-
Die
vorliegende Erfindung hat 1,3-Dihydro-2H-indol-2-on-Derivate, ihre
Herstellung und ihre Anwendung in der Therapeutik zum Gegenstand.
-
Die
Verbindungen gemäß der vorliegenden
Erfindung weisen eine Affinität
für die
Rezeptoren V1b von Arginin-Vasopressin (AVP)
und/oder für
die Rezeptoren von Oxytocin (OT) auf und außerdem bei einigen unter ihnen
eine Affinität
für die
Rezeptoren Via von AVP.
-
Das
AVP ist ein Hormon, das für
seine antidiuretische Wirkung und für seine Wirkung bei der Regulierung
des arteriellen Druckes bekannt ist. Es stimuliert mehrere Typen
von Rezeptoren: V1 (V1a,
V1b), V2. Diese Rezeptoren
finden sich insbesondere in der Leber, den Gefäßen (koronaren, renalen, cerebralen),
den Plättchen,
der Niere, dem Uterus, den Nebennieren-Drüsen, dem Pankreas, dem zentralen
Nervensystem, der Hypophyse. Das AVP übt somit cardiovasculäre, hepatische,
pankreatische, antidiuretische Wirkungen, Plättchenaggregations-Wirkungen
und Wirkungen auf das zentrale und periphere Nervensystem und auf
den Uterusbereich aus.
-
Das
OT ist ein neurohypophysäres
Hormon mit einer cyclischen Nonapeptid-Struktur, ähnlich der
des AVP. Die Rezeptoren von OT befinden sich im wesentlichen im
glatten Muskel des Uterus und in den myoepithelialen Zellen der
Brustdrüsen.
So spielt das OT eine wichtige Rolle beim Geburtsvorgang, da es
in die Kontraktion des Uterusmuskels und in die Laktation eingreift.
Außerdem
finden sich die Rezeptoren von OT ebenfalls in den anderen peripheren
Geweben und im Zentralnervensystem. Das OT kann somit Wirkungen
in den cardiovasculären,
renalen, endokrinen oder comportementalen Gebieten besitzen.
-
Die
Lokalisierung der verschiedenen Rezeptoren ist beschrieben in: S.
JARD et al., Vasopressin and Oxytocin Receptors: an overview, in
Progreß in
Endocrinology, H. IMURA and K. SHIZURNE ed., Experta Medica, Amsterdam,
1988, 1183–1188,
sowie in den folgenden Artikeln: L. Lab. Clin. Med., 1989, 114(6),
617–632 und
Pharmacol. Rev., 1991, 43(1), 73–108.
-
Die
Rezeptoren Via von AVP finden sich ganz besonders in zahlreichen
peripheren Organen und im Gehirn. Sie wurden geklont, insbesondere
bei der Ratte und beim Menschen, und sie regulieren die Mehrzahl der
für AVP
bekannten Wirkungen: die Plättchenaggregation;
die Uteruskontraktionen; die Kontraktionen der Gefäße; die
Sekretion von Aldosteron, von Cortisol, von CRF (engl. corticotropin-releasing
factor) und vom adrenocorticotrophischen Hormon (ACTH, engl. adrenocorticotrophic
hormon); die hepatische Glycogenolyse, die zellulare Proliferation
und die hauptsächlichen
zentralen Wirkungen von AVP (Hypothermie, Gedächtnis, ...).
-
Die
Rezeptoren V1b wurden ebenfalls in der Adenohypophyse
von verschiedenen Tierarten (Ratte, Schwein, Rind, Schaf ...) einschließlich des
Menschen (S. JARD et al., Mol. Pharmacol., 1986, 30, 171–177; Y.
ARSENIJEVIC et al., J. Endocrinol., 1994, 141, 383–391; J.
SCHWARTZ et al., Endocrinology, 1991, 129(2), 1107–1109; Y.
DE KEYSER et al., FEBS Letters, 1994, 356, 215–220) identifiziert, wo sie
die Freisetzung des adrenocorticotrophischen Hormons durch AVP stimulieren
und die Wirkungen von CRF auf die Freisetzung von ACTH potentialisieren
(G. E. GILLIES et al., Nature, 1982, 299, 355). Im Hypothalamus
induzieren die Rezeptoren V1b außerdem eine
direkte Freisetzung von CRF (Neuroendocrinology, 1994, 60, 503–508) und
sind bei diesen verschiedenen Fällen
in Streßsituationen
einbezogen.
-
Diese
Rezeptoren V1b wurden ebenfalls bei der
Ratte, beim Menschen und bei der Maus geklont (Y. DE KEYSER, FEBS
Letters, 1994, 356, 215–220;
T. SUGIMOTO et al., J. Biol. Chem., 1994, 269(43), 27088–27092;
M. SAITO et al., Biochem. Biophys. Res. Commun., 1995, 212(3), 751–757; S.
J. LOLAIT et al., Neurobiology, 1996, 92, 6783–6787; M. A. VENTURA et al.,
Journal of Molecular endocrinology, 1999, 22, 251–260) und
verschiedene Untersuchungen (Hybridisierung in situ, PCR, engl.
Polymerase Chain Reaction, ...) haben eine ubiquitäre Lokalisierung
dieser Rezeptoren in unterschiedlichen zentralen (insbesondere Gehirn,
Hypothalamus und Adenohypophyse) und peripheren Geweben (Niere,
Pankreas, Nebenniere, Herz, Lunge, Darm, Magen, Leber, Dünndarm,
Blase, Thymus, Milz, Uterus, Netzhaut, Schilddrüse ...) aufgedeckt, und bei
einigen Tumoren (hypophysären,
pulmonalen ...) regen sie eine biologische und/oder pathologische Rolle
an, die sich auf diese Rezeptoren und eine potentielle Einbeziehung
bei verschiedenen Erkrankungen erstreckt.
-
Als
Beispiel haben Arbeiten an der Ratte gezeigt, daß das AVP über die Rezeptoren V1b den endokrinen Pankreas reguliert und
die Sekretion von Insulin und Glucagon (B. LEE et al., Am. J. Physiol.
269 (Endocrinol. Metab. 32): E1095–E1100, 1995) oder die Produktion
von Catecholaminen in der medullären
Nebenniere stimuliert, die der Ort einer lokalen Synthese von AVP
ist (E. GRAZZINI et al., Endocrinology, 1996, 137(a), 3906–3914).
So würde
in diesem letzteren Gewebe das AVP über diese Rezeptoren eine ausschlaggebende Rolle
bei einigen Typen von surrenalen Pheochromocytomen besitzen, die
AVP absondern und aus diesem Grunde eine gleichbleibende Produktion
von Catecholaminen bei ursprünglicher
Hypertension induzieren, die gegenüber den Antagonisten der Rezeptoren
von Angiotensin II und gegenüber
den Inhibitoren des Conversions-Enzyms resistent ist. Die Nebennierenrinde
ist ebenfalls reich an Rezeptoren Via, die bei der Produktion von
Gluco- und Mineralcorticoiden
(Aldosteron und Cortisol) einbezogen sind. Über diese Rezeptoren kann das
AVP (zirkulierend oder lokal synthetisiert) eine Produktion von
Aldosteron mit einer Effektivität
induzieren, die mit der von Angiotensin II vergleichbar ist (G.
GUILLON et al., Endocrinology, 1995, 136(3) 1285–1295). Das Cortisol ist ein
wirksamer Regulator der Produktion von ACTH, des Streßhormons.
-
Neuere
Arbeiten haben gezeigt, daß die
Nebennieren fähig
sind, direkt CRF und/oder ACTG über
die Aktivierung der Rezeptoren V1b und/oder
V1a freizusetzen, die von den Zellen des
Knochenmarks getragen werden (G. MAZZOCCHI et al., peptides, 1997,
18(2), 191–195;
E. GRAZZINI et al., J. Clin. Endocrinol. Metab., 1999, 84(6), 2195–2203).
-
Die
Rezeptoren V1b werden außerdem als ein Tumormarker
betrachtet. So überexprimieren
beispielsweise die Tumore, die ACTH absondern, das sind einige pituitäre Tumore,
einige Bronchialkarzinome (Lungenkrebs mit kleinen Zellen, engl.
Small Cell Lung Cancers), pankreatische, surrenale und thyroide
Karzinome, die in einigen Fällen
ein Cushing-Syndrom induzieren (J. BERTHERAT et al., Eur. J. Endocrinol.,
1996, 135, 173; G. A. WITTERT et al., Lancet, 1990, 335, 991–994; G.
DICKSTEIN et al., J. Clin. Endocrinol. Metab., 1996, 81(8), 2934–2941),
die Rezeptoren V1b. Die Rezeptoren V1a sind ihrerseits ein spezifischerer Marker
für Lungenkrebs
mit kleinen Zellen (SCLC) (P. J. WOLL et al., Biochem. Biophys.
Res. Commun., 1989, 164(1), 66–73).
So sind die Verbindungen gemäß der vorliegenden
Erfindung offenkundige Werkzeuge für die Diagnose, die eine neue
therapeutische Betrachtungsweise bei der Proliferation und beim
Nachweis dieser Tumore anbieten, sogar in einem Früherkennungsstadium
(Radiomarkierung; SPECT, engl. Single Photon Emission Computed Tomography;
PET Scan, engl. Positron Emission Tomography Scanner).
-
Die
reichliche Anwesenheit des Boten für die Rezeptoren V1b im
Magen- und Darmbereich legt eine Implikation des AVP über diesen
Rezeptor auf die Freisetzung von gastro-intestinalen Hormonen wie
Cholecystokinin, Gastrin oder Sekretin nahe (T. SUGIMOTO et al.,
Molecular cloning and functional expression of V1b receptor
gene, in Neurohypophysis: Recent Progress of Vasopressin and Oxytocin
Research; T. SAITO, K. KUROKAWA and S. YOSHIDA ed., Elsevier Science,
1995, 409–413).
-
Derivate
von 1,3-Dihydro-2H-indol-2-on wurden in einigen Patentanmeldungen
als Liganden der Rezeptoren von Arginin-Vasopressin und/oder Oxytocin beschrieben.
Es können
hierzu die Patentanmeldungen WO 93/15051,
EP 636608 ,
EP 636609 , WO 95/18105, WO 97/15556
und WO 98/25901 genannt werden.
-
Es
wurden jetzt neue Derivate von 1,3-Dihydro-2H-indol-2-on gefunden,
die eine Affinität
und Selektivität
für die
Rezeptoren V1b von Arginin-Vasopressin und/oder
für die
Rezeptoren von Oxytocin aufweisen, und außerdem bei einigen Verbindungen
für die
Rezepto ren Via. Diese Derivate besitzen keine Affinität für die Rezeptoren
V2 von AVP.
-
So
hat die vorliegende Erfindung gemäß einem ihrer Aspekte Verbindungen
der Formel (I)
zum Gegenstand, in der
- – n
gleich 1 oder 2 ist,
- – X
eine Gruppe -CH2-; -O-; -NH-; -O-CH2-; -NH-CH2-; -NH-CH2-CH2- darstellt;
- – R1 ein Halogenatom; ein (C1-C4)-Alkyl; ein (C1-C4)-Alkoxy darstellt;
- – R2 ein Wasserstoffatom; ein Halogenatom; ein
(C1-C4)-Alkyl; ein
(C1-C4)-Alkoxy;
einen Rest Trifluormethyl darstellt;
- – R3 ein Halogenatom; ein (C1-C3)-Alkyl; ein (C1-C3)-Alkoxy; einen Rest Trifluormethyl; einen
Rest Trifluormethoxy darstellt;
- – R4 ein Wasserstoffatom; ein Halogenatom; ein
(C1-C3)-Alkyl; ein
(C1-C3)-Alkoxy darstellt;
- – R5 einen Rest darstellt, ausgewählt unter
- – R6 ein (C1-C4)-Alkoxy darstellt;
- – R7 ein (C1-C4)-Alkoxy darstellt,
sowie ihre
Salze mit Mineralsäuren
oder organischen Säuren,
ihre Solvate und/oder ihre Hydrate.
-
Die
Verbindungen der Formel (I) können
ein oder mehrere asymmetrische Kohlenstoffatome umfassen. Sie können daher
in Form von Enantiomeren oder Diastereoisomeren existieren. Diese
Enantiomeren oder Diastereoisomeren sowie ihre Mischungen, einschließlich der
racemischen Mischungen, bilden einen Teil der Erfindung.
-
Die
Verbindungen der Formel (I) können
im Zustand von Basen oder von Additionssalzen mit Säuren existieren.
Derartige Additionssalze bilden einen Teil der Erfindung.
-
Die
Salze werden vorteilhafterweise mit pharmazeutisch akzeptablen Säuren hergestellt,
aber die Salze mit anderen Säuren,
die für
die Reinigung oder Isolierung der Verbindungen der Formel (I) verwendet
werden, bilden ebenfalls einen Teil der Erfindung.
-
Die
Verbindungen der Formel (I) können
ebenfalls in Form von Hydraten oder Solvaten existieren, nämlich in
Form von Assoziationen oder Kombinationen mit einem oder mehreren
Molekülen
von Wasser oder mit einem Lösungsmittel.
Derartige Hydrate und Solvate bilden ebenfalls einen Teil der Erfindung.
-
Unter
Halogen versteht man ein Atom von Chlor, Brom, Fluor oder Iod.
-
Unter
Alkyl versteht man einen linearen oder verzweigten Rest Alkyl mit
ein bis drei Kohlenstoffatomen oder ein bis vier Kohlenstoffatomen
wie den Rest Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl,
sek.-Butyl oder tert.-Butyl.
-
Unter
Alkoxy versteht man einen linearen oder verzweigten Rest Alkoxy
mit ein bis drei Kohlenstoffatomen oder ein bis vier Kohlenstoffatomen
wie den Rest Methoxy, Ethoxy, Propoxy, Isopropoxy, Butoxy, Isobutoxy,
sek.-Butoxy oder tert.-Butoxy.
-
Vorteilhafterweise
betrifft die Erfindung Verbindungen der Formel (I), in denen R5 einen Rest darstellt, ausgewählt unter:
-
-
Gemäß der vorliegenden
Erfindung bevorzugt man die Verbindungen der Formel (I), in der
R1 ein Chloratom oder ein Rest Methyl ist.
-
Gemäß der vorliegenden
Erfindung bevorzugt man die Verbindungen der Formel (I), in der
R2 ein Wasserstoffatom, ein Chloratom, ein
Rest Methyl, ein Rest Methoxy oder ein Rest Trifluormethyl ist.
-
Gemäß der vorliegenden
Erfindung bevorzugt man die Verbindungen der Formel (I), in der
R3 ein Chloratom, ein Fluoratom, ein Rest
Methoxy, ein Rest Ethoxy, ein Rest Isopropoxy, ein Rest Trifluormethoxy
oder ein Rest Trifluormethyl ist.
-
Gemäß der vorliegenden
Erfindung bevorzugt man die Verbindungen der Formel (I), in der
R4 ein Wasserstoffatom oder ein Rest Methoxy
ist.
-
Gemäß der vorliegenden
Erfindung bevorzugt man die Verbindungen der Formel (I), in der
R5 ein 2-Pyridinyl, ein 3-Pyridinyl, ein
4-Pyridinyl, ein 2-Pyrimidinyl, ein 2-Pyrazinyl, ein 3-Pyridazinyl,
ein 1,3-Thiazol-2-yl ist.
-
Gemäß der vorliegenden
Erfindung bevorzugt man die Verbindungen der Formel (I), in der
R6 sich in Position -2- des Phenyls befindet
und einen Rest Methoxy darstellt.
-
Gemäß der vorliegenden
Erfindung bevorzugt man die Verbindungen der Formel (I), in der
R7 einen Rest Methoxy darstellt.
-
Ganz
besonders bevorzugt man die Verbindungen der Formel (I), in der
- – n
und X wie bei einer Verbindung der Formel (I) definiert sind;
- – R1 ein Chloratom oder ein Rest Methyl ist;
- – R2 ein Wasserstoffatom darstellt oder sich
in Position -4- oder -6- des Indol-2-on befindet und ein Chloratom,
einen Rest Methyl, einen Rest Methoxy oder einen Rest Trifluormethyl
darstellt;
- – R3 ein Chloratom, ein Fluoratom, einen Rest
Methoxy, einen Rest Ethoxy, einen Rest Isopropoxy, einen Rest Trifluormethyl
oder einen Rest Trifluormethoxy darstellt;
- – R4 ein Wasserstoffatom oder einen Rest Methoxy
darstellt;
- – R5 ein 2-Pyridinyl, ein 3-Pyridinyl, ein 4-Pyridinyl,
ein 2-Pyrimidinyl, ein 2-Pyrazinyl, ein 3-Pyridazinyl, ein 1,3-Thiazol-2-yl darstellt;
- – R6 sich in Position -2- des Phenyls befindet
und einen Rest Methoxy darstellt;
- – R7 einen Rest Methoxy darstellt;
sowie
ihre Salze mit Mineralsäuren
oder organischen Säuren,
ihre Solvate und/oder ihre Hydrate.
-
Ganz
besonders bevorzugt man die Verbindungen der Formel (I), in der
- – n
gleich 1 oder 2 ist;
- – X
eine Gruppe -CH2-; -O-; -NH- darstellt;
- – R1 ein Chloratom darstellt;
- – R2 ein Wasserstoffatom darstellt;
- – R3 einen Rest Methoxy, einen Rest Ethoxy,
einen Rest Isopropoxy darstellt;
- – R4 ein Wasserstoffatom darstellt;
- – R5 einen Rest darstellt, ausgewählt unter
- – R6 sich in Position -2- des Phenyls befindet
und einen Rest Methoxy darstellt;
- – R7 einen Rest Methoxy darstellt;
sowie
ihre Salze mit Mineralsäuren
oder organischen Säuren,
ihre Solvate und/oder ihre Hydrate.
-
In
noch mehr bevorzugter Weise bevorzugt man die Verbindungen der Formel
(I), in der
- – n gleich 1 ist;
- – X
eine Gruppe -CH2-; -O-; -NH- darstellt;
- – R1 ein Chloratom oder einen Rest Methyl darstellt;
- – R2 ein Wasserstoffatom darstellt oder sich
in Position -4- oder -6- des Indol-2-on befindet und ein Chloratom,
einen Rest Methyl, einen Rest Methoxy darstellt;
- – R3 ein Chloratom, ein Fluoratom oder einen
Rest Methoxy darstellt;
- – R4 ein Wasserstoffatom darstellt;
- – R5 einen Rest darstellt, ausgewählt unter
- – R6 sich in Position -2- des Phenyls befindet
und einen Rest Methoxy darstellt;
- – R7 einen Rest Methoxy darstellt;
sowie
ihre Salze mit Mineralsäuren
oder organischen Säuren,
ihre Solvate und/oder ihre Hydrate.
-
Schließlich bevorzugt
man noch mehr ganz besonders die Verbindungen der Formel (I), in
der
- – n
gleich 1 ist;
- – X
eine Gruppe -O-; -NH-; -NH-CH2-CH2- darstellt;
- – R1 ein Chloratom darstellt;
- – R2 ein Wasserstoffatom darstellt;
- – R3 ein Chloratom oder einen Rest Methoxy darstellt;
- – R4 ein Wasserstoffatom darstellt;
- – R5 einen Rest darstellt;
- – R6 sich in Position -2- des Phenyls befindet
und einen Rest Methoxy darstellt;
- – R7 einen Rest Methoxy darstellt;
sowie
ihre Salze mit Mineralsäuren
oder organischen Säuren,
ihre Solvate und/oder ihre Hydrate.
-
Die
folgenden Verbindungen
- – 5-Chlor-3-(2-ethoxyphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on;
- – 5-Chlor-3-(2-isopropoxyphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on;
- – 4-(2-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl;
- – 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl;
- – N-{5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl}-4-(4-pyridinyl)-homopiperazin-1-carboxamid;
- – N-{5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl}-4-(3-pyridinyl)-piperazin-1-carboxamid;
- – N-{5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl}-4-(4-pyridinyl)-piperazin-1-carboxamid;
- – 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-3-{[3-oxo-3-[4-(2-pyridinyl)-1-piperazinyl]-propyl]-amino}-1,3-dihydro-2H-indol-2-on;
- – 5,6-Dichlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on;
- – 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-methyl-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on;
- – 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl;
- – 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-6-methoxy-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl;
- – N-{5-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-2-oxo-2,3-dihydro-1H-indol-3-yl}-4-(2-pyridinyl)-piperazin-1-carboxamid;
- – N-{5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl}-4-(4-pyridinyl)-piperazin-1-carboxamid;
- – N-{6-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-5-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl}-4-(4-pyridinyl)-piperazin-1-carboxamid;
- – 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-fluorphenyl)-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on;
- – 5,6-Dichlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-fluorphenyl)-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on;
- – 5-Chlor-3-(2,3-dimethoxyphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on;
- – 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-ethoxyphenyl)-3-{2-oxo-2-[4-(3-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on;
- – 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-3-{2-oxo-2-[4-(3-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on;
- – 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-3-{2-oxo-2-[4-(3-pyridinyl)-1-homopiperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on;
- – 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-3-{2-oxo-2-[4-(1,3-thiazol-2-yl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on;
- – 4-(3-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-ethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl;
- – 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-ethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl;
in
Form von reinen optischen Isomeren oder in Form von Mischung sowie
ihre Salze mit Mineralsäuren
oder organischen Säuren,
ihre Solvate und/oder ihre Hydrate sind ganz besonders bevorzugt.
-
Gemäß einem
weiteren ihrer Aspekte hat die vorliegende Erfindung ein Verfahren
zur Herstellung der Verbindungen der Formel (I) zum Gegenstand,
das dadurch gekennzeichnet ist, daß man: eine Verbindung der Formel
(II)
in der
R
1,
R
2, R
3, R
4 und X wie bei einer Verbindung der Formel
(I) definiert sind und:
- – Y ein Hydroxy oder ein Chloratom
darstellt, wenn X eine Gruppe -CH2-; -OCH2-; -NH-CH2-; -NH-CH2-CH2- bedeutet;
oder
- – Y
ein Phenoxy darstellt, wenn X eine Gruppe -O-; -NH- bedeutet;
- – W
ein Wasserstoffatom darstellt, wenn X eine Gruppe -CH2-;
-OCH2- bedeutet; oder
- – W
eine Gruppedarstellt, in der R6 und R7 wie bei
einer Verbindung der Formel (I) definiert sind, wenn X eine Gruppe
-O-; -NH-; -NH-CH2-; -NH-CH2-CH2- bedeutet
mit einer Verbindung
der Formel (III) zur Reaktion bringt, in der
n und R5 wie bei einer Verbindung der Formel
(I) definiert sind; - – wenn W eine Gruppe darstellt,
erhält man die
erwartete Verbindung der Formel (I); oder
- – wenn
W ein Wasserstoffatom darstellt, bringt man die auf diese Weise
erhaltene Verbindung der Formel (IV) in Anwesenheit einer Base
mit einem Sulfonylhalogenid der Formel (V) zur Reaktion, in der R6 und R7 wie bei
einer Verbindung der Formel (I) definiert sind und Hal ein Halogenatom
darstellt.
-
Gegebenenfalls überführt man
die Verbindung der Formel (I) mit Mineralsäuren oder organischen Säuren in
ein Salz.
-
Wenn
Y ein Hydroxy darstellt, erfolgt die Reaktion der Verbindung der
Formel (II) mit der Verbindung der Formel (III) in Anwesenheit eines
in der Peptidchemie verwendeten Kupplungsmittels wie Benzotriazol-1-yl-oxytris-(dimethylamino)-phosphonium-hexafluorphosphat
oder Benzotriazol-1-yloxytripyrrolidino-phosphonium-hexafluorphosphat
und in Anwesenheit einer Base wie Triethylamin oder N,N-Diisopropylethylamin
in einem Lösungsmittel
wie Dichlormethan oder N,N-Dimethylformamid sowie bei einer Temperatur zwischen
0°C und
der Umgebungstemperatur.
-
Wenn
Y ein Chloratom darstellt, erfolgt die Reaktion der Verbindung der
Formel (II) mit der Verbindung der Formel (III) bei Abwesenheit
oder in Anwesenheit einer Base wie Triethylamin oder N,N-Diisopropylethylamin
in einem Lösungsmittel
wie Dichlormethan oder Chloroform sowie bei einer Temperatur zwischen –60°C und der
Umgebungstemperatur. Bei Abwesenheit von Base verwendet man einen Überschuß der Verbindung (III).
-
Wenn
Y ein Phenoxy darstellt, erfolgt die Reaktion der Verbindung der
Formel (II) mit der Verbindung der Formel (III) in einem Lösungsmittel
wie Dichlormethan, Chloroform oder Tetrahydrofuran oder in einer
Mischung dieser Lösungsmittel,
sowie bei einer Temperatur zwischen der Umgebungstemperatur und
der Rückflußtemperatur
des Lösungsmittels.
-
Man
erhält
auf diese Weise entweder direkt eine Verbindung der Formel (I) oder
eine Verbindung der Formel (IV). Die Reaktion der Verbindung der
Formel (IV) mit dem Sulfonylhalogenid (V) erfolgt in Anwesenheit einer
starken Base wie einem Metallhydrid, beispielsweise Natriumhydrid
oder ein Alkalialkoholat wie Kalium-tert.-Butylat in einem Lösungsmittel
wie N,N-Dimethylformamid oder Tetrahydrofuran sowie bei einer Temperatur
zwischen –70°C und der
Umgebungstemperatur. Die Reaktion erfolgt vorzugsweise unter Verwendung einer
Verbindung der Formel (V), in der Hal ein Chloratom darstellt.
-
Die
auf diese Weise erhaltenen Verbindungen der Formel (I) können später vom
Reaktionsmedium abgetrennt und nach klassischen Methoden gereinigt
werden, beispielsweise durch Kristallisation oder Chromatographie.
-
Die
auf diese Weise erhaltenen Verbindungen der Formel (I) werden nach
klassischen Techniken in Form der freien Base oder als Salz isoliert.
-
Die
Verbindungen der Formel (II) werden nach verschiedenen Verfahrensweisen
hergestellt.
-
Die
Verbindungen der Formel (II), in der -X- eine Gruppe -CH2- darstellt,
Y ein Hydroxy bedeutet und W Wasserstoff ist, werden gemäß dem nachfolgenden
SCHEMA 1 hergestellt, worin R1, R2, R3 und R4 wie bei einer Verbindung der Formel (I)
definiert sind und Alk ein (C1-C2)-Alkyl darstellt.
-
-
Bei
der Stufe a1 von SCHEMA 1 stellt man eine Verbindung der Formel
(VIII) durch Dehydroxylierung einer entsprechenden Verbindung der
Formel (VI) mittels Einwirkung von Triethylsilan her, gemäß Bioorganic and
Medicinal Letters, 1997, 7(10), 1255–1260.
-
Man
kann ebenfalls eine Verbindung der Formel (VIII) herstellen durch
Cyclisierungsreaktion (Stufe b1) einer Verbindung der Formel (VII)
im stark sauren Medium, beispielsweise Schwefelsäure oder Polyphosphorsäure, gemäß der in
WO 95/18105 oder in J. Org. Chem., 1968, 33, 1640–1643 beschriebenen
Methode.
-
Man
bringt in der Stufe c1 die Verbindung der Formel (VIII) mit einer
Verbindung der Formel Hal-CH2COOAlk zur
Reaktion, in der Hal ein Halogenatom, vorzugsweise Brom oder Chlor
darstellt und Alk ein (C1-C2)-Alkyl
ist, und zwar in Anwesenheit einer Base wie einem Alkalimetallcarbonat
(beispielsweise Kaliumcarbonat) und einem Alkalimetalliodid (beispielsweise
Kaliumiodid), oder in Anwesenheit einer starken Base wie einem Alkalimetallhydrid
(beispielsweise Natriumhydrid) oder einem Alkalimetallalkoholat
(beispielsweise Natriumethylat), um die Verbindung der Formel (IX)
zu erhalten. Die Reaktion erfolgt in einem Lösungsmittel wie Aceton oder
N,N-Dimethylformamid sowie bei einer Temperatur zwischen 0°C und der
Rückflußtemperatur
des Lösungsmittels.
-
In
der Stufe d1 erhält
man die erwartete Verbindung der Formel (II) durch Hydrolyse der
Verbindung der Formel (IX) im alkalischen Medium, unter Verwendung
eines Alkalimetallhydroxides wie Natriumhydroxid, Kaliumhydroxid
oder Lithiumhydroxid in einem Lösungsmittel
wie Wasser, Methanol, Ethanol, Tetrahydrofuran, Dioxan oder einer
Mischung dieser Lösungsmittel
sowie bei einer Temperatur zwischen 0°C und der Rückflußtemperatur des Lösungsmittels.
-
Die
Verbindung der Formel (VI) sind bekannt und lassen sich nach bekannten
Methoden wie in WO 95/18105 beschrieben herstellen.
-
So
stellt man beispielsweise eine Verbindung der Formel (VI) durch
Reaktion eines 1H-Indol-2,3-dion-Derivates der Formel (X)
in der R
1 und
R
2 wie bei einer Verbindung der Formel (I)
definiert sind, mit einem Organomagnesiumderivat der Formel (XI)
in der R
3 und
R
4 wie bei einer Verbindung der Formel (I)
definiert sind und Hal ein Halogenatom, vorzugsweise Brom oder Iod
darstellt, in einem Lösungsmittel
wie Tetrahydrofuran oder Diethylether sowie bei einer Temperatur
zwischen 0°C
und der Rückflußtemperatur
des Lösungsmittels
her.
