ES2822606T3 - Derivado de amina cíclica y uso farmacéutico del mismo - Google Patents
Derivado de amina cíclica y uso farmacéutico del mismo Download PDFInfo
- Publication number
- ES2822606T3 ES2822606T3 ES17744386T ES17744386T ES2822606T3 ES 2822606 T3 ES2822606 T3 ES 2822606T3 ES 17744386 T ES17744386 T ES 17744386T ES 17744386 T ES17744386 T ES 17744386T ES 2822606 T3 ES2822606 T3 ES 2822606T3
- Authority
- ES
- Spain
- Prior art keywords
- group
- compound
- atoms
- mmol
- atom
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CCC(*(CCC1)C1C(CCC(CC1NC)=CC=C1C(C=CCC1)=C1O)=C)=O Chemical compound CCC(*(CCC1)C1C(CCC(CC1NC)=CC=C1C(C=CCC1)=C1O)=C)=O 0.000 description 2
- QHDXTYZYBWOZIO-UHFFFAOYSA-N CC(C)(C)OC(NCCC(N(CCCC1)C1C(Nc(cc1)cc(Cl)c1-c1ccccc1OC(F)(F)F)O)=O)=O Chemical compound CC(C)(C)OC(NCCC(N(CCCC1)C1C(Nc(cc1)cc(Cl)c1-c1ccccc1OC(F)(F)F)O)=O)=O QHDXTYZYBWOZIO-UHFFFAOYSA-N 0.000 description 1
- AVLUIFVEEQEWOT-UHFFFAOYSA-N CC(C1Cl)C(NC(C(CCCC2)N2C(CN(C)S(C)(=O)=O)=O)=O)=CC=C1Br Chemical compound CC(C1Cl)C(NC(C(CCCC2)N2C(CN(C)S(C)(=O)=O)=O)=O)=CC=C1Br AVLUIFVEEQEWOT-UHFFFAOYSA-N 0.000 description 1
- QYYWXAIBQNUTJP-UHFFFAOYSA-N CC(CC=C1)C(c(ccc(NC(C(CCCC2)N2C(C(F)(F)F)O)=O)c2)c2Cl)=C1OC(F)(F)F Chemical compound CC(CC=C1)C(c(ccc(NC(C(CCCC2)N2C(C(F)(F)F)O)=O)c2)c2Cl)=C1OC(F)(F)F QYYWXAIBQNUTJP-UHFFFAOYSA-N 0.000 description 1
- DIHZBWSTAFQABW-UHFFFAOYSA-N CNCCC(N(CCCC1)C1C(Nc1ccc(-c2ccccc2OC(F)(F)F)c(Cl)c1)=O)=O Chemical compound CNCCC(N(CCCC1)C1C(Nc1ccc(-c2ccccc2OC(F)(F)F)c(Cl)c1)=O)=O DIHZBWSTAFQABW-UHFFFAOYSA-N 0.000 description 1
- IKPHBEBYVCCZJR-BYYHNAKLSA-N COC(CC(N(CCCC1)C1/N=N/c(cc1)cc(Cl)c1-c1ccccc1OC(F)(F)F)O)=O Chemical compound COC(CC(N(CCCC1)C1/N=N/c(cc1)cc(Cl)c1-c1ccccc1OC(F)(F)F)O)=O IKPHBEBYVCCZJR-BYYHNAKLSA-N 0.000 description 1
- RTNNYPIDWCISIH-OCEACIFDSA-N COC(N(CCCC1)C1/N=N/c1ccc(-c2ccccc2OC(F)(F)F)c(Cl)c1)O Chemical compound COC(N(CCCC1)C1/N=N/c1ccc(-c2ccccc2OC(F)(F)F)c(Cl)c1)O RTNNYPIDWCISIH-OCEACIFDSA-N 0.000 description 1
- JBIQUQJZBUVAGE-UHFFFAOYSA-N COCC(N(CCCC1)C1C(Nc1ccc(-c2ccccc2OC(F)(F)F)c(Cl)c1)=O)=O Chemical compound COCC(N(CCCC1)C1C(Nc1ccc(-c2ccccc2OC(F)(F)F)c(Cl)c1)=O)=O JBIQUQJZBUVAGE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/451—Non condensed piperidines, e.g. piperocaine having a carbocyclic group directly attached to the heterocyclic ring, e.g. glutethimide, meperidine, loperamide, phencyclidine, piminodine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4525—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/38—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/92—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with a hetero atom directly attached to the ring nitrogen atom
- C07D211/96—Sulfur atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Immunology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Dermatology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Transplantation (AREA)
- Biomedical Technology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Toys (AREA)
- Portable Nailing Machines And Staplers (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
Un derivado de amina cíclica representado por la siguiente fórmula general (I): **(Ver fórmula)** donde R1 representa un grupo alquiloxi que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno; R2 representa un átomo de halógeno; R3 representa un átomo de hidrógeno, un átomo de halógeno o un grupo hidroxi; R4 representa un átomo de hidrógeno o un átomo de halógeno; X representa -C(=O)-(CH2)n-R5 o -S(=O)2-R6; n es un número entero de 0 a 5; R5 representa un átomo de hidrógeno, -OR7, -SR7, -S(=O)2-R7, -C(=O)-OR7, -N(R7)R8, un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno o un grupo heteroarilo, cualquiera de cuyos átomos de hidrógeno están opcionalmente sustituidos con uno o más grupos alquilo que tienen de 1 a 3 átomos de carbono; R6 representa un grupo alquilo que tiene de 1 a 5 átomos de carbono; R7 representa un átomo de hidrógeno o un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno; y R8 representa un átomo de hidrógeno, un grupo alquilo que tiene de 1 a 3 átomos de carbono, un grupo acilo que tiene de 2 a 4 átomos de carbono, o un grupo alquilsulfonilo que tiene de 1 a 3 átomos de carbono, o una sal farmacéuticamente aceptable de los mismos.
Description
DESCRIPCIÓN
Derivado de amina cíclica y uso farmacéutico del mismo
ANTECEDENTES DE LA INVENCIÓN
Campo de la invención
[0001] La presente invención se refiere a un derivado de amina cíclica y al uso médico del mismo.
ANTECEDENTES DE LA TÉCNICA
[0002] Una enfermedad autoinmunitaria es un término general que designa enfermedades en las que una respuesta inmunitaria excesiva ataca las propias células y tejidos normales de un individuo, produciéndole síntomas. Los ejemplos de las mismas incluyen esclerosis múltiple, psoriasis, artritis reumatoide, lupus eritematoso sistémico, espondilitis anquilosante, uveítis o polimialgia reumática.
[0003] Una enfermedad alérgica es una enfermedad derivada de una respuesta inmunitaria excesiva a antígenos específicos, y los ejemplos incluyen dermatitis alérgica, dermatitis atópica, rinitis alérgica (polinosis), conjuntivitis alérgica, gastroenteritis alérgica, asma bronquial, asma infantil o alergia alimentaria.
[0004] Se han propuesto diversos mecanismos para la aparición y el progreso de las enfermedades autoinmunitarias y alérgicas. Como uno de estos mecanismos, se sabe que las células Th17, que forman parte de un subconjunto de células T auxiliares, y la IL-17, una citocina inflamatoria producida por las células Th17, desempeñan un papel importante en la aparición y el progreso de enfermedades autoinmunitarias y enfermedades alérgicas (Documentos no de patente 1 y 2).
[0005] La IL-17 actúa sobre diversas células, tales como fibroblastos, células epiteliales, células endoteliales vasculares y macrófagos, y participa en la inducción de citocinas inflamatorias, quimiocinas, metaloproteasas y otros mediadores inflamatorios y en la migración de neutrófilos. Por lo tanto, se considera que se demuestran efectos antiinflamatorios potentes si se puede suprimir la producción o función de la IL-17, y se han llevado a cabo estudios clínicos de anticuerpos anti IL-17 con indicaciones para diversas enfermedades autoinmunitarias.
[0006] Recientemente, quedó claro que el receptor y huérfano relacionado con retinoides (en adelante denominado RORy), que es un receptor nuclear, funciona como un factor de transcripción esencial para la diferenciación y proliferación de células Th17 y la expresión de IL-17 (Documento no de patente 3), y se demostró que la supresión de la expresión o función de RORy provoca la supresión de la diferenciación y activación de las células Th17 y la producción de IL-17 (Documento no de patente 4).
[0007] Se ha informado de que el nivel de expresión de RORy en células mononucleares de sangre periférica o tejido cutáneo en pacientes con enfermedades autoinmunitarias (esclerosis múltiple, psoriasis, lupus eritematoso sistémico, etc.) o pacientes con enfermedades alérgicas (dermatitis alérgica, etc.) es mayor que el de los individuos sanos (Documentos no de patente 5 a 7). Se ha informado de que, en un ratón knockout de RORy, se suprime el estado patológico de un modelo experimental de encefalomielitis autoinmunitaria de ratón, que es un modelo animal de esclerosis múltiple, y de que se suprimen los síntomas de enfermedades autoinmunitarias, tales como colitis, y los síntomas de enfermedades alérgicas, tales como asma (Documentos no de patente 3, 8 y 9).
[0008] Además, se sugiere que la unión entre RORy y un coactivador es necesaria para que RORy funcione como un factor de transcripción (Documento no de patente 10). Por lo tanto, se espera que un antagonista de RORy, que es un compuesto que inhibe la unión entre RORy y un coactivador, sea útil como agente terapéutico o agente preventivo para enfermedades autoinmunitarias o enfermedades alérgicas.
[0009] Por otro lado, como antagonista de RORy, se ha informado anteriormente de N-(5-(N-(4-(1,1,1,3,3,3-hexafluoro-2-hidroxipropan-2-il)fenil)sulfamoil)-4-metiltiazol-2-il)acetamida (Documento no de patente 11), derivados azólicos sustituidos (Documento de patente 1), tales como ácido 6-(2-cloro-4-metilfenil)-3-(4-ciclopropil-5-(3-neopentilciclo butil)isoxazol-3-il)-5-oxohexanoico y derivados de sulfonilbenceno (Documento de patente 2), tales como N-(5-(2-clorobenzoil)-4-(3-clorofenil) tiazol-2-il)-2-(4-(etilsulfonil)fenil)acetamida, pero no se han descrito compuestos que tienen una estructura de amina cíclica, tal como 1-piperidina sustituida-2-carboxamida.
[0010] Se ha informado de que el compuesto que tiene una estructura de amina cíclica, tal como 1 -piperidina sustituida-2-carboxamida, se ha reportado (S)-1-(2-(3,3-difluoropirrolidina-1-il)acetil)-N-(1-etil-5-fenil-1 H-1,2,4-triazol-3-il)piperidina-2-carboxamida, etc. es un agonista del receptor cannabinoide 2 (Documento de patente 3), y se ha informado de que (R)-N-(5-bencil-4-feniltiazol-2-il)-1-(2-ciclopenta-lacetil)piperidina-2-carboxamida, etc. es un inhibidor de acil-coenzima A: diacilglicerol aciltransferasa 1 (Documento de patente 4), pero los efectos de estos compuestos en RORy no se han descrito ni sugerido.
[0011] Como agente terapéutico o agente preventivo para la esclerosis múltiple, se ha informado de que (S)-(3-(5-(4-fluorobencil)-1,2,4-oxadiazol-3-il)piperidina-1-il)(4-fluorofenil)metanona, etc. es un modulador alostérico positivo de un receptor de glutamato metabotrópico (Documento de patente 5), pero no se han mostrado datos concretos sobre la eficacia farmacológica de estos compuestos en la esclerosis múltiple, y su utilidad no se ha demostrado en absoluto. En el Documento de patente 5, no se han descrito ni sugerido los efectos de estos compuestos sobre RORy, y no se han descrito los compuestos que tienen una estructura de 1 -piperidina sustituida-2-carboxamida.
Documento de técnica anterior
[Documento de patente]
[0012]
[Documento de patente 1] JP 2012-236822 A
[Documento de patente 2] WO 2012/027965
[Documento de patente 3] WO 2010/096371
[Documento de patente 4] WO 2010/007046
[Documento de patente 5 ] WO 2006/129199
[Documento de patente 6] WO 2015/129853
[Documento no de patente]
[0013]
[Documento no de patente 1] Chen y col., International Immunopharmacology, 2011, Vol. 11, p.536-542
[Documento no de patente 2] Hofmann y col., Current Opinion in Allergy and Clinical Immunology, 2016, vol. 16, p.451-457
[Documento no de patente 3] Ivanov y col., Cell, 2006, Vol. 126, p.1121-1133
[Documento no de patente 4] Jetten, Nuclear Receptor Signaling, 2009, Vol. 7, e003
[Documento no de patente 5 ] Hamzaoui y col., Medical Science Monitor, 2011, Vol. 17, p.CR227-23
[Documento no de patente 6] Ma y col., Journal of the European Academy of Dermatology and Venereology, 2014, Vol. 28, p.1079-1086
[Documento no de patente 7] Zhao y col., British Journal of Dermatology, 2009, Vol. 161, p.1301-1306
[Documento no de patente 8] Leppkes y col., Gastroenterology, 2009, Vol. 136, p.257-267
[Documento no de patente 9] Jetten y col., The Journal of Immunology, 2007, Vol. 178, p. 3208-3218
[Documento no de patente 10] Li y col. molecular Endocrinology, 2010, Vol. 24. p.923-929
[Documento no de patente 11] Burris y col., Nature, 2011, Vol. 472, p.491-494
RESUMEN DE LA INVENCIÓN
[0014] Para el tratamiento real de enfermedades autoinmunes y alérgicas, se utilizan esteroides o agentes inmunosupresores que actúan sobre todo el sistema inmunitario como medicamentos internos. Sin embargo, debido a los posibles efectos secundarios graves, tales como infecciones, actualmente hay muchos casos clínicos en los que se debe interrumpir la administración antes de obtener suficiente eficacia del fármaco. Por lo tanto, se desea desarrollar un nuevo medicamento dirigido a una molécula que desempeña un papel importante en el mecanismo de la aparición y el progreso de enfermedades autoinmunes y enfermedades alérgicas.
[0015] Por lo tanto, un objetivo de la presente invención es proporcionar un compuesto novedoso que tiene actividad antagonista de RORy. Otro objetivo de la presente invención es proporcionar un agente terapéutico o agente preventivo para una enfermedad autoinmunitaria o enfermedad alérgica basado en el efecto de suprimir la función de RORy mediante la actividad antagonista de RORy.
[0016] Los presentes inventores han estudiado intensamente para resolver los problemas mencionados anteriormente y han encontrado un derivado de amina cíclica novedoso que tiene actividad antagonista de RORy, completando así la presente invención.
[0017] Es decir, la presente invención proporciona un derivado de amina cíclica representado por la siguiente fórmula (I):
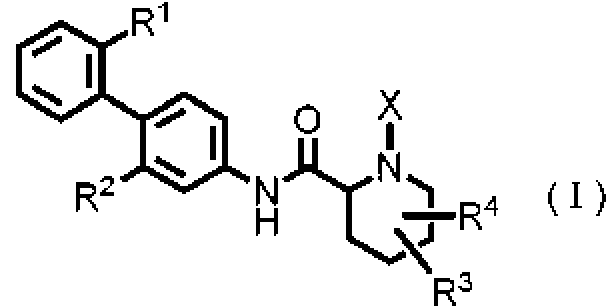
donde
R1 representa un grupo alquiloxi que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno;
R2 representa un átomo de halógeno;
R3 representa un átomo de hidrógeno, un átomo de halógeno o un grupo hidroxi;
R4 representa un átomo de hidrógeno o un átomo de halógeno;
X representa -C(=O)-(CH2)n-R5 o -S(=O)2-R6;
n es un número entero de 0 a 5;
R5 representa un átomo de hidrógeno, -OR7, -SR7, -S(=O)2-R7, -C(=O)-OR7, -N(R7)R8, un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno o un grupo heteroarilo, cualquiera de cuyos átomos de hidrógeno están opcionalmente sustituidos con uno o más grupos alquilo que tienen de 1 a 3 átomos de carbono;
R6 representa un grupo alquilo que tiene de 1 a 5 átomos de carbono;
R7 representa un átomo de hidrógeno o un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno; y R8 representa un átomo de hidrógeno, un grupo alquilo que tiene de 1 a 3 átomos de carbono, un grupo acilo que tiene de 2 a 4 átomos de carbono, o un grupo alquilsulfonilo que tiene de 1 a 3 átomos de carbono, o una sal farmacéuticamente aceptable de los mismos.
[0018] En el derivado de amina cíclica representado por la fórmula general (I) mencionada anteriormente, es preferible que R1 sea un grupo alquiloxi que tenga de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de flúor o uno o más átomos de cloro, que R2 sea un átomo de flúor o un átomo de cloro, que R3 sea un átomo de hidrógeno, un átomo de flúor, un átomo de cloro o un grupo hidroxi, que R4 sea un átomo de hidrógeno, un átomo de flúor, o un átomo de cloro, que R5 sea un átomo de hidrógeno, -OR7, -SR7, -S(=O)2-R7, -C (=O)-OR7, -N(R7)R8, un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno estando opcionalmente sustituidos con uno o más átomos de flúor o uno o más átomos de cloro, o un grupo heteroarilo, cualquiera de cuyos átomos de hidrógeno estando opcionalmente sustituidos con un grupo o grupos metilo, que R6 sea un grupo alquilo que tiene de 1 a 3 átomos de carbono, y que R7 sea un átomo de hidrógeno o un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno estando opcionalmente sustituidos con uno o más átomos de flúor o uno o más átomos de cloro.
[0019] En este caso, se puede esperar una mayor actividad antagonista de RORy.
[0020] En el derivado de amina cíclica representado por la fórmula general (I) mencionada anteriormente, es más preferible que R1 sea un grupo metoxi, cualquiera de cuyos 1 a 3 átomos de hidrógeno estando opcionalmente sustituidos con uno o más átomos de flúor, que R2 sea un átomo de flúor o un átomo de cloro, que R3 sea un átomo de hidrógeno, un átomo de flúor o un grupo hidroxi, que R4 sea un átomo de hidrógeno o un átomo de flúor, que n sea un número entero de 0 a 4, que R5 sea un átomo de hidrógeno, -OR7, -N(R7)R8, un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno estando opcionalmente sustituidos con uno o más átomos de flúor o un grupo heteroarilo de anillo de 5 miembros, cualquiera de cuyos átomos de hidrógeno estando opcionalmente sustituidos con uno o más grupos metilo, que R6 sea un grupo metilo o un grupo etilo, que R7 sea un átomo de hidrógeno o un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno estando opcionalmente sustituidos con uno o más átomos de flúor, y que R8 sea un átomo de hidrógeno, un grupo metilo, un grupo acilo que tiene de 2 a 4 átomos de carbono o un grupo alquilsulfonilo que tiene de 1 a 3 átomos de carbono.
[0021] En este caso, se puede esperar una mayor actividad antagonista de RORy y, además, se puede esperar un excelente efecto terapéutico o efecto preventivo en enfermedades autoinmunitarias, tales como esclerosis múltiple o psoriasis, o enfermedades alérgicas, tales como dermatitis alérgica.
[0022] En el derivado de amina cíclica representado por la fórmula general (I) mencionada anteriormente, es todavía más preferible que R1 sea un grupo trifluorometoxi, que R2 sea un átomo de cloro, que R3 sea un átomo de hidrógeno, que R4 sea un átomo de hidrógeno, que X sea -C(=O)-(CH2)n-R5, que n sea un número entero de 0 a 3, que R5 sea un grupo metilo, un grupo trifluorometilo, -N (R7)R8, o un grupo imidazolilo, triazolilo o tetrazolilo, cualquiera de cuyos átomos de hidrógeno estando opcionalmente sustituidos con un grupo o grupos metilo, que R7 sea un átomo de hidrógeno, un grupo metilo o un grupo etilo, y que R8 sea un átomo de hidrógeno, un grupo metilo, un grupo acetilo,
un grupo propionilo, un grupo metilsulfonilo o un grupo etilsulfonilo.
[0023] En este caso, se puede esperar una mayor actividad antagonista de RORy y, además, se puede esperar un excelente efecto terapéutico o efecto preventivo en enfermedades autoinmunitarias, tales como esclerosis múltiple o psoriasis, o enfermedades alérgicas, tales como dermatitis alérgica.
[0024] La presente invención proporciona un medicamento y un antagonista de RORy, cada uno de los cuales contiene el derivado de amina cíclica representado por la fórmula general (I) mencionada anteriormente o una sal farmacológicamente aceptable del mismo como principio activo.
[0025] El medicamento mencionado anteriormente es preferentemente un agente terapéutico o agente preventivo para una enfermedad autoinmunitaria o una enfermedad alérgica, más preferentemente un agente terapéutico o agente preventivo para la esclerosis múltiple o psoriasis como el agente terapéutico o agente preventivo mencionado anteriormente para una enfermedad autoinmunitaria, más preferentemente un agente terapéutico o agente preventivo para la dermatitis alérgica como el agente terapéutico o agente preventivo mencionado anteriormente para una enfermedad alérgica, y más preferentemente un agente terapéutico o agente preventivo para la dermatitis de contacto o dermatitis atópica como el agente terapéutico o agente preventivo mencionado anteriormente para la dermatitis alérgica.
[0026] Dado que el derivado de amina cíclica de la presente invención o una sal farmacológicamente aceptable del mismo tiene actividad antagonista de RORy, puede suprimir eficazmente la función de RORy y puede utilizarse como agente terapéutico o agente preventivo para enfermedades autoinmunitarias o enfermedades alérgicas.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
[0027]
La figura 1 es una gráfica que muestra el efecto supresor del compuesto del Ejemplo 20 sobre el aumento de la puntuación de síntomas neurológicos en un modelo experimental de encefalomielitis autoinmunitaria de ratón. La figura 2 es una gráfica que muestra el efecto supresor del compuesto del Ejemplo 29 sobre el aumento en la puntuación de síntomas neurológicos en un modelo experimental de encefalomielitis autoinmunitaria de ratón. La figura 3 es una gráfica que muestra el efecto supresor del compuesto del Ejemplo 57 sobre el aumento de la puntuación de síntomas neurológicos en un modelo experimental de encefalomielitis autoinmunitaria de ratón. La figura 4 es una gráfica que muestra el efecto supresor del compuesto del Ejemplo 20 sobre el aumento del grosor de la oreja en un modelo de psoriasis de ratón inducida por imiquimod.
La figura 5 es una gráfica que muestra el efecto supresor del compuesto del Ejemplo 29 sobre el aumento del grosor de la oreja en un modelo de psoriasis de ratón inducida por imiquimod.
La figura 6 es una gráfica que muestra el efecto supresor del compuesto del Ejemplo 57 sobre el aumento del grosor de la oreja en un modelo de psoriasis de ratón inducida por imiquimod.
La figura 7 es una gráfica que muestra el efecto supresor del compuesto del Ejemplo 20 en el aumento de la tasa de hinchazón del oído en un modelo de dermatitis alérgica de ratón inducida por dinitrofluorobenceno.
La figura 8 es una gráfica que muestra el efecto supresor del compuesto del Ejemplo 29 en el aumento de la tasa de hinchazón del oído en un modelo de dermatitis alérgica de ratón inducida por dinitrofluorobenceno.
La figura 9 es una gráfica que muestra el efecto supresor del compuesto del Ejemplo 57 en el aumento de la tasa de hinchazón del oído en un modelo de dermatitis alérgica de ratón inducida por dinitrofluorobenceno.
La figura 10 es una gráfica que muestra el efecto supresor del compuesto del Ejemplo 20 sobre el aumento del grosor de la oreja en un modelo de dermatitis atópica de ratón inducida por oxazolona.
La figura 11 es una gráfica que muestra el efecto supresor del compuesto del Ejemplo 29 sobre el aumento del grosor de la oreja en un modelo de dermatitis atópica de ratón inducida por oxazolona.
La figura 12 es una gráfica que muestra el efecto supresor del compuesto del Ejemplo 57 sobre el aumento del grosor de la oreja en un modelo de dermatitis atópica de ratón inducida por oxazolona.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
[0028] El derivado de amina cíclica de la presente invención se caracteriza por estar representado por la siguiente fórmula general (I):
donde
R1 representa un grupo alquiloxi que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno;
R2 representa un átomo de halógeno;
R3 representa un átomo de hidrógeno, un átomo de halógeno o un grupo hidroxi;
R4 representa un átomo de hidrógeno o un átomo de halógeno;
X representa -C(=O)-(CH2)n-R5 o -S(=O)2-R6;
n representa un número entero de 0 a 5;
R5 representa un átomo de hidrógeno, -OR7, -SR7, -S(=O)2-R7, -C (=O)-OR7, -N(R7)R8, un grupo alquilo que tiene de 1 a 3 átomos de carbono,
cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno o un grupo heteroarilo, cualquiera de cuyos átomos de hidrógeno están opcionalmente sustituidos con un grupo alquilo que tiene de 1 a 3 átomos de carbono;
R6 representa un grupo alquilo que tiene de 1 a 5 átomos de carbono;
R7 representa un átomo de hidrógeno o un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno; y R8 representa un átomo de hidrógeno, un grupo alquilo que tiene de 1 a 3 átomos de carbono, un grupo acilo que tiene de 2 a 4 átomos de carbono o un grupo alquilsulfonilo que tiene de 1 a 3 átomos de carbono.
[0029] Los siguientes términos que se utilizan en el presente documento se definen de la siguiente manera, a menos que se especifique lo contrario.
[0030] El término "grupo alquilo que tiene de 1 a 3 átomos de carbono" significa un grupo metilo, un grupo etilo, un grupo propilo o un grupo isopropilo.
[0031] El término "grupo alquilo que tiene de 1 a 5 átomos de carbono" significa un grupo hidrocarburo saturado lineal que tiene de 1 a 5 átomos de carbono o un grupo hidrocarburo saturado ramificado que tiene de 3 a 5 átomos de carbono, y los ejemplos de los mismos incluyen un grupo metilo, un grupo etilo, un grupo propilo, un grupo isopropilo, un grupo butilo, un grupo isobutilo, un grupo sec-butilo, un grupo terc-butilo, un grupo pentilo, un grupo isopentilo, un grupo neopentilo o un grupo terc-pentilo.
[0032] El término "grupo alquiloxi que tiene de 1 a 3 átomos de carbono" significa un grupo metoxi, un grupo etoxi, un grupo propiloxi o un grupo isopropiloxi.
[0033] El término "grupo acilo que tiene de 2 a 4 átomos de carbono" significa un grupo acetilo, un grupo propionilo, un grupo butanoilo o un grupo 2-metilpropanoilo.
[0034] El término "grupo alquilsulfonilo que tiene de 1 a 3 átomos de carbono" significa un grupo metilsulfonilo, un grupo etilsulfonilo, un grupo propilsulfonilo o un grupo isopropilsulfonilo.
[0035] El término "grupo heteroarilo" significa un grupo aromático heterocíclico que contiene de 1 a 4 heteroátomos seleccionados independientemente del grupo compuesto por un átomo de nitrógeno, un átomo de oxígeno y un átomo de azufre, y los ejemplos de los mismos incluyen un grupo tienilo, un grupo pirrolilo, un grupo furilo, un grupo tiazolilo, un grupo imidazolilo, un grupo oxazolilo, un grupo pirazolilo, un grupo isotiazolilo, un grupo isoxazolilo, un grupo triazolilo, un grupo oxadiazolilo, un grupo tetrazolilo, un grupo piridilo, un grupo piridazinilo, un grupo pirimidinilo, un grupo pirazinilo o un grupo triazinilo.
[0036] El término "grupo heteroarilo de anillo de 5 miembros" significa un grupo aromático heterocíclico que tiene 5 átomos que constituyen el anillo que contienen de 1 a 4 heteroátomos seleccionados independientemente del grupo compuesto por un átomo de nitrógeno, un átomo de oxígeno y un átomo de azufre, y los ejemplos de los mismos incluyen un grupo tienilo, un grupo pirrolilo, un grupo furilo, un grupo tiazolilo, un grupo imidazolilo, un grupo oxazolilo, un grupo pirazolilo, un grupo isotiazolilo, un grupo isoxazolilo, un grupo triazolilo, un grupo oxadiazolilo o un grupo tetrazolilo.
[0037] El término "átomo de halógeno" significa un átomo de flúor, un átomo de cloro, un átomo de bromo o un átomo de yodo.
[0038] El término "grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno" significa un grupo alquilo que tiene de 1 a 3 átomos de carbono como se definió anteriormente, cada uno de cualquiera de cuyos 1 a 3 átomos de hidrógeno pudiendo estar independientemente sustituido con un átomo de halógeno como se definió anteriormente, y los ejemplos de los mismos incluyen un grupo metilo, un grupo etilo, un grupo propilo, un grupo isopropilo, un grupo fluorometilo, un grupo difluorometilo, un trifluorometilo, un grupo 2-fluoroetilo, un grupo trifluoroetilo, un grupo triclorometilo o un grupo tricloroetilo.
[0039] El término "grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de flúor o uno o más átomos de cloro" significa un grupo alquilo que tiene de 1 a 3 átomos de carbono como se definió anteriormente, cada uno de cualquiera de cuyos 1 a 3 átomos de hidrógeno pudiendo estar independientemente sustituido con un átomo de flúor o un átomo de cloro, y los ejemplos de los mismos incluyen un grupo metilo, un grupo etilo, un grupo propilo, un grupo isopropilo, un grupo fluorometilo, un grupo difluorometilo, un grupo trifluorometilo, un grupo 2-fluoroetilo, un grupo trifluoroetilo, un grupo triclorometilo o un grupo tricloroetilo.
[0040] El término "grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de flúor" significa un grupo alquilo que tiene de 1 a 3 átomos de carbono como se definió anteriormente, cualquiera de cuyos 1 a 3 átomos de hidrógeno puede estar sustituidos con uno o más átomos de flúor, y los ejemplos de los mismos incluyen un grupo metilo, un grupo etilo, un grupo propilo, un grupo isopropilo, un grupo fluorometilo, un grupo difluorometilo, un grupo trifluorometilo, un grupo 2-fluoroetilo o un grupo trifluoroetilo.
[0041] El término "grupo alquiloxi que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno" significa un grupo alquiloxi que tiene de 1 a 3 átomos de carbono como se definió anteriormente, cada uno de cualquiera de cuyos 1 a 3 átomos de hidrógeno pudiendo estar independientemente sustituido con un átomo de halógeno como se definió anteriormente, y los ejemplos de los mismos incluyen un grupo metoxi, un grupo etoxi, un grupo propiloxi, un grupo isopropiloxi, un grupo fluorometoxi, un grupo difluorometoxi, un grupo trifluorometoxi, un grupo 2-fluoroetoxi, un grupo trifluoroetoxi, un grupo triclorometoxi o un grupo tricloroetoxi.
[0042] El término "grupo alquiloxi que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de flúor o uno o más átomos de cloro" significa un grupo alquiloxi que tiene de 1 a 3 átomos de carbono como se definió anteriormente, cada uno de cualquiera de cuyos 1 a 3 átomos de hidrógeno pudiendo estar sustituido independientemente con un átomo de flúor o un átomo de cloro, y los ejemplos de los mismos incluyen un grupo metoxi, un grupo etoxi, un grupo propiloxi, un grupo isopropiloxi, un grupo fluorometoxi, un grupo difluorometoxi, un grupo trifluorometoxi, un grupo 2-fluoroetoxi, un grupo trifluoroetoxi, un grupo triclorometoxi o un grupo tricloroetoxi.
[0043] El término "grupo metoxi, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de flúor" significa un grupo metoxi, un grupo fluorometoxi, un grupo difluorometoxi o un grupo trifluorometoxi.
[0044] El término "grupo heteroarilo, cualquiera de cuyos átomos de hidrógeno están opcionalmente sustituidos con uno o más grupos alquilo que tienen de 1 a 3 átomos de carbono" significa un grupo heteroarilo tal como se definió anteriormente, cada uno de cualquiera de cuyos átomos de hidrógeno puede estar sustituido independientemente con un grupo alquilo que tiene de 1 a 3 átomos de carbono tal como se definió anteriormente, y los ejemplos de los mismos incluyen un grupo tienilo, un grupo pirrolilo, un grupo furilo, un grupo tiazolilo, un grupo imidazolilo, un grupo oxazolilo, un grupo pirazolilo, un grupo isotiazolilo, un grupo isoxazolilo, un grupo triazolilo, un grupo oxadiazolilo, un grupo tetrazolilo, un grupo piridilo, un grupo piridazinilo, un grupo pirimidinilo, un grupo pirazinilo, un grupo triazinilo, un grupo metiltienilo, un grupo dimetiltienilo, un grupo etiltienilo, un grupo metilpirrolilo, un grupo dimetilpirrolilo, un grupo etilpirrolilo, un grupo metilfurilo, un grupo dimetilfurilo, un grupo etilfurilo, un grupo metiltiazolilo, un grupo dimetiltiazolilo, un grupo etiltiazolilo, un grupo metilimidazolilo, un grupo dimetillimidazolilo, un grupo etilimidazolilo, un grupo grupo metiloxazolilo, un grupo dimetiloxazolilo, un grupo etiloxazolilo, un grupo metilpirazolilo, un grupo dimetilpirazolilo, un grupo etilpirazolilo, un grupo metili-sotiazolilo, un grupo dimetilisotiazolilo, un grupo etilisotiazolilo, un grupo metilisoxazolilo, un grupo dimetilisoxazolilo, un grupo etilisoxazolilo, un grupo metiltriazolilo, un grupo dimetiltriazolilo, un grupo etiltriazolilo, un grupo metiloxa-diazolilo, un grupo dimetiloxdiazolilo, un grupo etiloxadiazolilo, un grupo metiltetrazolilo, un grupo etiltetrazolilo, un grupo metilpiridilo, un grupo dimetilpiridilo, un grupo etilpiridilo, un grupo metilpiridazinilo, un grupo dimetilpiridazinilo, un grupo etilpiridazinilo, un grupo metilpirimidinilo, un grupo dimetilpirimidinilo, un grupo etilpirimidinilo, un grupo metilpirazinilo, un grupo dimetilpirazinilo, un grupo etilpirazinilo, un grupo etilpirazinilo, un grupo metiltriazinilo, un grupo dimetiltriazinilo, un grupo etiltriazinilo o un grupo etiltriazinilo.
[0045] El término "grupo heteroarilo, cualquiera de cuyos átomos de hidrógeno están opcionalmente sustituidos con uno o más grupos metilo" significa un grupo heteroarilo tal como se definió anteriormente, cada uno de cualquiera de cuyos átomos de hidrógeno pudiendo estar sustituido independientemente con un grupo metilo, y los ejemplos de los mismos incluyen un grupo tienilo, un grupo pirrolilo, un grupo furilo, un grupo tiazolilo, un grupo imidazolilo, un grupo oxazolilo, un grupo pirazolilo, un grupo isotiazolilo, un grupo isoxazolilo, un grupo triazolilo, un grupo oxadiazolilo, un grupo tetrazolilo, un grupo piridilo, un grupo piridazinilo, un grupo pirimidinilo, un grupo pirazinilo, un grupo triazinilo, un grupo metiltienilo, un grupo dimetiltienilo, un grupo metilpirrolilo, un grupo dimetilpirrolilo, un grupo metilfurilo, un grupo dimetilfurilo, un grupo metiltiazolilo, un grupo dimetiltiazolilo, un grupo metilimidazolilo, un grupo dimetililimidazolilo, un grupo metiloxazolilo, un grupo dimetiloxazolilo, un grupo metilpirazolilo, un grupo dimetilpirazolilo, un grupo metilisotiazolilo, un grupo dimetilisotiazolilo, un grupo metilisoxazolilo, un grupo dimetilisoxazolilo, un grupo dimetilisoxazolilo, un grupo metiltriazolilo grupo, grupo dimetiltriazolilo, un grupo metiloxadiazolilo, un grupo
dimetiloxdiazolilo, un grupo metiltetrazolilo, un grupo metilpiridilo, un grupo dimetilpiridilo, un grupo metilpiridazinilo, un grupo dimetilpiridazinilo, un grupo metilpirimidinilo, un grupo dimetilpirimidinilo, un grupo metilpirazinilo, un grupo dimetilpirazinilo, un grupo metiltriazinilo o un grupo dimetiltriazinilo.
[0046] El término "grupo heteroarilo de anillo de 5 miembros, cualquiera de cuyos átomos de hidrógeno está opcionalmente sustituido con un grupo o grupos metilo" significa un grupo heteroarilo de anillo de 5 miembros tal como se definió anteriormente, cada uno de cualquiera de cuyos átomos de hidrógeno puede estar independientemente sustituido con un grupo metilo, y los ejemplos de los mismos incluyen un grupo tienilo, un grupo pirrolilo, un grupo furilo, un grupo tiazolilo, un grupo imidazolilo, un grupo oxazolilo, un grupo pirazolilo, un grupo isotiazolilo, un grupo isoxazolilo, un grupo triazolilo, un grupo oxadiazolilo, un grupo tetrazolilo, un grupo metiltienilo, un grupo dimetiltienilo, un grupo metilpirrolilo, un grupo dimetilpirrolilo, un grupo metilfurilo, un grupo dimetilfurilo, un grupo metiltiazolilo, un grupo dimetiltiazolilo, un grupo metilimidazolilo, un grupo dimetilimidazolilo, un grupo metiloxazolilo, un grupo dimetililoxazolilo, un grupo metilpirazolilo, un grupo dimetilpirazolilo, un grupo dimetilpirazolilo, un grupo metilisotiazolilo, un grupo dimetilisotiazolilo, un grupo metilisoxazolilo, un grupo dimetilisoxazolilo, un grupo metiltriazolilo, un grupo dimetiltriazolilo, un grupo metiloxadiazolilo, un grupo dimetiloxadiazolilo o un grupo metiltetrazolilo grupo.
[0047] Con respecto al derivado de amina cíclica mencionado anteriormente, en la fórmula general (I), R1 es preferentemente un grupo alquiloxi que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno de está opcionalmente sustituido con uno o más átomos de flúor o uno o más átomos de cloro, más preferentemente un grupo metoxi, cualquiera de cuyos 1 a 3 átomos de hidrógeno está opcionalmente sustituido con uno o más átomos de flúor, y aún más preferentemente un grupo trifluorometoxi.
[0048] R2 es preferentemente un átomo de flúor o un átomo de cloro, y más preferentemente un átomo de cloro.
[0049] R3 es preferentemente un átomo de hidrógeno, un átomo de flúor, un átomo de cloro o un grupo hidroxi, más preferentemente un átomo de hidrógeno, un átomo de flúor o un grupo hidroxi, y aún más preferentemente un átomo de hidrógeno.
[0050] R4 es preferentemente un átomo de hidrógeno, un átomo de flúor o un átomo de cloro, más preferentemente un átomo de hidrógeno o un átomo de flúor y aún más preferentemente, un átomo de hidrógeno.
[0051] X es preferentemente -C(=O)-(CH2)n-R5.
[0052] n es un número entero de 0 a 4, y n es un número entero de 0 a 3.
[0053] R5 es preferentemente un átomo de hidrógeno, -OR7, -SR7, -S(=O)2-R7, -C (=O)-OR7, -N(R7)R8, un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno está opcionalmente sustituido con uno o más átomos de flúor o uno o más átomos de cloro, o un grupo heteroarilo, cualquiera de cuyos átomos de hidrógeno está opcionalmente sustituido con uno o más grupos metilo, más preferentemente un átomo de hidrógeno, -OR7, -N(R7)R8, un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno está opcionalmente sustituido con uno o más átomos de flúor o un grupo heteroarilo de anillo de 5 miembros, cualquiera de cuyos átomos de hidrógeno está opcionalmente sustituido con uno o más grupos metilo y aún más preferentemente un grupo metilo, un grupo trifluorometilo, -N(R7)R8 o un grupo imidazolilo, triazolilo o tetrazolilo, donde cualquier átomo o átomos de hidrógeno están opcionalmente sustituidos con uno o más grupos metilo.
[0054] R6 es preferentemente un grupo alquilo que tiene de 1 a 3 átomos de carbono, y más preferentemente un grupo metilo o un grupo etilo.
[0055] R7 es preferentemente un átomo de hidrógeno o un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno está opcionalmente sustituido con uno o más átomos de flúor o uno o más átomos de cloro, más preferentemente un átomo de hidrógeno o un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno está opcionalmente sustituido con uno o más átomos de flúor y aún más preferentemente un átomo de hidrógeno, un grupo metilo o un grupo etilo.
[0056] R8 es más preferentemente un átomo de hidrógeno, un grupo metilo, un grupo acilo que tiene de 2 a 4 átomos de carbono, o un grupo alquilsulfonilo que tiene de 1 a 3 átomos de carbono, y aún más preferentemente un átomo de hidrógeno, un grupo metilo, un grupo acetilo, un grupo propionilo, un grupo metilsulfonilo o un grupo etilsulfonilo.
[0057] En las Tablas 1-1 a 1-3 se muestran ejemplos específicos del compuesto preferido del derivado de amina cíclica representado por la fórmula general (I), pero la presente invención no se limita a estos.
[Tabla 1-1]
Ċ


