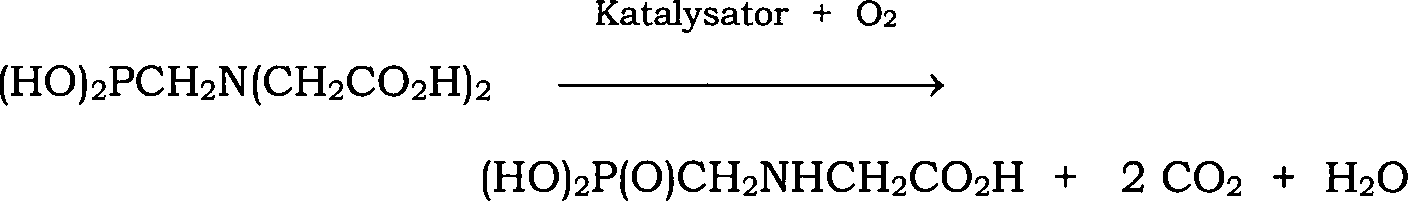-
Gebiet der
Erfindung
-
Die
vorliegende Erfindung betrifft Verfahren der Flüssigphasenoxidation zur Herstellung
von N-(Phosphonomethyl)glycin (in der agrikulturchemischen Industrie
auch als Glyphosat bekannt) und von verwandten Verbindungen. Diese
Erfindung betrifft zum Beispiel insbesondere Verfahren, bei denen
ein N-(Phosphonomethyl)iminodiessigsäuresubstrat (NPMIDA) (d. h.
N-(Phosphonomethyl)iminodiessigsäure,
ein Salz der N-(Phosphonomethyl)iminodiessigsäure oder ein Ester der N-(Phosphonomethyl)iminodiessigsäure kontinuierlich
oxidiert wird, um ein N-(Phosphonomethyl)glycinprodukt (d. h. N-(Phosphonomethyl)glycin,
ein Salz von N-(Phosphonomethyl)glycin oder einen Ester von N-(Phosphonomethyl)glycin)
zu bilden. Diese Erfindung betrifft insbesondere auch Verfahren,
bei denen ein N-(Phosphonomethyl)iminodiessigsäuresubstrat oxidiert wird,
um ein N-(Phosphonomethyl)glycinprodukt zu bilden, welches wiederum
(wenigstens teilweise) in einem adiabatischen Kristallisator kristallisiert
wird.
-
Hintergrund
der Erfindung
-
N-(Phosphonomethyl)glycin
wurde von Franz in US-A 3,799,758 beschrieben. N-(Phosphonomethyl)glycin
und seine Salze werden einfacher Weise als Nachauflaufherbizide
in wässriger
Formulierung angewendet. Es ist ein hochwirksames und kommerziell
wichtiges Herbizid mit einem Breitbandspektrum, das bei der Abtötung oder
zur Kontrolle des Wachstums einer breiten Vielfalt von Pflanzen
nützlich
ist, einschließend keimende
Samen, aufgehende Samensprösslinge,
reifende und bestehende Holz- und Unkrautvegetation, sowie von Wasserpflanzen.
-
Eines
der weiterverbreiteten Verfahren zur Herstellung von N-(Phosphonomethyl)glycinverbindungen umfasst
die oxidative Abspaltung eines Carboxymethylsubstituenten aus einem
N-(Phosphonomethyl)iminodiessigsäuresubstrat.
Im Verlauf der Jahre ist eine breite Vielfalt von Verfahren zur
Durchführung dieser
Oxidation offen gelegt worden. Vergleiche allgemein; Franz et al., "Glyphosate: A Unique
Global Herbicide",
ACS Monograph 189: 233–62
(1997) (und die darin zitierten Referenzen); Franz (US-A 3,950,402);
Hershman (US-A 3,969,398); Chou (US-A 4,624.937); Chou (US-A 4,696,772);
Ramon et al. (US-A 5,179,228); Felthouse (US-A 4,582,650); Siebenhaar
et al. (PCT/EP 99/04587) und Ebner et al. (Internationale Veröffentlichung
Nr. WO 99/43430). US-A 5,948,938 legt ein Verfahren zur Herstellung
von N-(Phosphonomethyl)glycin offen, umfassend das Behandeln von
N-(Phosphonomethyl)iminodiessigsäure
in Gegenwart von Wasser, Aktivkohle und Wasserstoffperoxid. Das
N-(Phosphonomethyl)glycinprodukt wird isoliert (und die Aktivkohle
entfernt) und anschließend
(durch Zugabe von Methanol, Aceton oder Acetonitril) kristallisiert.
Es werden Ausbeuten von bis zu 86,4% erzielt. Obwohl viele dieser
Verfahren geeignete Ausbeuten an N-(Phosphonomethyl)glycinprodukten
liefern, besteht nach wie vor Bedarf an einem verbesserten Verfahren
zur Oxidation von N-(Phosphonomethyl)iminodiessigsäuresubstraten.
Die erwünschten
Verbesserungen schließen
einen erhöhten
Durchsatz, verringerte Kosten pro Einheit N-(Phosphonomethyl)glycinprodukt und geringere
Konzentrationen an Nebenprodukten ein (z. B. Formaldehyd, Ameisensäure, N-Methyl-N-(Phosphonomethyl)glycin
(NMG) und Aminomethylphosphonsäure
(AMPA)).
-
Zusammenfassung
der Erfindung
-
Diese
Erfindung sieht zum Teil wirtschaftliche Verfahren zur Oxidation
von N-(Phosphonomethyl)iminodiessigsäure, Salzen
der N-(Phosphonomethyl)iminodiessigsäure und Ester vor, um N-(Phosphonomethyl)glycin,
Salze von N-(Phosphonomethyl)glycin oder Ester von N-(Phosphonomethyl)glycin)
zu bilden. Diese Erfindung sieht ebenfalls wirksame Verfahren zur
Reinigung und/oder Konzentrierung des in der Reaktionsmischung erhaltenen
N-(Phosphonomethyl)glycinprodukts vor.
-
Kurz
gesagt, ist die vorliegende Erfindung daher auf ein Verfahren zur
Herstellung eines N-(Phosphonomethyl)glycinprodukts gerichtet. Das
Verfahren umfasst das Einführen
eines wässrigen
Einspeisestroms, umfassend ein N-(Phosphonomethyl)iminodiessigsäuresubstrat
in ein Oxidations-Reaktorsystem, in dem das N-(Phosphonomethyl)iminodiessigsäuresubstrat
in Gegenwart eines Oxidationskatalysators zu einer ein N-(Phosphonomethyl)glycinprodukt
umfassenden Reaktionsproduktlösung
oxidiert wird. Die Reaktionsproduktlösung wird in mehrere Fraktionen
aufgeteilt, umfassend eine primäre
Fraktion und eine sekundäre Fraktion.
Die primäre
Fraktion wird durch Reduzieren des Druck bei gleichzeitigem Verdampfen
von Wasser unter im Wesentlichen adiabatischen Bedingungen unter
Ausfällung
von N-(Phosphonomethyl)glycinproduktkristallen abgekühlt, um
eine primäre
Produktaufschlämmung
zu erzeugen, umfassend ausgefällte
N-(Phosphonomethyl)glycinproduktkristalle und eine primäre Mutterlauge,
während
N-(Phosphonomethyl)glycinproduktkristalle auch aus einer wässrigen
sekundären
Kristallisationseinspeisemischung, umfassend in der sekundären Fraktion
enthaltenes N-(Phosphonomethyl)glycinprodukt ausgefällt werden,
um eine sekundäre
Produktaufschlämmung
zu erzeugen, welche ausgefällte
N-(Phosphonomethyl)glycinproduktkristalle und eine sekundäre Mutterlauge
umfasst.
-
Bei
einer anderen Ausführungsform
umfasst das Verfahren zur Herstellung eines N-(Phosphonomethyl)glycinprodukts
das Einführen
eines wässrigen
Einspeisestroms, umfassend ein N-(Phosphonomethyl)iminodiessigsäuresubstrat,
in ein Oxidations-Reaktorsystem und Oxidieren des N-(Phosphonomethyl)iminodiessigsäuresubstrats
in dem Oxidations-Reaktorsystem in Gegenwart eines Oxidationskatalysators,
um eine N-(Phosphonomethyl)glycinprodukt umfassende Reaktionsproduktlösung zu
erzeugen. Aus der Reaktionsproduktlösung werden N-(Phosphonomethyl)glycinproduktkristalle
ausgefällt,
um eine primäre
Produktaufschlämmung
zu erzeugen, welche ausgefällte
N-(Phosphonomethyl)glycinproduktkristalle und eine primäre Mutterlauge
umfasst. Das ausgefällte
N-(Phosphonomethyl)glycinprodukt wird dann aus der primären Mutterlauge abgetrennt,
welche durch Erwärmen
einer Verdampfungskristallisation unterworfen wird, um dadurch Wasser aus
der primären
Mutterlauge zu verdampfen und zusätzliche N-(Phosphonomethyl)glycinproduktkristalle
auszufällen
und eine sekundäre
Mutterlauge zu erzeugen.
-
Bei
einer anderen Ausführungsform
umfasst das Verfahren zur Herstellung eines N-(Phosphonomethyl)glycinprodukts
das Einführen
eines wässrigen
Einspeisestroms, umfassend ein N-(Phosphonomethyl)iminodiessigsäuresubstrat,
in ein katalytisches Oxidations-Reaktorsystem; das Oxidieren des
N-(Phosphonomethyl)iminodiessigsäuresubstrats,
in dem katalytischen Oxidations-Reaktorsystem in Gegenwart eines
ein Edelmetall auf Kohlenstoff umfassenden heterogenen Oxidationskatalysators
zur Erzeugung einer Reaktionssproduktmischung, umfassend das N-(Phosphonomethyl)glycinprodukt;
das Ausfällen
von N-(Phosphonomethyl)glycinproduktkristallen aus einer primären Kristallisationseinspeisemischung,
umfassend N-(Phosphonomethyl)glycinprodukt in dieser Reaktionsproduktmischung,
um eine primäre
Produktaufschlämmung
zu erzeugen, um fassend ausgefällte
N-(Phosphonomethyl)glycinproduktkristalle und eine primäre Mutterlauge;
und das Rezyklieren der primären
Mutterlauge und Einführen
in das katalytische Oxidations-Reaktorsystem, in dem das N-(Phosphonomethyl)iminodiessigsäuresubstrat
zum N-(Phosphonomethyl)glycinprodukt oxidiert wird.
-
Die
Verdampfungskühlung
der primären
Fraktion umfasst vorteilhafterweise:
Einführen einer, die primäre Fraktion
unfassenden, wässrigen
Verdampfungseinspeisemischung in eine Verdampfungszone;
Verdampfen
von Wasser aus der wässrigen
Verdampfungseinspeisemischung in der Verdampfungszone in Gegenwart
von teilchenförmigem
N-(Phosphonomethyl)glycinfeststoffprodukt unter Erzeugung einer
Wasserdampf umfassenden Dampfphase, das Ausfällen von N-(Phosphonomethyl)glycinproduktkristallen
aus der wässrigen
Phase und die Erzeugung eines Verdampfungsprodukt, umfassend N-(Phosphonomethyl)glycinfeststoffprodukt
und primäre
Mutterlauge, welche mit N-(Phosphonomethyl)glycinprodukt im Wesentlichen
gesättigt
oder übersättigt ist;
und
Beibehalten eines Verhältnisses
von N-(Phosphonomethyl)glycinfeststoffprodukt zu primärere Mutterlauge
in der Verdampfungszone, welches größer ist als das Verhältnis der
durch die Verdampfungswirkung erzeugten Zunahme an N-(Phosphonomethyl)glycinproduktkristallen
zu der dadurch erzeugten Zunahme an primärer Mutterlauge.
-
Das
geschilderte Verfahren umfasst vorzugsweise das Einführen der
die primäre
Fraktion umfassenden wässrigen
Verdampfungseinspeisemischung in eine Dampf/Flüssigkeits-Trennzone der Verdampfungszone,
in welcher der Druck unter dem Dampfdruck der Mischung liegt, was
die Verdampfung von Wasser aus der wässrigen Verdampfungeinspeisemischung
erlaubt, wodurch eine Wasserdampf umfassende Dampfphase erzeugt
und das Ausfällen
von N-(Phosphonomethyl)glycinproduktkristallen aus der wässrigen
flüssigen
Phase ermöglicht
wird, um einen ersten Aufschlämmungsstrom
zu erzeugen, umfassend N-(Phosphonomethyl)glycinfeststoffprodukt
und primäre
Mutterlauge, die mit N-(Phosphonomethyl)glycinprodukt
im Wesentlichen gesättigt
oder übersättigt ist;
Abtrennen
der Dampfphase vom ersten Aufschlämmungsstrom;
Einführen des
ersten Aufschlämmungsstroms
in eine Retentionszone, in welcher die überstehende Flüssigkeit, umfassend
eine Fraktion aus der primären
Mutterlauge, einen sekundären
Aufschämmungsstrom,
umfassend ausgefällte
N-(Phosphonomethyl)glycinproduktkristalle
und primäre
Mutterlauge, abgetrennt wird, wobei die Retentionszone einen Einlass
für die
erste Aufschlämmung,
einen Auslass für
die Dekanationsflüssigkeit
aufweist und die überstehende
Flüssigkeit über dem
Einlass angeordnet ist, und ein Auslass für die zweite Aufschlämmung oberhalb
des Einlasses aber unterhalb des Auslasses für die Dekantationsflüssigkeit
angeordnet ist; und
Beibehalten der relativen Raten mit denen
die erste Aufschlämmung
in die Retentionszone eingeführt
wird, die zweite Aufschlämmung
durch den zweiten Aufschlämmungsauslass
abgezogen wird und die überstehende Flüssigkeit
durch den Flüssigkeitsdekantationsauslass
abgezogen wird, in der Weise, dass die Strömungsgeschwindigkeit stromaufwärts in einem
unteren Bereich der Rückhaltezone
unterhalb des zweiten Aufschlämmunngsauslass
ausreicht, um ausgefällte
N-(Phosphonomethyl)glycinproduktkristalle in der flüssigen Phase in
Suspension zu halten, während
die Strömungsgeschwindigkeit
stromaufwärts
in einer oberen Rückhaltezone
oberhalb des zweiten Aufschlämmungsauslasses
geringer ist als die Sedimentationsgeschwindigkeit von mindestens
80 Gew.-% der ausgefällten
N-(Phosphonomethyl)glycinproduktkristalle im unteren Bereich.
-
Das
Oxidations-Reaktorsystem, in welches der wässrige Einspeisestrom, welcher
das N-(Phosphonomethyl)iminodiessigsäuresubstrat enthält, eingeführt wird,
ist vorteilhafterweise ein Primäroxidations-Reaktorsystem,
umfassend eine oder mehrere Oxidationsreaktionszonen, wobei das
N-(Phosphonomethyl)iminodiessigsäuresubstrat
in dem Primäroxidationsreaktorsystem
oxidiert wird, um die Reaktionsproduktlösung zu erzeugen, welche das
N-(Phosphonomethyl)glycinprodukt und nicht umgesetztes N-(Phosphonomethyl)iminodiessigsäuresubstrat
umfasst, wobei das Verfahren weiterhin das Einführen der sekundären Fraktion
in ein sekundäres
Oxidations-Reaktorsystem umfasst, welches eine oder mehrere Oxidationsreaktionszonen
und das Oxidieren von nicht umgesetztem N-(Phosphonomethyl)-iminodiessigsäuresubstrat
im zweiten Oxidations-Reaktorsystem umfasst, um einen sekundären Oxidationsreaktorauslass
zu erzeugen, umfassend das N-(Phosphonomethyl)glycinprodukt, die
wässrige
sekundäre
Kristallisationseinspeisemischung, welche das im sekundären Oxidationsreaktorausfluss
enthaltene N-(Phosphonomethyl)glycinprodukt umfasst.
-
Andere
Merkmale dieser Erfindung werden zum Teil ersichtlich und zum Teil
hierin nachstehend herausgestellt.
-
Kurze Beschreibung
der Zeichnungen
-
1 zeigt
ein Beispiel eines Querschnitts durch einen Honigwaben-Kata lysatorträger.
-
2 ist
ein schematisches Fließbild
eines kontinuierlichen Oxidations-Reaktorsystems für die Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
zur Bildung eines N-(Phosphonomethyl)glycinprodukts. Das Reaktorsystem
umfasst eine Oxidationsreaktionszone mit Rückvermischung unter Verwendung
einer Aufschlämmung
aus einem heterogenen teilchenförmigen
Katalysator, welche in einem vom Wärmeübertragungskreislauf unabhängigen Kreislauf
zurückgeführt wird.
-
2A ist ein schematisches Fließbild eines kontinuierlichen
Oxidations-Reaktorsystems
für die
Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats zur Bildung eines
N-(Phosphonomethyl)glycinprodukts. Das Reaktorsystem umfasst eine
Oxidationsreaktionszone mit Rückvermischung
unter Verwendung einer Aufschlämmung
aus einem heterogenen teilchenförmigen
Katalysator, welche in einem vom Wärmeübertragungskreislauf unabhängigen Kreislauf
zurückgeführt wird,
und einschließend
einen Spülbehälter und Katalysatorrückführbehälter.
-
2B ist ein schematisches Fließbild eines kontinuierlichen
Oxidations-Reaktorsystems
für die
Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats zur Bildung eines
N-(Phosphonomethyl)glycinprodukts. Das Reaktorsystem umfasst eine
Oxidations-Reaktionszone mit Rückvermischung
unter Verwendung einer Aufschlämmung
aus einem heterogenen teilchenförmigen
Katalysator, welche über
einen Wärmeübertragungskreislauf
zurückgeführt wird.
-
3 ist
ein schematisches Fließbild
eines kontinuierlichen Oxidations-Reaktorsystems für die Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
zur Bildung eines N-(Phosphonomethyl)glycinprodukts. Das Reaktorsystem
umfasst zwei in Reihe angeordnete Oxidationsreaktionszonen mit Rückvermischung
unter Verwendung einer Aufschlämmung
aus einem heterogenen teilchenförmigen
Katalysator, welche von der ersten Reaktionszone zur zweiten Reaktionszone
strömt
und zur ersten Reaktionszone zurückgeführt wird.
-
4 ist
ein schematisches Fließbild
eines kontinuierlichen Oxidationsreaktorsystems für die Oxidation
eines N-(Phosphonomethyl)iminodiessigsäuresubstrats zur Bildung eines
N-(Phosphonomethyl)glycinprodukts. Das Reaktorsystem umfasst zwei
in Reihe angeordnete Oxidationsreaktionszonen mit Rückvermischung
unter Verwendung einer Aufschlämmung
aus einem heterogenen teilchenförmigen
Katalysator, welche von der ersten Reaktionszone zur zweiten Reaktionszone
strömt
und zur ersten Reaktionszone zurückgeführt wird,
und welche zu beiden Reaktionszonen zurückgeführt wird.
-
5 ist
ein schematisches Fließbild
eines kontinuierlichen Oxidations-Reaktorsystems für die Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
zur Bildung eines N-(Phosphonomethyl)glycinprodukts. Das Reaktorsystem
umfasst zwei in Reihe angeordnete Oxidationsreaktionszonen mit Rückvermischung
unter Verwendung zweier unabhängiger
Aufschlämmungsmassen
aus einem heterogenen teilchenförmigen
Katalysator, dermaßen,
dass der Katalysator aus der ersten Reaktionszone zur ersten Reaktionszone zurückgeführt wird
und der Katalysator aus der zweiten Reaktionszone zur zweiten Reaktionszone
zurückgeführt wird.
-
6 ist
ein schematisches Fließbild
eines kontinuierlichen Oxidationsreaktorsystems für die Oxidation
eines N-(Phosphonomethyl)iminodiessigsäuresubstrats zur Bildung eines
N-(Phosphonomethyl)glycinprodukts. Das Reaktorsystem umfasst zwei
in Reihe angeordnete Oxidationsreaktionszonen mit Rückvermischung
unter Verwendung einer Aufschlämmung
aus einem heterogenen teilchenförmigen
Katalysator, welche aus der ersten Reaktionszone zur ersten Reaktionszone
und aus der zweiten Reaktionszone zu beiden Reaktionszonen rezirkuliert
wird.
-
7 ist
eine schematische Darstellung eines Kreislaufreaktors mit einer
Einspritzdüse,
welcher im kontinuierlichen Oxidations-Reaktorsystem der vorliegenden
Erfindung zur Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
zur Bildung eines N-(Phosphonomethyl)glycinprodukts verwendet werden kann.
-
8 ist
eine schematische Darstellung eines Festbettreaktors welcher im
kontinuierlichen Oxidations-Reaktorsystem der vorliegenden Erfindung
zur Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
zur Bildung eines N-(Phosphonomethyl)glycinprodukts verwendet werden
kann.
-
9 ist
eine schematische Darstellung eines zirkulierenden Fließbettreaktors,
welcher im kontinuierlichen Oxidations-Reaktorsystem der vorliegenden
Erfindung zur Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
zur Bildung eines N-(Phosphonomethyl)glycinprodukts verwendet werden
kann.
-
10 ist ein schematisches Fließbild eines kontinuierlichen,
aufgeteilten Reaktorsystems zur Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
zur Bildung eines N-(Phosphonomethyl)glycinprodukts. Das Reaktorsystem
umfasst mehrere Reaktoren in denen die Reaktionsmischung nacheinander
vom jeweiligen Reaktor zum nachfolgenden Reaktor in der Reihe fortschreitet.
-
11 ist ein schematisches Fließbild eines integrierten Verfahrens
zur Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
in einem Reak torsystem, um eine Oxidationsreaktionsmischung zu bilden,
umfassend ein N-(Phosphonomethyl)glycinprodukt
und zur Rückgewinnung
des N-(Phosphonomethyl)glycinprodukts aus der Oxidationsreaktionsmischung
die Verwendung eines nicht-adiabatischen wärmegesteuerten Verdampfungskristallisators.
-
12 ist ein schematisches Fließbild eines integrierten Verfahrens
zur Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
in einem Reaktorsystem, um eine Oxidationsreaktionsmischung zu bilden,
umfassend ein N-(Phosphonomethyl)glycinprodukt
und zur Rückgewinnung
des N-(Phosphonomethyl)glycinprodukts aus der Oxidationsreaktionsmischung
die Verwendung eines adiabatischen Kristallisators.
-
12A ist ein schematisches Fließbild eines adiabatischen Kristallisatorsystems
wie es zur Rückgewinnung
eines N-(Phosphonomethyl)glycinprodukts aus einer Oxidationsreaktionsmischung
verwendet wird.
-
13 ist ein schematisches Fließbild eines integrierten Verfahrens
zur Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
in einem Reaktorsystem, um eine Oxidationsreaktionsmischung zu bilden,
umfassend ein N-(Phosphonomethyl)glycinprodukt
und zur Rückgewinnung
des N-(Phosphonomethyl)glycinprodukts aus der Oxidationsreaktionsmischung
die Verwendung einer Kombination aus einem in Reihe angeordneten
adiabatischen Kristallisator und einem nicht-adiabatischen wärmegesteuerten
Verdampfungskristallisator.
-
14 ist ein schematisches Fließbild eines integrierten Verfahrens
zur Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
in einem Reaktorsystem, um eine Oxidationsreaktionsmischung zu bilden,
umfassend ein N-(Phosphonomethyl)glycinprodukt
und zur Rückgewinnung
des N-(Phosphonomethyl)glycinprodukts aus der Oxidationsreaktionsmischung
die Verwendung einer Kombination aus einem semi-parallel angeordneten
adiabatischen Kristallisator und einem nicht-adiabatischen wärmegesteuerten
Verdampfungskristallisator.
-
14A ist ein schematisches Fließbild eines integrierten Verfahrens
zur Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
unter Bildung eines N-(Phosphonomethyl)glycinprodukts unter Verwendung
einer Kombination aus einem semi-parallel angeordnetem adiabatischen
Kristallisator und einem nicht-adiabatischen wärmegesteuerten Verdampfungskristallisator.
Das N-(Phosphonomethyl)iminodiessigsäuresubstrat wird in einem primären Reaktorsystem
oxidiert, um eine Oxidationsreaktionsmischung zu bilden, umfassend
das N-(Phosphonomethyl)glycinprodukt
und nicht umgesetztes N-(Phosphonomethyl)iminodiessigsäuresubstrat.
Eine primäre
Fraktion aus der Oxidationsreaktionsmischung aus dem primären Reaktorsystem
wird in den adiabatischen Kristal lisator eingespeist, während nicht
umgesetztes N-(Phosphonomethyl)iminodiessigsäuresubstrat in einer Einspeisefraktion
für den
sekundären
Oxidationsreaktor in einem sekundären Reaktorsystem oxidiert
wird, um zusätzliches
N-(Phosphonomethyl)glycinprodukt
zu bilden, bevor es zum nicht-adiabatischen Kristallisator weitergeleitet
wird.
-
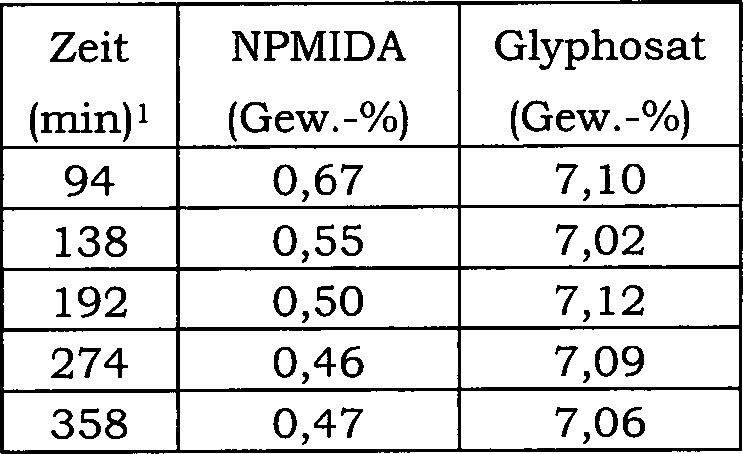
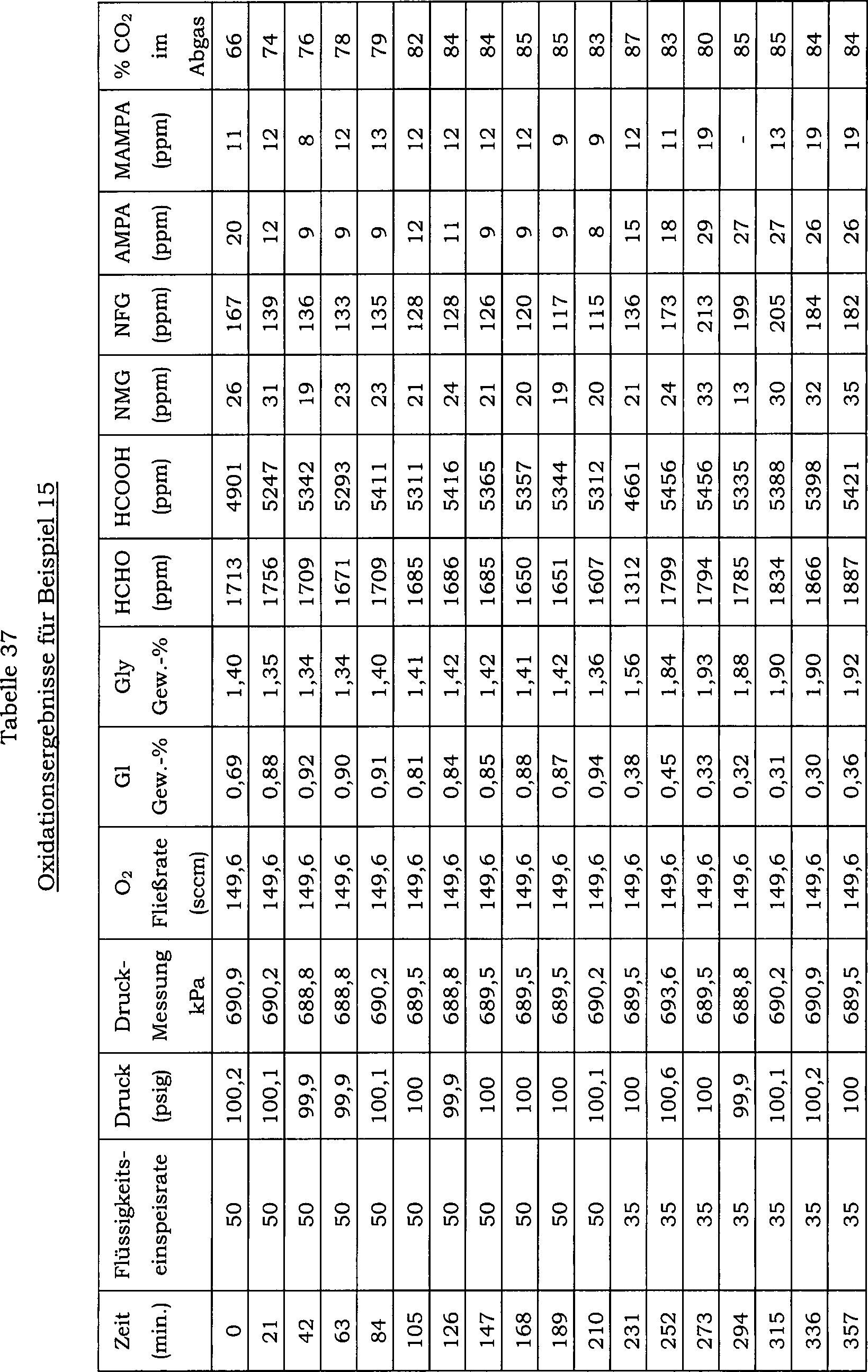
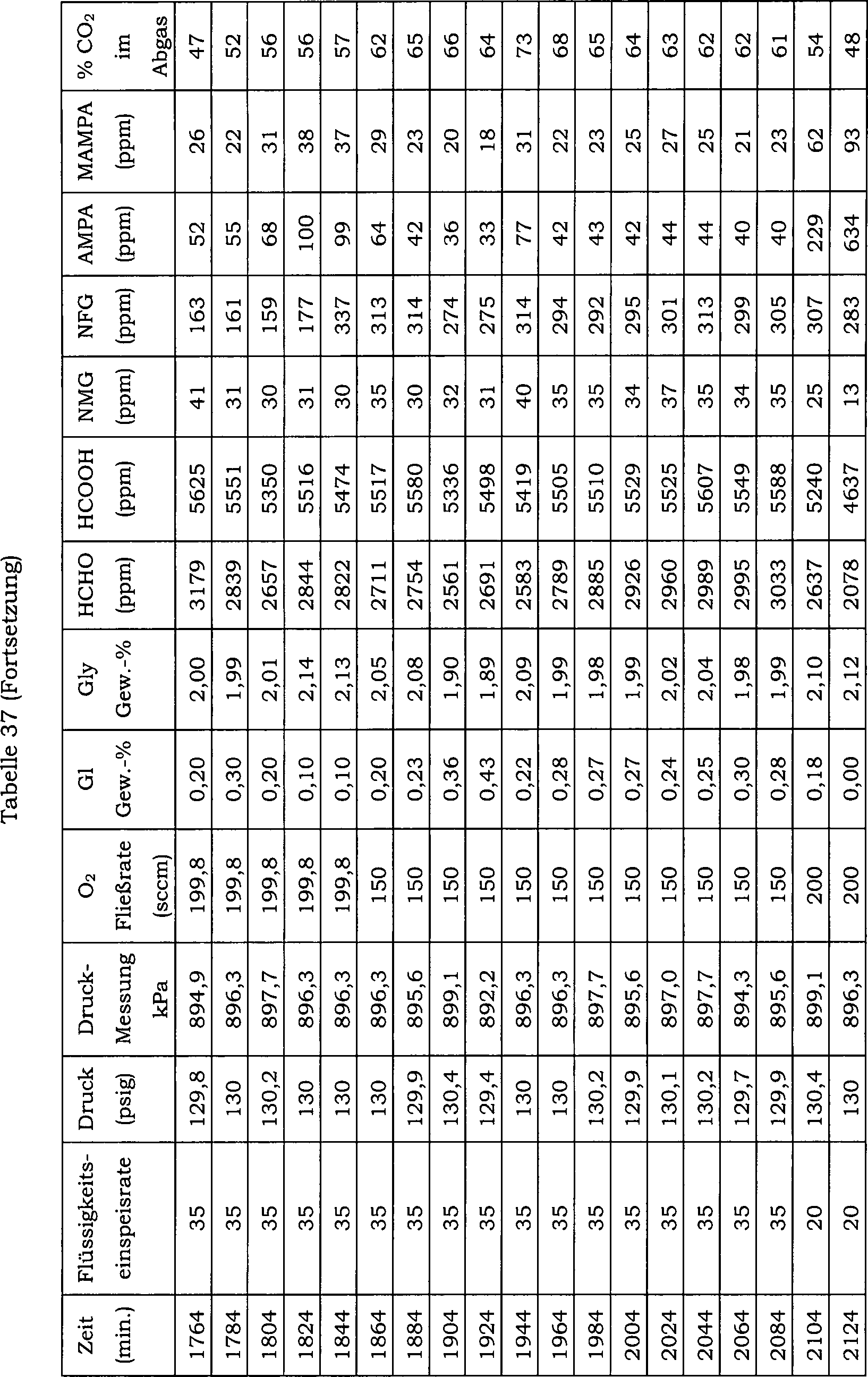
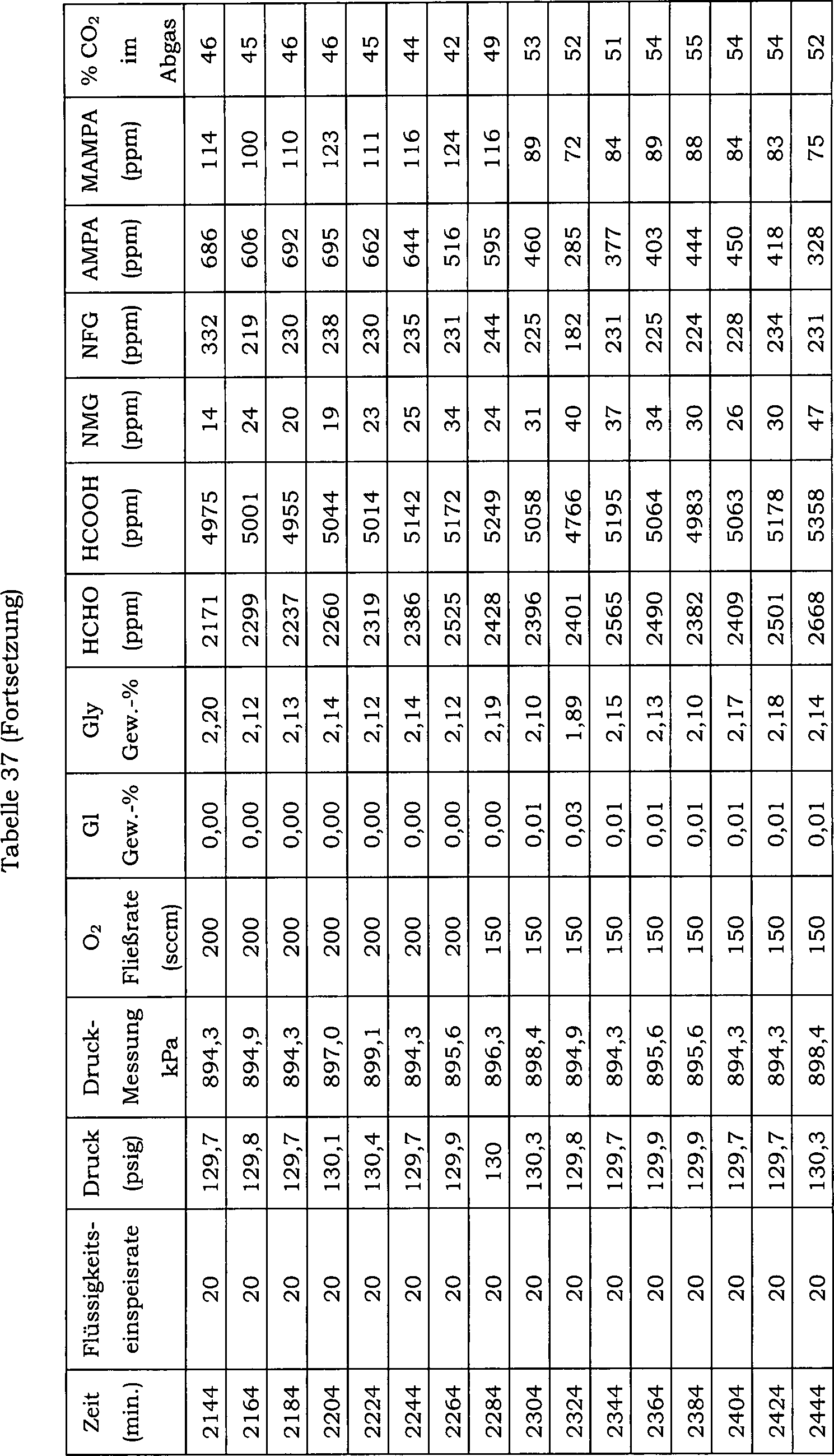
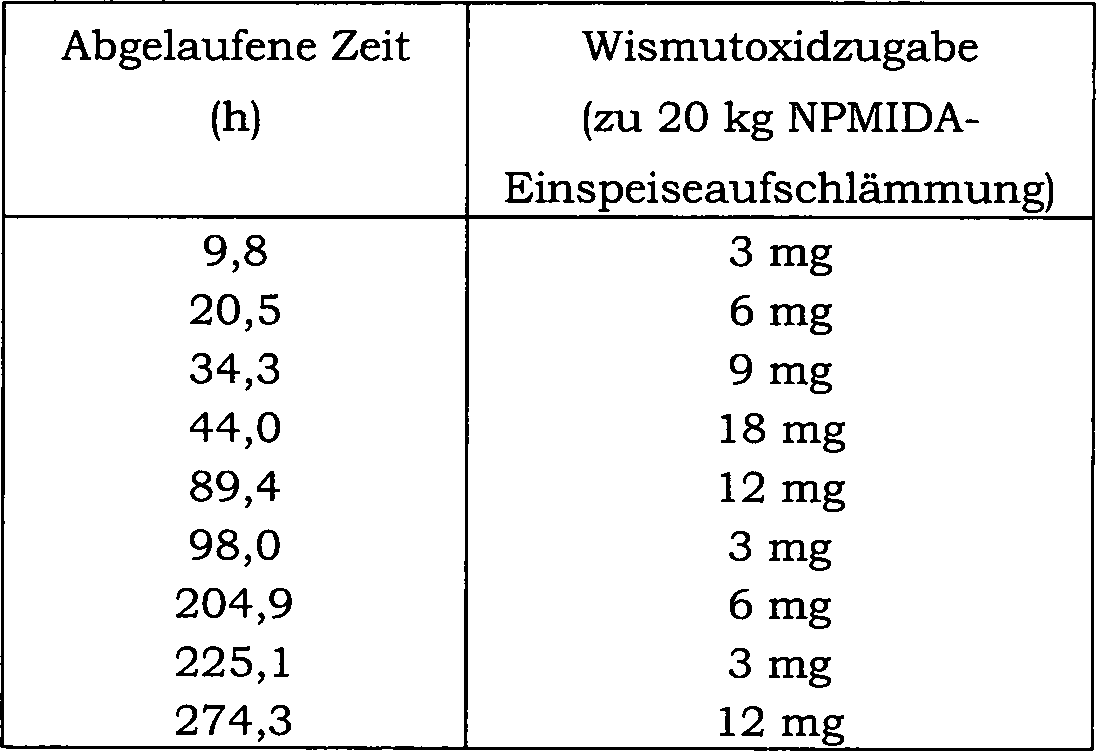
15 zeigt die Auswirkung einer direkten Einmalzugabe
von Wismutoxid zu einer N-(Phosphonomethyl)iminodiessigsäure-Reaktionsmischung über 20 Reaktionschargen
auf den Konzentrationsverlauf von Ameisensäure als Nebenprodukt. Hier
betrug die Katalysatorkonzentration in der Reaktionsmischung 0,5 Gew.-%
und der Katalysator enthielt 5 Gew.-% Platin und 0,5 Gew.-% Eisen.
-
16 zeigt die Auswirkung einer direkten Einmalzugabe
von Wismutoxid zu einer N-(Phosphonomethyl)iminodiessigsäure-Reaktionsmischung über 30 Reaktionschargen
auf den Konzentrationsverlauf von Ameisensäure als Nebenprodukt. Hier
betrug die Katalysatorkonzentration in der Reaktionsmischung 0,75 Gew.-%
und der Katalysator enthielt 5 Gew.-% Platin und 1 Gew.-% Zinn.
-
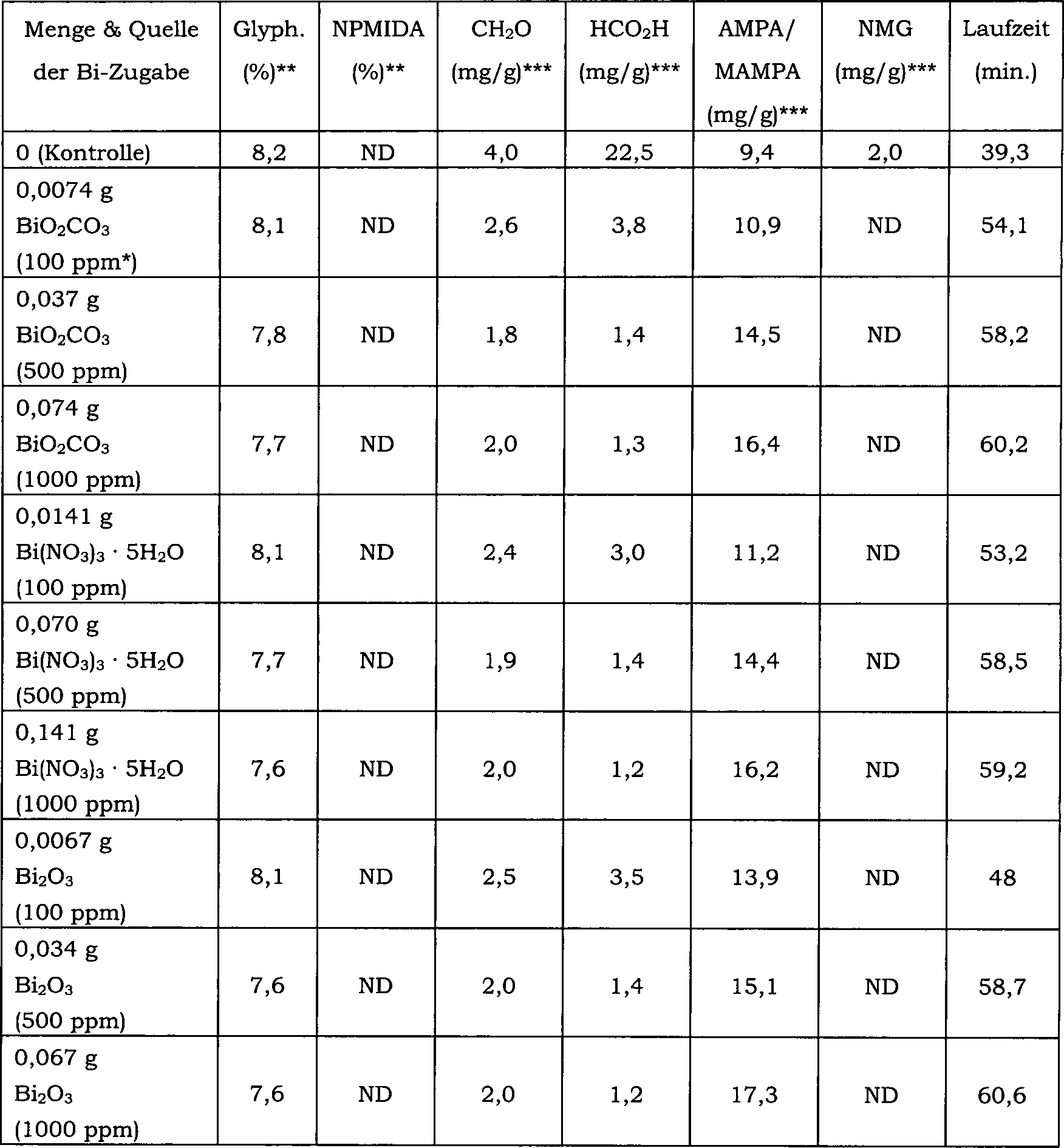
17 zeigt die Auswirkung einer direkten Einmalzugabe
von Wismutoxid zu einer N-(Phosphonomethyl)iminodiessigsäureoxidations-Reaktionsmischung über 30 Reaktionschargen
auf den Konzentrationsverlauf von Formaldehyd als Nebenprodukt.
Hier betrug die Katalysatorkonzentration in der Reaktionsmischung 0,75
Gew.-% und der Katalysator enthielt 5 Gew.-% Platin und 1 Gew.-%
Zinn.
-
18 zeigt die Auswirkung einer direkten Einmalzugabe
von Wismutoxid zu einer N-(Phosphonomethyl)iminodiessigsäureoxidations-Reaktionsmischung über 30 Reaktionschargen
auf den Konzentrationsverlauf von N-Methyl-N-(phosphonomethyl)glycin
(NMG) als Nebenprodukt. Hier betrug die Katalysatorkonzentration
in der Reaktionsmischung 0,75 Gew.-% und der Katalysator enthielt
5 Gew.-% Platin und Gew.-% Zinn.
-
19 zeigt die Auswirkung des Mischens von Wismutoxid
mit einem Oxidationskatalysator, welcher vorher zur Herstellung
von 133 Chargen N-(Phosphonomethyl)iminodiessigsäureoxidations-Reaktionen verwendet
worden war, auf die Ameisensäure-,
Formaldehyd- und N-Methyl-N-(phosphonomethyl)glycinerzeugung (NMG).
Hier umfasste der Katalysator 5 Gew.-% Platin und 0,5 Gew.-% Eisen
auf einem Kohlenstoffträger.
-
20 zeigt die Auswirkung des Mischens von Wismutoxid
mit einem Oxidationskatalysator, welcher vorher zur Herstellung
von 30 Chargen N-(Phosphonomethyl)iminodiessigsäure-Oxidationsreaktionen verwendet
worden war, auf die Ameisensäure-,
Formaldehyd- und N-Methyl-N-(phosphonomethyl)glycinerzeugung (NMG).
Hier umfasste der Katalysator 5 Gew.-% Platin und 1 Gew.-% Zinn
auf einem Kohlenstoffträger.
-
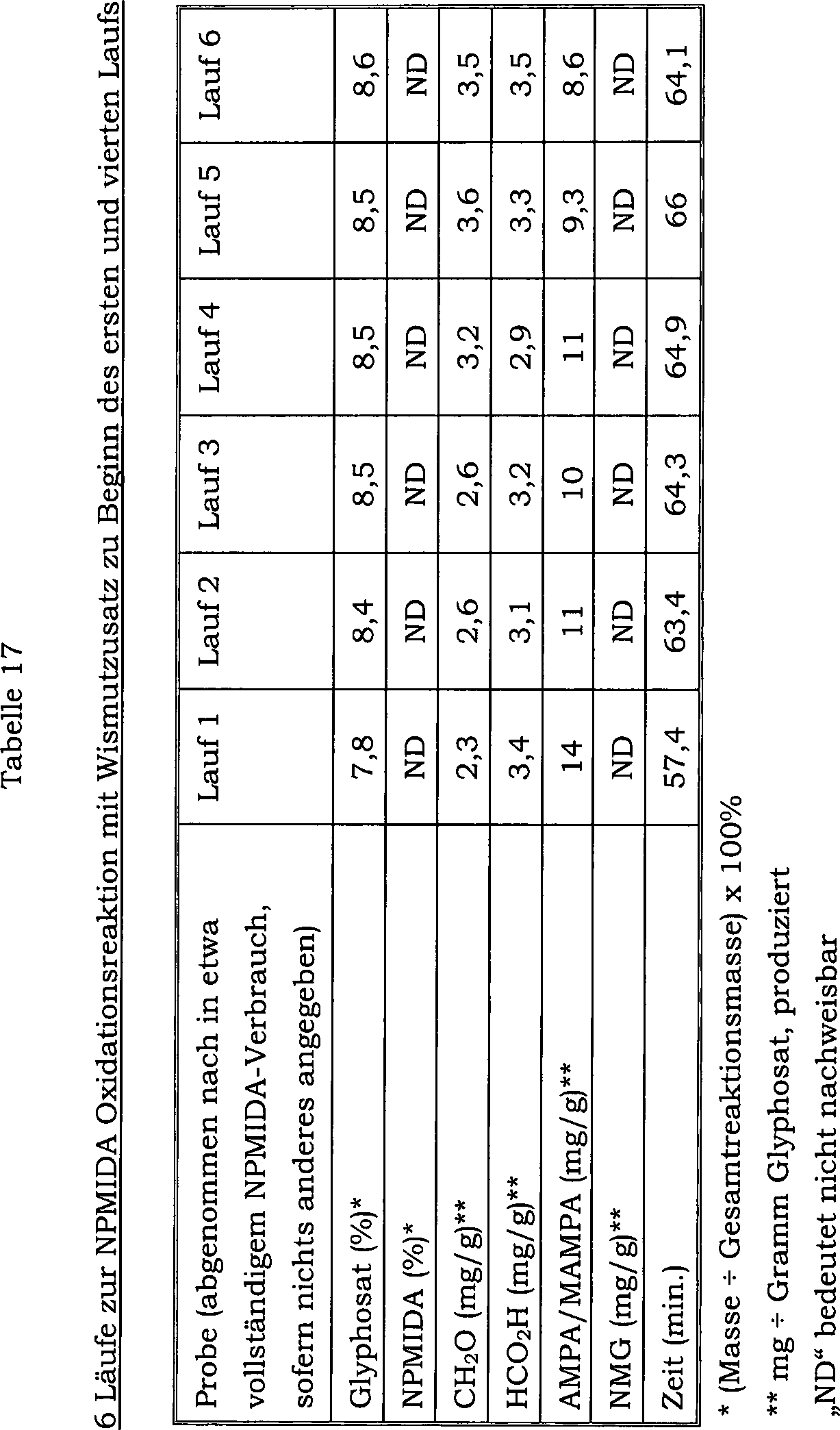
21 zeigt die Auswirkung auf den Konzentrationsverlauf
von Ameisensäure
als Nebenprodukt über 107
Chargen, welche durch einmaliges Mischen von Wismutoxid mit einem
Katalysator verursacht wird, welcher 5 Gew.-% Platin und 1 Gew.-%
Zinn enthält.
-
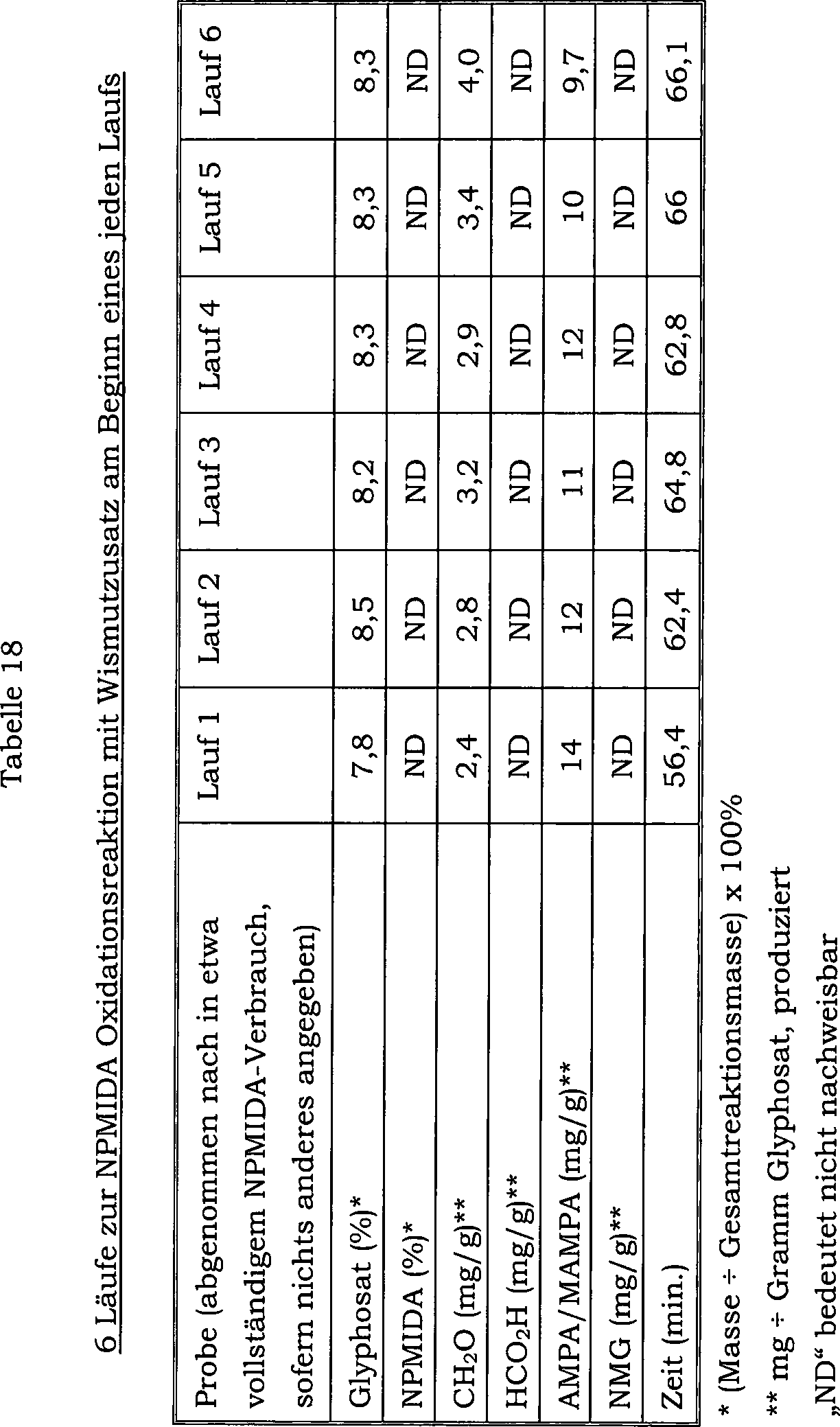
22 zeigt die Auswirkung auf den Konzentrationsverlauf
von Formaldehyd als Nebenprodukt über 107 Chargen, welche durch
einmaliges Mischen von Wismutoxid mit einem Katalysator verursacht
wird, welcher 5 Gew.-% Platin und 1 Gew.-% Zinn enthält.
-
23 zeigt die Auswirkung auf den Konzentrationsverlauf
von N-Methyl-N-(phosphonomethyl)glycin (NMG)
als Nebenprodukt über
107 Chargen, welche durch einmaliges Mischen von Wismutoxid mit
einem Katalysator verursacht wird, welcher 5 Gew.-% Platin und 1
Gew.-% Zinn enthält.
-
24 zeigt den Verlauf von Formaldehyd und Ameisensäure in der
Produktflüssigkeit
von Beispiel 21.
-
25 zeigt den Verlauf von Glyphosat und N-(Phosphonomethyl)iminodiessigsäure in der
Produktflüssigkeit
von Beispiel 22.
-
26 zeigt den Verlauf von Glyphosat und N-(Phosphonomethyl)iminodiessigsäure in der
Produktflüssigkeit
von Beispiel 23.
-
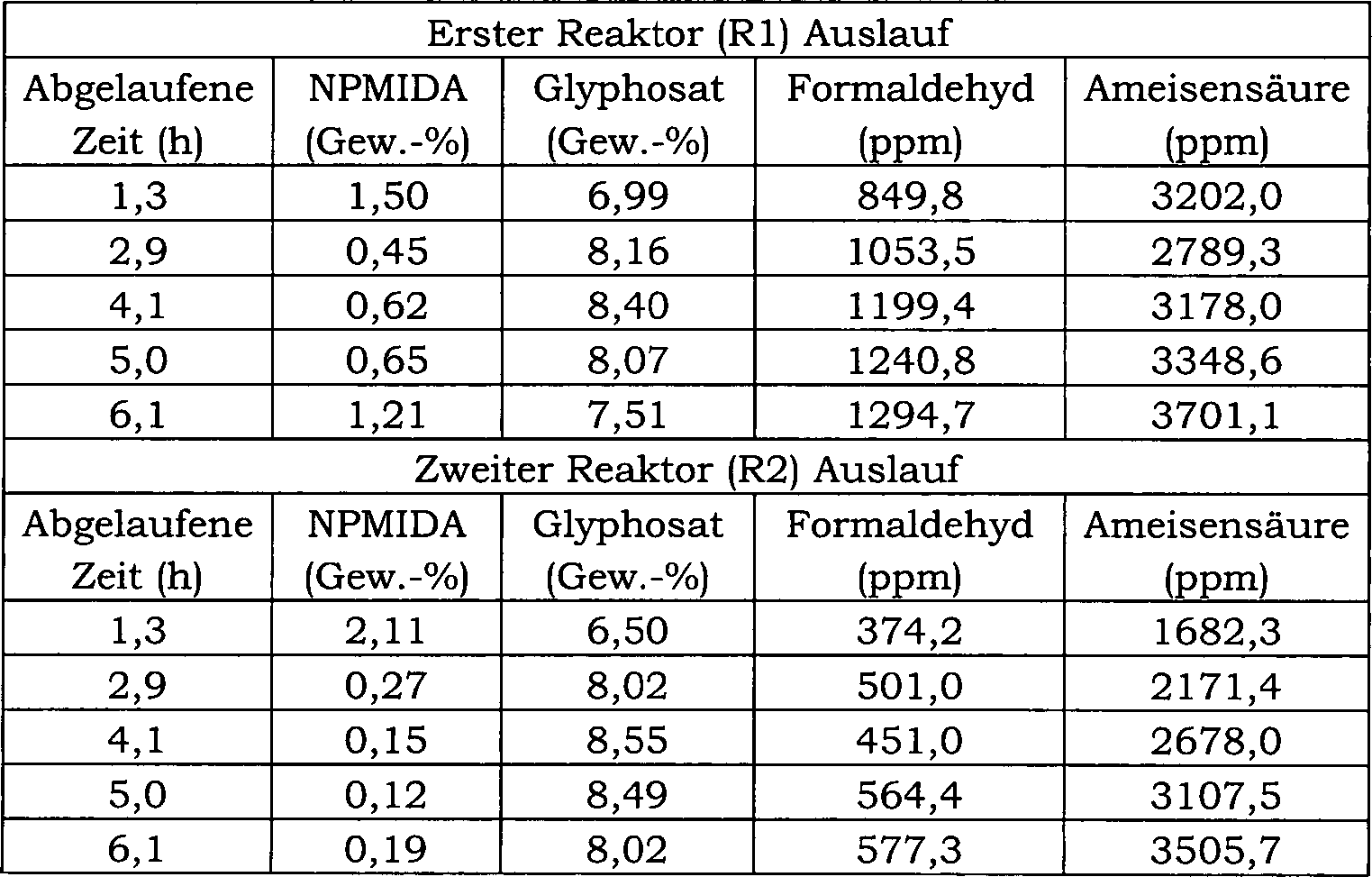
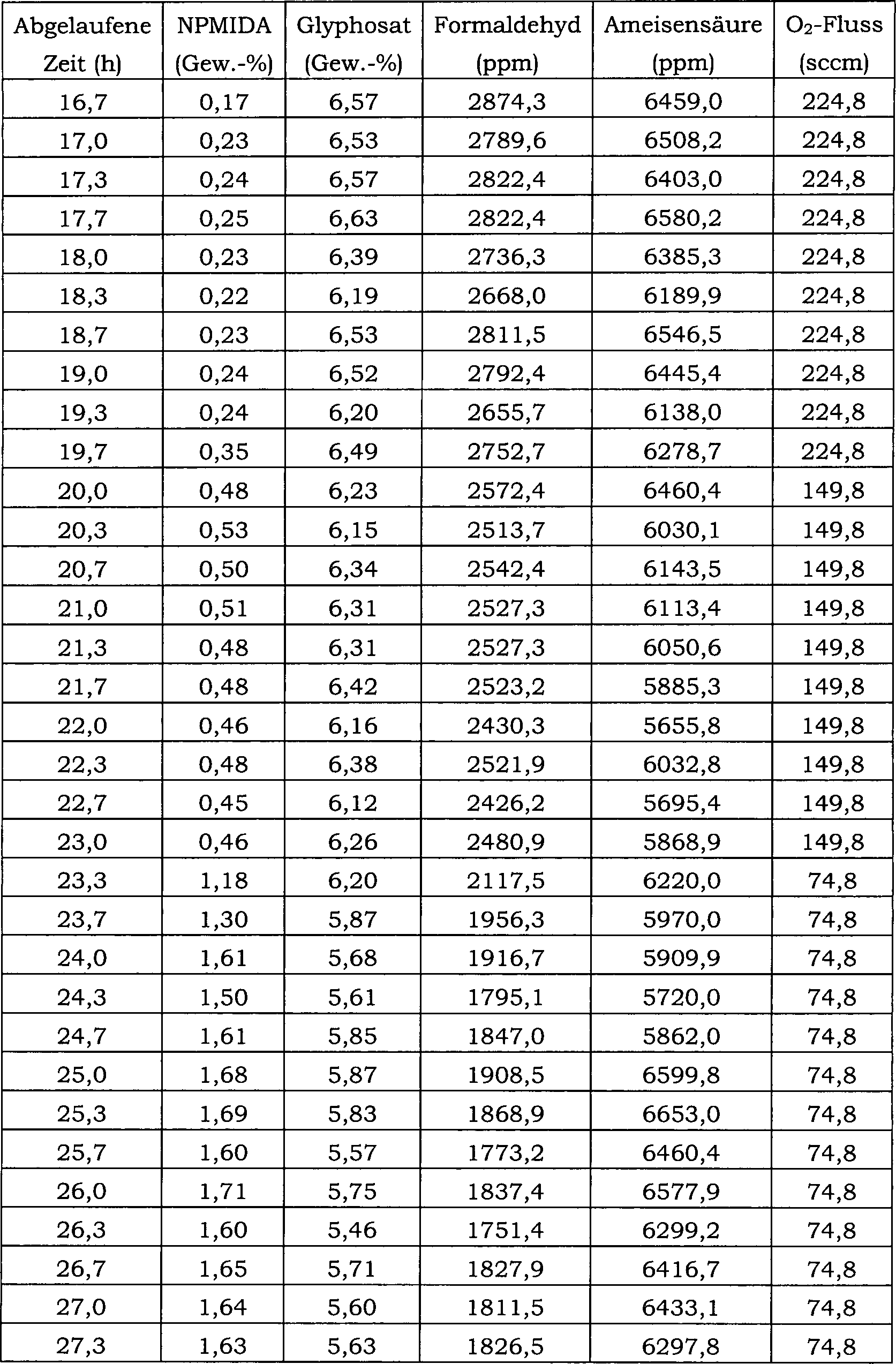
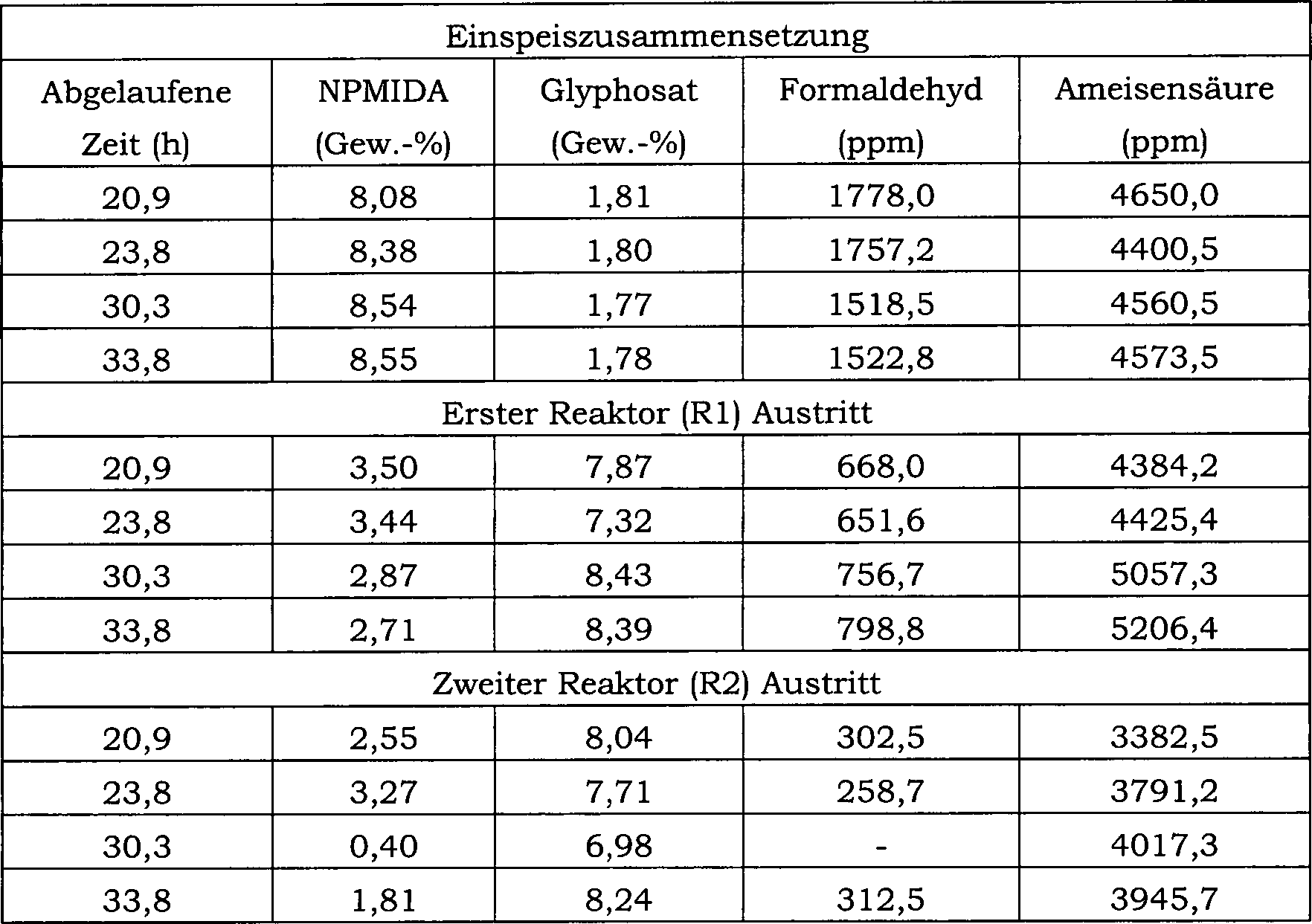
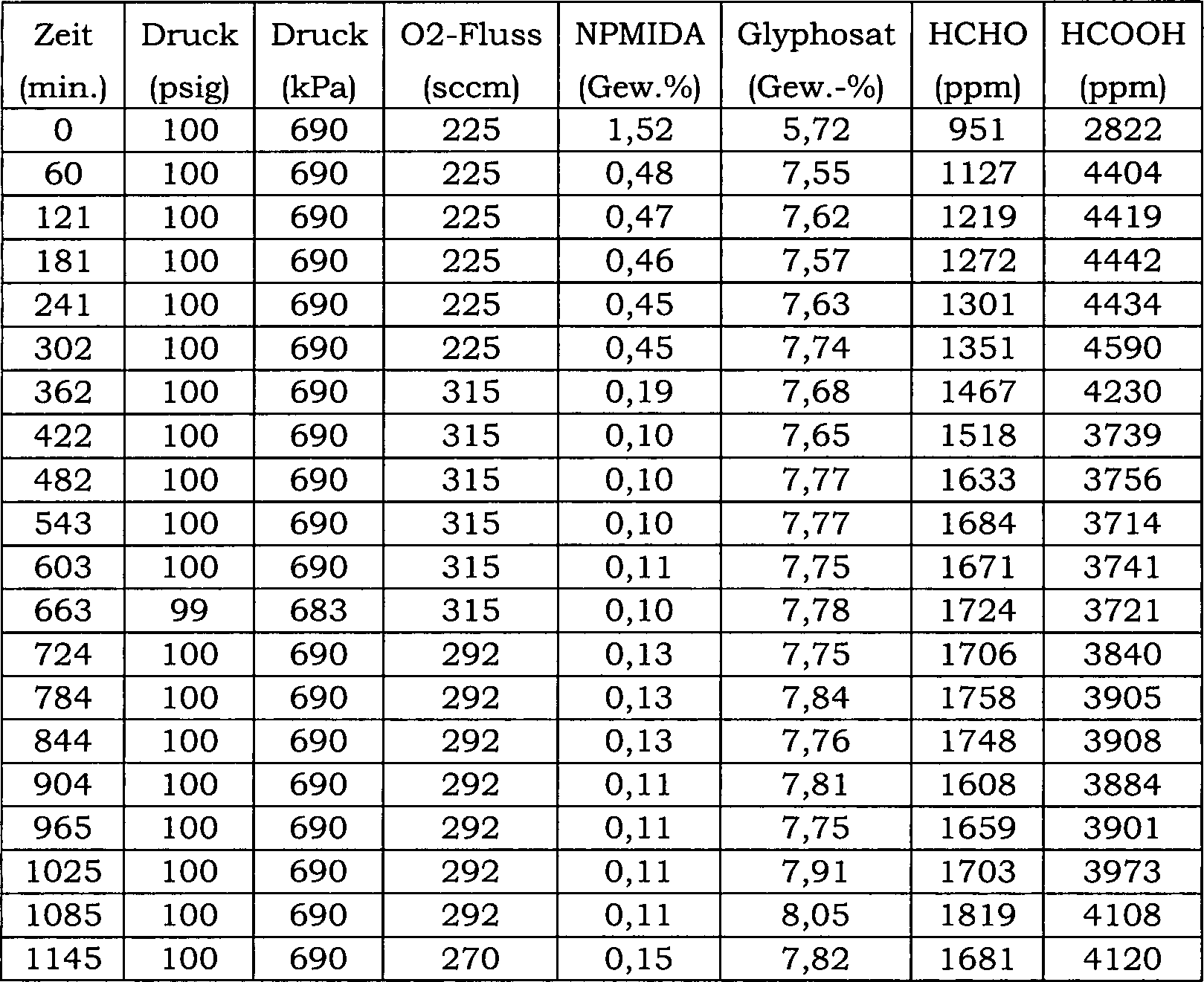
27 ist ein Blockfließbild für das in Beispiel 24 verwendete
kontinuierliche Reaktorsystem.
-
28 ist ein Blockfließbild für das in Beispiel 25 verwendete
kontinuierliche Reaktorsystem.
-
29 ist ein Blockfließbild für das in Beispiel 28 verwendete
kontinuierliche Reaktorsystem.
-
30 ist ein Blockfließbild für das in Beispiel 35 verwendete
kontinuierliche Reaktorsystem.
-
31 ist ein Blockfließbild für das in Beispiel 36 verwendete
kontinuierliche Reaktorsystem.
-
Detaillierte
Beschreibung der Erfindung
-
Generell
umfassen die Verfahren dieser Erfindung (1) das Oxidieren eines
N-(Phosphonomethyl)iminodiessigsäuresubstrats
in einer oder mehreren Oxidationsreaktionszonen unter Bildung eines
N-(Phosphonomethyl)glycinprodukts, und/oder (2) das Konzentrieren
und/oder Reinigen des N-(Phosphonomethyl)glycinprodukts. Diese Schritte
werden zusammen mit verschiedenen anderen Merkmalen der bevorzugten
Ausführungsformen
nachstehend erläutert.
-
Das
N-(Phosphonomethyl)iminodiessigsäuresubstrat
wird durch Einführen
des Substrats und eines Oxidationsmittels (d. h. eine Sauerstoffquelle)
in ein Reaktorsystem oxidiert, umfassend eine oder mehrere Oxidationsreaktionszonen,
welche einen Oxidationskatalysator enthalten. Die Oxidationsreaktion
verläuft
im Allgemeinen nach der folgenden Gleichung:
worin R
1,
R
2, R
3 und R
4 jeweils unabhängig Wasserstoff, ein landwirtschaftlich
annehmbares Kation, Hydrocarbyl oder substituiertes Hydrocarbyl
sind.
-
Ein
Hydrocarbyl ist eine beliebige Gruppe, welche ausschließlich aus
Kohlenstoff und Wasserstoff besteht. Das Hydrocarbyl kann verzweigt
oder unverzweigt sein, gesättigt
oder ungesättigt
sein und einen oder mehrere Ringe umfassen. Geeignete Hydrocarbylgruppen
schließen
Alkyl-, Alkenyl-, Alkynyl und Arylgruppen ein. Sie schließen auch
Alkyl-, Alkenyl-, Alkynyl und Arylgruppen ein, welche mit anderen
aliphatischen oder cyclischen Hydrocarbylgruppen substituiert sind,
wie Alkary, Alkenaryl und Alkynaryl.
-
Ein
substituiertes Hydrocarbyl ist ein Hydrocarbyl, worin mindestens
ein Wasserstoffatom substituiert worden ist mit (a) einem anderen
Atom als Wasserstoff, oder (b) mit einer Gruppe von Atomen, welche
mindestens ein anderes Atom als Wasserstoff enthält. Das Wasserstoffatom kann
zum Beispiel durch ein Halogenatom wie ein Chlor- oder Fluoratom
substituiert sein. Das Wasserstoffatom kann alternativ durch ein
Sauerstoffatom oder eine Gruppe substituiert sein, welche ein Sauerstoffatom
enthält,
um zum Beispiel eine Hydroxygruppe, einen Ether, einen Ester, ein
Anhydrid, einen Aldehyd, ein Keton oder eine Carbonsäure zu bilden. Das
Wasserstoffatom kann auch durch eine Gruppe ersetzt werden, welche
ein Stickstoffatom enthält,
um zum Beispiel ein Amid oder eine Nitrogruppe zu bilden. Zusätzlich kann
das Wasserstoffatom durch eine Gruppe ersetzt werden, die ein Schwefelatom
enthält,
um zum Beispiel -SO3H zu bilden.
-
Ein
landwirtschaftlich annehmbares Kation ist ein Kation, welches eine
landwirtschaftlich und wirtschaftlich nützliche herbizide Aktivität des N-(Phosphonomethyl)glycinanions
zulässt.
Ein solches Kation kann zum Beispiel ein Alkalimetallkation (z.
B. ein Natrium- oder Kaliumion), ein Ammoniumion, ein Isopropylammoniumion,
ein Tetraalkylammoniumion, ein Trialkylsulfoniumion, ein protoniertes
primäres
Amin, ein protoniertes sekundäres
Amin oder ein protoniertes tertiäres
Amin sein.
-
Bei
einer besonders bevorzugten Ausführungsform
sind R1, R2, R3 und R4 jeweils
unabhängig
Wasserstoff oder ein landwirtschaftlich annehmbares Kation, wobei
Wasserstoff oft am meisten bevorzugt ist.
-
Es
können
in Übereinstimmung
mit dieser Erfindung verschiedene Oxidationsmittel verwendet werden.
Diese schließen
zum Beispiel Peroxide (z. B. H2O2, Benzoylperoxid), Hydroperoxide, Peroxysäuren, O2-haltige Gase, und Flüssigkeiten ein, welche gelösten Sauerstoff
umfassen. O2-haltige Gase sind typischerweise
besonders bevorzugt. O2-haltiges Gas ist,
wie hierin verwendet, jede Gasmischung, welche O2 umfasst und
wahlweise ein oder mehrere Verdünner,
welche unter den Reaktionsbedingungen nicht mit Sauerstoff oder dem
Substrat oder dem Produkt reagieren. Beispiele für solche Gase sind Luft, reines
O2, oder mit He, Ar, N2 und/oder
nicht-oxidierenden Gasen verdünntes
O2. Die Sauerstoffquelle ist am meisten
vorzugsweise ein O2-haltiges Gas, welches
mindestens etwa 95 Mol.-% O2, weiter vorzugsweise
mindestens etwa 98 Mol.-% O2 enthält und der
Rest aus einem oder mehreren nicht-oxidierenden Gasen (insbesondere
N2 und/oder AR) besteht.
-
Bevorzugte
Oxidationskatalysatoren
-
Es
kann in Übereinstimmung
mit dieser Erfindung eine breite Vielfalt von Oxidationskatalysatoren
verwendet werden.
-
Es
können
zum Beispiel verschiedene Wolframsalze verwendet werden, um die
Oxidation von N-(Phosphonomethyl)iminodiessigsäuresubstraten mit H2O2 zu katalysieren.
N-(Phosphonomethyl)iminodiessigsäure
kann mit H2O2 in
Anwesen heit einer Säure
(z. B. H2SO4) und
Wärme zu
einem N-Oxid-Zwischenprodukt oxidiert werden. dieses N-Oxid-Zwischenprodukt
kann in Anwesenheit von Wärme
und verschiedenen wasserlöslichen
Eisen(II)-, Kupfer(I)-, Wolfram-, Molybdän- und Vanadinsalzkatalysatoren
wiederum unter Bildung von N-(Phosphonomethyl)glycin zersetzt werden.
Eine allgemeine Erörterung
zur Verwendung solcher homogener Katalysatoren zur Umwandlung von
N-(Phosphonomethyl)iminodiessigsäure
in N-(Phosphonomethyl)glycin findet sich zum Beispiel in Franz,
et al., "Glyphosate:
A Unique Global Herbicide",
ACS Monograph 189: 240–41
(1997).
-
Typischerweise
ist die Verwendung eines heterogenen Katalysators weiter bevorzugt.
Diese Bevorzugung beruht wenigstens teilweise auf der Leichtigkeit,
mit der heterogene Katalysatoren im Anschluss an die Oxidation normalerweise
aus der Reaktionsmischung abgetrennt werden können. Die Literatur ist voll
von geeigneten heterogenen Katalysatoren.
-
Einer
der ersten heterogenen Katalysatoren, welche zur oxidativen Spaltung
von N-(Phosphonomethyl)iminodiessigsäure verwendet wurde, wurde
von Franz in US-A 3,950,402 offen gelegt, Franz gibt an, dass N-(Phosphonomethyl)glycin
durch oxidative Spaltung von N-(Phosphonomethyl)iminodiessigsäure in flüssiger Phase
mit O2 in Gegenwart eine Katalysators, umfassend
ein auf der Oberfläche
eines Aktivkohleträgers
abgeschiedenen Edelmetalls, hergestellt werden kann.
-
-
Obwohl
das Franz'sche Verfahren
sogar im Allgemeinen eine annehmbare Ausbeute und Reinheit an N-(Phosphonomethyl)glycin
liefert, leidet es auch unter einer Reihe von Problemen:
- 1. Das teuere Edelmetall in den Franz'schen Katalysatoren
neigt dazu in der Reaktionslösung
verloren zu gehen (d. h. Herauslösung).
Dieses Herauslösen
des Edelmetalls ist das Ergebnis von mindestens zwei Faktoren: (a)
Unter den Oxidationsbedingungen der Reaktion wird etwas Edelmetall
zu einer löslicheren Form
oxidiert; und (b) fungieren sowohl das N-(Phosphonomethyl)iminodiessigsäuresubstrat
als auch das N-(Phosphonomethyl)glycinprodukt als Liganden, welche
das Edelmetall solubilisieren.
- 2. Das N-(Phosphonomethyl)glycinprodukt oxidiert sich oft unter
Bildung von Aminomethylphosphonsäure (AMPA),
insbesondere, wenn die Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats
abnimmt. Dies verringert offensichtlich die Ausbeute des gewünschten
N-(Phosphonomethyl)glycinprodukts.
-
In
US-A 3,969,398 legt Hershman offen, dass aktivierter Kohlenstoff
allein, ohne die Anwesenheit eines Edelmetalls, verwendet werden
kann, um die oxidative Spaltung von N-(Phosphonomethyl)iminodiessigsäure unter
Bildung von N-(Phosphonomethyl)glycin zu bewirken. In US-A 4,624,937
legt Chou weiterhin offen, dass die Aktivität des von Hershman offen gelegten
Kohlenstoffkatalysators durch die Entfernung der Oxide aus der Oberfläche des
Kohlenstoffkatalysators vor seiner Verwendung bei der Oxidationsreaktion
gesteigert werden kann. Vergleiche auch US-A 4,696,772 (bietet eine
separate Erörterung
durch Chou was die Erhöhung der
Aktivität
des Kohlenstoffkatalysators durch Entfernung von Oxiden aus der
Oberfläche
des Kohlenstoffkatalysators betrifft). Obwohl diese Verfahren offensichtlich
nicht unter dem Herauslösen
von Edelmetall leiden, neigen sie dazu, größere Konzentrationen Formaldehyd
und Ameisensäure
als Nebenprodukte zu bilden, wenn sie dazu verwendet werden, die
oxidative Spaltung von N-(Phosphonomethyl)iminodiessigsäure zu bewirken.
-
Es
ist optimal, wenn der Formaldehyd und die Ameisensäure gleichzeitig
zu Kohlendioxid und Wasser oxidiert werden, wenn das N-(Phosphonomethyl)iminodiessigsäuresubstrat
zu N-(Phosphonomethyl)glycin oxidiert wird, entsprechend der folgenden
Gleichung:
-
-
Katalysatoren,
welche ein Edelmetall auf einem Kohlenstoffträger umfassen haben aus zwei
Gründen viel
Aufmerksamkeit auf sich gezogen. Bei solchen Katalysatoren bewirkt
die Kohlenstoffkomponente hauptsächlich
die Oxidation der N-(Phosphonomethyl)iminodiessigsäure, um
N-(Phosphonomethyl)glycin und Formaldehyd zu bilden, während die
Edelmetallkomponente hauptsächlich
die Oxidation von Formaldehyd und Ameisensäure bewirkt, um Kohlendioxid
und Wasser zu bilden. Die Edelmetallkomponente neigt auch dazu die
Geschwindigkeit der Deaktivierung des Kohlenstoffs zu verringern.
Genauer gesagt neigt der aktivierte Kohlenstoff bei alleiniger Verwendung
dazu 10% pro Zyklus oder mehr zu deaktivieren. Ohne an irgendeine spezielle
Theorie gebunden sein zu wollen wird angenommen, dass die Deaktivierung
des aktivierten Kohlenstoffs als solchem auftritt, weil sich die
Oberfläche
des Kohlenstoffträgers
unter den Reaktionsbedingungen oxidiert. Vergleiche Chou, US-A 4,624,937.
Vergleiche auch Chou US-A 4,696,772 (772 (bietet eine separate Erörterung
durch Chou was die Deaktivierung von aktiviertem Kohlenstoff durch
die Oxidation der Oberfläche des
Kohlenstoffs betrifft). In Gegenwart des Edelmetalls wird die Geschwindigkeit
der Deaktivierung des aktivierten Kohlenstoffs herabgesetzt. Es
wird angenommen, dass das Edelmetall mit dem Oxidationsmittel mit größerer Geschwindigkeit
reagiert als die aktivierte Kohlenstoffoberfläche und dass es demzufolge
das Oxidationsmittel aus der Lösung
entfernt, ehe eine ausgedehnte Oxidation der Kohlenstoffoberfläche erfolgen
kann. Des Weiteren werden Oxidspezies, welche sich auf der Oberfläche eines
Edelmetalls bilden, anders als viele Oxidspezies, welche sich auf
aktivierten Kohlenstoffoberflächen
bilden und zu ihrer Reduzierung Hochtemperaturbehandlungen erfordern,
durch die in der Reaktionsmischung vorhandenen oder dieser zugesetzte
Reduktionsmittel (z. B. abgespaltene Aminbruchstücke, Formaldehyd, Ameisensäure, H2 etc.) typischerweise leicht reduziert,
um so den reduzierten Zustand der Edelmetalloberfläche wiederherzustellen.
Auf diese Weise weist der Katalysator vorteilhafterweise eine beträchtlich
längere
Lebensdauer auf, weil das Edelmetall nicht durch Auslaugen oder
Sintern (d. h. unerwünscht
dicke Schichten oder Klumpen bildet) infolge von Vorgängen wie
Auflösung
und Wiederablagerung oder Edelmetallagglomeration verloren geht.
-
Ramon
et al. (US-A 5,179,228) legen ein Beispiel zur Verwendung eines
auf der Oberfläche
eines Kohlenstoffträgers
abgeschiedenen Edelmetalls offen. Um das Problem des Auslaugens
zu reduzieren (von dem Ramon et al. berichten, dass es den Umfang
von 30% Edelmetallverlust pro Zyklus erreicht), legen Ramon et al.
das Spülen
der Reaktionsmischung mit N2 unter Druck
offen, nachdem die Oxidationsreaktion abgeschlossen ist, um die
Wiederablagerung des Edelmetalls auf der Oberfläche des Kohlenstoffträgers zu
veranlassen. Nach Ramon et al. reduziert die N2-Spülung den
Edelmetallverlust auf weniger als 1%.
-
Felthouse
(US-A 4,582,650) legt die Verwendung von 2 Katalysatoren offen:
(i) einen aktivierten Kohlenstoff, um die Oxidation von N-(Phosphonomethyl)iminodiessigsäure zu N-(Phosphonomethyl)glycin
zu bewirken, und (ii) einen Co-Katalysator,
um parallel hierzu die Oxidation von Formaldehyd zu Kohlendioxid und Wasser
zu bewirken. Der Co-Katalysator ist ein Aluminiumsilicatträger mit
Edelmetall innerhalb seiner Poren. Die Porengröße ist so ausgelegt, dass sie
N-(Phosphonomethyl)glycin
ausschließt
und dadurch das Edelmetall des Co-Katalysators daran hindert vom N-(Phosphonomethyl)glycin
vergiftet zu werden. Nach Felthouse erlaubt die gemeinsame Verwendung
dieser 2 Katalysatoren die gleichzeitige Oxidation von N-(Phosphonomethyl)iminodiessigsäure zu N-(Phosphonomethyl)glycin
und von Formaldehyd zu Kohlendioxid und Wasser.
-
Ebner
et al., Internationale Veröffentlichung
Nr. WO/43430, legen das Oxidieren des N-(Phosphonomethyl)iminodiessigsäuresubstrats
unter Verwendung eines tiefenreduzierten Katalysators offen, welcher
ein Edelmetall auf einem Kohlenstoffträger umfasst. Ein derartiger
Katalysator neigt dazu eine bessere Widerstandsfähigkeit gegen das Auslaugen
von Edelmetall und einen verbesserten Abbau von unerwünschten
Nebenprodukten (z. B. Formaldehyd) aufzuweisen. Die Vorteile solcher
Katalysatoren machen sie besonders bevorzugt. Ein Großteil der
folgenden Erörterung
wird sich folglich auf solche Katalysatoren konzentrieren. Nichtsdestoweniger
sollte man berücksichtigen,
dass die Merkmale dieser Erfindung generell auf die Verwendung einer
breiten Vielfalt homogener und heterogener Katalysatoren angewendet
werden kann, wie vorstehend beschrieben.
-
Sauerstoffhaltige
funktionelle Gruppen (z. B. Carbonsäuren, Ether, Alkohole, Aldehyde,
Lactone, Ester, Aminoxide und Amide) auf der Oberfläche des
Kohlenstoffträgers
neigen dazu das Auslaugen des Edelmetalls zu erhöhen und erhöhen möglicherweise das Sintern des
Edelmetalls während
der Flüssigphasenoxidationsreaktionen
und verringern demzufolge die Fähigkeit
des Katalysators während
der N-(Phosphonomethyl)iminodiessigsäureoxidationsreaktion oxidierbare
Substrate zu oxidieren, insbesondere Formaldehyd und Ameisensäure. Eine
sauerstoffhaltige funktionelle Gruppe liegt, wie hierin verwendet,
auf der Oberfläche
des Kohlenstoffträgers
vor, wenn sie an ein Atom des Kohlenstoffträgers gebunden ist und fähig ist,
mit Zusammensetzungen innerhalb der Reaktionsmischung oder mit dem
auf dem Kohlenstoffträger
abgeschiedenen Metallatom chemisch oder physikalisch in Wechselwirkung
zu treten.
-
Viele
der sauerstoffhaltigen funktionellen Gruppen, welche die Beständigkeit
von Edelmetallen gegen Auslaugen und Sintern reduzieren, reduzieren
auch die Aktivität
der Katalysatordesorption aus dem Kohlenstoffträger als Kohlenmonoxid, wenn
der Katalysator in einer inerten Atmosphäre (z. B. Helium oder Argon)
auf eine hohe Temperatur (z. B. 900°C) erhitzt wird. Folglich ist
die Messung der desorbierten CO-Menge aus einem frischen Katalysator
(d. h. einem Katalysator, welcher vorher nicht bei einer Oxidationsreaktion
in flüssiger Phase
verwendet worden ist) unter hohen Temperaturen ein Verfahren, das
verwendet werden kann, um die Oberfläche des Katalysators zu analysieren,
um die Edelmetallretention und den Erhalt der Katalysatoraktivität vorherzusagen.
Ein Weg, um die CO-Desorption zu messen ist die Verwendung der thermographischen
Analyse durch In-line Massenspektroskopie (TGA-MS). Vorzugsweise
desorbier nicht mehr als 1,2 mMol Kohlenmonoxid pro Gramm Katalysator
aus dem Katalysator, wenn eine trockene, frische Katalysatorprobe
in einer Heliumatmosphäre
einer Temperatur unterworfen wird, welche mit 10°C/min von 20 auf 900°C aufgeheizt
und dann 30 min bei 900°C
konstant gehalten wird. Unter diesen Bedingungen desorbiert weiter
vorzugsweise nicht mehr als 0,7 mMol Kohlenmonoxid pro Gramm frischen
Katalysators, sogar weiter vorzugsweise desorbiert nicht mehr als
0,5 mMol Kohlenmonoxid pro Gramm frischen Katalysators und am meisten
vorzugsweise desorbiert nicht mehr als etwa 0,3 mMol Kohlenmonoxid
pro Gramm frischen Katalysators. Ein Katalysator wird als trocken
betrachtet, wenn sein Feuchtigkeitsgehalt weniger als 1 Gew.-% beträgt. Ein
Katalysator kann typischerweise getrocknet werden, indem er bei
einem N2-gespülten Vakuum
von 85 kPa (25 inches Hg) und einer Temperatur von 120°C 16 Stunden
konditioniert wird.
-
Das
Messen der Zahl der Sauerstoffatomen auf der Oberfläche eines
frischen Katalysatorträgers
ist ein anderes Verfahren, welches verwendet werden kann, um den
Katalysator zu analysieren, um die Edelmetallretention und Aufrechterhaltung
der katalytischen Aktivität
vorherzusagen. Verwendet man zum Beispiel die Röntgen-Photoelektronen-Spektroskopie,
kann eine Oberflächenschicht
von etwa 50 Å Dicke
analysiert werden. Die Zurzeit verfügbaren Geräte zur Röntgen-Photoelektronen-Spektroskopie
weisen typischerweise eine Genauigkeit innerhalb von ±20% auf.
Typischerweise sind diese zur Messungen von Kohlenstoffatom/Sauerstoffatom-Verhältnissen
(gemessen mit den gegenwärtig
verfügbaren
Geräten
zur Röntgen-Photoelektronen-Spektroskopie)
auf der Oberfläche
von mindestens etwa 20:1 (Kohlenstoffatome:Sauerstoffatome) geeignet.
Vorzugsweise beträgt
das Verhältnis
jedoch mindestens etwa 30:1, weiter vorzugsweise mindestens etwa 40:1,
sogar weiter vorzugsweise mindestens etwa 50:1 und am meisten vorzugsweise
etwa 60:1. Zusätzlich beträgt das Verhältnis der
Sauerstoffatome zu den Metallatomen auf der Oberfläche (wiederum
gemessen mit den gegenwärtig
verfügbaren
Geräten
zur Röntgen-Photoelektronen- Spektroskopie) vorzugsweise
weniger als etwa 8:1 (Sauerstoffatome:Metallatome). Weiter vorzugsweise
beträgt
das Verhältnis
weniger als 7:1. sogar weiter vorzugsweise weniger als etwa 6:1
und am meisten vorzugsweise weniger als etwa 5:1.
-
Die
bei der vorliegenden Erfindung verwendeten Kohlenstoffträger sind
auf dem Fachgebiet allgemein bekannt. Aktivierte, nicht-graphitierte
Kohlenstoffträger
sind bevorzugt. Diese Träger
sind durch ein hohes Adsorptionsvermögen für Gase, Dämpfe und kolloidale Feststoffe
sowie relativ hohe spezifische Oberflächen gekennzeichnet. Der Träger kann
geeigneterweise eine Kohle, synthetische Kohle oder Holzkohle sein,
hergestellt nach auf dem Fachgebiet bekannten Verfahren, wie zum
Beispiel durch zersetzende Destillation von Holz, Torf, Braunkohle,
Steinkohle, Nussschalen, Knochen, Pflanzen oder von anderen natürlichen
oder synthetischen kohlenstoffhaltigen Materialien, der jedoch vorzugsweise
aktiviert wird, um ein Adsorptionsvermögen zu entwickeln. Die Aktivierung
erfolgt üblicherweise
durch Erhitzen auf hohe Temperaturen (800–900°C) mit Dampf oder mit Kohlendioxid,
was eine poröse
Teilchenstruktur und eine größere spezifische
Oberfläche mit
sich bringt. In manchen Fällen
werden vor der zersetzenden Destillation oder Aktivierung hygroskopische Substanzen
wie Zinkchlorid und/oder Phosphorsäure oder Natriumsulfat zugegeben,
um das Adsorptionsvermögen
zu erhöhen.
Der Kohlenstoffgehalt des Kohlenstoffträgers reicht von etwa 10% bei
Knochenkohle bis etwa 98% bei einigen Holzkohlen und beinahe bis
100% bei von organischen Polymeren abgeleiteten aktivierten Kohlen.
Das nicht-kohlenstoffhaltige Material von im Handel erhältlichen
aktivierten Kohlenstoffmaterialien schwankt in Abhängigkeit
von solchen Faktoren wie dem Ursprung des Vorläufers, der Herstellung und
dem Aktivierungsverfahren. Viele im Handel erhältliche Kohlenstoffträger enthalten
geringe Metallmengen. Kohlenstoffträger mit den wenigsten sauerstoffhaltigen
funktionellen Gruppen auf ihren Oberflächen sind am meisten bevorzugt.
-
Die
Form der in Festbettreaktoren verwendeten Träger kann beträchtlich
schwanken. Der Kohlenstoffträger
kann zum Beispiel in Form eines monolithischen Trägers vorliegen.
Geeignete monolithische Träger können eine
breite Vielfalt von Formen aufweisen. Ein monolithischer Träger kann
zum Beispiel die Form eines Reaktorlaufrades besitzen. Weiter vorzugsweise
kann ein solcher Träger
sogar zum Beispiel auch die Form eines Netzes oder einer Honigwabe
mit parallelen Kanälen
aufweisen, durch welche die Einspeisemischung geleitet wird. 1 zeigt
zum Beispiel den Querschnitt durch einen Honigwabenträger. Obwohl
die Kanalquerschnitte im Honigwabenträger von 1 eine
hexagonale Ge stalt aufweisen, kann ein wie hierin definierter Honigwabenträger alternativ
(oder zusätzlich)
Kanäle
mit anderen Querschnittsformen (z. B. kreisrund, oval, quadratisch,
dreieckig, rechteckig und dergleichen) umfassen. Die Kanäle des Honigwabenträgers sind
vorzugsweise gerade und/oder besitzen einen Querschnitt, welcher
groß genug
ist, so dass sie durch eine Aufschlämmung, welche festes N-(Phosphonomethyl)iminodiessigsäuresubstrat
enthält,
nicht verstopft werden. Alternativ können die Strömungskanäle in einem
monolithischen Träger
unregelmäßig und
ohne eine einheitliche Strömungsrichtung
sein (z. B. ein ungeordnetes Netz aus miteinander verbundenen Strömungskanälen).
-
Bei
einer besonders bevorzugten Ausführungsform
liegt der Träger
teilchenförmig
vor. Weil teilchenförmige
Träger
besonders bevorzugt sind, konzentriert sich der Hauptteil der anschließenden Diskussion
auf Ausführungsformen,
welche einen teilchenförmigen
Träger
verwenden. Diese Erfindung ist jedoch selbstverständlich nicht
auf die Verwendung teilchenförmiger
Träger
beschränkt.
-
Geeignete
teilchenförmige
Träger
können
eine breite Vielfalt von Formen aufweisen. Solche Träger können zum
Beispiel in Form von Pellets, Granalien oder Pulvern vorliegen.
Pelletträger
besitzen typischerweise eine Teilchengröße von etwa 1 mm bis etwa 10
mm. Der Träger
liegt vorzugsweise in Form eines Pulvers vor. Diese teilchenförmigen Träger können in
einem Reaktorsystem als freie Teilchen vorliegen oder alternativ an
eine Struktur im Reaktorsystem, wie an ein Netz oder an ein Laufrad
gebunden sein.
-
Ein
teilchenförmiger
Träger
umfasst typischerweise eine breite Teilchengrößenverteilung. Bei Pulvern weisen
vorzugsweise mindestens 95% der Teilchen eine größte Abmessung von etwa 2 bis
etwa 300 μm
auf, weiter vorzugsweise besitzen mindestens etwa 98% der Teilchen
eine größte Abmessung
von etwa 2 bis etwa 200 μm,
wobei etwa 95% der Teilchen eine größte Abmessung von etwa 3 bis
etwa 100 μm
besitzen. Teilchen, die eine größte Abmessung
von größer als
etwa 200 μm
besitzen, neigen dazu in superfeine Teilchen (d. h. weniger als
2 μm größte Abmessung)
zu zerbrechen, welche schwierig wiederzugewinnen sind.
-
Die
spezifische Oberfläche
des Kohlenstoffträgers,
gemessen nach dem BET (Brunauer-Emmett-Teller)-Verfahren unter Verwendung
von N2 beträgt vorzugsweise etwa 10 bis
etwa 3000 m2/g (Oberfläche des Kohlenstoffträgers pro
Gramm Kohlenstoffträger),
weiter vorzugsweise etwa 500 bis etwa 2100 m2/g
und noch weiter vorzugsweise etwa 750 bis etwa 2100 m2/g.
Bei manchen Ausführungs formen
beträgt
die am meisten bevorzugte Oberfläche
etwa 750 bis etwa 1750 m2/g.
-
Das
Porenvolumen des Trägers
kann stark schwanken. Bei Verwendung des in Beispiel 1 beschriebenen
Messverfahrens beträgt
das Porenvolumen vorzugsweise etwa 0,1 bis etwa 2,5 ml/g (Porenvolumen
pro Gramm Katalysator), weiter vorzugsweise etwa 0,2 bis etwa 2,0
ml/g und am meisten vorzugsweise etwa 0,4 bis etwa 1,7 ml/g. Katalysatoren,
welche Träger
mit einem Porenvolumen größer als
etwa 2,5 g/ml umfassen, neigen leicht zum Brechen. Andererseits
neigen Katalysatoren, welche Träger
mit einem Porenvolumen von weniger als 0,1 mg/g umfassen dazu, kleine
Oberflächen
und daher eine geringe Aktivität
zu besitzen.
-
Kohlenstoffträger zur
Verwendung bei der vorliegenden Erfindung sind aus einer Anzahl
von Quellen kommerziell verfügbar.
Das Folgende ist eine Aufstellung von einigen Aktivkohlen, welche
bei dieser Erfindung verwendet werden können: Darco G-60 Spec und Darco
X (ICI-America, Wilmington, DE); Norit SG Extra, Norit EN4, Norit
EXW, Norit A, Norit Ultra-C, Norit ACX und Norit 4 × 14 mesh
(Amer. Norit Co., Inc., Jacksonville, PL); GL-9615, VG-8408, VG-8590,
NB-9377, NW, und JV (Barnebey-Cheney, Columbus. OH); BL Pulv., PWA Pulv.,
Calgon C 450 und PCB Fines (Pittsburgh Activated Carbon, Div. of
Calgon Corporation, Pittsburgh, PA); P-100 (No. Amer. Carbon, Inc.,
Columbus, OH); Nuchar CN, Nuchar C-1000 N, Nuchar C-190 A, Nuchar
C-115 A und Nuchar SA-30 (Westvaco Corp., Carbon Department, Covinghton,
Virginia); Code 1551 (Baker and Adamson, Division of Allied Amer.
Norit Co., Inc., Jacksonville, FL); Grade 235, Grade 337, Grade
517 und Grade 256 (Witco Chemical Corp., Activated Carbon Division,
New York, NY) und Columbia SXAC (U(nion Carbide New York, NY).
-
Der
Kohlenstoffträger
weist vorzugsweise ein oder mehrere Edelmetall(e) auf seiner Oberfläche auf. Das/die
Edelmetall(e) ist/sind gewählt
aus der Gruppe, bestehend aus Platin (Pt), Palladium (Pd), Ruthenium (Ru),
Rhodium (Rh), Iridium (Ir), Silber (Ag), Osmium Os) und Gold (Au).
Im Allgemeinen sind Platin und Palladium weiter bevorzugt, und Platin
ist am meisten bevorzugt. Weil Platin gegenwärtig das am meisten bevorzugte
Edelmetall ist, ist die folgende Diskussion vorzugsweise auf Ausführungsformen
gerichtet, welche Platin verwenden. Die gleiche Diskussion ist selbstverständlich allgemein
auf andere Edelmetalle und Kombinationen davon anwendbar. Der Ausdruck
Edelmetall bedeutet, wie hierin verwendet, selbstverständlich auch
das Edelmetall sowohl in seinem elementaren Zustand als auch das
Edelmetall in allen seinen verschiedenen Oxidationszuständen.
-
Die
Konzentration des auf der Oberfläche
des Kohlenstoffträgers
abgeschiedenen Edelmetalls kann innerhalb weiter Grenzen liegen.
Sie liegt vorzugsweise im Bereich von etwa 0,5 bis etwa 20 Gew.-%
([Masse des Edelmetalls ÷ Katalysatorgesamtmasse] × 100%),
weiter vorzugsweise bei etwa 2,5 bis etwa 10 Gew.-% und am meisten
vorzugsweise bei etwa 3 bis etwa 7,5 Gew.-%. Werden während der
N-(Phosphonomethyl)iminodiessigsäureoxidationsreaktion
weniger als 0,5 Gew.-% verwendet, besteht die Tendenz, dass weniger
Formaldehyd oxidiert und daher eine größere Menge N-Methyl-N-(phosphonomethyl)glycin
erzeugt wird, wodurch die N-(phosphonomethyl)glycinausbeute reduziert
wird. Andererseits besteht bei Konzentrationen größer als
20 Gew.-% die Tendenz, dass sich Ablagerungen und Klumpen von Edelmetall
bilden. Demzufolge werden auf der Oberfläche weniger Metallatome verwendet,
bezogen auf den Edelmetallgesamtgehalt. Dies neigt dazu, die Katalysatoraktivität zu reduzieren
und stellt eine unwirtschaftliche Verwendung des teuren Edelmetalls
dar.
-
Die
Verteilung des Edelmetalls auf der Oberfläche des Kohlenstoffträgers ist
derart, dass die Konzentration der Oberflächenedelmetallatome etwa 10
bis etwa 400 μMol/g
(μMol Oberflächenedelmetallatome
pro Gramm Katalysator), weiter vorzugsweise etwa 10 bis etwa 150 μMol/g und
am meisten vorzugsweise etwa 100 μMol/g
beträgt.
Dies kann zum Beispiel durch Messung der Chemisorption von Hz oder
CO unter Verwendung einer Micromeritics ASAP 2010C (Micromeritics,
Norcross, GA) oder einer Altamira AMI 100 (Zeton Altamira, Pittsburgh,
PA) bestimmt werden.
-
Das
Edelmetall liegt auf der Oberfläche
des Kohlenstoffträgers
vorzugsweise in Form von Metallteilchen vor. Mindestens etwa 90%
(Zahlendichte) der Edelmetallteilchen auf der Oberfläche des
Kohlenstoffträgers
weisen vorzugsweise eine größte Abmessung
von etwa 0,5 bis etwa 35 nm auf, weiter vorzugsweise eine größte Abmessung
von etwa 1 bis etwa 20 nm und am meisten vorzugsweise eine größte Abmessung
von etwa 1,5 bis etwa 10 nm auf. Bei einer besonders bevorzugten
Ausführungsform
besitzen mindestens etwa 80% der Edelmetallteilchen auf der Oberfläche des
Kohlenstoffträgers
eine größte Abmessung
von etwa 1 bis etwa 15 nm, weiter vorzugsweise eine größte Abmessung
von etwa 1,5 bis etwa 10 nm und am meisten vorzugsweise eine größte Abmessung
von etwa 1,5 bis etwa 7 nm. Sind die Edelmetallteilchen zu klein,
besteht die Tendenz zu einer erhöhten
Auslaugung wenn der Katalysator in einer Umgebung verwendet wird,
welche dazu neigt, Edelmetalle zu solubilisieren, wie dies der Fall
ist, wenn N-(Phosphonomethyl)iminodiessigsäure unter Bildung von N-(Phospohonmomethyl)glycin
oxidiert wird. Nimmt andererseits die Teilchengröße zu, besteht die Tendenz,
dass weniger Oberflächenedelmetallatome
pro Edelmetallgesamtkonzentration verwendet werden. Wie vorstehend
erörtert,
tendiert dies dazu, die Katalysatoraktivität zu vermindern und stellt
eine unwirtschaftliche Verwendung des teueren Edelmetalls dar.
-
Zusätzlich zum
Edelmetall kann auf der Oberfläche
des Kohlenstoffträgers
mindestens ein Aktivator vorliegen. Ein Aktivator ist, wie hierin
definiert, ein Metall, das dazu neigt, die Selektivität, Aktivität und/oder Stabilität des Katalysators
zu erhöhen.
Ein Aktivator kann zusätzlich
das Auslaugen des Edelmetalls reduzieren. Obwohl der Aktivator üblicherweise
in einer Aktivatorabscheidungsstufe auf der Oberfläche des
Kohlenstoffträgers
abgeschieden wird, kann der Kohlenstoffträger natürlich selbst ebenfalls (oder
alternativ) einen Aktivator enthalten. Ein Aktivator, welcher auf
der Katalysatoroberfläche
abgeschieden wird oder von Haus aus auf der Katalysatoroberfläche vorliegt,
ehe die Kohlenstoffträgeroberfläche schließlich reduziert
wird (wie nachstehend beschrieben), wird hierin als ein Katalysatoroberflächenaktivator
bezeichnet.
-
Der/die
Katalysatoroberflächenaktivator(en)
kann/können
zum Beispiel ein weiteres/weitere Edelmetall(e) auf der Oberfläche des
Kohlenstoffträgers
sein. In Abhängigkeit
von der Anwendung können
zum Beispiel Ruthenium und Palladium auf einem Kohlenstoffträger, welcher
abgeschiedenes Platin umfasst, als Katalysatoroberflächenaktivatoren
wirken. Der/die Katalysatoroberflächenaktivator(en) kann/können zum
Beispiel alternativ Metall(e) sein, gewählt aus der Gruppe, bestehend
aus Zinn (Sn), Cadmium (Cd), Magnesium (Mg), Mangan Mn), Nickel
(Ni), Aluminium (Al), Kobalt (Co), Wismut (Bi), Blei (Pb), Titan
(Ti), Antimon (Sb), Selen (Se), Eisen (Fe), Rhenium (Re), Zink (Zn),
Cer (Ce), Zirkonium (Zr), Tellur (Te) und Germanium (Ge). Der Katalysatoroberflächenaktivator
wird vorzugsweise gewählt
aus der Gruppe, bestehend aus Wismut, Eisen, Zinn, Tellur und Titan.
Bei einer besonders bevorzugten Ausführungsform ist der Katalysatoroberflächenaktivator
Zinn. Bei einer anderen besonders bevorzugten Ausführungsform
ist der Katalysatoroberflächenaktivator
Eisen. Bei einer weiteren bevorzugten Ausführungsform ist der Katalysatoroberflächenaktivator
Titan. Bei einer weiteren bevorzugten Ausführungsform umfasst der Katalysator
auf seiner Oberfläche
sowohl Eisen als auch Zinn. Die Verwendung von Eisen, Zinn, oder von
beiden, reduziert im Allgemeinen (1) die Edelmetallauslaugung bei
einem über
mehre Zyklen verwendeten Katalysator, und neigt (2) dazu, die Aktivität des Katalysators
zu erhöhen
und/oder aufrechtzuerhalten, wenn der Katalysator dazu verwendet
wird, die Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
zu bewirken. Katalysatoren, welche Eisen umfassen, sind generell
am meisten bevorzugt, weil sie dazu neigen, die größte Aktivität und Stabilität im Hinblick
auf die Formaldehyd- und Ameisensäureoxidation aufzuweisen.
-
Bei
einer bevorzugten Ausführungsform
wird der Katalysatoroberfächenaktivator
leichter oxidiert als das Edelmetall (in den Fällen, in denen der Katalysatoroberflächenaktivator
auch ein Edelmetall ist, wird das Edelmetall des Katalysatoroberflächenaktivators
vorzugsweise leichter oxidiert als das Edelmetall des Nicht-Aktivators). Ein
Aktivator wird leichter oxidiert, wenn er ein niedrigeres Ionisationspotential
aufweist als das Edelmetall. Die ersten Ionisationspotentiale der
Elemente sind auf dem Fachgebiet hinreichend bekannt und finden
sich zum Beispiel im "CRC
Handbook of Chemistry and Physics", (CRC Press, Inc., Boca Raton, Florida).
-
Die
Menge Katalysatoroberflächenaktivator
auf der Oberfläche
des Kohlenstoffträgers
(ob mit der Kohlenstoffoberfläche
selbst, dem Metall oder einer Kombination hiervon vereinigt) kann
zum Beispiel in Abhängigkeit
des/der verwendeten Edelmetall(e) und Katalysatoroberflächenaktivator(en)
innerhalb weiter Grenzen schwanken. Der Gewichtsprozentanteil des
Katalysatoroberflächenaktivators
beträgt
typischerweise mindestens etwa 0,05% ([Masse des Katalysatoroberflächenaktivators ÷ Katalysatorgesamtmasse] × 100%).
Die Gewichtsprozente des Katalysatoroberflächenaktivators betragen vorzugsweise
etwa 0,05 bis etwa 10%, weiter vorzugsweise etwa 0,1 bis etwa 10%,
noch weiter vorzugsweise etwa 0,1 bis 2% und am meisten vorzugsweise
etwa 0,2% bis etwa 1,5%. Ist der Katalysatoroberflächenaktivator
Zinn, betragen die Gewichtsprozente vorzugsweise etwa 0,5 bis etwa
1,5%. Betragen die Gewichtsprozente des Katalysatoroberflächenaktivators weniger
als 0,05%, erhöhen
sie in der Regel die Aktivität
des Katalysators über
längere
Zeiträume
nicht. Andererseits erniedrigen Gewichtsprozente von mehr als 10%
die Aktivität
des Katalysators.
-
Das
Molverhältnis
des Edelmetalls zum Katalysatoroberflächenaktivator (und in den Fällen, in
denen der Katalysatoroberflächenaktivator
ebenfalls ein Edelmetall ist, das Molverhältnis des Edelmetall des Nicht-Aktivators
zum Edelmetall des Katalysatoroberflächenaktivators) kann zum Beispiel
in Abhängigkeit des/der verwendeten
Edelmetalls(e) und des/der Katalysatoroberflächenaktivators(en) ebenfalls
weit schwanken. Das Verhältnis
beträgt
vorzugsweise etwa 1000:1 bis etwa 0,01:1, weiter vorzugsweise etwa
150:1 bis etwa 0,05:1, noch weiter vorzugsweise etwa 50:1 bis etwa
0,05:1 und am meisten vorzugsweise etwa 10:1 bis etwa 0,05:1. Ein
Katalysator, welcher zum Beispiel Platin und Eisen umfasst, besitzt
vorzugsweise ein Platin/Eisen-Verhältnis von etwa 3:1.
-
Bei
einer besonders bevorzugten Ausführungsform
dieser Erfindung ist das Edelmetall (z. B. Pt) mit mindestens einem
Katalysatoraktivator (z. B. Sn, Fe oder beiden) legiert, um legierte
Metallteilchen zu bilden (und in den Fällen, in denen der Katalysatoroberflächenaktivator
ebenfalls ein Edelmetall ist, ist das Edelmetall des Nicht-Aktivators
mit dem Edelmetall des Katalysatoroberflächenaktivators legiert). Ein
Katalysator, welcher ein Edelmetall umfasst, welches mit mindestens
einem Katalysatoroberflächenaktivator
legiert ist, neigt dazu, alle Vorteile zu besitzen, welche vorstehend
im Hinblick auf Katalysatoren erörtert
wurden, welche generell einen Katalysatoroberflächenaktivator umfassen. Katalysatoren,
welche ein mindestens mit einem Katalysatoroberflächenaktivator
legiertes Edelmetall umfassen, neigen auch zu einer größeren Widerstandsfähigkeit
gegen das Auslaugen des Katalysatoroberflächenaktivators und weiterhin
eine größerer Stabilität von Zyklus
zu Zyklus bezüglich
der Formaldehyd- und Ameisensäureoxidation
(Vgl. z. B. Beispiel 1).
-
Der
Ausdruck Legierung beinhaltet jedes Metallteilchen, welches ein
Edelmetall und mindestens einen Katalysatoroberfächenaktivator umfasst, ungeachtet
der genauen Art und Weise, in der das Edelmetall und der Katalysatoroberflächenaktivator
innerhalb des Teilchens angeordnet sind (obwohl es im Allgemeinen
bevorzugt ist, wenn ein Teil der Edelmetallatome auf der Oberfläche des
legierten Metallteilchens vorliegt). Die Legierung kann zum Beispiel
irgendeine der folgenden sein:
- 1. Eine intermetallische
Verbindung. Eine Intermetallische Verbindung ist eine Verbindung,
welche ein Edelmetall und einen Aktivator umfasst (z. B. Pt3Sn);
- 2. Eine Substitutionslegierung. Eine Substitutionslegierung
besitzt eine einzelne kontinuierliche Phase, ungeachtet der Konzentrationen
von Edelmetall- und Aktivatoratomen. Eine Substitutionslegierung
enthält
typischerweise Edelmetall- und Aktivatoratome ähnlicher Größe (z. B. Platin und Silber
oder Platin und Palladium). Substitutionslegierungen werden auch
als einphasige Legierungen bezeichnet.
- 3. Eine Mehrphasenlegierung. Eine Mehrphasenlegierung ist eine
Legierung, welche mindestens zwei diskrete Phasen enthält. Eine
solche Legierung kann zum Beispiel Pt3Sn
in einer Phase und in Platin gelöstes Zinn
in einer getrennten Phase enthalten.
- 4. Eine Segregationslegierung. Eine Segregationslegierung ist
ein Metallteilchen, in dem die Teilchenstöchiometrie mit der Entfernung
von der Oberfläche
des Metallteilchens schwankt.
- 5. Eine Einlagerungslegierung. Eine Einlagerungslegierung ist
ein Metallteilchen, in dem Edelmetall- und Aktivatoratome mit Nichtmetallatomen
wie Bor, Kohlenstoff, Silizium, Stickstoff, Phosphor etc. kombiniert sind.
-
Vorzugsweise
mindestens etwa 80% (Zahlendichte) der legierten Metallteilchen
besitzen eine größte Abmessung
von etwa 0,5 bis etwa 35 nm, weiter vorzugsweise eine größte Abmessung
von etwa 1 bis etwa 20 nm, noch weiter vorzugsweise eine größte Abmessung
von etwa 1 bis etwa 15 nm und am meisten vorzugsweise eine größte Abmessung
von etwa 1,5 bis etwa 7 nm.
-
Die
legierten Metallteilchen brauchen keine einheitliche Zusammensetzung
aufzuweisen; Die Zusammensetzungen können von Teilchen zu Teilchen
schwanken oder sogar innerhalb des Teilchens selbst. Zusätzlich kann
der Katalysator weiterhin Teilchen umfassen, welche nur aus dem
Edelmetall oder nur aus dem Katalysatoroberflächenaktivator bestehen. Nichtsdestoweniger
ist es bevorzugt, dass die Zusammensetzung Metallteilchen von Teilchen
zu Teilchen und innerhalb eines jeden Teilchens im Wesentlichen
einheitlich ist und dass die Zahl der Edelmetallatome, welche sich
in innigem Kontakt mit den Atomen des Katalysatoroberflächenaktivators
befinden, maximiert ist. Es ist auch bevorzugt, wenn auch nicht
unbedingt erforderlich, dass die Mehrheit der Edelmetallatome mit
einem Katalysatoroberflächenaktivator
legiert ist, und weiter bevorzugt, dass im Wesentlichen alle Edelmetallatome
mit einem Katalysatoroberflächenaktivator
legiert sind. Es ist des Weiteren bevorzugt, wenn auch nicht unbedingt
erforderlich, dass die legierten Metallteilchen auf der Oberfläche des
Kohlenstoffträgers
einheitlich verteilt sind.
-
Ungeachtet
dessen, ob der Katalysatoroberflächenaktivator
an das Edelmetall legiert ist oder nicht, wird gegenwärtig angenommen,
dass der Katalysatoroberflächenaktivator
dazu neigt oxidiert zu werden, wenn der Katalysator während längerer Zeit
einem Oxidationsmittel ausgesetzt wird. Ein Katalysatoroberflächenaktivator
mit elementarem Zinn neigt zum Beispiel dazu sich unter Bildung
von Sn(II)O zu oxidieren, und Sn(II)O dazu neigt sich unter Bildung
von Sn(IV)O2 zu oxidieren. Diese Oxidation
kann zum Beispiel erfolgen, wenn der Katalysator länger als
1 Stunde der Luft ausgesetzt wird. Obwohl nicht beobachtet worden
ist, dass eine solche Oxidation des Katalysatoroberflächenaktivators
eine bedeutendere schädliche
Wirkung auf die Edelmetallauslaugung, Edelmetallsinterung, Katalysatoraktivität oder Katalysatorstabilität ausübt, macht
sie die Analyse der Konzentration schädlicher sauerstoffhaltiger
funktioneller Gruppen auf der Oberfläche des Kohlenstoffträgers schwieriger.
Wie vorstehend erörtert,
kann zum Beispiel die Konzentration schädlicher sauerstoffhaltiger
funktioneller Gruppen (d. h. sauerstoffhaltige funktionelle Gruppen,
welche die Widerstandsfähigkeit
des Edelmetalls gegen Auslaugen und Sintern herabsetzen und die
Katalysatoraktivität
reduzieren) durch Messung (unter Verwendung von zum Beispiel einer
TGA-MS) der CO-Menge bestimmt werden, welche unter hohen Temperaturen
in einer inerten Atmosphäre
vom Katalysator desorbiert wird. Es wird jedoch gegenwärtig angenommen,
dass dann, wenn ein oxidierter Katalysatoroberflächenaktivator auf der Oberfläche vorliegt,
die Sauerstoffatome aus dem oxidierten Katalysatoroberflächenaktivator
dazu neigen mit Kohlenstoffatomen des Trägers bei hohen Temperaturen
in einer inerten Atmosphäre
CO zu erzeugen, wodurch sie die Vorstellung schaffen, dass mehr
schädliche
sauerstoffhaltige funktionelle Gruppen auf der Oberfläche des
Trägers
vorliegen als tatsächlich
vorhanden sind. Solche Sauerstoffatome von einem oxidierten Katalysatoroberflächenaktivator
können
auch dabei stören,
eine verlässliche
Voraussage der Edelmetallauslaugung, der Edelmetallsinterung und
der Katalysatoraktivität
aus einer einfachen Messung (zum Beispiel über eine Röntgen-Photoelektronen-Spektroskopie) von
Sauerstoffatomen auf der Katalysatoroberfläche zu erhalten.
-
Ist
der Katalysator, welcher mindestens einen Katalysatoroberflächenaktivator
umfasst, demzufolge einem Oxidationsmittel ausgesetzt und dadurch
oxidiert worden (z. B. wenn der Katalysator länger als etwa 1 Stunde der
Luft ausgesetzt worden ist), wird der Katalysatoroberflächenaktivator
vorzugsweise zuerst im Wesentlichen reduziert (wodurch die Sauerstoffatome
vom oxidierten Katalysatoroberflächenaktivator
aus der Oberfläche
des Katalysators entfernt werden), ehe versucht wird, die Menge
an schädlichen
sauerstoffhaltigen funktionellen Gruppen auf der Oberfläche des
Kohlenstoffträgers
zu messen. Diese Reduktion erfolgt vorzugsweise durch Erhitzen des
Katalysators während
1 Stunde auf eine Temperatur von 500°C in einer Atmosphäre, welche
im Wesentlichen aus H2 besteht. Die Messung
der schädlichen
sauerstoffhaltigen funktionellen Gruppen auf der Oberfläche erfolgt
vorzugsweise (a) nach dieser Reduktion und (b) ehe die Oberfläche nach
der Reduktion an ein Oxidationsmittel ausgesetzt wird. Am meisten
vorzugsweise erfolgt die Messung sofort nach der Reduktion.
-
Die
bevorzugte Konzentration der Metallteilchen auf der Oberfläche des
Kohlenstoffträgers
hängt zum Beispiel
von der Größe der Metallteilchen,
der spezifischen Oberfläche
des Kohlenstoffträgers
und der Konzentration des Edelmetalls auf dem Katalysator ab. Es
wird gegenwärtig
allgemein angenommen, dass die bevorzugte Konzentration der Metallteilchen
ungefähr
etwa 3 bis etwa 1500 Teilchen/μm2 beträgt
(d. h. die Zahl der Metallteilchen pro μm2 Kohlenstoffträgeroberfläche), insbesondere
wenn: (a) mindestens etwa 80% (Teilchendichte) der Metallteilchen
eine größte Abmessung
von etwa 1,5 bis etwa 7 nm aufweisen, (b) der Kohlenstoffträger eine
spezifische Oberfläche
von etwa 750 bis etwa 2100 m2/g (d. h. m2 Oberfläche
des Kohlenstoffträgers
pro Gramm Kohlenstoffträger)
aufweist und (c) die Konzentration des Edelmetalls auf dem Kohlenstoffträger etwa
1 bis etwa 10 Gew.-% ([Masse des Edelmetalls ÷ Katalysatorgesamtmasse] × 100) beträgt. Bei weiter
bevorzugten Ausführungsformen
sind engere Bereiche der Metallteilchenkonzentrationen und Edelmetallkonzentrationen
erwünscht.
Bei einer solchen Ausführungsform
beträgt
die Konzentration der Metallteilchen etwa 15 bis etwa 800 Teilchen/μm2 und die Konzentration des Edelmetalls auf
der Kohlenstoffträgeroberfläche etwa
2 bis etwa 10 Gew.-%. Bei einer sogar weiter bevorzugten Ausführungsform
beträgt
die Konzentration der Metallteilchen etwa 15 bis etwa 600 Teilchen/μm2 und die Konzentration des Edelmetalls auf
der Kohlenstoffträgeroberfläche etwa
2 bis etwa 7,5 Gew.-%. Bei der am meisten bevorzugten Ausführungsform beträgt die Konzentration
der Metallteilchen etwa 15 bis etwa 400 Teilchen/μm2 und die Konzentration des Edelmetalls auf
der Kohlenstoffträgeroberfläche etwa
5 Gew.-%. Die Konzentration der Metallteilchen auf der Oberfläche des
Kohlenstoffträgers
kann mithilfe auf dem Fachgebiet bekannter Verfahren gemessen werden.
-
Die
Oberfläche
des Kohlenstoffträgers
wird vorzugsweise vom Sauerstoff befreit, ehe das Edelmetall darauf
abgeschieden wird. Die Oberfläche
wird vorzugsweise unter Verwendung einer Hochtemperaturdesoxidationsbehandlung
vom Sauerstoff befreit. Eine solche Behandlung kann in einem einstufigen
oder mehr stufigen Vorgang bestehen, welcher in jedem Fall zu einer
chemischen Reduktion aller sauerstoffhaltigen funktionellen Gruppen
auf der Oberfläche
des Kohlenstoffträgers
führt.
-
Bei
einer zweistufigen Hochtemperaturdesoxidationsbehandlung wird der
Kohlenstoffträger
zuerst mit einem Oxidationsmittel in der gasförmigen oder flüssigen Phase
behandelt, um in relativ niedrigen Oxidationszuständen vorliegende
sauerstoffhaltige Funktionalitäten
(z. B. Ketone, Aldehyde und Alkohole) in vergleichsweise höhere Funktionalitäten (z.
B. Carbonsäuren)
umzuwandeln, welche bei hohen Temperaturen leichter von der Katalysatoroberfläche abzuspalten
sind. Repräsentative
Flüssigphasenoxidationsmittel
schließen
Salpetersäure,
H2O2, Chromsäure und
Hypochlorit ein, wobei konzentrierte Salpetersäure mit etwa 10 bis etwa 80 Gramm
HNO3 pro 100 Gramm wässrige Lösung bevorzugt ist. Repräsentative
gasförmige
Oxidationsmittel schließen
molekularen Sauerstoff, Ozon, Stickstoffdioxide und Salpetersäuredämpfe ein.
Salpetersäuredämpfe sind
die bevorzugten Oxidationsmittel. Bei flüssigen Oxidationsmitteln sind
Temperaturen von etwa 60 bis etwa 90°C geeignet, während es
bei gasförmigen
Oxidationsmitteln oft von Vorteil sein kann Temperaturen von etwa
50 bis etwa 500°C
oder sogar höher
zu verwenden. Die Behandlungsdauer des Kohlenstoffs mit dem Oxidationsmittel
kann innerhalb von etwa 5 Minuten bis etwa 10 Stunden weit schwanken.
Die Reaktionsdauer beträgt
vorzugsweise etwa 30 Minuten bis etwa 6 Stunden. Versuchsergebnisse
zeigen, dass die Kohlenstoffbeladung, Temperatur, Oxidatorkonzentration
etc, in der ersten Behandlungsstufe nicht besonders kritisch sind,
um die gewünschte
Oxidation des Kohlenstoffmaterials zu erzielen und nach Gutdünken über einen
breiten Beriech geregelt werden können. Aus wirtschaftlichen
Gründen
ist die höchstmögliche Kohlenstoffbeladung
bevorzugt.
-
In
zweiter Stufe wird der oxidierte Kohlenstoffträger in einer Stickstoff-, Argon-,
Helium oder einer anderen nicht oxidierenden Umgebung (d. h. einer
im Wesentlichen sauerstofffreien Umgebung) bei einer Temperatur
von vorzugsweise im Bereich von etwa 500 bis etwa 1500°C und weiter
vorzugsweise von etwa 600 bis etwa 1200°C pyrolysiert (d. h. erhitzt),
um die sauerstoffhaltigen funktionellen Gruppen aus der Kohlenstoffoberfläche auszutreiben.
Bei Temperaturen größer als
500°C kann
eine Umgebung verwendet werden, welche eine geringe Menge Ammoniak
(oder eine andere chemische Einheit, welche während der Pyrolyse NH3 erzeugt), Dampf oder Kohlendioxid umfasst,
welche die Pyrolyse unterstützen.
Wird die Temperatur des Kohlenstoffträgers auf Temperaturen von weniger
als 500°C
abgekühlt,
kann die Anwesenheit von sauerstoffhaltigen Gasen wie Dampf und
Kohlendioxid jedoch zur Rückbildung
von Oberflächenoxiden
führen
und wird demzufolge vorzugsweise vermieden. Folglich erfolgt die
Pyrolyse vorzugsweise in einer nicht-oxidierenden Atmosphäre (z. B.
Stickstoff, Argon oder Helium). Bei einer Ausführungsform umfasst die nicht-oxidierende
Atmosphäre
Ammoniak, welcher dazu neigt, in kürzerer Zeit einen aktiveren
Katalysator zu erzeugen, verglichen mit der Pyrolyse in den anderen
Atmosphären.
Die Pyrolyse kann zum Beispiel in einem Drehrohrofen, einen Wirbelschichtreaktor
oder einem herkömmlichen
Ofen erfolgen.
-
Der
Kohlenstoffträger
wird im Allgemeinen während
einer Zeitspanne von etwa 5 Minuten bis etwa 60 Stunden, vorzugsweise
etwa 10 Minuten bis etwa 6 Stunden pyrolysiert. Kürzere Zeiten
sind bevorzugt, weil längeres
Aussetzen des Kohlenstoffs an erhöhte Temperaturen dazu neigt,
die Aktivität
des Katalysators zu verringern. Ohne Bindung an eine spezielle Theorie
wird gegenwärtig
angenommen, dass längeres
Aussetzen bei Pyrolysetemperaturen die Bildung von Graphit fördert, welcher
eine weniger bevorzugte Form eines Kohlenstoffträgers ist, weil er normalerweise
eine kleinere Oberfläche
aufweist. Wie vorstehend erörtert,
kann ein höher
aktiver Katalysator typischerweise in kürzerer Zeit durch Verwendung
einer Atmosphäre
hergestellt werden, welche Ammoniak umfasst.
-
Bei
einer bevorzugten Ausführungsform
dieser Erfindung erfolgt die Hochtemperaturdesoxidation in einem
Schritt. Diese einstufige Behandlung kann darin bestehen, dass lediglich
der Pyrolyseschritt der vorstehend erörterten zweistufigen Hochtemperaturdesoxidationsbehandlung
erfolgt. Weiter vorzugsweise besteht jedoch die einstufige Behandlung
in der Pyrolyse des Kohlenstoffträgers, wie vorstehend beschrieben,
bei gleichzeitigem Überleiten
eines N2-, NH3-Gasstroms
(oder irgendeiner andern chemischen Einheit, welche während der
Pyrolyse NH3 erzeugt) und Dampf über den
Kohlenstoff. Obwohl es kein kritisches Merkmal dieser Erfindung
ist, ist die Strömungsgeschwindigkeit
des Gasstrom vorzugsweise groß genug
um einem entsprechenden Kontakt zwischen den Frischgasreaktanten
und der Kohlenstoffoberfläche
zu erzielen, jedoch langsam genug, um die Abnahme des Kohlenstoffgewichts
und eine Materialverschwendung zu verhindern. Um einen ernsthaften
Gewichtsverlust des Kohlenstoffs zu verhindern, kann ein nicht reaktives
Gas als Verdünner
verwendet werden.
-
Verfahren
zur Abscheidung des Edelmetalls auf der Oberfläche des Kohlenstoffträgers sind
auf dem Fachgebiet allgemein bekannt und schließen ein: Flüssig phasenverfahren wie Reaktionsabscheidungsmethoden
(z. B. Abscheidung über
die Reduktion von Edelmetallverbindungen und Abscheidung über die
Hydrolyse von Edelmetallverbindungen), Ionenaustauschverfahren,
Imprägnierung
aus überschüssiger Lösung und
beginnende Nassimprägnierung;
Dampfphasenverfahren, wie Verfahren zur physikalischen und chemischen
Abscheidung; Fällung;
elektrochemische Abscheidung und stromlose Abscheidung. Vergleiche
allgemein Cameron, D. S., Copper, S. J., Dodgson, I. L., Harrison,
B. und Jenkins, J. W. "Carbons
as Supports für
Precious Metal Catalysts",
Catalysis Today 7: 113–137
(1990). Katalysatoren, welche Edelmetalle auf der Oberfläche eines
Kohlenstoffträgers
umfassen, sind im Handel ebenfalls erhältlich, z. B. Aldrich Katalog
Nr. 20.593-1,5% Platin auf Aktivkohle (Aldrich Chemical Co., Inc.,
Milwaukee, WI); Aldrich Katalog Nr. 20.568-0,5% Palladium auf Aktivkohle.
-
Das
Edelmetall wird vorzugsweise über
ein Reaktionsabscheidungsverfahren abgeschieden, umfassend das Inberührungbringen
des Kohlenstoffträgers
mit einer Lösung,
welche ein Salz des Edelmetalls umfasst und die anschließende Hydrolyse
des Salzes. Ein Beispiel eines geeigneten Platinsalzes, welches
relativ preiswert ist, ist Hexachloroplatinsäure (H2PtCl6). Die Verwendung dieses Salzes zur Abscheidung
von Platin auf einem Kohlenstoffträger über die hydrolytische Abscheidung
ist in Beispiel 3 veranschaulicht.
-
Bei
einer Ausführungsform
dieser Erfindung wird das Edelmetall auf der Oberfläche des
Kohlenstoffträgers
unter Verwendung einer Lösung
abgeschieden, die ein Edelmetallsalz in einem seiner stärker reduzierten
Oxidationszuständen
umfasst. Anstelle zum Beispiel ein Pt(IV)-Salz (z. B. H2PtCl6) zu verwenden, wird ein Pt(II)-Salz verwendet.
Bei einer anderen Ausführungsform
wird Platin in seinem elementaren Zustand (z. B. kolloidales Platin)
verwendet. Die Verwendung dieser stärker reduzierten Metallvorläufer führt zu einer
geringeren Oxidation des Kohlenstoffträges, weswegen auf der Oberfläche des
Trägers
weniger sauerstoffhaltige funktionelle Gruppen gebildet werden,
während
das Edelmetall auf der Oberfläche
abgeschieden wird. Ein Beispiel eines Pt(II)-Salzes ist H2PtCl4. Ein anderes
potentiell verwendbares Pt(II)-Salz ist Diaminodinitriloplatin(II).
Beispiel 11 zeigt, dass bei Verwendung dieses Salzes als Metallvorläufer zur
Abscheidung des Edelmetalls einen Katalysator liefert, welcher gegen
Auslaugen widerstandsfähiger
ist als ein unter Verwendung von H2PtCl6 hergestellter Katalysator. Ohne Bindung
an irgendeine Theorie wird angenommen, dass dies auf der Tatsache
beruht, dass Diaminodinitriloplatin(II) während der Reduktion in situ
Ammoniak erzeugt, welcher die Entfernung der sauerstoffhaltigen
funktionellen Gruppen auf der Oberfläche des Kohlenstoffträgers weiter
fördert.
Dieser Vorteil sollte jedoch gegen die mögliche Explosionsgefahr abgewogen
werden, welche mit der Verwendung von Diaminodinitriloplatin(II)
verbunden ist.
-
Katalysatoroberflächenaktivator(en)
kann/können
auf der Oberfläche
des Kohlenstoffträgers
vor, gleichzeitig mit, oder nach der Abscheidung des Edelmetalls
auf der Oberfläche
abgeschieden werden. Verfahren zur Abscheidung eines Aktivators
auf der Oberfläche
des Kohlenstoffträgers
sind auf dem Fachgebiet allgemein bekannt und schließen die
gleichen Verfahren ein, wie sie vorstehend für die Abscheidung von Edelmetall
erörtert
wurden. Bei einer Ausführungsform
wird zur Abscheidung des Katalysatoroberflächenaktivators eine Salzlösung verwendet,
welche einen Aktivator umfasst. Ein geeignetes Salz, welches zur
Abscheidung von Wismut verwendet werden kann, ist Bi(NO3)3·5H2O; ein geeignetes Salz, welches zur Abscheidung
von Eisen verwendet werden kann, ist FeCl3·6H2O und ein geeignetes Salz zur Abscheidung
von Zinn ist SnCl2·2H2O.
Es kann selbstverständlich
mehr als ein Katalysatoroberflächenaktivator
auf der Oberfläche
des Kohlenstoffträgers
abgeschieden werden. Die Beispiele 13, 14, 15 und 17 zeigen das
Abscheiden eines Aktivators auf einer Kohlenstoffoberfläche mit
einer Salzlösung,
welche einen Aktivator umfasst. Beispiel 8 zeigt das Abscheiden
von mehr als einem Aktivator (d. h. Eisen und Zinn) auf der Kohlenstoffoberfläche unter
Verwendung von Salzlösungen,
welche die Aktivatoren umfassen.
-
Ein
Katalysator, welcher ein Edelmetall umfasst, welches mit mindestens
einem Katalysatoroberflächenaktivator
legiert ist, ist wie vorstehend vermerkt, besonders bevorzugt. Auf
dem Fachgebiet ist eine Vielfalt von präparativen Methoden bekannt,
welche verwendet werden können,
um eine multimetallische Legierung auf einer Trägeroberfläche zu bilden. Vgl. z. B. V.
Ponec & G. C.
Bond, "Catalysis
by Metals and Alloys",
Studies in Surface Science and Catalysis, Band 95 (B. Delmon & J. T. Yates,
Beratende Hrsg., Elsevier Science B. V., Amsterdam, Niederlande).
-
Bei
einer der weiter bevorzugten Ausführungsformen wird die reaktive
Abscheidung verwendet, um Metallteilchen abzuscheiden, welche ein
mit einem Katalysatoroberflächenaktivator
legiertes Edelmetall enthalten. Die reaktive Abscheidung kann zum
Beispiel die reduktive Abscheidung umfassen, bei der eine Oberfläche eines
Kohlenstoffträgers
mit einer Lösung
in Berührung
gebracht wird, umfassend: (a) ein Reduktionsmittel und (b) (i) eine
Verbindung, umfassend das Edelmetall und eine Verbindung, umfassend
den Aktivator, oder (ii) eine Verbindung, umfassend sowohl das Edelmetall
als auch den Aktivator. Es kann eine breite Vielfalt von Reduktionsmitteln
verwendet werden wie Natriumborhydrid, Formaldehyd, Ameisensäure, Natriumformiat, Hydrazinhydrochlorid,
Hydroxylamin und unterphosphorige Säure. Verbindungen, welche ein
Edelmetall und/oder einen Aktivator umfassen schließen zum
Beispiel ein:
- 1. Halogenidverbindungen: Diese
schließen
zum Beispiel H2PtCl6,
K2PtCl4, Pt2Br6 2–,
K2PdCl4, AuCl4 1–, RuCl3,
RhCl3·3H2O, K2RuCl6, FeCl3·6H2O, (SnCl3)1–,
SnCl4, FeCl2 und
TiCl4 ein.
- 2. Oxide und Oxychloridverbindungen: Diese schließen zum
Beispiel RuO4 2– und
M2SnO4 ein.
- 3. Nitratverbindungen: Diese schließen zum Beispiel Fe(NO3)3 ein.
- 4. Aminkomplexe: Diese schließen zum Beispiel [Pt(NH3)4]Cl2,
[Pd(NH3)4]Cl2, Pt(NH3)2Cl2, [Pt(NH3)4]PtCl4, Pd(NH2CH2CH2NH2)Cl2, Pt(NH2CH2CH2NH2)Cl2 und [Ru(NH3)5Cl]Cl2 ein.
- 5. Phosphinkomplexe: Diese schließen zum Beispiel Pt(P(CH3)3)2Cl2, IrClCO(P(C6H5)3)2,
PtClH(PR3)2 ein, worin
jedes R unabhängig
ein Hydrocarbyl wie Methyl, Ethyl, Propyl, Phenyl etc. ist.
- 6. Metallorganische Komplexe: Diese schließen zum Beispiel Pt2(C3H6)2Cl4; Pd(C2H4)2C14; Pt(CH3COO)2, Pd(CH3COO)2; K[Sn(HCOO)3];
Fe(CO)5; Fe3(CO)12; Fe4(CO)16; Sn3(CH3)4 und Ti(OR)4 ein, worin jedes R unabhängig ein
Hydrocarbyl wie Methyl, Ethyl, Propyl, Phenyl etc. ist.
- 7. Edelmetall/Aktivator-Komplexe: Diese schließen zum
Beispiel Pt3(SnCl3)2(C8H12)3 und [Pt(SnCl3)5]3– ein.
-
Bei
einer besonders bevorzugten Ausführungsform
werden Hydrolysereaktionen zur Abscheidung eines mit einem Katalysatoroberflächenaktivator
legierten Edelmetalls verwendet. In diesem Fall werden Liganden
gebildet, welche das Edelmetall und den Aktivator enthalten und
dann hydrolysiert, um auf der Oberfläche des Kohlenstoffträgers gut
gemischte Metalloxid- und Metallhydroxidcluster zu bilden. Die Liganden
können zum
Beispiel durch Inberührungbringen der
Oberfläche
des Trägers
mit einer Lösung
gebildet werden, umfassend (a) eine Verbindung, welche das Edelmetall
und eine Verbindung umfasst, welche den Aktivator umfasst, oder
(b) eine Verbindung, welche sowohl das Edelmetall als auch den Aktivator
umfasst. Geeignete Verbindungen, welche ein Edelmetall und/oder
einen Aktivator umfassen, sind vorstehend im Hinblick auf die reduktive
Abscheidung aufgeführt.
Die Hydrolyse der Liganden kann zum Beispiel durch Erhitzen der
Mischung (z. B. auf eine Temperatur von mindestens etwa 60°C) erreicht
werden. Beispiel 1 veranschaulicht weiterhin die Verwendung von
Hydrolysereaktionen zur Abscheidung eines Edelmetalls (d. h. Platin),
das mit dem Katalysatoroberflächenaktivator
(d. h. Eisen) legiert ist.
-
Zusätzlich zu
den vorstehend beschriebenen reaktiven Abscheidungsmethoden gibt
es viele andere Methoden, welche zur Legierungsbildung verwendet
werden können.
Diese schließen
zum Beispiel ein:
- 1. Bilden der Legierung durch
Einführen
von Meallverbindungen (welche einfach oder komplex und kovalent
oder ionisch sein können)
auf die Oberfläche
des Trägers über Imprägnierung,
Adsorption aus einer Lösung
und/oder Ionenaustausch.
- 2. Bilden der Legierung durch gemeinsame Vakuumabscheidung von
Metalldämpfen,
welche das Edelmetall und den Aktivator enthalten, auf der Oberfläche.
- 3. Bilden der Legierung durch Abscheidung eines Erzmetalls auf
einem vorher abgeschiedenen Metall, welches zur Gruppe 8, 9 oder
10 des Periodischen Systems der Elemente gehört (d. h. Fe, Co, Ni, Ru, Rh,
Pd, Os, Ir und Pt) über
zum Beispiel elektrolytische oder stromlose Galvanisierung.
- 4. Bilden der Legierung durch: (a) Abscheiden von Metallkomplexen,
welche Metalle im Valenzzustand Null (d. h. Carbonyl-, π-Allyl- oder
Cyclopentadienylkomplexe aus Edelmetall und Aktivator) enthalten
auf der Oberfläche
des Kohlenstoffträgers;
und (b) Entfernen der Liganden durch zum Beispiel Erhitzen oder
Reduzieren, um auf der Oberfläche
Legierungsteilchen zu bilden.
- 5. Bilden der Legierung durch Inberührungbringen einer eine Metallver bindung
(z. B. Metallchlorid oder Metallalkylverbindung) enthalten-den Lösung mit
einem vorher abgeschiedenen Metallhydrid, welches ein zur Gruppe
8, 9 oder 10 des Periodischen Systems der Elemente gehörendes Metall
enthält.
- 6. Bilden der Legierung durch gemeinsame Abscheidung (entweder
gleichzeitig oder nacheinander) von Metallkomplexen (entweder vorgeformt
oder in situ, welche Edelmetall(e) und Aktivator(en) enthalten,
auf der Oberfläche
des Kohlenstoffträgers.
- 7. Bilden der Legierung durch Vorformen von Legierungsteilchen
als Kolloide oder Aerosole und anschließendes Abscheiden der vorgeformten
Legierungsteilchen auf der Oberfläche des Kohlenstoffträgers. Kolloidale
Teilchen, welche Platin und Zinn enthalten, können leicht durch Kochen einer
verdünnten
Lösung von
H2PtCl6 und SnCl2·2H2O mit einer Natriumcitratlösung gebildet
werden, um dies zu erläutern.
Schutzstoffe (Kohlenhydrate, Polymere, lipophile quaternäre Stickstoffsalze)
können
verwendet werden, um das Wachstum der Metalllegierungsteilchen wirksam
zu steuern. Diese Methode ist daher oft nützlich, um eine enge Verteilung
der Legierungsteilchengröße zu bilden.
-
Die
vorstehend erörterten
Verfahren zur Legierungsbildung dienen selbstverständlich lediglich
der Veranschaulichung und sind nicht erschöpfend. Ein Fachmann kann unter
Verwendung der Lehre dieser Spezifikation und der allgemeinen Kenntnis
auf dem Fachgebiet routinemäßig bestimmen,
welches der zahlreichen auf dem Fachgebiet bekannten Verfahren zur
Legierungsbildung für
eine spezielle Anwendung geeignet ist.
-
Ungeachtet
des zur Legierungsbildung verwendeten Verfahrens ist es nach Abscheidung
der Metalle auf der Oberfläche
des Kohlenstoffträgers
oft vorzuziehen, den Träger
unter Verwendung von zum Beispiel Unterdruck, nicht-oxidierender
Umgebung (vorzugsweise N2, einem Edelgas
oder beidem) zu trocknen. Die Verwendung eines Trocknungsschrittes
ist besonders bevorzugt, wenn die Oberfläche des Trägers anschließend durch
Erhitzen der Oberfläche
(und sogar weiter bevorzugt, wenn das Erhitzen in einer nicht-oxidierenden
Umgebung erfolgt) reduziert wird. Der Träger wird getrocknet um den
Feuchtigkeitsgehalt des Trägers
vorzugsweise auf weniger als etwa 5 Gew.-% zu reduzieren.
-
Das
Reduzieren der Oberfläche
des Kohlenstoffträgers
nach der Abscheidung des/der Edelmetalls(e) und des/der Katalysatoroberflächenaktivators(en)
erhöht
selbstverständlich
den Umfang des mit dem Katalysatoroberflächenaktivator legierten Edelmetalls.
Eine solche Reduktion tendiert auch oft dazu, die in den bevorzugten
Größenbereich
fallende Teilchenzahl zu erhöhen.
-
Nachdem
der Kohlenstoffträger
mit dem/den Edelmetall(en) (und Katalysatoroberflächenaktivator(en), sofern
vorliegend) imprägniert
worden ist, wird die Oberfläche
des Katalysators vorzugsweise reduziert. Die Oberfläche des
Katalysators kann geeigneterweise zum Beispiel durch Erhitzen der
Oberfläche
auf eine Temperatur von mindestens 400°C reduziert werden. Es ist besonders
bevorzugt, dieses Erhitzen in einer nicht-oxidierenden Umgebung
(z. B. Stickstoff, Argon oder Helium) durchzuführen. Es ist auch weiter bevorzugt,
dass die Temperatur größer als
etwa 500°C
ist. Die Temperatur beträgt
noch weiter vorzugsweise etwa 550 bis etwa 1200°C und am meisten vorzugsweise
etwa 550 bis etwa 900°C.
Temperaturen von weniger als 400°C
neigen dazu, die sauerstoffhaltigen funktionellen Gruppen von der
Oberfläche
des Kohlenstoffträgers
nicht zufriedenstellend zu entfernen. Andererseits neigen Temperaturen
größer 1200°C dazu, die
Aktivität
des Katalysators zu reduzieren. Temperaturen von etwa 400 bis etwa
500°C werden
vorzugsweise nur verwendet, wenn die Oberfläche des Kohlenstoffträgers ein
Kohlenstoffatom/Sauerstoffatom-Verhältnis von mindestens etwa 20:1
aufweist, ehe das Edelmetall auf der Oberfläche abgeschieden wird.
-
Bei
einer besonders bevorzugten Ausführungsform
wird die Oberfläche
des Katalysators durch ein Verfahren reduziert, welches das Aussetzen
der Oberfläche
an eine reduzierende Umgebung umfasst. Zum Beispiel kann die Katalysatorprobe
vor dem Erhitzen mit einem Flüssigphasenreduktionsmittel
wie Formaldehyd oder Ameisensäure
vorbehandelt werden. Das Erhitzen erfolgt sogar weiter vorzugsweise
in Gegenwart eines Gasphasenreduktionsmittels (Das Verfahren des
Erhitzens des Katalysators in Gegenwart eines Gasphasenreduktionsmittels
wird manchmal als Hochtemperaturgasphasenreduktion bezeichnet).
Während
des Erhitzens können
verschiedene Gasphasenreduktionsmittel verwendet werden, einschließend, jedoch
ohne Beschränkung
darauf, Ha, Ammoniak und Kohlenmonoxid. Wasserstoffgas ist am meisten
bevorzugt, weil die kleine Molekülgröße von Wasserstoff
eine bessere Penetration in die tiefsten Poren des Kohlenstoffträgers erlaubt.
Der Rest des Gases besteht vorzugsweise im Wesentlichen aus einem
nicht-oxidierenden Gas wie Stickstoff, Argon oder Helium. Das Gas
kann eine begrenzte H2-Konzentration umfassen,
obwohl H2-Konzentrationen von weniger als
1,0% wegen des Zeitbedarfs, der benötigt wird, um die Oberfläche des
Trägers
zu reduzieren, nachteilig sind. Das Gas umfasst vorzugsweise etwa
5 bis etwa 50 Vol.-% H2 und am meisten vorzugsweise
etwa 5 bis etwa 25 Vol.-% H2.
-
Die
bevorzugte Zeitdauer, während
der die Katalysatoroberfläche
erwärmt
wird, hängt
vom Stoffübergang
des Reduktionsmittels auf die Katalysatoroberfläche ab. Ist das Reduktionsmittel
ein nicht-oxidierendes Gas, welches etwa 10 bis etwa 20 Vol.-% Ha
umfasst, wird die Oberfläche
vorzugsweise von 15 Minuten bis 24 Stunden auf etwa 550 bis etwa
900°C bei
einer Raumgeschwindigkeit von etwa 1 bis etwa 5000 h–1 erhitzt. Die
Raumgeschwindigkeit beträgt
weiter vorzugsweise etwa 10 bis etwa 2500 h–1,
und sogar weiter vorzugsweise etwa 50 bis etwa 750 h–1.
Bei der am meisten bevorzugten Ausführungsform erfolgt die Wärmebehandlung
bei den vorstehend bevorzugten Temperaturen und Raumgeschwindigkeiten
von etwa 1 bis 10 Stunden. Das Erhitzen der Oberfläche bei
Raumgeschwindigkeiten von weniger als 1 h–1,
ist von Nachteil, weil die sauerstoffhaltigen funktionellen Gruppen
auf der Oberfläche
des Kohlenstoffträgers
nicht ausreichend zerstört werden
können.
Andererseits ist das Erhitzen der Oberfläche auf Raumgeschwindigkeiten
größer als
5000 h–1 unwirtschaftlich.
-
Sauerstoffhaltige
funktionelle Gruppen, welche von vornherein auf der Oberfläche des
Kohlenstoffträgers
vorhandenen sind, sind im Allgemeinen nicht erforderlich oder sogar
erwünscht,
um eine angemessene Verteilung und Retention des Edelmetalls zu
erhalten. Ohne Bindung an irgendeine spezielle Theorie wird angenommen,
dass dieser Aufheizschritt die Platin/Kohlenstoff-Wechselwirkung
auf dem Katalysator durch Entfernung sauerstoffhaltiger funktioneller
Gruppen auf der Oberfläche
des Kohlenstoffträgers
verstärkt
wird, einschließend
jene, welche durch Abscheidung des Edelmetalls auf der Oberfläche gebildet
werden. Es wird angenommen, das diese sauerstoffhaltigen funktionellen
Gruppen instabile Verankerungsstellen für das Edelmetall darstellen,
weil sie dazu neigen, die möglicherweise
stärkeren π-Wechselwirkungen
zwischen dem Edelmetall und dem Kohlenstoffträger zu beeinträchtigen.
Das Erhitzen allein wird viele der sauerstoffhaltigen funktionellen
Gruppen auf der Oberfläche
des Kohlenstoffträgers
zersetzen und dadurch entfernen. Durch Erhitzen der Oberfläche in Gegenwart
eines Reduktionsmittels (z. B. H2) können jedoch
mehr sauerstoffhaltige funktionelle Gruppen entfernt werden.
-
Beträgt das Kohlenstoffatom/Sauerstoffatom-Verhältnis auf
der Oberfläche
des Kohlenstoffträgers
weniger als etwa 20:1, ehe das Edelmetall auf der Oberfläche des
Trägers
abgeschieden wird, wird die Oberfläche vorzugsweise unter Verwendung
der vorstehend beschriebenen Hochtemperaturgasphasenreduktionsbehandlung
bei einer Temperatur größer als
500°C reduziert,
obwohl die Oberfläche
wahlweise zusätzlich
zur Hochtemperaturgasphasenreduktion mit anderen Reduktionsumgebungen
behandelt wird. Beträgt
das Kohlenstoffatom/Sauerstoffatom-Verhältnis auf der Oberfläche des
Kohlenstoffträgers
andererseits mindestens 20:1, ehe das Edelmetall auf der Oberfläche des
Trägers
abgeschieden wird, können
anstelle der Hochtemperaturgasphasenreduktion verschiedene alternative
Reduktionsumgebungen verwendet werden.
-
Die
Oberfläche
des Katalysators kann wenigstens teilweise dadurch reduziert werden,
dass sie mit einem Amin wie Harnstoff, einer Ammonimumionen umfassenden
Lösung
(z. B. Ammoniumformiat oder Ammoniumoxalat) oder Ammoniakgas behandelt
wird, wobei Ammoniakgas oder eine Ammoniumionen umfassende Lösung am
meisten bevorzugt sind. Diese Aminbehandlung wird vorzugsweise zu
anderen Reduktionsbehandlungen verwendet und wird am meisten vorzugsweise
vor der Hochtemperaturgasphasenreduktion verwendet. Bei einer solchen
Ausführungsform
wird das Edelmetall auf der Oberfläche durch Behandeln mit einer Ammoniumionen
umfassenden Edelmetallvorläuferlösung abgeschieden.
Alternativ kann der Träger,
nachdem das Edelmetall auf der Oberfläche des Trägers abgeschieden worden ist,
mit einer Ammoniumionen umfassenden Lösung gewaschen oder mit einem
Ammoniak umfassenden Gas in Kontakt gebracht werden. Am meisten
vorzugsweise wird die Katalysatoroberfläche nach der Abscheidung des
Edelmetalls mit verdünnter wässriger
Ammoniaklösung
gewaschen. In diesem Fall wird der Katalysator zu reinem Wasser
zugegeben und einige Stunden gerührt,
um die Oberfläche
des Katalysators zu benetzen. Als Nächstes wird unter Rühren der Katalysatoraufschlämmung eine
Ammoniumionen umfassende Lösung
zu der Katalysatoraufschlämmung
in einer Menge zugesetzt, welche ausreicht, einen pH von größer als
7, weiter vorzugsweise von etwa 8 bis etwa 12 und am meisten vorzugsweise
von etwa 9,5 bis etwa 11,0 zu liefern. Weil die Temperatur und der
Druck nicht kritisch sind, erfolgt dieser Schritt vorzugsweise bei
Raumtemperatur und Atmosphärendruck.
Beispiel 10 veranschaulicht diese Reduktionsbehandlung ausführlicher.
-
Natriumborhydrid
(NaBH4) kann ebenfalls zur Reduktion der
Katalysatoroberfläche
verwendet werden. Diese Behandlung wird, wie die Aminbehandlung,
vorzugsweise zusätzlich
zu anderen Reduktionsbehandlungen verwendet und am meisten vorzugsweise
vor der Hochtemperaturgasphasenreduktion. Vorzugsweise wird nach
Abscheidung des Edelmetalls auf der Oberfläche des Trägers der Träger mit einer NaBH4-Lösung in
Gegenwart von NaOH bei einem pH von etwa 8 bis etwa 14 etwa 15 bis
etwa 180 Minuten gewaschen. Die eingesetzte NaBH4-Menge reicht vorzugsweise
aus, um das gesamte Edelmetall zu reduzieren. Weil die Temperatur
und der Druck nicht kritisch sind, erfolgt dieser Schritt vorzugsweise
bei Raumtemperatur und Atmosphärendruck.
Beispiel 12 veranschaulicht diese Reduktionsbehandlung ausführlicher.
-
Alle
vorstehenden Behandlungen, welche dazu verwendet werden können, die
Oberfläche
des Katalysators zu reduzieren, können selbstverständlich auch
dazu verwendet werden, die Oberfläche des Kohlenstoffträgers vor
der Abscheidung des Edelmetalls auf der Oberfläche von Sauerstoff zu befreien.
-
Bei
vielen Verfahren, bei denen es erwünscht ist, dass der Katalysator
einen Aktivator enthält,
wird der Aktivator zum Beispiel mittels der vorstehend beschriebenen
Aktivatorabscheidungsmethoden vorher abgeschieden (Dieser Abscheidungsschritt
erfolgt oft durch den Hersteller des Katalysators). Dieser Aktivatorabscheidungsschritt
neigt jedoch dazu die Katalysatorherstellungskosten zu erhöhen. Um
diese zusätzlichen Kosten
zu vermeiden wurde gefunden, dass die Vorteile eines Aktivators
(z. B. erhöhte
Selektivität,
Aktivität und/oder
Katalysatorstabilität)
allein durch direktes Mischen eines Aktivators (d. h. eines zusätzlichen
Aktivators) mit einem Kohlenstoff-getragenen edelmetallhaltigen
Katalysator (insbesondere mit einem der vorstehend beschriebenen
reduzierten Katalysatoren) erhalten werden können. Dieses Mischen kann zum
Beispiel direkt in der Oxidationsreaktionszone erfolgen, in welcher
das N-(Phosphonomethyl)iminodiessigssäuresubstrat oxidiert wird.
Dieses Mischen kann zum Beispiel alternativ getrennt von der Oxidationsrektion
erfolgen, wie in einem Katalysatorvorratsbehälter.
-
Es
wurde insbesondere festgestellt, dass bestimmte Metalle und/oder
Metallverbindungen als Ergänzungsaktivatoren
bei der Oxidation eines durch einen Kohlenstoff getragenen edelmetallhaltigen
Katalysator katalysierten N-(Phosphonomethyl)iminodiessigssäuresubstrats
fungieren. Es wurde gefunden, dass derartige Ergänzungsaktivatoren bei der Verstärkung des
Oxidationsvermögens
des Edelmetalls auf Kohlenstoffkatalysatoren für die Oxidation eines N-(Phosphonomethyl)iminodiessigssäuresubstrats
zu einem N-(Phosphonomethyl)glycinprodukt wirksam sind, wobei sie
die Katalyse der erwünschten
Umwandlung zu N- (Phosphonomethyl)glycin,
die Oxidation des Nebenprodukts Formaldehyd zu Ameisensäure und
die Oxidation des Nebenprodukts Ameisensäure zu Kohlendioxid verstärken.
-
Der/die
Zusatzaktivator(en) können
je nach Anwendung zum Beispiel Zinn, Cadmium, Magnesium, Mangan,
Ruthenium, Nickel, Kupfer, Aluminium, Kobalt, Wismut, Blei, Titan,
Antimon, Selen, Eisen, Rhenium, Zink, Cer, Zirkon, Tellur, Natrium,
Kalium, Vanadin, Gallium, Tantal, Niob, Rubidium, Lanthan und/oder
Germanium sein. Wismut, Blei, Germanium, Tellur, Titan, Kupfer und/oder
Nickel sind als Zusatzaktivator(en) oft weiter bevorzugt.
-
Bei
einer besonders bevorzugten Ausführungsform
ist der Zusatzaktivator Wismut. In Übereinstimmung mit dieser Erfindung
wurde gefunden, dass die Anwesenheit von Wismut zur Erhöhung der
Selektivität eines
kohlenstoffhaltigen, edelmetallhaltigen Katalysators (insbesondere
des vorstehend beschriebenen reduzierten Katalysators) besonders
wirksam ist, wenn es dazu verwendet wird, die Oxidation eines N-(Phosphonomethyl)iminodiessigssäuresubstrats
zu einem N-(Phosphonomethyl)glycinprodukt zu katalysieren. Noch spezieller
ist gefunden worden, dass die Anwesenheit von Wismut eine Erhöhung der
Ameisensäuremenge als
Nebenprodukt verursacht, die katalytisch oxidiert wird. In einigen
Fällen
(insbesondere in denen der Katalysator einen Katalysatoroberflächenaktivator
umfasst) wurde auch gefunden, dass die Anwesenheit von Wismut eine
Erhöhung
der Formaldehydmenge als Nebenprodukt verursacht, die katalytisch
oxidiert wird. Dieser erhöhte
Abbau eines oder beider dieser Nebenprodukte führt umgekehrt dazu, dass weniger
N-Methyl-N-(phosphonomethyl)glycin als Nebenprodukt gebildet wird
(Es wird angenommen, dass dies auf der Tatsche beruht, dass die
Bildung von jeweils einem Molekül
N-Methyl-N-(phosphonomethyl)glycin
als Nebenprodukt entweder (a) zwei Formaldehydmoleküle, oder
(b) ein Ameisensäuremolekül und ein
Formaldehydmolekül
erfordert). Es wurde weiterhin gefunden, dass in manchen Fällen (insbesondere,
in denen mehr als ein Zusatzaktivator verwendet wird) die Anwesenheit
von Wismut auch die Menge Edelmetall reduzieren kann, welche aus
dem Kohlenstoffträger
des Katalysators während
der Oxidation eines N-(Phosphonomethyl)iminodiessigssäuresubstrats
ausgelaugt wird.
-
Bei
einer anderen bevorzugten Ausführungsform
dieser Erfindung wird Tellur als Zusatzaktivator verwendet. Wie
bei der vorstehenden Ausführungsform,
bei der Wismut als Zusatzaktivator eingebaut wird, ist gefunden
worden, dass die Anwesenheit von Tellur ebenfalls wirksam ist, um
die Selektivität
eines Kohlenstoff-getragenen, edelmetallhaltigen Katalysators (insbesondere
des vorstehend beschriebenen reduzierten Katalysators) zu erhöhen, wenn
er zur Katalyse der Oxidation eines N-(Phosphonomethyl)iminodiessigssäuresubstrats
verwendet wird, um ein N-(Phosphonomethyl)glycinprodukt zu bilden.
Insbesondere ist gefunden worden, dass Tellur die Aktivität des Katalysators
bei der Oxidation eines N-(Phosphonomethyl)iminodiessigssäuresubstrats
erhöhen
kann. Es ist weiterhin gefunden worden, dass das Auslaugen von Edelmetall
aus dem Kohlenstoffträger
des Katalysators während
der Oxidation eines N-(Phosphonomethyl)iminodiessigssäuresubstrats
in Anwesenheit von Tellur im Reaktionsmedium (insbesondere wenn
auch Wismut anwesend ist) reduziert werden kann.
-
Bei
einer am meisten bevorzugten Ausführungsform werden beide, Wismut
und Tellur, als Zusatzaktivatoren verwendet.
-
Das
Mischen des Zusatzaktivators und des Katalysators erfolgt vorzugsweise
in einem flüssigen
Medium. Das Mischen kann, wie vorstehend angegeben, zum Beispiel
direkt in einem flüssigen
Reaktionsmedium erfolgen, in dem das N-(Phosphonomethyl)iminodiessigssäuresubstrat
oxidiert wird. Erfolgt jedoch die Oxidationsreaktion unter Druck,
ist der Reaktionsbehälter
normalerweise abgedichtet, und es ist folglich oft weiter bevorzugt,
den Katalysator mit dem Zusatzaktivator getrennt vom Reaktionsbehälter zu
mischen, wie in einen Katalysatorvorratsbehälter oder -rezirkulationsbehälter.
-
Der
Zusatzaktivator wird in die Mischflüssigkeit typischerweise in
Form einer anorganischen oder organischen Verbindung eingeführt, welche
den Zusatzaktivator enthält.
Die aktivatorhaltige Verbindung kann in der Flüssigkeit löslich oder unlöslich sein,
ist am typischsten aber wenigstens teilweise löslich. Die am Zusatzaktivatoratom
befindliche funktionelle Gruppe ist generell nicht kritisch (obwohl
sie vorzugsweise eine landwirtschaftlich annehmbare funktionelle
Gruppe ist. Geeignete Verbindungen schließen zum Beispiel typischerweise
Oxide, Hydroxide, Salze anorganischer sauerstofffreier Säuren, Salze
organischer Oxysäuren,
Salze aliphatischer oder aromatischer organischer Säuren, und
Phenate ein.
-
Geeignete
wismuthaltige Verbindungen schließen zum Beispiel anorganische
oder organische Verbindungen ein, in denen das/die Wismutatom(e)
einen höheren
Oxidationsgrad als 0 (z. B. 2, 3, 4, oder 5), am meisten vorzugsweise
3 aufweisen. Beispiele derartiger geeigneter Wismutverbindungen
schließen
ein:
- 1. Wismutoxide: Diese schließen zum
Beispiel BiO, Bi2O3,
Bi2O4, Bi2O5 und ähnliche
ein
- 2. Wismuthydroxide: Diese schließen zum Beispiel Bi(OH)3 und ähnliche
ein.
- 3. Wismutsalze anorganischer sauerstofffreier Säuren: Diese
schließen
zum Beispiel Wismutchlorid (z. B. BiCl3),
Wismutbromid (z. B. BiBr3), Wismut-iodid
(z. B. BiI3), Wismuttellurid (z. B. Bi2Te3) und ähnliche
ein. Wismuthalogenide sind typischerweise weniger bevorzugt, weil
sie dazu neigen, die Prozessausrüstung
zu korrodieren.
- 4. Wismutsalze anorganischer Oxysäuren: Diese schließen zum
Beispiel Wismutsulfit (z. B. Bi2(SO3)3·Bi2O3·5H2O), Wismutsulfat (z. B. Bi2(SO4)3), Wismutylsulfat
(z. B. (Bi)HSO4), Wismutylnitrit (z. B. (BiO)NO2·0,5H2O), Wismutnitrat (z. B. Bi(NO3)3·5H2O, auch als Wismutnitratpentahydrat bekannt),
Wismutylnitrat (z. B. (BiO)NO3, auch als
Wismutsubnitrat, Wismutnitratoxid und Wismutoxynitrat bekannt) Wismutmagnesiumdoppelnitrat
(z. B. 2Bi(NO3)3·3Mg(NO3)2·24H2O), Wismutphosphit (z. B. Bi2(PO3H)3·3H2O), Wismutphosphat (z. B. BiPO4),
Wismutpyrophosphat (z. B. Bi4(P2O7)3), Wismutylcarbonat
(z. B. (BiO)2CO3, auch
als Wismutsubcarbonat bekannt), Wismutperchlorat (z. B. Bi(ClO4)3·5H2O), Wismut-antimonat (z. B. BiSbO4), Wismutarsenat (z. B. Bi(AsO4)3, Wismutselenit (z. B. Bi2(SeO2)3), Wismuttitanat
(z. B. Bi2O3·2TiO2) und ähnliche.
Diese Salze schließen
auch Oxysäuren
ein, welche sich von Übergangsmetallen
ableiten, wie zum Beispiel Wismutvanadat (z. B. BiVO4),
Wismutniobat (z. B. BiNbO4), Wismuttantalat
(BiTaO4), Wismutchromat (Bi2(CrO4), Wismutyl-dichromat (z. B. (BiO)2Cr2O7),
Wismutylchromat (z. B. H(BiO)CrO4), Wismutylkaliumdoppelchromat
(z. B. K(BiO)CrO4), Wismutmolybdat (z. B.
Bi2(MoO4)3), Wismutnatriumdoppelmolybdat (z. B. NaBi(MoO4)2), Wismutwolframat
(z. B. Bi2(WO4)3), Wismutpermanganat (z. B. Bi2O2(OH)MnO4), Wismutzirkonat
(z. B. 2Bi2O3·3ZrO2) und ähnliche.
- 5. Wismutsalze aliphatischer oder aromatischer organischer Säuren: Die se
schließen
zum Beispiel Wismutacetat (z. B. Bi(C2H3O2)3),
Wismutylpropionat (z. B. (BiO)CH3H5O2), Wismutbenzoat
(z. B. C6H5CO2Bi(OH)2), Wismutylsalicylat
(z. B. C6H4CO2(BiO)(OH)), Wismut-oxalat (z. B. (C2O4)3Bi2), Wismuttartrat (z. B. Bi2(C4H4O6)3·6H2O), Wismutlactat (z. B. (C6H9O5)OBi·7H2O), Wismutcitrat (z. B. C6H5O7Bi) und ähnliche
ein.
- 6. Wismutphenate: Diese schließen zum Beispiel Wismutgallat
(z. B. C7H7O7Bi), Wismutpyrogallat (z. B. C6H3(OH)2(OBi)(OH) und ähnliche
ein.
- 7. Verschiedenartige andere organische und anorganische Wismutverbindungen:
Diese schließen
zum Beispiel Wismutphosphid (z. B. BiP), Wismutarsenid (Bi3As4), Natriumwismutat
(z. B. NaBiO3), Wismutthiocyanessigsäure (z.
B. H2(Bi(BNS)5)·H3(Bi(CNS)6)), Natriumsalz
der Wismutthiocyanessigsäure,
Kaliumsalz der Wismutthiocyanessigsäure, Trimethylwismuthin (z.
B. Bi(CH3)3), Triphenylwismuthin
(z. B. Bi(C6H5)3), Wismutoxychlorid (z. B. BiOCl), Wismutoxyiodid
(z. B. BiOI) und ähnliche.
-
Bei
einer bevorzugten Ausführungsform
ist die Wismutverbindung ein Wismutoxid, Wismuthydroxid oder Wismutsalz
einer anorganischen Oxysäure.
Die Wismutverbindung ist weiter vorzugsweise Wismutnitrat (z. B.
Bi(NO3)3·5H2O, Wismutylcarbonat (z. B. (BiO)2CO3) oder Wismutoxid
(z. B. Bi2O3), wobei
Wismut(III)-oxid (d. h. Bi2O3)
am meisten bevorzugt ist, weil es keine Gegenionen enthält, welche
das Reaktionsendprodukt verunreinigen können.
-
Geeignete
tellurhaltige Verbindungen schließen zum Beispiel anorganische
oder organische Verbindungen ein, in denen das/die Telluratom(e)
in einer höheren
Oxidationsstufe als 0 (z. B. 2, 3, 4, 6, oder 6), am meisten vorzugsweise
4 vorliegen.
- 1. Telluroxide: diese schließen zum
Beispiel TeO2, Te2O3, Te2O5,
TeO3 und ähnliche ein.
- 2. Tellursalze anorganischer Sauerstofffreier Säuren: Diese
schließen
zum Beispiel Tellurtetrachlorid (z. B. TeCl4),
Tellurtetrabromid (z. B. TeBr4), Tellurtetraiodid
(z. B. TeI4) und ähnliche ein.
- 3. Tellursalze anorganischer Oxysäuren: Diese schließen zum
Beispiel Tellurige Säure
(z. B. H2TeO3),
Tellursäure
(z. B. H2TeO4 oder
Te(OH)6), Tellurnitrat (z. B. Te2O4·HNO3) und ähnliche
ein.
- 4. Verschiedenartige andere organische und anorganische Tellurverbindungen:
Diese schließen
zum Beispiel Dimethyltellurdichlorid, Bleitelluroxid, Tellurisopropoxid,
Ammoniumtellurat, Tellurthioharnstoff und ähnliche ein.
-
Bei
einer bevorzugten Ausführungsform
ist die Tellurverbindung ein Telluroxid oder ein Tellursalz einer anorganischen
sauerstofffreien Säure.
Die Tellurverbindung ist weiter vorzugsweise Tellurdioxid (z. B.
TeO2), Tellurtetrachlorid (z. B. TeCl4) oder Tellursäure (z. B. Te(OH)4),
wobei Tellurtetrachlorid am meisten bevorzugt ist.
-
Die
in die Reaktionszone eingeführte
bevorzugte Menge Zusatzaktivator hängt zum Beispiel von der Masse
des Kohlenstoff-getragenen edelmetallhaltigen Katalysators ab (d.
h. die Gesamtmasse aus Kohlenstoffträger, Edelmetall und irgendwelchen
anderen Katalysatorbestandteilen); der Gesamtmasse der in die Reaktion
eingespeisten Mischung; und der Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats.
-
Im
Allgemeinen beträgt
das Verhältnis
der in den/die Reaktor(en) eingespeisten Masse des Zusatzaktivators
zur Masse des Kohlenstoff-getragenen edelmetallhaltigen Katalysators
vorzugsweise mindestens etwa 1:15.000, weiter vorzugsweise mindestens
etwa 1:5000, sogar weiter vorzugsweise mindestens etwa 1:2500 und
am meisten vorzugsweise mindestens 1:1000. Obwohl es machbar ist,
die vorliegende Erfindung ohne Schädigung der Oxidationsreaktion
zu praktizieren, wenn das Verhältnis
der Masse des Zusatzaktivators zur Masse des Kohlenstoff-getragenen,
edelmetallhaltigen Katalysators in der Größe von etwa 1:750, etwa 1:500,
etwa 1:300 und sogar in der Größe von etwa
1:50 oder 1:40 liegt, haben sich die bevorzugten, vorstehend beschriebenen
kleineren Verhältnisse
für die
meisten Anwendungen, und insbesondere für die Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats,
als wirksam erwiesen.
-
Das
Verhältnis
der Masse des Zusatzaktivators zur der in den Reaktor eingespeisten
die Reaktionsgesamtmasse beträgt
mindestens etwa 1:1.000.000, weiter vorzugsweise mindestens etwa
1:100.000, sogar weiter vorzugsweise mindestens etwa 1:40.000 und
am meisten vorzugsweise etwa 1:40000 bis etwa 1:15.000.
-
Obwohl
Verhältnisse
größer als
1:8000 normalerweise ohne Schädigung
der Oxidationsreaktion verwendet werden können, wird im Allgemeinen ein
Verhältnis
kleiner als 1:8000 bevorzugt (insbesondere mit Wismut als Zusatzaktivator).
-
Das
Verhältnis
der Masse des Zusatzaktivators zur der in den Reaktor eingespeisten
Masse des N-(Phosphonomethyl)iminodiessigsäuresubstrats beträgt vorzugsweise
mindestens etwa 1:100.000, weiter vorzugsweise 1:10.000, sogar weiter
vorzugsweise wenigstens etwa 1:4000 und am meisten vorzugsweise etwa
1:4000 bis etwa 1:2000. Obwohl Verhältnisse größer als 1:1000 normalerweise
ohne Schädigung
der Oxidationsreaktion verwendet werden können, wird im Allgemeinen ein
Verhältnis
kleiner als 1:1000 bevorzugt (insbesondere wenn der Zusatzaktivator
Wismut ist).
-
Wo
ein besonderes Edelmetall auf einem Kohlenstoffkatalysator zur Oxidation
eines N-(Phosphonomethyl)iminodiessigsäuresubstrats verwendet wird,
kann sowohl der Katalysator als auch der Zusatzaktivator in ein
wässriges
Reaktionsmedium eingespeist werden, welches das N-(Phosphonomethyl)iminodiessigsäuresubstrat
und Sauerstoff enthält.
Der Zusatzaktivator kann in einem Massenverhältnis von wenigstens etwa 1:15.000,
vorzugsweise von wenigstens etwa 1:5000, weiter vorzugsweise von
wenigstens etwa 1:2500 und am meisten vorzugsweise von mindestens
1:1000 eingespeist werden. Mit fortschreitender Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats
werden Formaldehyd und Ameisensäure
als Nebenprodukte erzeugt. Der Katalysator oxidiert nicht nur das
N-(Phosphonomethyl)iminodiessigsäuresubstrat
wirksam, sondern auch die weitere Oxidation von Formaldehyd zu Ameisensäure sowie
die von Ameisensäure
zu Kohlendioxid. Die Anwesenheit des Zusatzaktivators wirkt sich
auf die katalytische Oxidation dieser Nebenprodukte, insbesondere
auf die Umwandlung von Ameisensäure
zu CO2 fördernd
aus.
-
Wo
die Oxidationsreaktion in einem Rührbehälter als Reaktor erfolgt, in
dem der Katalysator im Reaktionsmedium aufgeschlämmt ist, wird der Katalysator
vorzugsweise durch Filtration aus der Reaktionsmischung abgetrennt
und für
die weitere Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats
und die oben erwähnten
Nebenprodukte in den Reaktor zurückgeführt. Ein
solches Rührbehälterreaktorsystem
kann entweder chargenweise oder kontinuierlich betrieben werden.
Alternativ kann ein Fest- oder Fließbettkatalysator verwendet
werden. Bei einem kontinuierlichen Verfahren wird das N-(Phosphonomethyl)iminodiessigsäuresubstrat,
der Formaldehyd und die Ameisensäure
gemeinsam in einer kontinuierlichen Reaktionszone oxidiert, die
mit einem wässrigen
Reaktionsmedium, welches das N-(Phosphonomethyl)iminodiessigsäuresubstrat
umfasst, kontinuierlich oder diskontinuierlich mit einer Reaktionsmischung
beschickt wird und eine Reaktionsmischung, welche ein N-(Phosphonomethyl)glycinprodukt
umfasst, kontinuierlich oder diskontinuierlich abgezogen wird und
der Zusatzaktivator kontinuierlich oder diskontinuierlich in die
Reaktionszone eingespeist wird.
-
Es
ist beobachtet worden, dass die Zugabe einer einzelnen Charge von
Zusatzaktivator zum ersten Ansatz einer Reihe aufeinander folgender
Chargenreaktorzyklen den Katalysators für die Oxidation von Formaldehyd
und Ameisensäure über eine
Reihe von Reaktionszyklen ohne weitere Zugabe von Zusatzaktivator aus
einer externen Quelle verstärkt.
Es ist weiterhin beobachtet worden, dass der im rezyklierten Katalysator vorliegende
Zusatzaktivator darauf offensichtlich durch Adsorption auf dem Edelmetall
und/oder dem Kohlenstoffträger
abgeschieden worden ist. Es kann nach mehrfachen Zyklen nur ein
Bruchteil des Zusatzaktivators, welcher der ersten Charge der Reihe
zugesetzt wurde, auf dem Katalysator wiedergefunden werden. Wird
jedoch der Zusatzaktivator in den vorstehend beschriebenen Mengen
in die erste Charge eingeführt,
reicht der auf dem Katalysator verbleibende Bruchteil offensichtlich
aus, die Oxidation von Formaldehyd und Ameisensäure über die ganze Serie von Chargen
hinweg zu bewerkstelligen, in denen der Katalysator aus einem früheren Ansatz
im Wesentlichen die einzige Quelle für den Zusatzaktivator für die Ansätze der
Reaktionszyklusserie ist. Es ist gefunden worden, dass eine Einmalzugabe
des Zusatzaktivators im Massenverhältnis zum Katalysator von ungefähr 1:2500
wirksam ist, um die Nebenproduktoxidation in einer Reihe von 20
oder mehr, typischerweise 50 oder mehr, noch typischer 100 Chargenreaktionszyklen
zu bewerkstelligen. Danach kann dem Reaktionsmedium wahlweise eine
weitere diskrete Charge Zusatzaktivator zugegeben werden, um eine nachfolgende
Charge zu einer ersten einer anderen Serie von Oxidationsreaktionschargenzyklen
zu machen, in denen der rezyklierte Katalysator aus einem vorangehenden
Ansatz einer solchen weiteren Serie zur im Wesentlichen einzigen
Aktivatorquelle für
die anschließenden
Chargenreaktionszyklen der weiteren Serie von Chargenreaktionen
wird.
-
Wird
der Zusatzaktivator in ähnlicher
Weise dem Reaktionsmedium in einem kontinuierlichen Rührbehälterreaktor
zugesetzt, so ist die Zugabe des Zusatzaktivators in einer einzigen
diskreten Menge wirksam, um die Effizienz des Katalysator zur Formaldehyd-
und Ameisensäureoxidation
während
mehrfacher Umläufe
eines kontinuierlichen Laufs zu verstärken. Es erfolgt keine weitere
Zugabe von Zusatzaktivator bis zum Beginn eines zweiten Reaktionslaufs.
Aus diesem Grunde besteht ein Reaktionslauf aus der Zeitspanne für die Oxidation
von Formaldehyd und Ameisensäure,
beginnend mit dem Zeitpunkt der Zugabe irgendeiner diskreten Zugabe
von Zusatzaktivator zur Reaktionszone bis zum Zeitpunkt der folgenden
Zusatzaktivatorzugabe zur Reaktionszone, welche aus 50 oder mehr,
typischerweise 100 oder mehr, Umläufen des Arbeitsvolumens des Reaktors
bestehen kann.
-
Wie
erwähnt,
verbleibt nach mehrfachen Zyklen einer Serie von Chargenreaktionsläufen, oder
nach mehrfachen Umläufen
eines kontinuierlichen Reaktionslaufs nur ein Bruchteil des dem
ersten Ansatz des Zyklus zugesetzten Zusatzaktivators auf dem Katalysator.
Der Zusatzaktivator behält
jedoch seine Wirksamkeit, um die Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats,
des Formaldehyds und der Ameisensäure zu verstärken, wenn
das Substrat mit dem Oxidationsmittel in einer Reaktionszone in
Berührung
gebracht wird, welche das flüssige
Reaktionsmedium umfasst, wenn das Massenverhältnis des Zusatzaktivators zum
Katalysator in einer solchen Reaktionszone mindestens etwa 1:200.000,
vorzugsweise mindestens etwa 1:70.000, weiter vorzugsweise mindestens
etwa 1:30.000, am meisten vorzugsweise mindestens etwa 1:15.000
beträgt.
Insofern als rezyklierter Katalysator die im Wesentlichen einzige
Zusatzaktivatorquelle für den
Reaktor sein kann, ist es weiter bevorzugt, dass der Zusatzaktivator
im gleichen Massenverhältnis
auf oder im rezyklierten Katalysator vorliegt, d. h. mindestens
etwa 1.200.000, vorzugsweise mindestens etwa 1:70.000, weiter vorzugsweise
mindestens etwa 1:30.000, am meisten vorzugsweise mindestens etwa 1:15.000
beträgt.
-
Der
Zusatzaktivatorgehalt in der Reaktionszone kann auch als Massenverhältnis zur
Edelmetallkomponente des Katalysators ausgedrückt werden. Zum Beispiel sollte
für 5%
Edelmetall auf einem Kohlenstoffkatalysator das Verhältnis von
Zusatzaktivator zu Edelmetall mindestens etwa 1:10.000, weiter vorzugsweise 1:3500,
weiter vorzugsweise 1:1800, am meisten vorzugsweise 1:700 betragen.
Diese Bevorzugung des Edelmetallgehalts auf Kohlenstoffkatalysatoren
gilt typischerweise für
den Bereich von etwa 0,5 bis 20% Edelmetall. Wo jedoch der Edelmetallgehalt
relativ hoch ist, z. B. näherungsweise
20%, kann der Zusatzaktivator bei relativ niedrigeren Massenverhältnissen
gegenüber
der Edelmetallkomponente, sogar bis herab zu 1:40.000, wirksam sein.
-
Wird
ein Zusatzaktivator zu einer diskreten Charge zu Beginn einer Reihe
von Chargenreaktionszyklen oder zu Beginn eines kontinuierlichen
Reaktionslaufs zugegeben, wie vorstehend definiert, wird er im Massenverhältnis zur
Edelmetallkomponente des Katalysators von mindestens etwa 1:750,
vorzugsweise von mindestens etwa 1:250, weiter vorzugsweise von
mindestens etwa 1:125, am meisten vorzugsweise von mindestens 1:50
zugegeben. Das bevorzugte Verhältnis
von Zusatzaktivator zu Edelmetall kann, wie vorstehend angegeben,
mit dem Edelmetallgehalt des Katalysators stark schwanken. Nähert sich
der Edelmetallgehalt des Katalysators z. B. 20 Gew.-% an, kann der
Zusatzaktivator folglich wirksam sein, wenn er in einem Massenverhältnis zum
Edelmetall von 1:3000 oder höher,
weiter vorzugsweise von mindestens etwa 1:1000, 1:500 oder 1:200
zugegeben wird.
-
Periodische
Einzelzugaben des Zusatzaktivators können von Vorteil sein, weil Überschussmengen
an Zusatzaktivator die Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats
verzögern
können,
während
sie die Wirksamkeit des Katalysators für die Oxidation von Formaldehyd
und Ameisensäure
maximieren. Wird der Zusatzaktivator nur periodisch zugegeben, können die
auf dem Katalysator abgeschiedenen und in der Reaktionszone vorliegenden
Zusatzaktivator-gehalte
ziemlich rasch bis zu einer quasi-stationären Restmenge abnehmen, bei
welcher der Zusatzaktivator wirksam bleibt, um die katalytische
Aktivität
für die
Oxidation von Formaldehyd oder Ameisensäure zu erhöhen ohne die Geschwindigkeit
oder den Grad der Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats
beträchtlich
zu verzögern.
Demzufolge kann der optimale Gehalt an Zusatzaktivator innerhalb
der Oxidationsreaktionszone für
die Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats
und auf dem rezyklierten Katalysator für eine solche Reaktion geringer
als 1:15.000 sein und zum Beispiel in einem Bereich von 1:65.000
bis 1:25.000 liegen.
-
Die
Abscheidung des Zusatzaktivators auf der Oberfläche eines Edelmetalls auf einem
Kohlenstoffkatalysator im Reaktionsmedium führt zur Bildung eines neuen
Katalysatorkomplexes, welcher den Katalysator und den Aktivator
umfasst. Die Katalysatorkomponente des Katalysatorkomplexes kann
weiterhin einen Oberflächenaktivator
umfassen, welcher ein vom Zusatzaktivator verschiedenes Metall umfasst,
oder in manchen Fällen,
das gleiche Metall umfasst. Es wird angenommen, dass der Zusatzaktivator
durch Adsorption aus dem Reaktionsmedium abgeschieden wird und von
der Katalysatoroberfläche
in das Katalysatormedium desorbierbar bleibt. Während eine operative Fraktion
des Zusatzaktivatorrests der Desorption widersteht, um auf dem Katalysator über mehrfache
Reaktionszyklen haften zu bleiben (oder durch einen ausgedehnten
Lauf eines kontinuierlichen Reaktionssystems), wie hierin vorstehend
erläutert,
ist der Zusatzaktivator typischerweise leichter desorbierbar als
der Oberflächenaktivator,
welcher beim Katalysatorherstellungsverfahren verwendet wird.
-
Der
Katalysator wird, wie vorstehend beschrieben, in erster Stufe durch
Abscheiden von Edelmetall und wahlweise eines Oberflächenaktivators
auf einem Kohlenstoffträger
hergestellt, um eine Katalysatorvorstufe zu bilden und die Katalysatorvorstufe
dann reduziert, um den Reaktionskatalysator zu erzeugen. Der neue
Katalysatorkomplex wird durch anschließende Abscheidung eines Zusatzaktivators
auf dem Oxidationskatalysator, typischerweise durch Adsorption auf
der Kohlenstoff- oder Edelmetalloberfläche, gebildet. Der Zusatzaktivator
wird vorteilhafterweise mit dem Oxidationskatalysator im Reaktionsmedium
gemischt, so dass der Aktivator aus dem Reaktionsmedium auf der
Katalysatoroberfläche
abgeschieden wird. Alternativ kann der Zusatzaktivator jedoch selbstverständlich mit
dem Oxidationskatalysator in einem anderen flüssigen Medium vorgemischt werden,
um den Katalysatorkomplex zu bilden, wonach der Katalysatorkomplex
in das Reaktionsmedium zur Verwendung bei der Durchführung der
Oxidationsreaktion eingeführt
werden kann.
-
In
Abhängigkeit
von der gewünschten
Wirkung kann selbstverständlich
mehr als ein Zusatzaktivator verwendet werden. Zusätzlich kann
jeder Zusatzaktivator aus mehr als einer Quelle stammen. Weiterhin
kann der Kohlenstoff-getragene, edelmetallhaltige Katalysator schon
eine Menge Metall auf seiner Oberfläche enthalten, welches das
gleiche Metall ist wie das des Zusatzaktivators, wie in den Fällen, in
denen (a) der Katalysator mit einem solchen Metall auf seiner Oberfläche hergestellt
wird, um als Katalysatoroberflächenaktivator zu
fungieren oder (b) der Katalysator ein gebrauchter Katalysator ist,
welcher aus einer früheren
Reaktionsmischung, in welcher das Metall vorlag (z. B. als Zusatzaktivator),
wiedergewonnen worden ist.
-
Bei
einer besonders bevorzugten Ausführungsform
umfasst der Kohlenstoff-getragene,
edelmetallhaltige Katalysator auch selbst auf seiner Oberfläche einen
oder mehrere Katalysatoroberflächenaktivatoren,
wie vorstehend beschrieben. In den Fällen, in denen der Katalysator
zur Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
verwendet wird und der Zusatzaktivator Wismut ist, ist besonders
bevorzugt, das der Katalysator Zinn und/oder Eisen enthält (Die Anwesenheit
von Zinn tendiert dazu die Oxidation des Formaldehyd-Nebenprodukts
zu erhöhen
und zusätzlich
die Oxidation des Ameisensäure-Nebenprodukts
zu erhöhen).
-
In
vielen Fällen
scheidet sich dann, wenn ein Zusatzaktivator (insbesondere Wismut)
und ein Kohlenstoff-getragener, edelmetallhaltiger Katalysator kombiniert
worden sind, wenigstens eine Teil des Zusatzaktivators auf der Oberfläche des
Kohlenstoffträgers
und/oder des Edelmetalls des Katalysators ab und wird folglich vom
Katalysator zurückgehalten.
Weil der Katalysator den Aktivator zurückhält, kann der Katalysator typischerweise
für die
Verwendung zur Katalyse der Oxidation nachfolgender Mengen des Oxidationssubstrats
rezykliert werden (z. B. kann der Katalysator verwendet werden,
um zusätzliche
Chargen des Oxidationssubstrats zu oxidieren, oder er kann bei einem
kontinuierlichen Oxidationsverfahren verwendet werden), während er
immer noch die Vorteile des Zusatzaktivators beibehält. Und
so wie die Wirkung des Zusatzkatalysators im Verlauf der Verwendung
abnimmt, können
Ersatzmengen frischen Zusatzaktivators periodisch mit dem Katalysator
gemischt werden, um die Wirkungen aufzufrischen und/oder andere
erwünschte
Ergebnisse zu erzielen (z. B. das Ameisensäureniveau zu senken). Dort
wo der Katalysator zum Beispiel bei mehrfachen Chargenreaktionen
verwendet wird, kann eine solche periodische Auffrischung zum Beispiel
erfolgen nachdem der Katalysator bei der Oxidation von mindestens
etwa 20 Chargenreaktionen verwendet worden ist (weiter vorzugsweise
nachdem er bei der Oxidation von mindestens etwa 30 Chargenreaktionen
verwendet worden ist, und am meisten vorzugsweise nachdem er bei
der Oxidation von mindestens etwa 100 Chargenreaktionen verwendet
worden ist). Dort wo ein Katalysator periodisch mit frischem Zusatzaktivator
aufgefrischt wird, kann das Mischen zur Auffrischung in der/den
Oxidationsreaktionszone(n) oder getrennt davon erfolgen.
-
Bei
einer besonders bevorzugten Ausführungsform
wird ein Zusatzaktivator mit einem gebrauchten Katalysator gemischt
(d. h. einem Katalysator, welcher bei einer oder mehreren vorangehenden
Oxidationsreaktionen benützt
worden war). Typischerweise nimmt die Aktivität und/oder die gewünschte Selektivität bei der Verwendung
ab. Demzufolge tendiert zum Beispiel die Aktivität eines Kohlenstoff-getragenen, edelmetallhaltigen
Katalysators für
die Oxidation von Nebenprodukten (z. B. Formaldehyd und/oder Ameisensäure) der
Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats dazu, bei der
Verwendung des Katalysators abzunehmen, und verursacht dadurch,
dass weniger Formaldehyd und/ oder Ameisensäure abgebaut und folglich eine
größere Menge
N-Methyl-N-(Phosphonomethyl)glycin erzeugt wird. Diese Aktivität nimmt
unter Umständen
in der Tat bis zu einem Grad ab, bei dem eine nicht annehmbare Menge
Formaldehyd und/oder Ameisensäure
oxidiert wird, was folglich oft die Erzeugung einer nicht annehmbare
Menge N-Methyl-N-(Phosphonomethyl)glycinverbindungen verursacht
(d. h. die Selektivität
des Katalysators für
die Herstellung von N-(Phosphonomethyl)glycinverbindungen aus N-(Phosphonomethyl)iminodiessigsäuresubstraten
nimmt auf ein nicht vertretbares Niveau ab). Wenn die Katalysatoraktivität für die Oxidation
der Nebenprodukte einen solchen Punkt erreicht, ist der Katalysator
traditionell für
unbrauchbar gehalten worden, und ist folglich entweder durch ein
zeitaufwendiges und manchmal teueres Verfahren rezykliert (d. h.
reaktiviert) oder als Ganzes verworfen worden. In Übereinstimmung
mit dieser Erfindung ist gefunden worden, dass ein solcher Katalysator
durch Mischen des Katalysators mit einem Zusatzaktivator, insbesondere
Wismut oder Tellur, aufgefrischt werden kann (d. h. die Selektivität des Katalysators
für die
Herstellung des N-(Phosphonomethyl)glycinprodukts kann bis auf ein
annehmbares Niveau angehoben werden). Der Zusatzaktivator kann mit
anderen Worten verwendet werden, um die Katalysatorleistung zu modifizieren
und die Lebensdauer des Katalysators zu verlängern.
-
Es
ist beobachtet worden, dass ein Zusatzaktivator (insbesondere Wismut)
einen leichten Abfall der Oxidationsgeschwindigkeit eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
verursachen kann. In einem solchen Fall kann die Oxidationsgeschwindigkeit
typischerweise zumindest teilweise erhöht werden, indem die in die
Reaktionsmischung eingespeiste Sauerstoffmenge erhöht wird
und während
eines längeren Zeitraums
während
der Reaktion eine relativ hohe Sauerstoffströmungsrate aufrechterhalten
und/oder der Druck erhöht
wird. Wird jedoch die Sauerstoffströmung erhöht, wird sie vorzugsweise nicht
bis zu einem Grad erhöht
der dazu führt,
dass die Katalysatoroberfläche
in schädlicher
Weise überoxidiert
wird. Die erhöhte
Sauerstoffeinspeiserate wird folglich vorzugsweise auf einem solchen
Niveau gehalten, dass mindestens etwa 40% (weiter vorzugsweise mindestens
etwa 60%, sogar weiter vorzugsweise mindestens 80% und am meisten vorzugsweise
mindestens etwa 90%) des eingespeisten Sauerstoffs verbraucht wird.
-
Bevorzugte
Oxidationsreaktorsysteme
-
Die
Oxidationsreaktionszone(n) kann/können eine breite Reihe von
Reaktorkonfigurationen umfassen, einschließend jene mit Rückvermischungseigenschaften, sowohl
in der flüssigen
Phase als auch wahlweise in der Gasphase, und jene, welche Pfropfströmungseigenschaften
aufweisen. Geeignete Reaktorkonfigurationen mit Rückvermischungseigenschaften
schließen
zum Beispiel Rührbehälterreaktoren,
Ausstoßdüsenkreislaufreaktoren
(auch als Venturi-Kreislaufreaktoren bekannt) und Wirbelbettreaktoren.
Geeignete Reaktorkonfigurationen mit Pfropfströmungseigenschaften schließen jene,
welche ein gepacktes oder festes Katalysatorbett (z. B. Rieselbettreaktoren
und gepackte Blasensäulenreaktoren)
und Blasenaufschlämmungssäulenreaktoren
aufweisen. Wirbelbettreaktoren können
ebenfalls in einer solchen Weise betrieben werden, dass sie Pfropfströmungseigenschaften
aufweisen.
-
In
einem breiten Sinn kann die Oxidationsreaktion in Übereinstimmung
mit der vorliegenden Erfindung in einem weiten Temperaturbereich
und bei Drücken
praktiziert werden, die von unteratmosphärisch bis überatmosphärisch reichen. Die Verwendung
milder Bedingungen (z. B. Raumtemperatur und Atmosphärendruck) hat
insofern wirtschaftliche Vorteile, als im Reaktorsystem eine kostengünstigere
Ausrüstung
verwendet werden kann. Der Betrieb bei höheren Temperaturen und überatmosphärischen
Drücken,
tendiert jedoch dazu den Stoffübergang
zwischen der flüssigen
Phase und der Gasphase (z. B. der Sauerstoffquelle) zu verbessern,
die Reaktionsgeschwindigkeit der N-(Phosphonomethyl)iminodiessigsäureoxidation
zu steigern und die Löslichkeit
des N-(Phosphonomethyl)glycinprodukts zu erhöhen, und dadurch die zur Trennung
durch Ausfällung
und zur Isolierung des Produkts erforderliche Wassermenge zu verringern.
Entsprechend kann die Verwendung aggressiverer Oxidationsbedingungen
die Anlagengesamtkosten tatsächlich
verringern und die Betriebskosten pro erzeugte Einheit N-(Phosphonomethyl)glycinprodukt
reduzieren. Die N-(Phosphonomethyl)iminodiessigsäureoxidation erfolgt vorzugsweise
bei einer Temperatur von etwa 20°C
bis etwa 180°C,
weiter vorzugsweise bei etwa 50°C
bis etwa 140°C,
noch weiter vorzugsweise bei etwa 80°C bis etwa 110°C und sogar
noch weiter vorzugsweise bei etwa 95°C bis etwa 105°C. Bei Temperaturen
größer als
etwa 180°C
neigt das Einspeisematerial dazu sich langsam zu zersetzen. Darüber hinaus
tendiert die Selektivität
im Hinblick auf das gewünschte
N-(Phosphonomethyl)glycinprodukt dazu sich zu verschlechtern, sobald
die Oxidationsreaktionstemperatur stark über etwa 90°C ansteigt. Zum Beispiel neigt
die Erzeugung des unerwünschten
Nebenprodukts N-Methyl-N-(Phosphonomethyl)glycin (NMG) bei über 90°C dazu, bei
jeder Erhöhung
der Reaktionstemperatur um 10°C
näherungsweise
um das 2- bis 4fache zuzunehmen. Niedrige Temperaturen (d. h. Temperaturen
von weniger als etwa 95°C)
neigen dazu weniger vorteilhaft zu sein, weil die Löslichkeit
mancher N-(Phosphonomethyl)iminodiessigsäuresubstrate und N-(Phosphonomethyl)glycinprodukte
bei solchen Temperaturen verringert ist. Der in der/den Oxidationsreaktionszone(n)
aufrechterhaltene Gesamtdruck hängt
im Allgemeinen von der verwendeten Temperatur und der Reaktorkonfiguration
ab. Der Gesamtdruck in jeder Oxidationsreaktionszone ist vorzugsweise
gleich dem Atmosphärendruck
und reicht aus, um das flüssige
Reaktionsmedium in der Oxidationszone am Sieden zu hindern. Bevorzugte
Oxidationsreaktionsbedingungen für spezielle
Reaktorsysteme werden nachstehend ausführlicher erörtert.
-
Bei
einer bevorzugten Ausführungsform
erfolgt die Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats
in einer oder mehreren kontinuierlichen Oxidationsreaktionszonen,
worin das Substrat kontinuierlich zum N-(Phosphonomethyl)glycinprodukt
oxidiert wird. Die kontinuierliche Oxidation bietet die Möglichkeit
zu größerem Verfahrensdurchsatz
und geringeren Herstellungskosten. Weil die Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats
exotherm verläuft,
ist darüber
hinaus nach Inbetriebnahme eines kontinuierlichen Oxidationsreaktorsystems
typischerweise keine Wärmezufuhr
zu dem in die Oxidationszone eingeführten wässrigen Einspeisestrom nötig, um
die gewünschte
Oxidationsreaktionstemperatur aufrechtzuerhalten.
-
Es
können
geeigneterweise verschiedene Reaktorkonfigurationen eingesetzt werden,
um die kontinuierliche(n) Oxidationsreaktionszone(n) bereitzustellen.
In Übereinstimmung
mit einer bevorzugten Ausführungsform
erfolgt die kontinuierliche Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats
in einer oder mehreren Oxidationsreaktionszonen mit Rückvermischung
(d. h. Rückvermischung
in wenigstens der flüssigen
Phase) unter Verwendung eines heterogenen teilchenförmigen Katalysators,
vorzugsweise des vorstehend beschriebenen Katalysator mit tiefenreduziertem
Edelmetall auf teilchenförmigem
Kohlenstoff, der in Berührung
mit dem flüssigen
Reaktionsmedium suspendiert ist. Die Praktizierung der vorliegenden
Erfindung ist jedoch selbstverständlich
weder auf die Verwendung eines solchen tiefenreduzierten Katalysators
noch auf einen teilchenförmigen
Katalysator beschränkt.
Darüber
hinaus kann der im Reaktorsystem der vorliegenden Erfindung verwendete
Katalysator selbstverständlich
eine Mischung unterschiedlicher Katalysatoren umfassen und/oder
der Katalysator kann im Reaktorsystem von einer Oxidationsreaktionszone
zur nächsten
variieren.
-
2 zeigt
ein Beispiel eines Reaktorsystems, welches verwendet werden kann,
die kontinuierlichen Oxidationsprozesse der vorliegenden Erfindung
durchzuführen.
Das in 2 dargestellte System umfasst
einen kontinuierlichen Rührbehälterreaktor 3,
welcher für
das mechanische Rühren
des darin enthaltenen flüssigen
Reaktionsmediums, typischerweise mit einem Flügelrührer, sorgt. Rührbehälterreaktoren,
welche für
das Rückvermischen
der flüssigen
Phase in de Reaktionszone geeignet sind, weisen eine relativ einfache
Auslegung auf und ihr Betrieb kann auf die gewünschte Prozesskapazität gesteigert
werden. Es können
verschiedene Rührflügelausführungen
eingesetzt werden, einschließend
Systeme mit mehreren Blätter
an einer gemeinsamer Welle. Der Reaktorbehälter kann innere Wellenbrecher
und/oder Leitrohre einschließen,
um die Mischungseigenschaften zu modifizieren und die Wirbelbildung
des flüssigen
Reaktionsmediums zu verhindern, was dem Fachmann wohlbekannt ist.
-
Obwohl
das in 2 dargestellte Reaktorsystem
einen einzelnen kontinuierlichen Rührbehälterreaktor umfasst, ist in
vielen Fällen
ein Reaktorsystem bevorzugt, welches zwei oder mehr Oxidationsreaktionszonen
mit Rückvermischung
in Reihe umfasst, wie nachstehend ausführlicher beschrieben. Die Oxidationsreaktionszone(n)
mit Rückvermischung
können
in geeigneter Weise mit anderen Reaktorkonfigurationen als kontinuierliche
Rührbehälterreaktoren
vorgesehen werden (z. B. Ausstoßdüsenkreislaufreaktoren
und Wirbelbettreaktoren). Darüber
hinaus können
in einem Reaktorsystem, welches mehrfache Oxidationsreaktionszonen
umfasst, unterschiedliche Reaktorkonfigurationen kombiniert werden.
Zum Beispiel kann/können
ein oder mehrere Reaktor(en) mit Rückvermischungseigenschaften
mit einer Reaktorkonfiguration mit Pfropfströmungseigenschaften, wie einem
Katalysatorfestbettreaktor, kombiniert werden.
-
Ein
wässriger
Einspeisestrom 1, der das N-(Phosphonomethyl)iminodiessigsäuresubstrat
umfasst, wird zusammen mit einer Sauerstoffquelle kontinuierlich
oder diskontinuierlich in ein im Rührbehälterreaktor 3 befindliches
flüssige
Reaktionsmedium eingespeist. Der heterogene teilchenförmige Katalysator,
welcher dazu verwendet wird, die Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats
im wässrigen
Ansatz zu katalysieren, liegt in der Oxidationsreaktionszone in
Berührung
mit dem flüssigen
Reaktionsmedium ebenfalls vor. CO2-haltiger Dampf, der
sich mit Fortschreiten der Oxidationsreaktion entwickelt, wird aus
dem Kopfraum über
der Reaktionsmischung im Rührbehälterreaktor 3 abgelüftet. Ein
Reaktionsmischungsablauf 7, welcher das N-(Phosphonomethyl)glycinprodukt
und den heterogenen teilchenförmigen
Katalysator enthält,
wird kontinuierlich oder diskontinuierlich aus dem Rührbehälterreaktor 3 abgezogen
und zu einem Katalysatorfilter 9 weitergeleitet, in dem
im Wesentlichen der gesamte Katalysator aus der Reaktionsmischung
abgetrennt wird unter Bildung von: (1) eines Katalysatorrücklaufstroms 11,
welcher im Wesentlichen den gesamten Katalysator und Restmengen
des N-(Phosphonomethyl)glycinprodukts umfasst, und (2) eines Filtrats 13,
welches den Hauptanteil des N-(Phosphonomethyl)glycinprodukts enthält. Der
Katalysatorrücklaufstrom 11 wird
wieder in den Rührbehälterreaktor 3 eingespeist,
während
das Filtrat 13 weitergeleitet wird, um das N-(Phosphonomethyl)glycinprodukt
zu konzentrieren und zu reinigen.
-
Die
Temperatur innerhalb der Oxidationsreaktionszone wird im Hinblick
auf die Konzentration des N-(Phosphonomethyl)glycinprodukts vorzugsweise
ausreichend hoch gehalten, dermaßen, dass im Wesentlichen das
gesamte, aus dem Rührbehälterreaktor 3 über den
Reaktionsmischungsauslass 7 abgezogene N-(Phosphonomethyl)glycinprodukt
gelöst
ist. Ist demzufolge zum Beispiel das N-(Phosphonomethyl)glycinprodukt die freie
Säure des
N-(Phosphonomethyl)glycins, welche in einer Konzentration von etwa
7 bis etwa 15 Gew.-% vorliegt, wird eine Temperatur des aus dem
Rührreaktor 3 abgezogenen
Reaktionsmischungsablauf von etwa 80°C bis etwa 180°C, weiter
vorzugsweise von etwa 90°C
bis etwa 150°C,
weiter vorzugsweise von etwa 90°C
bis etwa 135°C,
sogar weiter vorzugsweise von etwa 95°C bis etwa 125°C und noch
weiter von etwa 95°C
bis etwa 105°C
aufrechterhalten. Die Ausfällung
des N-(Phosphonomethyl)glycinprodukts im Reaktionsmischungsablauf 7 kann
jedoch selbstverständlich
toleriert werden und dennoch zufrieden stellende Ergebnisse erhalten
werden. Das ausgefällte
N-(Phosphonomethyl)glycinprodukt kann mit dem teilchenförmigen Katalysator
vom Rest des Reaktionsmischungsablaufs 7 abgetrennt (z.
B. gemeinsam filtriert) werden.
-
Beim
Anfahren kann die Reaktionsmischung in der Oxidationsreaktionszone 3 und/oder
der wässrige Einspeisestrom 1 aufgeheizt
werden, um die gewünschte
Temperatur für
die Oxidationsreaktion zu erhalten. Wenn Wärme zugeführt werden muss, kann die gesamte
oder wenigstens ein Teil der Wärmeenergie
durch das Pumpen der verschiedenen Einspeiseströme in den Rührbehälterreaktor 3 und
durch das übrige
Reaktorsystem in einer Weise bereitgestellt werden, dass ein separater
herkömmlicher
Zulaufvorerhitzer nicht erforderlich sein muss. Weil die Oxidationsreaktion
exotherm ist, wird es normalerweise nötig sein Wärmeenergie aus der Reaktionsmischung
abzuführen
sobald die Oxida tionsreaktion beginnt, beträchtliche Wärmemengen zu entwickeln, um
innerhalb der Reaktionszone die gewünschte Temperatur aufrechtzuerhalten. Überschüssige Reaktionswärme kann,
wie in 2 dargestellt, der Reaktionsmischung
im Rührbehälterreaktor 3 entzogen werden,
indem die Reaktionsmischung durch einen externen Wärmeübergangskreislauf 15 geleitet
wird, welcher einen Wärmetauscher 16 enthält, in dem
die Wärme
indirekt von der Reaktionsmischung auf ein Kühlmedium (z. B. Kühlwasser) übertragen
wird. Die Reaktionstemperatur wird zum Beispiel durch Regelung der
Kühlwasserzufuhr
zum Wärmetauscher 16 als
Reaktion des Signals von einem Temperaturregler geregelt. Reaktionswärme kann
aus der Oxidationsreaktionszone ebenso mittels anderer herkömmlicher
Mittel abgeführt
werden, wie in die Reaktionsmischung eingetauchte Kühlschlangen
oder einen Reaktorbehältermantel,
durch den ein Kühlmedium
im Kreislauf geführt
wird.
-
Der
Gesamtdruck im Rührbehälterreaktor 3 beträgt im Allgemeinen
etwa 0 bis etwa 3.500 kPa (500 psig) und wird vorzugsweise ausreichend
hoch gehalten, um das flüssige
Reaktionsmedium darin am Sieden zu hindern. Der Gesamtdruck im Rührbehälterreaktor 3 beträgt etwa
207 kPa (30 psig) bis etwa 3.500 kPa (500 psig). Wird die Oxidationsreaktionszone
im besonders bevorzugten Temperaturbereichs von etwa 95°C bis etwa
105°C betrieben,
beträgt
der im Rührreaktor 3 aufrechterhaltene
Druck etwa 207 kPa (30 psig) bis etwa 896 kPa (130 psig) und weiter
vorzugsweise etwa 612 kPa (90 psig) bis etwa 758 kPa (110 psig).
-
Im
wässrigen
Einspeisestrom 1 kann eine breite Vielfalt von N-(Phosphonomethyl)diessigsäuresubstraten
verwendet werden. Der wässrige
Einspeisestrom 1 schließt den Katalyatorrücklaufstrom 11 und
irgendwelche anderen Rezirkulationsströme von anderen Verfahrensteilen
ein, die in den Rührbehälterreaktor 3 eingeführt werden.
In Aufschlämmungsreaktoren,
wie den in 2 dargestellten Rührbehälterreaktor 3,
wird die Substratkonzentration im wässrigen Einspeisestrom 1 vorzugsweise
im Hinblick auf die Temperatur des Reaktionsmischungsablaufs 7 so
gewählt,
dass im Wesentlichen das gesamte erwünschte N-(Phosphonomethyl)glycinprodukt
gelöst
wird. Wie vorstehend erwähnt,
können
Substratkonzentrationen, welche N-(Phosphonomethyl)glycinprodukt
in Konzentrationen enthalten, welche die Löslichkeitsgrenze des Produkts überschreiten
ebenfalls eingesetzt werden, sind jedoch im Allgemeinen weniger
bevorzugt. Verglichen mit vielen anderen herkömmlich praktizierten kommerziellen
Verfahren erlaubt diese Erfindung die Verwendung von höheren Temperaturen
und N-(Phosphonomethyl)iminodiessigsäuresubstratkonzentrationen
zur Herstellung von N-(Phosphonomethyl)glycin bei gleichzeitiger
Beschränkung
der Bildung von Nebenprodukten auf ein Mindestmaß. Bei herkömmlich praktizierten kommerziellen
Verfahren unter ausschließlicher
Verwendung von Kohlenstoffkatalysatoren, ist es oft wirtschaftlich
vorzuziehen gewesen, diese bei niedrigen Substratkonzentrationen
und Temperaturen zu betreiben, um die Bildung von N-Methyl-N-(phosphonomethyl)glycin
als Nebenprodukt zu minimieren. Mit jenen Verfahren und Katalysatoren
werden typischerweise Temperaturen von etwa 60°C bis 90°C verwendet, um kostengünstige Ausbeuten
zu erzielen und die Erzeugung von Abfall zu minimieren. Bei solchen
Temperaturen beträgt
die maximale N-(Phosphonomethyl)glycinlöslichkeit
typischerweise weniger als 6,5% ([Masse von N-(Phosphonomethyl)iminodiessigsäuresubstrat ÷ Reaktionsgesamtmasse] × 100%).
Mit dem bevorzugten Oxidationskatalysator und Reaktionsverfahren
dieser Erfindung wird jedoch der Edelmetallverlust aus dem Katalysator
und die Katalysatordeaktivierung auf ein Mindestmaß beschränkt, wobei
der Formaldehyd effektiver oxidiert wird, was Reaktionstemperaturen
von 180°C
(oder höher)
erlaubt. Die Verwendung hoher Oxidationsreaktionstemperaturen und
Rektoreinspeisekonzentrationen erlaubt die Erhöhung des Reaktordurchsatzes,
reduziert die pro Pfund (lbs) hergestelltes N-(Phosphonomethyl)glycinprodukt zu
entfernende Wassermenge und reduziert die Herstellungskosten von
N-(Phosphonomethyl)glycin. Diese Erfindung sieht demzufolge wirtschaftliche
Vorteile gegenüber
vielen herkömmlich
praktizierten Verfahren vor.
-
Die
bevorzugte Obergrenze für
das N-(Phosphonomethyl)iminodiessigsäuresubstrat hängt vom
speziell eingesetzten Substrat ab. Im Fall eines Salzes der N-(Phosphonomethyl)iminodiessigsäure (z.
B Kaliumsalz) können
zum Beispiel Konzentrationen bis zu 70 Gew.-% eingesetzt werden.
Typischerweise ist jedoch eine N-(Phosphonomethyl)iminodiessigsäuresubstratkonzentration
von bis zu etwa 50 Gew.-% bevorzugt (insbesondere bei einer Reaktionstemperatur
von etwa 20 bis etwa 180°C).
Weiter vorzugsweise wird eine N-(Phosphonomethyl)iminodiessigsäuresubstratkonzentration
bis zu etwa 25 Gew.-% verwendet (insbesondere bei einer Reaktionstemperatur
von etwa 60 bis etwa 150°C).
Weiter vorzugsweise wird sogar eine N-(Phosphonomethyl)iminodiessigsäuresubstratkonzentration
von etwa 3 bis etwa 20 Gew.-% verwendet (insbesondere bei einer
Reaktionstemperatur von etwa 100 bis etwa 130°C). Bei den bevorzugten Reaktionstemperaturen
von etwa 95 bis etwa 105°C
beträgt
die N-(Phosphonomethyl)iminodiessigsäuresubstratkonzentration vorzugsweise
etwa 7 bis etwa 15 Gew.-%, weiter vorzugsweise etwa 7 bis etwa 12
Gew.-% und sogar weiter vorzugsweise etwa 9 bis etwa 10 Gew.-%.
-
In
manchen Fällen
enthält
die Quelle des N-(Phosphonomethyl)iminodiessigsäuresubstrats, des in das Verfahren
eingespeisten wässrigen
Einspeisestroms 1 Chloridionen (Cl–),
welche aus der Synthese des Substrats eingeschleppt wurden. Umfasst
der Katalysator ein Kohlenstoff-getragenes Edelmetall, neigen Chloridionen
dazu mit dem Katalysator in Wechselwirkung zu treten, um das Auslaugen
des Edelmetalls zu erhöhen
und inhibieren die Oxidation der Ameisensäure als Nebenprodukt. Darüber hinaus
können
Chloridionengehalte dazu neigen, sich in Reaktorsystemen anzusammeln,
in denen Strömen
(z. B. aus den das Produkt konzentrierenden und/oder reinigenden
Schritten des Verfahrens) rezykliert werden und in die Oxidationsreaktionszone(n),
eingeführt
werden, wie nachstehend beschrieben. Die Chloridionenkonzentration
in der flüssigen
Phase des Reaktionsmediums, welche innerhalb der Reaktionszone(n)
in Berührung
mit dem Katalysator steht, wird bei nicht mehr als 500 ppm des Gewichts,
weiter vorzugsweise bei nicht mehr als etwa 300 ppm des Gewichts
und sogar bei nicht mehr als 100 ppm des Gewichts gehalten. Die
Kontrolle der Chloridgehalte in der/den Oxidationsreaktionszone(n)
erfolgt vorteilhafterweise durch Verwendung einer N-(Phosphonomethyl)iminodiessigsäuresubstratquelle
mit einem relativ niedrigen Chloridgehalt, um den wässrigen
Einspeisestrom 1 zu bilden. Die Chloridionenkonzentration
in der N-(Phosphonomethyl)iminodiessigsäuresubstratquelle, welche über den
wässrigen
Einsspeisestrom in das Verfahren eingespeist wird, beträgt auf Trockenbasis vorzugsweise
weniger als etwa 5000 ppm pro Gewicht, weiter vorzugsweise weniger
als etwa 3000 ppm pro Gewicht, sogar weiter vorzugsweise weniger
als etwa 2000 ppm pro Gewicht und insbesondere weniger als etwa
1000 ppm pro Gewicht. Eine N-(Phosphonomethyl)iminodiessigsäuresubstratquelle,
welche solchen Anforderungen genügt,
kann zum Beispiel nach dem in den US-Patentnummern 4,724,103 und
4,775,498 beschriebenen Verfahren hergestellt werden. Es kann zusätzlich von
Vorteil sein, ein tiefenreduziertes Edelmetall (z. B. Platin) auf
einem Kohlenstoffträger
zu verwenden, welcher, wie vorstehend beschrieben, mit einem Rutheniumzusatz
modifiziert ist, um die Reaktionen in einem kontinuierlichen Oxidationsreaktorsystem
zu katalysieren. Solche mit Ruthenium modifizierten Katalysatoren
können
für eine
erhöhte
Widerstandsfähigkeit
bei Chloridangriff und Edelmetallauslaugung sorgen, und können besonders
zur Verwendung in einem kontinuierlichen Oxidationsreaktorsystem
geeignet sein, in dem die Chloridgehalte in der/den Oxidationszone(n)
aufgrund verschiedener Rezirkulationsströme erhöht sind.
-
Die
Sauerstoffquelle kann in die Reaktionsmischung innerhalb des Rührreaktors 3 in
irgendeiner herkömmlichen
Weise eingeführt
werden, welche die gelöste
Sauerstoffkonzentration in der Reaktionsmischung auf dem gewünschten
Niveau hält.
Die Sauerstoffquelle ist vorzugsweise ein O2-haltiges
Gas wie Luft, reines O2 oder mit einem oder
mehreren nicht-oxidierenden Gasen (z. B. He, Ar und N2)
verdünntes
O2. Die Sauerstoffquelle ist weiter vorzugsweise
ein O2-haltiges Gas, welches mindestens
etwa 95 Mol.-% O2, typischerweise ungefähr 98 Mol.-%
O2 umfasst. Das O2-haltige
Gas wird in einer Weise in die Reaktionsmischung eingeführt, welche
für einen
innigen Kontakt des Gases mit der Reaktionsmischung sorgt. Das O2-haltige Gas kann zum Beispiel durch eine
Begasungsleitung oder eine ähnliche
Verteilervorrichtung eingeführt
werden, welche auf dem Boden des Rührreaktorbehälters 3 unterhalb
des Rührflügels angeordnet
ist, so das die durch den rotierenden Rührflügel hervorgerufene Turbulenz
das O2-haltige Gas innige mischt und verteilt,
während
es durch das flüssige
Reaktionsmedium aufsteigt. Die Verteilung des O2-haltigen
Gases innerhalb der Reaktionsmischung kann des Weiteren verstärkt werden,
wenn man das Gas durch eine Verteilereinrichtungen leitet, wie eine
poröse
Fritte oder durch andere auf dem Fachgebiet wohlbekannte Mittel.
Alternativ kann das O2-haltige Gas in den
Kopfraum über
der Reaktionsmischung in den Rührreaktor 3 eingeführt werden.
-
Ist
die Konzentration an gelöstem
Sauerstoff in der Reaktionsmischung zu groß, neigt die Katalysatoroberfläche dazu
in schädlicher
Weise oxidiert zu werden, (was wiederum dazu führt, dass mehr N-Methyl-N-(phosphonomethyl)glycin
erzeugt wird). Um dieses Problem zu vermeiden ist es allgemein bevorzugt, eine
solche Sauerstoffeinspeisrate zu verwenden, dass mindestens etwa
40%, weiter vorzugsweise mindestens etwa 60%, sogar weiter vorzugsweise
mindestens 80% und noch weiter vorzugsweise mindestens etwa 90%
des Sauerstoffs genutzt werden. Der prozentuale Sauerstoffverbrauch
entspricht, wie hierin verwendet: (der Sauerstoffgesamtverbrauchsrate ÷ Sauerstoffeinspeiserate) × 100%.
Der Ausdruck Sauerstoffgesamtverbrauchsrate bedeutet die Summe von:
(1) der Sauerstoffverbrauchsrate (Ri) der
Oxidationsreaktion des N-(Phosphonomethyl)iminodiessigsäuresubstrats
zur Bildung von N-(Phosphonomethyl)glycin und Formaldehyd, (ii)
die Sauerstoffverbrauchsrate (Rii) der Oxidationsreaktion
von Formaldehyd zur Bildung von Ameisensäure und (iii) die Sauerstoffverbrauchsrate
(Riii) der Oxidationsreaktion von Ameisensäure zur
Bildung von Kohlendioxid und Wasser.
-
Der
Sauerstoffpartialdruck kann in unterschiedlichen Bereichen der Oxidationsreaktionszone(n) schwanken.
Der Sauerstoffpartialdruck beträgt
in einem Rühr behälterreaktor
im Kopfraum über
der flüssigen Reaktionsmischung
vorzugsweise etwa 690 Pa (0,1 psi) bis etwa 241 kPa (35 psi), weiter
vorzugsweise etwa 6,9 kPa (1 psia) bis etwa 69,0 kPa (10 psia).
-
Erfolgt
die Oxidationsreaktion in einem einzigen kontinuierlichen Rührreaktorsystem
kann die Verweilzeit im Reaktor 3 in Abhängigkeit
vom spezifischen Katalysator und den verwendeten Oxidationsreaktionsbedingungen
weit schwanken. Die Verweilzeit beträgt typischerweise etwa 3 bis
etwa 120 Minuten, weiter vorzugsweise etwa 5 bis etwa 90 Minuten,
noch weiter vorzugsweise etwa 5 bis etwa 60 Minuten und sogar noch weiter
vorzugsweise etwa 15 bis etwa 60 Minuten. Die Verweilzeit ist als
Verhältnis
der Strömungsrate
des Filtrats 13 zum Arbeitsvolumen des Rührbehälterreaktors
definiert.
-
Der
in einem kontinuierlichen Oxidationsreaktionssystem verwendete teilchenförmige Katalysator kann
einen Träger
in Form eines Pulvers umfassen, welches eine, wie vorher beschriebene,
Teilchengrößenverteilung
aufweist. Die durchschnittliche Teilchengröße des teilchenförmigen Katalysators
beträgt
etwa 15 bis etwa 40 μm,
weiter vorzugsweise etwa 25 μm.
Die Konzentration des teilchenförmigen
Katalysators in der Reaktionsmischung innerhalb des Rührbehälterreaktors 3 beträgt vorzugsweise
etwa 0,1 bis etwa 10 Gew.-% ([Masse des Katalysators ÷ Reaktionsgesamtmasse] × 100%).
Die Katalysatorkonzentration beträgt weiter vorzugsweise etwa
0,5 bis etwa 5 Gew.-%, sogar weiter vorzugsweise etwa 1 bis etwa
3 Gew.-% und sogar noch weiter vorzugsweise etwa 2 Gew.-%. Konzentrationen
größer als
etwa 10 Gew.-% sind schwierig vom N-(Phosphonomethyl)-glycinprodukt abzutrennen.
Andererseits liefern Konzentrationen von weniger als 0,1 Gew.-%
unannehmbar niedrige Reaktionsgeschwindigkeiten.
-
Der
Katalysatorfilter 9, welcher zur Abtrennung des teilchenförmigen Katalysators
aus der Reaktionsmischung 7 verwendet wird, welche aus
dem Rührbehälterreaktor 3 abgezogenen
wird, besteht vorzugsweise aus einem an die kontinuierliche Trennung
des Katalysators aus der Reaktionsmischung angepasstem Filter. Das
heißt,
dass der Katalysatorfilter 9 in der Lage ist, einen kontinuierlichen
Strom der Reaktionsmischung 7 aufzunehmen, und kontinuierlich
das Filtrat 13 und den Katalysatorrücklaufstrom 11 zu
bilden, ohne dass die Strömung
der in den Filter eingeführten
Reaktionsmischung unterbrochen werden muss. In Übereinstimmung mit einer speziell
bevorzugten Ausführungsform
ist der Katalysatorfilter 9 ein Kreuzstromfilter oder ein
kontinuierlicher Rück spülfilter.
Bei der Durchführung
des in 2 dargestellten kontinuierlichen
Oxidationsverfahren ist ein Rückspülfilter
im Allgemeinen gegenüber
einem Kreuzstromfilter bevorzugt, weil die derzeit im Handel verfügbaren Rückspülfilter
einen Katalysatorrücklaufstrom 11 bilden
können,
welcher eine große
Katalysatorkonzentration enthält,
oft mindestens eine um das 5Fache größere Katalysatorkonzentration
als die derzeit im Handel erhältlichen
Kreuzstromfilter.
-
2A ist ein schematisches Fließbild eines kontinuierlichen
Reaktorsystems, ähnlich
dem in 2 dargestellten, welches besonders
an die Verwendung eines kontinuierlichen Rückspülfilters als Katalysatorfilter 9 angepasst
ist. Ist der Gesamtbetriebsdruck in der/den Oxidationsreaktionszone(n),
wie bevorzugt, viel höher
als der Atmosphärendruck,
nimmt der Druck über
dem aus dem Rührbehälterreaktor 3 abgezogenen
Reaktionsmischungsablauf 7 in Verbindung mit dem Konzentrieren
und Reinigen des N-(Phosphonomethyl)glycinprodukts typischerweise
ab. Mindestens ein Teil dieser Druckverminderung kann in einem Entspannungsbehälter 17 stromaufwärts des
Katalysatorfilters 9 erfolgen. Der Entspannungsbehälter 17 erniedrigt
den Druck auf der Reaktionsmischung 7 bis zu einem gewissen
Grad, veranlasst das gelöste
CO2 aus der Mischung ausgetrieben und als
Dampf aus dem Entspannungsbehälter
abgelüftet
zu werden. Der Entspannungsbehälter 17 vermindert
den Druck, bei dem der kontinuierliche Rückspülkatalysatorfilter 9 betrieben
werden muss und reduziert damit die Kapitalkosten und die Komplexität des Filtersystems.
In den Entspannungsbehälter 17 kann eine
Sauerstoffquelle (z. B. ein O2-haltiges
Gas) eingespeist (z. B. verdüst)
werden, um sowohl das im Rührbehälterreaktor 3 nicht
oxidierte N-(Phosphonometyl)iminodiessigsäuresubstrat
weiter zu oxidieren als auch die in der Reaktionsmischung als Nebenprodukte
vorliegenden Formaldehyd und Ameisensäure weiter zu oxidieren. Auf
diese Weise kann der Entspannungsbehälter 17 als eine mit
dem Rührbehälterreaktor 3 in
Reihe abgeordnete zusätzliche
Oxidationsreaktionszone wirksam werden.
-
Das
kontinuierliche Rückspülfiltersystem
umfasst ein Filterelement und wird vorzugsweise adiabatisch betrieben,
kann jedoch auch mit einer Heiz- oder Kühlvorrichtung ausgerüstet sein.
Die zum Rückspülen des Filterelements
und zur Entfernung des abgetrennten Katalysators verwendete Flüssigkeit
ist ein Teil des Filtrats 13. Das Filtrat 13 wird
weitergeleitet, um das N-(Phosphonomethyl)glycinprodukt zu konzentrieren
und zu reinigen, während
der Katalysatorrücklaufstrom 11 kontinuierlich
aus dem Katalysatorfilter 9 abgezogen und in einen fakultativen
Katalysatorvorratsbehälter 5 (auch
als Katalysatorrücklaufbehälter oder
Katalysatoraufschlämmungsbehälter bezeichnet) übergeführt wird,
bevor der Katalysator in den Rührbehälterreaktor 3 zurückgeführt wird.
-
Obwohl
der Katalysatorfilter 9 in dem in 2 und 2A dargestellten
Oxidationsreaktorsystem vorzugsweise ein kontinuierliches Rückspülfilter
ist, sind in manchen Fällen
kontinuierliche Kreuzstromfilter selbstverständlich stärker bevorzugt. Das in 2B abgebildete System ist dem in 2 und 2A abgebildeten ähnlich,
außer
dass der Katalysatorfilter 9 innerhalb des externen Wärmeübertragungskreislaufs 15 angeordnet
ist und nicht in einen getrennten Katalysatorrücklaufkreislauf. Bei einer
solchen Ausführungsform ist
der Katalysatorfilter 9 vorzugsweise ein kontinuierlicher
Kreuzstromfilter. In Verbindung mit einem Kreuzstromfilter wird
vor dem Filter typischerweise kein Entspannungsbehälter eingesetzt.
Weiterhin wird wegen des relativ großen Volumens des aus dem kontinuierlichen
Kreuzstromfilter austretenden Katalysatorrücklaufstroms 11 typischerweise
auf einen Katalysatorvorratsbehälter
gleichfalls verzichtet.
-
Abgesehen
von Kreuzstrom- und Rückspülfiltern
kann ein bei einem kontinuierlichen Oxidationsreaktorsystem verwendeter
Katalysatorfilter 9 alternativ ein Vakuumfilter sein oder
er kann einen Satz von Scheibenfiltern umfassen, welche dazu verwendet
werden, einen kontinuierlichen Strom des Reaktionsmischungsablaufs 7 in
gestaffelten Filtrationszyklen zu behandeln. Als weitere Alternative
kann der Rührbehälterreaktor 3 einen
internen Katalysatorfilter (z. B. eine poröse Fritte) einschließen, die
den teilchenförmigen
Katalysator zurückhält, damit
er nicht mit dem Reaktionsmischungsablauf 7 abgezogen wird,
so dass der Katalysator im Wesentlichen in der Oxidationsreaktionszone
zurückgehalten
wird und der Reaktionsmischungsablauf im Wesentlichen frei von teilchenförmigem Katalysator
ist. Darüber
hinaus können
anstelle (oder zusätzlich
zum) Katalysatorfilter 9 selbstverständlich andere Methoden der
Katalysatortrennung verwendet werden. Der Katalysator könnte zum
Beispiel aus dem Oxidationsreaktionsmischungsablauf mithilfe einer
Zentrifuge abgetrennt werden.
-
Nachdem
der Katalysator bei der Verwendung deaktiviert wird, kann er mindestens
teilweise entweder kontinuierlich oder diskontinuierlich reaktiviert
werden. Das Reaktivieren kann das Reduzieren der Oberfläche umfassen,
nachdem der Katalysator stark oxidiert worden ist. In einem solchen
Fall kann die Oberfläche
zum Beispiel gewaschen werden, um die organischen Bestandteile zu
entfernen und dann unter Verwendung der vorstehend beschriebnen
Reduktionsbehandlungen reduziert werden. Eine solche reduzierende
Behandlung kann zum Bei spiel die kontinuierliche oder diskontinuierliche
Einführung
eines Reduktionsmittels in das Reaktorsystem umfassen. Das Reduktionsmittel
kann zum Beispiel Formaldehyd und/oder Ameisensäure umfassen und kann vorteilhafterweise
oft aus den verschiedenen, hierin beschriebenen, Rücklaufströmen erhalten werden.
Die Reaktivierung kann zum Beispiel auch durch Einführung eines
Zusatzaktivators, insbesondere von Wismutoxid, in das Reaktorsystem
erreicht werden, wie vorstehend beschrieben. In Übereinstimmung mit einer bevorzugten
Ausführungsform
der vorliegenden Erfindung wird ein Zusatzaktivator (z. B. Bi2O3) kontinuierlich
oder diskontinuierlich derart in das kontinuierliche Reaktorsystem
eingeführt,
dass die Ameisensäurekonzentration
in dem aus der letzten Oxidationsreaktionszone abgezogenen Reaktionsmischungsablauf
bei weniger als etwa 6000 ppm, weiter vorzugsweise bei weniger als
etwa 1000 ppm bis etwa 3000 ppm gehalten wird. In Übereinstimmung
mit einer besonders bevorzugt Art der Anwendung der vorliegenden
Erfindung wird die Ameisensäurekonzentration
in dem aus der letzten Oxidationsreaktionszone abgezogenen Reaktionsmischungsablauf überwacht.
Sobald die gemessene Konzentration etwa 6000 ppm, weiter vorzugsweise
etwa 3000 ppm, sogar weiter vorzugsweise etwa 2000 ppm überschreitet,
wird mit der kontinuierlichen oder diskontinuierlichen Einführung eines
Zusatzaktivators in das Reaktorsystem begonnen und diese aufrechterhalten, bis
die Ameisensäurekonzentration
in dem aus der letzten Oxidationsreaktionszone abgezogenen Reaktionsmischungsablauf
abzunehmen beginnt. Die Zugaberate des Zusatzaktivators zum Reaktorsystem
ist vorzugsweise so, dass die Ameisensäurekonzentration in dem aus
der letzten Oxidationsreaktionszone abgezogenen Reaktionsmischungsablauf
eine Zeit lang ansteigt, nachdem die Zugabe eines Zusatzaktivators
zum System begonnen hat. In dem Fall, in dem Bi2O3 dem Reaktorsystem als Zusatzaktivator zugesetzt
wird, beträgt
das Gewichtsverhältnis
von Bi2O3 zu dem
in das System eingespeisten N-(Phosphonomethyl)iminodiessigsäuresubstrat
etwa 1:20.000.000 bis etwa 1:200.000.
-
Obwohl
der Katalysatorvorratsbehälter 5,
welcher in dem in 2A dargestellt kontinuierlichen
Oxidationsreaktorsystem fakultativ ist, kann er von Vorteil sein,
wenn ein tiefenreduzierter teilchenförmiger Katalysator verwendet
wird, weil er einen Ort bietet, wo der Katalysator einheitlich reaktiviert
werden kann. Ein Reduktionsmittel 18 und/oder ein Zusatzaktivator 19 können, wie
in 2A dargestellt, in den Katalysatorvorratsbehälter 5 eingeführt werden,
welcher den rezyklierten Katalysator enthält. Das Reduktionsmittel und/oder
der Zusatzaktivator können
alternativ direkt der/den Oxidationsreaktionszone(n) oder dem Reaktorsystem
anderswo zugegeben werden. Es ist weiterhin selbstverständlich, dass
allein die Tatsache, dass der rezyklierte Katalysator die Möglichkeit
erhält,
sich zusammen mit der restlichen Reaktionsmischung im Katalysatorvorratstank 5 aufzuhalten,
sich auch auf die Reduktion der Katalysatoroberfläche günstig auswirken
kann (insbesondere bei einem Katalysator, welcher ein Kohlenstoff-getragenes Edelmetall
umfasst). Der Katalysatorvorratsbehälter ist im Wesentlichen frei
von O2 und anderen oxidierenden Gasen. Es
kann demzufolge vorteilhaft sein, in den Behälter 5 Stickstoff
oder ein anderes nicht-oxidierendes Gas einzuleiten (z. B. durchzuperlen),
um die O2-Entfernung zu unterstützen. Lässt man
sich die Aufschlämmung
des teilchenförmigen
Katalysators und die restliche Aufschlämmung außerhalb der/den Oxidationsreaktionszone(n)
in einer im Wesentlichen O2-freien Umgebung
eine gewisse Zeit aufhalten, ehe sie wieder in die Oxidationsreaktionszone(n)
eingeführt
werden, wird angenommen, dass dies die Katalysatoroberfläche reduziert,
einen Grad der Reaktivierung herbeiführt und die Gebrauchdauer des
Katalysators verlängert.
Der Katalysatorvorratsbehälter
oder Katalysatoraufschlämmungsbehälter 5 kann
verschiedene Konfigurationen aufweisen, ist jedoch typischerweise
ein Rührbehälter, in
dem die Katalysatoraufschlämmung,
welche den teilchenförmigen
Katalysator und die restliche Reaktionsmischung umfasst, mit einem
rotierenden Rührflügel bewegt
wird, um die Einheitlichkeit der Katalysatoraufschlämmung zu
verbessern, und so den Katalysator daran hindert auf dem Boden des
Behälters 5 abzusetzen
und ebenso die einheitliche Reaktivierung des Katalysators fördert. Die
Verweilzeit des Katalysators im Katalysatorvorratsbehälter 5 kann
durch Einstellung des Volumens der Katalysatoraufschlämmung im
Katalysatorvorratsbehälter
im Verhältnis
zum Arbeitsvolumen des Reaktionsmediums innerhalb der/den Oxidationsreaktionszone(n)
eingestellt werden. Längere
Katalysatorverweilzeiten im Katalysatorvorratsbehälter sind für die Katalysatorleistung
generell günstig.
Weil jedoch längere
Verweilzeiten einen größeren Katalysatoreinsatz
im Reaktorsystem bedingen, müssen
die Vorteile gegenüber
den höheren
Katalysatorkosten abgewogen werden, welche insbesondere im Fall
eines Katalysators, welcher ein Kohlenstoff getragenes Edelmetall
umfasst, von Bedeutung werden können.
Die Verweilzeit des rezyklierten Katalysators im Katalysatorvorratsbehälter beträgt mindestens
etwa 2 Minuten, weiter vorzugsweise mindestens etwa 5 Minuten, sogar
weiter vorzugsweise etwa 5 bis etwa 40 Minuten.
-
Geringere
Edelmetallverlust können
bei dieser Erfindung dann beobachtet werden, wenn in der Reaktionslösung ein
Opferreduktionsmittel aufrechterhalten oder eingeführt wird.
Geeignete Reduktionsmittel schließen Formaldehyd, Ameisensäure und
Acetaldehyd ein. Am meisten vorzugsweise werden Ameisensäure, Formaldehyd
oder deren Mischungen verwendet (z. B. erhalten aus den verschiedenen,
hierin beschriebenen, Rücklaufströmen).
-
Ein
Katalysator (z. B. ein Katalysator mit einer verringerten Aktivität und/oder
Selektivität)
kann auch kontinuierlich oder diskontinuierlich aus dem kontinuierlichen
Oxidationsreaktorsystem über
den Katalysatorabführstrom 20 ausgeschieden
und durch frischen Katalysator über
den Frischkatalysatoreinspeisestrom 21 ersetzt werden.
Wird der Katalysator diskontinuierlich ausgeschieden, kann die gesamte
Katalysatormasse auf einmal aus dem Verfahren ausgeschieden werden
(was typischerweise das weiter bevorzugte Verfahren ist), oder es
kann ein Bruchteil der Katalysatormasse zu verschiedenen Zeitintervallen
ausgeschieden werden. Das diskontinuierliche Ausscheiden schließt mit anderen
Worten jedes wiederholte Ausscheiden von Katalysator ein, welches
nicht kontinuierlich erfolgt.
-
In Übereinstimmung
mit einer weiter bevorzugten Ausführungsform der vorliegenden
Erfindung wird die kontinuierliche Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats
in Anwesenheit einer teilchenförmigen
heterogenen Katalysatoraufschlämmung
stufenweise in zwei oder mehreren in Reihe angeordneten Oxidationsreaktionszonen
mit im Wesentlichen Rückvermischung
betrieben (d. h. Rückvermischung
in mindestens der flüssigen
Phase). Eine Kombination von zwei oder mehreren in Reihe angeordneten
Oxidationsreaktionszonen ist vorteilhaft, weil ein solches Reaktorsystem
dazu neigt, sich eher wie ein Pfropfströmungsreaktor zu verhalten,
indem es weniger Nebenprodukte erzeugt und die Ausbeute an N-(Phosphonomethyl)glycinprodukt
verbessert. Darüber
hinaus bietet die Kombination von zwei oder mehr Reaktionszonen
die Möglichkeit
die Reaktionsbedingungen in Übereinstimmung
mit der effektiven Reaktionskinetik in unterschiedlichen Stadien
der Oxidationsreaktion zu variieren.
-
Die
Oxidation eines N-(Phosphonomethyl)iminodiessigsäuresubstrats gehorcht ungefähr einer
Reaktion nullter Ordnung in Bezug auf die Substratkonzentration
bis die Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats
auf nicht mehr als 4,5 Gew.-%, noch typischer bis auf nicht mehr
als etwa 2,7 Gew.-%, sogar noch typischer auf etwa 0,4% bis etwa
1,8 Gew.-%, sogar noch typischer auf etwa 0,4% bis etwa 1,3 Gew.-%
und sogar noch weiter typisch auf nicht mehr als 1 Gew.-% abnimmt.
Beträgt
zum Beispiel die Substratkonzentration in der wässrigen Einspeisung in die
erste Reaktionszone 9 Gew.-%, wird die Reaktion dazu tendieren nach
nullter Ordnung in Bezug auf das Substrat zu verlaufen, bis mindestens
50%, noch typischer mindestens 70%, sogar noch typischer etwa 80%
bis etwa 95% und sogar noch typischer etwa 85% bis etwa 95% des
Substrats verbraucht sind. An diesem Punkt wird die Oxidationsgeschwindigkeit
stärker
von der Substratkonzentration abhängig (d. h. die Oxidation nähert sich
in Bezug auf die Substratkonzentration einem Verlauf nach 1. Ordnung)
und tendiert folglich dazu mit weiter sinkender Substratkonzentration
abzunehmen. Mit stärker
werdender Abhängigkeit
der Oxidationsgeschwindigkeit von der Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats
neigt die Oxidation des Substrats dazu langsamer zu werden als die parallel
verlaufenden Oxidationsreaktionen der Nebenprodukte Formaldehyd
und Ameisensäure.
-
Bei
Verwendung eines kontinuierlichen Oxidationsreaktorsystems, welches
zwei Oxidationsreaktionszonen in Reihe umfasst, kann die Verweilzeit
und/oder die Sauerstoffeinsspeisung in die erste Reaktionszone so
gesteuert werden, dass die Reaktion in der ersten Reaktionszone
im Wesentlichen nach nullter Ordnung in Bezug auf die Substratkonzentration
verläuft
(d. h. die Verweilzeit im ersten Reaktor kann so gesteuert werden, dass
die Umwandlung des Substrats im ersten Reaktor ausreicht, um eine
Reaktionsmischung mit einer Substratkonzentration von nicht mehr
als 4,5 Gew.-%, weiter vorzugsweise von nicht mehr als etwa 2,7
Gew.-%, sogar weiter vorzugsweise von etwa 0,4% bis etwa 1,8 Gew.-%,
sogar noch weiter vorzugsweise von etwa 0,4% bis etwa 1,3 Gew.-%
und sogar noch weiter vorzugsweise von nicht mehr als 1 Gew.-% zu
bilden). Diese Reaktionsmischung kann dann zur zweiten und irgendwelchen
weiteren Reaktionszonen übergeführt werden, worin
die Reaktion im Wesentlichen nach einer Reaktion 1. Ordnung in Bezug
auf die Substratkonzentration verläuft. Auf diese Weise kann/können die
Reaktorauslegung und/oder die Reaktionsbedingungen (z. B. Art des
Katalysators, die Reaktorauslegung und/oder die Reaktionsbedingungen,
Temperatur, Druck etc.) in jeder Reaktionszone unabhängig genau
geregelt werden, um die Reaktionsstufen und die Oxidation der Nebenprodukte
Formaldehyd und Ameisensäure
zu optimieren.
-
3 zeigt
ein bevorzugtes Oxidationsreaktorsystem in Übereinstimmung mit der vorliegenden
Erfindung, welches zwei in Reihe angeordnete Oxidationsreaktionszonen
mit Rückvermischung
umfasst. Die Oxidationsreaktionszonen mit Rückvermischung werden vorzugsweise
in Form von zwei kontinuierlichen Rührbehälterreaktoren 3 und 40 vorgesehen.
Ein wässriger
Einspeisestrom 1, der ein N-(Phosphonomethyl)iminodiessigsäuresubstrat
enthält,
wird zusammen mit einer Sauerstoffquelle, vorzugsweise ein O2-haltiges Gas, in den ersten Rühr behälterreaktor 3 kontinuierlich
oder diskontinuierlich eingeführt.
Das N-(Phosphonomethyl)iminodiessigsäuresubstrat wird im ersten
Rührbehälterreaktor 3 in
Anwesenheit des heterogenen teilchenförmigen Katalysator kontinuierlich
oxidiert, um eine wässrige
Reaktionszwischenproduktmischung 41 zu bilden, die ein
N-(Phosphonomethyl)glycinprodukt und nicht umgesetztes N-(Phosphonomethyl)iminodiessigsäuresubstrat
umfasst, welche kontinuierlich oder diskontinuierlich aus dem ersten
Rührbehälterreaktor
abgezogen wird. Ein wässriger
Zwischenprodukteinspeisestrom 42, welcher (a) das N-(Phosphonomethyl)glycinprodukt
aus der wässrigen
Zwischenproduktreaktionsmischung 41 umfasst und (b) nicht
umgesetztes N-(Phosphonomethyl)iminodiessigsäuresubstrat umfasst, welches
ebenfalls wenigstens teilweise aus der Zwischenproduktreaktionsmischung 41 stammt,
wird dann in den zweiten Rührbehälterreaktor 40 eingeführt. Typischerweise
wird auch Sauerstoff, vorzugsweise in Form eines O2-haltigen
Gases, in den zweiten Rührreaktorbehälter eingespeist.
Im zweiten Rührbehälterreaktor 40 wird
zusätzliches
N-(Phosphonomethyl)iminodiessigsäuresubstrat
in Anwesenheit des heterogenen teilchenförmigen Katalysators kontinuierlich
oxidiert, um einen Endreaktionsmischungsablauf 45 zu bilden,
welcher das N-(Phosphonomethyl)glycinprodukt
umfasst. Der Kopfraum über
der Reaktionsmischung innerhalb der Rührreaktorbehälter 3 und 40 wird
entlüftet,
um Dampf abzulassen, welcher CO2 aus dem
Fortschreiten der Oxidationsreaktion in den Oxidationsreaktionszonen
umfasst.
-
Obwohl
der wässrige
Zwischenprodukteinspeisestrom 42 in 3 so dargestellt
ist, als wie wenn er die gesamte wässrige Zwischenproduktreaktionsmischung 41 umfassen
würde,
enthält
der wässrige
Zwischenprodukteinspeisestrom 42 bei einigen Ausführungsformen
der vorliegenden Erfindung selbstverständlich weniger als die gesamte
wässrige
Zwischenproduktreaktionsmischung 41. Zum Beispiel kann
der teilchenförmige
heterogene Katalysator teilweise oder vollständig aus der wässrigen
Reaktionszwischenproduktmischung 41 entfernt werden, wie
nachstehend beschrieben (5 und 6).
Des Weiteren muss die erste und zweite Oxidationsreaktionszone selbstverständlich nicht
in getrennten Rührbehälterreaktoren 3 und 40 enthalten
sein, wie in 3 gezeigt. Mehrere Oxidationsreaktionszonen
können
hintereinander stufenweise angeordnet und innerhalb eines einzelnen
Reaktorbehälters
enthalten sein, der in Kammern unterteilt oder mit Leitblechen oder
anderen Mitteln zur Trennung von Reaktionszonen voneinander ausgerüstet ist.
-
Bei
der in 3 wiedergegebenen Ausführungsform,
fließt
der teilchenförmige Katalysator
aus der Reaktionszone im ersten Rührbehälterreaktor zur Reaktionszone
im zweiten Rührbehälterreaktor 40.
Der teilchenförmige
Katalysator ist vorzugsweise der vorstehend beschriebene tiefenreduzierte
Oxidationskatalysator. Der Katalysator wird über den Katalysatoreinspeisestrom 39 kontinuierlich
oder diskontinuierlich in den ersten Rührbehälterreaktor 3 eingeführt. Der
Katalysatoreinspeisestrom ist, wie in 3 dargestellt,
Teil des wässrigen
Einspeisestroms 1, welcher ein N-(Phosphonomethyl)iminodiessigsäuresubstrat
enthält.
Der Katalysator wird aus dem ersten Rührbehälterreaktor 3 als
Teil der wässrigen
Zwischenproduktreaktionsmischung 41 kontinuierlich oder
diskontinuierlich abgezogen, kontinuierlich oder diskontinuierlich
in den zweiten Rührbehälterreaktor 40 als
Teil des wässrigen
Zwischenprodukteinspeisestroms 42 eingeführt und
schließlich
kontinuierlich oder diskontinuierlich aus dem zweiten Rührbehälterreaktor 40 als
Teil des Endreaktionsmischungsablaufs 45 abgezogen. Der
Endreaktionsmischungsablauf 45 wird wahlweise in einem
Entspannungsbehälter 17 entspannt
und zum Katalysatorfilter 9 weitergeleitet. Im Katalysatorfilter 9 wird
im Wesentlichen der gesamte teilchenförmige Katalysator aus dem Endreaktionsmischungsablauf 45 abgetrennt,
um (1) einen Katalysatorrücklaufstrom 11 zu
bilden, welcher im Wesentlichen den gesamten Katalysator und eine
Restmenge N-(Phosphonomethyl)glycinprodukt aus der Endreaktionsmischung 45 umfasst;
und (2) ein Filtrat 13, welches die Hauptmenge des N-(Phosphonomethyl)glycinprodukts
aus der Endreaktionsmischung 45 umfasst. Bei der in 3 dargestellten
Ausführungsform
ist der Katalysatorfilter 9 vorzugsweise ein kontinuierliches
Rückspülfiltersystem,
um das Volumen des Katalysatorrücklaufstroms
auf ein Mindestmaß zu
beschränken
und die Bodenwirkung im Reaktorsystem aufrechtzuerhalten. Der Katalysatorrücklaufstrom 11 wird
zum Katalysatorvoratsbehälter 5 geleitet
und über
den Katalysatoreinspeisestrom 39 wieder in den ersten Rührbehälterreaktor 3 eingeführt, während das
Filtrat 13 weitergeleitet wird, um das N-(Phosphonomethyl)glycinprodukt
zu konzentrieren und zu reinigen. Deaktiviert sich der Katalysator
bei der Verwendung, kann er wenigsten teilweise, durch kontinuierliches
oder diskontinuierliches Kontaktieren des teilchenförmigen Katalysators
mit einem Reduktionsmittel 18 (z. B. im Katalysatorvorratsbehälter 5)
und/oder Einführen
eines Zusatzaktivators 19 in das Verfahren (z. B. in den
Katalysatorvorratsbehälter 5 und/oder
direkt in den ersten und/oder zweiten Rührbehälterreaktor 3 und 40)
reaktiviert werden, wie vorstehend beschrieben. Der Katalysator
kann durch den Katalysatorreinigungsstrom 20 kontinuierlich
oder diskontinuierlich aus dem System ausgeschieden und über den
Katalysatoreinspeisestrom 21 mit frischem Katalysator aufgefüllt werden.
-
Bei
der Inbetriebnahme des Reaktorsystems von 3 kann
der in den ersten Rührbehälterreaktor 3 eingeführte Katalysatoreinspeisestrom 39 und/oder
der wässrige
Einsspeisestrom 1 erwärmt
werden, um in den Oxidationsreaktionszonen die gewünschte Temperatur
zu erhalten. Während
des stationären
oder quasi-stationären
Betriebszustandes, reicht die exotherme Reaktionswärme normalerweise
aus, um die Einspeisematerialien auf die gewünschte Reaktionstemperatur
zu bringen, wobei überschüssige Reaktionswärme aus dem
flüssigen
Reaktionsmedium im ersten Reaktor 3 über einen Wärmetauscher 16 in
einem externen Wärmeübertragungskreislauf 15 abgeführt wird.
Die Reaktionstemperatur wird zum Beispiel durch Regelung der Kühlwasserzufuhr
zum Wärmetauscher 15 als
Folge des Signals eines Temperatursteuergeräts geregelt. Ähnlich kann
die Temperatur des flüssigen
Reaktionsmediums in der zweiten Oxidationsreaktionszone im Reaktor 40 durch
die Wärmeabführrate über den
Wärmetauscher 48,
der mit dem zweiten Reaktor über
den externen Wärmeübertragungskreislauf 47 verbunden
ist, geregelt werden. Die zweite Oxidationsreaktionszone kann jedoch
ohne den Wärmeübertragungskreislauf 47 oder
andere Mittel zur Abführung
der Reaktionswärme
betrieben (z. B. adiabatisch betrieben) werden. In manchen Fällen ist
zum Beispiel die Zunahme der Umwandlung des N-(Phosphonomethyl)iminodiessigsäuresubstrats
und die Oxidation von Formaldehyd und Ameisensäure im zweiten Rührbehälterreaktor 40 derart
beschränkt,
dass die bei den Oxidationsreaktionen entwickelte Wärme keine
Kühlung
der Reaktionsmischung erforderlich macht. Ist es erwünscht, die
Reaktion im zweiten Reaktor 40 bei einer Temperatur zu
vervollständigen,
welche höher
ist als die im ersten Reaktor 3 herrschende, kann die autogene
Reaktionswärme
im zweiten Reaktor die ganze oder einen Teil der Wärme beitragen,
welche erforderlich ist, die Temperatur des wässrigen Einspeisestroms 42 zu
erhöhen
und den gewünschten
Temperaturunterschied zwischen dem ersten Reaktor und dem zweiten
Reaktor aufrechtzuerhalten.
-
Die
Temperatur des Reaktionsmediums im zweiten Rührbehälterreaktor 40 wird
im Hinblick auf die N-(Phosphonomethyl)glycinkonzentration vorzugsweise
derart hoch gehalten, dass im Wesentlichen das gesamte aus dem zweiten
Reaktor abgezogene N-(Phosphonomethyl)glycinprodukt im Endreaktionsmischungsablauf 45 gelöst bleibt.
Wahlweise kann im Endreaktionsmischungsablauf 45 ausgefallenes
N-(Phosphonomethyl)glycinprodukt mit dem teilchenförmigen Katalysator
als Teil des Katalysatorrücklaufstroms 11 abgetrennt werden.
Die Temperatur der Reaktionsmischung in den Rührbehälterreaktoren 3 und 40 kann
von Reaktor zu Reaktor unterschiedlich sein. Nachdem zum Beispiel
die wässrige Zwischenreaktionsproduktmischung 41 nicht
filtriert ist und auch eine niedrigere Konzentration an N-(Phosphonomethyl)glycinprodukt
enthält
als der Endreaktionsmischungsablauf 45, kann die Temperatur
der Reaktionsmischung innerhalb des ersten Rührbehälterreaktors 3 typischerweise
etwas niedriger sein als die bevorzugte Betriebstemperatur der Reaktionsmischung
im zweiten Rührbehälterreaktor 40.
Der erste Rührbehälterreaktor 3 wird
vorzugsweise bei einer Temperatur von etwa 80°C bis etwa 120°C, weiter
vorzugsweise von etwa 85°C
bis etwa 110°C
und sogar noch weiter vorzugsweise von etwa 95°C bis etwa 100°C betrieben,
während
der zweite Rührreaktorbehälter 40 vorzugsweise
bei einer Temperatur von etwa 80°C
bis etwa 120°C,
weiter vorzugsweise von etwa 85°C
bis etwa 110°C
und sogar weiter vorzugsweise bei etwa 100°C bis 105°C betrieben wird. Es ist oft
von Vorteil den ersten Rührbehälterreaktor 3 bei
einer niedrigeren Temperatur zu betreiben, um die Bildungsgeschwindigkeit
von N-Methyl-N-(phosphonomethyl)glycin
zu verringern, welche bei höheren
Temperaturen zunimmt.
-
Im
ersten und zweiten Rührbehälterreaktor 3 und 40 wird
vorzugsweise ein Gesamtdruck aufrechterhalten, welcher hoch genug
ist, das flüssige
Reaktionsmedium in den Oxidationsreaktionszonen am Sieden zu hindern
und der im Allgemeinen etwa 0 bis etwa 3447 kPa (0 bis etwa 500
psig) beträgt.
Der Gesamtdruck in den Rührbehälterreaktoren 3 und 40 beträgt etwa
207 bis etwa 3447 kPa (30 bis etwa 500 psig). Wird eine Temperatur
der Reaktionsmischung in der ersten und zweiten Oxidationsreaktionszone
innerhalb des vorstehend offen gelegten Temperaturbereichs aufrechterhalten,
beträgt
der innerhalb des ersten und zweiten Rührbehälterreaktor 3 und 40 aufrechterhaltene
Gesamtdruck vorzugsweise etwa 207 bis etwa 896 kPa (30 bis etwa
130 psig) und weiter vorzugsweise etwa 621 bis etwa 758 kPa (90
bis etwa 110 psig).
-
Der
Sauerstoffpartialdruck kann in unterschiedlichen Bereichen der Oxidationsreaktionszonen schwanken.
Der Sauerstoffpartialdruck im Kopfbereich über dem flüssigen Reaktionsmedium in den
Rührbehälterreaktoren 3 und 40 beträgt vorzugsweise
etwa 690 Pa bis etwa 241 kPa (0,1 bis 35 psia), weiter vorzugsweise
etwa 6,9 kPa bis etwa 69,0 kPa (1 bis etwa 10 psia).
-
Insbesondere
dann, wenn die Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats
im wässrigen
Einspeisestrom 1 (welcher den Katalysatorrücklaufstrom 11 und
irgendwelche andere Rezirkulationsströme aus anderen Verfahrensteilen
einschließt)
etwa 7 bis etwa 12 Gew.-% und sogar weiter vor zugsweise etwa 9 Gew.-%
beträgt,
ist typischerweise bevorzugt, dass die Verweilzeit im ersten Rührbehälterreaktor 3 so
ist, dass die Umwandlung des N-(Phosphonomethyl)iminodiessigsäuresubstrats
zu N-(Phosphonomethyl)glycin in der ersten Oxidationsreaktionszone
mindestens etwa 50%, weiter vorzugsweise mindestens etwa 70%, sogar
weiter vorzugsweise etwa 80% bis etwa 95%, sogar noch weiter vorzugsweise
etwa 85% bis etwa 95% und am meisten vorzugsweise etwa 90% beträgt. Die
zur Erzielung des gewünschten
Umwandlungsgrades erforderliche Verweilzeit wird mit den im ersten
Rührbehälterreaktor
eingesetzten Oxidationsreaktionsbedingungen schwanken. Die Verweilzeit
im ersten Rührbehälterreaktor 3 beträgt typischerweise
etwa 5 bis etwa 50 Minuten, vorzugsweise etwa 10 bis etwa 30 Minuten,
sogar weiter vorzugsweise etwa 14 bis etwa 24 Minuten und noch weiter
vorzugsweise etwa 20 Minuten. Die Verweilzeit im zweiten Rührbehälterreaktor 40 beträgt typischerweise
etwa 1 bis etwa 50 min, vorzugsweise etwa 1 bis etwa 30 min, sogar
weiter vorzugsweise etwa 3 bis etwa 20 min, weiter vorzugsweise
etwa 6 bis etwa 20 min, noch weiter vorzugsweise etwa 6 bis etwa
12 min und sogar noch weiter vorzugsweise etwa 8 min. Die Verweilzeit
im ersten Rührbehälterreaktorbehälter 3 ist
mit Bezug auf die Strömungsrate
der Zwischenproduktreaktionsmischung 41 und das Arbeitsvolumen
des Reaktors definiert. Die Verweilzeit im zweiten Rührbehälterreaktor 40 ist
mit Bezug auf die Strömungsrate
des Ablaufs der Endreaktionsmischung 45 und das Arbeitsvolumen
des Reaktors definiert. Die mit einer bestimmten Verweilzeit erzielte
Umsetzung tendiert dazu mit der im Verlauf der Verwendung abnehmenden
Katalysatoraktivität
abzunehmen, was die Verstärkung
der Katalysatoraktivität
durch Reaktivierung oder das Beschicken des Systems mit frischen
Katalysator oder eine Erhöhung
der O2-Einspeiserate erfordert.
-
Das
Verhältnis
des Arbeitsvolumens des flüssigen
Reaktionsmediums im ersten Rührbehälterreaktor 3 zum
Arbeitsvolumen des flüssigen
Reaktionsmediums im zweien Rührbehälterreaktor 40 ist
größer als
1, weiter vorzugsweise größer als
1 und beträgt
bis etwa 10, sogar weiter vorzugsweise etwa 1,1 bis etwa 5 und sogar
noch weiter vorzugsweise etwa 1,1 bis etwa 2,5.
-
Umfasst
das kontinuierliche Reaktorsystem zwei Rührbehälterreaktoren in Reihe, wird
die in das kontinuierliche Reaktorsystem eingespeiste Sauerstoffgesamteinspeisung
(d. h. die kombinierte Sauerstoffeinspeisung in beide Rührbehälterreaktoren 3 und 40)
und werden die jedem Rührreaktor
zugeteilten Anteile der Sauerstoffgesamteinspeisung normalerweise
so eingestellt, dass sie die Ausbeute und Qualität des N-(Phosphonomethyl)glycinprodukts
beein flussen. Bei einer Ausführungsform
wird der in das kontinuierliche Reaktorsystem eingespeiste Sauerstoff
pro Mol N-(Phosphonomethyl)iminodiessigsäuresubstrat im wässrigen
Einspeisestrom 12, der dem ersten Rührbehälterreaktor 3 zugeführt wird,
variiert, um die Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats
aus dem im zweiten Rührreaktorbehälter 40 abgezogenen Endreaktionsmischungsablauf 45 zu
regeln. Die Konzentration des nicht umgesetzten N-(Phosphonomethyl)iminodiessigsäuresubstrats
in der Endreaktionsmischung 45 wird im Allgemeinen minimiert,
um zu große Ausbeuteverluste
zu vermeiden. Die Konzentration an nicht umgesetztem N-(Phosphonomethyl)iminodiessigsäuresubstrat
im Endreaktionsmischungsablauf beträgt nicht mehr als etwa 2000
ppm. Die Konzentration an N-(Phosphonomethyl)iminodiessigsäuresubstrat
im Endreaktionsmischungsablauf 45 sollte jedoch ausreichend
hoch bleiben, um die Geschwindigkeit zu inhibieren, mit der sich
das N-(Phosphonomethyl)glycinprodukt unter Bildung Aminomethylphosphonsäure oxidiert.
Die Geschwindigkeit der Aminophosphonsäurebildung ist offensichtlich
der Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats
umgekehrt proportional. Es wird darüber hinaus angenommen, dass
die Anwesenheit von N-(Phosphonomethyl)iminodiessigsäuresubstrat
die Überoxidation
des Katalysators inhibieren und die Lebensdauer des Katalysators
verlängern
kann. Demzufolge ist es bevorzugt, dass die Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats
im Endreaktionsmischungsablauf 45 in einem Bereich von
etwa 200 bis etwa 2000 ppm, weiter vorzugsweise von etwa 500 bis
etwa 1500 ppm und am meisten vorzugsweise von etwa 700 ppm des Gewichts gehalten
wird. Eine geeignete Konzentration an N-(Phosphonomethyl)iminodiessigsäuresubstrat
in der Endreaktionsmischung 45 wird typischerweise erhalten,
wenn der in das kontinuierliche Reaktorsystem eingespeiste Gesamtsauerstoff
etwa 0,5 bis etwa 5, weiter vorzugsweise etwa 1 bis etwa 3, noch
weiter vorzugsweise etwa 1,5 bis etwa 2,5 Mol O2 pro
Mol N-(Phosphonomethyl)iminodiessigsäuresubstrat im wässrigen
Einspeisestrom 1 beträgt,
welcher in den ersten Rührbehälterreaktor 3 eingespeist
wird.
-
Zusätzlich wird
die Verteilung der Sauerstoffgesamteinspeisung in das kontinuierliche
Reaktorsystem zwischen den Rührbehälterreaktoren 3 und 40 gewählt, um
die Menge an Nebenprodukten im Endreaktionsmischungsablauf 45 zu
reduzieren. Der Anteil der Gesamtsauerstoffeinspeisung in das kontinuierliche
Reaktorsystem, welche in den ersten Rührbehälterreaktor 3 eingespeist
wird, beträgt
etwa 10% bis etwa 95%, weiter vorzugsweise etwa 30% bis etwa 95%,
noch weiter vorzugsweise 50% bis etwa 95% und am meisten vorzugsweise
etwa 70% bis etwa 90%, wobei die Restmenge der Gesamtsauerstoffeinspeisung
in den zweiten Rührbehälterreaktor 40 eingeführt wird.
-
Bei
der Praktizierung der vorliegenden Erfindung kann die Konzentration
an nicht umgesetztem N-(Phosphonomethyl)iminodiessigsäuresubstrat,
N-(Phosphonomethyl)glycinprodukt und/oder der Oxidationsnebenprodukte
in der wässrigen
Zwischenproduktreaktionsmischung 41, welche aus dem ersten
Rührbehälterreaktor 3 abgezogen
wird und/oder im Endreaktionsmischungsablauf 45, der aus
dem zweiten Rührbehälterreaktor 40 abgezogen
wird, gemessen werden. Auf der Grundlage dieser Messungen kann die
Gesamtsauerstoffeinspeisung in das kontinuierlicher Reaktorsystem
und/oder die Verteilung der Gesamtsauerstoffeinspeisung zwischen
dem ersten und zweiten Rührbehälterreaktor 3 und 40 eingestellt
werden, um die Ausbeute und Qualität des N-(Phosphonomethyl)glycinprodukts
günstig
zu beeinflussen. Die Konzentration an nicht umgesetztem N-(Phosphonomethyl)iminodiessigsäuresubstrat,
N-(Phosphonomethyl)glycinprodukt und/oder Oxidationsnebenprodukten
kann unter Verwendung der Hochdruckflüssigkeitschromatographie (HPLC)-
oder der Fourie Transformations Infrarotspektroskopie (FTIR)-Analyse
von Stromproben gemessen werden. Zusätzlich kann ein In-line FTIR-Spektrometer
verwendet werden, um Echtzeit-Analysenzusammensetzungen
der Reaktorablaufströme
zu liefern, wobei diese Werte zur Einstellung der Sauerstoffeinspeisepraxis
in das kontinuierliche Reaktorsystem verwendet werden können. Die
In-line-Verwendung der Infrarot Spektroskopie zur Messung der Konzentration
von Analyten in Oxidationsreaktionsmischungen, wie den in Übereinstimmung
mit der vorliegenden Erfindung hergestellten, zur Prozessregelung
und Endpunktsbestimmung sind in der am 22. Mai 2001 angemeldeten
vorläufigen
US-Patentanmeldung Nr. 6,029,265 mit dem Titel "Use of Infrared Spectrocopy for On-Line
Process Control and Endpoint Detection" beschrieben.
-
Umfasst
das kontinuierliche Reaktorsystem zwei kontinuierliche Rührbehälterreaktoren 3 und 40 in Reihe,
beträgt
die Sauerstoffeinspeiserate in die erste Reaktionszone im Normalfall
vorzugsweise etwa 0,5 bis etwa 10, weiter vorzugsweise etwa 0,5
bis etwa 5, noch weiter vorzugsweise etwa 1,0 bis etwa 4,0 Mole
O2 pro Mol N-(Phosphonomethyl)iminodiessigsäuresubstrat,
welches im wässrigen
Einspeisestrom 1 in den ersten Reaktor 3 enthalten
ist. Die Sauerstoffeinspeiserate in die zweite Reaktionszone beträgt vorzugsweise etwa
0,5 bis etwa 10, weiter vorzugsweise etwa 0,5 bis etwa 5, noch weiter
vorzugsweise etwa 2 bis etwa 4 Mole O2 pro
Mol N-(Phosphonomethyl)iminodiessigsäuresubstrat, welches im Einspeisestrom
zur zweiten Reaktionszone enthalten ist.
-
Verwendet
das Verfahren zwei Rührbehälterreaktoren
in Reih, wird im ersten Reaktor vorzugsweise ein solches Molverhältnis von
N-(Phosphonomethyl)iminodiessigsäuresubstrat
zum N-(Phosphonomethyl)glycinprodukt aufrechterhalten, dass die
molare Oxidationsgeschwindigkeit des N-(Phosphonomethyl)iminodiessigsäuresubstrats
mindestens etwa 10-mal, weiter vorzugsweise mindestens etwa 20-mal,
sogar weiter vorzugsweise mindestens etwa 100-mal, sogar noch weiter
vorzugsweise mindestens etwa 150-mal und am meisten vorzugsweise
mindestens etwa 200-mal so schnell ist wie die molare Oxidationsgeschwindigkeit
des N-(Phosphonomethyl)glycinprodukts.
-
Es
können
verschiedene Alternativen zum Fließbild von 3 verwendet
werden, um den teilchenförmigen
heterogenen Katalysator durch die Oxidationsreaktionszonen mit Rückvermischung
innerhalb des kontinuierlichen Reaktorsystem zu zirkulieren. Beispiele
für solche
alternative Fließbilder
sind in 4–6 dargestellt.
In jedem der in 3–7 dargestellten
Fließbilder
kann die Katalysatorlebensdauer innerhalb des gewünschten
Bereichs aufrechterhalten oder nahe bei einem spezifischen Niveau
durch kontinuierliche oder diskontinuierliche Zugabe von frischem
Katalysator zu dem/den Katalysatorrückführstrom(strömen) oder direkt in die jeweiligen
Reaktionszonen eingeregelt werden. Die Katalysatoralterung kann
wahlweise weiterhin auch durch kontinuierliche oder diskontinuierliche
Abnahme eines Teils des Katalysators aus dem/den Katalysatorrücklaufstrom/strömen geregelt
werden. Die abgenommene Katalysatormenge ist oft gleich der dem
System zugesetzten Menge an frischem Katalysator. Die diskontinuierliche
Abnahme und Zugabe des Katalysators schließt jede wiederholte Abnahme
und Zugabe von Katalysator ein, welche nicht kontinuierlich ist.
Die diskontinuierliche Abnahme und Zugabe schließt zum Beispiel den periodischen
Abzug von Katalysator aus einem Katalysatorrücklaufstrom ein, mit einer
Zugabe von frischem Katalysator an einem Punkt stromabwärts des Abnahmepunkts
innerhalb eines Katalysatorkreislaufs. Die diskontinuierliche Abnahme
und Zugabe schließt zum
Beispiel auch den gleichzeitigen Abzug des gesamten Katalysators
aus weniger als allen Reaktionszonen, und die anschließende Zugabe
einer ganz frischen Katalysatorcharge zu weniger als allen Reaktionszonen
ein. Die diskontinuierliche Abnahme und Zugabe schließt zum Beispiel
ferner das gleichzeitige Abziehen des gesamten Katalysators aus
dem kontinuierlichen Katalysatorsystem und die anschließende Zugabe
einer ganz frischen Katalysatorcharge ein (z. B. sobald die Erzeugung
des N-(Posphonomethyl)glycinprodukts aus dem Reaktorsystem einen
vorgegebenen Sollwert auf der Grundlage einer berechneten Gebrauchsdauer
für die
Katalysatorbeladung erreicht hat oder sobald die Katalysatoraktivität bis zu
einem Grad abgenommen hat, der einen wirtschaftlichen Betrieb verhindert).
Das letztere Verfahren ist typischerweise stärker bevorzugt. Dies beruht
zum Beispiel auf der Tatsache, dass es oft schwierig ist das System
zu stabilisieren, wenn zu einem bestimmten Zeitpunkt nur Anteile
der Katalysatorladung abgeführt
und zugegeben werden. Es ist zum Beispiel auch schwierig irgendwelche Änderungen
an einem Katalysator zu analysieren (z. B. neue Verbesserungen) ohne
zuerst den gesamten, nicht modifizierten, Katalysator zu entfernen.
Es sei ferner darauf hingewiesen, dass es bei der Inbetriebnahme
des kontinuierlichen Oxidationsreaktorsystems vorteilhaft sein kann,
das System mit deutlich weniger als der vorgesehen Katalysatorbeladung
(z. B. 75% der vorgesehenen Katalysatorbeladung) zu betreiben und
das System anschließend
allmählich
mit zusätzlichem
Katalysator zu beschicken, um bei den vorgesehenen Betriebsbedingungen
eine optimale Katalysatorbeladung zu erreichen.
-
4 gibt
eine Ausführungsform
wieder, welche eine größer Flexibilität bietet,
weil sie es erlaubt, die Beschickung des ersten und zweiten Rührbehälterreaktors 3 und 40 mit
Katalysator so zu manipulieren, dass im zweiten Rührbehälterreaktor 40 auf
Wunsch eine größere Katalysatorladung
aufrechterhalten werden kann, um die verminderte Triebkraft infolge
der reduzierten Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats
zu kompensieren, welche infolge der geringeren Substratkonzentration
in der zweiten Reaktionszone typischerweise vorliegt. Der Katalysator
wird über
den Katalysatoreinspeisestrom 39 kontinuierlich oder diskontinuierlich
in den ersten Rührbehälterreaktor 3 eingespeist.
Der Katalysator wird dann aus dem ersten Rührbehälterreaktor 3 als
Teil der wässrigen
Reaktionszwischenproduktmischung 41 abgezogen, kontinuierlich
oder diskontinuierlich als Teil der wässrigen Reaktionszwischenproduktmischung 42 in
den zweiten Rührbehälterreaktor 40 eingeführt und
schließlich
kontinuierlich oder diskontinuierlich als Teil der wässrigen Endreaktionsmischung 45 aus
dem zweiten Rührreaktorsbehälter abgezogen.
Der Katalysator wird dann aus der wässrigen Endreaktionsmischung 45 durch
dem Katalysatorfilter 9 im Wesentlichen entfernt, um (1)
einen Katalysatorrücklaufstrom 11 zu
bilden, umfassend in der Hauptsache den gesamten Katalysator und
eine Restmenge N-(Phosphonomethyl)glycinprodukt aus der wässrigen
Endreaktiosmischung 45; und (2) ein Filtrat 13,
umfassend die Hauptmenge N-(Phosphono methyl)glycinprodukt aus der
wässrigen
Endreaktionsmischung 45. Der Katalysatorrücklaufstrom 11 wird
in den Katalysatoreinspeisestrom 39 und einen Katalysatorzwischenprodukteinspeisestrom 50 aufgeteilt.
Der Katalysatoreinspeisestrom 39 wird zum ersten Rührbehälterreaktor 3 rezykliert,
während
der Katalysatorzwischenprodukteinspeisestrom 50 zum zweiten
Rührbehälterreaktor 40 rezykliert
wird. Der Katalysator wird aus dem kontinuierlichen Reaktorsystem
zum Beispiel vorzugsweise durch den Katalysatorsablaufstrom 51 und/oder
den Katalysatorablaufstrom 53 kontinuierlich oder diskontinuierlich
abgenommen und zum Beispiel durch den Katalysatoreinspeisestrom 55 und/oder
Katalysatoreinspeisestrom 57 wieder aufgefüllt. Der
Katalysator könnte
alternativ oder zusätzlich
aus dem Katalysatorrücklaufrstrom 11 abgenommen
werden, und ebenso könnte
frischer Katalysator alternativ oder zusätzlich dem Katalysatorrücklaufstrom 11 vor
der Teilung des Rücklaufstroms 11 in
die Katalysatorrücklaufströme 39 und 50 zugegeben
werden. Der Katalysator kann, wie vorstehend beschrieben, wenigstens
teilweise durch diskontinuierliche oder kontinuierliche Einführung eines
Reduktionsmittels und/oder eines Zusatzaktivators in das kontinuierliche
Reaktorsystem reaktiviert werden, insbesondere wo der Katalysator
den vorstehend beschriebenen tiefenreduzierten Katalysator umfasst.
Das Reduktionsmittel und/oder der Zusatzaktivator kann zum Beispiel
in die Katalysatorrezirkulationsströme 11, 39 und/oder 50 eingeführt werden.
Eine derartige Reaktivierung kann wahlweise in einem oder mehreren
Katalysatorvorratsbehältern
(nicht dargestellt) erfolgen.
-
5 gibt
eine Ausführungsform
wieder, in der jede Oxidationsreaktionszone ihre eigene unabhängige teilchenförmige Katalysatormasse
verwendet. Bei einer solchen Ausführungsform wird ein wässriger
Einspeisestrom 1, welcher das N-(Phosphonomethyl)iminodiessigsäuresubstrat
umfasst, in den ersten Rührbehälterreaktor 3 eingespeist,
in dem er in Gegenwart einer ersten Katalysatormasse kontinuierlich
oxidiert wird, um eine wässrige
Zwischenreaktionsmischung 41 zu bilden. Diese wässrige Zwischenreaktionsmischung 41 wird
im Katalysatorfilter 9a filtriert, um in der Hauptsache
die gesamte erste Katalysatormasse aus der wässrigen Zwischenreaktionsmischung 41 abzutrennen
und (1) einen Katalysatorrücklaufstrom 11a zu
bilden, umfassend in der Hauptsache den gesamten Katalysator aus
der wässrigen
Zwischenproduktmischung 41; und (2) einen wässrigen
Zwischenprodukteinspeisestrom 60, das Filtrat aus dem Filter 9a,
umfassend den Hauptanteil an N-(Phosphonomethyl)glycinprodukt und
nicht umgesetzte N-(Phosphonomethyl)iminodiessigsäure aus
der wässrigen
Reaktionszwischenproduktmischung 41. Der erste Katalysatorrücklaufstrom 11a wird über den
Katalysatorrücklaufstrom 39a in
den ersten Rührbehälterreaktor 3 rezykliert,
während
der wässrige
Zwischenprodukteinspeisestrom 60 in den zweiten Rührbehälterreaktor 40 eingeführt wird,
in dem die weitere Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrat
(und von C1-Molekülen wie Formaldehyd und Ameisensäure) in
Anwesenheit einer zweiten teilchenförmigen Katalysatormasse erfolgt,
um den Endreaktionsmischungsablauf 45 zu bilden. Der Endreaktionsmischungsablauf 45 wird,
gegebenenfalls nach Entfernung des Drucks in einem Entlüftungsbehälter 17b im
Katalysatorfilter 9b filtriert, um die zweite Katalysatormasse
aus der wässrigen
Endreaktionsmischung 45 abzutrennen, um (1) einen Katalysatorrücklaufstrom 11b zu
bilden, umfassend in der Hauptsache den gesamten Katalysator aus
der wässrigen
Endreaktionsmischung 45; und (2) ein Filtrat 13,
umfassend den Hauptanteil an N-(Phosphonomethyl)glycinprodukt aus
der wässrigen Endreaktionsmischung 45.
Der Katalysatorrücklaufstrom 11b wird
dann über
den Katalysatoreinspeisestrom 39b in den zweiten Rührbehälterreaktor 40 rezykliert.
Die im ersten Rührbehälterreaktor
verwendete Katalysatormasse wird durch den Katalysatorauslasstrom 20a vorzugsweise
kontinuierlich oder diskontinuierlich abgelassen und durch den Katalysatoreinspeisestrom 21a wieder
aufgefüllt.
Desgleichen wird die im zweiten Rührbehälterreaktor 40 verwendete
Katalysatormasse durch den Katalystorauslasstrom 20b vorzugsweise
kontinuierlich oder diskontinuierlich abgelassen und durch den Katalysatoreinspeisestrom 21b wieder
aufgefüllt. Die
teilchenförmigen
Katalysatormassen für
den ersten und zweiten Rührbehälterreaktor 3 und 40 können auch,
wie vorstehend beschrieben, durch das kontinuierliche oder diskontinuierliche
Einführen
eines Reduktionsmittels 18a und 18b und/oder eines
Zusatzaktivators 19a und 19b in die jeweiligen
Katalysatorvorratsbehälter 5a und 5b oder
an anderen Stellen des kontinuierlichen Reaktorsystems wenigsten
teilweise reaktiviert werden. Der Zusatzaktivator kann zum Beispiel
einem oder beiden Rührbehälterreaktoren
direkt zugesetzt werden.
-
Das
in 5 dargestellte Katalysatorrücklaufschema ist vorteilhaft,
weil es unabhängig
von der Manipulation des Katalysatortyps, der Alterung und der Beladung
in jeder Reaktionszone für
Flexibilität
sorgt. Der im ersten Rührbehälterreaktor 3 eingesetzte
Katalysator kann zum Beispiel maßgeschneidert werden, um in der
ersten Oxidationsreaktionszone unter ausgewählten Betriebsbedingungen eine
hohe Umwandlung des N-(Phosphonomethyl)iminodiessigsäuresubstrats
zu erhalten, während
der im zweiten Rührbehälterreaktor 40 eingesetzte
Katalysator optimiert sein kann, um eine verbesserte Oxidation der
Nebenprodukte Formaldehyd und Ameisensäure und eine minimale Überoxidation
des N-(Phosphono methyl)glycinprodukts zu erhalten. Auch können zwei
Filterreaktorsysteme, wie das in 5 dargestellte,
ein Filter tolerieren, welches einen Katalysatorrücklaufstrom
erzeugt, welcher weniger konzentriert ist als wie die bei einem
Einzelfilterreaktorsystem gewünschte
Konzentration, wie bei dem in 3 dargestellten
System.
-
Bei
manchen Ausführungsformen
können
die Vorteile eines frischeren Katalysators in einer Reaktionszone
gegenüber
einer anderen größer sein.
Die Wirkung eines alternden Katalysators in der ersten Reaktionszone
(in welcher die Masse des N-(Phosphonomethyl)iminodiessigsäuresubstrats
normalerweise oxidiert wird) kann zum Beispiel bei manchen Ausführungsformen
nicht so schädlich
sein, wie die Wirkung eines alternden Katalysators in der zweiten
Reaktionszone, und die Wirkung eines frischen Katalysator kann ebenso
in der zweiten Reaktionszone größer sein
als in der ersten Reaktionszone. Dies kann zum Beispiel auf Ausführungsformen
zutreffen, bei denen die Masse des N-(Phosphonomethyl)iminodiessigsäuresubstrats
in der ersten Reaktionszone oxidiert wird und die resultierende
geringe Substratkonzentration in der zweiten Reaktionszone eine
geringere Reaktionsgeschwindigkeit verursacht. In einem solchen
Fall kann es manchmal vorzuziehen sein ein Reaktorsystem mit einem
Fließbild
zu verwenden, wie dem in 6 dargestellten.
Bei dieser Ausführungsform
kann der Katalysator aus der im zweiten Rührbehälterreaktor 40 verwendeten
Katalysatormasse kontinuierlich oder diskontinuierlich aus dem Katalysatorrücklaufstrom 11b über den
Strom 65 abgenommen und in den ersten Rührbehälterreaktor 3 über den
Katalysatorrücklaufstrom 11a eingeführt werden,
um dadurch die Gebrauchsdauer des Katalysators im Gesamtverfahren
zu verlängern.
Ein solches Schema ist besonders vorteilhaft, wo der Katalysator
ein teueres Material wie ein Edelmetall umfasst. Bei dieser Ausführungsform
wird frischer Katalysator normalerweise über den Katalysatorseinspeisestrom 21b nur
in die zweite Reaktionszone eingeführt, während der Katalysator aus dem
Verfahren nur über
den Katalysatorabflusstrom 20a entnommen wird. Das durchschnittliche
Katalysatoralter (d. h. die aufaddierte Zeit, während welcher der Katalysator
verwendet worden ist, um die Oxidationsreaktion zu katalysieren)
beträgt
im zweiten Rührbehälterreaktor 40 vorzugsweise
etwa 20 bis etwa 65% des durchschnittlichen Alters des Katalysators,
während
welcher der Katalysator im ersten Rührbehälterreaktor 3 verwendet
worden ist. Die durchschnittliche Menge N-(Phosphonomethyl)glycinprodukt,
welche pro Pfund (lbs) Katalysator im zweiten Rührbehälterreaktor 40 erzeugt
wird, beträgt
vorzugsweise etwa 5 bis etwa 30% der durchschnittlichen Menge des
N-(Phosphonomethyl)glycin produkts, welches pro Pfund Katalysator
im ersten Rührbehälterreaktor 3 erzeugt
wird.
-
Bei
manchen Ausführungsformen
ist es selbstverständlich
weiter vorzuziehen, den Katalysator in der Gegenrichtung zu rezyklieren,
wie in 6 dargestellt (d. h. der Katalysator
fließt
im Gleichstrom mit dem Substrat). In solchen Fällen wird frischer Katalysator
kontinuierlich oder diskontinuierlich in die erste Reaktionszone
eingeführt,
Katalysator aus der im ersten Rührbehälterreaktor 40 verwendeten
teilchenförmigen
Katalysatormasse wird kontinuierlich oder diskontinuierlich aus
dem Katalysatorrücklaufstrom 11a abgenommen
und kontinuierlich oder diskontinuierlich in die zweite Reaktionszone übergeführt, wobei
der Katalysator in der zweiten Reaktionszone kontinuierlich oder
diskontinuierlich aus dem Reaktorsystem abgezogen wird. Bei einer solchen
Ausführungsform
beträgt
das durchschnittliche Katalysatoralter im ersten Rührbehälterreaktor 3 vorzugsweise
etwa 33 bis etwa 80% des durchschnittlichen Alters des Katalysators
im zweiten Rührbehälterreaktor 40.
Die durchschnittliche Menge N-(Phosphonomethyl)glycinprodukt,
welche pro Pfund (lbs) verbrauchter Katalysator im ersten Rührbehälterreaktor 3 erzeugt
wird, beträgt
vorzugsweise etwa 75 bis etwa 90% der durchschnittlich im zweiten
Rührbehälterreaktor 40 erzeugten
Menge N-(Phosphonomethyl)glycinprodukt pro Pfund verbrauchter Katalysator.
-
Bei
der in den 5 und 6 dargestellten
Ausführungsformen
kann der externe Wärmeübertragungskreislauf 15 oder 47 anstatt
eines Katalysatorrezirkulationskreislaufs ebenso auch ein unabhängiger Katalysatorrücklaufstrom 11a bzw. 11b sein,
wie in 2B dargestellt. Bei einem solchen
kombinierten Kreislauf sind die Katalysatorfilter 9a und 9b vorzugsweise
kontinuierliche Kreuzstromfilter.
-
Bei
Verfahren, welche den Betrieb von zwei Oxidationsreaktionszonen
in Reihe, insbesondere zwei Rührbehälterreaktoren 3 und 40 in
Reihe einschließen,
ist in der ersten Oxidationsreaktionszone ein hoher Grad an Massenübergang
erwünscht.
Es ist daher bevorzugt, das O2-haltige Gas,
vorzugsweise ein Gas, welches mindestens etwa 95 Mol.-% O2, typischerweise etwa 98 Mol.-% O2 enthält,
direkt in die Reaktionsmischung im ersten Rührbehälterreaktor 3 über einen
Verteiler einzuführen,
welcher unmittelbar oder nahe beim Flügelrührer angeordnet ist, und auch
die Rückvermischung
des Gases zu minimieren, um die Triebkraft der Sauerstoffkonzentration
für einen
hohen Massenübergang
in der ersten Oxidationsreaktionszone zu maximieren. Im Hinblick
auf äquivalente
Drücke
und Sauerstoffumwandlung, wird erwartet, dass die mittlere räumliche Sauerstoffkonzentration
in Reaktionsumgebungen mit einer minimalen Gasphasenrückvermischung
höher ist. In
der Nähe
des Verteilers ist zum Beispiel der Sauerstoffpartialdruck der ungelösten Gase
zum Beispiel größer als
in anderen Reaktorbereichen, wie nahe der Grenzfläche zwischen
dem flüssigen
Reaktionsmedium und dem Kopfraum. In der zweiten Reaktionszone,
in welcher die Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats
jedoch typischerweise viel niedriger ist, sind die Anforderungen
an den Massenübergang
und der Bedarf an einer hohen Sauerstoffkonzentrationstriebkraft
beträchtlich
geringer. Die Rückvermischung
von Gasen wird daher in der zweiten Oxidationsreaktionszone leichter
toleriert und in manchen Fällen bevorzugt.
Der bei der Praktizierung der vorliegenden Erfindung bevorzugte,
tiefenreduzierte Edelmetall-auf-Kohlenstoff-Katalysator
ist gegen eine Überoxidation
in Reaktionsumgebungen mit Taschen hohen Sauerstoffpartialdrucks
an den ungelösten
Gasen, insbesondere bei geringen Konzentrationen von N-(Phosphonomethyl)iminodiessigsäuresubstrat,
stärker
empfindlich als jene, welche an der zweiten Oxidationsreaktionszone
beteiligt sind. Durch das Rückvermischen
der Gasphase im flüssigen
Reaktionsmedium innerhalb der zweiten Reaktionszone wird die mittlere
Sauerstoffraumkonzentration erniedrigt und die Stabilität eines solchen
Katalysators erhöht.
-
Es
können
verschiedene Reaktormodifikationen eingesetzt werden, um einen einheitlicheren
niedrigen Sauerstoffpartialdruck der ungelösten Gasen in der in der zweiten
Reaktionszone enthaltenen Reaktionsmischung aufrechtzuerhalten.
Eine bevorzugte Alternative besteht darin, für den zweiten Rührbehälterreaktor 40 ein
Rührflügelsystem
zu wählen,
welches darauf ausgelegt ist, für
ein hohe Gasübergangsrate
aus der Kopfraumgrenzfläche
in die Reaktionsmischung zu sorgen, wie ein von Lightnin (Rochester,
New York, U.S.A) erhältliches,
aufwärts
förderndes
A340 Axialstromrührflügelsystem.
Ein solches Rührflügelsystem
zieht Gas aus dem Kopfraum in die flüssige Reaktionsmischung, so
dass der Unterschied zwischen dem Sauerstoffpartialdruck des in
das flüssige
Reaktionsmedium gezogenen Gases und dem Sauerstoffpartialdruck des
Gases im Kopfraum kleiner wird, wodurch die mittlere Sauerstoffraumkonzentration
an ungelösten
Gasen in der Reaktionsmischung geringer wird. Zusätzlich kann
der zweite Rührbehälterreaktor 40 so
modifiziert werden, dass das O2-haltige
Gas vorrangig in den Kopfraum über
der Reaktionsmischung eingespeist wird, anstatt direkt durch die
flüssige
Reaktionsmischung hindurchgeperlt zu werden. Dies reduziert sogar
das Auftreten von Taschen mit hoher Sauerstoffkonzentration noch
weiter. Alternativ kann die mittlere Sauerstoffraumkonzentration durch
Einführen
des Kopfraumgases innerhalb des zweiten Rührbehälterreaktors 40 in
die flüssige
Reaktionsmischung durch den Flügelrührer reduziert
werden. Ein im Handel erhältliches
Beispiel eines solchen Rührflügelsystems,
welches eine hohe Welle für
den Gastransport einschließt,
ist das DISPERSIMAX-System, vertrieben von Autoclave France (Nogent-sur-Oise
Cedex, Frankreich). Eine andere Möglichkeit besteht darin, die O2-Konzentration im O2-haltigen
Gas herabzusetzen, welches in den zweiten Rührbehälterreaktor 40 eingespeist
wird (z. B. kann der zweiten Oxidationsreaktionszone als Sauerstoffquelle
Luft zugeführt
werden).
-
Bei
einer weiteren Modifikation wird der zweite kontinuierliche Rührbehälterreaktor 40 durch
einen Ejektordüsenkreislaufreaktor
ersetzt. 7 zeigt ein schematisches Fließbild eines
solchen Reaktors. Hier wird ein wässriger Einspeisestrom 901,
welcher mindestens einen Teil der wässrigen Reaktionszwischenproduktmischung 41 umfasst,
aus der ersten Oxidationsreaktionszone in den Zulauf 903 gepumpt
und durch eine Düse 907 in
die Mischkammer ausgestoßen,
in die über
den Einlass 911 auch ein O2-haltiges
Gas eingeführt wird
(d. h. das O2-haltige Gas wird in die Verengung
der Venturidüse 907 eingeführt). Dies
schafft einen hohen Massenübergangskoeffizienten
für den
Sauerstoffübergang
in die wässrige
Einspeisung 901. Wegen dieses hohen Massenübergangskoeffizienten
und die starke, durch die Düse 907 verursachte
Bewegung innerhalb des Reaktorbehälters 913, ist die
mittlere Sauerstoffraumkonzentration an ungelöstem Gas in der flüssigen Reaktionsmischung 915 gering.
Der Reaktionsmischungsablauf 917 wird aus dem Auslass 919 nahe
dem Boden des Reaktorbehälters 913 abgezogen,
in einem Wärmetrauscher 921 gekühlt und
mittels eines Filters 922, vorzugsweise einem Kreuzstromfilter,
filtriert. Der aus dem Reaktionsmischungsablauf 917 abgetrennte
Katalysator wird über
den Katalysatorrücklaufstrom 923 unter
Verwendung einer Pumpe 925 in den Reaktor 913 rezirkuliert.
Das Filtrat 927, welches die Hauptmenge des N-(Phosphonomethyl)glycinprodukts
enthält,
wird weitergeleitet, um in zusätzlichen
Schritten gereinigt und/oder konzentriert zu werden. Der Betrieb
und die Auslegung von Ejektordüsenkreislaufreaktoren
wurde von Dierendonck et al. in "Loop
Venturi Reactor – A
Feasible Alternative to Stired Tank Reactors?", Ind. Eng. Chem. Res. 37: 734–738 (1998)
beschrieben. Ein im Handel erhältliches
Beispiel eines Ejektordüsenkreislaufreaktors
ist der von Kvaerner Buss CPS (Pratteln, Schweiz) vertriebene BUSS-Kreislaufreaktor.
Es versteht sich, dass zusätzlich
zum Vorsehen einer zweiten oder weiterer Oxidationsreaktionszonen in
einem kontinuierlichen Reaktorsystem, welches mehrere Oxidationsreaktionszonen in
Reihe umfasst, ein Ejektrodüsenkreislaufreaktor
eine gleichermaßen
geeignete erste Oxidationsreaktionszone bereitstellen könnte. Die
Oxidationsreaktionsbedingungen und Betriebsparameter für einen
Ejektrodüsenkreislaufreaktor
sind denen ähnlich,
die vorstehend für
die von einem Rührbehälterreaktor
bereitgestellten Oxidationsreaktionszonen beschrieben wurden.
-
Die
vorangehende Diskussion hat sich hauptsächlich auf kontinuierliche
Reaktorsysteme konzentriert, welche eine wässrige Katalysatoraufschlämmung verwenden
und mindestens zwei hintereinander geschaltete Rührreaktorbehälter umfassen,
welche Oxidationsreaktionszonen vorsehen, bei denen im Wesentlichen
die Rückvermischung
von wenigstens der flüssigen
Phase erfolgt. Es können
selbstverständlich
andere Reaktorkonfigurationen als Rührreaktorbehälter gleichermaßen oder
besser geeignet sein als Rührbehälterreaktoren für eine oder
mehrere der Oxidationsreaktionszonen oder es könnten in Kombination mit mehrstufigen
Rührbehälterreaktoren
verwendet werden. Des Weiteren sind viele derartige alternative
Reaktorkonfigurationen ebenso für
die Verwendung in kontinuierlichen Reaktorsystemen geeignet, welche
nur eine Oxidationsreaktionszone einschließen. Einer der Nachteile eines
kontinuierlichen Reaktorsystems, welches einen oder mehrere Rührbehälterreaktoren
einschließt,
welche eine teilchenförmige
Katalysatoraufschlämmung
verwenden, sind die Kapital- und
Betriebskosten, welche mit einem, Katalysatorrücksführmechanismus verbunden sind, einschließlich eines
Katalysatorfilters oder andere Katalysatorabtrennvorrichtungen,
welche zur Rückgewinnung
des N-(Phosphonomethyl)glycinprodukts erforderlich sind. Folglich
können
Reaktorkonfigurationen, in denen der Katalysator in der Oxidationsreaktionszone
verbleiben kann, bei manchen Anwendungen einen wirtschaftlichen
Vorteil bieten. Zwei Beispiele solcher Reaktorkonfigurationen sind
Festbettkatalysatorreaktoren und Wirbelbettreaktoren. Ein weitere
Vorteil von Festbettreaktoren und Wirbelbettreaktoren besteht darin,
dass sie in einer Weise betrieben werden können, dass sie Pfropfströmungseigenschaften
aufweisen, welche dazu neigen, niedrigere Konzentrationen an unerwünschten
Nebenprodukten (z. B. N-Methyl-N-(phosphonomethyl)glycin) und folglich
eine größeres Ausbeute
an N-(Phosphonomethyl)glycinprodukt zu erzeugen.
-
8 zeigt
ein Beispiel eines Festbettreaktors 500 in Übereinstimmung
mit einer Ausführungsform
der vorliegenden Erfindung. In einem Reaktor 500 ist eine
primäre
Oxidationszone angeordnet, welche ein primäres Festbett 501 umfasst, welches
einen Oxidationskatalysator enthält,
vorzugsweise einen vorstehend beschriebenen tiefenreduzierten Katalysator.
Innerhalb des Reaktors ist ein Festbettträger 502 angeordnet,
um eine obere Kammer 503 und eine untere Kammer 504 oberhalb
bzw. unterhalb des Festbetts 501 zu bilden. Ein wässriger
Einspeisestrom 505, welcher das N-(Phosphonomethyl)iminodiessigsäuresubstrat
umfasst, wird kontinuierlich oder diskontinuierlich in die obere
Kammer 503 eingespeist und mittels der Sprühdüse 506 oder einem
anderen herkömmlichen
Flüssigkeitsverteilungssystem über das
Festbett 501 verteilt. Desgleichen wird in die obere Kammer 503 ein
O2-haltiges Gas eingespeist. Indem das O2-haltige Gas im Gleichstrom mit dem absteigenden
Strom der flüssigen
Reaktionsmischung durch das Festbett 501 strömt, wird
das N-(Phosphonomethyl)iminodiessigsäuresubstrat kontinuierlich
oxidiert. Ein primärer
Reaktorablauf 507, welcher das N-(Phosphonomethyl)glycinprodukt umfasst,
wird aus der unteren Kammer 504 zusammen mit einem CO2 umfassenden Dampfstrom abgezogen.
-
Obwohl
in 8 eine abwärts
gerichtete parallele Strömung
der flüssigen
Reaktionsmischung und des O2-haltigen Gases
durch das Festbett 501 dargestellt ist, sind selbstverständlich verschiedene
Strömungskombinationen
möglich.
Zum Beispiel könnten
der wässrige
Einspeisestrom 505 und das O2-haltige
Gas in die untere Kammer 504 des Reaktors 500 eingeführt werden
und im Gleichstrom aufwärts
durch das Festbett 501 strömen. Alternativ können die
flüssige
Reaktionsmischung und das O2-haltige Gas
im Gegenstrom durch das Festbett 501 strömen, wobei
das O2-haltige Gas in die untere Kammer 504 und
der wässrige
Einspeisestrom 505 in die obere Kammer 503 eingespeist
wird und umgekehrt.
-
Die
Temperatur innerhalb der Oxidationsreaktionszone des Festbettreaktors 500 liegt
vorzugsweise im Bereich von etwa 20 bis etwa 180°C, weiter vorzugsweise von etwa
60 bis etwa 140°C,
noch weiter vorzugsweise von etwa 80 bis etwa 130°C und sogar
noch weiter vorzugsweise von etwa 90 bis etwa 120°C. Obwohl das
Reaktionssystem wahlweise adiabatisch betrieben werden kann, können in
der primären
Oxidationsreaktionszone, d. h. einer Zone, in die eine beträchtliche
Fraktion an nicht umgesetztem Substrat eingeführt wird, infolge überhöhter Temperaturen,
die mit dem adiabatischen Betrieb zusammenhängen, negative Wirkungen auf
den Katalysator und eine übermäßige Bildung
von Nebenprodukten resultieren. Wenn das Substrat ein Substrat mit
beschränkter
Säurelöslichkeit
ist, muss am Reaktoreinlass eine Temperatur aufrechterhalten werden,
welche mindestens der Sättigungstemperatur
entspricht, um zu verhin dern, dass Substratfeststoffe im Bett abgeschieden
werden. Die Auswirkung auf die Nebenproduktbildung und die Katalysatorschädigung erfordern
jedoch, dass die Maximaltemperatur innerhalb des vorstehend skizzierten
Bereichs liegt. Dies beschränkt
aus praktischen Gründen
den in einem adiabatischen Festbett erzielbaren Umwandlungsgrad
auf nicht mehr als etwa 10 Gew.-%, bezogen auf die Reaktionsgesamtmischung,
vorzugsweise auf nicht mehr als etwa 7%, noch typischer auf den
Bereich von etwa 3% bis etwa 5%. Ist das Substrat ein Salz, wird
die Umwandlung nicht durch die Löslichkeit
des Substrats eingeschränkt,
sondern wird weiterhin durch die vorerwähnte Reihe von Auswirkungen
auf den Katalysator und die Nebenproduktbildung beschränkt. Um
eine bessere Umwandlung in einem einzelnen Festbett zu erreichen,
muss die exotherme Reaktionswärme
aus dem Reaktionssystem abgeführt
werden. Obwohl die Reaktionszone als solche adiabatisch betrieben
werden kann, muss von irgendwoher Wärme aus dem Reaktionssystem
abgeführt
werden, so dass der Unterschied an messbarer Wärme pro Gewichtseinheit zwischen
der Reaktionsmischung und dem wässrigen
Einspeisestrom auf einem Wert gehalten wird, der kleiner ist als
die in der Reaktionszone pro Gewichtseinheit des wässrigen Einspeisestroms
erzeugte exotherme Reaktionswärme.
Maßnahmen
zur Abführung
von Reaktionswärme
können,
wie nachstehend beschrieben, das Kühlen der Reaktionszone oder
das Einführen
eines gekühlten
Rezirkulationsstroms mit wässriger
Einspeisemischung einschließen.
Durch Anwendung einer derartigen Kühlung kann die Umwandlung,
ausgedrückt
als Differenz zwischen der Reaktionsmischungszusammensetzung und der
Einspeisezusammensetzung auf über
10% oder sogar über
15% gesteigert werden. Ist das Substrat und sind die Produkte wasserlösliche Salze,
kann die Umwandlung auf 20%, 30% oder sogar 50% gesteigert werden.
-
Die
Regelung der Temperatur innerhalb der Oxidationsreaktionszone eines
Festbettreaktors ist typischerweise schwieriger als die Temperaturregelung
in einem Reaktorsystem mit Rückvermischung.
Der primäre
Reaktorablauf 507 kann, wie in 8 dargestellt,
in eine primäre
Produktfraktion 508 und eine Reazikulationsfraktion 509 geteilt
werden, welche außerhalb
der Reaktionszone gekühlt
und zum Reaktoreinlass zurückgeführt wird.
Typischerweise wird mindestens etwa 5%, vorzugsweise mindestens
etwa 33%, weiter vorzugsweise etwa 50% bis etwa 90% und sogar weiter
vorzugsweise etwa 60% bis etwa 80% des aus dem Reaktor austretenden
primären
Reaktorablaufs 507 als Rezirkulationsfraktion abgezweigt.
Anders ausgedrückt,
beträgt
das Verhältnis
der volumetrischen Strömungsrate
der Rezirkulationsfraktion 509 zur volumetrischen Strömungs rate
der primären
Reaktionsproduktfraktion 508 typischerweise mindestens
etwa 0,05:1, vorzugsweise mindestens etwa 0,5:1, weiter vorzugsweise
etwa 1:1 bis etwa 10:1 und am meisten vorzugsweise etwa 1,5:1 bis
etwa 5:1. Die Rezirkulationsfraktion wird außerhalb des Festbetts gekühlt, ehe
sie zum Reaktor zurückgeführt wird,
wobei die Kühlung
in einem Wärmetauscher 510 erfolgt.
Bei einer in 8 dargestellten Ausführungsform
wird die aus dem Reaktor austretende primäre Reaktionsmischung in eine
primäre
Produktfraktion und eine Rezirkulationsfraktion geteilt und die
Produktfraktion entfernt, bevor die Rezirkulationsfraktion den Wärmetauscher
durchströmt.
Diese Alternative kann bei bestimmten Ausführungsformen vorteilhaft sein,
wie zum Beispiel dann, wenn die primäre Produktfraktion nicht umgesetztes
N-(Phosphonomethyl)iminodiessigsäuresubstrat
oder C1-Verbindungen als Nebenprodukte enthält, die
in einer weiteren Reaktionszone oxidiert werden sollen. Bei einer
in 8 dargestellten Alternative kann es vorzuziehen
sein, eine gekühlte
Produktfraktion zu erzeugen, indem man die gesamte primäre Reaktionsmischung,
oder die im Wesentlichen gesamte primäre Reaktionsmischung durch
einen externen Wärmetauscher
leitet, wie er als 510 dargestellt ist, und den gekühlten Strom
der primären
Reaktionsmischung in einen Rezirkulationsstrom und eine gekühlte Primärfraktion
teilt.
-
Die
gekühlte
Rezirkulationsfraktion 509 und der wässrige Einspeisestrom 505 werden
gemischt, um einen kombinierten Einlassstrom für die primäre Reaktionszone zu erzeugen.
Infolge der Reaktion armt die Rezirkulationsfraktion an N-(Phosphonomethyl)iminodiessigsäuresubstrat
ziemlich aus, ein Faktor, der dazu genutzt werden kann, die Produktivität durch
Einführen
einer wässrigen
Einspeisemischung mit einem hohen Substratgehalt zu maximieren,
einschließend
Substratkonzentrationen, welche die Löslichkeitsgrenze des Substrats
in der wässrigen
Phase der Einspeisemischung überschreiten.
Nachdem die Rezirkulationsfraktion an Substrat ziemlich ausgearmt
ist, bewirkt das Mischen die Erzeugung eines kombinierten Einlassstroms
mit einem beträchtlich
niedrigeren Substratgehalt als dem der wässrigen Einspeisemischung.
Diese Verdünnungswirkung
macht eine Einspeisemischung möglich,
welche konzentrierter ist als dies anderweitig möglich wäre. Die Einspeisemischung kann
zum Beispiel eines Aufschlämmung
von N-(Phosphonomethyl)iminodiessigsäure in einer gesättigten
oder im Wesentlichen gesättigten
Lösung
davon umfassen, welche andernfalls dazu neigen würde, das Verstopfen des Katalysatorfestbetts
zu verursachen. Das Mischen mit der Rezirkulationsfraktion reduziert
den N-(Phosphonomethyl)iminodiessigsäuregehalt ausreichend, um die
Feststoffe der Auf schlämmung
zu lösen
und einen kombinierten Einlassstrom vorzusehen, der im Wesentlichen
frei von festem Substrat ist. Typischerweise gibt die Wärme aus
der Rezikulationsfraktion Anlass dafür, dass die Temperatur des
kombinierten Stroms diejenige der wässrigen Einspeisemischung überschreitet,
wodurch sie zusätzlich zur
Auflösung
der Substratfeststoffe beiträgt.
Darüber
hinaus beeinflusst die Verdünnungswirkung
die Reaktionsgeschwindigkeit nicht negativ, weil die Oxidation von
N-(Phosphonomethyl)iminodiessigsäure
zu N-(Phosphonomethyl)glycin bis zu hohen Umsetzungsgraden im Wesentlichen
nach nullter Ordnung verläuft.
Wahlweise kann die wässrige
Einspeisemischung und die Rezikulationsfraktion zu einem Mischbehälter geleitet
werden, um sicherzustellen, dass die Feststoffe gelöst worden
sind, ehe der kombinierte Einlassstrom in das Katalysatorfestbett
eingeleitet wird. Auf diese Weise kann eine wässrige Einspeisemischung eingeführt werden, welche
zwischen etwa 8% und etwa 15 Gew.-% N-(Phosphonomethyl)iminodiessigsäure umfasst,
und durch Mischen dieser wässrigen
Einspeisemischung mit einer primären
Reaktorrezirkulationsfraktion ein kombinierter Einlassstrom erzeugt
werden, welcher zwischen etwa 0,5% und etwa 5 Gew.-% N-(Phosphonomethyl)glycin
umfasst. Es können,
wie hierin nachstehend beschrieben, beträchtlich höhere Konzentrationen verarbeitet werden,
wenn das Substrat ein wasserlösliches
Salz von N-(Phosphonomethyl)iminodiessigsäure und das Produkt ein wasserlösliches
Salz von N-(Phosphonomethyl)glycin ist.
-
Es
versteht sich, dass bei Verringerung des Substratgehalts im kombinierten
Einlassstrom die Verdünnung
des wässrigen
Einspeisestroms mit dem Rezirkulationsstrom dazu dient, den Unterschied
des Substratgehalts zwischen dem kombinierten flüssigen Katalysatorbetteinlasstrom
und dem flüssigen
Katalysatorbettauslasstrom zu verringern, wobei diese Differenz
in einem Bereich aufrechterhalten werden kann, der in einer adiabatischen
Reaktionszone toleriert werden kann, wir vorstehend beschrieben.
Die Aufrechterhaltung solcher Grenzen bezüglich der anteiligen Umwandlung
des Substrats innerhalb der Reaktionszone bleibt im System von 8 insofern
wichtig, als das Katalysatorbett selbst im Wesentlichen adiabatisch
betrieben werden kann, obwohl dies beim Gesamtreaktionssystem einschließlich des
Rezirkulationskreislaufs nicht der Fall ist.
-
Bei
einer alternativen Ausführungsform
der Erfindung (nicht dargestellt) kann ein kontinuierliches Katalysatorsystem
eine zweite Oxidationsreaktionszone umfassen, in die ein Teil oder
die gesamte primäre
Reaktionsproduktfraktion 508 zur weiteren Umwandlung des
N-(Phosphonomethyl)iminodiessigsäuresubstrats und
die Oxidation der C1-Nebenprodukte kontinuierlich
eingeführt
werden kann. Bei einer solchen Ausführungsform kann der gesamte
primäre
Reaktorablauf 507 in den Wärmetauscherrücklaufkreislauf
und die primäre
Produktfraktion 508 geteilt werden und aus der Rezikulationsfraktion 509,
welche stromabwärts
des Wärmetauschers 510 abgenommen
wird. Auf diese Weise wird ein Teil der exothermen Reaktionswärme aus
der primären
Produktfraktion 508 abgeführt werden, bevor sie in die
zweite Oxidationsreaktionszone eintritt. Die zweite Reaktionszone
enthält
einen Katalysator und kann, wie innerhalb des kontinuierlichen Rührbehälterreaktors
vorgesehen, rückvermischt
werden, oder kann ein zweites Katalysatorfestbett umfassen. Das
restliche N-(Phosphonomethyl)iminodiessigsäuresubstrat in der primären Reaktionsproduktfraktion
wird in der zweiten Reaktionszone kontinuierlich zu N-(Phosphonomethyl)glycin
oxidiert. Bei einer bevorzugten Ausführungsform, welche zwei oder
mehrere Festbettreaktoren in Reihe umfasst, wird bei der Reaktion
von N-(Phosphonomethyl)iminodiessigsäuresubstrat zu N-(Phosphonomethyl)glycinprodukt
in der primären
Oxidationsreaktionszone eine hohe Umwandlung herbeigeführt (z.
B. mindestens etwa 95%, vorzugsweise mindestens etwa 98%), was ganz
einfach ist, weil die Oxidation nach pseudo-erster Ordnung verläuft, bis
nur noch ein geringer Bruchteil des ursprünglichen N-(Phosphonomethyl)iminodiessigsäuresubstrats,
z. B. nur 0,2 ppm oder weniger, in der flüssigen Phase verbleibt. Bei
einem derartigen Betrieb wird die Wärmeinhalt der Reaktion ganz
hauptsächlich über den
Wärmetauscher
510 im Rücklaufkreislauf
des Primärreaktors
abgeführt,
so dass die zweite Oxidationsreaktionszone mit nur mäßiger Temperaturerhöhung im
Wesentlichen adiabatisch betrieben werden kann. Wahlweise kann die
Reaktionswärme über eine
Kühlflüssigkeit
in einem internen Wärmetauscher
(z. B. Kühlschlangen)
abgeführt
werden, welcher innerhalb der zweiten Reaktionszone angeordnet ist.
Eine derartige Anordnung ohne Rezirkulation ermöglicht es, die Flüssigphasenreaktionsmischung
durch ein zweites Festbett im Wesentlichen in Pfropfströmung (d.
h. im Wesentlichen ohne Rückvermischung
der flüssigen
Phase) strömen
zu lassen. Der Pfropfströmungsbetrieb
ist in der zweiten Reaktionszone erwünscht, weil die Oxidation des
Substrats zum N-(Phosphonomethyl)glycinprodukt bei hohen Umwandlungen
im Wesentlichen nach erster Ordnung verläuft. Der Pfropfströmungsbetrieb
maximiert die kinetische Triebkraft für die Beseitigung des restlichen
N-(Phosphonomethyl)iminodiessigsäuresubstrats
und reduziert die Nebenproduktmenge aus der Überoxidation.
-
Vorzugsweise
enthalten beide, das primäre
und sekundäre
Katalysatorbett ein Edelmetall auf einem Kohlenstoffkatalysators,
welches sowohl für
die Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats
als auch der C1-Nebenprodukte Formaldehyd
und Ameisensäure
wirksam ist.
-
Nachdem
das Edelmetall hauptsächlich
die Katalyse der C1-Nebenprodukte bewirkt,
während
die Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats
hauptsächlich
durch den Kohlenstoff katalysiert wird, umfasst eine alternative
Ausführungsform
der Erfindung die Verwendung eines primären Festbetts, welches in der
Hauptsache aus dem Kohlenstoffkatalysator besteht oder einen beträchtlich
geringeren Metallgehalt aufweist als der im zweiten Festbett verteilte
Katalysator. Der zweite Reaktor umfasst ein Edelmetall auf einem,
Kohlenstoffkatalysator, um die Oxidation der C1-Nebenprodukte
sicherzustellen. Nachdem die C1-Oxidation
in jedem Fall im Wesentlichen nach erster Ordnung verläuft, verläuft sie
unter im Wesentlichen Pfropfströmungsbedingungen,
welche sich im zweiten Reaktor infolge des viel geringeren Anfalls
an exothermer Wärme
in diesem Stadium leicht aufrechterhalten lassen, effektiver. Der
Wärmeinhalt
darf im zweiten Reaktor so mäßig sein,
dass er ohne externen Wärmetauscher
und ohne Rückvermischung
oder Rezirkulation betrieben werden kann. Bei einem Festbettsystem
kann es machbar sein einen Katalysator mit einer geringeren Edelmetallbeladung
pro Katalysatorgewichtseinheit zu verwenden, wie sie für ein kontinuierliches
Reaktionssystem mit Rückvermischung
optimal wäre.
-
Bei
einer noch weiteren Ausführungsform
kann eine dritte Festbettreaktionszone vorgesehen werden, welche
ebenfalls ein Festbett umfasst, welches ein Edelmetall auf Kohlenstoffkatalysator
enthält,
und die im Wesentlichen in Pfropfströmung oder wahlweise, in der
Tat vorzugsweise, unter adiabatischen Bedingungen betrieben werden
kann. Diese Option kann besonders wertvoll sein, wenn der erste
Reaktor nur einen Kohlenstoffkatalysator verwendet. Folglich verläuft die
Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats zu N-(Phosphonomethyl)glycin
in Anwesenheit des Kohlenstoffkatalysators in der primären Oxidationsreaktionszone
gut, C1-Nebenprodukte neigen aber dazu sich
im primären
Reaktorablauf anzusammeln. Die zweite Reaktionszone fördert sowohl
die Beseitigung des Substrats als auch die Oxidation der C1-Nebenprodukte, welche beide hauptsächlich Reaktionen
erster Ordnung sind, welche hauptsächlich auf dem Pfropfströmungsbetrieb
des zweiten Festbetts beruhen. Die restlichen C1-Verbindungen
werden in der dritten Festbettoxidationsreaktionszone wirksam beseitigt.
-
Um
eine hohe Umwandlung in der primären
Oxidationsreaktionszone zu erzielen, wird jede der Festbettreaktionszonen,
insbesondere die primäre
Reaktionszone, vorzugsweise unter einem relativ hohen Sauerstoffpartialdruck
betrieben, um die Sauerstoffübertragung
in die flüssige
Phase zu fördern.
Der integrierte mittlere Sauerstoffpartialdruck entlang des Strömungsweges
der flüssigen
Phase in der primären
Oxidationsreaktionszone beträgt
vorzugsweise mindestens etwa 345 kPa (50 psia), vorzugsweise mindestens
etwa 690 kPa (100 psia), weiter vorzugsweise mindestens etwa 1379
kPa (200 psia). Bei manchen Ausführungsformen
können
integrierte mittlere Sauerstoffpartialdrücke im Bereich von etwa 2068
kPa (300 psia) bis etwa 3447 kPa (500 psia) geeignet sein. Der Sauerstoffgehalt
der Gasphase am Gaseinlass in den Reaktor kann im Bereich von 20%
bis 30% oder sogar darunter liegen. Der Sauerstoffübergang
kann in einem Festbettreaktor auch durch das hohe Verhältnis von
Katalysatoroberfläche
zum Volumen der Flüssigphasenreaktionsmischung
gefördert
werden, wie hierin nachstehend beschrieben. Der Sauerstoffverbrauch
in der primären
Reaktionszone liegt vorzugsweise zwischen etwa 50% und etwa 95%.
Der Sauerstoff wird typischerweise in den Reaktor in einer Menge
von etwa 1.5 bis etwa 10 Mole O2/Mol N-(Phosphonomethyl)iminodiessigsäuresubstrat
eingespeist. Der Gesamtbetriebsdruck im Festbettreaktor 500 ist
typischerweise höher
als in einem Rührbehälterreaktor
und beträgt
vorzugsweise etwa 0 bis etwa 6895 kPa (0–1000 psig), weiter vorzugsweise
etwa 2068 bis etwa 6895 kPa (300–1000 psig) und sogar weiter
vorzugsweise 6909 bis etwa 2068 kPa (100–300 psig).
-
Im
Allgemeinen können
in einer zweiten und/oder dritten Festbettoxidationsreaktionszone
etwas niedrigere Sauerstoffpartialdrücke bevorzugt sein, um die Überoxidation
des Katalysators und die Schädigung
seiner Wirksamkeit bei der Oxidation von C1-Nebenprodukten
zu vermeiden. Demzufolge beträgt
in der zweiten oder dritten Reaktionszone der integrierte mittlere
Sauerstoffpartialdruck entlang des Flüssigkeitsströmungsweges
vorzugsweise zwischen etwa 207 kPa (30 psia) und etwa 2068 kPa (300
psia), weiter vorzugsweise zwischen etwa 207 kPa (30 psia) und etwa
690 kPa (100 psia). Alternativ kann/können der primäre Festbettreaktor
von 8 und/oder der/die nachgeschaltete(n) zweite(n)
oder dritte(n) Festbettreaktor(en) unter Verwendung eines anderen
Oxidationsmittel als molekularer Sauerstoff, wie zum Beispiel H2O2, betrieben werden,
wobei in diesem Fall der Reaktionsgesamtdruck und der Sauerstoffpartialdruck
wesentlich niedriger sein können als
wie vorstehend beschrieben.
-
Um
den Katalysator gegen eine Überoxidation
zu schützen
ist es generell bevorzugt, das der Sauerstoffpartialdruck am Flüssigkeitsauslass
eines jeden Festbett reaktors nicht größer als etwa 690 kPa (100 psia) beträgt und vorzugsweise
zwischen etwa 86,9 kPa (10 psia) und etwa 345 kPa (50 psia) liegt.
Es ist ebenfalls bevorzugt, dass der Sauerstoffpartialdruck an keiner
Stelle im Festbett, in dem der N-(Phosphonomethyl)iminodiessigsäuresubstratgehalt
der flüssigen
Phase weniger als 0,2 ppm beträgt,
345 kPa (50 psia) überschreitet,
und dass an allen Stellen im Bett, an denen der Substratgehalt der
flüssigen
Phase kleiner als etwa 0,1 ppm ist, vorzugsweise ein Sauerstoffpartialdruck
unter etwa 345 kPa (50 psia) aufrechterhalten wird.
-
Bei
noch einer weiteren Ausführungsform
kann das kontinuierliche Reaktorsystem eine Vielzahl kürzerer (flacherer)
Festbettreaktoren in Reihe dermaßen umfassen, dass der aus
einer Stufe austretende Reaktionszwischenproduktmischungsablauf
die nachfolgende Stufe durchströmt.
Diese Ausführungsform weicht
vom vorstehend beschriebenen System mit zwei- oder drei Reaktoren
insofern ab, als in jeder der Reihe von relativ flachen Festbettreaktorstufen
nur eine mäßige Umwandlung
erzielt werden würde.
Nachdem die Substratumwandlung in jedem einzelnen Bett relativ beschränkt ist,
kann jedes Bett im Wesentlichen adiabatisch betrieben werden, wobei
zwischen jedem der kürzeren
Festbettreaktoren und dem unmittelbar folgenden Reaktor ein Wärmetauscher
angeordnet wird, um die Reaktionsmischung so zu kühlen, dass
die Temperatur der Reaktionsmischung in keinem der Festbettreaktoren
die gewünschte
Betriebstemperatur überschreitet. Umfasst
die Serie mehr als zwei Reaktoren, kann es erforderlich sein, nur
die aus der ersten Stufe austretende Reaktionszwischenproduktmischung
zu kühlen,
worauf die restlichen zwei oder drei Festbettreaktionszonen leichter
adiabatisch betrieben werden können.
Die Reaktionstemperatur kann in einem Festbettreaktor zum Beispiel
auch dadurch geregelt werden, dass in das Festbett separate Kanäle oder
Leitungen eingebaut werden, durch die ein Kühlmittel geleitet werden kann.
Es sei darauf hingewiesen, dass bei dieser Ausführungsform nicht alle Reaktoren
in der Reihe notwendigerweise Festbettreaktoren sein müssen. Der
erste Reaktor der Reihe könnte
zum Beispiel ein kontinuierlicher Rührbehälterreaktor sein, in dem eine
Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats zu N-(Phosphonomethyl)glycin
nach im Wesentlichen erster Ordnung mit weitestgehender Umwandlung
erfolgen kann, um eine Reaktionszwischenproduktmischung zu erzeugen,
und die Reaktionszwischenproduktmischung dann zu einem Festbettreaktor
oder einer Reihe von Festbettreaktoren weitergeleitet wird, um die
Umwandlung zu vervollständigen
und die restlichen C1-Nebenprodukte zu oxidieren.
-
Festbettkatalysatoren,
welche einen Katalysator mit monolithischer Form (z. B. umfassend
einen Honigwabenträger
wie den in 1 dargestellten) enthalten,
sind manchmal stärker
bevorzugt als Reaktoren, welche ein Festbett mit einzelnen Katalysatorteilchen
enthalten. Dies beruht auf der Tatsache, dass ein Festbett mit Katalysatorteilchen
verstopfen kann, wenn das im wässrigen
Einspeisestrom 505 enthaltene N-(Phosphonomethyl)iminodiessigsäuresubstrat
in der Oxidationsreaktionszone zu einem beträchtlichen Grad ausfällt. Folglich
wird typischerweise gefordert, dass die Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats
im wässrigen
Einspeisesstrom 505 die Sättigungskonzentration bei der
Reaktoreinspeisetemperatur nicht übersteigt, was den Durchsatz
beträchtlich
beschränken
kann. Umfasst das Festbett 501 jedoch einen Katalysator
in Bienenwabenform oder einen ähnlichen
Monolithen, können
die Kanäle
darin im Wesentlichen gerade und mit einem Querschnitt hergestellt
werden, welcher groß genug
ist, dass sie von einer Reaktionsmischung, welche eine Aufschlämmung von
festem N-(Phosphonomethyl)iminodiessigsäuresubstrat enthält, nicht
verstopft werden. Auch wenn ein Reaktor mit gepacktem Bett nicht
verstopft, so kann der Monolith doch mit wesentlich geringerem Druckabfall
betrieben werden. Dieser allfällige
Vorteil der Verwendung eines monolithischen Katalysators in einem
Festbettreaktor muss gegenüber
den höheren
Kosten abgewogen werden, die mit der Herstellung des monolithischen
Trägers
verbunden sind, verglichen mit den oft beträchtlich preiswerteren Pellets
oder teilchenförmigen
Trägern,
die bei der Nutzung der vorliegenden Erfindung im Allgemeinen bevorzugt
sind. Dies trifft insbesondere dann zu, wenn mehrere Festbettstufen
mit getrennten N-(Phosphonomethyl)iminodiessigsäuresubstrateinspeiseströmen in jeder
Stufe eingesetzt werden, wodurch die Notwendigkeit einer hohen Substratkonzentration
im Einspeisestrom zur ersten Festbettstufe vermieden wird, um den gewünschten
Durchsatz zu erhalten.
-
Ein
weiterer Vorteil eines Festbettreaktors ist, dass durch Kombinieren
unterschiedlicher Katalysatoren die Katalysatoraktivität über die
Länge der
Festbettreaktorstufe oder von einer Stufe zur nächsten in Richtung der Reaktionsmischungsströmung selektiv
variiert werden kann. Ein weniger aktiver Katalysator (z. B. ein nur
aus Kohlenstoff bestehender Katalysator) kann zum Beispiel im Aufwärtsstromteil
einer Festbettreaktorstufe oder in den vorderen Stufen eines mehrstufigen
Festbettreaktorsystems verteilt werden und ein aktiverer Katalysator
(z. B. ein Katalysator mit tiefenreduziertem Edelmetall auf Kohlenstoff)
kann im Abwärtsstromteil einer
Festbettreaktorstufe oder den nachfolgenden Stufen eines mehrstufigen
System verteilt werden. Alternativ kann das Festbett eine Kombination
von Oxidationskatalysatorkörpern
und andere Mittel zur Förderung der
Gas/Flüssigkeits-Massenübertragung
umfassen, wie Ringe, Sattelkörper
oder strukturierte Füllungen.
Die Ringe, Sattelkörper
oder strukturierten Füllungen
fungieren als Verdünnungsmittel
für den
Katalysator, indem sie die Aktivität des Katalysatorbetts modulieren.
Auf diese Weise kann das Katalysatorbett in Richtung der Flüssigkeitsströmung in
Abhängigkeit
von der Veränderung
der Oberfläche
der Katalysatorkörper
verändert werden,
verglichen mit der Oberfläche
der inerten Füllung.
Eine solche Veränderung
der Katalysatoraktivität dient
dazu, den Konzentrationsabfall des N-(Phosphonomethyl)iminodiessigsäuresubstrats
in der Reaktionsmischung auszugleichen und gleichzeitig die Katalysatorkosten
und die Edelmetallverluste aus dem Verfahren zu verringern.
-
Die
Neigung der Festbettreaktoren infolge der Pfropfströmungseigenschaften
niedrigere Konzentrationen an unerwünschten Nebenprodukten zu erzeugen,
kann durch Verwendung eines Verhältnisses
von wirksamer Katalysatoroberfläche
zur Flüssigkeit
im Arbeitsvolumen, welches beträchtlich
größer ist
als das in typischen Reaktoren mit Rückvermischung (d. h. gut gemischten
Reaktoren), erhöht
werden. Die Notwendigkeit die Reaktionsmischung zu kühlen, um
die Bildung von Verunreinigungen zu verringern kann durch Verwendung
eines solchen Verhältnisses
in der Tat verringert werden. Dies beruht auf der Tatsache, dass
die große wirksame
Katalysatoroberfläche
die Reaktionsgeschwindigkeit erhöht
und folglich die Verweilzeit der Flüssigkeit verkürzt. Die
verkürzte
Verweilzeit tendiert wiederum dazu, die Bildung von Verunreinigungen,
welche durch homogene Reaktionen gebildet werden, insbesondere N-Methyl-N-(phosphonomethyl)glycin
zu vermindern. Bei dieser Ausführungsform
beträgt
das Verhältnis
der Katalysator-BET-Oberfläche
zum Flüssigkeitsvolumen
(Betriebsinhalt) im Arbeitsvolumen des Festbettreaktors vorzugsweise
mindestens etwa 3 m2/cm3,
weiter vorzugsweise etwa 100 bis etwa 6000 m2/cm3 und sogar weiter vorzugsweise etwa 200
bis etwa 2000 m2/cm3.
Bei manchen Anwendungen kann eine Katalysator-BET-Oberfläche zum
flüssigen
Betriebsinhalt des Reaktors am meisten vorzugsweise im Bereich von
etwa 400 bis etwa 1500 m2/cm3 liegen.
Das volumetrische Volumen zum Gesamtbettvolumen im Festbett liegt
vorzugsweise zwischen 0,1 und 0,7. Bei bestimmten Ausführungsformen
kann es die niedrige Flüssigkeitsverweilzeit
und das hohe Verhältnis
von Oberfläche
zu Volumen vorteilhaft machen, einen Festbettreaktor in einem relativ
hohen Temperaturbereich, z. B. 150°C, zu betreiben, wobei die integrierte
mittlere Temperatur der flüssigen
Phase über
den Flüssigkeitsströmungsweg
im primären
Festbett zwischen etwa 80°C
und etwa 130°C,
vorzugsweise 105°C
bis 120°C
liegt.
-
Ein
Festbettreaktor kann in Übereinstimmung
mit der Erfindung mit einem hohen Durchsatz betrieben werde, vorausgesetzt,
dass eine angemessene Wärmeübertragungskapazität bereitgestellt
wird, wie zum Beispiel in 8 abgebildet
oder hierin nachstehend beschrieben ist. Ein Festbettreaktor kann
im Allgemeinen mit einer stündlichen
Flüssigkeitsraumgeschwindigkeit
zwischen etwa 0,5 h–1 und etwa 20 h–1 betrieben
werden, berechnet auf der Grundlage des Katalysatorbettgesamtvolumens
bei N-(Phosphonomethyl)iminodiessigsäuresubstratumwandlungen von
mehr als 50%. Höhere
Umwandlungen von mehr als 95% oder 98%, können bei stündlichen Flüssigkeitsraumgeschwindigkeiten
im Bereich von etwa 0,5 bis etwa 5 h–1 erzielt
werden. Die stündliche
Flüssigkeitsraumgeschwindigkeit
beruht selbstverständlich
auf dem gesamten Flüssigphaseneinspeisestrom.
In dem in 8 abgebildeten Reaktionssystem
umfasst der Flüssigphaseneinspeisestrom folglich
sowohl den kombinierten Einlassstrom, welcher durch Mischen des
wässrigen
Einspeisemischungsstroms mit dem Rezirkulationsstrom erzeugt wird,
als auch irgendeinen anderen Rücklaufstrom
oder einen Querflussstrom, welche in Übereinstimmung mit einem speziellen
Vefahrensfließbild
in einen Festbettreaktor eingeführt
werden können.
Auf der Basis dieser stündlichen
Raumgeschwindigkeiten, verbunden mit den vorstehend erörterten
Umwandlungen, wurde festgestellt, dass ein N-(Phosphonomethyl)iminodiessigsäuresubstrat
in einem einzelnen Festbettreaktor mit Produktivitäten im Bereich
von etwa 0,05 bis etwa 4, weiter vorzugsweise von etwa 0,2 bis etwa
2 gMol/1 h N-(Phosphonomethyl)glycin pro Pfund wässrige Reaktionsmischung erzeugt.
-
In
Festbettreaktoren verwendete Katalysatoren können, wie vorstehend beschrieben,
eine Vielfalt von Formen annehmen, einschließend sowohl Pelletträger als
auch monolithische Träger.
Es ist, wie in 8 dargestellt, allgemein bevorzugt,
dass der im Festbett 501 enthaltende Oxidationskatalysator
in Form von Pellets vorliegt (z. B. der vorstehend beschriebene
tiefenreduzierte Katalysator, welcher einen Pelletträger mit
einem darauf abgeschiedenes Edelmetall umfasst). Solche Pelletkatalysatoren
besitzen typischerweise eine Teilchengröße von etwa 1 mm bis etwa 10
mm auf, weiter vorzugsweise von etwa 1,5 bis 5 mm. Es wurde weiterhin
festgestellt, dass die Edelmetallbeladung bei einem Katalysator
vom Typ Edelmetall auf Kohlenstoff, welcher in einem Festbett verwendet
wird, verglichen mit der Beladung eines Katalysators zur Verwendung
in einem Ausschlämmungsreaktor,
relativ niedrig sein kann. Der wirksame Betrieb eines Festbetts
ist zum Beispiel durch Verwendung eines Katalysators mit 2 Gew.-%
auf Kohlenstoff bei einem Verhältnis
von 200 bis 2000 m2/cm3 BET-Oberfläche zu dem
vorstehend aufgeführten
flüssigen
Betriebsvolumen gezeigt worden. Generell kann eine Beladung kleiner
als 35 Gew.-% zufrieden stellend sein. Umfasst der Katalysator Platin
auf Kohlenstoff, kann die Platinbeladung auf dem Katalysator weniger
als 70% derjenigen Ladung betragen, welche erforderlich ist, um
eine äquivalente
Produktivität
in Pfund N-(Phosphonomethyl)glycinprodukt pro Stunde und Pfund Katalysator
bei gleicher Temperatur in einem kontinuierlichen Rührbehälterreaktor
unter Verwendung eines Aufschlämmungskatalysators
aus Platin auf Kohlenstoff-Basis vorzusehen.
-
Es
ist in einem Festbettreaktor schwierig über längere Zeit eine konstante Katalysatoraktivität und -selektivität aufrechtzuerhalten.
Unter Umständen
fällt die
Aktivität
und Selektivität
des Katalysators auf ein unannehmbares Niveau ab, so dass das Reaktorsystem
abgeschaltet werden muss, um den Ersatz und/oder die Reaktivierung
des Katalysators zu ermöglichen.
Dies ist verglichen mit einem kontinuierlichen Reaktorsystem ein
Nachteil, welches einen oder mehrere der vorstehend beschriebenen
Rührbehälterreaktoren
einschließt, wo
der Ersatz und/oder die Reaktivierung des Katalysators erfolgen
kann, während
das Reaktorsystem in Betrieb bleibt. Das Problem der Katalysatorentfernung
und -rückgewinnung
kann gelöst
werden, indem Zwei Festbettreaktoren vorgesehen werden, welche entgegen
dem Rest des Reaktorsystems parallel geschaltet sind und wechselseitig
betrieben werden. Der Katalysator kann aus dem Reaktor, welcher
außer
Betrieb ist entfernt und durch frischen Katalysator ersetzt werden,
oder die Katalysatorreaktivierung kann in dem Reaktor in situ erfolgen,
welcher nicht angeschlossen ist.
-
In Übereinstimmung
mit einer anderen Ausführungsform
der vorliegenden Erfindung erfolgt die Oxidation des N-(Phosphonomethyl)iminodiessigsäuresubstrats
in einem Wirbelbettkreislaufreaktor unter Verwendung eines teilchenförmigen heterogenen
Katalysators, vorzugsweise dem vorstehend beschriebenen teilchenförmigen tiefenreduzierten
Katalysator. Ein Wirbelbettkreislaufreaktor sorgt typischerweise
für einen
größeren Massenübergangskoeffizienten
als ein Rührbehälterreaktor
und kann in einer Weise betrieben werden. die den teilchenförmigen Katalysator
im Wesentlichen innerhalb der Oxidationsreaktionszone derart zurückhält, dass
ein Katalysatorfilter oder eine andere Katalysatortrennvorrichtung überhaupt
nicht benötigt
wird oder wenigstens in geringerer Größe und geringeren Anforderungen
an die Dekompression. 9 gibt ein Beispiel eines Wirbelbettkreislaufreaktors 400 mit
einer darin definierten Oxidationsreaktionszone wieder. Ein das N-(Phosphonomethyl)iminodiessigsäuresubstrat
umfassender wässriger
Einspeisestrom 401 wird oben in den Einlass 403 der
Einspeiseröhre 405 gepumpt
und in der Nähe
des Bodens des Reaktors in das flüssige Reaktionsmedium 406 in
Kontakt mit den Katalysatorteilchen 407 ausgestoßen. Durch
die Reaktionsmischung kann durch eine Düse 409 am Boden des
Reaktors 400 ein O2-haltiges Gas
durchgeperlt werden. Die Reaktionslösung 412 wird über eine Überlauföffnung 411 abgezogen
und der CO2-haltige Dampf wird durch das
Oberteil des Reaktors abgelüftet.
Der Reaktor weist eine Reaktionsmischungszirkulationsabzugsöffnung 413 auf,
welche reichlich oberhalb der Ausstoßmündung der Einspeiseröhre 405 aber
unterhalb der Überlauföffnung 411 angeordnet
ist. Die Reaktionsmischung mit dem darin suspendierten teilchenförmigen Katalysator
wird über die Öffnung 413 abgezogen,
zur Abführung
der Reaktionswärme über einen
externen Kreislauf 420 durch einen Wärmetauscher 421 zirkuliert
und dann mit dem Einspeisestrom 401 zur Wiedereinspeisung
in den Reaktor über
die Einspeiseröhre 405 kombiniert.
Hält man
im Kreislauf 420 eine hohe Zirkulationsrate aufrecht, verglichen
mit der Einspeiseergänzung 401 und
zieht die Reaktionslösung 412 ab,
bildet sich in einer unteren Region der Oxidationsreaktionszone
eine Aufwärtsstromgeschwindigkeit
aus, insbesondere unterhalb der Öffnung 413,
welche viel höher
ist als die Aufwärtsstromgeschwindigkeit
im oberen Dekantationsbereich, im Allgemeinen oberhalb der Öffnung 413 der
Oxidationsreaktionszone. Die Größe der Ausrüstung wird
so ausgelegt und der Rezikulationsfluss so gesteuert, dass die Aufwärtsstromgeschwindigkeit
in der unteren Aufschlämmungsregion
reichlich über
der Sedimentationsgeschwindigkeit der Katalysatorteilchen 407 liegt
und daher für
die Aufrechterhaltung des Katalysators in Suspension (d. h. Rückhaltung)
im Reaktionsmedium innerhalb der Aufschlämmungsregion wirksam ist. Die
Aufwärtsgeschwindigkeit
in der Dekantationsregion oberhalb der Öffnung 413 liegt reichlich
unterhalb der Sedimentationsgeschwindigkeit der Katalysatorteilchen 407, was
die Abtrennung eines relativ klaren Reaktionslösungsdekantats 412 ermöglicht,
welches durch die Öffnung 411 austritt.
Die Größe der in
einem. wie in 9 dargestellten Reaktor, verwendeten
Katalysator-teilchen 407 beträgt etwa 200 μm bis etwa
1000 μm.
Kleinere Katalysatorteilchen, welche zum Beispiel bei der Inbetriebnahme
in das Dekantat 412 eingeschlossen werden können, können mit
einem Klärfilter
(nicht dargestellt) entfernt werden. Der Aufschlämmungskatalysator wird folglich
im Reaktor gehalten, was die Notwendigkeit der Filtration erübrigt, oder
wenigsten eines Filters, welches die Kapazität besitzt, den Katalysator
mit einer Geschwindigkeit zu entfernen, wie sie erforder lich wäre, wenn
die Katalysatorkonzentration in der stromaufwärts fließenden Reaktionslösung 412 mit
der Konzentration in der Aufschlämmungsregion
vergleichbar wäre.
-
Zur
Entfernung des Katalysators kann der zirkulierende Wirbelbettreaktor
auch einen Katalysatortrennkreislauf 414 einschließen, wie
in 9 dargestellt. In diesem Kreislauf wird ein Aufschlämmungsnebenstrom
aus der Öffnung 415 in
der Aufschlämmungsregion
der Reaktionszone abgezweigt und zur Entfernung des Katalysators 418 durch
einen Katalysatorfilter 417 geleitet. Der filtrierten Reaktionslösung kann frischer
Katalysator 419 zugesetzt werden, welcher der Einfachheit
halber zum Reaktor durch Mischen mit dem frischen Einspeisestrom 401 und
dem Rezirkulationsstrom 420 in die Einspeiseröhre 403 zurückgeführt wird. Der
Katalysatortrennkreislauf 414 kann je nach Bedarf (wenn
z. B. die Aktivität
und/oder Selektivität
des Katalysators abgenommen hat) kontinuierlich oder diskontinuierlich
betrieben werden und erübrigt
die Notwendigkeit den Reaktor für
den Ersatz des teilchenförmigen
Katalysators periodisch abzuschalten. Die Kapazität des Katalysatorfilters 417 braucht
jedoch bei weitem nicht so groß zu
sein wie die Filter, welche zur Abtrennung des Katalysators aus
der Reaktionsaufschlämmung
verwendet werden, die aus einen, wie vorstehend beschriebenen Rührbehälterreaktor
austritt. Folglich können
beträchtliche
Einsparungen bei Kapital-, Betriebs- und Wartungskosten verwirklicht
werden.
-
An
dem in 9 dargestellten Wirbelbettreaktor 400 können verschiedene Änderungen
vorgenommen werden. So kann zum Beispiel anstatt das O2-haltige
Gas vom Boden des Reaktors 400 aus durch die Reaktionsmischung
durchzuperlen, eine Düse
im Kopf des Reaktors vorgehen werden, ähnlich der in 7 dargestellten,
durch die sowohl der wässrige
Einspeisestrom 406 als auch das O2-haltige
Gas kombiniert und in die Reaktionsmischung abgegeben werden. Alternativ
kann für
die Zirkulation der Reaktionsmischung, welche den teilchenförmigen Katalysator
enthält
ein Rührflügel vorgesehen
werden, welcher in der Einspeiseröhre 405 in einer Weise
rotiert, dass er die Reaktionsmischung durch die Einspeiseröhre nach
unten in den unteren Bereich der Oxidationsreaktionszone zieht.
Weiterhin kann der Katalysatortrennkreislauf 414 wahlweise
in den Wäremeübertragungszirkulationskreiselauf 420 integriert
werden. Die Oxidationsreaktionsbedingungen und Betriebsparameter
für einen
Wirbelbettreaktor sind jenen ähnlich,
welche vorstehend für
Oxidationsreaktionszonen beschrieben wurden, welche von einem Rührbehälterreaktor
bereitgestellt werden.
-
Eine
noch andere alternative Ausführungsform
der Erfindung ist in 10 dargestellt, worin die Oxidation
von N-(Phosphonomethyl)iminodiessigsäuresubstrat zum N-(Phosphonomethyl)glycinprodukt
in einem mehrteiligen Reaktorsystem erfolgt, welches eine Vielfalt
von Reaktoren 800A, 800B, 800C, ... 800n umfasst, in
dem sich die Reaktionsmischung nacheinander von einem Reaktor zum
nächsten
Reaktor in der Reihe fortbewegt. Die primäre Einspeisemischung 814,
die N-(Phosphonomethyl)iminodiessigsäuresubstrat
umfasst, wird in den Reaktor 800A eingespeist und die Ergänzungseinspeisemischung
wird in parallele Komponenteneinspeiseströme 802B, 802C, 802D,
... 802n aufgeteilt, welche auf die Reihe von Reaktoren
aufgeteilt werden. Jeder Reaktor erhält einen Anteil an der Sauerstoffversorgung
oder eines anderen oxidierenden Mittels über die Einspeiseleitungen 804A, 804B, 804C,
... 804n. Wahlweise wird ein Wärmetauscher 806A, 806B, 806C etc.
zwischen dem jeweils vorangehenden Reaktor und dem unmittelbar folgenden
Reaktor dazwischengeschaltet, um die Reaktionswärme aus der Reaktionszwischenmischung 810A, 810B, 810C etc.
abzuführen, welche
aus dem unmittelbar vorausgehenden Reaktor 800A, 800B, 800C etc.
austritt, was es ermöglicht,
jeden Reaktor auf Wunsch adiabatisch zu betreiben. Alternativ kann
zu jedem Reaktor ein gekühlter
Rezikulationsstrom 808A, 808B, 808C, 808D etc.
zurückgeführt werden,
um die exotherme Reaktionswärme
abzuführen und
für die
Kühlung
der Reaktionsmischung im Reaktor zu sorgen. In jedem der auf den
ersten Reaktor 800A folgenden Reihe von Reaktoren besteht
der kombinierte Einlassstrom 812B, 812C, 812D,
... 812n aus einer Kombination der Einspeisesstromkomponente 802B, 802C, 802D etc.
der Reaktionszwischenproduktmischung, welche aus dem unmittelbar
vorhergehenden Reaktor 810A, 810B, 810C etc.
austritt, minus irgendwelcher Rezirkulationen 808A, 808B, 808C, 808D etc.
und irgendwelcher Rezirkulationsströme 808B, 808C, 808D etc.
Jeder der Reaktoren 800A, 800B etc. kann irgendeine
der hierin beschriebenen Konfigurationen annehmen, liegt jedoch
vorzugsweise in Form eines Reaktors vor, in dem der Katalysator
zurückgehalten
wird (z. B. in einem Festbett- oder Wirbelbettreaktor). Das Reaktionsendprodukt 810n wird
aus der letzten Reaktionszone 800n der Reaktionszonenreihe
abgezogen.
-
Jeder
Einspeisestrom 802B, 802C etc. der Komponenten
eines mehrteiligen Reaktorsystems kann hochkonzentriert sein, wodurch
er zu einer hohen Produktivität
des Verfahren beiträgt.
Es können
in der Tat Komponenteneinspeiseströme verwendet werden, welche
Komponenteneinspeiseströme
aus dichten Aufschlämmungen
oder Pasten von N-(Phosphonomethyl)iminodiessigsäuresubstrat umfassen. In jedem
auf den ersten Reaktor 800A folgenden Reaktor (z. B.
-
Reaktor 800B)
kann eine Aufschlämmung
oder Paste der Komponenteneinspeisemischung eingeführt werden,
obwohl insbesondere im Fall von Festbettreaktoren bevorzugt ist,
dass die Kombination aus der Einspeisezusammensetzung der Komponente 802B,
die Einspeiserate der Komponente 802B, die Zusammensetzung
und Strömungsrate
der Zwischenproduktreaktionsmischung 810A, welche aus dem
unmittelbar vorangehenden Reaktor 800A austritt (minus
irgendeiner Rezirkulation 808A), irgendeine Rezirkulation 808B von Zwischenreaktionsproduktmischung
aus dem Reaktor 800B so ist, dass der kombinierte Einlassstrom 812B im
Wesentlichen frei von Substratfeststoffen oder Feststoffen von N-(Phosphonomethyl)glycinprodukt
ist. Es wird jedoch für
den Fachmann selbstverständlich
sein, das bei bestimmten Ausführungsformen
der Erfindung die Komponenteneinspeise- und Zwischenproduktreaktionsmischungen
durchgehend in Aufschlämmungsform
vorliegen, wenn der Oxidationskatalysator ein homogener Katalysator
ist oder wenn ein monolithischer Katalysator verwendet wird, wie
ein Festbett in Honigwabenform.
-
Obwohl
sowohl ein zusätzlicher
Komponenteneinspeisestrom 802B, 802C etc. als
auch ein Oxidationsmittel vorzugsweise in jede der auf die erste
Reaktionszone folgende Reihe von Reaktionszonen 800B, 800C etc.
eingeführt
wird, kann es bei einer besonderen Anwendung selbstverständlich notwendig
oder erwünscht
sein, nur einige, aber nicht alle, der nachfolgenden Reaktionszonen
mit einer Reaktionsmischung der Komponenten zu versorgen. In manchen
Fällen
muss es nicht erforderlich sein alle Reaktionszonen mit Sauerstoff
zu versorgen, obwohl in den meisten Fällen die Zufuhr von Oxidationsmittel
zu jeder Zone bevorzugt ist.
-
Das
Festbett und die Aufführungsformen
zur verteilten Einspeisung der Erfindung sind in einzigartiger Weise
für die
Umwandlung von wasserlöslichen
Salzen der N-(Phosphonomethyl)iminodiessigsäure in wasserlösliche Salze
von N-(Phosphonomethyl)glycin
geeignet. Wegen der allgemein hohen Löslichkeit von Alkalimetall-
und Aminsalzen, z. B. Kalium-, Ammonium-, Isopropylamin- und Alkanolaminsalze,
sowohl der Substratsäure
als auch der Produktsäure,
kann entweder ein Festbett oder ein Rührbehälterreaktor bei viel höheren Substrat- und Produktkonzentrationen
betrieben werden, als dies bei Säureverfahren
machbar ist, bei denen die Produktivität durch die relativ geringe
Löslichkeit
eingeschränkt
ist. Im Fall von Salzen kann ein Festbettverfahren in der Tat besonders
vorteilhaft sein, weil es weder zur Entfernung kristalliner Produkte
noch zur Entfernung von Katalysatoren ohne die Notwendigkeit irgendwelcher
Filtrations- oder Zentrifugiervorgänge betrieben werden kann.
Eine N-(Phospho nomethyl)glycinsalzlösung kann mit verschiedenen
Streckmitteln formuliert werden, wie sie bei der kommerziellen Anwendung
von N-(Phosphonomethyl)glycin üblicherweise
verwendet werden, so dass es mit minimaler Weiterverarbeitung löslich ist.
Um die gewünschten
Konzentrate für den
Handel herzustellen sind nur mäßige Konzentrierungschritte
erforderlich. Eine umfangreiche Abtrennung von Verunreinigungen
muss nicht erforderlich sein.
-
Ein
Rührbehälterreaktionssystem,
insbesondere ein kontinuierliches Rührbehälterreaktionssystem, kann wegen
der höheren
Reaktionswärme,
welche mit der Oxidation hoher Substratkonzentrationen verbunden
ist und der begleitenden exothermen Oxidation relativ großer Anteile
von C1-Nebenprodukten, wie Formaldehyd und
Ameisensäure,
bei der Synthese von Salzen vorteilhaft sein. Ein kontinuierlicher
Rührbehälterreaktor
bietet gegenüber
einem Chargenreaktor bei der Verwendung der Reaktionswärme zum
Vorheizen der wässrigen
Einspeisung in den Reaktor beträchtliche
Vorteile. Kombinationen aus einem primären kontinuierlichen Rührbehälter zur
die anfängliche
Umwandlung mit einem Festbettreaktor zur Fertigbehandlung können ebenfalls
von Vorteil sein.
-
Ein
Festbett, hauptsächlich
ein Pfropfströmungsreaktor,
bietet nichtsdestoweniger besondere Vorteile, insbesondere wo das
Katalysatorbett Edelmetall auf Kohlenstoff umfasst, weil der Pfropfströmungsbetrieb dazu
dient, die Oxidation von C1-Nebenprodukten
zu fördern,
eine Reaktion, welche hauptsächlich
nach 1. Ordnung in Bezug auf das C1-Substrat
verläuft.
Aus dem gleichen Grund verschlimmert jedoch die Pfropfströmung die
hohe Wärmebelastung,
welche mit der Oxidation einer wässrigen
Einspeisemischung verbunden ist, welche eine hohe Konzentration
Substratsalz enthält.
Obwohl das Rezirkulationssystem von 8 verwendet werden
kann, um einen angemessenen Wärmeübergang
einzustellen, weist es eine ungünstige
Auswirkung auf die Kinetik der Abbaus von Formaldehyd und Ameisensäure auf,
selbst wenn je nach Rezirkulationsrate die Auswirkung auf den C1-Abbau gegenüber einem Reaktor mit vollständiger Rückvermischung überlegen bleibt.
-
In
manchen Fällen
kann es demzufolge vorteilhaft sein, die Oxidationsreaktion in einem
Reaktionssystem durchzuführen,
in dem das Festbett durch indirekten Wärmeübergang auf eine Kühlflüssigkeit
gekühlt wird,
umfassend einen Wärmeübergang
oder eine Prozessflüssigkeit,
welche durch eine Leitung innerhalb eines oder in Kontakt mit einem
Katalysatorbett strömt.
Das Festbett kann zum Beispiel mantel- oder rohrseitige in einem
Rohrbündelwärmetauscher
ange ordnet sein, wobei die Kühlflüssigkeit
durch die andere Seite des Austauschers geleitet wird. Bei einer
solchen Ausführungsform
kann das Festbett Mehrkomponentenbetten umfassen, welche in den
Rohren des Wärmetauschers
getrennt angeordnet sind, wobei die wässrige Einspeisemischung und
das Oxidationsmittel zwischen den Komponentenbetten verteilt sind
und die Kühlflüssigkeit durch
die Mantelseite des Wärmetauschers
strömt.
Bei einer alternativen Ausführungsform
kann das Festbett innerhalb des Mantels des Wärmetauschers enthalten sein,
wobei auf der Mantelseite wahlweise Leitbleche verwendet werden
können,
um im Wesentlichen eine Pfropfströmung der flüssigen Phase durch das Bett
sicherzustellen.
-
Alternativ
können
in einer Reihe von Reaktoren, welche durch Wärmetauscher zur Kühlung der
Zwischenproduktreaktionslösung
getrennt sind, Salze von N-(Phosphonomethyl)glycin
hergestellt werden, wie vorstehend beschrieben. Das aufgeteilte
Einspeisereaktionssystem von 10 kann
beim Umgang mit der bei der Oxidation von N-(Phosphonomethyl)iminodiesigsäuresalzen
zu N-(Phosphonomethyl)glycinsalzen erzeugte Wärme, besonders vorteilhaft
sein. Wie bemerkt können
besonders hohe Umsätze
erzielt werden, wenn sowohl das Substrat als auch das Produkt wasserlösliche Salze
sind. Wenn zum Beispiel die wässrige Einspeisemischung
mindestens etwa 15 Gew.-% Substratsalz enthält, kann das Endreaktionsgemisch
mindestens etwa 12 Gew.-% wasserlösliches Produktsalz enthalten;
Wenn die wässrige
Einspeisemischung mindestens etwa 25 Gew.-% wasserlösliches
Substratsalz enthält,
kann die Oxidationsreaktionsendmischung mindestens etwa 20 Gew.-%
wasserlösliches
Produktsalz enthalten; Und wenn die wässrige Einspeisemischung mindestens
etwa 35 Gew.-% wasserlösliches
Substratsalz enthält,
kann die Oxidationsreaktionsendmischung mindestens 28 Gew.-% wasserlösliches
Produktsalz enthalten, alle bezogen auf Säureäquivalentbasis. Es können in
der Tat hohe Produktsalzkonzentrationen von über 35%, vorzugsweise von über 40%
oder sogar 50 Gew.-% realisiert werden. Das Reaktionsendprodukt
kann, wie vorstehend beschrieben, die in einem einzelnen Reaktor
erhaltene primäre
Reaktionsmischung, die primäre
Produktfraktion aus einem einzelnen Festbettsystem mit Rückführung, wie
in 8 abgebildet, oder der Ablauf aus dem letzten
von einer Reihe von Reaktoren sein, wie vorstehend weiter beschrieben.
-
Das
Reaktionsendprodukt wird weiter konzentriert, indem ihm Wasser entzogen
wird. Die Reaktionsendmischung kann zum Beispiel in eine Schnellverdampfungszone
eingeführt
werden, worin der Druck niedriger ist als der Dampfdruck der Oxidationsendmischung
bei der Temperatur, bei der sie im Reaktor oder im letzten einer
Reihe von Reaktoren vorliegt. Es kann aus dem Reaktionsendprodukt
mit einem relativ geringen Energieaufwand genügend Wasser entzogen werden,
um eine konzentrierte Lösung
herzustellen, welche mindestens etwa 40 Gew.-% eines wasserlöslichen
Salzes von N-(Phosphonomethyl)glycin auf Säureäquivalentbasis enthält.
-
Die
Konzentration an N-(Phosphonomethyl)glycinprodukt im Oxidationsreaktionsmischungsablauf, welcher
aus dem Reaktorsystem der vorliegenden Erfindung austritt, kann
40 Gew.-% oder mehr betragen. Die N-(Phosphonomethyl)glycinkonzentration
beträgt
vorzugsweise etwa 5 bis etwa 40%, weiter vorzugsweise etwa 8 bis
etwa 30% und noch weiter vorzugsweise etwa 9 bis etwa 15%. Die Formaldehydkonzentration
in der Produktmischung beträgt
vorzugsweise weniger als etwa 5000 ppm, weiter vorzugsweise weniger
als etwa 4000 ppm, noch weiter vorzugsweise weniger als 2800 ppm
des Gewichts und sogar noch weiter vorzugsweise weniger als etwa
1500 ppm. Die Ameisensäurekonzentration
in der Produktmischung beträgt
vorzugsweise weniger als etwa 12.000 ppm, weiter vorzugsweise weniger
als etwa 4000 ppm, noch weiter vorzugsweise weniger als 2000 ppm
des Gewichts und sogar noch weiter vorzugsweise weniger als etwa
1500 ppm. Die Konzentrationen der Aminomethylphosphonsäure (AMPA),
N-Methylaminomethylphosphonsäure
(MAMPA), N-Methyl-N-(phosphonomethyl)glycin(NMG) in der Produktmischung
werden auf jeweils weniger als 9000 ppm eingestellt, können üblicherweise
auf weniger als 4500 ppm eingestellt und oft unterhalb von 1500
ppm gehalten werden. Diese Nebenproduktkonzentrationen beruhen selbstverständlich auf
einem Betrieb mit einem einmaligen Durchgang, bei dem die einzige
Einspeisung in das Reaktorsystem eine wässrige Mischung ist, welche
N-(Phosphonomethyl)iminodiessigsäure
oder deren Salz ist, wie es bei der Phosphonomethylierung von Iminodiessigsäure erhalten
wird. Wird irgendein Rücklaufstrom
in das Reaktorsystem eingeführt,
wie das Dekantat aus einem, wie nachstehend beschriebenen, adiabatischen
Kristallisator, tendiert die damit verbundene Rezyklisierung von
Nebenprodukten dazu, den Nebenproduktgehalt der Reaktionsproduktmischung
zu erhöhen.
-
Reinigung und/oder Konzentrierung
des N-(Phosphonomethyl)glycinprodukts
-
Ein
anderer Aspekt dieser Erfindung betrifft die Reinigung und/oder
Konzentrierung des im Oxidationsreaktionsmischungsablauf erhaltenen
N-(Phosphonomethyl)glycinprodukts. Die verschiedenen Verbesserungen
bei der Gewinnung des N-(Phosphonomethyl)glycinprodukts durch die
vorliegende Erfindung findet breite Anwendung und kann zum Beispiel
bei der Gewinnung des N-(Phosphonomethyl)glycinprodukts aus der
Reaktionsmischung verwendete werden, welche durch die hierin beschriebenen,
verschiedenen kontinuierlichen Oxidationsreaktorsysteme erzeugt
werden. Dieser weitere Aspekt der vorliegenden Erfindung ist jedoch
nicht auf solche Anwendungen oder die Verwendung in Zusammenhang
mit kontinuierlichen Oxidationsreaktorsystemen allgemein beschränkt. Wie
für den
Fachmann ersichtlich, können
die hierin aufgeführten
Konzepte vorteilhaft zur Gewinnung des N-(Phosphonomethyl)glycinprodukts
ebenso gut auf Oxidationsreaktionsmischungsabläufe anderer Reaktorsysteme
angewendet werden.
-
Die
Reaktionsmischung enthält
normalerweise neben dem gewünschten
N-(Phosphonomethyl)glycinprodukt Wasser und verschiedene Verunreinigungen.
Diese Verunreinigungen können
zum Beispiel einschließen:
verschiedene Nebenprodukte und nicht umgesetzte Ausgangsmaterialien
wie nicht umgesetztes N-(Phosphonomethyl)iminodiessigsäuresubstrat,
N-Formyl-N-(phosphonomethyl)glycin, Phosphorsäure, phosphorige Säure, Hexamethylentetramin,
Aminomethylphosphonsäure,
N-Methylaminomethylphosphonsäure,
Formaldehyd, Ameisensäure
und dergleichen. Der Wert des N-(Phosphonomethyl)glycinprodukts
verlangt normalerweise eine maximale Gewinnung des Produkts aus
der Reaktionsmischung und sorgt oft für Anreiz zum Rezyklieren wenigstens
eines Teils des Produkts der ausgearmten Reaktionsmischung zur/zu
den Oxidationsreaktionszone(n) zur weiteren Umwandlung von nicht
umgesetztem Substrat und Gewinnung von nicht isoliertem Produkt.
-
Wirtschaftliche Überlegungen
bedingen manchmal auch, dass die Konzentration des N-(Phosphonomethyl)glycinprodukts
in kommerziell verkauften Mischungen wesentlich höher sein
müssen,
als die Konzentrationen in den Reaktionsmischungen, die typischerweise
bei Verwendung der vorstehend beschriebenen Oxidationsreaktionssysteme
gebildet werden, insbesondere, wenn das N-(Phosphonomethyl)glycinprodukt
für landwirtschaftliche
Zwecke verwendet wird. Wird zum Beispiel ein heterogener Katalysator
zur Herstellung der freien Säure
des N-(Phosphonomethyl)glycins
bei den weiter bevorzugten Betriebstemperaturen (d. h. von etwa
95 bis etwa 105°C)
hergestellt, beträgt
die Maximalkonzentration des N-(Phosphonomethyl)glycinprodukts in
der Reaktionsmischung vorzugsweise nicht mehr als etwa 9 Gew.-%
um in Lösung
zu bleiben. Manchmal ist es jedoch erwünscht, dass handelsübliche Mischungen
eine N-(Phosphonomethyl)glycinkonzentration besitzen, welche deutlich
größer ist.
-
Nachdem
das N-(Phosphonomethyl)glycinprodukt gebildet und sofern erforderlich
vom Katalysator getrennt worden ist, ist es folglich typischerweise
vorzuziehen das Produkt zu konzentrieren und das Produkt von den
verschiedenen Verunreinigungen in der Reaktionsmischung zu trennen.
-
Smith
beschreibt (in US-A 5,087,740) ein Verfahren zur Reinigung und Konzentrierung
eines N-(Phosphonomethyl)glycinprodukts. Smith legt das Durchleiten
einer Reaktionsmischung, welche N-(Phosphonomethyl)glycin enthält, durch
eine erste Ionenaustauscherharzsäule
offen, um Verunreinigungen zu entfernen, die stärker sauer sind als N-(Phosphonomethyl)glycin,
das Durchleiten des Ablaufs aus der ersten Ionenaustauscherharzsäule durch
eines zweite Ionenaustauscherharzsäule, welche das N-(Phosphonomethyl)glycin
adsorbiert und die Gewinnung des N-(Phosphonomethyl)glycins durch
Durchleiten einer Base oder einer starken Mineralsäure durch
die zweite Ionenaustauscherharzsäule
offen.
-
Viele
andere Methoden zur Reinigung und Konzentrierung eines N-(Phosphonomethyl)glycinprodukts schließen eine
Kristallisationsstufe ein, wobei das N-(Phosphonomethyl)glycinprodukt kristallisiert
wird, um es wenigstens von einem Teil der restlichen Reaktionsmischung
zu trennen.
-
Die
in 11–14A dargestellten und nachstehend beschriebenen
Produktgewinnungsverfahren finden insbesondere bei der Konzentrierung
und Gewinnung von Produkten aus Oxidationsreaktionsmischungen Anwendung,
welche N-(Phosphonomethyl)glycinprodukt enthalten, welches der Kristallisation
unterworfen werden kann, und insbesondere bei jenen, welche die
freie Säure
von N-(Phosphonomethyl)glycin enthalten. Die konzentrierte freie
Säure von
N-(Phosphonomethyl)glycin wird typischerweise zur Herstellung anderer N-(Phosphonomethyl)glycinprodukte
verwendet, wie den vorstehend beschriebenen.
-
Bei
einer besonders bevorzugten Ausführungsform
wird mindestens ein Teil der Reaktionsendmischung (vorzugsweise
ohne irgendeinen Katalysator und insbesondere ohne irgendeinen heterogenen
Katalysator oder eines homogenen Katalysators, der gemeinsam mit
dem N-(Phosphonomethyl)glycinprodukt kristallisiert) in einen nicht-adiabatischen,
mit Wärme
betriebenen Verdampfungskristallisator eingeführt, in dem die Oxidationsreaktionsmischung
mit Wärme
beaufschlagt wird, um aus der Reaktionsmischung Wasser zu verdampfen
und dadurch das N-(Phosphonomethyl)glycinprodukt zu konzentrieren
und zu kristallisieren. Die im nicht-adiabatischen Kristallisator
verwendete Wärme
stammt normalerweise aus Dampf. Im nicht-adiabatischen Kristallisationssystem
wird vorzugsweise mindestens etwa 30%, weiter vorzugsweise mindestens
etwa 50%, sogar weiter vorzugsweise etwa 80% bis etwa 100%, sogar
noch weiter vorzugsweise etwa 90% bis beinahe 100% des Wassers der
Reaktionsmischung verdampft. Die Verdampfungskristallisation ist
besonders vorteilhaft, weil sie das Produkt auch von niedermolekularen
Verunreinigungen trennt, wovon hauptsächlich Formaldehyd und Ameisensäure zu nennen
sind, welche dazu neigen zusammen mit Wasser aus der Reaktionsmischung
zu verdampfen.
-
Der
Druck im wärmegesteuerten
Verdampfungskristallisator beträgt
vorzugsweise nicht mehr als etwa 6,9 kPa bis etwa 69,0 kPa (1 bis
etwa 10 psia), sogar weiter vorzugsweise etwa 6,9 kPa bis etwa 34,5
kPa (1 bis etwa 5 psia, sogar noch weiter vorzugsweise etwa 13,8
kPa bis etwa 20,7 kPa (2 bis etwa 3 psia) und sogar noch weiter
vorzugsweise etwa 18,3 kPa (2,8 psia). Die Betriebstemperatur des
wärmegesteuerten
Verdampfungskristallisators beträgt
vorzugsweise nicht mehr als 80°C,
weiter vorzugsweise etwa 40°C
bis etwa 80°C, sogar
weiter vorzugsweise etwa 50°C
bis etwa 70°C
und sogar noch weiter vorzugsweise etwa 60°C.
-
11 zeigt ein Beispiel eines Systems, das einen
Verdampfungskristallisator einsetzt. Eine wässrige Einspeisung 201,
welche ein N-(Phosphonomethyl)iminodiessigsäuresubstrat umfasst, wird in
ein Oxidationsreaktorsystem 203 eingeführt, welches eine oder mehrere
Oxidationsreaktionszone(n) umfasst, worin das Substrat oxidiert
wird, um eine Oxidationsreaktionsmischung 205 zu bilden,
welche N-(Phosphonomethyl)glycinprodukt umfasst. In 11, 12, 13 und 14 sind
Einzelheiten des Oxidationsreaktorsystems, einschließend Katalysatortrenn- und -rezyklisierungsmechanismen
(z. B. Katalysatorfilter, Katalysatorvorratsbehälter, Vorfilterspülbehälter und
dergleichen), welche vorliegen können,
weggelassen, wobei die aus dem Reaktorsystem abgezogene Oxidationsreaktionsmischung
je nach der/den eingesetzten spezifischen Reaktorkonfiguration(en)
selbstverständlich,
wie erforderlich, vom Katalysator befreit worden ist. Die Oxidationsreaktionsmischung 205 kann
wahlweise durch einen Vorkristallisationsentlüftungsbehälter 206 geleitet
werden. Der Vorkristallisationsentlüftungsbehälter 206 erniedrigt
den Druck über
dem Reaktionsgemisch 205 bis zu einem gewissen Grad und
veranlasst gelöstes
CO2 aus der Mischung zu entweichen und aus
dem Behälter
abgelüftet
zu werden. In den Vorkristallisationsentlüftungsbehälter 206 kann eine
Sauerstoffquelle (z. B. ein O2-haltiges
Gas) eingeleitet werden, um sowohl das N-(Phosphonomethyl)iminodiessigsäuresubstrat
in der Reaktionsmischung als auch die in der Reaktionsmischung 205 vorliegenden
Nebenprodukte Formaldehyd und Ameisensäure weiter zu oxidieren, welche
in der/den Oxidationsreaktionszone(n) des Reaktorsystems 203 nicht
oxidiert worden sind. Auf diese Weise fungiert der Vorkristallisationsentlüftungbehälter als
eine dem Reaktorsystem 203 nachgeschaltete Oxidationsreaktionszone.
-
Im
wärmegesteuerten
Verdampfungskristallisator 207 wird dann ein Verdampfungskristallisatoreinspeisestrom 239 eingeführt, in
dem Wärme
auf den Verdampfungskristallisatoreinspeisestrom 239 übertragen wird,
um Wasser (und niedermolekulare Verunreinigungen wie Formaldehyd
und Ameisensäure)
zu verdampfen und den nicht-adiabatischen Kristallisatorüberkopfdampfstrom 209 zu
bilden. Ein großer
Anteil des N-(Phosphonomethyl)glycinprodukts fällt aus (typischerweise etwa
50% bis etwa 60% auf Durchflussbasis), um eine Verdampfungskristallisationsaufschlämmung 211 zu
bilden. Die Aufschlämmung 211 wird
aus dem nicht-adiabatischen Kristallisator 207 abgezogen
und in ein Hydrozyklon (oder eine Serie von Hydrozyklone) 213 eingeführt, welches
eine konzentrierte, an ausgefälltem
N-(Phosphonomethyl)glycinprodukt angereicherte konzentrierte Aufschlämmung 215 und
einen an Feststoff ausgearmten Strom 221 bildet. Die konzentrierte Aufschlämmung 215 wird
in eine Feststofftrennvorrichtung, vorzugsweise in eine Zentrifuge,
eingeführt,
welche ein Zentrat 223 bildet (welches an ausgefälltem N-(Phosphonomethyl)glycinprodukt
weiter ausgearmt ist) und einen N-(Phosphonomethyl)glycinproduktnasskuchen 219.
-
Die
Konzentration des N-(Phosphonomethyl)glycinprodukts im Nasskuchen 219 beträgt normalerweise
mindestens etwa 95% (bezogen auf das Gewicht aller Verbindungen
außer
Wasser). Eine niedrigere Konzentration kann toleriert werden, wenn
der Nasskuchen 219 anschließend mit Wasser gewaschen oder
mit einem Produkt höherer
Reinheit verschnitten wird, wie nachstehend beschrieben.
-
Wenigstens
ein Teil des Überkopfprodukts 209 aus
dem wärmegesteuerten
Kristallisator kann zur (zu den) Oxidationsreaktionszone(n) des
Reaktorsystems 203 zurückgeführt werden.
Bei der in 11 dargestellten Ausführungsform
wird ein Anteil 243 kondensiert und zur Verwendung als
Wasserquelle zum Lösen
des N-(Phosphonomethyl)iminodiessigsäuresubstrats
verwendet, um den Einspeisestrom 201 für das Reaktorsystem 203 zu
bilden. Das Kondensat 243 wird vorzugsweise in die am meisten
stromaufwärts
gelegene Oxidationsreaktionszone eingeführt, wenn das Reaktorsystem
zwei oder mehr Oxidationsreaktionszonen in Reihe umfasst. Der Strom 243 kann,
wie beinahe alle Rezirkulationsströme dieser Erfindung, alternativ
(oder zusätzlich)
direkt in die Oxidationsreaktionszone(n) eingeführt werden anstatt mit anderen
Inhaltsstoffen (z. B. im wässrigen
Einspeisestrom 201) kombiniert zu werden, bevor er in die
Oxidationsreaktionszone(n) eingeführt wird. Insbesondere dort,
wo der Katalysator ein kohlenstoffhaltiger Katalysator ist und der
Katalysator weiter vorzugsweise ein Kohlenstoff-getragenes Edelmetall umfasst, kann
ein Teil des Überkopfprodukts 209 des nicht-adiabatischen
Kristallisators vorteilhafterweise auch dazu verwendet werden, die
Katalysatoroberfläche zu
reduzieren. Dies beruht auf der Tatsache, dass das Überkopfprodukt 209 des
wärmegesteuerten
Verdampfungskristallisators typischerweise Formaldehyd und Ameisensäure enthält, welche
beide als Reduktionsmittel fungieren, insbesondere gegenüber kohlenstoffhaltigen
Katalysatoren und insbesondere gegenüber Katalysatoren, welche Kohlenstoff-getragenes
Edelmetall umfassen. Der Anteil an Überkopfprodukt 209 des
nicht-adiabatischen Kristallisators, welcher bei einer solchen Reduktionsbehandlung
verwendet wird, wird zuerst kondensiert und das Kondensat kann in
einen oder mehrere Katalysatorvorratsbehälter innerhalb des Reaktorsystems 203 eingeführt werden,
wo die Reduktionsbehandlung stattfindet. Zusätzlich zur Reduktion des Katalysators
bewirkt eine solche Behandlung das Waschen des Katalysators und
macht vom Vorteil der Verweilzeit des Katalysators in dem/den Katalysatorvorratsbehälter(n)
Gebrauch. Bei einer besonders bevorzugten Ausführungsform wird ein Teil des Überkopfprodukts 209 des
nicht-adiabatischen Kristallisators des Weitem rektifiziert oder
destilliert, um einen Dampfstrom zu erhalten, dessen Konzentration
an Formaldehyd und/oder Ameisensäure
angereichert ist. Dieser angereicherte Dampfstrom kann wiederum
kondensiert und mit einem kohlenstoffhaltigen Katalysator in Kontakt
gebracht werden.
-
Wenigstens
ein anderer Teil 241 des Überkopfprodukt 209 des
wärmegesteuerten
Verdampfungskristallisators wird typischerweise aus dem System als
Ausscheidestrom ausgetrieben (d. h. verworfen). Bei einem kontinuierlichen
System hilft dieser Ausscheidestrom 241 dabei die sich
im System aufbauende Abfallmenge zu verringern (insbesondere den
Aufbau von niedermolekularen Verunreinigungen) und hilft das Wassergleichgewicht
zu handhaben. Der ausgeschiedene Abfall 241 kann umgekehrt
weiterbehandelt werden, um Verunreinigungen zu entfernen. Eine solche
Behandlung kann zum Beispiel das Kontaktieren des Ausscheidestroms 241 mit
O2-haltigem Gas und einem Katalysator einschließen, welcher
ein Gruppe VIII-Metall (insbesondere Platin, Palladium und/oder
Rhodium) umfasst und wahlweise einen Kohlenstoffträger, um
dadurch Formaldehyd und Ameisensäure
zu oxidieren und umweltverträgliches
CO2 und Wasser zu bilden. Die Reaktion erfolgt vorzugsweise
bei einer Temperatur von etwa Raumtemperatur bis etwa 90°C (weiter
vorzugsweise von etwa 50°C
bis etwa 90°C),
einem Druck von etwa Atmosphärendruck
bis etwa 690 kPa (200 psi), einer Konzentration an gelöstem Sauerstoff
von etwa 1 bis etwa 7 ppm und einem Gruppe VIII-Metall, bezogen
auf ein Reaktorbetriebsvolumen von etwa 0,00015:1 bis etwa 0,0040:1.
Dieses Verfahren ist von Smith im Detail in US-A 5,606,107 beschrieben.
Das aus der Oxidation des Überkopfprodukts 209 aus
dem wärmegesteuerten
Verdampfungskristallisator resultierende Produkt kann zur (zu den)
Oxidationsreaktionszone(n) des Reaktorsystems 203 rezykliert
und als Quelle für
die Bereitstellung von Wasser verwendet werden.
-
Der
an Feststoff ausgearmte Hydrozyklonstrom 221 wird zur weiteren
Gewinnung des N-(Phosphonomethyl)glycinprodukts vorzugsweise zum
wärmegesteuerten
Verdampfungskristallisator 207 rezykliert.
-
Wenigsten
ein Teil 231 des Zentrats 223 aus der Zentrifuge 217 wird
zur weiteren Gewinnung des N-(Phosphonomethyl)glycinprodukts vorzugsweise
zum wärmegesteuerten
Verdampfungskristallisator 207 rezykliert. Ein Teil 233 des
Zentrats 223 kann alternativ (oder zusätzlich) zur (zu den) Oxidationsreaktionszone(n) des
Reaktorsystems 203 rezykliert werden, um nicht umgesetztes
N-(Phosphonomethyl)iminodiessigsäuresubstrat
im Zentrat 223 zum N-(Phosphonomethyl)glycinprodukt umzuwandeln.
Alternativ (oder zusätzlich) kann
ein Teil 227 des Zentrats 223 aus dem System ausgeschieden
werden.
-
Das
Ausscheiden eines Teils 227 des Zentrats 223 aus
der Zentrifuge 217 hilft in einem kontinuierlichen System
die Menge an sich bildenden Verunreinigungen (insbesondere den Aufbau
höhermolekularer Verunreinigungen)
im System und folglich im Nasskuchen 217 zu verringern.
Eine solche Behandlung kann zum Beispiel einschließen:
- 1. Der Ausscheidestrom 227 kann mit
O2 und einem Gruppe VIII-Metall-Katalysator kontaktiert
werden, um Formaldehyd und Ameisensäure im Ausscheidestrom 227 zu
oxidieren, wie vorstehend für
die Überkopfausscheidung 241 bei
einem nicht-adiabatischen Kristallisator beschrieben.
- 2. Der ausgeschiedene Abfall kann mit O2 und
einem edelmetallhaltigen Katalysators kontaktiert werden, um irgendwelches
N-substituiertes N- (Phosphonomethyl)glycin
(am häufigsten
nämlich
N-Methyl-N-(phosphonomethyl)glycin oxidativ zu spalten, um zusätzliches
N-(Phosphonomethyl)glycinprodukt zu bilden, welches wiederum in
einem Kristallisator gesammelt werden kann, indem es zu dem nicht-adiabatischen
Kristallisator 207 zurückgeführt wird.
Diese Reaktion erfolgt vorzugsweise bei einem Druck von mindestens
Atmosphärendruck
(weiter vorzugsweise von etwa 207 kPa bis etwa 690 kPa (30 bis etwa
200 psig), einer Temperatur von etwa 50°C bis etwa 200°C (weiter
vorzugsweise von etwa 70°C
bis etwa 150°C
und sogar weiter vorzugsweise von etwa 125°C bis etwa 150°C), einer
Konzentration an gelöstem
Sauerstoff von nicht größer als
etwa 2 ppm und einem Gewichtsverhältnis von Edelmetall zu N-substituiertem
N-(Phosphonomethyl)glycinnebenprodukt(en) von etwa 1:500 bis etwa
1:5 (vorzugsweise von etwa 1:200 bis etwa 1:10 und sogar weiter
vorzugsweise von etwa 1:50 bis etwa 1:10). Diese Behandlungsmethode
ist von Morgenstern et al. in US-A 6,005,140 im Detail beschrieben.
- 3. Der ausgeschiedene Abfall 227 kann mit Formaldehyd
im stöchiometrischen Überschuss,
bezogen auf die N-(Phosphonomethyl)glycinverbindungen und deren
Derivate, kombiniert und dann in Anwesenheit eines Übergangsmetallkatalysators
(z. B. Mangan, Kobalt, Nickel, Chrom, Ruthenium, Aluminium, Molybdän, Vanadin,
Kupfer, Zink oder Cer) erhitzt werden, um besser umweltverträgliche Verbindungen
zu bilden. Dieses Verfahren ist von Grabiak et al. im Detail in
US-A 4,851,131 beschrieben.
- 4. Der ausgeschiedene Abfall 227 kann durch einen anderen
Kristallisator geleitet werden, um weiteres N-(Phosphonomethyl)glycinprodukt
zu gewinnen.
-
Bei
einer anderen, besonders bevorzugten Ausführungsform wird wenigstens
ein Teil des Oxidationsreaktionsmischungsablaufs (vorzugsweise ohne
irgendeinen Katalysator, insbesondere ohne irgendeinen heterogenen
Katalysator oder einen homogenen Katalysator, der zusammen mit dem
N-(Phosphonomethyl)glycinprodukt ausfällt) in einen Kristallisator
eingeführt,
welcher im Wesentlichen adiabatisch arbeitet (d. h. jede Wärmezufuhr
oder -abführung
zum/vom Kristallisator nicht mehr als etwa 200 kcal pro kg dem Kristallisator zugeführte Oxidationsreaktionsmischung
beträgt).
Anders als das, wie vorstehend beschrie ben, in einem nicht-adiabatischen
Kristallisator durchgeführte
Verfahren, führt
das Trennverfahren in einem adiabatischen Kristallisator hauptsächlich zur
Verminderung der Löslichkeit
infolge der Kühlung
anstatt zu einer Konzentierungswirkung infolge der Entfernung von
Wasser. Bei einer bevorzugten Ausführungsform wird die Trennung der
Mutterlauge von den ausgefallenen Feststoffen teilweise durch Dekantation
erreicht. Weil die bei der adiabatischen Kristallisation entfernte
Wassermenge relativ gering ist, weist die Mutterlauge einen relativ
geringen Gehalt an Verunreinigungen auf. Es wurde in Übereinstimmung
mit der Erfindung festgestellt, dass diese Mutterlauge direkt in
das Oxidationsreaktorsystem als Quelle für Prozesswasser zurückgeführt werden
kann. Die adiabatische Oxidation ist vorteilhaft, weil sie keine
Energie erfordert (typischerweise in Form von Dampf), wie sie für die Verdampfung
in einen nicht-adiabatischen Kristallisator benötigt wird.
-
In
einem besonders bevorzugten adiabatischen Kristallisatorsystem wird
die Reaktionsendmischung in einem Entlüftungsabschnitt einem plötzlichen
Druckabfall unterworfen, welcher einen Teil des Wassers in der Reaktionsmischung
verdampfen lässt.
Diese Verdampfung lässt
die verbleibende Reaktionsmischung wiederum abkühlen. Die Abkühlung führt zur
Ausfällung
des N-(Phosphonomethyl)glycinprodukts. Die Mutterlauge kann dann
dekantiert werden, um die Aufschlämmung des N-(Phosphonomethyl)glycinprodukts
zu konzentrieren. Die adiabatische Kristallisation ist vorteilhaft,
weil sie keine Energie erfordert (typischerweise in Form von Dampf),
wie sie für
die Verdampfung in einen nicht-adiabatischen
Kristallisator benötigt
wird.
-
12 gibt die Ausführungsform eines Systems wieder,
welches einen adiabatischen Kristallisator 115 umfasst.
Eine wässrige
Einspeisung 101, welche ein N-(Phosphonomethyl)iminodiessigsäuresubstrat
umfasst, wird in ein Oxidationsreaktorsystem 103 eingeführt, welches
eine oder mehrere Oxidationsreaktionszone(n) umfasst, in der/denen
das Substrat oxidiert wird, um die Oxidationsreaktionsmischung 105 zu
bilden, welche das N-(Phosphonomethyl)glycinprodukt umfasst. Die
vom Reaktorsystem 103 abgezogene Oxidationsreaktionsmischung 105 kann
gegebenenfalls durch einen Vorkristallisatorentlüftungsbehälter 107 geleitet werden.
Der Vorkristallisatorentlüftungsbehälter 107 erniedrigt
den Druck über
der Reaktionsmischung 105 bis zu einem gewissen Grad und
veranlasst gelöstes
CO2 aus der Mischung und aus dem Behälter zu
entweichen. Der bevorzugte Druckabfall hängt von dem Druck ab, bei dem
die Oxidationsreaktion im Reaktorsystem 103 durchgeführt wird.
Wenn zum Beispiel der Druck in der/den Oxidationsreaktionszone(n)
normalerweise nicht größer als
793 kPa (115 psia) beträgt,
ist der Druckabfall im Vorkristallisatorentlüftungsbehälter 107 normalerweise
nicht größer als
etwa 689 kPa (100 psig), weiter vorzugsweise etwa 138 bis etwa 552
kPa (20 bis etwa 80 psig), sogar weiter vorzugsweise etwa 414 bis
etwa 552 kPa (60 bis etwa 80 psig) und sogar noch weiter vorzugsweise
etwa 517 kPa (75 psig), wobei der bevorzugte Druckabfall dann, wenn
der Druck in der/den Reaktionszone(n) 1482 kPa (215 psia) beträgt, nicht
größer als
etwa 1379 kPa (1379 psig), weiter vorzugsweise etwa 827 bis etwa
1241 kPa (120 bis 180 psig), sogar noch weiter vorzugsweise etwa
1103 bis etwa 1241 kPa (160 bis 180 psig) und sogar noch weiter
vorzugsweise etwa 1207 kPa (175 psig). Dies veranlasst typischerweise
bis zu etwa 1,5% (noch typischer etwa 0,2 bis etwa 1%, sogar noch
typischer etwa 0,2% bis etwa 0,5% und sogar noch weiter typisch
etwa 0,25%) des Gewichts der Reaktionsendmischung 105 in
die Dampfphase überzugehen.
Der Druck über
dem resultierenden Kristallisatoreinspeisestrom 114, welcher
den Vorkristallisatorentlüftungsbehälter 107 verlässt, beträgt mindestens
etwa 103 kPa (15 psia), weiter vorzugsweise etwa 172 bis etwa 690
kPa (100 psia), sogar weiter vorzugsweise etwa 207 bis etwa 414
kPa (30 bis etwa 60 psia) und sogar noch weiter vorzugsweise etwa
276 kPa (40 psia).
-
In
den Vorkristallisatorentlüftungsbehälter 107 kann
eine Sauerstoffquelle (z. B. ein O2-haltiges
Gas) eingeführt
werden, um sowohl das N-(Phosphonomethyl)iminodiessigsäuresubstrat
in der Reaktionsmischung 105, das im Oxidationsreaktorsystem 103 nicht
oxidiert worden ist, als auch die in der Reaktionsmischung 105 vorliegenden
Nebenprodukte Formaldehyd und Ameisensäure, weiter zu oxidieren. Auf
diese Weise fungiert der Vorkristallisatorentlüftungsbehälter 107 in Reihe
mit dem Reaktorsystem 103 als Oxidationsreaktionszone.
-
Der
Kristallisatoreinspeisestrom 114 wird in den adiabatischen
Kristallisator 115 eingespeist. Eine detaillierte Beschreibung
des Betriebs eines mit der vorliegenden Erfindung übereinstimmenden
adiabatischen Kristallisatorsystems wird nachstehend im Zusammenhang
mit 12 angegeben. Der Betrieb des
adiabatischen Kristallisators 115 erzeugt Dampf 117 (d.
h. ein Überkopfprodukt
des adiabatischen Kristallisators) der oben aus dem Kristallisator
abgelassen wird, einen Dekantatstrom 124 (d. h. eine Mutterlauge),
der vom Kristallisator abgezogen wird und eine Kristallisationsproduktaufschlämmung 125,
welche ausgefälltes
kristallines N-(Phosphonomethyl)glycinprodukt umfasst, welches vom
Boden des Kristallisators ausgetragen wird.
-
Wenigstens
ein Teil 146 des Überkopfprodukts
des adiabatischen Kristallisators 117 und/oder wenigstens
ein Teil 132 des abgezogenen Dekantats 124 kann
zur (zu den) Oxidationsreaktionszone(n) des Reaktorsystems 103 zurückgeführt werden.
Das rezyklierte Überkopfprodukt 117 des
adiabatischen Kristallisators und/oder des abgezogenen Dekantats 124 wird
zur (zu den) Oxidationsreaktionszone(n) rezykliert und als Wasserquelle
zum Lösen
des N-(Phosphonomethyl)iminodiessigsäuresubstrats verwendet, um
den Einspeisestrom 101 für das Reaktorsystem 103 zu
bilden. Das rezyklierte Überkopfprodukt 117 des
adiabatischen Kristallisators und/oder das abgezogene Dekantat 124 wird/werden
in die in der Reihe am meisten stromaufwärts gelegene Oxidationsreaktionszone(n)
eingeführt,
wobei das Reaktorsystem 103 zwei oder mehr Oxidationsreaktionszonen
in Reihe umfasst. Die Rückführung wenigstens
eines Teils 132 des Dekantats 124 in das Reaktorsystem
ist vorteilhaft, weil es den Wasserbedarf und das Abfallvolumen
aus dem System verringert. Sie erlaubt oft auch die Gewinnung von
zusätzlichem
N-(Phosphonmomethyl)glycinprodukt aus dem nicht umgesetzten N-(Phosphonomethyl)iminodiessigsäuresubstrat
im Dekantat 124. Diese Rückführung ist zusätzlich von
Vorteil, weil sie oft die Oxidation zusätzlicher Nebenprodukte wie
Formaldehyd und Ameisensäure
erlaubt. Der Rücklaufstrom 132 ist
weiterhin vorteilhaft, weil er die direkte Rückführung von Wasser zur (zu den)
Oxidationsreaktionszone(n) aus dem Kristallisator 115 erlaubt,
ohne dass Energie für
die Verdampfung des Wassers aufgewendet werden muss (wie dies der
Fall bei der Rückführung des Überkopfprodukts
aus dem vorstehend diskutierten, wärmegesteuerten Verdampfungskristallisator
ist). Nachdem das rezyklierte Dekantat 132 auch eine relativ
hohe Temperatur beibehält
(am meisten vorzugsweise 60°C),
kann das rezyklierte Dekantat 132 dazu verwendet werden,
den wässrigen
Einspeisestrom 101 zu erwärmen. Wird ein Edelmetall auf
Kohlenstoffkatalysator im Reaktorsystem 103 verwendet,
kann ein weiterer Vorteil des Dekantatrückführstroms 132 genutzt
werden, insofern als aus dem Katalysator ausgelaugtes Edelmetall
in das Reaktorsystem zurückgeführt wird.
Es wird angenommen, dass das Rezyklieren von edelmetallhaltigen
Strömen
wie den Strom 132 zum Reaktorsystem 103 den Nettoverlust
an Edelmetall aus dem System verringert. Ein Teil des in einem solchen
Rückführstrom
enthaltenen ausgelaugten Edelmetalls kann sich auf der Oberfläche des
heterogenen Katalysators im katalytischen Reaktorsystem abscheiden.
-
Insbesondere
dann, wenn der Katalysator ein kohlenstoffhaltiger Katalysator ist
(und sogar noch spezieller, wenn der Katalysator ein Kohlenstoff-getragenes
Edelmetall umfasst), ist es vorzuziehen, wenigstens einen Teil des Überkopf produkts 117 des
adiabatischen Kristallisators indirekt zurückzuführen, indem es kondensiert
wird und das Kondensat dann mit dem Katalysator gemischt wird. Dies
ist oft vorteilhaft, weil das Überkopfprodukt 117 des
adiabatischen Kristallisators oft Formaldehyd und/oder Ameisensäure enthält, welche, wie
vorstehend bemerkt, beide als Reduktionsmittel wirken. Bei einer
besonders bevorzugten Ausführungsform
wird ein Teil des Überkopfprodukts 117 des
adiabatischen Kristallisators rektifiziert oder kondensiert und weiterhin
destilliert, um ein mit Formaldehyd und/oder Ameisensäure angereichertes
Kondensat zu erhalten. Diese angereicherte Lösung ist wiederum der Teil
des Überkopfprodukts
des adiabatischen Kristallisators, welcher mit dem kohlenstoffhaltigen
Katalysator kontaktiert wird. Wie vorstehend bemerkt, kann die Reduktionsbehandlung
in einem oder mehreren Katalystorvorratsbehälter(n) innerhalb des Reaktorsystems 103 erfolgen.
-
Wenigsten
ein anderer Teil 149 des Überkopfprodukts 117 des
adiabatischen Kristallisators und/oder wenigsten eine Teil 151 des
abgezogenen Dekantats 124 kann als Abfall aus dem System
ausgeschieden (d. h. verworfen) werden. In einem kontinuierlichen
System hilft dieses Ausscheiden den Aufbau von Verunreinigungen
im System zu verringern. Dieser ausgeschiedene Abfall kann wiederum
weiterbehandelt werden, um die Verunreinigungen nach auf dem Fachgebiet
bekanten Verfahren zu entfernen, wie die vorstehend für den ausgeschiedenen
Abfallstrom der Zentrifuge stromabwärts eines nicht-adiabatischen
Kristallisators beschriebenen. Der ausgeschiedene Abfall kann zum
Beispiel mit einem O2-haltigen Gas und einem
Gruppe VIII-Metall-Katalysator kontaktiert werden, um Formaldehyd
und Ameisensäure
zu CO2 und Wasser zu oxidieren. Das Produkt
einer solchen Oxidationsbehandlung kann zur (zu den) Oxidationsreaktionszone(n)
des Rektorsystems 103 zurückgeführt und als Quelle zur Lieferung
von Wasser verwendet werden.
-
Die
vom Boden des adiabatischen Kristallisators 115 abgezogene
N-(Phosphonomethyl)glycinproduktaufschlämmung 125 enthält den Hauptanteil
des N-(Phosphonomethyl)glycinprodukts. Die Aufschlämmung 125 durchströmt typischerweise
eine Zentrifuge 155 um die Aufschlämmung 125 weiter zu
konzentrieren und einen Nasskuchen 157 zu bilden, welcher
das N-(Phosphonomethyl)glycinprodukt enthält. Die Konzentration des N-(Phosphonomethyl)glycinprodukts
im Nasskuchen 157 beträgt
normalerweise mindestens etwa 95% (bezogen auf das Gewicht aller
Verbindungen außer
Wasser). Der an Feststoff ausgearmte Strom 161 (d. h. das
Zentrat) aus der Zentrifuge 155 kann zum Beispiel über den
Strom 165 zum adiabatischen Kristallisator zurückgeführt werden
oder über
den Strom 169 zur (zu den) Oxidationsreaktionszone(n) des
Reaktorsystems 103 zurückgeführt werden,
um als Quelle für
Wasser im wässrigen
Einspeisestrom 101 verwendet zu werden. Um die Konzentration
an Verunreinigungen auf einem annehmbaren Niveau zu halten und die
vorteilhafte Verwendung des zurückgeführten Dekantatstroms 132 zu
ermöglichen,
kann wenigstens ein Teil des an Feststoff ausgearmten Stroms 161 über den
Strom 173 entfernt werden. Der Strom 173 kann
anschließend
zum Beispiel durch die Abfallbehandlungsverfahren behandelt werden,
welche vorstehend für
den Ausscheidestrom der Zentrifuge stromabwärts eines nicht-adiabatischen
wärmegesteuerten
Kristallisators beschrieben sind. Bei einer weiteren Ausführungsform
wird der Strom 173 zur zusätzlichen Produktgewinnung in
einer Weise, ähnlich der
in 13 dargestellten, zu einem wärmegesteuerten Verdampfungskristallisator
geleitet.
-
12A ist eine schematische Darstellung eines bevorzugten
adiabatischen Kristallisationssystems zur Verwendung bei der Praktizierung
der vorliegenden Erfindung. Wie gezeigt, umfasst das System 115 die Dampf/Flüssigkeits-Trennvorrichtung 703,
welche eine Dampf/Flüssigkeits-Trennzone
definiert, die generell oberhalb und in Flüssigkeitskommunikation mit
einer Rückhaltekammer 705 angeordnet
ist. Die Dampf/Flüssigkeits-Trennvorrichtung 703 ist
von der direkten Kommunikation mit dem unteren Bereich der Rückhaltekammer 705 getrennt,
befindet sich jedoch mit dem unteren Bereich der Rückhaltekammer über ein
Beschickungsrohr 706 in Flüssigkeitskommunikation, wobei
die Mündung 708 des
Beschickungsrohrs 706 vom Boden der Rückhaltekammer nur durch einen
relativ kurzen Abstand getrennt ist. Kristallisationsapparate dieser
allgemeinen Bauweise sind von der HPD Products Division of U.S.
Filter (Plainfield, Illinois, U.S.A.) erhältlich. Ein Kristallisationszirkulationseinlass 709 ist
auf der Dampf/Flüssigkeits-Trennvorrichtung 703 angeordnet,
während die
Rückhaltekammer 705 mit
einem Dekantationsflüssigkeitsauslass 711 für die Kristallisation
der Mutterlauge oberhalb der Mündung 708 des
Beschickungsrohrs 706 vorgesehen ist, ein Zwischenproduktrezikulationsaufschlämmungsauslass
oberhalb der Mündung
des Beschickdungsrohrs und unterhalb des Dekantationsflüssigkeitsauslasses 711 angeordnet
ist und ein tiefer gelegener Produktaufschlämmungsauslass 713 am
Boden der Kammer 705 angeordnet ist. Während des Betriebs ist die
Rückhaltekammer 705 im
Wesentlichen mit Flüssigkeit
gefüllt,
während
der Flüssigkeitsspiegel 715 in
der Dampf/Flüssigkeits-Trennvorrichtung 703 etwas unterhalb
des Kristallisatorrezirkulationseinlasses 709 eingestellt
wird.
-
Eine
wässrige
Kristallisatoreinspeisemischung 716, welche eine aus dem
Reaktionsmischungsauslass 114 erhaltenes N-(Phosphonomethyl)glycinprodukt
umfasst, wird aus der/den Oxidationsreaktionszone(n) des Oxidationsreaktorsystems
(zusammen mit verschiedenen Rezirkulationsströmen, wie nachstehend beschrieben)
durch den Rezirkulationseinlass 709 in die Dampf/Flüssigkeits-Trennvorrichtung
eingespeist. Die Dampf/Flüssigkeits-Trennvorrichtung
definiert eine Verdampfungszone, welche von einem Vakuumsystem (nicht
dargestellt) unter Atmosphärendruck
und unter dem Dampfdruck der Kristallisatoreinspeisemischung 716 aufrechterhalten
wird. Das Flüssigkeitsniveau 715 in
der Dampf/Flüssigkeits-Trennvorrichtung 703 wird durch
Druckausgleich mittels Löchern
im oberen Abschnitt des Beschickungsrohres 706 aufrechterhalten,
welches mit der Rückhaltekammer 705 in
Verbindung steht. Die Kristallisatoreinspeisemischung 716 umfasst:
(a) den von der/den Oxidationsreaktionszone(n) des Reaktorsystems
abgezogenen Oxidationsreaktionsmischungsablauf 114 (d.
h. die Ausgangslösung),
welche filtriert worden sein kann, um den Katalysator zu entfernen;
(b) einen rezyklierten Aufschlämmungsstrom 723,
welcher wenigstens einen Teil der Aufschlämmung umfasst, die aus dem
Zwischenproduktrezikulationsaufschlämmungsauslass 712 austritt,
wie nachstehend beschrieben; und typischerweise auch (c) ein Zentrat 165,
welches Kristallisationsmutterlauge umfasst, welche vom Zentrifugensystem 155 rezykliert
wird, dem eine Kristallisationsproduktsaufschlämmung 125 aus dem Auslauf 713 zur
Gewinnung von festem N-(Phosphonomethyl)glycinprodukt zugeführt wird,
wie nachstehend weiter beschrieben. Der in der Dampf/Flüssigkeits-Trennvorrichtung
aufrecht erhaltene Druck ist generell nicht größer als etwa 55 kPa (8 psia)
und beträgt
vorzugsweise etwa 10 bis etwa 28 kPa (1,5 bis etwa 4 psia), sogar weiter
vorzugsweise etwa 17 bis etwa 24 kPa (2,5 bis etwa 3,5 psia) und
sogar noch weiter vorzugsweise etwa 31 kPa (3 psia). Der Druck der
Kristallisatoreinspeisemischung 716 ist unmittelbar stromaufwärts der Dampf/Flüssigkeits-Trennvorrichtung
typischerweise so, dass die Einspeisemischung einer Druckerniedrigung von
mindestens etwa 690 kPa (100 psig), vorzugsweise von etwa 69 kPa
bis etwa 552 kPa (10 bis etwa 80 psig), weiter vorzugsweise von
etwa 207 bis etwa 414 kPa (30 bis etwa 60 psig) und sogar weiter
vorzugsweise etwa 262 kPa (38 psig) nach Eintritt in die Dampf/Flüssigkeits-Trennvorrichtung
ausgesetzt wird. Der plötzliche Druckabfall
veranlasst Wasser und niedermolekulare Verunreinigungen (z. B. Formaldehyd
und Ameisensäure)
sich aus der Einspeisemischung 716 in der Dampf/Flüssigkeits-Trennvorrichtung
zu verflüchtigen
(d. h. zu verdampfen). Der produzierte Dampf 117 (d. h.
das Kopfprodukt) wird aus dem Kopf der Trennvorrichtung 703 abgetrennt
und verworfen, indem er zu einer Kondensa tionseinheit (nicht dargestellt)
geleitet wird. Normalerweise wird nicht mehr als etwa 30 Gew.-%,
weiter vorzugsweise etwa 5% bis etwa 30 Gew.-% und sogar weiter vorzugsweise
etwa 5% bis etwa 10 Gew.-% der Oxidationsreaktionsmischung 114 als
Dampf 117 verworfen. Als Folge der Verdampfung kühlt sich
der verbleibende Anteil der kondensierte Phase der Kristallisatoreinspeisemischungen 716 beträchtlich
ab, was zur Ausfällung
von N-(Phosphonomethyl)glycinprodukt und zur Erzeugung einer Verdampfungsproduktaufschlämmung 718 führt, welche
kristallines N-(Phosphonomethyl)glycinprodukt als Feststoff 719 umfasst,
welcher in einer Mutterlauge suspendiert ist, die im Wesentlichen
an N-(Phosphonomethyl)glycinprodukt gesättigt oder übersättigt ist. Die Kühlwirkung,
welche aus der Druckminderung resultiert, welche in der Dampf/Flüssigkeits-Trennvorrichtung 703 erfolgt,
reicht aus, damit die Temperatur der Verdampfungsproduktaufschlämmung 718 um
etwa 30°C
bis etwa 40°C
niedriger ist als die Temperatur der Oxidationsreaktionsmischung 114,
welche in das adiabatische Kristallisationssystem eingeführt wird. Die
Temperatur der Verdampfungsproduktaufschlämmung 718 beträgt nicht
mehr als 80°C,
weiter vorzugsweise etwa 45°C
bis etwa 80°C,
sogar weiter vorzugsweise etwa 55°C
bis etwa 70°C
und insbesondere etwa 60°C bis
etwa 70°C.
-
Die
Verdampfungsproduktaufschlämmung 718 tritt
aus der Trennvorrichtung 703 durch Absenken des Beschickungsrohrs 706 aus
und wird in den tiefer gelegenen Bereich der Rückhaltekammer 705 eingeführt. Die
Rückhaltezone
ist in einen unteren Kristallisationsbereich, im Allgemeinen unterhalb
des Niveaus 720 und in einen oberen Dekanationsbereich,
im Allgemeinen oberhalb des Niveaus 720 unterteilt. In
der Rückhaltezone
wird die Verdampfungsproduktaufschlämmung 718 in eine überstehende
Flüssigkeit 722,
welche eine Fraktion der Mutterlauge (typischerweise deren Nettoerzeugung)
umfasst und einen zweiten Aufschlämmungsstrom 723 aufgeteilt,
welcher ausgefällte
N-(Phosphonomethyl)glycinproduktkristalle und Mutterlauge umfasst, welche
aus der Rückhaltekammer 705 durch
den Zwischenproduktaufschlämmungsauslauf 712 abgezogen wird.
Eine Dekantatstrom 124, welche die überstehende Flüssigkeit 722 umfasst,
wird aus der Rückhaltekammer 705 durch
den Dekantationsauslass 711 im Dekantationsbereich in der
Nähe des
Kopfs der Rückhaltekammer
abgezogen.
-
Die
Kristallisationsproduktaufschlämmung 125,
welche die N-(Phosphonomethyl)glycinproduktaufschlämmung umfasst,
wird vom Boden der Rückhaltekammer 705 durch
den Auslass 713 im Kristallisationsbereich abgezogen. Die Kristallisationsproduktaufschlämmung wird
zum Zentrifugensystem 155 weitergeleitet, in dem die N-(Phosphonomethyl)glycinproduktkristalle
als Nasskuchen abgetrennt werden. Das N-(Phosphonomethyl)glycinprodukt
im Nasskuchen beträgt
normalerweise mindestens etwa 95% (bezogen auf das Gewicht aller
Verbindungen außer
Wasser). Das resultierender Zentrat 165 wird zurückgeführt und
mit einem zweiten Teil der Produktaufschlämmung mit dem Aufschlämmungsstrom 723 kombiniert,
der aus der Rückhaltekammer 705 an
der Grenzfläche
(d. h. Trübungszone)
zwischen dem Dekantationsbereich und dem Kristallisationsbereich
der Rückhaltezone
abgezogen wird. Der kombinierte Strom wird zusammen mit der Oxidationsreaktionsmsichung 114 als
Kristallisatoreinspeisemischung 716in die Dampf/Flüssigkeits-Trennvorrichtung 703 eingeführt.
-
Mindestens
ein Hauptanteil, vorzugsweise im Wesentlichen der gesamte, aus dem
Ablauf 712 abgezogene sekundäre Aufschlämmungsstrom 723 wird
zur Dampf/Flüssigkeits-Trennvorrichtung 703 zurückgeführt, indem
er mit dem Reaktionsmischungsstrom 114 und dem Zentrat 165 aus
der Zentrifuge 155 gemischt wird, um die Einspeisemischung 716 in
die Dampf/Flüssigkeits-Trennvorrichtung
zu bilden. Die Mutterlauge 722 wird von der Verdampfungsproduktaufschlämmung 718 im
Dekantationsbereich abgetrennt (d. h. abdekantiert). Die Dekantation
erfolgt durch die Aufrechterhaltung der relativen Raten, mit denen
die Reaktionsmischung 114 durch den Einlass 709 eingeführt und
das Dekantat 124 über
den Auslass 711 abgezogen wird, wobei und die gesamte oder
ein Teil der sekundären
Aufschlämmung 723 vom
Zwischenproduktaufschlämmungsauslass 712 über den
Kristallisatoreinspeisestrom 716 (wodurch die Geschwindigkeit,
mit der die Verdampfungsproduktaufschlämmung in die Rückhaltezone
eingeführt
wird, gesteuert wird) rezirkuliert wird, dermaßen, dass die Aufwärtsstromgeschwindigkeit
im unteren Kristallisationsbereich der Rückhaltezone unterhalb des Zwischenproduktaufschlämmungsauslaufs 712 ausreicht,
die ausgefällten
N-(Phosphonomethyl)glycinproduktkristalle 719 in der flüssigen Phase
in Suspension (d. h. eingeschlossen) zu halten, während die
Aufwärtsstromgeschwindigkeit
im oberen Dekantationsbereich der Rückhaltezone oberhalb des Zwischenproduktaufschlämmungsauslaufs 712 unterhalb
der Absetzgeschwindigkeit wenigstens etwa 80 Gew.-%, vorzugsweise
unterhalb der Absetzgeschwindigkeit von 95 Gew.-%, am meisten vorzugsweise
unterhalb der Absetzgeschwindigkeit von 98 Gew.-% der N-(Phosphonomethyl)glycinproduktkristalle 719 im
Kristallisationsbereich beträgt.
Es bildet sich demzufolge ungefähr
auf dem Niveau des Zwischenproduktaufschlämmungsauslaufs 712 zwischen
dem oberen Dekantationsbereich der Rückhaltezone, welcher hauptsäch lich Mutterlauge
enthält,
und dem unteren Kristallisationsbereich der Rückhaltezone, der eine Kristallisationsaufschlämmung enthält, eine
Grenzfläche
aus.
-
Die
relativen Raten, mit denen die Oxidationsreaktionsmischung 114 in
das adiabatische Kristallisationssystem 115 eingeführt, das
Dekantat 124 aus dem Auslauf 711 abgezogen, die
Produktaufschlämmung 125 aus
dem Auslauf 713 abgezogen und das Zentrat 165 aus
der Zentrifuge rezykliert wird, werden vorzugsweise so gesteuert,
dass das Verhältnis
von N-(Phosphonomethyl)glycinproduktfeststoffen zu Mutterlauge im unteren
Kristallisationsbereich der Rückhaltezone
höher ist
als das inkrementelle Verhältnis
von N-(Phosphonomethyl)glycinprodukt zu Mutterlauge, das sich aus
der Verdampfungswirkung ergibt, wobei ein solches inkrementelles
Verhältnis
das Verhältnis
von inkrementell erzeugten N-(Phosphonomethyl)glycinproduktfeststoffen
zu dadurch inkrementell erzeugter Mutterlauge ist, d. h. die Nettoerzeugung
von kristallinem N-(Phosphonomethyl)glycinprodukt. Das inkrementelle
Verhältnis
ist, anders ausgedrückt,
das Verhältnis,
welches erhalten werden würde,
wenn die Oxidationsreaktionsmischung in Abwesenheit von Feststoffen
in der Kristallisatoreinspeisemischung (d. h. in Abwesenheit der
rezirkulierten Sekundäraufschlämmung) abgedampft
werden würde.
Die Wirkung der Verdampfung schließt selbstverständlich sowohl
Konzentrierungseffekte als auch Kühlwirkungen ein; Wo jedoch
der Betrieb des Kristallisators im Wesentlichen adiabatisch erfolgt,
ist es bevorzugt, dass die Kristallisation hauptsächlich aus
der Kühlung
der flüssigen
Phase auf eine Temperatur resultiert, bei der die Löslichkeit
des N-(Phosphonomethyl)glycinprodukts wesentlich geringer ist, als
die bei der Temperatur der Oxidationsreaktionsmischung. Das Feststoff/Mutterlauge-Verhältnis im
unteren Bereich der Rückhaltezone
beträgt
vorzugsweise das Doppelte des aus der Verdampfungswirkung resultierenden
inkrementellen Verhältnisses,
und die Konzentration an Produktfeststoffen im Kristallisationsbereich
beträgt
mindestens das Doppelte der inkrementell erzeugten Konzentration.
Das N-(Phosphonomethyl)glycinfeststoffprodukt im unteren Kristallisationsbereich
der Rückhaltezone
beträgt,
anders ausgedrückt,
wenigstens etwa 12 Gew.-%, vorzugsweise wenigstens etwa 15 Gew.-%
und liegt weiter vorzugsweise im Bereich von etwa 18% bis etwa 25 Gew.-%.
Im dargestellten System ist die Abscheidungsrate in der Zentrifuge 155 und
der Mutterlauge als Dekantat 124 letztendlich durch das
Materialgleichgewicht im System gegeben, wobei jedoch die Feststoffgehalte im
unteren Kristallisationsbereich der Rückhaltezone durch eine vorübergehende
Erhöhung
oder Erniedrigung der Rate eingestellt werden können, mit der die Produktaufschlämmung aus
dem Auslauf 713 im Verhältnis zur
Dekantations rate über
den Ablauf 711 entfernt wird. Wie hierin nachstehend weiter
erörtert,
wurde gefunden, dass die Regelung des Feststoffgehalts im Kristallisationsbereich
der Rückhaltezone
die Steuerung der mittleren Teilchengröße des N-(Phosphonomethyl)glycinprodukts beim
Kristallisationsverfahren vorsieht.
-
Der
stationäre
Zustand der Aufwärtsstromgeschwindigkeit
im oberen Dekantationsbereich der Rückhaltezone wird durch die
Dimensionierung des Querschnitts der Rückhaltekammer 705 auf
der Basis des Materialgleichgewichts beim Verfahren und der Feststoffabscheidungsgeschwindigkeit
eingestellt. Vorzugsweise wird eine relativ hohe Aufwärtsgeschwindigkeit
im unteren Kristallisationsbereich der Rückhaltezone eingehalten, indem
eine hohe Rezirkulationsrate der zweiten Aufschlämmung 723 zwischen
dem Zwischenproduktaufschlämmungsauslass 712 und
dem Rezirkulationseinlass 709 gegenüber der Dampf/Flüssigkeits-Trennvorrichtung
aufrechterhalten wird (z. B. im Bereich von 20:1 bis 100:1, bezogen
auf Rate des Oxidationsreaktionsmedium 114, welches in
das Kristallisationssystem eingeführt oder das Dekantat 124,
welches daraus entfernt wird). Die Fraktion des Zentrats 165 aus
der Zentrifuge 155, welches als Teil der Kristallisatoreinspeisemischung 716 zurückgeführt wird,
erhöht
auch die Rezirkulationsrate und Stromaufwärtsströmungsgeschwindigkeit, neigt
jedoch andererseits dazu, die Aufschlämmung im Kristallisationsbereich
zu verdünnen.
Durch Kombinieren einer hohen sekundären Aufschlämmungsrezirkulationsrate mit
einer geeigneten Dimensionierung der Rückhaltekammer 705,
kann die Aufwärtsstromgeschwindigkeit
im unteren Kristallisationsbereich der Rückhaltezone auf das mindestens
4Fache der Sedimentationsgeschwindigkeit bei mindestens 80 Gew.-% darin
enthaltenem Feststoff eingestellt werden, während die Aufwärtsstromgeschwindigkeit
im unteren Dekantationsbereich der Rückhaltezone auf weniger als
ein Viertel der im zweiten Aufschlämmungsstrom enthaltenen Feststoffe
eingestellt werden kann.
-
Der
Betrieb bei hohen Feststoffgehalten im Kristallisationsbereich,
verbunden mit einer hohen Rezirkulationsrate der zweiten Aufschlämmung 723 aus
dem Zwischenproduktaufschlämmungsauslass 712 zur Dampf/Flüsigkeits-Trennvorrichtung 703,
sorgt weiterhin für
eine hohe Feststoffkonzentration in der gesamten Verdampfungszone.
Es wurde gefunden, dass diese Art des Betriebs sowohl auf die Leistungsfähigkeit
des Kristallisationsverfahrens als auch auf die Teilchengröße und die
Auslasseigenschaften des erhaltenen kristallinen N-(Phosphonomethyl)glycinprodukts
eine ausgesprochen günstige
Wirkung ausübt.
Es besteht eine Diskrepanz zwischen der Teilchengröße und der
Produktivität,
weil die Produktivität
positiv mit dem Übersättigungsgrad
zusammenhängt,
der Übersättigungsgrad
jedoch generell negativ mit der Teilchengröße zusammenhängt. Selbst
bei einer relativ hohen mittleren Teilchengröße erhöht jedoch der Betrieb mit hohen
Feststoffgehalten die Oberfläche
der festen kristallisierenden N-(Phosphonomethyl)glycinproduktkristalle
wirksam, und erlaubt es dem Kristallisationsverfahren mit hoher
Produktivität
bei relativ geringfügiger Übersättigung
der erforderlichen flüssigen
Phase abzulaufen sowie die Triebkraft für die Kristallisation bereitzustellen.
Die Kristallisation bei niedrigen Übersättigungsgraden fördert wiederum
die Bildung relativ großer
Kristalle. Folglich liefert bei einer gegebenen Kapazität der Verdampfungszone
das Kristallisationsverfahren der Erfindung eine mittlere Teilchengröße, welche
wesentlich größer ist
als die mittlere Teilchengröße, welche
bei einem Bezugsverdampfer mit vollständiger Rückvermischung erhalten wird,
wobei das Verhältnis
der inkrementell erzeugten N-(Phosphonomethyl)glycinproduktfeststoffe
zur Mutterlauge gleich dem Verhältnis
der inkrementell erzeugten N-(Phosphonomethyl)glycinproduktfeststoffe
ist, welche durch die Verdampfungswirkung auf die Mutterlauge inkrementell
erzeugt werden. Das Kristallisationsverfahren der Erfindung kann
zum Beispiel mit hoher Kapazität
betrieben werden, um ein Produkt zu erhalten, mit (1) einer mittleren
Würfelgewichtteilchengröße von mindestens
etwa 200 μm,
vorzugsweise mindestens etwa 225 μm,
weiter vorzugsweise mindestens etwa 250 μm, sogar weiter vorzugsweise
mindestens etwa 275 μm,
noch weiter vorzugsweise mit mindestens etwa 300 μm und insbesondere
mit mindestens etwa 350 μm;
(2) einer mittlere Längengewichtsteilchengröße von mindestens
etwa 85 μm,
vorzugsweise mindestens etwa 90 μm,
weiter vorzugsweise mindestens etwa 95 μm, sogar weiter vorzugsweise
mindestens etwa 100 μm,
noch weiter vorzugsweise mindestens etwa 105 μm und insbesondere mindestens
etwa 110 μm;
und (3) eine BET (Brunauer-Emmett-Teller)-Oberfläche von nicht größer als
etwa 0,11 m2/g, weiter vorzugsweise nicht
größer als
etwa 0,09 m2/g, sogar weiter vorzugsweise
nicht größer als
etwa 0,07 m2/g und sogar noch weiter vorzugsweise
von nicht mehr als 0,06 m2/g. Die vorstehend angegebenen
mittleren Würfelgewichtsteilchengrößen und
mittleren Längengewichtsteilchengrößen können unter
Verwendung einer Strahlenbündelreflexionsvorrichtung
(FBRM) bestimmt werden, wie dem LASSENTEC Modell M100F, das von
Laser Sensor Technology Inc. (Redmond, Washington, U.S.A.) erhältlich ist.
-
Bei
den hohen Strömungsraten,
welche entlang des Rezirkulationsweges zwischen dem Zwischenproduktaufschlämmungsauslass 712 und
der Mündung 708 des
Beschickungsrohres 706 herrschen, arbeitet das Kristallisationssystem
im Wesentlichen nach Pfropfströmungsart
(d. h. ohne wesentliche axiale Rückvermischung).
Als Folge davon nimmt der Gradient des Übersättigungsgrades entlang dieses
Weges ab, wodurch er die zur Kristallisation verfügbare integrierte
mittlere Triebkraft maximiert und einen niedrigeren Übersättigungsgrad
am stromabwärts
gelegenen Ende des Pfropfströmungswegen
(d. h. der Mündungen
des Beschickdungsrohres) ermöglicht
als er bei einem System mit Rückvermischung
verwirklicht werden könnte.
Kombiniert man die Wirkung der Pfropfströmung mit dem allgemein niedrigen Übersättigungsgrad,
welcher durch die hohe Kristalloberfläche, die vom hohen innerhalb
der Rezikulationsaufschlämmung
vorliegenden Feststoffgehalt ermöglicht
wird, wird das Nettoergebnis bei irgendeiner gegebenen Herstellungsrate
den Übersättigungsgrad
in der flüssigen
Phase innerhalb des unteren Kristallisationsbereichs der Rückhaltezone
weiter verringern und daher die dekantierte Mutterlauge 124,
welche aus dem System entfernt wird.
-
Der
Pfropfströmungsbetrieb
kann in Kombination mit hohem Feststoffgehalt auch im Hinblick auf
die Produktivität
ausgenutzt werden. Es ist gefunden worden, dass eine hohe Produktivität sogar
dann verwirklicht werden kann, wenn die maximale Übersättigung,
d. h. die Triebkraft für
die Kristallisation, ausgedrückt
als Differenz zwischen der N-(Phosphonomethyl)glycinkonzentration
in der wässrigen
Phase an irgendeiner Stelle innerhalb des Rezirkulationsweges und
der Sättigungskonzentration
des N-(Phosphonomethyl)glycinprodukts in der wässrigen flüssigen Phase an einer solchen
Stelle, nicht größer als
etwa 0,7 Gew.-% beträgt,
bezogen auf die wässrige
flüssige
Phase; oder wo der integrierte mittlere Übersättigungsgrad gegenüber dem
des Rezirkulationswegen nicht größer als
0,5% ist. Betrachtet man die Beziehung zwischen der Übersättigung
und der Produktivität
noch unter einem anderen Gesichtspunkt, kann das beschriebene Verfahren
bei einer integrierten mittleren Übersättigung über der im Rezirkulationsweg
effektiv arbeiten, wenn sie mindestens 0,2% unter dem Übersättigungsrad
liegt, der erforderlich ist, um eine äquivalente Kristallisationsproduktivität pro Arbeitsvolumen
eines Bezugsverdampfers bereitzustellen, welcher aus einer Verdampfungszone
mit vollständiger
Rückvermischung
besteht, in welchem das Verhältnis
von N-(Phosphomomethyl)glycinfeststoffen zu Mutterlauge gleich dem
Verhältnis
von durch die Verdampfungswirkung inkrementell erzeugten N-(Phosphomomethyl)glycinfeststoffen
zu dadurch inkrementell erzeugter Mutterlauge ist.
-
Wegen
der groben Teilchengröße der in Übereinstimmung
mit dem in 12A dargestellten Verfahren hergestellten
Kristalle, wird die Kapazität
einer Zentrifuge oder eines Filters zur Entwässerung der Produktaufschlämmung 125 in
Verbindung mit Einsparungen bei den Kapital- und Wartungskosten
wesentlich erhöht.
Bei Verwendung eines vertikalen Korbzentrifugensystems oder einer
anderen Entwässerungsvorrichtung
wird zum Beispiel ein kristallines N-(Phosphonomethyl)glycinprodukt mit einem
relativ niedrigen Wassergehalt erhalten, welches z. B. beim Trocknen
einen Verlust von nicht mehr als etwa 15 Gew.-%, weiter vorzugsweise von
nicht mehr als etwa 8 Gew.-% aufweist. Eine niedrigere Feuchte des
Zentrifugenkuchen führt
direkt zu einer geringeren Verunreinigung des Zenrifugenkuchens
mit Chloriden, NMG, nicht umgesetztem N-(Phosphonomethyl)iminodiessigsäuresubstrat
etc. Die Verwendung des adiabatischen Kristallisationssystems bietet folglich
die Gelegenheit ein N-(Phosphonomethyl)glycinprodukt mit einem außerordentlich
hohem Reinheitsgrad herzustellen. Um die Zerreibung der N-(Phosphonomethyl)glycinkristalle
auf ein Mindestmaß zu
beschränken
wird eine Propellerpumpe verwendet, um das Material im adiabatischen
Kristallisatorsystem umzupumpen.
-
Das
Kristallisationsverfahren von 12A erfolgt
vorteilhafterweise adiabatisch, d. h. es gibt keinen nennenswerten
Wärmeübergang
in das oder aus dem System infolge des Wärmeübergangs in die oder aus der
Dampf/Flüssigkeits-Trennzone,
Rückhaltezone,
Einspeismischung in die Dampf/Flüssigkeits-Trennzone, der
sekundären
Aufschlämmung,
welche in die Dampf/-Flüssigkeits-Trennzone
rezikuliert wird oder dem Zentrat, welches aus dem Zentrifugensystem
zurückgeführt wird.
Durch Verminderung des Drucks der Einspeisemischung wird, wie vorstehend
beschrieben, eine ausreichende Verdampfung erzielt, um die flüssige Phase bis
zu einem Grad abzukühlen,
dass eine wesentliche Kristallisation von N-(Phosphonomethyl)glycinprodukt erfolgt.
Kapital- und Energieeinsparungen werden durch den Wegfall von Verdampferwärmetauschern
realisiert und desgleichen durch Wegfall von Prozessausfallzeiten,
welche benötigt
werden, um den Belag von Wärmetauschern
periodisch auszuheizen.
-
Außerdem kann
ohne wesentliche Energiekosten für
die Trennung ein Dekanatationsstrom 124 vorgesehen werden,
welcher unschwer als Quelle für
das Prozesswasser zum Rezyklieren zur (zu den) Oxidationszone(n)
des Oxidationsreaktorsystems dienen kann. Nachdem die Kristallisation
mit hoher Produktivität
bei relativ begrenztem Übersättigungsgrad
bewirkt werden kann, weist das zur (zu den) Oxidationsreaktionszone(n)
rezirkulierte Dekantat beinahe die theoretische Mindestkonzentration
des N-(Phosphonomethyl)glycinprodukts auf. Weil die Produktivität des Oxidationsreaktionssystems
typischerweise durch die Löslichkeit
des N-(Phosphonomethyl)glycinprodukts im Reaktionsmischungsausfluss
begrenzt sein kann, insbesondere wenn bei der Herstellung der freien
Säureform
von N-(Phosphonomethyl)glycin ein teilchenförmiger Katalysator verwendet
wird, kann das N-(Phosphonomethyl)glycinprodukt im Rezirkulationsdekantat
wenigsten geringfügig von
der Nettoproduktivität
des Oxidationsreaktorsystems durch die Begrenzung der Geschwindigkeit
abweichen, mit der das N-(Phosphonomethyl)iminodiessigsäuresubstrat
in das N-(Phosphonomethyl)glycinprodukt darin umgewandelt werden
kann, ohne die Löslichkeit
des N-(Phosphonomethyl)glycinprodukts zu überschreiten. Dieser mäßige Nachteil,
welcher mit der Rückführung des
Dekantats verbunden ist, wird dadurch uriniert, dass beinahe die
gesamte theoretische Maximalausbeute des N-(Phosphonomethyl)glycinprodukts
im Kristallisator gewonnen wird, wodurch der Gehalt an N-(Phosphonomethyl)glycinprodukt
im Wasserstrom, welcher zurückgeführt wird,
auf das Mindestmaß herabgesetzt
wird.
-
Obwohl
das in 12A abgebildete System bevorzugt
ist, wird der Fachmann erkennen, dass andere Optionen zur Einstellung
und Aufrechterhaltung der hohen Verhältnisse von N-(Phosphonomethyl)glycinprodukt
zu Mutterlauge im Kristallisationsbereich der Rückhaltezone existieren, welche
die Bereitstellung relativ grober Kristalle bewirken. Das Verfahren
von 12A bewirkt die Zurückhaltung
von Feststoffen in der Verdampfungszone; Das Verfahren könnte alternativ
betrieben werden, um Feststoffe zur Verdampfungszone zurückzuführen. Wenn
die Kristallisation zum Beispiel in einem Verdampfungkristallisator
mit vollständiger
Rückvermischung
erfolgt, ist es möglich
im Kristallisator einen hohen und disproportionierten Feststoffgehalt
durch Zurückführen von
kristallinem Produkt aus dem Filter oder der Zentrifuge, welche
zur Gewinnung der N-(Phosphonomethyl)glycinfeststoffprodukte
verwendet werden, einzustellen und aufrechtzuerhalten, indem entweder kein
Filtrat/Zentrat zurückgeführt wird
oder ein geringerer Anteil an Filtrat/Zentrat in Bezug auf die Feststoffe zurückgeführt wird.
Wie der Fachmann jedoch erkennen wird, ist letzteres Betriebsschema
mit dem Nachteil einer kapitalintensiven Filter- oder Zentrifugenkapazität verbunden.
Ein beträchtlicher
Vorteil des bevorzugten Verfahrens von 12A ist
die Erzielung eines hohen Feststoffgehalts durch Dekantation anstatt
teuerer mechanischer Mittel zur Feststoff/Flüssigkeits-Trennung.
-
Überaschenderweise
ist gefunden worden, dass sich beim Betrieb des adiabatischen Kristallisators
in der bevorzugten, vorstehend beschriebenen Weise, die Konzentrierung
der Produktaufschlämmung
bei der Dekantation (wie durch Verwendung eines Hydrocyklons) vor
der Einführung
der Aufschlämmung
in eine Zentrifuge erübrigt.
-
13 gibt ein Beispiel einer bevorzugten Ausführungsform
wieder, bei der das Verfahren einen adiabatischen Kristallisator
in Reihe mit einem nicht-adiabatischen Kristallisator umfasst.
-
Viele
der verschiedenen in 13 dargestellten Ströme entsprechen
denen, welche vorstehend für nicht-adiabatische
Kristallisatoren und adiabatische Kristallisatoren allein beschrieben
worden sind. Ein wässriger
Einspeisestrom 601, welcher das N-(Phosphonomethyl)iminiodiessigsäuresubstrat
umfasst, wird in ein Oxidationsreaktorsystem eingeführt, welches
eine oder mehrere Oxidationsreaktionszone(n) umfasst, worin das
Substrat unter Bildung einer N-(Phosphonomethyl)glycinprodukt umfassenden
Oxidationsreaktionsmischung 605 oxidiert wird. Die Oxidationsreaktionsmischung 605 kann
wahlweise durch einen Vorkristallisatorentlüftungsbehälter 607 geleitet
werden. Der Vorkristallisatorentlüftungsbehälter 607 erniedrigt
den Druck über
der Reaktionsmischung 605 bis zu einem gewissen Grad und
veranlasst gelöstes
CO2 aus der Mischung zu entweichen und aus
dem Entlüftungsbehälter zu
entweichen. In den Vorkristallisatorentlüftungsbehälter kann eine Sauerstoffquelle
(z. B. O2-haltiges Gas) eingeführt werden,
um in der Reaktionsmischung 605 sowohl weiteres N-(Phosphonomethyl)iminodiessigsäuresubstrat
zu oxidieren, welches in der/den Oxidationsreaktionszone(n) des
Reaktorsystems 603 nicht oxidiert worden ist, als auch
in der Reaktionsmischung 605 vorliegenden Formaldehyd und
sauere Nebenprodukte weiter zu oxidieren. Auf diese Weise fungiert
der Vorkristallisatorentlüftungsbehälter 607 als
eine dem Reaktorsystem 603 nachgeschaltete Oxidationsreaktionszone.
-
Ein
Kristallisatoreinspeisestrom 614, welcher den Hauptanteil
des N-(Phosphonomethyl)glycinprodukts umfasst, wird in den adiabatischen
Kristallisator 615 eingeführt. Der Betrieb des adiabatischen
Kristallisators 615 liefert den Dampf 617 (d.
h. das Überkopfprodukt
des adiabatischen Kristallisators), der aus dem Kopf des Kristallisators
abgeführt
wird, einen Dekantatstrom 624 (d. h. eine primäre Mutterlauge),
welcher aus dem Kristallisator abgezogen wird und ein primäres Kristallisationsaufschlämmungsprodukt 625,
welches ausgeschiedenes kristallines N-(Phosphonomethyl)glycinprodukt
umfasst, das vom Boden des Kristallisators ausgetragen wird.
-
Wenigstens
ein Teil des Überkopfprodukts 646 des
adiabatischen Kristallisators und/oder wenigstens ein Teil 632 des
abgezogenen Dekantats kann zur (zu den) Oxidationsreaktionszone(n)
des Reaktorsystems 603 zurückgeführt werden. Typischerweise
wird das rezirkulierte Überkopfprodukt 617 des
adiabatischen Kristallisators und/oder das abgezogene Dekantat 624 zur
(zu den) Oxidationsreaktionszone(n) zurückgeführt und als Quelle für Wasser
zum Lösen
des N-(Phosphonomethyl)iminodiessigsäuresubstrats
verwendet, um den Einspeisestrom 601 für das Reaktorsystem 603 zu
bilden. Das rezirkulierte Überkopfprodukt 617 des
adiabatischen Kristallisators 617 und/oder das abgezogene
Dekantat 624 wird/werden in die am meisten stromaufwärts gelegene
Oxidationsreaktionszone eingeführt,
wenn das Reaktorsystem 603 zwei oder mehr Oxidationsreaktionszonen
in Reihe umfasst. Das Zurückführen wenigstens
eines Teils 632 des Dekantats 624 zum Oxidationsreaktorsystem
ist vorteilhaft, weil es den Wasserbedarf und das Abfallvolumen
aus dem System verringert. Es erlaubt oft auch die Gewinnung von
zusätzlichem
N-(Phosphonomethyl)glycinprodukt aus dem im Dekantat 624 nicht
umgesetzten N-(Phosphonomethyl)iminodiessigsäuresubstrat. Diese Rückführung ist
zusätzlich
vorteilhaft, weil es oft das Oxidieren weiterer Nebenprodukte wie
Formaldehyd und Ameisensäure
erlaubt. Das Zurückführen des
Stroms 632 ist weiter vorteilhaft, weil es die Rückführung von
Wasser direkt zur (zu den) Oxidationsreaktionszone(n) aus dem Kristallisator 615 erlaubt,
ohne dass zuerst Energie aufgewendet werden muss um das Wasser zu
verdampfen (wie dies bei einem nicht-adiabatischen wärmegesteuerten
Kristallisator der Fall ist, wie vorstehend erörtert). Weil das rezirkulierte
Dekantat 632 auf einer relativ erhöhten Temperatur bleibt (am
meisten vorzugsweise 60°C),
kann das rezirkulierte Dekantat 632 dazu verwendet werden,
den wässrigen
Einspeisestrom 601 vorzuwärmen.
-
Insbesondere
wenn der Katalysator ein kohlenstoffhaltiger Katalysator ist (und
insbesondere, wenn ein solcher Katalysator auch ein Edelmetall enthält), ist
es vorzuziehen, wenigstens einen Teil des Überkopfprodukts 617 des
adiabatischen Kristallisators durch Mischen mit dem Katalysator
indirekt zurückzuführen. Dies
ist vorteilhaft, weil das Überkopfprodukt 617 des
adiabatischen Kristallisators, wie vorstehend bemerkt, oft Formaldehyd
und/oder Ameisensäure
enthält,
die beide als Reduktionsmittel wirken. Bei einer besonders bevorzugten
Ausführungsform
wird ferner ein Teil des Überkopfprodukts 617 des
adiabatischen Kristallisators destilliert, um eine Lösung zu
erhalten, deren Formaldehyd- und/oder Ameisensäurekonzentration(en) angereichert
ist/sind. Diese angereicherte Lösung
wird wiederum mit dem kohlenstoffhaltigen Katalysator in Berührung gebracht.
Diese Reduktionsbehandlung kann, wie vorstehend bemerkt, in einem
oder mehreren Katalysatorvorratsbehältern im Reaktorsystem 603 erfolgen.
-
Wenigstens
eine Teil 649 des Überkopfprodukts 617 des
adiabatischen Kristallisators und/oder wenigstens ein Teil 651 des
abgezogenen Dekantats 624 kann aus dem System als Abfall
ausgeschieden (d. h. verworfen) werden. Bei einem kontinuierlichen
System unterstützt
dieses Ausscheiden die Verringerung des Aufbaus von Verunreinigungen
im System. Dieser ausgeschiedene Abfall kann wiederum weiterbehandelt werden,
um die Verunreinigungen nach auf dem Fachgebiet bekannten Verfahren
zu entfernen, wie vorstehend für
die ausgeschiedenen Abfallströme
aus der Zentrifuge stromabwärts
des nicht-adiabatischen Kristallisators beschrieben. Wenigsten ein
Teil 652 des abgezogenen Dekantats 624 kann alternativ
zum nicht-adiabatischen Verdampfungskristallisator 663 weitergeleitet
werden.
-
Die
vom Boden des adiabatischen Kristallisators 615 abgezogene
primäre
N-(Phosphonomethyl)glycinproduktaufschlämmung 625 enthält den Hauptanteil
des N-(Phosphonomethyl)glycinprodukts. Die Aufschlämmung 625 durchströmt typischerweise
eine Zentrifuge 655, um die Aufschlämmung 625 weiter zu
konzentrieren und einen Nasskuchen 657 zu bilden, welcher
das N-(Phosphonomethyl)glycinprodukt enthält. Die Konzentration des N-(Phosphonomethyl)glycinprodukts
im Nasskuchen 657 beträgt
normalerweise etwa 95% (bezogen auf das Gewicht aller Verbindungen
außer
Wasser).
-
Andererseits
wird wenigstens ein Teil (vorzugsweise wenigstens etwa 1%, weiter
vorzugsweise etwa 1% bis etwa 67%, sogar weiter vorzugsweise etwa
20% bis etwa 50%, sogar noch weiter vorzugsweise etwa 33%) des Zentrats 661 (d.
h. der primären
Mutterlauge) aus der Zentrifuge 655 zu einem wärmegesteuerten Verdampfungskristallisator 663 geleitet,
welcher Wärme
an das Zentrat 661 abgibt, um Wasser und niedermolekulare
Verunreinigungen zu verdampfen und den Überkopfdampfstrom 665 des
Verdampfungskristallisators zu bilden. Ein Großteil des N-(Phosphonomethyl)glycinprodukts
fällt im
flüssigen
Medium 667 aus. Dieses flüssige Medium wird aus dem nicht-adiabatischen
Verdampfungskristallisator 663 abgezogen und in ein Hydrozyklon 669 eingeführt, welches
einen an ausgefallenem N-(Phosphonomethyl)glycinprodukt angereicherten produktreichen
Strom 673 und einen an Feststoff ausgearmten Strom 671 liefert.
Der produktreiche Strom 673 wird in eine Zentrifuge 675 eingeführt, welche
ein Zentrat 677 bildet (welches an N-(Phosphonomethyl)glycinprodukt weiter
ausgearmt ist) und einen N-(Phosphonomethyl)glycinproduktnasskuchen 679.
In den Fällen,
in denen nicht das gesamte Zentrat 661 aus der Zentrifuge 655 in
den nicht-adiabatischen Kristallisator 663 eingeführt wird,
kann ein Teil 695 des Zentrats 661 zum adiabatischen
Kristallisator 615 zurückgeführt und/oder über den
Ausscheidestrom 693 ausgeschieden und zum Beispiel unter
Verwendung der verschiedenen. vorstehend erörterten Behandlungsmethoden
für Flüssigabfall
behandelt werden.
-
Bei
der in 13 dargestellten Ausführungsform
kann wenigstens ein Teil des Überkopfprodukts 665 des
wärmegesteuerten
Verdampfungskristallisators zum Reaktorsystem 603 zurückgeführt werden.
Oft wird ein Teil 685 zum Reaktorsystem 603 zurückgeführt und
als Quelle für
Wasser zum Lösen
des N-(Phosphonomethly)iminodiessigsäuresubstrats verwendet, um
den Einspeisestrom 601 für die Oxidationsreaktionszone(n)
zu bilden. Insbesondere wenn der Katalysator ein kohlenstoffhaltiger
Katalysator ist, kann ein Teil des Überkopfprodukts 665 des
wärmegesteuerten
Verdampfungskristallisators vorteilhafterweise auch dazu verwendet
werden, die Katalysatoroberfläche
zu reduzieren. Dies beruht auf der Tatsache, dass das Überkopfprodukt 665 des
Verdampfungskristallisators oft Formaldehyd und/oder Ameisensäure enthält, die
beide als Reduktionsmittel wirken, insbesondere gegenüber kohlenstoffhaltigen
Katalysatoren. Die Reduktionsbehandlung kann in einem oder mehreren
Katalysatorvorratsbehälter(n)
des Reaktorsystems 603 erfolgen.
-
Wenigstens
ein anderer Teil des Überkopfprodukts 665 des
wärmegesteuerten
Verdampfungskristallisators wird normalerweise als Ausscheidestrom 683 aus
dem System ausgeschieden. Bei einem kontinuierlichen System hilft
diese Ausscheidung die Menge an sich bildenden Verunreinigungen
(insbesondere den Aufbau niedermolekularer Verunreinigungen) im
System zu reduzieren. Der ausgeschiedene Abfall 683 kann
wiederum behandelt werden, um die Verunreinigungen zu entfernen,
wie vorstehend für
den ausgeschiedenen Überkopfstrom
aus adiabatischen Kristallisatoren und nicht-adiabatischen Kristallisatoren
erörtert.
-
Vorzugsweise
wird wenigstens ein Teil 689 des Zentrats 677 aus
der Zentrifuge 675 zum wärmegesteuerten Verdampfungskristallisator 663 (und/oder
dem adiabatischen Kristallisator 615) zur weiteren Gewinnung
von N-(Phosphonomethyl)glycinprodukt zurückgeführt. Alternativ (oder zusätzlich)
wird ein Teil 691 des Zentrats 677 zur (zu den)
Oxidationsreaktionszone(n) des Reaktorsystems 603 zurückgeführt, um
nicht umgesetztes N-(Phosphonomethyl)iminodiessigsäuresubstrat
im Zentrat 677 in N-(Phosphonomethyl)glycinprodukt umzuwandeln.
Ein Teil 687 des Zentrats 677 wird normalerweise
auch aus dem System ausgeschieden. Bei einem kontinuierlichen System
hilft diese Ausscheidung 687 die Menge an sich bildenden
Verunreinigungen (insbesondere den Aufbau höhermolekularer Verunreinigungen)
im System zu reduzieren. Der ausgeschiedene Abfall 687 kann
weiterbehandelt werden, um die Verunreinigungen zu entfernen, wie
zum Beispiel nach den gleichen Verfahren, wie vorstehend für die ausgeschiedenen
flüssigen
Abfälle
aus adiabatischen und nicht-adiabatischen Kristallisatoren erörtert.
-
Bei
einer alternativen Ausführungsform
wird vorzugsweise (oder zusätzlich)
zum Einspeisen des Zentrats 661 aus der Zentrifuge 655 in
den nicht-adiabatischen Kristallisator 663, wenigstens
eine Teil 652 (vorzugsweise wenigstens etwa 1%, weiter
vorzugsweise etwa 1% bis etwa 67%, sogar weiter vorzugsweise etwa 20%
bis etwa 50%, sogar noch weiter vorzugsweise etwa 30 bis etwa 40%
und sogar noch weiter vorzugsweise etwa 33%) des aus dem adiabatischen
Kristallisator 615 abgezogenen Dekantats 624 in
den nicht-adiabatischen Kristallisator 663 eingeführt. In
diesem Fall kann das Zentrat 661 zum Beispiel (über den
Strom 695) zum adiabatischen Kristallisator 615 des
Reaktorsystems 603 zurückgeführt werden,
aus dem System (über den
Strom 693) ausgeschieden und/oder in den nicht-adiabatischen
Kristallisator 663 eingeführt werden.
-
14 zeigt ein Beispiel einer besonders bevorzugten
Ausführungsform,
worin das Verfahren einen adiabatischen Kristallisator 319 und
einen nicht-adiabatischen
Kristallisator 343 umfasst. Hier arbeiten der adiabatische
Kristallisator 319 und der nicht-adiabatische Kristallisator 343 in
semi-paralleler Weise.
-
Viele
der in 14 dargestellten Ströme sind
jenen analog, welche vorstehend für nicht-adiabatische Kristallisatoren
und adiabatische Kristallisatoren allein beschrieben wurden. Ein
wässriger
Einspeisestrom 301, welcher ein N-(Phosphonomethyl)iminoidiessigsäuresubstrat
umfasst, wird gemeinsam mit Sauerstoff in ein Oxidationsreaktorsystem 303 eingespeist,
welches eine oder mehrere Oxidationsreaktionszone(n) umfasst, worin
das N-(Phosphonomethyl)iminodiessigsäuresubstrat in Anwesenheit
eines Katalysators oxidativ gespalten wird, um eine Oxidationsreaktionsproduktlösung 305 zu
bilden. Die aus der letzten Oxidationsreaktionszone des Reaktorsystems 303 abgezogene
Oxidationsreaktionsproduktlösung 305 wird
dann in einen Vorkristallisatorentlüftungsbehälter 306 eingeführt, um
den Druck zu vermindern und den Großteil des gelösten CO2 abzulüften.
Bei der in 14 dargestellten Ausführungsform
wird der resultierende Flüssigkeitsstrom 308 mit
einem Katalysatorfilter 307 filtriert, um den im Flüssigkeitsstrom 308 suspendierten
heterogenen teilchenförmigen
Katalysator zu entfernen und einen Katalysatorrezirkulationsstrom 309 und
ein Filtrat 311 zu bilden. Das Filtrat 311 wird
in mehrere Fraktionen geteilt und ein Teil 317 (d. h. eine
erste Fraktion der Reaktionsproduktlösung) wird in den adiabatischen
Kristallisator 319 eingeführt, um eine primäre Produktaufschlämmung zu
bilden, welche ausgefällte
N-(Phosphonomethyl)glycinproduktkristalle und eine primäre Mutterlauge
umfasst, während
ein anderer Teil 315 (d. h. eine zweite Fraktion der Reaktionsproduktlösung) in
den nicht-adiabatischen wärmegesteuerten
Verdampfungskristallisator 343 eingeführt wird, um eine Verdampfungskristallisationsaufschlämmung 344 (d.
h. eine sekundäre
Produktaufschlämmung)
herzustellen, welche ausgefällte N-(Phosphonomethyl)glycinproduktkristalle
und eine sekundäre
Mutterlauge umfasst. Bei einer solchen Ausführungsform kann der Teil 315 des
Filtrats 311, welcher in den Verdampfungskristallisator 343 eingeführt wird, zuerst
in einen Kristallisatoreinspeisebehälter (nicht dargestellt) eingeführt werden,
wo er mit dem an Feststoff ausgearmten Hydrozyklonstrom 351 und/oder
dem rezirkulierten Zentrat 365 gemischt wird. Zusätzlich zur
Bereitstellung einer Mischgelegenheit kann ein solcher Behälter auch
einen temporären
Puffer vorsehen, um zum Beispiel Material während der Inbetriebnahme oder
dem Abschalten aufzunehmen.
-
Der
Betrieb des adiabatischen Kristallisators 319 liefert einen
aus dem Kopf des Kristallisators entweichenden Dampf (d. h. das Überkopfprodukt
des adiabatischen Kristallisators), einen aus dem Kristallisator
abgezogenen Dektantstrom 232 (d. h. die primäre Mutterlauge)
und eine primäre
Kristallisationsproduktaufschlämmung 321,
welche ausgefallenes kristallines N-(Phosphonomethyl)glycinprodukt
und primäre
Mutterlauge umfasst, welche vom Boden des Kristallisators abgezogen
werden. Vorzugsweise wird wenigsten ein Teil (und vorzugsweise das
gesamte) Überkopfprodukt 369 des
adiabatischen Kristallisators und/oder das aus dem adiabatischen
Kristallisator 319 abgezogene Dekantat 323 zur
(zu den) Oxidationsreaktionszone(n) des Reaktorsystems 303 zurückgeführt. Typischerweise
wird wenigstens ein Teil 377 des Überkopfprodukts 369 des
adiabatischen Kristallisators und/oder ein Teil 324 des
abgezogenen Dekantats 323 zum Reaktorsystem 303 zurückgeführt und
als Wasserquelle für
die Oxidationsreaktionszone(n) verwendet. Alternativ kann wenigstens
ein Teil 325 des abgezogenen Dekantats 323 dem
nicht-adiabatischen Verdamp fungskristallisator 343 zugeführt werden.
Wenigstens ein Teil des Überkopfprodukts 369 des
adiabatischen Kristallisators kann (über den Strom 379)
indirekt zum Reaktorsystem 303 zurückgeführt werden, indem er zur Reduktion
des Katalysators verwendet wird. Diese Reduktionsbehandlung erfolgt,
wie vorstehend bemerkt, oft in (einem) Katalysatorvoratsbehälter(n) 373.
-
Weil
die an Feststoff ausgearmten Flüssigkeitsströme aus dem
adiabatischen Kristallisator 319 (d. h. dem Dekantatstrom 323)
und aus der nachfolgenden Zentrifuge 331 (d. h. dem Konzentratstrom 335)
typischerweise eine niedrigere Konzentration an Verunreinigungen
(insbesondere an höhermolekularen
Verunreinigungen) aufweisen als der an Feststoff ausgearmte Strom 357 (d.
h. das Zentrat) aus der Zentrifuge 355 (d. h. eine Feststoffkorbzentrifuge)
stromabwärts
des wärmegesteuerten
Verdampfungskristallisators 343, ist es normalerweise weiter
vorzuziehen, das gesamte, aus dem adiabatischen Kristallisator 319 abgezogenen
Dekantat 323 und wahlweise den gesamten an Feststoff ausgearmten
Strom 335 aus der Zentrifuge 331 stromabwärts des
adiabatischen Kristallisators 319 und wahlweise den gesamten
an Feststoff ausgearmten Strom 335 aus der Zentrifuge 331 stromabwärts des
adiabatischen Kristallisators 319 zum Reaktorsystem 303 zurückzuführen, indem
der feststoffarme Strom 357 (d. h. das Zentrat) der Zentrifuge 355 stromabwärts des
wärmegesteuerten
Verdampfungskristallisators 343 (über den Ausscheidestrom 361)
zum Ausscheiden höhermolekularer
Verunreinigungen aus dem System verwendet wird. Das Ausscheiden
ist bei kontinuierlichen Systemen von Vorteil, weil es die Geschwindigkeit
des Aufbaus von Verunreinigungen im System verringert, was die Rückführung der
feststoffarmen Ströme
(d. h. der Ströme 323 und 335)
aus dem adiabatischen Kristallisator 319 leichter durchführbar macht.
Dieser ausgeschiedene Abfall 361 kann, ähnlich den vorstehend erörterten ausgeschiedenen
Abfallströmen,
zum Beispiel durch weitere Kristallisation behandelt werden. Er
kann auch durch Verfahren behandelt werden, wie sie von Smith in
US-A 5,606,107 beschrieben wurden. Er kann zum Beispiel zusätzlich durch
ein Verfahren behandelt werden, welches im Detail von Morgernstern
et al. in US-A 6,005,140 beschrieben wurde. Er kann ferner nach
einem Verfahren behandelt werden, welches im Detail von Grabiak
et al. in US-A 4,851,131 beschrieben wurde.
-
Andererseits
wird der an Feststoff ausgearmte Strom 335 aus der Zentrifuge 331 stromabwärts des adiabatischen
Kristallisators 319 typischerweise vollständig, zum
Beispiel (über
den Strom 337), zum adiabatischen Kristallisator 319 oder (über den
Strom 341) zum Reaktorsystem 303 zurückgeführt. Der
an Feststoff ausgearmte Strom 351 aus dem Hydrozyklon 349 stromabwärts des
wärmegesteuerten
Verdampfungskristallisators 343 kann zum Verdampfungskristallisator
zurückgeführt werden.
Irgendwelche nicht ausgeschiedenen Teile 365 des an Feststoff
ausgearmten Stroms 357 aus der Zentrifuge 355 stromabwärts des
Verdampfungskristallisators 343 werden typischerweise zum
Verdampfungskristallisator zurückgeführt. Wenigstens
ein Teil des Überkopfprodukts 345 des
wärmegesteuerten
Verdampfungskristallisators wird aus dem System typischerweise über den
Strom 347 abgezogen, obwohl ein Teil 350 wahlweise
zum Oxidationsreaktorsystem 303 direkt über den Strom 383 oder
indirekt über
den Strom 381 zurückgeführt wird,
indem er als Reduktionsmittel für
den Katalysator verwendet wird.
-
Vorzugsweise
werden etwa 30% bis etwa 85%, weiter vorzugsweise etwa 50% bis etwa
80% und sogar weiter vorzugsweise etwa 65% bis 75% der Oxidationsreaktionsmischung
in Abwesenheit des Katalysators (d. h. Strom 311) in den
adiabatischen Kristallisator 319 über den Strom 317 als
die primäre
Fraktion eingeführt,
während
der restliche Teil in den nicht-adiabatischen, wärmegesteuerten Kristallisator 343 über den Strom 315 als
sekundäre
Fraktion eingeführt
wird. Das Gewichtsverhältnis
der sekundären
Fraktion 315 zum in das System eingespeisten N-(Phosphonomethyl)iminodiessigsäuresubstrat
beträgt
vorzugsweise etwa 0,1 bis etwa 9, weiter vorzugsweise etwa 2 bis
etwa 7, sogar weiter vorzugsweise etwa 2,5 bis 4.
-
Ausführungsformen,
welche einen adiabatischen Kristallisator und einen wärmegesteuerten
Verdampfungskristallisator in semi-paralleler Weise betreiben (wie
der in 14 gezeigte) sind typischerweise
stärker bevorzugt
als Ausführungsformen,
welche einen adiabatischen Kristallisator und einen Verdampfungskristallisator
in Reihe betreiben. Dies beruht zum Beispiel auf der Tatsache, dass
bei einer gegebenen Größe des Verdampfungskristallisators
generell eine größere Kristallisationskapazität erhalten
werden kann, wenn die Kristallisatoren parallel angeordnet sind.
Dies bietet eine größere Flexibilität bei der
Umrüstung
bestehender Anlagen.
-
Das
System in 14 liefert zwei Ströme von N-(Phosphonomethyl)glycinnasskuchen:
einen Nasskuchenstrom 333 aus der Zentrifuge 331 stromabwärts des
adiabatischen Kristallisators 319, und einen Nasskuchenstrom 359 aus
der Zentrifuge 355 stromabwärts des wärmegesteuerten Verdampfungskristallisa tors 343. Normalerweise
besitzt der Nasskuchenstrom 333 aus dem adiabatischen Kristallisator 319 vorzugsweise
eine N-(Phosphonomethyl)glycinproduktkonzentration von mindestens
90% (des Gewichts aller Verbindungen außer Wasser), weiter vorzugsweise
mindestens 95% (des Gewichts aller Verbindungen außer Wasser)
und sogar weiter vorzugsweise mindestens etwa 99% (des Gewichts
aller Verbindungen außer
Wasser), während
der Nasskuchenstrom 359 aus dem Verdampfungskristallisator 343 eine
N-(Phosphonomethyl)glycinproduktkonzentration von mindestens etwa
85% (des Gewichts aller Verbindungen außer Wasser), weiter vorzugsweise mindestens
90% (des Gewichts aller Verbindungen außer Wasser) und sogar weiter
vorzugsweise mindestens etwa 95% (des Gewichts aller Verbindungen
außer
Wasser) aufweist. Die Reinheit des Nasskuchens 333 aus dem
adiabatischen Kristallisatorabschnitt 319 ist typischerweise
größer als
die Reinheit des Nasskuchens 359 aus dem wärmegesteuerten
Verdampfungskristallisator 343.
-
Es
ist oft vorteilhaft diese beiden Nasskuchen 333 und 359 zu
kombinieren. Diese Kombination erlaubt aufgrund der größeren Reinheitsgrade,
welche normalerweise aus dem vom adiabatischen Kristallisator 319 gelieferten
Nasskuchen 333 erhalten werden, geringere Reinheitsgrade
im Nasskuchen 359 aus dem Verdampfungskristallisator 343 zu
tolerieren. Stammt demzufolge zum Beispiel 45% des kombinierten
Nasskuchens vom Verdampfungskristallisator 343 und 55%
des kombinierten Nasskuchens vom adiabatischen Kristallisator 319,
kann der Reinheitsgrad des Nasskuchens 359 aus dem Verdampfungskristallisator 343 nur
bis zu 90,2 Gew.-% betragen, um einen kombinierten Reinheitsgrad
von mindestens 95 Gew.-% zu erzielen, wenn der Nasskuchen 333 aus
dem adiabatischen Kristallisator 319 eine Reinheit von
99 Gew.-% aufweist. Normalerweise ist es erwünscht, dass ein kombinierter
Nasskuchen eine N-(Phosphonomethyl)glycinkonzentration von
mindestens etwa 95% (des Gewichts aller Verbindungen außer Wasser)
und weiter vorzugsweise etwa 96% (des Gewichts aller Verbindungen
außer
Wasser) aufweist.
-
14A zeigt ein Beispiel einer anderen bevorzugten
Ausführungsform,
wobei das Verfahren einen adiabatischen Kristallisator und einen
nicht-adiabatischen, mit Wärme
betriebenen Verdampfungskristallisator umfasst, die in einer semi-parallelen Weise
betrieben werden, wie vorstehend bei 14 beschrieben.
Dieses Reaktionssystem schließt
jedoch weiterhin ein sekundäres
Reaktorsystem ein, welches eine oder mehrere Oxidationsreaktionszone(n)
umfasst, die in Verbindung mit der Fraktion der Reaktionsproduktmischung
aus dem primären Oxidationsreaktorsystem
verwendet wird/werden, die zum mit Wärme angetriebenen Verdampfungskristallisator
geleitet wird.
-
Viele
der in 14A dargestellten Ströme sind
jenen analog, welche vorstehend für das in 14 dargestellte Reaktionssystem beschrieben sind,
in dem der adiabatische Kristallisator 319 und der wärmegesteuerte
Verdampfungskristallisator 343 in semi-paralleler Weise
betrieben werden. Ein wässriger
Einspeisestrom 302, welcher ein N-(Phosphonomethyl)iminodiessigsäuresubstrat
umfasst, wird gemeinsam mit Sauerstoff in ein primäres Oxidationsreaktorsystem 303 eingeführt, welches
eine oder mehrere Oxidationsreaktionszone(n) umfasst, worin das
N-(Phosphonomethyl)iminodiessigsäuresubstrat
in Anwesenheit eines Katalysators oxidativ gespalten wird, um eine
Reaktionsproduktlösung 305 zu
bilden, welche N-(Phosphonomethyl)glycinprodukt und nicht umgesetztes
N-(Phosphonomethyl)iminodiessigsäuresubstrat
umfasst. Die Reaktionsproduktlösung 305 aus
dem primären
Reaktorsystem 303 wird nach der Abtrennung des Katalysators
(z. B. durch Filtration), sofern erforderlich, in mehrere Fraktionen
geteilt, umfassend eine primäre
Fraktion 317 und eine sekundäre Oxidationsreaktoreinspeisefraktion 315.
Die primäre
Fraktion 317 wird in den adiabatischen Kristallisator 319 eingeführt und
durch Schnellverdampfung von Wasser daraus unter verminderten Druckbedingungen
gekühlt, um
das N-(Phosphonomethyl)glycinprodukt zu gewinnen, wie vorstehend
beschrieben. Die sekundäre
Oxidationsreaktoreinspeisefraktion 315 wird in ein sekundäres Oxidationsreaktorsystem 316 eingeführt, welches eine
oder mehrere Oxidationsreaktionszone(n) umfasst, in der/denen nicht
umgesetztes N-(Phosphonomethyl)iminodiessigsäuresubstrat oxidiert wird,
um einen sekundären
Oxidationsreaktorausfluss 318 zu bilden, welcher N-(Phosphonomethyl)glycinprodukt
umfasst. Die sekundäre
Reaktoreinspeisefraktion 315 kann vor der Einführung in
das sekundäre
Oxidationsreaktorsystem 316 gekühlt werden, um im primären Oxidationsreaktorsystem 303 erzeugte
exotherme Wärme
abzuführen
und die Bildung von Nebenprodukten zu verringern. Der Reaktorausfluss 318 wird
in den nicht-adiabatischen, wärmegesteuerten
Verdampfungskristallisator eingeführt, worin daraus Wasser verdampft
wird, um das N-(Phosphonomethyl)glycinprodukt, wie vorstehend beschrieben,
auszufällen
und zu gewinnen.
-
Im
Reaktionssystem von 14A kann das primäre und sekundäre Reaktorsystem 303 bzw. 315 selbstverständlich eine
oder mehrere Oxidationsreaktionszone(n) einschließen, die
von verschiedenen Reaktorkonfigurationen bereitgestellt werden,
einschließend
zum Beispiel die hierin vorstehend beschriebenen konti nuierlichen
Reaktorsysteme. Zum Zwecke der Erläuterung kann das primäre Reaktorsystem 303 einen
einzelnen Rührbehälterreaktor
umfassen, wie in den 2, 2A und 2B dargestellt,
zwei Rührbehälterreaktoren
in Reihe, wie in den 3–6 dargestellt,
oder einen oder mehrere Festbettreaktoren, wie in 8 dargestellt.
Bei einer Ausführungsform
umfasst das primäre
Reaktorsystem 303 eine oder mehrere Oxidationsreaktionszone(n)
in Reihe und die Reaktionsproduktlösung 305 wird aus
der letzten Oxidationsreaktionszone in der Reihe abgezogen und bei
Bedarf filtriert. Die Reaktionsproduktlösung 305 kann jedoch
selbstverständlich
vor der letzten Oxidationsreaktionszone des primären Reaktorsystems 303 so
geteilt werden, das die primäre
Fraktion 317 die restliche(n) Oxidationsreaktionszone(n)
des primären
Reaktorsystems durchströmt,
ehe es in den adiabatischen Kristallisator 319 eingeführt wird.
-
Das
sekundäre
Reaktorsystem 316 umfasst vorzugsweise eine oder mehrere
Oxidationsreaktionszone(n), welche von einem oder mehreren Festbettreaktoren
oder Rührbehälterreaktoren
bereitgestellt werden, welche eine teilchenförmige Katalysatoraufschlämmung oder
Kombinationen hiervon verwenden. Festbettreaktoren sind jedoch im
Allgemeinen stärker
bevorzugt, weil sich im sekundären
Reaktorsystem 316 ein Katalysatorrezirkulationsmechanismus,
einschließlich
eines Katalysatorsfilters, erübrigt.
Außerdem
werden Probleme, welche die Abführung
der exothermen Reaktionswärme
und die Temperaturregelung betreffen, welche auftreten, wenn ein
Festbettreaktor als erste Oxidationsreaktionszone im primären Reaktorsystem 303 dient, im
sekundären
Reaktorsystem 316 weitgehend umgangen, weil der Hauptanteil
des N-(Phosphonomethyl)iminodiessigsäuresubstrats vorzugsweise im
primären
Reaktorsystem 303 oxidiert wird. Folglich kann/können die Oxidationsreaktionszone(n)
im sekundären
Reaktorsystem adiabatisch betrieben werden. In Übereinstimmung mit einer besonders
bevorzugten Ausführungsform
umfasst das sekundäre
Reaktorsystem 316 eine einzelne Oxidationsreaktionszone,
welche von einem Festbettreaktor bereitgestellt wird. Ein solcher
Festbettreaktor wird vorzugsweise mit Parallelströmung von
Gas und Flüssigkeit
in der Oxidationsreaktionszone betrieben.
-
Die
Anwesenheit des sekundären
Reaktorsystems 316 in dem in 14A dargestellten
Reaktionssystems ermöglicht
es, das primäre
Reaktorsystem 303 in einer solchen Weise auszulegen und
zu betreiben, dass höhere
Konzentrationen an nicht umgesetztem N-(Phosphonomethyl)iminodiessigsäuresubstrat
in der Reaktionsmischung 305 erlaubt sind. Die zweite oder
folgenden Oxidationsreaktionszone(n) des primären Reaktorsystems 303 können zum
Beispiel beträchtlich
kleiner dimensioniert oder vollständig weggelassen werden (d. h.
das primäre
Reaktorsystem 303 kann eine einzige Oxidationsreaktionszone
umfassen). Die Konzentration an nicht umgesetztem N-(Phosphonomethyl)imonodiessigsäuresubstrat
in der Reaktionsproduktlösung 305, und
folglich in der primären
Fraktion 317, welche dem adiabatischen Kristallisator 319 zugeführt wird,
wird nichtsdestoweniger vorzugsweise ausreichend niedrig gehalten,
um die Ausfällung
von nicht umgesetztem N-(Phosphonomethyl)iminodiessigsäuresubstrat
bei der herrschenden Stromtemperatur zu vermeiden. Bei typischen
Betriebstemperaturen des adiabatischen Kristallisators (z. B. 60°C) ist die
Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats
in der Reaktionsproduktlösung 305 nicht
größer als
etwa 2 Gew.-%. Um jedoch den Vorteil des Vorliegens des sekundären Reaktorsystems 316 zu
nutzen, welches es erlaubt, die Dimension und den Betrieb des primären Reaktorsystems 303 wirtschaftlicher
zu gestalten, beträgt
die Konzentration des N-(Phosphonomethyl)iminodiessigsäuresubstrats
in der Reaktionsproduktlösung 305 vorzugsweise
wenigstens 0,2 Gew.-% und weiter vorzugsweise wenigstens etwa 0,5
Gew.-%.
-
Nicht
umgesetztes N-(Phosphonomethyl)iminodiessigsäuressubstrat in der primären Fraktion 317 zur Einführung in
den adiabatischen Kristallisator 319 wird vorzugsweise
wiedergewonnen und über
das aus dem adiabatischen Krtstallisator 319 abgezogene
Dekantat 323 in das primäre Reaktorsystem 303 zurückgeführt, ebenso
wie durch wahlweise Rückführung wenigstens
eines Teils des an Feststoff ausgearmten Stroms 335 aus
der Zentrifuge 331 stromabwärts des adiabatischen Kristallisators 319 über den
Strom 341 in das primäre Reaktorsystem.
Bei Einsatz einer stark entwässernden
Zentrifuge (z. B. einer Vertikalkorbzentrifuge) als Zentrifuge 331,
wird die Rückgewinnung
von nicht umgesetztem N-(Phosphonomethyl)iminodiessigsäuresubstrat aus
dem an Feststoff ausgearmten Strom 335 erhöht, während die
Fraktion des im Nasskuchenstrom 333 enthaltenen, nicht
umgesetzten N-(Phosphonomethyl)iminodiessigsäuresubstrats vorteilhafterweise
auf das Mindestmaß beschränkt wird.
-
Bei
einer weiteren Ausführungsform
der vorliegenden Erfindung kann das in 13 dargestellte
System durch Erweiterung durch ein sekundäres Reaktorsystem modifiziert
werden, um nicht umgesetztes N-(Phosphonomethyl)iminodiessigsäuresubstrat
im Zentrat 661 aus der Zentrifuge 655 stromaufwärts des nicht-adiabatischen Verdampfungskristallisators 663 weiter
zu oxidieren. Es ist seit langen bekannt gewesen, dass insbesondere
dann, wenn das N-(Phosphonomethyl)glycinprodukt das N-(Phosphonomethyl)glycin selbst
ist, das Produkt zu verschiedenen Salzen oder Estern umgewandelt
werden kann, um seine Löslichkeit in
Wasser zu erhöhen,
so dass es für
die kommerzielle Verwendung leichter zugänglich ist. Dies wird zum Beispiel
von Franz in den US-Patenten 3,977,860 und 4,405,531 allgemein erörtert. Bevorzugte
N-(Phosphonomethyl)glycinsalze schließen zum Beispiel Alkalimetallsalze
(insbesondere das Kaliumsalz), Alkanolaminsalze (insbesondere das
Monoethanolaminsalz), Alkylaminsalze (insbesondere das Isopropylaminsalz)
und Alkylsulfoniumsalze (insbesondere das Trimethylsulfoniumsalz)
ein. Das Isopropylaminsalz von N-(Phosphonomethyl)glycin ist besonders
bevorzugt. Vergleiche z. B. Bugg et al., US-A 5,994,269. Dieses
Salz weist typischerweise eine größere Aktivität auf als
die freie Säure
und ist zum Beispiel bei 25°C
näherungsweise
40-mal löslicher
als die freie Säure.
-
Bei
einigen Ausführungsformen
dieser Erfindung umfasst das in der/den Oxidationsreaktionszone(n) gebildete
N-(Phosphonomethyl)glycinprodukt einen Ester oder ein Salz, deren
Konzentration groß genug
ist, um eine Mischung mit der kommerziell gewünschten Konzentration zu bilden.
In jenen Fällen
kann der Wunsch nach Verfahrensschritten (z. B. Kristallisation,
Hydrozyklonierung, Zentrifugieren und dergleichen) zur Konzentrierung
des Produkts beträchtlich
vermindert sein oder vollkommen wegfallen. Dies trifft insbesondere
dann zu, wenn der Katalysator der vorstehend erörterte tiefenreduzierte Katalysator
ist, der typischerweise eine Reaktionsmischung mit einer niedrigen
Konzentration an Verunreinigungen bildet und folglich eine geringe
oder keine Reinigung erfordert.
-
Bei
manchen Ausführungsformen
ist zum Beispiel das in der/den Oxidationsreaktionszone(n) gebildete
N-(Phosphonomethyl)glycinprodukt das Isopropylaminsalz des N-(Phosphonomethyl)glycins.
Bei den weiter bevorzugten Oxidationsbetriebstemperaturen (d. h.
von etwa 95 bis etwa 105°C),
bleibt ein solches Produkt bei Konzentrationen von bis zu etwa 50
Gew.-% oder mehr löslich.
Das Salzprodukt kann in der/den Oxidationsreaktionszone(n) gebildet
werden durch (a) Verwendung des Isoproylaminsalzes von N-(Phosphonomethyl)iminodiessigsäure als
Substrat, (b) Einführen
von Isopropylamin in die Oxidationsreaktionszone(n) um das Oxidationsprodukt
zu Aminieren während
es sich in der/den Oxidationsreaktionszone(n) befindet, und (c)
Einführen
von Isopropylamin in einen Behälter
stromabwärts
der/den Oxidationsreaktionszone(n) und vor der Katalysatorfiltration.
Wenn es mehr als eine Oxidationsreaktionszone in Reihe gibt, ist
es normalerweise vorzuziehen (obwohl nicht unbedingt erforderlich),
das Isopropylaminsalz der N-(Phosphonomethyl)iminodiessigsäure als
Substrat zu verwenden und/oder das Isopropylamin in die erste der
Oxidationsreaktionszonen einzuführen.
Ungeachtet dessen liegt vorzugsweise mindestens ein Äquivalent
(oder weiter vorzugsweise geringfügig mehr als ein Äquivalent)
Isopropylaminkationen pro Mol gebildetes N-(Phosphonomethyl)glycinprodukt vor.
Diese die Bildung des Isopropylaminsalz betreffenden Grundsätze gelten
selbstverständlich
allgemein für die
Bildung anderer Salze, z. B. der Alkalimetallsalze (insbesondere
das Kaliumsalz), die Alkanolaminsalze (insbesondere das Monoethanolaminsalz),
die Alkylaminsalze, abgesehen vom Isoproylaminsalz, und die Alkylsulfoniumsalze
(insbesondere das Trimethylsulfoniumsalz).
-
Beispiele
-
Die
folgenden Beispiele sind lediglich dafür vorgesehen die vorliegende
Erfindung weiter zu veranschaulichen und zu erklären. Diese Erfindung sollte
daher nicht auf irgendwelche Details in diesen Beispielen beschränkt werden.
-
Beispiel 1
-
Herstellung eines Katalysators,
welcher mit Eisen legiertes Platin umfasst
-
Zur
Herstellung des Katalysators, welcher mit Eisen legiertes Platin
umfasst, werden ungefähr
10 g Aktivkohle in etwa 180 ml Wasser aufgeschlämmt. Als Nächstes werden 0,27 g FeCl3·6H2O und 1,39 g H2PtCl6-Hydrat gemeinsam in etwa 60 ml Wasser gelöst. Diese
Lösung
wird der Kohlenstoffaufschlämmung während eines
Zeitraums von etwa 30 min tropfenweise zugesetzt. Während der
Zugabe fiel der pH der Aufschlämmung
ab und wurde unter Verwendung von verdünnter NaOH-Lösung (d.
h. eine 1,0 bis 2,5 M NaOH-Lösung)
bei etwa 4,4 bis etwa 4,8 gehalten. Danach wurde die Aufschlämmung weitere
30 min bei einem pH von etwa 4,7 gerührt. Die Aufschlämmung wurde
dann unter Aufrechterhaltung eines pHs von etwa 4,7 unter N2 mit einer Rate von etwa 2°C/min auf
70°C erwärmt. Nach
Erreichen von 70°C
wurde der pH durch Zugabe der verdünnten NaOH-Lösung während eines
Zeitraums von etwa 30 min langsam auf 6,0 angehoben. Das Rühren wurde
während
eines Zeitraums von etwa 10 min fortgesetzt, bis sich der pH bei
etwa 6,0 stabilisiert hatte. Die Aufschlämmung wurde dann unter N2 auf etwa 35°C abgekühlt. Die Aufschlämmung wurde
anschließend
filtriert und der Kuchen 3-mal mit ungefähr 800 ml Wasser gewaschen.
Der Kuchen wurde dann bei 125°C
unter Vakuum getrocknet. Dies lieferte beim Tempern bei 690°C in 20%
H2 und 80% AR während 1–6 h einen Katalysator, welcher
5 Gew.-% Platin und 0,5 Gew.-% Eisen auf Kohlenstoff enthält.
-
Dieser
Katalysator wurde mittels Elektronenmikroskopie analysiert, wie
in Beispiel 3 in größerem Detail
beschrieben. Ein mittels TEM erhaltendes Abbild des Kohlenstoffträgers zeigte,
dass die legierten Metallteilchen durch den Kohlenstoffträger hindurch
hoch dispergiert und einheitlich verteilt waren (Die weißen Punkte
repräsentieren
die Metallteilchen; Und von den Unterschieden der Hintergrundintensität wird angenommen, dass
sie die Änderungen
der lokalen Dichte des porösen
Kohlenstoffs wiederspiegeln). Die mittlere Teilchengröße betrug
etwa 3,5 nm und der mittlere Abstand zwischen den Teilchen betrug
etwa 20 nm. Ein hochauflösendes
Röntgenspektrum
eines einzelnen Metallteilchens des Katalysators zeigte, dass sowohl
Platin- als auch Eisenpeaks vorlagen (Die Kupferpeaks stammten von
der Beugung des Kupfergitters). Die quantitative Analyse der hochauflösenden Röntgenspektren
von unterschiedlichen einzelnen Metallteilchen zeigte, dass die
Zusammensetzung der Teilchen innerhalb der Fehlergrenzen mit der
Größe oder
der Lage der Metallteilchen auf der Katalysatoroberfläche nicht
schwankten.
-
Beispiel 2
-
Herstellung eines Pt/Fe/Sn
auf Kohlenstoffkatalysators
-
Ungefähr 10 g
Aktivkohle wurden in etwa 90 ml Wasser aufgeschlämmt. Als Nächstes wurden etwa 0,2 g SnCl2·2H2O in 250 ml 0,025 M HCl gelöst. gelöst. Diese
Lösung
wurde der Kohlenstoffaufschlämmung tropfenweise
zugesetzt. Nach erfolgter Zugabe der gesamten Lösung wurde die Aufschlämmung 3
h gerührt. Dann
wurde der pH langsam mit einer verdünnten NaOH-Lösung (d.
h. eine 1,0 bis 2,5 M NaOH-Lösung)
auf 9,0 eingestellt und die Aufschlämmung noch einige Stunden gerührt. Als
Nächstes
wurde die Aufschlämmung filtriert
und mit einer reichlichen Menge Wasser gewaschen bis das Filtrat
eine konstante Leitfähigkeit
erreichte. Der Nasskuchen wurde bei 125°C unter Vakuum getrocknet. Dies
lieferte 0,9 Gew.-% Zinn auf Kohlenstoff. Etwa 6 g von diesem 0,9
Gew.-% Zinn auf Kohlenstoff wurden in etwa 500 ml Wasser aufgeschlämmt. Dann wurden
0,23 g Fe(NO3)3·9H2O und 0,85 g H2PtCl6 gemeinsam in etwa 150 ml Wasser gelöst und der
Aufschlämmung
tropfenweise zugesetzt. Nach Zugabe der gesamten Lösung wurde
die Aufschlämmung
4 h gerührt
und dann filtriert, um das überschüssige Eisen
(~80 Gew.-%) zu entfernen. Der Nasskuchen wurde erneut in 480 ml
Wasser aufgeschlämmt.
Nachdem der pH der Aufschlämmung
mit verdünnter
NaOH-Lösung
auf 9–10
eingestellt worden war, wurde die Aufschlämmung noch einige Stunden weitergerührt. Als
Nächstes
wurde die Aufschlämmung
filtriert und mit einer reichlichen Menge Wasser gewaschen, bis
das Filtrat eine konstante Leitfähigkeit
aufwies. Der Nasskuchen wurde bei 125°C unter Vakuum getrocknet. Dieser
lieferte bei der Hochtemperaturreduktion durch Erhitzen aus 700–750°C in 20%
H2 und 80% AR während 1–6 h einen Katalysator, welcher
4,9 Gew.-% Pt, und 0,9 Gew.-% Zinn und 0,1 Gew.-% Eisen auf Kohlenstoff
enthält.
-
Beispiel 3
-
Elektronenmikroskopische
Charakterisierung von Katalysatoren
-
Es
wurden elektronenmikroskopische Verfahren verwendet, um die Größe, die
räumliche
Verteilung und die Zusammensetzung der Metallteilchen des in Beispiel
1 hergestellten Katalysators zu analysieren. Vor der Analyse des
Katalysators wurde der Katalysator zuerst in ein EM Bed 812-Harz
(Electron Microscopy Sciences, Fort Washington, PA) eingebettet.
Das Harz wurde dann bei etwa 60°C
während
ungefähr
14 h polymerisiert. Der resultierende gehärtete Block wurde mittels Ultramikrotom
in Scheiben mit einer Dicke von etwa 50 nm geschnitten. Diese Scheiben
wurden kann für
die elektronenmikroskopische Beobachtung auf ein 200 mesh Kupfergitter
transferiert.
-
Es
erfolgten hochauflösende
analytische elektronenmikroskopische Versuche in einem auf ein Transmissionsrasterelektronenmikroskop
ausgelegten Vakuumgenerator (Modell Nr. VG HB501, Vacuum Generators,
East Brinstead, England) mit einer Bildauflösung von weniger als 0,3 nm.
Das Mikroskop wurde mit 100 kV betrieben. Das Vakuum im Bereich
der Probenkammer betrug weniger als 10–6 Pa.
Es wurde ein digitales Bildverarbeitungssystem (ES Vison Data Acquisition
Systems, EmiSpec Sys., Inc., Tempe, AT) verwendet, um hochaufgelöste elektronenmikroskopische
Abbildungen zu erhalten. Es wurde ein fensterloses energiedispersives
Röntgenspektrometer
(Link LZ-5 EDS Windowless Detector, Modell E5863, High Wycomb, Bucks,
England) verwendet, um von einzelnen Metallteilchen Röntgenspektren
mit hoher Energieauflösung
zu gewinnen. Zur Beobachtung der Metallteilchen wurde wegen der
Empfindlichkeit bei hohen Atomzahlen die anulare Hochwinkeldunkelfeldmikroskopie
(HAADF) verwendet. Es wurde eine Elektronenprobengröße von weniger
als etwa 0,5 nm verwendet, um die HAADF-Abbildungen zu erhalten
und eine Probengröße von weniger
als 1 nm, um die Röntgenspektren
mit hoher Energieauflösung
zu erhalten.
-
Beispiel 4
-
Wirkung eines
Zusatzaktivators
-
Dieses
Beispiel zeigt die Verwendung und die Vorteile des Mischens eines
Zusatzaktivators mit einem Kohlenstoff getragenen edelmetallhaltigen
Oxidationskatalysator.
-
A. Vergleich der Wirkungen
auf eine NPMIDA-Oxidationsreaktion, verursacht durch Mischen eines
Kohlenstoff-getragenen edelmetallhaltigen Katalysators mit verschiedenen
Mengen und Quellen von Wismut
-
Es
erfolgten mehrere N-(Phosphonomethyl)iminodiessigsäureoxidationsreaktionen
in Einzelchargen. Bei jeder Reaktion wurde dem Reaktionsmedium eine
unterschiedliche Menge Wismut zugesetzt. Die Wismutquelle war entweder
(Bi2CO3, Bi(NO3)3·5H2O oder Bi2O3. Die verwendete Wismutmenge entsprach einem
Wismut zu N-(Phosphonomethyl)iminodiessigsäure-Verhältnis von 1:10.000; 1:2000
oder 1:1000. Es wurde auch eine Kontrolle ohne Zusatz von Wismut
durchgeführt.
-
Jede
N-(Phosphonomethyl)iminodiessigsäureoxidation
erfolgte in Anwesenheit eines Katalysators, welcher 5 Gew.-% Platin
und 0,5 Gew.-% Eisen enthielt (Dieser Katalysator wurde nach einem
Verfahren ähnlich
dem für
Beispiel 1 beschriebenen hergestellt). Die Reaktion erfolgte in
einem 1000 ml Edelstahlreaktor (Autoclave Engineers, Pittsburgh,
PA) unter Verwendung von 2,5 g Katalysator (0,5 Gew.-% der gesamten
Reaktionsmasse), 60,5 g N-(Phosphonomethyl)iminodiessigsäure (12,1
Gew.-% der gesamten Reaktionsmasse), 1000 ppm Formaldehyd, 5000
ppm Ameisensäure,
einer Gesamtreaktionsmasse von 500 g, einem Druck von 758 kPa (110
psig), einer Temperatur von 100°C
und einer Rührgeschwindigkeit
von 1000 Upm. Die Sauerstoffeinspeiserate betrug während der
ersten 22 min 392 ml/min und dann 125 ml/min, bis die N-(Phosphonomethyl)iminodiessigsäure im Wesentlichen
verbraucht war.
-
Tabelle
14 gibt die Ergebnisse wieder. Bei allen Versuchen, bei denen eine
Wismutverbindung verwendet wurde, betrugen die Formaldehyd-, Ameisensäure- und
NMG-Gehalte weniger als die in der Kontrolle. Tabelle
14 Direkte
Zugabe von Wismut aus verschiedenen Quellen und Anteilen
- * ppm bedeutet ein Bi/N-(Phosphonomethyl)iminodiessigsäure Verhältnis von
1:1.000.000
- ** (Masse ÷ Gesamtreaktionsmasse) × 100%
- *** mg = Gramm Glyphosat, produziert
- „ND"
- bedeutet nicht nachweisbar
-
B. Wirkung der Wismutzugabe
auf die folgenden, mit dem Katalysator in Berührung gebrachten NPMIDA-Chargen
-
Es
erfolgten vier Versuche mit jeweils 6 Prüfläufen (d. h. bei jedem der 4
Versuche erfolgten 6 Chargenreaktionen in Folge), um die Wirkung
von (1) der anfänglichen
Wismutzugabe auf die folgenden Reaktionschargen gegenüber der
anfänglichen
Wismutzugabe, und (2) die zusätzliche
Zugabe zu einer oder mehreren der nachfolgenden Reaktionsläufen zu
bestimmen.
-
Alle
4 Untersuchungen erfolgten unter Verwendung eines Katalysators,
welcher 5 Gew.-% Platin und 0,5 Gew.-% Eisen enthielt (Dieser Katalysator
wurde nach einem Verfahren ähnlich
dem für
Beispiel 1 beschriebenen hergestellt). Bei allen Versuchen mit 6
Prüfläufen wurde
in allen 6 Läufen
der gleiche Katalysator verwendet (d. h. am Ende eines jeden Prüflaufs wurde
die Reaktionsproduktlösung
abgetrennt und der Katalysators entfernt und ein neuer Ansatz von
N-(Phosphonomethyl)iminodiessigsäure
mit dem Katalysator kombiniert, um einen neuen Lauf zu starten.
Die Reaktion erfolgte in einem 1000 ml Edelstahlreaktor (Autoclave Engineers)
unter Verwendung von 2,5 g Katalysator (0,5 Gew.-% der gesamten
Reaktionsmasse), 60,5 g N-(Phosphonomethyl)iminodiessigsäure (12,1
Gew.-% der gesamten Reaktionsmasse), 1000 ppm Formaldehyd, 5000
ppm Ameisensäure,
einer Gesamtreaktionsmasse von 500 g, einem Druck von 758 kPa (110
psig), einer Temperatur von 100°C
und einer Rührgeschwindigkeit
von 1000 Upm. Die Sauerstoffeinspeiserate betrug während der
ersten 22 min 392 ml/min und dann 125 ml/min, bis die N-(Phosphonomethyl)iminodiessigsäure im Wesentlichen
verbraucht war.
-
Beim
Kontrollversuch wurde während
keinem der 6 Läufe
Wismut in die Reaktionszone eingeführt. Bei den drei anderen Versuchen
wurde zu Beginn des ersten Reaktionslaufs 0,034 g Wismut(III)-oxid
(d. h. Bi2O3) in
das Reaktionsmedium eingeführt.
Bei einem anderen Versuch wurde zu Beginn des ersten und des vierten Reaktionslaufs
0,034 g Wismut(III)-oxid in das Reaktionsmedium eingeführt. Beim
letzten Versuch wurde zu Beginn aller 6 Reaktionsläufe 0,034
g Wismut(III)-oxid (d. h. Bi2O3)
in das Reaktionsmedium eingeführt.
-
Die
Tabellen 15, 16, 17 und 18 geben die Ergebnisse wieder. Die einmalige
Zugabe on Wismutoxid (Werte in Tabelle 16 angegeben) tendiert dazu,
die gleichen günstigen
Wirkungen auszuüben
wie die Zugabe des Wismutoxids bei allen 3 Läufen (Werte in Tabelle 17)
oder sogar bei jedem Lauf (Werte in Tabelle 18).
-
-
-
-
-
C. Wirkung der einmaligen
Wismutzugabe auf 20 NPMIDA-Oxidationsläufe unter Verwendung eines
Platin/Eisen/Kohlenstoff-Katalysators
-
Es
erfolgten zwei Versuche mit jeweils 20 Läufen, um die Wirkung einer
einmaligen Wismutzugabe auf 20 N-(Phosphonomethyl)iminodiessigsäureoxidationsreaktionsläufe zu bestimmen.
-
Beide
Versuche erfolgten unter Verwendung eines Katalysators, welcher
5 Gew.-% Platin und 0,5 Gew.-% Eisen enthielt (Dieser Katalysator
wurde nach einem Verfahren ähnlich
dem für
Beispiel 1 beschriebenen hergestellt). Während eines jeden Versuchs
wurde bei jedem der 20 Läufe
der gleiche Katalysator verwendet. Die Reaktion erfolgte in einem
1000 ml Edelstahlreaktor (Autoclave Engineers) unter Verwendung
von 2,5 g Katalysator (0,5 Gew.-% der gesamten Reaktionsmasse),
60,5 g N-(Phosphonomethyl)iminodiessigsäure (12,1 Gew.-% der gesamten
Reaktionsmasse), 1000 ppm Formaldehyd, 5000 ppm Ameisensäure, einer Gesamtreaktionsmasse
von 500 g, einem Druck von 758 kPa (110 psig), einer Temperatur
von 100°C
und einer Rührgeschwindigkeit
von 1000 Upm. Die Sauerstoffeinspeiserate betrug während der
ersten 22 min 392 ml/min und dann 125 ml/min, bis die N-(Phosphonomethyl)iminodiessigsäure im Wesentlichen
verbraucht war. Beim Kontrollversuch wurde während keinem der 20 Läufe Wismut
in die Reaktionszone eingeführt.
Bei einem anderen Versuchen wurde zu Beginn des ersten Reaktionslaufs
0,034 g Wismut(III)-oxid in das Reaktionsmedium eingeführt.
-
15 vergleicht die resultierenden Ameisensäurekonzentrationsprofile.
Die einmalige Einführung von
Wismut in die Reaktionszone verringert die Ameisensäurekonzentration
bei allen 20 Läufen.
-
D. Wirkung der einmaligen
Wismutzugabe auf 30 NPMIDA-Oxidationsläufe unter Verwendung eines
Platin/Zinn/Kohlenstoff-Katalysators
-
Es
erfolgten zwei Versuche mit 30 Läufen,
um die Wirkung einer einmaligen Wismutzugabe auf 30 N-(Phosphonomethyl)iminodiessigsäureoxidationsreaktionsläufe zu bestimmen.
-
Beide
Versuche erfolgten unter Verwendung eines Katalysators, welcher
5 Gew.-% Platin und 1 Gew.-% Zinn enthielt (Dieser Katalysator wurde
nach einem Verfahren ähnlich
dem für
Beispiel 2 beschriebenen hergestellt). Während eines jeden Versuchs
wurde bei jedem der 30 Läufe
der gleiche Katalysator verwendet. Jeder Lauf erfolgte in einem
300 ml Reaktor (hergestellt aus einer Metalllegierung, Hastelloy
C, Autoclave Engineers) unter Verwendung von 1,35 g Katalysator
(0,75 Gew.-% der gesamten Reaktionsmasse), 21,8 g N-(Phosphonomethyl)iminodiessigsäure (12,1
Gew.-% der gesamten Reaktionsmasse), 1000 ppm Formaldehyd, 5000
ppm Ameisensäure,
einer Gesamtreaktionsmasse von 180 g, einem Druck von 621 kPa (90
psig), einer Temperatur von 100°C
und einer Rührgeschwindigkeit
von 900 Upm. Die Sauerstoffeinspeiserate betrug während der
ersten 26 min 141 ml/min und dann 45 ml/min, bis die N-(Phosphonomethyl)iminodiessigsäure im Wesentlichen
aufgebraucht war. Beim Kontrollversuch wurde während keinem der 30 Läufe Wismut
in die Reaktionszone eingeführt.
Bei einem anderen Versuch wurde zu Beginn des ersten Reaktionslaufs
0,012 g Wismut(III)-oxid in das Reaktionsmedium eingeführt.
-
16 vergleicht die resultierenden Ameisensäurekonzentrationsprofile. 17 vergleicht die resultierenden Formaldehydkonzentrationsprofile
und 18 vergleicht die resultierenden
NMG-Konzentrationsprofile. Die einmalige Einführung von Wismut in die Reaktionszone
verringerte selbst nach 30 Läufen
die Ameisensäurekonzentration
um 98%, die Formaldehydkonzentration um 50% und die NMG-Konzentration
um 90%.
-
E. Wirkung der Zugabe
eines vorher bei 132 NPMIDA-Oxidationsreaktionschargen verwendeten
Pt/Fe/C-Katalysators
-
Es
erfolgte ein Versuch mit 14 Läufen,
um die Auswirkung des Mischens von Wismut mit einem gebrauchten
Pt/Fe/C-Katalysator zu bestimmen. Vor diesem Versuch war der Katalysator
dazu verwendet worden 129 N-(Phosphonomethyl)iminodiessigsäureoxidationsreaktionschargen
zu katalysieren. Der frische Katalysator (d. h. der Katalysator
ehe er bei den vorausgehenden 129 N-(Phosphonomethyl)iminodiessigsäureoxidationsreaktionsläufen verwendet
wurde) wurde nach einem Verfahren ähnlich dem für Beispiel
1 beschriebenen, hergestellt und enthielt 5 Gew.-% Platin und 0,5
Gew.-% Eisen.
-
Die
14 N-(Phosphonomethyl)iminodiessigsäureoxidationsreaktionsläufe erfolgten
in einem 300 ml Reaktor (hergestellt aus einer Metalllegierung,
Hastelloy C, Autoclave Engineers) unter Verwendung von 0,9 g erschöpftem Katalysator
(0,5 Gew.-%), 21,8 g N-(Phosphonomethyl)iminodiessigsäure (12,1
Gew.-%), 1000 ppm Formaldehyd, 5000 ppm Ameisensäure, einer Gesamtreaktionsmasse
von 180 g, einem Druck von 621 kPa (90 psig), einer Temperatur von
100°C und
einer Rührgeschwindigkeit
von 900 Upm. Die Sauerstoffeinspeiserate betrug während der
ersten 26 min 141 ml/min und dann 45 ml/min, bis die N-(Phosphonomethyl)iminodiessigsäure im Wesentlichen
verbraucht war. Zu Beginn des 4. Laufs wurden 0,012 g Wismut(III)-oxid
in die Reaktionszone eingeführt.
-
19 gibt die Wirkung der Wismutzugabe beim 4. Lauf
auf die Erzeugung von Ameisensäure,
Formaldehyd und des NMG-Nebenprodukts wieder.
-
F. Wirkung der Zugabe
von Wismut zu einem vorher bei 30 NPMIDA-Oxidationsreaktionschargen
verwendeten Pt/Sn/C-Katalysators
-
Es
erfolgte ein Versuch mit 11 Läufen,
um die Auswirkung des Mischens von Wismut mit einem gebrauchten
Pt/Sn/C-Katalysator zu bestimmen. Der Katalysator war vor diesem
Versuch dazu verwendet worden 30 N-(Phosphonomethyl)iminodiessigsäureoxidationsreaktionschargen
zu katalysieren. Der frische Katalysator (d. h. der Katalysator
ehe er bei den vorangehenden 30 N-(Phosphonomethyl)iminodiessigsäureoxidationsreaktionsläufen verwendet
wurde) wurde nach einem Verfahren ähnlich dem für Beispiel
2 beschriebenen, hergestellt und enthielt 5 Gew.-% Platin und 1
Gew.-% Zinn.
-
Die
11 N-(Phosphonomethyl)iminodiessigsäureoxidationsreaktionsläufe erfolgten
in einem 300 ml Reaktor (hergestellt aus einer Metalllegierung,
Hastelloy C, Autoclave Engineers) unter Verwendung von 1,35 g gebrauchtem
Katalysator (0,75 Gew.-% der gesamten Reaktionsmasse), 21,8 g N-(Phosphonomethyl)iminodiessigsäure (12,1
Gew.-% der gesamten Reaktionsmasse), 1000 ppm Formaldehyd, 5000
ppm Ameisensäure,
einer Gesamtreaktionsmasse von 180 g, einem Druck von 621 kPa (90
psig), einer Temperatur von 100°C und
einer Rührgeschwindigkeit
von 900 Upm. Die Sauerstoffeinspeiserate betrug während der
ersten 26 min 141 ml/min und dann 45 ml/min, bis die N-(Phosphonomethyl)iminodiessigsäure im Wesentlichen
verbraucht war. Zu Beginn des 4. Laufs wurden 0,012 g Wismut(III)-oxid
in die Reaktionszone eingeführt.
-
20 gibt die Wirkung der Wismutzugabe beim 4. Lauf
auf die Erzeugung von Ameisensäure,
Formaldehyd und des NMG-Nebenprodukts wieder.
-
G. Wirkung der Zugabe
von Wismut auf mehr als 100 nachfolgende, mit diesem Katalysator
kontaktierte, NPMIDA-Oxidationschargen.
-
Es
erfolgten zwei Versuch mit 125 Läufen,
um die Auswirkung der Zugabe von Wismut auf über 100 nachfolgende Reaktionsläufe bei
Verwendung des gleichen Katalysators zu bestimmen.
-
Beide
Versuche erfolgten unter Verwendung eines Katalysators, welcher
5 Gew.-% Platin und 1 Gew.-% Zinn enthielt (Dieser Katalysator wurde
nach einem Verfahren ähnlich
dem für
Beispiel 2 beschriebenen hergestellt). Bei jedem Versuch wurde für alle Läufe der
gleiche Katalysator verwendet. Die Reaktion erfolgte in einem Rührbehälterreaktor
unter Verwendung von 0,75% Katalysator (des Gewichts der gesamten Reaktionsmasse),
12,1% N-(Phosphonomethyl)iminodiessigsäure (des Gewichts der gesamten
Reaktionsmasse), einem Druck von 883 kPa (128 psig) und einer Temperatur
von 100°C.
Die Sauerstoffeinspeiserate betrug für den ersten Teil einer jeden
Chargenreaktion (Der genaue Zeitbedarf schwankte von Ansatz zu Ansatz
von 14,9 bis 20,3 min, wobei die Zeit für die anfänglichen Chargen näher bei
14,9 min und die Zeit für
die späteren
Chargen näher
bei 20,3 min lag), 1,3 mg/min pro Gramm Gesamtreaktionsmasse, und
dann 0,35 mg/min für
die Gesamtreaktionsmasse bis die N-(Phosphonomethyl)iminodiessigsäure im Wesentlichen
verbraucht war. Ein Teil des Reaktionsprodukts aus jeder Charge
wurde abgedampft, um in den Reaktor als Quelle von Formaldehyd und
Ameisensäure
zurückgeführt und
als Reduktionsmittel bei der nächsten
Chargenreaktion verbraucht zu werden. Die in den Reaktor zurückgeführten Mengen
Formaldehyd und Ameisensäure reichten
von 100 bis 300 ppm bzw. von 0 ppm bis 2300 ppm (0 bis 200 ppm Ameisensäure nach
den auf die Zugabe von Wismut(III)-oxid folgenden 25 Chargen).
-
Beim
Kontrollversuch wurde während
keinem der 125 Läufe
Wismut in die Reaktionszone eingeführt. Bei einem anderen Versuch
wurde der Katalysator zuerst dazu verwendet 17 Chargen von N-(Phosphonomethyl)iminodiessigsäure zu katalysieren.
Nach der Katalyse der 17 Chargen wurde der Katalysator im Wesentlichen
aus dem Reaktionsprodukt abgetrennt und die resultierende Katalysatormischung
in einen Katalysatorvorratsbehälter übergeführt, wo
9,0 g Wismut(III)-oxid pro Gramm Katalysator in die Katalysatormischung
eingeführt
wurden. Der Katalysator wurde dann dazu verwendet, um die Oxidation
von weiteren 107 Chargen N-(Phosphonomethyl)iminodiessigsäure zu katalysieren.
-
21 vergleicht die resultierenden Ameisensäurekonzentrationsprofile. 22 vergleicht die resultierenden Formaldehydkonzentrationsprofile
und 23 vergleicht die resultierenden
NMG-Konzentrationsprofile. Die einmalige Einführung von Wismut in die Mischung
mit dem Katalysator verringerte selbst nach 107 Läufen die
Ameisensäure-
und NMG-Konzentrationen um ungefähr
90%.
-
Beispiel 5
-
Kontinuierliche
Oxidation von NPMIDA zu Glyphosat mit einem teilweise verbrauchten
Katalysator und die Verwendung eines Zusatzaktivators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von N-(Phosphonomethyl)iminodiessigsäure ("NPMIDA") zu N-(Phosphonomethyl)glycin
("Glyphosat") in einem kontinuierlichen
Oxidationsreaktorsystem unter Verwendung eines bereits gebrauchten
Katalysators und eines zusätzlichen
Aktivators. Der Versuch war darauf ausgelegt, die Bedingungen zu
simulieren, welche in der ersten Reaktionszone auftreten könnten, insbesondere
wenn die Mutterlauge, die das Reaktionsprodukt enthält, zur
ersten Reaktionszone zurückgeführt wird.
-
Die
Reaktion erfolgte in einem kontinuierlichen Reaktorsystem unter
Verwendung eines 2-Liter Hastelloy C-Autoklav (Autoclave Engineers
Inc., Pittsburgh, PA). Der Reaktor war mit einem Rührwerk mit
einer Turbine mit 6 Laufschaufeln von 3,18 cm (1,25'') Durchmesser ausgerüstet, welches mit 1600 Upm
betrieben wurde. Der Flüssigkeitsstand
im Reaktor wurde mithilfe eines Drexelbrook Univesal IIITMSmart LevelTM mit
einem Teflon-beschichteten Fühler überwacht.
Es wurde eine Innenkühlschlange
verwendet, um die Temperatur innerhalb des Reaktors während des
Reaktionsverlaufs zu regeln.
-
Die
Reaktion begann mit der Beschickung des Reaktors mit einem wässrigen
Einspeisematerial (1420 g) und dem Katalysators (29,3 g oder etwa
2 Gew.-% Katalysator in der Reaktionsmasse). Die wässrige Einspeisematerialaufschlämmung enthielt
NPMIDA (78,53 Gew.-%), Glyphosat (2,87 Gew.-%), Formaldehyd (2127
ppm des Gewichts) und Ameisensäure
(3896 ppm des Gewichts). Die Einspeiseaufschlämmung enthielt auch NaCl (etwa
450 ppm des Gewichts) um den geringen Gehalt an Chloridverunreinigungen
zu simulieren, der typischerweise in kommerziell erhältlichem
NPMIDA vorliegt. Der Katalysator, welcher nach einem Verfahren, ähnlich dem
vorstehend für
Beispiel 1 beschriebenen, hergestellt wurde, umfasste Platin (5
Gew.-%) und Eisen (0,5 Gew.-%) auf einem speziellen Kohlenstoffträger. Der
Katalysator war bereits unter Bedingungen verwendet worden, ähnlich den
in Beispiel 4 beschriebenen.
-
Der
Reaktor wurde abgedichtet, um jeden Flüssigkeitszufluss oder -abfluss
zu unterbinden. Die Reaktionsmischung wurde auf etwa 105°C erwärmt und
mit Stickstoff ein Druck von etwa 690 kPa (100 psig) aufgebracht.
Es wurde der Sauerstoffstrom (1000 sccm) angestellt und die Reaktion
etwa 15 min ohne Flüssigkeitszufluss
oder -abfluss laufen lassen. Nach diesen anfänglichen 15 min wurde mit der
Einspeisung der Aufschlämmung
(70,4 g/min) begonnen und die Reaktionsflüssigkeit kontinuierlich abgezogen,
um einen konstantes Reaktorfüllstand
aufrechtzuerhalten, wie vom vorstehend beschriebenen Drexelbrook
Füllstandsanzeiger angezeigt.
Nach etwa 55 min wurde die Sauerstoffströmung leicht auf 800 sccm reduziert.
Nach einem Betrieb von etwa 280 min bei einer Sauerstoffströmung von
800 sccm wurde Bi
2O
3 (0,0336
g) in den Reaktor als zusätzlicher
Aktivator eingespritzt. Das flüssige
Produkt wurde mittels HPLC analysiert. Die Analysenergebnisse der
kontinuierlichen Oxidationsreaktion sind in Tabelle 19 wiedergegeben.
Desgleichen zeigt
24 die Profile für Formaldehyd
und Ameisensäure
im flüssigen
Produkt bei einer Sauerstoffströmung
von 800 sccm. Tabelle
19 Oxidationsergebnisse
aus HPLC für
Beispiel 21
- 1 Zeit nach Beginn
der Aufschlämmungseinspeisung
-
Beispiel 6
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in Anwesenheit eines bereits verwendeten
Pt/Fe/C-Katalysators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat unter Verwendung eines bereits gebrauchten heterogenen
teilchenförmigen
Katalysators. Der Versuch war darauf ausgelegt, die Bedingungen
zu simulieren, welche in der ersten Reaktionszone eines kontinuierlichen
Reaktorsystems auftreten könnten,
insbesondere wenn die Mutterlauge, welche das Reaktionsprodukt enthält, zur
Reaktionszone zurückgeführt wird.
-
Der
Versuch erfolgte in einem kontinuierlichen Reaktorsystem, ähnlich dem
in Beispiel 5 vorstehend beschriebenen. Die Reaktion begann mit
der Beschickung des Reaktors mit einer wässrigen Einspeisematerialaufschlämmung (1424
g) und einem heterogenen Katalysator (29,3 g oder etwa 2 Gew.-%
Katalysator in der Reaktionsmasse). Die wässrige Einspeisematerialaufschlämmung enthielt
NPMIDA (7,01 Gew.-%), Glyphosat (2,88 Gew.-%), Formaldehyd (2099,9
ppm des Gewichts) und Ameisensäure
(4690 ppm des Gewichts). Die Einspeiseaufschlämmung enthielt auch NaCl (etwa
450 ppm des Gewichts) um den geringen Gehalt an Chloridverunreinigungen
zu simulieren, der typischerweise in kommerziell erhältlichem
NPMIDA vorliegt. Der Katalysator, welcher nach einem Verfahren hergestellt
wurde, ähnlich
dem vorstehend für
Beispiel 1 beschriebenen, umfasste Platin (5 Gew.-%) und Eisen (0,5
Gew.-%) auf einem teilchenförmigen
Kohlenstoffträger.
Der Katalysator war bereits unter Bedingungen verwendet worden, ähnlich den
in Beispiel 4 beschriebenen.
-
Der
Reaktor wurde abgedichtet, um jeden Flüssigkeitszufluss oder -abfluss
zu auszuschließen.
Die Reaktionsmischung wurde auf etwa 107°C erwärmt und mit Stickstoff ein
Druck von etwa 690 kPa (100 psig) aufgebracht. Der Sauerstoffstrom
(1000 sccm) wurde angestellt und die Reaktion etwa 13 min ohne Flüssigkeitszufluss
oder -abfluss laufen lassen. Nach diesen anfänglichen 13 min wurde mit der
Einspeisung der Aufschlämmung
(70,4 g/min) begonnen und die Reaktionsflüssigkeit kontinuierlich abgezogen,
um einen konstanten Reaktorfüllstand
aufrechtzuerhalten, wie er von dem in Beispiel 5 vorstehend beschriebenen
Drexelbrook Füllstandsanzeiger
angezeigt wurde. Das flüssige
Produkt wurde mittels HPLC analysiert. Die Analysenergebnisse der
kontinuierlichen Oxidationsreaktion sind nachstehend in Tabelle
20 wiedergegeben.
25 zeigt den Verlauf für das erzeugte
Glyphosat und den im Flüssigprodukt
verbleibenden NPMIDA-Reaktionsteilnehmer. Tabelle
20 Oxidationsergebnisse
von Beispiel 6
- 1 Zeit nach Beginn
der Aufschlämmungseinspeisung
-
Beispiel 7
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in Anwesenheit eines Pt/Fe/C-Katalysators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat unter Verwendung eines frischen heterogenen teilchenförmigen Pt/Fe/C-Katalysators
(bei relativ niedriger Katalysatorkonzentration) über einen
längeren
Zeitraum. Der Versuch war darauf ausgelegt, die Bedingungen zu simulieren,
welche in der ersten Reaktionszone auftreten könnten, insbesondere wenn die
Mutterlauge, welche das Reaktionsprodukt enthält, zur Reaktionszone rezykliert
wird.
-
Der
Versuch erfolgte in einem kontinuierlichen Reaktorsystem, ähnlich dem
vorstehend in Beispiel 5 beschriebenen. Die Reaktion begann mit
der Beschickung des Reaktors mit einer wässrigen Einspeisematerialaufschlämmung (1447
g) und einem heterogenen Katalysator (3,63 g oder etwa 0,25 Gew.-%
Katalysator in der Reaktionsmasse). Die wässrige Einspeisematerialaufschlämmung enthielt
NPMIDA (3,45 Gew.-%), Glyphosat (1,55 Gew.-%), Formaldehyd (1140
ppm des Gewichts) und Ameisensäure
(2142 ppm des Gewichts). Die Einspeiseaufschlämmung enthielt auch NaCl (etwa
450 ppm des Gewichts) um den geringen Gehalt an Chloridverunreinigungen
zu simulieren, der typischerweise in kommerziell erhältlichem
NPMIDA vorliegt. Der Katalysator, welcher nach einem Verfahren,
hergestellt wurde, ähnlich
dem vorstehend für
Beispiel 1 beschriebenen, umfasste Platin (5 Gew.-%) und Eisen (0,5
Gew.-%) auf einem teilchenförmigen
Kohlenstoffträger.
Der Katalysator war vorher nicht verwendet worden.
-
Der
Reaktor wurde abgedichtet, um jeden Flüssigkeitszufluss oder -abfluss
zu auszuschließen.
Die Reaktionsmischung wurde auf etwa 100°C erwärmt und mit Stickstoff ein
Druck von etwa 690 kPa (100 psig) aufgebracht. Der Sauerstoffstrom
(300 sccm) wurde angestellt und die Reaktion etwa 22 min ohne Flüssigkeitszufluss
oder -abfluss laufen lassen. Nach diesen anfänglichen 22 min wurde mit der
Einspeisung der Aufschlämmung
(70,4 g/min) begonnen und die Reaktionsflüssigkeit kontinuierlich abgezogen,
um einen konstantes Reaktorfüllstand
aufrechtzuerhalten, wie vom in Beispiel 5 vorstehend beschriebenen
Drexelbrook Füllstandsanzeiger
angezeigt. Das Reaktorsystem wurde etwa 4300 min laufen lassen,
wonach die Flüssigkeitszulaufrate
verdoppelt wurde, um die Verweilzeit der Reaktorflüssigkeit
um den Faktor 2 wirksam zu erniedrigen. Das flüssige Produkt wurde mittels
HPLC analysiert. Die Analysenergebnisse der kontinuierlichen Oxidationsreaktion
sind nachstehend in Tabelle 21 wiedergegeben.
26 zeigt den Verlauf der Glyphosaterzeugung und
der im Flüssigprodukt
verbleibenden NPMIDA-Reaktionsteilnehmer. Tabelle
21 Oxidationsergebnisse
von Beispiel 7
- 1 Zeit nach Beginn
der Aufschlämmungseinspeisung
-
Beispiel 8
-
Kontinuierliche
Oxidation von NPMIDA zu Glyphosat in zwei In Reihe angeordneten
Rührbehälterreaktoren
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in einem kontinuierlichen Reaktorsystem, welches zwei
in Reihe angeordnete Rührbehälterreaktoren
umfasst.
-
Nach 27 erfolgte der Versuch in einem kontinuierlichen
Reaktorsystem, welches zwei Reaktoren und einen Kristallisator umfasste.
Die zwei Reaktoren (jeder 3,8 l (1 Gallone), Edelstahlautoklaven
von Autoclaves Engineers, Inc., Pittsburgh, PA) wurden als Rührbehälterreaktoren
in Reihe kontinuierlich betrieben. Das kontinuierliche Reaktorsystem
war so ausgelegt, dass die wässrige
Einspeiseaufschlämmung
kontinuierlich in die erste Reaktionszone (Reaktor R1) eingeführt wurde.
Der flüssige
Ablauf wurde kontinuierlich aus R1 abgezogen und in die zweite Reaktionszone
(Reaktor R2) eingeführt.
Der flüssige
Ablauf wurde kontinuierlich aus R2 abgezogen und zur Gewinnung des
Produkts und einer Glyphosataufschlämmung in den Kristallisator eingeführt. Sauerstoff
wurde unabhängig
in jede Reaktionszone eingespeist, während das Produktgas aus jedem
Reaktor unabhängig
abgelüftet
wurde. Sauerstoffgas wurde in R1 durch eine in der Nähe des Flügelrührers (6,35
cm (2'') Turbinenblattrührer) angeordnete
Fritte eingespeist. In R2 wird der Sauerstoff in den Kopfraum über dem
Flüssigkeitsspiegel
eingespeist und ein 6,35 cm (2,5'')-Flügelrührer vom
Typ DISPSERSIMAX verwendet, um das Gas im Kopfraum wirksam in die
Reaktionszone einzumischen. Die Temperatur der Reaktionsmasse wurde
in jedem Reaktor durch interne Kühlschlangen
geregelt. Der flüssige
Ablauf aus R1 wurde über
eine Fritte abgelassen, welche es erlaubte den heterogenen Katalysator
in R1 zurückzuhalten.
Auf ähnliche
Weise wurde der flüssige
Ablauf aus R2 über
eine Fritte abgelassen, um den heterogenen Katalysator im Innern
von R2 zurückzuhalten.
Das Reaktionsmasse/Volumen-Verhältnis
wurde in jedem Reaktor konstant gehalten.
-
Das
kontinuierliche Reaktorsystem wurde auf ähnliche Weise angefahren wie
das vorstehend in Beispiel 5 beschriebene, indem die Reaktoren nach
Chargenart angestellt und kurz danach mit der Flüssigkeitsströmung durch
das System begonnen wurde. Das Einspeisematerial war eine wässrige Aufschlämmung, welche
NPMIDA (etwa 7,6 Gew.-%), Glyphosat (etwa 2,8 Gew.-%), Formaldehyd
(etwa 2200 ppm des Gewichts) und Ameisensäure (etwa 4500 ppm des Gewichts) enthielt.
Der Einspeisung wurde auch eine geringe Menge NaCl (etwa 450 ppm)
zugesetzt, um den geringen Gehalt an Chloridverunreinigungen zu
simulieren, der typischerweise in kommerziell erhältlichem
NPMIDA vorliegt. Der Katalysator, der nach einem Verfahren hergestellt
wurde, ähnlich
dem vorstehend in Beispiel 1 beschriebenen, umfasste Platin (5 Gew.-%)
und Eisen (0,5 Gew.-%) auf einem teilchenförmigen Kohlenstoffträger. Jeder
Reaktor wurde mit dem wässrigen
Einspeiseaufschlämmungsmaterial
und dem Katalysator so beschickt, dass in jedem Reaktor eine Katalysatorkonzentration von
etwa 2 Gew.-% vorlag, wobei die gesamte Reaktor-Sollmasse für R1 und
R2 insgesamt 2693 g bzw. 1539 g betrug.
-
Die
Betriebsbedingungen sind nachstehend in Tabelle 22 zusammengefasst.
Die Analysenergebnisse der Zusammensetzung der wässrigen Einspeisung, des Flüssigkeits-
und Gasaustrags von R1 und des Flüssigkeits- und Gasaustrags
von R2, welche mittels HPLC analysiert wurden, sind in nachstehender
Tabelle 23 wiedergegeben.
-
Tabelle
22 Zusammenfassung
der Betriebsbedingungen von Beispiel 8
-
Tabelle
23 Oxidationsergebnisse
für Beispiel
8
-
Beispiel 9
-
Kontinuierliche
Oxidation von NPMIDA zu Glyphosat in zwei hintereinander angeordneten
Rührbehälterreaktoren
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in einem kontinuierlichen Reaktorsystem, welches zwei
hintereinander angeordnete Rührbehälterreaktoren
umfasst, wobei der flüssige
Ausfluss aus dem zweiten Reaktor an einen Kristallisator zur Glyphosatgewinnung
weitergeleitet und die resultierende Mutterlauge aus dem Kristallisator
als Teil der Reaktoreinspeisung in den ersten Reaktor rezirkuliert
wurde.
-
Nach 28 wurde Beispiel 9 in einem kontinuierlichen
Reaktorsystem durchgeführt, ähnlich dem
in Beispiel 5 beschriebenen, außer
das die Mutterlauge aus dem Kristallisator in den ersten Reaktor
R1 zurückgeführt wurde.
Das kontinuierliche Reaktorsystem wurde auf ähnliche Weise angefahren wie
in Beispiel 5 beschrieben, indem die Reaktoren nach Chargenart angestellt
und kurz danach mit der Flüssigkeitsströmung durch
das System begonnen wurde. Der Kristallisator (30 L) wurde anfänglich mit
einer wässrigen
Einspeisematerialaufschlämmung
beschickt, welche NPMIDA (0,16 Gew.-%), Glyphosat (2,0 Gew.-%),
Formaldehyd (2754 ppm des Gewichts) und Ameisensäure (5637 ppm des Gewichts)
umfasste und bei etwa 60°C
und einem Druck von 100 kPa (1 atm) betrieben. Das Aufschlämmungseinspeisesystem
wurde mit einer wässrigen Einspeisematerialaufschlämmung beschickt,
welche NPMIDA (etwa 25 Gew.-%) umfasste. Der verwendete Katalysator
war dem in Beispiel 8 verwendeten heterogenen teilchenförmigen Katalysator ähnlich.
-
Jeder
Reaktor wurde mit dem wässrigen
Einspeiseaufschlämmungsmaterial
und dem Katalysator so beschickt, dass in jedem Reaktor eine Katalysatorkonzentration
von etwa 2 Gew.-% vorlag, wobei die gesamte Reaktor-Sollmasse für R1 und
R2 insgesamt 2693 g bzw. 1539 g betrug. Der Flüssigkeitszulauf zu R1 umfasste
die wässrige
Einspeisematerialaufschlämmung
(etwa 40 ml/min) und den Mutterlaugenrücklauf (etwa 80 ml/min) aus
dem Kristallisator. Das Flüssigkeitsniveau
in jedem Reaktor wurde während
des Laufs geregelt, um in jedem Reaktor eine konstante Reaktionsmasse
aufrechtzuerhalten, wobei die angestrebte hydraulische Verweilzeit
in R1 und R2 21 min bzw. 12,2 min betrug, was eine Gesamtflüssigkeitsströmung durch
das System von etwa 120 ml/min ergab.
-
Die
Betriebsbedingungen sind nachstehend in Tabelle 24 zusammengefasst.
Das Flüssigprodukt
wurde mittels HPLC analysiert. Die Analysenergebnisse der Zusammensetzungen
der wässrigen
Einspeiseaufschlämmungen,
des R1-Flüssigkeitsablauf
und des R2-Flüssigkeitsablauf
sind in nachstehender Tabelle 25 wiedergegeben.
-
Die
abgelaufene Zeit bezieht sich auf den Zeitraum ab dem die kontinuierliche
Flüssigkeitszuführung einsetzte.
-
Tabelle
24 Zusammenfassung
der Betriebsbedingungen von Beispiel 9
-
Tabelle
25 Oxidationsergebnisse
für Beispiel
9
-
Beispiel 10
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in Anwesenheit eines Pt/Sn/C-Katalysators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in Anwesenheit eines heterogenen teilchenförmigen Pt/Sn(C-Katalysators
in einem Rührbehälterreaktor.
Der Versuch war darauf ausgelegt, die Bedingungen zu simulieren,
welche in der zweiten Reaktionszone eines kontinuierlichen Reaktorsystems
auftreten könnten.
-
Der
Versuch erfolgte in einem 500 ml Hastelloy C-Autoklav (Autoclave
Engineers Inc., Pittsburgh, PA). Der Reaktor war mit einem Rührwerk mit
einer Turbine mit 6 Laufschaufeln von 3,18 cm (1,25'') Durchmesser ausgerüstet. Der Flüssigkeitsstand
im Reaktor wurde mithilfe eines Füllstandsanzeigers überwacht, ähnlich dem
vorstehend in Beispiel 5 beschriebenen. Es wurde eine Innenkühlschlange
verwendet, um die Temperatur innerhalb des Reaktors während des
Reaktionsverlaufs zu regeln.
-
Während des
Betriebs wurde der Reaktor kontinuierlich mit einem Sauerstoffgasstrom
und einer wässrigen,
NPMIDA-haltigen Einspeisematerialaufschlämmung beschickt. Der Sauerstoff
wurde in das Reaktionsmedium durch eine in der Nähe des Rührwerks angeordnete Fritte
eingespeist. Ein flüssiger,
Glyphosat-haltiger Produktstrom wurde aus dem Reaktor durch eine
Fritte abgezogen, welche es erlaubte, jede Art von dem Reaktor zugeführten Katalysator
im Reaktionsmedium zurückzuhalten.
Der abgezogene flüssige
Produktstrom wurde In-line mit einer basischen Lösung gemischt, welche Glyphosat
lösen kann.
Der Produktgasstrom (enthaltend CO2 und
nicht umgesetzten Sauerstoff) wurde kontinuierlich aus dem Kopfraum
des Reaktors abgelüftet.
-
Das
kontinuierliche Reaktorsystem wurde in einer Weise ähnlich der
vorstehend in Beispiel 5 beschriebenen, angefahren, indem der Reaktor
nach Chargenart angefahren und kurz danach die Flüssigkeitsströmung durch
das System angestellt wurde. Die wässrige Einspeisematerialaufschlämmung umfasste
NPMIDA (2,46 Gew.-%), Glyphosat (3,72 Gew.-%), Formaldehyd (1381
ppm des Gewichts) und Ameisensäure
(6458 ppm des Gewichts). Der Katalysator wurde nach einem Verfahren, ähnlich dem
vorstehend für
Beispiel 14 beschriebenen, hergestellt und umfasste Platin (5 Gew.-%)
und Zinn (1,0 Gew.-%) auf einem teilchenförmigen Kohlenstoffträger.
-
Die
Betriebsbedingungen sind nachstehend in Tabelle 26 zusammengefasst.
Das Flüssigprodukt
wurde mittels HPLC analysiert. Die Analysenergebnisse des Oxidationslaufs
sind in nachstehender Tabelle 27 wiedergegeben.
-
Tabelle
26 Zusammenfassung
der Betriebsbedingungen von Beispiel 10
-
Tabelle
27 Oxidationsergebnisse
für Beispiel
10
-
Beispiel 11
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in Anwesenheit eines Pt/Sn/C-Katalysators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in Anwesenheit eines heterogenen teilchenförmigen Pt/Sn/C-Katalysators
in einem Rührbehälterreaktor.
Der Versuch war darauf ausgelegt, die Bedingungen zu simulieren,
die in der zweiten Reaktionszone eines kontinuierlichen Reaktorsystems
auftreten könnten.
Es wurde auch die Sauerstoffströmungsrate verändert, um
die Auswirkung verschiedener Sauerstoffströmungsraten auf die Umwandlung
zu veranschaulichen.
-
Der
Versuch erfolgte in einem kontinuierlichen Reaktorsystem, ähnlich dem
vorstehend unter Beispiel 10 beschriebenen Reaktorsystem. Während des
Betriebs wurde der Reaktor kontinuierlich mit einem Sauerstoffgasstrom
und einer wässrigen,
NPMIDA-haltigen Einspeisematerialaufschlämmung beschickt. Der Sauerstoff
wurde in das Reaktionsmedium durch eine in der Nähe des Rührwerks angeordnete Fritte
eingespeist. Ein flüssiger,
Glyphosat-haltiger Produktstrom wurde aus dem Reaktor über eine
Fritte abgezogen, welche es erlaubte, irgendeinen dem Reaktor zugeführten Katalysator
im Reaktionsmedium zurückzuhalten.
Der abgezogene flüssige
Produktstrom wurde In-line mit einer basischen Lösung gemischt, welche Glyphosat
lösen kann.
Der Produktgasstrom (enthaltend CO2 und
nicht umgesetzten Sauerstoff) wurde kontinuierlich aus dem Kopfraum
des Reaktors abgelüftet.
-
Das
kontinuierliche Reaktorsystem wurde in ähnlicher Weise angefahren wie
vorstehend in Beispiel 5 beschriebenen, indem der Reaktor nach Chargenart
angestellt und kurz danach mit der Flüssigkeitsströmung durch
das System begonnen wurde. Die wässrige
Einspeisematerialaufschlämmung
umfasste NPMIDA (etwa 2,8 Gew.-%), Glyphosat (etwa 4,2 Gew.-%),
Formaldehyd (etwa 1425 ppm des Gewichts) und Ameisensäure (etwa
6570 ppm des Gewichts). Der Katalysator wurde nach einem Verfahren
hergestellt, ähnlich
dem vorstehend für
Beispiel 14 beschriebenen, und umfasste Platin (5 Gew.-%) und Zinn
(1,0 Gew.-%) auf einem teilchenförmigen
Kohlenstoffträger.
-
Die
Betriebsbedingungen sind nachstehend in Tabelle 28 zusammengefasst.
Im Verlauf dieses Versuchs wurde die Sauerstoffströmungsrate
in den Reaktor im Bereich von 75 bis 300 sccm hoch- und zurückgefahren.
Das Flüssigprodukt
wurde mittels HPLC analysiert. Die Analysenergebnisse der kontinuierlichen Oxidation
sind nachstehend in Tabelle 29 wiedergegeben.
-
Tabelle
28 Zusammenfassung
der Betriebsbedingungen von Beispiel 11
-
Tabelle
29 Oxidationsergebnisse
für Beispiel
11
-
-
-
Beispiel 12
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in zwei In Reihe angeordneten Rührbehälterreaktoren
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in einem kontinuierlichen Reaktorsystem, welches zwei
in Reihe angeordnete Rührbehälterreaktoren
umfasst. Bei diesem Beispiel wurde der teilchenförmige heterogene Katalysator
aus dem ersten Reaktor in den zweiten Reaktor transportiert. Der
Katalysator trat aus dem zweiten Reaktor mit der aus dem zweiten
Reaktor austretenden Flüssigkeit
aus, wurde durch Filtration abgetrennt und in die erste Reaktionszone
zurückgeführt.
-
Nach 29 erfolgte die Reaktion in einem kontinuierlichen
Reaktorsystem, welches zwei Rührbehälterreaktoren,
ein Einspeiseaufschlämmungssystem
und ein Katalysatorfiltrationssystem umfasste. Die zwei Reaktoren
(jeder 3,8 l (1 Gallone), Edelstahlautoklaven von Autoclaves Engineers,
Inc., Pittsburgh, PA) wurden als in Reihe angeordnete Rührbehälterreaktoren
kontinuierlich betrieben. In jeden der Reaktoren wurde Sauerstoff
eingespeist. Der flüssige
Ablauf wurde aus R1 mittels eines Tauchrohrs abgezogen, welches
es dem Katalysator erlaubte aus dem ersten Reaktor mit dem flüssigen Auslauf
und in den zweiten Reaktor (R2) übergeführt zu werden.
Ein gewisser Anteil an gasförmigem
Reaktionsprodukt wurde durch das Tauchrohr ebenfalls von R1 nach
R2 übergeführt, während andere
gasförmige
Reaktionsprodukte aus dem Reaktor abgelüftet wurden. Auf ähnliche
Weise wurde flüssiger
Ausfluss aus R2 durch ein Tauchrohr abgezogen, welches es dem Katalysator
und einen gewissen Anteil an gasförmigem Reaktionsprodukt erlaubte
mit dem Ausfluss entfernt zu werden. Der katalysatorhaltige flüssige Ausfluss
aus R2 wurde zu einem Katalysator-filtrationssystem übergeführt. Das
Katalysatorfiltrationssystem erzeugte einen kontinuierlichen Strom
an katalysatorfreien Filtrat als Produkt. In das Katalysatorfiltrationssystem
wurde ein Filterrückspülmittel
eingespeist, um den abfiltrierten Katalyators auf kontinuierliche
Weise zu R1 zurückzuspülen. In
R1 und R2 wurde durch Fritten, welche in der Nähe des Flügelrührers (6,35 cm (2'') Turbinenblattrührer, sowohl in R1 als auch
in R2) angeordnet waren, Sauerstoff eingeleitet. Zur Regelung der
Temperatur wurde in jedem Reaktor eine Innnenkühlschlange verwendet.
-
Eine
wässrige
Einspeisematerialaufschlämmung,
die etwa 25 Gew.-% NPMIDA enthielt, wurde mit einer Rate von etwa
50 ml/min in den Reaktor eingespeist.
-
Das
Filterrückspülmittel
enthielt NPMIDA (etwa 3 Gew.-%), Glyphosat (etwa 0,1 Gew.-%), Formaldehyd
(etwa 3000 ppm des Gewichts) und Ameisensäure (etwa 7000 ppm des Gewichts).
Das Filterrückspülmittel
wurde mit einer Rate von etwa 100 ml/min zu R1 zurückgeführt. Der
Katalysator war nach einem Verfahren hergestellt worden, ähnlich dem
vorstehend in Beispiel 1 beschriebenen, und umfasste Platin (5 Gew.-%)
und Eisen (0,5 Gew.-%) auf einem teilchenförmigen Kohlenstoffträger. Das
Reaktorsystem wurde so mit Katalysator beschickt, dass eine Anfangskonzentration
von etwa 1 Gew.-% Katalysator vorlag. Das Reaktorsystem wurde in
einer Weise ähnlich
der vorstehend in Beispiel 8 beschriebenen, angefahren, indem der
Reaktor nach Chargenart angestellt und kurz danach mit der Flüssigkeitsströmung durch
das System begonnen wurde.
-
Die
Betriebsbedingungen sind nachstehend in Tabelle 30 zusammengefasst.
Das Flüssigprodukt
wurde mittels HPLC analysiert. Die Oxidationsergebnisse sind nachstehend
in Tabelle 31 wiedergegeben. Tabelle 31 gibt die Werte an, welche
die Zusammensetzung des Einlassstroms in R1 beschreiben (einschließlich des kombinierten
aus der Katalysatorfiltrationsrückspülung und
der wässrigen
Einspeiseaufschlämmung)
und der flüssigen
Ausflusszusammensetzungen für
R1 und R2.
-
Tabelle
30 Zusammenfassung
der Betriebsbedingungen von Beispiel 12
-
Tabelle
31 Oxidationsergebnisse
für Beispiel
12
-
Beispiel 13
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in Anwesenheit eines Pt/Sn/C-Katalysators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in Anwesenheit eines heterogenen teilchenförmigen Pt/Sn/C-Katalysators.
Der Versuch war darauf ausgelegt, die Reaktionsbedingungen zu simulieren,
welche in der ersten Rührbehälterreaktionszone
auftreten könnten.
-
Der
Versuch erfolgte in einem kontinuierlichen Reaktorsystem, ähnlich dem
vorstehend in Beispiel 10 beschriebenen, außer das ein 1000 ml Hastelloy
C Autoklav verwendet wurde. Der Reaktor war mit einem Rührwerk mit
einer Turbine mit 6 Laufschaufeln von 3,18 cm (1,25'') Durchmesser ausgerüstet. Der Flüssigkeitsstand
im Reaktor wurde mithilfe eines Füllstandsanzeigers überwacht, ähnlich dem
vorstehend in Beispiel 5 beschriebenen. Es wurde eine Innenkühlschlange
verwendet, um die Temperatur innerhalb des Reaktors während des
Reaktionsverlaufs zu regeln.
-
Während des
Betriebs wurde der Reaktor kontinuierlich mit einem Sauerstoffgasstrom
und einer wässrigen,
NPMIDA-haltigen Einspeisematerialaufschlämmung beschickt. Der Sauerstoff
wurde in das Reaktionsmedium durch eine in der Nähe des Rührwerks angeordnete Fritte
eingespeist. Ein flüssiger,
Glyphosat-haltiger Produktstrom wurde aus dem Reaktor kontinuierlich über eine
Fritte abgezogen, welche es erlaubte, jeden dem Reaktor zugeführten Katalysator
im Reaktionsmedium zurückzuhalten.
Der abgezogene flüssige
Produktstrom wurde dann In-line
mit einer basischen Lösung
gemischt, welche Glyphosat lösen
kann. Der Produktgasstrom (enthaltend CO2 und
nicht umgesetzten Sauerstoff) wurde kontinuierlich aus dem Kopfraum
des Reaktors abgelüftet.
-
Die
wässrige
Einspeisematerialaufschlämmung
umfasste NPMIDA (etwa 7,7 Gew.-%), Formaldehyd (etwa 3000 ppm des
Gewichts) und Ameisensäure
(etwa 6100 ppm des Gewichts). Der Katalysator wurde nach einem Verfahren
hergestellt, ähnlich
dem vorstehend in Beispiel 14 beschriebenen, und umfasste Platin (5
Gew.-%) und Zinn (1,0 Gew.-%) auf einem teilchenförmigen Kohlenstoffträger. Das
kontinuierliche Reaktorsystem wurde in einer Weise ähnlich der
vorstehend in Beispiel 5 beschriebenen, angefahren, indem der Reaktor
nach Chargenart angestellt und kurz danach mit der Flüssigkeitsströmung durch
das System begonnen wurde. Die Betriebsbedingungen sind nachstehend
in Tabelle 32 zusammengefasst. Die Analysenergebnisse der kontinuierlichen
Oxidation sind nachstehend in Tabelle 33 wiedergegeben.
-
Tabelle
32 Zusammenfassung
der Betriebsbedingungen von Beispiel 13
-
Tabelle
33 Oxidationsergebnisse
für Beispiel
13
-
Beispiel 14
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in Anwesenheit eines Pt/Sn/C-Katalysators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in Anwesenheit eines heterogenen teilchenförmigen Pt/Sn/C-Katalysators
in einem kontinuierlichen Reaktorsystem mit einer einzigen Reaktionszone.
-
Der
Versuch erfolgte in einem kontinuierlichen Reaktorsystem, ähnlich dem
vorstehend in Beispiel 11 beschriebenen, welches einen 500 ml Hastelloy
C Autoklav (Autoclave Engineers, Inc., Pittsburgh, PA) umfasste.
Der Reaktor war mit einem Rührwerk
mit einer Turbine mit 6 Laufschaufeln von 3,18 cm (1,25'') Durchmesser ausgerüstet. Der Flüssigkeitsstand
im Reaktor wurde mithilfe eines Füllstandsanzeigers überwacht, ähnlich dem
vorstehend in Beispiel 5 beschriebenen. Es wurde eine Innenkühlschlange
verwendet, um die Temperatur innerhalb des Reaktors während des
Reaktionsverlaufs zu regeln.
-
Während des
Betriebs wurde der Reaktor kontinuierlich mit einem Sauerstoffgasstrom
und einer wässrigen,
NPMIDA-haltigen Einspeisematerialaufschlämmung beschickt. Der Sauerstoff
wurde in das Reaktionsmedium durch eine in der Nähe des Rührwerks angeordnete Fritte
eingespeist. Ein flüssiger,
Glyphosat-haltiger Produktstrom wurde aus dem Reaktor kontinuierlich über eine
Fritte abgezogen, welche es erlaubte, jeden dem Reaktor zugeführten Katalysator
im Reaktionsmedium zurückzuhalten.
Der (CO2 und nicht umgesetzten Sauerstoff
enthal tende) Produktgasstrom wurde kontinuierlich aus dem Kopfraum
des Reaktors abgelüftet.
-
Das
kontinuierliche Reaktorsystem wurde in einer Weise ähnlich der
vorstehend in Beispiel 5 beschriebenen, angefahren, indem der Reaktor
nach Chargenart angestellt und kurz danach mit der Flüssigkeitsströmung durch
das System begonnen wurde. Die wässrige
Einspeisematerialaufschlämmung
umfasste NPMIDA (etwa 2,9 Gew.-%). Der Katalysator wurde nach einem
Verfahren hergestellt, ähnlich
dem vorstehend in Beispiel 14 beschriebenen, und umfasste Platin
(5 Gew.-%) und Zinn (1,0 Gew.-%) auf einem teilchenförmigen Kohlenstoffträger. Die
Betriebsbedingungen sind nachstehend in Tabelle 34 zusammengefasst.
Die Analysenergebnisse der kontinuierlichen Oxidation sind nachstehend
in Tabelle 35 wiedergegeben.
-
Tabelle
34 Zusammenfassung
der Betriebsbedingungen von Beispiel 14
-
Tabelle
35 Oxidationsergebnisse
für Beispiel
14
-
-
Beispiel 15
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in Anwesenheit eines Pt/Sn/C-Katalysators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in Anwesenheit eines heterogenen teilchenförmigen Pt/Sn/C-Katalysators.
Der Versuch war darauf ausgelegt die Auswirkung von Druck, Flüssigkeits- und Gasströmung auf
die Umwandlung von NPMIDA zu Glyphosat zu veranschaulichen.
-
Der
Versuch erfolgte in einem kontinuierlichen Reaktorsystem, ähnlich dem
vorstehend in Beispiel 14 beschriebenen, welches einen 500 ml Hastelloy
C Autoklav (Autoclave Engineers) umfasste. Der Reaktor war mit einem
Rührwerk
mit einer Turbine mit 6 Laufschaufeln von 3,18 cm (1,25'') Durchmesser ausgerüstet. Der Flüssigkeitsstand
im Reaktor wurde mithilfe eines Füllstandsanzeigers überwacht, ähnlich dem
vorstehend in Beispiel 5 beschriebenen. Es wurde eine Innenkühlschlange
verwendet, um die Temperatur innerhalb des Reaktors während des
Reaktionsverlaufs zu regeln.
-
Während des
Betriebs wurde der Reaktor kontinuierlich mit einem Sauerstoffgasstrom
und einer wässrigen,
NPMIDA-haltigen Einspeisematerialaufschlämmung beschickt. Der Sauerstoff
wurde in das Reaktionsmedium durch eine in der Nähe des Rührwerks angeordnete Fritte
eingespeist. Ein flüssiger,
Glyphosat-haltiger Produktstrom wurde aus dem Reaktor kontinuierlich über eine
Fritte abgezogen, welche es erlaubte, jeden dem Reaktor zugeführten Katalysator
im Reaktionsmedium zurückzuhalten.
Der Produktgasstrom (enthaltend CO2 und
nicht umgesetzten Sauerstoff) wurde kontinuierlich aus dem Kopfraum
des Reaktors abgelüftet.
-
Die
wässrige
Einspeisematerialaufschlämmung
umfasste NPMIDA (etwa 3,0 Gew.-%), Formaldehyd (etwa 1000 ppm des
Gewichts) und Ameisensäure
(etwa 5100 ppm des Gewichts). Der Katalysator wurde nach einem Verfahren
hergestellt, ähnlich
dem nachstehend beschriebenen, und umfasste Platin (5 Gew.-%) und
Zinn (1,0 Gew.-%) auf einem teilchenförmigen Kohlenstoffträger:
-
Abscheidung eines Zinn-Aktivators
auf einem Kohlenstoffträger.
-
Aktivkohle
(20 g) wurde in etwa 2 l Wasser aufgeschlämmt. Als Nächstes wurde 0,39 g SnCl2·2H2O in 500 g 0,5% HNO3 gelöst. Nach
Zugabe der gesamten Lösung
wurde die Aufschlämmung
2 h gerührt.
Dann wurde der pH auf 9,5 eingestellt und die Aufschlämmung noch
ein paar Stunden gerührt.
Als Nächstes
wurde die Aufschlämmung
filtriert und der Rückstand
bis zum Erreichen einer konstanten Leitfähigkeit mit viel Wasser gewaschen.
Der Nasskuchen wurde bei 125°C
unter Vakuum getrocknet und lieferte 1% Zinn auf Kohlenstoff. Nach
dem Trocknen wurde das Material aus 1% Zinn auf Kohlenstoff 6 h
in Argon bei 500°C
gebrannt.
-
Zur
Abscheidung von Platin auf dem Kohlenstoffträger wurde zuerst 5 g des Materials
aus 1% Zinn auf Kohlenstoff in etwa 500 ml Wasser aufgeschlämmt. Dann
wurde 0,705 g H2PtCl6 in
etwa 125 ml Wasser gelöst und
tropfenweise zugegeben. Nachdem die gesamte H2PtCl6-Lösung
zugegeben worden war, wurde die Aufschlämmung 2,5 h gerührt. Dann
wurde der pH mit verdünntem
NaOH auf 9,5 eingestellt und noch einige Stunden weitergerührt. Anschließend wurde
die Aufschlämmung
filtriert und der Rückstand
bis zum Erreichen einer konstanten Leitfähigkeit mit viel Wasser gewaschen.
Der Nasskuchen wurde bei 125°C
unter Vakuum getrocknet.
-
Diese
Verfahren lieferte einen Katalysator, welcher 5% Platin und 1% Zinn
auf Kohlenstoff umfasste.
-
Das
kontinuierliche Reaktorsystem wurde in einer Weise, ähnlich der
vorstehend in Beispiel 5 beschriebenen, angefahren, indem der Reaktor
nach Chargenart angestellt und kurz danach mit der Flüssigkeitsströmung durch
das System begonnen wurde. Während
des Versuchsverlaufs wurde die Sauerstoffströ mungsrate über den Bereich von 75 bis
300 sccm hoch- und zurückgefahren.
Die Betriebsbedingungen sind nachstehend in Tabelle 36 zusammengefasst.
Die Analysenergebnisse der kontinuierlichen Oxidation sind nachstehend
in Tabelle 37 wiedergegeben.
-
Tabelle
36 Zusammenfassung
der Betriebsbedingungen von Beispiel 15
-
-
-
-
-
-
-
Beispiel 16
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in Anwesenheit eines Pt/Fe/C-Katalysators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat unter Verwendung eines frischen heterogenen teilchenförmigen Pt/Fe/C-Katalysators
in einem Rührbehälterreaktor. Der
Versuch war darauf ausgelegt, die Bedingungen zu simulieren, welche
in der ersten Reaktionszone eines kontinuierlichen Oxydationsreaktorsystems
auftreten könnten.
-
Der
Versuch erfolgte in einem kontinuierlichen Reaktorsystem, ähnlich dem
vorstehend in Beispiel 13 beschriebenen, welches einen 500 ml Hastelloy
C Autoklav umfasste. Der Reaktor war mit einem Rührwerk mit einer Turbine mit
6 Laufschaufeln von 3,18 cm (1,25'')
Durchmesser ausgerüstet.
Der Flüssigkeitsstand
im Reaktor wurde mithilfe eines Füllstandsanzeigers überwacht, ähnlich dem
vorstehend in Beispiel 5 beschriebenen. Es wurde eine Innenkühlschlange
verwendet, um die Temperatur innerhalb des Reaktors während des Reaktionsverlaufs
zu regeln.
-
Während des
Betriebs wurde der Reaktor kontinuierlich mit einer wässrigen,
NPMIDA-haltigen Einspeisematerialaufschlämmung und einem Sauerstoffgasstrom
beschickt. Der Sauerstoff wurde in das Reaktionsmedium durch eine
in der Nähe
des Rührwerks
angeordnete Fritte eingespeist. Ein flüssiger, Glyphosat-haltiger
Produktstrom wurde aus dem Reaktor kontinuierlich über eine
Fritte abgezogen, welche es erlaubte, jeden dem Reaktor zugeführten Katalysator
im Reaktionsmedium zurückzuhalten.
Der Produktgasstrom (enthaltend CO2 und
nicht umgesetzten Sauerstoff) wurde kontinuierlich aus dem Kopfraum
des Reaktors abgelüftet.
-
Die
in den Reaktor eingespeiste wässrige
Einspeisematerialaufschlämmung
umfasste NPMIDA (etwa 9,9 Gew.-%), Glyphosat (1,3 Gew.-%), Formaldehyd
(3600 ppm des Gewichts) und Ameisensäure (etwa 6200 ppm des Gewichts).
Der Katalysator wurde nach einem Verfahren hergestellt, ähnlich dem
vorstehend für
Beispiel 1 beschriebenen, und umfasste Platin (5 Gew.-%) und Eisen
(0,5 Gew.-%) auf einem teilchenförmigen Kohlenstoffträger. Das
kontinuierliche Reaktorsystem wurde in einer Weise angefahren, ähnlich der
vorstehend in Beispiel 5 beschriebenen, indem der Reaktor nach Chargenart
angestellt und kurz danach mit der Flüssigkeitsströmung durch
das System begonnen wurde.
-
Die
Betriebsbedingungen sind nachstehend in Tabelle 38 zusammengefasst.
Die Analysenergebnisse der kontinuierlichen Oxidation sind nachstehend
in Tabelle 39 wiedergegeben.
-
Tabelle
38 Zusammenfassung
der Betriebsbedingungen von Beispiel 16
-
Tabelle
39 Oxidationsergebnisse
von Beispiel 16
-
Beispiel 17
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in Anwesenheit eines Pt/Fe/C-Katalysators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in Anwesenheit eines heterogenen teilchenförmigen Pt/Fe/C-Katalysators
in einem Rührbehälterreaktor.
Der Versuch war darauf ausgelegt, die Bedingungen zu simulieren,
welche in einer zweiten Reaktionszone eines kontinuierlichen Reaktorsystems
auftreten könnten.
-
Der
Versuch erfolgte in einem kontinuierlichen Reaktorsystem, ähnlich dem
vorstehend in Beispiel 13 beschriebenen, welches einen 1000 ml Hastelloy
C Autoklav umfasste. Der Reaktor war mit einem Rührwerk mit einer Turbine mit
6 Laufschaufeln von 3,18 cm (1,25'')
Durchmesser ausgerüstet.
Der Flüssigkeitsstand
im Reaktor wurde mithilfe eines Füllstandsanzeigers überwacht, ähnlich dem
vorstehend in Beispiel 5 beschriebenen. Es wurde eine Innenkühlschlange
verwendet, um die Temperatur innerhalb des Reaktors während des Reaktionsverlaufs
zu regeln.
-
Während des
Betriebs wurde der Reaktor kontinuierlich mit einer wässrigen,
NPMIDA-haltigen Einspeisematerialaufschlämmung und einem Sauerstoffgasstrom
beschickt. Der Sauerstoff wurde in das Reaktionsmedium durch eine
in der Nähe
des Rührwerks
angeordnete Fritte eingespeist. Ein flüssiger, Glyphosat-haltiger
Produktstrom wurde aus dem Reaktor kontinuierlich über eine
Fritte abgezogen, welche es erlaubte, jeden dem Reaktor zugeführten Katalysator
im Reaktionsmedium zurückzuhalten.
Der Produktgasstrom (enthaltend CO2 und
nicht umgesetzten Sauerstoff) wurde kontinuierlich aus dem Kopfraum
des Reaktors abgelüftet.
-
Die
wässrige
Einspeisematerialaufschlämmung
enthielt NPMIDA (1,9 Gew.-%), Glyphosat (6,7 Gew.-%), Formaldehyd
(2400 ppm des Gewichts) Ameisensäure
(4600 ppm des Gewichts), NMG (280 ppm des Gewichts), AMPA (400 ppm
des Gewichts) und MAMPA (200 ppm des Gewichts). Der Katalysator
wurde nach einem Verfahren hergestellt, ähnlich dem vorstehend in Beispiel
1 beschriebenen, und umfasste Platin (5 Gew.-%) und Eisen (0,5 Gew.-%)
auf einem teilchenförmigen
Kohlenstoffträger.
Das kontinuierliche Reaktorsystem wurde in einer Weise in Betrieb
genommen, ähnlich
der vorstehend in Beispiel 5 beschriebenen, indem der Reaktor nach
Chargenart angefahren und kurz danach die Flüssigkeitsströmung durch
das System angestellt wurde. Die Betriebsbedingungen sind nachstehend
in Tabelle 40 zusammengefasst. Das Flüssigprodukt wurde mittels HPLC
analysiert und die Analysenergebnisse der kontinuierlichen Oxidation
sind nachstehend in Tabelle 41 wiedergegeben.
-
Tabelle
40 Zusammenfassung
der Betriebsbedingungen von Beispiel 17
-
Tabelle
41 Oxidationsergebnisse
für Beispiel
17
-
-
Beispiel 18
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in Anwesenheit eines Pt/Fe/C-Katalysators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in Anwesenheit eines heterogenen teilchenförmigen Pt/Fe/C-Katalysators
in einem Festbettreaktor. Der Versuch war darauf ausgelegt einen
kleinen anfänglichen
Abschnitt eines Festbettreaktors zu simulieren, in den eine gasförmige und
flüssige
Einspeisung im Gleichstrom eintritt.
-
Der
Versuch erfolgte in einem kontinuierlichen Reaktorsystem, welches
einen vertikalen Rohrreaktors aus Edelstahl (2,2 cm Innendurchmesser;
61,5 cm Länge;
215 ml Volumen) umfasste. Die gasförmige und flüssige Einsspeisung
trat oben in den Rohrreaktor ein und strömte durch den Reaktor nach
unten. Der Reaktor war mit einer Mischung aus Katalysator (50 g)
und Raschig-Ringen aus Glas (6 mm) gefüllt. Der Katalysator umfasste
2 Gew.-% Platin und 0,2 Gew.-% Eisen auf einem granulären 3,28
mm (1/8-inch) Kohlenstoffträger. Der
Reaktor wurde mit warmem Wasser auf etwa 90°C erwärmt und mit Stickstoff mit
einem Druck von etwa 690 kPa (100 psig) beaufschlagt. Nachdem der
Reaktor eine Temperatur von 90°C
erreicht hatte, wurde der Wasser- und Stickstoffstrom abgestellt
und mit der Flüssigkeits-
und Sauerstoffeinspeisung begonnen.
-
Die
flüssige
Einspeisung wurde oben auf den Reaktor bei 90°C aufgegeben und umfasste eine
wässrige
Einspeisematerialaufschlämmung,
welche NPMIDA (3,00 Gew.-%) und Ameisensäure (0,54 Gew.-%) enthielt.
Der Sauerstoff wurde oben in den Reaktor unter Aufrechterhaltung
eines Drucks von 690 kPa (100 psig) eingespeist. Die Flüssigkeits-
und Sauerstoffeinspeiseraten wurden in einer Reihe von 4 Versuchen
variiert, wie nachstehend in Tabelle 42 angegeben. Bei jedem Versuch
ließ man
sich das Gleichgewicht im System mindestens eine halbe Stunde einstellen,
ehe am Säulenausfluss
Proben abgenommen und auf Ameisensäure und Glyphosat analysiert
wurden.
-
Tabelle
42 Ausflussanalyse
unter verschiedenen Betriebsbedingungen für Beispiel 18
-
Beispiel 19
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in zwei in Reihe angeordneten Rührbehälterreaktoren
mir Katalysatorrückführung und
Kristallisatorrückführung
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in Anwesenheit einer heterogenen teilchenförmigen Katalysatoraufschlämmung in
einem kontinuierlichen Reaktorsystem, welches zwei Rührbehälterreaktoren
in Reihe umfasst. Die zwei Rührbehälterreaktoren
(R1 und R2) entsprachen den in Beispiel 8 beschriebenen, außer dass
die Rührwerksanordnung
nicht nach DISPERSIMAX-Art betrieben wurde. Der aus dem Reaktionsmi schungsauslass
von R2 abgezogene Katalysator wurde kontinuierlich unter Verwendung
eines Rückspülfiltersystems
filtriert, welches parallele Filterkörper umfasste und der abgetrennte
Katalysator zu R1 zurückgeführt. Das
kristalline N-(Phosphonomethyl)glycinprodukt wurde aus dem Filtrat
in einen Kristallisator (30 L) gewonnen und die Mutterlauge aus
dem Kristallisator zu R1 zurückgeführt.
-
Der
Katalysator wurde nach einem Verfahren hergestellt, ähnlich dem
vorstehend in Beispiel 1 beschriebenen, und umfasste Platin (5 Gew.-%)
und Eisen (0,5 Gew.-%) auf einem teilchenförmigen Kohlenstoffträger. Bei
diesem Beispiel wurde der teilchenförmige heterogene Katalysator
mit dem Ausfluss aus R1 über R1
nach R2 transportiert, einschließlich einer geringen Menge
eingeschlossenen Gases. Der mit dem Reaktorausfluss aus R2 ausgetragene
Katalysator wurde im Rückspülfilter
abgetrennt und zu R1 zurückgeführt. Der Rückspülkatalysatorfilter
wirkte auch als Flüssigkeits/Gas-Trennvorrichtung
für den
Ausfluss aus R2. Das Filtrat aus dem Rückspülkatalysatorfilter wurde zur
Gewinnung von kristallinem N-(Phosphonomethyl)glycinprodukt zu einem
Kristallisator weitergeleitet. Die aus dem Kristallisator resultierende
Mutterlauge wurde dazu verwendet, die Katalysatorfilterkörper zurückzuspülen und
wurde mit dem abgetrennten Katalysator zu R1 zurückgeführt.
-
Die
Betriebsbedingungen für
R1 und R2 sind in Tabelle 43 zusammengefasst. R1 und R2 wurden anfänglich wie
in Tabelle 43 angegeben, beschickt und Sauerstoff im Gleichstrom
mit der NPMIDA-Einspeisung eingeleitet. Die NPMIDA-Einspeisung umfasste
eine wässrige
Einspeisematerialaufschlämmung,
welche etwa 12,5% bis etwa 15% NPMIDA sowie Muterlauge aus dem Rückfluss
des Katalysatorfilters enthielt, was eine wirksame vereinigte Einspeisung
in R1 ergab. Die wirksame vereinigte Einspeisung wurde anfänglich mit
4,3 Gew.-% eingeführt
und später
auf 5,2 Gew.-% erhöht.
Während
des gesamten Laufs wurde Wismutoxid zugegeben, um die die Ameisensäureabbaurate
zu erhöhen.
Wismutoxid wurde diskontinuierlich zu R1 (~5 mg pro Zugabe) zugegeben
und auch kontinuierlich durch Zugabe zur NPMIDA-Einspeiseaufschlämmung (4–25 mg pro
20 kg NPMIDA-Aufschlämmung).
Die der Einspeiseaufschlämmung
zugesetzte Häufigkeit
und Menge Wismutoxid ist in Tabelle 44 aufgeführt. Die wässrige Einspeiseaufschlämmung in
R1 (einschließend
die Komponenten aus der mit dem Katalysator zurückgeführten Kristallisatormutterlauge),
der Reaktorsauslauf aus R1 und der Reaktorauslauf aus R2 wurden
mittels HPLC analysiert. Die HPLC-Analysenwerte sind in Tabelle
45 wiedergegeben. Tabelle
43 Betriebsbedingungen
für das
kontinuierliche Oxidationsreaktorsystem von Beispiel 19
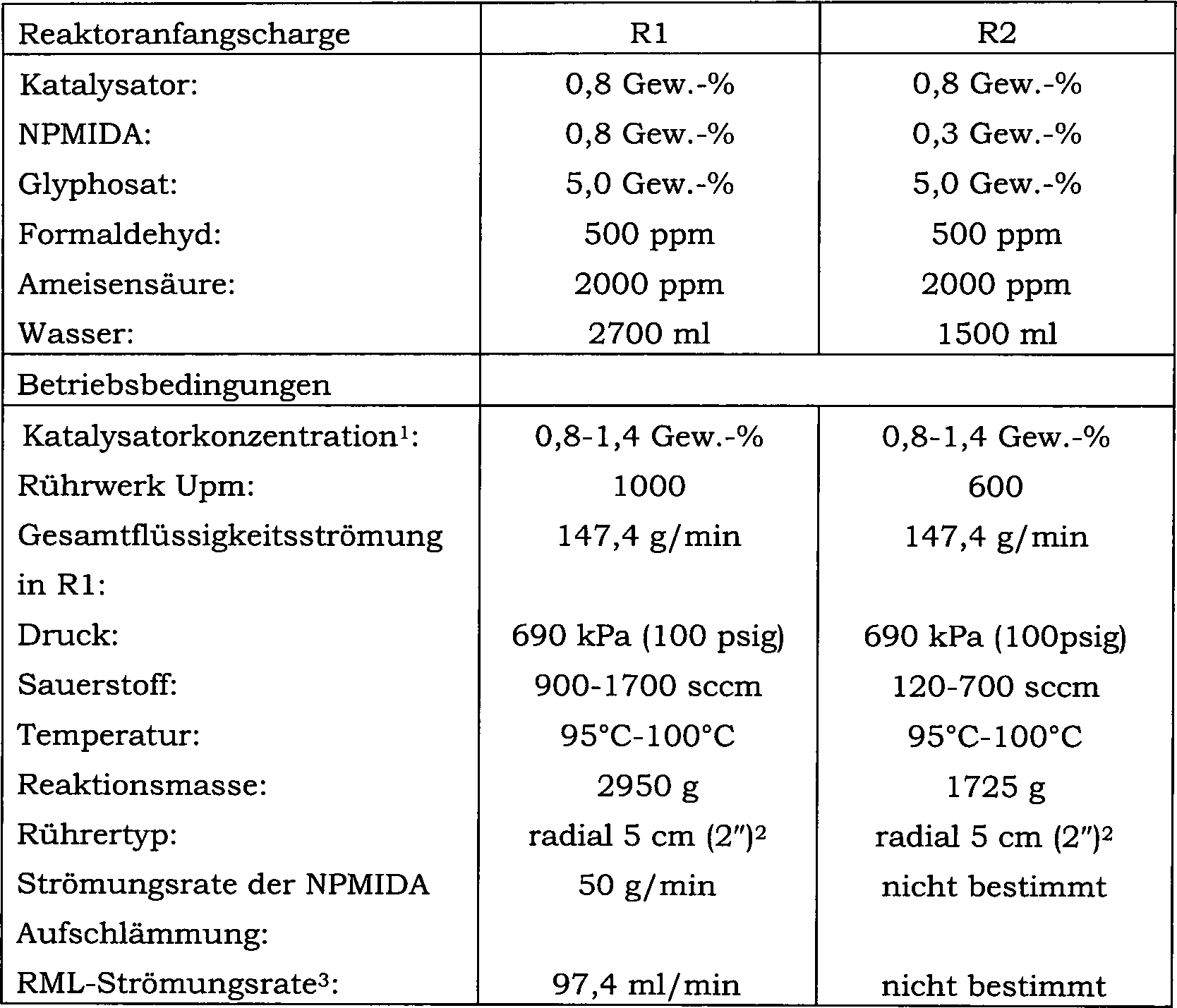
- 1. Die Katalysatorscharge betrug anfänglich 0,8
Gew.-%. Während
des Laufs wurde die Katalysatorbeladung nach 69 h auf 1,0 Gew.-%,
nach 119 h auf 1,2 Gew.-% und nach 143 h auf 1,4 Gew.-% gesteigert.
- 2. Es wurde ein stromabwärts
fördernder
Flügelrührer auf
der Rührwelle
etwa in der Mitte der Flüssigkeitssäule installiert.
- 3. Die Kristallisatormutterlauge (RML) wurde dazu verwendet,
den Katalysatorfilter rückzuspülen und
den abfiltrierten Katalysator mit RML zu R1 zurückzufördern.
-
Tabelle
44 Häufigkeit
und Menge der Wismutoxidzugabe zum Reaktorsystem
-
-
-
-
-
-
-
Beispiel 20
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in zwei in Reihe angeordneten Rührbehälterreaktoren
mir Katalysatorrückführung und
Kristallisatorrückführung
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in einem kontinuierlichen Reaktorsystem, welches zwei
Rührbehälterreaktoren
in Reihe umfasst, unter Verwendung einer heterogenen teilchenförmigen Katalysatoraufschlämmung. Das
Reaktorsystem war dem in 31 dargestellten ähnlich.
Die zwei Rührbehälterreaktoren
(R1 und R2) entsprachen den im vorstehenden Beispiel 19 beschriebenen.
Der aus dem Reaktionsmischungsauslass von R2 abgezogene Katalysator
wurde kontinuierlich unter Verwendung eines Rückspülfiltersystems filtriert, welches
parallele Filterkörper
umfasste, und der abgetrennte Katalysator zu R1 zurückgeführt. Das
kristalline N-(Phosphonomethyl)glycinprodukt wurde aus dem Filtrat
in einen Kristallisator (30 L) gewonnen und die Mutterlauge aus
dem Kristallisator zu R1 zurückgeführt. Zusätzlich wurde
ein Teil der Mutterlauge aus dem Kristallisator dem Ausfluss aus
R2 zugesetzt, um einen verdünnten
Ausflusstrom (75–100
ml/min) zu bilden und die Glyphosatkonzentration im R2-Abfluss bei
der Einführung
in den Rückspülfilter
zu verringern, um mögliche
Kristallisationsprobleme zu reduzieren. Das Reaktionssystem umfasste
weiterhin einen Katalysatorpufferbehälter (500 ml Hastelloy C Autoklav
mit aufwärts
pumpendem Flügelrührer), welcher
den abgetrennten Katalysator vor seine Wiedereinführung in
R1 sammelte. Der Katalysatorpufferbehälter wurde ohne Füllstandkontrolle
betrieben und die Katalysatoraufschlämmung am oberen Ende abfließen lassen.
-
Der
heterogene teilchenförmige
Katalysator wurde nach einem Verfahren hergestellt, ähnlich dem
vorstehend in Beispiel 1 beschriebenen, und umfasste Platin (5 Gew.-%)
und Eisen (0,5 Gew.-%) auf einem teilchenförmigen Kohlenstoff träger. Bei
diesem Beispiel wurde der teilchenförmige heterogene Katalysator
mit dem Ausfluss aus R1 über
R1 nach R2 transportiert, einschließlich einer geringen eingeschlossenen
Gasmenge. Der mit dem Reaktorausfluss aus R2 ausgetragene Katalysator
wurde im Rückspülfilter
abgetrennt und zum Katalysatorpufferbehälter weitergeleitet und zu
R1 zurückgeführt. Der
Rückspülkatalysatorfilter
wirkte auch als Flüssigkeits/Gas-Trennvorrichtung
für den
Ausfluss aus R2. Das Filtrat aus dem Rückspülkatalysatorfilter wurde zur
Gewinnung von kristallinem N-(Phosphonomethyl)glycinprodukt zu einem
Kristallisator weitergeleitet. Die aus dem Kristallisator resultierende
Mutterlauge wurde zum Zurück spülen der
Katalysatorfilterkörper,
als Verdünnungsmittel
für den
Ausfluss R2, welcher den Katalysatorfilterkörper durchströmt und zur Rückführung des
abgetrennten Katalysator R1 verwendet.
-
Die
Betriebsbedingungen für
R1 und R2 sind in Tabelle 46 zusammengefasst. R1 und R2 wurden anfänglich wie
in Tabelle 46 angegeben, beschickt und Sauerstoff im Gleichstrom
mit der NPMIDA-Einspeisung eingeleitet. Die NPMIDA-Einspeisung umfasste
eine wässrige
Einspeisematerialaufschlämmung,
welche etwa 20% bis etwa 20,5% NPMIDA sowie Muterlauge aus dem Rückfluss
des Katalysatorfilters enthielt, was eine wirksame vereinigte Einspeisung
in R1 ergab. Die wirksame vereinigte Einspeisung wurde anfänglich mit
7 Gew.-% eingeführt
und später
auf 7,7 Gew.-% erhöht.
Während
des gesamten Laufs wurde Wismutoxid zugegeben, um die Geschwindigkeit
des Ameisensäureabbaus
zu erhöhen.
Wismutoxid wurde zur NPMIDA-Einspeiseaufschlämmung (3–12 mg pro 20 kg NPMIDA-Aufschlämmung) kontinuierlich
zugegeben. Die der Einspeiseaufschlämmung zugesetzte Häufigkeit
und Menge Wismutoxid ist in Tabelle 47 aufgeführt. Die wässrige Einspeiseaufschlämmung in
R1 (einschließend
die Komponenten aus der mit dem Katalysator zurückgeführten Kristallisatormutterlauge),
der Reaktorsauslauf aus R1 und der Reaktorauslauf aus R2 wurden
mittels HPLC analysiert. Die HPLC-Analysenwerte sind in Tabelle
48 wiedergegeben. Tabelle
46 Betriebsbedingungen
für Beispiel
20
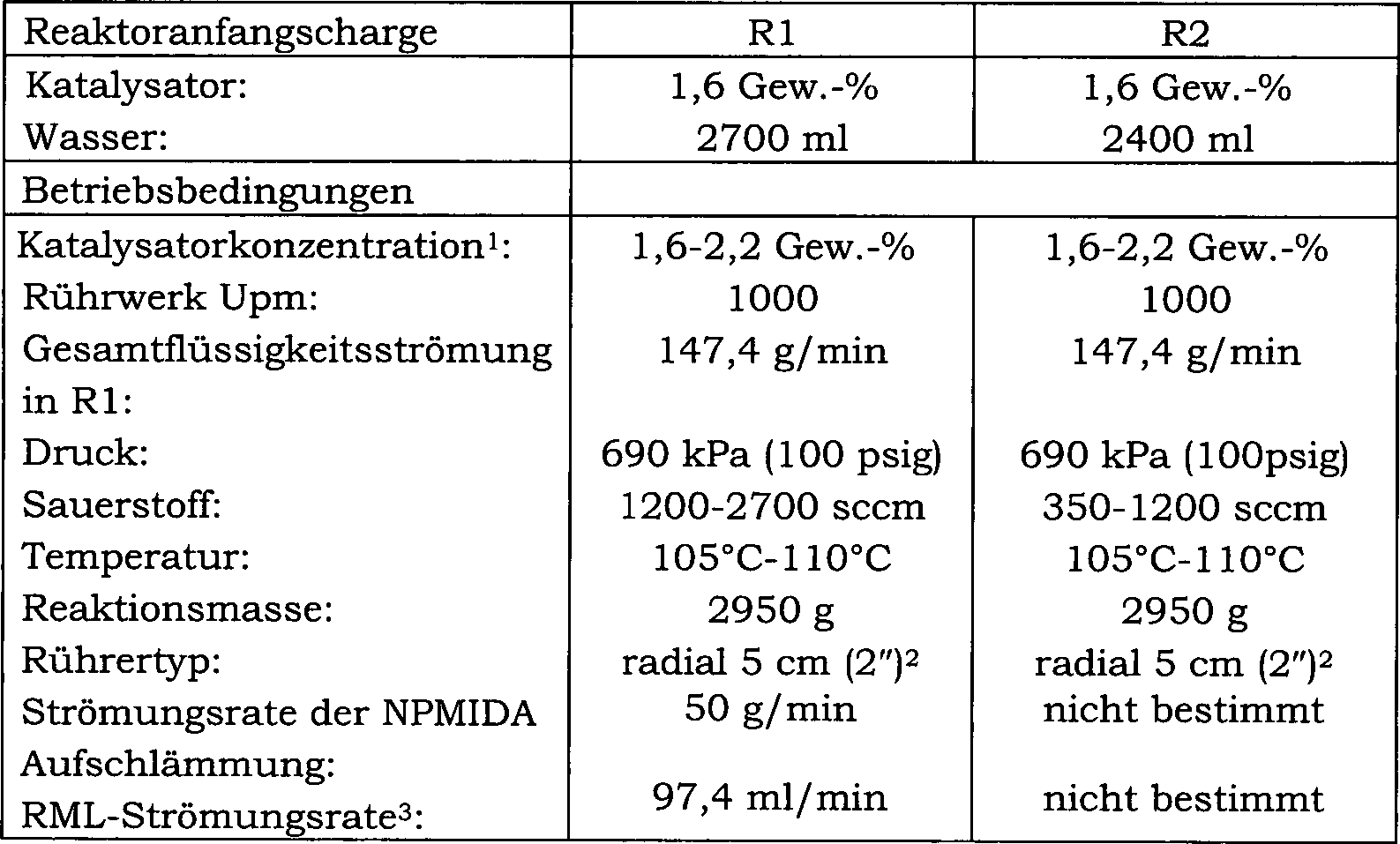
- 1. Die Katalysatorscharge betrug anfänglich 0,8
Gew.-%. Während
des Laufs wurde die Katalysatorbeladung nach 69 h auf 1,0 Gew.-%,
nach 119 h auf 1,2 Gew.-% und nach 143 h auf 1,4 Gew.-% gesteigert.
- 2. Es wurde ein stromabwärts
fördernder
Flügelrührer auf
der Rührwelle
etwa in der Mitte der Flüssigkeitssäule installiert.
- 3. Die Kristallisatormutterlauge (RML) wurde dazu verwendet,
den Katalysatorfilter zurückzuspülen und
den abfiltrierten Katalysator mit RML zu R1 zurückzufördern
-
Tabelle
47 Häufigkeit
und Menge der Wismutoxidzugabe zur NPMIDA-Einspeiseaufschlämmung
-
-
-
-
-
-
-
Beispiel 21
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in einem Festbettreaktor unter Verwendung
eines Pt/Fe/C-Katalysators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in Anwesenheit eines heterogenen teilchenförmigen Pt/Fe/C-Katalysators in einem
Festbettreaktor. Der Versuch war darauf ausgelegt, einen kleinen
anfänglichen
Abschnitt eines Festbettreaktors bei gasförmiger und flüssiger Einspeisung
im Gleichstrom zu simulieren.
-
Der
Versuch erfolgte in einem kontinuierlichen Reaktorsystem, welches
einen vertikalen Rohrreaktors aus Edelstahl (1,56 cm ID; 60,5 cm
Länge;
116 ml Volumen) umfasste. Die gasförmige und flüssige Einsspeisung
trat oben in den Rohrreaktor ein und strömte durch den Reaktor nach
unten. Der Reaktor enthielt einen Pt/Fe/C-Katalysator (42,38), welcher
Platin (2 Gew.-%) und Eisen (0,2 Gew.-%) auf einem extrudierten
Kohlenstoffträger
mit 1,2 mm Durchmesser umfasste, dessen Länge von etwa 1 mm bis etwa
9 mm reichte. Der Reaktor wurde mit warmem Wasser auf etwa 90°C erwärmt und
mit Stickstoff ein Druck von etwa 1034 kPa (150 psig) aufgebracht.
Nachdem der Reaktor eine Temperatur von 90°C erreicht hatte, wurde der
Wasser- und Stickstoffstrom abgestellt und mit der Flüssigkeits-
und Sauerstoffeinspeisung begonnen.
-
Die
flüssige
Einspeisung (50 ml/min) umfasste eine wässrige Einspeisematerialaufschlämmung, welche
NPMIDA (1,92 Gew.-%), Glyphosat (1,89 Gew.-%), Ameisensäure (0,26
Gew.-%) und Formaldehyd (0,15 Gew.-%) enthielt. Der Sauerstoff (200
sccm) wurde oben in den Reaktor unter Aufrechterhaltung eines Überdrucks
von 1034 kPa (150 psig) eingespeist. Nach 10 Tagen Dauerbetrieb
zeigte die Analyse des Reaktorprodukts 0,15% Formaldehyd, 0,26%
Ameisensäure
und 2,53% Glyphosat.
-
Beispiel 22
-
Kontinuierliche Oxidation
von NPMIDA zu Glyphosat in einem Festbettreaktor unter Verwendung
eines Pt/Fe/C-Katalysators
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA
zu Glyphosat in Anwesenheit eines heterogenen teilchenförmigen Pt/Fe/C-Katalysators
in einem Festbettreaktor bei stromaufwärts gerichtetem Gleichstrom
der gasförmigen
und flüssigen
Reaktanten.
-
Der
Versuch erfolgte in einem kontinuierlichen Oxidationsreaktorsystem,
das einen, wie in Beispiel 21 beschriebenen, vertikalen Rohrreaktors
aus Edelstahl umfasste, außer
dass die Reaktanten die Reaktionszone nach oben durchströmten. Der
Reaktor enthielt einen Pt/Fe/C-Katalysator (42,3 g), welcher Platin
(2 Gew.-%) und Eisen (0,2 Gew.-%) auf einem extrudierten Kohlenstoffträger mit
1,2 mm Durchmesser umfasste, dessen Länge von etwa 1 mm bis etwa
9 mm reichte. Der Reaktor wurde mit warmem Wasser auf etwa 90°C erwärmt und
mit Stickstoff ein Druck von etwa 1034 kPa (150 psig) aufgebracht
Nachdem der Reaktor eine Temperatur von 90°C erreicht hatte, wurde der
Wasser- und Stickstoffstrom abgestellt und mit der Flüssigkeits- und
Sauerstoffeinspeisung begonnen.
-
Die
flüssige
Einspeisung (50 ml/min) umfasste eine wässrige Einspeisematerialaufschlämmung, welche
NPMIDA (1,80 Gew.-%), Glyphosat (2,19 Gew.-%), Ameisensäure (0,26
Gew.-%) und Formaldehyd (0,14 Gew.-%) enthielt, die am Boden des
Reaktors bei 90°C
eingespeist wurde. Der Sauerstoff (200 sccm) wurde unter Aufrechterhaltung
eines Überdrucks
von 1034 kPa (150 psig) am Boden in den Reaktor eingespeist. Nach
19 h kontinuierlichem Betrieb zeigte die Analyse des Reaktorprodukts
0,13% Formaldehyd, 0,16% Ameisensäure und 2,42% Glyphosat.
-
Beispiel 23
-
Kontinuierliche Oxidation
von NPMIDA-Kaliumsalz zu Glyphosat-Kaliumsalz in einem Festbettreaktor
unter Verwendung eines Pt/Fe/C-Katalysators bei stromaufwärts berichtetem
Gleichstrom der gasförmigen
und flüssigen
Reaktanten.
-
Dieses
Beispiel veranschaulicht die kontinuierliche Oxidation von NPMIDA-Kaliumsalz zu Glyphosat-Kaliumsalz
in Anwesenheit eines heterogenen teilchenförmigen Pt/Fe/C-Katalysators
in einem Festbettreaktor bei stromaufwärts gerichtetem Gleichstrom
der gasförmigen
und flüssigen
Reaktanten.
-
Der
Versuch erfolgte in einem kontinuierlichen Oxidationsreaktorsystem,
das einen, wie in Beispiel 21 beschriebenen, vertikalen Rohrreaktors
aus Edelstahl umfasste, außer
dass die Reaktanten die Reaktionszone nach oben durchströmten. Der
Reaktor enthielt einen Pt/Fe/C-Katalysator (42,38), welcher Platin
(2 Gew.-%) und Eisen (0,2 Gew.-%) auf einem extrudierten Kohlenstoffträger mit
1,2 mm Durchmesser umfasste, dessen Länge von etwa 1 mm bis etwa
9 mm reichte. Der Reaktor wurde mit warmem Wasser auf etwa 90°C erwärmt und
mit Stickstoff ein Überdruck
von etwa 1034 kPa (150 psig) aufgebracht. Nach Erreichen von 90°C wurde der
Wasser- und Stickstoffstrom abgestellt und mit der Flüssigkeits-
und Sauerstoffeinspeisung begonnen.
-
Die
flüssige
Einspeisung (50 ml/min) umfasste eine wässrige Einspeisematerialaufschlämmung, welche
NPMIDA als Kaliumsalz (22,9 Gew.-%), Glyphosat (0,09 Gew.-%), Ameisensäure (0,20
Gew.-%) und Formaldehyd (0,14 Gew.-%) enthielt, die am Boden des
Reaktors bei 90°C
eingespeist wurde. Der Sauerstoff (500 sccm) wurde am Boden in den
Reaktor unter Aufrechterhaltung eines Drucks von 1034 kPa (150 psig) eingespeist.
Die Analyse des Reaktorprodukts lieferte 0,35% Formaldehyd, 0,20%
Ameisensäure
und 1,56% Glyphosat-Kaliumsalz.
-
Beispiel 24
-
Vergleich des Pt/Fe-Katalysators
mit eine Mischung aus Pt/Fe- und Pt/Fe/Te-Katalysatoren
-
Dieses
Beispiel vergleicht die Umwandlung von NPMIDA zu Glyphosat in einem
kontinuierlichen Oxidationsreaktorsystem unter Verwendung eines
heterogenen teilchenförmigen
Pt/Fe-Katalysators mit der Umwandlung von NPMIDA zu Glyphosat in
einem kontinuierlichen Oxidationsreaktorsystem unter Verwendung
einer Kombination von heterogenen teilchenförmigen Pt/Fe- und Pt/Fe/Te-Katalysatoren.
-
Die
Reaktionen erfolgten in einem kontinuierlichen Reaktorsystem unter
Verwendung eines 2 l Hastelloy C Autoklav (Autoclave Enigneers Inc.,
Pittsburgh, PA). Der Reaktor war mit einem Rührwerk mit einer Turbine mit
6 Laufschaufeln von 3,18 cm (1,25'')
Durchmesser ausgerüstet,
das mit 1600 Upm betrieben wurde. Der Flüssigkeitsstand im Reaktor wurde
mithilfe eines Drexelbrook Univesal IIITMSmart
LevelTM mit einem Teflon-beschichteten Fühler überwacht.
Es wurde eine Innenkühlschlange
verwendet, um die Temperatur innerhalb des Reaktors während des
Reaktionsverlaufs zu regeln.
-
Beim
ersten Versuch wurde der Reaktor mit einem heterogenen teilchenförmigen Pt/Fe-Katalysator (2,18
g) und einer wässrigen
Einspeisematerialaufschlämmung
(1448 g) beschickt. Der Katalysator umfasste Platin (5 Gew.-%) und
Eisen (0,5 Gew.-%). Die wässrige
Einspeisematerialaufschlämmung
umfasste NPMIDA (3,5 Gew.-%), Glyphosat (1,5 Gew.-%), Formaldehyd
(1200 ppm des Gewichts) und Ameisensäure (2500 ppm des Gewichts).
Die Einspeisaufschlämmung
enthielt auch NaCl (580 ppm des Gewichts), um die NaCl-Verunreinigung
zu simulieren. Der Reaktor wurde mit Stickstoff unter einen Druck
von 690 kPa (100 psi) gesetzt und auf 100°C erwärmt. Nach Erreichen der Temperatur
wurde ohne irgendeine Flüssigkeitsströmung ein
kontinuierlicher Strom gasförmigen
Sauerstoffs in den Reaktor eingespeist. Nach 9 min wurde mit der
kontinuierlichen Einspeisung der Aufschlämmung mit einer Rate von 70,4
g/min begonnen und die Sauerstoffdurchfluss fortgesetzt, wie nachstehend
in Tabelle 49 beschrieben. Aus dem Reaktor wurde kontinuierlich
ein flüssiger,
das Glyphosatprodukt enthaltender, Produktstrom abgezogen und mittels
HPLC analysiert. Die Oxidationsergebnisse sind in Tabelle 49 ebenfalls
wiedergegeben.
-
Beim
zweiten Versuch wurde der Reaktor mit einem heterogenen teilchenförmigen Pt/Fe-Katalysator (1,09
g), einem heterogenen teilchenförmigen
Pt/Fe/Te-Katalysator
(1,09 g) und einer wässrigen
Einspeisematerialaufschlämmung
(1455 g) beschickt. Der Pt/Fe-Katalysator umfasste Platin (5 Gew.-%)
und Eisen (0,5 Gew.-%), und der Pt/Fe/Te-Katalysator umfasste Platin
(5 Gew.-%) und Eisen (0,5 Gew.-%), sowie Tellur (0,2 Gew.-%). Die
wässrige
Einspeisematerialaufschlämmung
umfasste NPMIDA (3,5 Gew.-%), Glyphosat (1,5 Gew.-%), Formaldehyd
(1200 ppm des Gewichts) und Ameisensäure (2500 ppm des Gewichts).
Die Einspeisaufschlämmung
enthielt auch NaCl (580 ppm des Gewichts), um die Verunreinigung
mit NaCl zu simulieren.
-
Der
Reaktor wurde mit Stickstoff unter einen Druck von 690 kPa (100
psi) gesetzt und auf 100°C
erwärmt.
Nach Erreichen der Temperatur wurde ohne irgendeine Flüssigkeitsströmung ein
kontinuierlicher Strom gasförmigen
Sauerstoffs in den Reaktor eingespeist. Nach 19 min wurde mit der
kontinuierlichen Einspeisung der Aufschlämmung mit einer Rate von 70,4
g/min begonnen und die Sauerstoffdurchfluss fortgesetzt, wie nachstehend
in Tabelle 50 beschrieben. Aus dem Reaktor wurde kontinuierlich
ein flüssiger,
das Glyphosatprodukt enthaltender, Produktstrom abgezogen und mittels
HPLC analysiert. Die Oxidationsergebnisse sind in Tabelle 50 ebenfalls
wiedergegeben.
-
-