-
Man
kann ebenfalls eine Verbindung der Formel (VI), in der R
3 wie bei einer Verbindung der Formel (I) definiert
ist und R
4 von Wasserstoff verschieden ist
und sich in Position -3- oder -6- des Phenyls befindet, herstellen
durch Reaktion einer Verbindung der Formel (XXXIV)
in der R
3 wie
bei einer Verbindung der Formel (I) definiert ist und R
4 sich
in Position -2- oder -5- des Phenyls befindet, mit einem Lithiumderivat
wie n-Butyllithium, und anschließend das auf diese Weise erhaltene
Lithium-Zwischenprodukt mit einer Verbindung der Formel (X) zur
Reaktion bringen. Die Reaktion erfolgt in einem Lösungsmittel
wie Diethylether, Tetrahydrofuran, Hexan oder einer Mischung dieser
Lösungsmittel
sowie bei einer Temperatur zwischen –70°C und der Umgebungstemperatur.
-
Die
1H-Indol-2,3-dion-Derivate (X) sind im Handel verfügbar oder
lassen sich nach in Tetrahedron Letters, 1998, 39, 7679–7682; Tetrahedron
Letters, 1994, 35, 7303–7306;
J. Org. Chem., 1977, 42(8), 1344–1348; J. Org. Chem., 1952,
17, 149–156;
J. Am. Chem. Soc., 1946, 68, 2697–2703; Organic Synthèses, 1925,
V, 71–74
und Advances in Heterocyclic Chemistry, A. R. Katritzky und A. J.
Boulton, Academic Press, New-York, 1975, 18, 2–58, beschriebenen Methoden
herstellen.
-
Die
Organomagnesiumderivate (XI) werden nach klassischen, dem Fachmann
bekannten Methoden hergestellt.
-
In
besonderer Weise kann man die Verbindung der Formel (VI), in der
R3 = (C1-C2)-Alkoxy und R4 =
H, oder auch R3 = R4 =
(C1-C2)-Alkoxy mit
R4 in Position -3 oder -6 des Phenyls sind,
R2 von einem Halogenatom verschieden ist
und R1 wie bei einer Verbindung der Formel
(I) definiert ist, herstellen, indem nach dem folgenden SCHEMA 2
verfährt.
-
-
In
Stufe a2 von SCHEMA 2 bringt man zuerst eine Verbindung der Formel
(XII) mit einem Lithiumderivat wie n-Butyllithium zur Reaktion und
setzt anschließend
das auf diese Weise erhaltene Lithium-Zwischenprodukt mit einem
Diethyloxalat um, um die Verbindung der Formel (XII) zu ergeben.
Die Reaktion erfolgt in einem Lösungsmittel
wie Diethylether, Tetrahydrofuran, Hexan oder einer Mischung dieser
Lösungsmittel
sowie bei einer Temperatur zwischen –70°C und der Umgebungstemperatur.
-
In
Stufe b2 bringt man zuerst eine Verbindung der Formel (XIV) mit
zwei Äquivalenten
eines Lithiumderivates wie tert.-Butyllithium zur Reaktion und anschließend das
auf diese Weise erhaltene Lithium-Zwischenprodukt mit der Verbindung
der Formel (XIII), um die erwartete Verbindung der Formel (VI) zu
ergeben. Die Reaktion erfolgt in einem Lösungsmittel wie Diethylether,
Tetrahydrofuran, Pentan oder einer Mischung dieser Lösungsmittel
sowie bei einer Temperatur zwischen –70°C und der Umgebungstemperatur.
-
Die
Verbindungen der Formel (XII) sind im Handel verfügbar oder
werden in klassischer Weise synthetisiert.
-
Die
Verbindungen der Formel (XIV) werden durch Reaktion der entsprechenden
Anilinderivate mit Di-tert.-Butyldicarbonat nach klassischen Methoden
hergestellt.
-
Die
Verbindungen der Formel (VII) sind bekannt und lassen sich nach
bekannten Methoden wie in WO 95/18105 oder in J. Org. Chem., 1968,
33, 1640–1643
beschrieben herstellen.
-
Die
Verbindungen der Formel (II), in der X = -CH2-
ist, Y ein Chloratom darstellt und W = H ist, lassen sich ausgehend
von den entsprechenden Verbindungen der Formel (II), in der Y =
OH ist, durch Reaktion mit Thionylchlorid in einem Lösungsmittel
wie Toluol sowie bei einer Temperatur zwischen 0°C und der Rückflußtemperatur des Lösungsmittels
herstellen.
-
Die
Verbindungen der Formel (II), in der -X- eine Gruppe -O- darstellt,
Y ein Phenoxy bedeutet und W =
ist, lassen sich nach dem
folgenden SCHEMA 3 herstellen, worin R
1,
R
2, R
3, R
4, R
6 und R
7 wie bei einer Verbindung der Formel (I)
definiert sind.
-
-
-
In
der Stufe a3 von SCHEMA 3 schützt
man selektiv das Hydroxy einer Verbindung der Formel (VI), indem
man beispielsweise Hexamethyldisilazan gemäß der in Synthetic Communications,
1993, 23(12), 1633–1641
beschriebenen Methode verwendet.
-
Die
auf diese Weise erhaltene Verbindung der Formel (XV) wird in Stufe
b3 mit einem Sulfonylhalogenid der Formel (V) in Anwesenheit einer
starken Base nach den vorstehend beschriebenen Bedingungen zur Reaktion
gebracht.
-
In
der Stufe c3 ermöglicht
die Abspaltung der Trimethylsilylgruppe von der auf diese Weise
erhaltenen Verbindung der Formel (XVI) die Verbindung der Formel
(XVIII) zu erhalten. Die Reaktion erfolgt durch Einwirkung einer
starken Säure
wie Chlorwasserstoffsäure
oder Trifluoressigsäure
in einem Lösungsmittel
wie Dichlormethan, Aceton, Tetrahydrofuran, Wasser oder einer Mischung
dieser Lösungsmittel
sowie bei einer Temperatur zwischen der Umgebungstemperatur und
der Rückflußtemperatur
des Lösungsmittels.
-
Man
kann ebenfalls eine Verbindung der Formel (XVIII) erhalten durch
Reaktion in Stufe d3 von einer Verbindung der Formel (X) mit einem
Sulfonylhalogenid der Formel (V) nach den vorstehend beschriebenen Bedingungen,
gefolgt von der auf diese Weise erhaltenen Verbindung der Formel
(XVII) mit einem Organomagnesiumderivat der Formel (XI) nach den
vorstehend beschriebenen Bedingungen.
-
In
der Stufe f3 bringt man die auf diese Weise erhaltene Verbindung
der Formel (XVIII) mit Phenylchlorformiat in Anwesenheit einer Base
wie Pyridin und in einem Lösungsmittel
wie Dichlormethan oder auch ohne Lösungsmittel bei einer Temperatur
zwischen 0°C
und 100°C
zur Reaktion und erhält
die erwartete Verbindung der Formel (II).
-
Die
Verbindung der Formel (VI) läßt sich
nach den vorstehend beschriebenen Bedingungen herstellen. Man kann
ebenfalls eine Verbindung der Formel (VI) durch Oxidation einer
Verbindung der Formel (VIII) mit Hilfe von Luft in Anwesenheit einer
Base wie Natriumhydrid und in Anwesenheit von Dimethyldisulfid herstellen.
Man kann ebenfalls eine Verbindung der Formel (VI) durch Hydrolyse
eines Halogenides der Formel (XIX)
herstellen, in der R
1, R
2, R
3 und
R
4 wie bei einer Verbindung der Formel (I)
definiert sind und Hal ein Halogenatom, vorzugsweise Brom oder Chlor,
darstellt. Die Reaktion erfolgt in einem Lösungsmittel wie Tetrahydrofuran
sowie bei einer Temperatur zwischen der Umgebungstemperatur und
der Rückflußtemperatur
des Lösungsmittels.
-
Die
Verbindungen der Formel (XIX) sind bekannt und lassen sich nach
bekannten Methoden wie in WO 95/18105 beschrieben herstellen.
-
Die
Verbindungen der Formel (II), in der -X- eine Gruppe -NH- darstellt, Y ein
Phenoxy bedeutet und W eine Gruppe
ist, lassen sich nach den
in WO 95/18105 beschriebenen Verfahren herstellen.
-
Die
Verbindungen der Formel (II), in der -X- eine Gruppe -OCH2- darstellt,
Y ein Hydroxy bedeutet und W Wasserstoff ist, lassen sich nach dem
folgenden SCHEMA 4 herstellen, worin R1,
R2, R3 und R4 wie bei einer Verbindung der Formel (I)
definiert sind.
-
-
In
der Stufe a4 von SCHEMA 4 bringt man eine Verbindung der Formel
(XIX) mit Methylglycolat in Anwesenheit einer starken Base wie Natriumhydrid
in einem Lösungsmittel
wie Tetrahydrofuran oder Dichlormethan sowie bei einer Temperatur
zwischen 0°C
und der Rückflußtemperatur
des Lösungsmittels
zur Reaktion.
-
Die
die auf diese Weise erhaltene Verbindung (XX) wird in Stufe b4 im
alkalischen Medium nach den vorstehend in Stufe d1 von SCHEMA 1
beschriebenen Methoden hydrolysiert, und man erhält die erwartete Verbindung
der Formel (II).
-
Die
Verbindungen der Formel (II), in der X = -O-CH2-
darstellt, Y ein Chloratom bedeutet und W = H ist, lassen sich ausgehend
von den entsprechenden Verbindungen der Formel (II), in der Y =
OH ist, durch Reaktion mit Thionylchlorid nach der vorstehend angegebenen
Methode herstellen.
-
Die
Verbindungen der Formel (II), in der X eine Gruppe -NH-CH
2- darstellt,
Y ein Hydroxy bedeutet und W eine Gruppe
ist, lassen sich nach dem
folgenden SCHEMA 5 herstellen, worin R
1,
R
2, R
3, R
4, R
6 und R
7 wie bei einer Verbindung der Formel (I)
definiert sind.
-
-
In
der Stufe a5 von SCHEMA 5 bringt man eine Verbindung der Formel
(XIX) mit dem tert.-Butylester von Glycin in Anwesenheit einer Base
wie Triethylamin oder N,N-Diisopropylethylamin in einem Lösungsmittel wie
Dichlormethan, Chloroform, Tetrahydrofuran oder einer Mischung dieser
Lösungsmittel
sowie bei einer Temperatur zwischen 0°C und der Umgebungstemperatur
zur Reaktion.
-
Die
die auf diese Weise erhaltene Verbindung der Formel (XXI) wird in
Stufe b5 mit einem Sulfonylhalogenid der Formel (V) in Anwesenheit
einer starken Base nach den vorstehend beschriebenen Bedingungen zur
Reaktion gebracht.
-
In
der Stufe c5 wird die auf diese Weise erhaltene Verbindung der Formel
(XXII) im sauren Medium hydrolysiert, indem man eine starke Säure wie
Chlorwasserstoffsäure
oder Trifluoressigsäure
in einem Lösungsmittel
wie Dichlormethan, Tetrahydrofuran, Aceton, Wasser oder in einer
Mischung dieser Lösungsmittel oder
auch ohne Lösungsmittel
sowie bei einer Temperatur zwischen 0°C und der Umgebungstemperatur
verwendet. Man erhält
auf diese Weise die erwartete Verbindung der Formel (II).
-
Die
Verbindungen der Formel (II), in der X = -NH-CH
2-
darstellt, Y ein Chloratom bedeutet und
ist, lassen sich ausgehend
von den entsprechenden Verbindungen der Formel (II), in der Y =
OH ist, nach den vorstehend beschriebenen Methoden herstellen.
-
Die
Verbindungen der Formel (I), in der X eine Gruppe -NH-CH
2-CH
2- darstellt,
Y ein Hydroxy bedeutet und W eine Gruppe
ist, lassen sich nach dem
folgenden SCHEMA 6 herstellen, worin R
1,
R
2, R
3, R
4, R
6 und R
7 wie bei einer Verbindung der Formel (I)
definiert sind.
-
-
In
der Stufe a6 von SCHEMA 6 bringt man eine Verbindung der Formel
(XIX) mit dem tert.-Butylester von β-Alanin in Anwesenheit einer
Base wie Triethylamin oder N,N-Diisopropylethylamin in einem Lösungsmittel
wie Dichlormethan, Chloroform, Tetrahydrofuran oder einer Mischung
dieser Lösungsmittel
sowie bei einer Temperatur zwischen 0°C und der Umgebungstemperatur
zur Reaktion.
-
Die
die auf diese Weise erhaltene Verbindung der Formel (XXIII) wird
in Stufe b6 mit einem Sulfonylhalogenid der Formel (V) in Anwesenheit
einer starken Base nach den vorstehend beschriebenen Bedingungen
zur Reaktion gebracht.
-
In
der Stufe c6 wird die auf diese Weise erhaltene Verbindung der Formel
(XXIV) im sauren Medium nach den vorstehend in Stufe c5 von SCHEMA
5 beschriebenen Bedingungen hydrolysiert. Man erhält auf diese
Weise die erwartete Verbindung der Formel (II).
-
Die
Verbindungen der Formel (II), in der X = -NH-CH
2-CH
2- darstellt, Y ein Chloratom bedeutet und
ist, lassen sich ausgehend
von den entsprechenden Verbindungen der Formel (II), in der Y =
-OH ist, nach den vorstehend beschriebenen Methoden herstellen.
-
Die
Verbindungen der Formel (III) sind im Handel verfügbar oder
können
nach bekannten Methoden wie in J. Org. Chem., 1953, 18, 1484–1488; J.
Med. Chem., 1978, 21(6), 536–542;
Chem. Pharm. Bull., 1991, 39(9), 2288–2300; Tetrahedron Letters,
1998, 39, 617–620
oder in WO 97/28129 beschrieben hergestellt werden. Beispielsweise
stellt man eine Verbindung der Formel (III) durch Reaktion einer
Verbindung der Formel (XXV)
in der n wie bei einer Verbindung
der Formel (I) definiert ist und Z Wasserstoff oder eine N-Schutzgruppe
darstellt, mit einer Verbindung der Formel (XXVI)
Hal-R5 (XXVI)her,
in der R
5 wie bei einer Verbindung der Formel
(I) definiert ist und Hal ein Halogenatom, vorzugsweise Chlor, Brom
oder Iod darstellt.
-
Die
Reaktion erfolgt in Anwesenheit oder bei Abwesenheit einer Base,
in einem inerten Lösungsmittel wie
Ethanol, Propan-2-ol, n-Butanol oder Acetonitril sowie bei einer
Temperatur zwischen 0°C
und der Rückflußtemperatur
des Lösungsmittels.
-
Wenn
man eine Base verwendet, so wird diese ausgewählt unter den organischen Basen
wie Diisopropylethylamin oder unter den Alkalimetallcarbonaten wie
Natriumcarbonat oder Kaliumcarbonat. Bei Ab wesenheit einer Base
erfolgt die Reaktion unter Anwendung eines Überschusses der Verbindung
der Formel (XXV). Die Reaktion kann ebenfalls ohne Lösungsmittel
durchgeführt
werden durch Erhitzen der Mischung der Verbindungen (XXV) und (XXVI)
auf Temperaturen in der Größenordnung
von 140°C
und 180°C.
-
Wenn
Z eine N-Schutzgruppe darstellt, so entfernt man diese gegebenenfalls
nach klassischen Methoden und erhält die erwarteten Verbindungen
der Formel (III).
-
Die
Verbindungen der Formel (XXV) oder der Formel (XXVI) sind bekannt
oder lassen sich nach bekannten Methoden herstellen.
-
Die
Verbindungen der Formel (V) sind bekannt oder werden nach bekannten
Methoden wie in EP-0 469 984 B und WO 95/18105 beschrieben hergestellt.
Die Verbindungen der Formel (V) können beispielsweise durch Halogenierung
der entsprechenden Benzolsulfonsäuren
oder ihrer Salze, beispielsweise ihrer Natriumsalze oder ihrer Kaliumsalze,
hergestellt werden. Die Reaktion erfolgt in Anwesenheit eines Halogenierungsmittels
wie Phosphoroxychlorid, Thionylchlorid, Phosphortrichlorid, Phosphortribromid
oder Phosphorpentachlorid, ohne Lösungsmittel oder in einem inerten
Lösungsmittel
wie einem halogenierten Kohlenwasserstoff oder N,N-Dimethylformamid
sowie bei einer Temperatur zwischen –10°C und 200°C.
-
Das
2,4-Dimethoxybenzolsulfonylchlorid wird gemäß J. Am. Chem. Soc., 1952,
74, 2008 hergestellt. Das 3,4-Dimethoxybenzolsulfonylchlorid ist
handelsüblich
oder wird gemäß J. Med.
Chem., 1977, 20(10), 1235–1239
hergestellt.
-
Um
die Verbindungen der Formel (I) in Form von optisch reinen Isomeren
zu erhalten, kann man die klassischen Techniken zur Auftrennung
anwenden: beispielsweise fraktionierte Rekristallisationen eines
ausgehend von der racemischen Base oder mit einer optisch aktiven
Säure gebildeten
Salzes, deren Prinzip bekannt ist, oder die klassischen Techniken
der Chromatographie über
eine chirale Phase.
-
Man
kann ebenfalls die optisch reinen Verbindungen der Formel (I) ausgehend
von einer optisch reinen Zwischenverbindung herstellen, die für die Herstellung
der Verbindungen der Formel (I) verwendet wird.
-
Wenn
man also wünscht,
eine optisch reine Verbindung der Formel (I) herzustellen, in der
-X- = -CH2- ist, so führt man die optische Auflösung einer
Verbindung der Formel (II) durch, in der -X- = -CH2-
darstellt, Y = OH bedeutet und W = H ist, gemäß dem in J. Am. Chem. Soc.,
1989, 111, 7650–7651
beschriebenen Verfahren, unter Verwendung von (R)-Pantolacton als
chiralem Reagens oder auch unter Verwendung eines chiralen Amins
wie (+)-Cinchonin.
-
Man
kann eine optisch reine Verbindung der Formel (I) herstellen, in
der -X- = -O- ist, gemäß dem in dem
folgenden SCHEMA 7 beschriebenen Verfahren, worin R1,
R2, R3, R4, n und R5 wie bei
einer Verbindung der Formel (I) definiert sind und R ein Phenyl
oder Isobutyl darstellt.
-
-
-
In
der Stufe a7 von SCHEMA 7 bringt man eine Verbindung der Formel
(VI) mit Phenylchlorformiat in Anwesenheit einer Base wie Pyridin
und in einem Lösungsmittel
wie Dichlormethan oder auch ohne Lösungsmittel sowie bei einer
Temperatur zwischen 0°C
und 100°C
zur Reaktion.
-
Die
die auf diese Weise erhaltene Verbindung der Formel (XXVII) wird
in Stufe b7 mit einer Verbindung der Formel (III) in einem Lösungsmittel
wie Dichlormethan, Chloroform oder Tetrahydrofuran oder einer Mischung
dieser Lösungsmittel
sowie bei einer Temperatur zwischen der Umgebungstemperatur und
der Rückflußtemperatur
des Lösungsmittels
zur Reaktion gebracht, um eine Verbindung der Formel (XXVIII) zu
erhalten.
-
Durch
Reaktion der Verbindung (XXVIII) mit L-Leucinol (R = Isobutyl) oder
D-Leucinol oder mit (R)-(–)-α-Phenylglycinol
(R = Phenyl) oder (S)-(+)-α-Phenylglycinol
erhält
man in Stufe c7 eine Mischung der Diastereoisomeren einer Verbindung
der Formel (XXIX), die man beispielsweise durch Chromatographie
oder Kristallisation auftrennen kann.
-
Durch
Reaktion in Stufe d7 von einem Diastereoisomeren der Verbindung
der Formel (XXIX) mit einer starken Base wie Natriummethylat in
einem Lösungsmittel
wie Methanol oder Tetrahydrofuran oder einer Mischung dieser Lösungsmittel
bei einer Temperatur zwischen 0°C
und der Rückflußtemperatur
des Lösungsmittels
erhält
man eine optisch reine Verbindung der Formel (XXX).
-
Die
Reaktion der Verbindung (XXX) mit einem Sulfonylhalogenid der Formel
(V) nach den vorstehend beschriebenen Methoden ermöglicht es,
eine optisch reine Verbindung der Formel (I) zu erhalten, in der
X = -O- ist.
-
Wenn
man wünscht,
eine optisch reine Verbindung der Formel (I) herzustellen, in der
-X- = -NH- darstellt, stellt man zunächst eine optisch reine Verbindung
der Formel (II) her, in der -X- = -NH- darstellt, Y = Phenoxy bedeutet
und
ist, nach dem folgenden SCHEMA
8, worin R
1, R
2,
R
3, R
4, R
6 und R
7 wie bei
einer Verbindung der Formel (I) definiert sind und Hal ein Halogen
darstellt, vorzugsweise Chlor oder Brom.
-
-
In
der Stufe a8 von SCHEMA 8 bringt man eine Verbindung der Formel
(XIX) mit (S)-(+)-α-Phenylglycinol
oder (R)-(–)-α-Phenylglycinol
zur Reaktion und erhält
eine Mischung von Diastereoisomeren einer Verbindung der Formel
(XXXI), die man durch Chromatographie oder Kristallisation auftrennen
kann.
-
In
der Stufe b8 erhält
man durch Oxidation von einem der Diastereoisomeren der Verbindung
(XXXI) mit Bleitetraacetat und Hydro lyse im sauren Medium eine optisch
reine Verbindung der Formel (XXXII).
-
In
der Stufe c8 ermöglicht
die Reaktion der auf diese Weise erhaltenen Verbindung (XXXII) mit
einem Sulfonylhalogenid der Formel (V), gefolgt von der Reaktion
der auf diese Weise erhaltenen Verbindung (XXXIII) mit Phenylchlorformiat
(Stufe d8) nach den in WO 95/18105 beschriebenen Methoden, die erwartete
optisch reine Verbindung (II) zu erhalten.
-
Im
Verlauf von irgendeiner der Stufen zur Herstellung der Verbindungen
der Formel (I) oder der Zwischenverbindungen der Formel (II), (III)
oder (IV) kann es notwendig und/oder wünschenswert sein, die reaktiven
oder sensiblen funktionellen Gruppen zu schützen, wie die Gruppen Amin,
Hydroxyl oder Carboxy, die die in irgendeinem der betreffenden Moleküle anwesend
sind. Dieser Schutz kann unter Verwendung von üblichen Schutzgruppen erfolgen,
wie sie in Protective Groups in Organic Chemistry, J. F. W. McOmie,
Ed. Plenum Press, 1973, in Protective Groups in Organic Synthesis,
T. W. Greene und P. G. M. Wutts, Ed. John Wiley et Sons, 1991, oder
in Protecting Groups, Kcienski P. J., 1994, Georg Thieme Verlag,
beschrieben sind.
-
Die
Entfernung des Schutzes kann in einer späteren Stufe zweckmäßig sein,
und zwar unter Anwendung von dem Fachmann bekannten Methoden, die
den Rest des betreffenden Moleküls
nicht beeinträchtigen.
-
Die
gegebenenfalls verwendeten N-Schutzgruppen sind klassische, dem
Fachmann bekannte N-Schutzgruppen, wie beispielsweise die Gruppe
tert.-Butoxycarbonyl, Fluorenylmethoxycarbonyl, Benzyl, Benzhydryliden
oder Benzyloxycarbonyl.
-
Die
Verbindungen der Formel (IV) sind neu und bilden einen Teil der
Erfindung.
-
So
hat gemäß einem
ihrer Aspekte die Erfindung Verbindungen der Formel (IV) zum Gegenstand:
in der
- – n gleich
1 oder 2 ist;
- – X
eine Gruppe -CH2-; -O-CH2-
darstellt;
- – R1 ein Halogenatom; ein (C1-C4)-Alkyl; ein (C1-C4)-Alkoxy darstellt;
- – R2 ein Wasserstoffatom; ein Halogenatom; ein
(C1-C4)-Alkyl; ein
(C1-C4)-Alkoxy;
einen Rest Trifluormethyl darstellt;
- – R3 ein Halogenatom; ein (C1-C3)-Alkyl; ein (C1-C3)-Alkoxy; einen Rest Trifluormethyl; einen
Rest Trifluormethoxy darstellt;
- – R4 ein Wasserstoffatom; ein Halogenatom; ein
(C1-C3)-Alkyl; ein
(C1-C3)-Alkoxy darstellt;
- – R5 einen Rest darstellt, ausgewählt unter sowie
ihre Salze mit Mineralsäuren
oder organischen Säuren,
in Form von optisch reinen Isomeren oder in Form von Mischung.
-
Die
obenstehenden Verbindungen der Formel (I) umfassen ebenfalls diejenigen,
in denen ein oder mehrere Atome von Wasserstoff oder Kohlenstoff
durch ihr radioaktives Isotop ersetzt wurden, wie beispielsweise
Tritium oder Kohlenstoff-14. Derartige markierte Verbindungen sind
in biochemischen Untersuchungen und als Rezeptor-Liganden bei Forschungsarbeiten
zum Metabolismus und zur Pharmakokinetik nützlich.
-
Die
folgenden HERSTELLUNGEN und BEISPIELE veranschaulichen die Erfindung,
ohne sie jedoch einzuschränken.
-
In
den Herstellungen und Beispielen werden die folgenden Abkürzungen
verwendet:
Ether: Diethylether
Isoether: Diisopropylether
DMF:
N,N-Dimethylformamid
THF: Tetrahydrofuran
DCM: Dichlormethan
AcOEt:
Ethylacetat
TMEDA: N,N,N',N'-Tetramethylethylendiamin
DIPEA:
Diisopropylethylamin
TFA: Trifluoressigsäure
HMDS: Hexamethyldisilazan
BOP:
Benzotriazol-1-yl-oxy-tris-(dimethylamino)-phosphonium-hexafluorphosphat
PyBOP:
Benzotriazol-1-yl-oxy-tripyrrolidino-phosphonium-hexafluorphosphat
DCC: 1,3-Dicyclohexylcarbodiimid
HOBT:
1-Hydroxybenzotriazol-Hydrat
Chlorwasserstoff-Ether: gesättigte Lösung von
Chlorwasserstoffsäure
in Diethylether
F: Schmelzpunkt
TA: Umgebungstemperatur
Eb:
Siedetemperatur
CLHP: Hochleistungs-Flüssig-Chromatographie
-
Die
magnetischen Resonanz-Spektren des Protons (NMR 1H)
werden bei 200 MHz in DMSO-d6 unter Verwendung
des Pic von DMSO-d6 als Referenz aufgezeichnet.
Die chemischen Verschiebungen δ werden
in Teilen pro Million (ppm) ausgedrückt. Die beobachteten Signale
werden so ausgedrückt:
s: Singulett; se: Singulett erweitert, d: Dublett; dd: aufgespaltetes
Dublett; t: Triplett; td: aufgespaltetes Triplett; q: Quadruplett;
m: massiv; mt: Multiplett.
-
Die
NMR-Spektren bestätigen
die Strukturen der Verbindungen.
-
HERSTELLUNGEN
-
Herstellungen der Verbindungen
der Formel (II)
-
Herstellung 1.1
-
2-[5-Chlor-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
(II): R1 =
Cl; R2 = H; R3 =
OCH3; R4 = H; X
= -CH2-; Y = OH; W = H
-
A) 5-Chlor-3-hydroxy-3-(2-methoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
WO 95/18105 beschriebenen Verfahrensweise her. Man bereitet zunächst eine
Lösung
von 2-Methoxyphenylmagnesiumbromid ausgehend von 16 g Magnesium
in 35 ml Ether und einer Lösung
von 124 g 1-Brom-2-methoxybenzol in 175 ml Ether. Dann gibt man
tropfenweise unter Atmosphäre
von Argon diese Lösung
zu einer Mischung von 30 g 5-Chlor-1H-indol-2,3-dion in 250 ml THF, zuvor im Eisbad
gekühlt,
und läßt anschließend unter
Rühren
die Temperatur auf TA ansteigen. Nach 1 Stunde Rühren bei TA gießt man die
Reaktionsmischung langsam in eine gesättigte Lösung von NH4Cl
und verdampft das THF unter Vakuum. Danach zentrifugiert man den
gebildeten Rückstand
und wäscht
ihn mit Isoether. Man erhält
auf diese Weise 42 g des erwarteten Produktes, das man so wie es
ist bei der folgenden Stufe verwendet.
-
B) 5-Chlor-3-(2-methoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 5,8 g der in der vorstehenden Stufe erhaltenen
Verbindung, 25 ml TFA und 10 ml Triethylsilan 24 Stunden lang unter
Rückfluß. Danach
konzentriert man die Reaktionsmischung bei 100°C unter Vakuum, nimmt den Rückstand
in 30 ml Isoether auf und läßt das Ganze
15 Stunden lang bei 20°C stehen.
Anschließend
zentrifugiert man den gebildeten Niederschlag und wäscht ihn
mit Ether und dann mit Heptan. Man erhält auf diese Weise 4,3 g des
erwarteten Produktes.
