ES2841308T3 - Compuesto de pirrolidina novedoso y aplicación como agonista del receptor de melanocortina - Google Patents
Compuesto de pirrolidina novedoso y aplicación como agonista del receptor de melanocortina Download PDFInfo
- Publication number
- ES2841308T3 ES2841308T3 ES15799731T ES15799731T ES2841308T3 ES 2841308 T3 ES2841308 T3 ES 2841308T3 ES 15799731 T ES15799731 T ES 15799731T ES 15799731 T ES15799731 T ES 15799731T ES 2841308 T3 ES2841308 T3 ES 2841308T3
- Authority
- ES
- Spain
- Prior art keywords
- group
- optionally substituted
- alkyl
- independently selected
- aliphatic heterocyclic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 pyrrolidine compound Chemical class 0.000 title claims abstract description 352
- RWRDLPDLKQPQOW-UHFFFAOYSA-N tetrahydropyrrole Natural products C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 title description 19
- 239000000336 melanocortin receptor agonist Substances 0.000 title description 3
- 229940117029 Melanocortin receptor agonist Drugs 0.000 title description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 285
- 125000005843 halogen group Chemical group 0.000 claims abstract description 264
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 233
- 125000004433 nitrogen atom Chemical group N* 0.000 claims abstract description 167
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 164
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims abstract description 156
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 154
- 125000001931 aliphatic group Chemical group 0.000 claims abstract description 152
- 125000004093 cyano group Chemical group *C#N 0.000 claims abstract description 151
- 125000005842 heteroatom Chemical group 0.000 claims abstract description 132
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 124
- 125000004043 oxo group Chemical group O=* 0.000 claims abstract description 117
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims abstract description 113
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 111
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 104
- 239000001301 oxygen Substances 0.000 claims abstract description 104
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 101
- 239000011593 sulfur Substances 0.000 claims abstract description 100
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims abstract description 93
- 229910052757 nitrogen Chemical group 0.000 claims abstract description 92
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims abstract description 91
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims abstract description 76
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 72
- 125000001424 substituent group Chemical group 0.000 claims abstract description 61
- 125000004739 (C1-C6) alkylsulfonyl group Chemical group 0.000 claims abstract description 52
- 125000003277 amino group Chemical group 0.000 claims abstract description 47
- 125000003118 aryl group Chemical group 0.000 claims abstract description 47
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 claims abstract description 42
- 125000005529 alkyleneoxy group Chemical group 0.000 claims abstract description 41
- 125000004434 sulfur atom Chemical group 0.000 claims abstract description 33
- 125000004737 (C1-C6) haloalkoxy group Chemical group 0.000 claims abstract description 31
- 125000004183 alkoxy alkyl group Chemical group 0.000 claims abstract description 30
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 claims abstract description 28
- 125000000392 cycloalkenyl group Chemical group 0.000 claims abstract description 28
- 125000004454 (C1-C6) alkoxycarbonyl group Chemical group 0.000 claims abstract description 26
- 125000006577 C1-C6 hydroxyalkyl group Chemical group 0.000 claims abstract description 23
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims abstract description 21
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 claims abstract description 17
- 125000002252 acyl group Chemical group 0.000 claims abstract description 13
- 125000004429 atom Chemical group 0.000 claims abstract description 11
- FKCMADOPPWWGNZ-YUMQZZPRSA-N [(2r)-1-[(2s)-2-amino-3-methylbutanoyl]pyrrolidin-2-yl]boronic acid Chemical compound CC(C)[C@H](N)C(=O)N1CCC[C@H]1B(O)O FKCMADOPPWWGNZ-YUMQZZPRSA-N 0.000 claims abstract description 3
- 125000004966 cyanoalkyl group Chemical group 0.000 claims abstract description 3
- 150000003463 sulfur Chemical class 0.000 claims abstract 6
- 150000002926 oxygen Chemical class 0.000 claims abstract 2
- 150000001875 compounds Chemical class 0.000 claims description 600
- 125000000217 alkyl group Chemical group 0.000 claims description 141
- 125000001072 heteroaryl group Chemical group 0.000 claims description 104
- 239000002253 acid Substances 0.000 claims description 92
- 150000003839 salts Chemical class 0.000 claims description 82
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 79
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 73
- 125000003386 piperidinyl group Chemical group 0.000 claims description 71
- 229910052799 carbon Inorganic materials 0.000 claims description 66
- 125000002950 monocyclic group Chemical group 0.000 claims description 57
- 125000003545 alkoxy group Chemical group 0.000 claims description 54
- 125000004193 piperazinyl group Chemical group 0.000 claims description 50
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 claims description 49
- 125000002393 azetidinyl group Chemical group 0.000 claims description 48
- 125000002757 morpholinyl group Chemical group 0.000 claims description 48
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 45
- 125000004568 thiomorpholinyl group Chemical group 0.000 claims description 44
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 claims description 40
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims description 38
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 37
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 36
- 125000004076 pyridyl group Chemical group 0.000 claims description 36
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 35
- 125000003831 tetrazolyl group Chemical group 0.000 claims description 35
- 125000002636 imidazolinyl group Chemical group 0.000 claims description 33
- 125000001984 thiazolidinyl group Chemical group 0.000 claims description 33
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 32
- YNPNZTXNASCQKK-UHFFFAOYSA-N Phenanthrene Natural products C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 claims description 30
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 claims description 30
- 125000002971 oxazolyl group Chemical group 0.000 claims description 30
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 30
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 30
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 28
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 28
- 125000001715 oxadiazolyl group Chemical group 0.000 claims description 28
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 28
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 claims description 27
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 claims description 27
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 26
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 25
- 125000004306 triazinyl group Chemical group 0.000 claims description 25
- 125000002883 imidazolyl group Chemical group 0.000 claims description 23
- 125000000335 thiazolyl group Chemical group 0.000 claims description 23
- 125000004305 thiazinyl group Chemical group S1NC(=CC=C1)* 0.000 claims description 21
- 125000002541 furyl group Chemical group 0.000 claims description 19
- 125000001544 thienyl group Chemical group 0.000 claims description 18
- 125000003107 substituted aryl group Chemical group 0.000 claims description 14
- 125000001188 haloalkyl group Chemical group 0.000 claims description 13
- 229910052736 halogen Inorganic materials 0.000 claims description 12
- 125000004356 hydroxy functional group Chemical group O* 0.000 claims description 12
- 239000008194 pharmaceutical composition Substances 0.000 claims description 11
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 claims description 10
- 230000002265 prevention Effects 0.000 claims description 10
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 claims description 10
- 201000008220 erythropoietic protoporphyria Diseases 0.000 claims description 9
- 125000001624 naphthyl group Chemical group 0.000 claims description 9
- 201000001441 melanoma Diseases 0.000 claims description 8
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 claims description 7
- 206010029164 Nephrotic syndrome Diseases 0.000 claims description 6
- 206010047642 Vitiligo Diseases 0.000 claims description 6
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 6
- 208000002780 macular degeneration Diseases 0.000 claims description 6
- 125000006727 (C1-C6) alkenyl group Chemical group 0.000 claims description 5
- 206010016654 Fibrosis Diseases 0.000 claims description 5
- 208000007014 Retinitis pigmentosa Diseases 0.000 claims description 5
- 201000009594 Systemic Scleroderma Diseases 0.000 claims description 5
- 206010042953 Systemic sclerosis Diseases 0.000 claims description 5
- 125000003342 alkenyl group Chemical group 0.000 claims description 5
- 230000004761 fibrosis Effects 0.000 claims description 5
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 5
- 125000005346 substituted cycloalkyl group Chemical group 0.000 claims description 5
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 claims description 5
- 238000002560 therapeutic procedure Methods 0.000 claims description 5
- 201000004681 Psoriasis Diseases 0.000 claims description 4
- 208000000453 Skin Neoplasms Diseases 0.000 claims description 4
- 206010064930 age-related macular degeneration Diseases 0.000 claims description 4
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 claims description 4
- 150000002367 halogens Chemical class 0.000 claims description 4
- 201000004384 Alopecia Diseases 0.000 claims description 3
- 125000005115 alkyl carbamoyl group Chemical group 0.000 claims description 3
- 208000024963 hair loss Diseases 0.000 claims description 3
- 230000003676 hair loss Effects 0.000 claims description 3
- 201000000849 skin cancer Diseases 0.000 claims description 3
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 2
- 208000014644 Brain disease Diseases 0.000 claims description 2
- 208000015943 Coeliac disease Diseases 0.000 claims description 2
- 201000005569 Gout Diseases 0.000 claims description 2
- 206010018634 Gouty Arthritis Diseases 0.000 claims description 2
- 208000031886 HIV Infections Diseases 0.000 claims description 2
- 208000002193 Pain Diseases 0.000 claims description 2
- 206010036186 Porphyria non-acute Diseases 0.000 claims description 2
- 206010040047 Sepsis Diseases 0.000 claims description 2
- 206010040070 Septic Shock Diseases 0.000 claims description 2
- 206010046851 Uveitis Diseases 0.000 claims description 2
- 206010047115 Vasculitis Diseases 0.000 claims description 2
- 230000005713 exacerbation Effects 0.000 claims description 2
- 208000015181 infectious disease Diseases 0.000 claims description 2
- 230000002757 inflammatory effect Effects 0.000 claims description 2
- 230000009545 invasion Effects 0.000 claims description 2
- 201000008482 osteoarthritis Diseases 0.000 claims description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 2
- 230000036303 septic shock Effects 0.000 claims description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 claims description 2
- 238000002054 transplantation Methods 0.000 claims description 2
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims 1
- 206010063837 Reperfusion injury Diseases 0.000 claims 1
- 239000004480 active ingredient Substances 0.000 claims 1
- 208000006454 hepatitis Diseases 0.000 claims 1
- 231100000283 hepatitis Toxicity 0.000 claims 1
- 208000028867 ischemia Diseases 0.000 claims 1
- 230000000813 microbial effect Effects 0.000 claims 1
- 201000008383 nephritis Diseases 0.000 claims 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical group N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 abstract description 23
- 125000004438 haloalkoxy group Chemical group 0.000 abstract description 2
- 238000006243 chemical reaction Methods 0.000 description 185
- 239000002904 solvent Substances 0.000 description 155
- 125000006239 protecting group Chemical group 0.000 description 102
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 96
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 88
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 81
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 78
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 76
- 238000004519 manufacturing process Methods 0.000 description 69
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 62
- 239000000203 mixture Substances 0.000 description 54
- 239000002585 base Substances 0.000 description 49
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 45
- 239000000543 intermediate Substances 0.000 description 41
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 39
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 39
- 239000003054 catalyst Substances 0.000 description 38
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 36
- 238000007796 conventional method Methods 0.000 description 33
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 31
- 229910052763 palladium Inorganic materials 0.000 description 31
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 30
- 238000001308 synthesis method Methods 0.000 description 30
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 29
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 26
- 238000006722 reduction reaction Methods 0.000 description 25
- 229910052731 fluorine Inorganic materials 0.000 description 22
- 238000003756 stirring Methods 0.000 description 22
- 125000001153 fluoro group Chemical group F* 0.000 description 20
- 239000003638 chemical reducing agent Substances 0.000 description 17
- 230000000694 effects Effects 0.000 description 17
- 102000008314 Type 1 Melanocortin Receptor Human genes 0.000 description 16
- 108010021428 Type 1 Melanocortin Receptor Proteins 0.000 description 16
- 125000004432 carbon atom Chemical group C* 0.000 description 16
- 239000003795 chemical substances by application Substances 0.000 description 16
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 15
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 14
- 102000004378 Melanocortin Receptors Human genes 0.000 description 13
- 108090000950 Melanocortin Receptors Proteins 0.000 description 13
- 238000005859 coupling reaction Methods 0.000 description 13
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 13
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 12
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 12
- 239000000556 agonist Substances 0.000 description 12
- 150000001408 amides Chemical class 0.000 description 12
- 229910052801 chlorine Inorganic materials 0.000 description 12
- 150000002148 esters Chemical class 0.000 description 12
- 150000002825 nitriles Chemical class 0.000 description 12
- 230000004913 activation Effects 0.000 description 10
- 229910052739 hydrogen Inorganic materials 0.000 description 10
- 125000000547 substituted alkyl group Chemical group 0.000 description 10
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 9
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 9
- 239000001257 hydrogen Substances 0.000 description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- 239000000047 product Substances 0.000 description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- 238000007259 addition reaction Methods 0.000 description 8
- 239000002168 alkylating agent Substances 0.000 description 8
- 229940100198 alkylating agent Drugs 0.000 description 8
- 125000001309 chloro group Chemical group Cl* 0.000 description 8
- 238000000034 method Methods 0.000 description 8
- 238000007363 ring formation reaction Methods 0.000 description 8
- 239000012279 sodium borohydride Substances 0.000 description 8
- 229910000033 sodium borohydride Inorganic materials 0.000 description 8
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 7
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 7
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 7
- 239000003153 chemical reaction reagent Substances 0.000 description 7
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 7
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 7
- 229910000027 potassium carbonate Inorganic materials 0.000 description 7
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 7
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 6
- 150000001721 carbon Chemical group 0.000 description 6
- 238000006482 condensation reaction Methods 0.000 description 6
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 6
- 150000003951 lactams Chemical group 0.000 description 6
- 102000005962 receptors Human genes 0.000 description 6
- 108020003175 receptors Proteins 0.000 description 6
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- 239000000275 Adrenocorticotropic Hormone Substances 0.000 description 5
- 101800000414 Corticotropin Proteins 0.000 description 5
- 102400000739 Corticotropin Human genes 0.000 description 5
- 102400000740 Melanocyte-stimulating hormone alpha Human genes 0.000 description 5
- 101710200814 Melanotropin alpha Proteins 0.000 description 5
- 230000001270 agonistic effect Effects 0.000 description 5
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 5
- 150000008041 alkali metal carbonates Chemical class 0.000 description 5
- 125000002947 alkylene group Chemical group 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 description 5
- 229960000258 corticotropin Drugs 0.000 description 5
- USVZFSNDGFNNJT-UHFFFAOYSA-N cyclopenta-1,4-dien-1-yl(diphenyl)phosphane (2,3-dichlorocyclopenta-1,4-dien-1-yl)-diphenylphosphane iron(2+) Chemical compound [Fe++].c1cc[c-](c1)P(c1ccccc1)c1ccccc1.Clc1c(cc[c-]1Cl)P(c1ccccc1)c1ccccc1 USVZFSNDGFNNJT-UHFFFAOYSA-N 0.000 description 5
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 5
- 239000011737 fluorine Substances 0.000 description 5
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 5
- 238000006268 reductive amination reaction Methods 0.000 description 5
- WHNFPRLDDSXQCL-UAZQEYIDSA-N α-msh Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(N)=O)NC(=O)[C@H](CO)NC(C)=O)C1=CC=C(O)C=C1 WHNFPRLDDSXQCL-UAZQEYIDSA-N 0.000 description 5
- USVVENVKYJZFMW-ONEGZZNKSA-N (e)-carboxyiminocarbamic acid Chemical class OC(=O)\N=N\C(O)=O USVVENVKYJZFMW-ONEGZZNKSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- IVOMOUWHDPKRLL-UHFFFAOYSA-N UNPD107823 Natural products O1C2COP(O)(=O)OC2C(O)C1N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-UHFFFAOYSA-N 0.000 description 4
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 229940095074 cyclic amp Drugs 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 4
- 201000010099 disease Diseases 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- 150000003003 phosphines Chemical class 0.000 description 4
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 4
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 4
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 3
- 102100022455 Adrenocorticotropic hormone receptor Human genes 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 3
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 3
- 101000678419 Homo sapiens Adrenocorticotropic hormone receptor Proteins 0.000 description 3
- 101000978418 Homo sapiens Melanocortin receptor 4 Proteins 0.000 description 3
- 206010061218 Inflammation Diseases 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- 102000030612 Melanocortin 5 receptor Human genes 0.000 description 3
- 108010088565 Melanocortin 5 receptor Proteins 0.000 description 3
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 3
- 238000005804 alkylation reaction Methods 0.000 description 3
- 230000003110 anti-inflammatory effect Effects 0.000 description 3
- 230000033228 biological regulation Effects 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 238000009833 condensation Methods 0.000 description 3
- 230000005494 condensation Effects 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 239000012025 fluorinating agent Substances 0.000 description 3
- 238000003682 fluorination reaction Methods 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 3
- 125000005415 substituted alkoxy group Chemical group 0.000 description 3
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 125000004665 trialkylsilyl group Chemical group 0.000 description 3
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 description 2
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 2
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- LULAYUGMBFYYEX-UHFFFAOYSA-N 3-chlorobenzoic acid Chemical compound OC(=O)C1=CC=CC(Cl)=C1 LULAYUGMBFYYEX-UHFFFAOYSA-N 0.000 description 2
- VJFMSYZSFUWQPZ-WXPXUSHHSA-N 4-[(s)-[(2r,4s,5r)-5-ethenyl-1-azabicyclo[2.2.2]octan-2-yl]-hydroxymethyl]quinolin-6-ol Chemical compound C1=C(O)C=C2C([C@@H]([C@@H]3N4CC[C@H]([C@H](C4)C=C)C3)O)=CC=NC2=C1 VJFMSYZSFUWQPZ-WXPXUSHHSA-N 0.000 description 2
- OJOFMLDBXPDXLQ-UHFFFAOYSA-N 4-benzyl-1,3-oxazolidin-2-one Chemical compound C1OC(=O)NC1CC1=CC=CC=C1 OJOFMLDBXPDXLQ-UHFFFAOYSA-N 0.000 description 2
- QDMNNMIOWVJVLY-UHFFFAOYSA-N 4-phenyl-1,3-oxazolidin-2-one Chemical compound C1OC(=O)NC1C1=CC=CC=C1 QDMNNMIOWVJVLY-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 2
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 2
- 101000978431 Homo sapiens Melanocortin receptor 3 Proteins 0.000 description 2
- 101001134060 Homo sapiens Melanocyte-stimulating hormone receptor Proteins 0.000 description 2
- 101150078994 La gene Proteins 0.000 description 2
- 102100023726 Melanocortin receptor 3 Human genes 0.000 description 2
- 102100023724 Melanocortin receptor 4 Human genes 0.000 description 2
- 238000006845 Michael addition reaction Methods 0.000 description 2
- 102000003979 Mineralocorticoid Receptors Human genes 0.000 description 2
- 108090000375 Mineralocorticoid Receptors Proteins 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- 108010069820 Pro-Opiomelanocortin Proteins 0.000 description 2
- 239000000683 Pro-Opiomelanocortin Substances 0.000 description 2
- 102100027467 Pro-opiomelanocortin Human genes 0.000 description 2
- 208000034189 Sclerosis Diseases 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 208000027418 Wounds and injury Diseases 0.000 description 2
- 150000008065 acid anhydrides Chemical class 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- NNTOJPXOCKCMKR-UHFFFAOYSA-N boron;pyridine Chemical compound [B].C1=CC=NC=C1 NNTOJPXOCKCMKR-UHFFFAOYSA-N 0.000 description 2
- 125000001589 carboacyl group Chemical group 0.000 description 2
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 2
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 238000006114 decarboxylation reaction Methods 0.000 description 2
- VJFMSYZSFUWQPZ-UHFFFAOYSA-N demethyl quinine Natural products C1=C(O)C=C2C(C(C3N4CCC(C(C4)C=C)C3)O)=CC=NC2=C1 VJFMSYZSFUWQPZ-UHFFFAOYSA-N 0.000 description 2
- 230000003635 deoxygenating effect Effects 0.000 description 2
- 239000012351 deprotecting agent Substances 0.000 description 2
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- WDAXFOBOLVPGLV-UHFFFAOYSA-N ethyl isobutyrate Chemical compound CCOC(=O)C(C)C WDAXFOBOLVPGLV-UHFFFAOYSA-N 0.000 description 2
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 2
- 125000004672 ethylcarbonyl group Chemical group [H]C([H])([H])C([H])([H])C(*)=O 0.000 description 2
- 125000006125 ethylsulfonyl group Chemical group 0.000 description 2
- 230000002140 halogenating effect Effects 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 102000054812 human MC1R Human genes 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 208000014674 injury Diseases 0.000 description 2
- 238000006192 iodination reaction Methods 0.000 description 2
- 229910052740 iodine Chemical group 0.000 description 2
- 239000011630 iodine Chemical group 0.000 description 2
- 238000004811 liquid chromatography Methods 0.000 description 2
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 2
- 238000001465 metallisation Methods 0.000 description 2
- OIRDBPQYVWXNSJ-UHFFFAOYSA-N methyl trifluoromethansulfonate Chemical compound COS(=O)(=O)C(F)(F)F OIRDBPQYVWXNSJ-UHFFFAOYSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 238000006452 multicomponent reaction Methods 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 210000005036 nerve Anatomy 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 235000011056 potassium acetate Nutrition 0.000 description 2
- 150000003235 pyrrolidines Chemical class 0.000 description 2
- 238000007348 radical reaction Methods 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 229930195734 saturated hydrocarbon Natural products 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 238000010898 silica gel chromatography Methods 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- MFRIHAYPQRLWNB-UHFFFAOYSA-N sodium tert-butoxide Chemical compound [Na+].CC(C)(C)[O-] MFRIHAYPQRLWNB-UHFFFAOYSA-N 0.000 description 2
- 125000003003 spiro group Chemical group 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- JIAARYAFYJHUJI-UHFFFAOYSA-L zinc dichloride Chemical compound [Cl-].[Cl-].[Zn+2] JIAARYAFYJHUJI-UHFFFAOYSA-L 0.000 description 2
- BZMMRNKDONDVIB-UHFFFAOYSA-N (1-ethoxycyclopropyl)oxy-trimethylsilane Chemical compound CCOC1(O[Si](C)(C)C)CC1 BZMMRNKDONDVIB-UHFFFAOYSA-N 0.000 description 1
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- 125000004767 (C1-C4) haloalkoxy group Chemical group 0.000 description 1
- YRIZYWQGELRKNT-UHFFFAOYSA-N 1,3,5-trichloro-1,3,5-triazinane-2,4,6-trione Chemical compound ClN1C(=O)N(Cl)C(=O)N(Cl)C1=O YRIZYWQGELRKNT-UHFFFAOYSA-N 0.000 description 1
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical group CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- NQRCAVZHOLYEBJ-ZIAGYGMSSA-N 1-[3,5-bis(trifluoromethyl)phenyl]-3-[(1r,2r)-2-(dimethylamino)cyclohexyl]thiourea Chemical compound CN(C)[C@@H]1CCCC[C@H]1NC(=S)NC1=CC(C(F)(F)F)=CC(C(F)(F)F)=C1 NQRCAVZHOLYEBJ-ZIAGYGMSSA-N 0.000 description 1
- VUZNLSBZRVZGIK-UHFFFAOYSA-N 2,2,6,6-Tetramethyl-1-piperidinol Chemical group CC1(C)CCCC(C)(C)N1O VUZNLSBZRVZGIK-UHFFFAOYSA-N 0.000 description 1
- GPIQOFWTZXXOOV-UHFFFAOYSA-N 2-chloro-4,6-dimethoxy-1,3,5-triazine Chemical compound COC1=NC(Cl)=NC(OC)=N1 GPIQOFWTZXXOOV-UHFFFAOYSA-N 0.000 description 1
- 125000004777 2-fluoroethyl group Chemical group [H]C([H])(F)C([H])([H])* 0.000 description 1
- MWFMGBPGAXYFAR-UHFFFAOYSA-N 2-hydroxy-2-methylpropanenitrile Chemical compound CC(C)(O)C#N MWFMGBPGAXYFAR-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- BMTZEAOGFDXDAD-UHFFFAOYSA-M 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholin-4-ium;chloride Chemical compound [Cl-].COC1=NC(OC)=NC([N+]2(C)CCOCC2)=N1 BMTZEAOGFDXDAD-UHFFFAOYSA-M 0.000 description 1
- SBNYNTYNEJTMQO-UHFFFAOYSA-N 6655-27-2 Chemical compound CC(C)=NNS(=O)(=O)C1=CC=CC=C1[N+]([O-])=O SBNYNTYNEJTMQO-UHFFFAOYSA-N 0.000 description 1
- 208000009304 Acute Kidney Injury Diseases 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- 102100032257 E3 ubiquitin-protein ligase Mdm2 Human genes 0.000 description 1
- 108050002772 E3 ubiquitin-protein ligase Mdm2 Proteins 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical group CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 239000012448 Lithium borohydride Substances 0.000 description 1
- 108010008364 Melanocortins Proteins 0.000 description 1
- 239000000637 Melanocyte-Stimulating Hormone Substances 0.000 description 1
- 102400000744 Melanotropin gamma Human genes 0.000 description 1
- 101800000520 Melanotropin gamma Proteins 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 102000014415 Muscarinic acetylcholine receptor Human genes 0.000 description 1
- 108050003473 Muscarinic acetylcholine receptor Proteins 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 208000004550 Postoperative Pain Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 208000033626 Renal failure acute Diseases 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- ZVQOOHYFBIDMTQ-UHFFFAOYSA-N [methyl(oxido){1-[6-(trifluoromethyl)pyridin-3-yl]ethyl}-lambda(6)-sulfanylidene]cyanamide Chemical compound N#CN=S(C)(=O)C(C)C1=CC=C(C(F)(F)F)N=C1 ZVQOOHYFBIDMTQ-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 201000011040 acute kidney failure Diseases 0.000 description 1
- 208000012998 acute renal failure Diseases 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 210000004404 adrenal cortex Anatomy 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 230000037424 autonomic function Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical group C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 206010009887 colitis Diseases 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000004850 cyclobutylmethyl group Chemical group C1(CCC1)C* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 1
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- ZFTFAPZRGNKQPU-UHFFFAOYSA-N dicarbonic acid Chemical compound OC(=O)OC(O)=O ZFTFAPZRGNKQPU-UHFFFAOYSA-N 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 1
- 125000004786 difluoromethoxy group Chemical group [H]C(F)(F)O* 0.000 description 1
- 125000005046 dihydronaphthyl group Chemical group 0.000 description 1
- 125000004655 dihydropyridinyl group Chemical group N1(CC=CC=C1)* 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- RCRYEYMHBHPZQD-UHFFFAOYSA-N ditert-butyl-[2,3,4,5-tetramethyl-6-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane Chemical group CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=C(C)C(C)=C(C)C(C)=C1P(C(C)(C)C)C(C)(C)C RCRYEYMHBHPZQD-UHFFFAOYSA-N 0.000 description 1
- SACNIGZYDTUHKB-UHFFFAOYSA-N ditert-butyl-[2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane Chemical group CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C(C)(C)C)C(C)(C)C SACNIGZYDTUHKB-UHFFFAOYSA-N 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 230000009986 erectile function Effects 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 210000003499 exocrine gland Anatomy 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 102000057094 human MC4R Human genes 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000036737 immune function Effects 0.000 description 1
- 230000003832 immune regulation Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 125000004129 indan-1-yl group Chemical group [H]C1=C([H])C([H])=C2C(=C1[H])C([H])([H])C([H])([H])C2([H])* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 210000004969 inflammatory cell Anatomy 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 230000026045 iodination Effects 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 210000002510 keratinocyte Anatomy 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- LVKCSZQWLOVUGB-UHFFFAOYSA-M magnesium;propane;bromide Chemical compound [Mg+2].[Br-].C[CH-]C LVKCSZQWLOVUGB-UHFFFAOYSA-M 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 210000002752 melanocyte Anatomy 0.000 description 1
- 230000003061 melanogenesis Effects 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 230000002906 microbiologic effect Effects 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- RLKHFSNWQCZBDC-UHFFFAOYSA-N n-(benzenesulfonyl)-n-fluorobenzenesulfonamide Chemical compound C=1C=CC=CC=1S(=O)(=O)N(F)S(=O)(=O)C1=CC=CC=C1 RLKHFSNWQCZBDC-UHFFFAOYSA-N 0.000 description 1
- 125000004370 n-butenyl group Chemical group [H]\C([H])=C(/[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 210000004498 neuroglial cell Anatomy 0.000 description 1
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 description 1
- 150000002829 nitrogen Chemical group 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 125000005476 oxopyrrolidinyl group Chemical group 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 230000002633 protecting effect Effects 0.000 description 1
- 230000001012 protector Effects 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 230000010410 reperfusion Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 208000013223 septicemia Diseases 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000009518 sodium iodide Nutrition 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- VNFWTIYUKDMAOP-UHFFFAOYSA-N sphos Chemical group COC1=CC=CC(OC)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 VNFWTIYUKDMAOP-UHFFFAOYSA-N 0.000 description 1
- 238000011916 stereoselective reduction Methods 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 229950009390 symclosene Drugs 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 125000006173 tetrahydropyranylmethyl group Chemical group 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 125000006168 tricyclic group Chemical group 0.000 description 1
- 125000005951 trifluoromethanesulfonyloxy group Chemical group 0.000 description 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000011592 zinc chloride Substances 0.000 description 1
- 235000005074 zinc chloride Nutrition 0.000 description 1
- GZWUQPQBOGLSIM-VOOUCTBASA-N γ msh Chemical compound C([C@H](N)C(=O)N[C@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)NCC(O)=O)C(C)C)C1=CC=C(O)C=C1 GZWUQPQBOGLSIM-VOOUCTBASA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Dermatology (AREA)
- Neurosurgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pyrrole Compounds (AREA)
Abstract
Un derivado de pirrolidina representado por la fórmula [I]: **(Ver fórmula)** donde el anillo A es un grupo fenilo opcionalmente sustituido donde los sustituyentes en el grupo fenilo opcionalmente sustituido son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7, un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6; R1 es un grupo alquilo C1-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo adamantilo opcionalmente sustituido, un grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno, un grupo arilo de 6 a 10 miembros opcionalmente sustituido que puede ser parcialmente hidrogenado, un grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6, donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno y un grupo alquilenoxi C1- C6 y sustituyentes en cada uno del grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno, el grupo arilo de 6 a 10 miembros opcionalmente sustituido que puede ser parcialmente hidrogenado y el grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6; un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo hidroxialquilo C1-C6; un grupo alcoxialquilo C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno y un grupo alquilenoxi C1-C6; R2 es un átomo de halógeno, un grupo alquilo C1-C6 o un grupo alcoxi C1-C6; R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II]: **(Ver fórmula)** donde el anillo B representa un grupo heterocíclico alifático de 4 a 8 miembros que contenían nitrógeno que puede contener parcialmente un doble enlace; el anillo C representa un grupo arilo de 6 a 10 miembros o un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno; R5 y R6 representan cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1- C6 sustituido con de uno a tres heteroátomos; un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6; R7 representa un grupo alquilo C1-C6 opcionalmente sustituido, un grupo alquenilo C2-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo cicloalquenilo C3-C7 opcionalmente sustituido, un grupo arilo de 6 a 10 miembros opcionalmente sustituido, un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido, un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido, un grupo alcoxi C1-C6 opcionalmente sustituido, un grupo amino opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo, o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo, donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo hidroxi o un grupo oxo; un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con uno o dos grupos oxo; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (la porción alquilo C1-C6 es opcionalmente sustituida con un grupo hidroxilo, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1- C6 y un grupo alquilsulfonilo C1-C6 y los sustituyentes en cada uno del grupo alquenilo C2-C6 opcionalmente sustituido, el grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo cicloalquenilo C3-C7 opcionalmente sustituido, el grupo arilo de 6 a 10 miembros opcionalmente sustituido, el grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido, el grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno y el grupo alcoxi C1-C6 opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo hidroxi o un grupo oxo; un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con uno o dos grupos oxo; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (la porción alquilo C1-C6 es opcionalmente sustituida con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1- C6 y un grupo alquilsulfonilo C1-C6 y R8 y R9 representan cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
Description
DESCRIPCIÓN
Compuesto de pirrolidina novedoso y aplicación como agonista del receptor de melanocortina
Campo técnico
La presente invención se refiere a un compuesto de pirrolidina novedoso que tiene actividad agonística (actividad agonista) del receptor de melanocortina (MCR).
Técnica antecedente
La hormona estimuladora de a-melanocitos (a-MSH), es una hormona derivada de la pro-opiomelanocortina (POMC) (literatura no de patentes 1) y es referida como un péptido de melanocortina, junto con la p-MSH y la y-MSH y la hormona adrenocorticotrópica (ACTH). Se sabe que la a-MSH exhibe acción inhibidora sobre la producción de mediadores asociados con la fibrosis y la inflamación implicados en la ocurrencia de varias condiciones patológicas y muestra eficacia en modelos de enfermedades autoinmunes tales como colitis, uveorretinitis y artritis (literatura no de patentes 2). Se han desarrollado también análogos de a-MSH para su uso en el tratamiento de la protoporfiria, la insuficiencia renal aguda y el dolor postoperatorio.
Los receptores de melanocortina (MCR), los cuales son receptores de la a-MSH, son siete receptores acoplados a la proteína G (GPCR) de transmembrana e incrementan el AMP cíclico (AMPc) intracelular a través de su activación (literatura no de patentes 3). Existen 5 subtipos de MCR, es decir, del MC1R al MC5R.
El MC1R es un receptor que es activado principalmente por la a-MSH y es expresado en melanocitos, células inmunes e inflamatorias, fibroblastos, queratinocitos, células endoteliales y células gliales. De esta manera, se sabe que la activación del MC1R incrementa el nivel de AMPc en las células en las cuales el MC1R es expresado, llevando a efectos tales como la homeostasis en la piel contra la melanogénesis y estímulos externos (literatura no de patentes 4), acción antiinflamatoria y acción inhibidora sobre la fibrosis del tejido (literatura no de patentes 5). El MC2R es un receptor que tiene una baja respuesta a la a-MSH y es activado principalmente por la ACTH. El MC2R es expresado principalmente en la corteza suprarrenal. Se sabe que la activación del MC2R tiene un efecto sobre la producción de esteroides. El MC3R es un receptor que es activado principalmente por la gamma-MSH y la ACTH y es expresado en nervios centrales y macrófagos. Se sabe que la activación del MC3R produce efectos tales como la regulación de la función autónoma y acción antiinflamatoria. El MC4R es un receptor que es activado principalmente por la a-MSH y la ACTH y es expresado en nervios centrales. De esta manera, se sabe que la activación del MC4R produce efectos tales como supresión de la ingestión de alimento y mejora de la función eréctil. El MC5R es un receptor que es activado principalmente por la alfa-MSH y es expresado en las glándulas exocrinas y los linfocitos. Se sabe que la activación del MC5R produce efectos tales como la regulación de los fluidos exocrinos y la regulación de la función inmune. Por lo tanto, se espera que la activación de estos receptores de melanocortina (MCR) provea efectos tales como regulación inmune, anti-inflamación y supresión de la fibrosis, a través de la formación de AMPc.
Las literaturas de patentes 1 y 2, por ejemplo, se conocen como documentos que describen compuestos de pirrolidina que tienen un grupo carbamoílo en la posición 3 de la pirrolidina. Sin embargo, los compuestos descritos en la literatura de patentes 1, son compuestos que está unidos a HDM2 que exhiben acción anticáncer, los cuales tienen también un grupo alquilo, arilo o heteroarilo como un sustituyente en la posición 2 de la pirrolidina. De esta manera, la literatura de patentes 1 no describe compuestos de pirrolidina que tienen sustituyentes en las posiciones 1, 3 y 4, como los compuestos de la presente invención.
Los compuestos descritos en la literatura de patentes 2 son compuestos sustituidos con sólo un grupo carbamoílo en la posición 3 de la pirrolidina. De esta manera, la literatura de patentes 2 no describe compuestos de pirrolidina 3,3-di-sustituidos que tienen dos sustituyentes en la posición 3 de la pirrolidina, como los compuestos de la presente invención.
Lista de citas de referencia
Literatura de patentes
Literatura de patentes 1: Documento WO2010/028862
Literatura de patente 2: Documento WO2008/007930
Los documentos WO 2005/077935, WO 2004/078717, WO 02/068388 y WO 2005/040109 divulgan derivados de pirrolidina sustituidos en la posición 3 con un grupo heterocíclico CO-N como moduladores del receptor muscarínico 4.
Literatura no de patentes
Literatura no de patentes 1: Annals ofthe New York Academy of Science, 1999; 885: p. 1-21
Literatura no de patentes 2: Pharmacological Review, 2004; 56: p. 1-29
Literatura no de patentes 3: Endocrinology, 1996; 137: p. 1627-1633
Literatura no de patentes 4: Physiological Reviews, 2000; 80: p. 979-1020
Literatura no de patentes 5: Neuroimmunomodulation, 1994; 1: p. 28-32.
Herpin, T.F. et al.: Discovery of tyrosine- based potent and selective melanocortin-1 receptor small-molecule agonists with anti-inflammatory properties, J of Medicinal Chemistry, vol. 46, n.° 7, 2003, págs. 1123-1126 y Tian, X. et al.: Design, synthesis, and evaluation of proline and pyrrolidine based melanocortin receptor agonists. A conformationally restricted dipeptide mimic approach, Journal of Medicinal chemistry, vol. 49, n.° 15, 2006, págs. 4745-4761 divulgan agonistas de MC1R y/o moduladores de MCR estructuralmente diferentes.
Breve descripción de la invención
(Problemas que se resolverán por la invención)
La presente invención se refiere a un compuesto de pirrolidina novedoso que tiene actividad agonística (actividad agonista) del receptor de melanocortina (MCR), en particular, actividad agonística (actividad agonista) del receptor de melanocortina 1 (MC1R), o una sal farmacéuticamente aceptable del mismo. El compuesto de la presente invención es por lo tanto útil para la prevención o el tratamiento de varias enfermedades o síntomas en los cuales la activación de los MCRs, en particular, el MC1R, está implicado.
(Medios para resolver los problemas)
La presente invención se refiere a un derivado de pirrolidina representado por la fórmula [I]:

donde el anillo A representa un grupo fenilo opcionalmente sustituido;
R1 representa un grupo alquilo opcionalmente sustituido, un grupo cicloalquilo opcionalmente sustituido, un grupo adamantilo opcionalmente sustituido, un grupo heterocíclico alifático opcionalmente sustituido, un grupo arilo opcionalmente sustituido que puede ser parcialmente hidrogenado, un grupo heteroarilo opcionalmente sustituido o un grupo carbamoílo opcionalmente sustituido;
R2 representa un átomo de halógeno, un grupo alquilo o un grupo alcoxi opcionalmente sustituido;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un anillo heterocíclico alifático que contenía nitrógeno opcionalmente sustituido que puede contener parcialmente un doble enlace;
o una sal farmacéuticamente sal aceptable del mismo.
La presente invención se refiere también a una composición farmacéutica de acuerdo con la modalidad anterior para su uso en un método para la prevención o terapia de varias enfermedades y/o síntomas en los cuales la activación de los MCR (en particular, MC1R) está implicada, que comprende administrar a un paciente una cantidad efectiva del compuesto representado por la fórmula general [I] anterior, o una sal farmacéuticamente aceptable del mismo.
(Efecto de invención)
El compuesto de la presente invención exhibe actividad agonística (actividad agonista) del receptor de melanocortina (MCR), en particular, actividad agonística (actividad agonista) del MC1R. El compuesto de la presente invención es por lo tanto útil en la prevención o el tratamiento de varias enfermedades o síntomas en los cuales la activación de los MCRs, en particular, el MC1R, está implicada.
Modo para llevar a cabo la invención
Las definiciones de los grupos como se usan en la presente pueden combinarse según se desee, a menos que se especifique de otra manera.
Como se usa en la presente, el término "alquilo" se refiere a un grupo de cadena de hidrocarburo saturado recta o ramificada que tiene uno a seis átomos de carbono (C-i-a). En particular, es preferible el grupo alquilo que tiene uno a cuatro átomos de carbono (C1-4). Ejemplos específicos incluyen metilo, etilo, n-propilo, i-propilo, n-butilo, t-butilo, 2-metil-n-butilo, i-amilo (3- metil-n-butilo) y 2-metil-n-pentilo. En particular, es preferible metilo, etilo, i-propilo o t-butilo. El término "alquenilo" se refiere a un grupo de cadena de hidrocarburo recta o ramificada que tiene dos a seis átomos de carbono (C2-6), que tiene por lo menos un doble enlace. En particular, el "alquenilo" puede ser grupos alquenilo que tienen dos a cuatro átomos de carbono (C2-4). Ejemplos específicos incluyen vinilo, propenilo y butenilo.
El término "cicloalquilo" se refiere a un grupo hidrocarburo monocíclico saturado que tiene tres a siete átomos de carbono (C3-7) y adamantilo y ejemplos específicos incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo y adamantilo.
El término "cicloalquenilo" se refiere a un grupo cíclico que tiene tres a siete átomos de carbono (C3-7) que tiene por lo menos un doble enlace. Ejemplos específicos incluyen ciclopropenilo, ciclobutenilo, ciclopentenilo y ciclohexenilo. El término "alcoxi" se refiere a un grupo monovalente en el cual el alquilo descrito anteriormente está unido a un átomo de oxígeno, por ejemplo, un alquil-O- que tiene uno a seis átomos de carbono (C1-6). Es preferible el alquil O- que tiene uno a cuatro átomos de carbono (C1-4). Ejemplos específicos incluyen metoxi, etoxi, n-propoxi, ipropoxi, n-butoxi, t-butoxi, 2-metil-n-propoxi y 3-metil-n-butoxi.
El término "alcanoílo" se refiere a un grupo en el cual el carbonilo (C = O) está unido al alquilo descrito anteriormente, por ejemplo, un alquil-C(=O)- recto o ramificado que tiene uno a seis átomos de carbono (Ci-a). Es preferible el alquil-C(=O)- que tiene uno a cuatro átomos de carbono (C1-4). Ejemplos específicos incluyen acetilo, propionilo y butirilo.
El término "alquileno" se refiere a un grupo hidrocarburo saturado divalente recto o ramificado que tiene uno a seis átomos de carbono (Ci-a), siendo preferible el grupo alquileno que tiene uno a cuatro átomos de carbono (C1-4). Ejemplos específicos incluyen metileno, etileno, trimetileno (propileno) y tetrametileno (n-butileno).
El término "alquilenoxi" se refiere a un grupo divalente en el cual un átomo de oxígeno está unido al alquileno descrito anteriormente. El "alquilenoxi" puede ser específicamente alquilen-O- que tiene uno a seis átomos de carbono (Ci-a) y es de preferencia alquilen-O- que tiene uno a cuatro átomos de carbono (C1-4). Un grupo alquilenoxi puede estar unido como un sustituyente a dos átomos diferentes (por ejemplo, átomos de carbono), o puede estar unido como un sustituyente al mismo átomo (por ejemplo, átomo de carbono) para formar un anillo de espiro.
Ejemplos de halógeno o halo incluyen átomos de flúor, cloro, bromo y yodo. En particular, el halógeno o halo puede ser un átomo de flúor o cloro.
El término "haloalquilo" se refiere a un alquilo C1-C6 sustituido con uno a tres átomos de halógeno y ejemplos específicos incluyen difluorometilo, trifluorometilo, 1-fluorometilo y 2- fluoroetilo.
El término "haloalcoxi" se refiere a un alquil C1-C6-O- sustituido con uno a tres átomos de halógeno y un ejemplo específico puede ser trifluorometoxi.
El término "hidroxialquilo" se refiere a un alquilo C1-C6 sustituido con un grupo hidroxi y ejemplos específicos incluyen hidroximetilo, hidroxietilo, 2-hidroxi-1,1-dimetiletilo y 4-hidroxi-4-metil- n-pentilo.
El término "cianoalquilo" se refiere a un alquilo C1-C6 sustituido con un grupo ciano y un ejemplo específico puede ser cianometilo.
El término "alcoxialquilo" se refiere a un alquilo C1-C6 sustituido con un grupo alcoxi C1-C6 y ejemplos específicos incluyen metoximetilo, metoxietilo, 2-metoxi-1,1-dimetiletilo y 4-metoxi-4-metil-n-pentilo.
El término "arilo" puede ser un grupo cíclico de hidrocarburo aromático de 6 a 10 miembros. Es preferible el arilo monocíclico o bicíclico y ejemplos específicos incluyen fenilo y naftilo, siendo preferible, en particular, fenilo.
El "arilo que puede ser parcialmente hidrogenado" incluye el arilo descrito anteriormente y el arilo descrito anteriormente que está parcialmente hidrogenado e incluye, por ejemplo, un grupo cíclico formado por la condensación de un grupo fenilo y un grupo cicloalquilo C3-C7 y un grupo cíclico formado por la condensación de un grupo fenilo y un grupo cicloalquenilo C3-C7. Ejemplos específicos incluyen fenilo, naftilo, dihidrofenilo, indanilo, dihidronaftilo y tetrahidronaftilo.
El término "heteroanlo" se refiere a un grupo monocíclico o bicíclico de 5 a 10 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno. Un ejemplo preferible puede ser heteroarilo monocíclico de 5 o 6 miembros que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno. Otro ejemplo preferible es un grupo heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de nitrógeno, oxígeno y azufre. Ejemplos específicos incluyen pirrolilo, furanilo, tienilo, imidazolilo, pirazolilo, oxazolilo, isoxazolilo, tiazolilo, tetrazolilo, oxadiazolilo, piridinilo, pirazinilo, pirimidinilo, piridazinilo, tiazinilo, triazinilo, indolilo, isoindolilo y benzoimidazolilo.
El término "anillo heterocíclico alifático" se refiere a un grupo cíclico saturado de 4 a 8 miembros que contenía uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de nitrógeno, oxígeno y azufre. El anillo heterocíclico alifático también puede ser un grupo en el cual dos átomos de carbono que forman el anillo están puenteados por un grupo alquileno para formar un grupo bicíclico o tricíclico y puede contener un doble enlace en el anillo. Preferible es un anillo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado del grupo que consiste en átomos de oxígeno, azufre y nitrógeno. Otro ejemplo preferible es un anillo heterocíclico alifático monocíclico de 5 o 6 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en átomos de nitrógeno, oxígeno y azufre. Ejemplos específicos incluyen azetidinilo, pirrolidinilo, tetrahidrofuranilo, imidazolinilo, tiazolidinilo, isotiazolidinilo, piperidinilo, piperazinilo, morfolinilo, tiomorfolinilo, tetrahidropiranilo, dihidropiridinilo, homopiperazinilo, homomorfolinilo, 3-azabiciclo[3.1.0]hexilo y octahidropirrolo[3,4-c]pirrolilo. Son preferibles azetidinilo pirrolidinilo, piperidinilo, morfolinilo, tiomorfolinilo, piperazinilo, 3-azabiciclo[3.1.0]hexilo. Además, son preferibles pirrolidinilo, piperidinilo, piperazinilo, morfolinilo y son particularmente preferibles pirrolidinilo, piperidinilo y morfolinilo. Son también preferibles tetrahidrofuranilo, tetrahidropiranilo, piperidinilo.
El término "carbonilo heterocíclico alifático " se refiere a un grupo en el cual el grupo carbonilo está unido al anillo heterocíclico alifático descrito anteriormente. Preferible es un anillo heterocíclico alifático monocíclico --C(=O)- de 4 a 7 miembros que contenía uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno. Más preferible es un carbonilo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado del grupo que consiste en átomos de azufre, oxígeno y nitrógeno. Particularmente preferible es un carbonilo heterocíclico alifático monocíclico de 5 o 6 miembros que contenía por lo menos un átomo de nitrógeno, en el cual un grupo carbonilo está unido al átomo de nitrógeno en el anillo.
El término "sulfonilo heterocíclico alifático" se refiere a un grupo en el cual el grupo sulfonilo está unido a los anillos heterocíclicos alifáticos descritos anteriormente y puede ser, por ejemplo, un anillo heterocíclico alifático monocíclico -(SO2)- de 4 a 7 miembros que contenía uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno. En particular, es preferible un anillo heterocíclico alifático monocíclico -(SO2)- de 4 a 7 miembros que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado del grupo que consiste en átomos de azufre, oxígeno y nitrógeno. En particular, es preferible un anillo heterocíclico alifático monocíclico -(SO2)- de 5 o 6 miembros que contenía por lo menos un átomo de nitrógeno, en el cual un grupo sulfonilo está unido al átomo de nitrógeno.
Cada uno de los símbolos usados en el compuesto [I] de la presente invención se describe en lo sucesivo.
El anillo A representa un grupo fenilo opcionalmente sustituido.
Los sustituyentes en el "grupo fenilo opcionalmente sustituido" representados por el anillo A son de uno a tres grupos que se seleccionan cada uno independientemente y ejemplos incluyen un átomo de halógeno, grupo alquilo C1-C6, grupo haloalquilo C1-C6, grupo cicloalquilo C3-C7, grupo alcoxi C1-C6, grupo haloalcoxi C1-C6 y grupo alquilenoxi C1-C6. Ejemplos específicos incluyen un átomo de flúor, átomo de cloro, grupo metilo, grupo etilo, grupo ipropilo, grupo trifluorometilo, grupo ciclopropilo, grupo metoxi, grupo etoxi, grupo trifluorometoxi y grupo etilenoxi. De manera preferible es un grupo fenilo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, grupo alquilo C1-C6, grupo haloalquilo C1-C6, grupo cicloalquilo C3-C7 , grupo alcoxi C1-C6, grupo haloalcoxi C1-C6 y grupo alquilenoxi C1-C6. Más preferible es un grupo fenilo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, grupo alquilo C1-C4, grupo haloalquilo C1-C4, grupo cicloalquilo C3-C7, grupo alcoxi C1-C4, grupo haloalcoxi C1-C4 y grupo alquilenoxi C1-C4 y particularmente preferible es un grupo fenilo opcionalmente sustituido con un grupo alcoxi C1-C4.
Es más preferible es un grupo fenilo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de flúor, átomo de cloro, grupo metilo, grupo etilo, grupo ipropilo, grupo trifluorometilo, grupo ciclopropilo, grupo metoxi, grupo etoxi y grupo trifluorometoxi y lo más preferible
es un grupo fenilo opcionalmente sustituido con un grupo metoxi.
El alquilo C1-C6 del "grupo alquilo opcionalmente sustituido" representado por R1 es de preferencia t-butilo, en particular.
El cicloalquilo C3-C7 del "grupo cicloalquilo opcionalmente sustituido" representado por R1 es de preferencia ciclopentilo o ciclohexilo, en particular.
El anillo heterocíclico alifático de 4-8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno del "grupo heterocíclico alifático opcionalmente sustituido" representado por R1 es de preferencia un grupo heterocíclico alifático monocíclico de 5 o 6 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno. Específicamente, es preferible tetrahidrofuranilo, tetrahidropiranilo o piperidinilo.
El arilo de 6 a 10 miembros que puede ser parcialmente hidrogenado del "grupo arilo opcionalmente sustituido que puede ser parcialmente hidrogenado" representado por R1 puede ser fenilo, naftilo o indanilo. En particular, es preferible el indanilo.
El heteroarilo de 5 a 10 miembros del "grupo heteroarilo opcionalmente sustituido" representado por R1 es de preferencia un grupo heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno. En particular, es preferible un heteroarilo monocíclico que contenía nitrógeno de 5 o 6 miembros que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado del grupo que consiste en átomos de oxígeno, azufre y nitrógeno. Ejemplos específicos incluyen pirrolilo, imidazolilo, pirazolilo, oxazolilo, isoxazolilo, tiazolilo, oxadiazolilo, piridinilo, pirazinilo, pirimidinilo, piridazinilo, tiazinilo y triazinilo. Particularmente preferibles son piridinilo, piridazinilo, pirimidinilo.
Los sustituyentes en cada uno del "grupo alquilo opcionalmente sustituido", "grupo cicloalquilo opcionalmente sustituido", "grupo heterocíclico alifático opcionalmente sustituido", "grupo arilo opcionalmente sustituido que puede ser parcialmente hidrogenado", "grupo heteroarilo opcionalmente sustituido" y "grupo carbamoílo opcionalmente sustituido" tal como se han definido anteriormente, representados por R1 pueden ser uno a tres grupos y de preferencia uno o dos grupos, que son los mismos o diferentes. Ejemplos incluyen un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6; un grupo hidroxialquilo C1-C6; un grupo alcoxialquilo C1-C6; un grupo haloalquilo C1-C6; un grupo cicloalquilo C3-C7 ; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo heterocíclico alifático de 4 a 8 miembros tal como se ha definido anteriormente; un grupo carbonilo heterocíclico alifático tal como se ha definido anteriormente opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, grupo alquilo C1-Ca, grupo haloalquilo C1-C6 y grupo alcoxialquilo C1-C6; un grupo sulfonilo heterocíclico alifático tal como se ha definido anteriormente; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-Ca y un grupo alquilenoxi CrCa.
Más particularmente, ejemplos de los sustituyentes incluyen un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-Ca; un grupo hidroxialquilo C1-Ca; un grupo alcoxialquilo C1-Ca; un grupo haloalquilo C1-Ca; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-Ca; un grupo alcanoílo C1-Ca; un grupo alquilsulfonilo C1-Ca; un grupo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, grupo alquilo C1-Ca, grupo haloalquilo C1-Ca y grupo alcoxialquilo C1-Ca (donde el anillo heterocíclico alifático es un anillo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado del grupo que consiste en átomos de oxígeno, azufre y nitrógeno); un grupo sulfonilo heterocíclico alifático (donde el anillo heterocíclico alifático es un anillo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado del grupo que consiste en átomos de oxígeno, azufre y nitrógeno); un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-Ca y un grupo alquilenoxi C1-Ca.
En la presente, el "grupo cicloalquilo sustituido con un grupo alquilenoxi" tal como se ha definido anteriormente incluye un grupo en el cual un grupo alquilenoxi C1-Ca está unido a cualquier átomo de carbono común en el grupo cicloalquilo C3-C7 (a saber, un anillo de espiro). Por ejemplo, un grupo ciclohexilo sustituido con un grupo trimetilenoxi (propilenoxi) incluye un grupo de la fórmula siguiente:
a

