ES2717536T3 - Cromatografía de sobrecarga y elución - Google Patents
Cromatografía de sobrecarga y elución Download PDFInfo
- Publication number
- ES2717536T3 ES2717536T3 ES17157670T ES17157670T ES2717536T3 ES 2717536 T3 ES2717536 T3 ES 2717536T3 ES 17157670 T ES17157670 T ES 17157670T ES 17157670 T ES17157670 T ES 17157670T ES 2717536 T3 ES2717536 T3 ES 2717536T3
- Authority
- ES
- Spain
- Prior art keywords
- antibody
- chromatography
- polypeptide
- buffer
- antibodies
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/10—Selective adsorption, e.g. chromatography characterised by constructional or operational features
- B01D15/14—Selective adsorption, e.g. chromatography characterised by constructional or operational features relating to the introduction of the feed to the apparatus
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/42—Selective adsorption, e.g. chromatography characterised by the development mode, e.g. by displacement or by elution
- B01D15/424—Elution mode
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
- C07K1/165—Extraction; Separation; Purification by chromatography mixed-mode chromatography
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
- C07K1/20—Partition-, reverse-phase or hydrophobic interaction chromatography
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
- C07K1/22—Affinity chromatography or related techniques based upon selective absorption processes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/32—Bonded phase chromatography
- B01D15/325—Reversed phase
- B01D15/327—Reversed phase with hydrophobic interaction
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/36—Selective adsorption, e.g. chromatography characterised by the separation mechanism involving ionic interaction, e.g. ion-exchange, ion-pair, ion-suppression or ion-exclusion
- B01D15/361—Ion-exchange
- B01D15/363—Anion-exchange
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/38—Selective adsorption, e.g. chromatography characterised by the separation mechanism involving specific interaction not covered by one or more of groups B01D15/265 and B01D15/30 - B01D15/36, e.g. affinity, ligand exchange or chiral chromatography
- B01D15/3804—Affinity chromatography
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/38—Selective adsorption, e.g. chromatography characterised by the separation mechanism involving specific interaction not covered by one or more of groups B01D15/265 and B01D15/30 - B01D15/36, e.g. affinity, ligand exchange or chiral chromatography
- B01D15/3847—Multimodal interactions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
- C07K1/18—Ion-exchange chromatography
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Analytical Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Immunology (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Description
DESCRIPCIÓN
Cromatografía de sobrecarga y elución
SOLICITUDES RELACIONADAS
CAMPO DE LA INVENCIÓN
La presente invención proporciona procedimientos para purificar un producto a partir de una composición que comprende el producto y al menos un contaminante y formulaciones que comprenden el producto purificado con los procedimientos.
ANTECEDENTES DE LA INVENCIÓN
La cromatografía de intercambio aniónico (AEX) se usa ampliamente en un modo de flujo continuo como una etapa de refinado de plataforma para anticuerpos monoclonales (MAb). Ciertos MAb que se unen a la resina AEX en condiciones normales de flujo pueden plantear problemas de ajuste de la planta. Para el desarrollo clínico en etapa temprana en la que el requerimiento de masa es típicamente bajo, estos MAb sin plataforma se han purificado usando AEX o resina de modo mixto en un modo de unión y elución. Sin embargo, debido a la baja capacidad de unión dinámica (DBC) de estas resinas, la etapa tardía de implementación requeriría columnas que tienen aproximadamente 1000 l de tamaño o múltiples ciclos en una columna más pequeña limitando así el rendimiento de la planta.
La purificación de MAb se realiza típicamente usando cromatografía de unión y elución (B/E) o cromatografía de flujo continuo (F/T). Se han introducido recientemente cromatografía de reparto débil (Kelley, BD et al., 2008 Biotechnol Bioeng 101(3):553-566; publicación de solicitud de patente de EE. UU. n.° 2007/0060741) y cromatografía de sobrecarga (WO2011150110) sobre resinas AEX y resinas de intercambio catiónico (CEX), respectivamente, para mejorar la purificación de MAb. El mecanismo general y las limitaciones de cada uno de estos modos de cromatografía se destacan a continuación.
Cromatografía de unión y elución: Con cromatografía B/E, el producto se carga usualmente para maximizar la DBC para el material de cromatografía y luego se identifican las condiciones de lavado y elución de modo que se alcanza la máxima pureza del producto en el eluido. Una limitación de la cromatografía B/E es la restricción de la densidad de carga a la DBC real de la resina.
Cromatografía de flujo continuo: Usando cromatografía F/T, las condiciones de carga se identifican cuando las impurezas se unen fuertemente al material de cromatografía mientras el producto fluye. La cromatografía F/T permite una alta densidad de carga para MAb convencionales, pero no puede ser implementable para MAb sin plataforma o las condiciones de solución que permiten el funcionamiento F/T para estos MAb sin plataforma pueden ser tales que no son implementables en plantas de producción existentes.
Cromatografía de reparto débil: Este modo de funcionamiento mejora el modo F/T identificando condiciones de solución en las que hay una unión débil de MAb a la resina (2 a 20 g/l). En estas condiciones, las impurezas se unen más fuerte que en el modo F/T y se obtiene así una purificación mejorada. Sin embargo, las condiciones de carga se dirigen a tener un bajo coeficiente de reparto del producto (Kp) en el intervalo de 0,1-20.
Cromatografía de sobrecarga: En este modo de cromatografía, el producto de interés se carga más allá de la capacidad de unión dinámica del material de cromatografía para el producto, por lo que se denomina sobrecarga. Se ha demostrado que el modo de funcionamiento proporciona la purificación de MAb con medios de intercambio catiónico (CEX) y particularmente con membranas. Sin embargo, una limitación de este enfoque es que podría haber rendimientos bajos con resina ya que no hay fase de elución.
La purificación a gran escala y rentable de un polipéptido con una pureza suficiente para su uso como agente terapéutico humano sigue siendo un desafío formidable.
El documento WO2012068134A1 se refiere al enriquecimiento y concentración de determinadas isoformas del producto por cromatografía de unión con sobrecarga y elución. El documento WO2006116064A2 se refiere a un aparato y a procedimientos para la purificación dirigida por espectrometría de masas de biopolímeros. Liu, HF et al., 2011 J Chrom A 1218(39):6943-6952 se refiere a la exploración de la cromatografía de intercambio catiónico con sobrecarga para la purificación de anticuerpos monoclonales. El documento WO9508574A1 se refiere al procedimiento de cromatografía de desplazamiento y producto de hemoglobina purificada. El documento WO2011035282A1 se refiere a la separación por captura doble. El documento US2011065901A1 se refiere a procedimientos para purificar una proteína diana a partir de una o más impurezas en una muestra.
BREVE SUMARIO
La invención proporciona procedimientos para purificar un anticuerpo a partir de una composición que comprende el anticuerpo y uno o más contaminantes, comprendiendo dicho procedimiento a) cargar la composición en un material de cromatografía de modo mixto, de intercambio iónico, de interacción hidrófoba o de afinidad en una cantidad en exceso de la capacidad de unión dinámica del material de cromatografía para el anticuerpo usando un tampón de carga, en el que el coeficiente de reparto del material de cromatografía para el polipéptido es mayor que 30, b) eluir el anticuerpo del material de cromatografía en condiciones en las que el uno o más contaminantes permanecen unidos al material de cromatografía usando un tampón de elución, en el que el tampón de elución tiene una conductividad menor que la conductividad del tampón de carga y c) agrupar fracciones que comprenden el anticuerpo en el efluente de cromatografía de las etapas a) y b).
En algunos modos de realización, el anticuerpo es un anticuerpo monoclonal; por ejemplo, pero sin limitarse a un anticuerpo quimérico, anticuerpo humanizado o anticuerpo humano.
En algunos modos de realización, el anticuerpo es un fragmento de unión al antígeno; por ejemplo, pero sin limitarse a estos, un fragmento Fab', un fragmento F(ab')2, un scFv, un di-scFv, un bi-scFv, un (di, tri)-scFv en tándem, un Fv, un sdAb, un anticuerpo trifuncional, un BiTE, un diacuerpo y un triacuerpo.
En algunos modos de realización de la divulgación, el polipéptido es una enzima, una hormona, una proteína de fusión, una proteína que contiene Fc, un inmunoconjugado, una citocina o una interleucina.
En algunos modos de realización, el polipéptido se purifica a partir de una composición que comprende uno o más contaminantes; por ejemplo, proteína de ovario de hámster chino (CHOP), una proteína de célula huésped (HCP), proteína A lixiviada, ácido nucleico, ADN, variantes de producto, proteína agregada, componente de medio de cultivo celular, gentamicina, fragmentos polipeptídicos, endotoxinas y contaminante vírico.
En algunos modos de realización de la invención, el material de cromatografía se selecciona de un material de modo mixto, un material de intercambio aniónico, un material de interacción hidrófoba y un material de afinidad.
En algunos modos de realización, la composición se carga sobre el material de cromatografía a aproximadamente las capacidades de unión dinámica de los materiales de cromatografía para el uno o más contaminantes.
En algunos modos de realización, el coeficiente de reparto del material de cromatografía para el polipéptido es mayor que 30 o mayor que 100.
En algunos modos de realización, los procedimientos proporcionan OEC en la que el tampón de elución tiene una conductividad menor que la conductividad del tampón de carga. En algunos modos de realización, los procedimientos proporcionan OEC en la que el tampón de elución tiene un pH menor que el pH del tampón de carga. En otros modos de realización, el tampón de elución tiene un pH mayor que el pH del tampón de carga.
En algunos modos de realización, el polipéptido de los procedimientos está en un eluyente de una cromatografía de afinidad, una cromatografía de intercambio catiónico, una cromatografía de intercambio aniónico, una cromatografía de modo mixto y una cromatografía de interacción hidrófoba. En algunos modos de realización, el polipéptido está en un eluyente de una cromatografía de proteína A.
En algunos modos de realización, el polipéptido de los procedimientos se purifica adicionalmente; por ejemplo, por filtración de virus, cromatografía de afinidad, cromatografía de intercambio catiónico, cromatografía de intercambio aniónico, cromatografía en modo mixto y/o cromatografía de interacción hidrófoba. En algunos modos de realización, el polipéptido se concentra adicionalmente; por ejemplo, por ultrafiltración, diafiltración o una combinación de ultrafiltración y diafiltración. En algunos modos de realización, el polipéptido de los procedimientos se combina adicionalmente con un vehículo farmacéuticamente aceptable.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
La figura 1 muestra resultados de cribado ultrarrápido (HTS) sobre la resina Capto Adhere en condiciones de unión por lotes para MAb 3. La figura 1A muestra contornos de valores constantes de Kp para MAb 3. La figura 1B muestra la capacidad de unión real del MAb 3 en el sobrenadante a 80 g/l de exposición del producto a la resina. La figura 1C muestra la capacidad de unión real de la impureza (proteína de la célula huésped) en el sobrenadante a 80 g/l de exposición de producto a la resina. Todos los gráficos de contorno se generaron usando un modelo de superficie de respuesta derivado de los datos sin procesar.
La figura 2 muestra un cromatograma para un modo de funcionamiento OEC optimizado. El MAb 3 se usó para este desarrollo cromatográfico con una densidad de carga de 180 g/l.
La figura 3 muestra la optimización de carga para un modo OEC usando MAb 3.
La figura 4 muestra un cromatograma con condiciones de carga objetivo para un modo de funcionamiento OEC. Condiciones similares de carga y elución dan como resultado residuos y, por lo tanto, un aumento del 45 % en el volumen de la agrupación. El MAb 3 se usó para este desarrollo cromatográfico con una densidad de carga de 180 g/l.
La figura 5 muestra la optimización de la elución para el modo OEC usando MAb 3.
La figura 6 muestra el análisis de impurezas en fracciones y el análisis acumulativo de impurezas.
La figura 7 muestra el análisis de avance de MAb 3 CHOP de la resina Capto Adhere en el modo OEC a una densidad de carga de 1000 g/l.
La figura 8 muestra el análisis del rendimiento entre desarrollos cromatográficos a escala piloto en un amplio intervalo de densidades de carga de 70 g/l a 180 g/l.
La figura 9 muestra una comparación del modo de funcionamiento de la cromatografía de reparto débil y el modo de funcionamiento de la cromatografía de sobrecarga y elución. El producto era MAb 3 y el material de cromatografía era una resina Capto Adhere.
La figura 10 muestra el análisis de MAb 3 CHOP en resina QMA en modo de funcionamiento OEC a una densidad de carga de 150 g/l.
La figura 11 muestra el análisis de MAb 4 CHOP en resina Capto Adhere en el modo de funcionamiento OEC a una densidad de carga de 150 g/l.
La figura 12 muestra el análisis de MAb 4 CHOP en resina Capto MMC en el modo de funcionamiento OEC con una densidad de carga de 150 g/l.
La figura 13 muestra el análisis de avance de MAb 3 CHOP en la resina Capto Adhere en el modo OEC a una densidad de carga de 200 g/l. Se cargó una agrupación de MAb 3 proteína A en resina Capto Adhere a 200 g/l (que está más allá de su capacidad de unión al producto de 50 g/l). El análisis de avance de CHOP demostró que la CHOP no avanzaba hasta el procesamiento a 200 g/l de MAb.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
I. Definiciones
El término "producto" como se describe en el presente documento es la sustancia a purificar por OEC; por ejemplo, un polipéptido.
El término "polipéptido" o "proteína" se usan de manera intercambiable en el presente documento para referirse a polímeros de aminoácidos de cualquier longitud. El polímero puede ser lineal o ramificado, puede comprender aminoácidos modificados y puede estar interrumpido por moléculas distintas a aminoácidos. Los términos también engloban un polímero de aminoácidos que se ha modificado naturalmente o mediante intervención; por ejemplo, formación de enlaces disulfuro, glucosilación, lipidación, acetilación, fosforilación o cualquier otra manipulación o modificación, tal como conjugación con un componente de marcaje. También se incluyen dentro de la definición, por ejemplo, polipéptidos que contienen uno o más análogos de un aminoácido (incluyendo, por ejemplo, aminoácidos no naturales, etc.), así como otras modificaciones conocidas en la técnica. Los términos "polipéptido" y "proteína" como se usan en el presente documento engloban específicamente anticuerpos.
Polipéptido "purificado" (por ejemplo, anticuerpo o inmunoadhesina) significa que el polipéptido se ha incrementado en pureza, de modo que exista en una forma que sea más pura de la que existe en su entorno natural y/o cuando inicialmente se sintetiza y/o amplifica en condiciones de laboratorio. La pureza es un término relativo y no significa necesariamente pureza absoluta.
El término "epítopo marcado" cuando se usa en el presente documento se refiere a un polipéptido quimérico que comprende un polipéptido fusionado con un "polipéptido marcador". El polipéptido marcador tiene suficientes residuos para proporcionar un epítopo contra el que se puede producir un anticuerpo, pero es suficientemente corto de modo que no interfiere en la actividad del polipéptido al que está fusionado. El polipéptido marcador también es preferentemente bastante único de manera que el anticuerpo no reacciona sustancialmente de forma cruzada con otros epítopos. Los polipéptidos marcadores adecuados generalmente tienen al menos seis residuos de aminoácidos y habitualmente entre aproximadamente 8 y 50 residuos de aminoácidos (preferentemente, entre aproximadamente 10 y 20 residuos de aminoácidos).
"Activo" o "actividad" para los propósitos del presente documento se refiere a la forma o formas de un polipéptido que retienen una actividad biológica y/o inmunológica de un polipéptido natural o de origen natural, en el que actividad "biológica" se refiere a una función biológica (inhibidora o estimuladora) causada por un polipéptido natural o de origen
natural que no sea la capacidad para inducir la producción de un anticuerpo contra un epítopo antigénico que posee un polipéptido natural o de origen natural, y una actividad "inmunológica" se refiere a la capacidad para inducir la producción de un anticuerpo contra un epítopo antigénico que posee un polipéptido natural o de origen natural.
El término "antagonista" se usa en el sentido más amplio e incluye cualquier molécula que bloquee, inhiba o neutralice parcial o completamente una actividad biológica de un polipéptido natural. De una manera similar, el término "agonista" se usa en el sentido más amplio e incluye cualquier molécula que imite una actividad biológica de un polipéptido natural. Las moléculas agonistas o antagonistas adecuadas incluyen específicamente anticuerpos o fragmentos de anticuerpos agonistas o antagonistas, fragmentos o variantes de secuencia de aminoácidos de polipéptidos naturales, etc. Los procedimientos para identificar agonistas o antagonistas de un polipéptido pueden comprender poner en contacto un polipéptido con una molécula candidata a agonista o antagonista y medir un cambio detectable en una o más actividades biológicas normalmente asociadas al polipéptido.
"Citotoxicidad dependiente del complemento" o "CDC" se refiere a la capacidad de una molécula de efectuar la lisis de una diana en presencia del complemento. La ruta de activación del complemento se inicia mediante la unión del primer componente del sistema del complemento (C1q) a una molécula (por ejemplo, un polipéptido (por ejemplo, un anticuerpo)) que forma complejos con un antígeno afín. Para evaluar la activación del complemento, se puede realizar un ensayo de CDC, por ejemplo, como se describe en Gazzano-Santoro et al., J. Immunol. Methods 202:163 (1996).
Un polipéptido "que se une a" un antígeno de interés, por ejemplo, una diana de antígeno polipeptídico asociado a tumor, es uno que se une al antígeno con afinidad suficiente, de modo que el polipéptido sea útil como un agente de diagnóstico y/o terapéutico en la selección como diana de una célula o tejido que exprese el antígeno, y significativamente no reacciona de forma cruzada con otros polipéptidos. En dichos modos de realización, el grado de unión del polipéptido a un polipéptido "no diana" será de menos de aproximadamente un 10 % de la unión del polipéptido a su polipéptido diana particular, como se determina mediante análisis de clasificación celular activada por fluorescencia (FACS) o radioinmunoprecipitación (RIA).
Con respecto a la unión de un polipéptido a una molécula diana, el término "unión específica a" o "se une específicamente a" o "específico para" un polipéptido particular o un epítopo en una diana polipeptídica particular significa que la unión es diferente de forma mensurable de una interacción no específica. Se puede medir la unión específica, por ejemplo, determinando la unión de una molécula en comparación con la unión de una molécula de control que, en general, es una molécula de estructura similar que no tiene actividad de unión. Por ejemplo, se puede determinar la unión específica mediante competencia con una molécula de control que sea similar a la diana, por ejemplo, un exceso de diana no marcada. En este caso, se indica la unión específica si la unión de la diana marcada con respecto a una sonda se inhibe de forma competitiva mediante la diana no marcada en exceso.
El término "anticuerpo" en el presente documento se usa en el sentido más amplio y cubre específicamente anticuerpos monoclonales, anticuerpos policlonales, anticuerpos multiespecíficos (por ejemplo, anticuerpos biespecíficos) formados a partir de al menos dos anticuerpos intactos y fragmentos de anticuerpo, siempre que presenten la actividad biológica deseada. El término "inmunoglobulina" (Ig) se usa de manera intercambiable con anticuerpo en el presente documento.
Los anticuerpos son moléculas de inmunoglobulina naturales que tienen estructuras variables, todas basadas en el plegamiento de inmunoglobulina. Por ejemplo, los anticuerpos IgG tienen dos cadenas "pesadas" y dos cadenas "ligeras" que se unen por enlaces disulfuro para formar un anticuerpo funcional. Cada cadena pesada y ligera comprende por sí misma una región "constante" (C) y una región "variable" (V). Las regiones V determinan la especificidad de unión a antígeno del anticuerpo, mientras que las regiones C proporcionan soporte y función estructurales en las interacciones no específicas de antígeno con efectores inmunitarios. La especificidad de unión a antígeno de un anticuerpo o fragmento de unión a antígeno de un anticuerpo es la capacidad de un anticuerpo de unirse específicamente a un antígeno particular.
La especificidad de unión a antígeno de un anticuerpo se determina por las características estructurales de la región V. La variabilidad no se distribuye uniformemente entre el tramo de 110 aminoácidos de los dominios variables. En cambio, las regiones V consisten en tramos relativamente invariables denominados regiones estructurales (FR) de 15 30 aminoácidos separados por regiones más cortas de variabilidad extrema denominadas "regiones hipervariables" que tienen cada una 9-12 aminoácidos de longitud. Los dominios variables de las cadenas pesada y ligera naturales comprenden cada uno cuatro FR, que adoptan en gran medida una configuración de lámina p, conectadas mediante tres regiones hipervariables que forman bucles que conectan y, en algunos casos, forman parte de, la estructura de lámina p. Las regiones hipervariables de cada cadena se mantienen juntas en estrecha proximidad por medio de las FR y, con las regiones hipervariables de la otra cadena, contribuyen a la formación del sitio de unión a antígeno de los anticuerpos (véase Kabat et al., Sequences of Proteins of Immunological Interest, 5.a ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)). Los dominios constantes no están implicados directamente en la unión de un anticuerpo a un antígeno, pero presentan diversas funciones efectoras, tales como la participación del anticuerpo en la citotoxicidad celular dependiente de anticuerpos (ADCC).
Cada región V comprende típicamente tres regiones determinantes de la complementariedad ("CDR", cada una de las cuales contiene un "bucle hipervariable") y cuatro regiones estructurales. Un sitio de unión de anticuerpo, la unidad estructural mínima requerida para unirse con afinidad sustancial a un antígeno deseado particular, por lo tanto, incluirá típicamente las tres CDR y al menos tres, preferentemente cuatro, regiones estructurales intercaladas entre ellas para mantener y presentar las CDR en la conformación apropiada. Los anticuerpos clásicos de cuatro cadenas tienen sitios de unión a antígeno que están definidos por los dominios V h y VL en cooperación. Ciertos anticuerpos, tales como anticuerpos de camello y tiburón, carecen de cadenas ligeras y se basan en sitios de unión formados únicamente por cadenas pesadas. Se pueden preparar inmunoglobulinas genomanipuladas de dominio único en las que los sitios de unión se forman por cadenas pesadas o cadenas ligeras solas, en ausencia de cooperación entre VH y VL.
El término "variable" se refiere al hecho de que determinadas porciones de los dominios variables difieren ampliamente en secuencia entre anticuerpos y se usan en la unión y especificidad de cada anticuerpo particular por su antígeno particular. Sin embargo, la variabilidad no está distribuida uniformemente por los dominios variables de los anticuerpos. Se concentra en tres segmentos llamados regiones hipervariables, tanto en los dominios variables de la cadena ligera como de la cadena pesada. Las porciones más altamente conservadas de los dominios variables se llaman regiones estructurales (FR). Los dominios variables de las cadenas pesada y ligera naturales comprenden cada uno cuatro FR, que adoptan en gran medida una configuración de lámina p, conectadas mediante tres regiones hipervariables que forman bucles que conectan y, en algunos casos, forman parte de, la estructura de lámina p. Las regiones hipervariables de cada cadena se mantienen juntas en estrecha proximidad por medio de las FR y, con las regiones hipervariables de la otra cadena, contribuyen a la formación del sitio de unión a antígeno de los anticuerpos (véase Kabat et al., Sequences of Proteins of Immunological Interest, 5.a ed. Public Health Service, National Institutes of Health, Bethesda, MD. (1991)). Los dominios constantes no están implicados directamente en la unión de un anticuerpo a un antígeno, pero presentan diversas funciones efectoras, tales como la participación del anticuerpo en la citotoxicidad celular dependiente de anticuerpos (ADCC).
El término "región hipervariable", cuando se usa en el presente documento, se refiere a los residuos de aminoácido de un anticuerpo que son responsables de la unión al antígeno. La región hipervariable puede comprender residuos de aminoácidos de una "región determinante de la complementariedad" o "CDR" (por ejemplo, en torno a aproximadamente los residuos 24-34 (L1), 50-56 (L2) y 89-97 (L3) del dominio VL y en torno a aproximadamente los residuos 31-35B (H1), 50-65 (H2) y 95-102 (H3) del dominio VH (Kabat et al., Sequences of Proteins of Immunological Interest, 5.a ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)) y/o los residuos de un "bucle hipervariable" (por ejemplo, residuos 26-32 (L1), 50-52 (L2) y 91-96 (L3)) en el VL, y 26-32 (H1), 52A-55 (H2) y 96-101 (H3) en el VH (Chothia y Lesk J. Mol. Biol. 196:901-917 (1987)).
Los residuos de la "región estructural" o "FR" son los residuos del dominio variable distintos de los residuos de la región hipervariable, como se define en el presente documento.
Los "fragmentos de anticuerpo" comprenden una porción de un anticuerpo intacto, que comprende preferentemente la región de unión a antígeno del mismo. Los ejemplos de fragmentos de anticuerpo incluyen fragmentos Fab, Fab', F(ab')2 y Fv; diacuerpos; diacuerpos en tándem (taDb), anticuerpos lineales (por ejemplo, la patente de EE. UU. n.° 5.641.870, ejemplo 2; Zapata et al., Protein Eng. 8(10):1057-1062 (1995)); anticuerpos de un solo brazo, anticuerpos de dominio variable único, minicuerpos, moléculas de anticuerpo monocatenario; anticuerpos multiespecíficos formados a partir de fragmentos de anticuerpo (porejemplo, incluyendo pero sin limitarse a, Db-Fc, taDb-Fc, taDb-CH3, (scFV)4-Fc, di-scFv, bi-scFv o (di, tri)-scFv en tándem); y captadores biespecíficos de linfocitos T (BiTE).
La digestión con papaína de los anticuerpos produce dos fragmentos de unión a antígeno idénticos, llamados fragmentos "Fab", cada uno con un sitio de unión a antígeno único, y un fragmento "Fc" residual, cuyo nombre refleja su capacidad de cristalizar fácilmente. El tratamiento con pepsina proporciona un fragmento F(ab')2 que tiene dos sitios de unión a antígeno y todavía puede reticular el antígeno.
"Fv" es el fragmento de anticuerpo mínimo que contiene un sitio de reconocimiento de antígeno y de unión a antígeno completo. Esta región consiste en un dímero de un dominio variable de una cadena pesada y de una cadena ligera en estrecha asociación no covalente. Es en esta configuración en la que las tres regiones hipervariables de cada dominio variable interaccionan para definir un sitio de unión a antígeno en la superficie del dímero VH-VL. Colectivamente, las seis regiones hipervariables confieren especificidad de unión a antígeno al anticuerpo. Sin embargo, incluso un dominio variable único (o la mitad de un Fv que comprenda solo tres regiones hipervariables específicas para un antígeno) tiene la capacidad de reconocer y unirse al antígeno, aunque con una afinidad más baja que todo el sitio de unión.
El fragmento Fab también contiene el dominio constante de la cadena ligera y el primer dominio constante (CH1) de la cadena pesada. Los fragmentos Fab' difieren de los fragmentos Fab en la adición de unos pocos residuos en el extremo carboxílico del dominio CH1 de la cadena pesada, incluyendo una o más cisteínas de la región de bisagra del anticuerpo. Fab'-SH es la denominación en el presente documento para Fab' en el que el/los residuo(s) de cisteína de los dominios constantes tienen al menos un grupo tiol libre. Los fragmentos de anticuerpo F(ab')2 se producían originalmente como pares de fragmentos Fab' que tenían cisteínas de bisagra entre ellos. También son conocidos otros acoplamientos químicos de fragmentos de anticuerpo.
Se pueden asignar las "cadenas ligeras" de los anticuerpos (inmunoglobulinas) de cualquier especie de vertebrado a uno de dos tipos claramente distintos, llamados kappa (k) y lambda (A), basados en las secuencias de aminoácidos de sus dominios constantes.
Dependiendo de la secuencia de aminoácidos del dominio constante de sus cadenas pesadas, se pueden asignar anticuerpos a diferentes clases. Existen cinco clases principales de anticuerpos intactos: IgA, IgD, IgE, IgG e IgM, y varias de ellas se pueden dividir, además, en subclases (isotipos), por ejemplo, IgG1, IgG2, IgG3, IgG4, IgA e IgA2. Los dominios constantes de la cadena pesada que se corresponden con las diferentes clases de anticuerpos se llaman a, 6, £, y y M, respectivamente. Son bien conocidas las estructuras de las subunidades y las configuraciones tridimensionales de las clases de inmunoglobulinas diferentes.
Los fragmentos de anticuerpo "Fv monocatenario" o "scFv" comprenden los dominios VH y VL del anticuerpo, en los que estos dominios están presentes en una única cadena de polipéptido. En algunos modos de realización, el polipéptido Fv comprende además un conector de polipéptido entre los dominios VH y VL que posibilita que el scFv forme la estructura deseada para la unión a antígeno. Para una revisión de los scFv, véase, por ejemplo, Plückthun, en The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg y Moore eds., Springer-Verlag, Nueva York, pág.
269-315 (1994).
El término "diacuerpos" se refiere a pequeños fragmentos de anticuerpo con dos sitios de unión a antígeno, comprendiendo los fragmentos un dominio variable de la cadena pesada (VH) conectado con un dominio variable de la cadena ligera (VL) en la misma cadena de polipéptido (VH-VL). Usando un conector que sea demasiado corto para permitir el emparejamiento entre los dos dominios en la misma cadena, se obliga a que los dominios se emparejen con los dominios complementarios de otra cadena y creen dos sitios de unión a antígeno. Los diacuerpos se describen más completamente, por ejemplo, en el documento EP 404.097; documento WO 93/11161; y Hollinger et al., Proc. Natl. Acad. Sci. USA, 90:6444-6448 (1993).
El término "anticuerpo multiespecífico" se usa en el sentido más amplio y cubre específicamente un anticuerpo que tiene especificidad poliepitópica. Dichos anticuerpos multiespecíficos incluyen, pero no se limitan a, un anticuerpo que comprende un dominio variable de cadena pesada (VH) y un dominio variable de cadena ligera (VL), donde la unidad VHVL tiene especificidad poliepitópica, anticuerpos que tienen dos o más dominios V l y VH, uniéndose cada unidad VHVL a un epítopo diferente, anticuerpos que tienen dos o más dominios variables individuales, uniéndose cada dominio variable individual a un epítopo diferente, anticuerpos de longitud completa, fragmentos de anticuerpo tales como Fab, Fv, dsFv, scFv, diacuerpos, diacuerpos biespecíficos, triacuerpos, anticuerpos trifuncionales, fragmentos de anticuerpos que se han unido covalentemente o no covalentemente. "Especificidad poliepitópica" se refiere a la capacidad de unirse específicamente a dos o más epítopos diferentes en la(s) misma(s) o diferente(s) diana(s). "Monoespecífico" se refiere a la capacidad de unirse solo a un epítopo. De acuerdo con un modo de realización, el anticuerpo multiespecífico es un anticuerpo IgG que se une a cada epítopo con una afinidad de 5 pM a 0,001 pM, de 3 pM a 0,001 pM, de 1 pM a 0,001 pM, de 0,5 pM a 0,001 pM o de 0,1 pM a 0,001 pM.
La expresión "anticuerpos de dominio único" (sdAb) o "anticuerpos de dominio variable único (SVD)" se refiere, en general, a anticuerpos en los que un dominio variable único (VH o VL) puede conferir unión a antígeno. En otras palabras, el dominio variable único no necesita interactuar con otro dominio variable para reconocer el antígeno diana. Ejemplos de anticuerpos de dominio único incluyen los derivados de camélidos (llamas y camellos) y peces cartilaginosos (por ejemplo, tiburones nodriza) y los derivados de procedimientos recombinantes de anticuerpos humanos y murinos (Nature (1989) 341:544-546; Dev Comp Immunol (2006) 30:43-56; Trend Biochem Sci (2001) 26:230-235; Trends Biotechnol (2003):21:484-490; documento WO 2005/035572; documento WO 03/035694; Febs Lett (1994) 339:285-290; documento WO 00/29004; documento WO 02/051870).
El término "anticuerpo monoclonal" como se usa en el presente documento se refiere a un anticuerpo obtenido a partir de una población de anticuerpos sustancialmente homogénea, es decir, los anticuerpos individuales que comprenden la población son idénticos y/o se unen al mismo epítopo, excepto las posibles variantes que puedan surgir durante la producción del anticuerpo monoclonal, estando en general presentes dichas variantes en cantidades menores. A diferencia de las preparaciones de anticuerpos policlonales que típicamente incluyen anticuerpos diferentes dirigidos contra determinantes (epítopos) diferentes, cada anticuerpo monoclonal se dirige contra un determinante único en el antígeno. Además de su especificidad, los anticuerpos monoclonales son ventajosos en tanto que no están contaminados por otras inmunoglobulinas. El modificador "monoclonal" indica el carácter del anticuerpo que se obtiene de una población sustancialmente homogénea de anticuerpos, y no se debe interpretar que requiere la producción del anticuerpo mediante ningún procedimiento particular. Por ejemplo, los anticuerpos monoclonales que se van a usar de acuerdo con los procedimientos proporcionados en el presente documento se pueden preparar mediante el procedimiento de hibridoma descrito por primera vez por Kohler et al., Nature 256:495 (1975), o se pueden preparar mediante procedimientos de ADN recombinante (véase, por ejemplo, la patente de EE. UU. n.° 4.816.567). Los "anticuerpos monoclonales" también se pueden aislar de colecciones de anticuerpos de fagos usando las técnicas descritas en Clackson et al., Nature, 352:624-628 (1991) y Marks et al., J. Mol. Biol., 222:581-597 (1991), por ejemplo.
Los anticuerpos monoclonales en el presente documento incluyen específicamente anticuerpos "quiméricos" (inmunoglobulinas) en los que una porción de la cadena pesada y/o ligera es idéntica u homóloga a las secuencias correspondientes en anticuerpos derivados de una especie particular o que pertenecen a una clase o subclase de anticuerpos particular, mientras que el resto de la(s) cadena(s) es idéntico u homólogo a las secuencias correspondientes en anticuerpos derivados de otra especie o que pertenecen a otra clase o subclase de anticuerpos, así como fragmentos de dichos anticuerpos, siempre que presenten la actividad biológica deseada (patente de EE. UU. n.° 4.816.567; Morrison et al., Proc. Natl. Acad. Sci. USA 81:6851-6855 (1984)). Los anticuerpos quiméricos de interés en el presente documento incluyen anticuerpos "primatizados" que comprenden secuencias de unión a antígeno del dominio variable derivadas de un primate no humano (por ejemplo, mono del viejo mundo, tal como babuino, macaco de la India o macaco cangrejero) y secuencias de la región constante humanas (patente de EE. UU. n.° 5.693.780).
Las formas "humanizadas" de anticuerpos no humanos (por ejemplo, murinos) son anticuerpos quiméricos que contienen una secuencia mínima derivada de inmunoglobulina no humana. En su mayor parte, los anticuerpos humanizados son inmunoglobulinas humanas (anticuerpo receptor) en las que los residuos de una región hipervariable del receptor se reemplazan por residuos de una región hipervariable de una especie no humana (anticuerpo donador), tal como ratón, rata, conejo o primate no humano, que tiene la especificidad, afinidad y capacidad deseadas. En algunos casos, los residuos de la región estructural (FR) de la inmunoglobulina humana se reemplazan por los residuos no humanos correspondientes. Además, los anticuerpos humanizados pueden comprender residuos que no se encuentran en el anticuerpo receptor ni en el anticuerpo donador. Se pueden realizar estas modificaciones para refinar adicionalmente el funcionamiento del anticuerpo. En general, el anticuerpo humanizado comprenderá sustancialmente todos de al menos uno, y típicamente dos, dominios variables en los que todos o sustancialmente todos los bucles hipervariables corresponden a los de una inmunoglobulina no humana y todas o sustancialmente todas las FR son las de una secuencia de inmunoglobulina humana, excepto por la(s) sustitución/sustituciones en FR como se indica anteriormente. El anticuerpo humanizado también comprenderá opcionalmente al menos una porción de una región constante de inmunoglobulina, típicamente la de una inmunoglobulina humana. Para obtener más detalles, véase Jones et al., Nature 321:522-525 (1986); Riechmann et al., Nature, 332:323-329 (1988); y Presta, Curr. Op. Struct. Biol. 2:593-596 (1992).
Para los propósitos del presente documento, un "anticuerpo intacto" es uno que comprende dominios variables pesados y ligeros, así como una región Fc. Los dominios constantes pueden ser dominios constantes de secuencia natural (por ejemplo, dominios constantes de secuencia natural humana) o una variante de secuencia de aminoácidos de los mismos. Preferentemente, el anticuerpo intacto tiene una o más funciones efectoras.
Los "anticuerpos naturales" son normalmente glucoproteínas heterotetraméricas de aproximadamente 150 000 dalton, compuestas por dos cadenas ligeras (L) idénticas y dos cadenas pesadas (H) idénticas. Cada cadena ligera se enlaza a una cadena pesada mediante un enlace disulfuro covalente, aunque el número de enlaces disulfuro varía entre las cadenas pesadas de isotipos de inmunoglobulina diferentes. Cada cadena pesada y ligera también tiene puentes disulfuro intracatenarios espaciados regularmente. Cada cadena pesada tiene en un eXtremo un dominio variable (VH) seguido de una serie de dominios constantes. Cada cadena ligera tiene un dominio variable en un extremo (VL) y un dominio constante en su otro extremo; el dominio constante de la cadena ligera se alinea con el primer dominio constante de la cadena pesada y el dominio variable de la cadena ligera se alinea con el dominio variable de la cadena pesada. Se cree que residuos aminoacídicos particulares forman una superficie de contacto entre los dominios variables de la cadena ligera y de la cadena pesada.
Un "anticuerpo no marcado" es un anticuerpo (como se define en el presente documento) que no se conjuga con una molécula heteróloga, tal como un resto citotóxico o radiomarcador.
En algunos modos de realización, "funciones efectoras" de anticuerpo se refieren a las actividades biológicas atribuibles a la región Fc (una región Fc de secuencia natural o una región Fc de variante de secuencia de aminoácidos) de un anticuerpo y varían con el isotipo de anticuerpo. Los ejemplos de funciones efectoras de anticuerpos incluyen: unión a C1q y citotoxicidad dependiente del complemento; unión al receptor de Fc; citotoxicidad celular dependiente de anticuerpos (ADCC); fagocitosis; regulación por disminución de receptores de superficie celular.
"Citotoxicidad celular dependiente de anticuerpos" y "ADCC" se refieren a una reacción celular en la que las células citotóxicas no específicas que expresan los receptores Fc (FcR) (por ejemplo, linfocitos citolíticos naturales (NK), neutrófilos y macrófagos) reconocen un anticuerpo unido en una célula diana y posteriormente provocan la lisis de la célula diana. Las células principales para mediar en la ADCC, los linfocitos NK, expresan solo FcyRIII, mientras que los monocitos expresan FcyRI, FcyRII y FcyRIII. La expresión de FcR en células hematopoyéticas se resume en la Tabla 3 de la página 464 de Ravetch y Kinet, Annu. Rev. Immunol 9:457-92 (1991). Para evaluar la actividad de ADCC de una molécula de interés se puede realizar un ensayo de ADCC in vitro, tal como el descrito en la patente de EE. UU. n.° 5.500.362 o 5.821.337. Las células efectoras útiles para dichos ensayos incluyen leucocitos monomorfonucleares en la sangre periférica (PBMC) y linfocitos citolíticos naturales (NK). De forma alternativa, o adicionalmente, la actividad de ADCC de la molécula de interés se puede evaluar in vivo, por ejemplo, en un modelo animal tal como el divulgado en Clynes et al., Proc. Natl. Acad. Sci. (USA) 95:652-656 (1998).
Las "células efectoras humanas" son leucocitos que expresan uno o más FcR y realizan funciones efectoras. En algunos modos de realización, las células expresan al menos FcyRIII y llevan a cabo la función efectora ADCC. Los ejemplos de leucocitos humanos que median en la ADCC incluyen leucocitos monomorfonucleares en la sangre periférica (PBMC), linfocitos citolíticos naturales (NK), monocitos, linfocitos T citotóxicos y neutrófilos; siendo preferentes los PBMC y los linfocitos NK.
Los términos "receptor de Fc" o "FcR" se usan para describir un receptor que se une a la región Fc de un anticuerpo. En algunos modos de realización, el FcR es un FcR humano de secuencia natural. Además, un FcR preferente es uno que se une a un anticuerpo IgG (un receptor gamma) e incluye receptores de las subclases FcyRI, FcyRII y FcyRIII, incluyendo variantes alélicas y formas empalmadas de forma alternativa de estos receptores. Los receptores FcyRII incluyen FcyRIIA (un "receptor de activación") y FcyRIIB (un "receptor de inhibición"), que tienen secuencias de aminoácidos similares que difieren principalmente en los dominios citoplásmicos de los mismos. El receptor de activación FcyRIIA contiene un motivo de activación del inmunorreceptor basado en tirosi na (ITAM) en su dominio citoplásmico. El receptor de inhibición FcyRIIB contiene un motivo de inhibición del inmunorreceptor basado en tirosina (ITIM) en su dominio citoplasmático (véase Daeron, Annu. Rev. Immunol. 15:203-234 (1997)). Los FcR se revisan en Ravetch y Kinet, Annu. Rev. Immunol 9:457-92 (1991); Capel et al., Immunomethods 4:25-34 (1994); y de Haas et al., J. Lab. Clin. Med. 126:330-41 (1995). Otros FcR, incluyendo los que se vayan a identificar en el futuro, se engloban por el término "FcR" en el presente documento. El término también incluye el receptor neonatal, FcRn, que es responsable de la transferencia de las IgG maternas al feto (Guyer et al., J. Immunol. 117:587 (1976) y Kim et al., J. Immunol. 24:249 (1994)).
El término "secuencial" como se usa en el presente documento con respecto a la cromatografía se refiere a que presenta una primera cromatografía seguida por una segunda cromatografía. Pueden incluirse etapas adicionales entre la primera cromatografía y la segunda cromatografía.
El término "continuo" como se usa en el presente documento con respecto a la cromatografía se refiere a que presenta un primer material de cromatografía y un segundo material de cromatografía directamente conectado o algún otro mecanismo que permita un flujo continuo entre los dos materiales de cromatografía.
"Contaminantes" se refiere a materiales que son diferentes del producto de polipéptido deseado. El contaminante incluye, sin limitación: materiales de la célula huésped, tales como CHOP; proteína A lixiviada; ácido nucleico; una variante, fragmento, agregado o derivado del polipéptido deseado; otro polipéptido; endotoxina; contaminante vírico; componente de medio de cultivo celular, etc. En algunos ejemplos, el contaminante puede ser una proteína de célula huésped (HCP), por ejemplo, pero no limitada a, una célula bacteriana tal como una célula de E. coli, una célula de insecto, una célula procariota, una célula eucariota, una célula de levadura, una célula de mamífero, una célula aviar, una célula fúngica.
La "capacidad de unión dinámica" de un material de cromatografía es la cantidad de producto, por ejemplo, polipéptido, del material que se unirá en condiciones de flujo real antes de que se produzca un avance significativo del producto no unido.
El "coeficiente de reparto", Kp, como se usa en el presente documento se refiere a la concentración molar del producto, por ejemplo, polipéptido, en la fase estacionaria dividida entre la concentración molar del producto en la fase móvil.
"Densidad de carga" se refiere a la cantidad, por ejemplo, gramos, de composición puesta en contacto con un volumen de material de cromatografía, por ejemplo, litros. En algunos ejemplos, la densidad de carga se expresa en g/l.
La referencia a "aproximadamente" un valor o parámetro en el presente documento incluye (y describe) variaciones que se refieren a ese valor o parámetro per se. Por ejemplo, la descripción que se refiere a "aproximadamente X" incluye la descripción de "X".
Como se usa en el presente documento y en las reivindicaciones adjuntas, las formas singulares "un/a", "o" y "el/la" incluyen las referencias plurales a menos que el contexto indique claramente lo contrario. Se entiende que los aspectos y variaciones de la invención descrita en el presente documento incluyen "que consisten en" y/o "que consisten esencialmente en" aspectos y variaciones.
II. Procedimientos de purificación
Se proporcionan en el presente documento procedimientos para purificar un producto, tal como un polipéptido, a partir de una composición que comprende el producto y al menos un contaminante usando cromatografía de sobrecarga y elución (OEC). OEC proporciona un modo de funcionamiento en el que las ventajas proporcionadas por diferentes modos de cromatografía se realizan en un modo de cromatografía única. OEC se puede implementar en múltiples resinas, a la vez que proporciona una mayor retirada de impurezas y ventajas significativas de producción, como columnas más pequeñas, mejor ajuste de planta y menor costo. El modo de funcionamiento con OEC se puede dividir en tres componentes diferentes.
1. Sobrecarga - La composición se carga sobre el material de cromatografía de modo que el producto, por ejemplo, un polipéptido, se carga sobre el material de cromatografía en una cantidad que excede la capacidad de unión dinámica (DBC) del material para el producto. En algunos modos de realización, se determinan condiciones de carga, tales como pH y conductividad, donde las impurezas se unen fuertemente al material de cromatografía. En algunos modos de realización, las condiciones de cromatografía se eligen de modo que, incluso si el producto avanza después de unirse, la mayor parte de las impurezas, si no todas, no lo hacen. En algunos modos de realización la composición se carga a o cerca de la DBC del material para el uno o más contaminantes. El modo de sobrecarga permite utilizar el material de cromatografía más allá de la DBC típica del material para el producto.
2. Agrupamiento - El agrupamiento del producto, por ejemplo, un polipéptido, en el eluyente comienza en el avance del producto. Puesto que las condiciones de carga son de modo que las impurezas continúan uniéndose durante la fase de avance, se puede obtener una agrupación de producto limpia en el eluyente durante la fase de carga de la cromatografía.
3. Elución - Una vez completada la carga de la composición sobre el material de cromatografía, el producto, por ejemplo, polipéptido, se eluye del material de cromatografía usando condiciones de elución que se identifican de modo que el producto unido se eluye mientras que la mayoría de las impurezas permanecen unidas al material de cromatografía.
En algunos aspectos, OEC incrementa la utilización de material de cromatografía significativamente más allá de la DBC del material para el producto, proporcionando así una ventaja en comparación con otros procedimientos de cromatografía. Por ejemplo, una utilización del material de cromatografía 10 veces más alta puede dar como resultado un costo significativamente menor.
A diferencia de la cromatografía de unión y elución tradicional donde la carga del material de cromatografía se optimiza para maximizar la unión al producto, por ejemplo, polipéptido, para la resina de cromatografía, con condiciones de carga de OEC se puede optimizar para maximizar la unión de contaminantes al material de cromatografía y no la unión del producto al material de cromatografía. En algunos aspectos, la composición se carga sobre un material de cromatografía en una cantidad que excede la capacidad de unión dinámica del material de cromatografía para el producto. En el procedimiento de carga del material de cromatografía, parte del producto avanzará en el lavado y parte del producto permanecerá unido al material de cromatografía. Una vez completada la carga, el producto restante unido al material de cromatografía se puede eluir del material de cromatografía. En algunos modos de realización de lo anterior, el material de cromatografía es una columna de cromatografía. En algunos modos de realización de lo anterior, el material de cromatografía es una membrana cromatográfica.
En algunos aspectos de la invención, se carga una composición sobre un material de cromatografía a aproximadamente la capacidad de unión dinámica del material de cromatografía para uno o más de los contaminantes en la composición. En algunos modos de realización, se carga una composición sobre un material de cromatografía en una cantidad que excede la capacidad de unión del material de cromatografía para el producto. En algunos modos de realización, se carga una composición sobre un material de cromatografía a aproximadamente la capacidad de unión dinámica del material de cromatografía para uno o más de los contaminantes y se supera la capacidad de unión del material de cromatografía para el producto. En algunos modos de realización, la composición se carga sobre el material de cromatografía a 20 veces la DBC del material de cromatografía para el producto. En algunos modos de realización, la composición se carga sobre el material de cromatografía a 100 veces la DBC del material de cromatografía para el producto. En algunos modos de realización, la composición se carga sobre un material de cromatografía a aproximadamente la capacidad de unión dinámica del material de cromatografía para todos los contaminantes en la composición. En algunos modos de realización, se carga una composición sobre un material de cromatografía a aproximadamente la capacidad de unión dinámica del material de cromatografía para todos los contaminantes y superando la capacidad de unión del material de cromatografía para el producto. En algunos modos de realización, se carga una composición sobre un material de cromatografía a menos de la capacidad de unión dinámica del material de cromatografía para todos los contaminantes y superando la capacidad de unión del material de cromatografía para el producto. En algunos modos de realización de lo anterior, el material de cromatografía está en una columna de cromatografía. En algunos modos de realización, la columna de cromatografía es una columna de cromatografía a escala industrial. En algunos modos de realización de lo anterior, el material de cromatografía es una membrana cromatográfica.
La capacidad de unión dinámica de un material de cromatografía para un producto y para uno o más contaminantes se puede estimar determinando el coeficiente de reparto (Kp) para el producto o contaminantes en función del pH y la concentración de contraión para un material de cromatografía particular. Por ejemplo, la capacidad de unión dinámica de un material de cromatografía, por ejemplo, una resina de modo mixto, se puede determinar para un polipéptido. Las capacidades de unión reales de un material de cromatografía para un producto o contaminante en una combinación específica de pH y concentración de contraión se pueden determinar exponiendo la unión a un exceso del producto y/o contaminante.
En algunos modos de realización, la OEC se lleva a cabo cuando el Kp del producto, por ejemplo, polipéptido, es mayor que aproximadamente 30. En algunos modos de realización, la OEC se lleva a cabo cuando el Kp del producto es
mayor que aproximadamente 50. En algunos modos de realización, la OEC se lleva a cabo cuando el Kp del producto es mayor que aproximadamente 75. En algunos modos de realización, la OEC se lleva a cabo cuando el Kp del producto es mayor que aproximadamente 100.
Las condiciones para OEC de una composición particular que comprende un producto, tal como un polipéptido, y un contaminante se pueden determinar midiendo el Kp y la capacidad de unión dinámica de un material de cromatografía particular a diferentes pH y concentración de contraión. Se puede llevar a cabo un cribado ultrarrápido para determinar las condiciones de la Oe C en las que la unión de contaminantes es alta y donde el producto se puede eluir sin eluir la mayor parte, si no todos, los contaminantes. Por ejemplo, la composición se puede incubar con un material de cromatografía en tampón a varios pH y concentraciones de contraión en un sistema ultrarrápido; por ejemplo, en pocillos de una placa de múltiples pocillos. Después de un período de incubación, el sobrenadante se separa del material de cromatografía y se determina la cantidad de producto o contaminante en el sobrenadante. En algunos modos de realización, se usan concentraciones bajas de composición para determinar el Kp. En algunos modos de realización, se utilizan altas concentraciones de la composición para determinar las capacidades de unión dinámica.
Además de proporcionar información sobre el Kp y la capacidad de unión dinámica de un material de cromatografía para productos y contaminantes particulares, el cribado ultrarrápido proporciona una guía para condiciones de carga y elución en términos de pH y concentración de contraión. Por ejemplo, en algunos modos de realización, el tampón de carga se selecciona mediante un cribado ultrarrápido para un pH y concentración de contraión para maximizar la unión del contaminante al material de cromatografía, pero también para maximizar la cantidad de producto, por ejemplo, polipéptido, en el eluyente mientras se minimiza la cantidad de contaminante, por ejemplo, proteína de la célula huésped, en el eluyente. En algunos modos de realización, la composición se carga sobre un material de cromatografía a un pH y conductividad determinados mediante cribado ultrarrápido en el que aproximadamente todos los contaminantes en la composición se unen al material de cromatografía. En algunos modos de realización, el producto se eluye del material de cromatografía a un pH y una conductividad determinados mediante un cribado ultrarrápido, en el que aproximadamente todo el producto eluye del material de cromatografía y todos los contaminantes permanecen unidos al material de cromatografía.
En algunos modos de realización, la invención proporciona procedimientos para identificar las condiciones de funcionamiento (por ejemplo, utilizando técnicas de cribado ultrarrápido) que hacen que el material de cromatografía se una a una cantidad máxima de contaminantes con independencia de la cantidad de producto unido por ml de material de cromatografía. La etapa de cribado se utiliza para identificar las condiciones de elución de modo que el producto unido eluye del material de cromatografía y las impurezas permanecen estrechamente unidas al material de cromatografía.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el material de cromatografía es un material de modo mixto que comprende grupos funcionales que pueden tener una o más de las siguientes funcionalidades: intercambio aniónico, intercambio catiónico, enlace de hidrógeno e interacciones hidrófobas. En algunos modos de realización, el material de modo mixto comprende grupos funcionales con capacidad de intercambio aniónico e interacciones hidrófobas. El material de modo mixto puede contener N-bencil-N-metiletanolamina, 4-mercapto-etil-piridina, hexilamina o fenilpropilamina como ligando o contener polialilamina reticulada. Los ejemplos de materiales de modo mixto incluyen resina Capto Adhere, resina QMA, resina Capto MMC, resina MEP HyperCel, resina HEA HyperCel, resina PPA HyperCel o membrana ChromaSorb o Sartobind STIC. En algunos modos de realización, el material de modo mixto es una resina Capto Adhere. En algunos modos de realización de lo anterior, el material de modo mixto es una columna de cromatografía de modo mixto. En algunos modos de realización de lo anterior, el material de modo mixto es una membrana de modo mixto.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el material de cromatografía es un material de cromatografía de intercambio iónico; por ejemplo, un material de cromatografía de intercambio aniónico o un material de cromatografía de intercambio catiónico. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el material de cromatografía es un material de intercambio aniónico. En algunos modos de realización, el material de cromatografía de intercambio aniónico es una fase sólida que está cargada positivamente y tiene aniones libres para el intercambio con aniones en una solución acuosa que pasa por o a través de la fase sólida. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el material de intercambio aniónico puede ser una membrana, un monolito o resina. En un modo de realización, el material de intercambio aniónico puede ser una resina. En algunos modos de realización, el material de intercambio aniónico puede comprender una amina primaria, una amina secundaria, una amina terciaria o un grupo funcional de ion amonio cuaternario, un grupo funcional poliamina o un grupo funcional dietilaminoetilo. En algunos modos de realización de lo anterior, el material de cromatografía de intercambio aniónico es una columna de cromatografía de intercambio aniónico. En algunos modos de realización de lo anterior, el material de cromatografía de intercambio aniónico es una membrana de cromatografía de intercambio aniónico.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el material de cromatografía es un material de intercambio catiónico. En algunos modos de realización, el material de intercambio catiónico es una fase sólida que está cargada negativamente y tiene cationes libres para el intercambio con cationes en una solución acuosa que pasa por o a través de la fase sólida. En algunos modos de realización de cualquiera de
los procedimientos descritos en el presente documento, el material de intercambio catiónico puede ser una membrana, monolito o resina. En algunos modos de realización, el material de intercambio catiónico puede ser una resina. El material de intercambio catiónico puede comprender un grupo funcional ácido carboxílico o un grupo funcional ácido sulfónico tal como, pero sin limitarse a, sulfonato, carboxílico, ácido carboximetilsulfónico, sulfoisobutilo, sulfoetilo, carboxilo, sulfopropilo, sulfonilo, sulfoxietilo u ortofosfato. En algunos modos de realización de lo anterior, el material de cromatografía de intercambio catiónico es una columna de cromatografía de intercambio catiónico. En algunos modos de realización de lo anterior, el material de cromatografía de intercambio catiónico es una membrana de cromatografía de intercambio catiónico. En algunos modos de realización de la invención, el material de cromatografía no es un material de cromatografía de intercambio catiónico.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el material de intercambio iónico puede utilizar un material de cromatografía convencional o un material de cromatografía convectivo. Los materiales de cromatografía convencionales incluyen, por ejemplo, materiales perfusivos (por ejemplo, resina de poli(estireno-divinilbenceno)) y materiales difusivos (por ejemplo, resina de agarosa reticulada). En algunos modos de realización, la resina de poli(estireno-divinilbenceno) puede ser resina Poros. En algunos modos de realización, la resina de agarosa reticulada puede ser resina de sulfopropil-Sepharose Fast Flow ("SPSFF"). El material de cromatografía convectivo puede ser una membrana (por ejemplo, polietersulfona) o un material monolítico (por ejemplo, polímero reticulado). La membrana de polietersulfona puede ser Mustang. El material monolítico de polímero reticulado puede ser poli(metacrilato de glicidilo-co-dimetacrilato de etileno) reticulado.
En la técnica son conocidos ejemplos de materiales de intercambio aniónico e incluyen, pero no se limitan a, Poros HQ 50, Poros PI 50, Poros D, Mustang Q, Q Sepharose FF y DEAE Sepharose.
En la técnica son conocidos ejemplos de materiales de intercambio catiónico e incluyen, pero no se limitan a Mustang S, Sartobind S, SO3 Monolith, S Ceramic HyperD, Poros XS, Poros HS50, Poros Hs 20, SPSFF, SP-Sepharose XL (SPXL), CM Sepharose Fast Flow, Capto S, Fractogel Se HiCap, Fractogel SO3 o Fractogel COO. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el material de intercambio catiónico es Poros HS50. En algunos modos de realización, la resina Poros HS puede ser partículas Poros HS de 50 pm o Poros HS de 20 pm.
En algunos aspectos de la invención, el material de cromatografía es un material de cromatografía de interacción hidrófoba. La cromatografía de interacción hidrófoba (HIC) es una técnica de cromatografía de líquidos que separa las biomoléculas según la hidrofobia. Ejemplos de materiales de cromatografía HIC incluyen, pero no se limitan a, Toyopearl hexilo 650, Toyopear butilo 650, Toyopearl fenilo 650, Toyopearl éter 650, Source, Resource, Sepharose Hi-Trap, octil Sepharose, fenil Sepharose. En algunos modos de realización de lo anterior, el material de cromatografía HIC es una columna de cromatografía HIC. En algunos modos de realización de lo anterior, el material de cromatografía HIC es una membrana de cromatografía HIC.
En algunos aspectos de la invención, el material de cromatografía es un material de cromatografía de hidroxiapatita (HAP). Ejemplos de material de cromatografía de hidroxiapatita incluyen, pero están limitados a HA Ultrogel e hidroxiapatita CHT. En algunos modos de realización de lo anterior, el material de cromatografía de HAP es una columna de cromatografía de HAP. En algunos modos de realización de lo anterior, el material de cromatografía de HAP es una membrana de cromatografía de HAP.
En algunos aspectos de la invención, el material de cromatografía es un material de cromatografía de afinidad. Ejemplos de materiales de cromatografía de afinidad incluyen, pero no se limitan a materiales de cromatografía derivatizados con proteína A o proteína G. Ejemplos de material de cromatografía de afinidad incluyen, pero no se limitan a, Prosep-VA, Prosep-VA Ultra Plus, proteína A Sepharose Fast Flow, Tyopearl proteína A, MAbSelect, MAbSelect SuRe y MAbSelect SuRe LX. En algunos modos de realización de lo anterior, el material de cromatografía de afinidad es una columna de cromatografía de afinidad. En algunos modos de realización de lo anterior, el material de cromatografía de afinidad es una membrana de cromatografía de afinidad.
La carga de la composición sobre el material de cromatografía se puede optimizar para la separación del producto de los contaminantes por OEC. En algunos modos de realización, el producto es un polipéptido. En algunos modos de realización, la carga sobre la composición sobre el material de cromatografía se optimiza para la unión de los contaminantes al material de cromatografía. Por ejemplo, la composición se puede cargar sobre el material de cromatografía, por ejemplo, una columna de cromatografía, en un tampón de carga a una pluralidad de pH diferentes mientras que la conductividad del tampón de carga es constante. De forma alternativa, la composición se puede cargar sobre el material de cromatografía en un tampón de carga a varias conductividades diferentes mientras que el pH del tampón de carga es constante. Una vez completada la carga de la composición sobre el material de cromatografía y la elución del producto del material de cromatografía en una fracción de agrupación, la cantidad de contaminante en la fracción de agrupación proporciona información relativa a la separación del producto de los contaminantes para un pH o conductividad dado. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, la composición se carga sobre un material de cromatografía a una densidad de carga del polipéptido de más de aproximadamente cualquiera de 30 g/l, 40 g/l, 50 g/l, 60 g/l, 70 g/l, 80 g/l, 90 g/l, 100 g/l, 110 g/l, 120 g/l, 130 g/l, 140 g/l, 150 g/l, 160 g/l, 170 g/l, 180 g/l, 190 g/l, 200 g/l, 300 g/l, 400 g/l, 500 g/l, 550 g/l, 600 g/l, 650 g/l,
700 g/l, 800 g/l, 900 g/l, 1000 g/l, 2000 g/l o 5000 g/l de material de cromatografía. En algunos modos de realización, la composición se carga sobre un material de cromatografía a una densidad de carga del polipéptido de aproximadamente cualquiera de 30 g/l, 40 g/l, 50 g/l, 60 g/l, 70 g/l , 80 g/l, 90 g/l, 100 g/l, 110 g/l, 120 g/l, 130 g/l,
140 g/l, 150 g/l, 160 g/l, 170 g/l, 180 g/l, 190 g/l, 200 g/l, 300 g/l, 400 g/l, 500 g/l, 550 g/l, 600 g/l, 650 g/l, 700 g/l, 800 g/l,
900 g/l, 1000 g/l o 2000 g/l de material de cromatografía. La composición se puede cargar en un material de cromatografía a una densidad de carga del polipéptido de entre aproximadamente cualquiera de 30 g/l y 2000 g/l,
30 g/l y 1000 g/l, 30 g/l y 200 g/l, 30 g/l y 180 g/l, 50 g/l y 2000 g/l, 50 g/l y 1000 g/l, 50 g/l y 200 g/l, 50 g/l y 180 g/l,
150 g/l y 2000 g/l, 150 g/l y 1500 g/l, 150 g/l y 1000 g/l, 200 g/l y 1000 g/l, 200 g/l y 1500 g/l, 300 g/l y 1500 g/l, 400 g/l
y 1000 g/l, o 500 g/l y 1000 g/l de material de cromatografía.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, la composición se carga sobre un material de cromatografía de modo mixto a una densidad de carga del polipéptido de
más de aproximadamente cualquiera de 30 g/l, 40 g/l, 50 g/l, 60 g/l, 70 g/l, 80 g/l, 90 g/l, 100 g/l, 110 g/l, 120 g/l, 130 g/l,
140 g/l, 150 g/l, 160 g/l, 170 g/l, 180 g/l, 190 g/l, 200 g/l, 300 g/l, 400 g/l, 500 g/l, 550 g/l, 600 g/l, 650 g/l, 700 g/l, 800 g/l, 900 g/l, 1000 g/l, 2000 g/l o 5000 g/l de material de cromatografía de modo mixto. En algunos modos de realización,
la composición se carga sobre un material de cromatografía de modo mixto a una densidad de carga del polipéptido
de aproximadamente cualquiera de 30 g/l, 40 g/l, 50 g/l, 60 g/l, 70 g/l, 80 g/l, 90 g/l, 100 g/l, 110 g/l, 120 g/l, 130 g/l,
140 g/l, 150 g/l, 160 g/l, 170 g/l, 180 g/l, 190 g/l, 200 g/l, 300 g/l, 400 g/l, 500 g/l, 550 g/l, 600 g/l, 650 g/l, 700 g/l, 800 g/l, 900 g/l, 1000 g/l o 2000 g/l de material de cromatografía de modo mixto. La composición se puede cargar en un material de cromatografía de modo mixto a una densidad de carga del polipéptido de entre aproximadamente cualquiera de 30 g/l y 2000 g/l, 30 g/l y 1000 g/l, 30 g/l y 200 g/l, 30 g/l y 180 g/l, 50 g/l y 2000 g/l, 50 g/l y 1000 g/l,
50 g/l y 200 g/l, 50 g/l y 180 g/l, 150 g/l y 2000 g/l, 150 g/l y 1500 g/l, 150 g/l y 1000 g/l, 200 g/l y 1000 g/l, 200 g/l y
1500 g/l, 300 g/l y 1500 g/l, 400 g/l y 1000 g/l o 500 g/l y 1000 g/l de material de cromatografía de modo mixto. En
algunos modos de realización, el polipéptido se carga sobre un material de cromatografía a una densidad de 70 g/l a
180 g/l. En algunos modos de realización de la invención, la cromatografía de modo mixto es una resina Capto Adhere.
En algunos modos de realización, el polipéptido se carga sobre un material de cromatografía Capto Adhere a una densidad de 70 g/l a 180 g/l. En otros modos de realización de los modos de realización anteriores, el polipéptido es
un anticuerpo o un fragmento del mismo.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, la composición se carga sobre un material de cromatografía de intercambio aniónico a una densidad de carga del polipéptido de más de aproximadamente cualquiera de 30 g/l, 40 g/l, 50 g/l, 60 g/l, 70 g/l, 80 g/l, 90 g/l, 100 g/l, 110 g/l,
120 g/l, 130 g/l, 140 g/l, 150 g/l, 160 g/l, 170 g/l, 180 g/l, 190 g/l, 200 g/l, 300 g/l, 400 g/l, 500 g/l, 550 g/l, 600 g/l, 650 g/l,
700 g/l, 800 g/l, 900 g/l, 1000 g/l, 2000 g/l o 5000 g/l de material de cromatografía de intercambio aniónico. En algunos
modos de realización, la composición se carga sobre un material de cromatografía de intercambio aniónico a una densidad de carga del polipéptido de aproximadamente cualquiera de 30 g/l, 40 g/l, 50 g/l, 60 g/l, 70 g/l, 80 g/l, 90 g/l,
100 g/l, 110 g/l, 120 g/l, 130 g/l, 140 g/l, 150 g/l, 160 g/l, 170 g/l, 180 g/l, 190 g/l, 200 g/l, 300 g/l, 400 g/l, 500 g/l, 550 g/l,
600 g/l, 650 g/l, 700 g/l, 800 g/l, 900 g/l, 1000 g/l o 2000 g/l de material de cromatografía de intercambio aniónico. La composición se puede cargar sobre un material de cromatografía de intercambio aniónico a una densidad de carga
del polipéptido entre aproximadamente cualquiera de 30 g/l y 2000 g/l, 30 g/l y 1000 g/l, 30 g/l y 200 g/l, 30 g/l y 180 g/l,
50 g/l y 2000 g/l, 50 g/l y 1000 g/l, 50 g/l y 200 g/l, 50 g/l y 180 g/l, 150 g/l y 2000 g/l, 150 g/l y 1500 g/l, 150 g/l y
1000 g/l, 200 g/l y 1000 g/l, 200 g/l y 1500 g/l, 300 g/l y 1500 g/l, 400 g/l y 1000 g/l o 500 g/l y 1000 g/l de material de cromatografía de intercambio aniónico.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, la composición se carga sobre un material de cromatografía de intercambio catiónico a una densidad de carga del polipéptido de más de aproximadamente cualquiera de 30 g/l, 40 g/l, 50 g/l, 60 g/l, 70 g/l, 80 g/l, 90 g/l, 100 g/l, 110 g/l,
120 g/l, 130 g/l, 140 g/l, 150 g/l, 160 g/l, 170 g/l, 180 g/l, 190 g/l, 200 g/l, 300 g/l, 400 g/l, 500 g/l, 550 g/l, 600 g/l, 650 g/l,
700 g/l, 800 g/l, 900 g/l, 1000 g/l, 2000 g/l o 5000 g/l de material de cromatografía de intercambio catiónico. En algunos
modos de realización, la composición se carga sobre un material de cromatografía de intercambio catiónico a una densidad de carga de aproximadamente cualquiera de 30 g/l, 40 g/l, 50 g/l, 60 g/l, 70 g/l, 80 g/l, 90 g/l, 100 g/l, 110 g/l,
120 g/l, 130 g/l, 140 g/l, 150 g/l, 160 g/l, 170 g/l, 180 g/l, 190 g/l, 200 g/l, 300 g/l, 400 g/l, 500 g/l, 550 g/l, 600 g/l, 650 g/l,
700 g/l, 800 g/l, 900 g/l, 1000 g/l o 2000 g/l de material de cromatografía de intercambio catiónico. La composición se
puede cargar sobre un material de cromatografía de intercambio catiónico a una densidad de carga del polipéptido de
entre aproximadamente cualquiera de 30 g/l y 2000 g/l, 30 g/l y 1000 g/l, 30 g/l y 200 g/l, 30 g/l y 180 g/l, 50 g/l y
2000 g/l, 50 g/l y 1000 g/l, 50 g/l y 200 g/l, 50 g/l y 180 g/l, 150 g/l y 2000 g/l, 150 g/l y 1500 g/l, 150 g/l y 1000 g/l,
200 g/l y 1000 g/l, 200 g/l y 1500 g/l, 300 g/l y 1500 g/l, 400 g/l y 1000 g/l o 500 g/l y 1000 g/l de material de cromatografía de intercambio catiónico.
Los procedimientos descritos anteriormente pueden comprender además la etapa de cargar la composición en un material de cromatografía de afinidad de proteína A. En algunos modos de realización, el producto polipeptídico es un anticuerpo o fragmento del mismo que se purifica en primer lugar mediante cromatografía de afinidad de proteína A
antes que por OEC. La etapa de carga de la composición en un material de cromatografía de afinidad de proteína A
se efectúa generalmente, pero no necesariamente, antes de la otra u otras etapas de cromatografía. En algunos modos
de realización, la etapa de carga sobre un material de cromatografía de afinidad de proteína A se puede combinar con
las etapas secuenciales cromatografía de intercambio con sobrecarga y de elución. En algunos modos de realización,
las etapas secuenciales son continuas. En algunos modos de realización, la purificación continua utiliza el mismo caudal, conductividad y/o pH.
La elución del producto tal como un polipéptido del material de cromatografía en modo OEC se puede optimizar para el rendimiento del producto con contaminantes mínimos y con un volumen de agrupación mínimo. Por ejemplo, la composición se puede cargar sobre el material de cromatografía, por ejemplo, una columna de cromatografía, en un tampón de carga. Una vez completada la carga, el producto se eluye con tampones a varios pH diferentes mientras que la conductividad del tampón de elución es constante. De forma alternativa, el producto se puede eluir del material de cromatografía en un tampón de elución a varias conductividades diferentes mientras que el pH del tampón de elución es constante. Una vez completada la elución del producto del material de cromatografía, la cantidad de contaminante en la fracción de agrupación proporciona información relativa a la separación del producto de los contaminantes para un pH o conductividad dado. La elución del producto en un número elevado de fracciones (por ejemplo, ocho volúmenes de columna) indica "residuos" del perfil de elución. En algunos modos de realización de la invención, los residuos de la elución se minimizan.
Se pueden emplear diversos tampones dependiendo, por ejemplo, del pH deseado del tampón, de la conductividad deseada del tampón, de las características de la proteína de interés y del procedimiento de purificación. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, los procedimientos comprenden el uso de un tampón. El tampón puede ser un tampón de carga, un tampón de equilibrado o un tampón de lavado. En algunos modos de realización, uno o más del tampón de carga, el tampón de equilibrado y/o el tampón de lavado son iguales. En algunos modos de realización, el tampón de carga, el tampón de equilibrado y/o el tampón de lavado son diferentes. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el tampón comprende una sal. El tampón de carga puede comprender cloruro de sodio, acetato de sodio o una mezcla de los mismos. En algunos modos de realización, el tampón de carga es un tampón de cloruro de sodio. En algunos modos de realización, el tampón de carga es un tampón de acetato de sodio.
La carga, como se usa en el presente documento, es la composición cargada sobre un material de cromatografía. El tampón de carga es el tampón usado para cargar la composición que comprende el producto de interés sobre un material de cromatografía. El material de cromatografía se puede equilibrar con un tampón de equilibrado antes de cargar la composición que se va a purificar. En algunos ejemplos, el tampón de lavado se usa después de cargar la composición sobre un material de cromatografía y antes de la elución del polipéptido de interés de la fase sólida. Sin embargo, parte del producto de interés, por ejemplo, un polipéptido, se puede retirar del material de cromatografía mediante el tampón de lavado (por ejemplo, similar a un modo de flujo continuo).
La elución, como se usa en el presente documento, es la retirada del producto, por ejemplo, polipéptido, del material de cromatografía. El tampón de elución es el tampón usado para eluir el polipéptido u otro producto de interés de un material de cromatografía. En muchos casos, un tampón de elución tiene una característica física diferente del tampón de carga. Por ejemplo, el tampón de elución puede tener una conductividad diferente del tampón de carga o un pH diferente del tampón de carga. En algunos modos de realización, el tampón de elución tiene una conductividad más baja que el tampón de carga. En algunos modos de realización de la divulgación, el tampón de elución tiene una conductividad más alta que el tampón de carga. En algunos modos de realización, el tampón de elución tiene un pH más bajo que el tampón de carga. En algunos modos de realización, el tampón de elución tiene un pH más alto que el tampón de carga. En algunos modos de realización, el tampón de elución tiene una conductividad diferente y un pH diferente del tampón de carga. El tampón de elución puede tener cualquier combinación de conductividad menor y pH más alto o más bajo.
Conductividad se refiere a la capacidad de una solución acuosa de conducir una corriente eléctrica entre dos electrodos. En solución, la corriente fluye mediante transporte iónico. Por lo tanto, con una cantidad creciente de iones presentes en la solución acuosa, la solución tendrá una conductividad más alta. La unidad de medida básica para la conductividad es el siemensio (o mho), mho (mS/cm), y se puede medir usando un conductímetro, tal como diversos modelos de conductímetros Orion. Puesto que la conductividad electrolítica es la capacidad de los iones en una solución de transportar corriente eléctrica, se puede alterar la conductividad de una solución cambiando la concentración de iones en la misma. Por ejemplo, se puede alterar la concentración de un agente de tamponamiento y/o la concentración de una sal (por ejemplo, cloruro de sodio, acetato de sodio o cloruro de potasio) en la solución para lograr la conductividad deseada. Preferentemente, se modifica la concentración de sal de los diversos tampones para lograr la conductividad deseada.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el tampón de carga tiene una conductividad superior a aproximadamente cualquiera de 4,0 mS/cm, 4,5 mS/cm, 5,0 mS/cm, 5.5 mS/cm, 6,0 mS/cm, 6,5 mS/cm, 7,0 mS/cm, 7,5 mS/cm, 8,0 mS/cm, 8,5 mS/cm, 9,0 mS/cm, 9,5 mS/cm o 10 mS/cm. La conductividad puede ser de entre aproximadamente cualquiera de 4 mS/cm y 17 mS/cm, 4 mS/cm y 10 mS/cm, 4 mS/cm y 7 mS/cm, 5 mS/cm y 17 mS/cm, 5 mS/cm y 10 mS/cm o 5 mS/cm y 7 mS/cm. En algunos modos de realización, la conductividad es de aproximadamente 4 mS/cm, 4,5 mS/cm, 5,0 mS/cm, 5,5 mS/cm, 6,0 mS/cm, 6.5 mS/cm, 7,0 mS/cm, 7,5 mS/cm, 8,0 mS/cm, 8,5 mS/cm, 9,0 mS/cm, 9,5 mS/cm o 10 mS/cm. En un aspecto, la conductividad es la conductividad del tampón de carga, el tampón de equilibrado y/o el tampón de lavado. En algunos modos de realización, la conductividad de uno o más del tampón de carga, el tampón de equilibrado y el tampón de
lavado son iguales. En algunos modos de realización, la conductividad del tampón de carga es diferente de la conductividad del tampón de lavado y/o del tampón de equilibrado. En algunos modos de realización, la composición se carga sobre un material de cromatografía de modo mixto en un tampón con una conductividad de aproximadamente 5.5 mS/cm. En algunos modos de realización, el polipéptido es un anticuerpo o fragmento del mismo.
En algunos modos de realización, el tampón de elución tiene una conductividad menor que la conductividad del tampón de carga. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el tampón de elución tiene una conductividad inferior a aproximadamente cualquiera de 0,0 mS/cm, 0,5 mS/cm, 1.0 mS/cm, 1,5 mS/cm, 2,0 mS/cm, 2,5 mS/cm, 3,0 mS/cm, 3,5 mS/cm, 4,0 mS/cm, 4,5 mS/cm, 5,0 mS/cm, 5.5 mS/cm, 6,0 mS/cm, 6,5 mS/cm o 7,0 mS/cm. La conductividad puede ser de entre aproximadamente cualquiera de 0 mS/cm y 7 mS/cm, 1 mS/cm y 7 mS/cm, 2 mS/cm y 7 mS/cm, 3 mS/cm y 7 mS/cm o 4 mS/cm y 7 mS/cm, 0 mS/cm y 5,0 mS/cm, 1 mS/cm y 5 mS/cm, 2 mS/cm y 5 mS/cm, 3 mS/cm y 5 mS/cm o 4 mS/cm y 5 mS/cm. En algunos modos de realización, la conductividad del tampón de elución es aproximadamente cualquiera de 0,0 mS/cm, 0,5 mS/cm, 1,0 mS/cm, 1,5 mS/cm, 2,0 mS/cm, 2,5 mS/cm, 3,0 mS/cm, 3,5 mS/cm, 4 mS/cm, 4,5 mS/cm, 5,0 mS/cm, 5.5 mS/cm, 6,0 mS/cm, 6,5 mS/cm o 7,0 mS/cm. En algunos modos de realización, los tampones de elución descritos anteriormente se usan en OEC de modo mixto, OEC de intercambio aniónico, OEC de intercambio catiónico, OEC de afinidad u OEC HIC.