[Tabla 1-2]

[Tabla 1-3]

[0058] Los compuestos mencionados en las Tablas 1-1 a 1-3 también incluyen sales farmacológicamente aceptables de los mismos.
[0059] El derivado de amina cíclica representado por la fórmula general (I) puede incluir isómeros conformacionales, rotámeros, tautómeros, isómeros ópticos, diastereómeros y similares, e incluir no solo un único isómero, sino también racematos y mezclas de diastereómeros.
[0060] El derivado de amina cíclica representado por la fórmula general (I) puede estar marcado con uno o más isótopos, y los ejemplos del isótopo marcado incluyen 2H, 3H, 13C, 14C, 15N, 15O, 18O y/o 125I.
[0061] Los ejemplos de la "sal farmacológicamente aceptable" del derivado de amina cíclica representado por la fórmula general (I) incluyen una sal con una base inorgánica, una sal con una base orgánica, una sal con un ácido inorgánico y una sal con un ácido orgánico. Los ejemplos de la sal con una base inorgánica incluyen una sal de metal alcalino, tal como una sal de sodio o una sal de potasio, una sal de metal alcalinotérreo, tal como una sal de calcio o una sal de magnesio, una sal de amonio, una sal de aluminio o una sal de zinc, y los ejemplos de la sal con una base orgánica incluyen una sal con una amina orgánica, tal como trietilamina, etanolamina, morfolina, piperidina o diciclohexilamina, o una sal con un aminoácido básico, tal como arginina o lisina. Los ejemplos de la sal con un ácido inorgánico incluyen un clorhidrato, un sulfato, un nitrato, un hidrobromuro, un hidroyoduro, un fosfato o similares, y los ejemplos de la sal con un ácido orgánico incluyen un oxalato, un malonato, un citrato, un fumarato, un lactato, un malato, un succinato, un tartrato, un acetato, un trifluoroacetato, un maleato, un gluconato, un benzoato, un ascorbato, un glutarato, un mandelato, un ftalato, un metanosulfonato, un etanosulfonato, un bencenosulfonato, un ptoluenosulfonato, un alcanforsulfonato, un aspartato, un glutamato, un cinamato o similares.
[0062] El derivado de amina cíclica representado por la fórmula general anterior (I) o una sal farmacológicamente aceptable del mismo puede ser un anhídrido o un solvato, tal como un hidrato. En este caso, el solvato es preferentemente un solvato farmacéuticamente aceptable. El solvato farmacéuticamente aceptable puede ser un hidrato o un no hidrato y es preferentemente un hidrato. Los ejemplos del disolvente que constituye el solvato incluyen disolventes a base de alcohol, tales como metanol, etanol o n-propanol, N,N-dimetilformamida (en adelante abreviado como DMF), dimetilsulfóxido (en adelante abreviado como DMSO), o agua.
[0063] El derivado de amina cíclica representado por la fórmula general anterior (I) (en adelante, denominado un derivado de amina cíclica (I)) se puede producir mediante un procedimiento adecuado basado en características derivadas de un esqueleto básico y tipos de sustituyentes del mismo. Un material de partida y un reactivo utilizados en la producción de estos compuestos generalmente pueden estar comercialmente disponibles o producirse mediante métodos conocidos.
[0064] El derivado de amina cíclica (I) y el material intermedio y de partida que se utilizará en la producción del mismo se pueden aislar y purificar por medios conocidos. Los ejemplos de medios conocidos para el aislamiento y purificación incluyen extracción de disolventes, recristalización o cromatografía.
[0065] Cuando el derivado de amina cíclica (I) contiene un isómero óptico o un estereoisómero, cada isómero se puede obtener como un único compuesto mediante un procedimiento conocido. Los ejemplos del procedimiento conocido incluyen cristalización, resolución enzimática o cromatografía quiral.
[0066] En cada una de las reacciones de los procedimientos de producción que se mencionan a continuación, cuando el compuesto de partida tiene un grupo amino o un grupo carboxilo, se puede introducir un grupo protector en estos grupos y después de la reacción, el grupo protector se puede desproteger según corresponda para obtener el compuesto diana.
[0067] Los ejemplos del grupo protector del grupo amino incluyen un grupo alquilcarbonilo que tiene de 2 a 6 átomos de carbono (por ejemplo, un grupo acetilo), un grupo benzoilo, un grupo alquiloxicarbonilo que tiene de 2 a 8 átomos de carbono (por ejemplo, un grupo terc-butoxicarbonilo o un grupo benciloxicarbonilo), un grupo aralquilo que tiene de 7 a 10 átomos de carbono (por ejemplo, un grupo bencilo), o un grupo ftaloilo.
[0068] Los ejemplos del grupo protector del grupo carboxilo incluyen un grupo alquilo que tiene de 1 a 6 átomos de carbono (por ejemplo, un grupo metilo, un grupo etilo o un grupo terc-butilo), o un grupo aralquilo que tiene de 7 a 10 átomos de carbono (por ejemplo, un grupo bencilo).
[0069] La desprotección del grupo protector varía en función del tipo de grupo protector, pero la desprotección se puede realizar según un procedimiento conocido (por ejemplo, Greene, TW, "Greene 's Protective Groups in Organic Synthesis", Wiley-Interscience) o un método análogo.
[0070] Tal como se muestra, por ejemplo, en el Esquema 1, el derivado de amina cíclica (I) se puede obtener mediante una reacción de acoplamiento (primera etapa) de un derivado de ácido borónico (II) con un derivado de haluro de arilo (III) en presencia de un catalizador metálico y una base, seguida de una reacción de condensación (segunda etapa) del derivado de bifenilamina (IV) obtenido en la primera etapa con un derivado de ácido pipecólico (V) en presencia de un agente condensante y una base, seguida de una reacción de desprotección (tercera etapa) del derivado de amida de ácido N-terc-butoxicarbonilpipercólico (VI) obtenido en la segunda etapa en presencia de un ácido, y seguida de una reacción de condensación (cuarta etapa) del derivado de amida de ácido pipecólico (VII) obtenido en la tercera etapa con un derivado de anhídrido de ácido orgánico (VIII) en presencia de una base. El derivado de amina cíclica (I) también se puede obtener mediante una reacción de condensación del derivado de amida de ácido pipecólico (VII) con un derivado de éster de ácido orgánico (IX). El derivado de amina cíclica (I) también se puede obtener mediante una reacción de condensación del derivado de amida de ácido pipecólico (VII) con un derivado de cloruro de ácido orgánico (X) en presencia de una base. El derivado de amina cíclica (I) también se puede obtener
mediante una reacción de condensación del derivado de amida de ácido pipecólico (VII) y un derivado de ácido orgánico (XI) en presencia de un agente de condensación y una base. El derivado de amina cíclica (I) también se puede obtener mediante una reacción de condensación del derivado de amida de ácido pipecólico (VII) con isocianato de trimetilsililo en presencia de una base.
[0071] Cuando el derivado de amina cíclica (I) contiene, por ejemplo, un grupo amino, el grupo amino se puede convertir en un grupo amida, un grupo sulfonamida o similares, o un derivado de N-alquilo mediante una reacción de condensación, una reacción de aminación reductora o similares. Cuando el derivado de amina cíclica (I) contiene un grupo sulfuro, el grupo sulfuro puede convertirse en un grupo sulfonilo mediante una reacción de oxidación. Cuando el derivado de amina cíclica (I) contiene un grupo éster, el grupo éster puede convertirse en un grupo carboxilo mediante una reacción de hidrólisis.

[0072] En el Esquema 1, Q representa un átomo de halógeno y R1 a R4 y X son como se definió anteriormente. (Primera etapa)
[0073] La cantidad del derivado de haluro de arilo (III) utilizado en la reacción de acoplamiento es preferentemente de 0,5 a 10 equivalentes, y más preferentemente de 0,7 a 3 equivalentes, en función del derivado de ácido borónico (II).
[0074] Los ejemplos del catalizador metálico utilizado en la reacción de acoplamiento incluyen aducto de dicloropaladio (II) de 1,1'-bis(difenilfosfino) ferroceno, cloruro de paladio (II), bis (dibencilidenoacetona)paladio(O), tetrakist-rifenilfosfina paladio(0) o diclorobitrifenilfosfina paladio(0), y aducto de dicloropaladio (II) de 1,1'-bis(difenilfosfino)ferroceno.
[0075] La cantidad del catalizador metálico utilizado para la reacción de acoplamiento es preferentemente de 0,01 a 5 equivalentes, y más preferentemente de 0,05 a 0,5 equivalentes, en función del derivado de ácido borónico (II).
[0076] Los ejemplos de la base utilizada en la reacción de acoplamiento incluyen una base orgánica, tal como trietilamina o diisopropiletilamina, una base inorgánica, tal como carbonato de sodio o carbonato de potasio, una amida de litio, tal como hexametildisilazida de litio o diisopropilamida de litio, o un alcóxido metálico, tal como terc-butiloxi sodio, terc-butiloxi potasio, o una mezcla de estos, y una base inorgánica, tal como carbonato de sodio o carbonato de potasio, es preferible.
[0077] La cantidad de la base utilizada en la reacción de acoplamiento es preferentemente de 0,5 a 10 equivalentes, y más preferentemente de 1 a 3 equivalentes, en función del derivado de ácido borónico (II).
[0078] El disolvente de reacción utilizado para la reacción de acoplamiento se selecciona apropiadamente según el tipo de reactivo a utilizar o similares, pero no se limita particularmente, siempre y cuando no inhiba la reacción, y los ejemplos de los mismos incluyen disolventes a base de éter, tales como tetrahidrofurano, 1,4-dioxano, etilenglicol dimetil éter o dimetoxietano,
disolventes a base de nitrilo, tales como acetonitrilo o propionitrilo, disolventes de hidrocarburos aromáticos, tales como benceno o tolueno, disolventes polares apróticos, tales como DMF o DMSO, agua o una mezcla de los mismos. Es preferible un disolvente mixto de disolventes a base de nitrilo, tales como acetonitrilo o propionitrilo, y agua.
[0079] La temperatura de reacción de la reacción de acoplamiento es preferentemente de 0 a 200 °C, y más preferentemente de 50 a 150 °C.
[0080] El tiempo de reacción de la reacción de acoplamiento se selecciona adecuadamente en función de las condiciones, tal como la temperatura de reacción, y el tiempo de reacción es preferentemente de 1 a 30 horas.
[0081] La concentración del derivado de ácido borónico (II) utilizado en la reacción de acoplamiento al inicio de la reacción es preferentemente de 1 mmol/L a 1 mol/L.
[0082] El derivado de ácido borónico (II) y el derivado de haluro de arilo (III) utilizados en la reacción de acoplamiento se pueden comprar o producir mediante un procedimiento conocido.
(Etapa 2)
[0083] La cantidad del derivado de ácido pipecólico (V) utilizado en la reacción de condensación es preferentemente de 0,1 a 10 equivalentes, y más preferentemente de 0,5 a 3 equivalentes, en función del derivado de bifenilamina (IV).
[0084] Los ejemplos del agente de condensación utilizado en la reacción de condensación incluyen N,N'-diciclohexilcarbodiimida, clorhidrato de N-etil-N-3-dimetilaminopropilcarbodiimida, N,N'-carbodiimidazol, {{[(1-ciano-2-etoxi-2-oxoetil-ideno)amino]oxi}-4-morfolinometileno}dimetilamonio hexafluorofosfato (en adelante abreviado como COMU), hexafluorofosfato de O-(7-azabenzotriazol-1-il)-1,1,3,3-tetrametiluronio (en adelante abreviado como HATU) o hexafluorofosfato de O-(benzotriazol-1-il)-N,N,N',N'-tetrametiluronio (en adelante abreviado como HBTU), y es preferible HATU o HBTU.
[0085] La cantidad del agente de condensación utilizado en la reacción de condensación es preferentemente de 0,5 a 10 equivalentes, y más preferentemente de 1 a 3 equivalentes, en función del derivado de bifenilamina (IV).
[0086] Los ejemplos de la base utilizada en la reacción de condensación incluyen una base orgánica, tal como trietilamina o diisopropiletilamina, una base inorgánica, tal como carbonato de hidrógeno de sodio o carbonato de potasio, un compuesto metálico hidrogenado, tal como hidruro de sodio, hidruro de potasio o hidruro de calcio, un alquillitio, tal como metillitio o butilo litio, una amida de litio, tal como hexametildisilazida de litio o diisopropilamida de litio, o una mezcla de los mismos, y es preferible una base orgánica, tal como trietilamina o diisopropiletilamina.
[0087] La cantidad de la base utilizada en la reacción de condensación es preferentemente de 0,5 a 10 equivalentes, y más preferentemente de 1 a 5 equivalentes, en función del derivado de bifenilamina (IV).
[0088] El derivado de bifenilamina (IV) utilizado en la reacción de condensación puede ser una forma libre o una sal, tal como un clorhidrato.
[0089] El disolvente de reacción utilizado en la reacción de condensación se selecciona apropiadamente según el tipo de reactivo a utilizar o similares, pero no está particularmente limitado, siempre que no inhiba la reacción, y los ejemplos de los mismos incluyen disolventes a base de éter, tales como tetrahidrofurano, 1,4-dioxano, etilenglicol dimetil éter, o dimetoxietano, disolventes a base de halógeno, tales como diclorometano, cloroformo, o 1,2-dicloroetano, disolventes polares apróticos, tales como DMF o DMSO, o disolventes a base de nitrilo, tales como acetonitrilo o propionitrilo, y disolventes a base de halógeno, tales como diclorometano, cloroformo, o 1,2-dicloroetano, o son preferibles disolventes polares apróticos, tales como DMF o DMSO.
[0090] La temperatura de reacción de la reacción de condensación es preferentemente de 0 a 200 °C, y más preferentemente de 20 a 100 °C.
[0091] El tiempo de reacción de la reacción de condensación se selecciona adecuadamente en función de las condiciones, tal como la temperatura de reacción, y es preferentemente de 0,5 a 100 horas.
[0092] La concentración del derivado de bifenilamina (IV) utilizado en la reacción de condensación al inicio de la reacción es preferentemente de 1 mmol/L a 1 mol/L.
[0093] El derivado de ácido pipecólico (V) utilizado en la reacción de condensación puede adquirirse o producirse mediante un procedimiento conocido o un procedimiento análogo al mismo.
(Tercera etapa)
[0094] Los ejemplos del ácido utilizado en la reacción de desprotección incluyen ácido clorhídrico, ácido trifluoroacético o ácido fluorhídrico, y es preferible el ácido clorhídrico o el ácido trifluoroacético.
[0095] La cantidad de ácido utilizada en la reacción de desprotección es preferentemente de 0,5 a 100 equivalentes, y más preferentemente de 1 a 30 equivalentes, en función del derivado de amida de ácido N-tercbutoxicarbonilpipercólico (VI).
[0096] El disolvente de reacción utilizado en la reacción de desprotección se selecciona apropiadamente según el tipo de reactivo que se va a usar, pero no está particularmente limitado, siempre que no inhiba la reacción, y los ejemplos de los mismos incluyen disolventes a base de éter, tales como éter dietílico, tetrahidrofurano, dimetoxietano o 1,4-dioxano, disolventes a base de éster, tales como acetato de etilo o acetato de propilo, disolventes a base de cloro, tales como diclorometano, cloroformo o 1,2-dicloroetano, disolventes a base de alcohol, tales como metanol o etanol, disolventes polares apróticos, tales como DMF o DMSO, o disolventes mixtos de los mismos, y disolventes a base de halógeno, tales como diclorometano, cloroformo o 1,2-dicloroetano, o son preferibles disolventes polares apróticos, tales como DMF o DMSO.
[0097] La temperatura de reacción de la reacción de desprotección es preferentemente de -78°C a 200°C, y más preferentemente de -20°C a 100°C.
[0098] El tiempo de reacción de la reacción de desprotección se selecciona apropiadamente en función de las condiciones, tal como la temperatura de reacción, y el tiempo de reacción es preferentemente de 1 a 50 horas.
[0099] La concentración del derivado de amida de ácido N-terc-butoxicarbonilpipercólico (VI) utilizado en la reacción de desprotección al inicio de la reacción es preferentemente de 1 mmol/L a 1 mol/L.
(Etapa 4)
[0100] La cantidad del derivado de anhídrido ácido orgánico (VIII), el derivado de éster de ácido orgánico (IX), el derivado de cloruro de ácido orgánico (X), el derivado de ácido orgánico (XI) o isocianato de trimetilsililo utilizado en la reacción de condensación es preferentemente de 1 a 200 equivalentes, y más preferentemente de 1 a 80 equivalentes, en función del derivado de amida de ácido pipecólico (VII).
[0101] Los ejemplos del agente de condensación utilizado en la reacción de condensación incluyen N,N'-diciclohexilcarbodiimida, clorhidrato de N-etil-N'-3-dimetilaminopropilcarbodiimida, N,N'-carbodiimidazol, COMU, HATU o HBTU, y es preferible HATU o HBTU.
[0102] La cantidad del agente de condensación utilizado en la reacción de condensación es preferentemente de 0 a 10 equivalentes, y más preferentemente de 0 a 3 equivalentes, en función del derivado de amida de ácido pipecólico (VII).
[0103] Los ejemplos de la base utilizada en la reacción de condensación incluyen una base orgánica, tal como trietilamina o diisopropiletilamina, una base inorgánica, tal como carbonato de hidrógeno de sodio o carbonato de potasio, un compuesto metálico hidrogenado, tal como hidruro de sodio, hidruro de potasio o hidruro de calcio, un alquillitio, tal como metillitio o butilo litio, amida de litio, tal como hexametildisilazida de litio o diisopropilamida de litio, o una mezcla de los mismos, y es preferible una base orgánica, tal como trietilamina o diisopropiletilamina.
[0104] La cantidad de la base utilizada en la reacción de condensación es preferentemente de 0 a 10 equivalentes, y más preferentemente de 0 a 5 equivalentes, en función del derivado de amida de ácido pipecólico (VII).
[0105] El derivado de amida de ácido pipecólico (VII) utilizado en la reacción de condensación puede ser una forma libre o una sal, tal como un clorhidrato.
[0106] El disolvente de reacción utilizado en la reacción de condensación se selecciona apropiadamente según el tipo de reactivo a utilizar o similares, pero no está particularmente limitado, siempre que no inhiba la reacción, y los ejemplos de los mismos incluyen disolventes a base de éter, tales como tetrahidrofurano, 1,4-dioxano, etilenglicol dimetil éter, o dimetoxietano, disolventes a base de cloro, tales como diclorometano, cloroformo, o 1,2-dicloroetano, disolventes polares apróticos, tales como DMF o DMSO, o disolventes a base de nitrilo, tales como acetonitrilo o propionitrilo, y disolventes a base de halógeno, tales como diclorometano, cloroformo, o 1,2-dicloroetano, o son preferibles disolventes polares apróticos, tales como DMF o DMSO.
[0107] La temperatura de reacción de la reacción de condensación es preferentemente de -78°C a 200°C, y más preferentemente de -20°C a 100°C.
[0108] El tiempo de reacción de la reacción de condensación se selecciona adecuadamente en función de las condiciones, tal como la temperatura de reacción, y el tiempo de reacción es preferentemente de 0,5 a 100 horas.
[0109] La concentración del derivado de amida de ácido pipecólico (VII) utilizado en la reacción de acoplamiento al inicio de la reacción es preferentemente de 1 mmol/L a 1 mol/L.
[0110] El derivado de anhídrido ácido orgánico (VIII), el derivado de éster de ácido orgánico (IX), el derivado de cloruro de ácido orgánico (X), el derivado de ácido orgánico (XI) y el isocianato de trimetilsililo utilizado en la reacción de condensación se pueden comprar o producir mediante un procedimiento conocido o un procedimiento análogo al mismo.
[0111] El medicamento, el antagonista de RORy y el agente terapéutico o agente preventivo para una enfermedad autoinmunitaria o enfermedad alérgica de la presente invención se caracterizan por contener el derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo como principio activo. La enfermedad autoinmunitaria mencionada anteriormente es preferentemente esclerosis múltiple o psoriasis, y la enfermedad alérgica mencionada anteriormente es preferentemente dermatitis alérgica, y más preferentemente dermatitis de contacto o dermatitis atópica.
[0112] "Antagonista de RORy" significa un compuesto que tiene un efecto para suprimir la función de RORy, eliminando o atenuando la actividad del mismo.
[0113] "Enfermedad autoinmunitaria" es un término general que designa enfermedades en las que una respuesta inmunitaria excesiva ataca las propias células y tejidos normales de un individuo, produciéndole síntomas. Los ejemplos de las mismas incluyen esclerosis múltiple, psoriasis, artritis reumatoide, lupus eritematoso sistémico, espondilitis anquilosante, uveítis, polimialgia reumática, esclerodermia, vasculitis, pénfigo, pénfigo o dermatomiositis. Además, la enfermedad autoinmunitaria de la presente invención incluye acné o vitíligo.
[0114] "Enfermedad alérgica" es una enfermedad derivada de una respuesta inmunitaria excesiva a antígenos específicos, y los ejemplos de la misma incluyen dermatitis alérgica, dermatitis de contacto, dermatitis atópica, rinitis alérgica (polinosis), conjuntivitis alérgica, gastroenteritis alérgica, asma bronquial, asma infantil o alergia alimentaria.
[0115] "Esclerosis múltiple" es una enfermedad caracterizada por la desmielinización que destruye que la vaina de mielina que recubre las fibras nerviosas, tales como el cerebro, la médula espinal y el nervio óptico; y se caracteriza por la progresión del trastorno con recaídas y remisiones repetidas. Los síntomas varían en función del lugar de la lesión y representan diversos síntomas neurológicos, tales como discapacidad visual, cuadriplejia, alteración sensorial y alteraciones de la marcha. Los ejemplos de esclerosis múltiple incluyen esclerosis múltiple remitente-recidivante, esclerosis múltiple primaria progresiva y esclerosis múltiple secundaria progresiva.
[0116] "Psoriasis" es una enfermedad inflamatoria de la piel asociada con la invasión y activación de células inmunitarias y la acantosis resultante. Típicamente, se produce un síntoma llamado descamación, por el cual una capa espesa de escamas blancas se adhiere a erupciones rojas en diversas partes de todo el cuerpo y posteriormente, se desprenden. Los ejemplos de psoriasis incluyen psoriasis en placas, psoriasis pustulosa, psoriasis artropática,
psoriasis intestinal y psoriasis eritrodérmica.
[0117] "Dermatitis alérgica" es un término general que designa enfermedades de la piel causadas por reacciones alérgicas y se caracteriza por picazón crónica y erupciones en la cara, cuello, codos y/o rodillas. Los ejemplos de dermatitis alérgica incluyen dermatitis de contacto, dermatitis atópica y similares.
[0118] "Dermatitis de contacto" es una enfermedad inflamatoria eczematosa que se desarrolla cuando un antígeno exógeno entra en contacto con la piel, y los ejemplos de la misma incluyen dermatitis alérgica de contacto, dermatitis por fotocontacto, dermatitis de contacto sistémica y urticaria de contacto. Los ejemplos del antígeno incluyen alérgenos metálicos (cobalto, níquel, etc.), alérgenos vegetales (roble venenoso, onagra, etc.) y alérgenos alimentarios (mango, nuez de ginkgo, etc.).
[0119] La "dermatitis atópica" es una enfermedad de la piel por la cual muchos pacientes tienen predisposición atópica. Se caracteriza por un eczema sistémico simétrico con repetidas exacerbaciones y remisiones. Los ejemplos de la misma incluyen neurodermatitis difusa, eczema atópico, neurodermatitis atópica, prurigo de Besnier, eczema infantil agudo, eczema flexural, eczema pediátrico en las extremidades, eczema atópico pediátrico, eczema seco pediátrico, eczema pediátrico, dermatitis atópica en adultos, eczema endógeno, dermatitis infantil y eczema infantil crónico.
[0120] El derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo se caracteriza por suprimir la función de RORy mediante la inhibición de la unión entre RORy y un coactivador. Dado que se sabe que RORy está implicado en diversas enfermedades y que cabe esperar una mejora en el estado patológico o una remisión de los síntomas mediante la supresión de la función de RORy, el derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo puede utilizarse como medicamento para enfermedades en las que quepa esperar una mejora en el estado patológico o una remisión de los síntomas mediante la supresión de la función de RORy, particularmente como agente terapéutico o agente preventivo para enfermedades autoinmunitarias o enfermedades alérgicas. El agente terapéutico o agente preventivo para enfermedades autoinmunitarias mencionado anteriormente se puede utilizar preferentemente como agente terapéutico o agente preventivo para esclerosis múltiple, psoriasis, artritis reumatoide, lupus eritematoso sistémico, espondilitis anquilosante, uveítis, polimialgia reumática, esclerodermia, vasculitis, pénfigo, pénfigo, dermatomiositis, acné o vitíligo, y más preferentemente, se puede utilizar como agente terapéutico o agente preventivo para esclerosis múltiple o psoriasis. El agente terapéutico o agente preventivo para enfermedades alérgicas mencionado anteriormente puede utilizarse preferentemente como agente terapéutico o agente preventivo para dermatitis alérgica, dermatitis atópica, rinitis alérgica (polinosis), conjuntivitis alérgica, gastroenteritis alérgica, asma bronquial, asma infantil o alergia alimentaria, y más preferentemente como agente terapéutico o agente preventivo para la dermatitis de contacto o dermatitis atópica.
[0121] Es posible evaluar que el derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo tiene actividad antagonista de RORy que inhibe la unión entre RORy y un coactivador, utilizando un estudio in vitro. Los ejemplos del estudio in vitro incluyen un procedimiento para evaluar la unión entre RORy y un agonista (por ejemplo, colesterol) (WO 2012/158784, w O 2013/018695) y un procedimiento para evaluar la unión entre un dominio de unión al ligando de RORy y un coactivador (WO 2012/064744, WO 2013/018695). El efecto inhibidor sobre la actividad de transcripción de RORy se puede evaluar utilizando diversos ensayos genéticos reporteros (WO 2012/158784, WO 2012/064744, WO 2013/018695).
[0122] El hecho de que el derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo suprima la función de RORy se puede evaluar utilizando células linfocíticas derivadas de diversos órganos, tales como el bazo o la sangre periférica, con producción de IL-17 o diferenciación celular de Th17 como índice. Los ejemplos del procedimiento que utiliza la producción de IL-17 como índice incluyen un procedimiento para medir la producción de IL-17 mediante estimulación de IL-23 utilizando esplenocitos de ratón (The Journal of Biological Chemistry, 2003, Vol.
278, No. 3, p. 1910-1914). Los ejemplos del procedimiento que utiliza la diferenciación de células Th17 como índice incluyen un procedimiento para medir la cantidad de producción de IL-17 o la proporción de células positivas en IL-17, etc. mediante la estimulación de linfocitos T sin tratamiento previo CD4 derivados de esplenocitos de ratón o PBMC humana con diversas citocinas (por ejemplo, IL-1p, IL-6, IL-23 y/o TGF-p) y diversos anticuerpos (por ejemplo, anticuerpo anti-CD3, anticuerpo anti-CD28, anticuerpo anti-IL-4, anticuerpo anti-IFN-y y/o anticuerpo anti-IL-2) que se diferenciarán en Th17 (WO 2012/158784, WO 2013/018695).
[0123] El hecho de que el derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo sea eficaz para el tratamiento o prevención de enfermedades autoinmunitarias se puede evaluar utilizando un modelo de enfermedad. Los ejemplos del modelo de enfermedad incluyen un modelo experimental de encefalomielitis autoinmunitaria (Journal of Neuroscience Research, 2006, Vol. 84, p. 1225-1234), un modelo de psoriasis inducida por imiquimod (Journal of Immunology, 2009, Vol. 182, p. 5836-5845), un modelo de artritis de colágeno (Annual Review of Immunology, 1984, Vol. 2, p. 199-218), un modelo espontáneo de lupus eritematoso sistémico (Nature, 2000, Vol.
404, p. 995-999), un modelo de espondilitis anquilosante (Arthritis Research & Therapy, 2012, Vol. 14, p.253-265), un modelo de uveítis autoinmunitaria experimental (Journal of Immunology, 2006, Vol. 36, p.3071-3081), un modelo de esclerodermia (Journal of Investigative Dermatology, 1999, Vol. 112, p.456-462), un modelo de vasculitis (The Journal
of Clinical Investigation, 2002, Vol. 110, p.955-963), un modelo de pénfigo (The Journal of Clinical Investigation, 2000, Vol. 105, p.625-631), un modelo de penfigoide (Experimental Dermatology, 2012, Vol. 21, p.901-905), un modelo de dermatomiositis (American Journal of Pathology, 1985, Vol. 120, p.323-325), un modelo espontáneo de acné (European Journal of Dermatology, 2005, Vol. 15, p.459-464), o un modelo de vitíligo (Pigment Cell & Melanoma Research, 2014, Vol. 27, p,1075-1085). El modelo experimental de encefalomielitis autoinmunitaria se utiliza generalmente como modelo de esclerosis múltiple. El modelo de psoriasis inducida por imiquimod se utiliza generalmente como modelo de psoriasis.
[0124] El hecho de que el derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo sea eficaz para el tratamiento o prevención de enfermedades alérgicas se puede evaluar utilizando un modelo de enfermedad. Los ejemplos del modelo de enfermedad incluyen un modelo de dermatitis alérgica inducida por dinitrofluorobenceno (en adelante abreviado como DNFB) (Pharmacological Reports, 2013, Vol. 65, p. 1237-1246), un modelo de dermatitis atópica inducida por oxazolona (Journal of Investigative Dermatology, 2014, Vol. 134, p. 2122 2130), un modelo de rinitis alérgica inducida por ovoalbúmina (Journal of Animal Science, 2010, Vol. 81, p. 699-705), un modelo de conjuntivitis alérgica inducida por IgE (British Journal of Ophthalmology, 2012, Vol. 96, p. 1332-1336), un modelo de gastroenteritis alérgica (Gastroenterology, 1997, Vol. 113, p. 1560-1569), un modelo de asma inducida por ovoalbúmina (American Journal of Respiratory and Critical Care Medicine, 1997, Vol. 156, p.766-775) o un modelo de alergia alimentaria inducida por ovoalbúmina (Clinical & Experimental Allergy, 2005, Vol. 35, p.461-466). El modelo de dermatitis alérgica inducida por DNFB se utiliza generalmente como modelo de dermatitis alérgica, particularmente como modelo de dermatitis de contacto. El modelo de dermatitis atópica inducida por oxazolona se utiliza generalmente como modelo de dermatitis atópica.
[0125] La eficacia del derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo para el tratamiento o prevención de enfermedades autoinmunitarias o enfermedades alérgicas se puede evaluar utilizando el estudio in vitro mencionado anteriormente, por ejemplo, con disminución en la cantidad de unión entre un dominio de unión al ligando de RORy y un coactivador, o disminución en la cantidad de producción de IL-17, que es un índice de la función de RORy, como índice. La eficacia para el tratamiento o la prevención de la esclerosis múltiple se puede evaluar utilizando el modelo experimental de encefalomielitis autoinmunitaria mencionado anteriormente, por ejemplo, con disminución en la puntuación de síntomas neurológicos, que es un índice característico de esclerosis múltiple, como índice. La eficacia para el tratamiento o prevención de la psoriasis se puede evaluar utilizando el modelo de psoriasis inducida por imiquimod mencionado anteriormente, por ejemplo, con disminución en el grosor de la piel, tal como la aurícula, que aumenta con la progresión de los síntomas del modelo de psoriasis, como índice. La eficacia para el tratamiento o prevención de la dermatitis alérgica, particularmente la dermatitis de contacto, se puede evaluar utilizando el modelo de dermatitis alérgica inducida por DNFB mencionado anteriormente, por ejemplo, con disminución en el grosor de la piel, tal como la aurícula, que aumenta con la progresión de los síntomas de la dermatitis, como índice. La eficacia para el tratamiento o prevención de la dermatitis atópica se puede evaluar utilizando el modelo de dermatitis atópica inducida por oxazolona mencionado anteriormente, por ejemplo, con disminución en el grosor de la piel, tal como la aurícula, que aumenta con la progresión de los síntomas de dermatitis, como índice.
[0126] El derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo se puede utilizar como un medicamento útil (particularmente, un agente terapéutico o agente preventivo para una enfermedad autoinmunitaria o una enfermedad alérgica) cuando se administra a un mamífero (por ejemplo, ratón, rata, hámster, conejo, perro, mono, bovino, ovino o humano), particularmente humano. Cuando el derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo se utiliza clínicamente como medicamento, el derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo se puede utilizar por sí solo o se puede mezclar adecuadamente con aditivos, tales como excipientes, estabilizantes, conservantes, amortiguadores, solubilizantes, emulsionantes, diluyentes o agentes isotonizantes. El medicamento mencionado anteriormente se puede fabricar mediante un procedimiento habitual utilizando estos portadores de fármacos de manera adecuada. Los ejemplos de la forma de dosificación del medicamento mencionado anteriormente incluyen preparaciones orales, tales como comprimidos, cápsulas, gránulos, polvos o jarabes; preparaciones parenterales, tales como inhalantes, inyecciones, supositorios o líquidos; o ungüentos, cremas o parches para administración tópica. Además, se pueden utilizar preparaciones conocidas de acción prolongada.
[0127] El medicamento mencionado anteriormente contiene preferentemente del 0,00001 al 90% en peso, y más preferentemente del 0,01 al 70% en peso del derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo. La dosis se selecciona apropiadamente en función de los síntomas, la edad y el peso corporal del paciente, y el método de administración. Como cantidad de ingredientes eficaces para un adulto, son preferibles de 0,1 |jg a 1 g por día para inyecciones, de 1 jg a 10 g por día para preparaciones orales y de 1 jg a 10 g por día para parches, y cada uno se puede administrar una o varias veces en dosis divididas.
[0128] Los ejemplos del portador o diluyente farmacológicamente aceptable del medicamento mencionado anteriormente incluyen un aglutinante (jarabe, gelatina, goma arábiga, sorbitol, cloruro de polivinilo, tragacanto o similares), un excipiente (azúcar, lactosa, almidón de maíz, fosfato de calcio, sorbitol, glicina o similares) o un lubricante (estearato de magnesio, polietilenglicol, talco, sílice o similares).
[0129] Para complementar o mejorar el efecto terapéutico o preventivo o para reducir la dosificación, el medicamento se puede utilizar en mezcla o combinación con otros medicamentos en cantidades adecuadas.
[0130] La presente invención se describirá en más detalle mediante los siguientes Ejemplos y Ejemplos de referencia, pero la presente invención no está limitada a los mismos.
EJEMPLOS
[0131] Se utilizaron compuestos comercialmente disponibles para los compuestos utilizados en la síntesis de los compuestos de los Ejemplos y Ejemplos de referencia, sin mencionar el procedimiento de síntesis de los mismos. "Temperatura ambiente" en los siguientes Ejemplos y Ejemplos de referencia generalmente indica la temperatura en un intervalo de aproximadamente 10°C a aproximadamente 35 °C. El porcentaje (%) es mol/mol% de rendimiento, porcentaje en volumen para disolventes utilizados en cromatografía en columna y cromatografía líquida de alto rendimiento, y porcentaje en peso para otros, a menos que se especifique lo contrario. El nombre del disolvente que se muestra en los datos de RMN indica el disolvente utilizado para la medición. El espectro de RMN de 400 MHz se midió utilizando el espectrómetro de resonancia magnética nuclear JNM-AL 400 (JEOL Ltd.) o el espectrómetro de resonancia magnética nuclear JNM-ECS 400 (JEOL Ltd.). El cambio químico se indicó por 8 (unidad: ppm) con tetrametilsilano como estándar, las señales se indicaron por s (singlete), d (doblete), t (triplete), q (cuarteto), quint (quinteto), sept (septeto), m (multiplete), br (ancho), dd (doblete doble), dt (triplete doble), ddd (doble doble doblete), dq (doble cuarteto), td (doblete triplete) y tt (triplete triplete). Cuando los protones de un grupo hidroxilo, un grupo amino o similares tienen un pico muy suave, no se menciona. Los espectros ESI-MS se midieron utilizando Agilent Technologies 1200 Series, G6130A (Agilent Technologies). Se utilizó gel de sílice 60 (Merck) como gel de sílice, se utilizó gel de sílice de amina DM 1020 (Fuji Silysia Chemical Ltd.) como gel de sílice de amina y se utilizó YFLC W-prep2XY (Yamazen Corporation) como cromatografía.
(Ejemplo de referencia 1) Síntesis de
2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-amina:
[0132]