Los sustituyentes en el "grupo alquilo C1-C6 opcionalmente sustituido" representado por R1 pueden ser uno a tres grupos y de preferencia uno o dos grupos, que son los mismos o diferentes. Ejemplos de los sustituyentes incluyen un átomo de halógeno; un grupo hidroxi; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo heterocíclico alifático tal como se ha definido anteriormente; un grupo carbonilo heterocíclico alifático tal como se ha definido anteriormente opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, grupo alquilo C1-C6, grupo haloalquilo C1-C6 y grupo alcoxialquilo C1-C6; un grupo sulfonilo heterocíclico alifático tal como se ha definido anteriormente y un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6.
Más específicamente, ejemplos de los sustituyentes incluyen un átomo de flúor; un grupo hidroxi; un grupo ciclopropilo; un grupo ciclobutilo; un grupo metoxi; un grupo tetrahidropiranilo; un grupo pirrolidinilcarbonilo opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un átomo de flúor, grupo metilo, grupo trifluorometilo y grupo metoximetilo; un grupo piperidinilcarbonilo opcionalmente sustituido con uno o dos átomos de flúor; un grupo morfolinilcarbonilo; un grupo pirrolidinilsulfonilo y un grupo dimetilcarbamoílo.
Ejemplos del "grupo alquilo C1-C6 opcionalmente sustituido" representado por R1 incluyen los grupos metilo, etilo, ipropilo, i-amilo (3-metil-n-butilo), 3-metil-n-butilo, 1-fluorometil-2-fluoroetilo, t-butilo, ciclopropilmetilo, ciclobutilmetilo, 2-hidroxietilo, 1,1-dimetil-2-N,N-dimetilcarbamoil-etilo, 4-hidroxi-4-metil-n-pentilo, 1,1-dimetil-2-metoxietilo, 4-metoxi-4-metil-n-pentilo, N,N-dimetilaminocarbonilmetilo, 2-N,N-dimetilaminocarboniletilo, 1,1-dimetil-2-N,N-dimetilaminocarbonil-etilo, 3-N,N-dimetilaminocarbonil-n-propilo, tetrahidropiranilmetilo, piperazinilcarbonilmetilo, pirrolidinilcarbonilo, 2-metil-pirrolidinilcarbonilmetilo, 2-trifluorometil-pirrolidinilcarbonilmetilo, 2-metoximetilpirrolidinilcarbonilo, 3-fluoro-pirrolidinilcarbonilmetilo, morfolinilcarbonilmetilo, 4,4-difluoro-piperidinilcarbonilmetilo y 3-pirrolidinilsulfonilpropilo. Un grupo t-butilo, en particular, es preferible.
Los sustituyentes en el "grupo cicloalquilo C3-C7 opcionalmente sustituido" representado por R1 pueden ser uno a tres grupos, que son los mismos o diferentes y ejemplos incluyen un átomo de halógeno, grupo hidroxi, grupo oxo, grupo ciano, grupo alquilo C1-C6, grupo alcoxi C1-C6, grupo alcoxialquilo C1-C6 y grupo alquilenoxi C1-C6. En la presente, el grupo alquilenoxi puede ser un sustituyente en el carbono común en el cicloalquilo.
Más particularmente, los sustituyentes pueden ser uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, grupo hidroxi, grupo oxo, grupo ciano, grupo alquilo C1-C6, grupo alcoxi C1-Ca, grupo alcoxialquilo C1-C6 y grupo alquilenoxi C1-C6, por ejemplo. Más específicamente, los sustituyentes son uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de flúor, grupo hidroxi, grupo oxo, grupo ciano, grupo metilo, grupo metoxi, grupo etoxi, grupo isopropoxi, grupo metoximetilo y grupo trimetilenoxi (grupo propilenoxi). En particular, son preferibles los grupos metoxi, etoxi y ciano.
El "grupo cicloalquilo C3-C7 opcionalmente sustituido" representado por R1 puede ser un grupo cicloalquilo C3-C7 opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, grupo hidroxi, grupo oxo, grupo ciano, grupo alquilo C1-Ca, grupo alcoxi C1-Ca, grupo alcoxialquilo C1-Ca y grupo alquilenoxi C1-Ca.
Más particularmente, ejemplos incluyen un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, grupo hidroxi, grupo oxo, grupo ciano, grupo alquilo C1-Ca, grupo alcoxi C1-Ca, grupo alcoxialquilo C1-Ca y grupo alquilenoxi C1-Ca y un grupo adamantilo opcionalmente sustituido con un grupo hidroxi.
Más específicamente, ejemplos incluyen un grupo ciclopropilo; un grupo ciclobutilo; un grupo ciclopentilo opcionalmente sustituido con uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de flúor, grupo hidroxi, grupo oxo, grupo ciano, grupo metilo, grupo metoxi, grupo etoxi, grupo i-propoxi y grupo metoximetilo; un grupo ciclohexilo opcionalmente sustituido con uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de flúor, grupo hidroxi, grupo oxo, grupo ciano, grupo metilo, grupo metoxi, grupo etoxi, grupo i-propoxi y grupo trimetilenoxi (grupo propilenoxi) y un grupo cicloheptilo. En particular, son preferibles un grupo ciclopentilo opcionalmente sustituido con un grupo seleccionado del grupo que consiste en grupos metoxi, etoxi y ciano y un grupo ciclohexilo opcionalmente sustituido con un grupo seleccionado del grupo que consiste en grupos metoxi, etoxi y ciano.
Ejemplos de los sustituyentes en el "grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido" tal como se ha definido anteriormente representado por R1 incluyen grupos alquilo C1-Ca, hidroxialquilo C1-Ca,
haloalquilo C1-C6, alcanoílo C1-C6 y alquilsulfonilo C1-C6. Más específicamente, ejemplos incluyen grupos metilo, hidroximetilo, 2-fluoro-1-fluorometil-etilo, acetilo, etilcarbonilo y etilsulfonilo.
Ejemplos del "grupo heterocíclico alifático opcionalmente sustituido" tal como se ha definido anteriormente representado por R1 incluyen un grupo tetrahidrofuranilo opcionalmente sustituido con un grupo alquilo C1-C6, hidroxi o hidroxialquilo C1-C6; un grupo tetrahidropiranilo opcionalmente sustituido con un grupo hidroxi, alquilo C1-C6 o hidroxialquilo C1-C6 y un grupo piperidinilo opcionalmente sustituido con un grupo seleccionado del grupo que consiste en grupos haloalquilo C1-C6, alcanoílo C1-C6 y alquilsulfonilo C1-C6.
Más específicamente, ejemplos incluyen un grupo tetrahidrofuranilo; un grupo tetrahidropiranilo opcionalmente sustituido con un grupo metilo o hidroximetilo y un grupo piperidinilo opcionalmente sustituido con un grupo seleccionado del grupo que consiste en grupos 1-fluorometil-2-fluoroetilo, acetilo, etilcarbonilo y etilsulfonilo; siendo preferible un grupo tetrahidropiranilo.
El "grupo arilo de 6 a 10 miembros opcionalmente sustituido que puede ser parcialmente hidrogenado" tal como se ha definido anteriormente representado por R1 es de preferencia un grupo indanilo (en particular, un grupo 1-indanilo, un grupo 2-indanilo).
Los sustituyentes en el "grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido" tal como se ha definido anteriormente representado por R1 pueden ser uno a tres grupos, que son los mismos o diferentes y ejemplos incluyen grupos ciano, alquilo C1-C6, alcoxi C1-C6 y carbamoílo. Más específicamente, ejemplos incluyen grupos ciano, metilo, metoxi y carbamoílo. Un grupo metilo, en particular, es preferible.
Ejemplos del "grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido" tal como se ha definido anteriormente representado por R1 incluyen un grupo piridazinilo opcionalmente sustituido con un grupo seleccionado del grupo que consiste en grupos ciano, alquilo C1-C6, alcoxi C1-C6 y carbamoílo; un grupo piridinilo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en grupos ciano, alquilo C1-C6 y alcoxi C1-C6 y un grupo pirimidinilo opcionalmente sustituido con un grupo alquilo C1-C6.
Más específicamente, ejemplos incluyen un grupo piridazinilo opcionalmente sustituido con un grupo seleccionado del grupo que consiste en grupos ciano, metilo, metoxi y carbamoílo; un grupo piridinilo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en grupos ciano, metilo y metoxi y un grupo pirimidinilo opcionalmente sustituido con un grupo metilo. En particular, son preferibles un grupo pirimidinilo opcionalmente sustituido con un grupo metilo y un grupo piridinilo sustituido con un grupo metilo.
Un sustituyente en el "grupo carbamoílo opcionalmente sustituido" representado por R1 puede ser un grupo alquilo C1-C6, por ejemplo. Más específicamente, ejemplos incluyen grupos metilo, etilo e i-propilo, siendo preferible, en particular, un grupo metilo.
Ejemplos del "grupo carbamoílo opcionalmente sustituido" representado por R1 incluyen grupos carbamoílo, monometilcarbamoílo y dimetilcarbamoílo, siendo preferibles los grupos carbamoílo y monometilcarbamoílo.
R1 es de preferencia un grupo alquilo C1-C6, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo heterocíclico alifático tal como se ha definido anteriormente o un grupo heteroarilo de 5-10 miembros opcionalmente sustituido tal como se ha definido anteriormente. Más particularmente, R1 es de preferencia un grupo alquilo C1-C6; un grupo cicloalquilo C3-C7 opcionalmente sustituido con un grupo ciano o alcoxi C1-C6; un grupo heterocíclico alifático monocíclico de 5 o 6 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno o un grupo heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno sustituido con un grupo alquilo C1-C6. Específicamente, R1 es de preferencia un grupo t-butilo; un grupo ciclopentilo; un grupo ciclohexilo opcionalmente sustituido con un grupo metoxi, etoxi o ciano; un grupo tetrahidropiranilo; o un grupo piridinilo opcionalmente sustituido con un grupo metilo.
R1 es de preferencia un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo ciano o alcoxi C1-C6 o un grupo heterocíclico alifático de 4 a 8 miembros tal como se ha definido anteriormente opcionalmente sustituido con un grupo alquilo C1-C6, haloalquilo C1-C6 o hidroxialquilo C1-C6 (donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en grupos tetrahidrofuranilo, tetrahidropiranilo, pirrolidinilo y piperidinilo). Específicamente, R1 es de preferencia un grupo ciclopentilo; un grupo ciclohexilo opcionalmente sustituido con un grupo metoxi, etoxi o ciano; un grupo piperidinilo opcionalmente sustituido con un grupo haloalquilo; o un grupo tetrahidropiranilo opcionalmente sustituido con un grupo alquilo o hidroxialquilo.
Más particularmente, R1 es de preferencia un grupo cicloalquilo opcionalmente sustituido con un grupo ciano o alcoxi C1-C6. Específicamente, R1 es de preferencia un grupo ciclopentilo; o un grupo ciclohexilo opcionalmente sustituido con un grupo seleccionado del grupo que consiste en grupos metoxi, etoxi y ciano.
El alquilo del "grupo alquilo C1-C6" representado por R2 es de preferencia alquilo de C1-3 y específicamente, el alquilo C1-C6 es de preferencia metilo, etilo, i-propilo.
El alcoxi del "grupo alcoxi C1-C6 opcionalmente sustituido" representado por R2 es de preferencia alcoxi de C1-4 y es específicamente metoxi, etoxi, i-propoxi, n-butoxi.
Ejemplos de sustituyentes en el "grupo alcoxi C1-C6 opcionalmente sustituido" representado por R2 incluyen un átomo de halógeno, grupo hidroxi y grupo alcoxi C1-C6. Más específicamente, ejemplos incluyen un átomo de flúor, átomo de cloro, grupo hidroxi y grupo metoxi.
Ejemplos del "grupo alcoxi C1-C6 opcionalmente sustituido" representado por R2 incluyen grupos metoxi, etoxi, trifluorometoxi y difluorometoxi, siendo preferible un grupo metoxi.
Ejemplos adecuados de R2 incluyen un átomo de halógeno, un grupo alquilo C1-C6 y un grupo alcoxi C1-C6. En particular, es preferible un átomo de flúor o grupo metoxi.
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II]:

donde el anillo B representa un grupo heterocíclico alifático de 4-8 miembros que contenía nitrógeno que puede contener parcialmente un doble enlace,
el anillo C representa un grupo arilo de 6 a 10 miembros o heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno,
R5 y R6 representan cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, átomo de halógeno, grupo ciano, grupo alquilo C1-C6, grupo haloalquilo C1-C6, grupo cianoalquilo C1-C6, grupo hidroxialquilo C1-C6, grupo alcoxialquilo C1-C6, grupo carboxilo C1-C6, grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
R7 representa un grupo alquilo C1-C6 opcionalmente sustituido, un grupo alquenilo C2-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo cicloalquenilo C3-C7 opcionalmente sustituido, un grupo arilo de 6 a 10 miembros opcionalmente sustituido tal como se ha definido anteriormente, un grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido tal como se ha definido anteriormente, un grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido tal como se ha definido anteriormente, un grupo alcoxi C1-C6 opcionalmente sustituido, un grupo amino opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo, o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo y
R8 y R9 representan cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, átomo de halógeno, grupo ciano, grupo alquilo C1-C6, grupo haloalquilo C1-C6 y grupo haloalcoxi C1-C6.
Más preferiblemente, el grupo representado por la fórmula general [II] anterior puede ser un grupo donde R7 representa un grupo alquilo C1-C6 opcionalmente sustituido, un grupo alquenilo C2-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo cicloalquenilo C3-C7 opcionalmente sustituido, un grupo arilo de 6 a 10 miembros opcionalmente sustituido tal como se ha definido anteriormente, un grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido tal como se ha definido anteriormente, un grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido tal como se ha definido anteriormente, un grupo alcoxi C1-C6 opcionalmente sustituido, un grupo amino opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo, o un grupo carbamoílo el cual es opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo,
donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido son uno o dos grupos seleccionados
independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alcoxi Ci-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros tal como se ha definido anteriormente opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido tal como se ha definido anteriormente con un grupo hidroxi u oxo; un grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido con uno o dos grupos oxo; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (la porción alquilo es opcionalmente sustituida con un grupo hidroxi, alcoxi C1-C6 o carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en grupos alquilo C1-C6, alcanoílo C1-C6 y alquilsulfonilo C1-C6 y
los sustituyentes en cada uno del grupo alquenilo C2-C6 opcionalmente sustituido, el grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo cicloalquenilo C3-C7 opcionalmente sustituido, el grupo arilo de 6 a 10 miembros opcionalmente sustituido tal como se ha definido anteriormente, el grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido tal como se ha definido anteriormente, el grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido tal como se ha definido anteriormente y el grupo alcoxi C1-C6 opcionalmente sustituido, son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4-8 miembros tal como se ha definido anteriormente opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros tal como se ha definido anteriormente opcionalmente sustituido con un grupo hidroxi u oxo; un grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido con uno o dos grupos oxo; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (la porción alquilo es opcionalmente sustituida con un grupo hidroxi, alcoxi C1-C6 o carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en grupos alquilo C1-C6, alcanoílo C1-C6 y alquilsulfonilo C1-C6.
El "grupo heterocíclico alifático que contenía nitrógeno que puede contener parcialmente un doble enlace" representado por el anillo B, es de preferencia un anillo heterocíclico alifático monocíclico o bicíclico de 4 a 8 miembros que puede contener adicionalmente, además del átomo de nitrógeno mostrado en la fórmula [II], un heteroátomo seleccionado del grupo que consiste en átomos de oxígeno, azufre y nitrógeno. Específicamente, ejemplos incluyen grupos azetidinilo, pirrolidinilo, imidazolinilo, tiazolidinilo, isotiazolidinilo, piperidinilo, piperazinilo, morfolinilo, tiomorfolinilo, tetrahidropiridinilo, homopiperazinilo, homomorfolinilo, 3-azabiciclo[3.1.0]hexilo y octahidropirrolo[3,4-c]pirrolilo. Ejemplos preferibles incluyen grupos azetidinilo, pirrolidinilo, piperidinilo, tetrahidropiridinilo, piperazinilo, homopiperazinilo y octahidropirrolo[3,4-c]pirrolilo. Ejemplos más adecuados incluyen grupos pirrolidinilo y piperidinilo y en particular, es preferible un grupo pirrolidinilo.
El "grupo arilo de 6 a 10 miembros" tal como se ha definido anteriormente representado por el anillo C puede ser un así definido arilo monocíclico o bicíclico y puede ser específicamente fenilo o naftilo y es de preferencia fenilo.
El "grupo heteroarilo de 5 a 10 miembros" tal como se ha definido anteriormente representado por el anillo C puede ser, por ejemplo, un heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de nitrógeno, oxígeno y azufre. Más específicamente, es preferible un grupo piridinilo.
R5 y R6 pueden ser cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, átomo de halógeno, grupo ciano, grupo alquilo C1-C6, grupo haloalquilo C1-C6, grupo cianoalquilo C1-C6, grupo hidroxialquilo C1-C6, grupo alcoxialquilo C1-C6, grupo carboxilo, grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y grupo alcoxi C1-C6, por ejemplo. Más específicamente, R5 y R6 pueden ser cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, átomo de flúor, grupo ciano, grupo metilo, grupo fluorometilo, grupo cianometilo, grupo hidroximetilo, grupo 1 -hidroxi-1 -metiletilo, grupo metoximetilo, grupo etoximetilo, grupo carboxilo, grupo dimetilcarbamoílo y grupo metoxi, por ejemplo.
Ejemplos adecuados incluyen aquellos en los cuales R5 es un grupo seleccionado del grupo que consiste en un átomo de halógeno, grupo ciano, grupo alquilo C1-C6, grupo haloalquilo C1-C6, grupo cianoalquilo C1-C6, grupo hidroxialquilo C1-C6, grupo alcoxialquilo C1-C6, grupo carboxilo, grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y grupo alcoxi C1-C6 y R6 es un átomo de hidrógeno; R5 es un grupo alcoxialquilo C1-C6 y R6 es un átomo de halógeno, grupo alquilo C1-C6 o grupo alcoxi C1-C6; R5 y R6 son átomos de halógeno; R5 y R6 son grupos alquilo C1-C6 y R5 y R° son átomos de hidrógeno. Más específicamente, ejemplos incluyen aquellos en los cuales R5 es un grupo seleccionado del grupo que consiste en un átomo de flúor, grupo ciano, grupo metilo, grupo fluorometilo, grupo cianometilo, grupo hidroximetilo, grupo 1 -hidroxi-1 -metiletilo, grupo metoximetilo, grupo
etoximetilo, grupo carboxilo, grupo dimetilcarbamoílo y grupo metoxi y R6 es un átomo de hidrógeno; R5 es un grupo metoximetilo y R6 es un átomo de flúor, grupo metilo o grupo metoxi; R5 y R6 son átomos del flúor; R5 y R° son grupos metilo y R5 y R6 son átomos de hidrógeno.
El alquilo del "grupo alquilo C1-C6 opcionalmente sustituido" representado por R7 es de preferencia etilo, n-butilo. El alquenilo del "grupo alquenilo C1-C6 opcionalmente sustituido" representado por R7 es de preferencia n-butenilo. El cicloalquilo del "grupo cicloalquilo C3-C7 opcionalmente sustituido" representado por R7 es de preferencia ciclohexilo.
El cicloalquenilo del "grupo cicloalquenilo C3-C7 opcionalmente sustituido" representado por R7 es de preferencia ciclohexenilo.
La porción arilo del "grupo arilo de 6 a 10 miembros opcionalmente sustituido" tal como se ha definido anteriormente representado por R7 puede ser fenilo, naftilo y es de preferencia fenilo.
La porción heteroarilo del "grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido" tal como se ha definido anteriormente representado por R7 es de preferencia un heteroarilo monocíclico de 5 o 6 miembros que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado de átomos de oxígeno, azufre y nitrógeno. Específicamente, son preferibles pirrolilo, imidazolilo, pirazolilo, oxazolilo, isoxazolilo, tiazolilo, tetrazolilo, oxadiazolilo, piridinilo, pirazinilo, pirimidinilo, piridazinilo, tiazinilo y triazinilo. En particular, son preferibles los grupos pirazolilo y oxazolilo.
El anillo heterocíclico alifático del "grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido" tal como se ha definido anteriormente representado por R7 es de preferencia un anillo heterocíclico alifático monocíclico o bicíclico de 4 a 8 miembros que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado del grupo que consiste en átomos de oxígeno, azufre y nitrógeno. Específicamente, son preferibles los grupos azetidinilo, pirrolidinilo, piperidinilo, piperazinilo, morfolinilo, tiomorfolinilo y grupo 3-azabiciclo[3.1.0]hexilo, por ejemplo. Un ejemplo particularmente adecuado es un grupo piperidinilo.
El alcoxi del "grupo alcoxi C1-C6 opcionalmente sustituido" representado por R7 es de preferencia metoxi, n-propoxi, 2-metil-n-propoxi, 3-metil-n-butoxi.
El "grupo amino opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo" representado por R7, puede ser específicamente un grupo N-metil-N-3-carboxil-n-propilamino. El "grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo" representado por R7, es de preferencia un grupo carbamoílo.
Los sustituyentes en cada uno del "grupo alquilo C1-C6 opcionalmente sustituido", el "grupo alquenilo C1-C6 opcionalmente sustituido", el "grupo cicloalquilo C3-C7 opcionalmente sustituido", el "grupo cicloalquenilo C3-C7 opcionalmente sustituido", el "grupo arilo de 6 a 10 miembros opcionalmente sustituido" tal como se ha definido anteriormente, el "grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido" tal como se ha definido anteriormente, el "grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido" tal como se ha definido anteriormente y el "grupo alcoxi C1-C6 opcionalmente sustituido" representado por R7 pueden ser uno o dos grupos, que son los mismos o diferentes y pueden ser uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros tal como se ha definido anteriormente opcionalmente sustituido con un grupo carboxilo (donde el anillo heterocíclico alifático es de preferencia un anillo heterocíclico alifático monocíclico de 4 a 7 miembros y más preferiblemente un anillo heterocíclico alifático monocíclico de 5 o 6 miembros, que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado del grupo que consiste en átomos de oxígeno, azufre y nitrógeno); un grupo heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno y opcionalmente sustituido con un grupo hidroxi u oxo, un grupo heterocíclico alifático monocíclico de 4 a 7 miembros opcionalmente sustituido con uno dos grupos oxo y más preferiblemente un grupo heterocíclico alifático monocíclico de 5 o 6 miembros, que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en grupos alquilo C1-C6 e hidroxi (donde el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, alcoxi C1-C6 o carboxilo); un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en grupos alquilo C1-C6, alcanoílo C1-C6 y alquilsulfonilo C1-C6.
Más específicamente, ejemplos de los sustituyentes incluyen grupos hidroxi, oxo, ciano, metilo, carboximetilo, metoxi, carboxilo, carbamoílo, N-metilcarbamoílo, N-etilcarbamoílo, N-1-carboxilmetilcarbamoílo, N-2-hidroxietilcarbamoílo, N-2-metoxietilcarbamoílo, N-hidroxicarbamoílo, N- metil-N-1-carboxilmetilcarbamoílo, N-metil-N-2-hidroxietilcarbamoílo, N-metil-N-2-metoxietilcarbamoílo, N,N-dimetilcarbamoílo, t-butoxicarbonilo, acetilo, N,N-dimetilaminosulfonilaminocarbonilo, metilsulfonilo, metilsulfonilaminocarbonilo, aminosulfonilo, N,N-dimetilaminosulfonilo, N,N-dimetilamino, N-metil-N-acetilamino, N-metil-N-metilsulfonilamino, tetrazolilo, 4H-[1,2,4]oxadiazol-5-ona-2-ilo, 5-hidroxi-isoxazolilo, pirrolidincarbonilo, 2-carboxilpirrolidincarbonilo, 2-oxopirrolidinilo y 1,1'-dioxoisotiazolidin-2-ilo. En particular, son preferibles los grupos carbamoílo y carboxilo y un grupo carboxilo es particularmente preferible.
R7 puede ser
(1) un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo,
(2) un grupo alquenilo C2-C6 opcionalmente sustituido con un grupo carboxilo,
(3) un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo, (4) un grupo cicloalquenilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo, (5) un grupo fenilo opcionalmente sustituido con un grupo carboxilo,
(6) un grupo heteroarilo de 5 a 10 miembros tal como se ha definido anteriormente el cual es opcionalmente sustituido con un grupo carboxilo o un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo, (7) un grupo heterocíclico alifático de 4 a 8 miembros tal como se ha definido anteriormente opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en grupos alquilo C1-C6 e hidroxi (donde el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, alcoxi o carboxilo); un grupo alquilsulfonilaminocarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4 a o miembros tal como se ha definido anteriormente opcionalmente sustituido con un grupo carboxilo; un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en grupos alquilo C1-C6, alcanoílo C1-C6 y alquilsulfonilo C1-C6; un grupo heterocíclico alifático de 4 a 8 miembros tal como se ha definido anteriormente opcionalmente sustituido con uno o dos grupos oxo; un grupo alquilsulfonilo C1-C6; un grupo tetrazolilo y un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6,
(8) un grupo alcoxi C1-C6 opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo ciano; un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros tal como se ha definido anteriormente opcionalmente sustituido con un grupo hidroxi u oxo; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alquilsulfonilaminocarbonilo C1-C6,
(9) un grupo amino el cual es opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo o
(10) un grupo carbamoílo.
Específicamente, el grupo heteroarilo del "(6) grupo heteroarilo opcionalmente sustituido con un grupo carboxilo o un grupo alquilo opcionalmente sustituido con un grupo carboxilo", es un grupo heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno. Este grupo heteroarilo es, por ejemplo, un grupo pirrolilo, imidazolilo, pirazolilo, oxazolilo, isoxazolilo, tiazolilo, tetrazolilo, oxadiazolilo, piridilo, pirazinilo, pirimidinilo, piridazinilo, tiazinilo y triazinilo; de preferencia, un grupo pirrolilo, pirazolilo, oxazolilo, piridilo o pirimidinilo y más preferiblemente un grupo oxazolilo o pirazolilo.
La porción heterocíclica alifática del "(7) grupo heterocíclico alifático de 4 a 8 miembros tal como se ha definido anteriormente opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 y un grupo hidroxi (donde el grupo alquilo es opcionalmente sustituido con un grupo hidroxi, alcoxi C1-C6 o carboxilo); un grupo alquilsulfonilaminocarbonilo C1-C6; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con un grupo carboxilo; un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en grupos alquilo C1-C6, alcanoílo C1-C6 y alquilsulfonilo C1-C6; un grupo heterocíclico alifático de 4 a 8 miembros tal como se ha definido anteriormente opcionalmente sustituido con uno o dos grupos oxo; un grupo alquilsulfonilo C1-C6 y un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 ", es de preferencia un grupo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno. Ejemplos específicos incluyen grupos azetidinilo, pirrolidinilo, tetrahidrofuranilo, imidazolinilo, tiazolidinilo, isotiazolidinilo, piperidinilo, piperazinilo, morfolinilo, tiomorfolinilo, tetrahidropiranilo, homopiperazinilo, homomorfolinilo, 3-azabiciclo[3.1.0]hexilo y octahidropirrolo[3,4-c]pirrolilo. Son más preferibles los grupos azetidinilo, pirrolidinilo, piperidinilo, piperazinilo, morfolinilo, tiomorfolinilo y grupo 3
azabiciclo [3.1.0]hexilo.
En la presente, el anillo heterocíclico alifático del "grupo carbonilo heterocíclico alifático de 4-8 miembros tal como se ha definido anteriormente opcionalmente sustituido con un grupo carboxilo", es de preferencia un anillo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado del grupo que consiste en átomos de oxígeno, azufre y nitrógeno. Ejemplos incluyen grupos azetidinilo, pirrolidinilo, imidazolinilo, tiazolidinilo, isotiazolidinilo, piperidinilo, piperazinilo, morfolinilo, tiomorfolinilo, homopiperazinilo y homomorfolinilo. Un grupo pirrolidinilo es particularmente preferible. El grupo heterocíclico alifático del "grupo heterocíclico alifático de 4-8 miembros tal como se ha definido anteriormente opcionalmente sustituido con uno o dos grupos oxo", es de preferencia un grupo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en átomos de nitrógeno, oxígeno y azufre y puede ser específicamente un grupo azetidinilo, pirrolidinilo, tetrahidrofuranilo, imidazolinilo, tiazolidinilo, isotiazolidinilo, piperidinilo, piperazinilo, morfolinilo, tiomorfolinilo, tetrahidropiranilo, homopiperazinilo u homomorfolinilo. Un grupo pirrolidinilo o isotiazolidinilo es particularmente preferible.
El grupo heteroarilo de 5 a 10 miembros del "(8) grupo alcoxi opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo ciano; un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido con un grupo hidroxi u oxo; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alquilsulfonilaminocarbonilo C1-C6", es de preferencia un grupo heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno. Ejemplos específicos incluyen grupos pirrolilo, imidazolilo, pirazolilo, oxazolilo, isoxazolilo, tiazolilo, tetrazolilo, oxadiazolilo, piridinilo, pirazinilo, pirimidinilo, piridazinilo, tiazinilo y triazinilo. Los grupos tetrazolilo, oxadiazolilo e isoxazolilo son particularmente preferibles.
Ejemplos particularmente adecuados de R7 incluyen un grupo alquilo C1-C6 sustituido con un grupo carboxilo, un grupo alquenilo C2-C6 sustituido con un grupo carboxilo, un grupo cicloalquilo C3-C7 sustituido con un grupo carboxilo, un grupo cicloalquenilo sustituido con un grupo carboxilo, un grupo arilo de 6 a 10 miembros tal como se ha definido anteriormente sustituido con un grupo carboxilo, un grupo heteroarilo de 5 a 10 miembros tal como se ha definido anteriormente sustituido con un grupo carboxilo, un grupo heterocíclico alifático de 4 a 8 miembros tal como se ha definido anteriormente sustituido con un grupo carboxilo, un grupo alcoxi C1-C6 sustituido con un grupo carboxilo, un grupo amino el cual es opcionalmente sustituido con uno o dos grupos alquilo C1-C6 sustituidos con un grupo carboxilo y un grupo carbamoílo. En particular, es preferible un grupo heterocíclico alifático de 4 a 8 miembros tal como se ha definido anteriormente sustituido con un grupo carboxilo y es especialmente preferible un grupo piperidinilo sustituido con un grupo carboxilo.
El átomo de halógeno del "átomo de hidrógeno, átomo de halógeno, grupo ciano, grupo alquilo C1-C6, grupo haloalquilo C1-C6 y grupo haloalcoxi C1-C6" representado por R8 o R9, es de preferencia un átomo de flúor o cloro. Ejemplos adecuados del "grupo alquilo C1-C6" incluyen grupos metilo, etilo e i-propilo. El "grupo haloalquilo C1-C6" es de preferencia un grupo trifluorometilo o difluorometilo, por ejemplo. El "grupo haloalcoxi C1-C6" es de preferencia un grupo trifluorometoxi, por ejemplo.
Ejemplos preferibles de R8 y R9 incluyen un átomo de hidrógeno, átomo de flúor, átomo de cloro, grupo metilo, grupo etilo, grupo i-propilo, grupo trifluorometilo, grupo difluorometilo y grupo trifluorometoxi.
Particularmente de preferencia, R8 es un átomo de flúor, átomo de cloro, grupo metilo, grupo i-propilo, grupo trifluorometilo, grupo difluorometilo o grupo trifluorometoxi y R9 es un átomo de hidrógeno. En particular de preferencia, R8 es un átomo de flúor átomo de cloro, grupo metilo o grupo trifluorometilo y R9 es un átomo de hidrógeno. Además, de preferencia, R8 y R9 son átomos del flúor.1
[1] En una primera modalidad, la presente invención se refiere a un compuesto representado por la fórmula general [I]

a sal farmacéuticamente aceptable del mismo, donde
el anillo A es un grupo fenilo opcionalmente sustituido, donde los sustituyentes en el grupo fenilo son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7, un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6;
R1 es un grupo alquilo C1-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo adamantilo opcionalmente sustituido, un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente entre el grupo que consiste en átomos de oxígeno, azufre y nitrógeno, un grupo arilo de 6 a 10 miembros opcionalmente sustituido que puede ser parcialmente hidrogenado, un grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido que contiene de uno a cuatro heteroátomos seleccionados independientemente entre el grupo que consiste en átomos de azufre, oxígeno y nitrógeno o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6,
donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, grupo alquilo C1-C6, grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno o a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno y un grupo alquilenoxi C1-C6 y
los sustituyentes en cada uno del grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido que contiene de uno a tres heteroátomos seleccionados independientemente entre el grupo que consiste en átomos de oxígeno, azufre y nitrógeno, el grupo arilo de 6 a 10 miembros opcionalmente sustituido que puede ser parcialmente hidrogenado y el grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido que contiene de uno a cuatro heteroátomos seleccionados independientemente entre el grupo que consiste en átomos de azufre, oxígeno y nitrógeno es/son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6; un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo hidroxialquilo C1-C6; un grupo alcoxialquilo C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente entre el grupo que consiste en átomos de oxígeno, azufre y nitrógeno; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contienen de uno a tres heteroátomos seleccionados independientemente entre el grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno y un grupo alquilenoxi C1-C6;
R2 es un átomo de halógeno, grupo alquilo C1-C6 o grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II]:

donde el anillo B representa un grupo heterocíclico alifático de 4 a 8 miembros que contenía nitrógeno que puede contener parcialmente un doble enlace;
el anillo C representa un grupo arilo de 6 a 10 miembros o un grupo heteroarilo de 5 a 10 miembros que contienen de uno a cuatro heteroátomos seleccionados independientemente entre el grupo que consiste en átomos de azufre, oxígeno y nitrógeno;
R5 y R6 representan cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
R7 representa un grupo alquilo C1-C6 opcionalmente sustituido, un grupo alquenilo C2-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo cicloalquenilo C3-C7 opcionalmente sustituido, un grupo arilo de 6 a 10 miembros opcionalmente sustituido, un grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido que contiene de uno a cuatro heteroátomos seleccionados del grupo que consiste en átomos de azufre, oxígeno y nitrógeno, un grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno, un grupo alcoxi C1-C6 opcionalmente sustituido, un grupo amino opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo, donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente entre el grupo que consiste de átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo hidroxi u oxo; un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente entre el grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con uno o dos grupos oxo; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente entre el grupo que consiste en un grupo alquilo C1-C6 (la porción alquilo C1-C6 está opcionalmente sustituida con un grupo hidroxilo, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6 y
los sustituyentes en cada uno del grupo alquenilo C2-C6 opcionalmente sustituido, el grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo cicloalquenilo C3-C7 opcionalmente sustituido, el grupo arilo de 6 a 10 miembros opcionalmente sustituido, el grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno, el grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno y el grupo alcoxi C1-C6 opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste de un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos
seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo hidroxi o un oxo; un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con uno o dos grupos oxo; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente entre el grupo que consiste en un grupo alquilo C1-C6 (la porción alquilo C1-C6 es opcionalmente sustituida con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6 y
R8 y R9 representan cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
[2] En una segunda modalidad, la presente invención se refiere a un compuesto representado por la fórmula general [I]

a sal farmacéuticamente aceptable del mismo de acuerdo con la modalidad [1], donde
el anillo A es un grupo fenilo opcionalmente sustituido,
donde los sustituyentes en el grupo fenilo opcionalmente sustituidos son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, grupo alquilo C1-C6, grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7 , un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6;
R1 es un grupo alquilo C1-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo adamantilo opcionalmente sustituido, un grupo heterocíclico alifático opcionalmente sustituido, un grupo arilo de 6 a 10 miembros opcionalmente sustituido que puede ser parcialmente hidrogenado, un grupo heteroarilo opcionalmente sustituido o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6,
donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático y un grupo alquilenoxi C1-C6, los sustituyentes en cada uno del grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo heterocíclico alifático opcionalmente sustituido, el grupo arilo de 6 a 10 miembros opcionalmente sustituido que puede ser parcialmente hidrogenado y el grupo heteroarilo opcionalmente sustituido es/son de uno a tres grupos seleccionados independientemente entre el grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6; un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; a grupo hidroxialquilo C1-C6; un grupo alcoxialquilo C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o
dos grupos alquilo C1-C6; un grupo heterocíclico alifático monocíclico de 4 a 7 miembros que contiene uno o dos heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático y un grupo alquilenoxi C1-C6,
la porción heterocíclica alifática de cada uno del grupo carbonilo heterocíclico alifático y el grupo sulfonilo heterocíclico alifático con el cual R1 es sustituido, es un anillo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía por lo menos un átomo de nitrógeno y uno que opcionalmente contenía además un heteroátomo seleccionado del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno,
la porción heterocíclica alifática del grupo heterocíclico alifático opcionalmente sustituido representado por R1, es un anillo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, uno átomo de azufre y un átomo de nitrógeno y
la porción heteroarilo del grupo heteroarilo opcionalmente sustituido representado por R1, es un heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno;
R2 es un átomo de halógeno, un grupo alquilo C1-C6 o grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II],
donde el anillo B es un grupo heterocíclico alifático monocíclico o bicíclico de 4 a 8 miembros que puede contener adicionalmente, además del átomo de nitrógeno mostrado en la fórmula [II], un heteroátomo seleccionado del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno y puede contener parcialmente un doble enlace;
el anillo C es un grupo arilo monocíclico de 6 a 10 miembros o un grupo heteroarilo monocíclico de 5 o 6 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno;
R5 y R6 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
R7 es un grupo alquilo C1-C6 opcionalmente sustituido, un grupo alquenilo C2-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo cicloalquenilo C3-C7 opcionalmente sustituido, un grupo arilo de 6 a 10 miembros opcionalmente sustituido, un grupo heteroarilo opcionalmente sustituido, un grupo heterocíclico alifático opcionalmente sustituido, un grupo alcoxi C1-C6 opcionalmente sustituido, un grupo amino opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo,
donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido es/son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo hidroxi o un oxo; un grupo heterocíclico alifático monocíclico de 4 a 7 miembros opcionalmente sustituido con uno o dos grupos oxo y que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un alcoxi C1-C6 o un carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6,
el o los sustituyentes en cada uno del grupo alquenilo C2-C6 opcionalmente sustituido, el grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo cicloalquenilo C3-C7 opcionalmente sustituido, el grupo arilo de 6 a 10 miembros opcionalmente sustituido, el grupo heteroarilo opcionalmente sustituido, el grupo heterocíclico alifático opcionalmente sustituido y el grupo alcoxi C1-C6 opcionalmente sustituido es/son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático
de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo hidroxi o un oxo; un grupo heterocíclico alifático monocíclico de 4 a 7 miembros opcionalmente sustituido con uno o dos grupos oxo y que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6,
la porción heterocíclica alifática del grupo carbonilo heterocíclico alifático con el cual R7 es sustituido, es un anillo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno,
el grupo heteroarilo con el cual R7 es sustituido, es un grupo heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno,
la porción heteroarilo del grupo heteroarilo opcionalmente sustituido representado por R7, es un heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno y
la porción heterocíclica alifática del grupo heterocíclico alifático opcionalmente sustituido representado por R7, es un anillo heterocíclico alifático monocíclico o bicíclico de 4 a 8 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno y
R8 y R9 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
[3] En una modalidad más la presente invención se refiere al compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las modalidades [1] o [2], donde
el anillo A es un grupo fenilo opcionalmente sustituido,
donde el o los sustituyentes en el grupo fenilo opcionalmente sustituido, es/son uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7, un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6; R1 es un grupo alquilo C1-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo adamantilo opcionalmente sustituido, un grupo heterocíclico alifático opcionalmente sustituido, un grupo arilo opcionalmente sustituido que puede ser parcialmente hidrogenado, un grupo heteroarilo opcionalmente sustituido o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6,
donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido, son uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático y un grupo alquilenoxi C1-C6, los sustituyentes en cada uno del grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo heterocíclico alifático opcionalmente sustituido, el grupo arilo opcionalmente sustituido que puede ser parcialmente hidrogenado y el grupo heteroarilo opcionalmente sustituido, son uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6; un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo hidroxialquilo C1-C6; un grupo alcoxialquilo C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático y un
grupo alquilenoxi C1-C6,
el grupo heterocíclico alifático con el cual R1 es sustituido, es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo y un grupo homomorfolinilo, la porción heterocíclica alifática de cada uno del grupo carbonilo heterocíclico alifático y el grupo sulfonilo heterocíclico alifático con el cual R1 es sustituido, es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo homopiperazinilo y un grupo homomorfolinilo,
la porción arilo del grupo arilo opcionalmente sustituido que puede ser parcialmente hidrogenado representada por R1, es un grupo seleccionado del grupo que consiste en un grupo fenilo, un grupo naftilo, un grupo dihidrofenilo, un grupo indanilo y un grupo tetrahidronaftilo,
la porción heterocíclica alifática del grupo heterocíclico alifático opcionalmente sustituido representado por R1, es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo y un grupo homomorfolinilo y
la porción heteroarilo del grupo heteroarilo opcionalmente sustituido representado por R , es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, tetrazolilo, oxadiazolilo, piridinilo, grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo;
R2 es un átomo de halógeno, un grupo alquilo C1-C6 o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II],
donde el anillo B es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiridinilo, un grupo homopiperazinilo, un grupo homomorfolinilo, un grupo 3-azabiciclo[3.1.0]hexilo y un grupo octahidropirrolo[3,4-c]pirrolilo;
el anillo C es un grupo seleccionado del grupo que consiste en un grupo fenilo, un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo;
R5 y R6 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
R7 es un grupo alquilo C1-C6 opcionalmente sustituido, un grupo alquenilo C2-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo cicloalquenilo C3-C7 opcionalmente sustituido, un grupo arilo de 6 a 10 miembros opcionalmente sustituido, un grupo heteroarilo opcionalmente sustituido, un grupo heterocíclico alifático opcionalmente sustituido, un grupo alcoxi C1-C6 opcionalmente sustituido, un grupo amino opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo,
donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido, son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo opcionalmente sustituido con un grupo hidroxi o un grupo oxo; un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos oxo; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6.
los sustituyentes en cada uno del grupo alquenilo C2-C6 opcionalmente sustituido, el grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo cicloalquenilo C3-C7 opcionalmente sustituido, el grupo arilo de 6 a 10 miembros opcionalmente sustituido, el grupo heteroarilo opcionalmente sustituido, el grupo heterocíclico alifático opcionalmente sustituido y el grupo alcoxi C1-C6 opcionalmente sustituido es/son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo
alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo opcionalmente sustituido con un grupo hidroxi o un grupo oxo; un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos oxo; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6 ,
la porción heterocíclica alifática del grupo carbonilo heterocíclico alifático con el cual R7 es sustituido, es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo homopiperazinilo y un grupo homomorfolinilo, el grupo heteroarilo con el cual R7 es sustituido, es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo;
el grupo heterocíclico alifático con el cual R7 es sustituido, es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo y un grupo homomorfolinilo, la porción heteroarilo del grupo heteroarilo opcionalmente sustituido representado por R7, es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo y
la porción heterocíclica alifática del grupo heterocíclico alifático opcionalmente sustituido representado por R7, es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo, un grupo homomorfolinilo, un grupo 3-azabiciclo[3.1.0]hexilo y un grupo octahidropirrolo[3,4-c]pirrolilo y
R8 y R9 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
[4] En una modalidad más la presente invención se refiere al compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las modalidades [1] a [3], donde
el anillo A es un grupo fenilo opcionalmente sustituido cada vez con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7, un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6;
R1 es 12
(1) un grupo alquilo C1-C6 opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo heterocíclico alifático; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo sulfonilo heterocíclico alifático y un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo y un grupo homomorfolinilo y la porción heterocíclica alifática de cada uno del grupo carbonilo heterocíclico alifático y el grupo sulfonilo heterocíclico alifático, es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo homopiperazinilo y un grupo homomorfolinilo),
(2) un grupo cicloalquilo C3-C7 monocíclico opcionalmente sustituido con uno o dos grupos seleccionados
independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6, un grupo alcoxialquilo C1-C6 y un grupo alquilenoxi C1-C6,
(3) un grupo adamantilo opcionalmente sustituido con un grupo hidroxi,
(4) un grupo heterocíclico alifático opcionalmente sustituido con un grupo seleccionado entre el grupo que consiste en un grupo alquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6 (donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, grupo un imidazolinilo, grupo un tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo y un grupo homomorfolinilo), (5) un grupo seleccionado del grupo que consiste en un grupo fenilo, un grupo naftilo, un grupo dihidrofenilo, un grupo indanilo y un grupo tetrahidronaftilo,
(6) un grupo heteroarilo el cual es opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6 y un grupo carbamoílo (donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo),
(7) un grupo carbamoílo o
(8) un grupo mono-alquilcarbamoílo C1-C6;
R2 es un átomo de halógeno, un grupo alquilo C1-C6 o un grupo alcoxi C1-C6;
R123 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II],
donde el anillo B es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiridinilo, un grupo homopiperazinilo, un grupo homomorfolinilo, un grupo 3-azabiciclo[3.1.0]hexilo y un grupo octahidropirrolo[3,4-c]pirrolilo y R5 y R6 representan átomos de hidrógeno, o el anillo B es un grupo piperidinilo y R5 y R6 son cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, un grupo ciano y grupo alcoxialquilo C1-C6 o
el anillo B es un grupo pirrolidinilo y R5 y R6 son cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
el anillo C es un grupo seleccionado entre el grupo que consiste en un grupo fenilo, un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo triazinilo y un grupo triazinilo;
R7 es
(1) un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo,
(2) un grupo alquenilo C1-C6 opcionalmente sustituido con un grupo carboxilo,
3) un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo, (4) un grupo cicloalquenilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo,
(5) un grupo fenilo opcionalmente sustituido con un grupo carboxilo,
(6) un grupo heteroarilo el cual es opcionalmente sustituido con un grupo carboxilo o un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo
(donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo),
(7) un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilaminocarbonilo C1-C6; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con un grupo carboxilo (donde el anillo heterocíclico alifático es un grupo seleccionado entre el grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo pireridinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo homopiperazinilo y un grupo
homomorfolinilo);un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6; un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos oxo (donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo y un grupo homomorfolinilo); un grupo alquilsulfonilo C1-C6; un grupo heteroarilo (donde el grupo heteroarilo es un grupo seleccionado entre el grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo) y un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo un grupo tetrahidropiranilo, un grupo homopiperazinilo, un grupo homomorfolinilo, un grupo 3-azabiciclo[3.1.0]hexilo) y un grupo octahidropirrolo[3,4-c]pirrolilo),
(8) un grupo alcoxi C1-C6 opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo ciano; un grupo carboxilo; un grupo heteroarilo opcionalmente sustituido con un grupo hidroxi o un grupo oxo (donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo , un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo); un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alquilsulfonilaminocarbonilo C1-C6,
(9) un grupo amino el cual es opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo o
(10) un grupo carbamoílo y
R8 y R9 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno
[5] En una modalidad más la presente invención se refiere al compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las modalidades [1] a [4], donde
el anillo A es un grupo fenilo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7, un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6, R1 es 123456
(1) un grupo alquilo C1-C6 opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi; un grupo cicloalquilo monocíclico de 3 a 7 miembros; un grupo alcoxi C1-C6; un grupo tetrahidropiranilo; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo alquilo C1-C6; un grupo haloalquilo C1-C6; sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6 (donde el anillo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo pirrolidinilo, un grupo piperidinilo y un grupo morfolinilo); un grupo pirrolidinilsulfonilo y un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6,
(2) un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6, un grupo alcoxialquilo C1-C6 y un grupo alquilenoxi C1-C6,
(3) un grupo adamantilo opcionalmente sustituido con un grupo hidroxi,
(4) un grupo heterocíclico alifático opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo alquilo C1-C6, grupo hidroxialquilo C1-C6, grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo tetrahidrofuranilo, un grupo tetrahidropiranilo y un grupo piperidinilo),
(5) un grupo indanilo,
(6) un grupo heteroarilo el cual es opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo ciano, grupo alquilo C1-C6, grupo alcoxi C1-C6 y grupo carbamoílo
(donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo piridazinilo, un grupo piridinilo y un grupo pirimidinilo),
(7) un grupo carbamoílo o
(8 ) un grupo mono-alquilcarbamoílo;
R2 es un átomo de halógeno, un grupo alquilo C1-3 o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II],
donde el anillo B es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo tetrahidropiridinilo, un grupo piperazinilo, un grupo homopiperazinilo y un grupo octahidropirrolo[3,4-c]pirrolilo y R5 y R6 representan átomos de hidrógeno o
el anillo B es un grupo piperidinilo y R5 y R6 son cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, un grupo ciano y un grupo alcoxialquilo C1-C6 o
el anillo B es un grupo pirrolidinilo y R5 y R6 son cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
el anillo C es un grupo fenilo o un grupo piridinilo;
R7 es
(1) un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo,
(2) un grupo alquenilo C2-C6 opcionalmente sustituido con un grupo carboxilo,
(3) un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo, (4) un grupo cicloalquenilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo,
(5) un grupo fenilo opcionalmente sustituido con un grupo carboxilo,
(6) un grupo heteroarilo el cual es opcionalmente sustituido con un grupo carboxilo o un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo
(donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo oxazolilo y un grupo pirazolilo),
(7) un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilaminocarbonilo C1-C6; un grupo pirrolidinilcarbonilo opcionalmente sustituido con un grupo carboxilo; un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6; un grupo pirrolidinilo y un grupo isotiazolidinilo opcionalmente sustituido con uno o dos grupos oxo; un grupo alquilsulfonilo C1-C6; un grupo tetrazolilo y un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 (donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo piperidinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo piperazinilo y un grupo 3-azabiciclo[3.1.0]hexilo),
(8) un grupo alcoxi C1-C6 opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo ciano; un grupo carboxilo; un grupo heteroarilo opcionalmente sustituido con un grupo hidroxi o un grupo oxo (donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo isoxazolilo, un grupo oxadiazolilo y un grupo tetrazolilo); un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alquilsulfonilaminocarbonilo C1-C6,
(9) un grupo amino el cual es opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo o
(10) un grupo carbamoílo y
R8 y R9 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
[6] en una modalidad más la presente invención se refiere al compuesto o sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las modalidades [1] a [5], donde
R1 es 1
(1) un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con uno o dos grupos
seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6, un grupo alcoxialquilo C1-C6 y un grupo alquilenoxi C1-C6,
(2) un grupo adamantilo opcionalmente sustituido con un grupo hidroxi; o
(3) un grupo heterocíclico alifático opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo alquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo tetrahidrofuranilo, un grupo tetrahidropiranilo y un grupo piperidinilo).
[7] En una modalidad más la presente invención se refiere al compuesto o sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las modalidades [1] a [6], donde
R3 y R4 están unidos terminalmente entre sí y junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II]

donde el anillo B es un grupo pirrolidinilo.
[8] En una modalidad más la presente invención se refiere al compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las modalidades [1] a [7], donde
R1 es un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6, un grupo alcoxialquilo C1-C6 y un grupo alquilenoxi C1-C6.
[9] En una modalidad más la presente invención se refiere al compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las modalidades [1] a [8], donde
el anillo A es un grupo fenilo opcionalmente sustituido con uno o dos grupos seleccionados independientemente entre el grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7, un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6;
R1 es un grupo cicloalquilo monocíclico de 3 a 7 miembros sustituido opcionalmente con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6, un grupo alcoxialquilo C1-C6 y un grupo alquilenoxi C1-C6;
R1234es un átomo de halógeno o un grupo alcoxi C1-C6;
R3 y R4 están Unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la formula [II],
donde el anillo B es un grupo pirrolidinilo y R5 y R6 son cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
el anillo C es un grupo fenilo;
R7 es
(1) un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo,
(2) un grupo alquenilo C2-C6 opcionalmente sustituido con un grupo carboxilo,
(3) un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo, (4) un grupo cicloalquenilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo,
(5) un grupo fenilo opcionalmente sustituido con un grupo carboxilo,
(6) un grupo heteroarilo el cual es opcionalmente sustituido con un grupo carboxilo o un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo
(donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo oxazolilo y un grupo pirazolilo),
(7) un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilaminocarbonilo C1-C6; un grupo pirrolidinilcarbonilo opcionalmente sustituido con un grupo carboxilo; un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6; un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos oxo (donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo pirrolidinilo y un grupo isotiazolidinilo); un grupo alquilsulfonilo C1-C6; un grupo tetrazolilo y un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo piperidinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo piperazinilo y un grupo 3-azabiciclo[3.1.0]hexilo),
(8) un grupo alcoxi C1-C6 opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo ciano; un grupo carboxilo; un grupo heteroarilo opcionalmente sustituido con un grupo hidroxi o un grupo oxo (donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo isoxazolilo, un grupo oxadiazolilo y un grupo tetrazolilo); un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alquilsulfonilaminocarbonilo C1-C8,
(9) un grupo amino el cual es opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo o
(10) un grupo carbamoílo y
R8 y R9 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
[10] En una modalidad más la presente invención se refiere al compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con la modalidad [9], donde
el anillo A es un grupo fenilo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7, un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6;
R1 es un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6, un grupo alcoxialquilo C1-C6 y un grupo alquilenoxi C1-C6;
R2 es un átomo de halógeno o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II],
donde el anillo B es un grupo pirrolidinilo y
R5 y R6 son cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
el anillo C es un grupo fenilo;
R7 es un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilaminocarbonilo C1-C6; un grupo pirrolidinilcarbonilo
opcionalmente sustituido con un grupo carboxilo; un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6; un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos oxo (donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo pirrolidinilo y un grupo isotiazolidinilo); un grupo alquilsulfonilo C1-C6; un grupo tetrazolilo y un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo piperidinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo piperazinilo y un grupo 3-azabiciclo[3.1.0]hexilo) y
R8 y R9 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
[11] En todavía otra modalidad preferida, la presente invención abarca un compuesto representado por la fórmula general [I] de acuerdo con cualquiera de las modalidades [1] a [6] o una sal farmacéuticamente aceptable del mismo, donde el anillo A es un grupo fenilo opcionalmente sustituido con un grupo alcoxi C1-C6;
R1 es un grupo alquilo C1-C6; un grupo tetrahidrofuranilo, un grupo tetrahidropiranilo, un grupo piperidinilo; un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo alcoxi C1-C6 y un grupo ciano o un grupo heteroarilo opcionalmente sustituido con un grupo alquilo C1-C6 (donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en los grupos piridazinilo, piridinilo y pirimidinilo);
R2 es un átomo de halógeno o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II],

donde el anillo B se selecciona del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo piperidinilo, un grupo tetrahidropiridinilo y un grupo piperazinilo y R5 y R6 son átomos de hidrógeno o
el anillo B es un grupo pirrolidinilo, R5 es un grupo alcoxialquilo C1-C6 y R6 es un átomo de hidrógeno o de halógeno;
el anillo C es un grupo fenilo;
R7 es un grupo heterocíclico alifático sustituido con un grupo carboxilo (donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, grupo pirrolidinilo, grupo piperidinilo, grupo morfolinilo, grupo tiomorfolinilo, grupo piperazinilo y grupo 3-azabiciclo[3.1.0]hexilo), R8 es un átomo de halógeno o un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y
R9 es un átomo de hidrógeno.
[12] En todavía otra modalidad preferida, la presente invención abarca un compuesto representado por la fórmula general [I] o una sal farmacéuticamente aceptable del mismo de acuerdo con la modalidad [11], donde el anillo A es un grupo fenilo opcionalmente sustituido con un grupo alcoxi C1-C6;
R1 es un grupo tetrahidropiranilo o un grupo cicloalquilo monocíclico de 5 o 6 miembros opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo alcoxi C1-C6 y ciano;
R2 es un átomo de halógeno o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II],

donde el anillo B es un grupo pirrolidinilo, R5 es un grupo alcoxialquilo C1-C6 y R6 es un átomo de hidrógeno o de halógeno;
el anillo C es un grupo fenilo;
R7 es un grupo piperidinilo sustituido con un grupo carboxilo;
R8 es un átomo de halógeno o un grupo haloalquilo C1-C6 y
R9 es un átomo de hidrógeno.
[13] En todavía otra modalidad preferida, la presente invención se refiere a un compuesto representado por la fórmula general [1] o una sal farmacéuticamente aceptable del mismo de acuerdo con la modalidad [12], donde el anillo A es un grupo fenilo opcionalmente sustituido con un grupo alcoxi C1-C6;
R1 es un grupo cicloalquilo monocíclico de 5 o 6 miembros opcionalmente sustituido con un grupo seleccionado del grupo que consiste en alcoxi C1-C6 y grupos ciano;
R2 es un átomo de halógeno o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la formula [II]:

donde el anillo B es un grupo pirrolidinilo, R5 es un grupo alcoxialquilo C1-C6 y R6 es un átomo de hidrógeno o de halógeno;
el anillo C es un grupo fenilo;
R7 es un grupo piperidinilo sustituido con un grupo carboxilo;
R8 es un átomo de halógeno o un grupo haloalquilo y
R9 es un átomo de hidrógeno.
[14] En todavía otra modalidad preferida, la presente invención puede proveer un compuesto de acuerdo con la modalidad [1] seleccionado del grupo que consiste en:
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-etoxiciclohexil)-3-metoxi-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-5-(etoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico;
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-4-(4-metoxifenil)-1-(2-metilpiridin-4-il)pirrolidin-3-il]carbonil}-5-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico;
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-4-(4-metoxifenil)-1-(2-metilpirimidin-4-il)pirrolidin-3-il]carbonil}-5
(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico;
ácido 1-{5-fluoro-2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-il]carbonil}-5-(metoximetil)pirrolidin-3-il]fenil}piperidin-4-carboxílico;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-etox¡c¡clohex¡l)-3-metox¡-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-c¡anoc¡clohex¡l)-3-fluoro-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4S)-1-{[(3R,4R)-1-(frans-4-etox¡c¡clohex¡l)-3-metox¡-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-fluoro-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-1-(frans-4-etox¡c¡clohex¡l)-3-metox¡-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-c¡clohex¡l-3-fluoro-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-4-(4-metox¡fen¡l)-1-(tetrah¡dro-2H-p¡ran-4-¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tnfluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-c¡clopent¡l-3-fluoro-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-4-(4-metox¡fen¡l)-1-(2-met¡lp¡r¡d¡n-4-¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4S)-4-fluoro-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{5-cloro-2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-(frans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-4-(c¡anomet¡l)-1-{[(3R,4R)-1-c¡clopent¡l-3-fluoro-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tnfluoromet¡l)fen¡l}p¡perid¡n-4-carboxíl¡co;
ác¡do H2-(1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}p¡pend¡n-4-¡l)-5-(tr¡fluoromet¡l)fen¡l]p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-1-c¡clopent¡l-3-fluoro-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co y
ác¡do 1-{2-[(3S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co
o una sal farmacéut¡camente aceptable del mismo.
[15] En todavía otra modalidad preferida, la presente invención puede proveer un compuesto de acuerdo con la modal¡dad
[14] seleccionado entre el grupo que consiste en:
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-etox¡ddohex¡l)-3-metox¡-4-(4-metox¡fen¡l)p¡rrolid¡n-3-¡l]carbon¡l}-5-(etox¡met¡l)p¡rrol¡d¡n-3-il]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{5-fluoro-2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrolid¡n-3-¡l]fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-etox¡c¡clohex¡l)-3-metox¡-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-il]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-c¡anoc¡clohex¡l)-3-fluoro-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(trifluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{2-[(3S,4S)-1-{[(3R,4R)-1-(frans-4-etox¡c¡clohex¡l)-3-metox¡-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-fluoro-4-(metox¡met¡l)p¡rrol¡d¡n-3-il]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{2-[(3S,4R)-1-{[(3R,4R)-1-(frans-4-etox¡c¡clohex¡l)-3-metox¡-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-il]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-1-c¡clohex¡l-3-fluoro-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-il]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{2-[(3S,5S)-H[(3R,4R)-3-fluoro-1-(frans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-1-cidopent¡l-3-fluoro-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-il]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{2-[(3S,4S)-4-fluoro-1-{[(3R,4R)-3-fluoro-1frans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-il]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{5-cloro-2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrolid¡n-3-¡l]fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{2-[(3S,4R)-4-(c¡anomet¡l)-1-{[(3R,4R)-1-c¡clopent¡l-3-fluoro-4-(4-metox¡fen¡l)p¡rrolid¡n-3-¡l]carbon¡l}p¡rrol¡d¡n-3-il]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-1-(trans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico;
ác¡do 1-[2-(1-{[(3R,4R)-3-fluoro-1-(fra/75-4-metox¡c¡dohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}p¡per¡d¡n-4-¡l)-5-(tr¡fluoromet¡l)fen¡l]p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-1-c¡clopent¡l-3-fluoro-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co y
ác¡do 1-{2-[(3S)-1-{[(3R,4R)-3-fluoro-1-(trans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co
o una sal farmacéut¡camente aceptable del m¡smo.
[16] En todavía otra modal¡dad prefer¡da, la presente ¡nvenc¡ón puede proveer un compuesto de acuerdo con la modal¡dad
[15] selecc¡onado entre el grupo que cons¡ste en:
ác¡do 1-{5-fluoro-2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-1-(trans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(trans-4-etox¡c¡clohex¡l)-3-metox¡-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-í(3S,5S)-1-{[(3R,4R)-3-fluoro-1-(trans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-c¡clopent¡l-3-fluoro-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4S)-4-fluoro-1-{[(3R,4R)-3-fluoro-1-trans-4-metox¡c¡dohex¡l)-4-(4-fen¡lmetox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{5-cloro-2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-1-(trans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-1-(trans-4-metox¡c¡clohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co y
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-1-c¡clopent¡l-3-fluoro-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
o una sal farmacéut¡camente aceptable del m¡smo.
Cuando el compuesto [I] de la presente ¡nvendón t¡ene un átomo de carbono as¡métr¡co en la molécula, puede ex¡st¡r como una plural¡dad de estereo¡sómeros (es dec¡r, d¡aestereo¡sómeros o ¡sómeros ópt¡cos) con base en el átomo de carbono as¡métr¡co. La presente ¡nvenc¡ón abarca cualqu¡era de estos estereo¡sómeros y una mezcla de los m¡smos. Para ¡nd¡car que el compuesto [I] de la presente ¡nvenc¡ón es una mezcla de una plural¡dad de estereo¡sómeros, el compuesto [I] puede representarse usando un enlace mostrado por la s¡gu¡ente línea ondulada:
[Fórmula 10]
El compuesto [I] de la presente ¡nvenc¡ón tamb¡én puede contener ¡sómeros cis- y trans- como ¡sómeros geométr¡cos. Además, cuando el compuesto [I] de la presente ¡nvenc¡ón t¡ene qu¡ral¡dad ax¡al en la molécula, puede contener ¡sómeros con base en la qu¡ral¡dad ax¡al. La presente ¡nvenc¡ón abarca cualqu¡era de estos ¡sómeros y una mezcla de los m¡smos.
El compuesto [I] de la presente ¡nvenc¡ón abarca compuestos marcados con ¡sótopos (por ejemplo, H, C, C, N, 18F, 32P, 35S y 125I) y productos deuterados.
El compuesto [I] de la presente ¡nvenc¡ón o una sal farmacéut¡camente aceptable del mismo t¡ene act¡v¡dad agon¡sta del MC1R sobresal¡ente.
[17] En todavía otra modal¡dad prefer¡da, la presente ¡nvenc¡ón puede proporc¡onar una compos¡c¡ón farmacéut¡ca de acuerdo con las modal¡dades [1] a [16] que comprende, como pr¡nc¡p¡o act¡vo, el compuesto de acuerdo con una cualqu¡era de las modal¡dades anter¡ormente menc¡onadas o una sal farmacéut¡camente aceptable del m¡smo.
[18] En todavía otra modal¡dad prefer¡da, la presente ¡nvenc¡ón puede proporc¡onar una compos¡c¡ón farmacéut¡ca de acuerdo con la modal¡dad [17] para su uso en la prevenc¡ón oterap¡a de
artr¡t¡s reumato¡de, artr¡t¡s gotosa, osteoartros¡s, enfermedad ¡nflamator¡a del ¡ntest¡no, escleros¡s s¡stém¡ca, psor¡as¡s, f¡bros¡s, protoporf¡r¡a (por ejemplo, protoporf¡r¡a er¡tropoyét¡ca), lupus er¡tematoso s¡stém¡co, melanoma, cáncer de p¡el, v¡t¡l¡go, pérd¡da del cabello, dolor, daño por ¡squem¡a/reperfus¡ón, enfermedad ¡nflamator¡a cerebral, hepat¡t¡s, choque sépt¡co/sept¡cem¡a, nefr¡t¡s, trasplante, exacerbac¡ón de la enfermedad por el VIH, vascul¡t¡s, uveít¡s, ret¡n¡t¡s p¡gmentosa, degenerac¡ón macular relac¡onada con la edad, ¡nfecc¡ón m¡crob¡ana,
enfermedad celíaca, síndrome nefrótico e invasión por melanoma.
[19] En todavía otra modalidad preferida, la presente invención puede proporcionar una composición farmacéutica de acuerdo con la modalidad [18] para su uso en la prevención o terapia de
esclerosis sistémica, psoriasis, protoporfiria, melanoma, cáncer de piel, vitiligo, pérdida del cabello, síndrome nefrótico, retinitis pigmentosa o degeneración macular relacionada con la edad.
[20] En todavía otra modalidad preferida, la presente invención puede proporcionar una composición farmacéutica de acuerdo con la modalidad [19] para su uso en la prevención o terapia de
esclerosis sistémica, protoporfiria, melanoma, vitiligo, síndrome nefrótico, retinitis pigmentosa o degeneración macular relacionada con la edad.
[21] En todavía otra modalidad preferida, la presente invención puede proporcionar una composición farmacéutica de acuerdo con la modalidad [20] para su uso en la prevención o terapia de protoporfiria eritropoyética.
Como se describió anteriormente, el compuesto [I] de la presente invención o una sal farmacéuticamente aceptable del mismo tiene actividad agonista excelente para el MC1R y como resultado del examen de la actividad agonista para el MC1R humano de acuerdo con el método de prueba descrito en el ejemplo experimental 1 descrito más adelante, todos los compuestos descritos en los presentes ejemplos tuvieron un valor de EC50 de 1000 nM o menos. Se encontró también que el compuesto [I] de la presente invención o una sal farmacéuticamente aceptable del mismo tiene eficacia farmacológica en modelos de inflamación inducida por la bleomicina (de acuerdo con el método descrito en Arthritis and Rheumatology, 2009; 60: p. 592-603). Además, el compuesto [I] de la presente invención abarca compuestos que tienen alta selectividad por el MC1R. Por ejemplo, el compuesto que se describe en el ejemplo 147 (nombre químico: diclorhidrato de ácido 1-{5-fluoro-2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metoxiciclohexil)-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-5-(metoximetil)pirrolidin-3-il]fenil}piperidin-4-carboxílico), muestra que la relación entre el valor de la EC50 para la actividad agonista del MC1R humano (1,7 nM) y el valor de la EC50 para la actividad agonista del MC4R humano (341 nM) es de aproximadamente 1:200 y es de esta manera uno de los compuestos con alta selectividad por el MC1R.
El compuesto [I] de la presente invención puede usarse para propósitos farmacéuticos ya sea en forma libre o en forma de una sal farmacéuticamente aceptable del mismo. Ejemplos de sales farmacéuticamente aceptables incluyen sales de ácidos inorgánicos tales como clorhidrato, sulfato, fosfato y bromhidrato y sales de ácidos orgánicos tales como acetato, fumarato, oxalato, citrato, metansulfonato, bencensulfonato, tosilato y maleato.
El compuesto [I] de la presente invención o una sal farmacéuticamente aceptable del mismo incluye la totalidad de una sal interior o producto de adición del mismo, un solvato o hidrato del mismo, un co-cristal del mismo.
Uno o más de los compuestos de fórmula [I] de la presente invención o sales farmacéuticamente aceptables de los mismos pueden administrarse directamente a un paciente; de preferencia, sin embargo, el compuesto de fórmula [I] de la presente invención o una sal farmacéuticamente aceptable del mismo puede mezclarse con aditivos farmacéutica y farmacológicamente aceptables y puede proveerse como una preparación farmacéutica en una forma bien conocida por los expertos en la técnica.
Como aditivos farmacéutica y farmacológicamente aceptables, pueden usarse aditivos adecuados que se usan generalmente en la fabricación de productos farmacéuticos, tales como excipientes, desintegradores, aglutinantes, lubricantes, agentes de revestimiento, colorantes, diluyentes, bases y agentes isotónicos.
Entonces, el compuesto de la presente invención, junto con los aditivos descritos anteriormente, pueden prepararse en una forma de dosificación apropiada (por ejemplo, un polvo, inyección, tableta, cápsula o preparación tópica) y entonces administrados a un paciente (un humano o animal) usando un método de administración adecuado de acuerdo con la forma de dosificación (por ejemplo, por vía intravenosa, oral, percutánea o tópica).
La dosis puede determinarse dependiendo de la edad, peso corporal, condición de salud general, sexo, dieta, hora de administración, método de administración, tasa de excreción, combinación de fármacos y la severidad de la condición de la enfermedad del paciente bajo tratamiento durante la aplicación, en consideración de estos u otros factores. El compuesto de la presente invención o una sal farmacéuticamente aceptable del mismo tiene baja toxicidad y puede usarse de manera segura. La dosis diaria del mismo puede variar, dependiendo de la condición o el peso corporal del paciente, el tipo del compuesto o la vía de administración. Por ejemplo, en el caso de la administración parenteral, el compuesto de la presente invención se administra deseablemente por vía subcutánea, intravenosa, intramuscular o intrarrectal a una dosis de aproximadamente 0,0001 a 1000 mg/persona/día, de preferencia aproximadamente 0,001 a 1000 mg/persona/día y particularmente de preferencia 0,01 a 500 mg/persona/día y en el caso de la administración oral, el compuesto de la presente invención se administra deseablemente a una dosis de aproximadamente 0,0001 a 1000 mg/persona/día y más preferiblemente de aproximadamente 0,01 a 500 mg/persona/día.
El compuesto de la presente invención o una sal farmacéuticamente aceptable del mismo puede producirse como sigue, por ejemplo. Nótese que cada una de las abreviaturas usadas en esta especificación, significa lo siguiente:
Me: metilo
Et: etilo.
Método de síntesis A-1
[Fórmula 11]

