DE60214713T2 - Arylborsäuren für die behandlung von fettsucht - Google Patents
Arylborsäuren für die behandlung von fettsucht Download PDFInfo
- Publication number
- DE60214713T2 DE60214713T2 DE60214713T DE60214713T DE60214713T2 DE 60214713 T2 DE60214713 T2 DE 60214713T2 DE 60214713 T DE60214713 T DE 60214713T DE 60214713 T DE60214713 T DE 60214713T DE 60214713 T2 DE60214713 T2 DE 60214713T2
- Authority
- DE
- Germany
- Prior art keywords
- sub
- group
- substituted
- amine
- ammonium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 239000002253 acid Substances 0.000 title 1
- 150000007513 acids Chemical class 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 59
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims abstract description 40
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 34
- -1 phenyl boronic acid compound Chemical class 0.000 claims abstract description 32
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 31
- 125000001183 hydrocarbyl group Chemical group 0.000 claims abstract description 24
- 150000001412 amines Chemical class 0.000 claims abstract description 22
- 208000008589 Obesity Diseases 0.000 claims abstract description 17
- 235000020824 obesity Nutrition 0.000 claims abstract description 17
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 claims abstract description 16
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 14
- 125000003118 aryl group Chemical group 0.000 claims abstract description 14
- 125000006575 electron-withdrawing group Chemical group 0.000 claims abstract description 12
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 12
- 125000001931 aliphatic group Chemical group 0.000 claims abstract description 10
- 229910052736 halogen Inorganic materials 0.000 claims abstract description 9
- 150000002367 halogens Chemical class 0.000 claims abstract description 9
- 150000003568 thioethers Chemical class 0.000 claims abstract description 8
- 125000005647 linker group Chemical group 0.000 claims abstract description 7
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 claims abstract description 7
- 125000000547 substituted alkyl group Chemical group 0.000 claims abstract description 7
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 5
- 239000003085 diluting agent Substances 0.000 claims abstract description 4
- 125000005496 phosphonium group Chemical group 0.000 claims abstract description 4
- 125000003107 substituted aryl group Chemical group 0.000 claims abstract description 4
- 150000003573 thiols Chemical class 0.000 claims abstract description 4
- 239000003814 drug Substances 0.000 claims description 18
- 210000001035 gastrointestinal tract Anatomy 0.000 claims description 15
- 229940079593 drug Drugs 0.000 claims description 14
- 150000003839 salts Chemical class 0.000 claims description 9
- 125000004432 carbon atom Chemical group C* 0.000 claims description 8
- 230000005764 inhibitory process Effects 0.000 claims description 8
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical group CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 claims description 8
- 230000002401 inhibitory effect Effects 0.000 claims description 7
- 150000002632 lipids Chemical class 0.000 claims description 6
- 125000005208 trialkylammonium group Chemical group 0.000 claims description 5
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 claims description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims description 3
- 238000004519 manufacturing process Methods 0.000 claims description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 claims description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 claims description 2
- 208000024172 Cardiovascular disease Diseases 0.000 claims description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 claims description 2
- 206010011091 Coronary artery thrombosis Diseases 0.000 claims description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 claims description 2
- 208000002705 Glucose Intolerance Diseases 0.000 claims description 2
- 201000005569 Gout Diseases 0.000 claims description 2
- 208000034991 Hiatal Hernia Diseases 0.000 claims description 2
- 206010020028 Hiatus hernia Diseases 0.000 claims description 2
- 208000031226 Hyperlipidaemia Diseases 0.000 claims description 2
- 206010020772 Hypertension Diseases 0.000 claims description 2
- 102000004877 Insulin Human genes 0.000 claims description 2
- 108090001061 Insulin Proteins 0.000 claims description 2
- 208000000913 Kidney Calculi Diseases 0.000 claims description 2
- 206010028980 Neoplasm Diseases 0.000 claims description 2
- 206010029148 Nephrolithiasis Diseases 0.000 claims description 2
- 208000006011 Stroke Diseases 0.000 claims description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 2
- 229940072107 ascorbate Drugs 0.000 claims description 2
- 235000010323 ascorbic acid Nutrition 0.000 claims description 2
- 239000011668 ascorbic acid Substances 0.000 claims description 2
- 201000001883 cholelithiasis Diseases 0.000 claims description 2
- 208000002528 coronary thrombosis Diseases 0.000 claims description 2
- 230000001419 dependent effect Effects 0.000 claims description 2
- 206010012601 diabetes mellitus Diseases 0.000 claims description 2
- 125000001153 fluoro group Chemical group F* 0.000 claims description 2
- 208000001130 gallstones Diseases 0.000 claims description 2
- 208000021302 gastroesophageal reflux disease Diseases 0.000 claims description 2
- 201000001421 hyperglycemia Diseases 0.000 claims description 2
- 208000006575 hypertriglyceridemia Diseases 0.000 claims description 2
- 230000036512 infertility Effects 0.000 claims description 2
- 208000000509 infertility Diseases 0.000 claims description 2
- 231100000535 infertility Toxicity 0.000 claims description 2
- 229940125396 insulin Drugs 0.000 claims description 2
- 201000008482 osteoarthritis Diseases 0.000 claims description 2
- 201000009104 prediabetes syndrome Diseases 0.000 claims description 2
- 208000002815 pulmonary hypertension Diseases 0.000 claims description 2
- 201000002859 sleep apnea Diseases 0.000 claims description 2
- 208000011580 syndromic disease Diseases 0.000 claims description 2
- 230000004584 weight gain Effects 0.000 claims description 2
- 235000019786 weight gain Nutrition 0.000 claims description 2
- 150000003841 chloride salts Chemical group 0.000 claims 1
- 208000000689 peptic esophagitis Diseases 0.000 claims 1
- 125000002467 phosphate group Chemical class [H]OP(=O)(O[H])O[*] 0.000 claims 1
- HXITXNWTGFUOAU-UHFFFAOYSA-N dihydroxy-phenylborane Natural products OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 abstract description 23
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 abstract description 19
- 238000000034 method Methods 0.000 abstract description 15
- 239000003937 drug carrier Substances 0.000 abstract description 4
- 125000002947 alkylene group Chemical group 0.000 abstract description 3
- 239000000243 solution Substances 0.000 description 61
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 60
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 57
- 239000011541 reaction mixture Substances 0.000 description 50
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 39
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 36
- 230000015572 biosynthetic process Effects 0.000 description 35
- 238000003786 synthesis reaction Methods 0.000 description 34
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 30
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 30
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 30
- 239000002904 solvent Substances 0.000 description 29
- 239000000203 mixture Substances 0.000 description 28
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 27
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 25
- 229920000642 polymer Polymers 0.000 description 22
- 238000003756 stirring Methods 0.000 description 22
- 238000006243 chemical reaction Methods 0.000 description 19
- 239000000047 product Substances 0.000 description 19
- 239000007787 solid Substances 0.000 description 19
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 17
- 150000001543 aryl boronic acids Chemical class 0.000 description 15
- 230000027455 binding Effects 0.000 description 15
- 238000009739 binding Methods 0.000 description 15
- 125000001424 substituent group Chemical group 0.000 description 15
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 15
- 102000004882 Lipase Human genes 0.000 description 14
- 108090001060 Lipase Proteins 0.000 description 14
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 14
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 13
- 230000002829 reductive effect Effects 0.000 description 12
- 239000004367 Lipase Substances 0.000 description 11
- 235000019421 lipase Nutrition 0.000 description 11
- 239000003921 oil Substances 0.000 description 11
- 235000019198 oils Nutrition 0.000 description 11
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 10
- 229940127470 Lipase Inhibitors Drugs 0.000 description 10
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 9
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 125000005620 boronic acid group Chemical class 0.000 description 9
- 239000012044 organic layer Substances 0.000 description 9
- 238000001816 cooling Methods 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 8
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 7
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 7
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 7
- 229910052794 bromium Inorganic materials 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 229940040461 lipase Drugs 0.000 description 7
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 7
- 235000019341 magnesium sulphate Nutrition 0.000 description 7
- 239000012299 nitrogen atmosphere Substances 0.000 description 7
- SIAVMDKGVRXFAX-UHFFFAOYSA-N 4-carboxyphenylboronic acid Chemical compound OB(O)C1=CC=C(C(O)=O)C=C1 SIAVMDKGVRXFAX-UHFFFAOYSA-N 0.000 description 6
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- 241000700159 Rattus Species 0.000 description 6
- 239000007864 aqueous solution Substances 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- JPTNTBSBJPCXGI-UHFFFAOYSA-N 1-(4-bromo-2,5-difluorophenyl)ethanone Chemical compound CC(=O)C1=CC(F)=C(Br)C=C1F JPTNTBSBJPCXGI-UHFFFAOYSA-N 0.000 description 5
- 239000012267 brine Substances 0.000 description 5
- 239000012043 crude product Substances 0.000 description 5
- 239000008367 deionised water Substances 0.000 description 5
- 229910021641 deionized water Inorganic materials 0.000 description 5
- 239000000194 fatty acid Substances 0.000 description 5
- 238000010438 heat treatment Methods 0.000 description 5
- 238000001727 in vivo Methods 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- 238000002390 rotary evaporation Methods 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 239000011780 sodium chloride Substances 0.000 description 5
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 5
- 150000003626 triacylglycerols Chemical class 0.000 description 5
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 4
- 229940086609 Lipase inhibitor Drugs 0.000 description 4
- 230000009102 absorption Effects 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- 150000001642 boronic acid derivatives Chemical class 0.000 description 4
- 125000001246 bromo group Chemical group Br* 0.000 description 4
- 239000013058 crude material Substances 0.000 description 4
- 235000014113 dietary fatty acids Nutrition 0.000 description 4
- 150000002009 diols Chemical group 0.000 description 4
- 229930195729 fatty acid Natural products 0.000 description 4
- 150000004665 fatty acids Chemical class 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 230000004130 lipolysis Effects 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 229910052938 sodium sulfate Inorganic materials 0.000 description 4
- 235000011152 sodium sulphate Nutrition 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 4
- 238000004809 thin layer chromatography Methods 0.000 description 4
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- ULGGZAVAARQJCS-UHFFFAOYSA-N 11-sulfanylundecan-1-ol Chemical compound OCCCCCCCCCCCS ULGGZAVAARQJCS-UHFFFAOYSA-N 0.000 description 3
- QJJKXFKEZBWZHN-UHFFFAOYSA-N B1OSO1 Chemical class B1OSO1 QJJKXFKEZBWZHN-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 3
- BXVHKBNJUHSCRF-UHFFFAOYSA-N [4-(2-bromoacetyl)-3-fluorophenyl]boronic acid Chemical compound OB(O)C1=CC=C(C(=O)CBr)C(F)=C1 BXVHKBNJUHSCRF-UHFFFAOYSA-N 0.000 description 3
- BQZTUNUYGFMPBU-UHFFFAOYSA-N [4-(2-bromoacetyl)phenyl]boronic acid Chemical compound OB(O)C1=CC=C(C(=O)CBr)C=C1 BQZTUNUYGFMPBU-UHFFFAOYSA-N 0.000 description 3
- 229960000583 acetic acid Drugs 0.000 description 3
- 230000029087 digestion Effects 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 238000003818 flash chromatography Methods 0.000 description 3
- 125000001072 heteroaryl group Chemical group 0.000 description 3
- 230000002209 hydrophobic effect Effects 0.000 description 3
- 235000012054 meals Nutrition 0.000 description 3
- SLCVBVWXLSEKPL-UHFFFAOYSA-N neopentyl glycol Chemical compound OCC(C)(C)CO SLCVBVWXLSEKPL-UHFFFAOYSA-N 0.000 description 3
- 125000004433 nitrogen atom Chemical group N* 0.000 description 3
- 239000004006 olive oil Substances 0.000 description 3
- 235000008390 olive oil Nutrition 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 3
- 239000013585 weight reducing agent Substances 0.000 description 3
- YHEPWJRNHLTYSL-UHFFFAOYSA-N (4-acetyl-3-fluorophenyl)boronic acid Chemical compound CC(=O)C1=CC=C(B(O)O)C=C1F YHEPWJRNHLTYSL-UHFFFAOYSA-N 0.000 description 2
- 244000215068 Acacia senegal Species 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 229920000084 Gum arabic Polymers 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 206010033307 Overweight Diseases 0.000 description 2
- 102000019280 Pancreatic lipases Human genes 0.000 description 2
- 108050006759 Pancreatic lipases Proteins 0.000 description 2
- 235000010489 acacia gum Nutrition 0.000 description 2
- 239000000205 acacia gum Substances 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 2
- 229910052796 boron Inorganic materials 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- 239000002026 chloroform extract Substances 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- NCQDQONETMHUMY-UHFFFAOYSA-N dichloro(phenyl)borane Chemical compound ClB(Cl)C1=CC=CC=C1 NCQDQONETMHUMY-UHFFFAOYSA-N 0.000 description 2
- 210000002249 digestive system Anatomy 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 230000008014 freezing Effects 0.000 description 2
- 238000007710 freezing Methods 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 210000000936 intestine Anatomy 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- AHLBNYSZXLDEJQ-FWEHEUNISA-N orlistat Chemical compound CCCCCCCCCCC[C@H](OC(=O)[C@H](CC(C)C)NC=O)C[C@@H]1OC(=O)[C@H]1CCCCCC AHLBNYSZXLDEJQ-FWEHEUNISA-N 0.000 description 2
- 229940116369 pancreatic lipase Drugs 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- KIDHWZJUCRJVML-UHFFFAOYSA-N putrescine Chemical compound NCCCCN KIDHWZJUCRJVML-UHFFFAOYSA-N 0.000 description 2
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 210000000813 small intestine Anatomy 0.000 description 2
- ATHGHQPFGPMSJY-UHFFFAOYSA-N spermidine Chemical compound NCCCCNCCCN ATHGHQPFGPMSJY-UHFFFAOYSA-N 0.000 description 2
- PFNFFQXMRSDOHW-UHFFFAOYSA-N spermine Chemical compound NCCCNCCCCNCCCN PFNFFQXMRSDOHW-UHFFFAOYSA-N 0.000 description 2
- AKEJUJNQAAGONA-UHFFFAOYSA-N sulfur trioxide Chemical compound O=S(=O)=O AKEJUJNQAAGONA-UHFFFAOYSA-N 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 150000003613 toluenes Chemical class 0.000 description 2
- RYBVDDUJULLYDW-UHFFFAOYSA-N trimethyl(10-sulfanyldecyl)azanium;bromide Chemical compound [Br-].C[N+](C)(C)CCCCCCCCCCS RYBVDDUJULLYDW-UHFFFAOYSA-N 0.000 description 2
- XFNJVJPLKCPIBV-UHFFFAOYSA-N trimethylenediamine Chemical compound NCCCN XFNJVJPLKCPIBV-UHFFFAOYSA-N 0.000 description 2
- 229940002552 xenical Drugs 0.000 description 2
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 description 1
- OBQRODBYVNIZJU-UHFFFAOYSA-N (4-acetylphenyl)boronic acid Chemical compound CC(=O)C1=CC=C(B(O)O)C=C1 OBQRODBYVNIZJU-UHFFFAOYSA-N 0.000 description 1
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- GTQHJCOHNAFHRE-UHFFFAOYSA-N 1,10-dibromodecane Chemical compound BrCCCCCCCCCCBr GTQHJCOHNAFHRE-UHFFFAOYSA-N 0.000 description 1
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 1
- YPFDHNVEDLHUCE-UHFFFAOYSA-N 1,3-propanediol Substances OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 1
- WYECURVXVYPVAT-UHFFFAOYSA-N 1-(4-bromophenyl)ethanone Chemical compound CC(=O)C1=CC=C(Br)C=C1 WYECURVXVYPVAT-UHFFFAOYSA-N 0.000 description 1
- IGBZCOWXSCWSHO-UHFFFAOYSA-N 1-(5-bromothiophen-2-yl)ethanone Chemical compound CC(=O)C1=CC=C(Br)S1 IGBZCOWXSCWSHO-UHFFFAOYSA-N 0.000 description 1
- MYMSJFSOOQERIO-UHFFFAOYSA-N 1-bromodecane Chemical compound CCCCCCCCCCBr MYMSJFSOOQERIO-UHFFFAOYSA-N 0.000 description 1
- WSULSMOGMLRGKU-UHFFFAOYSA-N 1-bromooctadecane Chemical compound CCCCCCCCCCCCCCCCCCBr WSULSMOGMLRGKU-UHFFFAOYSA-N 0.000 description 1
- ZNJOCVLVYVOUGB-UHFFFAOYSA-N 1-iodooctadecane Chemical compound CCCCCCCCCCCCCCCCCCI ZNJOCVLVYVOUGB-UHFFFAOYSA-N 0.000 description 1
- XFGANBYCJWQYBI-UHFFFAOYSA-N 11-bromoundecan-1-ol Chemical compound OCCCCCCCCCCCBr XFGANBYCJWQYBI-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 1
- JYWKEVKEKOTYEX-UHFFFAOYSA-N 2,6-dibromo-4-chloroiminocyclohexa-2,5-dien-1-one Chemical compound ClN=C1C=C(Br)C(=O)C(Br)=C1 JYWKEVKEKOTYEX-UHFFFAOYSA-N 0.000 description 1
- MDNDJMCSXOXBFZ-UHFFFAOYSA-N 2-(5,5-dimethyl-1,3,2-dioxaborinan-2-yl)-5,5-dimethyl-1,3,2-dioxaborinane Chemical compound O1CC(C)(C)COB1B1OCC(C)(C)CO1 MDNDJMCSXOXBFZ-UHFFFAOYSA-N 0.000 description 1
- NSODVVYOWINKAG-UHFFFAOYSA-N 2-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethoxy]ethyl 4-methylbenzenesulfonate Chemical compound CC1=CC=C(S(=O)(=O)OCCOCCOCCOCCOCCO)C=C1 NSODVVYOWINKAG-UHFFFAOYSA-N 0.000 description 1
- VKRHTRDIFQXUSJ-UHFFFAOYSA-N 2-borono-5-(11-hydroxyundecyl)benzoic acid Chemical compound OCCCCCCCCCCCC1=CC=C(B(O)O)C(C(O)=O)=C1 VKRHTRDIFQXUSJ-UHFFFAOYSA-N 0.000 description 1
- XCRCSPKQEDMVBO-UHFFFAOYSA-N 2-bromo-1,4-difluorobenzene Chemical compound FC1=CC=C(F)C(Br)=C1 XCRCSPKQEDMVBO-UHFFFAOYSA-N 0.000 description 1
- WNIFCLWDGNHGMX-UHFFFAOYSA-N 3-borono-5-nitrobenzoic acid Chemical compound OB(O)C1=CC(C(O)=O)=CC([N+]([O-])=O)=C1 WNIFCLWDGNHGMX-UHFFFAOYSA-N 0.000 description 1
- DBVFWZMQJQMJCB-UHFFFAOYSA-N 3-boronobenzoic acid Chemical compound OB(O)C1=CC=CC(C(O)=O)=C1 DBVFWZMQJQMJCB-UHFFFAOYSA-N 0.000 description 1
- NNZGNZHUGJAKKT-UHFFFAOYSA-M 3-bromopropyl(trimethyl)azanium;bromide Chemical compound [Br-].C[N+](C)(C)CCCBr NNZGNZHUGJAKKT-UHFFFAOYSA-M 0.000 description 1
- YUHCOUPKUNJQIC-UHFFFAOYSA-N 4-(3-chloropropyl)octadecyl-dimethylazanium bromide Chemical compound [Br-].CCCCCCCCCCCCCCC(CCCCl)CCC[NH+](C)C YUHCOUPKUNJQIC-UHFFFAOYSA-N 0.000 description 1
- HGXWRDPQFZKOLZ-UHFFFAOYSA-N 4-bromo-2-fluorobenzonitrile Chemical compound FC1=CC(Br)=CC=C1C#N HGXWRDPQFZKOLZ-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 239000007992 BES buffer Substances 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 125000006539 C12 alkyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- JLVVSXFLKOJNIY-UHFFFAOYSA-N Magnesium ion Chemical compound [Mg+2] JLVVSXFLKOJNIY-UHFFFAOYSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- OAGKSROKYKIALY-UHFFFAOYSA-N OB(c(cc1)ccc1C(OCCOC/C=[O]/CCOCCO)=O)O Chemical compound OB(c(cc1)ccc1C(OCCOC/C=[O]/CCOCCO)=O)O OAGKSROKYKIALY-UHFFFAOYSA-N 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 239000005700 Putrescine Substances 0.000 description 1
- 238000004617 QSAR study Methods 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- LEHOTFFKMJEONL-UHFFFAOYSA-N Uric Acid Chemical compound N1C(=O)NC(=O)C2=C1NC(=O)N2 LEHOTFFKMJEONL-UHFFFAOYSA-N 0.000 description 1
- TVWHNULVHGKJHS-UHFFFAOYSA-N Uric acid Natural products N1C(=O)NC(=O)C2NC(=O)NC21 TVWHNULVHGKJHS-UHFFFAOYSA-N 0.000 description 1
- UWAOJIWUVCMBAZ-UHFFFAOYSA-N [1-[1-(4-chlorophenyl)cyclobutyl]-3-methylbutyl]-dimethylazanium;chloride Chemical compound Cl.C=1C=C(Cl)C=CC=1C1(C(N(C)C)CC(C)C)CCC1 UWAOJIWUVCMBAZ-UHFFFAOYSA-N 0.000 description 1
- YANFFNKCIXBHDU-UHFFFAOYSA-N [4-(dioctadecylcarbamoyl)phenyl]boronic acid Chemical compound CCCCCCCCCCCCCCCCCCN(CCCCCCCCCCCCCCCCCC)C(=O)C1=CC=C(B(O)O)C=C1 YANFFNKCIXBHDU-UHFFFAOYSA-N 0.000 description 1
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 229910001420 alkaline earth metal ion Inorganic materials 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000000118 anti-neoplastic effect Effects 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- OGBUMNBNEWYMNJ-UHFFFAOYSA-N batilol Chemical class CCCCCCCCCCCCCCCCCCOCC(O)CO OGBUMNBNEWYMNJ-UHFFFAOYSA-N 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910001424 calcium ion Inorganic materials 0.000 description 1
- 235000019577 caloric intake Nutrition 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 210000004534 cecum Anatomy 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000002178 crystalline material Substances 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 125000003963 dichloro group Chemical group Cl* 0.000 description 1
- BSHICDXRSZQYBP-UHFFFAOYSA-N dichloromethane;palladium(2+) Chemical compound [Pd+2].ClCCl BSHICDXRSZQYBP-UHFFFAOYSA-N 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- TVAVIKKWTBSOPJ-UHFFFAOYSA-N docosyl methanesulfonate Chemical compound CCCCCCCCCCCCCCCCCCCCCCOS(C)(=O)=O TVAVIKKWTBSOPJ-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 239000002024 ethyl acetate extract Substances 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000000105 evaporative light scattering detection Methods 0.000 description 1
- 238000013213 extrapolation Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 210000003736 gastrointestinal content Anatomy 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000004519 grease Substances 0.000 description 1
- 230000005802 health problem Effects 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 230000000055 hyoplipidemic effect Effects 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 229940060367 inert ingredients Drugs 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 229910052740 iodine Chemical group 0.000 description 1
- 239000011630 iodine Chemical group 0.000 description 1
- 125000003010 ionic group Chemical group 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 235000019626 lipase activity Nutrition 0.000 description 1
- 229910001425 magnesium ion Inorganic materials 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 229940045623 meridia Drugs 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- SNVLJLYUUXKWOJ-UHFFFAOYSA-N methylidenecarbene Chemical compound C=[C] SNVLJLYUUXKWOJ-UHFFFAOYSA-N 0.000 description 1
- IFYDWYVPVAMGRO-UHFFFAOYSA-N n-[3-(dimethylamino)propyl]tetradecanamide Chemical compound CCCCCCCCCCCCCC(=O)NCCCN(C)C IFYDWYVPVAMGRO-UHFFFAOYSA-N 0.000 description 1
- HKUFIYBZNQSHQS-UHFFFAOYSA-N n-octadecyloctadecan-1-amine Chemical compound CCCCCCCCCCCCCCCCCCNCCCCCCCCCCCCCCCCCC HKUFIYBZNQSHQS-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920000166 polytrimethylene carbonate Polymers 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 229910001414 potassium ion Inorganic materials 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000035935 pregnancy Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- JAJWGJBVLPIOOH-IZYKLYLVSA-M sodium taurocholate Chemical compound [Na+].C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(=O)NCCS([O-])(=O)=O)C)[C@@]2(C)[C@@H](O)C1 JAJWGJBVLPIOOH-IZYKLYLVSA-M 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 229940063673 spermidine Drugs 0.000 description 1
- 229940063675 spermine Drugs 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 238000005809 transesterification reaction Methods 0.000 description 1
- WRECIMRULFAWHA-UHFFFAOYSA-N trimethyl borate Chemical compound COB(OC)OC WRECIMRULFAWHA-UHFFFAOYSA-N 0.000 description 1
- 229940116269 uric acid Drugs 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 210000000707 wrist Anatomy 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/025—Boronic and borinic acid compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/74—Synthetic polymeric materials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/74—Synthetic polymeric materials
- A61K31/785—Polymers containing nitrogen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Diabetes (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
- HINTERGRUND DER ERFINDUNG
- Fettsucht bei Menschen ist ein bekanntes Gesundheitsproblem, wobei etwa siebenundneunzig Millionen Personen in den Vereinigten Staaten klinisch als übergewichtig betrachtet werden. Verschiedene chemische Ansätze wurden zur Behandlung von Fettsucht verwendet. In einem derartigen Ansatz wird ein Medikament, das Lipasen hemmt, an den fettsüchtigen Patienten verabreicht. Lipasen sind Schlüsselenzyme im Verdauungssystem, die Diglyceride und Triglyceride in Monoglyceride und Fettsäuren abbauen. Diglyceride und Triglyceride weisen einen hohen Kaloriengehalt auf, werden jedoch vom Dünndarm erst absorbiert, wenn sie durch die Lipasen abgebaut sind. Daher führt die Hemmung von Lipasen im Verdauungssystem zu einer Verringerung der Fettabsorption und folglich einer Verringerung der Kalorienaufnahme. XENICAL ist ein Beispiel für einen im Handel erhältlichen Lipaseinhibitor, der zur Behandlung von Fettsucht verwendet wird.
- Environ. Health Perspect. Suppl. (1994), 102 (Suppl. 7), 21–30, Hall et al., offenbart hypolipidämische, gegen Fettsucht wirkende, entzündungshemmende, gegen Osteoporose wirkende und antineoplastische Eigenschaften von Amincarboxyboranen.
- Jedoch besteht immer noch Bedarf an verbesserten Lipaseinhibitoren. Beispielsweise führt die Verabreichung von Lipaseinhibitoren zu Stühlen mit einem hohen Fett- oder Ölgehalt aufgrund der unverdauten Diglyceride und Triglyceride. Das Austreten von Öl aus dem Stuhl ist eine unangenehme Nebenwirkung, die häufig auftritt, wenn Stühle einen hohen Fett- oder Ölgehalt aufweisen. Dieser Zustand wird als "Ölstuhl" oder "Leckstuhl" bezeichnet. In der US-Anmeldung 09/166453 wurde berichtet, dass fettbindende Polymere, wenn sie mit Lipaseinhibitoren coverabreicht werden, das Öl binden oder "stabilisieren" können und dadurch das Austreten von Öl aus dem Stuhl verringern oder beseitigen können. Es wäre günstig, eine einzige Verbindung, die sowohl ein Lipaseinhibitor als auch ein Fettbindemittel ist, zu entwickeln. Ferner sollte ein Lipaseinhibitor durch den Darm minimal absorbiert werden, um systemische Nebenwirkungen zu verhindern. Weitere erwünschte Merkmale umfassen Einfachheit und Wirtschaftlichkeit der Herstellung.
- ZUSAMMENFASSUNG DER ERFINDUNG
- Die vorliegende Erfindung ist auf neue Arylboronsäuren und Derivate derselben, die wirksame Lipaseinhibitoren sind, gerichtet (Beispiele 11 und 12). Viele dieser Verbindungen sind an fettbindenden Polymeren, die Alkohol- oder Diolfunktionalitäten umfassen, mittels Boronatester-, Boronatthioester- und/oder Boronamidbindungen gebunden bzw. befestigt. Es wird angenommen, dass diese Bindungen in vivo hydrolysiert werden, wodurch es zur Abgabe eines Lipaseinhibitors und eines fettbindenden Polymers an den Gastrointestinaltrakt kommt. Auf der Basis dieser Ermittlungen sind hierin neue Arylboronsäuren und Derivate derselben, pharmazeutische Zusammensetzungen, die diese Arylboronsäuren und Derivate umfassen, und Verfahren zur Behandlung von Fettsucht mit einer neuen Arylboronsäure oder einem Derivat derselben offenbart.
- Die Erfindung betrifft eine Verbindung gemäß der Definition in Anspruch 1. Weitere Ausführungsformen sind in den Ansprüchen 2 bis 14 definiert.
- Ein Aspekt der vorliegenden Erfindung ist die Verwendung einer Verbindung der Strukturformel I gemäß der Definition in Anspruch 15:

- Z und Z' stehen unabhängig voneinander für -O-, -NH- oder -S-. Vorzugsweise sind Z und Z' beide -O-.
- Ar ist eine substituierte (beispielsweise monosubstituierte oder polysubstituierte) oder unsubstituierte Arylgruppe.
- X ist eine elktronenziehende Gruppe.
- R steht für eine substituierte oder unsubstituierte geradkettige Hydrocarbylgruppe, die optional eine oder mehrere Amin-, Ammonium-, Ether-, Thioether- oder Phenylenverknüpfungsgruppen umfasst, und Y steht für -H, ein Amin, -[NH-(CH2)q]r-NH2, Halogen, -CF3, Thiol, Ammonium, -OH, -COOH, -SO3H, -OSO3H oder eine Phosphoniumgruppe, die kovalent an die terminale Position von R gebunden ist. Vorzugsweise weist, wenn Y -H ist und R eine geradkettige Hydrocarbylgruppe ist, dann R dann 1 bis 30 Kohlenstoffatome, vorzugsweise 4 bis 30 Kohlenstoffatome (noch günstiger 6 bis 30 Kohlenstoffatome, noch besser 8 bis 30 Kohlenstoffatome und noch besser 10 bis 30 Kohlenstoffatome) auf. Jedes -NH- in -[NH-(CH2)q]r-NH2 ist optional N-alkyliert oder N,N-dialkyliert und -NH2 in -[NH-(CH2)q]r-NH2 ist optional N-alkyliert, N,N-dialkyliert oder N,N,N-trialkyliert.
- R1 und R1' stehen unabhängig voneinander für -H, eine aliphatische Gruppe, eine substituierte aliphatische Gruppe, eine Arylgruppe oder eine substituierte Arylgruppe oder sie bilden zusammengenommen eine substituierte oder unsubstituierte C2-C5-Alkylengruppe, die optional eine Aminverknüpfungsgruppe [-N+(R1a)-] umfasst. Vorzugsweise sind R1 und R1' in der Strukturformel (I) beide -H.
- R1a steht für -H, Alkyl, substituiertes Alkyl, Phenyl oder substituiertes Phenyl.
- q ist eine ganze Zahl von 2 bis 10 und r ist eine ganze Zahl von 1 bis 5.
- Eine weitere Ausführungsform der vorliegenden Erfindung ist eine pharmazeutische Zusammensetzung gemäß der Definition in Anspruch 27; weitere Ausführungsformen sind in den Ansprüchen 28 und 29 definiert. Die pharmazeutische Zusammensetzung umfasst die oben beschriebene Verbindung und einen pharmazeutisch akzeptablen Träger oder ein pharmazeutisch akzeptables Verdünnungsmittel. Vorzugsweise umfasst die pharmazeutische Zusammensetzung eine wirksame Konzentration der Verbindung.
- Eine weitere Ausführungsform der vorliegenden Erfindung ist ein Verfahren zur Entfernung von Fett aus dem Gastrointestinaltrakt (oder zur Hemmung der Aufnahme von Fett im Gastrointestinaltrakt) eines Subjekts, das eine derartige Behandlung (beispielsweise eine Behandlung eines Subjekts wegen Fettsucht) benötigt. Das Verfahren umfasst die Stufe der Verabreichung einer wirksamen Menge der oben beschriebenen Verbindung an das Subjekt.
- Die Arylboronsäuren und Arylboronsäurederivate der vorlie genden Erfindung sind starke Lipaseinhibitoren. Daher sind sie zur Behandlung von Fettsucht wirksam. Darüber hinaus können viele dieser Verbindungen an fettbindende Polymere gebunden werden. Diese borfunktionalisierten Polymere können ebenfalls zur Behandlung von Fettsucht verwendet werden, weisen jedoch den Vorteil auf, dass sie nicht die "Ölstühle", die normalerweise mit Lipaseinhibitoren in Verbindung stehen, verursachen. Die hierin offenbarten Arylboronsäuren und Arylboronsäurederivate sind daher Vorstufen für diese verbesserten Polymerarzneistoffe.
- KURZE BESCHREIBUNG DER ZEICHNUNGEN
-
1 ist ein Schema, das die Synthese von 4-(14'-Trimethylammonium-3'-thia-1'-ketotetradecyl)-3-fluorphenylboronsäurechlorid (6) zeigt. -
2 ist ein Schema, das die Synthese von 4-(14'-Hydroxy-3'-thia-1'-ketotetradecyl)-2,5-difluorphenylboronsäure (11) zeigt. -
3 ist ein Schema, das die Synthese von (Neopentylglykolato)-4-(14'-trimethylammonium-3'-thia-1'-ketotridecyl)-2,5-difluorphenylboronatesterchlorid (14) zeigt. - Die
4A –4F sind eine Zusammenfassung der Strukturformeln für Boronsäuren der vorliegenden Erfindung. R in4 ist eine geradkettige C12-Alkylgruppe. - DETAILLIERTE BESCHREIBUNG DER ERFINDUNG
- Die Erfindung wird im folgenden unter Bezug auf Arylboronsäuren und Arylboronatester, d.h., worin Z und Z' beide -O- sind, beschrieben. Dies ist so zu verstehen, dass diese Beschreibungen für die entsprechenden Boronamide und Boronatthioester, d.h., worin einer oder beide Reste Z und Z' für -NH- oder -S- stehen, gelten.
- Ar in der Strukturformel (I) ist substituiert oder unsubstituiert. Ar ist "substituiert", wenn es mindestens einen Substituenten zusätzlich zu der Boronsäuregruppe und der -X-R-Y-Gruppe umfasst. Geeignete Substituenten sind im folgenden für Arylgruppen beschrieben.
- -X- ist eine elektronenziehende Gruppe. Die hierin verwendete "elektronenziehende Gruppe" ist ein Substituent, der zu einem Phenylring führt, der eine geringere Elektronendichte, wenn die Gruppe vorhanden ist, als wenn sie nicht vorhanden ist, aufweist. Elektronenziehende Gruppen weisen einen Hammet-sigma-Wert von größer als 1 auf (siehe beispielsweise C. Hansch. A. Leo und D. Hoeckman, "Exploring QSAR Hydrophobic, Electronic and Steric Constants", American Chemical Society (1995), S. 217–32). Beispiele für geeignete Werte für X umfassen -CHZ''-, -CZ''2-, -COO-, -CON(R1b)-, -CO- oder -SO2-. Andere geeignete Werte für X umfassen -S(O)- und -S(O)2O-, Z'' ist ein Halogen und R1b ist -H, Alkyl oder substituiertes Alkyl (vorzugsweise -R-Y). In den Verbindungen der vorliegenden Erfindung ist der Phenylring A vorzugsweise mit einer oder mehreren elektronenziehenden Gruppen zusätzlich zu -X- substituiert. Geeignete Beispiele umfassen Halogene, -NO2 und -CN; Fluor ist ein bevorzugtes Beispiel.
- Eine "geradkettige Hydrocarbylgruppe" ist eine Alkylengruppe, d.h. -(CH2)x-, worin x eine positive ganze Zahl (beispielsweise von 1 bis etwa 30), vorzugsweise zwischen 6 und etwa 30, noch besser zwischen etwa 6 und etwa 15, ist. Eine "Verknüpfungsgruppe" bezeichnet eine funktionelle Gruppe, die ein Methylen in einem geradkettigen Hydrocarbyl ersetzt. Beispiele für geeignete Verknüpfungsgruppen umfassen ein Alken, Alkin, Phenylen, einen Ether (-O-), Thioether (-S-), ein Amin [-N+(Ra)-] oder Ammonium [-N+(RaRb)-]. Ra und Rb stehen unabhängig voneinander für -H, Alkyl, substituiertes Alkyl, Phenyl, substituiertes Phenyl oder zusammengenommen mit dem Stickstoffatom, an das sie gebunden sind, für eine nichtaromatische stickstoffhaltige heterocyclische Gruppe. Vorzugsweise sind Ra und Rb nicht -H. Noch günstiger sind Ra und Rb beide Alkylgruppen und noch besser beide Methyl. Ra und Rb können gleich oder verschieden sein, sind jedoch vorzugsweise gleich.
- Die Ausdrücke "terminale Position" oder "Terminus" bezeichnen den Methylenkohlenstoff der geradkettigen Hydrocarbylgruppe, der von Ar am weitesten entfernt ist. Substituenten an der terminalen Position einer geradkettigen Hydrocarbylgruppe werden hierin als "terminale Substituenten" bezeichnet. Wie oben angegeben, weisen eine Zahl von Verbindungen der vorliegenden Erfindung eine Amin-(-NRcRd) oder Ammonium(-N+RcRdRe)gruppe als terminalen Substituenten der durch R dargestellten Hydrocarbylgruppe auf. Rc, Rd und Re in einer Ammoniumgruppe stehen unabhängig voneinander für -H, Alkyl, substituiertes Alkyl, Phenyl, substituiertes Phenyl oder zusammengenommen mit dem Stickstoffatom, an das sie gebunden sind, eine stickstoffhaltige nichtaromatische heterocyclische Gruppe. Vorzugsweise sind Rc, Rd und Re nicht -H. Noch günstiger sind Rc, Rd und Re alle Alkylgruppen (d.h. eine Trialkylammoniumgruppe) und noch besser alle Methyl (d.h. eine Trimethylammoniumgruppe). Rc, Rd und Re können gleich oder verschieden sein, sind jedoch vorzugsweise alle gleich.
- In einem Beispiel ist Y derart gewählt, dass YH ein Polyamin eines kleinen Moleküls (H-[NH-CH2)q]r-NH2), wie Spermin, Spermidin, 1,2-Diaminoethan, 1,3-Diaminopropan oder 1,4-Diaminobutan, ist. Optional können eines oder mehrere der sekundären Amine optional N-alkyliert oder N,N-dialkyliert sein; das primäre Amin ist optional N-alkyliert, N,N-dialkyliert oder N,N,N-trialkyliert.
- Eine "substituierte Hydrocarbylgruppe" weist einen oder mehrere Substituenten auf, die an einer oder mehreren Positionen außer am Terminus gebunden sind. Geeignete Substituenten sind diejenigen, die das Lipasehemmvermögen oder Fettbindevermögen des Polymers nicht signifikant verringern, beispielsweise keine der Aktivitäten um mehr als einen Faktor von etwa zwei verringern. Beispiele für geeignete Substituenten umfassen geradkettiges oder verzweigtes C1-C3-Alkyl, geradkettiges oder verzweigtes C1-C3-Halogenalkyl, -OH, Halogen (-Br, -Cl, -I und -F), -O(geradkettiges oder verzweigtes C1-C3-Alkyl) oder -O(geradkettiges oder verzweigtes C1-C3-Halogenalkyl).
- In einer bevorzugten Ausführungsform wird die Verbindung der vorliegenden Erfindung durch die Strukturformel (II) dargestellt:

- Der Phenylring A ist substituiert oder unsubstituiert. Der Phenylring A ist "substituiert", wenn er mindestens einen Substituenten zusätzlich zu der Boronsäuregruppe und der -X-R-Y-Gruppe umfasst. geeignete Substituenten sind im fol genden für Arylgruppen beschrieben.
- R, R1, R'1, X und Y in der Strukturformel (II) sind wie für die Strukturformel I beschrieben. Vorzugsweise ist Y-R-X-para zu -B(OR1)(OR'1).
- In einer stärker bevorzugten Ausführungsform wird die Verbindung der vorliegenden Erfindung durch die Strukturformel (III) dargestellt:

- Der Phenylring A, R und Y in der Strukturformel (III) sind wie oben für die Strukturformel (II) beschrieben. Der Phenylring A ist vorzugsweise mit null, einer oder mehreren unabhängig voneinander ausgewählten elektronenziehenden Gruppen, die durch R2 dargestellt sind, substituiert.
- R in den Strukturformeln (II) und (III) ist eine substituierte oder unsubstituierte geradkettige Hydrocarbylgruppe, die optional eine oder mehrere Ether-, Thioether-, Phenylen-, Amin- oder Ammoniumverknüpfungsgruppen umfasst. Bevorzugte Verknüpfungsgruppen für R in den Strukturformeln (II) und (III) sind Ether oder Thioether. Alternativ ist R in den Strukturformeln (II) und (III) -CH2-O[-(CH2)pO]m-(CH2)p- oder -CH2-S[-(CH2)pO]m-; (CHz)p-; p 2 oder 3 und m eine ganze Zahl von 1 bis 8.
- Y in den Strukturformeln (II) und (III) ist vorzugsweise eine Amin- oder Ammoniumgruppe, die kovalent an der terminalen Position von R gebunden ist, noch günstiger eine Trialkylammoniumgruppe, die an der terminalen Position von R gebunden ist, und noch besser eine Trimethylammoniumgruppe, die an der terminalen Position von R gebunden ist.
- In einer stärker bevorzugten Ausführungsform wird die Verbindung der vorliegenden Erfindung durch die Strukturformeln (IV) und (V) dargestellt

