ES2699644T3 - Método para preparar penams - Google Patents
Método para preparar penams Download PDFInfo
- Publication number
- ES2699644T3 ES2699644T3 ES15726552T ES15726552T ES2699644T3 ES 2699644 T3 ES2699644 T3 ES 2699644T3 ES 15726552 T ES15726552 T ES 15726552T ES 15726552 T ES15726552 T ES 15726552T ES 2699644 T3 ES2699644 T3 ES 2699644T3
- Authority
- ES
- Spain
- Prior art keywords
- formula
- compound
- alkyl
- independently selected
- halogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims abstract description 44
- 150000002959 penams Chemical class 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 74
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 33
- 125000003342 alkenyl group Chemical group 0.000 claims abstract description 16
- 125000000304 alkynyl group Chemical group 0.000 claims abstract description 16
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 16
- 150000002367 halogens Chemical class 0.000 claims abstract description 16
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims abstract description 16
- 125000006239 protecting group Chemical group 0.000 claims abstract description 9
- 125000003709 fluoroalkyl group Chemical group 0.000 claims abstract description 7
- 238000006243 chemical reaction Methods 0.000 claims description 15
- OBETXYAYXDNJHR-UHFFFAOYSA-N 2-Ethylhexanoic acid Chemical compound CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 claims description 12
- 125000003545 alkoxy group Chemical group 0.000 claims description 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 10
- SHZIWNPUGXLXDT-UHFFFAOYSA-N caproic acid ethyl ester Natural products CCCCCC(=O)OCC SHZIWNPUGXLXDT-UHFFFAOYSA-N 0.000 claims description 9
- 125000001183 hydrocarbyl group Chemical group 0.000 claims description 7
- 229910052751 metal Inorganic materials 0.000 claims description 6
- 239000002184 metal Substances 0.000 claims description 6
- 150000003839 salts Chemical class 0.000 claims description 6
- 150000001450 anions Chemical class 0.000 claims description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 3
- 239000003513 alkali Substances 0.000 claims description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 2
- 239000000010 aprotic solvent Substances 0.000 claims 1
- 125000005010 perfluoroalkyl group Chemical group 0.000 claims 1
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 20
- -1 methyl penam derivatives Chemical class 0.000 description 17
- 239000000243 solution Substances 0.000 description 17
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 14
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 11
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 239000000203 mixture Substances 0.000 description 9
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 230000029936 alkylation Effects 0.000 description 8
- 238000005804 alkylation reaction Methods 0.000 description 8
- 239000003112 inhibitor Substances 0.000 description 8
- 239000008194 pharmaceutical composition Substances 0.000 description 8
- 239000000047 product Substances 0.000 description 7
- 239000007787 solid Substances 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- 239000003782 beta lactam antibiotic agent Substances 0.000 description 6
- 230000003115 biocidal effect Effects 0.000 description 6
- OIRDBPQYVWXNSJ-UHFFFAOYSA-N methyl trifluoromethansulfonate Chemical compound COS(=O)(=O)C(F)(F)F OIRDBPQYVWXNSJ-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 5
- 239000003242 anti bacterial agent Substances 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 239000000499 gel Substances 0.000 description 5
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 5
- 238000006884 silylation reaction Methods 0.000 description 5
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 4
- 229940088710 antibiotic agent Drugs 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- 238000002425 crystallisation Methods 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 230000011987 methylation Effects 0.000 description 4
- 238000007069 methylation reaction Methods 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 229930182555 Penicillin Natural products 0.000 description 3
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 3
- 230000000844 anti-bacterial effect Effects 0.000 description 3
- 239000012296 anti-solvent Substances 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- VUQUOGPMUUJORT-UHFFFAOYSA-N methyl 4-methylbenzenesulfonate Chemical compound COS(=O)(=O)C1=CC=C(C)C=C1 VUQUOGPMUUJORT-UHFFFAOYSA-N 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 3
- VYPDUQYOLCLEGS-UHFFFAOYSA-M sodium;2-ethylhexanoate Chemical compound [Na+].CCCCC(CC)C([O-])=O VYPDUQYOLCLEGS-UHFFFAOYSA-M 0.000 description 3
- 239000012265 solid product Substances 0.000 description 3
- 238000001228 spectrum Methods 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- LPQZKKCYTLCDGQ-WEDXCCLWSA-N tazobactam Chemical compound C([C@]1(C)S([C@H]2N(C(C2)=O)[C@H]1C(O)=O)(=O)=O)N1C=CN=N1 LPQZKKCYTLCDGQ-WEDXCCLWSA-N 0.000 description 3
- SIOVKLKJSOKLIF-HJWRWDBZSA-N trimethylsilyl (1z)-n-trimethylsilylethanimidate Chemical compound C[Si](C)(C)OC(/C)=N\[Si](C)(C)C SIOVKLKJSOKLIF-HJWRWDBZSA-N 0.000 description 3
- UQSHKMPMRUUOJP-BAFYGKSASA-N (5r)-3-methyl-4-thia-1-azabicyclo[3.2.0]heptan-7-one Chemical class S1C(C)CN2C(=O)C[C@H]21 UQSHKMPMRUUOJP-BAFYGKSASA-N 0.000 description 2
- MMRINLZOZVAPDZ-LSGRDSQZSA-N (6r,7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-methoxyiminoacetyl]amino]-3-[(1-methylpyrrolidin-1-ium-1-yl)methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid;chloride Chemical compound Cl.S([C@@H]1[C@@H](C(N1C=1C([O-])=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1C[N+]1(C)CCCC1 MMRINLZOZVAPDZ-LSGRDSQZSA-N 0.000 description 2
- WZPBZJONDBGPKJ-UHFFFAOYSA-N Antibiotic SQ 26917 Natural products O=C1N(S(O)(=O)=O)C(C)C1NC(=O)C(=NOC(C)(C)C(O)=O)C1=CSC(N)=N1 WZPBZJONDBGPKJ-UHFFFAOYSA-N 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- 229930186147 Cephalosporin Natural products 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 238000010268 HPLC based assay Methods 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- 238000001237 Raman spectrum Methods 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 229940126575 aminoglycoside Drugs 0.000 description 2
- WZPBZJONDBGPKJ-VEHQQRBSSA-N aztreonam Chemical compound O=C1N(S([O-])(=O)=O)[C@@H](C)[C@@H]1NC(=O)C(=N/OC(C)(C)C(O)=O)\C1=CSC([NH3+])=N1 WZPBZJONDBGPKJ-VEHQQRBSSA-N 0.000 description 2
- 229960003644 aztreonam Drugs 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 229960000603 cefalotin Drugs 0.000 description 2
- 229960002100 cefepime Drugs 0.000 description 2
- 229960002588 cefradine Drugs 0.000 description 2
- 229960004755 ceftriaxone Drugs 0.000 description 2
- VAAUVRVFOQPIGI-SPQHTLEESA-N ceftriaxone Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC1=NC(=O)C(=O)NN1C VAAUVRVFOQPIGI-SPQHTLEESA-N 0.000 description 2
- 229940106164 cephalexin Drugs 0.000 description 2
- ZAIPMKNFIOOWCQ-UEKVPHQBSA-N cephalexin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=CC=C1 ZAIPMKNFIOOWCQ-UEKVPHQBSA-N 0.000 description 2
- 229940124587 cephalosporin Drugs 0.000 description 2
- 150000001780 cephalosporins Chemical class 0.000 description 2
- VUFGUVLLDPOSBC-XRZFDKQNSA-M cephalothin sodium Chemical compound [Na+].N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC(=O)C)C([O-])=O)C(=O)CC1=CC=CS1 VUFGUVLLDPOSBC-XRZFDKQNSA-M 0.000 description 2
- RDLPVSKMFDYCOR-UEKVPHQBSA-N cephradine Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CCC=CC1 RDLPVSKMFDYCOR-UEKVPHQBSA-N 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 238000005828 desilylation reaction Methods 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 238000010253 intravenous injection Methods 0.000 description 2
- 229960002260 meropenem Drugs 0.000 description 2
- DMJNNHOOLUXYBV-PQTSNVLCSA-N meropenem Chemical compound C=1([C@H](C)[C@@H]2[C@H](C(N2C=1C(O)=O)=O)[C@H](O)C)S[C@@H]1CN[C@H](C(=O)N(C)C)C1 DMJNNHOOLUXYBV-PQTSNVLCSA-N 0.000 description 2