Entre los compuestos objetivo [I] de la presente invención, un compuesto donde R1 es un alquilo opcionalmente sustituido, cicloalquilo, o grupo heterocíclico alifático, el cual es representado por la fórmula general [Ia] (donde R11 representa un alquilo opcionalmente sustituido, cicloalquilo, o grupo heterocíclico alifático y los otros símbolos tienen los mismos significados como los descritos anteriormente), puede producirse como sigue, por ejemplo.
El compuesto objetivo [Ia] o una sal farmacéuticamente aceptable del mismo puede obtenerse sometiendo un compuesto representado por la fórmula general [a] (donde los símbolos tienen los mismos significados como los descritos anteriormente) o una sal del mismo a una reacción de aminación reductiva con un compuesto representado por la fórmula general [b] (donde Ra1 representa un grupo alquilo opcionalmente sustituido) o un equivalente de carbonilo del mismo, o con un compuesto representado por la fórmula general [c] (donde Ra2 y Ra3 representan cada uno independientemente un grupo alquilo opcionalmente sustituido, o Ra2 y Ra3 están unidos terminalmente entre sí y, junto con el átomo de carbono al cual están unidos, forman un cicloalquilo opcionalmente sustituido o un grupo heterocíclico alifático) o un equivalente de carbonilo del mismo y, según se desee, convirtiendo el producto resultante en una sal farmacéuticamente aceptable del mismo.
Por ejemplo, el equivalente de carbonilo puede ser un cetal y puede ser específicamente alcoxi-trialquilsililoxi-cetal. El equivalente de carbonilo del compuesto [c] puede ser de preferencia 1-etiloxi-1-(trimetilsililoxi)ciclopropano, por ejemplo.
Como la sal del compuesto [a], puede usarse una sal con un ácido inorgánico tal como ácido clorhídrico o un ácido carboxílico tal como ácido acético.
La reacción de aminación reductiva del compuesto [a] o una sal del mismo con el compuesto [b] o [c] o un equivalente de carbonilo del mismo puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de un agente reductor y un ácido, por ejemplo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo alifático halogenado tal como cloruro de metileno, un alcohol tal como metanol, un éter tal como tetrahidrofurano, un hidrocarburo aromático tal como tolueno y mezclas de los mismos. Ejemplos de agentes reductores incluyen triacetoxiborohidruro de sodio hidrogenado, borohidruro de sodio, así como catalizadores de hidrógeno y paladio (por ejemplo, un catalizador de paladio soportado sobre carbón activado). El ácido puede ser ácido acético, por ejemplo. La cantidad del compuesto [b], compuesto [c], o un equivalente de carbonilo del mismo usado puede ser 0,1 a 10 equivalentes, de preferencia 1 a 5 equivalentes, respecto al compuesto [a]. La cantidad del agente reductor usado puede ser 1 a 10 equivalentes, de preferencia 1 a 3 equivalentes, respecto al compuesto [a]. La cantidad del ácido usado puede ser 1 a 10 equivalentes, de preferencia 1 a 3 equivalentes, respecto al compuesto [a]. Esta reacción puede realizarse a -10 a 100 °C, de preferencia 10 a 50 °C.
Método de síntesis A-2
[Fórmula 12]

Entre los compuestos objetivo [I], un compuesto donde R1 es un grupo alquilo opcionalmente sustituido, el cual es representado por la fórmula general [Ib] (donde R12 representa un grupo alquilo opcionalmente sustituido y los otros símbolos tienen los mismos significados como los descritos anteriormente), puede producirse como sigue, por ejemplo.
El compuesto objetivo [Ib] puede obtenerse por la reacción del compuesto [a], o una sal del mismo, con un compuesto representado por la fórmula general [d] (donde Ra4 representa un grupo alquilo opcionalmente sustituido y X1 representa un grupo saliente) y, según se desee, convirtiendo el producto resultante en una sal farmacéuticamente aceptable del mismo.
En forma alternativa, el compuesto objetivo [Ib] o una sal farmacéuticamente aceptable del mismo puede obtenerse sometiendo el compuesto [a], o una sal del mismo, a una reacción de adición de Michael con un compuesto representado por la fórmula general [e] (donde Ra5 representa un grupo ávido de electrones (por ejemplo, un grupo alcoxicarbonilo, ciano o sulfonilo)) y, según se desee, convirtiendo el producto resultante en una sal farmacéuticamente aceptables del mismo.
Ejemplos del grupo saliente representado por X1 incluyen átomos de halógeno (por ejemplo, átomos de flúor, cloro, bromo y yodo), un grupo metilsulfoniloxi y un grupo p-toluensulfoniloxi. En particular, es adecuado un átomo de halógeno.
La reacción del compuesto [a] o una sal del mismo con el compuesto [d] puede realizarse en un solvente apropiado en presencia de una base, por ejemplo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un nitrilo tal como acetonitrilo, un hidrocarburo alifático halogenado tal como cloruro de metileno, un éter tal como tetrahidrofurano, un hidrocarburo aromático tal como tolueno, una amida tal como N,N-dimetilformamida, sulfóxido de dimetilo y mezclas de los mismos. Ejemplos de bases incluyen una amina tal como diisopropiletilamina y un carbonato de metal alcalino tal como carbonato de potasio. En esta reacción, puede añadirse un adyuvante de reacción para acelerar la reacción. Ejemplos de adyuvantes de reacción incluyen sales inorgánicas tales como yoduro de sodio y yoduro de potasio. La cantidad del compuesto [d] usado puede ser 0,1 a 10 equivalentes, de preferencia 0,8 a 2 equivalentes, respecto al compuesto [a]. La cantidad de la base usada puede ser 1 a 10 equivalentes, de preferencia 1 a 3 equivalentes, respecto al compuesto [a]. La cantidad del ayudante de reacción usado puede ser 0,01 a 10 equivalentes, de preferencia 0,1 a 1 equivalente, respecto al compuesto [a]. Esta reacción puede realizarse a 0 a 150 °C, de preferencia 20 a 100 °C.
La reacción de adición de Michael del compuesto [a] o una sal del mismo con el compuesto [e] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de una base, por ejemplo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un alcohol tal como etanol, un hidrocarburo aromático tal como tolueno, un éter tal como tetrahidrofurano, un hidrocarburo alifático halogenado tal como cloruro de metileno, una amida tal como N,N-dimetilformamida, sulfóxido de dimetilo, un nitrilo tal como acetonitrilo y mezclas de los mismos. Ejemplos de bases incluyen una amina tal como trietilamina y un carbonato de metal alcalino tal como carbonato de potasio. La cantidad del compuesto [e] usado puede ser 1 a 10 equivalentes, de preferencia 1 a 3 equivalentes, respecto al compuesto [a]. La cantidad de la base usada puede ser 1 a 10 equivalentes, de preferencia 1 a 3 equivalentes, respecto al compuesto [a]. Esta reacción puede realizarse a 0 a 150 °C, de preferencia 20 a 100 °C.
Método de síntesis A-3
[Fórmula 13]

Entre los compuestos objetivo [I], un compuesto donde R1 es un grupo arilo opcionalmente sustituido o un grupo heteroarilo opcionalmente sustituido, el cual es representado por la fórmula general [Ic] (donde R13 representa un grupo arilo opcionalmente sustituido o un grupo heteroarilo opcionalmente sustituido y los otros símbolos tienen los mismos significados como los descritos anteriormente), puede producirse como sigue, por ejemplo.
El compuesto objetivo [Ic] o una sal farmacéuticamente aceptable del mismo puede obtenerse sometiendo el compuesto [a], o una sal del mismo, a una reacción de acoplamiento con un compuesto representado por la fórmula general [f] (donde X2 representa un grupo saliente y el otro símbolo tiene el mismo significado que el descrito anteriormente) y, según se desee, convirtiendo el producto resultante en una sal farmacéuticamente aceptable del mismo.
Ejemplos del grupo saliente representado porX2 incluyen átomos de halógeno (por ejemplo, átomos de flúor, cloro y bromo) y un grupo trifluorometilsulfoniloxi. En particular, es adecuado un átomo de halógeno.
La reacción de acoplamiento del compuesto [a] o una sal del mismo con el compuesto [f] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de una base, por ejemplo y además en presencia de un catalizador de paladio, según se desee. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen éteres tales como 1,4-dioxano y tetrahidrofurano, una amida tal como N,N-dimetilformamida, un hidrocarburo aromático tal como tolueno, un alcohol tal como t-butanol y mezclas de los mismos. Ejemplos de catalizadores de paladio incluyen tris(dibencilidenoacetona)dipaladio, acetato de paladio, tetraquis(trifenilfosfina)paladio, cloruro de paladio y aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio (II). Ejemplos de bases incluyen t-butóxido de sodio, t-butóxido de potasio, carbonato del cesio, fosfato tripotásico y diisopropiletilamina. En esta reacción, puede añadirse un ligando para acelerar la reacción. Ejemplos de ligandos incluyen 2,2'-bis(difenilfosfino)-1,1'-binaftilo, 2-di-t-butilfosfino-2',4',6'-triisopropilbifenilo, 2-diciclohexil-fosfino-2',6'-dimetoxibifenilo, 2-di-t-butilfosfino-3,4,5,6-tetrametil-2',4',6'-triisopropil-1,1'-bifenilo y 1,1'-bis(difenilfosfino)ferroceno. La cantidad del compuesto [f] usado puede ser 1 a 10 equivalentes, de preferencia 1 a 3 equivalentes, respecto al compuesto [a]. La cantidad del catalizador de paladio usado puede ser 0,001 a 0,5 equivalentes, de preferencia 0,01 a 0,3 equivalentes, respecto al compuesto [a]. La cantidad de la base usada puede ser 1 a 10 equivalentes, de preferencia 1 a 3 equivalentes, respecto al compuesto [a]. La cantidad del ligando usado puede ser 0,001 a 0,5 equivalentes, de preferencia 0,01 a 0,3 equivalentes, respecto al compuesto [a]. Esta reacción puede realizarse a 0 a 200 °C, de preferencia 50 a 150 °C.
Método de síntesis A-4
[Fórmula 14]

Entre los compuestos objetivo [I], un compuesto donde R1 es un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo, el cual es representado por la fórmula general [Id] (donde R14 representa un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo y los otros símbolos tienen los mismos significados como los descritos anteriormente), puede producirse como sigue, por ejemplo.
El compuesto objetivo [Id] o una sal farmacéuticamente aceptable del mismo puede obtenerse por la reacción del compuesto [a], o una sal del mismo, con un compuesto representado por la fórmula general [g] (donde Ra6 representa un grupo alquilo o trialquilsililo) o con un compuesto representado por la fórmula general [h] (donde Ra7 y Ra8 son cada uno independientemente un grupo alquilo, o uno de ellos representa un grupo alquilo y el otro representa un átomo de hidrógeno) y, según se desee, convirtiendo el producto resultante en una sal farmacéuticamente aceptable del mismo.
La reacción del compuesto [a] o una sal del mismo con el compuesto [g] o [h] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de una base. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo alifático halogenado tal como cloruro de metileno, un éter tal como tetrahidrofurano, un hidrocarburo aromático tal como tolueno y mezclas de los mismos. Ejemplos de bases incluyen aminas tales como trietilamina y diisopropiletilamina y un carbonato de metal alcalino tal como carbonato de potasio. La cantidad del compuesto [g] o [h] usado puede ser 0,5 a 30 equivalentes, de preferencia 1 a 20 equivalentes, respecto al compuesto [a]. La cantidad de la base usada puede ser 1 a 10 equivalentes, de preferencia 1 a 5 equivalentes, respecto al compuesto [a]. Esta reacción puede realizarse a -20 a 100 °C, de preferencia 10 a 50 °C.
Método de síntesis B
[Fórmula 15]

El compuesto objetivo [I] (donde los símbolos tienen los mismos significados como los descritos anteriormente) de la presente invención puede producirse también como sigue, por ejemplo.
El compuesto objetivo [I] o una sal farmacéuticamente aceptable del mismo puede producirse por la condensación de un compuesto representado por la fórmula general [i] (donde los símbolos tienen los mismos significados como los descritos anteriormente), una sal del mismo, o un cloruro de ácido del mismo, con un compuesto representado por la fórmula general [j] (donde los símbolos tienen los mismos significados como los descritos anteriormente) o una sal del mismo y, según se desee, convirtiendo el producto resultante en una sal farmacéuticamente aceptable del mismo.
La reacción de condensación del compuesto [i] o una sal del mismo con el compuesto [j] o una sal del mismo puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de un agente de condensación, por ejemplo. Como la sal del compuesto [i], una sal con sodio o potasio es utilizable, por ejemplo. Como la sal del compuesto [j], una sal con un ácido inorgánico tal como clorhidrato o sulfato es utilizable, por ejemplo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen una amida tal como N,N-dimetilformamida, un hidrocarburo alifático halogenado tal como cloruro de metileno, un éter tal como tetrahidrofurano, agua y mezclas de los mismos. Ejemplos de agentes de condensación incluyen clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (EDAC); hexafluorofosfato de o-(7-azabenzotriazol-1-il)-N,N,N',N-tetrametiluronio (HATU) y cloruro de 4-(4,6-dimetoxi-1,3,5-triazin-2-il)-4-metilmorfolinio (DMT-MM). En esta reacción, puede añadirse una base para acelerar la reacción. Ejemplos de bases incluyen aminas tales como trietilamina y diisopropiletilamina y un carbonato de metal alcalino tal como carbonato de potasio. Un adyuvante de reacción puede añadirse también para acelerar la reacción. Ejemplos de dichos adyuvantes de reacción incluyen 1 -hidroxi-7-azabenzotriazol (HOAt), 1-hidroxibenzotriazol (HOBt) y 4-dimetilaminopiridina. La cantidad del compuesto [j] usado puede ser 0,1 a 10 equivalentes, de preferencia 0,5 a 5 equivalentes, respecto al compuesto [i]. La cantidad del
agente de condensación usado puede ser 0,5 a 10 equivalentes, de preferencia 1 a 3 equivalentes, respecto al compuesto [i]. La cantidad de la base usada puede ser 0 a 10 equivalentes, de preferencia 1 a 5 equivalentes, respecto al compuesto [i]. La cantidad del ayudante de reacción usado puede ser 0,5 a 10 equivalentes, de preferencia 1 a 3 equivalentes, respecto al compuesto [i]. Esta reacción puede realizarse a --10 a 100 °C, de preferencia 10 a 80 °C. La reacción de condensación de un cloruro de ácido del compuesto [i] con el compuesto [j] o una sal del mismo puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de una base.
Puede producirse un cloruro de ácido del compuesto [i] tratando el compuesto [i] con un reactivo convencional tal como cloruro de tionilo o cloruro de oxalilo, de acuerdo con un método convencional. Como el solvente y la base, pueden usarse convenientemente los mencionados para la reacción de condensación del compuesto [i] o una sal del mismo con el compuesto [j] o una sal del mismo.
Método de producción a del intermediario
[Fórmula 16]

El compuesto [a] descrito anteriormente usado en los métodos de síntesis A-1 a A-4 puede producirse como sigue, por ejemplo.
Un compuesto representado por la fórmula general [1] (donde X3 representa un grupo protector de amino y los otros símbolos tienen los mismos significados como los descritos anteriormente) se somete a una reacción de condensación con el compuesto [j] para obtener un compuesto representado por la fórmula general [2] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
El compuesto [a] puede obtenerse bajo agitación el grupo protector de amino del compuesto obtenido [2].
Ejemplos del grupo protector de amino representado por X3 incluyen grupos bencilo y t-butoxicarbonilo.
La reacción de condensación del compuesto [1] con el compuesto [j] puede realizarse como en la reacción de condensación del compuesto [i] con el compuesto [j] descrita en el método de síntesis B anterior.
La reacción de remoción del grupo protector de amino del compuesto [2] puede realizarse de acuerdo con un método convencional. Donde X3 es un grupo bencilo, por ejemplo, la reacción de remoción puede realizarse en un solvente apropiado en presencia de hidrógeno y un catalizador de paladio. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un alcohol tal como metanol, un éster tal como acetato de etilo y mezclas de los mismos. El catalizador de paladio puede ser un catalizador de paladio soportado sobre carbón activado, por ejemplo. Donde X3 es un grupo t- butoxicarbonilo, por ejemplo, la reacción de remoción puede realizarse en un solvente apropiado en presencia de un ácido. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo alifático halogenado tal como cloruro de metileno, un éter tal como 1,4-dioxano y mezclas de los mismos. Ejemplos de ácidos incluyen ácido clorhídrico y ácido trifluoroacético.
Método de producción b-1 del intermediario
[Fórmula 17]

Un compuesto representado por la fórmula general [3] (donde el símbolo tiene el mismo significado como el descrito anteriormente) se somete a una reacción de adición con un compuesto representado por la fórmula general [4] (donde X4 y X5 representan grupos protectores de carboxilo y el otro símbolo tiene el mismo significado como el descrito anteriormente), para obtener un compuesto representado por la fórmula general [5] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
El compuesto [5] se somete a una reacción de reducción y una reacción de cierre de anillo para obtener un compuesto representado por la fórmula general [6] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
El compuesto [6] se somete a una reacción de reducción para obtener un compuesto representado por la fórmula general [7] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
El sustituyente R1 se introduce en el grupo amino del compuesto [7] para obtener un compuesto representado por la fórmula general [8] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
El compuesto [i] puede obtenerse bajo agitación el grupo protector de carboxilo del compuesto [8] obtenido.
Los grupos protectores de carboxilo representados por X4 y X5 pueden ser grupos alquilo, por ejemplo.
La reacción de adición del compuesto [3] con el compuesto [4] puede realizarse en un solvente apropiado de acuerdo con un método convencional. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo aromático tal como tolueno, un hidrocarburo alifático halogenado tal como cloruro de metileno, un nitrilo tal como acetonitrilo y mezclas de los mismos. En esta reacción, la reacción de adición puede realizarse estereoselectivamente añadiendo un catalizador quiral en el sistema de reacción. Ejemplos de catalizadores quirales incluyen 1-[3,5-bis(trifluorometil)fenil]-3-[(1R,2R)-(-)-2-(dimetilamino)ciclohexil]tiourea y 6'-hidroxicinconina.
La reacción de reducción y la reacción de cierre de anillo del compuesto [5] pueden realizarse en un solvente apropiado de acuerdo con un método convencional, realizando el tratamiento con un agente reductor, seguido de tratamiento con una base, según se desee. Ejemplos de solventes incluyen un alcohol tal como metanol, un hidrocarburo alifático halogenado tal como cloruro de metileno, un éter tal como tetrahidrofurano, un hidrocarburo aromático tal como tolueno y mezclas de los mismos. Como los agentes reductores, son utilizables, por ejemplo, hidrógeno y un catalizador de paladio (por ejemplo, un catalizador de paladio soportado sobre carbón activado) o borohidruro de sodio y cloruro de níquel. En esta reacción, puede añadirse un ácido para acelerar la reacción de reducción. Ejemplos de ácidos incluyen ácido clorhídrico y ácido acético. Después, según se desee, puede realizarse la reacción de cierre de anillo por la acción de una base. La base puede ser 1,8-diazabiciclo[5.4.0]-7-undeceno, por ejemplo.
Puede realizarse la reacción de reducción del compuesto [6], tratando el compuesto [6] con un agente alquilizante, según se desee y posteriormente tratando el producto resultante con un agente reductor en un solvente apropiado, junto con un ácido, según se requiera. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo alifático halogenado tal como cloruro de metileno, un alcohol tal como metanol, un éter tal como tetrahidrofurano, un hidrocarburo aromático tal como tolueno y mezclas de los mismos. Ejemplos de agentes reductores incluyen cianoborohidruro de sodio, borohidruro de sodio y un complejo de piridinaborano. Ejemplos de ácidos incluyen ácido acético y ácido trifluoroacético. Ejemplos de agentes alquilizantes incluyen de tetrafluoroborato de trimetiloxonio y trifluorometansulfonato de metilo.
La reacción de introducción del sustituyente (R1) en el grupo amino del compuesto [7] puede realizarse, por ejemplo,
como en las reacciones de conversión del compuesto [a] en el compuesto [la], compuesto [Ib], compuesto [Ic] y compuesto [Id] mostradas en los métodos de síntesis A-1 a A-4 anteriores.
La reacción de remoción del grupo protector de carboxilo del compuesto [8] para producir el compuesto [i] puede realizarse de acuerdo con un método convencional, en un solvente apropiado, junto con una base, por ejemplo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un alcohol tal como metanol, agua y mezclas de los mismos. La base puede ser hidróxido de sodio, por ejemplo.
En forma alternativa, esta reacción puede realizarse en un solvente apropiado en presencia de un ácido. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo alifático halogenado tal como cloruro de metileno, un éter tal como 1,4-dioxano y mezclas de los mismos. Ejemplos de ácidos incluyen ácido clorhídrico y ácido trifluoroacético.
Método de producción b-2 del intermediario
[Fórmula 18]

Donde un compuesto ópticamente activo se requiere para el compuesto [i] usado en el método de síntesis B descrito anteriormente, dicho compuesto ópticamente activo puede producirse como sigue, por ejemplo.
En primer lugar, un compuesto representado por la fórmula general [9] (donde X6 representa un grupo auxiliar asimétrico y los otros símbolos tienen los mismos significados como los descritos anteriormente) se hace reaccionar con un compuesto representado por la fórmula general [10] (donde el símbolo tiene el mismo significado como el descrito anteriormente), para obtener un compuesto representado por la fórmula general [11] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
El compuesto [i] puede obtenerse bajo agitación el grupo auxiliar asimétrico del compuesto [11].
Ejemplos del grupo auxiliar asimétrico representado por X6 incluyen 4-bencil-2-oxazolidinona quiral, 4-fenil-2-oxazolidinona quiral y 10,2-alcanforsultam quiral.
La reacción del compuesto [9] con el compuesto [10] puede realizarse en un solvente apropiado en presencia de un ácido. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo alifático halogenado tal como cloruro de metileno, un hidrocarburo aromático tal como tolueno, un nitrilo tal como acetonitrilo, un éter tal como tetrahidrofurano y mezclas de los mismos. El ácido puede ser ácido trifluoroacético, por ejemplo.
La reacción de remoción del grupo auxiliar asimétrico del compuesto [11] puede realizarse de acuerdo con un método convencional, en un solvente apropiado, en presencia de agua junto con una base, por ejemplo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un alcohol tal como metanol y mezclas de los mismos. Ejemplos de bases incluyen hidróxido de sodio e hidróxido de litio.
Método de producción b-3 del intermediario
[Fórmula 19]

Donde un compuesto ópticamente activo se requiere para el compuesto [7] usado en el método de producción b-1 del intermediario descrito anteriormente, dicho compuesto ópticamente activo puede producirse también como sigue, por ejemplo.
El compuesto [9] se hace reaccionar con un compuesto representado por la fórmula general [12] (donde X7 representa un grupo protector de amino), para obtener un compuesto representado por la fórmula general [13] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
El compuesto [13] es convertido en un compuesto representado por la fórmula general [14] (donde X8 representa un grupo protector de amino diferente de X7 y los otros símbolos tienen el mismo significado como los descritos anteriormente), intercambiando el grupo protector de amino del compuesto [13] con otro grupo protector de amino. El grupo auxiliar asimétrico del compuesto [14] es removido para obtener un compuesto representado por la fórmula general [15] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
El grupo protector de amino es removido del compuesto [15] obtenido para obtener el compuesto [7], siendo protegido el grupo carboxilo.
El grupo protector de amino representado por X7 es de preferencia un grupo protector de amino que no es removido bajo condiciones ácidas, por ejemplo, un grupo bencilo.
El grupo protector de amino representado porX8 puede ser un grupo protector de amino diferente del grupo protector de amino X7 descrito anteriormente y en particular, es preferible un grupo capaz de ser removido con un ácido. Un ejemplo específico puede ser un grupo t-butoxicarbonilo.
La reacción del compuesto [9] con el compuesto [12] puede realizarse como en la reacción del compuesto [9] con el compuesto [10] en el método de producción b-2 del intermediario descrito anteriormente.
La reacción de sustitución del grupo protector de amino del compuesto [13] en el compuesto [14] puede realizarse de acuerdo con un método convencional. Donde X7 es un grupo bencilo, por ejemplo, la reacción de sustitución puede realizarse en un solvente apropiado en presencia de hidrógeno, un catalizador de paladio y un donador del grupo protector de amino. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un alcohol tal como etanol y mezclas de los mismos. El catalizador de paladio puede ser un catalizador de paladio soportado sobre carbón activado, por ejemplo. Donde X8 es un grupo t-butoxicarbonilo, por ejemplo, el donador del grupo protector de amino puede ser dicarbonato de di-tbutilo, por ejemplo.
La reacción de remoción del grupo auxiliar asimétrico del compuesto [14] puede realizarse como en la reacción de remoción del grupo auxiliar asimétrico del compuesto [11] en el método de producción b-2 del intermediario descrito anteriormente.
La reacción de conversión del compuesto [15] en el compuesto [7] puede realizarse en un solvente apropiado en presencia de un ácido. El solvente puede ser un alcohol tal como metanol, por ejemplo. El ácido puede ser ácido clorhídrico, por ejemplo. Esta reacción puede realizarse también usando una combinación de un alcohol tal como metanol y cloruro de tionilo.
Método de producción c del intermediario
[Fórmula 20]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto donde R3 y R4 son grupos que están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un anillo heterocíclico alifático que contenía nitrógeno sustituido con un grupo arilo o heteroarilo, cuyo compuesto es representado por la fórmula general [20] (donde cada r y s representa independientemente un entero de 1 o 2, la suma de r y s es 3 y los otros símbolos tienen los mismos significados como los descritos anteriormente), puede producirse como sigue, por ejemplo.
Un compuesto representado por la fórmula general [16] (donde X9 representa un grupo protector de amino y los otros símbolos tienen los mismos significados como los descritos anteriormente) se somete a una reacción de acoplamiento con un compuesto representado por la fórmula general [17] (donde X10 representa un grupo saliente y los otros símbolos tienen los mismos significados como los descritos anteriormente), para obtener un compuesto representado por la fórmula general [18] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
El compuesto [18] se somete a una reacción de hidrogenación catalítica para obtener un compuesto representado por la fórmula general [19] (donde los símbolos tienen los mismos significados como los descritos anteriormente). Un compuesto [20] puede obtenerse bajo agitación el grupo protector de amino del compuesto [19].
Ejemplos del grupo protector de amino representado porX9 incluyen grupos bencilo y t-butoxicarbonilo.
El grupo saliente representado por X10 puede ser un átomo de halógeno, por ejemplo.
La reacción de acoplamiento del compuesto [16] con el compuesto [17] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de un catalizador de paladio y una base, por ejemplo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen una amida tal como N,N-dimetilformamida, un éter tal como dioxano, un hidrocarburo aromático tal como tolueno, un alcohol tal como t-butanol y mezclas de los mismos. El catalizador de paladio puede ser aducto de diclorometano de dicloro[1,1'- bis(difenilfosfino)ferroceno]paladio (II), por ejemplo. La base puede ser carbonato de sodio, por ejemplo. La reacción de hidrogenación catalítica del compuesto [18] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de hidrógeno gaseoso y un catalizador de paladio, por ejemplo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un alcohol tal como etanol, un éter tal como tetrahidrofurano, un éster tal como acetato de etilo y mezclas de los mismos. El catalizador de paladio puede ser un catalizador de paladio soportado sobre carbón activado, por ejemplo.
La reacción de remoción del grupo protector de amino del compuesto [19] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción del intermediario descrito anteriormente.
Método de producción d del intermediario
[Fórmula 21]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [24] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
Un compuesto representado por la fórmula general [23] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse por la reacción de un compuesto representado por la fórmula general [21] (donde X11 representa un grupo protector de amino y los otros símbolos tienen los mismos significados como los descritos anteriormente) con un compuesto representado por la fórmula general [22] (donde X12 representa un grupo saliente y los otros símbolos tienen los mismos significados como los descritos anteriormente).
El compuesto [24] puede obtenerse bajo agitación el grupo protector de amino del compuesto [23] obtenido.
El grupo protector de amino representado por X11 puede ser un grupo t- butoxicarbonilo, por ejemplo.
El grupo saliente representado porX12 puede ser un átomo de halógeno (por ejemplo, un átomo de cloro o flúor), por ejemplo.
La reacción del compuesto [21] con el compuesto [22] puede realizarse en un solvente apropiado en presencia de una base. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo aromático tal como tolueno, un éter tal como tetrahidrofurano y mezclas de los mismos. La base puede ser bis(trimetilsilil)amida de potasio, por ejemplo.
La reacción de remoción del grupo protector de amino del compuesto [23] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción del intermediario descrito anteriormente.
Método de producción e-1 del intermediario [Fórmula 22]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [32] (donde Re1 representa un átomo de hidrógeno, átomo de halógeno, grupo alquilo o grupo alcoxi, Re2 representa un grupo alcoxi y los otros símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
Un compuesto representado por la fórmula general [25] (donde los símbolos tienen los mismos significados como los descritos anteriormente) se somete a una reacción de adición con un compuesto representado por la fórmula general [26] (donde X13 y X14 representan cada uno independientemente un grupo protector de carboxilo y el otro símbolo tiene el mismo significado como el descrito anteriormente), para producir un compuesto representado por la fórmula general [27] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
Entonces, el compuesto [27] se somete a una reacción de reducción y una reacción de cierre de anillo para producir un compuesto representado por la fórmula general [28] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
El compuesto [28] obtenido se somete a una reacción de reducción para producir un compuesto representado por la fórmula general [29] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
El compuesto [29] obtenido es convertido en un compuesto representado por la fórmula general [30] (donde X15 representa un grupo protector de amino y los otros símbolos tienen los mismos significados como los descritos anteriormente).
El grupo hidroxi del compuesto [30] obtenido es alquilizado para producir un compuesto representado por la fórmula general [31] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
Un compuesto [32] puede obtenerse bajo agitación el grupo protector de amino del compuesto [31].
Los grupos protectores de carboxilo representados por X13 y X14 pueden ser grupos alquilo, por ejemplo. Ejemplos específicos incluyen grupos metilo y etilo.
El grupo protector de amino representado porX15 puede ser un grupo t- butoxicarbonilo, por ejemplo.
La reacción de adición del compuesto [25] con el compuesto [26] puede realizarse como en la reacción del compuesto [3] con el compuesto [4] en el método de producción b-1 del intermediario descrito anteriormente. Del mismo modo, en esta reacción, la reacción de adición puede realizarse estereoselectivamente añadiendo un
catalizador quiral en el sistema de reacción. Ejemplos de catalizadores quirales incluyen 1 -[3,5-bis(trifluorometil)fenil]-3-[(1R,2R)-(-)-2-(dimetilamino)cidohexil]tiourea; 1-[3,5-bis(trifluorometil)fenil]-3-[(1S,2SH-)-2-(dimetilamino)cidohexil]tiourea y 6'-hidroxicinconina.
La reacción de reducción y la reacción de cierre de anillo del compuesto [27] pueden realizarse como en la reacción del compuesto [5] en el compuesto [6] en el método de producción b -1 del intermediario descrito anteriormente. La reacción de reducción del compuesto [28] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de un agente reductor. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un alcohol tal como etanol, un éter tal como tetrahidrofurano y mezclas de los mismos. El agente reductor puede ser borohidruro de sodio, por ejemplo.
La reacción de conversión del compuesto [29] en el compuesto [30] puede realizarse en un solvente apropiado en presencia de un agente alquilizante, un agente reductor y un donador del grupo protector. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo alifático halogenado tal como cloruro de metileno, un alcohol tal como metanol, un éter tal como tetrahidrofurano, un hidrocarburo aromático tal como tolueno y mezclas de los mismos. Ejemplos de agentes alquilizantes incluyen un tetrafluoroborato de trialquiloxonio tal como tetrafluoroborato de trimetiloxonio y trifluorometansulfonato de metilo. Ejemplos de agentes reductores incluyen borohidruro de sodio y un complejo de piridina-borano. El donador del grupo protector puede ser dicarbonato de di-t-butilo, por ejemplo.
La reacción de alquilación del grupo hidroxi en el compuesto [30] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de una base y un agente alquilizante, por ejemplo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen una amida tal como N,N-dimetilformamida, un éter tal como tetrahidrofurano y mezclas de los mismos. Ejemplos de bases incluyen hidruro de sodio, hidróxido de sodio y carbonato de potasio. El agente alquilizante puede ser un halogenuro de alquilo (por ejemplo yoduro de metilo o yoduro de etilo), por ejemplo.
La reacción de remoción del grupo protector de amino del compuesto [31] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [23] en el método de producción d del intermediario descrito anteriormente.
Método de producción e-2a del intermediario
[Fórmula 23]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, los compuestos representados por las fórmulas generales [39a] y [39a'] (donde Re3 representa un átomo de hidrógeno, grupo ciano o grupo alcoxi y los otros símbolos tienen los mismos significados como los descritos anteriormente) pueden producirse como sigue, por ejemplo.
Por la reacción de un compuesto representado por la fórmula general [33a] (donde X16 representa un grupo auxiliar asimétrico y los otros símbolos tienen los mismos significados como los descritos anteriormente) con un compuesto representado por la fórmula general [34] (donde X17 es un grupo protector de amino), puede obtenerse un compuesto representado por fórmula general [35a] o [35a'] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
Un compuesto representado por la fórmula general [36a] o [36a'] (donde X18 representa un grupo protector de amino diferente de X17 y los otros símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse convirtiendo el grupo protector de amino del compuesto [35a] o [35a'] obtenido.
Un compuesto representado por la fórmula general [37a] o [37a'] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo el compuesto [36a] o [36a'] obtenido a una reacción de reducción.
Un compuesto representado por la fórmula general [38a] o [38a'] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse convirtiendo de los compuestos [37a] o [37a'] obtenidos.
El compuesto representado por la fórmula [39a] o [39a'] puede producirse desprotegiendo el grupo protector de amino del compuesto [38a] o [38a'] obtenido.
Ejemplos del grupo auxiliar asimétrico representado por X16 incluyen 4-bencil-2- oxazolidinona quiral, 4-fenil-2-oxazolidinona quiral y 10,2-alcanforsultam quiral.
El grupo protector de amino representado porX17 puede ser un grupo bencilo, por ejemplo.
El grupo protector de amino representado por X18 puede ser un grupo protector de amino diferente de X17 y es de preferencia un grupo t-butoxicarbonilo, por ejemplo.
Cada una de las reacciones en el método de producción e-2 del intermediario, puede realizarse como sigue.
La reacción del compuesto [33a] con el compuesto [34] puede realizarse como en la reacción del compuesto [9] con el compuesto [12] en el método de producción b-3 del intermediario descrito anteriormente.
La reacción de conversión del compuesto [35a] en el compuesto [36a] o el compuesto [35a'] en el compuesto [36a'] puede realizarse como en la reacción de conversión del compuesto [13] en el compuesto [14] en el método de producción b-3 del intermediario descrito anteriormente.
La reacción de reducción del compuesto [36a] o [36a'] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de un agente reductor. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un alcohol tal como etanol, un éter tal como tetrahidrofurano, agua y mezclas de los mismos. El agente reductor puede ser borohidruro de sodio, por ejemplo.
Donde Re3 es un grupo alcoxi, la reacción de conversión del compuesto [37a] en el compuesto [38a] o el compuesto [37a'] en el compuesto [38a'] puede realizarse como en la reacción de conversión del compuesto [30] en el compuesto [31] en el método de producción e-1 del intermediario descrito anteriormente.
Donde Re3 es un grupo ciano, la reacción de conversión del compuesto [37a] en el compuesto [38a] o el compuesto [37a'] en el compuesto [38a'] puede realizarse en un solvente apropiado en presencia de un derivado de ácido azodicarboxílico, un derivado de fosfina y un donador del grupo ciano. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un hidrocarburo aromático tal como tolueno, un hidrocarburo alifático halogenado tal como cloruro de metileno y mezclas de los mismos. El derivado de ácido azodicarboxílico puede ser azodicarboxilato de dietilo, por ejemplo. El derivado de fosfina puede ser trifenilfosfina, por ejemplo. El donador del grupo ciano puede ser acetona cianohidrina, por ejemplo.
Donde Re3 es un átomo de hidrógeno, la reacción de conversión del compuesto [37a] en el compuesto [38a] o el compuesto [37a'] en el compuesto [38a'] puede realizarse en un solvente apropiado en presencia de un derivado de ácido azodicarboxílico, un derivado de fosfina y un agente de desoxigenación. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un hidrocarburo aromático tal como tolueno, un hidrocarburo alifático halogenado tal como cloruro de metileno y mezclas de los mismos. El derivado de ácido azodicarboxílico puede ser azodicarboxilato de dietilo, por ejemplo. El derivado de fosfina puede ser trifenilfosfina, por ejemplo. El agente de desoxigenación puede ser N'-isopropiliden-2-nitrobencensulfonohidrazida, por ejemplo.
La reacción de remoción del grupo protector de amino del compuesto [38a] o [38a'] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción del intermediario descrito anteriormente.
Método de producción e-2b del intermediario
[Fórmula 24]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [39b] (donde los símbolos tienen los mismos significados como los descritos anteriormente) y un compuesto representado por la fórmula general [39b'] (donde los símbolos tienen los mismos significados como los descritos anteriormente), pueden producirse a partir de un compuesto representado por la fórmula general [33b] (donde los símbolos tienen los mismos significados como los descritos anteriormente). Específicamente, esto puede realizarse tratando el compuesto [33b] como en la reacción de conversión del compuesto [33a] en el compuesto [39a] o compuesto [39a'], en el método de producción e-2a del intermediario descrito anteriormente. Donde sea necesario producir selectivamente el compuesto [39b] y el compuesto [39b'], un grupo auxiliar asimétrico apropiado puede seleccionarse como X4 del compuesto [33b]. En forma alternativa, después de que el compuesto [39b] y el compuesto [39b'] se obtienen como una mezcla, cada uno de los diastereómeros puede separarse en cualquier paso
subsiguiente. En este caso, la separación puede realizarse usando un método común, por ejemplo, cromatografía en columna de gel de sílice o cromatografía de líquidos.
Método de producción e-3 del intermediario
[Fórmula 25]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [42] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
Un compuesto representado por la fórmula general [40] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse bajo agitación el grupo protector de carboxilo del compuesto [28] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
Un compuesto representado por la fórmula general [41] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo el compuesto [40] a una reacción de descarboxilación.
El compuesto [42] puede obtenerse reduciendo el compuesto [41].
La reacción de conversión del compuesto [28] en el compuesto [40] puede realizarse como en la reacción de remoción del grupo protector de carboxilo del compuesto [8] en el método de producción b-1 del intermediario. La reacción de descarboxilación del compuesto [40] puede realizarse con calentamiento en un solvente apropiado. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo aromático tal como tolueno, una amida como N,N-dimetilformamida, sulfóxido de dimetilo y mezclas de los mismos. En esta reacción, puede añadirse un adyuvante de reacción para acelerar la reacción. El adyuvante de reacción puede ser ácido acético, por ejemplo.
La reacción de reducción del compuesto [41] puede realizarse como en la reacción de reducción del compuesto [6] en el método de producción b-1 del intermediario.
Método de producción f del intermediario
[Fórmula 26]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [50] (donde Rf representa un átomo de halógeno, grupo ciano o grupo alcoxi y los otros símbolos tienen los mismos significados como los descritos anteriormente) o fórmula general [50'] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
Por el tratamiento de un compuesto representado por la fórmula general [43] (donde X19 representa un grupo protector de amino, X20 representa un grupo saliente, X21 representa un grupo protector de hidroxi y n representa 1 o 2) con un donador de éster borónico, puede obtenerse un compuesto representado por la fórmula general [44] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
Un compuesto representado por la fórmula general [46] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo el compuesto [441 obtenido a una reacción de acoplamiento con un compuesto representado por la fórmula general [45] (donde X22 representa un grupo saliente y los otros símbolos tienen los mismos significados como los descritos anteriormente).
Un compuesto representado por la fórmula general [47] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse bajo agitación el grupo protector de hidroxi del compuesto [46] obtenido.
U n c o m p u e s to re p re s e n ta d o p o r la fó rm u la g e n e ra l [48 ] (d o n d e lo s s ím b o lo s t ie n e n lo s m is m o s s ig n if ic a d o s c o m o lo s
descritos anteriormente) o un compuesto representado por la fórmula general [48'] (donde los símbolos tienen los mismos significados como los descritos anteriormente), puede obtenerse reduciendo estereoselectivamente el compuesto [47] obtenido.
Un compuesto representado por la fórmula general [49] (donde los símbolos tienen los mismos significados como los descritos anteriormente) o un compuesto representado por la fórmula general [49'] (donde los símbolos tienen los mismos significados como los descritos anteriormente), pueden obtenerse convirtiendo el grupo hidroxi del compuesto [48] o [48'] obtenido en cualquiera de los varios sustituyentes.
El compuesto [50] o [50'] puede obtenerse bajo agitación el grupo protector de amino del compuesto [49] o [49'] obtenido.
El grupo protector de amino representado porX19 puede ser un grupo t- butoxicarbonilo, por ejemplo.
El grupo saliente representado por X20 puede ser un grupo trifluorometilsulfoniloxi, por ejemplo.
El grupo protector de hidroxi representado por X21 puede ser un grupo trialquilsililo tal como un grupo tbutildimetilsililo.
El grupo saliente representado por X22 puede ser un átomo de halógeno o grupo trifluorometilsulfoniloxi, por ejemplo. La reacción de conversión del compuesto [43] en el compuesto [44] puede realizarse por reacción con un donador de éster borónico en un solvente apropiado en presencia de un catalizador de paladio, una base y un ligando. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como 1,4-dioxano, una amida tal como N,N-dimetilformamida, un hidrocarburo aromático tal como tolueno, un alcohol tal como t-butanol y mezclas de los mismos. El catalizador de paladio puede ser aducto de diclorometano de dicloro[1,1'- bis(difenilfosfino)ferroceno]paladio (II), por ejemplo. El ligando puede ser 1,1'-bis(difenilfosfino)ferroceno, por ejemplo. La base puede ser acetato de potasio, por ejemplo. El donador de éster borónico puede ser bis(pinacolato)diboro, por ejemplo.
La reacción de acoplamiento del compuesto [44] con el compuesto [45] puede realizarse como en la reacción de acoplamiento del compuesto [16] con el compuesto [17] en el método de producción c del intermediario descrito anteriormente.
El grupo protector de hidroxi del compuesto [46] puede removerse de acuerdo con un método convencional, en un solvente apropiado en presencia de un agente desprotector, por ejemplo, dependiendo del tipo de grupo protector que será removido. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un hidrocarburo alifático halogenado tal como cloruro de metileno, un alcohol tal como metanol y mezclas de los mismos. El agente desprotector puede ser fluoruro de tetra-n-butilamonio, por ejemplo.
La reacción de reducción estereoselectiva del compuesto [47] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de hidrógeno gaseoso y un catalizador. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo aromático tal como tolueno, un hidrocarburo alifático halogenado tal como cloruro de metileno, un alcohol tal como metanol, un éter tal como tetrahidrofurano, un éster tal como acetato de etilo y mezclas de los mismos. Como el catalizador, un catalizador de paladio soportado sobre carbón activado, por ejemplo, puede usarse para obtener una forma cis tal como el compuesto [48] (por ejemplo, la forma (2R,4S) o la forma (2S,4r ) y el catalizador de Crabtree, por ejemplo, puede usarse para obtener una forma trans tal como el compuesto [48'] (por ejemplo, la forma (2S,4S) o la forma (2R,4R)).
Donde Rf es un grupo ciano o alcoxi, la reacción de conversión del compuesto [48] en el compuesto [49] o el compuesto [48'] en el compuesto [49'] puede realizarse como en la reacción de conversión del compuesto [37] en el compuesto [38] o el compuesto [37'] en el compuesto [38'] descrita en el método de producción e-2 del intermediario anterior.
Donde Rf es un átomo de flúor, la reacción de conversión del compuesto [48] en el compuesto [49] o el compuesto [48'] en el compuesto [49'] puede realizarse por reacción con un agente de fluoración en un solvente apropiado. El solvente puede ser cualquiera que no impida la reacción y puede ser un hidrocarburo alifático halogenado tal como cloruro de metileno, por ejemplo. Como el agente de fluoración puede usarse, por ejemplo, trifluoruro de (dietilamino)azufre.
La reacción de remoción del grupo protector de amino del compuesto [49] o [49'] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción a del intermediario descrito anteriormente.
Nótese que donde los compuestos [48] y [48'] se obtienen como una mezcla, pueden ser separados unos de otros en o después de este paso. La separación puede realizarse usando un método común, por ejemplo, cromatografía en columna de gel de sílice o cromatografía de líquidos.
Método de producción g del intermediario
[Fórmula 27]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [55] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
Por la yodación de un compuesto representado por la fórmula general [51] (donde X23 representa un grupo protector de amino), puede obtenerse un compuesto representado por la fórmula general [52] (donde el símbolo tiene el mismo significado como el descrito anteriormente).
Un compuesto representado por la fórmula general [54] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo el compuesto obtenido [52] a una reacción de acoplamiento con un compuesto representado por la fórmula general [53] (donde X24 representa un grupo saliente y los otros símbolos tienen los mismos significados como los descritos anteriormente).
El compuesto [55] puede obtenerse bajo agitación el grupo protector de amino del compuesto [54].
El grupo protector de amino representado por X23 puede ser un grupo t- butoxicarbonilo, por ejemplo.
El grupo saliente representado por X24 puede ser un átomo de halógeno (por ejemplo, un átomo de bromo), por ejemplo.
La reacción de yodación del compuesto [51] en el compuesto [52] puede realizarse en un solvente apropiado en presencia de yodo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo aromático tal como tolueno, un éter tal como tetrahidrofurano, un hidrocarburo alifático halogenado tal como cloruro de metileno y mezclas de los mismos. En esta reacción, puede añadirse un adyuvante de reacción para acelerar la reacción. Ejemplos de adyuvantes de reacción incluyen trifenilfosfina e imidazol.
La reacción de acoplamiento del compuesto [52] con el compuesto [53] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de un reactivo de zinc y un catalizador de paladio, por ejemplo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen una amida tal como dimetilacetamida, un hidrocarburo aromático tal como tolueno, un éter tal como tetrahidrofurano y mezclas de los mismos. Ejemplos de reactivos de zinc incluyen zinc en polvo y cloruro de zinc (II). El catalizador de paladio puede ser aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio (II), por ejemplo. En esta reacción, puede añadirse un adyuvante de reacción para acelerar la reacción. El adyuvante de reacción puede ser yoduro de cobre, por ejemplo.
La reacción de remoción del grupo protector de amino del compuesto [54] puede realizarse como en la reacción de
remoción del grupo protector de amino del compuesto [2] en el método de producción a del intermediario descrito anteriormente.
Método de producción h del intermediario
[Fórmula 28]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [59] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
Al someter un compuesto representado por la fórmula general [56] (donde X25 representa un grupo protector de amino) a una reacción de sustitución con un compuesto representado por la fórmula general [57] (donde los símbolos tienen los mismos significados como los descritos anteriormente), puede obtenerse un compuesto representado por la fórmula general [58] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
El compuesto [59] puede obtenerse bajo agitación el grupo protector de amino del compuesto [58].
El grupo protector de amino representado porX25 puede ser un grupo t- butoxicarbonilo, por ejemplo.
La reacción de sustitución del compuesto [56] con el compuesto [57] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de una base. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen una amida tal como N-metilpirrolidona, un nitrilo tal como acetonitrilo, un éter tal como 1,4- dioxano, sulfóxido de dimetilo y mezclas de los mismos. La base puede ser carbonato de potasio, por ejemplo.
La reacción de remoción del grupo protector de amino del compuesto [58] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción a del intermediario descrito anteriormente.
Método de producción i del intermediario
[Fórmula 29]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto donde R3 es un grupo alquilo sustituido con un grupo arilo opcionalmente sustituido, o un grupo alquilo sustituido con un grupo heteroarilo opcionalmente sustituido y R4 es un átomo de hidrógeno o un grupo alquilo, cuyo compuesto es representado por la fórmula general [63] (donde Ri representa un grupo arilo opcionalmente sustituido o un grupo heteroarilo opcionalmente sustituido y t representa un entero de 1 a 3), puede producirse como sigue, por ejemplo.
A l s o m e te r un c o m p u e s to re p re s e n ta d o p o r la fó rm u la g e n e ra l [60 ] (d o n d e lo s s ím b o lo s t ie n e n lo s m is m o s
significados como los descritos anteriormente) a una reacción de aminación reductiva con un compuesto representado por la fórmula general [61] (donde X26 representa un grupo protector de amino o hidrógeno y el otro símbolo tiene el mismo significado como el descrito anteriormente), puede obtenerse un compuesto representado por la fórmula general [62] (donde los símbolos tienen los mismos significados como los descritos anteriormente). El compuesto [63] puede obtenerse bajo agitación posteriormente el grupo protector de amino del compuesto [62] obtenido, según se requiera.
El grupo protector de amino representado por X26 puede ser un grupo bencilo, por ejemplo.
La reacción de aminación reductiva del compuesto [60] con el compuesto [61] puede realizarse como en la reacción de aminación reductiva del compuesto [a] con el compuesto [b] o [c] en el método de síntesis A-1 descrito anteriormente.
La reacción de remoción del grupo protector de amino del compuesto [62] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción a del intermediario descrito anteriormente.
Método de producción j del intermediario
[Fórmula 30]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [69] (donde Rj representa un grupo alcoxi opcionalmente sustituido y los otros símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
Un compuesto representado por la fórmula general [64] (donde X27 representa un grupo saliente, X28 representa un grupo protector de hidroxi y los otros símbolos tienen los mismos significados como los descritos anteriormente) se hace reaccionar con un compuesto representado por la fórmula general [65] (donde X29 representa un grupo protector de amino), para obtener un compuesto representado por la fórmula general [66] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
Un compuesto representado por la fórmula general [67] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse convirtiendo el compuesto [66].
Un compuesto representado por la fórmula general [68] (donde Rj representa un grupo alcoxi opcionalmente sustituido y los otros símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse alquilizando el grupo hidroxi del compuesto [67].
El compuesto [69] puede obtenerse bajo agitación del grupo protector de amino del compuesto [68].
El grupo saliente representado por X27 puede ser un átomo de halógeno, por ejemplo.
El grupo protector de hidroxi representado porX28 es de preferencia un grupo bencilo.
El grupo protector de amino representado porX29 puede ser un grupo t- butoxicarbonilo o bencilo, por ejemplo. La reacción del compuesto [64] con el compuesto [65] puede realizarse en un solvente apropiado en presencia de un reactivo de metalización. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un hidrocarburo alifático tal como hexano y mezclas de los mismos. El reactivo de metalización se puede n-butil litio, por ejemplo.
La reacción de conversión del compuesto [66] en el compuesto [67] puede realizarse en un solvente apropiado en presencia de un trialquilsilano, un ácido, hidrógeno y un catalizador de paladio. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo alifático halogenado tal como cloruro de metileno, un hidrocarburo aromático tal como tolueno y mezclas de los mismos. El trialquilsilano puede ser trietilsilano, por ejemplo. El ácido puede ser ácido trifluoroacético, por ejemplo. El catalizador de paladio puede ser un catalizador de paladio soportado sobre carbón activado, por ejemplo.
La reacción de alquilación del grupo hidroxi del compuesto [67] puede realizarse como en la reacción de alquilación del compuesto [30] en el método de producción e-1 del intermediario descrito anteriormente.
La reacción de remoción del grupo protector de amino del compuesto [68] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción a del intermediario descrito anteriormente.
Método de producción k del intermediario
[Fórmula 31]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [72] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
Un compuesto representado por la fórmula general [71] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse por la reacción de un compuesto representado por la fórmula general [70] (donde los símbolos tienen los mismos significados como los descritos anteriormente) con el compuesto representado por la fórmula general [12] (donde el símbolo tiene el mismo significado como el descrito anteriormente).
El compuesto [72] puede obtenerse bajo agitación el grupo protector de amino del compuesto [71].
La reacción del compuesto [70] con el compuesto [12] puede realizarse en un solvente apropiado en presencia de un ácido. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo alifático halogenado tal como cloruro de metileno, un hidrocarburo aromático tal como tolueno, un nitrilo tal como acetonitrilo, un éter tal como tetrahidrofurano y mezclas de los mismos. El ácido puede ser ácido trifluoroacético, por ejemplo.
La reacción de remoción del grupo protector de amino del compuesto [72] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción a del intermediario descrito anteriormente.
Método de producción l del intermediario
[Fórmula 32]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [77] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
Por la protección de un grupo lactama de un compuesto representado por la fórmula general [73] (donde los símbolos tienen los mismos significados como los descritos anteriormente), puede obtenerse un compuesto representado por la fórmula general [74] (donde X30 representa un grupo protector de lactama y los otros símbolos tienen los mismos significados como los descritos anteriormente).
Un compuesto representado por la fórmula general [75] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse por la fluoración del compuesto [74].
Un compuesto representado por la fórmula general [76] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse bajo agitación el grupo protector de lactama del compuesto [75] obtenido. El compuesto [77] puede obtenerse sometiendo el compuesto [76] a una reacción reductiva.
El grupo protector de lactama representado porX30 puede ser un grupo t- butoxicarbonilo, por ejemplo.
La reacción de introducción del grupo protector de lactama en el compuesto [73] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de un donador del grupo protector de lactama. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un nitrilo tal como acetonitrilo, un hidrocarburo alifático halogenado tal como cloruro de metileno, un hidrocarburo aromático tal como tolueno, un éter tal como tetrahidrofurano y mezclas de los mismos. El donador del grupo protector puede ser dicarbonato de di-t-butilo, por ejemplo. En esta reacción, puede añadirse un adyuvante de reacción para acelerar la reacción. El adyuvante de reacción puede ser 4-dimetilaminopiridina, por ejemplo.
La reacción de fluoración del compuesto [74] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de un agente de fluoración y una base, por ejemplo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un hidrocarburo alifático halogenado tal como cloruro de metileno y mezclas de los mismos. El agente de fluoración puede ser N- fluorobencensulfonimida, por ejemplo. La base puede ser bis(trimetilsilil)amida de litio, por ejemplo. La reacción de remoción del grupo protector de amino del compuesto [75] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción a del intermediario descrito anteriormente.
La reacción de reducción del compuesto [76] puede realizarse como en la reacción de reducción del compuesto [6] en el método de producción b-1 del intermediario.
Método de producción m del intermediario
[Fórmula 33]
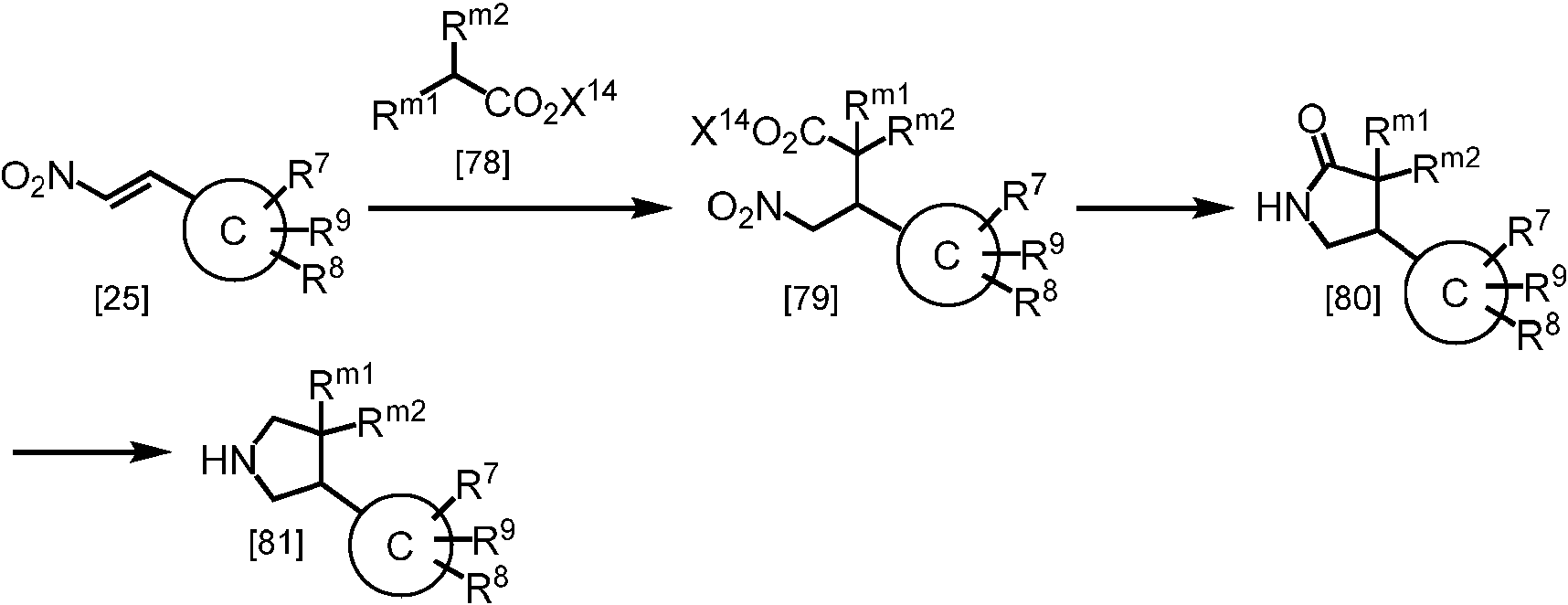
Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [81] (donde Rm1 y Rm2 representan cada uno independientemente un grupo alquilo o alcoxialquilo) puede producirse como sigue, por ejemplo.
Un compuesto representado por la fórmula general [79] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo el compuesto representado por la fórmula general [25] (donde los símbolos tienen los mismos significados como los descritos anteriormente) a una reacción de adición con un compuesto representado por la fórmula general [78] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
Un compuesto representado por la fórmula general [80] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo el compuesto [79] a una reacción de reducción y una reacción de cierre de anillo.
El compuesto representado por la fórmula general [81] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo el compuesto [80] obtenido a una reacción de reducción. La reacción de adición del compuesto [25] con el compuesto [78] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de un agente alquilizante y una base, por ejemplo. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un hidrocarburo alifático tal como hexano y mezclas de los mismos. El agente alquilizante puede ser isobutirato de etilo, por ejemplo. La base puede ser diisopropilamida de litio, por ejemplo.
La reacción de reducción y la reacción de cierre de anillo del compuesto [79] pueden realizarse como en la reacción de cierre de anillo del compuesto [5] en el método de producción b-1 del intermediario descrito anteriormente.
La reacción de reducción del compuesto [80] puede realizarse como en la reacción de reducción del compuesto [6] en el método de producción b-1 del intermediario descrito anteriormente.
Método de producción n del intermediario
[Fórmula 34]
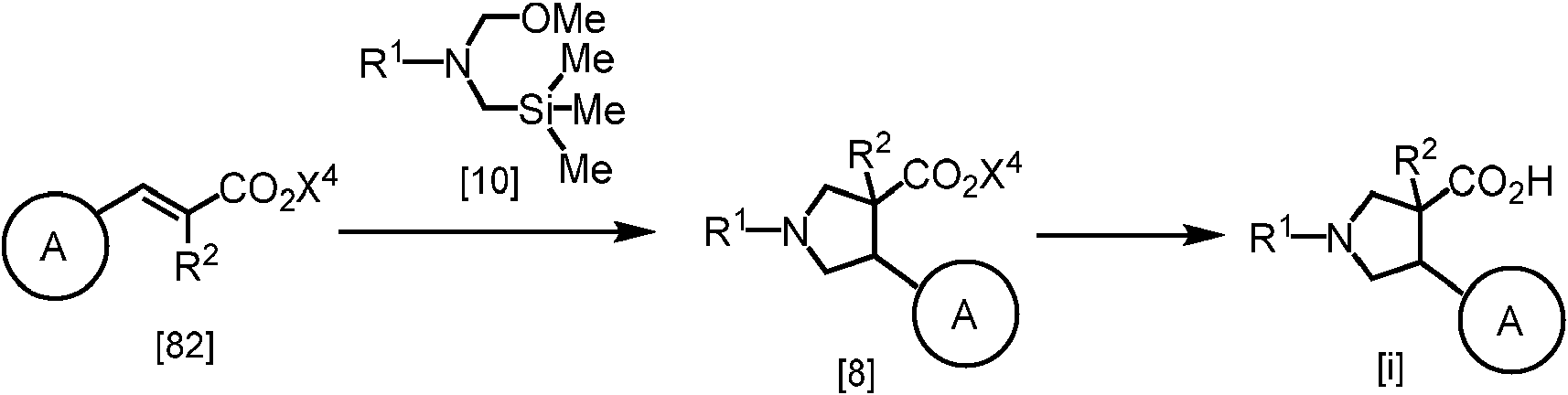
El compuesto [i] usado en el método de síntesis B descrito anteriormente puede producirse también como sigue, por ejemplo.
El c o m p u e s to re p re s e n ta d o p o r la fó rm u la g e n e ra l [8 ] (d o n d e lo s s ím b o lo s t ie n e n lo s m is m o s s ig n if ic a d o s c o m o lo s
descritos anteriormente) puede obtenerse por la reacción de un compuesto representado por la fórmula general [82] (donde los símbolos tienen los mismos significados como los descritos anteriormente) con el compuesto representado por la fórmula general [10] (donde el símbolo tiene el mismo significado como el descrito anteriormente).
El compuesto [i] puede obtenerse bajo agitación el grupo protector de carboxilo del compuesto [8] obtenido.
La reacción del compuesto [82] con el compuesto [10] puede realizarse como en la reacción del compuesto [9] con el compuesto [10] en método de producción b-2 del intermediario descrito anteriormente.
La reacción de remoción del grupo protector de carboxilo del compuesto [8] para formar el compuesto [i] puede realizarse como en la reacción del compuesto [8] en el compuesto [i] en el método de producción b-1 del intermediario descrito anteriormente.
Método de producción o del intermediario
[Fórmula 35]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [88] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
Un compuesto representado por la fórmula general [84] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo el compuesto representado por la fórmula general [64] (donde los símbolos tienen los mismos significados como los descritos anteriormente) a una reacción de acoplamiento con un compuesto representado por la fórmula general [83] (donde el símbolo tiene el mismo significado como el descrito anteriormente).
Un compuesto representado por la fórmula general [85] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse bajo agitación el grupo protector de hidroxi del compuesto [84] obtenido. Un compuesto representado por la fórmula general [86] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse convirtiendo el grupo hidroxi del compuesto [85] obtenido en un grupo saliente (por ejemplo, un grupo trifluorometansulfoniloxi).
Un compuesto representado por la fórmula general [87] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse por la reacción del compuesto [86] obtenido con un compuesto de ácido borónico deseado o un éster del mismo.
El compuesto representado por la fórmula general [88] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse bajo agitación el grupo protector de amino del compuesto [87] obtenido. La reacción de acoplamiento del compuesto [64] con el compuesto [83] puede realizarse como en la reacción del compuesto [a] en el compuesto [Ic] en el método de síntesis A-3 descrito anteriormente.
La reacción de conversión del compuesto [84] en el compuesto [85] puede realizarse de acuerdo con un método convencional, por ejemplo, en un solvente apropiado en presencia de hidrógeno y un catalizador de paladio. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un alcohol tal como metanol, un éster tal como acetato de etilo y mezclas de los mismos. El catalizador de paladio puede ser un catalizador de paladio soportado sobre carbón activado, por ejemplo.
La reacción de conversión del compuesto [85] en el compuesto [86] puede realizarse de acuerdo con un método convencional, por ejemplo, por reacción con un anhídrido de ácido en un solvente apropiado en presencia de una base. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un hidrocarburo alifático halogenado tal como cloruro de metileno, un nitrilo tal como acetonitrilo, un hidrocarburo aromático tal como tolueno y mezclas de los mismos. Ejemplos de bases incluyen una amina tal como diisopropiletilamina y un carbonato de metal alcalino tal como carbonato de potasio. El anhídrido de ácido puede ser anhídrido trifluorometansulfónico.
El paso de conversión del compuesto [86] en el compuesto [87] puede realizarse de acuerdo con un método convencional, por ejemplo, por reacción con un compuesto de ácido borónico deseado o un éster del mismo en un solvente apropiado en presencia de un catalizador de paladio y una base. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como 1,4-dioxano, una amida tal como N,N-dimetilformamida, un hidrocarburo aromático tal como tolueno, un alcohol tal como t-butanol y mezclas de los mismos. El catalizador de paladio puede ser aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio (II), por ejemplo. La base puede ser carbonato de sodio o acetato de potasio, por ejemplo.
La reacción de remoción del grupo protector de amino del compuesto [87] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción a del intermediario descrito anteriormente.
Método de producción p del intermediario
[Fórmula 36]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [94] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
Un compuesto representado por la fórmula general [91] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo un compuesto representado por la fórmula general [89] (donde los símbolos tienen los mismos significados como los descritos anteriormente) a una reacción de acoplamiento con un compuesto representado por la fórmula general [90] (donde el símbolo tiene el mismo significado como el descrito anteriormente).
U n c o m p u e s to re p re s e n ta d o p o r la fó rm u la g e n e ra l [92 ] (d o n d e X 31 re p re s e n ta un á to m o d e h a ló g e n o y lo s o tro s s ím b o lo s t ie n e n lo s m is m o s s ig n if ic a d o s c o m o lo s d e s c r ito s a n te r io rm e n te ) p u e d e o b te n e rs e h a lo g e n a n d o el
compuesto [91] obtenido.
Un compuesto representado por la fórmula general [93] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse por la reacción del compuesto [92] obtenido con un compuesto de ácido borónico deseado o un éster del mismo.
El compuesto representado por la fórmula general [94] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse bajo agitación el grupo protector de amino del compuesto [93] obtenido. La reacción de acoplamiento del compuesto [89] con el compuesto [90] puede realizarse como en la reacción del compuesto [a] en el compuesto [Ic] en el método de síntesis A-3 descrito anteriormente.
La reacción de halogenación del compuesto [91] en el compuesto [92] puede realizarse de acuerdo con un método convencional, por reacción con un agente halogenante en un solvente apropiado. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un alcohol tal como metanol, un hidrocarburo alifático halogenado tal como cloroformo y mezclas de los mismos. El agente halogenante puede ser N-bromosuccinimida, por ejemplo.
La reacción de conversión del compuesto [92] en el compuesto [93] por reacción con un compuesto de ácido borónico deseado o un éster del mismo puede realizarse como en la reacción del compuesto [86] en el compuesto [87] en el método de producción o del intermediario descrito anteriormente.
La reacción de remoción del grupo protector de amino del compuesto [93] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción a del intermediario descrito anteriormente.
Método de producción q del intermediario
[Fórmula 37]