[0133] A una solución de acetonitrilo (9,0 mL) de ácido 2-trifluorometoxifenilborónico (1,10 g, 5,33 mmol), aducto de diclorometano de 4-bromo-3-cloroanilina (1,00 g, 4,84 mmol), carbonato de potasio (1,00 g, 7,27 mmol), 1,1'-bis(difenilfosfino)ferroceno diclorometano(II) (0,396 g, 0,484 mmol) y agua destilada (3,0 mL) se agregaron a temperatura ambiente y la temperatura se elevó a 90 °C, seguido de agitación durante 18 horas. La solución de reacción se filtró a través de gel de sílice y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 85/15 a 67/33) para obtener 2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-amina (en adelante, el compuesto del Ejemplo de referencia 1) (1,03 g, 3,57 mmol, 73,6%) como un producto oleoso amarillo.
1H-NMR (400MHz, CDCla) 8: 3,79(s, 2H), 6,62(dd, J=8,3, 2,3Hz, 1H), 6,80(d, J=2,3Hz, 1H), 7,05(d, J=8,3Hz, 1H), 7,30-7,41(m, 4H). ESI-MS:m/z = 288(M+H)+.
(Ejemplo de referencia 2) Síntesis de
2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil) piperidina-1-carboxilato de terc-butilo:
[0134]

[0135] A una solución de DMF (2,0 mL) de ácido 1-(terc-butoxicarbonil) piperidina-2-carboxílico (0,263 g, 1,15 mmol), se agregó una solución de DMF (2,0 mL) del compuesto del Ejemplo de referencia 1 (0,300 g, 1,04 mmol), HATU (0,436 g, 1,15 mmol) y diisopropiletilamina (0,273 mL, 1,56 mmol) a temperatura ambiente, seguido de agitación a la misma temperatura durante 16 horas. A la solución de reacción, se agregó agua destilada y la solución se extrajo con un disolvente mixto de n-hexano/acetato de etilo = 20/80(v/v). La capa orgánica se lavó con salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El
residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 90/10 a 67/33) para obtener 2-((2-doro-2'-(trifluor-ometoxi)-[1,1'-bifenil]-4-il)carbamoil) piperidina-1-carboxilato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 2) (0,483 g, 0,968 mmol, 92,8%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8:1,43-1,51(m, 2H), 1,53(s, 9H), 1,60-1,75(m, 3H), 2,35(d, J=12,7Hz, 1H), 2,80-2,89(m, 1H), 4,03-4,13(m, 1H), 4,86-4,89(m, 1H), 7,22(d, J=8,3Hz, 1H), 7,29-7,45(m, 6H), 7,80(br, 1H).
ESI-MS:m/z = 499(M+H)+.
(Ejemplo de referencia 3) Síntesis de
N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0136]

[0137] A una solución de diclorometano (5,0 mL) del compuesto del Ejemplo de referencia 2 (0,483 g, 0,968 mmol), se agregó ácido trifluoroacético (0,522 mL, 6,78 mmol) a temperatura ambiente, seguido de agitación a la misma temperatura durante 20 horas. La solución de reacción se concentró a presión reducida, se neutralizó con una solución acuosa de carbonato de potasio y a continuación, se extrajo con cloroformo. La capa orgánica se secó sobre sulfato de magnesio anhidro y se filtró y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice de amina, n-hexano/acetato de etilo = 60/40 a 20/80) para obtener N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo de referencia 3) (0,309 g, 0,775 mmol, 80,0%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,53(ddd, J=36,8, 17,9, 8,8Hz, 4H), 1,78-1,86(m, 1H), 2,00-2,07(m, 1H), 2,74-2,82(m, 1H), 3,03-3,10(m, 1H), 3,38(dd, J=9,6, 3,5Hz, 1H), 7,23(d, J=8,3Hz, 1H), 7,31-7,37(m, 3H), 7,40-7,45(m, 1H), 7,53(dd, J=8,3, 2,0Hz, 1H), 7,82(d, J=2,2Hz, 1H), 9,02(br, 1H).
ESI-MS:m/z = 399(M+H)+.
(Ejemplo 1) Síntesis de 1-acetil-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0138]
[0139] A una solución de diclorometano (3,0 mL) del compuesto del Ejemplo de referencia 3 (0,0700 g, 0,176 mmol), se agregaron trietilamina (0,0367 mL, 0,263 mmol) y anhídrido acético (0,0182 mL, 0,193 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 1 hora. A la solución de reacción, se añadió agua destilada y la solución se extrajo con cloroformo. La capa orgánica se lavó con agua destilada y salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, cloroformo/metanol = 95/5) para obtener 1-acetil-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 1) (0,0730 g, 0,166 mmol, 94,3%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,46-1,63(m, 1H), 1,67(d, J=7,8Hz, 1H), 1,89-2,02(m, 2H), 2,22(s, 3H), 2,29(d, J=12,9Hz, 1H), 3,22(t, J=13,2Hz, 1H), 3,78(d, J=12,7Hz, 1H), 5,29(d, J=5,1Hz, 1H), 7,20(d, J=8,3Hz, 1H), 7,29-7,37(m, 3H), 7,40-7,44(m, 2H), 7,80(br, 1H), 8,65(br, 1H).
ESI-MS:m/z = 441(M+H)+.
(Ejemplo 2) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1 -(2,2,2-trif luoroacetil)piperidina-2-carboxamida:
[0140]

[0141] Según el mismo procedimiento que en el ejemplo 1, salvo que se utilizó anhídrido trifluoroacético en lugar de anhídrido acético, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenM]-4-il)-1-(2,2,2-trif luoroacetil)piperidina-2-carboxamida (en adelante, el compuesto del ejemplo 2) (0,0500 g, 0,101 mmol, 99,0%) como un sólido blanco. 1H-NMR (400MHz, CDCls) 8: 1,56-1,86(m, 4H), 1,98(dt, J=11,2, 4,6Hz, 1H), 2,36(d, J=14,1Hz, 1H), 3,37(td, J=13,4, 2,6Hz, 1H), 4,01(d, J=13,9Hz, 1H), 5,18(d, J=5,4Hz, 1H), 7,21(d, J=8,3Hz, 1H), 7,30-7,46(m, 5H), 7,79(br, 1H), 7,89(br, 1H).
ESI-MS:m/z = 495(M+H)+.
(Ejemplo 3) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1 -propionil piperidina-2-carboxamida:
[0142]

[0143] A una solución de diclorometano (2,0 mL) del compuesto del Ejemplo de referencia 3 (0,0300 g, 0,0752 mmol), se agregaron trietilamina (0,0157 mL, 0,113 mmol) y cloruro de propionilo (0,00719 mL, 0,0828 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 30 minutos. A la mezcla de reacción, se agregó metanol, y la solución se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, cloroformo/metanol = 100/0 a 90/10) para obtener N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1 -pro-pionanil piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 3) (0,0340 g, 0,0747 mmol, 99,4%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,22(t, J=7,3Hz, 3H), 1,55(br, 2H), 1,76(br, 2H), 1,97(t, J=13,2Hz, 1H), 2,30(d, J=12,7Hz, 1H), 2,48(dq, J=6,6, 2,0Hz, 2H), 3,12(td, J=13,2, 2,8Hz, 1H), 3,83(d, J=13,2Hz, 1H), 5,29(d, J=5,4Hz, 1H), 7,20(d, J=8,3Hz, 1H), 7,29-7,36(m, 4H), 7,39-7,45(m, 1H), 7,84(br, 1H), 8,56(br, 1H).
ESI-MS:m/z = 455(M+H)+.
(Ejemplo 4) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-metoxiacetil)piperidina-2-carboxamida:
[0144]

[0145] A una solución de DMF (0,5 mL) de ácido 2-metoxiacético (0,00693 ml, 0,0903 mmol), se agregó una solución de DMF (0,5 mL) del compuesto del Ejemplo de referencia 3 (0,0300 g, 0,0752 mmol), Ha TU (0,0343 g, 0,0902 mmol) y diisopropiletilamina (0,0197 mL, 0,113 mmol) a temperatura ambiente, seguido de agitación a la misma temperatura durante 3 horas. A la solución de reacción, se agregó agua destilada y la solución se extrajo con un disolvente mixto de n-hexano/acetato de etilo = 20/80. La capa orgánica se lavó con salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 50/50 a 0/100) para obtener N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-metoxiacetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 4) (0,0266 g, 0,0565 mmol, 74,6%) como un sólido blanco.
1H-NMR (400MHz, CDCfa) 8: 1,58-2,00(m, 5H), 2,33(d, J=14,4Hz, 0,8H), 2,48(d, J=12,7Hz, 0,2H), 2,63(t, J=12,7Hz, 0,2H), 3,14(t, J=13,0Hz, 0,8H), 3,48(s, 2,4H), 3,51(s, 0,6H), 3,82(d, J=12,7Hz, 0,8H), 4,12(d, J=11,7Hz, 0,2H), 4,18(d, J=13,9Hz, 0,8H), 4,26(d, J=13,9Hz, 0,8H), 4,34(d, J=11,7Hz, 0,2H), 4,52-4,60(m, 0,2H), 4,64-4,68(m, 0,2H), 5,23(d, J=6,1Hz, 0,8H), 7,20(d, J=8,3Hz, 1H), 7,28-7,45(m, 5H), 7,65-7,90(m, 1H), 8,46(br, 0,8H), 8,57(br, 0,2H). ESI-MS:m/z = 471(M+H)+.
(Ejemplo 5) Síntesis de N-(2-cloro-2'(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-hidrox yacetil) piperidina-2-carboxamida:
[0146]

[0147] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido glicólico en lugar de ácido 2-metoxiacético, se obtuvo N-(2-doro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-hidroxiacetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 5) (0,0114 g, 0,0250 mmol, 33,2%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,48-1.,55(m, 1H), 1,63-1,71(m, 1H), 1,75-1,85(m, 2H), 1,91-2,02(m, 1H), 2,32(d, J=13,4Hz, 1H), 3,17-3,25(m, 1H), 3,43-3,53(m, 2H), 4,28-4,32(m, 2H), 5,26(d, J=5,6Hz, 1H), 7,22(d, J=8,3Hz, 1H), 7,30-7,45(m, 5H), 7,77(br, 1H), 8,14(br, 1H).
ESI-MS:m/z = 457(M+H)+.
(Ejemplo 6) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(dimetilamino)acetil)piperidina-2-carboxamida:
[0148]

[0149] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó clorhidrato de N,N-dimetilglicina en lugar de ácido 2-metoxiacético, se obtuvo N-(2-cloro-2'-(trifluorometoxiH1,1'-bifenil]-4-il)-1-(2-(dimetilamino)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 6) (0,0273 g, 0,0564 mmol, 90,0%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,40-2,20(m, 6H), 2,34(s, 3H), 2,47-2,50(m, 3,4H), 2,56-2,64(m, 0,4H), 2,92(d, J=12,6Hz, 0,6H), 3,06-3,14(m, 0,4H), 3,24(s, 0,6H), 3,67(d, J=12,6Hz, 0,6H), 4,03-4,07(m, 0,4H), 4,54-4,62(m, 1,2H), 5,24-5,27(m, 0,4H), 7,19-7,23(m, 1H), 7,30-7,46(m, 5H), 7,73-7,75(m, 1H), 8,53(br, 0,4H), 10,69(br, 0,6H).
ESI-MS:m/z = 484(M+H)+.
(Ejemplo 7) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2,2-difluoroacetil)piperidina-2-carboxamida:
[0150]

[0151] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido difluoroacético en lugar de ácido 2-metoxiacético, se obtuvo N-(2-cloro-2'-(trifluorometoxiH1,1'-bifenil]-4-il)-1-(2,2-difluoroacetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 7) (0,0212 g, 0,0444 mmol, 59,1%) como un sólido blanco. 1H-NMR (400MHz, DMSO-Da) 8: 1,40-1,80(m, 5,0H), 2,21-2,24(m, 0,7H), 2,29-2,34(m, 0,3H), 2,65-2,68(m, 0,3H), 3,46-3,55(m, 0,7H), 3,82-3,88(m, 0,7H), 4,28-4,34(m, 0,3H), 4,79-4,81(m, 0,3H), 5,07-5,10(m, 0,7H), 6,73(t, J=52,6Hz, 0,3H), 6,83(t, J=52,7Hz, 0,7H), 7,32(d, J=8,3Hz, 1H), 7,39-7,43(m, 1H), 7,45-7,51(m, 2H), 7,53-7,60(m, 2H), 7,90-7,93(m, 1H), 10,19(br, 0,3H), 10,25(br, 0,7H).
ESI-MS:m/z = 477(M+H)+.
(Ejemplo 8) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(trifluo rometoxi)acetil)piperidina-2-carboxamida:
[0152]

[0153] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 2-trifluorometoxiacético en lugar de ácido 2-metoxiacético, se obtuvo N-(2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)-1-(2-(trifluorometoxi)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 8) (0,00890 g, 0,0170 mmol, 16,9%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,55-1,71(m, 2H), 1,76-1,84(m, 2H), 1,94-2,03(m, 1H), 2,32(d, J=14,5Hz, 1H), 3,29(td, J=13,1,2,7Hz, 1H), 3,67(d, J=12,7Hz, 1H), 4,68-4,76(m, 2H), 5,22(d, J=5,4Hz, 1H), 7,21-7,45(m, 6H), 7,81(br, 1H), 8,26(s, 1H).
ESI-MS:m/z = 523(M-H)-.
(Ejemplo de referencia 4) Síntesis de (2-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-2-oxoetil)carbamato de terc-butilo:
[0154]

[0155] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 2-((tercbutoxicarbonil)amino)acético en lugar de ácido 2-metoxiacético, se obtuvo (2-(2-((2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)carbamoil)piperidina-1-il)-2-oxoetil)carbamato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 4) (0,116 g, 0,208 mmol, cuantitativo) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,46(s, 9H), 1,45-1,90(m, 5H), 2,38(d, J=13,2Hz, 1H), 3,21(t, J=12,1Hz, 1H), 3,75(d, J=13,9Hz, 1H), 3,95(dd, J=16,7, 5,0Hz, 1H), 4,09-4,15(m, 1H), 5,32(d, J=4,9Hz, 1H), 5,42(br, 1H), 7,21(d, J=8,3Hz, 1H), 7,29-7,36(m, 3H), 7,40-7,45(m, 1H), 7,52(br, 1H), 7,80(br, 1H), 8,31(br, 1H).
ESI-MS:m/z = 556(M+H)+.
(Ejemplo de referencia 5) Síntesis de 1-(2-aminoacetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil ]-4-il)piperidina-2-carboxamida:
[0156]

[0157] A una solución de diclorometano (1,0 mL) del compuesto del Ejemplo de referencia 4 (0,115 g, 0,207 mmol), se agregó ácido trifluoroacético (0,112 mL, 1,45 mmol) a temperatura ambiente, seguido de agitación a la misma temperatura durante 15 horas. La solución de reacción se concentró a presión reducida, se neutralizó con una solución acuosa de carbonato de potasio y a continuación, se extrajo con cloroformo. La capa orgánica se secó sobre sulfato de magnesio anhidro y se filtró y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice de amina, cloroformo/metanol = 100/0 a 96/4) para obtener 1-(2-aminoacetil)-N-(2-cloro-2'-(trifluor-ometoxi)-[1 ,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo de referencia 5) (0,0613 g, 0,134 mmol, 65,0%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,45-1,63(m, 2H), 1,70-1,81(m, 2H), 1,89-1,99(m, 1H), 2,31(d, J=14,0Hz, 1H), 3,16(td, J=14,0,2,3Hz, 1H), 3,60(d, J=1,0Hz, 2H), 3,68(d, J=14,0Hz, 1H), 5,27(d, J=5,4Hz, 1H), 7,21(d, J=8,3Hz, 1H), 7,30-7,45(m, 5H), 7,77(br, 1H), 8,39(br, 1H).
ESI-MS:m/z = 456(M+H)+.
(Ejemplo de referencia 6) Síntesis de (2-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-2-oxoetil)(metil)carbamato de terc-butilo:
[0158]
[0159] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó N-(terc-butoxicarbonil)-N-metilglicina en lugar de ácido 2-metoxiacético, se obtuvo (2-(2-((2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)carbamoil)piperidina-1-il)-2-oxoetil)(metil)carbamato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 6) (0,132 g, 0,232 mmol, 92,6%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,57(s, 9H), 1,38-1,80(m, 5H), 2,46-2,53(m, 1H), 3,05(s, 3H), 3,15-3,22(m, 1H), 3,66(d, J=15,7Hz, 1H), 3,77-3,84(m, 1H), 4,41(d, J=15,7Hz, 1H), 5,41-5,44(m, 1H), 7,19(d, J=8,3Hz, 1H), 7,28-7,44(m, 4H), 7,70(br, 1H), 7,88(br, 1H), 8,61(br, 1H).
ESI-MS:m/z = 571(M+H)+.
(Ejemplo 9) Síntesis de 1-(2-acetamideacetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0160]

[0161] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó el compuesto del Ejemplo de referencia 5 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de acetilo en lugar de cloruro de propionilo, se obtuvo 1-(2-acetamideacetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 9) (0,0274 g, 0,0550 mmol, 80,9%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,50-1,90(m, 5H), 2,09(s, 3H), 2,37(d, J=14,4Hz, 1H), 3,25(td, J=13,0,2,4Hz, 1H), 3,75(d, J=12,4Hz, 1H), 4,11(dd, J=17,2, 4,0Hz, 1H), 4,21(dd, J=17,2, 4,0Hz, 1H), 5,29(d, J=5,1Hz, 1H), 6,53(br, 1H), 7,21(d, J=8,5Hz, 1H), 7,29-7,37(m, 3H), 7,40-7,48(m, 2H), 7,80(br, 1H), 8,26(br, 1H).
ESI-MS:m/z = 498(M+H)+.
(Ejemplo 10) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(metilsulfonamida)acetil)piperidina-2-carboxamida:
[0162]