-
C) 2-[5-Chlor-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure-ethylester
-
Man
erhitzt eine Mischung von 2,85 g der in der vorstehenden Stufe erhaltenen
Verbindung, 2,05 g Bromessigsäure-ethylester,
2,05 g KI und 3,1 g K2CO3 in
15 ml Aceton 20 Stunden lang unter Rückfluß. Dann filtriert man die Mineralsalze
und konzentriert das Filtrat unter Vakuum. Anschließend extrahiert
man den Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Danach
verreibt man den Rückstand
in Isoether und zentrifugiert den gebildeten Niederschlag. Dann
chromatographiert man den Rückstand über Kieselgel,
indem man mit DCM und danach mit AcOEt eluiert. Man erhält auf diese
Weise 1,55 g des erwarteten Produktes nach Rekristallisation in
Isoether, F = 184–187°C.
-
D) 2-[5-Chlor-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
Zu
einer Mischung von 1,5 g der in der vorstehenden Stufe erhaltenen
Verbindung in 30 ml MeOH gibt man bei TA eine Lösung von 0,5 g NaOH in Pastillen
in 10 ml Wasser und beläßt das Ganze
20 Stunden lang bei 20°C
unter Rühren.
Danach konzentriert man die Reaktionsmischung unter Vakuum, nimmt
den Rückstand in
20 ml Wasser auf, wäscht
die wäßrige Phase
mit AcOEt, säuert
die wäßrige Phase
durch Zugabe von konzentrierter Chlorwasserstoffsäure auf
pH = 1 an und zentrifugiert den gebildeten Niederschlag. Man erhält auf diese
Weise 1,15 g des erwarteten Produktes, F = 135–139°C.
-
Herstellung 1.2
-
2-[5-Chlor-3-(2-ethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
(II): R1 =
Cl; R2 = H; R3 =
OCH2CH3; R4 = H; X = -CH2-;
Y = OH; W = H
-
A) 1-Brom-2-ethoxybenzol
-
Man
erhitzt eine Mischung von 17,5 g 2-Bromphenol, 66 ml Diethylsulfat
und 170 ml einer Lösung
von NaOH (10%) 2 Stunden lang unter Rückfluß. Nach dem Abkühlen der
Reaktionsmischung auf TA extrahiert man mit AcOEt, wäscht die
organische Phase mit einer 2 N Lösung
von NaOH, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 19,6 g des erwarteten Produktes.
-
B) 5-Chlor-3-(2-ethoxyphenyl)-3-hydroxy-1,3-dihydro-2H-indol-2-on
-
Man
stellt eine Lösung
von 2-Ethoxyphenylmagnesiumbromid ausgehend von 2,2 g Magnesium
in 10 ml Ether und einer Lösung
von 16,5 g der in der vorstehenden Stufe erhaltenen Verbindung in
40 ml Ether her. Dann gibt man tropfenweise und unter Atmosphäre von Stickstoff
diese Lösung
zu einer Mischung von 5 g 5-Chlor-1H-indol-2,3-dion in 20 ml THF, indem die
Temperatur des Reaktionsmediums unter 35°C gehalten wird. Nach 2 Stunden
Rühren
bei TA gießt
man die Reaktionsmischung in 200 ml 2 N HCl, extrahiert mit AcOEt, trocknet
die organische Phase über
Na2SO4 und verdampft
die Lösungsmittel
unter Vakuum. Anschließend nimmt
man den Rückstand
in warmem Isoether auf und läßt kristallisieren.
Dann zentrifugiert man das gebildete kristalline Produkt, wäscht es
mit Isoether und trocknet. Man erhält auf diese Weise 5,7 g des
erwarteten Produktes, F = 251°C.
-
C) 5-Chlor-3-(2-ethoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe B) der Herstellung 1.1 beschriebenen Verfahrensweise her,
ausgehend von 4 g der in der vorstehenden Stufe erhaltenen Verbindung,
15 ml TFA und 5,6 ml Triethylsilan. Man erhält auf diese Weise 3,6 g des
erwarteten Produktes.
-
D) 2-[5-Chlor-3-(2-ethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure-ethylester
-
Man
stellt diese Verbindung gemäß der in
Stufe C der Herstellung 1.1 beschriebenen Verfahrensweise her, ausgehend
von 3,5 g der in der vorstehenden Stufe erhaltenen Verbindung, 2,2
g Bromessigsäure-ethylester,
2,2 g KI, 3,5 g K2CO3 in
20 ml Aceton. Man erhält
auf diese Weise 1,7 g des erwarteten Produktes nach Fällung in
Isoether, F = 181°C.
-
E) 2-[5-Chlor-3-(2-ethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
Man
beläßt eine
Mischung von 1,65 g der in der vorstehenden Stufe erhaltenen Verbindung
und 2 ml einer Lösung
von NaOH (30%) in 50 ml Wasser und 20 ml THF 24 Stunden lang bei
TA unter Rühren.
-
Dann
konzentriert man unter Vakuum, nimmt den Rückstand in 100 ml Wasser auf,
wäscht
die wäßrige Phase
mit AcOEt, säuert
die wäßrige Phase
durch Zugabe von konzentrierter HCl auf pH = 1 an, extrahiert mit AcOEt,
wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 1,2 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 212°C.
-
Herstellung 1.3
-
2-[5-Chlor-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure, rechtsdrehendes
Isomer
-
(II): R1 =
Cl; R2 = H; R3 =
OCH(CH3)2; R4 = H; X = -CH2-;
Y = OH; W = H
-
A) 1-Brom-2-isopropoxybenzol
-
Man
stellt eine Lösung
von Natriumethylat ausgehend von 280 ml EtOH und 7,3 g Natrium her,
gibt 50 g 2-Bromphenol hinzu und beläßt das Ganze 30 Minuten lang
bei TA unter Rühren.
Anschließend
setzt man 43 g Isopropylbromid hinzu und erhitzt 15 Stunden lang
unter Rückfluß. Dann
konzentriert man die Reaktionsmischung unter Vakuum, nimmt den Rückstand
in einer 1 N Lösung
von NaOH auf, extrahiert mit Ether, trocknet die organische Phase über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Anschließend
destilliert man das resultierende Öl unter reduziertem Druck und
erhält
49,3 g des erwarteten Produktes in Form einer farblosen Flüssigkeit,
Eb = 113–115°C unter 3366
Pa.
-
B) 5-Chlor-3-hydroxy-3-(2-isopropoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
stellt eine Lösung
von 2-Isopropoxyphenylmagnesiumbromid ausgehend von 6,3 g Magnesium in
20 ml Ether und 49,3 g der in der vorstehenden Stufe erhaltenen
Verbindung her. Dann gibt man innerhalb von 3 Minuten diese Lösung zu
einer Mischung von 13,3 g 5-Chlor-1H-indol-2,3-dion in 30 ml THF
und erhitzt anschließend
90 Minuten lang unter Rückfluß. Nach
dem Abkühlen
auf TA gießt
man die Reaktionsmischung auf eine Mischung von Eiskonzentrierter
HCl, extrahiert mit AcOEt, wäscht
die organische Phase mit einer 1 N Lösung von NaOH und mit Wasser,
trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Anschließend
nimmt man den Rückstand
in warmem Isoether auf und nach dem Verreiben zentrifugiert man den
gebildeten Niederschlag. Man erhält
auf diese Weise 17,4 g des erwarteten Produktes.
-
C) 5-Chlor-3-(2-isopropoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 3,5 g der in der vorstehenden Stufe erhaltenen
Verbindung, 15 ml TFA und 5,5 ml Triethylsilan 8 Stunden lang unter
Rückfluß. Dann
konzentriert man unter Vakuum und chromatographiert den Rückstand über Kieselgel,
indem man mit Isoether und danach mit DCM eluiert. Man erhält auf diese
Weise 2,2 g des erwarteten Produktes nach Kristallisation in Heptan,
F = 154°C.
-
D) 2-[5-Chlor-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure-ethylester
-
Man
stellt diese Verbindung gemäß der in
Stufe C der Herstellung 1.1 beschriebenen Verfahrensweise her, ausgehend
von 2,55 g der in der vorstehenden Stufe erhaltenen Verbindung,
1,67 g Bromessigsäure-ethylester,
1,66 g KI und 2,52 g K2CO3 in
12 ml Aceton. Dann chromatographiert man das erhaltene Produkt über Aluminiumoxid,
indem man mit DCM und danach mit Aceton eluiert. Man erhält auf diese
Weise 1,6 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 193°C.
-
E) 2-[5-Chlor-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
Zu
einer Lösung
von 1,6 g der in der vorstehenden Stufe erhaltenen Verbindung in
10 ml EtOH gibt man bei 40°C
eine Lösung
von 1 g NaOH in Pastillen in 10 ml Wasser und beläßt das Ganze
20 Stunden lang bei 20°C
unter Rühren.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in 30 ml Wasser auf,
erhitzt auf 40°C
und filtriert den unlöslichen
Anteil. Danach säuert
man das Filtrat durch Zugabe von konzentrierter HCl auf pH = 1 an,
zentrifugiert den gebildeten Niederschlag und trocknet. Man erhält auf diese
Weise 1,15 g des erwarteten Produktes, F = 146–148°C.
-
F) 2-[5-Chlor-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-acetat von
(3R)-4,4-Dimethyl-2-oxotetrahydro-3-furanyl, das am meisten polare
Isomer
-
Man
beläßt eine
Mischung von 2 g der in der vorstehenden Stufe erhaltenen Verbindung
und 10 ml Thionylchlorid 20 Stunden lang bei 20°C unter Rühren. Anschließend konzentriert
man unter Vakuum, nimmt den Rückstand
in DCM auf, konzentriert das Lösungsmittel
bei TA unter Vakuum und erhält
2 g Säurechlorid. Dann
löst man
das Säurechlorid
in 20 ml DCM, gibt diese Lösung
zu einer Mischung von 0,8 g (R)-Pantolacton und 1 g N-Methylpyrrolidin
in 10 ml DCM, zuvor gekühlt
auf 10°C,
und beläßt 2 Stunden
lang bei TA unter Rühren.
Danach konzentriert man die Reaktionsmischung unter Vakuum, nimmt
den Rückstand
in Wasser auf, extrahiert mit AcOEt, wäscht die organische Phase mit
Wasser, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Dann chromatographiert man den Rückstand über Kieselgel H, indem man
mit DCM eluiert. Man trennt die Diastereoisomeren und gewinnt die
am meisten polare Verbindung. Man erhält auf diese Weise 0,6 g des
erwarteten Produktes.
-
G) 2-[5-Chlor-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure, rechtsdrehendes
Isomer
-
Zu
einer Mischung von 0,8 g der in der vorstehenden Stufe erhaltenen
Verbindung in 30 ml MeOH gibt man eine Lösung von 0,8 g Lithiumhydroxid-Monohydrat
in 10 ml Wasser und beläßt das Ganze
20 Stunden lang bei 20°C
unter Rühren.
Dann konzentriert man die Reaktionsmischung unter Vakuum, extrahiert
den Rückstand
mit Wasser auf, wäscht
die wäßrige Phase
mit Ether, säuert
die wäßrige Phase
durch Zugabe von konzentrierter HCl auf pH = 1 an, extrahiert mit
DCM, trocknet die organische Phase über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 0,6 g des erwarteten Produktes.
α20 D = +84° (c
= 0,25; Chloroform).
-
Herstellung 1.4
-
2-[3-(2-Chlorphenyl)-5-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
(II): R1 =
CH3; R2 = H; R3 = Cl; R4 = H; X
= -CH2-; Y = OH; W = H
-
A) 3-(2-Chlorphenyl)-5-methyl-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
WO 95/18105 in den Stufen A und B der Herstellung 65 beschriebenen
Verfahrensweise her.
-
B) 2-[3-(2-Chlorphenyl)-5-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure-methylester
-
Man
kühlt eine
Mischung von 5 g der in der vorstehenden Stufe erhaltenen Verbindung
in 10 ml DMF auf eine Temperatur von unter 10°C, gibt in kleinen Fraktionen
0,87 g Natriumhydrid (60% in Öl)
hinzu und beläßt 30 Minuten
lang unter Rühren.
Anschließend
setzt man 3,15 ml Bromessigsäure-methylester
hinzu, beläßt 4 Stunden
lang bei TA unter Rühren,
gibt danach wiederum 1 ml Bromessigsäure-methylester hinzu und beläßt 16 Stunden
lang bei TA unter Rühren.
Dann gießt
man die Reaktionsmischung in Wasser, extrahiert mit AcOEt, wäscht die
organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Anschließend chromatographiert
man den Rückstand über Kieselgel,
indem man mit DCM und danach mit einer Mischung von DCM/AcOEt (95/5;
V/V) eluiert. Man erhält
auf diese Weise 0,57 g des erwarteten Produktes.
-
C) 2-[3-(2-Chlorphenyl)-5-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
Zu
einer Mischung von 0,57 g der in der vorstehenden Stufe erhaltenen
Verbindung in 10 ml Dioxan gibt man eine Lösung von 0,16 g NaOH in Pastillen
in 40 ml Wasser und beläßt das Ganze
24 Stunden lang bei TA unter Rühren.
Dann gibt man 50 ml Wasser zu der Reaktionsmischung wäscht die
wäßrige Phase
mit AcOEt, säuert
die wäßrige Phase
durch Zusatz von 1 N HCl auf pH = 1 an, extrahiert mit AcOEt, trocknet
die organische Phase über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/ AcOEt (95/5; V/V) eluiert. Man erhält auf diese
Weise 0,27 g des erwarteten Produktes.
-
Herstellung 1.5
-
2-[5,6-Dichlor-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
(II): R1 =
Cl; R2 = 6-Cl; R3 =
OCH3; R4 = H; X
= -CH2-; Y = OH; W = H
-
A) 5,6-Dichlor-1H-indol-2,3-dion
-
Man
stellt diese Verbindung gemäß der in
J. Am. Chem. Soc., 1946, 68, 2697–2703 beschriebenen Verfahrensweise
oder gemäß der in
J. Org. Chem., 1952, 17, 149–156
beschriebenen Verfahrensweise her.
-
B) 5,6-Dichlor-3-hydroxy-3-(2-methoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Zu
einer Suspension von 1,1 g Magnesium in 20 ml Ether, die einige
Kristalle Iod enthält,
gibt man tropfenweise 7,5 g 1-Brom-2-methoxybenzol, wobei man den Rückfluß aufrechterhält, wenn
er begonnen hat. Am Ende der Zugabe erhitzt man noch 2 Stunden lang
unter Rückfluß. Anschließend setzt
man eine Suspension von 4,3 g 5,6-Dichlor-1H-indol-2,3-dion in 20
ml THF hinzu und erhitzt 30 Minuten lang unter Rückfluß. Nach dem Abkühlen auf
TA gießt
man die Reaktionsmischung auf eine Mischung von Wasser/Eis/konzentrierter
HCl, extrahiert mit AcOEt, trocknet die organische Phase über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Dann reibt man den Rückstand in warmem Isoether
an, zentrifugiert den gebildeten Niederschlag und wäscht ihn
mit Ether. Man erhält
auf diese Weise 5,2 g des erwarteten Produktes, F = 245–246°C.
-
C) 5,6-Dichlor-3-(2-methoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 5,1 g der in der vorstehenden Stufe erhaltenen
Verbindung, 25 ml TFA und 10 ml Triethylsilan 15 Stunden lang unter
Rückfluß. Dann
konzentriert man unter Vakuum, verreibt den Rückstand in Heptan und zentrifugiert
den gebildeten Niederschlag. Man erhält auf diese Weise 4,8 g des
erwarteten Produktes nach Kristallisation in Isoether, F = 195–197°C.
-
D) 2-[5,6-Dichlor-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure-ethylester
-
Man
erhitzt eine Mischung von 4,8 g der in der vorstehenden Stufe erhaltenen
Verbindung, 2,7 g Bromessigsäure-ethylester,
2,7 g KI und 4,4 g K2CO3 in
25 ml Aceton 16 Stunden lang unter Rückfluß. Anschließend filtriert man die Mineralsalze
und konzentriert das Filtrat unter Vakuum. Dann chromatographiert
man den Rückstand über Kieselgel,
indem man mit DCM und danach mit AcOEt eluiert. Man erhält auf diese
Weise 2,6 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 214–219°C.
-
E) 2-[5,6-Dichlor-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
Zu
einer Mischung von 2,5 g der in der vorstehenden Stufe erhaltenen
Verbindung in 50 ml MeOH gibt man 3 ml einer Lösung von 2 N NaOH und beläßt das Ganze
48 Stunden lang bei 20°C
unter Rühren.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in 30 ml Wasser auf,
filtriert einen unlöslichen
Anteil, säuert
das Filtrat durch Zugabe von konzentrierter HCl auf pH = 1 an, extrahiert
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 2 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 222–224°C.
-
Herstellung 1.6
-
2-[5-Chlor-3-(2-methoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
(II): R1 =
Cl; R2 = 6-CH3;
R3 = OCH3; R4 = H; X = -CH2-;
Y = OH; W = H
-
A) 2-(2-Methoxyphenyl)-2-oxoessigsäure-ethylester
-
Man
kühlt eine
Lösung
von 27 g 1-Brom-2-methoxybenzol in 270 ml Ether unter Atmosphäre von Argon
auf eine Temperatur von –70°C, setzt
tropfenweise 90 ml einer 1,6 M Lösung
von n-Butyllithium in Pentan hinzu und beläßt das Ganze 45 Minuten lang
unter Rühren.
Anschließend
gibt man schnell 78 ml Oxalsäure-diethylester
zu und läßt die Temperatur
unter Rühren
wieder auf TA ansteigen. Nach 1 Stunde Rühren bei TA gibt man zu der
Reaktionsmischung eine ge sättigte
Lösung
von NH4Cl, dekantiert, extrahiert die wäßrige Phase
mit Ether, wäscht
die vereinigten organischen Phasen mit Wasser, mit einer gesättigten
Lösung
von NaCl, trocknet über
Na2SO4 und verdampft
die Lösungsmittel
unter Vakuum. Dann entfernt man den überschüssigen Oxalsäure-diethylester
durch Destillation unter Vakuum (Eb = 87°C unter 2000 Pa). Anschließend chromatographiert
man das resultierende Produkt über
Kieselgel, indem man mit einer Mischung von DCM/Hexan (50/50; V/V)
und danach mit DCM eluiert. Das erhaltene Produkt wird durch Destillation
unter Vakuum gereinigt. Man erhält
auf diese Weise 13 g des erwarteten Produktes, Eb = 110°C unter 3
Pa.
-
B) 5-Chlor-3-hydroxy-3-(2-methoxyphenyl)-6-methyl-1,3-dihydro-2H-indol-2-on
-
a) 4-Chlor-3-methylphenylcarbamat
von tert.-Butyl
-
Man
beläßt eine
Mischung von 10 g 4-Chlor-3-methylanilin und 15,26 g Di-tert.-Butyldicarbonat
in 50 ml Dioxan 24 Stunden lang bei TA unter Rühren. Dann konzentriert man
die Reaktionsmischung unter Vakuum und chromatographiert den Rückstand über Kieselgel,
indem man durch den Gradienten der Mischung von DCM/Hexan von (50/50;
V/V) bis (70/30; V/V) eluiert. Man erhält auf diese Weise 5,6 g des
erwarteten Produktes, das so wie es ist weiter verwendet wird.
-
b)
Man kühlt
eine Lösung
von 5 g 4-Chlor-3-methylphenylcarbamat von tert.-Butyl in 45 ml
Ether unter Atmosphäre
von Argon auf eine Temperatur von –70°C, setzt tropfenweise 30 ml
einer 1,5 M Lösung
von tert.-Butyllithium in Pentan hinzu, beläßt 1 Stunde lang unter Rühren, indem
die Temperatur auf –10°C ansteigt und
rührt dann
1 Stunde und 45 Minuten lang bei –10°C. Danach kühlt man die Reaktionsmischung
wieder auf –70°C, setzt
tropfenweise eine Lösung
von 5 g der in Stufe A erhaltenen Verbindung in 25 ml THF hinzu,
rührt 1
Stunde lang, indem man die Temperatur auf –30°C ansteigen läßt, und
anschließend über Nacht,
indem man die Temperatur auf TA zurückkehren läßt. Danach gibt man zu der
Reaktionsmischung eine gesättigte
Lösung von
NH4Cl, verdampft das THF, extrahiert die
resultierende wäßrige Phase
dreimal mit AcOEt, wäscht
die organische Phase mit Wasser und mit einer gesättigten Lösung von
NaCl, trocknet über
Na2SO4, verdampft
teilweise das Lösungsmittel
und zentrifugiert das kristallisierte Produkt. Man erhält auf diese
Weise 2,6 g des erwarteten Produktes, F = 254–256°C.
-
C) 5-Chlor-3-(2-methoxyphenyl)-6-methyl-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 3 g der in der vorstehenden Stufe erhaltenen
Verbindung, 15 ml TFA und 6 ml Triethylsilan 15 Stunden lang unter
Rückfluß. Dann
konzentriert man unter Vakuum, verreibt den Rückstand in Pentan und erhält auf diese
Weise 2,85 g des erwarteten Produktes, F = 193°C.
-
D) 2-[5-Chlor-3-(2-methoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure-ethylester
-
Man
erhitzt eine Mischung von 2,8 g der in der vorstehenden Stufe erhaltenen
Verbindung, 1,7 g Bromessigsäure-ethylester,
1,7 g KI und 2,7 g K2CO3 in
15 ml Aceton 16 Stunden lang unter Rückfluß. Anschließend filtriert man die Mineralsalze
und konzentriert das Filtrat unter Vakuum. Man erhält auf diese
Weise 2,35 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 170°C.
-
E) 2-[5-Chlor-3-(2-methoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
Man
beläßt eine
Mischung von 2,3 g der in der vorstehenden Stufe erhaltenen Verbindung
und 3,5 g einer 12 N Lösung
von NaOH in 100 ml MeOH und 5 ml Wasser 20 Stunden lang bei 20°C unter Rühren. Dann konzentriert
man unter Vakuum, nimmt den Rückstand
in 30 ml Wasser auf, säuert
durch Zugabe von konzentrierter HCl auf pH = 1 an, extrahiert mit
AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 1,3 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 214–216°C.
-
Herstellung 1.7
-
2-[5-Chlor-3-(2-isopropoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
(II): R1 =
Cl; R2 = 6-CH3;
R3 = OCH(CH3)2; R4 = H; X = -CH2-; Y = OH; W = H
-
A) 2-{2[(tert.-Butoxycarbonyl)-amino]-5-chlor-4-methylphenyl}-2-oxoessigsäure-ethylester
-
Man
kühlt eine
Lösung
von 5,9 g 4-Chlor-3-methylphenylcarbamat von tert.-Butyl in 60 ml
Ether unter Atmosphäre
von Stickstoff auf eine Temperatur von –50°C, setzt tropfenweise 42 ml
einer Lösung
von tert.-Butyllithium in Pentan hinzu und beläßt das Ganze 2 Stunden lang
unter Rühren,
indem man die Temperatur auf –20°C ansteigen
läßt. Dann
kühlt man
die Reaktionsmischung auf –70°C, gibt tropfenweise
eine Lösung
von 4,5 g Oxalsäure-di-ethylester in 30
ml THF zu und rührt
1 Stunde lang, wobei man die Temperatur wieder auf 20°C ansteigen
läßt. Anschließend gießt man die
Reaktionsmischung in eine gesättigte
Lösung
von NH4Cl, dekantiert, extrahiert die wäßrige Phase
mit AcOEt, wäscht
die vereinigten organischen Phasen mit 1 N HCl, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Anschließend
chromatographiert man den Rückstand über Kieselgel,
indem man mit einer Mischung von Isoether/Pentan (50/50; V/V) eluiert.
Man erhält
auf diese Weise 8 g des erwarteten Produktes in Form eines Öles, das
kristallisiert, F = 111°C.
-
B) 5-Chlor-6-methyl-1H-indol-2,3-dion
-
Man
erhitzt eine Mischung von 8 g der in der vorstehenden Stufe erhaltenen
Verbindung und 60 ml 3 N HCl in 80 ml THF 2 Stunden lang unter Rückfluß. Dann
konzentriert man die Reaktionsmischung unter Vakuum, nimmt den Rückstand
in EtOH auf und konzentriert von neuem das Lösungsmittel unter Vakuum. Danach
nimmt man den Rückstand
wieder in EtOH auf, zentrifugiert den gebildeten Niederschlag und
wäscht
ihn mit Isoether. Man erhält
auf diese Weise 1,9 g des erwarteten Produktes.
-
C) 5-Chlor-3-hydroxy-3-(2-isopropoxyphenyl)-6-methyl-1,3-dihydro-2H-indol-2-on
-
Zu
einer Suspension von 0,36 g Magnesium in 10 ml Ether gibt man tropfenweise
3,2 g 1-Brom-2-isopropoxybenzol, wobei man den Rückfluß aufrechterhält, wenn
er begonnen hat. Am Ende der Zugabe er hitzt man noch 2 Stunden lang
unter Rückfluß. Anschließend setzt
man auf einmal eine Lösung
von 1,8 g der in der vorstehenden Stufe erhaltenen Verbindung in
30 ml THF hinzu und erhitzt 30 Minuten lang unter Rückfluß. Nach
dem Abkühlen
auf TA gießt
man die Reaktionsmischung auf eine Mischung von Eis/konzentrierter
HCl, extrahiert mit AcOEt, trocknet die organische Phase über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Dann reibt man den Rückstand in warmem Isoether
an, zentrifugiert den gebildeten Niederschlag und wäscht ihn
mit Ether. Man erhält
auf diese Weise 1,8 g des erwarteten Produktes, das so wie es ist
weiter verwendet wird.
-
D) 5-Chlor-3-(2-isopropoxyphenyl)-6-methyl-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 2,3 g der in der vorstehenden Stufe erhaltenen
Verbindung, 15 ml TFA und 3,8 ml Triethylsilan 10 Stunden lang unter
Rückfluß. Dann
konzentriert man unter Vakuum (13 Pa), verreibt den Rückstand
in Pentan und zentrifugiert den gebildeten Niederschlag. Danach
chromatographiert man den Niederschlag über Kieselgel, indem man mit
DCM eluiert. Man erhält
auf diese Weise 0,85 g des erwarteten Produktes, F = 145–146°C.
-
E) 2-[5-Chlor-3-(2-isopropoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure-ethylester
-
Man
erhitzt eine Mischung von 0,8 g der in der vorstehenden Stufe erhaltenen
Verbindung, 0,53 g Bromessigsäure-ethylester,
0,53 g KI und 0,8 g K2CO3 in
6 ml Aceton 15 Stunden lang unter Rückfluß. Anschließend filtriert man die Mineralsalze
und konzentriert das Filtrat unter Vakuum, extrahiert den Rückstand
mit DCM, wäscht
die organische Phase mit Wasser und filtriert einen unlöslichen
Anteil. Dann chromatographiert man das Filtrat über Kieselgel, indem man mit
DCM und danach mit einer Mischung von DCM/AcOEt (70/30; V/V) eluiert.
Man erhält
auf diese Weise 0,55 g des erwarteten Produktes, F = 152–154°C.
-
F) 2-[5-Chlor-3-(2-isopropoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
Man
beläßt eine
Mischung von 0,53 g der in der vorstehenden Stufe erhaltenen Verbindung
und 0,25 g NaOH in Pastillen in 20 ml MeOH und 5 ml Wasser 20 Stunden
lang bei 25°C
unter Rühren.
Dann konzentriert man unter Vakuum, löst den Rückstand in 20 ml Wasser, säuert durch
Zugabe von konzentrierter HCl auf pH = 1 an und zentrifugiert den
gebildeten Niederschlag. Man erhält
auf diese Weise 0,45 g des erwarteten Produktes nach Kristallisation
in einer Mischung von Isoether/Pentan (50/50; V/V), F = 148–150°C.
-
Herstellung
1.8 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat (II):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = H; X = -O-;
-
A) 5-Chlor-3-(2-methoxyphenyl)-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 2,9 g der in der Stufe A der Herstellung
1.1 erhaltenen Verbindung und 0,16 g wasserfreies Zinkchlorid in
40 ml Acetonitril unter Rückfluß, setzt
1,7 g HMDS hinzu und erhitzt 1 Stunde lang unter Rückfluß. Dann
konzentriert man unter Vakuum, extrahiert den Rückstand mit AcOEt, wäscht die organische
Phase mit Wasser, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 2,4 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 185–187°C.
-
B) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Zu
einer Mischung von 2,4 g der in der vorstehenden Stufe erhaltenen
Verbindung, in 30 ml THF gibt man 0,17 g Natriumhydrid (60% in Öl) und beläßt 20 Minuten
lang unter Rühren
bei TA. Anschließend
setzt man 1,7 g 2,4-Dimethoxybenzol-sulfonylchlorid hinzu und rührt 1 Stunde
lang bei TA. Danach konzentriert man unter Vakuum, extrahiert den
Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 2,65 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 157–158°C.
-
C) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-hydroxy-3-(2-methoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
beläßt eine
Mischung von 2,4 g der in der vorstehenden Stufe erhaltenen Verbindung
und 10 ml TFA in 20 ml DCM 4 Stunden lang bei 20°C unter Rühren. Dann konzentriert man
unter Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit einer Lösung
von Na2CO3 (10%),
trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 1,75 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 153–160°C.
-
D) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat
-
Man
beläßt eine
Mischung von 1,6 g der in der vorstehenden Stufe erhaltenen Verbindung
und 1 g Phenylchlorformiat in 20 ml Pyridin 3 Tage lang bei 20°C unter Rühren. Dann
konzentriert man unter Vakuum, extrahiert den Rückstand mit AcOEt, wäscht die
organische Phase mit einer Lösung
von AcOH (10%), trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
Isoether eluiert. Man erhält
auf diese Weise 1,05 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 212–213°C.
-
Herstellung
1.9 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat (II):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -O-;
-
A) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-1H-indol-2,3-dion
-
Zu
einer Mischung von 3,6 g 5-Chlor-1H-indol-2,3-dion in 20 ml DMF
gibt man 0,5 g Natriumhydrid (60% in Öl) und beläßt 30 Minuten lang bei 20°C unter Rühren. Danach
setzt man 4,8 g 2,4-Dimethoxybenzol-sulfonylchlorid hinzu und rührt 1 Stunde
lang bei 20°C.
Dann konzentriert, man unter Vakuum (1,3 Pa), extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Anschließend verreibt
man den Rückstand
in Isoether und zentrifugiert den gebildeten Niederschlag. Man erhält auf diese
Weise 2,9 g des erwarteten Produktes nach Kristallisation in warmen
AcOEt, F = 194, 5°C.
-
B) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-hydroxy-3-(2-isopropoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
stellt eine Lösung
von 2-Isopropoxyphenylmagnesiumbromid ausgehend von 0,5 g Magnesium in
20 ml THF und 4 g 1-Brom-2-isopropoxybenzol her. Dann gibt man tropfenweise
und bei TA diese Lösung zu
einer Mischung von 4,8 g der in der vorstehenden Stufe erhaltenen
Verbindung in 50 ml THF und erhitzt anschließend 1 Stunde lang unter Rückfluß. Nach
dem Abkühlen
auf TA gießt
man die Reaktionsmischung auf eine Mischung von Eis/HCl 6 N, extrahiert
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Dann
chromatographiert man den Rückstand über Kieselgel,
indem man mit einer Mischung von DCM/MeOH (99/1; V/V) eluiert. Man
erhält
auf diese Weise 3,8 g des erwarteten Produktes.
-
C) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat
-
Man
beläßt eine
Mischung von 1 g der in der vorstehenden Stufe erhaltenen Verbindung
und 1,4 g Phenylchlorformiat in 10 ml Pyridin 48 Stunden lang bei
TA unter Rühren.
Dann konzentriert man unter Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser, mit einer 1 N Lösung von HCl und mit Wasser,
trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 0,86 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 197°C.
-
Herstellung 1.10
-
5-Chlor-3-(2-isopropoxyphenyl)-2-oxo-3-[(phenoxycarbonyl)-oxy]-1-indolincarboxylat
von Phenyl
-
(XXVII): R1 =
Cl; R2 = H; R3 =
OCH(CH3)2; R4 = H
-
Man
erhitzt eine Mischung von 6,34 g der in der Stufe B der Herstellung
1.3 erhaltenen Verbindung in 60 ml Pyridin auf 70°C, setzt
tropfenweise 7 g Phenylchlorformiat hinzu und beläßt 3 Stunden
lang bei 70°C sowie über Nacht
bei TA unter Rühren.
Dann gießt
man die Reaktionsmischung in Wasser, extrahiert mit DCM, wäscht die
organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Danach
chromatographiert man den Rückstand über Kieselgel,
indem man mit DCM eluiert. Anschließend verreibt man das erhaltene
Produkt in Isoether und zentrifugiert den erhaltenen Niederschlag.
Man erhält
auf diese Weise 8 g des erwarteten Produktes.
-
Herstellung
1.11 5-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat (II):
R
1 = Cl; R
2 = H;
R
3 = Cl; R
4 = H;
X = -O-;
-
A) 5-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-hydroxy-1,3-dihydro-2H-indol-2-on
-
Man
stellt eine Lösung
von 2-Chlorphenylmagnesiumbromid ausgehend von 0,4 g Magnesium,
0,01 g Iod, 20 ml Ether und 2,9 g 1-Brom-2-chlorbenzol her. Dann gibt man zu dieser
Lösung
unter Rückfluß und innerhalb
von 1 Minute eine Mischung von 2,8 g der in der Stufe A der Herstellung
1.9 erhaltenen Verbindung in 30 ml THF und hält das Ganze 20 Minuten lang
unter Rückfluß. Nach
dem Abkühlen
auf TA gießt
man die Reaktionsmischung auf eine Mischung von Eis/konzentrierter
HCl, extrahiert mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Dann
chromatographiert man den Rückstand über Kieselgel,
indem man mit Isoether und danach mit DCM eluiert. Man erhält auf diese
Weise 1,55 g des erwarteten Produktes in Form von Schaum, den man
so wie er ist weiter verwendet.
-
B) 5-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat
-
Man
beläßt eine
Mischung von 0,65 g der in der vorstehenden Stufe erhaltenen Verbindung
und 0,3 g Phenylchlorformiat in 5 ml Pyridin 20 Stunden lang bei
20°C unter
Rühren.
Dann konzentriert man unter Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit einer 1 N Lösung von HCl, mit einer 1 N
Lösung
von NaOH und mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 0,6 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 222–223°C.
-
Herstellung
1.12 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-fluorphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat (II):
R
1 = Cl; R
2 = H;
R
3 = F; R
4 = H;
X = -O-;
-
-
A) DL-2-Fluormandelsäure
-
Man
stellt diese Verbindung gemäß dem in
J. Org. Chem., 1968, 33, 2565–2566
beschriebenen Verfahren her. Man kann ebenfalls diese Verbindung
nach der folgenden Verfahrensweise herstellen.
-
Man
kühlt eine
Mischung von 17,4 g 2-Fluorbenzaldehyd und 9,6 g Kaliumcyanid in
30 ml Ether auf eine Temperatur von unter 10°C, setzt innerhalb von 30 Minuten
15 ml konzentrierte HCl hinzu und rührt 2 Stunden lang bei TA.
Nach dem Dekantieren trocknet man die organische Phase über Na2SO4 und verdampft das
Lösungsmittel
unter Vakuum. Den auf diese Weise erhaltenen rohen Rückstand
nimmt man in 20 ml konzentrierter HCl auf und erhitzt 5 Stunden
lang unter Rückfluß. Nach
dem Abkühlen
auf TA extrahiert man die Reaktionsmischung mit Ether, wäscht die
organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 17,5 g des erwarteten Produktes nach Kristallisation
in Isoether.
-
B) N-p-Chlorphenyl-DL-2-fluormandelamid
-
Man
erhitzt eine Mischung von 17,5 g der in der vorstehenden Stufe erhaltenen
Verbindung und 13 g p-Chloranilin in 100 ml 1,2-Dichlorbenzol 3 Stunden lang unter Rückfluß, indem
man das gebildete Wasser mit Hilfe einer Dean-Stark-Apparatur entfernt.
Nach dem Abkühlen
auf TA läßt man kristallisieren.
Danach zentrifugiert man den gebildeten Niederschlag, löst ihn in
AcOEt, wäscht
die organische Phase zweimal mit einer 4 N Lösung von HCl, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 16,2 g des erwarteten Produktes nach Kristallisation
in Isoether.
-
C) 5-Chlor-3-(2-fluorphenyl)-1,3-dihydro-2H-indol-2-on
-
Zu
einer Mischung von 64 ml konzentrierter H2SO4 (95%) und 16 ml rauchender Schwefelsäure (Oleum
zu 30%) gibt man bei TA 16,1 g der in der vorstehenden Stufe erhaltenen
Verbindung und rührt
an schließend
8 Stunden lang. Dann gießt
man die Reaktionsmischung auf eine Mischung von Eis/Wasser, extrahiert mit
AcOEt, wäscht
die organische Phase zweimal mit Wasser, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 12,2 g des erwarteten Produktes nach Kristallisation
in Isoether.
-
D) 3-Brom-5-chlor-3-(2-fluorphenyl)-1,3-dihydro-2H-indol-2-on
-
Zu
einer Lösung
von 4 g der in der vorstehenden Stufe erhaltenen Verbindung in 100
ml Chloroform gibt man bei TA und langsam eine Lösung von 0,78 ml Brom in 20
ml Chloroform. Dann konzentriert man die Reaktionsmischung unter
Vakuum und erhält
4 g des erwarteten Produktes nach Kristallisation in Isoether.
-
E) 5-Chlor-3-(2-fluorphenyl)-5-hydroxy-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 2 g der in der vorstehenden Stufe erhaltenen
Verbindung in 5 ml Wasser und 20 ml THF 20 Stunden lang unter Rückfluß. Dann
konzentriert man unter Vakuum, nimmt den Rückstand in einer Lösung von
Na2CO3 (10%) auf
und zentrifugiert den gebildeten Niederschlag. Man erhält auf diese
Weise 1,45 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 265°C
(Zers.).
-
F) 5-Chlor-3-(2-fluorphenyl)-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 1,4 g der in der vorstehenden Stufe erhaltenen
Verbindung und 0,12 g Zinkchlorid in 32 ml Acetonitril unter Rückfluß, gibt
6 ml HMDS hinzu und erhitzt weitere 8 Stunden lang unter Rückfluß. Dann
konzentriert man unter Vakuum, extrahiert den Rückstand mit AcOEt, wäscht die
organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 1,35 g des erwarteten Produktes nach Kristallisation
in Heptan, F = 125–127°C.
-
G) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-fluorphenyl)-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe B der Herstellung 1.8 beschriebenen Verfahrensweise her, ausgehend
von 1,3 g der in der vorstehenden Stufe erhaltenen Verbindung, 0,1
g Natriumhydrid (60% in Öl),
15 ml THF und 1 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 1,75 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 184–186°C.
-
H) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-fluorphenyl)-3-hydroxy-1,3-dihydro-2H-indol-2-on
-
Man
beläßt eine
Mischung von 1,7 g der in der vorstehenden Stufe erhaltenen Verbindung,
2 ml 12 N HCl und 2 ml Wasser in 30 ml Aceton 4 Stunden lang unter
Rühren.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in MeOH auf und konzentriert
von neuem unter Vakuum. Anschließend verreibt man den Rückstand
in Isoether und zentrifugiert den gebildeten Niederschlag. Man erhält auf diese
Weise 1,3 g des erwarteten Produktes, F = 144–146°C.
-
I) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-fluorphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat
-
Zu
einer Mischung von 1 g der in der vorstehenden Stufe erhaltenen
Verbindung und 0,5 ml Pyridin in 20 ml DCM gibt man bei TA 0,7 g
Phenylchlorformiat und beläßt 48 Stunden
lang unter Rühren.
Danach wäscht man
die Reaktionsmischung zweimal mit Wasser, trocknet die organische
Phase über
Na2SO4 und verdampft das
Lösungsmittel
unter Vakuum. Dann chromatographiert man den Rückstand über Kieselgel, indem man mit einer
Mischung von DCM/MeOH (99,5/0,5; V/V) eluiert. Man erhält auf diese
Weise 0,93 g des erwarteten Produktes.
-
Herstellung
1.13 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-trifluormethylphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat (II):
R
1 = Cl; R
2 = H;
R
3 = CF
3; R
4 = H; X = -O-;
-
-
A) 5-Chlor-3-hydroxy-3-(2-trifluormethylphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 2,3 g Magnesium, 0,01 g Iod in 20 ml Ether
unter Rückfluß, setzt
tropfenweise eine Lösung
von 26 g 1-Brom-2-trifluormethylbenzol in 20 ml Ether zu und erhitzt
60 Minuten lang unter Rückfluß. Dann
gibt man eine Lösung
von 9 g 5-Chlor-1H-indol-2,3-dion in 40 ml THF hinzu und hält den Rückfluß 30 Minuten
lang aufrecht. Nach dem Abkühlen
auf TA gießt
man die Reaktionsmischung auf eine Mischung von Eis/konzentrierter
HCl, extrahiert mit Ether, wäscht
die organische Phase mit einer Lösung
von 1 N NaOH und mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält auf
diese Weise 5,3 g des erwarteten Produktes nach Kristallisation
in Heptan, F = 205–208°C.
-
B) 5-Chlor-3-(2-trifluormethylphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 3 g der in der vorstehenden Stufe erhaltenen
Verbindung und 0,1 g Zinkchlorid in 20 ml Acetonitril unter Rückfluß, setzt
6 ml HMDS hinzu und erhitzt 1 Stunde lang unter Rückfluß. Dann
konzentriert man unter Vakuum, extrahiert den Rückstand mit Ether, wäscht die
organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 3,9 g des erwarteten Produktes in Form eines Öles, das
kristallisiert, F = 175–176°C.
-
C) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-trifluormethylphenyl)-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe B der Herstellung 1.8 beschriebenen Verfahrensweise her, ausgehend
von 3,8 g der in der vorstehenden Stufe erhaltenen Verbindung 0,45
g Natriumhydrid (60% in Öl),
30 ml THF und 2,5 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 5 g des erwarteten Produktes nach Kristallisation in Heptan,
F = 144–145°C.
-
D) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-hydroxy-3-(2-trifluormethylphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
beläßt eine
Mischung von 4,9 g der in der vorstehenden Stufe erhaltenen Verbindung,
3 ml 12 N HCl und 5 ml Wasser in 50 ml THF 15 Stunden lang bei 20°C unter Rühren. Dann
konzentriert man unter Vakuum bei einer Temperatur von unter 40°C, nimmt
den Rückstand
in 30 ml einer Lösung
von NaHCO3 (10%) auf, extrahiert mit AcOEt,
trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält auf
diese Weise 3,8 g des erwarteten Produktes nach Kristallisation
in einer Mischung von Isoether/Heptan (80/20; V/V), F = 143°C.
-
E) 5-Chlor-1-[(2,4-dirnethoxyphenyl)-sulfonyl]-3-(2-trifluormethylphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat
-
Zu
einer Mischung von 1,7 g der in der vorstehenden Stufe erhaltenen
Verbindung in 20 ml Pyridin gibt man tropfenweise und bei 20°C 2 g Phenylchlorformiat
und rührt
7 Stunden lang bei 20°C.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in 30 ml einer Lösung von
AcOH (10%) auf, extrahiert mit Ether, wäscht die organische Phase mit
einer Lösung
von NaHCO3 (10%) und mit Wasser, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 1,8 g des erwarteten Produktes nach Kristallisation
in Heptan, F = 191°C.
-
Herstellung
1.14 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-trifluormethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat (II):
R
1 = Cl; R
2 = H;
R
3 = OCF
3; R
4 = H; X = -O-;
-
A) 5-Chlor-3-hydroxy-3-(2-trifluormethoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Zu
einer Mischung von 2,8 g Magnesium in 20 ml Ether gibt man tropfenweise
eine Lösung
von 25 g 1-Brom-2-trifluormethoxybenzol in 130 ml Ether, wobei man
den Rückfluß aufrechterhält, wenn
er begonnen hat. Am Ende der Zugabe erhitzt man noch 1 Stunde lang
unter Rückfluß. Dann
gibt man bei einer Temperatur von unter 40°C eine Mischung von 7,5 g 5-Chlor-1H-indol-2,3-dion
in 100 ml THF hinzu und erhitzt 1 Stunde lang unter Rückfluß. Nach
dem Abkühlen
auf TA gießt
man die Reaktionsmischung auf eine Mischung von Eis/konzentrierter
HCl, extrahiert mit AcOEt, wäscht
die organische Phase mit einer Lösung
von 1 N NaOH, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 6,5 g des erwarteten Produktes nach Kristallisation
in einer Mischung von DCM/Isoether (20/80; V/V); F = 214°C.
-
B) 5-Chlor-3-(2-trifluormethoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 2 g der in der vorstehenden Stufe erhaltenen
Verbindung und 0,05 g Zinkchlorid und 1,4 g HMDS in 100 ml Acetonitril
2 Stunden lang unter Rückfluß. Dann
konzentriert man unter Vakuum, extrahiert den Rückstand mit AcOEt, wäscht die
organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 2,5 g des erwarteten Produktes, das man so wie es
ist weiter verwendet.
-
C) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-trifluormethoxyphenyl)-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe B der Herstellung 1.8 beschriebenen Verfahrensweise her, ausgehend
von 2,5 g der in der vorstehenden Stufe erhaltenen Verbindung 0,3
g Natriumhydrid (60% in Öl),
50 ml THF und 1,7 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 2,7 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 181°C.
-
D) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-hydroxy-3-(2-trifluormethoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
beläßt eine
Mischung von 2,7 g der in der vorstehenden Stufe erhaltenen Verbindung
und 1 ml konzentrierte HCl in 50 ml Aceton 1 Stunde lang bei einer
Temperatur von unter 40°C
unter Rühren.
Dann konzentriert man unter Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 1,66 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 81°C.
-
E) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-trifluormethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat
-
Zu
einer Mischung von 1,6 g der in der vorstehenden Stufe erhaltenen
Verbindung und 0,8 ml Pyridin in 20 ml DCM gibt man bei TA 1,15
g Phenylchlorformiat und rührt
1 Stunde lang bei TA. Dann wäscht
man die Reaktionsmischung mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man erhält auf diese
Weise 1,34 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 203–204°C.
-
Herstellung
1.15 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat (II):
R
1 = Cl; R
2 = 6-CH
3; R
3 = OCH
3; R
4 = H; X = -O-;
-
A) 5-Chlor-3-(2-methoxyphenyl)-6-methyl-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 0,65 g der in Stufe B der Herstellung
1.6 erhaltenen Verbindung und 0,05 g Zinkchlorid in 10 ml Acetonitril
unter Rückfluß, setzt
2,1 ml HMDS hinzu und hält
den Rückfluß noch 1 Stunde
lang aufrecht. Dann konzentriert man unter Vakuum, extrahiert den
Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 0,7 g des erwarteten Produktes nach Kristallisation
in Heptan, F = 227°C.
-
B) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-methyl-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe B der Herstellung 1.8 beschriebenen Verfahrensweise her, ausgehend
von 0,85 g der in der vorstehenden Stufe erhaltenen Verbindung,
0,06 g Natriumhydrid (60% in Öl),
20 ml THF und 0,6 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 0,95 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 190–191°C.
-
C) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-hydroxy-3-(2-methoxyphenyl)-6-methyl-1,3-dihydro-2H-indol-2-on
-
Zu
einer Lösung
von 0,85 g der in der vorstehenden Stufe erhaltenen Verbindung in
20 ml Aceton gibt man 0,5 ml einer 12 N Lösung von HCl und rührt das
Ganze 4 Stunden lang bei 20°C.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in 50 ml DCM auf und
konzentriert von neuem das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 0,75 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 207–208°C.
-
D) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat
-
Zu
einer Lösung
von 0,7 g der in der vorstehenden Stufe erhaltenen Verbindung in
10 ml Pyridin gibt man 1 g Phenylchlorformiat und rührt 48 Stunden
lang bei 20°C.
Dann konzentriert man unter Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit einer 1 N Lösung von HCl, mit Wasser, mit einer
1 N Lösung
von NaOH und mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 0,75 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 196°C
(Zers.).
-
Herstellung
1.16 5-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-6-methoxy-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat (II):
R
1 = Cl; R
2 = 6-OCH
3; R
3 = Cl; R
4 = H; X = -O-;
-
A) 4-Chlor-3-methoxyanilin
-
Man
hydriert eine Mischung von 36 g 2-Chlor-5-nitroanisol und Raney®-Nickel
in 150 ml MeOH und 200 ml THF bei 35°C und unter einem Druck von
1,3 bar in einer Parr-Apparatur 4 Stunden lang. Dann filtriert man
den Katalysator über
Celite® und
konzentriert das Filtrat unter Vakuum. Man erhält auf diese Weise 28 g des
erwarteten Produktes, das man so wie es ist weiter verwendet.
-
B) N-(4-Chlor-3-methoxyphenyl)-DL-2-chlormandelamid
-
Man
erhitzt eine Mischung von 28 g der in der vorstehenden Stufe erhaltenen
Verbindung und 33,13 g DL-2-Chlormandelsäure in 128 ml 1,2-Dichlorbenzol
4 Stunden lang auf 230°C,
indem man das gebildete Wasser mit Hilfe einer Dean-Stark-Apparatur
entfernt. Dann konzentriert man teilweise die Reaktionsmischung unter
Vakuum und läßt kristallisieren.
Anschließend
zentrifugiert man das gebildete kristalline Produkt und wäscht es
mit Isoether. Man erhält
auf diese Weise 40 g des erwarteten Produktes.
-
C) 5-Chlor-3-(2-chlorphenyl)-6-methoxy-1,3-dihydro-2H-indol-2-on
-
Man
gibt schnell 40 g der in der vorstehenden Stufe erhaltenen Verbindung
zu 550 g Polyphosphorsäure,
erhitzt dann 8 Stunden lang auf 60°C und läßt über Nacht unter Rühren stehen,
wobei die Temperatur wieder auf TA zurückkehrt. Anschließend gibt
man Eiswasser zu der Reaktionsmischung, zentrifugiert den gebildeten
Niederschlag und wäscht
ihn mit Wasser. Danach nimmt man den Nie derschlag in AcOEt auf,
zentrifugiert das erhaltene weiße
Produkt nach dem Verreiben und wäscht
es mit Isoether. Man erhält
auf diese Weise 17,2 g des erwarteten Produktes, F = 243–247°C.
-
D) 5-Chlor-3-(2-chlorphenyl)-3-hydroxy-6-methoxy-1,3-dihydro-2H-indol-2-on
-
Zu
einer Lösung
von 17,2 g der in der vorstehenden Stufe erhaltenen Verbindung in
220 ml THF gibt man bei TA und unter Atmosphäre von Argon 2,56 g Natriumhydrid
(60% in Öl).
Nach dem Aufhören
der Gasentwicklung setzt man 6,85 g Dimethyldisulfid hinzu, leitet
in die Reaktionsmischung Luft ein und rührt 72 Stunden lang bei TA.
Dann versetzt man die Reaktionsmischung mit Wasser, verdampft das
THF unter Vakuum, extrahiert die verbleibende wäßrige Phase mit AcOEt, wäscht die
organische Phase mit Wasser und mit einer gesättigten Lösung von NaCl, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Anschließend löst
man das erhaltene Produkt in DCM, konzentriert teilweise das Lösungsmittel,
läßt kristallisieren
und zentrifugiert das gebildete kristalline Produkt. Man erhält auf diese
Weise 6 g des erwarteten Produktes, F = 237–240°C.
-
E) 5-Chlor-3-(2-chlorphenyl)-6-methoxy-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 1 g der in der vorstehenden Stufe erhaltenen
Verbindung und 0,07 g Zinkchlorid in 10 ml Acetonitril unter Rückfluß, setzt
3 ml HMDS hinzu und hält
den Rückfluß noch 1
Stunde lang aufrecht. Dann konzentriert man unter Vakuum, extrahiert
den Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 1,25 g des erwarteten Produktes nach Kristallisation
in Heptan, F = 211–212°C.
-
F) 5-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-6-methoxy-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe B der Herstellung 1.8 beschriebenen Verfahrensweise her, ausgehend
von 1,2 g der in der vorstehenden Stufe erhaltenen Verbindung, 0,08
g Natriumhy drid (60% in Öl),
15 ml THF und 0,8 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 1,6 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 217–219°C.
-
G) 5-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-hydroxy-6-methoxy-1,3-dihydro-2H-indol-2-on
-
Man
beläßt eine
Mischung von 1,55 g der in der vorstehenden Stufe erhaltenen Verbindung
und 2 ml 12 N HCl in 30 ml Aceton 4 Stunden lang bei 20°C unter Rühren. Dann
konzentriert man unter Vakuum, nimmt den Rückstand in Aceton auf und konzentriert
von neuem unter Vakuum. Danach nimmt man den Rückstand in DCM auf und konzentriert
unter Vakuum. Anschließend
verreibt man den Rückstand
in Isoether und zentrifugiert den gebildeten Niederschlag. Man erhält auf diese
Weise 1,3 g des erwarteten Produktes, F = 233–235°C.
-
H) 5-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-6-methoxy-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat
-
Man
kühlt einer
Mischung von 1,3 g der in der vorstehenden Stufe erhaltenen Verbindung
in 10 ml Pyridin auf 10°C,
setzt 0,8 g Phenylchlorformiat hinzu und rührt 20 Minuten lang bei 20°C. Danach
setzt man wiederum 0,7 g Phenylchlorformiat hinzu und rührt 20 Stunden
lang bei 20°C.
Dann konzentriert man unter Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit einer 1 N Lösung von HCl, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Anschließend
chromatographiert man den Rückstand über Kieselgel,
indem man mit DCM eluiert. Man erhält auf diese Weise 0,7 g des
erwarteten Produktes nach Kristallisation in einer Mischung von
Pentan/Isoether, F = 162–163°C.
-
Herstellung
1.17 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2,5-dimethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat (II):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = 5-OCH
3; X = -O-;
-
A) 5-Chlor-3-hydroxy-3-(2,5-dimethoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
stellt eine Lösung
von 2,5-Dimethoxyphenylmagnesiumbromid ausgehend von 2,2 g Magnesium, 18
g 1-Brom-2,5-dimethoxybenzol und 50 ml Ether her. Dann gibt man
diese Lösung
bei einer Temperatur von unter 30°C
tropfenweise zu einer Mischung von 5 g 5-Chlor-1H-indol-2,3-dion in 50 ml THF
und erhitzt anschließend
3 Stunden lang unter Rückfluß. Nach
dem Abkühlen
auf TA gießt
man die Reaktionsmischung in eine 1 N Lösung von HCl, extrahiert mit
AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 7,1 g des erwarteten Produktes nach Kristallisation
in warmem Isoether.
-
B) 5-Chlor-3-(2,5-dimethoxyphenyl)-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 4 g der in der vorstehenden Stufe erhaltenen
Verbindung und 0,085 g Zinkchlorid in 45 ml Acetonitril unter Rückfluß, setzt
2,8 ml HMDS hinzu und hält
den Rückfluß noch 1
Stunde lang aufrecht. Dann konzentriert man unter Vakuum, nimmt
den Rückstand
in Wasser auf, extrahiert mit Ether, wäscht die organische Phase mit
Wasser, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 5 g des erwarteten Produktes nach Kristallisation
in Isoether.
-
C) 5-Chlor-3-(2,5-dimethoxyphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe B der Herstellung 1.8 beschriebenen Verfahrensweise her, ausgehend
von 2 g der in der vorstehenden Stufe erhaltenen Verbindung, 0,135
g Natriumhydrid (60% in Öl),
40 ml THF und 1,3 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 2,2 g des erwarteten Produktes nach Kristallisation in Isoether.
-
D) 5-Chlor-3-(2,5-dimethoxyphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-hydroxy-1,3-dihydro-2H-indol-2-on
-
Man
beläßt eine
Mischung von 1 g der in der vorstehenden Stufe erhaltenen Verbindung
und 0,5 ml 12 N HCl in 20 ml Aceton 4 Stunden lang bei TA unter
Rühren.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in DCM auf und konzentriert
von neuem unter Vakuum. Anschließend verreibt man den Rückstand
in Isoether und zentrifugiert den gebildeten Niederschlag. Man erhält auf diese
Weise 0,84 g des erwarteten Produktes.
-
E) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2,5-dimethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat
-
Man
beläßt eine
Mischung von 0,8 g der in der vorstehenden Stufe erhaltenen Verbindung
und 0,4 ml Phenylchlorformiat in 5 ml Pyridin über Nacht bei TA unter Rühren. Danach
versetzt man die Reaktionsmischung mit Wasser, extrahiert mit DCM,
wäscht
die organische Phase mit einer 2 N Lösung von HCl, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 0,84 g des erwarteten Produktes nach Kristallisation
in Isoether.
-
Herstellung
1.18 A)
5-Chlor-l-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat von
Phenyl (II):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = H; X = -NH-;
-
A) 3-Amino-5-chlor-3-(2-methoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
WO 95/18105 bei der Herstellung 7 beschriebenen Verfahrensweise
her.
-
B) 3-Amino-5-chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-1,3-dihydro-1H-indol-2-an
-
Man
kühlt eine
Lösung
von 1,64 g der in der vorstehenden Stufe erhaltenen Verbindung in
20 ml DMF auf 4°C,
setzt 0,25 g Natriumhydrid (60% in Öl) hinzu und rührt 30 Minuten
lang bei 4°C.
Dann gibt man 1,34 g 2,4-Dimethoxybenzolsulfonylchlorid hinzu und
rührt 4
Stunden lang bei TA. Anschließend
versetzt man die Reaktionsmischung mit 50 ml Wasser, extrahiert
mit AcOEt, wäscht
die organische Phase mit einer gesättigten Lösung von NaCl, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/AcOEt (97/3; V/V) eluiert. Man erhält auf diese
Weise 2 g des erwarteten Produktes nach Kristallisation in einer
Mischung DCM/Isoether.
-
C) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat von
Phenyl
-
Man
kühlt eine
Lösung
von 2 g der in der vorstehenden Stufe erhaltenen Verbindung und
10 ml Pyridin in 10 ml DCM auf 4°C,
setzt 0,77 ml Phenylchlorformiat hinzu und rührt 1 Stunde lang bei TA. Dann
konzentriert man unter Vakuum, extrahiert den Rückstand mit AcOEt, wäscht die
organische Phase mit einer gesättigten Lösung von
NaCl, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Anschließend
chromatographiert man den Rückstand über Kieselgel,
indem man mit einer Mischung von DCM/AcOEt (96/4; V/V) eluiert.
Man erhält
auf diese Weise 2,6 g des erwarteten Produktes nach Kristallisation
in einer Mischung DCM/Isoether, F = 191°C.
-
Herstellung
1.19 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat von
Phenyl, einziges Isomer (II):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = H; X = -NH-;
-
A) 3,5-Dichlor-3-(2-methoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
kühlt eine
Mischung von 9 g der in Stufe A der Herstellung 1.1 erhaltenen Verbindung
und 3,74 ml Pyridin in 100 ml DCM auf eine Temperatur von 0°C, setzt
dann tropfenweise innerhalb von 3 Minuten eine Lösung von 3,45 ml Thionylchlorid
in 3 ml DCM hinzu und rührt
30 Minuten lang. Dann versetzt man die Reaktionsmischung mit Wasser
und verdampft das DCM unter Vakuum. Anschließend zentrifugiert man den
gebildeten Feststoff, wäscht
ihn viermal mit Wasser und danach mit kaltem Isoether und trocknet
ihn. Man erhält auf
diese Weise 8,8 g des erwarteten Produktes.
-
B) 5-Chlor-3-{[(1S)-2-hydroxy-1-phenylethyl]-amino}-3-(2-methoxyphenyl)-1,3-dihydro-2H-indol-2-on,
Isomer A und B
-
Man
beläßt eine
Mischung von 7 g der in der vorstehenden Stufe erhaltenen Verbindung
und 7,65 g (S)-(+)-α-Phenylglycinol
in 100 ml Chloroform 2 Stunden lang bei TA unter Rühren. Dann
setzt man 2,9 g DIPEA hinzu und rührt 48 Stunden lang bei TA.
Anschließend
zentrifugiert man den gebildeten Niederschlag und gewinnt das Isomer
A. Danach wäscht
man die Laugen aus der Zentrifugierung mit einer Lösung von
K2CO3 (5%), trocknet
die organische Phase über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Dann chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/AcOEt (70/30; V/V) eluiert.
-
Man
trennt die zwei Diastereoisomeren:
- – das am
wenigsten polare, Isomer A: man erhält 4,55 g (Niederschlag und
Chromatographie); α20 D = +193° (c = 0,16;
Chloroform)
- – das
am meisten polare, Isomer B.
-
C) 3-Amino-5-chlor-3-(2-methoxyphenyl)-1,3-dihydro-2H-indol-2-on,
rechtsdrehendes Isomer
-
Man
beläßt eine
Mischung von 4,55 g der in der vorstehenden Stufe erhaltenen Verbindung
(Isomer A) und 5,5 g Bleitetraacetat in 75 ml DCM und 35 ml MeOH
1 Stunde und 30 Minuten lang bei TA unter Rühren. Dann konzentriert man
unter Vakuum, nimmt den Rückstand
in einer gesättigten
Lösung
von NaHCO3 auf, extrahiert mit AcOEt, wäscht die
organische Phase mit Wasser und mit einer gesättigten Lösung von NaCl, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Das erhaltene Öl
nimmt man in 100 ml einer 3 N Lösung
von HCl auf, setzt 10 ml MeOH hinzu und rührt 2 Stunden lang bei TA.
Dann konzentriert man die organischen Lösungsmittel unter Vakuum, wäscht die
wäßrige Phase
zweimal mit Ether, alkalisiert die wäßrige Phase durch Zugabe von
KOH in Pastillen, extrahiert mit AcOEt, wäscht die organische Phase mit
Wasser und mit einer gesättigten
Lösung
von NaCl, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 1,178 g des erwarteten Produktes nach Kristallisation
in einer Mischung von DCM/Isoether/THF, F = 202°C.
α20 D = +83,3° (c
= 0,16; Chloroform).
-
D) 3-Amino-5-chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-1,3-dihydro-2H-indol-2-on, rechtsdrehendes
Isomer
-
Man
kühlt eine
Lösung
von 1,178 g der in der vorstehenden Stufe erhaltenen Verbindung
in 10 ml DMF auf eine Temperatur von 0°C, setzt unter Atmosphäre von Argon
0,188 g Natriumhydrid (60% in Öl)
hinzu und rührt
bis zum Aufhören
der Gasentwicklung. Danach gibt man 1,02 g 2,4-Dimethoxybenzolsulfonylchlorid
hinzu und rührt
3 Stunden lang bei TA. Anschließend
gießt
man die Reaktionsmischung in eine Lösung von K2CO3 (5%), extrahiert mit AcOEt, wäscht die
organische Phase mit Wasser, mit einer Lösung von K2CO3 (5%) und mit einer gesättigten Lösung von NaCl, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Dann chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/AcOEt (95/5; V/V) eluiert. Man erhält auf diese
Weise 1,254 g des erwarteten Produktes nach Kristallisation in einer Mischung
von DCM/Ether/Isoether, F = 172–173°C.
α20 D = +113° (c
= 0,18; Chloroform).
-
E) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat
von Phenyl, einziges Isomer
-
Man
kühlt im
Eisbad eine Lösung
von 1,1 g der in der vorstehenden Stufe erhaltenen Verbindung in 10
ml DCM, setzt 1,8 ml Pyridin und anschließend tropfenweise 0,37 g Phenylchlorformiat
hinzu und läßt das Ganze über Nacht
in der Kälte
stehen. Dann verdünnt
man die Reaktionsmischung mit DCM, wäscht die organische Phase dreimal
mit Wasser, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/AcOEt (98/2; V/V) eluiert. Man erhält auf diese
Weise 1,136 g des erwarteten Produktes.
-
Herstellung
1.20 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-carbarnat von
Phenyl, einziges Isomer (II):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -NH-;
-
A) 3,5-Dichlor-3-(2-isopropoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
kühlt eine
Mischung von 12 g der in Stufe B der Herstellung 1.3 erhaltenen
Verbindung und 8,35 ml Pyridin in 165 ml DCM auf eine Temperatur
von 0°C,
setzt dann tropfenweise 7,62 ml Thionylchlorid hinzu und rührt 15 Minuten
lang. Dann konzentriert man unter Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht die
organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Danach
chromatographiert man den Rückstand über Kieselgel,
indem man mit einer Mischung von DCM/AcOEt (90/10; V/V) eluiert.
Man erhält
auf diese Weise 15,22 g des erwarteten Produktes.
-
B) 5-Chlor-3-{[(1S)-2-hydroxy-1-phenylethyl]-amino}-3-(2-isopropoxyphenyl)-1,3-dihydro-2H-indol-2-on,
Isomer A und B
-
Man
beläßt eine
Mischung von 8,17 g der in der vorstehenden Stufe erhaltenen Verbindung
und 3,66 g (S)-(+)-α-Phenylglycinol
in 265 ml Chloroform 1 Stunde lang bei TA unter Rühren. Dann
setzt man 4,66 ml DIPEA hinzu, rührt
3 Stunden lang bei TA und erhitzt danach 18 Stunden lang auf 50°C. Dann konzentriert
man unter Vakuum, extrahiert den Rückstand mit AcOEt, wäscht die
organische Phase mit einer Lösung
von K2CO3 (5%) und
mit Wasser, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/AcOEt (90/10; V/V) eluiert, und trennt:
- – das
am wenigsten polare, Isomer A: man erhält 4,98 g nach Kristallisation
in Isoether, F = 212,7°C
α20 D = +212,4° (c
= 0,2; Chloroform).
Dann setzt man die Chromatographie fort,
indem man mit einer Mischung von DCM/AcOEt (70/30; V/V) eluiert,
und trennt:
- – das
am meisten polare, Isomer B: man erhält 3,49 g nach Kristallisation
in Isoether, F = 241,3°C
α20 D = –6,6° (c = 0,2;
Chloroform).
-
C) 3-Amino-5-chlor-3-(2-isopropoxyphenyl)-1,3-dihydro-2H-indol-2-on, rechtsdrehendes
Isomer
-
Man
kühlt eine
Lösung
von 3,9 g der in der vorstehenden Stufe erhaltenen Verbindung (Isomer
A) in 75 ml DCM und 37 ml MeOH auf eine Temperatur von 0°C, setzt
4,35 g Bleitetraacetat hinzu und beläßt 2 Stunden lang unter Rühren. Dann
gibt man 3 Tropfen einer Lösung
von NaHCO3 (5%) hinzu, konzentriert unter
Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser und mit einer gesättigten Lösung von
NaCl, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Anschließend
nimmt man das erhaltene Produkt in 120 ml konzentrierter HCl auf, wäscht mit
Ether, alkalisiert die wäßrige Phase durch
Zusatz von K2CO3,
extrahiert mit AcOEt und läßt kristallisieren.
Danach zentrifugiert man die gebildeten Kristalle und trocknet sie.
Dann konzentriert man die Laugen aus der Zentrifugierung, chromatographiert
den Rückstand über Kieselgel,
indem man mit einer Mischung von DCM/AcOEt (65/35; V/V) eluiert,
und kristallisiert das erhaltene Produkt in Isoether. Man erhält auf diese
Weise insgesamt 2,03 g des erwarteten Produktes, F = 193°C.
α20 D = +44° (c
= 0,18; Chloroform).
-
D) 3-Amino-5-chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-1,3-dihydro-2H-indol-2-on, rechtsdrehendes
Isomer
-
Man
stellt diese Verbindung gemäß der in
Stufe D der Herstellung 1.19 beschriebenen Verfahrensweise her,
ausgehend von 1,86 g der in der vorstehenden Stufe erhaltenen Verbindung,
0,282 g Natriumhydrid (60% in Öl),
20 ml DMF und 1,395 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 2,68 g des erwarteten Produktes nach Kristallisation in Hexan.
α20 D = +79,5° (c
= 0,2; Chloroform).
-
E) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat
von Phenyl, einziges Isomer
-
Man
kühlt eine
Mischung von 2,68 g der in der vorstehenden Stufe erhaltenen Verbindung
und 4,5 ml Pyridin in 25 ml DCM auf eine Temperatur von 0°C, setzt
anschließend
innerhalb von 30 Sekunden 1,06 g Phenylchlorformiat hinzu und läßt das Ganze
18 Stunden lang in der Kälte
stehen. Dann chromatographiert man direkt die Reaktionsmischung über Kieselgel,
hergestellt in der Mischung DCM/Hexan (80/20; V/V), indem man mit
einer Mischung von DCM/AcOEt (95/5; V/V) eluiert. Man erhält auf diese
Weise 2,88 g des erwarteten Produktes, das man so wie es ist weiter
verwendet.
-
Herstellung
1.21 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat von
Phenyl, einziges Isomer (II):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -NH-;
-
A) 3-Amino-5-chlor-3-(2-isopropoxyphenyl)-1,3-dihydro-2H-indol-2-on, linksdrehendes
Isomer
-
Man
kühlt eine
Lösung
von 3,4 g der in Stufe B der Herstellung 1.20 erhaltenen Verbindung
(Isomer B) in 65 ml DCM und 32,4 ml MeOH auf eine Temperatur von
0°C, setzt
dann 3,8 g Bleitetraacetat hinzu und rührt 40 Minuten lang. Dann gibt
man 3 Tropfen einer Lösung
von NaHCO3 (5%) hinzu, konzentriert unter
Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser und mit einer gesättigten Lösung von
NaCl, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Das erhaltene Produkt nimmt man in 120 ml 0,5 N HCl
auf, wäscht
mit Ether, alkalisiert die wäßrige Phase
durch Zusatz von K2CO3 und
zentrifugiert den gebildeten Niederschlag. Man erhält auf diese
Weise 1,9 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 192°C.
α20 D = –42° (c = 0,2;
Chloroform).
-
B) 3-Amino-5-chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-1,3-dihydro-2H-indol-2-on, linksdrehendes
Isomer
-
Man
stellt diese Verbindung gemäß der in
Stufe D der Herstellung 1.19 beschriebenen Verfahrensweise her,
ausgehend von 1,67 g der in der vorstehenden Stufe erhaltenen Verbindung,
0,253 g Natriumhydrid (60% in Öl),
20 ml DMF und 1,249 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 2,453 g des erwarteten Produktes nach Kristallisation in Hexan.
α20 D = –79,8
(c = 0,2; Chloroform).
-
C) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat
von Phenyl, einziges Isomer
-
Man
kühlt eine
Mischung von 2,103 g der in der vorstehenden Stufe erhaltenen Verbindung
und 3,5 ml Pyridin in 20 ml DCM auf eine Temperatur von 0°C, setzt
anschließend
0,665 ml Phenylchlorformiat hinzu und läßt das Ganze 36 Stunden lang
in der Kälte
stehen. Dann konzentriert man unter Vakuum, nimmt den Rückstand
in einer Lösung
von K2CO3 (5%) auf,
extrahiert mit AcOEt, wäscht
die organische Phase mit Wasser und mit einer gesättigten
Lösung
von NaCl, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/Hexan (80/20; V/V) eluiert. Man erhält auf diese
Weise 1,88 g des erwarteten Produktes, das man so wie es ist weiter
verwendet.
-
Herstellung
1.22 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2,5-dimethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat
von Phenyl (II):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = 5-OCH
3; X = -NH-;
-
A) 3,5-Dichlor-3-(2,5-dimethoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
kühlt eine
Lösung
von 3 g der in Stufe A der Herstellung 1.17 erhaltenen Verbindung
und 1,2 ml Pyridin in 50 ml DCM auf eine Temperatur von unter 20°C, setzt
dann 0,8 ml Thionylchlorid hinzu und rührt 1 Stunde lang. Dann wäscht man
die Reaktionsmischung mit Wasser, trocknet die organische Phase über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
DCM eluiert. Man erhält
auf diese Weise 1,9 g des erwarteten Produktes, das man so wie es
ist weiter verwendet.
-
B) 3-Amino-5-chlor-3-(2,5-dimethoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
kühlt eine
Mischung von 1,25 g der in der vorstehenden Stufe erhaltenen Verbindung
in 7 ml THF auf eine Temperatur von 0°C, leitet anschließend innerhalb
von 6 Stunden viermal 10 Minuten lang Ammoniakgas ein und rührt 24 Stunden
lang bei TA. Dann konzentriert man unter Vakuum, nimmt den Rückstand
in DCM auf und verdampft das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
DCM und danach mit einer Mischung von DCM/AcOEt (60/40; V/V) eluiert.
Man erhält
auf diese Weise 0,808 g des erwarteten Produktes, das man so wie
es ist weiter verwendet.
-
C) 3-Amino-5-chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2,5-dimethoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe D der Herstellung 1.19 beschriebenen Verfahrensweise her,
ausgehend von 0,749 g der in der vorstehenden Stufe erhaltenen Verbindung,
0,113 g Natriumhydrid (60% in Öl),
7 ml DMF und 0,612 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 0,9 g des erwarteten Produktes nach Kristallisation in einer
Mischung von DCM/Isoether, F = 191°C.
-
D) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2,5-dimethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat
von Phenyl
-
Man
kühlt eine
Mischung von 0,52 g der in der vorstehenden Stufe erhaltenen Verbindung
und 1 ml Pyridin in 5 ml DCM auf eine Temperatur von 0°C, setzt
anschließend
0,16 ml Phenylchlorformiat hinzu und rührt 16 Stunden lang. Dann versetzt
man die Reaktionsmischung mit Wasser, verdampft das DCM unter Vakuum,
extrahiert mit AcOEt, wäscht
die organische Phase mit einer Lösung
von K2CO3 (5%),
mit Wasser und mit einer gesättigten
Lösung
von NaCl, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
DCM eluiert. Man erhält
auf diese Weise 0,46 g des erwarteten Produktes, das man so wie
es ist weiter verwendet.
-
Herstellung
1.23 5-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat
von Phenyl (II):
R
1 = Cl; R
2 = H;
R
3 = Cl; R
4 = H;
X = -NH-;
-
A) 3-Amino-5-chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
WO 95/18105 in Beispiel 3 beschriebenen Verfahrensweise her.
-
B) 5-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat von
Phenyl
-
Man
kühlt eine
Lösung
von 3 g der in der vorstehenden Stufe erhaltenen Verbindung in 25
ml Pyridin auf eine Temperatur von 0°C, setzt anschließend tropfenweise
eine Lösung
von 1,25 g Phenylchlorformiat in 2 ml DCM hinzu und rührt 3 Stunden
lang bei 0°C
sowie über
Nacht bei TA. Dann kühlt
man von neuem auf 0°C
ab, setzt 0,96 g Phenylchlorformiat hinzu und läßt das Ganze 18 Stunden lang
in der Kälte
stehen. Anschließend
konzentriert man unter Vakuum, nimmt den Rückstand in einer Lösung von
K2CO3 (5%) auf,
extrahiert mit AcOEt, trocknet die organische Phase über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/AcOEt (95/5; V/V) eluiert. Man erhält auf diese
Weise 2,88 g des erwarteten Produktes.
-
Herstellung
1.24 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat
von Phenyl (II):
R
1 = Cl; R
2 = 6-CH
3; R
3 = OCH
3; R
4 = H; X = -NH-;
-
A) 3,5-Dichlor-3-(2-methoxyphenyl)-6-methyl-1,3-dihydro-2H-indol-2-on
-
Man
kühlt eine
Mischung von 2,0 g der in Stufe B der Herstellung 1.6 erhaltenen
Verbindung in 45 ml DCM auf eine Temperatur von 0°C, setzt
anschließend
0,77 ml Pyridin und danach 1,17 g Thionylchlorid hinzu und rührt 2 Stunden
lang, nachdem man die Temperatur wieder auf TA angestiegen war.
Dann versetzt man die Reaktionsmischung mit Wasser und DCM, wäscht nach
dem Dekantieren die organische Phase viermal mit Wasser, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise das erwartete Produkt, das man so wie es ist weiter
verwendet.
-
B) 3-Amino-5-chlor-3-(2-methoxyphenyl)-6-methyl-1,3-dihydro-2H-indol-2-on
-
In
eine Mischung von 3,4 g der in der vorstehenden Stufe erhaltenen
Verbindung in 25 ml DCM leitet man bei TA 30 Minuten lang Ammoniakgas
ein und rührt
anschließend
18 Stunden lang. Dann konzentriert man unter Vakuum, extrahiert
den Rückstand
mit AcOEt, wäscht
die organische Phase mit einer gesättigten Lösung von NaCl, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/MeOH (95/5; V/V) eluiert. Man erhält auf diese
Weise 2 g des erwarteten Produktes.
-
C) 3-Amino-5-chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-methyl-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe B der Herstellung 1.18 beschriebenen Verfahrensweise her,
ausgehend von 2 g der in der vorstehenden Stufe erhaltenen Verbindung,
0,29 g Natriumhydrid (60% in Öl),
15 ml DMF und 1,54 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 3,2 g des erwarteten Produktes.
-
D) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat
von Phenyl
-
Man
kühlt eine
Lösung
von 3,2 g der in der vorstehenden Stufe erhaltenen Verbindung und
10 ml Pyridin in 30 ml DCM auf eine Temperatur von 4°C, setzt
anschließend
tropfenweise 1,2 ml Phenylchlorformiat hinzu und rührt 2 Stunden
lang bei TA. Dann konzentriert man unter Vakuum, extrahiert den
Rückstand
mit AcOEt, wäscht
die organische Phase mit einer Lösung
von KHSO4 (5%), mit einer Lösung von
Na2CO3 (5%), mit
Wasser und mit einer gesättigten
Lösung
von NaCl, trocknet über
Na2SO4 und verdampft
das Lösungsmittel unter
Vakuum. Man erhält
auf diese Weise 2,3 g des erwarteten Produktes nach Kristallisation
in einer Mischung von DCM/Isoether.
-
Herstellung
1.25 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-trifluormethyl-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat
von Phenyl, einziges Isomer (II):
R
1 = Cl; R
2 = 6-CF
3; R
3 = OCH
3; R
4 = H; X = -NH-;
-
A) 5-Chlor-3-hydroxy-3-(2-methoxyphenyl)-6-trifluormethyl-1,3-dihydro-2H-indol-2-on
-
a) 4-Chlor-3-trifluormethyl-phenylcarbamat
von tert.-Butyl
-
Man
stellt diese Verbindung gemäß der in
Stufe B der Herstellung 1.6 beschriebenen Verfahrensweise her, ausgehend
von 4-Chlor-3-trifluormethyl-anilin
und Di-tert.-Butyldicarbonat in Dioxan. Man erhält auf diese Weise das erwartete
Produkt in Form von Öl,
das sich verfestigt, F = 90°C.
-
b)
Man kühlt
eine Lösung
von 4 g 4-Chlor-3-trifluormethyl-phenylcarbamat von tert.-Butyl
in 30 ml Ether unter Atmosphäre
von Argon auf eine Temperatur von –70°C, setzt anschließend tropfenweise
22 ml einer 1,5 M Lösung
von tert.-Butyllithium in Pentan hinzu, rührt 1 Stunde lang, wobei man
die Temperatur auf –10°C ansteigen
läßt, und
rührt dann
noch 2 Stunden und 30 Minuten lang bei –10°C. Dann kühlt man die Reaktionsmischung
wieder auf –70°C ab, gibt
tropfenweise eine Lösung
von 3,05 g der in Stufe A der Herstellung 1.6 erhaltenen Verbindung
in 15 ml THF hinzu und rührt
1 Stunde lang, wobei man die Temperatur auf –30°C ansteigen läßt, und
anschließend
noch 16 Stunden lang, wobei man die Temperatur auf TA ansteigen
läßt. Dann
versetzt man die Reaktionsmischung mit einer gesättigten Lösung von NH4Cl,
verdampft den Ether und das THF, extrahiert die resultierende wäßrige Phase
mit AcOEt, wäscht
die organische Phase mit Wasser und mit einer gesättigten
Lösung
von NaCl, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
DCM und danach mit einer Mischung von DCM/AcOEt (90/10; V/V) eluiert.
Man erhält
auf diese Weise 1,48 g des erwarteten Produktes nach Kristallisation
in einer Mischung von Isoether/Hexan, F = 230–231°C.
-
B) 3,5-Dichlor-3-(2-methoxyphenyl)-6-trifluormethyl-1,3-dihydro-2H-indol-2-on
-
Man
kühlt eine
Suspension von 1,3 g der in Stufe A erhaltenen Verbindung in 8 ml
DCM auf eine Temperatur von 0°C,
setzt anschließend
0,43 ml Pyridin und danach 0,4 ml Thionylchlorid hinzu und rührt 15 Minuten
lang. Dann wäscht
man die Reaktionsmischung dreimal mit Wasser, trocknet die organische
Phase über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 1,2 g des erwarteten Produktes, das man so wie es
ist weiter verwendet.
-
C) 5-Chlor-3-{[(1S)-2-hydroxy-1-phenylethyl]-amino}-3-(2-methoxyphenyl)-6-trifluormethyl-1,3-dihydro-2H-indol-2-on,
Isomer A und Isomer B
-
Man
beläßt eine
Mischung von 1,29 g der in der vorstehenden Stufe erhaltenen Verbindung
und 0,47 g (S)-(+)-α-Phenylglycinol
in 35 ml Chloroform 4 Stunden lang bei TA unter Rühren. Dann
setzt man 0,6 ml DIPEA hinzu, konzentriert das Lösungsmittel auf die Hälfte und
rührt 22
Stunden lang bei TA. Dann konzentriert man die Reaktionsmischung
unter Vakuum und chromatographiert den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/AcOEt (85/15; V/V) eluiert. Man trennt ein
Diastereoisomer:
- – das am wenigsten polare,
Isomer A: man erhält
0,712 g nach Kristallisation in einer Mischung von DCM/Isoether,
F = 205°C
α20 D = +164° (c
= 0,16; Chloroform).
Dann setzt man die Chromatographie fort,
indem man mit einer Mischung von DCM/AcOEt (70/30; V/V) eluiert,
und das andere Diastereoisomer trennt:
- – das
am meisten polare, Isomer B: man erhält 0,45 g, F = 242°C
α20 D = –55° (c = 0,15;
Chloroform).
-
D) 3-Amino-5-chlor-3-(2-methoxyphenyl)-6-trifluormethyl-1,3-dihydro-2H-indol-2-on,
einziges Isomer
-
Man
kühlt eine
Lösung
von 0,7 g der in der vorstehenden Stufe erhaltenen Verbindung (Isomer
A) in 15 ml DCM und 7 ml MeOH auf eine Temperatur von 0°C, setzt
anschließend
0,907 g Bleitetraacetat hinzu und rührt 40 Minuten lang. Dann gibt
man einige Tropfen einer Lösung
von NaHCO3 (5%) hinzu und konzentriert unter
Vakuum. Danach nimmt man den Rückstand
in Wasser auf, extrahiert dreimal mit AcOEt, wäscht die organische Phase mit
Wasser und mit einer gesättigten
Lösung
von NaCl, trocknet über
Na2SO4 und verdampft das
Lösungsmittel
unter Vakuum. Das erhaltene Produkt nimmt man in 30 ml Wasser auf,
setzt tropfenweise 30 ml einer 5 N Lösung von HCl und danach THF
bis zur Auflösung
hinzu und rührt
1 Stunde lang. Anschließend
verdünnt
man die Reaktionsmischung mit 50 ml Wasser, wäscht die wäßrige Phase mit 150 ml Ether, alkalisiert
die wäßrige Phase
durch Zugabe von K2CO3 und
anschließend
einer konzentrierten Lösung
von NaOH auf pH = 12, extrahiert mit AcOEt, trocknet die organische
Phase über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/MeOH (92/8; V/V) eluiert. Man erhält auf diese
Weise 0,19 g des erwarteten Produktes.
-
E) 3-Amino-5-chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-trifluormethyl-1,3-dihydro-2H-indol-2-on,
einziges Isomer
-
Man
stellt diese Verbindung gemäß der in
Stufe B der Herstellung 1.18 beschriebenen Verfahrensweise her,
ausgehend von 0,19 g der in der vorstehenden Stufe erhaltenen Verbindung,
0,023 g Natriumhydrid (60% in Öl),
2 ml DMF und 0,138 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 0,22 g des erwarteten Produktes.
-
F) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-trifluormethyl-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat
von Phenyl, einziges Isomer
-
Man
stellt diese Verbindung gemäß der in
Stufe C der Herstellung 1.18 beschriebenen Verfahrensweise her,
ausgehend von 0,215 g der in der vorstehenden Stufe erhaltenen Verbindung,
0,31 ml Pyridin, 5 ml DCM und 0,079 g Phenylchlorformiat. Man erhält auf diese
Weise 0,208 g des erwarteten Produktes.
-
Herstellung
1.26 6-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-5-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat
von Phenyl (II):
R
1 = CH
3; R
2 = 6-Cl; R
3 = Cl;
R
4 = H; X = -NH-;
-
A) N-(3-Chlor-4-methylphenyl)-DL-2-chlormandelamid
-
Man
erhitzt eine Mischung von 52,72 g DL-2-Chlormandelsäure und
40 g 3-Chlor-4-methylanilin in 400 ml 1,2-Dichlorbenzol 6 Stunden
lang unter Rückfluß, indem
man das gebildete Wasser mit Hilfe einer Dean-Stark-Apparatur entfernt.
Nach dem Abkühlen
auf TA läßt man kristallisieren
und zentrifugiert den gebildeten Niederschlag. Man erhält auf diese
Weise das erwartete Produkt nach Kristallisation in einer Mischung von
DCM/Isoether, F = 164°C.
-
B) 6-Chlor-3-(2-chlorphenyl)-5-methyl-1,3-dihydroindol-2-on
-
Man
kühlt 102
ml konzentrierte H2SO4 (95%)
auf 5°C
und setzt tropfenweise 23 ml rauchende Schwefelsäure (Oleum zu 30%) hinzu. Dann
gibt man in kleinen Fraktionen und bei einer Temperatur von unter
5°C 25 g
der in der vorstehenden Stufe erhaltenen Verbindung hinzu und rührt anschließend 48
Stunden lang bei TA. Danach gießt
man die Reaktionsmischung auf eine Mischung von Eis/Wasser, zentrifugiert
den gebildeten Niederschlag und wäscht ihn mit Wasser bis zu
einem pH = 7. Dann löst
man den Niederschlag in AcOEt, wäscht
die organische Phase mit einer gesättigten Lösung von NaCl, trocknet über Na2SO4, verdampft teilweise
das Lösungsmittel
unter Vakuum und zentrifugiert den gebildeten Niederschlag. Man
erhält
auf diese Weise das erwartete Produkt, F = 186°C.
-
C) 3-Brom-6-chlor-3-(2-chlorphenyl)-5-methyl-1,3-dihydro-2H-indol-2-on
-
Zu
einer Suspension von 16,36 g der in der vorstehenden Stufe erhaltenen
Verbindung in 200 ml DCM gibt man tropfenweise bei TA und langsam
eine Lösung
von 2,6 ml Brom in 10 ml DCM. Dann setzt man wiederum eine Lösung von
0,26 ml Brom in 5 ml DCM hinzu und konzentriert die Reaktionsmischung
unter Vakuum. Anschließend
nimmt man den Rückstand
zweimal in DCM auf und verdampft jedesmal das Lösungsmittel unter Vakuum. Danach
löst man
den Rückstand
in AcOEt, wäscht
die organische Phase mit einer gesättigten Lösung von NaCl, trocknet über MgSO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise das erwartete Produkt.
-
D) 3-Amino-6-chlor-3-(2-chlorphenyl)-5-methyl-1,3-dihydro-2H-indol-2-on
-
Zu
einer Lösung
von 4,25 g der in der vorstehenden Stufe erhaltenen Verbindung in
30 ml THF gibt man 30 ml einer konzentrierten Lösung von Ammoniak und rührt 24 Stunden
lang bei TA. Dann setzt man 10 ml einer konzentrierten Lösung von
Ammoniak hinzu und läßt das Ganze über Nacht
und unter Rühren
stehen. Danach konzentriert man unter Vakuum, extrahiert den Rückstand
mit DCM, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 3,02 g des erwarteten Produktes nach Kristallisation
in DCM, F = 233°C.
-
E) 3-Amino-6-chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-5-methyl-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe B der Herstellung 1.18 beschriebenen Verfahrensweise her,
ausgehend von 0,54 g der in der vorstehenden Stufe erhaltenen Verbindung,
0,077 g Natriumhydrid (60% in Öl),
5 ml DMF und 0,435 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 0,592 g des erwarteten Produktes nach Kristallisation in einer
Mischung von DCM/Isoether.
-
F) 6-Chlor-3-(2-chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-5-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl-carbamat
von Phenyl
-
Man
stellt diese Verbindung gemäß der in
Stufe C der Herstellung 1.18 beschriebenen Verfahrensweise her,
ausgehend von 0,592 g der in der vorstehenden Stufe erhaltenen Verbindung,
1 ml Pyridin, 6 ml DCM und 0,19 ml Phenylchlorformiat. Man erhält auf diese
Weise 0,690 g des erwarteten Produktes, das man so wie es ist weiter
verwendet.
-
Herstellung 1.27
-
2-{[5-Chlor-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-oxy}-essigsäure
-
(II): R1 =
Cl; R2 = H; R3 =
-OCH(CH3)2; R4 = H; X = -O-CH2-;
Y = OH; W = H.
-
A) 2-{[5-Chlor-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-oxy}-essigsäure-methylester
-
Zu
einer Lösung
von 1,05 g Methylglycolat in 20 ml THF gibt man 0,29 g Natriumhydrid
(60% in Öl) und
rührt 10
Minuten lang bei TA. Dann setzt man tropfenweise eine Lösung von
1,88 g der in Stufe A der Herstellung 1.20 erhaltenen Verbindung
hinzu und rührt
10 Minuten lang bei TA. Danach konzentriert man unter Vakuum, extrahiert
den Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser und mit einer 1 N Lösung von
HCl, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Anschließend
chromatographiert man den Rückstand über Kieselgel,
indem man mit DCM und danach mit AcOEt eluiert. Man erhält auf diese
Weise 0,8 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 210–213°C.
-
B) 2-{[5-Chlor-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-oxy}-essigsäure
-
Zu
einer Lösung
von 0,8 g der in der vorstehenden Stufe erhaltenen Verbindung in
40 ml MeOH gibt man bei 20°C
eine Lösung
von 0,24 g NaOH in Pastillen in 5 ml Wasser und rührt 15 Stunden
lang bei TA. Dann konzentriert man unter Vakuum, löst den Rückstand
in 25 ml Wasser, säuert
durch tropfenweise Zugabe von 1 ml 12 N HCl an und zentrifugiert
den gebildeten Niederschlag. Man erhält auf diese Weise 0,8 g des erwarteten
Produktes, F = 225–228°C.
-
Herstellung
1.28 2-{[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-amino}-essigsäure (II):
R
1 = Cl; R
2 = H;
R
3 = -OCH
3; R
4 = H; X = -NH-CH
2-;
Y = OH;
-
A) 2-{[5-Chlor-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-amino}-essigsäure-tert.-butylester
-
Man
kühlt eine
Lösung
von 2,4 g Glycin-tert.-butylester-Hydrochlorid in 50 ml Chloroform
und 50 ml THF im Eisbad, setzt 2,3 g Triethylamin und danach 3,5
g der in Stufe A der Herstellung 1.19 erhaltenen Verbindung hinzu
und rührt über Nacht
bei TA. Dann konzentriert man unter Vakuum, nimmt den Rückstand
in Wasser auf, extrahiert mit DCM, trocknet die organische Phase über Na2SO4 und verdampft
das Lösungsmittel unter
Vakuum. Anschließend
verreibt man den Rückstand
in Isoether und zentrifugiert den gebildeten Niederschlag. Man erhält auf diese
Weise 2,2 g des erwarteten Produktes.
-
B) 2-{[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-lH-indol-3-yl]-amino}-essigsäure-tert.-butylester
-
Zu
einer Mischung von 2,1 g der in der vorstehenden Stufe erhaltenen
Verbindung in 10 ml DMF gibt man 0,225 g Natriumhydrid (60% in Öl) und rührt 30 Minuten
lang bei TA. Dann setzt man 1,2 g 2,4-Dimethoxybenzolsulfonylchlorid
hinzu und rührt
18 Stunden lang bei TA. Anschließend gießt man die Reaktionsmischung
in Wasser, extrahiert mit AcOEt, trocknet die organische Phase über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit DCM
eluiert. Man erhält
auf diese Weise 2,5 g des erwarteten Produktes nach Kristallisation
in Isoether.
-
C) 2-{[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-amino}-essigsäure
-
Man
beläßt eine
Mischung von 2,5 g der in der vorstehenden Stufe erhaltenen Verbindung
und 10 ml TFA 3 Stunden lang unter Rühren. Dann konzentriert man
unter Vakuum, nimmt den Rückstand
in Hexan auf und zentrifugiert den gebildeten Niederschlag. Man
erhält
auf diese Weise 2 g des erwarteten Produktes.
-
Herstellung
1.29 3-{[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-amino}-propionsäure (II):
R
1 = Cl; R
2 = H;
R
3 = -OCH
3; R
4 = H; X = -NH-CH
2-CH
2-; Y = OH;
-
A) 3-{[5-Chlor-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-amino}-propionsäure-tert.-butylester
-
Zu
einer Mischung von 3 g der in Stufe A der Herstellung 1.19 erhaltenen
Verbindung und 2,1 g β-Alanin-tert.-butylester-Hydrochlorid
in 50 ml DCM und 50 ml THF gibt man 2,15 g Triethylamin und rührt 18 Stunden
lang bei TA. Dann konzentriert man unter Vakuum, extrahiert den
Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Danach chromatographiert
man den Rückstand über Kieselgel,
indem man mit einer Mischung von DCM/AcOEt (80/20; V/V) eluiert.
Man erhält
auf diese Weise 3,2 g des erwarteten Produktes nach Kristallisation
in einer Mischung von DCM/Isoether, F = 170°C.
-
B) 3-{[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-amino}-propionsäuretert.-butylester
-
Man
stellt diese Verbindung gemäß der in
Stufe B der Herstellung 1.28 beschriebenen Verfahrensweise her,
ausgehend von 2,16 g der in der vorstehenden Stufe erhaltenen Verbindung,
0,225 g Natriumhydrid (60% in Öl),
15 ml DMF und 1,21 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 2,5 g des erwarteten Produktes nach Kristallisation in einer
Mischung von Heptan/AcOEt, F = 175°C.
-
C) 3-{[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-amino}-propionsäure
-
Man
beläßt eine
Mischung von 2,4 g der in der vorstehenden Stufe erhaltenen Verbindung
und 15 ml TFA 18 Stunden lang unter Rühren. Dann konzentriert man
unter Vakuum, nimmt den Rückstand
in Ether auf und zentrifugiert den gebildeten Niederschlag. Man
erhält
auf diese Weise 2,2 g des erwarteten Produktes, F > 250°C.
-
Herstellung 1.30
-
2-[5-Chlor-3-(2-fluorphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure, rechtsdrehendes
Isomer
-
(II): R1 =
Cl; R2 = H; R3 =
-F; R4 = H; X = -CH2-;
Y = OH; W = H.
-
A) 2-[5-Chlor-3-(2-fluorphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure-ethylester
-
Man
erhitzt eine Mischung von 2,6 g der in Stufe C der Herstellung 1.12
erhaltenen Verbindung, 1,84 g Bromessigsäure-ethylester, 1,66 g KI und
2 g K2CO3 in 10
Ml Aceton 20 Stunden lang unter Rückfluß. Dann filtriert man die Mineralsalze
und konzentriert das Filtrat unter Vakuum. Danach chromatographiert
man den Rückstand über Kieselgel,
indem man mit DCM und dann mit AcOEt eluiert. Man erhält auf diese
Weise 2,1 g des erwarteten Produktes nach Fällung in Isoether.
-
B) 2-[5-Chlor-3-(2-fluorphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
Man
beläßt eine
Mischung von 2 g der in der vorstehenden Stufe erhaltenen Verbindung
und 2 ml einer Lösung
von NaOH (30%) in 1 ml Wasser und 50 ml EtOH 20 Stunden lang bei
TA unter Rühren.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in 40 ml Wasser auf,
säuert
durch Zugabe von konzentrierter HCl auf pH = 1 an, zentrifugiert
den gebildeten Niederschlag und wäscht ihn mit Wasser. Man erhält auf diese Weise
1,9 g des erwarteten Produktes.
-
C) 2-[5-Chlor-3-(2-fluorphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure, rechtsdrehendes
Isomer
-
Man
erhitzt eine Mischung von 0,97 g der in der vorstehenden Stufe erhaltenen
Verbindung und 0,89 g (+)-Cinchonin (α20 D = +225°;
c = 0,5; EtOH) in 31,5 ml MeOH auf eine Temperatur von 40°C und anschließend unter
Rückfluß. Dann
zentrifugiert man den gebildeten Niederschlag in der Wärme, wäscht ihn
mit warmem Methanol und danach mit Ether und erhält 0,485 g des Salzes mit (+)-Cinchonin.
Das auf diese Weise gebildete Salz nimmt man in einer Mischung von
Wasser/AcOEt auf, säuert
durch Zusatz von 3 N HCl auf pH = 0 an, beläßt unter Rühren, extrahiert mit AcOEt,
wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 0,22 g des erwarteten Produktes in Form des rechtsdrehenden
Isomers. α20 D = +61,3° (c = 0,15;
Chloroform).
-
Herstellung 1.31
-
2-[5,6-Dichlor-3-(2-fluorphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
(II): R1 =
Cl; R2 = 6-Cl; R3 =
-F; R4 = H; X = -CH2-;
Y = OH; W = H.
-
A) N-(3,4-Dichlorphenyl)-DL-2-fluormandelamid
-
Man
erhitzt eine Mischung von 7,5 g der in Stufe A der Herstellung 1.12
erhaltenen Verbindung und 7,5 g 3,4-Dichloranilin in 40 ml 1,2-Dichlorbenzol
3 Stunden lang unter Rückfluß, indem
man das gebildete Wasser mit Hilfe einer Dean-Stark-Apparatur entfernt.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in Isoether auf und
zentrifugiert den gebildeten Niederschlag. Man erhält auf diese
Weise 9 g des erwarteten Produktes.
-
B) 5,6-Dichlor-3-(2-fluorphenyl)-1,3-dihydro-2H-indol-2-on
-
Zu
einer Mischung von 36 ml konzentrierter H2SO4 (95%) und 9 ml rauchender Schwefelsäure (Oleum zu
30%) gibt man bei TA 8,9 g der in der vorstehenden Stufe erhaltenen
Verbindung und rührt
anschließend
8 Stunden lang. Dann gießt
man die Reaktionsmischung auf eine Mischung von Eis/Wasser, zentrifugiert
den gebildeten Niederschlag und wäscht ihn mit Wasser. Danach
chromatographiert man den Rückstand über Kieselgel,
indem man mit DCM und dann mit Isoether eluiert. Man trennt die
zwei Isomeren:
- – das am wenigsten polare,
Verbindung von Stufe B, und erhält
3,7 g, F = 204–205°C
- – das
am meisten polare, 4,5-Dichlor-3-(2-fluorphenyl)-1,3-dihydro-2H-indol-2-on,
und erhält
1,5 g, F = 244–247°C.
-
B) 2-[5,6-Dichlor-3-(2-fluorphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure-ethylester
-
Man
erhitzt eine Mischung von 3 g der in der vorstehenden Stufe erhaltenen
Verbindung, 1,7 g Bromessigsäure-ethylester,
1,8 g und 1,4 g KI von K2CO3 in
20 ml Aceton 10 Stunden lang unter Rückfluß. Dann konzentriert man unter
Vakuum, nimmt den Rückstand
in Wasser auf, extrahiert mit AcOEt, trocknet die organische Phase über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
Isoether eluiert. Man erhält
auf diese Weise 2,1 g des erwarteten Produktes nach Verreiben in
Pentan und danach in Isoether, F = 152–158°C.
-
C) 2-[5,6-Dichlor-3-(2-fluorphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
Man
beläßt eine
Mischung von 2 g der in der vorstehenden Stufe erhaltenen Verbindung
und 3 ml einer Lösung
von NaOH (30%) in 5 ml Wasser und 40 ml EtOH 20 Stunden lang bei
TA unter Rühren.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in Wasser auf, wäscht die
wäßrige Phase
mit Ether, säuert
die wäßrige Phase
durch Zusatz von konzentrierter HCl auf pH = 1 an, extrahiert mit
AcOEt, trocknet die organische Phase über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 1,5 g des erwarteten Produktes nach verreiben in
Isoether, F = 245°C.
-
Herstellung 1.32
-
2-[5-Chlor-3-(2,3-dimethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
(II): R1 =
Cl; R2 = H; R3 =
OCH3; R4 = 3-OCH3; X = -CH2-; Y =
OH; W = H.
-
A) 2-(2,3-Dimethoxyphenyl)-2-oxoessigsäure-ethylester
-
Man
kühlt eine
Mischung von 27,6 g 1,2-Dimethoxybenzol in 160 ml Ether auf eine
Temperatur von –40°C, setzt
tropfenweise 250 ml einer 1,6 M Lösung von n-Butyllithium in
Hexan hinzu und rührt
24 Stunden lang, indem man die Temperatur wieder auf TA ansteigen
läßt. Dann
kühlt man
die Reaktionsmischung auf –20°C, gibt schnell
136 ml Oxalsäure-diethylester
hinzu und rührt,
indem man die Temperatur wieder auf TA ansteigen läßt. Nach
30 Minuten Rühren
bei TA gießt
man die Reaktionsmischung in eine gesättigte Lösung von NH4Cl,
dekantiert, extrahiert die wäßrige Phase
mit Ether, wäscht
die vereinigten organischen Phasen zweimal mit Wasser, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Dann entfernt man den überschüssigen Oxalsäure-diethylester
durch Destillation unter Vakuum (Eb = 90°C unter 2400 Pa). Anschließend chromatographiert
man das resultierende rohe Produkt über Kieselgel, indem man mit
einer Mischung von Heptan/Isoether (90/10; V/V) eluiert. Man erhält auf diese
Weise 25 g des erwarteten Produktes, das man so wie es ist in der
folgenden Stufe verwendet.
-
B) 5-Chlor-3-hydroxy-3-(2,3-dimethoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
a) 4-Chlorphenylcarbamat
von tert.-Butyl
-
Man
beläßt eine
Mischung von 12,7 g 4-Chloranilin und 22 g Di-tert.-Butyldicarbonat in 60 ml Dioxan 24
Stunden lang bei TA unter Rühren.
Dann konzentriert man die Reaktionsmischung unter Vakuum, nimmt den
Rückstand
in Pentan auf, zentrifugiert den gebildeten Niederschlag und trocknet
ihn. Man erhält
auf diese Weise 22,5 g des erwarteten Produktes.
-
b)
Man kühlt
eine Mischung von 11,4 g 4-Chlorphenylcarbamat von tert.-Butyl in
100 ml Ether unter Atmosphäre
von trockenem Stickstoff auf eine Temperatur von –40°C, gibt tropfenweise
80 ml einer 1,5 M Lösung
von tert.-Butyllithium in Pentan hinzu und rührt 3 Stunden lang bei –20°C. Dann kühlt man
die Reaktionsmischung wieder auf –40°C ab, setzt innerhalb von 1
Stunde eine Lösung
von 14 g der in der Stufe A erhaltenen Verbindung in 50 ml THF hinzu
und rührt
4 Tage lang bei TA. Danach gießt
man die Reaktionsmischung in eine gesättigte Lösung von NH4Cl,
zentrifugiert den gebildeten Niederschlag und trocknet ihn. Man
erhält auf
diese Weise 10,2 g des erwarteten Produktes, das man so wie es ist
in der folgenden Stufe verwendet.
-
C) 5-Chlor-3-(2,3-dimethoxyphenyl)-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 8,5 g der in der vorstehenden Stufe erhaltenen
Verbindung, 40 ml TFA und 11,5 ml Triethylsilan 5 Stunden lang unter
Rückfluß. Dann
konzentriert man unter Vakuum, nimmt den Rückstand in Wasser auf, extrahiert
mit AcOEt, zentrifugiert den gebildeten Niederschlag, wäscht ihn
mit AcOEt und dann mit Isoether und erhält 5,1 g des erwarteten Produktes.
Dann dekantiert man die Laugen von der Zentrifugierung, trocknet
die organische Phase über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise zusätzlich
1,4 g des erwarteten Produktes nach Kristallisation in AcOEt, F
= 193°C.
-
D) 2-[5-Chlor-3-(2,3-dimethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure-ethylester
-
Man
erhitzt eine Mischung von 6,2 g der in der vorstehenden Stufe erhaltenen
Verbindung, 3,6 g Bromessigsäure-ethylester,
3,5 g KI und 6,2 g K2CO3 in
20 ml DMF 8 Stunden lang auf eine Temperatur von 60°C. Dann konzentriert
man unter Vakuum, nimmt den Rückstand
in Wasser auf, extrahiert mit AcOEt, wäscht die organische Phase mit
Wasser, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/MeOH (99/1; V/V) eluiert. Man erhält auf diese
Weise 3 g des erwarteten Produktes, F = 155°C.
-
E) 2-[5-Chlor-3-(2,3-dimethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
Man
beläßt eine
Mischung von 3 g der in der vorstehenden Stufe erhaltenen Verbindung
und 0,5 g NaOH in Pastillen in 40 ml Wasser und 10 ml Dioxan 24
Stunden lang bei TA unter Rühren.
Dann setzt man 150 ml Wasser hinzu, wäscht die wäßrige Phase mit AcOEt, säuert die
wäßrige Phase
durch Zusatz von konzentrierter HCl auf pH = 1 an, zentrifugiert
den gebildeten Niederschlag, wäscht
ihn mit Wasser und trocknet ihn. Man erhält auf diese Weise 2,5 g des
erwarteten Produktes, F = 235°C.
-
Herstellung 1.33
-
2-[5-Chlor-3-(2-ethoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
(II): R1 =
Cl; R2 = 6-CH3;
R3 = OCH2CH3; R4 = H; X = -CH2-; Y = OH; W = H.
-
A) 5-Chlor-3-(2-ethoxyphenyl)-3-hydroxy-6-methyl-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe C der Herstellung 1.7 beschriebenen Verfahrensweise her, ausgehend
von der in Stufe A der Herstellung 1.2 erhaltenen Verbindung und
von der in Stufe B der Herstellung 1.7 erhaltenen Verbindung. Man
erhält
auf diese Weise 1,05 g des erwarteten Produktes, F = 253°C.
-
B) 5-Chlor-3-(2-ethoxyphenyl)-6-methyl-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 1,05 g der in der vorstehenden Stufe erhaltenen
Verbindung, 5,5 ml TFA und 2,2 ml Triethylsilan 5 Stunden lang unter
Rückfluß. Dann
konzentriert man unter Vakuum und chromatographiert den Rückstand über Kieselgel,
indem man mit DCM und danach mit einer Mischung von DCM/MeOH (99,5/0,5;
V/V) eluiert. Man erhält
auf diese Weise 0,5 g des erwarteten Produktes, F = 228°C.
-
C) 2-[5-Chlor-3-(2-ethoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure-ethylester
-
Man
stellt diese Verbindung gemäß der in
Stufe C der Herstellung 1.1 beschriebenen Verfahrensweise her, ausgehend
von 0,5 g der in der vorstehenden Stufe erhaltenen Verbindung, 0,29
g Bromessigsäure-ethylester,
0,29 g KI und 0,46 g K2CO3 in
5 ml Aceton. Man erhält
auf diese Weise 0,25 g des erwarteten Produktes.
-
D) 2-[5-Chlor-3-(2-ethoxyphenyl)-6-methyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-essigsäure
-
Man
stellt diese Verbindung gemäß der in
Stufe E der Herstellung 1.2 beschriebenen Verfahrensweise her, ausgehend
von 0,25 g der in der vorstehenden Stufe erhaltenen Verbindung,
0,3 ml einer Lösung
von NaOH (30%) in 2,8 ml Wasser und 7 ml THF. Man erhält auf diese
Weise 0,2 g des erwarteten Produktes.
-
Herstellung
1.34 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-ethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat (II):
R
1 = Cl; R
2 = H;
R
3 = -OCH
2CH
3; R
4 = H; X = -O-;
-
A) 5-Chlor-3-(2-ethoxyphenyl)-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe A der Herstellung 1.8 beschriebenen Verfahrensweise her, ausgehend
von 2 g der in Stufe B der Herstellung 1.2 erhaltenen Verbindung,
0,1 g Zinkchlorid, 6,6 ml HMDS und 30 ml Acetonitril. Man erhält auf diese
Weise 1,5 g des erwarteten Produktes nach Verreiben in Heptan.
-
B) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-ethoxyphenyl)-3-[(trimethylsilyl)-oxy]-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe B der Herstellung 1.8 beschriebenen Verfahrensweise her, ausgehend
von 1,45 g der in der vorstehenden Stufe erhaltenen Verbindung,
20 ml THF, 0,05 g Natriumhydrid (60% in Öl) und 1 g 2,4-Dimethoxybenzolsulfonylchlorid.
Man erhält
auf diese Weise 1,6 g des erwarteten Produktes nach Verreiben in
Isoether.
-
C) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-ethoxyphenyl)-3-hydroxy-1,3-dihydro-2H-indol-2-on
-
Man
beläßt eine
Mischung von 1,55 g der in der vorstehenden Stufe erhaltenen Verbindung
und 3 ml 12 N HCl in 100 ml Aceton 3 Stun den lang bei TA unter Rühren. Dann
konzentriert man unter Vakuum und chromatographiert den Rückstand über Kieselgel,
indem man mit DCM eluiert. Man erhält auf diese Weise 1,4 g des
erwarteten Produktes nach Verreiben in Isoether.
-
D) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-ethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl-phenylcarbonat
-
Man
kühlt eine
Mischung von 1,35 g der in der vorstehenden Stufe erhaltenen Verbindung
in 20 ml Pyridin auf eine Temperatur von 10°C, setzt 2 g Phenylchlorformiat
hinzu und rührt
20 Stunden lang, indem man die Temperatur wieder auf TA ansteigen
läßt. Dann
konzentriert man unter Vakuum, nimmt den Rückstand in einer 1 N Lösung von
HCl auf, extrahiert mit Isoether, zentrifugiert den gebildeten Niederschlag
und wäscht ihn
mit Isoether. Man erhält
auf diese Weise 1,8 g des erwarteten Produktes, F = 210–211°C (Zers.).
-
Herstellung
der Verbindung der Formel (III) Herstellung
2.1 1-(2-Pyrazinyl)-piperazin (III):
n = 1
-
Man
erhitzt eine Mischung von 3 g Piperazin, 1,04 ml 2-Chlorpyrazin
und 1,85 g K2CO3 in
100 ml EtOH 48 Stunden lang unter Rückfluß. Dann konzentriert man die
Reaktionsmischung unter Vakuum, nimmt den Rückstand in Wasser auf, alkalisiert
durch Zusatz von NaOH (10%) auf pH = 10, extrahiert mit Chloroform, wäscht die
organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise 1,8 g des erwarteten Produktes nach Kristallisation
in Hexan.
-
Herstellung
2.2 1-(3-Pyridinyl)-piperazin (III):
n = 1
-
Man
stellt diese Verbindung gemäß dem in
Tetrahedron Letters, 1998, 39, 617–620 beschriebenen Verfahren
her.
-
Herstellung
2.3 1-(2-Pyridinyl)-homopiperazin (III):
n = 2
-
Man
erhitzt eine Mischung von 2-Brompyridin und 12 g Homopiperazin 6
Stunden lang auf eine Temperatur von 100°C. Dann versetzt man die Reaktionsmischung
mit 50 ml Wasser, extrahiert mit AcOEt, wäscht die organische Phase mit
einer gesättigten
Lösung
von NaCl, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 1,28 g des erwarteten Produktes.
-
Herstellung
2.4 1-(4-Pyridinyl)-homopiperazin (III):
n = 2
-
Man
erhitzt eine Mischung von 2 g 4-Brompyridin und 10 g Homopiperazin
4 Stunden lang auf eine Temperatur von 100°C. Dann versetzt man die Reaktionsmischung
mit 100 ml Wasser, alkalisiert durch Zugabe einer Lösung von
NaOH (10%) auf pH = 10, extrahiert dreimal mit 100 ml Chloroform,
trocknet die organische Phase über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Man erhält
auf diese Weise 0,9 g des erwarteten Produktes.
-
Herstellung
2.5 1-(3-Pyridazinyl)-piperazin-Trihydrochlorid (III):
3HCl: n = 1
-
A) 4-(6-Chlor-3-pyridazinyl)-1-piperazincarbonsäure-tert.-butylester
-
Man
erhitzt eine Mischung von 13,52 g 1-Piperazincarbonsäure-tert.-butylester,
10,81 g 3,6-Dichlorpyridazin und 20 ml Triethylamin in 100 ml n-Butanol
5 Stunden lang unter Rückfluß. Dann
konzentriert man unter Vakuum und chromatographiert den Rückstand über Kieselgel,
indem man mit einer Mischung von DCM/AcOEt (90/10; V/V) eluiert.
Man erhält
auf diese Weise 14 g des erwarteten Produktes, das man so wie es
ist weiter verwendet.
-
B) 4-(3-Pyridazinyl)-1-piperazincarbonsäure-tert.-butylester
-
Man
hydriert eine Mischung von 10,5 g der in der vorstehenden Stufe
erhaltenen Verbindung und 2,5 g Palladium auf Kohle (10%) in 30
ml DMF und 250 ml EtOH bei atmosphärischem Druck und bei TA über Nacht.
Dann filtriert man den Katalysator und konzentriert das Filtrat
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/MeOH (97/3; V/V) bis (90/10; V/V) eluiert.
Man erhält
auf diese Weise 9,1 g des erwarteten Produktes, das man so wie es
ist weiter verwendet.
-
C) 1-(3-Pyridazinyl)-piperazin-Trihydrochlorid
-
Man
beläßt eine
Mischung von 3,8 g der in der vorstehenden Stufe erhaltenen Verbindung,
50 ml einer 2 N Lösung
von HCl in Ether und 20 ml MeOH über
Nacht bei TA unter Rühren.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in Ether auf und zentrifugiert
den gebildeten Niederschlag. Man erhält auf diese Weise 3 g des
erwarteten Produktes.
-
Herstellung
2.6 1-(1,3-Thiazol-2-yl)-piperazin-Dihydrochlorid (III):
2HCl: n = 1
-
A) 4-(1,3-Thiazol-2-yl)-piperazincarbonsäure-tert.-butylester
-
Man
erhitzt eine Mischung von 5 g 1-Piperazincarbonsäure-tert.-butylester, 4,4 g 2-Brom-1,3-thiazol und
7,4 g K2CO3 in 50
ml EtOH 4 Tage lang unter Rückfluß. Dann
versetzt man die Reaktionsmischung mit Wasser, verdampft das EtOH
unter Vakuum, extrahiert die resultierende wäßrige Phase mit AcOEt, wäscht die organische
Phase mit einer gesättigten
Lösung
von K2CO3 und mit
einer gesättigten
Lösung
von NaCl, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/MeOH (98/2; V/V) eluiert. Man erhält auf diese
Weise 5 g des erwarteten Produktes nach Fällung in der Kälte in der
Mischung DCM/Hexan und Zentrifugieren, F = 114–116°C.
-
B) 1-(1,3-Thiazol-2-yl)-piperazin-Dihydrochlorid
-
Man
beläßt eine
Mischung von 2,8 g der in der vorstehenden Stufe erhaltenen Verbindung
und 50 ml einer 2 N Lösung
von HCl in Ether, der zuvor ein Minimum von DCM und danach MeOH
bis zur Auflösung
der Reaktionsmischung zugesetzt wurden, 7 Stunden lang bei TA unter
Rühren.
Dann konzentriert man unter Vakuum und erhält 2,35 g des erwarteten Produktes.
-
BEISPIEL
1 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on (I):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = H; X = -CH
2-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
A) 5-Chlor-3-(2-methoxyphenyl)-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on
-
Zu
einer Mischung von 1,1 g der in der Herstellung 1.1 erhaltenen Verbindung
in 20 ml DCM gibt man bei 20°C
1,7 g BOP, 2,5 ml DIPEA und danach 0,6 g 1-(4-Pyridinyl)-piperazin
und rührt
2 Stunden lang bei 20°C.
Dann setzt man 30 ml 2 N NaOH hinzu und rührt 15 Minuten lang. Anschließend extrahiert
man die Reaktionsmischung mit AcOEt, wäscht die organische Phase mit
Wasser, trocknet über
Na2SO4 und verdampft die
Lösungsmittel
unter Vakuum. Dann verreibt man den Rückstand in Isoether und zentrifugiert
den gebildeten Niederschlag. Danach chromatographiert man den Rückstand über Kieselgel,
indem man mit DCM und dann mit Aceton eluiert. Man erhält auf diese
Weise 1,4 g des erwarteten Produktes nach Kristallisation in Isoether, F
= 111°C.
-
B) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on
-
Zu
einer Mischung von 0,7 g der in der vorstehenden Stufe erhaltenen
Verbindung in 15 ml THF gibt man bei 20°C 0,08 g Natriumhydrid (60%
in Öl)
und rührt
20 Minuten lang. Dann setzt man 0,44 g 2,4-Dimethoxybenzolsulfonylchlorid
hinzu und rührt
1 Stunde und 30 Minuten lang. Danach konzentriert man die Reaktionsmischung
unter Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Anschließend chromatographiert
man den Rückstand über Kieselgel,
indem man mit DCM, danach mit AcOEt und mit Aceton eluiert. Man
erhält
auf diese Weise 0,7 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 136–141°C (Zers.).
-
BEISPIEL
2 5-Chlor-3-(2-ethoxyphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on (I):
R
1 = Cl; R
2 = H;
R
3 = OCH
2CH
3; R
4 = H; X = -CH
2-; n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
A) 5-Chlor-3-(2-ethoxyphenyl)-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on
-
Man
beläßt eine
Mischung von 2,1 g der in der Herstellung 1.2 erhaltenen Verbindung,
1,52 g BOP, 2 ml DIPEA und 0,6 g 1-(4-Pyridinyl)-piperazin in 20
DCM 24 Stunden lang bei TA unter Rühren. Dann zentrifugiert man
das gebildete kristalline Produkt, trocknet es und erhält 0,81
g des erwarteten Produktes. Die Laugen von der Zentrifugierung extrahiert
man mit einer Lösung
von konzentrierter HCl, dekantiert, gibt zu der sauren Phase Eis,
alkalisiert die saure Phase durch Zusatz von 10 N NaOH, extrahiert
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft die Lösungsmittel unter Vakuum. Man
erhält
auf diese Weise zusätzliche
0,09 g des erwarteten Produktes, F = 185°C.
-
B) 5-Chlor-3-(2-ethoxyphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on
-
Zu
einer Mischung von 0,9 g der in der vorstehenden Stufe erhaltenen
Verbindung in 20 ml THF/DMF (90/10; V/V) gibt man 0,085 g Natriumhydrid
(60% in Öl)
und rührt
15 Minuten lang bei TA. Dann setzt man 0,45 g 2,4-Dimethoxybenzolsulfonylchlorid
hinzu und rührt
30 Minuten lang bei TA. Danach gießt man die Reaktionsmischung
in Wasser, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Anschließend chromatographiert
man den Rückstand über Kieselgel,
indem man mit einer Mischung von DCM/MeOH (96/4; V/V) eluiert. Man
erhält auf
diese Weise 0,42 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 225°C.
-
BEISPIEL
3 5-Chlor-3-(2-isopropoxyphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on,
linksdrehendes Isomer (I):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -CH
2-; n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
A) 5-Chlor-3-(2-isopropoxyphenyl)-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on, rechtsdrehendes
Isomer
-
Man
beläßt eine
Mischung von 0,43 g der in der Herstellung 1.3 erhaltenen Verbindung,
0, 6 g BOP, 0, 75 g DIPEA und 0,2 g 1-(4-Pyridinyl)-piperazin in 15 DCM 2 Stunden
lang bei 20°C
unter Rühren.
Dann extrahiert man die Reaktionsmischung mit AcOEt, wäscht die
organische Phase mit Wasser, trocknet über Na2SO4 und verdampft die Lösungsmittel unter Vakuum. Danach
chromatographiert man den Rückstand über Kieselgel,
indem man mit DCM und danach mit Aceton eluiert. Man erhält auf diese
Weise 0,33 g des erwarteten Produktes, α20 D = +37° (c
= 0,25; Chloroform).
-
B) 5-Chlor-3-(2-isopropoxyphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on,
linksdrehendes Isomer
-
Zu
einer Mischung von 0,3 g der in der vorstehenden Stufe erhaltenen
Verbindung in 10 ml THF gibt man 0,025 g Natriumhydrid (60% in Öl) und rührt 15 Minuten
lang bei 20°C.
Dann setzt man 0,19 g 2,4-Dimethoxybenzolsulfonylchlorid hinzu und
rührt 2
Stunden lang bei 20°C.
Dann konzentriert man die Reaktionsmischung unter Vakuum, nimmt
den Rückstand
in Wasser auf, extrahiert mit AcOEt, trocknet die organische Phase über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Anschließend
chromatographiert man den Rückstand über Kieselgel,
indem man mit DCM und danach mit Aceton eluiert. Man erhält auf diese
Weise 0,1 g des erwarteten Produktes.
α20 D = –21,3° (c = 0,1;
DCM).
NMR1H: DMSO-d6: δ (ppm): 0,6:
d: 3H; 1,2: d: 3H; 3,0 bis 4,0 m + 2s: 16H; 4,6: mt: 1H; 6,4 bis
7,2: mt: 9H; 7,4: de: 1H; 7,7: dd: 2H; 8,1: d: 2H.
-
BEISPIEL
4 3-(2-Chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-5-methyl-3-{2-oxo-2-[4-(2-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on (I):
R
1 = CH
3; R
2 = H; R
3 = Cl; R
4 = H; X = -CH
2-;
n = 1
-
A) 3-(2-Chlorphenyl)-5-methyl-3-{2-oxo-2-[4-(2-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on
-
Man
erhitzt eine Mischung von 0,26 g der in der Herstellung 1.4 erhaltenen
Verbindung und 0,2 ml Thionylchlorid in 10 ml Toluol 2 Stunden lang
unter Rückfluß und konzentriert
dann die Reaktionsmischung unter Vakuum. Das auf diese Weise erhaltene
Säurechlorid
löst man
in 10 ml DCM, gibt zu dieser Lösung
eine Mischung von 0,3 g 1-(2-Pyridinyl)-piperazin in 20 DCM und
rührt 2
Stunden lang bei TA. Dann konzentriert man die Reaktionsmischung
unter Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Danach
chromatographiert man den Rückstand über Kieselgel,
indem man mit einer Mischung von DCM/MeOH (98/2; V/V) eluiert. Man erhält auf diese
Weise 0,32 g des erwarteten Produktes.
-
B) 3-(2-Chlorphenyl)-1-[(2,4-dimethoxyphenyl)-sulfonyl]-5-methyl-3-{2-oxo-2-[4-(2-pyridinyl)-1-piperazinyl]-ethyl}-1,3-dihydro-2H-indol-2-on
-
Zu
einer Mischung von 0,3 g der in der vorstehenden Stufe erhaltenen
Verbindung in 10 ml THF gibt man bei TA 0,0335 g Natriumhydrid (60%
in Öl)
und rührt
30 Minuten lang. Dann setzt man 0,2 g 2,4-Dimethoxybenzolsulfonylchlorid
hinzu und rührt
1 Stunde lang bei TA. Danach versetzt man die Reaktionsmischung
mit 50 ml Wasser, extrahiert mit AcOEt, trocknet die organische
Phase über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Anschließend
chromatographiert man den Rückstand über Kieselgel,
indem man mit einer Mischung von DCM/MeOH (98/2; V/V) eluiert. Man
erhält
auf diese Weise 0,32 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 239°C.
-
BEISPIEL
5 4-(2-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl (I): R
1 = Cl; R
2 = H; R
3 = OCH
3; R
4 = H; X = -O-; n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
beläßt eine
Mischung von 0,5 g der in der Herstellung 1.8 erhaltenen Verbindung
und 0,3 g 1-(2-Pyridinyl)-piperazin in 10 ml DCM 20 Stunden lang
bei 20°C
unter Rühren.
Anschließend
chromatographiert man die Reaktionsmischung über Kieselgel, indem man mit
DCM und danach mit Aceton eluiert. Man erhält auf diese Weise 0,2 g des
erwarteten Produktes nach Kristallisation in Isoether, F = 210–215°C.
-
BEISPIEL
6 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl (I):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = H; X = -O-; n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
beläßt eine
Mischung von 0,32 g der in der Herstellung 1.8 erhaltenen Verbindung
und 0,32 g 1-(4-Pyridinyl)-piperazin in 15 ml DCM 20 Stunden lang
bei 20°C
unter Rühren.
Dann konzentriert man unter Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit einer 2 N Lösung von NaOH und mit Wasser,
trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Anschließend
chromatographiert man die Reaktionsmischung über Kieselgel, indem man mit
DCM, danach mit AcOEt und schließlich mit Aceton eluiert. Man
erhält
auf diese Weise 0,25 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 194–198°C.
-
BEISPIEL
7 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl (I):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -O-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
beläßt eine
Mischung von 0,66 g der in der Herstellung 1.9 erhaltenen Verbindung
und 0,45 g 1-(4-Pyridinyl)-piperazin in 20 ml DCM 24 Stunden lang
bei TA unter Rühren.
Dann wäscht
man die Reaktionsmischung mit Wasser, trocknet die organische Phase über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Anschließend
chromatographiert man den Rückstand über Kieselgel,
indem man mit einer Mischung von DCM/MeOH (99/1; V/V) eluiert. Man
erhält
auf diese Weise 0,41 g des erwarteten Produktes nach Kristallisation
in AcOEt, F = 253°C.
-
BEISPIEL
8 1,5-Fumarat
von 4-(4-Pyridinyl)-1-piperazincarboxylat von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl (I):
1,5 C
2H
2O
4: R
1 = Cl; R
2 = H; R
3 = OCH(CH
3)
2; R
4 =
H; X = -O=; n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
erhitzt eine Mischung von 0,3 g der in Beispiel 7 erhaltenen Verbindung
und 0,056 g Fumarsäure in
15 ml Acetonitril 3 Stunden lang unter Rückfluß. Dann zentrifugiert man den
gebildeten Niederschlag in der Wärme,
wäscht
ihn mit Ether und trocknet ihn. Man erhält auf diese Weise 0,24 g des
erwarteten Produktes, F = 235°C.
-
BEISPIEL
9 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl, linksdrehendes
Isomer (I):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -O-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
A) 5-Chlor-3-(2-isopropoxyphenyl)-2-oxo-3-{[[4-(4-pyridinyl)-1-piperazinyl]-carbonyl]-oxy}-1-indolincarboxylat von
Phenyl
-
Man
beläßt eine
Mischung von 6 g der in der Herstellung 1.10 erhaltenen Verbindung
und 1,8 g 1-(4-Pyridinyl)-piperazin in 60 ml DCM 24 Stunden lang
bei TA unter Rühren.
Dann konzentriert man teilweise das Lösungsmittel unter Vakuum und
chromatographiert direkt die resultierende Lösung über Kieselgel, indem man mit
einer Mischung von AcOEt/MeOH (95/5; V/V) eluiert. Man erhält auf diese
Weise 4,0 g des erwarteten Produktes.
-
B) 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-{[[(1S)1-hydroxymethyl)-3-methylbutyl]-amino]-carbonyl}-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl,
am wenigsten polares und am meisten polares Isomer
-
Man
beläßt eine
Mischung von 3,8 g der in der vorstehenden Stufe erhaltenen Verbindung
und 2,16 g L-Leucinol in 50 ml Chloroform 48 Stunden lang unter
Rühren.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in DCM auf und chromatographiert
die auf diese Weise erhaltene Verbindung über Aluminiumoxid, indem man
mit einer Mischung von DCM/MeOH (99/1; V/V) eluiert. Anschließend chromatographiert
man nochmals über
Kieselgel, indem man mit einer Mischung von DCM/MeOH (98,5/1,5;
V/V) eluiert. Man trennt die Diastereoisomeren:
- – das am
wenigsten polare Isomer: man erhält
0,53 g.
α20 D = –19,3° (c = 0,34;
Chloroform),
- – das
am meisten polare Isomer, das man in einer Mischung von DCM/Isoether
kristallisiert, und erhält: 0,548
g, F = 199–202°C, α20 D = –8,8° (c = 0,11;
Chloroform).
-
C) 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl, linksdrehendes
Isomer
-
Man
beläßt eine
Mischung von 0,5 g der in der vorstehenden Stufe erhaltenen Verbindung
(am meisten polares Isomer) und 0,042 g Natriummethylat in 5 ml
MeOH und 5 ml THF 18 Stunden lang bei TA unter Rühren. Dann versetzt man die
Reaktionsmischung mit Wasser, konzentriert die Lösungsmittel unter Vakuum, extrahiert
die resultierende wäßrige Phase
viermal mit DCM, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/MeOH (92/8; V/V) eluiert. Man erhält auf diese
Weise 0,225 g des erwarteten Produktes nach Kristallisation in einer
Mischung von DCM/Isoether, F = 195–245°C.
α20 D = –18,8° (c = 0,266;
Chloroform).
-
D) 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl,
linksdrehendes Isomer
-
Zu
einer Mischung von 0,213 g der in der vorstehenden Stufe erhaltenen
Verbindung in 3 ml DMF gibt man unter Atmosphäre von Argon 0,02 g Natriumhydrid
(60% in Öl)
und anschließend,
nach dem Aufhören
der Gasentwicklung, setzt man 0,119 g 2,4-Dimethoxybenzolsulfonylchlorid
hinzu und rührt
3 Stunden lang bei TA. Dann gießt
man die Reaktionsmischung in eine Lösung von K2CO3 (5%), extrahiert mit AcOEt, trocknet die
organische Phase über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/MeOH von (95/5; V/V) bis (93/7; V/V) eluiert.
Man erhält
auf diese Weise 0,161 g des erwarteten Produktes nach Kri stallisation
in einer Mischung von DCM/Hexan/Isoether, F = 160–164°C, α20 D = –71,8° (c = 0,18;
Chloroform).
-
BEISPIEL
10 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl, rechtsdrehendes
Isomer (I):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -O-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
A) 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl, rechtsdrehendes
Isomer
-
Man
stellt diese Verbindung gemäß der in
Stufe C von Beispiel 9 beschriebenen Verfahrensweise her, ausgehend
von 0,529 g der in Stufe B von Beispiel 9 erhaltenen Verbindung
(am wenigsten polares Isomer) und 0,043 g Natriummethylat in 5 ml
MeOH und 5 ml THF. Man erhält
auf diese Weise 0,198 g des erwarteten Produktes nach Kristallisation
in einer Mischung von DCM/Isoether, F = 196–198°C, α20 D = +20,7° (c
= 0,32; Chloroform).
-
B) 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl,
rechtsdrehendes Isomer
-
Man
stellt diese Verbindung gemäß der in
Stufe D von Beispiel 9 beschriebenen Verfahrensweise her, ausgehend
von 0,328 g der in der vorstehenden Stufe erhaltenen Verbindung,
0,03 g Natriumhydrid (60% in Öl),
4 ml DMF und 0,18 g 2,4-Dimethoxybenzolsulfonylchlorid, F = 161–167°C.
α20 D = +72,5° (c
= 0,14; Chloroform).
-
BEISPIEL
11 4-(2-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2,5-dimethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl (I):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = 5-OCH
3; X = -O-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
beläßt eine
Mischung von 0,4 g der in der Herstellung 1.17 erhaltenen Verbindung
und 0,4 g 1-(2-Pyridinyl)-piperazin in 5 ml DCM 72 Stunden lang
bei 20°C
unter Rühren.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in 30 ml Wasser auf
und zentrifugiert den gebildeten Niederschlag. Man erhält auf diese
Weise 0,4 g des erwarteten Produktes nach Kristallisation in MeOH,
F = 254°C.
-
BEISPIEL
12 4-(4-Pyridinyl)-1-piperazincarboxylat
von 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2,5-dimethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl (I):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = 5-OCH
3; X = -O-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
beläßt eine
Mischung von 0,4 g der in der Herstellung 1.17 erhaltenen Verbindung
und 0,4 g 1-(4-Pyridinyl)-piperazin in 5 ml DCM 72 Stunden lang
bei 20°C
unter Rühren.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in 20 ml Wasser auf
und zentrifugiert den gebildeten Niederschlag. Anschließend nimmt
man den Rückstand
in Aceton auf und konzentriert das Lösungsmittel unter Vakuum. Danach chromatographiert
man den Rückstand über Kieselgel,
indem man mit DCM, danach mit AcOEt und schließlich mit Aceton eluiert. Man
erhält
auf diese Weise 0,4 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 247–249°C.
-
BEISPIEL
13 N-[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-4-(4-pyridinyl)-piperazin-1-carboxamid, 0,25
H
2O (I): R
1 = Cl;
R
2 = H; R
3 = OCH
3; R
4 = H; X = -NH-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
erhitzt eine Mischung von 0,5 g der in der Herstellung 1.18 erhaltenen
Verbindung und 0,268 g 1-(4-Pyridinyl)-piperazin in 5 ml Chloroform
4 Stunden lang unter Rückfluß. Dann
konzentriert man unter Vakuum und chromatographiert den Rückstand über Kieselgel,
indem man mit DCM und danach durch den Gradienten einer Mischung
von DCM/AcOEt bis zu (85/15; V/V) eluiert. Man erhält auf diese
Weise 0,281 g des erwarteten Produktes nach Kristallisation in einer
Mischung von DCM/Isoether/Hexan.
-
BEISPIEL
14 N-[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-4-(4-pyridinyl)-piperazin-1-carboxamid, 0,75
H
2O, linksdrehendes Isomer (I):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = H; X = -NH-; n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
beläßt eine
Mischung von 1,136 g der in der Herstellung 1.19 erhaltenen Verbindung
und 0,608 g 1-(4-Pyridinyl)-piperazin in 10 ml Chloroform 48 Stunden
lang bei TA unter Rühren
und erhitzt danach 1 Stunde lang unter Rückfluß. Nach dem Abkühlen auf
TA chromatographiert man direkt die Reaktionsmischung über Kieselgel,
indem man mit einer Mischung von DCM/MeOH (95/5; V/V) bis (92/8;
V/V) eluiert. Dann kristallisiert man das erhaltene Produkt in einer
Mischung von DCM/Hexan/Isoether, und anschließend löst man das erhaltene Produkt
in einer minimalen Menge von MeOH und fällt durch Zusatz von Isoether.
Man erhält
auf diese Weise 0,65 g des erwarteten Produktes.
α20 D = –43° (c = 0,16;
Chloroform).
-
BEISPIEL
15 N-[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-4-(2-pyridinyl)-homopiperazin-1-carboxamid (I):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = H; X = -NH-; n = 2
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
beläßt eine
Mischung von 0,4 g der in der Herstellung 1.18 erhaltenen Verbindung
und 1,3 g der in der Herstellung 2.3 erhaltenen Verbindung in 20
ml DCM 18 Stunden lang bei TA unter Rühren. Dann konzentriert man
unter Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit einer gesättigten Lösung von NaCl, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/MeOH (94/6; V/V) eluiert. Man erhält auf diese
Weise 0,22 g des erwarteten Produktes nach Kristallisation in einer
Mischung von DCM/Isoether, F = 152°C.
-
BEISPIEL
16 N-[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-4-(4-pyridinyl)-homopiperazin-1-carboxamid,
0,8 H
2O (I): R
1 =
Cl; R
2 = H; R
3 =
OCH
3; R
4 = H; X
= -NH-; n = 2
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
erhitzt eine Mischung von 0,5 g der in der Herstellung 1.18 erhaltenen
Verbindung und 0,375 g der in der Herstellung 2.4 erhaltenen Verbindung
in 20 ml DCM 18 Stunden lang unter Rückfluß. Dann konzentriert man unter
Vakuum, extrahiert den Rückstand
mit AcOEt, wäscht
die organische Phase mit einer Lösung
von Na2CO3 (5%),
mit Wasser und mit einer gesättigten
Lösung
von NaCl, trocknet über
Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/MeOH (94/6; V/V) eluiert. Man erhält auf diese
Weise 0,386 g des erwarteten Produktes nach Kristallisation in einer
Mischung von DCM/Isoether, F = 168°C.
-
BEISPIEL
17 N-[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-ispropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-4-(3-pyridinyl)-piperazin-1-carboxamid, rechtsdrehendes
Isomer (I):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -NH-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
erhitzt eine Mischung von 0,639 g der in der Herstellung 1.20 erhaltenen
Verbindung und 0,325 g der in der Herstellung 2.2 erhaltenen Verbindung
in 10 ml Chloroform und 5 ml THF 36 Stunden lang unter Rückfluß. Dann
chromatographiert man direkt die Reaktionsmischung über Kieselgel,
indem man mit einer Mischung von DCM/MeOH (96/4; V/V) eluiert. Man
erhält
auf diese Weise 0,215 g des erwarteten Produktes nach Kristallisation
in einer Mischung von DCM/Isoether, F = 148°C.
α20 D = +25,6° (c
= 0,15; Chloroform).
NMR1H: DMSO-d6: δ (ppm):
1,0: d: 3H; 1,2: d: 3H; 3,0: mt: 4H; 3,4: mt: 4H; 3,5: s: 3H; 3,9:
s: 3H; 4,7: mt: 1H; 6,6 mt: 2H; 6,9: t: 1H; 7,1: d: 1H; 7,2 bis
7,4: m: 6H; 7,6 s: 1H; 7,8: dd: 1H; 7,9: d: 1H; 8,0: d: 1H; 8,3:
se: 1H.
-
BEISPIEL
18 N-[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-ispropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-4-(3-pyridinyl)-piperazin-1-carboxamid, linksdrehendes
Isomer (I):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -NH-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
stellt diese Verbindung gemäß der in
Beispiel 17 beschriebenen Verfahrensweise her, ausgehend von 0,5
g der in der Herstellung 1.21 erhaltenen Verbindung und 0,26 g der
in der Herstellung 2.2 erhaltenen Verbindung in 10 ml Chloroform
und 5 ml THF.
α20 D = –33° (c = 0,15;
Chloroform).
-
BEISPIEL
19 N-[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-4-(4-pyridinyl)-piperazin-1-carboxamid, rechtsdrehendes
Isomer (I):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -NH-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
erhitzt eine Mischung von 0,5 g der in der Herstellung 1.21 erhaltenen
Verbindung und 0,26 g 1-(4-Pyridinyl)-piperazin in 5 ml Chloroform
18 Stunden lang unter Rückfluß. Dann
chromatographiert man direkt die Reaktionsmischung über Kieselgel,
indem man mit einer Mischung von DCM/MeOH (95/5; V/V) eluiert. Man
kristallisiert das erhaltene Produkt durch Eindampfen einer Mischung
von DCM/Hexan/Isoether in der Kälte.
Man erhält
auf diese Weise 0,46 g des erwarteten Produktes.
α20 D = +7,9° (c
= 0,175; Chloroform).
-
BEISPIEL
20 N-[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-4-(4-pyridinyl)-piperazin-1-carboxamid, linksdrehendes
Isomer (I):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -NH-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
beläßt eine
Mischung von 0,9 g der in der Herstellung 1.20 erhaltenen Verbindung
und 0,463 g 1-(4-Pyridinyl)-piperazin in 10 ml Chloroform 48 Stunden
lang bei TA unter Rühren.
Dann chromatographiert man direkt die Reaktionsmischung über Kieselgel,
indem man mit einer Mischung von DCM/MeOH (95/5; V/V) eluiert. Man
kristallisiert das erhaltene Produkt durch Eindampfen einer Mischung
von DCM/Hexan/Isoether in der Kälte.
Man erhält
auf diese Weise 0,789 g des erwarteten Produktes.
α20 D = –10,3° (c = 0,17;
Chloroform).
NMR1H: DMSO-d6: δ (ppm): 1,0:
d: 6H; 3,2 bis 3,7: m + s: 11H; 3,8: s: 3H; 4,6: mt: 1H; 6,6: s
+ mt: 2H; 6,8: t: 1H; 6,9 d: 1H; 7,2: d: 2H; 7,3: mt: 4H; 7,6: s:
1H; 7,8: d: 1H; 8,2: d: 2H.
-
BEISPIEL
21 N-[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2,5-dimethoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl]-4-(2-pyridinyl)-piperazin-1-carboxamid (I):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = 5-OCH
3; X = -NH-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
stellt diese Verbindung gemäß der in
Beispiel 13 beschriebenen Verfahrensweise her, ausgehend von 0,46
g der in der Herstellung 1.22 erhaltenen Verbindung und 0,21 ml
1-(2-Pyridinyl)-pipe razin in 5 ml Chloroform. Man erhält auf diese
Weise 0,382 g des erwarteten Produktes nach Kristallisation in einer
Mischung von DCM/Isoether/Hexan.
NMR1H:
DMSO-d6: δ (ppm):
3,4 bis 3,8: m + 3s: 17H; 4,0: s: 3H; 6,8: mt: 2H; 7,0: mt: 4H;
7,4: mt: 3H; 7,8: d: 1H; 7,9 d: 1H; 8,2: mt: 2H.
-
BEISPIEL
22 N-[5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-6-trifluormethyl-2-oxo-2,3-dihydro-1H-indol-3-yl]-4-(4-pyridinyl)-piperazin-1-carboxamid,
einziges Enantiomer (I):
R
1 = Cl; R
2 = 6-CF
3; R
3 = OCH
3; R
4 = H; X = -NH-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
stellt diese Verbindung gemäß der in
Beispiel 15 beschriebenen Verfahrensweise her, ausgehend von 0,203
g der in der Herstellung 1.25 erhaltenen Verbindung und 0,098 g
1-(4-Pyridinyl)-piperazin
in 3 ml Chloroform und 3 ml THF. Man erhält auf diese Weise 0,098 g
des erwarteten Produktes nach Kristallisation in einer Mischung
von DCM/Isoether, F = 174°C.
-
BEISPIEL
23 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethoxy}-1,3-dihydro-2H-indol-2-on (I):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -O-CH
2-; n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
A) 5-Chlor-3-(2-isopropoxyphenyl)-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethoxy}-1,3-dihydro-2H-indol-2-on
-
Zu
einer Mischung von 1 g der in der Herstellung 1.27 erhaltenen Verbindung
in 20 ml DCM gibt man bei 20°C
1,2 g BOP, 2 ml DIPEA und danach 0,48 g 1-(4-Pyridinyl)-piperazin
und rührt
1 Stunde lang bei TA. Dann setzt man 20 ml 2 N NaOH hinzu, extrahiert
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft die Lösungsmittel unter Vakuum. Danach
chromatographiert man den Rückstand über Kieselgel,
indem man mit DCM, dann mit AcOEt und schließlich mit Aceton eluiert. Man
erhält
auf diese Weise 0,55 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 115–120°C.
-
B) 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-3-{2-oxo-2-[4-(4-pyridinyl)-1-piperazinyl]-ethoxy}-1,3-dihydro-2H-indol-2-on
-
Man
stellt diese Verbindung gemäß der in
Stufe B von Beispiel 1 beschriebenen Verfahrensweise her, ausgehend
von 0,5 g der in der vorstehenden Stufe erhaltenen Verbindung, 0,04
g Natriumhydrid (60% in Öl), 10
ml THF und 0,3 g 2,4-Dimethoxybenzolsulfonylchlorid. Man erhält auf diese
Weise 0,55 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 201–203°C.
-
BEISPIEL
24 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-3-{[2-oxo-2-[4-(2-pyridinyl)-1-piperazinyl]-ethyl]-amino}-1,3-dihydro-2H-indol-2-on (I):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = H; X = -NH-CH
2-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Zu
einer Lösung
von 0,33 g der in der Herstellung 1.28 erhaltenen Verbindung, 0,11
g 1-(2-Pyridinyl)-piperazin und 0,121 g Triethylamin in 5 ml DCM
gibt man 0,329 g PyBOP und rührt
3 Stunden lang bei TA. Dann konzentriert man unter Vakuum, extrahiert
den Rückstand
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum.
-
Danach
chromatographiert man den Rückstand über Kieselgel,
indem man mit einer Mischung von DCM/AcOEt (90/10; V/V) eluiert.
Man erhält
auf diese Weise 0,29 g des erwarteten Produktes nach Kristallisation
in Isoether, F = 155°C.
-
BEISPIEL
25 5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-methoxyphenyl)-3-{[3-oxo-3-[4-(2-pyridinyl)-1-piperazinyl]-propyl]-amino}-1,3-dihydro-2H-indol-2-on (I):
R
1 = Cl; R
2 = H;
R
3 = OCH
3; R
4 = H; X = -NH-CH
2-CH
2-; n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Zu
einer Lösung
von 0,4 g der in der Herstellung 1.29 erhaltenen Verbindung, 0,127
g 1-(2-Pyridinyl)-piperazin und 0,143 g Triethylamin in 10 ml DCM
gibt man 0,37 g PyBOP und rührt
2 Stunden lang bei TA. Dann verdünnt
man die Reaktionsmischung mit DCM, wäscht die organische Phase mit
Wasser, trocknet über Na2SO4 und verdampft
das Lösungsmittel
unter Vakuum. Danach chromatographiert man den Rückstand über Kieselgel, indem man mit
einer Mischung von DCM/AcOEt (90/10; V/V) eluiert. Man erhält auf diese
Weise 0,36 g des erwarteten Produktes nach Kristallisation in Isoether,
F = 214°C.
NMR1H: DMSO-d6: δ (ppm): 2,0
bis 2,5: 2mt: 4H; 3,1 bis 3,6 m + s: 11H; 3,7: s: 3H; 3,9: s: 3H;
6,6 bis 7,0: m: 6H; 7,1: t: 1H; 7,3: t: 1H; 7,5: dd: 1H; 7,6: mt:
1H; 7,7 bis 8,0: mt: 4H; 8,2: dd: 1H.
-
BEISPIEL
26 N-{5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl}-4-(3-pyridazinyl)-piperazin-1-carboxamid,
einziges Enantiomer (I):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -NH-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Zu
einer Lösung
von 0,054 g 1-(3-Pyridazinyl)-piperazin in 2 ml Chloroform gibt
man eine Lösung
von 0,07 g der in der Herstellung 1.20 erhaltenen Verbindung und
erhitzt anschließend
24 Stunden lang auf eine Temperatur von 60°C. Danach chromatographiert
man direkt die Reaktionsmischung über Kieselgel, indem man mit
einer Mischung von DCM/MeOH (95/5; V/V) eluiert. Man erhält auf diese
Weise das erwartete Produkt. Reinheit CLHP: 100%.
-
BEISPIEL
27 N-{5-Chlor-1-[(2,4-dimethoxyphenyl)-sulfonyl]-3-(2-isopropoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl}-4-(2-pyrimidinyl)-piperazin-1-carboxamid,
einziges Enantiomer (I):
R
1 = Cl; R
2 = H;
R
3 = OCH(CH
3)
2; R
4 = H; X = -NH-;
n = 1
R
6 = 2-OCH
3; R
7 = OCH
3.
-
Man
stellt diese Verbindung gemäß der in
Beispiel 26 beschriebenen Verfahrensweise her, ausgehend von 0,054
g 1-(2-Pyrimidinyl)-piperazin
in 2 ml Chloroform und 0,07 g der in der Herstellung 1.20 erhaltenen Verbindung.
Man erhält
auf diese Weise das erwartete Produkt.
-
Indem
man nach den in den obigen Beispielen beschriebenen Verfahrensweisen
arbeitet, stellt man ausgehend von den Verbindungen der Formel (II),
worin W = H ist, und den Verbindungen der Formel (V) die folgenden,
in der nachstehenden Tabelle I aufgeführten Verbindungen der Formel
(IV) her.
-
-
-
-
- (a) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Herstellung 1.1 und der Verbindung der Herstellung 2.2
- (b) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Herstellung 1.5 und 1-(4-Pyridinyl)-piperazin
- (c) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Herstellung 1.5 und der Verbindung der Herstellung 2.2
- (d) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
3 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Herstellung 1.6 und 1-(4-Pyridinyl)-piperazin
- (e) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
3 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Herstellung 1.6 und der Verbindung der Herstellung 2.2
- (f) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
3 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Herstellung 1.7 und 1-(4-Pyridinyl)-piperazin
- (g) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der in Stufe B der
Herstellung 1.30 erhaltenen Verbindung und 1-(4-Pyridinyl)-piperazin
- (h) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Herstellung 1.31 und 1-(4-Pyridinyl)-piperazin
- (i) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Herstellung 1.32 und 1-(4-Pyridinyl)-piperazin
- (j) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von den Verbindungen
der Herstellungen 1.2 und 2.2
- (k) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Herstellung 1.33 und 1-(4-Pyridinyl)-piperazin
- (l) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Stufe E der Herstellung 1.3 und der Verbindung der Herstellung 2.2
- (m) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Stufe E der Herstellung 1.3 und der Verbindung der Herstellung 2.4
- (n) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Stufe E der Herstellung 1.3 und 1-(2-Pyrimidinyl)-piperazin
- (o) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Stufe E der Herstellung 1.3 und 1-(3-Pyridazinyl)-piperazin
- (p) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Stufe E der Herstellung 1.3 und 1-(1,3-Thiazol-2-yl)-piperazin
- (q) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von den Verbindungen
der Herstellungen 1.7 und 2.2
- (r) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Herstellung 1.30 und 1-(4-Pyridinyl)-piperazin
- (s) Verbindung, hergestellt gemäß der in Stufe A von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung der
Stufe E der Herstellung 1.3 und 1-(4-Pyridinyl)-piperazin
-
Indem
man nach den in den obigen Beispielen beschriebenen Verfahrensweisen
arbeitet, stellt man ausgehend von den Verbindungen der Formel (II),
worin
ist, und den Verbindungen
der Formel (III), oder ausgehend von den Verbindungen der Formel
(IV) und 2,4-Dimethoxybenzolsulfonylchlorid die folgenden, in der
nachstehenden Tabelle II aufgeführten
Verbindungen gemäß der Erfindung
her.
-
-
-
-
-
-
-
-
- (a) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.1
- (b) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.2
- (c) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
I beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.3
- (d) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.4
- (e) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.5
- (f) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.6
- (g) Verbindung, hergestellt gemäß der in Beispiel 5 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.11
und 1-(2-Pyridinyl)-piperazin
- (h) Verbindung, hergestellt gemäß der in Beispiel 5 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.11
und 1-(4-Pyridinyl)-piperazin
- (i) Verbindung, hergestellt gemäß der in Beispiel 6 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.12
und 1-(4-Pyridinyl)-piperazin
- (j) Verbindung, hergestellt gemäß der in Beispiel 6 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.13
und 1-(4-Pyridinyl)-piperazin
- (k) Verbindung, hergestellt gemäß der in Beispiel 7 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.14
und 1-(4-Pyridinyl)-piperazin
- (l) Verbindung, hergestellt gemäß der in Beispiel 5 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.15
und 1-(4-Pyridinyl)-piperazin
- (m) Verbindung, hergestellt gemäß der in Beispiel 5 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.16
und 1-(4-Pyridinyl)-piperazin
- (n) Verbindung, hergestellt gemäß der in Beispiel 13 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.18
und 1-(2-Pyridinyl)-piperazin
- (o) Verbindung, hergestellt gemäß der in Beispiel 13 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.23
und 1-(2-Pyridinyl)-piperazin
- (p) Verbindung, hergestellt gemäß der in Beispiel 13 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.23
und 1-(4-Pyridinyl)-piperazin
- (q) Verbindung, hergestellt gemäß der in Beispiel 13 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.23
und 1-(2-Pyrimidinyl)-piperazin
- (r) Verbindung, hergestellt gemäß der in Beispiel 15 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.24
und 1-(4-Pyridinyl)-piperazin
- (s) Verbindung, hergestellt gemäß der in Beispiel 13 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.26
und 1-(4-Pyridinyl)-piperazin
- (t) Verbindung, hergestellt gemäß der in Beispiel 25 beschriebenen
Verfahrensweise, ausgehend von der Verbindung der Herstellung 1.29
und 1-(4-Pyridinyl)-piperazin
- (u) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.7
- (v) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.8
- (w) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.9
- (x) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.10
- (y) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.11
- (z) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.12
- (aa) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.13
- (ab) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.14
- (ac) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.15
- (ad) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.16
- (ae) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.17
- (af) Man beläßt eine
Mischung von 0,3 g der in der Herstellung 1.34 erhaltenen Verbindung,
0,3 g der Verbindung der Herstellung 2.2 und 2 g DIPEA in 15 ml
DMF 4 Tage lang bei TA unter Rühren.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in Wasser auf, extrahiert
mit AcOEt, wäscht
die organische Phase mit Wasser, trocknet über Na2SO4 und verdampft das Lösungsmittel unter Vakuum. Danach
chromatographiert man den Rückstand über Kieselgel,
indem man mit DCM und danach mit Aceton eluiert. Man erhält auf diese Weise
0,27 g des erwarteten Produktes nach Verreiben in Isoether.
- (ag) Man beläßt eine
Mischung von 0,4 g der in der Herstellung 1.34 erhaltenen Verbindung,
0,6 g 1-(4-Pyridinyl)-piperazin in 0,6 ml THF 5 Tage lang bei TA
unter Rühren.
Dann konzentriert man unter Vakuum, nimmt den Rückstand in 20 ml Wasser und
20 ml AcOEt auf, beläßt unter
Rühren,
zentrifugiert den gebildeten Niederschlag und wäscht ihn mit Isoether. Man
erhält
auf diese Weise 0,4 g des erwarteten Produktes.
- (ah) Verbindung, hergestellt gemäß der in Stufe B von Beispiel
1 beschriebenen Verfahrensweise, ausgehend von der Verbindung IV.18
- Verbindung, hergestellt gemäß der in
Stufe B von Beispiel 1 beschriebenen Verfahrensweise, ausgehend
von der Verbindung IV.19
- (aj) Zu einer Lösung
von 0,2 g der Verbindung von Beispiel 3 in 20 ml EtOH gibt man eine
Lösung
von 2 N HCl in Ether und konzentriert unter Vakuum. Dann nimmt man
den Rückstand
in Propan-2-ol auf, zentrifugiert den gebildeten Niederschlag, wäscht ihn
mit Ether und trocknet ihn. Man erhält auf diese Weise 0,12 g des
Hydrochlorides.
- (ak) Man erhitzt eine Mischung von 0,3 g der Verbindung von
Beispiel 3 und 0,059 g Fumarsäure
in 20 ml Acetonitril 10 Minuten lang unter Rückfluß. Nach dem Abkühlen zentrifugiert
man den gebildeten Niederschlag und erhält 0,24 g des Fumarates.
- (al) Man erhitzt eine Mischung von 0,3 g der Verbindung von
Beispiel 3 und 0,051 g H3PO4 (85%)
in 20 ml EtOH 10 Minuten lang auf eine Temperatur von 60°C. Nach dem
Abkühlen
zentrifugiert man den gebildeten Niederschlag und erhält 0,3 g
des erwarteten Produktes.
-
BEISPIEL 46
-
- NMR1H: DMSO-d6: δ (ppm): 2,3:
s: 3H; 3,2 bis 3,8: m + s: 1H; 3,9: s: 3H; 6,7: mt: 2H; 6,9: d:
2H; 7,2 bis 7,4: m: 5H; 7,8: s: 1H; 7,9: d: 1H; 8,0: s: 1H; 8,2:
d: 2H.
-
Die
Verbindung gemäß der Erfindung
bilden den Gegenstand von biochemischen Untersuchungen.
-
Die
Affinität
der Verbindungen der Formel (I) gemäß der Erfindung für die Rezeptoren
V1b von Arginin-Vasopressin wurde in vitro
unter Anwendung der von Y. DE KEYSER et al., Febs Letters, 1994,
356, 215–220
beschriebenen Methode untersucht. Diese Methode besteht darin, in
vitro die Bewegung von tritiertem Arginin-Vasopressin ([3H]-AVP) an den Rezeptoren V1b zu
untersuchen, die an den membranären
adenohypophysären
oder zellularen Präparationen
anwesend sind, die ihrerseits die Rezeptoren V1b von
Ratte oder Mensch tragen. Die Inhibitor-Konzentrationen 50% (CI50) der Fixierung von tritiertem Arginin-Vasopressin
der Verbindungen gemäß der Erfindung
sind gering und variieren von 10–6 bis
10–9 M.
-
Die
Affinität
der Verbindungen der Formel (I) gemäß der Erfindung für die Rezeptoren
Via von Arginin-Vasopressin wurde in vitro unter Anwendung der von
M. THIBONNIER et al., J. Biol. Chem., 1994, 269, 3304–3310 beschriebenen
Methode untersucht. Diese Methode besteht darin, in vitro die Bewegung
von tritiertem Arginin-Vasopressin ([3H]-AVP)
an den Rezeptoren V1a zu untersuchen, die
an den membranären
oder zellularen Präparationen
anwesend sind, die ihrerseits die Rezeptoren V1a von
Ratte oder Mensch tragen. Diese Verbindungen der Formel (I) weisen
eine Affinität
für die
Rezeptoren V1a von Arginin-Vasopressin mit
CI50 auf, die von 10–6 bis
10–9 M
variieren.
-
Die
Affinität
der Verbindungen der Formel (I) gemäß der Erfindung für die Rezeptoren
V2 von Vasopressin wurde ebenfalls untersucht
(von M. Birnbaumer et al., Nature (London), 1992, 357, 333–335 beschriebene
Methode). Die untersuchten Verbindungen sind wenig oder nicht für die Rezeptoren
V2 affin, mit CI50 von im
allgemeinen über
10–6 M.
-
Die
Affinität
der Verbindungen gemäß der Erfindung
für die
Rezeptoren von Ocytocin wurde in einem Bindungstest in vitro unter
Anwendung der von J. Elands et al. in Eur. J. Pharmacol., 1987,
147, 197–207
beschriebenen Methode untersucht. Diese Methode besteht darin, in
vitro die Bewegung eines radioiodierten Analogen von Ocytocin an
den Rezeptoren von Ocytocin in einer membranären Präparation von transfektierten
Zellen mit dem humanen uterinen Ocytocin-Rezeptor zu untersuchen.
Die IC50 (Inhibitor-Konzentration von 50%
der Bindung des radioiodierten Analogen von Ocytocin an seine Rezeptoren)
sind gering und variieren von 10–6 bis
10–9 M
bei diesem letzten Test.
-
Die
Verbindungen der vorliegenden Erfindung sind insbesondere Wirkstoffe
in pharmazeutischen Zusammensetzungen, deren Toxizität mit ihrer
Anwendung als Arzneimittel kompatibel ist.
-
Gemäß einem
anderen ihrer Aspekte betrifft die vorliegende Erfindung die Verwendung
der Verbindungen der Formel (I) oder von einem ihrer pharmazeutisch
akzeptablen Salze, Solvate und/oder Hydrate für die Herstellung von Arzneimitteln,
die zur Behandlung von jeder Erkrankung vorgesehen sind, wo Arginin-Vasopressin
und/oder seine Rezeptoren V1b und/oder seine
Rezeptoren Via und/oder Ocytocin und/oder seine Rezeptoren einbezogen
sind.
-
So
können
die Verbindungen gemäß der Erfindung
beim Menschen oder beim Tier für
die Behandlung oder Vorbeugung von verschiedenen von Vasopressin
abhängigen
Erkrankungen verwendet werden, beispielsweise kardiovaskuläre Erkrankungen
wie Hypertension, pulmonare Hypertension, Herzinsuffizienz, Myokardinfarkt,
oder koronare Vasospasmen, insbesondere beim Raucher, Raynaud-Erkrankung,
instabile Anginen und PTCA (engl. percutaneous transluminal coronary
angioplasty), Herz-Ischämie,
Unregelmäßigkeiten der
Hämostase;
Erkrankungen des Zentralnervensystems wie Migräne, cerebrale Vasospasmen,
cerebrale Hämorrhagie,
cerebrale Ödeme,
Depression, Angstzustände,
Streß,
emotionale Störungen,
krampfartige Besessenheitsstörungen,
Panikanfälle,
psychotische Zustände,
beispielsweise Störungen
des Gedächtnisses; Erkrankungen
des Nierensystems, wie Nieren-Vasospasmen, Nekrose der Nierenrinde; insipide
nephrogenische Diabetes; Erkrankungen des Verdauungssystems wie
gastrische Vasospasmen, Hepatozirrhose, Ulzera, Erkrankungen mit
Erbrechen, beispielsweise Übelkeit
einschließlich
der mit einer Chemotherapie einhergehenden Übelkeit, Reisekrankheit; diabetische
Nephropathie.
-
Die
Verbindungen gemäß der Erfindung
können
ebenfalls bei der Behandlung von Störungen des sexuellen Verhaltens
verwendet werden; die Verbindungen gemäß der Erfindung können bei
der Frau zur Behandlung von Dysmenorrhoe oder bei drohenden Frühgeburten
verwendet werden. Man kann die Verbindungen gemäß der Erfindung auch bei der
Behandlung von Lungenkrebs mit kleinen Zellen verwenden; bei hyponatremischen
Encephalopathien; beim pulmonaren Syndrom, bei der Ménière-Erkrankung;
bei Glaukom, beim grauen Star; bei Fettleibigkeit; bei Diabetes
vom Typ I und II; bei Atherosklerose; beim Cushing-Syndrom; bei
der Resistenz gegenüber
Insulin; bei Hypertriglyceridämie;
bei der Behandlung von postoperativen Zuständen, insbesondere nach einer
Unterleibschirurgie.
-
Die
Verbindungen gemäß der Erfindung
können
außerdem
bei der Behandlung oder Vorbeugung von allen Erkrankungen verwendet
werden, die auf Streß folgen
wie Müdigkeit
und ihre Syndrome, von ACTH abhängigen
Störungen,
von Herzstörungen,
bei Schmerzen, bei Veränderungen
bei der gastrischen Entleerung, bei der fäkalen Ausscheidung (Colitis,
Reizdarmsyndrom, Crohn-Erkrankung), bei der Säureausscheidung, bei Hyperglykämie, bei
Immunosuppression, bei inflammatorischen Prozessen (rheumatoide
Arthritis oder Osteoarthritis), bei multiplen Infektionen, bei Krebszuständen, Asthma,
Psoriasis, Allergien und verschiedenen neuropsychiatrischen Störungen wie
nervöser
Magersucht, Bulimie, bei Störungen
der Gemütsart,
bei Depression, Angst, Schlafstörungen,
bei Panikzuständen,
Phobien, Zwangsvorstellungen, bei Störungen der Wahrnehmung von
Schmerzen (Fibromyalgie), bei neurodegenerativen Erkrankungen (Alzheimer-Erkrankung,
Parkinson-Erkrankung, Huntington-Erkrankung), bei der Abhängigkeit
gegenüber
einer Substanz, bei Entzug bzw. der Entwöhnung, beim hämorrhagischen
Streß,
bei muskulären
Spasmen und Hypoglycämie.
-
Die
Verbindungen gemäß der Erfindung
können
ebenfalls bei der Behandlung oder Vorbeugung von chronischen Streßzuständen wie
Immu nodepression, bei Fruchtbarkeitsstörungen und bei Dysfunktionen
der hypothalamo-hypophysosurrenalen Achse verwendet werden.
-
Die
Verbindungen gemäß der Erfindung
können
auch als Psychostimulantien, zur Provozierung der Steigerung der
Aufmerksamkeit, der emotionalen Reaktivität gegenüber der Umwelt und der Vereinfachung der
Anpassung verwendet werden.
-
Die
Verbindungen gemäß der Erfindung,
die eine Affinität
für die
Rezeptoren von Ocytocin besitzen, sind besonders interessant, und
dies bei der Vorbeugung und/oder der Behandlung der von Ocytocin
abhängigen
Störungen.
Die Verbindungen gemäß der Erfindung
sind interessant bei der Narbenbildung, der Analgie, bei Angstzuständen, bei
der Vorbeugung von Schmerzen, bei der Vorbeugung von Angstzuständen, bei
Depression, Schizophrenie, Autismus, krampfartigen Besessenheitssyndromen,
beim mütterlichen
(Vereinfachung der Wiedererkennung und der Akzeptierung der Mutter
durch das Kind) und sozialen Verhalten, beim Gedächtnis; bei der Regulierung
der Aufnahme von Nahrung und Getränken, der Abhängigkeit
und der Entwöhnung
gegenüber
Drogen und der sexuellen Motivation. Sie können ebenfalls vorteilhafterweise
verwendet werden bei Störungen
der urogenitalen Sphäre,
insbesondere auf den Gebieten von Geburtshilfe und Gynäkologie,
vor allem als tokolytisches oder den Uterus entspannendes Mittel
oder zur Kontrolle der Uterus-Kontraktionen,
bevor die Schwangerschaft beendet ist, zur Kontrolle der pränatalen
Tätigkeit
oder auch zur Kontrolle der vorbereitenden Arbeiten im Hinblick
auf eine Entbindung durch Kaiserschnitt, zur Lösung der Probleme von Sterilität und Fruchtbarkeit,
zur Kontrolle von Geburten (insbesondere bei der veterinären Anwendung),
zur Kontrolle der Brunst, bei der Beendigung des Stillens, beim
Abstillen, bei der Überführung und
der Implantation von Embryonen bei der Befruchtung in vitro; bei
der Behandlung von Endometriose, von Dysmenorrhoe sowie bei dringender
Harninkontinenz oder bei Notfällen,
bei begniner Hypertrophie der Prostata und bei erektilen Dysfunktionen,
bei Hypertension, Hyponaträmie,
Herzinsuffizienz, Arteriosklerose, Angiogenese, bei der Proliferation
von Tumoren, bei Kaposi-Sarkom und zur Regulierung der Fettablagerung
bei fettleibigen Patienten.
-
Unter
der Maßgabe,
daß die
Rolle von Ocytocin auf die Kontrolle des Gelbkörperhormons (J. J. Evans, J.
Endocrin. 1996, 151, 169–174)
gegeben ist, können
die Verbindungen gemäß der Erfindung
außerdem
zur Induzierung der Kontrazeption verwendet werden.
-
Außerdem können die
Verbindungen gemäß der Erfindung
aufgrund ihrer antitumoralen Wirkungen bei Ocytocin absondernden
Tumoren verwendet werden, insbesondere bei Brustkrebs und Prostatakrebs.
-
Die
Verwendung der Verbindungen gemäß der Erfindung
für die
Vorbeugung und/oder Behandlung der vorstehend erwähnten Erkrankungen
sowie für
die Herstellung von Arzneimitteln, die zur Behandlung dieser Erkrankungen
vorgesehen sind, bildet einen wesentlichen Teil der Erfindung.
-
Die
Verbindungen der obigen Formel (I) oder eines ihrer pharmazeutisch
akzeptablen Salze, Solvate und/oder Hydrate können in täglichen Dosierungen von 0,01
bis 100 mg pro kg Körpergewicht
des zu behandelnden Säugetieres,
vorzugsweise in täglichen
Dosierungen von 0,1 bis 50 mg/kg, verwendet werden. Beim Menschen
kann die Dosis vorzugsweise von 0,1 bis 4000 mg pro Tag variieren,
spezieller von 0,5 bis 1000 mg, je nach Alter des zu behandelnden
Patienten oder dem Typ der Behandlung: prophylaktisch oder kurativ.
-
Für ihre Anwendung
als Arzneimittel werden die Verbindungen der Formel (I) im allgemeinen
in Einheitsdosen verabreicht. Die genannten Einheitsdosen werden
vorzugsweise in pharmazeutischen Zusammensetzungen formuliert, in
denen der Wirkstoff mit einem oder mehreren pharmazeutischen Trägerstoffen
vermischt ist.
-
So
betrifft die vorliegende Erfindung in einem weiteren ihrer Aspekte
die pharmazeutischen Zusammensetzungen, die als Wirkstoff eine Verbindung
der Formel (I) oder eines ihrer pharmazeutisch akzeptablen Salze,
Solvate und/oder Hydrate sowie einen oder mehre pharmazeutisch akzeptable
Trägerstoffe
umfassen.
-
In
den pharmazeutischen Zusammensetzungen der vorliegenden Erfindung
für eine
Verabreichung auf oralem, sublingualem, subkutanem, intramuskulärem, intravenösem, transdermalem,
lokalem, rektalem Wege oder dem Inhalationsweg können die Wirkstoffe in Form
von Einheitsverabreichungen in Mischung mit klassischen pharmazeuti schen
Trägern
an Tiere und Menschen verabfolgt werden. Die geeigneten Formen der
Einheitsverabreichung umfassen die Formulierungen für den oralen
Weg, wie Tabletten, Kapseln, Pulver, Granulate und orale Lösungen oder
Suspensionen, die Formulierungen für die sublinguale oder buccale
Verabreichung, die Aerosole, die Formulierungen für die topische
Verabreichung, die Implantate, die Formulierungen für die subkutane,
intramuskuläre,
intravenöse,
intranasale oder intraokulare Verabreichung und die Formulierungen
für die
rektale Verabreichung.
-
In
jeder Einheitsdosis liegt der Wirkstoff der Formel (I) in Mengen
vor, die an die vorgesehenen täglichen
Dosierungen angepaßt
sind. Im allgemeinen wird jede Einheitsdosis in geeigneter Weise
gemäß der Dosierung
und dem Typ der vorgesehenen Verabreichung eingestellt, beispielsweise
Tabletten, Kapseln und ähnliche,
Briefchen, Ampullen, Sirupe und ähnliche
und Tropfen, so daß eine
derartige Einheitsdosis 0,1 bis 1000 mg Wirkstoff, vorzugsweise
0,5 bis 250 mg Wirkstoff enthält,
die einmal bis viermal pro Tag verabreicht wird.
-
Obwohl
diese Dosierungen Beispiele für
durchschnittliche Situationen darstellen, können sie in besonderen Fällen, wo
höhere
oder niedrigere Dosierungen geeignet sind, derartige Dosierungen
aufweisen, die ebenfalls von der Erfindung stammen. Gemäß der üblichen
Praxis wird die für
jeden Patienten geeignete Dosis durch den Arzt gemäß der Verabreichungsweise,
dem Alter, dem Gewicht und der Ansprechreaktion des betreffenden
Patienten festgelegt.
-
Die
vorliegende Erfindung betrifft ebenfalls gemäß einem weiteren ihres Aspekte
eine Methode zur Behandlung der vorstehend genannten Erkrankungen,
welche die Verabreichung einer wirksamen Dosis einer Verbindung
gemäß der Erfindung
oder eines ihrer pharmazeutisch akzeptablen Salze an einen Patienten
umfaßt.









 zur Reaktion, in der R6 und R7 wie bei einer Verbindung der Formel (I) definiert sind und Hal ein Halogenatom darstellt.
zur Reaktion, in der R6 und R7 wie bei einer Verbindung der Formel (I) definiert sind und Hal ein Halogenatom darstellt.

 in der R3 und R4 wie bei einer Verbindung der Formel (I) definiert sind und Hal ein Halogenatom, vorzugsweise Brom oder Iod darstellt, in einem Lösungsmittel wie Tetrahydrofuran oder Diethylether sowie bei einer Temperatur zwischen 0°C und der Rückflußtemperatur des Lösungsmittels her.
in der R3 und R4 wie bei einer Verbindung der Formel (I) definiert sind und Hal ein Halogenatom, vorzugsweise Brom oder Iod darstellt, in einem Lösungsmittel wie Tetrahydrofuran oder Diethylether sowie bei einer Temperatur zwischen 0°C und der Rückflußtemperatur des Lösungsmittels her.








































































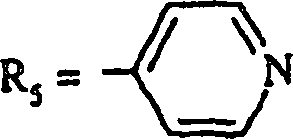
















 – R6 (C1-C4)-Alkoxy bedeutet; – R7 (C1-C4)-Alkoxy bedeutet; sowie deren Salze mit anorganischen oder organischen Säuren, deren Solvate und/oder deren Hydrate.
– R6 (C1-C4)-Alkoxy bedeutet; – R7 (C1-C4)-Alkoxy bedeutet; sowie deren Salze mit anorganischen oder organischen Säuren, deren Solvate und/oder deren Hydrate.
 bedeutet, in der R6 und R7 die in Anspruch 1 für die Verbindung der Formel (I) angegebenen Bedeutungen besitzen, wenn X eine Gruppe -O-; -NH-; -NH-CH2-; oder -NH-CH2-CH2- darstellt; mit einer Verbindung der Formel:
bedeutet, in der R6 und R7 die in Anspruch 1 für die Verbindung der Formel (I) angegebenen Bedeutungen besitzen, wenn X eine Gruppe -O-; -NH-; -NH-CH2-; oder -NH-CH2-CH2- darstellt; mit einer Verbindung der Formel: in der n und R5 die in Anspruch 1 für die Verbindung der Formel (I) angegebenen Bedeutungen besitzen; umsetzt, so daß man – wenn W eine Gruppe
in der n und R5 die in Anspruch 1 für die Verbindung der Formel (I) angegebenen Bedeutungen besitzen; umsetzt, so daß man – wenn W eine Gruppe bedeutet, die erwartete Verbindung der Formel (I) erhält; – oder wenn W ein Wasserstoffatom bedeutet, man die in dieser Weise erhaltene Verbindung der Formel:
bedeutet, die erwartete Verbindung der Formel (I) erhält; – oder wenn W ein Wasserstoffatom bedeutet, man die in dieser Weise erhaltene Verbindung der Formel: in Gegenwart einer Base mit einem Sulfonylhalogenid der Formel umsetzt:
in Gegenwart einer Base mit einem Sulfonylhalogenid der Formel umsetzt: in der R6 und R7 die in Anspruch 1 für die Verbindung der Formel (I) angegebenen Bedeutungen besitzen und Hal ein Halogenatom darstellt.
in der R6 und R7 die in Anspruch 1 für die Verbindung der Formel (I) angegebenen Bedeutungen besitzen und Hal ein Halogenatom darstellt.
 sowie deren Salze mit anorganischen oder organischen Säuren, in Form der optisch reinen Isomeren oder in Form einer Mischung.
sowie deren Salze mit anorganischen oder organischen Säuren, in Form der optisch reinen Isomeren oder in Form einer Mischung.