Entre los compuestos [j] usados en el método de síntesis B descrito anteriormente, un compuesto representado por la fórmula general [101] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
U n c o m p u e s to re p re s e n ta d o p o r la fó rm u la g e n e ra l [96 ] (d o n d e lo s s ím b o lo s t ie n e n lo s m is m o s s ig n if ic a d o s c o m o lo s
descritos anteriormente) puede obtenerse por la reacción de un compuesto representado por la fórmula general [95] (donde el símbolo tiene el mismo significado como el descrito anteriormente) con el compuesto representado por la fórmula general [64] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
Un compuesto representado por la fórmula general [97] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse por la conversión del grupo hidroxi del compuesto [96] obtenido.
Un compuesto representado por la fórmula general [98] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse bajo agitación el grupo protector de hidroxi del compuesto [97] obtenido. Un compuesto representado por la fórmula general [99] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse convirtiendo el grupo hidroxi del compuesto [98] obtenido en un grupo saliente.
Un compuesto representado por la fórmula general [100] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse por la reacción del compuesto [99] obtenido con un compuesto de ácido borónico deseado o un éster del mismo.
El compuesto representado por la fórmula general [101] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse bajo agitación el grupo protector de amino del compuesto [100] obtenido.
La reacción del compuesto [95] con el compuesto [64] puede realizarse en un solvente apropiado en presencia de un reactivo de metalización. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un hidrocarburo alifático tal como hexano y mezclas de los mismos. El reactivo de metalización puede ser bromuro de isopropilmagnesio, por ejemplo. En esta reacción, puede añadirse un adyuvante de reacción para acelerar la reacción. El adyuvante de reacción puede ser yoduro de cobre, por ejemplo. La reacción de conversión del compuesto [96] en el compuesto [97] puede realizarse como en la reacción de conversión del compuesto [30] en el compuesto [31] en el método de producción e-1 del intermediario descrito anteriormente.
La reacción de conversión del compuesto [97] en el compuesto [98] puede realizarse como en la reacción del compuesto [84] en el compuesto [85] en el método de producción o del intermediario descrito anteriormente.
La reacción de conversión del compuesto [98] en el compuesto [99] puede realizarse como en la reacción del compuesto [85] en el compuesto [86] en el método de producción o del intermediario descrito anteriormente.
La reacción de conversión del compuesto [99] en el compuesto [100] puede realizarse como en la reacción del compuesto [86] en el compuesto [87] en el método de producción o del intermediario descrito anteriormente.
La reacción de remoción del grupo protector de amino del compuesto [100] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción a del intermediario descrito anteriormente.
Método de producción r del intermediario
[Fórmula 38]

Una materia prima para el ejemplo 340 puede producirse como sigue de acuerdo con la reacción química mostrada anteriormente, por ejemplo.
Un compuesto representado por la fórmula general [103] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo un compuesto [102] (donde los símbolos tienen los mismos significados como los descritos anteriormente) a una reacción de reducción.
Un compuesto representado por la fórmula general [104] (donde Xr1 representa un grupo saliente y los otros símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse por la conversión del compuesto [103] obtenido.
Un compuesto representado por la fórmula general [105] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse por la conversión del compuesto [104] obtenido.
Un compuesto representado por la fórmula general [106] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo el compuesto [105] obtenido a una reacción de reducción. Un compuesto representado por la fórmula general [107] (donde el símbolo tiene el mismo significado como el descrito anteriormente) puede obtenerse bajo agitación el grupo protector de amino del compuesto [106] obtenido. El grupo saliente representado por X puede ser un grupo metilsulfoniloxi, por ejemplo.
La reacción de reducción del compuesto [102] puede realizarse de acuerdo con un método convencional, por reacción con un agente reductor en un solvente apropiado. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un alcohol tal como metanol, agua y mezclas de los mismos. El agente reductor puede ser borohidruro de sodio, por ejemplo.
La reacción de conversión del compuesto [103] en el compuesto [104] puede realizarse en un solvente apropiado en presencia de un agente de sulfonilación y una base. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un nitrilo tal como acetonitrilo, un hidrocarburo alifático halogenado tal como cloruro de metileno y mezclas de los mismos. El agente de sulfonilación puede ser cloruro de metansulfonilo, por ejemplo. La base puede ser diisopropiletilamina, por ejemplo.
La reacción de conversión del compuesto [104] en el compuesto [105] puede realizarse por calentamiento en un solvente apropiado en presencia de una base. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un nitrilo tal como acetonitrilo, un hidrocarburo alifático halogenado tal como cloruro de metileno, un hidrocarburo aromático tal como tolueno y mezclas de los mismos. La base puede ser 1,8-diazabiciclo[5.4.0]-7-undeceno, por ejemplo.
La reacción de hidrogenación catalítica del compuesto [105] puede realizarse como en la reacción de hidrogenación catalítica del compuesto [18] en el método de producción c del intermediario descrito anteriormente.
La reacción de remoción del grupo protector de amino del compuesto [106] puede realizarse como en la reacción de remoción del grupo protector de amino del compuesto [2] en el método de producción a del intermediario descrito
anteriormente.
Método de producción s del intermediario
[Fórmula 39]

El compuesto representado por la fórmula general [43] (donde los símbolos tienen los mismos significados como los descritos anteriormente) en el método de producción f del intermediario descrito anteriormente puede producirse como sigue, por ejemplo.
Un compuesto representado por la fórmula general [109] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo un compuesto [108] (donde los símbolos tienen los mismos significados como los descritos anteriormente) a una reacción de reducción.
Un compuesto representado por la fórmula general [110] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse por la conversión del compuesto [109] obtenido.
Un compuesto representado por la fórmula general [111] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse sometiendo el compuesto [110] obtenido a una reacción de oxidación. El compuesto representado por la fórmula general [43] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse por la conversión del compuesto [111] obtenido.
La reacción de reducción del compuesto [108] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de un agente reductor. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un alcohol tal como metanol y mezclas de los mismos. El agente reductor puede ser borohidruro de sodio o borohidruro de litio, por ejemplo.
La reacción de conversión del compuesto [109] en el compuesto [110] puede realizarse de acuerdo con un método convencional, dependiendo del tipo del grupo protector. Por ejemplo, la conversión en un compuesto que tiene un grupo trialquilsililo como X21 puede realizarse en un solvente apropiado en presencia de un agente sililante y una base. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo alifático halogenado tal como cloruro de metileno, un nitrilo tal como acetonitrilo, una amida tal como N,N-dimetilformamida, un éter tal como tetrahidrofurano y mezclas de los mismos. El agente sililante puede ser cloruro de t-butildimetilsililo, por ejemplo.
La reacción de oxidación del compuesto [110] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de un agente de reacción de radical y un agente oxidante. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un hidrocarburo alifático halogenado tal como cloruro de metileno, un nitrilo tal como acetonitrilo, agua y mezclas de los mismos. El agente de reacción de radical puede ser 2,2,6,6-tetrametilpiperidin-1-oxilo, por ejemplo. El agente oxidante puede ser ácido tricloroisocianúrico o ácido meta-clorobenzoico, por ejemplo.
La reacción de conversión del compuesto [111] en el compuesto [43] puede realizarse en un solvente apropiado en presencia de un agente de sulfonilación y una base. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un hidrocarburo aromático tal como tolueno y mezclas de los mismos. El agente de sulfonilación puede ser N-fenilbis(trifluorometansulfonimida), por ejemplo. La base puede ser bis(trimetilsilil)amida de sodio, por ejemplo.
Método de producción t del intermediario
[Fórmula 40]

Entre los compuestos representados por la fórmula general [109] en el método de producción s del intermediario descrito anteriormente, un compuesto representado por la fórmula general [114] (donde el símbolo tiene el mismo significado como el descrito anteriormente) puede producirse como sigue, por ejemplo.
Un compuesto representado por la fórmula general [113] (donde el símbolo tiene el mismo significado como el descrito anteriormente) puede obtenerse sometiendo un compuesto representado por la fórmula general [112] (donde el símbolo tiene el mismo significado como el descrito anteriormente) a una reacción de reducción.
El compuesto representado por la fórmula general [114] (donde el símbolo tiene el mismo significado como el descrito anteriormente) puede obtenerse por la conversión del grupo protector de amino del compuesto [113] obtenido.
La reacción de reducción del compuesto [112] puede realizarse de acuerdo con un método convencional, en un solvente apropiado en presencia de un agente reductor. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un alcohol tal como metanol y mezclas de los mismos. El agente reductor pueden ser borohidruro de sodio o borohidruro de litio, por ejemplo.
La reacción de conversión del compuesto [113] en el compuesto [114] puede realizarse como en la reacción de conversión del compuesto [13] en el compuesto [14] en el método de producción b-3 del intermediario descrito anteriormente.
Método de producción u del intermediario
[Fórmula 41]

Entre los compuestos representados por la fórmula [53] en el método de producción g del intermediario descrito anteriormente, un compuesto representado por la fórmula general [116] (donde X24a representa un átomo de halógeno; Ru1 es un grupo alquilo opcionalmente sustituido y Ru2 es un átomo de hidrógeno o un grupo alquilo, Ru1 y Ru2 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un anillo heterocíclico alifático opcionalmente sustituido que contenía nitrógeno y los otros símbolos tienen los mismos significados como los descritos anteriormente) puede producirse como sigue, por ejemplo.
El compuesto representado por la fórmula general [116] (donde los símbolos tienen los mismos significados como los descritos anteriormente) puede obtenerse halogenando un compuesto representado por la fórmula general [115] (donde los símbolos tienen los mismos significados como los descritos anteriormente).
La reacción de halogenación del compuesto [115] puede realizarse de acuerdo con un método convencional, por reacción con un agente halogenante en un solvente apropiado. El solvente puede ser cualquiera que no impida la reacción y ejemplos de dichos solventes incluyen un éter tal como tetrahidrofurano, un alcohol tal como metanol, un hidrocarburo alifático halogenado tal como cloroformo y mezclas de los mismos. El agente halogenante puede ser N-bromosuccinimida, por ejemplo.
Los compuestos como materia prima para su uso en los métodos descritos anteriormente pueden producirse como en los métodos existentes y/o los métodos descritos más adelante en los ejemplos.
Nótese que la introducción de grupos protectores en los grupos funcionales y la remoción de los grupos protectores de grupos funcionales pueden realizarse con relación a los métodos existentes (véase, por ejemplo, "PROTECTIVE GROUPS IN ORGANIC SYNTHESIS" (por Theodora W. Greene y Peter G. M. Wuts)).
Además, los compuestos de la presente invención y los compuestos intermediarios producidos usando los métodos descritos anteriormente pueden ser convertidos estructuralmente además en otros compuestos objetivo o intermediarios, usando los métodos descritos más adelante en los ejemplos y/o los métodos existentes, o combinaciones de los mismos. Ejemplos específicos incluyen los siguientes métodos.
(1) Conversión de un grupo alcoxicarbonilo en un grupo carboxilo
Un grupo alcoxicarbonilo puede ser convertido en un grupo carboxilo correspondiente por hidrólisis con una base de hidróxido de metal alcalino tal como hidróxido de sodio o hidróxido de potasio; un grupo benciloxicarbonilo puede ser convertido en un grupo carboxilo correspondiente por hidrogenólisis a través de tratamiento con carbón de paladio en una atmósfera de hidrógeno; o un grupo t-butoxicarbonilo puede ser convertido en un grupo carboxilo correspondiente por tratamiento con un ácido como ácido clorhídrico o ácido trifluoroacético.
(2) Conversión de un grupo carboxilo en un grupo carbamoílo
Un grupo carboxilo puede ser convertido en un grupo carbamoílo correspondiente, por ejemplo, por la reacción del grupo carboxilo o una sal del mismo con una amina en presencia de un agente de condensación, o por la conversión del grupo carboxilo o una sal del mismo en un halogenuro de acilo y entonces por la reacción del producto resultante con una amina.
(3) Conversión de un grupo ciano en un grupo tetrazolilo
Un grupo ciano puede ser convertido en un grupo tetrazolilo correspondiente por la reacción del grupo ciano con azida de tributilestaño.
(4) Conversión de un grupo ciano en un grupo oxadiazolilo
Un grupo ciano puede ser convertido en un grupo oxadiazolilo correspondiente por la reacción del grupo ciano con clorhidrato de hidroxilamina en presencia de una base, por ejemplo y entonces por la reacción del producto de reacción con 1,1-carbonildiimidazol.
(5) Conversión de un grupo amino en un grupo carbamoilamino
Un grupo amino puede ser convertido en un grupo carbamoilamino correspondiente por la reacción del grupo amino con un isocianato deseado, o por la reacción del grupo amino con un halogenuro de carbamoílo deseado.
(6) Conversión de un grupo amino en un grupo sulfonilamino
Un grupo amino puede ser convertido en un grupo sulfonilamino correspondiente por la reacción del grupo amino con un halogenuro de sulfonilo deseado.
(7) Conversión de un grupo amino en un grupo aminosulfonilamino
Un grupo amino puede ser convertido en un grupo aminosulfonilamino correspondiente por la reacción del grupo amino con un halogenuro de aminosulfonilo deseado.
(8) Conversión de un grupo hidroxi en un átomo de halógeno
Un grupo hidroxi puede ser convertido en un átomo de halógeno correspondiente por la reacción del grupo hidroxi con un agente de fluoración tal como trifluoruro de (dietilamino)azufre o un agente de cloración tal como cloruro de tionilo, por ejemplo.
(9) Conversión de un grupo hidroxi en un grupo ciano
Un grupo hidroxi puede ser convertido en un grupo ciano correspondiente por la reacción del grupo hidroxi con acetona cianohidrina, por ejemplo.
(10) Conversión de un grupo hidroxi en un grupo alcoxi
Un grupo hidroxi puede ser convertido en un grupo alcoxi correspondiente por la reacción del grupo hidroxi con un agente alquilizante deseado en presencia de una base. En forma alternativa, un grupo hidroxi puede ser convertido en un grupo alcoxi correspondiente por la reacción del grupo hidroxi con un alcohol deseado en presencia de azodicarboxilato de dietilo.
(11) Conversión de un grupo hidroxi en un grupo arilo
Un grupo hidroxi puede ser convertido en un grupo arilo correspondiente por la conversión del grupo hidroxi en un grupo saliente (por ejemplo, un grupo trifluorometilsulfoniloxi) de acuerdo con un método convencional y entonces acoplando el producto resultante con un halogenuro de arilo deseado.
(12) Conversión de un grupo formilo en un grupo hidroximetilo
Un grupo formilo puede ser convertido en un grupo hidroximetilo reduciendo el grupo formilo con borohidruro de sodio.
(13) Conversión de un átomo de halógeno en un grupo cicloalquilo
Un átomo de halógeno puede ser convertido en un grupo cicloalquilo correspondiente por el acoplamiento de un compuesto que tiene un átomo de halógeno con un ácido cicloalquilborónico o un éster borónico de cicloalquenilo, por ejemplo, seguido de reducción con hidrógeno y carbón de paladio, por ejemplo. En forma alternativa, un átomo de halógeno puede ser convertido en un grupo cicloalquilo correspondiente por la reacción de un compuesto que tiene un átomo de halógeno con alquil litio y por la reacción del producto de reacción con una cicloalcanona correspondiente y entonces reduciendo el grupo hidroxi producido con un trialquilsilano en presencia de un ácido. (14) Conversión de un grupo carbonilo en un grupo amino
Un grupo carbonilo puede ser convertido en un grupo amino por la reacción del grupo carbonilo con una amina deseada en presencia de un agente reductor.
(15) Conversión de un átomo de halógeno en un grupo alquilo
Un átomo de halógeno puede ser convertido en un grupo alquilo correspondiente por el acoplamiento de un compuesto que tiene un átomo de halógeno con un derivado de ácido alquilborónico o un derivado de ácido alquenilbórico, por ejemplo, seguido de reducción con hidrógeno y carbón de paladio, por ejemplo. En forma alternativa, un átomo de halógeno puede ser convertido en un grupo alquilo correspondiente por la reacción de un compuesto que tiene un átomo de halógeno con alquil litio y por la reacción del producto de reacción con una alcanona correspondiente y entonces reduciendo el grupo hidroxi producido con un trialquilsilano en presencia de un ácido.
(16) Conversión de un grupo formilo en un grupo dihalogenuro de alquilo
Un grupo formilo puede ser convertido en un grupo dihalogenuro de alquilo correspondiente por la reacción del grupo formilo con un agente de fluoración tal como trifluoruro de bis(2-metoxietil)aminoazufre, por ejemplo.
(17) Conversión de un grupo sulfuro en un grupo sulfonilo
Un grupo sulfuro puede ser convertido en un grupo sulfonilo correspondiente por la reacción del grupo sulfuro con un agente oxidante tal como Oxone, por ejemplo.
(18) Conversión de un grupo hidroximetilo en un grupo carboxilo
Un grupo hidroxialquilo puede ser convertido en un grupo carboxilo correspondiente por la reacción del grupo hidroxialquilo con un agente oxidante.
(19) Conversión de un grupo hidroxi en un grupo cicloalquilo
Un grupo hidroxi puede ser convertido en un grupo cicloalquilo correspondiente por la conversión del grupo hidroxi en un grupo saliente (por ejemplo, un grupo trifluorometilsulfoniloxi), acoplando el producto resultante con un derivado de ácido cicloalquenilborónico y entonces reduciendo el grupo alquenilo con hidrógeno y carbón de paladio, por ejemplo.
(20) Conversión de un grupo carbamoílo en un grupo ciano
Un grupo carbamoílo puede ser convertido en un grupo ciano correspondiente por la reacción del grupo carbamoílo con un agente deshidratante tal como cloruro cianúrico, por ejemplo.
(21) Conversión de un grupo alcoxicarbonilalquinilo en un grupo 3-hidroxiisoxazolilo
Un grupo alcoxicarbonilalquinilo puede ser convertido en un grupo 3-hidroxiisoxazolilo correspondiente por la reacción del grupo alcoxicarbonilalquinilo con clorhidrato de hidroxilamina en presencia de una base.
(22) Conversión de un grupo carbonilo en un grupo hidroxi
Un grupo carbonilo puede ser convertido en un grupo hidroxi por la reducción del grupo carbonilo con borohidruro de sodio.
(23) Conversión de un grupo carboxilo en un grupo hidroximetilo
Un grupo carboxilo puede ser convertido en un grupo hidroximetilo por la activación del grupo carboxilo con cloroformiato de isobutilo y entonces reduciendo el producto resultante con borohidruro de sodio. En forma alternativa, un grupo carboxilo puede ser convertido en un grupo hidroximetilo reduciendo el grupo carboxilo con hidruro de litio y aluminio.
(24) Conversión de un grupo alquenileno en un grupo alquileno
Un grupo alquenileno puede ser convertido en un grupo alquileno correspondiente por la reducción del grupo alquenileno con hidrógeno y carbón de paladio, por ejemplo.
(25) Conversión de un grupo ciano en un grupo formilo
Un grupo ciano puede ser convertido en un grupo formilo correspondiente por la reacción del grupo ciano con un agente reductor tal como hidruro de diisobutilaluminio, por ejemplo.
(26) Conversión de acetal en un grupo alcoxi
El acetal puede ser convertido en un grupo alcoxi correspondiente por la reacción del acetal con borano en presencia de trifluorometansulfonato de trimetilsililo, por ejemplo.
(27) Conversión de un átomo de halógeno en un grupo alquenilo
Un átomo de halógeno puede ser convertido en un grupo alquenilo correspondiente por el acoplamiento de un compuesto que tiene un átomo de halógeno con un compuesto que tiene un grupo alquenilo tal como un derivado de ácido alquenilcarboxílico o un derivado de ácido alquenilborónico, por ejemplo.
(28) Conversión de un átomo de halógeno en un grupo arilo
Un átomo de halógeno puede ser convertido en un grupo arilo correspondiente por el acoplamiento de un compuesto que tiene un átomo de halógeno con un ácido arilborónico, por ejemplo.
(29) Conversión de un grupo formilo en un grupo alcoxicarbonilalquenilo
Un grupo formilo puede ser convertido en un grupo alcoxicarbonilalquenilo correspondiente por la reacción del grupo formilo con un (alcoxicarbonilmetilen)trifenilfosforano deseado o un fosfonoacetato de trialquilo deseado.
Los compuestos de la presente invención producidos como se describió anteriormente o los compuestos como materia prima de los mismos son aislados y purificados en su forma libre o en la forma de sus sales. Las sales pueden producirse sometiendo los compuestos a un tratamiento de formación de la sal que se usa comúnmente. El aislamiento y la purificación pueden realizarse a través de la aplicación de operaciones químicas comunes tales como extracción, concentración, cristalización, filtración, recristalización y varios tipos de cromatografía.
Donde cualquiera de los compuestos de la presente invención o una sal farmacéuticamente aceptable de los mismos existen como un isómero óptico basado en el carbono asimétrico, el compuesto puede separarse en los isómeros ópticos individuales usando medios comunes para resolución óptica (por ejemplo, un método de cristalización fraccional o un método de resolución usando una columna quiral). Además, los isómeros ópticos pueden sintetizarse usando materiales de partida ópticamente puros. Los isómeros ópticos también pueden sintetizarse realizando estereoselectivamente cada una de las reacciones usando un grupo auxiliar asimétrico o un catalizador asimétrico.
Ejemplos
Ejemplo 1
[Fórmula química 42]

A una solución del compuesto 1 (142 mg) y el compuesto 2 (26 jl) en diclorometano (2 ml) se le añadió ácido acético (20 jl), la mezcla se agitó a temperatura ambiente por 10 minutos, se añadió a la misma triacetoxiborohidruro de sodio (74 mg) y se agitó por 16 horas. A la mezcla se le añadió una solución acuosa saturada de carbonato ácido de sodio y agua, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 80:20-50:50), para dar el 1-[2-(1-{[(3R,4R)-3-fluoro-4-(4-metoxifenil)-1-(tetrahidro-2H-piran-4-il)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperidin-4-carboxilato de etilo 3 (154 mg) como un polvo incoloro. MS (ESI): m/z 690 [M+H]+ Ejemplo 2
[Fórmula química 43]

A una solución del compuesto 1 (80 mg) y el compuesto 2 (528 jl) en diclorometano (2 ml) se le añadió ácido acético (76 jl) y triacetoxiborohidruro de sodio (336 mg) y se agitó y se calentó bajo reflujo por 16 horas. A la mezcla se le añadió una solución acuosa saturada de carbonato ácido de sodio y agua, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de
etilo = 95:5-70:30), para dar el 1-[2-(1-{[(3R,4R)-1-ciclopropil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperidin-4-carboxilato de etilo 3 (29 mg) como un polvo incoloro. MS (APCI): m/z 646 [M+H]+
Ejemplo 3
[Fórmula química 44]

Una suspensión del compuesto 1 (150 mg), bromoacetato de t-butilo (44 jl), diisopropiletilamina (66 jl) y yoduro de sodio (19 mg) en acetonitrilo (2 ml), se agitó bajo atmósfera de nitrógeno a 80 °C por 16 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se añadió agua a la misma, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 85:25-60:40), para dar el 1-[2-(1-{[(3R,4R)-1-(2-ferc-butoxi-2-oxoetil)-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperidin-4-carboxilato de etilo 2 (159 mg) como un polvo incoloro. MS (ESI): m/z 720 [M+H]+
Ejemplo 4
[Fórmula química 45]

Una solución mixta del compuesto 1 (150 mg), acrilato de t-butilo (44 jl) y trietilamina (53 jl) en etanol (2 ml) se agitó a 80 °C por 16 horas. A la mezcla se le añadió acrilato de t-butilo (88 jl) y trietilamina (106 jl) y se agitó por otras 4 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se añadió agua a la misma, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 90:10-60:40), para dar el 1-[2-(1-{[(3R,4R)-1-(3-ferc-butoxi-3-oxopropil)-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperidin-4-carboxilato de etilo 2 (89 mg) como un polvo incoloro. MS (ESI): m/z 734 [M+H]+
Ejemplo 5
[F ó rm u la q u ím ic a 46 ]

Una solución mixta del compuesto 1 (100 mg), el compuesto 2 (31 mg), tris(dibencilidenoacetona)dipaladio(0) (15 mg), (±)-2,2-bis(difenilfosfino)-1,1-binaftilo (20 mg) y t-butóxido de sodio (46 mg) en tolueno (2 ml) se agitó bajo atmósfera de nitrógeno a 110 °C por 21 horas. A la mezcla se le añadió el compuesto 2 (21 mg) y se agitó por otras 6 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se añadió agua a la misma, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 40:60-0:100), para dar el 1-{2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-4-(4-metoxifenil)-1-(2-metilpirimidin-4-il)pirrolidin-3-il]carbonil}-4-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxilato de metilo 3 (36 mg) como un polvo anaranjado pálido.
MS (APCI): m/z 714 [M+H]+
Ejemplo 6
[Fórmula química 47]

(1) Una solución del compuesto 1 (100 mg), el compuesto 2 (73 mg) y diisopropiletilamina (197 jl) en tetrahidrofurano (2 ml) se calentó bajo reflujo bajo atmósfera de nitrógeno por 4 horas. La solución de reacción se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 80:20-50:50), para dar el compuesto 3 (107 mg) como un material viscoso incoloro. MS (APCI): m/z 358 [M+H]+
(2) A una solución del compuesto 3 (106 mg) en etanol (2 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 297 jl) a temperatura ambiente y la mezcla se agitó a 70 °C por 6 horas. A la mezcla se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 297 jl) y entonces la solución de reacción se concentró bajo presión reducida para dar el compuesto 4 como un polvo incoloro (138 mg) que contenía cloruro de sodio. MS (APCI): m/z 344 [M+H]+
(3) El compuesto 4 (65 mg), el compuesto 5 (50 mg), clorhidrato de 1-etil-3-(3- dimetilaminopropil)carbodiimida (55 mg) y 1-hidroxi-7-azabenzotriazol (39 mg) se añadieron a N,N-dimetilformamida (2 ml) y la mezcla se agitó a temperatura ambiente por 13 horas. A la mezcla se le añadió una solución acuosa saturada de carbonato ácido de sodio y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 70:30-40:60), para dar el 1-{5-fluoro-2-[(3S,5S)-1-{[(3R,4R)-3-metoxi-4-(4-metoxifenil)-1-(2-metilpirimidin-4-il)pirrolidin-3-il]carbonil}-5-(metoximetil)pirrolidin-3-il]fenil}piperidin-4-carboxilato de metilo 6 (64 mg) como un polvo incoloro. MS (APCI): m/z 676 [M+H]+
Ejemplo 7
[F ó rm u la q u ím ic a 48 ]

(1) A una mezcla del compuesto 1 (300 mg) en cloroformo se le añadió una solución acuosa de hidróxido de sodio (1 mol/l, 1,03 ml) y agua, se agitó y entonces se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida. Una mezcla del residuo, el compuesto 2 (333 mg), tris(dibencilidenoacetona)dipaladio(0) (95 mg), (±)-2,2'-bis(difenilfosfino)-1,1 '-binaftilo (129 mg) y t-butóxido de sodio (299 mg) en tolueno (4 ml), se agitó bajo atmósfera de nitrógeno a 100 °C por 15 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se diluyó con agua, una solución acuosa de ácido clorhídrico (1 mol/l) se añadió a la misma para neutralizar la mezcla y se filtró. El filtrado se lavó con cloroformo y la capa acuosa resultante se concentró bajo presión reducida para dar el compuesto 3 como un material crudo (344 mg) que contenía otros materiales tales como cloruro de sodio.
(2) El compuesto 3 (68 mg), el compuesto 4 (68 mg), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (49 mg), 1-hidroxi-7-azabenzotriazol (35 mg) y trietilamina (53 pl) se añadieron a N,N-dimetilformamida (2 ml) y la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 70:30-40:60), para dar el 1-{2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-4-(4-metoxifenil)-1-(2-metilpirimidin-4-il)pirrolidin-3-il]carbonil}-5-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxilato de metilo 5 (28 mg) como un polvo incoloro. MS (APCI): m/z 714 [M+H]+
Ejemplo 8
[Fórmula química 49]

A una solución del compuesto 1 (90 mg) y diisopropiletilamina (40 pl) en diclorometano (2 ml) se le añadió gota a gota una suspensión de cloruro de metilaminoformilo (17 mg) en diclorometano (1 ml) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 1 hora. A la mezcla se le añadió tetrahidrofurano (1 ml), acetonitrilo (1 ml), cloruro de metilaminoformilo (51 mg) y diisopropiletilamina (120 pl) y se agitó por otras 2 horas. A la mezcla de reacción se le añadió agua, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 40:60-0:100), para dar el 1-[2-(1-{[(3R,4R)-1-carbamoil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperidin-4-carboxilato de etilo 2 (77 mg) como un polvo incoloro. MS (ESI): m/z 649 [M+H]+
Ejemplo 9
[F ó rm u la q u ím ic a 50 ]

A una solución del compuesto 1 (200 mg) y trietilamina (157 |jl) en diclorometano (8 ml) se le añadió isocianato de trimetilsililo (894 jl) bajo agitación y entonces la mezcla se agitó a temperatura ambiente por 17 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato de sodio (5 ml) y agua (80 ml), se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-90:10), para dar el 1-[2-(1-{[(3R,4R)-3-fluoro-4-(4-metoxifenil)-1-(metilcarbamoil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperidin-4-carboxilato de etilo 2 (192 mg) como un polvo incoloro. MS (ESI): m/z 663 [M+H]+
Ejemplo 10
[Fórmula química 51]

El compuesto 1 (116 mg), el compuesto 2 (120 mg), clorhidrato de 1-etil-3-(3- dimetilaminopropil)carbodiimida (85,5 mg), 1-hidroxi-7-azabenzotriazol (60,7 mg) y trietilamina (109,7 jl) se añadieron a N,N-dimetilformamida (2,4 ml) y la mezcla se agitó a temperatura ambiente por 5 horas. A la mezcla se le añadió una solución acuosa saturada de carbonato ácido de sodio y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-90:10), para dar el 1-[2-(1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperidin-4-carboxilato de etilo 3 (181 mg) como un material viscoso incoloro. MS (ESI): m/z 6 62 [M+H]+
Ejemplo 11
[Fórmula química 52]

A una solución del compuesto 1 (181 mg) en etanol (2 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 1,09 ml) a temperatura ambiente y la mezcla se agitó por 12 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 1,09 ml) y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice diol (gel de sílice esférico 30 g, SHOKO SCIENTIFIC Purif-Pack (marca registrada)) (cloroformo:metanol = 100:0-95:5). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 254 jl) y el solvente se evaporó bajo presión reducida. El residuo se pulverizó con el éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de ácido 1-[2-(1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperidin-4-carboxílico 2 (134 mg) como un polvo incoloro. MS (APCI): m/z 634 [M+H]+
Ejemplos 12 A 39
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 1 anterior y posteriormente el ejemplo 11, para dar cada compuesto incluido en el cuadro 1 siguiente.
[Los ejemplos que no están incluidos dentro del alcance de la presente invención están marcados con un "*"]
r 1

continuación

continuación

continuación

(continuación)

Ejemplo 40
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 2 anterior y posteriormente el ejemplo 11, para dar cada compuesto incluido en el cuadro 2 siguiente.
r 2

Ejemplo 41
[Fórmula química 53]

(1) El compuesto 1 (150 mg) y el compuesto 2 (45 mg) se trataron de una manera similar al ejemplo 1, para dar el compuesto 3 (180 mg) como un polvo incoloro. MS (APCI) m/z 762 [M+H]+
(2) A una solución del compuesto 3 (180 mg) en tetrahidrofurano (1,2 ml) se le añadió gota a gota una solución acuosa de ácido clorhídrico (2 moles/l, 1,2 ml) a temperatura ambiente y entonces la mezcla se agitó por 15 horas. A la mezcla se le añadió gota a gota una solución acuosa de hidróxido de sodio (2 moles/l, 2,4 ml) bajo enfriamiento en hielo y entonces se agitó por 2 horas a temperatura ambiente. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 1,2 ml) y se concentró bajo presión reducida. El residuo se suspendió en cloroformo, se filtró y el filtrado se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-85:15). A una solución del compuesto resultante en 1,4-dioxano (1 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 71 ^l) y el solvente se concentró bajo presión reducida. El residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de ácido 1-{2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-4-(4-metoxifenil)-1-(4-oxociclohexil)pirrolidin-3-il]carbonil}-4-(metoxifenil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico 4 (81 mg) como un polvo incoloro. MS (ESI): m/z 704 [M+H]+
Ejemplos 42 a 46
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 3 anterior y posteriormente el ejemplo 11, para dar cada compuesto incluido en el cuadro 3 siguiente.
r

Ejemplos 47 a 64
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 5 anterior y posteriormente el ejemplo 11, para dar cada compuesto incluido en el cuadro 4 siguiente.
r 4

continuación

(continuación)

Ejemplos 65 a 66
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 6 anterior y posteriormente el ejemplo 11, para dar cada compuesto incluido en el cuadro 5 siguiente.
r

Ejemplos 67 a 68
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 7 anterior y posteriormente el ejemplo 11, para dar cada compuesto incluido en el cuadro 6 siguiente.
ADR

Ejemplo 69
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 8 anterior y posteriormente el ejemplo 11, para dar cada compuesto incluido en el cuadro 7 siguiente.

Ejemplo 70
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 9 anterior y posteriormente el ejemplo 11, para dar cada compuesto incluido en el cuadro 8 siguiente.
[Cuadro 8]

Ejemplos 71 a 202
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 10 anterior y posteriormente el ejemplo 11, para dar cada compuesto incluido en el cuadro 9 siguiente.
r

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

(continuación)

Ejemplos 203 a 211
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 10 anterior. A una solución del compuesto resultante en diclorometano se le añadió 2 o 3 equivalentes o más de una solución de ácido clorhídrico en acetato de etilo (4 moles/l). La mezcla de reacción se concentró bajo presión reducida y el residuo se pulverizó con éter diisopropílico etc., se colectó por filtración y se secó bajo presión reducida para dar cada compuesto incluido en el cuadro 10 siguiente.
Cuadro 10

(continuación)

Ejemplo 212a y ejemplo 212b
[Fórmula química 54]

(1) El compuesto 1 (90 mg) y el compuesto 2 (85 mg) se trataron de una manera similar al ejemplo 10, para dar el compuesto 3 (106 mg). MS (ESI): m/z 716 [M+H]+
(2) El compuesto 3 (100 mg) se purificó con c La R QUIRAL (CHIRALPAK IE (20 x 250 mm) fabricada por dA iCEL CORPORATION, fase móvil: hexano/etanol/dietilamina= 75/25/0,1, magnitud de flujo: 10 ml/min), para dar los compuestos ópticamente activos 4 (43 mg) y 5 (42 mg) como polvos incoloros. Compuesto 4: MS (ESI): m/z 716 [M+H]+ (los tiempos de retención de los compuestos 4 y 5 en la CLAR fueron 10,23 minutos y 11,64 minutos, respectivamente, con CHIRALPAK IE-3 (4,6 x 150 mm) fabricada por DAICEL CORPORATION, bajo la condición de fase móvil: hexano/etanol/dietilamina= 75/25/0,1, magnitud de flujo: 0,500 ml/min, temperatura de la columna: 25 °C).
(3) A una solución del compuesto 4 (43 mg) en cloroformo (1 ml) se le añadió ácido trifluoroacético (300 pl) bajo enfriamiento en hielo y entonces la mezcla se agitó por 2 horas a temperatura ambiente. La mezcla de reacción se diluyó con cloroformo y entonces se añadió a la misma una solución acuosa de hidróxido de sodio (1 mol/l) para neutralizar la mezcla en pH = 7. La capa orgánica se separó, se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-85:15). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 25 pl) y entonces el solvente se concentró bajo presión reducida. El residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el estereoisómero 6 de diclorhidrato de ácido 1-[2-(1-{[(3R,4R)-1-ciclopentil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4,4-dimetilpirrolidin-3-il)-5-(trifluorometil)fenil]piperidin-4-carboxílico (30 mg) como un polvo incoloro (ejemplo 212a). MS (ESI): m/z 660 [M+H]+
(4) El compuesto 5 se trató de una manera similar al compuesto (3) anterior, para dar el otro estereoisómero 7 de diclorhidrato de ácido 1-[2-(1-{[(3R,4R)-1-ciclopentil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4,4-dimetilpirrolidin-3-il)-5-(trifluorometil)fenil]piperidin-4- carboxílico (ejemplo 212b). MS (ESI): m/z 660 [M+H]+
Ejemplos 213 a 221
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 212a anterior, para dar cada compuesto incluido en el cuadro 11 siguiente.
r 11

continuación

Ejemplo 222
[Fórmula química 55]

(1) El compuesto 1 y el compuesto 2 se trataron de una manera similar al ejemplo 10 anterior, para dar el compuesto 3 (138 mg). (ESI): m/z 727 [M+H]+
(2) A una solución del compuesto 3 en metanol (6 ml) se le añadió carbón de paladio a 10% (humedecido con aproximadamente 50% de agua, 41 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 3 horas. La mezcla de reacción se filtró y entonces el filtrado se concentró bajo presión reducida. A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 100 jl). La mezcla de reacción se concentró bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el clorhidrato de ácido 4-[2-(1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenoxi]-2,2-dimetilbutanoico 4 (127 mg) como un polvo incoloro. MS (APCI): m/z 637 [M+H]+ Ejemplo 223
[Fórmula química 56] 12

(1) El compuesto 1 y el compuesto 2 se trataron de una manera similar al ejemplo 10 anterior, para dar el compuesto 3 (300 mg). MS (ESl): m/z 879 [M+H]+
(2) A una solución del compuesto 3 en tetrahidrofurano (2 ml) se le añadió una solución acuosa (1 ml) de hidróxido de litio (monohidrato) (17 mg) a temperatura ambiente y la mezcla se agitó por 20 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (1 mol/l, 410 jl), entonces se añadieron agua y diclorometano a la misma y se agitó. La capa orgánica se separó, se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 98:2-80:20), para dar el ácido (3R,4S)-4-{2-[4-(terc-butoxicarbonil)piperidin-1-il]-4-(trifluorometil)fenil}-1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}pirrolidin-3-carboxílico 4 (204 mg) como un polvo incoloro. MS (ESl): m/z 720 [M+H]+
Ejemplo 224
[Fórmula química 57]

(1) A una solución del compuesto 1 (116 mg) en 1,2-dimetoxietano (2 ml) se le añadió N-metilmorfolina (20 |jl) y cloroformiato de isobutilo (23 jl) bajo atmósfera de nitrógeno y bajo enfriamiento en hielo y la mezcla se agitó a la misma temperatura por 1 hora. La materia insoluble precipitada se removió por filtración y al filtrado se le añadió una solución acuosa (1 ml) de borohidruro de sodio (9 mg) bajo atmósfera de nitrógeno y bajo enfriamiento en hielo y la mezcla se agitó a la misma temperatura por 2 horas. A la mezcla de reacción se le añadió agua y acetato de etilo, se agitó, entonces la capa orgánica se separó, se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 80:20-30:70), para dar el compuesto 2 (97 mg) como un polvo incoloro. MS (ESI): m/z 706 [M+H]+
(2) A una solución del compuesto 2 (94 mg) en diclorometano (1 ml) se le añadió ácido trifluoroacético (500 jl) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 3 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para neutralizar la mezcla en pH = 7 y se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 98:2-75:25). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 67 jl) y entonces el solvente se evaporó bajo presión reducida. El residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de ácido 1-{2-[(3S,4R)-1-{[(3R,4R)-1-terc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4-(hidroximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico 3 (84 mg) como un polvo incoloro. MS (ESl): m/z 650 [m H]+
Ejemplo 225
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 224 anterior, para dar el compuesto incluido en el cuadro 12 siguiente.
r 12

Ejemplo 226
[Fórmula química 58]

(1) A una solución del compuesto 1 (80 mg) en N,N-dimetilformamida (2 ml) se le añadió dimetilamina (2 moles/l de solución en tetrahidrofurano, 220 jl), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (84 mg), 1-hidroxi-7-azabenzotriazol (60 mg) y trietilamina (61 jl) y la mezcla se agitó a temperatura ambiente por 6 horas.
A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 70:30-20:80), para dar el compuesto 2 (55 mg) como un polvo incoloro. MS (ESI): m/z 747 [M+H]+
(2) A una solución del compuesto 2 (53 mg) en diclorometano (2 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 6 ml) y la mezcla se agitó a temperatura ambiente por 4 horas. La solución de reacción se concentró bajo presión reducida y el residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de ácido 1-{2-[(3S,4R)-1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4-(dimetilcarbamoil)pirrolidin-3-ii]-5-(trifluorometil)fenil}piperidin-4-carboxílico 3 (50 mg) como un polvo incoloro. Ms (ESI): m/z 691 [M+H]+
Ejemplo 227
[Fórmula química 59]

(1) A una solución del compuesto 1 (140 mg) en N,N-dimetilformamida (2 ml) se le añadió cloruro de amonio (20 mg), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (73 mg), 1-hidroxi-7-azabenzotriazol (52 mg) y trietilamina (106 pl) y la mezcla se agitó a temperatura ambiente por 64 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-94:6), para dar el compuesto 2 (129 mg) como un polvo incoloro. MS (ESI): m/z 719 [M+H]+
(2) A una solución del compuesto 2 (128 mg) en N,N-dimetilformamida (2 ml) se le añadió cloruro cianúrico bajo enfriamiento en hielo (99 mg) y la mezcla se agitó a la misma temperatura por 1 hora. A la mezcla de reacción se le añadió agua enfriada en hielo y acetato de etilo, se agitó y la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-93:7), para dar el compuesto 3 (118 mg) como un polvo incoloro. MS (ESI): m/z 701 [M+H]+
(3) A una solución del compuesto 3(117 mg) en diclorometano (1 ml) se le añadió ácido trifluoroacético (500 pl) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 3 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para neutralizar la mezcla en pH = 7 y se extrajo con diclorometano. La capa orgánica se separó, se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 98:2-85:15). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 84 pl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de ácido 1-{2-[(3S,4R)-1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4-cianopirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico 4 (100 mg) como un polvo incoloro. MS (APCI): m/z 645 [M+H]+
Ejemplo 228
[Fórmula química 60] 1

(1) A una solución del compuesto 1 (150 mg), la cual se obtuvo tratando un compuesto de partida correspondiente de una manera similar al ejemplo 223 anterior, en N,N-dimetilformamida (2 ml), se le añadió yoduro de metilo (19 pl) y carbonato de potasio (55 mg) y la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió agua y acetato de etilo, se agitó y entonces la capa orgánica se separó. La
capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 90:10-60:40), para dar el compuesto 2 (111 mg) como un polvo incoloro. MS (ESI): m/z 746 [M+H]+
(2) A una solución del compuesto 2 (110 mg) en tetrahidrofurano (2 ml) se le añadió gota a gota bromuro de metilmagnesio (3 moles/ml, 172 jl) bajo atmósfera de nitrógeno y bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 4 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de cloruro de amonio, se agitó y entonces se añadió acetato de etilo a la misma. La capa orgánica se separó, se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 80:20-50:50), para dar el compuesto 3 (30 mg) como un polvo incoloro. MS (ESI): m/z 746 [M+H]+
(3) A una solución del compuesto 3 (29 mg) en diclorometano (1 ml) se le añadió ácido trifluoroacético (500 jl) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 17 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para neutralizar la mezcla en pH = 7 y se extrajo con diclorometano. Las capas orgánicas se combinaron, se lavaron con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-85:15). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 20 jl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de ácido 1-{2-[(3S,4R)-1-{[(3R,4R)-1-ciclopentil-3- fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4-(2-hidroxipropan-2-il)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4- carboxílico 4 (18 mg) como un polvo incoloro. Ms (ESI): m/z 690 [M+H]+
Ejemplo 229
[Fórmula química 61] 1

(1) El compuesto 1 y el compuesto 2 se trataron de una manera similar al ejemplo 10 anterior, para dar el compuesto 3 (93 mg). MS (APCI) m/z 819 [M+H]+
(2) A una solución del compuesto 3 en 1,4-dioxano (1 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 500 jl). La mezcla se agitó a temperatura ambiente por 2 horas y entonces el solvente se evaporó bajo presión reducida. Al residuo se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizar la mezcla y se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-94:6), para dar el compuesto 4. MS (ESI): m/z 704 [M+H]+
(3) A una solución del compuesto 4 en etanol (1 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 220 jl) y la mezcla se agitó a temperatura ambiente por 2 horas. Una solución acuosa de ácido clorhídrico (2 moles/l, 220 jl) se añadió a la misma y entonces la solución de reacción se concentró bajo presión reducida. Al residuo se le añadió cloroformo y agua, la mezcla se agitó, entonces la capa orgánica se separó, se secó y se concentró bajo presión reducida. A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 55 jl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de ácido 1-[2-(1-{[(3R,4R)-3-fluoro-1-(trans-4-hidroxiciclohexil)-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperidin-4-carboxílico 5 (65 mg) como un polvo incoloro. MS (ESI): m/z 676 [M+H]+
Ejemplo 230
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 229 anterior, para dar el compuesto incluido en el cuadro 13 siguiente.
r 1

Ejemplo 231
[Fórmula química 62]

A una solución del compuesto 1 (101 mg) en diclorometano (2 ml) se le añadió clorhidrato de metilamina (21 mg), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (61 mg), 1-hidroxi-7-azabenzotriazol (65 mg) y trietilamina (111 Ml) y la mezcla se agitó a temperatura ambiente por 4 horas. A la mezcla se le añadió clorhidrato de metilamina (21 mg), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (61 mg), 1-hidroxi-7-azabenzotriazol (44 mg) y trietilamina (111 Ml) y se agitó a temperatura ambiente por 15 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-90:10). A una solución del compuesto resultante en cloroformo (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 100 Ml), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de 1-[2-(1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]-N-metilpiperidin-4-carboxamida 2 (50 mg) como un polvo incoloro. MS (ESI): m/z 647 [M+H]+
Ejemplos 232 a 267
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 231 anterior, para dar cada compuesto incluido en el cuadro 14 siguiente.
r 14

Ċ
continuación

(continuación)

(continuación)

(continuación)

continuación

Ejemplo 268
[Fórmula química 63]

(1) El compuesto 1 (60 mg), clorhidrato de éster metílico de glicina (15 mg), clorhidrato de 1 -etil-3-(3-dimetilaminopropil)carbodiimida (31 mg), 1-hidroxi-7-azabenzotriazol (22 mg) y trietilamina (45 pl) se añadieron a N,N-dimetilformamida (1 ml) y la mezcla se agitó a temperatura ambiente por 3 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 60:40-30:70), para dar el compuesto 2 (57 mg) como un material viscoso incoloro. MS (APCI): m/z 747 [M+H]+
(2) A una solución del compuesto 2 (56 mg) en metanol (1 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 300 jl) a temperatura ambiente y la mezcla se agitó por 1 hora. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 300 jl) y entonces se concentró bajo presión reducida. El residuo se suspendió en acetato de etilo, se filtró y el filtrado se concentró bajo presión reducida. A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídricoacetato de 1,4-etilo (4 moles/l, 94 jl) y entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de N-[(1-{2-[(3S,4R)-1-{[(3R,4R)-1-ciclopentil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-il)carbonil]glicina 3 (82 mg) como un polvo incoloro. MS (APCI): m/z 733 [M+H]+
Ejemplos 269 a 271
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 268 anterior, para dar cada compuesto incluido en el cuadro 15 siguiente.
r 1

Ejemplo 272
[Fórmula química 64]

dimetilsulfamida (62 mg), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (96 mg) y 4-dimetilaminopiridina (16 mg) y la mezcla se agitó a temperatura ambiente por 65 horas. La solución de reacción se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 98:2-91:9). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 63 jl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó en un solvente mixto de éter diisopropílico y heptano, se colectó por filtración y se secó bajo presión reducida para dar el clorhidrato de 4-[2-(1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenoxi]-N-(dimetilsulfamoil)butanamida 2 (159 mg) como un polvo incoloro. MS (APCI): m/z 715 [M+H]+
Ejemplos 273 a 276
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 272 anterior, para dar cada compuesto incluido en el cuadro 16 siguiente.
r 1

Ejemplo 277
[F ó rm u la q u ím ic a 65 ]

(1) Una solución del compuesto 1 (200 mg), carbazato de t-butilo (66 mg), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (127 mg), 1-hidroxi-7-azabenzotriazol (90 mg) y trietilamina (138 jl) en N,N-dimetilformamida (2 ml), se agitó a temperatura ambiente por 65 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y acetato de etilo, se agitó, entonces la capa orgánica se separó, se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-95:5), para dar el compuesto 2 (188 mg) como un polvo incoloro. MS (ESI): m/z 723 [M+H]+
(2) A una solución del compuesto 2 (180 mg) en diclorometano (1 ml) se le añadió ácido trifluoroacético (1 ml) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 1 hora. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizar la mezcla y se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se disolvió en tetrahidrofurano, se añadió 1,1-carbonildiimidazol (203 mg) a la misma y la mezcla se agitó a temperatura ambiente por 14 horas. La solución de reacción se concentró bajo presión reducida y entonces se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 98:2-90:10). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 63 jl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el clorhidrato de 5-{3-[2-(1-{[(3R,4R)-1-terc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenoxi]propil}-1,3,4-oxadiazol-2(3H)-ona 3 (93 mg) como un polvo incoloro. MS (APCI): m/z 649 [M+H]+
Ejemplo 278
[Fórmula química 66]

A una solución del compuesto 1 (310 mg) en N,N-dimetilformamida (4,5 ml) se le añadió cloruro cianúrico (166 mg) bajo enfriamiento en hielo y la mezcla se agitó a la misma temperatura por 1,5 horas. A la mezcla de reacción se le añadió agua enfriada en hielo y acetato de etilo, se agitó y la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 60:40-30:70) y posteriormente cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-94:6), para dar el compuesto 2 (185 mg) como un polvo incoloro. A una solución del compuesto 2 (35 mg) en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 26 ml), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de 1-{2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-4-(4-metoxifenil)-1-(tetrahidro-2H-piran-4-il)pirrolidin-3-il]carbonil}-4-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carbonitrilo 3 (34 mg) como un polvo incoloro. MS (ESI): m/z 673 [M+H]+
Ejemplo 279
[F ó rm u la q u ím ic a 67 ]

Una solución del compuesto 1 (80 mg) y azida de tri-n-butilestaño (331 |jl) en tolueno (2 ml) se calentó bajo reflujo por 17 horas y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 98:2-90:10). A una solución del compuesto resultante en diclorometano (2 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 60 jl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de [(3R,4R)-3-fluoro-4-(4-metoxifenil)-1-(tetrahidro-2H-piran-4-il)pirrolidin-3-il][(3R,4S)-3-(metoximetil)-4-{2-[4-(1H-tetrazol-5-il)piperidin-1-il]-4-(trifluorometií)fenil}pirrolidin-1-il]metanona 2 (66 mg) como un polvo incoloro. m S (Es I): m/z 716 [M+H]+
Ejemplo 280
[Fórmula química 68]

Una solución del compuesto 1 (193 mg) y azida de tri-n-butilestaño (901 jl) en tolueno (6 ml) se calentó bajo reflujo por 14 horas y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-95:5-80:20). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 100 jl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el clorhidrato de [(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il](4-(2-[3-(1H-tetrazol-5-il)propoxi]-4-(trifluorometil)fenil}piperidin-1-il)metanona 2 (66 mg) como un polvo incoloro. MS (ESI): m/z 633 [M+H]+
Ejemplo 281
[Fórmula química 69]

(1) El compuesto 1 y el compuesto 2 se trataron de una manera similar al ejemplo 10 anterior, para dar el compuesto 3 (500 mg).
(2) A una solución del compuesto 3 en metanol (10 ml)/tetrahidrofurano (10 ml) se le añadió carbón de paladio a 10% (humedecido con aproximadamente 50% de agua, 250 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) y a temperatura ambiente por 3 horas. A la mezcla de reacción se le añadió ácido acético (10 ml) y carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 125 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) y a temperatura ambiente por 1 hora. La materia insoluble se removió por filtración y entonces el filtrado se concentró bajo presión reducida. Al residuo se le añadió diclorometano y una solución acuosa saturada de carbonato ácido de sodio, la mezcla se agitó y la capa orgánica se separó. La capa orgánica resultante se secó y entonces se concentró bajo presión reducida para dar la [(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]{4-[2-(piperazin-1-il)-4-(trifluorometil)fenil]piperidin-1-il}metanona 4 (379 mg) como un polvo incoloro. MS (ESI): m/z 591 [M+H]+
Ejemplo 282
[Fórmula química 70]

A una solución del compuesto 1 (90 mg) y diisopropiletilamina (52 pl) en diclorometano (2 ml) se le añadió cloruro de acetilo (16 pl) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió agua y diclorometano, se agitó y la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 75:25-25:75). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 100 pl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de 1-{4-[2-(1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperazin-1-il}etanona 2 (86 mg) como un polvo incoloro. Ms (ESI): m/z 633 [M+h ]+
Ejemplo 283

A una solución del compuesto 1 (90 mg) y diisopropiletilamina (52 |jl) en diclorometano (2 ml) se le añadió cloruro de metansulfonilo (17 jl) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió agua y diclorometano, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 90:10-40:60). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 100 jl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de la [(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il](4-(2-[4-(metilsulfonil)piperazin-1-il]-4-(trifluorometil)fenil}piperidin-1-il)metanona 2 (93 mg) como un polvo incoloro. MS (ESI): m/z 669 [M+H]+
Ejemplo 284
[Fórmula química 72]

A una solución del compuesto 1 (90 mg) y trietilamina (71 jl) en diclorometano (3 ml) se le añadió isocianato de trimetilsililo (406 jl) bajo agitación y entonces la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió agua, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 98:2-88:12). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 100 jl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de la 4-[2-(1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperazin-1 -carboxamida 2 (87 mg) como un polvo incoloro. MS (ESI): m/z 634 [M+H]+
Ejemplo 285
[F ó rm u la q u ím ic a 73 ]

A una solución del compuesto 1 (90 mg) y el compuesto 2 (39 mg) obtenido por el método descrito en el documento US5192785 en cloroformo (1,5 ml)/acetonitrilo (1,5 ml) se le añadió trietilamina (105 jl) bajo agitación y entonces la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-93:7). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 75 jl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar, el diclorhidrato de la 4-[2-(1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperazin-1 -sulfonamida 3 (87 mg) como un polvo incoloro. MS (ESI): m/z 670 [M+H]+
Ejemplos 286a y 286b
[Fórmula química 74] 1

(1) El compuesto 1 (430 mg) y el compuesto 2 (343 mg) se trataron de una manera similar al ejemplo 10, para dar el compuesto 3 (492 mg) como una mezcla de diastereómeros. MS (APCI): m/z 648 [M+H]+
(2) El compuesto 3 (420 mg) se separó y se purificó con CLAR QUIRa L (CHIRALPAK IA (20 x 250 mm) fabricada por DAICEL CORPORATION, fase móvil: hexano/etanol/dietilamina = 60/40/0,1, magnitud de flujo: 10 ml/min), para dar el compuesto ópticamente activo 4 (184 mg) y el compuesto ópticamente activo 5 (169 mg) como polvos incoloros. Cada MS (APCI): m/z 648 [M+H]+ (los tiempos de retención del compuesto 4 y el compuesto 5 en la CLAR fueron 6,28 minutos y 7,52 minutos, respectivamente, con CHIRALPAK IA-3 (4,6 x 150 mm) fabricada por DAICEL CORPORATION, bajo las condiciones de la fase móvil: hexano/etanol/dietilamina= 60/40/0,1, magnitud de flujo: 0,500 ml/min, temperatura de la columna: 25 °C). Se confirmó que el compuesto de amida obtenido mediante la fusión de la amina ópticamente activa resultante, la cual se obtuvo por el método en el ejemplo de referencia 100, con el compuesto 1, es el compuesto 5 y se confirmó que el compuesto 4 y el compuesto 5 tienen, respectivamente, las configuraciones anteriores.
(3) El compuesto 4 y el compuesto 5 se trataron de una manera similar al ejemplo 11, para dar el diclorhidrato de ácido 1-{2-[(3S)-1-{[(3R,4R)-1-terc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico 6 (ejemplo 286a) y el diclorhidrato de ácido 1-{2-[(3R)-1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico 7 (ejemplo 286b) como polvos incoloros, respectivamente. Cada MS (APCI): m/z 620 [M+H]+
Ejemplos 287 a 294
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 286 anterior para dar un compuesto de éster como un intermediario y se trató posteriormente de una manera similar al ejemplo 11, para dar cada compuesto incluido en el cuadro 17 siguiente. El análisis por la CLAR del intermediario de éster se llevó a cabo con la columna de CLAR (4,6 x 150 mm) fabricada por DAICEL CORPORATION, bajo la magnitud de flujo: 0,500 ml/min, temperatura de la columna: 25 °C y las siguientes condiciones.
Condiciones del análisis:
A: CHIRALPAK IA-3, fase móvil:
hexano/isopropanol/tetrahidrofurano/dietilamina=80/10/10/0,1
B: CHIRALPAK IC-3, fase móvil:
etanol/dietilamina=100/0,1
C: CHIRALPAK IA-3, fase móvil:
hexano/etanol/dietilamina=80/20/0,1
D: CHIRALPAK IA-3, fase móvil:
hexano/isopropanol/tetrahidrofurano/dietilamina=80/10/10/0,1.
r 171

