, 4-diamino-6-pteridinil)metil]amino]benzoil] L-aspártico monohidratado; antifolato PT523 ((Na-(4-am¡no-4-desox¡pteroil)-Nv-hem¡ftalo¡l-L-orn¡t¡na)); antifol soluble de Baker (NSC 139105); dicloralil lawsona ((2-(3, 3-dicloroalil)-3-hidroxi-1,4-naftoquinona); brequinar; ftorafur ((profármaco; 5-fluoro-1-(tetrahidro-2-furil)-uracilo); 5,6-dihidro-5-azacitidina; metotrexato; derivado de metotrexato (ácido N-[[4-[[(2, 4-diamino-6-pteridinil)metil]metilamino]-1-naftalenil]carbonil] L-glutámico); PALA ((N-(fosfonoacetil)-L-aspartato); pirazofurina; trimetrexato.
Antimetabolitos de ADN: 3-HP; 2’-desoxi-5-fluorour¡d¡na; 5-HP; a-TGDR (a-2’-desoxi-6-tioguanos¡na); glicinato de afidicolina; ara C (arabinósido de citosina); 5-aza-2’-desox¡cit¡d¡na; (3-TGDR ((3-2’-desoxi-6-tioguanos¡na); ciclocitidina; guanazol; hidroxiurea; glicodialdehído de inosina; macbecina II; pirazoloimidazol; tioguanina; tiopurina.
Inhibidores de mitocondrias: pancratistatina; fenpanstatina; rodamina-123; edelfosina; succinato de d-alfatocoferol; compuesto 1113; aspirina; elipticina; berberina; cerulenina; GX015-070 (Obatoclax®; 1 H-indol, 2-(2-((3,5-dimetil-1H-pirrol-2-il)metileno)-3-metoxi-2H-pirrol-5-il)-); celastrol (tripterina); metformina; verde brillante; ME-344.
Agentes antimitóticos: alocolchicina; auristatinas, tales como MMAE (monometil auristatina E) y MMAF (monometil auristatina F); halicondrina B; cemadotin; colchicina; derivado de colchicina (N-benzoil-deacetil benzamida); dolastatina-10; dolastatina-15; maitansina; maitansinoides, tales como DM1 (A/2’-deacetil-A/2’-(3-mercapto-1-oxopropil)-maitansina); rozoxina; paclitaxel; derivado de paclitaxel ((2’-N-[3-(dimetilamino)propil]glutaramato paclitaxel); docetaxel; tiocolchicina; tritilcisteína; sulfato de vinblastina; sulfato de vincristina.
Inhibidores de exportación nuclear: calistatina A; delactonmicina; KPT-185 (propan-2-il(Z)-3-[3-[3-metoxi-5-(trifluorometil)fenil]-1,2,4-triazol-1-il]prop-2-enoato); kazusamicina A; leptolstatina; leptofuranina A; leptomicina B; ratjadone; verdinexor ((Z)-3-[3-[3,5-bis(trifluorometil)fenil]-1,2,4-triazol-1-il]-N’-piridin-2-ilprop-2-enohidrazida). Terapias hormonales: anastrozol; exemestano; arzoxifeno; bicalutamida; cetrorelix; degarelix; deslorelina; trilostano; dexametasona; flutamida; raloxifeno; fadrozol; toremifeno; fulvestrant; letrozol; formestano; glucocorticoides; doxercalciferol; carbonato de sevelámero; lasofoxifeno; acetato de leuprolida; megesterol; mifepristona; nilutamida; citrato de tamoxifeno; abarelix; prednisona; finasterida; rilostano; buserelina; hormona liberadora de la hormona luteinizante (LHRH); histrelina; trilostano o modrastano; fosrelina; goserelina.
Cualquiera de estos agentes que incluyen, o que pueden modificarse para incluir, un sitio de unión a un anticuerpo y/o fragmento de unión puede incluirse en un ADC anti-cMet.
Los expertos en la técnica apreciarán además que los mecanismos de acción anteriores no son mutuamente excluyentes, y que en algunas modalidades puede ser conveniente utilizar ADC anti-cMet capaces de ejercer actividad antitumoral contra la expresión de cMet (en la presente descripción denominados tumores cMet+) o tumores que sobreexpresan cMet a través de más de un mecanismo de acción. Como ejemplo específico, tal ADC anti-cMet puede incluir un agente citotóxico y/o citostático que penetra en las células que es citotóxico y/o citostático tanto para tumores cMet+/que sobreexpresan como para células tumorales negativas para cMet unidas a un anticuerpo anti-cMet por medio de un conector escindible.
Por consiguiente, en algunas modalidades, los agentes citotóxicos y o citostáticos incluidos en un ADC anti-cMet, tras la escisión del ADC, serán capaces de atravesar las membranas celulares ("agentes citostáticos y/o citotóxicos permeables a las células"). Los agentes citotóxicos y/o citostáticos específicos de interés, y/o los productos de escisión de los ADC que incluyen tales agentes, pueden analizarse para determinar la capacidad de atravesar las membranas celulares mediante el uso de métodos rutinarios conocidos por los expertos en la técnica. La permeabilidad (P) de las moléculas a través de una membrana se puede expresar como P = KD/Ax donde K es el coeficiente de partición, D es el coeficiente de difusión y Ax es el grosor de la membrana celular. El coeficiente de difusión (D) es una medida de la velocidad de entrada al citoplasma en dependencia del peso molecular o el tamaño de una molécula. K es una medida de la solubilidad de la sustancia en lípidos. Un valor bajo de K describe una
molécula como el agua que no es soluble en lípidos. Gráficamente, se espera que la permeabilidad (P) en función del coeficiente de partición (K) aumente linealmente cuando D y Ax son constantes. (Walter y Gutknecht, 1986, "Permeability of small nonelectrolytes through lipid bilayer membranes," Journal of Membrane Biology 90:207-217; Diamond & Katz, 1974, "Interpretaron of nonelectrolyte partition coefficients between dimyristoyl lecithin and water," Journal of Membrane Biology 17:121-154).
De acuerdo con la presente invención, el agente citotóxico y/o citostático es un agente antimitótico permeable a las células.
En la presente descripción se describe, que el agente citotóxico y/o citostático es una auristatina permeable a las células, tal como, por ejemplo, dolastatina-10 o MMAE.
En la presente descripción se describe, que el agente citotóxico y/o citostático es un agente de reticulación de ADN de unión a surcos menores que penetran en las células, tal como, por ejemplo, un dímero de pirrolobenzodiazepina ("PBD").
5.6.2. Conectores
En los ADC anti-cMet descritos en la presente descripción, los agentes citotóxicos y/o citostáticos están unidos a la porción de unión al antígeno por medio de conectores. Los conectores pueden ser cortos, largos, hidrófobos, hidrófilos, flexibles o rígidos, o pueden estar compuestos de segmentos que cada uno tiene independientemente una o más de las propiedades mencionadas anteriormente, de modo que el conector puede incluir segmentos que tienen propiedades diferentes. Los conectores pueden ser polivalentes de modo que unan covalentemente más de un agente a un único sitio en el anticuerpo, o monovalentes de modo que unan covalentemente un agente único a un único sitio en el anticuerpo.
Como apreciarán los expertos en la técnica, los conectores unen los agentes citotóxicos y/o citostáticos a la porción de unión al antígeno al formar un enlace covalente al agente citotóxico y/o citostático en una ubicación y un enlace covalente a la porción de unión al antígeno en otra. Los enlaces covalentes se forman por reacción entre grupos funcionales en el conector y grupos funcionales en los agentes y la porción de unión al antígeno. Como se usa en la presente descripción, la expresión "conector" pretende incluir (i) formas no conjugadas del conector que incluyen un grupo funcional capaz de unir covalentemente el conector a un agente citotóxico y/o citostático y un grupo funcional capaz de enlazar covalentemente el conector a la porción de unión al antígeno tal como un anticuerpo; (ii) formas parcialmente conjugadas del conector que incluyen un grupo funcional capaz de unir covalentemente el conector a una porción de unión al antígeno tal como un anticuerpo y que está unido covalentemente a un agente citotóxico y/o citostático, o viceversa; y (iii) formas completamente conjugadas del conector que está unido covalentemente tanto a un agente citotóxico y/o citostático como a una porción de unión al antígeno tal como un anticuerpo. En algunos conectores y ADC descritos en la presente descripción, así como los sintones usados para conjugar agentesconectores con anticuerpos, las porciones que comprenden los grupos funcionales en el conector y los enlaces covalentes formados entre el conector y el anticuerpo se ilustran específicamente como Rx y XY, respectivamente.
Los conectores que unen los agentes citotóxicos y/o citostáticos a la porción de unión al antígeno de un ADC anticMet pueden ser largos, cortos, flexibles, rígidos, de naturaleza hidrófila o hidrófoba, o pueden comprender segmentos que tienen características diferentes, tales como segmentos de flexibilidad, segmentos de rigidez, etc. El conector puede ser químicamente estable en entornos extracelulares, por ejemplo, químicamente estable en el torrente sanguíneo, o puede incluir enlaces que no son estables y liberan los agentes citotóxicos y/o citostáticos en el medio extracelular. En algunas modalidades, los conectores incluyen enlaces que se diseñan para liberar los agentes citotóxicos y/o citostáticos tras la internalización del ADC anti-cMet dentro de la célula. En algunas modalidades específicas, los conectores incluyen enlaces diseñados para escindirse y/o inmolarse o de cualquier otra manera descomponerse específicamente o no específicamente dentro de las células. En la técnica se conoce una amplia variedad de conectores útiles para unir fármacos a porciones de unión al antígeno tales como anticuerpos en el contexto de los ADC. Puede usarse cualquiera de estos conectores, así como también otros conectores, para unir los agentes citotóxicos y/o citostáticos a la porción de unión al antígeno de los ADC anti-cMet descritos en la presente descripción.
El número de agentes citotóxicos y/o citostáticos unidos a la porción de unión al antígeno de un ADC anti-cMet puede variar (denominado "relación fármaco-a-anticuerpo" o "DAR"), y estará limitado solo por el número de sitios de unión disponibles en la porción de unión al antígeno y el número de agentes unidos a un único conector. Típicamente, un conector unirá un único agente citotóxico y/o citostático a la porción de unión al antígeno de un ADC anti-cMet. En modalidades de ADC anti-cMet que incluyen más de un único agente citotóxico y/o citostático, cada agente puede ser igual o diferente. Mientras el ADC anti-cMet no exhiba niveles inaceptables de agregación en las condiciones de uso y/o almacenamiento, se contemplan los ADC anti-cMet con DAR de veinte, o incluso superior. En algunas modalidades, los ADC anti-cMet descritos en la presente descripción pueden tener un DAR en el intervalo de aproximadamente 1-10, 1-8, 1-6 o 1-4. En ciertas modalidades específicas, los ADC anti-cMet pueden tener un DAR de 2, 3 o 4. En ciertas modalidades, el ADC anti-cMet tiene un DAR promedio de 3,1.
Los conectores son preferentemente, pero no necesariamente, químicamente estables en las condiciones fuera de la célula, y pueden diseñarse para escindirse, inmolarse y/o de cualquier otra manera degradarse específicamente dentro de la célula. Alternativamente, pueden usarse conectores que no están diseñados para escindirse o degradarse específicamente dentro de la célula. La elección del conector estable frente al inestable puede depender de la toxicidad del agente citotóxico y/o citostático. En la técnica se conoce una amplia variedad de conectores útiles para unir fármacos a anticuerpos en el contexto de los ADC. Cualquiera de estos conectores, así como también, otros conectores, puede usarse para unir los agentes citotóxicos y/o citostáticos al anticuerpo de los ADC descritos en la presente descripción.
Los conectores polivalentes ilustrativos que pueden usarse para unir muchos agentes citotóxicos y/o citostáticos a una sola molécula de anticuerpo se describen, por ejemplo, en los documentos WO 2009/073445; WO 2010/068795; WO 2010/138719; WO 2011/120053; WO 2011/171020; WO 2013/096901; WO 2014/008375; WO 2014/093379; WO 2014/093394; WO 2014/093640. Por ejemplo, la tecnología de conector Fleximer desarrollada por Mersana y otros tiene el potencial de habilitar los ADC de alto DAR con buenas propiedades fisicoquímicas. Como se muestra más abajo la tecnología Mersana se basa en la incorporación de moléculas de fármaco en un esqueleto de poliacetal solubilizante a través de una secuencia de enlaces éster. La metodología proporciona ADC altamente cargados (DAR de hasta 20) mientras mantiene buenas propiedades fisicoquímicas.
Pueden encontrarse ejemplos adicionales de conectores de tipo dendrítico en los documentos US 2006/116422; US 2005/271615; de Groot y otros (2003) Angew. Chem. Int. Ed. 42:4490-4494; Amir y otros (2003) Angew. Chem. Int. Ed. 42:4494-4499; Shamis y otros (2004) J. Am. Chem. Soc. 126:1726-1731; Sun y otros (2002) Bioorganic & Medicinal Chemistry Letters 12:2213-2215; Sun y otros (2003) Bioorganic & Medicinal Chemistry 11:1761-1768; King y otros (2002) Tetrahedron Letters 43:1987-1990.
Los conectores monovalentes ilustrativos que pueden usarse se describen, por ejemplo, en Nolting, 2013, Antibody-Drug Conjugates, Methods in Molecular Biology 1045:71-100; Kitson y otros, 2013, CROs/CMOs - Chemica Oggi -Chemistry Today 31(4):30-38; Ducry y otros, 2010, Bioconjugate Chem. 21:5-13; Zhao y otros, 2011, J. Med. Chem 54:3606-3623; la patente de Estados Unidos núm. 7, 223,837; la patente de Estados Unidos núm. 8, 568,728; la patente de Estados Unidos núm. 8, 535,678; y el documento WO2004010957.
A modo de ejemplo y sin limitación, más abajo se describen algunos conectores escindibles y no escindibles que pueden incluirse en los ADC anti-cMet descritos en la presente descripción.
5.6.2.1Conectores escindibles
En ciertas modalidades, el conector seleccionado es escindible in vivo. Los conectores escindibles pueden incluir enlaces química o enzimáticamente inestables o degradables. Los conectores escindibles generalmente dependen de procesos dentro de la célula para liberar el fármaco, tal como la reducción del citoplasma, la exposición a condiciones ácidas en el lisosoma o la escisión por proteasas específicas u otras enzimas dentro de la célula. Los conectores escindibles generalmente incorporan uno o más enlaces químicos que pueden escindirse química o enzimáticamente mientras que el resto del conector no es escindible. En ciertos aspectos, un conector comprende un grupo químicamente lábil tal como grupos hidrazona y/o disulfuro. Los conectores que comprenden grupos químicamente lábiles explotan las propiedades diferenciales entre el plasma y algunos compartimentos citoplasmáticos. Las condiciones intracelulares para facilitar la liberación del fármaco para los conectores que contienen hidrazona son el entorno ácido de los endosomas y lisosomas, mientras que los conectores que contienen disulfuro se reducen en el citosol, que contiene altas concentraciones de tiol, por ejemplo, glutatión. En ciertas modalidades, la estabilidad plasmática de un conector que comprende un grupo químicamente lábil puede aumentarse al introducir impedimento estérico mediante el uso de sustituyentes cerca del grupo químicamente lábil. Los grupos lábiles al ácido, tales como la hidrazona, permanecen intactos durante la circulación sistémica en el entorno de pH neutro de la sangre (pH 7,3-7,5) y se someten a hidrólisis y liberan el fármaco una vez que el ADC se internaliza en los compartimentos levemente ácidos endosomales (pH 5,0-6,5) y lisosomales (pH 4,5-5,0) de la célula. Este mecanismo de liberación dependiente del pH se ha asociado con la liberación inespecífica del fármaco. Para aumentar la estabilidad del grupo hidrazona del conector, el conector puede variarse mediante modificación química, por ejemplo, sustitución, que permite la sintonización para lograr una liberación más eficiente en el lisosoma con una pérdida mínima en la circulación.
Los conectores que contienen hidrazona pueden contener sitios de escisión adicionales, tales como sitios de escisión lábiles a ácidos y/o sitios de escisión lábiles enzimáticamente, adicionales. Los ADC que incluyen conectores ilustrativos que contienen hidrazona incluyen las siguientes estructuras:
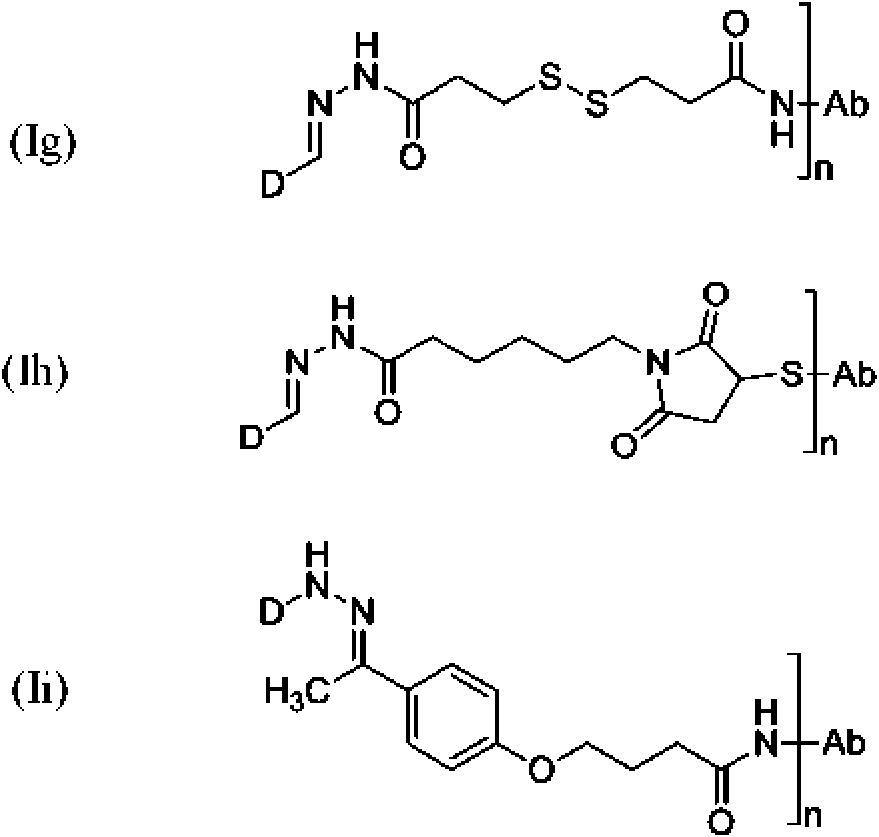
en donde D y Ab representan el agente citotóxico y/o citostático (fármaco) y el anticuerpo, respectivamente, y n representa el número de conectores de fármacos unidos al anticuerpo. En ciertos conectores como el conector (Ig), el conector comprende dos grupos escindibles: una porción disulfuro y una hidrazona. Para tales conectores, la liberación eficaz del fármaco libre no modificado requiere el pH ácido o reducción de disulfuro y un pH ácido. Se ha demostrado que los conectores tales como (Ih) y (Ii) son eficaces con un solo sitio de escisión de hidrazona.
Otros grupos lábiles a los ácidos que pueden incluirse en los conectores incluyen conectores que contienen cisaconitilo. La química de cis-aconitilo usa un ácido carboxílico yuxtapuesto a un enlace amida para acelerar la hidrólisis de la amida en condiciones ácidas.
Los conectores escindibles pueden incluir además un grupo disulfuro. Los disulfuros son termodinámicamente estables a pH fisiológico y se diseñan para liberar el fármaco tras la internalización dentro de las células, en donde el citosol proporciona un entorno significativamente más reductor en comparación con el entorno extracelular. La escisión de enlaces disulfuro generalmente requiere la presencia de un cofactor de tiol citoplasmático, como el glutatión (reducido) (GSH), de modo que los conectores que contienen disulfuro sean razonablemente estables en la circulación, al liberar selectivamente el fármaco en el citosol. La proteína enzimática intracelular disulfuro isomerasa, o enzimas similares capaces de escindir enlaces disulfuro, pueden contribuir además a la escisión preferencial de enlaces disulfuro dentro de las células. Se informa que el GSH está presente en las células en el intervalo de concentración de 0,5 a 10 mM en comparación con una concentración significativamente menor de GSH o cisteína, el tiol de bajo peso molecular más abundante, en circulación a aproximadamente 5 pM. Las células tumorales, donde el flujo sanguíneo irregular conduce a un estado hipóxico, resultan en una actividad mejorada de las enzimas reductoras y, por lo tanto, concentraciones de glutatión incluso superiores. En ciertos aspectos, la estabilidad in vivo de un conector que contiene disulfuro puede mejorarse mediante la modificación química del conector, por ejemplo, el uso de impedimento estérico adyacente al enlace disulfuro.
Los ADC que incluyen conectores que contienen disulfuro ilustrativos incluyen las siguientes estructuras:

en donde D y Ab representan el fármaco y el anticuerpo, respectivamente, n representa el número de conectores de fármacos unidos al anticuerpo, y R se selecciona independientemente en cada aparición de hidrógeno o alquilo, por ejemplo. En ciertos aspectos, el aumento del impedimento estérico adyacente al enlace disulfuro aumenta la estabilidad del conector. Las estructuras tales como (Ij) y (Il) muestran una mayor estabilidad in vivo cuando uno o más grupos R se seleccionan de un alquilo inferior tal como metilo.
Otro tipo de conector escindible que puede usarse es un conector que es escindido específicamente por una enzima. Tales conectores están típicamente basados en péptidos o incluyen regiones peptídicas que actúan como sustratos para enzimas. Los conectores basados en péptidos tienden a ser más estables en plasma y medio extracelular que los conectores químicamente lábiles. Los enlaces peptídicos generalmente tienen una buena estabilidad en suero, ya que las enzimas proteolíticas lisosomales tienen una actividad muy baja en la sangre debido a los inhibidores endógenos y al valor desfavorablemente alto del pH de la sangre en comparación con los lisosomas. La liberación de un fármaco a partir de un anticuerpo se produce específicamente debido a la acción de proteasas lisosomales, por ejemplo, catepsina y plasmina. Estas proteasas pueden estar presentes a niveles elevados en ciertas células tumorales.
Los péptidos escindibles ilustrativos se seleccionan de tetrapéptidos tales como Gly-Phe-Leu-Gly (SEQ ID NO:98), Ala-Leu-Ala-Leu (SEQ ID NO:99) o dipéptidos tales como Val-Cit, Val-Ala, Met-(D)Lys, Asn-(D)Lys, Val-(D)Asp, Phe-Lys, Ile-Val, Asp-Val, His-Val, NorVal-(D)Asp, Ala-(D)Asp, Met-Lys, Asn-Lys, Ile-Pro, Me3Lys-Pro, PhenylGly-(D)Lys, Met-(D)Lys, Asn-(D)Lys, Pro-(D)Lys, Met-(D)Lys, Asn-(D)Lys, Met-(D)Lys, Asn-(D)Lys. Los dipéptidos se prefieren a los polipéptidos más largos debido a la hidrofobicidad de los péptidos más largos.
Se han descrito una variedad de conectores escindibles basados en dipéptidos útiles para unir fármacos como doxorrubicina, mitomicina, campototecina, talisomicina y auristatina/miembros de la familia de auristatina a anticuerpos (véase, Dubowchik y otros, 1998, J. Org. Chem 67:1866-1872; Dubowchik y otros, 1998, Bioorg. Med. Chem. Lett. 8(21):3341-3346; Walker y otros, 2002, Bioorg. Med. Chem. Lett. 12:217-219; Walker y otros, 2004, Bioorg. Med. Chem. Lett. 14:4323-4327; y Francisco y otros, 2003, Blood 102:1458-1465, Dornina y otros, 2008, Bioconjugate Chemistry 19:1960-1963). Todos estos conectores de dipéptidos, o versiones modificadas de estos conectores de dipéptidos, pueden usarse en los ADC descritos en la presente descripción. Otros conectores de dipéptidos que pueden usarse incluyen los que se encuentran en los ADC como Brentuximab Vendotin SGN-35 de Seattle Genetics (Adcetris™), Seattle Genetics SGN-75 (anti-CD-70, Val-Cit-MMAF), Celldex Therapeutics glembatumumab (CDX-011) (anti-NMB, Val-Cit-MMAE) y Cytogen PSMA-ADC (PSMA-ADC-1301) (anti-PSMA, Val-Cit-MMAE).
Los conectores escindibles enzimáticamente pueden incluir un espaciador autoinmolativo para separar espacialmente el fármaco del sitio de escisión enzimática. La unión directa de un fármaco a un conector peptídico puede resultar en la liberación proteolítica de un aducto de aminoácidos del fármaco, lo que, por lo tanto, perjudica su actividad. El uso de un espaciador autoinmolativo permite la eliminación del fármaco completamente activo, químicamente no modificado, tras la hidrólisis del enlace amida.
Un espaciador autoinmolativo es el grupo alcohol bifuncional del para-aminobencilo, que está unido al péptido a través del grupo amino, lo que forma un enlace amida, mientras que los fármacos que contienen amina pueden estar unidos a través de funcionalidades carbamato con el grupo hidroxilo bencílico del conector (PABC). Los profármacos resultantes se activan tras la escisión mediada por proteasas, lo que conduce a una reacción de eliminación de 1,6 que libera el fármaco no modificado, dióxido de carbono y restos del grupo conector. El siguiente esquema muestra la fragmentación del éter de p-amidobencilo y la liberación del fármaco:

Se han descrito además variantes heterocíclicas de este grupo autoinmolativo. Véase, por ejemplo, la patente de Estados Unidos núm. US 7, 989,434.
En algunos aspectos, el conector enzimáticamente escindible es un conector basado en ácido (3-glucurónico. La liberación fácil del fármaco puede realizarse mediante la escisión del enlace glucosídico (3-glucurónido por la enzima lisosomal (3-glucuronidasa. Esta enzima está presente abundantemente dentro de los lisosomas y se sobreexpresa en algunos tipos de tumores, mientras que la actividad enzimática fuera de las células es baja. Los conectores basados en ácido (3-glucurónico pueden usarse para evadir la tendencia de un ADC a experimentar agregación debido a la naturaleza hidrófila de los (3-glucurónidos. En algunas modalidades, los conectores basados en ácido (3-glucurónico se prefieren como conectores para los ADC unidos a fármacos hidrófobos. El siguiente esquema
muestra la liberación del fármaco del ADC que contiene un conectar basado en ácido (3-glucurónico:

Se ha descrito una variedad de conectares escindibles basados en ácido (3-glucurónico útiles para unir fármacos tales como auristatinas, análogos de camptotecina y doxorrubicina, aglutinantes de surcos menores de CBI y psimberina a anticuerpos (véase, Nolting, Capítulo 5 "Linker Technology in Antibody-Drug Conjugates," En: Antibody-Drug Conjugates: Methods in Molecular Biology, vol. 1045, pp. 71-100, Laurent Ducry (Ed.), Springer Science & Business Medica, LLC, 2013; Jeffrey y otros, 2006, Bioconjug. Chem 17:831-840; Jeffrey y otros, 2007, Bioorg. Med. Chem. Lett. 17:2278-2280; y Jiang y otros, 2005, J. Am. Chem Soc. 127: 11254-11255). Todos estos conectares basados en ácido (3-glucurónico pueden usarse en los ADC anti-cMet descritos en la presente descripción.
Además, los agentes citotóxicos y/o citostáticos que contienen un grupo fenol pueden unirse covalentemente a un conector a través del oxígeno fenólico. Uno de estos conectores, descrito en el documento WO 2007/089149, se basa en una metodología en la que se usa un "separador" de diaminoetano junto con grupos autoinmolativos tradicionales basados en "PABO" para suministrar fenoles. La escisión del conector se representa esquemáticamente más abajo, donde D representa un agente citotóxico y/o citostático que tiene un grupo hidroxilo fenólico.

Los conectores escindibles pueden incluir porciones o segmentos no escindibles, y/o segmentos o porciones escindibles pueden incluirse de cualquier otra manera en un conector no escindible para hacerlo escindible. Solo a modo de ejemplo, el polietilenglicol (PEG) y los polímeros relacionados pueden incluir grupos escindibles en el esqueleto de polímero. Por ejemplo, un conector de polietilenglicol o polimérico pueden incluir uno o más grupos escindibles tales como un disulfuro, una hidrazona o un dipéptido.
Otros enlaces degradables que pueden emplearse en conectores incluyen, pero no se limitan a, enlaces éster formados por la reacción de ácidos carboxílicos de PEG o ácidos carboxílicos de PEG activados con grupos alcoholes en un agente biológicamente activo, en donde tales grupos éster generalmente se hidrolizan en condiciones fisiológicas para liberar el agente biológicamente activo. Los enlaces hidrolíticamente degradables incluyen, pero no se limitan a, enlaces carbonato; enlaces imina que resultan de la reacción de una amina y un aldehído; enlaces éster fosfato formados al reaccionar un alcohol con un grupo fosfato; enlaces acetal que son el producto de reacción de un aldehído y un alcohol; enlaces ortoéster que son el producto de reacción de un formato y un alcohol; y enlaces oligonucleotídicos formados por un grupo fosforamidita, que incluyen pero no se limitan, al final de un polímero, y un grupo 5’-hidroxilo de un oligonucleótido.
En ciertos aspectos, el conector comprende una porción peptídica enzimáticamente escindible, por ejemplo, un conector que comprende la fórmula estructural (IVa), (IVb), (IVc) o (IVd):

o una sal del mismo, en donde:
el péptido representa un péptido (¡lustrado C—>N y que no muestra los "terminales" carboxi y amino) escindible por una enzima lisosomal;
T representa un polímero que comprende una o más unidades de etilenglicol o una cadena de alquileno, o combinaciones de las mismas;
Ra se selecciona de hidrógeno, alquilo, sulfonato y sulfonato de metilo;
p es un número entero que varía de 0 a 5;
q es 0 o 1;
x es 0 o 1;
y es 0 o 1;
■ representa el punto de unión del conector a un agente citotóxico y/o citostático; y
* representa el punto de unión al resto del conector.
En ciertas modalidades, la enzima lisosomal se selecciona de catepsina B y (3-glucoronidasa.
En ciertos aspectos, el péptido se selecciona de un tripéptido o un dipéptido. En modalidades particulares, el dipéptido se selecciona de: Val-Cit; Cit-Val; Ala-Ala; Ala-Cit; Cit-Ala; Asn-Cit; Cit-Asn; Cit-Cit; Val-Glu; Glu-Val; Ser-Cit; Cit-Ser; Lys-Cit; Cit-Lys; Asp-Cit; Cit-Asp; Ala-Val; Val-Ala; Phe-Lys; Val-Lys; Ala-Lys; Phe-Cit; Leu-Cit; Ile-Cit; Phe-Arg y Trp-Cit. En ciertos aspectos, el péptido se selecciona de: Val-Cit; Cit-Val; Ala-Ala; Ala-Cit; Cit-Ala; Asn-Cit; Cit-Asn; Cit-Cit; Val-Glu; Glu-Val; Ser-Cit; Cit-Ser; Lys-Cit; Cit-Lys; Asp-Cit; Cit-Asp; Ala-Val; and Val-Ala y sales de los mismos.
Los conectores ilustrativos de acuerdo con la fórmula estructural (IVa) que pueden incluirse en los ADC descritos en la presente descripción incluyen los conectores ilustrados más abajo (como se ilustra, los conectores incluyen un grupo adecuado para unir covalentemente el conector a un anticuerpo):

Los conectores ilustrativos de acuerdo con la fórmula estructural (IVb) que pueden incluirse en los ADC descritos en la presente descripción incluyen los conectores ilustrados más abajo (como se ilustra, los conectores incluyen un grupo adecuado para unir covalentemente el conector a un anticuerpo):



Los conectores ilustrativos de acuerdo con la fórmula estructural (IVc) que pueden incluirse en los ADC descritos en la presente descripción incluyen los conectores ilustrados más abajo (como se ilustra, los conectores incluyen un
grupo adecuado para unir covalentemente el conector a un anticuerpo):

Los conectores ilustrativos de acuerdo con la fórmula estructural (IVd) que pueden incluirse en los ADC descritos en la presente descripción incluyen los conectores ilustrados más abajo (como se ilustra, los conectores incluyen un grupo adecuado para unir covalentemente el conector a un anticuerpo):



En ciertos aspectos, el conector que comprende la fórmula estructural (IVa), (IVb), (IVc) o (IVd) comprende además una porción de carbonato escindible por exposición a un medio ácido. En aspectos particulares, el conector está unido a través de un oxígeno a un agente citotóxico y/o citostático.
5.6.2.2Conectores no escindibles
Aunque los conectores escindibles pueden proporcionar ciertas ventajas, los conectores del ADC descritos en la presente descripción no necesitan ser escindibles. Para los conectores no escindibles, la liberación del fármaco no depende de las propiedades diferenciales entre el plasma y algunos compartimentos citoplasmáticos. Se postula que la liberación del fármaco se produce después de la internalización del ADC a través de la endocitosis mediada por antígenos y el suministro al compartimento lisosómico, donde el anticuerpo se degrada al nivel de aminoácidos a través de la degradación proteolítica intracelular. Este proceso libera un derivado del fármaco, que está formado por el fármaco, el conector y el residuo de aminoácido al que se unió covalentemente el conector. Los metabolitos aminoácidos del fármaco de los conjugados con conectores no escindibles son más hidrófilos y, en general, menos permeables a la membrana, lo que conduce a menos efectos en la vecindad y menos toxicidades inespecíficas en comparación con los conjugados con un conector escindible. En general, los ADC con conectores no escindibles tienen mayor estabilidad en la circulación que los ADC con conectores escindibles. Los conectores no escindibles pueden ser cadenas de alquileno, o pueden ser de naturaleza polimérica, tales como, por ejemplo, aquellos basados en polímeros de polialquilenglicol, polímeros de amida, o pueden incluir segmentos de cadenas de alquileno, polialquilenglicoles y/o polímeros de amida.
Se ha descrito una variedad de conectores no escindibles usados para unir fármacos a anticuerpos. Véase, Jeffrey y otros, 2006, Bioconjug. Chem. 17; 831-840; Jeffrey y otros, 2007, Bioorg. Med. Chem. Lett. 17:2278-2280; y Jiang y otros, 2005, J. Am. Chem Soc. 127: 11254-11255. Todos estos conectores pueden incluirse en los ADC descritos en la presente descripción.
En ciertos aspectos, el conector no es escindible in vivo, por ejemplo, un conector de acuerdo con la fórmula estructural (VIa), (VIb), (VIc) o (VId) (como se ilustra, los conectores incluyen un grupo adecuado para unir
covalentemente el conector a un anticuerpo:

o sales de los mismos, en donde:
Ra se selecciona de hidrógeno, alquilo, sulfonato y sulfonato de metilo;
Rx es una porción que incluye un grupo funcional capaz de unir covalentemente el conector a un anticuerpo; y
/
representa el punto de unión del conector a un agente citotóxico y/o citostático.
Los conectores ilustrativos de acuerdo con la fórmula estructural (VIa)-(VId) que pueden incluirse en los ADC descritos en la presente descripción incluyen los conectores ¡lustrados más abajo (como se ¡lustra, los conectores
incluyen un grupo adecuado para unir covalentemente el conector a un anticuerpo, y "«^"representa el punto de unión a un agente citotóxico y/o citostático):


5.6.2.3. Grupos usados para unir conectores a anticuerpos
Una variedad de grupos puede usarse para unir los sintones de conector-fármaco a los anticuerpos para producir los ADC. Los grupos de unión pueden ser de naturaleza electrofílica e incluyen: grupos maleimida, disulfuros activados, ésteres activos tales como ésteres de NHS y ésteres de HOBt, haloformatos, haluros de ácido, haluros de alquilo y bencilo tales como haloacetamidas. Como se discute más abajo, existen además tecnologías emergentes relacionadas con maleimidas "autoestabilizantes" y "disulfuros de puentes" que pueden usarse de acuerdo con la divulgación. El grupo específico usado dependerá, en parte, del sitio de unión al anticuerpo.
Un ejemplo de un grupo maleimida "autoestabilizante" que se hidroliza espontáneamente en condiciones de conjugación de anticuerpos para dar unas especies de ADC con estabilidad mejorada se representa en el esquema más abajo. Véase el documento US20130309256 A1; también Lyon y otros, Nature Biotech publicado en línea, doi: 10.1038/nbt.2968).

