ES2804510T3 - Composiciones y métodos para inhibir la expresión del gen ALAS1 - Google Patents
Composiciones y métodos para inhibir la expresión del gen ALAS1 Download PDFInfo
- Publication number
- ES2804510T3 ES2804510T3 ES14800179T ES14800179T ES2804510T3 ES 2804510 T3 ES2804510 T3 ES 2804510T3 ES 14800179 T ES14800179 T ES 14800179T ES 14800179 T ES14800179 T ES 14800179T ES 2804510 T3 ES2804510 T3 ES 2804510T3
- Authority
- ES
- Spain
- Prior art keywords
- dsrna
- alas1
- porphyria
- rnai
- nucleotides
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 230000014509 gene expression Effects 0.000 title claims abstract description 135
- 238000000034 method Methods 0.000 title claims description 100
- 101150038201 ALAS1 gene Proteins 0.000 title claims description 81
- 239000000203 mixture Substances 0.000 title claims description 72
- 125000003729 nucleotide group Chemical group 0.000 claims abstract description 366
- 239000002773 nucleotide Substances 0.000 claims abstract description 326
- 230000000692 anti-sense effect Effects 0.000 claims abstract description 224
- 101000843649 Homo sapiens 5-aminolevulinate synthase, nonspecific, mitochondrial Proteins 0.000 claims abstract description 215
- 102100030755 5-aminolevulinate synthase, nonspecific, mitochondrial Human genes 0.000 claims abstract description 208
- 108091081021 Sense strand Proteins 0.000 claims abstract description 118
- 229920002477 rna polymer Polymers 0.000 claims abstract description 23
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 claims abstract description 22
- 239000002342 ribonucleoside Substances 0.000 claims abstract description 16
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims abstract description 13
- 210000004027 cell Anatomy 0.000 claims description 159
- 206010036182 Porphyria acute Diseases 0.000 claims description 147
- 108091032973 (ribonucleotides)n+m Proteins 0.000 claims description 133
- 241000097929 Porphyria Species 0.000 claims description 131
- 208000010642 Porphyrias Diseases 0.000 claims description 131
- 108020004999 messenger RNA Proteins 0.000 claims description 129
- 239000003446 ligand Substances 0.000 claims description 93
- 208000005452 Acute intermittent porphyria Diseases 0.000 claims description 89
- 229960002749 aminolevulinic acid Drugs 0.000 claims description 78
- ZGXJTSGNIOSYLO-UHFFFAOYSA-N 88755TAZ87 Chemical compound NCC(=O)CCC(O)=O ZGXJTSGNIOSYLO-UHFFFAOYSA-N 0.000 claims description 76
- 150000004032 porphyrins Chemical class 0.000 claims description 66
- 230000001154 acute effect Effects 0.000 claims description 43
- 208000024891 symptom Diseases 0.000 claims description 42
- 210000002700 urine Anatomy 0.000 claims description 41
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 39
- 208000002193 Pain Diseases 0.000 claims description 35
- 230000007823 neuropathy Effects 0.000 claims description 31
- 201000001119 neuropathy Diseases 0.000 claims description 31
- 208000033808 peripheral neuropathy Diseases 0.000 claims description 31
- 208000033552 hepatic porphyria Diseases 0.000 claims description 30
- 208000035475 disorder Diseases 0.000 claims description 29
- 239000008194 pharmaceutical composition Substances 0.000 claims description 27
- MBLBDJOUHNCFQT-UHFFFAOYSA-N N-acetyl-D-galactosamine Natural products CC(=O)NC(C=O)C(O)C(O)C(O)CO MBLBDJOUHNCFQT-UHFFFAOYSA-N 0.000 claims description 25
- 150000001720 carbohydrates Chemical class 0.000 claims description 23
- 201000010275 acute porphyria Diseases 0.000 claims description 18
- 210000003494 hepatocyte Anatomy 0.000 claims description 18
- 230000002401 inhibitory effect Effects 0.000 claims description 18
- OOOQNKMJLOLMHC-UHFFFAOYSA-N 5-[[3,4-diethyl-5-[[5-formyl-3-(3-hydroxypropyl)-4-methyl-1h-pyrrol-2-yl]methyl]-1h-pyrrol-2-yl]methyl]-4-(3-hydroxypropyl)-3-methyl-1h-pyrrole-2-carbaldehyde Chemical compound N1C(CC2=C(C(C)=C(C=O)N2)CCCO)=C(CC)C(CC)=C1CC=1NC(C=O)=C(C)C=1CCCO OOOQNKMJLOLMHC-UHFFFAOYSA-N 0.000 claims description 17
- 208000003914 Acute hepatic porphyria Diseases 0.000 claims description 15
- 108020004414 DNA Proteins 0.000 claims description 15
- 238000003556 assay Methods 0.000 claims description 15
- 108700016481 Acute Hepatic Porphyria Proteins 0.000 claims description 14
- 230000001376 precipitating effect Effects 0.000 claims description 14
- 230000015556 catabolic process Effects 0.000 claims description 13
- 238000006731 degradation reaction Methods 0.000 claims description 13
- 230000002829 reductive effect Effects 0.000 claims description 13
- 230000003442 weekly effect Effects 0.000 claims description 11
- 208000000094 Chronic Pain Diseases 0.000 claims description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 7
- 150000004713 phosphodiesters Chemical class 0.000 claims description 6
- 102000053602 DNA Human genes 0.000 claims description 5
- 239000000126 substance Substances 0.000 claims description 5
- 150000003839 salts Chemical class 0.000 claims description 4
- 238000007920 subcutaneous administration Methods 0.000 claims description 4
- 239000008215 water for injection Substances 0.000 claims description 4
- 230000037396 body weight Effects 0.000 claims description 3
- 208000028389 Nerve injury Diseases 0.000 claims description 2
- 230000008764 nerve damage Effects 0.000 claims description 2
- 230000000069 prophylactic effect Effects 0.000 claims description 2
- OVRNDRQMDRJTHS-KEWYIRBNSA-N N-acetyl-D-galactosamine Chemical compound CC(=O)N[C@H]1C(O)O[C@H](CO)[C@H](O)[C@@H]1O OVRNDRQMDRJTHS-KEWYIRBNSA-N 0.000 claims 1
- 208000033685 pterin-4 alpha-carbinolamine dehydratase 1 deficiency Diseases 0.000 claims 1
- 230000009368 gene silencing by RNA Effects 0.000 description 256
- 108091030071 RNAI Proteins 0.000 description 254
- QSHWIQZFGQKFMA-UHFFFAOYSA-N porphobilinogen Chemical compound NCC=1NC=C(CCC(O)=O)C=1CC(O)=O QSHWIQZFGQKFMA-UHFFFAOYSA-N 0.000 description 140
- 230000004048 modification Effects 0.000 description 114
- 238000012986 modification Methods 0.000 description 114
- 239000003795 chemical substances by application Substances 0.000 description 89
- 108020004459 Small interfering RNA Proteins 0.000 description 77
- 125000005647 linker group Chemical group 0.000 description 75
- 108091034117 Oligonucleotide Proteins 0.000 description 69
- 239000000562 conjugate Substances 0.000 description 63
- 230000000295 complement effect Effects 0.000 description 61
- 238000011282 treatment Methods 0.000 description 60
- 108090000765 processed proteins & peptides Proteins 0.000 description 55
- 108010056651 Hydroxymethylbilane synthase Proteins 0.000 description 48
- 230000000694 effects Effects 0.000 description 47
- 102100034391 Porphobilinogen deaminase Human genes 0.000 description 45
- 150000003278 haem Chemical class 0.000 description 42
- DDBREPKUVSBGFI-UHFFFAOYSA-N phenobarbital Chemical compound C=1C=CC=CC=1C1(CC)C(=O)NC(=O)NC1=O DDBREPKUVSBGFI-UHFFFAOYSA-N 0.000 description 41
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 40
- -1 hexose nucleic acid Chemical class 0.000 description 40
- 108090000623 proteins and genes Proteins 0.000 description 38
- 150000007523 nucleic acids Chemical class 0.000 description 37
- 229960002695 phenobarbital Drugs 0.000 description 37
- 210000004185 liver Anatomy 0.000 description 35
- 208000000627 Hereditary Coproporphyria Diseases 0.000 description 34
- 208000033141 Porphyria variegata Diseases 0.000 description 34
- 102100029028 Protoporphyrinogen oxidase Human genes 0.000 description 34
- 201000011053 Variegate Porphyria Diseases 0.000 description 34
- 102000004190 Enzymes Human genes 0.000 description 33
- 108090000790 Enzymes Proteins 0.000 description 33
- 238000003776 cleavage reaction Methods 0.000 description 31
- 229940025294 hemin Drugs 0.000 description 31
- BTIJJDXEELBZFS-QDUVMHSLSA-K hemin Chemical compound CC1=C(CCC(O)=O)C(C=C2C(CCC(O)=O)=C(C)\C(N2[Fe](Cl)N23)=C\4)=N\C1=C/C2=C(C)C(C=C)=C3\C=C/1C(C)=C(C=C)C/4=N\1 BTIJJDXEELBZFS-QDUVMHSLSA-K 0.000 description 31
- 239000002777 nucleoside Substances 0.000 description 31
- 230000009467 reduction Effects 0.000 description 31
- 230000007017 scission Effects 0.000 description 31
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 30
- 239000011734 sodium Substances 0.000 description 30
- 125000002015 acyclic group Chemical group 0.000 description 27
- 150000002632 lipids Chemical class 0.000 description 26
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 25
- 102000039446 nucleic acids Human genes 0.000 description 25
- 108020004707 nucleic acids Proteins 0.000 description 25
- 239000002953 phosphate buffered saline Substances 0.000 description 25
- 239000003814 drug Substances 0.000 description 24
- 230000035772 mutation Effects 0.000 description 24
- 210000002381 plasma Anatomy 0.000 description 24
- 239000000523 sample Substances 0.000 description 24
- 239000002243 precursor Substances 0.000 description 23
- 210000002966 serum Anatomy 0.000 description 23
- 150000001875 compounds Chemical class 0.000 description 22
- 230000005764 inhibitory process Effects 0.000 description 22
- 235000014633 carbohydrates Nutrition 0.000 description 21
- 230000000875 corresponding effect Effects 0.000 description 21
- 230000008685 targeting Effects 0.000 description 21
- 241000700159 Rattus Species 0.000 description 20
- 230000001629 suppression Effects 0.000 description 20
- 210000004369 blood Anatomy 0.000 description 19
- 239000008280 blood Substances 0.000 description 19
- 230000007812 deficiency Effects 0.000 description 19
- 210000001519 tissue Anatomy 0.000 description 19
- 230000000750 progressive effect Effects 0.000 description 18
- 235000000346 sugar Nutrition 0.000 description 18
- 239000002253 acid Substances 0.000 description 17
- 229940079593 drug Drugs 0.000 description 17
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 16
- 108010016790 RNA-Induced Silencing Complex Proteins 0.000 description 16
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 16
- 230000001684 chronic effect Effects 0.000 description 16
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 16
- 230000000306 recurrent effect Effects 0.000 description 16
- 102000000574 RNA-Induced Silencing Complex Human genes 0.000 description 15
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 15
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol group Chemical group [C@@H]1(CC[C@H]2[C@@H]3CC=C4C[C@@H](O)CC[C@]4(C)[C@H]3CC[C@]12C)[C@H](C)CCCC(C)C HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 15
- 229940109239 creatinine Drugs 0.000 description 15
- 150000003833 nucleoside derivatives Chemical class 0.000 description 15
- 102000008100 Human Serum Albumin Human genes 0.000 description 14
- 108091006905 Human Serum Albumin Proteins 0.000 description 14
- 150000001413 amino acids Chemical group 0.000 description 14
- 239000008103 glucose Substances 0.000 description 14
- 230000037361 pathway Effects 0.000 description 14
- IYMAXBFPHPZYIK-BQBZGAKWSA-N Arg-Gly-Asp Chemical compound NC(N)=NCCC[C@H](N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(O)=O IYMAXBFPHPZYIK-BQBZGAKWSA-N 0.000 description 13
- 210000005229 liver cell Anatomy 0.000 description 13
- 208000004296 neuralgia Diseases 0.000 description 13
- 208000021722 neuropathic pain Diseases 0.000 description 13
- 125000003835 nucleoside group Chemical group 0.000 description 13
- 208000024624 porphyria due to ALA dehydratase deficiency Diseases 0.000 description 13
- 239000000047 product Substances 0.000 description 13
- 235000018102 proteins Nutrition 0.000 description 13
- 102000004169 proteins and genes Human genes 0.000 description 13
- 208000004998 Abdominal Pain Diseases 0.000 description 12
- 241001465754 Metazoa Species 0.000 description 12
- 125000000217 alkyl group Chemical group 0.000 description 12
- 230000027455 binding Effects 0.000 description 12
- 230000030279 gene silencing Effects 0.000 description 12
- 108091028043 Nucleic acid sequence Proteins 0.000 description 11
- 239000013060 biological fluid Substances 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 11
- 238000009472 formulation Methods 0.000 description 11
- 230000002440 hepatic effect Effects 0.000 description 11
- 238000000338 in vitro Methods 0.000 description 11
- 229920000768 polyamine Polymers 0.000 description 11
- 208000007209 Erythropoietic Porphyria Diseases 0.000 description 10
- 208000003591 Hepatoerythropoietic Porphyria Diseases 0.000 description 10
- 241000124008 Mammalia Species 0.000 description 10
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 10
- 229940024606 amino acid Drugs 0.000 description 10
- 235000001014 amino acid Nutrition 0.000 description 10
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 10
- 201000010099 disease Diseases 0.000 description 10
- 201000008220 erythropoietic protoporphyria Diseases 0.000 description 10
- 238000011156 evaluation Methods 0.000 description 10
- 102000056533 human ALAS1 Human genes 0.000 description 10
- 238000001727 in vivo Methods 0.000 description 10
- 230000003278 mimic effect Effects 0.000 description 10
- 239000000816 peptidomimetic Substances 0.000 description 10
- 206010010904 Convulsion Diseases 0.000 description 9
- 206010036186 Porphyria non-acute Diseases 0.000 description 9
- 101100016705 Rattus norvegicus Alas1 gene Proteins 0.000 description 9
- 230000006696 biosynthetic metabolic pathway Effects 0.000 description 9
- 150000002772 monosaccharides Chemical group 0.000 description 9
- 229910052758 niobium Inorganic materials 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- 102000004196 processed proteins & peptides Human genes 0.000 description 9
- 238000012384 transportation and delivery Methods 0.000 description 9
- 239000013598 vector Substances 0.000 description 9
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 8
- 229930024421 Adenine Natural products 0.000 description 8
- 208000034958 Congenital erythropoietic porphyria Diseases 0.000 description 8
- 101001067100 Homo sapiens Uroporphyrinogen-III synthase Proteins 0.000 description 8
- 102100034397 Uroporphyrinogen-III synthase Human genes 0.000 description 8
- 229960000643 adenine Drugs 0.000 description 8
- 150000002148 esters Chemical class 0.000 description 8
- 230000001976 improved effect Effects 0.000 description 8
- 230000001965 increasing effect Effects 0.000 description 8
- 238000004519 manufacturing process Methods 0.000 description 8
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 8
- 102000040430 polynucleotide Human genes 0.000 description 8
- 108091033319 polynucleotide Proteins 0.000 description 8
- 239000002157 polynucleotide Substances 0.000 description 8
- 229910052717 sulfur Inorganic materials 0.000 description 8
- 238000003786 synthesis reaction Methods 0.000 description 8
- 230000001052 transient effect Effects 0.000 description 8
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 7
- 241000024188 Andala Species 0.000 description 7
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical class C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 7
- 241000699666 Mus <mouse, genus> Species 0.000 description 7
- OVRNDRQMDRJTHS-CBQIKETKSA-N N-Acetyl-D-Galactosamine Chemical compound CC(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@H](O)[C@@H]1O OVRNDRQMDRJTHS-CBQIKETKSA-N 0.000 description 7
- 229910019142 PO4 Inorganic materials 0.000 description 7
- 150000007513 acids Chemical class 0.000 description 7
- 210000000267 erythroid cell Anatomy 0.000 description 7
- 238000013401 experimental design Methods 0.000 description 7
- BTIJJDXEELBZFS-UHFFFAOYSA-K hemin Chemical compound [Cl-].[Fe+3].[N-]1C(C=C2C(=C(C)C(C=C3C(=C(C)C(=C4)[N-]3)C=C)=N2)C=C)=C(C)C(CCC(O)=O)=C1C=C1C(CCC(O)=O)=C(C)C4=N1 BTIJJDXEELBZFS-UHFFFAOYSA-K 0.000 description 7
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 7
- 238000001802 infusion Methods 0.000 description 7
- 229910052760 oxygen Inorganic materials 0.000 description 7
- 229940055741 panhematin Drugs 0.000 description 7
- 235000021317 phosphate Nutrition 0.000 description 7
- 239000010452 phosphate Substances 0.000 description 7
- 230000008569 process Effects 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- 229940124597 therapeutic agent Drugs 0.000 description 7
- 230000001225 therapeutic effect Effects 0.000 description 7
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 6
- 101001021103 Homo sapiens Oxygen-dependent coproporphyrinogen-III oxidase, mitochondrial Proteins 0.000 description 6
- 102100036201 Oxygen-dependent coproporphyrinogen-III oxidase, mitochondrial Human genes 0.000 description 6
- 208000001871 Tachycardia Diseases 0.000 description 6
- 108010072041 arginyl-glycyl-aspartic acid Proteins 0.000 description 6
- 235000012000 cholesterol Nutrition 0.000 description 6
- 230000021615 conjugation Effects 0.000 description 6
- 238000009826 distribution Methods 0.000 description 6
- 239000003937 drug carrier Substances 0.000 description 6
- 230000006872 improvement Effects 0.000 description 6
- 210000003734 kidney Anatomy 0.000 description 6
- 239000002207 metabolite Substances 0.000 description 6
- 230000027758 ovulation cycle Effects 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 6
- 230000036470 plasma concentration Effects 0.000 description 6
- 229920001223 polyethylene glycol Polymers 0.000 description 6
- WDFJYRZCZIUBPR-UHFFFAOYSA-N preuroporphyrinogen Chemical compound OC(=O)CC1=C(CO)NC(CC2=C(C(CCC(O)=O)=C(CC3=C(C(CCC(O)=O)=C(CC4=C(C(CCC(O)=O)=CN4)CC(O)=O)N3)CC(O)=O)N2)CC(O)=O)=C1CCC(O)=O WDFJYRZCZIUBPR-UHFFFAOYSA-N 0.000 description 6
- 230000002265 prevention Effects 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- 230000006794 tachycardia Effects 0.000 description 6
- 229940088594 vitamin Drugs 0.000 description 6
- 229930003231 vitamin Natural products 0.000 description 6
- 235000013343 vitamin Nutrition 0.000 description 6
- 239000011782 vitamin Substances 0.000 description 6
- 108020005544 Antisense RNA Proteins 0.000 description 5
- 108090000371 Esterases Proteins 0.000 description 5
- 101100016698 Homo sapiens ALAS1 gene Proteins 0.000 description 5
- 206010028980 Neoplasm Diseases 0.000 description 5
- 208000037273 Pathologic Processes Diseases 0.000 description 5
- 239000002202 Polyethylene glycol Substances 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 238000009825 accumulation Methods 0.000 description 5
- 230000002378 acidificating effect Effects 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 239000002299 complementary DNA Substances 0.000 description 5
- 231100000673 dose–response relationship Toxicity 0.000 description 5
- 230000009977 dual effect Effects 0.000 description 5
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 5
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 5
- 230000003834 intracellular effect Effects 0.000 description 5
- 238000001990 intravenous administration Methods 0.000 description 5
- 208000014018 liver neoplasm Diseases 0.000 description 5
- 230000036961 partial effect Effects 0.000 description 5
- 230000009054 pathological process Effects 0.000 description 5
- 208000001297 phlebitis Diseases 0.000 description 5
- 238000011321 prophylaxis Methods 0.000 description 5
- 230000001105 regulatory effect Effects 0.000 description 5
- 208000031162 sideroblastic anemia Diseases 0.000 description 5
- 229940035893 uracil Drugs 0.000 description 5
- BHQCQFFYRZLCQQ-UHFFFAOYSA-N (3alpha,5alpha,7alpha,12alpha)-3,7,12-trihydroxy-cholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 BHQCQFFYRZLCQQ-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- KSFOVUSSGSKXFI-GAQDCDSVSA-N CC1=C/2NC(\C=C3/N=C(/C=C4\N\C(=C/C5=N/C(=C\2)/C(C=C)=C5C)C(C=C)=C4C)C(C)=C3CCC(O)=O)=C1CCC(O)=O Chemical compound CC1=C/2NC(\C=C3/N=C(/C=C4\N\C(=C/C5=N/C(=C\2)/C(C=C)=C5C)C(C=C)=C4C)C(C)=C3CCC(O)=O)=C1CCC(O)=O KSFOVUSSGSKXFI-GAQDCDSVSA-N 0.000 description 4
- 239000004380 Cholic acid Substances 0.000 description 4
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- 206010020772 Hypertension Diseases 0.000 description 4
- 206010065973 Iron Overload Diseases 0.000 description 4
- 102000007330 LDL Lipoproteins Human genes 0.000 description 4
- 108010007622 LDL Lipoproteins Proteins 0.000 description 4
- 108091005804 Peptidases Proteins 0.000 description 4
- 102000035195 Peptidases Human genes 0.000 description 4
- 108091093037 Peptide nucleic acid Proteins 0.000 description 4
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 4
- 108010039918 Polylysine Proteins 0.000 description 4
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 4
- 108091028664 Ribonucleotide Proteins 0.000 description 4
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 125000001931 aliphatic group Chemical group 0.000 description 4
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 4
- 229960002685 biotin Drugs 0.000 description 4
- 235000020958 biotin Nutrition 0.000 description 4
- 239000011616 biotin Substances 0.000 description 4
- 201000011510 cancer Diseases 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- BHQCQFFYRZLCQQ-OELDTZBJSA-N cholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 BHQCQFFYRZLCQQ-OELDTZBJSA-N 0.000 description 4
- 235000019416 cholic acid Nutrition 0.000 description 4
- 229960002471 cholic acid Drugs 0.000 description 4
- 239000003184 complementary RNA Substances 0.000 description 4
- 125000004122 cyclic group Chemical group 0.000 description 4
- 229940104302 cytosine Drugs 0.000 description 4
- 230000003111 delayed effect Effects 0.000 description 4
- KXGVEGMKQFWNSR-UHFFFAOYSA-N deoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 KXGVEGMKQFWNSR-UHFFFAOYSA-N 0.000 description 4
- 238000003745 diagnosis Methods 0.000 description 4
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 4
- 210000001163 endosome Anatomy 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 238000012226 gene silencing method Methods 0.000 description 4
- 230000002068 genetic effect Effects 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- 229910052739 hydrogen Inorganic materials 0.000 description 4
- 230000002209 hydrophobic effect Effects 0.000 description 4
- 230000006698 induction Effects 0.000 description 4
- 230000001939 inductive effect Effects 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 230000029849 luteinization Effects 0.000 description 4
- 210000000056 organ Anatomy 0.000 description 4
- 150000003904 phospholipids Chemical class 0.000 description 4
- 150000008300 phosphoramidites Chemical class 0.000 description 4
- 229920000656 polylysine Polymers 0.000 description 4
- 229950003776 protoporphyrin Drugs 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- 239000002336 ribonucleotide Substances 0.000 description 4
- 125000002652 ribonucleotide group Chemical group 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 125000001424 substituent group Chemical group 0.000 description 4
- 150000008163 sugars Chemical class 0.000 description 4
- 210000004881 tumor cell Anatomy 0.000 description 4
- AOTQGWFNFTVXNQ-UHFFFAOYSA-N 2-(1-adamantyl)acetic acid Chemical compound C1C(C2)CC3CC2CC1(CC(=O)O)C3 AOTQGWFNFTVXNQ-UHFFFAOYSA-N 0.000 description 3
- 102000009027 Albumins Human genes 0.000 description 3
- 108010088751 Albumins Proteins 0.000 description 3
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 3
- 208000019901 Anxiety disease Diseases 0.000 description 3
- 102000004506 Blood Proteins Human genes 0.000 description 3
- 108010017384 Blood Proteins Proteins 0.000 description 3
- 206010010774 Constipation Diseases 0.000 description 3
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 3
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 3
- 102000018832 Cytochromes Human genes 0.000 description 3
- 108010052832 Cytochromes Proteins 0.000 description 3
- 108010042407 Endonucleases Proteins 0.000 description 3
- 108091007413 Extracellular RNA Proteins 0.000 description 3
- 206010064571 Gene mutation Diseases 0.000 description 3
- 102100031181 Glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 108090000288 Glycoproteins Proteins 0.000 description 3
- 102000003886 Glycoproteins Human genes 0.000 description 3
- 102000016761 Haem oxygenases Human genes 0.000 description 3
- 108050006318 Haem oxygenases Proteins 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 108090001090 Lectins Proteins 0.000 description 3
- 102000004856 Lectins Human genes 0.000 description 3
- 101100230720 Mus musculus Alas1 gene Proteins 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 206010028817 Nausea and vomiting symptoms Diseases 0.000 description 3
- 108010077850 Nuclear Localization Signals Proteins 0.000 description 3
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical class OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 3
- 229930003427 Vitamin E Natural products 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 239000007853 buffer solution Substances 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 230000004700 cellular uptake Effects 0.000 description 3
- 239000013043 chemical agent Substances 0.000 description 3
- WIUGGJKHYQIGNH-UHFFFAOYSA-N coproporphyrinogen I Chemical compound C1C(=C(C=2C)CCC(O)=O)NC=2CC(=C(C=2C)CCC(O)=O)NC=2CC(N2)=C(CCC(O)=O)C(C)=C2CC2=C(CCC(O)=O)C(C)=C1N2 WIUGGJKHYQIGNH-UHFFFAOYSA-N 0.000 description 3
- NIUVHXTXUXOFEB-UHFFFAOYSA-N coproporphyrinogen III Chemical compound C1C(=C(C=2C)CCC(O)=O)NC=2CC(=C(C=2C)CCC(O)=O)NC=2CC(N2)=C(CCC(O)=O)C(C)=C2CC2=C(C)C(CCC(O)=O)=C1N2 NIUVHXTXUXOFEB-UHFFFAOYSA-N 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 201000008230 cutaneous porphyria Diseases 0.000 description 3
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 3
- 230000002354 daily effect Effects 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 238000012217 deletion Methods 0.000 description 3
- 230000037430 deletion Effects 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- NAGJZTKCGNOGPW-UHFFFAOYSA-N dithiophosphoric acid Chemical class OP(O)(S)=S NAGJZTKCGNOGPW-UHFFFAOYSA-N 0.000 description 3
- 210000003743 erythrocyte Anatomy 0.000 description 3
- 235000019152 folic acid Nutrition 0.000 description 3
- 239000011724 folic acid Substances 0.000 description 3
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 3
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 3
- 229940059489 heme arginate Drugs 0.000 description 3
- 125000000623 heterocyclic group Chemical group 0.000 description 3
- 229940088597 hormone Drugs 0.000 description 3
- 239000005556 hormone Substances 0.000 description 3
- 210000005260 human cell Anatomy 0.000 description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 239000002523 lectin Substances 0.000 description 3
- 230000007774 longterm Effects 0.000 description 3
- 210000004962 mammalian cell Anatomy 0.000 description 3
- 238000007726 management method Methods 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 238000002483 medication Methods 0.000 description 3
- 239000002679 microRNA Substances 0.000 description 3
- 239000000178 monomer Substances 0.000 description 3
- 230000002887 neurotoxic effect Effects 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 238000002515 oligonucleotide synthesis Methods 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- PTMHPRAIXMAOOB-UHFFFAOYSA-L phosphoramidate Chemical compound NP([O-])([O-])=O PTMHPRAIXMAOOB-UHFFFAOYSA-L 0.000 description 3
- 125000004437 phosphorous atom Chemical group 0.000 description 3
- 229920001282 polysaccharide Polymers 0.000 description 3
- 239000005017 polysaccharide Substances 0.000 description 3
- 150000004804 polysaccharides Chemical class 0.000 description 3
- 239000000186 progesterone Substances 0.000 description 3
- 235000019833 protease Nutrition 0.000 description 3
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 201000005060 thrombophlebitis Diseases 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- 238000002054 transplantation Methods 0.000 description 3
- 150000004043 trisaccharides Chemical class 0.000 description 3
- QTTNOSKSLATGQB-UHFFFAOYSA-N uroporphyrinogen I Chemical compound C1C(=C(C=2CCC(O)=O)CC(O)=O)NC=2CC(=C(C=2CCC(O)=O)CC(O)=O)NC=2CC(N2)=C(CC(O)=O)C(CCC(=O)O)=C2CC2=C(CC(O)=O)C(CCC(O)=O)=C1N2 QTTNOSKSLATGQB-UHFFFAOYSA-N 0.000 description 3
- HUHWZXWWOFSFKF-UHFFFAOYSA-N uroporphyrinogen-III Chemical compound C1C(=C(C=2CCC(O)=O)CC(O)=O)NC=2CC(=C(C=2CCC(O)=O)CC(O)=O)NC=2CC(N2)=C(CC(O)=O)C(CCC(=O)O)=C2CC2=C(CCC(O)=O)C(CC(O)=O)=C1N2 HUHWZXWWOFSFKF-UHFFFAOYSA-N 0.000 description 3
- 235000019165 vitamin E Nutrition 0.000 description 3
- 229940046009 vitamin E Drugs 0.000 description 3
- 239000011709 vitamin E Substances 0.000 description 3
- QGVQZRDQPDLHHV-DPAQBDIFSA-N (3s,8s,9s,10r,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1h-cyclopenta[a]phenanthrene-3-thiol Chemical compound C1C=C2C[C@@H](S)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 QGVQZRDQPDLHHV-DPAQBDIFSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- FZWGECJQACGGTI-UHFFFAOYSA-N 2-amino-7-methyl-1,7-dihydro-6H-purin-6-one Chemical compound NC1=NC(O)=C2N(C)C=NC2=N1 FZWGECJQACGGTI-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 2
- RYVNIFSIEDRLSJ-UHFFFAOYSA-N 5-(hydroxymethyl)cytosine Chemical compound NC=1NC(=O)N=CC=1CO RYVNIFSIEDRLSJ-UHFFFAOYSA-N 0.000 description 2
- 101710184696 5-aminolevulinate synthase, nonspecific, mitochondrial Proteins 0.000 description 2
- LRSASMSXMSNRBT-UHFFFAOYSA-N 5-methylcytosine Chemical compound CC1=CNC(=O)N=C1N LRSASMSXMSNRBT-UHFFFAOYSA-N 0.000 description 2
- UJBCLAXPPIDQEE-UHFFFAOYSA-N 5-prop-1-ynyl-1h-pyrimidine-2,4-dione Chemical compound CC#CC1=CNC(=O)NC1=O UJBCLAXPPIDQEE-UHFFFAOYSA-N 0.000 description 2
- QNNARSZPGNJZIX-UHFFFAOYSA-N 6-amino-5-prop-1-ynyl-1h-pyrimidin-2-one Chemical compound CC#CC1=CNC(=O)N=C1N QNNARSZPGNJZIX-UHFFFAOYSA-N 0.000 description 2
- HCGHYQLFMPXSDU-UHFFFAOYSA-N 7-methyladenine Chemical compound C1=NC(N)=C2N(C)C=NC2=N1 HCGHYQLFMPXSDU-UHFFFAOYSA-N 0.000 description 2
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical compound O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 2
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 2
- 108091032955 Bacterial small RNA Proteins 0.000 description 2
- VANGSNQGPRHWDS-UHFFFAOYSA-N CCCCCCCCCCCCCCCCC(C(C(CCCCCCCCCCCCCCCC)O)O)O.CCN(CC)CC Chemical compound CCCCCCCCCCCCCCCCC(C(C(CCCCCCCCCCCCCCCC)O)O)O.CCN(CC)CC VANGSNQGPRHWDS-UHFFFAOYSA-N 0.000 description 2
- 150000008574 D-amino acids Chemical class 0.000 description 2
- SHZGCJCMOBCMKK-UHFFFAOYSA-N D-mannomethylose Natural products CC1OC(O)C(O)C(O)C1O SHZGCJCMOBCMKK-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 2
- 208000005156 Dehydration Diseases 0.000 description 2
- 206010012735 Diarrhoea Diseases 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 206010014418 Electrolyte imbalance Diseases 0.000 description 2
- 102100031780 Endonuclease Human genes 0.000 description 2
- PNNNRSAQSRJVSB-SLPGGIOYSA-N Fucose Natural products C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C=O PNNNRSAQSRJVSB-SLPGGIOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- 208000004547 Hallucinations Diseases 0.000 description 2
- 102000001554 Hemoglobins Human genes 0.000 description 2
- 108010054147 Hemoglobins Proteins 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- 206010021036 Hyponatraemia Diseases 0.000 description 2
- 229930010555 Inosine Natural products 0.000 description 2
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 2
- 102100034349 Integrase Human genes 0.000 description 2
- 101710203526 Integrase Proteins 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- SHZGCJCMOBCMKK-DHVFOXMCSA-N L-fucopyranose Chemical compound C[C@@H]1OC(O)[C@@H](O)[C@H](O)[C@@H]1O SHZGCJCMOBCMKK-DHVFOXMCSA-N 0.000 description 2
- SMEROWZSTRWXGI-UHFFFAOYSA-N Lithocholsaeure Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)CC2 SMEROWZSTRWXGI-UHFFFAOYSA-N 0.000 description 2
- 108060001084 Luciferase Proteins 0.000 description 2
- 239000005089 Luciferase Substances 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 2
- OVRNDRQMDRJTHS-UHFFFAOYSA-N N-acelyl-D-glucosamine Natural products CC(=O)NC1C(O)OC(CO)C(O)C1O OVRNDRQMDRJTHS-UHFFFAOYSA-N 0.000 description 2
- OVRNDRQMDRJTHS-FMDGEEDCSA-N N-acetyl-beta-D-glucosamine Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O OVRNDRQMDRJTHS-FMDGEEDCSA-N 0.000 description 2
- MBLBDJOUHNCFQT-LXGUWJNJSA-N N-acetylglucosamine Natural products CC(=O)N[C@@H](C=O)[C@@H](O)[C@H](O)[C@H](O)CO MBLBDJOUHNCFQT-LXGUWJNJSA-N 0.000 description 2
- 208000008457 Neurologic Manifestations Diseases 0.000 description 2
- 206010060860 Neurological symptom Diseases 0.000 description 2
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical compound CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 2
- 206010033799 Paralysis Diseases 0.000 description 2
- 208000007542 Paresis Diseases 0.000 description 2
- KPKZJLCSROULON-QKGLWVMZSA-N Phalloidin Chemical compound N1C(=O)[C@@H]([C@@H](O)C)NC(=O)[C@H](C)NC(=O)[C@H](C[C@@](C)(O)CO)NC(=O)[C@H](C2)NC(=O)[C@H](C)NC(=O)[C@@H]3C[C@H](O)CN3C(=O)[C@@H]1CSC1=C2C2=CC=CC=C2N1 KPKZJLCSROULON-QKGLWVMZSA-N 0.000 description 2
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 2
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 2
- 239000004372 Polyvinyl alcohol Substances 0.000 description 2
- 101710149951 Protein Tat Proteins 0.000 description 2
- 208000001431 Psychomotor Agitation Diseases 0.000 description 2
- RADKZDMFGJYCBB-UHFFFAOYSA-N Pyridoxal Chemical compound CC1=NC=C(CO)C(C=O)=C1O RADKZDMFGJYCBB-UHFFFAOYSA-N 0.000 description 2
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 2
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 2
- 208000001647 Renal Insufficiency Diseases 0.000 description 2
- 208000004756 Respiratory Insufficiency Diseases 0.000 description 2
- 206010038743 Restlessness Diseases 0.000 description 2
- AUNGANRZJHBGPY-SCRDCRAPSA-N Riboflavin Chemical compound OC[C@@H](O)[C@@H](O)[C@@H](O)CN1C=2C=C(C)C(C)=CC=2N=C2C1=NC(=O)NC2=O AUNGANRZJHBGPY-SCRDCRAPSA-N 0.000 description 2
- 108091027568 Single-stranded nucleotide Proteins 0.000 description 2
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 206010046555 Urinary retention Diseases 0.000 description 2
- RLXCFCYWFYXTON-JTTSDREOSA-N [(3S,8S,9S,10R,13S,14S,17R)-3-hydroxy-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-16-yl] N-hexylcarbamate Chemical group C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC(OC(=O)NCCCCCC)[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 RLXCFCYWFYXTON-JTTSDREOSA-N 0.000 description 2
- 230000005856 abnormality Effects 0.000 description 2
- 229960001138 acetylsalicylic acid Drugs 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 125000004450 alkenylene group Chemical group 0.000 description 2
- 125000002877 alkyl aryl group Chemical group 0.000 description 2
- 125000002947 alkylene group Chemical group 0.000 description 2
- 125000000304 alkynyl group Chemical group 0.000 description 2
- 125000004419 alkynylene group Chemical group 0.000 description 2
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 2
- 125000003368 amide group Chemical group 0.000 description 2
- 230000010100 anticoagulation Effects 0.000 description 2
- 230000036506 anxiety Effects 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 206010003119 arrhythmia Diseases 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 230000002567 autonomic effect Effects 0.000 description 2
- 210000003403 autonomic nervous system Anatomy 0.000 description 2
- 239000003613 bile acid Substances 0.000 description 2
- 239000012472 biological sample Substances 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 239000008366 buffered solution Substances 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 210000003169 central nervous system Anatomy 0.000 description 2
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 230000007850 degeneration Effects 0.000 description 2
- 239000007857 degradation product Substances 0.000 description 2
- 230000018044 dehydration Effects 0.000 description 2
- 238000006297 dehydration reaction Methods 0.000 description 2
- 239000005549 deoxyribonucleoside Substances 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- 235000005911 diet Nutrition 0.000 description 2
- 230000037213 diet Effects 0.000 description 2
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 2
- 230000002526 effect on cardiovascular system Effects 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- 230000000913 erythropoietic effect Effects 0.000 description 2
- 229940071106 ethylenediaminetetraacetate Drugs 0.000 description 2
- 210000003608 fece Anatomy 0.000 description 2
- 229960000304 folic acid Drugs 0.000 description 2
- 229930182830 galactose Natural products 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 125000003827 glycol group Chemical group 0.000 description 2
- 244000144993 groups of animals Species 0.000 description 2
- 125000001475 halogen functional group Chemical group 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-UHFFFAOYSA-N hexane-1,2,3,4,5,6-hexol Chemical compound OCC(O)C(O)C(O)C(O)CO FBPFZTCFMRRESA-UHFFFAOYSA-N 0.000 description 2
- 230000003054 hormonal effect Effects 0.000 description 2
- 238000009396 hybridization Methods 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 208000008384 ileus Diseases 0.000 description 2
- 239000003701 inert diluent Substances 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 208000013810 inherited porphyria Diseases 0.000 description 2
- 229960003786 inosine Drugs 0.000 description 2
- 206010022437 insomnia Diseases 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 210000003292 kidney cell Anatomy 0.000 description 2
- 201000006370 kidney failure Diseases 0.000 description 2
- SMEROWZSTRWXGI-HVATVPOCSA-N lithocholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)CC1 SMEROWZSTRWXGI-HVATVPOCSA-N 0.000 description 2
- 238000012317 liver biopsy Methods 0.000 description 2
- 208000019423 liver disease Diseases 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 210000003712 lysosome Anatomy 0.000 description 2
- 230000001868 lysosomic effect Effects 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000003211 malignant effect Effects 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- YACKEPLHDIMKIO-UHFFFAOYSA-N methylphosphonic acid Chemical class CP(O)(O)=O YACKEPLHDIMKIO-UHFFFAOYSA-N 0.000 description 2
- 108091070501 miRNA Proteins 0.000 description 2
- 230000000813 microbial effect Effects 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- 210000003205 muscle Anatomy 0.000 description 2
- 229950006780 n-acetylglucosamine Drugs 0.000 description 2
- 230000003533 narcotic effect Effects 0.000 description 2
- 210000005036 nerve Anatomy 0.000 description 2
- 210000000653 nervous system Anatomy 0.000 description 2
- 230000000926 neurological effect Effects 0.000 description 2
- 230000002981 neuropathic effect Effects 0.000 description 2
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 2
- QTNLALDFXILRQO-UHFFFAOYSA-N nonadecane-1,2,3-triol Chemical compound CCCCCCCCCCCCCCCCC(O)C(O)CO QTNLALDFXILRQO-UHFFFAOYSA-N 0.000 description 2
- 125000002811 oleoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])/C([H])=C([H])\C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 229920001542 oligosaccharide Polymers 0.000 description 2
- 150000002482 oligosaccharides Chemical class 0.000 description 2
- 238000005457 optimization Methods 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 229940124583 pain medication Drugs 0.000 description 2
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- 210000001428 peripheral nervous system Anatomy 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 229920002451 polyvinyl alcohol Polymers 0.000 description 2
- 239000000955 prescription drug Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 229960003387 progesterone Drugs 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- ZCCUUQDIBDJBTK-UHFFFAOYSA-N psoralen Chemical compound C1=C2OC(=O)C=CC2=CC2=C1OC=C2 ZCCUUQDIBDJBTK-UHFFFAOYSA-N 0.000 description 2
- 238000011552 rat model Methods 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 239000003642 reactive oxygen metabolite Substances 0.000 description 2
- 238000011084 recovery Methods 0.000 description 2
- 239000013074 reference sample Substances 0.000 description 2
- 201000004193 respiratory failure Diseases 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000007790 solid phase Substances 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- ATHGHQPFGPMSJY-UHFFFAOYSA-N spermidine Chemical compound NCCCCNCCCN ATHGHQPFGPMSJY-UHFFFAOYSA-N 0.000 description 2
- PFNFFQXMRSDOHW-UHFFFAOYSA-N spermine Chemical compound NCCCNCCCCNCCCN PFNFFQXMRSDOHW-UHFFFAOYSA-N 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 125000000547 substituted alkyl group Chemical group 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 238000011285 therapeutic regimen Methods 0.000 description 2
- 150000003568 thioethers Chemical class 0.000 description 2
- 206010043554 thrombocytopenia Diseases 0.000 description 2
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- 125000002948 undecyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 230000002485 urinary effect Effects 0.000 description 2
- 238000001262 western blot Methods 0.000 description 2
- NOOLISFMXDJSKH-UTLUCORTSA-N (+)-Neomenthol Chemical compound CC(C)[C@@H]1CC[C@@H](C)C[C@@H]1O NOOLISFMXDJSKH-UTLUCORTSA-N 0.000 description 1
- DTGKSKDOIYIVQL-WEDXCCLWSA-N (+)-borneol Chemical compound C1C[C@@]2(C)[C@@H](O)C[C@@H]1C2(C)C DTGKSKDOIYIVQL-WEDXCCLWSA-N 0.000 description 1
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 description 1
- REPVLJRCJUVQFA-UHFFFAOYSA-N (-)-isopinocampheol Natural products C1C(O)C(C)C2C(C)(C)C1C2 REPVLJRCJUVQFA-UHFFFAOYSA-N 0.000 description 1
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- YIMATHOGWXZHFX-WCTZXXKLSA-N (2r,3r,4r,5r)-5-(hydroxymethyl)-3-(2-methoxyethoxy)oxolane-2,4-diol Chemical compound COCCO[C@H]1[C@H](O)O[C@H](CO)[C@H]1O YIMATHOGWXZHFX-WCTZXXKLSA-N 0.000 description 1
- HSINOMROUCMIEA-FGVHQWLLSA-N (2s,4r)-4-[(3r,5s,6r,7r,8s,9s,10s,13r,14s,17r)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid Chemical compound C([C@@]12C)C[C@@H](O)C[C@H]1[C@@H](CC)[C@@H](O)[C@@H]1[C@@H]2CC[C@]2(C)[C@@H]([C@H](C)C[C@H](C)C(O)=O)CC[C@H]21 HSINOMROUCMIEA-FGVHQWLLSA-N 0.000 description 1
- FYADHXFMURLYQI-UHFFFAOYSA-N 1,2,4-triazine Chemical class C1=CN=NC=N1 FYADHXFMURLYQI-UHFFFAOYSA-N 0.000 description 1
- ZIZMDHZLHJBNSQ-UHFFFAOYSA-N 1,2-dihydrophenazine Chemical compound C1=CC=C2N=C(C=CCC3)C3=NC2=C1 ZIZMDHZLHJBNSQ-UHFFFAOYSA-N 0.000 description 1
- YPFDHNVEDLHUCE-UHFFFAOYSA-N 1,3-propanediol Substances OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 1
- 229940035437 1,3-propanediol Drugs 0.000 description 1
- KIGHLLZHFAOEHO-PKPIPKONSA-N 1-[(2s)-4-hydroxy-2-(hydroxymethyl)pyrrolidin-1-yl]ethanone Chemical compound CC(=O)N1CC(O)C[C@H]1CO KIGHLLZHFAOEHO-PKPIPKONSA-N 0.000 description 1
- GHUBZYXDFSBGLI-UHFFFAOYSA-N 1-[[(2,3-dimethoxyphenyl)-diphenylmethoxy]-diphenylmethyl]-2,3-dimethoxybenzene Chemical compound COC1=CC=CC(C(OC(C=2C=CC=CC=2)(C=2C=CC=CC=2)C=2C(=C(OC)C=CC=2)OC)(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1OC GHUBZYXDFSBGLI-UHFFFAOYSA-N 0.000 description 1
- MZMNEDXVUJLQAF-UHFFFAOYSA-N 1-o-tert-butyl 2-o-methyl 4-hydroxypyrrolidine-1,2-dicarboxylate Chemical compound COC(=O)C1CC(O)CN1C(=O)OC(C)(C)C MZMNEDXVUJLQAF-UHFFFAOYSA-N 0.000 description 1
- TZMSYXZUNZXBOL-UHFFFAOYSA-N 10H-phenoxazine Chemical compound C1=CC=C2NC3=CC=CC=C3OC2=C1 TZMSYXZUNZXBOL-UHFFFAOYSA-N 0.000 description 1
- FPIPGXGPPPQFEQ-UHFFFAOYSA-N 13-cis retinol Natural products OCC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-UHFFFAOYSA-N 0.000 description 1
- NVKAWKQGWWIWPM-ABEVXSGRSA-N 17-β-hydroxy-5-α-Androstan-3-one Chemical compound C1C(=O)CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@H]21 NVKAWKQGWWIWPM-ABEVXSGRSA-N 0.000 description 1
- IHPYMWDTONKSCO-UHFFFAOYSA-N 2,2'-piperazine-1,4-diylbisethanesulfonic acid Chemical compound OS(=O)(=O)CCN1CCN(CCS(O)(=O)=O)CC1 IHPYMWDTONKSCO-UHFFFAOYSA-N 0.000 description 1
- VEPOHXYIFQMVHW-XOZOLZJESA-N 2,3-dihydroxybutanedioic acid (2S,3S)-3,4-dimethyl-2-phenylmorpholine Chemical compound OC(C(O)C(O)=O)C(O)=O.C[C@H]1[C@@H](OCCN1C)c1ccccc1 VEPOHXYIFQMVHW-XOZOLZJESA-N 0.000 description 1
- AZUHIVLOSAPWDM-UHFFFAOYSA-N 2-(1h-imidazol-2-yl)-1h-imidazole Chemical compound C1=CNC(C=2NC=CN=2)=N1 AZUHIVLOSAPWDM-UHFFFAOYSA-N 0.000 description 1
- QSHACTSJHMKXTE-UHFFFAOYSA-N 2-(2-aminopropyl)-7h-purin-6-amine Chemical compound CC(N)CC1=NC(N)=C2NC=NC2=N1 QSHACTSJHMKXTE-UHFFFAOYSA-N 0.000 description 1
- HTOVHZGIBCAAJU-UHFFFAOYSA-N 2-amino-2-propyl-1h-purin-6-one Chemical compound CCCC1(N)NC(=O)C2=NC=NC2=N1 HTOVHZGIBCAAJU-UHFFFAOYSA-N 0.000 description 1
- WROUWQQRXUBECT-UHFFFAOYSA-N 2-ethylacrylic acid Chemical compound CCC(=C)C(O)=O WROUWQQRXUBECT-UHFFFAOYSA-N 0.000 description 1
- USCCECGPGBGFOM-UHFFFAOYSA-N 2-propyl-7h-purin-6-amine Chemical compound CCCC1=NC(N)=C2NC=NC2=N1 USCCECGPGBGFOM-UHFFFAOYSA-N 0.000 description 1
- PYSRRFNXTXNWCD-UHFFFAOYSA-N 3-(2-phenylethenyl)furan-2,5-dione Chemical compound O=C1OC(=O)C(C=CC=2C=CC=CC=2)=C1 PYSRRFNXTXNWCD-UHFFFAOYSA-N 0.000 description 1
- BMUDPLZKKRQECS-UHFFFAOYSA-K 3-[18-(2-carboxyethyl)-8,13-bis(ethenyl)-3,7,12,17-tetramethylporphyrin-21,24-diid-2-yl]propanoic acid iron(3+) hydroxide Chemical compound [OH-].[Fe+3].[N-]1C2=C(C)C(CCC(O)=O)=C1C=C([N-]1)C(CCC(O)=O)=C(C)C1=CC(C(C)=C1C=C)=NC1=CC(C(C)=C1C=C)=NC1=C2 BMUDPLZKKRQECS-UHFFFAOYSA-K 0.000 description 1
- HIAJCGFYHIANNA-QIZZZRFXSA-N 3b-Hydroxy-5-cholenoic acid Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@@H](CCC(O)=O)C)[C@@]1(C)CC2 HIAJCGFYHIANNA-QIZZZRFXSA-N 0.000 description 1
- VXGRJERITKFWPL-UHFFFAOYSA-N 4',5'-Dihydropsoralen Natural products C1=C2OC(=O)C=CC2=CC2=C1OCC2 VXGRJERITKFWPL-UHFFFAOYSA-N 0.000 description 1
- OVONXEQGWXGFJD-UHFFFAOYSA-N 4-sulfanylidene-1h-pyrimidin-2-one Chemical compound SC=1C=CNC(=O)N=1 OVONXEQGWXGFJD-UHFFFAOYSA-N 0.000 description 1
- LMNPKIOZMGYQIU-UHFFFAOYSA-N 5-(trifluoromethyl)-1h-pyrimidine-2,4-dione Chemical compound FC(F)(F)C1=CNC(=O)NC1=O LMNPKIOZMGYQIU-UHFFFAOYSA-N 0.000 description 1
- 101710118512 5-aminolevulinate synthase 1 Proteins 0.000 description 1
- ZLAQATDNGLKIEV-UHFFFAOYSA-N 5-methyl-2-sulfanylidene-1h-pyrimidin-4-one Chemical compound CC1=CNC(=S)NC1=O ZLAQATDNGLKIEV-UHFFFAOYSA-N 0.000 description 1
- VPIAFVALSSSQJN-RGURZIINSA-N 6-amino-1-[(2s)-4-hydroxy-2-(hydroxymethyl)pyrrolidin-1-yl]hexan-1-one Chemical compound NCCCCCC(=O)N1CC(O)C[C@H]1CO VPIAFVALSSSQJN-RGURZIINSA-N 0.000 description 1
- DCPSTSVLRXOYGS-UHFFFAOYSA-N 6-amino-1h-pyrimidine-2-thione Chemical compound NC1=CC=NC(S)=N1 DCPSTSVLRXOYGS-UHFFFAOYSA-N 0.000 description 1
- OHILKUISCGPRMQ-UHFFFAOYSA-N 6-amino-5-(trifluoromethyl)-1h-pyrimidin-2-one Chemical compound NC1=NC(=O)NC=C1C(F)(F)F OHILKUISCGPRMQ-UHFFFAOYSA-N 0.000 description 1
- CKOMXBHMKXXTNW-UHFFFAOYSA-N 6-methyladenine Chemical compound CNC1=NC=NC2=C1N=CN2 CKOMXBHMKXXTNW-UHFFFAOYSA-N 0.000 description 1
- HRYKDUPGBWLLHO-UHFFFAOYSA-N 8-azaadenine Chemical compound NC1=NC=NC2=NNN=C12 HRYKDUPGBWLLHO-UHFFFAOYSA-N 0.000 description 1
- LPXQRXLUHJKZIE-UHFFFAOYSA-N 8-azaguanine Chemical compound NC1=NC(O)=C2NN=NC2=N1 LPXQRXLUHJKZIE-UHFFFAOYSA-N 0.000 description 1
- 229960005508 8-azaguanine Drugs 0.000 description 1
- RGKBRPAAQSHTED-UHFFFAOYSA-N 8-oxoadenine Chemical compound NC1=NC=NC2=C1NC(=O)N2 RGKBRPAAQSHTED-UHFFFAOYSA-N 0.000 description 1
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical compound NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 description 1
- 108010062307 AAVALLPAVLLALLAP Proteins 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- 235000001674 Agaricus brunnescens Nutrition 0.000 description 1
- 206010001497 Agitation Diseases 0.000 description 1
- 108090000531 Amidohydrolases Proteins 0.000 description 1
- 102000004092 Amidohydrolases Human genes 0.000 description 1
- RLFWWDJHLFCNIJ-UHFFFAOYSA-N Aminoantipyrine Natural products CN1C(C)=C(N)C(=O)N1C1=CC=CC=C1 RLFWWDJHLFCNIJ-UHFFFAOYSA-N 0.000 description 1
- RMMXTBMQSGEXHJ-UHFFFAOYSA-N Aminophenazone Chemical compound O=C1C(N(C)C)=C(C)N(C)N1C1=CC=CC=C1 RMMXTBMQSGEXHJ-UHFFFAOYSA-N 0.000 description 1
- 206010002198 Anaphylactic reaction Diseases 0.000 description 1
- 206010002942 Apathy Diseases 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 102000005427 Asialoglycoprotein Receptor Human genes 0.000 description 1
- 206010061666 Autonomic neuropathy Diseases 0.000 description 1
- 206010049612 Autonomic seizure Diseases 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 229940122361 Bisphosphonate Drugs 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 125000005865 C2-C10alkynyl group Chemical group 0.000 description 1
- KERQKKXVHHBJKM-RGURZIINSA-N CCCCCC(=O)N1CC(O)C[C@H]1CO Chemical compound CCCCCC(=O)N1CC(O)C[C@H]1CO KERQKKXVHHBJKM-RGURZIINSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 102000016938 Catalase Human genes 0.000 description 1
- 108010053835 Catalase Proteins 0.000 description 1
- 229920002101 Chitin Polymers 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- 206010008796 Chromaturia Diseases 0.000 description 1
- 206010009192 Circulatory collapse Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010053567 Coagulopathies Diseases 0.000 description 1
- 206010010071 Coma Diseases 0.000 description 1
- 206010010305 Confusional state Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- AUNGANRZJHBGPY-UHFFFAOYSA-N D-Lyxoflavin Natural products OCC(O)C(O)C(O)CN1C=2C=C(C)C(C)=CC=2N=C2C1=NC(=O)NC2=O AUNGANRZJHBGPY-UHFFFAOYSA-N 0.000 description 1
- NOOLISFMXDJSKH-UHFFFAOYSA-N DL-menthol Natural products CC(C)C1CCC(C)CC1O NOOLISFMXDJSKH-UHFFFAOYSA-N 0.000 description 1
- 230000009946 DNA mutation Effects 0.000 description 1
- 108010002069 Defensins Proteins 0.000 description 1
- 102000000541 Defensins Human genes 0.000 description 1
- 206010061619 Deformity Diseases 0.000 description 1
- 206010012218 Delirium Diseases 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical class S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 1
- IIUZTXTZRGLYTI-UHFFFAOYSA-N Dihydrogriseofulvin Natural products COC1CC(=O)CC(C)C11C(=O)C(C(OC)=CC(OC)=C2Cl)=C2O1 IIUZTXTZRGLYTI-UHFFFAOYSA-N 0.000 description 1
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- 108700006830 Drosophila Antp Proteins 0.000 description 1
- 102000004533 Endonucleases Human genes 0.000 description 1
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 102000006395 Globulins Human genes 0.000 description 1
- 108010044091 Globulins Proteins 0.000 description 1
- 229920001503 Glucan Polymers 0.000 description 1
- JMBQKKAJIKAWKF-UHFFFAOYSA-N Glutethimide Chemical compound C=1C=CC=CC=1C1(CC)CCC(=O)NC1=O JMBQKKAJIKAWKF-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 229920002527 Glycogen Polymers 0.000 description 1
- UXWOXTQWVMFRSE-UHFFFAOYSA-N Griseoviridin Natural products O=C1OC(C)CC=C(C(NCC=CC=CC(O)CC(O)C2)=O)SCC1NC(=O)C1=COC2=N1 UXWOXTQWVMFRSE-UHFFFAOYSA-N 0.000 description 1
- 206010018910 Haemolysis Diseases 0.000 description 1
- 101150034920 Hmbs gene Proteins 0.000 description 1
- 108010070875 Human Immunodeficiency Virus tat Gene Products Proteins 0.000 description 1
- 238000009015 Human TaqMan MicroRNA Assay kit Methods 0.000 description 1
- 108090001042 Hydro-Lyases Proteins 0.000 description 1
- 102000004867 Hydro-Lyases Human genes 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- 208000004356 Hysteria Diseases 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- 229920001202 Inulin Polymers 0.000 description 1
- 108010044467 Isoenzymes Proteins 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- 101710128836 Large T antigen Proteins 0.000 description 1
- 108091026898 Leader sequence (mRNA) Proteins 0.000 description 1
- LEROTMJVBFSIMP-UHFFFAOYSA-N Mebutamate Chemical compound NC(=O)OCC(C)(C(C)CC)COC(N)=O LEROTMJVBFSIMP-UHFFFAOYSA-N 0.000 description 1
- 108010007013 Melanocyte-Stimulating Hormones Proteins 0.000 description 1
- NPPQSCRMBWNHMW-UHFFFAOYSA-N Meprobamate Chemical compound NC(=O)OCC(C)(CCC)COC(N)=O NPPQSCRMBWNHMW-UHFFFAOYSA-N 0.000 description 1
- AJXPJJZHWIXJCJ-UHFFFAOYSA-N Methsuximide Chemical compound O=C1N(C)C(=O)CC1(C)C1=CC=CC=C1 AJXPJJZHWIXJCJ-UHFFFAOYSA-N 0.000 description 1
- 108060004795 Methyltransferase Proteins 0.000 description 1
- 108700011259 MicroRNAs Proteins 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- 102000015728 Mucins Human genes 0.000 description 1
- 108010063954 Mucins Proteins 0.000 description 1
- 208000010428 Muscle Weakness Diseases 0.000 description 1
- 206010028372 Muscular weakness Diseases 0.000 description 1
- 108010062374 Myoglobin Proteins 0.000 description 1
- 102100030856 Myoglobin Human genes 0.000 description 1
- BKAYIFDRRZZKNF-VIFPVBQESA-N N-acetylcarnosine Chemical compound CC(=O)NCCC(=O)N[C@H](C(O)=O)CC1=CN=CN1 BKAYIFDRRZZKNF-VIFPVBQESA-N 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- BCXBKOQDEOJNRH-UHFFFAOYSA-N NOP(O)=O Chemical class NOP(O)=O BCXBKOQDEOJNRH-UHFFFAOYSA-N 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- DDUHZTYCFQRHIY-UHFFFAOYSA-N Negwer: 6874 Natural products COC1=CC(=O)CC(C)C11C(=O)C(C(OC)=CC(OC)=C2Cl)=C2O1 DDUHZTYCFQRHIY-UHFFFAOYSA-N 0.000 description 1
- 108700019961 Neoplasm Genes Proteins 0.000 description 1
- 102000048850 Neoplasm Genes Human genes 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 206010029350 Neurotoxicity Diseases 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- KYRVNWMVYQXFEU-UHFFFAOYSA-N Nocodazole Chemical compound C1=C2NC(NC(=O)OC)=NC2=CC=C1C(=O)C1=CC=CS1 KYRVNWMVYQXFEU-UHFFFAOYSA-N 0.000 description 1
- 108091092724 Noncoding DNA Proteins 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 108010038807 Oligopeptides Proteins 0.000 description 1
- 102000015636 Oligopeptides Human genes 0.000 description 1
- 208000026251 Opioid-Related disease Diseases 0.000 description 1
- 101150053185 P450 gene Proteins 0.000 description 1
- 239000007990 PIPES buffer Substances 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 206010033864 Paranoia Diseases 0.000 description 1
- 208000027099 Paranoid disease Diseases 0.000 description 1
- 206010034620 Peripheral sensory neuropathy Diseases 0.000 description 1
- 108700020962 Peroxidase Proteins 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- 108010009711 Phalloidine Proteins 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- CXOFVDLJLONNDW-UHFFFAOYSA-N Phenytoin Chemical compound N1C(=O)NC(=O)C1(C=1C=CC=CC=1)C1=CC=CC=C1 CXOFVDLJLONNDW-UHFFFAOYSA-N 0.000 description 1
- 206010034972 Photosensitivity reaction Diseases 0.000 description 1
- 108010069381 Platelet Endothelial Cell Adhesion Molecule-1 Proteins 0.000 description 1
- 102100024616 Platelet endothelial cell adhesion molecule Human genes 0.000 description 1
- 229920002873 Polyethylenimine Polymers 0.000 description 1
- 108010020346 Polyglutamic Acid Proteins 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 101000781681 Protobothrops flavoviridis Disintegrin triflavin Proteins 0.000 description 1
- 208000003251 Pruritus Diseases 0.000 description 1
- 208000028017 Psychotic disease Diseases 0.000 description 1
- 239000004373 Pullulan Substances 0.000 description 1
- 229920001218 Pullulan Polymers 0.000 description 1
- 102000007615 Pulmonary Surfactant-Associated Protein A Human genes 0.000 description 1
- 108010007100 Pulmonary Surfactant-Associated Protein A Proteins 0.000 description 1
- 206010037660 Pyrexia Diseases 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 206010037714 Quadriplegia Diseases 0.000 description 1
- 238000011529 RT qPCR Methods 0.000 description 1
- 101000959515 Rattus norvegicus AH receptor-interacting protein Proteins 0.000 description 1
- 208000010476 Respiratory Paralysis Diseases 0.000 description 1
- 102000003661 Ribonuclease III Human genes 0.000 description 1
- 108010057163 Ribonuclease III Proteins 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 206010041349 Somnolence Diseases 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 229920000147 Styrene maleic anhydride Polymers 0.000 description 1
- 108091036066 Three prime untranslated region Proteins 0.000 description 1
- 108010061174 Thyrotropin Proteins 0.000 description 1
- 102000011923 Thyrotropin Human genes 0.000 description 1
- 206010044221 Toxic encephalopathy Diseases 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 102000004338 Transferrin Human genes 0.000 description 1
- 108090000901 Transferrin Proteins 0.000 description 1
- 101800001690 Transmembrane protein gp41 Proteins 0.000 description 1
- 206010044565 Tremor Diseases 0.000 description 1
- 206010046543 Urinary incontinence Diseases 0.000 description 1
- 102000008897 Uroporphyrinogen decarboxylase (URO-D) Human genes 0.000 description 1
- 108050000839 Uroporphyrinogen decarboxylase (URO-D) Proteins 0.000 description 1
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 1
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- FPIPGXGPPPQFEQ-BOOMUCAASA-N Vitamin A Natural products OC/C=C(/C)\C=C\C=C(\C)/C=C/C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-BOOMUCAASA-N 0.000 description 1
- 229930003270 Vitamin B Natural products 0.000 description 1
- 229930003779 Vitamin B12 Natural products 0.000 description 1
- 229930003448 Vitamin K Natural products 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- 108010046377 Whey Proteins Proteins 0.000 description 1
- 102000007544 Whey Proteins Human genes 0.000 description 1
- NRLNQCOGCKAESA-KWXKLSQISA-N [(6z,9z,28z,31z)-heptatriaconta-6,9,28,31-tetraen-19-yl] 4-(dimethylamino)butanoate Chemical compound CCCCC\C=C/C\C=C/CCCCCCCCC(OC(=O)CCCN(C)C)CCCCCCCC\C=C/C\C=C/CCCCC NRLNQCOGCKAESA-KWXKLSQISA-N 0.000 description 1
- 208000037919 acquired disease Diseases 0.000 description 1
- 150000001251 acridines Chemical class 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 235000013334 alcoholic beverage Nutrition 0.000 description 1
- PPQRONHOSHZGFQ-LMVFSUKVSA-N aldehydo-D-ribose 5-phosphate Chemical group OP(=O)(O)OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PPQRONHOSHZGFQ-LMVFSUKVSA-N 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000005217 alkenylheteroaryl group Chemical group 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000005083 alkoxyalkoxy group Chemical group 0.000 description 1
- 125000004948 alkyl aryl alkyl group Chemical group 0.000 description 1
- 125000005213 alkyl heteroaryl group Chemical group 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 125000005025 alkynylaryl group Chemical group 0.000 description 1
- FPIPGXGPPPQFEQ-OVSJKPMPSA-N all-trans-retinol Chemical compound OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-OVSJKPMPSA-N 0.000 description 1
- 150000001409 amidines Chemical class 0.000 description 1
- 125000005122 aminoalkylamino group Chemical group 0.000 description 1
- 229960000212 aminophenazone Drugs 0.000 description 1
- 229940051880 analgesics and antipyretics pyrazolones Drugs 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000036783 anaphylactic response Effects 0.000 description 1
- 208000003455 anaphylaxis Diseases 0.000 description 1
- 229960003473 androstanolone Drugs 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000012805 animal sample Substances 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 230000002429 anti-coagulating effect Effects 0.000 description 1
- 230000000573 anti-seizure effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- VEQOALNAAJBPNY-UHFFFAOYSA-N antipyrine Chemical compound CN1C(C)=CC(=O)N1C1=CC=CC=C1 VEQOALNAAJBPNY-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 230000006793 arrhythmia Effects 0.000 description 1
- 125000005018 aryl alkenyl group Chemical group 0.000 description 1
- 125000005015 aryl alkynyl group Chemical group 0.000 description 1
- 108010006523 asialoglycoprotein receptor Proteins 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 230000000386 athletic effect Effects 0.000 description 1
- 208000025261 autosomal dominant disease Diseases 0.000 description 1
- 210000003050 axon Anatomy 0.000 description 1
- 230000003376 axonal effect Effects 0.000 description 1
- RHISNKCGUDDGEG-UHFFFAOYSA-N bactenecin Chemical compound CCC(C)C1NC(=O)C(C(C)C)NC(=O)C(C(C)C)NC(=O)C(C(C)CC)NC(=O)C(CCCN=C(N)N)NC(=O)C(NC(=O)C(CC(C)C)NC(=O)C(N)CCCN=C(N)N)CSSCC(C(=O)NC(CCCN=C(N)N)C(O)=O)NC(=O)C(C(C)C)NC(=O)C(CCCN=C(N)N)NC1=O RHISNKCGUDDGEG-UHFFFAOYSA-N 0.000 description 1
- 108010016341 bactenecin Proteins 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 229940125717 barbiturate Drugs 0.000 description 1
- 230000037429 base substitution Effects 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 239000003139 biocide Substances 0.000 description 1
- 150000004663 bisphosphonates Chemical class 0.000 description 1
- 210000002449 bone cell Anatomy 0.000 description 1
- 229940116229 borneol Drugs 0.000 description 1
- CKDOCTFBFTVPSN-UHFFFAOYSA-N borneol Natural products C1CC2(C)C(C)CC1C2(C)C CKDOCTFBFTVPSN-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- UEJWRQDKIBEMCR-UHFFFAOYSA-L calcium;sodium;carbonate Chemical compound [Na+].[Ca+2].[O-]C([O-])=O UEJWRQDKIBEMCR-UHFFFAOYSA-L 0.000 description 1
- QXJJQWWVWRCVQT-UHFFFAOYSA-K calcium;sodium;phosphate Chemical compound [Na+].[Ca+2].[O-]P([O-])([O-])=O QXJJQWWVWRCVQT-UHFFFAOYSA-K 0.000 description 1
- 235000019577 caloric intake Nutrition 0.000 description 1
- 229960000623 carbamazepine Drugs 0.000 description 1
- FFGPTBGBLSHEPO-UHFFFAOYSA-N carbamazepine Chemical compound C1=CC2=CC=CC=C2N(C(=O)N)C2=CC=CC=C21 FFGPTBGBLSHEPO-UHFFFAOYSA-N 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- OFZCIYFFPZCNJE-UHFFFAOYSA-N carisoprodol Chemical compound NC(=O)OCC(C)(CCC)COC(=O)NC(C)C OFZCIYFFPZCNJE-UHFFFAOYSA-N 0.000 description 1
- 229960004587 carisoprodol Drugs 0.000 description 1
- 150000003943 catecholamines Chemical class 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000005859 cell recognition Effects 0.000 description 1
- 210000004671 cell-free system Anatomy 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- BHONFOAYRQZPKZ-LCLOTLQISA-N chembl269478 Chemical compound C([C@H](NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CCCNC(N)=N)[C@@H](C)CC)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(O)=O)C1=CC=CC=C1 BHONFOAYRQZPKZ-LCLOTLQISA-N 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 235000019504 cigarettes Nutrition 0.000 description 1
- 230000004087 circulation Effects 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- DGBIGWXXNGSACT-UHFFFAOYSA-N clonazepam Chemical compound C12=CC([N+](=O)[O-])=CC=C2NC(=O)CN=C1C1=CC=CC=C1Cl DGBIGWXXNGSACT-UHFFFAOYSA-N 0.000 description 1
- 229960003120 clonazepam Drugs 0.000 description 1
- AGVAZMGAQJOSFJ-WZHZPDAFSA-M cobalt(2+);[(2r,3s,4r,5s)-5-(5,6-dimethylbenzimidazol-1-yl)-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl] [(2r)-1-[3-[(1r,2r,3r,4z,7s,9z,12s,13s,14z,17s,18s,19r)-2,13,18-tris(2-amino-2-oxoethyl)-7,12,17-tris(3-amino-3-oxopropyl)-3,5,8,8,13,15,18,19-octamethyl-2 Chemical compound [Co+2].N#[C-].[N-]([C@@H]1[C@H](CC(N)=O)[C@@]2(C)CCC(=O)NC[C@@H](C)OP(O)(=O)O[C@H]3[C@H]([C@H](O[C@@H]3CO)N3C4=CC(C)=C(C)C=C4N=C3)O)\C2=C(C)/C([C@H](C\2(C)C)CCC(N)=O)=N/C/2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O AGVAZMGAQJOSFJ-WZHZPDAFSA-M 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000000306 component Substances 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 208000012839 conversion disease Diseases 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940099112 cornstarch Drugs 0.000 description 1
- 210000003792 cranial nerve Anatomy 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 238000011461 current therapy Methods 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- JVHIPYJQMFNCEK-UHFFFAOYSA-N cytochalasin Natural products N1C(=O)C2(C(C=CC(C)CC(C)CC=C3)OC(C)=O)C3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 JVHIPYJQMFNCEK-UHFFFAOYSA-N 0.000 description 1
- ZMAODHOXRBLOQO-UHFFFAOYSA-N cytochalasin-A Natural products N1C(=O)C23OC(=O)C=CC(=O)CCCC(C)CC=CC3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 ZMAODHOXRBLOQO-UHFFFAOYSA-N 0.000 description 1
- 210000004292 cytoskeleton Anatomy 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 229960000766 danazol Drugs 0.000 description 1
- POZRVZJJTULAOH-LHZXLZLDSA-N danazol Chemical compound C1[C@]2(C)[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=CC2=C1C=NO2 POZRVZJJTULAOH-LHZXLZLDSA-N 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 230000000368 destabilizing effect Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 229960002086 dextran Drugs 0.000 description 1
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 1
- 229960001259 diclofenac Drugs 0.000 description 1
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 208000037765 diseases and disorders Diseases 0.000 description 1
- DTGKSKDOIYIVQL-UHFFFAOYSA-N dl-isoborneol Natural products C1CC2(C)C(O)CC1C2(C)C DTGKSKDOIYIVQL-UHFFFAOYSA-N 0.000 description 1
- 239000013583 drug formulation Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 206010013990 dysuria Diseases 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 230000002124 endocrine Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 230000003511 endothelial effect Effects 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- CHNUOJQWGUIOLD-NFZZJPOKSA-N epalrestat Chemical compound C=1C=CC=CC=1\C=C(/C)\C=C1/SC(=S)N(CC(O)=O)C1=O CHNUOJQWGUIOLD-NFZZJPOKSA-N 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 239000003687 estradiol congener Substances 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- 229960002767 ethosuximide Drugs 0.000 description 1
- HAPOVYFOVVWLRS-UHFFFAOYSA-N ethosuximide Chemical compound CCC1(C)CC(=O)NC1=O HAPOVYFOVVWLRS-UHFFFAOYSA-N 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 210000001808 exosome Anatomy 0.000 description 1
- 210000000887 face Anatomy 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 238000005558 fluorometry Methods 0.000 description 1
- 229940014144 folate Drugs 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 210000001061 forehead Anatomy 0.000 description 1
- 235000019256 formaldehyde Nutrition 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 1
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 1
- 229920000370 gamma-poly(glutamate) polymer Polymers 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 230000004077 genetic alteration Effects 0.000 description 1
- 231100000118 genetic alteration Toxicity 0.000 description 1
- 238000010362 genome editing Methods 0.000 description 1
- 229960002972 glutethimide Drugs 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 229940096919 glycogen Drugs 0.000 description 1
- DDUHZTYCFQRHIY-RBHXEPJQSA-N griseofulvin Chemical compound COC1=CC(=O)C[C@@H](C)[C@@]11C(=O)C(C(OC)=CC(OC)=C2Cl)=C2O1 DDUHZTYCFQRHIY-RBHXEPJQSA-N 0.000 description 1
- 229960002867 griseofulvin Drugs 0.000 description 1
- 229960004198 guanidine Drugs 0.000 description 1
- 230000003779 hair growth Effects 0.000 description 1
- 229940109738 hematin Drugs 0.000 description 1
- 230000008588 hemolysis Effects 0.000 description 1
- 125000004447 heteroarylalkenyl group Chemical group 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 125000005312 heteroarylalkynyl group Chemical group 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 1
- 125000004449 heterocyclylalkenyl group Chemical group 0.000 description 1
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 1
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 230000006801 homologous recombination Effects 0.000 description 1
- 238000002744 homologous recombination Methods 0.000 description 1
- 108091008039 hormone receptors Proteins 0.000 description 1
- 238000002657 hormone replacement therapy Methods 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- 150000007857 hydrazones Chemical class 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 230000036033 hyponatraemia Effects 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- 238000010185 immunofluorescence analysis Methods 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000000126 in silico method Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- USSYUMHVHQSYNA-SLDJZXPVSA-N indolicidin Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N1CCC[C@H]1C(=O)N[C@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(N)=O)CC1=CNC2=CC=CC=C12 USSYUMHVHQSYNA-SLDJZXPVSA-N 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 239000000138 intercalating agent Substances 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 210000003963 intermediate filament Anatomy 0.000 description 1
- JYJIGFIDKWBXDU-MNNPPOADSA-N inulin Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)OC[C@]1(OC[C@]2(OC[C@]3(OC[C@]4(OC[C@]5(OC[C@]6(OC[C@]7(OC[C@]8(OC[C@]9(OC[C@]%10(OC[C@]%11(OC[C@]%12(OC[C@]%13(OC[C@]%14(OC[C@]%15(OC[C@]%16(OC[C@]%17(OC[C@]%18(OC[C@]%19(OC[C@]%20(OC[C@]%21(OC[C@]%22(OC[C@]%23(OC[C@]%24(OC[C@]%25(OC[C@]%26(OC[C@]%27(OC[C@]%28(OC[C@]%29(OC[C@]%30(OC[C@]%31(OC[C@]%32(OC[C@]%33(OC[C@]%34(OC[C@]%35(OC[C@]%36(O[C@@H]%37[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O%37)O)[C@H]([C@H](O)[C@@H](CO)O%36)O)[C@H]([C@H](O)[C@@H](CO)O%35)O)[C@H]([C@H](O)[C@@H](CO)O%34)O)[C@H]([C@H](O)[C@@H](CO)O%33)O)[C@H]([C@H](O)[C@@H](CO)O%32)O)[C@H]([C@H](O)[C@@H](CO)O%31)O)[C@H]([C@H](O)[C@@H](CO)O%30)O)[C@H]([C@H](O)[C@@H](CO)O%29)O)[C@H]([C@H](O)[C@@H](CO)O%28)O)[C@H]([C@H](O)[C@@H](CO)O%27)O)[C@H]([C@H](O)[C@@H](CO)O%26)O)[C@H]([C@H](O)[C@@H](CO)O%25)O)[C@H]([C@H](O)[C@@H](CO)O%24)O)[C@H]([C@H](O)[C@@H](CO)O%23)O)[C@H]([C@H](O)[C@@H](CO)O%22)O)[C@H]([C@H](O)[C@@H](CO)O%21)O)[C@H]([C@H](O)[C@@H](CO)O%20)O)[C@H]([C@H](O)[C@@H](CO)O%19)O)[C@H]([C@H](O)[C@@H](CO)O%18)O)[C@H]([C@H](O)[C@@H](CO)O%17)O)[C@H]([C@H](O)[C@@H](CO)O%16)O)[C@H]([C@H](O)[C@@H](CO)O%15)O)[C@H]([C@H](O)[C@@H](CO)O%14)O)[C@H]([C@H](O)[C@@H](CO)O%13)O)[C@H]([C@H](O)[C@@H](CO)O%12)O)[C@H]([C@H](O)[C@@H](CO)O%11)O)[C@H]([C@H](O)[C@@H](CO)O%10)O)[C@H]([C@H](O)[C@@H](CO)O9)O)[C@H]([C@H](O)[C@@H](CO)O8)O)[C@H]([C@H](O)[C@@H](CO)O7)O)[C@H]([C@H](O)[C@@H](CO)O6)O)[C@H]([C@H](O)[C@@H](CO)O5)O)[C@H]([C@H](O)[C@@H](CO)O4)O)[C@H]([C@H](O)[C@@H](CO)O3)O)[C@H]([C@H](O)[C@@H](CO)O2)O)[C@@H](O)[C@H](O)[C@@H](CO)O1 JYJIGFIDKWBXDU-MNNPPOADSA-N 0.000 description 1
- 229940029339 inulin Drugs 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- JEIPFZHSYJVQDO-UHFFFAOYSA-N iron(III) oxide Inorganic materials O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 1
- 230000007803 itching Effects 0.000 description 1
- 238000003367 kinetic assay Methods 0.000 description 1
- DDVBPZROPPMBLW-ZJBINBEQSA-N latrunculin a Chemical compound C([C@H]1[C@@]2(O)C[C@H]3C[C@H](O2)CC[C@@H](/C=C\C=C/CC\C(C)=C/C(=O)O3)C)SC(=O)N1 DDVBPZROPPMBLW-ZJBINBEQSA-N 0.000 description 1
- DDVBPZROPPMBLW-UHFFFAOYSA-N latrunculin-A Natural products O1C(=O)C=C(C)CCC=CC=CC(C)CCC(O2)CC1CC2(O)C1CSC(=O)N1 DDVBPZROPPMBLW-UHFFFAOYSA-N 0.000 description 1
- 238000001638 lipofection Methods 0.000 description 1
- 150000002634 lipophilic molecules Chemical class 0.000 description 1
- 229920006008 lipopolysaccharide Polymers 0.000 description 1
- 230000005976 liver dysfunction Effects 0.000 description 1
- 210000005228 liver tissue Anatomy 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 206010025482 malaise Diseases 0.000 description 1
- 125000002960 margaryl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 229960004119 mebutamate Drugs 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 230000003340 mental effect Effects 0.000 description 1
- 229940041616 menthol Drugs 0.000 description 1
- GMHKMTDVRCWUDX-UHFFFAOYSA-N mephenytoin Chemical compound C=1C=CC=CC=1C1(CC)NC(=O)N(C)C1=O GMHKMTDVRCWUDX-UHFFFAOYSA-N 0.000 description 1
- 229960000906 mephenytoin Drugs 0.000 description 1
- 229960004815 meprobamate Drugs 0.000 description 1
- 229960003729 mesuximide Drugs 0.000 description 1
- 230000006609 metabolic stress Effects 0.000 description 1
- 210000003632 microfilament Anatomy 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 230000002438 mitochondrial effect Effects 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 108091005601 modified peptides Proteins 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 208000022084 motor paralysis Diseases 0.000 description 1
- 208000018731 motor weakness Diseases 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- OKPYIWASQZGASP-UHFFFAOYSA-N n-(2-hydroxypropyl)-2-methylprop-2-enamide Chemical compound CC(O)CNC(=O)C(C)=C OKPYIWASQZGASP-UHFFFAOYSA-N 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- OZSVEZZAQGRTBE-PXYINDEMSA-N n-[6-[(2s)-4-hydroxy-2-(hydroxymethyl)pyrrolidin-1-yl]-6-oxohexyl]acetamide Chemical compound CC(=O)NCCCCCC(=O)N1CC(O)C[C@H]1CO OZSVEZZAQGRTBE-PXYINDEMSA-N 0.000 description 1
- QNILTEGFHQSKFF-UHFFFAOYSA-N n-propan-2-ylprop-2-enamide Chemical compound CC(C)NC(=O)C=C QNILTEGFHQSKFF-UHFFFAOYSA-N 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 230000007830 nerve conduction Effects 0.000 description 1
- 208000020470 nervous system symptom Diseases 0.000 description 1
- 230000036403 neuro physiology Effects 0.000 description 1
- 231100000189 neurotoxic Toxicity 0.000 description 1
- 231100000228 neurotoxicity Toxicity 0.000 description 1
- 230000007135 neurotoxicity Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229950006344 nocodazole Drugs 0.000 description 1
- 239000000820 nonprescription drug Substances 0.000 description 1
- 238000007899 nucleic acid hybridization Methods 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 229940005483 opioid analgesics Drugs 0.000 description 1
- 210000003463 organelle Anatomy 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- 229940000673 orphan drug Drugs 0.000 description 1
- 239000002859 orphan drug Substances 0.000 description 1
- 238000012261 overproduction Methods 0.000 description 1
- 239000003953 ovulation stimulant Substances 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 229940124641 pain reliever Drugs 0.000 description 1
- 210000004738 parenchymal cell Anatomy 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 230000007310 pathophysiology Effects 0.000 description 1
- MCYTYTUNNNZWOK-LCLOTLQISA-N penetratin Chemical compound C([C@H](NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CCCNC(N)=N)[C@@H](C)CC)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(N)=O)C1=CC=CC=C1 MCYTYTUNNNZWOK-LCLOTLQISA-N 0.000 description 1
- 230000000149 penetrating effect Effects 0.000 description 1
- 239000000863 peptide conjugate Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 229960005222 phenazone Drugs 0.000 description 1
- 125000001484 phenothiazinyl group Chemical class C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 229960002036 phenytoin Drugs 0.000 description 1
- 208000019899 phobic disease Diseases 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000036211 photosensitivity Effects 0.000 description 1
- SHUZOJHMOBOZST-UHFFFAOYSA-N phylloquinone Natural products CC(C)CCCCC(C)CCC(C)CCCC(=CCC1=C(C)C(=O)c2ccccc2C1=O)C SHUZOJHMOBOZST-UHFFFAOYSA-N 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229920002627 poly(phosphazenes) Polymers 0.000 description 1
- 108010064470 polyaspartate Proteins 0.000 description 1
- 125000005575 polycyclic aromatic hydrocarbon group Chemical group 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920002643 polyglutamic acid Polymers 0.000 description 1
- 229920000166 polytrimethylene carbonate Polymers 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 230000002117 porphyrinogenic effect Effects 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 238000011809 primate model Methods 0.000 description 1
- DQMZLTXERSFNPB-UHFFFAOYSA-N primidone Chemical compound C=1C=CC=CC=1C1(CC)C(=O)NCNC1=O DQMZLTXERSFNPB-UHFFFAOYSA-N 0.000 description 1
- 229960002393 primidone Drugs 0.000 description 1
- 150000003146 progesterones Chemical class 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 229940048914 protamine Drugs 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 235000019423 pullulan Nutrition 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 229960005206 pyrazinamide Drugs 0.000 description 1
- IPEHBUMCGVEMRF-UHFFFAOYSA-N pyrazinecarboxamide Chemical compound NC(=O)C1=CN=CC=N1 IPEHBUMCGVEMRF-UHFFFAOYSA-N 0.000 description 1
- JEXVQSWXXUJEMA-UHFFFAOYSA-N pyrazol-3-one Chemical class O=C1C=CN=N1 JEXVQSWXXUJEMA-UHFFFAOYSA-N 0.000 description 1
- 229960003581 pyridoxal Drugs 0.000 description 1
- 235000008164 pyridoxal Nutrition 0.000 description 1
- 239000011674 pyridoxal Substances 0.000 description 1
- 239000002265 redox agent Substances 0.000 description 1
- 210000005084 renal tissue Anatomy 0.000 description 1
- 230000001850 reproductive effect Effects 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 230000004202 respiratory function Effects 0.000 description 1
- 210000003019 respiratory muscle Anatomy 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 235000019192 riboflavin Nutrition 0.000 description 1
- 239000002151 riboflavin Substances 0.000 description 1
- 229960002477 riboflavin Drugs 0.000 description 1
- JQXXHWHPUNPDRT-WLSIYKJHSA-N rifampicin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C([O-])=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CC[NH+](C)CC1 JQXXHWHPUNPDRT-WLSIYKJHSA-N 0.000 description 1
- 229960001225 rifampicin Drugs 0.000 description 1
- COFLCBMDHTVQRA-UHFFFAOYSA-N sapphyrin Chemical compound N1C(C=2NC(C=C3N=C(C=C4NC(=C5)C=C4)C=C3)=CC=2)=CC=C1C=C1C=CC5=N1 COFLCBMDHTVQRA-UHFFFAOYSA-N 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 201000005572 sensory peripheral neuropathy Diseases 0.000 description 1
- 206010040560 shock Diseases 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 206010040882 skin lesion Diseases 0.000 description 1
- 231100000444 skin lesion Toxicity 0.000 description 1
- 230000005808 skin problem Effects 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 230000000391 smoking effect Effects 0.000 description 1
- 229910001467 sodium calcium phosphate Inorganic materials 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000010532 solid phase synthesis reaction Methods 0.000 description 1
- 230000003381 solubilizing effect Effects 0.000 description 1
- 229940063673 spermidine Drugs 0.000 description 1
- 229940063675 spermine Drugs 0.000 description 1
- 150000003408 sphingolipids Chemical class 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical class O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 1
- VNOYUJKHFWYWIR-ITIYDSSPSA-N succinyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)CCC(O)=O)O[C@H]1N1C2=NC=NC(N)=C2N=C1 VNOYUJKHFWYWIR-ITIYDSSPSA-N 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000035900 sweating Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- RJVBVECTCMRNFG-ANKJNSLFSA-N swinholide a Chemical compound C1[C@H](OC)C[C@H](C)O[C@H]1CC[C@H](C)[C@H](O)[C@H](C)[C@@H]1[C@@H](C)[C@H](O)C[C@H](O)[C@H](C)[C@@H](OC)C[C@H](CC=C2)O[C@@H]2C[C@@H](O)C/C=C(\C)/C=C/C(=O)O[C@H]([C@@H](C)[C@@H](O)[C@@H](C)CC[C@@H]2O[C@@H](C)C[C@H](C2)OC)[C@@H](C)[C@H](O)C[C@H](O)[C@H](C)[C@@H](OC)C[C@H](CC=C2)O[C@@H]2C[C@@H](O)C/C=C(\C)/C=C/C(=O)O1 RJVBVECTCMRNFG-ANKJNSLFSA-N 0.000 description 1
- GDACDJNQZCXLNU-UHFFFAOYSA-N swinholide-A Natural products C1C(OC)CC(C)OC1CCC(C)C(O)C(C)C1C(C)C(O)CC(O)C(C)C(OC)CC(CC=C2)OC2CC(O)CC=C(C)C=CC(=O)O1 GDACDJNQZCXLNU-UHFFFAOYSA-N 0.000 description 1
- 210000002437 synoviocyte Anatomy 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- TUNFSRHWOTWDNC-HKGQFRNVSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCC[14C](O)=O TUNFSRHWOTWDNC-HKGQFRNVSA-N 0.000 description 1
- 150000004044 tetrasaccharides Chemical class 0.000 description 1
- ZEMGGZBWXRYJHK-UHFFFAOYSA-N thiouracil Chemical compound O=C1C=CNC(=S)N1 ZEMGGZBWXRYJHK-UHFFFAOYSA-N 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 229940113082 thymine Drugs 0.000 description 1
- 229960000874 thyrotropin Drugs 0.000 description 1
- 230000001748 thyrotropin Effects 0.000 description 1
- 235000021476 total parenteral nutrition Nutrition 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 239000012581 transferrin Substances 0.000 description 1
- 230000005945 translocation Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- ZMANZCXQSJIPKH-UHFFFAOYSA-O triethylammonium ion Chemical compound CC[NH+](CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-O 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 1
- 229960000604 valproic acid Drugs 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 230000009278 visceral effect Effects 0.000 description 1
- 235000019155 vitamin A Nutrition 0.000 description 1
- 239000011719 vitamin A Substances 0.000 description 1
- 235000019156 vitamin B Nutrition 0.000 description 1
- 239000011720 vitamin B Substances 0.000 description 1
- 235000019163 vitamin B12 Nutrition 0.000 description 1
- 239000011715 vitamin B12 Substances 0.000 description 1
- 235000019168 vitamin K Nutrition 0.000 description 1
- 239000011712 vitamin K Substances 0.000 description 1
- 150000003721 vitamin K derivatives Chemical class 0.000 description 1
- 229940045997 vitamin a Drugs 0.000 description 1
- 150000003722 vitamin derivatives Chemical class 0.000 description 1
- 229940046010 vitamin k Drugs 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- 235000021119 whey protein Nutrition 0.000 description 1
- 229940075420 xanthine Drugs 0.000 description 1
- SFVVQRJOGUKCEG-OPQSFPLASA-N β-MSH Chemical compound C1C[C@@H](O)[C@H]2C(COC(=O)[C@@](O)([C@@H](C)O)C(C)C)=CCN21 SFVVQRJOGUKCEG-OPQSFPLASA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
- C12N15/1137—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing against enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/7105—Natural ribonucleic acids, i.e. containing only riboses attached to adenine, guanine, cytosine or uracil and having 3'-5' phosphodiester links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/713—Double-stranded nucleic acids or oligonucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y203/00—Acyltransferases (2.3)
- C12Y203/01—Acyltransferases (2.3) transferring groups other than amino-acyl groups (2.3.1)
- C12Y203/01037—5-Aminolevulinate synthase (2.3.1.37)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/10—Type of nucleic acid
- C12N2310/14—Type of nucleic acid interfering N.A.
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/31—Chemical structure of the backbone
- C12N2310/315—Phosphorothioates
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/32—Chemical structure of the sugar
- C12N2310/321—2'-O-R Modification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/32—Chemical structure of the sugar
- C12N2310/322—2'-R Modification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/34—Spatial arrangement of the modifications
- C12N2310/341—Gapmers, i.e. of the type ===---===
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/34—Spatial arrangement of the modifications
- C12N2310/345—Spatial arrangement of the modifications having at least two different backbone modifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/35—Nature of the modification
- C12N2310/351—Conjugate
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/35—Nature of the modification
- C12N2310/352—Nature of the modification linked to the nucleic acid via a carbon atom
- C12N2310/3521—Methyl
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2310/00—Structure or type of the nucleic acid
- C12N2310/30—Chemical structure
- C12N2310/35—Nature of the modification
- C12N2310/353—Nature of the modification linked to the nucleic acid via an atom other than carbon
- C12N2310/3533—Halogen
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/118—Prognosis of disease development
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Molecular Biology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Microbiology (AREA)
- Analytical Chemistry (AREA)
- Plant Pathology (AREA)
- Virology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Obesity (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Gastroenterology & Hepatology (AREA)
- Dermatology (AREA)
- Pathology (AREA)
Abstract
Un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde dicho ARNbc comprende una hebra sentido y una hebra antisentido, comprendiendo la hebra antisentido una región de complementariedad con un transcrito de ARN de ALAS1 (p. ej., SEQ ID NO: 1), cuya hebra antisentido comprende usAfsAfGfaUfgAfgAfcAfcUfcUfuUfcUfgsgsu (SEQ ID NO: 4161), donde c, a, g, u = ribonucleósidos 2'-OMe; Af, Cf, Gf, Uf = ribonucleósidos 2'F; s = fosforotioato, o una secuencia que difiere en no más de 1, 2 o 3 nucleótidos de esta.
Description
DESCRIPCIÓN
Composiciones y métodos para inhibir la expresión del gen ALAS1
Campo de la invención
La invención se refiere en general a la inhibición específica de la expresión del gen ALAS1.
Antecedentes
Las porfirias hereditarias constituyen una familia de trastornos provocados por la actividad deficiente de enzimas específicas en la vía biosintética del grupo hemo, que también se denomina en la presente vía de las porfirinas. La deficiencia de las enzimas de la vía de las porfirinas conlleva una producción insuficiente del grupo hemo y una acumulación de porfirinas y precursores de estas, que son tóxicos para los tejidos en concentraciones elevadas.
Entre las porfirias heredadas, la porfiria intermitente aguda (AIP, por sus siglas en inglés, p. ej., AIP dominante autosómica), la porfiria variegata (VP, por sus siglas en inglés, p. ej., VP dominante autosómica), coproporfiria hereditaria (coproporfiria o HCP, por sus siglas en inglés, p. ej., HCP dominante autosómica) y la porfiria por deficiencia de ácido 5’-aminolevulínico (también conocido como ácido 5-aminolevulínico o ALA) deshidratasa (ADP, por sus siglas en inglés, p. ej., ADP recesiva autosómica) se clasifican como porfirias hepáticas agudas y se manifiestan mediante ataques neurológicos agudos que pueden resultar mortales. Los ataques agudos se caracterizan por síntomas del sistema nervioso central, autonómico y periférico, que incluyen dolor abdominal intenso, hipertensión, taquicardias, estreñimiento, debilidad motriz, parálisis y convulsiones. Si no se tratan debidamente, pueden provocar tetraplegia, deficiencia respiratoria y defunción. Varios factores, que incluyen los fármacos que inducen el citocromo P450, la dieta y los cambios hormonales, pueden precipitar los ataques agudos mediante el incremento de la actividad del ácido 5’-aminolevulínico-sintasa 1 hepática (ALAS1), que es la primera enzima, y a la vez la enzima limitante de la velocidad, de la vía biosintética del grupo hemo. En las porfirias agudas, p. ej., AIP, VP, HCP y ADP, las deficiencias enzimáticas respectivas provocan la producción y la acumulación hepática de una o más sustancias (p. ej., porfirinas y/o precursores de porfirinas, p. ej., ALA y/o PBG) que pueden ser neurotóxicas y que pueden provocar la aparición de ataques agudos. Remítase, p. ej., a Balwani, M y Desnick, R.J., Blood, 120:4496-4504, 2012.
La terapia actual para los ataques neurológicos agudos consiste en la administración intravenosa de hemina (Panhematin®, Lundbeck o Normosang®, Orphan Europe), que proporciona el grupo hemo exógeno para la inhibición por retroalimentación negativa de ALAS1 y, por consiguiente, reduce la producción de ALA y PBG. La hemina se utiliza para el tratamiento durante un ataque agudo y para la prevención de ataques, particularmente en mujeres con porfirias agudas que experimentan ataques frecuentes con los cambios hormonales durante sus ciclos menstruales. Aunque generalmente los pacientes responden bien, su efecto es lento, normalmente lleva de dos a cuatro días o más normalizar las concentraciones de ALA y PBG en orina hasta niveles normales. Debido a que la hemina intravenosa se metaboliza rápidamente, normalmente son necesarias de tres a cuatro infusiones para tratar o prevenir eficazmente un ataque agudo. Además, las infusiones repetidas pueden provocar una sobrecarga de hierro y flebitis, las cuales pueden dificultar el acceso venoso periférico. Aunque el trasplante de hígado ortotrófico es curativo, este procedimiento presenta una morbidez y una mortalidad significativas y la disponibilidad de donantes de hígado es limitada. Por consiguiente, se necesita una estrategia terapéutica alternativa que sea más eficaz, que actúe rápido y que sea segura. Sería particularmente favorable si un tratamiento de este tipo se pudiera suministrar mediante una administración subcutánea, ya que esto eliminaría la necesidad de infusiones y de una hospitalización prolongada.
La AIP, también denominada deficiencia de porfobilinógeno-desaminasa (PBGD) o deficiencia de hidroximetilbilanosintasa (HMBS), es la porfiria hepática aguda más común. Se estima que la prevalencia de AIP es de 5-10 en 100 000, donde aproximadamente un 5-10% de los pacientes son sintomáticos. AIP es un trastorno dominante autosómico provocado por mutaciones en el gen HMBS que dan como resultado una actividad reducida, p. ej., seminormal de la enzima. Previamente, se ha generado por recombinación homóloga un modelo en ratones de AIP que presenta ~30% de actividad HMBS de origen natural. Del mismo modo que los pacientes humanos, estos ratones incrementan la actividad ALAS1 hepática y acumulan grandes cantidades de ALA y PBG urinarias y en plasma cuando se les administran fármacos porfirinogénicos tales como el fenobarbital. Por lo tanto, sirven como un modelo excelente para evaluar la eficacia de agentes terapéuticos novedosos para las porfirias hepáticas agudas.
Compendio
La presente invención está definida por las reivindicaciones. En consecuencia, la presente invención se refiere a un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde dicho ARNbc comprende una hebra sentido y una hebra antisentido, comprendiendo la hebra antisentido una región de complementariedad respecto a un transcrito de ARN de ALAS1 (p. ej., SEQ ID NO:1), cuya hebra antisentido comprende usAfsAfGfaUfgAfgAfcAfcUfcUfuUfcUfgsgsu (SEQ iD NO: 4161), donde c, a, g, u = 2'-OMe ribonucleósidos; Af, Cf, Gf, Uf = 2'F ribonucleósidos; s = fosforotioato, o una secuencia que difiere en no más de 1, 2 o 3 nucleótidos de esta.
En la presente se describen métodos y composiciones de ARNi para modular la expresión de un gen ALAS1. La expresión de un gen ALAS1 se puede reducir o inhibir utilizando un ARNi específico para ALAS1. Tal inhibición puede ser útil en el tratamiento de trastornos relacionados con la expresión de ALAS1 tales como las porfirias. Por consiguiente, en la presente se describen composiciones y métodos que producen la escisión mediada por el complejo de silenciamiento inducido por ARN (RISC, por sus siglas en inglés) de transcritos de ARN del gen ALAS1, por ejemplo, en una célula o en un sujeto (p. ej., en un mamífero tal como un sujeto humano). También se describen composiciones y métodos para tratar un trastorno relacionado con la expresión de un gen ALAS1, tal como una porfiria, p. ej., anemia sideroblástica relacionada con X (XLSA, por sus siglas en inglés), porfiria debida a la deficiencia de ALA-deshidratasa (porfiria de Doss o ADP, por sus siglas en inglés), porfiria intermitente aguda (AIP, por sus siglas en inglés), porfiria eritropoyética congénita (CEP, por sus siglas en inglés), porfiria cutánea tardía (PCT), coproporfiria hereditaria (coproporfiria o HCP), porfiria variegata (VP), protoporfiria eritropoyética (EPP, por sus siglas en inglés) o eritroporfiria transitoria de la infancia. El trastorno puede ser una porfiria hepática aguda, p. ej., una porfiria debida a la deficiencia de ALA-deshidratasa (ADP), AIP, HCP o VP. El trastorno puede ser una porfiria debida a la deficiencia de ALA-deshidratasa (ADP) o AIP.
La porfiria puede ser una porfiria hepática, p. ej., una porfiria seleccionada entre porfiria intermitente aguda (AIP), coproporfiria hereditaria (HCP), porfiria variegata (VP), porfiria debida a la deficiencia de ALA-deshidratasa (ADP) y porfiria hepatoeritropoyética. La porfiria puede ser una porfiria hepática dominante homozigótica (p. ej., AIP, HCP o VP dominante homozigótica) o una porfiria hepatoeritropoyética. La porfiria puede ser una porfiria dual.
Las expresiones "ARNi", ''iARN'', "agente de ARNi", "agente de iARN'' o “molécula de iARN”, tal como se utilizan en la presente, se refieren a un agente que contiene ARN, tal como se define este término en la presente, y el cual media la escisión dirigida de un transcrito de ARN, p. ej., mediante una vía del complejo de silenciamiento inducido por ARN (RISC). Un ARNi, tal como se describe en la presente, puede ejercer la inhibición de la expresión de ALAS1 en una célula o mamífero.
Los ARNi incluidos en las composiciones que se exponen en la presente engloban un ARNbc que tiene una hebra de ARN (la hebra antisentido) que tiene una región, p. ej., una región con una longitud de 30 nucleótidos o inferior, generalmente de 19-24 nucleótidos, que es sustancialmente complementaria respecto a al menos parte de un transcrito de ARNm de un gen ALAS1 (p. ej., un gen ALAS1 humano o de ratón) (también denominado en la presente "ARNi específico para ALAS1"). Como alternativa o de forma combinada, los ARNi engloban un ARNbc que tiene una hebra de ARN (la hebra antisentido) que tiene una región con una longitud de 30 nucleótidos o inferior, generalmente de 19-24 nucleótidos, que es sustancialmente complementaria respecto a al menos parte de un transcrito de ARNm de un gen ALAS1 (p. ej., una variante humana 1 o 2 de un gen ALAS1) (también denominado en la presente "ARNi específico para ALAS1").
El ARNi (p. ej., ARNbc) descrito en la presente puede comprender una hebra antisentido que tiene una región que es sustancialmente complementaria respecto a una región de un ALAS1 humano. El gen ALAS1 humano puede presentar la secuencia de NM_000688.4 (SEQ ID NO:1) o NM_000688.5 (SEQ ID NO:382). El ALAS1 humano puede tener la secuencia de NM_199166.1.
La secuencia antisentido del ARNi (p. ej., ARNbc) puede tener la diana dentro de la región de 871 a 895 (más o menos 5, 4, 3, 2 o 1 nucleótidos en cualquier o ambas direcciones en el extremo 5’ y/o 3’) en el transcrito de ALAS1 NM_000688.4. La secuencia antisentido puede tener como diana los nucleótidos 871 a 893, 871 a 892 u 873 a 895 en el transcrito de ALAS1 NM_000688.4. La secuencia antisentido puede comprender o estar constituida por una secuencia que es totalmente complementaria o sustancialmente complementaria a los nucleótidos 871 a 893, 871 a 892 u 873 a 895 en el transcrito de ALAS1 NM_000688.4.
En un aspecto, se proporciona un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde dicho ARNbc comprende una hebra sentido y una hebra antisentido, comprendiendo la hebra antisentido una región de complementariedad respecto a un transcrito de ARN de ALAS1, cuya hebra antisentido comprende al menos 15 (p. ej., al menos 16, 17, 18, 19, 20, 21,22 o 23) nucleótidos contiguos que difieren en no más de 3, 2 o 1 nucleótidos de la secuencia UAAGAUGAGACACUCUUUCUGGU (SEQ ID NO: 4153) o UAAGAUGAGACACUCTUUCUGGU (SEQ ID NO: 4154). La hebra antisentido puede comprender la secuencia de UAAGAUGAGACACUCUUUCUGGU (SEQ ID NO: 4153) o UAAGAUGAGACACUCTUUCUGGU (SEQ ID NO: 4154). La hebra sentido puede comprender la secuencia de CAGAAAGAGUGUCUCAUCUUA (SEQ ID NO: 4155). Uno o más nucleótidos de la hebra antisentido y/o hebra sentido están modificados como se describe en la presente. El ARNbc puede comprender (i) una hebra antisentido que comprende, o está constituida por, la secuencia antisentido de AD-60489, AD-60519, o AD-61193 y/o (ii) una hebra sentido que comprende, o está constituida por, la secuencia sentido de AD-60489, AD-60519, o AD-61193 (incluidas una o más (p. ej., la totalidad) de las modificaciones de la hebra antisentido y/o hebra antisentido de AD-60489, AD-60519 o AD-61193).
En un aspecto, se proporciona un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde dicho ARNbc comprende una hebra sentido y una hebra antisentido, comprendiendo la hebra antisentido una región de complementariedad respecto a un transcrito de ARN de ALAS1, cuya hebra antisentido comprende al menos 15 (p. ej., al menos 16, 17, 18, 19, 20, 21,22 o 23) nucleótidos contiguos que difieren en no más de 3 (p. ej., en no más
de 0, 1 o 2) nucleótidos de una secuencia antisentido enumerada en cualquiera de las Tablas 21 a 40, o una versión
no modificada de una secuencia antisentido (p. ej., una versión que tiene la misma secuencia de nucleótidos excepto
en que algunos o todos los nucleótidos no están modificados) enumerada en cualquiera de las Tablas 21 a 40. La secuencia antisentido puede comprender al menos 15 (p. ej., al menos 16, 17, 18, 19, 20, 21, 22 o 23) nucleótidos contiguos que difieren en no más de 3 (p. ej., no más de 0, 1 o 2) nucleótidos de (i) la secuencia antisentido de AD-60489, AD-60519 o AD-61193 o (ii) una versión no modificada de cualquiera de estas secuencias. La hebra antisentido puede comprender al menos 15 (p. ej., al menos 16, 17, 18, 19, 20, 21, 22 o 23) nucleótidos contiguos
que difieren en no más de 3 (p. ej., en no más de 0, 1 o 2) nucleótidos de la secuencia de UAAGAUGAGACACUCUUUCUGGU (SEQ ID NO: 4153) o UAAGAUGAGACACUCTUUCUGGU (SEQ ID NO:
4154). La secuencia antisentido puede tener como diana las posiciones 871-893 de NM_000688.4 (SEQ ID NO:1).
La hebra sentido puede comprender la secuencia de CAGAAAGAGUGUCUCAUCUUA (SEQ ID nO: 4155). Uno o
más nucleótidos de la hebra antisentido y/o hebra sentido están modificados como se describe en la presente.
El ARNbc puede no ser una secuencia sentido y/o antisentido enumerada en cualquiera de las Tablas 2, 3, 6, 7, 8, 9,
14, 15, 18 o 20.
También se proporciona en la presente un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde dicho ARNbc comprende una hebra sentido y una hebra antisentido, comprendiendo la hebra antisentido una región de complementariedad respecto a un transcrito de ARN de ALAS1, cuya hebra antisentido comprende al menos 15 (p. ej., al menos 16, 17, 18, 19, 20, 21, 22 o 23) nucleótidos contiguos que difieren en no
más de 3 nucleótidos, no más de 2 nucleótidos o no más de un nucleótido, de la secuencia antisentido de AD-60519.
Uno o más nucleótidos pueden estar modificados como se describe en la presente.
Se proporciona un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde dicho ARNbc comprende una hebra sentido y una hebra antisentido, comprendiendo la hebra antisentido una región de complementariedad respecto a un transcrito de ARN de ALAS1, cuya hebra antisentido comprende al menos 15 (p.
ej., al menos 16, 17, 18, 19, 20, 21,22, o 23) nucleótidos contiguos que difieren en no más de 3 (p. ej., en no más de
0, 1 o 2) nucleótidos de la secuencia antisentido de AD-60489, o un derivado de AD-60489 como se describe en la presente. Uno o más nucleótidos pueden estar modificados como se describe en la presente, p. ej., uno o más (o la totalidad) de los nucleótidos de AD-60489 están modificados como se describe en la presente. El derivado de AD-60489 puede ser AD-60501, AD-60519, AD-60901, AD-60495, AD-60900, AD-60935, AD-60879, AD-61190, AD-61191, AD-60865, AD-60861, AD-60876, AD-61193, AD-60519, AD-60519 o AD-60901. El derivado de AD-60489
puede ser AD-60519. El derivado de AD-60489 puede ser AD-61193.
Se puede proporcionar un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde dicho ARNbc comprende una hebra sentido y una hebra antisentido, comprendiendo la hebra antisentido una región de complementariedad respecto a un transcrito de ARN de ALAS1, cuya hebra antisentido comprende al menos 15 (p.
ej., al menos 16, 17, 18, 19, 20, 21,22 o 23) nucleótidos contiguos que difieren en no más de 3 (p. ej., no más de 0,
1 o 2) nucleótidos de un derivado de AD-58632 descrito en la presente. Uno o más nucleótidos pueden estar modificados como se describe en la presente, p. ej., uno o más (o la totalidad) de los nucleótidos de AD-58632 están modificados como se describe en la presente. El derivado de AD-58632 puede ser AD-60405, AD-60887, AD-60923,
AD-60434, AD-60892, AD-60419, AD-60924, AD-60445, AD-60925, y AD-60926, AD-60820, AD-60843, AD-60819,
AD-61140, AD-61141, AD-61142, AD-60835, AD-60839, AD-61143, AD-61144, AD-61145, AD-61146, AD-60892, o
AD-60419. El derivado de AD-58632 puede ser AD-60819.
El ARNbc puede tener una CI50 inferior a 1 nM. El ARNbc puede tener una CI50 en el intervalo de 0.01-1 nM. El ARNbc puede tener una CI50 inferior a 0.05 nM. El ARNbc puede tener una CI50 inferior a 0.02 nM. El ARNbc puede
tener una CI50 inferior a 0.01 nM. La CI50 se puede determinar como se describe en la presente en los Ejemplos.
El ARNbc puede tener una DE50 de dosis única inferior a aproximadamente 10 mg/kg. El ARNbc puede tener una
DE50 de dosis única inferior a aproximadamente 5 mg/kg. La CE50 se puede determinar como se describe en la presente en los Ejemplos.
El ARNbc puede mostrar una actividad mejorada en comparación con AD-58632. El ARNbc puede mostrar una actividad mejorada en comparación con AD-60489. El ARNbc puede mostrar una actividad mejorada en comparación con AD-58632 y AD-60489.
El ARNbc puede ser AD-60501, AD-60519, AD-60901, AD-60495, AD-60900, AD-60935, AD-60879, AD-61190, AD-61191, AD-60865, AD-60861, AD-60876, AD-61193, AD-60519, AD-60519, AD-60901, AD-60405, AD-60887, 60923, AD-60434, AD-60892, AD-60419, AD-60924, AD-60445,  AD-60925, AD-60926, AD-60820, AD-60843, 60819, AD-61140, AD-61141, AD-61142, AD-60835, AD-60839, AD-61143, AD-61144, AD-61145, AD-61146, 60892 o AD-60419 (p. ej., incluida la secuencia de nucleótidos y/o uno o más (p. ej. la totalidad) de las modificaciones de los ARNbc mencionados anteriormente). El ARNbc puede comprender una hebra antisentido que comprende, o está constituida por, una secuencia antisentido (y/o una o más (p. ej., la totalidad) de las modificaciones)) seleccionada de AD-60501, AD-60519, AD-60901, AD-60495, AD-60900, AD-60935, AD-60879,
AD-60925, AD-60926, AD-60820, AD-60843, 60819, AD-61140, AD-61141, AD-61142, AD-60835, AD-60839, AD-61143, AD-61144, AD-61145, AD-61146, 60892 o AD-60419 (p. ej., incluida la secuencia de nucleótidos y/o uno o más (p. ej. la totalidad) de las modificaciones de los ARNbc mencionados anteriormente). El ARNbc puede comprender una hebra antisentido que comprende, o está constituida por, una secuencia antisentido (y/o una o más (p. ej., la totalidad) de las modificaciones)) seleccionada de AD-60501, AD-60519, AD-60901, AD-60495, AD-60900, AD-60935, AD-60879,
AD-61190, AD-61191, AD-60865, AD-60861, AD-60876, AD-61193, AD-60519, AD-60519, AD-60901, AD-60405,
AD-60887, AD-60923, AD-60434, AD-60892, AD-60419, AD-60924, AD-60445, AD-60925, AD-60926, AD-60820,
AD-60843, AD-60819, AD-61140, AD-61141, AD-61142, AD-60835, AD-60839, AD-61143, AD-61144, AD-61145,
AD-61146, AD-60892 o AD-60419. El ARNbc puede comprender una hebra sentido que comprende, o está constituida por, una secuencia sentido (y/o una o más (por ejemplo, la totalidad) de las modificaciones)) seleccionada de AD-60501, AD-60519, AD-60901, AD-60495, AD-60900, AD-60935, AD-60879, AD-61190, AD-61191, AD-60865, AD-60861, AD-60876, AD-61193, AD-60519, AD-60519, AD-60901, AD-60405, AD-6088 60923, AD-60434, AD-60892, AD-60419, AD-60924, AD-60445, AD-60925, AD-60926, AD-60820, AD-60843, AD-60819, AD-61140, AD-61141, AD-61142, AD-60835, AD-60839, AD-61143, AD-61144, AD-61145, AD-61146, AD-60892 o AD-60419.
Se puede proporcionar un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde el ARNbc comprende (i) una hebra antisentido que comprende, o está constituida por, la secuencia de UAAGAUGAGACACUCUUUCUGGU (SEQ ID NO: 4153) o UAAGAUGAGACACUCTUUCUGGU (SEQ ID NO: 4154) y/o (ii) una hebra sentido que comprende, o está constituida por, la secuencia de CAGAAAGAGUGUCUCAUCUUA
(SEQ ID NO: 4155). Uno o más nucleótidos de la hebra antisentido y/o la hebra sentido pueden estar modificados como se describe en la presente.
Se puede proporcionar un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde el ARNbc comprende (i) una hebra antisentido que comprende, o está constituida por, la secuencia antisentido de AD-60489 y/o (ii) una hebra sentido que comprende, o está constituida por, la secuencia sentido de AD-60489 (donde la secuencia sentido y/o antisentido incluye una o más (por ejemplo, la totalidad) de las modificaciones de la hebra sentido y/o hebra antisentido de AD-60489).
Se puede proporcionar un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde el ARNbc comprende (i) una hebra antisentido que comprende, o está constituida por, la secuencia antisentido de AD-60519 y/o (ii) una hebra sentido que comprende, o está constituida por, la secuencia sentido de AD-60519 (donde la secuencia sentido y/o antisentido incluye una o más (por ejemplo, la totalidad) de las modificaciones de la hebra sentido y/o hebra antisentido de AD-60519).
Se puede proporcionar un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde el ARNbc comprende (i) una hebra antisentido que comprende, o está constituida por, la secuencia antisentido de AD-61193 y/o (ii) una hebra sentido que comprende, o está constituida por, la secuencia sentido de AD-61193 (donde la secuencia sentido y/o antisentido incluye una o más (por ejemplo, la totalidad) de las modificaciones de la hebra sentido y/o hebra antisentido de AD-61193).
Se puede proporcionar un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde el ARNbc comprende (i) una hebra antisentido que comprende, o está constituida por, la secuencia antisentido de AD-60819 y/o (ii) una secuencia sentido que comprende, o está constituida por, la secuencia sentido de AD-60819
(donde la secuencia sentido y/o antisentido incluye una o más (por ejemplo, la totalidad) de las modificaciones de la hebra sentido y/o hebra antisentido de AD-60819).
Se puede proporcionar un ARNbc para inhibir la expresión de ALAS1, donde el ARNbc comprende (i) una hebra antisentido que comprende, o está constituida por, la secuencia antisentido de AD-60489, AD-60519, AD-61193 o
AD-60819 (o una secuencia antisentido no modificada correspondiente) y/o (ii) una hebra sentido que comprende, o está constituida por, la secuencia sentido de AD-60489, Ad-60519, AD-61193 o AD-60819 (o una secuencia antisentido no modificada correspondiente). El ARNbc puede comprender (i) una hebra antisentido que está constituida por la secuencia antisentido de Ad-60489, AD-60519, AD-61193 o Ad-60819 y/o (ii) una hebra sentido que está constituida por la secuencia sentido de AD-60489, AD-60519, AD-61193 o AD-60819, excepto en que la hebra antisentido y/o hebra sentido del ARNbc difiere en 1, 2 o 3 nucleótidos de la correspondiente secuencia antisentido y/o sentido de AD-60489, AD-60519, AD-61193 o Ad-60819.
Las secuencias y modificaciones de AD-60489, AD-60519, AD-61193 y AD-60819 se muestran en la Tabla 44 a continuación.