Ejemplo 295
[F ó rm u la q u ím ic a 75 ]

Una solución en etanol (4 ml) del compuesto 1 (300 mg), clorhidrato de hidroxilamina (42 mg) y trietilamina (106 |jl) se agitó a temperatura ambiente por 62 horas. A la mezcla de reacción se le añadió agua y acetato de etilo, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-90:10). Una solución de la mezcla resultante (165 mg) y 1,1-carbonildiimidazol (215 mg) en tetrahidrofurano (3 ml) se agitó a 70 °C por 6 horas. La solución de reacción se concentró bajo presión reducida, al residuo se le añadió agua y cloroformo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó, se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-93:7). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 66 jl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el clorhidrato de la 3-{3-[2-(1-{[(3R,4R)-1-terc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenoxi]propil}-1,2,4-oxadiazol-5(4H)-ona 2 (57 mg) como un polvo incoloro. MS (ESI): m/z 649 [M+H]+
Ejemplos 296a, 296b y 296c
[Fórmula química 76] 1

(1) A una solución del compuesto 1 (302 mg) en cloroformo (7 ml) se le añadió ácido trifluoroacético (3,5 ml) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 1,5 horas. A la mezcla de reacción se le añadió una solución acuosa de hidróxido de sodio (2 moles/ml) bajo enfriamiento en hielo para ajustarla en pH = 8 y se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. A una solución del compuesto resultante (compuesto 2) en N,N-dimetilformamida (7,1 ml) se le añadió el compuesto 3 (314 mg), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (272 mg), 1-hidroxi-7-azabenzotriazol (193 mg) y trietilamina (200 ml) y la mezcla se agitó a temperatura ambiente por 16 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-90:10), para dar el compuesto 4 (403 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 604 [M+H]+
(2) A una solución del compuesto 4 (88 mg) en diclorometano (1,5 ml) se le añadió una solución de ácido
clorhídrico en acetato de etilo (4 moles/l, 70 ml), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de la 1-[2-(1-{[(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperidin-4-ona 5 (ejemplo 296a, 69 mg) como un polvo incoloro. MS (ESI): m/z 604 [M+H]+ (3) A una solución del compuesto 4 (314 mg) en metanol (5 ml) se le añadió borohidruro de sodio (24 mg) bajo enfriamiento en hielo y la mezcla se agitó a la misma temperatura por 1,5 horas. A la mezcla de reacción se le añadió agua y entonces se extrajo con cloroformo. La capa orgánica se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-90:10), para dar el compuesto 6 (202 mg) como un material viscoso incoloro. MS (ESI): m/z 606 [M+H]+
(4) A una solución del compuesto 6 (100 mg) en diclorometano (1,7 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 83 pl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de la [(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]{4-[2-(4-hidroxipiperidin-1-il)-4-(trifluorometil)fenil]piperidin-1-il}metanona 7 (ejemplo 296b, 105 mg) como un polvo incoloro. MS (ESI): m/z 606 [M+H]+
(5) A una solución del compuesto 6 (150 mg) en N,N-dimetilformamida (1,2 ml) se le añadió hidruro de sodio (60% en aceite, 15 mg) bajo enfriamiento en hielo, la mezcla se agitó por 10 minutos, entonces se añadió yoduro de metilo (31 pl) a la misma y se agitó a temperatura ambiente por 24 horas.
A la mezcla de reacción se le añadió hidruro de sodio (60 % en aceite, 50 mg) y yoduro de metilo (160 pl) y se agitó a 60 °C por 2 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, entonces se añadieron a la misma agua y acetato de etilo, se agitó y la capa orgánica se separó. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó, se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-90:10). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 50 pl) y entonces el solvente se evaporó bajo presión reducida. El residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de la [(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]{4-[2-(4-metoxipiperidin-1-il)-4-(trifluorometil)fenil]piperidin-1-il}metanona 8 (ejemplo 296c, 62 mg) como un polvo incoloro. MS (ESI): m/z 620 [M+H]+
Ejemplos 297a, 297b y 297c
[Fórmula química 77] 1

(1) A una solución del compuesto 1 (348 mg) en N,N-dimetilformamida (15 ml) se le añadió trietilamina (422 pl) y N,O-bis(trimetilsilil)acetamida (204 pl) a temperatura ambiente y la mezcla se agitó por 30 minutos. A la mezcla se le añadió el compuesto 2 (349 mg), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (290 mg) y 1-hidroxi-7-azabenzotriazol (206 mg) y se agitó a temperatura ambiente por 15 horas. A la mezcla de reacción se le añadió agua y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 90:10-50:50), para dar el 1-{2-[(3S,5S)-1-{[(3R,4R)-1-ciclopentil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-5-(hidroximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxilato de metilo 3 (ejemplo 297a, 406 mg). Ms (ESI): m/z 676 [M+H]+ (2) Bajo atmósfera de nitrógeno, a una solución del compuesto 3 (101 mg) en diclorometano (3 ml) se le añadió
trifluoruro de (dietilamino) azufre (59 pl) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 15 horas. A la solución de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 95:5-60:40) y posteriormente cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-30:70), para dar el diclorhidrato de ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-1-ciclopentil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carboil)-5-(fluorometil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico 4 (ejemplo 297b, 45 mg). MS (ESI): m/z678 [M+H]+
(3) A una solución del compuesto 3 (119 mg) y trifenilfosfina (370 mg) en tetrahidrofurano (4 ml) se le añadió acetona cianohidrina (129 pl) y azodicarboxilato de dietilo (solución de 2,2 moles/l de tolueno, 640 pl) a temperatura ambiente y entonces la mezcla se agitó por 14 horas. La solución de reacción se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-30:70), para dar el 1-{2-[(3S,5S)-5-(cianometil)-1-{[(3R,4R)-1-ciclopentil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxilato de metilo 5 (ejemplo 297c, 90 mg). MS (ESI): m/z 685 [M+H]+
Ejemplos 298 a 300
Cada compuesto en el ejemplo 297a, ejemplo 297b y ejemplo 297c anterior se trató de una manera similar al ejemplo 11 anterior, para dar cada compuesto incluido en el cuadro 18 siguiente.
r 1

Ejemplo 301
[Fórmula química 78]
(1) Una suspensión del compuesto 1 (500 mg), bromuro de propargilo (114 |jl) y carbonato de potasio (400 mg) en N,N-dimetilformamida (5 ml) se agitó bajo calentamiento bajo atmósfera de nitrógeno a 70 °C por 2 horas. Después de ser enfriada hasta temperatura ambiente, la mezcla de reacción se vertió agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-85:15), para dar el compuesto 2 (553 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 284[M-Boc+H]+
(2) A una solución del compuesto 2 (340 mg) en 1,4-dioxano (2 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 2 ml) y la mezcla se agitó a temperatura ambiente por 1 hora. La mezcla de reacción se concentró bajo presión reducida y el compuesto 3 resultante se usó en el siguiente paso sin purificación. (3) A una solución mixta del compuesto 3 y el compuesto 4 (316 mg) en N,N-dimetilformamida (3 ml) se le añadió trietilamina (496 jl), clorhidrato de 1-etil-3-(3- dimetilaminopropil)carbodiimida (341 mg) y 1 -hidroxi-7-azabenzotriazol (242 mg) a temperatura ambiente y la mezcla se agitó por 15 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, agua y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 90:10-60:40), para dar el compuesto 5 (417 mg) como un polvo incoloro. MS (ESI): m/z 561 [M+H]+
(4) A una solución del compuesto 5 (410 mg) en éter dietílico (3 ml) se le añadió gota a gota una solución de éter dietílico de metil litio (1,14 moles/ml, 772 jl) bajo atmósfera de nitrógeno a -78 °C, la mezcla se agitó por 1,5 horas, entonces una solución de cloroformiato de metilo (138 mg) en éter dietílico (1 ml) se añadió a la misma y se agitó a la misma temperatura por 1 hora. Después de ser agitada a temperatura ambiente por otras 3 horas, a la mezcla de reacción se le añadió agua y acetato de etilo y se agitó. La capa orgánica se separó y la capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-95:5), para dar el compuesto 6 (416 mg) como un material viscoso incoloro. MS (ESI): m/z 619 [M+H]+
(5) A una solución de clorhidrato de hidroxilamina (410 mg) en metanol (4 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/ml, 860 jl) bajo enfriamiento en hielo, entonces una solución del compuesto 6 (410 mg) en metanol (2 ml) se añadió gota a gota a la misma y la mezcla se agitó a temperatura ambiente por 20 horas. La mezcla de reacción se neutralizó con una solución de ácido cítrico acuoso, entonces agua y acetato de etilo se añadieron a la misma y se agitó. La capa orgánica se separó y la capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-90:10). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 165 jl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida, para dar el clorhidrato de la [(3R,4R)-1-ferc-butil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il](4-{2-[(3-hidroxiisoxazol-5-il)metoxi]-4-(trifluorometil)fenil}piperidin-1-il)metanona 7 (140 mg) como un polvo incoloro. MS (APCI): m/z 620 [M+H]+
Ejemplo 302
[Fórmula química 79] 1

(1) A una solución del compuesto 1 (152 mg) en 1,4-dioxano (1,5 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 1,5 ml) y entonces la mezcla se agitó a temperatura ambiente por 22 horas. La mezcla de reacción se concentró bajo presión reducida, al residuo se le añadió N,N-dimetilformamida (2 ml), una solución acuosa de dimetilamina (50 %, 38 jl), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (81 mg), 1-hidroxi-7-azabenzotriazol (57 mg) y trietilamina (117 jl) y la mezcla se agitó a temperatura ambiente por 18 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, agua y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 40:60-0:100).
(2) A una solución del compuesto 2 (73 mg) en etanol (2 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 636 jl) a temperatura ambiente y la mezcla se agitó por 1 hora. A la mezcla de reacción se añadió una solución acuosa de ácido clorhídrico (2 moles/l, 650 jl) y se concentró bajo presión reducida. Al
residuo se le añadió agua y diclorometano, se agitó, entonces la capa orgánica se separó, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-85:15). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 60 jl), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de ácido 1-[2-(1-{[(3R,4R)-1-[2-(dimetilamino)-2-oxoetil]-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}piperidin-4-il)-5-(trifluorometil)fenil]piperidin-4-carboxílico 3 (72 mg) como un polvo incoloro. MS (APCI): m/z 66 3 [M+H]+
Ejemplos 303 a 318
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 302 anterior, para dar cada compuesto incluido en el cuadro 19 siguiente.
Cuadro 19

comtinuación

(comtinuación)

Ejemplo 319
[Fórmula química 80]

(1) A una solución de ciclopentancarboxaldehído (51 |jl) en éter dietílico (1 ml) se le añadió bromo (18 |jl) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 1 hora. A la mezcla se le añadió bromo (18 j l) y se agitó por otros 30 minutos. A la solución de reacción se le añadió agua enfriada en hielo, la mezcla se agitó, se extrajo con acetato de etilo, se secó y se concentró bajo presión reducida. Al residuo se le añadió 1,2-dimetoxietano (2 ml), carbonato de potasio (100 mg) y el compuesto 1 (150 mg) y la mezcla se agitó a 80 °C por 15 horas. Otro material crudo de 1-bromo-ciclopentancarboxaldehído preparado de una manera similar a partir de ciclopentancarboxaldehído (51 m) se añadió a la misma y se agitó a 80 °C por 4 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, entonces agua y acetato de etilo se añadieron a la misma, se agitó y la capa orgánica se separó. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó, se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-40:60), para dar el compuesto 2 (111 mg) como un polvo incoloro. MS (ESI): m/z 718 [M+H]+
(2) A una solución del compuesto 2 (110 mg) en tetrahidrofurano (1 ml)/metanol (1 ml) se le añadió borohidruro de sodio (7 mg) bajo enfriamiento en hielo y la mezcla se agitó por 20 minutos. A la mezcla de reacción se le añadió agua, se agitó y entonces se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 60:40-20:80), para dar el compuesto 3 (98 mg) como un polvo incoloro. MS (ESI): m/z 720 [M+H]+
(3) A una solución del compuesto 3 (96 mg) en N,N-dimetilformamida (1 ml) se le añadió hidruro de sodio (60 % en aceite, 6,4 mg) bajo enfriamiento en hielo, la mezcla se agitó por 5 minutos, entonces se añadió a la misma yoduro de metilo (12,5 ml) y se agitó a temperatura ambiente por 1 hora. Se añadieron a la misma hidruro de sodio (60 % en aceite, 6,4 mg) y yoduro de metilo (12,5 ml) y la mezcla se agitó a temperatura ambiente por 1 hora. A la solución de reacción se le añadió agua y ácido clorhídrico 1 N para ajustarla en pH = 7 y entonces se extrajo con acetato de etilo. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se disolvió en metanol (1 ml), una solución acuosa de hidróxido de sodio (2 moles/l, 266 ml) se añadió a la misma a temperatura ambiente y la mezcla se agitó por 14 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 266 ml), se agitó y entonces se concentró bajo presión reducida. El residuo se suspendió en cloroformo, la materia insoluble se removió por filtración y entonces el filtrado se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-90:10). A una solución del compuesto resultante en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 50 ml), entonces el solvente se evaporó bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el diclorhidrato de ácido 1-{2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-1-[1-(metoximetil)ciclopentil]-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico 4 (70 mg) como un polvo incoloro. MS (ESI): m/z 720 [M+H]+
Ejemplos 320 a 322
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 319 anterior, para dar cada compuesto incluido en el cuadro 20 siguiente.
r 2

Ejemplo 323
[Fórmula química 81]

(1) A una solución del compuesto 1 (312 mg) en cloroformo (3,9 ml) se le añadió ácido trifluoroacético (1,9 ml) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió cloroformo para diluirla, entonces se añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizar la mezcla y la capa orgánica se separó. La capa acuosa se extrajo con cloroformo, las capas orgánicas se combinaron, se lavó con una solución acuosa saturada de carbonato ácido de sodio, se secó y se concentró bajo presión reducida para dar el compuesto 2 (280 mg) como un material viscoso amarillo. MS (ESI): m/z 705 [M+H]+
(2) A una solución del compuesto 2 (100 mg) y trietilamina (60 jl) en diclorometano se le añadió cloruro de acetilo (20 j l) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 2 horas. La mezcla de reacción se diluyó con cloroformo, una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 50:50-100:0), para dar el 1-{2-[(3S,4R)-1-{[(3R,4R)-1-(1-acetilpiperidin-4-il)-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxilato de metilo 3 (62 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 747 [M+H]+
Ejemplos 324 y 325
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 323 anterior, para dar cada compuesto incluido en el cuadro 21 siguiente.
r 21

Ejemplos 326 a 328
Cada compuesto en el ejemplo 323, ejemplo 324 y ejemplo 325 anterior se trató de una manera similar al ejemplo 11 anterior, para dar cada compuesto incluido en el cuadro 22 siguiente.
r 22

Ejemplos 329 A 331
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 1 anterior y posteriormente el ejemplo 11, para dar cada compuesto incluido en el cuadro 23 siguiente.
r 2

Ejemplos 332 a 345
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 10 anterior y posteriormente el ejemplo 11, para dar cada compuesto incluido en el cuadro 24 siguiente.
r 24

continuación

continuación

Ejemplos 346 y 347
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 286 anterior para dar un compuesto de éster como intermediario y se trató posteriormente de una manera similar al ejemplo 11, para dar cada compuesto incluido en el cuadro 25 siguiente. Se usó una columna de CLAR (CHIRALPAK IF (4,6 x 150 mm)) fabricada por DAICEL CORPORATION en el análisis por CLAR del intermediario de éster, en las condiciones de magnitud de flujo: 0,500 ml/min, temperatura de la columna: 25 °C, fase móvil: hexano/metanol/tetrahidrofurano/dietilamina=83/5/12/0,1.
r 2

Ejemplo 348
[Fórmula química 82]

(1) A una solución mixta del compuesto 1 (100 mg) y el compuesto 2 (124 mg) en N,N-dimetilformamida (3 ml) se le añadió clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (96 mg) y 1-hidroxi-7-azabenzotriazol (68 mg) y la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió una solución
acuosa saturada de carbonato ácido de sodio y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 85:15-65:35), para dar una mezcla de diastereómeros del compuesto 3 como un polvo incoloro (118 mg). MS (APCI): m/z 760 [M+H]+
(2) A una solución del compuesto 3(115 mg) en metanol (2 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 605 jl) y la mezcla se agitó a temperatura ambiente por 4 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 605 jl), se agitó, se concentró bajo presión reducida, se añadieron diclorometano y agua a la misma, se agitó, entonces la capa orgánica se separó y se concentró bajo presión reducida. El residuo resultante se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-90:10), para dar el compuesto 4 (58 mg) y el compuesto 5 (49 mg) como polvos incoloros, respectivamente. Cada Ms (APCI): m/z 746 [M+H]+ TLC (cloroformo:metanol= 95:5): valor de Rf del compuesto 4 “ 0,30, valor de Rf del compuesto 5 = 0,15 (placa de t Lc : gel de sílice para TLC 1.05715.0001 60 F254, fabricado por Merck KGaA). Los datos de 1H-RMN del ejemplo 146 y el compuesto 5 fueron los mismos entre sí que confirman que el compuesto 4 tenía la configuración anterior.
(3) A una solución del compuesto 4 (54 mg) en diclorometano (1 ml) se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l, 90 jl) y el solvente se evaporó bajo presión reducida. El residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el compuesto 6 (45 mg) como un polvo de color rosa pálido. MS (APCI): m/z 746 [M+H]+
Ejemplos 349 y 350
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo 348 anterior, para dar cada compuesto incluido en el cuadro 26 siguiente.

Ejemplos de referencia
Ejemplo de referencia 1
[Fórmula química 83]

(1) Bajo atmósfera de nitrógeno, a una solución de 2-fluoro-2-fosfonoacetato de trietilo (55,6 g) en tetrahidrofurano (100 ml) se añadieron complejo de bromuro de magnesio-éter dietílico (71,3 g) y trietilamina (35,3 ml) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 1,5 horas. A la mezcla de reacción se le añadió gota a gota una solución del compuesto 1 (25 g) en tetrahidrofurano (375 ml) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 15 horas. La mezcla de reacción se concentró hasta que el volumen llegara a ser aproximadamente la mitad, se añadieron a la misma agua (400 ml) y acetato de etilo (500 ml) y se agitó. La materia insoluble se removió por filtración y la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. A una solución del residuo en tetrahidrofurano (210 ml)/etanol (210 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 188 ml) a temperatura ambiente. A la mezcla de reacción se le añadió etanol (300 ml) y tetrahidrofurano (100 ml), se agitó, una solución acuosa de ácido clorhídrico (1 mol/l, 390 ml) se añadió después de 1 hora y entonces se evaporó el tetrahidrofurano bajo presión reducida. A una suspensión del compuesto resultante se le añadió agua (300 ml), la mezcla se agitó a temperatura ambiente por 1 hora y entonces el precipitado se colectó por filtración. El precipitado se lavó con agua y posteriormente éter diisopropílico y se secó bajo una corriente de aire a 60 °C por 12 horas, para dar el compuesto 2 (26,63 g) como un polvo incoloro. MS (ApCi): m/z 197 [M+H]+
(2) A una suspensión del compuesto 2 (26,6 g) en diclorometano (270 ml) se le añadió cloruro de tionilo (29,7 ml) y N,N-dimetilformamida (1,04 ml) y la mezcla se calentó bajo reflujo por 4 horas. La solución de reacción se concentró bajo presión reducida y entonces el residuo se concentró azeotrópicamente con tolueno dos veces. A una solución del residuo en diclorometano (532 ml) se le añadió el compuesto 3 (26,3 g) y cloruro de litio (12,5 g), se añadió trietilamina (65,9 ml) gota a gota a la misma bajo atmósfera de nitrógeno bajo enfriamiento en hielo y entonces se agitó a temperatura ambiente por 15 horas. La suspensión de reacción se vertió en una solución acuosa de ácido cítrico (ácido cítrico: 102 g agua: 950 ml) bajo enfriamiento en hielo y se extrajo con cloroformo (300 ml). La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se pulverizó con éter diisopropílico, se colectó por filtración y entonces se secó para dar el compuesto 4 (44,7 g) como un polvo incoloro. m S (APCI): m/z 356 [M+H]+
(3) A una suspensión del compuesto 4 (10 g) y el compuesto 5 (13,3 g) en diclorometano (100 ml) se le añadió ácido trifluoroacético (215 pl) a temperatura ambiente y la mezcla se calentó bajo reflujo bajo atmósfera de nitrógeno por 1 hora. A la solución de reacción se le añadió el compuesto 5 (3,34 g) y ácido trifluoroacético (54 pl) a temperatura ambiente y la mezcla se calentó bajo reflujo por otros 30 minutos. La solución de reacción se vertió en una solución acuosa de ácido cítrico (ácido cítrico: 35 g agua: 350 ml) bajo enfriamiento en hielo y se extrajo con diclorometano. La capa orgánica resultante se lavó con una solución acuosa saturada de carbonato ácido de sodio y solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-40:60) y se cristalizó con éter diisopropílico, para dar el compuesto 6 (6,06 g) y el compuesto 7 (3,3 g) como polvos incoloros, respectivamente. Cada MS (APCI): m/z 489 [M+H]+ t Lc (hexano:acetato de etilo = 80:20): valor de Rf del compuesto 6 “ 0,45, valor de Rf del compuesto 7 “ 0,2 (placa de TLC: gel de sílice para TLC 1.05715.0001 60 F254, fabricado por Merck KGaA). El compuesto 6 se disolvió en una pequeña cantidad de una solución mixta de diclorometano y acetato de etilo y se añadió éter dietílico a la misma para recristalizarla. El cristal resultante se
sometió a cristalografía de rayos X para confirmar que el compuesto 6 y el compuesto 7 tienen las configuraciones anteriores respectivamente.
(4) A una solución del compuesto 6 (6,05 g) y dicarbonato de di-t-butilo (2,84 g) en etanol (85 ml)/tetrahidrofurano (150 ml) se le añadió carbón de paladio a 10% (humedecido con aproximadamente 50% de agua, 1,8 g) bajo agitación y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) por 24 horas. El precipitado se disolvió en cloroformo (100 ml) y metanol (20 ml) y se removió el carbón de paladio por filtración. El filtrado se concentró, el residuo se pulverizó con hexano, se colectó por filtración y se secó bajo presión reducida para dar el compuesto 8 (6,28 g) como un polvo incoloro. MS (APCI): m/z 399 [M-Boc H]+. (5) A una suspensión del compuesto 8 (6,28 g) en tetrahidrofurano (45 ml) se le añadió una solución acuosa de hidróxido de litio (hidróxido de litio (monohidrato): 635 mg agua: 20 ml) bajo enfriamiento en hielo, la mezcla se agitó a temperatura ambiente por 1 hora y entonces se evaporó el tetrahidrofurano. Al residuo se le añadió agua (20 ml) y entonces se lavó con una solución mixta (30 ml) de acetato de etilo y éter dietílico (1:1) cuatro veces. A la capa acuosa resultante se le añadió una solución acuosa de ácido clorhídrico (1 mol/l, 15,5 ml) para acidificarla y entonces se extrajo con cloroformo dos veces. Las capas orgánicas resultantes se combinaron, se lavaron con solución salina saturada, se secaron y el solvente se evaporó, para dar el compuesto 9 (4,11 g) como un polvo incoloro. MS (APCI): m/z 338 [M-H]-.
Ejemplo de referencia 2
[Fórmula química 84]

(1) A una solución del compuesto 1 (6 g) en metanol (45 ml) se le añadió gota a gota cloruro de tionilo (5,16 ml) bajo enfriamiento en hielo. Después de agitada a temperatura ambiente por 3 horas, la mezcla de reacción se concentró bajo presión reducida. El residuo se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el compuesto 2 (4,906 g) como un polvo incoloro. MS (APCI): m/z 254 [M+H]+ (2) A una suspensión del compuesto 2 (579 mg) y ciclohexanona (248 ml) en diclorometano (15 ml) se le añadió acetato de sodio (197 mg), la mezcla se agitó a temperatura ambiente por 30 minutos, entonces se añadió a la misma triacetoxiborohidruro de sodio (509 mg) y la mezcla se agitó por 15 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y agua, se agitó y entonces se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de n H (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 3 (678 mg) como un material viscoso incoloro. MS (ESI): m/z 336 [M+H]+
(3) A una solución del compuesto 3 (655 mg) en metanol (6 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 1,27 ml) a temperatura ambiente y la mezcla se agitó por 2 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 1,27 ml), se agitó y entonces la solución de reacción se concentró bajo presión reducida para dar el compuesto 4 como un polvo incoloro (780 mg) que contenía cloruro de sodio. m S (ESI): m/z 322 [M+H]+
Ejemplos de referencia 3 a 16
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 2 anterior, para dar cada compuesto incluido en el cuadro 27 siguiente.
r 27

continuación

continuación

Ejemplo de referencia 17
[Fórmula química 85]

(1) A una suspensión del compuesto 1 (500 mg) y acetato de sodio (142 mg) en diclorometano (20 ml) se le añadió el compuesto 2 (413 mg), ácido acético (99 jl) y triacetoxiborohidruro de sodio (585 mg) a temperatura ambiente y la mezcla se agitó por 15 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y agua, se agitó y entonces se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-60:40), para dar el compuesto 3 (720 mg) como un material viscoso incoloro. MS (APCI): m/z 437 [M+H]+
(2) A una solución del compuesto 3 (719 mg) en diclorometano (14 ml) se le añadió ácido trifluoroacético (4 ml) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 3 horas. A la mezcla de reacción se le añadió diclorometano para diluirla, entonces se añadió una solución acuosa saturada de carbonato ácido de sodio bajo agitación para alcalinizar la mezcla y la capa orgánica se separó. La capa orgánica resultante se secó y entonces se concentró bajo presión reducida. A una solución del residuo en diclorometano (24 ml) se le añadió el compuesto 4 (741 mg), ácido acético (101 jl) y triacetoxiborohidruro de sodio (967 mg) y la mezcla se agitó por 15 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y agua, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 85:15-65:35), para dar el compuesto 5 (283 mg) como un material viscoso incoloro. MS (APCI): m/z 415 [M+H]+
(3) A una solución del compuesto 5 (280 mg) en metanol (6 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 405 jl) a temperatura ambiente y la mezcla se agitó por 2 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 405 jl), se agitó y entonces la solución de reacción se concentró bajo presión reducida para dar el compuesto 6 como un polvo incoloro (320 mg) que contenía cloruro de sodio. m S (APCI): m/z 401 [M+H]+
Ejemplo de referencia 18a y ejemplo de referencia 18b
[Fórmula química 86]

(1) A una solución del compuesto 1 (200 mg) y el compuesto 2 (128 mg) en diclorometano (7 ml) se le añadió triacetoxiborohidruro de sodio (219 mg) y la mezcla se agitó a temperatura ambiente por 16 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y agua, se agitó y entonces se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 70:30-0:100), para dar el compuesto 3 (cis) (81 mg) y el compuesto 4 (trans) (10 mg) como polvos incoloros. TLC (eluyente: acetato de etilo): valor de Rf del compuesto 3 “ 0,35, valor de Rf del compuesto 4 “ 0,45 (placa de TLC: gel de sílice para TLC 1.05715.0001 60 F254, fabricado por Merck KGaA). Un cristal obtenido de una solución del compuesto 3 en éter diisopropílico se sometió a cristalografía de rayos X para confirmar que el compuesto 3 y el compuesto 4 tienen las configuraciones anteriores, respectivamente. Cada MS (ESI): m/z 392 [M+H]+
(2) A una solución del compuesto 3 (40 mg) en metanol (1 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 220 pl) a temperatura ambiente y la mezcla se agitó por 2,5 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 220 pl), se agitó y entonces la solución de reacción se concentró bajo presión reducida para dar el compuesto 5 como un polvo incoloro (ejemplo de referencia 18a, 52 mg) que contenía cloruro de sodio. MS (ESI): m/z 378 [M+H]+
(3) El compuesto 4 (110 mg) se trató de una manera similar al paso (2) anterior, para dar el compuesto 6 como un polvo incoloro (ejemplo de referencia 18b, 125 mg) que contenía cloruro de sodio. MS (ESI): m/z 378 [M+H]+
Ejemplo de referencia 19
[Fórmula química 87] 1

(1) El compuesto 1 (7,45 g) y el compuesto 2 (8,54 g) obtenidos mediante el método descrito en el documento WO2004/089307 se trataron de una manera similar al ejemplo de referencia 1, para dar el compuesto 3 (3,58 g) como un polvo amarillo pálido. MS (ESI): m/z 455 [M+H]+
(2) A una solución del compuesto 3 (27,9 g) en tetrahidrofurano (140 ml) se le añadió una solución acuosa de hidróxido de litio (hidróxido de litio (monohidrato): 3,09 g agua: 70 ml) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (1 mol/l, 73,7 ml), se agitó y se evaporó el tetrahidrofurano bajo presión reducida. La solución acuosa resultante se lavó con acetato de etilo tres veces y la capa acuosa se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna (cloroformo:metanol = 97:3-80:20) usando gel de sílice diol (FUJI SILYSIA CHEMICAL LTD.), pulverizado con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el compuesto 4 (18,04 g) como un polvo incoloro. MS (ESI): m/z 296 [M+H]+
Ejemplos de referencia 20 a 31
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 19 anterior, para dar cada compuesto incluido en el cuadro 28 siguiente.
r 21


Ejemplo de referencia 32
[Fórmula química 88] 1

(1) A una solución del compuesto 1 (1,3 g) en tetrahidrofurano (20 ml) se le añadió gota a gota una solución de diisopropilamida de litio-tetrahidrofurano (2 moles/ml, 14 ml) bajo atmósfera de nitrógeno bajo enfriamiento en hielo y entonces la mezcla se agitó a 50 °C por 3 horas. A la mezcla de reacción se le añadió gota a gota yoduro de metilo (1,87 ml) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 19 horas. A la mezcla de reacción se le añadió una solución acuosa a 10 % de ácido cítrico (20 ml), se agitó, entonces se evaporó el tetrahidrofurano bajo presión reducida y se extrajo con acetato de etilo. La capa orgánica se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-50:50), para dar el compuesto 2 (1,05 g) como un polvo café pálido. A una solución del compuesto 2 (896 mg) en tolueno (37 ml) se le añadió trietilamina (1,12 ml) y el compuesto 3 (1,39 ml) y la mezcla se agitó bajo atmósfera de nitrógeno a 85 °C por 17 horas. A la solución de reacción se le añadió gota a gota una solución acuosa de hidróxido de sodio (1 mol/ml, 27 ml) bajo enfriamiento en hielo, acetato de
etilo y agua se añadieron a la misma y la capa orgánica se separó. La capa orgánica resultante se secó y se concentró bajo presión reducida para dar un material crudo del compuesto 4 (671 mg) como un material viscoso café. A una solución del compuesto 4 (665 mg) en tetrahidrofurano (55 ml) se le añadió una solución mixta de ácido clorhídrico concentrado (3,05 ml) y agua (5,5 ml) y la mezcla se agitó a temperatura ambiente por 48 horas. La solución de reacción se concentró bajo presión reducida para dar un material crudo del compuesto 5 (558 mg) como un polvo café pálido. MS (ESI): m/z 116 [M+H]+
(2) Una suspensión del compuesto 5 (630 mg), (clorometil)trimetilsilano (695 jl) yoduro de sodio (746 mg) y carbonato de potasio (1,38 g) en acetonitrilo (10 ml), se agitó bajo atmósfera de nitrógeno a 80 °C por 88 horas. Después de ser enfriada hasta temperatura ambiente, la materia insoluble se removió por filtración y el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 98:2-90:10), para dar el compuesto 6 (500 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 202 [M+H]+
(3) Al compuesto 6 (400 mg) se le añadió una solución de formaldehído (37 %, 216 jl) bajo enfriamiento en hielo y posteriormente metanol (488 jl) y la mezcla se agitó a temperatura ambiente por 12 horas. A la mezcla de reacción se le añadió carbonato de potasio (720 mg), se agitó a temperatura ambiente por 12 horas, entonces la materia insoluble se removió por filtración y el filtrado se concentró bajo presión reducida para dar un material crudo que contenía el compuesto 7. Todo el material crudo se usó en el siguiente paso sin purificación. MS (ESI): m/z 202 [M+H]+
(4) El compuesto 8 (283 mg) y el compuesto 7 se trataron de una manera similar al ejemplo de referencia 1, para dar el compuesto 9 (59 mg) como un polvo incoloro. MS (ESI): m/z 497 [M+H]+
(5) A una solución del compuesto 9 (59 mg) en tetrahidrofurano (1 ml) se le añadió una solución acuosa de hidróxido de litio (hidróxido de lítio (monohidrato): 7 mg agua: 500 jl) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 4 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 83,5 jl), se agitó, entonces se lavó tres veces con una solución mixta de acetato de etilo y éter dietílico (1:2) y la capa acuosa se concentró bajo presión reducida para dar el compuesto 10 como un material crudo incoloro (48 mg) que contenía cloruro de litio. MS (ESI): m/z 338 [M+H]+
Ejemplo de referencia 33
[Fórmula química 89] 1

(1) El compuesto 5 se preparó de acuerdo con el método descrito en una literatura (J. Am. Chem. Soc. 2005, 127, 119-125). A saber, a una solución del compuesto 1 (1 g) y el compuesto 2 (1534 jl) en tolueno (10 ml) se le añadió H3,5-bis(trifluorometil)fenil]-3-[(1R,2RH-)-2-(dimetilamino)ciclohexil]tiourea (231 mg) y la mezcla se agitó a temperatura ambiente por 24 horas. La solución de reacción se concentró bajo presión reducida, el residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 95:5-80:20), entonces se pulverizo con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el compuesto 3 (1294 mg) como un polvo incoloro. MS (ApCi): m/z 342 [M+H]+ pureza óptica 92,18 % de e.e.
(2) A una solución del compuesto 3 (1200 mg) en metanol (36 ml) se le añadió una solución acuosa de ácido clorhídrico (1 mol/l, 3867 jl) y carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 240 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 21 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró bajo presión reducida. Al residuo se le añadió tolueno, se concentró bajo presión reducida de nuevo, el residuo resultante se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el compuesto 4 (1174 mg) como un polvo incoloro. MS (APCI): m/z 312 [M+H]+
(3) A una suspensión del compuesto 4 (1170 mg) en diclorometano (23 ml) se le añadió 1,8-diazabiciclo[5.4.0]-7-undeceno (1258 jl) a -70 °C y la mezcla se agitó a la misma temperatura por 4 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (1 mol/l, 10 ml) y agua, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 70:30-40:60), para dar el compuesto 5 (577 mg) como un polvo incoloro. MS (APCI): m/z 280 [M+H]+
(4) A una solución del compuesto 5 (570 mg) en diclorometano (11 ml) se le añadió tetrafluoroborato de trimetiloxonio (604 mg) y la mezcla se agitó a temperatura ambiente por 6 horas. Se añadió a la misma tetrafluoroborato de trimetiloxonio (604 mg) y la mezcla se agitó a temperatura ambiente por otras 22 horas (solución de reacción A). Por separado, a una solución de cianoborohidruro de sodio (1026 mg) en metanol (11 ml) se le añadió ácido acético (1167 jl) y la mezcla se enfrió hasta 0 °C (solución de reacción B). La solución de reacción A se añadió gota a gota a la solución de reacción B a 0 °C y entonces la mezcla se agitó a temperatura ambiente por 1 hora. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y agua, se agitó y entonces se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-93:7), para dar el compuesto 6 (367 mg) como un material viscoso incoloro. MS (APCl): m/z 266 [M+H]+
(5) A una suspensión del compuesto 6 (320 mg) y ciclohexanona (131 jl) en diclorometano (6 ml) se le añadió triacetoxiborohidruro de sodio (383 mg) y la mezcla se agitó a temperatura ambiente por 7 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y agua, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 7 (345 mg) como un material viscoso incoloro. MS (APCI): m/z 348 [M+H]+
(6) A una solución del compuesto 7 (343 mg) en etanol (7 ml) se añadió una solución acuosa de hidróxido de sodio (2 moles/l, 987 jl) a temperatura ambiente y la mezcla se agitó a 70 °C por 13 horas. Después de ser enfriada hasta temperatura ambiente, a la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 987 jl), se agitó y se concentró bajo presión reducida para dar el compuesto 8 como un polvo incoloro (423 mg) que contenía cloruro de sodio. MS (APCI): m/z 334 [M+H]+
Ejemplos de referencia 34 a 40
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 32 anterior, para dar cada compuesto incluido en el cuadro 29 siguiente.
r 2

(continuación)

Ejemplo de referencia 41
[Fórmula química 90]

(1) Una solución del compuesto 1 (100 mg), el compuesto 2 (73 mg) y diisopropiletilamina (197 |ji) en tetrahidrofurano (2 ml) se calentó bajo reflujo bajo atmósfera de nitrógeno por 3 horas. A la solución de reacción se le añadió el compuesto 2 (24 mg) y diisopropiletilamina (66 pl) y la mezcla se calentó bajo reflujo por 1 hora. La solución de reacción se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 80:20-50:50), para dar el compuesto 3 (107 mg) como un material viscoso incoloro. MS (APCI): m/z 358 [M+H]+
(2) El compuesto 3 (106 mg) se trató de una manera similar al paso (6) del ejemplo de referencia 33, para dar el compuesto 4 como un polvo incoloro (138 mg) que contenía cloruro de sodio. MS (APCI): m/z 344 [M+H]+ Ejemplo de referencia 42
[Fórmula química 91]

(1) A una solución del compuesto 1 (265 mg) en metanol (2 ml) se le añadió ácido sulfúrico concentrado (20 jl) y la mezcla se agitó a 50 °C por 30 minutos. A la mezcla de reacción se le añadió ácido sulfúrico concentrado (200 jl) y se agitó a 70 °C por 3 horas. Después de ser enfriada hasta temperatura ambiente, a la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-40:60), para dar el compuesto 2 (217 mg) como un material viscoso incoloro. MS (ESI): m/z 358/360 [M+H]+
(2) Una solución mixta del compuesto 2 (105 mg), ácido ciclopropilborónico (38 mg) triciclohexilfosfina (8,1 mg) y fosfato de potasio (123 mg) en tolueno (2 ml) / agua (100 jl) se desgasificó, entonces a la mezcla se le añadió acetato de paladio (3,3 mg) y se agitó bajo atmósfera de nitrógeno a 100 °C por 14 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, entonces la materia insoluble se removió por filtración y el filtrado se concentró bajo presión reducida. El residuo se purificó secuencialmente con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-50:50) y cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 95:5-75:25), para dar el compuesto 3 (78 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 320 [M+H]+
(3) A una solución del compuesto 3 (77 mg) en metanol (2 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 480 jl) a temperatura ambiente y la mezcla se agitó por 15 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 480 jl), se agitó y entonces se concentró bajo presión reducida para dar el compuesto 4 como un polvo incoloro (130 mg) que contenía cloruro de sodio. MS (APCI): m/z 306 [M+H]+
Ejemplo de referencia 43
[Fórmula química 92]

(1) A una solución del compuesto 1 (700 mg) en metanol (5 ml) se le añadió cloruro de tionilo (205 jl) y la mezcla se agitó a temperatura ambiente por 6 horas. La solución de reacción se concentró bajo presión reducida, una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma para alcalinizarla y se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida para dar el compuesto 2 (585 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 314/316 [M+H]+
(2) A una solución del compuesto 2 (580 mg) en 1,2-dicloroetano (5 ml) se le añadió cloroformiato de 1 -cloroetilo (300 jl) y entonces la mezcla se agitó a 70 °C. A la mezcla de reacción se le añadió gota a gota una solución de diisopropiletilamina (484 jl) en 1,2-dicloroetano (5 ml) y se agitó a la misma temperatura por 1 hora. A la mezcla de reacción se le añadió gota a gota una solución de cloroformiato de 1 -cloroetilo (300 jl) en 1,2-dicloroetano (5 ml), entonces se añadió gota a gota una solución de diisopropiletilamina (484 jl) en 1,2-dicloroetano (5 ml) y la mezcla se agitó a la misma temperatura por 19 horas. A la mezcla de reacción se le añadió metanol (5 ml), se agitó a 70 °C por 1 hora y entonces la solución de reacción se concentró bajo presión reducida. Al residuo se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-93:7), para dar el compuesto 3 (165 mg) como un material viscoso café. MS (ESI): m/z 258/260 [M+H]+
(3) A una suspensión del compuesto 3 (80 mg) y el compuesto 4 (34 jl) en diclorometano (2 ml) se le añadió ácido acético (27 jl), la mezcla se agitó a temperatura ambiente por 30 minutos, entonces se añadió a la misma triacetoxiborohidruro de sodio (100 mg) y se agitó a temperatura ambiente por 17 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 90:10-60:40), para dar el compuesto 5 (93 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 342/344 [M+H]+
(4) A una solución del compuesto 5 (93 mg) en metanol (2 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 540 jl) y la mezcla se agitó a 40 °C por 1 hora. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 540 jl), se agitó y entonces se concentró bajo presión reducida para dar el compuesto 6 como un polvo incoloro (151 mg) que contenía cloruro de sodio. MS (ESI): m/z 328/330 [M+H]+
Ejemplo de referencia 44
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 43 anterior, para dar el compuesto incluido en el cuadro 30 siguiente.
r

Ejemplo de referencia 45
[Fórmula química 93]

(1) A una solución mixta del compuesto 1 (167 mg) y el compuesto 2 (150 mg) en N,N-dimetilformamida (3 ml) se le añadió clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (126 mg), 1-hidroxi-7-azabenzotriazol (89 mg) y trietilamina (183 jl) y la mezcla se agitó a temperatura ambiente por 2,5 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 85:15-70:30), para dar el compuesto 3 (218 mg) como un polvo incoloro. MS (APCI): m/z 706 [M+H]+ (2) A una solución del compuesto 3 (218 mg) en diclorometano (4,4 ml) se le añadió ácido trifluoroacético (1,1 ml) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 4 horas. La mezcla de reacción se diluyó con diclorometano, entonces una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma bajo agitación hasta que la mezcla llegara a ser alcalina y entonces la capa orgánica se separó. La capa orgánica resultante se secó y entonces se concentró bajo presión reducida para dar el compuesto 4 (186 mg) como un polvo incoloro. MS (ESI): m/z 606 [M+H]+
Ejemplos de referencia 46 a 48
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 45 anterior, para dar cada compuesto incluido en el cuadro 31 siguiente.
r 1

Ejemplo de referencia 49
[Fórmula química 94]

(1) A una solución del compuesto 1 (735 mg) obtenida tratando un compuesto de partida correspondiente de una manera similar al ejemplo de referencia 1 en tetrahidrofurano (8 ml) se le añadió una solución acuosa de hidróxido de litio (hidróxido de litio (monohidrato): 78 mg agua: 4 ml) bajo enfriamiento en hielo, la mezcla se agitó a temperatura ambiente por 2 horas y entonces el tetrahidrofurano se evaporó. Al residuo se le añadió agua (20 ml) y entonces se lavó tres veces con una solución mixta (20 ml) de acetato de etilo y éter dietílico (1:3). A la capa acuosa resultante se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 935 jl) y entonces se concentró bajo presión reducida para dar el compuesto 2 como un polvo incoloro (610 mg) que contenía cloruro de litio. MS (ESI): m/z 314 [M+H]+
(2) El compuesto 2 (200 mg), el compuesto 3 (303 mg), clorhidrato de 1-etil-3-(3- dimetilaminopropil)carbodiimida (184 mg), 1-hidroxi-7-azabenzotriazol (131 mg) y trietilamina (357 jl) se añadieron a N,N-dimetilformamida (5 ml) y la mezcla se agitó a temperatura ambiente por 17 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de Nh (hexano:acetato de etilo = 90:10-65:35), para dar el compuesto 4 (280 mg) como un material viscoso incoloro. MS (ESI): m/z 696 [M+H]+ (3) A una solución del compuesto 4 (280 mg) en metanol (5 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 80 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) por 2 horas. El carbón de paladio se removió por filtración y el filtrado se concentró bajo presión reducida para dar el compuesto 5 (240 mg) como un material viscoso incoloro. MS (ESI): m/z 606 [M+H]+
Ejemplos de referencia 50 y 51
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 49 anterior, para dar cada compuesto incluido en el cuadro 32 siguiente.
r 2

Ejemplo de referencia 52
[Fórmula química 95]

(1) Una suspensión del compuesto 1 (590 |jl), el compuesto 2 (3162 |jl) y carbonato de potasio (1191 mg) en N-metilpirrolidona (3 ml) se calentó bajo radiación de microondas a 180 °C por 2 horas. La solución de reacción se enfrió hasta temperatura ambiente, se vertió en agua y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 99:1-90:10), para dar el compuesto 3 (3,336 g) como un material viscoso incoloro. MS (ESI): m/z 380/382 [M+H]+
(2) A una solución del compuesto 3 (3336 mg) y el compuesto 4 (3256 mg) en N,N-dimetilformamida (67 ml) se le añadió una solución acuosa de carbonato de sodio (2 moles/l, 3,16 ml) y aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (358 mg) y la mezcla se agitó bajo calentamiento bajo atmósfera de nitrógeno a 80 °C por 4 horas. Después de ser enfriada hasta temperatura ambiente, la mezcla de reacción se vertió en agua y se extrajo con acetato de etilo. La capa orgánica se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-75:25), para dar el compuesto 5 (3483 mg) como un material viscoso amarillo pálido. MS (APCI): m/z 483 [M+H]+
(3) A una solución del compuesto 5 (3000 mg) en etanol (30 ml) se le añadió carbón de paladio a 10% (humedecido con aproximadamente 50% de agua, 600 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 4,5 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-70:30), para dar el compuesto 6 (2880 mg) como un material viscoso incoloro. MS (APCI): m/z 485 [M+H]+
(4) A una solución del compuesto 6 (2880 mg) en 1,4-dioxano (57,6 ml)/etanol (10 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 14,86 ml) y la mezcla se agitó a temperatura ambiente por 3 horas. La mezcla de reacción se concentró bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y entonces se secó bajo presión reducida para dar el compuesto 7 (2454 mg) como un polvo amarillo pálido. MS (ESI): m/z 385 [M+H]+
Ejemplos de referencia 53 a 63
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 52 anterior, para dar cada compuesto incluido en el cuadro 33 siguiente.

continuación

Ejemplo de referencia 64
Un material de partida del ejemplo de referencia 55 se preparó de acuerdo con el siguiente método.
[Fórmula química 96]

(1) A una solución del compuesto 1 (1,296 mg) en diclorometano (12 ml) se le añadió gota a gota trifluoruro de bis(2-metoxietil)aminoazufre (1,294 ml) bajo enfriamiento, la mezcla se agitó a temperatura ambiente por 12 horas y entonces la solución de reacción se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-92:8), para dar el compuesto 2 (1,38 g) como un líquido incoloro.
(2) Una solución del compuesto 2 (1,38 g), el compuesto 3 (1,6 ml) y carbonato de potasio (1,61 g) en 1,3-dimetil-2-imidazolidinona (6 ml) se calentó bajo radiación de microondas a 180 °C por 24 horas, entonces se añadieron a la misma yoduro de etilo (1,43 ml) y carbonato de potasio (924 mg) y la mezcla se agitó a 50 °C por 4 horas. La solución de reacción se enfrió hasta temperatura ambiente, se vertió en agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 97:3-80:20), para dar el compuesto 4 (1 g) como un material viscoso amarillo pálido. MS (ESI): m/z 362/364 [M+H]+
Ejemplo de referencia 65
Un material de partida del ejemplo de referencia 56 se preparó de acuerdo con el siguiente método.
[Fórmula química 97] 1

(1) A una solución del compuesto 1 (1574 mg) en diclorometano (10 ml) se le añadió una solución acuosa de dimetilamina (solución acuosa a 50%, 2 ml) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (0,5 moles/l) y cloroformo, se agitó, entonces la capa orgánica se separó, se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida para dar el compuesto 2 (1,609 g) como un material viscoso incoloro. MS (APCI): m/z 327 [M+H]+
(2) A una solución del compuesto 2 (1,57 g) en metanol (5 ml)/tetrahidrofurano (20 ml) se le añadió carbón de paladio a 10% (humedecido con aproximadamente 50% de agua, 300 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 2 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró bajo presión reducida para dar el compuesto 3 (906 mg) como un polvo incoloro. MS (APCI): m/z 193 [M+H]+
(3) Una solución del compuesto 3 (880 mg), el compuesto 4 (556 mg) y carbonato del potasio (665 mg) en 1,3-dimetil-2-imidazolidinona (5 ml) se calentó bajo atmósfera de nitrógeno a 190 °C por 5 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se vertió en agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-60:40), para dar el compuesto 5(119 mg) como un sólido incoloro. MS (ESI): m/z 415/417 [M+H]+
Ejemplo de referencia 66
Un material de partida del ejemplo de referencia 57 se preparó de acuerdo con el siguiente método.
[Fórmula química 98]

Una solución del compuesto 1 (2 g), el compuesto 2 (2,01 g), sulfato de magnesio (950 mg) y trietilamina (2,75 ml) en tetrahidrofurano (40 ml) se agitó a temperatura ambiente por 15 horas. La materia insoluble se removió por filtración y entonces el filtrado se concentró bajo presión reducida. A una solución del residuo en diclorometano (40 ml) se le añadió bromotriclorometano (2,33 ml) y posteriormente 1,8-diazabiciclo[5.4.0]-7-undeceno (3,55 ml) bajo enfriamiento en hielo y la mezcla se agitó por 1 hora. La mezcla se agitó a temperatura ambiente por 6 horas y entonces la solución de reacción se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-60:40), se pulverizó con hexano, se colectó por filtración y entonces se secó bajo presión reducida para dar el compuesto 3 (936 mg) como un polvo incoloro. MS (APCI): m/z 327 [M+H]+
Ejemplo de referencia 67
Un material de partida del ejemplo de referencia 58 se preparó de acuerdo con el siguiente método.
[Fórmula química 99] 1

(1) El compuesto 1 (3 g) y el compuesto 2 (2,47 ml) se trataron de una manera similar al ejemplo de referencia 52, para dar el compuesto 3 (1,71 g) como un material viscoso amarillo.
(2) A una solución del compuesto 3 (1,7 g) en N-metilpirrolidona (30 ml) se le añadió oxona (Sigma-Aldrich, 4,8 g) y la mezcla se agitó a temperatura ambiente por 5 horas. A la mezcla de reacción se le añadió agua y acetato de etilo, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 4 (1055 mg) como un polvo incoloro. MS (ESI): m/z 358/360 [M+H]+
Ejemplo de referencia 68
Un material de partida del ejemplo de referencia 60 se preparó de acuerdo con el siguiente método.
[Fórmula química 100]

(1) A una solución del compuesto 1 (1 g), el compuesto 2 (794 |jl) y trifenilfosfina (2,177 g) en tetrahidrofurano (20 ml) se le añadió gota a gota azodicarboxilato de dietilo (solución a 40 % en peso de tolueno, 3,722 ml) bajo agitación. La mezcla se calentó a 70 °C por 4 horas y entonces el solvente se evaporó bajo presión reducida. Al residuo se le añadió éter diisopropílico, se agitó y entonces la materia insoluble precipitada se removió por filtración. El filtrado se concentró y entonces el mismo procedimiento se repitió de nuevo. El filtrado se concentró y el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-90:10), para dar el compuesto 3 (1319 mg) como un material viscoso amarillo pálido.
(2) A una solución del compuesto 3 (1310 mg) y el compuesto 4 (1369 mg) en N,N-dimetilformamida (26 ml) se le añadió una solución acuosa de carbonato de sodio (2 moles/l, 5,533 ml) y aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (151 mg) y la mezcla se calentó bajo atmósfera de nitrógeno a 80 °C por 5 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se vertió en agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-85:15), para dar el compuesto 5 (1423 mg) como un material viscoso incoloro. MS (APCI): m/z 458 [M+H]+
Ejemplo de referencia 69
[Fórmula química 101]

(1) El compuesto 1 y el compuesto 2 se trataron de una manera similar al ejemplo de referencia 52, para dar el compuesto 3 (305 mg).
(2) A una solución del compuesto 3 en cloroformo (6,5 ml) se le añadió ácido trifluoroacético (3,2 ml) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 1,5 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para que llegara a ser alcalina y se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida para dar el compuesto 4 racémico (235 mg) como un material viscoso incoloro. MS (ESI): m/z 371 [M+H]+
Ejemplos de referencia 70 a 73
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 69 anterior, para dar cada compuesto incluido en el cuadro 34 siguiente.
r 4

continuación

Ejemplo de referencia 74
Un material de partida del ejemplo de referencia 73 se preparó de acuerdo con el siguiente método.
[Fórmula química 102]

(1) El compuesto 1 (1385 mg) y el compuesto 2 (740 mg) se trataron de una manera similar al ejemplo de referencia 52, para dar el compuesto 3 (1104 mg) como un polvo incoloro. MS (ESI) m/z 310/312 [M+H]+ (2) A una solución de cloruro de oxalilo (525 jl) en diclorometano (20 ml) se le añadió gota a gota una solución de sulfóxido de dimetilo (652 jl) en diclorometano (15 ml) a -78 °C y entonces la mezcla se agitó a la misma temperatura por 10 minutos. A la mezcla de reacción se le añadió gota a gota una solución del compuesto 3 (950 mg) en diclorometano (10 ml) y entonces se agitó a la misma temperatura por 1 hora. A la mezcla de reacción se le añadió gota a gota trietilamina (2,56 ml) y entonces agitó a temperatura ambiente por 10 minutos. La mezcla de reacción se diluyó con diclorometano, se lavó entonces con una solución acuosa saturada de carbonato ácido de sodio, se secó y entonces el solvente se evaporó bajo presión reducida. A una solución del residuo en tbutanol (15 ml)/agua (3 ml) se le añadió fosfato diácido de sodio (807 mg) y 2-metil-2-buteno (6,5 ml). La mezcla de reacción se enfrió en hielo, se añadió clorito de sodio (609 mg) a la misma, se agitó a temperatura ambiente por 1 hora, entonces agua y solución salina saturada se añadieron a la misma y se extrajo con acetato de etilo. La capa orgánica resultante se lavó secuencialmente con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 98:2-80:20), para dar el compuesto 4 (788 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 324/326 [M+H]+
(3) A una solución del compuesto 4 (780 mg) en N,N-dimetilformamida (15 ml) se le añadió carbonato de potasio (500 mg) y yoduro de metilo (180 jl) y la mezcla se agitó a temperatura ambiente por 12 horas. La mezcla de reacción se diluyó con acetato de etilo, entonces se lavó con agua y solución salina saturada, se secó y entonces el solvente se evaporó bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 98:2-85:15), para dar el compuesto 5 (766 mg) como un material viscoso incoloro. MS (ESI): m/z 338/340 [M+H]+
Ejemplo de referencia 75
Como material de partida del ejemplo de referencia 59, un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 74 anterior, para dar el compuesto incluido en el cuadro 35 siguiente.
r '

Ejemplo de referencia 76
[Fórmula química 103]

(1) El compuesto 1 obtenido por el método descrito en el documento WO2006/47277 se trató de una manera similar al ejemplo de referencia 52, para dar el compuesto 2 (250 mg).
(2) El compuesto 2 (250 mg) se disolvió en una solución de ácido clorhídrico en metanol (2 moles/l, 12 ml) y la mezcla se agitó a 50 °C por 14 horas. La mezcla de reacción se concentró bajo presión reducida, al residuo se le añadió tolueno y se concentró de nuevo para dar el compuesto 3 racémico (193 mg) como un polvo incoloro. MS (ESI): m/z 371 [M+H]+
Ejemplo de referencia 77
[Fórmula química 104] 1

(1) Una suspensión del compuesto 1 (3 g), el compuesto 2 (3,07 ml) y carbonato de potasio (2,76 g) en N-metilpirrolidona (3 ml) se calentó bajo radiación de microondas a 190 °C por 2 horas, entonces se enfrió hasta
temperatura ambiente y se vertió en agua. La mezcla de reacción se extrajo con acetato de etilo, la capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 97:3-93:7), para dar el compuesto 3 (2,449 g) como un material viscoso incoloro. MS (ESI): m/z 438/440 [M+H]+
(2) A una solución del compuesto 3(1 g) y el compuesto 4 (450 jl) en N,N-dimetilformamida (30 ml) se le añadió una solución acuosa de carbonato de sodio (2 moles/l, 3,42 ml) y aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (93 mg) y la mezcla se calentó a 80 °C bajo atmósfera de nitrógeno por 3 horas. Después de ser enfriada hasta temperatura ambiente, la mezcla de reacción se vertió en agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano: acetato de etilo = 95: 5-85: 15), para dar el compuesto 5 (768 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 353 [M+H]+
(3) El compuesto 5 se trató de una manera similar al paso (2) del ejemplo de referencia 52, para dar el compuesto 6. MS (APCI) m/z 359 [M+H]+
Ejemplo de referencia 78
[Fórmula química 105]

(1) Una suspensión del compuesto 1 (1,5 g), el compuesto 2 (1618 jl) y carbonato de potasio (2073 mg) en N-metilpirrolidona (15 ml) se calentó a 190 °C bajo radiación de microondas por 1,5 horas y entonces se enfrió hasta temperatura ambiente y se vertió en agua. La mezcla de reacción se extrajo con acetato de etilo, la capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 98:2-85:15), para dar el compuesto 3 (731 mg) como un material viscoso incoloro. MS (APCI): m/z 337/339 [M+H]+
(2) A una solución del compuesto 3 (730 mg) en ácido trifluoroacético (4 ml) se le añadió ácido sulfúrico concentrado (1 ml) y la mezcla se agitó a temperatura ambiente por 7 días. La solución de reacción se añadió gota a gota a agua enfriada en hielo, entonces una solución acuosa de hidróxido de sodio 1 N se añadió a la misma para neutralizarla y se extrajo con acetato de etilo. La capa orgánica se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el compuesto 4 (664 mg) como un polvo incoloro. MS (APCl): m/z 355/357 [M+H]+
(3) El compuesto 4 (640 mg) y el compuesto 5 (613 mg) se trataron de una manera similar al paso (2) del ejemplo de referencia 52, para dar el compuesto 6 (631 mg) como un polvo incoloro. MS (APCI) m/z 458 [M+H]+ (4) El compuesto 6 (620 mg) se trató de una manera similar al paso (3) del ejemplo de referencia 52, para dar el compuesto 7 (621 mg) como un polvo incoloro. MS (APCI) m/z 460 [M+H]+
(5) A una solución del compuesto 7 (500 mg) en N,N-dimetilformamida (10 ml) se le añadió cloruro cianúrico (590 mg) bajo enfriamiento en hielo y la mezcla se agitó por 1,5 horas. A la solución de reacción se le añadió agua enfriada en hielo y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con una solución acuosa saturada de carbonato ácido de sodio y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-75:25), para dar el compuesto 8 (459 mg) como un material viscoso incoloro. MS (ESI): m/z 442 [M+H]+ (6) El compuesto 8 (458 mg) se trató de una manera similar al paso (4) del ejemplo de referencia 52, para dar el compuesto 9 (458 mg) como un polvo incoloro. MS (ESI) m/z 342 [M+H]+
Ejemplo de referencia 79
[Fórmula química 106]

(1) El compuesto 1 se trató de una manera similar al paso (1) del ejemplo de referencia 52, para dar el compuesto 2 (393 mg).
(2) A una solución del compuesto 2 (393 mg) en etanol (4 ml) / ácido acético (4 ml) se le añadió óxido de platino(IV) (99 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 16 horas. La materia insoluble se removió por filtración, entonces al filtrado se le añadió agua y acetato de etilo, posteriormente se añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-80:20), para dar el compuesto 3 (202 mg) como un material viscoso incoloro. MS (ESI): m/z 451/453 [M+H]+
(3) El compuesto 3 (200 mg) se trató de una manera similar al paso (4) del ejemplo de referencia 52, para dar el compuesto 4 (181 mg) como un polvo incoloro. MS (ESI) m/z 351/353 [M+H]+
Ejemplo de referencia 80
[Fórmula química 107] 1