- Der Phenylring A in den Strukturformeln (IV) und (V) ist wie für die Strukturformeln (II)–(III) beschrieben.
- Y in den Strukturformeln (IV) und (V) ist eine Trialkylammoniumgruppe.
- n ist eine ganze Zahl von etwa 6 bis etwa 30, vorzugsweise etwa 6 bis etwa 15.
- Vorzugsweise ist in den Strukturformeln (IV) und (V) Y Trimethylammonium, der Phenylring A mit einer oder zwei Fluorgruppen substituiert und n wie oben definiert. Beispiele für geeignete Substitutionsmuster für den Phenylring A umfassen 3-Fluor und 2,5-Difluor, wobei der an Bor gebundene Kohlenstoff als Kohlenstoff eins betrachtet wird.
- Von der vorliegenden Erfindung werden auch Boronatester der Boronsäuren der Strukturformeln (III)–(V) umfasst. Ein Boronatester wird durch Ersetzen von einem oder beiden Boronsäurewasserstoffatomen durch R1 gemäß der Beschreibung in den Strukturformeln (I) und (II) erhalten. Es wird angenommen, dass Boronatester in dem Gastrointestinaltrakt hydrolysiert werden, wobei Boronsäuren gebildet werden, die dann als Lipaseinhibitoren fungieren.
- Ebenfalls von der vorliegenden Erfindung umfasst werden die Boronamide oder Boronatthioester, die den im vorhergehenden Absatz beschriebenen Boronatestern entsprechen. Das Boronamid oder der Boronatthioester wird durch unabhängiges Ersetzen von einem oder beiden Boronatestersauerstoffatomen durch -S- oder -NH- erhalten.
- Die hierin verwendeten aliphatischen Gruppen umfassen geradkettige, verzweigte oder cyclische C1-C30(vorzugsweise C1-C15)-Kohlenwasserstoffe, die vollständig gesättigt sind oder die eine oder mehrere Nichtsättigungseinheiten enthalten. Bevorzugte aliphatische Gruppen sind vollständig gesättigt und acyclisch, d.h. geradkettige oder verzweigte Alkylgruppen oder Alkylengruppen. Geeignete Substituenten für eine aliphatische Gruppe sind diejenigen, die das Lipasehemmvermögen der Erfindung nicht signifikant verringern, beispielsweise keine der Aktivitäten um mehr als einen Faktor von etwa zwei verringern. Beispiele umfassen -OH, Halo gen (-Br, -Cl, -I und -F), -O(R'), -O-CO-(R'), -CN, -NO2, -COOH, =O, -NH2, -NH(R'), -N(R')2, -COO(R'), -CONH2, -CONH(R'), -CON(R')2, -SH und -S(R'). Jedes R' ist unabhängig voneinander eine Alkylgruppe oder eine Arylgruppe. Eine substituierte aliphatische Gruppe kann mehr als einen Substituenten aufweisen.
- Arylgruppen umfassen carbocyclische aromatische Gruppen, wie Phenyl und Naphthyl, Heteroarylgruppen, wie Imidazolyl, Thienyl, Furanyl, Pyridyl, Pyrimidyl, Pyranyl, Pyrazolyl, Pyrazinyl, Thiazol, Oxazolyl und kondensierte polycyclische aromatische Ringsysteme, worin ein carbocyclischer aromatischer Ring oder Heteroarylring an einen oder mehrere andere Heteroarylringe kondensiert ist (beispielsweise Benzothienyl, Benzofuranyl, Indolyl, Chinolinyl, Benzothiazol, Benzooxazol, Benzimidazol und Chinolinyl). Geeignete Substituenten für eine Arylgruppe sind diejenigen, die das Lipasehemmvermögen der Verbindung nicht signifikant verringern, beispielsweise keine der Aktivitäten um mehr als einen Faktor von etwa 2 verringern. Beispiele umfassen Alkyl, halogeniertes Alkyl, -OH, Halogen (-Br, -Cl, -I und -F), -O(R'), -O-CO-(R'), -CN, -NO2, -COOH, -NH2, -NH(R'), -N(R')2, -COO(R'), -CONH2, -CONH(R'), -CON(R')2, -SH und -S(R'). Jedes R' ist unabhängig voneinander eine Alkylgruppe oder eine Arylgruppe. Eine substituierte Arylgruppe kann mehr als einen Substituenten aufweisen.
- Nichtaromatische stickstoffhaltige heterocyclische Ringe sind nichtaromatische carbocyclische Ringe, die mindestens ein Stickstoffatom und optional ein oder mehrere weitere Heteroatome, wie Sauerstoff oder Schwefel, im Ring umfassen. Der Ring kann fünf-, sechs-, sieben- oder achtgliedrig sein. Beispiele umfassen Morpholino, Thiomorpholino, Pyrrolidinyl, Piperazinyl und Piperidinyl.
- Von der vorliegenden Erfindung werden auch pharmazeutisch akzeptable Salze der offenbarten Verbindungen umfasst. Beispielsweise können Verbindungen, die saure funktionelle Gruppen aufweisen, in der anionischen oder Form der konjugierten Base in Kombination mit einem Kation vorhanden sein. Geeignete Kationen umfassen Erdalkalimetallionen, wie Natrium- und Kaliumionen, Erdalkaliionen, wie Calcium- und Magnesiumionen, und unsubstituierte und substituierte (primäre, sekundäre, tertiäre und quaternäre) Ammoniumionen. Verbindungen, die basische Gruppen aufweisen, wie Amine, können in einer protonierten Form zusammen mit einem pharmazeutisch akzeptablen Gegenion, wie Chlorid, Bromid, Acetat, Formiat, Citrat, Ascorbat, Sulfat oder Phosphat, vorhanden sein. In ähnlicher Weise umfassen Ammoniumgruppen ein pharmazeutisch akzeptables Gegenion. Boronsäuregruppen können mit Anionen, wie Natrium- oder Kaliumhydroxid, -alkoxid oder -carboxylat unter Bildung eines Salzes wie -B-(OH)3Na+, -B-(OH)3K+, -B-(OH)2(OCH3)Na+, -B-(OH)2(OCH3)K+, -B-(OH)2(OCOCH3)Na+, -B-(OH)2(OCOCH3)K+ und dgl. reagieren.
- Ein "Subjekt" ist vorzugsweise ein Säuger, beispielsweise ein Mensch, doch können es auch ein Begleitertier (beispielsweise Hunde, Katzen und dgl.), Hoftiere (beispielsweise Kühe, Schafe, Schweine, Pferde und dgl.) oder Labortiere (beispielsweise Ratten, Mäuse, Meerschweinchen und dgl.), die eine Behandlung wegen Fettsucht benötigen, sein.
- Die Verbindungen der vorliegenden Erfindung sind als Medikament zur Förderung einer Gewichtsreduktion bei Subjekten geeignet, da sie Lipasen im Gastrointestinaltrakt hemmen. Als solche werden sie in einer Weise verabreicht, die zum Erreichen des Gastrointestinaltrakts während der Verdauung geeignet ist. Sie werden daher vorzugsweise oral frühestens bis zu etwa 1 h vor einer Mahlzeit und spätestens bis etwa 1 h nach einer Mahlzeit verabreicht. Alternative Verabrei chungsweisen sind auch möglich, die eine rektale, nasale, pulmonale und topische Verabreichung umfassen.
- Die Verbindungen der vorliegenden Erfindung werden zur Hemmung der Aufnahme von Fett im Gastrointestinaltrakt (oder zur Förderung der Entfernung von Fett aus dem Gastrointestinaltrakt) verabreicht. Daher können sie auch vorteilhafterweise bei der Behandlung von einem oder mehreren der folgenden Zustände: Fettsucht, Typ-II (nicht-insulinabhängigem)-Diabetes mellitus, beeinträchtigter Glucosetoleranz, Hypertonie, Koronarthrombose, Schlaganfall, Lipidsyndromen, Hyperglykämie, Hypertriglyceridämie, Hyperlipidämie, Schlafapnoe, Hiatushernie, Refluxösophagitis, Osteoarthritis, Gicht, mit Gewichtszunahme in Verbindung stehenden Krebserkrankungen, Gallensteinen, Nierensteinen, pulmonaler Hypertonie, Unfruchtbarkeit, einer kardiovaskulären Erkrankung, höheres als Normalgewicht und höheren als normalen Lipidspiegeln; oder wenn das Subjekt aus einer verringerten Plättchenadhäsion, Gewichtsabnahme nach Schwangerschaft, gesenkten Lipidspiegeln, gesenkten Harnsäurespiegeln oder gesenkten Oxalatspiegeln Nutzen ziehen würde, verwendet werden. Ein Subjekt mit einem oder mehreren dieser Zustände wird als "eine Behandlung benötigend" mit einem Mittel, das die Absorption von Fett aus dem Gastrointestinaltrakt hemmt, bezeichnet.
- Die Verbindungen können den Subjekten in Verbindung mit einem akzeptablen pharmazeutischen Träger als Teil einer pharmazeutischen Zusammensetzung zur Behandlung von Fettsucht verabreicht werden. Formulierungen variieren entsprechend dem gewählten Verabreichungsweg, sie sind jedoch typischerweise Kapseln, Tabletten oder Pulver zur oralen Verabreichung. Lösungen und Emulsionen sind ebenfalls möglich. Geeignete pharmazeutische Träger können inerte Bestandteile enthalten, die mit der Verbindung nicht inter agieren. Pharmazeutische Standardformulierungstechniken, beispielsweise diejenigen gemäß der Beschreibung in Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, können verwendet werden. Verfahren zur Verkapselung von Zusammensetzungen (beispielsweise in einem Überzug von harter Gelatine oder Cyclodextran) sind einschlägig bekannt (Baker et al., "Controlled Release of Biological Active Agents", John Wiley and Sons, 1986).
- Eine "wirksame Menge" ist die Menge einer Verbindung, die zu einer größeren Gewichtsverringerungsmenge über einen Zeitraum, während dem ein Subjekt mit dem Arylboronsäurearzneistoff wegen Fettsucht behandelt wird, im Vergleich zu dem entsprechenden Zeitraum bei Fehlen einer derartigen Behandlung führt. Typische Dosierungen reichen von etwa 5 mg/Tag bis etwa 10 g/Tag, vorzugsweise von etwa 5 mg/Tag bis etwa 5 g/Tag. Die Verbindung kann allein oder in einer pharmazeutischen Zusammensetzung, die die Verbindung, einen akzeptablen Träger oder ein akzeptables Verdünnungsmittel und optional einen oder mehrere zusätzliche Arzneistoffe, typischerweise einen oder mehrere zusätzliche Arzneistoffe, die zur Gewichtsreduktion verwendet werden, (beispielsweise XENICAL oder MERIDIA) umfasst, verabreicht werden. Die einem Subjekt zu verabreichende genaue Arzneistoffmenge wird auf individueller Basis bestimmt und hängt zumindest teilweise von den individuellen Eigenschaften eines Subjekts, beispielsweise dem allgemeinen Gesundheitszustand, dem Alter, Geschlecht, Körpergewicht und der Toleranz gegenüber Arzneistoffen und dem Grad, in dem das Subjekt übergewichtig ist, und der angestrebten Gewichtsverringerungsmenge ab.
- Eine "wirksame Konzentration" ist die Konzentration einer in einer pharmazeutischen Zusammensetzung vorhandenen Verbindung, die, wenn sie in Einheitsdosierungsform geteilt wird, eine wirksame Menge der Verbindung ergibt.
- Die Arylboronsäureverbindungen der vorliegenden Erfindung können auch mit pharmazeutisch akzeptablen Polymeren mit freien Alkohol- oder Diolgruppen unter Bildung von Boronatestern umgesetzt und als Polymerarzneistoff verabreicht werden. Reaktionen zur Bildung von Boronatesterbindungen sind einschlägig bekannt und umfassen das Refluxieren der Boronsäure und des Diols in einem geeigneten Lösemittel (beispielsweise Alkohol, Toluol, Methylenchlorid, Tetrahydrofuran (THF) oder Dimethylsulfoxid (DMSO)). Alternativ kann eine Arylboronsäure an ein Polymer mit freien Alkohol- oder Diolgruppen mittels einer Umesterungsreaktion gemäß der Beschreibung in D. H. Kinder und M. M. Ames, Journal or Organic Chemistry 52: 2452 (1987) und D. S. Matteson und R. Ray, Journal of American Chemical Society 102: 7590 (1980), deren gesamte Lehren hierin als Bezug aufgenommen sind, gebunden werden.
- Es wird angenommen, dass der Boronatester dieser Polymerarzneistoffe im Gastrointestinaltrakt hydrolysiert wird, wobei die Arylboronsäure freigesetzt wird, die dann eine Hemmung von Lipaseenzymen bewirken kann. Vorzugsweise ist das Polymer ein fettbindendes Polymer. Nach Hydrolyse des Arylboronatesters, wobei die Arylboronsäure freigesetzt wird, ist das fettbindende Polymer dann zur Absorption der Diglyceride und Triglyceride, die infolge der Hemmung der Lipaseenzyme durch die freigesetzte Arylboronsäure unverdaut bleiben, verfügbar. Die unerwünschte Nebenwirkung von "Ölstühlen" wird durch die Verwendung dieser Polymerarzneistoffe dadurch minimiert oder beseitigt. "Fettbindende Polymere" sind Polymere, die Fett absorbieren, binden oder in anderer Weise assoziieren, wodurch (partiell oder vollständig) Fettverdauung, -hydrolyse oder -absorption im Gastrointestinaltrakt gehemmt werden und/oder das Entfernen von Fett aus dem Körper vor der Verdauung erleichtert wird. Die fettbindenden Polymere umfassen allgemein eine oder mehrere fettbindende Regionen. "Fettbindende Regionen" umfassen eine positiv geladene Region und optional eine hydrophobe Region oder eine Region, die sowohl positiv geladen als auch hydrophob ist. Die fettbindende Region weist eine positive Ladung auf, wenn die Region eine ionische Gruppe, beispielsweise ein quaternäres Amin oder ein Atom, beispielsweise den Stickstoff eines Amins, der unter den im Gastrointestinaltrakt vorhandenen Bedingungen eine positive Ladung besitzt, umfasst. Die Befestigung bzw. Bindung von Arylboronsäure-Lipaseinhibitoren an fettbindenden Polymeren und die Verwendung dieser Polymere als Antifettsuchtarzneistoffe sind in der gleichzeitig anhängigen US Provisional Application des Aktenzeichens 60/302 221 mit dem Titel "Aryl Boronate Functionalized Polymers for Treating Obesity", eingereicht am 29. Juni 2001, und US Provisional Application des Aktenzeichens 60/359 473 mit dem Titel "Aryl Boronate Functionalized Polymers for Treating Obesity", eingereicht am 22. Februar 2002, beschrieben. Die gesamten Lehren dieser Anmeldungen sind hierin als Bezug aufgenommen.
- Die Herstellung repräsentativer Phenylboronsäureverbindungen ist in den Beispielen 1–10 beschrieben und schematisch in den
1 –3 angegeben. Der Fachmann ist zur Auswahl geeigneter Ausgangsmaterialien zur Gewinnung der gewünschten Arylboronsäure und bei Durchführung dieser Reaktionen mit verschiedenen Ausgangsmaterialien zur Modifizierung der Reaktionsbedingungen, falls nötig, unter Verwendung von nicht mehr als Routineversuchen fähig. Beispielsweise kann das 4-Bromacetophenon in2 (Verbindung 7) durch jede geeignete Arylverbindung, die mit Brom oder Iod und Acetyl substituiert ist, ersetzt werden. Beispielsweise ist 2-Acetyl-5-bromthiophen im Handel von Aldrich Chemical Co., Milwaukee, WI, erhältlich. Die Länge der Hydrocarbylgruppe in den Arylboronsäuren kann entsprechend der Länge des 1,ω-Alkanthiolalkohols variiert werden. - Repräsentative Boronsäuren der vorliegenden Erfindung, die gemäß in den Beispielen beschriebenen Verfahren hergestellt wurden, sind in
4 gezeigt. - Die Erfindung wird ferner durch die folgenden Beispiele, die in keinster Weise beschränkend sein sollen, erläutert.
- BEISPIELE
- Beispiel 1 -4-(14'-Trimethylammonium-3'-thia-1'-ketotetradecyl)-3-fluorphenylboronsäurechlorid (6)
- Die Synthese der Verbindung (6) ist schematisch in
1 angegeben. Eine detaillierte Beschreibung des Verfahrens ist im folgenden angegeben. - Stufe 1. Synthese von 4-Acetyl-3-fluorphenylboronsäure (1)
- Ein ofengetrockneter 3-1-Dreihalsrundkolben (der mit einem Stickstoffeinlass, Zugabetrichter und Überkopfrührer ausgestattet ist) wurde mit 50 g (0,25 mol) 4-Cyano-3-fluorphenylbromid beschickt. Wasserfreies Tetrahydrofuran (200 ml) wurde zu dem Kolben gegeben, wobei eine klare Lösung erhalten wurde. Die Lösung wurde unter Verwendung eines Eisbads auf 0 °C gekühlt. Bei dieser Temperatur wurden 125 ml einer 3,0 M Lösung von CH3MgBr in Ether (1,5 Äquivalente, 0,375 mol) langsam zu dem Reaktionskolben unter Verwendung eines Zugabetrichters gegeben. Das Reaktionsgemisch wurde sich langsam auf Raumtemperatur erwärmen gelassen und 48 h gerührt. Dünnschichtchromatographie (DC) zeigte, dass das Ausgangsmaterial verbraucht war. Nach 48 h wurde das Reaktionsgemisch auf –78 °C unter Verwendung ei nes Isopropanol/Trockeneisbads abgekühlt. Bei –78 °C wurden 50 ml einer 10,5 M Lösung von Butyllithium in Hexan (2,0 Äquivalente, 0,5 mol) zu dem Reaktionsgemisch unter Fortsetzen des Rührens gegeben. Weitere 400 ml THF wurden zugegeben, um sicherzustellen, dass das Reaktionsgemisch homogen war und gut gerührt wurde. Das Reaktionsgemisch wurde bei –78 °C 3 h gerührt. Zu dem Reaktionsgemisch wurden 170 ml Trimethylborat (6,0 Äquivalente, 1,5 mol) langsam unter Verwendung eines Zugabetrichters gegeben und die Temperatur wurde bei –78 °C gehalten. Unter Rühren wurde das Reaktionsgemisch sich über Nacht auf Raumtemperatur erwärmen gelassen. Das Fortschreiten der Reaktion wurde durch DC überwacht. Nach Kühlen des Reaktionsgemischs auf 0 °C (unter Verwendung eines Eisbads) wurde der Inhalt in ein 5-1-Becherglas überführt. Der Kolben wurde mit 100 ml Methanol gespült und die Waschflüssigkeit wurde mit dem Reaktionsgemisch vereinigt. Zu dem Reaktionsgemisch wurden 500 ml 1 N HCl langsam gegeben. Anschließend wurde der pH-Wert des Gemischs durch Zugabe von konzentrierter HCl auf 4 gebracht. Das Reaktionsgemisch wurde 3 h gerührt. Das organische Lösemittel wurde durch einen Rotationsverdampfer entfernt. Der konzentrierte wässrige Inhalt wurde mit Ether (250 ml × 6) exrahiert. Die vereinigte organische Schicht wurde mit Kochsalzlösung (200 ml × 2) gewaschen und über MgSO4 getrocknet. Nach Filtration wurde Ether durch den Rotationsverdampfer entfernt. Der Rückstand wurde aus heißem Wasser umkristallisiert, wobei ein weißlicher Feststoff erhalten wurde. Ausbeute: 22 g (50 %).
- Stufe 2. Synthese von 4-(2'-Bromacetyl)-3-fluorphenylboronsäure (2)
- Ein ofengetrockneter 500-ml-Dreihalsrundkolben wurde mit 5 g (27,4 mmol) 4-Acetyl-3-fluorphenylbromsäure und 25 ml Methanol unter Stickstoffatmosphäre beschickt. Die Lösung wurde unter Verwendung eines Eisbads auf 0 °C gekühlt. Zu dieser Lösung wurden 0,2 ml (0,55 Äquivalente) Eisessig gegeben. In einen 100-ml-Erlenmeyerkolben wurden 1,27 ml (3,95 g, 24 mmol, 0,9 Äquivalente) elementares Brom, das in 4 ml kaltem Methanol gelöst war, gegeben. Die Bromlösung wurde unter Verwendung eines Zugabetrichters tropfenweise zu der obigen Lösung bei 0 °C gegeben. Bei Zugabe von Br2 wurde die Lösung langsam hellorange und schließlich dunkelorange, als die Zugabe beendet war. Nach etwa 5–6 h wurde das Fortschreiten der Reaktion durch NMR überwacht. Abhängig vom Fortschreiten der Reaktion wurden weitere 10–20 Mol-% Brom nach Kühlen der Lösung auf 0 °C zugegeben. Die Gesamtreaktionszeit betrug etwa 24 h.
- Nach Beendigung der Reaktion wurde das Lösemittel unter Verwendung eines Rotationsverdampfers entfernt. Der Rückstand wurde in 200 ml Ethylacetat gelöst. Er wurde mit entionisiertem Wasser (50 ml × 3) und mit Kochsalzlösung (50 ml × 2) gewaschen. Die organische Schicht wurde gewonnen und über wasserfreiem Natriumsulfat 1 h getrocknet. Die Lösung wurde filtriert und das Lösemittel wurde unter Verwendung eines Rotationsverdampfers entfernt. Der Rückstand wurde aus heißem Ethylacetat umkristallisiert. Ausbeute = 7 g (97 %).
- Stufe 3. Synthese von 4-(14'-Hydroxy-3'-thia-1'-ketotetradecyl)-3-fluorphenylboronsäure (3)
- Ein ofengetrockneter 500-ml-Dreihalsrundkolben wurde mit 5 g (19,15 mmol) 4-(2'-Bromacetyl)-3-fluorphenylbromsäure (2) und 50 ml wasserfreiem THF beschickt. Die Lösung wurde mindestens 30 min mit N2 gespült. Zu dieser Lösung wurden 3,9 g (19,15 mmol, 1 Äquivalent) 11-Mercaptoundecanol gegeben. Unter Rühren unter N2 wurden 6,62 ml (38,3 mmol, 2 Äquivalente) Diisopropylethylamin langsam zugegeben. Das Reaktionsgemisch wurde 24 h bei Raumtemperatur unter Stickstoffatmosphäre gerührt. Das Fortschreiten der Reaktion wurde durch DC und NMR (nach Aufwaschen des Aliquots mit 1 N HCl) überwacht. Wenn die Reaktion nicht vollständig war, wurde weiteres 11-Mercaptoundecanol (nach Bedarf) zugegeben und die Reaktion weitere 24 h fortschreiten gelassen. Nach vollständiger Durchführung der Reaktion wurde das Lösemittel abgedampft. Der Rückstand wurde in 200 ml Ethylacetat gelöst und mit Wasser (50 ml × 3), 1 N HCl (50 ml × 3) und mit Kochsalzlösung (50 ml × 2) gewaschen. Die organische Schicht wurde 1 h über wasserfreiem Natriumsulfat getrocknet. Nach Filtration wurde das Lösemittel durch den Rotationsverdampfer entfernt. Der Rückstand wurde aus Ethylacetat umkristallisiert. Ausbeute = 5 g (72 %).
- Stufe 4. Synthese von Neopentylglykol-geschützter 4-(14'-Hydroxy-3'-thia-1'-ketotetradecyl)-3-fluorphenylboronsäure (4)
- Ein ofengetrockneter 500-ml-Dreihalsrundkolben wurde mit 5 g (13 mmol) von 3, das oben hergestellt wurde, beschickt. Die Zugabe von 100 ml wasserfreiem Dichlormethan ergab eine Dispersion. Unter Rühren wurden 1,42 g (13,65 mmol, 1,05 Äquivalente) Neopentylglykol zu dieser Dispersion gegeben. Nach wenigen Minuten wurde eine klare Lösung erhalten. Das gerührte Reaktionsgemisch wurde auf Rückflusstemperatur erhitzt. Eine Kühlvorrichtung und eine Dean-Stark-Vorrichtung wurden zur Entfernung des Dichlormethan/Wasser-Azeotrops verwendet. Das Erhitzen wurde etwa 3 h fortgesetzt.
- Am Ende des Refluxierens wurde das Reaktionsgemisch auf Raumtemperatur abkühlen gelassen und das Lösemittel unter Verwendung eines Rotationsverdampfers entfernt. Wasserfreies Toluol (50 ml) wurde zu dem Rückstand gegeben und das Toluol wurde unter Verwendung eines Rotationsverdamp fers entfernt. Dieses Toluolbehandlungsverfahren wurde noch einmal wiederholt. Der Rückstand wurde in 5 ml Dichlormethan gelöst und Hexan wurde zu dieser Lösung (unter Rühren) gegeben, bis eine Trübung erschien (etwa 150 ml). Die Lösung wurde zur Umkristallisation im Kühlschrank gehalten. Nach wenigen Stunden kristallisierte das Produkt und es wurde durch Filtration isoliert. Ausbeute = 5,13 g (87 %).
- Stufe 5. Synthese von Neopentylglykol-geschützter 4-(14'-Brom-3'-thia-1'-ketotetradecyl)-3-fluorphenylboronsäure (5)
- Die Reaktion wurde unter N2-Atmosphäre durchgeführt.
- Ein ofengetrockneter 500-ml-Dreihalsrundkolben wurde mit 5,13 g (11,33 mmol) der Neopentylglykol-geschützten Boronsäure (4) und 50 ml wasserfreiem Dichlormethan unter Stickstoffatmosphäre beschickt. Zu dieser Lösung wurden 7,52 g (22,67 mmol, 2 Äquivalente) Tetrabromkohlenstoff gegeben. Die gebildete Lösung wurde bei 0 °C unter Verwendung eines Eisbads gerührt. Eine Lösung von 5,95 g (22,67 mmol, 2 Äquivalente) Triphenylphosphin, das in 10 ml wasserfreiem Dichlormethan gelöst war, wurde langsam zu dem Reaktionsgemisch unter Verwendung eines Zugabetrichters gegeben. Das Reaktionsgemisch wurde bei 0 °C gerührt und sich langsam auf Raumtemperatur erwärmen gelassen. Die Gesamtreaktionszeit betrug etwa 24 h. Am Ende der Reaktion wurden 20 ml Methanol zu dem Reaktionsgemisch gegeben. Nach Rühren während 1 h wurde das Lösemittel durch den Rotationsverdampfer entfernt. Der Rückstand wurde mit 200 ml Diethylether behandelt und 30 min gerührt. Das Gemisch wurde filtriert und das Lösemittel wurde unter vermindertem Druck entfernt. Der Rückstand erhielt eine weitere Etherbehandlung auf die obige Weise und das Lösemittel wurde entfernt. Der gebildete Rückstand wurde unter Verwendung von Hexan/Ethylacetat (98/2) als Lösemittelsystem flashchromatographiert. Nach Entfernen des Lösemittels wurde das Produkt als weißlicher Feststoff isoliert. Ausbeute = 4,3 g (74 %).
- Stufe 6: Synthese von 4-(14'-Trimethylammonium-3'-thia-1'-ketotetradecyl)-3-fluorphenylboronsäurechlorid (6)
- Ein 100-ml-Rundkolben wurde mit 4,3 g (8,3 mmol) Boronsäurederivat (5) und 40 m 1 Ethanol beschickt. Zu dieser Lösung wurden 40 ml einer wässrigen Trimethylaminlösung (40 %, Aldrich) gegeben. Das Reaktionsgemisch wurde bei 70 °C 24 h gerührt. Nach Kühlen auf Raumtemperatur wurde das Ethanol durch den Rotationsverdampfer entfernt. Die verbliebene wässrige Lösung wurde auf 0 °C gekühlt und 180 ml 1 N HCl wurden langsam in die gerührte Lösung gegeben. Wenn eine Fällung auftritt, wird etwas Methanol zugegeben, bis sich eine klare Lösung bildet. Nach Rühren während 5 h wurde die Lösung (trüb) mit Chloroform (3 × 200 ml) extrahiert. Organische Schichten wurden gewonnen und über Natriumsulfat getrocknet. Das Chloroform wurde abgedampft und der Rückstand wurde in Methanol (20 ml) gelöst. Eine Natriumchloridlösung (10 % (Gew/Gew), 200 ml) wurde zu der Methanollösung gegeben und 1 h gerührt. An diesem Punkt wurde das organische Lösemittel unter Verwendung eines Rotationsverdampfers entfernt und die Verbindung aus der wässrigen Lösung in Chloroform (3 × 200 ml) extrahiert. Organische Schichte wurden gewonnen und über Natriumsulfat getrocknet. Nach Filtration wurde das Lösemittel unter Verwendung eines Rotationsverdampfers entfernt. Der Rückstand wurde zu 600 ml Ether gegeben und das Gemisch wurde 3 h im Tiefkühlgerät gehalten. Das Lösemittel wurde abdekantiert, wobei das Produkt isoliert wurde. Ausbeute = 2 g.
- Beispiel 2 - Synthese von 2,5-Difluor-4-(14'-hydroxy-3'-thia-1'-ketotetradecyl)phenylboronsäure (11)
- Die Synthese der Verbindung (11) ist schematisch in
2 angegeben. Eine detaillierte Beschreibung des Verfahrens wird im folgenden bereitgestellt. - Stufe 1 - Synthese von 4-Brom-2,5-difluoracetophenon (7)
- Wasserfreies Aluminiumchlorid (5 g, 37,5 mmol, 2,4 Äquivalente) wurde mit 1-Brom-2,5-difluorbenzol in einem trockenen Rundkolben, der mit Stickstoff gespült und mit einem Kühler ausgestattet war, gemischt. Das Gemisch wurde auf 60 °C erhitzt und Acetylchlorid (1,7 ml, 23,3 mmol, 1,5 Äquivalente) wurde durch eine Spritze zugegeben. Der feuchte gelbe Feststoff änderte sich zu einer scharlachroten Lösung und wurde 1 h bei 90 °C erhitzt. Das Reaktionsgemisch wurde auf 38 g Eis gegossen, HCl wurde zugegeben (3 ml, Konzentration 37 %) und das Gemisch wurde mit Ether extrahiert. Das rohe Material wurde über Magnesiumsulfat getrocknet und abgedampft. Das rohe Material wurde durch Säulenchromatographie gereinigt oder destilliert. Das Produkt (1,2 g, 31 %) wurde als gelbes Öl erhalten.
- Stufe 2 - Synthese von Neopentylglykol-geschützter 4-Acetyl-2,5-difluorfluorphenylboronsäure (8)
- Dichlor-[(1,1'-bis(diphenylphosphino)ferrocen]palladium(II)-Dichlormethan-Addukt (1,7 g, 2,3 mmol, 5 Mol-%) wurde zu einer Suspension von 4-Brom-2,5-difluoracetophenon (7) (10,5 g, 46,38 mmol, 1 Äquivalente), Bis(neopentylglykolato)dibor (12,57 g, 55,65 mmol, 1,2 Äquivalente) und Kaliumacetat (13,66 g, 139,13 mmol, 3 Äquivalente) in wasserfreiem DMSO (100 ml) gegeben. Die Suspension wurde 1 h unter Stickstoff auf 80 °C erhitzt (J. Org. Chem. 60: 7508 (1995)). Nach 1 h zeigte DC die vollständige Umwandlung des Ausgangsmaterials und das Reaktionsgemisch wurde sich abkühlen gelassen und mit Toluol extrahiert, dreimal mit Wasser gewaschen und über Magnesiumsulfat getrocknet. Flashsäulenchromatographie wurde zur Reinigung des Rohprodukts (4,2 g, 32 %) verwendet.
- Stufe 3 - Synthese von Neopentylglykol-geschützter 4-(2'-Bromacetyl-2,5-difluorphenylboronsäure (9)
- Der Boronester (8) (4,1 g, 14,93 mmol, 1 Äquivalent) wurde in Methylenchlorid (50 ml) gelöst und auf –10 °C abgekühlt. Essigsäure (0,82 ml, 14,32 mmol, 1 Äquivalent) und anschließend Brom (0,7 ml, 13,4 mmol, 0,9 Äquivalente) wurden zugegeben und das Reaktionsgemisch wurde sich auf Raumtemperatur erwärmen gelassen. Nach Rühren während 2 h wurde das Reaktionsgemisch mit weiterem Methylenchlorid verdünnt und einmal mit Wasser und einmal mit Kochsalzlösung gewaschen. Das Rohprodukt wurde über Magnesiumsulfat getrocknet, abgedampft und in der nächsten Stufe ohne weitere Reinigung verwendet.
- Stufe 4 - Synthese von Neopentylglykol-geschützter 2,5-Difluor-4-(14'-hydroxy-3'-thia-1'-ketotetradecyl)phenylboronsäure (10)
- Die rohe Verbindung (9) (14,93 mmol) wurde in wasserfreiem Methanol (50 ml) gelöst und Stickstoffgas wurde 20 min in die Lösung perlen gelassen, um das Gemisch gasfrei zu machen. 11-Mercaptoundecanol (3,1 g, 14,93 mmol, 1 Äquivalent) wurde zu dem Reaktionsgemisch gegeben und die Lösung wurde unter Stickstoff 5 min gerührt, bevor wasserfreies Diisopropylamin (5,2 ml, 29,9 mmol, 2 Äquivalente) zugegeben wurde. Das Reaktionsgemisch wurde über Nacht unter Stickstoff gerührt und das Rohprodukt wurde durch Eindampfen des Reaktionsgemischs zur Trockne und erneutes Lösen desselben in einem 10 %-igen Gemisch von THF in Ethylacetat (100 ml) aufgearbeitet. Diese organische Schicht wurde dann mit 200 ml Wasser gewaschen und die wässrige Schicht wurde abgetrennt und mit 3 neuen Fraktionen des gleichen THF/Ethylacetat-Gemischs (jeweils 100 ml) gewaschen. Die rohen organischen Schichten wurden vereinigt, über Magnesiumsulfat getrocknet und eingedampft. Flashchromatographie wurde zur Reinigung des Rohprodukts verwendet und es wurde ein weißlicher Feststoff erhatlen (3,5 g, 50 %).
- Stufe 5 - Synthese von 2,5-Difluor-4-(14'-hydroxy-3'-thia-1'-ketotetradecyl)phenylboronsäure (11)
- Das Entschützen der Neopentylgruppe in Verbindung (10) unter Bildung der Verbindung (11) wurde durch Lösen der Verbindung (10) in Methanol und Zugabe von einigen Tropfen HCl durchgeführt. Nach Rühren während etwa 1 h wurde das rohe Produkt auf einem Rotationsverdampfer eingeengt und das Endprodukt aus heißem Ethylacetat umkristallisiert.
- Beispiel 3 - Synthese von 2,5-Difluor-4-(13'-trimethylammonium-3'-thia-1'-ketotridecyl)phenyl(neopentylglykolato)borchlorid (14)
- Die Synthese der Verbindung (14) ist schematisch in
3 angegeben. Eine detaillierte Beschreibung des Verfahrens wird im folgenden bereitgestellt. - Stufe 1 - Synthese von 10-Bromdecyltrimethylammoniumbromid
- 1,10-Dibromdecan (20 g, 66,7 mmol) und THF (100 ml) wurden in einen 500-ml-Dreihalskolben gegeben. Die Lösung wurde mit einem Eis/Wasserbad auf 0 °C gekühlt. Wasserfreies Trimethylamin (3 g, 50,8 mmol) wurde zu dem Gemisch durch langsames Einperlen von Triethylamingas während etwa 15 min zugegeben. Dann wurde das Reaktionsgemisch sich auf Raum temperatur erwärmen gelassen und über Nacht bei Raumtemperatur gerührt. Das feste Material wurde abfiltriert und mit THF (5 × 30 ml) gewaschen. Nach Trocknen unter Vakuum über Nacht wurden 12,5 g (34,82 mmol, 69 %, bezogen auf das verwendete Amin) des Produkts als weißer Feststoff erhalten
- Stufe 2 - Synthese von 10-Mercaptodecyltrimethylammoniumbromid
- 10-Bromdecyltrimethylammoniumbromid (10 g, 27,9 mmol) in 50 ml Methanol wurde in einen 250-ml-Dreihalskolben gegeben. Das Gemisch wurde durch Durchperlen von Stickstoff während 30 min kräftig luftfrei gemacht. Kaliumthioacetat (3,8 g, 33,5 mmol, 1,2 Äquivalente) wurde zu dem Reaktionsgemisch gegeben. Das Gemisch wurde bei 50 °C 12 h unter Stickstoff erhitzt. Das Reaktionsgemisch wurde mit einem Eis/Wasserbad auf 0 °C gekühlt, luftfrei gemachtes Natriumhydroxid (50 %, 2,7 g, 33,5 mmol, 1,2 Äquivalente) wurde zugegeben und das Gemisch wurde 1 h bei Raumtemperatur gerührt. Das Gemisch wurde auf 0 °C gekühlt und luftfrei gemachte konzentrierte Salzsäure wurde tropfenweise zugegeben, um einen pH-Wert von 2 zu erreichen. Luftfrei gemachtes Methanol (100 ml) wurde zu dem Reaktionsgemisch gegeben, worauf die Zugabe von 40 g Magnesiumsulfat folgte. Magnesiumsulfat wurde abfiltriert und mit Methanol gewaschen. Die Methanollösung wurde auf etwa 20 ml eingeengt und Ether (300 ml) wurde zu dem Gemisch gegeben. Der Kolben wurde fest verschlossen und in ein Tiefkühlgerät gegeben. Das Produkt kristallisierte als weißer Feststoff aus. Das Produkt wurde filtriert, mit Ether gewaschen und unter Vakuum getrocknet. Produkt (7,5 g, 24,0 mmol, 86 %) wurde als weißer hydroskopischer Feststoff erhalten.
- Stufe 3 - Synthese von 2,5-Difluor-4-(13'-trimethylammonium-3'-thia-1'-ketotridecyl)fluorphenylboronsäurebromid
- 4-(2'-Bromacetyl)-2,5-difluorphenyl(neopentylglykolato)bor (Verbindung 9) (1 mmol) wurde in wasserfreiem Methanol (10 ml) gelöst und Stickstoffgas wurde 20 min in die Lösung perlen gelassen, um das Gemisch luftfrei zu machen. 10-Mercaptodecyltrimethylammoniumbromid (0,19 g, 0,8 mmol, 0,8 Äquivalente) wurde zu dem Reaktionsgemisch gegeben und die Lösung wurde 5 min unter Stickstoff gerührt, bevor wasserfreies Diisopropylamin (0,14 ml, 1 mmol, 1 Äquivalent) zugegeben wurde. Das Reaktionsgemisch wurde über Nacht unter Stickstoff gerührt und nach Einengen auf einem Rotationsverdampfer durch präparative Umkehrphasen-PLC gereinigt.
- Beispiel 4 Synthese von 4-(14'-Trimethylammonium-3'-thia-1'-keto-tetradecyl)phenylboronsäurechlorid

