ES2716550T3 - Formulación farmacéutica o nutracéutica gastrorresistente que comprende una o más sales de ácido algínico - Google Patents
Formulación farmacéutica o nutracéutica gastrorresistente que comprende una o más sales de ácido algínico Download PDFInfo
- Publication number
- ES2716550T3 ES2716550T3 ES11714754T ES11714754T ES2716550T3 ES 2716550 T3 ES2716550 T3 ES 2716550T3 ES 11714754 T ES11714754 T ES 11714754T ES 11714754 T ES11714754 T ES 11714754T ES 2716550 T3 ES2716550 T3 ES 2716550T3
- Authority
- ES
- Spain
- Prior art keywords
- coating
- suspension
- minutes
- pharmaceutical
- weight
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/284—Organic macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyvinyl pyrrolidone
- A61K9/2846—Poly(meth)acrylates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/28—Dragees; Coated pills or tablets, e.g. with film or compression coating
- A61K9/2806—Coating materials
- A61K9/2833—Organic macromolecular compounds
- A61K9/286—Polysaccharides, e.g. gums; Cyclodextrin
- A61K9/2866—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
Landscapes
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
DESCRIPCIÓN
Formulación farmacéutica o nutracéutica gastrorresistente que comprende una o más sales de ácido algínico
Campo técnico
El estudio de Gürsoy A. y Cevik S., J. Microencapsulation, 2000, vol. 17, n.° 5, 565-575 se centra en las propiedades de microesferas de alginato de sodio de diclofenaco (DNa) y microesferas de alginato de DNa en comprimidos usando diferentes polímeros como aditivos. Las microesferas de alginato de DNa se prepararon mediante el método de emulsificación y diferentes polímeros tales como EUDRAGIT® NE 30 D, EUDRAGIT® RS 30 D o diferentes tipos de celulosas, que se incorporaron en el gel de alginato para controlar la tasa de liberación del fármaco.
El documento US 2007/053698 divulga métodos de administración de liberación mantenida de opioides, incluyendo, aunque sin limitación, hidromorfona y oxicodona, que muestran propiedades mejoradas con respecto a la coingesta con alcohol acuoso. Las composiciones de comprimido de matriz resistente a etanol pueden comprender hidromorfona u oxicodona como ingredientes activos y mezclas de sustancias como EUDRAGIT® RS, lactosa, estearato de Mg, alcohol estearílico cera de carnauba y similares.
El documento US 2007/0104789 A1 describe formulaciones de liberación controlada gastrorresistentes y resistentes a etanol que comprenden hidromorfona. La gastrorresistencia y resistencia a etanol puede usarse en una mezcla, así como el recubrimiento de las formulaciones. El ácido algínico se menciona entre los ejemplos para sustancias gastrorresistentes y resistentes a etanol adecuadas. Pueden prepararse núcleos de pellas o gránulos mediante granulación anhidra, pueden recubrirse con las sustancias gastrorresistentes y resistente a etanol y después pueden llenarse en cápsulas o bolsas o comprimirse en comprimidos con la adición de sustancias auxiliares farmacéuticas o nutracéuticamente aceptables secas.
Problema y solución
Las composiciones farmacéuticas o nutracéuticas se diseñan para liberar el ingrediente activo de una manera de curvas de liberación reproducibles. Esto producirá perfiles de nivel en sangre deseables y fiables que proporcionarán un efecto terapéutico óptimo. Si las concentraciones de nivel en sangre son demasiado bajas, el ingrediente activo no causará un efecto terapéutico suficiente. Si las concentraciones de nivel en sangre son demasiado elevadas, esto puede causar efectos tóxicos. En ambos casos, concentraciones de nivel en sangre no óptimas de un ingrediente activo pueden ser peligrosos para el paciente y, por lo tanto, deben evitarse. Existe un problema de que las relaciones ideales asumidas para la liberación de ingrediente activo durante el diseño de una composición farmacéutica o nutracéutica pueden alterarse por los hábitos de vida generales, desconsideración o por el comportamiento adictivo de los pacientes con respecto al uso de etanol o bebidas que contienen etanol. En estos casos, la forma farmacéutica o nutracéutica que se diseña realmente para un medio exclusivamente acuoso se expone adicionalmente a un medio que contiene etanol de mayor o menor fuerza. Como las autoridades sanitarias como, por ejemplo, la Food and Drug Administration (FDA) de EE. UU. se centra cada vez más en el problema del etanol, la resistencia al etanol puede ser un requisito de registro importante en el futuro cercano.
Como no todos los pacientes son conscientes del riesgo de tomar simultáneamente una forma farmacéutica o nutracéutica de liberación controlada y bebidas que contienen etanol o no siguen o no son capaces de seguir las advertencias apropiadas, avisos o recomendaciones, existe una demanda de composiciones farmacéuticas o nutracéuticas de liberación controlada, especialmente para composiciones farmacéutica o nutracéuticas gastrorresistentes, de modo que su modo de acción se vea afectado lo menos posible por la presencia de etanol.
Las composiciones farmacéuticas o nutracéuticas gastrorresistentes convencionales, estén recubiertas o no recubiertas, habitualmente no son resistentes a alcohol en absoluto. Por lo tanto, un problema de la presente invención fue proporcionar composiciones farmacéuticas o nutracéuticas gastrorresistentes que fueran resistentes frente a la influencia del etanol.
Especialmente, hay un problema para las composiciones gastrorresistentes o de formulación entérica. Estos tipos de formulaciones habitualmente se recubren con una capa de recubrimiento gastrorresistentes (capa de recubrimiento entérica) sobre el núcleo que tiene la función de que la liberación del ingrediente activo farmacéutico o nutracéutico en el estómago, respectivamente a pH 1,2 durante 2 horas de acuerdo con USP no exceda un 10 %, preferiblemente menos de un 5 %. Esta función asegura que los ingredientes activos farmacéuticos o nutracéuticos sensibles a ácido se protegen contra la inactivación y que los ingredientes farmacéuticos o nutracéuticos que pueden irritar la mucosa del estómago no quedan libres en cantidades demasiado elevadas. Por otro lado, en muchos casos, la liberación del ingrediente activo farmacéutico o nutracéutico en el intestino, respectivamente a pH 6,8 durante una hora o menos de acuerdo con el método USP, se diseña para que exceda al menos un 70, 75 % o más. La presencia de etanol en concentraciones de un 5, 10, 20 o 40 % (volumen/volumen) en el líquido gástrico habitualmente da lugar a un aumento en las tasas de liberación en el estómago. Debido al efecto de distribución, el efecto del etanol ingerido es en el intestino, no de tanta importancia como en el estómago. Por tanto, una protección eficaz contra la influencia del etanol debe evitar dicho aumento indeseado de ingrediente activo farmacéutico o nutracéutico en el estómago en
primer lugar. Además, puede desearse que la protección contra la influencia del etano al menos no influya las tasas de liberación comparablemente rápidas a pH 6,8 en medios sin etanol.
El problema se resuelve mediante una composición farmacéutica o nutracéutica gastrorresistente, que comprende un núcleo, que comprende un ingrediente activo farmacéutico o nutracéutico y una capa de recubrimiento gastrorresistente sobre el núcleo, en la que la liberación del ingrediente activo farmacéutico o nutracéutico es de no más de un 15, preferiblemente no más de un 10 % en condiciones in vitro a pH 1,2 durante 2 horas en medio de acuerdo con USP con y sin la adición de un 40 % (v/v) de etanol, en la que la capa de recubrimiento gastrorresistente comprende de un 10 a un 100 % en peso de una o más sales de alginato de amonio con una viscosidad de 30 a 720 cP, preferiblemente de 40 a 450 cP, de una solución acuosa al 1 %.
Descripción detallada de la invención
La invención se refiere a una composición farmacéutica o nutracéutica gastrorresistente que comprende un núcleo, que comprende un ingrediente activo farmacéutico o nutracéutico y una capa de recubrimiento gastrorresistente sobre el núcleo.
Núcleo
El núcleo puede comprender o puede contener una pella de vehículo neutro, por ejemplo, una esfera o bolitas de azúcar, en cuya parte superior se une el ingrediente activo en un aglutinante, tal como lactosa o polivinilpirrolidona. El núcleo, como alternativa, puede comprender una pella en forma de una matriz polimérica en que se une el ingrediente activo. El núcleo puede comprender una pella no recubierta que consiste en un ingrediente activo cristalizado. El núcleo puede ser también un comprimido, un minicomprimido o una cápsula.
La capa de recubrimiento gastrorresistente (capa de recubrimiento entérica) sobre el núcleo tiene la función de que la liberación del ingrediente activo farmacéutico o nutracéutico es de no más de un 15 %, no más de un 10 %, no más de un 8 %, no más de un 5 % en condiciones in vitro a pH 1,2 durante 2 horas en medio de acuerdo con USP con y sin la adición de un 40 % (v/v) de etanol. La USP (USP = Farmacopea de Estados Unidos) que puede usarse preferiblemente es USP32/NF27 (NF = National Formulary (vademécum nacional)), aparato II, método de palas, 50 r.p.m. para comprimidos o método de palas o cesta de 50 a 100 r.p.m., dependiendo de la monografía, para pellas.
Capa de recubrimiento gastrorresistente
La capa de recubrimiento gastrorresistente comprende de un 10 a un 100 % en peso de alginato de amonio y puede comprender además polímeros insolubles en agua y/o excipientes farmacéutica o nutracéuticamente aceptable como se describe en este documento. La capa de recubrimiento gastrorresistente sobre el núcleo puede tener además preferiblemente la función de que la liberación del ingrediente activo farmacéutico o nutracéutico es de no más de un 15 %, no más de un 10 %, no más de un 8 %, no más de un 5 % en condiciones in vitro a pH 1,2 durante 2 horas en medio de acuerdo con USP con y sin la adición de un 5, 10, 20 o 40 % (v/v) de etanol.
Tasas de liberación a pH 6,8
La liberación del ingrediente activo farmacéutico o nutracéutico puede ser de al menos un 50, al menos un 70, preferiblemente al menos un 75 %, muchos más preferiblemente al menos un 80 % en condiciones in vitro a pH 6,8 durante una hora, preferiblemente durante 45 minutos, en un medio tamponado de acuerdo con USP sin la adición de un 40 % (v/v) de etanol.
Cantidades de capa de recubrimiento
El aumento de peso en seco de polímero de la capa de recubrimiento puede ser de al menos 2,5, al menos 3,5, al menos 4, preferiblemente de 4 a 30, preferiblemente de 4 a 20, más preferiblemente de 5 a 18 o muchos más preferiblemente de 10 a 18 mg/cm2 de área superficial. Esto puede correlacionarse con un 2-60 % de aumento de peso seco de polímero respecto al peso del núcleo. En el caso de comprimidos recubiertos, el aumento de peso seco de polímero respecto al peso del núcleo (núcleo de comprimido: aproximadamente 1-25 o 1-10 mm de diámetro o longitud) puede ser de 2-30 %. En el caso de pellas recubiertas, el aumento de peso seco de polímero respecto al peso del núcleo (núcleo de la pella: de 0,1 a 1,5 mm de diámetro) puede ser de un 10-60 %.
Son posibles recubrimientos muy delgados con aumentos de peso de polímero de menos de 4 mg/cm2, pero a veces pueden ser difíciles de lograr o de reproducir. Pueden conseguirse resultados prometedores en este caso especialmente cuando se emplea alginato de potasio como sal de ácido algínico. Sin embargo, en general, se recomiendan recubrimientos de al menos 4 mg/cm2 de aumento de peso de polímero por los autores de la invención y pueden conseguirse fácilmente con todos los tipos de sales de ácido algínico.
Sales de ácido algínico
La capa de recubrimiento gastrorresistente puede comprender, puede comprender esencialmente o puede contener de un 10 a un 100; de un 20 a un 100, de un 30 a un 100, de un 40 a un 100, de un 50 a un 100, preferiblemente de un 60 a un 95, más preferiblemente de un 70 a un 90 % en peso de una o más sales de ácido algínico.
Las sales de ácido algínico pueden seleccionarse de alginato de sodio, alginato de potasio, alginato de magnesio, alginato de litio o alginato de amonio o mezclas de los mismos.
Viscosidad
Las sales de ácido algínico pueden tener una viscosidad de 30 a 720, preferiblemente de 40 a 450, preferiblemente de 40 a 400 o preferiblemente de 50 a 300 centipoises (cP) de una solución acuosa al 1 % (peso/peso),
La metodología de determinación de la viscosidad de una solución polimérica, por ejemplo, una solución de una sal de ácido algínico, es bien conocida para los expertos en la materia. La viscosidad se determina referiblemente de acuerdo con la Farmacopea Europea 7.a edición, capitulo general 2, métodos de análisis, 2.2.8 y 2.2.10 página 27 y siguientes. El ensayo se realiza usando un viscosímetro de husillo.
La viscosidad de una solución de alginato al 1 % puede determinarse añadiendo 3 g de producto a 250 ml de agua destilada en un vaso de precipitados mientras se agita a 800 r.p.m. usando un agitador suspendido. Después, se añaden 47 ml de agua adicionales con aclarado de las paredes del vaso de precipitados. Después de agitar durante 2 horas y obtener una solución completa, se mide la viscosidad usando un modelo LV del viscosímetro Brookfield a 25 °C (77 °F) a 60 r.p.m. con husillo n.° 2 para muestras con una viscosidad de más de 100 cP y a 60 r.p.m. con husillo n.° 1 para muestras con viscosidad de menos de 100 cP. Como el peso de agua es casi exactamente un 1 g/ml incluso a 25 °C, "peso/peso" se considera igual o idéntico a "peso/volumen" en el sentido de la invención. Diferencias marginales teóricamente posibles se consideran insignificantes.
Adición de polímeros adicionales a la capa de recubrimiento gastrorresistente
La capa de recubrimiento gastrorresistente puede comprender, comprender esencialmente o contener opcionalmente de un 0 a un 400, de un 0 a un 300, de un 0 a un 200, de un 0 a un 100, de un 0 a un 70, de un 0 a un 50, preferiblemente de un 5 a un 80, de un 5 a un 40 o mucho más preferiblemente de un 15 a un 60 o de un 15 a un 30 % en peso de uno o más polímeros insolubles en agua o uno o más polímeros celulósicos o mezclas de los mismos basándose en el peso de la una o más sales de ácidos algínico contenidas.
El uno o más polímeros insolubles en agua o uno o más polímeros celulósicos pueden contener preferiblemente no más de un 12 % en peso de residuos monoméricos con grupos laterales iónicos, preferiblemente no más de un 12 % en peso de residuos monoméricos con grupos laterales catiónicos.
El uno o más polímeros insolubles en agua o uno o más polímeros celulósicos pueden contener preferiblemente menos de un 5 % en peso, preferiblemente no más de un 2 % en peso, más preferiblemente no más de un 1 o un 0,05 a un 1 % en peso, de residuos monoméricos con grupos laterales aniónicos.
Polímeros insolubles en agua
Los polímeros insolubles en agua, en el sentido de la invención son polímeros que no se disuelven en agua o únicamente se pueden hinchar en agua sobre el intervalo completo de pH 1-14. Los polímeros insolubles en agua pueden ser al mismo tiempo polímeros que contienen no más de un 12 % de residuos monoméricos con grupos secundarios laterales iónicos como, por ejemplo, polímeros EUDRAGIT® NE/NM o EUDRAGIT® RL/RS.
Otros tipos de polímeros insolubles en agua, en el sentido de la invención, pueden ser copolímeros de vinilo como poli(acetato de vinilo), incluyendo derivados de poli(acetato de vinilo). El poli(acetato de vinilo) puede estar presente en forma de una dispersión. Un ejemplo es el tipo Kollicoat® SR 30 D (BASF), dispersión de poli(acetato de vinilo), estabilizada con povidona y laurilsulfato de Na.
Los polímeros insolubles en agua muy preferiblemente pertenecen al grupo de copolímeros de (met)acrilato.
Polímeros de tipo EUDRAGIT® NE 30D/EUDRAGIT® NM 30D
La capa de recubrimiento gastrorresistente puede comprender un copolímeros insoluble en agua que es un copolímero compuesto de unidades polimerizadas de radicales libres de más de un 95 % en peso, en particular hasta un grado de al menos un 98 % en peso, preferiblemente hasta un grado de la menos un 99 % en peso, en particular hasta un grado de al menos un 99 % en peso, más preferiblemente hasta un grado de un 100 % en peso, de monómeros de (met)acrilato con radicales neutros, especialmente radicales alquilo C1 a C4. Estos tipos de polímeros no se disuelven en agua o solamente pueden hincharse en agua sobre el intervalo completo de pH 1-14.
Monómeros de (met)acrilato adecuados con radicales neutros son, por ejemplo, metacrilato de metilo, metacrilato de etilo, metacrilato de butilo, acrilato de metilo, acrilato de etilo, acrilato de butilo. Se da preferencia a metacrilato de metilo, acrilato de etilo y acrilato de metilo.
Los monómeros de metacrilato con radicales aniónicos, por ejemplo, ácido acrílico y/o ácido metacrílico, pueden estar presentes en pequeñas cantidades de menos de 5 % en peso, preferiblemente no más de un 2 % en peso, más preferiblemente no más de 1 o un 0,05 a un 1 % en peso.