[0163] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó el compuesto del Ejemplo de referencia 5 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de metanosulfonilo en lugar de cloruro de propionilo, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(metilsulfonamida)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 10) (0,0202 g, 0,0378 mmol, 79,2%) como un sólido blanco. 1H-NMR (400MHz, CDCh) 8: 1,52-1,95(m, 5H), 2,32(d, J=14,1Hz, 1H), 3,02(s, 3H), 3,33(t, J=12,8Hz, 1H), 3,64(d, J=13,0Hz, 1H), 4,08(d, J=4,6Hz, 2H), 5,25(d, J=4,6Hz, 1H), 5,48(br, 1H), 7,22(d, J=8,3Hz, 1H), 7,29-7,45(m, 5H), 7,81(br, 1H), 8,09(br, 1H).
ESI-MS:m/z = 534(M+H)+.
(Ejemplo 11) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(metilamino)acetil)piperidina-2-carboxamida:
[0164]

[0165] Según el mismo procedimiento que en el Ejemplo de referencia 5, salvo que se utilizó el compuesto del Ejemplo de referencia 6 en lugar del compuesto del Ejemplo de referencia 4, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(metilamino)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 11) (0,0799 g, 0,170 mmol, 73,4%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,26-1,76(m, 5H), 1,90-2,06(m, 1H), 2,28-2,42(m, 1H), 2,50(s, 2,4H), 2,60-2,63(m, 0,8H), 3,15(t, J=12,2Hz, 0,8H), 3,43(d, J=12,7Hz, 0,2H), 3,52(s, 1,6H), 3,70-3,76(m, 1H), 4,58-4,63(m, 0,4H), 5,28(d, J=4,9Hz, 0,8H), 7,19-7,23(m, 1H), 7,30-7,45(m, 5H), 7,75-7,77(m, 1H), 8,44(s, 0,8H), 10,49(s, 0,2H).
ESI-MS:m/z = 470(M+H)+.
(Ejemplo 12) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(N-metilacetamida)acetil)piperidina-2-carboxamida:
[0166]

[0167] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó el compuesto del Ejemplo 11 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de acetilo en lugar de cloruro de propionilo, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(N-metilacetamida)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 12) (0,0261 g, 0,0510 mmol, 95,8%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,48-1,76(m, 5H), 2,20(s, 3H), 2,50-2,65(m, 1H), 3,24(s, 2,1H), 3,30(s, 0,9H), 3,20-3,31(m, 1H), 3,34(d, J=15,0Hz, 0,3H), 3,63(d, J=15,0Hz, 0,7H), 3,83-3,89(m, 0,7H), 4,60(d, J=15,0Hz, 0,7H), 4,64-4,70(m, 0,6H), 4,78(d, J=15,0Hz, 0,3H), 5,41(d, J=4,5Hz, 0,7H), 7,20-7,22(m, 1H), 7,30-7,35(m, 3H), 7,39-7,44(m, 1H), 7,60-7,75(m, 0,7H), 7,77(dd, J=8,4, 2,0Hz, 0,3H), 7,98-8,07(m, 0,7H), 8,14(d, J=1,8Hz, 0,3H), 8,64(br, 0,7H), 9,63(br, 0,3H). ESI-MS:m/z = 512(M+H)+.
(Ejemplo 13) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(N-metilmetilsulfonamida)acetil)piperidina-2-carboxamida:
[0168]

[0169] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó el compuesto del Ejemplo 11 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de metanosulfonilo en lugar de cloruro de propionilo, se obtuvo N-(2-cloro--2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(N-metilsulfonamida)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 13) (0,0264 g, 0,0482 mmol, 90,6%) como un sólido blanco. 1H-NMR (400MHz, CDCh) 8: 1,52-1,89(m, 5H), 2,35-2,38(m, 1H), 3,03-3,07(m, 6H), 3,20-3,31(m, 1H), 3,67-3,76(m, 1H), 4,16-4,27(m, 2H), 5,25-5,26(m, 1H), 7,21-7,23(m, 1H), 7,30-7,45(m, 5H), 7,83(s, 1H), 8,22(br, 1H).
ESI-MS:m/z = 548(M+H)+.
(Ejemplo 14) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(etilsulfonil)piperidina-2-carboxamida:
[0170]

[0171] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó cloruro de etanosulfonilo en lugar de cloruro de propionilo, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(etilsulfonil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 14) (0,0660 g, 0,134 mmol, 99,3%) como un sólido blanco. 1H-NMR (400MHz, CDCls) 8: 1,47(t, J=7,4Hz, 3H), 1,58-1,68(m, 2H), 1,69-1,84(m, 3H), 2,59(d, J=12,4Hz, 1H), 3,06-3,21(m, 4H), 3,88(d, J=12,0Hz, 1H), 4,56(d, J=8,3Hz, 1H), 7,24(d, J=8,3Hz, 1H), 7,31-7,38(m, 3H), 7,40-7,50(m, 2H), 7,85(s, 1H), 8,53(br, 1H).
ESI-MS:m/z = 491(M+H)+.
(Ejemplo 15) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(metilsulfonil)piperidina-2-carboxamida:
[0172]

[0173] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó cloruro de metanosulfonilo en lugar de cloruro de propionilo, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(metilsulfonil)pipendina-2-carboxamida (en adelante, el compuesto del Ejemplo 15) (0,0800 g, 0,168 mmol, 66,9%) como un sólido blanco. 1H-NMR (400MHz, CDCla) 8: 1,47-1,61(m, 1H), 1,62-1,81(m, 4H), 2,45(d, J = 10,4Hz, 1H), 3,04(s, 3H), 3,23(td, J=13,3, 2,4Hz, 1H), 3,93(t, J=7,0Hz, 1H), 4,64(br, 1H), 7,25(d, J=8,5Hz, 1H), 7,30-7,38(m, 3H), 7,43(dt, J=10,8, 3,7Hz, 2H), 7,84(d, J=2,2Hz, 1H), 8,29(br, 1H).
ESI-MS:m/z = 477(M+H)+.
(Ejemplo 16) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-formilpiperidina-2-carboxamida:
[0174]

[0175] A una solución de diclorometano (1,0 mL) del compuesto del Ejemplo de referencia 3 (0,0400 g, 0,100 mmol), se agregó formiato de etilo (0,567 mL, 7,02 mmol) a 0 °C y la temperatura se elevó a 90 °C, seguido de agitación durante 18 horas. La solución de reacción se concentró a presión reducida y el residuo se purificó mediante cromatografía en columna (gel de sílice, cloroformo/metanol = 100/0 a 90/10) para obtener N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bi-fenil]-4-il)-1-formilpiperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 16) (0,0300 g, 0,0703 mmol, 70,1%) como un sólido blanco.
1H-NMR (400MHz, CDCh) 8: 1,42-1,64(m, 2H), 1,82(d, J=10,0Hz, 2H), 1,95(dt, J=8,8, 4,3Hz, 1H), 2,35(d, J=13,9Hz, 1H), 3,29(td, J=13,2, 2,8Hz, 1H), 3,63(d, J=9,5Hz, 1H), 5,12(d, J=5,6Hz, 1H), 7,21(d, J=8,3Hz, 1H), 7,28-7,45(m, 5H), 7,80(br, 1H), 8,21(d, J=9,2Hz, 1H), 8,32(br, 1H).
ESI-MS:m/z = 427(M+H)+.
(Ejemplo 17) Síntesis de N2-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-1 ,2-dicarboxamida:
[0176]

[0177] A una solución de diclorometano (3,0 mL) del compuesto del Ejemplo de referencia 3 (0,100 g, 0,251
mmol), se agregaron isocianato de trimetilsililo (0,0333 mL, 0,251 mmol) y trietilamina (0,0349 mL, 0,251 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 72 horas. A la mezcla de reacción, se agregó metanol, y la solución se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, cloroformo) para obtener N2-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-1 ,2 dicarboxamida (en adelante, el compuesto del Ejemplo 17) (0,0300 g, 0,0679 mmol, 27,1%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,50-1,68(m, 2H), 1,73(br, 2H), 1,81-1,92(m, 1H), 2,30(d, J=12,9Hz, 1H), 3,21(dt, J=12,8, 2,6Hz, 1H), 3,52(d, J=13,2Hz, 1H), 4,81(br, 2H), 5,03(d, J=4,6Hz, 1H), 7,20(d, J=8,3Hz, 1H), 7,29-7,37(m, 3H), 7,42(dt, J=10,8, 3,8Hz, 2H), 7,81(br, 1H), 8,95(br, 1H).
ESI-MS:m/z = 442(M+H)+.
(Ejemplo 18) Síntesis de N2-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-N1,N1-dimetilpiperidina-1,2-dicarboxamida:
[0178]

[0179] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó cloruro de dimetilcarbamoilo en lugar de cloruro de propionilo, se obtuvo N2-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-N1,N 1-dimetilpiperidina-1,2-dicarboxamida (en adelante, el compuesto del Ejemplo 18) (0,0231 g, 0,0492 mmol, 65,4%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,48-1,78(m, 4H), 1,92-2,05(m, 1H), 2,27-2,35(m, 1H), 2,94(s, 6H), 2,87-2,99(m, 1H), 3,40-3,46(m, 1H), 4,47-4,51(m, 1H), 7,20(d, J=8,6Hz, 1H), 7,30-8,00(m, 6H), 10,60(br, 1H).
ESI-MS:m/z = 470(M+H)+.
(Ejemplo 19) Síntesis de 2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil) piperidina-1-carboxilato de metilo:
[0180]

[0181] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó cloroformiato de metilo como cloruro de propionilo, se obtuvo 2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil) piperidina-1-carboxilato de metilo (en adelante, el compuesto del Ejemplo 19) (0,0316 g, 0,0692 mmol, 92,0%) como un sólido blanco.
1H-NMR (400MHz, CDCh) 8: 1,46-1,78(m, 5H), 2,30-2,41(m, 1H), 2,92(t, J=12,1Hz, 1H), 3,81(s, 3H), 4,05-4,20(br, 1H), 4,93(d, J=4,6Hz, 1H), 7,23(d, J=8,5Hz, 1H), 7,31-7,45(m, 5H), 7,74-7,86(m, 1H), 8,21(br, 1H).
ESI-MS:m/z = 457(M+H)+.
(Ejemplo de referencia 7) Síntesis de (R)-2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-carboxilato de terc-butilo:
[0182]

[0183] A una solución de DMF (18 mL) de (R)-(+)-1-(terc-butoxicarbonil)piperidina-2-ácido carboxílico (0,840 g, 3,66 mmol), se agregó el compuesto del Ejemplo de referencia 1 (1,05 g, 3,66 mmol), HATU (1,53 g, 4,03 mmol) y diisopropiletilamina (0,768 mL, 4,40 mmol) a temperatura ambiente, seguido de agitación a la misma temperatura durante 18 horas. A la solución de reacción, se añadió agua destilada y la solución se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, nhexano/acetato de etilo = 80/20) para obtener (R)-2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-carboxilato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 7) (1,60 g, 3,20 mmol, 87,3%) como un sólido blanco.
1H-NMR (400MHz, CDCI3) 8:1,43-1,51(m, 2H), 1,53(s, 9H), 1,60-1,75(m, 3H), 2,35(d, J=12,7Hz, 1H), 2,80-2,89(m, 1H), 4,03-4,13(m, 1H), 4,86-4,89(m, 1H), 7,22(d, J=8,3Hz, 1H), 7,29-7,45(m, 6H), 7,80(br, 1H).
ESI-MS:m/z = 499(M+H)+.
(Ejemplo de referencia 8) Síntesis de (R)-N-(2-doro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina e-2-carboxamida:
[0184]

[0185] A una solución de diclorometano (30 mL) del compuesto del Ejemplo de referencia 7 (1,60 g, 3,21 mmol), se agregó ácido trifluoroacético (8,02 mL, 104 mmol) a temperatura ambiente, seguido de agitación a la misma temperatura durante 2 horas. A la solución de reacción, se añadió agua destilada y la solución se extrajo con cloroformo. La capa acuosa se neutralizó mediante la adición de una solución acuosa de hidróxido de sodio 1m y a continuación, se extrajo con cloroformo. La capa orgánica se lavó con agua destilada y salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida para obtener (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina e-2-carboxamida (en adelante, el compuesto del Ejemplo de referencia 8) (1,13 g, 2,84 mmol, 88,6%) como un sólido blanco. 1H-NMR (400MHz, CDCh) 8: 1,53(ddd, J=36,8,17,9,8,8Hz, 4H), 1,78-1,86(m, 1H), 2,00-2,07(m, 1H), 2,74-2,82(m, 1H), 3,03-3,10(m, 1H), 3,38(dd, J=9,6, 3,5Hz, 1H), 7,23(d, J=8,3Hz, 1H), 7,31-7,37(m, 3H), 7,40-7,45(m, 1H), 7,53(dd, J=8,3, 2,0Hz, 1H), 7,82(d, J=2,2Hz, 1H), 9,02(br, 1H).
ESI-MS:m/z = 399(M+H)+.
(Ejemplo 20) Síntesis de (R)-1-acetil-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0186]

[0187] A una solución de diclorometano (36 mL) del compuesto del Ejemplo de referencia 8 (1,43 g, 3,59 mmol), se agregaron trietilamina (0,750 mL, 5,38 mmol) y anhídrido acético (0,338 mL, 3,59 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 30 minutos. A la solución de reacción, se añadió agua destilada y la solución se extrajo con cloroformo. La capa orgánica se lavó con agua destilada y salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, cloroformo/metanol = 95/5) para obtener (R)-1-acetil-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 20) (1,02 g, 2,32 mmol, 64,6%) como un sólido blanco. Como resultado del análisis mediante columna quiral, el tiempo de retención del compuesto del Ejemplo 20 obtenido de este modo fue de 32,8 minutos, y la pureza óptica en ese momento fue de 99,0%ee. Las condiciones de análisis utilizando la columna quiral son las siguientes.
Equipos de medición; Cromatógrafo líquido de alto rendimiento LC-2010CHT, fabricado por Shimadzu Corporation Column; CHIRALCEL OD-RH 0,46 cmcp x 15 cm, tamaño de partícula de 5 pm, fabricado por Daicel Chemical Industries Ltd. Temperatura de columna; 40 °C
Fase móvil; (Solución A) solución acuosa de dihidrógenofosfato de potasio 20 mM, (Solución B) acetonitrilo Composición de fase móvil; Solución A:Solución B = 60:40 a 50: 50 (0 a 40 minutos, gradiente lineal)
Solución A:Solución B = 50:50 a 60: 40 (40 a 41 minutos, gradiente lineal)
Solución A:Solución B = 60:40 (41 a 50 minutos)
Caudal; 0,5 ml/minuto;
Detección; UV (210 nm)
1H-NMR (400MHz, CDCla) 8: 1,46-1,63(m, 1H), 1,67(d, J=7,8Hz, 1H), 1,89-2,02(m, 2H), 2,22(s, 3H), 2,29(d, J=12,9Hz, 1H), 3,22(t, J=13,2Hz, 1H), 3,78(d, J=12,7Hz, 1H), 5,29(d, J=5,1Hz, 1H), 7,20(d, J=8,3Hz, 1H), 7,29-7,37(m, 3H), 7,40-7,44(m, 2H), 7,80(br, 1H), 8,65(br, 1H).
ESI-MS:m/z = 441(M+H)+.
(Ejemplo 21) Síntesis de (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1 -(metilsulfonil) piperidina-2-carboxamida:
[0188]

[0189] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de metanosulfonilo en lugar de cloruro de propionilo, se obtuvo (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(metilsulfonil)pipendina-2-carboxamida (en adelante, el compuesto del Ejemplo 21) (0,0600 g, 0,126 mmol, 99,0%) como un sólido blanco. 1H-NMR (400MHz, CDCls) 8: 1,47-1,61(m, 1H), 1,62-1,81(m, 4H), 2,45(d, J=10,4Hz, 1H), 3,04(s, 3H), 3,23(td, J=13,3, 2,4Hz, 1H), 3,93(t, J=7,0Hz, 1H), 4,64(br, 1H), 7,25(d, J=8,5Hz, 1H), 7,30-7,38(m, 3H), 7,43(dt, J=10,8, 3,7Hz, 2H), 7,84 (d, J=2,2Hz, 1H), 8,29(br, 1H).
ESI-MS:m/z = 477(M+H)+.
(Ejemplo 22) Síntesis de (R)-N2-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-1,2-dicarboxamida:
[0190]
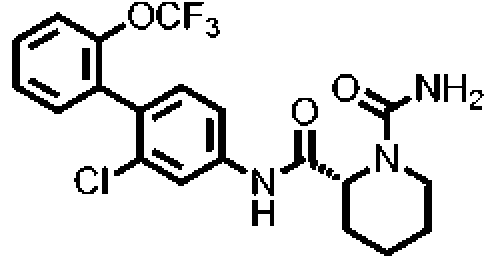
[0191] A una solución de diclorometano (30 mL) del compuesto del Ejemplo de referencia 8 (3,00 g, 7,52 mmol), se agregaron isocianato de trimetilsililo (2,00 mL, 15,04 mmol) y trietilamina (1,05 mL, 7,57 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 18 horas. A la mezcla de reacción, se agregó metanol, y la solución se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, cloroformo) para obtener (R)-N2-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-1,2-dicarboxamida (en adelante, el compuesto del Ejemplo 22) (2,50 g, 5,66 mmol, 75,2%) como un sólido blanco. 1H-NMR (400MHz, CDCla) 8: 1,50-1,68(m, 2H), 1,73(br, 2H), 1,81-1,92(m, 1H), 2,30(d, J=12,9Hz, 1H), 3,21(dt, J=12,8, 2,6Hz, 1H), 3,52(d, J=13,2Hz, 1H), 4,81(br, 2H), 5,03(d, J=4,6Hz, 1H), 7,20(d, J=8,3Hz, 1H), 7,29-7,37(m, 3H), 7,42(dt, J=10,8, 3,8Hz, 2H), 7,81(br, 1H), 8,95(br, 1H).
ESI-MS:m/z = 442(M+H)+.
(Ejemplo 23) Síntesis de (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2,2; 2-trifluoroacetil)piperidina-2-carboxamida:
[0192]

[0193] A una solución de diclorometano (75 mL) del compuesto del Ejemplo de referencia 8 (3,00 g, 7,52 mmol), se agregaron trietilamina (1,57 mL, 11,28 mmol) y anhídrido trifluoroacético (1,17 mL, 8,27 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 30 minutos. A la solución de reacción, se añadió agua destilada y la solución se extrajo con cloroformo. La capa orgánica se lavó con agua destilada y salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 20/80) para obtener (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2,2, 2-trifluoroacetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 23) (2,50 g, 5,05 mmol, 67,2%) como un sólido blanco. Como resultado del análisis mediante columna quiral, el tiempo de retención del compuesto del Ejemplo 23 obtenido de este modo fue de 33,6 minutos, y la pureza óptica en ese momento fue de 95,0%ee. Las condiciones de análisis utilizando la columna quiral son las siguientes. Equipos de medición; Cromatógrafo líquido de alto rendimiento LC-2010CHT, fabricado por Shimadzu Corporation
Columna; CHIRALCEL OD-RH 0,46 cmcp x 15 cm, tamaño de partícula de 5 pm, fabricado por Daicel Chemical Industries Ltd. Temperatura de columna; 40°C
Fase Móvil: (Solución A) solución acuosa de dihidrógenofosfato de potasio 20 mM, (Solución B) acetonitrilo Composición de fase móvil; Solución A:Solución B = 60:40 a 50:50 (0 a 40 minutos, gradiente lineal)
Solución A:Solución B = 50:50 a 60:40 (40 a 41 minutos, gradiente lineal)
Solución A:Solución B = 60:40 (41 a 50 minutos)
Caudal; 0,5 ml/minuto
Detección; UV (210 nm)
1H-NMR (400MHz, CDCls) 8: 1,56-1,86(m, 4H), 1,98(dt, J=11,2, 4,6Hz, 1H), 2,36(d, J=14,1Hz, 1H), 3,37(td, J=13,4, 2,6Hz, 1H), 4,01(d, J=13,9Hz, 1H), 5,18(d, J=5,4Hz, 1H), 7,21(d, J=8,3Hz, 1H), 7,30-7,46(m, 5H), 7,79(br, 1H), 7,89(br, 1H).
ESI-MS:m/z = 495(M+H)+.
(Ejemplo de referencia 9) Síntesis de (1R,5S)-2-((R)-1-feniletil)-6-oxa-2-azabiciclo[3,2,1]octano-7-o na:
[0194]

[0195] A una solución de DMF (30 mL) de (R)-a-metilbencilamina (3,77 mL, 29,6 mmol), se agregaron carbonato de potasio (4,09 g, 29,6 mmol) y 4-bromo-1-buteno (3,01 mL, 29,6 mmol) a temperatura ambiente, seguido de agitación a la misma temperatura durante 24 horas. A la solución de reacción, se añadió agua destilada y la solución se extrajo con éter dietílico. La capa orgánica se lavó con salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. A una solución de tetrahidrofurano (12 mL) del residuo, se añadió ácido glioxílico (4,09 mL, 36,8 mmol) a 0 °C y se elevó la temperatura a 60 °C, seguido de agitación durante 9 horas. A la solución de reacción, se añadió agua destilada y una solución acuosa de hidróxido de sodio 1M, y la solución se extrajo con acetato de etilo. La capa orgánica se lavó con una solución acuosa saturada de carbonato de hidrógeno de sodio, agua destilada y salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 91/9 a 85/15) para obtener (1R,5S)-2-((R)-1-feniletil)-6-oxa-2-azabiciclo[3,2,1]octano-7-o ona (en adelante, el compuesto del Ejemplo de referencia 9) (1,73 g, 7,48 mmol, 25,3%) como un producto oleoso amarillo pálido.
1H-NMR (400MHz, CDCla) 8: 1,33(d, J=6,6Hz, 3H), 1,82(d, J=11,7Hz, 1H), 1,87-1,95(m, 1H), 2,03-2,12(m, 2H), 2,47(td, J=11,8, 5,1Hz, 1H), 3,19(d, J=5,1Hz, 1H), 3,35 (dd, J=12,0, 6,6Hz, 1H), 3,70(q, J=6,6Hz, 1H), 4,78 (t, J=5,1Hz, 1H), 7,23-7,27(m, 1H), 7,31-7,35(m, 2H), 7,39-7,41(m, 2H).
ESI-MS:m/z = 232(M+H)+.
(Ejemplo de referencia 10) Síntesis de (1R,5S)-2-(terc-butoxicarbonil)-6-oxa-2-azabiciclo[3,2,1]-octano-7 -ona:
[0196]

[0197] A una solución de acetato de etilo (25 mL) del compuesto del Ejemplo de referencia 9 (1,73 g, 7,48 mmol), se agregaron un 20% en peso de hidróxido de paladio carbono (que contenía un 50% en peso de agua, 0,210 g) y dicarbonato de di-terc-butilo (1,80 g, 8,23 mmol) a temperatura ambiente, seguido de agitación en una atmósfera de hidrógeno a la misma temperatura durante 36 horas. La mezcla de reacción se filtró a través de una almohadilla Celite y a continuación, el filtrado se concentró a presión reducida. El residuo se suspendió en éter dietílico/n-hexano = 1/9(v/v) y el sólido obtenido se recogió mediante filtración y se secó para obtener (1R,5S)-2-(terc-butoxicarbonil)-6-oxa-2-azabiciclo[3,2,1]-octano-7-ona (en adelante, el compuesto del Ejemplo de referencia 10) (1,63 g, 7,17 mmol, 95,9%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,48(s, 9H), 1,84-1,93(m, 1H), 1,95(d, J=12,0Hz, 1H), 2,03-2,06(m, 1H), 2,29-2,32(m, 1H), 3,18-3,21(m, 1H), 4,06(m, 1H), 4,70-4,85(m, 1H), 4,97(t, J=5,1Hz, 1H). ESI-MS:m/z = 228(M+H)+.
(Ejemplo de referencia 11) Síntesis de (2R,4S)-2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)-4-hidroxipiperidina-1-carboxilato de terc-butilo:
[0198]

[0199] A una solución de tolueno (2,3 mL) del compuesto del Ejemplo de referencia 10 (0,320 g, 1,41 mmol), se agregó una solución de trimetilalumino-tolueno (1,4 M, 1,31 mL, 1,83 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 30 minutos. Se agregó una solución de tolueno (2,3 mL) del compuesto del Ejemplo de referencia 1 (0,486 g, 1,690 mmol) y se elevó la temperatura a 50 °C, seguido de agitación durante 4 horas. A la solución de reacción, se le añadió ácido clorhídrico 1M y la solución se extrajo con acetato de etilo. La capa orgánica se lavó con agua destilada y salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 80/20 a 50/50) para obtener (2R,4S)-2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)-4-hidroxipiperidina-1-carboxilato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 11) (0,654 g, 1,27 mmol, 90,2%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,54(s, 9H), 1,66-1,69(m, 1H), 1,78-1,82(m, 1H), 1,93-2,00(m, 1H), 2,40(d, J=13,4Hz, 1H), 3,25(td, J=13,2, 2,4Hz, 1H), 3,86-3,88(m, 1H), 4,12-4,14(m, 1H), 4,98-5,00(m, 1H), 5,20(br, 1H), 7,24 (d, J=8,3Hz, 1H), 7,30-7,46(m, 5H), 7,75-7,78(m, 1H), 9,08(br, 1H).
ESI-MS:m/z = 515(M+H)+.
(Ejemplo de referencia 12) Síntesis de clorhidrato de (2R,4S)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-4-hidroxipiperidina-2-carboxamida:
[0200]

[0201] A una solución de acetato de etilo (0,5 mL) del compuesto del Ejemplo de referencia 11 (0,0500 g, 0,0973 mmol), se agregó una solución de cloruro de hidrógeno-acetato de etilo (4,0 M, 0,486 mL, 1,94 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 3 horas. La solución de reacción se filtró y el sólido obtenido recogido por filtración se lavó con acetato de etilo y a continuación, se secó para obtener clorhidrato de (2R,4S)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bi-fenil]-4-il)-4-hidroxipiperidina-2-carboxamida (en adelante, el compuesto del Ejemplo de referencia 12) (0,0409 g, 0,0908 mmol, 93,3%) como un sólido blanco.
1H-NMR(DMSO-D6) 8: 1,46-1,62(m, 2H), 1,91-1,94(m, 1H), 2,42-2,45(m, 1H), 3,01(t, J=12,2Hz, 1H), 3,28-3,32(m, 1H), 3,69-3,78(m, 1H), 4,00(d, J=12,0Hz, 1H), 5,28 (d, J=4,9Hz, 1H), 7,38 (d, J=8,3Hz, 1H), 7,42(dd, J=7,8, 1,7Hz, 1H), 7,48-7,52(m, 2H), 7,56-7,64(m, 2H), 7,94(s, 1H), 8,93(br, 1H), 11,00(s, 1H). ESI-MS:m/z = 415(M+H)+.
(Ejemplo de referencia 13) Síntesis de (2R,4R)-2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)-4-fluoropiperidina-1-carboxilato de terc-butilo:
[0202]

[0203] A una solución de diclorometano (1,9 mL) del compuesto del Ejemplo de referencia 11 (0,100 g, 0,194 mmol), se agregó trifluoruro de (dietilamino)azufre (0,0380 mL, 0,291 mmol) a -78 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 24 horas. A la solución de reacción, se añadió una solución acuosa saturada de carbonato de hidrógeno de sodio, y la solución se extrajo con cloroformo. La capa orgánica se lavó con salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró
a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 90/10 a 80/20) para obtener (2R,4R)-2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)-4-fluoropipendina-1-carboxilato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 13) (0,0272 g, 0,0527 mmol, 27,2%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,48-1,56(m, 1H), 1,54(s, 9H), 1,59-1,86(m, 2H), 2,07-2,19(m, 1H), 2,65-2,71(m, 1H), 2,93(t, J=12,8Hz, 1H), 4,10-4,13(m, 1H), 5,04-5,06(m, 1H), 7,22(d, J=8,3Hz, 1H), 7,29-7,36(m, 4H), 7,41-7,45(m, 1H), 7,76(br, 1H). ESI-MS:m/z = 517(M+H)+.
(Ejemplo de referencia 14) Síntesis de clorhidrato de (2R,4R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-4-fluoropiperidina-2-carboxamida:
[0204 ]

[0205 ] Según el mismo procedimiento que en el Ejemplo 12, salvo que se utilizó el compuesto del Ejemplo de referencia 13 en lugar del compuesto del Ejemplo de referencia 11, se obtuvo clorhidrato de (2R,4R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-4-fluoropiperidina-2-carboxamida (en adelante, el compuesto del Ejemplo de referencia 14) (0,0186 g, 0,0410 mmol, 84,9%) como un sólido blanco.
1H-NMR(DMSO-Da) 8: 1,89-2,06(m, 2H), 2,24-2,33(m, 1H), 3,12-3,27(m, 2H), 4,18(d, J=12,4Hz, 1H), 5,16(d, J=47,1Hz, 1H), 7,38(d, J=8,5Hz, 1H), 7,42(dd, J=7,9, 1,8Hz, 1H), 7,48-7,52(m, 2H), 7,56-7,61(m, 2H), 7,95(br, 1H), 9,12(br, 1H), 10,97(s, 1H). ESI-MS:m/z = 417(M+H)+.
(Ejemplo de referencia 15) Síntesis de (2R,4R)-2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)-4-(formiloxi)piperidina-1-carboxilato de terc-butilo:
[0206 ]
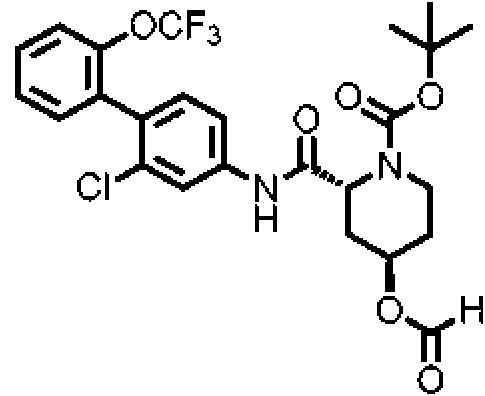
[0207 ] A una solución de tetrahidrofurano (1,0 mL) de trifenilfosfina (0,153 g, 0,583 mmol), se agregó azodicarboxilato de diisopropilo (0,113 mL, 0,583 mmol) a 0 °C y, después de agitar a la misma temperatura durante 1 hora, se agregó ácido fórmico (0,0220 mL, 0,583 mmol) y la mezcla se agitó a la misma temperatura durante 30 minutos. Se agregó una solución de tetrahidrofurano (1,00 mL) del compuesto del Ejemplo de referencia 11 (0,200 g, 0,388 mmol) gota a gota y se elevó la temperatura a temperatura ambiente, seguido de agitación durante 12 horas. La solución de reacción se concentró a presión reducida y el residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 85/15 a 70/30) para obtener (2R,4R)-2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)-4-(formi-loxi)piperidina-1-carboxilato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 15) (0,0939 g, 0,173 mmol, 44,5%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,28-1,37(m, 2H), 1,54(s, 9H), 1,67-1,74(m, 1H), 2,07-2,12(m, 1H), 2,61-2,63(m, 1H), 2,96-3,02(m, 1H), 4,12-4,14(m, 1H), 5,06(br, 1H), 5,43(br, 1H), 7,22(d, J=8,3Hz, 1H), 7,32-7,37(m, 3H), 7,41-7,45(m, 2H), 7,80(br, 1H), 8,06(s, 1H).
ESI-MS:m/z = 543(M+H)+.
(Ejemplo de referencia 16) Síntesis de (2R,4R)-2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)-4-hidroxipiperidina-1-carboxilato de terc-butilo:
[0208 ]