Los politéricos han divulgado un método para unir un par de grupos sulfhidrilo derivados de la reducción de un enlace nativo de disulfuro de bisagra. Véase, Badescu y otros, 2014, Bioconjugate Chem. 25:1124-1136. La reacción se representa en el esquema más abajo. Una ventaja de esta metodología es la capacidad de sintetizar los ADC DAR4 homogéneos mediante la reducción total de las IgG (para dar 4 pares de sulfhidrilos) seguido de la reacción con 4 equivalentes del agente alquilante. Los ADC que contienen "disulfuros en puente" se incorporan además para tener una mayor estabilidad.

De manera similar, como se representa más abajo, se ha desarrollado un derivado de maleimida (1, más abajo) que es capaz de unir un par de grupos sulfhidrilo. Véase el documento WO2013/085925.

5.6.2.4. Consideraciones de selección del conector
Como saben los expertos en la técnica, el conector seleccionado para un ADC particular puede estar influenciado por una variedad de factores, que incluyen, pero no se limitan a, el sitio de unión al anticuerpo (por ejemplo, Lys, Cys u otros residuos de aminoácidos), limitaciones del farmacóforo del fármaco y la lipofilicidad del fármaco. El conector específico seleccionado para un ADC debe buscar equilibrar estos factores diferentes para la combinación específica de anticuerpo/fármaco. Para una revisión de los factores que están influenciados por la elección de los conectores en los ADC, véase, Nolting, Capítulo 5 "Linker Technology in Antibody-Drug Conjugates," En: Antibody-Drug Conjugates: Methods in Molecular Biology, vol. 1045, pp. 71-100, Laurent Ducry (Ed.), Springer Science & Business Medica, LLC, 2013.
Por ejemplo, los ADC anti-cMet pueden efectuar la muerte de células tumorales negativas a cMet circunstantes presentes cerca de las células cancerosas que expresan cMet. El mecanismo de muerte celular circunstante por los ADC ha indicado que los productos metabólicos formados durante el procesamiento intracelular de los ADC pueden desempeñar un papel. Los metabolitos citotóxicos y/o citostáticos permeables a las células generados por el metabolismo de los ADC en las células que expresan cMet parecen desempeñar un papel en la muerte celular circunstante, mientras que los metabolitos no permeables a las células, que son incapaces de atravesar la membrana celular y difundirse en el medio no puede afectar a la muerte circunstante. En ciertos aspectos, el conector se selecciona para efectuar, mejorar o aumentar el efecto de muerte circunstante de los ADC anti-cMet.
Las propiedades del conector pueden afectar además la agregación del ADC en condiciones de uso y/o almacenamiento. Típicamente, los ADC informados en la literatura contienen no más de 3-4 moléculas de fármaco por porción de unión al antígeno, por ejemplo, por molécula de anticuerpo (véase, por ejemplo, Chari, 2008, Acc Chem Res 41:98-107). Los intentos de obtener relaciones más altas de fármaco-a-anticuerpo ("DAR") a menudo fallaban, particularmente si tanto el fármaco como el conector eran hidrófobos, debido a la agregación del ADC (King y otros, 2002, J Med Chem 45:4336-4343; Hollander y otros, 2008, Bioconjugate Chem 19:358-361; Burke y otros, 2009 Bioconjugate Chem 20:1242-1250). En muchos casos, los DAR superiores a 3-4 podrían ser beneficiosos como un medio para aumentar la potencia. En los casos en que el agente citotóxico y/o citostático es de naturaleza hidrófoba, puede ser conveniente seleccionar conectores que sean relativamente hidrófilos como un medio para reducir la agregación del ADC, especialmente en los casos donde se desean DARS mayores de 3-4. Así, en ciertos
aspectos, el conector incorpora porciones químicas que reducen la agregación de los ADC durante el almacenamiento y/o uso. Un conector puede incorporar grupos polares o hidrófilos, tales como grupos cargados o grupos que se cargan en pH fisiológico para reducir la agregación de los ADC. Por ejemplo, un conector puede incorporar grupos cargados tales como sales o grupos que desprotonan, por ejemplo, carboxilatos, o protonan, por ejemplo, aminas, a pH fisiológico.
Los conectores polivalentes ilustrativos que se ha informado que producen DAR tan altos como 20 que pueden usarse para unir numerosos agentes citotóxicos y/o citostáticos a un anticuerpo se describen en los documentos WO 2009/073445; WO 2010/068795; WO 2010/138719; WO 2011/120053; WO 2011/171020; WO 2013/096901; WO 2014/008375; WO 2014/093379; WO 2014/093394; WO 2014/093640.
En aspectos particulares, la agregación de los ADC durante el almacenamiento o uso es inferior a aproximadamente el 10 % según se determina por cromatografía de exclusión por tamaño (SEC). En aspectos particulares, la agregación de los ADC durante el almacenamiento o uso es inferior a 10 %, tal como inferior a aproximadamente el 5 %, inferior a aproximadamente el 4 %, inferior a aproximadamente el 3 %, inferior a aproximadamente el 2 %, inferior a aproximadamente el 1 %, inferior a aproximadamente el 0,5 %, inferior a aproximadamente el 0,1 %, o incluso más bajo, según se determina por cromatografía de exclusión por tamaño (SEC).
5.6.3. ABBV-399
Como se describe a lo largo de la descripción, ABBV-399 es un ADC compuesto por el anticuerpo dirigido a cMet ABT-700 (PR-1266688, h224G11) conjugado con la potente citotoxina monometil auristatina E (MMAE) a través de un conector de valina citrulina (vc). ABBV-399 se ha usado en un ensayo clínico de fase 1 (véase, ejemplo 16) con un DAR de 3,1.
En modalidades alternativas, el ABBV-399 puede usarse en una relación E2/E4 1:1, que corresponde a un DAR promedio de 3,0. En otras palabras, el ABBV-399 se usa como una composición que comprende una relación 1:1 de las fracciones purificadas E2 y E4 del conjugado de fármaco con anticuerpo.
5.6.4. ABT-700 PBD
ABT-700 (S238C)-PBD (numeración de Kabat) es lo mismo que ABT-700 (S239C)-PBD (numeración de Eu) y se compone de dos moléculas de fármaco PBD-conector conjugadas por cys con un mAb ABT-700 diseñado. El proceso de conjugación consiste en una reducción cuantitativa de los disulfuros diseñados genéticamente e intercatenarios. La mezcla de reducción luego se purifica para eliminar el exceso de reactivo y sus subproductos, seguido de la oxidación cuantitativa de los disulfuros intercatenarios y después la conjugación con un exceso de fármaco PBD-conector. Después de inactivar, la mezcla de reacción se purifica y se intercambia con tampón para producir ABT-700 (S238C)-PBD. Se han identificado parámetros de reacción para proporcionar un conjugado con >80 % de carga de fármaco DAR2.
La secuencia de ABT-700 PBD, que lleva una mutación S238C (numeración Kabat) (equivalente a la mutación S239C en la numeración Eu), es la siguiente (las CDR están subrayadas; el sistema de numeración es Kabat; y la mutación S238C está representada por C (negrita, subrayada y cursiva):
Secuencia de aminoácidos (10 AA por grupo, 5 grupos por línea)
Cadena pesada (SEQ ID NO: 171) (las secuencias de las CDR subrayadas divulgadas como las SEQ ID NOS 173-175, respectivamente, en orden de aparición):
QVQLVQSGAE VKKPGASVKV SCKASGYIFT AYTMHWYRQA PGQGLEWMGW 50
IKPNNGLANY AQKFQGRVTM TRDTSISTAY MELSRLRSDD TAVYYCARSE 100
ITGEFDYWGQ GTLVTVSSAS TKGPSVFPLA PSSKSTSGGT AALGCLVKDY 150
FPEPVTVSWN SGALTSGVHT FPAVLQSSGL Y S L SSW T V P SSSLGTQTYI 200
CNVNHKPSNT KVDKRVEPKS CDCHCPPCPA PELLGGPCVF LFPPKPKDTL 250
MISRTPEVTC WVDVSHEDP EVKFNWYVDG VEVHNAKTKP REEQYNSTYR 300
WSVLTVLHQ DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL 350
PPSREEMTKN QVSLTCLVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD 400
GSFFLYSKLT VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPG 445
Cadena ligera (SEQ ID NO: 172) (las secuencias de las CDR subrayadas divulgadas como SEQ ID NOS 176 178, respectivamente, en orden de aparición):
DIVMTQSPDS LAVSLGERAT IN CKSSESVD SYAHSFLHMY QQKPGQPPKL 50
LIYRASTRES GVPDRFSGSG SGTDFTLTIS SLQAEDVAVY YCQQSKEDPL 100
TFGGGTKVEI KRTVAAPSVF IFPPSDEQLK SGTASW CLL NNFYPREAKV 150
QWKVDNALQS GNSQESVTEQ DSKDSTYSLS STLTLSKADY EKHKVYACEV 200
THQGLSSPVT KSFNRGEC 218
5.7. Métodos de fabricación de conjugados de fármacos con anticuerpos anti-cMet
Los ADC descritos en la presente descripción pueden sintetizarse mediante el uso de productos químicos que son bien conocidos. Los productos químicos seleccionados dependerán, entre otras cosas, de la identidad del agente o agentes citotóxicos y/o citostáticos, el conector y los grupos usados para unir el conector al anticuerpo. Generalmente, los ADC de acuerdo con la fórmula (I) pueden prepararse de acuerdo con el siguiente esquema:

donde D, L, Ab, XY y n son como se definen anteriormente, y R Rx y Ry representan grupos complementarios capaces de formar uniones covalentes uno con el otro, como se discutió anteriormente.
Las identidades de los grupos Rx y Ry dependerán del producto químico usado para unir el sintón conector D-L-Rx al anticuerpo. Generalmente, el producto químico usado no debería alterar la integridad del anticuerpo, por ejemplo, su capacidad para unirse a su diana. Preferentemente, las propiedades de unión del anticuerpo conjugado se parecerán mucho a las del anticuerpo no conjugado. Una variedad de productos químicos y técnicas para conjugar moléculas a moléculas biológicas tales como anticuerpos se conocen bien en la técnica y, en particular, a anticuerpos. Véase, por ejemplo, Amon y otros, "Monoclonal Antibodies For Immunotargeting of Drugs in Cancer Therapy," en: Monoclonal Antibodies And Cancer Therapy, Reisfeld y otros Eds., Alan R. Liss, Inc., 1985; Hellstrom y otros, "Antibodies For Drug Delivery," en: Controlled Drug Delivery, Robinson y otros Eds., Marcel Dekker, Inc., 2da Ed. 1987; Thorpe, "Antibody Carriers Of Cytotoxic Agents In Cancer Therapy: A Review," en: Monoclonal Antibodies '84: Biological And Clinical Applications, Pinchera y otros,Eds., 1985; "Analysis, Results, and Future Prospective of the Therapeutic Use of Radiolabeled Antibody In Cancer Therapy," en: Monoclonal Antibodies For Cancer Detection And Therapy, Baldwiny otros, Eds., Academic Press, 1985; Thorpe y otros, 1982, Immunol. Rev. 62:119-58; Publicación de PCT WO 89/12624. Cualquiera de estos productos químicos puede usarse para unir los sintones a un anticuerpo.
Un número de grupos funcionales Rx y productos químicos útiles para unir sintones a residuos de lisina accesibles se conocen e incluyen a modo de ejemplo y sin limitación, ésteres de NHS e isotiocianatos.
Un número de grupos funcionales Rx y productos químicos útiles para unir sintones a grupos sulfhidrilos libres accesibles de residuos de cisteína se conocen e incluyen a modo de ejemplo y sin limitación, haloacetilos y maleimidas.
Sin embargo, los productos químicos de conjugación no se limitan a los grupos de cadenas laterales disponibles. Las cadenas laterales tales como las aminas pueden convertirse en otros grupos útiles, tales como los hidroxilos, al unir una molécula pequeña apropiada a la amina. Esta estrategia puede usarse para aumentar el número de sitios de enlace disponibles en el anticuerpo mediante la conjugación de pequeñas moléculas multifuncionales con cadenas laterales de residuos de aminoácidos accesibles del anticuerpo. Los grupos funcionales Rx adecuados para unir covalentemente los sintones a estos grupos funcionales "convertidos" se incluyen después en los sintones. Un anticuerpo puede diseñarse además para incluir residuos de aminoácidos para la conjugación. Un enfoque para diseñar anticuerpos que incluyen residuos de aminoácidos no codificados genéticamente útiles para conjugar fármacos en el contexto de los ADC se describe por Axup y otros, 2012, Proc Natl Acad Sci EE.UU. 109(40):16101-16106, como lo son los productos químicos y los grupos funcionales útiles para unir los sintones a los aminoácidos no codificados.
Típicamente, los sintones están unidos a las cadenas laterales de residuos de aminoácidos del anticuerpo, que incluyen, por ejemplo, el grupo amino primario de residuos de lisina accesibles o el grupo sulfhidrilo de residuos de cisteína accesibles. Pueden obtenerse grupos sulfhidrilo libres mediante reducción de los enlaces disulfuro intercatenarios.
Para enlaces donde Ry es un grupo sulfhidrilo (por ejemplo, cuando Rx es una maleimida), el anticuerpo es generalmente primero reducido total o parcialmente para interrumpir puentes disulfuro intercatenarios entre residuos de cisteína. Los residuos de cisteína específicos y los puentes disulfuro intercatenarios que pueden reducirse para la unión de los sintones fármaco-conector que incluyen un grupo adecuado para la conjugación a un grupo sulfhidrilo para el anticuerpo ilustrativo ABT-700, incluyen a modo de ejemplo y sin limitación, los residuos C221, C223, C225 y C228 en la cadena pesada de IgG1 humana, y residuo C218 sobre la cadena ligera de Ig kappa humana del ABT
700 divulgado en la presente descripción.
Los residuos de cisteína para la unión del sintón que no participan en puentes disulfuro pueden diseñarse en un anticuerpo mediante la mutación de uno o más codones. Estas cisteínas no apareadas proporcionan un grupo sulfhidrilo adecuado para la conjugación. Las posiciones preferidas para incorporar cisteínas diseñadas incluyen, a modo de ejemplo y sin limitación, las posiciones S112C, S113C, A114C, S115C, A176C, S180C, S252C, V286C, V292C, S357C, A359C, S398C, S428C (numeración de Kabat) en la cadena pesada de IgG1 humana y las posiciones V110C, S114C, S121C, S127C, S168C, V205C (numeración de Kabat) sobre la cadena ligera de Ig kappa humana (véase, por ejemplo, la patente de Estados Unidos núm. 7,521,541, la patente de Estados Unidos núm. 7,855,275 y la patente de Estados Unidos núm. 8,455,622).
Como apreciarán los expertos en la técnica, el número de agentes citotóxicos y/o citostáticos unidos a una molécula de anticuerpo puede variar, de modo que una preparación de ADC puede ser de naturaleza heterogénea, donde algunos anticuerpos en la preparación contienen un agente unido, algunos dos, algunos tres, etc. (y algunos ninguno). El grado de heterogeneidad dependerá, entre otras cosas, de los productos químicos usados para unir los agentes citotóxicos y/o citostáticos. Por ejemplo, cuando los anticuerpos se reducen para producir grupos sulfhidrilo para la unión, a menudo se producen mezclas heterogéneas de anticuerpos que tienen cero, 2, 4, 6 u 8 agentes unidos por molécula. Además, al limitar la relación molar del compuesto de unión, a menudo se producen anticuerpos que tienen cero, 1, 2, 3, 4, 5, 6, 7 u 8 agentes unidos por molécula. Por lo tanto, se entenderá que, en dependencia del contexto, las relaciones fármaco-a-anticuerpo (DAR) establecidas pueden ser promedios para una colección de anticuerpos. Por ejemplo, "DAR4" se refiere a una preparación de ADC que no se ha sometido a purificación para aislar picos de DAR específicos y comprende una mezcla heterogénea de moléculas de ADC que tienen diferentes números de agentes citostáticos y/o citotóxicos unidos por anticuerpo (por ejemplo, 0, 2, 4, 6, 8 agentes por anticuerpo), pero tiene una relación promedio de fármaco-a-anticuerpo de 4. De forma similar, "DAR8" se refiere a una preparación de ADC heterogénea en la que la relación promedio de fármaco-a-anticuerpo es 8.
Las preparaciones heterogéneas de ADC pueden procesarse, por ejemplo, mediante cromatografía de interacción hidrófoba ("HIC") para producir preparaciones enriquecidas en un ADC que tenga un DAR de interés especificado (o una mezcla de dos o más DARS especificados). Dichas preparaciones enriquecidas se diseñan en la presente descripción como "EX," donde "E" indica que la preparación de ADC se ha procesado y está enriquecida en un ADC que tiene un DAR específico y "X" representa el número de agentes citostáticos y/o citotóxicos unidos por molécula de ADC. Las preparaciones enriquecidas en una mezcla de ADC que tienen dos DAR específicos se denominan "EX/EY," tres DAR específicos "EX/EY/EZ" etc., donde "E" indica que la preparación de ADC se ha procesado para enriquecer los DAR especificados y "X," "Y" y "Z" representan los DAR enriquecidos. Como ejemplos específicos, "E2" se refiere a una preparación de ADC que se ha enriquecido para contener principalmente ADC que tienen dos agentes citostáticos y/o citotóxicos unidos por molécula de ADC. "E4" se refiere a una preparación de ADC que se ha enriquecido para contener principalmente ADC que tienen cuatro agentes citostáticos y/o citotóxicos unidos por molécula de ADC. "E2/E4" se refiere a una preparación de ADC que se ha enriquecido para contener principalmente dos poblaciones de ADC, una que tiene dos agentes citostáticos y/o citotóxicos unidos por molécula de ADC y otra que tiene cuatro agentes citostáticos y/o citotóxicos unidos por molécula de ADC.
Como se usa en la presente descripción, las preparaciones "E" enriquecidas generalmente serán al menos aproximadamente 80 % puras en los ADC DAR indicados, aunque niveles más altos de pureza, tales como purezas de al menos aproximadamente 85 %, 90 %, 95 %, 98 %, o incluso más alto, pueden ser obtenibles y convenientes. Por ejemplo, una preparación "EX" generalmente será al menos aproximadamente 80 % pura en ADC que tienen agentes citostáticos y/o citotóxicos X unidos por molécula de ADC. Para preparaciones enriquecidas de "orden superior", tales como, por ejemplo, preparaciones "EX/EY", la suma total de ADC que tienen agentes citostáticos y/o citotóxicos X e Y unidos por molécula de ADC generalmente comprenderán al menos aproximadamente 80 % de los ADC totales en la preparación. De manera similar, en una preparación enriquecida "EX/EY/EZ", la suma total de los ADC que tienen agentes citostáticos y/o citotóxicos X, Y Z unidos por molécula de ADC comprenderá al menos aproximadamente 80 % del total de ADC en la preparación.
La pureza puede evaluarse mediante una variedad de métodos, como se conoce en la técnica. Como ejemplo específico, una preparación de ADC puede analizarse mediante HPLC u otra cromatografía y la pureza puede evaluarse al analizar áreas bajo las curvas de los picos resultantes. Los métodos de cromatografía específicos que pueden emplearse para evaluar la pureza de las preparaciones de ADC se proporcionan en el ejemplo 6.
La figura 2 ilustra el Proceso I, que se usa para obtener un DAR de 3,1. La figura 3 ilustra el Proceso II, que se usó para obtener una relación E2/E41:1.
En la sección de ejemplos se proporcionan métodos específicos para obtener mezclas heterogéneas de ADC que comprenden el anticuerpo humanizado huM25 que tiene un DAR promedio de 4, así como también, preparaciones altamente purificadas que contienen 2 y 4 agentes unidos. Estos métodos específicos pueden modificarse mediante el uso de habilidades rutinarias para obtener ADC heterogéneos y/u homogéneos que comprenden otros anticuerpos anti-cMet, conectores y/o agentes citotóxicos y/o citostáticos.
Después de la conjugación de vcMMAE con ABT-700, se usa una etapa de proceso adicional para reducir la relación promedio de fármaco-a-anticuerpo (DAR) de aproximadamente 5 a aproximadamente 3, lo que resulta en un producto farmacológico más homogéneo con menos moléculas de MMAE conjugadas con el anticuerpo. Esta estrategia se implementó para reducir el número de moléculas de fármacos unidas al ABBV-399, lo que puede mejorar su tolerabilidad, ya que las moléculas de fármacos de alto orden pueden contribuir de manera desproporcionada a la toxicidad.
5.8. Composiciones
Los ADC descritos en la presente descripción pueden estar en forma de composiciones que comprenden el ADC y uno o más portadores, excipientes y/o diluyentes. Las composiciones pueden formularse para usos específicos, tales como para usos veterinarios o usos farmacéuticos en humanos. La forma de la composición (por ejemplo, polvo seco, formulación líquida, etc.) y los excipientes, diluyentes y/o portadores usados dependerán de los usos previstos del anticuerpo y/o ADC y, para usos terapéuticos, el modo de administración.
Para usos terapéuticos, las composiciones pueden suministrarse como parte de una composición farmacéutica estéril que incluye un portador farmacéuticamente aceptable. Esta composición puede estar en cualquier forma adecuada (en dependencia del método deseado de administrar a un paciente). La composición farmacéutica puede administrarse a un paciente por una variedad de rutas tales como oral, transdérmica, subcutánea, intranasal, intravenosa, intramuscular, intratumoral, intratecal, tópica o local. La ruta más adecuada para la administración en cualquier caso dependerá del anticuerpo particular y/o ADC, el sujeto, y la naturaleza y gravedad de la enfermedad y la condición física del sujeto. Típicamente, la composición farmacéutica se administrará por vía intravenosa o subcutánea.
Las composiciones farmacéuticas pueden presentarse convenientemente en formas de dosificación unitarias que contienen una cantidad predeterminada de un anticuerpo y/o ADC descritos en la presente descripción por dosis. La cantidad de anticuerpo y/o ADC incluida en una dosis unitaria dependerá de la enfermedad que se trate, así como también de otros factores que son bien conocidos en la técnica. Dichas dosis unitarias pueden estar en forma de un polvo seco liofilizado que contiene una cantidad de anticuerpo y/o ADC adecuada para una sola administración, o en forma de un líquido. Las formas de dosificación unitaria de polvo seco pueden empaquetarse en un estuche con una jeringa, una cantidad adecuada de diluyente y/u otros componentes útiles para la administración. Las dosis unitarias en forma líquida pueden suministrarse convenientemente en forma de una jeringa precargada con una cantidad de anticuerpo y/o ADC adecuada para una única administración.
Las composiciones farmacéuticas también pueden suministrarse en forma masiva que contiene cantidades de ADC adecuadas para múltiples administraciones.
Las composiciones farmacéuticas pueden prepararse para el almacenamiento como formulaciones liofilizadas o soluciones acuosas al mezclar un anticuerpo y/o ADC que tenga el grado deseado de pureza con portadores, excipientes o estabilizadores farmacéuticamente aceptables típicamente empleados en la técnica (todos los cuales se mencionan en la presente descripción como "portadores"), es decir, agentes tamponantes, agentes estabilizantes, conservantes, isotonificadores, detergentes no iónicos, antioxidantes y otros aditivos misceláneos. Véase, Remington's Pharmaceutical Sciences, 16ta edición (Osol, ed. 1980) y Remington: The Science and Practice of Pharmacy, 22da edición (Editado por Allen, Loyd V. Jr., 2012). Dichos aditivos no deben ser tóxicos para los receptores a las dosis y concentraciones empleadas.
Los agentes tamponantes ayudan a mantener el pH en el intervalo que estabiliza la proteína. Pueden estar presentes en una amplia variedad de concentraciones, pero típicamente estarán presentes en concentraciones que varían de aproximadamente 2 mM a aproximadamente 50 mM. Los agentes tamponantes adecuados para usar con la presente divulgación incluyen ácidos orgánicos e inorgánicos y sales de los mismos tales como tampones de citrato (por ejemplo, mezcla de citrato monosódico-citrato disódico, mezcla de ácido cítrico-citrato trisódico, mezcla de ácido cítrico-citrato monosódico, etc.), tampones de succinato (por ejemplo, mezcla de ácido succínico-succinato monosódico, mezcla de ácido succínico-hidróxido de sodio, mezcla de ácido succínico-succinato disódico, etc.), tampones de tartrato (por ejemplo, mezcla de ácido tartárico-tartrato de sodio, mezcla de ácido tartárico-tartrato de potasio, mezcla de ácido tartárico-hidróxido de sodio, etc.), tampones de fumarato (por ejemplo, mezcla de ácido fumárico-fumarato monosódico, mezcla de ácido fumárico-fumarato disódico, mezcla de fumarato monosódicofumarato disódico, etc.), tampones de gluconato (por ejemplo, mezcla de ácido glucónico-gluconato de sodio, mezcla de ácido glucónico-hidróxido de sodio, mezcla de ácido glucónico-gluconato de potasio, etc.), tampón de oxalato (por ejemplo, mezcla de ácido oxálico-oxalato de sodio, mezcla de ácido oxálico-hidróxido de sodio, mezcla de ácido oxálico-oxalato de potasio, etc.), tampones de lactato (por ejemplo, mezcla de ácido láctico-lactato de sodio, mezcla de ácido láctico-hidróxido de sodio, mezcla de ácido láctico-lactato de potasio, etc.) y tampones de acetato (por ejemplo, mezcla de ácido acético-acetato de sodio, mezcla de ácido acético-hidróxido de sodio, etc.). Además, pueden usarse tampones de fosfato, tampones de histidina y sales de trimetilamina tales como Tris.
Pueden añadirse conservantes para retardar el crecimiento microbiano, y pueden añadirse en cantidades que varían de aproximadamente 0,2 %-1 % (p/v). Los conservantes adecuados para usar con la presente divulgación incluyen
fenol, alcohol bencílico, meta-cresol, metil parabeno, propil parabeno, cloruro de octadecildimetilbencilamonio, haluros de benzalconio (por ejemplo, cloruro, bromuro y yoduro), cloruro de hexametonio y alquil parabenos tales como metil o propil parabeno, catecol, resorcinol, ciclohexanol y 3-pentanol. Pueden añadirse isotonicificadores a veces conocidos como "estabilizadores" para asegurar la isotonicidad de las composiciones líquidas de la presente divulgación e incluyen alcoholes de azúcar polihídricos, por ejemplo, alcoholes de azúcar trihídricos o superiores, tales como glicerina, eritritol, arabitol, xilitol, sorbitol y manitol. Los estabilizantes se refieren a una amplia categoría de excipientes que pueden variar en función de un agente de carga a un aditivo que solubiliza el agente terapéutico o ayuda a prevenir la desnaturalización o la adherencia a la pared del recipiente. Los estabilizadores típicos pueden ser alcoholes de azúcar polihídricos (enumerados anteriormente); aminoácidos tales como arginina, lisina, glicina, glutamina, asparagina, histidina, alanina, ornitina, L-leucina, 2-fenilalanina, ácido glutámico, treonina, etc., azúcares orgánicos o alcoholes de azúcar, tales como lactosa, trehalosa, estaquiosa, manitol, sorbitol, xilitol, ribitol, mioinositol, galactitol, glicerol y similares, incluidos ciclitoles tales como inositol; polietilenglicol; polímeros de aminoácidos; agentes reductores que contienen azufre, tales como urea, glutatión, ácido tióctico, tioglicolato de sodio, tioglicerol, a-monotioglicerol y tiosulfato de sodio; polipéptidos de bajo peso molecular (por ejemplo, péptidos de 10 residuos o menos); proteínas tales como albúmina de suero humano, albúmina de suero bovino, gelatina o inmunoglobulinas; polímeros hidrófilos, tales como polivinilpirrolidona, monosacáridos, tales como xilosa, manosa, fructosa, glucosa; disacáridos tales como lactosa, maltosa, sacarosa y trehalosa; y trisacáridos tales como rafinosa; y polisacáridos tales como dextrano. Los estabilizadores pueden estar presentes en cantidades que varían de 0,5 a 10 % en peso, por peso de ADC.
Pueden añadirse surfactantes o detergentes no iónicos (también conocidos como "agentes humectantes") para reducir la adsorción a las superficies y ayudar a solubilizar la glucoproteína, así como también, para proteger la glucoproteína contra la agregación inducida por la agitación, lo que permite además que la formulación quede expuesta para cortar el estrés superficial sin causar desnaturalización de la proteína. Los tensioactivos no iónicos adecuados incluyen polisorbatos (20, 80, etc.), poloxámeros (184, 188 etc.) y polioles plurónicos. Los surfactantes no iónicos pueden estar presentes en un intervalo de aproximadamente 0,05 mg/mL a aproximadamente 1,0 mg/mL, por ejemplo, aproximadamente 0,07 mg/mL a aproximadamente 0,2 mg/mL.
Los excipientes misceláneos adicionales incluyen agentes de carga (por ejemplo, almidón), agentes quelantes (por ejemplo, EDTA), antioxidantes (por ejemplo, ácido ascórbico, metionina, vitamina E) y codisolventes.
Las composiciones acuosas ilustrativas adecuadas para la administración mediante infusión intravenosa comprenden 20 mg/mL de ADC anti-cMet, tampón de histidina 10 mM, pH 6,0, sacarosa al 7 % (p/v), polisorbato 80 al 0,03 % (p/v). La composición puede estar en forma de un polvo liofilizado que, tras la reconstitución con 5,2 mL de agua estéril u otra solución adecuada para inyección o infusión (por ejemplo, solución salina al 0,9 %, solución de Ringer, solución de Ringer lactato, etc.) proporciona la composición acuosa anterior. Esta, u otras modalidades de las composiciones, también pueden estar en forma de una jeringa u otro dispositivo adecuado para inyección y/o infusión precargado con una cantidad de composición adecuada para una sola administración de ADC anti-cMet. En un aspecto, la composición comprende ABBV-399 en una relación 1:1 de fracciones E1 y E4 purificadas. Dichas fracciones pueden obtenerse por cualquier método conocido en la técnica para purificar ADC, que incluyen el método de los ejemplos 2 y 3. En un aspecto, la composición comprende ABBV-399 con un DAR en el intervalo de 0-10. En otro aspecto, la composición comprende ABBV-399 con un DAR en el intervalo de 1-4. En otro aspecto, la composición comprende ABBV-399 con un DAR en el intervalo de 2-4. En otro aspecto, la composición comprende ABBV-399 con un DAR de aproximadamente 3,1. En otro aspecto, la composición comprende ABBV-399 con un DAR de aproximadamente 3,0.
5.9. Métodos de uso
Como se discutió anteriormente, para una variedad de tumores sólidos, cMet se expresa/sobreexpresa. Los datos proporcionados en la presente descripción demuestran que los ADC anti-cMet ejercen una potente actividad antitumoral contra estos tumores que expresan/sobreexpresan cMet in vivo. Por consiguiente, los ADC y/o composiciones farmacéuticas que comprenden los ADC pueden usarse terapéuticamente para tratar tumores que expresan cMet (es decir, tumores cMet+) y que sobreexpresan cMet (es decir, tumores cMet+/que sobreexpresan). Generalmente, los métodos implican administrar a un paciente humano que tiene un tumor que expresa cMet o que sobreexpresa cMet, una cantidad de ADC anti-cMet eficaz para proporcionar un beneficio terapéutico. Puede usarse cualquier método conocido por un experto en la técnica para evaluar la presencia y/o el nivel de expresión de la proteína del receptor cMet en una célula. En una modalidad, los niveles de cMet son membranosos. En otra modalidad, los niveles de cMet son citoplasmáticos. En otra modalidad, se mide el nivel de expresión global de cMet. Un método preferido para determinar los niveles de expresión de cMet se describe en detalle en el ejemplo 17 y se denomina en la presente descripción como el "protocolo de tinción cMet ABBV-ADC." Las puntuaciones H (0-300) y la puntuación IHC (0, 1+, 2+ y 3+) se evalúan en base a métodos conocidos por un patólogo experto en la técnica. En una modalidad, los pacientes con puntuaciones H <150 y/o puntuaciones IHC 0 y 1+ se seleccionan para el tratamiento. En una modalidad, los pacientes con puntuaciones H S150 y/o puntuaciones IHC 2+ y 3+ se seleccionan para el tratamiento.
Los pacientes seleccionados para los tratamientos de ADC de esta divulgación incluyen aquellos con tumores que expresan cMet y aquellos que sobreexpresan cMet, que incluyen, pero no se limitan a, cualquier tumor sólido (incluidos también aquellos que sobreexpresan HGF y/o tienen activación anormal de señalización o expresión de HGF/cMet). Los ejemplos más específicos incluyen: cánceres de pulmón; cánceres de mama (por ejemplo, carcinoma ductal invasivo); cánceres de cabeza y cuello; cánceres de páncreas; carcinomas gástricos; cánceres colorrectales (incluidas metástasis pulmonares de cáncer colorrectal); cánceres de ovario (por ejemplo, adenocarcinoma seroso); cánceres de estómago; cánceres de riñón (por ejemplo, cáncer de células renales tales como carcinoma de células renales papilares, cánceres de células claras, carcinomas hereditarios de células renales papilares); cánceres suprarrenales; cánceres gastroesofágicos; meduloblastomas; gliomas; cánceres de hígado (por ejemplo, carcinomas hepatocelulares (incluido HCC avanzado, irresecable)); cáncer de próstata (metastásico o no metastásico); melanomas; tumores de las glándulas salivales; sarcomas; cánceres de cuello uterino; mixoidliposarcomas; adenocarcinomas de la glándula paratiroidea; cánceres de endometrio; mesoteliomas epitelioides; carcinomas de apéndice; carcinomas de células caliciformes; adenocarcinoma gástrico metastásico de tipo difuso con características de anillo de sello; linfoma anaplásico de células grandes (ALCL); cualquier neoplasia maligna avanzada que incluyen, pero no se limitan a, subtipos avanzados, reincidentes y refractarios de los cánceres enumerados en la presente descripción.
El cáncer de pulmón puede clasificarse mediante el uso de diferentes sistemas. En un sistema, el cáncer de pulmón incluye adenocarcinoma (mixto, acinar, papilar, sólido, micropapilar, lepídico no mucinoso y lepídico mucinoso), carcinoma de células escamosas, carcinoma de células grandes (por ejemplo, cáncer de pulmón de células no pequeñas o NSCLC (por ejemplo, avanzado o no avanzado, LCNEC, LCNEM, NSCLC no especificado de cualquier otra manera (NOS)/carcinoma adenoescamoso, carcinoma sarcomatoide, carcinoma adenoescamoso y carcinoma neuroendocrino de células grandes) y cáncer/carcinoma de pulmón de células pequeñas o SCLC)).
Alternativamente, en un sistema diferente, el cáncer de pulmón puede clasificarse en lesiones preinvasivas, adenocarcinoma mínimamente invasivo y adenocarcinoma invasivo (adenocarcinoma mucinoso invasivo, BAC mucinoso, coloide, fetal (bajo y alto grado) y entérico).
Con mayor frecuencia, el cáncer de pulmón puede categorizarse como cáncer de pulmón de células pequeñas ("SCLC") o cáncer de pulmón de células no pequeñas ("NSCLC"). Los NSCLC pueden categorizarse además como escamosos o no escamosos. Un ejemplo de NSCLC no escamoso es el adenocarcinoma.
El cáncer puede ser recién diagnosticado y sin tratamiento previo, o puede ser reincidente, refractario, o reincidente y refractario, o una metástasis o forma metastásica de tumores que expresan cMet o que sobreexpresan cMet. Como se demostró en el ejemplo 14 de esta divulgación, los tumores que sobreexpresan cMet que exhiben resistencia a otras quimioterapias dirigidas o no dirigidas, retienen sensibilidad al ABBV-399.
Además, como se muestra en la figura 12C, un tumor que sobreexpresa cMet que eventualmente creció de nuevo después del tratamiento con el anticuerpo anti-cMet ABT-700 permaneció sensible al retratamiento con el ADC anticMet, ABBV-399. Por consiguiente, los ADC anti-cMet descritos en la presente descripción proporcionan beneficios significativos sobre los enfoques actuales dirigidos y no dirigidos hacia el tratamiento de tumores que sobreexpresan cMet.
Los ADC anti-cMet pueden administrarse solos (monoterapia) o complementarios a, o con, otras terapias anticancerígenas y/o agentes anticancerígenos dirigidos o no dirigidos. Cuando se administra como monoterapia de ADC anti-cMet, pueden usarse uno o más ADC anti-cMet. En ciertas modalidades, un ADC anti-cMet se administra junto con un anticuerpo anti-cMet que reconoce un epítopo diferente en cMet que el reconocido por el ADC. Esto podría hacerse, por ejemplo, para estimular la internalización del receptor cMet. Alternativamente, ABT-700 puede administrarse antes que ABBV-399 (u otro ADC anti-cMet) para "bloquear" el cMet endógeno en tejidos normales en un esfuerzo por reducir la posible toxicidad asociada con la actividad de ABBV-399 en tejidos normales.
En otra modalidad, el ADC anti-cMet reconoce dos epítopos no superpuestos diferentes dentro de cMet. Tales ADC, también conocidos como ADC que portan un anticuerpo biparatópico bivalente, pueden tener varias ventajas sobre los anticuerpos monovalentes. Por ejemplo, pueden inducir la agrupación de receptores cMet, lo que a su vez podría promover una internalización robusta, tráfico lisosomal y degradación, lo que mejora así la liberación de la porción de fármaco del ADC en el citoplasma, así como también su disponibilidad para el efecto circunstante.
Ya sea que se administre como monoterapia o complementarios a, o con, otras terapias o agentes, se administra una cantidad de anti-cMet ADC de manera que el régimen de tratamiento general proporcione beneficio terapéutico. Por beneficio terapéutico se entiende que el uso de ADC anti-cMet para tratar el cáncer en un paciente, resulta en cualquier beneficio clínico demostrado en comparación sin terapia (cuando sea apropiado) o con un estándar de atención conocido. El beneficio clínico puede evaluarse mediante cualquier método conocido por un experto en la técnica. En una modalidad, el beneficio clínico se evalúa en base a la tasa de respuesta objetiva (ORR) (determinada mediante el uso de RECIST versión 1.1), la duración de la respuesta (DOR), la supervivencia libre de progresión (PFS) y/o la supervivencia global (OS). En algunas modalidades, una respuesta completa indica beneficio terapéutico. En algunas modalidades, una respuesta parcial indica beneficio terapéutico. En algunas modalidades, la
enfermedad estable indica beneficio terapéutico. En algunas modalidades, un aumento en la supervivencia global indica beneficio terapéutico. En algunas modalidades, el beneficio terapéutico puede constituir una mejora en el tiempo hasta la progresión de la enfermedad y/o una mejora en los síntomas o la calidad de vida. En otras modalidades, el beneficio terapéutico puede no traducirse en un mayor período de control de la enfermedad, sino más bien en una carga de síntomas marcadamente reducida que resulta en una calidad de vida mejorada. Como será evidente para los expertos en la técnica, puede observarse un beneficio terapéutico mediante el uso de los ADC anti-cMet solos (monoterapia) o complementarios a, o con, otras terapias anticancerígenas y/o agentes anticancerígenos dirigidos o no dirigidos. Los métodos preferenciales para evaluar el beneficio terapéutico se describen en detalle en los ejemplos, tal como se usan en un ensayo clínico de fase 1 con ABBV-399.
Típicamente, el beneficio terapéutico se evalúa mediante pruebas clínicas estándar diseñadas para medir la respuesta a un nuevo tratamiento para el cáncer. Para evaluar los beneficios terapéuticos de los ADC anti-cMet descritos en la presente descripción, puede usarse uno o una combinación de las siguientes pruebas: (1) criterios de evaluación de respuesta en tumores sólidos (RECIST) versión 1.1 (para más detalles, véase el ejemplo 16), (2) el estado de rendimiento del Grupo de Oncología Cooperativa del Este (ECOG), (3) criterios de respuesta relacionados con la inmunidad (irRC), (4) enfermedad evaluable mediante la evaluación de antígenos tumorales, (5) escalas de resultados informadas por el paciente validadas, y/o (6) estimaciones de Kaplan-Meier para la supervivencia general y la supervivencia libre de progresión.
La escala ECOG del estado de rendimiento que se muestra en la TABLA 3 se usa para describir el nivel de funcionamiento de un paciente en términos de su capacidad para cuidarse a sí mismo, la actividad diaria y la capacidad física. La escala fue desarrollada por el Grupo de Oncología Cooperativa del Este (ECOG), que ahora forma parte del Grupo de Investigación sobre el Cáncer ECOG-ACRIN, y se publicó en 1982.

