ES2732552T3 - Anticuerpos neutralizantes del virus de la influenza A y usos de los mismos - Google Patents
Anticuerpos neutralizantes del virus de la influenza A y usos de los mismos Download PDFInfo
- Publication number
- ES2732552T3 ES2732552T3 ES11784765T ES11784765T ES2732552T3 ES 2732552 T3 ES2732552 T3 ES 2732552T3 ES 11784765 T ES11784765 T ES 11784765T ES 11784765 T ES11784765 T ES 11784765T ES 2732552 T3 ES2732552 T3 ES 2732552T3
- Authority
- ES
- Spain
- Prior art keywords
- antibody
- seq
- amino acid
- binding fragment
- antigen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 241000712431 Influenza A virus Species 0.000 title claims abstract description 75
- 230000003472 neutralizing effect Effects 0.000 title description 28
- 230000027455 binding Effects 0.000 claims abstract description 148
- 239000000427 antigen Substances 0.000 claims abstract description 113
- 108091007433 antigens Proteins 0.000 claims abstract description 113
- 102000036639 antigens Human genes 0.000 claims abstract description 113
- 239000012634 fragment Substances 0.000 claims abstract description 111
- 208000015181 infectious disease Diseases 0.000 claims abstract description 49
- 125000003275 alpha amino acid group Chemical group 0.000 claims abstract description 39
- 150000007523 nucleic acids Chemical class 0.000 claims description 87
- 239000000178 monomer Substances 0.000 claims description 82
- 108020004707 nucleic acids Proteins 0.000 claims description 49
- 102000039446 nucleic acids Human genes 0.000 claims description 49
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 45
- 125000000539 amino acid group Chemical group 0.000 claims description 34
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 33
- 229920001184 polypeptide Polymers 0.000 claims description 30
- 239000013638 trimer Substances 0.000 claims description 28
- 239000008194 pharmaceutical composition Substances 0.000 claims description 26
- 239000013598 vector Substances 0.000 claims description 23
- 150000001413 amino acids Chemical class 0.000 claims description 22
- 208000037797 influenza A Diseases 0.000 claims description 21
- 238000011282 treatment Methods 0.000 claims description 21
- 229960005486 vaccine Drugs 0.000 claims description 20
- 206010022000 influenza Diseases 0.000 claims description 17
- 101710154606 Hemagglutinin Proteins 0.000 claims description 12
- 101710093908 Outer capsid protein VP4 Proteins 0.000 claims description 12
- 101710135467 Outer capsid protein sigma-1 Proteins 0.000 claims description 12
- 101710176177 Protein A56 Proteins 0.000 claims description 12
- 239000000185 hemagglutinin Substances 0.000 claims description 12
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 10
- 108091033319 polynucleotide Proteins 0.000 claims description 7
- 102000040430 polynucleotide Human genes 0.000 claims description 7
- 239000002157 polynucleotide Substances 0.000 claims description 7
- 238000003745 diagnosis Methods 0.000 claims description 5
- 239000003085 diluting agent Substances 0.000 claims description 2
- 210000004027 cell Anatomy 0.000 description 97
- 238000000034 method Methods 0.000 description 56
- 239000000203 mixture Substances 0.000 description 52
- 102100035360 Cerebellar degeneration-related antigen 1 Human genes 0.000 description 42
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 37
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 37
- 241000700605 Viruses Species 0.000 description 34
- 108090000623 proteins and genes Proteins 0.000 description 33
- 210000003719 b-lymphocyte Anatomy 0.000 description 32
- 210000004180 plasmocyte Anatomy 0.000 description 29
- 102000004169 proteins and genes Human genes 0.000 description 24
- 241000699670 Mus sp. Species 0.000 description 22
- 238000004519 manufacturing process Methods 0.000 description 20
- 230000001225 therapeutic effect Effects 0.000 description 19
- 239000003814 drug Substances 0.000 description 18
- 230000003993 interaction Effects 0.000 description 16
- 230000014509 gene expression Effects 0.000 description 15
- 239000003937 drug carrier Substances 0.000 description 13
- 238000006386 neutralization reaction Methods 0.000 description 13
- 101100112922 Candida albicans CDR3 gene Proteins 0.000 description 10
- 239000002202 Polyethylene glycol Substances 0.000 description 10
- 230000002209 hydrophobic effect Effects 0.000 description 10
- 229920001223 polyethylene glycol Polymers 0.000 description 10
- 239000000243 solution Substances 0.000 description 10
- 230000003612 virological effect Effects 0.000 description 10
- 238000002965 ELISA Methods 0.000 description 9
- 101000737793 Homo sapiens Cerebellar degeneration-related antigen 1 Proteins 0.000 description 9
- 239000013604 expression vector Substances 0.000 description 9
- 230000004927 fusion Effects 0.000 description 9
- 239000012528 membrane Substances 0.000 description 9
- 241001112090 Pseudovirus Species 0.000 description 8
- 150000001720 carbohydrates Chemical group 0.000 description 8
- 238000003776 cleavage reaction Methods 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- 230000001681 protective effect Effects 0.000 description 8
- 230000007017 scission Effects 0.000 description 8
- HVLSXIKZNLPZJJ-TXZCQADKSA-N HA peptide Chemical compound C([C@@H](C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 HVLSXIKZNLPZJJ-TXZCQADKSA-N 0.000 description 7
- 241000282412 Homo Species 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 102000005348 Neuraminidase Human genes 0.000 description 7
- 108010006232 Neuraminidase Proteins 0.000 description 7
- 230000037396 body weight Effects 0.000 description 7
- PEDCQBHIVMGVHV-UHFFFAOYSA-N glycerol group Chemical group OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 238000002560 therapeutic procedure Methods 0.000 description 7
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 230000000295 complement effect Effects 0.000 description 6
- 238000012258 culturing Methods 0.000 description 6
- 238000001514 detection method Methods 0.000 description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 6
- 229940088598 enzyme Drugs 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 239000001257 hydrogen Substances 0.000 description 6
- 229910052739 hydrogen Inorganic materials 0.000 description 6
- 230000002163 immunogen Effects 0.000 description 6
- 230000002458 infectious effect Effects 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 239000011159 matrix material Substances 0.000 description 6
- 229920000136 polysorbate Polymers 0.000 description 6
- 230000002265 prevention Effects 0.000 description 6
- 238000012163 sequencing technique Methods 0.000 description 6
- 101000737796 Homo sapiens Cerebellar degeneration-related protein 2 Proteins 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 5
- 230000008859 change Effects 0.000 description 5
- 238000010367 cloning Methods 0.000 description 5
- 239000013078 crystal Substances 0.000 description 5
- 238000002425 crystallisation Methods 0.000 description 5
- 230000008025 crystallization Effects 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 210000004408 hybridoma Anatomy 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- 210000004072 lung Anatomy 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 230000001404 mediated effect Effects 0.000 description 5
- 239000000825 pharmaceutical preparation Substances 0.000 description 5
- 229940127557 pharmaceutical product Drugs 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 230000004044 response Effects 0.000 description 5
- 230000001932 seasonal effect Effects 0.000 description 5
- 230000004083 survival effect Effects 0.000 description 5
- 229940124597 therapeutic agent Drugs 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 4
- 108010032595 Antibody Binding Sites Proteins 0.000 description 4
- 239000002033 PVDF binder Substances 0.000 description 4
- 206010035226 Plasma cell myeloma Diseases 0.000 description 4
- 239000004365 Protease Substances 0.000 description 4
- 230000000890 antigenic effect Effects 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 210000004899 c-terminal region Anatomy 0.000 description 4
- 238000012512 characterization method Methods 0.000 description 4
- 230000029087 digestion Effects 0.000 description 4
- 238000012377 drug delivery Methods 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- 230000013595 glycosylation Effects 0.000 description 4
- 238000006206 glycosylation reaction Methods 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- 230000028993 immune response Effects 0.000 description 4
- 238000003018 immunoassay Methods 0.000 description 4
- 238000001802 infusion Methods 0.000 description 4
- 238000002955 isolation Methods 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 201000000050 myeloid neoplasm Diseases 0.000 description 4
- 238000011275 oncology therapy Methods 0.000 description 4
- 238000005457 optimization Methods 0.000 description 4
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 230000000069 prophylactic effect Effects 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 238000006467 substitution reaction Methods 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 230000002195 synergetic effect Effects 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 241000712461 unidentified influenza virus Species 0.000 description 4
- 238000002255 vaccination Methods 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 3
- 206010069754 Acquired gene mutation Diseases 0.000 description 3
- 241000283707 Capra Species 0.000 description 3
- 108020004705 Codon Proteins 0.000 description 3
- 108020004414 DNA Proteins 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 3
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 3
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 3
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 3
- 108090000190 Thrombin Proteins 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 210000004556 brain Anatomy 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 150000001875 compounds Chemical class 0.000 description 3
- 239000012228 culture supernatant Substances 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- 210000004602 germ cell Anatomy 0.000 description 3
- 150000004676 glycans Chemical class 0.000 description 3
- 210000005260 human cell Anatomy 0.000 description 3
- 230000016784 immunoglobulin production Effects 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 229960003971 influenza vaccine Drugs 0.000 description 3
- 231100000518 lethal Toxicity 0.000 description 3
- 230000001665 lethal effect Effects 0.000 description 3
- 239000002502 liposome Substances 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 238000003752 polymerase chain reaction Methods 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 230000002285 radioactive effect Effects 0.000 description 3
- 238000003127 radioimmunoassay Methods 0.000 description 3
- 238000006722 reduction reaction Methods 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 230000037439 somatic mutation Effects 0.000 description 3
- 229960004072 thrombin Drugs 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- 238000001890 transfection Methods 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 238000013519 translation Methods 0.000 description 3
- 239000012588 trypsin Substances 0.000 description 3
- 230000003442 weekly effect Effects 0.000 description 3
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 2
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 2
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 2
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 101800001415 Bri23 peptide Proteins 0.000 description 2
- 101800000655 C-terminal peptide Proteins 0.000 description 2
- 102400000107 C-terminal peptide Human genes 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 208000032163 Emerging Communicable disease Diseases 0.000 description 2
- 241000287828 Gallus gallus Species 0.000 description 2
- 241000702620 H-1 parvovirus Species 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- MQUQNUAYKLCRME-INIZCTEOSA-N N-tosyl-L-phenylalanyl chloromethyl ketone Chemical compound C1=CC(C)=CC=C1S(=O)(=O)N[C@H](C(=O)CCl)CC1=CC=CC=C1 MQUQNUAYKLCRME-INIZCTEOSA-N 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 108090000526 Papain Proteins 0.000 description 2
- 241000702437 Parvovirus H3 Species 0.000 description 2
- 108010090804 Streptavidin Proteins 0.000 description 2
- 108090000631 Trypsin Proteins 0.000 description 2
- 102000004142 Trypsin Human genes 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 238000001042 affinity chromatography Methods 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 210000004102 animal cell Anatomy 0.000 description 2
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 2
- 229960002685 biotin Drugs 0.000 description 2
- 235000020958 biotin Nutrition 0.000 description 2
- 239000011616 biotin Substances 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 230000002301 combined effect Effects 0.000 description 2
- 239000002299 complementary DNA Substances 0.000 description 2
- 231100000599 cytotoxic agent Toxicity 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 238000002405 diagnostic procedure Methods 0.000 description 2
- 229910003460 diamond Inorganic materials 0.000 description 2
- 239000010432 diamond Substances 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- 239000003640 drug residue Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 210000003527 eukaryotic cell Anatomy 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 150000002314 glycerols Polymers 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 229940022353 herceptin Drugs 0.000 description 2
- 230000005847 immunogenicity Effects 0.000 description 2
- 230000004957 immunoregulator effect Effects 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 239000006193 liquid solution Substances 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 238000013507 mapping Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 210000001806 memory b lymphocyte Anatomy 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- 238000010172 mouse model Methods 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 229940055729 papain Drugs 0.000 description 2
- 235000019834 papain Nutrition 0.000 description 2
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 229920001583 poly(oxyethylated polyols) Polymers 0.000 description 2
- 229920002401 polyacrylamide Polymers 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 238000003259 recombinant expression Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000003757 reverse transcription PCR Methods 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 238000007619 statistical method Methods 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 238000004448 titration Methods 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 241000701447 unidentified baculovirus Species 0.000 description 2
- 238000002424 x-ray crystallography Methods 0.000 description 2
- 210000005253 yeast cell Anatomy 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- AXAVXPMQTGXXJZ-UHFFFAOYSA-N 2-aminoacetic acid;2-amino-2-(hydroxymethyl)propane-1,3-diol Chemical compound NCC(O)=O.OCC(N)(CO)CO AXAVXPMQTGXXJZ-UHFFFAOYSA-N 0.000 description 1
- CJIJXIFQYOPWTF-UHFFFAOYSA-N 7-hydroxycoumarin Natural products O1C(=O)C=CC2=CC(O)=CC=C21 CJIJXIFQYOPWTF-UHFFFAOYSA-N 0.000 description 1
- 108010066676 Abrin Proteins 0.000 description 1
- 102000012440 Acetylcholinesterase Human genes 0.000 description 1
- 108010022752 Acetylcholinesterase Proteins 0.000 description 1
- 108010000239 Aequorin Proteins 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 206010003445 Ascites Diseases 0.000 description 1
- 101100335652 Autographa californica nuclear polyhedrosis virus GP64 gene Proteins 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 239000007989 BIS-Tris Propane buffer Substances 0.000 description 1
- 241000193830 Bacillus <bacterium> Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 108010004032 Bromelains Proteins 0.000 description 1
- 241000282836 Camelus dromedarius Species 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000251730 Chondrichthyes Species 0.000 description 1
- 101800004419 Cleaved form Proteins 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- 108700010070 Codon Usage Proteins 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 102100025698 Cytosolic carboxypeptidase 4 Human genes 0.000 description 1
- 101710112752 Cytotoxin Proteins 0.000 description 1
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Polymers OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- XPDXVDYUQZHFPV-UHFFFAOYSA-N Dansyl Chloride Chemical compound C1=CC=C2C(N(C)C)=CC=CC2=C1S(Cl)(=O)=O XPDXVDYUQZHFPV-UHFFFAOYSA-N 0.000 description 1
- CYCGRDQQIOGCKX-UHFFFAOYSA-N Dehydro-luciferin Natural products OC(=O)C1=CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 CYCGRDQQIOGCKX-UHFFFAOYSA-N 0.000 description 1
- 102000016607 Diphtheria Toxin Human genes 0.000 description 1
- 108010053187 Diphtheria Toxin Proteins 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- BJGNCJDXODQBOB-UHFFFAOYSA-N Fivefly Luciferin Natural products OC(=O)C1CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 BJGNCJDXODQBOB-UHFFFAOYSA-N 0.000 description 1
- 101150010917 GP67 gene Proteins 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108060003393 Granulin Proteins 0.000 description 1
- 101000932590 Homo sapiens Cytosolic carboxypeptidase 4 Proteins 0.000 description 1
- 101001037139 Homo sapiens Immunoglobulin heavy variable 3-30 Proteins 0.000 description 1
- 101000604674 Homo sapiens Immunoglobulin kappa variable 4-1 Proteins 0.000 description 1
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 1
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 1
- 102100040219 Immunoglobulin heavy variable 3-30 Human genes 0.000 description 1
- 102100038198 Immunoglobulin kappa variable 4-1 Human genes 0.000 description 1
- 241000713196 Influenza B virus Species 0.000 description 1
- 241000371980 Influenza B virus (B/Shanghai/361/2002) Species 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- DDWFXDSYGUXRAY-UHFFFAOYSA-N Luciferin Natural products CCc1c(C)c(CC2NC(=O)C(=C2C=C)C)[nH]c1Cc3[nH]c4C(=C5/NC(CC(=O)O)C(C)C5CC(=O)O)CC(=O)c4c3C DDWFXDSYGUXRAY-UHFFFAOYSA-N 0.000 description 1
- 241000701076 Macacine alphaherpesvirus 1 Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 101001033003 Mus musculus Granzyme F Proteins 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102000057297 Pepsin A Human genes 0.000 description 1
- 108090000284 Pepsin A Proteins 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 108010067902 Peptide Library Proteins 0.000 description 1
- 108010004729 Phycoerythrin Proteins 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 101000762949 Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Exotoxin A Proteins 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 1
- 102000004389 Ribonucleoproteins Human genes 0.000 description 1
- 108010081734 Ribonucleoproteins Proteins 0.000 description 1
- 108010039491 Ricin Proteins 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- FBPFZTCFMRRESA-NQAPHZHOSA-N Sorbitol Polymers OCC(O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-NQAPHZHOSA-N 0.000 description 1
- 238000000692 Student's t-test Methods 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 230000024932 T cell mediated immunity Effects 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 229940022698 acetylcholinesterase Drugs 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000001919 adrenal effect Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 238000001261 affinity purification Methods 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 1
- 235000011130 ammonium sulphate Nutrition 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- HHKZCCWKTZRCCL-UHFFFAOYSA-N bis-tris propane Chemical compound OCC(CO)(CO)NCCCNC(CO)(CO)CO HHKZCCWKTZRCCL-UHFFFAOYSA-N 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 235000019835 bromelain Nutrition 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 239000005515 coenzyme Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 239000000599 controlled substance Substances 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 230000037029 cross reaction Effects 0.000 description 1
- 230000009260 cross reactivity Effects 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- 239000002619 cytotoxin Substances 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 210000001163 endosome Anatomy 0.000 description 1
- 239000002532 enzyme inhibitor Substances 0.000 description 1
- 238000003114 enzyme-linked immunosorbent spot assay Methods 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 1
- -1 for example Substances 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 150000002303 glucose derivatives Polymers 0.000 description 1
- 150000002304 glucoses Polymers 0.000 description 1
- 125000002791 glucosyl group Polymers C1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000003324 growth hormone secretagogue Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 239000012510 hollow fiber Substances 0.000 description 1
- 230000028996 humoral immune response Effects 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 229940127121 immunoconjugate Drugs 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000037798 influenza B Diseases 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 238000012804 iterative process Methods 0.000 description 1
- 238000005304 joining Methods 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 231100000636 lethal dose Toxicity 0.000 description 1
- 231100000225 lethality Toxicity 0.000 description 1
- 230000021633 leukocyte mediated immunity Effects 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- HWYHZTIRURJOHG-UHFFFAOYSA-N luminol Chemical compound O=C1NNC(=O)C2=C1C(N)=CC=C2 HWYHZTIRURJOHG-UHFFFAOYSA-N 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 150000002690 malonic acid derivatives Chemical class 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000001823 molecular biology technique Methods 0.000 description 1
- 230000004001 molecular interaction Effects 0.000 description 1
- 229940126619 mouse monoclonal antibody Drugs 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- ZTLGJPIZUOVDMT-UHFFFAOYSA-N n,n-dichlorotriazin-4-amine Chemical compound ClN(Cl)C1=CC=NN=N1 ZTLGJPIZUOVDMT-UHFFFAOYSA-N 0.000 description 1
- 210000004898 n-terminal fragment Anatomy 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 229920001542 oligosaccharide Polymers 0.000 description 1
- 150000002482 oligosaccharides Chemical class 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- PGZUMBJQJWIWGJ-ONAKXNSWSA-N oseltamivir phosphate Chemical compound OP(O)(O)=O.CCOC(=O)C1=C[C@@H](OC(CC)CC)[C@H](NC(C)=O)[C@@H](N)C1 PGZUMBJQJWIWGJ-ONAKXNSWSA-N 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 239000006174 pH buffer Substances 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 229940111202 pepsin Drugs 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 230000001817 pituitary effect Effects 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 108010052044 polyclonal B cell activator Proteins 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229950008882 polysorbate Drugs 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- 230000001323 posttranslational effect Effects 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000019419 proteases Nutrition 0.000 description 1
- 230000001012 protector Effects 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- ZLIBICFPKPWGIZ-UHFFFAOYSA-N pyrimethanil Chemical compound CC1=CC(C)=NC(NC=2C=CC=CC=2)=N1 ZLIBICFPKPWGIZ-UHFFFAOYSA-N 0.000 description 1
- 239000002510 pyrogen Substances 0.000 description 1
- 239000012857 radioactive material Substances 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- 238000005096 rolling process Methods 0.000 description 1
- 210000003079 salivary gland Anatomy 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 239000004017 serum-free culture medium Substances 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 235000020183 skimmed milk Nutrition 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- PRWXGRGLHYDWPS-UHFFFAOYSA-L sodium malonate Chemical compound [Na+].[Na+].[O-]C(=O)CC([O-])=O PRWXGRGLHYDWPS-UHFFFAOYSA-L 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 239000000021 stimulant Substances 0.000 description 1
- 238000005728 strengthening Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 150000005846 sugar alcohols Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 229940036185 synagis Drugs 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 229940061367 tamiflu Drugs 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229960000814 tetanus toxoid Drugs 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 230000010474 transient expression Effects 0.000 description 1
- 238000003146 transient transfection Methods 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 238000005829 trimerization reaction Methods 0.000 description 1
- ORHBXUUXSCNDEV-UHFFFAOYSA-N umbelliferone Chemical compound C1=CC(=O)OC2=CC(O)=CC=C21 ORHBXUUXSCNDEV-UHFFFAOYSA-N 0.000 description 1
- HFTAFOQKODTIJY-UHFFFAOYSA-N umbelliferone Natural products Cc1cc2C=CC(=O)Oc2cc1OCC=CC(C)(C)O HFTAFOQKODTIJY-UHFFFAOYSA-N 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/08—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from viruses
- C07K16/10—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from viruses from RNA viruses
- C07K16/1018—Orthomyxoviridae, e.g. influenza virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
- G01N33/56983—Viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/10—Immunoglobulins specific features characterized by their source of isolation or production
- C07K2317/14—Specific host cells or culture conditions, e.g. components, pH or temperature
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/21—Immunoglobulins specific features characterized by taxonomic origin from primates, e.g. man
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/33—Crossreactivity, e.g. for species or epitope, or lack of said crossreactivity
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/34—Identification of a linear epitope shorter than 20 amino acid residues or of a conformational epitope defined by amino acid residues
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2760/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses negative-sense
- C12N2760/00011—Details
- C12N2760/16011—Orthomyxoviridae
- C12N2760/16111—Influenzavirus A, i.e. influenza A virus
- C12N2760/16122—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/005—Assays involving biological materials from specific organisms or of a specific nature from viruses
- G01N2333/08—RNA viruses
- G01N2333/11—Orthomyxoviridae, e.g. influenza virus
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2469/00—Immunoassays for the detection of microorganisms
- G01N2469/10—Detection of antigens from microorganism in sample from host
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Virology (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Immunology (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Communicable Diseases (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Gastroenterology & Hepatology (AREA)
- Physics & Mathematics (AREA)
- Microbiology (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- Tropical Medicine & Parasitology (AREA)
- Analytical Chemistry (AREA)
- Food Science & Technology (AREA)
- Biotechnology (AREA)
- Cell Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Veterinary Medicine (AREA)
- Oncology (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
Anticuerpo aislado, o un fragmento de unión a antígeno del mismo, que neutraliza la infección de un subtipo del grupo 1 y un subtipo del grupo 2 del virus de la influenza A y que comprende: i) las secuencias de cadena pesada de CDR1, CDR2 y CDR3, tal como se establecen en las SEQ ID NO: 1, 41 y 43, respectivamente, o tal como se establecen en las SEQ ID NO: 1, 41 y 42, respectivamente; y ii) las secuencias de cadena ligera de CDR1, CDR2 y CDR3, tal como se establecen en las SEQ ID NO: 4, 5 y 6, respectivamente, o como se establecen en las SEQ ID NO: 44, 5 y 6, respectivamente, donde dicho anticuerpo o fragmento de unión al antígeno del mismo comprende una región variable de cadena pesada que tiene al menos un 90% de identidad de secuencia con la secuencia de aminoácidos tal como se establece en las SEQ ID NO: 59 o 55; y una región variable de cadena ligera que tiene una identidad de secuencia de al menos el 90% con la secuencia de aminoácidos según se establece en las SEQ ID NO: 57 o 61 y donde dicho anticuerpo o fragmento de unión a antígeno del mismo se produce en células transfectadas a títulos al menos 3 veces más altos que aquellos a los que se produce la variante 2 de FI6.
