-
TECHNISCHES
GEBIET
-
Diese
Erfindung ist eine pharmazeutische eine oder mehrere 2-Thienyl-imidazolo[4,5]pyridin-Verbindungen
enthaltende Zusammensetzung, die bei der Behandlung von Viren wirksam
ist. Die Zusammensetzung kann zum Behandeln von Virusinfektionen,
insbesondere Hepatitis, einschließlich des Hepatitis-C-Virus (HCV),
des Hepatitis-B-Virus (HBV), des menschlichen Immunschwächesyndroms
(HIV) und des Kaposi-Sarkom-Virus
verwendet werden.
-
ALLGEMEINER
STAND DER TECHNIK
-
HIV
und andere Virusinfektionen wie Hepatitis sind einige der Hauptursachen
von Todesfällen.
HIV ist das Virus, von dem bekannt ist, dass es das erworbene Immunschwächesyndrom
(AIDS) beim Menschen hervorruft. HIV ist eine Krankheit, bei der
ein Virus im Körper
oder in Wirtszellen repliziert wird. Das Virus greift das Immunsystem
des Körpers
an.
-
Mehrere
Medikamente sind zur Behandlung dieser verheerenden Krankheit zugelassen,
einschließlich
Azidovudin (AZT), Didanosin (Dedeoxyinosin, ddI), d4T, Zalcitabin
(Dideoxycytosin, ddC), Nevirapin, Lamivudin (Epivir, 3TC), Saquinavir
(Invirase), Ritonavir (Norvir), Indinavir (Crixivan) und Delavirdin
(Rescriptor). Man vergleiche M. I. Johnston & D.F. Hoth, Science, 260 (5112),
1286–1293
(1993) und D.D. Richman, Science, 272 (5270), 1886–1888 (1996).
Ein Impfstoff gegen AIDS (Salk-Impfstoff) ist getestet worden und
es wurde entdeckt, dass mehrere Proteine, bei denen es sich um Chemokine
aus CD8 handelt, als HIV-Hemmer
wirken. Zusätzlich
zu den obigen synthetischen Nukleosidanalogen, Proteinen und Antikörpern hat
es sich erwiesen, dass mehrere Pflanzen und aus Pflanzen gewonnene
Substanzen eine Anti-HIV-Aktivität
in vitro besitzen. Jedoch wird das HIV-Virus nicht ohne Weiteres
vernichtet und es besteht auch kein guter Mechanismus, um die Wirtszellen
davon abzuhalten, das Virus zu replizieren.
-
So
suchen Fachleute in der Medizin auch weiterhin nach Medikamenten,
die HIV-Infektionen verhindern, die Träger des HIV-Virus zum Verhindern,
dass ihre Krankheiten bis zu vollständigem tödlichem AIDS fortschreiten,
behandeln und den AIDS-Patienten behandeln können.
-
Die
Herpes-simplex-Viren (HSV) der Typen 1 und 2 sind persistente Viren,
die Menschen häufig
infizieren; sie verursachen eine Reihe verschiedener schwerer Erkrankungen.
Der HSV-Typ 1 verursacht „Fieberbläschen", (wiederholt auftretender
Herpes labialis) im Mund und der HSV-Typ 2 verursacht Herpes genitalis, das
in vielen Teilen der Welt zu einer Hauptgeschlechtskrankheit geworden
ist. Zur Zeit gibt es keine vollständig befriedigende Behandlung
des Herpes genitalis. Außerdem
kann das HSV auch Gehirnentzündung,
eine lebensbedrohende Infektion des Gehirns, hervorrufen, obwohl
dies ungewöhnlich
ist (The Merck Manual, Holvey, Ausgabe 1972; Whitley, Herpes Simplex
Viruses (Herpes-simplex-Viren),
in: Virology, 2. Ausgabe, Raven Press (1990)). Eine äußerst ernsthafte
durch HSV hervorgerufene Erkrankung ist die Keratitis dendrica,
eine Augeninfektion, die eine verzweigte Läsion der Hornhaut verursacht,
die wiederum zu permanenter Narbenbildung und Verlust an Sehvermögen führen kann.
Augeninfektionen durch HSV sind eine Hauptursache von Blindheit.
Das HSV ist auch ein Virus, das schwierig, wenn nicht unmöglich, zu
heilen ist.
-
Die
Hepatitis ist eine Erkrankung der menschlichen Leber. Sie tritt
in Form einer Entzündung
der Leber auf und wird gewöhnlich
durch Virusinfektionen und manchmal durch toxische Agentien verursacht.
Die Hepatitis kann bis zur Zirrhose der Leber, Leberkrebs und schließlich zum
Tod fortschreiten. Mehrere Viren, wie beispielsweise Hepatitis A,
B, C, D, E und G sind dafür
bekannt, dass sie verschiedene Typen von Virushepatitis hervorrufen.
Darunter sind HBV und HCV die schlimmsten. HBV ist ein DNA-Virus
mit einer Viruspartikelgröße von 42
nm. HCV ist ein RNA-Virus mit einer Viruspartikelgröße von 30–60 nm.
Man vergleiche D.S. Chen, J. Formos. Med. Assoc. 95(1), 6–12 (1996).
-
Die
Hepatitis C infiziert 4- bis 5-mal die Anzahl von Menschen, die
mit HIV infiziert werden. Die Hepatitis C ist schwierig zu behandeln
und es wird geschätzt,
dass es 500 Millionen Menschen weltweit gibt, die damit infiziert
sind (etwa 15-mal die Anzahl derjenigen, die mit HIV infiziert sind).
Zur Zeit steht keine wirksame Impfung zur Verfügung und die Hepatitis C kann
nur durch andere Vorbeugungsmaßnahmen,
wie beispielsweise bessere Hygiene und Sanitärbedingungen und Unterbrechung
des Übertragungswegs
unter Kontrolle gebracht werden. Zur Zeit besteht die einzig annehmbare
Behandlungsmöglichkeit
bei chronischer Hepatitis C aus Interferon, das eine mindestens
sechs (6) monatige Behandlung erfordert, und/oder Ribavarin, das
die Virusreplikation in infizierten Zellen hemmen und außerdem bei
einigen Menschen die Leberfunktion verbessern kann. Die Behandlung
mit Interferon mit oder ohne Ribavarin ist jedoch mit einer Reaktionsrate
von etwa 25 % in ihrer Wirksamkeit über längere Zeit beschränkt.
-
Die
Infektion durch das Hepatitis-B-Virus führt zu einer umfangreichen
Reihe von Leberschäden.
Außerdem
ist die chronische Infektion durch Hepatitis-B mit der darauffolgenden
Entwicklung von hepatozellulärem
Karzinom, einer wichtigen Todesursache, in Verbindung gebracht worden.
Die gegenwärtige
Verhinderungsmöglichkeit
der HBV-Infektion ist eine Hepatitis-B-Impfung, die unbedenklich
und wirksam ist. Jedoch ist eine Impfung zum Behandeln derjeniger,
die schon infiziert sind (d.h. den Trägern und Patienten) nicht wirksam.
Viele Medikamente sind zum Behandeln der chronischen Hepatitis B
verwendet worden und mit Ausnahme des Interferons haben sich keine
als wirksam erwiesen.
-
Die
Behandlung von HCV und HBV mit Interferon ist nur beschränkt erfolgreich
und ist häufig
mit negativen Nebenwirkungen wie Ermüdung, Fieber, Erkältungen,
Kopfweh, Muskelschmerzen, Gelenkschmerzen, mildem Haarausfall, psychiatrischen
Auswirkungen und damit verbundenen Beschwerden, Autoimmunphänomenen
und damit verbundenen Beschwerden und Funktionsstörung der
Schilddrüse
in Verbindung gebracht worden.
-
Da
die Interferontherapie eine begrenzte Wirksamkeit und oft negative
Auswirkungen hat, ist eine effizientere Behandlungsweise erforderlich.
-
Bei
der vorliegenden Erfindung hat man entdeckt, dass die oben beschriebenen
Verbindungen zur Behandlung des Hepatitis-C-Virus, des Hepatitis-B-Virus,
des Herpes simplex und bei der Behandlung der HIV-Infektion und
anderen Virusinfektionen nützlich
sind.
-
KURZFASSUNG
DER ERFINDUNG
-
Die
Verwendung zum Herstellen eines Medikaments für die Behandlung von Virusinfektionen
bei Patienten, die diese benötigen,
und insbesondere warmblütigen
Tieren und Menschen, eines pharmazeutisch akzeptablen Trägers und
einer thera peutisch wirksamen Menge eines 2-Thienylimidazolo[4,5]pyridin-Derivats der
Formel:
wobei n 1 bis 4 ist, R ausgewählt ist
aus der Gruppe bestehend aus Wasserstoff, Alkyl mit 1 bis 7 Kohlenstoffatomen,
Chlor, Brom oder Fluor, Oxychlor, Hydroxy, Sulfhydryl und Alkoxy
mit der Formel -O(CH
2)
yCH
3, wobei y von 0 bis 6 beträgt, dessen
Prodrug oder eines pharmazeutisch akzeptablen Additionssalzes desselben.
-
Das
bevorzugte Material ist
oder dessen pharmazeutische
Additionssalze, insbesondere das Hydrochloridsalz.
-
Bei
der vorliegenden Erfindung hat man entdeckt, dass die antiviralen
2-Thienyl-imidazolo[4,5]pyridin-Verbindungen zum Hemmen des HIV
und der Behandlung der HIV-Infektion sowie zum Behandeln von Hepatitis-B-Infektionen
nützlich
sind. Die vorliegende Erfindung bietet auch Verfahren zum Behandeln
von HIV-Infektion, umfassend das Verabreichen einer pharmazeutisch
oder therapeutisch wirksamen Menge einer antiviralen Verbindung,
wie hier beschrieben, an einen mit HIV infizierten Wirt.
-
Diese
Materialien sind gegen Cryptococus neoformas und Curvularia lunata
aktiv. Es handelt sich bei beiden um Pilze, die bei AIDS-Patienten
anzutreffen sind.
-
Die
Zusammensetzungen können
in Verbindung mit anderen Behandlungen bei Virusinfektionen verwendet
werden.
-
Das
Arzneimittel kann täglich
in einer Dosis oder in mehreren Dosen oder 1 bis 4-mal wöchentlich
verabreicht werden.
-
GENAUE BESCHREIBUNG DER
ERFINDUNG
-
A. Definitionen:
-
Wie
hier verwendet, ist eine „pharmazeutisch
akzeptable" Komponente
eine, die zur Verwendung bei Menschen und/oder Tieren ohne übermäßige negative
Nebenwirkungen (wie Toxizität,
Reizung und allergische Reaktion) einem vernünftigen Nutzen/Risikoverhältnis entsprechend
geeignet ist.
-
Wie
hier verwendet, betrifft der Ausdruck „unbedenkliche und wirksame
Menge" die Menge
einer Komponente, die ausreicht, um eine erwünschte therapeutische Reaktion
ohne übermäßige negative
Nebenwirkungen (wie Toxizität,
Reizung und allergische Reaktion) einem vernünftigen Nutzen/Risikoverhältnis entsprechend,
wenn sie erfindungsgemäß verwendet
wird, zu erreichen.
-
Mit „therapeutisch
wirksamer Menge" ist
eine Menge einer erfindungsgemäßen Verbindung
gemeint, die zum Erreichen der erwünschten therapeutischen Reaktion
wirksam ist, beispielsweise zum Hemmen der HIV-Infektion oder der
Behandlung der Infektionssymptome bei einem Wirt. Die spezifische
unbedenkliche und wirksame Menge oder die therapeutisch wirksame
Menge ist offensichtlich je nach Faktoren wie dem spezifischen Zustand,
der behandelt wird, dem körperlichen
Zustand des Patienten, dem Typ des behandelten Säugers oder Tiers, der Behandlungsdauer,
der Natur der gleichzeitigen Therapie (falls eine solche stattfindet)
und den spezifischen angewendeten Rezepturen und der Struktur der
antiviralen Verbindungen oder ihrer Derivate verschieden.
-
Wie
hier verwendet, ist ein „pharmazeutisches
Additionssalz oder -salze" Salz
der Thienyl-imidazolo[4,5]pyridin-Verbindung, die durch Herstellen saurer
oder basischer Salze der antiviralen Verbindungen modifiziert wird.
Beispiele pharmazeutisch akzeptabler Salze umfassen, sind jedoch
nicht darauf beschränkt,
Salze von Mineral- oder organischen Säuren basischer Rückstände, beispielsweise
Amine; Alkali- oder organische Salze saurer Rückstände, wie Carbonsäuren. Bevorzugt
werden die Salze unter Anwendung einer organischen oder anorganischen
Säure hergestellt.
Diese bevorzugten sauren Additionssalze sind Chloride, Bromide,
Sulfate, Nitrate, Phosphate, Sulfonate, Formiate, Tartrate, Maleate,
Malate, Citrate, Benzoate, Salicylate, Ascorbate und dergleichen.
-
Wie
hier verwendet, ist ein „pharmazeutischer
Träger" ein pharmazeutisch
akzeptables Lösungsmittel, Suspendiermittel
oder Vehikel für
die Abgabe des Thienylimidazolo[4,5]pyridin-Derivats an das Tier
oder den Menschen. Der Träger
kann flüssig
oder fest sein und wird auf die geplante Verabreichungsart hin ausgewählt.
-
Wie
hier verwendet, sind die „Thienyl-imidazolo[4,5]pyridin-Derivate" oder „2-Thienyl-imidazolo[4,5]pyridin-Verbindungen" oder „2-(2-Thienyl)imidazolo[4,5-b]pyridin-Verbindungsderivate" Mitglieder der Gruppe
von Verbindungen mit der Formel:
wobei n 1–4 ist, R ausgewählt ist
aus der Gruppe bestehend aus Wasserstoff, Alkyl mit 1 bis 7 Kohlenstoffatomen,
Chlor, Brom oder Fluor, Oxychlor, Hydroxy, Sulfhydryl und Alkoxy
mit der Formel -O(CH
2)
yCH
3, wobei y von 1 bis 6 beträgt, dessen
Prodrugs und pharmazeutisch akzeptable Additionssalze.
-
„Alkyl", wie es hier verwendet
wird, umfasst geradkettige, verzweigtkettige und cyclische Alkane.
Als „Prodrugs" werden irgendwelche
kovalent gebundene Träger
betrachtet, die das aktive Muttermedikament der Formel der Thienylimidazolo[4,5]pyridin-Verbindungen,
die oben beschrieben worden sind, entsprechend in vivo freisetzen,
wenn ein derartiges Prodrug einem Säugerpatienten verabreicht wird.
Prodrugs der Thienyl-imidazolo[4,5]pyridin-Verbindungen werden durch
Modifizieren funktioneller, in den Verbindungen anwesender Gruppen
derart hergestellt, dass die Modifikationen entweder bei einer Routinemanipulation
oder in vivo zu den Mutterverbindungen gespalten werden. Prodrugs
umfassen Verbindungen, in den Hydroxy-, Amin- oder Sulfhydrylgruppen
zu irgendeiner Gruppe verbunden werden, die, wenn einem Säugerpatienten
verabreicht, sich unter Bildung einer freien Hydroxyl-, Amino- bzw.
Sulfhydrylgruppe spaltet. Beispiele von Prodrugs umfassen, sind
jedoch nicht darauf beschränkt,
Acetat-, Formiat- oder Benzoatderivate von alkohol- und aminfunktionellen
Gruppen in den Thienylimidazolo[4,5]pyridin-Derivaten; Formamid-,
Acetamid- und Benzamidderivate der Aminogruppe; Phosphatester, Dimethylglycinester,
Aminoalkylbenzylester, Aminoalkylester und Carboxyalkylester von
alkohol- und phenolfunktionellen Gruppen in den Thienyl-imidazolo[4,5]pyridin-Derivaten und
dergleichen. Weitere Schutzgruppen umfassen Carboxyl-Schutzgruppen, die
in „Protective
Groups in Organic Synthesis (Schutzgruppen in der organischen Synthese)" (von Green & Woods, 1999,
3. Ausgabe); „Protecting
Groups (Tieme Foundations Organic Chemistry Series N Group" (von Kocienskie;
Tieme Medical Publishers, 1994) offenbart sind, deren relevanten
Offenbarungen hier summarisch eingefügt sind.
-
„Antivirale
Verbindungen" wie
hier verwendet, sind Thienylimidazolo[4,5]pyridin-Derivate und bevorzugt
2-(2-Thienyl)-Imidazolo[4,5]Pyridin
oder die pharmazeutisch akzeptablen sauren Additionssalze oder Prodrugs
derselben.
-
„Viren", wie hier verwendet,
umfassen Viren, die Tiere oder Säuger,
einschließlich
Menschen, infizieren. Viren umfassen Retroviren, HIV, Grippe, Polioviren,
Herpes simplex, Hepatitis B, Hepatitis C, andere Hepatitisviren,
Kaposi-Sarkom-Virus,
Rhinoviren, den Rinderdiarrhövirus
und dergleichen. HIV und AIDS sind immununtertrückende Krankheiten.
-
„Kombinationstherapie", wie hier verwendet,
bedeutet, dass der Patient, der das Arzneimittel benötigt, mit
einem anderen Arzneimittel für
die Krankheit in Verbindung mit 2-Thienyl-imidazolo[4,5]pyridin-Derivaten behandelt
wird oder dass ihm diese verabreicht werden. Diese Kombinationstherapie
kann eine sequentielle Therapie sein, wenn der Patient zuerst mit
einem oder mehreren Arzneimitteln behandelt wird und dann das andere
oder zwei oder mehrere Arzneimittel gleichzeitig verabreicht werden.
-
B. DIE ANTIVIRALEN VERBINDUNGEN
-
Die
2-Thienyl-imidazolo[4,5]pyridin-Verbindungen, die hier nützlich sind,
haben die Formel:
wobei n 1–4 ist, R ausgewählt ist
aus der Gruppe bestehend aus Wasserstoff, Alkyl mit 1 bis 7 Kohlenstoffatomen,
Chlor, Brom oder Fluor, Oxychlor, Hydroxy, Sulfhydryl, Alkoxy mit
der Formel -O(CH
2)
yCH
3, wobei y von 0 bis 6, bevorzugt von 1 bis
6 beträgt.
-
Bevorzugt
ist das 2-(2-Thienyl)-imidazolo-[4,5-b]pyridin mit einem Alkyl von
weniger als 4 Kohlenstoffen, einem Halogen, bevorzugt einem Chlor,
Nitro, Hydroxy oder Oxychlor in Position 7 oder 8 substituiert und die übrigen Substituenten
des Pyridinrings sind Wasserstoff.
-
Das
bevorzugte antivirale Mittel ist 2-(2-Thienyl)imidazolo-[4,5-b]pyridin:
oder dessen pharmazeutische
Additionssalze.
-
C. SYNTHESE
-
Die
Thienyl-imidazolo[4,5]pyridin-Derivate können auf eine Reihe verschiedener
Arten und Weisen zubereitet werden, die einem mit dem Stand der
Technik der organischen Synthese vertrauten Fachmann allgemein bekannt
sind. Thienylimidazolo[4,5]pyridin-Derivate können unter Anwendung der unten
beschriebenen Methoden in Verbindung mit Synthesemethoden, die im
Stand der Technik der organischen Synthesechemie bekannt sind, oder
Variationen derselben, wie sie von den mit dem Stand der Technik
vertrauten Fachleuten verstanden wird, synthetisiert werden. Bevorzugte
Methoden umfassen diejenigen, die unten beschrieben sind, sind jedoch
nicht darauf beschränkt.
Jeder der unten angegebenen Literaturhinweise wird hier summarisch
eingefügt.
-
Ein
allgemeiner Syntheseweg beginnt mit 2-Chlorpyridin, das in Gegenwart
von Schwefelsäure
unter Bildung von 2-Chlor-3-nitropyridin
nitriert wird. Dieses Material wird mit Ammoniumacetat in Gegenwart
von Diglyme bei etwa 160°C
unter Bildung von 2-Amino-3-nitropyridin reagiert, das unter Bildung
von 2,3-Diaminopyridin reduziert wird. Das 2,3-Diaminopyridin wird mit 2-Thiophencarbonsäure in Gegenwart
von Polyphosphorsäure
bei etwa 125 ºC
zum Zubereiten von 2-(2-Thienyl)-imidazolo[4,5]pyridin
reagiert.
-
Die
Zubereitung von 2-(2-Thienyl)-imidazolo[4,5]pyridin wird bei Germaise
et al. J. Org. Chem., 1964, Band 29, 3403 und Vanden Eynde et al.,
Bull. Soc. Chem. Belg., Band 2, Nr 5 (1993) beschrieben. Coates,
J. Medicine, 1993, Band 36, Seite 1387–1392 beschreibt die Synthese
anderer Imidazo-lo[4,5]pyridin-Derivate.
-
Die
erfindungsgemäßen pharmazeutisch
akzeptablen Salze können
aus den Thienyl-imidazolo[4,5]pyridin-Derivaten, die einen basischen
oder sauren Anteil enthalten, durch herkömmliche chemische Methoden
synthetisiert werden. Im Allgemeinen können derartige Salze durch
Reagieren der freien sauren oder basischen Formen dieser antiviralen
Verbindun gen mit einer stöchiometrischen
Menge der entsprechenden Base oder Säure in Wasser oder in einem
organischen Lösungsmittel
oder in einer Mischung der beiden zubereitet werden; im Allgemeinen
werden nichtwässrige
Medien wie Ether, Ethylacetat, Ethanol, Isopropanol oder Acetonitril
vorgezogen. Listen geeigneter Salze sind in Remington's Pharmaceutical
Sciences, 17. Ausgabe, Mack Publishing Company, Easton, Pa, 1985,
Seite 1418 zu finden, deren Offenbarung hier summarisch eingefügt wird.
-
Die
pharmazeutisch akzeptablen Salzen der Thienylimidazolo[4,5]pyridin-Verbindungen
umfassen die herkömmlichen
nicht toxischen Salze oder die quartären Ammoniumsalze der Thienyl-imidazolo[4,5]pyridin-Derivate,
die beispielsweise aus nicht toxischen anorganischen oder organischen
Säuren
gebildet werden. Beispielsweise umfassen derartige herkömmliche
nicht toxische Salze diejenigen, die von anorganischen Säuren wie
Salz-, Bromwasserstoff-, Schwefel-, Sulfam-, Phosphor-, Stickstoffsäure und
dergleichen deriviert sind und die Salze, die aus organischen Säuren wie
Essig-, Propion-, Bernstein-, Glykol-, Stearin-, Milch-, Äpfel-, Wein-,
Zitronen-, Ascorbin-, Malein-, Hydroxymalein-, Phenylessig-, Glutamin-,
Benzoe-, Salicyl-, Sulfanil-, 2-Acetoxybenzoe-,
Fumar-, Toluolsulfon-, Methansulfon-, Ethandisulfon-, Oxal-, Isethionsäure und
dergleichen deriviert sind.
-
D. DOSIS
-
Irgendeine
geeignete Dosis kann bei der erfindungsgemäßen Methode verabreicht werden.
Der Typ des Trägers
und die Menge wird je nach der Spezies des warmblütigen Tiers
oder Menschen und des zu behandelnden Virus äußerst verschieden sein. Die
verabreichte Dosis wird jedoch je nach bekannten Faktoren wie den
pharmakodynamischen Eigenschaften des spe zifischen Mittels und seinem
Verabreichungsmodus und -weg, dem Alter, der Gesundheit und/oder
dem Gewicht des Empfängers,
der Natur und dem Ausmaß der Symptome,
den metabolischen Eigenschaften des Arzneimittels und Patienten,
der Art der gleichzeitigen Behandlung, der Häufigkeit der Behandlung oder
der erwünschten
Auswirkung unterschiedlich sein.
-
Bevorzugt
wird das Thienyl-imidazolo[4,5]pyridin mikronisiert oder pulverförmig gemahlen,
so dass es sich im Körper
leichter dispergiert und löslich
wird. Verfahren zum Mahlen oder Pulverisieren von Arzneimitteln sind
im Stand der Technik allgemein bekannt. Beispielsweise kann eine
Hammermühle
oder ein ähnliches Mahlgerät verwendet
werden. Die bevorzugte Teilchengröße ist weniger als etwa 100 μm und bevorzugt
weniger als 50 μm.
-
Im
Allgemeinen ist eine Dosis, die so gering wie etwa ein Milligramm
(mg) pro Kilogramm (kg) Körpermasse
ist, geeignet, jedoch kann bevorzugt nur 10 mg/kg und bis zu etwa
10.000 mg/kg verwendet werden. Bevorzugt wird 10 mg/kg bis etwa
5.000 mg/kg verwendet. Am bevorzugtesten liegen die Dosen zwischen
250 mg/kg und etwa 5.000 mg/kg. Dosen, die bei der Behandlung von
Vireninfektionen nützlich
sind, sind 250 mg/kg, 500 mg/kg, 2500 mg/kg, 3500 mg/kg, 4000 mg/kg,
5000 mg/kg und 6000 mg/kg. Irgendein Dosisbereich kann angewendet
werden. Im Allgemeinen können
2-Thienylimidazolo[4,5]pyridin-Derivative auf täglicher Basis ein- oder zweimal täglich verabreicht
werden, oder 2-Thienylimidazolo[4,5]pyridin-Derivate können ein-
bis viermal wöchentlich
entweder in einer einzigen Dosis oder einzelnen Dosen während des
Tags verabreicht werden. Die zweimal wöchentliche Dosierung über einen
Zeitraum von mindestens mehreren Wochen wird vorgezogen und oft
wird das Dosieren über
längere
Zeiträume
fortgesetzt und kann über
die Le benszeit des Patienten hinweg erfolgen. Jedoch werden die
Dosis und das Dosisregime je nach der Fähigkeit des Patienten, die
erwünschten
und wirksamen Plasmaspiegel der antiviralen Mittel im Blut zu vertragen
verschieden sein.
-
Intravenös können die
bevorzugtesten Dosen im Bereich von etwa 1 bis etwa 10 mg/kg/Minute
während
einer Infusion mit konstanter Geschwindigkeit liegen.
-
Die
Dosierung beim Menschen ist geringer als diejenige, die bei Mäusen angewendet
wird und ist typischerweise etwa 1/12 der Dosis, die bei Mäusen wirksam
ist. So wäre,
wenn 500 mg/kg bei Mäusen
wirksam war, eine Dosis von 42 mg/kg bei Menschen angewendet werden.
Bei einem 60 kg schweren Mann wäre
diese Dosis 2520 mg.
-
Die
antiviralen Verbindungen sind im Allgemeinen unbedenklich. Die LD50 ist ziemlich hoch, etwa 1500 mg/kg bei
oraler Verabreichung bei Mäusen
und es bestehen keine speziellen Handhabungserfordernisse. Die antiviralen
Verbindungen können
oral verabreicht oder, da sie nicht sehr löslich sind, können sie
bevorzugt in Tablettenform oder als Suspension verabreicht werden.
-
E. VERABREICHUNGSMETHODE
UND FORMEN DER DOSISABGABE
-
Die
erfindungsgemäßen Verbindungen
können
auf irgendeine geeignete Weise einschließlich, jedoch nicht darauf
beschränkt,
beispielsweise oral, rektal, nasal, topisch (einschließlich transdermal,
aerosol, bukkal und sublingual), vaginal, parenteral (einschließlich subkutan,
intramuskulär,
intravenös
und intradermal), intravesikal oder durch Einspritzung in oder um
das Virus herum verabreicht werden.
-
Die
Dosismengen basieren auf der wirksamen Hemmungskonzentration, die
bei antiviralen Studien beobachtet worden sind. Der bevorzugte Weg
wird je nach (1) dem Zustand und Alter des Empfängers, (2) dem zu behandelnden
Virus, (3) der Natur der Infektion und (4) den erwünschten
Blutspiegeln verschieden sein. Man glaubt, dass eine parenterale
Behandlung durch intravenöses,
subcutanes oder intramuskuläres
Anwenden der erfindungsgemäßen Verbindungen,
die mit einem geeigneten Träger,
anderen antiviralen Mitteln oder Verbindungen oder Verdünnungsmitteln
formuliert sind, um die Anwendung zu erleichtern, die bevorzugte Verabreichungsmethode
für die
Verbindungen an warmblütige
Tiere sein wird.
-
Bevorzugt
werden die Thienyl-imidazolo[4,5]pyridin-Derivate mikronisiert oder
pulverförmig
gemahlen, so dass sie im Körper
leichter dispergiert und löslich
gemacht werden. Verfahren für
das Mahlen oder Pulverisieren von Arzneimitteln sind im Stand der
Technik allgemein bekannt. Beispielsweise kann eine Hammermühle oder
ein ähnliches
Mahlgerät
verwendet werden. Die bevorzugte Teilchengröße beträgt weniger als etwa 100 μm und bevorzugt
weniger als 50 μm.
Diese Verbindungen sind nicht sehr löslich und werden aus diesem Grund
bevorzugt in Tablettenform oder als Suspension verabreicht. Geeignete
Methoden zum Verabreichen der erfindungsgemäßen Verbindungen und Dosierformen
sind im Folgenden zu finden.
-
Die
erfindungsgemäßen Thienyl-imidazolo[4,5]pyridin-Derivate können als
Behandlung gegen Vireninfektionen durch irgendein Mittel, das den
Kontakt des aktiven Mittels mit der Einwirkungsstelle des Mittels
im Körper
herstellt, verabreicht werden. Sie können durch irgendein herkömmliches
Mittel verabreicht werden, das in Verbindung mit Arzneimitteln entweder
als einzelnen therapeutischen Mitteln oder in einer Kombination von
Therapeutika zur Verwendung zur Ver fügung steht. Bevorzugt werden
die erfindungsgemäßen Verbindungen
als pharmazeutische Rezeptur verabreicht, die mindestens eine erfindungsgemäße Verbindung,
wie oben definiert, zusammen mit einem oder mehreren pharmazeutisch
akzeptablen Trägern
umfasst. Sie kann in Form einer Tablette oder Kapsel, als agglomeriertes
Pulver oder in flüssiger
Form oder als Liposom verabreicht werden.
-
Die
erfindungsgemäßen Verbindungen
können
auch in Form von Liposomabgabesystemen wie beispielsweise als kleine
unilamellare Bläschen,
große
unilamellare Bläschen
und multilamellare Bläschen
verabreicht werden. Liposome können
aus einer Reihe verschiedener Phospolipide wie beispielsweise Cholesterin, Stearylamin
oder Phosphatidylcholin gebildet werden.
-
Die
erfindungsgemäßen Thienyl-imidazolo[4,5]pyridin-Verbindungenen oder
-Derivate können
auch mit löslichen
Polymeren als anzielbare Arzneimittelträger gekoppelt werden. Derartige
Polymere können
Polyvinylpyrrolidon, Pyrancopolymer, Polyhydroxylpropylmethacrylamidphenol,
Polyhydroxyethylaspartamidphenol oder Polyethylenoxidpolylysin,
das mit Palmitoylresten substituiert sind, umfassen. Des Weiteren
können die
erfindungsgemäßen Verbindungen
an eine Klasse biologisch abbaubarer Polymere, die zum Erzielen
einer gesteuerten Freisetzung eines Arzneimittels nützlich sind,
beispielsweise Polymilchsäure,
Polyglykolsäure, Copolymere
von Polymilch- und Polyglykolsäure,
Polyepsiloncaprolacton, Polyhydroxybuttersäure, Polyorthoestern, Polyacetale,
Polydihydropyrane, Polycyanoacylate und vernetzte oder amphipathische
Blockcopolymere von Hydrogelen, gekoppelt werden.
-
1. Kombinationstherapie
-
Die
erfindungsgemäßen Verbindungen
können
zusätzlich
mit anderen antiviralen Verbindungen kombiniert werden, um eine
funktionsfähige
Kombination bereitzustellen. „Kombinationstherapie", wie hier verwendet,
bedeutet, dass der Patient, der das Arzneimittel benötigt, mit
einem anderen Arzneimittel gegen die Krankheit in Verbindung mit
den Thienyl-imidazolo[4,5]pyridin-Derivaten behandelt oder diese
verabreicht wird. Bei dieser Kombinationstherapie kann es sich um
eine sequentielle Therapie handeln, wo der Patient zuerst mit mindestens
einem anderen Arzneimittel behandelt wird und ihm daraufhin die
anderen oder zwei oder mehrere Arzneimittel gleichzeitig verabreicht
werden. Die genaue Dosis und Verabreichungsmethode der Kombination hängen von
dem spezifischen zu behandelnden Virus und dem Typ und Ausmaß der Kombinationstherapie ab.
Es ist beabsichtigt, irgendeine chemisch verträgliche Kombination einer Verbindung
dieser erfindungsgemäßen Gruppe
mit anderen Verbindung der erfindungsgemäßen Gruppe oder anderen Verbindungen,
die außerhalb
der erfindungsgemäßen Gruppe
liegen, einzuschließen,
so lange die Kombination die antivirale Aktivität der Verbindung dieser erfindungsgemäßen Gruppe
nicht eliminiert. Beispielsweise können ein oder mehrere Thienylimidazolo[4,5]pyridin-Derivate
mit anderen antiviralen Mitteln oder Verstärkungsmitteln kombiniert werden.
Verstärkungsmittel
sind Materialien, die sich auf die Reaktion des Körpers gegen
das antivirale Mittel auswirken. Im Falle von HIV ist eine Kombinationstherapie
mit AZT, TC-3 oder Proteaseinhibitoren wirksam. Im Falle von Hepatitis
wird Cyclovier, Famiclovir oder Valacyclovir, Ribavirin, Interferon
oder eine Kombination von Ribavirin und Interferon oder Betaglobulin
als Kombinationstherapie verabreicht. Bei Herpes kann ein rekombinantes
Alpha-Interferon als Kombinationstherapie verwendet werden.
-
Bei
einigen Ausführungsformen
wird die 2-(2-Thienyl)imidazolo[4,5]pyridin-Verbindung in Kombination mit
einem oder mehreren Verstärkern
und/oder antiviralen Mitteln für
die Behandlung von Virusinfektionen angewendet. Ein beispielhafter
Verstärker
ist Triprolidin oder dessen Cis-Isomer,
die in Kombination mit chemotherapeutischen Mitteln und der 2-(2-Thienyl)-imidazolo[4,5]pyridin-Verbindung
verwendet werden. Triprolidin ist in
US
5,114,951 (1992) beschrieben. Ein weiterer Verstärker ist
Procodazol, 1H-Benzimidazolo-2-propansäure; [β-(2-Benzimidazolo)propionsäure, 2-(2-Carboxyethyl)benzimidazolo;
Propazol]. Procodazol ist ein nicht-spezifisches Immunschutzmittel,
das gegen Virus- und Bakterieninfektionen aktiv ist und mit den
hier beanspruchten Zusammensetzungen verwendet wird. Es ist mit
der 2-(2-Thienyl)-imidazolo[4,5]pyridin-Verbindung
beim Behandeln von Virusinfektionen wirksam. Procodazol kann auch
mit der 2-(2-Thienyl)-imidazolo[4,5]pyridin-Verbindung und anderen
antiviralen Mitteln kombiniert werden. Andere Verstärker, die
mit 2-(2-Thienyl)-imidazolo[4,5]Pyridin-Verbindungen verwendet werden
können,
umfassen Monensin, einen Antisenseinhibitor des RAD51-Gens, Bromodeoxyuridin,
Dipyridamol, Indomethacin, einen monoklonalen Antikörper, ein
Antitransferrinrezeptorimmuntoxin, Metoclopramid, 7-Thia-8-oxoguanosin, N-Solanesyl-N,N'-bis(3,4-dimethoxybenzyl)ethylendiamin,
Leucovorin, Heparin, N-[4-[(4-Fluorphenyl)sulfonyl]phenyl]acetamid,
Heparinsulfat, Cimetidin, einen Radiosensibilisator, einen Chemosensibilisator,
ein hypotoxisches zellzytotoxisches Mittel, Muramyldipeptid, Vitamin
A, 2'-Deoxycorfomycin,
ein Bisdiketopiperazinderivat und Dimethylsulfoxid.
-
Bei
einigen Ausführungsformen
der Erfindung wird eine 2-(2-Thienyl)-imidazolo[4,5]pyridin-Verbindung
in Kombination mit einem oder mehreren anderen therapeutischen Mitteln
wie entzündungshemmenden, antiviralen,
antifungalen, amöbizidalen,
trichomonozidalen, schmerzstillenden, antineoplasti-schen; antihypertensiven,
antimikrobiellen und/oder Steroidarzneimitteln zum Behandeln von
Virusinfektionen verwendet. Bei einigen bevorzugten Ausführungsformen
werden Patienten mit Virusinfektionen mit einer Kombination von
einem oder mehreren eine 2-(2-Thienyl)-imidazolo[4,5]pyridin-Verbindungen mit
einem oder mehreren Betalactam-Antibiotikum,
Tetracyclinen, Chloramphenicol, Neomycin, Gramicidin, Bacitracin,
Sulfonamiden, Nitrofurazon, Nalixidinsäure, Cortison, Hydrocortison,
Betamethason, Dexamethason, Fluorcortolon, Prednisolon, Triamcinolon,
Indomethacin, Sulindac, Acyclovir, Amantadin, Rimantadin, rekombinanter
löslicher
CD4 (rsCD4), Antirezeptorantikörpern
(bei Rhinoviren), Nevirapin, Cidofovir (VistideWZ),
Trinatriumphosphonoformiat (FoscarnetWZ),
Famcyclovir, Pencyclovir, Valacyclovir, Nucleinsäure-/Replikationsinhibitoren,
Interferon, Zidovudin (AZT, RetrovirWZ),
Didanosin (Dideoxyinosin, ddI, VidexWZ),
Stavudin (d4T, ZeritWZ), Zalcitabin (Dideoxycytosin, ddC,
HividWZ), Nevirapin (ViramuneWZ),
Lamivudin (EpivirWZ, 3TC), Proteaseinhibitoren,
Saquinavir (InviraseWZ, FortovaseWZ), Ritonavir (NorvirWZ)
, Nelvinavir (ViraceptWZ), Efavirenz (SustivaWZ), Abacavir (ZiagenWZ),
Amprenavir (AgeneraseWZ), Indinavir (CrixivanWZ), Ganciclovir, AzDU, Delavirdin (RescriptorWZ), Rifampin, Clathiromycin, Erythropoietin,
koloniestimulierenden Faktoren (G-CSF und GM-CSF), nicht-nukleoside
Reverstranskriptaseinhibitoren, Nukleosidinhibitoren, Adriamycin,
Fluoruracil, Methotrexat, Asparaginase und Kombinationen derselben
behandelt.
-
Die
Kombinationstherapie kann sequentiell sein, das heißt, die
Behandlung findet zuerst mit einem Mittel und dann mit dem zweiten
Mittel statt oder sie kann gleichzeitig mit beiden Mitteln stattfinden.
Die sequentielle Therapie kann innerhalb eines vernünftigen
Zeitraums nach Abschluss der ersten Therapie vor Beginn der zweiten
Therapie stattfinden. Die Behandlung mit beiden Mitteln zur gleichen
Zeit kann in der gleichen täglichen
Dosis oder in getrennten Dosen, beispielsweise durch Behandlung
mit einem Mittel an Tag 1 und dem anderen an Tag 2 erfolgen. Das
genau Behandlungssystem hängt
von der behandelten Krankheit, der Schwere der Infektion und der
Reaktion auf die Behandlung hin ab.
-
2. Einheitsdosis
-
Die
erfindungsgemäßen Verbindungen
können
in Form einer Einheitsdosis verabreicht und durch irgendeine im
Stand der Technik allgemein bekannte Methode zubereitet werden.
Derartige Methoden umfassen das Kombinieren der erfindungsgemäßen Verbindungen
mit einem Träger
oder Verdünnungsmittel,
das einen oder mehrere zusätzliche
Bestandteile darstellt. Typischerweise werden die Rezepturen durch
gleichmäßiges Mischen
des aktiven Bestandteils mit flüssigen
Trägern
oder feinverteilten festen Trägern
oder beiden, nötigenfalls
vom Formen des Produkts gefolgt, zubereitet. Ein pharmazeutischer
Träger
wird auf der Basis des gewählten
Verabreichungswegs und pharmazeutischen Standardpraktiken ausgewählt. Jeder
Träger
muss in dem Sinne „akzeptabel" sein, dass er mit
den anderen Bestandteilen der Rezeptur verträglich und für den Patienten nicht schädlich ist.
Bei dem Träger
kann es sich um einen Feststoff oder eine Flüssigkeit handeln und der Typ
wird im Allgemeinen auf der Basis des verwendeten Verabreichungstyps
gewählt.
Beispiele geeigneter fester Träger
umfassen Lactose, Saccharose, Gelatine, Agar und Füllpulver.
Beispiele geeigneter flüssiger Träger umfassen
Wasser, pharmazeutisch akzeptable Fette und Öle, Alkohole oder andere organische
Lösungsmittel
einschließlich
Ester, Emulsionen, Sirupe oder Elixiere, Sus pensionen, Lösungen und/oder
Suspensionen und Lösungen
und/oder Suspensionen, die aus nicht efferveszierenden Granulaten
rekonstituiert sind und efferveszierende Zubereitungen, die aus
efferveszierenden Granulaten rekonstituiert sind. Derartige flüssige Träger können beispielsweise
geeignete Lösungsmittel,
Konserviermittel, Emulgiermittel, Suspendiermittel, Verdünnungsmittel,
Süßstoffe,
Verdickungsmittel und Schmelzmittel enthalten. Bevorzugte Träger sind Speiseöle, beispielsweise
Mais- oder Canolaöle.
Polyethylenglykole, z.B. PEG, sind ebenfalls gute Träger.
-
Die
Dosierformen (für
die Verabreichung geeignete Zusammensetzungen) umfassen etwa 1 Milligramm
bis etwa 1000 Milligramm aktiver Bestandteil pro Dosiseinheit. Bevorzugt
enthalten die Dosierformen etwa 10 mg bis etwa 500 mg. In diesen
pharmazeutischen Zusammensetzungen liegt der aktive Bestandteil gewöhnlich in
einer Menge von etwa 0,5 bis etwa 95 Gewichtsprozent, auf das Gesamtgewicht
der Dosiseinheit bezogen, vor.
-
3. Pharmazeutische
Kits
-
Die
vorliegende Erfindung umfasst auch pharmazeutische Kits, die beispielsweise
bei der Behandlung einer Hepatitisinfektion nützlich sind, die einen oder
mehrere Behälter
umfassen, die eine pharmazeutische Zusammensetzung enthalten, die
eine therapeutisch wirksame Menge eines Thienylimidazolo[4,5]pyridin-Derivats
umfassen. Derartige Kits können
des Weiteren, falls erwünscht,
eine oder mehrere verschiedener herkömmlicher pharmazeutischer Kitkomponenten
wie beispielsweise Behälter
mit einem oder mehreren pharmazeutisch akzeptablen Trägern, zusätzliche
Behälter
usw. umfassen, wie den mit dem Stand der Technik vertrauten Fachleuten
ohne Weiteres offensichtlich ist. Gedruckte Anweisungen, entweder
als Beilageblätter
oder als Etikette, die die Mengen der zu verabreichenden Komponenten
angeben, Richtlinien für
die Verabreichung und/oder Richtlinien für das Mischen der Komponenten
können
ebenfalls in das Kit eingeschlossen werden. Bei der vorliegenden
Offenbarung sollte man sich im Klaren darüber sein, dass die angegebenen
Materialien und Bedingungen bei der praktischen Ausführung der
Erfindung wichtig sind, dass jedoch nicht spezifizierte Materialien
und Bedingungen nicht ausgeschlossen sind, solange sie die Vorteile
der Erfindung nicht daran hindern, erreicht zu werden.
-
Spezifische
Beispiele pharmazeutisch akzeptabler Träger und Vehikel, die zum Formulieren
erfindungsgemäßer oraler
Dosierformen verwendet werden können,
sind in der an Robert vergebenen, am 2. September 1975 herausgegebenen
Patentschrift Nr. 3,903,297 beschrieben.
-
Techniken
und Zusammensetzungen zum Herstellen von Dosierformen, die bei der
vorliegenden Erfindung nützlich
sind, werden hier weiter unten beschrieben.
-
Orale
Rezepturen, die zur Verwendung in der Praxis der vorliegenden Erfindung
geeignet sind, umfassen Kapseln, Gele, Oblatenkapseln, Tabletten,
efferveszierende oder nicht efferveszierende Pulver oder Tablette,
Pulver oder Granulate, als Lösung
oder Suspension in wässriger
oder nicht wässriger
Flüssigkeit
oder als flüssige Öl-in-Wasser-Emulsion
oder Wasser-in-Öl-Emulsion.
Die erfindungsgemäßen Verbindungen
können
auch als Bolus, Electuarium oder Paste vorliegen.
-
Die
Rezepturen für
die orale Verabreichung können
einen nicht-toxischen, pharmazeutisch akzeptablen, inerten Träger wie
Lactose, Stärke,
Saccharose, Glukose, Methylzellulose, Magnesiumstearat, Dicalciumphosphat,
Calciumsulfat, Mannit, Sorbit, Cyclodextrin und Cyclodextrinderivate
und dergleichen umfassen.
-
Kapseln
oder Tabletten können
ohne Weiteres formuliert und so hergestellt werden, dass sie leicht
zu schlucken oder kauen sind. Tabletten können geeignete Bindemittel,
Schmiermittel, Verdünnungsmittel, Sprengmittel,
Färbemittel,
Geschmacksstoffe, das Fließen
induzierende Mittel und Schmelzmittel enthalten. Eine Tablette kann
durch Komprimieren oder Verformen, wahlweise mit einem oder mehreren
zusätzlichen
Bestandteilen hergestellt werden. Komprimierte Tabletten können durch
Komprimieren des aktiven Bestandteils in freifließender Form
(z.B. Pulver, Granulate) wahlweise mit einem Bindemittel (z.B. Gelatine,
Hydroxypropylmethylzellulose) gemischt, Schmiermittel, inertem Verdünnungsmittel,
Konservierungsmittel, Sprengmittel (z.B. Natrium, Stärkeglykolat,
vernetzender Carboxymethylzellulose), oberflächenaktivem Mittel oder Dispergiermittel
zubereitet werden. Geeignete Bindemittel umfassen Stärke, Gelatine,
Naturzucker wie Glucose oder Betalactose, Maissüßungsmittel, natürliche und
synthetische Gummiarten wie Gummi arabicum, Tragacanth oder Natriumalginat,
Carboxymethylzellulose, Polyethylenglykol, Wachse und dergleichen.
Schmiermittel, die bei diesen Dosierformen verwendet werden, umfassen
Natriumoleat, Natriumstearat, Magnesiumstearat, Natriumbenzoat,
Natriumacetat, Natriumchlorid und dergleichen. Sprengmittel umfassen
ohne Einschränkung, Stärke, Methylzellulose,
Agar, Bentonit, Xanthangummi und dergleichen. Geformte Tabletten
können
durch Formen einer Mischung des pulverförmigen aktiven Bestandteils,
der mit einem inerten flüssigen
Verdünnungsmittel
befeuchtet ist, in einer geeigneten Maschine hergestellt werden.
-
Wahlweise
können
die Tabletten beschichtet oder eingeschnitten und so formuliert
werden, dass sie eine langsame oder gesteuerte Freisetzung des aktiven
Bestandteils bieten. Tabletten können
auch wahlweise mit einer enterischen Beschichtung versehen werden,
um die Freisetzung in Teilen des Darms, bei denen es sich nicht
um den Magen handelt, zu bieten.
-
Rezepturen,
die für
die topische Verabreichung im Mund geeignet sind, wobei der aktive
Bestandteil in einem geeigneten Träger gelöst oder suspendiert wird, umfassen
Pastillen, die den aktiven Bestandteil in einem mit Aromastoff versetzten
Träger – gewöhnlich Saccharose,
und Gummi arabicum oder Tragacanth – Gelatine, Glycerin oder Saccharose
und Gummi arabicum umfassen können;
und Mundwasser, die den aktiven Bestandteil in einem geeigneten
flüssigen
Träger
umfassen.
-
Topische
Anwendungen für
die Verabreichung der erfindungsgemäßen Methode entsprechend umfassen
Salben, Cremes, Suspensionen, Lotionen, Pulver, Lösungen,
Pasten, Gele, Spray, Aerosol oder Öl. Als Alternative kann eine
Rezeptur ein transdermales Pflaster oder einen Verband wie eine
mit einem aktiven Bestandteil und wahlweise einem oder mehreren
Trägern
oder Verdünnungsmitteln
imprägnierte
Binde umfassen. Um in Form eines transdermalen Abgabesystems verabreicht
zu werden, wird die Dosisverabreichung natürlich kontinuierlich anstatt
periodisch über
den Dosierzeitraum hindurch stattfinden.
-
Die
topischen Rezepturen können
wünschenswerterweise
eine Verbindung umfassen, die die Absorption oder Penetration des
aktiven Bestandteils durch die Haut oder andere betroffene Bereiche
verbessert. Beispiele derartiger dermaler Penetrationsverbesserer
umfassen Dimethylsulfoxid und verwandte Analoge.
-
Die Ölphase der
Emulsionen der Zusammensetzungen, die zum Behandeln von Patienten
bei der vorliegenden Erfindung verwendet werden, können auf
bekannte Weise aus bekannten Bestandteilen bestehen. Diese Phase
kann einen oder mehrere Emulgatoren umfassen. Beispielsweise umfasst
die Ölphase
zumindest einen Emulgator mit einem Fett oder einem Öl oder sowohl
mit einem Fett als auch einem Öl
oder ein hydrophiler Emulgator ist zusammen mit einem lipophilen
Emulgator, der als Stabilisator wirkt, eingearbeitet. Zusammen bildet
der Emulgator beziehungsweise die Emulgatoren mit oder ohne einem
Stabilisator bzw. Stabilisatoren ein emulgierendes Wachs und das
Wachs bildet zusammen mit dem Öl
und/oder Fett die emulgierende Salbenbase, die die dispergierte Ölphase der
Cremerezepturen bildet.
-
Emulgatoren
und Emulsionsstabilisatoren, die zur Verwendung in der Rezeptur
geeignet sind, umfassen Tween 60, Span 80, Cetosterylalkohol, Myristylalkohol,
Glycerylmonostearat und Natriumlaurylsulphat, Paraffin, geradkettige
oder verzweigtkettige, ein- oder zweibasige Alkylester, Mineralöl. Die Wahl
geeigneter Öle
oder Fette für
die Formulierung beruht auf dem Wunsch, erwünschte kosmetische Eigenschaften,
die erforderlichen Eigenschaften und Verträglichkeit mit dem aktiven Bestandteil,
zu erzielen.
-
Die
Verbindungen können
auch vaginal, beispielsweise als Vaginalzäpfchen, Tampons, Cremes, Gele, Pasten,
Schäume
oder Sprayrezepturen verabreicht werden, die zusätzlich zu dem aktiven Bestandteil
enthalten sind. Derartige Träger
sind im Stand der Technik bekannt.
-
Rezepturen
für die
rektale Verabreichung können
als Zäpfchen
mit einer geeigneten Base, die beispielsweise Kakaobutter oder Salicylat
umfasst, vorgelegt werden.
-
Rezepturen,
die für
die nasale Verabreichung geeignet sind, können in flüssiger Form, beispielsweise als
Nasenspray, Nasentropfen oder durch Aerosolverabreichung durch Zerstäuber, einschließlich wässrige oder ölhaltige
Lösungen
des aktiven Bestandteils verabreicht werden. Rezepturen für die nasale
Verabreichung, bei denen der Träger
ein Feststoff ist, umfassen ein grobes Pulver mit einer Teilchengröße von beispielsweise
weniger als etwa 100 Mikron, bevorzugt weniger als etwa 50 Mikron,
das auf die Art und Weise verabreicht wird, in der Schnupftabak
zu sich genommen wird, z.B. durch schnelles Einatmen durch den Nasengang
aus einem Behälter
des Pulvers, der nahe an die Nase gehalten wird.
-
Rezepturen,
die für
die parenterale Verabreichung geeignet sind, umfassen wässrige und
nichtwässrige
mit dem Blut des beabsichtigten Empfängers isotonische Substanzen
und wässrige
und nicht wässrige sterile
Suspensionen, die Suspendiersysteme enthalten können, die so konzipiert sind,
dass die Verbindung auf Blutkomponenten oder ein oder mehrere Organe
hin gezielt wird. Die Rezepturen können in dicht verschlossenen
Einheitsdosen- oder Mehrfachdosenbehältern, beispielsweise Ampullen
und Phiolen, vorliegen. Keine Vorbereitung benötigende Einspritzlösungen und
-suspensionen können
aus sterilen Pulvern, Granulaten und Tabletten der oben beschriebenen
Art zubereitet werden. Liposome werden für die intravenöse Verabreichung
der 2-Thienylimidazolo[4,5-b]pyridin-Verbindung bevorzugt.
-
Im
Allgemeinen sind Wasser, ein geeignetes Öl, physiologische Kochsalzlösung, wässrige Dextrose (Glucose)
und verwandte Zuckerlösungen
und Glykole wie Propylenglykole oder Polyethylenglykole geeignete Träger für parenterale
Lösungen.
Lösungen
für die
parenterale Verabreichung enthalten bevorzugt ein wasserlösliches
Salz des aktiven Bestandteils, geeignete Stabilisierungsmittel und
nötigenfalls Puffersubstanzen.
Antioxidationsmittel wie Natriumbisulfit, Natriumsulfit oder Ascorbinsäure sind
entweder als solche oder in Kombination geeignete Stabilisierungsmittel.
Zitronensäure
und deren Salze und Natrium-EDTA werden ebenfalls verwendet. Außerdem können parenterale
Lösungen
Konservierungsmittel wie Benzalkoniumchlorid, Methyl- oder Propylparaben
und Chlorbutanol enthalten. Geeignete pharmazeutische Träger sind
in Remington's Pharmaceutical
Sciences, Mack Publishing Company, einem Standardwerk auf diesem
Gebiet, beschrieben.
-
Intravenöse können die
bevorzugtesten Dosen im Bereich von etwa 1 bis etwa 10 mg/kg/Minute
während
einer Infusion mit konstanter Geschwindigkeit liegen. Thienylimidazolo[4,5]pyridin-Derivate
können
in Form einer einzigen täglichen
Dosis verabreicht werden oder die gesamte tägliche Dosis kann in aufgeteilten Dosen
zwei-, drei- oder viermal täglich
verabreicht werden. Die Thienylimidazolo[4,5]pyridin-Derivate können in
einer oder mehreren Dosen auf täglicher
Basis oder ein- bis dreimal wöchentlich
verabreicht werden.
-
Außerdem umfasst
die vorliegende Erfindung das Verabreichen von Verbindungen der
hier beschriebenen Rezeptur zur Verwendung in Form von Veterinärrezepturen,
die beispielsweise durch Methoden, die im Stand der Technik herkömmlich sind,
zubereitet werden können.
-
Nützliche
pharmazeutische Dosierformen zur Verabreichung der erfindungsgemäßen Verbindungen werden
wie folgt veranschaulicht:
-
Kapseln
-
Eine
große
Anzahl von Einheitskapseln wird durch Füllen von normalen zweiteiligen
harten Gelatinekapseln jeweils mit 100 Milligramm pulverförmigem aktivem
Bestandteil, 150 Milligramm Lactose, 50 Milligramm Zellulose und
6 Milligram Magnesiumstearat zubereitet.
-
Weichgelatinekapseln
-
Eine
Mischung von aktivem Bestandteil in einem verdaulichen Öl wie Sojaöl, Baumwollsamenöl oder Olivenöl wird zubereitet
und durch eine positive Verdrängungspumpe
in Gelatine unter Bildung weicher Gelatinekapseln injiziert, die
100 Milligramm des aktiven Bestandteils enthalten. Die Kapseln werden
gewaschen und getrocknet.
-
Tabletten
-
Eine
große
Anzahl von Tabletten werden durch herkömmliche Verfahrensweisen derart
zubereitet, dass die Dosiseinheit 100 Milligramm aktiver Bestandteil,
0,2 Milligramm colloidales Siliciumdioxid, 5 Milligramm Magnesiumstearat,
275 Milligramm mikrokristalline Zellulose, 11 Milligramm Stärke und
98,8 Milligramm Lactose betrug. Geeignete Beschichtungen können zum
Erhöhen
der Schmackhaftigkeit oder zum Verzögern der Absorption aufgebracht
werden.
-
Injizierbare
Substanz
-
Eine
parenterale Zusammensetzung, die zur Verabreichung durch Einspritzen
geeignet ist, wird durch Einrühren
von 1,5 Gewichtsprozent aktivem Bestandteil in 10 Volumenprozent
Polypropylenglykol und Wasser zubereitet. Die Lösung wird mit Natriumchlorid
isotonisch gemacht und sterilisiert.
-
Suspension
-
Eine
wässrige
Suspension wird für
die orale Verabreichung so zubereitet, dass jeweils 5 ml 100 mg feinverteilten
aktiven Bestandteil, 200 mg Natriumcarboxymethylzellulose, 5 mg
Natriumbenzoat, 1,0 g Sorbitlösung,
U.S.P., und 0,025 ml Vanillin enthalten.
-
Mechanismus
-
Der
Mechanismus der Wirkung der 2-(2-Thienyl)-imidazolo[4,5-b]pyridin-Derivate
ist nicht bekannt. Weder 2-(2-Thienyl)-imidazolo[4,5-b]pyridin
noch dessen Hydrochloridsalze haben eine Aktivität als Proteaseinhibitor, wenn
sie mit Hilfe einer fluorometrischen Methode gescreent worden sind,
oder als Integraseinhibitor gezeigt. Diese Ergebnisse sind in den
folgenden Tabellen zusammengefasst. 2-Thienylimidazolo[4,5]pyridinhydrochloridsalz
autofluoresziert und kann diesen Test in gewisser Weise stören.
-
Proteaseinhibitionsassay
-
Die
Proteaseinhibition wird mit Hilfe einer fluorometrischen Methode
beurteilt. Enzym (Bachem) wird mit 50 mM NaO-AC, 5 mM DTT, 2 mM EDTA, 10 % Glycerin
(pH 5,0) auf 116 μgm/ml
verdünnt
und als Proben von 10 μl
bei –20°C gelagert,
HIV Protease-Substrat I (Molecular Probes) wird auf eine Arbeitskonzentration von
0,32 nMol/μl
verdünnt.
Das Enzym (20 μl)
und das Arzneimittel (20 μl)
werden in jede Vertiefung einer Mikrotiterplatte wie zweckmäßig eingegeben.
Positive und negative Kontrollen werden parallel beurteilt. Die
Fluoreszenz wird auf Fluorskan II Laborsystemen unter Anwendung
von 355 nm/460 nm bei 37°C
bei einer Zeit von 0 und in Abständen
von 30 Minuten 2 Stunden lang quantitativ bestimmt. In Fällen, wo
die Autofluoreszenz die Verwendung des fluorometrischen HIV-1-Proteaseassays
ausschließt
oder eine Bestätigung
eines Ergebnisses erforderlich ist, kann ein Proteaseassay auf HPLC-Basis
angewendet werden.
-
Integraseinhibitionsassay
-
Es
handelt sich um ein biochemisches Integraseassay, das von Craigie
et al (HIV, Band 2: A practical Approach – ein praktischer Ansatz) Biochemistry,
Molecular Biology and Drug Discovery, Verfasser J. Karn 1995) beschrieben
worden ist, zum Screenen von Mitteln auf ihre Fähigkeit hin, die HIV-1-Integrase
zu hemmen. Bei diesem System dient ein kinasiertes Oligonukleotid
als Target des 3'-Verarbeitens
und der darauffolgenden Strangübertragungsreaktion.
Die 3'-Verarbeitungsreaktion
involviert die Entfernung von 2 Nukleotiden von den 3'-Enden des Substrats
und daraufhin folgt die Strangübertragungsreaktion,
bei der die 3'-Enden
an die ausgesetzten 5'-Enden
angeknüpft
werden. Die Reaktionsmischung von 20 μl enthält 25 mM MOPS (pH 7,2), 100
g/ml BSA, 10 mM β-Mercaptoethanol,
10 % Glycerin, 7,5 mM MnCl2, 25 nM (7 ng)
Substrat (Oligos usw., Wilsonville, OR) und 200 nM (128 ng) Integrase
(NIAID AIDS Forschungs- und Bezugsreagenzprogramm, Bethesda, MD).
Die Reaktion findet für
1–2 Stunden
bei 37°C
statt und wird durch Zugabe von 20 μl der Sequenzierstoplösung (USB
Amersham, Arlington Heights, IL) beendet. Die Reaktionsprodukte
werden durch Autoradiografie gefolgt von Elektrophorese in 15 %
Polyacrylamid 6M Harnstoffgel sichtbar gemacht. Das Substrat migriert
als 30Mer, das Produkt der 3'-Verarbeitung
migriert als N-2-Bande
und die Strangübertragungsprodukte
migrieren langsamer bei verschiedenen Größen, die größer als das Substrat sind.
-
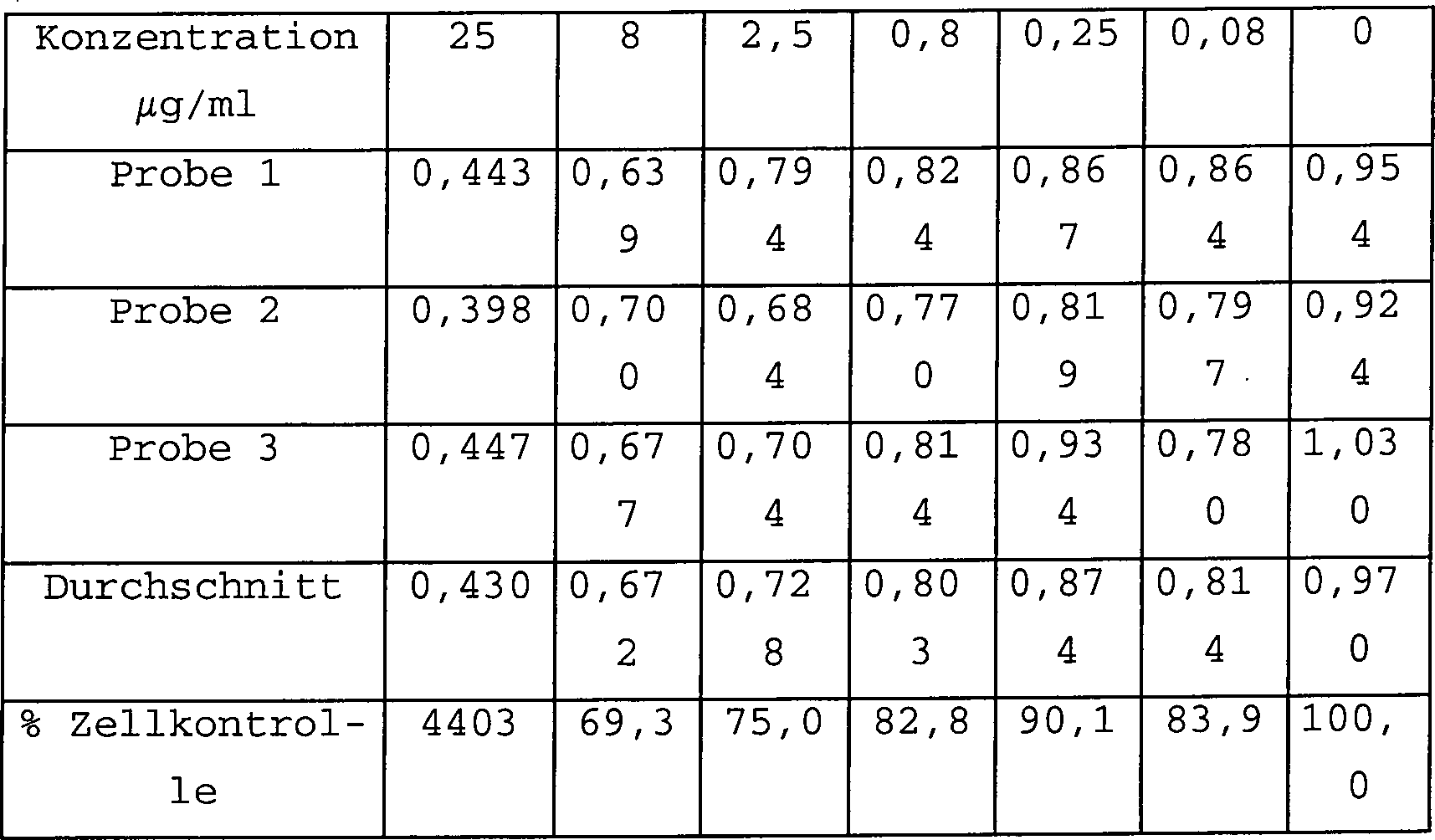
Proteaseinhibition
bei 654021F – einem
bekannten Proteaseinhibitor
-
Proteaseinhibition
durch 2-(2-Thienyl)-imidazolo[4,5-b]pyridin
-
Proteaseinhibition
durch 2-(2-Thienyl)-imidazolo[4,5-b]pyridinhydrochloridsalz
-
Der
EC50-Wert beträgt 0,699 μM bei 654021.
-
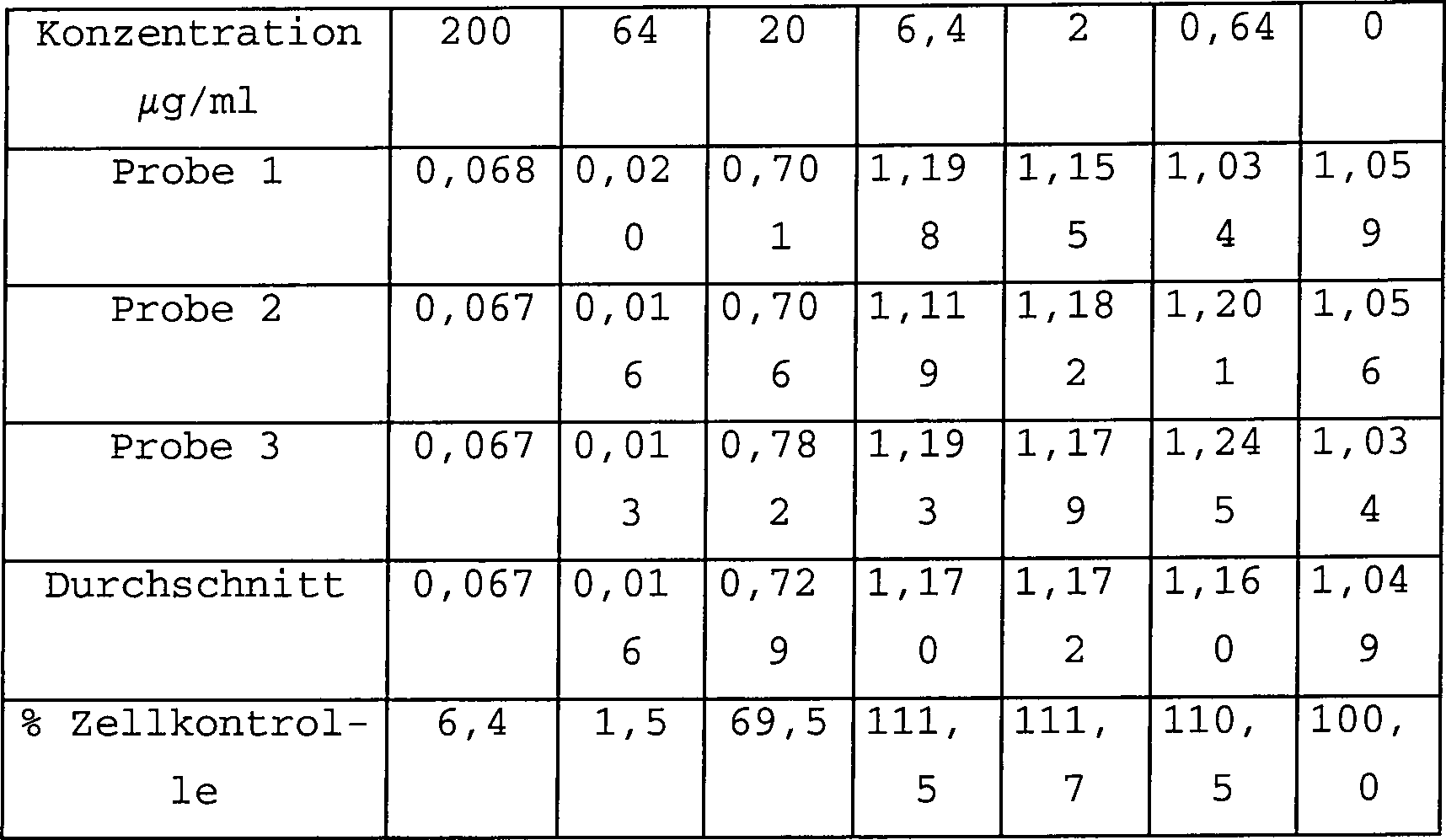
HIV-1-Integraseinhibition
2-(2-Thienyl)-imidazolo[4,5-b]pyridin
-
HIV-1-Integraseinhibition
2-(2-Thienyl)-imidazolo[4,5-b]pyridinhydrochloridsalz
-
HIV-1-Integraseinhibition
durch TPX – einem
bekannter Integraseinhibitor
-
Der
EC50-Wert beträgt 0,648 μM für TPX und > 100 μg/ml
sowohl für
das 2-(2-Thienyl)-imidazolo[4,5-b]pyridin als auch dessen Hydrochloridsalz.
-
Die
folgenden Beispiele sind veranschaulichend und sollen nicht auf
die Erfindung begrenzt sein. Folgende Methoden wurden bei diesen
Tests angewendet:
-
Viruszubereitung:
-
Ein
Aliquot eines Virus, dessen Konzentration vorbestimmt worden war,
wird aus der Gefriertruhe (–80°C) herausgenommen
und in einem biologisch unbedenklichen Schrank langsam auf Raumtemperatur aufgetaut.
Das Virus wird erneut suspendiert und in einem Gewebekulturmedium
derart verdünnt,
dass die Menge an Virus, die in jede Vertiefung in einem Volumen
von 50 μl
eingegeben wird, die Menge ist, die bestimmt worden ist, um eine
vollständige
Zellabtötung
am 6. Tag nach der Infektion zu ergeben. Im Allgemeinen erforderten
die mit IIIB-Isolat von HIV hergestellten Viruspools den Zusatz
von 5 μl
Virus pro Vertiefung. Die Pools von RF-Virus waren 5 – 10-mal
stärker
und erforderten 0,5 – 1 μl Virus pro
Vertiefung. Die TCID50-Berechnung durch
Endpunkttitrierung in CEM-SS-Zellen zeigte an, dass die Infektionsmultiplizität dieser
Assays im Bereich von 0,005 bis 2,5 lag.
-
Plattenformat:
-
Das
Format der Testplatte ist standardisiert worden. Jede Platte enthält Zellkontrollvertiefungen
(ausschließlich
Zellen), Viruskontrollvertiefungen (Zellen plus Virus), Arzneimitteltoxizitäts-Kontrollvertiefungen (Zellen
plus ausschließlich
Arzneimittel), kolorimetrische Arzneimittel-Kontrollvertiefungen (ausschließlich Arzneimittel)
sowie Versuchsvertiefungen (Arzneimittel plus Zellen plus Virus).
-
XTT-Färbung von Screenplatten:
-
Nach
einer Inkubation von 6 Tagen (oder der Versuchsinkubationszeit)
bei 37°C
in einem Inkubator mit 5 % Kohlendioxid werden die Testplatten durch
Färben
mit dem Tetrazoliumfarbstoff XTT analysiert. XTT-Tetrazolium wird
durch mitochondriale Enzyme der im Zusammenhang mit dem Stoffwechsel
aktiven Zellen unter Bildung eines löslichen Formazanprodukts metabolisiert,
was die schnelle quantitative Analyse der Inhibition der HIV-induzierten
Zellabtötung
durch Anti-HIV-Testsubstanzen
erlaubt. Am 6. Tag nach der Infektion werden die Platten aus dem
Inkubator entfernt und beobachtet. Die Verwendung von Mikrotiterrundplatten
erlaubt die schnelle makroskopische Analyse der Aktivität einer
vorgegebenen Testverbindung durch Bestimmung der Pelletgröße. Die
Ergebnisse der makroskopischen Beobachtungen wurden bestätigt und
durch weitere mikroskopische Analyse verbessert.
-
XTT-Lösung wird
täglich
als Stammlösung
von 1 mg/ml in PBS zubereitet. Phenazinmethosulfat-(PMS) Lösung wird
in einer Konzentration von 15 mg/ml in PBS zubereitet und im Dunkeln
bei –20°C gelagert.
XTT/PMS-Stammlösung
wird sofort vor der Anwendung durch Verdünnen des PMS im Verhältnis von 1:100
in PBS und Zusetzen von 40 μl
XTT-Lösung
pro ml zubereitet. Fünfzig
Mikroliter XTT/PMS werden in jede Vertiefung der Platte eingegeben
und die Platte wird wiederum 4 Stunden bei 37°C inkubiert. Haftfähige Plattenversiegler
werden anstatt der Deckel verwendet, die versiegelte Platte wird
mehrere-male umgedreht, um das lösliche
Formazanprodukt zu mischen und die Platte wird spectrofotometrisch
bei 450 nm mit einem Vmax-Plattenleser von Molecular Devices abgelesen.
Die Zellreduktion in Prozent, die Zelllebefähigkeit in Prozent, IC25,50&95,
und TC25,50&95 können dann
berechnet werden.
-
Analyse der Reverstranskriptaseaktivität:
-
Eine
Reverstranskriptase-(RT)-Reaktion auf Mikrotiterbasis wird verwendet
(Buckheit et al (1991) – AIDS
Research and Human Retroviruses (Aids Forschung und menschliche
Retroviren) 7:295–302).
Tritiiertes Thymindintriphosphat (NEN)(TTP) wird erneut in destilliertem
Wasser in einer Konzentration von 5 Ci/ml suspendiert. Poly rA und
Oligo dT werden als Stammlösung
zubereitet, die bei –20°C gelagert
wird. Der RT-Reaktionspuffer wird täglich frisch zubereitet und
besteht aus 125 μl
1M EGTA, 125 μm
Wasser, 125 μl
Triton X-100, 50 μl
Tris (pH 7,4), 50 μl
MDDT und 40 μl
1M MgCl2. Diese drei Lösungen werden miteinander im
Verhältnis
von 1 Teil TTP, 2,5 Teilen Poly rA:Oligo dT, 2,5 Teilen Reaktionspuffer
und 4 Teilen destilliertem Wasser gemischt. Zehn Mikroliter dieser
Reaktionsmischung werden in eine Mikrotiter-Rundplatte eingegeben
und 15 μl
virushaltige überstehende
Lösung
wird hinzugegeben und gemischt. Die Platte wird 60 Minuten bei 37°C inkubiert.
Auf die Reaktion hin wird das Reaktionsvolumen auf Filtermatten
getüpfelt,
6-mal jeweils 5 Minuten lang in einem 5 %igen Natriumphosphatpuffer,
2-mal jeweils 1 Minute lang in destilliertem Wasser, 2-mal jeweils
1 Minute lang in 70 % Ethanol gewaschen und dann getrocknet. Die
getrocknete Filtermatte wird in einen Kunststoffprobenbeutel eingegeben,
Betaplattenszintillationsfluid wird zugegeben und der Beutel hitzeversiegelt.
Die enthaltene Radioaktivität
wird unter Anwendung eines Wallac-Mikrobetaszintillationszählers quantifiziert.
-
Die
akute Infektion der meisten etablierten menschlichen Zelllinien
mit HIV-1-Virus führt
zum schließlichen
Herstellen einer konstitutiven, viruserzeugenden, chronisch infizierten
Zelllinie. Die Zellen können
ohne Verlust an Virusbildung für
längere
Zeitspannen in einer Kultur verweilen. Diese Zellen können zum
Beurteilen der Wirkungen von Anti-HIV-Verbindungen auf die Syncytium-Bildung
oder zum Beur teilen der Wirkungen von Anti-HIV-Verbindungen auf
die Viruserzeugungsspiegel aus diesen Zellen verwendet werden. Chronisch
infizierte Zelllinien weisen kaum eine oder keine CD4-Zelloberfläche auf
und können
mit anderen Isolaten von HIV-1 nicht überinfiziert werden. Jede dieser
Zellen enthält
ein integriertes HIV-Genom oder Provirus. Chonisch infizierte CEM-,
H9- und U937-Zelllinien sind durch Southern Research Institute,
Frederick, MD, zubereitet und kultiviert worden und sind dort erhältlich.
-
CEM-SS-Zellen,
die chronisch mit HIV-Isolat infiziert sind, beispielsweise SKI
(CEM-SKI) werden in RPMI1640-Gewebekulturmedium
kultiviert, das mit 10 % fötalem
Rinderserum und Antibiotika ergänzt
ist. Die Auswahl wird durch Kultivieren der Zellen in Gegenwart
der zu prüfenden
Verbindung in TZ5-Kolben durchgeführt. CEM-SKI oder andere infizierte
Zellen ohne zugesetztem Arzneimittel werden als Kontrollzellen verwendet.
Man lässt
die Zellen bis zu einer Dichte von etwa 1 × 106 Zellen/ml anwachsen und
sie werden dann in einer Verdünnung
von 1:10 aufbewahrt. Nach einer Zeitspanne, gewöhnlich einem einwöchigen Abstand
der Arzneimittelbehandlung, werden die Zellen beurteilt, um zu bestimmen,
ob die Hemmungsaktivität
der Verbindung durch die Behandlung der Zellen mit einer der Verbindungen
beeinflusst worden ist. Die Arzneimittelkonzentration in dem Kolben
wird dann zweifach erhöht
und die Zellen wie oben gehalten.
-
Die
Zellpopulationen enthalten integrierte Kopien des HIV-Genoms und erzeugen
konstitutiv HIV in einem relativ hohen Niveau oder sind latent infiziert
und erzeugen nur Virus nach Stimulierung mit Phorbolestern, Tumornekrosefaktor
oder IL6 (U1 und ACH2). Reduktionen der Virusprodukte werden beim
Quantifizieren der Aktivität
der überstehenden
Reverstranskriptase beobachtet.
-
Die
Toxizitätswerte
werden durch XTT gemessen und die Aktivität der Verbindung in den Tests
wird durch eine Reverstranskriptaseanalyse gemessen.
-
Beispiel 1
-
HIV
-
Bei
einem in vitro-Screentest von 2-(2-Thienyl)imidazolo[4,5-b]pyridin
gegen ein HIV-2-Virus, CEMROD, wurden die folgenden Daten erhalten.
Tabelle 1 zeigt die Ergebnisse eines Tests und Tabelle 2 gibt die Ergebnisse
einer Duplikationsstudie wider.
-
Tabelle
1 Reverstranskriptaseaktivität
-
-
-
Tabelle
2 Reverstranskriptaseaktivität
-
-
-
Wie
bei den höheren
Konzentrationen zu sehen ist, ist 2-(2-Thienyl)-imidazolo[4,5-b]pyridin nicht
toxisch und kann sich bei der Behandlung von HIV als wirksam erweisen.
-
Beispiel 2
-
HIV-1
-
Eine
Langzeit-in vitro-Studie von 2-(2-Thienyl)imidazolo[4,5-b]pyridin
gegen eine HIV-1-Zelllinie, CEMSKI, wurde bei drei verschiedenen
Niveaus durchgeführt.
Die Ergebnisse mit CEMSK-Zellen wurden in wöchentlichen Abständen aufgezeichnet.
Die Reverstranskriptasedaten sind unten zusammengefasst.
-
-
Dieser
Test wurde über
242 Tage durchgeführt
und die Daten blieben gleich.
-
Die
CEMSKI-Zelllinie ist ein Viralstamm der CEMSS-Zelllinie.
-
Als
das Hydrochloridsalz von 2-(2-Thienyl)-imidazolo[4,5-b]pyridin auf die
gleiche Weise untersucht wurde, wurden über 60 Tage ähnliche
Ergebnisse erhalten.
-
Beispiel 3
-
CFMRF
-
Eine
Langzeit-in vitro-Studie von 2-(2-Thienyl)imidazolo[4,5-b]pyridin
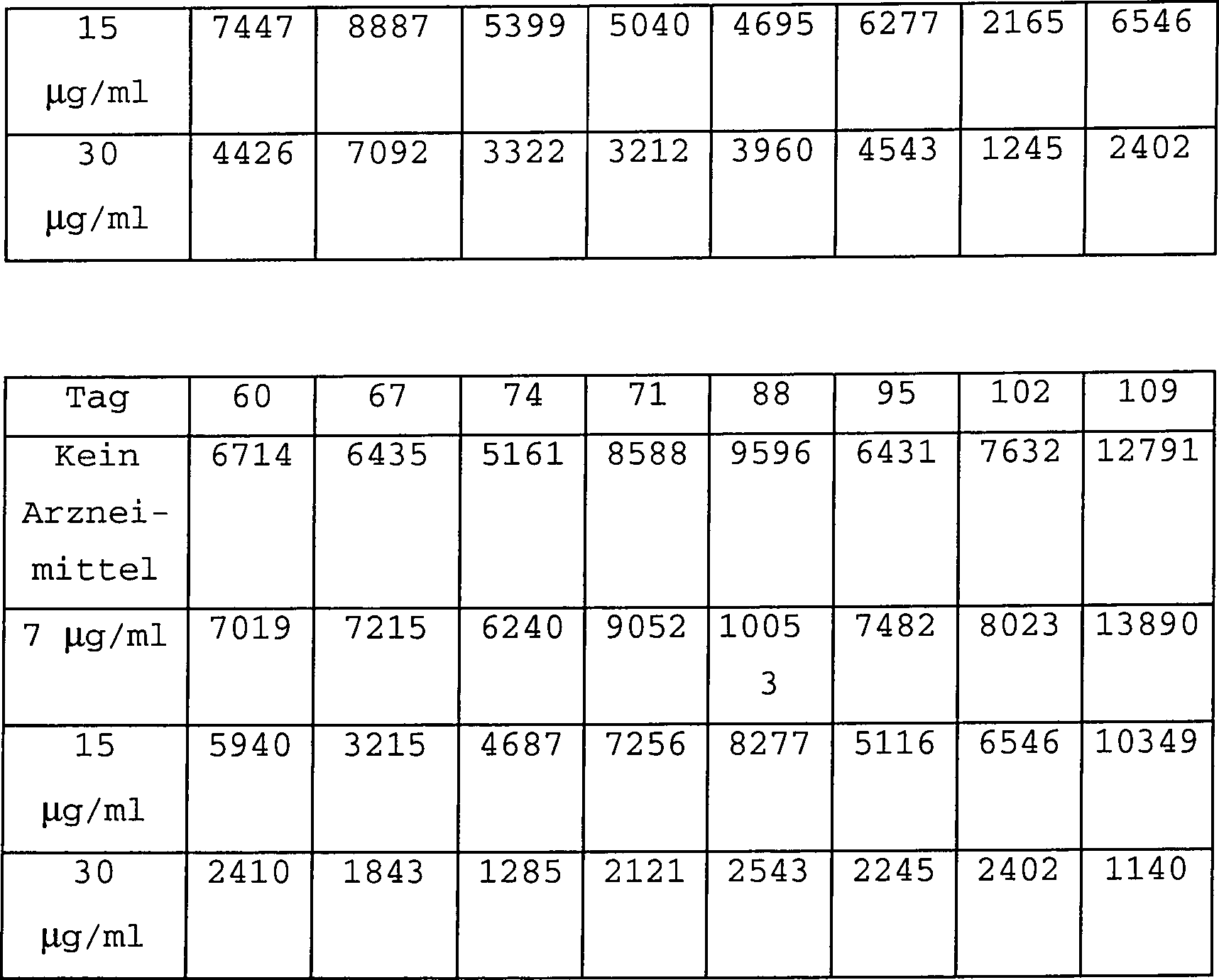
gegen eine HIV-1-Zelllinie, CEMRF, wurde bei drei verschiedenen
Niveaus durchgeführt.
Die Ergebnisse mit CEMRF-Zellen wurden in wöchentlichen Abständen aufgezeichnet.
Die Reverstranskriptasedaten sind unten zusammengefasst. Die mit 2-(2-Thienyl)-imidazolo[4,5-b]pyridin behandelten
Kulturen zeigten keine Hinweise darauf, dass sich bis zum 186. Tag
bei einem Niveau von 15 und 30 μg/ml
eine Resistenz entwickelte. CEMRF ist eine chronische HIV-Zelllinie.
-
-
-
Dieser
Test wurde über
242 Tage durchgeführt
und die Daten blieben gleich.
-
Beispiel 4
-
CEMIIIB
-
Eine
Langzeit-in vitro-Studie von 2-(2-Thienyl)imidazolo[4,5-b]pyridin
gegen eine HIV-1-Zelllinie, CEMIIIB, wurde bei drei verschiedenen
Niveaus durchgeführt.
Die Ergebnisse mit CEMIIIB-Zellen wurden in wöchentlichen Abständen aufgezeichnet.
Die Reverstranskriptasedaten sind unten zusammengefasst. CEMIIIB ist
ein Viralstamm der CEMSS-Zelllinie und eine chronische HIV-Zelllinie.
-
-
-
Dieser
Test wurde über
242 Tage durchgeführt
und die Daten blieben gleich.
-
Als
das Hydrochloridsalz von 2-(2-Thienyl)-imidazolo[4,5-b]pyridin auf die
gleiche Weise untersucht wurde, wurden über 60 Tage ähnliche
Ergebnisse erhalten.
-
Beispiel 5
-
CEMROD
-
Eine
Langzeit-in vitro-Studie von 2-(2-Thienyl)imidazolo[4,5-b]pyridin
gegen eine HIV-1-Zelllinie, CEMROD, wurde bei drei verschiedenen
Niveaus durchgeführt.
Die Ergebnisse mit CEMROD-Zellen wurden in wöchentlichen Abständen aufgezeichnet.
Die Reverstranskriptasedaten sind unten zusammengefasst.
-
-
-
Dieser
Test wurde über
242 Tage durchgeführt
und die Daten blieben gleich.
-
Beispiel 6
-
U937IIIB
-
Eine
Langzeit-in vitro-Studie von 2-(2-Thienyl)imidazolo[4,5-b]pyridin
gegen eine HIV-1-Zelllinie, U937IIIB, wurde bei drei verschiedenen
Niveaus durchgeführt.
Die Ergebnisse mit U937IIIB-Zellen wurden in wöchentlichen Abständen aufgezeichnet.
Die Reverstranskriptasedaten sind unten zusammengefasst.
-
-
-
Dieser
Test wurde über
242 Tage durchgeführt
und die Daten blieben gleich.
-
Beispiel 7
-
U937RF
-
Eine
Langzeit-in vitro-Studie von 2-(2-Thienyl)imidazolo[4,5-b]pyridin
gegen eine HIV-1-Zelllinie, U937RF, wurde bei drei verschiedenen
Niveaus durchgeführt.
Die Ergebnisse mit U937RF-Zellen wurden in wöchentlichen Abständen aufgezeichnet.
Die Reverstranskriptasedaten sind unten zusammengefasst.
-
-
-
Dieser
Test wurde über
242 Tage durchgeführt
und die Daten blieben gleich.
-
Als
das Hydrochloridsalz von 2-(2-Thienyl)-imidazolo[4,5-b]pyridin auf die
gleiche Weise untersucht wurde, wurden über 60 Tage ähnliche
Ergebnisse erhalten.
-
Ähnliche
Ergebnisse werden mit U937KN1272, einem proteaseresistenten Stamm
erhalten, wie unten berichtet.
-
-
-
Dieser
Test wurde über
242 Tage durchgeführt
und die Daten blieben gleich.
-
Als
das Hydrochloridsalz von 2-(2-Thienyl)-imidazolo[4,5-b]pyridin auf die
gleiche weise untersucht wurde, wurden über 60 Tage ähnliche
Ergebnisse erhalten.
-
Beispiel 8
-
Hepatitis
-
In
einem in vitro-Viruserzeugungstest von Hepatitis B, HEPG2 2,2,15,
wurden folgende Ergebnisse mit 2-(2-Thienyl)imidazolo[4,5-b]pyridin
erhalten.
-
DNA-Kopienummer
(pro 3 μl)
-
-
-
Die
IC50 beträgt 1,6 μg/ml; die TC50 beträgt 16,3 μg/ml und
der therapeutische Index oder TI beträgt 10,1.
-
In
einem Replikatversuch beträgt
die IC50 10,71 μg/ml, die TC50 beträgt 16,8 μg/ml und
der TI beträgt 23,4.
-
Zum
Vergleich wurde 3TC getestet und es wurden die folgenden Daten erhalten.
-
DNA-Kopienummer
(pro 3 μl)
-
-
Die
IC50 beträgt 0,089 μg/ml; die TC50 beträgt > 1 μg/ml und der TI beträgt 14,6.
-
In
einem Replikatversuch beträgt
die IC50 0,021 μg/ml; die TC50 beträgt > 1 μg/ml und der TI beträgt > 47,6.
-
2-(2-Thienyl)-imidazolo[4,5-b]pyridin
kann zum Behandeln von Hepatitis B verwendet werden.
-
Beispiel 9
-
Testen von
Herpes simplex
-
2-(2-Thienyl)-imidazolo[4,5-b]pyridin
wurde gegen HSV-2MS, einem Herpes simplex Virus-2 in vero Zellen
getestet und mit Acyclovir verglichen. Die IC50 für Acyclovir
beträgt
0,81 und 0,85 in einer Replikatstudie. Die TC50 beträgt >1 und der TI oder therapeutische
Index beträgt > 1,2. Für 2-(2-Thienyl)-imidazolo[4,5-b]pyridin
beträgt
die IC50 62,1, die TC50 beträgt 82,8
und der TI oder therapeutische Index beträgt 1,3.
-
Beispiel 10
-
Kaposi-Sarkom-Virus
-
2-(2-Thienyl)-imidazolo[4,5-b]pyridin
wurde gegen Kaposi-Sarkom,
einem Herpesvirus, in vitro unter Zuhilfenahme der HHV8-Zelllinie,
TPA-induzierten BCBL-1-Zellen, getestet. Die DNA-Kopie-Nummer und
der Toxizitätswert
wurden gemessen und mit Cidofovir verglichen.
-
Daten
für Cidofovir DNA-Kopienummer
(pro 3 il)
-
-
-
- IC50 μM = 1,1
- TC50 μM
= 21,1
- TI = 19,2
-
Daten
für 2-(2-Thienyl)-imidazolo[4,5-b]pyridin DNA-Kopienammer
(pro 3 μl)
-
-
-
- IC50 μM = 36,4
- TC50 μM
= 32,6
-
Beispiel 11
-
Antifungale Aktivität
-
2-(2-Thienyl)-imidazolo[4,5-b]pyridin
wurde gegen eine Reihe von Pilzen in vitro getestet. Es war gegen
Cryptococcus neoformans und Curvularia lunata aktiv. Die tödliche Aktivität gegen
C. neoformans ist stark genug, dass es eindeutig statisch gegen
diese Hefe ist. Dieser Test wurde mit Hilfe einer Methode durchgeführt, die
auf der NCCLS-Bezugsmethode
M-27A basiert, die 1997 veröffentlicht
worden ist. Lösungsmittel, Medium
und Wachstumskontrollen wurden mit dem Test angeordnet. Sobald diese
zum Validieren der Prüfleistung
abgelesen worden waren, wurden die QC-Pilze abgelesen, um sicherzustellen,
dass sie die erwarteten Er gebnisse boten. Diese Schritte validierten
das Prüfsystem.
DMSO wurde als chemisches Arzneimittellösungsmittel verwendet. Diese
Tests wurden auf die Inkubation bei 35 ºC hin abgelesen, wenn die QC-Organismen
(Candida spp.) ein gutes Wachstum zeigten. MIC-Werte waren Konzentrationen,
bei denen das Wachstum im Vergleich mit dem Kontrollwachstum gehemmt
oder um mindestens 90 % reduziert war. Der 90 %-Abbruch ist bei Azolen notwendig, die
statisch und nicht tödlich
sind. Das FMC- oder tötliche
Niveau wurde durch Subkultivieren einer Probe aus jedem Röhrchen bestimmt,
die kein Wachstum aufwies.
-
Curvularia
lunata verursacht eine Pilzinfektion der Hornhaut und tiefe Infektionen
der Organe. Bei immunkompromittierten Patienten ist es opportunistisch.
-
Cryptococcus
neoformans ist ein opportunistischer Erreger, der sich bei AIDS-Patienten
auf das Zentralnervensystem auswirkt und ist eine Hefe, die eine
Polysaccharidschutzkapsel aufweist, wobei es sich um einen Ständerpilz
handelt.
-
Die
für die
getesteten Verbindungen verwendeten Abkürzungen sind:
- AmB
- ist Amphotericin B
- Thia
- ist Thiabendazol
- Methyl
- ist Methyl-1,2-Benzimidazolcarbamat
oder Benomyl
- Itra
- ist Itraconazol
- THP
- ist 2-(2-Thienyl)-imidazolo[4,5-b]pyridin.
-
MIC-Daten
(ìg/ml) Culvularia
lunata
-
MIC-Daten
(ìg/ml) Cryptococcus
neoformans
-
MIC-Daten
(ìg/ml) Cryptococcus
neofromans