[0209 ] A una solución de metanol (1,1 mL) del compuesto del Ejemplo de referencia 15 (0,0900 g, 0,166 mmol), se agregó una solución de metóxido de sodio-metanol (4,0 M, 0,0207 mL, 0,0828 mmol) a 0 °C, seguido de agitación a la misma temperatura durante 15 minutos. A la solución de reacción, se le añadió ácido clorhídrico 1M y la solución se extrajo con acetato de etilo. La capa orgánica se lavó con salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 80/20 a 50/50) para obtener (2R,4R)-2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)-4-hidroxipiperidina-1-carboxilato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 16) (0,0844 g, 0,164 mmol, 99,3%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,46-1,54(m, 2H), 1,54(s, 9H), 1,68-1,71(m, 1H), 1,94-1,97(m, 1H), 2,54-2,56(m, 1H), 2,86-2,93(m, 1H), 4,13-4,22(m, 2H), 5,04(br, 1H), 7,22(d, J=8,3Hz, 1H), 7,30-7,45(m, 5H), 7,78(br, 1H), 8,52(s, 1H). ESI-MS:m/z = 515(M+H)+.
(Ejemplo de referencia 17) Síntesis de (R)-2-((2-cloro-2'-(trifluorometoxiH1,1'-bifenil]-4-il)carbamoil)-4-oxopiperidina-1-carboxilato de terc-butilo:
[0210]
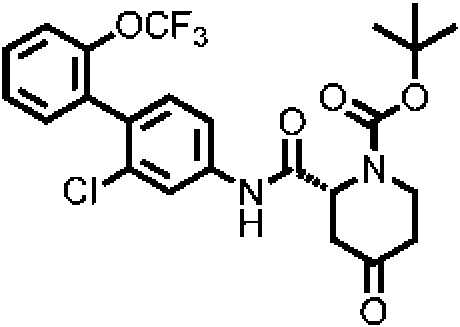
[0211 ] A una solución de diclorometano (2,0 mL) del compuesto del Ejemplo de referencia 11 (0,210 g, 0,408 mmol), se agregó periodinano Dess-Martin (0,190 g, 0,449 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 3 horas. A la solución de reacción, se añadió una solución acuosa de tiosulfato de sodio y la solución se extrajo con acetato de etilo. La capa orgánica se lavó con agua destilada y salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 90/10 a 60/40) para obtener (R)-2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)-4-oxopiperidina-1-carboxilato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 17) (0,190 g, 0,370 mmol, 90,9%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,56(s, 9H), 2,46-2,53(m, 1H), 2,61-2,73(m, 2H), 3,00(dd, J=16,5,3,3Hz, 1H), 3,66 3,73(m, 1H), 3,82(br, 1H), 5,05(s, 1H), 7,22(d, J=8,3Hz, 1H), 7,29-7,36(m, 3H), 7,41-7,45(m, 2H), 7,78(br, 1H), 9,14(br, 1H). ESI-MS:m/z = 513(M+H)+.
(Ejemplo de referencia 18) Síntesis de (R)-2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)-4,4-difluoropiperidina-1-carboxilato de terc-butilo:
[0212]

[0213 ] A una solución de diclorometano (1,9 mL) del compuesto del Ejemplo de referencia 17 (0,190 g, 0,370 mmol), se agregó trifluoruro de (dietilamino)azufre (0,108 mL, 0,815 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 24 horas. A la solución de reacción, se añadió una solución acuosa saturada de carbonato de hidrógeno de sodio, y la solución se extrajo con cloroformo. La capa orgánica se lavó con salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo
= 85/15 a 70/30) para obtener (R)-2-((2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)carbamoil)-4,4-difluoropiperidina-1-carboxilato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 18) (0,0393 g, 0,0735 mmol, 19,8%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,54(s, 9H), 1,86-2,15(m, 3H), 2,99-3,07(m, 1H), 3,21(td, J=13,3,2,7Hz, 1H), 4,22-4,25(m, 1H), 5,07(br, 1H), 7,24(d, J=8,5Hz, 1H), 7,30-7,37(m, 3H), 7,41-7,46(m, 2H), 7,76(s, 1H), 7,97(br, 1H).
ESI-MS:m/z = 535(M+H)+.
(Ejemplo 24) Síntesis de (2R,4S)-1-acetil-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-4-hidroxipiperidina-2-carboxamida:
[0214 ]

[0215 ] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó el compuesto del Ejemplo de referencia 12 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de acetilo en lugar de cloruro de propionilo, se obtuvo (2R,4S)-1-acetil-N-(2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)-4-hidroxipiperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 24) (0,0166 g, 0,0363 mmol, 91,1%) como un sólido blanco. 1H-NMR (400MHz, CDCla) 8: 1,68-1,76(m, 1H), 1,87-1,98(m, 2H), 2,24(s, 3H), 2,39(d, J=14,6Hz, 1H), 3,50-3,57(m, 1H), 3,61-3,66(m, 1H), 4,14-4,17(m, 1H), 5,42 (d, J=6,8Hz, 1H), 5,52(d, J=6,8Hz, 1H), 7,23(d, J=8,5Hz, 1H), 7,31 7,37(m, 3H), 7,41-7,46(m, 2H), 7,77(br, 1H), 9,15(s, 1H).
ESI-MS:m/z = 457(M+H)+.
(Ejemplo 25) Síntesis de (2R,4R)-1-acetil-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-4-fluoropiperidina-2-carboxamida:
[0216 ]

[0217 ] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó el compuesto del Ejemplo de referencia 14 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de acetilo en lugar de cloruro de propionilo, se obtuvo (2R,4R)-1-acetil-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-4-fluoropiperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 25) (0,0108 g, 0,0235 mmol, 62,8%) como un sólido blanco.
1H-NMR (400MHz, CDCh) 8: 1,69-1,85(m, 2H), 2,22-2,25(m, 1H), 2,25(s, 3H), 2,62-2,69(m, 1H), 3,21-3,28(m, 1H), 3,85(dd, J=13,4,2,9Hz, 1H), 5,24-5,43(m, 1H), 5,43 (d, J=5,4Hz, 1H), 7,21(d, J=8,3Hz, 1H), 7,30-7,36(m, 3H), 7,41 7,45(m, 2H), 7,70-7,82(m, 1H), 8,72(s, 1H).
ESI-MS:m/z = 459(M+H)+.
(Ejemplo 26) Síntesis de (2R,4S)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-4-hidroxi-1-(metilsulfo-nil)piperidina-2-carboxamida:
[0218 ]

[0219 ] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó el compuesto del Ejemplo de referencia 12 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de metanosulfonilo en lugar de
cloruro de propionilo, se obtuvo (2R,4S)-N-(2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)-4-hidroxi-1-(metilsulfonil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 26) (0,00841 g, 0,0171 mmol, 42,7%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,68-1,76(m, 1H), 1,85-1,88(m, 1H), 1,99(ddd, J=14,9,6,9,3,1Hz, 1H), 2,61(d, J=14,9Hz, 1H), 3,03(s, 3H), 3,55-3,62(m, 1H), 3,72(d, J=4,6Hz, 1H), 3,75-3,79(m, 1H), 4,22-4,24(m, 1H), 4,66(d, J=6,6Hz, 1H), 7,25-7,27(m, 1H), 7,31-7,37(m, 3H), 7,42-7,46(m, 2H), 7,81(d, J=2,0Hz, 1H), 8,58(s, 1H).
ESI-MS:m/z = 493(M+H)+.
(Ejemplo 27) Síntesis de (2R,4R)-1-acetil-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-4-hidroxipiperidina-2-carboxamida:
[0220]

[0221 ] A una solución de acetato de etilo (0,4 mL) del compuesto del Ejemplo de referencia 16 (0,0200 g, 0,0388 mmol), se agregó una solución de cloruro de hidrógeno-acetato de etilo (4,0 M, 0,194 mL, 1,94 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 3 horas. La solución de reacción se concentró a presión reducida y el residuo se disolvió en diclorometano (0,8 mL) y a continuación, se agregaron trietilamina (0,00135 mL, 0,0970 mmol) y cloruro de acetilo (0,00359 mL, 0,0504 mmol) a 0 °C, seguido de agitación a la misma temperatura durante 1 hora. A la mezcla de reacción, se agregó metanol, y la solución se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, acetato de etilo/metanol = 100/0 a 97/3) para obtener (2R,4R)-1-acetil-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-4-hidroxipiperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 27) (0,0106 g, 0,0232 mmol, 59,7%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,47-1,57(m, 2H), 1,81(d, J=3,6Hz, 1H), 2,04-2,09(m, 1H), 2,25(s, 3H), 2,50(ddt, J=13,1, 5,0, 1,8Hz, 1H), 3,22(td, J=13,4, 2,6Hz, 1H), 3,85(d, J=13,4Hz, 1H), 4,40-4,48(m, 1H), 5,43(d, J=5,9Hz, 1H), 7,21(d, J=8,2Hz, 1H), 7,30-7,37(m, 3H), 7,41-7,45(m, 2H), 7,69-7,83(m, 1H), 8,65(s, 1H).
ESI-MS:m/z = 457(M+H)+.
(Ejemplo 28) Síntesis de (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-4,4-difl uoro-1-(metilsulfonil)piperidina-2-carboxamida:
[0222]

[0223 ] Según el mismo procedimiento que en el Ejemplo 27, salvo que se utilizó el compuesto del Ejemplo de referencia 18 en lugar del compuesto del Ejemplo de referencia 16 y se utilizó cloruro de metanosulfonilo en lugar de cloruro de acetilo, se obtuvo (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-4,4-difl uoro-1-(metilsulfonil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 28) (0,0127 g, 0,0248 mmol, 77,0%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,95-2,12(m, 1H), 2,15-2,32(m, 2H), 2,95-3,02(m, 1H), 3,10(s, 3H), 3,60(td, J=13,5, 3,0Hz, 1H), 4,02-4,08(m, 1H), 4,89(d, J=7,2Hz, 1H), 7,26(d, J=8,2Hz, 1H), 7,30-7,37(m, 3H), 7,42-7,46(m, 2H), 7,76(s, 1H), 7,93 (s, 1H). ESI-MS:m/z = 513(M+H)+.
(Ejemplo de referencia 19) Síntesis de 2-(N-metilsulfonamida)acetato de terc-butilo:
[0224 ]

[0225 ] A una solución de diclorometano (2,0 mL) de clorhidrato de 2-(metilamino)acetato de terc-butilo (0,100 g, 0,550 mmol), se agregaron trietilamina (0,192 mL, 1,385 mmol) y cloruro de metanosulfonilo (0,0515 mL, 0,661
mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 3 horas. A la solución de reacción, se añadió una solución de cloruro de amonio saturado de acuoues y la solución se extrajo con cloroformo. La capa orgánica se secó sobre sulfato de magnesio anhidro y se filtró y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 90/10 a 70/30) para obtener 2-(N-metilmetilsulfonamida)acetato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 19) (0,117 g, 0,524 mmol, 95,2%) como un producto oleoso incoloro.
1H-NMR (400MHz, CDCla) 8: 1,48(s, 9H), 2,98(s, 3H), 3,00(s, 3H), 3,98(s, 2H).
(Ejemplo de referencia 20) Síntesis de ácido 2-(N-metilsulfonamida)acético:
[0226 ]

[0227 ] A una solución de acetonitrilo (1,5 mL) del compuesto del Ejemplo de referencia 19 (0,117 g, 0,524 mmol), se agregó una solución de cloruro de hidrógeno-acetato de etilo (4,0 M, 1,31 mL, 5,24 mmol) a 0 °C y se elevó la temperatura a temperatura ambiente, seguido de agitación durante 16 horas. La solución de reacción se concentró a presión reducida para obtener ácido 2-(N-metilmetilsulfonamida)acético bruto (en adelante, el compuesto del Ejemplo de referencia 20) (0,0855 g) como un producto oleoso incoloro. El compuesto del Ejemplo de referencia 20 se utilizó directamente para la reacción posterior.
1H-NMR (400MHz, CDCla) 8: 2,99(s, 3H), 3,01(s, 3H), 4,14(s, 2H). ESI-MS:m/z = 168(M+H)+.
(Ejemplo 29) Síntesis de (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(N-metilmetilsulfonamida)acetil)piperidina-2-carboxamida:
[0228 ]

[0229 ] A una solución de DMF (2,0 mL) del compuesto del Ejemplo de referencia 20 (0,0850 g, 0,508 mmol), se agregaron una solución de DMF (1,0 mL) del compuesto del Ejemplo de referencia 8 (0,184 g, 0,462 mmol), HATU (0,193 g, 0,508 mmol) y diisopropiletilamina (0,121 mL, 0,693 mmol) a temperatura ambiente, seguido de agitación a la misma temperatura durante 18 horas. A la solución de reacción, se agregó agua destilada y la solución se extrajo con un disolvente mixto de n-hexano/acetato de etilo = 20/80(v/v). La capa orgánica se lavó con salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 50/50 a 30/70) para obtener (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(N-metilmetilsulfonamida)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 29) (0,209 g, 0,380 mmol, 82,0%) como un sólido blanco. Como resultado del análisis mediante columna quiral, el tiempo de retención del compuesto del Ejemplo 29 obtenido de este modo fue de 34,5 minutos, y la pureza óptica en ese momento fue de 98,2%ee. Las condiciones de análisis utilizando la columna quiral son las siguientes.
Equipos de medición; Cromatógrafo líquido de alto rendimiento LC-2010CHT, fabricado por Shimadzu Corporation Columna; CHIRALCEL OD-RH 0,46 cmcp x 15 cm, tamaño de partícula de 5 |jm, fabricado por Daicel Chemical Industries Ltd. Temperatura de columna; 40°C
Fase Móvil: (Solución A) solución acuosa de dihidrógenofosfato de potasio 20 mM, (Solución B) acetonitrilo Composición de fase móvil; Solución A:Solución B = 60:40 a 50:50 (0 a 40 minutos, gradiente lineal)
Solución A:Solución B = 50:50 a 60:40 (40 a 41 minutos, gradiente lineal)
Solución A:Solución B = 60:40 (41 a 50 minutos)
Caudal; 0,5 ml/minuto
Detección; UV (210 nm)
1H-NMR (400MHz, CDOa) 8: 1,52-1,89(m, 5H), 2,35-2,38(m, 1H), 3,03-3,07(m, 6H), 3,20-3,31(m, 1H), 3,67-3,76(m, 1H), 4,16-4,27(m, 2H), 5,25-5,26(m, 1H), 7,21-7,23(m, 1H), 7,30-7,45(m, 5H), 7,83(s, 1H), 8,22(br, 1H).
ESI-MS:m/z = 548(M+H)+.
(E je m p lo d e re fe re n c ia 21 ) S ín te s is d e (3 -(2 -( (2 -c lo ro -2 '-( t r i f lu o ro m e to x i) -[1 ,1 '-b ife n il ] -4 - i l) c a rb a m o il)p ip e r id in a -1 - i l) -3 -o x o p ro p il) c a rb a m a to d e te rc -b u t ilo :
[0230 ]

[0231 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 3-((tercbutoxicarbonil)amino)propanoico en lugar de ácido 2-metoxiacético, se obtuvo (3-(2-((2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)carbamoil)piperidina-1-il)-3-oxopropil)carbamato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 21) (0,288 g, 0,505 mmol, cuantitativo) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,39(s, 9H), 1,50-1,90(5H,m)12,35(d, J=13,7Hz, 1H), 2,55-2,75(m, 2H), 3,20(t, J=12,8Hz, 1H), 3,41-3,49(m, 1H), 3,50-3,60(m, 1H), 3,80 (d, J=13,7Hz, 1H) 5,17(br, 1H), 5,33(d, J=4,9Hz, 1H), 7,20(d, J=8,3Hz, 1H)1 7,29-7,60(m1 5H)1 7,70-7,89(m1 1H)1 8,65(br1 1H).
ESI-MS:m/z = 570(M+H)+.
(Ejemplo 30) Síntesis de 1-(3-aminopropanoil)-N-(2-cloro-2'-(trifluorometoxi)-[111'-bifenil]-4-il)piperidina-2-carboxamida:
[0232 ]

[0233 ] Según el mismo procedimiento que en el Ejemplo de referencia 51, salvo que se utilizó el compuesto del Ejemplo de referencia 21 en lugar del compuesto del Ejemplo de referencia 41, se obtuvo 1-(3-aminopropanoil)-N-(2-cloro-2'-(trifluorometoxi)-[111'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 30) (0,155 g10,329 mmol165,6%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,46-1,82(m, 5H), 2,43(d, J=13,1Hz, 1H)2,56(dt, J=15,3, 6,2Hz, 1H), 2,71-2,79(m, 1H), 3,09-3,21(m1 3H)1 3,88(d1 J=13,1Hz1 1H)15,43(d1 J=5,0Hz1 1H)17,19(d1 J=8,2Hz1 1H)17,29-7,36(m1 3H)1 7,40-7,85(m1 3H)1 8,96(br1 1H).
ESI-MS:m/z = 470(M+H)+.
(Ejemplo 31) Síntesis de 1-(3-acetamidepropanoil)-N-(2-cloro-2'-(trifluorometoxi)-[111'-bifenil]-4-il)piperidina-2-carboxamida:
[0234 ]

[0235 ] Según el mismo procedimiento que en el Ejemplo 31, salvo que se utilizó el compuesto del Ejemplo 30 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de acetilo en lugar de cloruro de propionilo1, se obtuvo 1-(3-acetamida-propanoil)-N- (2-cloro-2'-(trifluorometoxi)-[111'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 31) (0,0218 g1 0,0420 mmol1 99,1%) como un sólido blanco.
1H-NMR (400MHz, CDCh) 8: 1,46-1,94(m, 5H), 1,98(s, 3H), 2,34(d, J=13,3Hz, 1H), 2,60-2,73(m, 2H), 3,20(td, J=13,3, 2,4Hz, 1H), 3,52-3,70(m, 2H), 3,79(d, J=13,3Hz, 1H), 5,30(d, J = 4,5Hz, 1H), 6,23(br, 1H), 7,21(d, J=8,2Hz, 1H), 7,30-7,90(m, 6H), 8,51(br, 1H). ESI-MS:m/z = 512(M+H)+.
(Ejemplo 32) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(metilsulfonamida)propanoil)piperidina-2-carboxamida:

[0237 ] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó el compuesto del Ejemplo 30 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de metanosulfonilo en lugar de cloruro de propionilo, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(metilsulfonamida)propanoil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 32) (0,0224 g, 0,0409 mmol, 96,1%) como un sólido blanco. 1H-NMR (400MHz, CDCls) 8: 1,50-1,90(m, 5H), 2,33(d, J=13,5Hz, 1H), 2,75-2,80(m, 2H)3,00(s, 3H), 3,22(t, J=13,5Hz, 1H), 3,45-3,51(m, 2H), 3,77(d, J=13,5Hz, 1H), 5,26-5,30(m, 2H), 7,22(d, J=8,3Hz, 1H), 7,30-7,44(m, 5H), 7,80(br, 1H), 8,22(br, 1H). ESI-MS:m/z = 548(M+H)+.
(Ejemplo 33) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(dimetilamino)propanoil)piperidina-2-carboxamida:
[0238 ]

[0239 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó clorhidrato de ácido 3-(dimetilamino)propanoico como ácido 2-metoxiacético, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(dimetillamino)propanoil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 33) (0,0274 g, 0,0550 mmol, 73,2%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,49-1,83(m, 5H), 2,27(s, 6H), 2,38-2,45(m, 1H), 2,56-2,61(m, 1H), 2,66-2,80(m, 3H), 3,12-3,20(m, 1H), 3,85-3,93(m, 1H), 5,42(d, J=5,4Hz, 1H), 7,21(d, J=8,3Hz, 1H), 7,27-7,45(m, 6H), 8,73(br, 1H). ESI-MS:m/z = 498(M+H)+.
(Ejemplo de referencia 22) Síntesis de (3-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-3-oxopropil)(metil)carbamato de terc-butilo:
[0240 ]

[0241 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 3-((tercbutoxicarbonil)(metil)amino)propanoico en lugar de ácido 2-metoxiacético, se obtuvo (3-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-3-oxopropil)(metil)carbamato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 22) (0,130 g, 0,223 mmol, 89,0%) como un sólido blanco
1H-NMR (400MHz, CDCh) 8: 1,35(s, 9H), 1,30-1,77(m, 5H), 2,37-2,80(m, 3H), 2,93 (s, 3H), 3,18-3,30(m, 2H)3,84(d, J=13,7Hz, 1H), 3,95-4,03(m, 1H), 5,38-5,42(m, 1H), 7,20(d, J=8,3Hz, 1H), 7,31-7,46(m, 4H), 7,66-7,72(m, 1H), 7,90-7,92(m, 1H), 9,10(br, 1H).
ESI-MS:m/z = 584(M+H)+.
(E je m p lo 34 ) S ín te s is d e N -(2 -c lo ro -2 '-( t r i f lu o ro m e to x i) -[1 ,1 '-b ife n il ] -4 - i l) -1 -(3 -(m e t i la m in o )p ro p a n o il)p ip e r id in a -2 -c a rb o x a m id a :
[0242]

[0243 ] Según el mismo procedimiento que en el Ejemplo de referencia 5, salvo que se utilizó el compuesto del Ejemplo de referencia 22 en lugar del compuesto del Ejemplo de referencia 4, se obtuvo N-(2-cloro-2-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(metilamino)propanoil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 34) (0,804 g, 0,166 mmol, 74,6%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,46-1,83(m, 5H), 2,47(s, 3H), 2,40-2,48(m, 1H), 2,75-2,83(m, 1H), 2,94-2,99(m, 2H), 3,16(td, J=13,1, 2,6Hz, 1H), 3,88(d, J=13,1Hz, 1H), 5,41(d, J=5,4Hz, 1H), 7,21 (d, J=8,3Hz, 1H), 7,30-7,81(m, 6H), 8,80(br, 1H). ESI-MS:m/z = 484(M+H)+.
(Ejemplo 35) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(N-metilacetamida)propanoil)piperidina-2-carboxamida:
[0244 ]

[0245 ] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó el compuesto del Ejemplo 34 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de acetilo en lugar de cloruro de propionilo, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(N-metilacetamida)propanoil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 35) (0,0316 g, 0,0601 mmol, 74,5%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,48-1,77(m, 5H), 2,09(s, 3H), 2,45(d, J=13,7Hz, 1H), 2,59-2,79(m, 2H), 3,11(s, 3H), 3,20-3,29(m, 2H), 3,84 (d, J=13,7Hz, 1H), 4,21-4,28(m, 1H), 5,35(d, J = 5,1Hz, 1H), 7,20(d, J=8,3Hz, 1H), 7,31-7,36(m, 3H), 7,38-7,44 (m, 1H), 7,70-7,80(m, 1H), 7,97-8,06(m, 1H), 9,09(br, 1H).
ESI-MS:m/z = 526(M+H)+.
(Ejemplo 36) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(N-metilmetilsulfonamida)propanoil)piperidina-2-carboxamida:
[0246 ]

[0247 ] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó el compuesto del Ejemplo 34 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de metanosulfonilo en lugar de cloruro de propionilo, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(N-metilmetilmetilsulfonamida)propanoil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 36) (0,0380 g, 0,0676 mmol, 86,2%) como un sólido blanco.
1H-NMR (400MHz, CDCh) 8: 1,50-1,98(m, 5H), 2,35(d, J=13,5Hz, 1H), 2,76(dt, J=15,9, 7,0Hz, 1H), 2,86 (s, 3H), 2,83-2,89(m, 1H), 2,95(s, 3H), 3,20(td, J=13,5, 2,7Hz, 1H), 3,50-3,57(m, 2H), 3,85(d, J=13,5Hz, 1H), 5,31 (d, J=5,0Hz, 1H), 7,21 (d, J=8,2Hz, 1H), 7,30-7,36(m, 3H), 7,40-7,52(m, 2H), 7,80-7,83(m, 1H), 8,42(br, 1H).
ESI-MS:m/z = 584(M+H)+.
(E je m p lo d e re fe re n c ia 23 ) S ín te s is d e N -(2 -c lo ro -2 '-( t r i f lu o ro m e to x i) -[1 ,1 '-b ife n il ] -4 - i l) -1 -(2 -(e t i la m in o )a c e t i l) p ip e r id in a -2 -c a rb o x a m id a :
[0248]

[0249 ] A una solución de diclorometano (1,0 mL) del compuesto del Ejemplo de referencia 5 (0,0400 g, 0,0877 mmol), se agregaron una solución de diclorometano (0,0600 mL) de acetaldehído (0,00464 g, 0,105 mmol), ácido acético (0,000502 mL, 0,00877 mmol) y triacetoxiborohidruro de sodio (0,0279 m g, 0,132 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 2,5 horas. A la solución de reacción, se añadió una solución acuosa saturada de carbonato de hidrógeno de sodio, y la solución se extrajo con cloroformo. La capa orgánica se lavó con salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice de amina, n-hexano/acetato de etilo = 40/60 a 0/100) para obtener N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(etilamino)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo de referencia 23) (0,0193 g, 0,00399 mmol, 45,5%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,17(t, J=7,1Hz, 3H), 1,38-1,80(m, 5H), 1,90-2,00(m, 1H), 2,30-2,45(m, 1H), 2,55-2,90(m, 2H), 3,16(t, J=13,3Hz, 1H), 3,56(s, 2H), 3,70-3,76(m, 1H), 5,29(d, J=4,9Hz, 1H), 7,20-7,45(m, 6H), 7,70-7,90(m, 1H), 8,46(s, 1H).
ESI-MS:m/z = 484(M+H)+.
(Ejemplo 37) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(N-etilmetilsulfonamida)acetil)piperidina-2-carboxamida:
[0250 ]

[0251 ] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó el compuesto del Ejemplo de referencia 23 en lugar del compuesto del Ejemplo de referencia 3 y se utilizó cloruro de metanosulfonilo en lugar de cloruro de propionilo, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(N-etilmetilsulfonamida)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 37) (0,0175 g, 0,0311 mmol, 78,1%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,25(t, J=7,1Hz, 3H), 1,50-1,90(m, 5H), 2,38(d, J=13,0Hz, 1H), 3,07(s, 3H), 3,25 (t, J=13,0Hz, 1H), 3,43(q, J=7,1Hz, 2H), 3,76(d, J=13,0Hz, 1H), 4,14(d, J=17,0Hz, 1H), 4,30(d, J=17,0Hz, 1H), 5,27(d, J=4,6Hz, 1H), 7,21(d, J=8,3Hz, 1H), 7,29-7,36(m, 3H), 7,40-7,45(m, 2H), 7,83-7,85(m, 1H), 8,23(br, 1H).
ESI-MS:m/z = 584(M+Na)+.
(Ejemplo de referencia 24) Síntesis de (R)-(3-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-3-oxopropil)carbamato de terc-butilo:
[0252 ]

[0253 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 3-((tercbutoxicarbonil)amino)propanoico en lugar de ácido 2-metoxiacético y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3, se obtuvo (R)-(3-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4
il)carbamoil)piperidina-1-il)-3-oxopropil)carbamato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 24) (0,104 g, 0,182 mmol, 96,7%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,39(s, 9H), 1,50-1,90(5H, m), 2,35(d, J=13,7Hz, 1H), 2,55-2,75(m, 2H), 3,20(t, J=12,8Hz, 1H), 3,41-3,49(m, 1H), 3,50-3,60(m, 1H), 3,80 (d, J=13,7Hz, 1H), 5,17(br, 1H), 5,33(d, J=4,9Hz, 1H), 7,20(d, J=8,3Hz, 1H), 7,29-7,60(m, 5H), 7,70-7,89(m, 1H), 8,65(br, 1H).
ESI-MS:m/z = 570(M+H)+.
(Ejemplo 38) Síntesis de (R)-1-(3-aminopropanoil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0254 ]

[0255 ] Según el mismo procedimiento que en el Ejemplo de referencia 5, salvo que se utilizó el compuesto del Ejemplo de referencia 24 en lugar del compuesto del Ejemplo de referencia 4, se obtuvo (R)-1-(3-aminopropanoil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 38) (0,497 g, 0,106 mmol, 58,5%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,46-1,82(m, 5H), 2,43(d, J=13,1Hz, 1H)2,56(dt, J=15,3, 6,2Hz, 1H), 2,71-2,79(m, 1H), 3,09-3,21(m, 3H), 3,88 (d, J=13,1Hz, 1H), 5,43 (d, J=5,0Hz, 1H), 7,19(d, J=8,2Hz, 1H), 7,29-7,36(m, 3H), 7,40-7,85(m, 3H), 8,96(br, 1H).
ESI-MS:m/z = 470(M+H)+.
(Ejemplo 39) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(metiltio)propanoil)piperidina-2-carboxamida:
[0256 ]

[0257 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 3-(metiltio)propanoico en lugar de ácido 2-metoxiacético, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(metiltio)propanoil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 39) (0,0489 g, 0,0976 mmol, 97,4%) como un sólido blanco.
1H-NMR (400MHz, CDCh) 8: 1,51-1,90(m, 5H), 2,18(s, 3H), 2,36(d, J=13,9Hz, 1H), 2,68-2,77(m, 1H), 2,80-2,99(m, 3H), 3,17(td, J=13,2, 2,6Hz, 1H), 3,86(d, J=12,4Hz, 1H), 5,37(d, J = 4,9Hz, 1H), 7,20(d, J=8,3Hz, 1H), 7,30-7,52(m, 5H), 7,66-7,90(m, 1H), 8,49(br, 1H).
ESI-MS:m/z = 501(M+H)+.
(Ejemplo 40) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(metilsulfonil)propanoil)piperidina-2-carboxamida:
[0258 ]

[0259 ] A una solución de diclorometano (1,0 mL) del compuesto del Ejemplo 39 (0,0480 g, 0,0958 mmol), se agregó ácido 3-cloroperbenzoico (0,0496 g, 0,287 mmol) a 0 °C y la temperatura se elevó a temperatura ambiente, seguido de agitación durante 17 horas. A la solución de reacción, se añadieron una solución acuosa saturada de tiosulfato de sodio y carbonato de hidrógeno de sodio saturado, y la solución se extrajo con cloroformo. La capa
orgánica se lavó con salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, nhexano/acetato de etilo = 50/50 a 25/75) para obtener N-(2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)-1-(3-(metilsulfonil)propanoil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 40) (0,0412 g, 0,0773 mmol, 80,6%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,50-1,82(m, 5H), 2,45-2,48(m, 1H), 2,79(td, J=11,0,5,9Hz, 1H), 3,06(s, 3H), 3,18-3,27(m, 2H), 3,40(dt, J=13,8, 5,5Hz, 1H), 3,77-3,79(m, 1H), 3,93-3,96(m, 1H), 5,41(d, J=5,4Hz, 1H), 7,20(d, J=8,5Hz, 1H), 7,30-7,36 (m, 3H), 7,40-7,44(m, 1H), 7,51-7,53(m, 1H), 7,86-7,89(m, 1H), 8,28(br, 1H).
ESI-MS:m/z = 533(M+H)+.
(Ejemplo de referencia 25) Síntesis de (R)-(3-(2-((2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)carbamoil)piperidina-1-il)-3-oxopropil)(metil)carbamato de terc-butilo:
[0260 ]

[0261 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 3-((tercbutoxicarbonil)(metil)amino)propanoico en lugar de ácido 2-metoxiacético y el compuesto del Ejemplo de referencia 8 se utilizó en lugar del compuesto del Ejemplo de referencia 3, se obtuvo (R)-(3-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-3-oxopropil)(metil)carbamato de terc-butilo (en adelante, el compuesto del Ejemplo de referencia 25) (0,117 g, 0,201 mmol, 91,5%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,35(s, 9H), 1,30-1,77(m5H,), 2,37-2,80(m, 3H), 2,93 (s, 3H), 3,18-3,30(m, 2H), 3,84 (d, J=13,7Hz, 1H), 3,95-4,03(m, 1H), 5,38-5,42(m, 1H), 7,20(d, J=8,3Hz, 1H), 7,31-7,46(m, 4H), 7,66-7,72(m, 1H), 7,90-7,92(m, 1H), 9,10(br, 1H).
ESI-MS:m/z = 584(M+H)+.
(Ejemplo 41) Síntesis de (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(metilamino)propanoil)piperidina-2-carboxamida:
[0262 ]