- Stufe 1. Synthese von 4-(2'-Bromacetyl)phenylboronsäure
- Ein ofengetrockneter 2-1-Dreihalsrundkolben wurde mit 4-Acetyl-phenylboronsäure (20 g, 0,152 mol) beschickt. Unter Rühren wurden 175 ml THF und anschließend 700 ml Chloroform zu dem Reaktionsgemisch gegeben. Zu der gebildeten Lösung wurden 5 ml Eisessig gegeben. Die Chloroformlösung von Brom (die durch Lösen von 7 ml Brom in 30 ml Chloroform hergestellt wurde) wurde langsam zu dem Reaktionsgemisch bei etwa 5 °C gegeben. Nach Beendigung der Zugabe von Brom wurde das Reaktionsgemisch sich auf Raumtemperatur erwärmen gelassen und 16 h bei Raumtemperatur gerührt. Das Lösemittel wurde durch Rotationsverdampfung entfernt und der Rückstand wurde in 1 1 Ethylacetat gelöst. Die gebildete Lösung wurde mit entionisiertem Wasser (3 × 200 ml) und Kochsalzlösung (2 × 100 ml) extrahiert. Die organische Schicht wurde über wasserfreiem Natriumsulfat 1 h getrocknet. Die Lösung wurde dann filtriert und auf etwa 1/3 von deren Volumen eingeengt. Die gebildete Lösung wurde in einem Tiefkühlgerät gehalten, um das Produkt zu kristallisieren. Der Feststoff wurde abfiltriert, wobei ein weißlicher Feststoff erhalten wurde. Ausbeute = 16 g.
- Stufe 2. 4-(14'-Hydroxy-3'-thia-1'-ketotetradecyl)phenylboronsäure
- Ein 500-ml-Dreihalsrundkolben wurde mit 15 g 4-(2'-Bromacetyl)phenylboronsäure und 300 ml wasserfreiem THF beschickt. Unter Rühren unter Stickstoffatmosphäre wurden 12,26 g 11-Mercaptoundecanol und anschließend 32,35 ml Diisopropylethylamin zu dem Reaktionsgemisch gegeben. Das Reaktionsgemisch wurde 48 h unter Stickstoffatmosphäre gerührt. Nach Entfernen des Lösemittels durch Rotationsverdampfung wurde der Rückstand in 500 ml Ethylacetat gelöst. Die organische Phase wurde mit entionisiertem Wasser (2 × 200 ml), 1 N HCl (3 × 200 ml), entionisiertem Wasser (200 ml) und Kochsalzlösung (200 ml) gewaschen. Die gewaschene organische Schicht wurde dann 15 min unter wasserfreiem Natriumsulfat getrocknet. Die Lösung wurde filtriert und auf 1/4 von deren Volumen eingeengt. Unter Rühren wurde Hexan langsam zu dieser Lösung gegeben, bis eine permanente Trübung auftrat. Die Lösung wurde im Tiefkühlgerät gehalten, um das Produkt zu kristallisieren. Nach Filtration wurde der Rückstand unter Vakuum bei Raumtempe ratur getrocknet, wobei 17 g des Produkts als weißlicher Feststoff erhalten wurden.
- Stufe 3. Synthese von (Neopentylglykolato)-4-(14'-hyxdroxy-3'-thia-1'-ketotetradecyl)phenylboronatester
- Ein ofengetrockneter 500-ml-Dreihalsrundkolben wurde mit 5 g 4-(14'-Hydroxy-3'-thia-1'-ketotetradecyl)phenylboronsäure und 100 ml wasserfreiem Dichlormethan beschickt. Unter Rühren wurde 1 g Neopentylglykol zugegeben und das Reaktionsgemisch unter Rühren auf Rückflusstemperatur erhitzt. Das Erhitzen wurde 3 h unter azeotroper Destillation von Wasser fortgesetzt. Das Reaktionsgemisch wurde sich auf Raumtemperatur abkühlen gelassen und das Lösemittel wurde unter Verwendung eines Rotationsverdampfers entfernt. Wasserfreies Toluol (50 ml) wurde zu dem Rückstand gegeben und das Toluol wurde unter Verwendung eines Rotationsverdampfers entfernt. Dieses Toluolbehandlungsverfahren wurde noch einmal wiederholt. Der Rückstand wurde in 5 ml Dichlormethan gelöst und Hexan wurde zu dieser Lösung (unter Rühren) gegeben, bis Trübung auftrat. Die Lösung wurde zur Umkristallisation in dem Tiefkühlgerät gehalten. Das Produkt wurde durch Filtration isoliert und beim Trocknen wurden 4,8 g der Verbindung als weißlicher Feststoff erhalten.
- Stufe 4. Synthese von (Neopentylglykolato)-4-(14'-brom-3'-thia-1'-ketotetradecyl)phenylboronatester
- Ein ofengetrockneter 500-ml-Dreihalsrundkolben wurde mit 5,13 g des (Neopentylglykolato)-4-(14'-hyxdroxy-3'-thia-1'-ketotetradecyl)phenylboronatesters und 50 ml wasserfreiem Dichlormethan beschickt. Zu dieser Lösung wurden 7,52 g Tetrabromkohlenstoff gegeben und das gebildete Reaktionsgemisch wurde unter Verwendung eines Eisbads bei 0 °C gerührt. Eine Lösung von 5,95 g Triphenylphosphin, das in 10 ml wasserfreiem Dichlormethan gelöst war, wurde unter Verwendung eines Zugabetrichters langsam zu dem Reaktionsgemisch gegeben. Das Reaktionsgemisch wurde bei 0 °C gerührt und dann sich langsam auf Raumtemperatur erwärmen gelassen. Nach 16 h wurden 20 ml Methanol zu dem Reaktionsgemisch gegeben. Nach Rühren während 1 h wurde das Lösemittel durch einen Rotationsverdampfer entfernt. Der Rückstand wurde mit 200 ml Diethylester behandelt und 30 min gerührt. Das Gemisch wurde filtriert und das Lösemittel wurde unter vermindertem Druck entfernt. Der Rückstand wurde erneut mit Ether behandelt und das Lösemittel wurde entfernt. Der gebildete Rückstand wurde unter Verwendung von Hexan/Ethylacetat (98/2) flashchromatographiert. Nach Entfernen des Lösemittels wurde das Produkt als weißlicher Feststoff isoliert (Ausbeute = 4,5 g).
- Stufe 5. 4-(14'-Trimethylammonium-3'-thia-1'-ketotetradecyl)phenylboronsäurechlorid
- Ein 100-ml-Rundkolben wurde mit 500 mg (Neopentylglykolato)-4-(14'-brom-3'-thia-1'-ketotetradecyl)phenylboronatester und 5 ml Ethanol beschickt. Zu dieser Lösung wurden 5 ml einer 40 %-igen wässrigen Triethylaminlösung gegeben. Das Reaktionsgemisch wurde 24 h bei 70 °C gerührt. Nach Abkühlen auf Raumtemperatur wurde das Lösemittel unter vermindertem Druck entfernt. Der Rückstand wurde in 5 ml Methanol und 20 ml 2 N HCl gelöst. Nach Rühren während 24 h wurde die Lösung mit Ethylacetat (2 × 100 ml) zum Entfernen des Neopentylglykols extrahiert. Die wässrige Lösung wurde mit Chloroform (3 × 50 ml) extrahiert. Die Chloroformextrakte wurden vereinigt und über MgSO4 getrocknet. Nach Filtration wurde das Lösemittel unter vermindertem Druck entfernt und der Rückstand unter Vakuum getrocknet, wobei 300 mg eines gummiartigen Feststoffs erhalten wurden.
- Beispiel 5 Synthese von (Neopentylglykolato)-4-(14'-dimethylamino-3'-thia-1'-ketotetradecyl)phenylboronatester