Ejemplos adecuados son copolímeros de (met)acrilato neutro o casi neutros compuestos de un 20 a un 40 % en peso de acrilato de etilo, de un 60 a un 80 % en peso de metacrilato de metilo y de un 0 a menos de un 5 % en peso, preferiblemente de un 0 a un 2 o de un 0,05 a un 1 % en peso de ácido metacrílico o cualquier ácido metacrílico (de tipo EUDRAGIT® NE 30D o EUDRAGIT® NM 30D).
EUDRAGIT® NE 30D y EUDRAGIT® NM 30D son dispersiones que contienen un 30 % en peso de copolímeros compuestos de unidades polimerizadas de radicales libres de un 30 % en peso de acrilato de etilo y un 70 % en peso de metacrilato de metilo.
Se da preferencia a copolímeros de acrilato de metilo neutros o esencialmente neutros, que, de acuerdo con el documento WO 01/68767, se han preparado como dispersiones usando un 1-10 % en peso de un emulsionante no iónico que tiene un valor de HLB de 15,2 a 17,3. El último ofrece la ventaja de que no hay separación de fases con formación de estructuras cristalinas por el emulsionante (de tipo EUDRAGIT® NM 30D).
De acuerdo con el documento EP 1571164 A2, también pueden prepararse copolímeros de (met)acrilato, prácticamente neutros correspondientes con pequeñas proporciones de un 0,05 a un 1 % en peso de ácidos carboxílicos C3-C8 monoolefínicamente insaturados, sin embargo, por polimerización por emulsión en presencia de cantidades comparativamente pequeñas de emulsionantes aniónicos, por ejemplo, de un 0,001 a un 1 % en peso.
Polímeros de tipo EUDRAGIT® RL/RS
La capa de recubrimiento gastrorresistente puede comprender un copolímero insoluble en agua que es un copolímero compuesto de unidades polimerizadas de radicales libres de una 85 a un 98 % en peso de ésteres alquilo C1 a C4 polimerizados de radicales libres de ácidos acrílico o metacrílico y un 15 a un 2 % en peso de monómeros de (met)acrilato con un grupo amino cuaternario en el radical alquilo. Estos tipos de polímeros no se disuelven en agua y solamente se pueden hinchar en agua sobre el intervalo completo de pH 1-14.
Polímeros celulósicos
Los polímeros adecuados también pueden pertenecer al grupo de polímeros celulósicos, preferiblemente al grupo de celulosas solubles en agua. El polímero celulósico es preferiblemente una celulosa soluble en agua. Un polímero celulósico adecuado es hidroxipropilmetilcelulosa (HPMC).
Ingrediente activo farmacéutico o nutracéutico
Nutracéuticos
La invención es preferiblemente útil para formas galénicas nutracéuticas. Los nutracéuticos pueden definirse como extractos de alimentos que se reivindica que tienen efectos médicos sobre la salud humana. El nutracéutico está contenido habitualmente en un formato médico tal como cápsula, comprimido o polvo en una dosis prescrita. Ejemplos para nutracéuticos son resveratrol de productos de uva como antioxidante, productos de fibra de la dieta solubles, tales como cáscaras de semilla de psilio para reducir la hipercolesterolemia, brécol (sulfano) como preservante del cáncer y soja o trébol (isoflavonoides) para mejorar la salud arterial. Otros ejemplos de nutracéuticos son flavonoides, antioxidantes, ácido alfa-linoleico de semilla de lino, beta-caroteno de pétalos de caléndula o antocianinas de bayas. A veces la expresión neutracéuticos se usa como sinónimo de nutracéuticos.
La composición farmacéutica o nutracéutica gastrorresistente comprende un núcleo, que comprende un ingrediente activo farmacéutico o nutracéutico. El ingrediente activo farmacéutico o nutracéutico puede ser un ingrediente activo farmacéutico o nutracéutico que puede inactivarse bajo la influencia de los líquidos gástricos a pH 1,2 o un ingrediente activo farmacéutico o nutracéutico que puede irritar la mucosa estomacal cuando queda libre en el estómago.
Ingredientes activos farmacéuticos
La invención también es útil preferiblemente para formas galénicas farmacéuticas con recubrimiento entérico.
Las clases terapéuticas y químicas de fármacos usadas en formas galénicas farmacéuticas de recubrimiento entérico son, por ejemplo, analgésicos, antibióticos o antiinfecciosos, anticuerpos, antiepilépticos, antígenos de
plantas, antirreumáticos, betabloqueantes, derivados de bencimidazol, bloqueantes beta, fármacos cardiovasculares, quimioterápicos, fármacos para el SNC, glucósidos de digitalis, fármacos gastrointestinales, por ejemplo, inhibidores de la bomba de protones, enzimas, hormonas, extractos naturales líquidos o sólidos, oligonucleótidos, proteínas de hormonas peptídicas, bacterias terapéuticas, péptidos, proteínas, inhibidores de la bomba de protones, sal(metálica), por ejemplo, aspartatos, cloruros, ortatos, fármacos para urología, vacunas.
Ejemplos de fármacos, que son inestables en ácido, que irritan o necesitan liberación controlada, pueden ser: Acamprosat, aescina, amilasa, ácido acetilsalicílico, adrenalina, ácido 5-amino salicílico, aureomicina, bacitracina, balsalacina, beta-caroteno, bicalutamida bisacodilo, bromelaína, bromelina, budesónida, calcitonina, carbamacipina, carboplatino, cefalosporinas, cetrorelix, claritromicina, cloromicetina, cimetidina, cisaprida, cladribina, cloracepato, cromalina, 1-desaminocisteina-8-D-arginina-vasopresina, deramciclano, detirelix, dexlansoprazol, diclofenaco, didanosina, digitoxina y otro glucósidos de digitalis, dihidroestreptomicina, dimeticona, divalproex, drospirenona, duloxetina, enzimas, eritromicina, esomeprazol, estrógenos, etopósido, famotidina, fluoruros, aceite de ajo, glucagón, factor estimulador de colonias de granulocitos (G-CSF), heparina, hidrocortisona, hormona del crecimiento humana (hGH), ibuprofeno, ilaprazol, insulina, interferón, interleucina, intrón A, cetoprofeno, lansoprazol, lipasa leuprolidacetat, ácido lipoico, litio, quinina, memantina, mesalacina, metenamina, milamelina, minerales, minoprazol, naproxeno, natamicina, nitrofurantiona, novobiocina, olsalacina, omeprazol, orotatos, pancreatina, pantoprazol, hormona paratiroidea, paroxetina, penicilina, perprazol, pindolol, polimixina, potasio, pravastatina, prednisona, preglumetacina progabide, prosomatostatina, proteasa, quinapril, rabeprazol, ranitidina, ranolacina, reboxetina, rutosid, somatostatina, estreptomicina, subtilina, sulfasalacina, sulfanilamida, tamsulosina, tenatoprazol, tripsina, ácido valproico, vasopresina, vitaminas, cinc, incluyendo sus sales, derivados, polimorfos, isomorfos o cualquier tipo de mezclas o combinaciones de los mismos.
Composición farmacéutica o nutracéutica gastrorresistente
La composición farmacéutica o nutracéutica gastrorresistente puede ser un comprimido recubierto, un minicomprimido recubierto, una pella recubierta, un gránulo recubierto, un sobrecito, una cápsula, rellena de pellas recubiertas o de polvo o de gránulos o una cápsula recubierta, rellena de pellas recubiertas o de polvo o de gránulos.
La expresión comprimido recubierto incluye comprimidos que contienen pellas o comprimidos por compresión y es bien conocida para los expertos en la materia. Dicho comprimido puede tener un tamaño de aproximadamente 5 a 25 mm, por ejemplo. Habitualmente, pluralidades definidas de pellas pequeñas que contienen ingrediente activo se comprimen en el mismo junto con excipientes aglutinantes para dar la forma de comprimido bien conocida. Después de la ingesta oral y el contacto con el líquido corporal, la forma del comprimido se altera y las pellas quedan libres. El comprimido por compresión combina la ventaja de la forma monodosis para ingesta con las ventajas de un múltiples formas, por ejemplo, la precisión de la dosificación.
La expresión minicomprimido recubierto es bien conocida para los expertos en la materia. Un minicomprimido es más pequeño que el comprimido tradicional y puede tener un tamaño de aproximadamente 1 a 4 mm. El minicomprimido es, como una pella, una forma monodosis a usar en múltiples dosificaciones. En comparación con las pellas, que pueden estar en el mismo tamaño, los minicomprimidos habitualmente tienen la ventaja de tener superficies más regulares que pueden recubrirse de forma más precisa y más uniforma. Los minicomprimidos pueden proporcionarse encerrados en cápsulas, tales como cápsulas de gelatina. Dichas cápsulas se alteran después de la ingesta oral y el contacto con los líquidos gástricos o intestinales y los minicomprimidos quedan libres. Otra aplicación de los minicomprimidos es el ajuste fino individual de la dosificación de ingrediente activo. En este caso, el paciente puede ingerir una cantidad definida de minicomprimidos directamente que coincide con la gravedad de la enfermedad a curar, pero también a su peso corporal individual. Un minicomprimido es diferente del comprimido por compresión que contiene pellas como se analiza anteriormente.
El término sobrecito es bien conocido para los expertos en la materia. Se refiere a un envase precintado pequeño que contiene el ingrediente activo a menudo en forma de líquido que contiene pellas o también en forma de pella o polvo seco. El propio sobrecito es únicamente la forma de envase y no está destinado a ingerirse. El contenido del sobrecito puede disolverse en agua o como una característica ventajosa puede remojarse o ingerirse directamente sin líquido adicional. La última es una característica ventajosa para el paciente cuando la forma galénica debe ingerirse en una situación donde no hay agua disponible. El sobrecito es una forma galénica alternativa a los comprimidos, minicomprimidos o cápsulas.
Las pellas recubiertas pueden rellenarse en una cápsula, por ejemplo, cápsula de gelatina o HPMC. Una cápsula que contiene pellas también puede recubrirse con la capa de recubrimiento entérico de acuerdo con la invención.
La composición farmacéutica o nutracéutica gastrorresistente está presente preferiblemente en forma de una solución, suspensión o dispersión de recubrimiento acuosa. El contenido de peso seco de la solución, suspensión o dispersión puede estar en el intervalo de un 10 a un 50, preferiblemente de un 15 a un 35 %.
Excipientes farmacéutica o nutracéuticamente aceptables
Una composición farmacéutica o nutracéutica gastrorresistente puede comprender opcionalmente en la capa de recubrimiento gastrorresistente hasta un 90, hasta un 80, hasta un 70, hasta un 60, hasta un 50, hasta un 40, hasta un 30, hasta un 20 o hasta un 10 % en peso de excipientes farmacéutica o nutracéuticamente aceptables. Los excipientes farmacéutica o nutracéuticamente aceptables son diferentes de las sales de ácido algínico y diferentes de los polímeros insolubles en agua o los polímeros celulósicos mencionados anteriormente y pueden seleccionarse del grupo de antioxidantes, abrillantadores, agentes aglutinantes, agentes aromatizantes, auxiliares de flujo, fragancias, emolientes, agentes que promueven la penetración, polímeros (diferentes de las sales de ácido algínico y diferentes de los polímeros insolubles en agua o los polímeros celulósicos mencionados anteriormente; los polímeros excipientes pueden ser, por ejemplo, disgregantes como polivinilpirrolidona reticulada), pigmentos, plastificantes, agentes que forman poros o estabilizantes o combinaciones de los mismos.
Proceso para producir una forma farmacéutica o nutracéutica
La invención se refiere además a un proceso para producir la forma farmacéutica o nutracéutica de acuerdo con la invención formando el núcleo que comprende el ingrediente activo por compresión directa, compresión de gránulos secos, húmedos o sinterizados, extrusión y posterior redondeo, granulación en húmedo o seco y peletización directa o aglutinando polvos (estratificación de polvos) en microesferas sin ingrediente activo o núcleos neutros (bolitas) o partículas que contienen ingrediente activo y aplicando el recubrimiento polimérico en forma de una dispersión acuosa en un proceso de pulverización o por granulación por pulverización de lecho fluido sobre el núcleo.
Recubrimiento superior y subrecubrimientos
La composición farmacéutica o nutracéutica gastrorresistente de acuerdo con la invención puede recubrirse además con un subrecubrimiento o un recubrimiento superior o ambos.
Un subrecubrimiento puede estar ubicado entre el núcleo y la capa de recubrimiento gastrorresistente (entérica). Un subrecubrimiento puede tener la función de separar sustancias del núcleo de las sustancias de la capa de control que pueden ser incompatibles entre sí. El subrecubrimiento no tiene esencialmente influencia sobre las características de liberación de ingrediente activo. Un subrecubrimiento preferiblemente es esencialmente soluble en agua, por ejemplo, puede consistir en sustancias como hidroxipropilmetilcelulosa (HPMC) como formador de película. El grosor promedio de la capa de subrecubrimiento es muy delgado, por ejemplo, no más de 15 gm, preferiblemente no más de 10 gm.
Un recubrimiento superior preferiblemente también es esencialmente soluble en agua. Un recubrimiento superior puede tener la función de colorear la forma farmacéutica o nutracéutica o protegerla de las influencias ambientales, por ejemplo, de la humedad durante el almacenamiento. El recubrimiento superior puede consistir en un aglutinante, por ejemplo, un polímero soluble en agua como un polisacárido HPMC, o un compuesto de azúcar como sacarosa. El recubrimiento superior puede contener además excipientes farmacéuticos o nutracéuticos como pigmentos o emolientes en altas cantidades. El recubrimiento superior no tiene esencialmente influencia sobre las características de liberación.
Las expresiones subrecubrimiento y recubrimiento superior son bien conocidas para los expertos en la materia.
Pellas/comprimidos
Como una estimación burda, las pellas recubiertas pueden tener un tamaño en el intervalo de 50 a 1000 gm (diámetro promedio), mientras que los comprimidos recubiertos pueden tener un tamaño en el intervalo por encima de 1000 gm hasta 25 mm (diámetro o longitud). Como norma, se puede decir que cuanto más pequeño sea el tamaño de los núcleos de pella mayor, será el aumento de peso del recubrimiento de la pella necesario. Esto se debe al área superficial comparativamente mayor de las pellas en comparación con los comprimidos.
En recubrimientos para pellas, también pueden usarse cantidades comparativamente altas de excipientes, preferiblemente talco, en contraste con el recubrimiento para comprimidos. Pueden usarse cantidades de más de un 50 y hasta un 250 % en peso en relación con la cantidad de la sal de ácido algínico, que puede corresponder a más de un 50 y hasta un 90 % en peso de la capa de recubrimiento.
En recubrimientos de comprimidos, pueden usarse cantidades comparativamente bajas de excipientes, preferiblemente talco, pero también otros excipientes, en contraste con las pellas. Pueden usarse cantidades de más de un 50 a un 100 % en peso en relación con la cantidad de sal de ácido algínico que puede corresponder hasta un 50 % en peso de la capa de recubrimiento.
Alginato de amonio (alginato de NH4)
Una realización adicional de la presente invención es el uso de alginato de amonio como sustituto para o en combinación con alginato de sodio en situaciones donde hay cantidades considerables de iones calcio presentes en
el alimento ingerido junto con la composición farmacéutica o nutracéutica gastrorresistente de la invención. Esto puede suceder cuando se consumen productos lácteos tales como leche o yogur. Sorprendentemente, se ha descubierto que la presencia de iones calcio en tampón USP pH 6,8 casi no tiene influencia sobre la tasa de liberación de recubrimientos que se usa alginato de amonio como sal alginato. Sin embargo, la tasa de liberación a pH 6,8 de los recubrimientos en que usa alginato de sodio como sal alginato desciende casi totalmente (véanse los ejemplos 32-37).
Recubrimiento
Se aplican suspensiones de recubrimiento por procesos de recubrimiento por pulverización siguiendo procesos conocidos. Como norma, las composiciones recubiertas se curan a temperaturas elevadas, por ejemplo, 24 horas a 40 °C o 60 °C después del recubrimiento por pulverización para proporcionar una funcionalidad reproducible y estable. Sorprendentemente, se descubrió que los recubrimientos de alginato puro no necesitan ningún curado para alcanzar una funcionalidad reproducible y estable. Por lo tanto, no se curaron los recubrimientos de alginato puro en los ejemplos. Sin embargo, cuando se mezcla alginato con uno o más polímeros insolubles en agua o uno o más polímeros celulósicos, las formulaciones recubiertas tenían que curarse después del proceso de pulverización. Más de una capa de recubrimiento gastrorresistente (recubrimientos de capa doble o de múltiples capas) En determinadas realizaciones, puede ser útil tener dos o más capas de recubrimiento gastrorresistentes diferentes. En el ejemplo 40, un recubrimiento de pella muestra una capa de recubrimiento interior con un alto contenido de talco y un disgregante. El recubrimiento exterior tiene un contenido alto de talco, pero no disgregante. En este caso, el recubrimiento interior acelera la liberación del fármaco en tampón a pH 6,8 sin afectar a las propiedades entéricas a pH 1,2. El recubrimiento exterior en solitario no mostraría esta combinación de propiedades.
E m l 1 2 r m n