- 150000002960 penicillins Chemical class 0.000 description 2
- 229960002292 piperacillin Drugs 0.000 description 2
- WCMIIGXFCMNQDS-IDYPWDAWSA-M piperacillin sodium Chemical compound [Na+].O=C1C(=O)N(CC)CCN1C(=O)N[C@H](C=1C=CC=CC=1)C(=O)N[C@@H]1C(=O)N2[C@@H](C([O-])=O)C(C)(C)S[C@@H]21 WCMIIGXFCMNQDS-IDYPWDAWSA-M 0.000 description 2
- 238000000634 powder X-ray diffraction Methods 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 238000004626 scanning electron microscopy Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- 239000011343 solid material Substances 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- WDLWHQDACQUCJR-ZAMMOSSLSA-N (6r,7r)-7-[[(2r)-2-azaniumyl-2-(4-hydroxyphenyl)acetyl]amino]-8-oxo-3-[(e)-prop-1-enyl]-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)/C=C/C)C(O)=O)=CC=C(O)C=C1 WDLWHQDACQUCJR-ZAMMOSSLSA-N 0.000 description 1
- YWKJNRNSJKEFMK-PQFQYKRASA-N (6r,7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-methoxyiminoacetyl]amino]-8-oxo-3-(5,6,7,8-tetrahydroquinolin-1-ium-1-ylmethyl)-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate Chemical compound N([C@@H]1C(N2C(=C(C[N+]=3C=4CCCCC=4C=CC=3)CS[C@@H]21)C([O-])=O)=O)C(=O)\C(=N/OC)C1=CSC(N)=N1 YWKJNRNSJKEFMK-PQFQYKRASA-N 0.000 description 1
- XUBOMFCQGDBHNK-JTQLQIEISA-N (S)-gatifloxacin Chemical compound FC1=CC(C(C(C(O)=O)=CN2C3CC3)=O)=C2C(OC)=C1N1CCN[C@@H](C)C1 XUBOMFCQGDBHNK-JTQLQIEISA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- DVLFYONBTKHTER-UHFFFAOYSA-N 3-(N-morpholino)propanesulfonic acid Chemical compound OS(=O)(=O)CCCN1CCOCC1 DVLFYONBTKHTER-UHFFFAOYSA-N 0.000 description 1
- HGGAKXAHAYOLDJ-FHZUQPTBSA-N 6alpha-[(R)-1-hydroxyethyl]-2-[(R)-tetrahydrofuran-2-yl]pen-2-em-3-carboxylic acid Chemical compound S([C@@H]1[C@H](C(N1C=1C(O)=O)=O)[C@H](O)C)C=1[C@H]1CCCO1 HGGAKXAHAYOLDJ-FHZUQPTBSA-N 0.000 description 1
- 108700042778 Antimicrobial Peptides Proteins 0.000 description 1
- 102000044503 Antimicrobial Peptides Human genes 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- UQLLWWBDSUHNEB-CZUORRHYSA-N Cefaprin Chemical compound N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC(=O)C)C(O)=O)C(=O)CSC1=CC=NC=C1 UQLLWWBDSUHNEB-CZUORRHYSA-N 0.000 description 1
- QYQDKDWGWDOFFU-IUODEOHRSA-N Cefotiam Chemical compound CN(C)CCN1N=NN=C1SCC1=C(C(O)=O)N2C(=O)[C@@H](NC(=O)CC=3N=C(N)SC=3)[C@H]2SC1 QYQDKDWGWDOFFU-IUODEOHRSA-N 0.000 description 1
- 108010078777 Colistin Proteins 0.000 description 1
- JWCSIUVGFCSJCK-CAVRMKNVSA-N Disodium Moxalactam Chemical compound N([C@]1(OC)C(N2C(=C(CSC=3N(N=NN=3)C)CO[C@@H]21)C(O)=O)=O)C(=O)C(C(O)=O)C1=CC=C(O)C=C1 JWCSIUVGFCSJCK-CAVRMKNVSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- UIOFUWFRIANQPC-JKIFEVAISA-N Floxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=C(F)C=CC=C1Cl UIOFUWFRIANQPC-JKIFEVAISA-N 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- JUZNIMUFDBIJCM-ANEDZVCMSA-N Invanz Chemical compound O=C([C@H]1NC[C@H](C1)SC=1[C@H](C)[C@@H]2[C@H](C(N2C=1C(O)=O)=O)[C@H](O)C)NC1=CC=CC(C(O)=O)=C1 JUZNIMUFDBIJCM-ANEDZVCMSA-N 0.000 description 1
- GSDSWSVVBLHKDQ-JTQLQIEISA-N Levofloxacin Chemical compound C([C@@H](N1C2=C(C(C(C(O)=O)=C1)=O)C=C1F)C)OC2=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-JTQLQIEISA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- RJQXTJLFIWVMTO-TYNCELHUSA-N Methicillin Chemical compound COC1=CC=CC(OC)=C1C(=O)N[C@@H]1C(=O)N2[C@@H](C(O)=O)C(C)(C)S[C@@H]21 RJQXTJLFIWVMTO-TYNCELHUSA-N 0.000 description 1
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- TYMABNNERDVXID-DLYFRVTGSA-N Panipenem Chemical compound C([C@@H]1[C@H](C(N1C=1C(O)=O)=O)[C@H](O)C)C=1S[C@H]1CCN(C(C)=N)C1 TYMABNNERDVXID-DLYFRVTGSA-N 0.000 description 1
- 206010034133 Pathogen resistance Diseases 0.000 description 1
- 108010093965 Polymyxin B Proteins 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- URWAJWIAIPFPJE-UHFFFAOYSA-N Rickamicin Natural products O1CC(O)(C)C(NC)C(O)C1OC1C(O)C(OC2C(CC=C(CN)O2)N)C(N)CC1N URWAJWIAIPFPJE-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- WKDDRNSBRWANNC-UHFFFAOYSA-N Thienamycin Natural products C1C(SCCN)=C(C(O)=O)N2C(=O)C(C(O)C)C21 WKDDRNSBRWANNC-UHFFFAOYSA-N 0.000 description 1
- KLFSEZJCLYBFKQ-WXYNYTDUSA-N [(3s)-3-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-[(1,5-dihydroxy-4-oxopyridin-2-yl)methoxyimino]acetyl]amino]-2,2-dimethyl-4-oxoazetidin-1-yl] hydrogen sulfate Chemical compound O=C1N(OS(O)(=O)=O)C(C)(C)[C@@H]1NC(=O)C(\C=1N=C(N)SC=1)=N/OCC1=CC(=O)C(O)=CN1O KLFSEZJCLYBFKQ-WXYNYTDUSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000005037 alkyl phenyl group Chemical group 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 229940024554 amdinocillin Drugs 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229960004821 amikacin Drugs 0.000 description 1
- LKCWBDHBTVXHDL-RMDFUYIESA-N amikacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)[C@@H](O)[C@H](O)[C@H]1O LKCWBDHBTVXHDL-RMDFUYIESA-N 0.000 description 1
- 229960003022 amoxicillin Drugs 0.000 description 1
- LSQZJLSUYDQPKJ-NJBDSQKTSA-N amoxicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=C(O)C=C1 LSQZJLSUYDQPKJ-NJBDSQKTSA-N 0.000 description 1
- 229960000723 ampicillin Drugs 0.000 description 1
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 1
- 230000002141 anti-parasite Effects 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000003096 antiparasitic agent Substances 0.000 description 1
- 229950006334 apramycin Drugs 0.000 description 1
- XZNUGFQTQHRASN-XQENGBIVSA-N apramycin Chemical compound O([C@H]1O[C@@H]2[C@H](O)[C@@H]([C@H](O[C@H]2C[C@H]1N)O[C@@H]1[C@@H]([C@@H](O)[C@H](N)[C@@H](CO)O1)O)NC)[C@@H]1[C@@H](N)C[C@@H](N)[C@H](O)[C@H]1O XZNUGFQTQHRASN-XQENGBIVSA-N 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 229960005397 arbekacin Drugs 0.000 description 1
- MKKYBZZTJQGVCD-XTCKQBCOSA-N arbekacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)CC[C@H]1N MKKYBZZTJQGVCD-XTCKQBCOSA-N 0.000 description 1
- 229960003623 azlocillin Drugs 0.000 description 1
- JTWOMNBEOCYFNV-NFFDBFGFSA-N azlocillin Chemical compound N([C@@H](C(=O)N[C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C=1C=CC=CC=1)C(=O)N1CCNC1=O JTWOMNBEOCYFNV-NFFDBFGFSA-N 0.000 description 1
- 229960002699 bacampicillin Drugs 0.000 description 1
- PFOLLRNADZZWEX-FFGRCDKISA-N bacampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)[C@H](C(S3)(C)C)C(=O)OC(C)OC(=O)OCC)=CC=CC=C1 PFOLLRNADZZWEX-FFGRCDKISA-N 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229940041011 carbapenems Drugs 0.000 description 1
- UIMOJFJSJSIGLV-JNHMLNOCSA-N carumonam Chemical compound O=C1N(S(O)(=O)=O)[C@H](COC(=O)N)[C@@H]1NC(=O)C(=N/OCC(O)=O)\C1=CSC(N)=N1 UIMOJFJSJSIGLV-JNHMLNOCSA-N 0.000 description 1
- 229960000662 carumonam Drugs 0.000 description 1
- 229960005361 cefaclor Drugs 0.000 description 1
- QYIYFLOTGYLRGG-GPCCPHFNSA-N cefaclor Chemical compound C1([C@H](C(=O)N[C@@H]2C(N3C(=C(Cl)CS[C@@H]32)C(O)=O)=O)N)=CC=CC=C1 QYIYFLOTGYLRGG-GPCCPHFNSA-N 0.000 description 1
- 229960004841 cefadroxil Drugs 0.000 description 1
- NBFNMSULHIODTC-CYJZLJNKSA-N cefadroxil monohydrate Chemical compound O.C1([C@@H](N)C(=O)N[C@H]2[C@@H]3N(C2=O)C(=C(CS3)C)C(O)=O)=CC=C(O)C=C1 NBFNMSULHIODTC-CYJZLJNKSA-N 0.000 description 1
- 229960003012 cefamandole Drugs 0.000 description 1
- OLVCFLKTBJRLHI-AXAPSJFSSA-N cefamandole Chemical compound CN1N=NN=C1SCC1=C(C(O)=O)N2C(=O)[C@@H](NC(=O)[C@H](O)C=3C=CC=CC=3)[C@H]2SC1 OLVCFLKTBJRLHI-AXAPSJFSSA-N 0.000 description 1
- 229960004350 cefapirin Drugs 0.000 description 1
- 229960001139 cefazolin Drugs 0.000 description 1
- MLYYVTUWGNIJIB-BXKDBHETSA-N cefazolin Chemical compound S1C(C)=NN=C1SCC1=C(C(O)=O)N2C(=O)[C@@H](NC(=O)CN3N=NN=C3)[C@H]2SC1 MLYYVTUWGNIJIB-BXKDBHETSA-N 0.000 description 1
- 229960001817 cefbuperazone Drugs 0.000 description 1
- SMSRCGPDNDCXFR-CYWZMYCQSA-N cefbuperazone Chemical compound O=C1C(=O)N(CC)CCN1C(=O)N[C@H]([C@H](C)O)C(=O)N[C@]1(OC)C(=O)N2C(C(O)=O)=C(CSC=3N(N=NN=3)C)CS[C@@H]21 SMSRCGPDNDCXFR-CYWZMYCQSA-N 0.000 description 1
- 229960003719 cefdinir Drugs 0.000 description 1