- Ein ofengetrockneter 250-ml-Dreihalsrundkolben wurde mit 2,5 g des (Neopentylglykolato)-4-(14'-Brom-3'-thia-1'-ketotetradecyl)phenylboronatesters (hergestellt gemäß der Beschreibung in Beispiel 4, Stufe 4) und 25 ml wasserfreiem Tetrahydrofuran (THF) beschickt. Zu diesem Gemisch wurden 8 ml 2 M Dimethylamin in THF gegeben. Nach Rühren bei Raumtemperatur während 48 h wurde das Lösemittel unter vermindertem Druck entfernt. Der Rückstand wurde 1 h mit 100 ml einer 5 %-igen wässrigen Natriumbicarbonatlösung gerührt und dann mit Ethylacetat (2 × 200 ml) extrahiert. Nach Trocknen über wasserfreiem Natriumsulfat wurde das Lösemittel unter vermindertem Druck entfernt, wobei 1,7 g der Verbindung als gummiartiger Feststoff erhalten wurden.
- Beispiel 6 Synthese von 4-{14'-(3''-Chlortrimethylammonium)dimethylpropylammonium-3'-thia-1'-ketotetradecyl}phenylboronsäurechlorid

- Ein 100-ml-Rundkolben wurde mit 700 mg (Neopentylglykolato)-4-(14'-dimethylamino-3'-thia-1'-ketotetradecyl)phenylboronatester (hergestellt gemäß der Beschreibung in Beispiel 5), 400 mg 3-Brompropyltrimethylamminiumbromid und 10 ml Ethanol beschickt. Das Reaktionsgemisch wurde 24 h bei 70 °C gerührt. Nach Kühlen auf Raumtemperatur wurde das Lösemittel unter vermindertem Druck entfernt. Der Rückstand wurde in 5 ml Methanol und 40 ml 2 N HCl gelöst. Nach Rühren während 24 h wurde die Lösung mit Ethylacetat (2 × 100 ml) zur Entfernung von Neopentylglykol extrahiert. Die angesäuerte wässrige Lösung wurde in dem Tiefkühlgerät gehalten. Der ausgefallene Feststoff wurde dann durch Entfernen des Lösemittels isoliert und unter Vakuum getrocknet, wobei 400 mg eines niedrigschmelzenden Feststoffs erhalten wurden.
- Beispiel 7 Synthese von 4-(14'-Sulfato-3'-thia-1'-ketotetradecyl)phenylboronsäure-natriumsalz
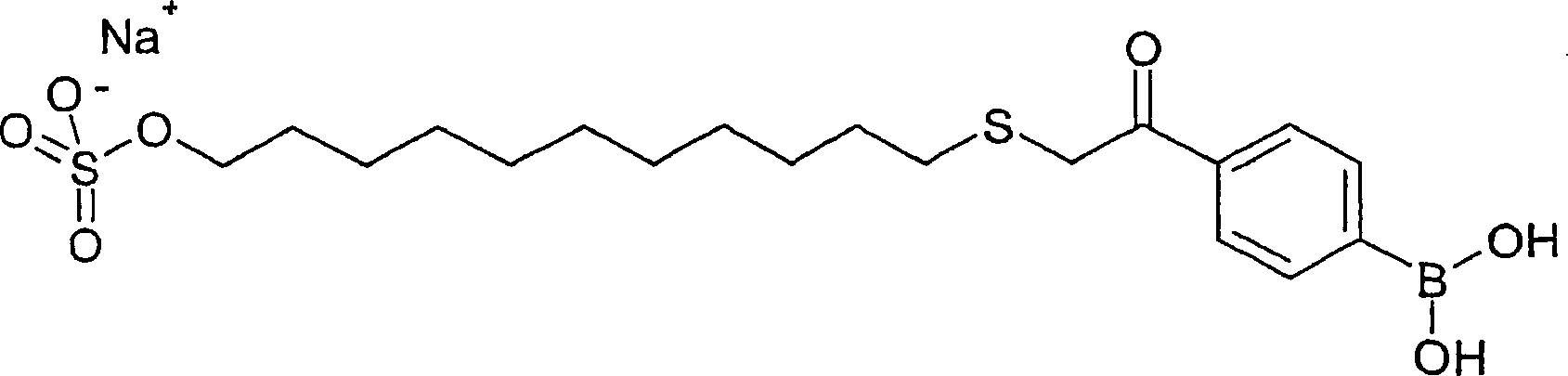
- Ein 100-ml-Rundkolben wurde mit 3 g 4-(14'-Hydroxy-3'-thia-1'-ketotetradecyl)phenylboronsäure (hergestellt gemäß der Beschreibung in Beispiel 4, Stufe 2) und 25 ml N,N-Dimethylformamid (DMF) beschickt. Zu dieser Lösung wurden 1,6 g Schwefeltrioxid: DMF-Komplex gegeben und das gebildete Reaktionsgemisch wurde 24 h bei Raumtemperatur gerührt. Zu dem Reaktionsgemisch wurde eine Lösung von 2 g NaOH, das in 100 ml eines Wasser: Methanol-Gemischs (1:1) gelöst war, gegeben und 1 h gerührt. Das Lösemittel wurde unter Druck entfernt und der Rückstand wurde mit 100 ml Methanol behandelt. Nach Rühren während 1 h wurde das Reaktionsgemisch abfiltriert. Das Filtrat wurde am Rotationsverdampfer zur Trockne eingedampft, wobei 1,5 g eines weißlichen Feststoffs erhalten wurden.
- Beispiel 8 Herstellung von 4-(11'-Hydroxyundecyl)carboxyphenylboronsäure

- Ein Gemisch von 4-Carboxyphenylboronsäure (1,0 g), Kaliumhydrogencarbonat (2,01 g), 11-Brom-1-undecanol und N,N-Dimethylformamid (60 ml) wurde 18 h bei 60 °C unter Stickstoffatmosphäre erhitzt. Nach der Heizperiode wurde das Gemisch sich auf Raumtemperatur abkühlen gelassen. Das Gemisch wurden dann filtriert und das Filtrat wurde auf einem Rotationsverdampfer eingeengt. Das eingeengte Filtrat wurde mit Ethylacetat (500 ml) verdünnt und das Ethylacetat wurde nacheinander mit gesättigtem wässrigem Natriumbicarbonat (3 × 300 ml) und anschließend gesättigtem wässrigem Natriumchlorid (300 ml) gewaschen. Nach Trocknen über Natriumsulfat wurde der Ethylacetatextrakt auf einem Rotationsverdampfer eingeengt und unter vermindertem Druck getrocknet, wobei 2,2 g des gewünschten Produkts als hellgelbes viskoses Öl, das sich bei Stehenlassen zu einem weißen Pulver verfestigte, erhalten wurden.
- Die folgenden Verbindungen wurden unter Verwendung ähnlicher Verfahren synthetisiert:aus 4-Carboxyphenylboronsäure und Iodoctadecan;
 aus 4-Carboxyphenylboronsäure und Docosylmethansulfonat;
aus 4-Carboxyphenylboronsäure und Docosylmethansulfonat; aus Bromoctadecan und (3-Carboxy-5-nitrophenyl)boronsäure;
aus Bromoctadecan und (3-Carboxy-5-nitrophenyl)boronsäure; aus 1-Bromdecan und (3-Carboxyphenyl)boronsäure;
aus 1-Bromdecan und (3-Carboxyphenyl)boronsäure; aus 4-Carboxyphenylboronsäure und (4-Chlorpropyl)dimethyloctadecylammoniumbromid und
aus 4-Carboxyphenylboronsäure und (4-Chlorpropyl)dimethyloctadecylammoniumbromid und aus 4-Carboxyphenylboronsäure und Pentaethylenglykolmonotosylat.
aus 4-Carboxyphenylboronsäure und Pentaethylenglykolmonotosylat.
- Beispiel 9 Synthese von [4-(N,N-Dioctadecylcarbamoyl)phenyl]boronsäure
- Stufe 1. Synthese von 2-(4-Carboxyphenyl)-1,3-dioxa-2-borinan

- Ein Gemisch von 4-Carboxyphenylboronsäure (5,0 g) und 1,3-Propandiol (2,5 g) in Toluol (300 ml) wurde 6 h mit einer Dean-Stark-Vorrichtung refluxiert. Nach der Heizperiode wurde die Reaktionslösung auf einem Rotationsverdampfer eingeengt und unter vermindertem Druck getrocknet, wobei 6,39 g des gewünschten Produkts als weißer Feststoff erhalten wurden.
- Stufe 2. Synthese von 2-(4-Carbonylchlorid)-1,3-dioxa-2-borinan
- Zu einer Lösung der obigen Propandiol-geschützten 4-Carboxyphenylboronsäure (1,0 g) in Chloroform (5 ml) wurden Thionylchlorid (3,0 ml) und Dimethylformamid (100 μl) gegeben. Die Lösung wurde 2 h auf Rückflusstemperatur erhitzt. Nach der Heizperiode wurde die Reaktionslösung sich auf Raumtemperatur abkühlen gelassen und auf einem Rotationsverdampfer unter vermindertem Druck eingeengt. Zu dem Rückstand wurde Chloroform (8 ml) gegeben und die gebildete Lösung wurde auf einem Rotationsverdampfer eingeengt. Die Zugabe von Chloroform (8 ml) und das Einengen der Lösung wurde zweimal wiederholt. Das rohe Material wurde unter Vakuum getrocknet, wobei 1,09 g des gewünschten Produkts als weißlicher Feststoff erhalten wurden.
- Stufe 3. Synthese von [4-(N, N-Dioctadecylcarbamoyl)phenyl]boronsäure
- Zu einer Lösung von 2-(4-Carbonylchlorid)-1,3-dioxa-2-borinan (0,8 g) in Chloroform (30 ml) unter Stickstoff wurden Dioctadecylamin (1,93 g), Triethylamin (1,0 ml) und Chloroform (10 ml) gegeben. Das Reaktionsgemisch wurde über Nacht gerührt, wonach es mit Chloroform (200 ml) verdünnt wurde. Die Chloroformlösung wurde in einem Scheidetrichter aufeinanderfolgend mit den folgenden wässrigen Lösungen: 10 %-ige HCl (3 × 100 ml), gesättigtes Natriumbicarbonat (3 × 100 ml) und gesättigtes Natriumchlorid (100 ml) gewaschen. Der Chloroformextrakt wurde über Natriumsulfat getrocknet, 2,41 g des rohen Materials wurden nach Filtration und Einengen auf einem Rotationsverdampfer unter vermindertem Druck isoliert. Das gewünschte Produkt wurde über Säulenchromatographie über Silicagel unter Verwendung eines Gemischs von Ethylacetat und Hexan als Elutionsmittel gereinigt.
- Beispiel 10 Synthese von 4-(13'-Carboxy-3'-thia-1'-ketotridecyl)phenylboronsäure

- Ein 100-ml-Dreihalskolben wurde mit 4-(2'-Bromacetyl)phenylboronsäure (0,95 g, 3,91 mmol) und 20 ml THF beschickt. Das Gemisch wurde durch Durchperlenlassen von Stickstoff durch das Reaktionsgemisch während etwa 20 min luftfrei gemacht. 11-Mercaptoundecansäure (0,9 g, 4,1 mmol) wurde zu dem Reaktionsgemisch unter Rühren unter Stickstoff gegeben. Diisopropylethylamin (1,52 g, 2,05 ml, 11,8 mmol) wurde dann über 5 min über eine Spritze zugegeben. Das Reaktionsgemisch wurde 72 h unter Stickstoff bei Raumtemperatur gerührt. Das Lösemittel wurde unter Vakuum entfernt und der Rückstand wurde zwischen Ethylacetat (100 ml) und Wasser (100 ml) verteilt. Der organische Extrakt wurde mit 1 N Salzsäure (3 × 100 ml), Wasser (100 ml) und Kochsalzlösung (100 ml) gewaschen. Der organische Extrakt wurde über Magnesiumsulfat getrocknet und dann filtriert. Das Filtrat wurde dann unter Vakuum eingeengt. Der Rückstand wurde in etwa 25 ml heißem Ethylacetat gelöst. Wenn das Gemisch auf Raumtemperatur gekühlt war, wurde es in ein Tiefkühlgerät gegeben. Produkt kristallisierte aus der Lösung. Das weiße kristalline Material wurde filtriert, mit kaltem Ethylacetat gewaschen und unter Vakuum getrocknet. 0,93 g (2,45 mmol) des reinen Produkts wurden erhalten. Ausbeute: 62,5 %.
- Beispiel 11 - Phenylboronsäuren der vorliegenden Erfindung hemmen Lipolyse in vitro
- Ein In-vitro-Assay der Pankreaslipaseaktivität wurde zur Ermittlung der Wirksamkeit von lipasehemmenden Verbindungen verwendet. Schweinepankreaslipase (23 Einheiten/ml) wurde 4 h bei 37 °C mit 72 mM Triglycerid (als Olivenöl/Gummiarabicum-Emulsion) in 5,5 ml eines 300 mM BES-Puffers, pH 7,0, der 10 mM CaCl2, 109 mM NaCl und 8 mM Natriumtaurocholat enthielt, inkubiert. Die Reaktion wurde durch Ansäuern mit HCl gestoppt und die Lipide wurden durch das Verfahren gemäß der Offenbarung in Folch et al., J. Biol. Chem. 226: 497 (1957) vor der Analyse durch HPLC extrahiert. Ein Aliquot der Chloroformschicht wurde eingedampft und in Hexan rekonstituiert und die Probe wurde auf einer Waters Alliance 2690 HPLC mit einem Sedex 55 Evaporative Light Scattering Detector unter Verwendung einer YMC PVA Sil 3 × 50 mm-Säule analysiert. Die mobile Phase bestand aus Hexan und Methyl-tert-butylether, die in einem linearen Gradienten mit einer Durchflussrate von 0,5 ml/min zugeführt wurde. Externe Standards wurden zur quantitativen Bestimmung von Triglyceriden, Diglyceriden und Fettsäuren verwendet und die prozentuale Lipolyse wurde bestimmt. Zur Beurteilung der Lipaseinhibitorwirksamkeit wurden Verbindungen in DMSO oder einem anderen geeigneten Lösemittel gelöst und vor der Inkubation direkt zu dem Testgemisch gegeben. Eine Hemmung wurde bezogen auf eine Kontrollinkubation bestimmt und IC50-Werte wurden aus einer Auftragung % Hemmung gegen Inhibitorkonzentration berechnet. Die Ergebnisse sind in der Tabelle angegeben. Es ist ersichtlich, dass die Boronsäureverbindungen der vorliegenden Erfindung wirksame Lipaseinhibitoren sind.
- Beispiel 12 - Phenylboronsäuren der vorliegenden Erfindung hemmen Lipolyse in vivo
- Verbindungen wurden bei Ratten zur Bestimmung von deren In-vivo-Wirksamkeit zur Hemmung der Fettabsorption durch Lipasehemmung beurteilt. Ratten werden etwa 1 Woche in individuellen Drahtbodenkäfigen an die Einrichtung akklimatisieren gelassen und mit einer Standardkaunahrung und Wasser nach Belieben versorgt. Die Ratten wurden dann willkürlich Gruppen von 4 zugeordnet. Sie erhielten durch Zwangsernährung um 7–8 Uhr vormittags 4 ml Olivenöl, das mit Gummiarabicum emulgiert war, mit oder ohne Arzneistoff nach 18 h ohne Nahrungsaufnahme. Testverbindungen wurden in DMSO oder entionisiertem Wasser gelöst. Arzneistofflösungen wurden unmittelbar vor der Verabreichung sorgfältig in die Olivenölemulsion eingemischt. Nach 8 h wurden die Ratten mit CO2 euthanasiert und der Darm entfernt. Der Darminhalt wurde aus der unteren Hälfte des Dünndarms und dem Zökum gewonnen. Der Inhalt wurde in getrennte, vorgewogene konische 15-ml-Schraubkappenröhrchen in einem Trockeneis/Alkohol-Bad zum Beibehalten von Gefriertemperatur bis zum endgültigen Einfrieren aller Proben gegeben. Die Proben wurden bei –80 °C bis zur Gefriertrocknung aufbewahrt.
- Die Proben wurde gefriergetrocknet und gemahlen, und dann auf Triglycerid und Fettsäure analysiert.
- Ein 20-mg-Aliquot jeder Probe wurde abgewogen und in ein konisches Röhrchen von 15 ml überführt. 3 ml Hexan wurden zu jedem Röhrchen gegeben. Dieses wurde dann verschlossen und 15 s mit hoher Geschwindigkeit verwirbelt. 3 ml 1 N HCl wurden zugegeben und dann wurden die Proben 1 h einer Handgelenkschüttelwirkung ausgesetzt. Die Proben wurden dann 5 min mit 3500 rpm zentrifugiert und die Hexanschicht wurde gewonnen. Ein Aliquot der Hexanschicht wurde in Hexan verdünnt und auf Triglycerid, Diglycerid und Fettsäure durch HPLC wie oben beschrieben verdünnt.
- Die Daten wurden wie im folgenden ausgedrückt. Die Milligramm des Darminhalts, die extrahiert wurden, und die Gesamtzahl der gesammelten Milligramm wurden aufgezeichnet. Die durch die HPLC-Analyse erhaltenen Milligramm/Milliliter-Werte wurden eingetragen. Die individuellen Lipidkomponenten wurden berechnet und als die gesamten gewonnenen Milligramm ausgedrückt. Dosierungseinheiten sind als Milligramm Arzneistoff pro Gramm Öl, die jeder Ratte verabreicht wurden, ausgedrückt. Die ED50-Werte wurden durch Extrapolation des Dosiswerts bei der Hälfte der maximal erreichbaren Triglyceridmenge, die in dem Test rückgewinnbar ist, bestimmt. Die Ergebnisse sind in der Tabelle angegeben. Wie ersichtlich ist, sind die Boronsäureverbindungen der vorliegenden Erfindung wirksame Lipaseinhibitoren in vivo.
- Tabelle: Hemmung von Lipolyse in vitro und in vivo

