EP0000727B1 - 3-(4-(1,3-Diazacycloalken-2-yl)-phenyl)-1,2-benzisothiazole, Verfahren zu ihrer Herstellung und diese enthaltende Arzneimittel. - Google Patents
3-(4-(1,3-Diazacycloalken-2-yl)-phenyl)-1,2-benzisothiazole, Verfahren zu ihrer Herstellung und diese enthaltende Arzneimittel. Download PDFInfo
- Publication number
- EP0000727B1 EP0000727B1 EP78100498A EP78100498A EP0000727B1 EP 0000727 B1 EP0000727 B1 EP 0000727B1 EP 78100498 A EP78100498 A EP 78100498A EP 78100498 A EP78100498 A EP 78100498A EP 0000727 B1 EP0000727 B1 EP 0000727B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- phenyl
- benzisothiazole
- general formula
- compound
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
- 238000000034 method Methods 0.000 title claims description 4
- 238000002360 preparation method Methods 0.000 title description 8
- 239000003814 drug Substances 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims description 25
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 16
- 239000002253 acid Substances 0.000 claims description 12
- 150000003839 salts Chemical class 0.000 claims description 11
- 239000011593 sulfur Substances 0.000 claims description 10
- 229910052717 sulfur Inorganic materials 0.000 claims description 10
- 239000004480 active ingredient Substances 0.000 claims description 7
- 230000003288 anthiarrhythmic effect Effects 0.000 claims description 7
- 125000004432 carbon atom Chemical group C* 0.000 claims description 4
- 239000001257 hydrogen Substances 0.000 claims description 4
- 229910052739 hydrogen Inorganic materials 0.000 claims description 4
- YMHMAHGCSMSXGV-UHFFFAOYSA-N 3-[4-(1,2,3,4-tetrahydropyrimidin-2-yl)phenyl]-1,2-benzothiazole Chemical compound N1C=CCNC1C1=CC=C(C=2C3=CC=CC=C3SN=2)C=C1 YMHMAHGCSMSXGV-UHFFFAOYSA-N 0.000 claims description 3
- 150000004985 diamines Chemical class 0.000 claims description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 3
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 3
- 239000003960 organic solvent Substances 0.000 claims description 3
- 150000007513 acids Chemical class 0.000 claims 3
- 239000008194 pharmaceutical composition Substances 0.000 claims 3
- 239000008024 pharmaceutical diluent Substances 0.000 claims 3
- 239000000546 pharmaceutical excipient Substances 0.000 claims 3
- 229940124531 pharmaceutical excipient Drugs 0.000 claims 3
- HKKJFRYNQZNGIL-UHFFFAOYSA-N 3-[4-(5-methyl-4,5-dihydro-1h-imidazol-2-yl)phenyl]-1,2-benzothiazole Chemical compound N1C(C)CN=C1C1=CC=C(C=2C3=CC=CC=C3SN=2)C=C1 HKKJFRYNQZNGIL-UHFFFAOYSA-N 0.000 claims 2
- 125000003545 alkoxy group Chemical group 0.000 claims 1
- 125000000217 alkyl group Chemical group 0.000 claims 1
- 229910052736 halogen Inorganic materials 0.000 claims 1
- 150000002367 halogens Chemical class 0.000 claims 1
- 238000004519 manufacturing process Methods 0.000 claims 1
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 24
- UHOVQNZJYSORNB-UHFFFAOYSA-N monobenzene Natural products C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 13
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 12
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- -1 alkyl radical Chemical class 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 11
- 239000003826 tablet Substances 0.000 description 11
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- LOUPRKONTZGTKE-LHHVKLHASA-N quinidine Chemical compound C([C@H]([C@H](C1)C=C)C2)C[N@@]1[C@H]2[C@@H](O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-LHHVKLHASA-N 0.000 description 8
- 238000002844 melting Methods 0.000 description 7
- 230000008018 melting Effects 0.000 description 7
- 239000011541 reaction mixture Substances 0.000 description 7
- MOOHKKXJCXFGQI-UHFFFAOYSA-N 3-[4-(chloromethyl)phenyl]-1,2-benzothiazole Chemical compound C1=CC(CCl)=CC=C1C1=NSC2=CC=CC=C12 MOOHKKXJCXFGQI-UHFFFAOYSA-N 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- 229910021529 ammonia Inorganic materials 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 239000000203 mixture Substances 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 5
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Chemical compound OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- REQCZEXYDRLIBE-UHFFFAOYSA-N procainamide Chemical compound CCN(CC)CCNC(=O)C1=CC=C(N)C=C1 REQCZEXYDRLIBE-UHFFFAOYSA-N 0.000 description 5
- 229960000244 procainamide Drugs 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- UKTBPPGWIBWGRH-UHFFFAOYSA-N 3-(4-methylphenyl)-1,2-benzothiazole Chemical class C1=CC(C)=CC=C1C1=NSC2=CC=CC=C12 UKTBPPGWIBWGRH-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 4
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- LOUPRKONTZGTKE-UHFFFAOYSA-N cinchonine Natural products C1C(C(C2)C=C)CCN2C1C(O)C1=CC=NC2=CC=C(OC)C=C21 LOUPRKONTZGTKE-UHFFFAOYSA-N 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 4
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 4
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 4
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 229960001404 quinidine Drugs 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 235000000346 sugar Nutrition 0.000 description 4
- 239000000454 talc Substances 0.000 description 4
- 229910052623 talc Inorganic materials 0.000 description 4
- 235000012222 talc Nutrition 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- HRPXFIPBQCHNFN-UHFFFAOYSA-N 5-chloro-3-[4-(4,5-dihydro-1h-imidazol-2-yl)phenyl]-1,2-benzothiazole Chemical compound C12=CC(Cl)=CC=C2SN=C1C(C=C1)=CC=C1C1=NCCN1 HRPXFIPBQCHNFN-UHFFFAOYSA-N 0.000 description 3
- NEGTVZMOBHKLHP-UHFFFAOYSA-N 5-chloro-3-[4-(chloromethyl)phenyl]-1,2-benzothiazole Chemical compound C1=CC(CCl)=CC=C1C1=NSC2=CC=C(Cl)C=C12 NEGTVZMOBHKLHP-UHFFFAOYSA-N 0.000 description 3
- XFSBVAOIAHNAPC-UHFFFAOYSA-N Aconitin Natural products CCN1CC(C(CC2OC)O)(COC)C3C(OC)C(C(C45)(OC(C)=O)C(O)C6OC)C1C32C4CC6(O)C5OC(=O)C1=CC=CC=C1 XFSBVAOIAHNAPC-UHFFFAOYSA-N 0.000 description 3
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 239000003416 antiarrhythmic agent Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 125000005843 halogen group Chemical group 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 2
- XFSBVAOIAHNAPC-XTHSEXKGSA-N 16-Ethyl-1alpha,6alpha,19beta-trimethoxy-4-(methoxymethyl)-aconitane-3alpha,8,10alpha,11,18alpha-pentol, 8-acetate 10-benzoate Chemical compound O([C@H]1[C@]2(O)C[C@H]3[C@@]45C6[C@@H]([C@@]([C@H]31)(OC(C)=O)[C@@H](O)[C@@H]2OC)[C@H](OC)[C@@H]4[C@]([C@@H](C[C@@H]5OC)O)(COC)CN6CC)C(=O)C1=CC=CC=C1 XFSBVAOIAHNAPC-XTHSEXKGSA-N 0.000 description 2
- YIWUKEYIRIRTPP-UHFFFAOYSA-N 2-ethylhexan-1-ol Chemical compound CCCCC(CC)CO YIWUKEYIRIRTPP-UHFFFAOYSA-N 0.000 description 2
- SZNYYWIUQFZLLT-UHFFFAOYSA-N 2-methyl-1-(2-methylpropoxy)propane Chemical compound CC(C)COCC(C)C SZNYYWIUQFZLLT-UHFFFAOYSA-N 0.000 description 2
- SVTBMSDMJJWYQN-UHFFFAOYSA-N 2-methylpentane-2,4-diol Chemical compound CC(O)CC(C)(C)O SVTBMSDMJJWYQN-UHFFFAOYSA-N 0.000 description 2
- CKZBFHFNBCKQLR-UHFFFAOYSA-N 3-[2-(2,3-dihydro-1h-imidazol-2-yl)phenyl]-1,2-benzothiazole Chemical compound N1C=CNC1C1=CC=CC=C1C1=NSC2=CC=CC=C12 CKZBFHFNBCKQLR-UHFFFAOYSA-N 0.000 description 2
- BABFRNZMMZTRBK-UHFFFAOYSA-N 3-[4-(1,2,3,4-tetrahydropyrimidin-2-yl)phenyl]-1,2-benzothiazole;hydrochloride Chemical compound Cl.N1C=CCNC1C1=CC=C(C=2C3=CC=CC=C3SN=2)C=C1 BABFRNZMMZTRBK-UHFFFAOYSA-N 0.000 description 2
- ZWVHREKHKSPSFL-UHFFFAOYSA-N 3-[4-(1-methyl-4,5-dihydroimidazol-2-yl)phenyl]-1,2-benzothiazole Chemical compound CN1CCN=C1C1=CC=C(C=2C3=CC=CC=C3SN=2)C=C1 ZWVHREKHKSPSFL-UHFFFAOYSA-N 0.000 description 2
- KTDZARVWRWCEND-UHFFFAOYSA-N 3-[4-(4,5-dihydro-1h-imidazol-2-yl)phenyl]-1,2-benzothiazole Chemical compound N1CCN=C1C1=CC=C(C=2C3=CC=CC=C3SN=2)C=C1 KTDZARVWRWCEND-UHFFFAOYSA-N 0.000 description 2
- TVAIEBGOEMVBPK-UHFFFAOYSA-N 3-[4-(4,5-dihydro-1h-imidazol-2-yl)phenyl]-5-nitro-1,2-benzothiazole Chemical compound C12=CC([N+](=O)[O-])=CC=C2SN=C1C(C=C1)=CC=C1C1=NCCN1 TVAIEBGOEMVBPK-UHFFFAOYSA-N 0.000 description 2
- FTOAOBMCPZCFFF-UHFFFAOYSA-N 5,5-diethylbarbituric acid Chemical compound CCC1(CC)C(=O)NC(=O)NC1=O FTOAOBMCPZCFFF-UHFFFAOYSA-N 0.000 description 2
- PEQVRQOGSVFQJT-UHFFFAOYSA-N 5-nitro-3-[4-(1,2,3,4-tetrahydropyrimidin-2-yl)phenyl]-1,2-benzothiazole Chemical compound C12=CC([N+](=O)[O-])=CC=C2SN=C1C(C=C1)=CC=C1C1NCC=CN1 PEQVRQOGSVFQJT-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- YNQLUTRBYVCPMQ-UHFFFAOYSA-N Ethylbenzene Chemical compound CCC1=CC=CC=C1 YNQLUTRBYVCPMQ-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- URLKBWYHVLBVBO-UHFFFAOYSA-N Para-Xylene Chemical group CC1=CC=C(C)C=C1 URLKBWYHVLBVBO-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 229940039750 aconitine Drugs 0.000 description 2
- STDXGNLCJACLFY-UHFFFAOYSA-N aconitine Natural products CCN1CC2(COC)C(O)CC(O)C34C5CC6(O)C(OC)C(O)C(OC(=O)C)(C5C6OC(=O)c7ccccc7)C(C(OC)C23)C14 STDXGNLCJACLFY-UHFFFAOYSA-N 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 150000001346 alkyl aryl ethers Chemical class 0.000 description 2
- RDOXTESZEPMUJZ-UHFFFAOYSA-N anisole Chemical compound COC1=CC=CC=C1 RDOXTESZEPMUJZ-UHFFFAOYSA-N 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 206010003119 arrhythmia Diseases 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- RWGFKTVRMDUZSP-UHFFFAOYSA-N cumene Chemical compound CC(C)C1=CC=CC=C1 RWGFKTVRMDUZSP-UHFFFAOYSA-N 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- AOHJOMMDDJHIJH-UHFFFAOYSA-N propylenediamine Chemical compound CC(N)CN AOHJOMMDDJHIJH-UHFFFAOYSA-N 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- XFNJVJPLKCPIBV-UHFFFAOYSA-N trimethylenediamine Chemical compound NCCCN XFNJVJPLKCPIBV-UHFFFAOYSA-N 0.000 description 2
- IMBKKGRPNHVURM-UHFFFAOYSA-N (2,5-dichlorophenyl)-(4-methylphenyl)methanone Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC(Cl)=CC=C1Cl IMBKKGRPNHVURM-UHFFFAOYSA-N 0.000 description 1
- WIKUXHHNKRKVGJ-UHFFFAOYSA-N (2-chlorophenyl)-(4-methylphenyl)methanone Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC=CC=C1Cl WIKUXHHNKRKVGJ-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- OTPDWCMLUKMQNO-UHFFFAOYSA-N 1,2,3,4-tetrahydropyrimidine Chemical class C1NCC=CN1 OTPDWCMLUKMQNO-UHFFFAOYSA-N 0.000 description 1
- CSNIZNHTOVFARY-UHFFFAOYSA-N 1,2-benzothiazole Chemical compound C1=CC=C2C=NSC2=C1 CSNIZNHTOVFARY-UHFFFAOYSA-N 0.000 description 1
- PZHIWRCQKBBTOW-UHFFFAOYSA-N 1-ethoxybutane Chemical compound CCCCOCC PZHIWRCQKBBTOW-UHFFFAOYSA-N 0.000 description 1
- NVJUHMXYKCUMQA-UHFFFAOYSA-N 1-ethoxypropane Chemical compound CCCOCC NVJUHMXYKCUMQA-UHFFFAOYSA-N 0.000 description 1
- ANFXTILBDGTSEG-UHFFFAOYSA-N 1-methyl-4,5-dihydroimidazole Chemical class CN1CCN=C1 ANFXTILBDGTSEG-UHFFFAOYSA-N 0.000 description 1
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 1
- ZWDNAGUIXUFRMK-UHFFFAOYSA-N 3-[4-(5-methyl-4,5-dihydro-1h-imidazol-2-yl)phenyl]-1,2-benzothiazole;hydrochloride Chemical compound Cl.N1C(C)CN=C1C1=CC=C(C=2C3=CC=CC=C3SN=2)C=C1 ZWDNAGUIXUFRMK-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- OOYPAIOHSSEZHA-UHFFFAOYSA-N 5-chloro-3-(4-methylphenyl)-1,2-benzothiazole Chemical compound C1=CC(C)=CC=C1C1=NSC2=CC=C(Cl)C=C12 OOYPAIOHSSEZHA-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N Acrylic acid Chemical group OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 0 CC(C)CNC(c1ccc(C=C2C(S*)=CC=CC2)cc1)N Chemical compound CC(C)CNC(c1ccc(C=C2C(S*)=CC=CC2)cc1)N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 229920000623 Cellulose acetate phthalate Polymers 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- AQZGPSLYZOOYQP-UHFFFAOYSA-N Diisoamyl ether Chemical compound CC(C)CCOCCC(C)C AQZGPSLYZOOYQP-UHFFFAOYSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 239000004129 EU approved improving agent Substances 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- XFSBVAOIAHNAPC-NPVHKAFCSA-N aconitin Chemical compound O([C@H]1[C@]2(O)C[C@H]3[C@@]45[C@H]6[C@@H]([C@@]([C@H]31)(OC(C)=O)[C@@H](O)[C@@H]2OC)[C@H](OC)[C@@H]4[C@]([C@@H](C[C@@H]5OC)O)(COC)CN6CC)C(=O)C1=CC=CC=C1 XFSBVAOIAHNAPC-NPVHKAFCSA-N 0.000 description 1
- 230000007059 acute toxicity Effects 0.000 description 1
- 231100000403 acute toxicity Toxicity 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 230000006793 arrhythmia Effects 0.000 description 1
- 230000003126 arrythmogenic effect Effects 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 229960002319 barbital Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000007963 capsule composition Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 229940105329 carboxymethylcellulose Drugs 0.000 description 1