(1) A una solución del compuesto 1 (4 g) en metanol (80 ml) se le añadió ácido sulfúrico concentrado (1,94 ml) bajo enfriamiento en hielo y la mezcla se calentó bajo reflujo por 15 horas. El metanol se evaporó bajo presión
reducida, al residuo se le añadió agua enfriada en hielo y entonces la mezcla se extrajo con acetato de etilo. La capa orgánica resultante se lavó con una solución acuosa saturada de carbonato ácido de sodio y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 98:2-85:15), para dar el compuesto 2 (3,68 g) como un material viscoso incoloro. MS (APCI): m/z 283 y 285 [M+H]+
(2) El compuesto 2 se trató de una manera similar al ejemplo de referencia 52, para dar el compuesto 3 como un polvo incoloro. MS (ESI) m/z 288 [M+H]+
(3) El compuesto 3 se trató de una manera similar al ejemplo 10, para dar el compuesto 4 como un polvo incoloro. MS (ESI) m/z 565 [M+H]+
(4) El compuesto 4 se trató de una manera similar al ejemplo 11, para dar el compuesto 5 como un polvo incoloro. MS (ESI) m/z 551 [M+H]+
Ejemplo de referencia 81
[Fórmula química 108] 1

(1) A una solución del compuesto 1 (2 g) y bromuro de bencilo (1086 jl) en N,N-dimetilformamida (40 ml) se le añadió hidruro de sodio (60 % en aceite, 398 mg) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 3 horas. La mezcla de reacción se vertió en agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 98:2-90:10), para dar el compuesto 2 (2,7 g) como un polvo incoloro.
(2) A una solución del compuesto 2 (1 g) y el compuesto 3 (1027 mg) en N,N-dimetilformamida (20 ml) se le añadió una solución acuosa de carbonato de sodio (2 moles/l, 4,53 ml) y aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (123 mg) y la mezcla se calentó a 80 °C bajo atmósfera de nitrógeno por 4 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se vertió en agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-85:15), para dar el compuesto 4 (935 mg) como un material viscoso amarillo pálido. MS (APCI): m/z 334 [M-Boc+H]+.
(3) A una solución del compuesto 4 (930 mg) en metanol (19 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 186 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 4 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró, para dar el compuesto 5 (723 mg) como un polvo incoloro.
(4) A una solución del compuesto 5 (300 mg) en diclorometano (3,6 ml) se le añadió anhídrido trifluorometansulfónico (183 jl) y posteriormente trietilamina (182 jl) bajo enfriamiento en hielo, la mezcla se agitó por 10 minutos, entonces se calentó hasta temperatura ambiente y se agitó por 1 hora. A la mezcla de reacción se le añadió anhídrido trifluorometansulfónico (43 jl) y posteriormente trietilamina (43 jl) bajo enfriamiento en hielo y se agitó a temperatura ambiente por 14 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y diclorometano, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se secó y se concentró bajo presión reducida. A una solución del residuo en N,N-dimetilformamida (2 ml) se le añadió ácido 4-metoxicarbonilfenilborónico (45 mg), una solución acuosa de carbonato de sodio (2 moles/l, 251 jl) y aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (7 mg) y la mezcla se agitó bajo atmósfera de nitrógeno a 80 °C por 2 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se vertió en agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo =
90:10-75:25), para dar el compuesto 6 (62 mg) como un polvo incoloro. MS (APCI): m/z 464 [M+H]+
(5) A una solución del compuesto 6 (60 mg) en 1,4-dioxano (1,2 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 324 jl) y la mezcla se agitó a temperatura ambiente por 23 horas. La mezcla de reacción se concentró bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y entonces se secó para dar el compuesto 7 (49 mg) como un polvo incoloro. Ms (APCI): m/z 364 [M+H]+
Ejemplo de referencia 82
[Fórmula química 109]

(1) A una solución del compuesto 1 (800 mg) en diclorometano (23 ml) se le añadió anhídrido trifluorometansulfónico (487 jl) y posteriormente trietilamina (484 jl) bajo enfriamiento en hielo, la mezcla se agitó por 10 minutos, entonces se calentó hasta temperatura ambiente y se agitó por 5 horas. A la mezcla de reacción se le añadió agua enfriada en hielo y se extrajo con diclorometano. La capa orgánica resultante se secó y se concentró bajo presión reducida. A una solución de la mitad de la cantidad del residuo y el compuesto 2 (361 mg) obtenida por el método descrito en el documento WO2013/187496 en N,N-dimetilformamida (8 ml), se le añadió una solución acuosa de carbonato de sodio (2 moles/l, 1759 jl) y aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (48 mg) y la mezcla se calentó a 60 °C bajo atmósfera de nitrógeno por 2,5 horas. A la mezcla de reacción se le añadió el compuesto 2 (164 mg) y aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (48 mg) y se agitó a 60 °C por 16 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se vertió en agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 3 racémico (502 mg) como un material viscoso incoloro. MS (ESI): m/z 482 [M+H]+
(2) A una solución del compuesto 3 (100 mg) en 1,4-dioxano (2 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 519 jl) y la mezcla se agitó a temperatura ambiente por 24 horas. La mezcla de reacción se concentró bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y entonces se secó para dar el compuesto 4 (81 mg) como un polvo incoloro. MS (APCI): m/z 382 [M+H]+
Ejemplo de referencia 83
[Fórmula química 110]
(1) El compuesto 1 (173 mg) y el compuesto 2 se trataron de una manera similar al ejemplo de referencia 82, para dar el compuesto 3 (102 mg) como un material viscoso incoloro. MS (ESI): m/z 482 [M+H]+
(2) A una solución del compuesto 3 (95 mg) en diclorometano (2 ml) se le añadió ácido trifluoroacético (1 ml) y la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con diclorometano. La capa orgánica resultante se secó y se concentró bajo presión reducida para dar el compuesto 4 (72 mg) como un material viscoso incoloro. MS (ESI): m/z 382 [M+H]+
Ejemplo de referencia 84
[Fórmula química 111]

(1) A una solución del compuesto 1 (400 mg) en etanol (8 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 80 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 3,5 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró bajo presión reducida para dar el compuesto 2 (390 mg) como un material viscoso incoloro. MS (ESI): m/z 384 [M-Boc+H]+.
(2) A una solución del compuesto 2 (290 mg) en t-butanol (6 ml) se le añadió t-butóxido de potasio (74 mg) y entonces la mezcla se calentó a 40 °C por 4 horas. A la mezcla de reacción se le añadió agua (30 jl), se agitó por 1,5 horas, entonces se añadió más agua (50 jl) y se agitó a la misma temperatura por 16 horas. A la mezcla de reacción se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 900 jl), se agitó a 30 °C por 0,5 horas y entonces se dejó que reposara a temperatura ambiente por 2 días. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 1200 jl), entonces se vertió en agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se pulverizó con hexano, se colectó por filtración y se secó bajo presión reducida para dar el compuesto 3 (trans) (210 mg) como un polvo incoloro. MS (APCI): m/z 454 [M-H]-.
(3) A una solución del compuesto 3 (205 mg) en metanol (3 ml) se le añadió cloruro de tionilo (174 jl) bajo enfriamiento en hielo y entonces la mezcla se calentó hasta temperatura ambiente por 16 horas. La mezcla de reacción se concentró bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y se secó bajo presión reducida para dar el compuesto 4 (180 mg) como un polvo incoloro. MS (APCl): m/z 370 [M+H]+
Ejemplo de referencia 85
[Fórmula química 112] 1

(1) Una suspensión del compuesto 1 (200 mg), el compuesto 2 (100 jl) y carbonato del potasio (160 mg) en N,N-dimetilformamida (4 ml) se calentó a 70 °C bajo atmósfera de nitrógeno por 2,5 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se vertió en agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 97:3-80:20), para dar el compuesto 3 (263 mg) como un material viscoso incoloro. MS (APCI): m/z 460 [M+H]+
(2) A una solución del compuesto 3 (263 mg) en 1,4-dioxano (5 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 1431 jl) y la mezcla se agitó a temperatura ambiente por 22 horas. La mezcla de reacción se concentró bajo presión reducida, el residuo se pulverizó con éter dietílico, se colectó por filtración y
entonces se secó para dar el compuesto 4 (184 mg) como un polvo incoloro. MS (APCI): m/z 360 [M+H]+ Ejemplo de referencia 86
[Fórmula química 113]

(1) Una suspensión del compuesto 1 (200 mg), el compuesto 2 (70 jl) y carbonato del potasio (160 mg) en N,N-dimetilformamida (4 ml) se calentó a 70 °C bajo atmósfera de nitrógeno por 2 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se vertió en agua y se extrajo con acetato de etilo. La capa orgánica se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-60:40), para dar el compuesto 3 (245 mg) como un material viscoso incoloro. MS (ESI): m/z 413 [M+H]+
(2) A una solución del compuesto 3 (240 mg) en diclorometano (3 ml) se le añadió ácido trifluoroacético (2 ml) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con cloroformo. La capa orgánica se secó y se concentró bajo presión reducida para dar el compuesto 4 (175 mg) como un material viscoso incoloro. MS (ESI): m/z 313 [M+H]+
Ejemplo de referencia 87
[Fórmula química 114]

(1) A una solución del compuesto 1 (200 mg), el compuesto 2 (193 mg) y trifenilfosfina (304 mg) en tetrahidrofurano (4 ml) se le añadió azodicarboxilato de dietilo (solución a 40 % en peso de tolueno, 526 jl) a temperatura ambiente y entonces se calentó a 70 °C por 2 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se concentró bajo presión reducida, se añadió éter diisopropílico a la misma, se agitó, la materia insoluble precipitada se removió por filtración y entonces el filtrado se concentró. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 99:1-85:15), para dar el compuesto 3 (124 mg) como un material viscoso incoloro. MS (ESI): m/z 550 [M+H]+
(2) A una solución del compuesto 3 (123 mg) en 1,4-dioxano (2,5 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 1120 jl) y la mezcla se agitó a temperatura ambiente por 14 horas. La mezcla de reacción se concentró bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y entonces se secó para dar el compuesto 4 (103 mg) como un polvo incoloro. MS (APCI): m/z 450 [M+H]+
Ejemplo de referencia 88
[F ó rm u la q u ím ic a 115 ]

(1) Una solución del compuesto 1 (3 g), el compuesto 2 (3,54 g) y carbonato de potasio (2,56 g) en 1,3-dimetil-2-imidazolidinona (6 ml) se calentó a 120 °C por 16 horas, entonces se enfrió hasta temperatura ambiente y se vertió en agua. La mezcla de reacción se extrajo con acetato de etilo, la capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 99:1-80:20), para dar el compuesto 3 (1,14 g) como un material viscoso incoloro. MS (ESI): m/z 366/368 [M+H]+
(2) A una solución del compuesto 3 (769 mg) en tetrahidrofurano (4 ml) se le añadió gota a gota una solución acuosa de ácido clorhídrico (2 moles/ml, 1,05 ml) y entonces la mezcla se agitó a temperatura ambiente por 5 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para neutralizarla y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 99:1-80:30), para dar el compuesto 4 (494 mg) como un material viscoso incoloro. Ms (ESI): m/z 322/324 [M+H]+
(3) A una solución del compuesto 4 (494 mg) y el compuesto 5 (570 mg) en N,N-dimetilformamida (12 ml) se le añadió una solución acuosa de carbonato de sodio (2 moles/l, 1,5 ml) y aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (125 mg) y la mezcla se calentó a 80 °C bajo atmósfera de nitrógeno por 15 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se vertió en agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-60:40), para dar el compuesto 6 (641 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 425 [M+H]+
(4) A una solución del compuesto 6 (641 mg) en etanol (15 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 213 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 17 horas. El carbón de paladio se removió por filtración, entonces el filtrado se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 70:30-30:70), para dar el compuesto 7 (213 mg) como un material viscoso incoloro. MS (ESI): m/z 427 [M+H]+
Ejemplo de referencia 89
[Fórmula química 116] 1

(1) A una solución del compuesto 1 (150 mg) y una solución de dimetilamina-tetrahidrofurano (2 moles/ml, 210 pl) en 1,2-dicloroetano (2 ml) se le añadió triacetoxiborohidruro de sodio (112 mg) y la mezcla se agitó por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y agua, se agitó y entonces se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de n H (hexano:acetato de etilo = 80:20-0:100), para dar el compuesto 2 (134 mg) como un material viscoso incoloro. MS (ESI): m/z 456 [M+H]+
(2) A una solución del compuesto 2 (134 mg) en cloroformo (3 ml) se le añadió ácido trifluoroacético (1,5 ml) y la mezcla se agitó a temperatura ambiente por 1,5 horas. A la solución de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con cloroformo. La capa orgánica se secó y se concentró para dar el compuesto 3 (102 mg) como un material viscoso amarillo pálido. MS (ESI): m/z
356 [M+H]+
Ejemplo de referencia 90
[Fórmula química 117]

(1) A una solución del compuesto 1 (300 mg) y una solución de metilamina-metanol (40 %, 90 |jl) en 1,2-dicloroetano (3,5 ml) se le añadió triacetoxiborohidmro de sodio (224 mg) y la mezcla se agitó por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y agua, se agitó y entonces se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida para dar el compuesto 2 (307 mg) como un material viscoso incoloro. MS (ESI): m/z 442 [M+H]+
(2) A una solución del compuesto 2 (100 mg) en cloroformo (1 ml) se le añadió trietilamina (40 jl) y cloruro de acetilo (20 jl) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 1,5 horas. A la mezcla de reacción se le añadió agua y cloroformo, se agitó, entonces la capa orgánica se separó, se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 70:30-20:80), para dar el compuesto 3 (86 mg) como un material viscoso incoloro. MS (ESI): m/z 484 [M+H]+
(3) A una solución del compuesto 3 (85 mg) en cloroformo (2 ml) se le añadió ácido trifluoroacético (1,8 ml) y la mezcla se agitó a temperatura ambiente por 1 hora. A la solución de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida para dar el compuesto 4 (63 mg) como un sólido incoloro. MS (ESI): m/z 384 [M+H]+
(4) A una solución del compuesto 2 (100 mg) y trietilamina (80 jl) en cloroformo (2 ml) se le añadió cloruro de metansulfonilo (40 jl) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 1 hora. A la mezcla de reacción se le añadió agua y diclorometano, se agitó, entonces la capa orgánica se separó, se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 85:15-40:60), para dar el compuesto 5 (64 mg) como un material viscoso incoloro. MS (ESI): m/z 520 [M+H]+
(5) A una solución del compuesto 5 (64 mg) en cloroformo (1,2 ml) se le añadió ácido trifluoroacético (0,6 ml) y la mezcla se agitó a temperatura ambiente por 1,5 horas. A la solución de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida para dar el compuesto 6 (48 mg) como un sólido incoloro. MS (ESI): m/z 420 [M+H]+
Ejemplo de referencia 91
[Fórmula química 118]

(1) A una solución del compuesto 1 (400 mg) y bencilamina (120 |jl) en 1,2-dicloroetano (5 ml) se le añadió triacetoxiborohidruro de sodio (298 mg) y la mezcla se agitó por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y agua, se agitó y entonces se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida para dar el compuesto 2 (408 mg) como un sólido incoloro. MS (ESI): m/z 518 [M+H]+
(2) A una solución del compuesto 2 (400 mg) en tetrahidrofurano (6 ml)/éter diisopropílico (2 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 80 mg) e hidróxido de carbón de paladio (humedecido con aproximadamente 50% de agua, 80 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 5 horas. La materia insoluble se removió por filtración y entonces el filtrado se concentró. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 50:50-0:100), para dar el compuesto 3 (222 mg) como un material viscoso incoloro. MS (ESI): m/z 428 [M+H]+
(3) A una solución del compuesto 3 (100 mg) en cloroformo (2,3 ml) se le añadió trietilamina (40 jl) y cloruro de 4-clorobutirilo (30 jl) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y cloroformo, se agitó, entonces la capa orgánica se separó, se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. A una solución del residuo en N,N-dimetilformamida (2,3 ml) se le añadió hidruro de sodio (60 % en aceite, 11 mg) bajo enfriamiento en hielo, la mezcla se agitó por 2 horas, entonces más hidruro de sodio (60 % en aceite, 11 mg) se añadió a la misma bajo enfriamiento en hielo y se agitó a temperatura ambiente por otras 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de cloruro de amonio y acetato de etilo, se agitó, entonces la capa orgánica se separó, se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 30:70-0:100), para dar el compuesto 4 (87 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 496 [M+H]+
(4) A una solución del compuesto 4 (87 mg) en cloroformo (1,8 ml) se le añadió ácido trifluoroacético (0,88 ml) y la mezcla se agitó a temperatura ambiente por 4 horas. A la solución de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con cloroformo. La capa orgánica se secó y se concentró para dar el compuesto 5 (65 mg) como un material viscoso incoloro. MS (ESI): m/z 396 [M+H]+
(5) A una solución del compuesto 3 (80 mg) en diclorometano (1,9 ml) se le añadió piridina (100 jl) y cloruro de 3-cloropropansulfonilo (60 jl) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 15 horas. A la mezcla de reacción se le añadió agua y cloroformo, se agitó, entonces la capa orgánica se separó, se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. A una solución del residuo en N,N-dimetilformamida (1,9 ml) se le añadió hidruro de sodio (60 % en aceite, 8,2 mg) a temperatura ambiente, la mezcla se agitó por 2 horas, entonces se calentó a 60 °C por 1,5 horas y se agitó a temperatura ambiente por otras 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de cloruro de amonio y acetato de etilo, se agitó, entonces la capa orgánica se separó, se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-90:10), para dar el compuesto 6 (25 mg) como un material viscoso incoloro. MS (ESI): m/z 532 [M+H]+
(6) A una solución del compuesto 6 (25 mg) en cloroformo (0,5 ml) se le añadió ácido trifluoroacético (0,5 ml) y la mezcla se agitó a temperatura ambiente por 3 horas. A la solución de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con cloroformo. La capa orgánica se secó y se concentró bajo presión reducida para dar el compuesto 7 (18 mg) como un material viscoso incoloro. MS (ESI): m/z 432 [M+H]+
Ejemplo de referencia 92
[F ó rm u la q u ím ic a 119 ]

(1) A una solución del compuesto 1 (4 g) en tolueno (31 ml) se le añadió el compuesto 2 (6,06 g) y entonces la mezcla se calentó bajo reflujo por 1 hora. La solución de reacción se concentró, entonces al residuo se le añadió éter diisopropílico y se agitó. La materia insoluble se removió por filtración, entonces el filtrado se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 98:2-80:20), para dar el compuesto 3 (4,7 g) como un material viscoso amarillo pálido. MS (APCI): m/z 323/325 [M+H]+
(2) El compuesto 3 se trató de una manera similar al ejemplo de referencia 52, para dar el compuesto 4 como un polvo incoloro. MS (APCI) m/z 330 [M+H]+
Ejemplo de referencia 93
[Fórmula química 120] 1

(1) A una solución del compuesto 1 (1 g) en diclorometano (10 ml) se le añadió gota a gota una solución de hidruro de diisobutilaluminio en tolueno (1 mol/l, 9,6 ml) bajo atmósfera de nitrógeno a -78 °C y la mezcla se agitó a 0 °C por 2 horas. A la mezcla de reacción se le añadió agua enfriada en hielo, entonces se añadió tetrahidrato de tartrato de sodio y potasio, se añadió acetato de etilo y se agitó a temperatura ambiente por 15 horas. La materia insoluble se removió por filtración y entonces la capa orgánica del filtrado se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 67:33-40:60), para dar un alcohol (252 mg) como un material viscoso incoloro.
(2) A una solución de cloruro de oxalilo (111 pl) en diclorometano (5 ml) se le añadió gota a gota una solución de sulfóxido de dimetilo (137 pl) en diclorometano (1 ml) a -78 °C y la mezcla se agitó por 10 minutos. A la mezcla de reacción se le añadió gota a gota una solución del alcohol (250 mg) obtenido en el paso (1) anterior en diclorometano (5 ml) a la misma temperatura y se agitó por 30 minutos. A la mezcla de reacción se le añadió gota a gota trietilamina (540 pl) a la misma temperatura y entonces se agitó a temperatura ambiente por 30 minutos. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se evaporó bajo presión reducida para dar el compuesto 2 (263 mg) como un material viscoso amarillo pálido. (3) A una solución del compuesto 2 (262 mg) en tolueno (1,4 ml) se le añadió el compuesto 3 (262 mg) y entonces la mezcla se calentó bajo reflujo a 70 °C por 2,5 horas. La solución de reacción se concentró bajo presión reducida, entonces al residuo se le añadió éter diisopropílico y la mezcla se agitó. La materia insoluble precipitada se removió por filtración y entonces el filtrado resultante se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-75:25), para dar el compuesto 4 (208 mg) como un material viscoso incoloro. MS (ESI): m/z 456 [M+H]+
(4) El compuesto 4 (60 mg) se trató de una manera similar al ejemplo de referencia 52, para dar el compuesto 5 (51 mg) como un polvo incoloro. MS (APCI) m/z 356 [M+H]+
(5) A una solución del compuesto 4 (145 mg) en etanol (3 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 29 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1
atm) a temperatura ambiente por 18 horas. La mezcla de reacción se filtró y el filtrado se concentró bajo presión reducida para dar el compuesto 6 (140 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 458 [M+H]+ (6) El compuesto 6 (139 mg) se trató de una manera similar al ejemplo de referencia 52, para dar el compuesto 7 (118 mg) como un polvo incoloro. MS (APCI) m/z 358 [M+H]+
Ejemplo de referencia 94
[Fórmula química 121]

(1) A una solución del compuesto 1 (15 g) y bromuro de bencilo (7 ml) en N,N-dimetilformamida (120 ml) se le añadió carbonato de potasio (12,9 g) y entonces la mezcla se agitó a temperatura ambiente por 17 horas. La mezcla de reacción se vertió en agua y se extrajo con acetato de etilo. La capa orgánica se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de n H (hexano:acetato de etilo = 100:0-90:10), para dar el compuesto 2 (19,67 g) como un material viscoso incoloro.
(2) A una solución del compuesto 2 (19,66 g) en tetrahidrofurano (60 ml) se le añadió gota a gota una solución de n-butil litio en hexano (1,59 moles/l, 39,2 ml) bajo atmósfera de nitrógeno a -78 °C y la mezcla se agitó por 1 hora. A la mezcla de reacción se le añadió gota a gota una solución del compuesto 3 (3,39 g) en tetrahidrofurano (60 ml) y entonces se agitó a temperatura ambiente por 17 horas. A la mezcla de reacción se le añadió gota a gota una solución acuosa de ácido clorhídrico (1 mol/l, 80 ml) y entonces se extrajo con acetato de etilo. La capa orgánica se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-90:10), para dar el compuesto 4 (2,76 g) como un material viscoso anaranjado. MS (ESI): m/z 365 [M-H]-.
(3) A una solución del compuesto 4 (2,75 g) en metanol (25 ml) se le añadió gota a gota una solución de trimetilsilildiazometano en hexano (2 moles/l, 30 ml) y la mezcla se agitó a temperatura ambiente por 17 horas. A la mezcla de reacción se le añadió ácido acético gota a gota hasta que no se formara una burbuja de gas y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-75:25), para dar el compuesto 5 (2,019 g) como un material viscoso amarillo pálido. S (ESI): m/z 379 [M-H]-.
(4) Una mezcla del compuesto 5 (2,01 g), trietilsilano (4,2 ml) y ácido trifluoroacético (3,9 ml) se agitó a temperatura ambiente por 5 horas. La mezcla de reacción se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-85:15), para dar el compuesto 6 (1,31 g) como un material viscoso amarillo pálido. MS (ESI): m/z 379 [M-H]-.
(5) A una solución del compuesto 6 (1,3 g) en metanol (20 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 650 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 2 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-70:30), para dar el compuesto 7 (865 mg) como un polvo incoloro. MS (APCI): m/z 275 [M+H]+
(6) Una solución del compuesto 7 (855 mg) en diclorometano (15 ml) se enfrió en hielo, trietilamina (652 pl) y anhídrido trifluorometansulfónico (629 pl) se añadieron a la misma bajo atmósfera de nitrógeno y entonces se agitó a la misma temperatura por 1 hora. A la mezcla de reacción se le añadió una solución acuosa saturada de cloruro de amonio y diclorometano, se agitó y entonces se extrajo con diclorometano. La capa orgánica se secó y se concentró bajo presión reducida. A una solución del residuo resultante en N,N-dimetilformamida (15 ml) se le
añadió el compuesto 8 (1,06 g), una solución acuosa de carbonato de sodio (2 moles/l, 4,68 ml) y aducto de diclorometano de didoro[1,1'-bis(difenilfosfino)ferroceno]paladio(N) (127 mg) y la mezcla se agitó bajo calentamiento a 80 °C bajo atmósfera de nitrógeno por 16 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, entonces se vertió en agua y se extrajo con acetato de etilo. La capa orgánica se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 9 (cis) (465 mg) y el compuesto 10 (trans) (164 mg) como materiales viscosos incoloros. Cada MS (ApCI): m/z 440 [M+H]+
(7) A una solución del compuesto 9 (460 mg) en metanol (3 ml)/ácido acético (3 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 230 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 2 horas. El carbón de paladio se removió por filtración con Celite y entonces el filtrado resultante se concentró. El residuo se disolvió en diclorometano, se lavó con una solución acuosa saturada de carbonato ácido de sodio, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-75:25). A una solución del compuesto resultante (405 mg) en diclorometano (2 ml) se le añadió ácido trifluoroacético (1 ml) y la mezcla se agitó a temperatura ambiente por 1 hora. A la solución de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con diclorometano. La capa orgánica se secó y se concentró bajo presión reducida para dar el compuesto 11 (314 mg) como un material viscoso incoloro. MS (ESI): m/z 342 [M+H]+
(8) El compuesto 10 se trató de una manera similar al paso 7, para dar el compuesto 12 como un material viscoso incoloro. MS (ESI): m/z 342 [M+H]+
Ejemplo de referencia 95
[Fórmula química 122]

(1) A una solución del compuesto 1 (1 g) y el compuesto 2 (870 mg) en tolueno (2,5 ml) se le añadió una solución de bis(trimetilsilil)amida de potasio en tolueno (0,5 moles/l, 8,23 ml) bajo atmósfera de nitrógeno a temperatura ambiente y entonces la mezcla se calentó bajo reflujo por 15 minutos. A la mezcla de reacción se le vertió una solución acuosa saturada de cloruro de amonio bajo enfriamiento en hielo y se extrajo con acetato de etilo. La capa orgánica se lavó con una solución acuosa de ácido clorhídrico (1 mol/l), una solución acuosa saturada de carbonato ácido de sodio y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-80:20), para dar el compuesto 3 (1,08 g) como un material viscoso amarillo pálido. MS (APCI): m/z 333/335 [M-Boc+H]+
(2) A una solución del compuesto 3 (160 mg) en N,N-dimetilformamida (3 ml) se le añadió ácido 4-metoxicarbonilfenilborónico (93 mg), una solución acuosa de carbonato de sodio (2 moles/l, 554 pl) y aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (14 mg) y la mezcla se calentó a 80 °C bajo atmósfera de nitrógeno por 2 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se vertió en agua y se extrajo con acetato de etilo. La capa orgánica se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 85:15-67:33), para dar el compuesto 4 (164 mg) como un polvo incoloro.
(3) A una solución del compuesto 4 (164 mg) en 1,4-dioxano (3 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 1680 pl) y la mezcla se agitó a temperatura ambiente por 14 horas. La mezcla de reacción se concentró bajo presión reducida, el residuo se pulverizó con éter diisopropílico, se colectó por filtración y entonces se secó para dar el compuesto 5 (134 mg) como un polvo incoloro. MS (ESI): m/z 389 [M+H]+
Ejemplo de referencia 96
[F ó rm u la q u ím ic a 123 ]

A una solución del compuesto 1 (200 mg) en cloroformo (4,1 ml) se le añadió ácido trifluoroacético (2 ml) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 1 hora. La mezcla de reacción se diluyó con cloroformo, entonces una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma para alcalinizarla y la capa orgánica se separó. La capa orgánica resultante se secó y entonces se concentró bajo presión reducida para dar el compuesto 2 (186 mg) como un polvo incoloro. MS (ESI): m/z 383 [M+H]+
Ejemplo de referencia 97
Un compuesto de partida correspondiente se hizo reaccionar y se trató de una manera similar al ejemplo de referencia 52 anterior y el ejemplo de referencia 96 anterior, para dar el compuesto incluido en el cuadro 36 siguiente.
r

Ejemplo de referencia 98
[Fórmula química 124]

El compuesto 1 obtenido por el método descrito en el documento WO2006/47277 se trató de una manera similar al ejemplo de referencia 52, para dar el compuesto 2 (106 mg). El compuesto 2 se disolvió en una solución de ácido clorhídrico en metanol (2 moles/l, 5,2 ml), se agitó a 50 °C por 18 horas, entonces la solución de reacción se concentró bajo presión reducida, se añadió tolueno a la misma y la mezcla se concentró de nuevo bajo presión reducida para dar el compuesto 3 (180 mg). MS (ESI): m/z 369 [M+H]+
Ejemplo de referencia 99
[F ó rm u la q u ím ic a 125 ]

(1) Una suspensión del compuesto 1 (3,05 g), el compuesto 2 (3,25 g) y carbonato de potasio (3,3 g) en N-metilpirrolidona (40 ml) se agitó a 80 °C por 19 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, se añadieron a la misma agua y acetato de etilo, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 98:2-88:12), para dar el compuesto 3 (4,61 g) como un material viscoso amarillo. MS (ESI): m/z 358 [M+H]+
(2) A una solución del compuesto 3 (4,61 g) en diclorometano (16 ml) se le añadió una solución del compuesto 4 (4,43 g) en diclorometano (16 ml) y la mezcla se agitó a temperatura ambiente por 16 horas. La solución de reacción se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 98:2-85:15), para dar el compuesto 5 (5,16 g) como un polvo amarillo pálido a la relación isomérica de E:Z “ 88:12. MS (ESI): m/z 414 [M+H]+
(3) A una solución del compuesto 5 (3,8 g) en metanol (20 ml) / tetrahidrofurano (10 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 5,05 ml) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 16 horas. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 5,05 ml), se agitó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-96:4), para dar el compuesto 6 (2,07 g) como un polvo amarillo pálido a la relación isomérica de E:Z “ 95:5. MS (ESI): m/z 398 [M-H]-(4) A una solución del compuesto 6 (2,06 g) y N,N-dimetilformamida (10 gotas) en diclorometano (30 ml) se le añadió cloruro de oxalilo (873 pl) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 1 hora. La solución de reacción se concentró bajo presión reducida, entonces el residuo se disolvió en diclorometano (30 ml), se añadieron a la misma cloruro de litio (437 mg), el compuesto 7 (1 g) y trietilamina (1,58 ml) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 16 horas. La solución de reacción se vertió en una solución acuosa de ácido cítrico enfriada en hielo (ácido cítrico: 10 g y agua: 100 ml) y se extrajo con diclorometano. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-75:25), para dar el compuesto 8 (2,21 g) como un polvo amarillo. MS (APCI): m/z 559 [M+H]+
(5) A una solución del compuesto 8 (2,21 g) y el compuesto 9 (2,82 g) en diclorometano (20 ml) se le añadió ácido trifluoroacético (30 pl) bajo enfriamiento en hielo y la mezcla se agitó a 40 °C por 1 hora. A la mezcla de reacción se le añadió agua, una solución acuosa saturada de carbonato ácido de sodio y diclorometano bajo enfriamiento en hielo, se agitó y la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-60:40), para dar el compuesto 10 (1,19 g) y el compuesto 11 (1,15 g) como polvos incoloros. Cada MS (APCI): m/z 692 [M+H]+ TLC (hexano:acetato de etilo = 2:1): valor de Rf del compuesto 10 ~ 0,8, valor de Rf del compuesto 11 “ 0,4 (placa de TLC: gel de sílice para TLC 1.05715.0001 60 F254, fabricado por Merck KGaA). A una solución del compuesto 10 en acetato de etilo se le añadió una solución de ácido clorhídrico en acetato de etilo (4 moles/l) en 1,2 equivalentes para dar un cristal del clorhidrato 12 y el cristal se sometió a cristalografía de rayos X para confirmar que los compuestos 10 y 11 tienen las configuraciones anteriores, respectivamente.
(6) A una solución del compuesto 11 (288 mg) en metanol (6 ml)/tetrahidrofurano (6 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 150 mg) y la mezcla se agitó a temperatura ambiente bajo atmósfera de hidrógeno (1 atm) por 2 horas. El carbón de paladio se removió por filtración con Celite y el filtrado se concentró bajo presión reducida para dar el compuesto 13 (261 mg) como un polvo incoloro. MS (ESI): m/z 602 [M+H]+
Ejemplo de referencia 100
[Fórmula química 126]

(1) A una solución del compuesto 1 (500 mg) en tetrahidrofurano (7,2 ml) se le añadió gota a gota una solución acuosa de hidróxido de litio (monohidrato) (36 mg) (0,7 ml) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 3 horas. El tetrahidrofurano se evaporó bajo presión reducida, al residuo se le añadió cloroformo y agua y entonces la capa orgánica se separó. La capa acuosa se extrajo dos veces con cloroformo, las capas orgánicas resultantes se combinaron, se lavaron con solución salina saturada, se secaron y se concentraron bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-85:15), para dar el compuesto 2 (328 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 533 [M+H]+
(2) A una solución del compuesto 2 (260 mg) en diclorometano (2,5 ml) se le añadió N-hidroxipiridin-2-tiona (75 mg) bajo luz-sombreado, entonces la mezcla se enfrió en hielo, se añadió N,N'-diciclohexilcarbodiimida (102 mg) a la misma y se agitó a la misma temperatura por 10 minutos y a temperatura ambiente por 3 horas. A la mezcla se le añadió 1,4-dioxano (5,0 ml), 2,2'-azobis(isobutironitrilo) (12 mg) e hidruro de tributilestaño (0,40 ml) y se agitó a 100 °C por 2 horas. La solución de reacción se filtró y el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 100:0-90:10), para dar el compuesto 3 (125 m) como un material viscoso incoloro. MS (ESI): m/z 489 [M+H]+
(3) A una solución del compuesto 3 (120 mg) en cloroformo (2,5 ml) se le añadió ácido trifluoroacético (1,2 ml) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 2 horas. La solución de reacción se enfrió en hielo, se diluyó con cloroformo y entonces se añadió a la misma una solución acuosa saturada de carbonato ácido de sodio para neutralizarla. La capa orgánica se separó, se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. A una solución del residuo en N,N-dimetilformamida (2,5 ml) se le añadió N,N'-dicidohexilcarbodiimida (61 mg) y etanol (0,15 ml) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 16 horas. A la mezcla de reacción se le añadió agua y acetato de etilo y la capa orgánica se separó. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-90:10), para dar el compuesto 4 (70 mg) como un polvo incoloro. MS (ESI): m/z 461 [M+H]+
(4) A una solución del compuesto 4 (70 mg) en etanol (1,5 ml) se le añadió ácido acético (0,6 ml) y carbón de paladio a 10% (humedecido con aproximadamente 50% de agua, 42 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 15 horas. El carbón de paladio se removió por filtración y el filtrado se concentró bajo presión reducida. El residuo se diluyó con acetato de etilo, entonces se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (cloroformo:metanol = 100:0-90:10), para dar el compuesto 5 (16 mg) como un material viscoso incoloro. MS (ESI): m/z 371 [M+H]+
Ejemplo de referencia 101
[Fórmula química 127]

(1) Una suspensión del compuesto 1 (30,5 g), el compuesto 2 (25 g) y carbonato de potasio (33 g) en N-metilpirrolidona (150 ml) se agitó a 130 °C por 15 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, entonces agua y acetato de etilo se añadieron a la misma, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 98:2-85:15), para dar el compuesto 3 (48,28 g) como un material viscoso amarillo. MS (ESI): m/z 316 [M+H]+ (2) A una solución de dietilfosfonoacetato de t-butilo (2,08 g) en tetrahidrofurano (40 ml) se le añadió complejo de bromuro de magnesio-éter dietílico (2,56 g) y trietilamina (1,26 ml) bajo enfriamiento en hielo, la mezcla se agitó a temperatura ambiente por 1 hora, entonces el compuesto 3 (2,00 g) se añadió a la misma y se agitó a temperatura ambiente por 23 horas. A la mezcla de reacción se le añadió hidruro de sodio (60 % en aceite, 507 mg), se agitó a temperatura ambiente por 23 horas, entonces una solución acuosa saturada de cloruro de amonio se añadió a la misma, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida para dar el compuesto 4 (3,17 g) como un material viscoso amarillo de un material crudo. Dicho compuesto se usó en el siguiente paso sin purificación. MS (APCI): m/z 414 [M+H]+
(3) A una solución del compuesto 4 (53,0 g) en diclorometano (500 ml) se le añadió ácido trifluoroacético (160 ml) y la mezcla se agitó a temperatura ambiente por 5 horas. A la mezcla de reacción se le añadió agua, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se secó y se concentró bajo presión reducida. Al residuo se le añadió éter diisopropílico, se agitó y entonces el precipitado se colectó por filtración para dar el compuesto 5 (26,3 g) como un polvo amarillo pálido. MS (ESI): m/z 358 [M+H]+
(4) El compuesto 5 (34 g) se trató de una manera similar al ejemplo de referencia 98 anterior, para dar el compuesto 6 (45,6 g) como un polvo amarillo. MS (APCI): m/z 517 [M+H]+
(5) El compuesto 6 (45 g) se trató de una manera similar al ejemplo de referencia 98 anterior, para dar el compuesto 7 (26,89 g) y el compuesto 8 (22,34 g) como polvos de color amarillo pálido. Cada MS (APCI): m/z 650 [M+H]+
(6) A una solución del compuesto 7 (24 g) y dicarbonato de di-t-butilo (8,45 g) en metanol (200 ml)/tetrahidrofurano (200 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 4,8 g) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 17 horas. El carbón de paladio se removió por filtración y el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 85:15-60:40), para dar el compuesto 9 (23,06 g) como un polvo incoloro. MS (ESI): m/z 660 [M+H]+
(7) A una solución del compuesto 9 (10,0 g) en tetrahidrofurano (100 ml)/agua (20 ml) se le añadió borohidruro de sodio (1,15 g) bajo enfriamiento en hielo y la mezcla se agitó a la misma temperatura por 3,5 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de cloruro de amonio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-50:50), para dar el compuesto 10 (6,93 g) como un polvo incoloro. MS (APCI): m/z 487 [M+H]+
(8) A una solución del compuesto 10 (3,82 g) en N,N-dimetilformamida (80 ml) se le añadió yoduro de metilo (4,9
ml), la mezcla se enfrió hasta -15 °C, se añadió a la misma hidruro de sodio (60 % en aceite, 411 mg) y se agitó a -10 °C por 1 hora y a 0 ° C por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de cloruro de amonio, se agitó, entonces se añadió agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-70:30), para dar el compuesto 11 (3,19 g) como un material viscoso incoloro. MS (ESI): m/z 501 [M+H]+
(9) A una solución del compuesto 11 (3,18 g) en diclorometano (30 ml) se le añadió ácido trifluoroacético (15 ml) y la mezcla se agitó a temperatura ambiente por 19 horas. La solución de reacción se concentró bajo presión reducida, entonces se diluyó con cloroformo, entonces una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma para alcalinizarla y se extrajo con cloroformo. La capa orgánica resultante se secó y entonces se concentró bajo presión reducida para dar el compuesto 12 (3,16 g) como un material viscoso amarillo pálido. MS (ESI): m/z 401 [M+H]+
(10) El compuesto 8 se trató de una manera similar a los pasos (6) a (9) anteriores, para dar el compuesto 13 como un material viscoso amarillo pálido. MS (APCI): m/z 401 [M+H], m S (ESI): m/z 401 [M+H]+
Ejemplos de referencia 102 a 104
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 101 anterior, para dar cada compuesto incluido en el cuadro 37 siguiente.
r 7

Ejemplo de referencia 105
El intermediario del ejemplo de referencia 104 se preparó de acuerdo con el siguiente método.
[Fórmula química 128]

(1) Una suspensión del compuesto 1 (2 g), el compuesto 2 (1,78 g), el ligando de fosfina 3 (483 mg), tris(dibencilidenoacetona)dipaladio(0) (474 mg) y carbonato de cesio (10,1 g) en 1,4-dioxano (103 ml), se agitó bajo calentamiento a 100 °C bajo atmósfera de nitrógeno por 16 horas. La materia insoluble se removió por filtración, el filtrado se concentró bajo presión reducida y entonces el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-75:25), para dar el compuesto 4 (1266 mg) como un material viscoso amarillo. MS (ESI): m/z 256 [M+H]+
(2) A una solución del compuesto 4 (700 mg) en cloroformo (27 ml)/metanol (13 ml) se le añadió N-bromosuccinimida (488 mg) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 5 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio y agua y entonces se extrajo con cloroformo. La capa orgánica resultante se lavó con agua y solución salina saturada y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-80:20), para dar el compuesto 5 (747 mg) como un polvo amarillo pálido. MS (ESI): m/z 334/336 [M+H]+
(3) Una suspensión del compuesto 5 (1,5 g), el compuesto 6 (2,88 g), tri-o-tolilfosfina (273 mg), acetato de paladio (100 mg) y diisopropiletilamina (1,53 ml) en N,N-dimetilformamida (15 ml), se calentó a 140 °C por radiación de microondas bajo atmósfera de nitrógeno por 2 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, entonces acetato de etilo y agua se añadieron a la misma y se agitó. La capa orgánica se separó, se lavó con agua y solución salina saturada y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-80:20), para dar el compuesto 7 (1,455 g) como un material viscoso amarillo. MS (ESI): m/z 382 [M+H]+
Ejemplo de referencia 106
[Fórmula química 129]

(1) Una solución del compuesto 1 (400 mg) y yoduro de etilo (656 ^l) en N,N-dimetilformamida (4 ml) se enfrió en hielo, se añadió a la misma hidruro de sodio (60 % en aceite, 39 mg) y la mezcla se agitó a la misma temperatura por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de cloruro de amonio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 2 (148 mg) como un material viscoso incoloro. MS (ESI): m/z 515 [M+H]+
(2) A una solución del compuesto 2 (140 mg) en diclorometano (2 ml) se le añadió ácido trifluoroacético (1 ml) y la mezcla se agitó a temperatura ambiente por 1 hora. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con diclorometano. La capa orgánica se secó y se concentró bajo presión reducida para dar el compuesto 3 (110 mg) como un material viscoso incoloro. MS (ESI): m/z 415 [M+H]+
Ejemplo de referencia 107
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 106 anterior, para dar el compuesto incluido en el cuadro 38 siguiente.
r 1

Ejemplo de referencia 108
[Fórmula química 130]

(1) A una solución del compuesto 1 (600 mg), acetona cianohidrina (228 jl) y trifenilfosfina (648 mg) en tetrahidrofurano (2 ml) se le añadió azodicarboxilato de dietilo (solución a 40 % en peso de tolueno, 1,12 ml) bajo enfriamiento en hielo, la mezcla se agitó a temperatura ambiente por 30 minutos, entonces acetona cianohidrina (228 jl), trifenilfosfina (648 mg) y azodicarboxilato de dietilo (solución a 40 % en peso de tolueno, 1,12 ml) se añadieron a la misma y se agitó a temperatura ambiente por 14 horas. La solución de reacción se concentró bajo presión reducida, éter diisopropílico se añadió a la misma y la mezcla se agitó. El precipitado se removió por filtración y entonces el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 84:16-70:30), para dar el compuesto 2 (838 mg) como un material viscoso incoloro. MS (ESI): m/z 496 [M+H]+
(2) A una solución del compuesto 2 (120 mg) en diclorometano (2 ml) se le añadió ácido trifluoroacético (0,5 ml) y la mezcla se agitó a temperatura ambiente por 30 minutos. La solución de reacción se concentró bajo presión reducida, se diluyó con diclorometano, entonces una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma, la mezcla se agitó y se extrajo la capa orgánica. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (acetato de etilo:metanol = 100:0-90:10), para dar el compuesto 3 (55 mg) como un polvo incoloro. MS (ESI): m/z 396 [M+H]+
Ejemplo de referencia 109
[Fórmula química 131]

(1) Bajo atmósfera de nitrógeno, a una solución del compuesto 1 (342 mg) en tetrahidrofurano (1,5 ml) se le añadió una solución de bis(trimetilsilil)amida de litio en tetrahidrofurano (1,09 moles/l, 1,26 ml) a -40 °C, la mezcla se agitó a la misma temperatura por 1 hora, entonces yoduro de metilo (85 ml) se añadió a la misma y se agitó a temperatura ambiente por 15 horas. A la mezcla de reacción se le añadió solución salina saturada y una solución
acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 93:780:20), para dar el compuesto 2 (167 mg) como un material viscoso incoloro. MS (APCI): m/z 515 [M+H]+
(2) A una solución del compuesto 2 (162 mg) en cloroformo (4 ml) se le añadió ácido trifluoroacético (4 ml) y la mezcla se agitó a temperatura ambiente por 1 hora. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida para dar el compuesto 3 (146 mg) como un material viscoso amarillo pálido. MS (APCI): m/z 415 [M+H]+
Ejemplo de referencia 110
[Fórmula química 132]

(1) A una solución del compuesto 1 (500 mg) en tetrahidrofurano (10 ml) se le añadió trifenilfosfina (484 mg), N'-isopropiliden-2-nitrobencensulfonohidrazida (474 mg) y azodicarboxilato de dietilo (solución a 40 % en peso de tolueno, 833 pl) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 3 horas. A la solución de reacción se le añadió fenilhidrazina (564 pl) y la mezcla se agitó a temperatura ambiente por 15 horas. La solución de reacción se concentró bajo presión reducida, acetato de etilo y una solución acuosa saturada de carbonato ácido de sodio se añadieron a la misma y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con una solución acuosa saturada de cloruro de amonio, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 85:15-60:40) y cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 90:10-65:35), para dar el compuesto 2 (168 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 421 [M+H]+
(2) A una solución del compuesto 2 (165 mg) en diclorometano (1,6 ml) se le añadió ácido trifluoroacético (0,8 ml) y la mezcla se agitó a temperatura ambiente por 6 horas. La solución de reacción se concentró, entonces acetato de etilo y una solución acuosa saturada de carbonato ácido de sodio se añadieron a la misma para alcalinizarla y la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida para dar el compuesto 3 (149 mg) como un material viscoso anaranjado pálido. MS (APCI): m/z 321 [M+H]+
Ejemplo de referencia 111
[Fórmula química 133]

(1) A una solución de 2-fluoro-2-fosfonoacetato de trietilo (1,01 g) en tetrahidrofurano (18 ml) le añadió complejo de éter dietílico - bromuro de magnesio (1,29 g) y trietilamina (638 jl) bajo enfriamiento en hielo, la mezcla se agitó a temperatura ambiente por 1 hora, entonces una solución del compuesto 1 (1,14 g) en tetrahidrofurano (5 ml) se añadió a la misma y se agitó a temperatura ambiente por 6 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de cloruro de amonio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida para dar el compuesto 2 (1,41 g) como un polvo amarillo a la relación isomérica de Z:E “ 85:15. Dicho compuesto se usó en el siguiente paso sin purificación. MS (APCI): m/z 446 [M+H]+
(2) A una solución del compuesto 2 (2,65 g) en etanol (13 ml) / tetrahidrofurano (13 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 5,95 ml) y la mezcla se agitó a temperatura ambiente por 1 hora. A la solución de reacción se le añadió una solución acuosa de ácido clorhídrico (1 mol/l, 10 ml), se agitó y entonces el solvente orgánico se evaporó bajo presión reducida. El precipitado se colectó por filtración, se disolvió en una solución mixta de acetato de etilo y una solución acuosa de ácido clorhídrico (0,1 moles/l) y la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se pulverizó con éter diisopropílico, se colectó por filtración y entonces se secó para dar el compuesto 3 (1,79 g) como un polvo incoloro. MS (APCI): m/z 418 [M+H]+
(3) El compuesto 3 (500 mg) se trató de una manera similar al ejemplo de referencia 99 anterior, para dar el compuesto 4 (123 mg) como un material viscoso amarillo. MS (APCI): m/z 577 [M+H]+
(4) El compuesto 4 (400 mg) se trató de una manera similar al ejemplo de referencia 99 anterior, para dar el compuesto 5 (107 mg) como un material viscoso anaranjado. MS (APCI): m/z 710 [M+H]+
(5) El compuesto 5 (105 mg) se trató de una manera similar al ejemplo de referencia 101, para dar el compuesto 6(119 mg) como un material viscoso amarillo. MS (APCI): m/z 720 [M+H]+
(6) El compuesto 6 (100 mg) se trató de una manera similar al ejemplo de referencia 101, para dar el compuesto 7 (57,9 mg) como un material viscoso incoloro. MS (APCI): m/z 547 [M+H]+
(7) El compuesto 7 (56 mg) se trató de una manera similar al ejemplo de referencia 101, para dar el compuesto 8 (50,3 mg) como un material viscoso incoloro. MS (APCI): m/z 561 [M+H]+
(8) A una solución del compuesto 8 (48 mg) en metanol (1,9 ml) se le añadió una solución de ácido clorhídrico en metanol (2 moles/l, 1,9 ml) y la mezcla se agitó a 50 °C por 5 horas. La mezcla de reacción se enfrió hasta temperatura ambiente y entonces se concentró bajo presión reducida. Al residuo se le añadió diclorometano y una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se secó y entonces se concentró bajo presión reducida para dar el compuesto 9 (37,9 mg) como un material viscoso amarillo pálido. MS (APCI): m/z 419 [M+H]+
Ejemplo de referencia 112
[Fórmula química 134]

(1) Una suspensión del compuesto 1 (10,0 g), el compuesto 2 (14,0 g) y carbonato de potasio (13,1 g) en 1,3-dimetil-2-imidazolidinona (80 ml), se agitó a 100 °C por 15 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, entonces se añadió agua a la misma y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 99:1-90:10), para dar el compuesto 3 (19,0 g) como un material viscoso amarillo. MS (APCI): m/z 324/326 [M+H]+
(2) A una solución del compuesto 3 (15,3 g) y nitrometano (12,8 ml) en metanol (76,5 ml) se le añadió una solución de metóxido de sodio en metanol (5 moles/l, 0,94 ml), la mezcla se agitó a temperatura ambiente por 4 horas, entonces se añadió nitrometano (25,6 ml) a la misma y se agitó a temperatura ambiente por 15 horas. A la solución de reacción se le añadió una solución acuosa de ácido clorhídrico (1 mol/l, 4,7 ml) para neutralizarla y se concentró bajo presión reducida. El residuo se diluyó con acetato de etilo y agua, una solución acuosa de ácido clorhídrico (1 mol/l) se añadió a la misma para acidificarla y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. A una solución del residuo en tetrahidrofurano (76,5 ml) se le añadió gota a gota cloruro de metansulfonilo (4,39 ml) y trietilamina (16,46 ml) bajo enfriamiento en hielo y la mezcla se agitó a la misma temperatura por 40 minutos. El tetrahidrofurano se evaporó bajo presión reducida, entonces al residuo se le añadió agua (200 ml) y la mezcla se agitó. El precipitado se colectó por filtración, se disolvió en acetato de etilo, entonces se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-85:15), se pulverizó con hexano enfriado, se colectó por filtración y se secó bajo presión reducida para dar el compuesto 4 (11,12 g) como un polvo amarillo.MS (ESI): m/z 367/369 [M+H]+
Como se describe en los pasos (3) y (4) siguientes, el compuesto 4 se trató de una manera similar a un método descrito en una literatura (J. Am. Chem. Soc. 2005, 127, 119-125), para preparar el compuesto 6.
(3) A una solución del compuesto 4 (11,12 g) en tolueno (55,6 ml) se le añadió 1-[3,5-bis(trifluorometil)fenil]-3-[(1S,2S)-(-)-2-(dimetilamino)ciclohexil]tiourea (630 mg) y malonato de dietilo (9,2 ml) y la mezcla se agitó a temperatura ambiente por 62 horas. La solución de reacción se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-90:10), para dar el compuesto 5 (16,13 g) como un material viscoso anaranjado pálido. MS (ESI): m/z 527/529 [M+H]+
(4) A una solución del compuesto 5 (16,13 g) y hexahidrato de cloruro de níquel (7,27 g) en metanol (161,3 ml) se le añadió borohidruro de sodio (6,95 g) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 2 horas. La solución de reacción se enfrió en hielo, una solución acuosa saturada de cloruro de amonio se añadió a la misma, se agitó y entonces el metanol se evaporó bajo presión reducida. Al residuo se le añadió cloroformo y agua, se agitó y la capa orgánica se separó. La capa orgánica resultante se lavó con agua, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 67:33-40:60), para dar el compuesto 6 (3,52 g) como un polvo anaranjado pálido. MS (ESI): m/z 451/453 [M+H]+
(5) A una solución del compuesto 6 (2,31 g) en etanol (45 ml) se le añadió borohidruro de sodio (775 mg) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 10 minutos y a 40 °C por 4 horas. La solución de reacción se enfrió en hielo, una solución acuosa saturada de cloruro de amonio se añadió a la misma, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-93:7), entonces se pulverizó con éter diisopropílico, se colectó por filtración y entonces se secó para dar el compuesto 7 (1,84 g) como un polvo incoloro. MS (APCI): m/z 409/411 [M+H]+
(6) A una solución de tetrafluoroborato de trimetiloxonio (1,32 g) en diclorometano (36 ml) se le añadió el compuesto 7 (1,82 g), la mezcla se agitó a temperatura ambiente por 2 horas, entonces el tetrafluoroborato de trimetiloxonio (330 mg) se añadió a la misma y se agitó a temperatura ambiente por 1 hora. El diclorometano se evaporó bajo presión reducida hasta que el volumen de la solución de reacción llegara a ser aproximadamente un tercio, entonces se añadieron a la misma metanol (36 ml) y dicarbonato de di-t-butilo (1,07 g), la mezcla se enfrió en hielo, se añadió borohidruro de sodio (505 mg) a la misma y se agitó a la misma temperatura por 30
minutos. A la mezcla de reacción se le añadió una solución acuosa de cloruro de amonio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 85:15-60:40), para dar el compuesto 8 (1,18 g) como un polvo incoloro. MS (APCI): m/z 495/497 [M+H]+
(7) Una solución del compuesto 8 (1,16 g) y yoduro de metilo (1,46 ml) en N,N-dimetilformamida (23,2 ml) se enfrió hasta -20 °C, se añadió a la misma hidruro de sodio (60 % en aceite, 123 mg) y la mezcla se agitó bajo atmósfera de nitrógeno a -15 °C por 1 hora y a -8 °C por 3 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de cloruro de amonio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-60:40), para dar el compuesto 9 (1,21 g) como un polvo incoloro. MS (APCI): m/z 509/511 [M+H]+
(8) A una solución del compuesto 9 (1,16 g) en metanol (2 ml) se le añadió una solución de ácido clorhídrico en metanol (2 moles/l, 17,6 ml) y la mezcla se agitó a 50 °C por 15 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, una solución acuosa saturada de carbonato de sodio se añadió para alcalinizarla, se evaporó el metanol bajo presión reducida y la mezcla se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida para dar el compuesto 10 (839 mg) como un material viscoso incoloro. MS (APCI): m/z 367/369 [M+H]+
Ejemplos de referencia 113 y 114
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 112 anterior, para dar cada compuesto incluido en el cuadro 39 siguiente.
r

Ejemplo de referencia 115
[Fórmula química 135]

(1) Una mezcla del compuesto 2 (100 mg) preparada de acuerdo con el ejemplo de referencia 112 anterior a partir del compuesto 1, ácido 4-metoxicarbonilfenilborónico (47 mg), aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (20 mg), una solución acuosa de carbonato de sodio (2 moles/l, 300 pl) y N,N-dimetilformamida (2 ml), se agitó bajo atmósfera de nitrógeno a 80 °C por 20 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, entonces se añadieron a la misma agua y acetato de etilo, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice
(hexano:acetato de etilo = 70:30-50:50), para dar el compuesto 3 (52,7 mg) como un polvo incoloro. MS (ESI): m/z 480 [M+H]+
(2) Una solución del compuesto 3 (51 mg), ácido acético (18 jl), clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (30 mg) y 4-dimetilaminopiridina (6,5 ml) en N,N-dimetilformamida (2 ml), se agitó a temperatura ambiente por 3 horas. A la mezcla de reacción se le añadió acetato de etilo y una solución acuosa saturada de carbonato ácido de sodio, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 85:15-50:50), para dar el compuesto 4 (47 mg) como un polvo incoloro. MS (ESI): m/z 422 [M-Boc+H]+
(3) A una solución del compuesto 4 (45 mg) en diclorometano (2 ml) se le añadió ácido trifluoroacético (1 ml) y la mezcla se agitó a temperatura ambiente por 17 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con diclorometano. La capa orgánica se secó y se concentró bajo presión reducida para dar el compuesto 5 (35 mg) como un material viscoso incoloro. MS (ESI): m/z 422 [M+H]+
(4) A una solución del compuesto 2 (250 mg) y yoduro de metilo (110 jl) en N,N-dimetilformamida (2 ml) se le añadió hidruro de sodio (60 % en aceite, 31 mg) bajo enfriamiento en hielo y la mezcla se agitó a la misma temperatura por 2 horas. A la mezcla de reacción se le añadió agua, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-70:30), para dar el compuesto 6 (166 mg) como un material viscoso incoloro. MS (ESI): m/z 382/384 [M-tBu]+ (5) El compuesto 6 (160 mg) se trató de una manera similar a la reacción que dio el compuesto 3 a partir del compuesto 2 en el paso (1) anterior, para dar el compuesto 7 (171 mg) como un polvo incoloro. MS (ESI): m/z 394 [M-Boc+H]+.
(6) A una solución del compuesto 7 (165 mg) en diclorometano (2 ml) se le añadió ácido trifluoroacético (1 ml) y la mezcla se agitó a temperatura ambiente por 17 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla y se extrajo con diclorometano. La capa orgánica se secó y se concentró bajo presión reducida para dar el compuesto 8 (130 mg) como un material viscoso incoloro. MS (ESI): m/z 394 [M+H]+
Ejemplo de referencia 116
[Fórmula química 136]

(1) El compuesto 1 se trató de una manera similar al ejemplo de referencia 112, para dar el compuesto 2 (36,2 g).
(2) A una solución del compuesto 2 en etanol (360 ml) se le añadió una solución acuosa de hidróxido de sodio (1 mol/l, 82,2 ml) y la mezcla se agitó a temperatura ambiente por 1 hora. El etanol se evaporó bajo presión reducida, el residuo se diluyó con agua, entonces una solución acuosa de ácido clorhídrico (1 mol/l) se añadió a la misma hasta que la mezcla llegara a tener un pH = 4 bajo enfriamiento en hielo y la mezcla se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. Al residuo se le añadió tolueno (180 ml), se calentó bajo reflujo por 2 horas, entonces la solución de reacción se concentró bajo presión reducida, el residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-95:5), entonces se pulverizó con hexano, se colectó por filtración y entonces se secó para dar el compuesto 3 (27,5 g) como un sólido incoloro. MS (ESI): m/z 413 [M+H]+
(3) A una solución del compuesto 3 (4,12 g) en tetrahidrofurano (82 ml) se le añadió tetrafluoroborato de trimetiloxonio (1,77 g) y la mezcla se agitó a temperatura ambiente por 1,5 horas. La mezcla de reacción se concentró bajo presión reducida y se añadió metanol (82 ml) al residuo y ácido acético (4,46 ml) y borohidruro de sodio (3,04 g) se añadieron a la misma bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 30 minutos. El solvente se evaporó bajo presión reducida, al residuo se le añadió una solución acuosa saturada de carbonato ácido del sodio y entonces se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (acetato de etilo:metanol = 100:0-80:20), para dar el compuesto 4 (2,95 g) como un material viscoso amarillo pálido. MS (ESI): m/z 399 [M+H]+
Ejemplo de referencia 117
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 116 anterior, para dar el compuesto incluido en el cuadro 40 siguiente.
r 41

Ejemplo de referencia 118
[Fórmula química 137]

(1) El compuesto 1 (2 g) obtenido en el ejemplo de referencia 112 se trató de una manera similar al ejemplo de referencia 116, para dar el compuesto 2 (829 mg). MS (ESI): m/z 379/381 [M+H]+
(2) A una solución del compuesto 2 (300 mg) en 1,4-dioxano (6 ml) se le añadió borohidruro de sodio (300 mg) y ácido acético (454 jl) y la mezcla se agitó a 100 °C por 30 minutos. La solución de reacción se enfrió en hielo, se añadió metanol (3 ml) a la misma, la mezcla se agitó, entonces una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma para alcalinizarla y se extrajo con cloroformo. La capa orgánica resultante se lavó con agua, se secó y se concentró bajo presión reducida. Una solución del residuo en metanol (2 ml) se enfrió en hielo, una solución de ácido clorhídrico en metanol (2 moles/l, 16 ml) se añadió a la misma y la mezcla se calentó bajo reflujo por 18 horas. A la mezcla de reacción se le añadió acetato de etilo y agua a temperatura ambiente, se agitó, entonces una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma para alcalinizarla y se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (acetato de etilo:metanol = 100:0-90:10), para dar el compuesto 3 (128 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 323/325 [M+H]+
Ejemplo de referencia 119
[Fórmula química 138]

g).
(2) A una solución del compuesto 2 (3,39 g) en etanol (39 ml) se le añadió una solución acuosa de hidróxido de sodio (1 mol/l, 8,58 ml) y la mezcla se agitó a temperatura ambiente por 3 horas. El etanol se evaporó bajo presión reducida, el residuo se diluyó con agua, entonces a la mezcla se le añadió una solución acuosa de ácido clorhídrico (1 mol/l, 8,8 ml) bajo enfriamiento en hielo y se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. Al residuo se le añadió tolueno (34 ml), se calentó bajo reflujo por 1 hora, entonces la solución de reacción se concentró bajo presión reducida y
el residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-97:3), para dar el compuesto 3 (2,8 g) como un polvo incoloro. MS (APCI): m/z 363 [M+H]+
(3) A una solución del compuesto 3 (2,63 g) en 1,4-dioxano (53 ml) se le añadió borohidruro de sodio (1,37 g) y ácido acético (2,08 ml) y la mezcla se agitó a 100 °C por 25 horas. La solución de reacción se enfrió hasta temperatura ambiente y se añadió agua (5 gotas) a la misma, se agitó y entonces se añadió metanol (19 ml) a la misma. A la mezcla se le añadió carbón de paladio a 10% (humedecido con aproximadamente 50% de agua, 530 mg), se agitó a temperatura ambiente por 3 días, entonces el carbón de paladio se removió por filtración y se lavó con metanol. El filtrado se concentró y entonces el residuo se purificó con cromatografía en columna de gel de sílice de NH (acetato de etilo:metanol = 100:0-98:2), para dar el compuesto 4 (1,226 g) como un polvo incoloro. MS (APCI): m/z 349 [M+H]+
Ejemplo de referencia 120
[Fórmula química 139] 1