La evaluación del cambio en la carga tumoral es una característica importante de la evaluación clínica de las terapéuticas del cáncer. Tanto la contracción del tumor (respuesta objetiva) como el tiempo para el desarrollo de la progresión de la enfermedad son criterios de valoración importantes en los ensayos clínicos sobre el cáncer. Los criterios de respuesta estandarizados, conocidos como RECIST (criterios de evaluación de respuesta en tumores sólidos), se publicaron en 2000. Una actualización (RECIST 1.1) se lanzó en 2009. Los criterios RECIST se usan típicamente en ensayos clínicos donde la respuesta objetiva es el criterio de valoración del estudio primario, así como en los ensayos donde se llevan a cabo evaluaciones de enfermedad estable, progresión tumoral o tiempo de progresión porque estas medidas de resultado se basan en una evaluación de la carga tumoral anatómica y su cambio en el transcurso del ensayo. La TABLA 4 proporciona las definiciones de los criterios de respuesta usados para determinar la respuesta tumoral objetiva a un fármaco del estudio, tales como los ADC anti-cMet descritos en la presente descripción.

Las medidas de resultado secundarias que pueden usarse para determinar el beneficio terapéutico de los ADC anticMet descritos en la presente descripción incluyen: tasa de respuesta objetiva (ORR), supervivencia libre de progresión (PFS), duración de la respuesta general (DOR) y profundidad de la respuesta (DpR). La ORR se define como la proporción de los participantes que logran una respuesta completa (CR) o una respuesta parcial (PR). La PFS se define como el tiempo desde la fecha de la primera dosis de los ADC anti-cMet hasta la progresión de la enfermedad o la muerte, lo que ocurra primero. La DOR se define como el tiempo desde la CR o PR inicial del participante hasta el momento de la progresión de la enfermedad. La DpR se define como el porcentaje de encogimiento del tumor observado en el punto de respuesta máxima en comparación con la carga tumoral inicial. Los criterios de valoración clínicos para ORR y PFS pueden determinarse en base a los criterios RECIST 1.1 descritos anteriormente.
Otro conjunto de criterios que pueden usarse para caracterizar completamente y determinar la respuesta a los agentes inmunoterapéuticos, tales como las terapias contra el cáncer basadas en anticuerpos, es el criterio de respuesta relacionado con la inmunidad (irRC), que se desarrolló para la medición de tumores sólidos en 2009, y actualizado en 2013 (Wolchok, y otros Clin. Cancer Res. 2009; 15(23): 7412-7420 y Nishino, y otros Clin. Cancer Res. 2013; 19(14):3936-3943). Los criterios de irRC actualizados se usan típicamente para evaluar el efecto de un agente inmunoterapéutico (por ejemplo, un anticuerpo anti-PD1) y define la respuesta de acuerdo con la TABLA 5.