- 229940096529 carboxypolymethylene Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 229940081734 cellulose acetate phthalate Drugs 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940109275 cyclamate Drugs 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 210000003298 dental enamel Anatomy 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 239000007941 film coated tablet Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 238000004508 fractional distillation Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 210000002837 heart atrium Anatomy 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 239000003978 infusion fluid Substances 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 231100000636 lethal dose Toxicity 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- UZKWTJUDCOPSNM-UHFFFAOYSA-N methoxybenzene Substances CCCCOC=C UZKWTJUDCOPSNM-UHFFFAOYSA-N 0.000 description 1
- GHDIHPNJQVDFBL-UHFFFAOYSA-N methoxycyclohexane Chemical compound COC1CCCCC1 GHDIHPNJQVDFBL-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 238000001050 pharmacotherapy Methods 0.000 description 1
- DLRJIFUOBPOJNS-UHFFFAOYSA-N phenetole Chemical compound CCOC1=CC=CC=C1 DLRJIFUOBPOJNS-UHFFFAOYSA-N 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229920002689 polyvinyl acetate Polymers 0.000 description 1
- 239000011118 polyvinyl acetate Substances 0.000 description 1
- 229940075065 polyvinyl acetate Drugs 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 238000011158 quantitative evaluation Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 239000013558 reference substance Substances 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000012453 sprague-dawley rat model Methods 0.000 description 1
- 239000002700 tablet coating Substances 0.000 description 1
- 238000009492 tablet coating Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- HNKJADCVZUBCPG-UHFFFAOYSA-N thioanisole Chemical compound CSC1=CC=CC=C1 HNKJADCVZUBCPG-UHFFFAOYSA-N 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 235000010215 titanium dioxide Nutrition 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 235000012141 vanillin Nutrition 0.000 description 1
- MWOOGOJBHIARFG-UHFFFAOYSA-N vanillin Chemical group COC1=CC(C=O)=CC=C1O MWOOGOJBHIARFG-UHFFFAOYSA-N 0.000 description 1
- FGQOOHJZONJGDT-UHFFFAOYSA-N vanillin Natural products COC1=CC(O)=CC(C=O)=C1 FGQOOHJZONJGDT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D275/00—Heterocyclic compounds containing 1,2-thiazole or hydrogenated 1,2-thiazole rings
- C07D275/04—Heterocyclic compounds containing 1,2-thiazole or hydrogenated 1,2-thiazole rings condensed with carbocyclic rings or ring systems
Definitions
- the invention relates to new 3- (4- (1,3-diazacycloalken-2-yl) phenyl] -1,2-benzisothiazoles and their acid addition salts, their preparation and pharmaceutical preparations containing these compounds.
- Hydrogen is particularly preferred for R and Y is preferred for Y. to emphasize.
- the compounds of the invention can be prepared by using a compound of general formula II in which R has the meanings given above, with a diamine of the general formula III, in which Y has the meanings given above, and elemental sulfur is advantageously reacted in an inert organic solvent and, if appropriate, the compound obtained in one transferred physiologically acceptable acid addition salt.
- the reaction is expediently carried out at temperatures from 40 to 150 ° C., preferably at temperatures from 70 to 120 ° C.
- Appropriate solvents for the reaction are aromatic hydrocarbons, especially benzene hydrocarbons, such as benzene or toluene, lower alcohols, such as methanol, ethanol, propanol, isopropanol, butanol or isobutanol, saturated cyclic or aliphatic ethers, such as dibutyl ether or dioxane, glycol ether, especially monoalkyl ether of glycol , such as glycol monomethyl ether or glycol monoethyl ether, or mixtures of the solvents mentioned.
- aromatic hydrocarbons especially benzene hydrocarbons, such as benzene or toluene
- lower alcohols such as methanol, ethanol, propanol, isopropanol, butanol or isobutanol
- saturated cyclic or aliphatic ethers such as dibutyl ether or dioxane
- glycol ether especially monoalkyl ether of glyco
- the benzene hydrocarbons in particular benzene and toluene, and monoalkyl ethers of glycol, in particular glycol monomethyl ether, are preferred.
- the diamine of the general formula III calculated on the compound of the general formula 11, is used in a stoichiometric or in excess amount, optionally up to 3 times the stoichiometric amount.
- the elemental sulfur calculated on the chloromethyl compound used, is used in stoichiometric or in excess amount up to 1.2 times.
- the stoichiometric amount of elemental sulfur is preferably used.
- the starting compounds of the general formula II can be obtained, for example, by side chain chlorination of 3- (4-methylphenyl) -1,2-benzisothiazoles with chlorine at about 170 ° C. and under UV radiation.
- the starting material IV, ammonia and elemental sulfur are used in approximately stoichiometric amounts, but a ratio of 2 to 10 moles of ammonia and 0.9 to 1.1 gram atom of sulfur per mole of starting material IV is preferably used.
- This reaction is generally carried out at a temperature of 20 to 250 ° C., advantageously from 20 to 200 ° C., preferably from 40 to 180 ° C., in particular from 40 to 120 ° C., without pressure or under pressure, continuously or batchwise.
- the reaction pressure is generally determined by the total vapor pressure of the components at the reaction temperature. If appropriate, organic solvents which are inert under the reaction conditions can be used, e.g.
- aromatic hydrocarbons such as toluene, ethylbenzene, o-, m-, p-xylene, isopropylbenzene; Alkanols and cycloalkanols, such as ethanol, n-butanol, isobutanol, methylglycol, cyclohexanol, propanol, methanol, 2-ethylhexanol; Ether, e.g.
- the solvent is expediently used in an amount of 200 to 10,000 percent by weight, preferably 300 to 1,000 percent by weight, based on starting material IV.
- the reaction can be carried out as follows: starting material IV, elemental sulfur and ammonia, optionally in the presence of a solvent, are combined in one Pressure reactor reacted with one another at the aforementioned temperature for 3 to 10 hours.
- the 1,2-benzisothiazole is obtained from the reaction mixture by the customary methods, for example by fractional distillation, filtration and, if appropriate, subsequent recrystallization from a suitable solvent, for example ligroin.
- the reaction mixture can also be poured into water, the mixture formed extracted with a suitable solvent, for example methylene chloride, benzene, and the extract worked up.
- the compounds of general formula 1 according to the invention are optionally converted in a conventional manner into the acid addition salt of a physiologically tolerated acid.
- suitable physiologically compatible organic or inorganic acids are hydrochloric acid, hydrobromic acid, phosphoric acid or sulfuric acid, and organic acids are, for example, oxalic acid, maleic acid, fumaric acid, lactic acid, tartaric acid, malic acid, citric acid, salicylic acid, adipic acid or benzoic acid or can be based on advances in drug research 10, pages 224 to 225, Birkhäuser Verlag, Basel and Stuttgart, 1966.
- the compounds according to the invention and their physiologically tolerated acid addition salts are distinguished by a strong antriarrhythmic effect and are particularly suitable for the pharmacotherapy of cardiac arrhythmias.
- the substances were administered orally to rats (strain: Sprague Dawley, weight: 180-240 g) 45 min before the start of anesthesia (thiobuteal barbital 100 mg / kg i.p.).
- Aconitin was used as the arrhythmogenic substance, which i.v. 60 min after the substance application. was infused (dosing rate: 0.005 mg / kg. min).
- arrhythmias occur after an average of 3.7 ⁇ 0.9 min, the onset of which can be delayed by antiarrhythmics depending on the dose.
- the acute toxicity was determined in groups of 10 or 20 female Swiss mice, weight 20-26 g, with intraperitoneal application.
- the LD 50 was calculated as the dose (probit analysis) after which 50% of the animals died within 7 days.
- Table 1 shows that the compounds of Examples 2 and 3, compared to the antiarrhythmicum procainamide, are around 5 times more antiarrhythmic. Another advantage is that the effect of the maximum dose reaches 114 (example 2) or 73% (example 3) higher values than that of procainamide; i.e. that the aconitine antagonism of the tested compounds is significantly more pronounced than that of procainamide.
- the therapeutic range as the quotient of the lethal dose (LD 50) and the antiarrhythmic dose (ED 50%) is 2 times (example 2) and 1.9 times (example 3) greater than that of procainamide.
- the invention accordingly also relates to therapeutic agents or preparations which, in addition to conventional carriers and diluents, contain a compound of the general formula I or its physiologically tolerable acid addition salt as active ingredient, and the use of the new compounds for therapeutic purposes.
- the therapeutic agents or preparations are produced in a known manner with the usual carriers or diluents and the commonly used pharmaceutical-technical auxiliaries according to the desired type of application with a suitable dosage.
- the preferred preparations are in a dosage form which is suitable for oral administration.
- Dosage forms of this type are, for example, tablets, film-coated tablets, coated tablets, capsules, pills, powders, solutions or suspensions or depot forms.
- parenteral forms of preparation such as injection solutions or additives to infusion solutions, can also be used.
- Suppositories may also be mentioned as preparations.
- the corresponding tablets can, for example, by mixing the active ingredient with known auxiliaries, for example inert diluents such as dextrose, sugar, sorbitol, mannitol, polyvinylpyrrolidone, calcium carbonate, calcium phosphate or milk sugar, disintegrants such as corn, starch, alginic acid or polyvinylpyrrolidone, binders such as starch or Gelatin, lubricants such as magnesium stearate or talc and / or agents for achieving the depot effect, such as carboxypolymethylene, carboxymethyl cellulose, cellulose acetate phthalate or polyvinyl acetate can be obtained.
- auxiliaries for example inert diluents such as dextrose, sugar, sorbitol, mannitol, polyvinylpyrrolidone, calcium carbonate, calcium phosphate or milk sugar, disintegrants such as corn, starch, alginic acid or polyvinylpyrrol
- coated tablets can be produced by coating cores produced analogously to the tablets with agents conventionally used in coated tablet coatings, for example collidone or shellac, gum arabic, talc, titanium dioxide or sugar.
- the coated tablet cover can also consist of several layers, wherein the auxiliaries mentioned above for the tablets can be used.
- Solutions or suspensions with the active compounds according to the invention can additionally contain taste-improving agents such as saccharin, cyclamate or sugar and, for example, flavorings such as vanillin or orange extract. They can also contain suspending aids such as sodium carboxymethyl cellulose or preservatives such as parahydroxybenzoates.
- Capsules containing active ingredients can be produced, for example, by mixing the active ingredient with an inert carrier, such as milk sugar or sorbitol, and encapsulating it in gelatin capsules.
- Suitable suppositories can be produced, for example, by mixing with the carrier material provided, such as neutral fats or polyethylene glycols or their derivatives.
- the single dose of a substance according to the invention in humans is 5 to 100 mg, preferably 10 to 80 mg.
- the hydrochloride of the compound has a melting point of 318 ° C.
- the active ingredient is moistened with polyvinylpyrrolidone in 10% strength aqueous solution, passed through a sieve with a mesh size of 1.0 mm and dried at 50.degree. These granules are mixed with polyethylene glycol (average M.G. 4000), hydroxypropylmethyl cellulose, talc and magnesium stearate and pressed into 280 mg tablets.
- the mixture of the active ingredient with lactose and corn starch is granulated with an 8% aqueous solution of the polyvinylpyrrolidone through a 1.5 mm sieve, dried at 50 ° C. and rubbed again through a 1.0 mm sieve.
- the granules thus obtained are mixed with magnesium stearate and pressed to dragee cores.
- the dragee cores obtained are coated in a conventional manner with a casing which consists essentially of sugar and talc.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
Description
- Die Erfindung betrifft neue 3-(4-(1,3-Diazacycloalken-2-yl)-phenyl]-1,2-benzisothiazole und ihre Säureadditionssalze, ihre Herstellung und diese Verbindungen enthaltende pharmazeutische Zubereitungen.
- Es wurde gefunden, daß Verbindungen der allgemeinen Formel I