[0263 ] Según el mismo procedimiento que en el Ejemplo de referencia 5, salvo que se utilizó el compuesto del Ejemplo de referencia 25 en lugar del compuesto del Ejemplo de referencia 4, se obtuvo (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(metilamino)propanoil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 41) (0,748 g, 0,155 mmol, 77,1%) como un sólido blanco.
1H-NMR (400MHz, CDCh) 8: 1,46-1,83(m, 5H), 2,47(s, 3H), 2,40-2,48(m, 1H), 2,75-2,83(m, 1H), 2,94-2,99(m, 2H), 3,16(td, J=13,1, 2,6Hz, 1H), 3,88(d, J=13,1Hz, 1H), 5,41(d, J=5,4Hz, 1H), 7,21 (d, J=8,3Hz, 1H), 7,30-7,81(m, 6H), 8,80(br, 1H).
ESI-MS:m/z = 484(M+H)+.
(E je m p lo 42 ) S ín te s is d e N -(2 -c lo ro -2 '-( t r i f lu o ro m e to x i) -[1 ,1 '-b ife n il ] -4 - i l) -1 -(3 -h id ro x ip ro p a n o il)p ip e r id in a -2 -c a rb o x a m id a :
[0264 ]

[0265 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 3-hidroxipropanoico en lugar de ácido 2-metoxiacético, se obtuvo N-(2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)-1-(3-hidroxipropanoil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 42) (0,212 g, 0,450 mmol, 59,9%) como un sólido blanco.
1H-NMR (400MHz, CDCh) 8: 1,50-1,92(m, 4H), 2,35(d, J=14,1Hz, 1H), 2,64-2,80(m, 2H), 3,06(t, J=6,3Hz, 1H), 3,19(td, J=13,2, 2,4Hz, 1H), 3,83(d, J=14,1Hz, 1H), 3,98 (q, J=5,4Hz, 2H), 5,34 (d, J=5,4Hz, 1H), 7,21(d, J=8,0Hz, 1H), 7,30-7,44(m, 6H), 7,70-7,90(brm,1H), 8,39(br, 1H).
ESI-MS:m/z = 471(M+H)+.
(Ejemplo 43) Síntesis de 3-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-3-oxopropanoato de metilo:
[0266 ]

[0267 ] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó 3-cloro-3-oxopropanoato de metilo en lugar de cloruro de propionilo, se obtuvo 3-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-3-oxopropanoato de metilo (en adelante, el compuesto del Ejemplo 43) (0,0500 g, 0,100 mmol, 80,0%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,50-1,80(m, 5H), 2,57-2,62(m, 1H), 3,16-3,25(m, 1H), 3,57(d, J=17,2Hz, 1H), 3,59-3,65(m, 1H), 3,84 (s, 3H), 3,85(d, J=17,2Hz, 2H), 5,49(s, 1H), 7,22(d, J=8,2Hz, 1H), 7,30-7,37(m, 3H), 7,40-7,44(m, 1H), 7,92-7,95(m, 1H), 8,87(br, 1H).
ESI-MS:m/z = 499(M+H)+.
(Ejemplo 44) Síntesis de 4-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-4-oxobutanoato de metilo:
[0268 ]

[0269 ] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó 4-cloro-4-oxobutanoato de metilo en lugar de cloruro de propionilo, se obtuvo 4-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-4-oxobutanoato de metilo (en adelante, el compuesto del Ejemplo 44) (0,0390 g, 0,0760 mmol, cuantitativo) como un sólido blanco.
1H-NMR (400MHz, CDCh) 8: 1,52-1,60(m, 5H), 2,42-2,60(m, 2H), 2,62-2,70(m, 1H), 2,88-2,96(m, 1H), 2,99-3,08(m, 1H), 3,22(d, J=14,6Hz, 1H), 3,74(s, 3H), 3,97 (d, J=14,6Hz, 1H), 5,46(d, J=5,4Hz, 1H), 7,21(d, J=8,3Hz, 1H), 7,28-7,44(m, 6H), 8,49(br, 1H). ESI-MS:m/z = 513(M+H)+.
(Ejemplo 45) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-meto xipropanoil)piperidina-2-carboxamida:
[0270 ]
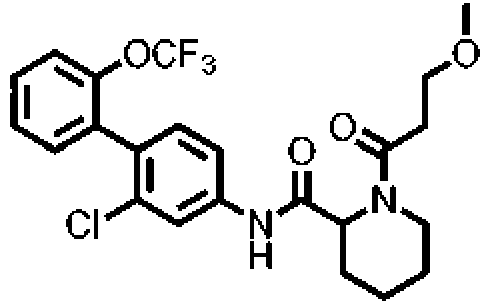
[0271 ] Según el mismo procedimiento que en el Ejemplo de referencia 2, salvo que se utilizó ácido 1-(3-metoxipropanoil)piperidina-2-carboxílico en lugar de ácido 1-(terc-butoxicarbonil)piperidina-2-carboxílico, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-metoxipropanoil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 45) (0,0467 g, 0,0963 mmol, 50,6%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,46-1,87(m, 5H), 2,39(d, J=13,9Hz, 1H), 2,64(dt, J=15,0, 5,7Hz, 1H), 2,85-2,92(m, 1H), 3,14(td, J=13,1,2,3Hz, 1H), 3,38(s, 3H), 3,67-3,78(m, 1H), 3,81-3,86 (m, 1H), 3,92(d, J=13,9Hz, 1H), 5,40(d, J=4,9Hz, 1H), 7,21(d, J=8,3Hz, 1H), 7,29-7,80(m, 6H), 8,46(s, 1H).
ESI-MS:m/z = 483(M-H)-.
(Ejemplo 46) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(1-metil-1H-pirazol-4-carbonil)piperidina-2-carboxamida:
[0272 ]
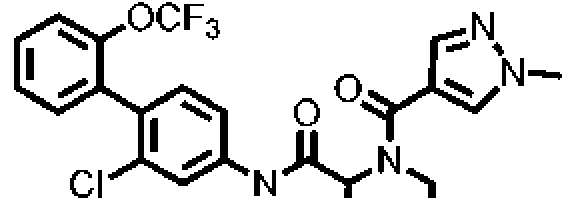
HU
[0273 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 1-metil-1H-pirazol-4-carboxílico en lugar de ácido 2-metoxiacético, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(1 -metil-1H-pirazol-4-carbonil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 46) (0,0314 g, 0,0619 mmol, 82,4%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,62-1,71(m, 2H), 1,78-1,87(m, 2H), 1,99-2,11(m, 1H), 2,37(d, J=12,9Hz, 1H), 3,08 3,21(m, 1H), 3,96(s, 3H), 4,13-4,23(m, 1H), 5,18-5,20(m, 1H), 7,22(d, J=8,3Hz, 1H), 7,27-7,90(m, 6H), 7,70 (s, 1H), 7,81(s, 1H), 9,18(br, 1H). ESI-MS:m/z = 507(M+H)+.
(Ejemplo 47) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(1-metil-1H-imidazol-4-carbonil)piperidina-2-carboxamida:
[0274 ]
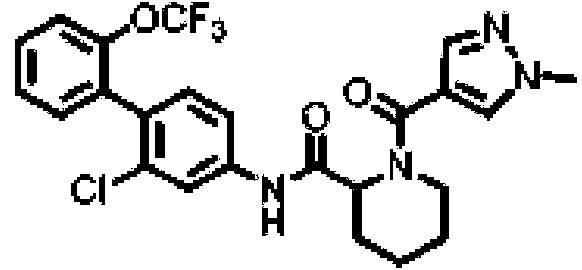
[0275 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 1-metil-1H-imidazol-4-carboxílico en lugar de ácido 2-metoxiacético, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(1-metil-1H-imidazol-4-carbonil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 47) (0,0369 g, 0,0728 mmol, 96,9%) como un sólido blanco.
1H-NMR (400MHz, CDCh) 8: 1,45-1,83(m, 4H), 2,15-2,32(m, 2H), 2,75-2,87(m, 1H), 3,79(s, 3H), 4,55-4,65(m, 1H) 5,31- 5,37(m, 1H), 7,21(d, J=8,3Hz, 1H), 7,31-7,63(m, 7H), 7,75-7,90(m, 1H), 11,47(br, 1H).
ESI-MS:m/z = 507(M+H)+.
(E je m p lo 48 ) S ín te s is d e N -(2 -c lo ro -2 '-( t r i f lu o ro m e to x i) -[1 ,1 '-b ife n il ] -4 - i l) -1 -(1 H -p ira z o l-4 -c a rb o n il)p ip e r id in a -2 -c a rb o x a m id a :
[0276 ]

[0277 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 1H-pirazol-4-carboxílico en lugar de ácido 2-metoxiacético, se obtuvo N-(2-cloro-2'-(trifluorometoxiH1,1'-bifenil]-4-il)-1-(1H-pirazol-4-carbonil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 48) (0,0163 g, 0,0331 mmol, 44,0%) como un sólido blanco.
1H-NMR (400MHz, COCls) 8: 1,45-1,90(m, 4H), 1,95-2,17(m, 1H), 2,33-2,43(m, 1H), 3,15-3,26(m, 1H), 4,09-4,21(m, 1H), 5,20-5,27(m, 1H), 7,22(d, J=8,5Hz, 1H), 7,28-7,52(m, 5H), 7,70-8,00(m, 3H), 9,16(br, 1H), 10,79(br, 1H). ESI-MS:m/z = 493(M+H)+.
(Ejemplo de referencia 26) Síntesis de 1-(2-(N-metilsulfonamida)acetil)piperidina-2-carboxilato de metilo:
[0278 ]

[0279 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó el compuesto del Ejemplo de referencia 20 en lugar de ácido 2-metoxiacético y se utilizó clorhidrato de metilpiperidina-2-carboxilato en lugar del compuesto del Ejemplo de referencia 3, se obtuvo ácido 1-(2-(N-metilsulfonamida)acetil)piperidina-2-carboxílico metilo (en adelante, el compuesto del Ejemplo de referencia 26) (0,934 g, 3,19 mmol, 82,0) como un sólido blanco.
1H-NMR (400MHz, CDCh) 8: 1,24-1,76(m, 5H), 2,25-2,32(m, 1H), 2,99(s, 3H), 3,00(s, 3H), 3,25(td, J=13,0, 3,2Hz, 1H), 3,58-3,64(m, 1H), 3,75(d, J=4,6Hz, 3H), 4,11(d, J=17,1Hz, 1H), 4,30 (d, J=17,1Hz, 1H), 5,25(d, J=5,6Hz, 1H).
ESI-MS:m/z = 293(M+H)+.
(Ejemplo de referencia 27) Síntesis de ácido 1-(2-(N-metilsulfonamida)acetil)piperidina-2-carboxílico:
[0280 ]

[0281 ] A una solución de metanol (10,0 mL) del compuesto del Ejemplo de referencia 26 (0,933 g, 3,19 mmol), se agregó una solución acuosa de hidróxido de sodio 1M (3,83 mL, 3,83 mmol) a 0 °C y se elevó la temperatura a temperatura ambiente, seguido de agitación durante 17 horas. A la solución de reacción, se añadió ácido clorhídrico 1M y la solución se extrajo con cloroformo. La capa orgánica se lavó con salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida para obtener ácido 1-(2-(N-metilsulfonamida)acetil)piperidina-2-carboxílico bruto (en adelante, el compuesto del Ejemplo de referencia 27) (0,812g) como un sólido blanco. El compuesto del Ejemplo de referencia 27 se utilizó directamente para la reacción posterior.
1H-NMR (400MHz, CDCls) 8: 1,18-1,75(m, 5H), 2,30(d, J=13,1Hz, 1H), 2,98(s, 3H), 2,99(s, 3H), 3,24(t, J=12,0Hz, 1H), 3,63(d, J=13,1Hz, 1H), 4,13(d, J=17,2Hz, 1H), 4,27(d, J=17,2Hz, 1H), 5,24(d, J=4,1Hz, 1H).
ESI-MS:m/z = 279(M+H)+.
(Ejemplo de referencia 28) Síntesis de N-(4-bromo-3-clorofenil)-1-(2-(N-metilsulfonamida)acetil)piperidina-2-carboxamida:
[0282]

[0283 ] Según el mismo procedimiento que en el Ejemplo de referencia 2, salvo que se utilizó el compuesto del Ejemplo de referencia 27 en lugar de ácido 1-(terc-butoxicarbonil)piperidina-2-carboxílico y se utilizó 4-bromo-3-cloroanilina en lugar del compuesto del Ejemplo de referencia 1, se obtuvo N-(4-bromo-3-clorofenil)-1-(2-(N-metilsulfonamida)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo de referencia 28) (0,296 g, 0,634 mmol, 58,8%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,49-1,85(m, 5H), 2,34(d, J=12,8Hz, 1H), 3,03(s, 3H), 3,03(s, 3H), 3,20(t, J=12,8Hz, 1H), 3,71(d, J=12,8Hz, 1H), 4,12(d, J=16,7Hz, 1H), 4,23(d, J=16,7Hz, 1H), 5,22(d, J=4,9Hz, 1H), 7,20-7,24(m, 1H), 7,50(dd, J=8,5, 2,0Hz, 1H), 7,84(t, J=2,3Hz, 1H), 8,20(br, 1H).
ESI-MS:m/z = 467(M+H)+.
(Ejemplo 49) Síntesis de N-(2-cloro-2'-isopropoxi-[1,1'-bifenil]-4-il)-1-(2-(N-metilmetilsulfonamida)acetil)piperidina-2-carboxamida:
[0284 ]
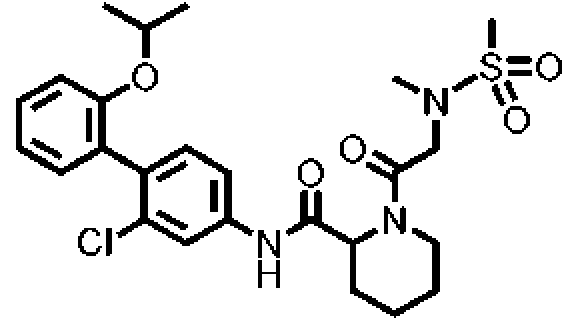
[0285 ] Según el mismo procedimiento que en el Ejemplo de referencia 1, salvo que se utilizó ácido 2 isopropoxifenilborónico en lugar de ácido 2-trifluorometoxifenilborónico y se utilizó el compuesto del Ejemplo de referencia 28 en lugar de 4-bromo-3-cloroanilina, se obtuvo N-(2-cloro-2-isopropoxi-[1,1-bifenil]-4-il)-1-(2-(N-metilmetilsulfonamida)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 49) (0,0253 g, 0,0485 mmol, 75,3%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,20(d, J=6,0Hz, 6H), 1,50-1,88(m, 5H), 2,31-2,39(m, 1H), 3,05(s, 3H), 3,05(s, 3H), 3,21 3,28(m, 1H), 3,69-3,74(m, 1H), 4,19(d, J=16,8Hz, 1H), 4,25(d, J=16,8Hz, 1H), 4,41(t, J = 6,0Hz, 1H), 5,23-5,26(m, 1H), 6,95-7,00(m, 2H), 7,17(dd, J=7,4,2,0Hz, 1H), 7,22-7,37(m, 3H), 7,76 (d, J=2,0Hz, 1H), 8,11(s, 1H).
ESI-MS:m/z = 523(M+H)+.
(Ejemplo 50) Síntesis de N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(1-metil-1H-imidazol-2-il)acetil)piperidina-2-carboxamida:
[0286 ]

[0287 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 2-(1-metil-1H-imidazol-2-il)acético en lugar de ácido 2-metoxiacético, se obtuvo N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(1-metil-1H-imidazol-2-il)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 50) (0,0341 g, 0,0654 mmol, 87,0%) como un sólido blanco.
1H-NMR (400MHz, COCh) 8: 1,49-1,80(m, 5H), 2,67-2,74(m, 1H), 3,20-3,27(m, 1H), 3,54-3,62(m, 1H), 3,63(s, 3H), 3,73(d, J=15,9Hz, 1H), 4,05(d, J=15,9Hz, 1H), 5,59-5,63(m, 1H), 6,89-6,98(m, 2H), 7,20-7,29(m, 1H), 7,32-7,37(m, 3H), 7,39-7,45(m, 1H), 7,71-7,99(m, 2H), 10,64(br, 1H).
ESI-MS:m/z = 521(M+H)+.
(Ejemplo de referencia 29) Síntesis de N-(4-bromo-3-fluorofenil)-1-(2-(N-metilsulfonamida)acetil)piperidina-2-carboxamida:
[0288]

[0289 ] Según el mismo procedimiento que en el Ejemplo de referencia 2, salvo que se utilizó el compuesto del Ejemplo de referencia 27 en lugar de ácido 1-(terc-butoxicarbonil)piperidina-2-carboxílico y se utilizó 4-bromo-3-fluoroanilina en lugar del compuesto del Ejemplo de referencia 1, se obtuvo N-(4-bromo-3-fluorofenil)-1-(2-(N-metilsulfonamida)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo de referencia 29) (0,0253 g, 0,0562 mmol, 52,1%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,48-1,83(m, 5H), 2,31-2,39(m, 1H), 3,03(s, 3H), 3,03(s, 3H), 3,16-3,23(m, 1H), 3,68 3,75(m, 1H), 4,10(d, J=16,6Hz, 1H), 4,24(d, J=16,6Hz, 1H), 5,21-5,24(m, 1H), 7,06(dd, J=9,0, 2,0Hz, 1H), 7,43(t, J=8,2Hz, 1H), 7,63 (dd, J=10,5, 2,4Hz, 1H), 8,24(br, 1H).
ESI-MS:m/z = 451(M+H)+.
(Ejemplo 51) Síntesis de N-(2-fluoro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(N-metilmetilsulfonamida)acetil)piperidina-2-carboxamida:
[0290 ]
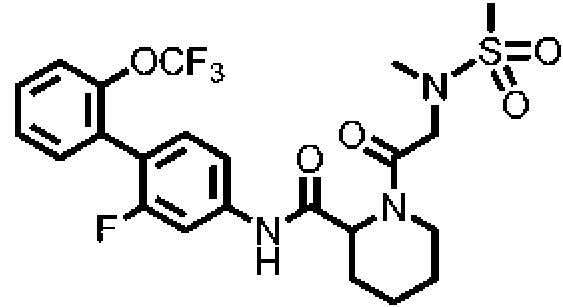
[0291 ] Según el mismo procedimiento que en el Ejemplo de referencia 1, salvo que el compuesto del Ejemplo de referencia 29 se utilizó como 4-bromo-3-cloroanilina, se obtuvo N-(2-fluoro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(N-metilsulfonamida)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 51) (0,0132 g, 0,0248 mmol, 44,7%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,45-1,90(m, 5H), 2,33-2,41(m, 1H), 3,05(s, 6H), 3,20-3,29(m, 1H), 3,69-3,76(m, 1H), 4,17(d, J=16,8Hz, 1H), 4,25(d, J=16,8Hz, 1H), 5,23-5,27(m, 1H), 7,20-7,44(m, 6H), 7,62(dd, J=11,7, 2,0Hz, 1H), 8,24(br, 1H).
(Ejemplo 52) Síntesis de 1-(2-(1H-imidazol-1-il)acetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0292 ]

[0293 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 1-imidazoleacético en lugar de ácido 2-metoxiacético, se obtuvo 1-(2-(1H-imidazol-1-il)acetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 52) (0,0189 g, 0,0373 mmol, 49,6%) como un sólido blanco.
1H-NMR (400MHz, COCls) 8: 1,44-2,05(m, 5H), 2,23-2,31(m, 1H), 3,37-3,47(m, 1H), 3,67-3,74(m, 1H), 4,86(d, J=16,6Hz, 1H), 4,91(d, J=16,6Hz, 1H), 5,16-5,22(m, 1H), 6,97(s, 1H), 7,13(s, 1H), 7,20(d, J=8,3Hz, 1H), 7,29-7,37(m, 4H), 7,40- 7,45(m, 1H), 7,53(s, 1H), 7,70-7,87(m, 1H), 8,41(br, 1H).
ESI-MS:m/z = 507(M+H)+.
(Ejemplo 53) Síntesis de 1-(2-(1H-tetrazol-1-il)acetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0294 ]

[0295 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 1H-tetrazol-1-acético en lugar de ácido 2-metoxiacético, se obtuvo 1-(2-(1H-tetrazol-1-il)acetil)-N-(2-cloro-2'-(trifluorometoxiH1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 53) (0,0244 g, 0,0479 mmol, 38,2%) como un sólido blanco.
1H-NMR (400MHz, COCls) 8: 1,59-2,02(m, 5H), 2,29-2,32(m, 1H), 3,49-3,57(m, 1H), 3,73-3,77(m, 1H), 5,18-5,19(m, 1H), 5,40(d, J=16,8Hz, 1H), 5,48(d, J=16,8Hz, 1H), 7,21-7,46(m, 6H), 7,81(br, 1H), 8,02(s, 1H), 8,86(s, 1H). ESI-MS:m/z = 509(M+H)+.
(Ejemplo 54) Síntesis de 1-(2-(furano-2-il)acetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0296 ]
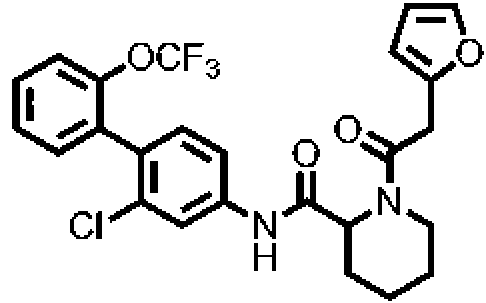
[0297 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 2-furanoacético en lugar de ácido 2-metoxiacético, se obtuvo 1-(2-(furano-2-il)acetil)-N-(2-cloro-2'-(trifluorometoxiH1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 54) (0,0605 g, 0,119 mmol, 95,2%) como un amorfo blanco.
1H-NMR (400MHz, CDCla) 8: 1,41-1,95(m, 5H), 2,33-2,38(m, 1H), 3,08-3,16(m, 1H), 3,85-3,97(m, 3H), 5,34-5,36(m, 1H), 6,22-6,23(m, 1H), 6,35-6,37(m, 1H), 7,19-7,44(m, 8H), 8,33(br, 1H).
ESI-MS:m/z = 505(M-H)-.
(Ejemplo 55) Síntesis de 1-(2-(3,5-dimetil-1H-pirazol-1-il)acetil)-N-(2-cloro-2'- (trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0298 ]

[0299 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 3,5-dimetil-1H-pirazol-1-acético en lugar de ácido 2-metoxiacético, se obtuvo 1-(2-(3,5-dimetil-1H-pirazol-1-il)acetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 55) (0,0579 g, 0,108 mmol, 86,3%) como un amorfo blanco.
1H-NMR (400MHz, CDCls) 8: 1,46-1,66(m, 2H), 1,74-1,79(m, 3H), 2,08(s, 3H), 2,27(s, 3H), 2,47-2,50(m, 1H), 3,11 3,19(m, 1H), 3,69-3,74(m, 1H), 4,83(d, J=15,2Hz, 1H), 4,96(d, J=15,2Hz, 1H), 5,35-5,36(m, 1H), 5,89 (s, 1H), 7,21-7,45(m, 7H), 8,86(br, 1H). ESI-MS:m/z = 535(M+H)+.
(E je m p lo 56 ) S ín te s is d e 1 -(2 -(3 -m e t i l is o x a z o l-5 - i l) a c e t i l) -N -(2 -c lo ro -2 '-( t r i f lu o ro m e to x i) -[1 ,1 '-b ife n il ] -4 - i l) p ip e r id in a -2 -c a rb o x a m id a :
[0300]

[0301 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 3-metil-5-isoxazoleacético en lugar de ácido 2-metoxiacético, se obtuvo 1-(2-(3-metilisoxazol-5-il)acetil)-N-(2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 56) (0,0652 g, 0,125 mmol, 99,6%) como un amorfo blanco.
1H-NMR (400MHz, CDCls) 8: 1,47-1,96(m, 5H), 2,29(s, 3H), 2,33-2,39(m, 1H), 3,21-3,28(m, 1H), 3,82-3,87(m, 1H), 3,95(d, J=16,1Hz, 1H), 4,01(d, J=16,1Hz, 1H), 5,32-5,33(m, 1H), 6,08(s, 1H), 7,21-7,45(m, 7H), 8,27(br, 1H).
ESI-MS:m/z = 520(M-H)-.
(Ejemplo 57) Síntesis de (R)-1-(2-(1H-tetrazol-1-il)acetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0302 ]

[0303 ] A una solución de DMF (30 mL) de ácido 1H-tetrazol-1-acético (1,67 g, 13,04 mmol), se agregaron una solución de DMF (10 mL) del compuesto del Ejemplo de referencia 8 (4,00 g, 10,03 mmol), HATU (4,96 g, 13,04 mmol) y diisopropiletilamina (2,63 mL, 15,04 mmol) a temperatura ambiente, seguido de agitación a la misma temperatura durante 17 horas. A la solución de reacción, se añadió agua destilada y la solución se extrajo con tolueno. La capa orgánica se concentró a presión reducida y el residuo se purificó mediante cromatografía en columna (gel de sílice de amina, n-hexano/acetato de etilo = 40/60 a 0/100) para obtener (R)-1-(2-(1H-tetrazol-1-il)acetil)-N-(2-cloro-2-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 57) (3,87 g, 7,60 mmol, 75,9%) como un amorfo blanco. Como resultado del análisis mediante columna quiral, el tiempo de retención del compuesto del Ejemplo 57 obtenido de este modo fue de 55,3 minutos, y la pureza óptica en ese momento fue de 99,4%ee. Las condiciones de análisis utilizando la columna quiral son las siguientes.
Equipos de medición; Cromatógrafo líquido de alto rendimiento LC-2010CHT, fabricado por Shimadzu Corporation Columna; CHIRALCEL OD-RH 0,46 cmcp x 15 cm, tamaño de partícula de 5 pm, fabricado por Daicel Chemical Industries Ltd. Temperatura de columna; 40°C
Fase Móvil: (Solución A) agua destilada, (Solución B) acetonitrilo Composición de fase móvil; Solución A:Solución B = 60:40 (0 a 75 minutos)
Caudal; Detección de 0,5 ml/minuto; UV (210 nm)
1H-NMR (400MHz, CDCla) 8: 1,62-2,00(m, 5H), 2,27-2,31(m, 1H), 3,52-3,58(m, 1H), 3,73-3,76(m, 1H), 5,18-5,19(m, 1H), 5,40(d, J=16,5Hz, 1H), 5,48(d, J=16,5Hz, 1H), 7,21-7,45(m, 6H), 7,81(br, 1H), 8,15(s, 1H), 8,86(s, 1H). ESI-MS:m/z = 509(M+H)+.
(Ejemplo 58) Síntesis de (R)-1-(3-(1H-tetrazol-1-il)propanoil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0304 ]

[0305 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 3-(tetrazol-1-il)propiónico en lugar de ácido 2-metoxiacético y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del
Ejemplo de referencia 3, se obtuvo (R)-1-(3-(1H-tetrazol-1-il)propanoil)-N-(2-cloro-2'-(trifluorometoxiH1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 58) (0,117 g, 0,224 mmol, 89,1%) como un amorfo blanco.
1H-NMR (400MHz, CDCls) 8: 1,43-1,97(m, 5H), 2,25-2,29(m, 1H), 3,04-3,19(m, 2H), 3,25(td, J=13,0, 2,7Hz, 1H), 3,70-3,74(m, 1H), 4,79-4,92(m, 2H), 5,18-5,19(m, 1H), 7,22-7,44(m, 6H), 7,79(br, 1H), 8,13(br, 1H), 8,84(s, 1H).
ESI-MS:m/z = 521(M-H)-.
(Ejemplo 59) Síntesis de (R)-1-(3-(1H-imidazol-1-il)propanoil)-N-(2-doro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0306 ]

[0307 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 3-(imidazol-1-il)propiónico en lugar de ácido 2-metoxiacético y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3, se obtuvo (R)-1-(3-(1H-imidazol-1-il)propanoil)-N-(2-cloro-2'-(trifluorometoxiH1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 59) (0,0796 g, 0,153 mmol, 60,9%) como un amorfo blanco.
1H-NMR (400MHz, CDCla) 8: 1,37-1,99(m, 5H), 2,26-2,29(m, 1H), 2,87-2,91(m, 2H), 3,17(td, J=13,3, 2,7Hz, 1H), 3,67-3,71(m, 1H), 4,34-4,47(m, 2H), 5,24-5,25(m, 1H), 6,99(s, 1H), 7,06(s, 1H), 7,21-7,45(m, 6H), 7,58(br, 1H), 7,71-7,82(m, 1H), 8,33(br, 1H). ESI-MS:m/z = 521(M+H)+.
(Ejemplo 60) Síntesis de (R)-1-(3-(3-metil-1H-pirazol-1-il)propanoil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0308 ]

[0309 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 3-(3-metil-pirazol-1-il)propiónico en lugar de ácido 2-metoxiacético y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3, se obtuvo (R)-1-(3-(3-metil-1H-pirazol-1-il)propanoil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 60) (0,135 g, 0,252 mmol, cuantitativo) como un amorfo blanco. 1H-NMR (400MHz, CDCh) 8: 1,37-1,89(m, 5H), 2,12(s, 3H), 2,35-2,38(m, 1H), 2,85-3,12(m, 3H), 3,67-3,70(m, 1H), 4,38-4,44(m, 1H), 4,51-4,58(m, 1H), 5,30-5,32(m, 1H), 5,96-5,97(m, 1H), 7,19-7,45(m, 8H), 8,59(br, 1H).
ESI-MS:m/z = 535(M+H)+.
(Ejemplo 61) Síntesis de (R)-1-(2-(1H-pirazol-1-il)acetil)-N-(2-cloro-2'-(trifluorometoxi) -[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0310 ]

[0311 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 2-(1H-pirazol-1-il)acético
en lugar de ácido 2-metoxiacético y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3, se obtuvo (R)-1-(2-(1H-pirazoM-il)acetil)-N-(2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 61) (0,0623 g, 0,123 mmol, 98,0%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8:1,46-1,74(m, 5H), 2,52-2,55(m, 1H), 3,04-3,12(m, 1H), 3,65-3,69(m, 1H), 5,01(d, J=14,5Hz, 1H), 5,22(d, J=14,5Hz, 1H), 5,43-5,44(m, 1H), 6,39(dd, J=2,3,2,0Hz, 1H), 7,24(d, J=8,2Hz, 1H), 7,32-7,37(m, 3H), 7,41- 7,45(m, 1H), 7,52-7,61(m, 1H), 7,53 (d, J=2,3Hz, 1H), 7,58(d, J=2,0Hz, 1H), 7,80-7,87(brm, 1H), 9,02(s, 1H). ESI-MS:m/z = 507(M+H)+.
(Ejemplo 62) Síntesis de (R)-1-(2-(4H-1,2,4-triazol-4-il)acetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0312 ]

[0313 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 2-(4H-1,2,4-triazol-4-il)acético en lugar de ácido 2-metoxiacético y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3, se obtuvo (R)-1-(2-(4H-1,2,4-triazol-4-il)acetil)-N-(2-cloro-2-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 62) (0,0575 g, 0,113 mmol, 90,3%) como un sólido blanco.
1H-NMR (400MHz, CDCla) 8: 1,55-1,90(m, 5H), 2,29-2,32(m, 1H), 3,57-3,71(m, 2H), 4,93 (d, J=16,8Hz, 1H), 5,02 (d, J=16,8Hz, 1H), 5,22-5,23(m, 1H), 7,19(d, J=8,2Hz, 1H), 7,28-7,37(m, 4H), 7,40-7,45(m, 1H), 7,81(s, 1H), 8,21(s, 2H), 8,84(s, 1H). ESI-MS:m/z = 508(M+H)+.
(Ejemplo 63) Síntesis de (R)-1-(2-(1H-1,2,4-triazol-1-il)acetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0314 ]

