DE69720979T2 - Thermostabilisiertes kontrastmittel - Google Patents
Thermostabilisiertes kontrastmittel Download PDFInfo
- Publication number
- DE69720979T2 DE69720979T2 DE69720979T DE69720979T DE69720979T2 DE 69720979 T2 DE69720979 T2 DE 69720979T2 DE 69720979 T DE69720979 T DE 69720979T DE 69720979 T DE69720979 T DE 69720979T DE 69720979 T2 DE69720979 T2 DE 69720979T2
- Authority
- DE
- Germany
- Prior art keywords
- stabilizers
- mixture
- stabilizer
- freeze
- gas
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 239000002872 contrast media Substances 0.000 title claims description 24
- 239000003381 stabilizer Substances 0.000 claims abstract description 61
- 238000004108 freeze drying Methods 0.000 claims abstract description 28
- 239000002961 echo contrast media Substances 0.000 claims abstract description 22
- 239000000203 mixture Substances 0.000 claims description 50
- 239000006185 dispersion Substances 0.000 claims description 23
- 238000000034 method Methods 0.000 claims description 21
- KAVGMUDTWQVPDF-UHFFFAOYSA-N perflubutane Chemical group FC(F)(F)C(F)(F)C(F)(F)C(F)(F)F KAVGMUDTWQVPDF-UHFFFAOYSA-N 0.000 claims description 15
- 229950003332 perflubutane Drugs 0.000 claims description 11
- 229930006000 Sucrose Natural products 0.000 claims description 10
- 239000005720 sucrose Substances 0.000 claims description 10
- SFZCNBIFKDRMGX-UHFFFAOYSA-N sulfur hexafluoride Chemical compound FS(F)(F)(F)(F)F SFZCNBIFKDRMGX-UHFFFAOYSA-N 0.000 claims description 10
- 229960000909 sulfur hexafluoride Drugs 0.000 claims description 10
- -1 H 2 O Chemical compound 0.000 claims description 9
- 238000003860 storage Methods 0.000 claims description 9
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 claims description 8
- 239000000945 filler Substances 0.000 claims description 8
- 239000002243 precursor Substances 0.000 claims description 8
- 238000004519 manufacturing process Methods 0.000 claims description 7
- 230000008569 process Effects 0.000 claims description 7
- 239000003795 chemical substances by application Substances 0.000 claims description 6
- 229910018503 SF6 Inorganic materials 0.000 claims description 5
- 150000008282 halocarbons Chemical class 0.000 claims description 5
- 239000012528 membrane Substances 0.000 claims description 5
- 229920005862 polyol Polymers 0.000 claims description 5
- QIYZKVMAFMDRTP-UHFFFAOYSA-N pentafluoro(trifluoromethyl)-$l^{6}-sulfane Chemical compound FC(F)(F)S(F)(F)(F)(F)F QIYZKVMAFMDRTP-UHFFFAOYSA-N 0.000 claims description 4
- NJCBUSHGCBERSK-UHFFFAOYSA-N perfluoropentane Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F NJCBUSHGCBERSK-UHFFFAOYSA-N 0.000 claims description 4
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 claims description 3
- 229960004692 perflenapent Drugs 0.000 claims description 3
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 claims description 2
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 claims description 2
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 claims description 2
- MUPFEKGTMRGPLJ-RMMQSMQOSA-N Raffinose Natural products O(C[C@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O[C@@]2(CO)[C@H](O)[C@@H](O)[C@@H](CO)O2)O1)[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 MUPFEKGTMRGPLJ-RMMQSMQOSA-N 0.000 claims description 2
- UQZIYBXSHAGNOE-USOSMYMVSA-N Stachyose Natural products O(C[C@H]1[C@@H](O)[C@H](O)[C@H](O)[C@@H](O[C@@]2(CO)[C@H](O)[C@@H](O)[C@@H](CO)O2)O1)[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@H](CO[C@@H]2[C@@H](O)[C@@H](O)[C@@H](O)[C@H](CO)O2)O1 UQZIYBXSHAGNOE-USOSMYMVSA-N 0.000 claims description 2
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 claims description 2
- MUPFEKGTMRGPLJ-UHFFFAOYSA-N UNPD196149 Natural products OC1C(O)C(CO)OC1(CO)OC1C(O)C(O)C(O)C(COC2C(C(O)C(O)C(CO)O2)O)O1 MUPFEKGTMRGPLJ-UHFFFAOYSA-N 0.000 claims description 2
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 claims description 2
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 claims description 2
- 238000001816 cooling Methods 0.000 claims description 2
- MUPFEKGTMRGPLJ-ZQSKZDJDSA-N raffinose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)O)O1 MUPFEKGTMRGPLJ-ZQSKZDJDSA-N 0.000 claims description 2
- UQZIYBXSHAGNOE-XNSRJBNMSA-N stachyose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@@H]3[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O3)O)O2)O)O1 UQZIYBXSHAGNOE-XNSRJBNMSA-N 0.000 claims description 2
- 239000008365 aqueous carrier Substances 0.000 claims 1
- 229910052698 phosphorus Inorganic materials 0.000 claims 1
- 239000011574 phosphorus Substances 0.000 claims 1
- 239000007789 gas Substances 0.000 description 28
- 239000000047 product Substances 0.000 description 23
- 239000000463 material Substances 0.000 description 15
- 150000003904 phospholipids Chemical class 0.000 description 13
- 239000012530 fluid Substances 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 10
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 7
- 229930195725 Mannitol Natural products 0.000 description 7
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 7
- 239000000594 mannitol Substances 0.000 description 7
- 235000010355 mannitol Nutrition 0.000 description 7
- 238000002604 ultrasonography Methods 0.000 description 7
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 description 6
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 6
- 238000009472 formulation Methods 0.000 description 6
- 230000009477 glass transition Effects 0.000 description 6
- 239000008103 glucose Substances 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- ZWZWYGMENQVNFU-UHFFFAOYSA-N Glycerophosphorylserin Natural products OC(=O)C(N)COP(O)(=O)OCC(O)CO ZWZWYGMENQVNFU-UHFFFAOYSA-N 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 150000001875 compounds Chemical class 0.000 description 5
- 238000011084 recovery Methods 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 125000004432 carbon atom Chemical group C* 0.000 description 4
- 229940039231 contrast media Drugs 0.000 description 4
- 238000009826 distribution Methods 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 238000012512 characterization method Methods 0.000 description 3
- 238000013016 damping Methods 0.000 description 3
- 125000001924 fatty-acyl group Chemical group 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 3
- ZJIJAJXFLBMLCK-UHFFFAOYSA-N perfluorohexane Chemical class FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F ZJIJAJXFLBMLCK-UHFFFAOYSA-N 0.000 description 3
- 150000008106 phosphatidylserines Chemical class 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 150000003077 polyols Chemical class 0.000 description 3
- 235000013772 propylene glycol Nutrition 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 3
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 2
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- QQONPFPTGQHPMA-UHFFFAOYSA-N Propene Chemical compound CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 230000036760 body temperature Effects 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000002577 cryoprotective agent Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 238000002592 echocardiography Methods 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 210000003743 erythrocyte Anatomy 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- 229960004624 perflexane Drugs 0.000 description 2
- 150000008103 phosphatidic acids Chemical class 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 125000000185 sucrose group Chemical group 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 229960003853 ultrasound contrast media Drugs 0.000 description 2
- COQIQRBKEGPRSG-UHFFFAOYSA-N 1,1,1,2,3,3,3-heptafluoro-2-(trifluoromethyl)propane Chemical compound FC(F)(F)C(F)(C(F)(F)F)C(F)(F)F COQIQRBKEGPRSG-UHFFFAOYSA-N 0.000 description 1
- WSJULBMCKQTTIG-UHFFFAOYSA-N 1,1,1,2,3,4,4,4-octafluorobut-2-ene Chemical class FC(F)(F)C(F)=C(F)C(F)(F)F WSJULBMCKQTTIG-UHFFFAOYSA-N 0.000 description 1
- FNVLGCVAWPSVSK-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5,6,6,7,7-tetradecafluorocycloheptane Chemical compound FC1(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F FNVLGCVAWPSVSK-UHFFFAOYSA-N 0.000 description 1
- RKIMETXDACNTIE-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5,6,6-dodecafluorocyclohexane Chemical compound FC1(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F RKIMETXDACNTIE-UHFFFAOYSA-N 0.000 description 1
- QIROQPWSJUXOJC-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5,6-undecafluoro-6-(trifluoromethyl)cyclohexane Chemical compound FC(F)(F)C1(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F QIROQPWSJUXOJC-UHFFFAOYSA-N 0.000 description 1
- PWMJXZJISGDARB-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5,5-decafluorocyclopentane Chemical compound FC1(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F PWMJXZJISGDARB-UHFFFAOYSA-N 0.000 description 1
- BCNXQFASJTYKDJ-UHFFFAOYSA-N 1,1,2,2,3,3,4,4,5-nonafluoro-5-(trifluoromethyl)cyclopentane Chemical compound FC(F)(F)C1(F)C(F)(F)C(F)(F)C(F)(F)C1(F)F BCNXQFASJTYKDJ-UHFFFAOYSA-N 0.000 description 1
- CIWUYWQUYMZILR-UHFFFAOYSA-N 1,1,2,2,3,3,4,4-octafluoro-5,5-bis(trifluoromethyl)cyclopentane Chemical class FC(F)(F)C1(C(F)(F)F)C(F)(F)C(F)(F)C(F)(F)C1(F)F CIWUYWQUYMZILR-UHFFFAOYSA-N 0.000 description 1
- ZVXOHSHODRJTCP-UHFFFAOYSA-N 1,1,2,2,3,3,4-heptafluoro-4-(trifluoromethyl)cyclobutane Chemical compound FC(F)(F)C1(F)C(F)(F)C(F)(F)C1(F)F ZVXOHSHODRJTCP-UHFFFAOYSA-N 0.000 description 1
- TXGPGHBYAPBDAG-UHFFFAOYSA-N 1,1,2,2,3,3-hexafluoro-4,4-bis(trifluoromethyl)cyclobutane Chemical class FC(F)(F)C1(C(F)(F)F)C(F)(F)C(F)(F)C1(F)F TXGPGHBYAPBDAG-UHFFFAOYSA-N 0.000 description 1
- YUFJLVUCHXMKKM-UHFFFAOYSA-N 1,1,2,2,3-pentafluoro-3,4,4-tris(trifluoromethyl)cyclobutane Chemical class FC(F)(F)C1(F)C(F)(F)C(F)(F)C1(C(F)(F)F)C(F)(F)F YUFJLVUCHXMKKM-UHFFFAOYSA-N 0.000 description 1
- ZVJOQYFQSQJDDX-UHFFFAOYSA-N 1,1,2,3,3,4,4,4-octafluorobut-1-ene Chemical class FC(F)=C(F)C(F)(F)C(F)(F)F ZVJOQYFQSQJDDX-UHFFFAOYSA-N 0.000 description 1