- Von den genannten Bedeutungen sind als bevorzugt für R Wasserstoff, ein Halogenatom, insbesondere Chlor oder Brom, oder eine Nitrogruppe und für Y 1-Methyläthylen-t,2 und Trimethylen-1,3 zu nennen.
- Als besonders bevorzugt sind für R Wasserstoff, und für Y

- Dementsprechend sind als erfindungsgemäße Verbindungen, die summarisch als 3-[4-(1,3-Diazacyclo-2-yl)]-1,2-benzoisothiazole bezeichnet werden können, der allgemeinen Formel 1 beispielsweise zu nennen:
- 3-[4-(Imidazolin-2-yl)-phenyl]-1,2-benzisothiazol
- 3-[4-(Methylimidazolin-2-yl)-phenyl]-1,2-benzisothiazol
- 3-[4-(Tetrahydropyrimidin-2-yl)-phenyl]-1,2-benzisothiazol
- 5-Chlor-3-[4-(imidazolin-2-yl)-phenyl]-1,2-benzisothiazol
- 5-Chlor-3-[4-(methylimidazolin-2-yl)-phenyll-1,2-benzisothiazol
- 4-Methoxy-3-[(4-(methylimidazolin-2-yl)-phenyl]-1,2-benzisothiazol
- 5-Nitro-3-[4-(imidazolin-2-yl)-phenyl]-1,2-benzisothiazol
- 5-Nitro-3-[4-(tetrahydropyrimidin-2-yl)-phenyl]-1,2-benzisothiazol
- Die erfindungsgemäßen Verbindungen können hergestellt werden, indem man eine Verbindung der allgemeinen Formel II