Ejemplos
Todos los ejemplos que no comprenden alginato de amonio se consideran ejemplos de referencia.
Todos los excipientes cumplen las especificaciones de la farmacopea o equivalentes.
Preparación de núcleos de comprimido
Fórmula para preparación de comprimidos:
1) Todos los ingredientes se tamizaron a través de tamiz de malla 40 (425 micrómetros) y se presaron de forma precisa.
2) La celulosa microcristalina (Avicel® 101 y Avicel® 200) y Povidona K-30 se mezclaron juntas en una bolsa de polietileno.
3) El fármaco tamizado se mezcló gradualmente con la mezcla anterior.
4) Se añadió talco a la mezcla anterior y se mezcló durante 5 minutos en una mezcladora de cono.
5) Se comprobó la pérdida en secado de la mezcla en una balanza de humedad. (Si la LOD era de más de un 2 % p/p, entonces la mezcla debe secarse en un secador de bandeja a 40 °C hasta que la LOD esté por debajo de un 2 % p/p.)
6) La mezcla de la etapa 4 se lubricó con estearato de magnesio en una mezcladora de cono durante 2 minutos.
7) La mezcla se comprimió en una máquina de compresión giratoria de 16 estaciones usando punzones cóncavos convencionales circulares de 11 mm.
Parámetros de comprimido:

Proceso de recubrimiento


Ácido algínico y sales usadas en los ejemplos

Metodología analítica para comprimidos
1. Ensayo de disolución:
a) Fase de ácido
Aparato USP tipo II
Medio de disolución HCl 0,1 N
Volumen del medio 750 ml
Velocidad 50 r.p.m.
Temperatura 37 °C ± 0,5 °C
Volumen de extracción 10 ml
Tiempo 120 minutos
b) Fase de tampón
Aparato: USP tipo II
Medio de disolución Medio de fase de tampón pH 6,8 Volumen del medio 1000 ml
Velocidad 50 r.p.m.
Temperatura 37 °C ± 0,5 °C
Volumen de extracción 10 ml
Puntos temporales 60 minutos (30, 45, 60 minutos) Medio de fase de tampón:
Se pesan de forma precisa y se transfieren 19,01 g de fosfato de trisodio y 6,37 ml de ácido clorhídrico concentrado a 1000 ml de agua. Se disuelven y se completa el volumen hasta un litro y se mezcla bien. Se ajusta el pH a 6,8 ± 0,05 usando NaOH 2 N o HCl 2 N.
2. Método de detección-HPLC
Condiciones cromatográficas
Columna : Agilent Zorbax Eclipse XDB C8 column, 150 x 4,6 mm, 5 gm o equivalente Fase móvil : Agua:acetonitrilo (80:20)
Longitud de onda : 273 nm
Temperatura de la columna : 30 °C
Volumen de inyección : 10 gl
Caudal : 1 ml/minuto
Tiempo de ejecución : 5 minutos
Preparación de patrón:
Preparación de solución madre de patrón: Se pesan de forma precisa y se transfieren aproximadamente 50 mg de patrón de cafeína en un matraz volumétrico de 100 ml. Se disuelven y se completa el volumen con agua.
Patrón de la fase de ácido: Se diluyen 5 ml de solución madre a 50 ml con HCl 0,1 N.
Patrón de la fase de tampón: Se diluyen 5 ml de solución madre a 50 ml con medio de fase de tampón.
Procedimiento:
Fase de ácido: Se pesa y se transfiere comprimido de cafeína en seis tarros de disolución diferentes y después se realiza el ensayo de disolución según los parámetros dados en el método anterior (fase de ácido). Después de 2 h se retiran 10 ml de alícuota y se analizan como la solución de muestra de fase de ácido.
Fase de tampón: Se transfiere comprimido a medio de fase de tampón pH 6,8. Se continúa el ensayo de disolución según los parámetros dados en el método anterior (fase de tampón). Se filtran las alícuotas de cada intervalo a través de un filtro de jeringa de membrana de nailon de 0,45 gm desechando los primeros ml del filtrado. Se analiza la solución de muestra de fase de tampón.
3. Método de disgregación
1) Fase de ácido
Aparato Medidor de disgregación
Medio de disolución SGF con pepsina, USP/NF
Volumen del medio 900 ml
Temperatura 37 °C ± 0,5 °C
Tiempo 60 minutos
2) Fase de tampón
Aparato : Medidor de disgregación
Medio de disolución : SIF con pancreatina, USP/NF
Volumen del medio : 900 ml
Temperatura : 37 °C ± 0,5 °C
Tiempo : 60 minutos
Preparación de líquido gástrico simulado (SGF) con pepsina, USP/NF
Se pesan de forma precisa y se transfieren 2,0 g de cloruro de sodio y 2,67 g de pepsina purificada (que se obtiene de la mucosa estomacal porcina, con una actividad de 3000 unidades por mg de proteína) a 500 ml de agua. Se disuelven las sales y a estas se añaden 7,0 ml de ácido clorhídrico y se completa el volumen hasta 1000 ml con agua. Esta solución de ensayo tiene un pH de aproximadamente 1,2.
Preparación de líquido intestinal simulado (SIF) con pancreatina, USP/NF:
Se pesan de forma precisa y se transfieren 6,8 g de fosfato de potasio monobásico y 0,61 g de hidróxido de sodio y se disuelven en 500 ml de agua. A esto se añaden 10 g de pancreatina, se mezclan y se ajusta la solución resultante con hidróxido de sodio 0,2 N o ácido clorhídrico 0,2 N hasta un pH de 6,8 ± 0,1. Se diluye con agua hasta 1000 ml.
Estudio de alcohol (etanol):
1) Parámetros de disolución
Aparato : USP de tipo II
Medio de disolución : Alcohol (etanol) al 5 %, 10 %, 20 % y 40 % en HCl 0,1 N
Volumen del medio : 750 ml
Velocidad : 50 r.p.m.
Temperatura : 37 °C ± 0,5 °C
Volumen de extracción : 10 ml
Tiempo : 2 horas
Método de detección-HPLC: igual que se da para el análisis.
Preparación de patrón:
Preparación de solución madre de patrón: Se pesan de forma precisa y se transfieren aproximadamente 50 mg de patrón de cafeína en un matraz volumétrico de 100 ml. Se disuelven y se completa el volumen con agua.
Patrón de alcohol: Se diluyen 5 ml de solución madre a 50 ml con medio alcohólico respectivo.
Procedimiento:
Se pesa y se transfiere comprimido de cafeína en seis tarros de disolución diferentes y después se realiza el ensayo de disolución según los parámetros dados en el método anterior (alcohol). Después de 2 h se retiran 10 ml de alícuota y se analizan como solución de muestra de alcohol. Se filtran las alícuotas con filtro de jeringa de membrana de nailon de 0,45 pm descartando los primeros ml del filtrado. Se analiza la solución de muestra de alcohol.
Metodología analítica para pellas de lansoprazol
1. Ensayo de disolución
a) Fase de ácido
Aparato : USP de tipo II
Medio de disolución : HCl 0,1 N
Volumen del medio : 500 ml
Velocidad : 75 r.p.m.
Temperatura : 37 °C ± 0,5 °C
Volumen de extracción : 25 ml
Tiempo : 60 minutos
Longitud de onda de detección : 306 nm
b) Fase de tampón
Aparato : USP de tipo II
Medio de disolución : Medio de fase de tampón pH 6,8 (remítase a la nota a continuación) Volumen del medio : 900 ml
Velocidad : 75 r.p.m.
Temperatura : 37 °C ± 0,5 °C
Volumen de extracción : 10 ml
Puntos temporales : 75 minutos (30, 45, 60, 75 minutos)
Longitud de onda de detección : Diferencia entre la absorbancia a 286 nm y 650 nm
Medio de fase de tampón:
El medio de fase de tampón es una mezcla de medio de fase de ácido (475 ml) y concentrado de tampón fosfato (425 ml) con pH ajustado a 6,8.
Preparación de concentrado de tampón fosfato:
Se pesan de forma preciso 16,3 g de fosfato de sodio monobásico, 7,05 g de hidróxido de sodio, 3,0 g de dodecilsulfato de sodio y se disuelve en agua y se completa el volumen hasta un litro y se mezcla bien.
Procedimiento:
Fase de ácido: Se pesan y se transfieren pellas de lansoprazol (equivalente a 30 mg) en seis tarros de disolución diferentes y después se realiza el ensayo de disolución según los parámetros dados en el método anterior (fase de ácido). Después de 1 h, se retiran 25 ml de alícuota y se analizan como la solución de muestra de fase de ácido.
Fase de tampón: Se añaden 425 ml de concentrado de tampón fosfato al medio de fase de ácido (fase de tampón, esto proporcionará un total de 900 ml de medio de pH 6,8). Se continúa el ensayo de disolución según los parámetros dados en el método anterior. Se filtran las alícuotas de cada intervalo a través de filtro de jeringa de membrana de nailon de 0,45 pm descartando los primeros ml del filtrado. Se analiza la solución de muestra de fase de tampón.
Metodología analítica para pellas de cafeína
Se ha usado el método analítico para comprimidos para las pellas de cafeína.
Ejemplo 1C (comparativo): Alginato de sodio (menos de 10 cP en solución acuosa al 1 %)
Recubrimiento de alginato de sodio simple polimérico de 8 mg/cm2
Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 30 minutos en un agitador suspendido para preparar solución al 7 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a solución de alginato y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
El recubrimiento continuó hasta 8 mg/cm2 de polímero
Suspensión aplicada: 639,8 g
Parámetro de curado: Sin curado
Resultados:
• Comprimidos de color amarillo con superficie lisa
• No se consiguió protección entérica hasta un nivel de recubrimiento de 8 mg/cm2 con un 94 % de liberación de fármaco en HCl 0,1 N.
Ejemplo 2C (comparativo): Alginato de sodio (menos de 10 cP en solución acuosa al 1 %)
Recubrimiento de 16 mg/cm2 de polímero (EUDRAGIT® NM 30D 4 mg/cm2 alginato de sodio 12 g/cm2)
Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 30 minutos en un agitador suspendido para preparar solución al 10 %.
• El pH del alginato de sodio se elevó hasta 10 mediante la adición de 25 ml de NaOH 0,1 N.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D se añadieron a solución de alginato de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
El recubrimiento continuó hasta 32 mg/cm2 de polímero.
Suspensión aplicada: 956,76 g
Parámetro de curado: 24 h a 60 °C en un secador de bandeja.
Resultados:
• Comprimidos de color amarillo con superficie lisa
• No se consiguió protección entérica hasta un nivel de recubrimiento de 32 mg/cm2 con un 96,1 % de liberación de fármaco en HCl 0,1 N.
Ejemplo 3: Alginato de sodio (40-90 cP en solución acuosa al 1 %)
Recubrimiento de 6 mg/cm2 de alginato de sodio simple polimérico
Fórmula para la suspensión de recubrimiento en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
Se pesó alginato de sodio y se mantuvo en agitación con agua durante 30 minutos en un agitador suspendido para preparar solución al 5 %.
Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
La suspensión de talco homogeneizada se añadió a solución de alginato de la etapa 2 y se continuó la agitación durante 30 min más.
La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
Recubrimiento hecho hasta un nivel de recubrimiento de 6 mg/cm2
Suspensión aplicada: 564,5 g
Parámetro de curado: Sin curado
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 6,1 % de liberación de fármaco en HCl 0,1 N para un nivel de recubrimiento de 5 mg/cm2
• Se consiguió protección entérica con un 6,4 % de liberación de fármaco en HCl 0,1 N para un nivel de recubrimiento de 6 mg/cm2
• Se observó liberación de fármaco de un 82 %, 89 % y 91 % en 30, 45 y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 5 mg/cm2.
• Se observó liberación de fármaco de un 82 %, 90 % y 92 % en 30, 45 y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 6 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 5 mg/cm2, así como de 6 mg/cm2 a niveles de alcohol de un 5 %, 10 %, 20 % y 40 %.



• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 4,1-5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 7 y 10 minutos para niveles de recubrimiento de 5 mg/cm2 y 6 mg/cm2 respectivamente en SIF.
Ejemplo 4: Alginato de sodio (40-90 cP en solución acuosa al 1 %)
Recubrimiento de 16 mg/cm2 de polímero (HPMC 4 mg/cm2 alginato de sodio 12 mg/cm2)
Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
Se pesó alginato de sodio y se mantuvo en agitación con agua durante 2 horas en un agitador suspendido para preparar solución al 5 %.
La cantidad pesada de HPMC se añadió a 135 g de agua y se agitó durante 60 minutos usando un agitador suspendido.
Se disolvió PEG 6000 en 15 g de agua caliente (70-75 °C) y se añadió a la etapa 2.
Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
La suspensión de la etapa 2 se añadió a la solución de etapa 1.
La suspensión de talco homogeneizada se añadió a solución de alginato y se continuó la agitación durante 30 min más.
La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
El recubrimiento se hizo hasta un nivel de recubrimiento de 16 mg/cm2.
Suspensión aplicada: 1223 g
Parámetro de curado: 24 h a 60 °C en un secador de bandeja.
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 4 % de liberación de fármaco en HCl 0,1 N, se observó liberación de fármaco de un 36 %, 88 % y 92 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 16 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 16 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol

• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 4,5-5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 30 minutos en SIF.
Ejemplo 5: Alginato de sodio (40-90 cP en solución acuosa al 1 %)
Recubrimiento de 16 mg/cm2 de polímero (EUDRAGIT® NM 30D 2,7 mg/cm2 alginato de sodio 13,3 mg/cm2) Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 30 minutos en un agitador suspendido para preparar solución al 5 %.
• El pH del alginato de sodio se elevó hasta 10 mediante la adición de 50 ml de NaOH 0,1 N.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D se añadieron a solución de alginato de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
El recubrimiento se hizo hasta un nivel de recubrimiento de 16 mg/cm2.
Suspensión aplicada: 1151,94 g
Parámetro de curado: 24 h a 60 °C en un secador de bandeja.
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 4,3 % de liberación de fármaco en HCl 0,1 N, se observó liberación de fármaco de un 5,3 %, 85 % y 90 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 16 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 16 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol

• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 20 minutos en SIF.
Ejemplo 6 : Alginato de sodio (40-90 cP en solución acuosa al 1 %)
Recubrimiento de 16 mg/cm2 de polímero (EUDRAGIT® NM 30D 3,2 mg/cm2 alginato de sodio 12,8 mg/cm2) Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g


Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 30 minutos en un agitador suspendido para preparar solución al 5 %.
• El pH del alginato de sodio se elevó hasta 10 mediante la adición de 50 ml de NaOH 0,1 N.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D se añadieron a solución de alginato de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
El recubrimiento se hizo hasta un nivel de recubrimiento de 16 mg/cm2.
Suspensión aplicada: 1187,55 g
Parámetro de curado: 24 h a 60 °C en un secador de bandeja.
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 5,8 % de liberación de fármaco en HCl 0,1 N, se observó liberación de fármaco de un 44,6 %, 89,7 % y 93,8 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 16 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 16 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol

• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 17 minutos en SIF.
Ejemplo 7: Alginato de sodio (40-90 cP en solución acuosa al 1 %)
Recubrimiento de 16 mg/cm2 de polímero (EUDRAGIT® NM 30D 4 mg/cm2 alginato de sodio 12 mg/cm2)
Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 30 minutos en un agitador suspendido para preparar solución al 5 %.
• El pH del alginato de sodio se elevó hasta 10 mediante la adición de 25 ml de NaOH 0,1 N.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D se añadieron a solución de alginato de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
Suspensión aplicada: 1196 g
Parámetro de curado: 24 h a 60 °C en un secador de bandeja.
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 4,2 % de liberación de fármaco en HCl 0,1 N, se observó liberación de fármaco de un 73,7 %, 92,9 % y 94,5 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 16 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 16 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol


• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 36 minutos en SIF
Ejemplo 8 : Alginato de sodio (40-90 cP en solución acuosa al 1 %)
Recubrimiento de 16 mg/cm2 de polímero (EUDRAGIT® NE 30D 4 mg/cm2 alginato de sodio 12 mg/cm2)
Fórmula para el recubrimiento polimérico en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 30 minutos en un agitador suspendido para preparar solución al 5 %.
• El pH del alginato de sodio se elevó hasta 10 mediante la adición de 25 ml de NaOH 0,1 N.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D se añadieron a solución de alginato de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
Suspensión aplicada: 1196 g
Parámetro de curado: 24 h a 60 °C en un secador de bandeja.
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 5 % de liberación de fármaco en HCl 0,1 N
• Se observó liberación de fármaco de un 7 %, 81 % y 89 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 16 mg/cm2
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 16 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol

• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 25 minutos en SIF.
Ejemplo 9: Alginato de sodio (40-90 cP en solución acuosa al 1 %)
Recubrimiento de 16 mg/cm2 de polímero (Kollicoat® SR 30 D 4 mg/cm2 alginato de sodio 12 mg/cm2)
Fórmula para el recubrimiento polimérico en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 2 horas en un agitador suspendido para preparar solución al 5 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• Se añadió propilenglicol a Kollicoat® SR 30 D y se mezclaron durante 30 minutos en un agitador magnético. • La suspensión de la etapa 3 se añadió a la solución de la etapa 1
• La suspensión de talco homogeneizada se añadió a solución de alginato y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
El recubrimiento se hizo hasta un nivel de recubrimiento de 16 mg/cm2
Suspensión aplicada: 1470 g
Parámetro de curado: 24 h a 60 °C en un secador de bandeja.
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 5 % de liberación de fármaco en HCl 0,1 N
• Se observó liberación de fármaco de un 82 %, 89 % y 91 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 16 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 16 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol

• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 20 minutos en SIF.
Ejemplo 10: Alginato de sodio (40-90 cP en solución acuosa al 1 %)
Recubrimiento de 16 mg/cm2 de polímero (EUDRAGIT® RS 30 D 4 mg/cm2 alginato de sodio 12 mg/cm2)
Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 2 horas en un agitador suspendido para preparar solución al 5 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® RS 30D se añadieron a solución de alginato y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
El recubrimiento se hizo hasta un nivel de recubrimiento de 16 mg/cm2
Suspensión aplicada: 1470 g
Parámetro de curado: 24 h a 60 °C en un secador de bandeja.
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 4 % de liberación de fármaco en HCl 0,1 N
• Se observó liberación de fármaco de un 7 %, 84 % y 91 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 16 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 16 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol

• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 25 minutos en SIF.
Ejemplo 11: Formulación de pellas de alginato de sodio (40-90 cP en solución acuosa al 1 %)
Recubrimiento de EUDRAGIT® NM 30D:MANUCOL® DH: 1:3
Fórmula para la suspensión de recubrimiento polimérica en pellas de lansoprazol de 600 g (tamaño de 1-14 mm, 11,7-14,3 % p/p de carga de fármaco)

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 30 minutos en un agitador suspendido para preparar solución al 5 %.
• El pH del alginato de sodio se elevó hasta 10 mediante la adición de 90 ml de NaOH 0,1 N.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D se añadieron a solución de alginato de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre pellas en FBP (GPCG 3.1)
Recubrimiento:
El recubrimiento se hizo hasta un nivel polímero de un 50 %
Suspensión aplicada: 6750 g
Parámetro de curado: Fluidización durante 2 horas a 50 °C
Resultados:
• Aspecto: Pellas de color crema
• No se consiguió protección entérica con un 17,5 % y un 10,1 % de liberación de fármaco en HCl 0,1 N después de 60 minutos con niveles de recubrimiento de polímero de un 40 % y un 45 %.
• Se consiguió protección entérica con un 8,9 % de liberación de fármaco en HCl 0,1 N después de 60 minutos para un nivel de recubrimiento de polímero de un 50 % p/p
• Se observó liberación de fármaco de un 71,6 % en 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de polímero de un 50 % p/p
• Se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de polímero de un 50 % p/p a un 5 %, 10 %, 20 % y 40 % de niveles de alcohol (etanol).