- LGPPATCNSOSOQH-UHFFFAOYSA-N 1,1,2,3,4,4-hexafluorobuta-1,3-diene Chemical compound FC(F)=C(F)C(F)=C(F)F LGPPATCNSOSOQH-UHFFFAOYSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- PORPENFLTBBHSG-MGBGTMOVSA-N 1,2-dihexadecanoyl-sn-glycerol-3-phosphate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(O)=O)OC(=O)CCCCCCCCCCCCCCC PORPENFLTBBHSG-MGBGTMOVSA-N 0.000 description 1
- YFWHNAWEOZTIPI-DIPNUNPCSA-N 1,2-dioctadecanoyl-sn-glycerol-3-phosphate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(O)=O)OC(=O)CCCCCCCCCCCCCCCCC YFWHNAWEOZTIPI-DIPNUNPCSA-N 0.000 description 1
- RFCAUADVODFSLZ-UHFFFAOYSA-N 1-Chloro-1,1,2,2,2-pentafluoroethane Chemical compound FC(F)(F)C(F)(F)Cl RFCAUADVODFSLZ-UHFFFAOYSA-N 0.000 description 1
- ABQLAMJAQZFPJI-UHFFFAOYSA-N 3-heptyloxolan-2-one Chemical compound CCCCCCCC1CCOC1=O ABQLAMJAQZFPJI-UHFFFAOYSA-N 0.000 description 1
- 241000700112 Chinchilla Species 0.000 description 1
- VOPWNXZWBYDODV-UHFFFAOYSA-N Chlorodifluoromethane Chemical compound FC(F)Cl VOPWNXZWBYDODV-UHFFFAOYSA-N 0.000 description 1
- 239000004340 Chloropentafluoroethane Substances 0.000 description 1
- PMPVIKIVABFJJI-UHFFFAOYSA-N Cyclobutane Chemical compound C1CCC1 PMPVIKIVABFJJI-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- KLFKZIQAIPDJCW-HTIIIDOHSA-N Dipalmitoylphosphatidylserine Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCC KLFKZIQAIPDJCW-HTIIIDOHSA-N 0.000 description 1
- 102000002322 Egg Proteins Human genes 0.000 description 1
- 108010000912 Egg Proteins Proteins 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 229920002560 Polyethylene Glycol 3000 Polymers 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 208000003455 anaphylaxis Diseases 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 210000001772 blood platelet Anatomy 0.000 description 1
- 238000009530 blood pressure measurement Methods 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- MEXUFEQDCXZEON-UHFFFAOYSA-N bromochlorodifluoromethane Chemical compound FC(F)(Cl)Br MEXUFEQDCXZEON-UHFFFAOYSA-N 0.000 description 1
- RJCQBQGAPKAMLL-UHFFFAOYSA-N bromotrifluoromethane Chemical compound FC(F)(F)Br RJCQBQGAPKAMLL-UHFFFAOYSA-N 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 210000001715 carotid artery Anatomy 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 235000019406 chloropentafluoroethane Nutrition 0.000 description 1
- AFYPFACVUDMOHA-UHFFFAOYSA-N chlorotrifluoromethane Chemical compound FC(F)(F)Cl AFYPFACVUDMOHA-UHFFFAOYSA-N 0.000 description 1
- 230000001268 conjugating effect Effects 0.000 description 1
- 150000001924 cycloalkanes Chemical class 0.000 description 1
- 238000002059 diagnostic imaging Methods 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 239000012470 diluted sample Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 235000013345 egg yolk Nutrition 0.000 description 1
- 210000002969 egg yolk Anatomy 0.000 description 1
- 235000013601 eggs Nutrition 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 150000002321 glycerophosphoglycerophosphoglycerols Chemical class 0.000 description 1
- 125000003976 glyceryl group Chemical group [H]C([*])([H])C(O[H])([H])C(O[H])([H])[H] 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 230000000004 hemodynamic effect Effects 0.000 description 1
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 1
- VBZWSGALLODQNC-UHFFFAOYSA-N hexafluoroacetone Chemical compound FC(F)(F)C(=O)C(F)(F)F VBZWSGALLODQNC-UHFFFAOYSA-N 0.000 description 1
- WMIYKQLTONQJES-UHFFFAOYSA-N hexafluoroethane Chemical compound FC(F)(F)C(F)(F)F WMIYKQLTONQJES-UHFFFAOYSA-N 0.000 description 1
- HCDGVLDPFQMKDK-UHFFFAOYSA-N hexafluoropropylene Chemical compound FC(F)=C(F)C(F)(F)F HCDGVLDPFQMKDK-UHFFFAOYSA-N 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 229910052743 krypton Inorganic materials 0.000 description 1
- DNNSSWSSYDEUBZ-UHFFFAOYSA-N krypton atom Chemical compound [Kr] DNNSSWSSYDEUBZ-UHFFFAOYSA-N 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 210000000865 mononuclear phagocyte system Anatomy 0.000 description 1
- JVKAWJASTRPFQY-UHFFFAOYSA-N n-(2-aminoethyl)hydroxylamine Chemical compound NCCNO JVKAWJASTRPFQY-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- BCCOBQSFUDVTJQ-UHFFFAOYSA-N octafluorocyclobutane Chemical compound FC1(F)C(F)(F)C(F)(F)C1(F)F BCCOBQSFUDVTJQ-UHFFFAOYSA-N 0.000 description 1
- 235000019407 octafluorocyclobutane Nutrition 0.000 description 1
- QYSGYZVSCZSLHT-UHFFFAOYSA-N octafluoropropane Chemical class FC(F)(F)C(F)(F)C(F)(F)F QYSGYZVSCZSLHT-UHFFFAOYSA-N 0.000 description 1
- 230000014207 opsonization Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- LGUZHRODIJCVOC-UHFFFAOYSA-N perfluoroheptane Chemical class FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F LGUZHRODIJCVOC-UHFFFAOYSA-N 0.000 description 1
- 150000003905 phosphatidylinositols Chemical class 0.000 description 1
- 229940067626 phosphatidylinositols Drugs 0.000 description 1
- 229920000233 poly(alkylene oxides) Polymers 0.000 description 1
- 229920000151 polyglycol Polymers 0.000 description 1
- 239000010695 polyglycol Substances 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 1
- 150000004756 silanes Chemical class 0.000 description 1
- 238000002603 single-photon emission computed tomography Methods 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 125000003696 stearoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 150000005846 sugar alcohols Chemical class 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- TXEYQDLBPFQVAA-UHFFFAOYSA-N tetrafluoromethane Chemical compound FC(F)(F)F TXEYQDLBPFQVAA-UHFFFAOYSA-N 0.000 description 1
- 238000003325 tomography Methods 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 238000012285 ultrasound imaging Methods 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 229910052724 xenon Inorganic materials 0.000 description 1
- FHNFHKCVQCLJFQ-UHFFFAOYSA-N xenon atom Chemical compound [Xe] FHNFHKCVQCLJFQ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/22—Echographic preparations; Ultrasound imaging preparations ; Optoacoustic imaging preparations
- A61K49/222—Echographic preparations; Ultrasound imaging preparations ; Optoacoustic imaging preparations characterised by a special physical form, e.g. emulsions, liposomes
- A61K49/223—Microbubbles, hollow microspheres, free gas bubbles, gas microspheres
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/22—Echographic preparations; Ultrasound imaging preparations ; Optoacoustic imaging preparations
- A61K49/222—Echographic preparations; Ultrasound imaging preparations ; Optoacoustic imaging preparations characterised by a special physical form, e.g. emulsions, liposomes
- A61K49/227—Liposomes, lipoprotein vesicles, e.g. LDL or HDL lipoproteins, micelles, e.g. phospholipidic or polymeric
Description
- Die vorliegende Erfindung betrifft thermisch stabilisierte gefriergetrocknete vesikelhaltige Ultraschallkontrastmittel sowie ein Verfahren zu deren Herstellung.
- Vesikel (dieser Begriff wird hierin verwendet, um unilamellare und multilamellare Strukturen zu bezeichnen, z. B. Strukturen, welche verschiedentlich als Liposome, Micellen, Mikrobläschen und Mikroballone bezeichnet werden) werden häufig als Mittel zur Gabe beziehungsweise Verabreichung von therapeutisch oder diagnostisch wirksamen Stoffen verwendet. In dem Gebiet der Ultraschallbildgebungskontrastmedien können Vesikel, welche Materialien enthalten (hierin als vesikuläre Materialien bezeichnet), die bei Körpertemperatur gasförmig sind, als echogene Kontrastmittel verwendet werden, insbesondere zur Verabreichung in das Gefäßsystem.
- Vesikuläre Kontrastmedien werden im Allgemeinen in der Form einer wässrigen Dispersion verabreicht, welche eine niedrige Konzentration der Vesikel relativ zu dem wässrigen Trägermedium enthält. Dementsprechend wird die Lagerung und der Transport solcher vesikulären Kontrastmittel deutlich effizienter, wenn die Vesikel in getrockneter Form gelagert werden können.
- Das Gefriertrocknen von vesikulären Zusammensetzungen ist möglich, und daher werden im Allgemeinen Formulierungshilfsmittel in die Zusammensetzung eingeschlossen, um die Trockentechnik zu unterstützen. Solche Hilfsmittel dienen im Allgemeinen zu einer von zwei Funktionen. Es werden Füllmittel zugesetzt, um den Gesamtfeststoffgehalt zu erhöhen, so dass ein mechanisch robusteres Produkt erreicht wird. Stabilisatoren, anders auch als Kryoprotektanden oder Lyoprotektanden bezeichnet, werden zugesetzt, um die Bildung des Glaszustandes zu unterstützen, welcher während der Entwässerung gebildet wird, und um dem getrockneten Produkt physikalische Festigkeit zu verleihen. Beispiele für auf diese Weise verwendete Stabilisatoren umschließen Mannit und Glukose.
- Gefriergetrocknete vesikuläre Ultraschallkontrastmittel vermitteln ebenso Probleme, obwohl sie Vorteile hinsichtlich des Transports und der Lagerung auf Grund der Verminderung in der Masse relativ zu den wässrigen zur Verwendung fertigen Dispersionen zu Verfügung stellen, da das gefriergetrocknete Produkt in dem Bereich von Umgebungstemperaturen normalerweise im Zusammenhang mit dem Transport und der Lagerung thermisch nicht stabil ist, und als ein Ergebnis muss es vor der sekundären Bildung beziehungsweise Herstellung in einer Umgebung gehalten werden, in welcher die Temperatur unterhalb der Umgebungstemperatur gehalten wird (z. B. bei 5 bis 10°C).
- Es wurde nun überraschenderweise gefunden, dass durch die geeignete Wahl des Stabilisators, welcher zum Gefriertrocknen verwendet wird, es möglich ist, gefriergetrocknete vesikuläre Ultraschallkontrastmittel herzustellen, welche bei Umgebungstemperaturen und oberhalb davon und in der Tat bei allen Temperaturen, welche im Normalfall während des Transports und der Lagerung auftreten, thermisch stabil sind.
- Das thermisch stabile gefriergetrocknete Produkt kann anschließend ohne den Bedarf einer Temperaturkontrolle von dessen Umgebung gelagert und transportiert werden und kann insbesondere an Krankenhäuser und Ärzte für die Vor-Ort-Formulierung in eine verabreichbare Dispersion geliefert werden, ohne das es erforderlich ist, dass solche Verwender spezielle Lagerungsvorrichtungen besitzen.