Los antígenos tumorales que pueden usarse para evaluar el beneficio terapéutico de los ADC anti-cMet descritos en la presente descripción incluyen ApoE, CD11c, CD40, CD45 (PTPRC), CD49D (ITGA4), CD80, CSF1R, CTSD, GZMB, Ly86, MS4A7, PIK3AP1, PIK3CD, CD74, CCL5, CCR5, CXCL10, IFNG, IL10RA1, IL-6, ACTA2, COL7A1, LOX, LRRC15, MCPT8, MMP10, NOG, SERPINE1, STAT1, TGFBR1, CTSS, PGF, VEGFA, C1QA, C1QB, ANGPTL4, EGLN, ANGPTL4, EGLN3, BNIP3, AIF1, CCL5, CXCL10, CXCL11, IFI6, PLOD2, KISS1R, STC2, DDIT4, PFKFB3, PGK1, PDK1, AKR1C1, AKR1C2, CADM1, CDH11, COL6A3, CTGF, HMOX1, KRT33A, LUM, WNT5A, IGFBP3, MMP14, CDCP1, PDGFRA, TCF4, TGF, TGFB1, TGFB2, CD11b, ADGRE1 (EMR1, F4/80), CD86, CD68, MHC-Class II, CD3, HLA-DR, CD4, CD3, CD5, CD19, CD7, CD8, CD16, TCRap, TCRyS, PD-1, PDL-1, CTLA-4, fosfatasa ácida, ACTH, fosfatasa alcalina, alfa-fetoproteína CA-125, CA15-3, CA19-9, CA-195, C-212, CA-549, calcitonina, catecolaminas, catepsina-D, CEA, ERBB2 (HER2/neu), cromagranina-A, c-Myc, EGFR, ERA (ensayo de receptor de estrógenos), ferritina, gastrina, 5-HIAA, hCG, alfa-HCG, beta-HCG, HVA, LDH1-5, NSE (enolasa específica de neuronas), polipéptido pancreático, PLAP, PLP, PRA (receptor de progesterona A), péptido de
proinsulina C, PSA, SMA, SCC, tiroglobulina, TDT, TPA y alfa-TSH. Estos antígenos pueden evaluarse a nivel de ADN, ARN o proteína mediante el uso de técnicas de secuenciación de ADN, técnicas de secuenciación de ARN, microarreglos de chips genéticos, métodos basados en PCR, citometría de flujo o métodos de inmunohistoquímica como los expertos en la técnica conocen.
Un beneficio terapéutico ilustrativo que resulta del uso de los ADC anti-cMet descritos en la presente descripción para tratar tumores que expresan cMet y que sobreexpresan cMet, ya sea administrados como monoterapia o complementarios a, o con, otras terapias o agentes, es una respuesta completa. Otro beneficio terapéutico ilustrativo que resulta del uso de ADC anti-cMet descritos en la presente descripción para tumores que expresan cMet y que sobreexpresan cMet, ya sea administrados como monoterapia o complementarios a, o con, otras terapias o agentes, es una respuesta parcial.
Las escalas de resultados informadas por el paciente validadas pueden usarse además para denotar la respuesta proporcionada por cada paciente a través de un sistema de informe específico. En lugar de centrarse en la enfermedad, tales escalas de resultados se refieren a la función retenida mientras se maneja una condición crónica. Un ejemplo no limitante de una escala de resultados informada por el paciente validada es PROMIS® (sistema de información de medición de resultados informados por el paciente) de los Institutos Nacionales de Salud de Estados Unidos. Por ejemplo, el instrumento de función física PROMIS® para pacientes adultos con cáncer puede evaluar las capacidades autorreportadas para el funcionamiento de las extremidades superiores (por ejemplo, la destreza), las extremidades inferiores (por ejemplo, caminar o movilidad) y las regiones centrales (por ejemplo, la movilidad del cuello, de la espalda), y también incluye actividades diarias de rutina, tales como hacer mandados.
Las curvas de Kaplan-Meier (Kaplan y Meier, J. Am. Stat. Assoc. 1958; 53(282): 457-481) pueden usarse además para estimar la supervivencia general y la supervivencia libre de progresión para pacientes con cáncer sometidos a terapia con anticuerpos anti-cMet o ADC en comparación con el estándar de atención.
5.9.1. Terapias complementarias
Los ADC anti-cMet pueden usarse complementarios a, o con, otros agentes o tratamientos con propiedades anticancerígenas. Cuando se usa de forma complementaria, el anti-cMet y otros agentes pueden formularse juntos en una sola formulación farmacéutica, o pueden formularse y administrarse por separado, ya sea en un único régimen de dosificación coordinada o en diferentes regímenes de dosificación. Los agentes administrados de forma complementaria con ADC anti-cMet típicamente tendrán actividades complementarias a los ADC anti-cMet de tal manera que los ADC y otros agentes no se afecten negativamente entre sí.
Los agentes que pueden usarse de forma complementaria con los ADC anti-cMet incluyen, pero no se limitan a, agentes alquilantes, inhibidores de la angiogénesis, anticuerpos, antimetabolitos, antimitóticos, antiproliferativos, antivirales, inhibidores de la quinasa Aurora, inhibidores de la quinasa ALK (por ejemplo, crizotinib (XALKORI®), ceritinib (ZYKADIA®) y alectinib (ALECENSA®), promotores de la apoptosis (por ejemplo, inhibidores de la familia Bcl-2), activadores de la vía del receptor de muerte, inhibidores de la quinasa Bcr-Abl, anticuerpos BiTE (acopladores biespecíficos de células T), conjugados de fármacos con anticuerpos, modificadores de la respuesta biológica, inhibidores de la quinasa dependiente de ciclina, inhibidores del ciclo celular, inhibidores de la ciclooxigenasa-2, DVD, inhibidores del receptor homólogo del oncogén viral de leucemia (ErbB2), inhibidores del factor de crecimiento, inhibidores de la proteína de choque térmico (HSP) -90, inhibidores de histona deacetilasa (HDAC), terapias hormonales, inmunológicas, inhibidores de inhibidores de proteínas de apoptosis (IAP), antibióticos intercalantes, inhibidores de quinasas, inhibidores de cinesina, inhibidor de Jak2, inhibidores de rapamicina diana de mamíferos, microARN, inhibidores de quinasa regulada por señales extracelulares activados por mitógenos, proteínas de unión multivalentes, fármacos antiinflamatorios no esteroideos (NSAID), inhibidores de poli ADP (difosfato de adenosina)-ribosa polimerasa (PARP), quimioterapéuticos de platino, inhibidores de la quinasa tipo polo (Plk), inhibidores de la fosfoinositida-3 quinasa (PI3K), inhibidores del proteasoma, análogos de purina, análogos de pirimidina, inhibidores de la tirosina quinasa del receptor, alcaloides vegetales de retinoides/deltoides, ácidos ribonucleicos inhibidores pequeños (ARNip), inhibidores de topoisomerasas, inhibidores de ubiquitina ligasa y similares, así como combinaciones de uno o más de estos agentes.
Los anticuerpos BiTE son anticuerpos biespecíficos que dirigen a las células T a atacar las células cancerosas al unir simultáneamente las dos células. La célula T luego ataca a la célula cancerosa diana. Los ejemplos de anticuerpos BiTE incluyen adecatumumab (Micromet MT201), blinatumomab (BLINCYTO®, Amgen and Onyx Pharmaceuticals) y similares. Sin estar limitado por la teoría, uno de los mecanismos por los cuales las células T provocan la apoptosis de la célula cancerosa diana es por la exocitosis de los componentes del gránulo citolítico, que incluyen perforina y granzima B.
Los ARNip son moléculas que tienen bases de ARN endógenas o nucleótidos modificados químicamente. Las modificaciones no eliminan la actividad celular, sino que imparten mayor estabilidad y/o mayor potencia celular. Los ejemplos de modificaciones químicas incluyen grupos fosforotioato, 2’-desoxinucleótidos, ribonucleótidos que contienen 2’-OCH3, 2’-F-ribonucleótidos, 2’-metoxiet¡lo ribonucleótidos, combinaciones de los mismos y similares. El ARNip puede tener diferentes longitudes (por ejemplo, 10-200 pb) y estructuras (por ejemplo, horquillas, cadenas
simples/dobles, protuberancias, mellas/huecos, desapareamientos) y se procesan en las células para proporcionar un silenciamiento génico activo. Un ARNip de doble cadena (ARNdc) puede tener el mismo número de nucleótidos en cada cadena (extremos romos) o extremos asimétricos (salientes). El saliente de 1-2 nucleótidos puede estar presente en la cadena sentido y/o antisentido, así como también presente en los extremos 5’ y/o 3’ de una cadena dada.
Las proteínas de unión multivalentes son proteínas de unión que comprenden dos o más sitios de unión al antígeno. Las proteínas de unión multivalentes están diseñadas para tener los tres o más sitios de unión al antígeno y, en general, no son anticuerpos naturales. El término "proteína de unión multiespecífica" se refiere a una proteína de unión capaz de unir dos o más dianas relacionadas o no relacionadas. Las proteínas de unión de dominio variable dual (DVD) son proteínas de unión tetravalente o multivalente que se unen a proteínas que comprenden dos o más sitios de unión al antígeno. Dichos DVD pueden ser monoespecíficos (es decir, capaces de unirse a un antígeno) o multiespecíficos (es decir, capaces de unirse a dos o más antígenos). Las proteínas de unión a DVD que comprenden dos polipéptidos de DVD de la cadena pesada y dos polipéptidos de DVD de la cadena ligera se denominan Ig de DVD. Cada mitad de una Ig de DVD comprende un polipéptido de DVD de la cadena pesada y un polipéptido de DVD de la cadena ligera y dos sitios de unión a antígeno. Cada sitio de unión comprende un dominio variable de la cadena pesada y un dominio variable de la cadena ligera con un total de 6 CDR involucradas en la unión al antígeno por sitio de unión al antígeno.
Los agentes alquilantes incluyen, pero no se limitan a, altretamina, AMD-473, AP-5280, apaziquona, bendamustina, brostalicina, busulfano, carboquona, carmustina (BCNU), clorambucilo, CLORETAZINE® (laromustina, VNP 40101M), ciclofosfamida, dacarbazina, estramustina, fotemustina, glufosfamida, ifosfamida, KW-2170, lomustina (CCNU), mafosfamida, melfalán, mitobronitol, mitolactol, nimustina, N-óxido de mostaza nitrogenada, ranimustina, temozolomida, tiotepa, TREANDA® (bendamustina), treosulfán y trofosfamida.
Los inhibidores de la angiogénesis incluyen, pero no se limitan a, inhibidores del receptor de tirosina quinasa (Tie-2) específicos del endotelio, inhibidores del receptor del factor de crecimiento epidérmico (EGFR), inhibidores del receptor del factor de crecimiento de insulina-2 (IGFR-2), inhibidores de la metaloproteinasa de matriz-2 (MMP-2), inhibidores de la metaloproteinasa de matriz-9 (MMP-9), inhibidores del receptor del factor de crecimiento derivado de plaquetas (PDGFR), análogos de trombospondina e inhibidores de la tirosina quinasa del receptor del factor de crecimiento endotelial vascular (VEGFR).
Los antimetabolitos incluyen, pero no se limitan a, ALIMTA® (pemetrexed disódico, LY231514, MTA), 5-azacitidina, XELODA® (capecitabina), carmofur, LEUSTAT® (cladribina), clofarabina, citarabina, ocfosfato de citarabina, citosina arabinosina, decitabina, deferoxamina, doxifluridina, eflomitina, EICAR (5-etinil-1 -|3-D-ribofuranosilimidazol-4-carboxamida), enocitabina, etilcitidina, fludarabina, 5-fluorouracilo solo o en combinación con leucovorina, GEMZAR® (gemcitabina) hidroxiurea, ALKERAN® (melfalán), mercaptopurina, 6-mercaptopurina ribosida, metotrexato, ácido micofenólico, nelarabina, nolatrexed, ocfosfato, pelitrexol, pentostatina, raltitrexed, ribavirina, triapin, trimetrexato, S-1, tiazofurina, tegafur, TS-1, vidarabina y UFT.
Los antivirales incluyen, pero no se limitan a, ritonavir, aciclovir, cidofovir, ganciclovir, foscarnet, zidovudina, ribavirina e hidroxicloroquina.
Los inhibidores de quinasa Aurora incluyen, pero no se limitan a, ABT-348, AZD-1152, MLN-8054, VX-680, inhibidores de quinasa específicos de Aurora A; inhibidores de quinasa específicos de Aurora B e inhibidores de quinasa pan-Aurora.
Los inhibidores de la proteína Bcl-2 incluyen, pero no se limitan a, AT-101 ((-)gosipol), GENASENSE® (G3139 o oblimersen (oligonucleótido antisentido dirigido a Bcl-2)), IPI-194, IPI-565, N-(4-(4-((4’-cloro( 1,1 ’-bifenil)-2-yl)metil)piperazin-1 -il)benzoil)-4-((( 1 R)-3-(dimetilamino)-1 -((fenilsulfanil)metil)propil)amino)-3-nitrobenzenosulfonamida), N-(4-(4-((2-(4-clorofenil)-5,5-dimetiM-ciclohex-1-en-1-il)metil)piperazin-1-il)benzoil)-4-(((1R)-3-(morpholin-4-yl)-1-((phenylsulfanyl)methyl)propyl)amino)-3-((trifluorometil)sulfonil)benzenosulfonamida, venetoclax y GX-070 (obatoclax).
Los inhibidores de la quinasa Bcr-Abl incluyen, pero no se limitan a, DASATINIB® (BMS-354825) y GLEEVEC® (imatinib).
Los inhibidores de CDK incluyen, pero no se limitan a, AZD-5438, BMI-1040, BMS-032, BMS-387, CVT-2584, flavopiridol, GPC-286199, MCS-5A, PD0332991, PHA-690509, seliciclib (CYC-202, R-roscovitina) y ZK-304709. Los inhibidores de COX-2 incluyen, pero no se limitan a, ABT-963, ARCOXIA® (etoricoxib), BEXTRA® (valdecoxib), BMS347070, CELEBREX® (celecoxib), COX-189 (lumiracoxib), CT-3, DERAMAXX® (deracoxib), JTE-522, 4-metil-2-(3,4-dimetilfenil)-1-(4-sulfamoilfenil-1H-pirrol), MK-663 (etoricoxib), NS-398, parecoxib, RS-57067, SC-58125, SD-8381, SVT-2016, S-2474, T-614 y VIOXX® (rofecoxib).
Los inhibidores de EGFR incluyen, pero no se limitan a, afatinib (GILOTRIF®), ABX-EGF, inmunoliposomas antiEGFR, vacuna EGF, EMD-7200, ERBITUX® (cetuximab), HR3, anticuerpos IgA, IRESSA® (gefitinib), TARCEVA® (erlotinib u OSI-774), TP-38, proteína de fusión EGFR, Po RTRAZZA® (necitumumab), TAGRISSO® (osimertinib), TYKERB® (lapatinib), TARCEVA® (erlotinib) y TAGRISSO® (osimertinib).
Los inhibidores del receptor de ErbB2 incluyen, pero no se limitan a, CP-724-714, CI-1033 (canertinib), HERCEPTIN® (trastuzumab), TYKERB® (lapatinib), OMNITARG® (2C4, pertuzumab), TAK-165, GW- 572016 (ionafarnib), GW-282974, EKB-569, PI-166, dHER2 (vacuna HER2), APC-8024 (vacuna HER-2), anticuerpo anti-HER/2neu biespecífico, B7.her2IgG3, anticuerpos biespecíficos trifuncionales AS HER2, mAB AR-209 y mAB 2B-1. Los inhibidores de histona deacetilasa incluyen, pero no se limitan a, depsipéptido, LAQ-824, MS-275, trapoxina, ácido hidroxámico suberoilanilida (SAHA), TSA y ácido valproico.
Los inhibidores de HSP-90 incluyen, pero no se limitan a, 17-AAG-nab, 17-AAG, CNF-101, CNF-1010, CNF-2024, 17-DMAG, geldanamicina, IPI-504, KOS-953, MYCOGRAB® (anticuerpo recombinante humano a HSP-90), NCS-683664, PU24FC1, PU-3, radicicol, SNX-2112, STA-9090 y VER49009.
Los inhibidores de las proteínas de apoptosis incluyen, pero no se limitan a, HGS1029, GDC-0145, GDC-0152, LCL-161 y LBW-242.
Los activadores de la vía del receptor de muerte incluyen, pero no se limitan a, TRAIL, anticuerpos u otros agentes que se dirigen a TRAIL o receptores de muerte (por ejemplo, DR4 y DR5) tales como Apomab, conatumumab, ETR2-ST01, GDC0145 (lexatumumab), HGS-1029, LBY-135, PRO-1762 y trastuzumab.
Los inhibidores de cinesina incluyen, pero no se limitan a, inhibidores de Eg5 tales como AZD4877, ARRY-520; e inhibidores de CENPE tales como GSK923295A.
Los inhibidores de JAK-2 incluyen, pero no se limitan a, CEP-701 (lesaurtinib), XL019 e INCB018424.
Los inhibidores de MEK incluyen, pero no se limitan a, ARRY-142886, ARRY-438162, PD-325901, PD-98059 y trametinib.
Los inhibidores de mTOR incluyen, pero no se limitan a, AP-23573, CCI-779, everolimus, RAD-001, rapamicina, temsirolimus, inhibidores TORC1/TORC2 competitivos por el ATP, que incluyen PI-103, PP242, PP30 y Torin 1. Los fármacos antiinflamatorios no esteroideos incluyen, pero no se limitan a, AMIGESIC® (salsalato), DOLOBID® (diflunisal), MOTRIN® (ibuprofeno), ORUDIS® (ketoprofeno), RELAFEN® (nabumetona), FELDENE® (piroxicam), crema de ibuprofeno, ALEVE® (naproxeno) y NAPROSYN® (naproxeno), VOLTAREN® (diclofenaco), INDOCIN® (indometacina), CLINORIL® (sulindac), To LeCTIN® (tolmetina), LODINE® (etodolaco), t OrADOL® (ketorolaco) y DAYPRO® (oxaprozina).
Los inhibidores de PDGFR incluyen, pero no se limitan a, C-451, CP-673 y CP-868596.
Los agentes quimioterapéuticos de platino incluyen, pero no se limitan a, cisplatino, ELOXATIN® (oxaliplatino) eptaplatino, lobaplatino, nedaplatino, PARAPLATIN® (carboplatino), satraplatino y picoplatino.
Los inhibidores de la quinasa tipo polo incluyen, pero no se limitan a, BI-2536.
Inhibidores de BRAF vemurafenib, dabrafenib, cobimetinib.
Los inhibidores de la fosfoinositida-3 quinasa (PI3K) incluyen, pero no se limitan a, wortmannin, LY294002, XL-147, CAL-120, ONC-21, AEZS-127, ETP-45658, PX-866, GDC-0941, BGT226, BEZ235 y XL765.
Los análogos de trombospondina incluyen, pero no se limitan a, ABT-510, ABT-567, ABT-898 y TSP-1.
Los inhibidores de VEGFR incluyen, pero no se limitan a, AVASTIN® (bevacizumab), ABT-869, AEE-788, ANGIOZYME ™ (una ribozima que inhibe la angiogénesis (Ribozyme Pharmaceuticals (Boulder, CO) y Chiron (Emeryville, CA)), axitinib (AG-13736), AZD-2171, CP-547,632, IM-862, MACUGEN® (pegaptamib), NEXAVAR® (sorafenib, BAY43-9006), pazopanib (GW-786034), vatalanib (PTK-787, ZK-222584), SUTENT® (sunitinib, SU-11248), trampa de VEGF y ZACTIMA™ (vandetanib, ZD-6474), cabozantanib (inhibidor de VEGFR2 y cMet), ramucirumab (mAb inhibidor anti-VEGFR2).
Los antibióticos incluyen, pero no se limitan a, antibióticos intercalantes, aclarrubicina, actinomicina D, amrubicina, annamicina, adriamicina, BLENOXANE® (bleomicina), daunorrubicina, CAELYX® o MYOCET® (doxorrubicina liposomal), elsamitrucina, epirbucina, glarbuicina, ZAVEDOS® (idarrubicina), mitomicina C, nemorrubicina, neocarzinostatina, peplomicina, pirarrubicina, rebeccamicina, stimalamer, estreptozocina, VALSTAR® (valrrubicina) y zinostatina.
Los inhibidores de la topoisomerasa incluyen, pero no se limitan a, aclarrubicina, 9-aminocamptotecina, amonafida, amsacrina, becatecarina, belotecán, BN-80915, CAMPTOSAR® (clorhidrato de irinotecán), camptotecina, CARDIOXANE® (dexrazoxina), diflomotecan, edotecarina, ELLENCE® o PHARMORUBICIN® (epirrubicina), etopósido, exatecan, 10-hidroxiycamptotecina, gimatecan, lurtotecan, mitoxantrona, Onivyde™ (irinotecán liposomal), oratecina, pirarbucina, pixantrona, rubitecan, sobuzoxano, SN-38, taflupósido y topotecan.
Los anticuerpos incluyen, pero no se limitan a, AVASTIN® (bevacizumab), anticuerpos específicos de CD40, chTNT-1/B, denosumab, e Rb ITUX® (cetuximab), HUMAX-CD4® (zanolimumab), anticuerpos específicos de IGF1R, lintuzumab, PANOREX ® (edrecolomab), RENCAREX® (WX G250), RITUXAN® (rituximab), ticilimumab, trastuzumab, pertuzumab, VECTIBIX® (panitumumab) y anticuerpos CD20 tipos I y II.
Las terapias hormonales incluyen, pero no se limitan a, ARIMIDEX® (anastrozol), AROMASIN® (exemestano), arzoxifeno, CASODEX® (bicalutamida), CETROTIDE® (cetrorelix), degarelix, deslorelina, DESOPAN® (trilostano), dexametasona, DROGENIL, DROGENIL (flutamida), EVISTA® (raloxifeno), AFEMA™ (fadrozol), FARESTON® (toremifeno), FASLODEX® (fulvestrant), FEMARA® (letrozol), formestano, glucocorticoides, HECTOROL® (doxercalciferol), RENAGEL® (carbonato de sevelámero), lasofoxifeno, acetato de leuprolida, MEGACE® (megesterol), MIFEPREX® (mifepristona), NILANDRON™ (nilutamida), NOLVADEX® (citrato de tamoxifen), PLENAXIS™ (abarelix), prednisona, PROPECIA® (finasterida), rilostano, SUPREFACT® (buserelin), TRELSTAR® (hormona liberadora de la hormona luteinizante (LHRH)), VANTAS® (implante de Histrelina), VETORYL® (trilostano o modrastano), XTANDI® (enzalutamida), ZOLADEX® (fosrelina, goserelina) y ZYTIGA® (abiratenona).
Los deltoides y retinoides incluyen, pero no se limitan a, seocalcitol (EB 1089, CB 1093), lexacalcitrol (KH1060), fenretinida, PANRETIN® (aliretinoína), ATRAGEN® (tretinoína liposomal), TARGRETIN® (bexaroteno) y lGd -1550. Los inhibidores de PARP incluyen, pero no se limitan a, ABT-888 (veliparib), olaparib, KU-59436, AZD-2281, AG-014699, BSI-201, BGP-15, INO-1001 y ONO-2231.
Los alcaloides vegetales incluyen, pero no se limitan a, vincristina, vinblastina, vindesina y vinorelbina.
Los inhibidores del proteasoma incluyen, pero no se limitan a, VELCADE® (bortezomib), KYPROLIS® (carfilzomib), MG132, NPI-0052 y PR-171.
Los ejemplos de inmunológicos incluyen, pero no se limitan a, interferones, inhibidores del punto de control inmunitario, agentes coestimuladores y otros agentes potenciadores de la inmunidad. Los interferones incluyen interferón alfa, interferón alfa-2a, interferón alfa-2b, interferón beta, interferón gamma-la, ACTIMMUNE® (interferón gamma-lb) o interferón gamma-nl, combinaciones de los mismos y similares. Los inhibidores del punto de control inmunitario incluyen anticuerpos que se dirigen a PD-1 (por ejemplo, pembrolizumab, nivolumab y pidilizumab), PD-L1 (por ejemplo, durvalumab, atezolizumab, avelumab, MEDI4736, MSB0010718C y MPDL3280A) y CTLA4 (antígeno linfocito citotóxico 4; por ejemplo, ipilimumab, tremelimumab). Los agentes coestimuladores incluyen, pero no se limitan a, anticuerpos contra CD3, CD40, CD40L, CD27, CD28, CSF1R, CD137 (por ejemplo, Urelumab), B7H1, GITR, ICOS, CD80, CD86, OX40, OX40L, CD70, HLA-DR, LIGHT, LIGHT-R, TIM3, A2AR, NKG2A, TIGIT (inmunorreceptor de células T con dominios Ig e ITIM), VISTA (supresor de Ig de dominio V de la activación de células T), B7-H3, B7-H4, CD47, CD73, CD39, KIR (por ejemplo, lirilumab), TGF-p (por ejemplo, fresolimumab) y combinaciones de los mismos.
Otros agentes incluyen, pero no se limitan a, ALFAFERONE® (IFN-a), BAM-002 (glutatión oxidado), BEROMUN® (tasonermina), BEXXAR® (tositumomab), CAMPATH® (alemtuzumab), dacarbazina, denileucina, epratuzumab, GRANOCYTF® (lenograstim), lentinan, interferón alfa leucocitario, imiquimod, vacuna contra el melanoma, mitumomab, molgramostim, MYLOTARG ™ (gemtuzumab ozogamicin), NEUPOGEN® (filgrastim), OncoVAC-CL, OVAREX® (oregovomab), pemtumomab (Y-muHMFG1), PROVENGE® (sipuleucel-T), sargaramostim, sizofilano, teceleucina, THERACYS® (Bacillus Calmette-Guerin), ubenimex, VIRULIZIN® (inmunoterapéutico, Lorus Pharmaceuticals), Z-100 (Sustancia específica de Maruyama (SSM)), WF- 10 (tetraclorodecaóxido (TCDO)), PROLEUKIN® (aldesleucina), ZADAXIN® (timalfasina), ZINBRYTA® (proceso de alto rendimiento con daclizumab) y ZEVALIN® (90Y-Ibritumomab tiuxetan).
Los modificadores de la respuesta biológica son agentes que modifican los mecanismos de defensa de los organismos vivos o las respuestas biológicas, tales como la supervivencia, el crecimiento o la diferenciación de las células tisulares para que tengan actividad antitumoral e incluyen, pero no se limitan a, krestin, lentinano, sizofiran, picibanil PF-3512676 (CpG-8954) y ubenimex.
Los análogos de pirimidina incluyen, pero no se limitan a, citarabina (ara C o arabinósido C), citosina arabinósido, doxifluridina, FLUDARA® (fludarabina), 5-FU (5-fluorouracilo), floxuridina, GEMZAR® (gemcitabina), TOMUDEX® (ratitrexed) y TROXATYL™ (triacetiIuridina troxacitabina).
Los análogos de purina incluyen, pero no se limitan a, LANVIS® (tioguanina) y PURI-NETHOL® (mercaptopurina).
Los agentes antimitóticos incluyen, pero no se limitan a, batabulina, epotilona D (KOS-862), N-(2-((4-hidroxifenil)amino)piridin-3-il)-4-metoxibencenosulfonamida, ixabepilona (BMS 247550), TAXOL® (paclitaxel), TAXOTERE® (docetaxel), PNU100940 (109881), patupilona, XRP-9881 (larotaxel), vinflunina y ZK-EPO (epotilona sintética).
Los inhibidores de la ubiquitina ligasa incluyen, pero no se limitan a, inhibidores de MDM2, tales como nutlinsa, e inhibidores de NEDD8 tales como MLN4924.
Los inhibidores de la tirosina quinasa incluyen (GLEEVEC®), dasatinib (SPRYCE®), nilotinib (TASIGNA®), bosutinib (BOSULIF®), ponatinib (ICLUSIG®), afatinib (GIOTRIF®), axitinib (INLYTA®), crizotinib (XALKORI®), erlotinib (TARCFVA®), gefitinib (IRESSA®), lapatinib (TYVERB®), nilotinib (TASIGNA®), pazopanib (VOTRIENT®), regorafenib (STIVARGA®), sorafenib (NEXAVa R®), sunitinib (SUTENT®), toceranib (PALLADIA®), vatalanib y radotinib (SUPECT®).
Los ADC anti-cMet pueden usarse además para mejorar la eficacia de la radioterapia. Los ejemplos de radioterapia incluyen radioterapia de haz externo, radioterapia interna (es decir, braquiterapia) y radioterapia sistémica.
Los ADC anti-cMet se pueden administrar junto con otros agentes quimioterapéuticos como ABRAXANE™ (ABI-007), ABT-100 (inhibidor de la famesil transferasa), ADVEXIN® (vacuna Ad5CMV-p53), ALTOCOR® o MEVACOR® (lovastatina), AMPLIGEN® (poli I:poli C12U, un ARN sintético), APTOSYN® (exisulind), AREDIA® (ácido pamidrónico), arglabina, L-asparaginasa, atamestano (1-metil-3,17-diona-androsta-1,4-dieno), AVAGE® (tazaroteno), AVE-8062 (derivado de la combreastatina) BEC2 (mitumomab), caquectina o caquexina (factor de necrosis tumoral), canvaxina (vacuna), CEAVAC® (vacuna contra el cáncer), CELEUK® (celmoleucina), CEPLENE® (diclorhidrato de histamina), CERVARIX® (vacuna contra el virus del papiloma humano), CHOP® (C: CYTOXAN® (ciclofosfamida); H: ADRIAMYCIN® (hidroxidoxorrubicina); O: Vincristina (ONCOVIN®); P: prednisona), CYPAT ™ (acetato de ciproterona), combrestatina A4P, DAB (389)EGF (dominios catalíticos y de translocación de la toxina diftérica fusionados mediante un conector His-Ala al factor de crecimiento epidérmico humano) o TransMID-107R™ (toxinas diftéricas), dacarbazina, dactinomicina, ácido 5,6-dimetilxantenona-4-acét¡co (DMXAA), eniluracilo, EVIZON™ (lactato de escualamina), DIMERICINE® (loción de liposomas T4N5), discodermolida, DX-8951Í (mesilato de exatecano), enzastaurina, EPO906 (epitilona B), GARDASIL® (vacuna recombinante contra el virus del papiloma humano tetravalente (tipos 6, 11, 16, 18)), GASTRIMMUNE®, GENASENSE®, GMK (vacuna conjugada de gangliósidos), GVAX® (vacuna contra el cáncer de próstata), halofuginona, histrelina, hidroxicarbamida, ácido ibandrónico, IGN-101, IL-13-PE38, IL-13-PE38QQR (cintredecina besudotox), IL-13-pseudomonas exotoxina, interferón-a, interferón-y, JUNOVAN™ o MEPACT™ (mifamurtida), lonafarnib, 5,10-metilentetrahidrofolato, miltefosina (hexadecilfosfocolina), NEOVASTAT® (AE-941), NEUTREXIN® (glucuronato de trimetrexato), NIPENT® (pentostatina), ONCONASE® (una enzima ribonucleasa), ONCOPHAGE® (vacuna para el tratamiento de melanoma), ONCOVAX® (vacuna IL-2), ORATHECIN™ (rubitecan), OSIDEM® (fármaco celular basado en anticuerpos), OVAREX® MAb (anticuerpo monoclonal murino), paclitaxel, PANDIMEX™ (saponinas agliconas de ginseng que comprenden 20(S)protopanaxadiol (aPPD) y 20(S)protopanaxatriol (aPPT)), panitumumab, PANVAC®-VF (vacuna contra el cáncer en investigación), pegaspargasa, PEG Interferón A, fenoxodiol, procarbazina, rebimastato, REMOVAB® (catumaxomab), REVLIMID® (lenalidomida), RSR13 (efaproxiral), SOMATULINE® LA (lanreotida), SORIATANE® (acitretina), estaurosporina (estaurosporas de Streptomyces), talabostato (PTT100), TARGRETIN® (bexaroteno), TAXOPREXIN® (DHA-paclitaxel), TELCYTA® (canfosfamida, TLK286), temilifeno, TEMODAR® (temozolomida), tesmilifeno, talidomida, THERATOPE® (STn-KLH), timitaq (diclorhidrato de 2-amino-3,4-dihidro-6-metil-4-oxo-5-(4-piridiltio)quinazolina), TNFERADE™ (adenovector: portador de ADN que contiene el gen del factor de necrosis tumoral-a), TRACLEER® o ZAVESCA® (bosentán), tretinoína (Retin-A)), tetrandrina, TRISENOX® (trióxido de arsénico), VIRULIZIN®, ucrania (derivado de alcaloides de la planta celidonia mayor), vitaxina (anticuerpo anti-alfavbeta3), XCYTRIN® (motexafina gadolinio), XINLAY™ (atrasentan), XYOTAX ™ (paclitaxel poliglumex), YONDELIS® (trabectedina), ZD-6126, ZINECARD® (dexrazoxano), ZOMETA® (ácido zolendrónico) y zorrubicina así como también combinaciones de cualquiera de estos agentes.
Las terapias complementarias y/o los agentes terapéuticos se usarán, típicamente, a su dosis, vía de administración y frecuencia de administración aprobadas, pero pueden usarse a dosis más bajas y/o con menos frecuencia. Cuando se administra como monoterapia, el ADC anti-cMet típicamente se administrará en un programa que genera un beneficio terapéutico. Se contempla que los ADC anti-cMet administrados una vez por semana, una vez cada dos semanas, una vez cada tres semanas, una vez cada cuatro semanas, una vez cada cinco semanas, una vez cada seis semanas, una vez cada siete semanas o una vez cada ocho semanas proporcionarán beneficio terapéutico, aunque la administración más o menos frecuente puede ser beneficiosa. Cuando se administra de forma complementaria a, o con otra terapia y/o agente, el ADC anti-cMet puede administrarse antes del tratamiento, después del tratamiento o simultáneamente con el tratamiento con la otra terapia o agente.
5.10. Dosis y regímenes de administración
La cantidad de ADC anti-cMet administrada dependerá de una variedad de factores, que incluyen, pero no se limitan, al tipo particular de tumores cMet+/que sobreexpresan, la etapa de los tumores cMet+/que sobreexpresan que se trata, el modo de administración, la frecuencia de administración, el beneficio terapéutico deseado, el
componente farmacológico del ADC (por ejemplo, MMAE frente a PBD) y otros parámetros tales como la edad, el peso y otras características del paciente, etc. La determinación de las dosis efectivas para proporcionar un beneficio terapéutico para modos específicos y la frecuencia de administración está dentro de las capacidades de los expertos en la técnica.
Las dosis efectivas para proporcionar un beneficio terapéutico pueden estimarse inicialmente a partir de modelos animales o clínicos in vivo. Los modelos animales adecuados para una amplia variedad de enfermedades se conocen en la técnica.
Los ADC anti-cMet pueden administrarse por cualquier ruta apropiada para la afección a tratar. Un ADC anti-cMet se administrará típicamente por vía parenteral, es decir, infusión, subcutánea, intramuscular, intravenosa (IV), intradérmica, intratecal, en bolo, inyección intratumoral o epidural ((Shire y otros, 2004, J. Pharm. Sciences 93(6):1390-1402)). En una modalidad, se proporciona un ADC anti-cMet como un polvo liofilizado en un vial. Los viales pueden contener, por ejemplo, 0,5 mg, 1 mg, 5 mg, 10 mg, 50 mg, 100 mg o 200 mg de ADC anti-cMet. En una modalidad, antes de la administración, el polvo liofilizado se reconstituye con agua estéril para inyección (SWFI) u otro medio adecuado para proporcionar una solución que contiene 20 mg/mL de ADC anti-cMet. La solución reconstituida resultante se diluye adicionalmente con solución salina u otro medio adecuado y se administra mediante una infusión IV una vez cada 7 días, una vez cada 14 días, una vez cada 21 días o una vez cada 28 días. En algunas modalidades, para el primer ciclo, la infusión ocurre durante 180 minutos, las infusiones posteriores duran más de 90 minutos. En otras modalidades, la infusión se produce durante 60 minutos. En algunas modalidades, todas las infusiones para cada ciclo ocurren durante 30 minutos.
En una modalidad ejemplar, se administra un ADC anti-cMet una vez cada 14 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,6 mg/kg, 1,8 mg/kg, 1,9 mg/kg, 2,1 mg/kg, 2,2 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg o 6,0 mg/kg del peso corporal del sujeto. En una modalidad, el ADC anti-cMet se administra una vez cada 14 días a 1,6 mg/kg. En una modalidad, el ADC anti-cMet se administra una vez cada 14 días a 1,9 mg/kg. En una modalidad, el ADC anti-cMet se administra una vez cada 14 días a 2,2 mg/kg. En una modalidad, el ADC anti-cMet se administra una vez cada 14 días a 2,5 mg/kg. En una modalidad, la administración continúa hasta la progresión de la enfermedad o toxicidad inaceptable.
En una modalidad, el cáncer es un adenocarcinoma de NSCLC, el ADC anti-cMet es ABBV-399, administrado a 1,6 o 1,9 mg/kg cada 14 días, y el paciente tiene una puntuación H de 225 o más o una puntuación IHC de 3+. En otra modalidad, el cáncer es un carcinoma de células escamosas de NSCLC, el ADC anti-cMet es ABBV-399, administrado a 1,6 o 1,9 mg/kg cada 14 días, y el paciente tiene una puntuación H entre 150 a 224 o una puntuación IHC de 2+.
En otra modalidad ilustrativa, se administra un ADC anti-cMet una vez cada 7 días a 0,15 mg/kg, 0,3 mg/kg, 0,45 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg o 3,0 mg/kg. En una modalidad, la administración continúa hasta la progresión de la enfermedad o toxicidad inaceptable.
En otra modalidad ilustrativa, se administra un ADC anti-cMet una vez cada 28 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,.8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg. En una modalidad, la administración continúa hasta la progresión de la enfermedad o toxicidad inaceptable.
En otra modalidad ilustrativa, se administra un ADC anti-cMet una vez cada 28 días a 2,7 mg/kg. En una modalidad, la administración continúa hasta la progresión de la enfermedad o toxicidad inaceptable.
En otra modalidad ilustrativa, se administra un ADC anti-cMet una vez cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg. En una modalidad, la administración continúa hasta la progresión de la enfermedad o toxicidad inaceptable.
En otra modalidad ilustrativa, se administra un ADC anti-cMet (por ejemplo, ABBV-399) una vez cada 21 días a 2,7 mg/kg. En una modalidad, la administración continúa hasta la progresión de la enfermedad o toxicidad inaceptable. En una modalidad, el cáncer es un adenocarcinoma de NSCLC, el ADC anti-cMet es ABBV-399, administrado a 2,7 mg/kg cada 21 días, y el paciente tiene una puntuación H de 225 o más o una puntuación IHC de 3+. En otra modalidad, el cáncer es un carcinoma de células escamosas de NSCLC, el ADC anti-cMet es ABBV-399, administrado a 2,7 mg/kg cada 21 días, y el paciente tiene una puntuación H de al menos 150 o más y al menos una puntuación IHC de 2+.
En otra modalidad ilustrativa, se administra un ADC PBD anti-cMet (por ejemplo, ABT-700 PBD) una vez cada 14 días, una vez cada 21 días o una vez cada 28 días, a una dosis entre 1,0 pg/kg a 1,0 mg/kg, 1,0 pg/kg a 500,0 pg/kg o 5,0 pg/kg a 200,0 pg/kg del peso corporal del sujeto. Como para cualquier otro ADC, la dosificación depende, por ejemplo, de la frecuencia de administración, el estado del paciente y la respuesta al tratamiento previo, si
corresponde. La concentración del ADC en una formulación líquida puede ser, por ejemplo, 0,01-10 mg/mL, tal como 1,0 mg/mL.
En una modalidad, se administra un ADC PBD anti-cMet (por ejemplo, ABT-700 PBD) una vez cada 14 días, una vez cada 21 días o una vez cada 28 días a 10 pg/kg, 50 pg/kg, 75 pg/kg, 100 pg/kg, 110 pg/kg, 120 pg/kg, 130 pg/kg, 140 pg/kg, 150 pg/kg, 160 pg/kg, 170 pg/kg, 180 pg/kg, 190 pg/kg, 200 pg/kg, 250 pg/kg, 300 pg/kg, 350 pg/kg, 400 pg/kg, 450 pg/kg o 500 pg/kg. En una modalidad, el ADC PBD anti-cMet (por ejemplo, ABT-700 PBD) se administra a 100 pg/kg. En una modalidad, el ADC PBD anti-cMet (por ejemplo, AbT-700 PBD) se administra a 200 pg/kg. En una modalidad, el ADC PBD anti-cMet (por ejemplo, ABT-700 PBD) se administra a 300 pg/kg. En una modalidad, el ADC PBD anti-cMet (por ejemplo, ABT-700 PBD) se administra a 400 pg/kg.
Cuando se administran complementarios a, o con, otros agentes quimioterapéuticos, los ADC pueden administrarse en el mismo programa que el(los) otro(s) agente(s) o en un programa diferente. Cuando se administra en el mismo horario, el ADC puede administrarse antes, después o simultáneamente con el otro agente. En algunas modalidades donde se administra un ADC complementario a, o con, los estándares de atención, el ADC puede iniciarse antes del comienzo de la terapia estándar, por ejemplo, un día, varios días, una semana, varias semanas, un mes o incluso varios meses antes del comienzo de la terapia estándar de atención.
En un conjunto de modalidades ilustrativas, el agente anticancerígeno adicional se selecciona del grupo que consiste en cabazitaxel, colcemid, colchicina, criptoficina, democolcina, docetaxel, nocodazol, paclitaxel, taccalonolida, taxano y vinblastina.
En una modalidad ilustrativa, se usa un ADC anti-cMet complementario a afatinib (GILOTRIF®) para tratar el NSCLC. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. GILOTRIF® se administra a 40 mg por vía oral una vez al día hasta la progresión de la enfermedad o ya no es tolerado por el paciente. En una modalidad, los pacientes se seleccionan para el tratamiento de primera línea de NSCLC metastásico con GILOTRIF® en base a la presencia de deleciones del exón 19 de EGFR o mutaciones de sustitución del exón 21 (L858R) en muestras tumorales.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a TARCEVA® (erlotinib) para tratar el cáncer de pulmón de células no pequeñas (NSCLC). El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión intravenosa una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La dosis y el programa recomendados para erlotinib es de 150 mg por vía oral, una vez al día. La terapia complementaria anti-cMet ADC/erlotinib se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En una modalidad, el cáncer es NSCLC, el ADC anti-cMet es ABBV-399, administrado a 2,7 mg/kg cada 21 días, y el erlotinib se administra a 150 mg por vía oral, una vez al día. La terapia complementaria anti-cMet ADC/erlotinib se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente. En una modalidad, el cáncer es un adenocarcinoma de NSCLC con EGFR mutado, el ADC anti-cMet es ABBV-399, administrado a 2,7 mg/kg cada 21 días, el erlotinib se administra a 150 mg por vía oral, una vez al día, y el paciente tiene una puntuación H de 225 o más o una puntuación IHC de 3+.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a IRESSA® (gefitinib) para tratar el cáncer de pulmón de células no pequeñas (NSCLC). El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La dosis y el programa recomendados para gefitinib es de 250 mg por vía oral, una vez al día. La terapia complementaria anti-cMet ADC/gefitinib se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a afatinib para tratar el cáncer de pulmón de células no pequeñas (NSCLC). El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La dosis y el programa recomendados para afatinib es de 40 mg por vía oral, una vez al día. La terapia complementaria anticMet ADC/afatinib se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a OPDIVO® (nivolumab) para tratar el
cáncer de pulmón de células no pequeñas (NSCLC). El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión intravenosa una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. A Nivolumab se le administra una infusión intravenosa a 3 mg/kg durante 60 minutos cada dos semanas. El tratamiento complementario anti-cMet ADC/nivolumab se continúa hasta la progresión de la enfermedad o ya no es tolerado por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a OPDIVO® (nivolumab) y YERVOY® (ipilimumab) para tratar el cáncer de pulmón no microcítico (NSCLC). El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión intravenosa una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg, para cuatro dosis con ipilimumab, luego cada 14 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, sin ipilimumab. Nivolumab se administra como una infusión intravenosa a 3 mg/kg durante 60 minutos cada dos semanas. Ipilimumab se administra por vía intravenosa a 3 mg/kg durante 90 minutos cada tres semanas en las primeras cuatro dosis. El tratamiento complementario anti-cMet ADC/nivolumab se continúa hasta la progresión de la enfermedad o ya no es tolerado por el paciente.
En otra modalidad ilustrativa más, se puede usar un ADC anti-cMet complementario a pembrolizumab (KEYTRUDA®) para tratar NSCLC. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Pembrolizumab se administra como una infusión intravenosa a 2 mg/kg durante 30 minutos cada 3 semanas. La terapia complementaria anti-cMet ADC y pembrolizumab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a cisplatino para tratar NSCLC. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. El cisplatino se administra a 20 mg/m2 o más, una vez cada 3 a 4 semanas. La terapia complementaria anti-cMet ADC/cisplatino se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a carboplatino para tratar el NSCLC. El ADC anti-cMet se administra por infusión IV una vez cada 14 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg. El carboplatino se administra a 300 mg/m2 o más, una vez cada 4 semanas. La terapia complementaria anti-cMet ADC/carboplatino se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a veliparib para tratar NSCLC. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Veliparib se administra por vía oral, dos veces al día. La terapia complementaria anti-cMet ADC/veliparib continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a veliparib y pemetrexed para tratar el NSCLC. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Veliparib se administra por vía oral, dos veces al día. Pemetrexed se administra a 500 mg/m2 por vía intravenosa cada 21 días. La terapia complementaria anti-cMet ADC/veliparib/pemetrexed se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a cetuximab para tratar NSCLC. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg,
preferentemente una vez cada 21 días a 2,7 mg/kg. Cetuximab se administra a una dosis inicial de 400 mg/m2 durante una infusión intravenosa de 120 minutos, seguida de una infusión semanal de 250 mg/m2 durante 60 minutos. La terapia complementaria anti-cMet ADC/cetuximab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a ipilimumab (YERVOY®) para tratar el NSCLC. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Ipilimumab se administra a 3 mg/kg por vía intravenosa durante 90 minutos cada 3 semanas durante 3 meses. La terapia anti-cMet ADC continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a la radiación para tratar NSCLC. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Típicamente, la radioterapia de haz externo se aplica durante unos minutos hasta 5 días a la semana durante 5 a 7 semanas, pero esto variará según el tipo de radioterapia de haz externo que se use. La terapia complementaria anti-cMet ADC/radioterapia se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a AVASTIN® (bevacizumab) para tratar el NSCLC. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La dosis y el programa recomendados para bevacizumab son de 10 mg/kg cada 14 días o de 15 mg/kg cada 21 días. La terapia complementaria anti-cMet ADC/bevacizumab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En una modalidad ilustrativa, se usa un ADC anti-cMet complementario a gemcitabina (GEMZAR®) para cáncer de NSCLC. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La gemcitabina se administra por infusión intravenosa a una dosis de 1000 mg/m2 durante 30 minutos los días 1, 8 y 15 durante un programa de cada 4 semanas. Administrar cisplatino por vía intravenosa a 100 mg/m2 el día 1 después de la infusión de gemcitabina. En otra modalidad, la gemcitabina se administra por infusión intravenosa a una dosis de 1250 mg/m2 durante 30 minutos los días 1 y 8 durante un programa de cada 3 semanas. Administrar cisplatino por vía intravenosa a 100 mg/m2 el día 1 después de la infusión de gemcitabina. Si se observa mielosupresión, se pueden usar modificaciones de dosis según se proporciona en la información de prescripción de gemcitabina. La terapia complementaria anticMet ADC/gemcitabina se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En una modalidad ilustrativa, se usa un ADC anti-cMet complementario a la gemcitabina (GEMZAR®) para tratar el cáncer de páncreas, ovario, mama o NSCLC. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. En el tratamiento del cáncer de páncreas, la gemcitabina se administra por infusión intravenosa a una dosis de 1000 mg/m2 durante 30 minutos una vez por semana durante un máximo de 7 semanas, seguido de una semana de descanso del tratamiento. Después de la semana 8: dosificación semanal en los días 1, 8 y 15 de ciclos de 28 días.
En el tratamiento del cáncer de ovario, la gemcitabina se administra por infusión intravenosa a una dosis de 1000 mg/m2 durante 30 minutos los días 1 y 8 de cada ciclo de 21 días, en combinación con carboplatino AUC 4 por vía intravenosa después de la administración de Gemzar el día 1 de cada ciclo de 21 días. Consulte la información de prescripción de carboplatino para obtener información adicional. En el tratamiento del cáncer de mama, la gemcitabina se administra por infusión intravenosa a una dosis de 1250 mg/m2 por vía intravenosa durante 30 minutos los días 1 y 8 de cada ciclo de 21 días que incluye paclitaxel. El paclitaxel debe administrarse a 175 mg/m2 el día 1 como una infusión intravenosa de 3 horas antes de la administración de Gemzar. Si se observa mielosupresión, se pueden usar modificaciones de dosis según se proporciona en la información de prescripción de gemcitabina. Los ciclos posteriores deben consistir en infusiones una vez por semana durante 3 semanas consecutivas de cada 4 semanas. La terapia complementaria anti-cMet ADC/gemcitabina se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa, se usa un ADC anti-cMet complementario a la formulación de nanopartículas estabilizadas con albúmina de paclitaxel (ABRAXANE®) para tratar el cáncer de mama o de pulmón. El ADC anticMet (por ejemplo, ABBV-399) se administra por infusión intravenosa una vez cada 14 días o cada 21 días a 0,15
mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La dosis y el programa recomendados para la formulación de nanopartículas estabilizadas con albúmina de paclitaxel son de 125 mg/m2 administrados como una infusión intravenosa durante 30-40 minutos los días 1, 8 y 15 de cada ciclo de 28 días. La terapia complementaria anti-cMet ADC/ABRAXANE® continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa se usa un ADC anti-cMet complementario a la formulación de nanopartículas estabilizadas con albúmina de paclitaxel (ABRAXANE®) más gemcitabina (GEMZAR®) para tratar el cáncer de páncreas. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La dosis y el programa recomendados para la formulación de nanopartículas estabilizadas con albúmina de paclitaxel son de 125 mg/m2 administrados como una infusión intravenosa durante 30-40 minutos los días 1, 8 y 15 de cada ciclo de 28 días. La gemcitabina se administra por infusión intravenosa a una dosis de 1000 mg/m2 durante 30 minutos una vez por semana durante hasta 7 semanas (o hasta que la toxicidad reduzca o mantenga una dosis), seguido de una semana de descanso del tratamiento. Los ciclos posteriores deben consistir en infusiones una vez por semana durante 3 semanas consecutivas de cada 4 semanas. La terapia complementaria anti-cMet ADC/ABRAX-ANE®/GEMZAR® se continúa hasta que la enfermedad progresa o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a AVASTIN® (bevacizumab) para tratar el cáncer colorrectal o el pulmón o el ovario. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/ mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La dosis y el programa recomendados para bevacizumab son de 10 mg/kg cada 14 días o de 15 mg/kg cada 21 días. La terapia complementaria anti-cMet ADC/bevacizumab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a FOLFIRINOX (o FOLFIRI o FOLFOX o irinotecán o 5-FU o capecitabina) para tratar el cáncer colorrectal. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a
2,7 mg/kg. FOLFIRINOX es una combinación de cuatro agentes de quimioterapia: fluorouracilo [5-FU], leucovorina, irinotecán y oxaliplatino. En algunas modalidades, FOLFIRINOX se administra como sigue: oxaliplatino, 85 mg/m2; irinotecán, 180 mg/m2; leucovorina, 400 mg/m2 y fluorouracilo, 400 mg/m2 administrados como un bolo seguido de
2400 mg/m2 administrados como una infusión continua de 46 horas, cada 2 semanas. La terapia complementaria anti-cMet ADC/FOLFIRINOX continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a Onivyde® para tratar el cáncer de páncreas. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Onivyde® es una formulación liposomal de irinotecán. En algunas modalidades, Onivyde® se administra a 70 mg/m2 por infusión intravenosa durante 90 minutos cada 2 semanas. La terapia complementaria anti-cMet ADC/Onivyde® continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a Onivyde®, fluorouracilo y leucovorina para tratar el páncreas. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión intravenosa una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Onivyde® es una formulación liposomal de irinotecán. En algunas modalidades, Onivyde® se administra a 70 mg/m2 por infusión intravenosa durante 90 minutos cada 2 semanas, con leucovorina 400 mg/m2 y fluorouracilo 2400 mg/m2 durante 46 horas cada 2 semanas. La terapia complementaria anti-cMet ADC/Onivyde®/leucovorin/fluorouracilo continúa hasta la progresión de la enfermedad o ya no es tolerado por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a nivolumab (OPDIVO®) para tratar el cáncer de pulmón y otros cánceres donde se utiliza nivolumab. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión intravenosa una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. A Nivolumab se le administra una infusión intravenosa a 3 mg/kg durante 60 minutos cada
dos semanas. La terapia complementaria anti-cMet ADC/nivolumab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se puede usar un ADC anti-cMet como complemento de pembrolizumab (KEYTRUDA®) para tratar el cáncer colorrectal. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión intravenosa una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Pembrolizumab se administra como una infusión intravenosa a 2 mg/kg durante 30 minutos cada 3 semanas. La terapia complementaria anti-cMet ADC/pembrolizumab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En una modalidad, el cáncer es cáncer de páncreas, el ADC anti-cMet es ABBV-399, administrado a 2,7 mg/kg cada 21 días, y el erlotinib se administra a 150 mg por vía oral, una vez al día. La terapia complementaria anti-cMet ADC/erlotinib se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a doxorrubicina para tratar el cáncer de mama. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3.0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Cuando se usa de forma complementaria con otros fármacos s, la dosis más utilizada de doxorrubicina es de 40 a 60 mg/m2 administrada como una inyección intravenosa única cada 21 a 28 días. La terapia complementaria anti-cMet ADC/doxorrubicina se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a AVASTIN® (bevacizumab) para tratar el cáncer de mama. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2.7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La dosis y el programa recomendados para bevacizumab son de 10 mg/kg cada 14 días o de 15 mg/kg cada 21 días. La terapia complementaria anti-cMet ADC/bevacizumab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a gemcitabina para tratar el cáncer de mama. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3.0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La gemcitabina se administra por infusión intravenosa a una dosis de 1000 mg/m2 durante 30 minutos una vez por semana durante hasta 7 semanas (o hasta que la toxicidad reduzca o mantenga una dosis), seguido de una semana de descanso del tratamiento. Los ciclos posteriores deben consistir en infusiones una vez por semana durante 3 semanas consecutivas de cada 4 semanas. La terapia complementaria anti-cMet ADC/gemcitabina se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a trastuzumab (HERCEPTIN®) para tratar el cáncer de mama. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La dosis de carga inicial recomendada para trastuzumab es de 4 mg/kg administrada como una infusión de 90 minutos. La dosis de mantenimiento semanal recomendada para trastuzumab es de 2 mg/kg que se puede administrar como una infusión de 30 minutos si la dosis de carga inicial fue bien tolerada. La terapia complementaria anti-cMet ADC/trastuzumab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a capecitabina (XELODA®) para tratar el cáncer de mama. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2.7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Capecitabina puede administrarse a 1250 mg/m2 dos veces al día durante 2 semanas seguido de un período de descanso de una semana en ciclos de 3 semanas. La terapia complementaria anti-cMet ADC/capecitabina se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a nivolumab (OPDIVO®) para tratar el cáncer de mama. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg,
2.7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. A Nivolumab se le administra una infusión intravenosa a 3 mg/kg durante 60 minutos cada dos semanas. La terapia complementaria anti-cMet ADC/nivolumab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, puede usarse un ADC anti-cMet como complemento de pembrolizumab (KEYTRUDA®) para tratar el cáncer de mama. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión intravenosa una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Pembrolizumab se administra como una infusión intravenosa a 2 mg/kg durante 30 minutos cada 3 semanas. La terapia complementaria anti-cMet ADC/pembrolizumab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a TARCEVA® (erlotinib) para tratar el cáncer de cabeza y cuello. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión intravenosa una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2.1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La dosis y el programa recomendados para erlotinib es de 150 mg por vía oral, una vez al día. La terapia complementaria anticMet ADC/erlotinib se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En una modalidad, el cáncer es cáncer de cabeza y cuello, el ADC anti-cMet es ABBV-399, administrado a 2,7 mg/kg cada 21 días, y el erlotinib se administra a 150 mg por vía oral, una vez al día. La terapia complementaria anti-cMet ADC/erlotinib se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet de forma complementaria a la radiación para tratar el cáncer de cabeza y cuello. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Típicamente, la radioterapia de haz externo se aplica durante unos minutos hasta 5 días a la semana durante 5 a 7 semanas, pero esto variará según el tipo de radioterapia de haz externo que se use. La terapia complementaria anti-cMet ADC/radioterapia se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a AVASTIN® (bevacizumab) para tratar el cáncer de cabeza y cuello. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión intravenosa una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2.1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La dosis y el programa recomendados para bevacizumab son de 10 mg/kg cada 14 días o de 15 mg/kg cada 21 días. La terapia complementaria anti-cMet ADC/bevacizumab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a cetuximab para tratar el cáncer de cabeza y cuello. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2.7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Cetuximab se administra a una dosis inicial de 400 mg/m2 durante una infusión intravenosa de 120 minutos, seguida de una infusión semanal de 250 mg/m2 durante 60 minutos. La terapia complementaria anti-cMet ADC/cetuximab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a carboplatino para tratar el cáncer de cabeza y cuello. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2.7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. El carboplatino se administra a 300 mg/m2 o más, una vez cada 4 semanas. La terapia complementaria anti-cMet ADC/carboplatino se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a nivolumab (OPDIVO®) para tratar el cáncer de cabeza y cuello. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión intravenosa una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2.1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. A Nivolumab se le
administra una infusión intravenosa a 3 mg/kg durante 60 minutos cada dos semanas. La terapia complementaria anti-cMet ADC/nivolumab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente. Aún en otra modalidad ilustrativa, puede usarse un ADC anti-cMet como adyuvante de pembrolizumab (KEYTRUDA®) para tratar el cáncer de cabeza y cuello. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. Pembrolizumab se administra como una infusión intravenosa a 2 mg/kg durante 30 minutos cada 3 semanas. La terapia complementaria anti-cMet ADC/pembrolizumab se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet como adyuvante del cisplatino para tratar el cáncer de cabeza y cuello. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión IV una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La terapia complementaria anti-LRRC15 ADC/cisplatino se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En otra modalidad ilustrativa más, se usa un ADC anti-cMet complementario a TARCEVA® (erlotinib) para tratar el cáncer de cabeza y cuello. El ADC anti-cMet (por ejemplo, ABBV-399) se administra por infusión intravenosa una vez cada 14 días o cada 21 días a 0,15 mg/kg, 0,3 mg/kg, 0,6 mg/kg, 0,9 mg/kg, 1,2 mg/kg, 1,5 mg/kg, 1,8 mg/kg, 2,1 mg/kg, 2,4 mg/kg, 2,7 mg/kg, 3,0 mg/kg, 3,3 mg/kg, 3,6 mg/kg, 3,9 mg/kg, 4,2 mg/kg, 4,5 mg/kg, 4,8 mg/kg, 5,1 mg/kg, 5,4 mg/kg, 5,7 mg/kg o 6,0 mg/kg, preferentemente una vez cada 21 días a 2,7 mg/kg. La dosis y el programa recomendados para erlotinib es de 150 mg por vía oral, una vez al día. La terapia complementaria anticMet ADC/erlotinib se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
En una modalidad, el cáncer es cáncer de cabeza y cuello, el ADC anti-cMet es ABBV-399, administrado a 2,7 mg/kg cada 21 días, y el erlotinib se administra a 150 mg por vía oral, una vez al día. La terapia complementaria anti-cMet ADC/erlotinib se continúa hasta la progresión de la enfermedad o ya no es tolerada por el paciente.
Como apreciarán los expertos en la técnica, las dosis recomendadas para los diversos agentes descritos anteriormente pueden necesitar ajustarse para optimizar la respuesta del paciente y maximizar el beneficio terapéutico.
En modalidades alternativas, todos los números que expresan cantidades de ingredientes, % de pureza, etc., usados en esta divulgación, se modifican por el término "aproximadamente."
5.11. Selección de pacientes
Los pacientes seleccionados para los tratamientos de ADC de esta divulgación incluyen aquellos con tumores que expresan cMet y aquellos con tumores que sobreexpresan cMet, que incluyen, entre otros, cualquier tumor sólido (incluidos también aquellos que sobreexpresan HGF y/o tienen una activación anormal de señalización o expresión de HGF/cMet). Los pacientes pueden seleccionarse para el tratamiento con los tratamientos de ADC de esta divulgación en función de su nivel de cMet, que se clasifica en términos de una puntuación H de inmunohistoquímica (IHC). Los detalles sobre cómo cuantificar y calificar el nivel de sobreexpresión de cMet se presentan en la Descripción detallada (sección 5.3.) Y en el ejemplo 17. La sobreexpresión de cMet se puede definir mediante una puntuación H de IHC mayor o igual a 150 cuando se mide de acuerdo con el ensayo del ejemplo 17 "protocolo de tinción cMet ABBV-ADC." Brevemente, el protocolo de tinción IHC para la sobreexpresión de cMet se ha desarrollado mediante el uso del estuche Ventana cMet CONFIRM (SP44). Las muestras de tejido se tiñen con el anticuerpo Ventana y después se evalúan mediante determinación de los porcentajes de tinción de células de tejidos diana a diversos niveles de intensidad de bajo a alto. La figura 20 representa puntuaciones H representativas mediante el uso del ensayo descrito en el ejemplo 17. Alternativamente, se describe en el ejemplo 21 el tejido tumoral que sobreexpresa cMet mediante el uso de una puntuación IHC de 0 a 3+. La figura 19 representa puntuaciones representativas de IHC mediante el uso del ensayo descrito en el ejemplo 21.
Para los propósitos de esta divulgación, una puntuación H entre 150 y 224 es equivalente a una puntuación IHC de 2+ y una puntuación H de 225 y superior es equivalente a una puntuación IHC de 3+. En un ejemplo, los pacientes con carcinoma de células escamosas de NSCLC pueden seleccionarse para el tratamiento cuando su cáncer tiene una puntuación H de al menos entre 150 y 224, o una puntuación IHC de 2+. En otro ejemplo, los pacientes con adenocarcinoma de NSCLC pueden seleccionarse para el tratamiento cuando su cáncer tiene una puntuación H de 225 o más, o una puntuación IHC de 3+.
El cáncer puede ser recién diagnosticado y sin tratamiento previo, o puede ser reincidente, refractario, o reincidente y refractario, o una metástasis o forma metastásica de un tumor que expresa cMet (en la presente descripción denominado tumores cMet ) o un tumor que sobreexpresa cMet, es decir, tumores cMet+/que sobreexpresan.
Como se demuestra en los ejemplos de esta divulgación, los tumores cMet+/que sobreexpresan que exhiben resistencia a otras quimioterapias dirigidas o no dirigidas, conservan la sensibilidad a ABBV-399.
Los ADC anti-cMet tienen innumerables usos, y en una modalidad son útiles terapéuticamente para el tratamiento de tumores que sobreexpresan cMet en humanos, tumores en donde se ha amplificado el gen MET; y tumores portadores de mutaciones en o alrededor del exón 14 del gen MET, entre otros. En otra modalidad, los ADC anticMet son útiles terapéuticamente para el tratamiento de tumores que expresan cMet en humanos, donde cMet no se sobreexpresa, pero aún se expresa.
Los tumores que llevan deleciones del exón 19 de EGFR o mutaciones del exón 21 de EGFR (L858R) también están dentro del alcance de esta divulgación. La amplificación del gen MET se considera una de las causas más comunes de resistencia adquirida en el NSCLC mutante de EGFR.
La respuesta a ABBV-399 y otros cMet-ADC divulgados en la presente descripción pueden correlacionarse con la expresión de cMet tanto a nivel proteico como genómico (por ejemplo, amplificación, mutaciones del exón 14). Los métodos preferenciales para medir ambos biomarcadores se describen en detalle en los ejemplos. Sin embargo, un experto en la técnica sabría cómo usar otros métodos para evaluar el mismo y aquellos métodos están dentro del alcance de esta divulgación.
Si se obtienen resultados diferentes con métodos diferentes, entonces los resultados obtenidos con los métodos descritos en los ejemplos son los que se utilizarán para determinar si una modalidad particular cae dentro del alcance de las modalidades. Por ejemplo, para evaluar la expresión de la proteína cMet se usaría el "protocolo de tinción cMet ABBV-ADC." Si los reactivos de Ventana usados en este protocolo ya no están disponibles, se puede usar otro protocolo aprobado por la FDA para la evaluación de los niveles de expresión de cMet por IHC. Para evaluar el número de copias del gen MET uno usaría el "método de amplificación de cMET MET/CEP7."
MET está sujeto a empalmes alternativos. Se han identificado múltiples transcriptos de MET de diferente tamaño en líneas y tejidos celulares humanos. Se han descrito al menos tres variantes de 8 kb y se supone que se generan mediante empalmes alternativos. Se describió una isoforma de cMet que carece de 18 aminoácidos en la región extracelular (exón 10) y es la forma más abundante en una variedad de tejidos y líneas celulares. El empalme alternativo del exón 14 genera otra variante que tiene una deleción en el marco de 47 aminoácidos en el dominio de yuxtamembrana citoplasmática del receptor. Un posible mecanismo de empalme alternativo podría estar en el origen de una forma de MET truncada en el extremo N de 85 kDa que se encuentra en los tumores musculoesqueléticos malignos, aunque esta forma corta también podría originarse desde el inicio de la transcripción alternativa o la escisión proteolítica.
Se ha demostrado que los mutantes MET que implican la deleción del exón 14 estabilizan el receptor cMet, lo que resulta en una ganancia de actividad de la función. El exón 14 de MET contiene el sitio de ubiquitina ligasas Cbl en el residuo de tirosina 1003 (Y1003) donde la ubiquitina se une normalmente de cualquier otra manera al residuo de tirosina y conduce a la degradación lisosomal de la proteína cMet. Por lo tanto, la mutación sin sentido del residuo Y1003 o la "omisión" de la región de la proteína que está codificada por el exón 14 de MET resulta en una sobreexpresión relativa de la proteína MET, activación de cMet mejorada y posterior oncogénesis. La inhibición por los inhibidores de la tirosina quinasa MET (TKI) puede dar lugar a un beneficio clínico en al menos pacientes con NSCLC que albergan estas alteraciones del exón 14 de MET. Los pacientes que portan cualquiera de estas mutaciones pueden beneficiarse de los tratamientos divulgados en la presente descripción.
Por consiguiente, los pacientes pueden además seleccionarse para el tratamiento si portan células con una mutación en el exón 14 del gen MET, cuyo resultado es un mayor nivel de proteína cMet en esas células cancerosas. Los ejemplos proporcionan además varios métodos para evaluar este biomarcador.
La amplificación de MET es reconocida como uno de los mecanismos moleculares potenciales de resistencia adquirida en NSCLC con EGFR mutado para EGFR-TKI. La decisión de seleccionar o no a un paciente en particular para el tratamiento con los ADC divulgados en la presente descripción también puede abarcar la determinación de si el cáncer del paciente lleva una deleción en el exón 19 del receptor del factor de crecimiento epidérmico (EGFR), una sustitución en el exón 21 (L858R), o ambos. Los pacientes cuyo cáncer lleva una o ambas de estas alteraciones genómicas en al menos algunas de sus células se seleccionan preferentemente para el tratamiento con ABBV-399 o cualquier otro ADC divulgado en la presente descripción. Los métodos para evaluar estos dos biomarcadores se proporcionan en los ejemplos más abajo.
6. Ejemplos
Los siguientes ejemplos, que resaltan ciertas características y propiedades de modalidades ilustrativas de ADC anticMet y métodos para usar estos ADS para tratar pacientes, se proporcionan con fines ilustrativos y no limitativos. Ejemplo 1. Preparación de ABT-700
El ABBV-399 (ABT-700-vcMMAE) es conjugado de fármaco con anticuerpo (ADC) compuesto por el anticuerpo ABT-700 conjugado con el inhibidor de microtúbulos citotóxicos monometilauristatina E (MMAE) a través de un conector de valina-citrulina (ve) escindible. El ABT-700 es una inmunoglobulina G kappa recombinante "humanizada" (IgG-iK) que se dirige a un epítopo único de cMet que resulta en el bloqueo de la señalización de cMet tanto dependiente de HGF como independiente de HGF.
El ABT-700 es un anticuerpo monoclonal recombinante humanizado dirigido contra cMet. El anticuerpo consta de 2 cadenas pesadas de IgG1 idénticas de 445 aminoácidos emparejadas con 2 cadenas ligeras idénticas de 218 aminoácidos. La cadena pesada se diseñó por ingeniería genética para introducir una cisteína adicional en la posición 223, así como también, la eliminación de un residuo de lisina que precede a la Cys-223 y la eliminación de 2 residuos de treonina que flanquean a la His-224. Además, el aminoácido lisina C-terminal en la cadena pesada se diseñó por ingeniería genética para eliminar la heterogeneidad en el extremo C debido a la escisión incompleta de la lisina. El anticuerpo se glicosila en la asparagina 296 en cada cadena pesada.
La cadena pesada contiene 12 residuos de cisteína y la cadena ligera contiene 5 residuos de cisteína. Cada cadena pesada contiene 4 puentes disulfuro intracatenarios y cada cadena ligera contiene 2 puentes disulfuro intracatenarios. Además, las 2 cadenas pesadas están unidas covalentemente por 3 puentes disulfuro intercatenarios. Cada cadena ligera participa en 1 enlace disulfuro con una cadena pesada.
El ABT-700 para los estudios in vitro descritos más abajo se preparó mediante técnicas de rutina, esencialmente como se describe en la patente de Estados Unidos núm. 8, 741,290. Brevemente, las células HEK293 EBNA adaptadas a la suspensión (InVitrogen, Estados Unidos) se cultivaron rutinariamente en frascos de 250 mL en 50 mL de medio libre de suero Excell 293 (SAFC Biosciences) suplementado con glutamina 6 mM en un agitador orbital (velocidad de rotación de 110 rpm). La transfección transitoria se realizó con 2 x 106 células/mL mediante el uso de polietilenimina lineal de 25 kDa (PEI) (Polysciences) preparada en agua a una concentración final de mezclado de 1 mg/mLy ADN plasmídico (concentración final de 1,25 pg/mL para la relación de plásmidos cadena pesada a ligera de 1:1). A las 4 horas después de la transfección, el cultivo se diluyó con un volumen de medio de cultivo fresco para lograr una densidad celular final de 106 células/mL. El proceso de cultivo se controló sobre la base de la viabilidad celular y la producción de Mab. Típicamente, los cultivos se mantuvieron durante 4 a 5 días. El ABT-700 se purificó mediante el uso de un enfoque de cromatografía convencional en una resina de proteína A (GE Healthcare, Estados Unidos).
El ABT-700 para los estudios clínicos descritos más abajo se preparó esencialmente como se describe a continuación. Primero, el plásmido pConPlusy1fAK/K-hz224G11 [TH7] se construyó para la expresión de alto nivel del anticuerpo monoclonal ABT-700 en células CHO mediante el uso de la tecnología de glutamina Sintetasa GS-CHO. Las secuencias de la cadena pesada y ligera se clonaron en los vectores pConPlusy1fAK-hz224G11/TH7VH0 y pConPlusK2-hz224G11A/L4 (4-39-84) respectivamente, mediante creación de vectores de un solo gen (SGV). A continuación, se combinaron los SGV que contenían los genes de la cadena pesada y la cadena ligera, junto con el gen de selección de glutamina sintetasa (GS), para generar el vector final de doble gen (DGV): pConPlusy1fAK/K-hz224G11 [TH7], Los componentes principales de pConPlusy1fAK/K-hz224G11 [TH7] incluyen los siguientes genes o elementos reguladores en el siguiente orden: promotor hCMV-MIE, 5’ UTR con intrón, secuencia codificante de la cadena ligera ABT-700 [224G11 (HzVL)], SV40 secuencia de poliadenilación, promotor de hCMV-MIE, 5’ UTR con intrón, secuencia de codificación de cadena pesada ABT-700 [224G11 (HzVH)], secuencia de poliadenilación de SV40, origen de replicación del plásmido, beta-lactamasa y ADNc de glutamina sintetasa con sus secuencias reguladoras.
El sistema de expresión usado para la producción de la sustancia farmacológica ABT-700 fue el sistema de expresión génica de glutamina sintetasa (GS) patentado por Lonza Biologics en células de ovario de hámster chino (CHO). La línea celular huésped se obtuvo del banco de células de trabajo anfitrión CHO-K1SV designado 269-W3 (preparado a partir del banco de células maestro anfitrión 269-M).
El vector de doble gen pConPlusy1fAK/K-hz224G11[TH7] se transfectó en células CHO-K1SV mediante electroporación y luego se distribuyó en placas de 96 pocillos. Las células que expresan GS y, por lo tanto, las que contienen el vector de expresión se seleccionaron mediante el crecimiento en medio sin proteínas y sin glutamina. Las placas se incubaron hasta que comenzaron a aparecer focos de células transfectadas. Solo progresaron las líneas celulares que provenían de pocillos que contenían colonias individuales (según se determina por evaluación visual). Los sobrenadantes de cultivo de los pocillos que contenían colonias individuales se seleccionaron para la producción de anticuerpos mediante el uso de un ELISA para el anticuerpo ensamblado. Se establecieron varias líneas celulares clonales y se seleccionó la que muestra el rendimiento más consistente para la producción de ABT-700. Las células se probaron para confirmar la calidad del ARNm y la fidelidad de las transcriptos de codificación.
Un solo vial congelado de células se expande mediante cultivo agitador o bolsas de células. Un mayor volumen de medio de cultivo se inocula con los cultivos expandidos y los cultivos se expandieron adicionalmente en un biorreactor (que comprende medio de crecimiento suplementado con sulfoximida metionina) en un 5 % de CO2, incubadora a 36 °C. Los cultivos se recolectaron y filtraron para la eliminación de células y residuos celulares. El ABT-700 se purifica a través de una columna de proteína A, seguido de cromatografía en membrana de intercambio
aniónico, cromatografía en columna de intercambio catiónico, filtración viral, ultrafiltración y filtración final en masa. Todas las soluciones se preparan de acuerdo con cGMP.
Ejemplo 2. Preparación de ADC heterogéneos DAR ABT700-vcMMAE
El ABBV-399 es un ADC compuesto por ABT-700 (un anticuerpo anti-cMet IgG1) conjugado con MMAE a través de un conector vc.
El ABBV399 se deriva de la conjugación de vcMMAE a enlaces disulfuro intercatenarios en ABT-700 después de una reducción leve a los grupos sulfhidrilo. Después de una etapa de proceso adicional para eliminar las especies DAR de orden superior, el DAR promedio para ABBV-399 es aproximadamente 3.
Se usaron dos procesos diferentes, el Proceso I (figura 2A y 2B) y el Proceso II (figura 3A y 3B) para preparar composiciones DAR heterogéneas de ABBV-399.
Se preparó una composición ABBV399 heterogénea en DAR mediante un proceso químico de dos etapas: reducción con disulfuro de ABT-700 seguido de alquilación (conjugación) con maleimidocaproil valina-citrulina ("val-cit") alcohol para-aminobencilo ("PABA") monometil auristatina E (denominada en la presente descripción "vcMMAE"), ilustrada más abajo:
En la primera etapa, un número limitado de enlaces disulfuro intercatenarios de ABT700 se reduce con tris(2-carboxietil)fosfina ("TCEP") (50,8 equiv.). El ABT700 parcialmente reducido se conjuga después a vcMMAE (51,8 equiv) en DMSO. El vcMMAE residual sin reaccionar se desactiva con N-acetil-L-cisteína.
Las figuras 2A y 3A muestran resoluciones cromatográficas de las preparaciones de ADC crudas resultantes obtenidas del proceso I (figura 2A) o el proceso II (figura 3A). Como puede verse, la preparación de ADC resultante es una mezcla heterogénea que contiene anticuerpos que tienen cero moléculas de MMAE unidas (pico "E0"), dos moléculas de MMAE unidas (pico "E2"), cuatro moléculas de MMAE unidas (pico "E4"), seis moléculas de MMAE unidas (pico "E6"), ocho moléculas de MMAE unidas (pico "E8") y diez moléculas de MMAE unidas (pico "E10"). Para el proceso I, el DAR promedio de la preparación del producto crudo es aproximadamente 4,3. Para el proceso II, el DAR promedio de la preparación del producto crudo es aproximadamente 3,2.
Ejemplo 3. Preparación de los ADC ABT700-vcMMAE enriquecidos en DAR 3,1 y ABBV-399 enriquecido en una relación E2/E41:1
Preparación de ABBV-399 enriquecido en DAR 3,1 mediante el uso del Proceso I
Para obtener un DAR promedio de 3,1, como se representa en la figura 2B, se usó un proceso cromatográfico por lotes. La solución de producto crudo ABBV-399 (figura 2A) se diluye con un tampón de fosfato de potasio y se trata con una resina HIC para reducir el DAR a aproximadamente 3. La resina HIC se elimina por filtración, se lava con una solución salina tamponada con fosfato y el lavado se combina opcionalmente con la solución del producto ABBV-399 DAR 3,1.
La figura 2B muestra un cromatograma HIC analítico del producto final del proceso I después del tratamiento con la resina HIC (como puede verse, la preparación de ADC resultante es una mezcla heterogénea que contiene anticuerpos que tienen cero moléculas de MMAE unidas (pico "E0"), dos moléculas de MMAE unidas (pico "E2"), cuatro moléculas de MMAE unidas (pico "E4") y seis moléculas de MMAE unidas (pico "E6") y tiene un DAR promedio de 3,1.
Preparación de ABBV-399 enriquecido en una relación E2/E41:1
Para obtener una relación E2/E4 1:1, como se representa en la figura YB, se usó un proceso cromatográfico en columna. La solución de producto crudo ABBV-399 (figura 3A) se diluye con una solución de sulfato de amonio/fosfato de sodio a la concentración diana de unión. Este material se carga en la columna y se une a la resina HIC. Se usa una elución en gradiente escalonado mediante el uso de un tampón de sulfato de amonio/fosfato de sodio para enriquecer los conjugados de fármacos con anticuerpos y aislar las especies de ADC con dos o cuatro moléculas de vcMMAE unidas. Estos se eluyen de la columna en un pico.
La figura 3B muestra un cromatograma HIC analítico del producto final del Proceso II después del enriquecimiento mediante el uso de la columna de cromatografía HIC. Como puede verse, la preparación de ADC resultante es una mezcla heterogénea mezcla heterogénea que contiene anticuerpos que tienen cero moléculas de MMAE unidas (pico "E0"), dos moléculas de MMAE unidas (pico "E2") y cuatro moléculas de MMAE unidas (pico "E4") y tiene un promedio DAR de 3,0.
Como se mostrará más abajo en el ejemplo 16, ABBV-399 ha mostrado efectos anticancerígenos en un ensayo clínico de fase I a una dosis de Q3W de 2,7 mg/kg. También se propone un escalado de la dosis a 3 mg/kg para
identificar la dosis máxima tolerada para los estudios de Fase II, al tener en cuenta que brentuximab vedotin y DCDT2980S (un CD22 dirigido a ADC MMAE) se han tolerado a 1,8 y 2,4 mg/kg, pero no 2,7 o 3,2 mg/kg, respectivamente. Según las consideraciones de la relación fármaco-a-anticuerpo (DAR; carga de MMAE por molécula de anticuerpo), ABBV-399 con un DAR de 3,1 puede ser potencialmente más tolerable que el brentuximab vedotin que tiene un DAR aproximado de 4.
Ejemplo 4. Preparación del conjugado de fármaco con anticuerpo ABT700-PBD
El ABT-700 (S238C)-PBD se compone de dos moléculas de fármaco PBD-conector conjugadas con el mAb de ingeniería cys ABT-700. El sintón de PBD, vaPBD, se conjugó con el anticuerpo ABT-700 (S238C si se usa Kabat, S239C si se usa el sistema de numeración de EU). El proceso de conjugación consiste en una reducción cuantitativa de los disulfuros diseñados genéticamente e intercatenarios. Esto se lleva a cabo mediante la reducción de los disulfuros intercatenarios, la oxidación cuantitativa y la conjugación con un exceso de conector de fármacos PBD. La mezcla de reducción luego se purifica para eliminar el exceso de reactivo y sus subproductos, seguido de la oxidación cuantitativa de los disulfuros intercatenarios y después la conjugación con un exceso de fármaco PBD-conector. Después de inactivar, la mezcla de reacción se purifica y se intercambia con tampón para producir ABT-700 (S238C)-PBD. Se han identificado parámetros de reacción para proporcionar un conjugado con >80 % de carga de fármaco DAR2.