En algunos modos de realización de la divulgación, el tampón de elución tiene una conductividad mayor que la conductividad del tampón de carga. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el tampón de elución tiene una conductividad mayor que aproximadamente cualquiera de 5.5 mS/cm, 6,0 mS/cm, 6,5 mS/cm, 7,0 mS/cm, 7,5 mS/cm, 8,0 mS/cm, 8,5 mS/cm, 9,0 mS/cm, 9,5 mS/cm, 10 mS/cm, 11 mS/cm, 12 mS/cm, 13 mS/cm, 14 mS/cm, 15 mS/cm, 16 mS/cm, 17,0 mS/cm, 18,0 mS/cm, 19,0 mS/cm, 20.0 mS/cm, 21,0 mS/cm, 22,0 mS/cm, 23,0 mS/cm, 24,0 mS/cm o 25,0 mS/cm. La conductividad puede ser de entre aproximadamente cualquiera de 5,5 mS/cm y 17 mS/cm, 6,0 mS/cm y 17 mS/cm, 7 mS/cm y 17 mS/cm, 8 mS/cm y 17 mS/cm, 9 mS/cm y 17 mS/cm o 10 mS/cm y 17 mS/cm. En algunos modos de realización, la conductividad del tampón de elución es de aproximadamente cualquiera de 5,5 mS/cm, 6,0 mS/cm, 6,5 mS/cm, 7,0 mS/cm, 7,5 mS/cm, 8.0 mS/cm, 8,5 mS/cm, 9,0 mS/cm, 9,5 mS/cm, 10 mS/cm, 11 mS/cm, 12 mS/cm, 13 mS/cm, 14 mS/cm, 15 mS/cm, 16 mS/cm o 17,0 mS/cm. En algunos modos de realización de la divulgación, los tampones de elución descritos anteriormente se usan en OEC de modo mixto, OEC de intercambio aniónico, OEC de intercambio catiónico, OEC de afinidad u OEC HIC. En algunos modos de realización de la divulgación, el polipéptido se eluye de una cromatografía de modo mixto a una conductividad de aproximadamente 4 hasta aproximadamente 1 mS/cm. En algunos modos de realización de la divulgación, el polipéptido se eluye de una cromatografía Capto Adhere a una conductividad de aproximadamente 4 hasta aproximadamente 1 mS/cm. En algunos modos de realización de la divulgación, un anticuerpo o fragmento del mismo se eluye de una cromatografía Capto Adhere a una conductividad de aproximadamente 4 mS/cm hasta aproximadamente 1 mS/cm.
En algunos aspectos de la divulgación, la conductividad del tampón de elución cambiaba del tampón de carga y/o el tampón de lavado por gradiente escalonado o por gradiente lineal.
En algunos modos de realización de la invención, la composición que comprende un polipéptido se carga sobre el material de cromatografía en un tampón con una conductividad de aproximadamente 5,5 mS/cm y el polipéptido se eluye del material de cromatografía en un tampón de elución con una conductividad de aproximadamente 4 mS/cm. En algunos modos de realización, el tampón de carga tiene una conductividad de aproximadamente 5,5 mS/cm y el tampón de elución tiene una conductividad de aproximadamente 3 mS/cm. En algunos modos de realización, el tampón de carga tiene una conductividad de aproximadamente 5,5 mS/cm y el tampón de elución tiene una conductividad de aproximadamente 2 mS/cm. En algunos modos de realización, el tampón de carga tiene una conductividad de aproximadamente 5,5 mS/cm y el tampón de elución tiene una conductividad de aproximadamente 1 mS/cm. En modos de realización adicionales de los modos de realización anteriores, el material de cromatografía es una resina Capto Adhere. En modos de realización adicionales de los modos de realización anteriores, el polipéptido es un anticuerpo o fragmento del mismo.
En algunos aspectos de la divulgación, la conductividad del tampón de elución cambiaba del tampón de carga y/o el tampón de lavado por gradiente escalonado o por gradiente lineal. En algunos modos de realización de la divulgación, la composición que comprende un polipéptido se carga en una cromatografía Capto Adhere a aproximadamente 5.5 mS/cm y el polipéptido de interés se eluye de una cromatografía Capto Adhere mediante un gradiente de conductividad lineal de aproximadamente 5,5 mS/cm hasta aproximadamente 1 mS/cm sobre aproximadamente 5 volúmenes de columna (VC). En algunos modos de realización de la divulgación, la composición que comprende un polipéptido se carga en una cromatografía Capto Adhere a aproximadamente 5,5 mS/cm y el polipéptido de interés se eluye de una cromatografía Capto Adhere mediante un gradiente de conductividad lineal de aproximadamente 5.5 mS/cm hasta aproximadamente 1 mS/cm sobre 10,0 VC. En algunos modos de realización de la divulgación, la composición que comprende un polipéptido se carga en una cromatografía Capto Adhere a aproximadamente 10 mS/cm y el polipéptido de interés se eluye de una cromatografía Capto Adhere mediante un gradiente de conductividad lineal de aproximadamente 10,0 mS/cm hasta aproximadamente 1 mS/cm sobre aproximadamente 5 VC. En algunos modos de realización de la divulgación, la composición que comprende un polipéptido se carga en una cromatografía Capto Adhere a aproximadamente 10 mS/cm y el polipéptido de interés se eluye de una
cromatografía Capto Adhere mediante un gradiente de conductividad lineal de aproximadamente 10,0 mS/cm hasta 1 mS/cm sobre aproximadamente 10 VC. En algunos modos de realización de la divulgación, la composición que comprende un polipéptido se carga en una cromatografía Capto Adhere a aproximadamente 10 mS/cm y el polipéptido de interés se eluye de una cromatografía Capto Adhere mediante un gradiente de conductividad lineal de aproximadamente 10,0 mS/cm hasta aproximadamente 1 mS/cm sobre aproximadamente 15 VC.
En algunos aspectos de cualquiera de la divulgación, la conductividad del tampón de elución cambiaba del tampón de carga y/o de lavado por gradiente escalonado o por gradiente lineal. En algunos modos de realización de la divulgación, la composición que comprende un polipéptido se carga sobre un material de cromatografía a aproximadamente 5.5 mS/cm y el polipéptido de interés se eluye de un material de cromatografía mediante un gradiente de conductividad lineal entre 5,5 mS/cm y 1 mS/cm sobre 5 volúmenes de columna, de 5,5 mS/cm a 1 mS/cm sobre 10,0 volúmenes de columna. En algunos modos de realización de la divulgación, la composición que comprende un polipéptido se carga sobre un material de cromatografía a aproximadamente 10 mS/cm y el polipéptido de interés se eluye de un material de cromatografía mediante un gradiente de conductividad lineal entre 10,0 mS/cm y 1 mS/cm sobre 5 volúmenes de columna, de 10,0 mS/cm a 1 mS/cm sobre 10 volúmenes de columna, de 10,0 mS/cm a 1 mS/cm sobre 15 volúmenes de columna.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el tampón de carga tiene un pH menor que aproximadamente cualquiera de 10, 9, 8, 7, 6 o 5. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el tampón de carga tiene un pH mayor que aproximadamente cualquiera de 4, 5, 6, 7, 8 o 9. El tampón de carga puede tener un pH de entre aproximadamente cualquiera de 4 y 9, 4 y 8, 4 y 7, 5 y 9, 5 y 8, 5 y 7, 5 y 6. En algunos modos de realización, el pH del tampón de carga es aproximadamente cualquiera de 4, 4,5, 5, 5,5, 6, 6,5, 7, 7,5 u 8. El pH puede ser el pH del tampón de carga, el tampón de equilibrado o el tampón de lavado. En algunos modos de realización, el pH de uno o más del tampón de carga, el tampón de equilibrado y/o el tampón de lavado son iguales. En algunos modos de realización, el pH del tampón de carga es diferente del pH del tampón de equilibrado y/o del tampón de lavado.
En algunos modos de realización, el tampón de elución tiene un pH menor que el pH del tampón de carga. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el tampón de elución tiene un pH menor que aproximadamente cualquiera de 8, 7, 6, 5, 4, 3 o 2. El pH del tampón de elución puede ser de entre aproximadamente 4 y 9, 4 y 8, 4 y 7, 4 y 6, 4 y 5, 5 y 9, 5 y 8, 5 y 7, 5 y 6, 6 y 9, 6 y 8, 6 y 7. En algunos modos de realización, el pH del tampón de elución es aproximadamente cualquiera de 4,0, 4,5, 5,0, 5,5, 6,0, 6,5, 7,0, 7,5, 8,0, 8.5 o 9,0.
En algunos modos de realización, el tampón de elución tiene un pH mayor que el pH del tampón de carga. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el tampón de elución tiene un pH mayor que aproximadamente cualquiera de 5, 6, 7, 8, o 9. El pH del tampón de elución puede ser de entre aproximadamente 4 y 9, 5 y 9, 6 y 9, 7 y 9, 8 y 9, 4 y 8, 5 y 8, 6 y 8, 7 y 8, 4 y 7, 5 y 7, y 6 y 7. En algunos modos de realización, el pH del tampón de elución es aproximadamente cualquiera de 4,0, 4,5, 5,0, 5,5, 6,0, 6,5, 7,0, 7,5, 8,0, 8.5 o 9,0.
En algunos aspectos de cualquiera de los modos de realización anteriores, el pH del tampón de elución cambiaba del tampón de carga y/o el tampón de lavado por gradiente escalonado o por gradiente lineal.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el caudal es inferior a aproximadamente cualquiera de 50 VC/h, 40 VC/h o 30 VC/h. El caudal puede ser de entre aproximadamente cualquiera de 5 VC/h y 50 VC/h, 10 VC/h y 40 VC/h o 18 VC/h y 36 VC/h. En algunos modos de realización, el caudal es aproximadamente cualquiera de 9 VC/h, 18 VC/h, 25 VC/h, 30 VC/h, 36 VC/h o 40 VC/h. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el caudal es inferior a aproximadamente cualquiera de 100 cm/h, 75 cm/h o 50 cm/h. El caudal puede ser de entre cualquiera de aproximadamente 25 cm/h y 150 cm/h, 25 cm/h y 100 cm/h, 50 cm/h y 100 cm/h o 65 cm/h y 85 cm/h.
La altura del lecho es la altura del material de cromatografía usado. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, la altura del lecho es mayor que aproximadamente cualquiera de 3 cm, 10 cm o 15 cm. La altura del lecho puede ser de entre aproximadamente cualquiera de 3 cm y 35 cm, 5 cm y 15 cm, 3 cm y 10 cm, o 5 cm y 8 cm. En algunos modos de realización, la altura del lecho es de aproximadamente cualquiera de 3 cm, 5 cm, 10 cm o 15 cm. En algunos modos de realización, la altura del lecho se determina basándose a la cantidad de polipéptido o contaminantes en la carga.
En algunos modos de realización, la cromatografía es en una columna de recipiente con un volumen de más de aproximadamente 1 ml, 2 ml, 3 ml, 4 ml, 5 ml, 6 ml, 7 ml, 8 ml, 9 ml, 10 ml, 15 ml, 20 ml, 25 ml, 30 ml, 40 ml, 50 ml, 75 ml, 100 ml, 200 ml, 300 ml, 400 ml, 500 ml, 600 ml, 700 ml, 800 ml, 900 ml, 1 l, 2 l, 3 l, 4 l, 5 l, 6 l, 7 l, 8 l, 9 l, 10 l, 25 l, 50 l, 100 l, 200 l, 300 l, 400 l, 500 l, 600 l, 700 l, 800 l, 900 l o 1000 l.
En algunos modos de realización de la invención, las fracciones se recogen de la cromatografía. En algunos modos de realización, las fracciones recogidas son mayores de aproximadamente 0,01 VC, 0,02 VC, 0,03 VC, 0,04 VC, 0,05 VC, 0,06 VC, 0,07 VC, 0,08 VC, 0,09 VC, 0,1 VC, 0,2 VC, 0,3 VC, 0,4 VC, 0,5 VC, 0,6 VC, 0,7 VC, 0,8 VC, 0,9 VC,
1,0 VC, 2,0 VC, 3,0 VC, 4,0 VC, 5,0 VC, 6,0 VC, 7,0 VC, 8,0 VC, 9,0 VC o 10,0 VC. En algunos modos de realización se agrupan fracciones que contienen el producto, por ejemplo, polipéptido. En algunos modos de realización, se agrupan fracciones que contienen el polipéptido de las fracciones de carga y de las fracciones de elución. La cantidad de polipéptido en una fracción se puede determinar por un experto en la técnica; por ejemplo, la cantidad de polipéptido en una fracción se puede determinar por espectroscopia UV. En algunos modos de realización, se agrupan fracciones que contienen fragmento de polipéptido detectable.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el al menos un contaminante es uno o más de materiales de la célula huésped, tales como CHOP; proteína A lixiviada; ácido nucleico; una variante, fragmento, agregado o derivado del polipéptido deseado; otro polipéptido; endotoxina; contaminante vírico; componente de medio de cultivo celular, gentamicina, etc. En algunos ejemplos, el contaminante puede ser una proteína de la célula huésped (HCP), por ejemplo, pero no limitada a, una célula bacteriana tal como una célula de E. coli, una célula de insecto, una célula procariota, una célula eucariota, una célula de levadura, una célula de mamífero, una célula aviar, una célula fúngica.
Las proteínas de la célula huésped (HCP) son proteínas de las células en las que se produjo el polipéptido. Por ejemplo, CHOP son proteínas de células huésped, es decir, proteínas de ovario de hámster chino. La cantidad de CHOP se puede medir mediante ensayo de inmunoadsorción enzimática ("ELISA") o Meso Scale Discovery ("MSD"). En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, la cantidad de HCP (por ejemplo, CHOP) se reduce en más de aproximadamente cualquiera de un 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 % %, 90 % o 95 %. La cantidad de HCP se puede reducir entre aproximadamente cualquiera de un 10 % y 99 %, 30 % y 95 %, 30 % y 99 %, 50 % y 95 %, 50 % y 99 %, 75 % y 99 % u 85 % y 99 %. En algunos modos de realización, la cantidad de HCP se reduce en aproximadamente cualquiera de un 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 85 %, 90 %, 95 % o 98 %. En algunos modos de realización, la reducción se determina comparando la cantidad de HCP en la composición recuperada de una etapa o etapas de purificación con la cantidad de HCP en la composición antes de la etapa o etapas de purificación.
El polipéptido agregado puede ser una proteína de alto peso molecular (HMW). En algunos modos de realización, el polipéptido agregado son multímeros del polipéptido de interés. La proteína HMW puede ser un dímero, hasta 8x monómero, o mayor del polipéptido de interés. Los procedimientos de medición de la proteína agregada (por ejemplo, proteína HMW) son conocidos en la técnica y se describen en la sección de ejemplos. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, la cantidad de proteína agregada se reduce en más de aproximadamente cualquiera de un 5 %, 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 % o 95 %. La cantidad de proteína agregada se puede reducir entre aproximadamente cualquiera de 10 % y 99 %, 30 % y 95 %, 30 % y 99 %, 50 % y 95 %, 50 % y 99 %, 75 % y 99 % u 85 % y 99 %. La cantidad de proteína agregada se puede reducir en aproximadamente cualquiera de un 5 %, 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 % o 95 %. En algunos modos de realización, la reducción se determina comparando la cantidad de proteína agregada (por ejemplo, proteína HMW) en la composición recuperada de una etapa o etapas de purificación con la cantidad de proteína agregada (por ejemplo, proteína HMW) en la composición antes de la etapa o etapas de purificación.
El polipéptido fragmentado puede ser proteína de bajo peso molecular (LMW). En algunos modos de realización, el polipéptido fragmentado es un fragmento del polipéptido de interés. Ejemplos de proteína LMW incluyen, pero no se limitan a, un Fab (fragmento de unión a antígeno), Fc (fragmento, cristalizable) o combinación de ambos o cualquier parte aleatoria fragmentada de un anticuerpo de interés. Los procedimientos para medir la proteína fragmentada (por ejemplo, proteína LMW) son conocidos en la técnica y se describen en la sección de ejemplos. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, la cantidad de proteína LMW se reduce en más de aproximadamente cualquiera de un 5 %, 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 % o 95 %. La cantidad de proteína LMW se puede reducir en aproximadamente entre cualquiera de un 10 % y 99 %, 30 % y 95 %, 30 % y 99 %, 50 % y 95 %, 50 % y 99 %, 75 % y 99 % u 85 % y 99 %. La cantidad de proteína LMW se puede reducir en aproximadamente cualquiera de un 5 %, 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 % o 95 %. En algunos modos de realización, la reducción se determina comparando la cantidad de proteína fragmentada (por ejemplo, proteína LMW) en la composición recuperada de una etapa o etapas de purificación con la cantidad de proteína fragmentada (por ejemplo, proteína LMW) en la composición antes de la etapa o etapas de purificación.
La proteína A lixiviada es proteína A separada o lavada de una fase sólida a la que está unida. Por ejemplo, la proteína A lixiviada se puede lixiviar de una columna de cromatografía de afinidad de proteína A. La cantidad de proteína A se puede medir, por ejemplo, mediante ELISA. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, la cantidad de proteína A lixiviada se reduce en más de aproximadamente cualquiera de un 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 % o 90 %. La cantidad de proteína A lixiviada se puede reducir en aproximadamente entre cualquiera de un 10 % y 99 %, 30 % y 95 %, 30 % y 99 %, 50 % y 95%, 50 % y 99 %, 75 % y 99 % u 85 % y 99 %. En algunos modos de realización, la cantidad de proteína A lixiviada se reduce en aproximadamente cualquiera de un 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 % o 95 %. En algunos modos de realización, la reducción se determina comparando la cantidad de proteína A lixiviada en la composición recuperada de una etapa o etapas de purificación con la cantidad de proteína A lixiviada en la composición antes de la etapa o etapas de purificación.
Los procedimientos de medición del ADN, tales como el ADN de la célula huésped, son conocidos en la técnica y se describen en la sección de ejemplos. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, la cantidad de ADN se reduce en más de aproximadamente cualquiera de un 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 % o 90 %. La cantidad de ADN se puede reducir en aproximadamente entre cualquiera de 10 % y 99 %, 30 % y 95 %, 30 % y 99 %, 50 % y 95 %, 50 % y 99 %, 75 % y 99 % u 85 % y 99%. La cantidad de ADN se puede reducir en aproximadamente cualquiera de un 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 %, 95 % o 99 %. En algunos modos de realización, la reducción se determina comparando la cantidad de ADN en la composición recuperada de una etapa o etapas de purificación con la cantidad de ADN en la composición antes de la etapa o etapas de purificación.
Componente de medio de cultivo celular se refiere a un componente presente en un medio de cultivo celular. Un medio de cultivo celular puede ser un medio de cultivo celular en el momento de la recolección de células. En algunos modos de realización, el componente de medio de cultivo celular es gentamicina. La cantidad de gentamicina se puede medir mediante ELISA. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, la cantidad de componente de medio de cultivo celular se reduce en más de aproximadamente cualquiera de un 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 % o 90 %. La cantidad de componente de medio de cultivo celular se puede reducir en aproximadamente entre cualquiera de 10 % y 99 %, 30 % y 95 %, 30 % y 99 %, 50 % y 95 %, 50 % y 99 %, 75 % y 99 % u 85 % y 99 %. En algunos modos de realización, la cantidad de componente de medio de cultivo celular se reduce en aproximadamente cualquiera de un 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, 90 %, 95 % o 98 %. En algunos modos de realización, la reducción se determina comparando la cantidad de componente de medio de cultivo celular en la composición recuperada de una etapa o etapas de purificación con la cantidad de componente de medio de cultivo celular en la composición antes de la etapa o etapas de purificación.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, los procedimientos pueden comprender además una o más etapas de purificación ya sea antes o después de cualquiera de las OEC descritas en el presente documento. Otros procedimientos de purificación incluyen, por ejemplo, cromatografía de intercambio iónico tal como cromatografía de intercambio aniónico y cromatografía de intercambio catiónico, cromatografía de afinidad tal como cromatografía de proteína A y cromatografía de proteína G, cromatografía de modo mixto, cromatografía de hidroxilapatita; cromatografía de filtración en gel; cromatografía de afinidad; electroforesis en gel; diálisis; precipitación con etanol; HPLC de fase inversa; cromatografía sobre sílice; cromatoenfoque; SDS-PAGE; precipitación con sulfato de amonio; y columnas quelantes de metales que se unen a las formas marcadas con epítopo del polipéptido.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, los procedimientos comprenden además la recuperación del polipéptido purificado. En algunos modos de realización, el polipéptido purificado se recupera de cualquiera de las etapas de purificación descritas en el presente documento. La etapa de cromatografía puede ser cromatografía de intercambio catiónico, cromatografía de modo mixto o cromatografía de afinidad de proteína A. En algunos modos de realización, la cromatografía OEC es una cromatografía de modo mixto y la cromatografía adicional es una cromatografía de intercambio aniónico. En algunos modos de realización, la cromatografía OEC es una cromatografía de modo mixto y la cromatografía adicional es una cromatografía de intercambio catiónico. En algunos modos de realización, la cromatografía OEC es una cromatografía de modo mixto y la cromatografía adicional es una cromatografía HIC. En algunos modos de realización, la cromatografía OEC es una cromatografía de intercambio aniónico y la cromatografía adicional es una cromatografía de intercambio catiónico. En algunos modos de realización, la cromatografía OEC es una cromatografía de intercambio aniónico y la cromatografía adicional es una cromatografía de modo mixto. En algunos modos de realización, la cromatografía OEC es una cromatografía de intercambio aniónico y la cromatografía adicional es una cromatografía HIC. En algunos modos de realización, la cromatografía OEC es una cromatografía de intercambio catiónico y la cromatografía adicional es una cromatografía de intercambio aniónico. En algunos modos de realización, la cromatografía OEC es una cromatografía de intercambio catiónico y la cromatografía adicional es una cromatografía de modo mixto. En algunos modos de realización, la cromatografía OEC es una cromatografía de intercambio catiónico y la cromatografía adicional es una cromatografía HIC. En algunos modos de realización, la cromatografía OEC es una cromatografía HIC y la cromatografía adicional es una cromatografía de modo mixto. En algunos modos de realización, la cromatografía OEC es una cromatografía HIC y la cromatografía adicional es una cromatografía de intercambio aniónico. En algunos modos de realización, la cromatografía OEC es una cromatografía HIC y la cromatografía adicional es una cromatografía de intercambio catiónico.
En algunos modos de realización, el polipéptido se purifica adicionalmente después de OEC por filtración vírica. La filtración vírica es la retirada de contaminantes víricos en una corriente de alimentación de purificación de polipéptidos. Los ejemplos de filtración vírica incluyen ultrafiltración y microfiltración. En algunos modos de realización, el polipéptido se purifica utilizando un filtro de parvovirus.
En algunos modos de realización, el polipéptido se concentra después de la cromatografía por el modo OEC. Son conocidos en la técnica ejemplos de procedimientos de concentración e incluyen, pero no se limitan a, ultrafiltración y diafiltración.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, los procedimientos comprenden además combinar el polipéptido purificado de los procedimientos de purificación con un vehículo farmacéuticamente aceptable.
En algunos modos de realización, la invención proporciona procedimientos para purificar un anticuerpo, que comprenden: a) cargar una composición que comprende el anticuerpo en una resina Capto Adhere a una densidad de carga de 150-200 g de anticuerpo por litro de resina Capto Adhere en un tampón de carga con un pH de aproximadamente 6,5 y una conductividad de aproximadamente 5,3 mS/cm hasta aproximadamente 5,6 mS/cm; b) eluir el anticuerpo de la resina con un tampón de elución que comprende ácido 2-(W-morfolino)etanosulfónico (MES) 100 mM con un pH de aproximadamente 6,5 y una conductividad de aproximadamente 1 mS/cm; y recoger una agrupación que comprende el anticuerpo.
En algunos modos de realización, la invención proporciona procedimientos para purificar un anticuerpo, que comprenden: a) cargar una composición que comprende el anticuerpo en una resina Capto Adhere en una columna de 1,6-10,8 l a una densidad de carga de 70-180 g de anticuerpo por litro de resina Capto Adhere en un tampón de carga con un pH de aproximadamente 6,5 y una conductividad de aproximadamente 5,3 mS/cm hasta aproximadamente 5,6 mS/cm; b) eluir el anticuerpo de la resina con un tampón de elución que comprende MES 100 mM con un pH de aproximadamente 6,5 y una conductividad de aproximadamente 1 mS/cm; y recoger una agrupación que comprende el anticuerpo.
En algunos modos de realización, la invención proporciona procedimientos para purificar un anticuerpo, que comprenden: a) cargar una composición que comprende el anticuerpo en una resina Capto Adhere a una densidad de carga de aproximadamente 200 g de anticuerpo por litro de resina Capto Adhere en un tampón de carga con un pH de aproximadamente 8,6 y una conductividad de aproximadamente menos de 6 mS/cm; b) eluir el anticuerpo de la resina con un tampón de elución de MES 20 mM con un pH de aproximadamente 6,5 y una conductividad de aproximadamente 1 mS/cm; y recoger una agrupación que comprende el anticuerpo.
En algunos modos de realización, la invención proporciona procedimientos para purificar un anticuerpo, que comprenden: a) cargar una composición que comprende el anticuerpo en una resina Capto Adhere a una densidad de carga de aproximadamente 200 g de anticuerpo por litro de resina Capto Adhere en un tampón de carga con un pH de aproximadamente 6,1 y una conductividad de aproximadamente menos de 6 mS/cm; b) eluir el anticuerpo de la resina con un tampón de elución de MES 20 mM con un pH de aproximadamente 6,0 y una conductividad de aproximadamente 0,65 mS/cm; y recoger una agrupación que comprende el anticuerpo.
En algunos modos de realización, la invención proporciona procedimientos para purificar un anticuerpo, que comprenden: a) cargar una composición que comprende el anticuerpo en una resina Capto Adhere a una densidad de carga de aproximadamente 200 g de anticuerpo por litro de resina Capto Adhere en un tampón de carga con un pH de aproximadamente 5,5 y una conductividad de aproximadamente menos de 6 mS/cm; b) eluir el anticuerpo de la resina con un tampón de elución de MES 20 mM con un pH de aproximadamente 4,9 y una conductividad de aproximadamente 1,1 mS/cm; y recoger una agrupación que comprende el anticuerpo.
En algunos modos de realización, la invención proporciona procedimientos para purificar un anticuerpo, que comprenden: a) cargar una composición que comprende el anticuerpo en una resina Capto Adhere a una densidad de carga de aproximadamente 200 g de anticuerpo por litro de resina Capto Adhere en un tampón de carga con un pH de aproximadamente 6,5 y una conductividad de aproximadamente menos de 6 mS/cm; b) eluir el anticuerpo de la resina con un tampón de elución de MES 20 mM con un pH de aproximadamente 6,5 y una conductividad de aproximadamente 1 mS/cm; y recoger una agrupación que comprende el anticuerpo.
En algunos modos de realización, la invención proporciona procedimientos para purificar un anticuerpo, que comprenden: a) cargar una composición que comprende el anticuerpo sobre una resina QMA a una densidad de carga de aproximadamente 103 g de anticuerpo por litro de resina QMA en un tampón de carga con un pH de aproximadamente 6,5 y una conductividad de aproximadamente menos de 5,5 mS/cm; b) eluir el anticuerpo de la resina con un tampón de elución de MES 20 mM con un pH de aproximadamente 6,5 y una conductividad de aproximadamente 1 mS/cm; y recoger una agrupación que comprende el anticuerpo.
En algunos modos de realización, la invención proporciona procedimientos para purificar un anticuerpo, que comprenden: a) cargar una composición que comprende el anticuerpo en una resina Poros XS a una densidad de carga de aproximadamente 200 g de anticuerpo por litro de resina Poros XS en un tampón de carga con un pH de aproximadamente 5,5 y una conductividad de aproximadamente menos de 6 mS/cm; b) eluir el anticuerpo de la resina con un tampón de elución de acetato 50-350 mM con un pH de aproximadamente 5,5; y recoger una agrupación que comprende el anticuerpo.
En algunos modos de realización, la invención proporciona procedimientos para purificar un anticuerpo, que comprenden: a) cargar una composición que comprende el anticuerpo en una resina Capto MMC a una densidad de carga de aproximadamente 147 g de anticuerpo por litro de resina Capto MMC en un tampón de carga con un pH de aproximadamente 7,0 y una conductividad de aproximadamente menos de 6 mS/cm; b) eluir el anticuerpo de la resina
con un tampón de elución de MES 20 mM con un pH de aproximadamente 6,5 y una conductividad de aproximadamente 1 mS/cm; y recoger una agrupación que comprende el anticuerpo.
III. Polipéptidos
Se proporcionan polipéptidos para su uso en cualquiera de los procedimientos de purificación de polipéptidos y formulaciones que comprenden los polipéptidos purificados por los procedimientos descritos en el presente documento.
En algunos modos de realización, la invención proporciona procedimientos para purificar un polipéptido usando cromatografía de sobrecarga y elución. En algunos modos de realización, el polipéptido es un polipéptido terapéutico. En algunos modos de realización de la divulgación, el polipéptido es un antagonista. En algunos modos de realización, el polipéptido es un agonista. En algunos modos de realización, el polipéptido es un anticuerpo. En algunos modos de realización, el polipéptido es un epítopo marcado. En algunos modos de realización, el polipéptido retiene una actividad biológica y/o inmunológica. En algunos modos de realización, el polipéptido es un antagonista. En algunos modos de realización, el polipéptido inicia la citotoxicidad dependiente del complemento. En algunos modos de realización, el polipéptido es un anticuerpo o inmunoadhesina. En modos de realización adicionales de los modos de realización anteriores, el polipéptido se purifica por OEC usando un medio de cromatografía de modo mixto. En modos de realización adicionales de los modos de realización anteriores, el polipéptido se purifica por OEC usando un medio de cromatografía de intercambio aniónico. En modos de realización adicionales de los modos de realización anteriores, el polipéptido se purifica por OEC usando un medio de cromatografía de intercambio catiónico. En modos de realización adicionales de los modos de realización anteriores, el polipéptido se purifica por OEC usando un medio de cromatografía HIC. En modos de realización adicionales de los modos de realización anteriores, el polipéptido se purifica por OEC usando un medio de cromatografía de HAP. En modos de realización adicionales de los modos de realización anteriores, el polipéptido se purifica por OEC usando un medio de cromatografía de afinidad. En modos de realización adicionales de los modos de realización anteriores, el polipéptido se purifica por OEC usando un medio de cromatografía que no es, o no incluye, una cromatografía de intercambio catiónico.
En algunos modos de realización, el polipéptido tiene un peso molecular de más de aproximadamente cualquiera de 5000 dalton, 10000 dalton, 15000 dalton, 25000 dalton, 50000 dalton, 75000 dalton, 100000 dalton, 125000 dalton o 150000 dalton. El polipéptido puede tener un peso molecular entre aproximadamente cualquiera de 50 000 dalton a 200000 dalton o 100000 dalton a 200000 dalton. De forma alternativa, el polipéptido para su uso en el presente documento puede tener un peso molecular de aproximadamente 120 000 dalton o aproximadamente 25 000 dalton.
El pI es el punto isoeléctrico y es el pH al que una molécula o superficie particular no tiene carga eléctrica neta. En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el pI del polipéptido puede ser de entre aproximadamente cualquiera de 6 y 10, de 7 a 9 o de 8 a 9. En algunos modos de realización, el polipéptido tiene un pI de aproximadamente cualquiera de 6, 7, 7,5, 8, 8,5, 9, 9,5 o 10.
Los polipéptidos a purificar usando los procedimientos descritos en el presente documento se producen generalmente utilizando técnicas recombinantes. Se describen procedimientos para producir proteínas recombinantes, por ejemplo, en las pat. de EE.UU. n.° 5.534.615 y 4.816.567. En algunos modos de realización, se produce la proteína de interés en una célula de CHO (véase, por ejemplo, el documento WO 94/11026). Cuando se usan técnicas recombinantes, los polipéptidos se pueden producir intracelularmente, en el espacio periplásmico, o segregar directamente al medio.
Los polipéptidos se pueden recuperar del medio de cultivo o de lisados de células huésped. Se pueden romper las células empleadas en la expresión de los polipéptidos mediante diversos medios físicos o químicos, tales como ciclos de congelación-descongelación, sonicación, ruptura mecánica o agentes de lisis de células. Si el polipéptido se produce intracelularmente, como una primera etapa, se retiran los restos de partículas, células huésped o bien fragmentos lisados, por ejemplo, mediante centrifugación o ultrafiltración. Carter et al., Bio/Technology 10:163-167 (1992) describen un procedimiento para aislar polipéptidos que se segregan al espacio periplásmico de E. coli. En resumen, se descongela la pasta de células en presencia de acetato de sodio (pH 3,5), EDTA y fluoruro de fenilmetilsulfonilo (PMSF) durante aproximadamente 30 min. Los restos celulares se pueden retirar mediante centrifugación. Cuando el polipéptido se segrega al medio, los sobrenadantes de dichos sistemas de expresión en primer lugar se concentran, en general, usando un filtro de concentración de polipéptidos disponible comercialmente, por ejemplo, una unidad de ultrafiltración Amicon o Millipore Pellicon. Se puede incluir un inhibidor de proteasa, tal como PMSF, en cualquiera de las etapas anteriores para inhibir la proteólisis y se pueden incluir antibióticos para prevenir el crecimiento de contaminantes casuales.
Ejemplos de polipéptidos que se pueden purificar mediante los procedimientos de la divulgación incluyen, pero no se limitan a inmunoglobulinas, inmunoadhesinas, anticuerpos, enzimas, hormonas, proteínas de fusión, proteínas que contienen Fc, inmunoconjugados, citocinas e interleucinas. Ejemplos de polipéptidos incluyen, pero no se limitan a, proteínas de mamífero, tales como, por ejemplo, renina; una hormona; una hormona de crecimiento, incluyendo la hormona de crecimiento humana y la hormona de crecimiento bovina; factor de liberación de la hormona de crecimiento; hormona paratiroidea; hormona estimulante del tiroides; lipoproteínas; alfa-1-antitripsina; cadena A de insulina; cadena B de insulina; proinsulina; hormona foliculoestimulante; calcitonina; hormona luteinizante; glucagón; factores de
coagulación tales como factor VIIIC, factor IX, factor tisular y factor de Von Willebrand; factores anticoagulación tales como proteína C; factor natriurético auricular; tensioactivo pulmonar; un activador de plasminógeno, tal como urocinasa o activador de plasminógeno de orina humana o de tipo tisular (t-PA); bombesina; trombina; factor de crecimiento hemopoyético; factor de necrosis tumoral alfa y beta; encefalinasa; RANTES (regulada por activación, normalmente expresada y segregada por linfocitos T); proteína inflamatoria de macrófagos humanos (MIP-1-alfa); una seroalbúmina tal como seroalbúmina humana; hormona antimuleriana; cadena A de relaxina; cadena B de relaxina; prorrelaxina; péptido asociado a gonadotropina de ratón; una enzima; una proteína microbiana, tal como betalactamasa; DNasa; IgE; un antígeno asociado a linfocitos T citotóxicos (CTLA), tal como CTLA-4; inhibina; activina; factor de crecimiento del endotelio vascular (VEGF); receptores para hormonas o factores de crecimiento; proteína A o D; factores reumatoides; un factor neurotrófico tal como el factor neurotrófico derivado del hueso (BDNF), neurotrofina-3, -4, -5 o -6 (NT-3, NT-4, NT-5 o NT-6) o un factor crecimiento de nervios tal como NGF-b; factor de crecimiento derivado de plaquetas (PDGF); factor de crecimiento de fibroblastos tal como aFGF y bFGF; factor de crecimiento epidérmico (EGF); factor de crecimiento transformante (TGF) tal como TGF-alfa y TGF-beta, incluyendo TGF-p1, TGF-p2, TGF-p3, TGF-p4 o TGF-p5; factor insulínico de crecimiento I y II (IGF-I e iGf -II); des(1-3)-IGF-I (IGF-I cerebral), proteínas de unión al factor insulínico de crecimiento (IGFBP); una citocina; proteínas CD tales como CD3, CD4, CD8, CD19 y CD20; eritropoyetina; factores osteoinductivos; inmunotoxinas; un polipéptido de fusión, es decir, un polipéptido compuesto por dos o más polipéptidos heterólogos o fragmentos de los mismos y codificado por un ácido nucleico recombinante; un polipéptido que contiene Fc, por ejemplo, una proteína de fusión que comprende una región Fc de inmunoglobulina, o fragmento de la misma, fusionada a un segundo polipéptido; un inmunoconjugado; una proteína morfogenética ósea (BMP); un interferón tal como interferón alfa, beta y gamma; factores estimulantes de colonias (CSF), por ejemplo, M-CSF, GM-CSF y G-CSF; interleucinas (IL), por ejemplo, IL-1 a IL-10; superóxido dismutasa; receptores de linfocitos T; proteínas de membrana de superficie; factor de aceleración de la descomposición; antígeno vírico tal como, por ejemplo, una porción de la envoltura de SIDA; proteínas de transporte; receptores de la migración dirigida; adresinas; proteínas reguladoras; integrinas tales como CD11a, CD11b, CD11c, CD18, ICAM, VLA-4 y VCAM; un antígeno asociado a un tumor tal como CA125 (antígeno de cáncer de ovario) o receptor HER2, HER3 o HER4; inmunoadhesinas; y fragmentos y/o variantes de cualquiera de las proteínas mencionadas anteriormente, así como anticuerpos, incluyendo fragmentos de anticuerpo, que se unen a una proteína, incluyendo, por ejemplo, cualquiera de las proteínas mencionadas anteriormente.
(A) Anticuerpos
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el polipéptido para su uso en cualquiera de los procedimientos de purificación de polipéptidos y formulaciones que comprenden los polipéptidos purificados por los procedimientos descritos en el presente documento es un anticuerpo.
Las dianas moleculares para anticuerpos incluyen las proteínas CD y sus ligandos, tales como, pero sin limitación: (i) CD3, CD4, CD8, CD19, CD11a, CD20, CD22, CD34, CD40, CD79a (CD79a) y CD79P (CD79b); (ii) miembros de la familia de receptores ErbB, tales como el receptor de EGF, el receptor HER2, HER3 o HER4; (iii) moléculas de adhesión celular, tales como LFA-1, Mac1, p150,95, VLA-4, ICAM-1, VCAM e integrina av/p3, incluyendo las subunidades alfa o bien beta de la misma (por ejemplo, anticuerpos anti-CD11 a, anti-CD18 o anti-CD11 b); (iv) factores de crecimiento, tales como VEGF; IgE; antígenos de grupos sanguíneos; receptor flk2/flt3; receptor de obesidad (OB); receptor mpl; CTLA-4; proteína C, BR3, c-met, factor tisular, p7, etc. y (v) antígenos asociados a tumor (TAA) de superficie celular y transmembranarios, tales como los descritos en la patente de EE. UU. n.° 7.521.541.
Otros anticuerpos ejemplares incluyen los seleccionados de, y sin limitación, anticuerpo antirreceptor de estrógenos, anticuerpo antirreceptor de progesterona, anticuerpo anti-p53, anticuerpo anti-HER-2/neu, anticuerpo anti-EGFR, anticuerpo anticatepsina D, anticuerpo anti-Bcl-2, anticuerpo anticadherina E, anticuerpo anti-CA125, anticuerpo anti-CA15-3, anticuerpo anti-CA19-9, anticuerpo anti-c-erbB-2, anticuerpo antiglucoproteína P, anticuerpo anti-CEA, anticuerpo antiproteína de retinoblastoma, anticuerpo antioncoproteína ras, anticuerpo anti-Lewis X, anticuerpo anti-Ki-67, anticuerpo anti-PCNA, anticuerpo anti-CD3, anticuerpo anti-CD4, anticuerpo anti-CD5, anticuerpo anti-CD7, anticuerpo anti-CD8, anticuerpo anti-CD9/p24, anticuerpo anti-CD10, anticuerpo anti-CD11a, anticuerpo anti-CD11c, anticuerpo anti-CD13, anticuerpo anti-CD14, anticuerpo anti-CD15, anticuerpo anti-CD19, anticuerpo anti-CD20, anticuerpo anti-CD22, anticuerpo anti-CD23, anticuerpo anti-CD30, anticuerpo anti-CD31, anticuerpo anti-CD33, anticuerpo anti-CD34, anticuerpo anti-CD35, anticuerpo anti-CD38, anticuerpo anti-CD41, anticuerpo anti-LCA/CD45, anticuerpo anti-CD45RO, anticuerpo anti-CD45RA, anticuerpo anti-CD39, anticuerpo anti-CD100, anticuerpo anti-CD95/Fas, anticuerpo anti-CD99, anticuerpo anti-CD106, anticuerpo antiubiquitina, anticuerpo anti-CD71, anticuerpo anti-c-myc, anticuerpo anticitoqueratinas, anticuerpo antivimentinas, anticuerpo antiproteínas HPV, anticuerpo anticadenas ligeras kappa, anticuerpo anticadenas ligeras lambda, anticuerpo antimelanosomas, anticuerpo antiantígeno específico de próstata, anticuerpo anti-S-100, anticuerpo antiantígeno tau, anticuerpo antifibrina, anticuerpo antiqueratinas y anticuerpo antiantígeno Tn.
(i) Anticuerpos policlonales
En algunos modos de realización, los anticuerpos son anticuerpos policlonales. Los anticuerpos policlonales se obtienen preferentemente en animales mediante múltiples inyecciones subcutáneas (s.c.) o intraperitoneales (i.p.) del antígeno pertinente y un adyuvante. Puede ser útil conjugar el antígeno pertinente con un polipéptido que sea
inmunógeno en la especie que se va a inmunizar, por ejemplo, hemocianina de lapa californiana, seroalbúmina, tiroglobulina bovina o inhibidor de tripsina de soja, usando un agente bifuncional o derivatizante, por ejemplo, éster sulfosuccinimida de maleimidobenzoílo (conjugación a través de residuos de cisteína), N-hidroxisuccinimida (a través de residuos de lisina), glutaraldehído, anhídrido succínico, SOCh o R1N=C=NR, donde R y R1 son grupos alquilo diferentes.
Los animales se inmunizan contra el antígeno, conjugados inmunógenos o derivados combinando, por ejemplo, 100 pg o 5 pg del polipéptido o conjugado (para conejos o ratones, respectivamente) con 3 volúmenes de adyuvante completo de Freund e inyectando la solución intradérmicamente en múltiples sitios. Un mes más tarde, se administra una dosis de refuerzo con 1/5 a 1/10 de la cantidad original de péptido o conjugado en adyuvante completo de Freund mediante inyección subcutánea en múltiples sitios. De siete a 14 días más tarde se extrae sangre de los animales y el suero se somete a ensayo para determinar el título de anticuerpos. Se administra una dosis de refuerzo a los animales hasta alcanzar una meseta del título. En algunos modos de realización, se administra una dosis de refuerzo al animal con el conjugado del mismo antígeno, pero conjugado con un polipéptido diferente y/o a través de un reactivo de reticulación diferente. Los conjugados también se pueden preparar en cultivo celular recombinante como fusiones polipeptídicas. Además, los agentes de agregación, como el alumbre, se usan adecuadamente para mejorar la respuesta inmunitaria.
(ii) Anticuerpos monoclonales
En algunos modos de realización, los anticuerpos son anticuerpos monoclonales. Se obtienen anticuerpos monoclonales de una población de anticuerpos sustancialmente homogénea, es decir, los anticuerpos individuales que comprenden la población son idénticos y/o se unen al mismo epítopo, excepto por posibles variantes que surjan durante la producción del anticuerpo monoclonal, estando presentes, en general, dichas variantes en cantidades menores. Por tanto, el modificador "monoclonal" indica que el carácter del anticuerpo no es una mezcla de anticuerpos discretos o policlonales.
Por ejemplo, se pueden preparar los anticuerpos monoclonales usando el procedimiento de hibridoma descrito por primera vez por Kohler et al., Nature 256:495 (1975), o se pueden preparar mediante procedimientos de ADN recombinante (patente de EE. UU n.° 4.816.567).
En el procedimiento de hibridoma, un ratón u otro animal huésped apropiado, tal como un hámster, se inmuniza como se describe en el presente documento para inducir linfocitos que producen o pueden producir anticuerpos que se unirán específicamente al polipéptido usado para la inmunización. De forma alternativa, los linfocitos se pueden inmunizar in vitro. Los linfocitos se fusionan entonces con células de mieloma utilizando un agente de fusión adecuado, tal como polietilenglicol, para formar una célula de hibridoma (Goding, Monoclonal Antibodies: Principies and Practice, pág. 59-103 (Academic Press, 1986)).
Las células de hibridoma preparadas de este modo se siembran y cultivan en un medio de cultivo adecuado que contenga preferentemente una o más sustancias que inhiban el crecimiento o la supervivencia de las células de mieloma originales no fusionadas. Por ejemplo, si las células de mieloma originales carecen de la enzima hipoxantina guanina fosforribosiltransferasa (HGPRT o HPRT), el medio de cultivo para los hibridomas incluirá típicamente hipoxantina, aminopterina y timidina (medio HAT), previniendo dichas sustancias el crecimiento de células carentes de HGPRT.
En algunos modos de realización, las células de mieloma son aquellas que se fusionan eficazmente, soportan una producción estable de alto nivel de anticuerpo mediante las células productoras de anticuerpos seleccionadas y son sensibles a un medio tal como medio HAT. Entre estas, en algunos modos de realización, las líneas de células de mieloma son líneas de mieloma murino, tales como las derivadas de tumores de ratón MOPC-21 y MPC-11 disponibles del Salk Institute Cell Distribution Center, San Diego, California EE. UU., y células SP-2 o X63-Ag8-653 disponibles de la American Type Culture Collection, Rockville, Maryland, EE. UU. También se han descrito líneas de células de heteromieloma de ratón-humano y mieloma humano para la producción de anticuerpos monoclonales humanos (Kozbor, J. Immunol. 133:3001 (1984); Brodeur et al., Monoclonal Antibody Production Techniques and Applications pág. 51-63 (Marcel Dekker, Inc., Nueva York, 1987)).
El medio de cultivo en el que se cultivan las células de hibridoma se somete a ensayo para determinar la producción de anticuerpos monoclonales dirigidos contra el antígeno. En algunos modos de realización, se determina la especificidad de unión de los anticuerpos monoclonales producidos por células de hibridoma mediante inmunoprecipitación o mediante un ensayo de unión in vitro, tal como radioinmunoanálisis (RIA) o ensayo de inmunoadsorción enzimática (ELISA).
La afinidad de unión del anticuerpo monoclonal se puede determinar, por ejemplo, mediante el análisis de Scatchard de Munson et al., Anal. Biochem. 107:220 (1980).
Después de que se identifiquen células de hibridoma que producen anticuerpos de la especificidad, afinidad y/o actividad deseadas, los clones se pueden subclonar mediante procedimientos de dilución limitante y cultivar mediante procedimientos estándar (Goding, Monoclonal Antibodies: Principles and Practice pág. 59-103 (Academic Press,
1986)). Los medios de cultivo adecuados para este propósito incluyen, por ejemplo, medio D-MEM o RPMI-1640. Además, las células de hibridoma se pueden cultivar in vivo como tumores ascíticos en un animal.
Los anticuerpos monoclonales segregados por los subclones se separan adecuadamente del medio de cultivo, líquido ascítico o suero mediante procedimientos de purificación de inmunoglobulinas convencionales, tales como, por ejemplo, polipéptido A-Sepharose, cromatografía con hidroxilapatita, electroforesis en gel, diálisis o cromatografía de afinidad.
El ADN que codifica los anticuerpos monoclonales se aísla y secuencia fácilmente usando procedimientos convencionales (por ejemplo, usando sondas de oligonucleótidos que se puedan unir específicamente a genes que codifican las cadenas pesada y ligera de anticuerpos murinos). En algunos modos de realización, las células de hibridoma sirven como una fuente de dicho ADN. Una vez aislado, se puede disponer el ADN en vectores de expresión que, a continuación, se transfectan en células huésped, tales como células de E. coli, células COS de simio, células de ovario de hámster chino (CHO) o células de mieloma, que de lo contrario no producen polipéptido inmunoglobulina, para obtener la síntesis de anticuerpos monoclonales en las células huésped recombinantes. Artículos de revisión sobre la expresión recombinante en bacterias de ADN que codifica el anticuerpo incluyen Skerra et al., Curr. Opinión in Immunol. 5:256-262 (1993) y Plückthun, Immunol. Revs., 130:151-188 (1992).
En otro modo de realización, se pueden aislar anticuerpos o fragmentos de anticuerpo a partir de colecciones de fagos de anticuerpos generadas usando las técnicas descritas en McCafferty et al., Nature 348:552-554 (1990). Clackson et al., Nature 352:624-628 (1991) y Marks et al., J. Mol. Biol. 222:581-597 (1991) describen el aislamiento de anticuerpos murinos y humanos, respectivamente, usando colecciones de fagos. Las publicaciones posteriores describen la producción de anticuerpos humanos de alta afinidad (intervalo nM) mediante reordenamiento de cadenas (Marks et al., Bio/Technology 10:779-783 (1992)), así como la infección combinatoria y la recombinación in vivo como una estrategia para construir colecciones de fagos muy grandes (Waterhouse et al., Nuc. Acids. Res. 21:2265-2266 (1993)). Por tanto, estas técnicas son alternativas viables a las técnicas tradicionales de hibridoma de anticuerpos monoclonales para el aislamiento de anticuerpos monoclonales.
También se puede modificar el ADN, por ejemplo, sustituyendo la secuencia codificante para los dominios constantes de la cadena pesada y ligera humana en lugar de las secuencias murinas homólogas (patente de EE. UU. n.° 4.816.567; Morrison et al., Proc. Natl Acad. Sci. USA 81:6851 (1984)) o uniendo de forma covalente a la secuencia codificante de inmunoglobulina toda o parte de la secuencia codificante para un polipéptido distinto a inmunoglobulina.
Típicamente, dichos polipéptidos distintos a inmunoglobulina se sustituyen con los dominios constantes de un anticuerpo, o se sustituyen con los dominios variables de un sitio de combinación a antígeno de un anticuerpo para crear un anticuerpo bivalente quimérico que comprenda un sitio de combinación a antígeno que tenga especificidad por un antígeno y otro sitio de combinación a antígeno que tenga especificidad por un antígeno diferente.
En algunos modos de realización de cualquiera de los procedimientos descritos en el presente documento, el anticuerpo es IgA, IgD, IgE, IgG o IgM. En algunos modos de realización, el anticuerpo es un anticuerpo monoclonal IgG.
(iii) Anticuerpos humanizados
En algunos modos de realización, el anticuerpo es un anticuerpo humanizado. Los procedimientos para humanizar anticuerpos no humanos se han descrito en la técnica. En algunos modos de realización, un anticuerpo humanizado tiene uno o más residuos de aminoácido introducidos en el mismo desde una fuente que es no humana. Estos residuos de aminoácido no humanos se denominan a menudo residuos de "importación", que se toman típicamente de un dominio variable de "importación". La humanización se puede realizar esencialmente siguiendo el procedimiento de Winter y sus colaboradores (Jones et al., Nature 321:522-525 (1986); Riechmann et al., Nature 332:323-327 (1988); Verhoeyen et al., Science 239:1534-1536 (1988)), sustituyendo las secuencias correspondientes de un anticuerpo humano por las secuencias de la región hipervariable. En consecuencia, dichos anticuerpos "humanizados" son anticuerpos quiméricos (patente de EE. UU. n.° 4.816.567) en los que se ha sustituido sustancialmente menos de un dominio variable humano intacto por la secuencia correspondiente de una especie no humana. En la práctica, los anticuerpos humanizados son típicamente anticuerpos humanos en los que algunos residuos de la región hipervariable y posiblemente algunos residuos de FR están sustituidos con residuos de sitios análogos en anticuerpos de roedor.
La elección de los dominios variables humanos, tanto ligeros como pesados, que se van a usar en la preparación de los anticuerpos humanizados es muy importante para reducir la antigenicidad. De acuerdo con el llamado procedimiento de "mejor ajuste", la secuencia del dominio variable de un anticuerpo de roedor se criba frente a toda la colección de secuencias de dominio variable humanas conocidas. A continuación, se acepta la secuencia humana que es la más próxima a la del roedor como la región estructural (FR) humana para el anticuerpo humanizado (Sims et al., J. Immunol. 151:2296 (1993); Chothia et al., J. Mol. Biol. 196:901 (1987)). Otro procedimiento usa una región estructural particular derivada de la secuencia consenso de todos los anticuerpos humanos de un subgrupo particular de regiones variables de la cadena ligera o pesada. Se puede usar la misma región estructural para varios anticuerpos
humanizados diferentes (Cárter et al., Proc. Natl. Acad. Sci. USA 89:4285 (1992); Presta et al., J. Immunol. 151:2623 (1993)).
Es importante además que los anticuerpos se humanicen con retención de alta afinidad por el antígeno y otras propiedades biológicas favorables. Para lograr este objetivo, en algunos modos de realización de los procedimientos, se preparan los anticuerpos humanizados mediante un procedimiento de análisis de las secuencias originales y diversos productos humanizados conceptuales usando modelos tridimensionales de las secuencias originales y humanizadas. Los modelos de inmunoglobulina tridimensionales están disponibles comúnmente y son conocidos por los expertos en la técnica. Están disponibles programas de ordenador que ilustran y presentan estructuras conformacionales tridimensionales probables de secuencias de inmunoglobulina candidatas seleccionadas. La inspección de estas presentaciones permite el análisis del papel probable de los residuos en el funcionamiento de la secuencia de inmunoglobulina candidata, es decir, el análisis de residuos que influyen en la capacidad de la inmunoglobulina candidata de unirse a su antígeno. De esta manera, los residuos de Fr se pueden seleccionar y combinar a partir de las secuencias receptoras y de importación de manera que se logre la característica de anticuerpo deseada, tal como una afinidad aumentada por el(los) antígeno(s) diana. En general, los residuos de la región hipervariable están directa y más sustancialmente implicados en la influencia en la unión a antígeno.
(iv) Anticuerpos humanos
En algunos modos de realización, el anticuerpo es un anticuerpo humano. De forma alternativa a la humanización, se pueden generar anticuerpos humanos. Por ejemplo, ahora es posible producir animales transgénicos (por ejemplo, ratones) que, tras su inmunización, puedan producir un repertorio completo de anticuerpos humanos en ausencia de producción de inmunoglobulina endógena. Por ejemplo, se ha descrito que la eliminación homocigótica del gen de la región de unión de la cadena pesada (JH) de anticuerpo en ratones mutantes quiméricos y de línea germinal da como resultado la inhibición completa de la producción de anticuerpos endógenos. La transferencia de la matriz genética de inmunoglobulina de línea germinal humana a dichos ratones mutantes de línea germinal dará como resultado la producción de anticuerpos humanos tras la exposición a antígeno. Véase, por ejemplo, Jakobovits et al., Proc. Natl. Acad. Sci. USA 90:2551 (1993); Jakobovits et al., Nature 362:255-258 (1993); Bruggermann et al., Year in Immuno.
7:33 (1993) y las patentes de EE. UU. n.° 5.591.669; 5.589.369 y 5.545.807.
De forma alternativa, se puede usar la tecnología de presentación en fagos (McCafferty et al., Nature 348:552-553 (1990)) para producir anticuerpos humanos y fragmentos de anticuerpo in vitro a partir de repertorios de genes de dominio variable (V) de inmunoglobulina de donadores no inmunizados. De acuerdo con esta técnica, los genes de dominio V de anticuerpo se clonan sin cambio de pauta de lectura en un gen de polipéptido de la cápside mayor o bien menor de un bacteriófago filamentoso, tal como M13 o fd, y se presentan como fragmentos de anticuerpo funcional en la superficie de la partícula de fago. Debido a que la partícula filamentosa contiene una copia de ADN monocatenario del genoma del fago, las selecciones basadas en las propiedades funcionales del anticuerpo también dan como resultado la selección del gen que codifica el anticuerpo que presenta esas propiedades. Por tanto, el fago imita algunas de las propiedades del linfocito B. La presentación en fagos se puede realizar en una variedad de formatos; para su revisión véase, por ejemplo, Johnson, Kevin S. y Chiswell, David J., Current Opinion in Structural Biology 3:564-571 (1993). Se pueden usar varias fuentes de segmentos de genes V para la presentación en fagos. Clackson et al., Nature 352:624-628 (1991) aislaron una diversidad de anticuerpos antioxazolona de una pequeña colección combinatoria aleatoria de genes V derivados de los bazos de ratones inmunizados. Se puede construir un repertorio de genes V de donadores humanos no inmunizados y se pueden aislar anticuerpos con respecto a una matriz diversa de antígenos (incluyendo autoantígenos) siguiendo esencialmente las técnicas descritas por Marks et al., J. Mol. Biol.
222:581-597 (1991) o Griffith et al., EMBO J. 12:725-734 (1993). Véanse también, las patentes de EE. UU. n.° 5.565.332 y 5.573.905.
Los anticuerpos humanos también se pueden generar por linfocitos B activados in vitro (véanse las patentes de EE. UU.
5.567.610 y 5.229.275).
(v) Fragmentos de anticuerpo
En algunos modos de realización, el anticuerpo es un fragmento de anticuerpo. Se han desarrollado diversas técnicas para la producción de fragmentos de anticuerpo. Tradicionalmente, estos fragmentos se derivaban por medio de digestión proteolítica de anticuerpos intactos (véase, por ejemplo, Morimoto et al., Journal of Biochemical and Biophysical Methods 24:107-117 (1992) y Brennan et al., Science 229:81 (1985)). Sin embargo, estos fragmentos se pueden producir ahora directamente mediante células huésped recombinantes. Por ejemplo, se pueden aislar los fragmentos de anticuerpo de las colecciones de fagos de anticuerpos analizadas anteriormente. De forma alternativa, se pueden recuperar directamente los fragmentos Fab'-SH de E. coli y acoplar químicamente para formar fragmentos F(ab')2 (Carter et al., Bio/Technology 10:163-167 (1992)). De acuerdo con otro enfoque, se pueden aislar directamente fragmentos F(ab')2 del cultivo de células huésped recombinantes. Otras técnicas para la producción de fragmentos de anticuerpo serán evidentes para el experto en la técnica. En otros modos de realización, el anticuerpo de elección es un fragmento Fv monocatenario (scFv). Véanse el documento WO 93/16185; la patente de EE. UU. n.° 5.571.894 y la patente de EE. UU. n.° 5.587.458. El fragmento de anticuerpo también puede ser un "anticuerpo lineal", por ejemplo,
como se describe en la patente de EE. UU. 5.641.870, por ejemplo. Dichos fragmentos de anticuerpo lineal pueden ser monoespecíficos o biespecíficos.
En algunos modos de realización, se proporcionan fragmentos de los anticuerpos descritos en el presente documento. En algunos modos de realización, el fragmento de anticuerpo es un fragmento de unión a antígeno. En algunos modos de realización, el fragmento de unión a antígeno se selecciona del grupo que consiste en un fragmento Fab, un fragmento Fab', un fragmento F(ab')2, un scFv, un Fv y un diacuerpo.
(vi) Anticuerpos biespecíficos
En algunos modos de realización, el anticuerpo es un anticuerpo biespecífico. Los anticuerpos biespecíficos son anticuerpos que tienen especificidades de unión por al menos dos epítopos diferentes. Los anticuerpos biespecíficos ejemplares se pueden unir a dos epítopos diferentes. De forma alternativa, se puede combinar un brazo de unión de anticuerpo biespecífico con un brazo que se una a una molécula desencadenante en un leucocito, tal como una molécula receptora de linfocitos T (por ejemplo, CD2 o CD3) o receptores de Fc para IgG (FcyR), tales como FcyRI (CD64), FcyRII (CD32) y FcyRIII (CD16) para centrar los mecanismos de defensa celular en la célula. Se pueden preparar anticuerpos biespecíficos como anticuerpos de longitud completa o fragmentos de anticuerpo (por ejemplo, anticuerpos biespecíficos F(ab')2).
En la técnica son conocidos procedimientos para preparar anticuerpos biespecíficos. La producción tradicional de anticuerpos biespecíficos de longitud completa se basa en la coexpresión de dos pares de cadena pesada-cadena ligera de inmunoglobulina, donde las dos cadenas tienen especificidades diferentes (Millstein et al., Nature 305:537-539 (1983)). Debido a la variedad aleatoria de las cadenas pesada y ligera de inmunoglobulina, estos hibridomas (cuadromas) producen una mezcla potencial de 10 moléculas de anticuerpo diferentes, de las que solo una tiene la estructura biespecífica correcta. La purificación de la molécula correcta, que se realiza normalmente mediante etapas de cromatografía de afinidad, es bastante engorrosa y los rendimientos de producto son bajos. Se divulgan procedimientos similares en el documento WO 93/08829, y en Traunecker et al., EMBO J., 10:3655-3659 (1991).
De acuerdo con un enfoque diferente, los dominios variables de anticuerpo con las especificidades de unión deseadas (sitios de combinación de anticuerpo-antígeno) se fusionan a secuencias de dominio constante de inmunoglobulina. En algunos modos de realización, la fusión es con un dominio constante de la cadena pesada de inmunoglobulina, que comprende al menos parte de las regiones de bisagra, CH2 y CH3. En algunos modos de realización, la primera región constante de la cadena pesada (CH1), que contiene el sitio necesario para la unión a la cadena ligera, está presente en al menos una de las fusiones. Los ADN que codifican las fusiones con la cadena pesada de inmunoglobulina y, si se desea, la cadena ligera de inmunoglobulina, se insertan en vectores de expresión separados y se cotransfectan en un organismo huésped adecuado. Esto proporciona una gran flexibilidad al ajustar las proporciones mutuas de los tres fragmentos de polipéptido en modos de realización en los que proporciones desiguales de las tres cadenas de polipéptido usadas en la construcción proporcionan los rendimientos óptimos. Sin embargo, es posible insertar las secuencias codificantes para dos o las tres cadenas de polipéptido en un vector de expresión cuando la expresión de al menos dos cadenas de polipéptido en proporciones iguales da como resultado altos rendimientos o cuando las proporciones no son de particular importancia.
En algunos modos de realización de este enfoque, los anticuerpos biespecíficos se componen de una cadena pesada de inmunoglobulina híbrida con una primera especificidad de unión en un brazo, y un par de cadena pesada-cadena ligera de inmunoglobulina híbrida (proporcionando una segunda especificidad de unión) en el otro brazo. Se descubrió que esta estructura asimétrica facilita la separación del compuesto biespecífico deseado de combinaciones indeseadas de cadenas de inmunoglobulina, ya que la presencia de una cadena ligera de inmunoglobulina en solo una mitad de la molécula biespecífica proporciona una manera fácil de separación. Este enfoque se divulga en el documento WO 94/04690. Para obtener más detalles de la generación de anticuerpos biespecíficos véase, por ejemplo, Suresh et al., Methods in Enzymology 121:210 (1986).
De acuerdo con otro enfoque descrito en la patente de EE. UU. n.° 5.731.168, se puede genomanipular la superficie de contacto entre un par de moléculas de anticuerpo para maximizar el porcentaje de heterodímeros que se recuperan del cultivo de células recombinantes. En algunos modos de realización, la superficie de contacto comprende al menos una parte del dominio Ch3 de un dominio constante de anticuerpo. En este procedimiento, una o más cadenas laterales de aminoácidos pequeños de la superficie de contacto de la primera molécula de anticuerpo se reemplazan por cadenas laterales más grandes (por ejemplo, tirosina o triptófano). Se crean "cavidades" compensatorias de tamaño idéntico o similar a la(s) cadena(s) lateral(es) grande(s) en la superficie de contacto de la segunda molécula de anticuerpo reemplazando las cadenas laterales de aminoácidos grandes por otras más pequeñas (por ejemplo, alanina o treonina). Esto proporciona un mecanismo para incrementar el rendimiento del heterodímero frente a otros productos finales indeseados, tales como homodímeros.
Los anticuerpos biespecíficos incluyen anticuerpos reticulados o "heteroconjugados". Por ejemplo, se puede acoplar uno de los anticuerpos en el heteroconjugado a avidina, el otro a biotina. Por ejemplo, se han propuesto dichos anticuerpos para dirigir células del sistema inmunitario a células indeseadas (patente de EE. UU. n.° 4.676.980) y para el tratamiento de la infección por el VIH (documentos WO 91/00360, WO 92/200373 y EP 03089). Se pueden preparar
anticuerpos heteroconjugados usando cualquier procedimiento de reticulación conveniente. Los agentes de reticulación adecuados son bien conocidos en la técnica y se divulgan en la patente de EE. UU. n.° 4.676.980, junto con varias técnicas de reticulación.
En la literatura también se han descrito técnicas para generar anticuerpos biespecíficos a partir de fragmentos de anticuerpo. Por ejemplo, se pueden preparar anticuerpos biespecíficos usando enlace químico. Brennan et al., Science 229: 81 (1985) describen un procedimiento en el que los anticuerpos intactos se escinden proteolíticamente para generar fragmentos F(ab')2. Estos fragmentos se reducen en presencia del agente arsenito de sodio que forma complejos con ditiol para estabilizar los ditioles vecinos y prevenir la formación de disulfuro intermolecular. A continuación, los fragmentos Fab' generados se convierten en derivados de tionitrobenzoato (TNB). A continuación, uno de los derivados Fab'-TNB se reconvierte en Fab'-tiol mediante reducción con mercaptoetilamina y se mezcla con una cantidad equimolar del otro derivado Fab'-TNB para formar el anticuerpo biespecífico. Los anticuerpos biespecíficos producidos se pueden usar como agentes para la inmovilización selectiva de enzimas.
También se han descrito diversas técnicas para preparar y aislar directamente fragmentos de anticuerpo biespecífico del cultivo de células recombinantes. Por ejemplo, se han producido anticuerpos biespecíficos usando cremalleras de leucina. Kostelny et al., J. Immunol. 148(5):1547-1553 (1992). Los péptidos de cremalleras de leucina de las proteínas Fos y Jun se enlazaron a las porciones Fab' de dos anticuerpos diferentes mediante fusión génica. Los homodímeros de anticuerpo se redujeron en la región de bisagra para formar monómeros y, a continuación, se reoxidaron para formar los heterodímeros de anticuerpo. Este procedimiento también se puede utilizar para la producción de homodímeros de anticuerpo. La tecnología de "diacuerpos" descrita por Hollinger et al., Proc. Natl. Acad. Sci. USA 90:6444-6448 (1993) ha proporcionado un mecanismo alternativo para preparar fragmentos de anticuerpo biespecífico. Los fragmentos comprenden un dominio variable de la cadena pesada (VH) conectado a un dominio variable de la cadena ligera (VL) mediante un conector que es demasiado corto para permitir el emparejamiento entre los dos dominios en la misma cadena. En consecuencia, se obliga a que los dominios VH y VL de un fragmento se emparejen con los dominios VL y VH complementarios de otro fragmento, formando de este modo dos sitios de unión a antígeno. También se ha informado de otra estrategia para preparar fragmentos de anticuerpo biespecífico mediante el uso de dímeros de Fv monocatenario (sFv). Véase Gruber et al., J. Immunol. 152:5368 (1994).
Se contemplan anticuerpos con más de dos valencias. Por ejemplo, se pueden preparar anticuerpos triespecíficos. Tutt et al., J. Immunol. 147: 60 (1991).
(vii) Anticuerpos multivalentes
En algunos modos de realización, los anticuerpos son anticuerpos multivalentes. Un anticuerpo multivalente se puede internalizar (y/o catabolizar) más deprisa que un anticuerpo bivalente por una célula que exprese un antígeno al que se unan los anticuerpos. Los anticuerpos proporcionados en el presente documento pueden ser anticuerpos multivalentes (que son distintos de la clase IgM) con tres o más sitios de unión a antígeno (por ejemplo, anticuerpos tetravalentes), que se pueden producir fácilmente mediante expresión recombinante del ácido nucleico que codifica las cadenas de polipéptido del anticuerpo. El anticuerpo multivalente puede comprender un dominio de dimerización y tres o más sitios de unión a antígeno. El dominio de dimerización preferente comprende (o consiste en) una región Fc o una región de bisagra. En esta situación, el anticuerpo comprende una región Fc y tres o más sitios de unión a antígeno en el extremo amínico con respecto a la región Fc. El anticuerpo multivalente preferente en el presente documento comprende (o consiste en) de tres hasta aproximadamente ocho, pero preferentemente cuatro, sitios de unión a antígeno. El anticuerpo multivalente comprende al menos una cadena de polipéptido (y preferentemente dos cadenas de polipéptido), en el que la(s) cadena(s) de polipéptido comprende(n) dos o más dominios variables. Por ejemplo, la(s) cadena(s) de polipéptido puede(n) comprender VD1-(X1)n-VD2-(X2) n-Fc, en el que VD1 es un primer dominio variable, VD2 es un segundo dominio variable, Fc es una cadena de polipéptido de una región Fc, X1 y X2 representan un aminoácido o polipéptido, y n es 0 o 1. Por ejemplo, la(s) cadena(s) de polipéptido puede(n) comprender: VH-CH1-conector flexible-VH-CH1-cadena de región Fc; o VH-CH1-VH-CH1-cadena de región Fc. El anticuerpo multivalente en el presente documento preferentemente comprende además al menos dos (y preferentemente cuatro) polipéptidos de dominio variable de la cadena ligera. El anticuerpo multivalente en el presente documento puede comprender, por ejemplo, de aproximadamente dos hasta aproximadamente ocho polipéptidos de dominio variable de la cadena ligera. Los polipéptidos de dominio variable de la cadena ligera contemplados aquí comprenden un dominio variable de la cadena ligera y, opcionalmente, comprenden además un dominio Cl .
En algunos modos de realización, el anticuerpo es un anticuerpo multiespecífico. Los ejemplos de anticuerpos multiespecíficos incluyen, pero no se limitan a, un anticuerpo que comprende un dominio variable de la cadena pesada (VH) y un dominio variable de la cadena ligera (VL), donde la unidad VHVL tiene especificidad poliepitópica, anticuerpos que tienen dos o más dominios VL y VH, uniéndose cada unidad VHVL a un epítopo diferente, anticuerpos que tienen dos o más dominios variables individuales, uniéndose cada dominio variable individual a un epítopo diferente, anticuerpos de longitud completa, fragmentos de anticuerpo tales como Fab, Fv, dsFv, scFv, diacuerpos, diacuerpos biespecíficos, triacuerpos, anticuerpos trifuncionales, fragmentos de anticuerpo que se han enlazado de forma covalente o no covalente. En algunos modos de realización, ese anticuerpo tiene especificidad poliepitópica; por ejemplo, la capacidad de unirse específicamente a dos o más epítopos diferentes en la(s) misma(s) o diferente(s) diana(s). En algunos modos de realización, los anticuerpos son monoespecíficos; por ejemplo, un anticuerpo que solo
se une a un epítopo. De acuerdo con un modo de realización, el anticuerpo multiespecífico es un anticuerpo IgG que se une a cada epítopo con una afinidad de 5 pM a 0,001 pM, de 3 pM a 0,001 pM, de 1 pM a 0,001 pM, de 0,5 pM a 0,001 pM o de 0,1 pM a 0,001 pM.
(viii) Otras modificaciones de los anticuerpos
Puede ser deseable modificar el anticuerpo proporcionado en el presente documento con respecto a la función efectora, por ejemplo, para potenciar la citotoxicidad celular dependiente de antígeno (ADCC) y/o la citotoxicidad dependiente del complemento (CDC) del anticuerpo. Esto se puede lograr introduciendo una o más sustituciones de aminoácido en una región Fc del anticuerpo. De forma alternativa o adicional, se puede(n) introducir (un) residuo(s) de cisteína en la región Fc, permitiendo de este modo la formación de enlaces disulfuro intercatenarios en esta región. El anticuerpo homodimérico generado así puede tener una capacidad de internalización mejorada y/o una destrucción celular mediada por el complemento y citotoxicidad celular dependiente de anticuerpos (ADCC) incrementadas. Véase Caron et al., J. Exp Med. 176:1191-1195 (1992) y Shopes, B. J., Immunol. 148:2918-2922 (1992). También se pueden preparar anticuerpos homodiméricos con actividad antitumoral mejorada utilizando agentes de reticulación heterobifuncionales como se describe en Wolff et al., Cancer Research 53:2560-2565 (1993). De forma alternativa, se puede genomanipular un anticuerpo que tenga regiones Fc dobles y, de este modo, pueda tener una lisis mediada por el complemento potenciada y capacidades de ADCC. Véase Stevenson et al., Anti-Cancer Drug Design 3:219-230 (1989).
Para incrementar la semivida en suero del anticuerpo, se pueden realizar alteraciones de aminoácido en el anticuerpo como se describe en el documento US 2006/0067930.
(B) Variantes y modificaciones de los polipéptidos
Se puede(n) usar la(s) modificación/modificaciones de los polipéptidos, incluyendo anticuerpos, descritos en el presente documento en los procedimientos de purificación de polipéptidos (por ejemplo, anticuerpos) descritos en el presente documento.
(i) Polipéptidos variantes
"Variante de polipéptido" significa un polipéptido, preferentemente un polipéptido activo, como se define en el presente documento, que tiene al menos aproximadamente un 80 % de identidad de secuencia de aminoácidos con una secuencia natural de longitud completa del polipéptido, una secuencia de polipéptido que carece del péptido señal, un dominio extracelular de un polipéptido, con o sin el péptido señal. Dichas variantes de polipéptido incluyen, por ejemplo, polipéptidos en los que se añaden o eliminan uno o más residuos de aminoácido en el extremo N o C de la secuencia de aminoácidos natural de longitud completa. Habitualmente, una variante de polipéptido TAT tendrá al menos aproximadamente un 80 % de identidad de secuencia de aminoácidos, de forma alternativa, al menos aproximadamente cualquiera de un 85 %, 90 %, 95 %, 96 %, 97 %, 98 % o 99 % de identidad de secuencia de aminoácidos con respecto a una secuencia de polipéptido de secuencia natural de longitud completa, una secuencia de polipéptido que carece del péptido señal, un dominio extracelular de un polipéptido, con o sin el péptido señal. Opcionalmente, los polipéptidos variantes no tendrán más de una sustitución aminoacídica conservadora en comparación con la secuencia de polipéptido natural, de forma alternativa, no más de aproximadamente cualquiera de 2, 3, 4, 5, 6, 7, 8, 9 o 10 sustituciones de aminoácido conservadoras en comparación con la secuencia de polipéptido natural.
El polipéptido variante se puede truncar en el extremo N o en el extremo C, o puede carecer de residuos internos, por ejemplo, cuando se compara con un polipéptido natural de longitud completa. Determinados polipéptidos variantes pueden carecer de residuos de aminoácido que no sean esenciales para una actividad biológica deseada. Estos polipéptidos variantes con truncamientos, eliminaciones e inserciones se pueden preparar mediante cualquiera de varias técnicas convencionales. Los polipéptidos variantes deseados se pueden sintetizar químicamente. Otra técnica adecuada implica aislar y amplificar un fragmento de ácido nucleico que codifique un polipéptido variante deseado mediante reacción en cadena de la polimerasa (PCR). Se emplean oligonucleótidos que definen los extremos deseados del fragmento de ácido nucleico en los cebadores 5' y 3' en la PCR. Preferentemente, los polipéptidos variantes comparten al menos una actividad biológica y/o inmunológica con el polipéptido natural divulgado en el presente documento.
Las inserciones en la secuencia de aminoácidos incluyen fusiones en el extremo amínico y/o carboxílico que varían en longitud desde un residuo a polipéptidos que contienen cien o más residuos, así como inserciones intrasecuenciales de residuos de aminoácido únicos o múltiples. Los ejemplos de inserciones en los extremos incluyen un anticuerpo con un residuo de metionilo en el extremo N o el anticuerpo fusionado a un polipéptido citotóxico. Otras variantes de inserción de la molécula de anticuerpo incluyen la fusión en el extremo N o C del anticuerpo a una enzima o un polipéptido que incrementa la semivida en suero del anticuerpo.
Por ejemplo, puede ser deseable mejorar la afinidad de unión y/u otras propiedades biológicas del polipéptido. Las variantes de secuencia de aminoácidos del polipéptido se preparan introduciendo cambios de nucleótidos apropiados
en el ácido nucleico del anticuerpo o mediante síntesis de péptidos. Dichas modificaciones incluyen, por ejemplo, eliminaciones de y/o inserciones en y/o sustituciones de residuos en las secuencias de aminoácidos del polipéptido. Se realiza cualquier combinación de eliminación, inserción y sustitución para llegar a la construcción final, siempre que la construcción final posea las características deseadas. Los cambios de aminoácido también pueden alterar los procesos postraduccionales del polipéptido (por ejemplo, anticuerpo), tales como el cambio del número o posición de los sitios de glucosilación.
Se pueden encontrar directrices para determinar el residuo de aminoácido que se puede insertar, sustituir o eliminar sin afectar negativamente a la actividad deseada comparando la secuencia del polipéptido con la de moléculas de polipéptido conocidas homólogas y minimizando el número de cambios en la secuencia de aminoácidos realizados en regiones de homología alta.
Un procedimiento útil para la identificación de determinados residuos o regiones del polipéptido (por ejemplo, anticuerpo) que sean localizaciones preferentes para la mutagénesis se llama "mutagénesis por barrido de alanina" como se describe por Cunningham y Wells, Science 244:1081-1085 (1989). Aquí, un residuo o grupo de residuos diana (por ejemplo, residuos cargados, tales como Arg, Asp, His, Lys y Glu) se identifica y reemplaza por un aminoácido neutro o cargado negativamente (lo más preferentemente alanina o polialanina) para que afecten a la interacción de los aminoácidos con el antígeno. Esas localizaciones de aminoácidos que demuestran sensibilidad funcional a las sustituciones se refinan, a continuación, introduciendo variantes adicionales u otras en, o para, los sitios de sustitución. Por tanto, aunque el sitio para introducir una variación de secuencia de aminoácidos está predeterminado, no es necesario que la naturaleza de la mutación per se esté predeterminada. Por ejemplo, para analizar el funcionamiento de una mutación en un sitio dado, se realiza una mutagénesis aleatoria o por barrido de Ala en el codón o región diana y se criban las variantes de anticuerpo expresadas para determinar la actividad deseada.
Otro tipo de variante es una variante de sustitución amioacídica. Estas variantes tienen al menos un residuo de aminoácido en la molécula de anticuerpo reemplazado por un residuo diferente. Los sitios de mayor interés para la mutagénesis por sustitución incluyen las regiones hipervariables, pero también se contemplan alteraciones en Fr . Las sustituciones conservadoras se muestran en la Tabla 1 bajo el encabezamiento de "sustituciones preferentes". Si tales sustituciones dar como resultado un cambio en la actividad biológica, entonces se pueden introducir cambios más sustanciales, denominados "sustituciones ejemplares" en la Tabla 1, o como se describe adicionalmente más adelante con referencia a las clases de aminoácidos, y se examinan los productos.
Tabla 1.