- Von einem Aspekt betrachtet stellt somit die vorliegende Erfindung ein gefriergetrocknetes, vesikelhaltiges Ultraschallkontrastmittel zur Verfügung, enthaltend einen Gefriertrocknungsstabilisator oder eine Mischung von Stabilisatoren, wobei (i) das Gewichtsverhältnis des Stabilisators oder der Mischung von Stabilisatoren zu den Vesikeln in dem Mittel mindestens 10 : 1 beträgt, (ii) der Stabilisator oder die Mischung von Stabilisatoren einen Tg-Wert oberhalb von 20°C (bevorzugt von mindestens 22°C, insbesondere bevorzugt von mindestens 25°C, ganz besonders bevorzugt von mindestens 30°C und äußerst bevorzugt von mindestens 40°C, z. B. bis zu 65°C oder höher) und einen Tg'-Wert von –37°C oder darüber (bevorzugt oberhalb von –36°C, besonders bevorzugt oberhalb von –35°C, z. B. von –10 bis –37°C) aufweist, und (iii) das Mittel bei Temperaturen oberhalb von 20°C (bevorzugt von mindestens 22°C, besonders bevorzugt von mindestens 25°C, ganz besonders bevorzugt von mindestens 30°C und äußerst bevorzugt von mindestens 40°C, z. B. von bis zu 65°C oder höher) thermisch stabil ist.
- Von einem weiteren Aspekt betrachtet stellt die vorliegende Erfindung ein Verfahren zur Herstellung eines thermisch stabilen gerfiergetrockneten vesikelhaltigen Ultraschallkontrastmittels zur Verfügung, welches bei Temperaturen oberhalb von 20°C (bevorzugt von mindestens 22°C, besonders bevorzugt von mindestens 25°C, ganzu besonders bevorzugt von mindestens 30°C und äußerst bevorzugt von mindestens 40°C, z. B. bis zu 65°C oder höher) thermisch stabil ist, wobei das Verfahren das Gefriertrocknen einer wässrigen Dispersion, umfassend ein vesikuläres Ultraschallkontrastmittel und einen Gefriertrocknungsstabilisator oder eine Mischung von Stabilisatoren, umfasst, welches dadurch gekennzeichnet ist, dass der Stabilisator oder die Mischung von Stabilisatoren in der Dispersion in einem Gewichtsverhältnis bezüglich der Vesikel darin von mindestens 10 : 1 vorliegt, und dass der Stabilisator oder die Mischung von Stabilisatoren einen Tg-Wert oberhalb von 20°C (bevorzugt von mindestens 22°C, besonders bevorzugt von mindestens 25°C, ganz besonders bevorzugt von mindestens 30°C und außerst bevorzugt von mindestens 40°C, z. B. bis zu 65°C oder höher) und einen Tg'-Wert von –37°C oder darüber (bevorzugt oberhalb von –36°C, besonders bevorzugt oberhalb von –35°C, z. B. von –10 bis –37°C) besitzt.
- Von noch einem weiteren Aspekt betrachtet stellt die vorliegende Erfindung die Verwendung eines gefriergetrockneten Stabilisators oder eine Mischung von Stabilisatoren mit einem Tg-Wert oberhalb von 20°C (bevorzugt von mindestens 22°C, besonders bevorzugt von mindestens 25°C, ganz besonders bevorzugt von mindestens 30°C und äußerst bevorzugt von mindestens 40°C, z. B. bis zu 65°C oder höher) und einen Tg'-Wert von –37°C oder darüber (bevorzugt oberhalb –36°C, besonders bevorzugt oberhalb –35°C, z. B. von –10 bis 37°C) in der Herstellung eines vesikelhaltigen Ultraschallkontrastmittels zur Verfügung, welches bei Temperaturen oberhalb von 20°C (bevorzugt von mindestens 22°C, besonders bevorzugt von mindestens 25°C, ganz besonders bevorzugt von mindestens 30°C und äußerst bevorzugt von mindestens 40°C, z. B. bis zu 65°C oder höher) thermisch stabil ist und welches den Stabilisator oder die Mischung von Stabilisatoren in einem Gewichtsverhältnis bezüglich des Vesikels darin von mindestens 10 : 1 enthält.
- Von einem weiteren Aspekt betrachtet stellt die vorliegende Erfindung ein Verfahren zur Lagerung und zum Transport eines gefriergetrockneten vesikelhaltigen Ultraschallkontrastmittels, wie es oben definiert wurde, zur Verfügung, worin die Lagerung oder der Transport ohne Kühlung stattfindet.
- Von noch einem weiteren Aspekt betrachtet stellt die vorliegende Erfindung ein Verfahren zur Herstellung eines vesikelhaltigen Ultraschallkontrastmittels zur Verfügung, welches das Dispergieren eines gefriergetrockneten vesikelhaltigen Ultraschallkontrastmittels, wie es oben definiert wurde, in einem physiologisch tolerierbaren wässrigen Dispersionsmedium umfasst.
- Für ein jedes Material ist Tg die Glasübergangstemperatur des getrockneten Materials, während Tg' die Glasübergangstemperatur der maximal gefrieraufkonzentrierten reinen wässrigen Lösung des Materials darstellt.
- Neben der verbesserten thermischen Stabilität erhöhen die gefriergetrockneten vesikulären Kontrastmittel gemäß der vorliegenden Erfindung ebenso überraschenderweise die Fähigkeit der Vesikel, Halocarbongase und Gasvorstufen, welche in herkömmlichen Ultraschallkontrastmitteln verwendet werden, zurückzuhalten.
- In der Erfindung können die Ultraschallkontrastmittel jegliche physiologisch tolerierbare echogene vesikuläre Mittel sein. Bevorzugt enthalten die Vesikel jedoch ein Gas oder eine Gasvorstufe beziehungsweise einen Gasvorläufer (z. B. eine Verbindung oder eine Verbindungsmischung, welche im wesentlichen gasförmig (einschlielilich Dampft bei normaler Temperatur des menschlichen Körpers (37°C) vorliegt). Ein jegliches biokompatibles Gas, Gasvorläufer oder Mischung kann eingesetzt werden. Das Gas kann daher zum Beispiel umfassen Luft; Stickstoff; Sauerstoff; Kohlendioxid; Wasserstoff; und Stickoxid; ein Inertgas, wie Helium, Argon, Xenon oder Krypton; ein Schwefelfluorid, wie Schwefelhexafluorid, Dischwefeldekafluorid, oder Trifluormethylschwefelpentafluorid; Selenhexafluorid; ein wahlweise halogeniertes Silan, wie Tetramethylsilan; einen niedermolekulargewichtigen Kohlenwasserstoff (z. B. enthaltend bis zu 7 Kohlenstoffatome), beispielsweise ein Alkan, wie Methan, Ethan, ein Propan, ein Butan oder ein Pentan, ein Cycloalkan, wie Cyclobutan oder Cyclopentan, ein Alken, wie Propen oder ein Beten, oder ein Alkin, wie Acetylen; einen Ether; ein Keton; einen Ester; einen halogenierten niedermolekulargewichtigen Kohlenwasserstoff (z. B. enthaltend bis zu 7 Kohlenstoffatome); oder eine Mischung irgendwelcher der vorstehend genannten Materialien. Mindestens einige der Halogenatome in den halogenierten Gasen sind vorteilhafterweise Fluoratome. Daher können beispielsweise biokompatible halogenierte Kohlenwasserstoffgase ausgewählt sein aus Bromchlordifluormethan, Chlordifluormethan, Dichlordifluormethan, Bromtrifluormethan, Chlortrifluormethan, Chlorpentafluorethan, Dichlortetrafluorethan und Perfluorkohlenstoffen, z. B. Perfluoralkanen, wie Perfluormethan, Perfluorethan, Perfluorpropanen, Perfluorbutanen (z. B. Perfluor-n-butan, wahlweise in Mischung mit anderen Isomeren, wie Perfluorisobutan), Perfluorpentanen, Perfluorhexanen und Perfluorheptanen; Perfluoralkenen, wie Perfluorpropen, Perfluorbutenen (z. B. Perfluorbut-2-en) und Perfluorbutadien; Perfluoralkinen, wie Perfluorbut-2-in; und Perfluorcycloalkanen, wie Perfluorcyclobutan, Perflu ormethylcyclobutan, Perfluordimethylcyclobutanen, Perfluortrimethylcyclobutanen, Perfluorcyclopentan, Perfluormethylcyclopentan, Perfluordimethylcyclopentanen, Perfluorcyclohexan, Perfluormethylcyclohexan und Perfluorcycloheptan. Andere halogenierte Gase umschliessen fluorierte, z. B. perfluorierte, Ketone, wie Perfluoraceton, und fluorierte, z. B. perfluorierte, Ether, wie Perfluordiethylether.
- Besonders bevorzugt enthalten die Vesikel ein Perfluoralkan, insbesondere ein Perfluorbutan, Perfluorpentan oder Perfluorhexan, insbesondere n-Perfluorbutan.
- In den Vesikeln kann die Membran aus jeglichen physiologisch tolerierbaren membranbildenden Materialien gebildet sein, insbesondere Phospholipiden, und kann entweder vernetzt oder nicht vernetzt sein. Die aus Mischungen von geladenen und nicht geladenen Phospholipiden gebildeten Membranen sind insbesondere bevorzugt, und es ist besonders bevorzugt, dass die Vesikel eine Nettooberflächenladung tragen sollten, bevorzugt eine negative Ladung. Solche Phospholipid-Vesikel haben besonders günstige Blutverweilzeiten.
- Die Vesikel können weiter mit einem Blutaufenthaltsverlängerungsmittel versehen sein, z. B. durch, Konjugieren solch eines Mittels an die Membran oder an eine lipophile Gruppe, welche sich in der Membran verankert. Solche Blutaufenthaltsverlängerungsmittel, z. B. Polyalkylenoxide, wie Polyethylenglykol, können als Opsonisationsinhibitoren wirken, wobei. die Aufnahme der Vesikel aus dem Gefäß. durch das Retikuloendothelialsystem verzögert wird.
- Wünschenswerterweise bestehen mindestens 75%, bevorzugt im wesentlichen das gesamte Phospholipidmaterial in dem Kontrastmittel gemäß der Erfindung aus Molekülen, welche einzeln eine Nettogesamtladung unter Bedingungen der Herstellung und/oder der Verwendung tragen, wobei die Ladung positiv oder, besonders bevorzugt, negativ sein kann. Repräsentative positiv geladene Phospholipide umschließen Ester von Phosphatidsäuren, wie Dipalmitoylphosphatidsäure oder Distearoylphosphatidsäure, mit Aminoalkoholen, wie Hydroxyethylendiamin. Beispiele für negativ geladene Phospholipide umschließen natürlich auftretende (z. B. abgeleitet von Soya oder Eidotter), semisynthetische (z. B. teilweise oder vollständig hydrierte) und synthetische Phosphatidylserine, Phosphatidylglycerine, Phosphatidylinositole, Phosphatidsäuren und Kardiolipine. Die Fettacylgruppen solcher Phospholipide enthalten typischerweise ungefähr 14–22 Kohlenstoffatome, wie beispielsweise in Palmitoyl- und Stearoyl-Gruppen. Lysoformen sol cher geladenen Phospholipide sind ebenso gemäß der vorliegenden Erfindung nützlich, wobei der Begriff "Lyso" Phospholipide bezeichnet, welche nur eine Fettacylgruppe enthalten, wobei diese bevorzugt an das Kohlenstoffatom der 1-Position der Glyceryleinhet estergebunden ist. Solche Lysoformen geladener Phospholipide können vorteilhafterweise in Mischung mit geladenen Phospholipiden, welche zwei Fettacylgruppen enthalten, verwendet werden.