- Diese Erfindung wurde zwar speziell unter Bezug auf bevorzugte Ausführungsformen derselben angegeben und beschrieben, doch ist dem Fachmann klar, dass verschiedene Änderungen im Hinblick auf Form und Einzelheiten hierin ohne Abweichen vom Schutzumfang der Erfindung, der von den angehängten Ansprüchen umfasst wird, durchgeführt werden können.
Claims (30)
- Verbindung der folgenden Strukturformel:oder ein pharmazeutisch akzeptables Salz derselben, worin: der Phenylring A substituiert oder unsubstituiert ist; X für -CZ''2-, -CHZ''-, -CO- oder -SO2- steht; R für eine substituierte oder unsubstituierte geradkettige Hydrocarbylgruppe steht, die optional eine oder mehrere Amin-, Ammonium-, Ether-, Thioether- oder Phenylenverknüpfungsgruppen umfasst; Y für -H, ein Amin, -[NH-(CH2)q]r-NH2, Halogen, -CF3, Thiol, Ammonium, -OH, -COOH, -SO3H, -OSO3H oder eine Phosphoniumgruppe, die kovalent an die terminale Position von R gebunden sind, steht, wobei, wenn Y -H ist und R eine geradkettige Hydrocarbylgruppe ist, R dann 1 bis 30 Kohlenstoffatome aufweist, wobei jedes -NH- in -[NH-(CH2)q]r-NH2 optional N-alkyliert oder N,N-dialkyliert ist und -NH2 in -[NH-(CH2)q]r-NH2 optional N-alkyliert, N,N-dialkyliert oder N,N,N-trialkyliert ist; Z'' für ein Halogen steht; q für eine ganze Zahl von 2 bis 10 steht und r für eine ganze Zahl von 1 bis 5 steht, mit der Maßgabe, dass: wenn der Phenylring A unsubstituiert ist, Y Br ist und R CH2 ist, dann X nicht -CHBr- ist; und wenn der Phenylring A unsubstituiert ist, Y H ist und R CH2 ist, dann X nicht -CO- ist.

- Verbindung nach Anspruch 1, wobei die substituierte oder unsubstituierte geradkettige Hydrocarbylgruppe -(CH2)x- ist und x zwischen 6 und 30 beträgt.
- Verbindung nach Anspruch 1 ode Anspruch 2, wobei X für -CO- steht, R für eine substituierte oder unsubstituierte geradkettige Hydrocarbylgruppe, die eine oder mehrere Amin- oder Ammoniumverknüpfungsgruppen umfasst, steht und Y für -H, eine Amin- oder eine Ammoniumgruppe steht.
- Verbindung nach Anspruch 3, wobei R für eine unsubstituierte geradkettige Hydrocarbylgruppe, die eine Ammoniumverknüpfungsgruppe umfasst, steht; Y -H ist; und der Phenylring A mit einer oder mehreren Gruppen R2, wobei jedes R2 eine elektronenziehende Gruppe ist und unabhängig voneinander ausgewählt ist, substituiert ist.
- Verbindung nach Anspruch 1 oder Anspruch 2 oder ein pharmazeutisch akzeptables Salz derselben, wobei X -CO- ist und R für eine substituierte oder unsubstituierte geradkettige Hydrocarbylgruppe, die optional eine oder mehrere Ether-, Thioether-, Phenylen-, Amin- oder Ammoniumverknüpfungsgruppen umfasst, steht; und Y für eine Amin- oder Ammoniumgruppe, die kovalent an die terminale Position von R gebunden ist, steht.
- Verbindung nach Anspruch 5, wobei R für -CH2-O[- (CH2)pO]m-(CH2)p- oder -CH2-S[-(CH2)pO]m-(CH2)p- steht; p für 2 oder 3 steht und m für eine ganze Zahl von 1–8 steht.
- Verbindung nach Anspruch 5, wobei R für eine geradkettige Hydrocarbylgruppe, die optional eine oder mehrere Ether- oder Thioetherverknüpfungsgruppen umfasst, steht.
- Verbindung nach Anspruch 7, wobei der Phenylring A optional mit einer oder mehreren Gruppen R2, wobei jedes R2 eine elektronenziehende Gruppe ist und unabhängig voneinander ausgewählt ist, substituiert ist.
- Verbindung nach Anspruch 8, wobei R für eine unsubstituierte geradkettige Hydrocarbylgruppe, die optional eine Ether- oder eine Thioetherverknüpfungsgruppe umfasst, steht und Y für eine Trialkylammoniumgruppe steht.
- Verbindung nach Anspruch 8, wobei der Phenylring A mit einer oder zwei Gruppen R2 substituiert ist und jedes R2 -F ist.
- Verbindung nach Anspruch 1 oder Anspruch 2 einer Strukturformel, die ausausgewählt ist, oder ein pharmazeutisch akzeptables Salz derselben, worin Y für eine Trialkylammoniumgruppe steht; n für eine ganze Zahl von 6 bis 30 steht; und der Phenylring A mit einer oder zwei Gruppen R2, wobei jedes R2 eine elektronenziehende Gruppe ist und unabhängig voneinander ausgewählt ist, substituiert ist.

- Verbindung nach Anspruch 11, wobei Y für eine Trimethylammoniumgruppe steht und der Phenylring A mit bis zu zwei Fluorgruppen substituiert ist.
- Verbindung nach Anspruch 12 der folgenden Strukturformel:worin R3 -H oder -F ist.

- Verbindung nach Anspruch 11 der folgenden Strukturformel:oder ein pharmazeutisch akzeptables Salz derselben, worin R3 -H oder -F ist; n eine ganze Zahl von 6 bis 15 ist; und Y eine Trimethylammoniumgruppe ist.

- Verwendung einer Verbindung der folgenden Strukturformel:oder eines pharmazeutisch akzeptablen Salzes derselben, worin: Z und Z' unabhängig voneinander für -O-, -NH- oder -S- stehen; Ar für eine substituierte oder unsubstituierte Arylgruppe steht; X für eine elktronenziehende Gruppe steht; R für eine substituierte oder unsubstituierte geradkettige Hydrocarbylgruppe steht, die optional eine oder mehrere Amin-, Ammonium-, Ether-, Thioether- oder Phenylenverknüpfungsgruppen umfasst; Y für -H, ein Amin, -[NH-(CH2)q]r-NH2, Halogen, -CF3, Thiol, Ammonium, -OH, -COOH, -SO3H, -OSO3H oder eine Phosphoniumgruppe, die kovalent an die terminale Position von R gebunden sind, steht, wobei, wenn Y -H ist und R eine geradkettige Hydrocarbylgruppe ist, R dann 1 bis 30 Kohlenstoffatome aufweist, und wobei jedes -NH- in -[NH-(CH2)q]r-NH2 optional N-alkyliert oder N, N-dialkyliert ist und -NH2 in -[NH-(CH2)q]r-NH2 optional N-alkyliert, N,N-dialkyliert oder N,N,N-trialkyliert ist; q für eine ganze Zahl von 2 bis 10 steht; r für eine ganze Zahl von 1 bis 5 steht; R1 und R1' unabhängig voneinander für -H, eine aliphatische Gruppe, eine substituierte aliphatische Gruppe, eine Arylgruppe oder eine substituierte Arylgruppe stehen oder zusammengenommen für eine substituierte oder unsubstituierte C2-C5-Alkylengruppe, die optional eine Aminverknüpfungsgruppe [-N+(R1a)-] umfasst, stehen; und R1a für -H, Alkyl, substituiertes Alkyl, Phenyl oder substituiertes Phenyl steht, bei der Herstellung eines Medikaments zur Behandlung eines Subjekts wegen Fettsucht oder zur Hemmung der Aufnahme von Fett im Gastrointestinaltrakt eines Subjekts.