Se consiguen modificaciones sustanciales en las propiedades biológicas del polipéptido seleccionando sustituciones que difieran significativamente en su efecto sobre el mantenimiento de (a) la estructura de la cadena principal del polipéptido en el área de sustitución, por ejemplo, como una conformación de lámina o hélice, (b) la carga o hidrofobia de la molécula en el sitio diana o (c) el volumen de la cadena lateral. Los aminoácidos se pueden agrupar de acuerdo con las similitudes en las propiedades de sus cadenas laterales (en A. L. Lehninger, Biochemistry segunda ed., pág.
73-75, Worth Publishers, Nueva York (1975)):
(1) no polares: Ala (A), Val (V), Leu (L), Ile (I), Pro (P), Phe (F), Trp (W), Met (M)
(2) polares no cargados: Gly (G), Ser (S), Thr (T), Cys (C), Tyr (Y), Asn (N), Gln (Q)
(3) ácidos: Asp (D), Glu (E)
(4) básicos: Lys (K), Arg (R), His (H)
De forma alternativa, los residuos naturales se pueden dividir en grupos basados en propiedades de cadena lateral comunes:
(1) hidrófobos: norleucina, Met, Ala, Val, Leu, Ile;
(2) hidrófilos neutros: Cys, Ser, Thr, Asn, Gln;
(3) ácidos: Asp, Glu;
(4) básicos: His, Lys, Arg;
(5) residuos que influyen en la orientación de la cadena: Gly, Pro;
(6) aromáticos: Trp, Tyr, Phe.
Las sustituciones no conservadoras supondrán intercambiar un miembro de una de estas clases por otra clase. Cualquier residuo de cisteína no implicado en el mantenimiento de la conformación apropiada del anticuerpo también se puede sustituir, en general, con serina, para mejorar la estabilidad oxidativa de la molécula y prevenir la reticulación anómala. Por el contrario, se puede(n) añadir (un) enlace(s) de cisteína al polipéptido para mejorar su estabilidad (en particular, si el anticuerpo es un fragmento de anticuerpo tal como un fragmento Fv).
Un tipo de variante de sustitución preferente en particular implica sustituir uno o más residuos de la región hipervariable de un anticuerpo original (por ejemplo, un anticuerpo humanizado). En general, la(s) variante(s) resultante(s) seleccionada(s) para su desarrollo adicional tendrá(n) propiedades biológicas mejoradas con respecto al anticuerpo original a partir del que se generan. Una manera conveniente de generar dichas variantes de sustitución implica la maduración de la afinidad usando presentación en fagos. En resumen, se mutan varios sitios de la región hipervariable (por ejemplo, 6-7 sitios) para generar todas las posibles sustituciones de aminoácido en cada sitio. Las variantes de anticuerpo generadas así se presentan de una forma monovalente a partir de partículas de fagos filamentosos como fusiones con el producto del gen III de M13 empaquetadas en cada partícula. A continuación, se criban las variantes de presentación en fagos para determinar su actividad biológica (por ejemplo, afinidad de unión) como se divulga en el presente documento. Para identificar los sitios de la región hipervariable candidatos para la modificación, se puede realizar mutagénesis por barrido de alanina para identificar los residuos de la región hipervariable que contribuyan significativamente a la unión a antígeno. De forma alternativa o adicional, puede ser beneficioso analizar una estructura cristalina del complejo antígeno-anticuerpo para identificar los puntos de contacto entre el anticuerpo y la diana. Dichos residuos de contacto y residuos cercanos son candidatos para la sustitución de acuerdo con las técnicas detalladas en el presente documento. Una vez que se generan dichas variantes, el panel de variantes se somete a cribado como se describe en el presente documento y se pueden seleccionar los anticuerpos con propiedades superiores en uno o más ensayos pertinentes para su desarrollo adicional.
Otro tipo de variante de aminoácidos del polipéptido altera el patrón de glucosilación original del anticuerpo. El polipéptido puede comprender restos distintos a aminoácidos. Por ejemplo, se puede glucosilar el polipéptido. Dicha glucosilación se puede producir de forma natural durante la expresión del polipéptido en la célula huésped u organismo huésped, o puede ser una modificación intencionada que surja de la intervención humana. Por alteración se entiende eliminar uno o más restos glucídicos encontrados en el polipéptido y/o añadir uno o más sitios de glucosilación que no están presentes en el polipéptido.
La glucosilación del polipéptido es típicamente con enlace N o bien con enlace O. Con enlace N se refiere al acoplamiento del resto glucídico a la cadena lateral de un residuo de asparagina. Las secuencias de tripéptido asparagina-X-serina y asparagina-X-treonina, donde X es cualquier aminoácido excepto prolina, son las secuencias de reconocimiento para el acoplamiento enzimático del resto glucídico a la cadena lateral de asparagina. Por tanto, la presencia de cualquiera de estas secuencias de tripéptido en un polipéptido crea un sitio de glucosilación potencial. La glucosilación con enlace O se refiere al acoplamiento de uno de los azúcares N-acetilgalactosamina, galactosa o xilosa a un hidroxiaminoácido, lo más comúnmente serina o treonina, aunque también se puede usar 5-hidroxiprolina o 5-hidroxilisina.
La adición de sitios de glucosilación al polipéptido se consigue convenientemente alterando la secuencia de aminoácidos de modo que contenga una o más de las secuencias de tripéptido descritas anteriormente (para sitios de glucosilación con enlace N). La alteración también se puede realizar mediante la adición de, o sustitución con, uno o más residuos de serina o treonina con respecto a la secuencia del anticuerpo original (para sitios de glucosilación con enlace O).
Se puede conseguir la retirada de los restos glucídicos presentes en el polipéptido química o enzimáticamente o mediante sustitución por mutación de codones que codifican residuos de aminoácido que sirven como dianas para la glucosilación. Se puede lograr la escisión enzimática de restos glucídicos en polipéptidos mediante el uso de una variedad de endo y exoglucosidasas.
Otras modificaciones incluyen la desamidación de residuos de glutaminilo y asparaginilo en los residuos de glutamilo y aspartilo correspondientes, respectivamente, la hidroxilación de prolina y lisina, la fosforilación de grupos hidroxilo de residuos de serilo o treonilo, la mutilación de los grupos a-amino de las cadenas laterales de lisina, arginina e histidina, la acetilación de la amina del extremo N y la amidación de cualquier grupo carboxilo del extremo C.
(ii) Polipéptidos quiméricos
El polipéptido descrito en el presente documento se puede modificar de manera que forme moléculas quiméricas que comprendan el polipéptido fusionado a otro polipéptido heterólogo o secuencia de aminoácidos. En algunos modos de realización, una molécula quimérica comprende una fusión del polipéptido con un polipéptido de marca que proporciona un epítopo al que se puede unir selectivamente un anticuerpo antimarca. La marca de epítopo se dispone, en general, en el extremo amínico o carboxílico del polipéptido. La presencia de dichas formas con marca de epítopo del polipéptido se puede detectar usando un anticuerpo frente al polipéptido de marca. Además, la provisión de la marca de epítopo posibilita que el polipéptido se purifique fácilmente mediante purificación por afinidad usando un anticuerpo antimarca u otro tipo de matriz de afinidad que se una a la marca de epítopo.
En un modo de realización alternativo, la molécula quimérica puede comprender una fusión del polipéptido con una inmunoglobulina o una región particular de una inmunoglobulina. Una forma bivalente de la molécula quimérica se denomina "inmunoadhesina".
Como se usa en el presente documento, el término "inmunoadhesina" indica moléculas similares a anticuerpo que combinan la especificidad de unión de un polipéptido heterólogo con las funciones efectoras de dominios constantes de inmunoglobulina. Estructuralmente, las inmunoadhesinas comprenden una fusión de una secuencia de aminoácidos con la especificidad de unión deseada que es distinta del sitio de reconocimiento y unión a antígeno de un anticuerpo (es decir, es "heteróloga"), y una secuencia de dominio constante de inmunoglobulina. La parte de adhesina de una molécula de inmunoadhesina es típicamente una secuencia de aminoácidos contiguos que comprende al menos el sitio de unión de un receptor o un ligando. Se puede obtener la secuencia del dominio constante de inmunoglobulina en la inmunoadhesina a partir de cualquier inmunoglobulina, tal como los subtipos IgG-1, IgG-2, IgG-3 o IgG-4, IgA (incluyendo IgA-1 e IgA-2), IgE, IgD o IgM.
Las fusiones con Ig incluyen preferentemente la sustitución de una forma soluble (dominio transmembranario eliminado o inactivado) de un polipéptido en lugar de al menos una región variable en una molécula de Ig. En un modo de realización preferente en particular, la fusión con inmunoglobulina incluye las regiones de bisagra, CH2 y CH3, o de bisagra, CH1, CH2 y CH3 de una molécula de IgG1.
(iii) Conjugados de polipéptido
El polipéptido para su uso en formulaciones de polipéptido se puede conjugar con un agente citotóxico, tal como un agente quimioterápico, un agente inhibidor del crecimiento, una toxina (por ejemplo, una toxina activa enzimáticamente de origen bacteriano, fúngico, vegetal o animal, o fragmentos de la misma) o un isótopo radioactivo (es decir, un radioconjugado).
Se pueden usar agentes quimioterápicos útiles en la generación de dichos conjugados. Además, las toxinas activas enzimáticamente y fragmentos de las mismas que se pueden usar incluyen cadena A de difteria, fragmentos activos que no se unen de la toxina diftérica, cadena A de exotoxina (de Pseudomonas aeruginosa), cadena A de ricina, cadena A de abrina, cadena A de modecina, alfa-sarcina, proteínas de Aleurites fordii, proteínas de diantina, proteínas de Phytolaca americana (PAPI, PAPII y PAP-S), inhibidor de Momordica charantia, curcina, crotina, inhibidor de Saponaria officinalis, gelonina, mitogelina, restrictocina, fenomicina, enomicina y los tricotecenos. Una variedad de radionúclidos está disponible para la producción de polipéptidos radioconjugados. Los ejemplos incluyen 212Bi, 131I, 131In, 90Y y 186Re. Se preparan conjugados del polipéptido y del agente citotóxico usando una variedad de agentes bifuncionales de acoplamiento de proteínas tales como N-succinimidil-3-(2-piridilditiol)propionato (SPDP), iminotiolano (IT), derivados bifuncionales de imidoésteres (tales como clorhidrato de adipimidato de dimetilo), ésteres activos (tales como suberato de disuccinimidilo), aldehídos (tales como glutaraldehído), compuestos bis-azido (tales como bis-(pazidobenzoil)hexanodiamina), derivados de bis-diazonio (tales como bis-(p-diazoniobenzoil)etilendiamina), diisocianatos (tales como 2,6-diisocianato de tolueno) y compuestos de flúor bis-activos (tales como 1,5-difluoro-2,4-dinitrobenceno). Por ejemplo, se puede preparar una inmunotoxina de ricina como se describe en Vitetta et al., Science 238:1098 (1987). El ácido 1-isotiocianatobencil-3-metildietilentriaminopentaacético (MX-DTPA) marcado con carbono 14 es un agente quelante ejemplar para la conjugación de radionucleótidos al polipéptido.
Los conjugados de un polipéptido y una o más toxinas de molécula pequeña, tales como calicheamicina, maitansinoides, un tricoteno y CC1065, y los derivados de estas toxinas que tienen actividad de toxina, también se contemplan en el presente documento.
Los maitansinoides son inhibidores mitóticos que actúan inhibiendo la polimerización de tubulina. La maitansina se aisló por primera vez a partir del arbusto africano Maytenus serrata. Posteriormente, se descubrió que determinados microbios también producen maitansinoides, tales como maitansinol y ésteres C-3 de maitansinol. También se contemplan maitansinol sintético y derivados y análogos del mismo. Existen muchos grupos de enlace conocidos en la técnica para preparar conjugados de polipéptido-maitansinoide, incluyendo, por ejemplo, los divulgados en la pat. de EE. UU. n.° 5.208.020. Los grupos de enlace incluyen grupos disulfuro, grupos tioéter, grupos lábiles en ácido, grupos fotolábiles, grupos lábiles en peptidasas o grupos lábiles en esterasas, como se divulgan en las patentes identificadas anteriormente, siendo preferentes los grupos disulfuro y tioéter.
El conector se puede acoplar a la molécula de maitansinoide en diversas posiciones, dependiendo del tipo de enlace. Por ejemplo, se puede formar un enlace éster mediante reacción con un grupo hidroxilo usando técnicas de acoplamiento convencionales. La reacción se puede producir en la posición C-3, que tiene un grupo hidroxilo, en la posición C-14, modificada con hidroximetilo, en la posición C-15, modificada con un grupo hidroxilo, y en la posición C-20, que tiene un grupo hidroxilo. En un modo de realización preferente, el enlace se forma en la posición C-3 de maitansinol o un análogo de maitansinol.
Otro conjugado de interés comprende un polipéptido conjugado con una o más moléculas de calicheamicina. Los antibióticos de la familia de calicheamicina pueden producir roturas en el ADN bicatenario en concentraciones subpicomolares. Para la preparación de conjugados de la familia de calicheamicina, véase, por ejemplo, la pat. de EE. UU. n.° 5.712.374. Los análogos estructurales de calicheamicina que se pueden usar incluyen, pero no se limitan a, Y1I, a2I, a3I, N-acetil-Y1I, PSAG y 01I. Otro fármaco antitumoral con el que se puede conjugar el anticuerpo es QFA, que es un antifolato. Tanto calicheamicina como QFA tienen sitios de acción intracelulares y no atraviesan fácilmente
la membrana plasmática. Por lo tanto, la captación celular de estos agentes a través de la internalización mediada por el polipéptido (por ejemplo, anticuerpo) potencia en gran medida sus efectos citotóxicos.
Otros agentes antitumorales con los que se pueden conjugar los polipéptidos descritos en el presente documento incluyen BCNU, estreptozoicina, vincristina y 5-fluorouracilo, la familia de agentes conocidos colectivamente como complejo LL-E33288, así como esperamicinas.
En algunos modos de realización, el polipéptido puede ser un conjugado entre un polipéptido y un compuesto con actividad nucleolítica (por ejemplo, una ribonucleasa o una ADN endonucleasa, tal como una desoxirribonucleasa, DNasa).
Aún en otro modo de realización, el polipéptido (por ejemplo, anticuerpo) se puede conjugar con un "receptor" (tal como estreptavidina) para utilizarse en la preselección como diana de un tumor, en el que el conjugado de polipéptidoreceptor se administra al paciente, seguido de la retirada del conjugado no unido de la circulación usando un agente de aclaramiento y, a continuación, la administración de un "ligando" (por ejemplo, avidina) que se conjuga con un agente citotóxico (por ejemplo, un radionucleótido).
En algunos modos de realización, el polipéptido se puede conjugar con una enzima activadora de profármacos que convierte un profármaco (por ejemplo, un agente quimioterápico peptidílico) en un fármaco antineoplásico activo. El componente enzimático del inmunoconjugado incluye cualquier enzima que pueda actuar sobre un profármaco de tal manera que lo encubra en su forma citotóxica más activa.
Las enzimas que son útiles incluyen, pero no se limitan a, fosfatasa alcalina, útil para convertir profármacos que contienen fosfato en fármacos libres; arilsulfatasa, útil para convertir profármacos que contienen sulfato en fármacos libres; citosina desaminasa, útil para convertir 5-fluorocitosina no tóxica en el antineoplásico, 5-fluorouracilo; proteasas, tales como proteasa de Serratia, termolisina, subtilisina, carboxipeptidasas y catepsinas (tales como catepsinas B y L), que son útiles para convertir profármacos que contienen péptidos en fármacos libres; D-alanilcarboxipeptidasas, útiles para convertir profármacos que contienen sustituyentes de D-aminoácidos; enzimas que escinden glúcidos, tales como p-galactosidasa y neuraminidasa, útiles para convertir profármacos glucosilados en fármacos libres; p-lactamasa, útil para convertir fármacos derivatizados con p-lactamas en fármacos libres; y penicilina amidasas, tales como penicilina V amidasa o penicilina G amidasa, útiles para convertir fármacos derivatizados en sus nitrógenos amínicos con grupos fenoxiacetilo o fenilacetilo, respectivamente, en fármacos libres. De forma alternativa, se pueden usar anticuerpos con actividad enzimática, también conocidos en la técnica como "abzimas", para convertir los profármacos en fármacos activos libres.
(iv) Otras
Otro tipo de modificación covalente del polipéptido comprende enlazar el polipéptido a uno de una variedad de polímeros no proteínicos, por ejemplo, polietilenglicol, polipropilenglicol, polioxialquilenos o copolímeros de polietilenglicol y polipropilenglicol. El polipéptido también se puede atrapar en microcápsulas preparadas, por ejemplo, mediante técnicas de coacervación o mediante polimerización en interfase (por ejemplo, microcápsulas de hidroximetilcelulosa o gelatina y microcápsulas de poli(metacrilato de metilo), respectivamente), en sistemas de administración de fármacos coloidales (por ejemplo, liposomas, microesferas de albúmina, microemulsiones, nanopartículas y nanocápsulas) o en macroemulsiones. Dichas técnicas se divulgan en Remington's Pharmaceutical Sciences, 18.a edición, Gennaro, A.R., Ed., (1990).
IV. Obtención de polipéptidos para su uso en las formulaciones y procedimientos
Los polipéptidos usados en los procedimientos de purificación descritos en el presente documento se pueden obtener usando procedimientos bien conocidos en la técnica, incluyendo los procedimientos de recombinación. Las siguientes secciones proporcionan directrices sobre estos procedimientos.
(A) Polinucleótidos
"Polinucleótido" o "ácido nucleico", como se usa de manera intercambiable en el presente documento, se refieren a polímeros de nucleótidos de cualquier longitud e incluyen ADN y ARN.
Se pueden obtener polinucleótidos que codifican polipéptidos a partir de cualquier fuente incluyendo, pero sin limitarse a, una colección de ADNc preparada a partir de tejido que se cree que posee el ARNm del polipéptido y lo expresa a un nivel detectable. En consecuencia, se pueden obtener convenientemente polinucleótidos que codifican polipéptidos a partir de una colección de ADNc preparada a partir de tejido humano. El gen que codifica el polipéptido también se puede obtener a partir de una colección genómica o por procedimientos sintéticos conocidos (por ejemplo, síntesis automatizada de ácidos nucleicos).
Por ejemplo, el polinucleótido puede codificar toda una cadena de molécula de inmunoglobulina, tal como una cadena ligera o una cadena pesada. Una cadena pesada completa no solo incluye una región variable de la cadena pesada
(VH ), sino también una región constante de la cadena pesada (CH), que típicamente comprenderá tres dominios constantes: CH1, CH2 y CH3; y una región de bisagra. En algunas situaciones, es deseable la presencia de una región constante.
Otros polipéptidos que se pueden codificar por el polinucleótido incluyen fragmentos de anticuerpo de unión a antígeno, tales como anticuerpos de dominio único ("dAb"), Fv, scFv, Fab' y F(ab')2 y "minicuerpos". Los minicuerpos son (típicamente) fragmentos de anticuerpo bivalente a partir de los que se ha extraído el dominio C h 1 y Ck o Cl . Como los minicuerpos son más pequeños que los anticuerpos convencionales, deberían lograr una mejor penetración en el tejido en el uso clínico/diagnóstico, aunque al ser bivalentes deberían retener una afinidad de unión más alta que los fragmentos de anticuerpo monovalentes, tales como dAb. En consecuencia, a menos que el contexto lo indique de otro modo, el término "anticuerpo" como se usa en el presente documento no solo engloba moléculas de anticuerpo entero, sino también fragmentos de anticuerpo de unión a antígeno del tipo analizado anteriormente. Preferentemente, cada región estructural presente en el polipéptido codificado comprenderá al menos una sustitución aminoacídica con respecto a la región estructural aceptadora humana correspondiente. Por tanto, por ejemplo, las regiones estructurales pueden comprender, en total, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, once, doce, trece, catorce o quince sustituciones de aminoácido con respecto a las regiones estructurales aceptadoras.
Convenientemente, los polinucleótidos descritos en el presente documento se pueden aislar y/o purificar. En algunos modos de realización, los polinucleótidos son polinucleótidos aislados.
El término "polinucleótido aislado" pretende indicar que la molécula se retira o se separa de su entorno normal o natural o se ha producido de tal manera que no está presente en su entorno normal o natural. En algunos modos de realización, los polinucleótidos son polinucleótidos purificados. El término purificado pretende indicar que se han retirado al menos algunas moléculas o sustancias contaminantes.
De forma adecuada, los polinucleótidos están sustancialmente purificados, de modo que los polinucleótidos pertinentes constituyen los polinucleótidos dominantes (es decir, más abundantes) presentes en una composición.
(B) Expresión de polinucleótidos
La descripción que sigue se refiere principalmente a la producción de polipéptidos cultivando células transformadas o transfectadas con un vector que contiene polinucleótidos que codifican polipéptidos. Se contempla, por supuesto, que se pueden emplear procedimientos alternativos, que son bien conocidos en la técnica, para preparar polipéptidos. Por ejemplo, la secuencia de aminoácidos apropiada, o partes de la misma, se puede producir mediante síntesis directa de péptidos usando técnicas en fase sólida (véase, por ejemplo, Stewart et al., Solid-Phase Peptide Synthesis W.H. Freeman Co., San Francisco, Calif. (1969); Merrifield, J. Am. Chem. Soc. 85:2149-2154 (1963)). Se puede realizar la síntesis de proteínas in vitro usando técnicas manuales o por automatización. La síntesis automatizada se puede realizar, por ejemplo, usando un sintetizador de péptidos Applied Biosystems (Foster City, California) usando las instrucciones del fabricante. Se pueden sintetizar varias partes del polipéptido químicamente por separado y combinar usando procedimientos químicos o enzimáticos para producir el polipéptido deseado.
Los polinucleótidos como se describen en el presente documento se insertan en un vector o vectores de expresión para la producción de los polipéptidos. El término "secuencias de control" se refiere a secuencias de ADN necesarias para la expresión de una secuencia codificante enlazada de forma funcional en un organismo huésped particular. Las secuencias de control incluyen, pero no se limitan a, promotores (por ejemplo, promotores asociados de forma natural o heterólogos), secuencias señal, elementos potenciadores y secuencias de terminación de la transcripción.
Un ácido nucleico está "enlazado de forma funcional" cuando se coloca en una relación funcional con otra secuencia de polinucleótido. Por ejemplo, los ácidos nucleicos de una presecuencia o secuencia líder secretora están enlazados de forma funcional a ácidos nucleicos de un polipéptido si se expresan como una preproteína que participa en la secreción del polipéptido; un promotor o potenciador está enlazado de forma funcional a una secuencia codificante si afecta a la transcripción de la secuencia; o un sitio de unión a ribosoma está enlazado de forma funcional a una secuencia codificante si se coloca de manera que facilite la traducción. En general, "enlazado de forma funcional" significa que las secuencias de ADN que están unidas son contiguas y, en el caso de una secuencia líder secretora, que son contiguas y están en la pauta de lectura. Sin embargo, los potenciadores no tienen que ser contiguos. La unión se logra mediante ligamiento en sitios de restricción convenientes. Si no existen dichos sitios, entonces se usan adaptadores o conectores oligonucleotídicos sintéticos de acuerdo con la práctica convencional.
Para anticuerpos, las cadenas ligera y pesada se pueden clonar en el mismo o en diferentes vectores de expresión. Los segmentos de ácido nucleico que codifican las cadenas de inmunoglobulina están enlazados de forma funcional a secuencias de control en el vector o vectores de expresión que aseguran la expresión de polipéptidos de inmunoglobulina.
Los vectores que contienen las secuencias polinucleotídicas, por ejemplo, las secuencias codificantes de cadena pesada variable y/o ligera variable y secuencias de control de la expresión opcionales) se pueden transferir a una célula huésped por procedimientos bien conocidos, que varían dependiendo del tipo de huésped celular. Por ejemplo,
la transfección con cloruro de calcio se utiliza comúnmente para células procariotas, mientras que el tratamiento con fosfato de calcio, electroporación, lipofección, biolística o transfección basada en virus se pueden usar para otros huéspedes celulares. (Véase, en general, Sambrook et al., Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Press, 2.a ed., 1989). Otros procedimientos usados para transformar células de mamífero incluyen el uso de polibreno, fusión de protoplastos, liposomas, electroporación y microinyección. Para la producción de animales transgénicos, los transgenes se pueden microinyectar en ovocitos fertilizados, o se pueden incorporar al genoma de células madre embrionarias, y los núcleos de dichas células se pueden transferir a ovocitos enucleados.
(C) Vectores
El término "vector" incluye vectores de expresión y vectores de transformación y vectores lanzadera.
El término "vector de expresión" significa una construcción que se puede expresar in vivo o in vitro.
El término "vector de transformación" significa una construcción que se puede transferir de una entidad a otra entidad, que puede ser de la especie o puede ser de una especie diferente. Si la construcción se puede transferir de una especie a otra, tal como de un plásmido de Escherichia coli a una bacteria, tal como del género Bacillus, entonces el vector de transformación se denomina a veces "vector lanzadera". Puede incluso ser una construcción que se pueda transferir de un plásmido de E. coli a una Agrobacterium a una planta.
Los vectores se pueden transformar en una célula huésped adecuada como se describe a continuación para proporcionar la expresión de un polipéptido. Diversos vectores están disponibles públicamente. El vector puede estar, por ejemplo, en forma de un plásmido, cósmido, partícula vírica o fago. La secuencia de ácido nucleico apropiada se puede insertar en el vector mediante varios procedimientos. En general, el ADN se inserta en un(os) sitio(s) de endonucleasa de restricción apropiado(s) usando técnicas conocidas en la técnica. La construcción de vectores adecuados que contienen uno o más de estos componentes emplea técnicas de unión estándar que son conocidos por el experto en la técnica.
Los vectores pueden ser, por ejemplo, vectores de plásmidos, virus o fagos provistos de un origen de replicación, opcionalmente un promotor para la expresión de dicho polinucleótido y opcionalmente un regulador del promotor. Los vectores pueden contener uno o más genes marcadores seleccionables que son bien conocidos en la técnica.
Estos vectores de expresión son típicamente replicables en los organismos huésped, bien como episomas o bien como parte integrante del ADN cromosómico del huésped.
(D) Células huésped
La célula huésped puede ser una bacteria, una levadura u otra célula fúngica, célula de insecto, una célula de planta, o una célula de mamífero, por ejemplo.
Se puede usar un organismo huésped multicelular transgénico que se ha manipulado genéticamente para producir un polipéptido. El organismo puede ser, por ejemplo, un organismo transgénico de mamífero (por ejemplo, una línea transgénica de cabra o de ratón).
Los procariotas adecuados incluyen, pero no se limitan a, eubacterias, tales como organismos gramnegativos o grampositivos, por ejemplo, Enterobacteriaceae tales como E. coli. Diversas cepas de E. coli están disponibles públicamente, tales como E. coli K12 cepa MM294 (ATCC 31.446); E. coli X1776 (ATCC 31.537); E. coli cepa W3110 (ATCC 27.325) y K5772 (ATCC 53.635). Otras células huésped procariotas adecuadas incluyen Enterobacteriaceae tales como Escherichia, por ejemplo, E. coli, Enterobacter, Erwinia, Klebsiella, Proteus, Salmonella, por ejemplo, Salmonella typhimurium, Serratia, por ejemplo, Serratia marcescans y Shigella, así como Bacillus tales como B. subtilis y B. licheniformis (por ejemplo, B. licheniformis 41P), Pseudomonas tales como P. aeruginosa y Streptomyces. Estos ejemplos son ilustrativos en lugar de limitantes. La cepa W3110 es un huésped o huésped original particularmente preferente porque es una cepa huésped común para fermentaciones de producto de polinucleótido recombinante. Preferentemente, la célula huésped segrega cantidades mínimas de enzimas proteolíticas. Por ejemplo, la cepa W3110 se puede modificar para efectuar una mutación genética en los genes que codifican polipéptidos endógenos para el huésped, incluyendo los ejemplos de dichos huéspedes E. coli W3110 cepa 1A2, que tiene el genotipo completo tonA; E. coli W3110 cepa 9E4, que tiene el genotipo completo tonA ptr3; E. coli W3110 cepa 27C7 (ATCC 55, 244), que tiene el genotipo completo tonA ptr3 phoA E15 (argF-lac) 169 degP ompT kan'; E. coli W3110 cepa 37D6, que tiene el genotipo completo tonA ptr3 phoA E15 (argF-lac) 169 degP ompT rbs7 ilvG kan'; E. coli W3110 cepa 40B4, que es la cepa 37D6 con una mutación de eliminación de degP no resistente a kanamicina y una cepa de E. coli que tiene proteasa periplásmica mutante. De forma alternativa, son adecuados procedimientos de clonación in vitro, por ejemplo, PCR u otras reacciones de polimerasa de ácido nucleico.
En estos huéspedes procariotas se pueden producir vectores de expresión, que típicamente contendrán secuencias de control de la expresión compatibles con la célula huésped (por ejemplo, un origen de replicación). Además, estarán presentes muchos de una variedad de promotores bien conocidos, tales como el sistema promotor de lactosa, un sistema promotor de triptófano (trp), un sistema promotor de beta-lactamasa o un sistema promotor del fago lambda.
Los promotores controlarán típicamente la expresión, opcionalmente con una secuencia operadora, y tendrán secuencias de sitios de unión a ribosoma y similares, para iniciar y completar la transcripción y la traducción.
Se pueden usar microbios eucariotas para la expresión. Los microbios eucariotas tales como hongos filamentosos o levaduras son huéspedes de clonación o expresión adecuados para vectores codificantes de polipéptidos. Saccharomyces cerevisiae es un microorganismo huésped eucariota inferior comúnmente usado. Otros incluyen Schizosaccharomyces pombe; huéspedes de Kluyveromyces tales como, por ejemplo, K. lactis (MW98-8C, CBS683, CBS4574), K. fragilis (ATCC 12.424), K. bulgaricus (ATCC 16.045 ), K. wickeramii (ATCC 24.178), K. waltii (ATCC 56.500), K. drosophilarum (ATCC 36.906), K. thermotolerans y K. marxianus; Yarrowia (documento EP 402.226); Pichia pastoris; Candida; Trichoderma reesia; Neurospora crassa; Schwanniomyces tales como Schwanniomyces occidentalis y hongos filamentosos tales como, por ejemplo, huéspedes de Neurospora, Penicillium, Tolypocladium, y Aspergillus tales como A. nidulans y A. niger. Las levaduras metilotrópicas son adecuadas en el presente documento e incluyen, pero no se limitan a, levaduras que pueden crecer sobre metanol, seleccionadas de los géneros que consisten en Hansenula, Candida, Kloeckera, Pichia, Saccharomyces, Torulopsis y Rhodotorula. Saccharomyces es un huésped de levadura preferente, con vectores adecuados que tienen secuencias de control de la expresión (por ejemplo, promotores), un origen de replicación, secuencias de terminación y similares según se desee. Los promotores típicos incluyen 3-fosfoglicerato cinasa y otras enzimas glucolíticas. Los promotores de levadura inducibles incluyen, entre otros, promotores de alcohol deshidrogenasa, isocitocromo C y enzimas responsables de la utilización de maltosa y galactosa.
Además de microorganismos, también se puede usar el cultivo de células de tejido de mamífero para expresar y producir los polipéptidos como se describe en el presente documento y, en algunos casos, es preferente (véase Winnacker, From Genes to Clones VCH Publishers, NY, NY (1987). Para algunos modos de realización, las células eucariotas pueden ser preferentes, porque se han desarrollado en la técnica una serie de líneas de células huésped adecuadas que pueden segregar polipéptidos heterólogos (por ejemplo, inmunoglobulinas intactas), e incluyen líneas celulares de c Ho , diversas líneas celulares Cos, células HeLa, preferentemente, líneas celulares de mieloma o linfocitos B transformados o hibridomas. En algunos modos de realización, la célula huésped de mamífero es una célula de CHO.
En algunos modos de realización, la célula huésped es una célula huésped de vertebrado. Son ejemplos de líneas de células huésped de mamífero la línea CV1 de riñón de mono transformada por SV40 (COS-7, ATCC CRL 1651); la línea de riñón embrionario humano (293 o células 293 subclonadas para crecimiento en cultivo de suspensión); células de riñón de cría de hámster (BHK, At CC CCL 10); células de ovario de hámster chino/DHFR (línea Ch O o CHO-DP-12); células de Sertoli de ratón; células de riñón de mono (CV1 ATCC CCL 70); células de riñón de mono verde africano (v Er O-76, ATCC CRL-1587); células de carcinoma de cuello uterino humano (HELA, ATCC CCL 2); células de riñón canino (MDCK, ATCC CCL 34); células de hígado de rata Buffalo (BRL 3A, ATCC CRL 1442); células de pulmón humano (W138, ATCC CCL 75); células de hígado humano (Hep G2, HB 8065); células de tumor mamario de ratón (MMT 060562, ATCC CCL51); células TRI; células MRC 5; células FS4 y una línea de hepatoma humano (Hep G2).
V. Formulaciones y procedimientos de producción de las formulaciones
En el presente documento también se proporcionan formulaciones y procedimientos para producir la formulación que comprende los polipéptidos (por ejemplo, anticuerpos) purificada por los procedimientos descritos en el presente documento. Por ejemplo, el polipéptido purificado se puede combinar con un vehículo farmacéuticamente aceptable.
Las formulaciones de polipéptido en algunos modos de realización se pueden preparar para almacenamiento mezclando un polipéptido que tiene el grado deseado de pureza con vehículos, excipientes o estabilizantes farmacéuticamente aceptables (Remington Pharmaceutical Sciences, 16.a edición, Osol, A. Ed. (1980)), en forma de formulaciones liofilizadas o soluciones acuosas.
Los "vehículos" como se usan en el presente documento incluyen vehículos, excipientes o estabilizantes farmacéuticamente aceptables que no son tóxicos para la célula o mamífero que se expone a los mismos a las dosificaciones y concentraciones empleadas. A menudo, el vehículo fisiológicamente aceptable es una solución acuosa de pH tamponado.
Los vehículos, excipientes o estabilizantes aceptables no son tóxicos para los destinatarios a las dosificaciones y concentraciones empleadas, e incluyen tampones tales como fosfato, citrato y otros ácidos orgánicos; antioxidantes incluyendo ácido ascórbico y metionina; conservantes (tales como cloruro de octadecildimetilbencilamonio; cloruro de hexametonio; cloruro de benzalconio, cloruro de bencetonio; fenol, alcohol butílico o bencílico; alquilparabenos tales como metilparabeno o propilparabeno; catecol; resorcinol; ciclohexanol; 3-pentanol y m-cresol); polipéptidos de bajo peso molecular (menos de aproximadamente 10 residuos); proteínas, tales como seroalbúmina, gelatina o inmunoglobulinas; polímeros hidrófilos tales como polivinilpirrolidona; aminoácidos tales como glicina, glutamina, asparagina, histidina, arginina o lisina; monosacáridos, disacáridos y otros glúcidos incluyendo glucosa, manosa o dextrinas; agentes quelantes tales como EDTA; azúcares tales como sacarosa, manitol, trehalosa o sorbitol; contraiones formadores de sales tales como sodio; complejos metálicos (por ejemplo, complejos de Zn-proteína) y/o tensioactivos no iónicos tales como TWEEN™, PLURONICS™ o polietilenglicol (PEG).
En algunos modos de realización, el polipéptido de la formulación de polipéptido mantiene la actividad funcional.
Las formulaciones que se vayan a usar para la administración in vivo deben ser estériles. Esto se consigue fácilmente por filtración a través de membranas de filtración estériles.
Las formulaciones del presente documento pueden contener también más de un compuesto activo según sea necesario para la indicación particular que se está tratando, preferentemente aquellos que tienen actividades complementarias que no resultan afectadas de manera adversa entre sí. Por ejemplo, además de un polipéptido, puede ser deseable incluir en la formulación un polipéptido adicional (por ejemplo, un anticuerpo). De forma alternativa o adicional, la composición puede comprender además un agente quimioterápico, un agente citotóxico, una citocina, un agente inhibidor del crecimiento, un agente antihormonal y/o un cardioprotector. Dichas moléculas están presentes de forma adecuada en combinación en cantidades que son eficaces para el propósito previsto.
VI. Artículos de fabricación
Los polipéptidos purificados por los procedimientos descritos en el presente documento y/o las formulaciones que comprenden los polipéptidos purificados por los procedimientos descritos en el presente documento pueden estar contenidos en un artículo de fabricación. El artículo de fabricación puede comprender un recipiente que contiene el polipéptido y/o la formulación de polipéptido. Preferentemente, el artículo de fabricación comprende: (a) un recipiente que comprende una composición que comprende el polipéptido y/o la formulación de polipéptido descrita en el presente documento dentro del recipiente y (b) un prospecto con instrucciones para administrar la formulación a un sujeto.
El artículo de fabricación comprende un recipiente y una ficha técnica o prospecto en el recipiente o asociado con el mismo. Los recipientes adecuados incluyen, por ejemplo, botellas, viales, jeringas, etc. Los recipientes se pueden obtener en una variedad de materiales, tales como vidrio o plástico. El recipiente retiene o contiene una formulación y puede tener una boquilla de acceso estéril (por ejemplo, el recipiente puede ser una bolsa de solución intravenosa o un vial que tiene un tapón perforable por una aguja de inyección hipodérmica). Al menos un agente activo de la composición es el polipéptido. La ficha técnica o prospecto indica el uso de la composición en un sujeto con directrices específicas en lo referente a las cantidades de dosificación y los intervalos de polipéptido y cualquier otro fármaco que se proporcione. El artículo de fabricación puede incluir además otros materiales deseables desde un punto de vista comercial y del usuario, incluyendo otros tampones, diluyentes, filtros, agujas y jeringas. En algunos modos de realización, el recipiente es una jeringa. En algunos modos de realización, la jeringa está además contenida dentro de un dispositivo de inyección. En algunos modos de realización, el dispositivo de inyección es un autoinyector.
El término "prospecto" se usa para hacer referencia a las instrucciones incluidas normalmente en envases comerciales de productos terapéuticos, que contienen información sobre las indicaciones, uso, dosificación, administración, contraindicaciones, otros productos terapéuticos para combinar con el producto envasado y/o advertencias sobre el uso de tales productos terapéuticos.
VII. Modos de realización ejemplares
En algunos modos de realización, la invención proporciona procedimientos para purificar un anticuerpo a partir de una composición que comprende el anticuerpo y uno o más contaminantes, comprendiendo dicho procedimiento a) cargar la composición en un material de cromatografía de modo mixto, de intercambio iónico, de interacción hidrófoba o de afinidad en una cantidad en exceso de la capacidad de unión dinámica del material de cromatografía para el anticuerpo usando un tampón de carga, en el que el coeficiente de reparto del material de cromatografía para el polipéptido es mayor que 30, b) eluir el anticuerpo del material de cromatografía en condiciones en las que el uno o más contaminantes permanecen unidos al material de cromatografía usando un tampón de elución, en el que el tampón de elución tiene una conductividad menor que la conductividad del tampón de carga y c) agrupar fracciones que comprenden el anticuerpo en el efluente de cromatografía de las etapas a) y b).
En otros modos de realización de la divulgación, el polipéptido es un anticuerpo o inmunoadhesina.
En otros modos de realización de la divulgación, el polipéptido es un inmunoadhesina.
En otros modos de realización del modo de realización anterior, el polipéptido es un anticuerpo.
En otros modos de realización del modo de realización anterior, el anticuerpo es un anticuerpo monoclonal.
Aún en otro modo de realización del modo de realización anterior, el anticuerpo monoclonal es un anticuerpo quimérico, un anticuerpo humanizado o un anticuerpo humano.
En otros modos de realización del modo de realización anterior, el anticuerpo monoclonal es un anticuerpo monoclonal IgG.
En otros modos de realización del modo de realización anterior, el anticuerpo es un fragmento de unión al antígeno. Aún en otros modos de realización del modo de realización anterior, el fragmento de unión al antígeno se selecciona del grupo que consiste en un fragmento Fab, un fragmento Fab', un fragmento F(ab')2, un scFv, un di-scFv, un bi-scFv, un (di, tri)-scFv en tándem, un Fv, un sdAb, un anticuerpo trifuncional, un BiTE, un diacuerpo y un triacuerpo.
En otros modos de realización del modo de realización anterior, el polipéptido se selecciona de una enzima, una hormona, una proteína de fusión, una proteína que contiene Fc, un inmunoconjugado, una citocina y una interleucina. En otros modos de realización del modo de realización anterior, el al menos un contaminante es uno cualquiera o más de proteína de ovario de hámster chino (CHOP), una proteína de célula huésped (HCP), proteína A lixiviada, ácido nucleico, ADN, variantes de producto, proteína agregada, componente de medio de cultivo celular, gentamicina, fragmento polipeptídico, endotoxina y contaminante vírico.
En otros modos de realización del modo de realización anterior, el material de cromatografía se selecciona de un material de modo mixto, un material de intercambio aniónico, un material de intercambio catiónico, un material de interacción hidrófoba y un material de afinidad.
En otros modos de realización del modo de realización anterior, la densidad de carga es de entre aproximadamente 50 g/l y aproximadamente 2000 g/l.
Aún en otros modos de realización del modo de realización anterior, la densidad de carga es de entre aproximadamente 200 g/l y aproximadamente 1000 g/l.
En otros modos de realización del modo de realización anterior, la composición se carga sobre el material de cromatografía a aproximadamente las capacidades de unión dinámica de los materiales de cromatografía para el uno o más contaminantes.
En otros modos de realización del modo de realización anterior, la composición se carga sobre el material de cromatografía a 20 veces la capacidad de unión dinámica del material de cromatografía para el polipéptido.
En otros modos de realización del modo de realización anterior, el coeficiente de reparto del material de cromatografía para el polipéptido es mayor que 30.
Aún en otros modos de realización del modo de realización anterior, el coeficiente de reparto del material de cromatografía para el polipéptido es mayor que 100.
En otros modos de realización del modo de realización anterior, el procedimiento comprende además el uso de un tampón de carga y un tampón de elución.
En otros modos de realización del modo de realización anterior, el tampón de elución tiene una conductividad menor que la conductividad del tampón de carga.
En otros modos de realización del modo de realización anterior, el tampón de carga tiene una conductividad de aproximadamente 4,0 mS hasta aproximadamente 7,0 mS.
En otros modos de realización del modo de realización anterior, el tampón de elución tiene una conductividad de aproximadamente 0,0 mS hasta aproximadamente 7,0 mS.
En otros modos de realización de la divulgación, el tampón de elución tiene una conductividad mayor que la conductividad del tampón de carga.
En otros modos de realización del modo de realización anterior, el tampón de carga tiene una conductividad de aproximadamente 4,0 mS hasta aproximadamente 7,0 mS.
En otros modos de realización del modo de realización anterior, el tampón de elución tiene una conductividad de aproximadamente 5,5 mS hasta aproximadamente 17,0 mS.
En otros modos de realización de la divulgación, la conductividad del tampón de elución disminuye en un gradiente de aproximadamente 5,5 mS hasta aproximadamente 1,0 mS sobre aproximadamente 10 volúmenes de columna (VC). En otros modos de realización de la divulgación, la conductividad del tampón de elución disminuye en un gradiente de aproximadamente 5,5 mS hasta aproximadamente 1,0 mS sobre aproximadamente 15 VC.
En otros modos de realización de la divulgación, la conductividad del tampón de elución disminuye en un gradiente de aproximadamente 10,0 mS hasta aproximadamente 1,0 mS sobre aproximadamente 5 VC.
En otros modos de realización de la divulgación, la conductividad del tampón de elución disminuye en un gradiente de aproximadamente 10,9 mS hasta aproximadamente 1,0 mS sobre aproximadamente 10 VC.
En otros modos de realización del modo de realización anterior, el tampón de elución tiene un pH menor que el pH del tampón de carga.
En otros modos de realización del modo de realización anterior, el tampón de carga tiene un pH de aproximadamente 4 hasta aproximadamente 9.
En otros modos de realización del modo de realización anterior, el tampón de elución tiene un pH de aproximadamente 4 hasta aproximadamente 9.
En otros modos de realización del modo de realización anterior, el tampón de elución tiene un pH mayor que el pH del tampón de carga.
En otros modos de realización del modo de realización anterior, el tampón de carga tiene un pH de aproximadamente 4 hasta aproximadamente 9.
En otros modos de realización del modo de realización anterior, el tampón de elución tiene un pH de aproximadamente 4 hasta aproximadamente 9.
En otros modos de realización del modo de realización anterior, la composición es un eluyente de una cromatografía de afinidad, una cromatografía de intercambio catiónico, una cromatografía de intercambio aniónico, una cromatografía de modo mixto y una cromatografía de interacción hidrófoba.
En otros modos de realización del modo de realización anterior, la cromatografía de afinidad es una cromatografía de proteína A.
En otros modos de realización del modo de realización anterior, el polipéptido se purifica adicionalmente.
Aún en otros modos de realización del modo de realización anterior, el polipéptido se purifica adicionalmente mediante filtración de virus.
En otros modos de realización adicionales del modo de realización anterior, el polipéptido se purifica adicionalmente mediante una o más de una cromatografía de afinidad, una cromatografía de intercambio catiónico, una cromatografía de intercambio aniónico, una cromatografía de modo mixto o una cromatografía de interacción hidrófoba.
En otros modos de realización del modo de realización anterior, el polipéptido se concentra adicionalmente.
Aún en otros modos de realización del modo de realización anterior, el polipéptido se concentra por ultrafiltración, diafiltración o una combinación de ultrafiltración y diafiltración.
En otros modos de realización del modo de realización anterior, los procedimientos comprenden además la combinación del polipéptido con un vehículo farmacéuticamente aceptable.
Todos los rasgos característicos divulgados en esta memoria descriptiva se pueden combinar en cualquier combinación. Cada rasgo característico divulgado en esta memoria descriptiva se puede reemplazar por un rasgo característico alternativo que cumpla un propósito igual, equivalente o similar. Por tanto, a menos que se establezca expresamente de otro modo, cada rasgo característico divulgado es solo un ejemplo de una serie genérica de rasgos característicos equivalentes o similares.
Se ilustran más detalles de la invención por los siguientes ejemplos no limitantes.
EJEMPLOS
Los ejemplos a continuación pretenden ser puramente ejemplares de la invención y, por lo tanto, no se debe considerar que limitan la invención de ningún modo. Se aportan los siguientes ejemplos y descripción detallada a modo de ilustración y no a modo de limitación.
Materiales y procedimientos
Los materiales y procedimientos para todos los ejemplos se realizaron como se indica a continuación a menos que se indique lo contrario en el Ejemplo.
Materia prima de MAb
La materia prima de MAb para todos los ejemplos se seleccionó de lotes de cultivo celular a escala industrial, piloto o pequeña en Genentech (South San Francisco, CA, EE. UU.). Después de un período de fermentación del cultivo celular, las células se separaron y el líquido depurado se purificó mediante cromatografía de proteína A. La proteína A se usó para investigar el mecanismo de depurado de impurezas. La Tabla 2 muestra las características de la materia prima para cada mAb usado en los ejemplos.
Tabla 2. Características de la materia prima de MAb.
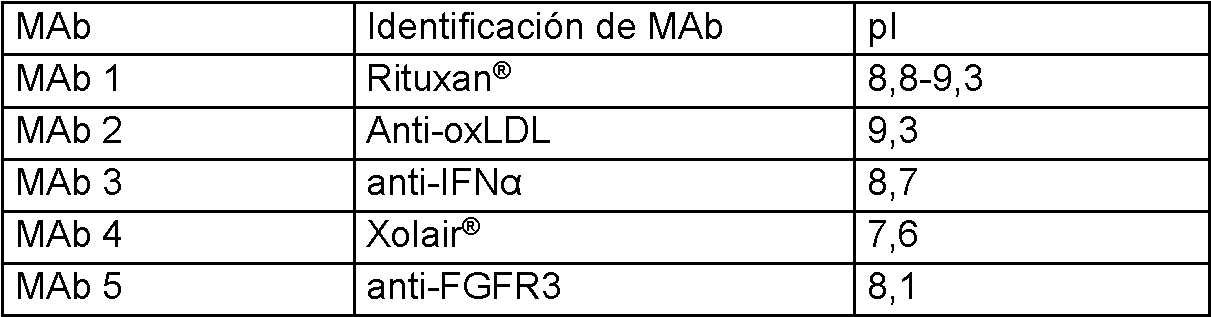
Cuantificación del MAb
La concentración de anticuerpo se determinó mediante absorbancia a 280 y 320 nm usando un espectrofotómetro UV-visible (modelo 8453 G1103A, Agilent Technologies, Santa Clara, CA, EE. UU.) o NanoDrop 1000 modelo ND-1000 (Thermo Fisher Scientific, Waltham, MA, EE. UU.). Especies distintas del anticuerpo (es decir, impurezas) eran demasiado bajas en concentración para tener un efecto apreciable sobre la absorbancia UV. Cuando era necesario, las muestras se diluyeron con un diluyente no interferente apropiado en el intervalo de 0,1-1,0 unidad de absorbancia. La preparación de la muestra y las mediciones de UV se realizaron por duplicado y se registró el valor promedio. Los coeficientes de absorción de MAb variaron de 1,42 a 1,645/mgmlcm.
Cuantificación de la proteína de la célula huésped CHO (CHOP)
Se usó un ELISA para cuantificar los niveles de la proteína de la célula huésped llamada CHOP. Se inmovilizaron anticuerpos anti-CHoP en pocillos de placas de microvaloración. Las diluciones de las muestras que contenían CHOP, patrones y controles se incubaron en los pocillos, seguido de incubación con anticuerpos anti-CHOP conjugados con peroxidasa de rábano picante (HRP). La actividad enzimática de HRP se detectó con o-fenilendiamina, y la CHOP se cuantificó por lectura de la absorbancia a 490 nm en un lector de placas de microvaloración. Basándose en los principios de ELISA de tipo sándwich, la concentración de peroxidasa correspondía a la concentración de CHOP. El intervalo de ensayo para el ELISA era típicamente 5-320 ng/ml con variabilidad intraensayo <10 %. Los valores de CHOP se informaron en unidades de ng/ml. De forma alternativa, los valores de CHOP se dividieron por la concentración de MAb y los resultados se informaron en PPM (partes por millón, por ejemplo, ng de CHOP/mg de MAb). El ELISA de CHOP se puede usar para cuantificar los niveles totales de CHOP en una muestra, pero no cuantifica la concentración de proteínas individuales.
Condiciones de funcionamiento de cromatografía
Las resinas Capto Adhere y Capto MMC Capto Adhere se adquirieron en GE Healthcare (Uppsala, Suecia). Se adquirieron resinas de intercambio catiónico fuertes (Poros XS y Poros 50 HS) en Applied Biosystems (Address). Todos los experimentos de cromatografía de laboratorio se llevaron a cabo usando un sistema cromatográfico AKLC FPLC de GE Healthcare (Uppsala, Suecia) usando el programa informático UNICORN. Las columnas de laboratorio tenían 0,66 cm de diámetro y 10-20 cm de altura. Las columnas se equilibraron a las condiciones de funcionamiento especificadas de pH y conductividad antes de la carga. A continuación, las agrupaciones de proteína A se cargaron en la columna seguido por el tampón de elución según se requiera para eluir la proteína unida. El flujo o eluido durante las fases de carga, sobrecarga y elución se recogieron como fracciones y después se analizaron las impurezas. La densidad de carga del MAb variaba de 100 a 1000 g por litro de resina.
Análisis de la agrupación cromatográfica
Las agrupaciones de flujo continuo durante las fases de carga, sobrecarga y elución se recogieron en fracciones (1 volumen de columna cada una) y se analizaron para determinar la concentración de MAb, la concentración de CHOP, los agregados, la proteína A lixiviada, el ADN de CHO y el rendimiento. Se generaron gráficos acumulativos en función de las fracciones de la agrupación de elución. El rendimiento acumulado se obtuvo usando la Ecuación 1.
donde, para la fracción i, Ci es la concentración de Mab (mg/ml), Vi es el volumen de la fracción (ml), Mp es la masa de proteína cargada (mg).