- Die Umsetzung wird zweckmäßig bei Temperaturen von 40 bis 150°C, bevorzugt bei Temperaturen von 70 bis 120°C, durchgeführt.
- Zweckmäßige Lösungsmittel für die Umsetzung sind aromatische Kohlenwasserstoffe, insbesondere Benzolkohlenwasserstoffe, wie Benzol oder Toluol, niedere Alkohole, wie Methanol, Äthanol, Propanol, Isopropanol, Butanol oder Isobutanol, gesättigte cyclische oder aliphatische Äther, wie Dibutyläther oder Dioxan, Glykoläther, insbesondere Monoalkyläther des Glykols, wie Glykolmonomethyläther oder Glykolmonoäthyläther, oder Mischungen der genannten Lösungsmittel.
- Von den genannten Lösungsmitteln sind die Benzolkohlenwasserstoffe, insbesondere Benzol und Toluol, and Monoalkyläther des Glykols, insbesondere Glykolmonomethyläther, bevorzugt.
- Das Diamin der allgemeinen Formel 111 wird, berechnet auf die Verbindung der allgemeinen Formel 11, in stöchiometrischer oder in überschüssiger Menge, gegebenenfalls bis zur 3-fachen stöchiometrischen Menge, verwendet.
- Der elementare Schwefel wird, berechnet auf die verwendete Chlormethylverbindung, in stöchiometrischer oder in überschüssiger Menge bis zum 1,2-fachen verwendet. Bevorzugt wird die stöchiometrische Menge elementarer Schwefel eingesetzt.
- Die Umsetzung von beispielsweise 3-(4-Chlormethylphenyl)-1,2-benzisothiazol mit Äthylendiamin und Schwefel kann durch die folgende Reaktionsgleichung beschrieben werden:

- Die Ausgangsverbindungen der allgemeinen Formel II können beispielsweise durch Seitenkettenchlorierung von 3-(4-Methylphenyl)-1,2-benzisothiazolen mit Chlor bei etwa 170°C und unter UV-Bestrahlung erhalten werden.
- Die 3-(4-Methylphenyl)-1,2-benzisothiazole, die den Verbindungen der allgemeinen Formel II zugrundeliegen, können erhalten werden, indem man ein o-Halogenarylketon der allgemeinen Formel IV

- Der Ausgangsstoff IV, Ammoniak und elementarer Schwefel werden in etwa stöchiometrischen Mengen verwendet, jedoch verwendet man vorzugsweise ein Verhältnis von 2 bis 10 Mol Ammoniak und von 0,9 bis 1,1 Grammatom Schwefel je Mol Ausgangsstoff IV.
- Diese Umsetzung wird in der Regel bei einer Temperatur von 20 bis 250°C, vorteilhaft von 20 bis 200°C, vorzugsweise von 40 bis 180°C, insbesondere von 40 bis 120°C, drucklos oder unter Druck, kontinuierlich oder diskontinuierlich durchgeführt. Der Reaktionsdruck wird im allgemeinen durch den Gesamtdampfdruck der Komponenten bei der Umsetzungstemperatur bedingt. Gegebenenfalls kann man unter den Reaktionsbedingungen inerte organische Lösungsmittel verwenden, z.B. aromatische Kohlenwasserstoffe, wie Toluol, Äthylbenzol, o-, m-, p-Xylol, Isopropylbenzol; Alkanole und Cycloalkanole, wie Äthanol, n-Butanol, Isobutanol, Methylglykol, Cyclohexanol, Propanol, Methanol, 2-Äthylhexanol; Äther, z.B. Äthylpropyläther, Diisobutyläther, Methyl-tert.-butyläther, n-Butyläthyläther, Di-n-butyläther, Dioxan, Diisoamyläther, Diisopropyläther, Anisol, Phenetol, Cyclohexylmethyläther, Diäthyläther, Tetrahydrofuran, Thioanisol; und entsprechende Gemische. Zweckmäßig verwendet man das Lösungsmittel in einer Menge von 200 bis 10 000 Gewichtsprozent, vorzugsweise von 300 bis 1 000 Gewichtsprozent, bezogen auf Ausgangsstoff IV.
- Im einzelnen kann die Reaktion wie folgt durchgeführt werden: Ausgangsstoff IV, elementarer Schwefel und Ammoniak, gegebenenfalls in Gegenwart eines Lösungsmittels, werden in einem Druckreaktor während 3 bis 10 Stunden bei der vorgenannten Temperatur miteinander umgesetzt. Aus dem Reaktionsgemisch erhält man das 1,2-Benzisothiazol nach den üblichen Verfahren, z.B. durch fraktionierte Destillation, Filtration und gegebenenfalls anschließender Umkristallisation aus einem geeigneten Lösungsmittel, z.B. Ligroin. Man kann das Reaktionsgemisch auch nach der Entfernung überflüssigen Ammoniaks und Lösungsmittels in Wasser gießen, das gebildete Gemisch mit einem geeigneten Lösungsmittel, z.B. Methylenchlorid, Benzol, extrahieren und den Extrakt aufarbeiten.
- Die erfindungsgemäßen Verbindungen der allgemeinen Formel 1 werden gegebenenfalls in an sich üblicher Weise in das Säureadditionssalz einer physiologisch verträglichen Säure überführt. Als übliche physiologisch verträgliche organische oder anorganische Säuren kommen beispielsweise in Betracht Salzsäure, Bromwasserstoffsäure, Phosphorsäure oder Schwefelsäure und als organische Säuren beispielsweise Oxalsäure, Maleinsäure, Fumarsäure, Milchsäure, Weinsäure, Apfelsäure, Zitronensäure, Salicylsäure, Adipinsäure oder Benzoesäure oder können aus Fortschritte der Arzneimittelforschung Band 10, Seiten 224 bis 225, Birkhäuser Verlag, Basel und Stuttgart, 1966 entnommen werden.
- Die erfindungsgemäßen Verbindungen und ihre physiologisch verträglichen Säureadditionssalze zeichnen sich durch eine starke antriarrhythmische Wirkung aus und sind besonders zur Pharmakotherapie von Herzrhythmusstörungen geeignet.
- Zur Bestimmung ihrer antiarrhythmischen Wirksamkeit wurden die Substanzen Ratten (Stamm: Sprague Dawley, Gewicht: 180-240 g) 45 min vor Beginn der Narkose (Thiobutäbarbital 100 mg/kg i.p.) oral appliziert.
- Als arrhythmogene Substanz diente Aconitin, das 60 min nach der Substanzapplikation i.v. infundiert wurde (Dosierungsgeschwindigkeit: 0,005 mg/kg. min). Bei nichtbehandelten Tieren (N = 30) treten nach durchschnittlich 3,7 ± 0,9 min Arrhythmien auf, deren Eintritt durch Antiarrhythmica dosisabhängig verzögert werden kann.
- Zur quantitativen Auswertung der linearen Beziehung zwischen log Dosis (mg/kg) der Prüfsubstanzen und der relativen Verlängerung der Aconitininfusionsdauer (A%) wurde die Dosis bestimmt, welche die Infusionsdauer um 50% verlängert (ED 50%). Als Vergleichssubstanz diente das bekannte Antiarrhythmicum Procainamid.
- Die akute Toxizität wurde an Gruppen von je 10 oder 20 weiblichen Swiss-Mäusen, Gewicht 20-26 g, bei intraperitonealer Applikation ermittelt. Als LD 50 wurde die Dosis berechnet (Probit-Analyse), nach der 50% der Tiere innerhalb von 7 Tagen starben.
- Die Tabelle 1 zeigt, daß die Verbindungen der Beispiele 2 und 3, verglichen mit dem Antiarrhythmicum Procainamid, rund 5 mal stärker antiarrhythmisch wirksam sind. Ein weiterer Vorteil besteht darin, daß die Wirkung der Maximaldosis um 114 (Beispiel 2) bzw. 73% (Beispiel 3) höhere Werte erreicht, als die von Procainamid; d.h., daß der Aconitinantagonismus der geprüften Verbindungen deutlich stärker ausgeprägt ist als bei Procainamid.
- Die therapeutische Breite als Quotient aus der letalen Dosis (LD 50) und der antiarrhythmisch wirkenden Dosis (ED 50%) ist 2 mal (Beispiel 2) bzw. 1,9 mal (Beispiel 3) größer als beim Procainamid.