- Phosphatidylserine repräsentieren besonders bevorzugte Phospholipide zur Verwendung in Kontrastmitteln gemäß der vorliegenden Erfindung und stellen bevorzugt einen wesentlichen Teil, z. B. mindestens 80% des anfänglichen Phospholipidgehalts davon, beispielsweise 85–92%, dar, obwohl dieser in der nachfolgenden Verarbeitung, wie der Wärmesterilisation, nachträglich leicht vermindert werden kann, z. B. auf ca. 70%. Ohne an irgendeine theoretische Betrachtung gebunden sein zu wollen, wird angenommen, dass das ionische Verbrücken zwischen den Carboxyl- und Aminogruppen der benachbarten Serin-Einheiten zu der Stabilität solcher Systeme beiträgt. Bevorzugte Phosphatidylserine umschließen gesättigtes (z. B. hydriertes oder synthetisches) natürliches Phosphatidylserin und synthetische oder semisynthetische Dialkanoylphosphatidylserine, wie Distearoylphosphatidylserin, Dipalmitoylphosphatidylserin und Diarachidoylphosphatidylserin.
- Ein wichtiger Vorteil der Verwendung solcher Phosphatidylserin-basierten Kontrastmittel liegt darin, dass der Körper gealterte rote Blutzellen und Blutplättchen durch hohe Konzentrationen von Phosphatidylserin auf deren Oberfläche erkennt und solche Kontrastmittel aus dem Blutkreislauf in einer ähnlichen Weise wie die Eliminierung von gealterten roten Blutzellen eliminieren kann. Darüberhinaus können sie, da die Oberfläche solcher Kontrastmittel als vom Körper endogen registriert werden kann, die Induktion von nachteilhaften systemischen Nebeneffekten, wie haemodynamischen Effekten und von anderern anaphylaktischen Reaktionen, vermeiden, welche die Verabreichung einiger Liposomzubereitungen begleiten können (siehe zum Beispiel die WO-A-95/12386).
- Liposomale Ultraschallkontrastmittel, welche zur Verwendung gemäß der vorliegenden Erfindung geeignet sind, können hergestellt werden, wie es in der Literatur beschrieben ist, siehe beispielsweise die WO-A-92/22247, WO-A-94/28780, WO-A-93/05819, WO-A-95/16467, die PCT/GB96/01361 und Unger et al. Invest. Radiol. 29 (Suppl. 2): S134–S136 (1994).
- Wie vorstehend angegeben wurde, ist der Stabilisator, welcher gemäß der vorligenden Erfindung verwendet wird, ein physiologisch tolerierbarer Gefriertrocknungsstabilisator (oder eine Mischung) mit einer Glasübergangstemperatur (Tg) oberhalb von 20°C, z. B. in dem Bereich von 25 bs 70°C, und mit. einen Tg'-Wert von –37°C oder darüber. Beispiele geeigneter Stabilisatoren umschließen Saccharose, Maltose H2O, Trihalose, Raffinose, und Stachyose. Ein besonders geeignetes Beispiel ist Saccharose, wahlweise in Mischung mit geringeren Mengen (z. B. bis zu 20 Gew.-%, bevorzugt bis zu 10 (Gew.-%) anderer Stabilisatoren.
- Wiederum, wie oben bereits verzeichnet wurde, liegt der Stabilisator in der Zusammensetzung in einem Gewichtsverhältnis von mindestens 10 : 1, bevorzugt von mindestens 20 : 1, optimalerweise in der Höhe von 200 : 1, z. B. bis zu 5000 : 1 oder sogar darüber, vor. Dementsprechend ist der Beitrag der Glasübergangstemperatur (Tg) des getrockneten Produkts relativ unabhängig von der vesikulären Komponente, und Stabilisatorkandidaten können leicht durch Routinetechniken gescreent werden, wobei bestimmt wird, ob sie in Kombination mit den anderen Bestandteilen, welche in dem wässrigen Trägermedium vorliegen, unter Bildung eines Produkts mit einer Glasübergangstemperatur (Tg) oberhalb von 20°C trocknen.
- Herkömmlich liegt der Stabilisator in 1 bis 50 Gew.-%, bevorzugt in 5 bis 30 Gew.-%, besonders bevorzugt in ungefähr 10 bis 20 Gew.-% in der Zusammensetzung vor, welche einer Gefriertrocknung unterzogen wird. Die Konzentration des Stabilisators kann, wenn dies gewünscht ist, ebenso oberhalb der isotonischen Konzentrationen liegen, da das Produkt unter Wiederherstellung nach dem Gefriertrocknen verdünnt werden kann. Die Vesikelkomponente liegt bevorzugt in 0,01 bis 5 Gew.-%, bevorzugt in 0,1 bis 3 Gew.-%, besonders bevorzugt in ungefähr 0,5 bis 1,5 Gew.-% vor (wobei angenommen wird, dass dessen Gewicht nur das Gewicht des membranbildenden Materials ist). Die Menge an Stabilisator in Bezug auf das Wiederherstellungsfluid, welches zur Umwandlung des gefriergetrockneten Produkts in eine verabreichbare Dispersion verwendet wird, wird in Abhängikeit der Körperregion oder des Organs, welche/welches einem Bildgebungsverfahren unterzogen werden soll, und in Abhängikeit des gewählten Verabreichungsmodus ausgewählt. Zum Beispiel kann sie mindestens zweimal die in der Zusammensetzung darstellen, welche der Gefriertrocknung unterzogen wurde.
- Für Ultraschallanwendungen, wie die Echocardiographie, kann es angenehm sein, um einen freien Durchgang durch das Lungensystem zu erlauben und um eine Resonanz mit den bevorzugten Bildgebungsfrequenzen von ungefähr 0,1 bis 15 MHz zu erreichen, dass Vesikel eingesetzt werden, welche eine mittlere Größe von 0,1 bis 10 μm, z. B. 1–7 μm, aufweisen. Die Vesikel können mit einer sehr schmalen Größenverteilung innerhalb des für die Echocardiographie bevorzugten Bereichs hergestellt werden, wodurch deren Echogenizität sowie deren in-vivo-Sicherheit stark verbessert wird, und wodurch den Kontrastmitteln ein besonderer Vorteil in Anwendungen verliehen wird, wie in Blutdruckmessungen, Blutflußüberwachung und Ultraschalltomographie. Somit können beispielsweise Produkte, in denen über 90% (z. B. mindestens 95%, bevorzugt mindestens 98%) der Vesikel Durchmesser in dem Bereich von 1–7 μm aufweisen und weniger als 5% (z. B. nicht mehr als 3%, bevorzgt nicht mehr als 2%) der Vesikel Durchmesser oberhalb von 7 μm aufweisen, leicht hergestellt werden.
- In Ultraschallanwendungen können die Kontrastmedien beispielsweise in Dosen verabreicht werden, so dass die Menge des membranbildenden Materials (z. B. Phospholipid) in dem Bereich von 0,1–10 μg/kg Körpergewicht, besonders bevorzugt 1–5 μg/kg eingespritzt wird. Es ist verständlich, dass die Verwendung solcher niedrigen Teile an membranbildendem Material von substantiellem Vorteil in der Minimierung möglicher toxischer Nebeneffekte ist.
- Die Gesamtkonzentration des membranbildenden Materials in den zur Verwendung fertigen Zusammensetzungen, welche unter Verwendung des getrockneten Produkts der Erfindung hergestellt sind, liegt wünschenswerterweise in dem Bereich von 0,01 bis 5 Gew.-%, bevorzugt von 0,05 bis 2,0 Gew.-%, und insbesondere bei ungefähr 0,5 Gew.-%.
- Die der Gefriertrocknung unterzogene Zusammensetzung enthält vorteilhafterweise mindestens einen Füllstoff, z. B. ein Polyol (z. B. ein C3-Polyol, wie Glycerin oder Propylenglykol) oder ein Polysaccharid wie Dextran, oder ein Polyglykol, wie Polyethylenglykol, oder Mischungen davon Typischerweise kann das Füllmittel in Konzentrationen verwendet werden, welche ähnlich zu denen oder geringfügig niedriger als die des Stabilisators sind, z. B. 3 bis 10 Gew.-%, bevorzugt ungefähr 5 Gew.-%. Die Füllmittel sollten dazu in der Lage sein, während des Gefriertrocknungsverfahrens zu kristallisieren, da sie nur in diesem Zustand einen neutralen Effekt auf die Produktstabilität ausüben. Sie unterscheiden sich somit von den Stabilisatoren, welche während des Gefriertrocknens in dem amorphen Zustand vorliegen sollten.
- Andere Bestandteile beziehungsweise Hilfsmittel können, wenn dieses erwünscht ist, in der Zusammensetzung, welche getrocknet werden soll, vorliegen oder können bei der Formulierung zur Verabreichung zugesetzt werden. Solche Bestandteile können beispielsweise pH-Regulatoren, Osmolalitätseinsteller, Viskositätssteigerungsmittel, Emulgatoren etc. einschließen und können in herkömlichen Mengen verwendet werden.
- Das getrocknete Produkt liegt im Allgemeinen in pulvriger Form vor und ist in Wasser, einer wässrigen Lösung, wie Kochsalzlösung (welche vorteilhafterweise derartig im Gleichgewicht gehalten wird, dass das fertige Produkt zur Injektion nicht hypotonisch ist) oder einer Lösung einer oder mehrerer Tonus- beziehungsweise Tonizität-einstellender Substanzen, wie Salze (z. B. von Plasmakationen mit physiologisch tolerierbaren Gegenionen) oder Zucker, Zuckeralkohole, Glykole und andere nichtionische Polyolmaterialien (z. B. Glukose, Saccharose, Sorbit, Mannit, Glycerin, Polyethylenglykole, Propylenglykole und ähnliches) leicht wiederherstellbar. Die Wiederherstellung beziehungsweise Rekonstitution erfordert im Allgemeinen nur minimale Bewegung, wie sie beispielsweise durch vorsichtiges Schütteln mit der Hand zur Verfügung gestellt wird. Die Größe der Vesikel, welche auf diese Weise gebildet werden, ist einheitlich reproduzierbar und ist in der Praxis unabhängig von der Menge der angewendeten Bewegungsenergie, welche durch die Größe der in der anfänglichen Vesikeldispersion gebildeten Vesikel bestimmt wird, wobei dieser Größenparameter überaschenderweise im wesentlichen in dem lyophilisierten und wiederhergestellten Produkt aufrecht erhalten wird. Da die Größe der Vesikel in der anfänglichen Dispersion durch die Verfahrensparameter, wie das Verfahren, die Geschwindigkeit und die Dauer der Bewegung, leicht reguliert beziehungsweise kontrolliert werden kann, kann somit die letztendliche Vesikelgröße leicht reguliert beziehungsweise konrolliert werden.