Cromatografía de exclusión molecular
La heterogeneidad de tamaño de los anticuerpos monoclonales se determinó mediante un ensayo de cromatografía de exclusión molecular de alto rendimiento. Una columna TSK G3000SWXL SEC (diámetro = 7,8 mm, altura = 300 mm, número de referencia 08541) fabricada por Tosoh Bioscience (Tokio, Japón) se hizo funcionar a temperatura ambiente en un equipo de HPLC de la serie 1200 (Agilent Technologies) y se usó para determinar los niveles relativos de monómero de MAb para las muestras recogidas. La columna se utilizó a un caudal de 0,3 ml/min usando una fase móvil de fosfato de potasio 200 mM, cloruro de potasio 250 mM y pH 6,2. Se inyectaron 20 pg de anticuerpo para cada muestra. Se usó absorbancia UV a 280 nm para monitorizar la separación de monómeros, proteínas LMW y proteínas HMW. Los porcentajes de monómero, proteínas LMW y proteínas HMW se analizaron manualmente utilizando el programa informático ChemStation (Agilent Technologies).
Cuantificación de ADN de CHO
El ADN de CHO en muestras de producto se cuantificó usando PCR en tiempo real (TaqMan PCR). El ADN de las muestras y los controles se extrajeron primero usando el kit Qiagen Virus Biorobot. Las muestras extraídas, los controles y el ADN de patrón, se sometieron a reacción en cadena de la polimerasa (PCR) en tiempo real TaqMan usando cebadores de PCR y sonda en una placa de 96 pocillos con el sistema de detección de secuencias de ABI. Los cebadores se definieron por un segmento de 110 pares de bases de una secuencia de ADN repetitiva en el genoma de Cricetulus griseus. La sonda se marcó con un tinte indicador fluorescente en el extremo 5' y un tinte de neutralizador en el extremo 3'. Cuando la sonda está intacta, el espectro de emisión del indicador se suprime por el neutralizador. La actividad 5' nucleasa de la polimerasa hidroliza la sonda y libera el indicador, lo que da como resultado un aumento en la emisión de fluorescencia. El detector de secuencia cuantificó el producto amplificado en proporción directa con el aumento de la emisión de fluorescencia medida continuamente durante la amplificación del ADN. Los números de ciclo en los que el ADN se había amplificado más allá del umbral (CT) se calcularon para la curva patrón. Se generó una curva patrón de 1 pg/ml-10000 pg/ml, que se utilizó para cuantificar ADN en muestras.
Cuantificación de proteína A lixiviada
El nivel de proteína A lixiviada en las agrupaciones de proteína A se determinó mediante un ELISA de tipo sándwich de proteína A. Se inmovilizaron anticuerpos antiproteína A estafilocócica de pollo sobre pocillos de placas de microvaloración. El procedimiento de tratamiento de la muestra incluía la dilución de la muestra y después la disociación del complejo de proteína A/IgG usando calentamiento asistido por microondas como una etapa de pretratamiento antes de someter las muestras a un ELISA de tipo sándwich. La proteína A, si está presente en la muestra, se une al anticuerpo recubierto. La proteína A unida se detectó usando anticuerpos antiproteína conjugados con peroxidasa de rábano picante. La actividad enzimática de la peroxidasa de rábano picante se cuantificó con una solución de sustrato TMB de 2 componentes que produce una señal colorimétrica.
Acondicionamiento de la materia prima
Las materias primas usadas para los experimentos en este estudio eran frescas o se retiraron del almacenamiento en frío (2-8 °C o -70 °C) y se dejaron equilibrar a temperatura ambiente. Posteriormente, se ajustaron el pH y/o la conductividad según se necesitaba usando un agente de valoración (base Tris 1,5 M o ácido acético 1 M) o diluyente (agua purificada, cloruro de sodio 5 M o acetato de sodio 5 M). Todas las materias primas se filtraron a 0,2 pm usando un Millipak 20 (Millipore), AcroPakTM 20 (Pall Corporation) o un filtro de vacío (Thermo Fisher Scientific, Rochester, NY, EE. UU.).
Cribado ultrarrápido
Se utilizó un robot Tecan Freedom Evo 200 (Tecan US, Research Triangle Park, NC) para la manipulación de líquidos y resinas. Se usó una placa de filtro de 96 pocillos (Seahorse 800 pl de polipropileno de 0,45 pm, E & K Scientific EK-2223) para incubar la resina con la proteína y el tampón. Después de incubar la solución de proteína con la resina, la placa de filtro se centrifugó a 1200 xg durante 3 minutos para separar la solución de la resina. Para cada etapa, se pusieron en contacto 300 ml de solución con 50 ml de resina, dando como resultado una proporción volumétrica de la fase de 6:1. Cada pocillo se equilibró hasta el pH y la concentración de acetato de sodio apropiados. El pH variaba de 5,00 a 7,5 a intervalos de 0,5 unidades de pH (se usó acetato para tamponar las condiciones de pH 5,00 a 5,5, se usó MES para tamponar las condiciones de pH 6,00 a 6,5 y se usó MOPS para tamponar las condiciones de 7,00 a 7,5). Todos los experimentos se realizaron a temperatura ambiente. Se usó como carga para estos experimentos una agrupación de proteína A parcialmente purificada, concentrada a 5 g/l o 97 mg/ml, y el tampón se intercambió a NaOAc 15 mM. La resina se expuso a 5 g/l durante los experimentos para determinar los valores Kp de producto. Sin embargo, para la capacidad de unión del producto real, la resina se expuso a 97 g/l. La solución de filtrado se capturó en una placa de recogida y después se analizó usando el lector de placas Infinite M200. La proteína unida se separó posteriormente de la resina usando dos etapas de un tampón de NaCl 2 M para aproximarse al equilibrio de masa.
Estudios de aclaramiento de virus
El objetivo de este estudio fue evaluar la capacidad de retirada de virus de la Capto Adhere para los 2 virus modelo (MMV y X-MuLV). Las columnas, de 0,66 cm de diámetro, se compactaron con resinas originales hasta una altura de lecho de 20 cm. Se hizo pasar la materia prima de MAb con un 1 % de virus y luego se procesó sobre la resina Capto Adhere. Las agrupaciones se recogieron de forma inmediata y las muestras de carga y de elución se ensayaron para determinar los recuentos víricos. Se hicieron múltiples diluciones de las agrupaciones con Medio Completo (1:10 y 1:100) para determinar cualquier interferencia potencial entre los componentes del tampón y los virus.
Ejemplo 1. Cribado ultrarrápido
Este ejemplo describe procedimientos de cribado ultrarrápido para determinar las capacidades de unión del material de cromatografía. Se realizó un cribado ultrarrápido en la resina Capto Adhere en condiciones de unión por lotes para el anticuerpo monoclonal MAb 3.
Los resultados del cribado ultrarrápido de las condiciones de unión se presentan en la figura 1. En las figuras de superficie de respuesta (figuras 1A y 1B), las regiones de unión al producto se indican en rojo (región 8 de la figura 1A y región 7 en la figura 1B) y el producto, por ejemplo, polipéptido, las regiones no unidas son verdes (indicadas como región 1). Los contenidos reales de proteína de la célula huésped (HCP) (ng/mg) en el sobrenadante se muestran en el gráfico de contorno de la figura 1C. El Kp del producto como una función del pH y la concentración de contraión para MAb 3 se muestra en la figura 1A. La resina se cargó a 5 g/l y los datos sin procesar se analizaron usando un modelo de superficie de respuesta. El modelo se utilizó entonces para estimar el Kp para cualquier combinación de pH y la concentración de contraión en el espacio experimental. Los datos muestran que, a medida que el pH aumenta y cuando la conductividad aumenta, el log Kp aumenta. Los incrementos en log Kp reflejan aumentos en el producto que se une a la resina. Para descubrir la capacidad real de unión del producto a la resina, la resina se expuso a 80 g/l en 48 condiciones diferentes combinando diferentes pH y concentraciones de contraión (figura 1B). El sobrenadante de la misma placa experimental se analizó también para HCP (ng/mg) y los datos se representaron en forma de contorno (figura 1C). Se usó agrupación de proteína A parcialmente purificada que contiene varios miles de ppm de CHOP como una carga para los experimentos de cribado ultrarrápido. Las regiones verdes (región 1) de la figura indican la menor cantidad de CHOP en el sobrenadante y las regiones rojas (región 8 de la figura 1B y región 9 de la figura 1C) indican una mayor cantidad de CHOP en el sobrenadante.
El espacio de diseño para las condiciones óptimas de carga y elución se puede determinar a partir de los gráficos de contorno de datos de CHOP y de unión. Estos gráficos permiten el diseño de un modo de funcionamiento de flujo o un modo de funcionamiento de unión y elución. Las condiciones de carga para una cromatografía de flujo continuo son, en general, condiciones en las que se minimiza la unión del producto, por ejemplo, polipéptido, y se maximiza la unión de impurezas (CHOP). Las regiones verdes de solapamiento completas (región 1) entre las figuras 1B y 1C sugieren que es posible un modo F/T. Sin embargo, la región verde (región 1) en este gráfico es una región donde la conductividad es realmente baja ~1 mS. Tales condiciones requieren aproximadamente 4 veces la dilución de la agrupación de proteína A, dando como resultado problemas de ajuste potenciales a escala de planta.
Las regiones rojas (región 7 en la figura 1B y región 8 en la figura 1C) indican regiones de unión para MAb y CHOP. Hay algunas regiones rojas de solapamiento en ambos gráficos. Sin embargo, la figura 1 muestra que, dentro de las concentraciones de pH y contraión funcionales, era posible una capacidad de unión máxima de 55 g/l. Para un procedimiento de título elevado con un título de cultivo celular de ~3,5 g/l, se necesitaría una columna de 1000 l para recuperar todo el producto, tal como un polipéptido, en un ciclo o ciclos múltiples que se realizarían en una columna más pequeña.
En un modo OEC, se puede elegir una condición de carga basada en el comportamiento de las impurezas (por ejemplo, CHOP) en la columna. La cantidad de MAb que se une a la columna es menos preocupante porque el MAb unido se puede recuperar mediante la fase de elución. Por lo tanto, existe un espacio experimental completo para diseñar la condición de carga de modo que se obtenga el máximo depurado de impurezas sin estar limitada por la capacidad de unión de producto de la resina. Mientras que el modo de unión y elución permite una densidad de carga de 55 g/l, OEC permite una capacidad de carga aproximadamente 10 veces superior permitiendo de este modo la implementación de una columna más pequeña que a su vez puede reducir el costo de la resina por gramo de prod ucto y ofrece un buen ajuste de planta.
Ejemplo 2. Modo OEC optimizado
Los datos de HTS se usaron para la determinación de parámetros para el funcionamiento en un modo OEC. Basándose en los requisitos de densidad de carga, ajuste de la planta y depurado de impurezas, se seleccionaron las condiciones de carga para el MAb 3 que son pH 6,5 y 5,5 mS/cm. La densidad de carga para el desarrollo cromatográfico optimizado mostrada en la figura 2 fue de 180 g/l. Aproximadamente 50 g/l de producto polipeptídico se une a la resina durante la fase de carga. La agrupación de producto se recogió comenzando a aproximadamente 0,5 DO, y el producto se sobrecargó en la resina hasta 180 g/l. Después de completarse la fase de carga, se usó la fase de elución con tampón de baja conductividad de 1 mS/cm (tampón de elución: MES 20 mM, pH 6,5) para eluir la proteína unida, dando como resultado una reducción del volumen de la agrupación del 10-15 % en comparación con
la cromatografía de flujo continuo. El rendimiento y el depurado de impurezas para este desarrollo cromatográfico se muestran en la figura 3.
Ejemplo 3. Optimización de carga
Este estudio se llevó a cabo para comparar los datos de HTS y los datos reales de rendimiento de la columna en el modo de funcionamiento de OEC. Las columnas se cargaron a tres pH diferentes. Se seleccionó pH 6,5 para ser la mejor condición basándose en el depurado inicial de CHOP y en los datos de rendimiento. Las concentraciones de CHOP en las agrupaciones variaron de 900 a 50 hasta 400 ppm en función del pH de la carga. A partir de este estudio, se determinó que el pH 6,5 era la mejor condición para cargar la composición. Además, aunque el rendimiento no se optimizó, el pH 6,5 proporcionó también el rendimiento máximo (figura 3, Tabla 3).
Tabla 3. Optimización de la condición de carga para un modo OEC