donde c, a, g, u = 2’-OMe riblonucleósidos; Af, Cf, G, Uf = 2’F ribonucleósidos; S = fosforotioato; L96 tiene la estructura representada en la Tabla 1.
Se puede proporcionar un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde el ARNbc comprende (i) una hebra antisentido que comprende, o está constituida por, la secuencia antisentido de AD-60489, AD-60519 o AD-61193 y/o (ii) una hebra sentido que comprende, o está constituida por, la secuencia sentido de AD-60489, AD-60519 o AD-61193 (incluida la secuencia de nucleótidos y una o más (por ejemplo, la totalidad) de las modificaciones de la hebra sentido y/o hebra antisentido de AD-60489, Ad-60519 o AD-61193).
Se puede proporcionar un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde el ARNbc es AD-60489, AD-60519, AD-61193 o AD-60819. Se puede proporcionar un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde el ARNbc es a D-60489, AD-60519 o AD-61193 (por ejemplo, incluida la secuencia de nucleótidos y/o una o más (por ejemplo, la totalidad) de las modificaciones de AD-60489, AD-60519 o AD-61193).
El ARNbc es, puede comprender, o puede estar constituido por, AD-60489 (por ejemplo, incluida la secuencia de nucleótidos y/o una o más (por ejemplo, la totalidad) de las modificaciones de AD-60489).
El ARNbc es, puede comprender, o puede estar constituido por, AD-60519 (por ejemplo, incluida la secuencia de nucleótidos y/o una o más (por ejemplo, la totalidad) de las modificaciones de AD-60519).
El ARNbc es, puede comprender, o puede estar constituido por, AD-61193 (por ejemplo, incluida la secuencia de nucleótidos y/o una o más (por ejemplo, la totalidad) de las modificaciones de AD-61193).
El ARNbc es, puede comprender, o puede estar constituido por, AD-60819 (por ejemplo, incluida la secuencia de nucleótidos y/o una o más (por ejemplo, la totalidad) de las modificaciones de AD-60819).
El ARNbc (por ejemplo, AD-60489, AD-60519, AD-61193, AD-60819, u otro ARNbc descrito en la presente en cualquiera de las Tablas 21 a 40) puede ser eficaz para suprimir el nivel hepático del ARNm de ALAS1, p. ej., para lograr el silenciamiento de al menos un 10%, 20%, 30%, 40%, 50%, 60%, 70% u 80% (p. ej., de modo que los niveles del ARNm de ALAS1 se reducen hasta un 90% o menos, 80% o menos, 70% o menos, 60% o menos, 50% o menos, 40% o menos, 30% o menos o 20% o menos de un nivel de control de ARNm de ALAS1 hepático, p. ej., el nivel de un individuo o grupo de individuos no tratados, p. ej., un individuo o grupo de individuos tratados solo con PBS). La eficacia del ARNbc en la supresión del nivel hepático del ARNm de ALAS1 se puede evaluar utilizando un modelo en primates no humanos, p. ej., como se describe en la presente en los Ejemplos.
El ARNbc (p. ej., AD-60489, AD-60519, AD-61193, AD-60819 u otro ARNbc descrito en la presente en cualquiera de las Tablas 21 a 40) puede ser eficaz para suprimir el nivel circulante del ARNm de ALAS1, p. ej., para lograr silenciar al menos un 10%, 20%, 30%, 40%, 50%, 60%, 70% u 80% (p. ej., de modo que los niveles de ARNm de ALAS1 se reduzcan hasta un 90% o menos, 80% o menos, 70% o menos, 60% o menos, 50% o menos, 40% o menos, 30% o menos o 20% o menos de un nivel de control de ARNm de ALAS1 circulante, p. ej., el nivel anterior al tratamiento con el ARNbc, o el nivel en un individuo o grupo de individuos no tratados). La eficacia del ARNbc en la supresión del nivel circulante de ARNm de ALAS1 se puede evaluar utilizando un modelo en primates no humanos, p. ej., como se describe en la presente en los Ejemplos. El nivel circulante de ARNm de ALAS1 se puede evaluar utilizando un ensayo de detección de ARN extracelular circulante (cERD, por sus siglas en inglés), p. ej., como se describe en la presente o en Sehgal, A. et al. Quantitation of tissue-specific target gene modulation using circulating RNA (Póster presentado el 9 de febrero de 2012 en el simposio Keystone Gene Silencing by small RNAs (Vancouver, 7-12 de febrero de 2012) o Sehgal, A. et al. Tissue-specific gene silencing monitored in circulating RNA, RNA, 20: 1 -7, con publicación electrónica el 19 de diciembre de 2013.
El método de cERD se puede aplicar a cualquier muestra biológica apropiada. El nivel circulante de ARNm de ALAS1 se puede evaluar utilizando una muestra de sangre, p. ej., una muestra de suero. El nivel circulante de ARNm de ALAS1 se puede evaluar utilizando una muestra de orina.
El ARNbc puede ser un derivado de AD-60489 que se describe en la presente, p. ej., en cualquiera de las tablas de la presente. El ARNbc puede mostrar una actividad mejorada en comparación con AD-60489. En algunos de tales aspectos, el ARNbc es AD-60519.
El ARNbc puede ser un derivado de AD-58632 que se describe en la presente, p. ej., en cualquiera de las tablas de la presente. El ARNbc puede mostrar una actividad mejorada en comparación con AD-58632.
La actividad mejorada puede estar indicada por una CI50 menor, p. ej., como se determina en función de ensayos in vitro, p. ej., como se describe en la presente, p. ej., en los Ejemplos.
La actividad mejorada puede estar indicada por una dosis eficaz menor. La dosis eficaz se puede determinar en función de la administración de una única dosis o de múltiples dosis repetidas. La dosis eficaz se puede determinar en función de la DE50 de dosis única. La dosis eficaz o la DE50 de dosis única se pueden determinar en función de un
ensayo in vivo. El ensayo in vivo se puede llevar a cabo en un animal no humano, p. ej., en una rata, en un primate no humano o en un ratón.
La dosis eficaz se puede determinar en función de la dosis requerida para obtener una reducción de un nivel del ARNm de ALAS1 (p. ej., un nivel hepático del ARNm de ALAS1 y/o un nivel circulante de ARNm de ALAS1), p. ej., como se describe en la presente en los Ejemplos. El ARNm circulante se puede evaluar utilizando el ensayo de cERD.
La dosis eficaz se puede determinar en función de la dosis requerida para obtener una reducción de un nivel (p. ej., un nivel en orina y/o plasma) de ALA y/o PBG.
La dosis eficaz se puede determinar en función de la dosis requerida para obtener un efecto de tratamiento particular descrito en la presente, p. ej., prevención o reducción de síntomas asociados con una porfiria.
La actividad mejorada puede estar indicada por el logro de un nivel hepático más elevado del ARNbc. Un nivel hepático más elevado se puede obtener después de una única dosis de ARNbc (p. ej., una dosis de 1,2.5, 3, 5 o 10 mg/kg). Un nivel hepático más elevado se puede obtener después de que se hayan administrado múltiples dosis de ARNbc (p. ej., 2-10 dosis diarias o semanales de 1,2.5, 3, 5 o 10 mg/kg).
El ARNi puede englobar un ARNbc que tiene una hebra de ARN (la hebra antisentido) que tiene una región que es sustancialmente complementaria respecto a una porción de un ARNm de ALAS1, p. ej., un ARNm de ALAS1 humano (p. ej., un ARNm de ALAS1 humano como el que se proporciona en la SEQ ID NO:1 o SEQ ID NO:382). Un ARNi para inhibir la expresión de un gen ALAS1 puede incluir al menos dos secuencias que son complementarias entre sí. El ARNi incluye una hebra sentido que tiene una primera secuencia y una hebra antisentido que tiene una segunda secuencia. La hebra antisentido incluye una secuencia de nucleótidos que es sustancialmente complementaria respecto a al menos parte de un ARNm que codifica un transcrito de ALAS1, y la región de complementariedad tiene una longitud de 30 nucleótidos o inferior, y de al menos 15 nucleótidos. Generalmente, el ARNi tiene una longitud de 19-24 nucleótidos.
El ARNi puede tener una longitud de 19-21 nucleótidos. El ARNi puede tener una longitud de 19-21 nucleótidos y se puede encontrar en una formulación lipídica, p. ej., una formulación de nanopartículas lipídicas (LNP, por sus siglas en inglés) (p. ej., una formulación LNP11).
El ARNi puede tener una longitud de 21-23 nucleótidos. El ARNi puede tener una longitud de 21-23 nucleótidos y se puede encontrarencuentra en forma de conjugado, p. ej., conjugado con uno o más derivados de tipo GalNAc según se describe en la presente.
Un ARNi (p. ej., un ARNbc) que se expone en la presente incluye una primera secuencia de un ARNbc que se selecciona a partir del grupo constituido por las secuencias sentido de las Tablas 21 a 40 y una segunda secuencia que se selecciona a partir del grupo constituido por las secuencias antisentido correspondientes de las Tablas 21 a 40.
Las moléculas de ARNi que se exponen en la presente pueden incluir nucleótidos de origen natural o pueden incluir al menos un nucleótido modificado. El al menos un nucleótido modificado puede incluir una o más de una modificación en el nucleótido elegido del grupo constituido por un ácido nucleico bloqueado (LNA, por sus siglas en inglés), un nucleótido acíclico, un hexitol o un ácido nucleico con hexosa (HNA, por sus siglas en inglés), un ácido nucleico con ciclohexeno (CeNA, por sus siglas en inglés), 2’-metoxietilo, 2’-O-alquilo, 2’-O-alilo, 2’-C-alilo, 2’-fluoro, 2’-desoxi, 2’-hidroxilo o cualquier combinación de estos. El al menos un nucleótido modificado puede incluir, sin carácter limitante, un nucleótido con una modificación de tipo 2’-O-metilo, nucleótido con modificación de tipo 2’-fluoro, un nucleótido que tiene un grupo 5’-fosforotioato y un nucleótido terminal ligado a un ligando, p. ej., una N-acetilgalactosamina (GalNAc) o un derivado de colesterilo. Como alternativa, el nucleótido modificado se puede seleccionar a partir del grupo constituido por: un nucleótido con una modificación de tipo 2'-desoxi-2'-fluoro, un nucleótido con una modificación de tipo 2'-desoxi, un nucleótido bloqueado, un nucleótido acíclico, un nucleótido que no sea básico, un nucleótido con una modificación de tipo 2’-amino, un nucleótido con una modificación de tipo 2’-alquilo, un nucleótido de tipo morfolino, un fosforamidato y un nucleótido que comprende una base que no sea natural. Una secuencia modificada de este tipo se puede basar, p. ej., en una primera secuencia de dicho ARNi seleccionada a partir del grupo constituido por las secuencias sentido divulgadas en las Tablas 21 a 40 y una segunda secuencia seleccionada a partir del grupo constituido por las secuencias antisentido correspondientes divulgadas en las Tablas 21 a 40.
Un ARNi según se describe en la presente puede tener como diana una variante de un transcrito de ARN de ALAS1 de origen natural y el ARNi puede tener además como diana un transcrito mutado (p. ej., un ARN de ALAS1 portador de una variante alélica). Por ejemplo, un ARNi mencionado en la presente puede tener como diana una variante polimórfica tal como un polimorfismo de un único nucleótido (SNP, por sus siglas en inglés) de ALAS1. El ARNi puede tener como diana tanto un transcrito de ALAS1 mutado como uno de origen natural. El ARNi puede además tener como diana una variante de un transcrito particular de ALAS1 (p. ej., la variante 1 de ALAS1 humano). El
agente de ARNi puede además tener como diana múltiples variantes del transcrito (p. ej., tanto la variante 1 como la variante 2 de ALAS1 humano).
Un ARNi mencionado en la presente puede tener como diana una región no codificante de un transcrito de ARN de ALAS1 tal como la región no traducida 5’ o 3’ de un transcrito.
Un ARNi según se describe en la presente se puede encuentrar en forma de conjugado, p. ej., un conjugado con un carbohidrato, el cual puede actuar como un resto y/o ligando de direccionamiento, según se describe en la presente. El conjugado puede estar unido al extremo 3’ de la hebra sentido del ARNbc. El conjugado puede estar unido mediante un conector, p. ej., mediante un conector ramificado bivalente o trivalente.
El conjugado puede comprender uno o más derivados de tipo W-acetilgalactosamina (GalNAc). Tal conjugado también se denomina en la presente conjugado de tipo GalNAc. El conjugado puede dirigir el agente de iARN hacia una célula particular, p. ej., una célula hepática, p. ej., un hepatocito. Los derivados de tipo GalNAc se pueden unir mediante un conector, p. ej., un conector ramificado bivalente o trivalente. El conjugado puede ser

El agente de iARN puede estar unido al conjugado de tipo carbohidrato mediante un conector, p. ej., un conector como el que se muestra en la siguiente representación esquemática, donde X es O o S

X puede ser O. X puede ser S.
El agente de iARN se puede conjugar a L96 según se define en la Tabla 1 y se muestra a continuación
El ARNbc puede tener uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce, quince, dieciséis o la totalidad de los siguientes:
(i) se sintetiza por medios químicos, p. ej., se sintetiza mediante síntesis de oligonucleótidos en fase sólida; (ii) todos los nucleótidos en el ARNbc están modificados, por ejemplo, todos los nucleótidos tienen una modificación de tipo 2’-OMe o 2’-F, o una combinación de modificaciones de tipo 2’-OMe y 2’-F;
(iii) todos los nucleótidos están conectados mediante uniones 3’-5’-fosfodiéster;
(iv) la hebra sentido comprende o está constituida por 21 nucleótidos;
(v) la hebra antisentido sentido comprende o está constituida por 23 nucleótidos;
(vi) tiene un extremo romo en el extremo 3’ de la hebra sentido;
(vii) tiene una protuberancia en 3’, p. ej., tiene una protuberancia de 2 nucleótidos, en el extremo 3’ de la hebra antisentido;
(viii) está unido covalentemente a un ligando que contiene tres restos de N-acetilgalactosamina (GalNAc); (ix) el extremo 3’ de la hebra sentido está conjugado con el resto de GalNAc de tres antenas (p. ej., denominado en la presente L96 como se define en la Tabla 1). El extremo 3’ puede estar unido al resto de GalNAc de tres antenas a través de una unión fosfodiéster;
(x) tiene una hebra antisentido que comprende una o más (p. ej., cuatro) uniones fosforotioato. Las uniones fosforotioato pueden estar ubicadas en el extremo 3’ y en el extremo 5’ de la hebra antisentido. Dos uniones fosforotioato pueden estar ubicadas en el extremo 3’ y dos uniones fosforotioato están ubicadas en el extremo 5’ de la hebra antisentido;
(xi) tiene una hebra sentido que comprende una o más (p. ej., dos) uniones fosforotioato.
La una o más (p. ej., dos) uniones fosforotioato pueden estar ubicadas en el extremo 5’ de la hebra sentido; (xii) 21 nucleótidos de la hebra sentido se hibridan con los 21 nucleótidos complementarios de la hebra antisentido;
(xiii) forma 21 pares de bases de nucleótidos y una protuberancia de dos bases en el extremo 3’ de la hebra antisentido;
(xiv) comprende, o está constituido por, una hebra sentido y antisentido que tiene la secuencia de AD-60519;
(xv) tiene una hebra sentido con 10, 12, 14, 16, 18, 19, 20 o la totalidad de las modificaciones de la hebra sentido de AD-60519;
(xvi) tiene una hebra antisentido con 10, 12, 14, 16, 18, 19, 20 o la totalidad de las modificaciones de la hebra antisentido de AD-60519; o
(xvii) tiene la secuencia dúplex y todas las modificaciones de AD-60519.
El ARNbc puede estar en forma de un conjugado que tiene la siguiente estructura (también denominada en la presente AD-60519 o ALN-60519) (SEQ ID nOs 5238-5239, respectivamente, en orden de aparición):

Af, Cf, Gf, Uf = 2’-F ribonucleósidos
Am, Cm, Gm, Um = 2’-OMe ribonucleósidos
S = fosforotioato

Un aspecto proporcionado en la presente es una composición, p. ej., una composición farmacéutica, que incluye uno o más de los ARNi descritos en la presente y un portador o vehículo de suministro farmacéuticamente aceptable. La composición se puede utilizar para inhibir la expresión de un gen de ALAS1 en un organismo, por lo general, un sujeto humano. La composición se puede utilizar para tratar una porfiria, p. ej., AIP.
En un aspecto, se proporciona en la presente un ARNi que es un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde dicho ARNbc comprende una hebra sentido y una hebra antisentido con una longitud de 15-30 pares de bases y la hebra antisentido es complementaria respecto a al menos 15 nucleótidos contiguos de la SEQ ID NO: 1 o 382.
En otro aspecto, se proporciona un ARNi que es un ARNi bicatenario (ARNbc) que comprende una hebra sentido complementaria respecto a una hebra antisentido, donde dicha hebra antisentido comprende una región de complementariedad respecto a un transcrito de ARN de ALAS1, donde cada hebra tiene de aproximadamente 14 a aproximadamente 30 nucleótidos, donde dicho agente de iARN bicatenario se representa mediante la fórmula (III):
sentido: 5' np -Na -(X X X)i-Nb -Y Y Y -Nb -(Z Z Z)j -Na - nq 3'
antisentido: 3' np '-Na '-(X'X'X')k-Nb '-Y'Y'Y'-Nb '-(Z'Z'Z')l-Na '- nq ' 5' (III) donde:
i, j, k, y l son cada uno independientemente 0 o 1;
p, p’, q y q' son cada uno independientemente 0-6;
cada Na y Na ' representa independientemente una secuencia oligonucleotídica que comprende 0-25 nucleótidos, los cuales están modificados o no modificados, o combinaciones de estos, comprendiendo cada secuencia al menos dos nucleótidos modificados de forma diferente;
cada Nb y Nb ' representa independientemente una secuencia oligonucleotídica que comprende 0-10 nucleótidos, los cuales están modificados o no modificados, o combinaciones de estos;
cada np , np ', nq y nq ' representa independientemente un nucleótido protuberante;
XXX, YYY, ZZZ, X'X'X', Y'Y'Y' y Z'Z'Z' representan cada uno independientemente un motivo de tres modificaciones idénticas en tres nucleótidos consecutivos;
las modificaciones en Nb difieren de la modificación en Y y las modificaciones en Nb ' difieren de la modificación en Y'.
La hebra sentido puede estar conjugada con al menos un ligando.
i puede ser 1; j puede ser 1; o tanto i como j pueden ser 1.
k puede ser 1; l puede ser 1; o tanto k como l pueden ser 1.
XXX es complementario respecto a X'X'X', YYY puede ser complementario respecto a Y'Y'Y' y ZZZ puede ser complementario respecto a Z'Z'Z'.
Y'Y'Y' se puede encontrar en las posiciones 11, 12 y 13 de la hebra antisentido del extremo 5'.
Y' puede ser 2'-O-metilo.
La región dúplex puede tener una longitud de 15-30 pares de nucleótidos.
La región dúplex puede tener una longitud de 17-23 pares de nucleótidos.
La región dúplex puede tener una longitud de 19-21 pares de nucleótidos.
La región dúplex puede tener una longitud de 21 -23 pares de nucleótidos.
La modificación en el nucleótido se puede seleccionar del grupo constituido por un ácido nucleico bloqueado (LNA), un nucleótido acíclico, un hexitol o ácido nucleico con hexosa (HNA), un ácido nucleico con ciclohexeno (CeNA), 2’-metoxietilo, 2’-O-alquilo, 2’-O-alilo, 2’-C-alilo, 2’-C-alilo, 2’-fluoro, 2’-desoxi, 2’-hidroxilo y cualquier combinación de estos.
Las modificaciones de los nucleótidos se pueden seleccionar a partir del grupo constituido por LNA, HNA, CeNA, 2'-metoxietilo, 2'-O-alquilo, 2'-O-alilo, 2'-C-alilo, 2'-fluoro, 2'-desoxi, 2’-hidroxilo y combinaciones de estas.
Las modificaciones de los nucleótidos son 2'-O-metilo, 2'-fluoro o ambas.
El ligando puede comprender un carbohidrato.
El ligando puede estar unido mediante un conector.
El conector puede ser un conector ramificado bivalente o trivalente.
El ligando puede ser

El ligando y el conector pueden ser como se muestran en la Fórmula XXIV:

El ligando puede estar unido al extremo 3’ de la hebra sentido.
El ARNbc puede consistir en o comprender una secuencia de nucleótidos seleccionada a partir del grupo de secuencias proporcionadas en las Tablas 21-40.
En un aspecto adicional, un ARNi proporcionado en la presente es un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde dicho ARNbc comprende una hebra sentido y una hebra antisentido, comprendiendo la hebra antisentido una región de complementariedad respecto a un transcrito de ARN de ALAS1, donde la hebra antisentido comprende al menos 15 nucleótidos contiguos que no difieren en más de 3 nucleótidos de una de las secuencias antisentido enumeradas en cualquiera de las Tablas 21-40. Los nucleótidos de la hebra antisentido pueden tener menos modificaciones, más modificaciones o modificaciones diferentes en comparación con las secuencias antisentido enumeradas en cualquiera de las Tablas 21-40.
Las secuencias sentido y antisentido pueden ser las de un dúplex descrito en la presente que suprime la expresión del ARNm de ALAS1 al menos un 50%, 60%, 70%, 80%, 85% o 90%, p. ej., según se evalúa utilizando un ensayo descrito en los Ejemplos que se proporcionan en la presente.
La expresión del ANRm de ALAS1 se puede evaluar en función de un nivel del ARNm de ALAS1 en el hígado, p. ej., como se evalúa utilizando una muestra de una biopsia hepática. La expresión del ARNm de ALAS1 se puede evaluar en función del nivel del ARNm de ALAS1 en un fluido biológico, p. ej., sangre, suero, plasma, líquido cefalorraquídeo u orina. La expresión del ARNm de ALAS1 se puede evaluar utilizando un ensayo de detección de ARN extracelular circulante (cERD), p. ej., un ensayo cERD como se describe en la presente o en Sehgal, A. et al.
Quantitation of tissue-specific target gene modulation using circulating RNA (Póster presentado el 9 de febrero de 2012 en el simposio Keystone Gene Silencing by small RNAs (Vancouver, 7-12 de febrero de 2012) o Sehgal, A. et al. Tissue-specific gene silencing monitored in circulating RNA, RNA, 20: 1-7, con publicación electrónica el 19 de diciembre de 2013.
El ARNbc puede comprender al menos un nucleótido modificado.
Al menos uno de los nucleótidos modificados se puede seleccionar a partir del grupo constituido por: un nucleótido con una modificación de tipo 2'-0-metilo, un nucleótido que comprende un grupo 5'-fosforotioato y un nucleótido terminal unido a un derivado de tipo colesterilo o un grupo de tipo bisdecilamida del ácido dodecanoico.
El nucleótido modificado se puede seleccionar a partir del grupo constituido por: un nucleótido con una modificación de tipo 2'-desoxi-2'-fluoro, un nucleótido con una modificación de tipo 2'-desoxi, un nucleótido bloqueado, un nucleótido acíclico, un nucleótido que no sea básico, un nucleótido con una modificación de tipo 2’-amino, un nucleótido con una modificación de tipo 2’-alquilo, un nucleótido de tipo morfolino, un fosforamidato y un nucleótido que comprende una base que no sea natural.
La región de complementariedad puede tener una longitud de al menos 17 nucleótidos.
La región de complementariedad puede tener una longitud comprendida entre 19 y 21 nucleótidos.
La región de complementariedad puede tener una longitud de 19 nucleótidos.
Cada hebra puede tener una longitud que no es superior a 30 nucleótidos.
Al menos una hebra puede comprender una protuberancia 3’ de al menos 1 nucleótido. La hebra antisentido puede comprender una protuberancia 3’ de al menos 1 nucleótido.
Al menos una hebra puede comprender una protuberancia 3’ de al menos 2 nucleótidos. La hebra antisentido puede comprender una protuberancia 3’ de al menos 2 nucleótidos. La hebra antisentido puede comprender una protuberancia 3’ de 2 nucleótidos.
Un ARNbc descrito en la presente puede comprender además un ligando.
El ligando puede ser un ligando de tipo GalNAc.
El ligando puede dirigir el ARNbc hacia los hepatocitos.
El ligando puede estar conjugado con el extremo 3’ de la hebra sentido del ARNbc.
La región de complementariedad puede estar constituida por una secuencia antisentido seleccionada de las secuencias antisentido enumeradas en las Tablas 21-40, o una secuencia antisentido correspondiente en la que algunos o la totalidad de los nucleótidos están modificados. La región de complementariedad puede estar constituida por la secuencia UAAGAUGAGACACUCUUUCUGGU (SEQ ID NO: 4153) o UAAGAUGAGACACUCTUUCUGGU (SEQ ID NO: 4154). La región de complementariedad puede estar constituida por la secuencia antisentido del AD-60489 dúplex. La región de complementariedad puede estar constituida por la secuencia antisentido del AD-60519 dúplex.
La región de complementariedad puede estar constituida por una secuencia antisentido seleccionada de un dúplex descrito en la presente que suprime la expresión del ARNm de ALAS1 en al menos un 50%, 60%, 70%, 80%, 85% o 90%, p. ej., según se evalúa utilizando un ensayo descrito en los Ejemplos proporcionados en la presente.
El ARNbc puede comprender una hebra sentido que está constituida por una secuencia de la hebra sentido seleccionada de las Tablas 21-40, y una hebra antisentido que está constituida por una secuencia antisentido seleccionada de las Tablas 21-40. El ARNbc puede comprender un par de las correspondientes secuencias sentido y antisentido seleccionadas de los dúplex descritos en las Tablas 21 -40.
También se proporciona en la presente una célula que contiene al menos uno de los ARNi (p. ej., ARNbc) mostrados en la presente. Por lo general, la célula es una célula de mamífero, tal como una célula humana. En algunas realizaciones, la célula es una célula eritroide. La célula también puede ser una célula hepática (p. ej., un hepatocito).
En un aspecto, se proporciona en la presente una composición farmacéutica para inhibir la expresión de un gen ALAS1, comprendiendo la composición un ARNi (p. ej. un ARNbc) descrito en la presente.
Las composiciones farmacéuticas descritas en la presente, el ARNi (p. ej., ARNbc) se puede administrar en una solución no tamponada. La solución no tamponada puede ser solución salina o agua, p. ej. Agua para inyección. La composición farmacéutica puede comprender AD-60519 y agua para inyección. La composición puede comprender aproximadamente de 100 a 300 mg/mL, p. ej., 200 mg/mL, de AD-60519. La composición puede tener un pH de 6.0-7.5, p. ej., de aproximadamente 7.0. La composición puede ser para inyección subcutánea. La composición farmacéutica se puede empaquetar en un envase (p. ej., un vial de vidrio, p. ej., un vial de vidrio de 2 mL), en un volumen de aproximadamente 0.3 a 1 mL, p. ej., 0.55 mL. La composición farmacéutica puede ser ALN-AS1 como se describe en la presente en los ejemplos.
En un aspecto de las composiciones farmacéuticas descritas en la presente, el ARNi (p. ej., ARNbc) se administra con una solución tampón. La solución tapón puede comprender acetato, citrato, prolamina, carbonato o fosfato o cualquier combinación de estos. La solución tampón puede ser una solución salina tamponada con fosfato (PBS). En un aspecto de las composiciones farmacéuticas descritas en la presente, el ARNi (p. ej., ARNbc) se dirige hacia los hepatocitos.
En un aspecto de las composiciones farmacéuticas descritas en la presente, la composición se administra por vía intravenosa.
En un aspecto de las composiciones farmacéuticas descritas en la presente, la composición se administra por vía subcutánea.
En un aspecto, una composición farmacéutica comprende un ARNi (p. ej., ARNbc) descrito en la presente que comprende un ligando (p. ej., un ligando de tipo GalNAc) que dirige el ARNi (p. ej., ARNbc) hacia los hepatocitos. Una composición farmacéutica puede comprender un ARNi (p. ej., ARNbc) descrito en la presente que comprende un ligando (p. ej., un ligando de tipo GalNAc) y la composición farmacéutica se puede administrar por vía subcutánea. El ligando puede dirigir el ARNi (p. ej., ARNbc) hacia los hepatocitos.
Una composición farmacéutica, p. ej., una composición descrita en la presente, puede incluir una formulación lipídica. El agente de iARN puede ser una formulación LNP, p. ej., una formulación MC3. La formulación LNP puede dirigir el agente de iARN hacia una célula particular, p. ej., una célula hepática, p. ej., un hepatocito. La formulación lipídica puede ser una formulación LNP11. La composición se puede administrar por vía intravenosa.
La composición farmacéutica se puede formular para ser administrada de acuerdo con una pauta posológica descrita en la presente, p. ej., no más de una vez cada cuatro semanas, no más de una vez cada tres semanas, no más de una vez cada dos semanas o no más de una vez por semana. La administración de la composición farmacéutica se puede mantener durante un mes o más, p. ej., uno, dos, tres o seis meses, o un año o más.
Una composición que contiene un ARNi que se expone en la presente, p. ej., un ARNbc que tiene ALAS1 como diana, se puede administrar con un agente terapéutico que no sea un ARNi tal como un agente conocido por tratar una porfiria (p. ej., AIP) o un síntoma de una porfiria (p. ej., dolor). Una composición que contiene un ARNi que se expone en la presente, p. ej., un ARNbc que tiene AIP como diana, se puede administrar junto con un régimen terapéutico que no sea un ARNi tal como hemina o glucosa (p. ej., una infusión de glucosa (p. ej., glucosa IV)). Por ejemplo, un ARNi que se expone en la presente se puede administrar antes, después o de forma simultánea con glucosa, dextrosa o un tratamiento similar que sirva para restablecer el equilibrio energético (p. ej., nutrición parenteral total). Un ARNi que se expone en la presente también se puede administrar antes, después o de forma simultánea con la administración de un producto de tipo hemo (p. ej., hemina, arginato de hemo o albumina que contenga hemo) y opcionalmente también se puede administrar combinado con glucosa (p. ej., glucosa IV) o similares.
Normalmente, la glucosa administrada para el tratamiento de una porfiria se administra por vía intravenosa (IV). La administración de glucosa por vía intravenosa se denomina en la presente "glucosa IV". Sin embargo, también se contemplan aspectos alternativos en las cuales la glucosa se administre por otros medios.
Un ARNi de ALAS1 se puede administrar a un paciente y a continuación se administra el agente o régimen terapéutico que no sea un ARNi (p. ej., glucosa y/o un producto de tipo hemo) al paciente (o viceversa). El ARNi de ALAS1 y el agente terapéutico o régimen terapéutico que no sea un ARNi se pueden administrar de forma simultánea.
En un aspecto de la presente se proporciona además un método para inhibir la expresión de ALAS1 en una célula, comprendiendo el método: (a) introducir en la célula un ARNi (p. ej., un ARNbc) descrito en la presente y (b) mantener la célula del paso (a) durante un tiempo suficiente para obtener la degradación del transcrito de ARNm del gen ALAS1, de este modo se inhibe la expresión del gen ALAS1 en la célula.
En un aspecto de la presente se proporciona además un método para reducir o inhibir la expresión de un gen ALAS1 en una célula (p. ej., una célula eritroide o una célula hepática tal como, p. ej., un hepatocito). El método incluye:
(a) introducir en la célula un ácido ribonucleico bicatenario (ARNbc), donde el ARNbc incluye al menos dos secuencias que son complementarias entre sí. El ARNbc tiene una hebra sentido que tiene una primera secuencia y una hebra antisentido que tiene una segunda secuencia; la hebra antisentido tiene una región de complementariedad que es sustancialmente complementaria respecto a al menos una parte de un ARNm que codifica ALAS1, y donde la región de complementariedad tiene una longitud de 30 nucleótidos o inferior, es decir, 15-30 nucleótidos, y generalmente tiene una longitud de 19-24 nucleótidos, y donde el ARNbc, cuando entra en contacto con una célula que expresa ALAS1, inhibe la expresión del gen ALAS1 al menos un 10%, p. ej., al menos un 20%, al menos un 30%, al menos un 40% o más; y
(b) mantener la célula del paso (a) durante un tiempo suficiente para obtener la degradación del transcrito de ARNm del gen ALAS1, de este modo se reduce o inhibe la expresión de un gen ALAS1 en la célula.
En aspectos de los métodos anteriores para inhibir la expresión de ALAS1 en una célula, la célula se trata ex vivo, in vitro o in vivo. La célula puede ser un hepatocito.
La célula puede estar presente en un sujeto que necesita tratar, prevenir y/o gestionar un trastorno relacionado con la expresión de ALAS1.
El trastorno puede ser una porfiria. La porfiria puede ser una porfiria intermitente aguda o una porfiria debida a la deficiencia de ALA-deshidratasa.
La porfiria puede ser una porfiria hepática, p. ej., una porfiria seleccionada entre porfiria intermitente aguda (AIP), coproporfiria hereditaria (HCP), porfiria variegata (VP), porfiria debida a la deficiencia de ALA-deshidratasa (ADP) y porfiria hepatoeritropoyética. La porfiria puede ser una porfiria hepática dominante homozigótica (p. ej., AIP, HCP o VP dominante homozigótica) o una porfiria hepatoeritropoyética. La porfiria puede ser una porfiria dual.
La expresión de ALAS1 se puede inhibir al menos un 30%.
El ARNi (p. ej., ARNbc) puede presentar una CI50 comprendida en el intervalo de 0.01 -1 nM.
La célula (p. ej., el hepatocito) puede ser una célula de mamífero (p. ej., una célula humana, de primate no humano o de roedor).
La célula se puede tratar ex vivo, in vitro o in vivo (p. ej., la célula está presente en un sujeto (p. ej., un paciente que necesita tratar, prevenir y/o gestionar un trastorno relacionado con la expresión de ALAS1)).
El sujeto puede ser un mamífero (p. ej., un ser humano) que corre el riesgo de parecer una porfiria o al cual se le ha diagnosticado una porfiria, p. ej., anemia sideroblástica relacionada con X (XLSA), porfiria debida a la deficiencia de ALA-deshidratasa (porfiria de Doss o ADP), porfiria intermitente aguda (AIP), porfiria eritropoyética congénita (CEP), porfiria cutánea tardía (PCT), coproporfiria hereditaria (coproporfiria o HCP), porfiria variegata (VP), protoporfiria
eritropoyética (EPP) o eritroporfiria transitoria de la infancia. El trastorno puede ser una porfiria hepática aguda, p. ej., una porfiria debida a la deficiencia de ALA-deshidratasa (ADP), AIP, HCP o VP. El trastorno puede ser una porfiria debida a la deficiencia de ALA-deshidratasa (ADP) o AIP.
La porfiria puede ser una porfiria hepática, p. ej., una porfiria seleccionada entre porfiria intermitente aguda (AIP), coproporfiria hereditaria (HCP), porfiria variegata (VP), porfiria debida a la deficiencia de ALA-deshidratasa (ADP) y porfiria hepatoeritropoyética. La porfiria puede ser una porfiria hepática dominante homozigótica (p. ej., AIP, HCP o VP dominante homozigótica) o una porfiria hepatoeritropoyética. La porfiria puede ser una porfiria dual.
El ARNbc introducido puede reducir o inhibir la expresión de un gen ALAS1 en la célula.
El ARNbc introducido puede reducir o inhibir la expresión de un gen ALAS1, o el nivel de una o más porfirinas o precursores de porfirinas (p. ej., ácido 5-aminolevulínico (ALA), porfopilinógeno (PBG), hidroximetilbilano (HMB), uroporfirinógeno I o III, coproporfirinógeno I o III, protoporfrinógeno IX y protoporfirina IX) o productos o metabolitos de porfirinas, al menos un 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50% o más en comparación con una referencia (p. ej., una célula no tratada o una célula tratada con un ARNbc de control que no actúe sobre la diana). Sin pretender vincularse a ninguna teoría, ALAS1 es la primera enzima de la vía de las porfirinas. Por lo tanto, es probable que la reducción de la expresión del gen ALAS1 reduzca el nivel de uno o más precursores de porfirinas, porfirinas o productos o metabolitos de porfirinas.
En otros aspectos, se proporcionan además en la presente métodos para tratar, prevenir o gestionar procesos patológicos relacionados con la expresión de ALAS1 (p. ej., procesos patológicos en los que participan porfirinas, precursores de porfirinas o defectos en la vía de las porfirinas tales como, por ejemplo, las porfirias). El método puede incluir administrar a un sujeto, p. ej., un paciente que necesite tal tratamiento, prevención o gestión, una cantidad eficaz (p. ej., terapéutica o profilácticamente eficaz) de uno o más de los ARNi que se exponen en la presente.
En un aspecto de la presente se proporciona además un método para tratar y/o prevenir un trastorno relacionado con la expresión de ALAS1 que comprende administrar a un sujeto que necesite tal tratamiento una cantidad terapéuticamente eficaz de un ARNi (p. ej., un ARNbc) descrito en la presente o una composición que comprenda un ARNi (p. ej., un ARNbc) descrito en la presente.
En un aspecto de la presente se proporciona además un método para tratar y/o prevenir una porfiria que comprende administrar a un sujeto que necesite tal tratamiento un ácido ribonucleico bicatenario (ARNbc), donde dicho ARNbc comprende una hebra sentido y una hebra antisentido con una longitud de 15-30 pares de bases y la hebra antisentido es complementaria respecto a al menos 15 nucleótidos contiguos de la SEQ ID NO:1 o la SEQ ID NO:382.
El sujeto (p. ej., el paciente) puede padecer una porfiria. El sujeto (p. ej., el paciente) puede correr el riesgo de desarrollar una porfiria. La administración del ARNi que tiene ALAS1 como diana puede aliviar o reducir la gravedad de al menos un síntoma de un trastorno relacionado con ALAS1 en el paciente.
El sujeto puede ser un mamífero (p. ej., un ser humano) que corre el riesgo de parecer o al cual se le ha diagnosticado un trastorno relacionado con la expresión de ALAS1, p. ej., una porfiria, p. ej., anemia sideroblástica relacionada con X (XLSA), porfiria debida a la deficiencia de ALA-deshidratasa (porfiria de Doss), porfiria intermitente aguda (AIP), porfiria eritropoyética congénita (CEP), porfiria cutánea tardía (PCT), coproporfiria hereditaria (coproporfiria o HCP), porfiria variegata (VP), protoporfiria eritropoyética (EPP) o eritroporfiria transitoria de la infancia. Asimismo, la porfiria puede ser una porfiria hepática aguda, p. ej., una porfiria debida a la deficiencia de ALA-deshidratasa (ADP), AIP, HCP o VP. Aquí, el trastorno puede ser una porfiria debida a la deficiencia de ALA-deshidratasa (ADP) o AIP.
El sujeto puede padecer o correr el riesgo de desarrollar una porfiria. La porfiria puede ser una porfiria hepática, p. ej., una porfiria seleccionada entre porfiria intermitente aguda (AIP), coproporfiria hereditaria (HCP), porfiria variegata (VP), porfiria debida a la deficiencia de ALA-deshidratasa (ADP) y porfiria hepatoeritropoyética. La porfiria puede ser una porfiria hepática dominante homozigótica (p. ej., AIP, HCP o VP dominante homozigótica) o una porfiria hepatoeritropoyética. La porfiria puede ser una porfiria dual.
Una porfiria, un síntoma de porfiria, un pródromo o un ataque de porfiria puede ser inducido por la exposición a un factor precipitante, según se describe en la presente. El factor precipitante puede ser la exposición a un agente químico. El factor precipitante puede ser un fármaco, p. ej., un fármaco con receta o un fármaco sin receta. El factor precipitante puede ser el ciclo menstrual, p. ej., una fase particular del ciclo menstrual, p. ej., la fase luteínica.
El ARNi (p. ej., ARNbc) o la composición que comprende el ARNi se puede administrar después de un ataque agudo de porfiria.
El ARNi (p. ej., ARNbc) o la composición que comprende el ARNi se puede administrar durante un ataque agudo de porfiria.
El ARNi (p. ej., ARNbc) o la composición que comprende el ARNi se puede administrar profilácticamente para prevenir un ataque agudo de porfiria.
El ARNi (p. ej., ARNbc) se puede formular como una formulación LNP.
El ARNi (p. ej., ARNbc) se puede encuentrar en forma de un conjugado de tipo GalNAc.
El ARNi (p. ej., ARNbc) se puede administrar con una dosis de 0.05-50 mg/kg.
El ARNi (p. ej., ARNbc) se puede administrar con una concentración de 0.01 mg/kg-5 mg/kg de peso corporal del sujeto.
El ARNi (p. ej., ARNbc) se puede formular como una formulación LNP y se administra con una dosis de 0.05-5 mg/kg.
El ARNi (p. ej., ARNbc) se puede encuentrar en forma de un conjugado de tipo GalNAc y se administra con una dosis de 0.5-50 mg/kg. El ARNi en el conjugado de GalNAc se puede administrar con una dosis inferior a 10 mg/kg (p. ej., 5 mg/kg o inferior), p. ej., una vez a la semana; p. ej., una dosis de 1 mg/kg o inferior, 2.5 mg/kg o inferior o 5 mg/kg o inferior, p. ej., una vez a la semana. El ARNi en el conjugado de GalNAc se puede administrar en una dosis de aproximadamente 2.5 mg/kg o inferior, p. ej., una vez a la semana. La administración del ARNi en el conjugado de GalNAc puede ser subcutánea.
El ARNi (p.ej., ARNbc) puede estar en forma de un conjugdo de GalNAc y se administra, p. ej., por vía subcutánea, en una dosis de 0-5 mg/kg, p. ej., 0-2.5 mg/kg o 1-2.5 mg/kg. El ARNi se puede administrar semanalmente. El ARNi se puede administrar como una composición que comprende el ARNi y agua para inyección. El ARNi puede ser AD-60519. La composición puede comprender el ARNi, p. ej., AD-60519, en una concentración de aproximadamente 200 mg/mL.
El método reduce el nivel de una porfirina o de un precursor de una porfirina en el sujeto.
El nivel se reduce al menos un 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80% o 90%. En una realización, el nivel se reduce al menos un 30%.
El precursor de porfirina es ácido 5-aminolevulínico (ALA) o porfopilinógeno (PBG).
El ARNi (p. ej., ARNbc) puede presentar una CI50 comprendida en el intervalo de 0.01 -1 nM.
Un método descrito en la presente
(i) puede mejorar un síntoma asociado con un trastorno relacionado con ALAS1 (p. ej., una porfiria),
(ii) puede inhibir la expresión de ALAS1 en el sujeto (p. ej., según se evalúa utilizando el ensayo cERD), (iii) puede reducir el nivel de un precursor de porfirina (p. ej., ALA o PBG) o una porfirina en el sujeto,
(iv) puede reducir la frecuencia de ataques agudos de síntomas asociados con una porfiria en el sujeto, o (v) puede reducir la incidencia de ataques agudos de síntomas asociados con una porfiria en el sujeto cuando el sujeto se expone a un factor precipitante (p. ej., la fase premenstrual o la fase luteínica).
El método puede mejorar el dolor y/o una neuropatía progresiva.
El ARNi (p. ej., ARNbc) o la composición que comprende el ARNi se puede administrar de acuerdo con una pauta posológica.
El ARNi (p. ej., ARNbc) o la composición que comprende el ARNi se puede administrar antes de un ataque agudo de porfiria o después de este. El ARNi se puede administrar antes de un ataque agudo de porfiria.
El ARNi (p. ej., ARNbc) o la composición que comprende el ARNi se puede administrar durante un pródromo. El pródromo se puede caracterizar por dolor abdominal, náusea, síntomas psicológicos (p. ej., ansiedad), inquietud y/o insomnio.
El ARNi (p. ej., ARNbc) o la composición que comprende el ARNi se puede administrar durante una fase particular del ciclo menstrual, p. ej., durante la fase luteínica.
El método puede mejorar o prevenir ataques cíclicos de porfiria, p. ej., reduciendo la intensidad, duración o frecuencia de los ataques. Los ataques cíclicos se asocian con un factor precipitante. El factor precipitante es el ciclo menstrual, p. ej., una fase particular del ciclo menstrual, p. ej., la fase luteínica.
El sujeto puede presentar un nivel elevado de ALA y/o PBG. El nivel de ALA y/o PBG puede estar elevado en el plasma u orina del sujeto. El sujeto puede padecer o correr el riesgo de desarrollar una porfiria, p. ej., una porfiria hepática. El sujeto puede no presentar síntomas. El sujeto puede ser portador de una alteración genética (p. ej., una mutación génica) asociada con una porfiria, según se describe en la presente.
El sujeto puede padecer o correr el riesgo de desarrollar una porfiria y sufre dolor (p. ej., dolor crónico, p. ej., dolor neuropático crónico) y/o una neuropatía (p. ej., una neuropatía progresiva). El sujeto puede no padecer ataques agudos pero puede sufrir dolor (p. ej., dolor crónico, p. ej., dolor neuropático crónico) y/o una neuropatía (p. ej., una neuropatía progresiva). El dolor puede ser dolor abdominal.
El sujeto (a) puede presentar un nivel elevado de ALA y/o PBG y (b) puede sufrir dolor (p. ej., dolor crónico, p. ej., dolor neuropático crónico) y/o una neuropatía (p. ej., una neuropatía progresiva). El dolor puede ser dolor abdominal. El sujeto puede presentar un nivel en plasma y/o un nivel en orina de ALA y/o PBG que es elevado. El nivel elevado de ALA y/o PBG puede venir acompañado de otros síntomas, p. ej., dolor (p. ej., dolor crónico, p. ej., dolor neuropático crónico) y/o una neuropatía (p. ej., una neuropatía progresiva). El dolor puede ser dolor abdominal. El sujeto puede no presentar síntomas. El sujeto puede tener una mutación genética asociada con una porfiria, p. ej., una mutación según se describe en la presente.
El sujeto puede presentar un nivel (p. ej., un nivel en plasma o un nivel en orina) de un precursor de porfirina, p. ej., ALA y/o PBG, que es elevado, p. ej., el nivel es superior a un valor de referencia, o superior o igual a un valor de referencia. El nivel puede ser superior al valor de referencia. El valor de referencia se puede encontrar dos desviaciones estándar por encima del nivel medio de una muestra de individuos sanos. El valor de referencia puede ser un límite de referencia superior.
El sujeto puede presentar un nivel en plasma y/o un nivel en orina de ALA y/o PBG superior a, o superior o igual a, 2 veces, 3 veces, 4 veces o 5 veces el valor del límite de referencia superior. La expresión "límite de referencia superior", tal como se utiliza en la presente, se refiere a un nivel que representa el límite superior del intervalo de confianza del 95% para una muestra de referencia, p. ej., una muestra de individuos sanos o normales (p. ej., de origen natural), p. ej., individuos que no son portadores de ninguna mutación genética asociada con una porfiria y/o individuos que no padecen ninguna porfiria. El sujeto puede presentar un nivel en orina de ALA y/o PBG superior a 2-4 veces el valor del límite de referencia superior. El sujeto puede presentar un nivel en orina de ALA y/o PBG superior a 4 veces el valor del límite de referencia superior.
El valor de referencia para PBG en plasma puede ser de 0.12 pmol/L. El sujeto puede ser un ser humano y presenta un nivel de PBG en plasma superior a, o superior o igual a, 0.12 pmol/L, 0.24 pmol/L, 0.36 pmol/L, 0.48 pmol/L o 0.60 pmol/L. El sujeto puede ser un ser humano y puede presentar un nivel de PBG en plasma superior a, o superior o igual a, 0.48 pmol/L.
El valor de referencia para PBG en orina puede ser de 1.2 mmol/mol de creatinina. El sujeto puede ser un ser humano y presenta un nivel de PBG en orina superior a, o superior o igual a, 1.2 mmol/mol de creatinina, 2.4 mmol/mol de creatinina, 3.6 mmol/mol de creatinina, 4.8 mmol/mol de creatinina o 6.0 mmol/mol de creatinina. El sujeto puede ser un ser humano y puede presentar un nivel de PBG en orina superior a, o superior o igual a, 4.8 mmol/mol de creatinina.
El valor de referencia para ALA en plasma puede ser de 0.12 pmol/L. El sujeto puede ser un ser humano y puede presentar un nivel de ALA en plasma superior a, o superior o igual a, 0.12 pmol/L, 0.24 pmol/L, 0.36 pmol/L, 0.48 pmol/L o 0.60 pmol/L. El sujeto puede ser un ser humano y puede presentar un nivel de ALA en plasma superior a, o superior o igual a, 0.48 pmol/L.
El valor de referencia para ALA en orina puede ser de 3.1 mmol/mol de creatinina. El sujeto puede ser un ser humano y puede presentar un nivel de ALA en orina superior a, o superior o igual a, 3.1 mmol/mol de creatinina, 6.2 mmol/mol de creatinina, 9.3 mmol/mol de creatinina, 12.4 mmol/mol de creatinina o 15.5 mmol/mol de creatinina. El método puede reducir uno o más signos o síntomas de porfiria. El método puede reducir un nivel elevado de ALA y/o PBG. El método puede reducir el dolor (p. ej., dolor crónico, p. ej., dolor neuropático crónico) y/o una neuropatía (p. ej., una neuropatía progresiva). El dolor puede ser dolor abdominal. El dolor puede ser dolor neuropático (p. ej., dolor asociado con la neuropatía progresiva de porfirias agudas). La reducción del dolor puede incluir, p. ej., la prevención del dolor, el retraso del inicio del dolor, la reducción de la frecuencia del dolor y/o la reducción de la intensidad del dolor. La reducción del dolor se puede evaluar en base al uso del sujeto de medicamentos contra el dolor.
El método puede mejorar o prevenir ataques agudos de porfiria, p. ej., reduciendo la intensidad, duración o frecuencia de los ataques.
El método puede reducir o prevenir lesiones nerviosas.
El método puede prevenir el deterioro (p. ej., previene el desarrollo de anomalías) de medidas clínicas o puede provocar una mejora de estas, p. ej., medidas clínicas de la función muscular y/o nerviosa, p. ej., EMG y/o velocidades de conducción nerviosa.
El método puede reducir el hemo utilizado por el sujeto.
El método puede reducir los medicamentos para el dolor utilizados por el sujeto.
El método puede reducir la hospitalización.
El método puede ser eficaz para reducir el nivel de ALA y/o PBG (p. ej., el nivel en plasma u orina de ALA y/o PBG). El método puede ser eficaz para producir una reducción predeterminada del nivel elevado de ALA y/o PBG.
La reducción predeterminada puede ser una reducción hasta un valor que es inferior o igual a un valor de referencia. El valor de referencia puede ser un límite de referencia superior. El valor de referencia puede ser el valor que corresponde a dos desviaciones estándar por encima del nivel medio de una muestra de referencia.
El método puede ser eficaz para reducir el nivel de ALA y/o PBG en un sujeto hasta un nivel que está por debajo de dos veces el límite de referencia superior. El método puede ser eficaz para reducir el nivel de ALA hasta por debajo de dos veces el límite de referencia superior. El método puede ser eficaz para reducir el nivel de PBG hasta por debajo de dos veces el límite de referencia superior.
El ARNi (p. ej., ARNbc) o la composición que comprende el ARNi se puede administrar como una dosis única o una dosis múltiple, p. ej., de acuerdo con una pauta posológica.
El ARNi (p. ej., ARNbc) o la composición que comprende el ARNi se puede administrar profilácticamente a un sujeto que corre el riesgo de desarrollar una porfiria. El ARNi (p. ej., ARNbc) o la composición que comprende el ARNi se administra profilácticamente empezando en la pubertad. El sujeto puede ser portador de una mutación genética asociada con una porfiria y/o presenta un nivel elevado de ALA y/o PBG (p. ej., un nivel elevado en plasma u orina de ALA y/o PBG). La mutación puede hacer que el individuo sea susceptible de padecer un ataque agudo (p. ej., tras la exposición a un factor precipitante, p. ej., un fármaco, el seguimiento de un régimen u otro factor precipitante, p. ej., un factor precipitante como los que se describen en la presente). La mutación se puede asociar con unos niveles elevados de una porfirina o un precursor de porfirina (p. ej., ALA y/o PBG). La mutación se asocia con dolor crónico (p. ej., dolor neuropático crónico) y/o una neuropatía (p. ej., una neuropatía progresiva).
La mutación es una mutación en el gen ALAS1. La mutación puede ser una mutación en el promotor del gen ALAS1 0 en regiones en dirección 5' o dirección 3' del gen ALAS1. La mutación puede ser una mutación en factores de transcripción u otros genes que interaccionan con ALAS1. La mutación puede ser una mutación en un gen que codifica una enzima en la vía biosintética del grupo hemo.
El ARNi (p. ej., ARNbc o un conjugado de este) o la composición que comprende el ARNi se puede administrar por vía subcutánea. El ARNi se puede encontrar en forma de un conjugado de tipo GalNAc. El ARNi (p. ej., el ARNbc) se puede administrar con una dosis de 0.5-50 mg/kg. El ARNi se puede administrar en una dosis inferior a 10 mg/kg (p. ej., 5 mg/kg o inferior) una vez a la semana; p. ej., una dosis de 1 mg/kg o inferior, 2.5 mg/kg o inferior o 5 mg/kg o inferior, p. ej., una vez a la semana. El ARNi se puede administrar en una dosis de aproximadamente 2.5 mg/kg o inferior, p. ej., una vez a la semana.
El sujeto que se va a tratar puede estar asintomático y puede tener un nivel elevado de ALA y/o PBG. El sujeto puede tener una porfiria, p. ej., AIP. El paciente puede padecer ataques porfíricos recurrentes.
El ARNi (p. ej., AD-60519) se puede administrar en una dosis inferior a 5 mg/kg, p. ej., de 0.1, 0.35, 1.0 o 2.5 mg/kg. El ARNi (p. ej., AD-60519) se puede administrar en dosis repetidas, p. ej., dosis semanales.
El sujeto puede estar asintomático y tiene un nivel elevado de ALA y/o PBG, y el ARNi (p. ej., AD-60519) se puede administrar en dosis únicas, p. ej., de 0.1,0.35, 1.0 o 2.5 mg/kg; o en dosificaciones semanales repetidas, p. ej., de 1 y 2.5 mg/kg durante varias semanas (p. ej., durante 4 semanas).
El sujeto puede tener AIP, p. ej., es un paciente con AIP, el ARNi (p. ej., AD-60519) se administra con una dosis de 1 -2.5 mg/kg a la semana.
Se puede emplear una pauta de tratamiento en la que el ARNi se administra inicialmente con más frecuencia, seguido por una administración menos frecuente. El ARNi se puede administrar inicialmente una vez al día durante múltiples días (p. ej., durante 2-14 días, p. ej., durante 2, 3, 4, 5, 6 o 7 días). El ARNi se puede administrar posteriormente una vez a la semana. El ARNi se puede administrar posteriormente una vez cada dos semanas. El ARNi se puede administrar posteriormente con una frecuencia que puede ser eficaz para reducir uno o más signos o síntomas de una porfiria.
En un aspecto de la presente se proporciona ademásun método para tratar a un sujeto con un nivel elevado de ALA y/o PBG, comprendiendo el método administrar al sujeto un ácido ribonucleico bicatenario (ARNbc), donde dicho
ARNbc comprende una hebra sentido y una hebra antisentido con una longitud de 15-30 pares de bases y la hebra antisentido es complementaria respecto a al menos 15 nucleótidos contiguos de la SEQ ID NO:1 o la SEQ ID NO:382.
En un aspecto de la presente se proporciona además un método para tratar a un sujeto con un nivel elevado de ALA y/o PBG, comprendiendo el método administrar al sujeto una cantidad terapéuticamente eficaz de un ARNbc o una composición que comprenda un ARNbc, según se describe la presente.
Los métodos descritos en la presente pueden ser eficaces para reducir el nivel de ALA y/o PBG. El nivel de ALA y/o PBG se puede reducir de modo tal que sea inferior a, o inferior o igual a, un valor de referencia, p. ej., un límite de referencia superior.
El sujeto que se va a tratar puede estar asintomático y tener un nivel elevado de ALA y/o PBG. El sujeto puede tener una porfiria, p. ej., AIP.
El ARNi se puede administrar en una dosis inferior a 5 mg/kg, p. ej., con 0.1, 0.35, 1.0 o 2.5 mg/kg. El ARNi se puede administrar en dosis repetidas, p. ej., dosis semanales.
En otro aspecto, en la presente se proporcionan además métodos para reducir el nivel de una porfirina o un precursor de porfirina en una célula (p. ej., una célula eritroide o una célula hepática tal como, p. ej., un hepatocito). La célula se puede tratar ex vivo, in vitro o in vivo (p. ej., la célula está presente en un sujeto (p. ej., un paciente que necesita tratar, prevenir y/o gestionar un trastorno relacionado con la expresión de ALAS1)). El método incluye poner en contacto la célula con una cantidad eficaz de uno o más de los ARNi que tienen ALAS1 como diana, p. ej., uno o más de los ARNi descritos en la presente, de este modo se reduce el nivel de una porfirina o un precursor de porfirina en la célula; o se reduce el nivel de una porfirina o un precursor de porfirina en otras células, tejidos o fluidos del sujeto en el cual se encuentra localizada la célula; con relación al nivel antes de la puesta en contacto. Tales métodos se pueden utilizar para tratar (p. ej., mejorar la gravedad) de trastornos relacionados con la expresión de ALAS1, tales como las porfirias, p. ej., AIP o una porfiria debida a la deficiencia de ALA-deshidratasa.
El paso de puesta en contacto se puede realizar ex vivo, in vitro o in vivo. Por ejemplo, la célula puede estar presente en un sujeto, p. ej., un mamífero (p. ej., un ser humano) que corre el riesgo de padecer o al cual se le ha diagnosticado una porfiria. La porfiria puede ser una porfiria hepática aguda. La porfiria puede ser una porfiria hepática, p. ej., una porfiria seleccionada entre porfiria intermitente aguda (AIP), coproporfiria hereditaria (HCP), porfiria variegata (VP), porfiria debida a la deficiencia de ALA-deshidratasa (ADP) y porfiria hepatoeritropoyética. La porfiria puede ser una porfiria hepática dominante homozigótica (p. ej., AIP, HCP o VP dominante homozigótica) o una porfiria hepatoeritropoyética. La porfiria puede ser una porfiria dual.
En un aspecto de la presente se proporciona un método para reducir el nivel de una porfirina o un precursor de porfirina (p. ej., ALA o PBG) en una célula, que comprende poner en contacto la célula con un ARNi (p. ej., un ARNbc), según se describe en la presente, en una cantidad eficaz para reducir el nivel de la porfirina o el precursor de porfirina en la célula. La célula es un hepatocito. La porfirina o el precursor de porfirina es ácido 5-aminolevulílico (ALA), porfopilinógeno (PBG), hidroximetilbilano (HMB), uroporfirinógeno I o III, coproporfirinógeno I o III, protoporfrinógeno IX o protoporfirina IX. El precursor de porfirina es ALA o PBG.
La célula puede ser una célula eritroide. La célula puede ser una célula hepática (p. ej., un hepatocito).
En un aspecto de la presente se proporciona un vector que codifica al menos una hebra de un ARNi (p. ej., un ARNbc) según se describe en la presente.
En un aspecto de la presente se proporciona un vector que codifica al menos una hebra de un ARNbc, donde dicho ARNbc comprende una región de complementariedad respecto a al menos una parte de un ARNm que codifica ALAS1, donde dicho ARNbc tiene una longitud de 30 pares de bases o inferior y donde dicho ARNbc tiene como diana la escisión de dicho ARNm.
La región de complementariedad puede tener una longitud de al menos 15 nucleótidos.
La región de complementariedad puede tener una longitud de 19-21 nucleótidos. En un aspecto, en la presente se proporciona además un vector para inhibir la expresión de un gen ALAS1 en una célula. El vector puede comprender un ARNi según se describe en la presente. El vector puede incluir al menos una secuencia reguladora unida operablemente a una secuencia de nucleótidos que codifica al menos una hebra de un ARNi según se describe en la presente. El vector puede comprender al menos una hebra de un ARNi de ALAS1.
En un aspecto de la presente se proporciona una célula que comprende un vector según se describe en la presente. En un aspecto de la presente se proporciona una célula que contiene un vector para inhibir la expresión de un gen ALAS1 en una célula. El vector incluye una secuencia reguladora unida operablemente a una secuencia de nucleótidos que codifica al menos una hebra de uno de los ARNi que se describen en la presente. La célula puede ser una célula hepática (p. ej., un hepatocito). La célula puede ser una célula eritroide.
En otro aspecto, se proporciona un método para someter a ensayo el nivel de ARNm de ALAS1 extracelular circulante en un sujeto, comprendiendo dicho método detectar (p. ej., medir) el nivel de ARNm de ALAS1 en una muestra de fluido biológico (p. ej., una muestra de sangre (p. ej., una muestra de suero o plasma), una muestra de líquido cefalorraquídeo, o una orina del sujeto, comprendiendo dicha muestra de fluido biológico el ARNm de ALAS1, para someter a ensayo de esta manera el nivel de ARNm de ALAS1 extracelular circulante en el sujeto.
En otro aspecto, se proporciona un método para someter a ensayo el nivel de ARNm de ALAS1 extracelular circulante en un sujeto, comprendiendo dicho método (i) proporcionar ARN (p. ej., ARN extracelular) de una muestra de fluido biológico (p. ej., muestra de sangre o plasma) del sujeto, comprendiendo dicha muestra de fluido biológico el ARNm de ALAS1; (ii) obtener un ADNc de a La S1 del ARNm de ALAS1; (iii) poner el contacto el ADNc de ALAS1 con un ácido nucleico complementario (p. ej., sonda y/o cebador) al ADNc de ALAS1 o una porción de este, para producir de esta manera una mezcla de reacción; y (iv) detectar (p. ej., medir) el nivel de ADNc de ALAS1 en la mezcla de reacción, donde el nivel de ADNc de ALAS1 es indicativo del nivel de ARN de ALAS1, para someter a ensayo de esta manera el nivel de ARNm de ALAS1 extracelular circulante en el sujeto.
Dicha muestra de fluido biológico puede ser una muestra de sangre. Dicha muestra de fluido biológico puede ser una muestra de suero. Dicha muestra de fluido biológico puede ser una muestra de orina.
El método puede comprender PCR, qPCR o 5’-RACE.
Dicho ácido nucleico puede ser una sonda o un cebador.
Dicho ácido nucleico puede comprender un resto detectable y el nivel de ARNm de ALAS1 se determina mediante la detección de la cantidad del resto detectable.
Dicho método puede comprender además obtener la muestra de fluido biológico del sujeto. La muestra de fluido biológico puede estar separada del tejido y contiene exosomas. En aspectos de estos métodos, la eficacia de un tratamiento de porfiria se puede someter a ensayo en función de una comparación con el nivel de ARNm de ALAS1 extracelular circulante en el sujeto respecto al valor de referencia.
Un descenso en el nivel de ARNm de ALAS1 extracelular circulante en el sujeto en respuesta al tratamiento de porfiria, respecto al valor de referencia, puede indicar que el tratamiento de la porfiria es eficaz. El valor de referencia puede ser el nivel de ARNm de ALAS1 extracelular circulante en el sujeto antes del tratamiento de la porfiria.
Descripción de los dibujos
La FIG. 1 representa la vía biosintética del grupo hemo.
Las FIG. 2A y FIG. 2B muestran una tabla que resume ciertas porfirias asociadas con errores genéticos en el metabolismo del grupo hemo.
Las FIG. 3A y FIG. 3B representan la variante 1 de un transcrito de la secuencia de ARNm de ALAS1 humano (Ref. Sec. n M_000688.4 (GI:40316942, registro con fecha de 19 de noviembre de 2011), SEQ ID NO: 1).
Las FIG. 4A y 4B representan el transcrito de la secuencia de ARNm de ALAS1 humano (Ref. Sec. NM_000688.5 (GI: 362999011, registro con fecha de 1 de abril de 2012), SEQ ID NO: 382).
La FIG. 5 muestra la dosis-respuesta del ARNip AD-53558 para la supresión del ARNm de ALAS1 de ratón (ALAS1r) con relación a un control de PBS. También se muestran los resultados para un control de luciferasa (LUC) AD-1955.
La FIG. 6 muestra la dosis-respuesta del ARNip AD-53558 para la supresión del ARNm de ALAS1 en ratas con relación a un control de PBS. También se muestran los resultados para un control de luciferasa (LUC) AD-1955. La FIG. 7 muestra la durabilidad de la supresión del ARNm de ALAS1 de ratón (ALAS1r) por parte del ARNip AD-53558 con relación a un control de PBS.
La FIG. 8 muestra las medias ± desviaciones estándar de los niveles de ALA en plasma (en pM) en el punto de referencia y después de un tratamiento con fenobarbital en los grupos experimentales (ARNip de ALAS1) y de control (ARNip de LUC).
La FIG. 9 muestra los niveles de ALA en plasma (en pM) de animales individuales en el punto de referencia y después del tratamiento con fenobarbital en animales que recibieron tratamiento con ARNip de ALAS1 y control (ARNip de LUC).
La FIG. 10 muestra las medias ± desviaciones estándar de los niveles de PBG en plasma (en pM) en el punto de referencia y después de un tratamiento con fenobarbital en animales que recibieron tratamiento con ARNip de ALAS1 y control (ARNip de LUC).
La FIG. 11 muestra los niveles de PBG en plasma (en pM) de animales individuales en el punto de referencia y después del tratamiento con fenobarbital en animales que recibieron tratamiento con ARNip de ALAS1 y control (ARNip de LUC).
La FIG. 12 muestra el nivel de ARNm de ALAS1r relativo en el hígado en el punto de referencia y después de un tratamiento con fenobarbial en animales experimentales representativos seleccionados (ARNip de ALAS1) y de control (PBS).
La FIG. 13 muestra los efectos de tres ARNip de ALAS1 r conjugados con GalNAc sobre la expresión de ALAS1 r (en comparación con un control de PBS) en tejido hepático de ratón.
La FIG. 14 muestra los niveles de ALA y PBG en plasma en función del tiempo después de la administración de fenobarbital y el tratamiento con ARNip de ALAS1 o ARNip de LUC como control.
La FIG. 15 muestra los efectos de un ARNip de ALAS1 conjugado con GalNAc sobre los niveles de ALA en plasma y PBG en plasma en el modelo de inducción con fenobarbital de AIP en ratón.
La FIG. 16 muestra los efectos dependientes de la dosis de un ARNip de ALAS1 en los niveles en plasma de ALA y PGB en el modelo de inducción con fenobarbital de AIP en ratón. Para los animales que recibieron ARNip de ALAS1, la dosis de ARNip administrada (0.05 mg/kg, 0.1 mg/kg, 0.5 mg/kg o 1.0 mg/kg) se muestra en el eje horizontal.
La FIG. 17, panel superior, muestra el diseño experimental utilizado para estudiar la supresión de ALA y PGB con ARNip de ALAS1. El panel inferior muestra los niveles en plasma de ALA y PBG en el punto de referencia, en la condición de control (Luc) y después del tratamiento con ARNip de ALAS1 en la semana 0, semana 2 y semana 4.
La FIG. 18 muestra el diseño experimental utilizado para comparar los efectos del tratamiento con ARNip de ALAS1 o hemina en un modelo en animales de AIP (parte superior) y los resultados para los niveles en plasma de ALA (pmol/L) (parte central) y niveles en plasma de PBG (pmol/L) (parte inferior).
La FIG. 19 muestra los niveles de ARNm relativos (ALAS1/GAPDH) en animales tratados con 30 mg/kg, 10 mg/kg o 3 mg/kg de AD-58632 en comparación con los animales tratados con control de PBS.
La FIG. 20 muestra el diseño experimental utilizado para investigar el efecto de respuesta a la dosis del conjugado AD-58632-ALAS1-GalNAc en un modelo de AIP en rata.
La FIG. 21 muestra los niveles relativos de ARNm de PBGD en el hígado (gráfico superior) y los niveles relativos de ARNm de ALAS1 en el hígado (gráfico inferior) en un modelo en rata de AIP. Los grupos de animales se sometieron a uno de cuatro tratamientos: (1) tratamiento con fenobarbital (PB) solo, (2) tratamiento con fenobarbital y ARNip de porfobilinógeno-desaminasa (PBGD), (3) fenobarbital, ARNip de PBGD y 30 mg/kg de ARNip de AlAs 1, (4 ) fenobarbital, ARNip de PBGD y 10 mg/kg de ARNip de ALAS1.
La FIG. 22 muestra los niveles en orina de PBG (panel superior) y ALA (panel inferior) respecto a los niveles de creatinina en un modelo en rata de AIP. Los grupos de animales se sometieron a uno de cuatro tratamientos: (1) tratamiento con fenobarbital (PB) solo, (2) tratamiento con fenobarbital y ARNip de porfobilinógeno-desaminasa (PBGD), (3) fenobarbital, ARNip de PbGD y 30 mg/kg de ARNip de ALAS1, (4) fenobarbital, ARNip de PBGD y 10 mg/kg de ARNip de ALAS1.
La FIG. 23 muestra la supresión de ARNm de ALAS-1 por AD-58632, en comparación con el control de PBS, en grupos de ratas que recibieron cinco dosis diarias de ARNip de 6 mg/kg, 2 mg/kg o 1 mg/kg frente a dosis en bolo único de ARNip de 30 mg/kg, 10 mg/kg o 5 mg/kg.
La FIG. 24 muestra la supresión de ARNm de ALAS-1 por AD-58632, en comparación con control de PBS, en grupos de ratas que recibieron cuatro dosis semanales de ARNip de 10 mg/kg, 5 mg/kg o 2.5 mg/kg.
La FIG. 25 muestra la supresión de ARNm de ALAS-1 por AD-58632 y por cinco dúplex de nonadecámero/nonadecámero.
La FIG. 26 muestra los resultados de una evaluación del efecto de la longitud de la hebra y en protuberancias en el mejor de los dos nonadecámeros.
La FIG. 27 es un gráfico que muestra los niveles de ARNm de ALAS1 en el hígado (barras de la izquierda) y en suero (barras de la derecha) para cada grupo de tratamiento en el estudio NHP descrito en el Ejemplo 34. La FIG. 28 muestra la supresión de ARNm de ALAS-1, en comparación con el control de PBS, en grupos de ratas que recibieron 3 mg/kg o 10 mg/kg de AD-58632 o AD-60489.
La FIG: 29 muestra el diseño experimental utilizado para investigar la eficacia de los ARNip de ALAS1 AD-58632 y AD-60489 en la supresión de ARNm hepático en primates no humanos.
La FIG. 30 muestra la supresión dependiente de la dosis de ARNm hepático en primates no humanos tras el tratamiento con 1.25 mg/kg, 2.5 mg/kg o 5 mg/kg de AD-58632 o AD-60489.
La FIG. 31 muestra una comparación de los resultados de supresión de ARNm obtenidos de biopsias hepáticas y del ensayo cERD en un estudio con primates no humanos.
La FIG. 32 muestra el transcurso temporal de la supresión de ARNm según se evalúa utilizando el ensayo cERD en un estudio con primates no humanos. El eje horizontal muestra el tiempo de acuerdo con el día de estudio. La FIG. 33 muestra la supresión de ARNm de ALAS1 en ratas que recibieron PBS o una única dosis de 5 mg/kg de uno de los dúplex de ARNip indicados.
La FIG. 34 muestra las concentraciones hepáticas del ARNip en ratas que recibieron una única dosis de 5 mg/kg del ARNip indicado.
La FIG. 35 (parte superior) muestra el diseño experimental utilizado para investigar la eficacia terapéutica de AD-60925 y AD-60926. La FIG. 35 (parte inferior) muestra los niveles relativos de ARNm de ALAS1/GAPDH de rata en ratas tratadas con (1) AF11 -PbGD, (2) a F11 -PBGD y PB, (3) AF-11PBGD, PB, y 3 mg/kg de AD-60925, o (4) AF11 -PBGD, PB, y AD-60926.
La FIG. 36 muestra los niveles relativos de PBG en orina (parte superior) y ALA en orina (parte inferior) en ratas tratadas con (1) AF11 -PBGD, (2) AF11 -PBGD y PB, (3) AF-11 PBGD, PB, y 3 mg/kg de AD-60925, o (4) AF11 -PBGD, PB, y AD-60926.
La FIG. 37 muestra los niveles relativos de PBG en orina (parte superior) y ALA en orina (parte inferior) en el tiempo en ratas tratadas con (1) AF11-PBGD, (2) AF11-PBGD y PB, (3) AF-11PBGD, PB, y 3 mg/kg de AD-60925, o (4) AF11-PBGD, PB, y AD-60926. Las flechas indican los puntos temporales en los que se administró PB.
La FIG. 38 muestra los niveles relativos de ARNm de ALAS1 de rata (rALAS1) en ratas que recibieron 4 dosis de PBS o 2.5 mg/kg de uno de los ARNip indicados.
La FIG. 39 muestra los niveles relativos de ARNm de ALAS1 de rata (rALAS1) en ratas que recibieron una única dosis de PBS o 2.5 mg/kg de uno de los ARNip indicados.
La FIG. 40 (parte superior) muestra los niveles relativos de ARNm de ALAS1 de rata (rALAS1) en ratas que recibieron una única dosis de PBS o 3 mg/kg de uno de los ARNip indicados. La FIG. 40 (parte inferior) muestra la concentración de ARNip en el hígado.
La FIG. 41 (parte superior) muestra la supresión de ARNm de ALAS1 de rata (rALAS1) por AD-60489, AD-60519 y AD-60901. La FIG. 41 (parte inferior) muestra la concentración de ARNip en el hígado.
La FIG. 42 muestra los niveles relativos de ARNm de ALAS1 de rata (rALAS1) en ratas que fueron tratadas con una única dosis de PBS o 2.5 mg/kg de uno de los ARNip indicados.
La FIG. 43 muestra los niveles relativos de ARNm de ALAS1 de rata (rALAS1) en ratas que fueron tratadas con PBS o uno de los ARNip indicados con una dosis de 2.5 mg/kg dos veces a la semana durante 2 semanas. La FIG. 44 (parte superior) muestra una representación esquemática del diseño experimental utilizado para investigar la eficacia terapéutica de múltiples dosis bisemanales de AD-60519. La FIG. 44 (parte inferior) muestra gráficos que presentan la supresión de PBG en orina y ALA en orina en ratas que fueron tratadas con (i) ARNip de PBGD y seis dosis de PBS, (ii) ARNip de PBGD, PB y seis dosis de PBS, (iii) ARNip de PBGD, PB y seis dosis de 2.5 mg/kg de AD-60519 o (iv) ARNip de PBGD, PB y seis dosis de 5 mg/kg de AD-60519.
La FIG. 45 muestra gráficos que presentan la supresión de PBG en suero (gráfico superior) y ALA en suero (gráfico inferior) en un modelo de AIP en ratones que fueron tratados con (i) ARNip de PBGD y seis dosis de PBS (punto de referencia), (ii) ARNip de PBGD, PB y seis dosis de PBS (solución salina), (iii) ARNip de PBGD, PB y seis dosis de 2.5 mg/kg de AD-60519 o (iv) ARNip de PBGD, PB y seis dosis de 5 mg/kg de AD-60519. La FIG. 46 (parte superior) muestra una representación esquemática del diseño experimental utilizado para investigar la eficacia terapéutica de múltiples dosis semanales de AD-60519. La FIG. 46 (parte inferior) muestra un gráfico que presenta los niveles relativos de ARNm de ALAS1 de rata (rALAS1/GAPDH) en ratas que fueron tratadas con (i) ARNip de PBGD y cuatro dosis de PBS, (ii) ARNip de PBGD, PB y cuatro dosis de PBS, (iii) ARNip de PBGD, PB y cuatro dosis de 3 mg/kg de AD-60519, (iv) ARNip de PBGD, PB y cuatro dosis de 1 mg/kg de AD-60519 o (v) ARNip de PBGD, PB y cuatro dosis de 0.3 mg/kg de AD-60519.
La FIG. 47 muestra gráficos que presentan los niveles de PBG en orina (gráfico superior) y ALA en orina (gráfico inferior) en ratas que fueron tratadas con (i) ARNip de PBGD y cuatro dosis de pBs , (ii) ARNip de PBGD, PB y cuatro dosis de pBs , (iii) ARNip de PBGD, PB y cuatro dosis de 3 mg/kg de AD-60519, (iv) ARNip de PBGD, pB y cuatro dosis de 1 mg/kg de AD-60519 o (v) ARNip de PBGD, PB y cuatro dosis de 0.3 mg/kg de AD-60519. La FIG. 48 es una representación esquemática que muestra el diseño de un estudio en primates no humanos en el que se investigaron los efectos de los conjugados de ARN ip de ALAS1-GalNAc en la supresión del ARNm de ALAS1 y ARNm de ALAS1 circulante.
La FIG. 49 es un gráfico que muestra la supresión de ARNm hepático en primates no humanos (NHP, por sus siglas en inglés) tras el tratamiento con conjugados de ARNip de ALAS1-GalNAc.
La FIG. 50 es un gráfico que muestra los niveles en suero normalizados de ARNm de ALAS1 en primates no humanos (NHP) en diversos momentos durante el transcurso de un estudio en el que se investigaron los efectos del tratamiento con conjugados de ARNip de ALAS1-GalNAc. Los días en el eje horizontal corresponden a los días en la representación esquemática en la FIG. 48.
La FIG. 51 muestra los niveles de ARNm de ALAS1 normalizados (mostrados como una fracción del nivel predosis) según se evalúa en un estudio de única dosis en ratas que utilizó cERD en orina para monitorizar la supresión de ALAS1.
La FIG. 52 es una representación esquemática que muestra el diseño de un estudio en primates no humanos en el que se investigaron los efectos de múltiples dosis y de una única dosis de AD-60519 en la supresión de ARNm de ALAS1 en hígado y ARNm de ALAS1 circulante.
La FIG. 53 es un gráfico de barras que muestra los niveles de ARNm de ALAS1 en hígado relativos promedio (% de control de PBS) en el día de estudio 24 (grupos de múltiples dosis) o en el día de estudio 4 (grupos de única dosis).
La FIG. 54 es un gráfico que muestra los niveles de ARNm de ALAS1 en suero normalizados (mostrados como una fracción del nivel predosis) según se evalúa utilizando cERD para los grupos de múltiples dosis (gráfico superior, que muestra los resultados hasta el día 24) y grupos de una única dosis (gráfico inferior, que muestra los resultados hasta el día 22).
La FIG. 55 es un gráfico que muestra los niveles de ARNm en hígado, ARNm en suero y ARNm en orina en el día de estudio 4 (en los grupos de una única dosis) o en el día de estudio 24 (en los grupos de múltiples dosis). Se muestran los datos para los animales individuales y los promedios para cada grupo.
La FIG. 56 es un gráfico que muestra los niveles normalizados de ARNm de ALAS1 en suero (mostrados como una fracción del nivel predosis) después de 8 semanas según se evaluó utilizando cERD para los grupos de múltiples dosis. Cada punto de datos del gráfico representa el ARNm de ALAS1 restante para el promedio del grupo de muestras de 3 animales ± la desviación estándar del grupo.
La FIG. 57 es una representación esquemática de la estructura de ALN-60519 (también denominada en la presente AD-60519). La FIG. 57 describe las SEQ ID Nos 5238-5239, respectivamente, en orden de aparición. La FIG. 58 muestra los niveles de ARNm de ALAS1 según se evalúan en muestras de suero o de orina coincidentes obtenidas de pacientes con AIP o voluntarios sanos normales (NHV, por sus siglas en inglés). Los niveles de ARNm de ALAS1 en suero u orina se midieron utilizando el método cERD. En los pacientes con AIP A y B, se recogió un segundo conjunto de muestras de suero y orina para evaluar la variabilidad de ARNm de ALAS1 en el tiempo.
Descripción detallada
El ARNi dirige la degradación de ARNm específica según la secuencia mediante un proceso conocido como interferencia por ARN (iARN). En la presente se describen ARNi y métodos para utilizarlos con el fin de inhibir la expresión de un gen ALAS1 en una célula o un mamífero, donde el ARNi tiene como diana un gen ALAS1. También se proporcionan composiciones y métodos para trastornos relacionados con la expresión de ALAS1 tales como las porfirias (p. ej., porfiria debida a la deficiencia de ALA-deshidratasa (porfiria de Doss o ADP), porfiria intermitente aguda, porfiria eritropoyética congénita, porfiria cutánea tardía, coproporfiria hereditaria (coproporfiria), porfiria variegata, protoporfiria eritropoyética (EPP), anemia sideroblástica relacionada con X (XLSA) y eritroporfiria transitoria de la infancia).
Las porfirias son trastornos heredados o adquiridos que pueden ser provocados por una actividad reducida o potenciada de enzimas específicas de la vía biosintética del grupo hemo, que también se denomina en la presente vía de las porfirinas (remítase a la FIG. 1). Las porfirinas son los precursores principales del grupo hemo. Las porfirinas y los precursores de porfirinas incluyen ácido 5-aminolevulínico (ALA), porfopilinógeno (PBG), hidroximetilbilano (HMB), uroporfirinógeno I o III, coproporfirinógeno I o III, protoporfrinógeno IX y protoporfirina IX. El
grupo hemo es una parte esencial de la hemoglobina, mioglobina, catalasas, peroxidasas y citocromos, incluyendo estos últimos los citocromos hepáticos P450 y respiratorios. El grupo hemo se sintetiza en la mayoría de las células humanas o en todas ellas. Aproximadamente un 85% del grupo hemo se produce en células eritroides, principalmente para la hemoglobina. La mayor parte del grupo hemo remanente se produce en el hígado, un 80% del cual se utiliza para la síntesis de los citocromos. La deficiencia de enzimas específicas de la vía de las porfirinas conlleva una producción insuficiente del grupo hemo y también una acumulación de porfirinas y/o precursores de porfirinas, que son tóxicos para la función de células u órganos en concentraciones elevadas.
Las porfirias se pueden manifestar con complicaciones neurológicas ("agudas"), problemas de la piel ("cutáneos") o ambos. Las porfirias se pueden clasificar en función del sitio principal de la sobreproducción y acumulación de porfirinas o sus precursores. En las porfirias hepáticas, las porfirinas y los precursores de porfirinas se sobreproducen principalmente en el hígado, mientras que en las porfirias eritropoyéticas, las porfirinas se sobreproducen en las células eritroides en el hueso. Las porfirias agudas o hepáticas conllevan la disfunción del sistema nervioso y manifestaciones neurológicas que pueden afectar tanto al sistema nervioso central como al periférico, lo cual provoca síntomas tales como, por ejemplo, dolor (p. ej., dolor abdominal y/o dolor neuropático crónico), vómitos, neuropatía (p. ej., neuropatía aguda, neuropatía progresiva), debilidad muscular, convulsiones, problemas mentales (p. ej., alucinaciones, depresión-ansiedad, paranoia), arritmias cardíacas, taquicardia, estreñimiento y diarrea. Las porfirias cutáneas o eritropoyéticas afectan principalmente a la piel y provocan síntomas tales como fotosensibilidad, la cual puede ser dolorosa, ampollas, necrosis, picores, hinchazón y un mayor crecimiento del pelo en áreas tales como la frente. La infección posterior de las lesiones de la piel puede provocar la pérdida de tejidos y huesos, así como también cicatrices, desfiguración y pérdida de dedos (p. ej., de las manos y de los pies). La mayoría de las porfirias están provocadas por mutaciones que codifican enzimas de la vía biosintética del grupo hemo. En la FIG. 2 se proporciona un resumen de las porfirias asociadas con errores genéticos en el metabolismo del grupo hemo.
No todas las porfirias son genéticas. Por ejemplo, los pacientes con una enfermedad hepática pueden desarrollar una porfiria como resultado de la disfunción hepática y se ha descrito una forma transitoria de eritroporfiria (eritroporfiria transitoria de la infancia) en la infancia (remítase a Crawford, R.I. et al., J Am Acad Dermatol. agosto de 1995; 33(2 Pt 2):333-6.) Los pacientes con PCT pueden adquirir una actividad deficiente de la uroporfirinógenodescarboxilasa (URO-D), debido a la formación de una enzima ORO-D con una actividad enzimática inferior a la normal (remítase a Phillips et al. Blood, 98:3179-3185, 2001).
La porfiria intermitente aguda (AIP) (también denominada deficiencia de porfobilinógeno (PBG)-desaminasa o deficiencia de hidroximetilbilano-sintasa (HMBS)) es el tipo más común de porfiria hepática aguda. Otros tipos de porfirias hepáticas agudas incluyen la coproporfiria hereditaria (HCP), porfiria variegata (VP) y porfiria debida a la deficiencia de ALA-deshidratasa (ADP). Las porfirias hepáticas agudas se describen, p. ej., en Balwani, M y Desnick, R.J., Blood , 120:4496-4504, 2012.
Normalmente, AIP es una enfermedad dominante autosómica que se caracteriza por una deficiencia de la enzima porfobilinógeno-desaminasa (PBG-desaminasa); esta enzima también se conoce como hidroximetilbilano-sintasa (HMB-sintasa o HMBS). La PBG-desaminasa es la tercera enzima de la vía biosintética del grupo hemo (remítase a la FIG. 1) y cataliza la condensación cabeza con cola de cuatro moléculas de porfobilinógeno para obtener el tetrapirrol lineal hidroximetilbilano (HMB). Como alternativa, se han descrito variantes de transcritos de corte y empalme que codifican diferentes isoformas de la PBG-desaminasa. Las mutaciones en el gen de la PBG-desaminasa se asocian con la AIP. Tales mutaciones pueden provocar una reducción de las cantidades de PBG-desaminasa y/o una reducción de la actividad de la PBG-desaminasa (los individuos afectados normalmente presentan una reducción de ~50% en la actividad de la PBG-desaminasa).
Existen al menos dos modelos diferentes de la patofisiología de AIP y otras porfirias hepáticas agudas (remítase, por ejemplo, a Lin CS-Y et al., Clinical Neurophysiology, 2011; 122:2336-44). De acuerdo con un modelo, la reducción de la producción del grupo hemo como resultado de la deficiencia de la PBG-desaminasa provoca una deficiencia energética y degeneración axónica. De acuerdo con el otro modelo, actualmente más favorecido, la acumulación de precursores de porfirinas (p. ej., ALA y PBG) provoca neurotoxicidad.
Se ha establecido que la AIP presenta una prevalencia de incluso 1 en 10000 en ciertas poblaciones (p. ej., en el norte de Suecia; remítase a Floderus Y et al. Clin Genet. 2002;62:288-97). Se estima que la prevalencia en la población general en los Estados Unidos y Europa, excluido el Reino Unido, es desde aproximadamente 1 en 10000 hasta 1 en 20000. La enfermedad clínica se manifiesta en sí únicamente en aproximadamente un 10-15% de los individuos que son portadores de las mutaciones que se sabe que están asociadas con la AIP. Sin embargo, la penetrancia es de incluso un 40% en individuos con ciertas mutaciones (p. ej., la mutación W198X). Normalmente, la AIP es latente antes de la pubertad. Los síntomas son más comunes en mujeres que en hombres. Probablemente la prevalencia de la enfermedad se subestima debido a su penetrancia incompleta y a los periodos prolongados de latencia. En los Estados Unidos, se estima que existen aproximadamente 2000 pacientes que han sufrido al menos un ataque. Se estima que existen aproximadamente 150 casos recurrentes activos en Francia, Suecia, el Reino Unido y Polonia; estos pacientes son principalmente mujeres jóvenes, con una edad media de 30. Remítase, p. ej., a Elder et al., J Inherit Metab Dis., publicado en línea el 1 de noviembre de 2012.
La AIP afecta, por ejemplo, al sistema nervioso central, autonómico, periférico y visceral. Los síntomas de la AIP son variables e incluyen síntomas gastrointestinales (p. ej., dolor abdominal intenso y poco localizado, náusea/vómitos, estreñimiento, diarrea, íleo), síntomas urinarios (disuria, retención/incontinencia urinaria u orina oscura, p. ej., orina roja oscura), síntomas neurológicos (p. ej., neuropatía sensorial, neuropatía motriz (p. ej., que afecta a los nervios craneales y/o que provoca debilidad en los brazos o las piernas), convulsiones, dolor neuropático (p. ej., dolor asociado con una neuropatía progresiva, p. ej., dolor neuropático crónico), síntomas neuropsiquiátricos (p. ej., confusión mental, ansiedad, agitación, alucinación, histeria, delirio, apatía, depresión, fobias, psicosis, insomnio, somnolencia, coma), participación del sistema nervioso autonómico (que provoca, p. ej., síntomas cardiovasculares tales como taquicardia, hipertensión y/o arritmias, así como también otros síntomas tales como, p. ej., un incremento de los niveles de catecolamina en circulación, sudoración, inquietud y/o temblores), deshidratación y anomalías electrolíticas. Los síntomas más comunes son dolor abdominal y taquicardia. Manifestaciones neurológicas incluyend neuropatías motoras y autonómicas y convulsiones. Los pacientes presentan frecuentemente dolor neuropático crónico y desarrollan una neuropatía progresiva. Los pacientes con ataques recurrentes a menudo presentan un pródromo. Se puede producir una parálisis permanente después de un ataque grave. La recuperación de ataques graves que no se tratan inmediatamente puede llevar semanas o meses. Un ataque agudo puede resultar mortal, p. ej., debido a la parálisis de músculos respiratorios o a un paro cardiovascular provocado por el desequilibrio de los electrolitos (remítase, p. ej., a Thunell S. Hydroxymethylbilane Synthase Deficiency. 27 de septiembre de 2005 [actualizado el 1 de septiembre de 2011]. En: Pagon RA, Bird TD, Dolan CR et al., editores. GeneReviews™ [Internet]. Seattle (WA): Universidad de Washington, Seattle; 1993- (en lo sucesivo en la presente Thunell (1993)). Antes de la existencia de tratamientos con hemina, hasta un 20% de los pacientes con AIP fallecían a causa de la enfermedad.
En los individuos que son portadores de genes para la AIP, el riesgo de cáncer hepatocelular es mayor. En aquellos que padecen ataques recurrentes, el riesgo de cáncer hepatocelular es particularmente grave: después de la edad de 50, el riesgo es casi 100 veces mayor que en la población general.
Los ataques de porfiria aguda se pueden precipitar debido a factores endógenos o exógenos. Los mecanismos mediante los cuales tales factores inducen ataques pueden incluir, por ejemplo, un incremento de la demanda de las enzimas P450 hepáticas y/o una inducción de la actividad de ALAS1 en el hígado. Un incremento de la demanda de las enzimas P450 hepáticas provoca una reducción del grupo hemo hepático libre, de este modo se induce la síntesis de ALAS1 hepático.
Los factores precipitantes incluyen ayuno (u otras formas de ingesta calórica reducida o inadecuada, debido a dietas extremas, atletismo de fondo, etc.), estrés metabólico (p. ej., infecciones, cirugía, viajes aéreos internacionales y estrés psicológico), hormonas endógenas (p. ej., progesterona), fumar cigarrillos, agentes químicos exógenos liposolubles (que incluyen, p. ej., agentes químicos presentes en el humo del tabaco, ciertos fármacos recetados, disolventes orgánicos, biocidas, componentes de bebidas alcohólicas), factores endocrinos (p. ej., hormonas reproductivas (las mujeres pueden experimentar exacerbaciones durante el periodo premenstrual), estrógenos sintéticos, progesteronas, estimulantes de la ovulación y terapia de reemplazo hormonal). Remítase, por ejemplo, a Thunell (1993).
Se han contraindicado más de 1000 fármacos para las porfirias hepáticas agudas (p. ej., AIP, HCP, ADP y VP), que incluyen, por ejemplo, alcohol, barbitúricos, Carbamazepina, Carisoprodol, Clonazepam (dosis elevadas), Danazol, Diclofenac y posiblemente otros NSAID (siglas en inglés referentes a los antiinflamatorios no esteroideos), hongos Ergot, estrógenos, Eticlorvinol, Glutetimida, Griseofulvina, Mefenitoína, Meprobamato (también mebutamato y tibutamato), Metiprilón, Metodopramida, Fenitoína, Primidona, progesterona y progestinas sintéticas, Pirazinamida, Pirazolonas (aminopirina y antipirina), Rifampina, Succinimidas (etosuximida y metsuximida), antibiótidos de tipo sulfonamida y ácido valproico.
Los signos objetivo de la AIP incluyen un cambio de color de la orina durante un ataque agudo (la orina puede aparecer de color rojo o rojo parduzco) y un incremento de las concentraciones de PBG y ALA en la orina durante un ataque agudo. La evaluación genética molecular ha identificado mutaciones en el gen de la PBG-desaminasa (también conocida como HMBS) en más de un 98% de individuos afectados. Thunell (1993).
El diagnóstico de la porfiria puede implicar la evaluación de los antecedentes familiares, evaluación de los niveles del precursor de la porfirina en orina, sangre o heces, y/o evaluación de la actividad enzimática y análisis de la mutación del ADN.El diagnóstico diferencial de las porfirias puede implicar la determinación del tipo de porfiria midiendo niveles individuales de porfirinas o precursores de porfirinas (p. ej., ALA, PBG) en la orina, las heces y/o el plasma (p. ej., mediante cromatografía y fluorometría) durante un ataque. El diagnóstico de la AIP se puede confirmar estableciendo que la actividad de la PBG-desaminasa en eritrocitos corresponda a un 50% del nivel normal o menos. En pacientes y miembros familiares de riesgo se pueden llevar a cabo evaluaciones del ADN en busca de mutaciones. El diagnóstico de la AIP se confirma normalmente mediante una evaluación del ADN para identificar una mutación génica causal específica (p. ej., una mutación en HMBS).
La gestión actual de ataques agudos de AIP incluye hospitalización, monitorización de síntomas y el cese de fármacos no seguros. Normalmente, el tratamiento de ataques agudos requiere hospitalización para controlar y tratar los síntomas agudos, que incluyen, p. ej., dolor abdominal, convulsiones, deshidratación/hiponatremia,
náuseas/vómitos, taquicardia/hipertensión, retención urinaria/íleo. Por ejemplo, el dolor abdominal se puede tratar, p. ej., con analgésicos narcóticos, las convulsiones se pueden tratar con precauciones para las convulsiones y posiblemente medicamentos (aunque muchos de los medicamentos contra las convulsiones están contraindicados), las náuseas/vómitos se pueden tratar, p. ej., con fenotiazinas y la taquicardia/hipertensión se puede tratar, p. ej., con bloqueadores beta. El tratamiento puede incluir la retirada de medicamentos no seguros, la monitorización de la función respiratoria, así como también del estado neurológico y la fuerza muscular. Los ataques moderados (p. ej., los que no presentan paresia ni hiponatremia) se pueden tratar con al menos 300 g de glucosa al 10% intravenosa por día, aunque cada vez con más frecuencia se proporciona hemina inmediatamente. Normalmente, los ataques graves se tratan tan pronto como sea posible con hemina intravenosa (3-4 mg/kg diariamente durante 4-14 días) y con glucosa IV mientras se espera que la hemina IV haga efecto. Normalmente, los ataques se tratan con hemina IV durante 4 días y con glucosa IV mientras se espera a que se administre la hemina IV. En un plazo de 3-4 días desde el comienzo de la administración de hemina, suele haber una mejora clínica acompañada por un descenso de los niveles de ALA y PBG.
La hemina (Panhematin® o hemina para inyección, previamente conocida como hematina) es el único producto de tipo hemo aprobado para ser utilizado en los Estados Unidos y fue el primer fármaco aprobado según el Orphan Drug Act. Panhematin® es hemina obtenida a partir de glóbulos rojos procesados (PRBC, por sus siglas en inglés) y consiste en Protoporfirina IX que contiene un ión de hierro férrico (Hemo B) con un ligando de tipo cloruro. El grupo hemo actúa limitando la síntesis hepática y/o medular de la porfirina. El mecanismo exacto mediante el cual la hemina produce una mejora de los síntomas en pacientes con episodios agudos de las porfirias hepáticas no ha sido elucidado; sin embargo, es probable que su acción sea debida a la inhibición (por retroalimentación) de la ácido 5-aminolevulínico (ALA)-sintasa, la enzima que limita la velocidad de la vía biosintética de las porfirinas/del grupo hemo. Remítase a la etiqueta del producto Panhematin®, Lundbeck, Inc., octubre de 2010. La inhibición de la ALA-sintasa debería provocar una reducción de la producción de ALA y PBG, así como también de las porfirinas y los intermedios de porfirinas.
Los inconvenientes de los productos de tipo hemo (p. ej., hemina) incluyen un impacto retrasado en los síntomas clínicos e incapacidad para prevenir la recurrencia de los ataques. Las reacciones adversas asociadas con la administración de hemo (p. ej., hemina) pueden incluir flebitis (p. ej., tromboflebitis), dificultad con el acceso venoso, anticoagulación (o coagulopatías), trombocitopenia, insuficiencia renal o sobrecarga de hierro, la cual es particularmente probable en pacientes que requieren múltiples programas de tratamiento con hemina para ataques recurrentes. Para prevenir la flebitis, se necesita una sonda venosa permanente de acceso en pacientes con ataques recurrentes. Puede ocurrir daño renal con dosis altas. Los efectos secundarios que se registran de forma poco habitual incluyen fiebre, dolor, malestar, hemólisis, anafilaxis e insuficiencia circulatoria. Remítase a Anderson, K.E., Approaches to Treatment and Prevention of Human Porphyrias, en The Porphyrin Handbook: Medical Aspects of Porphyrins, editado por Karl M. Kadish, Kevin M. Smith, Roger Guilard (2003) (en lo sucesivo en la presente Anderson).
Resulta difícil preparar el grupo hemo en una forma estable para la administración intravenosa. Este es insoluble a un pH neutro pero se puede preparar como el hidróxido de hemo a un pH de 8 o superior. Anderson. Panhematin es un preparado de hemina liofilizada. Cuando la hemina liofilizada se solubiliza para la administración intravenosa, se forman rápidamente productos de degradación; estos productos de degradación son los responsables de que se produzca un efecto anticoagulante transitorio y flebitis en el punto de la infusión. Anderson. La albúmina que contiene hemo y el arginato de hemo (Normosang, la versión europea de la hemina) son más estables y, potencialmente, pueden provocar menos tromboflebitis. Sin embargo, el uso del arginato de hemo no está aprobado en los Estados Unidos. Panhematin se puede estabilizar solubilizándolo para infusión en albúmina humana al 30% en lugar de en agua estéril; sin embargo, la albúmina añade efectos de expansión del volumen intravascular e incrementa el costo del tratamiento, así como también el riesgo de patógenos ya que se aísla a partir de sangre humana. Remítase, p. ej., a Anderson supra.
El tratamiento con éxito de un ataque agudo no evita ni retrasa su reaparición. Existe la duda de si la hemina en sí puede desencadenar ataques recurrentes debido a la inducción de la hemo-oxigenasa. A pesar de ello, en algunas áreas (especialmente Francia), se está tratando a mujeres jóvenes con múltiples ataques recurrentes con hemina semanalmente con el objetivo de alcanzar la profilaxis.
La experiencia limitada con trasplantes de hígado sugiere que, cuando tienen éxito, suponen un tratamiento eficaz para la AIP. Se han producido aproximadamente 12 trasplantes en pacientes humanos en Europa, con efectos curativos o variados. El trasplante de hígado puede restablecer la excreción normal de ALA y PBG y evitar los ataques agudos. Remítase, p. ej., a Dar, F.S. et al. Hepatobiliary Pancreat. Dis. Int., 9(1):93-96 (2010). Además, si el hígado de un paciente con AIP se trasplanta en otro paciente ("trasplante dominó"), el paciente que recibe el trasplante puede desarrollar AIP.
Entre los efectos clínicos a largo plazo de las porfirias agudas, se encuentra el dolor neuropático crónico que puede ser el resultado de una neuropatía progresiva debida a los efectos neurotóxicos, p. ej., de niveles elevados de precursores de porfirinas (p. ej., ALA y/o PBG). Los efectos neurotóxicos pueden estar asociados con la producción del intermedio hemo tóxico, por ejemplo, señalización de GABA alterada y/o producción de oxidación mediada por hierro y especies reactivas del oxígeno (ROS, por sus siglas en inglés).Los pacientes pueden sufrir dolor neuropático
antes de un ataque agudo o durante este. Los pacientes de edad más avanzada pueden experimentar un incremento del dolor neuropático con la edad, para el cual se recetan normalmente varios fármacos narcóticos. Se han registrado anomalías en el electromiograma y una reducción de los tiempos de conducción en pacientes con porfirias hepáticas agudas. Cabe destacar que ratones con AIP (deficiencia de PBG-desaminasa) no inducidos ni tratados desarrollan una neuropatía motriz progresiva que se ha demostrado que provoca la degeneración y la pérdida progresivas del axón del nervio de los cuádriceps debido a los niveles constitutivamente elevados de precursores de porfirinas (ALA y PBG), a la deficiencia del grupo hemo y/o porfirinas (Lindberg et al., J. Clin. Invest., 103(8): 1127—1134, 1999). En pacientes con porfiria aguda (p. ej., ADP, AlP, HCP o VP), los niveles de precursores de porfirinas (ALA y PBG) son a menudo elevados en pacientes sin síntomas y en pacientes con síntomas entre ataques. Por lo tanto, cabe esperar que la reducción de los precursores de porfirinas y el restablecimiento de la biosíntesis normal del grupo hemo mediante la reducción del nivel de actividad y/o expresión de ALAS1 evite y/o minimice el desarrollo de una neuropatía progresiva y crónica. Un tratamiento, p. ej., un tratamiento crónico (p. ej., un tratamiento periódico con ARNi según se describe en la presente, p. ej., un tratamiento de acuerdo con una pauta posológica según se describe en la presente, p. ej., un tratamiento semanal o cada dos semanas) puede reducir de forma continuada la expresión de ALAS1 en pacientes con porfiria aguda que presentan niveles elevados de precursores de porfirinas, porfirinas, productos de porfirinas o sus metabolitos. Tal tratamiento se puede proporcionar según convenga para prevenir o reducir la frecuencia o la gravedad de los síntomas de un paciente individual (p. ej., dolor y/o neuropatía) y/o para reducir el nivel de un precursor de porfirina, porfirina, producto o metabolito de porfirina.
Es necesario identificar agentes terapéuticos novedosos que se puedan utilizar para el tratamiento de las porfirias. Según se ha discutido anteriormente, los tratamientos existentes tales como los productos de tipo hemo (p. ej., hemina) presentan numerosas desventajas. Por ejemplo, el impacto de la hemina sobre los síntomas clínicos presenta un retraso, la hemina es cara y puede presentar efectos secundarios (p. ej., tromboflebitis, anticoagulación, trombocitopenia, sobrecarga de hierro, insuficiencia renal). Los agentes terapéuticos novedosos, tales como los que se describen en la presente, pueden resolver estas desventajas y atender las necesidades no cubiertas de los pacientes actuando más rápidamente, no induciendo flebitis, proporcionando la comodidad de una administración subcutánea, evitando con éxito ataques recurrentes, evitando o mejorando el dolor (p. ej., dolor neuropático crónico) y/o una neuropatía progresiva, y/o no provocando ciertos efectos adversos asociados con la hemina (p. ej., sobrecarga de hierro, incremento del riesgo de padecer un cáncer hepatocelular).
Los pacientes con AIA incluyen aquellos que padecen ataques recurrentes y aquellos que padecen ataques agudos esporádicos. En los pacientes que padecen ataques recurrentes, aproximadamente un 5-10% tienen ataques intermitentes recurrentes (2-3 ataques al año) o ataques recurrentes (>4 ataques al año). Estos pacientes son los que más probablemente consideren un trasplante de hígado o reciban infusiones de hemo (p. ej., hemina) profilácticas. Es probable que los pacientes con ataques recurrentes tengan una calidad de vida baja debido a largas estancias hospitalarias, adicción a opiáceos y/o toxicidad de la red venosa. La administración crónica de hemo puede inducir a la hemo-oxigenasa (HO-1). Por lo tanto, puede ser difícil que los pacientes dejen el hemo y algunos requieren un tratamiento más frecuente. Así pues, algunos facultativos restringen el uso de hemo a los ataques más serios. En consecuencia, hay una necesidad médica no cubierta de profilaxis y tratamientos convenientes y eficaces con una mejor tolerabilidad.
Para los pacientes que padecen ataques agudos, las directrices clínicas sugieren la administración de hemo tan pronto como sea posible. Sin embargo, dados los problemas del diagnóstico y la falta de una disponibilidad inmediata del fármaco, la administración puede verse retrasada. El inicio lento de los efectos del hemo (p. ej., hemina) y su mala tolerabilidad retrasan el momento de mejora. La persistencia de dolor abdominal grave, incluso después de la administración de hemo, puede requerir que los pacientes reciban opiáceos durante varios días.
El retraso en la administración del hemo o la exposición prolongada a factores desencadenantes puede conllevar complicaciones más serias, que incluyen neuropatía motora y síntomas concomitantes (p. ej., debilidad, paresia). En los casos graves pueden ocurrir insuficiencia respiratoria y parálisis. La recuperación de los síntomas neurológicos puede requerir mucho más tiempo hasta conseguir resolverlos. En consecuencia, en el contexto de ataques agudos, se necesitan tratamientos que tiene un inicio de acción más rápido y una mejor tolerabilidad.
La validación farmacológica de ALAS1 como una diana del silenciamiento del ARNm está respaldada por al menos los siguientes hallazgos: ARNm de ALAS1 presenta un marcado aumento regulado durante un ataque; la panhematina provoca una reducción modulada de ALAS-1; y la adición de hemo a las células hepáticas en cultivo conlleva una reducción del ARNm de ALAS-1. Varios hallazgos también indican que la supresión del ARNm de ALAS1 se puede lograr teniendo como diana el hígado. Por ejemplo, se ha mostrado que el trasplante de hígado es curativo; y los metabolitos derivados del hígado impulsan los ataques (remítase, p. ej., a Dar et al. Hepatobiliary Pancreat Dis Int. 9:93-6 (2010); Dowman et al. Ann Intern Med 154:571 -2 (2011); y Wu et al. Genes Dev 23:2201 -2209 (2009). Por lo tanto, la reducción de la expresión de ALAS1, p. ej., en el hígado, utilizando composiciones de ARNi se puede utilizar para tratar una porfiria. Las composiciones de ARNi se pueden utilizar para la profilaxis y tratamiento agudo de porfirias. Por ejemplo, las composiciones de ARNi se pueden utilizar de manera profiláctica en un contexto de ataques recurrentes para inducir la supresión crónica o a largo plazo de la expresión de ALAS1 (que conlleva una supresión crónica o a largo plazo de ALA/PBG) y, de esta manera, atenuar el aumento regulado recurrente de ALAS1 que impulsa los ataques. Un tratamiento profiláctico de este tipo puede reducir el número y
gravedad de los ataques. En un contexto de ataque agudo, la administración de una composición de ARNi puede tratar un ataque agudo, p. ej., al reducir los niveles de ALA/PBG.
La presente descripción proporciona métodos y composiciones de ARNi para modular la expresión de un gen ALAS1. La expresión de ALAS1 se puede reducir o inhibir utilizando un ARNi específico para ALAS1, de este modo se obtiene una reducción de la expresión de un gen ALAS1. La reducción de la expresión de un gen ALAS1 puede reducir el nivel de uno o más precursores de porfirinas, porfirinas, o productos o metabolitos de porfirinas. La reducción de la expresión de un gen ALAS1, así como también reducciones relacionadas del nivel de uno o más precursores de porfirinas y/o porfirinas, puede ser útil en el tratamiento de trastornos relacionados con la expresión de ALAS1, p. ej., las porfirias.
Los ARNi de las composiciones que se exponen en la presente incluyen una hebra de ARN (la hebra antisentido) que tiene una región con una longitud de 30 nucleótidos o inferior, es decir, una longitud de 15-30 nucleótidos, generalmente una longitud de 19-24 nucleótidos, donde dicha región es sustancialmente complementaria respecto a al menos parte de un transcrito de ARNm de un gen ALAS1 (también se denomina en la presente "ARNi específico para ALAS1"). El uso de un ARNi de este tipo permite la degradación dirigida de los ARNm de genes que participan en patologías asociadas con la expresión de ALAS1 en mamíferos, p. ej., porfirias tales como la porfiria debida a la deficiencia de ALA-deshidratasa (también conocida como porfiria de Doss o plumboporfiria) o la porfiria intermitente aguda. Unas dosis muy bajas de ARNi específicos para ALAS1 pueden mediar de forma específica y eficaz la iARN, lo cual provoca una inhibición significativa de la expresión de un gen ALAS1. Los ARNi que tienen como diana ALAS1 pueden mediar de forma específica y eficaz la iARN, lo cual provoca una inhibición significativa de la expresión de un gen ALAS1, p. ej., en ensayos basados en células. De este modo, los métodos y las composiciones que incluyen estos ARNi son útiles para tratar procesos patológicos relacionados con la expresión de ALAS1, tales como las porfirias (p. ej., anemia sideroblástica relacionada con X (XLSA), porfiria debida a la deficiencia de ALA-deshidratasa (porfiria de Doss), porfiria intermitente aguda (AIP), porfiria eritropoyética congénita, porfiria cutánea tardía, coproporfiria hereditaria (coproporfiria), porfiria variegata, protoporfiria eritropoyética (EPP) y eritroporfiria transitoria de la infancia).
La siguiente descripción describe cómo preparar y utilizar composiciones que contienen ARNi para inhibir la expresión de un gen ALAS1, así como también composiciones y métodos para tratar enfermedades y trastornos provocados o modulados por la expresión de este gen. Los aspectos de las composiciones farmacéuticas que se exponen en la presente incluyen un ARNi que tiene una hebra antisentido que comprende una región con una longitud de 30 nucleótidos o inferior, generalmente una longitud de 19-24 nucleótidos, donde dicha región es sustancialmente complementaria respecto a al menos parte de un transcrito de ARN de un gen ALAS1, junto con un portador farmacéuticamente aceptable. Los aspectos de composiciones que se exponen en la presente también incluyen un ARNi que tiene una hebra antisentido que tiene una región de complementariedad con una longitud de 30 nucleótidos o inferior, generalmente una longitud de 19-24 nucleótidos, y que es sustancialmente complementaria respecto a al menos parte de un transcrito de ARN de un gen ALAS1.
Por consiguiente, en algunos aspectos, se exponen en la presente composiciones farmacéuticas que contienen un ARNi de ALAS1 y un portador farmacéuticamente aceptable, métodos para utilizar las composiciones con el fin de inhibir la expresión de un gen ALAS1 y métodos para utilizar las composiciones farmacéuticas con el fin de tratar trastornos relacionados con la expresión de ALAS1.
I. Definiciones
Para una mayor comodidad, a continuación se proporciona el significado de ciertos términos y expresiones que se utilizan en la memoria descriptiva, los ejemplos y las reivindicaciones adjuntas. En el caso de que exista una discrepancia aparente entre el uso de un término en otras partes de esta memoria descriptiva y su definición proporcionada en esta sección, la definición de esta sección deberá prevalecer.
Las letras “G,” “C,” “A,” “T” y “U” se refieren en general, cada una de ellas, a un nucleótido que contiene guanina, citosina, adenina, timidina y uracilo como base, respectivamente. Sin embargo, se sobreentenderá que el término "ribonucleótido" o "nucleótido" también se puede referir a un nucleótido modificado, según se detalla adicionalmente más adelante, o a un resto de reemplazo alternativo. El experto es consciente de que los restos de guanina, citosina, adenina y uracilo se pueden reemplazar por otros restos sin alterar de forma sustancial las propiedades de apareamiento de bases de un oligonucleótido que comprenda un nucleótido que contenga tal resto de reemplazo. Por ejemplo, sin carácter limitante, un nucleótido que comprenda inosina como su base podrá aparear sus bases con nucleótidos que contengan adenina, citosina o uracilo. Por lo tanto, los nucleótidos que contengan uracilo, guanidina o adenina se podrán reemplazar en las secuencias de nucleótidos del ARNbc que se expone en la presente por un nucleótido que contenga, por ejemplo, inosina. En otro ejemplo, los restos de adenina y citosina en cualquier posición del oligonucleótido se pueden reemplazar por restos de guanina y uracilo, respectivamente, para formar un apareamiento de bases Wobble G-U con el ARNm diana. Las secuencias que contienen tales restos de reemplazo son adecuadas para las composiciones y los métodos que se exponen en la presente.
El término “ALAS1” (también conocido como ALAS-1; 5-aminolevulinato-sintasa 1; 5-ALA-sintasa 1; ácido 5’-aminolevulínico-sintasa 1; ALAS-H; ALASH; ALAS-N; ALAS3; EC2.3.1.37; 5-aminolevulinato-sintasa, no específica,
mitocondrial; ALAS; MIG4; 0TTHUMP00000212619; 0TTHUMP00000212620; 0TTHUMP00000212621; 0TTHUMP00000212622; proteína 4 inductora de migración; EC 2.3.1 ), tal como se utilizó en la presente, se refiere a una enzima mitocondrial codificada en el núcleo que es la primera enzima, y normalmente la enzima limitante de la velocidad, de la vía biosintética del grupo hemo en mamíferos. ALAS1 cataliza la condensación de la glicina con succinil-CoA para formar el ácido 5-aminolevulínico (ALA). El gen ALAS1 humano se expresa de forma ubicua, se encuentra en el cromosoma 3p21.1 y normalmente codifica una secuencia de 640 aminoácidos. Por el contrario, el gen ALAS-2, que codifica una isozima, se expresa solamente en los eritrocitos, se encuentra en el cromosoma Xp11.21 y normalmente codifica una secuencia de 550 aminoácidos. La expresión "proteína ALAS1", tal como se utiliza en la presente, se refiere a cualquier variante proteica de ALAS1 de cualquier especie (p. ej., de un ser humano, ratón, primate no humano), así como también a cualesquiera mutantes y fragmentos de esta que conserven actividad de ALAS1. De forma similar, la expresión "transcrito de ALAS1" se refiere a cualquier variante de un transcrito de ALAS1 de cualquier especie (p. ej., de un ser humano, ratón, primate no humano). Se puede consultar un transcrito de ARNm de la de ALAS1 humano en NM_000688.4 (FIG. 3A y FIG. 3B; SEQ iD NO:1). Se puede consultar otro transcrito de ARNm de ALAS1 humano, en NM_000688.5 (FIG. 4A y FIG. 4B; SEQ ID NO:382). El nivel de la proteína ALAS1 codificada madura está regulado por el grupo hemo: unos niveles elevados del grupo hemo reducen la expresión de la enzima madura en las mitocondrias, mientras que unos niveles bajos del grupo hemo incrementan su expresión. Se han identificado múltiples variantes de corte y empalme alternativas que codifican la misma proteína.
Las expresiones "ARNi", "iARN", "agente de ARNi" o "agente de iARN", tal como se utilizan en la presente, se refieren a un agente que contiene ARN, tal como se define este término en la presente, y el cual media la escisión dirigida de un transcrito de ARN, p. ej., mediante una vía del complejo de silenciamiento inducido por ARN (RISC). Un ARNi, tal como se describe en la presente, puede ejercer la inhibición de la expresión de ALAS1. La inhibición de la expresión de ALAS1 se puede evaluar basándose en la reducción del nivel del ARNm de ALAS1 o la reducción del nivel de la proteína ALAS1. La expresión "secuencia diana", tal como se utiliza en la presente, se refiere a una porción contigua de la secuencia de nucleótidos de una molécula de ARNm formada durante la transcripción de un gen ALAS1, incluido el ARNm que es un producto del procesamiento de ARN de un producto de transcripción primario. La porción diana de la secuencia tendrá una longitud que sea al menos suficiente para que sirva como sustrato para la escisión dirigida por el ARNi en esa porción o cerca de ella. Por ejemplo, la secuencia diana tendrá una longitud generalmente de 9-36 nucleótidos, p. ej., una longitud de 15-30 nucleótidos, incluidos todos los subintervalos comprendidos entre estos valores. A modo de ejemplos no limitantes, la secuencia diana puede tener 15-30 nucleótidos, 15-26 nucleótidos, 15-23 nucleótidos, 15-22 nucleótidos, 15-21 nucleótidos, 15-20 nucleótidos, 15-19 nucleótidos, 15-18 nucleótidos, 15-17 nucleótidos, 18-30 nucleótidos, 18-26 nucleótidos, 18-23 nucleótidos, 18- 22 nucleótidos, 18-21 nucleótidos, 18-20 nucleótidos, 19-30 nucleótidos, 19-26 nucleótidos, 19-23 nucleótidos, 19- 22 nucleótidos, 19-21 nucleótidos, 19-20 nucleótidos, 20-30 nucleótidos, 20-26 nucleótidos, 20-25 nucleótidos, 20- 24 nucleótidos, 20-23 nucleótidos, 20-22 nucleótidos, 20-21 nucleótidos, 21-30 nucleótidos, 21-26 nucleótidos, 21- 25 nucleótidos, 21-24 nucleótidos, 21-23 nucleótidos o 21-22 nucleótidos.
La expresión "hebra que comprende una secuencia", tal como se utiliza en la presente, se refiere a un oligonucleótido que comprende una cadena de nucleótidos descrita por la secuencia a la cual se hace referencia utilizando la nomenclatura estándar de nucleótidos.
El término "complementariedad", tal como se utiliza en la presente y a menos que se indique de otro modo, cuando se utiliza para describir una primera secuencia de nucleótidos en relación con una segunda secuencia de nucleótidos, se refiere a la capacidad de un oligonucleótido o polinucleótido que comprende la primera secuencia de nucleótidos para hibridarse y formar una estructura de tipo dúplex en ciertas condiciones con un oligonucleótido o polinucleótido que comprenda la segunda secuencia de nucleótidos, como sobreentenderá un experto. Tales condiciones pueden ser, por ejemplo, condiciones rigurosas, donde las condiciones rigurosas pueden incluir: NaCl 400 mM, PIPES 40 mM de pH 6.4, EDTA 1 mM, 50 oC o 70 oC durante 12-16 horas y a continuación un lavado. Se pueden aplicar otras condiciones tales como las condiciones fisiológicamente relevantes que se pueden encontrar dentro de un organismo. El experto será capaz de determinar el conjunto de condiciones que sean más adecuadas para una prueba de complementariedad de dos secuencias de acuerdo con la aplicación final de los nucleótidos hibridados.
Las secuencias complementarias dentro de un ARNi, p. ej., dentro de un ARNbc que se describe en la presente, incluyen el apareamiento de bases del oligonucleótido o polinucleótido que comprende una primera secuencia de nucleótidos con un oligonucleótido o polinucleótido que comprende una segunda secuencia de nucleótidos a lo largo de la longitud completa de una o ambas secuencias de nucleótidos. En la presente, se puede decir que tales secuencias son "totalmente complementarias" entre sí. Sin embargo, cuando en la presente se dice que la primera secuencia es "sustancialmente complementaria" con respecto a una segunda secuencia, las dos secuencias pueden ser totalmente complementarias o pueden formar uno o dos, pero generalmente no más de 5, 4, 3 o 2, pares de bases con apareamientos erróneos cuando se hibridan para un dúplex de hasta 30 pares de bases, a la vez que mantienen la capacidad de hibridarse en las condiciones más relevantes para su aplicación final, p. ej., la inhibición de la expresión génica mediante una vía de RISC. Sin embargo, cuando dos oligonucleótidos se diseñan para que formen, cuando se hibriden, una o más protuberancias de una hebra, tales protuberancias no se deberán considerar como apareamientos erróneos con respecto a la determinación de la complementariedad. Por ejemplo, se puede decir que un ARNbc que comprenda un oligonucleótido con una longitud de 21 nucleótidos y otro oligonucleótido con
una longitud de 23 nucleótidos, donde el oligonucleótido más largo comprende una secuencia de 21 nucleótidos que es totalmente complementaria respecto al oligonucleótido más corto, es "totalmente complementario" a los efectos descritos en la presente.
Las secuencias "complementarias", tal como se utilizan en la presente, también pueden incluir, o estar formadas totalmente por, pares de bases que no sean de Watson-Crick y/o pares de bases formados a partir de nucleótidos modificados y no naturales, siempre que se cumplan los requisitos anteriores respecto a su capacidad para hibridarse. Los pares de bases que no son de Watson-Crick incluyen, sin carácter limitante, el apareamiento de bases de Hoogstein o Wobble G:U.
Las expresiones "complementario/a", "totalmente complementario/a" y "sustancialmente complementario/a" en la presente se pueden utilizar con respecto a la coincidencia de bases entre la hebra sentido y la hebra antisentido de un ARNbc, o entre la hebra antisentido de un agente de ARNi y una secuencia diana, como se comprenderá a partir del contexto de su uso.
Un polinucleótido que sea "sustancialmente complementario respecto a al menos parte de" un ARN mensajero (ARNm), tal como se utiliza en la presente, se refiere a un polinucleótido que es sustancialmente complementario respecto a una porción contigua del ARNm de interés (p. ej., un ARNm que codifica una proteína ALAS1). Por ejemplo, un polinucleótido es complementario respecto a al menos una parte de un ARNm de ALAS1 si la secuencia es sustancialmente complementaria respecto a una porción no interrumpida de un ARNm que codifica ALAS1. Por ejemplo, un polinucleótido es complementario respecto a al menos una parte de un ARNm de ALAS1 si la secuencia es sustancialmente complementaria respecto a una porción no interrumpida de un ARNm que codifica ALAS1.
La expresión "ARN bicatenario" o "ARNbc", tal como se utiliza en la presente, se refiere a un ARNi que incluye una molécula de ARN o un complejo de moléculas con una región dúplex hibridada que comprende dos hebras de ácido nucleico antiparalelas y sustancialmente complementarias, que se dirá que tienen orientaciones "sentido" y "antisentido" respecto al ARN diana. La región dúplex puede tener cualquier longitud que permita una degradación específica de un ARN diana deseado, p. ej., mediante una vía de RISC, pero normalmente tendrá una longitud comprendida en un intervalo de 9 a 36 pares de bases, p. ej., una longitud de 15-30 pares de bases. Considerando un dúplex entre 9 y 36 pares de bases, el dúplex puede tener cualquier longitud dentro de este intervalo, por ejemplo, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 o 36 pares de bases y cualquier subintervalo comprendido entre estos valores, que incluye, sin carácter limitante, 15-30 pares de bases, 15-26 pares de bases, 15-23 pares de bases, 15-22 pares de bases, 15-21 pares de bases, 15-20 pares de bases, 15-19 pares de bases, 15-18 pares de bases, 15-17 pares de bases, 18-30 pares de bases, 18-26 pares de bases, 18-23 pares de bases, 18-22 pares de bases, 18-21 pares de bases, 18-20 pares de bases, 19-30 pares de bases, 19-26 pares de bases, 19-23 pares de bases, 19-22 pares de bases, 19-21 pares de bases, 19-20 pares de bases, 20-30 pares de bases, 20-26 pares de bases, 20-25 pares de bases, 20-24 pares de bases, 20-23 pares de bases, 20-22 pares de bases, 20-21 pares de bases, 21-30 pares de bases, 21-26 pares de bases, 21-25 pares de bases, 21-24 pares de bases, 21-23 pares de bases o 21-22 pares de bases. Los ARNbc generados en la célula mediante el procesamiento con Dicer y enzimas similares tienen generalmente una longitud comprendida en el intervalo de 19-22 pares de bases. Una hebra de la región dúplex de un ADNbc comprende una secuencia que es sustancialmente complementaria respecto a una región de un ARN diana. Las dos hebras que forman la estructura dúplex pueden ser de una única molécula de ARN que tenga al menos una región autocomplementaria o se pueden formar a partir de dos o más moléculas de ARN diferentes. Cuando la región dúplex se forma a partir de dos hebras de una única molécula, la molécula puede tener una región dúplex separada por una única cadena aislada de nucleótidos (que se denomina en la presente "bucle de horquilla") entre el extremo 3' de una hebra y el extremo 5' de la otra hebra respectiva que forma la estructura de dúplex. El bucle de horquilla puede comprender al menos un nucleótido no apareado; y el bucle de horquilla puede comprender al menos 3, al menos 4, al menos 5, al menos 6, al menos 7, al menos 8, al menos 9, al menos 10, al menos 20, al menos 23 o más nucleótidos no apareados. Cuando las dos hebras sustancialmente complementarias de un ARNbc comprenden moléculas de ARN diferentes, estas moléculas no tienen que estar necesariamente conectadas covalentemente, pero pueden estarlo. Cuando las dos hebras están conectadas covalentemente por medios que no sean un bucle de horquilla, la estructura conectora se denomina "conector". En la presente también se utiliza el término "ARNip" para referirse a un ARNbc según se ha descrito anteriormente.
El agente de ARNi puede ser además un "ARNip monocatenario" que se introduce en una célula u organismo para inhibir un ARNm diana. Los agentes que son ARNi monocatenarios se unen a la endonucleasa de RISC Argonauta 2, que a continuación escinde el ARNm diana. Los ARNip monocatenarios contienen generalmente 15-30 nucleótidos y están modificados químicamente. El diseño y la evaluación de ARNip monocatenarios se describe en la Patente de EE. UU. N.° 8.101.348 y en Lima et al., (2012) Cell 150: 883-894. Cualquiera de las secuencias de nucleótidos antisentido descritas en la presente (p. ej., las secuencias proporcionadas en las Tablas 2, 3, 6, 7, 8, 9, 14, 15, 18 y 20 o en las Tablas 21-40) se pueden utilizar como un ARNip monocatenario según se describe en la presente o se pueden modificar químicamente mediante los métodos descritos en Lima et al., (2012) Cell 150;:883-894.
En otro aspecto, el agente de ARN es una "molécula de ARN antisentido monocatenaria". Una molécula de ARN antisentido monocatenaria es complementaria respecto a una secuencia dentro del ARNm diana. Las moléculas de
ARN antisentido monocatenarias pueden inhibir la traducción de un modo estequiométrico apareando sus bases con el ARNm y obstruyendo físicamente la maquinaria de traducción, remítase a Dias, N. et al., (2002) Mol Cáncer Ther 1:347-355. Como alternativa, las moléculas antisentido monocatenarias inhiben un ARNm diana hibridándose con la diana y escindiendo la diana a través de un evento de escisión de tipo RNasaH. La molécula de ARN antisentido monocatenaria puede tener una longitud comprendida entre aproximadamente 10 y aproximadamente 30 nucleótidos y puede tener una secuencia que sea complementaria respecto a una secuencia diana. Por ejemplo, la molécula de ARN antisentido monocatenaria puede comprender una secuencia que tenga al menos aproximadamente 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 o más nucleótidos contiguos de cualquiera de las secuencias de nucleótidos antisentido descritas en la presente, p. ej., las secuencias proporcionadas en cualquiera de las Tablas 2, 3, 6, 7, 8, 9, 14, 15, 18 y 20 o en las Tablas 21-40.
El experto reconocerá que la expresión "molécula de ARN" o "molécula de ácido ribonucleico" engloba, no solo las moléculas de ARN tal como se expresan o encuentran en la naturaleza, sino también análogos y derivados de ARN que comprenden uno o más análogos o derivados de ribonucleótidos/ribonucleósidos según se describen en la presente o como se conocen en la técnica. En términos estrictos, un "ribonucleósido" incluye una base nucleosídica y un azúcar de tipo ribosa, y un "ribonucleótido" es un ribonucleósido con uno, dos o tres restos de tipo fosfato. Sin embargo, los términos "ribonucleósido" y "ribonucleótido" se pueden considerar equivalentes según se utilizan en la presente. El ARN se puede modificar en la estructura de la base nucleotídica, en la estructura de la ribosa o en la estructura del esqueleto ribosa-fosfato, p. ej., según se describe en la presente más adelante. Sin embargo, las moléculas que comprenden análogos o derivados de ribonucleósidos deben conservar la capacidad para formar un dúplex. A modo de ejemplos no limitantes, una molécula de ARN también puede incluir al menos un ribonucleósido modificado, que incluye, sin carácter limitante, un nucleósido con una modificación de tipo 2'-O-metilo, un nucleósido que comprende un grupo 5'-fosforotioato, un nucleósido terminal unido a un derivado de tipo colesterilo o un grupo de tipo bisdecilamida del ácido dodecanoico, un nucleósido bloqueado, un nucleósido que no sea básico, un nucleósido acíclico, un nucleósido con una modificación de tipo 2'-desoxi-2'-fluoro, un nucleósido con una modificación de tipo 2'-amino, un nucleósido con una modificación de tipo 2'-alquilo, un nucleósido de tipo morfolino, un nucleósido que comprende una base que no sea natural o un fosforamidato, o cualquier combinación de estos. Como alternativa, una molécula de ARN puede comprender al menos dos ribonucleósidos modificados, al menos 3, al menos 4, al menos 5, al menos 6, al menos 7, al menos 8, al menos 9, al menos 10, al menos 15, al menos 20 o más, hasta la longitud completa de la molécula de ARNbc. Las modificaciones no tienen que ser necesariamente las mismas para cada uno de esta pluralidad de ribonucleósidos modificados en una molécula de ARN. Los ARN modificados contemplados para ser utilizados en los métodos y las composiciones que se describen en la presente pueden ser ácidos nucleicos peptídicos (PNA, por sus siglas en inglés) que tienen la capacidad de formar la estructura de dúplex requerida y que permiten o median la degradación específica de un ARN diana, p. ej., mediante una vía de RISC.
En un aspecto, un ribonucleósido modificado incluye un desoxiribonucleósido. En un caso de este tipo, un agente de ARNi puede comprender uno o más desoxinucleósidos, que incluyen, por ejemplo, una o más protuberancias de desoxinucleósidos, o uno o más desoxinucleósidos dentro de la porción bicatenaria del ARNbc. La molécula de ARN puede comprender un porcentaje de desoxirribonucleosas de al menos un 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95% o superior (pero no un 100%) de desoxirribonucleósidos, p. ej., en una o ambas hebras. El término “ARNi” puede que no englobe una molécula de ADN bicatenario (p. ej., una molécula de ADN bicatenario de origen natural o una molécula de ADN que contiene un 100% de desoxinucleósidos). En un aspecto, un agente de interferencia por ARN incluye un ARN monocatenario que interacciona con una secuencia de ARN diana para dirigir la escisión del ARN diana. Sin pretender vincularse a ninguna teoría, el ARN bicatenario largo que es introducido en células es fragmentado para obtener ARNip por una endonucleasa de Tipo III conocida como Dicer (Sharp et al., Genes Dev. 2001, 15:485). Dicer, una enzima similar a la ribonucleasa III, procesa el ARNbc para obtener ARN interferentes pequeños de 19-23 pares de bases con protuberancias 3' de dos bases características (Bernstein et al., (2001) Nature 409:363). A continuación, los ARNip se incorporan en un complejo de silenciamiento inducido por ARN (RISC), donde una o más helicasas deshacen el dúplex de ARNip, lo cual permite que la hebra antisentido complementaria guíe el reconocimiento de la diana (Nykanen et al., (2001) Cell 107:309). Tras la unión al ARNm diana adecuado, una o más endonucleasas del RISC escinden la diana para inducir el silenciamiento (Elbashir et al., (2001) Genes Dev. 15:188). Por lo tanto, en un aspecto, la divulgación se refiere a un ARN monocatenario que propicia la formación de un complejo RISC para ejercer el silenciamiento del gen diana.
La expresión "protuberancia de nucleótidos", tal como se utiliza en la presente, se refiere a al menos un nucleótido no apareado que sobresale de la estructura del dúplex de un ARNi, p. ej., un ARNbc. Por ejemplo, cuando un extremo 3' de una hebra de un ARNbc se extiende más allá del extremo 5' de la otra hebra, o viceversa, se produce una protuberancia de nucleótidos. Un ARNbc puede comprender una protuberancia de al menos un nucleótido; como alternativa, la protuberancia puede comprender al menos dos nucleótidos, al menos tres nucleótidos, al menos cuatro nucleótidos, al menos cinco nucleótidos o más. Una protuberancia de nucleótidos puede comprender o estar constituida por un análogo de nucleótido/nucleósido, que incluye un desoxinucleótido/nucleósido. La o las protuberancias se pueden encontrar en la hebra sentido, la hebra antisentido o cualquier combinación de estas. Además, el o los nucleótidos de una protuberancia pueden estar presentes en el extremo 5', el extremo 3' o ambos extremos de la hebra sentido o antisentido de un ARNbc.
La hebra antisentido de un ARNbc puede tener una protuberancia de 1-10 nucleótidos en el extremo 3' y/o el extremo 5'. La hebra sentido de un ARNbc puede tener una protuberancia de 1-10 nucleótidos en el extremo 3' y/o el extremo 5'. Uno o más de los nucleótidos de la protuberancia se pueden reemplazar por un nucleósido-tiofosfato. Las expresiones "romo" o "de extremos romos", tal como se utilizan en la presente para hacer referencia a un ARNbc, se refieren a que no hay nucleótidos ni análogos de nucleótidos no apareados en un extremo terminal determinado de un ARNbc, es decir, no hay protuberancias de nucleótidos. Uno o ambos extremos de un ARNbc pueden ser romos. Cuando ambos extremos de un ARNbc son romos, se dice que el ARNbc es de extremos romos. Para que no haya lugar a dudas, un ARNbc "de extremos romos" es un ARNbc que es romo en ambos extremos, es decir, que no tiene protuberancia de nucleótidos en ninguno de los extremos de la molécula. En la mayoría de los casos, una molécula de este tipo será bicatenaria en toda su longitud.
La expresión "hebra antisentido" o "hebra guía" se refiere a la hebra de un ARNi, p. ej., un ARNbc, que incluye una región que es sustancialmente complementaria respecto a una secuencia diana. La expresión "región de complementariedad", tal como se utiliza en la presente, se refiere a la región de la hebra antisentido que es sustancialmente complementaria respecto a una secuencia, por ejemplo, una secuencia diana, según se define en la presente. Cuando la región de complementariedad no es totalmente complementaria respecto a la secuencia diana, los apareamientos erróneos se pueden producir en las regiones internas o terminales de la molécula. En general, los apareamientos erróneos más tolerados se encuentran en las regiones terminales, p. ej., en los 5, 4, 3 o 2 nucleótidos de los extremos 5' y/o 3'.
La expresión "hebra sentido" o "hebra pasajera", tal como se utiliza en la presente, se refiere a la hebra de un ARNi que incluye una región que es sustancialmente complementaria respecto a una región de la hebra antisentido, según se define este término en la presente.
El término "SNALP" (por sus siglas en inglés), tal como se utiliza en la presente, se refiere a una partícula de ácido nucleico-lípido estable. Una SNALP representa una vesícula de lípidos que recubre un interior acuoso reducido que comprende un ácido nucleico tal como un ARNi o un plásmido a partir del cual se transcribe un ARNi. Las SNALP se describen, p. ej., en las Publicaciones de Solicitud de Patente de EE. UU. N.°s 20060240093, 20070135372, y en la Solicitud Internacional N.° WO 2009082817.
La expresión "se introduce en una célula", cuando hace referencia a un ARNi, se refiere a que se facilita o se lleva a cabo su captación o absorción en la célula, según sobreentienden los expertos en la técnica. La absorción o captación de un ARNi se puede producir a través de procesos celulares activos o difusos sin ayuda, o mediante dispositivos o agentes auxiliares. El significado de este término no se limita a células in vitro ; un ARNi también se puede "introducir en una célula", donde la célula sea parte de un organismo vivo. En un caso de este tipo, la introducción en la célula incluirá el suministro al organismo. Por ejemplo, para un suministro in vivo, el ARNi se pueden inyectar en un punto tisular o se puede administrar por vía sistémica. El suministro in vivo también se puede llevar a cabo mediante un sistema de suministro de p-glucano tal como los que se describen en las Patentes de EE. UU. N.°s 5.032.401 y 5.607.677, y en la Publicación de EE. UU. N.° 2005/0281781. La introducción in vitro en una célula incluye métodos conocidos en la técnica tales como la electroporación y lipofección. Existen otras estrategias conocidas en la técnica o que se describen en la presente más adelante.
La expresión "modular la expresión de", tal como se utiliza en la presente, se refiere a una "inhibición" al menos parcial o una "activación" parcial de la expresión de un gen ALAS1 en una célula tratada con una composición de ARNi según se describe en la presente en comparación con la expresión de ALAS1 en una célula de control. Una célula de control incluye una célula no tratada o una célula tratada con un ARNi de control que no actúe sobre la diana.
Las expresiones "activar", "potenciar", "aumentar la expresión de", "incrementar la expresión de" y similares, siempre que hagan referencia a un gen ALAS1, se refieren en la presente a la activación al menos parcial de la expresión de un gen ALAS1, que se manifiesta por un incremento de la cantidad de ARNm de ALAS1, el cual se puede aislar o detectar a partir de una primera célula o grupo de células en las cuales se transcriba un gen ALAS1 y que se han o hayan tratado de un modo tal que se incremente la expresión de un gen ALAS1, en comparación con una segunda célula o grupo de células sustancialmente idénticas a la primera célula o grupo de células pero que no se han o hayan tratado de ese modo (células de control).
La expresión de un gen ALAS1 se puede activar al menos aproximadamente un 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45% o 50% mediante la administración de un ARNi según se describe en la presente. Un gen ALAS1 se puede activar al menos aproximadamente un 60%, 70% u 80% mediante la administración de un ARNi que se expone en la presente. La expresión de un gen ALAS1 se puede activar al menos aproximadamente un 85%, 90% o 95% o más mediante la administración de un ARNi según se describe en la presente. La expresión del gen ALAS1 se puede incrementar al menos 1 vez, al menos 2 veces, al menos 5 veces, al menos 10 veces, al menos 50 veces, al menos 100 veces, al menos 500 veces, al menos 1000 veces o más en células tratadas con un ARNi según se describe en la presente en comparación con la expresión en una célula no tratada. La activación de la expresión por parte de ARNbc pequeños se describe, por ejemplo, en Li et al., 2006 Proc. Natl. Acad. Sci. U.S.A. 103:17337-42, y en US20070111963 y US2005226848.
Las expresiones "silenciar", "inhibir la expresión de", "reducir la expresión de", "suprimir la expresión de" y similares, siempre que hagan referencia a un gen ALAS1, se refieren en la presente a la supresión al menos parcial de la expresión de un gen ALAS1, según se evalúa, p. ej., en función de la expresión del ARNm de ALAS1, la expresión de la proteína ALAS1 u otro parámetro relacionado funcionalmente con la expresión del gen ALAS1 (p. ej., las concentraciones de ALA o PBG en plasma u orina). Por ejemplo, la inhibición de la expresión de ALAS1 se puede manifestar por una reducción de la cantidad de ARNm de ALAS1 que se puede aislar o detectar a partir de una primera célula o grupo de células en las que se transcriba un gen ALAS1 y que se han o hayan tratado de un modo tal que se inhiba la expresión de un gen ALAS1, en comparación con un control. El control puede ser una segunda célula o grupo de células sustancialmente idénticas a la primera célula o grupo de células, con la excepción de que la segunda célula o grupo de células no se han tratado del mismo modo (células de control). El grado de inhibición se expresa normalmente como un porcentaje de un nivel de control, p. ej.,
(ARNmencélulasdecontrol)- (ARNmencélulas tratadas)^^ &
(ARNmen células de control) °
Como alternativa, el grado de inhibición se puede proporcionar en términos de una reducción de un parámetro que esté relacionado funcionalmente con la expresión del gen ALAS1, p. ej., la cantidad de proteína codificada por un gen ALAS1 o el nivel de una o más porfirinas. La reducción de un parámetro relacionado funcionalmente con la expresión del gen ALAS1 se puede expresar de forma similar como un porcentaje de un nivel de control. En principio, el silenciamiento de un gen ALAS1 se puede determinar en cualquier célula que exprese ALAS1, ya sea constitutivamente o mediante una modificación genómica, y mediante cualquier ensayo adecuado. Sin embargo, cuando se necesite una referencia para determinar si un ARNi determinado inhibe la expresión del gen ALAS1 en cierto grado y, por consiguiente, queda englobado en la presente divulgación, los ensayos proporcionados en los Ejemplos más adelante deberán servir como una referencia de este tipo.
Por ejemplo, en ciertos casos, la expresión de un gen ALAS1 se suprime al menos aproximadamente un 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45% o 50% mediante la administración de un ARNi que se expone en la presente. Un gen ALAS1 se puede suprimir al menos aproximadamente un 60%, 65%, 70%, 75% u 80% mediante la administración de un ARNi que se expone en la presente. Un gen ALAS1 se puede suprimir al menos aproximadamente un 85%, 90%, 95%, 98%, 99% o más mediante la administración de un ARNi según se describe en la presente.
Los términos "tratar", "que trata", "tratamiento" y similares, tal como se utilizan en la presente en el contexto de la expresión de ALAS1, se refieren al alivio o la reducción de procesos patológicos relacionados con la expresión de ALAS1 (p. ej., procesos patológicos que implican porfirinas o defectos en la vía de las porfirinas tales como, por ejemplo, las porfirias). En el contexto de la presente divulgación, y siempre que se refieran a cualquiera de las diferentes condiciones mencionadas en la presente más adelante (además de los procesos patológicos relacionados con la expresión de ALAS1), los términos "tratar", "tratamiento" y similares se refieren a prevenir, aliviar o reducir al menos un síntoma asociado con tal afección, o ralentizar o revertir la evolución o la evolución anticipada de tal afección. Por ejemplo, los métodos que se exponen en la presente, cuando se emplean para tratar una porfiria, pueden servir para reducir o prevenir uno o más síntomas asociados con la porfiria (p. ej., el dolor), para reducir la gravedad o frecuencia de los ataques asociados con la porfiria, para reducir la probabilidad de que se produzca un ataque de uno o más síntomas asociados con la porfiria tras la exposición a una condición precipitante, para reducir la duración de un ataque asociado con la porfiria y/o para reducir el riesgo de desarrollar afecciones asociadas con la porfiria (p. ej., un cáncer hepatocelular o una neuropatía (p. ej., una neuropatía progresiva)). De este modo, a menos que el contexto indique claramente lo contrario, se pretende que los términos "tratar", "tratamiento" y similares engloben la profilaxis, p. ej., la prevención de trastornos y/o síntomas de trastornos relacionados con la expresión de ALAS1.
El término "inferior", en el contexto de un síntoma o marcador de una enfermedad, se refiere a una reducción estadística o clínicamente significativa de dicho nivel. La reducción puede ser, por ejemplo, de al menos un 10%, al menos un 20%, al menos un 30%, al menos un 40% o más y normalmente disminuye hasta un nivel que se acepta como perteneciente al intervalo normal para un individuo sin tal trastorno.
Las expresiones "cantidad terapéuticamente eficaz" y "cantidad profilácticamente eficaz", tal como se utilizan en la presente, se refieren a una cantidad que proporciona un beneficio terapéutico en el tratamiento, la prevención o la gestión de procesos patológicos relacionados con la expresión de ALAS1. La cantidad específica que es terapéuticamente eficaz puede ser determinada fácilmente por un médico común y puede variar dependiendo de factores conocidos en la técnica tales como, por ejemplo, el tipo de proceso patológico, el historial y la edad del paciente, la etapa del proceso patológico y la administración de otros agentes.
La expresión "composición farmacéutica", tal como se utiliza en la presente, comprende una cantidad farmacológicamente eficaz de un ARNi y un portador farmacéuticamente aceptable. La expresión "cantidad farmacológicamente eficaz", "cantidad terapéuticamente eficaz" o simplemente "cantidad eficaz", tal como se utiliza en la presente, se refiere a aquella cantidad de un ARNi que es eficaz para producir el resultado farmacológico, terapéutico o preventivo esperado. Por ejemplo, en un método para tratar un trastorno relacionado con la expresión de ALAS1 (p. ej., en un método para tratar una porfiria), una cantidad eficaz incluye una cantidad eficaz para reducir
uno o más síntomas asociados con una porfiria, una cantidad eficaz para reducir la frecuencia de los ataques, una cantidad eficaz para reducir la probabilidad de que se produzca un ataque de uno o más síntomas asociados con una porfiria tras la exposición a un factor precipitante, o una cantidad eficaz para reducir el riesgo de desarrollar afecciones asociadas con una porfiria (p. ej., una neuropatía (p. ej., una neuropatía progresiva), un cáncer hepatocelular). Por ejemplo, si un tratamiento clínico determinado se considera eficaz cuando se produce una reducción de al menos un 10% en un parámetro medible asociado con una enfermedad o trastorno, una cantidad terapéuticamente eficaz de un fármaco para el tratamiento de esta enfermedad o trastorno será la cantidad necesaria para ejercer una reducción de al menos un 10% en ese parámetro. Por ejemplo, una cantidad terapéuticamente eficaz de un ARNi que tenga ALAS1 como diana puede reducir los niveles de la proteína ALAS1 en cualquier cantidad medible, p. ej., al menos un 10%, 20%, 30%, 40% o 50%.
La expresión "portador farmacéuticamente aceptable" se refiere a un portador para la administración de un agente terapéutico. Tales portadores incluyen, sin carácter limitante, solución salina, solución tamponada, dextrosa, agua, glicerol, etanol y combinaciones de estos. El término excluye específicamente el medio de cultivo celular. Para los fármacos que se administran por vía oral, los portadores farmacéuticamente aceptables incluyen, sin carácter limitante, excipientes farmacéuticamente aceptables tales como diluyentes inertes, agentes desintegrantes, agentes aglutinantes, agentes lubricantes, agentes edulcorantes, agentes saborizantes, agentes colorantes y conservantes. Los diluyentes inertes adecuados incluyen carbonato de sodio y calcio, fosfato de sodio y calcio, y lactosa, mientras que el almidón de maíz y el ácido algínico son agentes desintegrantes adecuados. Los agentes aglutinantes pueden incluir almidón y gelatina, mientras que el agente lubricante, si está presente, será generalmente estearato de magnesio, ácido esteárico o talco. Si se desea, los comprimidos se pueden recubrir con un material tal como monoestearato de glicerilo o diestearato de glicerilo, para retrasar la absorción en el aparato gastrointestinal. Los agentes incluidos en las formulaciones farmacológicas se describen con más detalle en la presente posteriormente.
El término "aproximadamente", cuando hace referencia a un número o a un intervalo, se refiere a que el número o intervalo mencionado es una aproximación dentro de la variabilidad experimental (o dentro del error experimental estadístico) y, por lo tanto, el número o intervalo numérico puede variar, por ejemplo, entre un 1% y un 15% del número o intervalo numérico mencionado.
II. Agentes de ARNi
En la presente se describen agentes de ARNi que inhiben la expresión de un gen ALAS1. El agente de ARNi puede incluir moléculas de ácido ribonucleico bicatenarias (ARNbc) para inhibir la expresión de un gen ALAS1 en una célula o en un sujeto (p. ej., en un mamífero, p. ej., en un ser humano que padezca una porfiria), donde el ARNbc incluye una hebra antisentido que tiene una región de complementariedad que es complementaria respecto a al menos una parte de un ARNm formado en la expresión de un gen ALAS1, y donde la región de complementariedad tiene una longitud de 30 nucleótidos o inferior, generalmente una longitud de 19-24 nucleótidos, y donde el ARNbc, cuando entra en contacto con una célula que expresa el gen ALAS1, inhibe la expresión del gen ALAS1 al menos un 10%, según se evalúa mediante, por ejemplo, un método basado en PCR o ADN ramificado (ADNr), o mediante un método basado en proteínas tal como la inmunotransferencia de Western. El agente de ARNi puede activar la expresión de un gen ALAS1 en una célula o mamífero. La expresión de un gen ALAS1 en un cultivo celular, tal como células COS, células HeLa, hepatocitos primarios, células HepG2, células cultivadas primarias, o en una muestra biológica procedente de un sujeto se puede evaluar midiendo los niveles de ARNm de ALAS1, por ejemplo, mediante un ensayo de ADNr o TaqMan, o midiendo los niveles de proteínas, por ejemplo, mediante un análisis de inmunofluorescencia, utilizando, por ejemplo, técnicas de citometría de flujo o inmunotransferencia de Western.
Un ARNbc incluye dos hebras de ARN que son suficientemente complementarias para hibridarse y formar una estructura de dúplex en las condiciones en las que se utilizará el ARNbc. Una hebra de un ARNbc (la hebra antisentido) incluye una región de complementariedad que es sustancialmente complementaria, y en general totalmente complementaria, respecto a una secuencia diana, derivada de la secuencia de un ARNm formado durante la expresión de un gen ALAS1. La otra hebra (la hebra sentido) incluye una región que es complementaria respecto a la hebra antisentido, de modo que las dos hebras se hibridan y forman una estructura de dúplex cuando se combinan en condiciones adecuadas. En general, la estructura de dúplex tiene una longitud comprendida entre 15 y 30 pares de bases inclusive, más generalmente entre 18 y 25 inclusive, aún más generalmente entre 19 y 24 inclusive, y de la forma más general entre 19 y 21 inclusive. De forma similar, la región de complementariedad respecto a la secuencia diana tiene una longitud comprendida entre 15 y 30 nucleótidos inclusive, más generalmente entre 18 y 25 inclusive, aún más generalmente entre 19 y 24 inclusive, y de la forma más general entre 19 y 21 inclusive. El ARNbc puede tener una longitud comprendida entre 15 y 20 nucleótidos inclusive, y además el ARNbc puede tener una longitud comprendida entre 25 y 30 nucleótidos inclusive. Como reconocerá el experto, la región que actúa como diana de un ARN que actúa como diana para ser escindido formará parte en la mayoría de los casos de una molécula de ARN más larga, a menudo una molécula de ARNm. Cuando sea relevante, una "parte" de una diana de ARNm es una secuencia contigua de una diana de ARNm de longitud suficiente para que sea un sustrato para la escisión dirigida por iARN (es decir, la escisión a través de una vía de RISC). Los ARNbc que tienen dúplex de tan solo 9 pares de bases pueden mediar, en algunas circunstancias, la escisión del ARN dirigida por iARN. En la mayoría de los casos, una diana tendrá una longitud de al menos 15 nucleótidos, p. ej., una longitud de 15-30 nucleótidos.
Un experto en la técnica también reconocerá que la región dúplex es una porción funcional principal de un ARNbc, p. ej., una región dúplex de 9 a 36, p. ej., de 15-30 pares de bases. De este modo, siempre que se procese hasta obtener un dúplex funcional de, p. ej., 15-30 pares de bases que tenga como diana un ARN deseado para escindirlo, una molécula de ARN o un complejo de moléculas de ARN que tengan una región dúplex con un tamaño superior a 30 pares de bases puede ser un ARNbc. De este modo, un experto reconocerá que, en un aspecto, un miARN será entonces un ARNbc. Un ARNbc puede no ser un miARN de origen natural. Un agente de ARNi útil para actuar sobre la expresión de ALAS1 puede no generarse en la célula diana mediante la escisión de un ARNbc más largo.
Un ARNbc según se describe en la presente puede incluir además una o más protuberancias de nucleótidos monocatenarias. El ARNbc se puede sintetizar mediante métodos estándar conocidos en la técnica, según se describe adicionalmente más adelante, p. ej., mediante el uso de un sintetizador de ADN automatizado tal como los comercializados por, por ejemplo, Biosearch, Applied Biosystems, Inc. Un gen de ALAS1 puede ser un gen de ALAS1 humano. El gen de ALAS1 puede ser un gen de ALAS1 de rata o ratón. La primera secuencia puede ser una hebra sentido de un ARNbc que incluye una secuencia sentido expuesta en la presente, p. ej., en las Tablas 21-40, y la segunda secuencia puede ser una hebra antisentido de un ARNbc que incluye una secuencia antisentido expuesta en la presente, p. ej., en las Tablas 21-40.
Específicamente, la primera secuencia puede ser una hebra sentido de un ARNbc que incluye una secuencia sentido de la Tabla 2 o la Tabla 3, y la segunda secuencia puede ser una hebra antisentido de un ARNbc que incluye una secuencia antisentido de la Tabla 2 o la Tabla 3. La primera secuencia puede ser una hebra sentido de un ARNbc que incluye una secuencia sentido de la Tabla 2, 3, 6, 7, 8, 9, 14 o 15, y la segunda secuencia es una hebra antisentido de un ARNbc que incluye una secuencia antisentido de la Tabla 2, 3, 6, 7, 8, 9, 14 o 15. La primera secuencia puede ser una hebra sentido de un ARNbc que incluye una secuencia sentido de la Tabla 2, 3, 6, 7, 8, 9, 14, 15, 18 o 20, y la segunda secuencia puede ser una hebra antisentido de un ARNbc que incluye una secuencia antisentido de la Tabla 2, 3, 6, 7, 8, 9, 14, 15, 18 o 20.
En un aspecto, un ARNbc puede incluir al menos secuencias de nucleótidos sentido y antisentido, donde la hebra sentido se selecciona a partir de las secuencias sentido que se proporcionan en la presente, p. ej., en las Tablas 21 40, y la hebra antisentido correspondiente de la hebra sentido se selecciona a partir de las secuencias antisentido que se proporcionan en la presente, p. ej., en las Tablas 21-40.
En un aspecto, un ARNbc puede incluir al menos secuencias de nucleótidos sentido y antisentido, donde la hebra sentido se selecciona a partir de los grupos de secuencias que se proporcionan en las Tablas 2 y 3, y la hebra antisentido correspondiente de la hebra sentido se selecciona a partir de las Tablas 2 y 3. En un aspecto adicional, un ARNbc puede incluir al menos secuencias de nucleótidos sentido y antisentido, donde la hebra sentido se selecciona a partir de los grupos de secuencias que se proporcionan en las Tablas 2, 3, 6, 7, 8, 9, 14 y 15, y la hebra antisentido correspondiente de la hebra sentido se selecciona a partir de las Tablas 2, 3, 6, 7, 8, 9, 14 y 15. En un aspecto adicional, el ARNbc puede incluir al menos secuencias de nucleótidos sentido y antisentido, en las cuales la hebra sentido se selecciona de los grupos de secuencias proporcionadas en las Tablas 2, 3, 6, 7, 8, 9, 14, 15, 18 y 20, y la hebra antisentido correspondiente de la hebra sentido se selecciona de las Tablas 2, 3, 6, 7, 8, 9, 14, 15, 18 y 20.
El ARNi puede ser AD-60501, AD-60519, AD-60901, AD-60495, AD-60900, AD-60935, AD-60879, AD-61190, AD-61191, AD-60865, AD-60861, AD-60876, AD-61193, AD-60519, AD-60519, AD-60901, AD-60405, AD-60887, AD-60923, AD-60434, AD-60892, AD-60419, AD-60924, AD-60445, AD-60925, AD-60926, AD-60820, AD-60843, AD-60819, AD-61140, AD-61141, AD-61142, AD-60835, AD-60839, AD-61143, AD-61144, AD-61145, AD-61146, AD-60892, o AD-60419 (p. ej., incluidas la secuencia de nucleótidos y/o una o más (p. ej., la totalidad) de las modificaciones de los ARNbc mencionados anteriormente). El ARNi puede comprender una hebra antisentido que comprende, o está constituida por, una secuencia antisentido (incluidas una o más (p. ej., la totalidad de las modificaciones)) seleccionada entre la secuencia antisentido de AD-60501, AD-60519, Ad-60901, AD-60495, AD-60900, AD-60935, AD-60879, AD-61190, AD-61191, AD-60865, AD-60861, AD-60876, AD-61193, AD-60519, AD-60519, AD-60901, AD-60405, AD-60887, AD-60923, AD-60434, AD-60892, AD-60419, AD-60924, AD-60445, AD-60925, AD-60926, AD-60820, AD-60843, AD-60819, AD-61140, AD-61141, AD-61142, AD-60835, AD-60839, AD-61143, AD-61144, AD-61145, AD-61146, AD-60892, o AD-60419. El ARNi puede comprender una hebra sentido que comprende, o está constituida por, una secuencia sentido (y/o una o más (p. ej., la totalidad) de las modificaciones)) seleccionada entre AD-60501, AD-60519, AD-60901, AD-60495, AD-60900, AD-60935, AD-60879, AD-61190, AD-61191, AD-60865, AD-60861, AD-60876, AD-61193, AD-60519, AD-60519, AD-60901, AD-60405, AD-60887, AD-60923, AD-60434, AD-60892, AD-60419, AD-60924, AD-60445, AD-60925, AD-60926, AD-60820, AD-60843, AD-60819, AD-61140, AD-61141, AD-61142, AD-60835, AD-60839, AD-61143, AD-61144, AD-61145, AD-61146, AD-60892, o AD-60419.
El ARNi puede comprender (i) una hebra antisentido que comprende, o está constituida por, la secuencia de UAAGAUGAGACACUCUUUCUGGU o UAAGAUGAGACACUCTUUCUGGU y/o (ii) una hebra sentido que comprende, o está constituida por, la secuencia de CAGAAAGAGUGUCUCAUCUUA. Uno o más nucleótidos de la hebra antisentido y/o hebra sentido pueden estar modificados como se describe en la presente.
El ARNi puede comprender (i) una hebra antisentido que comprende, o está constituida por, la secuencia antisentido de AD-60489 y/o (ii) una hebra sentido que comprende, o está constituida por, la secuencia sentido de AD-60489 (y/o una o más (p. ej., la totalidad) de las modificaciones de la hebra sentido y/o hebra antisentido de AD-60489). El ARNi puede comprender (i) una hebra antisentido que comprende, o está constituida por, la secuencia antisentido de AD-60519 y/o (ii) una hebra sentido que comprende, o está constituida por, la secuencia sentido de AD-60519 (y/o una o más (p. ej., la totalidad) de las modificaciones de la hebra sentido y/o hebra antisentido de AD-60489). El ARNi puede comprender (i) una hebra antisentido que comprende, o está constituida por, la secuencia antisentido de AD-61193 y/o (ii) una hebra sentido que comprende, o está constituida por, la secuencia sentido de AD-61193 (y/o una o más (p. ej., la totalidad) de las modificaciones de la hebra sentido y/o hebra antisentido de AD-60489). El ARNi puede comprender (i) una hebra antisentido que comprende, o está constituida por, la secuencia antisentido de AD-60819 y/o (ii) una secuencia sentido que comprende, o está constituida por, la secuencia sentido de AD-60819 (y/o una o más (p. ej., la totalidad) de las modificaciones de la hebra sentido y/o hebra antisentido de AD-60489).
Se puede proporcionar el ARNi para inhibir la expresión de ALAS1, donde el ARNbc comprende (i) una hebra antisentido que comprende, o está constituida por, la secuencia antisentido de AD-60489, AD-60519, AD-61193, o AD-60819 (o una secuencia antisentido no modificada correspondiente) y/o (ii) una hebra sentido que comprende, o está constituida por, la secuencia sentido de AD-60489, Ad-60519, AD-61193, o AD-60819 (o una secuencia antisentido no modificada correspondiente). El ARNi puede comprender (i) una hebra antisentido que está constituida por la secuencia antisentido de Ad-60489, AD-60519, a D-61193 o AD-60819 y/o (ii) una hebra sentido que está constituida por la secuencia sentido de AD-60489, AD-60519, AD-61193 o AD-60819, excepto en que la hebra antisentido y/o hebra sentido del ARNbc difieren en 1, 2 o 3 nucleótidos de la correspondiente secuencia antisentido y/o sentido de AD-60489, AD-60519, AD-61193 o AD-60819.
Las secuencias y modificaciones de AD-60489, AD-60519, AD-61193 y AD-60819 se muestran en la Tabla 44 descrita en la presente.
El ARNi puede ser ALN-60519. ALN-60519 puede ser un oligonucleótido bicatenario sintetizado por medios químicos ligado covalentemente a un ligando que contiene tres residuos de N-acetilgalactosamina (GalNAc) (mostrado en la FIG. 57). Todos los nucleótidos de ALN-60519 pueden tener modificaciones de tipo 2’-OMe o 2’-F y pueden estar conectados mediante uniones fosfodiéster 3’-5’, y formar de esta manera el esqueleto azúcar-fosfato del oligonucleótido. La hebra sentido y la hebra antisentido de ALN-60519 contienen 21 y 23 nucleótidos, respectivamente. El extremo 3’ de la hebra sentido de ALN-60519 está conjugado con el resto de GalNAc triantenario (denominado L96) mediante una unión fosfodiéster. La hebra antisentido contiene cuatro uniones fosforotioato, dos en el extremo 3’ y dos en el extremo 5’. La hebra sentido de ALN-60519 contiene dos uniones fosforotioato en el extremo 5’. Los 21 nucleótidos de la hebra sentido de ALN-60519 se hibridan con los 21 nucleótidos complementarios de la hebra antisentido, y forman de esta manera 21 pares de bases nucleotídicas y una protuberancia de dos bases en el extremo 3’ de la hebra antisentido. Las dos hebras sencillas, la hebra sentido y la hebra antisentido, de ALN-60519 se pueden sintetizar mediante síntesis de oligonucleótidos en fase sólida convencional, empleando química de fosforamiditos estándar con el grupo 5’-hidroxilo protegido como un éter de dimetoxitrifenilmetilo (DMT). Cada hebra se puede ensamblar desde el extremo 3’ al 5’ mediante la adición secuencial de nucleósidos fosforamiditos protegidos.
En estos aspectos, una de las dos secuencias es complementaria respecto a la otra de las dos secuencias, siendo una de las secuencias sustancialmente complementaria respecto a una secuencia de un ARNm generado mediante la expresión de un gen ALAS1. En este sentido, un ARNbc incluirá dos oligonucleótidos, donde un oligonucleótido se describe en la presente como la hebra sentido y el segundo oligonucleótido se describe como la hebra antisentido correspondiente de la hebra sentido. Según se describe en otras partes de la presente y se sabe en la técnica, las secuencias complementarias de un ARNbc también pueden estar contenidas como regiones autocomplementarias de una única molécula de ácido nucleico, en lugar de estar en oligonucleótidos separados.
El experto es muy consciente de que los ARNbc que tienen una estructura de dúplex con una longitud comprendida entre 20 y 23, pero específicamente de 21, pares de bases se consideran particularmente eficaces a la hora de inducir la interferencia por ARN (Elbashir et al., EMBO 2001, 20:6877-6888). Sin embargo, otros han observado que estructuras de dúplex de ARN más largas o más cortas también pueden ser eficaces. En los aspectos descritos anteriormente, debido a la naturaleza de las secuencias de oligonucleótido proporcionadas en las tablas de la presente, los ARNbc descritos en la presente pueden incluir al menos una hebra con una longitud de como mínimo 21 nucleótidos. Cabe esperar razonablemente que dúplex más cortos que tengan una de las secuencias divulgadas en la presentemenos solamente unos pocos nucleótidos en uno o ambos extremos puedan ser similarmente eficaces en comparación con los ARNbc descritos anteriormente. Por lo tanto, se contemplan ARNbc con una secuencia parcial de al menos 15, 16, 17, 18, 19, 20 o más nucleótidos contiguos de una de las secuencias divulgadas en la presente y que difieren en su capacidad para inhibir la expresión de un gen ALAS1 en no más de un 5, 10, 15, 20, 25 o 30% de inhibición en comparación con un ARNbc que comprenda toda la secuencia de acuerdo con la divulgación.
Además, los ARN proporcionados en las tablas de la presente identifican un sitio en un transcrito de ALAS1 que es susceptible de sufrir una escisión mediada por RISC. En este sentido, la presente divulgación expone además ARNi que actúan dentro de una de estas secuencias. Según se utiliza en la presente, se dice que un ARNi actúa dentro de un sitio particular de un transcrito de ARN si el ARNi fomenta la escisión del transcrito en cualquier lugar dentro de ese sitio particular. Un ARNi de este tipo incluye generalmente al menos 15 nucleótidos contiguos de una de las secuencias proporcionadas en la presente, p. ej., en las Tablas 2, 3, 6, 7, 8, 9, 14, 15, 18, 20 y en las Tablas 21-40 acoplados a secuencias de nucleótidos adicionales procedentes de la región contigua a la secuencia seleccionada en un gen ALAS1.
Aunque una secuencia diana tiene generalmente una longitud de 15-30 nucleótidos, existe una gran variación en la idoneidad de secuencias particulares en este intervalo para dirigir la escisión de cualquier ARN diana determinado. Varios paquetes de software y las directrices que se exponen en la presente proporcionan una guía para la identificación de secuencias diana óptimas para cualquier diana génica determinada, pero también se puede adoptar una estrategia empírica en la cual se coloca una "ventana" o "máscara" de un tamaño determinado (a modo de ejemplo no limitante, 21 nucleótidos) de forma literal o figurada (que incluye, p. ej., in silico) sobre la secuencia de ARN diana para identificar secuencias en el intervalo de tamaños que puedan servir como secuencias diana. Moviendo la "ventana" de la secuencia progresivamente un nucleótido hacia adelante o hacia atrás respecto a una localización de la secuencia diana inicial, se puede identificar la siguiente secuencia diana potencial, hasta que se identifica el conjunto completo de posibles secuencias para cualquier tamaño seleccionado de la diana determinada. Este proceso, acoplado con la síntesis y la evaluación sistemáticas de las secuencias identificadas (utilizando ensayos como los que se describen en la presente o se conocen en la técnica) para identificar las secuencias que presentan un comportamiento óptimo, permite identificar las secuencias de ARN que, cuando actúan como diana de un agente de ARNi, median la mejor inhibición de la expresión del gen diana. De este modo, aunque las secuencias identificadas, por ejemplo, en las de la presente, representan secuencias diana eficaces, se contempla que se puede conseguir una optimización adicional de la eficiencia de la inhibición "desplazando la ventana" progresivamente un nucleótido hacia adelante o hacia atrás en las secuencias determinadas para identificar secuencias con unas características de inhibición iguales o mejores.
Adicionalmente, se contempla que para cualquier secuencia identificada, p. ej., en las tablas de la presente, se puede conseguir una optimización adicional añadiendo o eliminando nucleótidos de forma sistemática para generar secuencias más largas o más cortas y evaluando estas secuencias y secuencias generadas desplazando una ventana del tamaño más largo o más corto hacia adelante o hacia atrás en el ARN diana a partir de ese punto. Nuevamente, el acoplamiento de esta estrategia para generar nuevas dianas candidato con la evaluación para determinar la eficacia de los ARNi basándose en estas secuencias diana en un ensayo de inhibición como los que se conocen en la técnica o como los que se describen en la presente puede proporcionar mejoras adicionales en la eficacia de la inhibición. Es más, tales secuencias optimizadas se pueden ajustar mediante, p. ej., la introducción de nucleótidos modificados como los que se describen en la presente o como los que se conocen en la técnica, la adición o la introducción de cambios en la protuberancia u otras modificaciones como las que se conocen en la técnica y/o se describen en la presente para optimizar adicionalmente la molécula (p. ej., incrementar la estabilidad en suero o la semivida en circulación, incrementar la estabilidad térmica, potenciar el suministro transmembranal, ejercer un direccionamiento hacia una localización o tipo celular particular, incrementar la interacción con las enzimas de la vía de silenciamiento, incrementar la liberación a partir de endosomas, etc.) como un inhibidor de la expresión.
Un ARNi según se describe en la presente puede contener uno o más apareamientos erróneos con la secuencia diana. Un ARNi, tal como se describe en la presente, puede no contener más de 3 apareamientos erróneos. Si la hebra antisentido del ARNi contiene apareamientos erróneos con una secuencia diana, es preferible que el área de apareamiento erróneo no se encuentre localizada en el centro de la región de complementariedad. Si la hebra antisentido del ARNi contiene apareamientos erróneos con la secuencia diana, es preferible que el apareamiento erróneo esté restringido a encontrarse dentro de los últimos 5 nucleótidos considerados desde el extremo 5' o 3' de la región de complementariedad. Por ejemplo, para una hebra de ARN de un agente de ARNi de 23 nucleótidos que sea complementaria respecto a una región de un gen ALAS1, la hebra de ARN generalmente no contendrá ningún apareamiento erróneo dentro de los 13 nucleótidos centrales. Los métodos descritos en la presente o los métodos conocidos en la técnica se pueden utilizar para determinar si un ARNi que contiene un apareamiento erróneo con una secuencia diana es eficaz a la hora de inhibir la expresión de un gen ALAS1. La consideración de la eficacia de ARNi con apareamientos erróneos a la hora de inhibir la expresión de un gen ALAS1 es importante, especialmente si existe constancia de que la región de complementariedad particular de un gen ALAS1 presente una variación de la secuencia polimórfica dentro de la población.
Al menos un extremo de un ARNbc puede tener una protuberancia de nucleótidos monocatenaria de 1 a 4, generalmente 1 o 2, nucleótidos. Los ARNbc que tienen al menos una protuberancia de nucleótidos presentan, inesperadamente, unas propiedades inhibitorias superiores con relación a sus homólogos de extremos romos. Aún más, el ARN de un ARNi, p. ej., un ARNbc, se puede modificar químicamente para mejorar su estabilidad u otras características beneficiosas. Los ácidos nucleicos que se exponen en la presente se pueden sintetizar y/o modificar mediante métodos muy establecidos en la técnica tales como los que se describen en “Current protocols in nucleic acid chemistry,” Beaucage, S.L. et al. (Eds.), John Wiley & Sons, Inc., Nueva York, NY, EE. UU. Las modificaciones incluyen, por ejemplo, (a) modificaciones de los extremos, p. ej., modificaciones del extremo 5' (fosforilación,
conjugación, uniones invertidas, etc.), modificaciones del extremo 3' (conjugación, nucleótidos de ADN, uniones invertidas, etc.) (b) modificaciones de las bases, p. ej., reemplazo por bases estabilizantes, bases desestabilizantes o bases que se apareen con bases con un mayor repertorio de parejas, eliminación de bases (nucleótidos sin bases), o bases conjugadas, (c) modificaciones del azúcar (p. ej., en la posición 2' o en la posición 4', o que tengan un azúcar acíclico) o reemplazo del azúcar, así como también (d) modificaciones del esqueleto, que incluyen la modificación o el reemplazo de las uniones de tipo fosfodiéster. Los ejemplos específicos de compuestos de ARN útiles en esta divulgación incluyen, sin carácter limitante, ARN que contienen esqueletos modificados o uniones entre nucleósidos no naturales. Los ARN que tienen esqueletos modificados incluyen, entre otros, aquellos que no contienen un átomo de fósforo en el esqueleto. A los efectos de esta memoria descriptiva y según se los denomina en ocasiones en la técnica, los ARN modificados que no contienen un átomo de fósforo en su esqueleto entre los nucleósidos también se pueden considerar oligonucleósidos. En particular, el ARN modificado puede contener un átomo de fósforo en su esqueleto entre los nucleósidos.
Los esqueletos de ARN modificados incluyen, por ejemplo, fosforotioatos, fosforotioatos quirales, fosforoditioatos, fosfotriésteres, aminoalquilfosfotriésteres, metilfosfonatos y fosfonatos con otros grupos alquilo que incluyen 3'-alquilenfosfonatos y fosfonatos quirales, fosfinatos, fosforamidatos que incluyen 3'-aminofosforamidato y aminoalquilfosforamidatos, tionofosforamidatos, tionoalquilfosfonatos, tionoalquilfosfotriésteres y boranofosfatos con uniones 3'-5' normales, análogos con uniones 2'-5' de estos y aquellos con polaridad invertida en los que los pares adyacentes de unidades de nucleósidos están unidos del siguiente modo: 3'-5' con 5'-3' o 2'-5' con 5'-2'. También se incluyen varias sales, sales mixtas y formas de ácido libre.
Las patentes de EE. UU. representativas que describen la preparación de las uniones que contienen fósforo anteriores incluyen, sin carácter limitante, las Pat. de EE. UU. N.°s 3.687.808, 4.469.863, 4.476.301, 5.023.243, 5.177.195, 5.188.897, 5.264.423, 5.276.019, 5.278.302, 5.286.717, 5.321.131, 5.399.676, 5.405.939, 5.453.496, 5.455.233, 5.466.677, 5.476.925, 5.519.126, 5.536.821, 5.541.316, 5.550.111, 5.563.253, 5.571.799, 5.587.361, 5.625.050, 6.028.188, 6.124.445, 6.160.109, 6.169.170, 6.172.209, 6.239.265, 6.277.603, 6.326.199, 6.346.614, 6.444.423, 6.531.590, 6.534.639, 6.608.035, 6.683.167, 6.858.715, 6.867.294, 6.878.805, 7.015.315, 7.041.816, 7.273.933, 7.321.029 y la Pat. de EE. UU. RE39464.
Los esqueletos de ARN modificados que no incluyen ningún átomo de fósforo en ellos tienen esqueletos que se forman mediante uniones entre nucleósidos de tipo alquilo o cicloalquilo de cadena corta, uniones entre nucleósidos de tipo alquilo o cicloalquilo y heteroátomos mixtas, o una o más uniones entre nucleósidos heteroaromáticas o heterocíclicas de cadena corta. Estos incluyen los que tienen uniones de tipo morfolino (formadas en parte a partir de la porción del azúcar de un nucleósido); esqueletos de tipo siloxano; esqueletos de tipo sulfuro, sulfóxido y sulfona; esqueletos de tipo formacetilo y tioformacetilo; esqueletos de tipo metilenformacetilo y tioformacetilo; esqueletos que contienen alquenos; esqueletos de tipo sulfamato; esqueletos de tipo metilenimino y metilenhidrazino; esqueletos de tipo sulfonato y sulfonamida; esqueletos de tipo amida; y otros que tienen partes de componentes N, O, S y CH2 mixtas.
Las patentes de EE. UU. representativas que describen la preparación de los oligonucleótidos anteriores incluyen, sin carácter limitante, las Pat. de EE. UU. N.2s 5.034.506, 5.166.315, 5.185.444, 5.214.134, 5.216.141, 5.235.033, 5.64.562, 5.264.564, 5.405.938, 5.434.257, 5.466.677, 5.470.967, 5.489.677, 5.541.307, 5.561.225, 5.596.086, 5.602.240, 5.608.046, 5.610.289, 5.618.704, 5.623.070, 5.663.312, 5.633.360, 5.677.437 y 5.677.439.
En otros miméticos de ARN adecuados o contemplados para su uso en ARNi, tanto el azúcar como la unión entre nucleósidos, es decir, el esqueleto, de las unidades nucleotídicas se reemplazan por grupos novedosos. Las unidades de las bases se mantienen para la hibridación con un compuesto diana de tipo ácido nucleico adecuado. Un compuesto oligomérico de este tipo, un mimético de ARN que se ha demostrado que presenta unas propiedades de hibridación excelentes, se denomina ácido nucleico peptídico (PNA). En los compuestos de tipo PNA, el esqueleto del azúcar de un ARN se ha reemplazado por un esqueleto que contiene amidas, en particular un esqueleto de tipo aminoetilglicina. Las bases nucleotídicas se conservan y están unidas directa o indirectamente a átomos de nitrógeno aza de la porción amídica del esqueleto. Las patentes de EE. UU. representativas que describen la preparación de compuestos de tipo PNA incluyen, sin carácter limitante, las Pat. de EE. UU. N.°s 5.539.082, 5.714.331 y 5.719.262. Otras descripciones de compuestos de tipo PNA se pueden encontrar, por ejemplo, en Nielsen etal., Science, 1991,254, 1497-1500.
Algunos aspectos expuestos en la presente incluyen ARN con esqueletos de tipo fosforotioato y oligonucleósidos con esqueletos que contienen heteroátomos, y en particular --CH2--NH--CH2--, --CH2--N(CH3)--O--CH2--[que se conoce como un esqueleto de tipo metilen(metilimino) o MMI], --CH2--O--N(CH3)--CH2--, --CH2--N(CH3)--N(CH3)--CH2-- y --N(CH3)--CH2--CH2--[donde el esqueleto de tipo fosfodiéster nativo se representa como --O--P--O--CH2--] de la Pat. de EE. UU. N.° 5.489.677 a la que se ha hecho referencia anteriormente y los esqueletos de tipo amida de la Pat. de EE. UU. N.° 5.602.240 a la que se ha hecho referencia anteriormente. Los ARN que se exponen en la presente pueden tener las estructuras del esqueleto de tipo morfolino de la Pat. de EE. UU. N.° 5.036.506 a la que se ha hecho referencia anteriormente.
Los ARN modificados también pueden contener uno o más restos de azúcar sustituidos. Los ARNi, p. ej., ARNbc que se exponen en la presente pueden incluir uno de los siguientes grupos en la posición 2': OH; F; O-, S- o Nalquilo; O-, S- o N-alquenilo; O-, S- o N-alquinilo; o O-alquil-O-alquilo, donde el alquilo, alquenilo y alquinilo pueden ser un alquilo C1-C10, alquenilo y alquinilo C2-C10 sustituido o no sustituido. Las modificaciones adecuadas ilustrativas incluyen O[(CH2)nO] mCH3, O(CH2).nOCH3, O(CH2)nNH2, O(CH2)nCH3, O(CH2)nONH2 y O(CH2)nON[(CH2)nCH3)]2, donde n y m están comprendidos entre 1 y aproximadamente 10. En otros aspectos, los ARNbc pueden incluir uno de los siguientes grupos en la posición 2': alquilo inferior C1-C10, alquilo inferior sustituido, alcarilo, aralquilo, O-alcarilo u O-aralquilo, SH, SCH3, o Cn , Cl, Br, CN, CF3, OCF3, SOCH3, SO2CH3, ONO2, NO2, N3, NH2, heterocicloalquilo, heterocicloalcarilo, aminoalquilamino, polialquilamino, sililo sustituido, un grupo que escinde ARN, un grupo marcador, un intercalador, un grupo para mejorar las propiedades farmacocinéticas de un ARNi o un grupo para mejorar las propiedades farmacodinámicas de un ARNi, y otros sustituyentes con propiedades similares. La modificación puede incluir un 2'-metoxietoxi (2'-O--CH2CH2OCH3, también conocido como 2'-O-(2-metoxietilo) o 2'-MOE) (Martin et al., Helv. Chim. Acta, 1995, 78:486-504), es decir, un grupo alcoxi-alcoxi. Otra modificación ilustrativa es 2'-dimetilaminooxietoxi, es decir, un grupo O(CH2)2ON(CH3)2, también conocido como 2'-DMAOE, según se describe en los ejemplos de la presente más adelante, y 2'-dimetilaminoetoxietoxi (que también se conoce en la técnica como 2'-O-dimetilaminoetoxietilo o 2'-DMAEOE), es decir, 2'-O--CH2--O--CH2--N(CH2)2, que también se describe en los ejemplos de la presente más adelante.
Un agente de ARNi puede comprender uno o más nucleótidos (o nucleósidos) acíclicos (p. ej., aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 o más). La hebra sentido o la hebra antisentido, o la hebra sentido y la hebra antisentido, pueden incluir menos de cinco nucleótidos acíclicos por hebra (p. ej., cuatro, tres, dos o un nucleótido acíclico por hebra). El uno o más nucleótidos acíclicos se puede encontrar, por ejemplo, en la región bicatenaria de la hebra sentido o antisentido o de ambas hebras; en el extremo 5’, el extremo 3’, ambos extremos 5’ y 3’ de la hebra sentido o antisentido, o ambas hebras, del agente de ARNi. Uno o más nucleótidos acíclicos pueden estar presentes en las posiciones 1 a 8 de la hebra sentido o antisentido, o ambas. Uno o más nucleótidos acíclicos se pueden encontrar en la hebra antisentido en las posiciones 4 a 10 (p. ej., posiciones 6-8) desde el extremo 5’ de la hebra antisentido. El uno o más nucleótidos acíclicos se pueden encontrar en una o ambas protuberancias terminales 3’ del agente de ARNi.
La expresión “nucleótido acíclico” o “nucleósido acíclico”, tal como se utiliza en la presente se refiere a cualquier nucleótido o nucleósido que tiene un azúcar acíclico, p. ej., una ribosa acíclica. Un nucleótido o nucleósido acíclico ejemplar puede incluir una base nuecleotídica, p. ej., una base nucleotídica modificada o de origen natural (p. ej., una base nucleotídica como se describe en la presente). Un enlace entre cualquiera de los carbonos de la ribosa (C1, C2, C3, C4 o C5) puede estar independientemente o en combinación ausente del nucleótido. El enlace entre los carbonos C2-C3 del anillo de ribosa puede estar ausente, p. ej., un monómero de 2’-3’-seco-nucleótido acíclico). Se describen nucleótidos acíclicos ejemplares en el documento US 8.314.227. Por ejemplo, un nucleótido acíclico puede incluir cualquiera de los monómeros D-J en las Figuras 1-2 del documento US 8.314.227. El nucleótido acíclico puede incluir el siguiente monómero:

donde Base es una nucleobase, p. ej., una nucleobase modificada o de origen natural (p. ej., una nucleobase como se describe en la presente).
El nucleótido acíclico puede estar modificado o derivatizado, p. ej., acoplando el nucleótido acíclico a otro resto, p. ej., un ligando (p. ej., un GalNAc, un ligando de colesterol), un alquilo, una poliamina, un azúcar, un polipéptido, entre otros.
El agente de ARNi puede incluir uno o más nucleótidos acíclicos y uno o más LNA (p. ej., un LNA como se describe en la presente). Por ejemplo, uno o más nucleótidos acíclicos y/o uno o más LNA pueden estar presentes en la hebra sentido, la hebra antisentido o ambas. El número de nucleótidos acíclicos en una hebra puede ser idéntico o diferente del número de LNA en la hebra opuesta. La hebra sentido y/o la hebra antisentido pueden comprender menos de cinco LNA (p. ej., cuatro, tres, dos o un LNA) ubicados en la región bicatenaria o una protuberancia 3’. Uno o dos LNA pueden estar ubicados en la región bicatenaria o la protuberancia 3’ de la hebra sentido. Como alternativa, o en combinación, la hebra sentido y/o la hebra antisentido comprende menos de cinco nucleótidos acíclicos (p. ej., cuatro, tres, dos o un nucleótido acíclico) en la región bicatenaria o protuberancia 3’. La hebra sentido del agente de ARNi puede comprender uno o dos LNA en la protuberancia 3’ de la hebra sentido y uno o dos nucleótidos acíclicos en la región bicatenaria de la hebra antisentido (p. ej., en las posiciones 4 a 10 (p. ej., posiciones 6-8) desde el extremo 5’ de la hebra antisentido) del agente de ARNi.
La inclusión de uno o más nucleótidos acíclicos (solos o además del uno o más LNA) en el agente de ARNi puede dar como resultado uno o más (o la totalidad) de: (i) una reducción en un efecto inespecífico; (ii) una reducción en la
participación de hebra pasajera en la iARN; (iii) un incremento en la especificidad de la hebra guía por su ARNm diana; (iv) una reducción en un efecto inespecífico del microARN; (v) un incremento en la estabilidad; o (vi) un incremento en la resistencia a la degradación, de la molécla de ARNi.
Otras modificaciones incluyen 2'-metoxi (2'-OCH3), 2'-aminopropoxi (2'-OCH2CH2CH2NH2) y 2'-fluoro (2'-F). También se pueden realizar modificaciones similares en otras posiciones del ARN de un ARNi, particularmente la posición 3' del azúcar en el nucleótido terminal 3' o en ARNbc con uniones 2'-5' y la posición 5' del nucleótido terminal 5'. Los ARNi también pueden tener miméticos de azúcares tales como restos de tipo ciclobutilo en lugar del azúcar de tipo pentofuranosilo. Las patentes de EE. UU. representativas que describen la preparación de tales estructuras de azúcares modificadas incluyen, sin carácter limitante, las Pat. de EE. UU. N.°s 4.981.957, 5.118.800, 5.319.080, 5.359.044, 5.393.878, 5.446.137, 5.466.786, 5.514.785, 5.519.134, 5.567.811, 5.576.427, 5.591.722, 5.597.909, 5.610.300, 5.627.053, 5.639.873, 5.646.265, 5.658.873, 5.670.633 y 5.700.920, algunas de las cuales son propiedad en general de la presente solicitud..
Un ARNi también puede incluir sustituciones o modificaciones en la base nucleotídica (a la cual se hace referencia a menudo en la técnica simplemente como "base"). Las bases nucleotídicas "naturales" o "no modificadas", según se utilizan en la presente, incluyen las bases púricas adenina (A) y guanina (G), y las bases pirimidínicas timina (T), citosina (C) y uracilo (U). Las bases nucleotídicas modificadas incluyen otras bases nucleotídicas naturales y sintéticas tales como 5-metilcitosina (5-me-C), 5-hidroximetilcitosina, xantina, hipoxantina, 2-aminoadenina, 6-metiladenina y 6-metilguanina y otros derivados alquílicos de la adenina y la guanina, 2-propiladenina y 2-propilguanina y otros derivados alquílicos de la adenina y la guanina, 2-tiouracilo, 2-tiotimina y 2-tiocitosina, 5-halouracilo y 5-halocitosina, 5-propiniluracilo y 5-propinilcitosina, 6-azouracilo, 6-azocitosina y 6-azotimina, 5-uracilo (pseudouracilo), 4-tiouracilo, 8-halo-, 8-amino-, 8-tiol-, 8-tioalquil-, 8-hidroxil-adenina y -guanina y adeninas y guaninas con otros sustituyentes en la posición 8, 5-halo-, particularmente 5-bromo-, 5-trifluorometil-uracilo y -citosina y uracilos y citosinas con otros sustituyentes en la posición 5, 7-metilguanina y 7-metiladenina, 8-azaguanina y 8-azaadenina, 7-desazaguanina y 7-desazaadenina y 3-desazaguanina y 3-desazaadenina. Otras bases nucleotídicas incluyen las que se describen en la Pat. de Ee . UU. N.° 3.687.808, las que se describen en Modified Nucleosides in Biochemistry, Biotechnology and Medicine, Herdewijn, P. ed. Wiley-VCH, 2008; las que se describen en The Concise Encyclopedia Of Polymer Science And Engineering, páginas 858-859, Kroschwitz, J. L, ed. John Wiley & Sons, 1990, las que se describen en Englisch et al., Angewandte Chemie, International Edition, 1991, 30, 613 y las que se describen en Sanghvi, Y S., Capítulo 15, dsRNA Research and Applications, páginas 289-302, Crooke, S. T. y Lebleu, B., Ed., CRC Press, 1993. Algunas de estas bases nucleotídicas son particularmente útiles para incrementar la afinidad de unión de los compuestos oligoméricos que se exponen en la presente. Estas incluyen pirimidinas sustituidas en la posición 5, 6-azapirimidinas y purinas sustituidas en la posición N-2, N-6 y 0-6, que incluyen 2-aminopropiladenina, 5-propiniluracilo y 5-propinilcitosina. Se ha demostrado que las sustituciones de tipo 5-metilcitosina incrementan la estabilidad del dúplex de ácido nucleico de 0.6 a 1.2 °C (Sanghvi, Y. S., Crooke, S. T. y Lebleu, B., Eds., dsRNA Research and Applications, CRC Press, Boca Ratón, 1993, págs. 276-278) y son sustituciones de las bases ilustrativas, aún más particularmente cuando se combinan con modificaciones del azúcar de tipo 2'-O-metoxietilo.
Las patentes de EE. UU. representativas que describen la preparación de algunas de las bases nucleotídicas modificadas mencionadas anteriormente, así como también de otras bases nucleotídicas modificadas incluyen, sin carácter limitante, la Pat. de EE. UU. mencionada anteriormente N.° 3.687.808, así como también las Pat. de EE. UU. N.°s 4.845.205, 5.130.30, 5.134.066, 5.175.273, 5.367.066, 5.432.272, 5.457.187, 5.459.255, 5.484.908, 5.502.177, 5.525.711, 5.552.540, 5.587.469, 5.594.121, 5.596.091, 5.614.617, 5.681.941, 6.015.886, 6.147.200, 6.166.197, 6.222.025, 6.235.887, 6.380.368, 6.528.640, 6.639.062, 6.617.438, 7.045.610, 7.427.672 y 7.495.088 y la Pat. de EE. UU. N.25.750.692.
El ARN de un ARNi también se puede modificar para que incluya uno o más (p. ej., aproximadamente 1,2, 3, 4, 5, 6, 7, 8, 9, 10 o más) ácidos nucleicos bloqueados (LNA, por sus siglas en inglés)(también denominados en la presente como “nucleótidos bloqueados”). Un ácido nucleico bloqueado puede ser un nucleótido que tiene un resto de ribosa modificado en el cual el resto de ribosa comprende un puente adicional que conecta, p. ej., los carbonos 2' y 4'. Esta estructura "bloquea" de forma eficaz la ribosa en la conformación estructural 3'-endo. Se ha demostrado que la adición de ácidos nucleicos bloqueados a los ARNip incrementa la estabilidad del ARNip en suero, incremente la estabilidad térmica y reduce los efectos laterales (Elmen, J. et al., (2005) Nucleic Acids Research 33(1):439-447; Mook, OR. et al., (2007) Mol Canc Ther 6(3):833-843; Grunweller, A. et al., (2003) Nucleic Acids Research 31 (12):3185-3193).
Las Patentes de EE. UU. representativas que describen la preparación de nucleótidos de ácidos nucleicos bloqueados incluyen, sin carácter limitante, las siguientes: Pat. de EE. UU. N.°s 6.268.490, 6.670.461, 6.794.499, 6.998.484, 7.053.207, 7.084.125, 7.399.845 y 8.314.227. Los LNA ejemplares incluyen, sin carácter limitante, un nucleótido 2’,4’-C-metileno biciclo (remítase, por ejemplo, a Wengel et al., Publicación Internacional PCT N.° WO 00/66604 y WO 99/14226).
Los agentes de ARNi pueden incluir uno o más (p. ej., aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 o más) nucleótidos con fijación de G (G-clamp). Un nucleótido con fijación de G es un análogo de citosina modificado donde las modificaciones confieren la capacidad de formar enlaces de hidrógeno tanto entre las caras de Watson-Crick
como las de Hoogsteen de una guanina complementaria dentro de un dúplex, remítase, por ejemplo, a Lin y Matteucci, 1998, J. Am. Chem. Soc., 120, 8531-8532. Una única sustitución de un análogo con fijación de G dentro de un oligonucleótido puede dar como resultado una mejora sustancial de estabilidad térmica helicoidal y discriminación de apareamientos erróneos cuando se hibrida con oligonucleótidos complementarios. La inclusión de tales nucleótidos en las moléculas de ARNi puede dar como resultado una mejora de la afinidad y especificidad por las dianas de ácido nucleico, secuencias complementarias o hebras molde.
Las modificaciones potencialmente estabilizantes en los extremos de las moléculas de ARN pueden incluir N-(acetilaminocaproil)-4-hidroxiprolinol (Hip-C6-NHAc), N-(caproil-4-hidroxiprolinol (Hip-C6), N-(acetil-4-hidroxiprolinol (Hip-NHAc), timidin-2'-0-desoxitimidina (éter), N-(aminocaproil)-4-hidroxiprolinol (Hip-C6-amino), 2-docosanoiluridin-3"- fosfato, base dT invertida (idT) y otras. La descripción de esta modificación se puede encontrar en la Publicación de PCT N.2 WO 2011/005861.
Motivos del ARNi
La secuencia de la hebra sentido se puede representar mediante la fórmula (I):
5' np-Na-(X X X )i-Nb-Y Y Y -Nb-(Z Z Z )j-Na-nq 3' (I)
donde:
i y j son cada uno independientemente 0 o 1;
p y q son cada uno independientemente 0-6;
cada Na representa independientemente una secuencia oligonucleotídica que comprende 0-25 nucleótidos modificados, comprendiendo cada secuencia al menos dos nucleótidos modificados de forma diferente; cada Nb representa independientemente una secuencia oligonucleotídica que comprende 0-10 nucleótidos modificados;
cada np y nq representa independientemente un nucleótido protuberante;
donde Nb e Y no tienen la misma modificación; y
XXX, YYY y ZZZ representan cada uno independientemente un motivo de tres modificaciones idénticas en tres nucleótidos consecutivos. Preferentemente, YYY son todos nucleótidos con la modificación 2’-F.
Na y/o Nb pueden comprender modificaciones de patrón alterno.
El motivo YYY se pueden encuentrar en el sitio de escisión de la hebra sentido o cerca de este. Por ejemplo, cuando el agente de iARN tiene una región dúplex con una longitud de 17-23 nucleótidos, el motivo YYY se puede encontrar en el sitio de escisión o cerca de este (p. ej.: se puede encontrar en las posiciones 6, 7, 8; 7, 8, 9; 8, 9, 10; 9, 10, 11; 10, 11, 12 u 11, 12, 13) de la hebra sentido, empezando a contar a partir del primer nucleótido del extremo 5' u, opcionalmente, empezando a contar en el primer nucleótido apareado dentro de la región dúplex, a partir del extremo 5'.
i puede ser 1 y j puede ser 0, o i puede ser 0 y j puede ser 1, o tanto i como j pueden ser 1. Por consiguiente, la hebra sentido se puede representar mediante las siguientes fórmulas:
5' np-Na-YYY-Nb-ZZZ-Na-nq 3' (Ib);
5' np-Na-XXX-Nb-YYY-Na-nq 3' (Ic); o
5' np-Na-XXX-Nb-YYY-Nb-ZZZ-Na-nq 3' (Id).
Cuando la hebra sentido se representa mediante la fórmula (Ib), Nb representa una secuencia oligonucleotídica que comprende 0-10, 0-7, 0-5, 0-4, 0-2 o 0 nucleótidos modificados. Cada Na puede representar independientemente una secuencia oligonucleotídica que comprende 2-20, 2-15 o 2-10 nucleótidos modificados.
Cuando la hebra sentido se representa como la fórmula (Ic), Nb representa una secuencia oligonucleotídica que comprende 0-10, 0-7, 0-5, 0-4, 0-2 o 0 nucleótidos modificados. Cada Na puede representar independientemente una secuencia oligonucleotídica que comprende 2-20, 2-15 o 2-10 nucleótidos modificados.
Cuando la hebra sentido se representa como la fórmula (Id), cada Nb representa independientemente una secuencia oligonucleotídica que comprende 0-10, 0-7, 0-5, 0-4, 0-2 o 0 nucleótidos modificados. Preferentemente, Nb es 0, 1,2, 3, 4, 5 o 6. Cada Na puede representar independientemente una secuencia oligonucleotídica que comprende 2-20, 2 15 o 2-10 nucleótidos modificados.
Cada uno de los grupos X, Y y Z puede ser igual o diferente de los demás.
i puede ser 0 y j puede ser 0, y la hebra sentido se puede representar mediante la fórmula:
5' np-Na-YYY- Na-nq 3' (Ia).
Cuando la hebra sentido se representa mediante la fórmula (Ia), cada Na puede representar independientemente una secuencia oligonucleotídica que comprende 2-20, 2-15 o 2-10 nucleótidos modificados.
La secuencia de la hebra antisentido del ARNi se puede representar mediante la fórmula (II):
5' nq’-Na'-(Z’Z'Z')k-Nb'-YTY'-Nb'-(X'XX)l-N a-np' 3' (II)
donde:
k y l son cada uno independientemente 0 o 1;
p' y q' son cada uno independientemente 0-6;
cada Na' representa independientemente una secuencia oligonucleotídica que comprende 0-25 nucleótidos modificados, comprendiendo cada secuencia al menos dos nucleótidos modificados de forma diferente; cada Nb' representa independientemente una secuencia oligonucleotídica que comprende 0-10 nucleótidos modificados;
cada np' y nq' representa independientemente un nucleótido protuberante;
donde Nb' e Y' no tienen la misma modificación;
y
X'X'X', Y'Y'Y' y Z'Z'Z' representan cada uno independientemente un motivo de tres modificaciones idénticas en tres nucleótidos consecutivos.
Na' y/o Nb' pueden comprender modificaciones de patrón alterno.
El motivo Y'Y'Y' se encuentra en el sitio de escisión de la hebra antisentido o cerca de este. Por ejemplo, cuando el agente de iARN tiene una región dúplex con una longitud de 17-23 nucleótidos, el motivo Y'Y'Y se puede encontrar en las posiciones 9, 10, 11; 10, 11, 12; 11, 12, 13; 12, 13, 14 o 13, 14, 15 de la hebra antisentido, empezando a contar a partir del primer nucleótido del extremo 5' u, opcionalmente, empezando a contar en el primer nucleótido apareado dentro de la región dúplex, a partir del extremo 5'. Preferentemente, el motivo Y'Y'Y' se encuentra en las posiciones 11, 12, 13.
El motivo Y'Y'Y' puede consistir en que todos los nucleótidos presentan la modificación 2’-OMe.
k puede ser 1 y l puede ser 0, o k puede ser 0 y l puede ser 1, o tanto k como l pueden ser 1.
Por consiguiente, la hebra antisentido se puede representar mediante las siguientes fórmulas:
5' nq-Na'-Z'Z'Z'-Nb'-Y'Y'Y'-Na'-np’ 3' (IIb);
5' nq-Na'-Y'Y'Y'-Nb'-X'X'X'-np’ 3' (IIc);
o
5' nq-Na'- Z'Z'Z'-Nb'-Y'Y'Y'-Nb'- X'X'X'-Na'-np’ 3' (IId).
Cuando la hebra antisentido se representa mediante la fórmula (IIb), Nb' representa una secuencia oligonucleotídica que comprende 0-10, 0-7, 0-5, 0-4, 0-2 o 0 nucleótidos modificados. Cada Na' representa independientemente una secuencia oligonucleotídica que comprende 2-20, 2-15 o 2-10 nucleótidos modificados.
Cuando la hebra antisentido se representa como la fórmula (IIc), Nb' representa una secuencia oligonucleotídica que comprende 0-10, 0-7, 0-5, 0-4, 0-2 o 0 nucleótidos modificados. Cada Na' representa independientemente una secuencia oligonucleotídica que comprende 2-20, 2-15 o 2-10 nucleótidos modificados.
Cuando la hebra antisentido se representa como la fórmula (IId), cada Nb' representa independientemente una secuencia oligonucleotídica que comprende 0-10, 0-7, 0-5, 0-4, 0-2 o 0 nucleótidos modificados. Cada Na' representa independientemente una secuencia oligonucleotídica que comprende 2-20, 2-15 o 2-10 nucleótidos modificados. Preferentemente, Nb es 0, 1, 2, 3, 4, 5 o 6.
k puede ser 0 y l puede ser 0, y la hebra antisentido se puede representar mediante la fórmula:
5' np-Na-Y’Y’Y’- Na-nq’ 3' (Ia).
Cuando la hebra antisentido se representa como la fórmula (Ila), cada Na' representa independientemente una secuencia oligonucleotídica que comprende 2-20, 2-15 o 2-10 nucleótidos modificados.
Cada uno de los grupos X', Y' y Z' puede ser igual o diferente de los demás.
Cada nucleótido de la hebra sentido y la hebra antisentido se puede modificar independientemente con LNA, HNA, CeNA, 2’-metoxietilo, 2’-O-metilo, 2’-O-alilo, 2’-C-alilo, 2’-hidroxilo o 2’-fluoro. Por ejemplo, cada nucleótido de la hebra sentido y la hebra antisentido se modifica independientemente con 2’-O-metilo o 2’-fluoro. Cada X, Y, Z, X', Y' y Z', en particular, puede representar una modificación de tipo 2’-O-metilo o una modificación de tipo 2’-fluoro.
La hebra sentido del agente de iARN puede contener un motivo YYY que se encuentre en las posiciones 9, 10 y 11 de la hebra cuando la región dúplex tiene 21 nt, empezando a contar a partir del primer nucleótido del extremo 5' u, opcionalmente, empezando a contar en el primer nucleótido apareado dentro de la región dúplex, a partir del extremo 5'; e Y representa una modificación de tipo 2’-F. La hebra sentido puede contener adicionalmente un motivo XXX o motivos ZZZ como modificaciones laterales en el extremo opuesto de la región dúplex; y cada motivo XXX y ZZZ representa independientemente una modificación de tipo 2’-OMe o una modificación de tipo 2’-F.
La hebra antisentido puede contener un motivo Y'Y'Y' que se encuentre en las posiciones 11, 12, 13 de la hebra, empezando a contar a partir del primer nucleótido del extremo 5' u, opcionalmente, empezando a contar en el primer nucleótido apareado dentro de la región dúplex, a partir del extremo 5'; e Y' representa una modificación de tipo 2’-O-metilo. La hebra antisentido puede contener adicionalmente un motivo X'X'X' o motivos Z'Z'Z' como modificaciones laterales en el extremo opuesto de la región dúplex; y cada motivo X'X'X' y Z'Z'Z' representa independientemente una modificación de tipo 2’-OMe o una modificación de tipo 2’-F.
La hebra sentido representada mediante cualquiera de las fórmulas anteriores (Ia), (Ib), (Ic) y (Id) forma un dúplex con una hebra antisentido representada mediante cualquiera de las fórmulas (IIa), (IIb), (IIc) y (IId), respectivamente. Por consiguiente, los agentes de iARN que se pueden utilizar en los métodos de la divulgación pueden comprender una hebra sentido y una hebra antisentido, teniendo cada hebra 14-30 nucleótidos, donde el dúplex de iARN está representado mediante la fórmula (III):
sentido:
antisentido: 
donde:
i, j, k, y l son cada uno independientemente 0 o 1;
p, p’, q y q' son cada uno independientemente 0-6;
cada Na y Na' representa independientemente una secuencia oligonucleotídica que comprende 0-25 nucleótidos modificados, comprendiendo cada secuencia al menos dos nucleótidos modificados de forma diferente; cada Nb y Nb' representa independientemente una secuencia oligonucleotídica que comprende 0-10 nucleótidos modificados;
donde
cada np’, np, nq’ y nq, cada uno de los cuales puede estar presente o no, representa independientemente un nucleótido protuberante; y
XXX, YYY, ZZZ, X'X'X', Y'Y'Y' y Z'Z'Z' representan cada uno independientemente un motivo de tres modificaciones idénticas en tres nucleótidos consecutivos.
i puede ser 0 y j puede ser 0; o i puede ser 1 y j puede ser 0; o i puede ser 0 y j puede ser 1; o tanto i como j pueden ser 0; o tanto i como j pueden ser 1. k puede ser 0 y l puede ser 0; o k puede ser 1 y l puede ser 0; o k puede ser 0 y l puede ser 1; o tanto k como l pueden ser 0; o tanto k como l pueden ser 1.
Las combinaciones ilustrativas de la hebra sentido y la hebra antisentido para formar un dúplex de iARN incluyen las fórmulas siguientes:

5' np-Na- X X X -Nb -Y Y Y - Na-nq 3'
3' np'-Na-X'X'X'-Nb'-Y'Y'Y'-Na-nq' 5' (IIIc)
5' np -Na -X X X -Nb-Y Y Y -Nb- Z Z Z -Na-nq 3'
3' np'-Na'-X'X'X'-Nb-Y'Y'Y'-Nb-Z'Z'Z'-Na-nq' 5' (IIId)
Cuando el agente de iARN se representa mediante la fórmula (IIIa), cada Na representa independientemente una secuencia oligonucleotídica que comprende 2-20, 2-15 o 2-10 nucleótidos modificados.
Cuando el agente de iARN se representa mediante la fórmula (IIIb), cada Nb representa independientemente una secuencia oligonucleotídica que comprende 1-10, 1-7, 1-5 o 1-4 nucleótidos modificados. Cada Na representa independientemente una secuencia oligonucleotídica que comprende 2-20, 2-15 o 2-10 nucleótidos modificados. Cuando el agente de iARN se representa como la fórmula (IIIc), cada Nb, Nb' representa independientemente una secuencia oligonucleotídica que comprende 0-10, 0-7, 0-5, 0-4, 0-2 o 0 nucleótidos modificados. Cada Na representa independientemente una secuencia oligonucleotídica que comprende 2-20, 2-15 o 2-10 nucleótidos modificados. Cuando el agente de iARN se representa como la fórmula (IIId), cada Nb, Nb' representa independientemente una secuencia oligonucleotídica que comprende 0-10, 0-7, 0-5, 0-4, 0-2 o 0 nucleótidos modificados. Cada Na, Na' representa independientemente una secuencia oligonucleotídica que comprende 2-20, 2-15 o 2-10 nucleótidos modificados. Cada uno de los grupos Na, Na', Nb y Nb' comprende independientemente modificaciones de patrón alterno.
Cada uno de los grupos X, Y y Z en las fórmulas (III), (IIIa), (IIIb), (IIIc) y (IIId) puede ser igual o diferente de los demás.
Cuando el agente de iARN se representa mediante la fórmula (III), (IIIa), (IIIb), (IIIc) y (IIId), al menos uno de los nucleótidos Y puede formar un par de bases con uno de los nucleótidos Y'. Como alternativa, al menos dos de los nucleótidos Y forman pares de bases con los nucleótidos Y' correspondientes; o los tres nucleótidos Y forman todos ellos pares de bases con los nucleótidos Y' correspondientes.
Cuando el agente de iARN se representa mediante la fórmula (IIIb) o (IIId), al menos uno de los nucleótidos Z puede formar un par de bases con uno de los nucleótidos Z'. Como alternativa, al menos dos de los nucleótidos Z forman pares de bases con los nucleótidos Z' correspondientes; o los tres nucleótidos Z forman todos ellos pares de bases con los nucleótidos Z' correspondientes.
Cuando el agente de iARN se representa mediante la fórmula (IIIc) o (IIId), al menos uno de los nucleótidos X puede formar un par de bases con uno de los nucleótidos X'. Como alternativa, al menos dos de los nucleótidos X forman pares de bases con los nucleótidos X' correspondientes; o los tres nucleótidos X forman todos ellos pares de bases con los nucleótidos X' correspondientes.
La modificación en el nucleótido Y puede ser diferente de la modificación en el nucleótido Y', la modificación en el nucleótido Z puede ser diferente de la modificación en el nucleótido Z' y/o la modificación en el nucleótido X puede ser diferente de la modificación el nucleótido X'.
Cuando el agente de iARN se representa mediante la fórmula (IIId), las modificaciones en Na pueden ser modificaciones de tipo 2,-O-metilo o 2'-fluoro. Asimismo, cuando el agente de iARN se representa mediante la fórmula (IIId), las modificaciones en Na pueden ser modificaciones de tipo 2,-O-metilo o 2'-fluoro, np' > 0 y al menos un np' puede estar unido a un nucleótido contiguo mediante una unión de tipo fosforotioato. Aún más, cuando el agente de iARN se representa mediante la fórmula (IIId), las modificaciones en Na pueden ser modificaciones de tipo 2,-O-metilo o 2'-fluoro, np' > 0 y al menos un np' puede estar unido a un nucleótido contiguo mediante una unión de tipo fosforotioato, y la hebra sentido puede estar conjugada con uno o más derivados de tipo GalNAc unidos mediante un conector ramificado bivalente o trivalente. Asimismo, cuando el agente de iARN se representa mediante la fórmula (IIId), las modificaciones en Na pueden ser modificaciones de tipo 2,-O-metilo o 2'-fluoro, np' > 0 y al menos un np' está unido a un nucleótido contiguo mediante una unión de tipo fosforotioato, la hebra sentido puede comprender al menos una unión de tipo fosforotioato y la hebra sentido puede estar conjugada con uno o más derivados de tipo GalNAc unidos mediante un conector ramificado bivalente o trivalente.
Cuando el agente de iARN se puede representar mediante la fórmula (IIIa), las modificaciones en Na son modificaciones de tipo 2,-O-metilo o 2'-fluoro, np' > 0 y al menos un np' está unido a un nucleótido contiguo mediante una unión de tipo fosforotioato, la hebra sentido comprende al menos una unión de tipo fosforotioato y la hebra sentido está conjugada con uno o más derivados de tipo GalNAc unidos mediante un conector ramificado bivalente o trivalente.
El agente de iARN puede ser un multímero que contiene al menos dos dúplex representados mediante la fórmula (III), (IIIa), (IIIb), (IIIc) y (IIId), donde los dúplex están conectados mediante un conector. El conector puede ser
escindible o no escindible. Opcionalmente, el multímero comprende además un ligando. Cada uno de los dúplex puede tener como diana el mismo gen o dos genes diferentes, o cada uno de los dúplex puede tener como diana el mismo gen en dos sitios diana diferentes.
El agente de iARN puede ser un multímero que contiene tres, cuatro, cinco, seis o más dúplex representados mediante la fórmula (III), (IIIa), (IIIb), (IIIc) y (IIId), donde los dúplex están conectados mediante un conector. El conector puede ser escindible o no escindible. Opcionalmente, el multímero comprende además un ligando. Cada uno de los dúplex puede tener como diana el mismo gen o dos genes diferentes, o cada uno de los dúplex puede tener como diana el mismo gen en dos sitios diana diferentes.
Dos agentes de iARN representados mediante la fórmula (III), (IIIa), (IIIb), (IIIc) y (IIId) pueden estar unidos el uno con el otro en el extremo 5’, y uno o ambos extremos 3’ pueden estar conjugados opcionalmente con un ligando. Cada uno de los agentes puede tener como diana el mismo gen o dos genes diferentes, o cada uno de los agentes puede tener como diana el mismo gen en dos sitios diana diferentes.
Conjugados de ARNi
Los agentes de ARNi descritos en la presente pueden adoptar la forma de conjugados. El conjugado se puede unir en cualquier posición adecuada de la molécula de ARNi, p. ej., en el extremo 3' o el extremo 5' de la hebra sentido o antisentido. Los conjugados se unen opcionalmente mediante un conector.
Un agente de ARNi descrito en la presente puede estar unido químicamente a uno o más ligandos, restos o conjugados, los cuales pueden conferir una funcionalidad, p. ej., ejerciendo un efecto sobre (p. ej., potenciando) la actividad, la distribución celular o la captación celular del ARNi. Tales restos incluyen, sin carácter limitante, restos lipídicos tales como un resto de colesterol (Letsinger et al., Proc. Nati. Acid. Sci. USA, 1989, 86: 6553-6556), ácido cólico (Manoharan et al., Biorg. Med. Chem. Let., 1994, 4:1053-1060), un tioéter, p. ej., beril-S-tritiltiol (Manoharan et al., Ann. N.Y. Acad. Sci., 1992, 660:306-309; Manoharan et al., Biorg. Med. Chem. Let., 1993, 3:2765-2770), un tiocolesterol (Oberhauser et al., Nucl. Acids Res., 1992, 20:533-538), una cadena alifática, p. ej., residuos de dodecandiol o undecilo (Saison-Behmoaras et al., EMBO J, 1991, 10:1111-1118; Kabanov et al., FEBS Lett., 1990, 259:327-330; Svinarchuk et al., Biochimie, 1993, 75:49-54), un fosfolípido, p. ej., dihexadecil-rac-glicerol o 1,2-di-O-hexadecil-rac-glicero-3-fosfonato de trietilamonio (Manoharan et al., Tetrahedron Lett., 1995, 36:3651-3654; Shea et al., Nucl. Acids Res., 1990, 18:3777-3783), una poliamina o una cadena de polietilenglicol (Manoharan et al., Nucleosides & Nucleotides, 1995, 14:969-973) o ácido adamantanacético (Manoharan et al., Tetrahedron Lett., 1995, 36:3651-3654), un resto de palmitilo (Mishra et al., Biochim. Biophys. Acta, 1995, 1264:229-237), o un resto de octadecilamina o hexilaminocarboniloxicolesterol (Crooke et al., J. Pharmacol. Exp. Ther., 1996, 277:923-937).
Un ligando puede alterar la distribución, la diana o la vida útil del agente de ARNi en el cual se incorpora. Un ligando puede proporcionar una afinidad mejorada para una diana seleccionada, p. ej., una molécula, una célula o tipo celular, un compartimento, p. ej., un compartimento celular o de un órgano, un tejido, un órgano o una región del cuerpo en comparación, p. ej., con una especie que carezca del ligando. Los ligandos habituales no formarán parte del apareamiento del dúplex en un ácido nucleico que forme un dúplex.
Los ligandos pueden incluir una sustancia de origen natural tal como una proteína (p. ej., albúmina de suero humano (HSA), una lipoproteína de baja densidad (LDL) o globulina); un carbohidrato (p. ej., un dextrano, pululano, chitina, chitosano, inulina, ciclodextrina o ácido hialurónico) o un lípido. El ligando también puede ser una molécula sintética o recombinante tal como un polímero sintético, p. ej., un ácido poliamínico sintético. Los ejemplos de ácidos poliamínicos incluyen un ácido poliamínico que es una polilisina (PLL), ácido poli-L-aspártico, ácido poli-L-glutámico, copolímero de estireno-anhídrido del ácido maleico, copolímero de poli(L-láctido-co-glicólido), copolímero de éter divinílico-anhídrido maleico, copolímero de N-(2-hidroxipropil)metacrilamida (HMPA), polietilenglicol (PEG), alcohol polivinílico (PVA), poliuretano, ácido poli(2-etilacrílico), polímeros de N-isopropilacrilamida o polifosfazina. Los ejemplos de poliaminas incluyen: polietilenimina, polilisina (PLL), espermina, espermidina, poliamina, pseudopéptidopoliamina, poliamina peptidomimética, poliamina dendrimérica, arginina, amidina, protamina, lípido catiónico, porfirina catiónica, sal cuaternaria de una poliamina o un péptido con hélice a.
Los ligandos también pueden incluir grupos de direccionamiento, p. ej., un agente de direccionamiento hacia una célula o tejido, p. ej., una lectina, glicoproteína, lípido o proteína, p. ej., un anticuerpo, que se une a un tipo celular especificado tal como una célula renal. Un grupo de direccionamiento puede ser una tirotropina, melanotropina, lectina, glicoproteína, proteína A surfactante, carbohidrato de mucina, lactosa multivalente, galactosa multivalente, N-acetilgalactosamina, N-acetilglucosamina, manosa multivalente, fucosa multivalente, poliaminoácidos glicosilados, galactosa multivalente, transferrina, bisfosfonato, poliglutamato, poliaspartato, un lípido, colesterol, un esteroide, ácido biliar, folato, vitamina B12, biotina, o un péptido RGD o un mimético de un péptido RGD.
El ligando puede ser un ligando de tipo GalNAc que comprende uno o más derivados de tipo N-acetilgalactosamina (GalNAc). En la sección titulada "Conjugados de carbohidratos" se proporciona una descripción adicional de los ligandos de tipo GalNAc.
Otros ejemplos de ligandos incluyen tintes (p. ej., acridinas), agentes reticulantes (p. ej., psoraleno, mitomicina C), porfirinas (TPPC4, texafirina, Sapfirina), hidrocarburos aromáticos policíclicos (p. ej., fenazina, dihidrofenazina),
endonucleasas artificiales (p. ej. EDTA), moléculas lipófilas, p. ej., colesterol, ácido cólico, ácido adamantanacético, ácido 1-pirenobutírico, dihidrotestosterona, 1,3-bis-O(hexadecil)glicerol, un grupo geraniloxihexilo, hexadecilglicerol, borneol, mentol, 1,3-propanodiol, un grupo heptadecilo, ácido palmítico, ácido mirístico, ácido O3-(oleoil)litocólico, ácido O3-(oleoil)colénico, dimetoxitritilo o fenoxazina) y conjugados de péptidos (p. ej., péptido antennapedia, péptido Tat), agentes alquilantes, fosfato, amino, mercapto, PEG (p. ej., PEG-40K), MPEG, [MPEG]2, poliamino, alquilo, alquilo sustituido, marcadores radiomarcados, enzimas, haptenos (p. ej., biotina), potenciadores del transporte/la absorción (p. ej., aspirina, vitamina E, ácido fólico), ribonucleasas sintéticas (p. ej., imidazol, bisimidazol, histamina, agregados de imidazol, conjugados de acridina-imidazol, complejos de tetraazamacrociclos con Eu3+), dinitrofenilo, HRP o AP.
Los ligandos pueden ser proteínas, p. ej., glicoproteínas, o péptidos, p. ej., macromoléculas con una afinidad específica por un coligando, o anticuerpos, p. ej., un anticuerpo que se una a un tipo celular especificado tal como una célula cancerosa, célula endotelial o célula ósea. Los ligandos también pueden incluir hormonas y receptores de hormonas. También pueden incluir especies no peptídicas tales como lípidos, lectinas, carbohidratos, vitaminas, cofactores, lactosa multivalente, galactosa multivalente, N-acetilgalactosamina, N-acetilglucosamina, manosa multivalente o fucosa multivalente. El ligando puede ser, por ejemplo, un lipopolisacárido, un activador de la cinasa MAP p38 o un activador de NF-kB.
El ligando puede ser una sustancia, p. ej., un fármaco, que puede incrementar la captación del agente de ARNi en la célula, por ejemplo, alterando el citoesqueleto de la célula, p. ej., alterando los microtúbulos, microfilamentos y/o filamentos intermedios de la célula. El fármaco puede ser, por ejemplo, taxon, vincristina, vinblastina, citocalasina, nocodazol, japlaquinolida, latrunculina A, faloidina, swinholida A, indanocina o mioservina.
Un ligando unido a un ARNi según se describe en la presente puede actuar como un modulador farmacocinético (modulador PK, por sus siglas en inglés). Los moduladores PK incluyen lipófilos, ácidos biliares, esteroides, análogos de fosfolípidos, péptidos, agentes de unión a proteínas, PEG, vitaminas, etc. Los moduladores PK ilustrativos incluyen, sin carácter limitante, colesterol, ácidos grasos, ácido cólico, ácido litocólico, dialquilglicéridos, diacilglicérido, fosfolípidos, esfingolípidos, naproxeno, ibuprofeno, vitamina E, biotina, etc. Existe constancia de que los oligonucleótidos que comprenden una serie de uniones de tipo fosforotioato también se unen a las proteínas del suero, por lo tanto, los oligonucleótidos cortos, p. ej., oligonucleótidos de aproximadamente 5 bases, 10 bases, 15 bases o 20 bases, que comprenden múltiples uniones de tipo fosforotioato en el esqueleto también se pueden utilizar en la presente divulgación como ligandos (p. ej., como ligandos moduladores PK). Además, los aptámeros que se unen a los componentes del suero (p. ej., proteínas del suero) también son adecuados para ser utilizados como ligandos moduladores PK en los aspectos descritos en la presente.
Los oligonucleótidos conjugados con ligandos de la divulgación se pueden sintetizar utilizando un oligonucleótido que contenga una funcionalidad reactiva como sustituyente, tal como la derivada de la unión de una molécula conectora en el oligonucleótido (que se describe más adelante). Este oligonucleótido reactivo se puede hacer reaccionar directamente con ligandos que se pueden adquirir de proveedores comerciales, ligandos que se sintetizan y que contienen un grupo protector cualquiera de entre una gama de grupos protectores, o ligandos que tienen un resto conector unido a ellos.
Los oligonucleótidos utilizados en los conjugados de la presente divulgación se pueden preparar de forma conveniente y rutinaria mediante la técnica bien establecida de síntesis en fase sólida. Existen varios proveedores que comercializan el equipo para realizar una síntesis de este tipo, los cuales incluyen, por ejemplo, Applied Biosystems (Foster City, Calif.). Adicionalmente o de forma alternativa, se pueden emplear otros medios para este tipo de síntesis conocidos en la técnica. También existe constancia del uso de técnicas similares para preparar otros oligonucleótidos, tales como los fosforotioatos y derivados alquilados.
En los oligonucleótidos conjugados con ligandos y los nucleósidos unidos específicamente a secuencias que contienen moléculas que son ligandos de la presente divulgación, los oligonucleótidos y oligonucleósidos se pueden acoplar en un sintetizador de ADN adecuado utilizando precursores de nucleótidos o nucleósidos estándar, o precursores de conjugados de nucleótidos o nucleósidos que ya contengan el resto conector, precursores de conjugados de nucleótidos- o nucleósidos-ligando que ya contengan la molécula de ligando, o bloques estructurales que no sean nucleósidos y que contengan el ligando.
Cuando se utilizan precursores de conjugados de nucleótidos que ya contienen un resto conector, normalmente se completa la síntesis de los nucleósidos unidos específicamente a secuencias y a continuación la molécula de ligando se hace reaccionar con el resto conector para formar el oligonucleótido conjugado con el ligando. Los oligonucleótidos o nucleósidos unidos de la presente divulgación se pueden sintetizar con un sintetizador automatizado utilizando fosforamiditos derivados de conjugados de nucleósido-ligando, además de los fosforamiditos estándar y fosforamiditos no estándar que se pueden adquirir de proveedores comerciales y que se utilizan de forma rutinaria en la síntesis de oligonucleótidos.
Conjugados de lípidos
El ligando puede ser un lípido o una molécula basada en un lípido. Este lípido o esta molécula basada en un lípido se puede unir normalmente a una proteína del suero tal como la albúmina de suero humano (HSA). Un ligando de unión a HSA permite distribuir el conjugado en un tejido diana, p. ej., un tejido diana no renal del cuerpo. Por ejemplo, el tejido diana puede ser el hígado, incluidas las células parenquimales del hígado. También se pueden utilizar como ligandos otras moléculas que se puedan unir a HSA. Por ejemplo, se puede utilizar la neproxina o la aspirina. Un lípido o un ligando basado en un lípido puede (a) incrementar la resistencia a la degradación del conjugado, (b) incrementar el direccionamiento o el transporte hacia una célula o membrana celular diana y/o (c) se puede utilizar para ajustar la unión a una proteína del suero, p. ej., HSA.
Un ligando basado en un lípido se puede utilizar para modular, p. ej., controlar (p. ej., inhibir) la unión del conjugado a un tejido diana. Por ejemplo, un lípido o un ligando basado en un lípido que se una a HSA con una unión más fuerte presentará una menor probabilidad de ser dirigido hacia el riñón y, por consiguiente, presentará una menor probabilidad de ser expulsado del cuerpo. Un lípido o un ligando basado en un lípido que se una a HSA con una unión menos fuerte se puede utilizar para dirigir el conjugado hacia el riñón.
El ligando basado en un lípido puede unirse a HSA. Por ejemplo, el ligando se puede unir a HSA con una afinidad suficiente de manera que la distribución del conjugado en un tejido no renal se vea potenciada. Sin embargo, normalmente la afinidad no es tan fuerte como para que la unión ligando-HSA no se pueda revertir.
El ligando basado en un lípido puede unirse a HSA débilmente o no se une en absoluto, de manera que la distribución del conjugado en el riñón se vea potenciada. También se pueden utilizar otros restos que ejerzan un direccionamiento hacia las células renales, en lugar o además del ligando basado en un lípido.
El ligando puede ser un resto, p. ej., una vitamina, que es absorbido por una célula diana, p. ej., una célula proliferante. Estos son particularmente útiles para tratar trastornos caracterizados por la proliferación de células no deseadas, p. ej., células cancerosas, p. ej., de tipo maligno o no maligno. Las vitaminas ilustrativas incluyen la vitamina A, E y K. Otras vitaminas ilustrativas incluyen la vitamina B, p. ej., ácido fólico, B12, riboflavina, biotina, piridoxal u otras vitaminas o nutrientes absorbidos por las células cancerosas. También se incluyen HSA y lipoproteínas de baja densidad (LDL).
Agentes de permeación celular
En otro aspecto, el ligando es un ligando de permeación celular tal como un ligando de permeación celular helicoidal. El agente puede ser anfifático. Un agente ilustrativo es un péptido tal como tat o antennopedia. Si el agente es un péptido, este puede ser un péptido modificado, que incluye un peptidomimético, invertómeros, conectores no peptídicos o pseudopeptídicos y el uso de aminoácidos D. El agente helicoidal es normalmente un agente de hélice a y puede tener una fase lipófila y lipófoba.
El ligando puede ser un péptido o peptidomimético. Un peptidomimético (también denominado en la presente como oligopeptidomimético) es una molécula capaz de plegarse en una estructura tridimensional definida similar a la de un péptido natural. La unión de péptidos y peptidomiméticos a agentes de ARNi puede afectar a la distribución farmacocinética del ARNi, por ejemplo, potenciando el reconocimiento y la absorción celular. El resto peptídico o peptidomimético puede tener una longitud de aproximadamente 5-50 aminoácidos, p. ej., una longitud de aproximadamente 5, 10, 15, 20, 25, 30, 35, 40, 45 o 50 aminoácidos.
Un péptido o peptidomimético puede ser, por ejemplo, un péptido de permeación celular, un péptido catiónico, un péptido anfifático o un péptido hidrófobo (p. ej., constituido principalmente por Tyr, Trp o Phe). El resto peptídico puede ser un péptido dendrimérico, un péptido de movimiento restringido o un péptido reticulado. En otra alternativa, el resto peptídico puede incluir una secuencia de translocación de membrana hidrófoba (MTS, por sus siglas en inglés). Un péptido que contiene una MTS hidrófoba a modo de ejemplo es RFGF, que tiene la secuencia de aminoácidos AAVALLPAVLLALLAP (SEQ ID NO:3367). Un análogo de RfGF (p. ej., la secuencia de aminoácidos AALLPVLLAAP (SEQ ID NO:3368)) que contenga una MTS hidrófoba también puede ser un resto de direccionamiento. El resto peptídico puede ser un péptido de "suministro", el cual puede transportar moléculas polares grandes, incluidos péptidos, oligonucleótidos y proteínas, a través de las membranas celulares. Por ejemplo, se ha demostrado que la proteína Tat del VIH (GRk KRRQRRRPPQ (SEQ ID NO:3369)) y una proteína de Drosophila Antennapedia (RQIKIWFQNRRMKWKK (SEQ ID NO: 3370)) son capaces de actuar como péptidos de suministro. Un péptido o peptidomimético puede ser codificado por una secuencia aleatoria de ADN, tal como un péptido identificado a partir de una colección de presentación en pagos o una colección combinatoria de unamicroesfera-un-compuesto (OBOC, por sus siglas en inglés) (Lam et al., Nature, 354:82-84, 1991). Normalmente, el péptido o peptidomimético unido a un agente de ARNbc mediante una unidad monomérica incorporada es un péptido de direccionamiento hacia una célula tal como el péptido arginina-glicina-ácido aspártico (RGD) o un mimético de RGD. Un resto peptídico puede tener una longitud comprendida entre aproximadamente 5 aminoácidos y aproximadamente 40 aminoácidos. Los restos peptídicos pueden presentar una modificación estructural, por ejemplo, para incrementar la estabilidad o dirigir propiedades conformacionales. Se puede utilizar cualquiera de las modificaciones estructurales que se describen más adelante.
Un péptido RGD para su uso en las composiciones y métodos de la divulgación puede ser lineal o cíclico y puede estar modificado, p. ej., glicosilado o metilado, para facilitar el direccionamiento hacia uno o más tejidos específicos. Los péptidos y peptidomiméticos que contienen RGD pueden incluir aminoácidos D, así como también miméticos de RGD sintéticos. Además de RGD, se pueden utilizar otros restos que tengan como diana el ligando integrina. Los conjugados preferidos de este ligando tienen como diana PECAM-1 o VEGF.
Se puede utilizar un resto peptídico RGD para que actúe sobre un tipo celular particular, p. ej., una célula tumoral tal como una célula tumoral endotelial o una célula tumoral de cáncer de mama (Zitzmann et al., Cancer Res., 62:5139-43, 2002). Un péptido RGD puede facilitar el direccionamiento de un agente de ARNbc hacia tumores de varios tejidos diferentes, que incluyen el pulmón, el riñón, el bazo o el hígado (Aoki et al., Cancer Gene Therapy 8:783-787, 2001). Normalmente, el péptido RGD facilitará el direccionamiento de un agente de ARNi hacia el riñón. El péptido RGD puede ser lineal o cíclico y puede estar modificado, p. ej., glicosilado o metilado, para facilitar el direccionamiento hacia tejidos específicos. Por ejemplo, un péptido RGD glicosilado puede suministrar un agente de ARNi a una célula tumoral que exprese aVB3 (Haubner et al., Jour. Nucl. Med., 42:326-336, 2001).
Un "péptido de permeación celular" es capaz de penetrar en una célula, p. ej., una célula microbiana, tal como una célula bacteriana o fúngica, o una célula de mamífero tal como una célula humana. Un péptido de permeación en células microbianas puede ser, por ejemplo, un péptido lineal de hélice a (p. ej., LL-37 o Ceropina P1), un péptido que contenga un enlace de tipo disulfuro (p. ej., a -defensina, p-defensina o bactenecina) o un péptido que contenga únicamente uno o dos aminoácidos dominantes (p. ej., PR-39 o indolicidina). Un péptido de permeación celular también puede incluir una señal de localización nuclear (NLS, por sus siglas en inglés). Por ejemplo, un péptido de permeación celular puede ser un péptido anfifático bipartito tal como MPG, el cual deriva del dominio del péptido de fusión de gp41 del VlH-1 y NLS del antígeno T grande SV40 (Simeoni et al., Nucl. Acids Res. 31:2717-2724, 2003).
Conjugados de carbohidratos
En las composiciones y los métodos de la divulgación, un oligonucleótido de tipo ARNi puede comprender además un carbohidrato. Los ARNi conjugados con carbohidratos son convenientes para el suministro in vivo de ácidos nucleicos, así como también para composiciones adecuadas para el uso terapéutico in vivo, según se describe en la presente. El término "carbohidrato", tal como se utiliza en la presente, se refiere a un compuesto que o bien es un carbohidrato de por sí constituido por una o más unidades de monosacáridos que contienen al menos 6 átomos de carbono (que pueden ser lineales, ramificados o cíclicos) con un átomo de oxígeno, nitrógeno o azufre unido a cada átomo de carbono; o un compuesto que tiene como parte de este un resto de carbohidrato constituido por una o más unidades de monosacáridos, teniendo cada una de ellas al menos seis átomos de carbono (que pueden ser lineales, ramificados o cíclicos), con un átomo de oxígeno, nitrógeno o azufre unido a cada átomo de carbono. Los carbohidratos representativos incluyen los azúcares (mono-, di-, tri- y oligosacáridos que contienen desde aproximadamente 4, 5, 6, 7, 8 o 9 unidades de monosacáridos) y polisacáridos tales como almidones, glicógeno, celulosa y gomas de polisacáridos. Los monosacáridos específicos incluyen azúcares C5 y los azúcares anteriores (p. ej., C5, C6, C7 o C8); los di- y trisacáridos incluyen azúcares que tienen dos o más unidades de monosacáridos (p. ej., C5, C6, C7 o C8).
Un conjugado de carbohidrato puede comprender un monosacárido. El monosacárido es una N-acetilgalactosamina (GalNAc). Los conjugados de tipo GalNAc se describen, por ejemplo, en la Patente de EE. UU. N.° 8.106.022. El conjugado de tipo GalNAc puede servir como ligando que dirige el ARNi hacia células particulares. El conjugado de tipo GalNAc dirige el ARNi hacia células hepáticas, p. ej., actuando como un ligando para el receptor de la asialoglicoproteína de las células hepáticas (p. ej., los hepatocitos).
El conjugado de carbohidrato puede comprender uno o más derivados de tipo GalNAc. Los derivados de tipo GalNAc se pueden unir mediante un conector, p. ej., un conector ramificado bivalente o trivalente. El conjugado de tipo GalNAc puede estar conjugado con el extremo 3’ de la hebra sentido. El conjugado de tipo GalNAc puede estar conjugado con el agente de ARNi (p. ej., con el extremo 3' de la hebra sentido) mediante un conector, p. ej., un conector según se describe en la presente.
El conjugado de tipo GalNAc puede ser

El agente de iARN puede estar unido al conjugado de carbohidrato mediante un conector, según se muestra en la siguiente representación esquemática, donde X es O o S

El agente de iARN se puede conjugar a L96 según se define en la Tabla 1 y se muestra a continuación

Un conjugado de carbohidrato para su uso en las composiciones y los métodos de la divulgación se puede seleccionar del grupo constituido por:




,

.
Otro conjugado de carbohidrato representativo para su uso en los aspectos descritos en la presente incluye, sin carácter limitante,

(Fórmula XXIII), donde uno de los grupos X o Y es un oligonucleótido y el otro es un hidrógeno.
El conjugado de carbohidrato puede comprender además uno o más ligandos adicionales como los que se describen en la presente, tales como, sin carácter limitante, un modulador PK y/o un péptido de permeación celular.
Un ARNi de la divulgación puede estarconjugado con un carbohidrato a través de un conector. Los ejemplos no limitantes de conjugados de carbohidrato y ARNi con conectores de las composiciones y los métodos de la divulgación incluyen, sin carácter limitante,

(Fórmula XXVI),

(Fórmula XXX), donde uno de los grupos X o Y es un oligonucleótido y el otro es un hidrógeno.
Conectores
El conjugado o ligando descrito en la presente se puede unir a un oligonucleótido de tipo ARNi con varios conectores que pueden ser escindibles o no escindibles.
El término ''conector'' o "grupo conector" se refiere a un resto orgánico que conecta dos partes de un compuesto, p. ej., une covalentemente dos partes de un compuesto. Los conectores comprenden normalmente un enlace directo o un átomo tal como oxígeno o azufre, una unidad tal como NR8, C(O), C(O)NH, SO, SO2, SO2NH o una cadena de átomos tal como, sin carácter limitante, alquilo sustituido o no sustituido, alquenilo sustituido o no sustituido, alquinilo
sustituido o no sustituido, arilalquilo, arilalquenilo, arilalquinilo, heteroarilalquilo, heteroarilalquenilo, heteroarilalquinilo, heterociclilalquilo, heterociclilalquenilo, heterociclilalquinilo, arilo, heteroarilo, heterociclilo, cicloalquilo, cicloalquenilo, alquilarilalquilo, alquilarilalquenilo, alquilarilalquinilo, alquenilarilalquilo, alquenilarilalquenilo, alquenilarilalquinilo, alquinilarilalquilo, alquinilarilalquenilo, alquinilarilalquinilo, alquilheteroarilalquilo, alquilheteroarilalquenilo, alquilheteroarilalquinilo, alquenilheteroarilalquilo, alquenilheteroarilalquenilo, alquenillheteroarilalquinilo, alquinilheteroarilalquilo, alquinilheteroarilalquenilo, alquinilheteroarilalquinilo, alquilheterociclilalquilo, alquilheterociclilalquenilo, alquilheterociclilalquinilo, alquenilheterociclilaquilo, alquenilheterociclilalquenilo, alquenilheterociclilalquinilo, alquinilheterociclilalquilo, alquinilheterociclilalquenilo, alquinilheterociclilalquinilo, alquilarilo, alquenilarilo, alquinilarilo, alquilheteroarilo, alquenilheteroarilo, alquinilheteroarilo, donde uno o más metilenos se pueden interrumpir o terminar con O, S, S(O), SO2, N(R8), C(O), arilo sustituido o no sustituido, heteroarilo sustituido o no sustituido, heterociclilo sustituido o no sustituido; donde R8 es hidrógeno, acilo, un resto alifático o un resto alifático sustituido. El conector puede tener aproximadamente 1-24 átomos, 2-24, 3-24, 4-24, 5-24, 6-24, 6-18, 7-18, 8-18 átomos, 7-17, 8-17, 6-16, 7-16 o 8-16 átomos.
Un ARNbc de la divulgación puede estar conjugado con un conector ramificado bivalente o trivalente seleccionado a partir del grupo de estructuras que se muestran en cualquiera de las fórmulas (XXXI)-(XXXIV):
Fórmula XXXI Fórmula XXXII

donde:
q2A, q2B, q3A, q3B, q4A, q4B, q5A, q5B y q5C representan independientemente en cada caso 0-20 y donde la unidad de repetición puede ser igual o diferente;
P2A, P2B, P3A, P3B, P4A, P4B, P5A, P5B, P5C, T2A, T2B, T3A, T3B, T4A, T4B, T4A, T5B, T5C, cada uno independientemente en cada caso, están ausentes o son CO, NH, O, S, OC(O), NHC(O), CH2, CH2NH, CH2O; Q2A, Q2B, Q3A, Q3B, Q4A, Q4B, Q5A, Q5B, Q5C, cada uno independientemente en cada caso, están ausentes o son al uileno al uileno sustituido donde uno o más metilenos ueden estar interrum idos o

o heterociclilo;
L2A, L2B, L3A, L3B, L4A, L4B, L5A, L5B y L5C representan el ligando, es decir, cada uno independientemente en cada caso representa un monosacárido (tal como GalNAc), disacárido, trisacárido, tetrasacárido, oligosacárido o polisacárido; y Ra es H o una cadena lateral aminoacídica. Los derivados de tipo GalNAc de
conjugación trivalente son particularmente útiles para ser utilizados con los agentes de iARN para inhibir la expresión de un gen diana, tales como los de fórmula (XXXV):
Fórmula XXXV

donde L5A, L5B y L5C representan un monosacárido tal como un derivado de tipo GalNAc.
Los ejemplos de grupos conectores ramificados bivalentes y trivalentes adecuados que se conjugan con derivados de tipo GalNAc incluyen, sin carácter limitante, las estructuras mencionadas anteriormente como fórmulas II, VII, XI, X y XIII.
Un grupo conector escindible es aquel que es suficientemente estable fuera de la célula pero que, al entrar en una célula diana, se escinde para liberar las dos partes que el conector mantenía unidas. Preferiblemente, el grupo conector escindible se escinde al menos aproximadamente 10 veces, 20 veces, 30 veces, 40 veces, 50 veces, 60 veces, 70 veces, 80 veces, 90 veces o más, o al menos aproximadamente 100 veces más rápido en una célula diana o en una primera condición de referencia (la cual se puede seleccionar, p. ej., para que imite o represente las condiciones intracelulares) que en la sangre de un sujeto, o en una segunda condición de referencia (la cual se puede seleccionar, p. ej., para que imite o represente las condiciones que se encuentran en la sangre o el suero). Los grupos conectores escindibles son sensibles a los agentes de escisión, p. ej., el pH, el potencial rédox o la presencia de moléculas que provocan degradación. En general, los agentes de escisión se encuentran o prevalecen en mayor grado en concentraciones o actividades más elevadas dentro de las células que en suero o sangre. Los ejemplos de tales agentes que provocan degradación incluyen: agentes rédox que se seleccionan para sustratos particulares o que no tienen especificidad por un sustrato, los cuales incluyen, p. ej., enzimas oxidantes o reductoras o agentes reductores tales como los mercaptanos, presentes en las células, que pueden degradar un grupo conector escindible rédox mediante una reducción; estearasas; endosomas o agentes que pueden crear un entorno ácido, p. ej., los que proporcionan un pH de cinco o inferior; enzimas que pueden hidrolizar o degradar un grupo conector escindible con ácido actuando como un ácido general, peptidasas (que pueden ser específicas para un sustrato) y fosfatasas.
Un grupo conector escindible, tal como un enlace de tipo disulfuro, puede ser sensible al pH. El pH del suero humano es de 7.4, mientras que el pH intracelular medio es ligeramente inferior, está comprendido entre aproximadamente 7.1 y 7.3. Los endosomas tienen un pH más ácido, comprendido en el intervalo de 5.5 a 6.0, y los lisosomas tienen un pH todavía más ácido de aproximadamente 5.0. Algunos conectores tendrán un grupo conector escindible que se escinde a un pH preferido, liberando de este modo un lípido catiónico a partir del ligando en el interior de la célula o en el compartimento deseado de la célula.
Un conector puede incluir un grupo conector escindible que pueda ser escindido por una enzima particular. El tipo de grupo conector escindible incorporado en un conector puede depender de la célula que se tenga como diana. Por ejemplo, un ligando que tenga el hígado como diana se puede unir a un lípido catiónico a través de un conector que incluya un grupo éster. Las células hepáticas son ricas en estearasas y, por consiguiente, el conector se escindirá de una forma más eficaz en las células hepáticas que en los tipos celulares que no sean ricos en estearasas. Otros tipos celulares ricos en estearasas incluyen las células del pulmón, la corteza renal y los testículos.
Se pueden utilizar conectores que contengan enlaces peptídicos cuando se tengan como diana tipos celulares ricos en peptidasas tales como las células hepáticas y los sinoviocitos.
En general, la idoneidad de un grupo conector escindible candidato se puede evaluar estudiando la capacidad de un agente (o condición) de degradación para escindir el grupo conector candidato. También será deseable estudiar además el grupo conector escindible candidato con el fin de determinar su capacidad para resistir la escisión en sangre o cuando entre en contacto con otro tejido que no sea la diana. Por lo tanto, se puede determinar la susceptibilidad relativa a la escisión entre una primera y una segunda condición, donde la primera se selecciona para que sea indicativa de la escisión en una célula diana y la segunda se selecciona para que sea indicativa de la escisión en otros tejidos o fluidos biológicos, p. ej., en sangre o suero. Las evaluaciones se pueden llevar a cabo en sistemas exentos de células, en células, en cultivos celulares, en cultivos tisulares o de órganos, o en animales enteros. Puede resultar útil realizar evaluaciones iniciales en condiciones de cultivo o exentas de células y confirmarlas mediante evaluaciones adicionales en animales enteros. Preferiblemente, los compuestos candidatos
útiles se pueden escindir al menos aproximadamente 2, 4, 10, 20, 30, 40, 50, 60, 70, 80, 90 o aproximadamente 100 veces más rápido en la célula (o en condiciones in vitro seleccionadas para imitar las condiciones intracelulares) en comparación con la sangre o el suero (o en condiciones in vitro seleccionadas para imitar las condiciones extracelulares).
Grupos conectores escindibles rédox
Un grupo conector escindible puede ser un grupo conector escindible rédox que se escinde tras una reducción u oxidación. Un ejemplo de un grupo conector escindible de forma reductiva es un grupo conector de tipo disulfuro (-S-S-). Para determinar si un grupo conector escindible candidato es un "grupo conector escindible de forma reductiva" adecuado o, por ejemplo, es adecuado para su uso con un resto de ARNi particular y un agente de direccionamiento particular, se pueden consultar los métodos descritos en la presente. Por ejemplo, un candidato se puede evaluar mediante la incubación con ditiotreitol (DTT) u otro agente reductor utilizando reactivos conocidos en la técnica que imitan la tasa de escisión que se observaría en una célula, p. ej. una célula diana. Los candidatos también se pueden evaluar en unas condiciones que se seleccionan para que imiten las condiciones en sangre o suero. Los compuestos candidatos se escinden como máximo aproximadamente un 10% en sangre. Los compuestos candidatos útiles se degradan al menos aproximadamente 2, 4, 10, 20, 30, 40, 50, 60, 70, 80, 90 o aproximadamente 100 veces más rápido en la célula (o en condiciones in vitro seleccionadas para imitar las condiciones intracelulares) en comparación con la sangre (o en condiciones in vitro seleccionadas para imitar las condiciones extracelulares). La tasa de escisión de los compuestos candidatos se puede determinar utilizando ensayos cinéticos enzimáticos estándar en condiciones seleccionadas para que imiten los medios intracelulares y comparándolos con unas condiciones seleccionadas para imitar los medios extracelulares.
Grupos conectores escindibles basados en fosfatos
Un conector escindible puede comprender un grupo conector escindible basado en un fosfato. Un grupo conector escindible basado en un fosfato es escindido por agentes que degradan o hidrolizan el grupo fosfato. Un ejemplo de un agente que escinde grupos fosfato en células son enzimas tales como las fosfatasas en células. Algunos ejemplos de grupos conectores basados en fosfatos son -O-P(O)(ORk)-O-, -O-P(S)(ORk)-O-, -O-P(S)(SRk)-O-, -S-P(O)(ORk)-O-, -O-P(O)(ORk)-S-, -S-P(O)(ORk)-S-, -O-P(S)(ORk)-S-, -S-P(S)(ORk)-O-, -O-P(O)(Rk)-O-, -O-P(S)(Rk)-O-, -S-P(O)(Rk)-O-, -S-P(S)(Rk)-O-, -S-P(O)(Rk)-S-, -O-P(S)( Rk)-S-. Las realizaciones preferidas son -O-P(O)(OH)-O-, -O-P(S)(OH)-O-, -O-P(S)(SH)-O-, -S-P(O)(OH)-O-, -O-P(O)(OH)-S-, -S-P(O)(OH)-S-, -O-P(S)(OH)-S-, -S-P(S)(OH)-O-, -O-P(O)(H)-O-, -O-P(S)(H)-O-, -S-P(O)(H)-O, -S-P(S)(H)-O-, -S-P(O)(H)-S-, -O-P(S)(H)-S-. Se prefiere -O-P(O)(OH)-O-. Estos candidatos se pueden evaluar utilizando métodos análogos a los descritos anteriormente.
Grupos conectores escindibles con ácido
Un conector escindible puede comprender un grupo conector escindible con ácido. Un grupo conector escindible con ácido es un grupo conector que se escinde en condiciones ácidas. Preferiblemente, los grupos conectores escindibles con ácido se escinden en un entorno ácido con un pH de aproximadamente 6.5 o inferior (p. ej., de aproximadamente 6.0, 5.75, 5.5, 5.25, 5.0 o inferior) o con agentes tales como enzimas que pueden actuar como un ácido general. En una célula, los orgánulos de pH bajo específicos, tales como los endosomas y lisosomas, pueden proporcionar un entorno de escisión para los grupos conectores escindibles con ácido. Los ejemplos de grupos conectores escindibles con ácido incluyen, sin carácter limitante, hidrazonas, ésteres, y ésteres de aminoácidos. Los grupos escindibles con ácido presentan la fórmula general -C=NN-, C(O)O o -OC(O). Una realización preferida se produce cuando el carbono unido al oxígeno del éster (el grupo alcoxi) es un grupo arilo, un grupo alquilo sustituido o un grupo alquilo terciario tal como dimetilpentilo o t-butilo. Estos candidatos se pueden evaluar utilizando métodos análogos a los descritos anteriormente.
Grupos conectores escindibles basados en ésteres
Un conector escindible puede comprender un grupo conector escindible basado en un éster. Un grupo conector escindible basado en un éster es escindido por enzimas tales como esterasas y amidasas en las células. Los ejemplos de grupos conectores escindibles basados en ésteres incluyen, sin carácter limitante, ésteres de grupos alquileno, alquenileno y alquinileno. Los grupos conectores escindibles basados en ésteres presentan la fórmula general -C(O)O- o -OC(O)-. Estos candidatos se pueden evaluar utilizando métodos análogos a los descritos anteriormente.
Grupos conectores escindibles basados en péptidos
Un conector escindible puede comprender un grupo conector escindible basado en péptidos. Un grupo conector escindible basado en péptidos es escindido por enzimas tales como peptidasas y proteasas en las células. Los grupos conectores escindibles basados en péptidos son enlaces peptídicos formados entre aminoácidos para proporcionar oligopéptidos (p. ej., dipéptidos, tripéptidos, etc.) y polipéptidos. Los grupos escindibles basados en péptidos no incluyen el grupo amida (-C(O)NH-). El grupo amida se puede formar entre cualquier alquileno, alquenileno o alquinileno. Un enlace peptídico es un tipo especial de enlace de tipo amida formado entre aminoácidos para proporcionar péptidos y proteínas. El grupo escindible basado en un péptido se limita en general
al enlace peptídico (es decir, el enlace de tipo amida) formado entre aminoácidos para proporcionar péptidos y proteínas y no incluye el grupo funcional amida entero. Los grupos conectores escindibles basados en péptidos presentan la fórmula general -NHCHRAC(O)NHCHRBC(O)-, donde RA y RB son los grupos R de los dos aminoácidos adyacentes. Estos candidatos se pueden evaluar utilizando métodos análogos a los descritos anteriormente.
Las patentes de EE. UU. representativas que describen la preparación de conjugados de ARN incluyen, sin carácter limitante, las Pat. de EE. UU. N.2s 4.828.979, 4.948.882, 5.218.105, 5.525.465, 5.541.313, 5.545.730, 5.552.538, 5.578.717, 5.580.731, 5.591.584, 5.109.124, 5.118.802, 5.138.045, 5.414.077, 5.486.603, 5.512.439, 5.578.718, 5.608.046, 4.587.044, 4.605.735, 4.667.025, 4.762.779, 4.789.737, 4.824.941, 4.835.263, 4.876.335, 4.904.582, 4.958.013, 5.082.830, 5.112.963, 5.214.136, 5.082.830, 5.112.963, 5.214.136, 5.245.022, 5.254.469, 5.258.506, 5.262.536, 5.272.250, 5.292.873, 5.317.098, 5.371.241, 5.391.723, 5.416.203, 5.451.463, 5.510.475, 5.512.667, 5.514.785, 5.565.552, 5.567.810, 5.574.142, 5.585.481, 5.587.371, 5.595.726, 5.597.696, 5.599.923, 5.599.928 y 5.688.941,6.294.664, 6.320.017, 6.576.752, 6.783.931,6.900.297, 7.037.646, 8.106.022.
No es necesario que todas las posiciones de un compuesto determinado se modifiquen de forma uniforme y, de hecho, más de una de las modificaciones mencionadas anteriormente se pueden incorporar en un único compuesto o incluso en un único nucleósido dentro de un ARNi. La presente divulgación también incluye compuestos de ARNi que son compuestos quiméricos.
Los compuestos de ARNi "quiméricos" o "quimeras", en el contexto de la presente divulgación, son compuestos de ARNi, p. ej., ARNbc, que contienen dos o más regiones químicamente diferentes, cada una de las cuales está constituida por al menos una unidad monomérica, es decir, un nucleótido en el caso de un compuesto de ARNbc. Estos ARNi contienen normalmente al menos una región en la cual el ARN está modificado para conferir al ARNi una mayor resistencia a la degradación por parte de nucleasas, una mayor captación celular y/o una mayor afinidad de unión para el ácido nucleico diana. Una región adicional del ARNi puede servir como sustrato para enzimas capaces de escindir híbridos de ARN:ADN o ARN:ARN. A modo de ejemplo, una RNasa H es una endonucleasa celular que escinde la hebra de ARN de un dúplex de ARN:ADN. Por consiguiente, la activación de una RNasa H provoca la escisión de la diana de ARN y de este modo potencia enormemente la eficacia de la inhibición de la expresión génica por parte del ARNi. Como consecuencia, a menudo se pueden obtener resultados comparables con ARNi más cortos cuando se utilizan ARNbc quiméricos, en comparación con fosforotioato desoxi ARNbc que se hibridan con la misma región diana. La escisión de la diana de ARN se puede detectar de forma rutinaria mediante electroforesis en gel y, cuando proceda, técnicas de hibridación de ácidos nucleicos asociados conocidas en la técnica.
En ciertos casos, el ARN de un ARNi se puede modificar con un grupo que no sea un ligando. Se han conjugado una serie de moléculas que no son ligandos a ARNi con el fin de potenciar la actividad, distribución celular o captación celular del ARNi, y los procedimientos para llevar a cabo tales conjugaciones se pueden consultar en la bibliografía científica. Tales restos que no son ligandos han incluido restos lipídicos, tales como el colesterol (Kubo, T. et al., Biochem. Biophys. Res. Comm., 2007, 365(1):54-61; Letsinger et al., Proc. Natl. Acad. Sci. USA, 1989, 86:6553), ácido cólico (Manoharan et al., Bioorg. Med. Chem. Lett., 1994, 4:1053), un tioéter, p. ej., hexil-S-tritiltiol (Manoharan et al., Ann. N.Y. Acad. Sci., 1992, 660:306; Manoharan et al., Bioorg. Med. Chem. Let., 1993, 3:2765), un tiocolesterol (Oberhauser et al., Nucl. Acids Res., 1992, 20:533), una cadena alifática, p. ej., residuos de dodecandiol o undecilo (Saison-Behmoaras et al., EMBO J, 1991, 10:111; Kabanov et al., FeBs Lett., 1990, 259:327; Svinarchuk et al., Biochimie, 1993, 75:49), un fosfolípido, p. ej., dihexadecil-rac-glicerol o 1,2-di-O-hexadecil-rac-glicero-3H-fosfonato de trietilamonio (Manoharan et al., Tetrahedron Lett., 1995, 36:3651; Shea et al., Nucl. Acids Res., 1990, 18:3777), una poliamina o una cadena de polietilenglicol (Manoharan et al., Nucleosides & Nucleotides, 1995, 14:969) o ácido adamantanacético (Manoharan et al., Tetrahedron Lett., 1995, 36:3651), un resto de palmitilo (Mishra et al., Biochim. Biophys. Acta, 1995, 1264:229), o un resto de octadecilamina o hexilaminocarboniloxicolesterol (Crooke et al., J. Pharmacol. Exp. Ther., 1996, 277:923). Las patentes de Estados Unidos representativas que describen la preparación de tales conjugados de ARN se han enumerado anteriormente. Los protocolos de conjugación habituales implican la síntesis de un ARN que contiene un aminoconector en una o más posiciones de la secuencia. A continuación, el grupo camino se hace reaccionar con la molécula que se ha de conjugar utilizando reactivos activantes o de acoplamiento adecuados. La reacción de conjugación se puede llevar a cabo o bien con el ARN todavía unido al soporte sólido o tras la escisión del ARN, en fase de solución. La purificación del conjugado de ARN mediante HPLC proporciona normalmente el conjugado puro.
Suministro de ARNi
El suministro de un ARNi a un sujeto que lo necesite se puede llevar a cabo de diferentes maneras. Se puede llevar a cabo un suministro in vivo directamente administrando una composición que comprenda un ARNi, p. ej. un ARNbc, a un sujeto. Como alternativa, el suministro se puede llevar a cabo de forma indirecta administrando uno o más vectores que codifiquen y dirijan la expresión del ARNi. Estas alternativas se exponen con más detalle a continuación.
Suministro directo
En general, cualquier método para suministrar una molécula de ácido nucleico se puede adaptar para ser utilizado con un ARNi (remítase, p. ej., a Akhtar S. y Julian RL. (1992) Trends Cell. Biol. 2(5):139-144 y WO94/02595. Sin embargo, existen tres factores importantes que se deben tener en cuenta para suministrar con éxito una molécula de ARNi in vivo: (a) la estabilidad biológica de la molécula suministrada, (2) la prevención de efectos no específicos y (3) la acumulación de la molécula suministrada en el tejido diana. Los efectos no específicos de un ARNi se pueden minimizar mediante una administración local, por ejemplo, mediante una inyección directa o un implante en un tejido (a modo de ejemplo no limitante, un tumor) o administrando el preparado por vía tópica. La administración local a un sitio de tratamiento maximiza la concentración local del agente, limita la exposición del agente a tejidos sistémicos que de otro modo podrían ser dañados por el agente o que podrían degradar el agente, y permite administrar una dosis total más baja de la molécula de ARNi. Varios estudios han mostrado una supresión con éxito de productos génicos cuando se administra un ARNi de forma local. Por ejemplo, se ha demostrado que el suministro intraocular de un ARNbc VEGF mediante una inyección intravítrea en monos cinomólogos (Tolentino, MJ., et al. (2004) Retina 24:132-138) e inyecciones subretinales en ratones (Reich, SJ., et al. (2003) Mol. Vis. 9:210-216) previenen en ambos casos la neovascularización en un modelo experimental de degeneración macular relacionada con la edad. Además, la inyección intratumoral directa de un ARNbc en ratones reduce el volumen tumoral (Pille, J. et al. (2005) Mol. Ther.11:267-274) y puede prolongar la supervivencia de ratones portadores de tumores (Kim, WJ. et al. (2006) Mol. Ther. 14:343-350; Li, S. et al. (2007) Mol. Ther. 15:515-523). La interferencia por ARN también ha tenido éxito con el suministro local al SNC mediante inyección directa (Dorn, G. et al. (2004) Nucleic Acids 32:e49; Tan, PH., et al. (2005) Gene Ther. 12:59-66; Makimura, H. et al. (2002) BMC Neurosci. 3:18; Shishkina, GT., et al. (2004) Neuroscience 129:521-528; Thakker, ER. et al. (2004) Proc. Natl. Acad. Sci. U.S.A. 101:17270-17275; Akaneya, Y. et al. (2005) J. Neurophysiol. 93:594-602) y a los pulmones mediante una administración intranasal (Howard, Ka . et al. (2006) Mol. Ther. 14:476-484; Zhang, X. et al. (2004) J. Biol. Chem. 279:10677-10684; Bitko, V. et al. (2005) Nat. Med. 11:50-55). Con el fin de administrar un ARNi por vía sistémica para el tratamiento de una enfermedad, el ARN se puede modificar o suministrar de forma alternativa utilizando un sistema de suministro farmacológico; ambos métodos actúan para prevenir la degradación rápida del ARNbc por parte de endo- y exonucleasas in vivo.
La modificación del ARN o el portador farmacéutico también puede permitir el direccionamiento de la composición de ARNi hacia el tejido diana y evitar efectos laterales no deseados. Las moléculas de ARNi se pueden modificar mediante una conjugación química con otros grupos, p. ej., un grupo que sea un lípido o un carbohidrato según se describe en la presente. Tales conjugados se pueden utilizar para dirigir el ARNi hacia células particulares, p. ej., células hepáticas, p. ej., hepatocitos. Por ejemplo, se pueden utilizar conjugados de tipo GalNAc o formulaciones lipídicas (p. ej., LNP) para dirigir el ARNi hacia células particulares, p. ej., células hepáticas, p. ej., hepatocitos.
Se pueden utilizar grupos lipófilos, tales como el colesterol, para potenciar la captación celular y evitar la degradación. Por ejemplo, se inyectó un ARNi dirigido contra ApoB conjugado con un resto de colesterol lipófilo por vía sistémica en ratones y provocó la supresión del ARNm de apoB tanto en el hígado como en el yeyuno (Soutschek, J. et al. (2004) Nature 432:173-178). Se ha demostrado que la conjugación de un ARNi con un aptámero inhibe el crecimiento tumoral y media la regresión tumoral en un modelo de cáncer de próstata en ratón (McNamara, JO. et al. (2006) Nat. Biotechnol. 24:1005-1015). De manera alternativa, el ARNi se puede suministrar utilizando sistemas de suministro farmacológico tales como una nanopartícula, un dendrímero, un polímero, liposomas o un sistema de suministro catiónico. Los sistemas de suministro catiónico de carga positiva facilitan la unión de una molécula de ARNi (cargada negativamente) y también potencian las interacciones en la membrana celular cargada negativamente para permitir una captación eficaz de un ARNi por parte de la célula. Los lípidos catiónicos, dendrímeros o polímeros o bien se pueden unir a un ARNi o pueden ser inducidos para que formen una vesícula o micela (remítase, p. ej., a Kim SH. et al. (2008) Journal of Controlled Release 129(2):107-116), la cual encierra un ARNi. La formación de vesículas o micelas previene adicionalmente la degradación del ARNi cuando se administra por vía sistémica. Un experto en la técnica estará familiarizado con los métodos para preparar y administrar complejos de ARNi catiónicos (remítase, p. ej., a Sorensen, DR. et al. (2003) J. Mol. Biol 327:761-766; Verma, UN. et al. (2003) Clin. Cancer Res. 9:1291-1300; Arnold, AS et al. (2007) J. Hypertens. 25:197-205). Algunos ejemplos no limitantes de sistemas de suministro farmacológico utilizados para el suministro sistémico de ARNi incluyen DOTAP (Sorensen, DR. et al. (2003), mencionado anteriormente; Verma, UN. et al. (2003), mencionado anteriormente), Oligofectamina, "partículas sólidas de ácido nucleico-lípido" (Zimmermann, TS. et al. (2006) Nature 441:111-114), cardiolipina (Chien, PY. et al. (2005) Cancer Gene Ther. 12:321-328; Pal, A. et al. (2005) Int J. Oncol.
26:1087-1091), polietilenimina (Bonnet ME. et al. (2008) Pharm. Res. 16 de agosto, publicación electrónica previa a la impresa; Aigner, A. (2006) J. Biomed. Biotechnol. 71659), péptidos Arg-Gly-Asp (Rg D) (Liu, S. (2006) Mol. Pharm.
3:472-487) y poliamidoaminas (Tomalia, DA. et al. (2007) Biochem. Soc. Trans. 35:61-67; Yoo, H. et al. (1999) Pharm. Res. 16:1799-1804). Un ARNi puede formar un complejo con una ciclodextrina para la administración sistémica. Se pueden consultar métodos de administración y composiciones farmacéuticas de ARNi y ciclodextrinas en la Patente de EE. UU. N.° 7. 427.605.
ARNi codificados en vectores
En otro aspecto, un ARNi que tiene como diana el gen ALAS1 se puede expresar a partir de unidades de transcripción insertadas en vectores de ADN o ARN (remítase, p. ej., a Couture, A et al., TIG. (1996), 12:5-10; Skillern, A. et al., Publicación de PCT Internacional N.° WO 00/22113, Conrad, Publicación de PCT Internacional N.° WO 00/22114 y Conrad, Pat. de EE. UU. N.° 6.054.299). La expresión puede ser transitoria (del orden de horas a semanas) o sostenida (de semanas a meses o más prolongada), dependiendo del constructo específico utilizado y
del tejido o tipo celular diana. Estos transgenes se pueden introducir como un constructo lineal, un plásmido circular o un vector vírico, el cual puede ser un vector integrador o no integrador. El transgén también se puede construir de modo que se permita que sea heredado como un plásmido extracromosómico (Gassmann et al., Proc. Natl. Acad. Sci. USA (1995) 92:1292).
La hebra individual o las hebras de un ARNi se pueden transcribir a partir de un promotor en un vector de expresión. Cuando se han de expresar dos hebras separadas para generar, por ejemplo, un ARNbc, se pueden introducir conjuntamente dos vectores de expresión separados (p. ej., mediante transfección o infección) en una célula diana. Como alternativa, cada hebra individual de un ARNbc puede ser transcrita por promotores que estén ambos localizados en el mismo plásmido de expresión. Un ARNbc se puede expresar como una repetición invertida unida mediante una secuencia polinucleotídica conectora tal como la de un ARNbc presenta una estructura de tallo y bucle.
Un vector de expresión de ARNi es normalmente un plásmido de ADN o un vector vírico. Para producir constructos recombinantes para la expresión de un ARNi según se describe en la presente, se puede utilizar un vector de expresión compatible con células eucariotas, p. ej., con células de vertebrados. Los vectores de expresión para células eucariotas son de uso común en la técnica y se pueden adquirir a partir de una serie de proveedores comerciales. Normalmente, tales vectores contienen sitios de restricción convenientes para la inserción del segmento de ácido nucleico deseado. El suministro de vectores de expresión de ARNi puede ser sistémico, por ejemplo, mediante una administración intravenosa o intramuscular, mediante una administración que tenga como diana células explantadas procedentes del paciente seguida de su reintroducción en el paciente, o mediante cualquier otro medio que permita la introducción en una célula diana deseada.
Se puede transfectar un plásmido de expresión de ARNi en una célula diana como un complejo con un portador lipídico catiónico (p. ej., Oligofectamina) o un portador basado en un lípido que no sea catiónico (p. ej., Transit-TKOTM). En la presente también se contemplan múltiples transfecciones de lípidos para supresiones mediadas por ARNi que tienen como diana diferentes regiones de un ARN diana durante un periodo de una semana o más. La introducción con éxito de vectores en células huésped se puede monitorizar utilizando varios métodos conocidos. Por ejemplo, la transfección transitoria se puede señalizar con un marcador, tal como un marcador fluorescente, tal como la proteína fluorescente verde (GFP, por sus siglas en inglés). La transfección estable de células ex vivo se puede garantizar utilizando marcadores que proporcionen a la célula infectada resistencia a factores medioambientales específicos (p. ej., antibióticos y fármacos), por ejemplo, resistencia a la higromicina B.
Los sistemas de vectores víricos que se pueden utilizar con los métodos y las composiciones que se describen en la presente incluyen, sin carácter limitante (a) vectores de adenovirus; (b) vectores de retrovirus, que incluyen, sin carácter limitante, vectores de lentivirus, virus de la leucemia de murinos de Moloney, etc.; (c) vectores de virus adenoasociados; (d) vectores del virus del herpes simple; (e) vectores de SV40; (f) vectores del virus del polioma; (g) vectores del virus del papiloma; (h) vectores de picornavirus; (i) vectores del virus de la varicela tal como un orthopox, p. ej., vectores del virus vaccinia o de la varicela aviar, p. ej., la varicela del canario o la varicela de aves de corral; y (j) un adenovirus "gutless" o dependiente de cooperadores. Los virus de replicación deficiente también pueden ser beneficiosos. Los diferentes vectores se incorporarán o no al genoma de las células. Los constructos pueden incluir secuencias víricas para la transfección, cuando proceda. Como alternativa, el constructo se puede incorporar en vectores capaces de llevar a cabo una replicación episomal, p. ej., vectores EPV y EBV. Los constructos para la expresión recombinante de un ARNi generalmente requerirán elementos reguladores, p. ej., promotores, potenciadores, etc., para garantizar la expresión del ARNi en las células diana. A continuación se describen adicionalmente otros aspectos que se deben considerar para los vectores y los constructos.
Los vectores útiles para el suministro de un ARNi incluirán elementos reguladores (un promotor, potenciador, etc.) suficientes para la expresión del ARNi en la célula o el tejido diana deseado. Los elementos reguladores se pueden seleccionar para que proporcionen una expresión constitutiva o regulada/inducible.
La expresión del ARNi se puede regular de forma exacta, por ejemplo, utilizando una secuencia reguladora inducible que sea sensible a ciertos reguladores fisiológicos, p. ej., los niveles de glucosa en circulación u hormonas (Docherty et al., 1994, FASEB J. 8:20-24). Tales sistemas de expresión inducible adecuados para el control de la expresión de ARNbc en células o en mamíferos incluyen, por ejemplo, la regulación por parte de ecdisona, por parte de un estrógeno, progesterona, tetraciclina, inductores químicos de dimerización e isopropil-p-D1-tiogalactopiranósido (IPTG). Un experto en la técnica sería capaz de seleccionar la secuencia promotora/reguladora adecuada en función del uso deseado del transgén de ARNi.
Específicamente, se pueden utilizar vectores víricos que contengan secuencias de ácido nucleico que codifiquen un ARNi. Por ejemplo, se puede utilizar un vector retrovírico (remítase a Miller et al., Meth. Enzymol. 217:581-599 (1993)). Estos vectores retrovíricos contienen los componentes necesarios para el empaquetamiento correcto del genoma vírico y su integración en el ADN de la célula huésped. Las secuencias de ácido nucleico que codifican un ARNi se clonan en uno o más vectores, lo cual facilita el suministro del ácido nucleico en un paciente. Se pueden consultar más detalles sobre los vectores retrovíricos, por ejemplo, en Boesen et al., Biotherapy 6:291-302 (1994), que describe el uso de un vector retrovírico para suministrar el gen mdr1 a células madre hematopoyéticas con el fin de hacer que las células madre sean más resistentes a la quimioterapia. Otras referencias que ilustran el uso de
vectores retrovíricos en la terapia génica son: Clowes et al., J. Clin. Invest. 93:644-651 (1994); Kiem et al., Blood 83:1467-1473 (1994); Salmons y Gunzberg, Human Gene Therapy 4:129-141 (1993); y Grossman y Wilson, Curr. Opin. in Genetics and Devel. 3:110-114 (1993). Los vectores lentivíricos contemplados para ser utilizados incluyen, por ejemplo, los vectores basados en el VIH que se describen en las Patentes de EE. UU. N.°s 6.143.520, 5.665.557 y 5.981.276.
También se contemplan los adenovirus para su uso en el suministro de ARNi. Los adenovirus son vehículos especialmente atractivos, p. ej., para suministrar genes a epitelios respiratorios. Los adenovirus infectan de forma natural los epitelios respiratorios, donde provocan una enfermedad leve. Otras dianas para los sistemas de suministro basados en adenovirus son el hígado, el sistema nervioso central, las células endoteliales y los músculos. Los adenovirus presentan la ventaja de que son capaces de infectar células que no se dividen. Kozarsky y Wilson, Current Opinion in Genetics and Development 3:499-503 (1993) presentan un artículo de revisión de la terapia génica basada en adenovirus. Bout et al., Human Gene Therapy 5:3-10 (1994) demostraron el uso de vectores de adenovirus para transferir genes a los epitelios respiratorios de monos rhesus. Se pueden encontrar otros ejemplos del uso de adenovirus en terapia génica en Rosenfeld et al., Science 252:431-434 (1991); Rosenfeld et al., Cell 68:143-155 (1992); Mastrangeli et al., J. Clin. Invest. 91:225-234 (1993); la Publicación de PCT WO94/12649; y Wang et al., Gene Therapy 2:775-783 (1995). En Xia H et al. (2002), Nat. Biotech. 20: 1006-1010 se describe un vector de AV adecuado para expresar un ARNi que se expone en la presente, un método para construir el vector de AV recombinante y un método para suministrar el vector en células diana.
También se contempla el uso de virus adenoasociados (AAV) (Walsh et al., Proc. Soc. Exp. Biol. Med. 204:289-300 (1993); Pat. de EE. UU. N.° 5.436.146). El ARNi se puede expresar como dos moléculas de ARN bicatenarias complementarias separadas a partir de un vector de AAV recombinante que tenga, por ejemplo, promotores de ARN de H1 o U6, o el promotor de citomegalovirus (CMV). Algunos vectores de AAV adecuados para expresar los ARNbc que se exponen en la presente, métodos para construir el vector de AV recombinante y métodos para suministrar los vectores en las células diana se describen en Samulski R et al. (1987), J. Virol. 61: 3096-3101; Fisher K J et al. (1996), J. Virol, 70: 520-532; Samulski R et al. (1989), J. Virol. 63: 3822-3826; Pat. de EE. UU. N.25.252.479; Pat. de EE. u U. N.° 5.139.941; Solicitud de Patente Internacional N.° WO 94/13788; y Solicitud de Patente Internacional N.° WO 93/24641.
Otro vector vírico típico es un virus de la varicela tal como un virus vaccinia, por ejemplo, un virus vaccinia atenuado tal como el virus modificado Ankara (MVA) o NYVAC, un virus de la varicela aviar tal como la varicela de aves de corral o la varicela del canario.
El tropismo de los vectores víricos se puede modificar mediante el pseudotipado de vectores con proteínas de la envoltura u otros antígenos de superficie procedentes de otros virus, o mediante la sustitución de diferentes proteínas de la cápside vírica, según sea conveniente. Por ejemplo, los vectores lentivíricos pueden ser pseudotipados con proteínas de superficie procedentes de virus de la estomatitis vesicular (VSV), rabias, Ebola, Mokola y similares. Se pueden preparar vectores de AAV que tengan como diana diferentes células modificando genéticamente los vectores para que expresen diferentes serotipos de proteínas de la cápside; remítase, p. ej., a Rabinowitz J E et al. (2002), J Virol 76:791 -801.
El preparado farmacéutico de un vector puede incluir el vector en un diluyente aceptable o puede incluir una matriz de liberación lenta en la cual se encuentre embebido el vehículo de suministro génico. Como alternativa, cuando el vector de suministro génico completo se pueda producir intacto a partir de células recombinantes, p. ej., vectores retrovíricos, el preparado farmacéutico podrá incluir una o más células que produzcan el sistema de suministro génico.
III. Composiciones farmacéuticas que contienen ARNi
Se proporciona además en la presente composiciones farmacéuticas que contienen un ARNi, según se describe en la presente, y un portador farmacéuticamente aceptable. La composición farmacéutica que contiene el ARNi es útil para tratar una enfermedad o trastorno relacionado con la expresión o la actividad de un gen ALAS1 (p. ej., un trastorno en el que participe la vía de las porfirinas). Tales composiciones farmacéuticas se formulan en función del modo de suministro. Por ejemplo, las composiciones se pueden formular para una administración sistémica mediante un suministro parenteral, p. ej., mediante un suministro intravenoso (IV). Una composición proporcionada en la presente (p. ej., una formulación LNP) se puede formular para un suministro intravenoso. Una composición proporcionada en la presente (p. ej., una composición que comprende un conjugado de tipo GalNAc) se puede formular para un suministro subcutáneo.
Las composiciones farmacéuticas que se exponen en la presente se administran con una dosis suficiente para inhibir la expresión de un gen ALAS1. En general, una dosis adecuada de ARNi estará comprendida en el intervalo de 0.01 a 200.0 miligramos por kilogramo de peso corporal del receptor por día, generalmente en el intervalo de 1 a 50 mg por kilogramo de peso corporal por día. Por ejemplo, se pueden administrar 0.05 mg/kg, 0.5 mg/kg, 1 mg/kg, 1.5 mg/kg, 2 mg/kg, 3 mg/kg, 10 mg/kg, 20 mg/kg, 30 mg/kg, 40 mg/kg o 50 mg/kg del ARNbc en cada dosis. La composición farmacéutica se puede administrar una vez al día o el ARNi se puede administrar como dos, tres o más subdosis en intervalos adecuados a lo largo del día o incluso utilizando una infusión continua o un suministro a
través de una formulación de liberación controlada. En este caso, la cantidad de ARNi contenida en cada subdosis debe ser correspondientemente menor con el fin de obtener la dosis diaria total. La unidad de dosificación también se puede componer para que se suministre durante varios días, p. ej., utilizando una formulación de liberación sostenida convencional que proporcione una liberación sostenida del ARNi durante un periodo de varios días. Las formulaciones de liberación sostenida son de uso común en la técnica y son particularmente útiles para suministrar agentes en un sitio particular, tal como se puede utilizar con los agentes descritos en la presente. En este aspecto, la unidad de dosificación contiene un múltiple correspondiente de la dosis diaria.
El efecto de una única dosis sobre los niveles de ALAS1 puede tener una duración prolongada, de manera que las dosis posteriores se administran en intervalos de no más de 3, 4 o 5 días, o en intervalos de no más de 1, 2, 3 o 4 semanas.
El experto apreciará que ciertos factores pueden ejercer un efecto sobre la dosis y el tiempo requerido para tratar de forma eficaz a un sujeto, que incluyen, sin carácter limitante, la gravedad de la enfermedad o trastorno, los tratamientos previos, la salud general y/o la edad del sujeto, y otras enfermedades presentes. Además, el tratamiento de un sujeto con una cantidad terapéuticamente eficaz de una composición puede incluir un tratamiento único o una serie de tratamientos. Las estimaciones de las dosis eficaces y las semividas in vivo para los ARNi individuales englobados en la divulgación se pueden realizar utilizando metodologías convencionales o en función de la evaluación in vivo utilizando un modelo animal adecuado, según se describe en otras partes de la presente. Los avances en la genética del ratón han generado una serie de modelos de ratón para el estudio de varias enfermedades humanas tales como los procesos patológicos relacionados con la expresión de ALAS1 (p. ej., procesos patológicos en los que participan las porfirinas o los defectos en la vía de las porfirinas tales como, por ejemplo, las porfirias). Tales modelos se pueden utilizar para la evaluación in vivo del ARNi, así como también para determinar una dosis terapéuticamente eficaz y/o una pauta posológica eficaz.
Un modelo de ratón adecuado es, por ejemplo, un ratón que contenga un transgén que exprese ALAS1 humano. Se pueden utilizar ratones que contienen mutaciones activantes (p. ej., mutaciones que se asocian con porfirias hepáticas agudas en humanos) para determinar la dosis terapéuticamente eficaz y/o la duración de la administración del ARNip de ALAS1. Se describen además en la presente composiciones farmacéuticas y formulaciones que incluyen los compuestos de ARNi que se exponen en la presente. Las composiciones farmacéuticas de la presente divulgación se pueden administrar de varias maneras dependiendo de si se desea un tratamiento local o sistémico y en función del área que se ha de tratar. La administración puede ser tópica (p. ej., mediante un parche transdérmico), pulmonar, p. ej., mediante inhalación o insuflación de polvos o aerosoles, que incluyen un nebulizador; intratraqueal, intranasal, epidérmica y transdérmica, oral o parenteral. La administración parenteral incluye la inyección o infusión intravenosa, intraarterial, subcutánea, intraperitoneal o intramuscular; la administración subdérmica, p. ej., mediante un dispositivo implantado; o intracraneal, p. ej., mediante administración intraparenquimal, intratecal o intraventricular.
El ARNi se puede suministrar de modo que tenga como diana un tejido particular tal como un tejido que produzca eritrocitos. Por ejemplo, el ARNi se puede suministrar a la médula ósea, el hígado (p. ej., los hepatocitos del hígado), los nódulos linfáticos, el bazo, los pulmones (p. ej., la pleura de los pulmones) o la columna vertebral. El ARNi se puede suministrar a la médula ósea.
Las composiciones farmacéuticas y las formulaciones para la administración tópica pueden incluir parches transdérmicos, ungüentos, lociones, cremas, geles, gotas, supositorios, aerosoles, líquidos y polvos. Pueden resultar necesarios o deseables portadores farmacéuticos convencionales, bases oleosas, acuosas o en polvo, espesantes y similares. También pueden resultar útiles condones recubiertos, guantes y similares. Las formulaciones tópicas adecuadas incluyen aquellas en las que los ARNi que se exponen en la presente se encuentran en una mezcla con un agente de suministro tópico tal como lípidos, liposomas, ácidos grasos, ésteres de ácidos grasos, esteroides, agentes quelantes y surfactantes. Los lípidos y liposomas adecuados incluyen los que son neutros (p. ej., dioleoilfosfatidiletanolamina DOPE, dimiristoilfosfatidilcolina DMPC, diestearoilfosfatidilcolina) negativos (p. ej., dimiristoilfosfatidilglicerol DMPG) y catiónicos (p. ej., dioleoiltetrametilaminopropilo DOTAP y dioleoilfosfatidiletanolamina DOTMA). Los ARNi que se exponen en la presente se pueden enapsular dentro de liposomas o pueden formar complejos con ellos, en particular con liposomas catiónicos. Como alternativa, los ARNi pueden formar complejos con lípidos, en particular con lípidos catiónicos. Los ácidos grasos y ésteres adecuados incluyen, sin carácter limitante, ácido araquidónico, ácido oleico, ácido eicosanoico, ácido láurico, ácido caprílico, ácido cáprico, ácido mirístico, ácido palmítico, ácido esteárico, ácido linoleico, ácido linolénico, dicaprato, tricaprato, monooleína, dilaurina, 1-monocaprato de glicerilo, 1-dodecilazacicloheptan-2-ona, una acilcarnitina, una acilcolina o un éster de alquilo C1-20 (p. ej., isopropilmiristato IPM), monoglicérido, diglicérido o una sal farmacéuticamente aceptable de estos. Las formulaciones tópicas se describen con detalle en la Patente de EE. UU. N.° 6.747.014. Formulaciones de liposomas
Existen muchas estructuras de surfactantes organizadas, además de las microemulsiones, que han sido estudiadas y utilizadas para la formulación de fármacos. Estas incluyen monocapas, micelas, bicapas y vesículas. Las vesículas, tales como los liposomas, han despertado mucho interés debido a su especificidad y duración de la
acción que ofrecen desde el punto de vista del suministro farmacológico. El término "liposoma", tal como se utiliza en la presente, se refiere a una vesícula compuesta por lípidos anfifílicos dispuestos en una bicapa o bicapas esféricas. Los liposomas son vesículas unilamelares o multilamelares que contienen una membrana formada de un material lipófilo y un interior acuoso. La porción acuosa contiene la composición que se ha de suministrar. Los liposomas catiónicos poseen la ventaja de que son capaces de fusionarse con la pared celular. Los liposomas no catiónicos, aunque no son capaces de fusionarse tan eficazmente con la pared celular, son captados por los macrófagos in vivo.
Con el fin de atravesar la piel de los mamíferos y mantenerse intactas, las vesículas lipídicas deben pasar a través de una serie de poros finos, cada uno de ellos con un diámetro inferior a 50 nm, bajo la influencia de un gradiente transdérmico adecuado. Por consiguiente, es deseable utilizar un liposoma que se pueda deformar en gran medida y que sea capaz de pasar a través de estos poros finos.
Otras ventajas de los liposomas incluyen: los liposomas obtenidos a partir de fosfolípidos naturales son biocompatibles y biodegradables; los liposomas pueden incorporar una amplia gama de fármacos solubles en agua y lípidos; los liposomas pueden proteger fármacos encapsulados en sus compartimentos internos frente al metabolismo y la degradación (Rosoff, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág. 245). Algunas consideraciones importantes en la preparación de formulaciones de liposomas son la carga superficial del lípido, el tamaño de la vesícula y el volumen acuoso de los liposomas.
Los liposomas son útiles para transferir y suministrar principios activos al sitio de acción. Debido a que la membrana de los liposomas es estructuralmente similar a las membranas biológicas, cuando los liposomas se aplican a un tejido, los liposomas se empiezan a fusionar con las membranas celulares y, a medida que la fusión de los liposomas y la célula avanza, los contenidos de los liposomas se vierten en la célula, donde el agente activo puede actuar.
Las formulaciones de liposomas han sido el centro de investigaciones exhaustivas como el modo de suministro para muchos fármacos. Cada vez existen más evidencias de que, para la administración tópica, los liposomas presentan varias ventajas en comparación con otras formulaciones. Tales ventajas incluyen una reducción de los efectos laterales relacionados con una absorción sistémica elevada del fármaco administrado, un incremento de la acumulación del fármaco administrado en la diana deseada y la capacidad de administrar una amplia variedad de fármacos, tanto hidrófilos como hidrófobos, en la piel.
Varios informes han detallado la capacidad de los liposomas para suministrar agentes, incluido el ADN de peso molecular elevado, en la piel. Se han administrado a la piel compuestos que incluyen analgésicos, anticuerpos, hormonas y ADN de peso molecular elevado. La mayoría de aplicaciones actuaron sobre la epidermis superior. Los liposomas se clasifican en dos grandes clases. Los liposomas catiónicos son liposomas de carga positiva que interaccionan con las moléculas de ADN de carga negativa para formar un complejo estable. El complejo de ADN/liposoma de carga positiva se une a la superficie celular de carga negativa y se interioriza en un endosoma. Debido al pH ácido del interior del endosoma, los liposomas se fracturan y liberan sus contenidos en el interior del citoplasma celular (Wang et al., Biochem. Biophys. Res. Commun., 1987, 147, 980-985).
Los liposomas que son sensibles al pH o de carga negativa, atrapan el ADN en lugar de complejarse con él. Debido a que tanto el ADN como el lípido tienen una carga similar, se produce una repulsión en lugar de la formación de un complejo. A pesar de ello, parte del ADN queda atrapado en el interior acuoso de estos liposomas. Los liposomas sensibles al pH han sido utilizados para suministrar ADN que codifica el gen de la timidina-cinasa a monocapas celulares en cultivo. Se detectó la expresión del gen exógeno en las células diana (Zhou et al., Journal of Controlled Release, 1992, 19, 269-274).
Un tipo principal de composición de liposomas incluye fosfolípidos distintos de la fosfatidilcolina de origen natural. Las composiciones de liposomas neutros se pueden formar, por ejemplo, a partir de dimiristoilfosfatidilcolina (DMPC) o dipalmitoilfosfatidilcolina (DPPC). Las composiciones de liposomas aniónicos se forman en general a partir de dimiristoilfosfatidilglicerol, mientras que los liposomas fusogénicos aniónicos se forman principalmente a partir de dioleoilfosfatidiletanolamina (DOPE). Otro tipo de composición de liposomas se forma a partir de fosfatidilcolina (PC) tal como, por ejemplo, PC de soja y PC de huevo. Otro tipo se forma a partir de mezclas de un fosfolípido y/o fosfatidilcolina y/o colesterol.
Varios estudios han evaluado el suministro tópico de formulaciones farmacológicas de liposomas en la piel. La aplicación de liposomas que contenían interferón a la piel de conejillos de Índias dio como resultado una reducción de úlceras de herpes en la piel, mientras que el suministro de interferón por otros medios (p. ej., como una solución o como una emulsión) no fue eficaz (Weiner et al., Journal of Drug Targeting, 1992, 2, 405-410). Además, un estudio adicional evaluó la eficacia del interferón administrado como parte de una formulación de liposomas en comparación con la administración del interferón utilizando un sistema acuoso, y llegó a la conclusión de que la formulación de liposomas era mejor que la administración acuosa (du Plessis et al., Antiviral Research, 1992, 18, 259-265).
También se han examinado sistemas de liposomas no iónicos para determinar su utilidad en el suministro de fármacos a la piel, en particular sistemas que comprenden un surfactante no iónico y colesterol. Se utilizaron formulaciones de liposomas no iónicos que comprendían NovasomeTM I (dilaurato de glicerilo/colesterol/éter polioxietilen-10-estearílico) y NovasomeTM II (diestearato de glicerilo/colesterol/éter polioxietilen-10-estearílico) para suministrar ciclosporina A en la dermis de piel de ratón. Los resultados indicaron que tales sistemas de liposomas no iónicos eran eficaces a la hora de facilitar la deposición de la ciclosporina A en diferentes capas de la piel (Hu et al. S.T.P.Pharma. Sci., 1994, 4, 6, 466).
Los liposomas también incluyen liposomas "estabilizados estéricamente", un término que, tal como se utiliza en la presente, se refiere a liposomas que comprenden uno o más lípidos especializados que, cuando se incorporan en los liposomas, proporcionan unos tiempos de vida en circulación mejorados en comparación con los liposomas que carecen de tales lípidos especializados. Los ejemplos de liposomas estabilizados estéricamente son aquellos en los que parte de la porción lipídica del liposoma que forma las vesículas (A) comprende uno o más glicolípidos, tales como el monosialogangliósido GM1, o (B) se derivatiza con uno o más polímeros hidrófilos, tales como un resto de polietilenglicol (PEG). Sin pretender vincularse a ninguna teoría particular, se cree en la técnica que, al menos para los liposomas estabilizados estéricamente que contienen gangliósidos, esfingomielina o lípidos derivatizados con PEG, la semivida en circulación mejorada de estos liposomas estabilizados estéricamente procede de una reducción de la captación en células del sistema reticuloendotelial (RES) (Allen et al., FEBS Letters, 1987, 223, 42; Wu et al., Cancer Research, 1993, 53, 3765).
En la técnica se conocen varios liposomas que comprenden uno o más glicolípidos. Papahadjopoulos et al. (Ann. N.Y. Acad. Sci., 1987, 507, 64) han descrito la capacidad del monosialogangliósido GM1, el sulfato de galactocerebrósido y el fosfatidilinositol para mejorar las semividas en sangre de los liposomas. Estos resultados fueron corroborados por Gabizon et al. (Proc. Natl. Acad. Sci. U.S.A., 1988, 85, 6949). La Pat. de EE. UU. N.° 4.837.028 y WO 88/04924, ambas de Allen et al., describen liposomas que comprenden (1) esfingomielina y (2) el gangliósido GM1 o un éster de tipo sulfato de un galactocerebrósido. La Pat. de EE. UU. N.° 5.543.152 (Webb et al.) describe liposomas que comprenden esfingomielina. En WO 97/13499 (Lim et al.) se describen liposomas que comprenden 1,2-sn-dimiristoilfosfatidilcolina.
En la técnica se han descrito muchos liposomas que comprenden lípidos derivatizados con uno o más polímeros hidrófilos y métodos para prepararlos. Sunamoto et al. (Bull. Chem. Soc. Jpn., 1980, 53, 2778) han descrito liposomas que comprenden un detergente no iónico, 2C1215G, que contiene un resto de PEG. Illum et al. (FEBS Lett., 1984, 167, 79) descubrieron que un recubrimiento hidrófilo de partículas de poliestireno con glicoles poliméricos proporcionaba unas semividas en sangre significativamente mejoradas. Sears (Pat. de EE. UU. N.°s 4.426.330 y 4.534.899) ha descrito fosfolípidos sintéticos modificados mediante la unión de grupos carboxílicos de polialquilenglicoles (p. ej., PEG). Klibanov et al. (FEBS Lett., 1990, 268, 235) han descrito experimentos que demuestran que los liposomas que comprenden fosfatidiletanolamina (PE) derivada con PEG o estearato de PEG presentan incrementos significativos de las semividas en circulación en sangre. Blume et al. (Biochimica et Biophysica Acta, 1990, 1029, 91) extendieron estas observaciones a otros fosfolípidos derivatizados con PEG, p. ej., DSPE-PEG, formados a partir de la combinación de diestearoilfosfatidiletanolamina (DSPE) y PEG. Se han descrito liposomas que tienen restos de PEG unidos covalentemente en su superficie externa en la Patente Europea N.° EP 0 445 131 B1 y en WO 90/04384 de Fisher. Woodle et al. (Pat. de EE. UU. N.2s 5.013.556 y 5.356.633) y Martin et al. (Pat. de EE. UU. N.° 5.213.804 y Patente Europea N.° EP 0 496 813 B1) han descrito composiciones de liposomas que contienen un porcentaje molar de 1-20 de PE derivatizado con PEG y métodos para utilizarlas. En Wo 91/05545 y la Pat. de EE. UU. N.° 5.225.212 (ambas de Martin et al.) y en WO 94/20073 (Zalipsky et al.) se describen liposomas que comprenden una serie de conjugados de lípido-polímero diferentes. En WO 96/10391 (Choi et al.) se describen liposomas que comprenden lípidos de ceramida modificados con PEG. La Pat. de EE. UU. N.° 5.540.935 (Miyazaki et al.) y la Pat. de EE. UU. N.° 5.556.948 (Tagawa et al.) describen liposomas que contienen PEG y que se pueden derivatizar adicionalmente con restos funcionales en sus superficies.
En la técnica se conocen varios liposomas que comprenden ácidos nucleicos. El documento WO 96/40062 de Thierry et al. describe métodos para encapsular ácidos nucleicos de peso molecular elevado en liposomas. La Pat. de EE. UU. N.° 5.264.221 de Tagawa et al. describe liposomas unidos a proteínas y afirma que los contenidos de tales liposomas pueden incluir un ARNbc. La Pat. de EE. UU. N.° 5.665.710 de Rahman et al. describe ciertos métodos para encapsular oligodesoxinucleótidos en liposomas. El documento WO 97/04787 de Love et al. describe liposomas que comprenden ARNbc que tienen como diana el gen raf.
Los transfersomas son otro tipo más de liposomas y son agregados lipídicos que se pueden deformar en gran medida, los cuales son candidatos atractivos para ser utilizados como vehículos de suministro farmacológico. Los transfersomas se pueden describir como gotículas lipídicas que se pueden deformar tanto que son capaces de penetrar fácilmente a través de poros que sean más pequeños que la gotícula. Los transfersomas se pueden adaptar al entorno en el cual se utilizan, p. ej., se pueden autooptimizar (se pueden adaptar a la forma de los poros en la piel), se pueden autoreparar, frecuentemente llegan a sus dianas sin sufrir fragmentación y a menudo se pueden autocargar. Para preparar transfersomas, es posible añadir activadores de los bordes superficiales, normalmente surfactantes, a la composición de liposomas estándar. Los transfersomas han sido utilizados para suministrar albúmina de suero a la piel. Se ha demostrado que el suministro mediado por transfersomas de la albúmina de suero es tan eficaz como la inyección subcutánea de una solución que contenga albúmina de suero.
Los surfactantes se aplican de forma generalizada en formulaciones tales como emulsiones (incluidas las microemulsiones) y liposomas. La manera más común de clasificar y puntuar las propiedades de los numerosos tipos diferentes de surfactantes, tanto naturales como sintéticos, es mediante el uso del equilibrio hidrófilo/lipófilo (HLB, por sus siglas en inglés). La naturaleza del grupo hidrófilo (que también se conoce como la "cabeza") proporciona el medio más útil para clasificar los diferentes surfactantes utilizados en las formulaciones (Rieger, en Pharmaceutical Dosage Forms, Marcel Dekker, Inc., Nueva York, N.Y., 1988, pág. 285).
Si la molécula de surfactante no está ionizada, se clasifica como un surfactante no iónico. Los surfactantes no iónicos se aplican de forma generalizada en productos cosméticos y farmacéuticos, y se pueden utilizar en un amplio intervalo de valores de pH. En general, sus valores de HLB están comprendidos entre 2 y aproximadamente 18, dependiendo de su estructura. Los surfactantes no iónicos incluyen ésteres no iónicos tales como ésteres de etilenglicol, ésteres de propilenglicol, ésteres de glicerilo, ésteres de poliglicerilo, ésteres de sorbitán, ésteres de sacarosa y ésteres etoxilados. Las alcanolamidas y los éteres no iónicos, tales como los alcoholes grasos etoxilados, alcoholes propoxilados, polímeros en bloque etoxilados/propoxilados, también se incluyen en esta clase. Los surfactantes de polioxietileno son los miembros más populares de la clase de surfactantes no iónicos.
Si la molécula de surfactante tiene una carga negativa cuando se disuelve o dispersa en agua, el surfactante se clasifica como aniónico. Los surfactantes aniónicos incluyen carboxilatos tales como jabones, acillactilatos, amidas acílicas de aminoácidos, ésteres del ácido sulfúrico tales como alquilsulfatos y alquilsulfatos etoxilados, sulfonatos tales como alquilbencenosulfonatos, acilisotionatos, aciltauratos y sulfosuccinatos, y fosfatos. Los miembros más importantes de la clase de surfactantes aniónicos son los alquilsulfatos y los jabones.
Si la molécula de surfactante tiene una carga positiva cuando se disuelve o dispersa en agua, el surfactante se clasifica como catiónico. Los surfactantes catiónicos incluyen sales de amonio cuaternario y aminas etoxiladas. Las sales de amonio cuaternario son los miembros más utilizados de esta clase.
Si la molécula de surfactante tiene la capacidad de tener tanto una carga positiva como negativa, el surfactante se clasifica como anfótero. Los surfactantes anfóteros incluyen derivados del ácido acrílico, alquilamidas sustituidas, N-alquilbetaínas y fosfátidos.
El uso de surfactantes en productos farmacológicos, formulaciones y en emulsiones ha sido objeto de artículos de revisión (Rieger, en Pharmaceutical Dosage Forms, Marcel Dekker, Inc., Nueva York, N.Y., 1988, pág. 285).
Partículas de ácido nucleico-lípido
Un ARNbc de ALAS1 mencionado en la presente puede estar totalmente encapsulado en la formulación lipídica, p. ej., para formar una partícula de SPLP, pSPLP, SNALP u otra partícula de ácido nucleico-lípido. El término "SNALP", tal como se utiliza en la presente, se refiere a una partícula de ácido nucleico-lípido estable, incluida una SPLP. El término "SPLP", tal como se utiliza en la presente, se refiere a una partícula de ácido nucleico-lípido que comprende ADN plasmídico encapsulado dentro de una vesícula lipídica. Las SNALP y SPLP contienen normalmente un lípido catiónico, un lípido no catiónico y un lípido que evita la agregación de la partícula (p. ej., un conjugado de PEG-lípido). Las SNALP y SPLP son extremadamente útiles para las aplicaciones sistémicas, ya que exhiben unos tiempos de vida en circulación extendidos tras su inyección intravenosa (i.v.) y se acumulan en sitios alejados (p. ej., sitios separados físicamente del sitio de administración). Las SPLP incluyen "pSPLP", las cuales incluyen un complejo de ácido nucleico-agente de condensación encapsulado, según se expone en la Publicación de PCT N.° WO 00/03683. Las partículas de la presente divulgación tienen normalmente un diámetro medio comprendido entre aproximadamente 50 nm y aproximadamente 150 nm, más habitualmente entre aproximadamente 60 nm y aproximadamente 130 nm, más habitualmente entre aproximadamente 70 nm y aproximadamente 110 nm, de la forma más habitual entre aproximadamente 70 nm y aproximadamente 90 nm y son sustancialmente atóxicas. Además, los ácidos nucleicos, cuando están presentes en las partículas de ácido nucleico-lípido de la presente divulgación, son resistentes en solución acuosa a la degradación por parte de una nucleasa. Las partículas de ácido nucleico-lípido y su método de preparación se describen, p. ej., en las Patentes de EE. UU. N.°s 5.976.567, 5.981.501,6.534.484, 6.586.410, 6.815.432 y la Publicación de PCT N.2 WO 96/40964.
La proporción de lípido frente a fármaco (proporción de masa/masa) (p. ej., proporción de lípido frente a ARNbc) puede estar comprendida en el intervalo de aproximadamente 1:1 a aproximadamente 50:1, de aproximadamente 1:1 a aproximadamente 25:1, de aproximadamente 3:1 a aproximadamente 15:1, de aproximadamente 4:1 a aproximadamente 10:1, de aproximadamente 5:1 a aproximadamente 9:1 o de aproximadamente 6:1 a aproximadamente 9:1.
El lípido catiónico puede ser, por ejemplo, cloruro de N,N-dioleil-N,N-dimetilamonio (DODAC), bromuro de N,N-diestearil-N,N-dimetilamonio (DDAB), cloruro de N-(I-(2,3- dioleoiloxi)propil)-N,N,N-trimetilamonio (DOTAP), cloruro de N-(I-(2,3-dioleiloxi)propil)-N,N,N-trimetilamonio (dOTmA), N,N-dimetil-2,3-dioleiloxi)propilamina (DOd Ma ), 1,2-dilinoleiloxi-N,N-dimetilaminopropano (DLinDMA), l,2-dilinoleniloxi-N,N-dimetilaminopropano (DLenDMA), 1,2-dilinoleilcarbamoiloxi-3-dimetilaminopropano (DLin-C-DAP), 1,2-dilinoleioxi-3-(dimetilamino)acetoxipropano (DLin-DAC), 1,2-dilinoleioxi-3-morfolinopropano (DLin-MA), 1,2-dilinoleoil-3-dimetilaminopropano (DLinDAP), 1,2-dilinoleiltio-3-dimetilaminopropano (DLin-S-DMA), 1-linoleoil-2-linoleiloxi-3-dimetilaminopropano (DLin-2-DMAP), sal
de tipo cloruro de 1,2-dilinoleiloxi-3-trimetilaminopropano (DLin-TMA.Cl), sal de tipo cloruro de 1,2-dilinoleoil-3-trimetilaminopropano (DLin-TAP.Cl), 1,2-dilinoleiloxi-3-(N-metilpiperazino)propano (DLin-MPZ) o 3-(N,N-dilinoleilamino)-1,2-propanodiol (DLinAP), 3-(N,N-dioleilamino)-1,2-propanodiol (Do AP), 1,2-dilinoleiloxo-3-(2-N,N-dimetilamino)etoxipropano (DLin-EG-DMA), l,2-dilinoleniloxi-N,N-dimetilaminopropano (DLinDMA), 2,2-dilinoleil-4-dimetilaminometil-[1,3]-dioxolano (DLin-K-DMA) o análogos de este, (3aR,5S,6aS)-N,N-dimetil-2,2-di((9Z,12Z)-octadeca-9,12-dienil)tetrahidro-3aH-ciclopenta[d][1,3]dioxol-5-amina (ALN100), 4-(dimetilamino)butanoato de (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-ilo (MC3), 1,1'-(2-(4-(2-((2-(bis(2-hidroxidodecil)amino)etil)(2-hidroxidodecil)amino)etil)piperazin-1-il)etilazanodiil)didodecan-2-ol (Tech G1) o una mezcla de estos. El lípido catiónico puede comprender de aproximadamente un 20% mol a aproximadamente un 50% mol o aproximadamente un 40% mol del lípido total presente en la partícula.
Se puede utilizar el compuesto 2,2-dilinoleil-4-dimetilaminoetil-[1,3]-dioxolano para preparar nanopartículas de lípido-ARNip.
La partícula de lípido-ARNip puede incluir un 40% de 2,2-dilinoleil-4-dimetilaminoetil-[1,3]-dioxolano: 10% de DSPC: 40% de colesterol: 10% de PEG-C-DOMG (porcentaje molar) con un tamaño de partícula de 63.0 ± 20 nm y una proporción de ARNip/lípido de 0.027.
El lípido no catiónico puede ser un lípido aniónico o un lípido neutro, que incluye, sin carácter limitante, diestearoilfosfatidilcolina (DSPC), dioleoilfosfatidilcolina (DOPC), dipalmitoilfosfatidilcolina (DPPC), dioleoilfosfatidilglicerol (DOPG), dipalmitoilfosfatidilglicerol (DPPG), dioleoilfosfatidiletanolamina (DOPE), palmitoiloleoilfosfatidilcolina (POPC), palmitoiloleoilfosfatidiletanolamina (POPE), 4-(N-maleimidometil)ciclohexan-lcarboxilato de dioleoilfosfatidiletanolamina (DOPE-mal), dipalmitoilfosfatidiletanolamina (DPPE), dimiristoilfosfoetanolamina (DMPE), diestearoilfosfatidiletanolamina (DSPE), 16-O-monometil-PE, 16-O-dimetil-PE, 18-1-trans-PE, 1-estearoil-2-oleoilfosfatidiletanolamina (SOPE), colesterol o una mezcla de estos. El lípido no catiónico puede constituir de aproximadamente un 5% mol a aproximadamente un 90% mol, aproximadamente un 10% mol o aproximadamente un 58% mol si se incluye colesterol, del lípido total presente en la partícula.
El conjugado de lípido que inhibe la agregación de partículas puede ser, por ejemplo, un polietilenglicol (PEG)-lípido, que incluye, sin carácter limitante, un PEG-diacilglicerol (DAG), un PEG-dialquiloxipropilo (DAA), un PEG-fosfolípido, un PEG-ceramida (Cer) o una mezcla de estos. El conjugado de PEG-DAA puede ser, por ejemplo, un PEG-dilauriloxipropilo (Ci2), un PEG-dimiristiloxipropilo (Ci4), un PEG-dipalmitiloxipropilo (Ci6) o un PEG-diesteariloxipropilo (C]8). El lípido conjugado que evita la agregación de partículas puede constituir desde un 0% mol hasta aproximadamente un 20% mol o aproximadamente un 2% mol del lípido total presente en la partícula.
La partícula de ácido nucleico-lípido puede incluir además colesterol con una concentración comprendida, p. ej., entre aproximadamente un 10% mol y aproximadamente un 60% mol o de aproximadamente un 48% mol del lípido total presente en la partícula.
El ARNi se puede formular en una nanopartícula lipídica (LNP).
LNP01
Se pueden utilizar el lipidoide ND984HCl (PM 1487) (remítase a la Solicitud de Patente de EE. UU. N.° 12/056.230, presentada el 26/3/2008), colesterol (Sigma-Aldrich) y PEG-ceramida C16 (Avanti Polar Lipids) para preparar nanopartículas de lípido-ARNbc (p. ej., partículas LNP01). Se pueden preparar soluciones patrón de cada uno de ellos en etanol como se indica a continuación: ND98, 133 mg/mL; colesterol, 25 mg/mL, PEG-ceramida C16, 100 mg/mL. A continuación, las soluciones patrón de ND98, colesterol y PEG-ceramida C16 se pueden combinar en una relación molar de, p. ej., 42:48:10. La solución lipídica combinada se puede mezclar con ARNbc acuoso (p. ej., en acetato de sodio de pH 5) de manera que la concentración final de etanol sea de aproximadamente un 35-45% y la concentración final de acetato de sodio sea de aproximadamente 100-300 mM. Normalmente, las nanopartículas de lípido-ARNbc se forman espontáneamente cuando se produce la mezcla. Dependiendo de la distribución de tamaños de partícula deseada, la mezcla de nanopartículas resultante se puede extruir a través de una membrana de policarbonato (p. ej., con un punto de corte de 100 nm) utilizando, por ejemplo, una extrusora de tambor térmico tal como la extrusora Lipex (Northern Lipids, Inc). En algunos casos, se puede omitir el paso de extrusión. Se puede conseguir eliminar el etanol e intercambiar el tampón de forma simultánea mediante, por ejemplo, diálisis o filtración de flujo tangencial. El tampón se puede intercambiar, por ejemplo, con una solución salina tamponada con fosfato (PBS) a un pH de aproximadamente 7, p. ej., un pH de aproximadamente 6.9, un pH de aproximadamente 7.0, un pH de aproximadamente 7.1, un pH de aproximadamente 7.2, un pH de aproximadamente 7.3 o un pH de aproximadamente 7.4.

s mero
Fórmula 1
Las formulaciones de LNP01 se describen, p. ej., en la Publicación de Solicitud Internacional N.° WO 2008/042973. En la tabla a continuación se proporcionan formulaciones de lípido-ARNbc ilustrativas adicionales.
Tabla 10: Formulaciones lipídicas ilustrativas

(continuación)

Síntesis de lípidos catiónicos
Cualquiera de los compuestos, p. ej., lípidos catiónicos y similares, utilizados en las partículas de ácido nucleicolípido que se exponen en la presente se pueden preparar mediante técnicas conocidas de síntesis orgánica, que incluyen los métodos que se describen con más detalle en los Ejemplos. Todos los sustituyentes son como se definen a continuación, a menos que se indique de otro modo.
El término "alquilo" se refiere a un hidrocarburo alifático saturado, cíclico o no cíclico, de cadena lineal o ramificada, que contiene de 1 a 24 átomos de carbono. Los alquilos saturados de cadena lineal representativos incluyen metilo, etilo, n-propilo, n-butilo, n-pentilo, n-hexilo y similares; mientras que los alquilos saturados ramificados incluyen isopropilo, sec-butilo, isobutilo, tert-butilo, isopentilo y similares. Los alquilos cíclicos saturados representativos incluyen ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo y similares; mientras que los alquilos cíclicos no saturados incluyen ciclopentenilo y ciclohexenilo, y similares.
El término "alquenilo" se refiere a un alquilo, como se ha definido anteriormente, que contiene al menos un doble enlace entre átomos de carbono adyacentes. Los alquenilos incluyen los isómeros tanto cis como trans. Los alquenilos de cadena lineal y ramificada representativos incluyen etilenilo, propilenilo, 1-butenilo, 2-butenilo, isobutilenilo, 1-pentenilo, 2-pentenilo, 3-metil-1-butenilo, 2-metil-2-butenilo, 2,3-dimetil-2-butenilo y similares.
El término "alquinilo" se refiere a cualquier alquilo o alquenilo, como se han definido anteriormente, que contiene adicionalmente al menos un triple enlace entre átomos de carbono adyacentes. Los alquinilos de cadena lineal y
ramificada representativos incluyen acetilenilo, propinilo, 1-butinilo, 2-butinilo, 1-pentinilo, 2-pentinilo, 3-metil-1 butinilo y similares.
El término "acilo" se refiere a cualquier alquilo, alquenilo o alquinilo donde el átomo de carbono en el punto de unión se ha sustituido con un grupo oxo, según se define más adelante. Por ejemplo, -C(=O)alquilo, -C(=O)alquenilo y -C(=O)alquinilo son grupos acilo.
El término "heterociclo" se refiere a un anillo heterocíclico monocíclico de 5 a 7 miembros o bicíclico de 7 a 10 miembros, el cual está saturado, no saturado o es aromático, y el cual contiene 1 o 2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno y azufre, y donde los heteroátomos de nitrógeno y azufre pueden estar opcionalmente oxidados y el heteroátomo de nitrógeno puede estar opcionalmente cuaternalizado, incluidos los anillos bicíclicos en los cuales cualquiera de los heterociclos anteriores está fusionado con un anillo de benceno. El heterociclo puede estar unido mediante cualquier heteroátomo o átomo de carbono. Los heterociclos incluyen los heteroarilos, según se definen más adelante. Los heterociclos incluyen morfolinilo, pirrolidinonilo, pirrolidinilo, piperidinilo, piperizinilo, hidantoinilo, valerolactamilo, oxiranilo, oxetanilo, tetrahidrofuranilo, tetrahidropiranilo, tetrahidropiridinilo, tetrahidropirimidinilo, tetrahidrotiofenilo, tetrahidrotiopiranilo, tetrahidropirimidinilo, tetrahidrotiofenilo, tetrahidrotiopiranilo y similares.
Las expresiones "alquilo opcionalmente sustituido", "alquenilo opcionalmente sustituido", "alquinilo opcionalmente sustituido", "acilo opcionalmente sustituido" y "heterociclo opcionalmente sustituido" se refieren a que, cuando están sustituidos, al menos un átomo de hidrógeno se ha reemplazado por un sustituyente. En el caso de un sustituyente oxo (=O), se han reemplazado dos átomos de hidrógeno. En este sentido, los sustituyentes incluyen oxo, halógeno, heterociclo, -CN, -ORx, -NRxRy, -NRxC(=O)Ry, -NRxSO2Ry, -C(=O)Rx, -C(=O)ORx, -C(=O)NRxRy, -SOnRx y -SOnNRxRy, donde n es 0, 1 o 2, Rx y Ry son iguales o diferentes y son independientemente hidrógeno, alquilo o heterociclo, y cada uno de dichos sustituyentes alquilo y heterociclo puede estar sustituido adicionalmente con uno o más de los siguientes: oxo, halógeno, -Oh , -CN, alquilo, -ORx, heterociclo, -NRxRy, -NRxC(=O)Ry, -NRxSO2Ry, -C(=O)Rx, -C(=O)ORx, -C(=O)NRxRy, -SOnRx y -SOnNRxRy.
El término "halógeno" se refiere a fluoro, cloro, bromo y yodo.
Los métodos que se exponen en la presente pueden requerir el uso de grupos protectores. Los expertos en la técnica estarán familiarizados con la metodología de grupos protectores (remítase, por ejemplo, a Protective Groups in Organic Synthesis, Green, T.W. et al., Wiley-Interscience, Ciudad de Nueva York, 1999). Resumiendo, los grupos protectores, en el contexto de la presente, son cualquier grupo que reduzca o elimine la reactividad no deseada de un grupo funcional. Se puede añadir un grupo protector a un grupo funcional para enmascarar su reactividad durante ciertas reacciones y a continuación se elimina para revelar el grupo funcional original. Se puede utilizar un "grupo protector de alcoholes". Un "grupo protector de alcoholes" es cualquier grupo que reduzca o elimine la reactividad no deseada de un grupo funcional de tipo alcohol. Los grupos protectores se pueden añadir y eliminar utilizando técnicas de uso común en la materia.
Síntesis de la Fórmula A
Las partículas de ácido nucleico-lípido que se exponen en la presente se formulan utilizando un líquido catiónico de fórmula A:

donde R1 y R2 son independientemente alquilo, alquenilo o alquinilo, pudiendo estar cada uno de ellos opcionalmente sustituido, y R3 y R4 son independientemente alquilo inferior o R3 y R4 se pueden considerar conjuntamente para formar un anillo heterocíclico opcionalmente sustituido. El lípido catiónico puede ser XTC (2,2-dilinoleil-4-dimetilaminoetil-[1,3]-dioxolano). En general, el lípido de fórmula A anterior se puede preparar mediante los siguientes Esquemas de reacción 1 o 2, donde todos los sustituyentes son como se han definido anteriormente, a menos que se indique de otro modo.
Esquema 1

Esquema 1
El lípido A, donde R1 y R2 son independientemente alquilo, alquenilo o alquinilo, pudiendo estar cada uno de ellos opclonalmente sustituido, y R3 y R4 son independientemente alquilo inferior o R3 y R4 se pueden considerar conjuntamente para formar un anillo heterocíclico opcionalmente sustituido, se puede preparar de acuerdo con el Esquema 1. La cetona 1 y el bromuro 2 se pueden adquirir o preparar de acuerdo con métodos conocidos por los expertos en la técnica. La reacción de 1 y 2 proporciona el cetal 3. El tratamiento del cetal 3 con la amina 4 proporciona los lípidos de fórmula A. Los lípidos de fórmula A se pueden convertir en la correspondiente sal de amonio con una sal orgánica de fórmula 5, donde X es un contraión seleccionado entre halógeno, hidróxido, fosfato, sulfato o similares.
Esquema 2

Como alternativa, la cetona 1 que actúa como material de partida se puede preparar de acuerdo con el Esquema 2. El reactivo de Grignard 6 y el cianuro 7 se pueden adquirir o preparar de acuerdo con métodos conocidos por los expertos en la técnica. La reacción de 6 y 7 proporciona la cetona 1. La conversión de la cetona 1 en los lípidos correspondientes de fórmula A se lleva a cabo como se ha descrito en el Esquema 1.
Síntesis de MC3
La preparación de DLin-M-C3-DMA (es decir, 4-(dimetilamino)butanoato de (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-ilo) se llevó a cabo como se indica a continuación. Una solución de (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-ol (0.53 g), clorhidrato del ácido 4-N,N-dimetilaminobutírico (0.51 g), 4-N,N-dimetilaminopiridina (0.61 g) y clorhidrato de 1-etil-3-(3-dimetilaminopropil)carbodiimida (0.53 g) en diclorometano (5 mL) se agitó a temperatura ambiente durante toda la noche. La solución se lavó con ácido clorhídrico diluido y a continuación con bicarbonato de sodio acuoso diluido. Las fracciones orgánicas se secaron con sulfato de magnesio anhidro, se filtraron y se eliminó el disolvente en un rotavapor. El residuo se hizo pasar a través de una columna de gel de sílice (20 g) utilizando un gradiente de elución de un 1-5% de metanol/diclorometano. Las fracciones que
contenían el producto purificado se combinaron y se eliminó el disolvente, para proporcionar un aceite incoloro (0.54 g).
Síntesis de ALNY-100
La síntesis del cetal 519 [ALNY-100] se llevó a cabo utilizando el siguiente esquema 3:

Síntesis de 515:
A una suspensión agitada de LiAlH4 (3.74 g, 0.09852 mol) en 200 mL de THF anhidro en un matraz de fondo redondo de dos bocas (1 L), se añadió una solución de 514 (10 g, 0.04926 mol) en 70 mL de THF lentamente a 0 oC en atmósfera de nitrógeno. Tras completar la adición, la mezcla de reacción se calentó hasta temperatura ambiente y a continuación se calentó a reflujo durante 4 h. La evolución de la reacción se monitorizó mediante TLC. Después de que se completara la reacción (mediante TLC), la mezcla se enfrió hasta 0 oC y se desactivó añadiendo cuidadosamente una solución saturada de Na2SO4. La mezcla de reacción se agitó durante 4 h a temperatura ambiente y se separó por filtración. El residuo se lavó bien con THF. El filtrado y los lavados se mezclaron, se diluyeron con 400 mL de dioxano y 26 mL de HCl conc. y se agitaron durante 20 minutos a temperatura ambiente. Los componentes volátiles se eliminaron al vacío para proporcionar la sal clorhídrica de 515 como un sólido blanco. Rendimiento: 7.12 g. 1H-RMN (DMSO, 400MHz): 5 = 9.34 (ancho, 2H), 5.68 (s, 2H), 3.74 (m, 1H), 2.66-2.60 (m, 2H), 2.50-2.45 (m, 5H).
Síntesis de 516:
A una solución agitada del compuesto 515 en 100 mL de DCM anhidro en un matraz de fondo redondo de dos bocas de 250 mL, se añadió NEt3 (37.2 mL, 0.2669 mol) y se enfrió hasta 0 oC en atmósfera de nitrógeno. Después de una adición lenta de N-(benciloxicarboniloxi)succinimida (20 g, 0.08007 mol) en 50 mL de DCM anhidro, se permitió que la mezcla de reacción se calentara hasta temperatura ambiente. Después de que se completara la reacción (2-3 h mediante TLC), la mezcla se lavó sucesivamente con una solución de HCl 1 N (1 x 100 mL) y una solución saturada de NaHCO3 (1 x 50 mL). A continuación, la fase orgánica se secó con Na2SO4 anhidro y se evaporó el disolvente para obtener un material crudo, el cual se purificó mediante cromatografía en columna de gel de sílice para obtener 516 como una masa pegajosa. Rendimiento: 11 g (89%). 1H-RMN (CDCl3, 400 MHz): 5 = 7.36-7.27 (m, 5H), 5.69 (s, 2H), 5.12 (s, 2H), 4.96 (a, 1H) 2.74 (s, 3H), 2.60 (m, 2H), 2.30-2.25 (m, 2H). LC-MS [M+H] -232.3 (96.94%).
Síntesis de 517A y 517B:
El ciclopenteno 516 (5 g, 0.02164 mol) se disolvió en una solución de 220 mL de acetona y agua (10:1) en un matraz de fondo redondo de 500 mL de una boca y a esto se le añadió N-óxido de N-metilmorfolina (7.6 g, 0.06492 mol) y a continuación 4.2 mL de una solución al 7.6% de OsO4 (0.275 g, 0.00108 mol) en tert-butanol a temperatura ambiente. Después de que se completara la reacción (~ 3 h), la mezcla se desactivó con la adición de Na2SO3 sólido y la mezcla resultante se agitó durante 1.5 h a temperatura ambiente. La mezcla de reacción se diluyó con DCM (300 mL) y se lavó con agua (2 x 100 mL), a continuación con una solución saturada de NaHCO3 (1 x 50 mL), agua (1 x 30 mL) y finalmente con salmuera (1x 50 mL). La fase orgánica se secó con Na2SO4 anhidro y se eliminó el disolvente al vacío. La purificación del material crudo mediante cromatografía en columna de gel de sílice proporcionó una mezcla de diastereómeros, los cuales se separaron mediante HPLC prep. Rendimiento: - 6 g de crudo
517A - Pico 1 (sólido blanco), 5.13 g (96%). 1H-RMN (DMSO, 400 MHz): 5 = 7.39-7.31 (m, 5H), 5.04 (s, 2H), 4.78 4.73 (m, 1H), 4.48-4.47 (d, 2H), 3.94-3.93 (m, 2H), 2.71 (s, 3H), 1.72- 1.67 (m, 4H). LC-MS - [m+H]-266.3, [M+NH4+]-283.5 presente, HPLC-97.86%. Estereoquímica confirmada mediante rayos X.
Síntesis de 518:
Utilizando un procedimiento análogo al descrito para la síntesis del compuesto 505, se obtuvo el compuesto 518 (1.2 g, 41%) como un aceite incoloro. 1H-RMN (CDCl3, 400 MHz): 5 = 7.35-7.33 (m, 4H), 7.30-7.27 (m, 1H), 5.37-5.27 (m, 8H), 5.12 (s, 2H), 4.75 (m, 1H), 4.58-4.57 (m, 2H), 2.78-2.74 (m, 7H), 2.06-2.00 (m, 8H), 1.96-1.91 (m, 2H), 1.62 (m, 4H), 1.48 (m, 2H), 1.37-1.25 (m a, 36 H), 0.87 (m, 6H). HPLC-98.65%.
Procedimiento general para la síntesis del compuesto 519:
Una solución del compuesto 518 (1 eq) en hexano (15 mL) se añadió gota a gota a una solución enfriada con hielo de LAH en THF (1 M, 2 eq). Tras completar la adición, la mezcla se calentó a 40 oC durante 0.5 h y a continuación se enfrió de nuevo en un baño de hielo. La mezcla se hidrolizó cuidadosamente con Na2SO4 acuoso saturado, a continuación se filtró a través de celite y se redujo hasta obtener un aceite. La cromatografía en columna proporcionó 519 puro (1.3 g, 68%), que se obtuvo como un aceite incoloro. 13C RMN = 130.2, 130.1 (x2), 127.9 (x3), 112.3, 79.3, 64.4, 44.7, 38.3, 35.4, 31.5, 29.9 (x2), 29.7, 29.6 (x2), 29.5 (x3), 29.3 (x2), 27.2 (x3), 25.6, 24.5, 23.3, 226, 14.1; Ms por electronebulización (modo positivo): Peso molecular para C44H80NO2 (M H)+ Calc. 654.6, Experimental 654.6.
Las formulaciones preparadas mediante el método estándar o un método exento de extrusión se pueden caracterizar de maneras similares. Por ejemplo, las formulaciones se caracterizan normalmente mediante una inspección visual. Deberían ser soluciones translúcidas blanquecinas exentas de agregados o sedimento. El tamaño de partícula y la distribución de tamaños de partícula de las nanopartículas lipídicas se pueden medir mediante dispersión de la luz utilizando, por ejemplo, un instrumento Malvern Zetasizer Nano ZS (Malvern, EE. UU.). Las partículas deberían de tener un tamaño de aproximadamente 20-300 nm, tal como de 40-100 nm. La distribución de tamaños de partícula debería ser unimodal. La concentración total de ARNbc en la formulación, así como también la fracción atrapada, se estima utilizando un ensayo de exclusión de tinte. Una muestra del ARNbc formulado se puede incubar con un tinte de unión a ARN, tal como Ribogreen (Molecular Probes), en presencia o ausencia de un surfactante que altere la formulación, p. ej., Triton-X100 al 0.5%. El ARNbc total de la formulación se puede determinar mediante la señal procedente de la muestra que contiene el surfactante, en relación con una curva patrón. La fracción atrapada se determina restando el contenido de ARNbc "libre" (que se mide mediante la señal en ausencia de surfactante) del contenido de ARNbc total. El porcentaje de ARNbc atrapado es normalmente > 85%. Para una formulación SNALP, el tamaño de partícula es de al menos 30 nm, al menos 40 nm, al menos 50 nm, al menos 60 nm, al menos 70 nm, al menos 80 nm, al menos 90 nm, al menos 100 nm, al menos 110 nm y al menos 120 nm. En el intervalo adecuado es normalmente de aproximadamente al menos 50 nm a aproximadamente al menos 110 nm, de aproximadamente al menos 60 nm a aproximadamente al menos 100 nm o de aproximadamente al menos 80 nm a aproximadamente al menos 90 nm.
Las composiciones y formulaciones para administración oral incluyen polvos o gránulos, micropartículas, nanopartículas, suspensiones o soluciones en agua o medios no acuosos, cápsulas, cápsulas de gel, sobres, comprimidos o minicomprimidos. Puede ser deseable utilizar espesantes, agentes saborizantes, diluyentes, emulsionantes, adyuvantes dispersantes o aglutinantes. Las formulaciones orales pueden ser aquellas en las que los ARNbc que se exponen en la presente se administran conjuntamente con uno o más agentes quelantes y surfactantes potenciadores de la penetración. Los surfactantes adecuados incluyen ácidos grasos y/o ésteres o sales de estos, ácidos biliares y/o sales de estos. Los ácidos/sales biliares adecuados incluyen ácido quenodesoxicólico (CDCA) y ácido ursodesoxiquenodesoxicólico (UDCA), ácido cólico, ácido deshidrocólico, ácido desoxicólico, ácido glucólico, ácido glicólico, ácido glicodesoxicólico, ácido taurocólico, ácido taurodesoxicólico, tauro-24,25-dihidrofusidato de sodio y glicodihidrofusidato de sodio. Los ácidos grasos adecuados incluyen ácido araquidónico, ácido undecanoico, ácido oleico, ácido láurico, ácido caprílico, ácido cáprico, ácido mirístico, ácido palmítico, ácido esteárico, ácido linoleico, ácido linolénico, dicaprato, tricaprato, monooleína, dilaurina, 1-monocaprato de glicerilo, 1-dodecilazacicloheptan-2-ona, una acilcarnitina, una acilcolina o un monoglicérido, un diglicérido o una sal farmacéuticamente aceptable de estos (p. ej., de sodio). Se pueden utilizar combinaciones de potenciadores de la penetración, por ejemplo, sales/ácidos grasos combinados con sales/ácidos biliares. Una combinación ilustrativa es la sal sódica de ácido láurico, ácido cáprico y UDCA. Otros potenciadores de la penetración incluyen el éter polioxietilen-9-laurílico y el éter polioxietilen-20-cetílico. Los ARNbc que se exponen en la presente se pueden suministrar por vía oral, en forma granular, que incluye partículas secas pulverizadas, o complejado para formar micro- o nanopartículas. Los agentes complejantes de ARNbc incluyen poliaminoácidos; poliiminas; poliacrilatos; polialquilacrilatos, polioxetanos, polialquilcianoacrilatos; gelatinas cationizadas, albúminas, almidones, acrilatos, polietilenglicoles (PEG) y almidones; polialquilcianoacrilatos; poliiminas derivatizadas con DEAE, polulanos, celulosas y almidones. Los agentes complejantes adecuados incluyen quitosano, N-trimetilquitosano, poli-L-lisina, polihistidina, poliornitina, poliesperminas, protamina, polivinilpiridina, politiodietilaminometiletileno P(TDAE), poliaminoestireno (p. ej., p-amino), poli(metilcianoacrilato), poli(etilcianoacrilato), poli(butilcianoacrilato), poli(isobutilcianoacrilato), poli(isohexilcianoacrilato), DEAE-metacrilato, DEAE-hexilacrilato, DEAE-acrilamida, DEAE-alúmina y DEAE-dextrano, polimetilacrilato, polihexilacrilato, poli(ácido D,L-láctico), poli(ácido DL-láctico-co-glicólico (PLGA), alginato y polietilenglicol (PEG). Se describen detalladamente formulaciones orales para ARNbc y su preparación en la Patente de EE. UU. 6.887.906, la Publicación de EE. UU. N.220030027780 y la Patente de EE. UU. N.26.747.014.
Las composiciones y formulaciones para administración parenteral, intraparenquimal (en el cerebro), intratecal, intraventricular o intrahepática pueden incluir soluciones acuosas estériles, las cuales también pueden contener
tampones, diluyentes y otros aditivos adecuados tales como, sin carácter limitante, potenciadores de la penetración, compuestos portadores y otros excipientes o portadores farmacéuticamente aceptables.
Las composiciones farmacéuticas de la presente divulgación incluyen, sin carácter limitante, soluciones, emulsiones y formulaciones que contienen liposomas. Estas composiciones se pueden generar a partir de varios componentes que incluyen, sin carácter limitante, líquidos preformados, sólidos autoemulsionantes y semisólidos autoemulsionantes.
Las formulaciones farmacéuticas que se exponen en la presente, las cuales se pueden presentar convenientemente en una forma farmacéutica unitaria, se pueden preparar de acuerdo con técnicas convencionales muy conocidas en la industria farmacéutica. Tales técnicas incluyen el paso de asociar los principios activos con el o los portadores o el o los excipientes farmacéuticos. En general, las formulaciones se preparan asociando de forma uniforme e íntima los principios activos con portadores líquidos o portadores sólidos finamente divididos o ambos y a continuación, cuando proceda, dando forma al producto.
Las composiciones que se exponen en la presente se pueden formular en cualquiera de las muchas formas farmacéuticas posibles tales como, sin carácter limitante, comprimidos, cápsulas, cápsulas de gel, jarabes líquidos, geles blandos, supositorios y enemas. Las composiciones también se pueden formular como suspensiones en medios acuosos, no acuosos o mixtos. Las suspensiones acuosas pueden contener además sustancias que incrementen la viscosidad de la suspensión, que incluyen, por ejemplo, carboximetilcelulosa de sodio, sorbitol y/o dextrano. La suspensión también puede contener estabilizantes.
Formulaciones adicionales
Emulsiones
Las composiciones descreitas en la presente se pueden preparar y formular como emulsiones. Las emulsiones son normalmente sistemas heterogéneos de un líquido dispersado en otro en forma de gotículas con un diámetro normalmente superior a 0.1pm (remítase, p. ej., a Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems, Allen, Lv ., Popovich NG. y Ansel HC., 2004, Lippincott Williams & Wilkins (8.a ed.), Nueva York, NY; Idson, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág. 199; Rosoff, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., Volumen 1, pág. 245; Block en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 2, pág. 335; Higuchi et al., en Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., 1985, pág. 301). Las emulsiones suelen ser sistemas bifásicos que comprenden dos fases líquidas inmiscibles mezcladas de forma íntima y dispersadas una en la otra. En general, las emulsiones pueden ser de la variedad agua en aceite (a/ac) o aceite en agua (ac/a). Cuando una fase acuosa se divide finalmente y se dispersa en forma de gotículas minúsculas en una masa de fase oleosa, la composición resultante se denomina emulsión de agua en aceite (a/ac). Como alternativa, cuando una fase oleosa se divide finalmente y se dispersa en forma de gotículas minúsculas en una masa de fase acuosa, la composición resultante se denomina emulsión de aceite en agua (ac/a). Las emulsiones pueden contener componentes adicionales además de las fases dispersadas y del fármaco activo, el cual puede estar presente como una solución tanto en la fase acuosa, la fase oleosa o por sí mismo como una fase separada. Cuando proceda, en las emulsiones también puede haber excipientes farmacéuticos presentes tales como emulsionantes, estabilizantes, tintes y antioxidantes. Las emulsiones farmacéuticas también pueden ser emulsiones múltiples que comprendan más de dos fases tales como, por ejemplo, en el caso de emulsiones de aceite en agua en aceite (ac/a/ac) y de agua en aceite en agua (a/ac/a). Tales formulaciones complejas suelen proporcionar ciertas ventajas que las emulsiones binarias simples no proporcionan. Las emulsiones múltiples en las cuales gotículas de aceite individuales de una emulsión de ac/a encierran pequeñas gotículas de agua constituyen una emulsión de a/ac/a. De modo similar, un sistema de gotículas de aceite encerradas en glóbulos de agua estabilizados en una fase continua oleosa proporcionan una emulsión de ac/a/ac.
Las emulsiones se caracterizan por tener una estabilidad dinámica escasa o nula. A menudo, la fase dispersada o discontinua de la emulsión está bien dispersada en la fase externa o continua y se mantiene en esta forma a través de los medios de emulsionantes o la viscosidad de la formulación. Cualquiera de las fases de la emulsión puede ser un semisólido o un sólido, como es el caso de las cremas y bases de ungüentos de tipo emulsión. Otros medios para estabilizar emulsiones implican el uso de emulsionantes que se pueden incorporar en cualquier fase de la emulsión. Los emulsionantes se pueden clasificar a grandes rasgos en cuatro categorías: surfactantes sintéticos, emulsionantes de origen natural, bases de absorción y sólidos finamente dispersados (remítase, p. ej., a Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems, Allen, LV., Popovich NG. y Anse1HC., 2004, Lippincott Williams & Wilkins (8.a ed.), Nueva York, NY; Idson, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág. 199).
Los surfactantes sintéticos, también conocidos como agentes tensioactivos, se aplican de forma generalizada en la formulación de emulsiones y han sido objeto de artículos de revisión en la bibliografía (remítase, p. ej., a Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems, Allen, LV., Popovich NG. y Anse1HC., 2004, Lippincott Williams & Wilkins (8.a ed.), Nueva York, NY; Rieger, en Pharmaceutical Dosage Forms, Lieberman, Rieger y
Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág. 285; Idson, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), Marcel Dekker, Inc., Nueva York, N.Y., 1988, volumen 1, pág. 199). Los surfactantes son normalmente anfifílicos y comprenden una porción hidrófila y una hidrófoba. La proporción de la naturaleza hidrófila frente a la hidrófoba del surfactante se ha denominado equilibrio hidrófilo/lipófilo (HLB) y es una herramienta valiosa para clasificar y seleccionar surfactantes en la preparación de formulaciones. Los surfactantes se pueden clasificar en diferentes clases en función de la naturaleza del grupo hidrófilo: no iónicos, aniónicos, catiónicos y anfóteros (remítase, p. ej., a Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems, Allen, LV., Popovich NG. y Ansel HC., 2004, Lippincott Williams & Wilkins (8.a ed.), Nueva York, nY; Rieger, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág. 285).
Los emulsionantes de origen natural utilizados en las formulaciones de emulsiones incluyen lanolina, cera de abejas, fosfátidos, lecitina y acacia. Las bases de absorción poseen propiedades hidrófilas, por ejemplo, pueden empaparse de agua para formar emulsiones de a/ac a la vez que conservan sus consistencias semisólidas, por ejemplo, lanolina anhidra y vaselina hidrófila. También se han utilizado sólidos finamente divididos como emulsionantes satisfactorios, especialmente combinados con surfactantes y en preparados viscosos. Estos incluyen sólidos inorgánicos polares, tales como hidróxidos de metales pesados, arcillas que no se expanden tales como bentonita, atapulgita, hectorita, caolín, montmorillonita, silicato de aluminio coloidal y silicato de aluminio y magnesio coloidal, pigmentos y sólidos no polares tales como carbón o triestearato de glicerilo.
En las formulaciones de emulsiones también se incluye una gran variedad de materiales no emulsionantes, los cuales contribuyen a definir las propiedades de las emulsiones. Estos incluyen grasas, aceites, ceras, ácidos grasos, alcoholes grasos, ésteres grasos, humectantes, coloides hidrófilos, conservantes y antioxidantes (Block, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág. 335; Idson, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág. 199).
Los coloides o hidrocoloides hidrófilos incluyen gomas de origen natural y polímeros sintéticos tales como polisacáridos (por ejemplo, acacia, agar, ácido algínico, carragenano, goma guar, goma karaya y tragacanto), derivados de la celulosa (por ejemplo, carboximetilcelulosa y carboxipropilcelulosa) y polímeros sintéticos (por ejemplo, carbómeros, éteres de celulosa y polímeros de carboxivinilo). Estos se dispersan o expanden en agua para formar soluciones coloidales que estabilizan las emulsiones formando películas interfaciales resistentes alrededor de las gotículas de la fase dispersadas e incrementando la viscosidad de la fase externa.
Debido a que las emulsiones suelen contener una serie de ingredientes, tales como carbohidratos, proteínas, esteroles y fosfátidos, que pueden propiciar fácilmente el crecimiento de microbios, estas formulaciones suelen incorporar conservantes. Los conservantes utilizados habitualmente que se incluyen en las formulaciones de emulsiones incluyen parabeno metílico, parabeno propílico, sales de amonio cuaternario, cloruro de benzalconio, ésteres del ácido p-hidroxibenzoico y ácido bórico. Habitualmente también se añaden antioxidantes a las formulaciones de emulsiones para evitar el deterioro de la formulación. Los antioxidantes utilizados pueden ser atrapadores de radicales libres tales como tocoferoles, galatos de alquilo, hidroxianisol butilado, hidroxitolueno butilado o agentes reductores tales como ácido ascórbico y metabisulfito de sodio, y singergistas antioxidantes tales como ácido cítrico, ácido tartárico y lecitina.
La aplicación de las formulaciones de emulsiones por vía dermatológica, oral y parenteral y los métodos para su elaboración han sido objeto de artículos de revisión en la bibliografía (remítase, p. ej., a Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems, Allen, LV., Popovich NG. y Ansel HC., 2004, Lippincott Williams & Wilkins (8.a ed.), Nueva York, nY; Idson, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág. 199). Las formulaciones de emulsiones para un suministro oral han sido utilizadas de forma generalizada debido a que su formulación resulta sencilla, así como también debido a su eficacia desde el punto de vista de la absorción y biodisponibilidad (remítase, p. ej., a Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems, Allen, LV., Popovich NG. y Anse1HC., 2004, Lippincott Williams & Wilkins (8.a ed.), Nueva York, NY; Rosoff, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág. 245; Idson, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág. 199). Los laxantes con una base de aceite mineral, las vitaminas solubles en aceites y los preparados nutritivos ricos en grasas se encuentran entre los materiales que se han administrado habitualmente por vía oral como emulsiones de ac/a.
Las composiciones de ARNi y los ácidos nucleicos se pueden formular como microemulsiones. Una microemulsión se puede definir como un sistema de agua, aceite y un material anfífilo que es una solución líquida termodinámicamente estable y ópticamente isotrópica estable (remítase, p. ej., a Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems, Allen, LV., Popovich NG. y Ansel HC., 2004, Lippincott Williams & Wilkins (8.a ed.), Nueva York, NY; Rosoff, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág. 245). Normalmente, las microemulsiones son sistemas que se preparan dispersando en primer lugar un aceite en una solución acuosa de surfactante y a continuación añadiendo una cantidad suficiente de un cuarto componente, generalmente un alcohol con una longitud de cadena intermedia para formar un sistema transparente. Por consiguiente, las microemulsiones también se han descrito como
dispersiones termodinámicamente estables e isotrópicamente transparentes de dos líquidos inmiscibles que se estabilizan mediante películas interfaciales de moléculas tensioactivas (Leung y Shah, en: Controlled Release of Drugs: Polymers and Aggregate Systems, Rosoff, M., Ed., 1989, VCH Publishers, Nueva York, páginas 185-215). Las microemulsiones se preparan normalmente mediante una combinación de tres a cinco componentes que incluyen aceite, agua, surfactante, cosurfactante y electrolito. El hecho de que la microemulsión sea del tipo agua en aceite (a/ac) o aceite en agua (ac/a) depende de las propiedades del aceite y el surfactante utilizados y de la estructura y la geometría del empaquetamiento de las cabezas polares y las colas hidrocarbonadas de las moléculas de surfactante (Schott, en Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., 1985, pág.
271).
La estrategia fenomenológica que utiliza diagramas de fase se ha estudiado exhaustivamente y ha proporcionado un conocimiento comprensivo, para el experto en la técnica, sobre cómo formular las microemulsiones (remítase, p. ej., a Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems, Allen, LV., Popovich NG. y Anse1HC., 2004, Lippincott Williams & Wilkins (8.a ed.), Nueva York, NY; Rosoff, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág. 245; Block, en Pharmaceutical Dosage Forms, Lieberman, Rieger y Banker (Eds.), 1988, Marcel Dekker, Inc., Nueva York, N.Y., volumen 1, pág.
335). En comparación con las emulsiones convencionales, las microemulsiones ofrecen la ventaja de que solubilizan fármacos insolubles en agua en una formulación de partículas termodinámicamente estables que se forman espontáneamente.
Los surfactantes utilizados en la preparación de microemulsiones incluyen, sin carácter limitante, surfactantes iónicos, surfactantes no iónicos, Brij 96, éteres oleílicos de polioxietileno, ésteres de poliglicerol y ácidos grasos, monolaurato de tetraglicerol (ML310), monooleato de tetraglicerol (MO310), monooleato de hexaglicerol (PO310), pentaoleato de hexaglicerol (PO500), monocaprato de decaglicerol (MCA750), monooleato de decaglicerol (MO750), sequioleato de decaglicerol (SO750), decaoleato de decaglicerol (DAO750), solos o combinados con cosurfactantes. El cosurfactante, normalmente un alcohol de cadena corta tal como etanol, 1-propanol y 1-butanol, sirve para incrementar la fluidez interfacial penetrando en la película surfactante y consecuentemente creando una película desordenada debido al espacio hueco generado entre las moléculas de surfactante. A pesar de ello, las microemulsiones se pueden preparar sin el uso de cosurfactantes y en la técnica se conocen sistemas de microemulsiones autoemulsionantes exentos de alcoholes. Normalmente, la fase acuosa es, sin carácter limitante, agua, una solución acuosa del fármaco, glicerol, PEG300, PEG400, poligliceroles, propilenglicoles y derivados del etilenglicol. La fase oleosa puede incluir, sin carácter limitante, materiales tales como Captex 300, Captex 355, Capmul MCM, ésteres de ácidos grasos, mono-, di- y triglicéridos de cadena media (C8-C12), ésteres de ácidos grasos glicerílicos polioxietilados, alcoholes grasos, glicéridos poliglicolixados, glicéridos C8-C10 poliglicolizados saturados, aceites vegetales y aceite de silicona.
Las microemulsiones tienen un interés particular desde el punto de vista de la solubilización de fármacos y la absorción mejorada de fármacos. Se ha postulado que las microemulsiones basadas en lípidos (tanto de ac/a como de a/ac) potencian la biodisponibilidad oral de los fármacos, incluidos los péptidos (remítase, p. ej., a las Patentes de EE. UU. N.°s 6.191.105, 7.063.860, 7.070.802, 7.157.099; Constantinides et al., Pharmaceutical Research, 1994, 11, 1385-1390; Ritschel, Meth. Find. Exp. Clin. Pharmacol., 1993, 13, 205). Las microemulsiones ofrecen las ventajas de una solubilización mejorada del fármaco, una protección del fármaco frente a la hidrólisis enzimática, una posible mejora de la absorción del fármaco debido a alteraciones inducidas por el surfactante en la fluidez y permeabilidad de la membrana, la sencillez de su preparación, la sencillez de su administración oral en comparación con las formas farmacéuticas sólidas, una potencia clínica mejorada y una menor toxicidad (remítase, p. ej., a las Patentes de EE. UU. N.°s 6.191.105, 7.063.860, 7.070.802, 7.157.099, Constantinides et al., Pharmaceutical Research, 1994, 11, 1385; Ho et al., J. Pharm. Sci., 1996, 85, 138-143). A menudo las microemulsiones se pueden formar espontáneamente cuando se combinan sus componentes a temperatura ambiente. Esto puede resultar particularmente conveniente cuando se formulan fármacos, péptidos o ARNi termolábiles. Las microemulsiones también han resultado ser eficaces en el suministro transdérmico de componentes activos en aplicaciones tanto cosméticas como farmacéuticas. Cabe esperar que las formulaciones y composiciones de microemulsiones de la presente divulgación propicien un incremento de la absorción sistémica de los ARNi y los ácidos nucleicos a partir del aparato gastrointestinal, así como también que mejoren la captación celular local de los ARNi y los ácidos nucleicos.
Las microemulsiones de la presente divulgación también pueden contener componentes y aditivos adicionales tales como monoestearato de sorbitán (malla 3), labrasol y potenciadores de la penetración para mejorar las propiedades de la formulación y para mejorar la absorción de los ARNi y los ácidos nucleicos de la presente divulgación. Los potenciadores de la penetración utilizados en las microemulsiones de la presente divulgación se pueden clasificar como pertenecientes a una de las cinco categorías generales siguientes: surfactantes, ácidos grasos, sales biliares, agentes quelantes y agentes no surfactantes ni quelantes (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, pág. 92). Cada una de estas clases ha sido expuesta anteriormente.
Potenciadores de la penetración
La presente divulgación puede emplear varios potenciadores de la penetración para ejercer un suministro eficaz de los ácidos nucleicos, particularmente los ARNi, en la piel de animales. La mayoría de los fármacos están presentes
en solución tanto en forma ionizada como no ionizada. Sin embargo, normalmente solo los fármacos lipófilos o solubles en lípidos atraviesan fácilmente las membranas celulares. Se ha descubierto que incluso los fármacos no lipófilos pueden atravesar las membranas celulares si la membrana que se ha de atravesar se trata con un potenciador de la penetración. Además de propiciar la difusión de los fármacos no lipófilos a través de las membranas celulares, los potenciadores de la penetración también potencian la permeabilidad de los fármacos lipófilos.
Los potenciadores de la penetración se pueden clasificar como pertenecientes a una de entre cinco categorías generales, es decir, surfactantes, ácidos grasos, sales biliares, agentes quelantes y agentes no surfactantes ni quelantes (remítase, p. ej., a Malmsten, M. Surfactants and polymers in drug delivery, Informa Health Care, Nueva York, NY, 2002; Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, pág. 92). Cada una de las clases mencionadas anteriormente de potenciadores de la penetración se describen a continuación con más detalle.
Surfactantes: En relación con la presente divulgación, los surfactantes (o "agentes tensioactivos") son entidades químicas que, cuando se disuelven en una solución acuosa, reducen la tensión superficial de la solución o la tensión interfacial entre la solución acuosa y otro líquido, con el resultado de que se potencia la absorción de los ARNi a través de la mucosa. Además de las sales biliares y los ácidos grasos, estos potenciadores de la penetración incluyen, por ejemplo, laurilsulfato de sodio, éter polioxietilen-9-laurílico y éter polioxietilen-20-cetílico (remítase, p. ej., a Malmsten, M. Surfactants and polymers in drug delivery, Informa Health Care, Nueva York, NY, 2002; Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, pág. 92) y emulsiones perfluoroquímicas tales como FC-43. Takahashi et al., J. Pharm. Pharmacol., 1988, 40, 252).
Ácidos grasos: Los diferentes ácidos grasos y sus derivados que actúan como potenciadores de la penetración incluyen, por ejemplo, ácido oleico, ácido láurico, ácido cáprico (ácido n-decanoico), ácido mirístico, ácido palmítico, ácido esteárico, ácido linoleico, ácido linolénico, dicaprato, tricaprato, monooleína (1-monooleoil-rac-glicerol), dilaurina, ácido caprílico, ácido araquidónico, 1-monocaprato de glicerol, 1-dodecilazacicloheptan-2-ona, acilcarnitinas, acilcolinas, sus ésteres de alquilo C1-20 (p. ej., metilo, isopropilo y t-butilo), y mono- y diglicéridos de estos (es decir, oleato, laurato, caprato, miristato, palmitato, estearato, linoleato, etc.) (remítase, p. ej., a Touitou, E. et al. Enhancement in Drug Delivery, CRC Press, Danvers, MA, 2006; Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, pág. 92; Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1 33; El Hariri et al., J. Pharm. Pharmacol., 1992, 44, 651-654).
Sales biliares: La función fisiológica de la bilis incluye facilitar la dispersión y la absorción de lípidos y de vitaminas solubles en grasas (remítase, p. ej., a Malmsten, M. Surfactants and polymers in drug delivery, Informa Health Care, Nueva York, NY, 2002; Brunton, Capítulo 38 en: Goodman & Gilman's The Pharmacological Basis of Therapeutics, 9.a Ed., Hardman et al. Eds., McGraw-Hill, Nueva York, 1996, págs. 934-935). Varias sales biliares naturales, y sus derivados sintéticos, actúan como potenciadores de la penetración. Por lo tanto, la expresión "sales biliares" incluye cualquiera de los componentes de origen natural de la bilis, así como también cualquiera de sus derivados sintéticos. Las sales biliares adecuadas incluyen, por ejemplo, ácido cólico (o su sal sódica farmacéuticamente aceptable, el colato de sodio), ácido deshidrocólico (deshidrocolato de sodio), ácido desoxicólico (desoxicolato de sodio), ácido glucólico (glucolato de sodio), ácido glicólico (glicolato de sodio), ácido glicodesoxicólico (glicodesoxicolato de sodio), ácido taurocólico (taurocolato de sodio), ácido taurodesoxicólico (taurodesoxicolato de sodio), ácido quenodesoxicólico (quenodesoxicolato de sodio), ácido ursodesoxicólico (UDCA), tauro-24,25-dihidrofusidato de sodio (STDHF), glicodihidrofusidato de sodio y éter polioxietilen-9-laurílico (POE) (remítase, p. ej., a Malmsten, M. Surfactants and polymers in drug delivery, Informa Health Care, Nueva York, NY, 2002; Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, página 92; Swinyard, Capítulo 39 en: Remington's Pharmaceutical Sciences, 18.a Ed., Gennaro, ed., Mack Publishing Co., Easton, Pa., 1990, páginas 782-783; Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33; Yamamoto et al., J. Pharm. Exp. Ther., 1992, 263, 25; Yamashita et al., J. Pharm. Sci., 1990, 79, 579-583).
Agentes quelantes: Los agentes quelantes, cuando se utilizan en relación con la presente divulgación, se pueden definir como compuestos que eliminan iones metálicos de la solución mediante la formación de complejos con ellos, con el resultado de que se potencia la absorción de los ARNi a través de la mucosa. En lo que respecta a su uso como potenciadores de la penetración en la presente divulgación, los agentes quelantes presentan la ventaja añadida de que también sirven como inhibidores de la DNasa, ya que la mayoría de ADN-nucleasas caracterizadas requieren un ion metálico bivalente para la catálisis y, por lo tanto, son inhibidas por los agentes quelantes (Jarrett, J. Chromatogr., 1993, 618, 315-339). Los agentes quelantes adecuados incluyen, sin carácter limitante, etilendiamintetraacetato de sodio (EDTA), ácido cítrico, salicilatos (p. ej., salicilato de sodio, 5-metoxisalicilato y homovanilato), derivados N-acílicos del colágeno, derivados de tipo lauret-9 y N-aminoacilo de p-dicetonas (enaminas)(remítase, p. ej., a Katdare, A. et al., Excipient development for pharmaceutical, biotechnology, and drug delivery, c Rc Press, Danvers, MA, 2006; Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, página 92; Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33; Buur et al., J. Control Rel., 1990, 14, 43-51).
Agentes no surfactantes ni quelantes: Los compuestos potenciadores de la penetración no surfactantes ni quelantes, tal como se utilizan en la presente, se pueden definir como compuestos que presentan una actividad insignificante como agentes quelantes o como surfactantes pero que, a pesar de ello, potencian la absorción de los ARNi a través
de la mucosa alimentaria (remítase, p. ej., a Muranishi, Critical Reviews in Therapeutic Drug Carrier Systems, 1990, 7, 1-33). Esta clase de potenciadores de la penetración incluyen, por ejemplo, ureas cíclicas insaturadas, derivados de tipo 1 -alquil- y 1 -alquenilazacicloalcanona (Lee et al., Critical Reviews in Therapeutic Drug Carrier Systems, 1991, page 92) y agentes antiinflamatorios no esteroideos tales como diclofenac sódico, indometacina y fenilbutazona (Yamashita et al., J. Pharm. Pharmacol., 1987, 39, 621-626).
También se pueden añadir agentes que potencian la captación de ARNi a nivel celular a las composiciones farmacéuticas u otras composiciones de la divulgación. Por ejemplo, también existe constancia de que lípidos catiónicos, tales como la lipofectina (Junichi et al, Pat. de EE. UU. N.° 5.705.188), derivados catiónicos del glicerol y moléculas policatiónicas tales como la polilisina (Lollo et al., Solicitud de PCT WO 97/30731) potencian la captación celular de ARNbc. Los ejemplos de reactivos de transfección que se pueden adquirir de proveedores comerciales incluyen, por ejemplo, Lipofectamine™ (Invitrogen; Carlsbad, CA), Lipofectamine 2000™ (Invitrogen; Carlsbad, CA), 293fectin™ (Invitrogen; Carlsbad, CA), Cellfectin™ (Invitrogen; Carlsbad, CA), DMRIE-C™ (Invitrogen; Carlsbad, CA), FreeStyle™ MAX (Invitrogen; Carlsbad, cA), Lipofectamine™ 2000 CD (Invitrogen; Carlsbad, CA), Lipofectamine™ (Invitrogen; Carlsbad, CA), RNAiMAx (Invitrogen; Carlsbad, CA), Oligofectamine™ (Invitrogen; Carlsbad, CA), Optifect™ (Invitrogen; Carlsbad, CA), el reactivo de transfección X-tremeGENE Q2 (Roche; Grenzacherstrasse, Suiza), el reactivo de transfección liposomal DOTAP (Grenzacherstrasse, Suiza), el reactivo de transfección liposomal DOSPER (Grenzacherstrasse, Suiza) o Fugene (Grenzacherstrasse, Suiza), el reactivo Transfectam® (Promega; Madison, WI), el reactivo de transfección TransFast™ (Promega; Madison, WI), el reactivo Tfx™-20 (Promega; Madison, WI), el reactivo Tfx™-50 (Promega; Madison, WI), DreamFect™ (Oz Biosciences; Marsella, Francia), EcoTransfect (OZ Biosciences; Marsella, Francia), el reactivo de transfección TransPass8 D1 (New England Biolabs; Ipswich, mA, EE. UU.), LyoVec™/LipoGen™ (Invivogen; San Diego, CA, EE. UU.), el reactivo de transfección PerFectin (Genlantis; San Diego, CA, EE. UU.), el reactivo de transfección NeuroPORTER (Genlantis; San Diego, CA, EE. UU.), el reactivo de transfección GenePORTER (Genlantis; San Diego, CA, EE. UU.), el reactivo de transfección GenePORTER 2 (Genlantis; San Diego, CA, EE. UU.), el reactivo de transfección Cytofectin (Genlantis; San Diego, CA, EE. UU.), el reactivo de transfección BaculoPORTER (Genlantis; San Diego, CA, EE. u U.), el reactivo de transfección TroganPORTER™ (Genlantis; San Diego, CA, EE. UU.), RiboFect (Bioline; Taunton, MA, EE. UU.), PlasFect (Bioline; Taunton, MA, e E. UU.), UniFECTOR (B-Bridge International; Mountain View, CA, EE. UU.), SureFECTOR (B-Bridge International; Mountain View, CA, EE. UU.) o HiFect™ (B-Bridge International, Mountain View, CA, EE. UU.), entre otros.
Se pueden utilizar otros reactivos para potenciar la penetración de los ácidos nucleicos administrados, que incluyen glicoles tales como etilenglicol y propilenglicol, pirroles tales como 2-pirrol, azonas y terpenos tales como limoneno y mentona.
Portadores
Ciertas composiciones de la presente divulgación también incorporan compuestos portadores en la formulación. La expresión "compuesto portador" o "portador", tal como se utiliza en la presente, se puede referir a un ácido nucleico o un análogo de este, que es inerte (es decir, no posee actividad biológica de por sí) pero que es reconocido como un ácido nucleico por procesos in vivo que reducen la biodisponibilidad de un ácido nucleico con actividad biológica, por ejemplo, degradando el ácido nucleico biológicamente activo o propiciando su eliminación de la circulación. La administración conjunta de un ácido nucleico y un compuesto portador, normalmente con un exceso de la última sustancia, puede dar como resultado una reducción sustancial de la cantidad de ácido nucleico que se recupera en el hígado, el riñón u otros depósitos extracirculatorios, probablemente debido a la competición entre el compuesto portador y el ácido nucleico por un receptor común. Por ejemplo, la recuperación de un ARNbc de tipo parcialmente fosforotioato en tejido hepático se puede reducir cuando se administra conjuntamente con ácido poliinosínico, sulfato de dextrano, ácido policitídico o ácido 4-acetamido-4'isotiocianoestilben-2,2'-disulfónico (Miyao et al., DsRNA Res. Dev., 1995, 5, 115-121; Takakura et al., DsRNA & Nucl. Acid Drug Dev., 1996, 6, 177-183.
Excipientes
A diferencia de un compuesto portador, un "excipiente" o "portador farmacéutico" es un agente de suspensión o un disolvente farmacéuticamente aceptable o cualquier otro vehículo farmacológicamente inerte para suministrar uno o más ácidos nucleicos a un animal. El excipiente puede ser líquido o sólido y se selecciona, teniendo en cuenta la vía de administración planeada, de modo que proporcione la masa, consistencia, etc. deseadas, cuando se combina con un ácido nucleico y los demás componentes de una composición farmacéutica determinada. Los portadores farmacéuticos habituales incluyen, sin carácter limitante, agentes aglutinantes (p. ej., almidón de maíz pregelatinizado, polivinilpirrolidona o hidroxipropilmetilcelulosa, etc.); rellenos (p. ej., lactosa u otros azúcares, celulosa microcristalina, pectina, gelatina, sulfato de calcio, etilcelulosa, poliacrilatos o hidrogenofosfato de calcio, etc.); lubricantes (p. ej., estearato de magnesio, talco, sílice, dióxido de sílice coloidal, ácido esteárico, estearatos metálicos, aceites vegetales hidrogenados, almidón de maíz, polietilenglicoles, benzoato de sodio, acetato de sodio, etc.); desintegrantes (p. ej., almidón, glicolato de almidón sódico, etc.) y agentes humectantes (p. ej., laurilsulfato de sodio, etc.).
Para formular las composiciones de la presente divulgación, también se pueden utilizar excipientes orgánicos o inorgánicos farmacéuticamente aceptables adecuados para la administración no parenteral que no reaccionen de
forma perjudicial con los ácidos nucleicos. Los portadores farmacéuticamente aceptables adecuados incluyen, sin carácter limitante, agua, soluciones salinas, alcoholes, polietilenglicoles, gelatina, lactosa, amilosa, estearato de magnesio, talco, ácido silícico, parafina viscosa, hidroximetilcelulosa, polivinilpirrolidona y similares.
Las formulaciones para la administración tópica de los ácidos nucleicos pueden incluir soluciones acuosas estériles y no estériles, soluciones no acuosas en disolventes habituales tales como alcoholes, o soluciones de los ácidos nucleicos en bases oleosas sólidas o líquidas. Las soluciones también pueden contener tampones, diluyentes u otros aditivos adecuados. Se pueden utilizar excipientes orgánicos o inorgánicos farmacéuticamente aceptables adecuados para la administración no parenteral que no reaccionen de forma perjudicial con los ácidos nucleicos. Los excipientes farmacéuticamente aceptables adecuados incluyen, sin carácter limitante, agua, soluciones salinas, alcohol, polietilenglicoles, gelatina, lactosa, amilosa, estearato de magnesio, talco, ácido silícico, parafina viscosa, hidroximetilcelulosa, polivinilpirrolidona y similares.
Otros componentes
Las composiciones de la presente divulgación pueden contener adicionalmente otros componentes adjuntos que se encuentran convencionalmente en las composiciones farmacéuticas, con sus niveles de uso establecidos en la técnica. De este modo, las composiciones pueden contener, por ejemplo, materiales farmacéuticamente activos compatibles adicionales tales como, por ejemplo, agentes antipruríticos, astringentes, anestésicos locales o antiinflamatorios, o pueden contener materiales adicionales útiles a la hora de formular físicamente varias formas farmacéuticas de las composiciones de la presente divulgación, tales como tintes, agentes saborizantes, conservantes, antioxidantes, opacificantes, agentes espesantes y estabilizantes. Sin embargo, tales materiales, cuando se añaden, no deberían de interferir indebidamente con las actividades biológicas de los componentes de las composiciones de la presente divulgación. Las formulaciones se pueden esterilizar y, cuando proceda, mezclar con agentes auxiliares, p. ej., lubricantes, conservantes, estabilizantes, agentes humectantes, emulsionantes, sales para ejercer un efecto sobre la presión osmótica, tampones, colorantes, saborizantes y/o sustancias aromáticas y similares, los cuales no interaccionan de forma perjudicial con el o los ácidos nucleicos de la formulación.
Las suspensiones acuosas pueden contener sustancias que incrementen la viscosidad de la suspensión, que incluyen, por ejemplo, carboximetilcelulosa de sodio, sorbitol y/o dextrano. La suspensión también puede contener estabilizantes.
Las composiciones farmacéuticas que se exponen en la presente pueden incluir (a) uno o más compuestos de ARNi y (b) uno o más agentes biológicos que actúan mediante un mecanismo que no es de iARN. Los ejemplos de tales agentes biológicos incluyen agentes que interfieren con una interacción de ALAS1 y al menos una pareja de unión a ALAS1.
La toxicidad y la eficacia terapéutica de tales compuestos se pueden determinar mediante procedimientos farmacéuticos estándar en cultivos celulares o animales de experimentación, p. ej., para determinar la DL50 (la dosis letal para un 50% de la población) y la DE50 (la dosis terapéuticamente eficaz en un 50% de la población). La proporción de dosis entre los efectos tóxico y terapéutico es el índice terapéutico y se puede expresar como la proporción de DL50/DE50. Es habitual encontrar compuestos que exhiben índices terapéuticos elevados.
Los datos obtenidos a partir de ensayos de cultivos celulares y estudios con animales se pueden utilizar para formular una serie de dosis para uso en humanos. La dosis de las composiciones que se exponen en la presente se encuentra generalmente dentro de un intervalo de concentraciones en circulación que incluye la DE50 con una toxicidad baja o nula. La dosis puede variar dentro de este intervalo dependiendo de la forma farmacéutica empleada y la vía de administración utilizada. Para cualquier compuesto utilizado en los métodos que se exponen en la presente, la dosis terapéuticamente eficaz se puede estimar inicialmente a partir de ensayos de cultivo celular. Se puede formular una dosis en modelos con animales para conseguir un intervalo de concentraciones en circulación en plasma del compuesto o, cuando proceda, del producto polipeptídico de una secuencia diana (p. ej., para conseguir una reducción de la concentración del polipéptido) que incluya la CI50 (es decir, la concentración del compuesto de prueba que proporciona una inhibición de los síntomas que es la mitad de la máxima) según se determina en un cultivo celular. Tal información se puede utilizar para determinar con más exactitud las dosis útiles en seres humanos. Los niveles en plasma se pueden medir, por ejemplo, mediante cromatografía líquida de alta resolución.
Además de su administración, según se ha discutido anteriormente, los ARNi que se exponen en la presente se pueden administrar combinados con otros agentes conocidos eficaces en el tratamiento de enfermedades o trastornos relacionados con la expresión de ALAS1. En cualquier caso, el médico que lo administre puede ajustar la cantidad y los intervalos de tiempo para la administración del ARNi en función de los resultados observados utilizando medidas estándar de la eficacia que se conocen en la técnica o se describen en la presente.
Métodos para tratar enfermedades relacionadas con la expresión de un gen ALAS1
En la presente se describe además el uso de un ARNi que tiene ALAS1 como diana para inhibir la expresión de ALAS1 y/o tratar una enfermedad, trastorno o proceso patológico que se relacione con la expresión de ALAS1.
La expresión "un trastorno relacionado con la expresión de ALAS1", un "trastorno relacionado con la expresión de ALAS1", un "proceso patológico relacionado con la expresión de ALAS1" o similares, tal como se utilizan en la presente, incluyen cualquier afección, trastorno o enfermedad en la cual se altera (p. ej., se incrementa) la expresión de ALAS1, se altera (p. ej., se incrementa) el nivel de una o más porfirinas, se altera el nivel o la actividad de una o más enzimas de la vía biosintética del grupo hemo (vía de las porfirinas) u otros mecanismos que produzcan cambios patológicos en la vía biosintética del grupo hemo. Por ejemplo, un ARNi que tenga un gen ALAS1 como diana, o una combinación de estos, se puede utilizar para el tratamiento de afecciones en las cuales los niveles de una porfirina o un precursor de una porfirina (p. ej., ALA o PBG) sean elevados (p. ej. ciertas porfirias) o afecciones en las que existan defectos en las enzimas de la vía biosintética del grupo hemo (p. ej., ciertas porfirias). Los trastornos relacionados con la expresión de ALAS1 incluyen, por ejemplo, anemia sideroblástica relacionada con X (XLSA), porfiria debida a la deficiencia de ALA-deshidratasa (porfiria de Doss), porfiria intermitente aguda (AIP), porfiria eritropoyética congénita, porfiria cutánea tardía, coproporfiria hereditaria (coproporfiria), porfiria variegata, protoporfiria eritropoyética (EPP) y eritroporfiria transitoria de la infancia.
Un "sujeto", tal como se utiliza en la presente, que se ha de tratar de acuerdo con los métodos descritos en la presente incluye un ser humano o un animal no humano, p. ej., un mamífero. El mamífero puede ser, por ejemplo, un roedor (p. ej., una rata o un ratón) o un primate (p. ej., un mono). El sujeto puede ser un ser humano.
El sujeto puede padecer un trastorno relacionado con la expresión de ALAS1 (p. ej., se le ha diagnosticado una porfiria o ha sufrido uno o más síntomas de porfiria y es portador de una mutación asociada con la porfiria) o corre el riesgo de desarrollar un trastorno relacionado con la expresión de ALAS1 (p. ej., un sujeto con un historial familiar de porfiria o un sujeto que es portador de una mutación genética asociada con la porfiria).
Las clasificaciones de las porfirias, incluidas las porfirias hepáticas agudas, se describen, p. ej., en Balwani, M. y Desnick, R.J., Blood, 120(23), publicado en línea como un artículo de la primera edición de Blood, 12 de julio, 102; DOI 10.1182/blood-2012-05-423186. Según describen Balwain y Desnick, la porfiria intermitente aguda, (AIP) la coproporfiria hereditaria (HCP) y la porfiria variegata (VP) son porfirias dominantes autosómicas y la porfiria debida a la deficiencia de ALA- deshidratasa (ADP) es recesiva autosómica. En raras ocasiones, AIP, HCP y VP aparecen como formas dominantes homozigóticas. Además, existe una forma recesiva homozigótica rara de la porfiria cutánea tardía (PCT), que es la única porfiria cutánea hepática, y también se conoce como porfiria hepatoeritropoyética. Las características clínicas y de laboratorio de estas porfirias se describen en la Tabla 11 a continuación.

El sujeto puede padecer o correr el riesgo de desarrollar una porfiria, p. ej., una porfiria hepática, p. ej., AIP, HCP, VP, ADP o una porfiria hepatoeritropoyética.
La porfiria puede ser una porfiria hepática aguda, p. ej., una porfiria hepática aguda seleccionada entre porfiria intermitente aguda (AIP), coproporfiria hereditaria (HCP), porfiria variegata (VP) y porfiria debida a la deficiencia de ALA-deshidratasa (Adp).
La porfiria puede ser una porfiria dual, p. ej., al menos dos porfirias. La porfiria dual puede comprender dos o más porfirias seleccionadas entre una porfiria intermitente aguda (AIP), coproporfiria hereditaria (HCP), porfiria variegata (VP) y porfiria debida a la deficiencia de ALA-deshidratasa (ADP).
La porfiria puede ser una porfiria hepática dominante homozigótica (p. ej., AIP, HCP o VP dominante homozigótica) o una porfiria hepatoeritropoyética. La porfiria puede ser AIP, HCP, VP o una porfiria hepatoeritropoyética, o una combinación de estas (p. ej., una porfiria dual). La AIP, HCP o VP es o bien dominante heterozigótica o dominante homozigótica.
El sujeto puede padecer o correr el riesgo de desarrollar una porfiria, p. ej., ADP y puede presentar un nivel elevado (p. ej., un nivel elevado en orina) de ALA y/o coproporfirina III. El sujeto puede padecer o correr el riesgo de desarrollar una porfiria, p. ej., ADP y puede presentar un nivel elevado del eritrocito Zn-protoporfirina.
El sujeto puede padecer o correr el riesgo de desarrollar una porfiria, p. ej., AIP y puede presentar un nivel elevado (p. ej., un nivel elevado en orina) de ALA, PBG y/o uroporfirina.
El sujeto puede padecer o correr el riesgo de desarrollar una porfiria, p. ej., HCP y presenta un nivel elevado (p. ej., un nivel elevado en orina) de ALA, PBG, y/o coproporfirina III. El sujeto puede padecer o correr el riesgo de desarrollar una porfiria, p. ej., HCP y puede presentar un nivel elevado (p. ej., un nivel elevado en las heces) de coproporfirina III.
El sujeto puede padecer o correr el riesgo de desarrollar una porfiria, p. ej., VP y puede presentar un nivel elevado (p. ej., un nivel elevado en orina) de ALA, PBG, y/o coproporfirina III.
El sujeto puede padecer o correr el riesgo de desarrollar una porfiria, p. ej., HCP y puede presentar un nivel elevado (p. ej., un nivel elevado en las heces) de coproporfirina III y/o protoporfirina.
El sujeto puede padecer o correr el riesgo de desarrollar una porfiria, p. ej., PCT (p. ej., porfiria hepatoeritropoyética) y puede presentar un nivel elevado (p. ej., un nivel elevado en orina) de uroporfirina y/o 7-carboxilatoporfirina. El sujeto puede padecer o correr el riesgo de desarrollar una porfiria, p. ej., PCT (p. ej., porfiria hepatoeritropoyética) y puede presentar un nivel elevado (p. ej., un nivel elevado en las heces) de uroporfirina y/o 7-carboxilatoporfirina. Una mutación asociada con la porfiria incluye cualquier mutación en un gen que codifique una enzima de la vía biosintética del grupo hemo (vía de las porfirinas) o un gen que altere la expresión de un gen de la vía biosintética del grupo hemo. El sujeto puede ser portador de una o más mutaciones en una enzima de la vía de las porfirinas (p. ej., una mutación en ALA-deshidratasa o PBG-desaminasa). El sujeto puede padecer una porfiria aguda (p. ej., AIP, porfiria debida a la deficiencia de ALA-deshidratasa).
En algunos casos, los pacientes con una porfiria hepática aguda (p. ej., AIP) o pacientes que son portadores de mutaciones asociadas con una porfiria hepática aguda (p. ej., AIP) pero los cuales no presentan síntomas, tienen unos niveles elevados de ALA y/o PBG en comparación con individuos sanos. Remítase, p. ej., a Floderus, Y. et al., Clinical Chemistry, 52(4): 701-707, 2006; Sardh et al., Clinical Pharmacokinetics, 46(4): 335-349, 2007. En tales casos, el nivel de ALA y/o PBG se puede incrementar incluso cuando el paciente no padece o nunca ha padecido un ataque. En algunos casos de este tipo, el paciente es por lo demás completamente asintomático. En algunos casos de este tipo, el paciente sufre dolor, p. ej., dolor neuropático, el cual puede ser un dolor crónico (p. ej., dolor neuropático crónico). En algunos casos, el paciente padece una neuropatía. En algunos casos, el paciente padece una neuropatía progresiva.
El sujeto que se ha de tratar de acuerdo con los métodos descritos en la presente puede tener un nivel elevado de una porfirina o un precursor de una porfirina, p. ej., ALA y/o PBG. Los niveles de una porfirina o un precursor de una porfirina se pueden evaluar utilizando métodos conocidos en la técnica o métodos que se describen en la presente. Por ejemplo, los métodos para evaluar los niveles de ALA y PBG en orina y en plasma, así como también los niveles de porfirinas en orina y en plasma, se describen en Floderus, Y. et al., Clinical Chemistry, 52(4): 701-707, 2006; y Sardh et al., Clinical Pharmacokinetics, 46(4): 335-349, 2007.
El sujeto puede ser un modelo animal de una porfiria, p. ej., un modelo de ratón de una porfiria (p. ej., un ratón mutado según se describe en Lindberg et al. Nature Genetics, 12: 195-199, 1996). El sujeto puede ser un ser humano, p. ej., un ser humano que padece o corre el riesgo de desarrollar una porfiria, según se describe en la presente. El sujeto puede no estar sufriendo un ataque agudo de porfiria. El sujeto puede que nunca haya sufrido un ataque. El paciente puede sufrir dolor crónico. El paciente puede presentar daños nerviosos. El sujeto puede presentar cambios en el EMG y/o cambios en la velocidad de conducción nerviosa. El sujeto puede no presentar
síntomas. El sujeto puede correr el riesgo de desarrollar una porfiria (p. ej., es portador de una mutación génica asociada con una porfiria) y no presenta síntomas. El sujeto puede haber sufrido previamente un ataque agudo pero no presentar síntomas en el momento del tratamiento.
El sujeto puede correr el riesgo de desarrollar una porfiria y se trata profilácticamente para prevenir el desarrollo de la porfiria. El sujeto puede presentar un nivel elevado de una porfirina o un precursor de una porfirina, p. ej., ALA y/o PBG. El tratamiento profiláctico puede empezar en la pubertad. El tratamiento puede reduce el nivel (p. ej., el nivel en plasma o el nivel en orina) de una porfirina o un precursor de una porfirina, p. ej., ALA y/o PBG. El tratamiento puede prevenir el desarrollo de un nivel elevado de una porfirina o un precursor de una porfirina, p. ej., ALA y/o PBG. El tratamiento puede prevenir el desarrollo, o reduce la frecuencia o la gravedad, de un síntoma asociado con una porfiria, p. ej., dolor o daños nerviosos.
El nivel de una porfirina o un precursor de una porfirina, p. ej., ALA o PBG, puede ser elevado, p. ej., en una muestra de plasma u orina del sujeto. El nivel de una porfirina o un precursor de una porfirina, p. ej., ALA o PBG, en el sujeto se puede evaluar en función del nivel absoluto de la porfirina o el precursor de la porfirina, p. ej., ALA o PBG en una muestra del sujeto. El nivel de una porfirina o un precursor de una porfirina, p. ej., ALA o PBG, en el sujeto se puede evaluar en función del nivel relativo de la porfirina o el precursor de la porfirina, p. ej., ALA o PBG, en una muestra del sujeto. El nivel relativo puede ser relativo respecto al nivel de otra proteína o compuesto, p. ej., el nivel de creatinina, en una muestra del sujeto. La muestra puede ser una muestra de orina. La muestra puede ser una muestra de plasma. La muestra puede ser una muestra de las heces.
Un nivel elevado de una porfirina o un precursor de una porfirina, p. ej., ALA y/o PBG, se puede establecer, p. ej., mostrando que el sujeto presenta un nivel de una porfirina o un precursor de una porfirina, p. ej., ALA y/o PBG (p. ej., un nivel en plasma u orina de ALA y/o PBG) que sea superior, o superior o igual, a un valor de referencia. Un médico con experiencia en el tratamiento de porfirias sería capaz de determinar si el nivel de una porfirina o un precursor de una porfirina (p. ej., ALA y/o PBG) es elevado, p. ej., con el fin de diagnosticar una porfiria o para determinar si un sujeto corre el riesgo de desarrollar una porfiria, p. ej., un sujeto puede estar predispuesto a sufrir un ataque agudo o una patología asociada con una porfiria tal como, p. ej., dolor crónico (p. ej., dolor neuropático) y una neuropatía (p. ej., una neuropatía progresiva).
La expresión "valor de referencia", tal como se utiliza en la presente, se refiere a un valor del sujeto cuando el sujeto no padece un estado patológico, o un valor de un sujeto normal o sano, o un valor de una muestra o población de referencia, p. ej., un grupo de sujetos normales o sanos (p. ej., un grupo de sujetos que no son portadores de ninguna mutación asociada con una porfiria y/o un grupo de sujetos que no padecen los síntomas asociados con una porfiria).
El valor de referencia puede ser un nivel previo a la enfermedad en el mismo individuo. El valor de referencia puede ser un nivel en una muestra o población de referencia. El valor de referencia puede ser el valor medio o mediano en una muestra o población de referencia. El valor de referencia puede ser el valor que corresponde a dos desviaciones estándar por encima de la media en una muestra o población de referencia. El valor de referencia puede ser el valor que corresponde a 2.5, 3, 3.5, 4, 4.5 o 5 desviaciones estándar por encima de la media en una muestra o población de referencia.
En algunos aspectos, donde el sujeto presenta un nivel elevado de una porfirina o un precursor de una porfirina, p. ej., ALA y/o pBg , el sujeto presenta un nivel de ALA y/o PBG que es al menos un 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80% o 90% superior respecto a un valor de referencia. El sujeto puede presenta un nivel de una porfirina o un precursor de una porfirina, p. ej., ALA y/o PBG, que es al menos 2, 3, 4, 5, 6, 7, 8, 9 o 10 veces superior respecto a un valor de referencia.
El valor de referencia puede ser un límite de referencia superior. La expresión "límite de referencia superior", tal como se utiliza en la presente, se refiere a un nivel que representa el límite superior del intervalo de confianza del 95% para una muestra o población de referencia, p. ej., un grupo de individuos sanos o normales (p. ej., de origen natural), p. ej., individuos que no son portadores de ninguna mutación genética asociada con una porfiria y/o individuos que no padecen ninguna porfiria. Por consiguiente, un límite de referencia inferior se refiere a un nivel que representa el límite inferior del mismo intervalo de confianza del 95%.
En algunos aspectos, cuando el sujeto presenta un nivel elevado, p. ej., un nivel en plasma o un nivel en orina, de una porfirina o un precursor de una porfirina, p. ej., ALA o PBG, el nivel es superior o igual a 2 veces, 3 veces, 4 veces o 5 veces el de un valor de referencia, p. ej., un valor de referencia superior. El sujeto puede presentar un nivel en orina de una porfirina o un precursor de una porfirina, p. ej., ALA o PBG que es superior a 4 veces el de un límite de referencia superior.
El valor de referencia puede ser un valor proporcionado en Floderus, Y. et al., Clinical Chemistry, 52(4): 701-707, 2006 o Sardh et al., Clinical Pharmacokinetics, 46(4): 335-349, 2007. El valor de referencia puede ser un valor proporcionado en la Tabla 1 de Sardh et al.
El sujeto puede ser un ser humano y presenta un nivel de PBG en orina superior o igual a 4.8 mmol/mol de creatinina. El sujeto puede ser un ser humano y presenta un nivel de PBG en orina superior a, o superior o igual a, aproximadamente 3, 4, 5, 6, 7 u 8 mmol/mol de creatinina.
El valor de referencia para PBG en plasma puede ser de 0.12 pmol/L. El sujeto puede ser un ser humano y presenta un nivel de PBG en plasma superior a, o superior o igual a, 0.10 gmol/L, 0.12 gmol/L, 0.24 gmol/L, 0.36 gmol/L, 0.48 gmol/L o 0.60 gmol/L. El sujeto puede ser un ser humano y presenta un nivel de PBG en plasma superior a, o superior o igual a, 0.48 gmol/L.
El valor de referencia para PBG en orina puede ser de 1.2 mmol/mol de creatinina. El sujeto puede ser un ser humano y puede presentar un nivel de PBG en orina superior a, o superior o igual a, 1.0 mmol/mol de creatinina, 1.2 mmol/mol de creatinina, 2.4 mmol/mol de creatinina, 3.6 mmol/mol de creatinina, 4.8 mmol/mol de creatinina o 6.0 mmol/mol de creatinina. El sujeto puede ser un ser humano y puede presentar un nivel de PBG en orina superior a, o superior o igual a, 4.8 mmol/mol de creatinina.
El valor de referencia para ALA en plasma puede ser de 0.12 gmol/L. El sujeto puede ser un ser humano y puede presentar un nivel de ALA en plasma superior a, o superior o igual a, 0.10 gmol/L, 0.12 gmol/L, 0.24 gmol/L, 0.36 gmol/L, 0.48 gmol/L o 0.60 gmol/L. El sujeto puede ser un ser humano y puede presentar un nivel de ALA en plasma superior a, o superior o igual a, 0.48 gmol/L.
El valor de referencia para ALA en orina puede ser de 3.1 mmol/mol de creatinina. El sujeto puede ser un ser humano y puede presentar un nivel de ALA en orina superior a, o superior o igual a, 2.5 mmol/mol de creatinina, 3.1 mmol/mol de creatinina, 6.2 mmol/mol de creatinina, 9.3 mmol/mol de creatinina, 12.4 mmol/mol de creatinina o 15.5 mmol/mol de creatinina.
El valor de referencia para la porfirina en plasma puede ser de 10 nmol/L. El sujeto puede ser un ser humano y puede presentar un nivel de porfirina en plasma superior a, o superior o igual a, 10 nmol/L. El sujeto puede ser un ser humano y puede presentar un nivel de porfirina en plasma superior a, o superior o igual a 8, 10, 15, 20, 25, 30, 35, 40, 45 o 50 nmol/L. El sujeto puede ser un ser humano y puede presentar un nivel de porfirina en plasma superior a, o superior o igual a, 40 nmol/L. El valor de referencia para la porfirina en orina puede ser de 25 gmol/mol de creatinina. ElEl sujeto es un ser humano y presenta un nivel de porfirina en orina superior a, o superior o igual a, 25 gmol/mol de creatinina. El sujeto puede ser un ser humano y puede presentar un nivel de porfirina en orina superior a, o superior o igual a, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75 u 80 gmol/mol de creatinina.
El sujeto puede presentar un nivel, p. ej., un nivel en plasma o un nivel en orina, de una porfirina o un precursor de una porfirina, p. ej., ALA o PBG, que es superior al de un 99% de los individuos en una muestra de individuos sanos. El sujeto puede presentar un nivel, p. ej., un nivel en plasma o un nivel en orina, de ALA o PBG que es superior a dos desviaciones estándar por encima del nivel medio en una muestra de individuos sanos.
El sujeto puede presentar un nivel de ALA en orina que es 1.6 o más veces el nivel medio en un sujeto normal (p. ej., un sujeto que no es portador de ninguna mutación asociada con una porfiria). El sujeto puede presentar un nivel de ALA en plasma que es 2 o 3 veces el nivel medio en un sujeto normal. El sujeto puede presentar un nivel de PBG en orina que es cuatro o más veces el nivel medio en un sujeto normal. El sujeto puede presentar un nivel de PBG en plasma que es cuatro o más veces el nivel medio en un sujeto normal.
El método puede ser eficaz para reducir el nivel de una porfirina o un precursor de una porfirina, p. ej., ALA y/o PBG. El método puede ser eficaz para producir una reducción predeterminada en el nivel elevado de la porfirina o el precursor de la porfirina, p. ej., ALA o PBG. La reducción predeterminada puede ser una reducción de al menos un 10%, 20%, 30%, 40% o 50%. La reducción predeterminada puede ser una reducción que es eficaz para prevenir o mejorar los síntomas, p. ej., el dolor o ataques recurrentes.
La reducción predeterminada puede ser una reducción que corresponde a 1, 2, 3 o más desviaciones estándar, donde la desviación estándar se determina en función de los valores de una muestra de referencia, p. ej., una muestra de referencia según se describe en la presente.
La reducción predeterminada puede ser una reducción que modifica el nivel de la porfirina o el precursor de la porfirina hasta un nivel que es inferior a, o hasta un nivel que es inferior o igual a, un valor de referencia (p. ej., un valor de referencia según se describe en la presente).
El sujeto que se ha de tratar de acuerdo con los métodos descritos puede sufrir dolor, p. ej., dolor crónico. El sujeto puede padece o correr el riesgo de desarrollar una porfiria, p. ej., una porfiria hepática aguda, p. ej., AIP. El método puede ser eficaz para tratar el dolor, p. ej., mediante la reducción de la intensidad del dolor o curando el dolor. El método puede ser eficaz para reducir o prevenir daños nerviosos.
El sujeto que se ha de tratar de acuerdo con los métodos descritos en la presente (a) puede presentar un nivel elevado de ALA y/o PBG y (b) puede sufrir dolor, p. ej., dolor crónico. El método puede ser eficaz para reducir un
nivel elevado de ALA y/o PBG y/o para tratar el dolor, p. ej., mediante la reducción de la intensidad del dolor o curando el dolor.
El sujeto puede ser un mamífero que sirve como modelo para un trastorno relacionado con la expresión de ALAS1. El sujeto puede ser un animal que sirve como modelo para una porfiria (p. ej., un animal modificado genéticamente con una o más mutaciones). La porfiria es AIP y el sujeto puede ser un modelo animal de AIP. El sujeto puede ser un ratón modificado genéticamente que es deficiente en porfobilinógeno-desaminasa tal como, por ejemplo, el ratón descrito en Lindberg et al., Nature Genetics, 12:195-199, 1996, o el ratón R167Q homozigótico descrito en Yasuda, M., Yu, C. Zhang, J., Clavero, S., Edelmann, W., Gan, L., Phillips, J.D. y Desnick, R.J. Acute intermittent porphyria: A severely affected knock-in mouse that mimics the human homozygous dominant phenotype (resumen de la presentación del 14 de octubre de 2011 en el Congreso American Society of Human Genetics ; Programa N.° 1308F; al cual se accedió en línea el 4 de abril de 2012 en ichg2011.org/cgi-bin/showdetail.pl?absno=21167). Se han generado varios modelos de activación para mutaciones que provocan AIP dominante homozigótica en seres humanos. Las mutaciones empleadas incluyen, p. ej., R167Q, R173Q y R173W en la PBG-desaminasa. Los casos homozigóticos viables incluyeron R167Q/R176Q y R167Q/R173Q, los cuales presentan niveles de ALA y PBG constitutivamente elevados análogos a los del genotipo en AIP dominante homozigótica en seres humanos; p. ej., un modelo de ratón de AIP homozigótica variable de este tipo puede ser el sujeto.
Un sujeto que se ha de tratar de acuerdo con los métodos descritos en la presente (p. ej., un paciente o sujeto humano) puede correr el riesgo de desarrollar, o puede que se le haya diagnosticado, un trastorno relacionado con la expresión de ALAS1, p. ej., una porfiria. El sujeto puede ser un sujeto que ha padecido uno o más ataques agudos de uno o más síntomas porfíricos. El sujeto puede ser un sujeto que ha padecido crónicamente uno o más síntomas de porfiria (p. ej., dolor, p. ej., dolor neuropático y/o una neuropatía, p. ej., una neuropatía progresiva). El sujeto puede ser portador de una alteración genética (p. ej., una mutación) según se describe en la presente, pero por lo demás no presenta síntomas. El sujeto puede haber sido tratado previamente con un producto de tipo hemo (p. ej., hemina, arginato de hemo o albúmina que contiene hemo), según se describe en la presente.
Un sujeto (p. ej., un sujeto con una porfiria tal como, p. ej., AIP) que se ha de tratar de acuerdo con los métodos descritos en la presente puede haber experimentado recientemente o estar experimentando actualmente un pródromo. En algunos aspectos, se puede administrar al sujeto un tratamiento combinado, p. ej., un ARNi según se describe en la presente y uno o más tratamientos adicionales que se sabe que son eficaces contra la porfiria (p. ej., glucosa y/o un producto de tipo hemo tal como la hemina, según se describe en la presente) o sus síntomas asociados.
Un ARNi según se describe en la presente se puede administrar combinado con glucosa o dextrosa. Por ejemplo, se puede proporcionar dextrosa al 10-20% en solución salina normal por vía intravenosa. Normalmente, cuando se administra glucosa, se administran al menos 300 g de glucosa al 10% por vía intravenosa diariamente. El ARNi (p. ej., un ARNi en una formulación LNP) también se puede administrar por vía intravenosa como parte de la misma infusión que se utiliza para administrar la glucosa o dextrosa, o como una infusión separada que se administra antes, después o de forma simultánea a la administración de la glucosa o dextrosa. El ARNi se puede administrar mediante una vía de administración diferente (p. ej., subcutáneamente). En aún otra realización, el ARNi se administra combinado con nutrición parenteral total. El ARNi se puede administrar antes, después o de forma simultánea con la administración de nutrición parenteral total.
El ARNi se puede administrar combinado con un producto de tipo hemo (p. ej., hemina, arginato de hemo o albúmina que contiene hemo). Asimismo, el ARNi se puede administrar combinado con un producto de tipo hemo y glucosa, un producto de tipo hemo y dextrosa o un producto de tipo hemo y nutrición parenteral total.
Un "pródromo", tal como se utiliza en la presente, incluye cualquier síntoma que el sujeto individual haya experimentado previamente justo antes de desarrollar un ataque agudo. Los síntomas típicos de un pródromo incluyen, p. ej., dolor abdominal, náusea, cefaleas, síntomas psicológicos (p. ej., ansiedad), inquietud y/o insomnio. El sujeto experimenta dolor (p. ej., dolor abdominal y/o una cefalea) durante el pródromo. El sujeto puede experimentar náusea durante el pródromo. El sujeto experimenta síntomas psicológicos (p. ej., ansiedad) durante el pródromo. El sujeto se puede volver inquieto y/o padecer insomnio durante el pródromo.
Un "ataque" agudo de porfiria implica el inicio de uno o más síntomas de porfiria, normalmente en un paciente que es portador de una mutación asociada con la porfiria (p. ej., una mutación en un gen que codifica una enzima de la vía de las porfirinas).
La administración de un ARNi de ALAS1 puede provocar una reducción del nivel de una o más porfirinas o precursores de porfirinas, según se describe la presente (p. ej., ALA y/o PBG). La reducción se puede medir con relación a cualquier valor de referencia o control adecuado. Por ejemplo, la reducción del nivel de una o más porfirinas o precursores de porfirinas se puede establecer en un sujeto individual, p. ej., como una reducción de al menos un 5%, 10%, 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50% o más en comparación con el nivel previo al tratamiento (p. ej., inmediatamente antes del tratamiento). La reducción del nivel de un precursor de una porfirina, una porfirina o un metabolito de una porfirina se puede medir utilizando cualquier método conocido en la técnica. Por
ejemplo, el nivel de PBG y/o ALA en orina o plasma se puede evaluar utilizando la prueba de Watson-Schwartz, cromatografía de intercambio iónico o cromatografía líquida de alta resolución-espectrometría de masas. Remítase, p. ej., a Thunell (1993).
La administración de un ARNip de ALAS1 puede ser eficaz para reducir el nivel de ALA y/o PBG en el sujeto. El nivel de ALA o PBG en el sujeto se puede evaluar, p. ej., basándose en el nivel absoluto de ALA o PBG, o basándose en el nivel relativo de ALA o PBG (p. ej., relativo respecto al nivel de otra proteína o compuesto, p. ej., el nivel de creatinina) en una muestra del sujeto. La muestra puede ser una muestra de orina. La muestra puede ser una muestra de plasma.
Un ARNi que tiene ALAS1 como diana se puede administrar combinado con uno o más tratamientos adicionales, p. ej., otro tratamiento que se sabe que es eficaz para tratar la porfiria o síntomas de la porfirina. Por ejemplo, el otro tratamiento puede ser glucosa (p. ej., glucosa IV) o un producto de tipo hemo (p. ej., hemina, arginato de hemo o albúmina que contiene hemo). El o los tratamientos adicionales se pueden administrar antes, después o de forma simultánea con la administración del ARNi.
El ARNi y un agente terapéutico adicional se pueden administrar combinados en la misma composición, p. ej., por vía intravenosa, o el agente terapéutico adicional se puede administrar como parte de una composición separada o mediante otro método descrito en la presente.
La administración de ARNi o la administración de ARNi combinado con uno o más tratamientos adicionales (p. ej., glucosa, dextrosa o similares) puede reducir la frecuencia de ataques agudos (p. ej., mediante la prevención de los ataques agudos de modo que ya no se produzcan, o puede reducir el número de ataques que se producen en un cierto periodo de tiempo, p. ej., se producen menos ataques por año). El ARNi se puede administrar de acuerdo con una pauta posológica regular, p. ej., diariamente, semanalmente, cada dos semanas o mensualmente.
El ARNi se administra después de un ataque agudo de porfiria. En algunas realizaciones de este tipo, el ARNi se encuentra en una composición, p. ej., una composición que comprende una formulación lipídica, p. ej., una formulación LNP.
El ARNi se puede administrar durante un ataque agudo de porfiria. El ARNi se puede encontrar en una composición, p. ej., una composición que comprende una formulación lipídica (p. ej., una formulación LNP) o una composición que comprende un conjugado de tipo GalNAc.
La administración de un ARNip de ALAS1 puede ser eficaz para reducir la gravedad del ataque (p. ej., mejora uno o más signos o síntomas asociados con el ataque). La administración de un ARNip de ALAS1 puede ser eficaz para reducir la duración de un ataque. La administración de un ARNip de ALAS1 es eficaz para detener un ataque. El ARNi se puede administrar profilácticamente para prevenir un ataque agudo de porfiria. El ARNi se puede encontrar en la forma de un conjugado de tipo GalNAc, p. ej., en una composición que comprende un conjugado de tipo GalNAc. La administración profiláctica se puede llevar a cabo antes, durante o después de la exposición a un factor precipitante o la aparición de este. El sujeto puede correr el riesgo de desarrollar una porfiria.
El ARNip se puede administrar durante un pródromo. El pródromo se puede caracterizar por dolor (p. ej., cefalea y/o dolor abdominal), náusea, síntomas psicológicos (p. ej., ansiedad), inquietud y/o insomnio.
El ARNi se puede administrar durante una fase particular del ciclo menstrual, p. ej., durante la fase luteínica.
La administración de un ARNip de ALAS1 puede ser eficaz para prevenir ataques (p. ej., ataques recurrentes que se asocian con un pródromo y/o con un factor precipitante, p. ej., con una fase particular del ciclo menstrual, p. ej., la fase luteínica). La administración de un ARNip de ALAS1 puede ser eficaz para reducir la frecuencia de los ataques. La administración de un ARNip de ALAS1 puede ser eficaz para reducir la gravedad del ataque (p. ej., mejora uno o más signos o síntomas asociados con el ataque). La administración de un ARNip de ALAS1 puede ser eficaz para reducir la duración de un ataque. La administración de un ARNip de ALAS1 puede ser eficaz para detener un ataque.
La administración de un ARNip de ALAS1 puede ser eficaz para prevenir o reducir la frecuencia o la intensidad del dolor, p. ej., el dolor neuropático.
La administración de un ARNip de ALAS1 puede ser eficaz para prevenir o reducir la frecuencia o la intensidad de una neuropatía.
Los efectos de la administración de un ARNip de ALAS1 se pueden establecer, por ejemplo, por comparación con un control adecuado. Por ejemplo, se puede establecer una reducción de la frecuencia de los ataques agudos, así como también una reducción del nivel de una o más porfirinas o precursores de porfirinas, por ejemplo, en un grupo de pacientes con AIP, como una reducción de la frecuencia en comparación con un grupo de control adecuado. Un grupo de control (p. ej., un grupo de individuos similares o el mismo grupo de individuos en un diseño cruzado) puede incluir, por ejemplo, una población no tratada, una población que ha sido tratada con un tratamiento convencional para la porfiria (p. ej., un tratamiento convencional para AIP puede incluir glucosa, hemina o ambas);
una población que ha sido tratada con placebo o un ARNi sin diana, opcionalmente combinado con uno o más tratamientos convencionales para la porfiria (p. ej., glucosa, p. ej., glucosa IV) y similares.
Un sujeto "que corre el riesgo" de desarrollar una porfiria, tal como se utiliza en la presente, incluye un sujeto con un historial familiar de porfiria y/o un historial de uno o más síntomas porfíricos recurrentes o crónicos, y/o un sujeto que es portador de una alteración genética (p. ej., una mutación) en un gen que codifica una enzima de la vía biosintética del grupo hemo, y un sujeto que es portador de una alteración genética, p. ej., una mutación que se sabe que está asociada con la porfiria.
La alteración, p. ej., la mutación puede hacer que el individuo sea susceptible de padecer un ataque agudo (p. ej., tras la exposición a un factor precipitante, p. ej., un fármaco, el seguimiento de un régimen u otro factor precipitante, p. ej., un factor precipitante como los que se describen en la presente). La alteración, p. ej., la mutación, se puede asociar con unos niveles elevados de una porfirina o un precursor de una porfirina (p. ej., ALA y/o PBG). La alteración, p. ej., la mutación, se puede asociar con dolor crónico (p. ej., dolor neuropático crónico) y/o una neuropatía (p. ej., una neuropatía progresiva). La alteración, p. ej., la mutación, se puede asociar con cambios en el EMG y/o en las velocidades de conducción nerviosa.
La alteración puede ser una mutación en el gen ALAS1. La alteración puede ser una mutación en el promotor del gen ALAS1 o en regiones en dirección 5' o dirección 3' del gen ALAS1. La alteración puede ser una mutación en factores de transcripción u otros genes que interaccionan con ALAS1. La alteración puede ser una alteración, p. ej., una mutación, en un gen que codifica una enzima en la vía biosintética del grupo hemo.
El sujeto puede tener una alteración genética según se describe en la presente (p. ej., una mutación genética que se sabe que está asociada con una porfiria). En algunos aspectos, el sujeto puede presentar un nivel elevado (p. ej., nivel en orina o plasma) de ALA y/o PBG. En algunos aspectos, el sujeto puede no presentar un nivel elevado de ALA y/o PBG. El sujeto puede tener una alteración genética según se describe en la presente y presenta otros síntomas, p. ej., dolor crónico, cambios en el EMG, cambios en la velocidad de conducción nerviosa y/u otros síntomas asociados con una porfiria. El sujeto puede tener una alteración genética pero no padece ataques agudos. El sujeto puede tener una mutación asociada con AIP, HCP, VP o ADP.
La porfiria puede ser AIP. En algunos de tales aspectos, el sujeto puede tener una alteración, p. ej., al menos una mutación, en el gen de la PBG-desaminasa. En la técnica se conocen muchas mutaciones de la PBG-desaminasa, por ejemplo, según se describe en Hrdinka, M. et al. Physiological Research, 55 (Supl. 2):S119-136 (2006). El sujeto puede ser heterozigótico respecto a una mutación de la PBG-desaminasa. El sujeto puede ser homozigótico respecto a una mutación de la PBG-desaminasa. Un sujeto homozigótico puede ser portador de dos mutaciones idénticas o dos mutaciones diferentes en el gen de la PBG-desaminasa.
La porfiria puede ser HCP. En algunos de tales aspectos, el sujeto puede tener una alteración, p. ej., al menos una mutación, en el gen que codifica la enzima coproporfirinógeno III-oxidasa.
La porfiria puede ser VP. En algunos de tales aspectos, el sujeto puede tener una alteración, p. ej., al menos una mutación, en el gen que codifica la protoporfrinógeno-oxidasa.
La porfiria puede ser ADP, p. ej., ADP recesiva autosómica. El sujeto puede tener una alteración, p. ej., al menos una mutación, en el gen que codifica la ALA-deshidratasa.
Los métodos de tratamiento que se proporcionan en la presente pueden servir para mejorar uno o más síntomas asociados con una porfiria, para reducir la frecuencia de los ataques asociados con la porfiria, para reducir la probabilidad de que se produzca un ataque de uno o más síntomas asociados con la porfiria tras la exposición a un factor precipitante o para reducir el riesgo de desarrollar afecciones asociadas con la porfiria (p. ej., una neuropatía (p. ej., una neuropatía progresiva), un cáncer hepatocelular). Además, los métodos que se proporcionan en la presente pueden servir para reducir el nivel de uno o más precursores de porfirinas, porfirinas y/o productos o metabolitos relacionados con las porfirinas. El nivel de una porfirina o un precursor de una porfirina se puede medir en cualquier muestra biológica tal como, p. ej., orina, sangre, heces, fluido cerebroespinal o una muestra tisular. La muestra puede estar presente dentro de un sujeto o se puede obtener o extraer a partir del sujeto. La porfiria puede ser AIP y se reduce el nivel de PBG y/o ALA. El producto o metabolito de la porfirina puede ser porfobilina, porfobilinógeno o uroporfirina. La reducción del nivel de un producto o metabolito de una porfirina se puede medir utilizando cualquier método conocido en la técnica. Por ejemplo, el nivel de PBG y/o ALA en orina o plasma se puede evaluar utilizando la prueba de Watson-Schwartz, cromatografía de intercambio iónico o cromatografía líquida de alta resolución-espectrometría de masas. Remítase, p. ej., a Thunell (1993).
Los métodos descritos en la presente también pueden servir para reducir niveles crónicamente elevados de precursores de porfirinas (p. ej. ALA y/o PBG) en sujetos que padecen una porfiria (p. ej., una porfiria hepática aguda, p. ej., AIP) o que corren el riesgo de desarrollar una porfiria. Los métodos para evaluar los niveles en plasma y orina (p. ej., niveles crónicamente elevados) de precursores de porfirinas incluyen, p. ej., HPLC-espectrometría de masas y cromatografía de intercambio iónico. Los niveles de precursores de porfirinas se pueden expresar como el
nivel relativo respecto a otra proteína o compuesto, p. ej., la creatinina. Remítase, p. ej., a Floderus, Y. et al., Clinical Chemistry, 52(4): 701-707, 2006; Sardh et al., Clinical Pharmacokinetics, 46(4): 335-349, 2007.
Un "factor precipitante", tal como se utiliza en la presente, se refiere a un factor endógeno o exógeno que puede inducir un ataque agudo de uno o más síntomas asociados con una porfiria. Los factores precipitantes incluyen ayuno (u otras formas de ingesta calórica reducida o inadecuada, debido a dietas extremas, atletismo de fondo, etc.), estrés metabólico (p. ej., infecciones, cirugía, viajes aéreos internacionales y estrés psicológico), hormonas endógenas (p. ej., progesterona), fumar cigarrillos, agentes químicos exógenos liposolubles (que incluyen, p. ej., agentes químicos presentes en el humo del tabaco, ciertos fármacos recetados, disolventes orgánicos, biocidas, componentes de bebidas alcohólicas), factores endocrinos (p. ej., hormonas reproductivas (las mujeres pueden experimentar exacerbaciones durante el periodo premenstrual), estrógenos sintéticos, progesteronas, estimulantes de la ovulación y terapia de reemplazo hormonal). Remítase, por ejemplo, a Thunell (1993). Los factores precipitantes comunes incluyen fármacos que inducen el citocromo P450 y fenobarbital.
Los síntomas asociados con una porfiria pueden incluir dolor o calambres abdominales, cefaleas, efectos provocados por anomalías del sistema nervioso y sensibilidad a la luz, que provoca sarpullidos, ampollas y aparición de cicatrices en la piel (fotodermatitis). La porfiria puede ser AIP. Los síntomas de la AIP incluyen síntomas gastrointestinales (p. ej., dolor abdominal intenso y poco localizado, náusea/vómitos, estreñimiento, diarrea, íleo), síntomas urinarios (disuria, retención/incontinencia urinaria u orina oscura), síntomas neurológicos (p. ej., neuropatía sensorial, neuropatía motriz (p. ej., que afecta a los nervios craneales y/o que provoca debilidad en los brazos o las piernas), convulsiones, dolor neuropático, neuropatía progresiva, cefaleas, síntomas neuropsiquiátricos (p. ej., confusión mental, ansiedad, agitación, alucinación, histeria, delirio, apatía, depresión, fobias, psicosis, insomnio, somnolencia, coma), participación del sistema nervioso autonómico (que provoca, p. ej., síntomas cardiovasculares tales como taquicardia, hipertensión y/o arritmias, así como también otros síntomas tales como, p. ej., un incremento de los niveles de catecolamina en circulación, sudoración, inquietud y/o temblores), deshidratación y anomalías electrolíticas.
Se puede administrar un ARNi que tiene ALAS1 como diana junto con (p. ej., antes, después o de forma simultánea) otro tratamiento que pueda servir para aliviar uno o más de los síntomas anteriores. Por ejemplo, el dolor abdominal se puede tratar, p. ej., con analgésicos narcóticos, las convulsiones se pueden tratar, p. ej., con medicamentos contra las convulsiones, las náuseas/vómitos se pueden tratar, p. ej., con fenotiazinas y la taquicardia/hipertensión se puede tratar, p. ej., con bloqueadores beta.
Se pretende que el término "reducir" (o "incrementar") se refiera a un cambio medible, p. ej., un cambio estadísticamente significativo. El cambio puede ser, por ejemplo, un cambio de al menos un 5%, 10%, 20%, 30%, 40%, 50% o más (p. ej., reducción (o incremento) respecto a un valor de referencia, p. ej., una referencia en la que no se proporcione ARNi).
Se describe además en la presente el uso de un ARNi o una composición farmacéutica de este, p. ej., para tratar un trastorno relacionado con la expresión de ALAS1, combinado con otros agentes farmacéuticos y/u otros métodos terapéuticos, p. ej., con agentes terapéuticos conocidos y/o métodos terapéuticos conocidos tales como, por ejemplo, los que se emplean actualmente para tratar el trastorno. El ARNi o la composición farmacéutica de este se puede administrar junto con un producto de tipo hemo (p. ej., hemina, arginato de hemo o albúmina que contiene hemo, según se describe en la presente) y/o junto con infusiones intravenosas de glucosa. El ARNi o la composición farmacéutica de este se puede utilizar profilácticamente, p. ej., para prevenir o mejorar los síntomas de un ataque anticipado de porfiria aguda. El uso profiláctico se puede hacer coincidir con la exposición o exposición anticipada del sujeto a un factor precipitante. Un factor precipitante, según se describe en la presente, puede ser cualquier factor endógeno o exógeno que se sepa que precipita un ataque agudo. Por ejemplo, la fase premenstrual es un factor precipitante endógeno y un fármaco que induce el citocromo P450 es un factor precipitante exógeno.
La cantidad eficaz para el tratamiento del trastorno relacionado con la expresión de ALAS1 (p. ej., una porfiria tal como AIP) depende del tipo de trastorno que se ha de tratar, la gravedad de los síntomas, el sujeto que se esté tratando, el sexo, la edad, el estado de salud general del sujeto, la vía de administración, etc. Para cualquier caso dado, un experto en la técnica podrá determinar la "cantidad eficaz" adecuada utilizando experimentación rutinaria. La monitorización de la eficacia del tratamiento o la prevención se encuentra dentro de las competencias de un experto en la técnica y se lleva a cabo midiendo cualquiera de tales parámetros o cualquier combinación de parámetros. En relación con la administración de un ARNi que tiene ALAS1 como diana o una composición farmacéutica de este, la expresión "eficaz contra" un trastorno relacionado con la expresión de ALAS1 indica que la administración de un modo clínicamente adecuado provoca un efecto beneficioso, p. ej., para un paciente individual o para al menos una fracción de los pacientes, p. ej., una fracción estadísticamente significativa de los pacientes. Los efectos beneficiosos incluyen, p. ej., la prevención o la reducción de los síntomas u otros efectos. Por ejemplo, los efectos beneficiosos incluyen, p. ej., una mejora (p. ej., reducción de la gravedad o la frecuencia) de los síntomas, una reducción de la gravedad o la frecuencia de los ataques, un menor riesgo de desarrollar una enfermedad asociada (p. ej., una neuropatía (p. ej., una neuropatía progresiva), cáncer hepatocelular), una capacidad mejorada para tolerar un factor precipitante, una mejora en la calidad de vida, una reducción de la expresión de ALAS1, una reducción del nivel (p. ej., el nivel en plasma u orina) de una porfirina o un precursor de
una porfirina (p. ej., ALA y/o PBG) u otro efecto que sea reconocido en general como positivo por los doctores médicos familiares con el tratamiento del tipo particular de trastorno.
Un efecto del tratamiento o la prevención se pone de manifiesto cuando se produce una mejora, p. ej., una mejora estadísticamente significativa en uno o más parámetros del estado patológico, o porque no empeoran o no se desarrollan síntomas que de otro modo cabría anticipar. A modo de ejemplo, un cambio favorable de al menos un 10% en un parámetro medible de la enfermedad, p. ej., de al menos un 20%, 30%, 40%, 50% o más, puede ser indicativo de un tratamiento eficaz. La eficacia de un fármaco de ARNi determinado o una formulación de este fármaco también se puede juzgar utilizando un modelo animal experimental para la enfermedad determinada según se conoce en la técnica. Cuando se utiliza un modelo animal experimental, la eficacia del tratamiento se pone de manifiesto cuando se observa una reducción estadísticamente significativa de un marcador (p. ej., ALA o PBG en plasma u orina) o síntoma.
Se puede administrar una cantidad terapéutica de ARNi a los pacientes. La cantidad terapéutica puede ser, p. ej., de 0.05-50 mg/kg. Por ejemplo, la cantidad terapéutica puede ser de 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.5, 2.0 o 2.5, 3.0, 3.5, 4.0, 4.5, 5, 10, 15, 20, 25, 30, 35, 40, 45 o 50 mg/kg de ARNbc.
El ARNi se puede formular como una formulación lipídica, p. ej., una formulación LNP según se describe en la presente. La cantidad terapéutica puede ser de 0.05-5 mg/kg, p. ej., de 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5 o 5.0 mg/kg de ARNbc. La formulación lipídica, p. ej., una formulación LNP, se puede administrar por vía intravenosa.
El ARNi se puede administrar mediante una infusión intravenosa durante un periodo de tiempo tal como durante un periodo de 5 minutos, 10 minutos, 15 minutos, 20 minutos o 25 minutos.
El ARNi se puede encontrar en forma de un conjugado de tipo GalNAc según se describe en la presente. La cantidad terapéutica puede ser de 0.5-50 mg, p. ej., de 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45 o 50 mg/kg de ARNbc. El conjugado de tipo GalNAc se puede administrar por vía subcutánea.
La administración se puede repitir, por ejemplo, de forma regular, tal como diariamente, cada dos semanas durante un mes, dos meses, tres meses, cuatro meses o más. Después de un régimen de tratamiento inicial, los tratamientos se pueden administrar con menos frecuencia. Por ejemplo, después de la administración cada dos semanas durante tres meses, la administración se puede repetir una vez al mes durante seis meses o un año o más.
El agente de ARNi se puede administrar en dos o más dosis. El número o la cantidad de dosis posteriores puede depender de la obtención del efecto deseado, p. ej., la supresión de un gen ALAS, la reducción del nivel de una porfirina o un precursor de una porfirina (p. ej., ALA y/o PBG), o la obtención de un efecto terapéutico o profiláctico, p. ej., la reducción o la prevención de uno o más síntomas asociados con una porfiria (p. ej., dolor, p. ej., dolor neuropático) y/o la prevención de ataques o la reducción de la frecuencia y/o la gravedad de los ataques asociados con una porfiria.
El agente de ARNi se puede administrar siguiendo un programa. Por ejemplo, el agente de ARNi se puede administrar una vez por semana, dos veces por semana, tres veces por semana, cuatro veces por semana o cinco veces por semana. El programa puede implicar administraciones espaciadas de forma regular, p. ej., cada hora, cada cuatro horas, cada seis horas, cada ocho horas, cada doce horas, diariamente, cada 2 días, cada 3 días, cada 4 días, cada 5 días, semanalmente, una vez cada dos semanas o mensualmente. El agente de ARNi se puede administrar semanalmente o cada dos semanas para conseguir un efecto deseado, p. ej., para reducir el nivel de ALA y/o PBG, para reducir el dolor y/o para prevenir ataques agudos.
El programa puede implicar administraciones poco espaciadas seguidas por un periodo de tiempo más prolongado durante el cual no se administra el agente. Por ejemplo, el programa puede implicar un conjunto inicial de dosis que se administran en un periodo de tiempo relativamente corto (p. ej., aproximadamente cada 6 horas, aproximadamente cada 12 horas, aproximadamente cada 24 horas, aproximadamente cada 48 horas o aproximadamente cada 72 horas) seguido de un periodo de tiempo más prolongado (p. ej., aproximadamente 1 semana, aproximadamente 2 semanas, aproximadamente 3 semanas, aproximadamente 4 semanas, aproximadamente 5 semanas, aproximadamente 6 semanas, aproximadamente 7 semanas o aproximadamente 8 semanas) durante el cual no se administra el agente de ARNi. El agente de ARNi se puede administrar inicialmente cada hora y posteriormente se administra en un intervalo más prolongado (p. ej., diariamente, semanalmente, cada dos semanas o mensualmente). El agente de ARNi se puede administrar inicialmente de forma diaria y posteriormente se puede administrar en un intervalo más prolongado (p. ej., semanalmente, cada dos semanas o mensualmente). El intervalo más prolongado aumenta con el tiempo o se determina en función de la obtención del efecto deseado. El agente de ARNi se administra una vez al día durante un ataque agudo y a continuación se administran dosis semanalmente empezando en el octavo día de administración. Específicamente, además, el agente de ARNi se puede administrar una vez cada dos días durante una primera semana y a continuación se administran dosis semanalmente empezando en el octavo día de administración.
El agente de ARNi se puede administrar para prevenir o reducir la gravedad o la frecuencia de ataques recurrentes, p. ej., ataques cíclicos asociados con un factor precipitante. El factor precipitante puede ser el ciclo menstrual. El ARNi se puede administrar de forma repetida, p. ej., en intervalos regulares para prevenir o reducir la gravedad o la frecuencia de ataques recurrentes, p. ej., ataques cíclicos asociados con un factor precipitante, p. ej., el ciclo menstrual, p. ej., una frase particular del ciclo menstrual, p. ej., la fase luteínica. El ARNi se puede administrar durante una fase particular del ciclo menstrual o en función de los niveles hormonales del paciente que se esté tratando (p. ej., en función de los niveles hormonales que se asocian con una fase particular del ciclo menstrual). El ARNi se puede administrar en uno o más días particulares del ciclo menstrual, p. ej., en el día 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,22, 23, 24, 25, 26, 27 o en el día 28 (o en un día posterior para sujetos que tienen un ciclo menstrual más prolongado). El ARNi se puede administrar durante la fase luteínica, p. ej., en uno o más días entre los días 14-28 del ciclo menstrual (o posteriormente en sujetos que tienen un ciclo menstrual de más de 28 días). Se puede evaluar la ovulación del sujeto (p. ej., utilizando una prueba en sangre u orina que detecta una hormona asociada con la ovulación, p. ej., lH) y el ARNi se administra en un intervalo predeterminado después de la ovulación. El ARNi se puede administrar inmediatamente después de la ovulación. El ARNi se puede administrar 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 o 18 días después de la ovulación. Cualquiera de estos programas se puede repetir opcionalmente durante una o más iteraciones. El número de iteraciones puede depender de la obtención del efecto deseado, p. ej., la supresión de un gen ALAS1 y/o la obtención de un efecto terapéutico o profiláctico, p. ej., reducir o prevenir uno o más síntomas asociados con una porfiria, para reducir la frecuencia de los ataques asociados con una porfiria.
Se describe que se puede administrar una dosis inicial del agente de ARNi y se puede evaluar el nivel de ALA o PBG, p. ej., 1-48 horas, p. ej., 2, 4, 8, 12 o 24 horas después de la administración de la dosis inicial. Si el nivel de ALA y/o PBG se ha reducido (p. ej. hasta conseguir una reducción predeterminada, p. ej., una normalización) y/o si los síntomas asociados con una porfiria (p. ej., dolor) han mejorado (p. ej., de manera que el paciente no presente síntomas), no se puede administrar ninguna dosis adicional, mientras que si el nivel de ALA y/o PBG no se ha reducido (p. ej., no ha alcanzado una reducción predeterminada, p. ej., no se ha normalizado), se administra una dosis adicional de ALA o PBG. La dosis adicional se puede administrar 12, 24, 36, 48, 60 o 72 horas después de la dosis inicial. Si la dosis inicial no es eficaz para reducir el nivel de ALA y/o PBG, la dosis adicional se puede modifica, p. ej., se incrementa para alcanzar una reducción deseada (p. ej., una reducción predeterminada, p. ej., una normalización) de los niveles de ALA o PBG.
La reducción predeterminada puede ser una reducción de al menos un 10%, 20%, 30%, 40% o 50%. La reducción predeterminada puede ser una reducción que es eficaz para prevenir o mejorar los síntomas, p. ej., el dolor, síntomas prodrómicos o ataques recurrentes.
La reducción predeterminada puede ser una reducción que corresponde a al menos 1, 2, 3 o más desviaciones estándar, donde la desviación estándar se determina en función de los valores de una muestra de referencia, p. ej., una muestra de referencia según se describe en la presente.
La reducción predeterminada puede ser una reducción que modifica el nivel de la porfirina o el precursor de la porfirina hasta un nivel que es inferior a, o hasta un nivel que es inferior o igual a, un valor de referencia (p. ej., un valor de referencia según se describe en la presente).
Una "normalización" de los niveles de ALA o PBG (o un nivel "normal" o "normalizado"), tal como se utiliza en la presente, se refiere a un nivel (p. ej., un nivel en orina y/o plasma) de ALA o PBG, o de ambos, que se encuentra dentro del intervalo esperado para un individuo sano, un individuo que no presenta síntomas (p. ej., un individuo que no experimenta dolor y/o no sufre ninguna neuropatía) o un individuo que no es portador de ninguna mutación asociada con una porfiria. Por ejemplo, un nivel normalizado se puede encontrar dentro de dos desviaciones estándar de la media normal. Un nivel normalizado se encuentra dentro de los límites de referencia normales, p. ej., dentro del intervalo de confianza del 95% para una muestra de control adecuada, p. ej., una muestra de individuos sanos o individuos que no son portadores de ninguna mutación génica asociada con una porfiria. El nivel de ALA y/o PBG del sujeto (p. ej., el nivel de ALA y/o PBG en orina y/o plasma) se puede monitorizar por intervalos y se puede administrar una dosis adicional del agente de ARNi cuando el nivel aumenta por encima del valor de referencia. La administración del ARNi puede reducir los niveles proteicos o de ARNm de ALAS1, p. ej., en una célula, tejido, sangre, orina u otro compartimento del paciente al menos un 10%, al menos un 15%, al menos un 20%, al menos un 25%, al menos un 30%, al menos un 40%, al menos un 50%, al menos un 60%, al menos un 70%, al menos un 80 % o al menos un 90% o más. La administración del ARNi puede reducir los niveles de productos asociados con la expresión del gen ALAS1, p. ej., los niveles de una o más porfirinas o precursores de porfirinas (p. ej., el nivel de ALA y/o PBG). La administración del agente de ARNi también puede inhibir o prevenir el aumento de los niveles proteicos o de ARNm de ALAS1 durante un ataque agudo de AIP.
Antes de la administración de una dosis completa del ARNi, se puede administrar a los pacientes una dosis más pequeña, tal como una dosis de infusión de un 5%, y monitorizar a los pacientes en busca de efectos adversos, tales como una reacción alérgica, o de una presión sanguínea o niveles lipídicos elevados. En otro ejemplo, se puede monitorizar al paciente en busca de efectos no deseados.
Métodos para modular la expresión de un gen ALAS1
En otro aspecto más, se describe además en la presente un método para modular (p. ej., inhibir o activar) la expresión de un gen ALAS1, p. ej., en una célula o en un sujeto. La célula se puede encontrar ex vivo, in vitro o in vivo. La célula puede ser una célula eritroide o un hepatocito. La célula se encuentra en un sujeto (p. ej., un mamífero tal como, por ejemplo, un ser humano). En algunas realizaciones, el sujeto (p. ej., el ser humano) corre el riesgo de padecer o ha sido diagnosticado con una enfermedad relacionada con la expresión de ALAS1, según se describe en la presente.
El método puede incluir poner en contacto la célula con un ARNi según se describe en la presente, en una cantidad eficaz para reducir la expresión de un gen ALAS1 en la célula. La expresión "poner en contacto", tal como se utiliza en la presente, incluye poner en contacto directamente una célula, así como también poner en contacto indirectamente una célula. Por ejemplo, una célula de un sujeto (p. ej., una célula eritroide o una célula hepática tal como un hepatocito) se puede poner en contacto cuando se administra una composición que comprende un ARNi (p. ej., por vía intravenosa o por vía subcutánea) al sujeto.
La expresión de un gen ALAS1 se puede evaluar en función del nivel de expresión de un ARNm de ALAS1, una proteína de ALAS1 o el nivel de un parámetro relacionado funcionalmente con el nivel de expresión de un gen ALAS1 (p. ej., el nivel de una porfirina o la incidencia o gravedad de un síntoma relacionado con una porfiria). La expresión de ALAS1 se puede inhibir al menos un 5%, al menos un 10%, al menos un 15%, al menos un 20%, al menos un 25%, al menos un 30%, al menos un 35%, al menos un 40%, al menos un 45%, al menos un 50%, al menos un 55%, al menos un 60%, al menos un 65%, al menos un 70%, al menos un 75%, al menos un 80%, al menos un 85%, al menos un 90% o al menos un 95%. El ARNi puede presentar una CI50 comprendida en el intervalo de 0.001-0.01 nM, 0.001-0.10 nM, 0.001-1.0 nM, 0.001-10 nM, 0.01-0.05 nM, 0.01-0.50 nM, 0.02-0.60 nM, 0.01-1.0 nM, 0.01-1.5 nM, 0.01-10 nM. El valor de CI50 se puede normalizar respecto a un valor de control adecuado, p. ej., la CI50 de un ARNi que no tenga diana.
El método puede incluir introducir en la célula un ARNi según se describe en la presente y mantener la célula durante un tiempo suficiente para obtener la degradación del transcrito de ARNm de un gen ALAS1, de este modo se inhibe la expresión del gen ALAS1 en la célula.
El método puede incluir administrar una composición descrita en la presente, p. ej., una composición que comprende un ARNi que tiene ALAS1 como diana, al mamífero de modo que la expresión del gen ALAS1 diana se reduzca, por ejemplo, durante un periodo prolongado, p. ej., al menos dos, tres, cuatro días o más, p. ej., una semana, dos semanas, tres semanas o cuatro semanas o más. La reducción de la expresión de ALAS1 se puede detectar en un periodo de 1 hora, 2 horas, 4 horas, 8 horas, 12 horas o 24 horas desde la primera administración.
El método puede incluir administrar una composición según se describe en la presente a un mamífero de modo que la expresión del gen ALAS1 se incremente, p. ej., al menos un 10% en comparación con un animal no tratado. La activación de ALAS1 puede tener lugar durante un periodo prolongado, p. ej., al menos dos, tres, cuatro días o más, p. ej., una semana, dos semanas, tres semanas, cuatro semanas o más. Sin pretender vincularse a ninguna teoría, un ARNi puede activar la expresión de ALAS1 estabilizando el transcrito de ARNm de ALAS1, interaccionando con un promotor en el genoma y/o inhibiendo un inhibidor de la expresión de ALAS1.
Los ARNi útiles para los métodos y las composiciones que se exponen en la presente tienen específicamente como diana ARN (primarios o procesados) de un gen ALAS1. Las composiciones y los métodos para inhibir la expresión de un gen ALAS1 utilizando ARNi se pueden preparar y aplicar según se describe en otras partes de la presente. El método puede incluir administrar una composición que contiene un ARNi, donde el ARNi incluye una secuencia de nucleótidos que es complementaria respecto a al menos una parte de un transcrito de ARN del gen ALAS1 del mamífero que se ha de tratar. Cuando el organismo que se ha de tratar es un mamífero tal como un ser humano, la composición se puede administrar mediante cualquier medio conocido en la técnica, que incluye, sin carácter limitante, la vía oral, intraperitoneal o parenteral, incluida la administración intracraneal (p. ej., intraventricular, intraparenquimal e intratecal), intravenosa, intramuscular, subcutánea, transdérmica, a través de las vías respiratorias (aerosol), nasal, rectal y tópica (incluida la bucal y sublingual).
Las composiciones se pueden administrar mediante una infusión o inyección intravenosa. Las composiciones comprenden un ARNip formulado en un líquido (p. ej., una formulación LNP tal como una formulación LNP11) para una infusión intravenosa. En particular, tales composiciones se pueden utilizar para tratar ataques agudos de porfiria y/o para la profilaxis (p. ej., para reducir la gravedad o la frecuencia de los ataques).
Las composiciones se pueden administrar además por vía subcutánea. En algunos aspectos de este tipo, las composiciones pueden comprender un ARNi conjugado con un ligando de tipo GalNAc. En particular, tales composiciones se pueden utilizar para tratar ataques agudos de porfiria o para la profilaxis (p. ej., para reducir la gravedad o la frecuencia de los ataques).
Métodos para reducir el nivel de una porfirina o un precursor de una porfirina
En otro aspecto, la divulgación proporciona un método para reducir el nivel de una porfirina o un precursor de una porfirina, p. ej., en una célula o en un sujeto.
La célula se puede encontrar ex vivo, in vitro o in vivo. La célula puede ser una célula eritroide o un hepatocito. La célula puede ser un hepatocito. La célula se puede encontrar en un sujeto (p. ej., un mamífero tal como, por ejemplo, un ser humano).
El sujeto (p. ej., el ser humano) puede correr el riesgo de padecer o ha sido diagnosticado con una porfiria, según se describe en la presente. El método puede ser eficaz para tratar una porfiria según se describe en la presente (p. ej., mejorando uno o más síntomas asociados con una porfiria, reduciendo la frecuencia de los ataques asociados con una porfiria, reduciendo la probabilidad de que se produzca un ataque de uno o más síntomas asociados con una porfiria tras la exposición a un factor precipitante o reduciendo el riesgo de desarrollar afecciones asociadas con una porfiria (p. ej., una neuropatía (p. ej., una neuropatía progresiva), un cáncer hepatocelular). El método puede incluir poner en contacto la célula con un ARNi, según se describe en la presente, en una cantidad suficiente para reducir el nivel de la porfirina o del precursor de la porfirina (p. ej., ALA o PBG) en la célula, o en otra célula o grupo de células relacionadas, o en el sujeto. La expresión "poner en contacto", tal como se utiliza en la presente, incluye poner en contacto directamente una célula, así como también poner en contacto indirectamente una célula. Por ejemplo, una célula de un sujeto (p. ej., una célula eritroide o una célula hepática tal como un hepatocito) se puede poner en contacto cuando se administra una composición que comprende un ARNi (p. ej., por vía intravenosa o por vía subcutánea) al sujeto. La expresión "otra célula o grupo de células relacionadas", tal como se utiliza en la presente, incluye cualquier célula o grupo de células en las que el nivel de la porfirina o el precursor de la porfirina se reduce como resultado de la puesta en contacto. Por ejemplo, la célula puede formar parte de un tejido presente en un sujeto (p. ej., una célula hepática presente en un sujeto) y el hecho de poner en contacto la célula del sujeto (p. ej., poner en contacto una o más células hepáticas presentes en un sujeto) con el ARNi puede provocar una reducción del nivel de la porfirina o del precursor de la porfirina en otra célula o grupo de células relacionadas (p. ej., células nerviosas del sujeto) o en un tejido o fluido del sujeto (p. ej., en la orina, sangre, plasma o fluido cerebroespinal del sujeto).
La porfirina o el precursor de porfirina se puede seleccionar del grupo constituido por ácido 5-aminolevulínico (ALA), porfopilinógeno (PBG), hidroximetilbilano (HMB), uroporfirinógeno III, coproporfirinógeno III, protoporfrinógeno IX y protoporfirina IX. El precursor de porfirina puede ser ALA. El precursor de porfirina puede ser PBG. El método puede reducir el nivel de ALA y PBG. El nivel de una porfirina o un precursor de una porfirina se puede medir según se describe en la presente y se conoce en la técnica.
Ensayos y métodos para monitorizar la actividad de iARN
En otro aspecto, también se proporcionan en la presente ensayos y métodos para monitorizar los niveles de ARNm de ALAS1. La actividad de iARN en el hígado se puede monitorizar detectando los niveles de ARNm o el producto 5’RACE en el tejido, o detectando el nivel de la proteína secretada circulante.
Como alternativa, o en combinación, los niveles extracelulares circulantes de ARNm de ALAS1 se pueden detectar, p. ej., mediante ensayos cERD (siglas en inglés de detección de ARN extracelular circulante, Circulating Extracellular RNA Detection). El nivel de ARNm de ALAS1 se puede detectar en una muestra de fluido corporal, p. ej., una muestra de suero u orina. Diferentes tipos de células pueden verter exosomas, que contienen ARNm y ARNmi derivados de un tejido de origen, en los fluidos corporales. Tales exosomas se pueden utilizar para monitorizar el nivel de iARN en circulación. Una muestra, p. ej., una muestra de suero u orina de un sujeto tratado con una ARNi descrita en la presente se puede purificar con una centrifugación a baja velocidad, seguido por una centrifugación a aproximadamente 160000g durante aproximadamente 2 horas para formar un sedimento. El ARN se puede extraer y analizar para medir los niveles de ARNm de ALAS1. Se describen métodos y ensayos ejemplares en el documento PCT/US2012/043584, publicado como WO 2012/177906.
En consecuencia, se proporciona un ensayo, o método, para detectar el nivel de ARNm de ALAS1 extracelular circulante en un sujeto. El ensayo, o método, incluye proporcionar ARN (p. ej., ARN extracelular) de una muestra de fluido biológico (p. ej., muestra de orina, sangre o plasma) del sujeto, comprendiendo dicha muestra de fluido biológico el ARNm de ALAS1; y detectar el nivel de ARNm de ALAS1 extracelular circulante en la muestra.
El ensayo o método puede incluir el paso de obtener un ADNc de ALAS1 a partir del ARNm de ALAS1; y poner en contacto el ADNc de ALAS1 con un ácido nucleico complementario (p. ej., sonda y/o cebador) con el ADNc de ALAS1 o una porción de este, para producir de esta manera una mezcla de reacción; y detectar (p. ej., medir) el nivel de ADNc de ALAS1 en la mezcla de reacción, donde el nivel de ADNc de ALAS1 es indicativo del nivel de ARNm de ALAS1, para analizar de esta manera el nivel de ARNm de ALAS1 extracelular circulante en el sujeto. El ensayo o método puede incluir adquirir una muestra de fluido biológico de un sujeto, donde la muestra biológica se separa del tejido, y donde la muestra biológica contiene exosomas. El ensayo o método puede incluir además detectar los niveles de ARN en la muestra biológica, donde el ARN se expresa a partir del gen en el tejido del sujeto, donde los exosomas no se purifican a partir de la muestra biológica antes de detectar los niveles de ARN en la muestra biológica.
Dicha muestra de fluido biológico puede ser una muestra de sangre. Dicha muestra de fluido biológico puede ser una muestra de suero. La muestra de fluido biológico puede ser una muestra de orina.
El método puede comprender PCR, qPCR o 5’-RACE.
Dicho ácido nucleico puede ser una sonda o cebador.
Dicho ácido nucleico puede comprender un resto detectable y el nivel de ARNm de ALAS1 se determina mediante la detección de la cantidad del resto detectable.
Dicho método puede comprender además obtener la muestra de fluido biológico del sujeto.
En algunos de estos métodos, se evalúa la eficacia de un tratamiento de la porfiria en función de una comparación del nivel de ARNm de ALAS1 extracelular circulante en el sujeto respecto a un valor de referencia.
Una reducción en el nivel de ARNm de ALAS1 extracelular circulante en el sujeto en respuesta al tratamiento de la porfiria, respecto al valor de referencia, puede indicar que el tratamiento de la porfiria es eficaz. El valor de referencia puede ser el nivel de ARNm de ALAS1 extracelular circulante en el sujeto antes del tratamiento de la porfiria.
A menos que se defina de otro modo, todos los términos técnicos y científicos utilizados en la presente tienen el mismo significado que interpreta habitualmente un experto en la técnica a la cual pertenece esta invención. Asimismo, los materiales, métodos y ejemplos son únicamente ilustrativos y no pretenden ser limitantes.
EJEMPLOS
Ejemplo 1. Síntesis de ARNip
Origen de los reactivos
Cuando en la presente no se proporcione específicamente el origen de un reactivo, tal reactivo se podrá obtener a partir de cualquier proveedor de reactivos para biología molecular con una calidad/pureza estándar para su aplicación en biología molecular.
Síntesis de oligonucleótidos
Todos los oligonucleótidos se sintetizan en un sintetizador AKTAoligopilot. Para la síntesis de los oligonucleótidos, se utilizaron soportes sólidos de vidrio con poros controlados que se pueden adquirir de proveedores comerciales (dT-CPG, 500 A, Prime Synthesis) y fosforamiditos de ARN con grupos protectores estándar, 5’-O-dimetoxitritil-N6-benzoil-2’-t-butildimetilsililadenosin-3’-O-N,N’-diisopropil-2-cianoetilfosforamidito, 5’-O-dimetoxitritil-N4-acetil-2’-tbutildimetilsililcitidin-3’-O-N,N’-diisopropil-2-cianoetilfosforamidito, 5’-O-dimetoxitritil-N2-isobutil-2’-tbutildimetilsililguanosin-3’-O-N,N’-diisopropil-2-cianoetilfosforamidito y 5’-O-dimetoxitritil-2’-t-butildimetilsililuridin-3’-O-N,N’-diisopropil-2-cianoetilfosforamidito (Pierce Nucleic Acids Technologies). Los fosforamiditos con la sustitución 2’-F, 5’-O-dimetoxitritil-N4-acetil-2’-flurocitidin-3’-O-N,N’-diisopropil-2-cianoetilfosforamidito y 5’-O-dimetoxitritil-2’-flurouridin-3’-O-N,N’-diisopropil-2-cianoetilfosforamidito, se adquieren de Promega. Todos los fosforamiditos se utilizan con una concentración de 0.2 M en acetonitrilo (CH3CN) excepto para la guanosina, que se utiliza con una concentración de 0.2 M en THF al 10%/ACN (v/v). Se utiliza un tiempo de acoplamiento/reciclaje de 16 minutos. El activador es 5-etiltiotetrazol (0.75 M, American International Chemicals); para la oxidación de PO se utiliza yodo/agua/piridina y para la oxidación de PS se utiliza PADS (2%) en 2,6-lutidina/ACN (1:1 v/v).
Las hebras conjugadas con el ligando 3' se sintetizan utilizando un soporte sólido que contiene el ligando correspondiente. Por ejemplo, la introducción de una unidad de colesterol en la secuencia se lleva a cabo partiendo de un fosforamidito de hidroxiprolinol-colesterol. El colesterol se une a trans-4-hidroxiprolinol mediante un conector de tipo 6-aminohexanoato para obtener un resto de hidroxiprolinol-colesterol. Los ARNi marcados con Cy-3 y Cy-5.5 (fluoróforo) en el extremo 5' se sintetizan a partir del fosforamidito Quasar-570 (Cy-3) correspondiente, que se adquiere de Biosearch Technologies. La conjugación de los ligandos en el extremo 5' y/o en una posición interna se consigue utilizando bloques estructurales de fosforamidito-ligando protegidos adecuadamente. Un acoplamiento prolongado de 15 min de una solución de fosforamidito 0.1 M en CH3CN anhidro en presencia del activador 5-(etiltio)-1 H-tetrazol a un oligonucleótido unido a un soporte sólido. La oxidación del fosfito entre nucleótidos para obtener el fosfato se lleva a cabo utilizando yodo-agua estándar según se ha descrito (1) o mediante el tratamiento con hidroperóxido de tert-butilo/acetonitrilo/agua (10: 87: 3) con un tiempo de espera para la oxidación de 10 min para el oligonucleótido conjugado. El fosforotioato se introduce mediante la oxidación del fosfito para obtener el fosforotioato utilizando un reactivo de transferencia de azufre tal como DDTT (se adquiere de AM Chemicals), PADS y/o el reactivo de Beaucage. El fosforamidito de colesterol se sintetiza dentro de la empresa y se utiliza con una concentración de 0.1 M en diclorometano. El tiempo de acoplamiento para el fosforamidito de colesterol es de 16 minutos.
Desprotección I (desprotección de bases nucleotídicas)
Tras completar la síntesis, el soporte se transfiere a una botella de vidrio de 100 mL (VWR). El oligonucleótido se escinde del soporte con la desprotección simultánea de la base y los grupos fosfato utilizando 80 mL de una mezcla de amoniaco en etanol [amoniaco: etanol (3:1)] durante 6.5 h a 55 oC. La botella se enfría brevemente en hielo y a continuación la mezcla de amoniaco en etanol se filtra en una nueva botella de 250 mL. El CPG se lava con 2 x 40 mL porciones de etanol/agua (1:1 v/v). A continuación, el volumen de la mezcla se reduce hasta ~ 30 mL en un rotavapor. A continuación, la mezcla se congela en nieve carbónica y se seca al vacío en un aparato SpeedVac. Desprotección II (eliminación del grupo 2’-TBDMS)
El residuo seco se vuelve a suspender en 26 mL de trietilamina, trihidrofluoruro de trietilamina (TEA^3HF) o piridina-HF y DMSO (3:4:6) y se calienta a 60 oC durante 90 minutos para eliminar los grupos tert-butildimetilsililo (TBDMS) de la posición 2’. A continuación, la reacción se desactiva con 50 mL de acetato de sodio 20 mM y el pH se ajusta hasta 6.5. El oligonucleótido se conserva en un congelador hasta su purificación.
Análisis
Los oligonucleótidos se analizan mediante cromatografía líquida de alta resolución (HPLC) antes de su purificación y la selección del tampón y la columna depende de la naturaleza de la secuencia y/o del ligando conjugado.
Purificación por HPLC
Los oligonucleótidos conjugados con ligandos se purifican mediante HPLC preparativa de fase inversa. Los oligonucleótidos no conjugados se purifican mediante HPLC de intercambio aniónico o en una columna de gel TSK empaquetada dentro de la empresa. Los tampones son fosfato de sodio 20 mM (pH 8.5) en CH3CN al 10% (tampón A) y fosfato de sodio 20 mM (pH 8.5) en CH3CN al 10%, NaBr 1 M (tampón B). Las fracciones que contienen los oligonucleótidos de longitud completa se agrupan, se eliminan las sales y se liofilizan. Aproximadamente 0.15 DO de los oligonucleótidos, una vez que se han eliminado las sales, se diluyen en agua hasta obtener 150 pL y a continuación se pipetean en viales especiales para el análisis de CGE y LC/MS. A continuación, los compuestos se analizan mediante LC-ESMS y CGE.
Preparación de los ARNip
Para la preparación general de los ARNip, se calientan cantidades equimolares de la hebra sentido y antisentido en 1xPBS a 95 °C durante 5 min y se enfrían lentamente hasta temperatura ambiente. La integridad del dúplex se confirma mediante análisis de HPLC.
Más adelante se representan las secuencias de los ácidos nucleicos utilizando la nomenclatura estándar y, específicamente, las abreviaciones de la Tabla 1.
Tabla 1: Abreviaciones de los monómeros nucleotídicos que se utilizan en la representación de las secuencias de los ácidos nucleicos. Se sobreentenderá que estos monómeros, cuando estén presentes en un oligonucleótido, estarán unidos mutuamente mediante enlaces 5'-3'-fosfodiéster.