• Se observó resistencia entérica seguida de liberación de fármaco de un 89 % en 60 minutos en tampón USP pH 5,5 con un nivel de recubrimiento de polímero de un 50 %.
Ejemplo 12: Alginato de sodio (no menos de 45 cP en solución acuosa al 1 %)
Recubrimiento de 4 mg/cm2 de alginato simple polimérico
Fórmula para el recubrimiento de alginato de sodio (calidad alimenticia) en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 1 hora en un agitador suspendido para preparar solución al 10 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a solución de alginato y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
Suspensión aplicada para nivel de recubrimiento 3 mg/cm2: 300 g
Suspensión aplicada para nivel de recubrimiento 4 mg/cm2: 400 g
Resultados
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 8,2 % de liberación de fármaco en HCl 0,1 N con un nivel de recubrimiento de 3 mg/cm2.
• Se observó liberación de fármaco de un 88,9 %, 91,6 % y 92,2 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 3 mg/cm2.
• Se consiguió protección entérica con un 7,0 % de liberación de fármaco en HCl 0,1 N con un nivel de recubrimiento de 4 mg/cm2.
• Se observó liberación de fármaco de un 86,2 %, 87,8 % y 88,7 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 4 mg/cm2.
• No se observó resistencia a absorción rápida de alcohol con 3 mg/cm2, a un 5 %, y 20 % de alcohol, pero se observó resistencia a alcohol a un 10 % y 40 % de alcohol
• Se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 4 mg/cm2 a un 5 %, 10 %, 20 % y 40 % de alcohol.

• El comprimido recubierto con 3 mg/cm2 estuvo intacto en SGF y se observó disgregación en 18 minutos en SIF • El comprimido recubierto con 4 mg/cm2 estuvo intacto en SGF y se observó disgregación en 20 minutos en SIF
Ejemplo 13: Alginato de sodio (no menos de 45 cP en solución acuosa al 1 %)
Recubrimiento de 6 mg/cm2 de polímero (EUDRAGIT® NM 30D 4 mg/cm2+ alginato de sodio 2 mg/cm2)
Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
Se pesó alginato de sodio y se mantuvo en agitación con agua durante 1 hora en un agitador suspendido para preparar solución a 10 %.
Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
La suspensión de talco homogeneizada y EUDRAGIT® NM 30D se añadieron a solución de alginato y se continuó la agitación durante 30 min más.
La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento
Recubrimiento
Suspensión aplicada: 239,6 g
Curado: 24 h a 60 °C en un secador de bandeja
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 3,8 % de liberación de fármaco en HCl 0,1 N
• Se observó liberación de fármaco de un 11,8 %, 30,5 % y 67 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 6 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 6 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol

El comprimido estuvo intacto después del ensayo de disgregación en SGF y se observó hinchamiento en 60 minutos en SIF
Ejemplo 14C (comparativo): Alginato de potasio (200-400 cP para solución acuosa al 1 %)
Recubrimiento de 2 mg/cm2 de alginato de potasio
Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de potasio y se mantuvo en agitación con agua durante 2 horas en un agitador suspendido para preparar solución al 3 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a solución de alginato de potasio de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
Suspensión aplicada para 2 mg/cm2: 199,22 g
Parámetro de curado: Sin curado
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• No se consiguió protección entérica con un 34,5 % de liberación de fármaco en HCl 0,1 N para un nivel de recubrimiento de 2 mg/cm2.
Ejemplo 15: Alginato de sodio (50-150 cP en solución acuosa al 1 %)
Recubrimiento de 4 mg/cm2 de alginato de sodio puro polimérico
Fórmula para la suspensión de recubrimiento en comprimidos de 300 g


Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 3 horas en un agitador suspendido para preparar solución al 6 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a solución de alginato y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
Suspensión aplicada: 319,8 g
Parámetro de curado: Sin curado
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 7,7 % de liberación de fármaco en HCl 0,1 N
• Se consiguió protección entérica con un 65,1 %, 82,6 % y 88,2 % de liberación de fármaco en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 4 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 4 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de niveles de alcohol.

• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 15 minutos en SIF.
Ejemplo 16: Alginato de sodio (50-150 cP en solución acuosa al 1 %)
Recubrimiento de 16 mg/cm2 de polímero (EUDRAGIT® NM 30D 4 mg/cm2+ alginato de sodio 12 mg/cm2)
Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g.
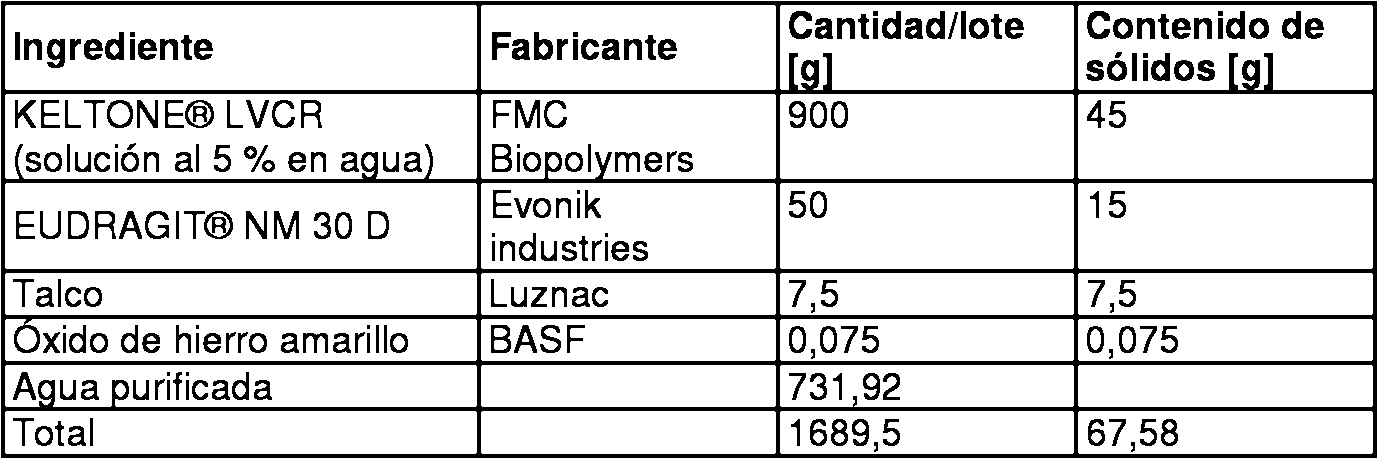
Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 2 horas en un agitador suspendido para preparar solución al 5 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D 30D se añadieron a solución de alginato y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
Suspensión aplicada: 1196 g
Parámetro de curado: 24 h a 60 °C en un secador de bandeja.
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 4 % de liberación de fármaco en HCl 0,1 N
• Se observó liberación de fármaco en un 20,6 %, 85,8 % y 92,0 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 16 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 16 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol.

• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 25 minutos en SIF.
Ejemplo 17: Alginato de sodio (70-200 cP en solución acuosa al 1 %)
Recubrimiento de 4 mg/cm2 de alginato de sodio simple polimérico
Fórmula para la suspensión de recubrimiento en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 30 minutos en un agitador suspendido para preparar solución al 6 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a solución de alginato de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
El recubrimiento se hizo hasta un nivel de recubrimiento de 4 mg/cm2
Suspensión aplicada: 399,75 g
Parámetro de curado: Sin curado
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 6,6 % de liberación de fármaco en HCl 0,1 N
• Se observó liberación de fármaco en un 59,5 %, 86,8 % y 91 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 4 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 4 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de niveles de alcohol.


• Se retuvo la resistencia entérica seguida de liberación de fármaco rápida en tampón USP pH 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 25 minutos en SIF.
Ejemplo 18C (comparativo): Alginato de sodio (70-200 cP en solución acuosa al 1 %)
Recubrimiento de 3 mg/cm2 de alginato de sodio simple polimérico
Fórmula para la suspensión de recubrimiento en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 30 minutos en un agitador suspendido para preparar solución al 6 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a solución de alginato de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
El recubrimiento se hizo hasta un nivel de recubrimiento de 3 mg/cm2
Suspensión aplicada: 300 g
Parámetro de curado: Sin curado
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• No se consiguió protección entérica con un 25 % de liberación de fármaco en HCl 0,1 N
Ejemplo 19: Alginato de sodio (70-200 cP en solución acuosa al 1 %)
Recubrimiento de 16 mg/cm2 de polímero (EUDRAGIT® NM 30D 4 mg/cm2+ alginato de sodio 12 mg/cm2)
Fórmula para la suspensión de recubrimiento polimérica de un 20 % p/p en comprimidos de 300 g.

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 2 horas en un agitador suspendido para preparar solución al 5 %.
• Se elevó el pH del alginato de sodio hasta 10 mediante la adición de 30 ml de NaOH 0,1 N.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D se añadieron a solución de alginato de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
Suspensión aplicada: 1196 g
Parámetro de curado: 24 h a 60 °C en un secador de bandeja.
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 4,1 % de liberación de fármaco en HCl 0,1 N
• Se observó liberación de fármaco de un 66,2 %, 92,5 % y 94,0 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 16 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 16 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol.

• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 25 minutos en SIF.
Ejemplo 20: Alginato de potasio (200-400 cP para solución acuosa al 1 %)
Recubrimiento de 3 mg/cm2 y 4 mg/cm2 de alginato de potasio
Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de potasio y se mantuvo en agitación con agua durante 2 horas en un agitador suspendido para preparar solución al 3 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a solución de alginato de potasio de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
Suspensión aplicada para 3 mg/cm2: 677,4 g
Suspensión aplicada para 4 mg/cm2: 903,2 g
Parámetro de curado: Sin curado
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 13,1 % de liberación de fármaco en HCl 0,1 N. Se observó liberación de fármaco de un 85,0 %, 91,3 % y 92,9 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 3 mg/cm2.
• Se consiguió protección entérica con un 5,2 % de liberación de fármaco en HCl 0,1 N. Se observó liberación de fármaco de un 47,9 %, 85,5 % y 90,6 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 4 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 3 mg/cm2 y 4 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol.

• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 5,1 - 5,5.
• El comprimido recubierto con 3 mg/cm2 estuvo intacto en SGF y se observó disgregación en 17 minutos en SIF • El comprimido recubierto con 4 mg/cm2 estuvo intacto en SGF y se observó disgregación en 24 minutos en SIF
Ejemplo 21: Alginato de potasio (200-400 cP para solución acuosa al 1 %)
Recubrimiento de 7 mg/cm2 de polímero (EUDRAGIT® NM 30D 1,75 mg/cm2 alginato de potasio 5,25 mg/cm2)
Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g
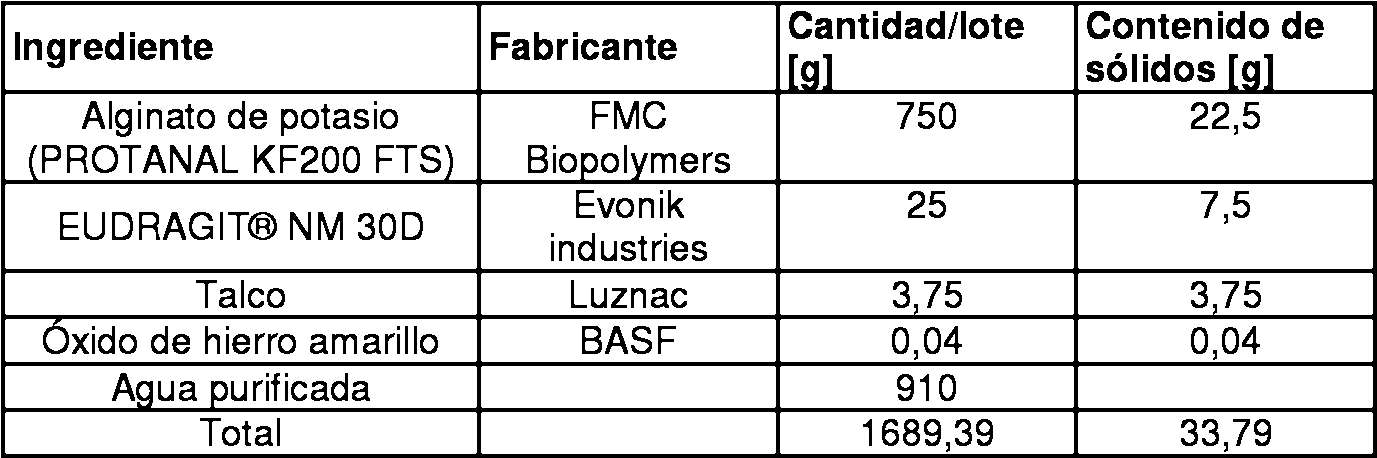
Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de potasio y se mantuvo en agitación con agua durante 2 horas en un agitador suspendido para preparar solución al 3 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D se añadieron a solución de alginato de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
El recubrimiento se hizo hasta un nivel de recubrimiento de 7 mg/cm2
Suspensión aplicada: 1047,21 g
Parámetro de curado: 60 °C durante 24 horas en secador de bandeja
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 7,3 % de liberación de fármaco en HCl 0,1 N. Se observó liberación de fármaco de un 66 %, 91,8 % y 94,9 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 4 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 7 mg/cm2 a un 5 %, 10 %, 20 % y 40 % de alcohol.


• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 5,1 - 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 25 minutos en SIF.
Ejemplo 22: Alginato de sodio (480-720 cP en solución acuosa al 1 %)
Recubrimiento de 6 mg/cm2 de alginato de sodio simple
Fórmula para la solución de suspensión de recubrimiento en comprimido de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 3 horas en un agitador suspendido para preparar solución al 6 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a solución de alginato y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
El recubrimiento se hizo hasta un nivel de recubrimiento de 6 mg/cm2
Suspensión aplicada: 1199,25 g
Parámetro de curado: Sin curado
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 7,2 % de liberación de fármaco en HCl 0,1 N
• Se observó liberación de fármaco de un 46,3 %, 78,3 % y 88,7 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 6 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 6 mg/cm2, a un 10 %, 20 % y 40 % de niveles de alcohol.

• El comprimido estuvo intacto en SGF y se observó disgregación en 13 minutos en SIF.
Ejemplo 23: Alginato de sodio (480-720 cP en solución acuosa al 1 %)
Recubrimiento de 8 mg/cm2 de alginato de sodio puro polimérico
Fórmula para la suspensión de recubrimiento en comprimidos de 300 g


Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 3 horas en un agitador suspendido para preparar solución al 6 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a solución de alginato y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
El recubrimiento se hizo hasta un nivel de recubrimiento de 8 mg/cm2
Suspensión aplicada: 1599 g
Parámetro de curado: Sin curado
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 5,7 % de liberación de fármaco en HCl 0,1 N
• Se observó liberación de fármaco de un 35,3 %, 89,5 % y 93,9 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 8 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 8 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de niveles de alcohol (etanol).

• Se retuvo la resistencia entérica seguida de un comportamiento de liberación de fármaco rápida en tampón USP pH 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 20 minutos en SIF.
Ejemplo 24: Alginato de sodio (480-720 cP en solución acuosa al 1 %)
Recubrimiento de 12 mg/cm2 de polímero (EUDRAGIT® NM 30D 3 mg/cm2 alginato de sodio 9 mg/cm2)
Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 2 horas en un agitador suspendido para preparar solución al 3 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D 30D se añadieron a solución de alginato y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
Suspensión aplicada: 1794,38 g
Parámetro de curado: 24 horas a 60 °C en un secador de bandeja.
Resultados:
• Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 4,5 % de liberación de fármaco en HCl 0,1 N
• Se observó liberación de fármaco de un 5,3 %, 86,9 % y 94,9 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 12 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 12 mg/cm2, a un 10 %, 20 % y 40 % de niveles de alcohol.

• El comprimido estuvo intacto en SGF y se observó disgregación en 20 minutos en SIF.
Ejemplo 25C (comparativo) carragenina
Recubrimiento de 10 mg/cm2 de carragenina
Fórmula para la suspensión de recubrimiento en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó carragenina y se mantuvo en agitación con agua durante 2 horas en un agitador suspendido para preparar solución al 1,5 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a solución de carragenina de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
Suspensión aplicada: 2258 g
Parámetro de curado: Sin curado
Resultados:
• Comprimidos de color amarillo con superficie lisa
• No se consiguió protección entérica con un 92,7 % de liberación de fármaco en HCl 0,1 N para un nivel de recubrimiento de 10 mg/cm2
Ejemplo 26C (comparativo): Ácido algínico
Recubrimiento de 4 mg/cm2 de ácido algínico simple
Fórmula para la suspensión de recubrimiento en comprimidos de 300 g


Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó ácido algínico y se mantuvo en agitación con agua durante 30 minutos en un agitador suspendido para preparar solución al 6 %.
• Se homogeneizaron el talco el citrato de trietilo y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a la suspensión de ácido algínico de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Resultados:
• No se observó formación de película
Ejemplo 27C (comparativo): Ácido algínico
Recubrimiento de 16 mg/cm2 de polímero (EUDRAGIT® NM 30D 4 mg/cm2 ácido algínico 12 mg/cm2) Fórmula para la suspensión de recubrimiento en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó ácido algínico y se mantuvo en agitación con agua durante 30 minutos en un agitador suspendido para preparar solución al 5 %.
• El pH del ácido algínico se elevó hasta 10 mediante la adición de 150 ml de NaOH 0,1 N.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D se añadieron a solución de ácido algínico de la etapa 2 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento.
Recubrimiento:
Suspensión pulverizada: 796 g
Parámetro de curado: Sin curado
Resultados:
• Comprimidos de color pardo con superficie ligeramente rugosa
• No se consiguió protección entérica con un 96,3 % p/p de liberación de fármaco en HCl 0,1 N a un nivel de recubrimiento de 16 mg/cm2
Ejemplo 28C (comparativo): EUDRAGIT® L30 D-55
Recubrimiento de 5 mg/cm2 de polímero seco EUDRAGIT® L 30 D-55
Fórmula para la suspensión de recubrimiento en comprimidos de 300 g


Procedimiento para la preparación de suspensión de recubrimiento:
• Se homogeneizaron el talco y el color con 202,85 g de agua durante 30 minutos.
• El citrato de trietilo se añadió a la dispersión de talco homogeneizada y se continuó la homogeneización durante 10 minutos más.
• La dispersión de EUDRAGIT® L30 D-55 se pesó de forma precisa y se mantuvo en agitación con un agitador magnético.
• La suspensión de talco homogeneizada se añadió a la dispersión de EUDRAGIT® L30 D-55 y se continuó la agitación durante 10 min más.
• La suspensión preparada final se pasó a través de un tamiz de 150 micrómetros (n.° 100).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento
Recubrimiento:
El recubrimiento se hizo hasta un nivel de recubrimiento de 5 mg/cm2
Suspensión aplicada: 142,1 g
Parámetro de curado: Sin curado
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 0 % de liberación de fármaco en HCl 0,1 N
• Se observó liberación de fármaco de un 93 %, 94 % y 95 en 30, 45 y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 5 mg/cm2.
• No se observó resistencia a absorción rápida de alcohol con alcohol superior

• Se observó resistencia entérica seguida de liberación de fármaco lenta en tampón USP pH 5,5 con un nivel de recubrimiento de 5 mg/cm2 con solamente un 46 %, 59 % y 66 % de liberación de fármaco observado en 30, 45 y 60 minutos, respectivamente.
Ejemplo 29C (comparativo): EUDRAGIT® NM 30D
Recubrimiento de 5 mg/cm2 de polímero seco EUDRAGIT® NM 30D
Fórmula para la suspensión de recubrimiento de EUDRAGIT® NM 30D en comprimidos de 300 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se homogeneizaron el talco y el color con 185,15 g de agua durante 30 minutos.
• La dispersión de EUDRAGIT® NM 30D se pesó de forma precisa y se mantuvo en agitación con un agitador magnético.
• La suspensión de talco homogeneizada se añadió a la dispersión de EUDRAGIT® NM 30D y se continuó la agitación durante 10 min más.
• La suspensión preparada final se pasó a través de un tamiz de 150 micrómetros (n.° 100).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento
Recubrimiento:
El recubrimiento se hizo hasta un nivel de recubrimiento de 4 mg/cm2
Suspensión aplicada: 106,5 g
Parámetro de curado: 24 horas a 60 °C en un secador de bandeja.
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se observó resistencia entérica con un 0,1 % de liberación de fármaco en HCl 0,1 N seguido de únicamente un 0,6 % y 0,9 % de liberación de fármaco en 45 y 60 minutos, respectivamente, en tampón USP pH 6,8 con un nivel de recubrimiento de 4 mg/cm2.

E m l - 7 r m n

E m l -4 r m n

Ejemplo 30: Recubrimiento de alginato de amonio simple en comprimido de cafeína
Formulación y metodología de procesamiento
Tamaño de lote : 300 g
Forma del comprimido : Circular
Tamaño del comprimido : 11 mm
Nivel de recubrimiento polimérico : 5 mg/cm2
Máquina usada : Bandeja de recubrimiento Ganson de 30,48 cm (12 pulgadas)
Fórmula para aumento de peso de un 5 % en 300 g

Procedimiento:
• Se homogeneizaron el taco y el color en 136 g de agua durante 30 minutos
• Se disolvió alginato de amonio en los 405 g de agua usando agitador suspendido durante 30 minutos
• L a suspensión de talco se añadió a la solución de alginato de amonio y se agitó en agitador suspendido durante 10 minutos.
• La suspensión resultante se pasó a través de un tamiz n.° 60.
pH de la suspensión final: 7,15
Cantidad requerida para comprimidos de 300 g
Para recubrimiento de 5 mg/cm2 suspensión de 663,47
Parámetros de la máquina:
Bandeja de recubrimiento 30,48 cm (12 pulgadas)
Deflectores Presentes
Tubo de silicona (de/di) 5/3 mm
Temperatura de entrada 50 ° - 55 °C
Temperatura del producto 29 ° - 32 °C
Escape Activo
Soplador Activo
Presión del aire de pulverización 1,2 bares
Peso inicial de 20 comprimidos 8,04 g
Aumento de peso requerido 8,57 g
P r m r n l r

Aumento de peso final 8,57 g
Intervalo de r.p.m. de la bandeja 22
Intervalo de r.p.m. de la bomba 2
Intervalo de la tasa pulverización 3,31 g/min/2 r.p.m.
Curado: 60 °C durante 24 h
Observación:
• El proceso fue suave sin ningún problema técnico
Resultados:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 5,4 % de liberación de fármaco en HCl 0,1 N, se observó liberación de fármaco de un 82,0 %, 86,1 % y 87,8 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 5 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 5 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol (etanol).

• Se retuvo la resistencia entérica seguida por un comportamiento de liberación de fármaco rápida en tampón USP pH 4,5 - 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 23 minutos en SIF.
Ejemplo 31: Recubrimiento de EUDRAGIT® NM 30D:alginato de amonio (1:3) en comprimido de cafeína
Formulación y metodología de procesamiento
Tamaño de lote : 300 g
Forma del comprimido : Circular
Tamaño del comprimido : 11 mm
Nivel de recubrimiento polimérico : 7 mg/cm2
Máquina usada : Bandeja de 30,48 cm (12 pulgadas)
F rm l r m n n 1 n

Procedimiento:
1. Se disolvió alginato de amonio en los 1400 g de agua lentamente mientras se agitaba usando agitador suspendido durante 30 minutos
2. Se homogeneizaron el taco y el color en el agua restante durante 30 minutos
3. La solución de alginato de amonio se añadió a dispersión de EUDRAGIT® NM 30D en agitación
4. La suspensión de talco se añadió a la suspensión de la etapa 3 y se agitó la suspensión final durante 10 minutos 5. La suspensión resultante se pasó a través de un tamiz n.° 60 y se usó para pulverización.
pH de la suspensión final: 7,18
Cantidad requerida para comprimidos de 300 g
Para recubrimiento de 7 mg/cm suspensión de 694,95 g
Parámetros de la máquina:
Bandeja de recubrimiento 30,48 cm (12 pulgadas)
Deflectores Presentes
Tubo de silicona (de/di) 5/3 mm
Temperatura de entrada 50 ° - 65 °C
Temperatura del producto 30 ° - 37 °C
Escape Activo
Soplador Activo
Presión del aire de pulverización 1.0 bares
Peso inicial de 20 comprimidos 8.0 g
Aumento de peso requerido : 8,57 g
P r m r n l r :

Aumento de peso final 8,56 g
Intervalo de r.p.m. de la bandeja 20
Intervalo de r.p.m. de la bomba 2-3
Intervalo de la tasa pulverización 3,42 g/min/2 r.p.m. - 5,04 g/min/3 r.p.m.
Curado: 60 °C durante 24 h
Observación:
• El proceso fue suave sin ningún problema técnico
Resultados para el ejemplo 31:
• Aspecto: Comprimidos de color amarillo con superficie lisa
• Se consiguió protección entérica con un 5,1 % de liberación de fármaco en HCl 0,1 N, se observó liberación de fármaco de un 86,8 %, 88,7 % y 89,9 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de 7 mg/cm2.
• También se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento de 7 mg/cm2, a un 5 %, 10 %, 20 % y 40 % de alcohol.