Ejemplo 4. Optimización de la elución
Este estudio se llevó a cabo para optimizar las condiciones de elución para OEC. Para recuperar el producto (MAb 3, ~50 g/l) que se unió durante la fase de carga, se pasaron unos cuantos volúmenes de columna de tampón de lavado (MES 50 mM, acetato de Na 30 mM, pH 6,5, ~5,5 mS/cm) a través de la columna después de la finalización de la fase de carga. Se observó una gran cantidad de residuos, que dan como resultado un aumento en el volumen de la agrupación al final de la fase de carga. El aumento en el volumen de la agrupación fue de aproximadamente un 45 % usando un tampón de lavado con un pH y conductividad similares al tampón de carga. Este aumento en el volumen de la agrupación puede dar como resultado problemas de ajuste de la planta (figura 4).
El objetivo del estudio de optimización de la elución fue obtener el máximo rendimiento con un mínimo de CHOP y una reducción máxima del volumen de la agrupación. La fase de elución se desarrolló para eluir los 50 g/l de producto unido de la columna. La figura 5 muestra la fase de elución de los cromatogramas. Un tampón de conductividad inferior eluye el producto de la columna en 2 volúmenes de columna dando como resultado un rendimiento más alto y un volumen de agrupación menor (Tabla 4). Por otro lado, los tampones de elución de mayor conductividad dieron como resultado residuos de más de 8 volúmenes de columna. Como se muestra en la Tabla 4, CHOP en la agrupación fue menor de 20 ppm en todos los tampones de elución probados. Por lo tanto, se seleccionó MES 20 mM como tampón de elución para aumentar el rendimiento y para minimizar los residuos.
Tabla 4. Optimización de la elución para OEC