__________________
(continuación)


Ejemplo 2. Diseño y síntesis de ARNip de ALAS1
Métodos experimentales
Bioinformática
Transcritos
Se llevó a cabo un diseño de ARNip para identificar ARNip que tuvieran como diana transcritos de ALAS1 humanos, de rhesus (Macaca mulatta), ratón y rata registrados en la base de datos de genes del NCBI (http://www.ncbi.nlm.nih.gov/gene/). El diseño utilizó los siguientes transcritos de la colección de RefSeq del NCBI: Humano - NM_000688.4 (remítase a la FIG.3), NM_199166.1; de rhesus - XM_001090440.2, XM_001090675.2; de ratón - NM_020559.2; de rata - NM_024484.2. Debido a la elevada divergencia de las secuencias de primate/roedor, los dúplex de ARNip se diseñaron en varios lotes separados, que incluyeron, sin carácter limitante, lotes que contenían dúplex que coincidían con transcritos humanos y de rhesus únicamente; transcritos humanos, de rhesus, ratón y rata únicamente; y transcritos de ratón y rata únicamente. La mayoría de los dúplex de ARNip se diseñaron de modo que compartieran una identidad del 100% con el transcrito humano enumerado y los transcritos de otras especies considerados en cada lote de diseño (anterior). En algunos casos (remítase a la Tabla 8), se permitieron apareamientos erróneos entre el dúplex y la diana de ARNm en la primera posición antisentido (la última posición sentido) cuando el par de bases complementario de la hebra antisentido:ARNm diana era un par GC o CG. En estos casos, los dúplex se designaron con pares UA o AU en el primer par antisentido:último par sentido. De este modo, los dúplex conservaron la complementariedad pero tenían apareamientos erróneos respecto a la diana (U:C, U:G, A:C o A:G). Dieciocho de estos dúplex de "intercambio de UA" fueron designados como parte del conjunto humano/rhesus/ratón/rata (remítase a los dúplex en la Tabla 8 con las marcas “C19U”, “G19U”, “C19A” o “G19A” en la columna de la Posición).
Diseño, especificidad y predicción de la eficacia de los ARNip
Se pronosticó la especificidad prevista para todos los nonadecámeros posibles a partir de cada secuencia. A continuación, se seleccionaron los nonadecámeros candidatos que carecían de repeticiones de más de 7 nucleótidos. Estos 1510 ARNip candidatos humanos/de rhesus, 114 humanos/de rhesus/ratón/rata, y 717 de ratón/rata se utilizaron en búsquedas exhaustivas frente a transcriptomas adecuados (definidos como el conjunto de registros NM_ y XM_ dentro de los conjuntos de Refseq del NCBI para humanos, rhesus, perros, ratones o ratas) utilizando un algoritmo exhaustivo de "fuerza bruta" implementado en el script Python ‘BruteForce.py’. A continuación, el script analizó las alineaciones de transcrito-oligo para generar una puntuación basada en la posición y el número de apareamientos erróneos entre el ARNip y cualquier transcrito "alejado de la diana" potencial. La puntuación alejada de la diana se pondera para enfatizar las diferencias en la región "semilla" de los ARNip, en las posiciones 2-9 partiendo del extremo 5' de la molécula. A cada par de oligo-transcrito de la búsqueda de fuerza bruta se le otorgó una puntuación de apareamientos erróneos sumando las puntuaciones de apareamientos erróneos individuales; los apareamientos erróneos en las posiciones 2-9 se contaron como 2.8, los apareamientos erróneos en las posiciones de los sitios de escisión 10-11 se contaron como 1.2 y los apareamientos erróneos en la región 12 19 se contaron como 1.0. Se realizó una predicción alejada de la diana adicional comparando la frecuencia de los heptámeros y octómeros derivados de 3 hexámeros diferentes derivados de la región semilla de cada oligo. Los hexámeros de las posiciones 2-7 con relación al inicio 5' se utilizan para crear 2 heptámeros y un octómero. Hemos creado el "heptámero 1" añadiendo una A 3' al hexámero; hemos creado el heptámero 2 añadiendo una A 5' al hexámero; hemos creado el octómero añadiendo una A a ambos extremos 5' y 3' del hexámero. Se precalculó la frecuencia de los octómeros y heptámeros en 3'UTRome humana, de rhesus, ratón o rata (que se define como la subsecuencia del transcriptoma de la base de datos de Refseq del NCBI en la que el extremo de la región codificante, la "CDS", está claramente definido). La frecuencia de los octómeros se normalizó respecto a la frecuencia de los heptámeros utilizando el valor mediano de la gama de frecuencias de los octómeros. A continuación, se calculó una puntuación "mirSeedScore" calculando la suma de ((3 X recuento de octómeros normalizado) (2 X recuento del heptámero 2) (1 X recuento del heptámero 1)).
Ambas hebras de ARNip fueron asignadas a una categoría de especificidad de acuerdo con las puntuaciones calculadas: una puntuación por encima de 3 se califica como altamente específica, igual a 3 como específica y entre 2.2 y 2.8 como moderadamente específica. Realizamos la clasificación según la especificidad de la hebra antisentido. A continuación, seleccionamos dúplex cuyos oligos antisentido carecían de GC en la primera posición, carecían de G en las posiciones 13 y 14 y tenían 3 o más Us o As en la región semilla (características de dúplex con una eficacia prevista elevada).
Se diseñaron dúplex conjugados con GalNAc candidatos, con una longitud de 21 y 23 nucleótidos para las hebras sentido y antisentido, respectivamente, extendiendo nonadecámeros antisentido 4 nucleótidos adicionales en la dirección 3' (conservando una complementariedad perfecta con el transcrito diana). Se especificó la hebra sentido como el complemento inverso de los primeros 21 nucleótidos del tricosámero antisentido. Se seleccionaron los dúplex que conservaron apareamientos perfectos para todos los transcritos de las especies seleccionadas en los 23 nucleótidos.
Selección de las secuencias de ARNip
Se sintetizaron oligos nonadecaméricos de ARNip humanos/de rhesus derivados de un total de 90 hebras sentido y 90 hebras antisentido, humanos/de rhesus/ratón/ratón/rata derivados de 40 hebras sentido y 40 hebras antisentido, y de ratón/rata derivados de 40 hebras sentido y 40 hebras antisentido y se utilizaron para formar los dúplex. Se sintetizaron oligos henicosaméricos/tricosaméricos humanos/de rhesus derivados de un total de 45 hebras sentido y 45 hebras antisentido para proporcionar 45 dúplex conjugados con GalNAc.
Las secuencias de las hebras sentido y antisentido de los dúplex modificados se muestran en la Tabla 2 y las secuencias de las hebras sentido y antisentido de los dúplex no modificados se muestran en la Tabla 3.
Síntesis de secuencias de ALAS1
Se sintetizaron secuencias de ALAS1 en un sintetizador MerMade 192 a escala de 1 o 0.2 pmol. Se prepararon hebras individuales con modificaciones de tipo 2’O-metilo para realizar un cribado in vitro utilizando reactivos de transfección. Se prepararon conjugados 3’ GalNAc con secuencias que contenían modificaciones de tipo 2’F y 2’-O-metilo en la hebra sentido en los diseños henicosaméricos/tricosaméricos para la captación libre en células. Para todas las secuencias henicosaméricas de la lista, se aplicó una química "endolight" según se detalla a continuación.
• Todas las pirimidinas (citosina y uridina) de la hebra sentido contenían bases con modificaciones de tipo 2’-O-metilo (C con modificación 2’-O-metilo y U con modificación 2’-O).
• En la hebra antisentido, las pirimidinas adyacentes (hacia la posición 5') a un ribonucleósido A se reemplazaron por sus correspondientes nucleósidos con modificaciones 2-O-metilo.
• Se introdujo una extensión de dos bases dTsdT en el extremo 3’ de ambas secuencias sentido y antisentido.
• El archivo de las secuencias se convirtió en un archivo de texto con el fin de que fuera compatible para cargarlo en el programa informático de síntesis MerMade 192.
Para las hebras sentido conjugadas con GalNAc y las secuencias antisentido complementarias, se introdujeron nucleósidos con modificaciones 2'F y con otras modificaciones combinados con ribo con nucleósidos de modificaciones 2'O-metilo. La síntesis se llevó a cabo en un soporte CPG modificado con GalNAc para la hebra sentido y CPG modificado con un soporte universal para la secuencia antisentido.
Síntesis, escisión y desprotección:
La síntesis de las secuencias de ALAS1 fue una síntesis de oligonucleótidos en soporte sólido basada en la química de los fosforamiditos. Para las secuencias "endolight" henicosaméricas, se utilizó desoxitimidina-CPG como soporte sólido, mientras que para los conjugados de GalNAc, se utilizó un soporte sólido de GalNAc para la hebra sentido y CPG universal para la hebra antisentido.
La síntesis de las secuencias anteriores se llevó a cabo a escala de 1 o 0.2 pm en placas de 96 pocillos. Las soluciones de los amiditos se prepararon con una concentración de 0.1 M y se utilizó etiltiotetrazol (0.6 M en acetonitrilo) como activador.
Las secuencias sintetizadas se escindieron y desprotegieron en placas de 96 pocillos, utilizando metilamina en el primer paso y un reactivo de fluoruro en el segundo paso. Para las secuencias que contenían GalNAc y nucleósidos con modificaciones 2'F, se modificaron las condiciones de desprotección. Después de la escisión y desprotección de las secuencias, se provocó su precipitación utilizando una mezcla de acetona:etanol (80:20) y el pellet se suspendió de nuevo en un tampón de acetato de sodio 0.2 M. Las muestras de cada secuencia se analizaron mediante LC-MS para confirmar su identidad y mediante UV para cuantificarlas, y un conjunto de muestras seleccionadas se analizó mediante cromatografía de intercambio iónico para determinar su pureza.
Purificación y eliminación de las sales:
Se provocó la precipitación de las secuencias de ALAS1 y estas se purificaron en un sistema purificador AKTA utilizando una columna Sephadex. El ALAS1ess se llevó a cabo a temperatura ambiente. La inyección y la recolección de las muestras se llevaron a cabo en placas de 96 pocillos (pocillos con una profundidad de 1.8 mL). En el eluyente se recolectó un único pico correspondiente a la secuencia de longitud completa. Las secuencias de ALAS1, una vez eliminadas las sales, se analizaron para determinar su concentración (mediante una medición de UV a A260) y pureza (mediante HPLC de intercambio iónico). A continuación, las hebras individuales complementarias se combinaron con una relación estequiométrica 1:1 para formar los dúplex de ARNip.





_



Ejemplo 3. Cribado in v itro de los dúplex de ARNip de ALAS1 para determinar la actividad de desactivación de ALAS1
Los dúplex de ARNip de ALAS1 se cribaron con el fin de determinar su capacidad para desactivar la expresión de ALAS1 in vitro.
Cribado in v itro
Cultivos celulares y transfecciones
Se cultivaron células Hep3B (ATCC, Manassas, VA) hasta casi llegar a la confluencia a 37 °C en una atmósfera de un 5% de CO2 en MEM (ATCC) suplementada con un 10% de FBS, antes de liberarlas de la placa mediante tripsinización. La transfección se llevó a cabo añadiendo 14.8 pl de Opti-MEM más 0.2 pl de Lipofectamine RNAiMax por pocillo (Invitrogen, Carlsbad CA, N.° de catálogo 13778-150) a 5 pl de los dúplex de ARNip por pocillo en una placa de 96 pocillos y se incubó a temperatura ambiente durante 15 minutos. A continuación, se añadieron 80 pl de medio de cultivo completo que contenían ~2 x 104 células Hep3B a la mezcla de ARNip. Las células se incubaron durante 24 o 120 horas antes de la purificación del ARN. Se llevaron a cabo experimentos de dosis individuales para una concentración final del dúplex de 10 nM y 0.1n M y se llevaron a cabo experimentos de dosis-respuesta para una concentración final del dúplex de 10, 1.67, 0.27, 0.046, 0.0077, 0.0013, 0.00021 y 0.00004 nM.
Aislamiento de ARN total utilizando el kit de aislamiento de ARNm DYNABEADS (Invitrogen, componente N.°: 610 12)
Las células se recolectaron y lisaron en 150 pl de tampón de lisis/unión, a continuación se mezclaron durante 5 minutos a 850 rpm utilizando un instrumento Eppendorf Thermomixer (la velocidad de mezclado fue la misma a lo largo de todo el proceso). Se añadieron diez microlitros de microesferas magnéticas y 80 pl de mezcla de tampón de lisis/unión a una placa de fondo redondo y se mezclaron durante 1 minuto. Las microesferas magnéticas se capturaron utilizando un soporte magnético y se retiró el sobrenadante sin que se alteraran las microesferas. Después de retirar el sobrenadante, las células lisadas se añadieron a las microesferas remanentes y se mezclaron durante 5 minutos. Después de retirar el sobrenadante, las microesferas magnéticas se lavaron 2 veces con 150 pl de tampón de lavado A y se mezclaron durante 1 minuto. Las microesferas se capturaron nuevamente y se retiró el sobrenadante. A continuación, las microesferas se lavaron con 150 pl de tampón de lavado B, se capturaron y se retiró el sobrenadante. A continuación, las microesferas se lavaron con 150 pl de tampón de elución, se capturaron y se retiró el sobrenadante. Se permitió que las microesferas se secaran durante 2 minutos. Después del secado, se añadieron 50 pl de tampón de elución y se mezclaron durante 5 minutos a 70 °C. Las microesferas se capturaron con un imán durante 5 minutos. Se retiraron 40 pl de sobrenadante y se añadieron a otra placa de 96 pocillos.
Síntesis de ADNc utilizando el kit de transcripción inversa de ADNc de alta capacidad de ABI (Applied Biosvstems. Foster City, CA, N.° de catálogo 4368813)
Una mezcla patrón de 2 pl de 10X tampón, 0.8 pl de 25X dNTPs, 2 pl de cebadores aleatorios, 1 pl de transcriptasa inversa, 1 pl de inhibidor de RNasa y 3.2 pl de H2O por reacción se añadió a 10 pl de ARN total. Se generó ADNc utilizando un ciclador térmico Bio-Rad C-1000 o S-1000 (Hercules, CA) mediante los pasos siguientes: 25 oC 10 min, 37 oC 120 min, 85 oC 5 s y se mantiene a 4 oC.
PCR a tiempo real
Se añadieron 2 pl de ADNc a una mezcla patrón que contenía 0.5 pl de una sonda TaqMan de GAPDH (Applied Biosystems, N.° de catálogo 4326317E), 0.5 pl de una sonda TaqMan de ALAS1 (Applied Biosystems, N.° de catálogo Hs00167441_m1) y 5 pl de una mezcla patrón para una sonda Lightcycler 480 (Roche, N.° de catálogo 04887301001) por pocillo en una placa de 384 pocillos (Roche, N.° de catálogo 04887301001). Se llevó a cabo una PCR a tiempo real en un sistema de PCR a tiempo real Roche LC480 (Roche) utilizando el ensayo de AACt(RQ). Cada dúplex se evaluó en dos transfecciones independientes con dos réplicas biológicas para cada uno de ellos, y cada transfección se evaluó por duplicado, a menos que se indique de otro modo en las tablas de resumen.
Para calcular el factor de cambio relativo, los datos a tiempo real se analizaron utilizando el método de AACt y se normalizaron respecto a los ensayos realizados con células transfectadas con AD-1955 10 nM o células transfectadas con vector vacío. Los valores de CI50 se calcularon utilizando un modelo de ajuste de 4 parámetros empleando XLFit y se normalizaron respecto a células transfectadas con AD-1955 o células vírgenes para el mismo intervalo de dosis, o respecto a su propia dosis más baja.
Desactivación in vitro de la expresión de ALAS1 endógeno mediante los dúplex de ARNip de ALAS1
La Tabla 4 ilustra la desactivación de ALAS1 en células Hep3B mediante los dúplex de ARNip modificados para ALAS1 (remítase a la Tabla 2). El silenciamiento se expresa como la fracción de mensaje de ARN remanente respecto al ARNip AD-1955 que actúa como control negativo (luciferasa). Los datos se generaron según se ha descrito anteriormente tras la transfección de una concentración 10 nM o 0.1 nM de cada ARNip. La qPCR se llevó a cabo utilizando la sonda TaqMan de ALAS1 Hs00167441_m1.
Tabla 4: Expresión de ALAS1 en células Hep3B tras la transfección con ARNip de ALAS1

(continuación)

(continuación)

Valores de CI50 de los dúplex de ARNip de ALAS1 seleccionados en un cribado in v itro
La Tabla 5 ilustra los valores de CI50 de los dúplex de ARNip de ALAS1 seleccionados determinados a partir de la desactivación de ALAS1 expresado de forma endógena en la línea celular Hep3B, mediante dúplex de ARNip modificados para ALAS1 (remítase a la Tabla 2). Los datos se generaron como se ha descrito anteriormente, 24 o 120 horas después de la transfección de cada dúplex de ARNip. En este ejemplo, el silenciamiento de ALAS1 se expresa como la fracción de mensaje de ARNm remanente respecto al ARNip AD-1955, un ARNip que no actúa sobre la diana y que se utiliza como control negativo. Los datos de experimentos de transfección replicados se utilizaron para ajustar una única línea con el fin de determinar la CI50. Varios dúplex (p. ej., AD-53541.1, AD-53542.1 y AD-53547.1) presentaron una CI50 de tan solo aproximadamente 0.03 nM a las 24 horas. Numerosos dúplex presentaron una CI50 inferior a 0.1 nM (p. ej., AD-53534.1, AD-53536.1, AD-53540.1, AD-53541.1, AD-53542.1, AD-53547.1, AD-53548.1, AD-53550.1, a D-53552.1) a las 24 horas y algunos de estos también presentaron una CI50 inferior a 0.1 nM (p. ej., AD-53534.1, AD-53540.1, AD-53541.1, AD-53542.1, AD-53547.1, AD-53552.1) a las 120 horas.
Tabla 5: Valores de CI50 de los dúplex de ARNip de ALAS1 normalizados respecto a AD-1955

Ejemplo 4. Silenciamiento in v iv o utilizando un ARNip de ALAS1 de ratón/rata formulado como un LNP En la Tabla 6 a continuación se muestran las secuencias del dúplex modificado AD-53558.

Este dúplex se formuló como una formulación LNP11 (remítase a la Tabla 10 mostrada anteriormente). El ARNip AD-53558 formulado en LNP se evaluó in vivo en ratones (N = 25 animales; 5 animales por grupo) y ratas (N = 20 animales; 4 animales por grupo) y se confirmó que silenciaba el ARNm de ALAS1 in vivo. Los resultados se muestran en la FIG. 5 y la FIG. 6.
La FIG. 5 muestra que el ARNip presentó un efecto de dosis-respuesta en ratones. La expresión de ARNm de ALAS1 de ratón (ALAS1r) se redujo aproximadamente un 78% cuando el ARNip se administró con una dosis de 1 mg/kg; el ARNm de ALAS1 de ratón se redujo aproximadamente un 60% cuando el ARNip se administró con una dosis de 0.3 mg/kg; y el ARNm de ALAS1 de ratón se redujo aproximadamente un 49% cuando el ARNip se administró con una dosis de 0.1 mg/kg. Estas reducciones se expresan respecto a un control de PBS. También se empleó un control de LUC AD-1955, según se muestra en la FIG. 5.
De modo similar, la FIG. 6 muestra que el ARNip presentó un efecto de dosis-respuesta en ratas. La expresión de ARN de ALAS1 se redujo aproximadamente un 70% cuando el ARNip se administró con una dosis de 1 mg/kg; el ARNm de ALAS1 se redujo aproximadamente un 62% cuando el ARNip se administró con una dosis de 0.3 mg/kg; y el ARNm de ALAS1 se redujo aproximadamente un 34% cuando el ARNip se administró con una dosis de 0.1 mg/kg.
También se evaluó la durabilidad del silenciamiento en ratones (N = 15; 3 animales por punto de evaluación). Los resultados se muestran en la FIG. 7, la cual muestra que AD-53558 suprimió el ARNm de ALAS1 r aproximadamente un 80% durante al menos 9 días. Una supresión de al menos aproximadamente un 50% perduró durante al menos 14 días.
Ejemplo 5. Eficacia del ARNip de ALAS1 en un modelo animal de AIP
Se investigaron los efectos de la formulación LNP11 de AD-53558 (un ARNip de ALAS1 de ratón/rata descrito en el ejemplo previo) en un modelo de ratón de AIP. La desactivación de PBGD no es viable (-/-, 0% de actividad). Existen ratones heterozigóticos con desactivación de PBGD (+/-, ~50% de actividad), pero no se dispone de su fenotipo bioquímico completo y, por lo tanto, no reproducen el fenotipo humano de la enfermedad. Por lo tanto, se ha desarrollado un modelo de ratón de AIP que es un caso heterozigótico compuesto con alelos T1/T2, que incluye la disrupción del promotor de T1 (+/-) y la alteración del sitio de corte y empalme de T2 (-/-). Se ha observado que estos ratones presentan una actividad de PBGD residual hepática que es aproximadamente un 30% del nivel de origen natural o presentan niveles de ALA y PBG en plasma de referencia ligeramente elevados o normales. Se ha observado que los ratones parecen normales en estadios iniciales de su vida y se vuelven ligeramente más lentos y atáxicos con la edad. Se ha descrito que, cuando los ratones tienen una edad de seis meses, desarrollan una coordinación motriz y un rendimiento muscular deteriorados, así como también una degeneración axonal cuando se someten a un examen patológico. La investigación de la patología del modelo de ratón ha mostrado degeneración axonal, así como coordinación motriz y rendimiento muscular deteriorados en ratones de edad más avanzada. Se ha observado que ALA y PBG en plasma y orina aumentan notablemente con la administración i.p. en serie de fenobarbital (remítase a Lindberg et al., (1996), Nature Genetics, 12:195-219 y Lindberg et al., (1999), Journal of Clinical Investigation, 103:1127-34). Los ratones se rescataron mediante la expresión mediada por AAV de PBGD en el hígado (Yasuda et al. (2010), Molecular Medicine, 1:17-22 y Unzu et al. (2011), Molecular Medicine, 2:243-50).
El día 1, se administró a los ratones 1 mg/kg de ARNip de ALAS1 (n = 5) o el control AD-1955 de LUC (n = 3) mediante inyección i.v. Se suministraron tres inyecciones de fenobarbital (1 inyección por día en los días 2, 3 y 4) para inducir ALAS1 hepático y los precursores de porfirinas, ALA y PBG. Se tomaron muestras de orina durante la noche y de plasma en el día 5 y se midieron los niveles de metabolitos mediante LC-MS. Los niveles de metabolitos se midieron en plasma mediante LC-MS y también se midieron en la orina. Se midieron los niveles de referencia de los metabolitos antes del primer tratamiento el día 1. Los resultados se muestran en las FIG. 8-12 y en las Tablas 12 y 13.
La FIG. 8 y la FIG. 9 muestran los niveles de ALA en plasma en pM. Los niveles de ALA de referencia fueron bajos (n = 4) y el tratamiento con fenobarbital indujo incrementos significativos en los niveles de ALA en plasma en los animales tratados con ARNip de LUC como control (n = 3). El tratamiento con ARNip de ALAS1 inhibió la inducción de ALA en plasma (n = 5), según se muestra en la FIG. 8. El ARNip de ALAS1 fue uniformemente eficaz a la hora de bloquear la inducción de ALA en plasma en cada uno de los animales individuales estudiados (remítase a la FIG. 9). Estos resultados indican que el tratamiento con ARNip de ALAS1 fue eficaz a la hora de prevenir los incrementos de ALA en plasma asociados con los ataques agudos inducidos por fenobarbital en este modelo animal de AIP.
La FIG. 10 y la FIG. 11 muestran los niveles de PBG en plasma en pM. Los niveles de PBG de referencia fueron bajos (n = 4) y el tratamiento con fenobarbital indujo incrementos significativos en los niveles de PBG en plasma en los animales tratados con ARNip de LUC como control (n = 3). El tratamiento con ARNip de ALAS1 inhibió la inducción de PBG en plasma (n = 5), según se muestra en la FIG. 10. El ARNip de ALAS1 fue uniformemente eficaz a la hora de bloquear la inducción de PBG en plasma en cada uno de los animales individuales estudiados (remítase a la FIG. 11). Estos resultados indican que el tratamiento con ARNip de ALAS1 fue eficaz a la hora de prevenir los incrementos de PBG en plasma asociados con los ataques agudos inducidos por fenobarbital en este modelo animal de AIP.
Las Tablas 12 y 13 muestran los niveles de ALA y PBG en orina en el punto de referencia y después del tratamiento con fenobarbital en animales tratados con ARNip de LUC (n = 2), control (CTR, que se refiere a un animal tratado con tampón de PBS, n = 1) y ARNip de ALAS1 (n = 5).
Tabla 12: Datos en orina de animales individuales que muestran la prevención de un ataque agudo inducido

Tabla 13: Datos medios en orina

El tratamiento con fenobarbital indujo incrementos marcados (incrementos de ~25-30 veces) de ALA en orina (~9 veces por encima de los niveles de referencia) y PBG en orina (~19 veces por encima de los niveles de referencia) en los ratones tratados con ARNip de LUC, el control, mientras que tales incrementos no se observaron en los animales tratados con ARNip de ALAS1. Por lo tanto, el ARNip de ALAS1 bloqueó los incrementos inducidos por fenobarbital de ALA y PBG en orina. Estos resultados son coherentes con las medidas en plasma y muestran que el tratamiento con ARNip de ALAS1 fue eficaz a la hora de prevenir los incrementos de metabolitos en orina (ALA y PBG) asociados con los ataques agudos inducidos por fenobarbital en este modelo animal de AIP.
En otros experimentos (FIG. 12), se observó que el tratamiento con fenobarbital indujo grandes incrementos (~25 veces) en la expresión de ARNm de ALAS1 en el hígado del modelo de ratón. La administración de ARNip de ALAS1 bloqueó completamente esta inducción de ARNm de ALAS1. Estos resultados proporcionan más pruebas de que el ARNip de ALAS1 es eficaz en un modelo animal de AIP.
En conjunto, los resultados proporcionados en este Ejemplo muestran que el ARNip de ALAS1 fue eficaz a la hora de tratar ataques agudos en un modelo animal de la porfiria hepática aguda AIP. Las medidas de múltiples parámetros respaldan esta conclusión, incluidos los niveles de ALA en plasma, los niveles de PBG en plasma, los niveles de ALA en orina, los niveles de PBG en orina y los niveles de expresión de ARNm de ALAS1 en el hígado. Ejemplo 6. Silenciamiento in v iv o utilizando ARNip de ALAS1 de ratón conjugado con GalNAc
Los experimentos descritos en este ejemplo investigaron la eficacia in vivo de tres ARNip conjugados con GalNAc (remítase a la Tabla 7). Estos ARNip se diseñaron y produjeron con métodos tales como los descritos en el Ejemplo 2.

Los ratones (n = 40; n = 4 por cada condición experimental) se dividieron en grupos que recibieron PBS o dosis de 3 mg/kg, 10 mg/kg o 30 mg/kg de ARNip administradas por vía subcutánea. Se determinó el nivel de ARNm de ALAS1r/GAPDHr, respecto al control de PBS, en células hepáticas 72 horas después de la administración. Los resultados se muestran en la FIG. 13. No se observó un efecto de dosis-respuesta claro para los ARNip AD-56211 y AD-56173. Por el contrario, el ARNip de ALAS1 AD-57929 presentó un efecto de dosis-respuesta para la inhibición de la expresión de ALAS1r. Estos resultados demuestran que un conjugado de GalNAc de ALAS1 fue eficaz a la hora de inhibir la expresión de ARNm de ALAS1 in vivo y presentó un efecto de dosis-respuesta.
Ejemplo 7. ARNip humanos
Se diseñaron y se produjeron ARNip humanos adicionales según se ha descrito en el Ejemplo 2. Se seleccionaron los 45 ARNip mejores en función de su eficacia prevista. Las secuencias de estos 45 ARNip se proporcionan en la Tabla 8 y la Lista de Secuencias adjunta a esta (p. ej., una secuencia sentido correspondiente a una de las secuencias con números impares identificadas como SEQ ID Nos: 391 a 551, y una secuencia antisentido correspondiente a una de las secuencias con números pares identificadas como SEQ ID Nos: 392 a 552, respectivamente). La Tabla 8 se describe en la Publicación Internacional N.° WO2013/155204A2.
Ejemplo 8. ARNip humanos
Se generaron ARNip humanos nonadecaméricos adicionales. Las secuencias de estos ARNip se proporcionan en la Tabla 9 y la Lista de Secuencias adjunta a esta (p. ej., una secuencia sentido correspondiente a una de las secuencias con números impares idenficadas como SEQ ID Nos: 553 a 3365, y una secuencia antisentido correspondiente a una de las secuencias con números pares identificadas como SEQ ID Nos: 554 a 3366, respectivamente). La Tabla 9 se describe en la Publicación Internacional N.° WO2013/155204A2. Estos ARNip se pueden evaluar para determinar su eficacia utilizando métodos descritos en la presente y/o métodos conocidos en la técnica.
Ejemplo 9. Supresión de precursores de porfirinas utilizando ARNip de ALAS1 en un paradigma de tratamiento agudo
Se utilizó el modelo de ratón de AIP (remítase al Ejemplo 5) para investigar si el ARNip de ALAS1 funcionaría en un paradigma de tratamiento agudo para reducir los niveles ya elevados de ALA y PBG que estarían presentes, por ejemplo, cuando un paciente humano con porfiria padece un ataque agudo. La administración del ARNip de la formulación LNP11 AD-53558 con una dosis de 1 mg/kg 12 horas después de la última dosis de fenobarbital redujo rápidamente los niveles de ALA y PBG en plasma de ratón, mientras que en los animales tratados con control de Luc los niveles siguieron subiendo (FIG. 14). Estos resultados indican que el ARNip de ALAS es eficaz a la hora de tratar un ataque agudo. El ARNip de ALAS1 fue eficaz a la hora de reducir y prevenir incrementos adicionales de los niveles de ALA y PBG.
Como se puede observar en la FIG. 14, el ARNip de ALAS tuvo un rápido inicio del efecto en la reducción de los niveles de ALA y PBG. El inicio del efecto ocurrió en las horas posteriores a la administración del ARNip. El efecto del plasma en ALA se pudo observar en las 4 horas posteriores a la administración del ARNip (remítase a la FIG. 14; el ARNip se administró 12 horas después de la última dosis de fenobarbital, y se puede observar una reducción en ALA en plasma respecto al control 16 horas después de la última dosis de fenobarbital). El efecto en PBG en plasma se pudo observar en las 8 horas siguientes a la administración del ARNip (remítase a la FIG. 14; el ARNip se administró 12 horas después de la última dosis de fenobarbital, y se puede observar una reducción en ALA en plasma respecto al control 20 horas después de la última dosis de fenobarbital).
Ejemplo 10. ARNip que tienen ALAS1 como diana
Se diseñaron y produjeron secuencias de ARNip modificadas y no modificadas adicionales que tenían como diana el ARNip de ALAS1, según se ha descrito en el Ejemplo 2. Se evaluó la actividad in vitro de los dúplex modificados según se describe a continuación.
Métodos
Transfección mediada por lípidos
Para Hep3B, PMH y hepatocitos de Cynomolgus primarios, la transfección se llevó a cabo añadiendo 14.8 pl de Opti-MEM más 0.2 pl de Lipofectamine RNAiMax por pocillo (Invitrogen, Carlsbad CA., número de catálogo 13778 150) a 5 pl de cada dúplex de ARNip en un pocillo individual en una placa de 96 pocillos. A continuación, la mezcla se incubó a temperatura ambiente durante 20 minutos. A continuación, se añadieron 80 pl de medio de cultivo completo sin antibiótico que contenían el número adecuado de células a la mezcla de ARNip. Las células se incubaron durante 24 horas antes de la purificación del ARN.
Se llevaron a cabo experimentos de dosis única con una concentración final del dúplex de 1 pM, 500 nM, 20 nM, 10 nM y 0.2 nM para GalNAc modificado.
Transfección de captación libre
Hepatocitos de Cynomolgus primarios conservados criológicamente (Celsis In Vitro Technologies, M003055-P) se descongelaron a 37 °C en un baño de agua inmediatamente antes de su uso y se suspendieron de nuevo con una concentración de 0.26x106 células/ml en medio InVitroGRO CP (para placas) (Celsis In Vitro Technologies, número de catálogo Z99029). Durante las transfecciones, las células se colocaron en una placa de colágeno de 96 pocillos BD BioCoat (BD, 356407) con una concentración de 25 000 células por pocillo y se incubaron a 37 °C en una atmósfera de un 5% de CO2. Se realizaron experimentos de captación libre añadiendo 10 pl de dúplex de ARNip en PBS por pocillo en una placa de 96 pocillos (96p). A continuación, se añadieron 90 pl de medio de cultivo completo que contenía el número adecuado de células para el tipo celular al ARNip. Las células se incubaron durante 24 horas antes de la purificación del ARN. Se llevaron a cabo experimentos de dosis única con una concentración final del dúplex de 1 pM, 500 nM, 20 nM y 10 nM.
Aislamiento de ARN total utilizando el kit de aislamiento de ARNm DYNABEADS (Invitrogen, componente N.°: 610 12)
Las células se recolectaron y lisaron en 150 pl de tampón de lisis/unión, a continuación se mezclaron durante 5 minutos a 850 rpm utilizando un instrumento Eppendorf Thermomixer (la velocidad de mezclado fue la misma a lo largo de todo el proceso). Se añadieron diez microlitros de microesferas magnéticas y 80 pl de mezcla de tampón de lisis/unión a una placa de fondo redondo y se mezclaron durante 1 minuto. Las microesferas magnéticas se capturaron utilizando un soporte magnético y se retiró el sobrenadante sin que se alteraran las microesferas. Después de retirar el sobrenadante, las células lisadas se añadieron a las microesferas remanentes y se mezclaron durante 5 minutos. Después de retirar el sobrenadante, las microesferas magnéticas se lavaron 2 veces con 150pl de tampón de lavado A y se mezclaron durante 1 minuto. Las microesferas se capturaron de nuevo y se retiró el sobrenadante. A continuación, las microesferas se lavaron con 150 pl de tampón de lavado B, se capturaron y se retiró el sobrenadante. A continuación, las microesferas se lavaron con 150 pl de tampón de elución, se capturaron y se retiró el sobrenadante. Finalmente, se permitió que las microesferas se secaran durante 2 minutos. Después del secado, se añadieron 50 pl de tampón de elución y se mezclaron durante 5 minutos a 70 °C. Las microesferas se capturaron con un imán durante 5 minutos. Se retiraron 45 pl de sobrenadante y se añadieron a otra placa de 96 pocillos.
Síntesis de ADNc utilizando el kit de transcripción inversa de ADNc de alta capacidad de ABI (Applied Biosystems, Foster City, CA, N.° de catálogo 4368813)
Se preparó una mezcla patrón de 2 pl de 10X tampón, 0.8 pl de 25X dNTPs, 2 pl de cebadores aleatorios, 1 pl de transcriptasa inversa, 1 pl de inhibidor de RNasa y 3.2 pl de H2O por reacción. Se mezclaron volúmenes iguales de mezcla patrón y ARN para obtener un volumen final de 12 pl para las muestras cribadas in vitro o 20 pl para las muestras cribadas in vivo. El ADNc se generó utilizando un ciclador térmico Bio-Rad C-1000 o S-1000 (Hercules, CA) mediante los pasos siguientes: 25 oC durante 10 minutos, 37 oC durante 120 minutos, 85 oC durante 5 segundos y se mantiene a 4 oC.
PCR a tiempo real
Se añadieron 2 pl de ADNc a una mezcla patrón que contenía 2 pl de H2O, 0.5 pl de una sonda TaqMan de GAPDH (Life Technologies, número de catálogo 4326317E para células Hep3B, número de catálogo 352339E para hepatocitos de ratón primarios o una sonda adaptada para hepatocitos primarios de cinomólogo), 0.5 pl de una sonda TaqMan C5 (Life Technologies, número de catálogo Hs00167441_m1 para células Hep3B o Mm00457879_m1 para hepatocitos de ratón primarios o una sonda adaptada para hepatocitos primarios de cinomólogo) y 5 pl de una mezcla patrón para una sonda Lightcycler 480 (Roche, número de catálogo 04887301001) por pocillo en una placa de 384 pocillos (384 p) (Roche, número de catálogo 04887301001). Se llevó a cabo una PCR a tiempo real en un sistema de PCR a tiempo real Roche LC480 (Roche) utilizando el ensayo AACt(RQ). Para el cribado in vitro, cada dúplex se evaluó con dos réplicas biológicas, a menos que se indique de otro modo, y cada PCR a tiempo real se llevó a cabo en réplicas técnicas por duplicado. Para el cribado in vivo, cada dúplex se evaluó en uno o más experimentos (3 ratones por grupo) y cada PCR a tiempo real se llevó a cabo en réplicas técnicas por duplicado.
Para calcular el factor de cambio relativo en los niveles de ARNm de ALAS1, los datos a tiempo real se analizaron utilizando el método de AACt y se normalizaron respecto a los ensayos realizados con células transfectadas con AD-1955 10 nM o células transfectadas con vector vacío. Los valores de CI50 se calcularon utilizando un modelo de ajuste de 4 parámetros empleando XLFit y se normalizaron respecto a células transfectadas con AD-1955 para el mismo intervalo de dosis o respecto a su propia dosis más baja.
Las secuencias sentido y antisentido de AD-1955 son:
SENTIDO: cuuAcGcuGAGuAcuucGAdTsdT (SEQ ID NO:3682)
ANTISENTIDO: UCGAAGuACUcAGCGuAAGdTsdT (SEQ ID NO:3683).
Las secuencias de los dúplex y de las hebras individuales de los ARNip modificados y no modificados se proporcionan en la Tabla 14 y la Tabla 15, respectivamente.






En la Tabla 16 se proporcionan los resultados de los ensayos in vitro. La Tabla 16 también indica las especies diana de cada uno de los ARNip.
Tabla 16: Resultados de los ensayos funcionales

(continuación)

La Tabla 17 ilustra los valores de CI50 de dúplex de ARNip de ALAS1 seleccionados. Los valores de CI50 se determinaron a partir de la desactivación de ALAS1 expresado de forma endógena en la línea celular Hep3B, 24 horas después de la transfección de cada dúplex de ARNip modificado para ALAS1 (remítase a la Tabla 14). Al menos siete dúplex, que incluyeron AD-58882, AD-58878, AD-58886, AD-58877, AD-59115, AD-58856 y AD-59129, presentaron uniformemente valores de CI50 inferiores a 0.1 nm, lo cual indica que estos dúplex fueron particularmente eficaces a la hora de suprimir la expresión de ALAS1.
Tabla 17: Valores de CI50 de dúplex de ARNip de ALAS1 seleccionados

Ejemplo 11. Actividad de ALAS1-GalNAc en un modelo de ratón de inducción de AIP con fenobarbital
Se utilizó el modelo de ratón de AIP para investigar el efecto de un ARNip que era un conjugado de ALAS1-GalNAc. El ARNip tenía la secuencia del dúplex AD-58632 (remítase a la Tabla 20).

El día 1, los ratones con AIP o bien no se trataron (referencia) o se les inyectó por vía subcutánea solución salina o el conjugado de ALAS1-GalNAc con una dosis de 20 mg/kg. Los días 2, 3 y 4, los ratones no se trataron (referencia) o se trataron con inyecciones IP de fenobarbital. El día 5, se tomaron muestras de plasma y se midieron los niveles de ALA y PBG utilizando un ensayo de LC-MS. Como se muestra en la FIG. 15, el conjugado de ALAS1-GalNAc mitigó la producción de ALA y PBG en plasma aproximadamente un 84 y 80%, respectivamente. Estos resultados indican que el tratamiento con un conjugado de ALAS1-GalNAc fue eficaz a la hora de prevenir los incrementos de ALA y PBG en plasma asociados con los ataques agudos inducidos por fenobarbital en este modelo animal de AIP.
Ejemplo 12. Otros ARNip que tienen ALAS1 como diana e inhiben la expresión de ALAS1
Se diseñaron y produjeron secuencias de ARNip modificadas que tenían como diana el ARNip de ALAS1, según se ha descrito en el Ejemplo 2. Las secuencias se proporcionan en la Tabla 18. Se evaluó la actividad in vitro de los dúplex modificados según se describe a continuación.







La actividad in vitro de los ARNip para suprimir el ARNm de ALAS1 se evaluó en un cribado de dosis única en células Hep3B que se habían transfectado utilizando Lipofectamine2000 como reactivo de transfección. Los experimentos de dosis única se llevaron a cabo con una concentración del dúplex 10 nM y se analizaron mediante un ensayo de ADN ramificado (ADNr). Los resultados se muestran en la Tabla 19 y se expresan como el porcentaje de ARNm remanente.
Tabla 19: Supresión del ARNm de ALAS1 evaluada mediante un ensayo de ADNr

(continuación)

(continuación)

(continuación)

Los doscientos treinta y dos dúplex que se evaluaron suprimieron el ARNm de ALAS1 en diferentes grados en este ensayo de dosis única. De acuerdo con este ensayo, al menos cuatro de los dúplex (AD-59453, AD-59395, AD-59477 y AD-59492) suprimieron el ARNm de ALAS1 un 85% o más, 39 de los dúplex suprimieron el ARNm de ALAS1 un 80% o más, 101 de los dúplex suprimieron el ARNm de ALAS1 un 70% o más y 152 de los dúplex suprimieron el ARNm de ALAS1 un 50% o más. Por el contrario, algunos dúplex no mostraron ninguna supresión apreciable en este ensayo.
Ejemplo 13: Inhibición en respuesta a la dosis de los precursores de porfirina ALA y PBG utilizando ARNip de ALAS1
Se investigaron los efectos de respuesta a la dosis de un ARNip de ALAS1 en un modelo en ratón de AIP (remítase al Ejemplo 5). Este modelo muestra ~30% de actividad de PBGD residual, un incremento de ~2 veces en los niveles de ALA y PBG del punto de referencia, un incremento de ~30-100 veces en los niveles de ALA y PBG tras la inducción con inyecciones de fenobarbital una vez al día durante 3-4 días. Los animales de más edad tienen degeneración axonal y deficiencia de la función motora.
El ARNip de ALAS1 utilizado en este ejemplo fue el dúplex AD-53558 en la formulación AF11. En el día 1, a los ratones se les administraron 1 mg/kg, 0.5 mg/kg, 0.1 mg/kg o 0.05 mg/kg de ARNip de ALAS1 o control de LUC AD-1955 mediante inyección i.v. Se administraron tres inyecciones de fenobarbital (1 inyección al día en los días 2, 3 y 4) para inducir ALAS1 hepática y los precursores de la porfirina, ALA y PBG. Se recogieron muestras de orina de toda la noche y de plasma en el día 5 y se midieron los niveles de metabolitos por LC-MS. Se midieron los niveles del punto de referencia de ALA y PBG antes del primer tratamiento en el día 1. Los resultados se muestran en la FIG. 16. El ARNip de ALAS1 inhibió los niveles de ALA y PBG en una manera dependiente de la dosis. Se observó el efecto inhibidor en los niveles de ALA en plasma con dosis de ARNip de ALAS1 de tan solo 0.05 mg/kg, y se observó el efecto inhbidor en los niveles de PBG en plasma con dosis de ARNip de tan solo 0.1 mg/kg.
Ejemplo 14: Inhibición duradera de los precursores de porfirina ALA y PBG utilizando ARNip de ALAS1 Se investigó la duración de los efectos de ARNip de ALAS1 en un modelo en ratón de AIP (remítase al Ejemplo 5). El ARNip de ALAS1 utilizado en este ejemplo fue el dúplex AD-53558 en la formulación AF11. El diseño experimental y los resultados de este experimento se muestran en la Fig. 17. En el día 1, a los ratones se les administró 1 mg/kg de ARNip de ALAS1 o control de LUC AD-1955 mediante inyección i.v. Se administraron tres inyecciones de fenobarbital en la semana 0 (1 inyección al día en los días 2, 3 y 4), semana 2 (1 inyección al día en los días 15, 16 y 17) y semana 4 (1 inyección al día en los días 29, 30 y 31) para inducir ALAS1 hepático y los precursores de porfirina ALA y PBG. Se recogieron muestras de orina de toda la noche y plasma en los días 5, 18 y 32 y se midieron los niveles de metabolitos por LC-MS. Se midieron los niveles del punto de referencia de ALA y PBG antes del primer tratamiento en el día 1.
Tal como se muestra en la FIG. 17, el ARNip de ALAS1 tuvo un efecto duradero en la reducción de los niveles de ALA y PBG en plasma. La administración de ARNip de ALAS1 suprimió los niveles de ALA y PBG en plasma durante al menos 2 semanas. Estos resultados indican que el ARNip de ALAS1 es un tratamiento eficaz para reducir los niveles elevados de ALA y PBG y, por lo tanto, se puede utilizar en la profilaxis, por ejemplo, para reducir los niveles crónicamente elevados de ALA y PBG y para prevenir ataques porfíricos recurrentes.
Ejemplo 15: ARNip de ALAS1 proporciona un inicio de acción más rápido en comparación con el tratamiento con hemina
Se compararon los efectos del tratamiento con ARNip de ALAS1 con los efectos del tratamiento con hemina en un modelo en ratón de AIP (remítase al Ejemplo 5). El ARNip de ALAS1 utilizado en este ejemplo fue el dúplex AD-53558 en la formulación AF11. El diseño experimental y los resultados de este experimento se muestran en la FIG.
18. Se administraron fenobarbital (PB) y dietilditiocarbamato (DDC) en los días 1, 2 y 3. DDC es otro inductor de p450 que, como el fenobarbital, incrementa la demanda de hemo y ayuda a propagar la inducción de los metabolitos ALA/PBG.
Se administró una dosis de hemina de 4 mg/kg, una dosis de ARNip de ALAS1 de 2 mg/kg o tratamiento de control por vía intravenosa 8 horas después de la última administración de PB y DDC.
Como se muestra en la FIG. 18, el inicio de los efectos del tratamiento fue más rápido con el tratamiento de ARNip de ALAS1 en comparación con el tratamiento de hemina. La rápida reducción de ALA y PBG con el tratamiento con ARNip indica que ARNip es un tratamiento eficaz para los ataques agudos, debido a que se espera que la reducción de los niveles de ALA y PBG esté acompañanada por una rápida mejora en los síntomas clínicos.
Ejemplo 16: Efectos del conjugado de ARNip de ALAS1-GalNAc AD-58632
AD-58632 es un heneicosámero/tricosámero descrito en el Ejemplo 11. AD-58632 tiene como diana el transcrito humano NM_000688.4 y también presenta reactividad cruzada con transcritos de ARNm de ratón, rata y mono cinomolgo. AD-58632 es el único heneicosámero/tricosámero con reactividad cruzada identificado en un cribado de aproximadamente 45 compuestos. Se describen experimentos adicionales con este dúplex en este ejemplo.
Efectos dependientes de la dosis de AD-58632 en la supresión de ARNm de ALAS1
Se investigaron en ratas los efectos de respuesta a la dosis de AD-58632 en la supresión de ARNm de ALAS1, respecto a ARNm de GAPDH. Se estudiaron dosis de 30 mg/kg, 10 mg/kg y 3 mg/kg. Se midieron los niveles de ARNm de ALAS1 en el hígado 72 horas después de la última dosis. AD-58632, comparado con el control de PBS, suprimió el ARNm de ALAS1 de manera dependiente de la dosis (remítase a la FIG. 19). AD-58632 tuvo una DE50 de dosis única de aproximadamente 10 mg/kg.
Efectos de AD-58632 en un modelo de AIP en rata
Se investigó en mayor medida el efecto de respuesta a la dosis del ARNip conjugado ALAS1-GalNAc AD-58632 en un modelo de AIP en rata. En este modelo, se utilizó ARNip en un LNP para reducir el nivel de PBGD específicamente en el hígado antes de inducir la demanda de hemo con fenobarbital. El modelo de AIP en rata muestra una reducción transitoria con ARNip de PBGD en el hígado, tiene ~15% ARNm de PBGD residual, y muestra un incremento de aproximadamente 10-50 veces en los niveles de ALA y PBG tras la inducción con inyecciones diarias de fenobarbital durante tres días.
El diseño experimental se muestra en la FIG. 20. Se estudiaron cuatro grupos de ratas. Un grupo se trató con solo fenobarbital (PB) en los puntos temporales indicados. Un segundo grupo se trató con fenobarbital y ARNip de porfobilinógeno-desaminasa (PBGD). Un tercer grupo recibió fenobarbital, ARNip de PBGD y una dosis de 30 mg/kg del ARNip de ALAS1. Un cuartro grupo recibió fenobarbital, ARNip de PBGD y una dosis de 10 mg/kg del ARNip de ALAS1. Como se muestra en la FIG. 20, el ARNip de PBGD se administró por vía intravenosa en el día 1. El ARNip ALAS1 -GalNAc se administró en el día 4. Las inyecciones de fenobarbital se administraron en los días 4, 5, 6 y 7. Se recogió la orina a lo largo de un periodo de 24 horas empezando en el día 7 y finalizando en el día 8. Se evaluaron los niveles de ARNm de PBGD, ARNm de GAPDH y ARNm de ALAS-1 en hígado en el día 8 utilizando un ensayo de ADNr. Se determinaron los niveles de PBG y ALA en orina utilizando LC-MS.
Los resultados del ARNm se muestran en la FIG. 21. El ARNip de PBGD disminuyó el nivel de ARNm de PBGD pero no disminuyó el nivel de ARNm de ALAS1. El ARNip de ALAS1 disminuyó los niveles de ARNm de ALAS1 de manera dependiente de la dosis (véase la FIG. 21). Los resultados de ALA y PBG se muestran en la FIG. 22. ARNip de ALAS1 disminuyó los niveles de ALA y PBG de manera dependiente de la dosis (véase la FIG. 22).
Ejemplo 17: Dosificación dividida con AD-58632
Se investigó la eficacia del conjugado ARNip de ALAS1-GalNAc AD-58632 en dos paradigmas de dosificación dividida separados. Para cada uno de estos estudios, se utilizaron ratas Sprague Dawley hembra. Las ratas se alojaron en SCLR (una habitación con ciclo de luz de 12 horas con luz y 12 horas sin luz) y se sacrificaron 72 horas después de la última inyección. Se midieron los niveles de ARNm de ALAS1 y GAPDH en el hígado utilizando un ensayo de ADN ramificado (ADNr).
Paradigma de cinco dosis diarias frente a una dosis en bolo
En el primer paradigma, se administraron a los grupos de ratas cinco dosis de ARNip (una dosis cada día) o una única dosis en bolo que tuvo la misma concentración total que la suma de las cinco dosis individuales. Específicamente, las ratas se asignaron a una de las siguientes condiciones de tratamiento: (1) inyección subcutánea de 6 mg/kg de ARNip una vez al día durante cinco días, (2) inyección subcutánea de 2 mg/kg de ARNip una vez al día durante cinco días, (3) inyección subcutánea de 1 mg/kg de ARNip una vez al día durante cinco días, (4) inyección subcutánea de una única dosis en bolo de 30 mg/kg de ARNip, (5) inyección subcutánea de una única dosis en bolo de 10 mg/kg de ARNip, (6) inyección subcutánea de una única dosis en bolo de 5 mg/kg de ARNip o (7) tratamiento de control con PBS.
Los resultados se muestran en la FIG. 23. En este paradigma, una única dosis en bolo de ARNip proporcionó una supresión mayor del ARNm de ALAS1 que la dosificación repetida de la misma concentración de ARNip a lo largo de un ciclo de cinco días. Esto fue cierto para todas las dosis estudiadas.
Dosificación de una vez a la semana durante cuatro semanas
En el segundo paradigma, se administraron a las ratas inyecciones subcutáneas de ARNip con una de tres dosis (10 mg/kg, 5 mg/kg o 2.5 mg/kg) una vez a la semana durante cuatro semanas. Un grupo de control recibió inyecciones de PBS.
Los resultados se muestran en la FIG. 24. En comparación con la dosificación única, proporcionar cuatro dosis semanales de 10 mg/kg mejoró la reducción máxima conseguida (DE50 es 10 mg/kg con una dosis única). Por el contrario, la dosificación múltiple de 5 y 2.5 mg/kg a la semana no mejoró el silenciamiento en este paradigma. Ejemplo 18: Identificación y estudio de ARNip de ALAS1 con hebras sentido y antisentido más cortas Se realizaron experimentos adicionales para explorar los efectos del acortamiento de dúplex de ARNip hasta nonadecámeros-nonadecámeros. Se identificaron cinco dúplex más nonadecámeros-nonadecámeros con
reactividad cruzada que se unen a transcritos de ARNm de ALAS1 humano (h) (NM_000688.4), mono Rhesus (rh) (XM_001090440.2), ratón (m) (NM_020559.2) y rata (r) (NM_024484.2). Ninguno de estos dúplex mostró resultados tan buenos como el heneicosámero/tricosámero AD-58632 (remítase a la FIG. 25).
Se investigaron los efectos de modificar la longitud y protuberancias de los dos mejores nonadecámerosnonadecámeros (AD-59115 y AD-59125) (FIG. 26 y 27). Las secuencias modificadas se muestran en la Tabla 21.

Las protuberancias mejoran la potencia. También proporcionan una secuencia derivada adicional (AD-60489, que se basó en AD-60095) para estudios adicionales de la relación estructura actividad (SAR) (1 apareamiento erróneo en la pos23 con roedor).
Ejemplo 19: Efectos de conjugados de ARNip de ALAS1-GalNAc AD-60489 y AD-58632
Se investigaron los efectos de un dúplex de ARNip de ALAS1 conjugado con GalNAc AD-60489 y se compararon con los efectos de AD-58632. Las secuencias de estos dúplex se muestran en la Tabla 22A. AD-60489 tuvo un único emparejamiento erróneo con ARNm de ALAS1 de roedor en el extremo 3’ de la secuencia antisentido. Por lo tanto, mientras que AD-58632 es totalmente complementario con las secuencias de ser humano, de mono cinomolgo, ratón y rata, AD-60489 es totalmente complemenario solo con secuencias de ser humano y mono cinomolgo.

La supresión de ARNm de ALAS1 se muestra en la FIG. 28. En comparación con AD-58632, AD-60489 proporcionó una supresión más eficaz con 3 mg/kg y 10 mg/kg y mostró una mejora de aproximadamente dos veces en la DE50. La DE50 de dosis única de AD-60489 fue de aproximadamente 5 mg/kg.
Ejemplo 20: Efectos de los conjugados ARNip de ALAS1-GalNAc AD-60489 y AD-58632 en estudios con primates no humanos
Se estudió la eficacia de AD-58632 y AD-60489 en la supresión del ARNm hepático en primates no humanos. El diseño experimental se muestra en la FIG. 29. Se administraron por vía subcutánea dosis de ARNip (5 mg/kg, 2.5 mg/kg o 1.25 mg/kg) o control de PBS en un volumen de 2 ml/kg a diario durante 5 días, después cada 2 días durante 3 semanas. Se evaluó el silenciamiento de ARNm de ALAS1 en tejido hepático obtenido de una biopsia hepática realizada en el día 15. La biopsia se realizó después de una extracción de suero y antes de la administración de la dosis 10 (remítase a la FIG. 29). Se extrajeron muestras de suero para el método de detección de ARN extracelular circulante (cERD) (remítase al Ejemplo 21) en los días -10, -3, 7, 15, 23, 31 y 43. Se extrajo suero para un panel de química clínica en los días -3, 6, 30 y 43. El panel de química clínica incluyó la evaluación de los niveles de alanina-aminotransferasa (ALT), aspartato-aminotransferasa (AST) y fosfatasa alcalina (ALP).
AD-58632 y AD-60489 suprimieron el ARNm de ALAS1 en el hígado en una manera dependiente de la dosis (remítase a la FIG. 30). AD-60489 mostró una eficacia mayor que AD-58632. Por ejemplo, en el nivel de dosis más bajo estudiado (1.25 mg/kg), AD-60489 suprimió el mensaje de ALAS1 relativo hasta aproximadamente un 42% del nivel de control, mientras que AD-58632 mostró poca supresión a esta dosis. Con 2.5 mg/kg, AD-60489 suprimió el mensaje de ALAS1 relativo hasta aproximadamente un 26% del nivel de control, mientras que AD-58632 suprimió el mensaje de ALAS1 relativo hasta aproximadamente un 64% del nivel de control. Con 5 mg/kg, AD-60489 suprimió el mensaje de ALAS1 relativo hasta aproximadamente un 21% del nivel de control, y AD-58632 suprimió el mensaje de ALAS1 relativo hasta aproximadamente un 55% del nivel de control.
Los resultados de la química clínica indicaron que la atenuación sostenida de ALAS1 utilizando los ARNip de ALAS1 fue segura y bien tolerada. No se observaron elevaciones de ALT, AST o ALP.
Ejemplo 21: Efectos de los conjguados de ARNip de ALAS1-GalNAc AD-60489 y AD-58632 en estudios con primates no humanos según se evalúa utilizando el ensayo cERD
Se evaluaron los efectos de los conjugados de ARNip de ALAS1-GalNAc AD-60489 y AD-58632 en primates no humanos utilizando el método de detección de ARN extraceluar circulante (cERD). Este método se describe, p. ej., en Sehgal, A. et al. Quantitation of tissue-specific target gene modulation using circulating RNA (Póster presentado en el 9 de febrero de 2012 en el simposio Keystone Gene Silencing by small RNAs (Vancouver, 7-12 de febrero de 2012) y en Sehgal, A. et al. Tissue-specific gene silencing monitored in circulating RNA, RNA, 20: 1-7, con publicación electrónica del 19 de diciembre de 2013. Como se muestra en la FIG. 29, se tomaron muestras de suero para el método de detección del ARN extracelular circulante (cERD) en los días -10, -3, 7, 15, 23, 31 y 43.
Para el ensayo cERD, se descongelaron las muestras de suero en hielo. Se añadieron 375-400 pl de LiCl 8 M a 3 3.5 ml de suero en tubos de ultracentrifugación (UC, por sus siglas en inglés), y se incubaron a una temperatura de 4 °C durante al menos 1 hora. Se añadió PBS en la parte superior de cada tubo de UC, dejando aproximadamente 1 cm de espacio seco en la parte superior del tubo para evitar que las paredes de los tubos se rompieran durante la centrifugación. Los tubos se secaron para evitar que cualquier condensación se incubara en el hielo. Las muestras se colocaron en un Rotor MC 55 en una campana y las muestras se centrifugaron a 150000-200000g durante 100 200 minutos. Se desechó el sobrenadante del sedimento. Se añadió 1 ml de de Trizol al sedimento en el tubo de UC, el tubo se agitó en vórtex y el contenido se transfirió a un tubo de microcentrifugación de 1.5 ml. Se añadieron 200 pl de cloroformo a cada tubo y el tubo se invirtió varias veces para mezclarlo. Se preparó una muestra de cada vez. Las muestras se centrifugaron a 13000 RPM durante 10-20 minutos a 4 °C. La fase acuosa superior se transfirió a un tubo de 1.5 ml nuevo (volumen de ~500 pl). Se añadió un volumen idéntico de isopropanol al 100%, 1 pl de acrilamida lineal (4°) y 1/10 volúmenes de NaOAc 3 M pH 5.5 o menos a cada muestra (normalmente 500 pl de isopropanol y 50 pl de NaOAc). La muestra se centrifugó a 13000 RPM durante 10 min a 4 °C. Se guardaron los sobrenadantes. El sedimento se lavó dos veces con EtOH al 70% enfriado en hielo (500 pl en cada lavado) y se centrifugó a 13000 RPM durante ~5 min a 4 °C después de cada lavado. Se permitió que el sedimento se secara al aire durante ~5 minutos y a continuación se resuspendió en 20 pl de NF H2O. Se utilizaron 10 pl en la reacción de ADNc. El ARN resuspendido se almacenó a -80 °C.
Resultados
Se evaluó la atenuación del ARNm en suero utilizando el ensayo cERD correlacionado con los resultados obtenidos de la biopsia hepática. Remítase a la FIG. 31. Este es un resultado sorprendente debido a que ALAS1 no es una proteína sérica. El ensayo cERD que se proporciona en la presente permite monitorizar el ARNm de ALAS1 circulante. Esto tiene la ventaja, por ejemplo, de que los niveles de ARNm de ALAS1 se pueden medir en el tiempo sin realizar biopsias hepáticas en serie, lo que sería caro y difícil desde un punto de vista técnico.
Se determinó la cinética de la atenuación del ARNm utilizando los resulados del ensayo cERD. Remítase a la FIG.
32. AD-60489 logró más de un 50% de atenuación, incluso con una dosis de tan solo 1.25 mg/kg.
Ejemplo 22: Estudios de seguridad de los ARNip de ALAS1
Los siguientes estudios de seguridad indican que la atenuación sostenida de ALAS1 es segura y se tolera bien. Estudios con primates no humanos
Como se ha descrito anteriormente (remítase al Ejemplo 20), en los estudios con primates no humanos, no se observaron elevaciones de ALT, AST o ALP después de la administración de AD-60489 y AD-58632.
Estudios en ratas
En ratas, se llevó a cabo un estudio de cuatro semanas con AD-58632. Se administraron 10 mg/kg del ARNip cada día durante 5 días en la primera semana y después 10 mg/kg en días alternos durante las semanas 2-4 del estudio. La exposición total fue de 140 mg. No se observaron señales clínicas adversas ni cambios en el peso corporal. No se observaron cambios relacionados con el artículo de prueba en los parámetros hematológicos o de coagulación. Además, no se observó una histopatología adversa. Hubo una vacuolación mínima y una fibrosis subcapsular mínima en el riñón.
Estudios en ratones
En ratones, se evaluaron los ARNm de P450 después de la atenuación de ALAS1. Se observaron incrementos dependientes de la dosis mínimos en Cyp2b10 48 horas después de la administración de una formulación de LNP de ALAS1. Esto se resolvió en 168 horas.
Ejemplo 23: Identificación de ARNip de ALAS1 más eficaces utilizando estudios de la relación estructuraactividad
Se llevaron a cabo estudios de la relación estructura-actividad (SAR, por sus siglas en inglés), incluidos estudios descritos en otros ejemplos de la presente, para identificar ARNip de ALAS1 más eficaces derivados de los que ya se habían identificado, p. ej., AD-58632 y AD-60489. Se estudiaron los efectos de las modificaciones químicas. Las modificaciones químicas incluyen 1) modificaciones 2’-O-metilo frente a 2’-fluoro, 2) descenso en 2’Uf (modificaciones 2’-fluoro), 3) adición de PS (fosforotioato), 4) utilización de dT internas y/o 5) ácidos glicolnucleicos (GNA). Sin desear ceñirse a la teoría, las modificaciones pueden mejorar la potencia, p. ej., mediante 1) mejor desplegamiento o mejora de la carga RISC, o 2) mejor acoplamiento catalítico con la diana. Las modificaciones también pueden mejorar la estabilidad de manera que los compuestos se pueden acumular y funcionar mejor cuando se administran múltiples dosis.
Se observó mejora de la actividad respecto a otros dúplex (p. ej., AD-58632 y/o AD-60489) en algunos casos (remítase a la Tabla 22B), mientras que se observó una actividad similar (remítase a la Tabla 23) o una actividad reducida (Tabla 24) en otros casos. Estos casos se presentan meramente como ejemplos basados en el cribado de más de 150 ARNip. En la presente se proporciona una ejemplificación adicional de los estudios SAR.
Tabla 22B: Actividad mejorada respecto al original

Tabla 23: Actividad similar respecto al original pero mayor estabilidad

Tabla 24: Actividad reducida respecto al original

Ejemplo 24: Estudios de la reación estructura-actividad in v itro de AD-58632
Se generaron AD-58632 y derivados de ARNip de AD-58632 y se cribaron algunos ARNip in vitro para determinar la actividad. En la Tabla 1 se proporcionan las abreviaturas para las modificaciones químicas.
Actividad in vitro con ARNip 10 nM y 0.1 nM
Se estudió la actividad in vitro de los ARNip para suprimir el ARNm de ALAS1 en células Hep3B que se transfectaron utilizando Lipofectamine2000 como un reactivo de transfección. Los experimentos se realizaron con las concentraciones de ARNip indicadas (p. ej., 0.1 nM, 10 nM) y se analizaron mediante el ensayo de ADN ramificado (ADNr) 24 horas después de la transfección. Los resultados se expresan como ARNm restante porcentual relativo al ARNip AD-1955, un ARNip no dirigido que se utilizó como control negativo.
Las secuencias de los ARNip y los resultados de las pruebas in vitro se proporcionan en la Tabla 25, Tabla 26 y Tabla 27.






Como se muestra en la tabla anterior, en este cribado in vitro, los ARNip que proporcionan la supresión de ARNm de ALAS1 mayor (más de un 80% de supresión, de modo que permanece menos de un 20% del ARNm) con la concentración de 10 nM incluyeron AD-58632, AD-60472, AD-60423, AD-60445, AD-60423, AD-60417, a D-60466, AD-60473, AD-60434, AD-60448, AD-60460, AD-60411, AD-60481, AD-60486, y AD-60453, AD-60480, AD-60405, AD-60477, AD-60461, AD-60470, AD-60467, AD-60482, AD-60446, AD-60555, AD-60454, AD-60469 y AD-60463. Además, en este cribado in vitro, los ARNip que proporcionan la mayor supresión de ARNm de ALAS1 (más de un 30% de supresión, de modo que permanece menos de un 70% del ARNm) con la concentración de 0.1 nM incluyeron AD-60423, AD-58632, AD-60434, AD-60423, AD-60466, AD-60419, AD-60438, AD-60448, AD-60460, AD-60473, AD-60411, AD-60405, AD-60472, AD-60477, AD-60417, AD-60480, AD-60482, AD-60421, AD-60560, AD-60433, AD-60481, AD-60475, AD-60555, AD-60437, AD-60550, AD-60415, AD-60463 y AD-60443.
Como se muestra en la siguiente tabla, las pruebas de más ARNm mostraron que los siguientes dúplex proporcionaron más de un 80% de supresión con la concentración de 10 nM: AD-58632, AD-60405, AD-60423, AD-60434, AD-60445, AD-60480, AD-60460 y AD-60466, y los siguientes dúplex proporcionaron más de un 30% de supresión con la concentración de 0.1 nM: AD-58632, AD-60405, AD-60423, a D-60434, AD-60419, AD-60480, AD-60460 y AD-60466.




CI50 basadas en la actividad in vitro
De manera similar a los experimentos descritos anteriormente, se realizaron más experimentos de dosis-respuesta con una concentración final del dúplex de 10 nM, 1.66667 nM, 0.277778 nM, 0.046296 nM, 0.007716 nM, 0.001286 nM, 0.000214 nM y 3.57E-05 nM, y se calcularon las CI50.



Como se muestra en la Tabla 28, los siguientes dúplex tuvieron una CI50 inferior a 0.01 nM: AD-60845, AD-60843, AD-60849, AD-60820, AD-60848, AD-60822, AD-60826, AD-60819 y AD-60460.
Los siguientes dúplex tuvieron una CI50 inferior a 0.02 nM: AD-60845, AD-60843, AD-60849, AD-60820, AD-60848, AD-60822, AD-60826, AD-60819, y AD-60460, AD-60841, AD-60842, AD-60846, AD-60847, AD-60838, AD-60419, AD-60839, AD-60835, AD-586320, AD-60844, AD-60850 y AD-60830.
Los siguientes dúplex tuvieron una CI50 inferior a 0.05 nM: AD-60845 , AD-60843, AD-60849, AD-60820, AD-60848, AD-60822, AD-60826, AD-60819, y AD-60460, AD-60841, AD-60842, AD-60846, AD-60847, AD-60838, AD-60419, AD-60839, AD-60835, AD-586320, AD-60844, AD-60850, AD-60830, AD-60423, AD-60834, AD-60419, AD-60434, AD-60825, AD-60837, AD-60823, AD-60824, AD-60840, AD-60829, AD-60893, AD-60832 y AD-60827.
Ejemplo 25: Estudios de la relación estructura-actividad in v ivo de AD-58632
Se generaron derivados del ARNip original AD-58632 y se cribaron in vivo en ratas. Las secuencias de ARNip que se cribaron se proporcionan en la siguiente tabla.

Se administró una única dosis de 5 mg/kg de ARNip. El día 5 tras la administración del ARNip, se realizaron medidas del ARNm de ARNm de ALAS1 de rata (rALAS1) y ARNm de GAPDH de rata (rGAPDH) utilizando un ensayo de ADNr, y se determinaron los niveles tisulares del fármaco utilizando qPCR. Los resultados se proporcionan en la FIG. 33 y FIG. 34. Como se muestra en la FIG. 33, al menos diez dúplex (AD-60405, AD-60887, AD-60923, AD-60434, AD-60892, AD-60419, AD-60924, AD-60445, AD-60925 y AD-60926) que se cribaron mostraron una mejora de la supresión del ARNm de ALAS1 en comparación con AD-58632. Además, como se muestra en la FIG. 34, estos dúplex (con la excepción de AD-60926) lograron concentraciones hepáticas más elevadas que AD-58632.
Ejemplo 26: Eficacia de AD-60925 y AD-60926 en un modelo de AIP en rata
Se estudió la eficacia terapéutica de AD-60925 y AD-60926 (descritos en el ejemplo anterior) en un modelo de AIP en rata. Se muestra el diseño experimental en la parte superior de la FIG. 35. Las ratas fueron tratadas con PBS o 3 mg/kg de ARNip de ALAS1 -GalNAc tres veces por semana, fenobarbital (PB) y ARNip de PBGD en una formulación de LNP AF11 (AF11-PBGD) en los momentos indicados en la FIG. 35. Las ratas de control recibieron el ARNip de PBGD solo, sin inducción con fenobarbital.
Los resultados se muestran en la FIG. 35, FIG. 36 y FIG. 37. Administrar fenobarbital indujo la expresión de ARNm de ALAS1 e incrementó los niveles de PBG y ALA en orina, en comparación con el control. El tratamiento con un total de ocho dosis de 3 mg/kg de AD-60925 o AD-60926 tres veces a la semana suprimió el ARNm de ALAS1 (FIG.
35), PBG en orina (FIG. 36 y FIG. 37, parte superior) y ALA en orina (FIG. 36 y FIG. 37, parte inferior). El fenobarbital indujo incrementos en ARNm de ALAS1, PBG y ALA en orina. La evolución temporal de los efectos del tratameinto se muestra en la FIG. 37. Las flechas indican los puntos temporales en los que se administró PB. El tratamiento con ARNip previno los incrementos inducidos por fenobarbital en ARNm de ALAS1, PBG y ALA en orina. Tanto AD-60925 como AD-60926 mostraron la eficacia terapéutica del tratamiento de AIP. AD-60925 fue aún más eficaz que AD-60926 al suprimir ARNm de ALAS1, ALA en orina y PBG en orina.
Ejemplo 27: Estudios adicionales de la relación estructura-actividad in v ivo de AD-58632
Se generaron y cribaron in vivo en ratas derivados del ARNip original AD-58632.
Cribado in vivo, parte I
Las secuencias de los ARNip que se cribaron se proporcionan en la siguiente tabla.

A las ratas se les administraron cuatro dosis de 2.5 mg/kg de ARNip bisemanalmente (dos veces por semana) durante dos semanas. 72 horas después de la administración de la última dosis de ARNip, se sacrificaron los animales y se realizaron medidas de los niveles de ARNm de ALAS1 de rata (rALAS1) y ARNm de GAPDH de rata (rGAPDH) utilizando el ensayo de ADNr.
Como se muestra en la FIG. 38, al menos cuatro de los ARNip (AD-60820, AD-60843, AD-60819 y AD-61140) que se estudiaron mostraron una mejora de la supresión del ARNm de ALAS1 en comparación con AD-58632.
Cribado in vivo, parte II
Las secuencias de los ARNip que se cribaron se proporcionan en la siguiente tabla.
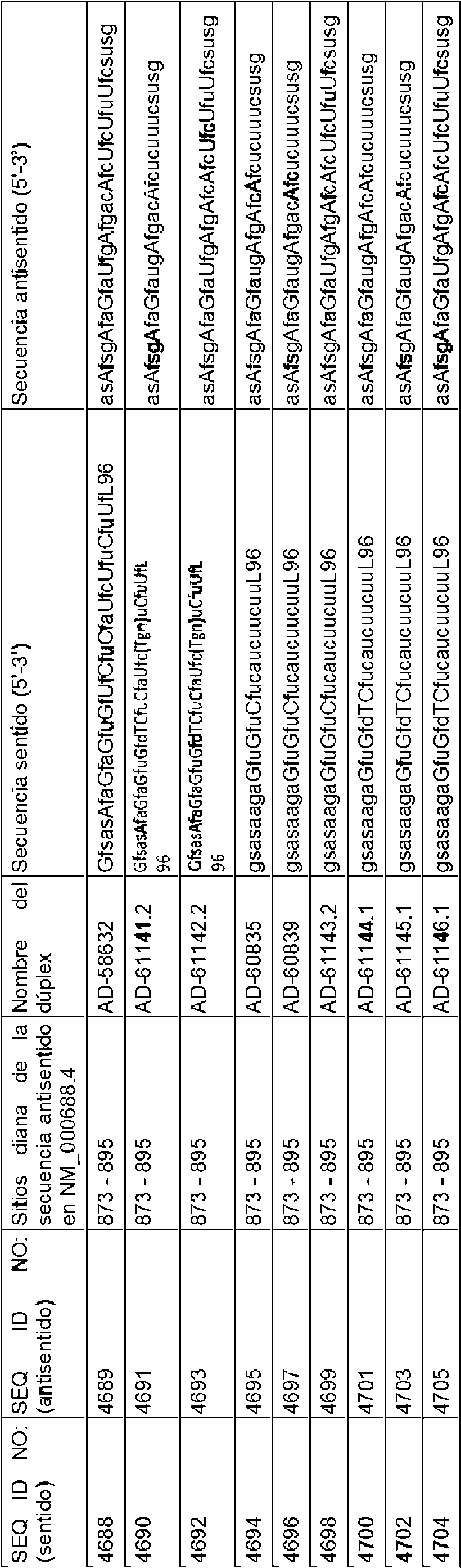
A las ratas se les administró una única dosis de 2.5 mg/kg de ARNip. 72 horas después de la administración del ARNip, se realizaron medidas de ARNm de los niveles de ARNm de a La S1 de rata (rALAS1) y ARNm de GAPDH de rata (rGAPDH) utilizando el ensayo de ADNr.
Como se muestra en la FIG. 39, los ARNip AD-61141, AD-61142, AD-60835, AD-60839, AD-61143, AD-61144, AD-61145 y AD-61146 mostraron una mejora de la supresión de ARNm de ALAS1 en comparación con AD-58632. El ARNip que proporcionó la supresión mayor en este experimento fue AD-60835.
Ejemplo 28: Estudios de la relación estructura-actividad in v itro de AD-60489
Se generaron AD-60489 y ARNip derivados de AD-60489 y algunos ARNip se cribaron in vitro para determinar su actividad. La actividad in vitro de los ARNip para suprimir el ARNm de ALAS1 se estudió tal como se ha descrito en el Ejemplo 24. Las secuencias de ARNip y los resultados de los estudios in vitro se proporcionan en las siguientes tablas.






En el cribado in vitro para el cual se muestran los resultados en la tabla anterior, los ARNip que proporcionaron la mayor supresión de ARNm de ALAS1 (más de un 80% de supresión, de modo que permaneció menos de un 20% del ARNm) con una concentración de 10 nM incluyeron AD-60501, AD-60592, AD-60591, AD-60513, AD-60507, AD-60587, AD-60519, AD-60593, AD-60583, AD-60524, AD-60489, AD-60495, AD-60506 y AD-60582.
En el cribado in vitro para el cual se muestran los resultados en la tabla anterior, los ARNip que proporcionaron la mayor supresión del ARNm de ALAS1 (más de un 30% de supresión, de modo que permaneció menos de un 70% de ARNm) con una concentración de 0.1 nM incluyeron AD-60592, AD-60591, AD-60593, AD-60587, AD-60583, AD-60589, AD-60501, AD-60507, AD-60585, AD-60489, AD-60513, AD-60582, AD-60519, AD-60541, AD-60570, AD-60584, AD-60569, AD-60558, AD-60573, AD-60556, AD-60495, AD-60523, AD-60566 y AD-60544.
Como se muestran en la siguiente tabla, el estudio de más ARNip reveló que los siguientes dúplex proporcionaron más de un 80% de supresión con una concentración de 10 nM: AD-60489, AD-60495, AD-60501, Ad-60507, AD-60513, AD-60519, AD-60583, AD-60591, AD-60592 y AD-60593, y los siguientes dúplex proporcionaron más de un 30% de supresión con una concentración de 0.1 nM: AD-60489, AD-60495, AD-60501, Ad-60507, AD-60513, AD-60519, AD-60583, AD-60591, AD-60592 y AD-60593.