- Das Volumen und die Konzentrationen der Wiederherstellungs- beziehungsweise Rekonstitutionsflüssigkeit kann wünschenswerterweise derartig ausbalanciert sein, dass die resultierenden zur Verwendung fertigen Formulierungen im wesentlichen isotonisch sind. Demnach hängt das Volumen und die Konzentration der gewählten Rekonstitutionsflüssigkeit von dem Typ und der Menge des Stabilisators (und anderer Füllmittel) ab, welche in dem gefriergetrockneten Produkt vorliegen.
- Die lyophilisierten Produkte gemäß der vorliegenden Erfindung haben bewiesen, dass sie innerhalb mehrerer Monate unter Umgebungsbedingungen lagerungsstabil sind. Die durch die Rekonstitution beziehungsweise Wiederherstellung in Wasser (oder anderen Rekonstitutionsflüssigkeiten, wie vorstehend diskutiert) gebildeten Vesikeldispersionen können innerhalb einer beachtlichen Zeitdauer, z. B. bis zu mindestens 12 Stunden, stabil sein, wobei eine beachtliche Flexibilität erlaubt wird, wenn das getrocknete Produkt vor der Injektion wiederhergestellt wird.
- Wenn die Wiederherstellungs- beziehungsweise Rekonstitutionsflüssigkeit als ein Tonuseinstellungsmittel dieselbe Verbindung enthält wie sie als ein Stabilisator in der Lyophilisierung verwendet wird, muss die Menge des in der Zusammensetzung zum Gefriertrocknen anwesenden Stabilisators nur derartig ausreichend sein, das sie die optimale Stabilisierung während des Gefriertrocknens ergibt. Die Isotonizität des fertigen Produkts kann somit durch Auswahl einer adäquaten Menge und Konzentration der Rekonstitutions- beziehungsweise Wiederherstellungsflüssigkeit erhalten werden. Somit wird eine beachtliche Flexibilität in Abhängigkeit der Konzentration und dem Typ der Verbindung(en), welche als Stabilisatoren) während des Gefriertrocknungsschritts verwendet werden, und der Konzentration und dem Typ der Verbindungen) in der Rekonstitutionsflüssigkeit erlaubt, während weiterhin ein stabiles wiederhergestelltes Produkt erreicht wird.
- Das Gefriertrocknen gemäß der vorliegenden Erfindung kann in einer konventionellen Weise bewirkt werden, obwohl die Verwendung von Stabilisatoren gemäß der Erfindung den zusätzlichen Vorteil aufweisen kann, dass kürzere Gefriertrocknungszyklen verwendet werden können, da die Zusammensetzungen vor dem Trocknen im Allgemeinen höhere Glastemperaturen (Tg') als gleichwertige Zusammensetzungen aufweisen, welche Kryoprotektanden, wie Glukose oder Mannit, enthalten.
- Die Erfindung wurde oben in Hinblick auf vesikuläre Ultraschallkontrastmittel beschrieben. Sie ist jedoch ebenso auf vesikuläre Kontrastmittel für andere diagnostische Bildgebungsmodalitäten anwendbar (z. B. MRI, Röntgenstrahlen, SPECT, PET, magnetographische Bildgebung etc.).
- Die Erfindung wird im Folgenden weiter durch Bezugnahme auf die folgenden nichtbegrenzenden Beispiele beschrieben:
- BEISPIEL 1
- Herstellung eines lyophilisierten Produkts
- Es wurden 500,4 mg hydriertes Ei-Phosphatidylserin zu 100 ml Wasser, enthaltend 5,4% (w/w) einer Mischung von Propylenglykol und Glycerin (3 : 10 w/w) gegeben. Die Mischung wurde geschüttelt und auf 80°C fünf Minuten lang erwärmt, auf Raumtemperatur abkühlen gelassen, wiederum geschüttelt und über Nacht vor der Verwendung stehen gelassen.
- Es wurden 50 ml der resultierenden Lösung in einem Rundbodenkolben mit einem konischen Hals transferiert. Der Kolben wurde mit einem Glasmantel mit einem Temperaturkontrolleingang und -ausgang versehen, verbunden mit einem Wasserbad, welches bei 25°C gehalten wurde. Ein Rotor-Stator-Mischschaft wurde in die Lösung eingeführt, und, um ein Austreten von Gas zu vermeiden, der Raum zwischen der Halswand und dem Mischschaft wurde mit einem speziell entworfenen Metallstopfen abgedichtet, ausgestattet mit einer Gaseinlass-/-auslassverbindung für die Einstellung des Gasgehalts und zur Druckkontrolle beziehungsweise -regulierung. Der Gasauslass wurde an eine Vakuumpumpe angeschlossen, und die Lösung wurde eine Minute lang entgast. Eine Atmosphäre von Perfluor-n-butan-Gas wurde anschließend durch den Gaseinlass eingefüllt.
- Die Lösung wurde bei 23000 U/min 10 Minuten lang homogenisiert, wobei der Rotor-Stator-Mischschaft derartig gehalten wurde, dass sich die Öffnungen leicht oberhalb der Oberfläche der Flüssigkeit befanden. Es wurde eine weiß gefärbte chremige Dispersion erhalten, welche in einen abschließbaren beziehungsweise versiegelbaren Behälter transferiert wurde und mit Perfluor-n-butan gespühlt wurde. Die Dispersion wurde anschließend in einen Scheidetrichter transferiert und bei 12000 U/min 30 Minuten lang zentrifugiert, wobei eine chremige Schicht von Bläschen am oberen Rand sowie eine trübe darunterliegende Schicht (infranatant) erhalten wurden. Die Unterschicht wurde entfernt und durch Wasser ersetzt. Das Zentrifugieren wurde anschließend zweimal wiederholt, allerdings jetzt bei 12000 U/min innerahlb von 15 Minuten. Nach dem letzten Zentrifugieren wurde der Überstand durch 10% (w/w) Saccharose ersetzt. Es wurden 2 ml-Portionen der resultierenden Dispersion auf 10 ml-Flachbodenfläschchen, welche speziell für die Lyophilisierung entworfen wurden, aufgeteilt, und die Fläschchen wurden auf –47°C abgekühlt und ungefähr 48 Stunden lang lyophilisiert, wobei eine weiße flockige Feststoffsubstanz erhalten wurde. Die Fläschchen wurden in eine Vakuumkammer gegeben, und die Luft wurde durch eine Vakuumpumpe entfernt und durch Perfluor-n-butan-Gas ersetzt. Vor der Verwendung wurde Wasser zugesetzt, und die Fläschchen wurden mehrere Sekunden lang vorsichtig mit der Hand geschüttelt, wobei Mikrobläschen erhalten wurden, welche als Ultraschallkontrastmittel geeignet waren.
- Charakterisierung
- Die Größenverteilung und Volumenkonzentration der Mikrobläschen wurde unter Verwendung eines Coulter-Counter-Mark-II-Apparates, ausgestattet mit einer 50 μm-Öffnung mit einem Meßbereich beziehungsweise einer -Blende von 1–30 μm, gemessen. Es wurden 20 μl-Proben in 200 ml Kochsalzlösung, gesättigt mit Luft bei Raumtemperatur, verdünnt, und man ließ 3 Minuten lang vor der Messung äquilibrieren.
- Die Ultraschallcharakterisierung wurde mit einem experimentellen Aufbau, welcher von dem von de Jong, N. und Hoff, "Ultrasonics", 31: 175–181 (1993) leicht modifiziert wurde, durchgeführt. Dieses Instrument misst die Ultraschalldämpfungswirksamkeit (ultrasound attenuation efficacy) in dem Frequenzbereich von 2–8 MHz einer verdünnten Suspension von Kontrastmittel. Während der Dämpfungsmessung wurde ein Druckstabilitätststest durch Aussetzten der Probe einem Überdruck von 120 mmHg für 90 Sekunden durchgeführt. Typischerweise wurden 2–3 μl der Probe in 55 ml Isoton II verdünnt, und die verdünnte Probensuspension wurde 3 Minuten lang vor der Analyse gerührt. Als primärer Antwortparameter wurde die Dämpfung bei 3,5 MHz verwendet, zusammen mit dem Verzögerungsdämpfungswert (recovery attenuation value) bei 3,5 MHz nach dem Entlassen des Überdrucks.
- Tabelle 1
- In vitro-Eigenschaften der Bläschchen-Dispersionen, hergestellt nach Beispiel 1. Anzahl- und Volumen-gewichtete Konzentrationen und mittlere Volumendurchmesser. Akustische Eigenschaften, gemessen gemäß der obigen Beschreibung.
-

- BEISPIEL 2
- Der Gasgehalt der fünf Proben, welche gemäß Beispiel 1 oben hergestellt wurden, wurde jeweils durch Luft, Perfluorbutan, Schwefelhexafluorid, Trifluormethylschfelpentafluorid und Tetramethylsilan gemäß der folgenden Prozedur ersetzt:
- Zwei Proben, enthaltend lyophilisiertes Produkt aus Beispiel 1, wurden in einen Exsikkator mit einem Gaseinlass und einem Gasauslass gegeben. Der Exsikkator wurde an einen Büchi 168 Vakuum/Destillations-Regulator angeschlossen, welcher die kontrollierte Evakuierung der Proben und den Einlass eines ausgewählten Gases erlaubt. Die Proben wurden fünf Minuten lang bei ungefähr 10 mbar evakuiert, woraufhin der Druck auf Atmosphärendruck durch Einlass des gewählten Gases erhöht wurde, gefolgt vom vorsichtigen Abdecken der Fläschchen. Das Verfahren wurde unter Verwendung weiterer Paare von Proben für jedes der gewählten Gase wiederholt.
- Es wurde 2 ml destilliertes Wasser zu einem. jeden Fläschchen zugesetzt, und die Fläschchen wurden vor der Verwendung vorsichtig mit der Hand geschüttelt. Die resultierenden Mikrobläschen-Dispersionen wurden im Hinblick auf die Größenverteilungsmessungen, wie im Beispiel 1 beschrieben, charakterisiert. Die Ergebnisse sind in Tabelle 2 zusammengefasst.
- Tabelle 2
- In vitro-Eigenschaften von Phosphatidylserin-stabilisierten Mikrobläschen-Dispersionen, hergestellt gemäß Beispiel 2 – Anzahl-, und Volumen-gewichtete Konzentrationen und mittlere Volumendurchmesser.