[0315 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 2-(1H-1,2,4-triazol-1-il)acetato de sodio en lugar de ácido 2-metoxiacético y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3, se obtuvo (R)-1-(2-(1H-1,2,4-triazoM-il)acetil)-N-(2-cloro-2-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 63) (0,0587 g, 0,116 mmol, 92,2%) como un sólido blanco.
1H-NMR (400MHz, CDCh) 8: 1,54-1,70(m, 2H), 1,77-1,90(m, 3H), 2,38-2,41(m, 1H), 3,31-3,39(m, 1H), 3,74-3,78(m, 1H), 5,13(d, J=15,4Hz, 1H), 5,22(d, J=15,4Hz, 1H), 5,29(d, J=5,0Hz, 1H), 7,23 (d, J=8,2Hz, 1H), 7,30-7,37 (m, 4H), 7,41- 7,45(m, 1H), 7,79(brs, 1H), 8,02(s, 1H), 8,26(s, 1H), 8,39(s, 1H).
ESI-MS:m/z = 508(M+H)+.
(Ejemplo 64) Síntesis de (R)-1-(2-(1H-1,2,3-triazol-1-il)acetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0316 ]

[0317 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 2-(1H-1,2,3-triazol-1-il)acético en lugar de ácido 2-metoxiacético y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3, se obtuvo (R)-1-(2-(1H-1,2,3-triazol-1-i¡)acetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 64) (0,0619 g, 0,122 mmol, 97,2%) como un sólido blanco.
1H-NMR (400MHz, CDCls) 8: 1,51-1,70(m, 2H), 1,76-1,88(m, 3H), 2,39-2,42(m, 1H), 3,32-3,39(m, 1H), 3,74-3,78(m, 1H), 5,30-5,31(m, 1H), 5,34(d, J=15,4Hz, 1H), 5,41 (d, J=15,4Hz, 1H), 7,23(d, J=8,6Hz, 1H), 7,30-7,37(m, 3H), 7,41-7,45(m, 1H), 7,52(brs, 1H), 7,76(d, J=0,9Hz, 1H), 7,82(d, J=0,9Hz, 1H), 7,91(brs, 1H), 8,43(s, 1H).
ESI-MS:m/z = 508(M+H)+.
(Ejemplo 65) Síntesis de (R)-1-(2-(1H-imidazol-1-il)acetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0318 ]
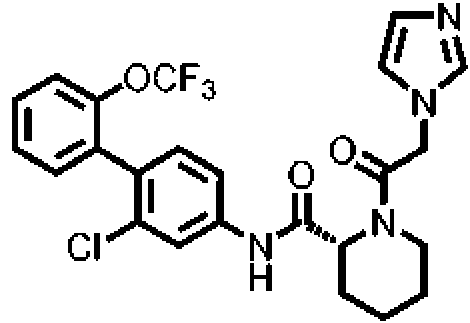
[0319 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 1-imidazoleacético en lugar de ácido 2-metoxiacético y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3, se obtuvo (R)-1-(2-(1H-imidazoM-il)acetil)-N-(2-cloro-2-(trifluorometoxiH1,1-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 65) (0,635 g, 1,25 mmol, 63,5%) como un amorfo blanco.
1H-NMR (400MHz, CDCla) 8: 1,44-2,05(m, 5H), 2,23-2,31(m, 1H), 3,37-3,47(m, 1H), 3,67-3,74(m, 1H), 4,86(d, J=16,6Hz, 1H), 4,91(d, J=16,6Hz, 1H), 5,16-5,22(m, 1H), 6,97(s, 1H), 7,13(s, 1H), 7,20(d, J=8,3Hz, 1H), 7,29-7,37(m, 4H), 7,40- 7,45(m, 1H), 7,53(s, 1H), 7,70-7,87(m, 1H), 8,41(brs, 1H).
ESI-MS:m/z = 507(M+H)+.
(Ejemplo 66) Síntesis de (R)-1-(2-(2H-1,2,3-triazol-2-il)acetil)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida:
[0320 ]
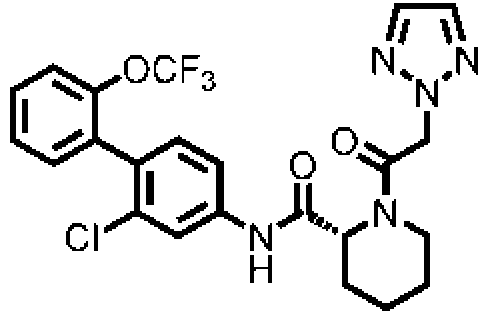
[0321 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó ácido 2-(2H-1,2,3-triazol-2-il)acético en lugar de ácido 2-metoxiacético y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3, se obtuvo (R)-1-(2-(2H-1,2,3-triazol-2-i¡)acetil)-N-(2-cloro-2-(trifluorometoxi)-[1,1'-bifenil]-4-il)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 66) (0,0321 g, 0,0632 mmol, 50,4%) como un amorfo blanco.
1H-NMR (400MHz, CDCh) 8: 1,47-1,65(m, 2H), 1,68-1,77(m, 3H), 2,49-2,52(m, 1H), 2,98-3,06(m, 1H), 3,55-3,58(m, 1H), 5,33(d, J=15,0Hz, 1H), 5,42(d, J=5,0Hz, 1H), 5,57(d, J=15,0Hz, 1H), 7,24 (d, J=8,2Hz, 1H), 7,32-7,37(m, 3H), 7,41-7,46(m, 1H), 7,42-7,88(brm, 2H), 7,73(s, 2H), 8,63(s, 1H).
ESI-MS:m/z = 530(M+Na)+.
(Ejemplo de referencia 30) Síntesis de 2-(5-metil-1,3,4-oxadiazol-2-il)acetato de etilo:
[0322 ]
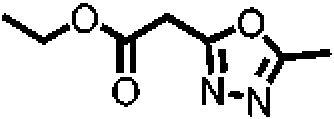
[0323 ] A una solución de dicloroetano (6,4 mL) de 2-(1H-tetrazol-5-il) acetato de etilo (0,500 g, 3,20 mmol), se agregó anhídrido acético (0,393 mL, 4,16 mmol) a temperatura ambiente y la temperatura se elevó a 100 °C, seguido
de agitación durante 11 horas. A la solución de reacción, se añadió una solución acuosa de hidróxido de sodio 1M y la solución se extrajo con acetato de etilo. La capa orgánica se lavó con una solución acuosa de hidróxido de sodio 1M y salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, n-hexano/acetato de etilo = 70/30 a 40/60) para obtener 2-(5-metil-1,3,4-oxadiazol-2-il)acetato de etilo (en adelante, el compuesto del Ejemplo de referencia 30) (0,0908 g, 0,534 mmol, 16,7%) como un producto oleoso incoloro.
1H-NMR (400MHz, CDCls) 8: 1,29(t, J=7,2Hz, 3H), 2,55(s, 3H), 3,92(s, 2H), 4,23(q, J=7,2Hz, 2H).
ESI-MS:m/z = 171(M+H)+.
(Ejemplo de referencia 31) Síntesis de 2-(5-metil-1,3,4-oxadiazol-2-il)acetato de sodio:
[0324 ]

[0325 ] A una solución de tetrahidrofurano (1,0 mL) del compuesto del Ejemplo de referencia 30 (0,0900 g, 0,529 mmol), se agregaron una solución acuosa de hidróxido de sodio 1M (1,06 mL, 1,06 mmol) y etanol (1,0 mL) a temperatura ambiente, seguido de agitación a la misma temperatura durante 2 horas. La solución de reacción se concentró a presión reducida para obtener 2-(5-metil-1,3,4-oxadiazol-2-il)acetato de sodio bruto (en adelante, el compuesto del Ejemplo de referencia 31) (0,0835 g) como un sólido blanco. El compuesto del Ejemplo de referencia 31 se utilizó directamente para la reacción posterior.
1H-NMR (400MHz, DMSO-D6) 8: 2,41(s, 3H), 3,38(s, 2H). ESI-MS:m/z = 143(M+H)+.
(Ejemplo 67) Síntesis de (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(5 -metil-1,3,4-oxadiazol-2-il)acetil)piperidina-2-carboxamida:
[0326 ]

[0327 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó el compuesto del Ejemplo de referencia 31 en lugar de ácido 2-metoxiacético y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3, se obtuvo (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(2-(5-metil-1,3,4-oxadiazol-2-il)acetil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 67) (0,0524 g, 0,0910 mmol, 90,8%) como un amorfo blanco.
1H-NMR (400MHz, CDCla) 8: 1,50-1,66(m, 3H), 1,74-1,77(m, 2H), 2,57(s, 3H), 2,62-2,65(m, 1H), 3,27-3,34(m, 1H), 3,61-3,64(m, 1H), 3,94 (d, J=17,4Hz, 1H), 4,21(d, J=17,4Hz, 1H), 5,54-5,55(m, 1H), 7,25-7,27(m, 1H), 7,33-7,36(m, 3H), 7,40- 7,44(m, 1H), 7,77(brs, 1H), 8,18(brs, 1H), 9,38(s, 1H).
ESI-MS:m/z = 545(M+Na)+.
(Ejemplo 68) Síntesis de (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(dimetilamino)propanoil)piperidina-2-carboxamida:
[0328 ]
[0329 ] Según el mismo procedimiento que en el Ejemplo 4, salvo que se utilizó clorhidrato de ácido 3-(dimetilamino)propanoico en lugar de ácido 2-metoxiacético y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3, se obtuvo (R)-N-(2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)-1-(3-(dimetilamino)propanoil)piperidina-2-carboxamida (en adelante, el compuesto del Ejemplo 68) (0,0826 g, 0,166 mmol, 66,2%) como un amorfo blanco.
1H-NMR (400MHz, CDCfa) 8: 1,46-1,83(m, 5H), 2,26(s, 0,6H), 2,28(s, 5,4H), 2,40-2,44(m, 1H), 2,55-2,62(m, 1H), 2,652,81(m, 3H), 2,99-3,05(m, 0,1H), 3,13-3,20(m, 0,9H), 3,88-3,91(m, 0,9H), 4,69 (d, J=5,0Hz, 0,1H), 4,73-4,76(m, 0,1H), 5,43(d, J=5,0Hz, 0,9H), 7,21(d, J=8,6Hz, 1H), 7,31-7,86(m, 6H), 8,76(br, 0,9H), 9,33(br, 0,1H).
ESI-MS:m/z = 498(M+H)+.
(Ejemplo 69) Síntesis de (R)-5-(2-((2-cloro-2'-(trifluorometoxiH1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-5-oxopentanoato de metilo:
[0330 ]
[0331 ] Según el mismo procedimiento que en el Ejemplo 3, salvo que se utilizó 4-(cloroformil)butirato de metilo en lugar de cloruro de propionilo y se utilizó el compuesto del Ejemplo de referencia 8 en lugar del compuesto del Ejemplo de referencia 3, se obtuvo tanoato de metilo (R)-5-(2-((2-cloro-2'-(trifluorometoxiH1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-5-oxopeno (en adelante, el compuesto del Ejemplo 69) (0,130 g, 0,247 mmol, 98,4%) como un amorfo blanco.
1H-NMR (400MHz, CDCh) 8:1,44-1,79(m, 4H), 1,84-1,95(m, 1H), 1,96-2,13(m, 2H), 2,35(d, J=13,7Hz, 1H), 2,40-2,62(m, 4H), 2,63-2,70(m, 0,1H), 3,14-3,21(m, 0,9H), 3,68(s, 2,7H), 3,69(s, 0,3H), 3,84-3,88(m, 0,9H), 4,66-4,69(m, 0,2H), 5,34(d, J=5,0Hz, 0,9H), 7,20(d, J=8,2Hz, 1H), 7,29-7,94(m, 6H), 8,68(s, 0,9H), 8,90(s, 0,1H).
(Ejemplo 70) Síntesis de ácido (R)-5-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-5-oxopentanoico:
[0332 ]

[0333 ] A una solución de metanol (2,5 mL) del compuesto del Ejemplo 69 (0,130 g, 0,247 mmol), se agregaron una solución acuosa de hidróxido de sodio 1M (2,47 mL, 2,47 mmol) y tetrahidrofurano (2,5 mL) a 0 °C y se elevó la temperatura a temperatura ambiente, seguido de agitación durante 5 horas. A la solución de reacción, se añadió ácido clorhídrico 1M y la solución se extrajo con cloroformo. La capa orgánica se lavó con salmuera saturada, se secó sobre sulfato de sodio anhidro y se filtró, y a continuación, el filtrado se concentró a presión reducida. El residuo se purificó mediante cromatografía en columna (gel de sílice, cloroformo/metanol = 98/2 a 90/10) para obtener ácido (R)-5-(2-((2-cloro-2'-(trifluorometoxi)-[1,1'-bifenil]-4-il)carbamoil)piperidina-1-il)-5-oxopentanoico (en adelante, el compuesto del Ejemplo 70) (0,0592 g, 0,115 mmol, 46,7%) como un amorfo blanco.
1H-NMR (400MHz, CDCla) 8: 1,49-1,79(m, 4H), 1,84-1,94(m, 1H), 2,00-2,10(m, 2H), 2,32(d, J=13,6Hz, 1H), 2,44 2,63(m, 4H), 3,20(td, J=13,3, 2,6Hz, 1H), 3,81-3,87(m, 1H), 5,30(d, J=4,5Hz, 1H), 7,20(d, J=8,6Hz, 1H), 7,29-7,36(m, 3H), 7,40- 7,45(m, 2H), 7,75-7,88(m, 1H), 8,62(s, 1H).
ESI-MS:m/z = 535(M+Na)+.
(Ejemplo 71) Efecto inhibidor sobre la unión del coactivador RORy:
[0334 ] El efecto inhibidor del derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo en la unión entre un dominio de unión al ligando de RORy (en adelante, RORy-LBD) y un coactivador se evaluó utilizando el kit de ensayo de coactivador gamma del receptor huérfano relacionado con retinoides TR-FRET LanthaScreen™ de Invitrogen utilizando transferencia de energía de fluorescencia resuelta en el tiempo (TR-FRET).
[0335 ] El compuesto de prueba se disolvió en DMSO y se diluyó con amortiguador de corregulador TR-FRET D (Invitrogen) que contenía 5 mmol/L de DTT para tener una concentración final de DMSO de 1% antes de su uso. A cada pocillo de una placa negra de 384 pocillos (Corning Inc.), se añadieron 4 nmol/L de RORy-LBD fusionado con GST (Invitrogen) diluido con el amortiguador mencionado anteriormente y el compuesto de prueba. Se preparó un pocillo sin adición del compuesto de prueba y sin adición de RORy-LBD fusionado con GST (fondo) y un pocillo sin
adición del compuesto de prueba y con adición de RORy-LBD fusionado con GST (control). A continuación, se añadieron 150 nmol/L de TRAP220/DRIP-2 marcado con fluoresceína (Invitrogen) diluido con el amortiguador mencionado anteriormente y 32 nmol/L de anticuerpo anti-GST marcado con terbio (Invitrogen) a cada pocillo. Después de incubar la placa a temperatura ambiente durante 16 a 24 horas, se midió la fluorescencia a 495 nm y 520 nm cuando se excitó a 320 nm para cada pocillo y se calculó la relación (valor de fluorescencia a 520 nm/valor de fluorescencia a 495 nm).
[0336] Se calculó el cambio de pliegue con la adición del compuesto de prueba (relación con la adición del compuesto de prueba/relación del fondo), el cambio de pliegue del control (relación del control/relación del fondo) y el cambio de pliegue del fondo (relación del fondo/relación del fondo), y a continuación, se calculó la tasa de inhibición de unión entre RORy- LBD y un coactivador (en adelante, velocidad de inhibición de unión del coactivador RORy) (%) a partir de la siguiente fórmula 1 :
[Fórmula 1]
Tasa de inhibición de unión del coactivador de R,ORy (%) = (1 -((Cambio de pliegue con la adición del compuesto de prueba) -(Cambio de pliegue del fondo))((Cambio de pliegue del control) -(Cambio de pliegue del fondo)) x 100 [0337] La tasa de inhibición de unión del coactivador de RORy (%) a 33 pmol/L del compuesto de prueba se muestra en las Tablas 2-1 y 2-2.
Tabla 2-1

continuación

T l 2-21
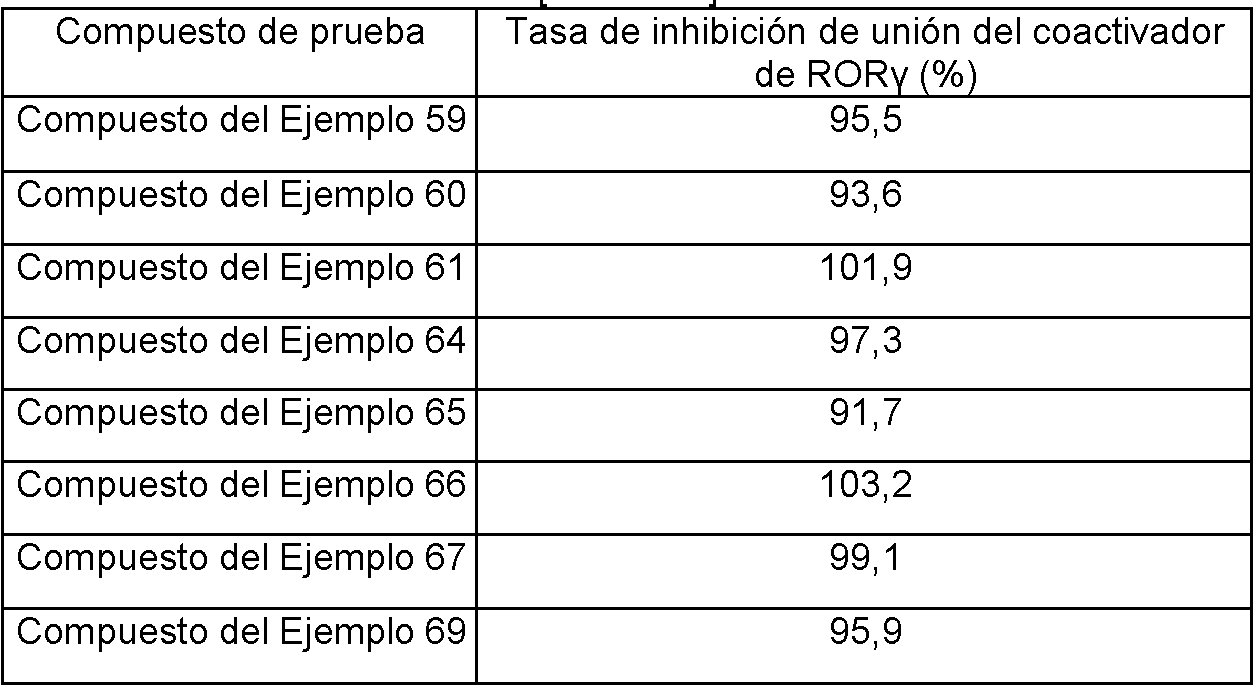
[0338 ] Estos resultados revelaron que el derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo inhibe notablemente la unión entre RORy-LBD y un coactivador.
(Ejemplo 72) Efecto supresor sobre la producción de IL-17 en esplenocitos de ratón:
[0339 ] Utilizando esplenocitos de ratón, se evaluó el efecto supresor del derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo sobre la producción de IL-17 mediante estimulación de IL-23 utilizando un método parcialmente modificado mencionado en The Journal of Biological Chemistry, 2003, Vol. 278, No. 3, p. 1910 1914.
[0340 ] Se preparó una suspensión celular única a partir del bazo de ratones C57BL/6J (macho de 7 a 23 semanas de edad) (Charles River Laboratories Japan, Inc.) y se prepararon esplenocitos usando Histopaque-1083 (Sigma-Aldrich Japan). El medio de cultivo se utilizó mediante la adición de FBS al 10% (Gibco), 50 U/ mL de
penicilina-50 |jg/ mL de estreptomicina (Gibco), 50 jmol/L de 2-mercaptoetanol (Gibco) y 100 U/ mL de IL-2 humana (Cell Science & Technology Institute, Inc.) al medio RPMI1640 (Gibco). El compuesto de prueba se disolvió en DMSO y a continuación, se diluyó con el medio de cultivo para tener una concentración final de DMSO de 0,1% antes de su uso. Los esplenocitos (33 105 células/pocillo) preparados en el medio de cultivo se sembraron en pocillos de una placa de fondo plano de 96 pocillos (Corning Incorporated), se le añadieron el compuesto de prueba y 10 ng/ mL de IL-23 humana (R & D systems, Inc.), y las células se cultivaron a 37 °C bajo CO2 al 5% durante 3 días. Se prepararon un pocillo sin adición de IL-23 humana y sin adición del compuesto de prueba y un pocillo con adición de iL-23 humana y sin adición del compuesto de prueba. Después de completar el cultivo, se recogió el sobrenadante de cultivo y se determinó la cantidad de producción de IL-17 en el sobrenadante mediante el método ELISA (R & D systems, Inc.).
[0341 ] La tasa de inhibición de la producción de IL-17 (%) se calculó a partir de la siguiente Fórmula 2.
[Fórmula 2]
Tasa de inhibición de la producción de IL-17 (%) = (1 -((cantidad de producción de IL-17 con adición de IL-23 y con adición del compuesto de prueba) -(cantidad de producción de IL-17 sin adición de IL-23 y sin adición del compuesto de prueba))((cantidad de producción de IL-17 con adición de IL-23 y sin adición del compuesto de prueba) -(cantidad de producción de IL-17 sin adición de IL-23 y sin adición del compuesto de prueba)) x 100 [0342 ] La tasa de inhibición de la producción de IL-17 (%) a 5 jmol/L del compuesto de prueba se muestra en las Tablas 3-1 y 3-2.
T l -1
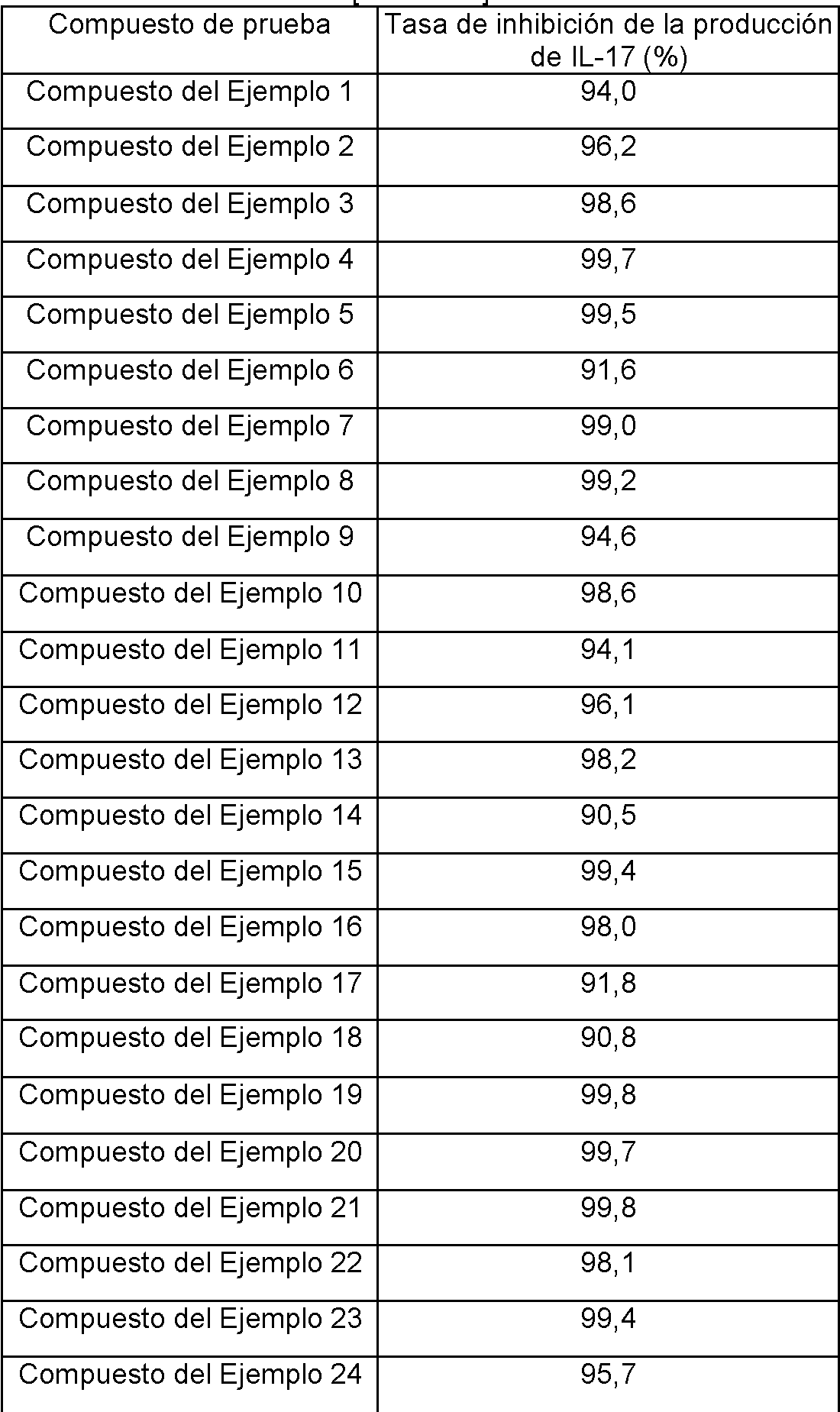
continuación