• Se retuvo la resistencia entérica seguida por un comportamiento de liberación de fármaco rápida en tampón USP pH 4,5 - 5,5.
• El comprimido estuvo intacto en SGF y se observó disgregación en 15 minutos en SIF.
Tampones y procedimiento para los ejemplos 32 a 37
Preparación del medio:
1) Medio de la fase de ácido: (HCl 0,1 N)
Para 1 l se añaden 85 ml de clorhidrato concentrado en 900 ml de agua destilada. Se completa el volumen hasta un litro y se mezcla bien.
2) Medio de la fase de tampón: (Simple pH 6,8)
Se pesan de forma precisa y se transfieren 19,01 g de fosfato de trisodio y 6,37 ml de ácido clorhídrico concentrado a 990 ml de agua. Se disuelven y se completa el volumen hasta un litro y se mezclan bien. Se ajusta el pH hasta 6,8 ± 0,05 usando NaOH 2 N o HCl 2 N.°
3) Medio de la fase de ácido: (HCl 0,1 N con iones Ca++)
Para 1 l se añaden 85 ml de clorhidrato concentrado en 900 ml de agua destilada. Se añaden 0,185 g de CaCl2 .2 H2O, se completa el volumen hasta un litro y se mezcla bien.
4) Medio de la fase de tampón: (Tampón pH 6,8 con iones Ca++)
Se pesan de forma precisa y se transfieren 19,01 g de fosfato de trisodio y 6,37 ml de ácido clorhídrico concentrado a 1000 ml de agua. Se añaden 0,185 g de CaCl2.2H2O y se disuelven y se completa el volumen total hasta un litro y se mezcla bien. Se ajusta el pH hasta 6,8 ± 0,05 usando NaOH 2 N o HCl 2 N.°
Procedimiento:
Fase de ácido: Se pesa y se transfiere el comprimido de cafeína en seis tarros de disolución diferentes y después se realiza el ensayo de disolución según los parámetros dados en el método anterior (fase de ácido). Después de 2 h se retiran 10 ml de alícuota y se analiza como la solución de muestra de la fase de ácido.
Fase de tampón: Se transfiere el comprimido al medio de la fase de tampón pH 6,8. Se continúa el ensayo de disolución según los parámetros datos en el método anterior (fase de tampón). Se filtran las alícuotas de cada intervalo a través de un filtro de jeringa de membrana de nailon de 0,45 pm descartando los primeros ml del filtrado. Se analiza la solución de muestra de fase de tampón.
Los estudios se hicieron de la siguiente manera:
1) Disolución en 0,1 N seguido de tampón pH 6,8 (simple)
2) Disolución en Ca++ 1 mM en HCl seguido de Ca++ 1 mM en tampón pH 6,8
3) Adición de Ca++ 1 mM en HCl seguido de tampón pH 6,8 (sin calcio)
Ejemplos 32 a 34: Comparación de alginato de sodio y alginato de amonio en presencia de iones calcio



Resultado: El recubrimiento de alginato de sodio es sensible a la presencia de calcio en tampón pH 6,8, mientras que el recubrimiento de alginato de amonio no.
Ejemplos 35 a 37: Comparación de alginato de sodio y alginato de amino con la adición de EUDRAGIT® NM en presencia de iones calcio




Resultado: El recubrimiento de alginato de sodio EUDRAGIT® NM es sensible a la presencia de calcio en tampón a pH 6,8, mientras que el recubrimiento de alginato de amonio EUDRAGIT® NM no.
Ejemplo 38: Formulación de pella de alginato de potasio (200-400 cP para solución acuosa al 1 %) con talco al 200 %
Recubrimiento de EUDRAGIT® NM 30D: alginato de potasio: 1:3
Fórmula para la suspensión de recubrimiento polimérica en pellas de cafeína de 600 g (tamaño de 1,0 a 1,4 mm, carga de fármaco de aproximadamente un 25 %)

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de potasio y se mantuvo en agitación con agua durante 60 minutos en un agitador suspendido para preparar solución al 2 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D se añadieron a solución de alginato de la etapa 1 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre pellas en un procesador de lecho fluido.
Recubrimiento:
El recubrimiento se hizo hasta un nivel de polímero de un 25 %
Curado de 24 horas a 60 °C en secador de bandeja
Resultados:
• Aspecto: Pellas de color crema
• Se consiguió protección entérica con un 13,9 % de liberación de fármaco en HCl 0,1 N después de 120 minutos para un nivel de recubrimiento polimérico de un 25 % p/p
• Se observó liberación de fármaco de un 50,8 % en 60 minutos en tampón USP pH 6,8 con un nivel de recubrimiento polimérico de un 25 % p/p
• Se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento polimérico de un 25 % p/p a un 5 %, 10 %, 20 % y 40 % de niveles de alcohol.
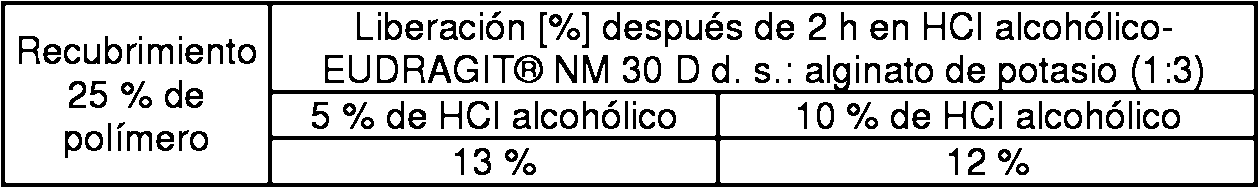

Ejemplo 39: Alginato de sodio (no menos de 45 cP en solución acuosa al 1 %)
Recubrimiento de 5,2 mg/cm2 de polímero (EUDRAGIT® NM 30D 4 mg/cm2 alginato de sodio 1,2 mg/cm2) Fórmula para la suspensión de recubrimiento polimérica en comprimidos de 500 g

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de sodio y se mantuvo en agitación con agua durante 1 hora en un agitador suspendido para preparar solución al 10 %.
• Se homogeneizaron el talco y el color con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada y EUDRAGIT® NM 30D se añadieron a solución de alginato y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre comprimidos en una bandeja de recubrimiento Recubrimiento:
Suspensión aplicada: 284,08 g
• Aspecto: Comprimidos de color amarillo con superficie lisa
Resultados de disolución:
• Se consiguió protección entérica con un 1 % de liberación de fármaco en HCl 0,1 N
• Se observó liberación de fármaco de un 5,4 %, 12,1 % y 29 % en 30, 45 minutos y 60 minutos respectivamente en tampón USP pH 6,8 con un nivel de recubrimiento de un 5,2 mg/cm2.
Resultados de disgregación
• El comprimido estuvo intacto en SGF y se observó disgregación en 50 minutos en SIF

Se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento polimérico de un 5,2 mg/cm2 a un 5 %, 10 %, 20 % y 40 % de niveles de alcohol.
Ejemplo 40: Formulación de pellas con el superdisgragante crospovidona (Polyplasdone XL)
Fórmula del núcleo:
F rm l ll f ín 1


Tamaño de la pella: 1 mm - 1,4 mm
Formulación para el recubrimiento interior
Recubrimiento de un 25 % de alginato de potasio
Alginato de potasio (200-400 cP para solución acuosa al 1 %) formulación de pella
F rm l r n i n r rimi n lim ri n ll

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de potasio y se mantuvo en agitación con agua durante 60 minutos en un agitador suspendido para preparar solución al 2 %.
• Se homogeneizaron el talco y Poliplasdone XL con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a solución de alginato de la etapa 1 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre pellas en FBP (GPCG 3.1)
Recubrimiento:
El recubrimiento se hizo hasta un nivel de polímero de un 25 %
Suspensión aplicada: 10468,75 g
Formulación para el recubrimiento exterior
Recubrimiento de un 10 % de alginato de potasio
Alginato de potasio (200-400 cP para solución acuosas al 1 %) formulación de pella
F rm l r l n i n r rimi n lim ri n^ ll

Procedimiento para la preparación de suspensión de recubrimiento:
• Se pesó alginato de potasio y se mantuvo en agitación con agua durante 60 minutos en un agitador suspendido para preparar solución al 2 %.
• Se homogeneizó el talco con la cantidad restante de agua durante 30 minutos.
• La suspensión de talco homogeneizada se añadió a solución de alginato de la etapa 1 y se continuó la agitación durante 30 min más.
• La suspensión preparada final se pasó a través de un tamiz de 300 micrómetros (n.° 60).
• Esta suspensión se pulverizó adicionalmente sobre pellas en FBP (GPCG 3.1)
Recubrimiento:
El recubrimiento se hizo hasta un nivel de polímero de un 10 %
Suspensión aplicada: 3750 g
Recubrimiento total realizado: recubrimiento interior de un 25 % recubrimiento exterior de un 10 % = recubrimiento total de un 35 %
Resultados:
• Aspecto: Pellas de color crema
• Se consiguió protección entérica con un 13 % de liberación de fármaco en HCl 0,1 N después de 120 minutos para un nivel de recubrimiento de polímero de un 35 % p/p
• Se observó liberación de fármaco de un 59 % y 70 % en 45 y 60 minutos en tampón USP pH 6,8 con un nivel de recubrimiento de polímero de un 35 % p/p
• Se observó resistencia a absorción rápida de alcohol con un nivel de recubrimiento polimérico de un 35 % p/p a un 5 %, 10 %, 20 % y 40 % de niveles de alcohol.