- RTXOFQZKPXMALH-GHXIOONMSA-N cefdinir Chemical compound S1C(N)=NC(C(=N\O)\C(=O)N[C@@H]2C(N3C(=C(C=C)CS[C@@H]32)C(O)=O)=O)=C1 RTXOFQZKPXMALH-GHXIOONMSA-N 0.000 description 1
- 229960004041 cefetamet Drugs 0.000 description 1
- MQLRYUCJDNBWMV-GHXIOONMSA-N cefetamet Chemical compound N([C@@H]1C(N2C(=C(C)CS[C@@H]21)C(O)=O)=O)C(=O)\C(=N/OC)C1=CSC(N)=N1 MQLRYUCJDNBWMV-GHXIOONMSA-N 0.000 description 1
- 229960002129 cefixime Drugs 0.000 description 1
- OKBVVJOGVLARMR-QSWIMTSFSA-N cefixime Chemical compound S1C(N)=NC(C(=N\OCC(O)=O)\C(=O)N[C@@H]2C(N3C(=C(C=C)CS[C@@H]32)C(O)=O)=O)=C1 OKBVVJOGVLARMR-QSWIMTSFSA-N 0.000 description 1
- HJJDBAOLQAWBMH-YCRCPZNHSA-N cefmenoxime Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC1=NN=NN1C HJJDBAOLQAWBMH-YCRCPZNHSA-N 0.000 description 1
- 229960004489 cefonicid Drugs 0.000 description 1
- DYAIAHUQIPBDIP-AXAPSJFSSA-N cefonicid Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)[C@H](O)C=2C=CC=CC=2)CC=1CSC1=NN=NN1CS(O)(=O)=O DYAIAHUQIPBDIP-AXAPSJFSSA-N 0.000 description 1
- 229960004682 cefoperazone Drugs 0.000 description 1
- GCFBRXLSHGKWDP-XCGNWRKASA-N cefoperazone Chemical compound O=C1C(=O)N(CC)CCN1C(=O)N[C@H](C=1C=CC(O)=CC=1)C(=O)N[C@@H]1C(=O)N2C(C(O)=O)=C(CSC=3N(N=NN=3)C)CS[C@@H]21 GCFBRXLSHGKWDP-XCGNWRKASA-N 0.000 description 1
- 229960004261 cefotaxime Drugs 0.000 description 1
- AZZMGZXNTDTSME-JUZDKLSSSA-M cefotaxime sodium Chemical compound [Na+].N([C@@H]1C(N2C(=C(COC(C)=O)CS[C@@H]21)C([O-])=O)=O)C(=O)\C(=N/OC)C1=CSC(N)=N1 AZZMGZXNTDTSME-JUZDKLSSSA-M 0.000 description 1
- 229960005495 cefotetan Drugs 0.000 description 1
- SRZNHPXWXCNNDU-RHBCBLIFSA-N cefotetan Chemical compound N([C@]1(OC)C(N2C(=C(CSC=3N(N=NN=3)C)CS[C@@H]21)C(O)=O)=O)C(=O)C1SC(=C(C(N)=O)C(O)=O)S1 SRZNHPXWXCNNDU-RHBCBLIFSA-N 0.000 description 1
- 229960001242 cefotiam Drugs 0.000 description 1
- ZJGQFXVQDVCVOK-MSUXKOGISA-N cefovecin Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)/C(=N/OC)C=2N=C(N)SC=2)CC=1[C@@H]1CCCO1 ZJGQFXVQDVCVOK-MSUXKOGISA-N 0.000 description 1
- 229960003391 cefovecin Drugs 0.000 description 1
- 229960005090 cefpodoxime Drugs 0.000 description 1
- WYUSVOMTXWRGEK-HBWVYFAYSA-N cefpodoxime Chemical compound N([C@H]1[C@@H]2N(C1=O)C(=C(CS2)COC)C(O)=O)C(=O)C(=N/OC)\C1=CSC(N)=N1 WYUSVOMTXWRGEK-HBWVYFAYSA-N 0.000 description 1
- 229960002580 cefprozil Drugs 0.000 description 1
- 229950009592 cefquinome Drugs 0.000 description 1
- 229960003202 cefsulodin Drugs 0.000 description 1
- SYLKGLMBLAAGSC-QLVMHMETSA-N cefsulodin Chemical compound C1=CC(C(=O)N)=CC=[N+]1CC1=C(C([O-])=O)N2C(=O)[C@@H](NC(=O)[C@@H](C=3C=CC=CC=3)S(O)(=O)=O)[C@H]2SC1 SYLKGLMBLAAGSC-QLVMHMETSA-N 0.000 description 1
- 229960000484 ceftazidime Drugs 0.000 description 1
- NMVPEQXCMGEDNH-TZVUEUGBSA-N ceftazidime pentahydrate Chemical compound O.O.O.O.O.S([C@@H]1[C@@H](C(N1C=1C([O-])=O)=O)NC(=O)\C(=N/OC(C)(C)C(O)=O)C=2N=C(N)SC=2)CC=1C[N+]1=CC=CC=C1 NMVPEQXCMGEDNH-TZVUEUGBSA-N 0.000 description 1
- 229960004366 ceftezole Drugs 0.000 description 1
- DZMVCVMFETWNIU-LDYMZIIASA-N ceftezole Chemical compound O=C([C@@H](NC(=O)CN1N=NN=C1)[C@H]1SC2)N1C(C(=O)O)=C2CSC1=NN=CS1 DZMVCVMFETWNIU-LDYMZIIASA-N 0.000 description 1
- 229960004086 ceftibuten Drugs 0.000 description 1
- UNJFKXSSGBWRBZ-BJCIPQKHSA-N ceftibuten Chemical compound S1C(N)=NC(C(=C\CC(O)=O)\C(=O)N[C@@H]2C(N3C(=CCS[C@@H]32)C(O)=O)=O)=C1 UNJFKXSSGBWRBZ-BJCIPQKHSA-N 0.000 description 1
- 229960005229 ceftiofur Drugs 0.000 description 1
- ZBHXIWJRIFEVQY-IHMPYVIRSA-N ceftiofur Chemical compound S([C@@H]1[C@@H](C(N1C=1C(O)=O)=O)NC(=O)\C(=N/OC)C=2N=C(N)SC=2)CC=1CSC(=O)C1=CC=CO1 ZBHXIWJRIFEVQY-IHMPYVIRSA-N 0.000 description 1
- 229960001991 ceftizoxime Drugs 0.000 description 1
- NNULBSISHYWZJU-LLKWHZGFSA-N ceftizoxime Chemical compound N([C@@H]1C(N2C(=CCS[C@@H]21)C(O)=O)=O)C(=O)\C(=N/OC)C1=CSC(N)=N1 NNULBSISHYWZJU-LLKWHZGFSA-N 0.000 description 1
- VOAZJEPQLGBXGO-SDAWRPRTSA-N ceftobiprole Chemical compound S1C(N)=NC(C(=N\O)\C(=O)N[C@@H]2C(N3C(=C(\C=C/4C(N([C@H]5CNCC5)CC\4)=O)CS[C@@H]32)C(O)=O)=O)=N1 VOAZJEPQLGBXGO-SDAWRPRTSA-N 0.000 description 1
- 229950004259 ceftobiprole Drugs 0.000 description 1
- 229960001668 cefuroxime Drugs 0.000 description 1
- JFPVXVDWJQMJEE-IZRZKJBUSA-N cefuroxime Chemical compound N([C@@H]1C(N2C(=C(COC(N)=O)CS[C@@H]21)C(O)=O)=O)C(=O)\C(=N/OC)C1=CC=CO1 JFPVXVDWJQMJEE-IZRZKJBUSA-N 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- BWWVAEOLVKTZFQ-ISVUSNJMSA-N chembl530 Chemical compound N(/[C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)=C\N1CCCCCC1 BWWVAEOLVKTZFQ-ISVUSNJMSA-N 0.000 description 1
- 229960004621 cinoxacin Drugs 0.000 description 1
- VDUWPHTZYNWKRN-UHFFFAOYSA-N cinoxacin Chemical compound C1=C2N(CC)N=C(C(O)=O)C(=O)C2=CC2=C1OCO2 VDUWPHTZYNWKRN-UHFFFAOYSA-N 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- 229960003326 cloxacillin Drugs 0.000 description 1
- LQOLIRLGBULYKD-JKIFEVAISA-N cloxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=CC=CC=C1Cl LQOLIRLGBULYKD-JKIFEVAISA-N 0.000 description 1
- 229960003346 colistin Drugs 0.000 description 1
- 239000008139 complexing agent Substances 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 229960003807 dibekacin Drugs 0.000 description 1
- JJCQSGDBDPYCEO-XVZSLQNASA-N dibekacin Chemical compound O1[C@H](CN)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N JJCQSGDBDPYCEO-XVZSLQNASA-N 0.000 description 1
- YFAGHNZHGGCZAX-JKIFEVAISA-N dicloxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=C(Cl)C=CC=C1Cl YFAGHNZHGGCZAX-JKIFEVAISA-N 0.000 description 1
- 229960001585 dicloxacillin Drugs 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 229960000895 doripenem Drugs 0.000 description 1
- AVAACINZEOAHHE-VFZPANTDSA-N doripenem Chemical compound C=1([C@H](C)[C@@H]2[C@H](C(N2C=1C(O)=O)=O)[C@H](O)C)S[C@@H]1CN[C@H](CNS(N)(=O)=O)C1 AVAACINZEOAHHE-VFZPANTDSA-N 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 229950007919 egtazic acid Drugs 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 229960002549 enoxacin Drugs 0.000 description 1
- IDYZIJYBMGIQMJ-UHFFFAOYSA-N enoxacin Chemical compound N1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNCC1 IDYZIJYBMGIQMJ-UHFFFAOYSA-N 0.000 description 1
- 229960002770 ertapenem Drugs 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 229960000379 faropenem Drugs 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229960002878 flomoxef Drugs 0.000 description 1
- UHRBTBZOWWGKMK-DOMZBBRYSA-N flomoxef Chemical compound O([C@@H]1[C@@](C(N1C=1C(O)=O)=O)(NC(=O)CSC(F)F)OC)CC=1CSC1=NN=NN1CCO UHRBTBZOWWGKMK-DOMZBBRYSA-N 0.000 description 1
- 229960004273 floxacillin Drugs 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 229960003923 gatifloxacin Drugs 0.000 description 1
- 229960003170 gemifloxacin Drugs 0.000 description 1
- ZRCVYEYHRGVLOC-HYARGMPZSA-N gemifloxacin Chemical compound C1C(CN)C(=N/OC)/CN1C(C(=C1)F)=NC2=C1C(=O)C(C(O)=O)=CN2C1CC1 ZRCVYEYHRGVLOC-HYARGMPZSA-N 0.000 description 1
- 229960002518 gentamicin Drugs 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- VKWVKRZCWUQLHV-UHFFFAOYSA-N hex-3-ene-1,6-diamine Chemical group NCCC=CCCN VKWVKRZCWUQLHV-UHFFFAOYSA-N 0.000 description 1
- 238000007327 hydrogenolysis reaction Methods 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 229960002182 imipenem Drugs 0.000 description 1
- ZSKVGTPCRGIANV-ZXFLCMHBSA-N imipenem Chemical compound C1C(SCC\N=C\N)=C(C(O)=O)N2C(=O)[C@H]([C@H](O)C)[C@H]21 ZSKVGTPCRGIANV-ZXFLCMHBSA-N 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229960000318 kanamycin Drugs 0.000 description 1
- 229930027917 kanamycin Natural products 0.000 description 1
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 1
- 229930182823 kanamycin A Natural products 0.000 description 1
- 229960000433 latamoxef Drugs 0.000 description 1
- ZKUKMWMSYCIYRD-ZXFNITATSA-N lenampicillin Chemical compound O1C(=O)OC(COC(=O)[C@H]2C(S[C@H]3N2C([C@H]3NC(=O)[C@H](N)C=2C=CC=CC=2)=O)(C)C)=C1C ZKUKMWMSYCIYRD-ZXFNITATSA-N 0.000 description 1
- 229950005831 lenampicillin Drugs 0.000 description 1
- 229960003376 levofloxacin Drugs 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 238000002514 liquid chromatography mass spectrum Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 229960001977 loracarbef Drugs 0.000 description 1
- JAPHQRWPEGVNBT-UTUOFQBUSA-N loracarbef Chemical compound C1([C@H](C(=O)N[C@@H]2C(N3C(=C(Cl)CC[C@@H]32)C([O-])=O)=O)[NH3+])=CC=CC=C1 JAPHQRWPEGVNBT-UTUOFQBUSA-N 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 239000012022 methylating agents Substances 0.000 description 1
- 229960003085 meticillin Drugs 0.000 description 1
- 229960000198 mezlocillin Drugs 0.000 description 1
- YPBATNHYBCGSSN-VWPFQQQWSA-N mezlocillin Chemical compound N([C@@H](C(=O)N[C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C=1C=CC=CC=1)C(=O)N1CCN(S(C)(=O)=O)C1=O YPBATNHYBCGSSN-VWPFQQQWSA-N 0.000 description 1
- 229940041009 monobactams Drugs 0.000 description 1
- 229960003702 moxifloxacin Drugs 0.000 description 1
- FABPRXSRWADJSP-MEDUHNTESA-N moxifloxacin Chemical compound COC1=C(N2C[C@H]3NCCC[C@H]3C2)C(F)=CC(C(C(C(O)=O)=C2)=O)=C1N2C1CC1 FABPRXSRWADJSP-MEDUHNTESA-N 0.000 description 1
- 230000036457 multidrug resistance Effects 0.000 description 1
- LULXBAGMGMJJRW-UHFFFAOYSA-N n,2-bis(trimethylsilyl)acetamide Chemical compound C[Si](C)(C)CC(=O)N[Si](C)(C)C LULXBAGMGMJJRW-UHFFFAOYSA-N 0.000 description 1
- JORAUNFTUVJTNG-BSTBCYLQSA-N n-[(2s)-4-amino-1-[[(2s,3r)-1-[[(2s)-4-amino-1-oxo-1-[[(3s,6s,9s,12s,15r,18s,21s)-6,9,18-tris(2-aminoethyl)-3-[(1r)-1-hydroxyethyl]-12,15-bis(2-methylpropyl)-2,5,8,11,14,17,20-heptaoxo-1,4,7,10,13,16,19-heptazacyclotricos-21-yl]amino]butan-2-yl]amino]-3-h Chemical compound CC(C)CCCCC(=O)N[C@@H](CCN)C(=O)N[C@H]([C@@H](C)O)CN[C@@H](CCN)C(=O)N[C@H]1CCNC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCN)NC(=O)[C@H](CCN)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CCN)NC1=O.CCC(C)CCCCC(=O)N[C@@H](CCN)C(=O)N[C@H]([C@@H](C)O)CN[C@@H](CCN)C(=O)N[C@H]1CCNC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCN)NC(=O)[C@H](CCN)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CCN)NC1=O JORAUNFTUVJTNG-BSTBCYLQSA-N 0.000 description 1
- GPXLMGHLHQJAGZ-JTDSTZFVSA-N nafcillin Chemical compound C1=CC=CC2=C(C(=O)N[C@@H]3C(N4[C@H](C(C)(C)S[C@@H]43)C(O)=O)=O)C(OCC)=CC=C21 GPXLMGHLHQJAGZ-JTDSTZFVSA-N 0.000 description 1
- 229960000515 nafcillin Drugs 0.000 description 1
- 229960000210 nalidixic acid Drugs 0.000 description 1
- MHWLWQUZZRMNGJ-UHFFFAOYSA-N nalidixic acid Chemical compound C1=C(C)N=C2N(CC)C=C(C(O)=O)C(=O)C2=C1 MHWLWQUZZRMNGJ-UHFFFAOYSA-N 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 229960000808 netilmicin Drugs 0.000 description 1
- ZBGPYVZLYBDXKO-HILBYHGXSA-N netilmycin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@]([C@H](NC)[C@@H](O)CO1)(C)O)NCC)[C@H]1OC(CN)=CC[C@H]1N ZBGPYVZLYBDXKO-HILBYHGXSA-N 0.000 description 1
- MGFYIUFZLHCRTH-UHFFFAOYSA-N nitrilotriacetic acid Chemical compound OC(=O)CN(CC(O)=O)CC(O)=O MGFYIUFZLHCRTH-UHFFFAOYSA-N 0.000 description 1
- 229960001180 norfloxacin Drugs 0.000 description 1
- OGJPXUAPXNRGGI-UHFFFAOYSA-N norfloxacin Chemical compound C1=C2N(CC)C=C(C(O)=O)C(=O)C2=CC(F)=C1N1CCNCC1 OGJPXUAPXNRGGI-UHFFFAOYSA-N 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 230000006911 nucleation Effects 0.000 description 1
- 125000001181 organosilyl group Chemical group [SiH3]* 0.000 description 1
- UWYHMGVUTGAWSP-JKIFEVAISA-N oxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=CC=CC=C1 UWYHMGVUTGAWSP-JKIFEVAISA-N 0.000 description 1
- 229960001019 oxacillin Drugs 0.000 description 1
- LSQZJLSUYDQPKJ-UHFFFAOYSA-N p-Hydroxyampicillin Natural products O=C1N2C(C(O)=O)C(C)(C)SC2C1NC(=O)C(N)C1=CC=C(O)C=C1 LSQZJLSUYDQPKJ-UHFFFAOYSA-N 0.000 description 1
- 229950011346 panipenem Drugs 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 229940056360 penicillin g Drugs 0.000 description 1
- 229960003330 pentetic acid Drugs 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 239000003880 polar aprotic solvent Substances 0.000 description 1
- 229920000024 polymyxin B Polymers 0.000 description 1
- XDJYMJULXQKGMM-UHFFFAOYSA-N polymyxin E1 Natural products CCC(C)CCCCC(=O)NC(CCN)C(=O)NC(C(C)O)C(=O)NC(CCN)C(=O)NC1CCNC(=O)C(C(C)O)NC(=O)C(CCN)NC(=O)C(CCN)NC(=O)C(CC(C)C)NC(=O)C(CC(C)C)NC(=O)C(CCN)NC1=O XDJYMJULXQKGMM-UHFFFAOYSA-N 0.000 description 1
- KNIWPHSUTGNZST-UHFFFAOYSA-N polymyxin E2 Natural products CC(C)CCCCC(=O)NC(CCN)C(=O)NC(C(C)O)C(=O)NC(CCN)C(=O)NC1CCNC(=O)C(C(C)O)NC(=O)C(CCN)NC(=O)C(CCN)NC(=O)C(CC(C)C)NC(=O)C(CC(C)C)NC(=O)C(CCN)NC1=O KNIWPHSUTGNZST-UHFFFAOYSA-N 0.000 description 1
- 229960005266 polymyxin b Drugs 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 150000007660 quinolones Chemical class 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- URWAJWIAIPFPJE-YFMIWBNJSA-N sisomycin Chemical compound O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H](CC=C(CN)O2)N)[C@@H](N)C[C@H]1N URWAJWIAIPFPJE-YFMIWBNJSA-N 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- HELHAJAZNSDZJO-OLXYHTOASA-L sodium L-tartrate Chemical compound [Na+].[Na+].[O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O HELHAJAZNSDZJO-OLXYHTOASA-L 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000017550 sodium carbonate Nutrition 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000011083 sodium citrates Nutrition 0.000 description 1
- 239000001433 sodium tartrate Substances 0.000 description 1
- 229960002167 sodium tartrate Drugs 0.000 description 1
- 235000011004 sodium tartrates Nutrition 0.000 description 1
- 238000011146 sterile filtration Methods 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 229960001114 temocillin Drugs 0.000 description 1
- BVCKFLJARNKCSS-DWPRYXJFSA-N temocillin Chemical compound N([C@]1(OC)C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C(C(O)=O)C=1C=CSC=1 BVCKFLJARNKCSS-DWPRYXJFSA-N 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 229960004659 ticarcillin Drugs 0.000 description 1
- OHKOGUYZJXTSFX-KZFFXBSXSA-N ticarcillin Chemical compound C=1([C@@H](C(O)=O)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)C=CSC=1 OHKOGUYZJXTSFX-KZFFXBSXSA-N 0.000 description 1
- 229950010206 tigemonam Drugs 0.000 description 1
- VAMSVIZLXJOLHZ-QWFSEIHXSA-N tigemonam Chemical compound O=C1N(OS(O)(=O)=O)C(C)(C)[C@@H]1NC(=O)C(=N/OCC(O)=O)\C1=CSC(N)=N1 VAMSVIZLXJOLHZ-QWFSEIHXSA-N 0.000 description 1
- 229960000707 tobramycin Drugs 0.000 description 1
- NLVFBUXFDBBNBW-PBSUHMDJSA-N tobramycin Chemical compound N[C@@H]1C[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N NLVFBUXFDBBNBW-PBSUHMDJSA-N 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1896—Compounds having one or more Si-O-acyl linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D499/00—Heterocyclic compounds containing 4-thia-1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. penicillins, penems; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D499/87—Compounds being unsubstituted in position 3 or with substituents other than only two methyl radicals attached in position 3, and with a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, e.g. an ester or nitrile radical, directly attached in position 2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/429—Thiazoles condensed with heterocyclic ring systems
- A61K31/43—Compounds containing 4-thia-1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula, e.g. penicillins, penems
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Steroid Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Feed For Specific Animals (AREA)
Abstract
Un método para formar un compuesto de fórmula (IIIa):**Fórmula** en donde: R1 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5; R2 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5; R4 es alquilo C1-5; y R6 es un fluoroalquilo C1-5; y PG es un grupo protector, el método comprende la etapa de hacer reaccionar un compuesto de fórmula (IIa) con un compuesto de fórmula (VIII)**Fórmula**
Description
DESCRIPCIÓN
Método para preparar penams
Campo de la invención
La presente invención se relaciona con métodos para formar derivados de metil penam, particularmente derivados de metil penam adecuados para su uso con antibióticos de p-lactamas como inhibidores de p-lactamasas.
Antecedentes de la invención
La aparición y diseminación de la resistencia es una consecuencia inevitable de la dinámica evolutiva puesta en marcha por la introducción de antibióticos, independientemente de la clase estructural o el modo de acción (Shapiro S. 2013. Speculative strategies for new antibacterials: all roads should not lead to Rome. J. Antibiot. 66: 371-386). La propagación de la resistencia entre patógenos clínicamente relevantes ha tenido un impacto especialmente fuerte en el valor de los antibióticos de p-lactamas, hasta ahora considerados como terapias muy seguras y eficaces para infecciones bacterianas graves. La aparición de nuevas y agresivas p-lactamasas, particularmente de p-lactamasas de espectro extendido (ESBL) y otras enzimas de clase A, ha comprometido la capacidad de las p-lactamas para combatir infecciones, lo que resalta la necesidad de desarrollar nuevos productos (Fisher JF, Meroueh SO, Mobashery S. 2005. Bacterial resistance to p-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 105: 395-424). Aunque varios inhibidores de la p-lactamasas, que protegen los antibióticos de p-lactamas de la hidrólisis, se han usado en combinación con algunas p-lactamas, la capacidad de estos inhibidores de p-lactamasas para conservar la actividad antibacteriana de las p-lactamas se ha erosionado severamente durante la pasada década, lo que requiere la búsqueda de nuevos y más potentes inhibidores de p-lactamasas para restaurar la utilidad terapéutica de sus socios de plactamas (Watkins RR, Papp-Wallace KM, Drawz SM, Bonomo RA. 2013. Novel p-lactamase inhibitors: a therapeutic hope againstthe scourge of multidrug resistance. Front. Microbiol. 4: 392).
El documento WO 2008/010048 describe inhibidores de p -lactamasas que tienen la siguiente fórmula:

Los inhibidores de p-lactamasas descritos en el documento WO 2008/010048 incluyen el compuesto 4,4-dióxido de (2S,3S,5R)-3-metil-3-((3-metil-1H-1,2,3-triazol-3-io-1-il)metil)-7-oxo-4-tia-1-azabiciclo[3.2.0]heptano-2-carboxilato (fórmula A):

El grupo R se forma mediante una reacción de sustitución, por ejemplo, mediante reacción con yoduro de metilo en el caso de la fórmula (A).
El documento WO 2008/010048 describe la formación de compuestos amorfos aislados por filtración y liofilización. Es un objeto de la invención proporcionar un proceso mejorado para la fabricación de derivados de 2-metilpenam. Es un objeto adicional de la invención proporcionar un proceso para la fabricación de derivados de 2-metilpenam que sea adecuado para la fabricación a escala industrial.
Resumen de la invención
En un primer aspecto la invención proporciona un método para formar un compuesto de fórmula (Illa):

Fórmula (IIIa)
en donde:
R1 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5; R2 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5; R4 es alquilo C1-5; y
R6 es un fluoroalquilo C1-5; y
PG es un grupo protector,
el método que comprende la etapa de hacer reaccionar un compuesto de fórmula (IIa) con un compuesto de fórmula (VIII):
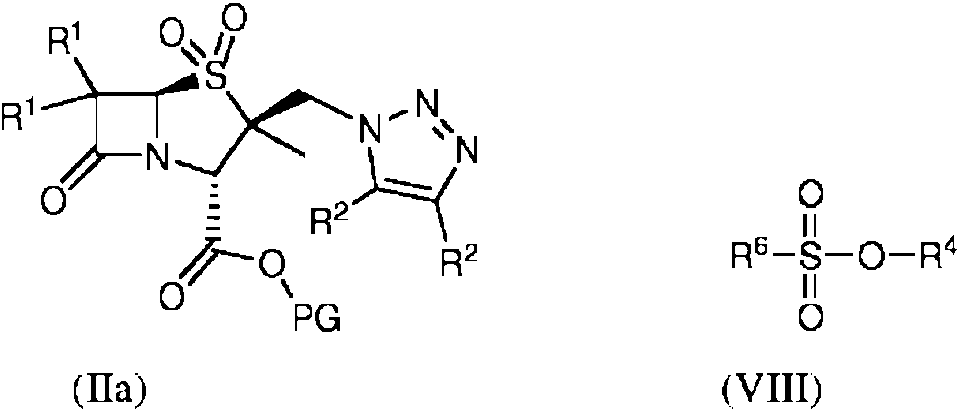
En un segundo aspecto, la invención proporciona el método del primer aspecto que comprende además un método para formar un compuesto de fórmula (IV):

en donde R1 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5; R2 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5; y R4 es alquilo C1-5; el método que comprende la etapa de hacer reaccionar un compuesto de fórmula (III) con una sal de 2-etilhexanoato:

en donde X- es un anión R6-SO3-; R6 es fluoroalquilo C1-5; y cada R3 se selecciona independientemente del grupo que consiste en hidrocarbilo C1-10 y alcoxi C1-5.
El compuesto de fórmula (III) que reacciona en el segundo aspecto de la invención se forma mediante el método descrito en el primer aspecto en el caso donde PG de fórmula (IIIa) es un grupo de fórmula SiR33.
En un tercer aspecto, la invención proporciona el método del primer o segundo aspectos que comprenden además un método para formar un compuesto de fórmula (II):

en donde R1 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5 ; R2 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5 , alquenilo C2-5 y alquinilo C2-5; y R3 en cada caso se selecciona independientemente del grupo que consiste en alcoxi C1-5 e hidrocarbilo C1-10; el método que comprende la etapa de hacer reaccionar un compuesto de fórmula (I) con menos de un equivalente molar de un compuesto de fórmula (V)

en donde R5 es alquilo C1-5.
El compuesto de fórmula (II) formado por el método del tercer aspecto se usa en la reacción del primer aspecto en el caso donde PG de fórmula (IIa) es un grupo de fórmula SiR33.
Descripción de las figuras
La Figura 1 es un espectro de XRPD de un compuesto cristalino preparado mediante un proceso de acuerdo con una modalidad de la invención;
La Figura 2 es un espectro Raman de un compuesto cristalino preparado mediante un proceso de acuerdo con una modalidad de la invención; y
La Figura 3 es una imagen de microscopía electrónica de barrido de un compuesto cristalino cristalino preparado mediante un proceso de acuerdo con una modalidad de la invención; y
La Figura 4 es un espectro de LCMS del producto de una reacción entre 4,4-dióxido de (2S,3S,5R)-3-metil-3-((3-metil-1H-1,2,3-triazol-3-io-1-il)metil)-7-oxo-4-tia-1-azabiciclo[3.2.0]heptano-2-carboxilato y N,O-bis trimetilsililacetamida.
Descripción detallada de la invención
Un proceso para preparar un compuesto de fórmula (IV) se ilustra en el Esquema 1.
Esquema 1

en donde:
R1 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5; R2 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5; R3 en cada caso se selecciona independientemente de hidrocarbilo C1-10y alcoxi C1-5, preferentemente alquilo C1-5, alcoxi C1-5, fenilo, y alquil fenil-C1-4;
R4 es alquilo C1-5; y;
X es un anión R6-SO3-; y R6 es fluoroalquilo C1-5.
Preferentemente, cada R1 es H.
Preferentemente, cada R2 es H.
Preferentemente, R4 es metilo.
Cada una de las etapas (i) -(iii) ahora se describirá con más detalle.
Etapa (i): sililación
El grupo carboxilo del compuesto de fórmula (I) se silila en la etapa (i). La sililación se lleva a cabo mediante el uso de una amida de fórmula (V):
R5
R3 J . R3
R3- S i —O * ' N -S i-R 3
R3 R3
(V)
en donde R3 en cada caso se selecciona independientemente de hidrocarbilo Cmoo alcoxi C1-5, preferentemente alquilo C1-5, alcoxi C1-5, fenilo, y alquil fenil- 01 - 4 y R5 se selecciona de alquilo C1-5.
Preferentemente, cada R3 es metilo. Preferentemente, cada R5 es metilo.
Un compuesto preferido de fórmula (V) es N,O-bis trimetilsilacetamida.
La reacción puede llevarse a cabo en un disolvente aprótico polar, opcionalmente un disolvente clorado tal como diclorometano.
El compuesto de fórmula (I) puede hacerse reaccionar con al menos un equivalente molar del compuesto de fórmula (V), opcionalmente un exceso molar del compuesto de fórmula (V). Sin embargo, los presentes inventores han encontrado sorprendentemente que ambos grupos sililo del compuesto de fórmula (V) pueden utilizarse en la sililación del compuesto de fórmula (I).
En consecuencia, en una modalidad el compuesto de fórmula (I) se hace reaccionar con menos de un equivalente molar del compuesto de fórmula (V), opcionalmente no más de 0,9 equivalentes molares, opcionalmente no más de 0,8, 0,7 o 0,6 equivalentes molares.
El compuesto de fórmula (V) puede adicionarse a la mezcla de reacción en una sola adición o puede adicionarse en dos o más porciones.
Etapa (ii): alquilación
La etapa de alquilación (ii) es una alquilación C1-5, preferentemente una metilación. La alquilación puede llevarse a cabo con un compuesto de fórmula (VIII):
R6-SO2-O-R4 (VIII)
En donde R4 es un grupo alquilo C1-5 y R6 es fluoroalquilo C1-5.
Preferentemente, R4 es metilo.
Preferentemente, R6 es trifluorometilo. La reacción puede llevarse a cabo a cualquier temperatura hasta el punto de ebullición de la mezcla de reacción a presión atmosférica. Sorprendentemente, los presentes inventores han encontrado que el uso de un R6 de alquilo fluorado puede permitir que la etapa de alquilación avance a baja temperatura, opcionalmente a una temperatura inferior a 20°C, opcionalmente inferior a 10°C, opcionalmente a aproximadamente 0°C. Los presentes inventores han encontrado además que el uso de un R6 de alquilo fluorado permite una reacción significativamente más rápida que el uso de grupos halógenos R6.
Mediante el uso de una reacción a baja temperatura usando un alquilo fluorado R6, la evaporación de agentes alquilantes volátiles, por ejemplo, yoduro de metilo, puede reducirse o eliminarse.
Preferentemente, el compuesto sililado formado en la etapa (i) no se aísla antes de la etapa de alquilación. Los compuestos (II) y (III) del Esquema 1 llevan un grupo protector sililo que protege el grupo carboxilo de estos compuestos, sin embargo, se apreciará que el grupo protector para la etapa de reacción (ii) puede ser otro grupo protector PG. El experto en la técnica conocerá otros grupos protectores adecuados para proteger el grupo carboxilo de los compuestos de fórmula (II) durante la alquilación de la etapa (ii). Los grupos protectores PG ilustrativos distintos del grupo de fórmula SiR33 incluyen alilo, que puede eliminarse después de la alquilación mediante el uso de un 2-etilhexanoato metálico y Pd(0); grupos que pueden eliminarse por hidrogenolisis, por ejemplo bencilo, bencidrilo y pnitrobencilo; y grupos que pueden eliminarse con una base, por ejemplo fluorenilmetilo. Otros grupos protectores para la protección de un carboxilo se describen en Theodora W. Greene y Peter G. M. Wuts, "Protective Groups in Organic Synthesis", segunda edición, John Wiley & Sons, Inc., cuyos contenidos se incorporan en la presente descripción como referencia.
Etapa (iii): Desprotección
Los presentes inventores han encontrado que el grupo sililo del compuesto de fórmula (III) puede eliminarse mediante tratamiento con 2-etilhexanoatos para producir un producto sólido y el tratamiento con un 2-etilhexanoato puede producir cristales del compuesto de fórmula (IV). Esto es sorprendente porque los presentes inventores han encontrado que el tratamiento con metanol, etanol o isopropanol o las bases de hidróxido de sodio o acetato de sodio produce un producto no sólido, tal como un aceite, una goma o un gel, que no puede convertirse fácilmente en una forma sólida. Los 2-etilhexanoatos ilustrativos son 2-etilhexanoatos metálicos. Los metales adecuados incluyen metales alcalinos y alcalinotérreos, por ejemplo, litio, sodio, potasio, calcio y magnesio.
Preferentemente, el compuesto de fórmula (III) no se aísla antes de la etapa de desililación.
El compuesto de fórmula (III) puede adicionarse a una solución de un 2-etilhexanoato metálico para producir cristales del compuesto de fórmula (IV). Los disolventes ilustrativos para la solución son alcoholes, preferentemente etanol. Cristalización
El compuesto de fórmula (IV) puede ser amorfo o cristalino. Los compuestos cristalinos de fórmula (IV) pueden ser más fáciles de manejar y más estables que los compuestos amorfos.
Los métodos para formar compuestos cristalinos de fórmula (IV) incluyen, sin limitación, disolver o dispersar un compuesto amorfo de fórmula (IV) en un disolvente o mezcla de disolventes e inducir la formación de cristales mediante la adición de uno o más antidisolventes a la solución o dispersión; enfriar la solución o dispersión; y/o adicionar un cristal de un compuesto de fórmula (IV) para proporcionar un punto de nucleación para la cristalización. Un "antidisolvente" como se usa en la presente descripción significa un líquido en el que el compuesto de fórmula (IV) tiene una solubilidad más baja que un disolvente de la solución o dispersión a la que se adiciona el antidisolvente.
Los métodos de cristalización se describen en el documento GB 1319776.9.
Aplicaciones
Los compuestos de fórmula (IV) pueden usarse en una composición farmacéutica con uno o más antibióticos, y pueden comprender uno o más excipientes farmacéuticamente aceptables convencionales.
Los compuestos de fórmula (IV) pueden administrarse en una composición con un antibiótico, o pueden administrarse por separado un antibiótico y un compuesto de fórmula (IV).
Una composición farmacéutica como se describe en la presente descripción puede estar en una forma inyectable para inyección intravenosa. La composición puede contener agentes estabilizantes. La composición puede estar en forma sólida estéril adecuada lista para reconstituirse para formar una solución inyectable, por ejemplo una solución salina. Los antibióticos ilustrativos son antibióticos de p-lactamas, particularmente penicilinas y cefalosporinas y pueden seleccionarse de Amoxicilina, Ampicilina, Apalcilina, Azlocilina, Bacampicilina, Cloxacilina, Dicloxacilina, Flucloxacilina, Lenampicilina, Mecillinam, Metacilina, Mezlocilina, Nafcilina, Oxacilina, Penicilina G, Penicilina V, Piperacilina, Temocilina, Ticarcilina, Aztreonam, BAL30072, Carumonam, PTX2416, Tigemonam, Cefaclor, Cefadroxil, Cefalexina, Cefalotina, Cefamandol, Cefapirina, Cefazolina, Cefbuperazona, Cefdinir, Cefepima, Cefetamet, Cefixima, Cefmenoxima, Cefmetazol, Cefrninox, Cefonicid, Cefoperazona, Cefotaxima, Cefotetan, Cefotiam, Ceftiofur, Cefovecin, Cefoxtina, Cefpodoxima, Cefprozil, Cefquinoma, Cefradina, Cefminox, Cefsulodin, Ceftarolina, Ceftazidima, Ceftezol, Ceftibuten, Ceftizoxima, Ceftobiprol, Ceftolozana, Ceftriaxona, Cefuroxima, Cefuzonama, Cefalexina, Cefalotina, Flomoxef, Latamoxef, Loracarbef Imipenem, Meropenem, Doripenem, Ertapenem, Biapenem, Panipenem, Faropenem o derivados de estos.
El antibiótico puede seleccionarse de aminoglucósidos: Amikacina, Arbekacina, Apramicina, Dibekacina, Gentamicina, Isepamicina, Kanamicina, Neomicina, Netilmicina, Plazomicina, Sisomicina, Espectinomina, Estreptomicina, Tobramicina o derivados de estos.
El antibiótico puede seleccionarse de quinolonas: Cinoxacina, Ciprofloxacina, Enofloxacina, Gatifloxacina, Gemifloxacina, Levofloxacina, Moxifloxacina, Ácido nalidíxico, Norfloxacina, Oxafloxacina, o derivados de estos.
El antibiótico puede seleccionarse de péptidos antimicrobianos, por ejemplo, Colistina, Polimixina B o derivados de estos.
Una composición farmacéutica como se describe en la presente descripción puede comprender solo uno o más de un antibiótico.
Una composición farmacéutica que contiene un compuesto cristalino de fórmula (I) puede contener o coadministrarse con un producto de proteína g bactericida o que aumenta la permeabilidad (BPI) o con inhibidores de la bomba de flujo de salida para mejorar la actividad contra bacterias gram negativas y bacterias resistentes a los agentes antimicrobianos. Los agentes antivirales, antiparasitarios, antifúngicos también pueden administrarse en combinación con los compuestos inhibidores.
La composición farmacéutica puede contener agentes acomplejantes o anticoagulantes, antioxidantes, estabilizantes, aminoglucósidos, sales farmacéuticamente aceptables o similares o mezclas de estos.
Particularmente la composición farmacéutica puede contener antibióticos p-lactámicos, preferentemente penicilinas, cefalosporinas, carbapenémicos, monobactámicos, con mayor preferencia piperacilina, cefepima; ceftriaxona; meropenem, aztreonam.
La composición farmacéutica puede contener tampones, por ejemplo, citrato de sodio, acetato de sodio, tartrato de sodio, carbonato de sodio, bicarbonato de sodio, ácido morfolinopropanosulfónico, otros tampones de fosfato y similares y agentes quelantes como el ácido etilendiaminotetracético (EDTA), ácido dietilentriaminopentacético, ácido hidroxietilendiametriacético, ácido nitrilotriacético, ácido 1,2-diaminociclohexanotetracético, ácido bis(2-aminoetil)etilenglicoltetraacético, ácido 1,6-hexametilendiaminotetraacético y similares o sales farmacéuticamente aceptables de estos.
Una composición farmacéutica como se describe en la presente descripción puede administrarse a un animal humano o de sangre caliente por cualquier método adecuado y preferentemente por inyección intravenosa.
Ejemplos
Síntesis de 4,4-dióxido de (2S,3S,5R)-3-metil-3-((3-metil-1H-1,2,3-triazol-3-io-1-il)metil)-7-oxo-4-tia-1-azabiciclo[3.2.0]heptano-2-carboxilato (4),
El compuesto (4) se preparó de acuerdo con el Esquema 2.
Esquema 2

¡) N,O-b¡s-tr¡met¡ls¡l¡lacetam¡da, CH2Cl2; ¡i) CHaOTf; ¡¡¡) Na 2- etilhexanoato
En un matraz de fondo redondo bajo flujo de nitrógeno, se cargan 100 g de ácido tazobactámico (1) y 500 mL de diclorometano. La temperatura se ajusta a 30/35°C y después se cargan 37 g de N,O-bis(trimetilsilil) acetamida en 15 20 minutos, mientras se mantiene la temperatura a 35/42°C. La mezcla se calienta a reflujo (+40/42°C) durante 60 minutos. Si la solución no es transparente, se carga N,O-bis(trimetilsilil) acetamida en pequeñas porciones (0,5-1,0 g cada una) y se espera 15 minutos cada vez hasta obtener una solución transparente que contenga el intermediario (2). Se usan 0,55 moles de N,O-bis(trimetilsilil) acetamida, con 0,1-0,2 equivalentes adicionales que se adicionan si la reacción no se completa.
Después la temperatura se enfría a 0/+5°C y se cargan 70 g de trifluorometanosulfonato de metilo en 60-90 minutos mientras se mantiene la temperatura a 0/+5°C. Después de 30 minutos, la reacción se monitorea por HPLC para controlar la desaparición del intermediario (2) y la formación del intermediario (3). La reacción se monitorea cada 30 minutos hasta la terminación.
En un matraz de fondo redondo, bajo nitrógeno, se cargan 500 mL de etanol y 55 g de 2-etilhexanoato de sodio y la temperatura se ajusta a 20/25°C, después se adiciona la solución de reacción que contiene el intermediario (3) en 60 90 minutos mientras se mantiene la temperatura de 20/25°C bajo agitación vigorosa. La suspensión se agita durante 30 minutos, después se filtra y se lava con 300 mL de etanol, seguido de 500 mL de diclorometano bajo nitrógeno. El producto en bruto (4) se seca bajo flujo de nitrógeno hasta que se obtiene un peso constante (150 g). El compuesto del producto en bruto (4) se aisló como un producto sólido (ensayo de HPLC = 70%, rendimiento = 80%).
Purificación de 4,4-dióxido de (2S,3S,5R)-3-metil-3-((3-metil-1H-1,2,3-triazol-3-io-1-il)metil)-7-oxo-4-tia-1-azabiciclo[3.2.0]heptano-2-carboxilato (4),
En un matraz de fondo redondo se cargan 800 mL de dimetilformamida, la temperatura se ajusta a 20/25°C después se carga el Compuesto 4 en bruto (150 g) obtenido anteriormente mediante el uso de 100 mL de dimetilformamida para facilitar la transferencia. La mezcla se agita durante 5 minutos y se obtiene una solución, a continuación y después de unos pocos minutos tiene lugar la cristalización. La suspensión se agita durante aproximadamente 3 horas, después se enfría a 0/+5°C y se agita durante otras 3 horas.
El sólido se filtra y se lava con 300 mL de dimetilformamida previamente enfriada a 0/+5°C. El compuesto 4 se suspende después en 700 mL de acetato de etilo y la temperatura se ajusta a 40/45°C. La suspensión se agita durante 30 minutos, después el sólido se filtra y se lava con 150 mL de acetato de etilo precalentado a 40/45°C. La suspensión con acetato de etilo se repite dos veces. Finalmente, el Compuesto 4 se seca al vacío a 40°C hasta alcanzar un peso constante (66 g, ensayo de HPLC = 99%, rendimiento = 76%).
Procedimiento estéril de filtración y recristalización del Compuesto 4
En un matraz de fondo redondo se cargan 350 mL de metanol, la temperatura se ajusta a 30/35°C después se cargan 100 g del Compuesto 4 y finalmente se lava el matraz con 60 mL de metanol. Después de 5-10 minutos se obtiene una solución. La solución se diluye con 330 mL de acetona mientras se ajusta la temperatura a 20/+25°C. La solución obtenida se trata con 2,2 g de carbón vegetal durante 20 minutos, después se filtra con un filtro de 0,22 microM y el filtro se lava con una mezcla de 13 mL de metanol y 110 mL de acetona. La temperatura de la solución se ajusta a 30/35°C y bajo agitación vigorosa se cargan 830 mL de acetona en aproximadamente 15-20 minutos. Después de agitar durante 60 minutos a una temperatura de 30/35°C se cargan 1170 mL de acetona en 45-60 minutos. Después, la temperatura se ajusta a 20/25°C en aproximadamente 30-60 minutos y se mantiene durante 30 minutos. El sólido cristalino obtenido se filtra y se lava con 430 mL de acetona. Finalmente, el producto se seca al vacío a 40°C hasta alcanzar un peso constante (se obtienen 83 g de Compuesto 4 con un ensayo de HPLC = 98-99%, rendimiento =t 80%).
La Figura 1 es un espectro de XRPD del Compuesto cristalino (4) adquirido en modo de transmisión en un Rigaku MiniFlex 600 con el uso de las siguientes condiciones:
Rayos X 40KVoltios, 15 mA
Longitud de onda CuKalfa => lambda 1,541862A
Eje de exploración Theta / 2-Theta
Intervalo de exploración 5.0000-60.0000 deg
Adquisición de tiempo 60 min
La Figura 2 es un espectro de Raman del Compuesto cristalino (4) que se ejecuta en un Jasco RFT-600: fuente de luz: Nd-YAG (1064 nm : longitud de onda de excitación).
La Figura 3 es una imagen de microscopía electrónica de barrido del Compuesto cristalino (4) con el uso de un microscopio electrónico de barrido JEOL JSM 5500 LV, que funciona a 30 kV en bajo vacío (30 Pa) con la técnica de electrones retrodispersados.
Ejemplo Comparativo 1
La etapa de sililación (i) del Esquema 2 se realizó mediante el uso de relaciones molares variables del agente de sililación N,0-bis (BSA).
La sililación con el uso de 1,2 equivalentes de BSA a 20-25°C como se describe en el documento WO 2008/010048 resulta en la formación de un producto secundario no identificado, observable en LC-MS como se ilustra en la Figura 4.
Con referencia a la Tabla 1, la cantidad de esta impureza puede reducirse en gran medida mediante el uso de un equivalente molar inferior de BSA.
Tabla 1
Equivalentes de BSA Temperatura Relación % de Impureza / producto sililado
1.2 30/35°C 24
1.8 0/5°C 73
0.5 40/42°C 1.5
Ejemplo comparativo 2
La etapa de metilación (ii) del Esquema 2 se realizó mediante el uso de yodometano, como se describe en el documento WO 2008/010048 y metil tosilato. Con referencia a la Tabla 2, las reacciones que usan triflato de metilo son mucho más rápidas, proporcionan un mayor rendimiento, pueden realizarse a temperaturas mucho más bajas y requieren una cantidad más pequeña de agente de metilación que el tosilato de metilo o el yodometano. Además, el uso de triflato de
metilo a una temperatura relativamente baja evita problemas de seguridad derivados de la toxicidad de los agentes de metilación usados a una temperatura relativamente alta, tal como el uso de yodometano en o por encima de su punto de ebullición.
Tabla 2
Agente de metilación Disolvente Equivalentes Tiempo de reacción Temperatura rendimiento Yodometano Acetona 7,2 22 h 45/48°C 44,3%
MeOTs Acetona 7,2 25 h 45/48°C 54,1%
MeOTf Acetona 1,4 30 min. 0/+5°C 98,0%
MeOTf Acetona 1,4 30 min. 10/15°C 68,0%
MeOTf THF 1,4 30 min. 15/20°C 67,0%
MeOTf CH2Cl2 1,4 30 min. 0/+5°C 98,0%
Ejemplo Comparativo 3
La etapa de desililación (iii) del Esquema 2 se intentó usar con una gama de alcoholes tales como metanol, etanol y 2-propanol. Este enfoque condujo a la recuperación del producto en forma de aceite o gel. Estos aceites o gel se trataron con diferentes disolventes, tales como ACN, THF y acetona, que dieron una disolución completa y no se pudo recuperar material sólido. El uso de éter dietílico, tolueno, hexano y heptano condujo a otros geles y no se pudo recuperar ningún sólido. Se intentaron bases para ajustar el pH a la neutralidad. Para este propósito se probaron la solución de NaOH, AcONa (como tal y en soluciones acuosas y orgánicas) y 2-etilhexanoato de sodio (como tal o en soluciones orgánicas). El uso de hidróxido de sodio en solución acuosa condujo al producto en gel y mucilaginoso. AcONa como tal, no originó precipitación de ningún sólido. Se obtuvo un material sólido que contiene el Compuesto 4 con el uso de una mezcla de 2-etilhexanoato de sodio y etanol como se describió en el Ejemplo anterior.
Claims (13)
1. Un método para formar un compuesto de fórmula (Illa):

Fórmula (IIIa)
en donde:
R1 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5;
R2 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5;
R4 es alquilo C1-5; y
R6 es un fluoroalquilo C1-5; y
PG es un grupo protector,
el método comprende la etapa de hacer reaccionar un compuesto de fórmula (IIa) con un compuesto de fórmula (VIII)

2. Un método de acuerdo con la reivindicación 1 en donde PG es un grupo de fórmula SiR33 en donde R3 en cada caso se selecciona independientemente de hidrocarbilo C1-10 o alcoxi C1-5.
3. Un método de acuerdo con la reivindicación 2 en donde R3 en cada caso se selecciona independientemente de alquilo C1-5, alcoxi C1-5, fenilo, y alquil fenilo-C1-4.
4. Un método de acuerdo con cualquier reivindicación anterior, en donde R4 es metilo.
5. Un método de acuerdo con cualquier reivindicación anterior, en donde R6 es un perfluoroalquilo C1-5.
6. Un método de acuerdo con la reivindicación 5 en donde R6 es trifluorometilo.
7. Un método de acuerdo con cualquier reivindicación anterior, en donde la reacción se realiza en un disolvente polar, aprótico.
8. Un método de acuerdo con cualquier reivindicación anterior, en donde la reacción se realiza a una temperatura de no más de 10°C.