(1) Una solución del compuesto 1 (10,0 g), el compuesto 2 (30,5 g) y carbonato de potasio (17,1 g) en 1,3-dimetil-2-imidazolidinona (50 ml), se agitó a 155 °C por 5 horas. La mezcla de reacción se diluyó con acetato de etilo a temperatura ambiente, la materia insoluble se removió por filtración y el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-90:10), para dar el compuesto 3 (8,70 g) como un material viscoso incoloro. MS (APCI): m/z 380/382 [M+H]+ (2) A una solución de trietilamina (4,18 ml) y ácido fórmico (0,384 ml) en N,N-dimetilformamida (20 ml) se le añadió aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (205 mg), el compuesto 3 (1,90 g), el compuesto 4 (3,98 g) y N,N-dimetilformamida (10 ml) y la mezcla se agitó a 95 °C por 2 horas. A la mezcla de reacción se le añadió acetato de etilo y agua a temperatura ambiente, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-50:50), para dar el compuesto 5 racémico (1,14 g) como un material viscoso amarillo pálido. MS (APCI): m/z 461 [M+H]+ (3) A una solución del compuesto 5 (611 mg) en etanol (12 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 611 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 16 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró bajo presión reducida para dar el compuesto 6 racémico (447 mg) como un material viscoso café oscuro. MS (APCI): m/z 371 [M+H]+
Ejemplo de referencia 121
[Fórmula química 140]

(1) Una suspensión del compuesto 1 (20 g), ácido piperidin-4-carboxílico (15,9 g) y carbonato de potasio (34,12 g) en N-metilpirrolidona (160 ml), se agitó bajo atmósfera de nitrógeno a 190 °C por 4 horas. La mezcla de reacción se vertió en una solución acuosa enfriada en hielo de ácido clorhídrico (1 mol/l, 480 ml), una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma para neutralizarla, entonces se diluyó con agua y se extrajo dos veces con acetato de etilo. Las capas orgánicas resultantes se combinaron, se lavaron con agua y solución salina saturada, sulfato de magnesio y carbón activo se añadieron a la misma, la mezcla se agitó, entonces se filtró y el filtrado se concentró bajo presión reducida. El residuo se pulverizó con éter isopropílico, se colectó por filtración y entonces se secó para dar el compuesto 2 (14,1 g) como un sólido incoloro. MS (APCI): m/z 352/354 [M+H]+
(2) A una suspensión del compuesto 2 (21,3 g) y dicarbonato de di-t-butilo (15,8 g) en acetonitrilo (200 ml) se le añadió 4-dimetilaminopiridina (1,48 g), la mezcla se agitó a temperatura ambiente por 1 hora, entonces se añadió trietilamina (8,42 ml) a la misma y la mezcla se agitó a temperatura ambiente por 15 horas. A la mezcla se le añadió trietilamina (4,21 ml) y dicarbonato de di-t-butilo (2,60 g) y se agitó a temperatura ambiente por 2 horas. La mezcla de reacción se concentró y entonces se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 99:1-96:4), para dar el compuesto 3 (20,4 g) como un sólido incoloro. MS (APCI): m/z 408/410 [M+H]+
(3) El compuesto 4 se preparó de acuerdo con un método descrito en una literatura (J. Org. Chem. 2003, 68, 3923-3931).
(4) Una solución de una solución de bis(trimetilsilil)amida de potasio en tolueno (2 moles/l, 18,14 ml) en tetrahidrofurano (110 ml) se enfrió hasta -78 °C, una solución del compuesto 4 (2,49 g) en tetrahidrofurano (20 ml) se añadió gota a gota a la misma, la mezcla se agitó a la misma temperatura por 25 minutos, entonces se añadió gota a gota a la misma una solución de N- fenilbis(trifluorometansulfonimida) (4,05 g) en tetrahidrofurano (20 ml) y la mezcla se agitó a temperatura ambiente por 15 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio a 0 °C, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5), para dar el compuesto 5 (3,39 g) como un material viscoso amarillo pálido.
(5) A una solución del compuesto 5 (1,5 g) en 1,4-dioxano (11 ml) se le añadió bis(pinacolato)diboro (991 mg), aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (133 mg), 1,1'-bis(difenilfosfino)ferroceno (90 mg) y acetato de potasio (958 mg) y la mezcla se agitó a 100 °C por 50 minutos. A la mezcla de reacción se le añadió agua a temperatura ambiente, se agitó y se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida. Al residuo se le añadió N,N-dimetilformamida (11 ml), agua (3 ml), el compuesto 3 (1,33 g), aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (133 mg) y carbonato de sodio (1,03 g) y la mezcla se agitó a 100 °C por 2,5 horas. A la mezcla de reacción se le añadió agua a temperatura ambiente, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 6 (1,63 g) como un material viscoso amarillo pálido.
(6) A una solución del compuesto 6 (1,63 g) en tetrahidrofurano (6 ml) se le añadió una solución de fluoruro de tetrabutilamonio en tetrahidrofurano (1 mol/l, 2,80 ml) y la mezcla se agitó a temperatura ambiente por 3 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó, entonces se extrajo con cloroformo, la capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-60:40), para dar el compuesto 7 (1,16 g) como un material viscoso incoloro. MS (ESI): m/z 527 [M+H]+
(7) A una solución del compuesto 7 (917 mg) en diclorometano (34,8 ml) se le añadió catalizador de Crabtree (14 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 19,5 horas. La mezcla de reacción se concentró bajo presión reducida y entonces el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-60:40), para dar el compuesto 8 (870 mg) como un polvo incoloro. MS (ESI): m/z 529 [M+H]+
(8) A una solución del compuesto 8 (870 mg) en tetrahidrofurano (8 ml) se le añadió yoduro de metilo (1,03 ml) e hidruro de sodio (60% en aceite, 99 mg) y la mezcla se agitó a temperatura ambiente por 2 horas y 15 minutos. A la mezcla de reacción se le añadió agua, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 9 (864 mg) como un material viscoso incoloro.
(9) A una solución del compuesto 9 (31,4 g) en metanol (314 ml) se le añadió una solución de ácido clorhídrico en metanol (2 moles/l, 314 ml) y la mezcla se agitó a 50 °C por 10 horas. El solvente se evaporó bajo presión reducida, entonces al residuo se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizar la mezcla y se extrajo con cloroformo. La capa orgánica resultante se secó, se concentró bajo presión reducida para dar el compuesto 10 (23,8 g) como un material viscoso café. MS (ESI): m/z 401 [M+H]+ Ejemplos de referencia 122 a 124
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 121, para dar cada compuesto incluido en el cuadro 41 siguiente.
r 41

Ejemplo de referencia 125
[Fórmula química 141]

Al compuesto 1 (529 mg) se le añadió una solución de ácido clorhídrico en metanol (2 moles/l, 25 ml) y la mezcla se agitó a 50 °C por 15 horas. El solvente se evaporó bajo presión reducida y al residuo se le añadió tolueno y la mezcla se concentró bajo presión reducida para dar el compuesto 2 (460 mg) como un polvo café pálido. MS (ESI): m/z 387 [M+H]+
Ejemplo de referencia 126
[Fórmula química 142] 1

(1) Una suspensión del compuesto 1 (2,25 g) preparada de acuerdo con el método descrito en una literatura (J. Org. Chem. 2012, 77, 5286-5296), bis(pinacolato)diboro (1,83 g), aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (245 mg), 1,1'-bis(difenilfosfino)ferroceno (166 mg) y acetato de potasio (1,77 g) en 1,4-dioxano (60 ml), se agitó bajo atmósfera de nitrógeno a 80 °C por 1,5 horas. A la mezcla de reacción se le añadió agua a temperatura ambiente, se agitó y se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida. Al residuo se le añadió el compuesto 2 (2,45 g), aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (490 mg), una solución acuosa 2 M de carbonato de sodio (9 ml) y N,N-dimetilformamida (60 ml) y la mezcla se agitó bajo atmósfera de nitrógeno a 80 °C por 30 minutos. A la mezcla de reacción se le añadió agua y solución salina saturada a temperatura ambiente, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 3 (1,23 g) como un polvo amarillo pálido. MS (ESI): m/z 555 [M+H]+
(2) A una solución del compuesto 3 (373 mg) en metanol (7 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 75 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 2 horas. A la mezcla se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 150 mg) y se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por otra hora. El carbón de paladio se removió por filtración y entonces el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 4 (293 mg) como un polvo incoloro. MS (APCI): m/z 485 [M+H]+
(3) A una solución del compuesto 4 (282 mg) en tetrahidrofurano (4 ml)/metanol (1 ml) se le añadió borohidruro de sodio (77 mg) y cloruro de litio (86 mg) y la mezcla se agitó a temperatura ambiente por 30 minutos. A la mezcla de reacción se le añadió agua, se agitó y se extrajo con cloroformo. La capa orgánica se secó, se concentró bajo presión reducida y entonces el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-70:30), para dar el compuesto 5 (254 mg) como un material viscoso incoloro. MS (ESI): m/z 529 [M+H]+
(4) A una solución del compuesto 5 (248 mg) en tetrahidrofurano (5 ml) se le añadió yoduro de metilo (292 pl) e
hidruro de sodio (60 % en aceite, 28 mg) y la mezcla se agitó a temperatura ambiente por 5 minutos y entonces a 50 °C por 20 minutos. A la mezcla de reacción se le añadió agua a temperatura ambiente, se agitó y se extrajo con cloroformo. La capa orgánica se secó, se concentró bajo presión reducida y entonces el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-75:25), para dar el compuesto 6 (167 mg) como un material viscoso amarillo pálido.
(5) Una mezcla del compuesto 6 (160 mg) y una solución de ácido clorhídrico en metanol (2 moles/l, 8 ml) se agitó a 60 °C por 6 horas. La mezcla de reacción se concentró bajo presión reducida para dar el compuesto 7 (140 mg) como un polvo café. MS (ESI): m/z 401 [M+H]+
Ejemplo de referencia 127
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 126, para dar el compuesto incluido en el cuadro 42 siguiente.
Cuadro 42

Ejemplo de referencia 128
[Fórmula química 143]

(1) Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 126, para dar el compuesto 1 (260 mg).
(2) A una solución del compuesto 1 (260 mg) en tetrahidrofurano (4 ml)/metanol (1 ml) se le añadió borohidruro de sodio (71 mg) y cloruro de litio (79 mg) y la mezcla se agitó a temperatura ambiente por 10 minutos. A la mezcla de reacción se le añadió agua, se agitó y se extrajo con cloroformo. La capa orgánica se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-70:30), para dar el compuesto 2 (270 mg) como un material viscoso incoloro. MS (ESI): m/z 527 [M+H]+
(3) A una solución del compuesto 2 (240 mg) en diclorometano (10 ml) se le añadió catalizador de Crabtree (7,3 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 15 horas. La mezcla de reacción se concentró bajo presión reducida y entonces el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-70:30), para dar el compuesto 3 (218 mg) como un polvo incoloro. MS (ESI): m/z 529 [M+H]+
(4) El compuesto 3 se trató de una manera similar al ejemplo de referencia 126, para dar el compuesto 4. MS (ESI): m/z 401 [M+H]+
Ejemplo de referencia 129
[F ó rm u la q u ím ic a 144 ]

(1) A una solución del compuesto 1 (2,0 g) en tolueno (115 ml) se le añadió imidazol (2,36 g), trifenilfosfina (6,06 g) y yodo (4,4 g) y la mezcla se agitó a 100 °C por 1 hora y 45 minutos. La mezcla de reacción se enfrió hasta temperatura ambiente, una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma, se agitó, entonces se añadió yodo a la misma, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-75:25), para dar el compuesto 2 (3,23 g) como un líquido incoloro.
(2) Bajo atmósfera de nitrógeno, a una suspensión de zinc (292 mg) en N,N- dimetilacetamida (0,9 ml) se le añadió 1,2-dibromoetano (0,04 ml), la mezcla se agitó a 65 °C por 10 minutos, entonces se añadió cloruro de trimetilsililo (0,09 ml) a la misma y se agitó a temperatura ambiente por 30 minutos. A la mezcla de reacción se le añadió gota a gota una solución del compuesto 2 (1,04 g) en N,N-dimetilacetamida (1,8 ml) y se agitó a temperatura ambiente por 1 hora. La materia insoluble se removió por filtración y el filtrado se añadió gota a gota a una suspensión del compuesto 3 (1 g) yoduro de cobre (50 mg) y aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (107 mg) en N,N-dimetilformacetamida (3,6 ml) y la mezcla se agitó bajo atmósfera de nitrógeno a 85 °C por 15 horas. La materia insoluble se removió por filtración a temperatura ambiente, el filtrado se diluyó con acetato de etilo, se lavó con agua, una solución acuosa de carbonato ácido de sodio y solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-50:50), para dar el compuesto 4 (543 mg) como un material viscoso amarillo. MS (ESI): m/z 457 [M+H]+
(3) A una solución del compuesto 4(110 mg) en diclorometano (2,4 ml) se le añadió 2,6-dimetilpiridina (0,11 ml), la mezcla se enfrió hasta 0 °C, se añadió gota a gota triflato de trimetilsililo (0,13 ml) a la misma y se agitó a la misma temperatura por 2 horas. La mezcla de reacción se diluyó con diclorometano y entonces se lavó con una solución acuosa diluida de carbonato ácido de sodio y solución salina saturada. La capa orgánica resultante se secó y se concentró bajo presión reducida para dar un material crudo del compuesto 5 (96,9 mg) como un material viscoso amarillo. MS (ESI): m/z 357 [M+H]+
Ejemplo de referencia 130
[Fórmula química 145] 1

(1) Una suspensión del compuesto 1 (5,52 g), carbonato de potasio (6,28 g) y el compuesto 2 (4,65 g) en N-metilpirrolidona (70 ml) se agitó a 190 °C por 3 horas, entonces dicarbonato de di-t-butilo (4,96 g) se añadió a la misma a temperatura ambiente y se agitó a la misma temperatura por 30 minutos. A la mezcla de reacción se le añadió agua y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. A la mezcla se le añadió hexano, se agitó, el precipitado se removió por filtración y entonces el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-90:10), para dar el compuesto 3 (3,80 g) como un material viscoso amarillo pálido. MS (ESI): m/z 409/411 [M+H]+
(2) Una suspensión del compuesto 3 (1,23 g), aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (122 mg), una solución acuosa de carbonato de sodio (2 moles/l, 4,5 ml) y
ácido 4-metoxicarbonilfenilborónico (649 mg) en N,N-dimetilformamida (30 ml), se agitó bajo atmósfera de nitrógeno a 80 °C por 1 hora. A la mezcla de reacción se le añadió agua a temperatura ambiente y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-80:20), para dar el compuesto 4 (1,19 g) como un polvo incoloro. MS (ESI): m/z 465 [M+H]+
(3) A una solución del compuesto 4 (1,16 g) en 1,4-dioxano (20 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 6,25 ml) y la mezcla se agitó a temperatura ambiente por 15 horas. La solución de reacción se concentró bajo presión reducida para dar el compuesto 5 (1,09 g) como un polvo incoloro. MS (ESI): m/z 365 [M+H]+
(4) A una suspensión del compuesto 3 (1,5 g), el compuesto 6 (732 mg) obtenido por el método descrito en el documento WO2013/187496 y una solución acuosa de carbonato de sodio (2 moles/l, 3,56 ml) en N,N-dimetilformamida (30 ml), se añadió aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (122 mg) bajo atmósfera de nitrógeno y la mezcla se agitó a 80 °C por 3,5 horas. A la mezcla de reacción se le añadió acetato de etilo y agua a temperatura ambiente, se agitó y la materia insoluble se removió por filtración. La capa orgánica del filtrado se separó, se lavó con solución salina, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-75:25), para dar el compuesto racémico 7 (1,15 g) como un material viscoso amarillo pálido. MS (APCI): m/z 483 [M+H]+
(5) A una solución del compuesto 7 (1,0 g) en etanol (20 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 200 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 6 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró bajo presión reducida para dar el compuesto 8 (943 mg, cis: trans = 2,1:1) como un polvo incoloro. MS (ESI): m/z 485 [M+H]+
(6) A una solución del compuesto 8 (943 mg) en t-butanol (19 ml) se le añadió t- butóxido de potasio (240 mg) y la mezcla se agitó a 35 °C por 2 horas. A la mezcla de reacción se le añadió agua (25 pl), se agitó a la misma temperatura por 10 minutos, entonces se añadió agua (25 pl) a la misma, se agitó por 5 horas, se añadió más agua (25 pl) a la misma y se agitó a la misma temperatura por 1 hora. La solución de reacción se concentró bajo presión reducida hasta que el volumen llegara a ser la mitad, entonces acetato de etilo y una solución acuosa saturada de cloruro de amonio se añadieron a la misma, se agitó y entonces la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se pulverizó con hexano, se colectó por filtración y entonces se secó para dar un compuesto trans, el compuesto 9 (747 mg) como un polvo incoloro. MS (APCI): m/z 457 [M+H]+
(7) Una solución del compuesto 9 (747 mg) en metanol (11 ml) se enfrió en hielo, se añadió cloruro de tionilo (597 mg) a la misma y la mezcla se agitó a la misma temperatura por 1 hora y entonces a temperatura ambiente por 15 horas. La solución de reacción se concentró bajo presión reducida, se pulverizó con éter diisopropílico, se colectó por filtración y entonces se secó para dar un compuesto trans, el compuesto 10 (702 mg) como un polvo incoloro. MS (APCI): m/z 371 [M+H]+
Ejemplo de referencia 131
[Fórmula química 146] 1

(1) A una solución del compuesto 1 (3,0 g) en N-metilpirrolidona (30 ml) se le añadió el compuesto 2 (2,64 ml) y carbonato de potasio (3,22 g) y la mezcla se agitó a 130 °C por 2 horas. A la mezcla de reacción se le añadió agua y acetato de etilo a temperatura ambiente, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-85:15), para dar el compuesto 3 (4,94 g) como un material viscoso amarillo. MS (ESI): m/z 330 [M+H]+
(2) A una solución del compuesto 3 (600 mg) y una solución de metilamina en etanol (33% en peso, 257 mg) en diclorometano (10 ml) se le añadió ácido acético (156 pl) y triacetoxiborohidruro de sodio (579 mg) y entonces la mezcla se agitó a temperatura ambiente por 1 hora. A la mezcla de reacción se le añadió una solución acuosa de carbonato ácido de sodio, se agitó, entonces se extrajo con diclorometano, la capa orgánica se secó y se
concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 80:20-50:50), para dar el compuesto 4 (435 mg) como un material viscoso de color rosa pálido. MS (ESI): m/z 345 [M+H]+
Ejemplos de referencia 132 y 133
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 131 anterior, para dar cada compuesto incluido en el cuadro 43 siguiente.

Ejemplo de referencia 134
[Fórmula química 147]

(1) Una solución del compuesto 1 (500 mg), que es un intermediario (compuesto 2) del ejemplo de referencia 81 anterior, en tetrahidrofurano (10 ml) se enfrió hasta -78 °C, una solución de n-butil litio en hexano (1,6 moles/l, 1,04 ml) se añadió gota a gota a la misma bajo atmósfera de nitrógeno, la mezcla se agitó a la misma temperatura por 5 minutos, entonces el compuesto 2 (308 mg) se añadió a la misma, se agitó por 40 minutos y se agitó a temperatura ambiente por 15 horas. A la mezcla de reacción se le añadió una solución acuosa de cloruro de amonio, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-65:35), para dar el compuesto 3 racémico (288 mg) como un polvo amarillo pálido. MS (ESI): m/z 438 [M+H]+
(2) Una solución del compuesto 3 (100 mg) en diclorometano (2 ml) se enfrió hasta -30 °C, se añadieron trietilsilano (197 pl) y ácido trifluoroacético (289 pl) a la misma y se agitó a la misma temperatura por 30 minutos y entonces a temperatura ambiente por 15 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó, entonces se extrajo con diclorometano, la capa orgánica resultante se secó y se concentró bajo presión reducida. Al residuo (73 mg) se le añadió dicarbonato de di-tbutilo (59 mg), trietilamina (63 pl) y tetrahidrofurano (1 ml) y la mezcla se agitó a temperatura ambiente por 15
horas. La solución de reacción se concentró bajo presión reducida y el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-82:18). A una solución del material crudo resultante (35 mg) en metanol (2,4 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 17,5 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 15 horas. El carbón de paladio se removió por filtración y el filtrado se concentró bajo presión reducida. El residuo se pulverizó con hexano, se colectó por filtración y entonces se secó para dar el compuesto 4 (21 mg) como un polvo incoloro.
(3) A una suspensión del compuesto 4 (21 mg) y carbonato de potasio (17,5 mg) en N,N-dimetilformamida (0,80 ml) se le añadió 4-bromobutirato de etilo (10,9 jl) y la mezcla se agitó a 70 °C por 15 horas. A la mezcla de reacción se le añadió acetato de etilo y solución salina a temperatura ambiente y la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina, se secó y el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-75:25), para dar el compuesto 5 (28 mg) como un material viscoso incoloro. MS (ESI): m/z 446 [M+H]+
(4) A una solución del compuesto 5 (25 mg) en 1,4-dioxano (2 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 140 jl) y la mezcla se agitó a temperatura ambiente por 15 horas. A la mezcla de reacción se le añadió una solución de ácido clorhídrico en 1,4- dioxano (4 moles/l, 840 jl), se agitó a temperatura ambiente por 8 horas y entonces se concentró bajo presión reducida para dar el compuesto racémico 6 (21 mg) como un material viscoso de color púrpura. MS (ESI): m/z 346 [M+H]+
Ejemplo de referencia 135
[Fórmula química 148]

(1) A una solución del compuesto 1 (315 mg), que es un intermediario (compuesto 2) del ejemplo de referencia 101, en tetrahidrofurano (10 ml), se le añadió dibromodifluorometano (365 mg) y tris(dimetilamino)fosfina (727 jl) bajo enfriamiento en hielo, la mezcla se agitó a la misma temperatura por 1 hora, entonces se añadió zinc (262 mg) a la misma y se calentó bajo reflujo por 1 hora. A la mezcla de reacción se le añadió agua a temperatura ambiente, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 98:2-85:15), para dar el compuesto 2 (159 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 350 [M+H]+
(2) A una solución del compuesto 2 (105 mg) y el compuesto 3 (214 mg) en diclorometano (2 ml) se le añadió ácido trifluoroacético (4,5 jl), la mezcla se calentó bajo reflujo por 16 horas, entonces el compuesto 3 (214 mg) y ácido trifluoroacético (4,5 j l) se añadieron a la misma y se calentó bajo reflujo por 2 horas. A la mezcla de reacción se le añadió ácido trifluoroacético (9,0 jl), se calentó bajo reflujo por 18 horas, entonces se enfrió hasta temperatura ambiente y se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 98:2-88:12), para dar el compuesto racémico 4 (86 mg) como un material viscoso incoloro. MS (ESI): m/z 483 [M+H]+
(3) A una solución del compuesto 4 (266 mg) en metanol (4 ml) se le añadió carbón de paladio a 10% (humedecido con aproximadamente 50% de agua, 80 mg) y ácido acético (5 ml) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 2 horas. El carbón de paladio se removió por filtración y el filtrado se concentró bajo presión reducida. Al residuo se le añadió acetato de etilo y una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces la mezcla se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-93:7), para dar el compuesto 5 racémico (184 mg) como un material viscoso incoloro. MS (ESI): m/z 393 [M+H]+
Ejemplo de referencia 136
[F ó rm u la q u ím ic a 149 ]

(1) Una solución del compuesto 1 (3,24 g), que es un intermediario (compuesto 3) del ejemplo de referencia 116, dicarbonato de di-t-butilo (2,06 g) y 4-dimetilaminopiridina (0,01 g) en acetonitrilo (32,4 ml), se agitó a temperatura ambiente por 6 horas. A la mezcla de reacción se le añadió agua y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 2 (3,78 g) como un polvo amarillo pálido. MS (APCI): m/z 530 [M+NH4]+
(2) Una solución del compuesto 2 (3,34 g) en tetrahidrofurano (25 ml) se enfrió hasta -78 °C, entonces una solución de bis(trimetilsilil)amida de litio en tetrahidrofurano (1,09 moles/l, 6,28 ml) se añadió gota a gota a la misma bajo atmósfera de nitrógeno, la mezcla se agitó a la misma temperatura por 1 hora, entonces una solución de N-fluorobencensulfonimida (4,11 g) en tetrahidrofurano (8 ml) se añadió gota a gota a la misma y se agitó a la misma temperatura por 6 horas. A la mezcla de reacción se le añadió una solución acuosa de cloruro de amonio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. Al residuo se le añadió acetato de etilo, la mezcla se agitó, entonces la materia insoluble se removió por filtración y el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-85:15), para dar el compuesto 3 (2,08 g) como un polvo incoloro. MS (APCI): m/z 548 [M+NH4]+
(3) A una solución del compuesto 3 (1,0 g) en metanol (10 ml) se le añadió una solución de ácido clorhídrico en metanol (2 moles/l, 10 ml) y la mezcla se agitó a 50 °C por 16 horas. La mezcla de reacción se enfrió hasta temperatura ambiente, entonces cloroformo y una solución acuosa saturada de carbonato ácido de sodio se añadieron a la misma y la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida para dar el compuesto 4 (711 g) como un polvo amarillo pálido. MS (APCI): m/z 389 [M+H]+
(4) A una solución del compuesto 4 (350 mg) en diclorometano (7 ml) se le añadió tetrafluoroborato de trimetiloxonio (1,11 g) y la mezcla se agitó a temperatura ambiente por 1,5 horas. La solución de reacción se añadió gota a gota a una solución de cianoborohidruro de sodio (630 mg), que se enfrió hasta 0 °C y ácido acético (717 pl) en metanol (7 ml) y la mezcla se agitó a temperatura ambiente por 1 hora. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-94:6), para dar el compuesto 5 (244 mg) como un material viscoso incoloro. MS (APCI): m/z 375 [M+H]+
(5) A una solución del compuesto 3 (710 mg) en tetrahidrofurano (10 ml) se le añadió gota a gota una solución de bis(trimetilsilil)amida de litio en tetrahidrofurano (1,09 moles/l, 1,29 ml) bajo atmósfera de nitrógeno a -78 °C, la mezcla se agitó a la misma temperatura por 1 hora, entonces se añadió una solución de 2,6-di-t-butilfenol (331 mg) en tetrahidrofurano (4 ml) gota a gota a la misma y se agitó a la misma temperatura por 4 horas. A la mezcla de reacción se le añadió una solución acuosa de cloruro de amonio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-85:15), para dar el compuesto 6 (308 mg) como un polvo incoloro. MS (APCI): m/z 548 [M+NH4]+
(6) El compuesto 6 se trató de una manera similar a los pasos (3) y (4) anteriores, para dar el compuesto 7 como un material viscoso incoloro. MS (APCI): m/z 375 [M+H]+
Ejemplo de referencia 137
[F ó rm u la q u ím ic a 150 ]

(1) A una solución de éster etílico de ácido difenilfosfonoacético (3,05 g) en tetrahidrofurano (143 ml) se le añadió hidruro de sodio (60 % en aceite, 381 mg) bajo enfriamiento en hielo y la mezcla se agitó bajo atmósfera de nitrógeno a la misma temperatura por 30 minutos. La solución de reacción se enfrió hasta -78 °C, una solución del compuesto 1 (3,40 g), que es un intermediario (compuesto 2) del ejemplo de referencia 99, en tetrahidrofurano (48 ml) se añadió gota a gota a la misma y la mezcla se agitó a la misma temperatura por 3 horas. A la mezcla de reacción se le añadió una solución acuosa de cloruro de amonio, se agitó a temperatura ambiente y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua, una solución acuosa saturada de carbonato ácido de sodio y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 98:2-90:10), para dar el compuesto 2 (3,65 g) como un polvo amarillo pálido. MS (ESI): m/z 428 [M+H]+
(2) A una solución del compuesto 2 (3,64 g) en etanol (30 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 4,69 ml) y la mezcla se agitó a temperatura ambiente por 3 horas. La solución de reacción se enfrió en hielo, una solución acuosa de ácido clorhídrico (2 moles/l, 4,69 ml) se añadió a la misma, la mezcla se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se pulverizó con un solvente mixto de éter diisopropílico y hexano (relación de la mezcla = 1:2), se colectó por filtración y entonces se secó para dar el compuesto 3 (1,84 g) como un polvo incoloro. MS (ESI): m/z 400 [M+H]+
(3) A una solución del compuesto 3 (300 mg) y carbonato de potasio (207 mg) en N,N-dimetilformamida (2 ml) se le añadió yoduro de metilo (70 pl) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió agua y acetato de etilo y la capa orgánica se separó. La capa orgánica resultante se lavó con agua y solución salina saturada y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 99:1-91:9), para dar el compuesto 4 (294 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 414 [M+H]+
(4) A una solución del compuesto 4 (288 mg) y el compuesto 5 (499 mg) en diclorometano (3 ml) se le añadió ácido trifluoroacético (5,4 pl) bajo enfriamiento en hielo, la mezcla se agitó a temperatura ambiente por 2 horas, entonces el compuesto 5 (499 mg) y ácido trifluoroacético (10,8 pl) se añadieron a la misma y se agitó a temperatura ambiente por otra hora más. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con cloroformo. La capa orgánica se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo resultante se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-75:25), para dar compuestos cis (racematos), el compuesto 6 (335 mg, 61 %) como un material viscoso incoloro. MS (ESI): m/z 547 [M+H]+
(5) A una solución del compuesto 6 (330 mg) y dicarbonato de di-t-butilo (137 mg) en metanol (5 ml)/tetrahidrofurano (5 ml) se le añadió carbón de paladio a 10% (humedecido con aproximadamente 50% de agua, 99 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 5 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-70:30), para dar el compuesto 7 (315 mg) como un material viscoso incoloro. MS (ESI): m/z 557 [M+H]+
(6) A una solución del compuesto 7 (240 mg) en metanol (1 ml) / tetrahidrofurano (4 ml) se le añadió borohidruro de litio (37 mg) bajo enfriamiento en hielo y la mezcla se agitó a la misma temperatura por 2 horas. A la mezcla de reacción se le añadió una solución acuosa de cloruro de amonio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 85:15-60:40), para dar el compuesto 8 (187 mg) como un material viscoso incoloro. MS (ESI): m/z 529 [M+H]+
(7) A una solución del compuesto 8 (210 mg) y yoduro de metilo (212 pl) en N,N-dimetilformamida (4 ml) se le añadió hidruro de sodio (60 % en aceite, 19 mg) bajo enfriamiento en hielo y la mezcla se agitó a la misma temperatura por 3 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de cloruro de amonio y agua, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 9 (200 mg) como un material viscoso incoloro. Ms (APCI): m/z 543 [M+H]+
(8) A una solución del compuesto 9 (195 mg) en metanol (1 ml) se le añadió una solución de ácido clorhídrico en metanol (2 moles/l, 2,7 ml) y la mezcla se agitó a 50 °C por 3 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla a temperatura ambiente y se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida para dar compuestos cis (racematos), el compuesto 10 (133 mg) como un material viscoso incoloro. MS (ESI): m/z 401 [M+H]+
Ejemplo de referencia 138
[Fórmula química 151]

(1) El compuesto 1 se trató de una manera similar al ejemplo de referencia 112, para dar el compuesto 2.
(2) Bajo atmósfera de nitrógeno, a tetrahidrofurano (1,1 ml) se le añadió una solución de n-butil litio en hexano (1,6 moles/l, 1,4 ml) a -78 °C, entonces diisopropilamina (0,32 ml) se añadió gota a gota a la misma y la mezcla se agitó bajo enfriamiento en hielo por 10 minutos. A la solución de reacción se le añadió gota a gota isobutirato de etilo (0,28 ml) a -78 °C y la mezcla se agitó a la misma temperatura por 30 minutos. A la solución de reacción se le añadió gota a gota una solución del compuesto 2 (750 mg) en tetrahidrofurano (2 ml) y la mezcla se agitó a -78 °C a -40 °C por 1 hora. A la mezcla de reacción se le añadió una solución acuosa de cloruro de amonio bajo enfriamiento en hielo, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 100:0-75:25), para dar el compuesto 3 (816 mg) como un material viscoso amarillo. MS (ESI): m/z 517 [M+H]+
(3) Una suspensión del compuesto 3 (1,23 g), formiato de amonio (1,31 g) y carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 410 mg) en metanol (24 ml), se calentó bajo radiación de microondas a 120 °C por 15 minutos. La mezcla de reacción se enfrió hasta temperatura ambiente, el carbón de paladio se removió por filtración y entonces el filtrado se concentró bajo presión reducida. Al residuo se le añadió acetato de etilo, la materia insoluble se removió por filtración y entonces el filtrado se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 80:20-0:100), para dar el compuesto 4 (428 mg) como un polvo incoloro (racematos). MS (ESI): m/z 441 [M+H]+ (4) Una solución del compuesto 4 (220 mg) y tetrafluoroborato de trimetiloxonio (148 mg) en diclorometano (5 ml), se agitó a temperatura ambiente por 1,5 horas. La solución de reacción se añadió gota a gota a una solución, que se enfrió hasta 0 °C, de cianoborohidruro de sodio (314 mg) y ácido acético (0,29 ml) en metanol (5 ml) y la mezcla se agitó a la misma temperatura por 30 minutos y entonces a temperatura ambiente por 5 horas. La solución de reacción se enfrió en hielo, una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma, la mezcla se agitó y entonces se extrajo con cloroformo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (cloroformo:metanol = 100:0-90:10), para dar el compuesto 5 (85 mg) como un material viscoso incoloro racémico. MS (ESI): m/z 427 [M+H]+
Ejemplo de referencia 139
[Fórmula química 152] 1

(1) A una solución del compuesto 1 (5,13 g) y el compuesto 2 (1,3 ml) en metanol (45 ml) se le añadió gota a gota ácido sulfúrico concentrado (350 pl) y la mezcla se agitó a 50 °C por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio para alcalinizarla a temperatura ambiente, se añadió cloroformo a la misma y se agitó. La capa orgánica se separó, se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice
(hexano:acetato de etilo = 97:3-85:15), para dar el compuesto 3 (6,2 g) como un líquido amarillo pálido.
(2) A una solución del compuesto 3 (6,2 g) en metanol (60 ml) se le añadió borohidruro de sodio (4,4 g) bajo enfriamiento en hielo y entonces la mezcla se agitó a temperatura ambiente por 5 horas. A la mezcla de reacción se le añadió agua, se agitó y entonces el metanol se evaporó bajo presión reducida a temperatura ambiente. A la solución acuosa resultante se le añadió cloroformo, la mezcla se agitó y la capa orgánica se separó. La capa orgánica resultante se lavó con solución salina saturada, se secó y se evaporó bajo presión reducida a temperatura ambiente. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-94:6), para dar el compuesto 4 (3,65 g) como un líquido incoloro. 1H-RMN (CDCh) 81,18 (s, 6H), 1,55-1,68 (m, 4H), 2,28 (t, J = 5,4 Hz, 1H), 3,20 (s, 3H), 3,64 (td, J = 5,7, 5,7 Hz, 2H).
(3) A una solución de sulfóxido de dimetilo (4,81 ml) en diclorometano (40 ml) se le añadió gota a gota una solución de cloruro de oxalilo (2,91 ml) en diclorometano (60 ml) a -78 °C, la mezcla se agitó por 2 minutos, entonces una solución del compuesto 4 (3,64 g) y piridina (4,97 ml) en diclorometano (30 ml) se añadió gota a gota a la misma a la misma temperatura y se agitó por 15 minutos. A la mezcla de reacción se le añadió gota a gota una solución de trietilamina (21,5 ml) en diclorometano (15 ml) a la misma temperatura y entonces se agitó bajo enfriamiento en hielo por 1 hora. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (1 mol/l, 240 ml), se agitó y se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se evaporó bajo presión reducida a temperatura ambiente. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-75:25), para dar el compuesto 5 (3,29 g) como un líquido amarillo pálido. 1H-Rm N (CDCh) 81,16 (s, 6H), 1,82 (t, J = 7,5 Hz, 2H), 2,49 (td, J = 7,5, 1,5 Hz, 2H), 3,15 (s, 3H), 9,78 (t, J = 1,8 Hz, 1H).
Ejemplo de referencia 140
[Fórmula química 153]

A una solución del compuesto 1 (3,54 g) en tetrahidrofurano (50 ml) se le añadió gota a gota una solución de pirrolidina (2,13 g) y trietilamina (1,9 ml) en tetrahidrofurano (20 ml) bajo enfriamiento en hielo bajo atmósfera de nitrógeno y entonces la mezcla se agitó a temperatura ambiente por 1 hora. La materia insoluble se removió por filtración y entonces el filtrado se concentró. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-70:30), para dar el compuesto 2 (3,99 g). MS (APCI): m/z 212/214 [M+H]+ Ejemplo de referencia 141
[Fórmula química 154]
B r ^ ^ ^ C 0 2Et

1 2
A una solución del compuesto 1 (1 ml) en éter dietílico (60 ml) se le añadió gota a gota bromuro de metilmagnesio (0,99 moles/l, 15,6 ml) bajo atmósfera de nitrógeno a -15 °C, entonces la mezcla se calentó hasta temperatura ambiente y se agitó por 18 horas. A la mezcla de reacción se le añadió secuencialmente una solución acuosa saturada de cloruro de amonio y una solución acuosa de ácido clorhídrico (1 mol/l, 10 ml), se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se evaporó bajo presión reducida a temperatura ambiente. El residuo se purificó parcialmente con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-75:25) y el compuesto 2 resultante (853 mg) se usó en el siguiente paso sin purificación.
Ejemplo de referencia 142
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 1 anterior, para dar el compuesto incluido en el cuadro 44 siguiente.
[Cuadro 44]

Ejemplo de referencia 143
[Fórmula química 155]

(1) El compuesto 1 se trató de una manera similar al ejemplo de referencia 1, para dar el compuesto 2 (1240 mg) como un polvo amarillo pálido. MS (APCI): m/z 484 [M+H]+
(2) A una solución del compuesto 2 (620 mg) en diclorometano (6,2 ml) se le añadió cloroformiato de 1 -cloroetilo (550 mg), la mezcla se agitó bajo reflujo de calor por 2 horas, entonces se añadió diisopropiletilamina (998 pl) a la misma y se agitó bajo reflujo de calor por otra hora más. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente, entonces se concentró bajo presión reducida, al residuo resultante se le añadió metanol (6,2 ml) y se agitó bajo reflujo de calor por 30 minutos. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente, entonces se añadió dicarbonato de di-t-butilo (336 mg) a la misma y se agitó a temperatura ambiente por 1 hora. A la mezcla se le añadió dicarbonato de di-t-butilo (672 mg) y diisopropiletilamina (998 pl) y se agitó a temperatura ambiente por 15 horas. A la mezcla de reacción se le añadió agua, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 85:15-70:30), para dar el compuesto 3 (374 mg) como un material viscoso anaranjado. MS (APCI): m/z 249 [M-Boc+H]+
(3) A una solución del compuesto 3 (365 mg) en metanol (3,7 ml) se le añadió una solución acuosa de hidróxido de sodio (1 mol/l, 1,3 ml) y la mezcla se agitó a temperatura ambiente por 30 minutos. La mezcla de reacción se enfrió en hielo, una solución acuosa de ácido clorhídrico (1 mol/l, 1,3 ml) se añadió a la misma, entonces acetato de etilo y agua se añadieron a la misma, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida para dar el compuesto 4 (331 mg) como un polvo amarillo pálido. MS (APCI): m/z 333 [M-H]-.
Ejemplo de referencia 144
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 33 anterior, para dar el compuesto incluido en el cuadro 45 siguiente.
r 4

Ejemplo de referencia 145
[Fórmula química 156]

(1) A una solución del compuesto 1 (1,19 g), que se obtuvo tratando un compuesto de partida correspondiente de una manera similar al ejemplo de referencia 33 anterior en tetrahidrofurano (11,9 ml) se le añadió una solución acuosa de ácido clorhídrico (1 mol/l, 11,9 ml) y la mezcla se agitó a temperatura ambiente por 16 horas y 15 minutos y entonces a 40 °C por 3 horas y 45 minutos. Se dejó que la mezcla se enfriara hasta temperatura ambiente, entonces a la mezcla de reacción se le añadió una solución acuosa de hidróxido de sodio (1 mol/l) para que sea alcalina (pH 8) y se extrajo con acetato de etilo. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 99:1-95:5), para dar el compuesto 2 (757 mg) como un líquido incoloro. MS (ESI): m/z 390 [M+H]+
(2) A una solución del compuesto 2 (376 mg) en etanol (3,76 ml) se le añadió ortoformiato de trietilo (715 mg) y fluoruro de boro y litio (109 mg) y la mezcla se agitó a temperatura ambiente por 1,5 horas y entonces a 80 °C por 43 horas y 30 minutos. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente, entonces una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 90:10-80:20), para dar el compuesto 3 (430 mg) como un líquido incoloro. MS (APCI): m/z 464 [M+H]+ (3) A una solución del compuesto 3 (425 mg) en diclorometano (4,3 ml) se le añadió una solución de complejo de borano-tetrahidrofurano en tetrahidrofurano (1 mol/l, 1 ml) a -63 °C, entonces trifluorometansulfonato de trimetilsililo (414 pl) se añadió a la misma y la mezcla se agitó a la misma temperatura por 1,5 horas. A la mezcla de reacción se le añadió etanol (1 ml), entonces se añadió una solución acuosa saturada de carbonato ácido de sodio bajo enfriamiento en hielo, se agitó y se extrajo con diclorometano. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se disolvió en metanol (8,5 ml), carbón de paladio a 10% (humedecido con aproximadamente 50 % de agua, 98 mg) se añadió a la misma, la mezcla se agitó, entonces se filtró a través de Celite y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 85:15-60:40), para dar el compuesto 4 (159 mg) como un líquido incoloro. MS (ESI): m/z 420 [M+H]+
(4) A una solución del compuesto 4 (425 mg) en etanol (1,5 ml) se le añadió una solución acuosa de hidróxido de sodio (2,0 moles/l, 906 pl) y la mezcla se agitó a temperatura ambiente por 30 minutos y a 60 °C por 1 hora. A la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (1 mol/l) para neutralizarla y entonces se concentró bajo presión reducida para dar el compuesto 5 (246 mg) como un sólido incoloro que contenía cloruro de sodio. m S (ESI): m/z 392 [M+H]+
Ejemplo de referencia 146
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 145 anterior, para dar el compuesto incluido en el cuadro 46 siguiente.
r 4

Ejemplo de referencia 147
[Fórmula química 157]

(1) El compuesto 1 se trató de una manera similar a un método descrito en una literatura (Tetrahedron 1994, 50, 7543-7556), para dar el compuesto 2 (47,6 g) como un líquido incoloro. MS (APCI): m/z 223 [M+H]+
(2) Una solución del compuesto 3 (984 mg), que se preparó de una manera similar a un método descrito en una literatura (J. Med. Chem. 1996, 39, 314-322, documento WO2007/006760, J. Med. Chem. 2013, 56, 10003 10015, documento W2004/089307), en diclorometano (6 ml) se añadió gota a gota a una solución, la cual se calentó hasta 45 °C, del compuesto 2 (400 mg) y ácido trifluoroacético (42 ^l) en diclorometano (6 ml) y la mezcla se agitó a la misma temperatura por 3 horas. A una solución acuosa enfriada en hielo de ácido cítrico (ácido cítrico: 10 g agua: 100 ml) se vertió la solución de reacción, la cual se dejó que se enfriara hasta temperatura ambiente y se extrajo con diclorometano. La capa orgánica resultante se lavó con carbonato ácido de sodio saturado, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 50:50-0:100), para dar racematos (cis) del compuesto 4 como un líquido amarillo pálido (522 mg). MS (APCI): m/z 392 [M+H]+
(3) A una solución del compuesto 4 (520 mg) en etanol (10 ml) se le añadió una solución acuosa de hidróxido de sodio (2 moles/l, 1328 ^l) y la mezcla se agitó a 70 °C por 20 horas. Después de ser enfriada hasta temperatura ambiente, a la mezcla de reacción se le añadió una solución acuosa de ácido clorhídrico (2 moles/l, 1328 ^l), se agitó y se concentró bajo presión reducida para dar racematos (cis) del compuesto 5 como un polvo incoloro (656 mg) que contenían cloruro de sodio. MS (APCI): m/z 378 [M+H]+
Ejemplo de referencia 148
[Fórmula química 158]

(1) El compuesto 1 (565 mg), que se preparó de una manera similar a un método descrito en una literatura (Chemistry Letters 2008, 37, 809-810), se trató de una manera similar al ejemplo de referencia 1, para dar el compuesto 2 (341 mg) como un polvo incoloro. MS (APCI): m/z 186 [M+H]+
(2) El compuesto 2 (320 mg) y el compuesto 3 (310 mg) se trataron de una manera similar al ejemplo de referencia 1, para dar el compuesto 4 (529 mg) como un polvo gris. MS (APCI): m/z 331 [M+H]+
(3) El compuesto 4 (520 mg) y el compuesto 5 (1121 mg) se trataron de una manera similar al ejemplo de referencia 1, para dar el compuesto 6 (439 mg) como un líquido amarillo pálido. MS (APCI): m/z 464 [M+H]+ (4) El compuesto 6 (435 mg) se trató de una manera similar al ejemplo de referencia 49, para dar el compuesto 7 como un polvo incoloro (307 mg) que contenía cloruro de litio. MS (APCI): m/z 319 [M+H]+
(5) El compuesto 7 (75 mg) y el compuesto 8 (80 mg) se trataron de una manera similar al ejemplo de referencia 49, para dar el compuesto 9 (116 mg) como un líquido incoloro. MS (APCI): m/z 701 [M+H]+
(6 ) El compuesto 9 (113 mg) se trató de una manera similar al ejemplo de referencia 49, para dar el compuesto 10 (85 mg) como un líquido incoloro. MS (APCI): m/z 611 [M+H]+
Ejemplo de referencia 149
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 45 anterior, para dar el compuesto incluido en el cuadro 47 siguiente.
r 47

Ejemplo de referencia 150
[Fórmula química 159]

(1) El compuesto 1 (108 mg) y el compuesto 2 (142 mg) se trataron de una manera similar al ejemplo de referencia 45 anterior, para dar el compuesto 3 (129 mg) como un material viscoso amarillo pálido. MS (ESI): m/z 718 [M+H]+
(2) Una mezcla del compuesto 3 (129 mg) y una solución de ácido clorhídrico en metanol (2 moles/l, 449 jl) se agitó a temperatura ambiente por 17 horas. La mezcla de reacción se concentró bajo presión reducida para dar el compuesto 4(117 mg) como un polvo amarillo pálido. MS (ESI): m/z 618 [M+H]+
Ejemplo de referencia 151
[Fórmula química 160] 1

(1) A una solución mixta del compuesto 1 (14,34 g), que se preparó de una manera similar a un método descrito en una literatura (Tetrahedron 2012, 68, 9578-9582), en tetrahidrofurano (80 ml)/metanol (20 ml) se le añadió poco a poco borohidruro de litio bajo enfriamiento en hielo y la mezcla se agitó a la misma temperatura por 1 hora. A la mezcla de reacción se le añadió una solución acuosa de carbonato ácido de sodio, se agitó, entonces se añadió cloruro de sodio a la misma y se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 40:60-0:100). A una solución del material crudo resultante (10,69 g) en diclorometano (166 ml) se le añadió trietilamina (20,55 ml) y cloruro de t-butildimetilsililo (8,16 g) en este orden y la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de cloruro de amonio, se agitó y entonces se extrajo con cloroformo. La capa orgánica se lavó con agua y solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-60:40), para dar el compuesto 3 (13,24 g) como un
material viscoso incoloro. MS (APCI): m/z 232 [M-Boc+H]+.
(2) Una solución del compuesto 3 (13,24 g) en diclorometano (201 ml) se enfrió en hielo, entonces ácido tricloroisocianúrico (9,23 g) y 2,2,6,6-tetrametilpiperidin-1-oxilo (624 mg) se añadieron a la misma y la mezcla se agitó a la misma temperatura por 1 hora. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con cloroformo. La capa orgánica resultante se lavó con una solución acuosa de ácido clorhídrico (0,5 moles/l), se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 97:3-85:15), para dar el compuesto 4 como un líquido incoloro racémico (13,22 g). m S (APCI): m/z 330 [M+H]+
(3) Una solución de una solución de bis(trimetilsilil)amida de sodio en tolueno (1 mol/l, 10 ml) en tetrahidrofurano (30 ml) se enfrió hasta -78 °C, una solución del compuesto 4 (3,0 g) en tetrahidrofurano (6 ml) se añadió gota a gota a la misma, la mezcla se agitó a la misma temperatura por 1 hora, entonces se añadió gota a gota una solución de N-fenilbis(trifluorometansulfonimida) (4,89 g) en tetrahidrofurano (12 ml) a la misma y se agitó a temperatura ambiente por 18 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo 100:0-95/5) y cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo 98:2-90/10), para dar el compuesto 5 (3,57 g) como un material viscoso incoloro. MS (APCI): m/z 462 [M+H]+
(4) A una solución del compuesto 5 (3,57 g) en 1,4-dioxano (36 ml) se le añadió bis(pinacolato)diboro (2,36 g), aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (318 mg), 1,1'-bis(difenilfosfino)ferroceno (216 mg) y acetato de potasio (2,28 g) y la mezcla se agitó a 100 °C por 1 hora. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente, el compuesto 6 (3,16 g) y una solución acuosa de carbonato de sodio (2 moles/l, 11,6 ml) se añadieron a la misma y se agitó a 100 °C por 1,5 horas. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente, se añadió agua, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20) y cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 7 (2,55 g) como un polvo incoloro. MS (APCI): m/z 641 [M+H]+ (5) A una solución del compuesto 7 (2,54 g) en tetrahidrofurano (10 ml) se le añadió una solución de fluoruro de tetrabutilamonio en tetrahidrofurano (1 mol/l, 4,36 ml) y la mezcla se agitó a temperatura ambiente por 15 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-60:40) y cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 80:20-50:50), para dar el compuesto 8 (2,01 g) como un polvo incoloro. MS (APCI): m/z 527 [M+H]+
(6) A una solución del compuesto 8 (1,0 g) en diclorometano (35 ml) se le añadió catalizador de Crabtree (31 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 16 horas. La mezcla de reacción se concentró bajo presión reducida y entonces el residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 85:15-65:35), para dar compuestos trans (racematos), el compuesto 9 (518 mg) como un polvo amarillo pálido. MS (APCI): m/z 529 [M+H]+
(7) A una solución del compuesto 9 (511 mg) en tetrahidrofurano (5,1 ml) se le añadió yoduro de metilo (604 pl) e hidruro de sodio (60% en aceite, 46 mg) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 4 horas. A la mezcla de reacción se le añadió agua, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar compuestos trans (racematos), el compuesto 10 (433 mg) como un material viscoso amarillo pálido. MS (APCI): m/z 543 [M+H]+
(8) Al compuesto 10 (427 mg) se le añadió una solución de ácido clorhídrico en metanol (2 moles/l, 8,5 ml) y la mezcla se agitó a 50 °C por 3 horas. El solvente se evaporó bajo presión reducida, entonces el residuo se diluyó con cloroformo, una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma para alcalinizar la mezcla y se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida para dar compuestos trans (racematos), el compuesto 11 (329 mg) como un material viscoso amarillo pálido. MS (APCI): m/z 401 [M+H]+
Ejemplo de referencia 152
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 101 anterior, para dar el compuesto incluido en el cuadro 48 siguiente.
r 4

Ejemplo de referencia 153
Un intermediario del ejemplo de referencia 152, se preparó de acuerdo con el siguiente método.
[Fórmula química 161]

(1) Una solución del compuesto 1 (15 g) en etanol (150 ml) se enfrió en hielo, se añadió borohidruro de sodio (2,0 g) a la misma y la mezcla se agitó a la misma temperatura por 10 minutos y entonces a temperatura ambiente por 30 minutos. La mezcla de reacción se enfrió en hielo, una solución acuosa de ácido clorhídrico (1 mol/l) se añadió a la misma, se agitó y entonces se concentró bajo presión reducida. La mezcla se extrajo con cloroformo, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-50:50), para dar compuestos cis (racematos), el compuesto 2 (6,44 g) como un polvo incoloro. MS (APCI): m/z 288 [M+H]+
(2) A una solución del compuesto 2 (6,43 g) en tetrahidrofurano (130 ml) se le añadió diisopropiletilamina, entonces se añadió gota a gota cloruro de metansulfonilo (3,84 g) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 30 minutos. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó, entonces se extrajo con acetato de etilo, se secó y se concentró bajo presión reducida. A una solución del residuo en tetrahidrofurano (130 ml) se le añadió 1,8-diazabiciclo[5.4.0]-7-undeceno (5,0 ml) y la mezcla se agitó a 70 °C por 30 minutos. La mezcla de reacción se concentró bajo presión reducida, entonces acetato de etilo y una solución acuosa saturada de carbonato ácido de sodio se añadieron a la misma, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 3 (5,59 g) como un líquido incoloro. MS (APCI): m/z 287 [M+NH4]+
(3) A una solución del compuesto 3 (5,58 g) en etanol (112 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 560 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 5 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró bajo presión reducida para dar el compuesto racémico 4 (5,37 g) como un líquido incoloro. MS (APCI): m/z 272 [M+H]+
(4) A una solución del compuesto 4 (5,36 g) en 1,4-dioxano (15 ml) se le añadió una solución de ácido clorhídrico en 1,4-dioxano (4 moles/l, 15 ml) y la mezcla se agitó a temperatura ambiente por 23 horas. La mezcla de reacción se concentró bajo presión reducida, se pulverizó con éter dietílico, se colectó por filtración y se secó bajo presión reducida para dar el compuesto 5 como un polvo incoloro. MS (APCI): m/z 172 [M+H]+
Ejemplo de referencia 154
Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 115 anterior, para dar el compuesto incluido en el cuadro 49 siguiente.
r 4

Ejemplo de referencia 155
[Fórmula química 162]

(1) A una suspensión de 4-yodobenzotrifluoruro (1015 mg), dibencilidenoacetona de paladio (II) (67 mg), (±)-2,2'-bis(difenilfosfino)-1,1'-binaftilo (92 mg) y t-butóxido de sodio (565 mg) en tolueno (20 ml) se le añadió el compuesto 1 (1000 mg) y la mezcla se agitó a 85 °C por 6 horas. Después de que se dejó que se enfriara hasta temperatura ambiente, a la mezcla de reacción se le añadió agua, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-70:30), para dar el compuesto 2 (644 mg) como un líquido anaranjado. MS (APCI): m/z 357 [M+H]+
(2) A una solución del compuesto 2 (376 mg) en cloroformo (5 ml) se le añadió N- bromosuccinimida (320 mg) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 14 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-75:25), para dar el compuesto 3 (663 mg) como un líquido amarillo pálido. MS (a Pc I): m/z 435/437 [M+H]+ (3) El compuesto 3 (660 mg) se trató de una manera similar al ejemplo de referencia 115 anterior, para dar el compuesto 4 (602 mg) como un polvo incoloro. MS (APCI): m/z 491 [M+H]+
(4) El compuesto 4 (590 mg) se trató de una manera similar al ejemplo de referencia 115 anterior, para dar el compuesto 5 (500 mg) como un líquido amarillo pálido. MS (APCI): m/z 391 [M+H]+
Ejemplo de referencia 156
[Fórmula química 163]

(1) Una suspensión del compuesto 1 (5,0 g), bromuro de bencilo (2,96 ml) y carbonato de potasio (4,3 g) en N,N-dimetilformamida (20,7 ml) se agitó a 80 °C por 90 minutos. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente y entonces una solución acuosa saturada de cloruro de amonio se añadió a la misma, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 98:2), para dar el compuesto 2 (6,34 g) como un polvo incoloro. MS (ESI): m/z 329/331 [M-H]-(2) Una solución del compuesto 2 (1000 mg), el compuesto 3 (714 jl), tris(dibencilidenoacetona)dipaladio(0) (138 mg), (±)-2,2'-bis(difenilfosfino)-1,1'-binaftilo (188 mg) y t-butóxido de sodio (581 mg) en tolueno (10 ml), se agitó bajo atmósfera de nitrógeno a 100 °C por 15 horas. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente, entonces se filtró por Celite, al filtrado resultante se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-70:30), para dar el compuesto 4 (1070 mg) como un líquido incoloro. MS (ESI): m/z 451 [M+H]+
(3) A una solución del compuesto 4 (1070 mg) en etanol (7,9 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50% de agua, 214 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) por 6,5 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró para dar el compuesto 5 (798 mg) como un material viscoso amarillo. MS (ESI): m/z 361 [M+H]+
(4) Una solución del compuesto 5 (400 mg) y piridina (449 jl) en diclorometano (5,5 ml) se enfrió en hielo, se añadió anhídrido trifluorometansulfónico (280 jl) a la misma y entonces la mezcla se agitó a la misma temperatura por 1 hora. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica se lavó con una solución acuosa saturada de cloruro de amonio y solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-70:30). A una solución del compuesto resultante (537 mg) en 1,4-dioxano (3,6 ml) / agua (364 jl) se le añadió ácido 4-metoxicarbonilfenilborónico (294 mg), carbonato de sodio (347 mg) y aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (45 mg) y la mezcla se agitó a 100 °C por 19,5 horas. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente, acetato de etilo y una solución acuosa saturada de carbonato ácido de sodio se añadieron a la misma, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-70:30), para dar el compuesto 6 (410 mg) como un líquido incoloro. MS (ESI): m/z 479 [M+H]+
(5) Una mezcla del compuesto 6 (101 mg) y una solución de ácido clorhídrico en metanol (2 moles/l, 1,01 ml), se agitó a temperatura ambiente por 13,5 horas. La mezcla de reacción se concentró bajo presión reducida para dar el compuesto 7 (88 mg) como un polvo incoloro. MS (ESI): m/z 379 [M+H]+
Ejemplo de referencia 157
[Fórmula química 164]

(1) Una suspensión del compuesto 1 (2,0 g), el compuesto 2 (1,8 ml) y carbonato de potasio (2,1 g) en acetonitrilo (26 ml), se agitó a 60 °C por 17 horas. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente, entonces se añadió agua a la misma, se agitó y se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-70:30), para dar el compuesto 3 (2,9 g) como un líquido incoloro. MS (APCI): m/z 381/383 [M+H]+
(2) El compuesto 3 (2,9 g) y el compuesto 4 (3,3 ml) se trataron de una manera similar al ejemplo de referencia 105 anterior, para dar el compuesto 5 (2,8 g) como un material viscoso amarillo. MS (ESI): m/z 429 [M+H]+ (3) El compuesto 5 se trató de una manera similar al ejemplo de referencia 101 anterior, para dar el compuesto 6 (48 mg) como un material viscoso incoloro. MS (ESI): m/z 502 [M+H]+
(4) Una mezcla del compuesto 6 (48 mg) y una solución de ácido clorhídrico en metanol (2 moles/l, 479 pl), se agitó a temperatura ambiente por 18 horas. La mezcla de reacción se concentró bajo presión reducida para dar el compuesto 7 (45 mg) como un material viscoso incoloro. MS (ESI): m/z 402 [M+H]+
Ejemplo de referencia 158
[Fórmula química 165] 1

(1) Un compuesto de partida correspondiente se trató de una manera similar al ejemplo de referencia 121, para dar el compuesto 1 (1108 mg) como un material viscoso incoloro. MS (ESI): m/z 466 [M+H]+
(2) A una solución del compuesto 1 (1070 mg) en etanol (7,9 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 222 mg) y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) por 3 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró para dar el compuesto 2 (496 mg) como un material viscoso incoloro. MS (ESI): m/z 374 [M-H]-.
(3) Una solución del compuesto 2 (200 mg), el compuesto 3 (126 mg), azodicarboxilato de diisopropilo (309 pl) y trifenilfosfina (419 mg) en tetrahidrofurano (2,7 ml) se agitó a temperatura ambiente por 24 horas. La mezcla de reacción se diluyó con acetato de etilo, se lavó con una solución acuosa saturada de carbonato ácido de sodio y una solución acuosa saturada de cloruro de amonio, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-70:30), para dar el compuesto 4 (183 mg) como un material viscoso incoloro. MS (ESI): m/z 516 [M+H]+
(4) Una mezcla del compuesto 4 (180 mg) y una solución de ácido clorhídrico en metanol (2 moles/l, 873 pl), se agitó a temperatura ambiente por 6,5 horas. La mezcla de reacción se concentró bajo presión reducida para dar
el compuesto 5 (157 mg) como un polvo incoloro. MS (ESI): m/z 416 [M+H]+
Ejemplo de referencia 159