Ejemplo 5. Análisis de impurezas en OEC
Las fracciones de OEC se analizaron en busca de impurezas. La figura 6A muestra las concentraciones de MAb y los niveles de CHOP en las fracciones recogidas durante la fase de carga y durante la fase de elución. Durante la fase de carga, CHOP permaneció entre 20-25 ppm y durante la fase de elución donde solo se eluye el MAb unido, el nivel de CHOP fraccionario disminuye. El análisis acumulativo demuestra que la CHOP y otras impurezas permanecen bastante constantes entre toda la fase de carga y de elución (figura 6b ). La densidad de carga para este desarrollo cromatográfico fue de 180 g/l. El aclaramiento de virus, utilizando X-MuLV y MMV como virus modelo, también se
estudió para la misma densidad de carga (Tabla 5). Para este experimento, el pH de carga fue 6,5 y la conductividad de carga fue de 5,5 mS/cm. Las condiciones de elución fueron MES 20 mM pH 6,5 y conductividad de ~1 mS/cm.
Tabla 5. Aclaramiento del virus para la cromatografía en modo OEC.

Ejemplo 6. Determinación de la capacidad de unión de impurezas máxima
Para hallar la máxima capacidad de unión de impurezas a la resina cargada con MAb 3 por el modo OEC, la resina Capto Adhere se expuso a 1000 g/l con la agrupación de proteína A. El tampón de carga fue de pH 6,5, ~5,5 mS/cm; el tampón de elución fue MES 20 mM, pH 6,5, ~1 mS/cm. La muestra en la agrupación se recogió cada 50 g/l y se analizó para CHOP y la concentración de proteínas (figura 7). Para densidades de carga de hasta 800 g/l, CHOP no avanzó y la CHOP en el eluido fue inferior a 20 ng/mg.
Ejemplo 7. Implementación a escala piloto
El modo de funcionamiento de OEC se implementó en columnas de escala piloto que varían en tamaño de 1,6 l a 10,8 l. Los diámetros de las columnas variaban de 10 cm a 25 cm con alturas de lecho que varían de 20-22 cm (Tabla 6). Las muestras se cargaron a pH 6,5, ~5,5 mS/cm y se eluyeron con MES 20 mM a pH 6,5, ~1 mS/cm. El rendimiento entre los desarrollos cromatográficos a escala piloto se muestra en la figura 8. Las densidades de carga en los desarrollos cromatográficos a escala piloto cubrían un intervalo de densidades de carga de 70 a 180 g/l. Entre todas las densidades de carga se alcanzó un rendimiento promedio de un 94 %. No hubo impacto en el rendimiento sobre el intervalo de densidades de carga ensayadas. Entre todos los desarrollos cromatográficos a escala piloto, la CHOP en la agrupación del modo OEC era inferior a 25 ppm, que se depuró posteriormente hasta <2 ppm de CHOP en el producto polipéptido final (agrupación de UFDF) (Tabla 7). En promedio, hubo una reducción de un 1,1 % en las proteínas HMW entre todos los desarrollos cromatográficos a escala piloto (Tabla 8) y una reducción del 0,19 % en las proteínas LMW entre todos los desarrollos cromatográficos a escala piloto (Tabla 9).
Tabla 6. Tamaño de columna Capto Adhere para los desarrollos cromatográficos a escala piloto