9. Un método de acuerdo con cualquier reivindicación anterior, que comprende además un método para formar un compuesto de fórmula (IV):

en donde R1 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5; R2 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5; y R4 es alquilo C1-5;
el método comprende la etapa de hacer reaccionar un compuesto de fórmula (III) con una sal de 2-etilhexanoato:
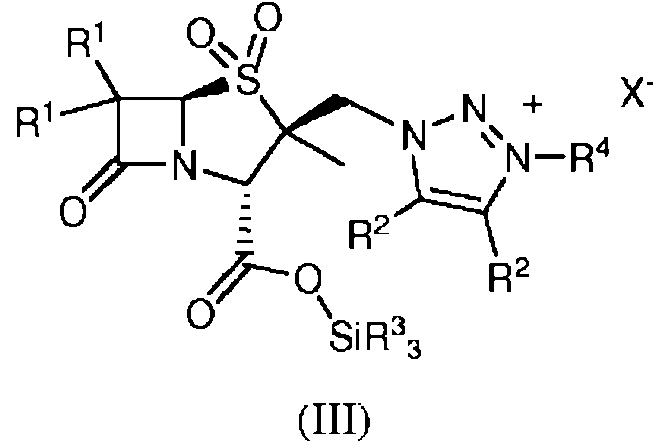
en donde X- es un anión R6-SO3-; R6 es fluoroalquilo C1-5; y cada R3 se selecciona independientemente del grupo que consiste en hidrocarbilo C1-10 y alcoxi C1-5; en donde el compuesto de fórmula (III) se forma mediante el método de cualquier reivindicación anterior y en donde PG de fórmula (IIIa) es un grupo de fórmula SiR33.
10. Un método de acuerdo con la reivindicación 9, en donde la sal de 2-etilhexanoato es un 2-etilhexanoato metálico.
11. Un método de acuerdo con la reivindicación 10, en donde el metal es un álcali.
12. Un método de acuerdo con cualquiera de las reivindicaciones 9-11 en donde el compuesto de fórmula (III) se adiciona a una solución de la sal de 2-etilhexanoato
13. Un método de acuerdo con cualquier reivindicación anterior, que comprende además un método para formar un compuesto de fórmula (II)

en donde R1 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5; R2 en cada caso se selecciona independientemente de H, halógeno, amino, alquilo C1-5, alquenilo C2-5 y alquinilo C2-5; y R3 en cada caso se selecciona independientemente del grupo que consiste en alcoxi C1-5 e hidrocarbilo C1-10; el método que comprende la etapa de hacer reaccionar un compuesto de fórmula (I) con menos de un equivalente molar de un compuesto de fórmula (V)

en donde R5 es alquil C1-5;
en donde el compuesto de fórmula (II) formado se usa en el método de cualquier reivindicación anterior y en donde PG de fórmula (IIa) es un grupo de fórmula SiR33.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1408649.0A GB201408649D0 (en) | 2014-05-15 | 2014-05-15 | Method |
| PCT/EP2015/060733 WO2015173378A2 (en) | 2014-05-15 | 2015-05-14 | Method |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2699644T3 true ES2699644T3 (es) | 2019-02-12 |
Family
ID=51134924
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17168966T Active ES2725675T3 (es) | 2014-05-15 | 2015-05-14 | Métodos para preparar penams |
| ES15726552T Active ES2699644T3 (es) | 2014-05-15 | 2015-05-14 | Método para preparar penams |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17168966T Active ES2725675T3 (es) | 2014-05-15 | 2015-05-14 | Métodos para preparar penams |
Country Status (29)
| Country | Link |
|---|---|
| US (1) | US9963465B2 (es) |
| EP (3) | EP3231806B1 (es) |
| JP (1) | JP6613303B2 (es) |
| KR (1) | KR102329186B1 (es) |
| CN (1) | CN106459086B (es) |
| AU (1) | AU2015261416B2 (es) |
| BR (1) | BR112016026074B1 (es) |
| CA (1) | CA2945900C (es) |
| CL (2) | CL2016002880A1 (es) |
| CR (1) | CR20160580A (es) |
| CY (2) | CY1120957T1 (es) |
| DK (2) | DK3143028T3 (es) |
| EA (2) | EA201991629A3 (es) |
| EC (1) | ECSP16085143A (es) |
| ES (2) | ES2725675T3 (es) |
| GB (1) | GB201408649D0 (es) |
| HR (2) | HRP20182065T1 (es) |
| HU (1) | HUE044511T2 (es) |
| IL (1) | IL248834B (es) |
| LT (2) | LT3231806T (es) |
| MX (1) | MX2016014644A (es) |
| PE (1) | PE20161433A1 (es) |
| PL (2) | PL3143028T3 (es) |
| PT (2) | PT3231806T (es) |
| RS (2) | RS58430B1 (es) |
| SG (1) | SG11201609494YA (es) |
| SI (2) | SI3143028T1 (es) |
| TR (1) | TR201906867T4 (es) |
| WO (1) | WO2015173378A2 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201319776D0 (en) * | 2013-11-08 | 2013-12-25 | Allecra Therapeutics Sas | Compound |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1319776A (en) | 1969-07-15 | 1973-06-06 | Yorkshire Chemicals Ltd | Basic dyes |
| SI2046802T1 (sl) * | 2006-07-12 | 2014-03-31 | Allecra Therapeutics Gmbh C/O Loeba Treuhand Gmbh | 2-substituirani metil penamski derivati |
| KR101933084B1 (ko) | 2010-11-25 | 2018-12-28 | 알레크라 테라퓨틱스 게엠베하 | 화합물 및 그의 용도 |
| KR20120067257A (ko) * | 2010-12-15 | 2012-06-25 | 서강대학교산학협력단 | 3-메틸-1,4-치환된 1,2,3-트라이아졸륨 기반의 이온성 액체 및 그의 제조방법과 응용 |
-
2014
- 2014-05-15 GB GBGB1408649.0A patent/GB201408649D0/en not_active Ceased
-
2015
- 2015-05-14 ES ES17168966T patent/ES2725675T3/es active Active
- 2015-05-14 MX MX2016014644A patent/MX2016014644A/es unknown
- 2015-05-14 EP EP17168966.4A patent/EP3231806B1/en active Active
- 2015-05-14 CR CR20160580A patent/CR20160580A/es unknown
- 2015-05-14 PT PT17168966T patent/PT3231806T/pt unknown
- 2015-05-14 HU HUE17168966 patent/HUE044511T2/hu unknown
- 2015-05-14 DK DK15726552.1T patent/DK3143028T3/da active
- 2015-05-14 SI SI201530509T patent/SI3143028T1/sl unknown
- 2015-05-14 SI SI201530738T patent/SI3231806T1/sl unknown
- 2015-05-14 CN CN201580024639.6A patent/CN106459086B/zh active Active
- 2015-05-14 PL PL15726552T patent/PL3143028T3/pl unknown
- 2015-05-14 BR BR112016026074-0A patent/BR112016026074B1/pt active IP Right Grant
- 2015-05-14 LT LTEP17168966.4T patent/LT3231806T/lt unknown
- 2015-05-14 AU AU2015261416A patent/AU2015261416B2/en active Active
- 2015-05-14 RS RS20181383A patent/RS58430B1/sr unknown
- 2015-05-14 EP EP18195989.1A patent/EP3441395A1/en not_active Withdrawn
- 2015-05-14 EP EP15726552.1A patent/EP3143028B1/en active Active
- 2015-05-14 DK DK17168966.4T patent/DK3231806T3/da active
- 2015-05-14 KR KR1020167032608A patent/KR102329186B1/ko active IP Right Grant
- 2015-05-14 EA EA201991629A patent/EA201991629A3/ru unknown
- 2015-05-14 TR TR2019/06867T patent/TR201906867T4/tr unknown
- 2015-05-14 PE PE2016002171A patent/PE20161433A1/es unknown
- 2015-05-14 RS RS20190558A patent/RS58748B1/sr unknown
- 2015-05-14 US US15/311,146 patent/US9963465B2/en active Active
- 2015-05-14 JP JP2017512434A patent/JP6613303B2/ja active Active
- 2015-05-14 EA EA201692306A patent/EA033494B1/ru unknown
- 2015-05-14 PL PL17168966T patent/PL3231806T3/pl unknown
- 2015-05-14 CA CA2945900A patent/CA2945900C/en active Active
- 2015-05-14 SG SG11201609494YA patent/SG11201609494YA/en unknown
- 2015-05-14 WO PCT/EP2015/060733 patent/WO2015173378A2/en active Application Filing
- 2015-05-14 PT PT15726552T patent/PT3143028T/pt unknown
- 2015-05-14 LT LTEP15726552.1T patent/LT3143028T/lt unknown
- 2015-05-14 ES ES15726552T patent/ES2699644T3/es active Active
-
2016
- 2016-10-29 EC ECIEPI201685143A patent/ECSP16085143A/es unknown
- 2016-11-08 IL IL248834A patent/IL248834B/en active IP Right Grant
- 2016-11-11 CL CL2016002880A patent/CL2016002880A1/es unknown
-
2018
- 2018-07-19 CL CL2018001961A patent/CL2018001961A1/es unknown
- 2018-12-05 CY CY181101299T patent/CY1120957T1/el unknown
- 2018-12-06 HR HRP20182065TT patent/HRP20182065T1/hr unknown
-
2019
- 2019-05-06 HR HRP20190830TT patent/HRP20190830T1/hr unknown
- 2019-05-09 CY CY20191100501T patent/CY1121756T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10201532B2 (en) | Compounds and their use | |
| WO2009138847A2 (en) | An improved process for the preparation of cefozopran | |
| ES2699644T3 (es) | Método para preparar penams | |
| KR870001440B1 (ko) | 세팔로스포린 화합물의 제조방법 | |
| JP7346485B2 (ja) | 結晶性β-ラクタマーゼ阻害剤 | |
| NZ725121B2 (en) | Methods for Preparing Penams | |
| OA16437A (en) | Compounds and their use. | |
| CA2411714A1 (en) | Novel cephalosporin compounds and process for preparing the same |