(1) A una suspensión de borohidruro de litio (622 mg) en tetrahidrofurano (25 ml) se le añadió una solución del compuesto 1 (2,0 g), que se preparó de una manera similar a un método descrito en una literatura (Bioorganic & Medicinal Chemistry Letters 1996, 6, 963-966), en tetrahidrofurano (15 ml), la mezcla se agitó a temperatura ambiente por 1 hora, entonces se añadió metanol (5 ml) a la misma y se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió agua, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se secó y se concentró bajo presión reducida para dar el compuesto 2 (2,09 g) como un material viscoso incoloro. MS (APCI): m/z 236 [M+H]+
(2) A una solución del compuesto 2 (2,09 g) y dicarbonato de di-t-butilo (2,03 g) en metanol (42 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 625 mg) bajo agitación y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 1,5 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró para dar el compuesto 3 (2,33 g) como un material viscoso amarillo pálido. MS (APCI): m/z 232 [M+H]+
(3) A una solución del compuesto 3 (2,33 g) y cloruro de t-butildimetilsililo (1,67 g) en N,N-dimetilformamida (47
ml) se le añadió trietilamina (2,10 ml) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 72 horas. A la mezcla de reacción se le añadió agua y acetato de etilo, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 90:10-70:30), para dar el compuesto 4 (1,99 g) como un material viscoso incoloro. MS (APCI): m/z 346 [M+H]+
(4) Una solución del compuesto 4 (1,99 g) en diclorometano (30 ml) se enfrió en hielo, entonces ácido tricloroisocianúrico (1,34 g) y 2,2,6,6-tetrametilpiperidin-1-oxilo (90 mg) se añadieron a la misma y la mezcla se agitó a la misma temperatura por 30 minutos. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 5 (1,67 g) como un material viscoso incoloro. MS (APCI): m/z 344 [M+H]+
(5) Una solución de una solución de bis(trimetilsilil)amida de sodio en tolueno (1 mol/l, 5,3 ml) en tetrahidrofurano (50 ml) se enfrió hasta -78 °C, una solución del compuesto 5 (1,67 g) en tetrahidrofurano (10 ml) se añadió gota a gota a la misma, la mezcla se agitó a la misma temperatura por 40 minutos, entonces una solución de N-fenilbis(trifluorometansulfonimida) (2,60 g) en tetrahidrofurano (7 ml) se añadió a la misma y la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio bajo enfriamiento en hielo, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo: 98:2-93/7), para dar una mezcla del compuesto 6 y el compuesto 7 como un material viscoso incoloro (2,43 g). MS (APCI): m/z 476 [M+H]+
(6) A una solución de una mezcla (2,43 g) del compuesto 6 y el compuesto 7 en 1,4- dioxano (24 ml) se le añadió bis(pinacolato)diboro (1,55 g), aducto de diclorometano de dicloro[1,1'-bis(difenilfosfino)ferroceno]paladio(II) (416 mg), 1,1'-bis(difenilfosfino)ferroceno (282 mg) y acetato de potasio (1,50 g) y la mezcla se agitó a 100 °C por 1 hora. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente, el compuesto 8 (2,08 g) y una solución acuosa de carbonato de sodio (2 moles/l, 7,63 ml) se añadieron a la misma y se agitó a 85 °C por 17 horas. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente, se añadió agua a la misma, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 98:2-92:8), para dar una mezcla del compuesto 9 y el compuesto 10 como un polvo incoloro (1,50 g). MS (APCI): m/z 655 [M+H]+
(7) A una solución de una mezcla (1,49 g) del compuesto 9 y el compuesto 10 en tetrahidrofurano (7,5 ml) se le añadió una solución de fluoruro de tetrabutilamonio en tetrahidrofurano (1 mol/l, 3,4 ml) y la mezcla se agitó a temperatura ambiente por 1,5 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de carbonato ácido de sodio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-60:40) y cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 80:20-60:40), para dar una mezcla del compuesto 11 y el compuesto 12 como un polvo incoloro (1,53 g). MS (APCI): m/z 541 [M+H]+
(8) A una solución de una mezcla (1,17 g) del compuesto 11 y el compuesto 12 en metanol (23 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 350 mg) bajo agitación y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 89 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró para dar el compuesto 13, que era una mezcla de diastereómeros, como un polvo incoloro (1,15 g). MS (APCI): m/z 543 [M+H]+
(9) A una solución del compuesto 13 (1,23 g) en N,N-dimetilformamida (25 ml) se le añadió yoduro de metilo (1,41 ml), entonces se añadió hidruro de sodio (60 % en aceite, 109 mg) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 21 horas. A la mezcla de reacción se le añadió agua, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar el compuesto 14 (683 mg) como un polvo incoloro y el compuesto 15 (164 mg) como un material viscoso incoloro. Cada MS (APCI): m/z 557 [M+H]+ TLC (hexano:acetato de etilo = 80:20): valor de Rf del compuesto 14 “ 0,55, valor de Rf del compuesto 15 “ 0,50 (placa de TLC: gel de sílice para TLC 1.05715.0001 60 F254 , fabricado por Merck KGaA). La configuración del compuesto 1 y el espectro 1D NOESY confirmaron que el compuesto 14 tenía la configuración anterior.
(10) Al compuesto 14 (500 mg) se le añadió una solución de ácido clorhídrico en metanol (2 moles/l, 5 ml) y la mezcla se agitó a 50 °C por 4 horas. A la mezcla se le añadió una solución de ácido clorhídrico en metanol (2 moles/l, 2 ml) y se agitó a 60 °C por 2 horas. El solvente se evaporó bajo presión reducida, entonces el residuo se diluyó con cloroformo, una solución acuosa saturada de carbonato ácido de sodio se añadió a la misma para alcalinizar la mezcla y se extrajo con cloroformo. La capa orgánica resultante se secó y se concentró bajo presión reducida para dar el compuesto 16 (367 mg) como un material viscoso incoloro. MS (APCI): m/z 415 [M+H]+ (11) Al compuesto 15 (149 mg) se le añadió una solución de ácido clorhídrico en metanol (2 moles/l, 4 ml) y la mezcla se agitó a 60 °C por 5 horas. A la mezcla se le añadió ácido sulfúrico (0,15 ml) y se agitó a 60 °C por 2 horas. El solvente se evaporó bajo presión reducida, entonces el residuo se diluyó con diclorometano, una solución acuosa saturada de carbonato ácido de sodio se añadió para alcalinizar la mezcla y se extrajo con
diclorometano. La capa orgánica resultante se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (cloroformo:metanol = 100:0-90:10), para dar el compuesto 17 (80,4 mg) como un material viscoso incoloro. MS (APCI): m/z 415 [M+H]+
Ejemplo de referencia 160
[Fórmula química 167] 1

(1) A una solución del compuesto 1 (3,0 g) en diclorometano (30 ml) se le añadió ácido 3-cloroperbenzoico (8,74 g) y la mezcla se agitó a temperatura ambiente por 22 horas. A la mezcla de reacción se le añadió una solución acuosa de hidróxido de sodio (1 mol/l), se agitó y entonces se extrajo con diclorometano. La capa orgánica resultante se lavó con una solución acuosa de hidróxido de sodio (1 mol/l), se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 85:15-60:40), para dar el compuesto 2 (2,52 g) como un líquido incoloro. MS (APCI): m/z 130 [M-tBu+2H]+ (2) A una solución del compuesto 3 (1,29 g) en tetrahidrofurano (9 ml) se le añadió una solución de bromuro de isopropilmagnesio en tetrahidrofurano (2 moles/l, 2,1 ml) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 2 horas. La mezcla de reacción se enfrió en hielo de nuevo yoduro de cobre (62 mg) y una solución del compuesto 2 (600 mg) en tetrahidrofurano (4 ml) se añadieron a la misma y la mezcla se agitó a temperatura ambiente por 17 horas. La mezcla de reacción se enfrió en hielo, una solución acuosa de ácido clorhídrico (1 mol/l) se añadió a la misma, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con una solución acuosa saturada de carbonato ácido de sodio, entonces se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice de NH (hexano:acetato de etilo = 70:30-40:60), para dar compuestos trans (racematos), el compuesto 4 (2,52 g) como un polvo amarillo pálido. MS (APCI): m/z 338 [M-Boc+H]+
(3) A una solución del compuesto 4 (460 mg) en N,N-dimetilformamida (4,6 ml) se le añadió yoduro de metilo (262 |jl) e hidruro de sodio (60 % en aceite, 50 mg) bajo enfriamiento en hielo y la mezcla se agitó a temperatura ambiente por 2 horas. A la mezcla de reacción se le añadió agua, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con agua y solución salina saturada, se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 95:5-80:20), para dar compuestos trans (racematos), el compuesto 5 (362 mg) como un material viscoso incoloro. MS (APCI): m/z 352 [M-Boc+H]+
(4) A una solución del compuesto 5 (355 mg) en metanol (4 ml) / tetrahidrofurano (2 ml) se le añadió carbón de paladio a 10 % (humedecido con aproximadamente 50 % de agua, 106 mg) bajo agitación y la mezcla se agitó bajo atmósfera de hidrógeno (1 atm) a temperatura ambiente por 15 horas. El carbón de paladio se removió por filtración y entonces el filtrado se concentró para dar compuestos trans (racematos), el compuesto 6 (280 mg) como un polvo incoloro. MS (APCI): m/z 262 [M-Boc+H]+
(5) El compuesto 6 (275 mg) y el compuesto 7 se trataron de una manera similar al ejemplo de referencia 82, para dar compuestos trans (racematos), el compuesto 8 (293 mg) como un polvo incoloro. MS (APCI): m/z 380 [M-Boc+H]+
(6) El compuesto 8 (287 mg) se trató de una manera similar al ejemplo de referencia 115 anterior, para dar compuestos trans (racematos), el compuesto 9 (215 mg) como un líquido amarillo pálido. MS (APCI): m/z 380 [M+H]+
Ejemplo de referencia 161
[Fórmula química 168]

(1) A una solución del compuesto 1 (150 mg) y trifenilfosfina (186 mg) en tetrahidrofurano (3 ml) se le añadió tetracloruro de carbono (68 jl) y la mezcla se agitó a 60 °C por 17 horas. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente y los materiales innecesarios fueron removidos por filtración. El filtrado se concentró bajo presión reducida, entonces acetonitrilo (3 ml), cianuro de trimetilsililo (133 jl) y una solución de fluoruro de tetrafluoroamonio en tetrahidrofurano (1 mol/l, 1,06 ml) se añadieron a la misma y la mezcla se agitó a 80 °C por 3 horas. A la mezcla se le añadió cianuro de trimetilsililo (44 jl) y una solución de fluoruro de tetrafluoroamonio en tetrahidrofurano (1 mol/l, 354 jl) y se agitó a 80 °C por otras 2 horas. Se dejó que la mezcla de reacción se enfriara hasta temperatura ambiente, se añadió agua a la misma, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se lavó con solución salina saturada, se secó y se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 80:20-60:40), para dar el compuesto 2 (120 mg) como un material viscoso incoloro. MS (APCI): m/z 333/335 [M-Boc+H]+
(2) Una solución del compuesto 2 (118 mg) en tolueno (3 ml) se enfrió hasta -78 °C, una solución de hidruro de diisobutilaluminio en tolueno (1,01 moles/l, 674 jl) se añadió a la misma y la mezcla se agitó a la misma temperatura por 1 hora y entonces a temperatura ambiente por 2 horas. La mezcla de reacción se enfrió en hielo, 1 N de una solución acuosa de ácido clorhídrico se añadió a la misma, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se secó y entonces se concentró bajo presión reducida. A una solución del material crudo resultante en tetrahidrofurano (4 ml)/agua (1 ml) se le añadió borohidruro de sodio (12 mg) bajo enfriamiento en hielo y la mezcla se agitó a la misma temperatura por 10 minutos. A la mezcla de reacción se le añadió una solución acuosa de cloruro de amonio, se agitó y entonces se extrajo con acetato de etilo. La capa orgánica resultante se secó y entonces se concentró bajo presión reducida. El residuo se purificó con cromatografía en columna de gel de sílice (hexano:acetato de etilo = 70:30-40:60), para dar el compuesto 3 (59 mg) como un polvo incoloro. MS (APCI): m/z 338/340 [M-Boc+H]+
(3) El compuesto 3 (87 mg) se trató de una manera similar al ejemplo de referencia 115 anterior, para dar el compuesto 4 (71 mg) como un material viscoso amarillo pálido. MS (APCI): m/z 352/354 [M-Boc+H]+
(4) El compuesto 4 (69 mg) se trató de una manera similar al ejemplo de referencia 115 anterior, para dar el compuesto 5 (79 mg) como un material viscoso amarillo pálido. MS (APCI): m/z 408 [M-Boc+H]+
(5) El compuesto 5 (78 mg) se trató de una manera similar al ejemplo de referencia 115 anterior, para dar el compuesto 6 (77 mg) como un material viscoso amarillo pálido. MS (APCI): m/z 408 [M+H]+
[Ejemplo experimental]
Medición del agonista del MCR del ejemplo experimental 1 (medición del AMPc)
(1) Método para el cultivo de células
Se llevó a cabo la medición de la actividad del agonista del MC1R humano usando la línea de células de melanoma humano HBL. Cultivo de HBL: Se usó la mezcla de nutrientes F-10 que contenía FCS a 10 % y penicilinaestreptomicina en el cultivo.
(2) Prueba del AMPc y cálculo de datos
Cada solución de compuestos con cada concentración se mezcló con regulador de pH de prueba de AMPc (HBSS (solución salina balanceada de Hank) que contenía HEPES 10 mM y BSA a 0,1 %) y se dispensó en la placa de 96
cavidades. Se suspendió HBL en regulador de pH de prueba de AMPc que contenía IBMX 0,5 mM de modo que el concentrado llegara a ser 5 x 104/ml, dispensado en la placa de 96 cavidades anterior, entonces se mezcló, se dejó reposar a 37 °C por 30 minutos y entonces la concentración intracelular de AMP se midió por el método de fluorescencia usando Envision (por ejemplo, 320 nm, 590 nm y 665 nm). El valor del cociente (valor de medición de 665 nm / valor de medición de 590 nm x 10000) se calculó a partir de los datos resultantes, entonces el valor cuantitativo de la concentración de AMPc se calculó usando Prism 5.02, se calculó el % del valor de inducción (% de cada muestra cuando la concentración promedio de AMPc del vehículo es 0 % y la concentración promedio de AMPc de aMSH a 10-6 M es 100 %) y se calcularon el valor de la EC50 y el % del valor de la actividad intrínseca (IA).
Cuadro 50

continuación

Campo de aplicación industrial
El compuesto objetivo [I] de la presente invención o una sal farmacéuticamente aceptable del mismo tiene excelente actividad agonista del MCR, en particular, actividad agonista del MC1R y de esta manera, puede usarse como un agente para prevenir o tratar, o para mejorar el pronóstico de, varias enfermedades y/o síntomas cuyas condiciones patológicas se espera sean mejoradas a través de la activación del MC1R, por ejemplo, artritis reumatoide, artritis gotosa, osteoartrosis, enfermedad inflamatoria del intestino, esclerosis sistémica, psoriasis, fibrosis, protoporfiria, lupus eritematoso sistémico, melanoma, cáncer de piel, vitiligo, pérdida del cabello, dolor, daño por isquemia/reperfusión, enfermedad inflamatoria cerebral, hepatitis, choque séptico/septicemia, nefritis, trasplante, exacerbación de la enfermedad por el VIH, vasculitis, uveítis, retinitis pigmentosa, degeneración macular relacionada con la edad, infección microbiana, enfermedad celíaca, síndrome nefrótico e invasión por melanoma.
Claims (21)
1. Un derivado de pirrolidina representado por la fórmula [I]:

donde
el anillo A es un grupo fenilo opcionalmente sustituido
donde los sustituyentes en el grupo fenilo opcionalmente sustituido son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7 , un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6; R1 es un grupo alquilo C1-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo adamantilo opcionalmente sustituido, un grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno, un grupo arilo de 6 a 10 miembros opcionalmente sustituido que puede ser parcialmente hidrogenado, un grupo heteroarilo de 5 a 10 miembros opcionalmente sustituido que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6, donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno y un grupo alquilenoxi C1-C6 y sustituyentes en cada uno del grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno, el grupo arilo de 6 a 10 miembros opcionalmente sustituido que puede ser parcialmente hidrogenado y el grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6; un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo hidroxialquilo C1-C6; un grupo alcoxialquilo C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno y un grupo alquilenoxi C1-C6;
R2 es un átomo de halógeno, un grupo alquilo C1-C6 o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II]:

donde el anillo B representa un grupo heterocíclico alifático de 4 a 8 miembros que contenían nitrógeno que puede contener parcialmente un doble enlace;
el anillo C representa un grupo arilo de 6 a 10 miembros o un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno;
R5 y R6 representan cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres heteroátomos; un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
R7 representa un grupo alquilo C1-C6 opcionalmente sustituido, un grupo alquenilo C2-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo cicloalquenilo C3-C7 opcionalmente sustituido, un grupo arilo de 6 a 10 miembros opcionalmente sustituido, un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido, un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido, un grupo alcoxi C1-C6 opcionalmente sustituido, un grupo amino opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo, o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo,
donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo hidroxi o un grupo oxo; un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con uno o dos grupos oxo; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (la porción alquilo C1-C6 es opcionalmente sustituida con un grupo hidroxilo, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6 y
los sustituyentes en cada uno del grupo alquenilo C2-C6 opcionalmente sustituido, el grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo cicloalquenilo C3-C7 opcionalmente sustituido, el grupo arilo de 6 a 10 miembros opcionalmente sustituido, el grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido, el grupo heterocíclico alifático de 4 a 8 miembros opcionalmente sustituido que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno y el grupo alcoxi C1-C6 opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del
grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo hidroxi o un grupo oxo; un grupo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con uno o dos grupos oxo; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (la porción alquilo C1-C6 es opcionalmente sustituida con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6 y
R8 y R9 representan cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
2. El compuesto o sal farmacéuticamente aceptable del mismo de conformidad con la reivindicación 1 , donde el anillo A es un grupo fenilo opcionalmente sustituido,
donde los sustituyentes en el grupo fenilo opcionalmente sustituido son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7, un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6;
R1 es un grupo alquilo C1-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo adamantilo opcionalmente sustituido, un grupo heterocíclico alifático opcionalmente sustituido, un grupo arilo de 6 a 10 miembros opcionalmente sustituido que puede ser parcialmente hidrogenado, un grupo heteroarilo opcionalmente sustituido o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6, donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático y un grupo alquilenoxi C1-C6,
los sustituyentes en cada uno del grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo heterocíclico alifático opcionalmente sustituido, el grupo arilo de 6 a 10 miembros opcionalmente sustituido que puede ser parcialmente hidrogenado y el grupo heteroarilo opcionalmente sustituido son uno a tres grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6; un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno; un grupo cicloalquilo C3-C7 ; un grupo alcoxi C1-C6; un grupo hidroxialquilo C1-C6; un grupo alcoxialquilo C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático y un grupo alquilenoxi C1-C6,
la porción heterocíclica alifática de cada uno del grupo carbonilo heterocíclico alifático y el grupo sulfonilo heterocíclico alifático con el cual R1 es sustituido es un anillo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía por lo menos un átomo de nitrógeno y uno que contenía además opcionalmente un heteroátomo seleccionado del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno, la porción heterocíclica alifática del grupo heterocíclico alifático opcionalmente sustituido representado por R1 es un anillo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno y la porción heteroarilo del grupo heteroarilo opcionalmente sustituido representado por R1 es un heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno;
R2 es un átomo de halógeno, un grupo alquilo C1-C6 o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II],
donde el anillo B es un grupo heterocíclico alifático monocíclico o bicíclico de 4 a 8 miembros que puede contener adicionalmente, además el átomo de nitrógeno mostrado en la fórmula [II], un heteroátomo seleccionado del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno y puede contener parcialmente un doble enlace;
el anillo C es un grupo arilo monocíclico de 6 a 10 miembros o un grupo heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno;
R5 y R6 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
R7 es un grupo alquilo C1-C6 opcionalmente sustituido, un grupo alquenilo C2-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo cicloalquenilo C3-C7 opcionalmente sustituido, un grupo arilo de 6 a 10 miembros opcionalmente sustituido, un grupo heteroarilo opcionalmente sustituido, un grupo heterocíclico alifático opcionalmente sustituido, un grupo alcoxi C1-C6 opcionalmente sustituido, un grupo amino opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo,
donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros que contenía de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo hidroxi u oxo; un grupo heterocíclico alifático monocíclico de 4 a 7 miembros opcionalmente sustituido con uno o dos grupos oxo y que contiene uno o dos heteroátomos grupos oxo y que contiene uno o dos heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6,
los sustituyentes en cada uno del grupo alquenilo C2-C6 opcionalmente sustituido, el grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo cicloalquenilo C3-C7 opcionalmente sustituido, el grupo arilo de 6 a 10 miembros opcionalmente sustituido, el grupo heteroarilo opcionalmente sustituido, el grupo heterocíclico alifático opcionalmente sustituido y el grupo alcoxi C1-C6 opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático de 4 a 8 miembros que contiene de uno a tres heteroátomos seleccionados independientemente del grupo que consiste en átomos de oxígeno, azufre y nitrógeno opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo de 5 a 10 miembros que contiene de uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en átomos de azufre, oxígeno y nitrógeno opcionalmente sustituido con un grupo hidroxi o un grupo oxo; un grupo heterocíclico alifático monocíclico de 4 a 7 miembros opcionalmente sustituido con uno o dos grupos oxo y que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6,
la porción heterocíclica alifática del grupo carbonilo heterocíclico alifático con el cual R7 es sustituido es un anillo heterocíclico alifático monocíclico de 4 a 7 miembros que contenía por lo menos un átomo de nitrógeno y opcionalmente que contenía además un heteroátomo seleccionado del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno, el grupo heteroarilo con el cual R7 es sustituido es un grupo heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno,
la porción heteroarilo del grupo heteroarilo opcionalmente sustituido representado por R7 es un heteroarilo monocíclico de 5 o 6 miembros que contenía uno a cuatro heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno y
la porción heterocíclica alifática del grupo heterocíclico alifático opcionalmente sustituido representado por R7 es un anillo heterocíclico alifático monocíclico o bicíclico de 4 a 8 miembros que contenía uno o dos heteroátomos seleccionados independientemente del grupo que consiste en un átomo de oxígeno, un átomo de azufre y un átomo de nitrógeno y
R8 y R9 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
3. El compuesto o sal farmacéuticamente aceptable del mismo de conformidad con cualquiera de las reivindicaciones 1 a 2, donde
el anillo A es un grupo fenilo opcionalmente sustituido
donde los sustituyentes en el grupo fenilo opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno un grupo cicloalquilo C3-C7, un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6,
R1 es un grupo alquilo C1-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo adamantilo opcionalmente sustituido, un grupo heterocíclico alifático opcionalmente sustituido, un grupo arilo opcionalmente sustituido que puede ser parcialmente hidrogenado, un grupo heteroarilo opcionalmente sustituido o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6,
donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático y un grupo alquilenoxi C1-C6,
los sustituyentes en cada uno del grupo cicloalquilo C3-C7 opcionalmente sustituido, el grupo heterocíclico alifático opcionalmente sustituido, el grupo arilo opcionalmente sustituido que puede ser parcialmente hidrogenado y el grupo heteroarilo opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6; un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno; un grupo cicloalquilo C3-C7 ; un grupo alcoxi C1-C6; un grupo hidroxialquilo C1-C6; un grupo alcoxialquilo C1-C6; un grupo alcanoílo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo heterocíclico alifático; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alcoxialquilo C1-C6; un grupo alquilsulfonilo C1-C6; un grupo sulfonilo heterocíclico alifático y un grupo alquilenoxi C1-C6,
el grupo heterocíclico alifático con el cual R1 es sustituido es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo y un grupo homomorfolinilo,
la porción heterocíclica alifática de cada uno del grupo carbonilo heterocíclico alifático y el grupo sulfonilo heterocíclico alifático con el cual R1 es sustituido es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo homopiperazinilo y un grupo homomorfolinilo,
la porción arilo del grupo arilo opcionalmente sustituido que puede ser parcialmente hidrogenado representado por R1 es un grupo seleccionado del grupo que consiste en un grupo fenilo, un grupo naftilo, un grupo dihidrofenilo, un grupo indanilo y un grupo tetrahidronaftilo,
la porción heterocíclica alifática del grupo heterocíclico alifático opcionalmente sustituido representado por R1 es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo y un grupo homomorfolinilo y
la porción heteroarilo del grupo heteroarilo opcionalmente sustituido representado por R es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo; R2 es un átomo de halógeno, un grupo alquilo C1-C6 o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II],
donde el anillo B es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiridinilo, un grupo homopiperazinilo, un grupo homomorfolinilo, un grupo 3-azabiciclo[3.1.0]hexilo y un grupo octahidropirrolo[3,4-c]pirrolilo;
el anillo C es un grupo seleccionado del grupo que consiste en un grupo fenilo, un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo;
R5 y R6 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
R7 es un grupo alquilo C1-C6 opcionalmente sustituido, un grupo alquenilo C2-C6 opcionalmente sustituido, un grupo cicloalquilo C3-C7 opcionalmente sustituido, un grupo cicloalquenilo C3-C7 opcionalmente sustituido, un grupo arilo de 6 a 10 miembros opcionalmente sustituido, un grupo heteroarilo opcionalmente sustituido, un grupo heterocíclico alifático opcionalmente sustituido, un grupo alcoxi C1-C6 opcionalmente sustituido, un grupo amino opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo o un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo,
donde los sustituyentes en el grupo alquilo C1-C6 opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo opcionalmente sustituido con un grupo hidroxi o un grupo oxo; un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos oxo; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6,
los sustituyentes en cada uno del grupo alquenilo C2-C6 opcionalmente sustituido, el grupo cicloalquilo C3-C7 opcionalmente sustituido, el cicloalquenilo C3-C7 opcionalmente sustituido, el grupo arilo de 6 a 10 miembros opcionalmente sustituido, el grupo heteroarilo opcionalmente sustituido, el grupo heterocíclico alifático opcionalmente sustituido y el grupo alcoxi C1-C6 opcionalmente sustituido son uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con un grupo carboxilo; un grupo heteroarilo opcionalmente sustituido con un grupo hidroxi o un grupo oxo; un grupo heterocíclico opcionalmente sustituido con uno o dos grupos oxo; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilo C1-C6; un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6; un grupo alquilsulfonilaminocarbonilo C1-C6 y un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6,
la porción heterocíclica alifática del grupo carbonilo heterocíclico alifático con la cual R7 es sustituida es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo homopiperazinilo y un grupo homomorfolinilo,
el grupo heteroarilo con el cual R7 se sustituye es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo,
el grupo heterocíclico alifático con el cual R7 se sustituye es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo y un grupo homomorfolinilo,
la porción heteroarilo del grupo heteroarilo opcionalmente sustituido representada por R7 es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo triazinilo y un grupo triazinilo y la porción heterocíclica alifática del grupo heterocíclico alifático opcionalmente sustituido representada por R7 es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo, un grupo homomorfolinilo, un grupo 3-azabiciclo[3,1,0]hexilo y un grupo octahidropirrolo[3,4-c]pirrolilo y
R8 y R9 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno,
un átomo de halógeno un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
4. El compuesto o la sal farmacéuticamente aceptable del mismo de acuerdo con una cualquiera de las reivindicaciones 1 a 3, donde
el anillo A es un grupo fenilo sustituido opcionalmente con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 opcionalmente sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7 , un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6;
R1 es
(1) un grupo alquilo C1-C6 opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo cicloalquilo C3-C7; un grupo alcoxi C1-C6; un grupo heterocíclico alifático; un grupo carbonilo heterocíclico alifático opcionalmente sustituido don uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un guro alcoxialquilo C1-C6; un grupo sulfonilo heterocíclico alifático y un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo y un grupo homomorfolinilo y
la porción heterocíclica alifática de cada uno del grupo carbonilo heterocíclico alifático y el grupo sulfonilo heterocíclico alifático es un grupo seleccionado entre el grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo homopiperazinilo y un grupo homomorfolinilo), (2) un grupo cicloalquilo C3-C7 monocíclico opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6, un grupo alcoxialquilo C1-C6 y un grupo alquilenoxi C1-C6, (3) un grupo adamantilo opcionalmente sustituido con un grupo hidroxi,
(4) un grupo heterocíclico alifático opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo alquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo y un grupo homomorfolinilo),
(5) un grupo seleccionado del grupo que consiste en un grupo fenilo, un grupo naftilo, un grupo dihidrofenilo, un grupo indanilo y un grupo tetrahidronaftilo,
(6) un grupo heteroarilo que es opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6 y un grupo carbamoílo
(donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo),
(7) un grupo carbamoílo o
(8) un grupo mono-alquilcarbamoílo C1-C6 ;
R2 es un átomo de halógeno, un grupo alquilo C1-C6 o un grupo alcoxi C1-C6;
R3 y R4 están terminalmente unidos entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la formula [II],
donde el anillo B es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiridinilo, un grupo homopiperazinilo, un grupo homomorfolinilo, un grupo 3-azabiciclo[3.10]hexilo y un grupo octahidropirrolo[3,4-c]pirrolilo y ambos R5 y R6 representan átomos de hidrógeno o
el anillo B es un grupo piperidinilo y R5 y R 6 es cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, un grupo ciano y un grupo alcoxialquilo C1-C6 o
el anillo B es un grupo pirrolidinilo y R5 y R6 es cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
el anillo C es un grupo seleccionado entre el grupo que consiste en un grupo fenilo, un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo
R7 es
(1) un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo,
(2) un grupo alquenilo C2-C6 opcionalmente sustituido con un grupo carboxilo,
(3) un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo, (4) un grupo cicloalquenilo de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo,
(5) un grupo fenilo opcionalmente sustituido con un grupo carboxilo,
(6) un grupo heteroarilos que es opcionalmente sustituido con un grupo carboxilo o un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo (donde el grupo heteroarilos es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo a piridazinilo un grupo tiazinilo y un grupo triazinilo), (7) un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6, un grupo alcanoílo C1-C6, un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 está opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carbonilo) y un grupo hidroxi; un grupo alquilsulfonilaminocarbonilo C1-C6; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con un grupo carboxilo (donde el anillo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo homopiperazinilo y un grupo homomorfolinilo); un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo; un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos oxo (donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo y un grupo homomorfolinilo); un grupo alquilsulfonilo C1-C6; un grupo heteroarilo (donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo y un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo tetrahidrofuranilo, un grupo imidazolinilo, un grupo tiazolidinilo, un grupo isotiazolidinilo, un grupo piperidinilo, un grupo piperazinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo tetrahidropiranilo, un grupo homopiperazinilo, un grupo homomorfolinilo, un grupo 3-azabiciclo[3.1.0]hexilo y un grupo octahidropirrolo[3,4-c])pirrolilo),
(8) un grupo alcoxi C1-C6 opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo ciano; un grupo carboxilo; un grupo heteroarilo opcionalmente sustituido con un grupo hidroxi o un grupo oxo (donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo pirrolilo, un grupo furanilo, un grupo tienilo, un grupo imidazolilo, un grupo pirazolilo, un grupo oxazolilo, un grupo isoxazolilo, un grupo tiazolilo, un grupo tetrazolilo, un grupo oxadiazolilo, un grupo piridinilo, un grupo pirazinilo, un grupo pirimidinilo, un grupo piridazinilo, un grupo tiazinilo y un grupo triazinilo); un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo C1-C6 alquilsulfonilaminocarbonilo, (9) un grupo amino el cual es opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo o
(10) un grupo carbamoílo y
R8 y R9 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
5. El compuesto o sal farmacéuticamente aceptable del mismo de conformidad con cualquiera de las reivindicaciones 1 a 4, donde
el anillo A es un grupo fenilo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7 , un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6, R1 es
(1) un grupo alquilo C1-C6 opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno; un grupo hidroxi; un grupo cicloalquilo monocíclico de 3 a 7 miembros; un grupo alcoxi C1-C6; un grupo tetrahidropiranilo; un grupo carbonilo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de
halógeno y un grupo alcoxialquilo C1-C6 (donde el anillo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo pirrolidinilo, un grupo piperidinilo y un grupo morfolinilo); un grupo pirrolidinilsulfonilo y un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6,
(2) un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6, un grupo alcoxialquilo C1-C6 y un grupo alquilenoxi C1-C6,
(3) un grupo adamantilo opcionalmente sustituido con un grupo hidroxi,
(4) un grupo heterocíclico alifático opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo alquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo tetrahidrofuranilo, un grupo tetrahidropiranilo y un grupo piperidinilo),
(5) un grupo indanilo,
(6) un grupo heteroarilo el cual es opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6 y un grupo carbamoílo
(donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo piridazinilo, un grupo piridinilo y un grupo pirimidinilo),
(7) un grupo carbamoílo o
(8) un grupo mono-alquilcarbamoílo;
R12 es un átomo de halógeno, un grupo alquilo de C1-3 o un grupo alcoxi C1-C6;
R3 y R4 están terminalmente unidos entre sí y, junto con el átomo de nitrógeno al que están unidos, forman un grupo representado por la formula [II],
donde el anillo B es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo tetrahidropiridinilo, un grupo piperazinilo, un grupo homopiperazinilo y un grupo octahidropirrolo[3,4-c]pirrolilo y R5 y R6 representan átomos de hidrógeno o
el anillo B es un grupo piperidinilo y R5 y R6 son cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, un grupo ciano y un grupo alcoxialquilo C1-C6 o
el anillo B es un grupo pirrolidinilo y R5 y R6 son cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
el anillo C es un grupo fenilo o un grupo piridinilo;
R78es
(1) un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo,
(2) un grupo alquenilo C1-C6 opcionalmente sustituido con un grupo carboxilo,
(3) un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo, (4) un grupo cicloalquenilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo, (5) un grupo fenilo opcionalmente sustituido con un grupo carboxilo,
(6) un grupo heteroarilo el cual es opcionalmente sustituido con un grupo carboxilo o un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo
(donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo oxazolilo y un grupo pirazolilo),
(7) un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilaminocarbonilo C1-C6; un grupo pirrolidinilcarbonilo opcionalmente sustituido con un grupo carboxilo; un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6; un grupo pirrolidinilo y un grupo isotiazolidinilo opcionalmente sustituido con uno o dos grupos oxo; un grupo alquilsulfonilo C1-C6; un grupo tetrazolilo y un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo piperidinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo piperazinilo y un grupo 3-azabiciclo[3.1.0]hexilo),
(8) un grupo alcoxi C1-C6 opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo ciano; un grupo carboxilo; un grupo heteroarilo opcionalmente sustituido con un grupo hidroxi o un grupo oxo (donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo isoxazolilo, un grupo oxadiazolilo y un grupo tetrazolilo); un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alquilsulfonilaminocarbonilo C1-C6,
(9) un grupo amino el cual es opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo o
(10) un grupo carbamoílo y
R8 y R9 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
6. El compuesto o sal farmacéuticamente aceptable del mismo de conformidad con cualquiera de las reivindicaciones 1 a 5, donde R1 es
(1) un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6, un grupo alcoxialquilo C1-C6 y un grupo alquilenoxi C1-C6;
(2) un grupo adamantilo opcionalmente sustituido con un grupo hidroxi o
(3) un grupo heterocíclico alifático opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo alquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo tetrahidrofuranilo, un grupo tetrahidropiranilo y un grupo piperidinilo).
7. El compuesto o sal farmacéuticamente aceptable del mismo de conformidad con cualquiera de las reivindicaciones 1 a 6, donde
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II]:

donde el anillo B es un grupo pirrolidinilo.
8. El compuesto o sal farmacéuticamente aceptable del mismo de conformidad con cualquiera de las reivindicaciones 1 a 7, donde
R1 es un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6, un grupo alcoxialquilo C1-C6 y un grupo alquilenoxi C1-C6.
9. El compuesto o sal farmacéuticamente aceptable del mismo de conformidad con una cualquiera de las reivindicaciones 1 a 8, donde
el anillo A es un grupo fenilo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7 , un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6;
R1 es un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6, un grupo alcoxialquilo C1-C6 y un grupo alquilenoxi C1-C6;
R2 es un átomo de halógeno o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo
representado por la fórmula [II],
donde el anillo B es un grupo pirrolidinilo y R5 y R6 son cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi C1-C6;
el anillo C es un grupo fenilo;
R7 es
(1) un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo,
(2) un grupo alquenilo C1-C6 opcionalmente sustituido con un grupo carboxilo,
(3) un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo, (4) un grupo cicloalquenilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo carboxilo, (5) un grupo fenilo opcionalmente sustituido con un grupo carboxilo,
(6) un grupo heteroarilo el cual es opcionalmente sustituido con un grupo carboxilo o un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo
(donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo oxazolilo y un grupo pirazolilo),
(7) un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilaminocarbonilo C1-C6; un grupo pirrolidinilcarbonilo opcionalmente sustituido con un grupo carboxilo; un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6; un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos oxo (donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo isotiazolidinilo y un grupo pirrolidinilo); un grupo alquilsulfonilo C1-C6; un grupo tetrazolilo y un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo piperidinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo piperazinilo y un grupo 3-azabiciclo[3,1,0]hexilo),
(8) un grupo alcoxi C1-C6 opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo ciano; un grupo carboxilo; un grupo heteroarilo opcionalmente sustituido con un grupo hidroxi o un grupo oxo (donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo isoxazolilo, un grupo oxadiazolilo y un grupo tetrazolilo); un grupo aminosulfonilaminocarbonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alquilsulfonilaminocarbonilo C1-C6,
(9) un grupo amino el cual es opcionalmente sustituido con uno o dos grupos alquilo C1-C6 opcionalmente sustituidos con un grupo carboxilo o
(10) un grupo carbamoílo y
R8 y R9 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
10. El compuesto o sal farmacéuticamente aceptable del mismo de conformidad con la reivindicación 9, donde el anillo A es un grupo fenilo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cicloalquilo C3-C7 , un grupo alcoxi C1-C6, un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo alquilenoxi C1-C6;
R1 es un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un átomo de halógeno, un grupo hidroxi, un grupo oxo, un grupo ciano, un grupo alquilo C1-C6, un grupo alcoxi C1-C6, un grupo alcoxialquilo C1-C6 y un grupo alquilenoxi C1-C6;
R2 es un átomo de halógeno o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II],
donde el anillo B es un grupo pirrolidinilo y
R5 y R6 son cada uno un grupo seleccionado independientemente del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno, un grupo cianoalquilo C1-C6, un grupo hidroxialquilo C1-C6, un grupo alcoxialquilo C1-C6, un grupo carboxilo, un grupo carbamoílo opcionalmente sustituido con uno o dos grupos alquilo C1-C6 y un grupo alcoxi; C1-C6
el anillo C es un grupo fenilo;
R7 es un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo hidroxi; un grupo oxo; un grupo ciano; un grupo alquilo C1-C6 opcionalmente sustituido con un grupo carboxilo; un grupo alcoxi C1-C6; un grupo alcanoílo C1-C6; un grupo carboxilo; un grupo alcoxicarbonilo C1-C6; un grupo carbamoílo opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6 (el grupo alquilo C1-C6 es opcionalmente sustituido con un grupo hidroxi, un grupo alcoxi C1-C6 o un grupo carboxilo) y un grupo hidroxi; un grupo alquilsulfonilaminocarbonilo C1-C6; un grupo pirrolidinilcarbonilo opcionalmente sustituido con un grupo carboxilo; un grupo amino opcionalmente sustituido con uno o dos grupos seleccionados independientemente del grupo que consiste en un grupo alquilo C1-C6, un grupo alcanoílo C1-C6 y un grupo alquilsulfonilo C1-C6; un grupo heterocíclico alifático opcionalmente sustituido con uno o dos grupos oxo (donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo pirrolidinilo y un grupo isotiazolidinilo); un grupo alquilsulfonilo C1-C6; un grupo tetrazolilo y un grupo aminosulfonilo opcionalmente sustituido con uno o dos grupos alquilo C1-C6
(donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo piperidinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo piperazinilo y un grupo 3-azabiciclo[3.1.0]hexilo) y
R8 y R9 son cada uno independientemente un grupo seleccionado del grupo que consiste en un átomo de hidrógeno, un átomo de halógeno, un grupo ciano, un grupo alquilo C1-C6, un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y un grupo haloalcoxi C1-C6 sustituido con de uno a tres átomos de halógeno.
11. El compuesto o sal farmacéuticamente aceptable del mismo de conformidad con cualquiera de las reivindicaciones 1 a 6, donde
el anillo A es un grupo fenilo opcionalmente sustituido con un grupo alcoxi C1-C6;
R1 es un grupo alquilo C1-C6; un grupo tetrahidrofuranilo, un grupo tetrahidropiranilo y un grupo piperidinilo; un grupo cicloalquilo monocíclico de 3 a 7 miembros opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo alcoxi C1-C6 y un grupo ciano; o un grupo heteroarilo opcionalmente sustituido con un grupo alquilo C1-C6 (donde el grupo heteroarilo es un grupo seleccionado del grupo que consiste en un grupo piridazinilo, un grupo piridinilo y un grupo pirimidinilo);
R2 es un átomo de halógeno o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II]:

donde el anillo B se selecciona del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo piperidinilo, un grupo tetrahidropiridinilo y un grupo piperazinilo y R5 y R6 son átomos de hidrógeno
el anillo B es un grupo pirrolidinilo, R5 es un grupo alcoxialquilo C1-C6 y R6 es un átomo de hidrógeno o un átomo de halógeno;
el anillo C es un grupo fenilo;
R7 es un grupo heterocíclico alifático sustituido con un grupo carboxilo (donde el grupo heterocíclico alifático es un grupo seleccionado del grupo que consiste en un grupo azetidinilo, un grupo pirrolidinilo, un grupo piperidinilo, un grupo morfolinilo, un grupo tiomorfolinilo, un grupo piperazinilo y un grupo 3- ,azabiciclo[3.1.0]hexilo);
R8 es un átomo de halógeno o un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y R9 es un átomo de hidrógeno.
12. El compuesto o sal farmacéuticamente aceptable del mismo de conformidad con la reivindicación 11, donde el anillo A es un grupo fenilo opcionalmente sustituido con un grupo alcoxi C1-C6;
R1 es un grupo tetrahidropiranilo o un grupo cicloalquilo monocíclico de 5 o 6 miembros opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo alcoxi C1-C6 y un grupo ciano;
R2 es un átomo de halógeno o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo
representado por la fórmula
donde el anillo B es un grupo pirrolidinilo, R5 es un grupo alcoxialquilo C1-C6 y R6 es un átomo de hidrógeno o un átomo de halógeno;
el anillo C es un grupo fenilo;
R7 es un grupo piperidinilo sustituido con un grupo carboxilo;
R8 es un átomo de halógeno o un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y R9 es un átomo de hidrógeno.
13. El compuesto o sal farmacéuticamente aceptable del mismo de conformidad con la reivindicación 12, donde el anillo A es un grupo fenilo opcionalmente sustituido con un grupo alcoxi C1-C6;
R1 es un grupo cicloalquilo monocíclico de 5 o 6 miembros opcionalmente sustituido con un grupo seleccionado del grupo que consiste en un grupo alcoxi C1-C6 y un grupo ciano;
R2 es un átomo de halógeno o un grupo alcoxi C1-C6;
R3 y R4 están unidos terminalmente entre sí y, junto con el átomo de nitrógeno al cual están unidos, forman un grupo representado por la fórmula [II]:

donde el anillo B es un grupo pirrolidinilo, R5 es un grupo alcoxialquilo C1-C6 y R6 es un átomo de hidrógeno o un átomo de halógeno;
el anillo C es un grupo fenilo;
R7 es un grupo piperidinilo sustituido con un grupo carboxilo;
R8 es un átomo de halógeno o un grupo haloalquilo C1-C6 sustituido con de uno a tres átomos de halógeno y R9 es un átomo de hidrógeno.
14. Un compuesto de acuerdo con la reivindicación 1 seleccionado del grupo que consiste en:
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-etoxiciclohexil)-3-metoxi-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-5-(etoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico;
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-4-(4-metoxifenil)-1-(2-metilpiridin-4-il)pirrolidin-3-il]carbonil}-5-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico;
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-4-(4-metoxifenil)-1-(2-metilpirimidin-4-il)pirrolidin-3-il]carbonil}-5-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico;
ácido 1-{5-fluoro-2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metoxiciclohexil)-4-(4-metoxifenil)pirrolidin-3il]carbonil}-5-(metoximetil)pirrolidin-3-il]fenil}piperidin-4-carboxílico;
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-etox¡ddohex¡l)-3-metox¡-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-danoddohex¡l)-3-fluoro-4-(4-metox¡feml)p¡rrol¡dm-3-¡l]carboml}-5-(metox¡met¡l)p¡n'ol¡dm-3-¡l]-5-(tnfluoromet¡l)feml}p¡peridm-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4S)-1-{[(3R,4R)-1-(frans-4-etox¡ddohex¡l)-3-metox¡-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carboml}-4-fluoro-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-1-(frans-4-etox¡ddohex¡l)-3-metox¡-4-(4-metox¡fen¡l)p¡n'ol¡d¡n-3-¡l]carboml}-4-(metox¡met¡l)p¡n'ol¡dm-3-¡l]-5-(trifluoromet¡l)feml}p¡pend¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-ddohex¡l-3-fluoro-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carboml}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-4-(4-metox¡fen¡l)-1-(tetrah¡dro-2H-p¡ran-4-¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carboml}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-ddopent¡l-3-fluoro-4-(4-metox¡feml)p¡n'ol¡d¡n-3-¡l]carboml}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-4-(4-metox¡fen¡l)-1-(2-met¡lp¡r¡d¡n-4-¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4S)-4-fluoro-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-metox¡feml)p¡n'ol¡dm-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{5-doro-2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-metox¡feml)p¡n'ol¡dm-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-4-(danomet¡l)-1-{[(3R,4R)-1-ddopent¡l-3-fluoro-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carbon¡l}p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-metox¡feml)p¡rrol¡dm-3-¡l]carboml}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-[2-(1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-metox¡feml)p¡n'ol¡dm-3-¡l]carboml}p¡pend¡n-4-¡l)-5-(tr¡fluoromet¡l)fen¡l]p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-1-c¡dopent¡l-3-fluoro-4-(4-fen¡lmetox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co
y ác¡do 1-{2-[(3S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-fen¡lmetox¡feml)p¡n'ol¡d¡n-3-¡l]carbon¡l}p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co
o una sal farmacéut¡camente aceptable del mismo.
15. Un compuesto de acuerdo con la re¡v¡nd¡cac¡ón 14, selecc¡onado del grupo que cons¡ste en:
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-etox¡ddohex¡l)-3-metox¡-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carboml}-5-(etox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{5-fluoro-2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡c¡dohex¡l)-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-etox¡c¡dohex¡l)-3-metox¡-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-c¡anoc¡dohex¡l)-3-fluoro-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4S)-1-{[(3R,4R)-1-(frans-4-etox¡ddohex¡l)-3-metox¡-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-fluoro-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-1-(frans-4-etox¡c¡dohex¡l)-3-metox¡-4-(4-metox¡fen¡l)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-ddohex¡l-3-fluoro-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carboml}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carboml}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,5S)-1-{[(3R,4R)-1-dclopent¡l-3-fluoro-4-(4-metox¡feml)p¡rrol¡dm-3-¡l]carboml}-5-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4S)-4-fluoro-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{5-doro-2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carbon¡l}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-4-(danomet¡l)-1-{[(3R,4R)-1-ddopent¡l-3-fluoro-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carbon¡l}p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-{2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-metox¡feml)p¡n'ol¡dm-3-¡l]carboml}-4-(metox¡met¡l)p¡rrol¡d¡n-3-¡l]-5-(tr¡fluoromet¡l)fen¡l}p¡per¡d¡n-4-carboxíl¡co;
ác¡do 1-[2-(1-{[(3R,4R)-3-fluoro-1-(frans-4-metox¡ddohex¡l)-4-(4-metox¡feml)p¡rrol¡d¡n-3-¡l]carboml}p¡pend¡n-4-¡l)-5-(tr¡fluoromet¡l)fen¡l]p¡per¡d¡n-4-carboxíl¡co;
ácido 1-{2-[(3S,4R)-1-{[(3R,4R)-1-ciclopentil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico y
ácido 1-{2-[(3S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metoxiciclohexil)-4-(4-metoxifenil)pirrolidin-3-il]carbonil}pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico
o una sal farmacéuticamente aceptable del mismo.
16. Un compuesto de acuerdo con la reivindicación 15 seleccionado del grupo que consiste en:
ácido 1-{5-fluoro-2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metoxiciclohexil)-4-(4-metoxifenil)pirrolidin-3-il]carboill}-5-(metoximetil)pirrolidin-3-il]fenil}piperidin-4-carboxílico;
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-1-(frans-4-etoxiciclohexil)-3-metoxi-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-5-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico;
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metoxiciclohexil)-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-5-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico;
ácido 1-{2-[(3S,5S)-1-{[(3R,4R)-1-ciclopentil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-5-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico;
ácido 1-{2-[(3S,4S)-4-fluoro-1-{[(3R,4R)-3-fluoro-1-(frans-4-metoxiciclohexil)-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico;
ácido 1-{5-cloro-2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metoxiciclohexil)-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4-(metoximetil)pirrolidin-3-il]fenil}piperidin-4-carboxílico;
ácido 1-{2-[(3S,4R)-1-{[(3R,4R)-3-fluoro-1-(frans-4-metoxiciclohexil)-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico y
ácido 1-{2-[(3S,4R)-1-{[(3R,4R)-1-ciclopentil-3-fluoro-4-(4-metoxifenil)pirrolidin-3-il]carbonil}-4-(metoximetil)pirrolidin-3-il]-5-(trifluorometil)fenil}piperidin-4-carboxílico
o una sal farmacéuticamente aceptable del mismo.
17. Una composición farmacéutica que comprende, como un ingrediente activo, el compuesto de cualquiera de las reivindicaciones 1 a 16 o una sal farmacéuticamente aceptable del mismo.
18. La composición farmacéutica de conformidad con la reivindicación 17 para su uso en la prevención o terapia de artritis reumatoide, artritis gotosa, osteoartrosis, enfermedad inflamatoria del intestino, esclerosis sistémica, psoriasis, fibrosis, protoporfiria, lupus eritematoso sistémico, melanoma, cáncer de piel, vitiligo, pérdida del cabello, dolor, daño por isquemia/reperfusión, enfermedad inflamatoria cerebral, hepatitis, choque séptico/septicemia, nefritis, trasplante, exacerbación de la enfermedad por el VIH, vasculitis, uveítis, retinitis pigmentosa, degeneración macular relacionada con la edad, infección microbiana, enfermedad celíaca, síndrome nefrótico o invasión por melanoma.
19. La composición farmacéutica para su uso de conformidad con la reivindicación 18, para la prevención o el tratamiento de esclerosis sistémica, psoriasis, protoporfiria, melanoma, cáncer de piel, vitiligo, pérdida del cabello, síndrome nefrótico, retinitis pigmentosa o degeneración macular relacionada con la edad.
20. La composición farmacéutica para su uso de conformidad con la reivindicación 19, para la prevención o el tratamiento de esclerosis sistémica, protoporfiria, melanoma, vitiligo, síndrome nefrótico, retinitis pigmentosa o degeneración macular relacionada con la edad.
21. La composición farmacéutica para su uso de conformidad con la reivindicación 20, caracterizada además porque la protoporfiria es protoporfiria eritropoyética.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014111378 | 2014-05-29 | ||
| PCT/JP2015/065469 WO2015182723A1 (ja) | 2014-05-29 | 2015-05-28 | 新規ピロリジン化合物およびメラノコルチン受容体作動薬としての用途 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2841308T3 true ES2841308T3 (es) | 2021-07-08 |
Family
ID=54699046
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15799731T Active ES2841308T3 (es) | 2014-05-29 | 2015-05-28 | Compuesto de pirrolidina novedoso y aplicación como agonista del receptor de melanocortina |
Country Status (19)
| Country | Link |
|---|---|
| US (1) | US9981960B2 (es) |
| EP (1) | EP3150578B1 (es) |
| JP (1) | JP6377736B2 (es) |
| KR (1) | KR101919680B1 (es) |
| CN (2) | CN112194649A (es) |
| AU (1) | AU2015268505C1 (es) |
| CA (1) | CA2950072C (es) |
| DK (1) | DK3150578T3 (es) |
| ES (1) | ES2841308T3 (es) |
| HU (1) | HUE052714T2 (es) |
| MX (1) | MX2016015641A (es) |
| MY (1) | MY193597A (es) |
| PH (1) | PH12016502358B1 (es) |
| PL (1) | PL3150578T3 (es) |
| PT (1) | PT3150578T (es) |
| RU (1) | RU2669938C2 (es) |
| SG (1) | SG11201609864UA (es) |
| TW (1) | TWI669301B (es) |
| WO (1) | WO2015182723A1 (es) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3333165B1 (en) | 2015-08-04 | 2019-09-18 | Astellas Pharma Inc. | Piperazine derivative |
| EP3381913B1 (en) * | 2015-11-27 | 2021-03-31 | Mitsubishi Tanabe Pharma Corporation | 2-(((pyrrolidin-3-yl)carbonyl)amino)-1h-imidazole derivatives as melanocortin receptor 1 (mcr1) agonists for treating rheumatoid arthritis |
| CN116854624A (zh) | 2017-01-31 | 2023-10-10 | 田边三菱制药株式会社 | 光学活性吡咯烷化合物的制造方法 |
| CN111094270B (zh) * | 2017-09-29 | 2023-09-22 | 田边三菱制药株式会社 | 光学活性吡咯烷化合物及其制造方法 |
| WO2020138481A1 (ja) | 2018-12-28 | 2020-07-02 | 田辺三菱製薬株式会社 | ピロリジン化合物の結晶 |
| ES2970510T3 (es) * | 2019-11-07 | 2024-05-29 | Lg Chemical Ltd | Agonistas del receptor de melanocortina-4 |
| KR20230016008A (ko) | 2020-06-10 | 2023-01-31 | 미쓰비시 타나베 파마 코퍼레이션 | 광선성 피부질환의 예방 또는 치료제 |
| US20230248713A1 (en) * | 2020-06-10 | 2023-08-10 | Mitsubishi Tanabe Pharma Corporation | Prophylactic or therapeutic agent for porphyria |
| TW202214249A (zh) | 2020-06-10 | 2022-04-16 | 日商田邊三菱製藥股份有限公司 | 紫質症之預防或治療劑 |
| JP2023540585A (ja) * | 2020-09-09 | 2023-09-25 | クリヌヴェル (ユーケー) リミテッド | 色素性乾皮症を治療するためのピロリジン化合物 |
| KR102394875B1 (ko) * | 2021-05-11 | 2022-05-06 | 주식회사 비드테크 | 피롤리딘을 링커로 포함하는 화합물 및 이를 포함하는 약학 조성물 |
| AU2023213357A1 (en) | 2022-01-31 | 2024-08-15 | Mitsubishi Tanabe Pharma Corporation | Novel use of melanocortin-1 receptor agonist |
| JP7527582B2 (ja) | 2022-08-03 | 2024-08-05 | 田辺三菱製薬株式会社 | 1-{2-[(3s,4r)-1-{[(3r,4r)-1-シクロペンチル-3-フルオロ-4-(4-メトキシフェニル)ピロリジン-3-イル]カルボニル}-4-(メトキシメチル)ピロリジン-3-イル]-5-(トリフルオロメチル)フェニル}ピペリジン-4-カルボン酸もしくはその医薬的に許容し得る塩又は共結晶を含有する医薬組成物 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6376489B1 (en) * | 1999-12-23 | 2002-04-23 | Icos Corporation | Cyclic AMP-specific phosphodiesterase inhibitors |
| US6294561B1 (en) * | 1999-12-23 | 2001-09-25 | Icos Corporation | Cyclic AMP-specific phosphodiesterase inhibitors |
| CA2395196C (en) | 1999-12-23 | 2006-02-21 | Icos Corporation | Cyclic amp-specific phosphodiesterase inhibitors |
| US6258833B1 (en) | 1999-12-23 | 2001-07-10 | Icos Corporation | Cyclic AMP-specific phosphodiesterase inhibitors |
| KR20030076716A (ko) | 2001-02-28 | 2003-09-26 | 머크 앤드 캄파니 인코포레이티드 | 멜라노코르틴-4 수용체 효능제로서 아실화 피페리딘 유도체 |
| AR043434A1 (es) | 2003-03-03 | 2005-07-27 | Merck & Co Inc | Derivados de piperizacina acilados como agonistas del receptor de melanocortina-4. composiciones farmaceuticas y usos |
| US20050192286A1 (en) | 2003-10-22 | 2005-09-01 | Neurocrine Biosciences, Inc. | Ligands of melanocortin receptors and compositions and methods related thereto |
| GB0402492D0 (en) * | 2004-02-04 | 2004-03-10 | Pfizer Ltd | Pharmaceutically active compounds |
| TWI332501B (en) | 2006-07-14 | 2010-11-01 | Lg Life Sciences Ltd | Melanocortin receptor agonists |
| KR20080007046A (ko) * | 2006-07-14 | 2008-01-17 | 주식회사 엘지생명과학 | 멜라노코틴 수용체의 항진제 |
| US20090253744A1 (en) * | 2006-09-27 | 2009-10-08 | Bakshi Raman K | Acylated piperidine derivatives as melanocortin-4 receptor modulators |
| AR073578A1 (es) | 2008-09-15 | 2010-11-17 | Priaxon Ag | Pirrolidin-2-onas |
-
2015
- 2015-05-28 CA CA2950072A patent/CA2950072C/en active Active
- 2015-05-28 KR KR1020167036300A patent/KR101919680B1/ko active IP Right Grant
- 2015-05-28 JP JP2016523563A patent/JP6377736B2/ja active Active
- 2015-05-28 HU HUE15799731A patent/HUE052714T2/hu unknown
- 2015-05-28 US US15/313,407 patent/US9981960B2/en active Active
- 2015-05-28 MX MX2016015641A patent/MX2016015641A/es unknown
- 2015-05-28 WO PCT/JP2015/065469 patent/WO2015182723A1/ja active Application Filing
- 2015-05-28 PT PT157997313T patent/PT3150578T/pt unknown
- 2015-05-28 MY MYPI2016704348A patent/MY193597A/en unknown
- 2015-05-28 ES ES15799731T patent/ES2841308T3/es active Active
- 2015-05-28 RU RU2016151381A patent/RU2669938C2/ru active
- 2015-05-28 DK DK15799731.3T patent/DK3150578T3/da active
- 2015-05-28 CN CN202011071876.7A patent/CN112194649A/zh active Pending
- 2015-05-28 SG SG11201609864UA patent/SG11201609864UA/en unknown
- 2015-05-28 TW TW104117133A patent/TWI669301B/zh active
- 2015-05-28 PL PL15799731T patent/PL3150578T3/pl unknown
- 2015-05-28 EP EP15799731.3A patent/EP3150578B1/en active Active
- 2015-05-28 CN CN201580028290.3A patent/CN106458887B/zh active Active
- 2015-05-28 AU AU2015268505A patent/AU2015268505C1/en active Active
-
2016
- 2016-11-25 PH PH12016502358A patent/PH12016502358B1/en unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2841308T3 (es) | Compuesto de pirrolidina novedoso y aplicación como agonista del receptor de melanocortina | |
| JP6411345B2 (ja) | RORγT阻害剤としてのN−アルキル化インドールおよびインダゾール化合物およびそれらの使用 | |
| ES2742651T3 (es) | Compuestos de 3-aminocicloalquilo como inhibidores de ROR-gamma-T y sus usos | |
| ES2311152T3 (es) | Derivados de piperazina y su uso para el tratamiento de enfermedades neurologicas y psiquiatricas. | |
| AU2019210624A1 (en) | Cyclopropylamines as lsd1 inhibitors | |
| ES2334517T3 (es) | Derivados de triazol sustituidos como antagonistas de oxitocina. | |
| KR100843848B1 (ko) | H3 리간드로서 유용한 3- 또는 4-단일치환된 페놀 및싸이오페놀 유도체 | |
| ES2314922T3 (es) | N-arilpirrolidinas sustituidas como moduladores selectivos del receptor de androgenos. | |
| ES2869229T3 (es) | Derivados de 2-(((pirrolidin-3-il)carbonil)amino)-1H-imidazol como agonistas del receptor de melanocortina 1 (MCR1) para tratar la artritis reumatoide | |
| CA2935880A1 (en) | Indazole compounds as irak4 inhibitors | |
| US20150328228A1 (en) | Novel 4-(indol-3-yl)-pyrazole derivatives, pharmaceutical compositions and methods for use | |
| WO2020216190A1 (zh) | 一种喹唑啉化合物及其在医药上的应用 | |
| BRPI0708264A2 (pt) | piperidinilpirrolidinas agonistas do receptor tipo 4 de melanocortina | |
| WO2017021879A1 (en) | Novel compounds as ror gamma modulators | |
| KR20230019496A (ko) | Cyp11a1 (시토크롬 p450 모노옥시게나제 11a1) 억제제로서 피란 유도체 | |
| ES2920359T3 (es) | Amidas de pirrolidinas sustituidas II | |
| JP6392297B2 (ja) | 医薬組成物 | |
| CN108137505A (zh) | 含氮杂环化合物 | |
| CA2999940A1 (en) | Substituted morpholine derivatives having activity against pain | |
| BR112016027352B1 (pt) | Composto de pirrolidina, composição farmacêutica compreendendo o referido composto e uso do mesmo | |
| JP2018199673A (ja) | 医薬組成物 | |
| BR122023020299A2 (pt) | Composto, uso de um composto, composição farmacêutica e processo para preparação de um composto | |
| JP2006282602A (ja) | メラノコルチン−4受容体アゴニスト作用を有する新規ピペリジン誘導体 |