- Verwendung nach Anspruch 15 mit der Maßgabe, dass, wenn Y -H ist und R eine geradkettige Hydrocarbylgruppe ist, R dann 4 bis 30 Kohlenstoffatome aufweist.
- Verwendung nach Anspruch 16, wobei Z und Z' beide -O- sind.
- Verwendung nach Anspruch 17, wobei Ar für eine substituierte oder unsubstituierte Phenylgruppe steht.
- Verwendung nach Anspruch 18, wobei R1 und R1' beide -H sind.
- Verwendung nach Anspruch 19, wobei X -CZ''Z-, -CHZ''-, -COO-, -CONR1b-, -CO-, -S(O)-, -S(O2)O- oder -SO2- ist; R1b für -H, Alkyl oder substituiertes Alkyl steht; Z'' für ein Halogen steht und -X-R-Y para zu -B(OH)2 ist.
- Verwendung nach Anspruch 20, wobei X -CZ''2-, -CHZ''-, -COO-, -CONR1b-, -CO- oder -SO2- ist.
- Verwendung nach Anspruch 20, wobei X -CO- ist, R für eine substituierte oder unsubstituierte geradkettige Hydrocarbylgruppe, die eine oder mehrere Amin- oder Ammoniumverknüpfungsgruppen umfasst, steht und Y für -H, eine Amin- oder Ammoniumgruppe steht.
- Verwendung nach Anspruch 22, wobei R für eine unsubstituierte geradkettige Hydrocarbylgruppe, die eine Ammoniumverknüpfungsgruppe umfasst, steht; Y -H ist; und der Phenylring A mit einer oder mehreren Gruppen R2, wobei jedes R2 eine elektronenziehende Gruppe ist und unabhängig voneinander ausgewählt ist, substituiert ist.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 14 ohne die Vorbehalte bei der Herstellung eines Medikaments zur Hemmung der Aufnahme von Fett im Gastrointestinaltrakt eines Subjekts.
- Verwendung nach Anspruch 24, wobei das Subjekt wegen Fettsucht behandelt wird.
- Verwendung nach Anspruch 24, wobei das Subjekt wegen Typ-II (nicht-insulinabhängigem)-Diabetes mellitus, beeinträchtigter Glucosetoleranz, Hypertonie, Koronarthrombose, Schlaganfall, Lipidsyndromen, Hyperglykämie, Hypertriglyceridämie, Hyperlipidämie, Schlafapnoe, Hiatushernie, Refluxösophagitis, Osteoarthritis, Gicht, mit Gewichtszunahme in Verbindung stehenden Krebserkrankungen, Gallensteinen, Nierensteinen, pulmonaler Hypertonie, Unfruchtbarkeit oder einer kardiovaskulären Erkrankung behandelt wird.
- Pharmazeutische Zusammensetzung, die einen pharmazeutisch akzeptablen Träger oder ein pharmazeutisch akzeptables Verdünnungsmittel und eine Verbindung nach einem der Ansprüche 1 bis 14 ohne die vorbehalte umfasst.
- Pharmazeutische Zusammensetzung nach Anspruch 27, wobei die Verbindung ein pharmazeutisch akzeptables Salz ist, das aus einem Chlorid-, Bromid-, Acetat-, Formiat-, Citrat-, Ascorbat-, Sulfat- oder Phosphatsalz ausgewählt ist.
- Pharmazeutische Zusammensetzung nach Anspruch 28, wobei das pharmazeutisch akzeptable Salz ein Chloridsalz ist.
- Verbindung nach einem der Ansprüche 1 bis 14 ohne die Vorbehalte zur Verwendung in der Medizin.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US30208101P | 2001-06-29 | 2001-06-29 | |
| US302081P | 2001-06-29 | ||
| US35946702P | 2002-02-22 | 2002-02-22 | |
| US359467P | 2002-02-22 | ||
| PCT/US2002/020923 WO2003002570A1 (en) | 2001-06-29 | 2002-07-01 | Aryl boronic acids for treating obesity |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE60214713D1 DE60214713D1 (de) | 2006-10-26 |
| DE60214713T2 true DE60214713T2 (de) | 2007-09-06 |
Family
ID=26972758
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE60214713T Expired - Fee Related DE60214713T2 (de) | 2001-06-29 | 2002-07-01 | Arylborsäuren für die behandlung von fettsucht |
Country Status (13)
| Country | Link |
|---|---|
| US (3) | US6858592B2 (de) |
| EP (1) | EP1404685B1 (de) |
| JP (1) | JP2004535452A (de) |
| AT (1) | ATE339426T1 (de) |
| AU (2) | AU2002316499B2 (de) |
| CA (1) | CA2489681A1 (de) |
| CY (1) | CY1105829T1 (de) |
| DE (1) | DE60214713T2 (de) |
| DK (1) | DK1404685T3 (de) |
| ES (1) | ES2275888T3 (de) |
| HK (1) | HK1065046A1 (de) |
| PT (1) | PT1404685E (de) |
| WO (1) | WO2003002570A1 (de) |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7041280B2 (en) * | 2001-06-29 | 2006-05-09 | Genzyme Corporation | Aryl boronate functionalized polymers for treating obesity |
| US6858592B2 (en) * | 2001-06-29 | 2005-02-22 | Genzyme Corporation | Aryl boronic acids for treating obesity |
| AU2003223930A1 (en) * | 2002-06-14 | 2003-12-31 | Novo Nordisk A/S | Pharmaceutical use of boronic acids and esters thereof |
| WO2004046211A1 (en) * | 2002-11-19 | 2004-06-03 | Genzyme Corporation | Polymeric boronic acid derivatives as lipase inhibitors |
| KR20070086076A (ko) * | 2004-11-12 | 2007-08-27 | 트러스티즈 오브 터프츠 칼리지 | 리파아제 억제제 |
| WO2006098270A1 (ja) * | 2005-03-14 | 2006-09-21 | Eisai R & D Management Co., Ltd. | オキシメチルホウ素化合物 |
| US7622492B2 (en) | 2005-08-31 | 2009-11-24 | Hoffmann-La Roche Inc. | Pyrazolones as inhibitors of 11β-hydroxysteroid dehydrogenase |
| US10157355B2 (en) | 2005-11-15 | 2018-12-18 | General Electric Company | Method to view schedule interdependencies and provide proactive clinical process decision support in day view form |
| US7801058B2 (en) * | 2006-07-27 | 2010-09-21 | Mobitrum Corporation | Method and system for dynamic information exchange on mesh network devices |
| JO3598B1 (ar) * | 2006-10-10 | 2020-07-05 | Infinity Discovery Inc | الاحماض والاسترات البورونية كمثبطات اميد هيدروليز الحامض الدهني |
| WO2009012596A1 (en) * | 2007-07-24 | 2009-01-29 | The University Of British Columbia | Substituted aryl-fluoroborates as imaging agents |
| TW201000107A (en) * | 2008-04-09 | 2010-01-01 | Infinity Pharmaceuticals Inc | Inhibitors of fatty acid amide hydrolase |
| EP2417115A4 (de) | 2009-04-07 | 2012-10-31 | Infinity Pharmaceuticals Inc | Fettsäureamidhydrolase-hemmer |
| JP2012523424A (ja) | 2009-04-07 | 2012-10-04 | インフイニトイ プハルマセウトイカルス インコーポレイテッド | 脂肪酸アミドヒドロラーゼの阻害薬 |
| US8709489B2 (en) | 2009-09-30 | 2014-04-29 | Surmodics, Inc. | Emulsions containing arylboronic acids and medical articles made therefrom |
| CA2788587C (en) | 2010-02-03 | 2020-03-10 | Infinity Pharmaceuticals, Inc. | Fatty acid amide hydrolase inhibitors |
| US8546617B1 (en) | 2012-03-23 | 2013-10-01 | Empire Technology Development Llc | Dioxaborinanes and uses thereof |
| US9120938B2 (en) | 2012-07-31 | 2015-09-01 | Empire Technology Development Llc | Polymerizable organoboron alkyd resin anti fouling coatings |
| WO2014021845A1 (en) * | 2012-07-31 | 2014-02-06 | Empire Technology Development Llc | Polymerizable organoboron alkyd resin anti fouling coatings |
| EP2909216B1 (de) * | 2012-10-17 | 2016-12-21 | Board of Supervisors of Louisiana State University and Agricultural and Mechanical College | Zusammensetzungen und verfahren zur verbesserung der glucoseaufnahme |
| EP2964658B1 (de) | 2013-03-08 | 2024-08-07 | The University Of British Columbia | Positronenemittierende substituierte organofluorborate als bildgebungsmittel |
| EP3913068B1 (de) | 2015-10-12 | 2023-08-02 | Advanced Cell Diagnostics, Inc. | In-situ-detektion von nukleotidvarianten bei proben mit hohem rauschen sowie zugehörige zusammensetzungen und verfahren |
| CN106243343B (zh) * | 2016-08-11 | 2018-04-03 | 江苏大学 | 一种苯硼酸功能化嵌段聚合物的合成及应用 |
| TW202304469A (zh) * | 2021-03-24 | 2023-02-01 | 美商杜夫特學院信託管理公司 | 酸化合物、組合物及方法 |
Family Cites Families (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4496722A (en) * | 1981-05-01 | 1985-01-29 | The Children's Medical Center Corporation | Reporter compounds |
| GB8819224D0 (en) | 1988-08-12 | 1988-09-14 | Merck Patent Gmbh | Process for preparation of rod-like compounds |
| DE3930663C1 (en) | 1989-09-14 | 1990-11-15 | Merck Patent Gmbh, 6100 Darmstadt, De | Cross-coupling metal organic cpds. and e.g. halogen cpds. - includes using transition metal-palladium catalyst and opt. metal alcoholate |
| EP0478050A1 (de) | 1990-09-24 | 1992-04-01 | Unilever N.V. | Detergentzusammensetzung |
| GB9024775D0 (en) | 1990-11-14 | 1991-01-02 | Axis Research | Chemical compounds |
| IE914573A1 (en) | 1991-01-17 | 1992-07-29 | Ici Plc | Boron compounds |
| CA2109526C (en) * | 1991-04-30 | 1998-01-20 | Dwight M. Peterson | Liquid detergents with an aryl boroic acid |
| GB9119920D0 (en) * | 1991-09-18 | 1991-10-30 | Glaxo Group Ltd | Chemical compounds |
| CH683522A5 (de) | 1992-03-13 | 1994-03-31 | Hoffmann La Roche | Verfahren zur Herstellung von Diarylen. |
| JP3257029B2 (ja) | 1992-04-22 | 2002-02-18 | 日本油脂株式会社 | フェニルボロン酸誘導体及び製造方法 |
| CA2096496A1 (en) | 1992-05-29 | 1993-11-30 | Bradley C. Pearce | Acyclic tocotrienol analogs in the treatment of hypercholesterolemia and hyperlipidemia |
| US5290817A (en) * | 1992-06-09 | 1994-03-01 | The Du Pont Merck Pharmaceutical Co. | Substituted 1-benzylindanes and their use as inhibitors of phospholipase A2 |
| EP0674635B1 (de) | 1992-12-21 | 2001-03-28 | Syngenta Limited | Chinuclidin derivate als squalen synthase inhibitoren |
| GB2276162A (en) | 1993-03-17 | 1994-09-21 | Glaxo Group Ltd | Aniline and benazilide derivatives |
| AU7006094A (en) | 1993-06-30 | 1995-01-24 | Wellcome Foundation Limited, The | Anti-atherosclerotic diaryl compounds |
| EP0707642A1 (de) * | 1993-07-09 | 1996-04-24 | Novo Nordisk A/S | Borsäure und borsäurederivate als enzymstabilisatoren |
| JP3412198B2 (ja) * | 1993-08-31 | 2003-06-03 | 日本油脂株式会社 | 変性マレイン酸共重合体および用途 |
| EP0724580A1 (de) | 1993-10-19 | 1996-08-07 | Smithkline Beecham Plc | Benzamilid-derivate als 5ht-1d-rezeptor antagonisten |
| MXPA96003015A (es) | 1994-01-27 | 2005-04-29 | Novartis Ag | Proceso para la preparacion de derivados de esterarilacetico por medio de una reaccion de acoplamiento cruzado catalizada por paladio. |
| US5631364A (en) * | 1994-03-31 | 1997-05-20 | Axis Biochemicals Asa | Labelled boronic acid derivatives |
| SI9400289A (en) | 1994-07-13 | 1996-02-29 | Ponikvar Jadranka Buturovic | A method for preventing blood coagulation in a haemodialysis circuit or haemodialysis analogy procedure in the extra-body circuit. |
| JPH11505509A (ja) | 1994-12-09 | 1999-05-21 | スミスクライン・ビーチャム・コーポレイション | 二環式フィブリノーゲン拮抗薬 |
| AU4328396A (en) | 1995-01-09 | 1996-07-31 | Novo Nordisk A/S | Stabilization of liquid enzyme compositions |
| US6008219A (en) | 1995-03-27 | 1999-12-28 | Smithkline Beech P.L.C. | Bicyclic amine derivatives and their use as anti-psychotic agents |
| UA58494C2 (uk) | 1995-06-07 | 2003-08-15 | Зенека Лімітед | Похідні n-гетероарилпіридинсульфонаміду, фармацевтична композиція, спосіб одержання та спосіб протидії впливам ендотеліну |
| DE69621131T2 (de) * | 1995-06-13 | 2002-11-28 | Novozymes A/S, Bagsvaerd | 4-substituierte-phenylboronsäuren als enzymstabilisatoren |
| US5631371A (en) * | 1995-11-22 | 1997-05-20 | Bayer Corporation | Method for the preparation of substituted 3-(phenylimino)-3H-phenothiazines and phenoxazines |
| GB9603137D0 (en) | 1996-02-15 | 1996-04-17 | Merck Sharp & Dohme | Therapeutic agents |
| WO1998022820A1 (en) | 1996-11-21 | 1998-05-28 | Lawrence Livermore National Laboratory | Detection of biological molecules using boronate-based chemical amplification and optical sensors |
| US6159979A (en) | 1997-04-18 | 2000-12-12 | Smithkline Beecham P.L.C. | Bicyclic aryl or a bicyclic heterocyclic ring containing compounds having a combined 5HT1A, 5HT1B and 5HT1D receptor antagonistic activity |
| JP2002504122A (ja) | 1997-06-13 | 2002-02-05 | ノースウエスタン ユニバーシティー | ベータラクタマーゼ阻害剤及びその使用方法 |
| US6184363B1 (en) * | 1997-06-13 | 2001-02-06 | Northwestern University | Inhibitors of β-lactamases and uses therefor |
| WO1999005107A1 (en) | 1997-07-25 | 1999-02-04 | Smithkline Beecham Corporation | Vitronectin receptor antagonist |
| CO5011067A1 (es) | 1997-11-03 | 2001-02-28 | Novartis Ag | Derivados de bifenilo como productos farmaceuticos, su pre- paracion y composiciones farmaceuticas que los contienen |
| US6264937B1 (en) * | 1998-01-09 | 2001-07-24 | Geltex Pharmaceuticals, Inc. | Fat-binding polymers |
| AU3144299A (en) | 1998-03-18 | 1999-10-11 | Ciba Specialty Chemicals Holding Inc. | Coupling reactions with palladium catalysts |
| WO2000006537A1 (en) | 1998-07-31 | 2000-02-10 | Eli Lilly And Company | N-substituted sulfonamide derivatives |
| JP4979157B2 (ja) | 1998-09-09 | 2012-07-18 | バイオリポックス アーべー | 置換されたγ−フェニル−Δ−ラクトンおよびそのアナログならびにこれらに関する使用 |
| GB9824310D0 (en) | 1998-11-05 | 1998-12-30 | Univ London | Activators of soluble guanylate cyclase |
| GB9824579D0 (en) | 1998-11-10 | 1999-01-06 | Novartis Ag | Organic compounds |
| DE19857765A1 (de) * | 1998-12-15 | 2000-06-21 | Clariant Gmbh | Verfahren zur Herstellung von para-Oxadiazolyl-phenyl-boronsäuren |
| AU3124300A (en) | 1998-12-16 | 2000-07-03 | Northwestern University | Inhibitors of beta-lactamases and uses therefor |
| DE60045474D1 (de) | 1999-01-13 | 2011-02-17 | Univ New York State Res Found | Neues verfahren zum erschaffen von proteinkinase-inhibitoren |
| DZ3265A1 (fr) | 1999-04-14 | 2000-10-19 | Pacific Corp | Derives de 4,5-diaryl-3(2h)-furanone comme inhibiteurs de cyclo-oxygenase-2 |
| JP4507294B2 (ja) | 1999-05-27 | 2010-07-21 | チッソ株式会社 | ビアリール誘導体の製造方法 |
| CA2315614C (en) | 1999-07-29 | 2004-11-02 | Pfizer Inc. | Pyrazoles |
| US6566372B1 (en) | 1999-08-27 | 2003-05-20 | Ligand Pharmaceuticals Incorporated | Bicyclic androgen and progesterone receptor modulator compounds and methods |
| KR100825183B1 (ko) | 2000-11-30 | 2008-04-24 | 캐논 가부시끼가이샤 | 발광 소자 및 표시 장치 |
| JP3589450B2 (ja) | 2001-03-13 | 2004-11-17 | 独立行政法人 科学技術振興機構 | アミド脱水縮合触媒として有用なパーフルオロアルキルフェニルホウ酸 |
| US6858592B2 (en) * | 2001-06-29 | 2005-02-22 | Genzyme Corporation | Aryl boronic acids for treating obesity |
| US7514579B2 (en) | 2002-06-13 | 2009-04-07 | Johns Hopkins University | Boronic chalcone derivatives and uses thereof |
| US7045652B2 (en) * | 2002-07-03 | 2006-05-16 | Pfizer Inc | Processes for preparing substituted aryl boronic acids |
-
2002
- 2002-06-27 US US10/187,397 patent/US6858592B2/en not_active Expired - Lifetime
- 2002-07-01 AU AU2002316499A patent/AU2002316499B2/en not_active Ceased
- 2002-07-01 CA CA002489681A patent/CA2489681A1/en not_active Abandoned
- 2002-07-01 DE DE60214713T patent/DE60214713T2/de not_active Expired - Fee Related
- 2002-07-01 JP JP2003508951A patent/JP2004535452A/ja active Pending
- 2002-07-01 WO PCT/US2002/020923 patent/WO2003002570A1/en active IP Right Grant
- 2002-07-01 AT AT02746808T patent/ATE339426T1/de not_active IP Right Cessation
- 2002-07-01 PT PT02746808T patent/PT1404685E/pt unknown
- 2002-07-01 DK DK02746808T patent/DK1404685T3/da active
- 2002-07-01 ES ES02746808T patent/ES2275888T3/es not_active Expired - Lifetime
- 2002-07-01 EP EP02746808A patent/EP1404685B1/de not_active Expired - Lifetime
-
2004
- 2004-10-07 HK HK04107699A patent/HK1065046A1/xx not_active IP Right Cessation
- 2004-12-30 US US11/027,643 patent/US7049304B2/en not_active Expired - Lifetime
-
2005
- 2005-10-05 AU AU2005220192A patent/AU2005220192A1/en not_active Abandoned
-
2006
- 2006-01-31 US US11/343,598 patent/US7456156B2/en not_active Expired - Fee Related
- 2006-12-05 CY CY20061101748T patent/CY1105829T1/el unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CA2489681A1 (en) | 2003-01-09 |
| DK1404685T3 (da) | 2007-01-22 |
| US20030064963A1 (en) | 2003-04-03 |
| EP1404685A1 (de) | 2004-04-07 |
| US6858592B2 (en) | 2005-02-22 |
| US7456156B2 (en) | 2008-11-25 |
| US20060128664A1 (en) | 2006-06-15 |
| WO2003002570A1 (en) | 2003-01-09 |
| ATE339426T1 (de) | 2006-10-15 |
| US20050107336A1 (en) | 2005-05-19 |
| AU2002316499B2 (en) | 2005-08-04 |
| HK1065046A1 (en) | 2005-02-08 |
| EP1404685B1 (de) | 2006-09-13 |
| DE60214713D1 (de) | 2006-10-26 |
| CY1105829T1 (el) | 2011-02-02 |
| JP2004535452A (ja) | 2004-11-25 |
| ES2275888T3 (es) | 2007-06-16 |
| PT1404685E (pt) | 2007-01-31 |
| US7049304B2 (en) | 2006-05-23 |
| AU2005220192A1 (en) | 2005-12-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE60214713T2 (de) | Arylborsäuren für die behandlung von fettsucht | |
| DE69310204T2 (de) | Salpetersäureester mit pharmazeutischer wirkung und verfahren zu deren herstellung | |
| DE60037313T2 (de) | Pharmazeutische verbindungen | |
| AU2002316499A1 (en) | Aryl Boronic Acids For Treating Obesity | |
| DE2414680C2 (de) | Substituierte 2-Furancarbonsäuren und 2-Furancarbonsäureester, Verfahren zu ihrer Herstellung und Arzneimittel, die diese Verbindungen enthalten | |
| DE3239172A1 (de) | Salze von sulfodehydroabietinsaeure und arzneimittel, welche diese enthalten | |
| DE60307799T2 (de) | Hemmer des phosphattransports | |
| EP0029247B1 (de) | Alkenyl-thienyl-alkancarbonsäuren und ihre Derivate, Verfahren zu deren Herstellung und diese enthaltende Arzneimittel | |
| DE3821317A1 (de) | Thioharnstoffderivate | |
| EP0810989B1 (de) | Neuartig substituierte dtpa-derivate, deren metallkomplexe, diese komplexe enthaltende pharmazeutische mittel, deren verwendung in der diagnostik und therapie, sowie verfahren zur herstellung der komplexe und mittel | |
| EP0030632B1 (de) | Benzothiazolderivate, diese enthaltende pharmazeutische Zubereitungen und Verfahren zu ihrer Herstellung | |
| DE60013818T2 (de) | Quaternäre ammoniumverbindungen, verfahren zu deren herstellung und deren pharmazeutische verwendung | |
| DE602005001918T2 (de) | S-tenatoprazolnatrium-monohydrat-salz und dessen verwendung in form eines protonenpumpenhemmers | |
| DE69208464T2 (de) | N-((4,5-dihydroxy- und 4,5,8-trihydroxy-9,10-dihydro-9,10-dioxo-2-anthracen-yl)carbonyl)aminosäure zur therapie osteoartikulärer leiden | |
| DE69907720T2 (de) | Phospholipid-derivate von nichtsteroiden entzündungshemmenden medikamenten | |
| BE1001251A4 (fr) | Composition pharmaceutique comprenant un complexe de zinc organique et procede pour preparer le produit actif. | |
| DE2434929A1 (de) | Acetaminophenester von arylsalicylsaeuren | |
| DE60116081T2 (de) | Bis-(n,n'-bis-(2-haloethyl)amino) phosphoramidate als antitumor-mittel | |
| DE2412388A1 (de) | Dibenzothiophenderivate, verfahren zu deren herstellung und sie enthaltende arzneimittel | |
| US4275068A (en) | Lipid lowering alkylene glycols and ester derivatives thereof | |
| DE2320945A1 (de) | Neue ester der acetylsalicylsaeure | |
| EP0498011B1 (de) | Neue Salze der 2-(2,6-Dichloranilino)-phenylessigsäure, Verfahren zu ihrer Herstellung und ihre Verwendung für topisch anwendbare pharmazeutische Zubereitungen | |
| DE3019834C2 (de) | Ester von 2-(4-Isobutylphenyl)-propanol-1, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| DE3420387A1 (de) | 2-(4-biphenylyl)-4-hexenonsaeure und ihre derivate mit entzuendungshemmender wirksamkeit | |
| DE2309100C2 (de) | N-Acylierte Aminophenolätherderivate, Verfahren zu ihrer Herstellung und sie enthaltende pharmazeutische Formulierung zur Behandlung von Leberegelinfektionen bei Säugern |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8339 | Ceased/non-payment of the annual fee |