Ejemplo 5. El ABBV-399 se une a ELISA de unión in vitro de cMet recombinante y celular, ensayo de unión celular y análisis de clasificación de células activadas por fluorescencia (FACS)
Se recubrieron placas de 96 pocilios (Costar# 3369) con 100 pL/pocillo de anticuerpo anti-His de ratón (Invitrogen # 37-2900) a 1 pg/mL en PBS pH 7,4 a 4 ° C durante la noche, y luego se bloquearon mediante el uso de Superblock (Pierce, # 37535) durante una hora a temperatura ambiente. Las placas se lavaron 4 veces con PBST y luego se incubaron con 100 pL de dominio extracelular de cMet humano recombinante (rh-cMet ECD-6His) ("6His" divulgado como SEQ ID NO: 100) a 2 pg/mL en Superblock al 10 % en PBST durante 1 h a temperatura ambiente. Las placas se lavaron 4 veces con PBST y luego se incubaron con ABT-700 o IgG humana control en diluciones seriadas en Superblock al 10 % en pocilios por triplicado a temperatura ambiente durante 1 h. Las placas se lavaron 4 veces con PBST y luego se incubaron con 100 pL de IgG de cabra anti-humana-HRP 1:15000 (Thermo-Scientific Pierce, Cat# 31412) a temperatura ambiente durante 1 h. Las placas se lavaron 4 veces en PBST y se añadieron 100 pL de TMB (Pierce, # 34028) a cada pocillo y se incubaron a temperatura ambiente hasta que se desarrolló el color (aproximadamente 10 min). Las reacciones se detuvieron mediante la adición de ácido sulfúrico 2 N (Mallinckrodt chemicals, Cat # H381-05) y se leyó la densidad óptica (OD) a 450 nm.
La unión de ABBV-399 a la superficie de cMet en un panel de células cancerosas humanas se determinó mediante análisis de clasificación celular asistida por fluorescencia (FACS). Para los estudios de unión celular de cMet, las células se recogieron de los frascos con aproximadamente 80 % de confluencia mediante el uso de Tampón de disociación celular (Invitrogen # 13151-014 o # 13150-016). Las células se lavaron una vez en PBS/1 % FBS (tampón FACS), se resuspendieron a 1,5-2 x 106 células/mL en tampón FACS y se transfirieron a una placa de 96 pocilios de fondo redondo (BD Falcon # 3910) a 100 pL/pocillo. Se agregaron diez pL de una concentración 10x de ABT-700, ABBV-399 o controles y las placas se incubaron a 4 °C durante dos horas. Los pocilios se lavaron dos veces con tampón FACS y se resuspendieron en 50 pL de Ab IgG anti-humano 1:500 (AlexaFluor 488, Invitrogen# 11013) diluido en tampón FACS. Las placas se incubaron a 4 °C durante una hora, se lavaron dos veces con tampón FACS. Las células se resuspendieron en 100 pL de PBS/formaldehído al 1 % y se analizaron en un citómetro de flujo Becton Dickinson LSRII.
El ABBV-399 es reactivo con la forma recombinante del dominio extracelular de cMet humano (ECD, residuos 25 932) según se determina por el ensayo inmunoabsorbente ligado a enzimas (ELISA), mediante el uso de un método
de rutina para la medición de la afinidad aparente, como se conoce y está disponible para un experto en la técnica. El ABBV-399 se une al ECD de cMet humano con una afinidad aparente (EC50) de 0,30 nM (TABLA 6), similar a ABT-700 (EC50 de 0,22 nM) (TABLA 6).
El ABBV-399 mostró una afinidad de unión de 0,2 a 1,5 nM (TABLA 6) a las células tumorales, que incluyen las células de cáncer de pulmón NCI-H441, NCI-H292 y NCI-H1650 y las líneas de cáncer gástrico Hs746T, IM-95 y SNU-5. Este ensayo se realizó mediante una rutina para la medición de la afinidad aparente, como se conoce y está disponible para un experto en la técnica.
TABLA 6. Afinidad de unión de ABBV-399 a cMet celular y recombinante
ABBV-399 (EC 50 nmol/L) ABT-700 (EC 50 nmol/L) ECDa de cMet por ELISAb 0,30 0,22
cMet celular por FACSc
Hs746T 0,4 /- 0,1 0,4 /-0,1
SNU-5 1,4 /- 0,4 1,6 /-1,1
IM-95 1,5 /- 0,9 1,8 /- 0,4
NCI-H820 0,2 /- 0,1 0,3 /- 0,2
NCI-H441 1,0 /- 0,6 1,1 /-1,1
NCI-H1573 0,6 /- 0,1 0,4 /-0,1
NCI-H1650 0,3 /- 0,2 0,4 /- 0,2
a Dominio extracelular (residuos 25-932 de cMet)
b Valores de EC50 derivados de ELISA en el que el ECD de cMet se capturó en la placa por medio de una etiqueta His. Los valores son el promedio de seis experimentos.
c Valores de EC50 derivados del análisis FACS de ABBV-399 en líneas celulares de cáncer. Los valores son el promedio de al menos dos experimentos, /- la desviación estándar.
Ejemplo 6. Potencia in vitro de ABBV-399 contra líneas celulares tumorales
Ensayo de citotoxicidad
Las células tumorales se sembraron a 2000-5000 células/pocillo en 180 |jL de medio de crecimiento que contenía FBS al 10 % en placas de 96 pocillos y se cultivaron a 37 °C en una incubadora humidificada con 5 % de CO2. Al día siguiente, se agregaron titulaciones de anticuerpos o ADC en 20 pL y las células se incubaron durante 6 días. La viabilidad celular se determinó mediante el uso de un ensayo de viabilidad celular luminiscente CellTiter-Glo (Promega) de acuerdo con las instrucciones del fabricante. También se incluyó un ADC irrelevante de control negativo sin unir, conjugado con MMAE en todos los ensayos para confirmar que la muerte celular era dependiente del antígeno.
El ABBV-399 inhibió la proliferación de células cancerosas que sobreexpresan cMet, que incluye las líneas celulares amplificadas por MET Hs746T y células de cáncer gástrico SNU-5 (figura 4). Como comparación, el ABT-700 inhibió la proliferación de células con amplificación de MET (figura 4A y figura 4B) pero no de líneas celulares sin amplificación de MET, es decir, las NCI-H820 y NCI-H441 (figura 4C y figura 4D).
Determinación de la densidad del receptor
La densidad en la superficie celular de cMet (capacidad de unión al antígeno por célula) se determinó mediante tinción de inmunofluorescencia indirecta de antígenos de la superficie celular en células cultivadas mediante el uso de QIFIKIT (Dako). Brevemente, las células se recolectaron de un frasco de cultivo como se describió anteriormente para el análisis FACS, se añadieron a una placa de 96 pocillos de fondo redondo a 100 pL/pocillo y se incubaron a 4 °C con 3 pg/mL del anticuerpo a cMet m224G11. Los pocillos, tratados con un anticuerpo monoclonal de ratón irrelevante del mismo isotipo mIgGI a 3 jg/mL, se incluyeron como controles. Después de una hora de incubación con el anticuerpo primario, las células se centrifugaron durante 3 minutos a 300 x g, se lavaron dos veces con tampón FACS y se incubaron durante una hora a 4 °C con 100 pL del anticuerpo conjugado con FITC proporcionado por QIFIT diluido 1:50 en tampón FACS. Las células se centrifugaron durante 3 minutos a 300 x g, se lavaron dos veces con tampón FACS y se fijaron con 100 pL/pocillo de formaldehído al 1% en PBS. Para la tinción de inmunofluorescencia indirecta de las perlas de QIFIKIT, se añadieron 100 pL de perlas resuspendidas del Vial 1 y Vial 2 a pocillos separados, se centrifugaron durante 3 min a 300 x g, se lavaron una vez con tampón FACS y se fijaron con 100 pL/pocillo de formaldehído al 1% en PBS. Los datos se obtuvieron en un citómetro de flujo LSRII de Becton Dickinson y se registraron los valores de Geomean para las 5 poblaciones de perlas y se usaron para calcular una curva estándar en base a las moléculas de anticuerpo específicas del lote por perla. La curva estándar se usó para asignar ABC (capacidad de unión a anticuerpos o número de receptores) a muestras de células teñidas.
El ABBV-399 es citotóxico para las células cancerosas que sobreexpresan cMet. Para determinar la correlación del nivel de expresión de cMet con la sensibilidad al ABBV-399, el análisis in vitro se expandió para incluir un panel de 16 líneas celulares. Estos incluyeron 6 líneas de NSCLC (A549, NCI-H1573, NCI-H820, NCI-H441 y NCI-H1650, 4 líneas de cáncer gastroesofágico (Hs746T, SNU-5, SNU-620 e IM-95), 2 líneas de CCR (SW-48 y Ht -29), 2 líneas de cáncer de mama (MDA-MB-231 y MCF-7), la línea de cáncer pancreático KP4 y la línea de cáncer de glioblastoma U-87 MG. También se analizaron líneas celulares de NSCLC adicionales (EBC-1, NCI-H226, SW900, HCC15, SK-MES-1 y NCI-H1702) y se muestran en la Tabla 7A.
Tabla 7A
receptores Citotoxicidad ICñn (nM)
Línea celular de NSCLC cMet/célula ABBV-399 ABT 700-PBD MMAE/PBD H820(Adeno) 320000 0 ,1 0 , 02 5 H441(Adeno) 197000 0,06 0,003 20 H1573(Adeno) 116000 18,3 0,07 261 H1650(Adeno) 55 000 47,9 0,4 120 A549(Adeno) 43000 1,6 0 ,1 16
EBC-1 (escamoso, amp) 233000 0,06 0,095 0,6 H226 (Escamoso) 114000 igual que el control 0,04 n/a SW900 (escamoso) 63000 7,5 0,02 375 HCC15 (escamoso) 59 000 igual que el control 0,003 n/a
SK-MES-1 (Escamoso) 39000 igual que el control 0,17 n/a H1703 (Escamoso) 23000 igual que el control 0,7 n/a Otras líneas celulares Hs746T
(Ga, amp) 350000 0,11 0,018 6,1
BT-20 (Br) 41000 0,23 0 ,1 2,3 U87MG (GBM) 22000 1,9 0,21 9 M059J (GBM) 87000 3,6 0,03 120 U118MG (GBM) 12500 0,54 0,2 2,7
KP4 (Pa) 15000 2,9 0,02 145 SW48 (CRC) 26 000 igual que el control 0,0029 >1000 NHBE (epitelial bronquial
normal) 40 000 ninguna ninguna n/a
El análisis FACS demostró que estas líneas celulares poseen un intervalo de niveles de expresión de cMet cuantificados mediante la capacidad de unión del anticuerpo a cMet que representa el número de moléculas de cMet de la superficie celular (TABLA 7B). La sensibilidad a ABBV-399 en el ensayo de proliferación celular se cuantificó como muerte máxima e IC50 (TABLA 7B). Estos datos sugieren que hay un nivel umbral de expresión de cMet requerido para una muerte significativa por ABBV-399. Las excepciones a esto fueron las líneas celulares conocidas por tener un lazo de HGF autocrino, tales como IM 95, KP4 y U-87 MG, en las que los niveles de expresión de cMet más bajos eran suficientes para que ABBV-399 ejerciera una citotoxicidad significativa.
TABLA 7B. Expresión de cMet en células tumorales in vitro y sensibilidad a ABBV-399______ Expresión de cMeta Muerte máximab ABBV-399 IC50c
Cáncer de pulmón
A549 43000 22 % 1,6 /- 1,1 NCI-H1573 115667 18 % 18 /- 14 NCI-H820 320000 87 % 0,20 /- 0,07 NCI-H441 197000 56 % 0,06 /- 0,05 NCI-H1650 4500 13 % 47,9 /- 8,5 EBC-1 233231 96 % 0,06 /- 0,03 Cáncer gástrico
Hs746T 350000 87 % 0,11 /- 0,06 SNU-620 230000 80 % 0,17 /- 0,08 SNU-5 291 000 85 % 0,28 /- 0,07 IM-95 21500 53 % 1,7 /- 0,9 Cáncer colorrectal
SW48 25500 0 % NA
HT-29 161438 70 % 9,0 /- 1,4 Cáncer de mama
MDA-MB-231 30500 0 % NA
MCF-7 8300 0 % NA
Cáncer de páncreas
KP4 15300 53 % 2,9 /- 1,9 Glioblastoma
U-87MG 22 000 30 % 1,9 /- 0,1 Líneas celulares no tumorales
NHBE (epitelial bronquial) 40085 10 % NA HUVEC (endotelial vascular 15790 6 % NA HMEC (epitelial mamario) ND 0 % NA PrEC (epitelial de próstata) 64853 0 % NA NHDF (fibroblastos dérmicos) 1602 0 % NA a Número aproximado de moléculas de cMet en la superficie celular determinada por análisis FACS como capacidad de unión de anticuerpos para la unión de m224G11 (parental murino de ABT-700) a 10 pg/mL.
b Relativo al control no tratado a s i pg/mL en una proliferación de seis días.
Ejemplo 7. El ADC ABT 700-PBD inhibe la proliferación de células tumorales en un amplio panel de líneas celulares
Las células tumorales se sembraron a 2000-5000 células/pocillo en 180 pL de medio de crecimiento que contenía FBS al 10 % en placas de 96 pocilios y se cultivaron a 37 °C en una incubadora humidificada con 5 % de C02. Al día siguiente, se agregaron titulaciones de ADC en 20 pL y las células se incubaron durante 6 días. La viabilidad celular se determinó mediante el uso de un ensayo de viabilidad celular luminiscente CellTiter-Glo (Promega) de acuerdo con las instrucciones del fabricante. También se incluyó un ADC irrelevante de control negativo sin unir, conjugado con MMAE en todos los ensayos para confirmar que la muerte celular era dependiente del antígeno.
Los resultados se muestran en la figura 5. Ambos ADC de cMet fueron activos contra un panel diverso de tipos de tumores, con niveles variables de expresión de cMet (alta/baja) y amplificación génica (amp). La columna MMAE/PBD indica cuánto más el ADC MMAE se requiere para dar la misma actividad citotóxica que la lograda con el ADC PBD. En la mayoría de las líneas celulares, el ADC PBD es significativamente más potente que el conjugado MMAE.
Ejemplo 8. El ADC ABT 700-PBD es activo in vitro contra líneas celulares de cáncer colorrectal humano
Las células tumorales se sembraron a 2000-5000 células/pocillo en 180 pL de medio de crecimiento que contenía FBS al 10 % en placas de 96 pocillos y se cultivaron a 37 °C en una incubadora humidificada con 5 % de CO2. Al
día siguiente, se añadieron titulaciones de ADC y fármaco libre (PBD y MMAE) en 20 |jL y las células se incubaron durante 6 días. La viabilidad celular se determinó mediante el uso de un ensayo de viabilidad celular luminiscente CellTiter-Glo (Promega) de acuerdo con las instrucciones del fabricante. También se incluyó un ADC irrelevante de control negativo sin unir, conjugado con MMAF (Ab095 MMAF) en todos los ensayos para confirmar que la muerte celular era dependiente del antígeno. El ADC cetuximab-MMAE es un control positivo. Los niveles de densidad del receptor se calcularon como se describe en el ejemplo 6.
Los resultados se muestran en las figuras 6A y 6B. El ABT700-PBD es activo contra una variedad de líneas celulares de cáncer colorrectal, incluidas aquellas con bajos niveles de receptores cMet en la superficie celular (por ejemplo, SW48, figura 6B). Una línea celular amplificada por el gen cMet en promedio tiene 200-300 K de receptores por célula. La actividad del ADC ABBV-399 se muestra además con fines comparativos. Donde no se ingresan resultados, no se observó actividad. En general, ABT700-PBD es más activo que ABBV-300 en líneas celulares de cáncer colorrectal.
Ejemplo 9. El ADC ABT 700-PBD es activo in vitro contra líneas celulares de cáncer de cerebro humano
Las células tumorales se sembraron a 2000-5000 células/pocillo en 180 |jL de medio de crecimiento que contenía FBS al 10 % en placas de 96 pocilios y se cultivaron a 37 °C en una incubadora humidificada con 5 % de C02. Al día siguiente, se añadieron titulaciones de ADC y fármaco libre (PBD y MMAE) en 20 pL y las células se incubaron durante 6 días. La viabilidad celular se determinó mediante el uso de un ensayo de viabilidad celular luminiscente CellTiter-Glo (Promega) de acuerdo con las instrucciones del fabricante. También se incluyó un ADC irrelevante de control negativo sin unir, conjugado con MMAF (Ab095 MMAF) en todos los ensayos para confirmar que la muerte celular era dependiente del antígeno. Los niveles de densidad del receptor se calcularon como se describe en el ejemplo 6. Una línea celular amplificada por el gen cMet en promedio tiene 200-300 K de receptores por célula. Los resultados se muestran en la figura 7. ABT700-PBD es activo contra una variedad de líneas celulares de cáncer de cerebro, que incluyen aquellas con bajos niveles de receptores cMet en la superficie celular (por ejemplo, SW48, figura 6B). La actividad del ADC ABBV-399 se muestra además con fines comparativos. Donde no se ingresan resultados, no se observó actividad. En general, ABT700-PBD es más activo que ABBV-300 en líneas celulares de cáncer de cerebro.
Ejemplo 10. El ADC ABT 700-PBD es activo in vivo contra xenoinjertos de tumores colorrectales humanos
Se evaluó la eficacia in vivo de ABT-700, ABBV-399 y ABT-700 PBD en ratones trasplantados con células colorrectales SW-48 (cMet IHC 1+). Los experimentos se realizaron esencialmente como se describe en el ejemplo 13 más abajo.
Los ADC o anticuerpos se administraron cada siete días a las dosis mostradas (mg/kg). ABT-700 PBD es superior a ABBV-399 en xenoinjertos SW-48 de baja expresión de cMet. Véase la figura 8.
Ejemplo 11. Los ADC ABBV-399 y ABT700-PBD son activos in vivo contra xenoinjertos derivados de pacientes con NSc Lc humano
La eficacia de los ABBV-399 ABT700-PBD se determinó en xenoinjertos derivados de pacientes con cáncer de pulmón de células no pequeñas y colorrectal. Se implantaron por vía subcutánea fragmentos tumorales de 3 a 5 mm3 en la etapa 3 (P3) en el flanco trasero derecho de ratones NSG (The Jackson Laboratory) con un trocar. ABBV-399 y ABT-700 PBD se administraron cada siete días para un total de seis dosis. Los números entre paréntesis representan la dosis administrada en mg/kg. Para todos los grupos, los volúmenes tumorales se trazaron solo por la duración que permitió que el conjunto completo de animales permaneciera en estudio. Si los animales tuvieran que retirarse del estudio, se monitoreó el crecimiento tumoral de los animales restantes hasta alcanzar los criterios de valoración definidos. Los resultados del retraso del crecimiento tumoral (TGD) se muestran en la tabla 8.