Como se muestra en la tabla anterior, una serie de dúplex mostraron eficacia para suprimir ARNm de ALAS1. Los siguientes dúplex tuvieron una CI50 de menos de 0.01 nM:_AD-60879, AD-60859, AD-60863, AD-60854, AD-60882, AD-60874, AD-60883, AD-60875, AD-60501, AD-60593, AD-60853, AD-60877, AD-60878, AD-60871 y AD-60873. Los siguientes dúplex tuvieron una CI50 de menos de 0.02 nM:_AD-60879, a D-60859, a D-60863, AD-60854, AD-60882, AD-60874 , AD-60883, AD-60875, AD-60501, AD-60593, AD-60853, AD-60877, AD-60878, AD-60871, AD-60873, AD-60489, AD-60592, AD-60894, AD-60489, AD-60870, AD-60862, AD-60858, AD-60592, AD-60591, AD-60872, AD-60866, AD-60905, AD-60857, AD-60513 y AD-60861. Los siguientes dúplex tuvieron una CI50 de menos de 0.05 nM:_AD-60879, AD-60859, AD-60863, AD-60854, AD-60882, AD-60874, AD-60883, AD-60875, AD-60501, AD-60593, AD-60853, AD-60877, AD-60878, AD-60871, AD-60873, AD-60489, AD-60592, AD-60894, AD-60489, AD-60870, AD-60862, AD-60858, AD-60592, AD-60591, AD-60872, AD-60866, AD-60905, AD-60857, AD-60513, AD-60861, AD-60583.2, AD-60902.1, AD-60881.1, AD-60519.2, AD-60507.2, AD-60591.3, AD-60851.1, AD-60896.1 y AD-60537.2.
Ejemplo 29: Estudios de la relación estructura-actividad in v ivo de AD-60489
Se generaron y cribaron in vivo en ratas derivados del ARNip original AD-60489.
Cribado in vivo 1 de derivados de AD-60489
Las secuencias de los ARNip que se cribaron se proporcionan en la siguiente tabla.

A las ratas se les administró una única dosis de 3 mg/kg de ARNip. 5 días después de la administración del ARNip, se realizaron medidas de ARNm de ARNm de ALAS1 de rata (rALAS1) y ARNm de GAPDH de rata (rGAPDH) utilizando un ensayo de ADNr, y se determinaron los niveles tisulares del fármaco (ARNip) utilizando qPCR.
Como se muestra en la FIG. 40 (parte superior), los ARNip AD-60501, AD-60519, AD-60901, AD-60495, AD-60900 y AD-60935 mostraron una mejora de la supresión del ARNm de ALAS1 en comparación con AD-60489. Los ARNip AD-60519, AD-60901, AD-60495 y AD-60935, lograron niveles hepáticos más elevados que AD-60489 (remítase a la FIG. 40, parte inferior). Por lo tanto, la mayoría de los dúplex que proporcionaron una mejora de la supresión del ARNm de ALAS1 también lograron niveles hepáticos más elevados.
Al menos para los dúplex AD-60489, AD-60519 y AD-60901, la eficacia se correlacionó con los niveles hepáticos del ARNip (remítase a la FIG. 41), de modo que un nivel más elevado de ARNip en el hígado se asoció con una mayor supresión del ARNm de ALAS1.
Cribado in vivo 2 de derivados de AD-60489
Las secuencias de los ARNip que se cribaron se proporcionan en la siguiente tabla.

A las ratas se les administró una única dosis de 2.5 mg/kg de ARNip. 5 días después de la administración del ARNip, se realizaron las medidas del ARNm de ARNm de ALAS1 de rata (rALAS1) y ARNm de GAPDH de rata (rGAPDH) utilizando el ensayo de ADNr.
Como se muestra en la FIG. 42, los ARNip AD-60879, AD-61190, AD-61191, AD-60865, AD-60861, AD-60876, AD-61193 y AD-60519 mostraron una mejora de la supresión del ARNm de ALAS1 en comparación con AD-60489.
Ejemplo 30: Múltiples dosis mejoran la potencia
Para estudiar los efectos de la administración de múltiples dosis de ARNip, se administró a ratas (n=3 por grupo) PBS o un ARNip (AD-58632, AD-60925, AD-60419, AD-60445, AD-60892, AD-60489, AD-60519 o AD-60901) en una dosis de 2.5 mg/kg dos veces a la semana durante 2 semanas. Los niveles de ARNm de ALAS1 de rata (rALAS1) y ARNm de GAPDH de rata (rGAPDH) se evaluaron utilizando el ensayo de ADNr.

Como se muestra en la FIG. 43, los ARNip derivados de AD-58632 AD-60892, AD-60419, AD-60445 y AD-60925, mostraron una mejora de la supresión de ARNm de ALAS1 en comparación con el AD-58632 original. Además, los ARNip derivados de AD-60489 AD-60519 y AD-60901 mostraron una mejora de la supresión del ARNm de ALAS1 en comparación con el AD-60489 original.
Ejemplo 31: Estudios con múltiples dosis con AD-60519 y AD-60489
Se estudió la eficacia terapéutica de AD-60519 en un modelo de AIP en rata. El diseño experimental se muestra en la FIG. 44 (parte superior). Las ratas se trataron con PBS o con 2.5 mg/kg o 5 mg/kg ARNip de ALS1-GalNAc dos veces a la semana durante tres semanas. Se administraron fenobarbital (Fenobarb) y un ARNip de PBGD en una formulación de LNP AF11 en los momentos indicados en la FIG. 44. Un grupo de control recibió PBS y el ARNip de PBGD solo, sin inducción con fenobarbital. Se recogió orina en los días 18-19 del estudio.
Los resultados se muestran en la FIG. 44 (parte inferior). La administración de fenobarbital y PBS indujo la expresión de ARNm de ALAS1 e incrementó los niveles de PBG y ALA en orina (remítase a la FIG. 44), en comparación con PBS solo. El tratamiento con un total de seis dosis de 2.5 o 5 mg/kg de AD-60519 dos veces a la semana suprimió los incrementos inducidos por fenobarbital en PBG en orina y ALA en orina (FIG. 44). Estos resultados demuestran que AD-60519 es eficaz para suprimir ALA y PBG cuando se administran dosis repetidas de tan solo 2.5 mg/kg. En particular, AD-60519 fue eficaz para reducir los incrementos en los niveles en orina de PBG y ALA asociados con ataques agudos en el model de AIP en rata.
En estudios adicionales que utilizan el mismo diseño experimental pero en un modelo en ratón (remítase a la representación esquemática en la parte superior de la FIG. 44), se estudió la eficacia terapéutica de AD-60519 y AD-60489 para suprimir los incrementos inducidos por fenobarbital de PGB y ALA en suero. En el grupo de control con PBS (“solución salina”), la administración de fenobarbital incrementó los niveles de PBG y ALA en suero (remítase a la FIG. 45), en comparación con PBS solo. El tratamiento con un total de seis dosis de 2.5 o 5 mg/kg de AD-60519 o AD-60489 dos veces a la semana suprimió los incrementos inducidos por fenobarbital de PBG en suero y ALA en suero (FIG. 44). Estos resultados demuestran que tanto AD-60519 como AD-60489 son eficaces para suprimir ALA y PBG cuando se administran dosis repetidas de tan solo 2.5 mg/kg. En particular, AD-60519 y AD-60489 son eficaces para reducir los incrementos de PBG y ALA en suero asociados con los ataques agudos.
Debido a que los tratamientos en este ejemplo se administraron antes de la inducción con fenobarbital, estos resultados indican que AD-60519 y AD-60489 tienen efectos profilácticos.
Ejemplo 32: Secuencias de ARNip adicionales
También se han generado las siguientes secuencias de ARNip derivadas de AD-58632 (Tabla 39) y derivadas de AD-60489 (Tabla 40).


Ejemplo 33: Estudios de múltiples dosis adicionales con AD-60519
Se estudió la eficacia terapéutica de AD-60519 en un modelo de AIP en rata como el utilizado en el Ejemplo 31. El diseño experimental se muestra en la FIG. 46 (parte superior). Las ratas se trataron con PBS o 3 mg/kg, 1 mg/kg o 0.3 mg/kg de ARNip de ALAS1-GalNAc una vez a la semana durante cuatro semanas (tratamiento en el día 0, día 7, día 14 y día 21). Se administraron fenobarbital (Fenobarb) y un ARNip de PBGD en una formulación de LNP AF11 en los momentos indicados en la FIG. 46. Un grupo de control recibió PBS y el ARNip de PBGD solo, sin inducción con fenobarbital. La orina se recogió en el día 25 del estudio.
Los resultados se muestran en la FIG. 46 (parte inferior) y en la FIG. 47. La administración de fenobarbital y PBS indujo la expresión de ARNm de ALAS1 e incrementó los niveles de PBG y ALA en orina, en comparación con PBS solo. El tratamiento con un total de cuatro dosis de 3 mg/kg, 1 mg/kg o 0.3 mg/kg de AD-60519 una vez a la semana suprimió los incrementos inducidos por fenobarbital de los niveles de ARNm de ALAS1 de rata en el hígado en una manera dependiente de la dosis (remítase a la FIG. 46). (Los niveles de ARNm de ALAS1 de rata (rALAS1) en el hígado se expresan en relación con los niveles de ARNm de GAPDH de rata). Los niveles de PBG en orina y ALA en orina también mostraron efectos del tratamiento dependientes de la dosis.
Las dosis semanales repetidas de AD-60519 fueron eficaces para suprimir la expresión de ARNm de ALAS1 y para reducir los niveles elevados de ALA y PBG asociados con ataques agudos inducidos en un modelo de AIP en rata. Estos efectos del tratamiento fueron dependientes de la dosis. Estos resultados ilustran que AD-60519 puede actuar de manera profiláctica cuando se administran dosis antes de un ataque.
Ejemplo 34: Efectos de múltiples dosis de conjugados de ARNip de ALAS1-GalNAc en primates no humanos
Se estudiaron los efectos de conjugados de ARNip de ALAS1-GalNAc para suprimir el ARNm de ALAS1 en el hígado y ARNm de ALAS1 circulante en un estudio en primates no humanos (NHP). Se emplearon los conjugados de GalNAc AD-58632, AD-60519, AD-61193 y AD-60819. El diseño del estudio se muestra en la Tabla 41 y en la FIG. 48.
Tabla 41: Diseño del estudio con NHP

(continuación)

Cada grupo recibió múltiples dosis subcutáneas de un conjugado de ANRip de ALASI-GalNAc en un volumen de dosis de 2 mg/ml. El Grupo 1 (n=3) recibió 2.5 mg/kg de AD-58632 de 1.25 mg/ml en los días 1, 2, 3, 4, 5, 8, 11, 15, 18, 22 y 25. El Grupo 2 (n=3) recibió 1.25 mg/kg de AD-60519 de 0.625 mg/ml en 1,2, 3, 4, 5, 8, 11, 15, 18, 22 y 25. El Grupo 3 (n=3) recibió 2.5 mg/kg de AD-60519 de 1.25 mg/ml en los días 1, 2, 3, 4, 5, 8, 11, 15, 18, 22 y 25. El Grupo 4 (n=3) recibió 2.5 mg/kg de AD-60519 de 1.25 mg/ml en los días 1, 2, 3, 4, 5, 11, 18 y 25. El Grupo 5 (n=3) recibió 5 mg/kg de AD-60519 de 2.5 mg/ml en los días 1, 2, 3, 4, 5, 11, 18, y 25. El Grupo 6 (n=3) recibió 2.5 mg/kg de AD-61193 de 1.25 mg/ml en los días 1, 2, 3, 4, 5, 8, 11, 15, 18, 22 y 25. El Grupo 7 (n=3) recibió 2.5 mg/kg de AD-60819 de 1.25 mg/ml en los días 1,2, 3, 4, 5, 8, 11, 15, 18, 22 y 25.
Las muestras de suero para un ensayo de detección de ARN extracelular circulante (cERD) (remítase al Ejemplo 21) se recogieron en los días -3, 7, 13, 21,27, 39, 46 y 60 (en la FIG. 48, “Extracciones PD” indica los días en los que se recogió el suero). El suero se recogió para un panel de química clínica en los días -3 y 6. El panel de química clínica incluyó la evaluación de los niveles de alanina-aminotransferasa (ALT), aspartato-aminotransferasa (AST) y fosfatasa alcalina (ALP). Se evaluó el silenciamiento del ARNm de ALAS1 en tejido hepático obtenido de una biopsia hepática realizada el día 21 (remítase a la FIG. 48). La biopsia se realizó después de una extracción de suero. Supresión de los niveles de ARNm de ALAS1 en el hígado
Los niveles de ARNm de ALAS 1 en el hígado en el día de estudio 21 se muestran en la FIG. 49. Los resultados se muestran como un porcentaje del nivel promedio observado en un grupo de control tratado con PBS. Los resultados se muestran como los valores promedio para cada grupo de tratamiento.
Estos resultados presentados en la FIG. 49 demuestran que en comparación con los animales de control que recibieron tratamiento con PBS, todas las condiciones de tratamiento fueron eficaces para suprimir los niveles en el hígado de ARNm de ALAS1. Los tratamientos lograron un silenciamiento de ARNm comprendido entre aproximadamente un 20% y un 80% (correspondiente a niveles de ARNm de ALAS1 comprendidos entre aproximadamente un 80% y un 20% de los niveles de control).
Los animales individuales que recibieron AD-58632 mostraron un silenciamiento de aproximadamente un 20-50%, siendo el nivel promedio de silenciamiento de aproximadamente un 40% (los niveles de ARNm de ALAS1 estuvieron
de promedio en aproximadamente un 60% de los niveles de control). Con todas las programaciones posológicas empleadas, AD-60519 fue sumamente eficaz para suprimir los niveles de ARNm de ALAS1. Los animales individuales que recibieron AD-60519 mostraron un silenciamiento de entre aproximadamente un 60% y un 80% (los niveles de ARNm de ALAS1 fueron de aproximadamente un 20% a un 40% de los niveles de control). De promedio, los regímenes de tratamiento AD-60519 lograron un silenciamiento de entre aproximadamente un 65% y un 75%. Como se describe en la presente, AD-60519 es un derivado de AD-60489. Se describen resultados similares para AD-60489 en el Ejemplo 20 y se muestran en la FIG. 30. Además, AD-61193 (un derivado de AD-60489) y AD-60819 (un derivado de AD-58632) también lograron un silenciamiento de más de un 50%. Cabe destacar que los niveles de silenciamiento mencionados en este ejemplo y en el Ejemplo 20 (p. ej., de aproximadamente un 20% a un 80%) se lograron incluso en un estado “no inducido”; se anticipa que en un estado inducido, p. ej., cuando los niveles de ALAS1 estén elevados de manera crónica o aguda (p. ej., en un paciente que tiene o está en riesgo de tener porfiria, p. ej., una porfiria hepática aguda, p. ej., AIP), niveles más bajos de silenciamiento, p. ej., reducción de los niveles de ARNm de ALAS1 hasta niveles normales o anteriores al ataque, puedan ser suficiente para lograr una eficacia terapéutica.
Supresión de los niveles de ARNm de ALAS1 extracelular circulante
La FIG. 50 muestra los niveles de ARNm de ALAS1 extracelular circulante (medias y desviaciones estándar) en cada punto temporal a lo largo del estudio cuando se obtuvieron las muestras de suero. Los resultados de ARNm de ALAS1 extracelular circulante demuestran eficacia del silenciamiento de ARNm tras el tratamiento multidosis con cada uno de los ARNip estudiados (AD60519, AD-61193, AD-60819 y AD-58632). En todos los grupos, el efecto de supresión mayor en el ARNm de ALAS1 circulante se observó en el día 27, tras la dosis final de ARNip en el día 25. En todos los grupos de tratamiento, durante las semanas después del cese del tratamiento, los niveles de ARNm de ALAS1 se incrementaron gradualmente y volvieron al punto de referencia en la medida final del día 60.
La supresión más pronunciada de ARNm de ALAS1 circulante (silenciamiento máximo de casi un 80%) se observó en el Grupo 3 (2.5 mg/kg de AD-60519 cada día x 5, dos veces a la semana x 3) y el Grupo 5 (5 mg/kg de AD-60519, cada día x 5, cada semana x 3). El Grupo 2 (1.25 mg/kg de AD-60519, cada día x 5, dos veces a la semana x 3), Grupo 4 (2.5 mg/kg de AD-60519, cada día x 5, cada semana x 3), Grupo 7 (2.5 mg/kg de AD-60819, cada día x 5, dos veces a la semana x 3) y el Grupo 6 (2.5 mg/kg de AD-61193, cada día x 5, dos veces a la semana x 3) también mostraron una supresión excelente, con un silenciamiento máximo (día 27) superior a un 50%. En el grupo 1, también se logró un silenciamiento notable (más de un 30% en el día 27).
Los resultados concuerdan con los resultados de ARNm de ALAS1 en el hígado y confirman la potente actividad de AD-60519. Con niveles de dosis de tan solo 1.25 mg/kg, AD-60519 proporcionó un 65-75% de silenciamiento.
Correlación entre los niveles de ARNm de ALAS1 circulante y en el hígado
La FIG. 27 muestra los niveles de ARNm de ALAS1 en el hígado (barras de la izquierda) y en suero (barras de la derecha). Existe una buena correlación entre los niveles de ARNm de ALAS1 relativo medidos en el hígado y suero, lo que indica que estas medidas proporcionan resultados coherentes.
Ejemplo 35: Estudios con dosis única en rata de AD-60519 y AD-60589 utilizando un ensayo cERD en orina para monitorizar la duración de la supresión de ARNm de ALAS1
Se llevó a cabo un estudio de dosis única en ratas utilizando los conjugados de ARNip de ALAS1-GalNAc AD-60489 y AD-60519. La eficacia de estos conjugados de GalNAc para inhibir la expresión de ARNm de ALAS1 se monitorizó utilziando evaluaciones de orina con un ensayo de detección de ARN extracelular circulante. El ensayo fue similar al ensayo utilizado en los Ejemplos 21 y 34, excepto en que se utilizaron muestras de orina. Las muestras de orina se liofilizaron para concentrarlas. La orina liofilizada se resuspendió en 4 ml de dH2O y se agitó en vórtex. A continuación, la muestra se centrifugó a 4000xg durante 10-20 minutos para sedimentar cualquier desecho. Los pasos restantes fueron similares a los descritos en el Ejemplo 21.
A los grupos de ratas se les administró una dosis única de 10 mg/kg de AD-60489 o AD-60519. Los niveles normalizados de ARNm de ALAS1 en diversos puntos temporales a lo largo del estudio se muestran en la FIG. 51. El punto temporal indicado como “0 horas” es para la muestra de orina anterior a la dosis de referencia extraída justo antes de la administración del ARNm de ALAS1. Los resultados para los puntos temporales posteriores se expresan como una fracción del nivel anterior a la dosis.
Como se puede apreciar a partir de los resultados mostrados en la FIG. 51, AD-60519 proporcionó una mejora de la potencia en comparación con AD-60489. En su máximo, la dosis única de AD-60519 proporcionó una supresión de hasta aproximadamente un 80%, mientras que la supresión proporcionada por AD-60489 fue de aproximadamente un 60%. El efecto de una única dosis de 10 mg/kg de los ARNip de ALAS1 en la supresión de ARNm de ALAS1 duró aproximadamente 21 días. Estos resultados demuestran la validez del ensayo cERD en orina para monitorizar los niveles de ARNm de ALAS1.
Ejemplo 36: Efectos farmacológicos de AD-60519 en primates no humanos
Se llevó a cabo un estudio adicional de los efectos del conjugado de ARNip de ALASI-GalNAc AD-60519 en primates no humanos. El estudio investigó los efectos de una dosificación semanal frente a bisemanal, el uso de una dosis de carga frente a ausencia de dosis de carga, y la cinética del silenciamiento de ARNm de ALAS1 tras una única dosis. El diseño del estudio se muestra en la Tabla 42 y en la FIG. 52.
Tabla 42: Diseño del estudio farmacológico para el estudio con AD-60519

Cada grupo recibió una o más dosis subcutáneas de AD-60519 como se proporciona en la Tabla 42. El Grupo 1 (n=3) recibió 2.5 mg/kg en un volumen de dosis de 0.125 ml/kg una vez a la semana durante 8 semanas (las dosis se administraron en los días de dosis 1, 8, 15, 22, 29, 36, 43 y 50). El Grupo 2 (n=3) recibió 5 mg/kg en un volumen de dosis de 0.25 mg/ml una vez a la semana durante 8 semanas (las dosis se administraron en los días de dosis 1, 8, 15, 22, 29, 36, 43 y 50). El Grupo 3 (n=3) recibió una dosis de carga de 5 mg/kg en un volumen de dosis de 0.25 ml/kg una vez al día durante tres días seguida por una dosis de manteniemiento de 5 mg/kg en un volumen de dosis de 0.25 ml/kg una vez a la semana durante 7 semanas (las dosis se administraron en los días 1, 2, 3, 8, 15, 22, 29, 36, 43 y 50). El Grupo 4 (n=3) recibió una dosis de carga de 5 mg/kg en un volumen de dosis de 0.25 ml/kg una vez al día durante tres días, seguida por una dosis de mantenimiento de 2.5 mg/kg en un volumen de dosis de 0.125 ml/kg una vez a la semana durante 7 semanas (las dosis se administraron en los días 1, 2, 3, 8, 15, 22, 29, 36, 43 y 50). El Grupo 5 (n=3) recibió 5 mg/kg en un volumen de dosis de 0.25 ml/kg dos veces a la semana durante 8 semanas (las dosis se administraron en los días de dosis 1,4, 8, 11, 15, 18, 22, 25, 29, 32, 36, 39, 43, 46, 50 y 53). El Grupo 6 (n=3) recibió una única dosis de 1 mg/kg en un volumen de dosis de 0.05 ml/kg en el día 1. El Grupo 7 (n=3) recibió una única dosis de 10 mg/kg en un volumen de dosis de 0.5 ml/kg en el día 1.
Las muestras de suero (enumeradas como “extracciones PD” en la FIG. 52), muestras de plasma (enumeradas como “extracciones PK” en la FIG 52) y las muestras de orina se recogieron como se indica en la FIG. 52 y la Tabla
43. Las muestras de orina y suerose sometieron al ensayo cERD. Todas las muestras de sangre y orina recogidas en el día de una biopsia hepática se recogieron antes de la biopsia hepática.
Tabla 43: Programación de la recogida de muestras

Los resultados de ARNm de ALAS1 se muestran en la FIG. 53. Se logró una supresión de ARNm de ALAS1 significativa en todas las condiciones de estudio. Se logró hasta un 75-80% de silenciamiento de ALAS1 en regímenes de múltiples dosis utilizando AD-60519. Tres días después de una única dosis, se logró un silenciamiento de aproximadamente un 15% con una única dosis de 1 mg/kg, y se logró un silenciamiento de aproximadamente un 70% con una única dosis de 10 mg/kg (remítase a la FIG. 53). La comparación de los datos de los grupos 1 y 7 revela una ligera diferencia en la cinética después de una única dosis (en el grupo 7) frente a múltiples dosis (grupo 1) de la misma cantidad acumulada (30 mg administrados), según se evalúa 3 días después de la dosis en el grupo 7 y dos días después de la cuarta dosis en el grupo 1. En particular, se observó un silenciamiento mayor después de una única dosis (silenciamiento de aproximadamente un 70% en promedio en el grupo 7 frente al silenciamiento de aproximadamente un 45% en promedio en el grupo 1). Remítase a la FIG. 54. Este tipo de resultado también se observó en los estudios en ratas.
Los resultados de ARNm de ALAS1 en suero a lo largo del día 22 se muestran en la FIG. 54 (parte superior). La correlación entre ARNm de ALAS1 en hígado, ARNm de ALAS1 en suero y ARNm de ALAS1 en plasma se muestra en la FIG. 55. Estos resultados demuestran una buena correlación entre los niveles de ARNm de ALAS1 en hígado, suero y orina. Los resultados también proporcionan evidencia adicional que demuestra una potente actividad de AD-60519. Se observó un silenciamiento de un 55-75% en todos los niveles de dosis en los regímenes posológicos de múltiples dosis. La administración de dosis de carga (una vez al día durante 3 días, como en los grupos 3 y 4) dio como resultado una disminución regulada ligeramente más rápida del ARNm de ALAS1. Los grupos que recibieron dosis semanales (grupos 1 y 2) o dosis bisemanales (grupo 5) mostraron en última instancia niveles comparables de supresión de ARNm de ALAS1, lo que indica que la acumulación a lo largo del tiempo proporciona una atenuación sostenida.
Los resultados que muestran la cinética del silenciamiento de ARNm de ALAS1 después de una única dosis se muestran en la FIG. 54 (parte inferior). En el grupo de 1 mg/kg, se observó una supresión de ARNm de ALAS1 de aproximadamente un 20% el día 6. En el grupo de 10 mg/kg, hubo una reducción rápida, de aproximadamente un 70% de ARNm de ALAS1 el día 4, con una recuperación de un 20% o menos del punto de referencia el día 22 (21 días después de la dosis). Los niveles de ARNm de ALAS1 en suero volvieron al punto de referencia aproximadamente 2 semanas o 4 semanas después de una única dosis de 1 mg/kg o 10 mg/kg, respectivamente.
El ciclo completo de ARNm de ALAS en suero de hasta 8 semanas después de la administración de AD-60519 se muestra en la FIG. 56. Todos los grupos alcanzaron un máximo de un 80% de la supresión de ARNm de ALAS1 en las semanas 5 a 8 después de la dosificación con ALN-AS1. Los grupos con tres dosis diarias en la semana 1 (cada día x 3) tuvieron un incio más rápido de supresión de ARNm de ALAS1 que los que simplemente recibieron una dosis en la primera semana (cada semana x 8). Todos los animales volvieron a los niveles de ALAS1 del punto de referencia, aproximadamente 30-40 días después de la última dosis.
Ejemplo 37: Producción de un producto farmacológico de ARNip
ALN-60519 (FIG. 57) es un oligonucleótido bicatenario sintetizado por medios químicos ligado covalentemente a un ligando que contiene tres residuos de N-acetilgalactosamina (GalNAc). Todos los nucleótidos tienen modificaciones 2’-OMe o 2’-F y están conectados a través de uniones fosfodiéster 3’-5’, para formar de esta manera el esqueleto de azúcar-fosfato del oligonucleótido. La hebra sentido y la hebra antisentido contienen 21 y 23 nucleótidos, respectivamente. El extremo 3’ de la hebra sentido está conjugado con el resto de GalNAc de tres antenas (denominado L96) mediante una unión fosfodiéster. La hebra antisentido contiene cuatro uniones fosforotioato, dos en el extremo 3’ y dos en el extremo 5’. La hebra sentido contiene dos uniones fosforotioato en el extremo 5’. Los 21 nucleótidos de la hebra sentido se hibridan con los 21 nucleótidos complementarios de la hebra antisentido y de esta manera forman pares de bases de 21 nucleótidos y una protuberancia de dos bases en el extremo 3’ de la hebra antisentido. Las dos hebras sencillas, la hebra sentido y la hebra antisentido, se sintetizaron mediante síntesis de oligonucleótidos en fase sólida convencional, empleando química de fosforamiditos estándar con el grupo 5’-hidroxilo protegido como éter dimetoxitrifenilmetílico (DMT). Cada hebra se ensambló desde el extremo 3’ al 5’ mediante la adición secuencial de nucleósidos fosforamiditos protegidos.
AD-60519, también denominado en la presente ALN-60519, se formuló como una solución para la inyección para el uso subcutáneo, denominada en la presente ALN-AS1. ALN-60519 se disolvió en agua para inyección (WFI, por sus siglas en inglés) y se ajustó el pH (objetivo 7.0). Se determinó la concentración de ALN-60519 y se ajustó añadiendo WFI. La solución con una concentración final de aproximadamente 200 mg/ml se esterilizó por filtración a continuación y se rellenaron viales de vidrio de Tipo I con 2 ml. Se escogió un volumen de relleno de aproximadamente 0.55 ml para permitir la extracción completa de 0.5 ml de producto farmacológico.
Ejemplo 38: Medida de los niveles de ARNm de ALAS1 en suero u orina en pacientes con AIP o voluntarios sanos utilizando el método cERD
Los estudios farmacológicos en primates no humanos con ALN-AS1 indicaron que el método de detección de ARN extracelular circulante (cERD) para medir el ARNm de ALAS1 en suero u orina era robusto y reproducible. El ensayo cERD también se utilizó para medir los niveles de ARNm de ALAS1 en suero y orina de pacientes con AIP y voluntarios sanos. Los niveles de ARNm de ALAS1 se incrementaron en general en pacientes con AIP respecto a los voluntarios sanos, lo que concuerda con la función que la inducción de ALAS1 desempeña en la patofisiología de la enfermedad (FIG. 58). De manera importante, los niveles de ALAS1 en suero y orina de un mismo paciente se correlacionaron entre sí. En dos pacientes en los que se recogieron repetidamente orina y suero el nivel del ARNm de ALAS1 fue coherente a lo largo del tiempo. En global, estos datos indican que el ARNm de ALAS1 se puede medir en muestras de suero y orina de sujetos humanos incluidos pacientes con AIP, y el método cERD es útil para rastrear la actividad farmacodinámica de ALN-AS1.
Ejemplo 39: Estudios clínicos ejemplares
Se puede llevar a cabo un estudio en seres humanos para determinar la seguridad y tolerabilidad de ALN-AS1 cuando se administra como una única dosis y múltiples dosis a pacientes con AIP que son excretores elevados asintomáticos (ASHE, por sus siglas en inglés) (pacientes que tienen niveles elevados de ALA y/o PBG, como se describe en la presente) o pacientes con AIP que padecen ataques recurrentes.
Los objetivos secundarios incluyen la caracterización de PK en plasma y orina para ALN-AS1 así como también la evaluación después de la dosis del impacto de ALN-AS1 en los niveles de ALA y PBG tanto en plasma como urinarios. Se utiliza el ensayo cERD que mide el ARNm en los exosomas para medir 5-aminolevulinato-sintasa (ARNm de ALAS-1) en suero (o en plasma) y urinario.
En los excretores elevados asintomáticos, se adminsitra ALN-AS1 en dosis únicas, p. ej., de 0.1, 0.35, 1.0 o 2.5 mg/kg, o en dosis semanales repetidas, p. ej., de 1 y 2.5 mg/kg, durante varias semanas (p. ej., durante 4 semanas). Como una comparación, se administra un tratamiento de control (p. ej., placebo). Se evalúan la seguridad, farmacocinética y efectos del fármaco en los niveles de ALA y PBG. Una dosis de ALN-AS1 que reduce ALA y PBG hasta dentro del intervalo de referencia normal (p. ej., una dosis que normaliza los niveles de ALA y/o PBG por debajo de 2x el valor de referencia superior) se puede seleccionar para estudios posteriores, p. ej., en pacientes con AIP.
En los pacientes con AIP, la tasa de ataques y los síntomas de referencia se evalúan durante un periodo de preinclusión anterior a la dosificación (p. ej., de 12 semanas). A los pacientes se les administra ALN-AS1, p. ej., con una dosis de 1-2.5 mg/kg a la semana. Se evalúan la seguridad, farmacocinética y efectos del fármaco en los niveles
de ALA y PBG. Además, se monitorizan los cambios en el número de ataques, uso de hemo, uso de medicamentos para el dolor y hospitalización.
Claims (16)
1. Un ácido ribonucleico bicatenario (ARNbc) para inhibir la expresión de ALAS1, donde dicho ARNbc comprende una hebra sentido y una hebra antisentido, comprendiendo la hebra antisentido una región de complementariedad con un transcrito de ARN de ALAS1 (p. ej., SEQ ID NO: 1), cuya hebra antisentido comprende usAfsAfGfaUfgAfgAfcAfcUfcUfuUfcUfgsgsu (SEQ ID NO: 4161), donde c, a, g, u = ribonucleósidos 2’-OMe; Af, Cf, Gf, Uf = ribonucleósidos 2’F; s = fosforotioato, o una secuencia que difiere en no más de 1,2 o 3 nucleótidos de esta.
2. El ARNbc de la reivinidicación 1, que comprende además una hebra sentido que comprende csasgaaaGfaGfuGfuCfuCfaucuuaL96 (SEQ ID NO: 4160), donde

3. El ARNbc de la reivindicación 1 o 2, donde dicho ARNbc tiene uno o más de los siguientes:
(a) comprende una región dúplex que tiene una longitud de 17-23 pares de nucleótidos;
(b) al menos una hebra comprende una protuberancia 3’ de al menos 2 nucleótidos; o
(c) cada hebra no tiene más de 26 nucleótidos de longitud.
4. El ARNbc de la reivindicación 3, donde la región dúplex tiene una longitud de 21-23 pares de nucleótidos o 21 pares de nucleótidos.
5. El ARNbc de cualquiera de las reivindicaciones anteriores, que comprende además un ligando.
6. El ARNbc de la reivindicación 5, donde el ligando tiene uno o más de los siguientes:
(a) se conjuga con el extremo 3’ de la hebra sentido del ARNbc;
(b) comprende un carbohidrato, opcionalmente donde el ligando es un ligando GalNAc, donde opcionalmente el ligando es:

(c) se une mediante un conector ramificado bivalente o trivalente, opcionalmente donde el ligando y conector son como se muestra en la Fórmula XXIV:

o
(d) dirige el ARNbc a los hepatocitos.
7. El ARNbc de cualquiera de las reivindicaciones precedentes, donde la hebra antisentido comprende o está constituida por usAfsAfGfaUfgAfgAfcAfcUfcUfuUfcUfgsgsu (SEQ ID NO: 4161), y/o la hebra sentido comprende o está constituida por csasgaaaGfaGfuGfuCfuCfaucuuaL96 (SEQ ID NO: 4160).
8. El ARNbc de cualquiera de las reivindicaciones anteriores, donde el ARNbc tiene uno, dos o todos los siguientes:
(i) se sintetiza por medios químicos;
(ii) tiene una protuberancia 3’; o
(iii) comprende o está constituido por una hebra sentido que tiene csasgaaaGfaGfuGfuCfuCfaucuuaL96 (SEQ ID NO: 4160) y una hebra antisentido que tiene usAfsAfGfaUfgAfgAfcAfcUfcUfuUfcUfgsgsu (SEQ ID NO: 4161).
9. El ARNbc de la reivindicación 1, donde dicho ARNbc está en forma de un conjugado que tiene la estructura de:

o una sal farmacéuticamente aceptable de este,
donde Af, Cf, Gf, Uf = ribonucleósidos 2’F; Am, Cm, Gm, Um = ribonucleósidos 2’-OMe;

= fosforotioato;
O
p ÍK
O*
= fosfodiéster, y donde
L96 =

10. Una célula que comprende el ARNbc de cualquiera de las reivindicaciones 1 a 9.
11. Una composición farmacéutica que comprende el ARNbc de cualquiera de las reivindicaciones 1 a 9, opcionalmente donde la composición comprende agua para inyección, opcionalmente donde la composición es adecuada para la administración subcutánea.
12. Un ARNbc de cualquiera de las reivindicaciones 1-9, o una composición farmacéutica de la reivindicación 11, para su uso en un método para inhibir la expresión de ALAS1 en una célula, comprendiendo el método:
(a) introducir en la célula el ARNbc de cualquiera de las reivindicaciones 1 a 9, o la composición farmacéutica de la reivindicación 11, y
(b) mantener la célula del paso (a) durante un tiempo suficiente para obtener degradación del transcrito de ARNm de un gen de ALAS1, para inhibir de esta manera la expresión del gen de ALAS1 en la célula, opcionalmente donde la expresión de ALAS1 se inhibe en la célula en al menos un 20%, o al menos un 30%, opcionalmente donde la expresión de ALAS1 se inhibe en la célula en al menos un 80% según se mide mediante un ensayo de ADN ramificado (ADNr).
13. Un ADNbc de cualquiera de las reividicaciones 1-9, o una composición farmacéutica de la reivindicaión 11, para su uso en un método para reducir el nivel de una porfirina o un precursor de porfirina (p. ej., ALA o PBG) en una célula (p. ej., un hepatocito), que comprende poner en contacto la célula con el ARNbc de cualquiera de las reivindicaciones 1 a 9, o con la composición farmacéutica de la reivindicación 11, en una cantidad eficaz para reducir el nivel de porfirina o del precursor de porfirina en la célula.
14. Un ARNbc de cualquiera de las reivindicaciones 1-9, o una composición farmacéutica de la reivindicación 11, para su uso en el tratamiento de una porfiria en un sujeto, opcionalmente donde la porfiria es una porfiria hepática o una porfiria intermitente aguda (AIP), opcionalmente donde la porfiria es porfiria hepática aguda.
15. Un ARNbc de la reivindicación 7 para su uso en el tratamiento de una porfiria hepática aguda.
16. El ARNbc, o una composición farmacéutica, para su uso de la reivindicación 14 o 15, donde el uso tiene uno o más de los siguientes:
(a) el sujeto está en riesgo de desarrollar, o se le ha diagnosticado, una porfiria;
(b) la porfiria es porfiria intermitente aguda o porfiria por deficiencia de ALA-deshidratasa;
(c) (i) el ARNbc o la composición se administra después de un ataque agudo de porfiria, (ii) el ARNbc o la composición se administra durante un ataque agudo de porfiria, o (iii) el ARNbc o la composición se administra de manera profiláctica para prevenir un ataque agudo de porfiria;
(d) el ARNbc se administra en una dosis de 0.05 a 50 mg/kg de peso corporal del sujeto, p. ej., en una dosis de 0.01 mg/kg a 5 mg/kg de peso corporal del sujeto;
(e) el método (i) reduce el nivel de una porfirina o un precursor de porfirina (p. ej., ácido 5-aminolevulínico (ALA) o porfopilinógeno (PBG)) en el sujeto, opcionalmente donde el nivel se reduce en al menos un 30%, y/o (ii) inhibe la expresión de ALAS1 en el sujeto;
(f) dicho método (i) mejora un síntoma asociado con un trastorno relacionado con ALAS1 (p. ej., una porfiria), (ii) reduce la frecuencia de ataques agudos de síntomas asociados con una porfiria en el sujeto, y/o (iii) reduce la incidencia de ataques agudos de síntomas asociados con una porfiria en el sujeto donde el sujeto está expuesto a un factor precipitante;
(g) el ARNbc o la composición que comprende el ARNbc se administra de acuerdo con un régimen posológico, p. ej., semanal, bisemanal o mensualmente;
(h) el ARNbc se administra antes de un ataque agudo de porfiria, p. ej., durante un pródromo;
(i) el sujeto tiene un nivel elevado (p. ej., nivel en plasma u orina) de ALA y/o PBG, y opcionalmente donde el sujeto padece de dolor crónico;
(j) el método reduce el nivel elevado de ALA y/o PBG;
(k) el método reduce o previene el dolor, neuropatía y/o daño nervioso;
(l) el método previene ataques agudos de porfiria; o
(m) el ARNbc o la composición que comprende el ARNbc se administra repetidamente.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361887288P | 2013-10-04 | 2013-10-04 | |
| US201461983720P | 2014-04-24 | 2014-04-24 | |
| PCT/US2014/059160 WO2015051318A1 (en) | 2013-10-04 | 2014-10-03 | Compositions and methods for inhibiting expression of the alas1 gene |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2804510T3 true ES2804510T3 (es) | 2021-02-08 |
Family
ID=51932569
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14800179T Active ES2804510T3 (es) | 2013-10-04 | 2014-10-03 | Composiciones y métodos para inhibir la expresión del gen ALAS1 |
Country Status (36)
| Country | Link |
|---|---|
| US (3) | US10119143B2 (es) |
| EP (2) | EP3693463A1 (es) |
| JP (3) | JP6613227B2 (es) |
| KR (3) | KR102307389B1 (es) |
| CN (1) | CN105980559B (es) |
| AU (3) | AU2014331604B2 (es) |
| BR (2) | BR122020001264B1 (es) |
| CA (2) | CA3227061A1 (es) |
| CL (2) | CL2016000772A1 (es) |
| CR (1) | CR20160195A (es) |
| CY (2) | CY1122975T1 (es) |
| DK (1) | DK3052628T3 (es) |
| DO (2) | DOP2016000073A (es) |
| EA (1) | EA036477B1 (es) |
| ES (1) | ES2804510T3 (es) |
| GT (1) | GT201600066A (es) |
| HK (1) | HK1221738A1 (es) |
| HR (1) | HRP20200822T1 (es) |
| HU (2) | HUE049227T2 (es) |
| IL (3) | IL282747B2 (es) |
| LT (2) | LT3052628T (es) |
| MX (2) | MX2016004319A (es) |
| MY (2) | MY183490A (es) |
| NL (1) | NL301061I2 (es) |
| NZ (2) | NZ757749A (es) |
| PE (2) | PE20211249A1 (es) |
| PH (1) | PH12016500574A1 (es) |
| PL (1) | PL3052628T3 (es) |
| PT (1) | PT3052628T (es) |
| SG (2) | SG11201602631XA (es) |
| SI (1) | SI3052628T1 (es) |
| TN (1) | TN2016000114A1 (es) |
| TW (3) | TWI768330B (es) |
| UA (1) | UA124961C2 (es) |
| WO (1) | WO2015051318A1 (es) |
| ZA (2) | ZA201602931B (es) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9133461B2 (en) | 2012-04-10 | 2015-09-15 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for inhibiting expression of the ALAS1 gene |
| TWI768330B (zh) | 2013-10-04 | 2022-06-21 | 美國西奈山伊坎醫學院 | 抑制alas1基因表現的組合物及方法 |
| MX2017002144A (es) | 2014-08-20 | 2017-08-15 | Alnylam Pharmaceuticals Inc | Agentes de ácido ribonucleico (arn) de cadena doble modificados. |
| WO2016061487A1 (en) | 2014-10-17 | 2016-04-21 | Alnylam Pharmaceuticals, Inc. | Polynucleotide agents targeting aminolevulinic acid synthase-1 (alas1) and uses thereof |
| TW201718857A (zh) | 2015-09-14 | 2017-06-01 | 艾爾妮蘭製藥公司 | 用於抑制alas1基因表現之組合物及方法 |
| AU2017363892B2 (en) | 2016-11-23 | 2023-06-15 | Alnylam Pharmaceuticals, Inc. | Modified RNA agents with reduced off-target effect |
| WO2019136432A1 (en) | 2018-01-08 | 2019-07-11 | Novartis Ag | Immune-enhancing rnas for combination with chimeric antigen receptor therapy |
| BR112022016370A2 (pt) * | 2020-02-21 | 2022-10-04 | Ionis Pharmaceuticals Inc | Métodos para reduzir a expressão de htt |
| WO2023060064A1 (en) | 2021-10-06 | 2023-04-13 | Alnylam Pharmaceuticals, Inc. | Renal injury biomarkers as biomarkers for acute hepatic porphyria (ahp) |
| WO2024148236A1 (en) * | 2023-01-06 | 2024-07-11 | Memorial Sloan-Kettering Cancer Center | Methods for enhancing the efficacy of rnai therapy by targeting alas1/alas2 |
| CN118460654A (zh) * | 2024-07-10 | 2024-08-09 | 凯莱英医药集团(天津)股份有限公司 | 一种Givosiran的制备方法 |
Family Cites Families (245)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US564562A (en) | 1896-07-21 | Joseph p | ||
| US513030A (en) | 1894-01-16 | Machine for waxing or coating paper | ||
| US3687808A (en) | 1969-08-14 | 1972-08-29 | Univ Leland Stanford Junior | Synthetic polynucleotides |
| US4469863A (en) | 1980-11-12 | 1984-09-04 | Ts O Paul O P | Nonionic nucleic acid alkyl and aryl phosphonates and processes for manufacture and use thereof |
| US4426330A (en) | 1981-07-20 | 1984-01-17 | Lipid Specialties, Inc. | Synthetic phospholipid compounds |
| US4534899A (en) | 1981-07-20 | 1985-08-13 | Lipid Specialties, Inc. | Synthetic phospholipid compounds |
| US5023243A (en) | 1981-10-23 | 1991-06-11 | Molecular Biosystems, Inc. | Oligonucleotide therapeutic agent and method of making same |
| US4476301A (en) | 1982-04-29 | 1984-10-09 | Centre National De La Recherche Scientifique | Oligonucleotides, a process for preparing the same and their application as mediators of the action of interferon |
| JPS5927900A (ja) | 1982-08-09 | 1984-02-14 | Wakunaga Seiyaku Kk | 固定化オリゴヌクレオチド |
| FR2540122B1 (fr) | 1983-01-27 | 1985-11-29 | Centre Nat Rech Scient | Nouveaux composes comportant une sequence d'oligonucleotide liee a un agent d'intercalation, leur procede de synthese et leur application |
| US4605735A (en) | 1983-02-14 | 1986-08-12 | Wakunaga Seiyaku Kabushiki Kaisha | Oligonucleotide derivatives |
| US4948882A (en) | 1983-02-22 | 1990-08-14 | Syngene, Inc. | Single-stranded labelled oligonucleotides, reactive monomers and methods of synthesis |
| US4824941A (en) | 1983-03-10 | 1989-04-25 | Julian Gordon | Specific antibody to the native form of 2'5'-oligonucleotides, the method of preparation and the use as reagents in immunoassays or for binding 2'5'-oligonucleotides in biological systems |
| US4587044A (en) | 1983-09-01 | 1986-05-06 | The Johns Hopkins University | Linkage of proteins to nucleic acids |
| US5118802A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | DNA-reporter conjugates linked via the 2' or 5'-primary amino group of the 5'-terminal nucleoside |
| US5118800A (en) | 1983-12-20 | 1992-06-02 | California Institute Of Technology | Oligonucleotides possessing a primary amino group in the terminal nucleotide |
| US5550111A (en) | 1984-07-11 | 1996-08-27 | Temple University-Of The Commonwealth System Of Higher Education | Dual action 2',5'-oligoadenylate antiviral derivatives and uses thereof |
| FR2567892B1 (fr) | 1984-07-19 | 1989-02-17 | Centre Nat Rech Scient | Nouveaux oligonucleotides, leur procede de preparation et leurs applications comme mediateurs dans le developpement des effets des interferons |
| US5367066A (en) | 1984-10-16 | 1994-11-22 | Chiron Corporation | Oligonucleotides with selectably cleavable and/or abasic sites |
| US5258506A (en) | 1984-10-16 | 1993-11-02 | Chiron Corporation | Photolabile reagents for incorporation into oligonucleotide chains |
| US5430136A (en) | 1984-10-16 | 1995-07-04 | Chiron Corporation | Oligonucleotides having selectably cleavable and/or abasic sites |
| US4828979A (en) | 1984-11-08 | 1989-05-09 | Life Technologies, Inc. | Nucleotide analogs for nucleic acid labeling and detection |
| FR2575751B1 (fr) | 1985-01-08 | 1987-04-03 | Pasteur Institut | Nouveaux nucleosides de derives de l'adenosine, leur preparation et leurs applications biologiques |
| US5166315A (en) | 1989-12-20 | 1992-11-24 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5405938A (en) | 1989-12-20 | 1995-04-11 | Anti-Gene Development Group | Sequence-specific binding polymers for duplex nucleic acids |
| US5034506A (en) | 1985-03-15 | 1991-07-23 | Anti-Gene Development Group | Uncharged morpholino-based polymers having achiral intersubunit linkages |
| US5235033A (en) | 1985-03-15 | 1993-08-10 | Anti-Gene Development Group | Alpha-morpholino ribonucleoside derivatives and polymers thereof |
| US5185444A (en) | 1985-03-15 | 1993-02-09 | Anti-Gene Deveopment Group | Uncharged morpolino-based polymers having phosphorous containing chiral intersubunit linkages |
| US4762779A (en) | 1985-06-13 | 1988-08-09 | Amgen Inc. | Compositions and methods for functionalizing nucleic acids |
| US5139941A (en) | 1985-10-31 | 1992-08-18 | University Of Florida Research Foundation, Inc. | AAV transduction vectors |
| US5317098A (en) | 1986-03-17 | 1994-05-31 | Hiroaki Shizuya | Non-radioisotope tagging of fragments |
| JPS638396A (ja) | 1986-06-30 | 1988-01-14 | Wakunaga Pharmaceut Co Ltd | ポリ標識化オリゴヌクレオチド誘導体 |
| US4920016A (en) | 1986-12-24 | 1990-04-24 | Linear Technology, Inc. | Liposomes with enhanced circulation time |
| US4837028A (en) | 1986-12-24 | 1989-06-06 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5276019A (en) | 1987-03-25 | 1994-01-04 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US5264423A (en) | 1987-03-25 | 1993-11-23 | The United States Of America As Represented By The Department Of Health And Human Services | Inhibitors for replication of retroviruses and for the expression of oncogene products |
| US4904582A (en) | 1987-06-11 | 1990-02-27 | Synthetic Genetics | Novel amphiphilic nucleic acid conjugates |
| JP2828642B2 (ja) | 1987-06-24 | 1998-11-25 | ハワード フローレイ インスティテュト オブ イクスペリメンタル フィジオロジー アンド メディシン | ヌクレオシド誘導体 |
| US5585481A (en) | 1987-09-21 | 1996-12-17 | Gen-Probe Incorporated | Linking reagents for nucleotide probes |
| US4924624A (en) | 1987-10-22 | 1990-05-15 | Temple University-Of The Commonwealth System Of Higher Education | 2,',5'-phosphorothioate oligoadenylates and plant antiviral uses thereof |
| US5188897A (en) | 1987-10-22 | 1993-02-23 | Temple University Of The Commonwealth System Of Higher Education | Encapsulated 2',5'-phosphorothioate oligoadenylates |
| US5525465A (en) | 1987-10-28 | 1996-06-11 | Howard Florey Institute Of Experimental Physiology And Medicine | Oligonucleotide-polyamide conjugates and methods of production and applications of the same |
| DE3738460A1 (de) | 1987-11-12 | 1989-05-24 | Max Planck Gesellschaft | Modifizierte oligonukleotide |
| US5082830A (en) | 1988-02-26 | 1992-01-21 | Enzo Biochem, Inc. | End labeled nucleotide probe |
| WO1989009221A1 (en) | 1988-03-25 | 1989-10-05 | University Of Virginia Alumni Patents Foundation | Oligonucleotide n-alkylphosphoramidates |
| US5278302A (en) | 1988-05-26 | 1994-01-11 | University Patents, Inc. | Polynucleotide phosphorodithioates |
| US5109124A (en) | 1988-06-01 | 1992-04-28 | Biogen, Inc. | Nucleic acid probe linked to a label having a terminal cysteine |
| US5216141A (en) | 1988-06-06 | 1993-06-01 | Benner Steven A | Oligonucleotide analogs containing sulfur linkages |
| US5175273A (en) | 1988-07-01 | 1992-12-29 | Genentech, Inc. | Nucleic acid intercalating agents |
| US5262536A (en) | 1988-09-15 | 1993-11-16 | E. I. Du Pont De Nemours And Company | Reagents for the preparation of 5'-tagged oligonucleotides |
| GB8824593D0 (en) | 1988-10-20 | 1988-11-23 | Royal Free Hosp School Med | Liposomes |
| US5512439A (en) | 1988-11-21 | 1996-04-30 | Dynal As | Oligonucleotide-linked magnetic particles and uses thereof |
| US5599923A (en) | 1989-03-06 | 1997-02-04 | Board Of Regents, University Of Tx | Texaphyrin metal complexes having improved functionalization |
| US5457183A (en) | 1989-03-06 | 1995-10-10 | Board Of Regents, The University Of Texas System | Hydroxylated texaphyrins |
| US5391723A (en) | 1989-05-31 | 1995-02-21 | Neorx Corporation | Oligonucleotide conjugates |
| US4958013A (en) | 1989-06-06 | 1990-09-18 | Northwestern University | Cholesteryl modified oligonucleotides |
| US5032401A (en) | 1989-06-15 | 1991-07-16 | Alpha Beta Technology | Glucan drug delivery system and adjuvant |
| US5451463A (en) | 1989-08-28 | 1995-09-19 | Clontech Laboratories, Inc. | Non-nucleoside 1,3-diol reagents for labeling synthetic oligonucleotides |
| US5134066A (en) | 1989-08-29 | 1992-07-28 | Monsanto Company | Improved probes using nucleosides containing 3-dezauracil analogs |
| US5436146A (en) | 1989-09-07 | 1995-07-25 | The Trustees Of Princeton University | Helper-free stocks of recombinant adeno-associated virus vectors |
| US5254469A (en) | 1989-09-12 | 1993-10-19 | Eastman Kodak Company | Oligonucleotide-enzyme conjugate that can be used as a probe in hybridization assays and polymerase chain reaction procedures |
| US5591722A (en) | 1989-09-15 | 1997-01-07 | Southern Research Institute | 2'-deoxy-4'-thioribonucleosides and their antiviral activity |
| US5356633A (en) | 1989-10-20 | 1994-10-18 | Liposome Technology, Inc. | Method of treatment of inflamed tissues |
| US5225212A (en) | 1989-10-20 | 1993-07-06 | Liposome Technology, Inc. | Microreservoir liposome composition and method |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5399676A (en) | 1989-10-23 | 1995-03-21 | Gilead Sciences | Oligonucleotides with inverted polarity |
| US5264564A (en) | 1989-10-24 | 1993-11-23 | Gilead Sciences | Oligonucleotide analogs with novel linkages |
| ATE269870T1 (de) | 1989-10-24 | 2004-07-15 | Isis Pharmaceuticals Inc | 2'-modifizierte oligonukleotide |
| US5292873A (en) | 1989-11-29 | 1994-03-08 | The Research Foundation Of State University Of New York | Nucleic acids labeled with naphthoquinone probe |
| US5177198A (en) | 1989-11-30 | 1993-01-05 | University Of N.C. At Chapel Hill | Process for preparing oligoribonucleoside and oligodeoxyribonucleoside boranophosphates |
| CA2029273A1 (en) | 1989-12-04 | 1991-06-05 | Christine L. Brakel | Modified nucleotide compounds |
| US5486603A (en) | 1990-01-08 | 1996-01-23 | Gilead Sciences, Inc. | Oligonucleotide having enhanced binding affinity |
| US5578718A (en) | 1990-01-11 | 1996-11-26 | Isis Pharmaceuticals, Inc. | Thiol-derivatized nucleosides |
| US5852188A (en) | 1990-01-11 | 1998-12-22 | Isis Pharmaceuticals, Inc. | Oligonucleotides having chiral phosphorus linkages |
| US6783931B1 (en) | 1990-01-11 | 2004-08-31 | Isis Pharmaceuticals, Inc. | Amine-derivatized nucleosides and oligonucleosides |
| US5587361A (en) | 1991-10-15 | 1996-12-24 | Isis Pharmaceuticals, Inc. | Oligonucleotides having phosphorothioate linkages of high chiral purity |
| US5646265A (en) | 1990-01-11 | 1997-07-08 | Isis Pharmceuticals, Inc. | Process for the preparation of 2'-O-alkyl purine phosphoramidites |
| US5587470A (en) | 1990-01-11 | 1996-12-24 | Isis Pharmaceuticals, Inc. | 3-deazapurines |
| US7037646B1 (en) | 1990-01-11 | 2006-05-02 | Isis Pharmaceuticals, Inc. | Amine-derivatized nucleosides and oligonucleosides |
| US5670633A (en) | 1990-01-11 | 1997-09-23 | Isis Pharmaceuticals, Inc. | Sugar modified oligonucleotides that detect and modulate gene expression |
| US5681941A (en) | 1990-01-11 | 1997-10-28 | Isis Pharmaceuticals, Inc. | Substituted purines and oligonucleotide cross-linking |
| US5459255A (en) | 1990-01-11 | 1995-10-17 | Isis Pharmaceuticals, Inc. | N-2 substituted purines |
| US5214136A (en) | 1990-02-20 | 1993-05-25 | Gilead Sciences, Inc. | Anthraquinone-derivatives oligonucleotides |
| WO1991013080A1 (en) | 1990-02-20 | 1991-09-05 | Gilead Sciences, Inc. | Pseudonucleosides and pseudonucleotides and their polymers |
| US5321131A (en) | 1990-03-08 | 1994-06-14 | Hybridon, Inc. | Site-specific functionalization of oligodeoxynucleotides for non-radioactive labelling |
| US5470967A (en) | 1990-04-10 | 1995-11-28 | The Dupont Merck Pharmaceutical Company | Oligonucleotide analogs with sulfamate linkages |
| US5665710A (en) | 1990-04-30 | 1997-09-09 | Georgetown University | Method of making liposomal oligodeoxynucleotide compositions |
| GB9009980D0 (en) | 1990-05-03 | 1990-06-27 | Amersham Int Plc | Phosphoramidite derivatives,their preparation and the use thereof in the incorporation of reporter groups on synthetic oligonucleotides |
| EP0745689A3 (en) | 1990-05-11 | 1996-12-11 | Microprobe Corporation | A dipstick for a nucleic acid hybridization assay |
| US5981276A (en) | 1990-06-20 | 1999-11-09 | Dana-Farber Cancer Institute | Vectors containing HIV packaging sequences, packaging defective HIV vectors, and uses thereof |
| US5618704A (en) | 1990-07-27 | 1997-04-08 | Isis Pharmacueticals, Inc. | Backbone-modified oligonucleotide analogs and preparation thereof through radical coupling |
| US5688941A (en) | 1990-07-27 | 1997-11-18 | Isis Pharmaceuticals, Inc. | Methods of making conjugated 4' desmethyl nucleoside analog compounds |
| BR9106702A (pt) | 1990-07-27 | 1993-06-08 | Isis Pharmaceuticals Inc | Analogo de oligonucleotideos e processos para modular a producao de uma proteina por um organismo e para tratar um organismo |
| US5677437A (en) | 1990-07-27 | 1997-10-14 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5541307A (en) | 1990-07-27 | 1996-07-30 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogs and solid phase synthesis thereof |
| US5138045A (en) | 1990-07-27 | 1992-08-11 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5218105A (en) | 1990-07-27 | 1993-06-08 | Isis Pharmaceuticals | Polyamine conjugated oligonucleotides |
| US5489677A (en) | 1990-07-27 | 1996-02-06 | Isis Pharmaceuticals, Inc. | Oligonucleoside linkages containing adjacent oxygen and nitrogen atoms |
| US5610289A (en) | 1990-07-27 | 1997-03-11 | Isis Pharmaceuticals, Inc. | Backbone modified oligonucleotide analogues |
| US5623070A (en) | 1990-07-27 | 1997-04-22 | Isis Pharmaceuticals, Inc. | Heteroatomic oligonucleoside linkages |
| US5602240A (en) | 1990-07-27 | 1997-02-11 | Ciba Geigy Ag. | Backbone modified oligonucleotide analogs |
| US5608046A (en) | 1990-07-27 | 1997-03-04 | Isis Pharmaceuticals, Inc. | Conjugated 4'-desmethyl nucleoside analog compounds |
| US5245022A (en) | 1990-08-03 | 1993-09-14 | Sterling Drug, Inc. | Exonuclease resistant terminally substituted oligonucleotides |
| BR9106729A (pt) | 1990-08-03 | 1993-07-20 | Sterling Winthrop Inc | Composto,processos para inibir a degradacao por nuclease de compostos e para estabilizar sequencias de nicleotideos ou oligonucleosideos,composicao utilizavel para inibir expressao de genes e processo para inibir expressao de genes em um mamifero necessitando de tal tratamento |
| US5512667A (en) | 1990-08-28 | 1996-04-30 | Reed; Michael W. | Trifunctional intermediates for preparing 3'-tailed oligonucleotides |
| US5214134A (en) | 1990-09-12 | 1993-05-25 | Sterling Winthrop Inc. | Process of linking nucleosides with a siloxane bridge |
| US5561225A (en) | 1990-09-19 | 1996-10-01 | Southern Research Institute | Polynucleotide analogs containing sulfonate and sulfonamide internucleoside linkages |
| JPH06505704A (ja) | 1990-09-20 | 1994-06-30 | ギリアド サイエンシズ,インコーポレイテッド | 改変ヌクレオシド間結合 |
| US5432272A (en) | 1990-10-09 | 1995-07-11 | Benner; Steven A. | Method for incorporating into a DNA or RNA oligonucleotide using nucleotides bearing heterocyclic bases |
| JP3172178B2 (ja) | 1990-11-08 | 2001-06-04 | ハイブライドン インコーポレイテッド | 合成オリゴヌクレオチドに対する多重リポータ基の組込み |
| GB9100304D0 (en) | 1991-01-08 | 1991-02-20 | Ici Plc | Compound |
| US7015315B1 (en) | 1991-12-24 | 2006-03-21 | Isis Pharmaceuticals, Inc. | Gapped oligonucleotides |
| JP3220180B2 (ja) | 1991-05-23 | 2001-10-22 | 三菱化学株式会社 | 薬剤含有タンパク質結合リポソーム |
| US5719262A (en) | 1993-11-22 | 1998-02-17 | Buchardt, Deceased; Ole | Peptide nucleic acids having amino acid side chains |
| US5714331A (en) | 1991-05-24 | 1998-02-03 | Buchardt, Deceased; Ole | Peptide nucleic acids having enhanced binding affinity, sequence specificity and solubility |
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US5371241A (en) | 1991-07-19 | 1994-12-06 | Pharmacia P-L Biochemicals Inc. | Fluorescein labelled phosphoramidites |
| US5571799A (en) | 1991-08-12 | 1996-11-05 | Basco, Ltd. | (2'-5') oligoadenylate analogues useful as inhibitors of host-v5.-graft response |
| EP0538194B1 (de) | 1991-10-17 | 1997-06-04 | Novartis AG | Bicyclische Nukleoside, Oligonukleotide, Verfahren zu deren Herstellung und Zwischenprodukte |
| US5594121A (en) | 1991-11-07 | 1997-01-14 | Gilead Sciences, Inc. | Enhanced triple-helix and double-helix formation with oligomers containing modified purines |
| US5252479A (en) | 1991-11-08 | 1993-10-12 | Research Corporation Technologies, Inc. | Safe vector for gene therapy |
| US5484908A (en) | 1991-11-26 | 1996-01-16 | Gilead Sciences, Inc. | Oligonucleotides containing 5-propynyl pyrimidines |
| US6235887B1 (en) | 1991-11-26 | 2001-05-22 | Isis Pharmaceuticals, Inc. | Enhanced triple-helix and double-helix formation directed by oligonucleotides containing modified pyrimidines |
| US5359044A (en) | 1991-12-13 | 1994-10-25 | Isis Pharmaceuticals | Cyclobutyl oligonucleotide surrogates |
| ATE204879T1 (de) | 1991-12-24 | 2001-09-15 | Isis Pharmaceuticals Inc | Antisense oligonukleotide |
| US6277603B1 (en) | 1991-12-24 | 2001-08-21 | Isis Pharmaceuticals, Inc. | PNA-DNA-PNA chimeric macromolecules |
| US5595726A (en) | 1992-01-21 | 1997-01-21 | Pharmacyclics, Inc. | Chromophore probe for detection of nucleic acid |
| US5565552A (en) | 1992-01-21 | 1996-10-15 | Pharmacyclics, Inc. | Method of expanded porphyrin-oligonucleotide conjugate synthesis |
| FR2687679B1 (fr) | 1992-02-05 | 1994-10-28 | Centre Nat Rech Scient | Oligothionucleotides. |
| DE4203923A1 (de) | 1992-02-11 | 1993-08-12 | Henkel Kgaa | Verfahren zur herstellung von polycarboxylaten auf polysaccharid-basis |
| US5633360A (en) | 1992-04-14 | 1997-05-27 | Gilead Sciences, Inc. | Oligonucleotide analogs capable of passive cell membrane permeation |
| US5434257A (en) | 1992-06-01 | 1995-07-18 | Gilead Sciences, Inc. | Binding compentent oligomers containing unsaturated 3',5' and 2',5' linkages |
| US5587308A (en) | 1992-06-02 | 1996-12-24 | The United States Of America As Represented By The Department Of Health & Human Services | Modified adeno-associated virus vector capable of expression from a novel promoter |
| EP0577558A2 (de) | 1992-07-01 | 1994-01-05 | Ciba-Geigy Ag | Carbocyclische Nukleoside mit bicyclischen Ringen, Oligonukleotide daraus, Verfahren zu deren Herstellung, deren Verwendung und Zwischenproduckte |
| US5272250A (en) | 1992-07-10 | 1993-12-21 | Spielvogel Bernard F | Boronated phosphoramidate compounds |
| JPH07509133A (ja) | 1992-07-17 | 1995-10-12 | リボザイム・ファーマシューティカルズ・インコーポレイテッド | 動物疾患の処置のための方法および剤 |
| US6346614B1 (en) | 1992-07-23 | 2002-02-12 | Hybridon, Inc. | Hybrid oligonucleotide phosphorothioates |
| EP0673431A1 (en) | 1992-12-03 | 1995-09-27 | Genzyme Corporation | Gene therapy for cystic fibrosis |
| US5478745A (en) | 1992-12-04 | 1995-12-26 | University Of Pittsburgh | Recombinant viral vector system |
| US5574142A (en) | 1992-12-15 | 1996-11-12 | Microprobe Corporation | Peptide linkers for improved oligonucleotide delivery |
| JP3351476B2 (ja) | 1993-01-22 | 2002-11-25 | 三菱化学株式会社 | リン脂質誘導体及びそれを含有するリポソーム |
| US5476925A (en) | 1993-02-01 | 1995-12-19 | Northwestern University | Oligodeoxyribonucleotides including 3'-aminonucleoside-phosphoramidate linkages and terminal 3'-amino groups |
| JP3189279B2 (ja) | 1993-02-19 | 2001-07-16 | 日本新薬株式会社 | 核酸コポリマー含有医薬組成物 |
| US5395619A (en) | 1993-03-03 | 1995-03-07 | Liposome Technology, Inc. | Lipid-polymer conjugates and liposomes |
| GB9304618D0 (en) | 1993-03-06 | 1993-04-21 | Ciba Geigy Ag | Chemical compounds |
| DK0691968T3 (da) | 1993-03-30 | 1998-02-23 | Sanofi Sa | Acykliske nukleosid-analoge og oligonukleotidsekvenser indeholdende disse |
| HU9501974D0 (en) | 1993-03-31 | 1995-09-28 | Sterling Winthrop Inc | Oligonucleotides with amide linkages replacing phosphodiester linkages |
| DE4311944A1 (de) | 1993-04-10 | 1994-10-13 | Degussa | Umhüllte Natriumpercarbonatpartikel, Verfahren zu deren Herstellung und sie enthaltende Wasch-, Reinigungs- und Bleichmittelzusammensetzungen |
| US6191105B1 (en) | 1993-05-10 | 2001-02-20 | Protein Delivery, Inc. | Hydrophilic and lipophilic balanced microemulsion formulations of free-form and/or conjugation-stabilized therapeutic agents such as insulin |
| US5955591A (en) | 1993-05-12 | 1999-09-21 | Imbach; Jean-Louis | Phosphotriester oligonucleotides, amidites and method of preparation |
| US6015886A (en) | 1993-05-24 | 2000-01-18 | Chemgenes Corporation | Oligonucleotide phosphate esters |
| US6294664B1 (en) | 1993-07-29 | 2001-09-25 | Isis Pharmaceuticals, Inc. | Synthesis of oligonucleotides |
| US5502177A (en) | 1993-09-17 | 1996-03-26 | Gilead Sciences, Inc. | Pyrimidine derivatives for labeled binding partners |
| CA2176256A1 (en) | 1993-11-16 | 1995-05-26 | Lyle John Arnold, Jr. | Synthetic oligomers having chirally pure phosphonate internucleosidyl linkages mixed with non-phosphonate internucleosidyl linkages |
| US5540935A (en) | 1993-12-06 | 1996-07-30 | Nof Corporation | Reactive vesicle and functional substance-fixed vesicle |
| US5457187A (en) | 1993-12-08 | 1995-10-10 | Board Of Regents University Of Nebraska | Oligonucleotides containing 5-fluorouracil |
| US5446137B1 (en) | 1993-12-09 | 1998-10-06 | Behringwerke Ag | Oligonucleotides containing 4'-substituted nucleotides |
| US5519134A (en) | 1994-01-11 | 1996-05-21 | Isis Pharmaceuticals, Inc. | Pyrrolidine-containing monomers and oligomers |
| US5596091A (en) | 1994-03-18 | 1997-01-21 | The Regents Of The University Of California | Antisense oligonucleotides comprising 5-aminoalkyl pyrimidine nucleotides |
| US5599922A (en) | 1994-03-18 | 1997-02-04 | Lynx Therapeutics, Inc. | Oligonucleotide N3'-P5' phosphoramidates: hybridization and nuclease resistance properties |
| US5627053A (en) | 1994-03-29 | 1997-05-06 | Ribozyme Pharmaceuticals, Inc. | 2'deoxy-2'-alkylnucleotide containing nucleic acid |
| US5625050A (en) | 1994-03-31 | 1997-04-29 | Amgen Inc. | Modified oligonucleotides and intermediates useful in nucleic acid therapeutics |
| US6054299A (en) | 1994-04-29 | 2000-04-25 | Conrad; Charles A. | Stem-loop cloning vector and method |
| US5525711A (en) | 1994-05-18 | 1996-06-11 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Pteridine nucleotide analogs as fluorescent DNA probes |
| US5543152A (en) | 1994-06-20 | 1996-08-06 | Inex Pharmaceuticals Corporation | Sphingosomes for enhanced drug delivery |
| US5597696A (en) | 1994-07-18 | 1997-01-28 | Becton Dickinson And Company | Covalent cyanine dye oligonucleotide conjugates |
| US5597909A (en) | 1994-08-25 | 1997-01-28 | Chiron Corporation | Polynucleotide reagents containing modified deoxyribose moieties, and associated methods of synthesis and use |
| US5580731A (en) | 1994-08-25 | 1996-12-03 | Chiron Corporation | N-4 modified pyrimidine deoxynucleotides and oligonucleotide probes synthesized therewith |
| US5820873A (en) | 1994-09-30 | 1998-10-13 | The University Of British Columbia | Polyethylene glycol modified ceramide lipids and liposome uses thereof |
| US6608035B1 (en) | 1994-10-25 | 2003-08-19 | Hybridon, Inc. | Method of down-regulating gene expression |
| US5665557A (en) | 1994-11-14 | 1997-09-09 | Systemix, Inc. | Method of purifying a population of cells enriched for hematopoietic stem cells populations of cells obtained thereby and methods of use thereof |
| JP3269301B2 (ja) | 1994-12-28 | 2002-03-25 | 豊田合成株式会社 | ガラスラン用ゴム配合物 |
| AU5359496A (en) | 1995-03-06 | 1996-09-23 | Isis Pharmaceuticals, Inc. | Improved process for the synthesis of 2'-o-substituted pyrimidines and oligomeric compounds therefrom |
| US6166197A (en) | 1995-03-06 | 2000-12-26 | Isis Pharmaceuticals, Inc. | Oligomeric compounds having pyrimidine nucleotide (S) with 2'and 5 substitutions |
| US7422902B1 (en) | 1995-06-07 | 2008-09-09 | The University Of British Columbia | Lipid-nucleic acid particles prepared via a hydrophobic lipid-nucleic acid complex intermediate and use for gene transfer |
| US5981501A (en) | 1995-06-07 | 1999-11-09 | Inex Pharmaceuticals Corp. | Methods for encapsulating plasmids in lipid bilayers |
| DE69634084T2 (de) | 1995-06-07 | 2005-12-08 | Inex Pharmaceuticals Corp. | Herstellung von lipid-nukleinsäure partikeln duch ein hydrophobische lipid-nukleinsäuree komplexe zwischenprodukt und zur verwendung in der gentransfer |
| US5756122A (en) | 1995-06-07 | 1998-05-26 | Georgetown University | Liposomally encapsulated nucleic acids having high entrapment efficiencies, method of manufacturer and use thereof for transfection of targeted cells |
| WO1997004787A1 (en) | 1995-08-01 | 1997-02-13 | Novartis Ag | Liposomal oligonucleotide compositions |
| US5858397A (en) | 1995-10-11 | 1999-01-12 | University Of British Columbia | Liposomal formulations of mitoxantrone |
| AU7435296A (en) | 1995-10-16 | 1997-05-07 | Dana-Farber Cancer Institute | Novel expression vectors and methods of use |
| US6160109A (en) | 1995-10-20 | 2000-12-12 | Isis Pharmaceuticals, Inc. | Preparation of phosphorothioate and boranophosphate oligomers |
| US5858401A (en) | 1996-01-22 | 1999-01-12 | Sidmak Laboratories, Inc. | Pharmaceutical composition for cyclosporines |
| US5994316A (en) | 1996-02-21 | 1999-11-30 | The Immune Response Corporation | Method of preparing polynucleotide-carrier complexes for delivery to cells |
| US6444423B1 (en) | 1996-06-07 | 2002-09-03 | Molecular Dynamics, Inc. | Nucleosides comprising polydentate ligands |
| US6172209B1 (en) | 1997-02-14 | 2001-01-09 | Isis Pharmaceuticals Inc. | Aminooxy-modified oligonucleotides and methods for making same |
| US6639062B2 (en) | 1997-02-14 | 2003-10-28 | Isis Pharmaceuticals, Inc. | Aminooxy-modified nucleosidic compounds and oligomeric compounds prepared therefrom |
| US6576752B1 (en) | 1997-02-14 | 2003-06-10 | Isis Pharmaceuticals, Inc. | Aminooxy functionalized oligomers |
| JP3756313B2 (ja) | 1997-03-07 | 2006-03-15 | 武 今西 | 新規ビシクロヌクレオシド及びオリゴヌクレオチド類縁体 |
| EP1012331B1 (en) | 1997-07-01 | 2006-03-29 | Isis Pharmaceuticals, Inc. | Compositions and methods for the delivery of oligonucleotides via the alimentary canal |
| DE04020014T1 (de) | 1997-09-12 | 2006-01-26 | Exiqon A/S | Bi-zyklische - Nukleosid,Nnukleotid und Oligonukleotid-Analoga |
| US6794499B2 (en) | 1997-09-12 | 2004-09-21 | Exiqon A/S | Oligonucleotide analogues |
| US6617438B1 (en) | 1997-11-05 | 2003-09-09 | Sirna Therapeutics, Inc. | Oligoribonucleotides with enzymatic activity |
| US6528640B1 (en) | 1997-11-05 | 2003-03-04 | Ribozyme Pharmaceuticals, Incorporated | Synthetic ribonucleic acids with RNAse activity |
| US6320017B1 (en) | 1997-12-23 | 2001-11-20 | Inex Pharmaceuticals Corp. | Polyamide oligomers |
| US7273933B1 (en) | 1998-02-26 | 2007-09-25 | Isis Pharmaceuticals, Inc. | Methods for synthesis of oligonucleotides |
| US7045610B2 (en) | 1998-04-03 | 2006-05-16 | Epoch Biosciences, Inc. | Modified oligonucleotides for mismatch discrimination |
| US6531590B1 (en) | 1998-04-24 | 2003-03-11 | Isis Pharmaceuticals, Inc. | Processes for the synthesis of oligonucleotide compounds |
| US6867294B1 (en) | 1998-07-14 | 2005-03-15 | Isis Pharmaceuticals, Inc. | Gapped oligomers having site specific chiral phosphorothioate internucleoside linkages |
| WO2000003683A2 (en) | 1998-07-20 | 2000-01-27 | Inex Pharmaceuticals Corporation | Liposomal encapsulated nucleic acid-complexes |
| US6537777B1 (en) | 1998-07-27 | 2003-03-25 | Hemebiotech A/S | Human porphobilinogen deaminase sequences |
| AU6298899A (en) | 1998-10-09 | 2000-05-01 | Ingene, Inc. | Production of ssdna (in vivo) |
| IL142490A0 (en) | 1998-10-09 | 2002-03-10 | Ingene Inc | ENZYMATIC SYNTHESIS OF ssDNA |
| US6465628B1 (en) | 1999-02-04 | 2002-10-15 | Isis Pharmaceuticals, Inc. | Process for the synthesis of oligomeric compounds |
| JP2002537343A (ja) | 1999-02-23 | 2002-11-05 | アイシス・ファーマシューティカルス・インコーポレーテッド | 多重粒子製剤 |
| US7084125B2 (en) | 1999-03-18 | 2006-08-01 | Exiqon A/S | Xylo-LNA analogues |
| PT1178999E (pt) | 1999-05-04 | 2007-06-26 | Santaris Pharma As | Análogos de l-ribo-lna |
| US6593466B1 (en) | 1999-07-07 | 2003-07-15 | Isis Pharmaceuticals, Inc. | Guanidinium functionalized nucleotides and precursors thereof |
| US6147200A (en) | 1999-08-19 | 2000-11-14 | Isis Pharmaceuticals, Inc. | 2'-O-acetamido modified monomers and oligomers |
| WO2001053307A1 (en) | 2000-01-21 | 2001-07-26 | Geron Corporation | 2'-arabino-fluorooligonucleotide n3'→p5'phosphoramidates: their synthesis and use |
| IT1318539B1 (it) | 2000-05-26 | 2003-08-27 | Italfarmaco Spa | Composizioni farmaceutiche a rilascio prolungato per lasomministrazione parenterale di sostanze idrofile biologicamente |
| ATE325806T1 (de) | 2000-10-04 | 2006-06-15 | Santaris Pharma As | Verbesserte synthese von purin-blockierten nukleinsäure-analoga |
| WO2003015698A2 (en) | 2001-08-13 | 2003-02-27 | University Of Pittsburgh | Application of lipid vehicles and use for drug delivery |
| EP2314690A1 (en) | 2002-07-10 | 2011-04-27 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | RNA-interference by single-stranded RNA molecules |
| US6878805B2 (en) | 2002-08-16 | 2005-04-12 | Isis Pharmaceuticals, Inc. | Peptide-conjugated oligomeric compounds |
| PT2284266E (pt) * | 2002-11-14 | 2013-12-17 | Thermo Fisher Scient Biosciences Inc | Siarn contra tp53 |
| WO2005001110A2 (en) | 2003-05-29 | 2005-01-06 | The Salk Institute For Biological Studies | Transcriptional regulation of gene expression by small double-stranded modulatory rna |
| JP5183064B2 (ja) | 2003-07-02 | 2013-04-17 | エムユーエスシー ファウンデイション フォー リサーチ デべロップメント | 甲殻類動物、及び他の無脊椎動物における、dsRNAで誘導された特異的、及び非特異的免疫、及びそこで使用する生物送達媒体 |
| EP1648519B1 (en) | 2003-07-16 | 2014-10-08 | Protiva Biotherapeutics Inc. | Lipid encapsulated interfering rna |
| EP1648914A4 (en) | 2003-07-31 | 2009-12-16 | Regulus Therapeutics Inc | OLIGOMERIC COMPOUNDS AND COMPOSITIONS USEFUL FOR MODULATING SMALL NON-CODING RNA |
| ATE555118T1 (de) | 2003-08-28 | 2012-05-15 | Takeshi Imanishi | Neue synthetische nukleidsäuren vom typ mit quervernetzter n-o-bindung |
| KR20070085113A (ko) | 2004-05-11 | 2007-08-27 | 가부시키가이샤 알파젠 | Rna간섭을 생기게 하는 폴리뉴클레오티드, 및 이를 이용한유전자발현억제 방법 |
| US7740861B2 (en) | 2004-06-16 | 2010-06-22 | University Of Massachusetts | Drug delivery product and methods |
| AU2006230436B2 (en) | 2005-03-31 | 2011-11-24 | Calando Pharmaceuticals, Inc. | Inhibitors of ribonucleotide reductase subunit 2 and uses thereof |
| JP5336853B2 (ja) | 2005-11-02 | 2013-11-06 | プロチバ バイオセラピューティクス インコーポレイティッド | 修飾siRNA分子およびその使用法 |
| DK2641970T3 (en) | 2005-11-17 | 2015-02-02 | Univ Texas | Modulation of gene expression by oligomers targeted to chromosomal DNA |
| PL1984381T3 (pl) | 2006-01-27 | 2011-03-31 | Isis Pharmaceuticals Inc | Zmodyfikowane w pozycji 6 analogi bicykliczne kwasów nukleinowych |
| TWI322690B (en) | 2006-05-11 | 2010-04-01 | Flysun Dev Co Ltd | Short interference ribonucleic acids for treating allergic dieases |
| EP2057465A4 (en) | 2006-08-09 | 2010-04-21 | Homestead Clinical Corp | SPECIFIC ORGAN PROTEINS AND METHODS OF USE |
| CA2927045A1 (en) | 2006-10-03 | 2008-04-10 | Muthiah Manoharan | Lipid containing formulations |
| EP2121923A1 (en) | 2007-03-02 | 2009-11-25 | MDRNA, Inc. | Nucleic acid compounds for inhibiting erbb family gene expression and uses thereof |
| CN104480112B (zh) | 2007-05-22 | 2018-06-12 | 阿克丘勒斯治疗公司 | 用于治疗的una寡聚体 |
| US20090247608A1 (en) | 2007-12-04 | 2009-10-01 | Alnylam Pharmaceuticals, Inc. | Targeting Lipids |
| EP2238251B1 (en) | 2007-12-27 | 2015-02-11 | Protiva Biotherapeutics Inc. | Silencing of polo-like kinase expression using interfering rna |
| CA2713379A1 (en) | 2008-01-31 | 2009-11-05 | Alnylam Pharmaceuticals, Inc. | Optimized methods for delivery of dsrna targeting the pcsk9 gene |
| US20110269814A1 (en) | 2008-03-26 | 2011-11-03 | Alnylam Pharamaceuticals, Inc. | 2'-f modified rna interference agents |
| EP2279254B1 (en) | 2008-04-15 | 2017-07-05 | Protiva Biotherapeutics Inc. | Novel lipid formulations for nucleic acid delivery |
| CN104651408A (zh) | 2009-06-15 | 2015-05-27 | 阿尔尼拉姆医药品有限公司 | 靶向pcsk9基因的脂质配制的dsrna |
| WO2011005861A1 (en) | 2009-07-07 | 2011-01-13 | Alnylam Pharmaceuticals, Inc. | Oligonucleotide end caps |
| EA024534B1 (ru) | 2010-02-24 | 2016-09-30 | Эрроухэд Рисерч Корпорейшн | КОНЪЮГИРОВАННАЯ СИСТЕМА ДОСТАВКИ И КОМПОЗИЦИЯ ДЛЯ НАПРАВЛЕННОЙ ДОСТАВКИ МАЛЫХ ИНТЕРФЕРИРУЮЩИХ РНК (миРНК) И СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИЦИИ |
| EP2723865B1 (en) | 2011-06-21 | 2019-03-27 | Alnylam Pharmaceuticals, Inc. | METHODS FOR DETERMINING ACTIVITY OF RNAi IN A SUBJECT |
| EP2780454A2 (en) | 2011-11-18 | 2014-09-24 | Alnylam Pharmaceuticals, Inc. | Modified rnai agents |
| US9133461B2 (en) | 2012-04-10 | 2015-09-15 | Alnylam Pharmaceuticals, Inc. | Compositions and methods for inhibiting expression of the ALAS1 gene |
| TWI768330B (zh) | 2013-10-04 | 2022-06-21 | 美國西奈山伊坎醫學院 | 抑制alas1基因表現的組合物及方法 |
| TW201718857A (zh) | 2015-09-14 | 2017-06-01 | 艾爾妮蘭製藥公司 | 用於抑制alas1基因表現之組合物及方法 |
-
2014
- 2014-10-03 TW TW109113729A patent/TWI768330B/zh active
- 2014-10-03 DK DK14800179.5T patent/DK3052628T3/da active
- 2014-10-03 MX MX2016004319A patent/MX2016004319A/es unknown
- 2014-10-03 UA UAA201604801A patent/UA124961C2/uk unknown
- 2014-10-03 SI SI201431579T patent/SI3052628T1/sl unknown
- 2014-10-03 CA CA3227061A patent/CA3227061A1/en active Pending
- 2014-10-03 TW TW103134649A patent/TWI694080B/zh active
- 2014-10-03 CR CR20160195A patent/CR20160195A/es unknown
- 2014-10-03 PL PL14800179T patent/PL3052628T3/pl unknown
- 2014-10-03 PE PE2021000554A patent/PE20211249A1/es unknown
- 2014-10-03 BR BR122020001264-1A patent/BR122020001264B1/pt active IP Right Grant
- 2014-10-03 TW TW111121394A patent/TW202310853A/zh unknown
- 2014-10-03 SG SG11201602631XA patent/SG11201602631XA/en unknown
- 2014-10-03 US US15/027,176 patent/US10119143B2/en active Active
- 2014-10-03 MY MYPI2016000562A patent/MY183490A/en unknown
- 2014-10-03 CN CN201480062979.3A patent/CN105980559B/zh active Active
- 2014-10-03 EP EP20154129.9A patent/EP3693463A1/en active Pending
- 2014-10-03 LT LTEP14800179.5T patent/LT3052628T/lt unknown
- 2014-10-03 PE PE2016000454A patent/PE20161130A1/es unknown
- 2014-10-03 EA EA201690685A patent/EA036477B1/ru unknown
- 2014-10-03 JP JP2016519974A patent/JP6613227B2/ja active Active
- 2014-10-03 SG SG10201910929QA patent/SG10201910929QA/en unknown
- 2014-10-03 IL IL282747A patent/IL282747B2/en unknown
- 2014-10-03 KR KR1020167011726A patent/KR102307389B1/ko active IP Right Grant
- 2014-10-03 TN TN2016000114A patent/TN2016000114A1/en unknown
- 2014-10-03 ES ES14800179T patent/ES2804510T3/es active Active
- 2014-10-03 WO PCT/US2014/059160 patent/WO2015051318A1/en active Application Filing
- 2014-10-03 HU HUE14800179A patent/HUE049227T2/hu unknown
- 2014-10-03 AU AU2014331604A patent/AU2014331604B2/en active Active
- 2014-10-03 NZ NZ757749A patent/NZ757749A/en unknown
- 2014-10-03 NZ NZ718995A patent/NZ718995A/en unknown
- 2014-10-03 CA CA2925357A patent/CA2925357C/en active Active
- 2014-10-03 PT PT148001795T patent/PT3052628T/pt unknown
- 2014-10-03 KR KR1020227040207A patent/KR20220159478A/ko not_active Application Discontinuation
- 2014-10-03 EP EP14800179.5A patent/EP3052628B1/en active Active
- 2014-10-03 MY MYPI2020006311A patent/MY197646A/en unknown
- 2014-10-03 KR KR1020217030663A patent/KR102469850B1/ko active IP Right Grant
- 2014-10-03 IL IL302726A patent/IL302726A/en unknown
- 2014-10-03 BR BR112016007226-0A patent/BR112016007226B1/pt active IP Right Grant
-
2016
- 2016-03-24 IL IL244749A patent/IL244749B/en active IP Right Grant
- 2016-03-29 PH PH12016500574A patent/PH12016500574A1/en unknown
- 2016-04-01 CL CL2016000772A patent/CL2016000772A1/es unknown
- 2016-04-04 GT GT201600066A patent/GT201600066A/es unknown
- 2016-04-04 DO DO2016000073A patent/DOP2016000073A/es unknown
- 2016-04-04 MX MX2022001017A patent/MX2022001017A/es unknown
- 2016-04-29 ZA ZA2016/02931A patent/ZA201602931B/en unknown
- 2016-08-18 HK HK16109909.3A patent/HK1221738A1/zh unknown
-
2018
- 2018-01-19 CL CL2018000158A patent/CL2018000158A1/es unknown
- 2018-05-04 ZA ZA2018/02919A patent/ZA201802919B/en unknown
- 2018-09-20 US US16/137,046 patent/US11028392B2/en active Active
-
2019
- 2019-11-01 JP JP2019199538A patent/JP7289254B2/ja active Active
-
2020
- 2020-05-21 HR HRP20200822TT patent/HRP20200822T1/hr unknown
- 2020-05-27 CY CY20201100482T patent/CY1122975T1/el unknown
- 2020-08-18 CY CY2020029C patent/CY2020029I1/el unknown
- 2020-08-26 LT LTPA2020527C patent/LTC3052628I2/lt unknown
- 2020-08-26 HU HUS2000034C patent/HUS2000034I1/hu unknown
- 2020-08-26 NL NL301061C patent/NL301061I2/nl unknown
- 2020-12-11 AU AU2020286311A patent/AU2020286311B2/en active Active
-
2021
- 2021-05-04 US US17/307,846 patent/US20230026968A1/en not_active Abandoned
-
2022
- 2022-04-19 DO DO2022000085A patent/DOP2022000085A/es unknown
-
2023
- 2023-05-30 JP JP2023088554A patent/JP2023120219A/ja active Pending
- 2023-11-16 AU AU2023266354A patent/AU2023266354A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7377246B2 (ja) | Alas1遺伝子の発現を阻害する組成物および方法 | |
| JP6933670B2 (ja) | Serpinc1 iRNA組成物およびその使用方法 | |
| ES2804510T3 (es) | Composiciones y métodos para inhibir la expresión del gen ALAS1 | |
| JP2020141685A (ja) | アポリポタンパク質C3(APOC3)iRNA組成物およびその使用方法 | |
| TW202223090A (zh) | 用於抑制hao1(羥酸氧化酶1(乙醇酸鹽氧化酶))基因表現的組合物及方法 | |
| WO2017048843A1 (en) | Compositions and methods for inhibiting expression of the alas1 gene | |
| BR122024013552A2 (pt) | Agente de ácido ribonucleico de filamento duplo, seus usos, composição farmacêutica e métodos para inibir a expressão de klkb1, f12 e kng1 em uma célula | |
| EA042137B1 (ru) | КОМПОЗИЦИИ НА ОСНОВЕ iRNA TMPRSS6 И СПОСОБЫ ИХ ПРИМЕНЕНИЯ |