-
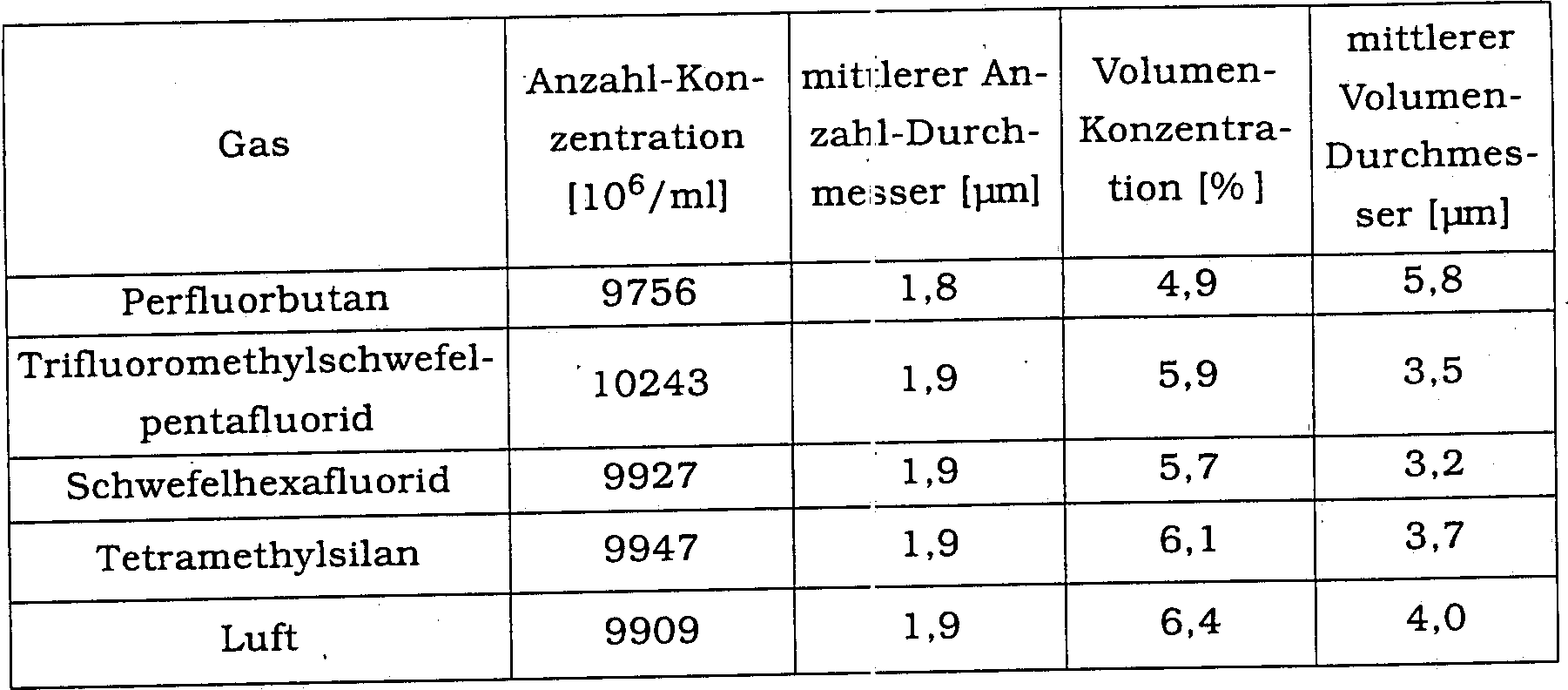
- Wie aus den obigen Ergebnissen ersichtlich ist, kommt es zu keiner deutlichen Änderung in der Größenverteilung durch den Gasaustausch, was zeigt, dass die vorgebildete Mikrobläschengröße im wesentlichen erhalten bleibt während sowohl der Lyophilisierung als auch der Rekonstitution.
- In vivo-Ergebnisse
- Ein Ansatz, welcher mit jedem der fünf Gase hergestellt wurde, wurde in vivo hinsichtlich der Doppler-Verstärkungs-Eigenschaften bei 10 MHz evaluiert. Die Dispersionen wurden in Chinchillahäschen über eine Ohrvene injiziert und unter Verwendung einer Doppler-Technik gemessen, wobei ein Ultraschallfühler direkt an die Halsschlagader angelegt wurde. Die Signalintensitäten und -Dauer wurden aufgezeichnet, und das Integral der Doppler-Kurve wurde berechnet. Die erhaltenen Ergebnisse (siehe Tabelle 3 unten) zeigen, dass Mikrobläschen, welche Perfluorbutan enthalten, die stärkste Doppler-Intensitäts-Verstärkung ergeben. Die Mikrobläschen, welche Schwefelhexafluorid, Trifluormethylschwefelpentafluorid oder Tetramethylsilan enthalten, sind nur geringfügig weniger wirksam als Doppler-Verstärker als jene, welche Perfluorbutan enthalten, wobei Integrale in dem Bereich von 60–80% der Abbildung für Perfluorbutan erhalten werden.
- Tabelle 3
- Die Ergebnisse für die I.V.-Injektionen von Beispiel 2-Produkten in Hasen. Die Werte werden für die Drift in der Basislinie eingestellt. Die Doppler-Einheit wird als die Erhöhung in dem Doppler-Signal relativ zu dem von Blut definiert.
-

- BEISPIEL 3
- Ein Fläschchen, enthaltend lyophilisiertes Material unter einer Atmosphäre von Perfluorbutan, wurde wie in Beispiel 1 beschrieben hergestellt. Es wurde direkt vor der Verwendung Wasser zugesetzt, so dass eine Mikrobläschensuspension erhalten wurde.
- Es wurden 200 ml Isoton II-Fluid mehrere Tage lang bei Raumtemperatur der Luft ausgesetzt, so dass eine vollständig Luft-gesättigte Lösung erhalten wurde. Es wurden weitere 200 ml des Fluids entgast in einem Vakuumkolben bei 60°C innerhalb von einer Stunde und auf Raumtemperatur unter Erhalt des Vakuums abgekühlt. Luft wurde direkt vor der Verwendung in den Kolben eingelassen.
- Es wurden 10 μl-Portionen der Mikrobläschen-Suspension zu jedem der Fluide zugegeben, und die resultierenden Mischungen wurden fünf Minuten lang vor der Größe-Charakterisierung inkubiert (Coulter Multisizer Mark II).
- In der entgasten Umgebung, wo keine Diffusion von Gasen aus dem Fluid in die Mikrobläschen erwartet wurde, betrug der mittlere Mikrobläschen-Durchmesser 1,77 μm, und 0,25% der Mikrobläschen waren größer als 5 μm. In dem Luftgesättigten Fluid betrugen die entsprechenden Werte 2,43 μm und 0,67%; wiederholte Messungen, welche nach weiteren 5 Minuten durchgeführt wurden, zeigten an, dass die Mikrobläschengrößen einen stabilen Wert erreicht hatten.
- Diese Ergebnisse zeigen, dass der mittlere Durchmesser der Mikrobläschen um nur 37% stieg, wenn sie einem Luft-gesättigten Fluid analog zu arteriellem Blut ausgesetzt wurden, wobei nur sehr wenige Mikrobläschen eine Größe erreichten, welche eine Blockade der kapillaren Blutgefäße hervorrufen könnte. Dieses könnte zu der Verdopplung in der Größe von Luft-/Perfluorhexan-enthaltenden Mikrobläschen in einer ähnlichen Umgebung einem Kontrast bilden (d. h. eine hochverdünnte Dispersion von Mikrobläschen in Wasser, enthaltend gelöste Luft), wie dies in Beispiel II der WO-A-95/03835 berichtet wird.
- BEISPIEL 4
- VERGLEICH
- Beispiel 1 wurde wiederholt, wobei der Überstand vor der Lyophilisierung an Stelle mit (a) 65 mg/mL mit Saccharose plus 65 mg/mL Mannit, (b) 100 mg/mL Mannit plus 50 mg/mL Glucose, (c) 20 mg/mL Saccharose, 76 mg/mL Mannit und 38 mg/mL Glucose sowie (d) 90 mg/mL Saccharose ersetzt wurde.
- Die Tg'- und Tg-Werte der nassen und getrockneten Zusammensetzungen wurden bestimmt und sind in Tabelle 4 unten dargestellt.
- Tabelle 4
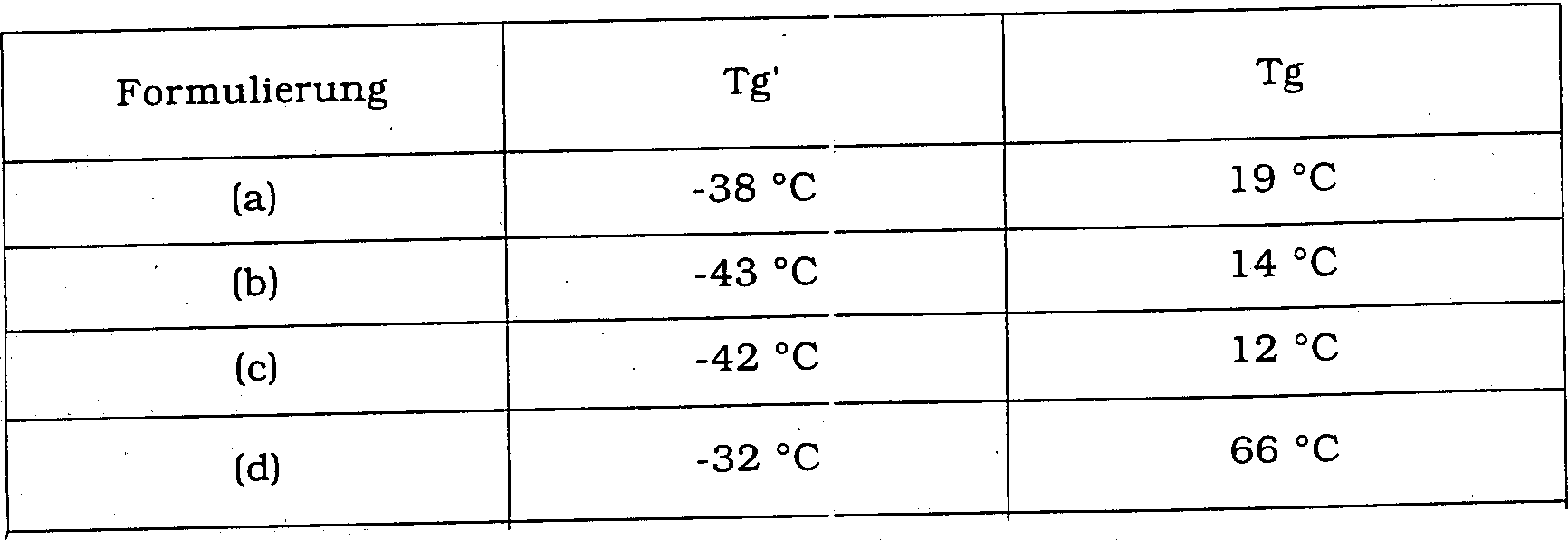
- Formulierungen (a) bis (c) benötigen längere Gefriertrocknungszyklen als die Formulierung (d) und müssen ungleich der Formulierung (d) unterhalb von Umgebungstemperatur zum Erhalt deren Integrität beziehungsweise Beständigkeit gelagert werden.