Claims (13)
1. Una composición farmacéutica o nutracéutica gastrorresistente, que comprende un núcleo, que comprende un ingrediente activo farmacéutico o nutracéutico y una capa de recubrimiento gastrorresistente en el núcleo, en la que la liberación del ingrediente activo farmacéutico o nutracéutico es de no más de un 15 % en condiciones in vitro a pH 1,
2 durante 2 horas en medio de acuerdo con USP con y sin la adición de etanol al 40 % (v/v), en la que la capa de recubrimiento gastrorresistente comprende de un 10 a un 10 0 % en peso de alginato de amonio con una viscosidad de 30 a 720 cP de una solución acuosa al 1 %.
2. Composición farmacéutico o nutracéutica gastrorresistente de acuerdo con la reivindicación 1 , en la que el aumento de peso de la capa de recubrimiento es de al menos 2,5 mg/cm2.
3. Composición farmacéutica o nutracéutica gastrorresistente de acuerdo con la reivindicación 1 o 2, en la que la capa de recubrimiento comprende hasta un 90 % en peso de excipientes farmacéutica o nutracéuticamente aceptables.
4. Composición farmacéutica o nutracéutica gastrorresistente de acuerdo con una o más de las reivindicaciones 1 a 3, en la que la liberación del ingrediente activo farmacéutico o nutracéutico es de al menos un 50 % en condiciones in vitro a pH 6 , 8 durante una hora en un medio tamponado de acuerdo con USP.
5. Composición farmacéutica o nutracéutica gastrorresistente de acuerdo con una o más de las reivindicaciones 1 a 4, en la que la capa de recubrimiento gastrorresistente comprende de un 0 a un 400 % en peso de uno o más polímeros insolubles en agua o uno o más polímeros celulósicos o mezclas de los mismos basado en el peso del alginato de amonio contenido.
6. Composición farmacéutica o nutracéutica gastrorresistente de acuerdo con la reivindicación 5, en la que el uno o más polímeros insolubles en agua pertenecen al grupo de copolímeros de (met)acrilato.
7. Composición farmacéutica o nutracéutica gastrorresistente de acuerdo con la reivindicación 5 o 6 , en la que el copolímero insoluble en agua es un copolímero compuesto de unidades polimerizadas de radicales libres de más de un 95 hasta un 100 % en peso de ésteres alquílicos C1 a C4 de ácido acrílico o metacrílico y menos de un 5 % en peso de ácido acrílico o metacrílico.
8. Composición farmacéutica o nutracéutica gastrorresistente de acuerdo con la reivindicación 6 o 7, en la que el polímero insoluble en agua está compuesto de un 20 a un 40 % en peso de acrilato de etilo, de un 60 a un 80 % en peso de metacrilato de metilo y de un 0 a menos de un 5 % en peso de ácido metacrílico.
9. Composición farmacéutica nutracéutica gastrorresistente de acuerdo con la reivindicación 5 o 6 , en la que el polímero insoluble en agua está compuesto de un 85 a un 98 % en peso de ésteres alquílicos C1 a C4 polimerizados de radicales libres de ácido acrílico o metacrílico y de un 15 a un 2 % en peso de monómeros de (met)acrilato de alquilo con un grupo amino cuaternario en el radical alquilo.
10. Composición farmacéutica o nutracéutica gastrorresistente de acuerdo con la reivindicación 5, en la que el polímero celulósico es hidroxipropilmetilcelulosa.
11. Composición farmacéutica o nutracéutica gastrorresistente de acuerdo con una o más de las reivindicaciones 1 a 1 0 , en la que el núcleo o la capa de recubrimiento gastrorresistente comprende excipientes farmacéutica o nutracéuticamente aceptables seleccionados del grupo de antioxidantes, abrillantadores, aglutinantes, agentes aromatizantes, auxiliares de flujo, fragancias, emolientes, agentes que promueven la penetración, pigmentos, plastificantes, polímeros, diferentes de sales de ácido algínico y diferentes de los polímeros insolubles en agua o polímeros celulósicos, agentes que forman poros o estabilizantes o combinaciones de los mismos.
12. Composición farmacéutica o nutracéutica gastrorresistente de acuerdo con una o más de las reivindicaciones 1 a 1 1 , en la que la composición farmacéutica o nutracéutica gastrorresistente es un comprimido recubierto, un minicomprimido recubierto, una pella recubierta, un gránulo recubierto, un sobrecito, una cápsula, rellena de pellas recubiertas o de polvo o de gránulos, o una cápsula recubierta.
13. Proceso para producir la forma farmaceuta o nutracéutica de una o más de las reivindicaciones 1 a 12, formando el núcleo que comprende el ingrediente activo por compresión directa, compresión de gránulos secos, húmedos o sinterizados, extrusión y posterior redondeo, granulación en húmedo o seco o peletización directa o por aglutinación de polvos en microesferas sin ingrediente activo o núcleos neutros o partículas que contienen ingrediente activo y aplicando el recubrimiento polimérico en forma de una dispersión acuosa en un proceso de pulverización o mediante granulación por pulverización de lecho fluido en el núcleo.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN2378CH2010 | 2010-08-18 | ||
| PCT/EP2011/055809 WO2012022498A1 (en) | 2010-08-18 | 2011-04-13 | Gastric resistant pharmaceutical or nutraceutical formulation comprising one or more salts of alginic acid |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2716550T3 true ES2716550T3 (es) | 2019-06-13 |
Family
ID=44168760
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES11714754T Active ES2716550T3 (es) | 2010-08-18 | 2011-04-13 | Formulación farmacéutica o nutracéutica gastrorresistente que comprende una o más sales de ácido algínico |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US9492394B2 (es) |
| EP (1) | EP2605759B1 (es) |
| JP (1) | JP5705319B2 (es) |
| KR (1) | KR101873075B1 (es) |
| CN (1) | CN103068376B (es) |
| AU (1) | AU2011290958B2 (es) |
| BR (1) | BR112013003581B8 (es) |
| CA (1) | CA2808546C (es) |
| ES (1) | ES2716550T3 (es) |
| HK (1) | HK1182003A1 (es) |
| MX (1) | MX360389B (es) |
| RU (1) | RU2013111605A (es) |
| WO (1) | WO2012022498A1 (es) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6161701B2 (ja) * | 2012-08-27 | 2017-07-12 | エボニック レーム ゲゼルシャフト ミット ベシュレンクテル ハフツングEvonik Roehm GmbH | 徐放特性及びエタノールの影響に対する耐性を有する医薬又は栄養補助組成物 |
| PL2887925T3 (pl) * | 2012-08-27 | 2017-07-31 | Evonik Röhm Gmbh | Odporna żołądkowo kompozycja farmaceutyczna lub nutraceutyczna z odpornością na wpływ etanolu |
| CA2888278A1 (en) * | 2012-10-15 | 2014-04-24 | Isa Odidi | Oral drug delivery formulations |
| CN104688775A (zh) * | 2013-12-04 | 2015-06-10 | 博晖(大连)实业有限公司 | 一种含海星成份的组合胃药 |
| EP3107529A1 (en) * | 2014-02-17 | 2016-12-28 | Evonik Röhm GmbH | Pharmaceutical or nutraceutical composition with sustained release characteristic and with resistance against the influence of ethanol |
| CA2910865C (en) | 2014-07-15 | 2016-11-29 | Isa Odidi | Compositions and methods for reducing overdose |
| CA2936741C (en) * | 2014-10-31 | 2018-11-06 | Purdue Pharma | Methods and compositions particularly for treatment of attention deficit disorder |
| BR112017013598A2 (pt) * | 2014-12-23 | 2018-03-06 | Fmc Corp | composições de revestimento de película entérico, método de revestimento, e formas revestidas |
| EP3288556A4 (en) * | 2015-04-29 | 2018-09-19 | Dexcel Pharma Technologies Ltd. | Orally disintegrating compositions |
| BR112017025148A2 (pt) | 2015-06-05 | 2018-08-07 | Evonik Röhm Gmbh | composição farmacêutica ou nutracêutica com resistência contra a influência do etanol |
| EP3167879A1 (en) * | 2015-11-10 | 2017-05-17 | Evonik Technochemie GmbH | Gastric retention active delivery systems |
| JP6801203B2 (ja) * | 2016-03-18 | 2020-12-16 | 日油株式会社 | pH応答溶出性被覆造粒物および食品 |
| US10076494B2 (en) | 2016-06-16 | 2018-09-18 | Dexcel Pharma Technologies Ltd. | Stable orally disintegrating pharmaceutical compositions |
| JP7111104B2 (ja) * | 2017-08-09 | 2022-08-02 | ライオン株式会社 | コーティング組成物、コーティング膜、コーティング製剤及びその製造方法 |
| EP3694491A1 (en) | 2017-10-10 | 2020-08-19 | Capsugel Belgium NV | Gelling multiparticulates |
| MX2022000619A (es) * | 2019-07-16 | 2022-03-11 | Astrazeneca Ab | Composiciones farmaceuticas resistentes a la descarga de dosis que comprenden venirunad. |
| CN117794518A (zh) | 2021-07-29 | 2024-03-29 | 营养与生物科学美国1有限责任公司 | 控释包衣剂 |
| EP4176874A1 (en) | 2021-11-09 | 2023-05-10 | Evonik Operations GmbH | Pharmaceutical or nutraceutical composition with an alcohol resistant single layer coating system |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0725663B2 (ja) | 1985-03-11 | 1995-03-22 | 大正製薬株式会社 | 腸溶性軟カプセルの製造方法 |
| US5882715A (en) | 1995-06-20 | 1999-03-16 | Pharma-Vinci A/S | Method of preparing an oral preparation provided on the outer side with an enteric coating, as well as an oral preparation obtained by the method |
| US5811126A (en) * | 1995-10-02 | 1998-09-22 | Euro-Celtique, S.A. | Controlled release matrix for pharmaceuticals |
| DE10011447A1 (de) | 2000-03-10 | 2001-09-20 | Roehm Gmbh | Dispersion mit nichtionischem Emulgator |
| US6544556B1 (en) * | 2000-09-11 | 2003-04-08 | Andrx Corporation | Pharmaceutical formulations containing a non-steroidal antiinflammatory drug and a proton pump inhibitor |
| US6620426B2 (en) * | 2001-06-05 | 2003-09-16 | Isp Investments Inc. | Tablet coating composition |
| US20040253309A1 (en) * | 2003-01-27 | 2004-12-16 | Yamanouchi Pharmaceutical Co., Ltd. | Enteric sustained-release fine particles of tamsulosin and its salt and manufacturing method thereof |
| JP2004249231A (ja) * | 2003-02-21 | 2004-09-09 | Takeda Chem Ind Ltd | 粒状物の製造方法とその装置 |
| GB0320020D0 (en) * | 2003-08-27 | 2003-10-01 | Mw Encap Ltd | Improved formulation for providing an enteric coating material |
| DE102004011349A1 (de) | 2004-03-05 | 2005-09-22 | Basf Ag | Wässrige Polymerdispersion auf Basis von Alkyl(meth)-acrylaten |
| TW200533391A (en) * | 2004-03-25 | 2005-10-16 | Sun Pharmaceutical Ind Ltd | Gastric retention drug delivery system |
| AU2005231145B8 (en) * | 2004-03-31 | 2010-11-11 | Bpsi Holdings, Inc. | Enteric coatings for orally ingestible substrates |
| CA2561700A1 (en) * | 2004-04-16 | 2005-12-15 | Santarus, Inc. | Combination of proton pump inhibitor, buffering agent, and prokinetic agent |
| KR100676075B1 (ko) * | 2005-02-17 | 2007-02-01 | 한국유나이티드제약 주식회사 | 경구투여용 클래리스로마이신 함유 펠렛 제제 |
| WO2006102964A2 (en) * | 2005-03-29 | 2006-10-05 | Evonik Röhm Gmbh | Multiparticulate pharmaceutical form comprising pellets with a substance having a modular effect in relation to active ingredient release |
| US7645802B2 (en) * | 2005-06-27 | 2010-01-12 | Biovail Laboratories International Srl. | Bupropion hydrobromide and therapeutic applications |
| US9048951B2 (en) | 2005-09-02 | 2015-06-02 | Georgios Margaritis | Free space optics photodetector and transceiver |
| US20070104789A1 (en) | 2005-11-04 | 2007-05-10 | Donald Spector | Gastro-resistant and ethanol-resistant controlled-release formulations comprising hydromorphone |
| WO2007063553A1 (en) * | 2005-11-29 | 2007-06-07 | Ideal Cures Pvt. Ltd. | Aqueous film coating composition containing sodium alginate and preparation thereof |
| MX2009010556A (es) * | 2007-03-29 | 2009-10-22 | Panacea Biotec Ltd | Formas de dosificacion modificada de tacrolimus. |
| MX2010001071A (es) * | 2007-07-27 | 2010-03-09 | Depomed Inc | Formas de dosificacion retentivas gastricas pulsatiles. |
| US9700520B2 (en) * | 2007-09-21 | 2017-07-11 | Evonik Roehm Gmbh | PH-dependent controlled release pharmaceutical opioid composition with resistance against the influence of ethanol |
-
2011
- 2011-04-13 KR KR1020137006647A patent/KR101873075B1/ko active IP Right Grant
- 2011-04-13 EP EP11714754.6A patent/EP2605759B1/en not_active Not-in-force
- 2011-04-13 US US13/086,079 patent/US9492394B2/en active Active
- 2011-04-13 MX MX2013001824A patent/MX360389B/es active IP Right Grant
- 2011-04-13 RU RU2013111605/15A patent/RU2013111605A/ru not_active Application Discontinuation
- 2011-04-13 CA CA2808546A patent/CA2808546C/en active Active
- 2011-04-13 JP JP2013524383A patent/JP5705319B2/ja not_active Expired - Fee Related
- 2011-04-13 WO PCT/EP2011/055809 patent/WO2012022498A1/en active Application Filing
- 2011-04-13 CN CN201180040052.6A patent/CN103068376B/zh not_active Expired - Fee Related
- 2011-04-13 AU AU2011290958A patent/AU2011290958B2/en not_active Ceased
- 2011-04-13 BR BR112013003581A patent/BR112013003581B8/pt not_active IP Right Cessation
- 2011-04-13 ES ES11714754T patent/ES2716550T3/es active Active
-
2013
- 2013-08-08 HK HK13109297.6A patent/HK1182003A1/zh not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| HK1182003A1 (zh) | 2013-11-22 |
| WO2012022498A1 (en) | 2012-02-23 |
| MX2013001824A (es) | 2013-03-08 |
| AU2011290958B2 (en) | 2014-06-19 |
| KR20130099944A (ko) | 2013-09-06 |
| US9492394B2 (en) | 2016-11-15 |
| AU2011290958A1 (en) | 2013-04-04 |
| BR112013003581B1 (pt) | 2021-07-13 |
| CA2808546A1 (en) | 2012-02-23 |
| CA2808546C (en) | 2018-02-20 |
| BR112013003581A2 (pt) | 2016-06-07 |
| JP5705319B2 (ja) | 2015-04-22 |
| US20120093926A1 (en) | 2012-04-19 |
| JP2013535509A (ja) | 2013-09-12 |
| RU2013111605A (ru) | 2014-09-27 |
| EP2605759B1 (en) | 2018-11-21 |
| MX360389B (es) | 2018-10-31 |
| BR112013003581B8 (pt) | 2022-07-05 |
| EP2605759A1 (en) | 2013-06-26 |
| KR101873075B1 (ko) | 2018-06-29 |
| CN103068376A (zh) | 2013-04-24 |
| CN103068376B (zh) | 2016-04-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2716550T3 (es) | Formulación farmacéutica o nutracéutica gastrorresistente que comprende una o más sales de ácido algínico | |
| ES2600860T3 (es) | Composición farmacéutica o nutracéutica con característica de liberación retardada y con resistencia contra la influencia de etanol | |
| ES2625017T3 (es) | Composición farmacéutica o nutracéutica gastrorresistente con resistencia contra la influencia de etanol | |
| ES2838816T3 (es) | Composición farmacéutica o nutracéutica con resistencia contra la influencia de etanol | |
| ES2786312T3 (es) | Composición farmacéutica o nutracéutica con resistencia contra la influencia de etanol | |
| JP6474421B2 (ja) | 持続放出特性を有するとともにエタノールの影響に対する耐性を有する医薬品組成物または栄養機能食品組成物 | |
| ES2909605T3 (es) | Polímero y forma de dosificación con propiedades de liberación mantenida y resistencia contra la influencia del etanol |