T l -21

continuación

[0343 ] Estos resultados revelaron que el derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo suprime la producción de IL-17.
(Ejemplo 73) Efecto supresor de síntomas en el modelo experimental de encefalomielitis autoinmunitaria de ratón:
[0344 ] Utilizando el aumento en la puntuación de síntomas neurológicos como un índice de exacerbación de los síntomas, se evaluó el efecto del derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo en un modelo experimental de encefalomielitis autoinmunitaria de ratón. El modelo experimental de encefalomielitis autoinmunitaria de ratón se preparó mediante un procedimiento parcialmente modificado por Hindinger y col. (Journal of Neuroscience Research, 2006, Vol. 84, p. 1225-1234).
[0345 ] Un total de 0,1 mL (0,05 mL en cada lado) de una solución de administración de MOG35-55 preparada mezclando una solución de PBS que contiene péptidos sintéticos parciales de glicoproteínas de oligodendrocitos de mielina (MOG35-55; CSBio, Inc.) preparada a una concentración de 4 mg/mL con adyuvante completo de Freund en la misma cantidad se inoculó por vía intradérmica a ambos lados del flanco de ratones C57BL/6J (macho, 8 semanas de edad) (Charles River Laboratories Japan, Inc.). Además, se administraron 200 pL de toxina pertussis (Sigma-Aldrich Japan) preparada a una concentración de 1 pg/ mL por vía intraperitoneal a los ratones el día de la inoculación y 2 días después de la inoculación de la solución de administración de MOG35-55.
[0346 ] Después de la inoculación de la solución de administración de MOG35-55, se administró el compuesto de prueba a los ratones. Como compuesto de prueba, se utilizaron el compuesto del Ejemplo 20, el compuesto del Ejemplo 29 y el compuesto del Ejemplo 57. El compuesto del Ejemplo 20 se suspendió en una solución de metilcelulosa al 0,5% p/v y se administró por vía oral una vez al día a una dosis de 3 mg/kg todos los días a partir de 2 días después de la inoculación de la solución de administración de MOG35-55. El compuesto del Ejemplo 29 y el compuesto del Ejemplo 57 se suspendieron en una solución de metilcelulosa al 0,5% p/v y se administraron por vía oral una vez al día a una dosis de 1 mg/kg todos los días a partir de 13 días después de la inoculación de la solución de administración de MOG35-55. El grupo en el que se administró el compuesto del Ejemplo 20 a ratones se definió como el grupo de administración del compuesto del Ejemplo 20, el grupo en el que se administró el compuesto del Ejemplo 29 se definió como el grupo de administración del compuesto del Ejemplo 29 y el grupo en el que se administró el compuesto del Ejemplo 57 se definió como el grupo de administración del compuesto del Ejemplo 57. En el grupo de administración del vehículo, se administró de manera similar un vehículo de cada compuesto de prueba (solución de metilcelulosa al 0,5% p/v).
[0347 ] Después de la inoculación de la solución de administración de MOG35-55, se calificaron las puntuaciones de los síntomas neurológicos en los grupos en los que se administró el compuesto de prueba y los grupos correspondientes de administración del vehículo (0: normal, 1 : debilidad de la cola o extremidad trasera flácida, 2: debilidad de la cola y extremidad trasera flácidas, 3: parálisis parcial de la extremidad trasera, 4: parálisis completa de la extremidad trasera, 5: estado moribundo). Se utilizó como método de puntuación el método mencionado en Current Protocols in Immunology (John Wiley & Sons. Inc, 2000, p. 15,1,1-15,1,20).
[0348 ] Los resu ltad os se m ue stra n en las f ig u ra s 1, 2 y 3. El eje v e rtica l rep rese n ta la pun tua c ió n de los s ín tom a s n e u ro ló g ico s (e rro r e s tá n d a r m ed io de 6, n = 8 a 9). "V e h ícu lo " en el eje ho rizon ta l rep rese n ta el g rupo de a d m in is tra c ió n de l ve h ícu lo , el "C o m p u e s to de l E je m p lo 20 " rep rese n ta el g ru p o de a d m in is tra c ió n de l c o m p u e s to del E je m p lo 20, el "C o m p u e s to del E je m p lo 29 " re p re se n ta el g ru po de ad m in is tra c ió n de l co m p u e s to del E je m p lo 29 y el "C o m p u e s to del E je m p lo 57" rep rese n ta el g ru po de a d m in is tra c ió n del co m p ue s to del E jem p lo 57. La figu ra 1 m uestra la pu n tua c ió n de s ín tom a s ne u ro lóg ico s el día 13 de spu és de la in ocu lac ión de la so luc ión de ad m in is tra c ió n de M O G 35-55 , la fig u ra 2 m ue s tra la pun tua c ió n de s ín to m a s n e u ro ló g ico s el día 23 d e sp u é s de la in ocu lac ión de la so luc ió n de ad m in is tra c ió n de M O G 35-55 y la fig u ra 3 m ue s tra la pu n tua c ió n de s ín to m a s ne u ro lóg ico s el d ía 30 de sp u é s de la in ocu lac ión de la so luc ió n de ad m in is tra c ió n de M O G 35-55. El a s te risco (*) ind ica la s ig n ifica c ió n e s ta d ís tica en co m p a ra c ió n con el g ru p o de a d m in is tra c ió n de l v e h íc u lo (p ru e b a de W ilc o x o n ) (*: P < 0,05).
[0349 ] La in o cu la c ió n de la so lu c ió n de a d m in is tra c ió n de M O G 35 -55 a u m e n tó la p u n tu a c ió n de los s ín to m a s n e u ro ló g ico s en el g ru po de ad m in is tra c ió n del v e h íc u lo a en tre 1,0 y 2,4. E ste au m e n to en la pun tua c ió n de los s ín to m a s ne u ro lóg ico s se sup rim ió e s ta d ís tica m e n te s ig n ifica tiva m e n te m ed ia n te la ad m in is tra c ió n de l co m p u e s to del E je m p lo 20, el co m p u e s to de l E je m p lo 29 o el co m p u e s to de l E je m p lo 57.
[0350 ] E stos resu ltad os reve la ron que el de riva d o de am ina c íc lica (I) o una sa l fa rm a c o ló g ic a m e n te ace p tab le de l m ism o m ue s tra un m a rca d o e fe c to su p re s o r de los s ín to m a s de la e sc le ro s is m últip le .
(E je m p lo 74 ) E fec to s u p re s o r de s ín to m a s en el m ode lo de pso ria s is de ra tón in du c ida po r Im iqu im od :
[0351 ] U tiliza n d o el au m e n to en el g ro s o r de la o re ja com o un índ ice de e x a ce rb a c ió n de los s ín tom a s, se eva luó el e fe c to de l de riva d o de am ina c íc lica (I) o una sal fa rm a co ló g ica m e n te a ce p tab le del m ism o en un m od e lo de pso rias is de ra tón in du c ida po r im iqu im od . El m od e lo de pso rias is de ratón induc ida po r im iqu im od se p reparó m ed ia n te un m é tod o p a rc ia lm e n te m o d ifica d o po r S c h a p e r y col. (T he Jo u rn a l o f D e rm a to lo g ica l S c ience , 2013 , V o l. 71, No. 1, p. 29 -36 ).
[0352 ] S e u tiliza ro n ra ton es B A L B /c (m acho , de 7 sem a na s de ed ad ) (C h a rle s R ive r L a b o ra to rie s Japan , Inc.) a las 8 sem a na s de edad de spu és de la rep rod ucc ió n p re lim ina r. Para in d u c ir s ín tom a s s im ila re s a la pso rias is , se ap lica ra 5 m g de B E S E L N A C R E A M al 5% una vez al d ía al e x te rio r de las au rícu las de recha e izqu ie rda de los ra tones d u ran te 8 d ías a p a rtir de l d ía de la p rim era ad m in is tra c ió n de im iqu im od (en ade lan te , d ía de la in d u cc ió n ) hasta 7 d ías d e sp u é s de la in du cc ión (do s is de im iqu im od , 0 ,5 m g /cue rpo /d ía ).
[0353 ] S e a d m in is tró el c o m p u e s to de p ru eb a a una do s is de 10 m g /kg a los ra to n e s una v e z al d ía d u ra n te 5 d ías desde 3 d ías a p a rtir de de la in du cc ión hasta 7 d ías de sp u é s de la in du cc ión . C om o co m p u e s to de p rueba , se u tiliza ron el co m p u e s to del E je m p lo 20, el co m p u e s to de l E je m p lo 29 y el co m p u e s to de l E je m p lo 57. El co m p ue s to de l E jem p lo 20, el co m p u e s to del E jem p lo 29 y el com p ue s to del E je m p lo 57 se susp en d ie ron en una so luc ió n de m e tilc e lu lo s a al 0 ,5 % p /v y se a d m in is tra ro n po r v ía ora l. El g ru p o en el que se a d m in is tró el c o m p u e s to de l E je m p lo 20 a ra tones se de fin ió com o el g rupo de adm in is trac ión del com p ue sto del E jem plo 20, el g rupo en el que se adm in is tró el c o m p u e s to de l E je m p lo 29 se d e fin ió com o el g ru po de a d m in is tra c ió n de l co m p u e s to de l E je m p lo 29 y el g ru p o en el que se ad m in is tró el co m p ue s to del E je m p lo 57 se de fin ió com o el g rupo de ad m in is tra c ió n del co m p ue s to del E je m p lo 57. En el g ru po de a d m in is tra c ió n del ve h ícu lo , se ad m in is tró de m an e ra s im ila r un v e h ícu lo de cada c o m p u e s to de p ru eb a (so luc ió n de m e tilce lu lo sa al 0 ,5 % p/v).
[0354 ] S e m id ie ron el g ro so r de la o re ja de rech a e izq u ie rda an tes de la ap licac ión de la so luc ió n de im iqu im od (a n te s de la in d u cc ió n ) el d ía de la in d u cc ió n y el g ro s o r de la o re ja d e re c h a e izq u ie rd a el o c ta vo d ía d e s p u é s de la in d u cc ió n con un m ic ró m e tro d ig ita l (M itu to yo C o rp o ra tio n ). El g ro s o r de la o re ja se d e fin ió co m o la m ed ia de l g ro so r de la o re ja de rech a e izqu ie rda , y se u tilizó el cam b io en el g ro so r de la o re ja (g ro so r de la o re ja el oc ta vo d ía después de la indu cc ión - g ro s o r de la o re ja an tes de la in du cc ión ) com o un índ ice de e va lu a c ió n de la e fica c ia de l fá rm aco .
[0355 ] Los re su lta d o s se m ue s tra n en las fig u ra s 4, 5 y 6. El e je v e rtic a l re p re se n ta el ca m b io en el g ro s o r de la o re ja (m m ) (e rro r e s tá n d a r m ed io de 6, n = 6). "V e h ícu lo " en el e je h o rizo n ta l rep rese n ta el g ru p o de ad m in is tra c ió n de l ve h ícu lo , e l "C o m p u e s to de l E je m p lo 20 " re p re se n ta el g ru po de a d m in is tra c ió n de l co m p u e s to de l E je m p lo 20, el "C o m p u e s to del E je m p lo 29 " rep rese n ta el g rupo de ad m in is tra c ió n de l co m p u e s to de l E je m p lo 29 y el "C o m p u e s to del E je m p lo 57" rep rese n ta el g ru po de ad m in is tra c ió n del co m p u e s to del E je m p lo 57. El as te risco (*) in d ica la s ig n ifica c ió n e s ta d ís tica en com p a ra c ió n con el g ru po de ad m in is tra c ió n de l v e h íc u lo (p ru eb a t de S tu d e n t) (*: P < 0 ,05).
[0356 ] La in d u cc ió n m e d ia n te im iq u im o d a u m e n tó el g ro s o r de la o re ja el o c ta vo d ía d e sp u é s de la inducc ión , en el g ru po de ad m in is tra c ió n de l veh ícu lo , en 0 ,23 m m a 0 ,27 m m en co m p a ra c ió n con el g ro s o r de la o re ja an tes de la in du cc ión . Este au m e n to en el g ro s o r de la o re ja se su p rim ió e s ta d ís tica m e n te s ig n ifica tiva m e n te m ed ia n te la ad m in is tra c ió n de l co m p u e s to de l E je m p lo 20, el c o m p u e s to de l E je m p lo 29 o el co m p u e s to de l E je m p lo 57.
[0357 ] E stos resu ltad os reve la ron que el de riva d o de am ina c íc lica (I) o una sa l fa rm a c o ló g ic a m e n te ace p tab le de l m ism o m ue s tra un m arca do e fe c to su p re so r de los s ín to m a s de la pso rias is .
(Ejemplo 75) Efecto supresor de síntomas en el modelo de dermatitis alérgica de ratón inducida por DNFB:
[0358 ] Utilizando el aumento en la velocidad de hinchazón de la oreja como un índice de exacerbación de los síntomas, se evaluó el efecto del derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo en un modelo de dermatitis alérgica de ratón inducida por DNFB. El modelo de dermatitis alérgica de ratón inducida por DNFB se preparó mediante un método parcialmente modificado por Curzytek y col. (Pharmacological Reports, 2013, Vol. 65, p. 1237-1246).
[0359 ] Se utilizaron ratones BALB/c (hembra, de 6 semanas de edad) (Charles River Laboratories Japan, Inc.) a las 7 semanas de edad después de la reproducción preliminar. A la espalda de los ratones, se aplicaron 25 pL de una solución de DNFB al 0,5 v/v% disuelta en acetona: aceite de oliva (4:1). Al día siguiente, se repitió la misma operación para sensibilizar a los ratones. Cuatro días después de la sensibilización, se aplicaron 10 pL de una solución de DNFB al 0,2 v/v% disuelta en acetona: aceite de oliva (4:1) a ambos lados de la aurícula derecha de los ratones sensibilizados para inducir la inflamación.
[0360 ] Una hora antes de la inducción, se administró el compuesto del Ejemplo 20, el compuesto del Ejemplo 29 o el compuesto del Ejemplo 57 a una dosis de 10 mg/kg a los ratones. El compuesto del Ejemplo 20, el compuesto del Ejemplo 29 y el compuesto del Ejemplo 57 se suspendieron en una solución de metilcelulosa al 0,5% p/v y se administraron por vía oral. El grupo en el que se administró el compuesto del Ejemplo 20 a ratones se definió como el grupo de administración del compuesto del Ejemplo 20, el grupo en el que se administró el compuesto del Ejemplo 29 se definió como el grupo de administración del compuesto del Ejemplo 29 y el grupo en el que se administró el compuesto del Ejemplo 57 se definió como el grupo de administración del compuesto del Ejemplo -57. En el grupo de administración del vehículo, se administró de manera similar un vehículo de cada compuesto de prueba (solución de metilcelulosa al 0,5% p/v).
[0361 ] Se midieron el grosor de la oreja derecha antes de la aplicación de la solución de DNFB (antes de la inducción) el día de la inducción y el grosor de la oreja derecha a las 24 horas después de la inducción con un micrómetro digital (Mitutoyo Corporation). La velocidad de hinchazón de la aurícula se calculó mediante la siguiente Fórmula 3 y se utilizó como índice de la evaluación de la eficacia del fármaco.
[Fórmula 3]
Velocidad de hinchazón de la oreja (%) = ((Grosor de la oreja derecha a las 24 horas después de la inducción) -(Grosor de la oreja derecha antes de la inducción)); Grosor de la oreja derecha antes de la inducción x 100
[0362 ] Los resultados se muestran en las figuras 7, 8 y 9. El eje vertical representa la velocidad de hinchazón de la oreja (%) (error estándar medio de ± n = 6 a 8). "Vehículo" en el eje horizontal representa el grupo de administración del vehículo, el "Compuesto del Ejemplo 20" representa el grupo de administración del compuesto del Ejemplo 20, el "Compuesto del Ejemplo 29" representa el grupo de administración del compuesto del Ejemplo 29 y el "Compuesto del Ejemplo 57" representa el grupo de administración del compuesto del Ejemplo 57. El asterisco (*) indica la significación estadística en comparación con el grupo de administración del vehículo (prueba t de Student) (*: P < 0,05).
[0363 ] La aplicación de la solución de DNFB a la aurícula aumentó la velocidad de hinchazón de la oreja en el grupo de administración del vehículo en un 30,9 % a 41,7 %. Este aumento en la velocidad de hinchazón de la oreja se suprimió estadísticamente significativamente mediante la administración del compuesto del Ejemplo 20, el compuesto del Ejemplo 29 o el compuesto del Ejemplo 57.
[0364 ] Este resultado reveló que el derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo muestra un marcado efecto supresor de síntomas de la dermatitis alérgica, particularmente la dermatitis de contacto.
(Ejemplo 76) Efecto supresor de síntomas en el modelo de dermatitis atópica de ratón inducida por oxazolona:
[0365 ] Utilizando el aumento en el grosor de la oreja como un índice de exacerbación de los síntomas, se evaluó el efecto del derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo en un modelo de dermatitis atópica de ratón inducida por oxazolona. El modelo de dermatitis atópica de ratón inducida por oxazolona se preparó mediante un método parcialmente modificado por Nakajima y col. (Journal of Investigative Dermatology, 2014, vol. 134, p.2122-2130).
[0366 ] Se utilizaron ratones BALB/c (hembra, de 7 semanas de edad) (Charles River Laboratories Japan, Inc.) a las 8 o 9 semanas de edad después de la reproducción preliminar. A la espalda de los ratones, se aplicaron 25 pL de una solución de oxazolona al 3% p/v disuelta en etanol para sensibilizar a los ratones. Cada dos días de los 5 días a los 13 días después de la sensibilización, se aplicaron 10 pL de una solución de oxazolona al 0,6% p/v disuelta en etanol a ambos lados de la aurícula derecha de los ratones sensibilizados para inducir la inflamación.
[0367 ] Se administró el compuesto de prueba a una dosis de 10 mg/kg a los ratones una vez al día durante 15 días desde el día de sensibilización hasta 14 días después de la sensibilización. Como compuesto de prueba, se utilizaron el compuesto del Ejemplo 20, el compuesto del Ejemplo 29 y el compuesto del Ejemplo 57. El compuesto del Ejemplo 20, el compuesto del Ejemplo
29 y el compuesto del Ejemplo 57 se suspendieron en una solución de metilcelulosa al 0,5% p/v y se administraron por vía oral. El grupo en el que se administró el compuesto del Ejemplo 20 a ratones se definió como el grupo de administración del compuesto del Ejemplo 20, el grupo en el que se administró el compuesto del Ejemplo 29 se definió como el grupo de administración del compuesto del Ejemplo 29 y el grupo en el que se administró el compuesto del Ejemplo 57 se definió como el grupo de administración del compuesto del Ejemplo -57. En el grupo de administración del vehículo, se administró de manera similar un vehículo de cada compuesto de prueba (solución de metilcelulosa al 0,5% p/v).
[0368 ] Se midieron el grosor de la oreja derecha antes de la aplicación de la solución de oxazolona (antes de la sensibilización) el día de la sensibilización y el grosor de la oreja derecha al día siguiente de la inducción final con un micrómetro digital (Mitutoyo Corporation). Se utilizó el cambio en el grosor de la oreja derecha (grosor de la oreja derecha al día siguiente de la inducción final - grosor de la oreja derecha antes de la sensibilización) como índice de evaluación de la eficacia del fármaco.
[0369 ] Los resultados se muestran en las figuras 10, 11 y 12. El eje vertical representa el cambio en el grosor de la oreja (mm) (error estándar medio de ± n = 7). "Vehículo" en el eje horizontal representa el grupo de administración del vehículo, el "Compuesto del Ejemplo -20" representa el grupo de administración del compuesto del Ejemplo 20, el "Compuesto del Ejemplo 29" representa el grupo de administración del compuesto del Ejemplo 29 y el "Compuesto del Ejemplo 57" representa el grupo de administración del compuesto del Ejemplo 57. El asterisco (*) indica la significación estadística en comparación con el grupo de administración del vehículo (prueba t de Student) (*: P < 0,05).
[0370 ] La aplicación de la solución de oxazolona a la aurícula aumentó el grosor de la oreja al día siguiente de la inducción final en el grupo de administración del vehículo en 0,60 mm a 0,70 mm en comparación con el grosor de la oreja antes de la sensibilización. Este aumento en el grosor de la oreja se suprimió estadísticamente significativamente mediante la administración del compuesto del Ejemplo 20, el compuesto del Ejemplo 29 o el compuesto del Ejemplo 57.
[0371 ] Este resultado reveló que el derivado de amina cíclica (I) o una sal farmacológicamente aceptable del mismo muestra un marcado efecto supresor de síntomas de la dermatitis alérgica, particularmente la dermatitis atópica. APLICABILIDAD INDUSTRIAL
[0372 ] Dado que el derivado de amina cíclica de la presente invención o una sal farmacológicamente aceptable del mismo tiene una excelente actividad antagonista de RORy, se puede utilizar como un medicamento para enfermedades en las que cabe esperar una mejora en el estado patológico o una remisión de los síntomas mediante la supresión de la función de RORy. Particularmente, el derivado de amina cíclica de la presente invención o una sal farmacológicamente aceptable del mismo se puede utilizar como agente terapéutico o agente preventivo para enfermedades autoinmunitarias, tales como esclerosis múltiple o psoriasis, o enfermedades alérgicas, que incluyen dermatitis alérgica o similares, tales como dermatitis de contacto o dermatitis atópica.
Claims (12)
1. Un derivado de amina cíclica representado por la siguiente fórmula general (I):

donde
R1 representa un grupo alquiloxi que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno;
R2 representa un átomo de halógeno;
R3 representa un átomo de hidrógeno, un átomo de halógeno o un grupo hidroxi;
R4 representa un átomo de hidrógeno o un átomo de halógeno;
X representa -C(=O)-(CH2)n-R5 o -S(=O)2-R6;
n es un número entero de 0 a 5;
R5 representa un átomo de hidrógeno, -OR7, -SR7, -S(=O)2-R7, -C(=O)-OR7, -N(R7)R8, un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno o un grupo heteroarilo, cualquiera de cuyos átomos de hidrógeno están opcionalmente sustituidos con uno o más grupos alquilo que tienen de 1 a 3 átomos de carbono;
R6 representa un grupo alquilo que tiene de 1 a 5 átomos de carbono;
R7 representa un átomo de hidrógeno o un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de halógeno; y R8 representa un átomo de hidrógeno, un grupo alquilo que tiene de 1 a 3 átomos de carbono, un grupo acilo que tiene de 2 a 4 átomos de carbono, o un grupo alquilsulfonilo que tiene de 1 a 3 átomos de carbono,
o una sal farmacéuticamente aceptable de los mismos.
2. El derivado de amina cíclica según la reivindicación 1, donde
R1 es un grupo alquiloxi que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de flúor o uno o más átomos de cloro;
R2 es un átomo de flúor o un átomo de cloro;
R3 es un átomo de hidrógeno, un átomo de flúor, un átomo de cloro o un grupo hidroxi;
R4 es un átomo de hidrógeno, un átomo de flúor o un átomo de cloro;
R5 es un átomo de hidrógeno, -OR7, -SR7, -S(=O)2-R7, -C (=O)-OR7, -N(R7)R8, un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de flúor o uno o más átomos de cloro, o un grupo heteroarilo, cualquiera de cuyos átomos de hidrógeno están opcionalmente sustituidos con uno o más grupos metilo;
R6 es un grupo alquilo que tiene de 1 a 3 átomos de carbono; y
R7 es un átomo de hidrógeno o un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de flúor o uno o más átomos de cloro,
o una sal farmacológicamente aceptable de los mismos.
3. El derivado de amina cíclica según la reivindicación 1, donde
R1 es un grupo metoxi, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de flúor;
R2 es un átomo de flúor o un átomo de cloro;
R3 es un átomo de hidrógeno, un átomo de flúor o un grupo hidroxi;
R4 es un átomo de hidrógeno o un átomo de flúor;
n es un número entero de 0 a 4;
R5 es un átomo de hidrógeno, -OR7, -N(R7)R8, un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de flúor o un grupo heteroarilo de anillo de 5 miembros, cualquiera de cuyos átomos de hidrógeno están opcionalmente sustituidos con uno o más grupos metilo;
R6 es un grupo metilo o etilo,
R7 es un átomo de hidrógeno o un grupo alquilo que tiene de 1 a 3 átomos de carbono, cualquiera de cuyos 1 a 3 átomos de hidrógeno están opcionalmente sustituidos con uno o más átomos de flúor; y
R8 es un átomo de hidrógeno, un grupo metilo, un grupo acilo que tiene de 2 a 4 átomos de carbono, o un grupo
alquilsulfonilo que tiene de 1 a 3 átomos de carbono,
o una sal farmacológicamente aceptable de los mismos.
4. El derivado de amina cíclica según la reivindicación 1, donde
R1 es un grupo trifluorometoxi;
R2 es un átomo de cloro;
R3 es un átomo de hidrógeno;
R4 es un átomo de hidrógeno;
X es -C(=O)-(CH2)n-R5;
n es un número entero de 0 a 3;
R5 es un grupo metilo, un grupo trifluorometilo, -N(R7)R8, o un grupo imidazolilo, triazolilo o tetrazolilo, cualquiera de cuyos átomos de hidrógeno están opcionalmente sustituidos con uno o más grupos metilo;
R7 es un átomo de hidrógeno, un grupo metilo o un grupo etilo; y
R8 es un átomo de hidrógeno, un grupo metilo, un grupo acetilo, un grupo propionilo, un grupo metilsulfonilo o un grupo etilsulfonilo,
o una sal farmacológicamente aceptable de los mismos.
5. El derivado de amina cíclica según cualquiera de las reivindicaciones 1 a 4, o una sal farmacológicamente aceptable del mismo, para su uso como medicamento.
6. Un medicamento que comprende el derivado de amina cíclica según cualquiera de las reivindicaciones 1 a 4, o una sal farmacológicamente aceptable del mismo, como principio activo.
7. El derivado de amina cíclica según cualquiera de las reivindicaciones 1 a 4, o una sal farmacológicamente aceptable del mismo, o el medicamento según la reivindicación 6, para su uso en el tratamiento o prevención de una enfermedad autoinmunitaria.
8. El derivado de amina cíclica según cualquiera de las reivindicaciones 1 a 4, o una sal farmacológicamente aceptable del mismo, o el medicamento según la reivindicación 6, para su uso en el tratamiento o prevención de esclerosis múltiple o psoriasis.
9. El derivado de amina cíclica según cualquiera de las reivindicaciones 1 a 4, o una sal farmacológicamente aceptable del mismo, o el medicamento según la reivindicación 6, para su uso en el tratamiento o prevención de una enfermedad alérgica.
10. El derivado de amina cíclica según cualquiera de las reivindicaciones 1 a 4, o una sal farmacológicamente aceptable del mismo, o el medicamento según la reivindicación 6, para su uso en el tratamiento o prevención de la dermatitis alérgica.
11. El derivado de amina cíclica según cualquiera de las reivindicaciones 1 a 4, o una sal farmacológicamente aceptable del mismo, o el medicamento según la reivindicación 6, para su uso en el tratamiento o prevención de dermatitis de contacto o dermatitis atópica.
12. Un antagonista del receptor y huérfano relacionado con retinoides, que comprende el derivado de amina cíclica según cualquiera de las reivindicaciones 1 a 4, o una sal farmacológicamente aceptable del mismo, como principio activo.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016015512 | 2016-01-29 | ||
| JP2016212629 | 2016-10-31 | ||
| PCT/JP2017/002924 WO2017131156A1 (ja) | 2016-01-29 | 2017-01-27 | 環状アミン誘導体及びその医薬用途 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2822606T3 true ES2822606T3 (es) | 2021-05-04 |
Family
ID=59398579
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17744386T Active ES2822606T3 (es) | 2016-01-29 | 2017-01-27 | Derivado de amina cíclica y uso farmacéutico del mismo |
Country Status (23)
| Country | Link |
|---|---|
| US (1) | US10364222B2 (es) |
| EP (1) | EP3409660B1 (es) |
| JP (1) | JP6807054B2 (es) |
| KR (1) | KR20180100573A (es) |
| CN (1) | CN108473426B (es) |
| AU (1) | AU2017210685B2 (es) |
| BR (1) | BR112018015125A2 (es) |
| CA (1) | CA3009971C (es) |
| DK (1) | DK3409660T3 (es) |
| ES (1) | ES2822606T3 (es) |
| HU (1) | HUE050744T2 (es) |
| IL (1) | IL260230B (es) |
| MX (1) | MX2018008454A (es) |
| MY (1) | MY195416A (es) |
| PH (1) | PH12018501434A1 (es) |
| PL (1) | PL3409660T3 (es) |
| PT (1) | PT3409660T (es) |
| RU (1) | RU2696862C1 (es) |
| SG (1) | SG11201805474PA (es) |
| TR (1) | TR201912580U5 (es) |
| TW (1) | TWI691486B (es) |
| WO (1) | WO2017131156A1 (es) |
| ZA (1) | ZA201804325B (es) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2019022234A1 (ja) * | 2017-07-27 | 2020-05-28 | 東レ株式会社 | 炎症性腸疾患の治療剤又は予防剤 |
| CA3071235A1 (en) * | 2017-07-27 | 2019-01-31 | Toray Industries, Inc. | Therapeutic agent or preventive agent for alopecia areata |
| TW201910312A (zh) * | 2017-07-28 | 2019-03-16 | 日商東麗股份有限公司 | 環狀胺衍生物及其醫藥用途 |
| WO2019044940A1 (ja) * | 2017-08-31 | 2019-03-07 | 東レ株式会社 | 環状アミン誘導体及びその医薬用途 |
| BR112020013438A2 (pt) | 2018-01-31 | 2020-12-01 | Toray Industries, Inc. | derivado de amina cíclico, medicamento, antagonista de receptor órfão gama relacionado a retinoide, e, agentes terapêuticos ou agentes preventivos para uma doença autoimune, para psoríase ou alopecia areata, para uma doença alérgica, para dermatite alérgica e para dermatite de contato ou dermatite atópica |
| US11230555B2 (en) | 2018-03-12 | 2022-01-25 | Escalier Biosciences B.V. | Bicyclic RORγ modulators |
| CN112118841A (zh) | 2018-03-12 | 2020-12-22 | 爱思凯利尔生物科学私人有限责任公司 | 螺环ROR-γ调节剂 |
| CN111548303A (zh) * | 2020-06-28 | 2020-08-18 | 山东美泰医药有限公司 | 一种盐酸左布比卡因的杂质及其制备和分析方法 |
| CN113252831B (zh) * | 2021-05-20 | 2022-08-26 | 京博农化科技有限公司 | N-氯甲酰基-n-[4-(三氟甲氧基)苯基]氨基甲酸甲酯超高效液相色谱分析方法 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101203512A (zh) | 2005-04-22 | 2008-06-18 | 第一三共株式会社 | 杂环化合物 |
| GB0510139D0 (en) | 2005-05-18 | 2005-06-22 | Addex Pharmaceuticals Sa | Novel compounds B1 |
| EA201000958A1 (ru) * | 2007-12-12 | 2010-12-30 | Астразенека Аб | Пептидилнитрилы и их применение в качестве ингибиторов дипептидилпептидазы i |
| US8703761B2 (en) | 2008-07-15 | 2014-04-22 | Novartis Ag | Organic compounds |
| JP5705748B2 (ja) * | 2009-02-18 | 2015-04-22 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Cb2受容体を変調する複素環化合物 |
| WO2012027965A1 (en) | 2010-09-01 | 2012-03-08 | Glaxo Group Limited | Novel compounds |
| WO2012064744A2 (en) | 2010-11-08 | 2012-05-18 | Lycera Corporation | Tetrahydroquinoline and related bicyclic compounds for inhibition of rorϒ activity and the treatment of disease |
| MX2013012542A (es) | 2011-04-28 | 2013-11-20 | Japan Tobacco Inc | Compuestos de amida y aplicacion farmaceutica para el mismo. |
| WO2012158784A2 (en) | 2011-05-16 | 2012-11-22 | Theodore Mark Kamenecka | Modulators of the nuclear hormone receptor ror |
| US9156837B2 (en) | 2011-07-29 | 2015-10-13 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| GB201207406D0 (en) * | 2012-04-27 | 2012-06-13 | Glaxo Group Ltd | Novel compounds |
| EA201791271A1 (ru) * | 2012-05-31 | 2018-01-31 | Фенекс Фармасьютикалз Аг | ТИАЗОЛЫ, ЗАМЕЩЕННЫЕ КАРБОКСАМИДОМ ИЛИ СУЛЬФОНАМИДОМ, И РОДСТВЕННЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ ДЛЯ ОРФАННОГО ЯДЕРНОГО РЕЦЕПТОРА RORγ |
| BR112015022226A2 (pt) * | 2013-03-14 | 2017-07-18 | Celtaxsys Inc | inibidores de leucotrieno a4 hidrolase |
| WO2015103508A1 (en) * | 2014-01-06 | 2015-07-09 | Bristol-Myers Squibb Company | CARBOCYCLIC SULFONE RORγ MODULATORS |
| ES2704460T3 (es) * | 2014-01-06 | 2019-03-18 | Bristol Myers Squibb Co | Moduladores de ROR gamma de ciclohexilsulfona |
| WO2015129853A1 (ja) * | 2014-02-27 | 2015-09-03 | 東レ株式会社 | 環状アミン誘導体及びその医薬用途 |
-
2017
- 2017-01-25 TW TW106102788A patent/TWI691486B/zh active
- 2017-01-27 CN CN201780008139.2A patent/CN108473426B/zh active Active
- 2017-01-27 MY MYPI2018000987A patent/MY195416A/en unknown
- 2017-01-27 ES ES17744386T patent/ES2822606T3/es active Active
- 2017-01-27 CA CA3009971A patent/CA3009971C/en active Active
- 2017-01-27 WO PCT/JP2017/002924 patent/WO2017131156A1/ja active Application Filing
- 2017-01-27 DK DK17744386.8T patent/DK3409660T3/da active
- 2017-01-27 MX MX2018008454A patent/MX2018008454A/es unknown
- 2017-01-27 AU AU2017210685A patent/AU2017210685B2/en active Active
- 2017-01-27 PT PT177443868T patent/PT3409660T/pt unknown
- 2017-01-27 US US16/072,482 patent/US10364222B2/en active Active
- 2017-01-27 HU HUE17744386A patent/HUE050744T2/hu unknown
- 2017-01-27 EP EP17744386.8A patent/EP3409660B1/en active Active
- 2017-01-27 RU RU2018126968A patent/RU2696862C1/ru active
- 2017-01-27 JP JP2017507454A patent/JP6807054B2/ja active Active
- 2017-01-27 SG SG11201805474PA patent/SG11201805474PA/en unknown
- 2017-01-27 PL PL17744386T patent/PL3409660T3/pl unknown
- 2017-01-27 KR KR1020187019685A patent/KR20180100573A/ko not_active Application Discontinuation
- 2017-01-27 BR BR112018015125-4A patent/BR112018015125A2/pt active Search and Examination
-
2018
- 2018-06-24 IL IL260230A patent/IL260230B/en active IP Right Grant
- 2018-06-27 ZA ZA2018/04325A patent/ZA201804325B/en unknown
- 2018-07-04 PH PH12018501434A patent/PH12018501434A1/en unknown
-
2019
- 2019-08-21 TR TR2019/12580U patent/TR201912580U5/tr unknown
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2017131156A1 (ja) | 2018-11-22 |
| PT3409660T (pt) | 2020-07-31 |
| EP3409660B1 (en) | 2020-07-01 |
| US10364222B2 (en) | 2019-07-30 |
| PH12018501434A1 (en) | 2019-03-04 |
| EP3409660A4 (en) | 2019-06-26 |
| CN108473426B (zh) | 2021-09-07 |
| PL3409660T3 (pl) | 2021-01-25 |
| CA3009971A1 (en) | 2017-08-03 |
| MX2018008454A (es) | 2018-08-14 |
| IL260230B (en) | 2020-06-30 |
| SG11201805474PA (en) | 2018-07-30 |
| JP6807054B2 (ja) | 2021-01-06 |
| KR20180100573A (ko) | 2018-09-11 |
| MY195416A (en) | 2023-01-20 |
| TW201731816A (zh) | 2017-09-16 |
| CN108473426A (zh) | 2018-08-31 |
| ZA201804325B (en) | 2019-09-25 |
| HUE050744T2 (hu) | 2021-01-28 |
| US20180370916A1 (en) | 2018-12-27 |
| WO2017131156A1 (ja) | 2017-08-03 |
| RU2696862C1 (ru) | 2019-08-07 |
| AU2017210685A1 (en) | 2018-07-05 |
| TR201912580U5 (tr) | 2020-09-21 |
| DK3409660T3 (da) | 2020-08-31 |
| BR112018015125A2 (pt) | 2019-02-26 |
| CA3009971C (en) | 2024-03-19 |
| AU2017210685B2 (en) | 2020-09-24 |
| TWI691486B (zh) | 2020-04-21 |
| EP3409660A1 (en) | 2018-12-05 |
| IL260230A (en) | 2018-07-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2822606T3 (es) | Derivado de amina cíclica y uso farmacéutico del mismo | |
| JP2022539538A (ja) | Il-17調節剤としての縮合イミダゾール誘導体 | |
| US10150762B2 (en) | Trifluoromethyl alcohols as modulators of RORγt | |
| BR112019024488A2 (pt) | inibidores de pirazol magl | |
| EP3448856A1 (en) | 6-aminopyridin-3-yl thiazoles as modulators of ror t | |
| US10881649B2 (en) | Therapeutic agent or preventive agent for alopecia areata | |
| JPWO2019022223A1 (ja) | 環状アミン誘導体及びその医薬用途 | |
| BR112020005174A2 (pt) | composto que possui estrutura cíclica | |
| RU2772507C2 (ru) | Терапевтическое средство или профилактическое средство против гнездной алопеции | |
| WO2019022234A1 (ja) | 炎症性腸疾患の治療剤又は予防剤 |