Se muestran gráficos para tres xenoinjertos tumorales humanos diferentes con niveles de expresión relativamente bajos (CTG-0363), intermedios (CTG-0159) y altos (CTG-0170) de ARNm de cMet, un sustituto de los niveles de proteína cMet en la superficie celular (figuras 9A, 9B y 9C, respectivamente). La respuesta tumoral a cada ADC depende de los niveles de cMet. El ADC ABT700-PBD es más activo que el ABBV-399, a aproximadamente 1/10 de la dosis.
Ejemplo 12. Los ABBV-399 son activos in vivo contra xenoinjertos derivados de pacientes con NSCLC humano Para los modelos de xenoinjerto derivados de pacientes LG0703 y LG1049 (The Jackson Laboratory, Sacramento, CA), se determinó la eficacia de ABBV-399 en xenoinjertos derivados de pacientes con cáncer de pulmón de células no pequeñas. Se implantaron por vía subcutánea fragmentos tumorales de 3 a 5 mm3 en la etapa 3 (P3) en el flanco trasero derecho de ratones NSG (The Jackson Laboratory) con un trocar. Para todos los grupos, los volúmenes tumorales se trazaron solo por la duración que permitió que el conjunto completo de animales permaneciera en estudio. Si los animales tuvieran que retirarse del estudio, se monitoreó el crecimiento tumoral de los animales restantes hasta alcanzar los criterios de valoración definidos. La eficacia se representa en un diagrama de Kaplan-Meier para los modelos (A) LG0703 y (B) LG1049 como fracciones que alcanzan los volúmenes de tumores indicados después de la terapia. En ambos modelos, se administraron ABBV-399 y agentes de control cada cuatro días para un total de seis dosis. En el modelo LG1049, se administró ABT-700 cada siete días para un total de seis dosis. Los números entre paréntesis representan la dosis administrada en mg/kg.
El ADC ABBV-399 es más activo que el ABT-700 solo. Véase la figura 10.
Ejemplo 13. El ABBV399, solo y en combinación, inhibe el crecimiento tumoral en tumores que sobreexpresan cMet en modelos animales
ABBV-399 ha demostrado efectos antitumorales robustos y reproducibles en una variedad de modelos de xenoinjerto, que incluyen los modelos multiformes de cáncer gástrico, NSCLC y glioblastoma. La actividad en los tumores se basa en parte en la entrega de la carga útil citotóxica de MMAE. Además, ABBV-399 puede tener
además actividad antitumoral mediante la inhibición de la señalización de cMet tanto dependiente de HGF como independiente de HGF y la función efectora mediada por anticuerpos.
La eficacia in vivo de ABBV-399 se evaluó en ratones trasplantados con (figura 11A) cáncer gástrico Hs746T, (figura 11B) células de cáncer de pulmón NCI-H441 y (figura 11C) células de cáncer colorrectal SW-40. Se obtuvieron ratones hembra SCID, SCID-Beige y desnudos de Charles River (Wilmington, MA) y se alojaron a diez ratones por jaula. El peso corporal tras la llegada fue de 20-22 g. La comida y agua estaban disponibles a voluntad. Los ratones se aclimataron a las instalaciones de animales durante un período de al menos una semana antes del comienzo de los experimentos. Los animales se probaron en la fase de luz de un programa de 12 horas de luz: 12 horas de oscuridad (luces encendidas a las 06:00 horas). Todos los experimentos se realizaron de conformidad con el Comité Institucional de Uso y Cuidado de Animales de AbbVie y las Guías de los Institutos Nacionales de Salud para el Cuidado y Uso de Animales de Laboratorio en una instalación acreditada por la Asociación para la Evaluación y Acreditación del Cuidado de Animales de Laboratorio.
Para generar xenoinjertos, se inyectó por vía subcutánea una suspensión de células tumorales viables mezcladas con una cantidad igual de Matrigel (BD Biosciences) en el costado de ratones de 6 a 8 semanas de edad. El volumen de inyección fue de 0,2 mL compuesto de una mezcla 1:1 de S-MEM y Matrigel (BD Biosciences). Los tumores se emparejaron en tamaño a aproximadamente 200-250 mm3 a menos que se indicara de cualquier otra manera. La terapia comenzó el día o 24 h después del emparejamiento del tamaño de los tumores. Los ratones pesaron aproximadamente 25 g al inicio de la terapia. Cada grupo experimental incluyó 8-10 animales. Los tumores se midieron dos o tres veces por semana. Las mediciones de la longitud (L) y el ancho (W) del tumor se obtuvieron mediante calibradores electrónicos y el volumen se calculó de acuerdo con la siguiente ecuación: V = L x W2/2. Los ratones se sacrificaron cuando el volumen del tumor alcanzó un máximo de 3000 mm3 o tras la presentación de ulceraciones cutáneas u otras morbilidades, lo que ocurriera primero. Para Hs746T, el ABT-700 se administró cada siete días, mientras que el ABBV-399 se administró cada cuatro días. Para los xenoinjertos de NCI-H441, se administraron ABT-700 y ABBV-399 cada cuatro días para un total de seis dosis. Los números entre paréntesis representan la dosis administrada en mg/kg y las flechas indican los días de administración. En ambos tipos de cáncer, el ADB ABBV-399 es más activo que el ABT-700 solo, y el efecto depende de la dosis (figuras 11A y 11B).
(figura 11C) La eficacia de combinación de ABBV-399 y FOLFIRI se determinó mediante el uso de xenoinjertos de cáncer colorrectal humano SW-48. Se obtuvieron 5-fluorouracilo (APP Pharmaceuticals, Schaumburg, IL), irinotecán (Hospira, Lake Forest, IL) como soluciones y se diluyeron con cloruro de sodio al 0,9 % para inyección (USP) y leucovorina cálcica (Fluka Chemical Corp., Milwaukee, WI) se obtuvo como una sal y se reconstituyó con solución salina antes de la dosificación. Los agentes estándar de atención 5-fluorouracilo (50 mg/kg) e irinotecán (30 mg/kg) se administraron por vía intravenosa y leucovorina (25 mg/kg) se administró por vía oral en régimen de Q7Dx5 (FOLFIRI). El control de IgG, Ig MMAE y ABBV-399 se administraron por vía intraperitoneal cada siete días. Los números entre paréntesis representan la dosis administrada en mg/kg y las flechas indican los días de administración. La combinación de ABBV-399 FOLFIRI es eficaz en los xenoinjertos de cáncer de colon SW-48.
Ejemplo 14. Eficacia de ABBV-399 contra modelos de xenoinjerto de tumores humanos refractarios a ABT-700
La eficacia de ABBV-399 se evaluó en ratones xenotrasplantados con Hs746T parental solo (figura 12B) o después de una recaída tras el tratamiento con ABT-700 (figuras 12A y 12B). La figura 12C evalúa la eficacia de ABBV=399 después de una recaída tras el tratamiento con ABT-700 en xenoinjertos de ratones trasplantados con tumores de xenoinjerto EBC-1. Los números entre paréntesis representan la dosis administrada en mg/kg y las flechas indican los días de administración. Los volúmenes tumorales se representan como media ± S.E.M.
La eficacia de ABBV-399 se evaluó en un modelo de carcinoma gástrico (Hs746T) y un modelo de carcinoma de células escamosas de pulmón (EBC-1) que se hicieron refractarios a ABT-700 por exposición repetida al anticuerpo in vivo (Hs746T ABT-700R y EBC -1 ABT-700R). Inicialmente, el tratamiento de xenoinjertos derivados de1Hs746T parental con ABT-700 resultó en estancamiento tumoral seguido de recaída (figura 12A; línea azul). El tratamiento de estos tumores recurrentes (línea roja) con ABBV-399 condujo a la regresión (figura 12A, línea roja). Por el contrario, los xenoinjertos Hs746T ABT-700R fueron refractarios al tratamiento con ABT-700 con rápido crecimiento tumoral en la terapia (figura 12B; línea azul). Cuando estos tumores refractarios alcanzaron un tamaño medio de cohorte de aproximadamente 1000 mm3, el tratamiento con ABBV-399 resultó en una regresión tumoral (figura 12B; línea roja) seguida de un rápido crecimiento eventual. El tratamiento de Hs746T ABT-700R de aproximadamente 300 mm3 con ABBV-399 resultó en una regresión tumoral completa (figura 12B). Se observaron resultados similares después del tratamiento de la línea celular resistente a ABT-700 EBC-1 con ABT-700 seguido de ABBV-399. Estos resultados sugieren que la eficacia de ABBV-399 es independiente de la respuesta a ABT-700, al menos para líneas celulares con cMet amplificado.
Ejemplo 15. Formulación de ABBV-399 para uso clínico
El producto farmacológico ABBV-399 se proporciona como un polvo liofilizado estéril para reconstitución. Cada vial contiene 100 mg de ABBV-399. Después de la reconstitución con 5,0 mL de agua estéril para inyección, la concentración final de ABBV-399 es de 20 mg/mL. Además de ABBV-399, la formulación contiene sacarosa,
polisorbato 80 y está en un tampón de histidina. Antes de la administración, el ABBV-399 se diluye adicionalmente en solución salina normal a un rango de concentración entre 1-10 mg/mL, en dependencia del peso del sujeto. Ejemplo 16. Estudio abierto de fase I, de escalado de dosis y de expansión de ABBV-399, un conjugado de fármacos con anticuerpos (ADC) dirigido a cMet, en pacientes (pts) con tumores sólidos avanzados
16.1. Resumen
Un estudio abierto de fase 1/1b en curso evalúa la seguridad, la farmacocinética (PK) y la eficacia preliminar de ABBV-399 en sujetos con tumores sólidos avanzados. El estudio consta de dos fases: (1) una Fase de Escalado/Expansión de dosis (Monoterapia) y una Fase de Terapia Combinada. Los sujetos con tumores sólidos avanzados con sobreexpresión de cMet, mutación del exón 14 de MET o amplificación de MET que posiblemente incluyen, pero no se limitan a, cáncer de NSCLC, esófago/gástrico, CCR o de cabeza y cuello pueden inscribirse en las fases de expansión de dosis y terapia combinada del estudio.
La fase de monoterapia del estudio evaluó la seguridad y el perfil farmacocinético de ABBV-399 cuando se administró por vía intravenosa en aproximadamente 24 a 42 sujetos al seguir el esquema de escalado de dosis representado en la figura 13. El ABBV-399 se administró a niveles de dosis crecientes que comienzan a partir de 0,15 mg/kg en ciclos de dosificación de 21 días. Según los datos de seguridad y PK a partir de la dosificación cada 21 días, el ABBV-399 también se administrará cada 14 días en un programa de 28 días. Se inscribirán de tres a 6 sujetos en cada cohorte y se dosificarán una vez cada 21 (una dosis por ciclo de 21 días) o 14 (2 dosis por ciclo de 28 días) hasta la progresión de la enfermedad o toxicidad inaceptable para determinar la dosis máxima tolerada (MTD) o la dosis máxima administrada (MAD). Las definiciones de toxicidad limitante de dosis (DLT) se usarán para tomar decisiones con respecto al escalado de la dosis. Según los datos disponibles de seguridad, PK y farmacodinámica (PDx), se inscribirán hasta 40 sujetos en una cohorte de expansión que evaluará además el ABBV-399 a un nivel de dosis que sea igual o inferior a la MTD o MAD. En la dosis de expansión, se inscribirán sujetos con tumores sólidos avanzados con sobreexpresión de cMet, mutación del exón 14 de MET o amplificación de MET. En la fase de terapia combinada, se inscribirán hasta 18 sujetos en cada uno de los grupos de terapia combinada como se describe más abajo:
• Cohorte de combinación A: sujetos elegibles para recibir ABBV-399 más erlotinib
• Cohorte de combinación B: sujetos elegibles para recibir ABBV-399 más cetuximab
• Cohorte de combinación C: sujetos elegibles para recibir ABBV-399 más bevacizumab
• Cohorte de combinación D: sujetos elegibles para recibir ABBV-399 más nivolumab
Todos los sujetos serán evaluados para la seguridad y la tolerabilidad del régimen, el perfil PK de ABBV-399 y la evidencia preliminar de eficacia. En los grupos de terapia combinada, se pueden inscribir sujetos con tumores sólidos avanzados con sobreexpresión de cMet, mutación del exón 14 de MET o amplificación de MET. Los sujetos en los grupos combinados A, B o C serán asignados a un programa de dosificación de ABBV-399 de 14 o 21 días, mientras que los sujetos en el grupo D recibirán ABBV-399 en un programa de 14 días para coincidir con nivolumab con dosificación cada 14 días.
Se requiere tejido tumoral de archivo para inscribirse en este estudio. El tejido tumoral se analizará para determinar la proteína cMet, el número de copias de MET y otros biomarcadores. La expresión de cMet se determinará mediante un ensayo de inmunohistoquímica; la amplificación de MET se determinará por hibridación fluorescente in situ (FISH) o secuenciación de ADN del tumor o ADN tumoral circulante.
16.2. Selección de pacientes: diagnóstico y criterios principales para la inclusión/exclusión
Algunos de los criterios para la inclusión en el escalado/expansión de dosis en monoterapia con ABBV-399:
• El sujeto debe tener s i 8 años
• Sujeto con tumor sólido avanzado que incluye, pero no se limita a, cáncer de pulmón de células no pequeñas (NSCLC), cáncer colorrectal, de mama, de ovario, esofágico/gástrico y de cabeza y cuello.
• El sujeto debe tener un tumor sólido avanzado que no sea susceptible de resección quirúrgica u otras opciones terapéuticas aprobadas que hayan demostrado beneficio clínico.
o Para la expansión de dosis: el sujeto debe tener tumor con sobreexpresión de cMet, mutación del exón 14 de MET o amplificación de MET.
• El sujeto tiene un estado de rendimiento del Grupo de Oncología Cooperativa del Este (ECOG) de 0 a 2.
• El sujeto debe tener una enfermedad medible según RECIST versión 1.1
Criterios de inclusión adicionales para sujetos inscritos en la fase de terapia combinada
• Los sujetos en los grupos de terapia combinada deben cumplir con los criterios de inclusión anteriores y ser elegibles para recibir erlotinib, cetuximab, bevacizumab o nivolumab según la información de prescripción más actualizada, o según el criterio del investigador.
Criterios principales de exclusión:
Para todas las cohortes:
• El sujeto ha recibido terapia contra el cáncer que incluye quimioterapia, inmunoterapia, radioterapia, inmunoterapia, terapia biológica o cualquier investigación dentro de un período de 21 días, o terapia herbal dentro de los 7 días anteriores a la primera dosis de ABBV-399.
• La radioterapia paliativa para o metástasis de huesos, pieles subcutáneas dolorosas durante 10 fracciones o menos no está sujeta a un período de lavado.
• Para moléculas pequeñas específicas aprobadas, un período de lavado de 5 semividas es adecuado (no se requiere un período de lavado para los sujetos que actualmente toman erlotinib).
• El sujeto ha conocido metástasis no controladas en el sistema nervioso central (SNC). Los sujetos con metástasis cerebrales son elegibles después de la terapia definitiva, siempre que estén asintomáticos sin esteroides y anticonvulsivos durante al menos 2 semanas antes de la primera dosis de ABBV-399.
• El sujeto tiene eventos adversos clínicamente significativos no resueltos sGrado 2 de la terapia contra el cáncer previa, excepto para alopecia o anemia.
• El sujeto ha tenido una cirugía mayor dentro de los 21 días previos a la primera dosis de ABBV-399.
Criterios de exclusión adicionales para sujetos inscritos en la fase de terapia combinada
• Los sujetos inscritos en la fase de terapia combinada deben satisfacer los criterios de exclusión anteriores y también los siguientes: Los sujetos no pueden recibir ABBV-399 en combinación con erlotinib, cetuximab, bevacizumab o nivolumab si tienen alguna condición médica que en opinión del investigador el sujeto tiene un riesgo inaceptablemente alto de toxicidad a partir de la combinación.
• Los sujetos pueden no recibir cetuximab si tienen mutación K-ras.
• Los sujetos pueden no recibir bevacizumab si tienen NSCLC escamoso.
También está previsto que, en ciertos estudios y para el uso clínico futuro de los ADC anti-cMet divulgados en la presente descripción, los pacientes se seleccionen en función de sus niveles de expresión de cMet (amplificación génica, cMet de membrana) y mutación del exón 14 de MET. Los métodos para evaluar cada uno de estos marcadores se proporcionan más abajo.
16.3 Régimen de dosificación
Fase de escalado/expansión de dosis:
El ABBV-399 se administró como una infusión intravenosa una vez cada 21 días hasta la progresión de la enfermedad o toxicidad intolerable. La dosificación comenzó a 0,15 mg/kg y se escaló a 0,3, 0,6, 1,2, 1,8, 2,4, 3,0 y 3.3 mg/kg en cohortes posteriores según lo tolerado. Pueden emplearse dosis alternativas (intermedias o superiores) o programas de dosificación en base a la seguridad clínica y los datos de PK. También se utilizó una dosis de 2,7 mg/kg en función de la seguridad clínica y los datos de PK. Según los datos de seguridad y PK de la dosificación cada 21 días, el ABBV-399 se administrará además cada 14 días en un programa de 28 días (dosis inicial de 1,6 mg/kg). El ABBV-399 se ha administrado durante 30 ± 10 minutos. No se administra como un impulso intravenoso o bolo.
Fase de terapia combinada:
El ABBV-399 se combinará con dosis estándar de erlotinib, cetuximab, bevacizumab o nivolumab a partir de un nivel
de dosis de ABBV-399 por debajo de la MTD o MAD y después se elevará no más que la MTD o MAD determinada en el escalado/expansión de dosis con monoterapia. Las definiciones de toxicidad limitante de la dosis se aplicarán a la porción de escalado de dosis de cada combinación.

16.4. Evaluaciones
Las visitas de estudio y las evaluaciones se realizarán en la selección, y al menos semanalmente durante el primer ciclo y el día 1 de cada ciclo posterior. Las evaluaciones incluirán un examen físico limitado, pruebas de hematología y química antes de la dosificación del fármaco del estudio y en la visita final. Los ECG se recolectarán en la selección, el día 1 del ciclo 1, el día 1 del ciclo 2 y en la visita final. Los eventos adversos, los datos de laboratorio y los signos vitales se evaluarán a lo largo del estudio.
Las evaluaciones radiográficas iniciales del tumor con CT (o MRI) de la cabeza, el tórax, el abdomen y la pelvis se obtendrán no más de 28 días antes del día 1 del ciclo 1. La exploración de CT (o MRI) se repetirá aproximadamente cada 6 semanas después del inicio de la terapia para evaluar el alcance de la carga tumoral. Las evaluaciones radiográficas del tumor continuarán hasta que la progresión de la enfermedad se documente mediante imágenes, el inicio de una nueva terapia contra el cáncer, la muerte o la retirada del consentimiento. La evaluación de la respuesta se basará en RECIST versión 1.1. Además, el investigador evaluará al sujeto en busca de evidencia de progresión clínica de la enfermedad en cada visita.
16.4.1.Evaluaciones de biomarcadores
Se requiere tejido tumoral de archivo (se prefiere la muestra más reciente) para inscribirse en este estudio. Si un sujeto tiene datos de laboratorio locales o centrales que muestran la sobreexpresión de cMet, mutación del exón 14 de MET o amplificación de MET y no hay tejido tumoral de archivo disponible, el sujeto puede ser elegible después de una discusión con el Monitor Médico. Puede obtenerse una biopsia opcional previa y durante el tratamiento (en cualquier momento después del inicio de la terapia) de los sujetos que consienten voluntariamente si es seguro hacerlo a juicio del investigador. Deben seguirse los procedimientos institucionales para fijar e incrustar tejido recién recolectado en parafina. El tejido tumoral se analizará para determinar la proteína cMet, el número de copias de MET y otros biomarcadores.
La expresión de cMet se determinará mediante un ensayo de inmunohistoquímica (véase el ejemplo 17); la amplificación de MET se determinará mediante hibridación fluorescente in situ (FISH) o secuenciación de ADN de tumor o ADN de tumor circulante (véase el ejemplo 18). Las muestras biológicas se recolectarán en puntos de tiempo designados durante todo el estudio para realizar investigaciones con la intención de identificar biomarcadores
asociados con el resultado del sujeto o para caracterizar mejor la enfermedad.
Criterios de evaluación

Eficacia
Todos los análisis de eficacia son de naturaleza exploratoria. Los criterios de valoración de eficacia exploratoria incluyen la tasa de respuesta objetiva (ORR) (determinada mediante el uso de RECIST versión 1.1) supervivencia libre de progresión (PFS) y duración de la respuesta (DOR).
Tasa de respuesta objetiva
La tasa de respuesta objetiva (ORR) se define como la proporción de sujetos con una respuesta parcial o completa confirmada al tratamiento. La ORR para cada cohorte de tratamiento se estimará con todos los sitios agrupados. Los intervalos de confianza del 80 % a 2 lados de ORR, así como también de las tasas de CR y PR, se proporcionarán según el método de Clopper-Pearson (exacto).
Supervivencia libre de progresión
Para cada sujeto, el tiempo de PFS se define como el tiempo desde la primera dosis de ABBV-399 del sujeto hasta la progresión de la enfermedad o la muerte del sujeto, lo que ocurra primero. En el caso de que ninguno de los dos eventos ocurra, el tiempo de PFS se censurará en la fecha de la última evaluación de la enfermedad. Todos los sujetos serán seguidos hasta la progresión de la enfermedad o hasta 24 meses para aquellos que continúan el estudio de fármacos.
El tiempo de PFS para las cohortes de tratamiento se resumirá mediante estimaciones de Kaplan-Meier. El tiempo medio y mediano con intervalos de confianza de 80 % a 2 lados se calculará para describir las distribuciones del tiempo hasta el evento.
Duración de la respuesta
La duración de la respuesta (DOR) para un sujeto se define como el tiempo desde la respuesta objetiva inicial del sujeto a la terapia con el fármaco del estudio hasta la progresión de la enfermedad o la muerte, lo que ocurra primero. Si las fechas de progresión de la enfermedad o muerte no están disponibles, el DOR será censurado en la fecha de la última evaluación del tumor. La DOR se analizará de la misma manera que para PFS.
Evaluaciones de tumores
La evaluación radiográfica inicial del tumor debe realizarse dentro de los 28 días anteriores al día 1 del ciclo 1y
consistirá en CT (o MRI o CT sin contraste en sujetos que no pueden tolerar el contraste) de la cabeza, el tórax, el abdomen y la pelvis (y otras regiones involucradas en tumores según lo clínicamente indicado). En general, las imágenes durante el tratamiento con ABBV-399 ocurrirán aproximadamente cada 6 semanas (las imágenes pueden obtenerse hasta 7 días antes de la siguiente dosis del fármaco). Para la combinación ABBV-399 con nivolumab, la primera imagen en tratamiento planificada ocurrirá aproximadamente a las 9 semanas con imágenes posteriores aproximadamente cada 6 semanas. Las imágenes deben realizarse antes de administrar la siguiente dosis programada de ABBV-399. Los sujetos que suspendan el fármaco del estudio por cualquier motivo que no sea la enfermedad progresiva demostrada por imagen serán seguidos hasta que tengan una enfermedad progresiva documentada por imagen o comiencen una nueva terapia anticancerígena, la muerte o retiren el consentimiento. Las imágenes se realizarán además en la visita final para los sujetos que no hayan tenido una progresión radiográfica documentada según los criterios RECIST 1.1 si se justifica clínicamente. Las imágenes pueden realizarse además en otros momentos si el investigador sospecha la progresión del tumor. Las imágenes del cerebro para la enfermedad metastásica solo se repetirán si está clínicamente indicado. La misma técnica de imagen debe usarse durante todo el estudio si es posible. La evaluación del tumor realizada en la selección servirá como base inicial para la evaluación clínica. Los cambios en las lesiones medibles durante el curso de la terapia se evaluarán mediante el uso de RECIST versión 1.1, como se describe más abajo.
RECIST (Versión 1.1) Criterios para la respuesta tumoral
Los criterios de respuesta se evaluarán mediante el uso de RECIST (versión 1.1). Los cambios en las lesiones medibles durante el curso de la terapia deben evaluarse mediante el uso de los criterios que se enumeran más abajo.
a. Elegibilidad
Los sujetos con enfermedad medible al inicio pueden tener una respuesta tumoral objetiva evaluada por los criterios RECIST. La enfermedad medible se define por la presencia de al menos una lesión medible. Si la enfermedad medible se limita a una lesión solitaria, su naturaleza neoplásica debe confirmarse mediante citología/histología si es posible.
b. Capacidad de medición

Todas las mediciones deben tomarse y registrarse en notación métrica, mediante el uso de calibradores si se evalúa clínicamente. Todas las evaluaciones de inicio deben realizarse lo más cerca posible al comienzo del tratamiento y no más de 4 semanas antes del comienzo del tratamiento.
Se debe utilizar el mismo método de evaluación y la misma técnica para caracterizar cada lesión identificada e informada al inicio y durante el seguimiento.
Las lesiones clínicas solo se considerarán medibles cuando sean superficiales (por ejemplo, nodulos cutáneos y ganglios linfáticos palpables) y tengan un diámetro S10 mm, según se evalúe con calibradores. Para el caso de las lesiones cutáneas, se recomienda la documentación mediante fotografía en color que incluya una regla para estimar el tamaño de la lesión.
c. Métodos de medición
La CT convencional debe realizarse con cortes de 5 mm o menos en el grosor de corte contiguo. Esto se aplica a los tumores de tórax y abdomen. Debe incorporarse una escala en todas las mediciones radiográficas.
La citología y la histología pueden usarse para diferenciar entre la respuesta parcial (PR) y la respuesta completa (CR) en casos raros.
d. Documentación de inicio de lesiones "diana" y "no diana"
Todas las lesiones medibles hasta un máximo de 2 lesiones por órgano y 5 lesiones en total, representativas de todos los órganos involucrados deben identificarse como lesiones diana y registrarse y medirse al inicio. Las lesiones tumorales situadas en un área previamente irradiada, o en un área sometida a otra terapia loco-regional, generalmente no se consideran medibles a menos que se haya demostrado progresión en la lesión.
Los ganglios linfáticos merecen una mención especial ya que son estructuras anatómicas normales que pueden ser visibles mediante imágenes, incluso si no están involucradas por el tumor. Los ganglios patológicos que se definen como medibles y pueden identificarse como lesiones diana deben cumplir el criterio de un eje corto de S15 mm mediante exploración de CT. Solo el eje corto de estos ganglios contribuirá a la suma del inicio. El eje corto del ganglio es el diámetro normalmente usado por los radiólogos para juzgar si un ganglio está afectado por un tumor sólido. El tamaño nodal se informa normalmente como dos dimensiones en el plano en el que se obtiene la imagen (para la exploración de CT, este es casi siempre el plano axial). La más pequeña de estas medidas es el eje corto. Por ejemplo, un ganglio abdominal que se informa como de 20 mm x 30 mm tiene un eje corto de 20 mm y califica como un ganglio maligno medible. En este ejemplo, 20 mm debe registrarse como la medición del ganglio. Todos los demás ganglios patológicos (aquellos con eje corto S10 mm, pero <15 mm) deben considerarse lesiones no diana. Los ganglios que tienen un eje corto <10 mm se consideran no patológicos y no deben registrarse ni seguirse. Se calculará una suma de diámetros para todas las lesiones diana y se informará como la suma inicial de los diámetros. Si se van a incluir ganglios linfáticos en la suma, entonces, como se señaló anteriormente, solo se agrega el eje corto a la suma. Los diámetros de la suma inicial se utilizarán como referencia para caracterizar la respuesta tumoral objetiva.
Todas las demás lesiones (o sitios de enfermedad), incluidos los ganglios linfáticos patológicos, deben identificarse como lesiones no diana y también deben registrarse al inicio del estudio. No se requiere mediciones de estas lesiones, pero la presencia (estable, creciente o decreciente) o la ausencia de cada una debe notarse durante el seguimiento.
e. Evaluación de lesiones diana
Respuesta completa (CR):
La desaparición de todas las lesiones diana. Cualquier ganglio linfático patológico (ya sea diana o no diana) debe tener una reducción en el eje corto a <10 mm.
Respuesta parcial (PR):
Al menos una disminución de 30 % en la suma de los diámetros de las lesiones diana, donde se toma como referencia la suma de los diámetros inicial.
Enfermedad progresiva (PD):
Al menos un aumento del 20 % en la suma de los diámetros de las lesiones diana, donde se toma como referencia la suma más pequeña de los diámetros registrada desde que comenzó el tratamiento (inicial o posterior) o la aparición de una o más lesiones nuevas. Además del aumento relativo del 20 %, la suma también debe demostrar un aumento absoluto de al menos 5 mm.
Enfermedad estable (DE):
Ni contracción suficiente para calificar para RP ni aumento suficiente para calificar para PD, donde se toma como referencia la suma más pequeña de los diámetros desde que comenzó el tratamiento (inicial o posterior).
Evaluación de lesiones diana:
Los ganglios linfáticos identificados como lesiones diana siempre deben tener la medición real del eje corto registrada (medida en el mismo plano anatómico que el examen inicial), incluso si los ganglios retroceden a menos de 10 mm en el estudio. Esto significa que cuando los ganglios linfáticos se incluyen como lesiones diana, la ’suma’de las lesiones puede no ser cero incluso si se cumplen los criterios de respuesta completa, ya que un ganglio linfático normal se define como el que tiene un eje corto de <10 mm. Para PR, SD y PD, la medición real del eje
corto de los ganglios debe incluirse en la suma de las lesiones diana.
Todas las lesiones (nodales y no nodales) registradas al inicio del estudio deben tener sus mediciones reales registradas en cada evaluación posterior, incluso cuando son muy pequeñas (<5 mm). Sin embargo, a veces las lesiones diana o los ganglios linfáticos se vuelven demasiado pequeños para medir. Si el radiólogo considera que la lesión probablemente ha desaparecido, la medición debe registrarse como 0 mm. Si se cree que la lesión está presente, pero es demasiado pequeña para medirla, se debe asignar un valor predeterminado de 5 mm (derivado del espesor de corte de CT de 5 mm). La medición de estas lesiones es potencialmente no reproducible; por lo tanto, proporcionar este valor predeterminado evitará respuestas falsas o progresos basados en errores de medición. f. Evaluación de lesiones no diana
Respuesta completa (CR):
La desaparición de todas las lesiones no diana y la normalización del nivel del marcador tumoral. Todos los ganglios linfáticos deben ser de tamaño no patológico (<10 mm de eje corto).
Sin CR/Sin PD:
Persistencia de una o más lesiones no diana y/o mantenimiento del nivel del marcador tumoral por encima de los límites normales.
Enfermedad progresiva (PD):
Progresión inequívoca de las lesiones no diana existentes.
En este contexto, para lograr una 'progresión inequívoca’ sobre la base de la enfermedad no diana, debe haber un nivel general de empeoramiento sustancial en la enfermedad no diana tal que, incluso en presencia de SD o PR en la enfermedad diana, la carga tumoral general ha aumentado lo suficiente como para merecer la interrupción del tratamiento. Un modesto 'aumento' en el tamaño de una o más lesiones no diana no suele ser suficiente para calificar para el estado de progresión inequívoca. Por lo tanto, la designación de la progresión general únicamente sobre la base del cambio en la enfermedad no diana frente a SD o PR de la enfermedad diana será extremadamente rara.
Nota: Si el sujeto interrumpe el tratamiento por deterioro sintomático, debe hacerse todo lo posible para documentar la progresión objetiva incluso después de la interrupción del tratamiento.
Nuevas lesiones
La aparición de nuevas lesiones malignas denota la progresión de la enfermedad. Si bien no existen criterios específicos para la identificación de nuevas lesiones radiográficas, los hallazgos de una nueva lesión deben ser inequívocos, es decir, no atribuibles a diferencias en la técnica de exploración, el momento de la exploración, la fase de administración de contraste, el cambio en la modalidad de imagen o la búsqueda de ideas para representar algo diferente a un tumor (por ejemplo, algunas lesiones óseas 'nuevas' pueden ser simplemente la curación o el brote de lesiones preexistentes). Una lesión identificada en un estudio de seguimiento en una ubicación anatómica que no se exploró al inicio se considera una lesión nueva e indicará la progresión de la enfermedad. Un ejemplo de esto es el sujeto que tiene enfermedad visceral al inicio y, mientras está en estudio, se le ordenó una CT o MRI cerebral que revela metástasis. Las metástasis cerebrales del sujeto se consideran evidencia de enfermedad progresiva incluso si él/ella no tenía imágenes cerebrales al inicio.
Si una nueva lesión es equívoca (es decir, demasiado pequeña para medirla), la terapia continua y la evaluación de seguimiento aclarará si representa una enfermedad realmente nueva. Si las exploraciones repetidas confirman que hay una nueva lesión, la progresión debe declararse mediante el uso de la fecha de la exploración inicial.
16.5. Resultados
16.5.1.Fase de escalado/expansión de dosis en monoterapia con ABBV-399 (Fase 1):
En el diseño de escalado de dosis 3+3, el ABBV-399 se administró en dosis que van desde 0,15 a 3,3 mg/kg una vez cada 21 días a pts con tumores sólidos metastásicos (NCT02099058). Como se representa en la figura 13, el ABBV-399 se administró como una infusión intravenosa una vez cada 21 días hasta la progresión de la enfermedad o toxicidad intolerable. La dosificación comenzó a 0,15 mg/kg y se escaló a 0,3, 0,6, 1,2, 1,8, 2,4, 3,0 y 3,3 mg/kg en cohortes posteriores según lo tolerado. Se evaluó además una dosis de 2,7 mg/kg de ABBV-399 administrada cada 21 días y, en función de la seguridad y la PK, se eligió como la dosis para la cohorte de expansión. Según los datos de seguridad y PK de la dosificación cada 21 días, el ABBV-399 se administrará además cada 14 días en un programa de 28 días (dosis inicial de 1,6 mg/kg a 2,5 mg/kg en aumentos incrementales de 0,3 mg/kg, es decir, 1,6,
1,9, 2,2 y 2,5 mg/kg). Para la administración a los 14 o 21 días, se administrará el ABBV-399 durante 30 ± 10 minutos. No se administra como un impulso intravenoso o bolo.
Al 31 de marzo de 2016, 48 pts recibieron al menos 1 dosis de ABBV-399. Se observaron aumentos proporcionales a la dosis del área bajo la curva para ABBV-399 y el anticuerpo total después de la administración de una dosis única. Las vidas medias para ABBV-399 y el anticuerpo total fueron de aproximadamente 2-4 días. La toxicidad limitante de la dosis de neutropenia febril ocurrió en 1 pt a 3 mg/kg y 1 pt (con shock séptico) a 3,3 mg/kg. El mejor cambio porcentual en las lesiones diana en pacientes con al menos 1 evaluación tumoral posterior al inicio se muestra en la figura 14. Como se muestra en la figura 14 (y de los datos que no se muestran en las figuras), las mejores respuestas a la monoterapia con ABBV-399 en todos los pacientes tratados fueron: 3/40 (7,5 %) de respuesta parcial, 20/40 (50 %) pacientes con enfermedad estable y 17/40 (42,5 %) pacientes con enfermedad progresiva. Los datos RECIST no estaban disponibles para ocho pacientes debido a la progresión clínica (4), los eventos adversos (2), la retirada del consentimiento (1) y la muerte por neumonía (1). Los tres pacientes con una respuesta parcial tenían cáncer de pulmón de células no pequeñas (NSCLC) con sobreexpresión de cMet.
Se eligió una dosis de 2,7 mg/kg para la expansión de la dosis en base principalmente a la seguridad y la tolerabilidad. Para la inscripción en esta fase del estudio, los sujetos de NSCLC se seleccionaron para la sobreexpresión de cMet mediante el uso de un ensayo IHC que utiliza el estuche de anticuerpo monoclonal primario de conejo anti-cMet total (SP44) CONFIRM adquirido de Ventana (REF # 790-4430). Las muestras de tejido se puntuaron mediante determinación de los porcentajes de tinción de células de tejido diana a diversos niveles de intensidad de bajo a alto, es decir, puntuación IHC de 0, 1+, 2+ o 3+ o una puntuación H de 0 a 149, 150-224, o 225 300. La puntuación puede hacerse de forma manual o con la ayuda de una computadora. Los detalles del ensayo de IHC y la puntuación se describen en el ejemplo 17. La siguiente tabla muestra el número de pacientes con NSCLC seleccionados prospectivamente y la puntuación H usada para evaluar la sobreexpresión de cMet:

No hubo muertes relacionadas con el tratamiento. Los eventos adversos relacionados con el tratamiento que ocurrieron en S10 % de los pts (incluidos todos los niveles de dosis y todos los grados) fueron fatiga (22,9 %), náuseas (20,8 %), neuropatía (14,6 %), disminución del apetito (12,5 %), vómitos (12,5 %) e hipoalbuminemia (10,4 %). Entre 16 pacientes con NSCLC cMet+ tratados con ABBV-399, los resultados de 11 se muestran en la figura 15. La figura 15 es un diagrama en cascada que muestra el mejor cambio porcentual en la lesión diana en respuesta a la monoterapia con ABBV-399 en base a los datos radiográficos. Como se muestra en la figura 15 (y de los datos no mostrados en la figura) 3/16 pacientes tratados con respuestas parciales (19 %), 6/16 pacientes tratados con enfermedad estable (37,5 %), 2/16 pacientes tratados con enfermedad progresiva radiográfica (12,5 %), y 5 pacientes sin imágenes disponibles debido a la progresión clínica (3), retirada del consentimiento (1) y muerte por neumonía (1).
La figura 16 muestra el número de semanas que los 16 pacientes estaban en estudio antes de la progresión clínica.
16.5.2.Fase de terapia combinada (Fase 1b):
Los resultados de un ensayo de terapia combinada de NSCLC mediante el uso de ABBV-399 a 2,7 mg/kg una vez cada 21 días y erlotinib 150 mg administrados por vía oral todos los días se muestran en las figuras 17 y 18. La figura 17 es un diagrama en cascada que muestra el mejor cambio porcentual en las lesiones diana para 6 pacientes tratados con ABBV-399 y erlotinib. Como se muestra en la figura 17, 2/6 pacientes lograron una respuesta parcial, 1/6 con enfermedad progresiva como lo demuestran las nuevas lesiones. La figura 18 muestra el número de semanas que los 6 pacientes estuvieron en estudio antes de la progresión clínica.
16.5.3 La selección previa al tratamiento para pacientes con tumores cMet+ con puntuaciones IHC2+/3+ o puntuaciones H S150 puede mejorar significativamente el resultado del tratamiento
Los resultados preclínicos con líneas celulares y modelos de xenoinjerto sugieren que aquellos con una puntuación IHC2+/IHC3+ de cMet responderán mejor que aquellos con IHC 0/1+. El uso de diagnósticos complementarios para ayudar en la selección previa al tratamiento de aquellos pacientes con cánceres cMet IHC2/3+ o puntuación HS150 mejorarían significativamente los resultados generales del tratamiento y evitaría que los pacientes recibieran un tratamiento que se predice que será ineficaz. Como se usa en la presente descripción, el término cMet+ abarca todos los tumores que expresan cMet, independientemente de si el cMet se sobreexpresa o no. En algunas modalidades de cMet+, el cMet se sobreexpresa. En algunas modalidades de cMet+, el cMet no se sobreexpresa. De manera similar, los resultados de este ensayo clínico en fase 1 en curso sugieren que las puntuaciones H de 150 y superiores están vinculadas y pueden predecir la respuesta al tratamiento con un ADC anti-cMet, que incluye
ABBV-399.
Sin estar sujetos a ninguna teoría, los resultados preliminares sugieren que la heterogeneidad tumoral puede ser un factor limitante en la eficacia de ABBV-399. Entre los tumores cMet+ con puntuaciones IHC2+ e IHC3+, hay células cancerosas que muestran de ninguna a baja expresión de cMet. De ellos, al menos algunas de las células que no son destruidas por un "efecto circunstante" podrían repoblar el tumor e impedir la respuesta tumoral. El ABBV-399 podría combinarse con tratamientos estándar de atención que inhiban o eliminen las células tumorales con baja expresión de cMet, no limitados a agentes dirigidos como erlotinib e inmunoterapias como nivolumab, sino también quimioterapia estándar de atención, preferentemente con toxicidad no superpuesta.
La tabla 9 proporciona resultados clínicos del ensayo de fase 1 en curso que correlaciona la respuesta general con la puntuación H en pacientes con NSCLC tratados con ABBV-399 como monoterapia una vez cada dos (Q2W) o tres (Q3W) semanas o con ABBV-399 en combinación con erlotinib. La puntuación IHC se obtuvo mediante el uso del protocolo descrito en el ejemplo 17.
TABLA 9

Como se muestra en la tabla 9, cuatro pacientes con adenocarcinomas de NSCLC tratados con 2,7 mg/kg de ABBV-399 una vez cada 3 semanas (Q3W) y erlotinib que tienen puntuaciones IHC de 225 o más lograron respuestas parciales (RP). Dos pacientes con adenocarcinomas de NSCLC tratados una vez cada dos semanas con ABBV-399 que tienen puntuaciones IHC de 225 o más lograron una respuesta parcial o una respuesta completa. Tres pacientes con carcinomas de células escamosas de NSCLC tratados con 2,7 mg/kg de ABBV-399 una vez cada 3 semanas que tienen puntuaciones de IHC entre 150 y 224 lograron respuestas parciales.
Ejemplo 17. Ensayo de inmunohistoquímica y la puntuación H de cMet: el "protocolo de tinción cMet ABBV-ADC" Hay varios métodos disponibles en la técnica para evaluar los niveles de expresión de la proteína cMet mediante inmunohistoquímica (IHC). Un experto en la técnica habría sabido habitualmente cómo usarlos y adaptarlos a su estudio particular. Varios proveedores proporcionan tinción de cMet como una tarifa por servicio (véase, por ejemplo, Flagship Biosciences L.L.C., ARUP Laboratories, PathGroup Inc.). En este estudio de Fase I, los niveles de expresión de cMet se evaluaron mediante el uso del mAb anti-cMet SP44 de Ventana Medical Systems, más específicamente el anticuerpo monoclonal de conejo anti-cMet total CONFIRM® de Ventana (Ventana Medical Systems, Inc; cat no. 790-4430), en combinación con una tinción automatizada de portaobjetos Ventana® (BenchMark ULTRA®) y un estuche ultraView® Universal DAB detection de Ventana (cat. no. 760-500). Las tinciones y los resultados se procesaron por Flagship Biosciences L.L.C. en colaboración con los Laboratorios ARUP. Los tejidos de control positivo incluyen adenocarcinomas de colon y adenocarcinoma de pulmón. Los tejidos de control negativo incluyen el control Breast ER100, el control Breast ER13781 y el control del linfoma de Hodgkin CD15-5. Para los fines de esta solicitud, incluidas las reivindicaciones, el ensayo particular usado en este estudio de Fase 1 se denomina en la presente descripción "protocolo de tinción cMet ABBV-ADC."
Las biopsias tumorales de los pacientes se fijaros en formalina en PBS y se embebieron en parafina. Los portaobjetos se cortaron a 4 micras, se dejaron secar y luego se hornearon durante 60 minutos a 60 °C. Los portaobjetos se usaron dentro de las 2 semanas posteriores al corte. Los portaobjetos se transfirieron a un instrumento BenchMark ULTRA® y se seleccionaron los siguientes parámetros:
Procedimiento: ultraView® DAB
Nombre: cMet CONFIRM®
Parafina [seleccionado]
Desparafinación [seleccionado]
Acondicionamiento celular [seleccionado]
Acondicionador # 1 [seleccionado]
[corto - 8 minutos de acondicionamiento]
CC1 Leve [seleccionado]
[severo - 95 minutos de acondicionamiento]
Temperaturas de incubación del Ab [seleccionado]
36 °C Ab [seleccionado
Anticuerpo [seleccionado]
ESTUCHE DE PREP # [4430] ** 0 H 16 min
Contratinción [seleccionado]
HEMATOXILINA [2021] 4 minutos
Contratinción posterior [seleccionado]
REACTIVO DE BLUING [2037] 4 minutos
Cuando terminó la tinción, los portaobjetos se retiraron del instrumento y se enjuagaron con agua corriente. Los portaobjetos se deshidrataron de la siguiente manera:
Sumergir los portaobjetos en etanol al 70 %, 2 cambios, 1-2 minutos cada uno.
Sumergir los portaobjetos en etanol al 95 %, 1-2 minutos.
Sumergir los portaobjetos en etanol al 99 % (o absoluto), 3-5 minutos.
Aclarar con xileno, 3 cambios, 3-5 minutos cada uno.
Después de la deshidratación, los portaobjetos se cubrieron con un medio de montaje no acuoso mediante el uso de cubreobjetos de vidrio.
Se usaron los siguientes reactivos en este sistema automatizado:
cMet CONFIRM® de Ventana® cat. no. 790-4430 (incubación aproximadamente 16 minutos a 36 °C)
Un dispensador de 5 mL de CONFIRM® anti-cMet Total contiene aproximadamente 48,75 pg del anticuerpo monoclonal de conejo recombinante SP44 (también disponible en otros proveedores comerciales). El anticuerpo se diluye en Tris-HCl 0,05 M con 1 % de la proteína transportadora y 0,10 % de ProClin 300® (conservante). La concentración de proteína total del reactivo es de aproximadamente 10 mg/mL. La concentración de anticuerpos específicos es de aproximadamente 9,75 pg/mL. No se conoce reactividad de anticuerpos no específicos observado en este producto.
Tampón Ultra CC1 de Ventana cat. no. 950-224
El acondicionamiento celular en solución Ultra CC1 se realizó a 64 °C durante 95 minutos.
Estuche de detección ultraView® Universal de Ventana cat. No. 760-500
Hematoxilina II de Ventana cat. Núm. 760-2021
Reactivo de Bluing de Ventana cat. Núm. 760-2037
Determinaciones de puntuaciones H y puntuaciones IHC
Los portaobjetos procesados se analizaron por un patólogo MD certificado por la junta. Se usó una guía de puntuación, según lo dispuesto por el fabricante (véase, por ejemplo, la figura 19). Se usaron de 10-12 áreas representativas de cada portaobjeto para deducir la puntuación. Tras la evaluación de la tinción de cMet, se
determinó que un enfoque de puntuación H sería el mejor enfoque para cuantificar la expresión de cMet. El enfoque de la puntuación H proporciona una resolución óptima de los datos para determinar la variación en la intensidad y el porcentaje tumoral de tinción dentro y entre los tipos de tumor. Se proporciona además una buena herramienta para determinar los umbrales para la tinción positiva. En este método, se proporciona el porcentaje de células (0-100) dentro de un tumor con intensidades de tinción que varían de 0-3+. Este protocolo da como resultado la tinción de la proteína cMet tanto en el citoplasma como en la superficie/membrana celular. Se determina la intensidad de tinción para cada célula en un campo fijo de la biopsia tumoral procesada, y se atribuye un valor individual a cada célula de la siguiente manera, en dependencia de la tinción de la superficie/membrana de la célula:
0 = sin tinción
1+ = tinción débil
2+ = tinción moderada
3+ = tinción fuerte
Para obtener una puntuación H, el porcentaje de células tumorales se multiplica por cada intensidad y se suman. La puntuación H máxima es 300 si el 100 % de las células tumorales se marcan con una intensidad de 3+. La puntuación H se calcula de la siguiente manera:
puntuación H = [1 x (% de células 1+) 2 x (% de células 2+) 3 x (% de células 3+)] Este protocolo da como resultado tanto la tinción de cMet citoplasmática como de membrana. Para los cálculos de la puntuación H mencionados en la presente descripción, se usó la tinción de membrana. La puntuación de la puntuación H final del tumor (0-300) da más peso relativo a la tinción de membrana de mayor intensidad (células 3+ > células 2+ > células 1+).
La figura 20 muestra resultados de tinción ilustrativos para diversas puntuaciones H de tumor (15, 90, 180 y 290) obtenidas con el "protocolo de tinción cMet ABBV-ADC."
Cada tumor puede recibir además una puntuación IHC de IHC 0, IHC 1+, IHC 2+ o IHC 3+. Si bien ambas puntuaciones IHC implican valores 0, 1+, 2+ y 3+, no deben confundirse. Para la puntuación H, los valores 0, 1+, 2+ y 3+ se refieren a la intensidad de la tinción de una célula individual en particular. Para la puntuación IHC, los valores 0, 1+, 2+ y 3+ se refieren a la tinción general de un área particular de la muestra tumoral. La figura 21 muestra resultados de tinción ilustrativos para diversas puntuaciones IHC0/1+/2+/3+ de tumor obtenidas con el "protocolo de tinción cMet ABBV-ADC."
Para los propósitos de esta divulgación, y al seguir el protocolo descrito en la presente descripción, si ninguna de las células en un campo fijo se tiñe, el valor atribuido al tumor es IHC 0. Si el nivel general de tinción en un campo fijo es bajo, el valor atribuido es IHC 1+. Si la mayoría de las células en un campo fijo exhiben una tinción moderada, el valor atribuido es IHC 2+. Si la mayoría de las células en un campo fijo exhiben una fuerte tinción, el valor atribuido es IHC 3+.
Para los propósitos de esta divulgación, y al seguir el protocolo descrito en la presente descripción, si ninguna de las células en un campo fijo se tiñe, el valor atribuido al tumor es IHC 0. Si el nivel general de tinción en un campo fijo es bajo, el valor atribuido es IHC 1+. Si al menos el 15 % de las células en un campo fijo exhiben una tinción moderada, el valor atribuido es IHC 2+. Si al menos 15 % de las células en un campo fijo exhiben una tinción fuerte, el valor atribuido es IHC 3+.
Ejemplo 18. Medición de la amplificación del número de copias del gen MET
La amplificación del gen MET puede mejorar la respuesta del paciente a los inhibidores de cMet, incluidos los tratamientos divulgados en la presente descripción. Se han descrito una variedad de métodos para medir la amplificación de genes MET en la técnica. Véase, por ejemplo, Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, y otros Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J Clin Oncol 2009; 27:1667-74; Koeppen H, Yu W, Zha J, Pandita A, Penuel E, Rangell L, y otros Biomarker analyses from a placebo-controlled phase II study evaluating erlotinib {+/-} onartuzumab in advanced non-small-cell lung cancer: MET expression levels are predictive of patient benefit. Clin Cancer Res 2014; 20:4488-98.
El método preferido se describe más abajo y se denomina en la presente descripción el "método de amplificación de cMET MET/CEP7." Brevemente, los bloques de tejido embebidos en parafina y fijados con formalina pueden someterse a ensayos de FISH de doble color mediante el uso de un cóctel de sonda MET/CEP7 preparado con una sonda de ADN MET (clon RP 11-95120 BAC), o mediante el uso de una sonda de 319 kb construida de 3 clones de cromosoma artificial bacteriano (BAC) que abarca todo el gen MET en 7q31.1, marcado con SpectrumRed y SpectrumGreen CEP7 (Abbott Molecular). Los ensayos FISH pueden realizarse, por ejemplo, de acuerdo con un protocolo descrito previamente (Cappuzzo F, Hirsch FR, Rossi E, y otros (2005) Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small cell lung cancer. J Natl Cancer Inst 97: 643-655), que incluye el
pretratamiento con tampón de cloruro de sodio-citrato de sodio 2* a 75 °C y digestión con proteinasa K durante 7 a 15 minutos cada una, codesnaturalización a 85 °C durante 15 minutos, hibridación durante aproximadamente 36 horas y lavados rápidos de poshibridación con tampón de cloruro de sodio-citrato de sodio 2*/0,4 nonil-fenoxilpolietoxiletanol. Las señales se enumeran en al menos 50 núcleos tumorales por núcleo, mediante el uso de un microscopio de epifluorescencia con conjuntos de filtros de interferencia única para filtros de paso de banda verde (FITC), rojo (Texas rojo) y azul (DAPI), así como dual (rojo/verde) y triple (azul, rojo, verde). Para cada núcleo, la media y la desviación estándar del número de copias por célula de cada secuencia de ADN analizada, el porcentaje de células con s2, 3 y s4 copias de los genes MET y la proporción de MET/CEP7 (un gen localizado cerca del centrosoma del mismo cromosoma). Cuando se detectaron resultados heterogéneos entre los tres núcleos probados, se usó el núcleo con el número medio de copias más alto para representar al paciente en los análisis estadísticos. Para la documentación, las imágenes se capturaron mediante el uso de una cámara CCD y se fusionaron mediante el uso de un software dedicado (CytoVision; Genetix USA, Boston, MA). MET puede considerarse amplificado cuando la relación de señal MET: CEP7 es s2,0 o cuando esta relación es <2,0 pero hay >20 copias de señales MET en más del 10 % de los núcleos tumorales contados, según los criterios establecidos por el Departamento de Patología de MD Anderson en base a estudios previos. Zeng, ZS, Weiser MR, Kuntz E, Chen CT, Khan SA, Forslund A, y otros cMet gene amplification is associated with advanced stage colorectal cancer and liver metastases. Cancer Lett 2008; 265:258-69. En algunos estudios, se ha informado que el número de copias del gen MET en relación con CEP7 osciló entre 2,05 y 16,14 (mediana 3,48).
Otra prueba de amplificación de cMET es una prueba a base de sangre. Esto se puede realizar con cualquiera de una variedad de reactivos disponibles comercialmente tales como, por ejemplo, Biocept Liquid Biopsy MET Amplication Test (Biocept), MET Detect-R® (Personal Genome Diagnostics) y Guardant360® (Guardant Health®).
Ejemplo 19. Evaluación de la presencia de mutación del exón 14/omisión del gen MET
El exón 14 de MET contiene el sitio de ubiquitina ligasas Cbl en el residuo de tirosina 1003 (Y1003) donde la ubiquitina se une normalmente de cualquier otra manera al residuo de tirosina y conduce a la degradación lisosomal de la proteína cMet. Por lo tanto, la mutación sin sentido del residuo Y1003 o la "omisión" de la región de la proteína que está codificada por el exón 14 de MET resulta en una sobreexpresión relativa de la proteína MET, activación de cMet mejorada y posterior oncogénesis. La inhibición por los inhibidores de tirosina quinasa MET (TKI) puede resultar en un beneficio clínico en al menos pacientes con NSCLC que albergan estas alteraciones del exón 14 de MET. Los pacientes que portan cualquiera de estas mutaciones pueden beneficiarse de los tratamientos divulgados en la presente descripción.
Hay varios métodos disponibles para un experto en la técnica para detectar mutaciones en el gen MET. Debido a que una mutación está presente o no (es decir, es un valor absoluto y no una cuestión de grado), su detección no depende del ensayo y puede usarse cualquier método para detectarla en muestras tumorales. Se han descrito múltiples mutaciones en el exón 14 del gen MET, muchas de las cuales se han resumido en Impaired cMet Receptor Degradation Mediated by MET Exon 14 Mutations in Non-Small-Cell Lung Cancer, Mark M. Awad JCO 10 de marzo de 2016: 879 -881; publicado en línea el 19 de enero de 2016; 10.1200/JCO.2015.64.2777. Estos métodos pueden usarse para identificar esas mutaciones particulares en muestras de cáncer. Además, esta divulgación está dirigida a cualquier mutación conocida en el gen exón 14 y no está limitada a las ejemplificadas en la presente descripción.
Se han identificado varias mutaciones de empalme del exón 14 en el adenocarcinoma pulmonar. Por ejemplo:
MET amplification, protein expression, and mutations in pulmonary adenocarcinoma. Park S, Koh J, Kim DW, Kim M, Keam B, Kim TM, Jeon YK, Chung DH, Heo DS. Lung Cancer. Diciembre de 2015;90(3):381-7. doi: 10.1016/j.lungcan.2015.10.022. Epub 27 de octubre de 2015. PMID: 26791796. Estos métodos pueden usarse para identificar esas mutaciones particulares en muestras de cáncer.
Responses to the multitargeted MET/ALK/ROS1 inhibitor crizotinib and co-occurring mutations in lung adenocarcinomas with MET amplification or MET exon 14 skipping mutation. Jorge SE, Schulman S, Freed JA, VanderLaan PA, Rangachari D, Kobayashi SS, Huberman MS, Costa DB. Lung Cancer. Diciembre de 2015; 90(3):369-74. doi: 10.1016/j.lung-can.2015.10.028. Epub 31 de octubre de 2015. Estos métodos pueden usarse para identificar esas mutaciones particulares en muestras de cáncer.
Se pueden detectar mutaciones adicionales del exón 14 en el NSCLC mediante los métodos descritos en las siguientes referencias: Next-Generation Sequencing of Pulmonary Sarcomatoid Carcinoma Reveals High Frequency of Actionable MET Gene Mutations Exon 14 Xuewen Liu, Yuxia Jia, Mark B. Stoopler, Yufeng Shen, Haiying Cheng, Jinli Chen, Mahesh Mansukhani, Sanjay Koul, Balazs Halmos, and Alain C. Borczuk, JCO 10 de marzo de 2016: 794-802; publicado en línea el 27 de julio de 2015. Estos métodos pueden usarse para identificar esas mutaciones particulares en muestras de cáncer.
MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and cMet Overexpression. Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, Heng JC, Dahlberg SE, Janne PA, Verma S, Christensen J, Hammerman PS, Sholl LM. J Clin Oncol.
1 de marzo de 2016; 34(7):721-30. doi: 10.1200/JC0.2015.63.4600. Epub 4 de enero 2016. Estos métodos pueden
Claims (8)
1. Un conjugado de fármaco con anticuerpo (“ADC”) anti-cMet, en donde el conjugado de fármaco es monometil auristatina E ("MMAE") y el ADC tiene la siguiente estructura:

en donde Ab es un anticuerpo anti-cMet que comprende una cadena VH que comprende tres CDR, denominadas VH CDR #1 (SEQ ID NO: 112), VH CDR #2 (SEQ ID NO: 113) y VH CDR #3 (SEQ ID NO: 114); una cadena VL que comprende tres CDR, denominadas VL CDR #1 (SEQ ID NO: 115), VL CDR #2 (SEQ ID NO: 116) y VL CDR #3 (SEQ ID NO: 117); y en donde n tiene un valor que varía de 2 a 4.
2. El ADC de la reivindicación 1 que comprende una cadena VH de SEQ ID NO: 78 y una cadena VL de SEQ ID NO: 79.
3. El ADC de la reivindicación 1 que comprende una cadena pesada de SEQ ID NO: 86 y una cadena ligera de SEQ ID NO: 87.
4. El ADC de una cualquiera de las reivindicaciones 1 a 3, en donde n es 2 o 4.
5. Una composición farmacéutica que comprende el ADC de una cualquiera de las reivindicaciones 1 a 4.
6. La composición farmacéutica de la reivindicación 5, en donde n es 2 y 4.
7. La composición farmacéutica de la reivindicación 5 o 6 que tiene una relación fármaco-a-anticuerpo ("DAR") de aproximadamente 3,1.
8. La composición farmacéutica de una cualquiera de las reivindicaciones 5 a 7, en donde el ADC está enriquecido hasta una pureza E2/E4 de al menos 95 %.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662337796P | 2016-05-17 | 2016-05-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2864150T3 true ES2864150T3 (es) | 2021-10-13 |
Family
ID=59014737
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19197371T Active ES2864150T3 (es) | 2016-05-17 | 2017-05-17 | Conjugados de fármacos con anticuerpos anti-cMet y métodos para su uso |
| ES17728311T Active ES2811351T3 (es) | 2016-05-17 | 2017-05-17 | Conjugados de fármacos con anticuerpos anti-cMet y métodos para su uso |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17728311T Active ES2811351T3 (es) | 2016-05-17 | 2017-05-17 | Conjugados de fármacos con anticuerpos anti-cMet y métodos para su uso |
Country Status (25)
| Country | Link |
|---|---|
| US (5) | US20170348429A1 (es) |
| EP (4) | EP3804765A1 (es) |
| JP (4) | JP2019521087A (es) |
| KR (3) | KR20230028574A (es) |
| CN (3) | CN109562189B (es) |
| AU (1) | AU2017268342B2 (es) |
| BR (1) | BR112018073574A2 (es) |
| CA (1) | CA3024386C (es) |
| CY (2) | CY1123256T1 (es) |
| DK (2) | DK3458102T3 (es) |
| ES (2) | ES2864150T3 (es) |
| HR (2) | HRP20201186T1 (es) |
| HU (2) | HUE050398T2 (es) |
| IL (3) | IL262869B (es) |
| LT (2) | LT3626273T (es) |
| MX (1) | MX2018014175A (es) |
| NZ (1) | NZ748314A (es) |
| PL (2) | PL3458102T3 (es) |
| PT (2) | PT3458102T (es) |
| RS (2) | RS60663B1 (es) |
| RU (1) | RU2740996C2 (es) |
| SG (2) | SG11201810034XA (es) |
| SI (2) | SI3458102T1 (es) |
| WO (1) | WO2017201204A1 (es) |
| ZA (1) | ZA201808386B (es) |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK3458102T3 (da) * | 2016-05-17 | 2020-07-27 | Abbvie Biotherapeutics Inc | Anti-cmet-antistoflægemiddelkonjugater og fremgangsmåder til anvendelse deraf |
| TWI782930B (zh) | 2016-11-16 | 2022-11-11 | 美商再生元醫藥公司 | 抗met抗體,結合met之雙特異性抗原結合分子及其使用方法 |
| PL3544634T3 (pl) * | 2016-11-23 | 2021-09-27 | Eli Lilly And Company | Koniugaty przeciwciało MET-lek |
| CN110144325A (zh) * | 2018-02-12 | 2019-08-20 | 深圳宾德生物技术有限公司 | 一种靶向性t淋巴细胞及其制备方法和应用 |
| HUE063489T2 (hu) | 2018-03-13 | 2024-01-28 | Zymeworks Bc Inc | Anti-HER2 biparatopikus antitest-gyógyszer konjugátumok és alkalmazási módszereik |
| WO2019183633A1 (en) | 2018-03-23 | 2019-09-26 | Case Western Reserve Univeristy | Psma targeted conjugate compounds and uses thereof |
| TW201946656A (zh) * | 2018-03-28 | 2019-12-16 | 日商田邊三菱製藥股份有限公司 | cMET單株抗體結合劑的藥物接合物及其用途 |
| EP3854816A4 (en) * | 2018-09-30 | 2022-09-07 | Jiangsu Hengrui Medicine Co., Ltd. | EXATECAN ANTI-B7H3-ANALOG ANTIBODY CONJUGATE AND ASSOCIATED MEDICAL USE |
| KR102353568B1 (ko) * | 2018-11-14 | 2022-01-20 | 주식회사 헬릭스미스 | 안정성이 향상된 항 c-Met 항체 또는 그의 항원 결합 단편 |
| US11896682B2 (en) | 2019-09-16 | 2024-02-13 | Regeneron Pharmaceuticals, Inc. | Radiolabeled MET binding proteins for immuno-PET imaging and methods of use thereof |
| CN112604004B (zh) * | 2019-09-19 | 2022-05-10 | 中国医学科学院医药生物技术研究所 | 一类抗人egfr抗体药物偶联物及其制备方法与应用 |
| CN110893236A (zh) * | 2019-10-09 | 2020-03-20 | 中山大学 | 溶酶体靶向的抗体药物偶联物及其应用 |
| CN115297889A (zh) * | 2020-09-01 | 2022-11-04 | 荣昌生物制药(烟台)股份有限公司 | 抗c-Met抗体药物偶联物及其应用 |
| CN116472064A (zh) * | 2020-10-12 | 2023-07-21 | 昆山新蕴达生物科技有限公司 | 抗体-药物偶联物及其应用 |
| CN114533891B (zh) * | 2020-11-24 | 2023-04-18 | 深圳先进技术研究院 | 一种靶向无痕释放药物缀合物及其制备方法与应用 |
| KR20230136758A (ko) * | 2021-02-03 | 2023-09-26 | 마이틱 테라퓨틱스, 인크. | 항체 및 이의 용도 |
| BR112023020562A2 (pt) * | 2021-04-06 | 2023-12-05 | Abbvie Biotherapeutics Inc | Métodos de tratamento de carcinoma pulmonar de células não pequenas com o uso de telisotuzumabe vedotina |
| IL306129A (en) | 2021-04-07 | 2023-11-01 | Mythic Therapeutics Inc | Structures of antigen-binding proteins and antibodies and their uses |
| WO2022214517A1 (en) | 2021-04-08 | 2022-10-13 | Byondis B.V. | Anti-c-met antibodies and antibody-drug conjugates |
| TW202304929A (zh) | 2021-04-29 | 2023-02-01 | 美商艾伯維有限公司 | 抗c-Met抗體藥物結合物 |
| US20230285394A1 (en) | 2022-03-11 | 2023-09-14 | Abbvie Biotherapeutics Inc. | Methods of treatment of non-small-cell lung carcinoma using telisotuzummab vedotin and osimertinib |
| WO2024065056A1 (en) * | 2022-09-28 | 2024-04-04 | Zymeworks Bc Inc. | Antibody drug conjugates targeting c-met and methods of use |
| WO2024081729A2 (en) | 2022-10-12 | 2024-04-18 | Mythic Therapeutics, Inc. | Lrrc-15-binding protein constructs and uses thereof |
Family Cites Families (84)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4444887A (en) | 1979-12-10 | 1984-04-24 | Sloan-Kettering Institute | Process for making human antibody producing B-lymphocytes |
| US4399216A (en) | 1980-02-25 | 1983-08-16 | The Trustees Of Columbia University | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US4634665A (en) | 1980-02-25 | 1987-01-06 | The Trustees Of Columbia University In The City Of New York | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US5179017A (en) | 1980-02-25 | 1993-01-12 | The Trustees Of Columbia University In The City Of New York | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US4716111A (en) | 1982-08-11 | 1987-12-29 | Trustees Of Boston University | Process for producing human antibodies |
| US4510245A (en) | 1982-11-18 | 1985-04-09 | Chiron Corporation | Adenovirus promoter system |
| GB8308235D0 (en) | 1983-03-25 | 1983-05-05 | Celltech Ltd | Polypeptides |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US5807715A (en) | 1984-08-27 | 1998-09-15 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and transformed mammalian lymphocyte cells for producing functional antigen-binding protein including chimeric immunoglobulin |
| US5168062A (en) | 1985-01-30 | 1992-12-01 | University Of Iowa Research Foundation | Transfer vectors and microorganisms containing human cytomegalovirus immediate-early promoter-regulatory DNA sequence |
| US4968615A (en) | 1985-12-18 | 1990-11-06 | Ciba-Geigy Corporation | Deoxyribonucleic acid segment from a virus |
| GB8607679D0 (en) | 1986-03-27 | 1986-04-30 | Winter G P | Recombinant dna product |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| EP0428534B1 (en) | 1988-06-14 | 1995-03-29 | Cetus Oncology Corporation | Coupling agents and sterically hindered disulfide linked conjugates prepared therefrom |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5413923A (en) | 1989-07-25 | 1995-05-09 | Cell Genesys, Inc. | Homologous recombination for universal donor cells and chimeric mammalian hosts |
| GB8928874D0 (en) | 1989-12-21 | 1990-02-28 | Celltech Ltd | Humanised antibodies |
| DE69133566T2 (de) | 1990-01-12 | 2007-12-06 | Amgen Fremont Inc. | Bildung von xenogenen Antikörpern |
| GB9015198D0 (en) | 1990-07-10 | 1990-08-29 | Brien Caroline J O | Binding substance |
| US5545806A (en) | 1990-08-29 | 1996-08-13 | Genpharm International, Inc. | Ransgenic non-human animals for producing heterologous antibodies |
| US5814318A (en) | 1990-08-29 | 1998-09-29 | Genpharm International Inc. | Transgenic non-human animals for producing heterologous antibodies |
| DK0814159T3 (da) | 1990-08-29 | 2005-10-24 | Genpharm Int | Transgene, ikke-humane dyr, der er i stand til at danne heterologe antistoffer |
| US5633425A (en) | 1990-08-29 | 1997-05-27 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5661016A (en) | 1990-08-29 | 1997-08-26 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| US5625126A (en) | 1990-08-29 | 1997-04-29 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| EP0519596B1 (en) | 1991-05-17 | 2005-02-23 | Merck & Co. Inc. | A method for reducing the immunogenicity of antibody variable domains |
| MX9204374A (es) | 1991-07-25 | 1993-03-01 | Idec Pharma Corp | Anticuerpo recombinante y metodo para su produccion. |
| US5565332A (en) | 1991-09-23 | 1996-10-15 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| ATE463573T1 (de) | 1991-12-02 | 2010-04-15 | Medimmune Ltd | Herstellung von autoantikörpern auf phagenoberflächen ausgehend von antikörpersegmentbibliotheken |
| US5639641A (en) | 1992-09-09 | 1997-06-17 | Immunogen Inc. | Resurfacing of rodent antibodies |
| EP0822830B1 (en) | 1995-04-27 | 2008-04-02 | Amgen Fremont Inc. | Human anti-IL-8 antibodies, derived from immunized xenomice |
| AU2466895A (en) | 1995-04-28 | 1996-11-18 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US5916771A (en) | 1996-10-11 | 1999-06-29 | Abgenix, Inc. | Production of a multimeric protein by cell fusion method |
| CA2722378C (en) | 1996-12-03 | 2015-02-03 | Amgen Fremont Inc. | Human antibodies that bind tnf.alpha. |
| JP3876002B2 (ja) | 1997-04-14 | 2007-01-31 | ミクロメート・アクチエンゲゼルシャフト | 抗ヒト抗原受容体を産生するための斬新な方法およびそれらの使用 |
| US6235883B1 (en) | 1997-05-05 | 2001-05-22 | Abgenix, Inc. | Human monoclonal antibodies to epidermal growth factor receptor |
| US6632926B1 (en) | 1998-11-18 | 2003-10-14 | Genentech, Inc. | Antibody variants |
| EP1243276A1 (en) | 2001-03-23 | 2002-09-25 | Franciscus Marinus Hendrikus De Groot | Elongated and multiple spacers containing activatible prodrugs |
| ES2544527T3 (es) | 2002-07-31 | 2015-09-01 | Seattle Genetics, Inc. | Conjugados de fármacos y su uso para tratar el cáncer, una enfermedad autoinmune o una enfermedad infecciosa |
| WO2004019993A1 (en) | 2002-08-30 | 2004-03-11 | Ramot At Tel Aviv University Ltd. | Self-immolative dendrimers releasing many active moieties upon a single activating event |
| US7217797B2 (en) | 2002-10-15 | 2007-05-15 | Pdl Biopharma, Inc. | Alteration of FcRn binding affinities or serum half-lives of antibodies by mutagenesis |
| EP1560599A1 (en) | 2002-11-14 | 2005-08-10 | Syntarga B.V. | Prodrugs built as multiple self-elimination-release spacers |
| ES2347959T3 (es) | 2003-02-20 | 2010-11-26 | Seattle Genetics, Inc. | Conjugados de anticuerpos anti-cd70-farmaco y su uso para el tratamiento del cancer. |
| EP2457586A1 (en) * | 2003-06-27 | 2012-05-30 | Amgen Fremont Inc. | Antibodies directed to the deletion mutants of epidermal growth factor receptor and uses thereof |
| SG149815A1 (en) * | 2003-11-06 | 2009-02-27 | Seattle Genetics Inc | Monomethylvaline compounds capable of conjugation to ligands |
| EP1718667B1 (en) | 2004-02-23 | 2013-01-09 | Genentech, Inc. | Heterocyclic self-immolative linkers and conjugates |
| JP4942643B2 (ja) * | 2004-03-02 | 2012-05-30 | シアトル ジェネティックス, インコーポレイテッド | 部分的に付加された抗体およびそれらの結合体化方法 |
| WO2005123780A2 (en) | 2004-04-09 | 2005-12-29 | Protein Design Labs, Inc. | Alteration of fcrn binding affinities or serum half-lives of antibodies by mutagenesis |
| CN101065151B (zh) | 2004-09-23 | 2014-12-10 | 健泰科生物技术公司 | 半胱氨酸改造的抗体和偶联物 |
| PL3248613T3 (pl) | 2005-07-18 | 2022-04-19 | Seagen Inc. | Koniugaty β-glukuronid-linker-lek |
| CA2641899A1 (en) | 2006-02-02 | 2007-08-09 | Syntarga B.V. | Water-soluble cc-1065 analogs and their conjugates |
| PT2099823E (pt) | 2006-12-01 | 2014-12-22 | Seattle Genetics Inc | Agentes de ligação ao alvo variantes e suas utilizações |
| EP2014681A1 (en) | 2007-07-12 | 2009-01-14 | Pierre Fabre Medicament | Novel antibodies inhibiting c-met dimerization, and uses thereof |
| EA020251B1 (ru) | 2007-11-28 | 2014-09-30 | Мерсана Терапьютикс, Инк. | Биосовместимые и биоразлагаемые конъюгаты аналогов фумагиллина |
| AR074439A1 (es) | 2008-12-02 | 2011-01-19 | Pf Medicament | Anticuerpo anti-cmet (receptor c-met) |
| US9469691B2 (en) | 2008-12-02 | 2016-10-18 | Pierre Fabre Medicament | Anti-cMET antibody |
| US8545839B2 (en) | 2008-12-02 | 2013-10-01 | Pierre Fabre Medicament | Anti-c-Met antibody |
| US20140112911A9 (en) | 2008-12-02 | 2014-04-24 | Liliane Goetsch | Novel anti-cmet antibody |
| WO2010064089A1 (en) | 2008-12-02 | 2010-06-10 | Pierre Fabre Medicament | Novel anti-cmet antibody |
| US20100152725A1 (en) | 2008-12-12 | 2010-06-17 | Angiodynamics, Inc. | Method and system for tissue treatment utilizing irreversible electroporation and thermal track coagulation |
| JP2012528240A (ja) | 2009-05-28 | 2012-11-12 | メルサナ セラピューティックス, インコーポレイテッド | 可変速度放出リンカーを含むポリアル−薬物コンジュゲート |
| EP2287197A1 (en) | 2009-08-21 | 2011-02-23 | Pierre Fabre Medicament | Anti-cMET antibody and its use for the detection and the diagnosis of cancer |
| JP5687206B2 (ja) | 2009-12-07 | 2015-03-18 | 株式会社Nttドコモ | 伝搬経路推定方法、プログラム及び装置 |
| WO2011111020A1 (en) | 2010-03-10 | 2011-09-15 | Novartis Ag | Streptococcus thermophilus bacterium |
| EP2553019A1 (en) | 2010-03-26 | 2013-02-06 | Mersana Therapeutics, Inc. | Modified polymers for delivery of polynucleotides, method of manufacture, and methods of use thereof |
| DE102010012915A1 (de) | 2010-03-26 | 2011-09-29 | Schaeffler Technologies Gmbh & Co. Kg | Vorrichtung und Verfahren zur Ermittlung eines Schädigungszustands eines Radlagers |
| EP2402370A1 (en) | 2010-06-29 | 2012-01-04 | Pierre Fabre Médicament | Novel antibody for the diagnosis and/or prognosis of cancer |
| RS59253B1 (sr) * | 2011-05-08 | 2019-10-31 | Legochem Biosciences Inc | Konjugati aktivnog agensa proteina i postupak za njihovo dobijanje |
| JP5926374B2 (ja) | 2011-06-10 | 2016-05-25 | メルサナ セラピューティクス,インコーポレイティド | タンパク質−高分子−薬剤コンジュゲート |
| KR101463098B1 (ko) * | 2011-11-28 | 2014-11-27 | 한국생명공학연구원 | c-Met에 대한 인간항체에 약물이 접합된 약물 복합체 및 이의 용도 |
| CN104244718A (zh) | 2011-12-05 | 2014-12-24 | 伊格尼卡生物治疗公司 | 抗体-药物缀合物以及相关化合物、组合物和方法 |
| TW201332574A (zh) | 2011-12-23 | 2013-08-16 | Mersana Therapeutics Inc | 煙曲黴素衍生物-phf共軛物之醫藥調配物 |
| MX353608B (es) * | 2012-02-24 | 2018-01-19 | Alteogen Inc | Anticuerpo modificado en el que el tema que comprende residuos de cisteina esta ligado, conjugado de farmaco de anticuerpo modificado que comprende el anticuerpo modificado y metodo de produccion para el mismo. |
| US9504756B2 (en) | 2012-05-15 | 2016-11-29 | Seattle Genetics, Inc. | Self-stabilizing linker conjugates |
| US20140017265A1 (en) | 2012-07-05 | 2014-01-16 | Mersana Therapeutics, Inc. | Terminally Modified Polymers and Conjugates Thereof |
| JP6334553B2 (ja) | 2012-12-10 | 2018-05-30 | メルサナ セラピューティクス,インコーポレイティド | タンパク質−高分子−薬剤コンジュゲート |
| US10226535B2 (en) | 2012-12-10 | 2019-03-12 | Mersana Therapeutics, Inc. | Auristatin compounds and conjugates thereof |
| US9872918B2 (en) | 2012-12-12 | 2018-01-23 | Mersana Therapeutics, Inc. | Hydroxyl-polymer-drug-protein conjugates |
| PT2968588T (pt) * | 2013-03-15 | 2019-05-08 | Abbvie Inc | Formulações de conjugado de fármaco-anticorpo anti-egfr |
| KR102150616B1 (ko) * | 2013-09-12 | 2020-09-03 | 삼성전자주식회사 | c-Met 표적 화합물-생체활성 물질 접합체 및 그 용도 |
| WO2016042412A1 (en) | 2014-09-16 | 2016-03-24 | Symphogen A/S | Anti-met antibodies and compositions |
| WO2016165580A1 (zh) * | 2015-04-17 | 2016-10-20 | 江苏恒瑞医药股份有限公司 | 抗c-Met抗体和抗c-Met抗体-细胞毒性药物偶联物及其医药用途 |
| CN106188293A (zh) * | 2015-04-17 | 2016-12-07 | 江苏恒瑞医药股份有限公司 | 抗c-Met抗体和抗c-Met抗体-细胞毒性药物偶联物及其医药用途 |
| DK3458102T3 (da) | 2016-05-17 | 2020-07-27 | Abbvie Biotherapeutics Inc | Anti-cmet-antistoflægemiddelkonjugater og fremgangsmåder til anvendelse deraf |
-
2017
- 2017-05-17 DK DK17728311.6T patent/DK3458102T3/da active
- 2017-05-17 SG SG11201810034XA patent/SG11201810034XA/en unknown
- 2017-05-17 CA CA3024386A patent/CA3024386C/en active Active
- 2017-05-17 WO PCT/US2017/033176 patent/WO2017201204A1/en unknown
- 2017-05-17 AU AU2017268342A patent/AU2017268342B2/en active Active
- 2017-05-17 SI SI201730337T patent/SI3458102T1/sl unknown
- 2017-05-17 KR KR1020237005212A patent/KR20230028574A/ko unknown
- 2017-05-17 NZ NZ748314A patent/NZ748314A/en unknown
- 2017-05-17 LT LTEP19197371.8T patent/LT3626273T/lt unknown
- 2017-05-17 EP EP20208371.3A patent/EP3804765A1/en not_active Withdrawn
- 2017-05-17 EP EP17728311.6A patent/EP3458102B1/en active Active
- 2017-05-17 PT PT177283116T patent/PT3458102T/pt unknown
- 2017-05-17 US US15/597,624 patent/US20170348429A1/en not_active Abandoned
- 2017-05-17 JP JP2018559845A patent/JP2019521087A/ja active Pending
- 2017-05-17 DK DK19197371.8T patent/DK3626273T3/da active
- 2017-05-17 EP EP23163423.9A patent/EP4233909A3/en active Pending
- 2017-05-17 CN CN201780044009.4A patent/CN109562189B/zh active Active
- 2017-05-17 LT LTEP17728311.6T patent/LT3458102T/lt unknown
- 2017-05-17 EP EP19197371.8A patent/EP3626273B1/en active Active
- 2017-05-17 PL PL17728311T patent/PL3458102T3/pl unknown
- 2017-05-17 HU HUE17728311A patent/HUE050398T2/hu unknown
- 2017-05-17 CN CN202111566314.4A patent/CN114177308A/zh active Pending
- 2017-05-17 ES ES19197371T patent/ES2864150T3/es active Active
- 2017-05-17 BR BR112018073574-4A patent/BR112018073574A2/pt unknown
- 2017-05-17 KR KR1020227033500A patent/KR102512597B1/ko active IP Right Grant
- 2017-05-17 RS RS20200927A patent/RS60663B1/sr unknown
- 2017-05-17 HU HUE19197371A patent/HUE053332T2/hu unknown
- 2017-05-17 KR KR1020187036427A patent/KR102449294B1/ko active IP Right Grant
- 2017-05-17 ES ES17728311T patent/ES2811351T3/es active Active
- 2017-05-17 CN CN202111593727.1A patent/CN114246953A/zh active Pending
- 2017-05-17 SI SI201730649T patent/SI3626273T1/sl unknown
- 2017-05-17 PT PT191973718T patent/PT3626273T/pt unknown
- 2017-05-17 RU RU2018144315A patent/RU2740996C2/ru active
- 2017-05-17 SG SG10201914095SA patent/SG10201914095SA/en unknown
- 2017-05-17 MX MX2018014175A patent/MX2018014175A/es unknown
- 2017-05-17 RS RS20210246A patent/RS61659B1/sr unknown
- 2017-05-17 IL IL262869A patent/IL262869B/en unknown
- 2017-05-17 PL PL19197371T patent/PL3626273T3/pl unknown
- 2017-05-17 IL IL294146A patent/IL294146B2/en unknown
-
2018
- 2018-03-02 US US15/910,788 patent/US10383948B2/en active Active
- 2018-12-12 ZA ZA2018/08386A patent/ZA201808386B/en unknown
-
2019
- 2019-05-28 US US16/424,324 patent/US10603389B2/en active Active
-
2020
- 2020-01-17 US US16/746,497 patent/US20200215200A1/en active Pending
- 2020-07-29 HR HRP20201186TT patent/HRP20201186T1/hr unknown
- 2020-08-10 CY CY20201100742T patent/CY1123256T1/el unknown
-
2021
- 2021-01-25 JP JP2021009320A patent/JP6870161B1/ja active Active
- 2021-02-25 HR HRP20210326TT patent/HRP20210326T1/hr unknown
- 2021-03-01 CY CY20211100168T patent/CY1123858T1/el unknown
- 2021-04-14 JP JP2021068144A patent/JP7150087B2/ja active Active
-
2022
- 2022-09-26 JP JP2022152185A patent/JP2022185003A/ja active Pending
-
2023
- 2023-02-22 US US18/172,769 patent/US20230321271A1/en active Pending
- 2023-07-25 IL IL304717A patent/IL304717A/en unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2864150T3 (es) | Conjugados de fármacos con anticuerpos anti-cMet y métodos para su uso | |
| ES2810755T3 (es) | Conjugados fármaco-anticuerpo anti-lrrc15 humano y métodos para su uso | |
| JP2019501124A (ja) | 抗huLRRC15抗体薬物コンジュゲート及びその使用方法 |