- BEISPIEL 5
- Gasretention
- Das analog Beispiel 1, allerdings unter Verwendung von (a) 10% (w/w) Saccharose, (b) 5% (w/w) PEG 3000, (c) 2% (w/w) Mannit und 1% (w/w) Glucose, sowie (d) 5% (w/w) Trehalose zum Ersatz des Überstands vor der Lyophilisierung hergestellte Material wurde einem ausgiebigen Spühlen mit N2, einer Aussetzung von wiederholten Vakuumcyklen und einem Zerdrücken zum Testen der Fähigkeit des Produkts, Perfluorbutan zurückzuhalten, unterzogen. Nach der Beanspruchungs-Behandlung wurde der verbleibende Perfluorbutan-Gehalt bestimmt. Die Ergebnisse sind in Tabelle 5 unten dargestellt.
- Tabelle 5

- In den vorstehenden Beispielen können höhere Prozentteile an Stabilisator (z. B. 20% eher als 10%) verwendet werden, und andere Rekonstitutionsfluide als Wasser, z. B. Kochsalzlösung oder Polyollösungen, auf die oben Bezug genommen wurde, können eingesetzt werden. In ähnlicher Weise können die Portionen, wel che lyophilisiert werden, größer sein (z. B. 4 mL eher als 2 mL), die Lyophilisierungs-Fläschen können größer sein (z. B. 20 mL) und die Lyophilisierung kann länger andauern (z. B. 60 Stunden).
Claims (20)
- Gefriergetrocknetes, vesikelhaltiges Ultraschallkontrastmittel, enthaltend einen Gefriertrocknungsstabilisator oder Mischung von Stabilisatoren, wobei (i) das Gewichtsverhältnis des Stabilisators oder der Mischung von Stabilisatoren zu den Vesikeln in dem Mittel mindestens 10 : 1 beträgt, (ii) der Stabilisator oder die Mischung von Stabilisatoren eine Tg-Wert oberhalb 20°C und einen Tg'-Wert von –37°C oder darüber aufweist, und (iii) das Mittel bei Temperaturen oberhalb 20°C thermisch stabil ist.
- Kontrastmittel nach Anspruch 1, wobei der Stabilisator oder die Mischung von Stabilisatoren aus einem oder mehreren aus Saccharose, Maltose, H2O, Trehalose, Raffinose und Stachyose gewählt ist.
- Kontrastmittel nach Anspruch 1 oder Anspruch 2, wobei der Stabilisator oder die Mischung von Stabilisatoren Saccharose umfasst.
- Kontrastmittel nach mindestens einem der vorangehenden Ansprüche, wobei das Gewichtsverhältnis des Stabilisators oder der Mischung, von Stabilisatoren zu den Vesikeln in dem Mittel mindestens 20 : 1 beträgt.
- Kontrastmittel nach mindestens einem der vorangehenden Ansprüche, wobei die Vesikel ein Halogenkohlenstoffgas oder Gasvorläufer oder ein Schwefelfluoridgas enthalten.
- Kontrastmittel nach Anspruch 5, wobei das Halogenkohlenstoffgas oder Gasvorläufer ein Perflouralkan ist.
- Kontrastmittel nach Anspruch 6, wobei das Perfluoralkan Perfluorbutan oder Perfluorpentan ist.
- Kontrastmittel nach Anspruch 6, wobei das Schwefelfluoridgas Schwefelhexafluorid, Dischwefeldecafluorid oder Trifluormethylschwefelpentafluorid ist.
- Kontrastmittel nach mindestens einem der vorangehenden Ansprüche, wobei die Vesikel Membranen besitzen, welche ein Phosphorlipid umfassen.
- Verfahren zur Herstellung eines gefriergetrockneten, vesikelhaltigen Ultraschallkontrastmittels, das bei Temperaturen oberhalb –20°C thermisch stabil ist, wobei das Verfahren das Gefriertrocknen einer wässrigen Dispersion, umfassend ein vesikuläres Ultraschallkontrastmittel und einen Gefriertrocknungsstabilisator oder eine Mischung von Stabilisatoren, umfasst, dadurch gekennzeichnet, dass der Stabilisator oder die Mischung von Stabilisatoren in der Dispersion in einem Gewichtsverhältnis bezüglich der Vesikel darin von mindestens 10 : 1 vorliegt, und dass den Stabilisator oder die Mischung von Stabilisatoren einen Tg-Wert oberhalb 20°C und einen Tg'-Wert von –37°C oder darüber besitzt.
- Verfahren nach Anspruch 10, wobei die wässrige Dispersion 1 bis 50 Gew.-% des Stabilisators oder der Mischung von Stabilisatoren enthält.
- Verfahren nach Anspruch 10 oder Anspruch 11, wobei das vesikuläre Kontrastmittel ein Halogenkohlenstoffgas oder Gasvorläufer oder ein Schwefelfluoridgas enthält.
- Verfahren nach Anspruch 12, wobei das Halogenkohlenstoffgas oder der Gasvorläufer ein Perfluorbutan oder Perfluorpentan ist.
- Verfahren nach Anspruch 12, wobei das Schwefelfluoridgas Schwefelhexafluorid, Dischwefeldecafluorid oder Trifluormethylschwefelpentafluorid ist.
- Verfahren nach mindestens einem Ansprüche 10 bis 14, wobei die wässrige Dispersion weiterhin ein Füllmittel enthält.
- Verfahren nach Anspruch 15, wobei das Füllmittel ein C3-Polyol ist.
- Verfahren nach Anspruch 15 oder Anspruch 16, wobei die wässrige Dispersion 3 bis 10 Gew.-% des Füllmittels enthält.
- Verfahren zum Lagern oder Transportieren eines gefriergetrockneten, vesikelhaltigen Ultraschallkontrastmittels wie in mindestens einem der Ansprüche 1 bis 9 definiert, wobei die Lagerung oder der Transport ohne Anwendung einer Kühlung stattfindet.
- Verfahren zur Herstellung eines injizierbaren, vesikelhaltigen Ultraschallkontrastmittels, umfassend das Dispergieren eines gefriergetrockneten, vesikel haltigen Ultraschallkontrastmittels wie in mindestens einem der Ansprüche 1 bis 9 definiert, in einem physiologisch annehmbaren, injizierbaren wässrigen Trägermedium.
- Verwendung eines Gefriertrocknungsstabilisators oder einer Mischung von Stabilisatoren mit einem Tg-Wert oberhalb 20°C und einem Tg'-Wert von –37°C oder darüber bei der Herstellung eines vesikelhaltigen Ultraschallkontrastmittels, das bei Temperaturen oberhalb 20°C thermisch stabil ist, und das den Stabilisator oder die Mischung von Stabilisatoren in einem Gewichtsverhältnis bezüglich den Vesikeln darin von mindestens 10 : 1 enthält.
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB9603466 | 1996-02-19 | ||
| GBGB9603466.5A GB9603466D0 (en) | 1996-02-19 | 1996-02-19 | Improvements in or relating to contrast agents |
| GBGB9611894.8A GB9611894D0 (en) | 1996-06-07 | 1996-06-07 | Improvements in or relating to contrast agents |
| GB9611894 | 1996-06-07 | ||
| GBGB9624919.8A GB9624919D0 (en) | 1996-11-29 | 1996-11-29 | Product |
| GB9624919 | 1996-11-29 | ||
| PCT/GB1997/000458 WO1997029782A1 (en) | 1996-02-19 | 1997-02-19 | Thermally stabilized contrast agent |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69720979D1 DE69720979D1 (de) | 2003-05-22 |
| DE69720979T2 true DE69720979T2 (de) | 2004-02-26 |
Family
ID=27268136
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69720979T Expired - Lifetime DE69720979T2 (de) | 1996-02-19 | 1997-02-19 | Thermostabilisiertes kontrastmittel |
Country Status (18)
| Country | Link |
|---|---|
| EP (1) | EP0885016B1 (de) |
| JP (1) | JP4250747B2 (de) |
| KR (1) | KR100501863B1 (de) |
| CN (1) | CN1092988C (de) |
| AT (1) | ATE237362T1 (de) |
| AU (1) | AU722735B2 (de) |
| CA (1) | CA2246778A1 (de) |
| CZ (1) | CZ262698A3 (de) |
| DE (1) | DE69720979T2 (de) |
| EA (1) | EA000900B1 (de) |
| EE (1) | EE9800248A (de) |
| ES (1) | ES2197331T3 (de) |
| HU (1) | HUP9900812A3 (de) |
| IL (1) | IL125800A0 (de) |
| NO (1) | NO313815B1 (de) |
| NZ (1) | NZ331372A (de) |
| PL (1) | PL328309A1 (de) |
| WO (1) | WO1997029782A1 (de) |
Families Citing this family (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0918546B1 (de) | 1996-08-02 | 2008-10-15 | GE Healthcare AS | Verbesserungen für oder in bezug auf kontrastmittel |
| GB9717542D0 (en) | 1997-08-19 | 1997-10-22 | Nycomed Imaging As | Process |
| BR0112417A (pt) * | 2000-07-13 | 2003-07-01 | Daiichi Seiyaku Co | Composições farmacêuticas contendo composto dds |
| US6793626B2 (en) | 2001-01-17 | 2004-09-21 | Fuji Photo Film Co., Ltd. | Ultrasonic scatterer, ultrasonic imaging method and ultrasonic imaging apparatus |
| DK1228770T3 (da) * | 2001-01-31 | 2005-11-07 | Bracco Int Bv | Törret kontrastmiddel |
| WO2004049950A1 (en) | 2002-11-29 | 2004-06-17 | Amersham Health As | Ultrasound triggering method |
| WO2004069284A2 (en) | 2003-02-04 | 2004-08-19 | Bracco International B.V. | Ultrasound contrast agents and process for the preparation thereof |
| JP5513708B2 (ja) | 2003-12-22 | 2014-06-04 | ブラッコ・シュイス・ソシエテ・アノニム | 造影イメージング用の気体封入マイクロベシクル・アセンブリー |
| CN101005858A (zh) | 2004-08-18 | 2007-07-25 | 伯拉考开发股份有限公司 | 用于反差成像的充气微泡组合物 |
| WO2006076826A1 (fr) * | 2005-01-18 | 2006-07-27 | Institute Of Pharmacology Toxicology, Academy Of Military Medical Sciences | Composition de contraste pour ultrasons utilisant un phospholipide filmogene et procede de preparation de celle-ci |
| US9446156B2 (en) | 2006-09-05 | 2016-09-20 | Bracco Suisse S.A. | Gas-filled microvesicles with polymer-modified lipids |
| WO2008075192A2 (en) | 2006-12-19 | 2008-06-26 | Bracco International Bv | Targeting and therapeutic compounds and gas-filled microvesicles comprising said com ounds |
| WO2009033687A1 (en) | 2007-09-11 | 2009-03-19 | Mondobiotech Laboratories Ag | Spantide ii and bfgf (119-126) for therapeutic applications |
| JP2010539027A (ja) | 2007-09-11 | 2010-12-16 | モンドバイオテック ラボラトリーズ アクチエンゲゼルシャフト | 治療剤としてのペプチドの使用 |
| RU2010114039A (ru) | 2007-09-11 | 2011-10-20 | Мондобайотек Лабораториз Аг (Li) | Применение октреотида в качестве терапевтического средства |