Description
DESCRIPCIÓN
Anticuerpos neutralizantes del virus de la influenza A y usos de los mismos
ANTECEDENTES
La respuesta de los anticuerpos neutralizantes del virus de la influenza A es típicamente específica para un subtipo viral dado. Existen 16 subtipos de influenza A definidos por sus proteínas hemaglutinina ("HA"). Los 16 HA, H1 - H16, se pueden clasificar en dos grupos. El grupo 1 consiste en los subtipos H1, H2, H5, H6, H8, H9, H11, H12, H13 y H16 y el grupo 2 incluye los subtipos H3, H4, H7, H10, H14 y H15. Si bien todos los subtipos están presentes en las aves, la mayoría de los subtipos H1, H2 y H3 causan enfermedades en los seres humanos. Los subtipos H5, H7 y H9 provocan infecciones esporádicas graves en los seres humanos y pueden generar nuevas pandemias. Los virus H1 y H3 evolucionan continuamente, generando nuevas variantes, un fenómeno denominado deriva antigénica. Como consecuencia, los anticuerpos producidos en respuesta a virus del pasado son poco o nada protectores contra nuevos virus derivados. Como consecuencia, se debe producir una nueva vacuna cada año contra los virus H1 y H3 que se prevé que surjan, un proceso que es muy costoso y no siempre eficiente. Lo mismo se aplica a la producción de nuevas vacunas contra la influenza H5. De hecho, no está claro si las vacunas H5 actuales basadas en aislados de los virus de influenza A de Vietnam o Indonesia protegerán frente a un futuro virus H5 pandémico.
Por estas razones, sería muy deseable disponer de una vacuna que indujera anticuerpos neutralizantes ampliamente, capaces de neutralizar todos los subtipos de virus de la influenza A, así como sus variantes anuales (revisado por Gerhard et al., 2006). Además, podrían administrarse anticuerpos heterosubtípicos ampliamente neutralizantes como medicamentos para la prevención o el tratamiento de la infección por influenza A. Para la fabricación de tales medicamentos es importante seleccionar anticuerpos que se producen a altas titulaciones para reducir los costes de producción.
Se han caracterizado anticuerpos que reconocen el virus de la influenza A. Los anticuerpos de M2, una pequeña proteína invariante expresada en las células infectadas pero no en los virus infecciosos, han mostrado cierto efecto protector in vivo, posiblemente señalando células infectadas para su destrucción por células NK o células T citotóxicas. También es posible atacar la proteína HA con anticuerpos neutralizantes. La HA se sintetiza como un polipéptido precursor homo-trimérico HA0. Cada monómero puede escindirse independientemente después de la traducción para formar dos polipéptidos, HA1 y HA2, unidos por un único enlace disulfuro. El fragmento N-terminal más grande (HA1, 320-330 aminoácidos) forma un dominio globular distal de membrana que contiene el sitio de unión al receptor y la mayoría de los determinantes reconocidos por anticuerpos neutralizantes de virus. El polipéptido HA1 de HA es responsable de la unión del virus a la superficie celular. La porción C-terminal más pequeña (HA2, ~180 aminoácidos) forma una estructura de tipo tallo que ancla el dominio globular a la membrana celular o viral. El polipéptido HA2 media la fusión de las membranas víricas y celulares en los endosomas, permitiendo la liberación del complejo de ribonucleoproteína en el citoplasma.
El grado de homología de secuencia entre subtipos es menor en los polipéptidos de HA1 (homología del 34%-59% entre subtipos) que en el polipéptido HA2 (51% - 80% de homología). La región más conservada es la secuencia alrededor del sitio de escisión, en particular los 11 aminoácidos HA N-terminales, denominados péptidos de fusión, que se conservan entre todos los subtipos del virus de la influenza A. Parte de esta región se muestra como un bucle superficial en la molécula precursora de HA (HAO), pero se vuelve inaccesible cuando HA0 se divide en HA1/HA2. En resumen, existen regiones conservadas entre los diferentes subtipos de HA, especialmente en la región de unión HA1-HA2 y en la región de HA2. Sin embargo, estas regiones pueden ser poco accesibles para los anticuerpos neutralizantes.
Solo ha habido un éxito limitado en la identificación de anticuerpos que neutralizan más de un subtipo de virus de la influenza A. Además, el rango de neutralización de los anticuerpos identificados hasta ahora es estrecho y su potencia es baja. Okuno et al. Inmunizaron ratones con el virus de la influenza A/Okuda/57 (H2N2) y aislaron un anticuerpo monoclonal (C179) que se une a un epítopo conformacional conservado en HA2 y neutraliza los virus de la influenza del subtipo A del Grupo 1 H2, H1 y H5 in vitro e in vivo en modelos animales (Okuno et al., 1993; Smirnov et al., 1999; Smirnov et al., 2000).
Gioia et al. describieron la presencia de anticuerpos neutralizantes del virus H5N1 en el suero de algunos individuos que recibieron una vacuna contra la gripe estacional convencional (Gioia et al., 2008). Los autores sugieren que la actividad neutralizadora podría deberse a anticuerpos parea la neuraminidasa (N1). Sin embargo, no se aislaron anticuerpos monoclonales y no se caracterizaron los epítopos diana. Además, no está claro si los anticuerpos séricos neutralizan otros subtipos del virus de la influenza A.
Se han aislado anticuerpos humanos heterosubtípicos que se unen a un epítopo en la región de tipo tallo de HA y capaces de neutralizar ciertos subtipos del virus de la influenza A del Grupo 1 o Grupo 2 a partir de células B de memoria y células plasmáticas de donantes inmunes. La WO 2010/010466 A2 describe la infección neutralizante de anticuerpos humanos de un subtipo del grupo 1 y un subtipo del grupo 2 del virus de la influenza A que se unen específicamente a un epítopo en la región del tallo de la hemaglutinina.
A pesar de décadas de investigación, no existen en el mercado anticuerpos que neutralicen o inhiban ampliamente la infección por el virus de la influenza A o que atenúen la enfermedad causada por el virus de la influenza A. Por tanto, existe una necesidad de identificar nuevos anticuerpos que neutralicen múltiples subtipos del virus de la influenza A y que puedan usarse como medicamentos para la prevención o el tratamiento de la infección por influenza A. Existe además la necesidad de identificar anticuerpos que se producen a altas titulaciones para reducir los costes de producción.
SUMARIO
La invención se refiere, en parte, al aislamiento, de individuos vacunados con la vacuna contra la gripe estacional, de anticuerpos monoclonales humanos que existen naturalmente que se unen a1 HA y neutralizan la infección de más de un subtipo del virus de la influenza A, así como a epítopos a los cuales se unen los anticuerpos de la descripción comprendida en la invención. Por consiguiente, en un aspecto de la invención, la invención comprende un anticuerpo y fragmentos de unión a antígeno del mismo que neutralizan la infección de más de un subtipo del virus de la influenza A seleccionados de los subtipos grupo 1 y grupo 2.
En una realización de la invención, la invención comprende un anticuerpo aislado o un fragmento de unión a antígeno del mismo que neutraliza la infección de un subtipo del grupo 1 y un subtipo del grupo 2 del virus de la influenza A y comprende: (i) las secuencias de la cadena pesada de CDR1, CDR2 y CDR3 tal como se establecen las SEQ ID NO: 1, 41 y 43, respectivamente, o como se establece en las SEQ ID NO: 1, 41 y 42, respectivamente; y (ii) las secuencias de la cadena ligera de CDR1, CDR2 y CDR3 tal como se establecen en las SEQ ID NO: 4, 5 y 6, respectivamente, o como se establecen en las SEQ ID NO: 44, 5 y 6, respectivamente, donde dicho anticuerpo o antígeno de unión a fragmento del mismo comprende una región variable de cadena pesada que tiene al menos un 90% de identidad de secuencia con la secuencia de aminoácidos tal como se establece en las SEQ ID NO: 59 o 55 y una región variable de cadena ligera que tiene al menos un 90% de identidad de secuencia con la secuencia de aminoácidos tal como se establece en las SEQ ID NO: 57 o 61, y donde dicho anticuerpo o fragmento de anticuerpo de unión de antígeno de la misma es producido en células transfectadas a títulos al menos 3 veces más altos que la titulación a la que se produce la variante 2 de FI6. Preferiblemente, comprende un anticuerpo aislado o un fragmento de unión a antígeno del mismo que neutraliza la infección de un subtipo del grupo 1 y de un subtipo del grupo 2 del virus de la influenza A y se une específicamente a un epítopo en la región del tallo de un trímero de hemaglutinina (HA) de la influenza A, donde la cadena pesada y la cadena ligera del anticuerpo o de un fragmento de unión al antígeno del mismo entra en contacto con los aminoácidos en un primer monómero proximal y un segundo monómero distal del trímero de HA.
En otra realización más de la invención, la invención comprende un anticuerpo aislado, o un fragmento de unión a antígeno del mismo, que neutraliza la infección de un subtipo del grupo 1 y un subtipo del grupo 2 del virus de la influenza A y se une específicamente a un epítopo en la región del tallo de un trímero HA de la influenza A, donde la cadena pesada y ligera del anticuerpo, o de su fragmento de unión a antígeno, entran en contacto con los aminoácidos en un primer monómero proximal y un segundo monómero distal derecho del trímero HA, y donde el anticuerpo, o un fragmento de unión a antígeno del mismo, es producido en células transfectadas a títulos al menos 3 veces más altos que la titulación a la que se produce la variante 2 de FI6, comprendiendo el anticuerpo o un fragmento de unión a antígeno del mismo: (i) las secuencias CDR1, CDR2 y CDR3 de cadena pesada como se establecen en las SEQ ID NO: 1, 41 y 43, respectivamente, o como se establecen en las SEQ ID NO: 1,41 y 42, respectivamente; y (ii) las secuencias de la cadena ligera CDR1, CDR2 y CDR3 como se establecen en las SEQ ID NO: 4, 5 y 6, respectivamente, o como se establecen en las SEQ ID NO: 44, 5 y 6, respectivamente.
La invención también comprende un anticuerpo aislado, o un fragmento de unión a antígeno del mismo, donde el anticuerpo comprende una región variable de cadena pesada que comprende la secuencia de aminoácidos de SEQ ID NO: 59 y una región variable de cadena ligera que comprende la secuencia de aminoácidos de SEQ ID NO: 57; o una región variable de cadena pesada que comprende la secuencia de aminoácidos de SEQ ID NO: 59 y una región variable de cadena ligera que comprende la secuencia de aminoácidos de SEQ ID NO: 61; o una región variable de cadena pesada que comprende la secuencia de aminoácidos de SEQ ID NO: 55 y una región variable de cadena ligera que comprende la secuencia de aminoácidos de SEQ ID NO: 57; o una región variable de cadena pesada que comprende la secuencia de aminoácidos de SEQ ID NO: 55 y una región variable de cadena ligera que comprende la secuencia de aminoácidos de SEQ ID NO: 61 y donde el anticuerpo neutraliza un subtipo del grupo 1 y un subtipo del grupo 2 del virus de la influenza A. La invención además comprende un anticuerpo, o un fragmento de unión
a antígeno del mismo, donde el anticuerpo es una variante 3 de FI6, una variante 4 de FI6 o una variante 5 de FI6.
En otro aspecto, la invención comprende una molécula de ácido nucleico que comprende un polinucleótido que codifica un anticuerpo o un fragmento de anticuerpo de la invención. En otro aspecto más, la invención comprende un vector que comprende una molécula de ácido nucleico de la invención o una célula que expresa un anticuerpo de la invención o un fragmento de unión a antígeno de la misma. En otra realización más, la invención comprende una célula que comprende un vector de la invención.
La invención comprende además una composición farmacéutica que comprende un anticuerpo de la invención o un fragmento de unión a antígeno del mismo, una molécula de ácido nucleico de la invención, un vector que comprende una molécula de ácido nucleico de la invención, una célula que expresa un anticuerpo o un fragmento de anticuerpo de la invención o una célula que comprende un vector de la invención, y un diluyente o vehículo farmacéuticamente aceptable. La invención también comprende una composición farmacéutica que comprende un primer anticuerpo o un fragmento de unión a antígeno del mismo, y un segundo anticuerpo, o un fragmento de unión a antígeno del mismo, donde el primer anticuerpo es un anticuerpo de la invención y el segundo anticuerpo es cualquier anticuerpo, o fragmento de unión a antígeno del mismo, que neutraliza la infección por el virus de la influenza A o la influenza B.
También se contempla dentro del alcance de la invención el uso de un anticuerpo de la invención, o de un fragmento de unión a antígeno del mismo, de un ácido nucleico de la invención, de un vector que comprende un ácido nucleico de la invención, de una célula que expresa un vector de la invención o de una composición farmacéutica de la invención (i) en la fabricación de un medicamento para el tratamiento de la infección por el virus de la influenza A, (ii) en una vacuna o (iii) en el diagnóstico de la infección por el virus de la influenza A.
DESCRIPCION DE LAS FIGURAS
Figura 1: muestra los títulos de producción de anticuerpos de células 293F transfectadas transitoriamente con vectores que expresan genes que codifican la variante 2, 3, 4 o 5 de FI6.
Figuras 2A y 2B: representaciones superficiales del subdominio F de H1 HA y H3 HA, respectivamente, complejados con la variante 3 de FI6 (la referencia en la figura es FI6). Las cadenas laterales seleccionadas en HA1 y HA2 que contribuyen al surco hidrófobo conservado están indicadas por las flechas, cuyos límites aproximados están indicados por la línea negra. Thr (40) y Thr (318) están en HA1 y IIe (45), Trp (21), Thr (41), Leu (38) y el bucle 18-21 están en HA2.
Figura 3: muestra la unión de la variante 3 de FI6 (referida en la figura como FI6) al subdominio F del trímero HA. La Figura 3A muestra el trímero de H3 HA que se une a tres anticuerpos de la variante 3 de FI6 en la representación en cintas. Uno de los monómeros HA es de color negro para HA1 y gris oscuro para HA2, mientras que los otros dos monómeros HA están en gris claro. Las Figuras 3B y 3C muestran una vista ampliada de la interacción del bucle LCDR1 con el péptido de fusión del monómero HA vecino en complejos de la variante 3 H1/FI6 y la variante 3 H3/FI6, respectivamente. Las Figuras 3D y 3E muestran las estructuras de los monómeros de H5 HA complejadas con CR6261 (pdb ID 2GBM) (D) y F10 (pdb 3FKU)
(E) que se unen a una región similar en comparación con la variante 3 de Fi6, pero con su dominio VH asentado 5 -10 A más bajo en la HA.
Figura 4: muestra las interacciones de la variante 3 de FI6 (referida en la figura como FI6-v3) con el subdominio F de H1 y H3 HA. Muestra una representación superficial del subdominio F de H1 HA y H3 HA con los bucles HCDR3 y LCDR1 de la variante 3 de FI6 con cadenas laterales seleccionadas. El monómero de HA proximal se representa en gris claro; el monómero distal derecho se representa en gris claro; y el glicano unido a N38 de H3 HA1 se representa como esferas de color gris oscuro.
Figura 5: muestra las diferencias específicas de grupo en los sitios de unión del anticuerpo con reactividad cruzada. Las Figuras 5A y 5B muestran el posicionamiento de la cadena lateral de carbohidrato a Asn-38 en la apoestructura H3 HA (A) y en la estructura unida de la variante 3 de FI6 (B). Las Figuras 5C y 5D muestran la orientación de HA1 Trp-21 en diferentes complejos de anticuerpos. El panel C muestra un Phe-100D del bucle HCDR3 de la variante 3 de FI6 (referido en la figura como FI6) interactuando con el subdominio F de H1 o H3 de HA. El panel D muestra el bucle HCDR2 de los anticuerpos F10 y CR6261, mostrando Phe-55 y Phe-54 respectivamente, hacia Trp-21 de H5 HA.
Figura 6: muestra la superficie de contacto de la variante 3 de FI6 en HA. Los cuatro paneles muestran las huellas (contorneadas con una línea negra) sobre HA de la variante 3 de FI6, anticuerpos CR6261 y F10; los tres monómeros HA se muestran en blanco, gris claro u oscuro. Figura 6A: huella de contacto para FI6 variante 3/H1 sin escindir; Figura 6B: FI6 variante 3/H3 escindida; Figura 6C: CR6261/H5; y Figura 6D: F10/H5. A diferencia de CR6261 y F10, la variante 3 de FI6 hace contacto con dos monómeros HA. Para los complejos de la variante 3 de FI6, se marcan los sitios de glicosilación en la interfaz anticuerpo/HA.
Figura 7: muestra los residuos (en negrita, numeración de Kabat) en contacto con HA en la variante 3 de FI6, cadenas VH y VK.
Figura 8: muestra que la variante 3 de FI6 confiere protección en modelos de ratón de infección por el virus de la influenza A. La Figura 8A muestra las curvas de supervivencia y la Figura 8B muestra la pérdida de peso corporal de ratones BALB/c (cinco por condición experimental) que recibieron diferentes dosis de FI6 variante 3 iv tres horas antes de infección intranasal con 10 MLD50 (50% de la dosis letal en ratones) del virus H1N1 A/PR/8/34. La Figura 8C muestra la pérdida de peso corporal de ratones (diez por condición experimental) que recibieron diferentes dosis de FI6 variante 3 iv tres horas antes de la infección con 3x105 pfu del virus H3N2 HK-x31. Se muestra un experimento representativo de los tres que se realizaron. También se muestran la curva de supervivencia (Figura 8D) y la pérdida de peso corporal (Figura 8E) de ratones (cinco por condición experimental) que recibieron 15 mg/kg de la variante 3 de FI6 iv en el día 0 (3 horas antes de la infección) o en los días 1, 2 y 3 después de la infección con 10 MLD50 de virus A/PR/8/34. Se muestra un experimento representativo de los dos realizados. Se muestran los valores medios SD. Las Figuras 8F y 8G muestran las curvas de supervivencia de ratones (diez por condición experimental) que recibieron 10 mg/kg (F) o 3 mg/kg (G) de la variante 2 de FI6 (FI6-v2), FI6-v2 KA (sin unión al complemento), FI6-v2 LALA (sin complemento y unión a FcR) o un control de anticuerpos un día antes de la infección con el virus A/PR/8/34.
Figura 9: muestra los títulos de virus pulmonares en ratones tratados con la variante 3 de FI6 después del desafío letal con H1N1 PR/8/34. Los ratones BALB/c (cuatro ratones por condición experimental) recibieron una inyección iv de 15 mg/kg de la variante 3 de FI6 o un anticuerpo de control (HJ16, específico para VIH-1) tres horas antes (día 0) o 1, 2 o 3 días después de la infección in con 10 MLD50 de H1N1 PR/8/34. Los títulos víricos se determinaron 4 días después de la infección en los pulmones. El virus era indetectable en el cerebro. Los datos se muestran en forma de cajas y bigotes, donde las cajas se extienden desde el percentil 25 al 75, con una línea horizontal en la mediana. Los bigotes arriba y debajo de las cajas indican los valores extremos. Los resultados del análisis estadístico de la prueba t de Student se indican como * para p <0,05, y como *** para p <0,001.
Figura 10: muestra que la unión de la variante 3 de FI6 a la región del tallo HA interfiere con la escisión de HA0 mediada por proteasa. Se incubó HA recombinante de aislado de H1 NC/99 con la variante 3 de FI6, FE17 (un anticuerpo humano que reconoce el sitio Ca2 en la cabeza globular de HA) o con un anticuerpo de control. La mezcla de anticuerpos HA se expuso luego a TPCK tratado con tripsina durante 5, 10 o 20 minutos a 37°C. Las muestras se procesaron luego en un gel de poliacrilamida y se realizaron transferencias Western utilizando un mAb humano biotinilado (F032) que reconoce HA2 y HA0 de todas las cepas de influenza A en condiciones de desnaturalización. Se muestra la banda HA0. Se muestra un experimento representativo de cada tres.
DESCRIPCIÓN DETALLADA
La invención se refiere, en parte, al descubrimiento y aislamiento, a partir de individuos que estaban vacunados con la vacuna contra la gripe estacional A, de anticuerpos humanos naturales que neutralizan ampliamente el virus de la influenza A de diferentes subtipos, así como a epítopos a los cuales los anticuerpos aquí descritos se unen. Dichos anticuerpos son deseables, ya que solo se requiere uno o unos pocos anticuerpos para neutralizar diferentes subtipos del virus de la influenza A. Además, los anticuerpos heterosubtípicos ampliamente neutralizantes se producen a títulos altos para reducir los costes de producción de los medicamentos que comprenden los anticuerpos. Además, los epítopos reconocidos por dichos anticuerpos pueden formar parte de una vacuna capaz de inducir una amplia protección contra ambos aislados pandémicos tanto estacionales como candidatos de diferentes subtipos.
Como se discutió anteriormente, la proteína HA se sintetiza como un polipéptido precursor trimérico HA0 que comprende tres monómeros idénticos (homo-trímero). Cada monómero puede o no estar separado independientemente de los otros dos monómeros. Con la escisión postraduccional, cada monómero forma dos polipéptidos, HA1 y HA2, que de otro modo están unidos por un solo enlace disulfuro. Las cadenas pesada y ligera de los anticuerpos de la invención entran en contacto con dos de los tres monómeros del trímero de HA. Los monómeros en contacto con los anticuerpos de la invención pueden estar escindidos o no escindidos. Con fines de claridad y para comprender las representaciones esquemáticas de las figuras, nos referimos a los dos monómeros contactados por los anticuerpos de la invención como monómero proximal y monómero derecho distal.
Tal como se usa aquí, los términos "fragmento de unión a antígeno", "fragmento" y "fragmento de anticuerpo" se emplean indistintamente para referirse a cualquier fragmento de un anticuerpo que retiene la actividad de unión a antígeno del anticuerpo. Ejemplos de fragmentos de anticuerpos incluyen un
anticuerpo de cadena simple, Fab, Fab', F(ab')2, Fv o scFv. Además, el término "anticuerpo" como se usa aquí incluye tanto anticuerpos como fragmentos de unión a antígeno de los mismos.
Tal como se usa aquí, un "anticuerpo neutralizante" es aquel que puede neutralizar, es decir, prevenir, inhibir, reducir, impedir o interferir con la capacidad de un patógeno para iniciar y/o perpetuar una infección en un huésped. Las expresiones "anticuerpo neutralizante" y "un anticuerpo que neutraliza" o "anticuerpos que neutralizan" se usan aquí indistintamente. Estos anticuerpos se pueden usar solos o en combinación, como agentes profilácticos o terapéuticos en la formulación apropiada, en asociación con la vacunación activa, como herramienta diagnóstica o como herramienta de producción como se describe aquí.
Los estudios de cristalografía de rayos X de los anticuerpos heterosubtípicos específicos del Grupo 1 co cristalizados con H1 y H5 de HA conocidos en la técnica muestran que los anticuerpos interactúan con un solo monómero del trímero HA. Además, los estudios muestran que los anticuerpos entran en contacto con el HA solo con los residuos CDR de la cadena pesada, pero no de la cadena ligera. En contraste, los anticuerpos o fragmentos de unión a antígeno de la invención entran en contacto, no con uno, sino con dos de los tres monómeros de HA. Además, los anticuerpos de la invención ponen en contacto e1HA con residuos de CDR tanto de cadena pesada como de cadena ligera. Además, la naturaleza de las interacciones creadas por los anticuerpos o los fragmentos de unión a antígeno de la invención con e1HA son marcadamente diferentes a las creadas mediante otros anticuerpos CR6261 y F10. La diferencia más notable es que la interacción de los anticuerpos de la invención con el surco hidrófobo de1HA está mediada únicamente por CDR3 de cadena pesada (HCDR3), mientras que para CR6261 y F10, los tres HCDR están involucrados en la unión.
En una realización, las cadenas pesadas de los anticuerpos o de los fragmentos de unión a antígeno de la invención entran en contacto con residuos aminoácidos en el monómero proximal y las cadenas ligeras de los anticuerpos o de los fragmentos de unión a antígeno de la invención entran en contacto con residuos aminoácidos tanto en el monómero proximal como en el monómero distal derecho del trímero HA. Los monómeros en contacto con los anticuerpos de la invención, es decir, el monómero proximal y el monómero distal derecho, pueden no escindirse o pueden escindirse para formar los polipéptidos HA1 y HA2. En una realización, el monómero proximal y el monómero distal derecho se escinden. En otra realización, el monómero proximal y el monómero distal derecho no están escindidos.
Los anticuerpos y los fragmentos de unión a antígeno de la invención se unen específicamente a un epítopo que se conserva entre los 16 HA diferentes de los 16 subtipos del virus de la influenza A. En una realización, los anticuerpos o los fragmentos de unión a antígeno de la invención se unen a un epítopo que comprende el residuo aminoácido en la posición 329 de HA1 y los residuos aminoácido en las posiciones 1,2, 3 y 4 de HA2, donde el HA1 y HA2 están presentes en un monómero no escindido del trímero de HA.
En otra realización, las cadenas pesadas de los anticuerpos o fragmentos de unión a antígeno de la invención entran en contacto con el residuo aminoácido en la posición 318 en HA1 y los residuos aminoácidos en las posiciones 18, 19, 20, 21, 38, 41, 42, 45, 49 , 53 y 57 en HA2 del monómero derecho proximal o distal. Los monómeros pueden ser escindidos o escindidos.
En otra realización más, las cadenas ligeras de los anticuerpos o fragmentos de unión a antígeno de la invención entran en contacto con los residuos aminoácidos en las posiciones 38, 39 y 43 en HA2 del
monómero proximal no escindido y con los residuos aminoácidos en las posiciones 327, 328 y 329 en HA1 y 1, 2, 3 y 4 en HA2 del monómero derecho distal no escindido.
En otra realización más, los anticuerpos y los fragmentos de unión a antígeno de la invención se unen específicamente a un epítopo que comprende el residuo aminoácido en la posición 318 de HA1 y los residuos aminoácidos en las posiciones 18, 19, 20, 21, 38, 39, 41, 42, 43, 45, 48, 49, 53, 56 y 57 de1HA2 del monómero proximal no escindido. Además, los anticuerpos se unen específicamente a un epítopo que comprende los residuos aminoácidos en las posiciones 327, 328, 329 de1HA1 y los residuos aminoácidos en las posiciones 1,2, 3 y 4 del polipéptido HA2 del monómero no escindido distal drecho.
En otra realización, las cadenas ligeras de los anticuerpos o fragmentos de unión a antígeno de la invención entran en contacto con los residuos aminoácidos en las posiciones 38, 39, 42 y 46 en HA2 del monómero proximal y los residuos aminoácidos en las posiciones 321 y 323 en HA1 y 7 y 11 en HA2 del monómero distal derecho. En esta realización, tanto los monómeros derechos proximales como los distales se escinden.
En otra realización más, los anticuerpos y los fragmentos de unión a antígeno de la invención se unen específicamente a un epítopo que comprende el residuo aminoácido en la posición 318 de HA1 y los residuos aminoácido en las posiciones 18, 19, 20, 21, 38, 39, 41, 42, 45, 46, 49, 52, 53 y 57 de HA2 del monómero proximal escindido, así como los residuos aminoácidos en las posiciones 321 y 323 de HA1 y los residuos aminoácidos en las posiciones 7 y 11 de HA2 del monómero derecho distal escindido.
Como se muestra aquí, los anticuerpos o fragmentos de unión a antígeno de la invención son capaces de unirse específicamente a las HA de todos los 16 subtipos del virus de la influenza A. En una realización, los anticuerpos de la invención se unen específicamente a un HA del virus de la influenza A de los subtipos H1, H2, H3, H4, H5, H6, H7, H8, H9, H10, HI1, H12, H13, H14, H15 y H16.
En otra realización de la invención, la invención proporciona anticuerpos que tienen altos títulos de producción. Como ejemplo, una vez que se aislaron dos anticuerpos muy similares, la variante 1 de FI6 y la variante 2 de FI6, se sintetizaron varias variantes de los anticuerpos (en particular, variantes de la variante 2 de FI6) para mejorar la producción en células transfectadas. En una realización, los anticuerpos o fragmentos de unión a antígeno de la invención se producen en células transfectadas a títulos al menos 1,5 veces más altos que el título al que se produce la variante 2 de FI6. En otra realización, los anticuerpos de la invención se producen a títulos al menos 1,8, 2, 2,2, 2,5, 2,7, 3, 3,2, 3,4, 3,6, 3,8, 4, 4,2, 4,4, 4,6, 4,8, 5, 5,3, 5,6 o 6 veces más altos que el título al que se produce la variante 2 de FI6. En algunas realizaciones, los anticuerpos o fragmentos de unión a antígeno de la invención se producen en células transfectadas a títulos al menos 3, al menos 4 o al menos 4,5 veces más altos que el título al que se produce la variante 2 de FI6.
Así, en una realización, la invención proporciona un anticuerpo aislado, o un fragmento de unión a antígeno del mismo, que neutraliza la infección de un subtipo del grupo 1 y un subtipo del grupo 2 del virus de la influenza A y se une específicamente a un epítopo en la región del tallo de un trímero HA de la influenza A, donde la cadena pesada y ligera del anticuerpo, o su fragmento de unión al antígeno, entran en contacto con los aminoácidos en un primer monómero proximal y un segundo monómero derecho distal del trímero HA, y donde el anticuerpo, o el fragmento de unión a antígeno del mismo, se produce en células
transfectadas a títulos superiores a, por ejemplo, al menos 3 veces más alto, que el título al que se produce la variante 2 de FI6, donde dicho anticuerpo, o fragmento de unión a antígeno del mismo, comprende: las secuencias CDR1, CDR2 y CDR3 de cadena pesada como se establecen en las SEQ ID NO: 1, 41 y 43, respectivamente, o como se establecen en las SEQ ID NO: 1,41 y 42, respectivamente; y (ii) las secuencias de la cadena ligera CDR1, CDR2 y CDR3 como se establecen en las SEQ ID NO: 4, 5 y 6, respectivamente, o como se establecen en las SEQ ID NO: 44, 5 y 6, respectivamente.
Como se describe aquí, las células transfectadas pueden ser cualquier célula ahora conocida o descubierta posteriormente por un experto en la técnica para expresar las secuencias de ácido nucleico que codifican los anticuerpos de la invención. Ejemplos de tales células incluyen células huésped de mamífero, como células CHO, HEK293T, PER.C6, NS0, células de mieloma o hibridoma. Además, las células pueden transfectarse de forma transitoria o estable. El tipo de transfección, así como el tipo de célula adecuado para su uso en la transfección, está dentro del alcance del experto en la técnica.
En otra realización, el anticuerpo o los fragmentos de unión a antígeno de la invención se unen específicamente a un polipéptido que comprende la secuencia de aminoácidos de cualquiera de las SEQ ID NO: 37, 38, 39 o 40.
Los anticuerpos monoclonales humanos, los clones de células B inmortalizadas o las células huésped transfectadas que secretan los anticuerpos de la invención, y el ácido nucleico que codifica los anticuerpos de la invención también se incluyen dentro del alcance de la invención. Tal como se usa aquí, el término "amplia especificidad" se emplea para referirse a un anticuerpo o un fragmento de unión a antígeno de la invención que puede unirse y/o neutralizar uno o más subtipos del grupo 1 y uno o más subtipos del grupo 2 del virus de la influenza A.
El anticuerpo o los fragmentos de unión a antígeno de la invención neutralizan uno o más virus de influenza A del grupo 1 (H1, H2, H5, H6, H8, H9, H11, H12, H13 y H16 y sus variantes) y uno o más subtipos del virus de la influenza A del grupo 2 (H3, H4, H7, H10, H14 y H15 y sus variantes). En una realización, subtipos ejemplares del grupo 1 incluyen H1, H2, H5, H6 y H9 y subtipos ejemplares del grupo 2 incluyen H3 y H7.
El anticuerpo y el fragmento de anticuerpo de la invención es capaz de neutralizar diversas combinaciones de subtipos de virus de la influenza A. En una realización, el anticuerpo puede neutralizar los subtipos H1 y H3 del virus de la influenza A o los subtipos H2 y H3 o los subtipos H3 y H5 o los subtipos H3 y H9 o los subtipos H1 y H7 o los subtipos H2 y H7 o los subtipos H5 y H7 o los subtipos H7 y H9.
En otra realización, el anticuerpo y el fragmento de anticuerpo de la invención pueden neutralizar los subtipos H1, H2 y H3 del virus de la influenza A o los subtipos H1, H3 y H5 o los subtipos H1, H3 y H9 o los subtipos H2, H3 y H5 o H2, H3 y H9 o los subtipos H3, H5 y H9 o los subtipos H1, H2 y H7 o los subtipos H1, H5 y H7 o los subtipos H1, H7 y H9 o los subtipos H2, H5 y H7 o los subtipos H2, H7 y H9 o los subtipos H5, H7 y H9 o los subtipos H1, H3 y H7 o los subtipos H2, H3 y H7 o los subtipos H3, H5 y H7 o los subtipos H3, H7 y H9.
En otra realización más, el anticuerpo puede neutralizar el virus de la influenza A de los subtipos H1, H2, H3 y H7 o los subtipos H1, H3, H5 y H7 o los subtipos H1, H3, H7 y H9 o los subtipos H2, H3, H5 y H7 o
los subtipos H2, H3, H7 y H9 o los subtipos H3, H5, H7 y H9 o los subtipos H1, H2, H3 y H5 o los subtipos H1, H2, H3 y H9 o los subtipos H1, H3, H5 y H9 o los subtipos H2, H3, H5 y H9 o los subtipos H1, H2, H5 y H7 o los subtipos H1, H2, H7 y H9 o los subtipos H1, H5, H7 y H9 o los subtipos H2, H5, H7 y H9.
En otra realización más, el anticuerpo de la invención puede neutralizar los subtipos H1, H2, H3, H5 y H7 del virus de la influenza A o los subtipos H1, H2, H3, H7 y H9 o los subtipos H1, H3, H5, H7 y H9 o los subtipos H2, H3, H5, H7 y H9 o los subtipos H1, H2, H3, H5 y H9 o los subtipos H1, H2, H5, H7 y H9 o los subtipos H1, H2, H3, H5, H7 y H9. En otra realización más, el anticuerpo y los fragmentos de unión a antígeno de la invención neutralizan una o más de las combinaciones anteriores, además de neutralizar el subtipo H6 del virus de la influenza A.
El anticuerpo y el fragmento de unión a antígeno de la invención tienen una alta potencia neutralizante. La concentración del anticuerpo de la invención requerida para una neutralización del 50% del virus de la influenza A puede ser, por ejemplo, aproximadamente 50 pg/ml o inferior. En una realización, la concentración del anticuerpo de la invención requerida para una neutralización del 50% del virus de la influenza A es aproximadamente 50, 45, 40, 35, 30, 25, 20, 17,5, 15, 12,5, 11, 10, 9, 8, 7, 6, 5, 4,5, 4, 3,5, 3, 2,5, 2, 1,5 o aproximadamente 1 pg/ml o inferior. En otra realización, la concentración del anticuerpo de la invención requerida para una neutralización del 50% del virus de la influenza A es aproximadamente 0,9, 0,8, 0,75, 0,7, 0,65, 0,6, 0,55, 0,5, 0,45, 0,4, 0,35, 0,3, 0,25, 0,2, 0,15, 0,1, 0,075, 0,05, 0,04, 0,03, 0,02, 0,01, 0,008, 0,006, 0,004, 0,003, 0,002 o aproximadamente 0,001 pg/ml o inferior. Esto significa que solo se requieren bajas concentraciones de anticuerpos para la neutralización del 50% del virus de la influenza A. La especificidad y la potencia se pueden medir utilizando un ensayo de neutralización estándar como es conocido por el experto en la técnica.
En una realización, el anticuerpo, por ejemplo un anticuerpo aislado o un anticuerpo purificado, se une específicamente a un epítopo conservado en la región del tallo de HA de los subtipos de virus de la influenza A del grupo 1 y del grupo 2 e interfiere con la replicación o la propagación viral. El anticuerpo, por ejemplo un anticuerpo aislado o un anticuerpo purificado, también puede unirse específicamente a un epítopo conservado en la región del tallo de HA de los subtipos de grupo 1 y grupo 2 e inhibir la entrada del virus en una célula. Sin estar ligado a ninguna teoría, en una realización ilustrativa, el anticuerpo o los fragmentos de unión a antígeno de la invención se unen a un epítopo conservado en la región del tallo del virus de la influenza A e inhiben la entrada del virus en una célula al interferir con la etapa de fusión. En una realización, el anticuerpo o los fragmentos de unión a antígeno de la invención limitan la propagación del virus al reclutar células asesinas de complemento y que expresan FcR y mediar en la citotoxicidad celular dependiente de anticuerpos (ADCC). Un epítopo o determinante antigénico de una proteína corresponde a aquellas partes de la molécula que son específicamente reconocidas por el sitio de unión (o parátopo) de un anticuerpo. Así, los epítopos son entidades relacionales que requieren parátopos complementarios para su reconocimiento operacional. Un epítopo que se conserva entre diferentes variantes de una proteína significa que el mismo parátopo puede reconocer específicamente estas diferentes variantes al contactar las mismas partes de las moléculas.
Los anticuerpos de la invención pueden ser monoclonales, por ejemplo anticuerpos monoclonales humanos, o anticuerpos recombinantes. La invención también proporciona fragmentos de los anticuerpos de la invención, en particular fragmentos que retienen la actividad de unión a antígeno de los anticuerpos.
Aunque la descripción, incluidas las reivindicaciones, puede, en algunos momentos, referirse explícitamente a fragmentos de unión a antígeno, fragmentos de anticuerpos, variantes y/o derivados de anticuerpos, se entiende que el término "anticuerpo" o "anticuerpo de la invención" incluye todas las categorías de anticuerpos, a saber, fragmento(s) de unión a antígeno, fragmento(s) de anticuerpo, variante(s) y derivado(s) de anticuerpos.
En una realización, los anticuerpos y fragmentos de anticuerpos de la invención neutralizan una combinación de dos o más subtipos de virus de la influenza A del grupo 1 y 2. Subtipos ilustrativos del virus de la influenza A incluyen, entre otros, H5N1 (A/Vietnam/1203/04), H1N1 (A/Nueva Caledonia/20/99), H1N1 (A/Salomon Island/3/2006), H3N2 (A/Wyoming/3/03) y H9N2 (A/chicken/Hong Kong/G9/97). En otra realización, los anticuerpos neutralizan y/o son específicos para una combinación de 3, 4, 5, 6, 7 o más subtipos de virus de la influenza A del grupo 1 y 2.
En una realización ilustrativa, la invención comprende un anticuerpo, o un fragmento de anticuerpo del mismo, que es específico para los subtipos H1 y H3 del virus de la influenza A (por ejemplo H1N1 y H3N2); o H1, H3, H5 y H9 (por ejemplo H1N1, H3N2, H5N1 y H9N2). En otra realización más, el anticuerpo o sus fragmentos de anticuerpo son específicos para H1, H3, H5, H7 y H9 (por ejemplo H1N1, H3N2, H5N1, H7N1, H7N7, H9N2). Otros ejemplos de combinaciones de subtipos del virus de la influenza A se proporcionan anteriormente en la solicitud.
Los números de SEQ ID para la secuencia de aminoácidos de la región variable de la cadena pesada (VH) y la región variable de la cadena ligera (VL) de los anticuerpos ilustrativos de la descripción que comprende la invención, así como los números de SEQ ID para las secuencias de ácido nucleico que los codifican se citan en Tabla 1.
Tabla 1: Números de SEQ ID para los polipéptidos y polinucleótidos Vh y Vl para anticuerpos neutralizantes del virus de la influenza A ilustrativos

En una realización, un anticuerpo o fragmento de anticuerpo de la descripción que comprende la invención incluye una región variable de cadena pesada con una secuencia de aminoácidos que es aproximadamente un 70%, 75%, 80%, 85%, 90%, 95%, 97%, 98%, 99% o 100% idéntica a la secuencia citada en cualquiera de las SEQ ID NO: 13, 33, 55, 59, 29 o 35. En otra realización, un anticuerpo o fragmento de anticuerpo de la descripción que comprende la invención incluye una región variable de cadena ligera con una secuencia de aminoácidos que es aproximadamente un 70%, 75%, 80%, 85%, 90%, 95%, 97%, 98%, 99% o 100% idéntica a la secuencia citada en las SEQ ID NO: 14, 57, 61 o 30.
En otra realización, la región variable de la cadena pesada de un anticuerpo de la descripción que comprende la invención puede estar codificada por un ácido nucleico que tiene una secuencia que es aproximadamente un 70%, 75%, 80%, 85%, 90%, 95%, 97%. , 98%, 99% o 100% idéntica a la secuencia citada en cualquiera de las SEQ ID NO: 15, 34, 56, 60, 31 o 36. En otra realización más, la región variable de la cadena ligera de un anticuerpo la descripción que comprende la invención puede estar codifica por un ácido nucleico con una secuencia que es aproximadamente un 70%, 75%, 80%, 85%, 90%, 95%, 97%, 98%, 99% o 100% idéntica a la secuencia citada en las SEQ ID NO: 16, 58, 62 o 32.
Las CDR de las cadenas pesadas de los anticuerpos se denominan CDRH1 (o HCDR1), CDRH2 (o HCDR2) y CDRH3 (o HCDR3), respectivamente. De manera similar, las CDR de las cadenas ligeras de anticuerpos se denominan CDRL1 (o LCDR1), CDRL2 (o LCDR2) y CDRL3 (o LCDR3), respectivamente. Las posiciones de los aminoácidos de laa CDR se definen de acuerdo con el sistema de numeración IMGT como: CDR1 - IMGT Posiciones 27 a 38, CDR2 - IMGT Posiciones 56 a 65 y CDR3 - IMGT Posiciones 105 a 117.
La Tabla 2 proporciona los números de SEQ ID para las secuencias de aminoácidos de las seis CDR de las cadenas pesada y ligera, respectivamente, de los anticuerpos ilustrativos de la descripción que comprende la invención.
Tabla 2: Números de SEQ ID para los polipéptidos de CDR de anticuerpos neutralizantes del virus de la influenza A ilustrativos

En una realización, un anticuerpo o fragmento de anticuerpo de descripción que comprende la invención incluye al menos una CDR con una secuencia que tiene al menos un 95% de identidad de secuencia con cualquiera de las SEC ID NO: 1-6, 41-44 o 17-22.
En otra realización, la descripción que comprende la invención proporciona un anticuerpo que comprende una cadena pesada que incluye una o más CDR de cadena pesada (es decir, una, dos o las tres) de la variante 1 de FI6, la variante 2 de FI6, la variante 3 de FI6, la variante 4 de FI6, la variante 5 de FI6, la variante 1 de FI28 o la variante 2 de FI28. En una realización ilustrativa, el anticuerpo o el fragmento de unión a antígeno de la descripción que comprende la invención incluye una CDR1 de cadena pesada con la secuencia de aminoácidos de SEQ ID NO: 1 o SEQ ID NO: 17; una CDR2 de cadena pesada con la SEQuencia de aminoácidos de SEQ ID NO: 2, SEQ ID NO: 41 o SEQ ID NO: 18; y una CDR3 de cadena pesada con la SEQuencia de aminoácidos de SEQ ID NO: 3, SEQ ID NO: 42, SEQ ID NO: 43 o SEQ ID
NO: 19. En ciertas realizaciones, un anticuerpo o fragmento de anticuerpo como se describe aquí comprende una cadena pesada que comprende (i) la SEQ ID NO: 1 para CDRH1, la SEQ ID NO: 2 para CDRH2 y la SEQ ID NO: 3 para CDRH3, (ii) la SEQ ID NO: 1 para CDRH1, la SEQ ID NO: 41 para CDRH2 y la SEQ ID NO: 42 para CDRH3, (iii) la SEQ ID NO: 1 para CDRH1, la SEQ ID NO: 41 para CDRH2 y la SEQ ID NO: 43 para CDRH3, o iv) la SEQ ID NO: 17 para CDRH1, la SEQ ID NO: 18 para CDRH2 y la SEQ ID NO: 19 para CDRH3.
La descripción que comprende la invención también proporciona un anticuerpo que comprende una cadena ligera que comprende una o más CDR de cadena ligera (es decir, una, dos o las tres) de la variante 1 de FI6, la variante 2 de FI6, la variante 3 de FI6, la variante 4 de FI6, la variante 5 de FI6, la variante 1 de FI28 o la variante 2 de FI28. En una realización ilustrativa, el anticuerpo o el fragmento de unión a antígeno de descripción que comprende la invención comprende una CDR1 de cadena ligera con la secuencia de aminoácidos de SEQ ID NO: 4, SEQ ID NO: 44 o SEQ ID NO: 20; una CDR2 de cadena ligera con la SEQuencia de aminoácidos de SEQ ID NO: 5 o SEQ ID NO: 21; y una CDR3 de cadena ligera con la SEQuencia de aminoácidos de SEQ ID NO: 6 o SEQ ID NO: 22. En ciertas realizaciones, un anticuerpo como se describe aquí comprende una cadena ligera que comprende (i) la SEQ ID NO: 4 para CDRL1, la SEQ ID NO: 5 para CDRL2 y la SEQ ID NO: 6 para CDRL3, (ii) la SEQ ID NO: 44 para CDRL1, SEQ ID NO: 5 para CDRL2 y SEQ ID NO: 6 para CDRL3 o (iii) SEQ ID NO: 20 para CDRL1, SEQ ID NO: 21 para CDRL2 y SEQ ID NO: 22 para CDRL3.
En una realización, un anticuerpo de la descripción no reivindicado, o un fragmento de unión a antígeno del mismo, comprende todas las CDR de la variante 1 del anticuerpo FI6 indicadas en la Tabla 2 y neutraliza la infección por el virus de la influenza A. En otra realización, un anticuerpo de la descripción no reivindicado, o un fragmento de unión a antígeno del mismo, comprende todas las CDR de la variante 2 del anticuerpo FI6 indicadas en la Tabla 2 y neutraliza la infección por el virus de la influenza A. En otra realización, un anticuerpo de la invención, o un fragmento de unión a antígeno del mismo, comprende todas las CDR de la variante 3 del anticuerpo FI6 indicadas en la Tabla 2 y neutraliza la infección por el virus de la influenza A. En otra realización, un anticuerpo de la invención, o un fragmento de unión a antígeno del mismo, comprende todas las CDR de la variante 4 del anticuerpo FI6 indicadas en la Tabla 2 y neutraliza la infección por el virus de la influenza A. En otra realización, un anticuerpo de la invención, o un fragmento de unión a antígeno del mismo, comprende todas las CDR de la variante 5 del anticuerpo FI6 indicadas en la Tabla 2 y neutraliza la infección por el virus de la influenza A.
En otra realización más, un anticuerpo de la descripción no reivindicado, o un fragmento de unión a antígeno del mismo, comprende todas las CDR de la variante 1 del anticuerpo FI28 indicadas en la Tabla 2 y neutraliza la infección por el virus de la influenza A. En otra realización más, un anticuerpo de descripción no reivindicado, o un fragmento de unión a antígeno del mismo, comprende todas las CDR de la variante 2 del anticuerpo FI28 indicadas en la Tabla 2 y neutraliza la infección por el virus de la influenza A.
Los ejemplos de anticuerpos de la descripción que comprende la invención incluyen, pero no se limitan a, variante 1 de FI6, variante 2 de FI6, variante 3 de FI6, variante 4 de FI6, variante 5 de FI6, variante 1 de FI28 y variante 2 de FI28.
La descripción además describe un anticuerpo, o fragmento del mismo, que se une al mismo epítopo que un anticuerpo de la invención, o un anticuerpo que compite con un anticuerpo o fragmento de unión a antígeno de la descripción que comprende la invención.
Los anticuerpos aquí descritos también incluyen moléculas de anticuerpos híbridas que comprenden una o más CDR de un anticuerpo de la invención y una o más CDR de otro anticuerpo para el mismo epítopo. En una realización, dichos anticuerpos híbridos comprenden tres CDR de un anticuerpo de la invención y tres CDR de otro anticuerpo para el mismo epítopo. Anticuerpos híbridos ilustrativos comprenden i) las tres CDR de cadena ligera de un anticuerpo de la invención y las tres CDR de cadena pesada de otro anticuerpo para el mismo epítopo, o ii) las tres CDR de cadena pesada de un anticuerpo de la invención y las tres CDR de cadena ligera de otro anticuerpo para el mismo epítopo.
Los anticuerpos variantes también están incluidos dentro del alcance de la invención. Así, las variantes de las secuencias citadas en la solicitud también están incluidas dentro del alcance de la invención. Tales variantes incluyen variantes naturales generadas por mutación somática in vivo durante la respuesta inmune o in vitro tras el cultivo de clones de células B inmortalizadas. Alternativamente, pueden surgir variantes debido a la degeneración del código genético, como se mencionó anteriormente, o pueden producirse debido a errores en la transcripción o la traducción.
Se pueden obtener variantes adicionales de las secuencias de anticuerpos de afinidad y/o potencia mejoradas usando métodos conocidos en la técnica y se incluyen dentro del alcance de la invención. Por ejemplo, pueden usarse sustituciones de aminoácidos para obtener anticuerpos con afinidad mejorada adicional. Alternativamente, puede emplearse la optimización de codón de la secuencia de nucleótidos para mejorar la eficiencia de la traducción en sistemas de expresión para la producción del anticuerpo. Además, los polinucleótidos que comprenden una secuencia optimizada para la especificidad del anticuerpo o la actividad neutralizante mediante la aplicación de un método de evolución dirigida a cualquiera de las secuencias de ácido nucleico de la invención también están dentro del alcance de la invención.
En una realización, las secuencias de anticuerpos variantes pueden compartir un 70% o más (es decir, 75%, 80%, 85%, 90%, 95%, 97%, 98%, 99% o más) de identidad de secuencia de aminoácidos con las secuencias citadas en la solicitud. En algunas realizaciones, dicha identidad de secuencia se calcula con respecto a la longitud completa de la secuencia de referencia (es decir, la secuencia citada en la solicitud). En otras realizaciones, el porcentaje de identidad, como se menciona aquí, se determina utilizando la versión 2.1.3 de BLAST utilizando los parámetros predeterminados especificados por el NCBI (National Center for Biotechnology Information; http: //www.ncbi.nlm.nih).gov/) [matriz Blosum 62; penalización de apertura de abertura = 11 y penalización de extensión de separación = 1].
En otro aspecto, la descripción que comprende la invención también incluye secuencias de ácido nucleico que codifican parte o la totalidad de las cadenas ligeras y pesadas y las CDR de los anticuerpos de la presente invención. Se proporcionan aquí secuencias de ácido nucleico que codifican parte o la totalidad de las cadenas ligeras y pesadas y las CDR de anticuerpos ilustrativos de la invención. La Tabla 1 proporciona los números de SEQ ID para las secuencias de ácido nucleico que codifican las regiones variables de cadena pesada y ligera de anticuerpos ilustrativos de la descripción que comprende la invención. Por ejemplo, las secuencias de ácido nucleico aquí descritas incluyen la SEQ ID NO: 15 (que codifica la región variable de la cadena pesada de la variante 1 de FI6), la SEQ ID NO: 16 (que codifica la
región variable de la cadena ligera de la variante 1 de FI6 y la variante 2 de FI6), la SEQ ID NO: 34 (que codifica la región variable de la cadena pesada de la variante 2 de FI6), la SEQ ID NO: 56 (que codifica la región variable de la cadena pesada de la variante 3 de FI6), la SEQ ID NO: 58 (que codifica la variante 3 de FI6 y la región variable de la cadena ligera de la variante 4 de FI6 ), la SEQ ID NO: 60 (que codifica la región variable de la cadena pesada de la variante 4 de FI6 y la variante 5 de FI6), la SEQ ID NO: 62 (que codifica la región variable de la cadena ligera de la variante 5 de FI6), la SEQ ID NO: 31 (que codifica la región variable de cadena pesada de la variante 1 de FI28), la SEQ ID NO: 36 (que codifica la región variable de cadena pesada de la variante 2 de FI28) y la SEQ ID NO: 32 (que codifica la región variable de la variante 1 y la variante 2 de FI28).
La Tabla 3 proporciona los números de SEQ ID para las secuencias de ácido nucleico que codifican las CDR de anticuerpos ilustrativos de la descripción que comprende la invención. Debido a la redundancia del código genético, existirán variantes de estas secuencias que codifican las mismas secuencias de aminoácidos.
Tabla 3: Números de SEQ ID para los polinucleótidos de CDR de anticuerpos neutralizantes del virus de la influenza ilustrativos

En una realización, las secuencias de ácido nucleico según descripción que comprende la invención incluyen secuencias de ácido nucleico que tienen al menos un 70%, al menos un 75%, al menos un 80%, al menos un 85%, al menos un 90%, al menos un 95%, al menos un 98%, o al menos el 99% de identidad con el ácido nucleico que codifica una cadena pesada o ligera de un anticuerpo de la descripción que comprende la invención. En otra realización, una secuencia de ácido nucleico de la descripción que comprende la invención tiene la secuencia de un ácido nucleico que codifica una CDR de cadena pesada o ligera de un anticuerpo de la descripción que comprende la invención. Por ejemplo, una secuencia de ácido nucleico según la descripción que comprende la invención comprende una secuencia que es al menos un 75% idéntica a las secuencias de ácido nucleico de SEQ ID NO: 7-12, 15, 16, 34, 23-28, 31, 32, 36, 45 54, 56, 58, 60 o 62.
Se incluyen además dentro del alcance de la invención los vectores, por ejemplo, vectores de expresión, que comprenden una secuencia de ácido nucleico de acuerdo con la invención. Las células transformadas con tales vectores también se incluyen dentro del alcance de la invención. Ejemplos de tales células incluyen células eucariotas, por ejemplo células de levadura, células animales o células vegetales. En una realización, las células son de mamífero, por ejemplo células humanas, CHO, HEK293T, PER.C6, NS0, mieloma o hibridoma.
Los anticuerpos monoclonales y recombinantes son particularmente útiles en la identificación y purificación de los polipéptidos individuales u otros antígenos contra los que se dirigen. Los anticuerpos de la invención tienen utilidad adicional en el sentido de que pueden emplearse como reactivos en inmunoensayos, radioinmunoensayos (RIA) o ensayos inmunoabsorbentes ligados a enzimas (ELISA). En estas aplicaciones, los anticuerpos pueden marcarse con un reactivo analíticamente detectable, como un radioisótopo, una molécula fluorescente o una enzima. Los anticuerpos también pueden usarse para la identificación y caracterización molecular (mapeo de epítopos) de antígenos.
Los anticuerpos de la invención pueden acoplarse a un fármaco para ser administrados a un sitio de tratamiento o acoplarse a una etiqueta detectable para facilitar la obtención de imágenes de un sitio que comprende células de interés, tales como células infectadas con el virus de la influenza A. Los métodos para acoplar anticuerpos a fármacos y etiquetas detectables son bien conocidos en la técnica, ya que son métodos para obtener imágenes usando etiquetas detectables. Los anticuerpos marcados pueden emplearse en una amplia variedad de ensayos, empleando una amplia variedad de marcadores. La detección de la formación de un complejo antígeno-anticuerpo entre un anticuerpo de la invención y un epítopo de interés (un epítopo del virus de la influenza A) puede facilitarse uniendo una sustancia detectable al anticuerpo. Medios de detección adecuados incluyen el uso de marcadores tales como radionúclidos, enzimas, coenzimas, fluorescentes, quimioluminiscentes, cromógenos, sustratos enzimáticos o cofactores, inhibidores enzimáticos, complejos de grupos prostéticos, radicales libres, partículas y colorantes. Ejemplos de enzimas adecuadas incluyen peroxidasa de rábano picante, fosfatasa alcalina, p-galactosidasa o acetilcolinesterasa; ejemplos de complejos de grupos prostéticos adecuados incluyen estreptavidina/biotina y avidina/biotina; ejemplos de materiales fluorescentes adecuados incluyen umbeliferona, fluoresceína, isotiocianato de fluoresceína, rodamina, diclorotriazinilamina-fluoresceína, cloruro de dansilo o ficoeritrina; un ejemplo de un material luminiscente es luminol; ejemplos de materiales bioluminiscentes incluyen luciferasa, luciferina y aequorina; y ejemplos de material radiactivo adecuado incluyen 125I, 131I, 35S o 3H. Dichos reactivos marcados pueden usarse en diversos ensayos bien conocidos, tales como radioinmunoensayos, inmunoensayos enzimáticos, por ejemplo ELISA, inmunoensayos fluorescentes, y similares. (Véase US 3.766.162, US 3.791.932, US 3.817.837 y US 4.233.402, por ejemplo).
Un anticuerpo de acuerdo con la invención puede estar conjugado con un residuo terapéutico tal como una citotoxina, un agente terapéutico o un ion metálico radioactivo o radioisótopo. Ejemplos de radioisótopos incluyen I-131, I-123, I-125, Y-90, Re-188, Re-186, At-211, Cu-67, Bi-212, Bi-213, Pd-109, Tc-99 e In-111. Dichos conjugados de anticuerpos pueden usarse para modificar una respuesta biológica dada; el residuo farmacológico no debe interpretarse como limitado a los agentes terapéuticos químicos clásicos. Por ejemplo, el residuo de fármaco puede ser una proteína o un polipéptido que posee una actividad biológica deseada. Dichas proteínas pueden incluir, por ejemplo, una toxina tal como abrina, ricina A, exotoxina de pseudomonas o toxina de la difteria.
Las técnicas para conjugar dicho residuo terapéutico con anticuerpos son bien conocidas. Ver, por ejemplo, Arnon et al. (1985) " Monoclonal Antibodies for Immunotargeting of Drugs in Cancer Therapy ", en Monoclonal Antibodies and Cancer Therapy, ed. Reisfeld et al. (Alan R. Liss, Inc.), pp. 243-256; ed. Hellstrom et al. ( 1987) " Antibodies for Drug Delivery ", en Controlled Drug Delivery, ed. Robinson et al. (2a ed. Marcel Dekker, Inc.), pp. 623-653; Thorpe (1985) " Antibody Carriers of Cytotoxic Agents in Cancer Therapy: A Review", en Monoclonal Antibodies '84: Biological and Clinical Applications, ed. Pinchera et al.,
pp. 475-506 (Editrice Kurtis, Milán, Italia, 1985); " Analysis, Results, and Future Prospective of the Therapeutic Use of Radiolabeled Antibody in Cancer Therapy", en Monoclonal Antibodies for Cancer Detection and Therapy, ed. Baldwin et al. (Academic Press, Nueva York, 1985), pp. 303-316; y Thorpe et al. (1982) Immunol. Rev. 62: 119-158.
Alternativamente, un anticuerpo, o fragmento de anticuerpo del mismo, puede estar conjugado con un segundo anticuerpo, o fragmento de anticuerpo del mismo, para formar un heteroconjugado de anticuerpo como se describe en la US 4.676.980. Además, se pueden usar enlazadores entre los marcadores y los anticuerpos de la invención (por ejemplo US 4.831.175). Los anticuerpos o los fragmentos de unión a antígeno de los mismos pueden marcarse directamente con yodo radiactivo, indio, ytrio u otra partícula radiactiva conocida en la técnica (por ejemplo US 5.595.721). El tratamiento puede consistir en una combinación de tratamiento con anticuerpos conjugados y no conjugados administrados simultáneamente o con posterioridad (por ejemplo WO00/52031; WO00/52473).
Los anticuerpos de la invención también pueden unirse a un soporte sólido. Además, los anticuerpos de la invención, o fragmentos de anticuerpos funcionales de los mismos, pueden modificarse químicamente mediante conjugación covalente con un polímero para, por ejemplo, aumentar su vida media circulante. Ejemplos de polímeros y métodos para unirlos a péptidos se encuentran en los documentos US 4.766.106, US 4.179.337, US 4.495.285 y US 4.609.546. En algunas realizaciones, los polímeros pueden seleccionarse entre polioles polioxietilados y polietilenglicol (PEG). El PEG es soluble en agua a temperatura ambiente y tiene la fórmula general: R(O-CH2-CH2)nO-R, donde R puede ser hidrógeno o un grupo protector tal como un grupo alquilo o alcanol. En una realización, el grupo protector puede tener entre 1 y 8 carbonos. En una realización adicional, el grupo protector es metilo. El símbolo n es un entero positivo. En una realización, n está entre 1 y 1.000. En otra realización, n está entre 2 y 500. En una realización, el PEG tiene un peso molecular promedio entre 1.000 y 40.000. En una realización adicional, el PEG tiene un peso molecular entre 2.000 y 20.000. En otra realización adicional, el PEG tiene un peso molecular entre 3.000 y 12.000. En una realización, el PEG tiene al menos un grupo hidroxilo. En otra realización, el PEG tiene un grupo hidroxilo terminal. En otra realización más, es el grupo hidroxilo terminal el que se activa para reaccionar con un grupo amino libre en el inhibidor. Sin embargo, se entenderá que el tipo y la cantidad de grupos reactivos pueden variar para lograr un conjugado covalente PEG/anticuerpo de la presente invención.
Los polioles polioxietilados solubles en agua también son útiles en la presente invención. Incluyen sorbitol polioxietilado, glucosa polioxietilada, glicerol polioxietilado (POG) y similares. En una realización, se usa POG. Sin estar limitado a ninguna teoría, debido a que el esqueleto glicerol del glicerol polioxietilado es el mismo esqueleto que ocurre naturalmente en, por ejemplo, animales y humanos en mono-, di-, triglicéridos, esta ramificación no se verá necesariamente como un agente extraño en el cuerpo. En algunas realizaciones, el POG tiene un peso molecular en el mismo intervalo que el PEG. Otro sistema de administración de fármacos que puede usarse para aumentar la vida media en circulación es el liposoma. En Gabizon et al. (1982), Cafiso (1981) y Szoka (1980) se describen métodos para preparar sistemas de administración de liposomas. Otros sistemas de administración de fármacos son conocidos en la técnica y se describen, por ejemplo, en Poznansky et al. (1980) y Poznansky (1984).
Los anticuerpos de la invención se pueden proporcionar en forma purificada. Típicamente, el anticuerpo estará presente en una composición que está esencialmente libre de otros polipéptidos, por ejemplo donde
menos del 90% (en peso), generalmente menos del 60% y más generalmente menos del 50% de la composición está formada por otros polipéptidos.
Los anticuerpos de la invención pueden ser inmunogénicos (o heterólogos) en huéspedes no humanos, por ejemplo en ratones. En particular, los anticuerpos pueden tener un idiotopo que es inmunogénico en huéspedes no humanos, pero no en un huésped humano. Los anticuerpos de la invención para uso humano incluyen aquellos que no pueden aislarse fácilmente de huéspedes tales como ratones, cabras, conejos, ratas, mamíferos no primates, etc. y generalmente no pueden obtenerse mediante humanización o a partir de xeno-ratones.
Los anticuerpos de la invención pueden ser de cualquier isotipo (por ejemplo IgA, IgG, IgM (es decir, una cadena pesada a, y o p), pero generalmente es IgG. Dentro del isotipo IgG, los anticuerpos pueden ser de las subclases IgG1, IgG2, IgG3 o IgG4. Los anticuerpos de la invención pueden tener una cadena ligera k o A.
Producción de anticuerpos
Los anticuerpos de acuerdo con la invención pueden prepararse mediante cualquier método conocido en la técnica. Por ejemplo, la metodología general para producir anticuerpos monoclonales utilizando la tecnología de hibridoma es bien conocida (Kohler, G. y Milstein, C., 1975; Kozbar et al., 1983). En una realización, se utiliza el método alternativo de inmortalización por EBV descrito en la WO2004/076677.
Utilizando el método descrito en la WO2004/076677, células B que producen el anticuerpo de la invención pueden transformarse con EBV en presencia de un activador de células B policlonales. La transformación con EBV es una técnica estándar y puede adaptarse fácilmente para incluir activadores de células B policlonales.
Se pueden añadir opcionalmente estimulantes adicionales del crecimiento y la diferenciación celular durante la etapa de transformación para mejorar aún más la eficiencia. Estos estimulantes pueden ser citoquinas como IL-2 e IL-15. En un aspecto, se agrega IL-2 durante la etapa de inmortalización para mejorar aún más la eficiencia de la inmortalización, pero su uso no es esencial. Las células B inmortalizadas producidas usando estos métodos se pueden cultivar luego mediante métodos conocidos en la técnica y aislarse entonces los anticuerpos.
Usando el método descrito en la Solicitud de Patente UK 0819376.5 se pueden cultivar células plasmáticas individuales en micropocillos de placas de cultivo. Los anticuerpos pueden aislarse de cultivos de células plasmáticas individuales. Además, a partir de cultivos de células plasmáticas individuales, se puede extraer ARN y se puede realizar una PCR de células individuales utilizando métodos conocidos en la técnica. Las regiones VH y VL de los anticuerpos pueden amplificarse por RT-PCR, secuenciarse y clonarse en un vector de expresión, que luego se transfecta en células HEK293T u otras células huésped. La clonación del ácido nucleico en vectores de expresión, la transfección de células huésped, el cultivo de las células huésped transfectadas y el aislamiento del anticuerpo producido pueden realizarse utilizando cualquier método conocido por los expertos en la técnica.
Los anticuerpos pueden además purificarse, si se desea, por filtración, centrifugación y diversos métodos cromatográficos, como HPLC o cromatografía de afinidad. Las técnicas para la purificación de anticuerpos,
por ejemplo anticuerpos monoclonales, que incluyen técnicas para producir anticuerpos de calidad farmacéutica, son bien conocidas.
Los fragmentos de los anticuerpos de la invención se pueden obtener a partir de los anticuerpos por métodos que incluyen la digestión con enzimas, tales como pepsina o papaína, y/o por escisión de los enlaces disulfuro mediante reducción química. Alternativamente, los fragmentos de los anticuerpos pueden obtenerse por clonación y expresión de parte de las secuencias de las cadenas pesadas o ligeras. Los "fragmentos" de anticuerpos pueden incluir fragmentos Fab, Fab', F(ab')2 y Fv. La invención también abarca fragmentos Fv de una sola cadena (scFv) derivados de las cadenas pesada y ligera de un anticuerpo de la invención, por ejemplo la invención incluye un scFv que comprende las CDR de un anticuerpo de la invención. También se incluyen los monómeros y dímeros de cadena pesada o ligera, los anticuerpos de cadena pesada de un solo dominio, los anticuerpos de cadena ligera de un solo dominio, así como los anticuerpos de cadena única, por ejemplo Fv de una sola cadena donde los dominios variables de las cadenas pesada y ligera se unen con un enlazador peptídico.
Los fragmentos de anticuerpos de la invención pueden impartir interacciones monovalentes o multivalentes y estar contenidos en diversas estructuras, como se describió anteriormente. Por ejemplo, las moléculas scFv se pueden sintetizar para crear un "triacuerpo" trivalente o un “tetracuerpo” tetravalente. Las moléculas scFv pueden incluir un dominio de la región Fc que resulta en minicuerpos bivalentes. Además, las secuencias de la invención pueden ser un componente de moléculas multiespecíficas donde las secuencias de la invención se dirigen a los epítopos de la invención y otras regiones de la molécula se unen a otras dianas. Moléculas ilustrativas incluyen Fab2 biespecífico, Fab3 triespecífico, scFv biespecífico y diacuerpos (Holliger y Hudson, 2005, Nature Biotechnology 9: 1126-1136).
Se pueden usar técnicas estándar de biología molecular para preparar secuencias de ADN que codifican los anticuerpos o los fragmentos de anticuerpos de la presente invención. Las secuencias de ADN deseadas se pueden sintetizar completa o parcialmente usando técnicas de síntesis de oligonucleótidos. Se pueden usar técnicas de mutagénesis dirigida y reacción en cadena de la polimerasa (PCR) según sea apropiado.
Se puede usar cualquier sistema célula huésped/vector adecuado para la expresión de las secuencias de ADN que codifican las moléculas de anticuerpo de la presente invención o fragmentos de las mismas. Se pueden usar sistemas bacterianos, por ejemplo E. coli, y otros sistemas microbianos, en parte, para la expresión de fragmentos de anticuerpos tales como los fragmentos Fab y F(ab')2, y especialmente los fragmentos Fv y los fragmentos de anticuerpos de cadena simple, por ejemplo de cadena Fvs. Se pueden emplear sistemas de expresión en células huésped eucariotas, por ejemplo de mamíferos, para la producción de moléculas de anticuerpos más grandes, incluidas moléculas de anticuerpos completas. Células huésped de mamífero adecuadas incluyen células CHO, HEK293T, PER.C6, NSO, mieloma o hibridoma.
La presente invención también proporciona un proceso para la producción de una molécula de anticuerpo de acuerdo con la presente invención, que comprende cultivar una célula huésped que incluye un vector que codifica un ácido nucleico de la presente invención en condiciones adecuadas para conducir a la expresión de una proteína a partir de un ADN que codifica la molécula de anticuerpo de la presente invención, y aislar la molécula de anticuerpo.
La molécula de anticuerpo puede comprender solo un polipéptido de cadena pesada o ligera, en cuyo caso solo es necesario usar una secuencia codificante de polipéptido de cadena pesada o cadena ligera para transfectar las células huésped. Para la producción de productos que comprenden tanto cadenas pesadas como ligeras, la línea celular puede transfectarse con dos vectores, un primer vector que codifica un polipéptido de cadena ligera y un segundo vector que codifica un polipéptido de cadena pesada. Alternativamente, se puede usar un solo vector, incluyendo el vector secuencias que codifican polipéptidos de cadena ligera y cadena pesada.
Alternativamente, los anticuerpos según la invención pueden producirse por i) expresión de una secuencia de ácido nucleico de acuerdo con la invención en una célula huésped, y ii) aislamiento del producto de anticuerpo expresado. Adicionalmente, el método puede incluir iii) purificar el anticuerpo.
Cribado de células B transformadas, células plasmáticas individuales cultivadas y células HEK293T transfectadas
Las células B transformadas y las células plasmáticas individuales pueden examinarse para detectar aquellas que producen anticuerpos con la especificidad o función deseada.
La etapa de cribado se puede llevar a cabo mediante cualquier inmunoensayo, por ejemplo ELISA, mediante tinción de tejidos o células (incluidas las células transfectadas), mediante un ensayo de neutralización o mediante uno de varios otros métodos conocidos en la técnica para identificar la especificidad o función deseadas. El ensayo se puede seleccionar en base a un simple reconocimiento de uno o más antígenos, o adicionalmente en base a una función deseada, por ejemplo para seleccionar anticuerpos neutralizantes en lugar de solo anticuerpos de unión a antígeno, para seleccionar anticuerpos que pueden cambiar las características de las células seleccionadas, como sus cascadas de señalización, su forma, su tasa de crecimiento, su capacidad de influir en otras células, su respuesta a la influencia de otras células o por otros reactivos o por un cambio en las condiciones, su estado de diferenciación.
Los clones de células B transformados individuales pueden producirse a partir del cultivo positivo de células B transformadas. La etapa de clonación para separar los clones individuales de la mezcla de células positivas puede llevarse a cabo utilizando dilución limitante, micromanipulación, deposición de células individuales por clasificación celular u otro método conocido en la técnica.
El ácido nucleico de las células plasmáticas individuales cultivadas puede aislarse, clonarse y expresarse en células HEK293T u otras células huésped mediante métodos conocidos en la técnica.
Los clones de las células B inmortalizadas o las células HEK293T transfectadas de la invención se pueden usar de diversas maneras, por ejemplo como fuente de anticuerpos monoclonales, como fuente de ácido nucleico (ADN o ARNm) que codifica un anticuerpo monoclonal de interés, para investigación.
La invención proporciona una composición que comprende células B de memoria inmortalizadas o células huésped transfectadas que producen anticuerpos que neutralizan al menos dos subtipos diferentes del virus de la influenza A seleccionados de los subtipos del grupo 1 y del grupo 2.
Epítopos
Como se mencionó anteriormente, los anticuerpos de la invención se pueden usar para mapear los epítopos a los que se unen. Los inventores han descubierto que los anticuerpos que neutralizan la infección por el virus de la influenza A se dirigen hacia los epítopos que se encuentran en la HA. En una realización, los anticuerpos se dirigen a uno o más epítopos en la región del tallo de HA conservados entre uno o más subtipos del grupo 1 y 2 del virus de la influenza A. Los epítopos a los que se unen los anticuerpos de la invención pueden ser lineales (continuos) o conformacionales (discontinuos). En una realización, los anticuerpos y los fragmentos de anticuerpos de la invención se unen a una región del polipéptido que comprende las SEQ ID NO: 37, 38, 39 o 40, como se describe aquí.
En otra realización, el epítopo al que se unen los anticuerpos de la invención comprende residuos de aminoácidos en los polipéptidos HA1 y HA2 de uno o dos monómeros de HA como se describe anteriormente. Los monómeros de HA pueden estar escindidos o escindidos.
Los epítopos reconocidos por los anticuerpos de la presente invención pueden tener varios usos. El epítopo y los mimotopos de los mismos en forma purificada o sintética se pueden usar para generar respuestas inmunitarias (es decir, como una vacuna, o para la producción de anticuerpos para otros usos) o para detectar sueros en busca de anticuerpos que inmunoreaccionen con el epítopo o mimotopos de los mismos. En una realización, tal epítopo o mimotopo, o tal antígeno que comprende tal epítopo o mimotopo, puede usarse como vacuna para provocar una respuesta inmune. Los anticuerpos y los fragmentos de anticuerpos de la invención también se pueden usar en un método para controlar la calidad de las vacunas. En particular, los anticuerpos pueden usarse para verificar que el antígeno de una vacuna contiene el epítopo inmunogénico correcto en la conformación correcta.
El epítopo también puede ser útil en la detección de ligandos que se unen a dicho epítopo. Tales ligandos incluyen anticuerpos, incluyendo aqulleos de camello, tiburón y otras especies, fragmentos de anticuerpos, péptidos, productos de tecnología de exhibición de fagos, aptámeros, adnectinas o fragmentos de otras proteínas virales o celulares, pueden bloquear el epítopo y prevenir la infección.
Expresión Recombinante
El clon de células B inmortalizado o la célula plasmática cultivada de la invención también se puede usar como fuente de ácido nucleico para la clonación de genes de anticuerpos para la expresión recombinante posterior. La expresión a partir de fuentes recombinantes es más común para fines farmacéuticos que la expresión de células B o hibridomas, por ejemplo por razones de estabilidad, reproducibilidad, facilidad de cultivo.
Así, se describe aquí un método para preparar una célula recombinante que comprende las etapas de: (i) obtener uno o más ácidos nucleicos (por ejemplo ARNm de cadena pesada y/o ligera) a partir del clon de células B o de la célula plasmática única que codifica para un anticuerpo de interés; (ii) insertar el ácido nucleico en un vector de expresión y (iii) transfectar el vector en una célula huésped para permitir la expresión del anticuerpo de interés en esa célula huésped.
De manera similar, se describe aquí un método para preparar una célula recombinante que comprende las etapas de: (i) secuenciar el o los ácidos nucleicos del clon de células B o de la célula plasmática única que codifica el anticuerpo de interés; y (ii) usar la información de secuencia de la etapa (i) para preparar ácido(s)
nucleico(s) para su inserción en una célula huésped con el fin de permitir la expresión del anticuerpo de interés en esa célula huésped. El ácido nucleico puede, pero no necesita, ser manipulado entre los pasos (i) y (ii) para introducir sitios de restricción, para cambiar el uso de codones y/o para optimizar la transcripción y/o las secuencias reguladoras de la traducción.
También se describe un método para preparar una célula huésped transfectada que comprende la etapa de transfectar una célula huésped con uno o más ácidos nucleicos que codifican un anticuerpo de interés, donde los ácidos nucleicos son ácidos nucleicos que se derivaron de un clon de células B inmortalizado o de una célula plasmática única cultivada tal como se describe aquí. Así, los procedimientos para preparar primero el (los) ácido(s) nucleico(s) y luego usarlo(s) para transfectar una célula huésped pueden realizarse en diferentes momentos por diferentes personas en diferentes lugares (por ejemplo, en diferentes países).
Estas células recombinantes se pueden usar para fines de expresión y cultivo. Son particularmente útiles para la expresión de anticuerpos en la producción farmacéutica a gran escala. También pueden usarse como ingrediente activo en una composición farmacéutica. Se puede utilizar cualquier técnica de cultivo adecuada, incluyendo cultivo estático, cultivo en frasco rodante, fluido ascítico, cartucho de biorreactor de tipo de fibra hueca, minifermentador modular, tanque agitado, cultivo de microportadores, perfusión de núcleos cerámicos.
Los métodos para obtener y secuenciar genes de inmunoglobulina de células B o de células plasmáticas son bien conocidos en la técnica (por ejemplo, veáse el Capítulo 4 de Kuby Immunology, 4a edición, 2000).
La célula huésped transfectada puede ser una célula eucariota, incluyendo células de levadura y animales, en particular células de mamíferos (por ejemplo células CHO, células NSO, células humanas como PER.C6 (Jones et al 2003) o HKB-11 (Cho et al. 2001; Cho et al. 2003), células de mieloma (US 5.807.715; US 6.300.104)), así como las células vegetales. Los huéspedes de expresión preferidos pueden glicosilar el anticuerpo de la invención, en particular con estructuras carbohidrato no inmunogénicas por sí mismas en humanos. En una realización, la célula huésped transfectada puede ser capaz de crecer en medios libres de suero. En una realización adicional, la célula huésped transfectada puede ser capaz de crecer en cultivo sin la presencia de productos derivados de animales. La célula huésped transfectada también puede cultivarse para dar una línea celular.
También se describe aquí un método para preparar una o más moléculas de ácido nucleico (por ejemplo, genes de cadena pesada y ligera) que codifican un anticuerpo de interés que comprende las etapas de:
(i) preparar un clon de células B inmortalizadas o cultivar una célula plasmática como se describe aquí;
(ii) obtener a partir del clon de células B o de las células plasmáticas individuales cultivadas un ácido nucleico que codifica el anticuerpo de interés.
También se describe aquí un método para obtener una secuencia de ácido nucleico que codifica un anticuerpo de interés que comprende las etapas de: (i) preparar un clon de células B inmortalizadas o cultivar una única célula de plasma de acuerdo con la invención; (ii) secuenciar el ácido nucleico del clon de células B o de la célula plasmática cultivada que codifica el anticuerpo de interés.
También se describe aquí un método para preparar una o más moléculas de ácido nucleico que codifican un anticuerpo de interés, comprendiendo el método la etapa de obtener el ácido nucleico que se obtuvo a partir de un clon de células B transformadas o de una célula plasmática cultivada de la invención. Así, los procedimientos para obtener primero el clon de células B o la célula plasmática cultivada y luego obtener el (los) ácido(s) nucleico(s) del clon de células B o la célula plasmática cultivada pueden llevarse a cabo en diferentes momentos, por diferentes personas, en diferentes lugares (por ejemplo, en diferentes países).
También se describe aquí un método para preparar un anticuerpo (por ejemplo para uso farmacéutico), que comprende las etapas de: (i) obtener y/o secuenciar uno o más ácidos nucleicos (por ejemplo, genes de cadena pesada y ligera) del clon de células B seleccionado o de células plasmáticas cultivadas que expresan el anticuerpo de interés; (ii) insertar el (los) ácido(s) nucleico(s) en o usar la(s) secuencia(s) de ácido(s) nucleico(s) para preparar un vector de expresión; (iii) transfectar una célula huésped que pueda expresar el anticuerpo de interés; (iv) cultivar o subcultivar las células huésped transfectadas en condiciones en las que se expresa el anticuerpo de interés; y, opcionalmente, (v) purificar el anticuerpo de interés.
También se describe aquí un método para preparar un anticuerpo que comprende las etapas de: cultivar o subcultivar una población de células huésped transfectadas en condiciones en las que se expresa el anticuerpo de interés y, opcionalmente, purificar el anticuerpo de interés, donde dicha población de células huésped transfectadas se ha preparado (i) proporcionando ácido(s) nucleico(s) que codifica(n) un anticuerpo seleccionado de interés que es producido por un clon de células B o una célula plasmática cultivada preparada como se describe anteriormente, (ii) insertando el (los) ácido(s) nucleico(s) en un vector de expresión, (iii) transfectando el vector en una célula huésped que puede expresar el anticuerpo de interés, y (iv) cultivando o subcultivando la célula huésped transfectada que comprende los ácidos nucleicos insertados para producir el anticuerpo de interés. Así, los procedimientos para preparar primero la célula huésped recombinante y luego cultivarla para expresar el anticuerpo pueden realizarse en momentos muy diferentes, por diferentes personas, en diferentes lugares (por ejemplo, en diferentes países).
Composiciones farmacéuticas
La invención proporciona una composición farmacéutica que contiene los anticuerpos y/o fragmentos de anticuerpos de la invención y/o el ácido nucleico que codifica dichos anticuerpos y/un péptido inmunogénico que comprende los epítopos reconocidos por los anticuerpos de la invención. Una composición farmacéutica también contiene un vehículo farmacéuticamente aceptable para permitir la administración. El vehículo no debe inducir por sí mismo la producción de anticuerpos dañinos para el individuo que recibe la composición y no debe ser tóxico. Los vehículos adecuados pueden ser macromoléculas grandes y de metabolismo lento, como proteínas, polipéptidos, liposomas, polisacáridos, ácidos polilácticos, ácidos poliglicólicos, aminoácidos poliméricos, copolímeros de aminoácidos y partículas víricas inactivas.
Se pueden usar sales farmacéuticamente aceptables, por ejemplo sales de ácidos inorgánicos, como clorhidratos, bromhidratos, fosfatos y sulfatos, o sales de ácidos orgánicos, como acetatos, propionatos, malonatos y benzoatos.
Los vehículos farmacéuticamente aceptables en composiciones terapéuticas pueden contener adicionalmente líquidos como agua, solución salina, glicerol y etanol. Además, en tales composiciones pueden estar presentes sustancias auxiliares, como agentes humectantes o emulsionantes o sustancias
tampón del pH. Dichos portadores permiten que las composiciones farmacéuticas se formulen en forma de comprimidos, píldoras, grageas, cápsulas, líquidos, geles, jarabes, suspensiones y suspensiones espesas, para su ingesta por parte del sujeto.
Dentro del alcance de la invención, las formas de administración pueden incluir aquellas adecuadas para la administración parenteral, por ejemplo por inyección o infusión, por ejemplo mediante inyección en bolo o infusión continua. Cuando el producto es para inyección o infusión, puede tener forma de suspensión, solución o emulsión en un vehículo oleoso o acuoso y puede contener agentes de formulación, como agentes de suspensión, conservantes, estabilizantes y/o dispersantes. Alternativamente, la molécula de anticuerpo puede estar en forma seca, para reconstituirse antes de su uso con un líquido estéril apropiado.
Una vez formuladas, las composiciones de la invención se pueden administrar directamente al sujeto. En una realización, las composiciones están adaptadas para la administración a sujetos humanos.
Las composiciones farmacéuticas de esta invención pueden administrarse por cualquier vía, incluyendo las vías oral, intravenosa, intramuscular, intraarterial, intramedular, intraperitoneal, intratecal, intraventricular, transdérmica, transcutánea, tópica, subcutánea, intranasal, intranasal, enteral, sublingual, intravaginal o rectal. También se pueden usar hipoespray para administrar las composiciones farmacéuticas de la invención. Típicamente, las composiciones terapéuticas pueden prepararse como inyectables, ya sea como soluciones líquidas o suspensiones. También se pueden preparar formas sólidas adecuadas para solución o suspensión en vehículos líquidos antes de la inyección.
El suministro directo de las composiciones generalmente se llevará a cabo mediante inyección subcutánea, intraperitoneal, intravenosa o intramuscular, o se administrará al espacio intersticial de un tejido. Las composiciones también se pueden administrar en una lesión. El régimen de dosificación puede ser un programa de dosis única o un programa multidosis. Los medicamentos basados en anticuerpos conocidos proporcionan una guía en relación a la frecuencia de administración, por ejemplo si un producto farmacéutico debe administrarse diariamente, semanalmente, mensualmente. La frecuencia y la dosis también pueden depender de la gravedad de los síntomas.
Las composiciones de la invención se pueden preparar en diversas formas. Por ejemplo, las composiciones se pueden preparar como inyectables, como soluciones líquidas o como suspensiones. También se pueden preparar formas sólidas adecuadas para la solución o suspensión en vehículos líquidos antes de la inyección (por ejemplo una composición liofilizada, como Synagis™ y Herceptin™, para la reconstitución con agua estéril que contiene un conservante). La composición se puede preparar para la administración tópica, por ejemplo en forma de pomada, crema o polvo. La composición se puede preparar para la administración oral, por ejemplo en forma de tableta o cápsula, como un aerosol o como un jarabe (opcionalmente aromatizado). La composición se puede preparar para la administración pulmonar, por ejemplo en forma de inhalador, utilizando un polvo fino o un spray. La composición puede prepararse como un supositorio o pesario. La composición puede prepararse para la administración nasal, aural u ocular, por ejemplo en forma de gotas. La composición puede estar en forma de kit diseñado de manera que una composición combinada es reconstituida justo antes de la administración a un sujeto. Por ejemplo, un anticuerpo liofilizado se puede proporcionar en forma de kit con agua estéril o con un tampón estéril.
Se apreciará que el ingrediente activo de la composición será una molécula de anticuerpo, un fragmento de anticuerpo o variantes y derivados de los mismos. Como tal, será susceptible a la degradación en el tracto gastrointestinal. Por tanto, si la composición se administra por una vía que utiliza el tracto gastrointestinal, la composición deberá contener agentes que protejan al anticuerpo de la degradación, pero que lo liberen una vez se haya absorbido del tracto gastrointestinal.
Una discusión detallada sobre los vehículos farmacéuticamente aceptables está disponible en Gennaro (2000) Remington: The Science and Practice of Pharmacy, edición 20, ISBN: 0683306472.
Las composiciones farmacéuticas de la invención generalmente tienen un pH entre 5,5 y 8,5, en algunas realizaciones éste puede estar entre 6 y 8, y en otras realizaciones ser aproximadamente 7. El pH puede mantenerse empleando un tampón. La composición puede ser estéril y/o libre de pirógenos. La composición puede ser isotónica con respecto a los humanos. En una realización, las composiciones farmacéuticas de la invención se suministran en recipientes herméticamente sellados.
Las composiciones farmacéuticas incluirán una cantidad efectiva de uno o más anticuerpos de la invención, es decir, una cantidad suficiente para tratar, mejorar o prevenir una enfermedad o condición deseada, o para exhibir un efecto terapéutico detectable. Los efectos terapéuticos también incluyen la reducción de los síntomas físicos. La cantidad efectiva precisa para cualquier sujeto particular dependerá de su tamaño y su salud, de la naturaleza y el alcance de la condición y de la terapia o combinación de terapias seleccionadas para la administración. La cantidad efectiva para una situación dada está determinada por la experimentación de rutina y dentro del criterio clínico. Para los fines de la presente invención, en general una dosis efectiva será de aproximadamente 0,01 mg/kg a aproximadamente 50 mg/kg, o de aproximadamente 0,05 mg/kg a aproximadamente 10 mg/kg de las composiciones de la presente invención en el individuo al que se administra Los productos farmacéuticos basados en anticuerpos conocidos proporcionan orientación a este respecto, por ejemplo Herceptin™ se administra mediante infusión intravenosa de una solución de 21 mg/ml, con una dosis de carga inicial de 4 mg/kg de peso corporal y una dosis de mantenimiento semanal de 2 mg/kg de peso corporal; Rituxan™ se administra semanalmente a 375 mg/m2.
En una realización, las composiciones pueden incluir más de un anticuerpo (por ejemplo 2, 3) de la invención para proporcionar un efecto terapéutico aditivo o sinérgico. En otra realización, la composición puede comprender uno o más (por ejemplo 2, 3) anticuerpos de la invención y uno o más (por ejemplo 2, 3) anticuerpos adicionales contra el virus de la influenza A o B. Por ejemplo, un anticuerpo puede unirse a un epítopo HA, mientras que otro puede unirse a un epítopo diferente en HA, o a un epítopo en la neuraminidasa y/o a proteínas de matriz. Además, la administración de anticuerpos de la invención junto con una vacuna contra la influenza A o con anticuerpos de especificidades distintas del virus de la influenza A, por ejemplo el virus de la influenza B, están dentro del alcance de la invención. Los anticuerpos de la invención pueden administrarse ya sea combinados/simultáneamente o en momentos separados de una vacuna contra la influenza o de anticuerpos de especificidades distintas del virus de la influenza A.
En otra realización, la invención proporciona una composición farmacéutica que comprende dos o más anticuerpos, donde el primer anticuerpo es un anticuerpo de la invención y es específico para un epítopo HA y el segundo anticuerpo es específico para un epítopo de neuraminidasa, un segundo epítopo HA y/o un epitopo de matriz. Por ejemplo, la invención proporciona una composición farmacéutica que comprende
dos o más anticuerpos, donde el primer anticuerpo es específico para un epítopo en el tallo de un virus de la influenza A HA y el segundo anticuerpo es específico para un epítopo de neuraminidasa, un segundo epítopo HA (por ejemplo un epítopo en el cabeza globular de HA, un segundo epítopo en el tallo de HA) y/o un epítopo de matriz. El segundo epítopo en el tallo o el epítopo en la cabeza globular del virus de la influenza A, HA, puede conservarse, pero no es necesario, entre más de un subtipo de virus de la influenza A.
En otra realización más, se describe aquí una composición farmacéutica que comprende dos o más anticuerpos donde el primer anticuerpo es específico para un epítopo de neuraminidasa y el segundo anticuerpo es específico para un segundo epítopo de neuraminidasa, un epítopo HA y/o un epítopo de matriz.
En otra realización más, se describe aquí una composición farmacéutica que comprende dos o más anticuerpos donde el primer anticuerpo es específico para un epítopo de matriz y el segundo anticuerpo es específico para un segundo epítopo de matriz, un epítopo en HA y/o neuraminidasa.
Anticuerpos ilustrativos de la invención específicos para una proteína diana del virus de la influenza A incluyen la variante 1 de FI6, la variante 2 de FI6, la variante 3 de FI6, la variante 4 de FI6, la variante 5 de FI6, la variante 1 de FI28 o la variante 2 de FI28.
En una realización, se describe aquí una composición farmacéutica que comprende la variante 1 del anticuerpo FI6 o un fragmento de unión a antígeno del mismo, y un vehículo farmacéuticamente aceptable. En otra realización, se describe aquí una composición farmacéutica que comprende la variante 2 del anticuerpo FI6 o un fragmento de unión a antígeno del mismo, y un vehículo farmacéuticamente aceptable. En otra realización, la invención proporciona una composición farmacéutica que comprende la variante 3 del anticuerpo FI6 o un fragmento de unión a antígeno del mismo y un vehículo farmacéuticamente aceptable. En otra realización, la invención proporciona una composición farmacéutica que comprende la variante 4 del anticuerpo FI6 o un fragmento de unión a antígeno del mismo y un vehículo farmacéuticamente aceptable. En otra realización, la invención proporciona una composición farmacéutica que comprende la variante 5 del anticuerpo FI6 o un fragmento de unión a antígeno del mismo y un vehículo farmacéuticamente aceptable. En otra realización más, se describe aquí una composición farmacéutica que comprende la variante 1 del anticuerpo FI28 o un fragmento de unión a antígeno del mismo y un vehículo farmacéuticamente aceptable. En otra realización más, se describe aquí una composición farmacéutica que comprende la variante 2 del anticuerpo FI28 o un fragmento de unión a antígeno del mismo y un vehículo farmacéuticamente aceptable.
Los anticuerpos de la invención se pueden administrar (combinados o por separado) con otros agentes terapéuticos, por ejemplo con compuestos quimioterapéuticos, con radioterapia. En una realización, los compuestos terapéuticos incluyen compuestos antivirales tales como Tamiflu™. Dicha terapia de combinación proporciona una mejora aditiva o sinérgica en la eficacia terapéutica en relación con los agentes terapéuticos individuales cuando se administran solos. El término "sinergia" se utiliza para describir un efecto combinado de dos o más agentes activos que es mayor que la suma de los efectos individuales de cada agente activo respectivo. Así, cuando el efecto combinado de dos o más agentes resulta en una "inhibición sinérgica" de una actividad o proceso, se entiende que la inhibición de la actividad o proceso sea mayor que la suma de los efectos inhibidores de cada agente activo respectivo. El término "efecto
terapéutico sinérgico" se refiere a un efecto terapéutico observado con una combinación de dos o más terapias, donde el efecto terapéutico (medido por cualquiera de diversos parámetros) es mayor que la suma de los efectos terapéuticos individuales observados con las respectivas terapias individuales.
Los anticuerpos se pueden administrar a aquellos sujetos que previamente no han tenido una respuesta al tratamiento para la infección por el virus de la influenza A, es decir, que se ha demostrado que son refractivos al tratamiento contra la influenza. Dicho tratamiento puede incluir un tratamiento previo con un agente antiviral. Esto puede deberse, por ejemplo, a una infección con una cepa del virus de la influenza A
En una realización, una composición de la invención puede incluir anticuerpos de la invención donde los anticuerpos pueden constituir al menos el 50% en peso (por ejemplo 60%, 70%, 75%, 80%, 85%, 90%, 95%, 97%, 98%, 99% o más) de la proteína total en la composición. En tal composición, los anticuerpos están en forma purificada.
Se describe aquí un método para preparar un producto farmacéutico que comprende las etapas de: (i) preparar un anticuerpo de la invención; y (ii) mezclar el anticuerpo purificado con uno o más vehículos farmacéuticamente aceptables.
También se describe aquí un método para preparar un producto farmacéutico que comprende la etapa de mezclar un anticuerpo con uno o más vehículos farmacéuticamente aceptables, donde el anticuerpo es un anticuerpo monoclonal que se obtuvo de una célula B transformada o una célula plasmática cultivada tal como se describe aquí. Así, los procedimientos para obtener primero el anticuerpo monoclonal y luego preparar el producto farmacéutico se pueden realizar en momentos muy diferentes, por diferentes personas, en diferentes lugares (por ejemplo, en diferentes países).
Como alternativa al suministro de anticuerpos o de células B con fines terapéuticos, es posible administrar un ácido nucleico (típicamente ADN) que codifica el anticuerpo monoclonal de interés (o un fragmento activo del mismo) derivado de la célula B o la célula plasmática cultivada a un sujeto, de modo que el ácido nucleico se pueda expresar in situ en el sujeto para proporcionar un efecto terapéutico deseado. La terapia génica adecuada y los vectores de suministro de ácido nucleico son conocidos en la técnica.
Las composiciones de la invención pueden ser composiciones inmunogénicas y, en algunas realizaciones, pueden ser composiciones de vacuna que comprenden un antígeno que comprende un epítopo reconocido por un anticuerpo de la invención. Las vacunas pueden ser profilácticas (es decir, para prevenir una infección) o terapéuticas (es decir, para tratar una infección). En una realización, se describe una vacuna que comprende un polipéptido que comprende la secuencia de aminoácidos de SEQ ID NO: 37, 38, 39 o 40. En otra realización, se describe una vacuna que comprende un polipéptido que comprende los residuos aminoácidos en los polipéptidos HA1 y HA2 de uno o dos monómeros de HA como se describe anteriormente. Los monómeros de HA pueden estar no escindidos o escindidos.
Las composiciones pueden incluir un agente antimicrobiano, en particular si están envasadas en un formato multidosis. Pueden comprender un tensioactivo, por ejemplo Tween (polisorbato), tal como Tween 80. Los tensioactivos generalmente están presentes en niveles bajos, por ejemplo <0,01. Las composiciones también pueden incluir sales de sodio (por ejemplo cloruro de sodio) para dar tonicidad. Una concentración típica es de 10 2 mg/ml de NaCl.
Además, las composiciones pueden comprender un alcohol azúcar (por ejemplo manitol) o un disacárido (por ejemplo sacarosa o trehalosa), por ejemplo aproximadamente 15-30 mg/ml (por ejemplo 25 mg/ml), en particular si van a ser liofilizadas o si incluyen material que ha sido reconstituido a partir de un material liofilizado. El pH de una composición para la liofilización se puede ajustar a alrededor de 6,1 antes de la liofilización.
Las composiciones de la invención también pueden comprender uno o más agentes inmunorreguladores. En una realización, uno o más de los agentes inmunorreguladores incluyen un adyuvante.
Las composiciones de epítopos de la invención pueden provocar tanto una respuesta inmune mediada por células como una respuesta inmune humoral para abordar de manera eficaz la infección por el virus de la influenza A. Esta respuesta inmune puede inducir anticuerpos de larga duración (por ejemplo neutralizantes) y una inmunidad mediada por células que puede responder rápidamente al exponerse al virus de la influenza A.
Tratamientos médicos y usos
Los anticuerpos y fragmentos de anticuerpos de la invención o sus derivados y variantes pueden usarse para el tratamiento de la infección por el virus de la influenza A, para la prevención de la infección por el virus de la influenza A o para el diagnóstico de la infección por el virus de la influenza A.
Los métodos de diagnóstico pueden incluir poner en contacto un anticuerpo o un fragmento de anticuerpo con una muestra. Dichas muestras pueden ser muestras de tejido tomadas de, por ejemplo, fosas nasales, cavidades sinusales, glándulas salivales, pulmón, hígado, páncreas, riñón, oído, placenta, tracto alimentario, corazón, ovarios, pituitaria, suprarrenales, tiroides, cerebro o piel. Los métodos de diagnóstico también pueden incluir la detección de un complejo antígeno/anticuerpo.
Así, la descripción que comprende la invención proporciona (i) un anticuerpo, un fragmento de anticuerpo o variantes y derivados del mismo de acuerdo con la invención, (ii) un clon de células B inmortalizadas de acuerdo con la invención, (iii) un epítopo capaz de unirse a un anticuerpo de la invención o (iv) un ligando, preferiblemente un anticuerpo, capaz de unirse a un epítopo que se une a un anticuerpo de la invención para uso en terapia.
También se describe un método para tratar a un sujeto, que comprende administrar al sujeto un anticuerpo, un fragmento de anticuerpo o sus variantes y derivados de acuerdo con la invención, o un ligando, preferiblemente un anticuerpo, capaz de unirse a un epítopo que se une a un anticuerpo de la invención. En una realización, el método da como resultado una infección reducida del virus de la influenza A en el sujeto. En otra realización, el método previene, reduce el riesgo o retrasa la infección por el virus de la influenza A en el sujeto.
También se proporciona el uso de (i) un anticuerpo, un fragmento de anticuerpo, o sus variantes y derivados según la invención, (ii) un clon de células B inmortalizadas según la invención, (iii) un epítopo capaz de unirse a un anticuerpo de la invención o (iv) un ligando, preferiblemente un anticuerpo, que se une a un epítopo capaz de unirse a un anticuerpo de la invención, en la fabricación de un medicamento para la prevención o el tratamiento de la infección por el virus de la influenza A.
La invención proporciona una composición de la invención para su uso como un medicamento para la prevención o el tratamiento de una infección por el virus de la influenza A. También proporciona el uso de un anticuerpo de la invención y/o de una proteína que comprende un epítopo al que dicho anticuerpo se une en la fabricación de un medicamento para el tratamiento y/o diagnóstico de un sujeto. También se describe un método para tratar a un sujeto, que comprende la etapa de administrar al sujeto una composición de la invención. En algunas realizaciones, el sujeto puede ser un humano. Una forma de verificar la eficacia del tratamiento terapéutico consiste en controlar los síntomas de la enfermedad después de la administración de la composición de la invención. El tratamiento puede ser un programa de dosis única o un programa multidosis.
En una realización, un anticuerpo, fragmento de anticuerpo, clon de células B inmortalizadas, epítopo o composición de acuerdo con la invención se administra a un sujeto que necesita tal tratamiento. Dicho sujeto incluye aquel que tiene un riesgo particular o es susceptible a una infección por el virus de la influenza A, incluyendo por ejemplo un sujeto inmunocomprometido. El anticuerpo o el fragmento de anticuerpo de la invención también puede usarse en la inmunización pasiva o la vacunación activa.
Los anticuerpos y los fragmentos de los mismos como se describen en la presente invención también se pueden usar en un kit para el diagnóstico de la infección por el virus de la influenza A. Además, los epítopos capaces de unirse a un anticuerpo de la invención se pueden usar en un kit para controlar la eficacia de los procedimientos de vacunación, detectando la presencia de anticuerpos protectores contra el virus de la influenza A. Los anticuerpos, fragmentos de anticuerpos o variantes y derivados de los mismos como se describen en la presente invención también se pueden usar en un kit para controlar la fabricación de vacunas con la inmunogenicidad deseada.
También se proporciona un método para preparar un producto farmacéutico, que comprende la etapa de mezclar un anticuerpo monoclonal con uno o más vehículos farmacéuticamente aceptables, siendo el anticuerpo monoclonal un anticuerpo monoclonal que se obtuvo de una célula huésped transfectada de la invención. Por tanto, los procedimientos para obtener primero el anticuerpo monoclonal (por ejemplo expresarlo y/o purificarlo) y luego mezclarlo con el (los) vehículo(s) farmacéutico(s) se pueden realizar en diferentes momentos, por diferentes personas, en diferentes lugares (por ejemplo, en diferentes países).
Comenzando con una célula B transformada o una célula plasmática cultivada tal como se describe aquí, se pueden realizar varias etapas de cultivo, subcultivo, clonación, subclonación, secuenciación, preparación de ácido nucleico para perpetuar el anticuerpo expresado por las células B transformadas o las células plasmáticas cultivadas, con optimización opcional en cada paso. En una realización preferente, los métodos anteriores comprenden además técnicas de optimización (por ejemplo maduración u optimización por afinidad) aplicadas a los ácidos nucleicos que codifican el anticuerpo. En todos estos métodos, el ácido nucleico usado en el huésped de expresión puede manipularse para insertar, eliminar o modificar ciertas secuencias de ácido nucleico. Los cambios a partir de dicha manipulación incluyen cambios para introducir sitios de restricción, para modificar el uso del codón, para agregar u optimizar la transcripción y/o las secuencias reguladoras de la traducción. También es posible cambiar el ácido nucleico para alterar los aminoácidos codificados. Por ejemplo, puede ser útil introducir uno o más (por ejemplo 1, 2, 3, 4, 5, 6, 7, 8, 9, 10) sustituciones, deleciones y/o inserciones en la secuencia de aminoácidos del anticuerpo. Dichas mutaciones puntuales pueden modificar las funciones efectoras, la afinidad de unión a antígeno, las
modificaciones postraduccionales, la inmunogenicidad, pueden introducir aminoácidos para la unión de grupos covalentes (por ejemplo etiquetas) o pueden introducir etiquetas (por ejemplo con fines de purificación). Las mutaciones se pueden introducir en sitios específicos o se pueden introducir al azar, seguidas de una selección (por ejemplo, evolución molecular). Por ejemplo, uno o más ácidos nucleicos que codifican cualquiera de las regiones CDR, regiones variables de cadena pesada o regiones variables de cadena ligera, de los anticuerpos de la invención pueden mutarse de forma aleatoria o direccional para introducir diferentes propiedades en los aminoácidos codificados. Dichos cambios pueden ser el resultado de un proceso iterativo en el que se mantienen los cambios iniciales y se introducen nuevos cambios en otras posiciones de nucleótidos. Además, los cambios logrados en pasos independientes pueden combinarse. Las diferentes propiedades introducidas en los aminoácidos codificados pueden incluir una afinidad mejorada.
Generalidades
El término "que comprende" abarca "que incluye", así como "que consiste", por ejemplo una composición "que comprende" X puede consistir exclusivamente en X o puede incluir algo adicional, por ejemplo X Y.
La palabra "esencialmente" no excluye "completamente", por ejemplo una composición que está "esencialmente libre" de Y puede estar completamente libre de Y. Cuando sea necesario, la palabra "esencialmente" puede omitirse de la definición de la invención.
El término "aproximadamente" en relación a un valor numérico x significa, por ejemplo, x 10%.
El término "enfermedad" tal como se usa aquí en general pretende ser sinónimo, y se usa indistintamente, con los términos "trastorno" y "condición" (como en la condición médica), en el sentido de que todos reflejan una condición anormal del cuerpo humano o animal. o de una de sus partes que dificulta el funcionamiento normal, se manifiesta típicamente al distinguir signos y síntomas y causa que el humano o el animal tengan una duración o calidad de vida reducida.
Tal como se usa aquí, la referencia al "tratamiento" de un sujeto o paciente pretende incluir la prevención, la profilaxis y la terapia. Los términos "sujeto" o "paciente" se usan indistintamente aquí para referirse a todos los mamíferos, incluidos los humanos. Ejemplos de sujetos incluyen humanos, vacas, perros, gatos, caballos, cabras, ovejas, cerdos y conejos. En una realización, el paciente es un humano.
EJEMPLOS
En los siguientes ejemplos se proporcionan realizaciones ilustrativas de la presente invención se proporcionan.
Ejemplo 1. Generación y caracterización de anticuerpos ampliamente neutralizantes del virus de la influenza A a partir de células plasmáticas
Para identificar aquellos individuos que pueden producir anticuerpos heterosubtípicos en respuesta a la vacuna contra la gripe estacional (que contiene HA de H1 y H3), se analizaron mediante ELISPOT células plasmáticas circulantes recolectadas el día 7 después del refuerzo de su capacidad para secretar anticuerpos que se unen a una vacuna o a H5 HA no relacionadas (A/VN/1203/04). Sorprendentemente,
mientras que en cuatro de los cinco donantes analizados no fueron detectables las células plasmáticas específicas de H5, en un donante el 14% de las células plasmáticas que secretan IgG produjo anticuerpos contra H5, mientras que el 57% produjo anticuerpos contra la vacuna. Las células plasmáticas CD138 se aislaron a partir de células mononucleares de sangre periférica (PBMC) recogidas 7 días después de la vacunación usando micro-perlas magnéticas, seguida de clasificación celular usando una máquina FACSAria. Se sembraron números limitantes de células plasmáticas en micropocillos de placas de cultivo. Los sobrenadantes de cultivo se analizaron en tres ELISA paralelos utilizando como antígenos H5 o H9 HA recombinantes y el antígeno irrelevante toxoide tetánico. De los 4.928 sobrenadantes de cultivo seleccionados, 12 se unieron a H5 pero no a H9 HA, 25 a H9 pero no a H5 HA y 54 a H5 y H9. Algunos de los 54 cultivos con la señal OD más alta se sometieron a RT-PCR y se recuperaron dos genes VH y VL pareados.
Los genes VH y VL se clonaron en vectores de expresión y los anticuerpos recombinantes se produjeron transfectando células HEK293T. Los dos anticuerpos monoclonales, FI6 variante 2 y FI28, compartían la mayoría de los fragmentos de los genes V, D y J (IGHV3-30*01, IGHD3-9*01, IGHJ4*02 e IGKV4-1*01), pero diferían en las regiones N, en el uso de IGKJ y en el patrón de mutaciones somáticas y, por tanto, no estaban relacionados clonalmente.
La especificidad de los anticuerpos recombinantes se investigó mediante ELISA utilizando un panel de HA pertenecientes a diferentes subtipos. Sorprendentemente, FI6 se unió a todos los subtipos de HA de la influenza A probados, incluidos el grupo 1 (H1, H5 y H9) y el grupo 2 (H3 y H7), mientras que no se unió a HA del virus de la influenza B. En contraste, FI28 se unió solo a los tres grupos 1 HA (H1, H5 y H9).
Tabla 4

Dada la homología de las secuencias VH y VL de los dos anticuerpos, los experimentos aleatorios se realizaron utilizando las cadenas H y L de la variante 2 de FI6, la variante 1 de FI28 y 7I13, un anticuerpo específico de hCMV que usa los mismos elementos V, D y J de la cadena H. Mientras que la unión a H7 requería el emparejamiento de las cadenas H y L de la variante 2 de FI6, la unión a H5 se mantuvo cuando la variante 2 de FI6 y las cadenas L de la variante 1 de FI28 se aleatorizaban. Además, también se observó unión a H5 cuando la cadena H de la variante 2 de FI6 se emparejaba con la cadena L de 7I13 no relacionada. Por el contrario, no se observó unión a H5 cuando la cadena homóloga de H de 7113 se emparejaba con la cadena L de la variante 2 de FI6. Sin limitarse a ninguna teoría particular, estos resultados sugieren que la contribución principal a la unión a H5 proviene de la cadena H, mientras que la unión a H7 requiere un emparejamiento preciso entre las cadenas H y L de la variante 2 de FI6.
Se ensayaron la variante 2 de FI6 y la variante 1 de FI28 para determinar su capacidad de neutralizar los subtipos de influenza A del grupo 1 y del grupo 2 utilizando virus pseudotipados (Tabla 5), así como virus infecciosos (Tabla 6). Notablemente, la variante 2 de FI6 neutralizó todos los pseudovirus analizados, incluidos seis aislados H5 pertenecientes a los clados antigénicamente divergentes 0, 1, 2.1, 2.2 y 2.3, y
dos aislados H7 aviares. Además, la variante 2 de FI6 neutralizó todos los virus infecciosos analizados, incluidos dos virus H3N2 y cuatro virus H1N1 expandidos durante varias décadas, hasta el reciente aislado pandémico A/CA/04/09 (Tabla 6). La variante 1 de FI28 neutralizó todos los pseudovirus H5, pero no neutralizó los pseudovirus H7 ni todos los virus infecciosos analizados. Los títulos neutralizantes en pseudovirus eran más altos que los títulos en virus infecciosos.
Tabla 5. Neutralización de pseudotipos de HA (IC90, pg/ml)

Tabla 6. Neutralización de virus infecciosos (IC90, pg/ml)

Ejemplo 2. Sitios de unión antigénicos de FI6 Variante 2 y FI28 Variante 1
Para identificar los sitios antigénicos a los que se unen los anticuerpos FI6 variante 2 y FI28 variante 1, primero se ensayó su capacidad para inhibir la unión de C179, un anticuerpo monoclonal de ratón que se asignó a una región conservada de la región del tallo HA (Y. Okuno, et al., J Virol 67, 2552 (1993)). Tanto la variante 2 de FI6 como la variante 1 de FI28 inhibieron completamente la unión de C179 a1H5 VN/1203/04 recombinante, lo que indica que reconocen un epítopo superpuesto. Por el contrario, la variante 2 de FI6 y la variante 1 de FI28 no compitieron con un panel de anticuerpos específicos de H5 aislados de donantes inmunes H5N1 que reconocen diferentes epítopos en la cabeza globular de la HA (C.P. Simmons et al., PLoS Med 4, e178 (2007); S. Khurana et al., PLoS Med 6, e1000049 (2009)). Los intentos de mapear el epítopo de la variante 2 de FI6 mediante la selección de mutantes de escape fallaron, lo que sugiere que su epítopo no se puede mutar fácilmente sin comprometer la adaptabilidad viral.
A continuación, se realizó el mapeo basado en péptidos utilizando librerías de péptidos lineales y ciclados de HA A/VN/1194/04, así como también el escaneo de hélices utilizando los sistemas de Pepscan Presto BV (Lelystad, Países Bajos). Este análisis identificó una región de unión de la variante 2 de FI6 que incluye el péptido de fusión HA2 FGAIAG (aminoácido 3-8, según la numeración H3; SEQ ID NO: 37), el péptido de la hélice A HA2 DGVTNKVNS (aminoácido 46-54; SEC. ID NO: 38), el péptido HA2 Helix B MENERTLDFHDSNVK (aminoácido 102-116; SEC ID NO: 39) y el péptido HA1 C-terminal LVLATGLRNSP (aminoácido 315-325; SEQ ID NO: 40). La región de unión de la variante 1 de FI28 era diferente de la de FI6, ya que este anticuerpo no reaccionó con el péptido C-terminal HA1 y el péptido Helix B de HA2.
Ejemplo 3. Generación y caracterización de variantes 3, 4 y 5 de FI6 con productividad mejorada
Se sintetizaron varias variantes de FI6 variante 2 VH y VL para mejorar la producción en células de mamíferos y eliminar mutaciones somáticas innecesarias y características no deseadas. Los genes VH y VL se clonaron en vectores de expresión que codifican la región constante de IgG1 y Ck, respectivamente. Las secuencias de la línea germinal de la variante 2 de FI6 se determinaron en referencia a la base de datos IMGT. Las variantes de anticuerpos en las que mutaciones únicas o múltiples de la línea germinal se revirtieron a la línea germinal se produjeron mediante síntesis (Genscript, Piscatawy, NJ) o mediante mutagénesis dirigida al sitio (Promega) y se confirmaron mediante secuenciación. Todas las secuencias de variantes fueron optimizadas en codón para la expresión en células humanas utilizando el sistema OptimumGeneTM de GenScript. Se produjeron anticuerpos monoclonales por transfección transitoria de células Freestyle 293 cultivadas en suspensión (Invitrogen) con PEI. Los sobrenadantes de las células transfectadas se recogieron después de 7 días de cultivo y las IgG se purificaron por afinidad mediante cromatografía de Proteína A (GE Healthcare) y se desalaron contra PBS. La productividad en el sistema de expresión transitoria se evaluó en varios experimentos independientes. Los valores medios se muestran en la Figura 1. Como se muestra en la Figura 1, la variante 2 de FI6 se produce a un título de 13,5 pg/ml; la variante 3 de FI6 se produce a un título de 46,3 pg/ml; la variante 4 de FI6 se produce a un título de 60,7 pg/ml; y la variante 5 de FI6 se produce a un título de 61,6 pg/ml. Así, se pudo lograr un aumento de 3,4-, 4,5- y 4,6- veces los títulos de producción de las variantes 3, 4 y 5 de FI6, respectivamente, en comparación con la variante 2 de FI6.
Los anticuerpos recombinantes también se caracterizaron por la unión mediante ELISA a H5 y H7 HA y la neutralización de los pseudovirus H5 y H7 (Tabla 7) en comparación con la variante 2 de FI6 original IgG. Las placas de ELISA de área media (Corning) se recubrieron con 5 pl de 1 pg/ml de HA recombinantes derivadas de baculovirus (Protein Sciences Corp.) en PBS. Después de bloquear con PBS al 1%/BSA, se agregaron anticuerpos y la unión se reveló usando IgG anti-humana de cabra F(ab')2 conjugada con fosfatasa alcalina (Southern Biotech). Luego se lavaron las placas, se añadió sustrato (p-NPP, Sigma) y se leyeron las placas a 405 nm. Las afinidades relativas de la unión del anticuerpo a los HA se determinaron con ELISA midiendo la concentración de anticuerpo requerida para lograr una unión máxima del 50% (EC50). Para los ensayos de neutralización de pseudovirus, las diluciones en serie del anticuerpo se incubaron con un sobrenadante de cultivo que contenía pseudovirus de concentración fija durante 1 hora a 37°C. Las mezclas se añadieron a células HEK 293T/17 y se incubaron durante 3 días a 37°C. Luego, las células se lisaron con reactivo Britelite (Perkin Elmer) y las unidades de luz relativas (RLU) en los lisados celulares se determinaron con un lector de microplacas luminómetro (Veritas, Turner Biosystems). La reducción de la infectividad se determinó comparando la RLU en presencia y ausencia de anticuerpos y se expresó como porcentaje de neutralización. La dosis inhibidora del 50% (IC50) se definió como la concentración de la muestra a la que se redujo la RLU en un 50% en comparación con los pocillos de control del virus después de la sustracción de la RLU de fondo en los pocillos de control de células solamente. La Tabla 7 muestra las variantes 3-5 de FI6 que se seleccionaron en base a unas características de secuencia mejoradas combinadas con una actividad de unión conservada o mejorada.
Tabla 7

La variante 2 de FI6 y la variante 3 de FI6 se unieron a todos los HA recombinantes o purificados ensayados pertenecientes al Grupo 1 (H1, H2, H5, H6, H8 y H9) y al Grupo 2 (H3, H4, H7 y H10), con valores EC50 en el rango de 10 a 270 ng/ml (Tabla 8). Además, la variante 2 de FI6 y la variante 3 de FI6 tiñeron las células transfectadas con genes HA pertenecientes al Grupo 1 (H11, H12, H13 y H16) y al Grupo 2 (H4, H10, H14 y H15 (Tabla 8).
Tabla 8
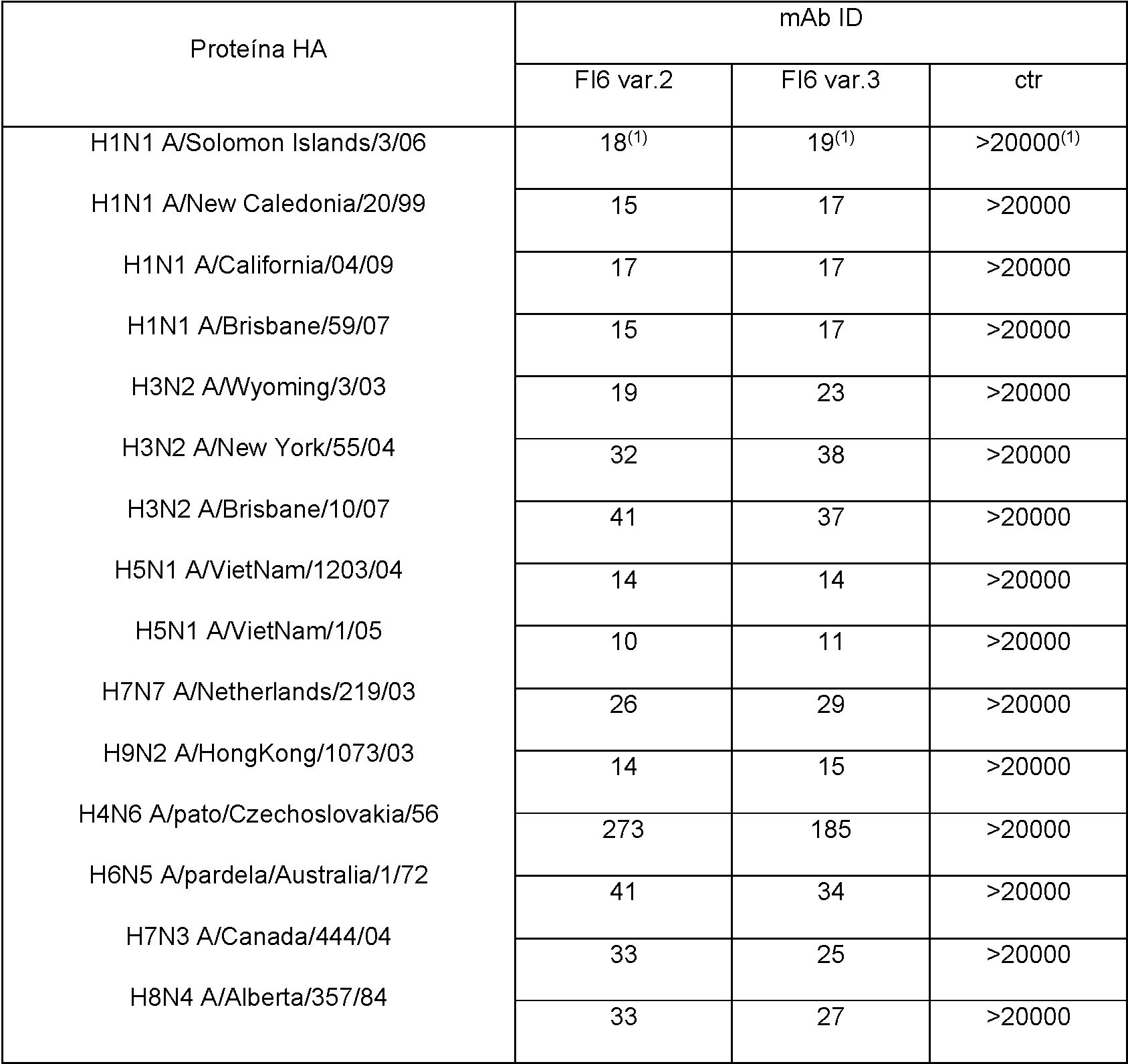

Ejemplo 4. Caracterización estructural de epítopos de la variante 3 de FI6 de HA H1 y H3
Para identificar el epítopo reconocido por la variante 3 de FI6 en las HA del Grupo 1 y 2 y para describir las interacciones moleculares entre el anticuerpo y su antígeno diana, se cristalizaron los complejos del fragmento Fab de la variante 3 de FI6 con homotrímeros de HA H1 (Grupo 1) y H3 (Grupo 2). Para la cristalización del complejo homotrímero de HA variante 3/H1 de FI6, el ectodominio de H1 HA0 se expresó en células de insecto Sf9. El ADNc correspondiente a los residuos 11-329 (HA1) y 1-176 (HA2) (basado en la numeración de H3) se clonó en un vector de expresión BioFocus que incorporaba la señal de secreción de GP67 para permitir la secreción de proteínas expresadas en el medio de cultivo. El ADNc clonado se fusionó a una secuencia de plegado de trimerización C-terminal para permitir la formación de la forma trimérica de H1 HA0. Se incluyó una secuencia de escisión de trombina entre la secuencia de plieque y el extremo C de HA2 para permitir la eliminación de la etiqueta de plegado antes de la cristalización y una etiqueta 6-His se incorporó al extremo C-terminal de la secuencia polipeptídica expresada para su uso en la purificación por afinidad.
Las células de insecto Sf9 se infectaron con baculovirus recombinante y e1H1 HA0 marcado con 6-His se recuperó del medio de cultivo mediante su paso sobre resina de Ni-NTA (Qiagen) y filtración en gel (columna S200). La proteína eluida correspondiente al H1 HA0 trimérico se concentró a 1 mg/ml antes de la eliminación de la etiqueta C-terminal mediante tratamiento con trombina (5 unidades de trombina por mg de HA0) durante dos horas a 20°C. La proteína escindida H1 HA0 purificada finalmente se fraccionó en una columna de intercambio aniónico Mono Q. Para permitir la formación de su complejo con variante 3 de FI6-Fab, se mezcló H1 HA0 purificado, entre 0,5 y 1 mg/ml, con un exceso molar de dos veces la variante 3 de FI6-Fab purificada. Se dejó que el complejo se formara por incubación a 4°C durante tres horas antes de la
separación del exceso de variante 3 de FI6-Fab mediante fraccionamiento en una columna de filtración con gel S200.
El complejo purificado de Fab-FI6 variante 3 y H1 HA0 trimérico sin procesar se concentró a 12 mg/ml para su uso en la cristalización. Los cristales del complejo Fab-FI6 variante 3 y H1 HA0 se hicieron crecer en gotas colgantes por difusión de vapor sobre una solución en pocillo que consistía en 0,1M de Bis Tris propano de pH 7,0, 2,2M de sulfato de amonio. Los cristales crecieron a 20°C durante un período de cuatro semanas y se recogieron de la gota en una mezcla 1:1 de solución en pocillo y malonato de sodio 3,4M, pH 7,0, para la protección criogénica antes de la congelación flash en nitrógeno líquido. El conjunto de datos se recopiló en la Diamond Light Source, línea de haz 103, y se indexó, integró y ajustó usando MOSFLM y SCALA, respectivamente.
El análisis estadístico de los parámetros celulares unitarios y los pesos moleculares de las proteínas sugirió un monómero de hemaglutinina y un fragmento Fab por unidad asimétrica; por tanto, la sustitución molecular se realizó utilizando modelos de búsqueda en sus estados monoméricos. Las fases iniciales se obtuvieron utilizando las coordenadas del H1 HA monomérico (ID de PDB 1RD8) como modelo de búsqueda con el programa CCP4 PHASER. Usando el programa de ajuste de modelo automatizado FFFEAR, el dominio variable de la cadena pesada se ajustó con éxito a la densidad y la comparación posterior con las estructuras de anticuerpos HA 3FKU y 3GBN permitió la colocación del dominio variable de la cadena ligera. Se realizaron rondas alternas de construcción y refinamiento de modelos usando COOT y REFMAC5, respectivamente. Esto se repitió hasta que se ajustó la mayor parte posible del mapa de densidad de electrones y los valores de R-work y R-free se habían nivelado.
Los aminoácidos en el archivo final de PDB están numerados siguiendo la convención de Kabat. El modelo final contiene todos los H1 HA y los dominios variables de cadena pesada y ligera. Para la cristalización de los complejos heterotriméricos de HA FI6 variante 3H3, se purificó el virus X-31 (H3N2) y e1HA liberado con bromelina (BHA) y los fragmentos Fab se prepararon por digestión con papaína. La variante 3 de FI6 Fab se purificó utilizando cromatografía de afinidad de proteína A sefarosa (HiTrap Protein A HP, 1 ml), seguida de columna de exclusión de tamaño S-200. 3,5 mg de Fab se mezclaron con 3 mg de X-31 BHA y se incubaron a 4°C durante la noche para la formación del complejo y el complejo se purificó utilizando una columna Superose 6 SEC. Las fracciones pico correspondientes al complejo Fab-HA se agruparon y se concentraron para la cristalización.
Los cristales del complejo variante 3 FI6-H3 se hicieron crecer por difusión de vapor en gotas que asentadas dispensadas por un robot de cristalización Oryx-6 de Douglas Instruments. Los cristales se protegieron criogénicamente por la adición de glicerol al 25% a la solución del reservorio. El conjunto de datos se recopiló en la Diamond Light Source, línea de haz 103, y se indexó, integró y ajustó usando Denzo y Scalepack. Los cristales, que contenían un trímero de H3 HA complejado con tres Fabs variante 3 de FI6 en la unidad asimétrica se resolvieron mediante sustitución molecular utilizando Amore. Los cálculos de reemplazo molecular se realizaron utilizando las coordenadas para la estructura de 2 A del trímero H3 HA, los dominios variables de cadena pesada y ligera de la variante 3 de FI6 de la variante 3 de FI6/H1 y los dominios constantes (ID de PDB 3HC0.pdb) como objetos de búsqueda independientes. La solución de reemplazo molecular se refinó con Refmac5 y Pheonix intercalados con rondas de ajustes manuales usando
Coot. Los mapas de densidad de electrones se mejoraron sustancialmente mediante promedios no cristalográficos utilizando DM.
La cristalografía de rayos X mostró que la variante 3 de FI6 se unía a un epítopo conservado en el subdominio F. Aunque las dos HA son filogenéticas y estructuralmente distintas y los complejos cristalizan con diferentes disposiciones de empaquetamiento, se encontró que las superficies de interacción eran muy similares (Figura 2, A y B). En ambos casos, cada monómero del trímero de HA se une a una molécula de la variante 3 de FI6 (Figura 3). El lazo HCDR3 de la variante 3 de FI6 se une a un surco poco profundo en el subdominio F de los HA, donde los lados del surco están formados por residuos de la hélice A de HA2 y partes de dos cadenas de HAl (38-42 y 318- 320), mientras que el fondo está formado por el bucle HA2 que abarca los residuos 18-21 (Figura 2, A y B).
El bucle HCDR3 cruza la hélice A en un ángulo de aproximadamente 45°, permitiendo a Leu-100A, Tyr-100C, Phe-100D y Trp-100F hacer un contacto hidrófobo con residuos en la ranura (Figura 4). Tyr-100C y Trp-100F también forman enlaces de hidrógeno potenciales con la cadena lateral de Thr-318 de HA1 y la cadena principal carbonilo del residuo 19 de HA1, respectivamente. Existen dos interacciones polares adicionales por los carbonilos de la cadena principal en los residuos 98 y 99 de HCDR3 con Asn-53 y Thr-49 en la hélice A. En conjunto, la interacción de HCDR3 con HA (H1 y H3) sepulta alrededor de 750 Á2 de la superficie del anticuerpo y aproximadamente 2/3 de esta interacción se explican por la interacción con la cadena HA2.
En general, las interacciones realizadas por la variante 3 de FI6 con el surco hidrófobo en H1 y H3 son notablemente similares. El lazo LCDR1 de la variante 3 de FI6 hace dos contactos con el lado de la hélice A, opuesto al lado que contribuye al surco hidrófobo; Phe-27D hace contacto hidrofóbico con la parte alifática de los enlaces de hidrógeno Lys-39 y Asn-28 a Asn-43, que en conjunto representan un área de superficie sepultada de aproximadamente 190 Á2 para H1 y H3. Con H1 HA, que se co-cristalizó en la forma no dividida, LCDR1 también hace contacto extenso con el "péptido de fusión" no dividido del monómero de HA derecho distal vecino (Figura 3, B y C y Figura. 4, A y B), lo que equivale a 320 Á2 adicionales de la superficie sepultada de la variante 3 de FI6.
Los residuos 28 y 29 de LCDR1 forman enlaces de hidrógeno de la amida de la cadena principal con los carbonilos de la cadena principal del residuo 329 de HA1 y el siguiente, pero un, residuo de HA2, Leu-2, abarcando así el sitio de escisión. Phe-27D de LCDR1 hace contactos hidrófobos con Leu-2 de1HA2 distal derecho vecino, mientras que el hidroxilo de la cadena lateral forma enlaces de hidrógeno en Tyr-29 con el carbonilo de la cadena principal del residuo 325 de la cadena HA1 derecha distal vecina. En contraste con la interacción muy similar entre las HA de H1 y H3 con HCDR3, la interacción de LCDR1 con el "péptido de fusión" del monómero de H3 HA distal derecho escindido vecino es significativamente menos extensa que la interacción formada por la H1 HA no escindida (Figura 3, inserto B). Aunque Phe-27D vuelve a hacer contacto con el resto alifático de Lys-39 de HA2, Tyr-29 hace contacto con un enlace de hidrógeno potencial con el carbonilo de la cadena principal de Ala-7 del HA2 distal derecho vecino (a diferencia del residuo 329 en H1 HA no escindido).
Por el contrario, no hay contactos de cadena principales entre el bucle LCDR1 y el "péptido de fusión" del H3 HA escindido, lo que representa el área de contacto más pequeña de 114 Á (cf 320Á2 en H1 HA). También parece que la escisión del precursor de HA para producir H3 HA, da como resultado una
orientación ligeramente diferente de la variante 3 de FI6 con respecto a la HA en los complejos variante 3 de FI6/H3 y variante 3 de FI6/H1. Los residuos de contacto en la interfaz entre las cadenas VH y VL de la variante 3 de FI6 y el HA homotrimérico H3 escindido se muestran en la Tabla 9. Los residuos de contacto en la interfaz entre las cadenas VH y VL de la variante 3 de FI6 y el HA homotrimérico no escindido se indican en la Tabla 10.
Tabla 9. Residuos de contacto en la interfaz entre FI6 Variante 3 VH y VL y el Trímero H3-HA escindido

Tabla 9 (continuación). Residuos de contacto en la interfaz entre FI6 Variante 3 VH y VL y Trímero H3-HA escindido

Tabla 10. Residuos de contacto en la interfaz entre FI6 Variante 3 VH y VL y Trímero H1-HA no escindido

Tabla 10 (continuación). Residuos de contacto en la interfaz entre FI6 Variante 3 VH y VL y Trímero H1-HA no escindido

Las estructuras de dos anticuerpos de reacción cruzada CR6261 y F10, que son específicos del Grupo 1, se han descrito previamente como complejos con H5 y H1 HA. Los anticuerpos CR6261 y F10 que se unen a HA están mediados solo por sus dominios VH orientados aproximadamente de la misma manera con respecto al HA, pero ambos anticuerpos están significativamente girados en relación con la variante 3 de FI6 y están 5-10 A más cerca del extremo proximal de la membrana de HA (Figura 3, D y E).
Las estructuras de la variante 3 de FI6/H1 y la variante 3 de FI6/H3 aquí mostradas también revelan que, aunque los sitios de unión en HA de los tres anticuerpos se superponen ampliamente, la naturaleza de las interacciones creadas por la variante 3 de FI6 son marcadamente diferentes de las creadas por CR6261 y
los anticuerpos F10. La diferencia más notable es que la interacción de la variante 3 de FI6 con el surco hidrófobo en HA está mediada únicamente por la longitud de HCDR3, mientras que para CR6261 y F10 los tres HCDR están involucrados en la unión.
Una diferencia importante entre los complejos FI6 variante 3/H1 y FI6 variante 3/H3 es que H3 HA está glicosilada en Asn-38 (HA1), al igual que las HA H7, H10 y H15 del Grupo 2, mientras que la de H1, al igual que todos las HA de Grupos 1, no lo está. En la estructura no unida de H3, esta cadena lateral de carbohidratos se proyecta desde la hebra beta de HA1, que contiene el residuo Asn-38, hacia la hélice A de HA2 de la misma subunidad HA, de manera que se superpone la huella de la variante 3 de FI6 (Figura 5A). Se sabe que las cadenas de carbohidratos laterales influyen en la antigenicidad de las glicoproteínas del virus, por lo que se ha sugerido que este solapamiento explica la falta de unión a las HA del Grupo 2 de otros anticuerpos reactivos cruzados del Grupo 1 que se dirigen a la región proximal de la membrana de HA. Sin embargo, la unión de la variante 3 de FI6 a H3 HA se habilita mediante la reorientación del oligosacárido, una rotación de aproximadamente 80° de la superficie de1HA, de modo que hace nuevos contactos con Asp-53 y Asn-55 del bucle HCDR2 (Figura 5B).
Dado que la flexibilidad de la cadena de carbohidratos lateral en Asn-38 permite adaptarse a la variante 3 de FI6 al H3 HA, preguntamos si este sitio de glicosilación probablemente sea la razón por la que e1H3 HA no se une a CR6261 o F10. El modelado simple sugiere que el mismo cambio en la orientación de la cadena carbohidrato lateral sería compatible con la unión del CR6261, pero no con F10, anticuerpos reactivos cruzados. El giro beta que abarca los residuos 73-77 de VH de F10 chocaría con el carbohidrato ligado a Asn-38 en la orientación que adopta en el complejo variante 3 de FI6/H3 y no está claro si el carbohidrato podría girar libremente fuera del sitio de unión para acomodar la unión de F10. Sin embargo, ni CR6261 ni F10 pudieron neutralizar un pseudovirus H7 (A/pollo/Italy/99) en el que se eliminó el sitio de glicosilación (Asn-38) (IC50 > 50 pg/ml), lo que indica que el impedimento estérico del glicano no es la única restricción estructural que impide la unión de los anticuerpos CR6261 y F10 a las HA del Grupo 2.
Además de la glicosilación de Asn-38, la diferencia más notable en la estructura del subdominio F entre el Grupo 1 y el Grupo 2 de HA implica el entorno y la orientación distintivos del Grupo de HA2 Trp-21. En las HA del Grupo 1, T rp-21 es aproximadamente paralelo a la superficie del subdominio F, mientras que en las HA del Grupo 2 está orientado aproximadamente perpendicular a la superficie (Figura 5, C y D). Los tres anticuerpos (FI6 variante 3, CR6261 y F10) hacen contacto con Trp-21, principalmente a través de una cadena lateral de fenilalanina; Phe-100D en FI6 variante 3, Phe-54 en CR6261 y Phe-55 en F10 (Figura 5, C y D). En el caso de FI6 variante 3, los reordenamientos locales en el bucle HCDR3 significan que Phe-100D se encuentra aproximadamente 2 A más profundo en el surco hidrófobo en el complejo H1 que en el complejo H3; por tanto, mantiene una distancia de contacto similar con Trp-21 en ambos casos.
Los dos anticuerpos específicos del Grupo 1 posicionan a Phe-54 (CR6261) y Phe-55 (F10) de manera similar a la variante 3 de FI6 en complejo con H1 HA. Sin embargo, como Phe-54 (CR6261) y Phe-55 (F10) están ubicados en el lazo corto HCDR2, que conecta dos hebras antiparalelas adyacentes, parece que hay menos flexibilidad que en la variante 3 de FI6 para que la fenilalanina se mueva más allá del surco hidrófobo con el fin de acomodar la orientación del Grupo-2 de Trp-21. Por tanto, la unión de CR6261 y F10 a las HA del Grupo 2 probablemente esté bloqueada por un choque estérico entre la fenilalanina HCDR2 y Trp-21.
En resumen, los datos estructurales obtenidos indican que, aunque el epítopo central en la hélice A es similar al reconocido por CR6261 y F10, la variante 3 de FI6 se une con un ángulo diferente, 5-10 A más de membrana distal y entra en contacto con un área más grande que abarca la hélice A y que se extiende al péptido de fusión del monómero derecho distal vecino, tanto en forma escindida como no escindida (Figura 6). La unión a la variante 3 de FI6 está mediada por CDR tanto de VH como de VL, con contribuciones destacadas del HCDR3 largo, que acomoda diferentes conformaciones del bucle Trp-21 específico del Grupo, y del LCDR1 fuertemente mutado. El uso de las cadenas VH y VK y e1HCDR3 largo son característicos de los anticuerpos seleccionados naturalmente y contrastan con la propiedad de los anticuerpos derivados de fagos, como CR6261 y F10, que se unen utilizando solo la cadena VH. Los residuos de contacto en la variante 3 de FI6 VH y VK se muestran en la Figura 7.
Ejemplo 5. Efecto profiláctico in vivo de la variante 3 de FI6
La eficacia protectora de la variante 3 de FI6 se probó in vivo en modelos de ratón de infección por el virus de la influenza A. Grupos de ratones BALB/c hembra de 6 a 8 semanas de edad fueron inyectados vía intravenosa (i.v.) con anticuerpos purificados en concentraciones que variaban de 1 a 16 mg/kg. Tres horas después, los ratones se anestesiaron profundamente y se testaron vía intranasal (i.n.) con 10 MLD50 (dosis letal del ratón del cincuenta por ciento) de H1N1 A/PR/8/34. En entorno terapéutico, los ratones recibieron el anticuerpo 1,2 o 3 días después de la infección. Los ratones se controlaron diariamente para determinar la supervivencia y la pérdida de peso hasta el día 14 post-infección (p.i.). Los animales que perdieron más del 25% de su peso corporal inicial fueron sacrificados de acuerdo con el protocolo de estudio en animales.
Para evaluar la influencia de la variante 3 de FI6 en la replicación viral, los ratones expuestos a 10 MLD50 de H1N1 A/PR/8/34 recibieron el anticuerpo en diferentes puntos de tiempo y se sacrificaron cuatro días después para recolectar pulmones y cerebros. Los tejidos se homogeneizaron en medio Leibovitz L-15 (Invitrogen) suplementado con una solución antibiótico-antimicótica (Invitrogen) para lograr una suspensión de órganos al 10% p/v. Los homogenados de órganos se valoraron en células MDCK y se determinaron las titulaciones virales. En entorno profiláctico, la variante 3 de FI6 fue totalmente protectora y, cuando se administró a 4 mg/kg, fue parcialmente protectora (80% de supervivencia) cuando se administró a 2 mg/kg a ratones infectados con el virus del Grupo 1 H1N1 A/PR/8/34 (Figura 8). Los títulos de virus pulmonar el día cuatro después de la infección se redujeron aproximadamente cien veces en ratones tratados con la variante 3 de FI6 el día 0 o 1 día después de la infección (Figura 9). Además, la variante 3 de FI6 previno la pérdida de peso corporal de ratones infectados con el virus H3N2 HK-x31 del Grupo 2 (Figura 8).
Ejemplo 6. Mecanismos de neutralización viral por la variante 3 de FI6
Para experimentos in vivo dirigidos a determinar la eficacia protectora de los anticuerpos FI6, se produjeron mutantes Fc de la variante 2 de FI6 que carecían de la unión al complemento (FI6-v2 KA) o del complemento y la unión a FcR (FI6-v2 LALA). Estos anticuerpos mostraron la misma unión y propiedades neutralizadoras in vitro que la variante 2 de FI6 y vidas medias comparables in vivo (valores medios 3,3, 3,4 y 3,5 días para FI6-v2, FI6-v2 KA y FI6-v2 LALA, respectivamente). Su eficacia protectora se probó en ratones con infección letal por el virus A/Puerto Rico/8/34 (H1N1). FI6.v2 protegió completamente a los ratones de la letalidad cuando se administró a 4 mg/kg y protegió al 80% de los ratones a 2 mg/kg (Figura 8F). Cuando se administraron a 10 mg/kg, FI6-v2 y FI6-v2 KA fueron totalmente protectores, mientras que FI6-v2 LALA mostró una pérdida sustancial de actividad, pudiendo proteger solo al 40% de los animales (Figura 8F).
Esta disminución de la eficacia era particularmente evidente cuando se administraban anticuerpos mutantes a la concentración límite de 3 mg/kg (Figura 8G).
Para investigar los mecanismos que contribuyen a la actividad neutralizadora de la variante 3 de FI6, se incubó el HA derivado de bacilo NC/99 (Protein Sciences Corporation) durante 40 minutos a 37°C con una cantidad molar 15 veces mayor de la variante 3 de FI6, FE17, mAb no específico (HBD85) o sin mAb, en solución de PBS. Se añadió tripsina tratada con TPCK a cada muestra hasta una concentración final de 2,5 pg/ml y la digestión se realizó a 37°C durante 5, 10 y 20 minutos. En cada punto de tiempo, la digestión se detuvo agregando un tampón que contenía SDS y DTT e hirviendo a 95°C durante 5 minutos. Las muestras se cargaron luego en un gel de poliacrilamida con tris-glicina al 12%. La transferencia de proteínas en una membrana de PVDF se realizó con el sistema de transferencia iBlot de Invitrogen. La membrana de PVDF se bloqueó durante 30 minutos con leche en polvo desnatada al 10% en TBS-Tween. La incubación con anticuerpo primario contra HA0 (F032 biotinilado producido en casa) se realizó a 0,5 pg/ml en TBS-Tween durante la noche a 4°C. El PVDF se lavó tres veces con TBS-Tween y se incubó durante 1 hora a temperatura ambiente con estreptavidina conjugada con HRP (Sigma).
La membrana de PVDF se lavó tres veces con TBS-Tween y se detectaron bandas positivas utilizando el reactivo de detección de transferencia Western ECL Plus™ (GE Healthcare) y el sistema de cámara CCD LAS4000. Los datos de la Figura 10 demuestran que la variante 3 de FI6 inhibe la escisión de HA0 por la TPCK-tripsina, lo que indica que la unión de la cadena ligera del anticuerpo a la HA0 no procesada bloquea la infectividad, al menos para aquellos virus en los que se produce la escisión extracelular.
Referencias
Okuno et al, (1993) Journal of Virology 67: 2552
Gerhard et al., (2006) Emerging Infectious Diseases 12: 569
Gioia et al., (2008) Emerging Infectious Diseases 14: 121
US 3.766162
US 3.791,932
US 3.817.837
US 4.233.402
US 4.676,980
US 4.831.175
US 5.595.721
WO00 /52031
WO00 /52473
US 4.766106
US 4.179.337
US 4.495.285
US 4.609.546
Gabizon et al., (1982) Cancer Research 42:4734
Cafiso (1981) Biochem Biophys Acta 649: 129
Szoka (1980) Ann. Rdo. Biofis Ing. 9: 467
Poznansky et al., (1980) Dmg Delivery Systems (R.L. Juliano, ed., Oxford, N.Y.) pp. 253-315
Poznansky (1984) Pharm Revs 36:277
Kohler, G. y Milstein, C 1975, Nature 256: 495-497.
Kozbar et al., 1983, Immunology Today 4:72.
WO2004 /076677
Capítulo 4 de Kuby Immunology (4a edición, 2000; ASIN: 0716733315 Jones et al., Biotechnol Prog 2003,19 (1): 163-8
Cho et al., Cytotechnology 2001, 37: 23-30
Cho et al., Biotechnol Prog 2003,19: 229-32
US 5.807.715
US 6.300.104
Rowe et al., (1999) J Clinic Microbiol 37 (4): 937-43.
Temperton, et al., (2005). Emerg Infect Dis 11, 411-416.
Smirnov et al., (2000). Arch Virol 145, 1733-1741.
Smirnov et al., (1999). Acta Virol 43, 237-244.
Simmons et al., (2007). PLoS Med 4, el78.
Traggiai et al., (2004). Nat Med 10, 871-875.
Claims (1)
- REIVINDICACIONESAnticuerpo aislado, o un fragmento de unión a antígeno del mismo, que neutraliza la infección de un subtipo del grupo 1 y un subtipo del grupo 2 del virus de la influenza A y que comprende: i) las secuencias de cadena pesada de CDR1, CDR2 y CDR3, tal como se establecen en las SEQ ID NO: 1, 41 y 43, respectivamente, o tal como se establecen en las SEQ ID NO: 1, 41 y 42, respectivamente; y ii) las secuencias de cadena ligera de CDR1, CDR2 y CDR3, tal como se establecen en las SEQ ID NO: 4, 5 y 6, respectivamente, o como se establecen en las SEQ ID NO: 44, 5 y 6, respectivamente, donde dicho anticuerpo o fragmento de unión al antígeno del mismo comprende una región variable de cadena pesada que tiene al menos un 90% de identidad de secuencia con la secuencia de aminoácidos tal como se establece en las SEQ ID NO: 59 o 55; y una región variable de cadena ligera que tiene una identidad de secuencia de al menos el 90% con la secuencia de aminoácidos según se establece en las SEQ ID NO: 57 o 61 y donde dicho anticuerpo o fragmento de unión a antígeno del mismo se produce en células transfectadas a títulos al menos 3 veces más altos que aquellos a los que se produce la variante 2 de FI6.Anticuerpo aislado, o un fragmento de unión a antígeno del mismo, de la reivindicación 1, que comprende una región variable de cadena pesada que comprende la secuencia de aminoácidos de SEQ ID NO: 59 y una región variable de cadena ligera que comprende la secuencia de aminoácidos de SEQ ID NO: 57; o una región variable de cadena pesada que comprende la secuencia de aminoácidos de SEQ ID NO: 59 y una región variable de cadena ligera que comprende la secuencia de aminoácidos de SEQ ID NO: 61; o una región variable de cadena pesada que comprende la secuencia de aminoácidos de SEQ ID NO: 55 y una región variable de cadena ligera que comprende la secuencia de aminoácidos de SEQ ID NO: 57; o una región variable de cadena pesada que comprende la secuencia de aminoácidos del SEQ ID NO: 55 y una región variable de cadena ligera que comprende la secuencia de aminoácidos de SEQ ID NO: 61, y donde el anticuerpo neutraliza un subtipo del grupo 1 y un subtipo del grupo 2 del virus de la influenza A.Anticuerpo aislado, o un fragmento de unión a antígeno del mismo, de la reivindicación 1, que neutraliza la infección de un subtipo del grupo 1 y de un subtipo del grupo 2 del virus de la influenza A y se une específicamente a un epítopo en la región del tallo de un trímero HA de la influenza A, donde la cadena pesada y ligera de dicho anticuerpo, o de su fragmento de unión a antígeno, entra en contacto con aminoácidos en un primer monómero proximal y en un segundo monómero distal derecho de dicho trímero y donde anticuerpo o su fragmento de unión a antígeno comprende: i) las secuencias de cadena pesada de CDR1, CDR2 y CDR3 tal como se establecen en las SEQ ID NO: 1, 41 y 43, respectivamente, o tal como se establecen en las SEQ ID NO: 1, 41 y 42, respectivamente; y ii) las secuencias de cadena ligera de CDR1, CDR2 y CDR3 tal como se establecen en las SEQ ID NO: 4, 5 y 6, respectivamente, o tal como se establecen en las SEQ ID NO: 44, 5 y 6, respectivamente.Anticuerpo aislado, o un fragmento de unión a antígeno del mismo, de la reivindicación 1, que neutraliza la infección de un subtipo del grupo 1 y de un subtipo del grupo 2 del virus de la influenza A y se une específicamente a un epítopo en la región del tallo de un trímero de hemaglutinina (HA) de la influenza A, donde la cadena pesada y la cadena ligera de dicho anticuerpo, o de un fragmento de unión a antígeno del mismo, entra en contacto aminoácidos en un primer monómero proximal y en un segundo monómero distal derecho de dicho trímero de HA, y donde dicho anticuerpo, o fragmento de unión a antígeno del mismo, se produce en células transfectadas a títulos al menos 3 veces más altos que el título al que se produce la variante 2 de FI6, donde dicho anticuerpo, o fragmento de unión a antígeno del mismo, comprende: i) las secuencias de cadena pesada de CDR1, CDR2 y CDR3 tal como se establecen en las SEQ ID NO: 1, 41 y 43, respectivamente, o tal como se establecen en las SEQ ID NO: 1, 41 y 42, respectivamente; y ii) las secuencias de cadena ligera de CDR1, CDR2 y CDR3 tal como se establecen en las SEQ ID NO: 4, 5 y 6, respectivamente, o tal como se establecen en las SEQ ID NO: 44, 5 y 6, respectivamente.Anticuerpo, o fragmento de unión a antígeno del mismo, de la reivindicación 3 o 4, seleccionado del grupo consistente en:a. un anticuerpo, o un fragmento de unión a antígeno del mismo donde la cadena pesada de dicho anticuerpo, o del fragmento de unión a antígeno del mismo, contacta con residuos aminoácidos en dicho monómero proximal y donde la cadena ligera de dicho anticuerpo, o del fragmento de unión a antígeno del mismo, contacta con residuos aminoácidos en dicho monómero proximal y en dicho monómero derecho distal del citado trímero de HA yb. un anticuerpo o un fragmento de unión a antígeno que se une específicamente a un epítopo en la región del tallo de un trímero HA de la influenza A y donde el primer o segundo monómero de dicho trímero HA de la influenza A está escindido o no escindido.Anticuerpo, o fragmento de unión a antígeno del mismo, de la reivindicación 3 o 4, seleccionado del grupo consistente en:a. un anticuerpo, o fragmento de unión a antígeno del mismo, donde la cadena pesada de dicho anticuerpo, o de su fragmento de unión a antígeno, entra en contacto con el aminoácido en la posición 318 en HA1 y con residuos aminoácidos en las posiciones 18, 19, 20, 21, 38, 41, 42, 45, 49, 53 y 57 en HA2 de dicho primer o segundo monómero, y donde dicho monómero está escindido o no escindido;b. un anticuerpo, o fragmento de unión a antígeno del mismo, donde la cadena ligera de dicho anticuerpo, o de su fragmento de unión a antígeno, entra en contacto con residuos aminoácidos en las posiciones 38, 39 y 43 en HA2 de dicho monómero proximal y con los residuos aminoácidos en las posiciones 327, 328 y 329 en HA1 y 1, 2, 3 y 4 en HA2 de dicho monómero distal derecho, y donde dichos monómeros proximal y distal derecho están no escindidos;c. un anticuerpo, o fragmento de unión a antígeno del mismo, donde la cadena ligera de dicho anticuerpo, o su fragmento de unión a antígeno, entra en contacto con residuos aminoácidos en las posiciones 38, 39, 42 y 46 en HA2 de dicho monómero proximal y con residuos aminoácidos en las posiciones 321 y 323 en HA1 y 7 y 11 en HA2 de dicho monómero distal derecho, y donde dichos monómeros proximal y distal derecho están escindidos;d. un anticuerpo, o fragmento de unión a antígeno del mismo, que se une específicamente a un epítopo que comprende el aminoácido en la posición 318 de HA1 y los residuos aminoácidos en las posiciones 18, 19, 20, 21, 38, 39, 41, 42, 43, 45, 48, 49, 53, 56, y 57 de HA2 de dicho monómero proximal, y los residuos aminoácidos en las posiciones 327, 328, 329 de HA1 y los residuos aminoácidos en las posiciones 1, 2, 3, y 4 del polipéptido HA2 de dicho monómero distal derecho, donde dichos monómeros proximal y distal están no escindidos;e. un anticuerpo, o fragmento de unión a antígeno del mismo, que se une específicamente a un epítopo que comprende el aminoácido en la posición 318 de HA1 y los residuos aminoácidos en las posiciones 18, 19, 20, 21,38, 39, 41,42, 45, 46, 49, 52, 53 y 57 de HA2 de dicho monómero proximal y los residuos aminoácidos en las posiciones 321 y 323 de HA1 y los residuos aminoácidos en las posiciones 7 y 11 de HA2 de dicho monómero distal derecho, donde dichos monómeros proximal y distal derecho están escindidos; yf. un anticuerpo, o fragmento de unión a antígeno del mismo, que se une específicamente a un epítopo que comprende el aminoácido en la posición 329 de HA1 y los residuos aminoácidos en las posiciones 1, 2, 3 y 4 de HA2, donde dichos HA1 y HA2 están presentes en un monómero no escindido de dicho trímero HA.7. Anticuerpo de cualquiera de las reivindicaciones anteriores, o un fragmento de unión a antígeno del mismo, donde el anticuerpo es un anticuerpo humano, un anticuerpo monoclonal, un anticuerpo purificado, un anticuerpo de cadena única, Fab, Fab', F(ab')2, Fv o scFv.8. Molécula de ácido nucleico que comprende un polinucleótido que codifica el anticuerpo, o un fragmento de unión a antígeno del mismo, de cualquiera de las reivindicaciones anteriores.9. Molécula de ácido nucleico según la declaración 8, donde el polinucleótido comprende una secuencia que es al menos un 75% idéntica a la secuencia de ácido nucleico establecida en cualquiera de las SEQ ID NO: 45-54, 56, 58, 60 o 62, o donde el polinucleótido comprende una secuencia tal como se establece en las SEQ ID NO: 60 y 58 o las SEQ ID NO: 60 y 62 o las SEQ ID NO: 56 y 58; o las SEQ ID NO: 56 y 62.10. Vector que comprende la molécula de ácido nucleico de la reivindicación 8 o 9.11. Célula que expresa el anticuerpo de cualquiera de las reivindicaciones 1-7, o un fragmento de unión a antígeno del mismo; o que comprende el vector de la reivindicación 8 o 9.12. Composición farmacéutica que comprende el anticuerpo de cualquiera de las reivindicaciones 1-7, o un fragmento de unión a antígeno del mismo, el ácido nucleico de la reivindicación 8 o 9, el vector de la reivindicación 10, o la célula de la reivindicación 11, y un diluyente o vehículo farmacéuticamente aceptable.13. Anticuerpo de cualquiera de las reivindicaciones 1-7, o un fragmento de unión a antígeno del mismo, ácido nucleico de la reivindicación 8 o 9, vector de la reivindicación 10, célula de la reivindicación 11 o composición farmacéutica de la reivindicación 12 para su uso (i) en el tratamiento de la infección por el virus de la influenza A, (ii) en una vacuna o (iii) en el diagnóstico de infección por virus de la influenza A.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/IB2011/002329 WO2013011347A1 (en) | 2011-07-18 | 2011-07-18 | Neutralizing anti-influenza a virus antibodies and uses thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2732552T3 true ES2732552T3 (es) | 2019-11-25 |
Family
ID=44993618
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES11784765T Active ES2732552T3 (es) | 2011-07-18 | 2011-07-18 | Anticuerpos neutralizantes del virus de la influenza A y usos de los mismos |
| ES18180926T Active ES2841899T3 (es) | 2011-07-18 | 2011-07-18 | Anticuerpos neutralizantes del virus de la influenza A y usos de los mismos |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18180926T Active ES2841899T3 (es) | 2011-07-18 | 2011-07-18 | Anticuerpos neutralizantes del virus de la influenza A y usos de los mismos |
Country Status (15)
| Country | Link |
|---|---|
| US (3) | US9587010B2 (es) |
| EP (3) | EP3418300B1 (es) |
| JP (1) | JP6035332B2 (es) |
| CN (1) | CN103717617B (es) |
| AU (1) | AU2011373387B2 (es) |
| CA (1) | CA2841551C (es) |
| DK (2) | DK3418300T3 (es) |
| ES (2) | ES2732552T3 (es) |
| HR (2) | HRP20190714T1 (es) |
| HU (2) | HUE051756T2 (es) |
| LT (2) | LT3418300T (es) |
| MX (1) | MX352338B (es) |
| PL (2) | PL2734545T3 (es) |
| SI (2) | SI3418300T1 (es) |
| WO (1) | WO2013011347A1 (es) |
Families Citing this family (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7960139B2 (en) | 2007-03-23 | 2011-06-14 | Academia Sinica | Alkynyl sugar analogs for the labeling and visualization of glycoconjugates in cells |
| ES2442024T3 (es) | 2008-07-15 | 2014-02-07 | Academia Sinica | Matrices de glucano sobre portaobjetos de vidrio revestidos con aluminio de tipo PTFE y métodos relacionados |
| US10087236B2 (en) | 2009-12-02 | 2018-10-02 | Academia Sinica | Methods for modifying human antibodies by glycan engineering |
| US11377485B2 (en) | 2009-12-02 | 2022-07-05 | Academia Sinica | Methods for modifying human antibodies by glycan engineering |
| WO2011130332A1 (en) | 2010-04-12 | 2011-10-20 | Academia Sinica | Glycan arrays for high throughput screening of viruses |
| KR101745386B1 (ko) | 2010-05-10 | 2017-06-09 | 아카데미아 시니카 | 항-인플루엔자 활성을 가진 자나미비르 포스포네이트 동족체 및 인플루엔자 바이러스의 오셀타미비르 감수성을 확인하는 방법 |
| CN107417789A (zh) * | 2011-07-18 | 2017-12-01 | 生物医学学会 | 中和抗甲型流感病毒抗体及其用途 |
| MX352338B (es) | 2011-07-18 | 2017-11-17 | Inst Res Biomedicine | Anticuerpos neutralizantes del virus de la influenza a y usos de estos. |
| US10130714B2 (en) | 2012-04-14 | 2018-11-20 | Academia Sinica | Enhanced anti-influenza agents conjugated with anti-inflammatory activity |
| US9969794B2 (en) | 2012-05-10 | 2018-05-15 | Visterra, Inc. | HA binding agents |
| AU2013306098A1 (en) | 2012-08-18 | 2015-02-12 | Academia Sinica | Cell-permeable probes for identification and imaging of sialidases |
| EP2888238A4 (en) | 2012-08-21 | 2016-01-27 | Academia Sinica | BENZOCYCLO-OCTYNE COMPOUNDS AND USES THEREOF |
| SG11201503734UA (en) | 2012-11-13 | 2015-06-29 | Genentech Inc | Anti-hemagglutinin antibodies and methods of use |
| KR20140118682A (ko) * | 2013-03-29 | 2014-10-08 | (주)셀트리온 | 2 이상의 인플루엔자 a 바이러스 중화 결합 분자를 포함하는 조성물 |
| US10086054B2 (en) | 2013-06-26 | 2018-10-02 | Academia Sinica | RM2 antigens and use thereof |
| WO2014210564A1 (en) | 2013-06-27 | 2014-12-31 | Academia Sinica | Glycan conjugates and use thereof |
| JP6486368B2 (ja) | 2013-09-06 | 2019-03-20 | アカデミア シニカAcademia Sinica | 改変されたグリコシル基を含む糖脂質を用いたヒトiNKT細胞の活性化 |
| CN113667013B (zh) * | 2013-10-02 | 2024-04-09 | 免疫医疗有限责任公司 | 中和抗甲型流感抗体及其用途 |
| US9982041B2 (en) | 2014-01-16 | 2018-05-29 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| US10150818B2 (en) | 2014-01-16 | 2018-12-11 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| US20170174751A1 (en) | 2014-03-19 | 2017-06-22 | Dana-Farber Cancer Institute, Inc. | Immunogenetic restriction on elicitation of antibodies |
| EP3129767B1 (en) | 2014-03-27 | 2021-09-01 | Academia Sinica | Reactive labelling compounds and uses thereof |
| WO2015166329A1 (en) | 2014-05-01 | 2015-11-05 | Indian Institute Of Science | Polypeptides for generating anti-influenza antibodies and uses thereof |
| CN106661099A (zh) | 2014-05-27 | 2017-05-10 | 中央研究院 | 抗her2醣抗体及其用途 |
| US10118969B2 (en) | 2014-05-27 | 2018-11-06 | Academia Sinica | Compositions and methods relating to universal glycoforms for enhanced antibody efficacy |
| CA2950423A1 (en) | 2014-05-27 | 2015-12-03 | Academia Sinica | Compositions and methods relating to universal glycoforms for enhanced antibody efficacy |
| KR20240096599A (ko) | 2014-05-27 | 2024-06-26 | 아카데미아 시니카 | 항-cd20 글리코항체 및 이의 용도 |
| WO2015184001A1 (en) | 2014-05-28 | 2015-12-03 | Academia Sinica | Anti-tnf-alpha glycoantibodies and uses thereof |
| EP3169407A4 (en) | 2014-07-15 | 2018-04-25 | Medimmune, LLC | Neutralizing anti-influenza b antibodies and uses thereof |
| KR102422375B1 (ko) | 2014-09-08 | 2022-07-18 | 아카데미아 시니카 | 당지질을 사용한 인간 iNKT 세포 활성화 |
| TWI701258B (zh) | 2014-12-19 | 2020-08-11 | 美商再生元醫藥公司 | 流行性感冒病毒血球凝集素之人類抗體 |
| US9975965B2 (en) | 2015-01-16 | 2018-05-22 | Academia Sinica | Compositions and methods for treatment and detection of cancers |
| US10495645B2 (en) | 2015-01-16 | 2019-12-03 | Academia Sinica | Cancer markers and methods of use thereof |
| AU2015378564A1 (en) | 2015-01-24 | 2017-07-13 | Academia Sinica | Novel glycan conjugates and methods of use thereof |
| BR112017016417A2 (pt) * | 2015-02-05 | 2018-03-27 | Janssen Vaccines & Prevention B.V. | moléculas de ligação dirigidas contra hemaglutinina da gripe e seus usos |
| GB201502137D0 (en) * | 2015-02-09 | 2015-03-25 | Ucl Business Plc | Treatment |
| WO2016164835A1 (en) * | 2015-04-08 | 2016-10-13 | Dana-Farber Cancer Institute, Inc. | Humanized influenza monoclonal antibodies and methods of use thereof |
| CN107667114B (zh) | 2015-06-01 | 2021-07-02 | 免疫医疗有限责任公司 | 中和抗流感结合分子及其用途 |
| EP3374390A1 (en) | 2015-11-13 | 2018-09-19 | Visterra, Inc. | Compositions and methods for treating and preventing influenza |
| IL260413B2 (en) * | 2016-01-13 | 2024-01-01 | Medimmune Llc | A method for treating type A influenza |
| AU2017222564A1 (en) | 2016-02-24 | 2018-09-06 | Visterra, Inc. | Formulations of antibody molecules to influenza virus |
| JP2019515876A (ja) | 2016-03-08 | 2019-06-13 | アカデミア シニカAcademia Sinica | N−グリカンおよびそのアレイのモジュール合成のための方法 |
| WO2017192589A1 (en) * | 2016-05-02 | 2017-11-09 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Neutralizing antibodies to influenza ha and their use and identification |
| KR20220035288A (ko) * | 2016-05-05 | 2022-03-21 | 더 트러스티스 오브 더 유니버시티 오브 펜실바니아 | 인플루엔자 바이러스를 표적으로 하는 dna 단일 클론 항체 |
| CA3034057A1 (en) | 2016-08-22 | 2018-03-01 | CHO Pharma Inc. | Antibodies, binding fragments, and methods of use |
| WO2018038096A1 (ja) * | 2016-08-23 | 2018-03-01 | 国立大学法人東京大学 | モノクローナル抗体又はその抗原結合フラグメント及びその使用 |
| CN107868121B (zh) * | 2016-09-28 | 2021-03-16 | 中国医学科学院病原生物学研究所 | H3亚型流感病毒血凝素特异性保守表位、其抗体及其应用 |
| CA3051377A1 (en) | 2017-01-27 | 2018-08-02 | National Research Council Of Canada | Hemagglutinin-specific antibodies and uses thereof |
| CN113924147A (zh) | 2019-03-25 | 2022-01-11 | 威特拉公司 | 用于治疗和预防流感的组合物和方法 |
| US20220340644A1 (en) * | 2019-09-19 | 2022-10-27 | Duke University | Influenza neutralizing antibodies and their uses |
| TWI817041B (zh) * | 2019-09-20 | 2023-10-01 | 中央研究院 | 嵌合血球凝集素蛋白質及包含其之疫苗組成物 |
| AU2020403133A1 (en) | 2019-12-11 | 2022-06-23 | Visterra, Inc. | Compositions and methods for treating and preventing influenza |
| IL312538A (en) * | 2021-11-05 | 2024-07-01 | Abviro Llc | Broadly reactive human monoclonal influenza antibodies and methods for their use |
| CN117430664B (zh) * | 2023-10-24 | 2024-04-09 | 暨南大学附属第六医院(东莞市东部中心医院) | 一种甲型流感病毒t细胞抗原表位肽及其应用 |
Family Cites Families (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB819376A (en) | 1956-10-20 | 1959-09-02 | Colne Switchgear K & W Ltd | Improvements in electro-magnetic overload units for electrical apparatus |
| NL154600B (nl) | 1971-02-10 | 1977-09-15 | Organon Nv | Werkwijze voor het aantonen en bepalen van specifiek bindende eiwitten en hun corresponderende bindbare stoffen. |
| US3817837A (en) | 1971-05-14 | 1974-06-18 | Syva Corp | Enzyme amplification assay |
| US3766162A (en) | 1971-08-24 | 1973-10-16 | Hoffmann La Roche | Barbituric acid antigens and antibodies specific therefor |
| US4179337A (en) | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| US4233402A (en) | 1978-04-05 | 1980-11-11 | Syva Company | Reagents and method employing channeling |
| JPS5896026A (ja) | 1981-10-30 | 1983-06-07 | Nippon Chemiphar Co Ltd | 新規ウロキナ−ゼ誘導体およびその製造法ならびにそれを含有する血栓溶解剤 |
| DE3380726D1 (en) | 1982-06-24 | 1989-11-23 | Japan Chem Res | Long-acting composition |
| US5807715A (en) | 1984-08-27 | 1998-09-15 | The Board Of Trustees Of The Leland Stanford Junior University | Methods and transformed mammalian lymphocyte cells for producing functional antigen-binding protein including chimeric immunoglobulin |
| US4766106A (en) | 1985-06-26 | 1988-08-23 | Cetus Corporation | Solubilization of proteins for pharmaceutical compositions using polymer conjugation |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US4831175A (en) | 1986-09-05 | 1989-05-16 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Backbone polysubstituted chelates for forming a metal chelate-protein conjugate |
| US5595721A (en) | 1993-09-16 | 1997-01-21 | Coulter Pharmaceutical, Inc. | Radioimmunotherapy of lymphoma using anti-CD20 |
| CA2229043C (en) | 1995-08-18 | 2016-06-07 | Morphosys Gesellschaft Fur Proteinoptimierung Mbh | Protein/(poly)peptide libraries |
| WO1998057993A1 (en) | 1997-06-19 | 1998-12-23 | The Regents Of The University Of California | Secretory immunoglobulin produced by single cells and methods for making and using same |
| MY133346A (en) | 1999-03-01 | 2007-11-30 | Biogen Inc | Kit for radiolabeling ligands with yttrium-90 |
| US20020102208A1 (en) | 1999-03-01 | 2002-08-01 | Paul Chinn | Radiolabeling kit and binding assay |
| WO2001062907A1 (en) | 2000-02-22 | 2001-08-30 | Medical & Biological Laboratories Co., Ltd. | Antibody library |
| AU2003251809A1 (en) | 2002-07-11 | 2004-02-02 | The General Hospital Corporation | Polynucleotide and polypeptide fat metabolism regulators and uses thereof |
| AU2004215125B2 (en) | 2003-02-26 | 2011-01-06 | Institute For Research In Biomedicine | Monoclonal antibody production by EBV transformation of B cells |
| CN1756562A (zh) * | 2003-03-07 | 2006-04-05 | 麦克公司 | 流感病毒疫苗 |
| AU2004263037B2 (en) | 2003-08-11 | 2011-02-10 | Japan As Represented By The Director-General Of National Institute Of Infectious Diseases | Novel vaccine containing adjuvant capable of inducing mucosal immunity |
| WO2006124269A2 (en) | 2005-05-16 | 2006-11-23 | Amgen Fremont Inc. | Human monoclonal antibodies that bind to very late antigen-1 for the treatment of inflammation and other disorders |
| CA2625664C (en) | 2005-10-21 | 2016-01-05 | Novartis Ag | Human antibodies against il13 and therapeutic uses |
| TWI477602B (zh) | 2006-02-09 | 2015-03-21 | Educational Foundation Jichi Medical Univ | Novel viral vector |
| AU2007249160B2 (en) | 2006-05-15 | 2013-09-12 | I2 Pharmaceuticals, Inc. | Neutralizing antibodies to influenza viruses |
| AU2007293662B2 (en) | 2006-09-07 | 2012-10-04 | Crucell Holland B.V. | Human binding molecules capable of neutralizing influenza virus H5N1 and uses thereof |
| CN101541832B (zh) * | 2006-09-07 | 2014-11-12 | 克鲁塞尔荷兰公司 | 能中和流感病毒h5n1的人结合分子及其应用 |
| MX2009003393A (es) | 2006-10-02 | 2009-05-11 | Regeneron Pharma | Anticuerpos humanos de alta afinidad para el receptor de il-4 humano. |
| CA2668800A1 (en) | 2006-11-08 | 2008-06-05 | Macrogenics West, Inc. | Tes7 and antibodies that bind thereto |
| EP1925318A1 (en) | 2006-11-20 | 2008-05-28 | Paul-Ehrlich-Institut | Recombinant modified vaccinia virus Ankara (MVA)-based vaccine for the avian flu |
| RU2448979C2 (ru) | 2006-12-14 | 2012-04-27 | Ридженерон Фармасьютикалз, Инк. | Антитела человека к дельта-подобному лиганду-4 человека |
| GB0700133D0 (en) | 2007-01-04 | 2007-02-14 | Humabs Llc | Human cytomegalovirus neutralising antibodies and use thereof |
| CN102046654A (zh) | 2007-03-13 | 2011-05-04 | 胡马斯有限公司 | 针对甲型流感病毒h5n1株系的抗体 |
| SG187500A1 (en) | 2008-01-21 | 2013-02-28 | Medicago Inc | Recombinant influenza virus-like particles (vlps) produced in transgenic plants expressing hemagglutinin |
| ITTO20080204A1 (it) | 2008-03-17 | 2009-09-18 | Pomona Biotechnologies Llc | Anticorpi monoclonali atti a reagire con una pluralita di sottotipi del virus influenzale a |
| JP5607620B2 (ja) * | 2008-07-25 | 2014-10-15 | インスティテュート・フォー・リサーチ・イン・バイオメディシン | 抗a型インフルエンザウイルス中和抗体およびその使用 |
| MX2011003855A (es) | 2008-10-22 | 2011-12-16 | Inst Research In Biomedicine | Metodos para producir anticuerpos a partir de celulas plasmaticas. |
| JP2012521786A (ja) | 2009-03-30 | 2012-09-20 | モウント シナイ スクール オフ メディシネ | インフルエンザウイルスワクチン及びその使用 |
| WO2010148511A1 (en) | 2009-06-24 | 2010-12-29 | Medicago, Inc. | Chimeric influenza virus-like particles comprising hemagglutinin |
| MX352338B (es) | 2011-07-18 | 2017-11-17 | Inst Res Biomedicine | Anticuerpos neutralizantes del virus de la influenza a y usos de estos. |
-
2011
- 2011-07-18 MX MX2014000749A patent/MX352338B/es active IP Right Grant
- 2011-07-18 SI SI201131938T patent/SI3418300T1/sl unknown
- 2011-07-18 US US14/233,719 patent/US9587010B2/en active Active
- 2011-07-18 EP EP18180926.0A patent/EP3418300B1/en active Active
- 2011-07-18 CA CA2841551A patent/CA2841551C/en active Active
- 2011-07-18 ES ES11784765T patent/ES2732552T3/es active Active
- 2011-07-18 DK DK18180926.0T patent/DK3418300T3/da active
- 2011-07-18 LT LTEP18180926.0T patent/LT3418300T/lt unknown
- 2011-07-18 SI SI201131700T patent/SI2734545T1/sl unknown
- 2011-07-18 AU AU2011373387A patent/AU2011373387B2/en active Active
- 2011-07-18 HU HUE18180926A patent/HUE051756T2/hu unknown
- 2011-07-18 PL PL11784765T patent/PL2734545T3/pl unknown
- 2011-07-18 CN CN201180072356.0A patent/CN103717617B/zh active Active
- 2011-07-18 HU HUE11784765A patent/HUE044089T2/hu unknown
- 2011-07-18 WO PCT/IB2011/002329 patent/WO2013011347A1/en active Application Filing
- 2011-07-18 EP EP11784765.7A patent/EP2734545B1/en active Active
- 2011-07-18 DK DK11784765.7T patent/DK2734545T3/en active
- 2011-07-18 PL PL18180926T patent/PL3418300T3/pl unknown
- 2011-07-18 LT LTEP11784765.7T patent/LT2734545T/lt unknown
- 2011-07-18 ES ES18180926T patent/ES2841899T3/es active Active
- 2011-07-18 JP JP2014520738A patent/JP6035332B2/ja active Active
- 2011-07-18 EP EP20200200.2A patent/EP3812397A1/en active Pending
-
2017
- 2017-01-20 US US15/411,677 patent/US20170204167A1/en not_active Abandoned
-
2018
- 2018-11-01 US US16/178,497 patent/US10815294B2/en active Active
-
2019
- 2019-04-16 HR HRP20190714TT patent/HRP20190714T1/hr unknown
-
2020
- 2020-12-10 HR HRP20201975TT patent/HRP20201975T1/hr unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10815294B2 (en) | Neutralizing anti-influenza A virus antibodies and uses thereof | |
| US8871207B2 (en) | Neutralizing anti-influenza A virus antibodies and uses thereof | |
| US9340603B2 (en) | Neutralizing anti-influenza A virus antibodies and uses thereof | |
| JP2022061986A (ja) | A型インフルエンザウイルス中和抗体及びその使用法 | |
| JP6622825B2 (ja) | A型インフルエンザウイルス中和抗体及びその使用法 | |
| CN107417789A (zh) | 中和抗甲型流感病毒抗体及其用途 | |
| JP2017086068A (ja) | A型インフルエンザウイルス中和抗体及びその使用法 |