- In Testmodellen auf antiarrhythmische Wirkung wurden folgende Befunde erhoben:
- Reizzeitspannungsbeziehung (isol. Vorhof):
- Beispiel 2 (Tetrahydropyrimidinderivat): 1,24-fache Chinidinwirkung
- Beispiel 3 (Methylimidazolinderivat): 1,5-fache Chinidinwirkung
- Flimmerschwelle am Meerschweinchen:
- Beispiel 2: i.v. = 1,8-fache Chinidinwirkung
- Maximale Folgefrequenz am unnarkotisierten Kaninchen:
- Beispiel 2: i.v. = 7-fache Chinidinwirkung.
- Gegenstand der Erfindung sind demnach auch therapeutische Mittel oder Zubereitungen, die neben üblichen Träger- und Verdünnungsmittel eine Verbindung der allgemeinen Formel I oder dessen physiologisch verträgliches Säureadditionssalz als Wirkstoff enthalten sowie die Verwendung der neuen Verbindungen zu therapeutischen Zwecken.
- Die therapeutischen Mittel bzw. Zubereitungen werden mit den üblichen Trägerstoffen oder Verdünnungsmitteln und den üblicherweise verwendeten pharmazeutisch-technischen Hilfsstoffen entsprechend der gewünschten Applikationsart mit einer geeigneten Dosierung in bekannter Weise hergestellt.
- Die bevorzugten Zubereitungen bestehen in einer Darreichungsform, die zur oralen Applikation geeignet ist. Solche Darreichungsformen sind beispielsweise Tabletten, Filmtabletten, Dragees, Kapseln, Pillen, Pulver, Lösungen oder Suspensionen oder Depotformen.
- Selbstverständlich kommen auch parenterale Zubereitungsformen, wie Injektionslösungen oder Zusätze zu Infusionslösungen, in Betracht. Weiterhin seien als Zubereitungen beispielsweise auch Suppositorien genannt.
- Die entsprechenden Tabletten können beispielsweise durch Mischen des Wirkstoffs mit bekannten Hilfsstoffen, beispielsweise inerte Verdünnungsmittel, wie Dextrose, Zucker, Sorbit, Mannit, Polyvinylpyrrolidon, Kalziumcarbonat, Kalziumphosphat oder Milchzucker, Sprengmitteln, wie Mais, Stärke, Alginsäure oder Polyvinylpyrrolidon, Bindemitteln, wie Stärke oder Gelatine, Gleitmitteln, wie Magnesiumstearat oder Talkum und/oder Mitteln zur Erzielung des Depoteffektes, wie Carboxypolymethylen, Carboxymethylcellulose, Celluloseacetatphthalat oder Polyvinylacetat erhalten werden. Die Tabletten können auch aus mehreren Schichten bestehen.
- Entsprechend können Dragees durch Überziehen von analog den Tabletten hergestellten Kernen mit üblicherweise in Drageeüberzügen verwendeten Mitteln, beispielsweise Kollidon oder Schellack, Gummi arabicum, Talk, Titandioxid oder Zucker, hergestellt werden. Dabei kann auch die Drageehülle aus mehreren Schichten bestehen, wobei die oben bei den Tabletten erwähnten Hilfsstoffe verwendet werden können.
- Lösungen oder Suspensionen mit den erfindungsgemäßen Wirkstoffen können zusätzlich geschmackverbessemde Mittel, wie Saccharin, Cyclamat oder Zucker sowie beispielsweise Aromastoffe, wie Vanillin oder Orangenextrakt, enthalten. Sie können außerdem Suspendierhilfsstoffe, Wie Natriumcarboxymethylcellulose oder Konservierungsstoffe, wie Parahydroxybenzoate, enthalten. Wirkstoffe enthaltende Kapseln können beispielsweise hergestellt werden, indem man den Wirkstoff mit einem inerten Träger, wie Milchzucker oder Sorbit, mischt und in Gelatinekapseln einkapselt.
- Geeignete Suppositorien lassen sich beispielsweise durch Vermischen mit dafür vorgesehenem Trägermaterial, wie Neutralfetten oder Polyäthylenglykolen bzw. dessen Derivaten, herstellen.
- Die Einzeldosis einer erfindungsgemäßen Substanz am Menschen liegt bei 5 bis 100 mg, vorzugsweise bei 10 bis 80 mg.
- Die Erfindung wird durch die nachfolgenden Ausführungsbeispiele näher erläutert.
-

- In einem Emailleautoklav werden 230,5 Teile 2-Chlor-4'-methyl-benzophenon, 32 Teile Schwefel und 100 Teile NH3 in 800 Teilen Methylglykol 6 Stunden bei 160°C umgesetzt. Man erhält 200 Teile 3-(4'-Methylphenyl)-1,2-benzisothiazol mit Fp 56°C. Die Ausbeute entspricht 89% der Theorie.
- In entsprechender Weise wird aus 2,5-Dichlor-4'-methylbenzophenon, im selben Molverhältnis und unter denselben Bedingungen mit Schwefel und Ammoniak umgesetzt, 5-Chlor-3-(4'-methylphenyl)-1,2-benzisothiazol vom Fp. 121°C in 85% Ausbeute erhalten.
- Aus 2-Chlor-5-nitro-4-methylbenzophenon wird 5-Nitro-3-(4'-methylphenyl)-1,2-benzisothiazol vom Fp. 179°C in 90% Ausbeute erhalten.
- 225 g 3-(4'-Methylphenyl)-1,2-benzisothiazol werden in einer Rührapparatur auf 170°C erhitzt und unter Bestrahlung mit einer UV-Lampe werden innerhalb von 2 Stunden 100 g Chlor eingeleitet. Der Endpunkt der Reaktion wird gaschromatographisch bestimmt (Verschwinden des Ausgangsprodukts). Anschließend wird das Reaktionsgemisch abgekühlt, abgesaugt und aus Methanol umkristallisiert. Man erhält 208 g 3-(4-Chlormethylphenyl)-1,2-benzisothiazol von einem Schmelzpunkt von 86 bis 89°C, was einer Ausbeute von 80% der Theorie entspricht.
- In entsprechender Weise wird 5-Chlor-3-(4'-Chlormethylphenyl)-1,2-benzisothiazol vom Fp. 116°C mit 78% Ausbeute hergestellt.
-

- 26 g 3-(4-Chlormethylphenyl)-1,2-benzisothiazol, 6,4 g Schwefel und 300 ml Toluol werden auf 50°C erwärmt und bei dieser Temperatur 12 g Äthylendiamin langsam zugegeben. Anschließend wird das Reaktionsgemisch 15 Stunden bei Rückflußtemperatur gerührt, heiß abfiltriert und das Filtrat auf 10° bis 15°C abgekühlt. Man erhält 21 g 3-(4-lmidazolin-2-yl-phenyl)-1,2-benzisothiazol mit einem Schmelzpunkt von 177°C. Das entspricht einer Ausbeute von 75% der Theorie.