| EP2205269A1 (de) | 2007-09-11 | 2010-07-14 | Mondobiotech Laboratories AG | Therapeutische anwendungen von intermedin 47 und 53 peptiden |
| RU2010113992A (ru) | 2007-09-11 | 2011-10-20 | Мондобайотек Лабораториз Аг (Li) | Применение урокортина и кортиколиберина в качестве терапевтических средств |
| ATE522225T1 (de) | 2007-09-11 | 2011-09-15 | Mondobiotech Lab Ag | Verwendung des peptids phpfhlfvy (renin- inhibitor) in anti-angiogenischer therapie von gewissen erkrankungen |
| WO2009033754A2 (en) | 2007-09-11 | 2009-03-19 | Mondobiotech Laboratories Ag | Use of a peptide as a therapeutic agent |
| WO2009039991A2 (en) | 2007-09-11 | 2009-04-02 | Mondobiotech Laboratories Ag | Use of aviptadil as a therapeutic agent |
| WO2009033762A2 (en) | 2007-09-11 | 2009-03-19 | Mondobiotech Laboratories Ag | Use of endothelin-3 as a therapeutic agent |
| KR20100056509A (ko) | 2007-09-11 | 2010-05-27 | 몬도바이오테크 래보래토리즈 아게 | 치료제로서의 펩티드의 용도 |
| RU2010114040A (ru) | 2007-09-11 | 2011-10-20 | Мондобайотек Лабораториз Аг (Li) | Применение дезлорелина и мастопана в качестве терапевтического средства |
| JP5385277B2 (ja) | 2007-09-11 | 2014-01-08 | モンドバイオテック ラボラトリーズ アクチエンゲゼルシャフト | 治療剤としての、プロアドレノメデュリンの単独またはビッグガストリンiとの組合せの使用 |
| EP2090322A1 (de) | 2008-02-18 | 2009-08-19 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Verwendung von FSH-Rezeptorliganden zur Diagnose und Behandlung von Krebs |
| WO2010040772A2 (en) | 2008-10-07 | 2010-04-15 | Bracco Research Sa | Targeting construct comprising anti-polymer antibody and liposomes or microvesicles binding to the same |
| CN101649293B (zh) * | 2009-09-02 | 2012-07-18 | 中国农业大学 | 一种微生物冷冻干燥保护剂及其应用 |
| EP2603242B1 (de) | 2010-08-09 | 2018-03-14 | Bracco Suisse SA | Gasgefüllte gerichtete mikrovesikel |
| EP2426202A1 (de) | 2010-09-03 | 2012-03-07 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Kinasen als Targets für antidiabetische Therapien |
| JP5992920B2 (ja) | 2010-12-24 | 2016-09-14 | ブラッコ・スイス・ソシエテ・アノニム | ワクチンとしての使用のためのガス入りの微小胞 |
| US10028912B2 (en) | 2011-10-21 | 2018-07-24 | Celator Pharmaceuticals, Inc. | Method of lyophilizing liposomes |
| EP2589383A1 (de) | 2011-11-06 | 2013-05-08 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. Berlin | FKBP-subtypspezifisches Rapamycin-Analogon zur Verwendung bei der Behandlung von Erkrankungen |
| GB201405735D0 (en) * | 2014-03-31 | 2014-05-14 | Ge Healthcare As | Ultrasound precursor preparation method |
| GB201411423D0 (en) | 2014-06-26 | 2014-08-13 | Ge Healthcare As | Lipid sterilisation method |
| EP3020813A1 (de) | 2014-11-16 | 2016-05-18 | Neurovision Pharma GmbH | Antisense-Oligonukleotide als Inhibitoren der TGF-R-Signalgebung |
| CN107206065A (zh) | 2014-12-22 | 2017-09-26 | 博莱科瑞士股份有限公司 | 用作疫苗的气体填充的微囊 |
| IL277165B1 (en) | 2018-03-07 | 2024-03-01 | Bracco Suisse Sa | Preparation of microbubbles with controlled size |
| JP2022538770A (ja) | 2019-06-25 | 2022-09-06 | ブラッコ・スイス・ソシエテ・アノニム | 校正されたガス充填微小胞を調製するための凍結乾燥組成物 |
| JP2022538769A (ja) * | 2019-06-25 | 2022-09-06 | ブラッコ・スイス・ソシエテ・アノニム | 校正されたガス充填微小胞を調製するための凍結乾燥組成物 |
| US10953023B1 (en) | 2020-01-28 | 2021-03-23 | Applaud Medical, Inc. | Phospholipid compounds and formulations |
| CN112891565A (zh) * | 2021-04-09 | 2021-06-04 | 上海师范大学 | 锌-二甲基吡啶胺在制备诊断血栓性疾病药物中的应用 |
| EP4105328A1 (de) | 2021-06-15 | 2022-12-21 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Antisense-oligonukleotide zur prävention von nierenfunktionsstörungen, die durch endotheliale dysfunktion gefördert werden, durch unterdrückung von ephrin-b2 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4857319A (en) * | 1985-01-11 | 1989-08-15 | The Regents Of The University Of California | Method for preserving liposomes |
| US4642903A (en) * | 1985-03-26 | 1987-02-17 | R. P. Scherer Corporation | Freeze-dried foam dosage form |
| JPH06211645A (ja) * | 1993-01-14 | 1994-08-02 | Mitsubishi Kasei Corp | リポソーム凍結乾燥製剤 |
-
1997
- 1997-02-19 EE EE9800248A patent/EE9800248A/xx unknown
- 1997-02-19 IL IL12580097A patent/IL125800A0/xx not_active IP Right Cessation
- 1997-02-19 AU AU18051/97A patent/AU722735B2/en not_active Ceased
- 1997-02-19 PL PL97328309A patent/PL328309A1/xx unknown
- 1997-02-19 CZ CZ982626A patent/CZ262698A3/cs unknown
- 1997-02-19 HU HU9900812A patent/HUP9900812A3/hu unknown
- 1997-02-19 ES ES97903507T patent/ES2197331T3/es not_active Expired - Lifetime
- 1997-02-19 JP JP52913097A patent/JP4250747B2/ja not_active Expired - Lifetime
- 1997-02-19 EP EP97903507A patent/EP0885016B1/de not_active Expired - Lifetime
- 1997-02-19 CA CA002246778A patent/CA2246778A1/en not_active Abandoned
- 1997-02-19 EA EA199800739A patent/EA000900B1/ru not_active IP Right Cessation
- 1997-02-19 DE DE69720979T patent/DE69720979T2/de not_active Expired - Lifetime
- 1997-02-19 CN CN97193181A patent/CN1092988C/zh not_active Expired - Fee Related
- 1997-02-19 WO PCT/GB1997/000458 patent/WO1997029782A1/en not_active Application Discontinuation
- 1997-02-19 AT AT97903507T patent/ATE237362T1/de not_active IP Right Cessation
- 1997-02-19 NZ NZ331372A patent/NZ331372A/xx unknown
- 1997-02-19 KR KR10-1998-0706416A patent/KR100501863B1/ko active IP Right Grant
-
1998
- 1998-08-05 NO NO19983584A patent/NO313815B1/no not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| EP0885016A1 (de) | 1998-12-23 |
| JP4250747B2 (ja) | 2009-04-08 |
| HUP9900812A2 (hu) | 1999-07-28 |
| KR100501863B1 (ko) | 2005-09-27 |
| EA000900B1 (ru) | 2000-06-26 |
| NO313815B1 (no) | 2002-12-09 |
| AU1805197A (en) | 1997-09-02 |
| PL328309A1 (en) | 1999-01-18 |
| EP0885016B1 (de) | 2003-04-16 |
| CA2246778A1 (en) | 1997-08-21 |
| EE9800248A (et) | 1999-02-15 |
| ATE237362T1 (de) | 2003-05-15 |
| HUP9900812A3 (en) | 1999-11-29 |
| WO1997029782A1 (en) | 1997-08-21 |
| DE69720979D1 (de) | 2003-05-22 |
| CN1092988C (zh) | 2002-10-23 |
| EA199800739A1 (ru) | 1999-02-25 |
| NZ331372A (en) | 2000-01-28 |
| AU722735B2 (en) | 2000-08-10 |
| NO983584L (no) | 1998-10-16 |
| ES2197331T3 (es) | 2004-01-01 |
| CN1213971A (zh) | 1999-04-14 |
| JP2000506122A (ja) | 2000-05-23 |
| IL125800A0 (en) | 1999-04-11 |
| KR19990082670A (ko) | 1999-11-25 |
| NO983584D0 (no) | 1998-08-05 |
| CZ262698A3 (cs) | 1999-03-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69720979T2 (de) | Thermostabilisiertes kontrastmittel | |
| DE69721235T2 (de) | Verbesserungen an (oder im bezug auf) kontrastmittel | |
| DE69632907T2 (de) | Neue zusammensetzungen von lipiden und stabilisierenden materialen | |
| DE69738223T3 (de) | Verbesserte verfahren zur diagnostischen bilderzeugung unter verwendung eines kontrastmittels und eines koronaren vasodilatators | |
| DE69111719T3 (de) | Stabile mikroblasensuspensionen zur injektion in lebewesen. | |
| DE69636486T2 (de) | Gasemulsionen, die durch fluorierte Ether mit niedrigen Ostwaldkoeffizienten stabilisiert sind | |
| DE69231950T3 (de) | Gasgefüllte liposome als ultraschall-kontrastmittel | |
| DE69432358T2 (de) | Gashaltige mikrosphären zur topischen und subkutanen anwendung | |
| DE69737915T2 (de) | Verfahren zur diagnostischen Bilderzeugung der Nierenregion unter Verwendung eines Kontrastmittels und eines Vasodilators | |
| EP0918546B1 (de) | Verbesserungen für oder in bezug auf kontrastmittel | |
| DE69819309T2 (de) | Verabreichbare formulierugen und ihre anwendung in mri | |
| US20060257321A1 (en) | Ultrasound contrast agents and methods of making and using them | |
| US6165442A (en) | Thermally stabilized ultrasound contrast agent | |
| DE60111917T2 (de) | Lyophilisierbares Kontrastmittel, gasgefüllte Mikrobläschen enthaltend | |
| US6217850B1 (en) | Method of making lyophilized microbubble compositions useful as contrast agents | |
| US6613306B1 (en) | Ultrasound contrast agents and methods of making and using them | |
| DE60006683T2 (de) | Methode um ein gasenthaltendes konstrastmittel mit einer spülflüssigkeit zu mischen vor der verabrechnung in einer kontinuierlichen infustion | |
| US20010008626A1 (en) | Ultrasound contrast agents and methods of making and using them | |
| US20030185759A1 (en) | Ultrasound contrast agents and methods of making and using them | |
| US20030194376A1 (en) | Ultrasound contrast agents and methods of making and using them | |
| US20010012507A1 (en) | Ultrasound contrast agents and methods of making and using them |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8332 | No legal effect for de | ||
| 8370 | Indication related to discontinuation of the patent is to be deleted | ||
| 8364 | No opposition during term of opposition | ||
| 8327 | Change in the person/name/address of the patent owner |
Owner name: GE HEALTHCARE AS, OSLO, NO |
|
| 8328 | Change in the person/name/address of the agent |
Representative=s name: HAMMONDS LLP, LONDON, GB |
|
| R082 | Change of representative |
Ref document number: 885016 Country of ref document: EP Representative=s name: J D REYNOLDS & CO., GB |