Tabla 7. Datos de CHOP para escala piloto

* CHOP después de la filtración en profundidad a pH 6,5
HCCF es el fluido de cultivo celular recolectado
Tabla 8. % promedio de reducción de proteínas HMW entre 12 desarrollos cromatográficos a escala piloto.

Tabla 9. % promedio de reducción de proteínas LMW entre 12 desarrollos cromatográficos a escala piloto.

El ADN de CHO y la proteína A lixiviada eran menos que detectables en la agrupación después de OEC.
Ejemplo 8. Implementación a escala de producción
El modo de funcionamiento de OEC se implementó en una columna a escala de producción con un tamaño de 157 l (100 cm de diámetro * 20 cm de altura). A escala de producción, la columna se cargó a ~96 g/l. El % de rendimiento entre los dos desarrollos cromatográficos a escala industrial fue >95 % para ambos desarrollos cromatográficos. Entre todos los desarrollos cromatográficos a escala de producción, la CHOP en la agrupación del modo OEC fue inferior a 17 ppm (Tabla 10), que se depuró posteriormente hasta un nivel menor que el detectable de CHOP. En promedio, hubo una reducción de un 1,1 % en las HMW, una reducción de un 0,1 % en las LMW y una reducción de un 1,65 % en ácidos entre los dos desarrollos cromatográficos a escala de producción.
Tabla 10. Datos de CHOP (ppm) a escala de producción

El ADN de CHO y la proteína A lixiviada eran menos detectables en la agrupación después del modo OEC.
Ejemplo 9. Aplicabilidad de OEC a otros MAb
Las agrupaciones de proteína A se usaron como cargas para estos desarrollos cromatográficos. Las condiciones de carga y elución se seleccionaron para permitir el modo OEC. Las densidades de carga, el % de rendimiento, el MAb unido a la resina (g/l), las impurezas en la carga y la agrupación se muestran en las siguientes tablas (Tabla 11, 12, 13 y 14).
Tabla 11. Aplicabilidad de OEC a otros MAb: CHOP
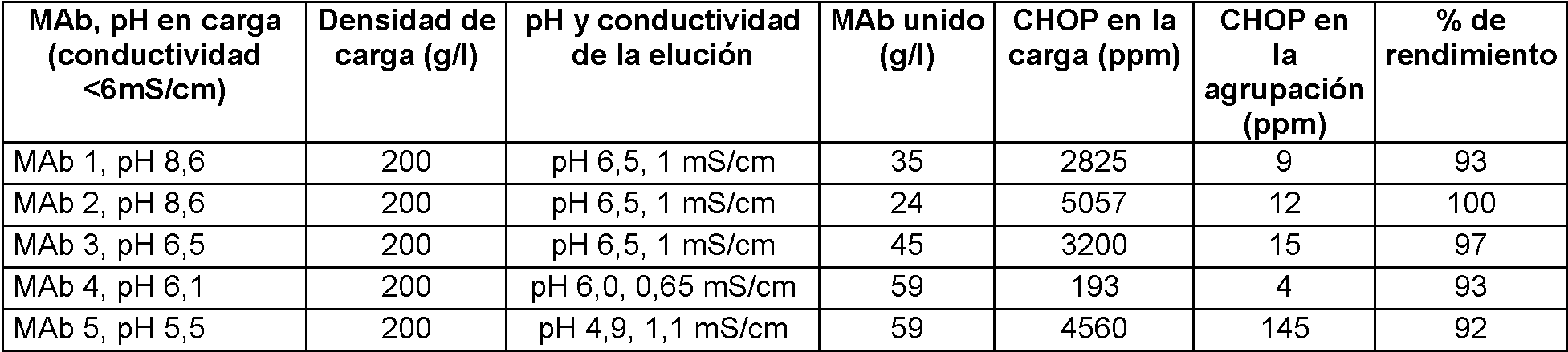
Tabla 12. Aplicabilidad de OEC a otros MAb: % de proteínas HMW

Tabla 13. Aplicabilidad de OEC a otros MAb: % de proteína A lixiviada

* Menos que detectable
Tabla 14. Aplicabilidad de OEC a otros MAb: ADN de CHO

Ejemplo 10. Modos de funcionamiento de cromatografía AEX basadas en valores de Kp
En cromatografía de flujo continuo (F/T), Kp es <0,1 y no hay unión de proteína a la resina. En la cromatografía de reparto débil (WPC), Kp es de 0,1 a 20 y hay un reparto débil entre el producto y los medios de cromatografía. En un modo de unión y elución, el producto se une estrechamente a la resina y el Kp es >100, pero la densidad de carga se limita a la capacidad de unión del producto. Sin embargo, en un modo de sobrecarga y elución de cromatografía (MAb 3), se encontraron condiciones de carga de modo que el Kp del producto y las impurezas era >100 y aunque el producto fluye después de alcanzar su capacidad de unión, las impurezas mantienen la unión a la resina y no avanzan hasta que alcanzan su capacidad de unión, que podría ser superior a la capacidad de unión del producto.
Se encontraron condiciones de elución de modo que el Kp del producto polipeptídico <2. El producto unido se recupera, pero la mayoría de las impurezas permanecen unidas (Kp de impureza >100). Por tanto, este modo permite una buena depuración de impurezas al tiempo que proporciona un rendimiento muy alto (por ejemplo, ~95 %) (Tabla 15). Tabla 15. MAb 3 como un anticuerpo modelo en la resina Capto Adhere
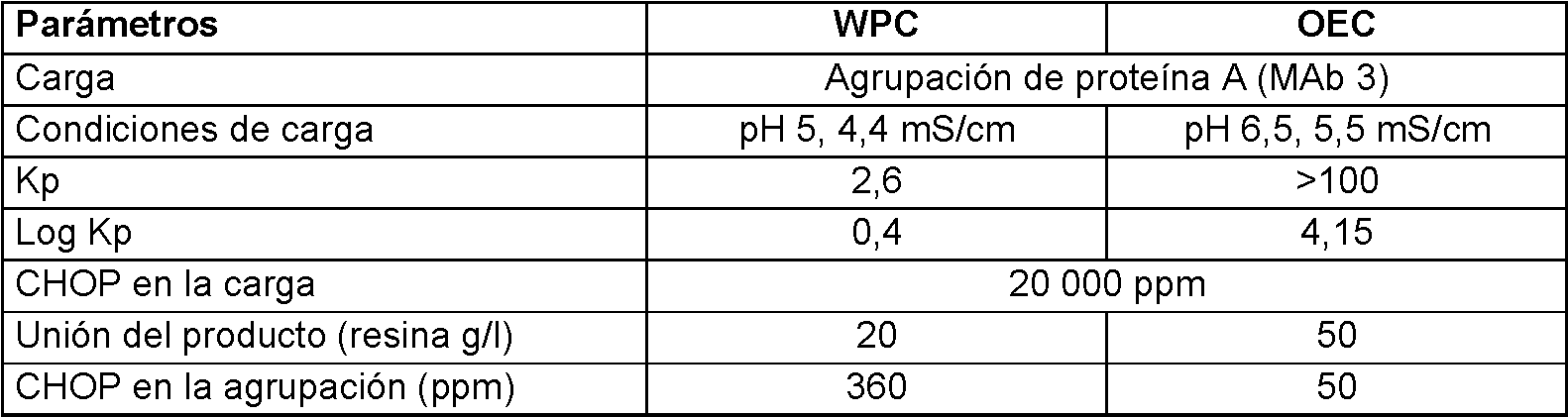

Los cromatogramas de desarrollos cromatográficos en columna en condiciones de reparto débiles (Kp = 2,4) y condiciones de sobrecarga y elución (Kp >100) muestran los efectos del aumento del Kp de mAb en las regiones de avance del producto del cromatograma (figura 9). Condiciones de carga de WPC: pH 5,5, 4,4 mS/cm; condiciones de lavado de WPC: Acetato 20 mM pH 5, 4,4 mS/cm. Condiciones de carga en OEC: pH 6,5, 5,5 mS/cm; condiciones de elución en OEC: MES 20 mM pH 6,5, ~1 mS/cm.
Tabla 16. Análisis de una etapa de refinado de AEX para un MAb que se une a la resina AEX a un flujo típico con las condiciones del procedimiento
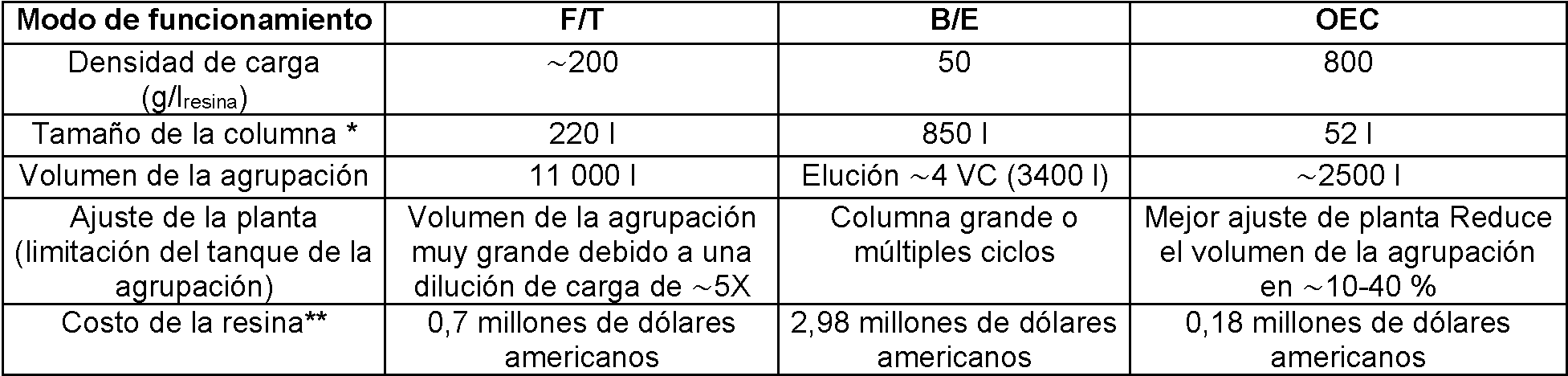
Anticuerpo modelo: MAb 3
* Se supone una recolección de 12500 l a un título de 3,5 g/l.
** Resina CA a 3500 dólares americanos/l
Ejemplo 11. Aplicabilidad de OEC para diferentes resinas
Dependiendo del pI de la molécula, se puede aplicar OEC a las siguientes resinas multimodo (Capto Adhere, QMA, MEP Hypercel, HEA Hypercel, PPA Hypercel, Capto MMC). La unión del producto polipeptídico a la resina Capto Adhere era de naturaleza más hidrófoba y el producto unido se podía eluir de la columna disminuyendo la conductividad del tampón de elución (figura 5). Sin embargo, el modo de unión del producto a la resina y la elución del producto de la resina no se limita a las interacciones hidrófobas y, por lo tanto, el modo de funcionamiento de la OEC se puede usar ampliamente también en otros materiales de cromatografía. La Tabla 17 demuestra que OEC se puede aplicar a otras resinas CEX y resinas IEX. Los tampones de elución eran MES 20 mM a menos que se indicara de otro modo. El potencial de OEC no se limita a las resinas anteriores y actualmente se está evaluando. El análisis de avance del MAb 3 en la resina QMA se muestra en la figura 10. El análisis de avance del MAb 4 en la resina Capto Adhere se muestra en la figura 11. El análisis de avance del MAb 4 en la resina Capto MMC se muestra en la figura 12. El análisis de avance del MAb 3 en la resina Capto Adhere se muestra en la figura 13.
Tabla 17. Aplicabilidad de OEC a diferentes resinas.

** Condiciones de carga para Poros XS MAb 3: pH 5,5, <6 mS/cm; condiciones de elución: Tampón A - acetato 50 mM pH 5,5 ~3 mS/cm, tampón B - acetato 350 mM pH 5,5, ~24 mS/cm, gradiente: 20-85 % sobre 10 VC, agrupamiento: 1-8 VC.
Ejemplo 12. Elución con gradiente en OEC
Otro objetivo del estudio de optimización de la elución fue evaluar el efecto de las condiciones de elución con gradiente en el modo OEC sobre la resina Capto Adhere. La fase de elución se desarrolló para eluir el producto unido (Tabla 18). En un desarrollo cromatográfico de elución con gradiente, se puede variar la fuerza iónica, el pH, la composición y la concentración de la fase móvil basándose en los requerimientos. La Tabla 18 muestra las condiciones de
funcionamiento: las condiciones de carga (pH y conductividad) y de elución (pH y gradiente de conductividad). Todos los datos mostrados en la tabla se obtuvieron a partir de desarrollos cromatográficos cargados a una densidad de carga de 150 g/l con MAb 3. Estos desarrollos cromatográficos se realizaron como estudio de viabilidad para demostrar que la elución con gradiente se puede realizar en modo de cromatografía OEC y se puede optimizar la pendiente de gradiente (concentración de la sal (mM)/volumen de columna). Se puede observar que el % de HMW se reduce en un 38 % en promedio cuando se compara con la carga (Tabla 18). La CHOP se redujo a <20 ppm en una conductividad de 5,5 mS/cm y la mayor conductividad funcionó a 10 mS/cm, lo que dio como resultado CHOP en la agrupación de —150 ppm (Tabla 19).
Tabla 18. Desarrollos cromatográficos de elución con gradiente en OEC: datos de % de HMW

Tabla 19. Desarrollos cromatográficos de elución con gradiente en OEC: Datos de CHOP:

Claims (15)
1. Un procedimiento para purificar un anticuerpo partir de una composición que comprende el anticuerpo y uno o más contaminantes, comprendiendo dicho procedimiento
a) cargar la composición en un material de cromatografía de modo mixto, de intercambio aniónico, de interacción hidrófoba o de afinidad en una cantidad en exceso de la capacidad de unión dinámica del material de cromatografía para el anticuerpo usando un tampón de carga, en el que el coeficiente de reparto del material de cromatografía para el anticuerpo es mayor que 30,
b) eluir el anticuerpo del material de cromatografía en condiciones en las que el uno o más contaminantes permanecen unidos al material de cromatografía usando un tampón de elución, en el que el tampón de elución tiene una conductividad menor que la conductividad del tampón de carga y
c) agrupar fracciones que comprenden el anticuerpo en el efluente de cromatografía de las etapas a) y b).
2. El procedimiento de la reivindicación 1, en el que el anticuerpo es un anticuerpo monoclonal.
3. El procedimiento de la reivindicación 2, en el que el anticuerpo monoclonal es un anticuerpo quimérico, anticuerpo humanizado o anticuerpo humano.
4. El procedimiento de la reivindicación 1, en el que el anticuerpo es un fragmento de unión al antígeno, opcionalmente en el que el fragmento de unión al antígeno es un fragmento Fab, un fragmento Fab', un fragmento F(ab')2, un scFv, un di-scFv, un bi-scFv, un (di, tri)-scFv en tándem, un Fv, un sdAb, un anticuerpo trifuncional, un BiTE, un diacuerpo o un triacuerpo.
5. El procedimiento de una cualquiera de las reivindicaciones 14, en el que el al menos un contaminante es uno cualquiera o más de proteína de ovario de hámster chino (CHOP), una proteína de célula huésped (HCP), proteína A lixiviada, ácidos nucleicos, ADN, variantes de producto, proteína agregada, componente de medio de cultivo celular, gentamicina, fragmento polipeptídico, endotoxina y contaminante vírico.
6. El procedimiento de una cualquiera de las reivindicaciones 1 a 5, en el que el material de cromatografía de intercambio iónico es un material de intercambio catiónico o un material de intercambio aniónico.
7. El procedimiento de una cualquiera de las reivindicaciones 1 a 6, en el que la densidad de carga es mayor que aproximadamente 200 g/l.
8. El procedimiento de una cualquiera de las reivindicaciones 1 a 7, en el que la composición se carga sobre el material de cromatografía a aproximadamente las capacidades de unión dinámica de los materiales de cromatografía para el uno o más contaminantes.
9. El procedimiento de una cualquiera de las reivindicaciones 1 a 8, en el que el tampón de carga tiene una conductividad de aproximadamente 4,0 mS hasta aproximadamente 7,0 mS y el tampón de elución tiene una conductividad de aproximadamente 0,0 mS hasta aproximadamente 7,0 mS.
10. El procedimiento de una cualquiera de las reivindicaciones 1 a 9, en el que el tampón de elución tiene un pH menor que el pH del tampón de carga.
11. El procedimiento de la reivindicación 10, en el que el tampón de carga tiene un pH de aproximadamente 4 hasta aproximadamente 9 y el tampón de elución tiene un pH de aproximadamente 4 hasta aproximadamente 9.
12. El procedimiento de una cualquiera de las reivindicaciones 1 a 9, en el que el tampón de elución tiene un pH mayor que el pH del tampón de carga.
13. El procedimiento de la reivindicación 12, en el que el tampón de carga tiene un pH de aproximadamente 4 hasta aproximadamente 9 y el tampón de elución tiene un pH de aproximadamente 4 hasta aproximadamente 9.
14. El procedimiento de una cualquiera de las reivindicaciones 1 a 13, en el que la composición es un eluyente de una cromatografía de afinidad, una cromatografía de intercambio catiónico, una cromatografía de intercambio aniónico, una cromatografía de modo mixto o una cromatografía de interacción hidrófoba.
15. El procedimiento de la reivindicación 14, en el que la cromatografía de afinidad es una cromatografía de proteína A.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161554898P | 2011-11-02 | 2011-11-02 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| ES2717536T3 true ES2717536T3 (es) | 2019-06-21 |
| ES2717536T5 ES2717536T5 (en) | 2025-02-04 |
Family
ID=48192803
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18206718T Active ES3062963T3 (en) | 2011-11-02 | 2012-11-02 | Overload and elute chromatography |
| ES12845437T Active ES2637423T5 (es) | 2011-11-02 | 2012-11-02 | Cromatografía de sobrecarga y elución |
| ES17157670T Active ES2717536T5 (en) | 2011-11-02 | 2012-11-02 | Overload and elute chromatography |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18206718T Active ES3062963T3 (en) | 2011-11-02 | 2012-11-02 | Overload and elute chromatography |
| ES12845437T Active ES2637423T5 (es) | 2011-11-02 | 2012-11-02 | Cromatografía de sobrecarga y elución |
Country Status (25)
| Country | Link |
|---|---|
| US (3) | US20140301977A1 (es) |
| EP (3) | EP2773438B2 (es) |
| JP (4) | JP6356607B2 (es) |
| KR (1) | KR20140091721A (es) |
| CN (2) | CN107163101B (es) |
| AU (1) | AU2012323995B2 (es) |
| BR (1) | BR112014010406A2 (es) |
| CA (1) | CA2854330A1 (es) |
| DK (2) | DK3257564T4 (es) |
| ES (3) | ES3062963T3 (es) |
| FI (1) | FI3257564T4 (es) |
| HR (2) | HRP20260140T1 (es) |
| HU (2) | HUE035685T2 (es) |
| IL (1) | IL232146A0 (es) |
| LT (1) | LT3257564T (es) |
| MX (1) | MX2014005330A (es) |
| PL (3) | PL3257564T5 (es) |
| PT (1) | PT3257564T (es) |
| RS (1) | RS58472B2 (es) |
| RU (1) | RU2014121884A (es) |
| SG (1) | SG11201402019TA (es) |
| SI (2) | SI3257564T2 (es) |
| TR (1) | TR201903707T4 (es) |
| WO (1) | WO2013067301A1 (es) |
| ZA (1) | ZA201402932B (es) |
Families Citing this family (44)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101619865B1 (ko) | 2010-07-30 | 2016-05-11 | 이엠디 밀리포어 코포레이션 | 크로마토그래피 매질 |
| US9062106B2 (en) | 2011-04-27 | 2015-06-23 | Abbvie Inc. | Methods for controlling the galactosylation profile of recombinantly-expressed proteins |
| HUE035685T2 (en) | 2011-11-02 | 2018-05-28 | Hoffmann La Roche | Overload and elute chromatography |
| WO2013158273A1 (en) | 2012-04-20 | 2013-10-24 | Abbvie Inc. | Methods to modulate c-terminal lysine variant distribution |
| WO2013158279A1 (en) | 2012-04-20 | 2013-10-24 | Abbvie Inc. | Protein purification methods to reduce acidic species |
| US9067990B2 (en) | 2013-03-14 | 2015-06-30 | Abbvie, Inc. | Protein purification using displacement chromatography |
| WO2013176754A1 (en) * | 2012-05-24 | 2013-11-28 | Abbvie Inc. | Novel purification of antibodies using hydrophobic interaction chromatography |
| US9512214B2 (en) | 2012-09-02 | 2016-12-06 | Abbvie, Inc. | Methods to control protein heterogeneity |
| WO2014066471A1 (en) | 2012-10-24 | 2014-05-01 | Genzyme Corporation | Elution of biomolecules from multi-modal resins using mes and mops as mobile phase modifiers |
| HK1207960A1 (en) | 2013-03-12 | 2016-02-19 | Abbvie Inc. | Human antibodies that bind human tnf-alpha and methods of preparing the same |
| US10023608B1 (en) | 2013-03-13 | 2018-07-17 | Amgen Inc. | Protein purification methods to remove impurities |
| US9017687B1 (en) | 2013-10-18 | 2015-04-28 | Abbvie, Inc. | Low acidic species compositions and methods for producing and using the same using displacement chromatography |
| US9499614B2 (en) | 2013-03-14 | 2016-11-22 | Abbvie Inc. | Methods for modulating protein glycosylation profiles of recombinant protein therapeutics using monosaccharides and oligosaccharides |
| US9598667B2 (en) | 2013-10-04 | 2017-03-21 | Abbvie Inc. | Use of metal ions for modulation of protein glycosylation profiles of recombinant proteins |
| US9085618B2 (en) | 2013-10-18 | 2015-07-21 | Abbvie, Inc. | Low acidic species compositions and methods for producing and using the same |
| US9181337B2 (en) | 2013-10-18 | 2015-11-10 | Abbvie, Inc. | Modulated lysine variant species compositions and methods for producing and using the same |
| US20150139988A1 (en) | 2013-11-15 | 2015-05-21 | Abbvie, Inc. | Glycoengineered binding protein compositions |
| JP6326499B2 (ja) | 2013-12-12 | 2018-05-16 | イー・エム・デイー・ミリポア・コーポレイシヨン | アクリルアミド含有フィルターを使用したタンパク質分離 |
| EP3079722B1 (en) * | 2013-12-13 | 2021-03-24 | Novo Nordisk Health Care AG | Method for thioether conjugation of proteins |
| KR20150083689A (ko) * | 2014-01-10 | 2015-07-20 | 삼성전자주식회사 | 항 c-Met 항체 정제 방법 |
| PL3116891T3 (pl) | 2014-03-10 | 2020-07-27 | Richter Gedeon Nyrt. | Oczyszczanie immunoglobuliny z zastosowaniem etapów wstępnego oczyszczania |
| CN106460027B (zh) * | 2014-03-24 | 2020-07-31 | 日东纺绩株式会社 | 使用阳离子性接枝聚合物的目标物分离浓缩方法 |
| US10280195B2 (en) * | 2014-05-28 | 2019-05-07 | Agency For Science, Technology And Research | Virus reduction method |
| RU2017101727A (ru) * | 2014-06-25 | 2018-07-25 | ДжейЭйчЭл БИОТЕК, ИНК. | Способы и реагенты для очистки белков |
| CN104208719B (zh) * | 2014-09-24 | 2017-05-03 | 北京天广实生物技术股份有限公司 | 一种抗体偶联药物的阳离子交换层析纯化方法 |
| EP3230299A1 (en) | 2014-12-08 | 2017-10-18 | EMD Millipore Corporation | Mixed bed ion exchange adsorber |
| HRP20192217T1 (hr) * | 2015-03-06 | 2020-02-21 | F. Hoffmann - La Roche Ag | ULTRA-PROČIŠĆENI DsbA I DsbC TE POSTUPCI NJIHOVE IZRADE I UPORABE |
| CN106290591B (zh) * | 2015-05-14 | 2019-07-26 | 上海医药工业研究院 | 一种庆大霉素C1a的检测方法 |
| GB2539420B (en) * | 2015-06-16 | 2021-01-13 | Cytiva Sweden Ab | Determination of chromatography conditions |
| SI3337812T1 (sl) | 2015-08-21 | 2021-08-31 | F. Hoffmann-La Roche Ag | Postopek za zmanjševanje beljakovin gostiteljske celice v afinitetni kromatografiji |
| EP3337818B1 (en) * | 2015-08-21 | 2026-03-18 | F. Hoffmann-La Roche AG | Affinity chromatography purification with low conductivity wash buffer |
| HRP20250718T1 (hr) * | 2016-08-15 | 2025-08-15 | F. Hoffmann-La Roche Ag | Kromatografski postupak za kvantifikaciju neionskog surfaktanta u sastavu koji sadrži neionski surfaktant i polipeptid |
| GB201702856D0 (en) * | 2017-02-22 | 2017-04-05 | Ge Healthcare Bio Sciences Ab | Method in continuous chromatography |
| US11339377B2 (en) * | 2017-11-08 | 2022-05-24 | Bluesky Immunotherapies Gmbh | SO3 chromatography for use in a method for virus purification |
| US11999975B2 (en) * | 2018-07-04 | 2024-06-04 | Probiogen Ag | Method for purifying an enveloped virus |
| US12306152B2 (en) * | 2018-12-21 | 2025-05-20 | Solventum Intellectual Properties Company | Method for testing a chromatography device used for ion exchange |
| US20220135619A1 (en) | 2019-02-24 | 2022-05-05 | Bristol-Myers Squibb Company | Methods of isolating a protein |
| AR119264A1 (es) | 2019-06-05 | 2021-12-09 | Genentech Inc | Método para reutilización de cromatografía |
| CN111351876A (zh) * | 2020-04-01 | 2020-06-30 | 上海中科新生命生物科技有限公司 | 一种抗体药中宿主细胞蛋白半绝对定量检测方法 |
| US11379651B1 (en) * | 2020-07-06 | 2022-07-05 | Turtl Surf & Immerse Limited | Methods and systems for interactive content creation |
| EP4217383A1 (en) | 2020-09-22 | 2023-08-02 | Bristol-Myers Squibb Company | Methods for producing therapeutic proteins |
| JP7620456B2 (ja) * | 2021-03-15 | 2025-01-23 | 株式会社カネカ | κ鎖を含む抗体の製造方法 |
| EP4408856A4 (en) * | 2021-09-28 | 2025-11-12 | Kashiv Biosciences Llc | IMPROVED PROCESS FOR PROTEIN PURIFICATION |
| EP4423109A4 (en) * | 2021-10-27 | 2025-12-24 | Plasma Tech Llc | COMPOSITIONS AND METHODS FOR ISOLATING PROTEINS |
Family Cites Families (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2413974A1 (fr) | 1978-01-06 | 1979-08-03 | David Bernard | Sechoir pour feuilles imprimees par serigraphie |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US5567610A (en) | 1986-09-04 | 1996-10-22 | Bioinvent International Ab | Method of producing human monoclonal antibodies and kit therefor |
| IL85035A0 (en) | 1987-01-08 | 1988-06-30 | Int Genetic Eng | Polynucleotide molecule,a chimeric antibody with specificity for human b cell surface antigen,a process for the preparation and methods utilizing the same |
| GB8823869D0 (en) | 1988-10-12 | 1988-11-16 | Medical Res Council | Production of antibodies |
| US5175384A (en) | 1988-12-05 | 1992-12-29 | Genpharm International | Transgenic mice depleted in mature t-cells and methods for making transgenic mice |
| EP0402226A1 (en) | 1989-06-06 | 1990-12-12 | Institut National De La Recherche Agronomique | Transformation vectors for yeast yarrowia |
| DE3920358A1 (de) | 1989-06-22 | 1991-01-17 | Behringwerke Ag | Bispezifische und oligospezifische, mono- und oligovalente antikoerperkonstrukte, ihre herstellung und verwendung |
| ES2096590T3 (es) | 1989-06-29 | 1997-03-16 | Medarex Inc | Reactivos biespecificos para la terapia del sida. |
| US5208020A (en) | 1989-10-25 | 1993-05-04 | Immunogen Inc. | Cytotoxic agents comprising maytansinoids and their therapeutic use |
| US5229275A (en) | 1990-04-26 | 1993-07-20 | Akzo N.V. | In-vitro method for producing antigen-specific human monoclonal antibodies |
| WO1992000373A1 (en) | 1990-06-29 | 1992-01-09 | Biosource Genetics Corporation | Melanin production by transformed microorganisms |
| US5571894A (en) | 1991-02-05 | 1996-11-05 | Ciba-Geigy Corporation | Recombinant antibodies specific for a growth factor receptor |
| WO1992020373A1 (en) | 1991-05-14 | 1992-11-26 | Repligen Corporation | Heteroconjugate antibodies for treatment of hiv infection |
| EP1400536A1 (en) | 1991-06-14 | 2004-03-24 | Genentech Inc. | Method for making humanized antibodies |
| MX9204374A (es) | 1991-07-25 | 1993-03-01 | Idec Pharma Corp | Anticuerpo recombinante y metodo para su produccion. |
| US5565332A (en) | 1991-09-23 | 1996-10-15 | Medical Research Council | Production of chimeric antibodies - a combinatorial approach |
| FI941572A7 (fi) | 1991-10-07 | 1994-05-27 | Oncologix Inc | Anti-erbB-2-monoklonaalisten vasta-aineiden yhdistelmä ja käyttömenete lmä |
| WO1993008829A1 (en) | 1991-11-04 | 1993-05-13 | The Regents Of The University Of California | Compositions that mediate killing of hiv-infected cells |
| ATE207080T1 (de) | 1991-11-25 | 2001-11-15 | Enzon Inc | Multivalente antigen-bindende proteine |
| CA2372813A1 (en) | 1992-02-06 | 1993-08-19 | L.L. Houston | Biosynthetic binding protein for cancer marker |
| WO1993016177A1 (en) | 1992-02-11 | 1993-08-19 | Cell Genesys, Inc. | Homogenotization of gene-targeting events |
| US5573905A (en) | 1992-03-30 | 1996-11-12 | The Scripps Research Institute | Encoded combinatorial chemical libraries |
| CA2140280A1 (en) | 1992-08-17 | 1994-03-03 | Avi J. Ashkenazi | Bispecific immunoadhesins |
| DK0752248T3 (da) | 1992-11-13 | 2000-11-13 | Idec Pharma Corp | Terapeutisk anvendelse af kimæriske og radioaktivt mærkede antistoffer mod humant B-lymfocytbegrænset differentieringsantig |
| CA2106612C (en) * | 1993-09-21 | 2001-02-06 | Diana Pliura | Displacement chromatography process |
| US5534615A (en) | 1994-04-25 | 1996-07-09 | Genentech, Inc. | Cardiac hypertrophy factor and uses therefor |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US5641870A (en) | 1995-04-20 | 1997-06-24 | Genentech, Inc. | Low pH hydrophobic interaction chromatography for antibody purification |
| US5712374A (en) | 1995-06-07 | 1998-01-27 | American Cyanamid Company | Method for the preparation of substantiallly monomeric calicheamicin derivative/carrier conjugates |
| MX340738B (es) * | 1996-09-13 | 2016-07-22 | Shire Human Genetic Therapies | Terapia para deficiencia de a-galactosidasa a. |
| DE69738075T2 (de) * | 1996-11-27 | 2008-05-21 | Genentech, Inc., South San Francisco | Affinitätsreinigung von polypeptiden an einer protein a-matrix |
| NZ507557A (en) * | 1998-05-06 | 2003-10-31 | Genentech Inc | Protein purification by ion exchange chromatography |
| IL127127A0 (en) | 1998-11-18 | 1999-09-22 | Peptor Ltd | Small functional units of antibody heavy chain variable regions |
| US8372954B2 (en) | 2000-12-22 | 2013-02-12 | National Research Council Of Canada | Phage display libraries of human VH fragments |
| DK1355936T3 (da) * | 2001-02-01 | 2007-10-29 | Biogen Idec Inc | Polymerkonjugater af neublastin og fremgangsmåder til anvendelse af samme |
| JP2005289809A (ja) | 2001-10-24 | 2005-10-20 | Vlaams Interuniversitair Inst Voor Biotechnologie Vzw (Vib Vzw) | 突然変異重鎖抗体 |
| US20070261962A1 (en) * | 2002-04-02 | 2007-11-15 | Ryszard Gajek | Separation Systems with Charge Mosaic Membrane |
| AU2003301399B2 (en) * | 2002-10-18 | 2006-07-06 | Asahi Kasei Medical Co., Ltd. | Microporous hydrophilic membrane |
| DE602004017726D1 (de) | 2003-06-30 | 2008-12-24 | Domantis Ltd | Pegylierte Single-domain-antikörper (dAb) |
| AU2005285347A1 (en) | 2004-08-19 | 2006-03-23 | Genentech, Inc. | Polypeptide variants with altered effector function |
| DK1791565T3 (en) | 2004-09-23 | 2016-08-01 | Genentech Inc | Cysteingensplejsede antibodies and conjugates |
| EP1869065B1 (en) | 2005-03-11 | 2020-05-06 | Wyeth LLC | A method of weak partitioning chromatography |
| WO2006116064A2 (en) | 2005-04-22 | 2006-11-02 | Waters Investments Limited | Apparatus and methods for mass-spectrometric directed purification of biopolymers |
| WO2008025747A1 (en) * | 2006-08-28 | 2008-03-06 | Ares Trading S.A. | Process for the purification of fc-fusion proteins |
| BRPI0716382B8 (pt) * | 2006-08-28 | 2021-05-25 | Ares Trading Sa | método para reduzir o teor de porções de fc livres em um fluido compreendendo uma proteína contendo fc, e uso de cromatografia de troca catiônica |
| CN101679509A (zh) | 2007-06-01 | 2010-03-24 | 弗·哈夫曼-拉罗切有限公司 | 免疫球蛋白纯化 |
| DE102009005479A1 (de) * | 2009-01-21 | 2010-07-22 | Sartorius Stedim Biotech Gmbh | Vorrichtung und Verfahren für die Stofftrennung |
| JP5614936B2 (ja) * | 2009-02-19 | 2014-10-29 | 旭化成ケミカルズ株式会社 | アニオン交換基が固定された多孔膜を用いた核酸の精製方法 |
| EP2462157B1 (en) * | 2009-08-07 | 2020-06-17 | EMD Millipore Corporation | Methods for purifying a target protein from one or more impurities in a sample |
| WO2011035282A1 (en) | 2009-09-21 | 2011-03-24 | Ge Healthcare Bio-Sciences Ab | Dual capture separation |
| CN102094034A (zh) * | 2009-12-09 | 2011-06-15 | 泰州新生源生物医药有限公司 | 一种重组人Fc融合长效干扰素的纯化工艺 |
| WO2012030512A1 (en) * | 2010-09-03 | 2012-03-08 | Percivia Llc. | Flow-through protein purification process |
| WO2012068134A1 (en) * | 2010-11-15 | 2012-05-24 | Biogen Idec Inc. | Enrichment and concentration of select product isoforms by overloaded bind and elute chromatography |
| HUE035685T2 (en) | 2011-11-02 | 2018-05-28 | Hoffmann La Roche | Overload and elute chromatography |
-
2012
- 2012-11-02 HU HUE12845437A patent/HUE035685T2/en unknown
- 2012-11-02 BR BR112014010406A patent/BR112014010406A2/pt not_active Application Discontinuation
- 2012-11-02 HR HRP20260140TT patent/HRP20260140T1/hr unknown
- 2012-11-02 HR HRP20190485TT patent/HRP20190485T4/hr unknown
- 2012-11-02 TR TR2019/03707T patent/TR201903707T4/tr unknown
- 2012-11-02 CA CA2854330A patent/CA2854330A1/en not_active Abandoned
- 2012-11-02 ES ES18206718T patent/ES3062963T3/es active Active
- 2012-11-02 EP EP12845437.8A patent/EP2773438B2/en active Active
- 2012-11-02 MX MX2014005330A patent/MX2014005330A/es unknown
- 2012-11-02 US US14/355,818 patent/US20140301977A1/en not_active Abandoned
- 2012-11-02 FI FIEP17157670.5T patent/FI3257564T4/fi active
- 2012-11-02 PL PL17157670.5T patent/PL3257564T5/pl unknown
- 2012-11-02 SI SI201231562T patent/SI3257564T2/sl unknown
- 2012-11-02 DK DK17157670.5T patent/DK3257564T4/da active
- 2012-11-02 WO PCT/US2012/063242 patent/WO2013067301A1/en not_active Ceased
- 2012-11-02 CN CN201710173196.8A patent/CN107163101B/zh active Active
- 2012-11-02 JP JP2014540118A patent/JP6356607B2/ja active Active
- 2012-11-02 CN CN201280065729.6A patent/CN104023804B/zh active Active
- 2012-11-02 ES ES12845437T patent/ES2637423T5/es active Active
- 2012-11-02 PT PT17157670T patent/PT3257564T/pt unknown
- 2012-11-02 EP EP18206718.1A patent/EP3527274B1/en active Active
- 2012-11-02 EP EP17157670.5A patent/EP3257564B2/en active Active
- 2012-11-02 RS RS20190358A patent/RS58472B2/sr unknown
- 2012-11-02 SI SI201231031T patent/SI2773438T2/sl unknown
- 2012-11-02 KR KR1020147014357A patent/KR20140091721A/ko not_active Withdrawn
- 2012-11-02 PL PL12845437T patent/PL2773438T5/pl unknown
- 2012-11-02 HU HUE17157670A patent/HUE043755T2/hu unknown
- 2012-11-02 RU RU2014121884/10A patent/RU2014121884A/ru not_active Application Discontinuation
- 2012-11-02 PL PL18206718.1T patent/PL3527274T3/pl unknown
- 2012-11-02 DK DK12845437.8T patent/DK2773438T4/da active
- 2012-11-02 LT LTEP17157670.5T patent/LT3257564T/lt unknown
- 2012-11-02 SG SG11201402019TA patent/SG11201402019TA/en unknown
- 2012-11-02 ES ES17157670T patent/ES2717536T5/es active Active
- 2012-11-02 AU AU2012323995A patent/AU2012323995B2/en active Active
-
2014
- 2014-04-22 IL IL232146A patent/IL232146A0/en unknown
- 2014-04-23 ZA ZA2014/02932A patent/ZA201402932B/en unknown
-
2018
- 2018-04-18 US US15/956,635 patent/US20190023768A1/en not_active Abandoned
- 2018-06-14 JP JP2018113263A patent/JP2018184405A/ja active Pending
-
2019
- 2019-11-20 JP JP2019209674A patent/JP6979442B2/ja active Active
-
2020
- 2020-10-20 US US17/075,238 patent/US20210206836A1/en active Pending
-
2021
- 2021-11-15 JP JP2021185350A patent/JP7203928B2/ja active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2717536T3 (es) | Cromatografía de sobrecarga y elución | |
| ES2991883T3 (es) | Procedimientos de purificación de polipéptidos | |
| TWI870412B (zh) | 過載層析管柱之再生方法 | |
| HK40061242A (en) | A method for regeneration of an overload chromatography column | |
| HK1240254B (zh) | 超载和洗脱层析 | |
| HK1240254A1 (en) | Overload and elute chromatography | |
| HK1201489B (en) | Overload and elute chromatography | |
| NZ624317B2 (en) | Overload and elute chromatography |