- Das Hydrochlorid der Verbindung hat einen Schmelzpunkt von 318°C.
-

- 26 g 3-(4-Chlormethylphenyl)-1,2-benzisothiazol, 6,4 g Schwefel und 300 ml Toluol werden auf 50°C erwärmt. Bei dieser Temperatur werden 15 g 1,3-Diaminopropan langsam zugegeben. Das Reaktionsgemisch wird noch 20 Stunden bei Rückflußtemperatur gerührt. Anschließend werden innerhalb 1 Stunde 20 g Chlorwasserstoffgas eingeleitet, es wird auf Raumtemperaturen abgekühlt und der entstandene Feststoff abgesaugt. Nach dem Umkristallisieren aus Wasser unter Zusatz von Aktivkohle erhält man 18 g 3-[4-(Tetrahydropyrimidin-2-yl)-phenyl]-1 ,2-benzisothiazol-hydrochlorid mit einem Schmelzpunkt von 314°C (Z). Die Ausbeute entspricht 54,6% der Theorie.

-

- 26 g 3-(4'-Chlormethylphenyl)-1,2-benzisothiazol, 6,4 g Schwefel und 15 g 1,2-Diaminopropan werden in 400 ml Glykolmonomethyläther 15 Stunden auf 110°C erwärmt. Nach dem Abdestillieren des Lösungsmittels wird der Rückstand in 50 ml Methanol gelöst und unter Kühlen in 400 ml ätherische Salzsäure (15 g HCI in Diäthyläther) eingerührt. Die gebildeten Kristalle werden abgesaugt, in 300 ml Wasser gelöst, filtriert, das Filtrat mit conc. NaOH alkalisch gestellt und mit Methylenchlorid extrahiert. Die Methylenchloridphase wird getrocknet und 20 g Chlorwasserstoffgas eingeleitet. Man erhält 17 g des gewünschten Endstoffs als farblose Kristalle vom Schmelzpunkt 243°C. Die Ausbeute entspricht 52% der Theorie.
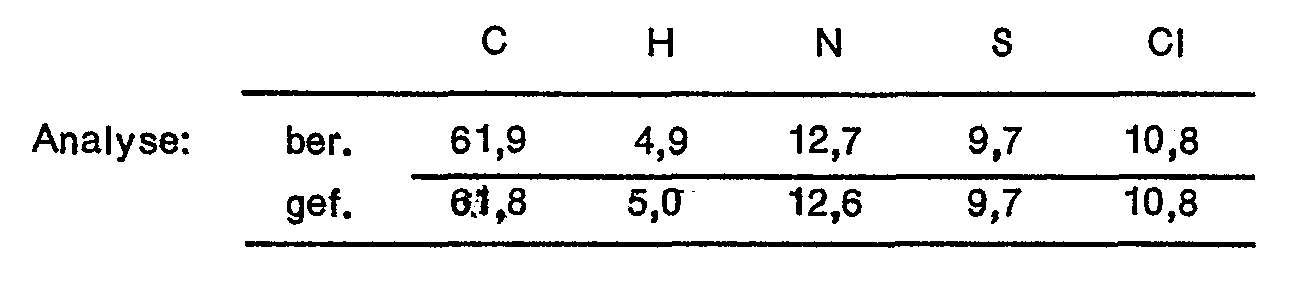
-

- 44 g 5-Chlor-3-(4'-chlormethylphenyl)-1,2-benzisothiazol, 9,6 g Schwefel und 18,0 g Äthylendiamin werden in 500 ml Toluol 20 Stunden unter Rückfluß gekocht. Das Reaktionsgemisch wird heiß filtriert, das Filtrat abgekühlt und die gebildeten Kristalle werden abgesaugt. Nach dem Umkristallisieren aus Toluol unter Zusatz von Aktivkohle werden 26 g β-Chlor-3-[4-(imidazolin-2-yl)-phenyl]-1,2-benzisothiazol vom Schmelzpunkt 195°C erhalten. Die Ausbeute entspricht 55% der Theorie.

-

- 44 g 5-Chlor-3-(4'-chlormethylphenyl)-1,2-benzisothiazol, 9,6 g Schwefel und 22,5 g 1,2-Diaminopropan werden in 600 ml Toluol 24 Stunden unter Rückfluß gekocht. Das Reaktionsgemisch wird heiß filtriert und am Rotationsverdampfer eingeengt. Der Rückstand wird in 500 ml Äther gelöst und 30 g Chlorwasserstoffgas unter Kühlen eingeleitet. Die gebildeten Kristalle werden abgesaugt und aus Wasser umkristallisiert. Man erhält 31 g, Schmelzpunkt 248°C (Z). Die Ausbeute entspricht 57% der Theorie.

- Formulierungsbeispiele, die in üblicher Weise hergestellt werden
-

- Der Wirkstoff wird mit Polyvinylpyrrolidon in 10%iger wäßriger Lösung befeuchtet, durch ein Sieb mit der lichten Maschenweite 1,0 mm getrieben und bei 50°C getrocknet. Dieses Granulat wird mit Polyäthylenglykol (mittl. M.G. 4000), Hydroxypropylmethylcellulose, Talkum und Magnesiumstearat vermischt und zu Tabletten ä 280 mg verpreßt.
-

- Die Mischung der Wirkstoffsubstanz mit Lactose und Maisstärke wird mit einer 8%igen wäßrigen Lösung des Polyvinylpyrrolidons durch Sieb 1,5 mm granuliert, bei 50°C getrocknet und nochmals durch Sieb 1,0 mm gerieben. Das so erhaltene Granulat wird mit Magnesiumstearat gemischt und zu Drageekernen verpreßt. Die erhaltenen Drageekerne werden in üblicher Weise mit einer Hülle überzogen, die im wesentlichen aus Zucker und Talkum besteht.
-

-

Claims (7)



Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19772734882 DE2734882A1 (de) | 1977-08-03 | 1977-08-03 | 3- eckige klammer auf 4-(1,3-diazacycloalken-2-yl)-phenyl eckige klammer zu -1,2-benzisothiazole, verfahren zu ihrer herstellung und diese enthaltende arzneimittel |
| DE2734882 | 1977-08-03 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0000727A1 EP0000727A1 (de) | 1979-02-21 |
| EP0000727B1 true EP0000727B1 (de) | 1980-08-20 |
Family
ID=6015470
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP78100498A Expired EP0000727B1 (de) | 1977-08-03 | 1978-07-26 | 3-(4-(1,3-Diazacycloalken-2-yl)-phenyl)-1,2-benzisothiazole, Verfahren zu ihrer Herstellung und diese enthaltende Arzneimittel. |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US4206217A (de) |
| EP (1) | EP0000727B1 (de) |
| AT (1) | AT364841B (de) |
| DE (2) | DE2734882A1 (de) |
| IT (1) | IT1105391B (de) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ182325A (en) * | 1975-11-18 | 1979-03-16 | Beecham Group Ltd | 2-substituted-1,2-benzisothiazol-3-ones |
| DE2734866A1 (de) * | 1977-08-03 | 1979-02-22 | Basf Ag | Neue 1,2-benzisothiazole und verfahren zu ihrer herstellung |
| US4957931A (en) * | 1987-07-08 | 1990-09-18 | Ciba-Geigy Corporation | Certain 1,2-benzisoxazole and 1,2-benzisothiazole derivatives |
| US20170233199A1 (en) * | 2016-01-20 | 2017-08-17 | Rehrig Pacific Company | Bakery tray stacker |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3549624A (en) * | 1967-08-17 | 1970-12-22 | Pfizer & Co C | 2-substituted-2-delta**2-tetrahydropyrimidines and delta**2-im |
| US3557115A (en) * | 1968-11-29 | 1971-01-19 | Sandoz Ag | 2 - (2 - imidazolin and pyrimidin substituted methylthio) - imidazoles and pyrimidines |
| US4147698A (en) * | 1978-07-13 | 1979-04-03 | E. R. Squibb & Sons, Inc. | 3-(Heterocyclicalkylamino)benzisothiazole-1,1-dioxides |
| US4148798A (en) * | 1978-02-03 | 1979-04-10 | E. R. Squibb & Sons, Inc. | [(1,1-dioxo-1,2-benzisothiazol-3-yl)amino]alkanoic acids and esters thereof |
-
1977
- 1977-08-03 DE DE19772734882 patent/DE2734882A1/de not_active Withdrawn
-
1978
- 1978-07-26 EP EP78100498A patent/EP0000727B1/de not_active Expired
- 1978-07-26 DE DE7878100498T patent/DE2860232D1/de not_active Expired
- 1978-07-28 US US05/928,815 patent/US4206217A/en not_active Expired - Lifetime
- 1978-07-31 IT IT50533/78A patent/IT1105391B/it active
- 1978-08-02 AT AT0559778A patent/AT364841B/de not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| EP0000727A1 (de) | 1979-02-21 |
| DE2734882A1 (de) | 1979-02-22 |
| DE2860232D1 (en) | 1980-12-04 |
| IT7850533A0 (it) | 1978-07-31 |
| ATA559778A (de) | 1981-04-15 |
| AT364841B (de) | 1981-11-25 |
| US4206217A (en) | 1980-06-03 |
| IT1105391B (it) | 1985-10-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE2430237C2 (de) | Äthanolaminoderivate und sie enthaltende pharmazeutische Zusammensetzungen | |
| AT363096B (de) | Verfahren zur herstellung von neuen phthalazinderivaten und deren salzen | |
| DE1964761B2 (de) | Fluorenon- und Fluorenverbindungen und Arzneimittel, welche diese enthalten | |
| EP0204269B1 (de) | Pyridin-Derivate, ihre Herstellung und Verwendung | |
| DE2420618C2 (de) | Aminoalkylanilide, Verfahren zu ihrer Herstellung sowie pharmazeutische Zubereitungen, die diese Aminoalkylanilide enthalten | |
| EP0101574B1 (de) | 2-Substituierte 1-Aminoalkyl-1,2,3,4-tetrahydro-beta-carboline, ihre Herstellung und Verwendung als Arzneimittel | |
| EP0000727B1 (de) | 3-(4-(1,3-Diazacycloalken-2-yl)-phenyl)-1,2-benzisothiazole, Verfahren zu ihrer Herstellung und diese enthaltende Arzneimittel. | |
| EP0018360B1 (de) | N-(5-Methoxybentofuran-2-ylcarbonyl)-N'-benzylpiperazin und Verfahren zu dessen Herstellung | |
| DE3025238A1 (de) | Basische cycloalkanonoximaether, verfahren zur herstellung derselben und diese enthaltende arzneimittel | |
| DD153550A5 (de) | Verfahren zur herstellung von neuer 1,2,4-oxadiazolin-5-on-derivaten | |
| DE1915230B2 (de) | Hydroxyphenylalkylaminderivate, verfahren zu deren herstellung und arzneimittel auf deren basis | |
| DD205682A5 (de) | Verfahren zur herstellung von (-)-2-(1-(2,6-dichlorphenoxy)-ethyl)-1,3-diazacyclopent-2-en und seinen salzen | |
| AT389872B (de) | Verfahren zur herstellung von neuen substituierten 2-phenylmethylen-1aminoalkyloximinocycloalkanen und deren saeureadditionssalzen | |
| DE2003744C2 (de) | In 3- und 4-Stellung disubstituierte 3-Amino-2-bicyclo[2,2,2]octan-2-ole, Verfahren zu deren Herstellung und dieselben enthaltende Arzneimittel | |
| DE2507429A1 (de) | Aminosaeureester, verfahren zu ihrer herstellung und sie enthaltende arzneimittel | |
| EP0027928A2 (de) | Piperidinderivate von 4,5-Dialkyl-3-hydroxy-pyrrol-2-carbonsäureestern, ihre Herstellung und diese enthaltende pharmazeutische Zubereitungen | |
| DE2624918A1 (de) | Arzneimittel mit antiarrhythmischer wirkung | |
| CH630895A5 (de) | Verfahren zur herstellung neuer oximaetherverbindungen. | |
| EP0000750A1 (de) | Neue 1,2-Benzisothiazole und Verfahren zu ihrer Herstellung | |
| DE2738131A1 (de) | Aminoketone, verfahren zu ihrer herstellung und sie enthaltende arzneimittel | |
| EP0152556A1 (de) | Aminopropanolderivate von substituierten 2-Hydroxy-propiophenonen, Verfahren zu ihrer Herstellung und diese enthaltende therapeutische Mittel | |
| DE2166270B2 (de) | Nicotinoylaminoäthansulfonyl-2amino-thiazol | |
| DE2530768C3 (de) | PhenoxyaUcylaminpyridyläther, Verfahren zu ihrer Herstellung sowie diese enthaltende pharmazeutische Präparate | |
| DE2442158A1 (de) | Neue substituierte n- eckige klammer auf l-(3,4-methylendioxyphenyl)propyl(2) eckige klammer zu -n'-subst. phenylpiperazine | |
| DE69523346T2 (de) | Substituierte 6-h-1,3,4-thiadiazin-2-amine, deren verwendung als anaesthetisierende cardiovaskulare und hypometabole mittel, und eine sie enthaltende pharmazeutische zubereitung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB NL |
|
| 17P | Request for examination filed | ||
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB NL |
|
| REF | Corresponds to: |
Ref document number: 2860232 Country of ref document: DE Date of ref document: 19801204 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19840622 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19840625 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 19840627 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 19840630 Year of fee payment: 7 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 19860731 Year of fee payment: 9 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19870727 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: CH Effective date: 19870731 |
|
| BERE | Be: lapsed |
Owner name: BASF A.G. Effective date: 19870731 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19880201 |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19880331 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee | ||
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 19881117 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BE Effective date: 19890731 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |