DE60008763T2 - Oxazol ppar antagonisten - Google Patents
Oxazol ppar antagonisten Download PDFInfo
- Publication number
- DE60008763T2 DE60008763T2 DE60008763T DE60008763T DE60008763T2 DE 60008763 T2 DE60008763 T2 DE 60008763T2 DE 60008763 T DE60008763 T DE 60008763T DE 60008763 T DE60008763 T DE 60008763T DE 60008763 T2 DE60008763 T2 DE 60008763T2
- Authority
- DE
- Germany
- Prior art keywords
- phenyl
- methyl
- ethyl
- benzoylphenyl
- ethoxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 102000003728 Peroxisome Proliferator-Activated Receptors Human genes 0.000 title claims abstract description 16
- 108090000029 Peroxisome Proliferator-Activated Receptors Proteins 0.000 title claims abstract description 16
- 239000005557 antagonist Substances 0.000 title abstract description 20
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 title description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims abstract description 69
- 150000001875 compounds Chemical class 0.000 claims abstract description 38
- 108010016731 PPAR gamma Proteins 0.000 claims abstract description 28
- 150000003839 salts Chemical class 0.000 claims abstract description 16
- 239000012453 solvate Substances 0.000 claims abstract description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims abstract description 6
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims abstract description 5
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims abstract description 5
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims abstract description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims abstract description 3
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 3
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 3
- 229910052717 sulfur Inorganic materials 0.000 claims abstract description 3
- -1 2-benzoylphenyl Chemical group 0.000 claims description 136
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 claims description 66
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 66
- 102100038825 Peroxisome proliferator-activated receptor gamma Human genes 0.000 claims description 19
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 15
- 238000011282 treatment Methods 0.000 claims description 14
- 201000010099 disease Diseases 0.000 claims description 13
- 238000002360 preparation method Methods 0.000 claims description 7
- 208000008589 Obesity Diseases 0.000 claims description 6
- 206010012601 diabetes mellitus Diseases 0.000 claims description 6
- 235000020824 obesity Nutrition 0.000 claims description 6
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims description 5
- 239000008103 glucose Substances 0.000 claims description 5
- 230000001404 mediated effect Effects 0.000 claims description 4
- 239000003873 peroxisome proliferator activated receptor gamma antagonist Substances 0.000 claims description 3
- 230000002265 prevention Effects 0.000 claims description 3
- 208000001145 Metabolic Syndrome Diseases 0.000 claims description 2
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 claims description 2
- 230000001575 pathological effect Effects 0.000 claims description 2
- 208000011580 syndromic disease Diseases 0.000 claims description 2
- 239000003814 drug Substances 0.000 claims 1
- 229940079593 drug Drugs 0.000 claims 1
- 238000000034 method Methods 0.000 abstract description 13
- 239000000556 agonist Substances 0.000 abstract description 11
- 102000005962 receptors Human genes 0.000 abstract description 10
- 108020003175 receptors Proteins 0.000 abstract description 10
- 102000000536 PPAR gamma Human genes 0.000 abstract description 9
- 229910052739 hydrogen Inorganic materials 0.000 abstract description 9
- 239000001257 hydrogen Substances 0.000 abstract description 9
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 abstract description 9
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 abstract description 8
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 abstract description 8
- 230000004913 activation Effects 0.000 abstract description 8
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 abstract description 8
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 abstract description 6
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 abstract description 6
- 102100038495 Bile acid receptor Human genes 0.000 abstract description 5
- 101150095442 Nr1h2 gene Proteins 0.000 abstract description 5
- 108090000865 liver X receptors Proteins 0.000 abstract description 5
- 102000004311 liver X receptors Human genes 0.000 abstract description 5
- 101000603876 Homo sapiens Bile acid receptor Proteins 0.000 abstract description 4
- 230000015572 biosynthetic process Effects 0.000 abstract description 4
- 229940125388 beta agonist Drugs 0.000 abstract 1
- 239000002876 beta blocker Substances 0.000 abstract 1
- 238000007385 chemical modification Methods 0.000 abstract 1
- 238000009510 drug design Methods 0.000 abstract 1
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 47
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 45
- 239000000203 mixture Substances 0.000 description 37
- 210000004027 cell Anatomy 0.000 description 29
- 239000003446 ligand Substances 0.000 description 27
- 238000001819 mass spectrum Methods 0.000 description 25
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 24
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 23
- 235000019439 ethyl acetate Nutrition 0.000 description 22
- 239000000243 solution Substances 0.000 description 22
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 18
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- 239000000741 silica gel Substances 0.000 description 17
- 229910002027 silica gel Inorganic materials 0.000 description 17
- 238000012360 testing method Methods 0.000 description 17
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 16
- 238000005160 1H NMR spectroscopy Methods 0.000 description 12
- ZZCHHVUQYRMYLW-HKBQPEDESA-N farglitazar Chemical compound N([C@@H](CC1=CC=C(C=C1)OCCC=1N=C(OC=1C)C=1C=CC=CC=1)C(O)=O)C1=CC=CC=C1C(=O)C1=CC=CC=C1 ZZCHHVUQYRMYLW-HKBQPEDESA-N 0.000 description 12
- 239000006260 foam Substances 0.000 description 12
- 239000007787 solid Substances 0.000 description 11
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 10
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 10
- 102100038831 Peroxisome proliferator-activated receptor alpha Human genes 0.000 description 10
- 108091008725 peroxisome proliferator-activated receptors alpha Proteins 0.000 description 10
- 238000003756 stirring Methods 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 9
- 238000009472 formulation Methods 0.000 description 9
- 229960004586 rosiglitazone Drugs 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 230000005764 inhibitory process Effects 0.000 description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 239000012267 brine Substances 0.000 description 7
- 230000000694 effects Effects 0.000 description 7
- 239000000543 intermediate Substances 0.000 description 7
- 239000003921 oil Substances 0.000 description 7
- 235000019198 oils Nutrition 0.000 description 7
- 238000007363 ring formation reaction Methods 0.000 description 7
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 7
- FKASFBLJDCHBNZ-UHFFFAOYSA-N 1,3,4-oxadiazole Chemical compound C1=NN=CO1 FKASFBLJDCHBNZ-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- 108010015181 PPAR delta Proteins 0.000 description 6
- 102100038824 Peroxisome proliferator-activated receptor delta Human genes 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 108091008765 peroxisome proliferator-activated receptors β/δ Proteins 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 238000013519 translation Methods 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- BYJQIHKCAGVCSP-HKBQPEDESA-N (2s)-2-(2-benzoylanilino)-3-[4-[2-(5-methyl-2-phenyl-1,3-oxazol-4-yl)ethoxy]phenyl]propanehydrazide Chemical compound N([C@@H](CC1=CC=C(C=C1)OCCC=1N=C(OC=1C)C=1C=CC=CC=1)C(=O)NN)C1=CC=CC=C1C(=O)C1=CC=CC=C1 BYJQIHKCAGVCSP-HKBQPEDESA-N 0.000 description 5
- BBVIDBNAYOIXOE-UHFFFAOYSA-N 1,2,4-oxadiazole Chemical compound C=1N=CON=1 BBVIDBNAYOIXOE-UHFFFAOYSA-N 0.000 description 5
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 5
- 241000282414 Homo sapiens Species 0.000 description 5
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 5
- 108010028924 PPAR alpha Proteins 0.000 description 5
- 102000023984 PPAR alpha Human genes 0.000 description 5
- 210000001789 adipocyte Anatomy 0.000 description 5
- 230000004069 differentiation Effects 0.000 description 5
- 229940088597 hormone Drugs 0.000 description 5
- 239000005556 hormone Substances 0.000 description 5
- 108020001756 ligand binding domains Proteins 0.000 description 5
- 239000002609 medium Substances 0.000 description 5
- 230000023603 positive regulation of transcription initiation, DNA-dependent Effects 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 4
- JZVKMPJMTAXOBC-UHFFFAOYSA-N 2-[4-[2-[(4-fluorophenyl)carbamoyl-heptylamino]ethyl]phenoxy]-2-methylpropanoic acid Chemical compound C=1C=C(F)C=CC=1NC(=O)N(CCCCCCC)CCC1=CC=C(OC(C)(C)C(O)=O)C=C1 JZVKMPJMTAXOBC-UHFFFAOYSA-N 0.000 description 4
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 4
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 4
- 208000001132 Osteoporosis Diseases 0.000 description 4
- URLKBWYHVLBVBO-UHFFFAOYSA-N Para-Xylene Chemical group CC1=CC=C(C)C=C1 URLKBWYHVLBVBO-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 230000009286 beneficial effect Effects 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 239000010410 layer Substances 0.000 description 4
- 238000000159 protein binding assay Methods 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- ITOFPJRDSCGOSA-KZLRUDJFSA-N (2s)-2-[[(4r)-4-[(3r,5r,8r,9s,10s,13r,14s,17r)-3-hydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H](CC[C@]13C)[C@@H]2[C@@H]3CC[C@@H]1[C@H](C)CCC(=O)N[C@H](C(O)=O)CC1=CNC2=CC=CC=C12 ITOFPJRDSCGOSA-KZLRUDJFSA-N 0.000 description 3
- MBIZXFATKUQOOA-UHFFFAOYSA-N 1,3,4-thiadiazole Chemical compound C1=NN=CS1 MBIZXFATKUQOOA-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 208000002874 Acne Vulgaris Diseases 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 101000741790 Homo sapiens Peroxisome proliferator-activated receptor gamma Proteins 0.000 description 3
- 101100189356 Mus musculus Papolb gene Proteins 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 206010000496 acne Diseases 0.000 description 3
- 239000012190 activator Substances 0.000 description 3
- 230000011759 adipose tissue development Effects 0.000 description 3
- WQZGKKKJIJFFOK-FPRJBGLDSA-N beta-D-galactose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-FPRJBGLDSA-N 0.000 description 3
- 108010005774 beta-Galactosidase Proteins 0.000 description 3
- 239000003610 charcoal Substances 0.000 description 3
- 229940125810 compound 20 Drugs 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 239000003925 fat Substances 0.000 description 3
- 235000019197 fats Nutrition 0.000 description 3
- 239000012091 fetal bovine serum Substances 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 230000037356 lipid metabolism Effects 0.000 description 3
- 230000004132 lipogenesis Effects 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 210000004378 sebocyte Anatomy 0.000 description 3
- 208000021070 secondary pulmonary alveolar proteinosis Diseases 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 3
- NSPMIYGKQJPBQR-UHFFFAOYSA-N 4H-1,2,4-triazole Chemical compound C=1N=CNN=1 NSPMIYGKQJPBQR-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- 244000215068 Acacia senegal Species 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- 208000032928 Dyslipidaemia Diseases 0.000 description 2
- 102100038595 Estrogen receptor Human genes 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 229920000084 Gum arabic Polymers 0.000 description 2
- 101000741788 Homo sapiens Peroxisome proliferator-activated receptor alpha Proteins 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- 208000017170 Lipid metabolism disease Diseases 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- 229940080774 Peroxisome proliferator-activated receptor gamma agonist Drugs 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 108091027981 Response element Proteins 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- 102000007451 Steroid Receptors Human genes 0.000 description 2
- 108010085012 Steroid Receptors Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 235000010489 acacia gum Nutrition 0.000 description 2
- 239000000205 acacia gum Substances 0.000 description 2
- 230000035508 accumulation Effects 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 210000000577 adipose tissue Anatomy 0.000 description 2
- 230000001270 agonistic effect Effects 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 210000001185 bone marrow Anatomy 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- BJDCWCLMFKKGEE-CMDXXVQNSA-N chembl252518 Chemical compound C([C@@](OO1)(C)O2)C[C@H]3[C@H](C)CC[C@@H]4[C@@]31[C@@H]2O[C@H](O)[C@@H]4C BJDCWCLMFKKGEE-CMDXXVQNSA-N 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 239000013058 crude material Substances 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 108010038795 estrogen receptors Proteins 0.000 description 2
- 235000019441 ethanol Nutrition 0.000 description 2
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 229940125753 fibrate Drugs 0.000 description 2
- 235000021588 free fatty acids Nutrition 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 2
- 102000054223 human PPARA Human genes 0.000 description 2
- 238000002513 implantation Methods 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 238000002329 infrared spectrum Methods 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 230000006372 lipid accumulation Effects 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 102000006255 nuclear receptors Human genes 0.000 description 2
- 108020004017 nuclear receptors Proteins 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- ZRSNZINYAWTAHE-UHFFFAOYSA-N p-methoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1 ZRSNZINYAWTAHE-UHFFFAOYSA-N 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 230000035479 physiological effects, processes and functions Effects 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 230000007115 recruitment Effects 0.000 description 2
- 102000027483 retinoid hormone receptors Human genes 0.000 description 2
- 108091008679 retinoid hormone receptors Proteins 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 150000001467 thiazolidinediones Chemical class 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 150000003626 triacylglycerols Chemical class 0.000 description 2
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 230000004584 weight gain Effects 0.000 description 2
- 235000019786 weight gain Nutrition 0.000 description 2
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- FGWYWKIOMUZSQF-UHFFFAOYSA-N 1,1,1-triethoxypropane Chemical compound CCOC(CC)(OCC)OCC FGWYWKIOMUZSQF-UHFFFAOYSA-N 0.000 description 1
- JAFMOTJMRSZOJE-UHFFFAOYSA-N 1,1,1-trimethoxybutane Chemical compound CCCC(OC)(OC)OC JAFMOTJMRSZOJE-UHFFFAOYSA-N 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- ROFVGYAMRSGUSQ-UHFFFAOYSA-N 1-(2-bromoethyl)piperazine;hydrobromide Chemical compound Br.BrCCN1CCNCC1 ROFVGYAMRSGUSQ-UHFFFAOYSA-N 0.000 description 1
- TUSDEZXZIZRFGC-UHFFFAOYSA-N 1-O-galloyl-3,6-(R)-HHDP-beta-D-glucose Natural products OC1C(O2)COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC1C(O)C2OC(=O)C1=CC(O)=C(O)C(O)=C1 TUSDEZXZIZRFGC-UHFFFAOYSA-N 0.000 description 1
- WGMHMVLZFAJNOT-UHFFFAOYSA-N 1-ethoxyethylideneazanium;chloride Chemical compound [Cl-].CCOC(C)=[NH2+] WGMHMVLZFAJNOT-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- OFJBYLCQNJHFMI-UHFFFAOYSA-N 2,5-dihydro-1,2-oxazole Chemical compound C1ONC=C1 OFJBYLCQNJHFMI-UHFFFAOYSA-N 0.000 description 1
- KZDCMKVLEYCGQX-UDPGNSCCSA-N 2-(diethylamino)ethyl 4-aminobenzoate;(2s,5r,6r)-3,3-dimethyl-7-oxo-6-[(2-phenylacetyl)amino]-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid;hydrate Chemical compound O.CCN(CC)CCOC(=O)C1=CC=C(N)C=C1.N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 KZDCMKVLEYCGQX-UDPGNSCCSA-N 0.000 description 1
- NWYYWIJOWOLJNR-UHFFFAOYSA-N 2-Amino-3-methyl-1-butanol Chemical compound CC(C)C(N)CO NWYYWIJOWOLJNR-UHFFFAOYSA-N 0.000 description 1
- JCBPETKZIGVZRE-UHFFFAOYSA-N 2-aminobutan-1-ol Chemical compound CCC(N)CO JCBPETKZIGVZRE-UHFFFAOYSA-N 0.000 description 1
- ULAXUFGARZZKTK-UHFFFAOYSA-N 2-aminopentan-1-ol Chemical compound CCCC(N)CO ULAXUFGARZZKTK-UHFFFAOYSA-N 0.000 description 1
- YZZDEYSCSPZMIG-UHFFFAOYSA-N 2-fluoro-1-methoxy-3-(trifluoromethyl)benzene Chemical compound COC1=CC=CC(C(F)(F)F)=C1F YZZDEYSCSPZMIG-UHFFFAOYSA-N 0.000 description 1
- JJKWHOSQTYYFAE-UHFFFAOYSA-N 2-methoxyacetyl chloride Chemical compound COCC(Cl)=O JJKWHOSQTYYFAE-UHFFFAOYSA-N 0.000 description 1
- PLNNJQXIITYYTN-UHFFFAOYSA-N 2-methylpropanehydrazide Chemical compound CC(C)C(=O)NN PLNNJQXIITYYTN-UHFFFAOYSA-N 0.000 description 1
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- 125000002373 5 membered heterocyclic group Chemical group 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- SHGAZHPCJJPHSC-ZVCIMWCZSA-N 9-cis-retinoic acid Chemical compound OC(=O)/C=C(\C)/C=C/C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-ZVCIMWCZSA-N 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 108020004414 DNA Proteins 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- QRLVDLBMBULFAL-UHFFFAOYSA-N Digitonin Natural products CC1CCC2(OC1)OC3C(O)C4C5CCC6CC(OC7OC(CO)C(OC8OC(CO)C(O)C(OC9OCC(O)C(O)C9OC%10OC(CO)C(O)C(OC%11OC(CO)C(O)C(O)C%11O)C%10O)C8O)C(O)C7O)C(O)CC6(C)C5CCC4(C)C3C2C QRLVDLBMBULFAL-UHFFFAOYSA-N 0.000 description 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 1
- 101100326341 Drosophila melanogaster brun gene Proteins 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 239000001263 FEMA 3042 Substances 0.000 description 1
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 108010001515 Galectin 4 Proteins 0.000 description 1
- 102100039556 Galectin-4 Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 241000951897 Mystrium mirror Species 0.000 description 1
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 102000016978 Orphan receptors Human genes 0.000 description 1
- 108070000031 Orphan receptors Proteins 0.000 description 1
- 229940126033 PPAR agonist Drugs 0.000 description 1
- LRBQNJMCXXYXIU-PPKXGCFTSA-N Penta-digallate-beta-D-glucose Natural products OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@@H]2[C@H]([C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-PPKXGCFTSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 102000034527 Retinoid X Receptors Human genes 0.000 description 1
- 108010038912 Retinoid X Receptors Proteins 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 229940123464 Thiazolidinedione Drugs 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- OFLXLNCGODUUOT-UHFFFAOYSA-N acetohydrazide Chemical compound C\C(O)=N\N OFLXLNCGODUUOT-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000002293 adipogenic effect Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 229960001445 alitretinoin Drugs 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 230000003388 anti-hormonal effect Effects 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- FCCCRBDJBTVFSJ-UHFFFAOYSA-N butanehydrazide Chemical compound CCCC(=O)NN FCCCRBDJBTVFSJ-UHFFFAOYSA-N 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 244000309466 calf Species 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003240 coconut oil Substances 0.000 description 1
- 235000019864 coconut oil Nutrition 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- ZOOSILUVXHVRJE-UHFFFAOYSA-N cyclopropanecarbonyl chloride Chemical compound ClC(=O)C1CC1 ZOOSILUVXHVRJE-UHFFFAOYSA-N 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 239000000857 delta opiate receptor antagonist Substances 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- UVYVLBIGDKGWPX-KUAJCENISA-N digitonin Chemical compound O([C@@H]1[C@@H]([C@]2(CC[C@@H]3[C@@]4(C)C[C@@H](O)[C@H](O[C@H]5[C@@H]([C@@H](O)[C@@H](O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)CO7)O)[C@H](O)[C@@H](CO)O6)O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O7)O)[C@@H](O)[C@@H](CO)O6)O)[C@@H](CO)O5)O)C[C@@H]4CC[C@H]3[C@@H]2[C@@H]1O)C)[C@@H]1C)[C@]11CC[C@@H](C)CO1 UVYVLBIGDKGWPX-KUAJCENISA-N 0.000 description 1
- UVYVLBIGDKGWPX-UHFFFAOYSA-N digitonine Natural products CC1C(C2(CCC3C4(C)CC(O)C(OC5C(C(O)C(OC6C(C(OC7C(C(O)C(O)CO7)O)C(O)C(CO)O6)OC6C(C(OC7C(C(O)C(O)C(CO)O7)O)C(O)C(CO)O6)O)C(CO)O5)O)CC4CCC3C2C2O)C)C2OC11CCC(C)CO1 UVYVLBIGDKGWPX-UHFFFAOYSA-N 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000008157 edible vegetable oil Substances 0.000 description 1
- 150000002066 eicosanoids Chemical class 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- YGBFFMMKLWVKGU-UHFFFAOYSA-N ethyl butanimidate;hydrochloride Chemical compound [Cl-].CCCC(=[NH2+])OCC YGBFFMMKLWVKGU-UHFFFAOYSA-N 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 238000010265 fast atom bombardment Methods 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- LRBQNJMCXXYXIU-QWKBTXIPSA-N gallotannic acid Chemical compound OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@H]2[C@@H]([C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-QWKBTXIPSA-N 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 239000006481 glucose medium Substances 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N hydroxymaleic acid group Chemical group O/C(/C(=O)O)=C/C(=O)O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 230000013190 lipid storage Effects 0.000 description 1
- 230000003520 lipogenic effect Effects 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229940099690 malic acid Drugs 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- ANSUDRATXSJBLY-VKHMYHEASA-N methyl (2s)-2-amino-3-hydroxypropanoate Chemical compound COC(=O)[C@@H](N)CO ANSUDRATXSJBLY-VKHMYHEASA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 210000003097 mucus Anatomy 0.000 description 1
- UNKQCVYVFHGUTQ-UHFFFAOYSA-N n'-hydroxy-2-methoxyethanimidamide Chemical compound COCC(N)=NO UNKQCVYVFHGUTQ-UHFFFAOYSA-N 0.000 description 1
- OPENCMFJZQABIY-UHFFFAOYSA-N n'-hydroxybutanimidamide Chemical compound CCCC(N)=NO OPENCMFJZQABIY-UHFFFAOYSA-N 0.000 description 1
- AEXITZJSLGALNH-UHFFFAOYSA-N n'-hydroxyethanimidamide Chemical compound CC(N)=NO AEXITZJSLGALNH-UHFFFAOYSA-N 0.000 description 1
- RLZPCFQNZGINRP-UHFFFAOYSA-N n'-hydroxypropanimidamide Chemical compound CCC(N)=NO RLZPCFQNZGINRP-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 235000020925 non fasting Nutrition 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 230000011164 ossification Effects 0.000 description 1
- 210000000963 osteoblast Anatomy 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000004031 partial agonist Substances 0.000 description 1
- 229940056360 penicillin g Drugs 0.000 description 1
- 239000002307 peroxisome proliferator activated receptor agonist Substances 0.000 description 1
- 239000002508 peroxisome proliferator activated receptor antagonist Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- DHRLEVQXOMLTIM-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum Chemical compound O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O DHRLEVQXOMLTIM-UHFFFAOYSA-N 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 210000000229 preadipocyte Anatomy 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical class CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 102000003998 progesterone receptors Human genes 0.000 description 1
- 108090000468 progesterone receptors Proteins 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical class CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 239000013014 purified material Substances 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000016438 regulation of fat cell differentiation Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000003571 reporter gene assay Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 238000011808 rodent model Methods 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 235000020374 simple syrup Nutrition 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 210000002325 somatostatin-secreting cell Anatomy 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 229960004274 stearic acid Drugs 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000015523 tannic acid Nutrition 0.000 description 1
- 229940033123 tannic acid Drugs 0.000 description 1
- 229920002258 tannic acid Polymers 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 102000004217 thyroid hormone receptors Human genes 0.000 description 1
- 108090000721 thyroid hormone receptors Proteins 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- IECKAVQTURBPON-UHFFFAOYSA-N trimethoxymethylbenzene Chemical compound COC(OC)(OC)C1=CC=CC=C1 IECKAVQTURBPON-UHFFFAOYSA-N 0.000 description 1
- 150000003668 tyrosines Chemical class 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 102000009310 vitamin D receptors Human genes 0.000 description 1
- 108050000156 vitamin D receptors Proteins 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/32—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Child & Adolescent Psychology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
- Die vorliegende Erfindung betrifft Verbindungen, die an PPAR-alpha, PPAR-gamma und PPAR-delta binden und diese beeinflussen. In einem anderen Aspekt betrifft die vorliegende Erfindung Verfahren zur Prävention und Behandlung von PPAR-gamma-vermittelten Erkrankungen und Zuständen.
- Peroxisomproliferator-aktivierte Rezeptoren (PPARs) sind Orphan-Rezeptoren, die zur Steroid/Retinoid-Rezeptor-Überfamilie der Ligandaktivierten Transkriptionsfaktoren gehören. Siehe z. B. T.M. Willson und w. Wahli, Curr. Opin. Chem. Biol. (1997), Bd. 1, S. 235–241.
- Drei Säugetier-PPARs wurden identifiziert, die als PPAR-alpha, PPARgamma und PPAR-delta bezeichnet werden. PPARs regulieren die Expression von Zielgenen durch Bindung an DNA-Response-Elemente (PPRE) als Heterodimere mit dem Retinoid-X-Rezeptor. Diese DNA-Response-Elemente wurden in den regulatorischen Regionen einer Anzahl von Genen identifiziert, die Proteine codieren, die am Lipidmetabolismus und Energiegleichgewicht beteiligt sind. Die biologische Rolle der PPARs in der Regulierung des Lipidmetabolismus und der Lipidlagerung wurde vor kurzem besprochen. Siehe z. B. B.M. Spiegelman, Diabetes (1998), Bd. 47, S. 507–514, K. Shoonjans, G. Martin, B. Staels und J. Auwerx, Curr. Opin. Lipidol. (1997), Bd. 8, S. 159–166 und R.P. Brun, J.B. Kim, E. Hu und B.M. Spiegelmann, Curr. Opin Lipidol. (1997), Band 8, S. 212–218.
- PPAR-gamma-Liganden der Thiazolidindion-Klasse (TZD) steigern die Wirkungen von Insulin im Menschen und reduzieren die Kreislaufglucosespiegel in Nagetiermodellen von Diabetes. Der PPAR-gamma-Rezeptor wird in Fettgewebe exprimiert und spielt eine Schlüsselrolle in der Regulierung der Adipozytendifferenzierung in vitro. TZD, wie z. B. Rosiglitazon, induzieren die Adipozytendifferenzierung in vitro durch Aktivierung des PPAR-gamma-Rezeptors. Obwohl es ersichtlich therapeutische Verwendungen für PPARgamma-Liganden in der Behandlung von Erkrankungen des Lipidmetabolismus und des Energiegleichgewichts gibt, ist es möglich, daß es Nebenwirkungen dieser Wirkstoffe geben wird. Z. B. könnten PPAR-gamma-Liganden, die die Adipozytendifferenzierung in vivo fördern, zu erhöhter Fettansammlung und Gewichtszunahme führen. Diese Nebenwirkung könnte die vorteilhaften Wirkungen eines PPAR-gamma-Liganden in der Behandlung von Diabetes oder anderen Erkrankungen aufwiegen, wenn Fettsucht ein Risikofaktor ist. Siehe z. B. die oben zitierten Artikel von Spiegelman und Brun.
- Essentielle Ernährungsfettsäuren und bestimmte ihrer Eicosanoid-Metaboliten sind natürlich vorkommende Hormone für die PPAR-Rezeptoren (Kliewer, 1997; Kliewer 1995). Diese Hormone können die Adipogenese durch Aktivierung des PPAR-gamma-Rezeptors fördern. Siehe z. B. S.A. Kliewer et al., Proc. Natl. Acad. Sci. USA (1997), Bd. 94, S. 4318–4323 und S.A. Kliewer, et al., Cell (1995), Bd. 83, S. 813–819. Moleküle, die die adipogenen Wirkungen von endogenen PPAR-gamma-Hormonen hemmen, können nützlich in der Behandlung von Erkrankungen sein, die durch erhöhte Fettansammlung oder Lipidansammlung verursacht werden. Siehe z. B. P. Tontonoz, E. Hu, B.M. Spiegelman, Curr. Opin. Genet. Dev. (1995), Bd. 5, S. 571–576. Beispiele für diese Erkrankungen sind Fettsucht, Osteoporose und Akne. Z. B. wurde ebenfalls angemerkt, daß TZD die Adipogenese im Knochenmark fördern und die Expression von Markern des Osteoblastenphänotyps hemmen, wie z. B. alkalische Phosphatase. Siehe z. B. M.A. Paulik und J.M. Lenhard, Cell Tissue Res. (1997), Bd. 290, S. 79–87. Diese Wirkungen können zu einer niedrigen Mineraliendichte im Knochen und Osteoporose führen. Verbindungen, die die Osteogeneseaktivität fördern, können nützlich in der Behandlung von Osteoporose sein. Ähnlich ist es bekannt, daß die TZDs die Lipidansammlung in Sebozyten fördern können. Siehe z. B. R.L. Rosenfield, D. Deplewski, A. Kentsis und N. Cliette, Dermatology (1998), Bd. 196, S. 43–46. Diese Wirkungen können zu Sebozyten-Differenzierung und Aknebildung führen. Somit können Moleküle, die die Adipogenese in Adipozyten, Präadipozyten, Knochenmark oder Sebozyten blockieren, vorteilhafte Wirkungen in der Behandlung von Fettsucht, Osteoporose oder Akne haben.
- Der PPAR-gamma-Rezeptor wurde in anderen als Fettgewebe gefunden, und es wird angenommen, daß synthetische PPAR-gamma-Liganden und natürliche PPAR-gamma-Hormone (natürliche Liganden) vorteilhafte Wirkungen bei vielen anderen Erkrankungen haben können, einschließlich Kreislauferkrankung, Entzündung und Krebs. Siehe z. B. den oben zitierten Schoonjans-Artikel, M. Ricote et al., Nature (1998), Bd. 391, S. 79–82, und E. Mueller et al., Mol. Cell (1998), Bd. 1, S. 465–470.
- Es gibt frühere Fälle unter anderen Mitgliedern der Steroid/Retinoid-Rezeptor-Überfamilie, daß synthetische Liganden identifiziert werden können, die viele der vorteilhaften Wirkungen nachahmen, aber einige der nachteiligen Nebenwirkungen der natürlichen Hormone hemmen. Siehe z. B. D.P. McDonnell, Biochem. Soc. Trans. (1998), Bd. 26, S. 54–60. Diesen synthetischen Liganden wurden verschiedene Kennzeichen gegeben, einschließlich Antagonisten, Antihormone, Partialagonisten, selektive Rezeptormodulatoren, gewebeselektiuve Liganden und andere. Siehe z. B. J.A. Katzenellenbogen, B.W. O'Malley und B.S. Katzenellenbogen, Mol. Endocinol. (1996), Bd. 10, S. 119–131.
- PPAR-alpha-Liganden der Fibratklasse reduzieren Kreislauf-Triglyceridspiegel und erhöhen HDL. PPAR-alpha-Liganden können nützlich zur Behandlung von Dyslipidämie und Kreislauferkrankungen sein, siehe J.-C. Fruchart, P. Duriez und B. Staels, Curr. Opin. Lipidol. (1999), Bd. 10, S. 245–257. Weniger ist über die Biologie von PPAR-delta-Liganden bekannt, obwohl berichtet wurde, daß sie HDL-Spiegel erhöhen, siehe J. Berger et al., J. Biol. Chem. (1999), Bd. 274, S. 6718–6725.
- Antagonisten von PPAR-alpha oder PPAR-delta würden nützlich zur Charakterisierung der Rolle dieser Rezeptoren in der Säugetierphysiologie sein. Z. B. würde die Verabreichung eines PPAR-alpha-Antagonisten oder PPARdelta-Antagonisten an ein ganzes Tier eine chemische Ausschaltung des Zielrezeptors darstellen. Die Charakterisierung des Phänotyps dieser chemischen Ausschaltung würde die Rolle des Zielrezeptors in der Säugetierphysiologie anzeigen. Dieses Wissen würde es erlauben, den Zielrezeptor mit einer besonderen Erkrankung zu verbinden.
- Die Aktivierung der Transkription durch nukleäre Rezeptoren beinhaltet die Rekrutierung von Co-Aktivatorproteinen. Agonistische Liganden fördern die Rekrutierung von Co-Aktivatorproteinen zum Rezeptor durch Stabilisierung der C-terminalen AF-2-Helix der Ligandenbindungsdomäne in einer Konformation, die eine "Ladungsklammer" bildet, siehe Nolte et al., Nature (1998) und A.K. Shiau et al., Cell (1998), Bd. 95, S. 927–937.
- PPAR-Agonisten, wie z. B. Thiazolidindione, Fibrate und Fettsäuren, teilen einen gemeinsamen Bindungsmodus an ihre Rezeptoren. Trotz Unterschiede in der chemischen Struktur dieser Agonisten akzeptieren die sauren Kopfgruppen dieser agonistischen Liganden eine Wasserstoffbindung von einem Tyrosin-Rest in der AF2-Helix und/oder einem Histidin- oder Tyrosin-Rest in Helix-5. Diese Wasserstoffbindungen stabilisieren die Ladungsklammer. Dies ist ein kritischer Schritt in der Aktivierung des Rezeptors durch einen agonistischen Liganden, siehe Xu et al., Mol. Cell (1999), Bd. 3, S. 397–403 und Oberfield et al., PNAS (1999), Bd. 96, S. 6102–6106. In PPAR-alpha sind diese Reste Tyrosin 464 bzw. Typrosin 314, wobei die Restenumerierung in Genbank S74349 verwendet wird (Translation G765240). In PPAR-gamma sind diese Reste Tyrosin 473 bzw. Histidin 323, wobei die Restenumerierung in Genbank X90563 verwendet wird (Translation G1490313). In PPAR-delta sind diese Reste Tyrosin 437 bzw. Histidin 287, wobei die Restenumerierung in Genbank L07592 verwendet wird (Translation G190230).
- Strukturuntersuchungen lassen vermuten, daß viele nukleäre Rezeptoren einen ähnlichen allgemeinen Mechanismus der Aktivierung teilen, wenn die Bindung des Liganden die AF2-Helix stabilisiert, wodurch die Ladungsklammer stabilisiert und die Bindung von Co-Aktivatoren erlaubt wird. Röntgenstrukturen des Östrogenrezeptors, Progesteronrezeptors, Schilddrüsenrezeptors, Reninolsäurerezeptors und Vitamin-D-Rezeptors zeigen, daß in diesen Fällen der Ligand allgemein lipophile Kontakte mit der AF2-Helix macht. In manchen Fällen, wie z. B. beim Östrogenrezeptor, sind diese Kontakte sehr schwach.
- Die PPARs sind ungewöhnlich, indem sie einen Typrosinrest in der AF2-Helix aufweisen, der zur Ausbildung einer direkten Wasserstoffbindung mit dem Liganden verfügbar ist. Die Wechselwirkung mit diesem Tyrosin scheint wesentlich zur vollen Aktivierung des PPAR zu sein. Sequenzanalyse und Homologiemodellierung zeigen an, daß FXR, LXR-alpha und LXR-beta ähnlich den PPARs sind, indem sie eine Aminosäure in der AF2-Helix aufweisen, die eine Wasserstoffbindung mit dem Liganden bilden kann. In humanem FXR ist dieser Rest Tryptophan 469, wobei die Restenumerierung in Genbank U68233 verwendet wird (Translation G1546084). In humanem LXR-alpha ist dieser Rest Tryptophan 443, wobei die Restenumerierung in Genbank U22662 verwendet wird (Translation G726513). In humanem LXR-beta ist dieser Rest Tryptophan 457, wobei die Restenumerierung in Genbank U07132 verwendet wird (Translation G641962). Die Homologiemodellierung läßt ferner vermuten, daß der Ligand eine Wasserstoffbindung mit dem Seitenketten-NH dieses AF2-Tryptophans ausbilden kann, und daß diese Wasserstoffbindung wesentlich für die volle Aktivierung von FXR, LXR-alpha und LXR-beta sein kann.
- Wie hier verwendet, ist ein "PPAR-gamma-Ligand" eine Verbindung, die humanes PPAR-gamma mit einem pKi von mehr als 5 bei Untersuchung im nachfolgend beschriebenen Bindungstest bindet. Wie hier verwendet, ist ein "PPAR-gamma-Antagonist" ein PPAR-gamma-Ligand, der eine mehr als 50%ige Inhibierung der Lipogenese bei Untersuchung im nachfolgend beschriebenen Adipozyten-Differenzierungstest und eine mehr als 50%ige Inhibierung der Transaktivierung durch 100 nM Rosiglitazon bei Untersuchung im nachfolgend beschriebenen zellbasierten Reportertest ergibt.
- Wie hier verwendet, ist ein "PPAR-alpha-Ligand" eine Verbindung, die an humanes PPAR-alpha mit einem pKi von mehr als 5 bei Untersuchung im nachfolgend beschriebenen Bindungstest bindet. Wie hier verwendet, ist ein "PPAR-alpha-Antagonist" ein PPAR-alpha-Ligand, der eine mehr als 50%ige Inhibierung der Transaktivierung durch 100 nM 2-(4-(2-(1-Heptyl-3-(4-fluorphenyl)ureido)ethyl)phenoxy)-2-methylpropionsäure bei Untersuchung im nachfolgend beschriebenen zellbasierten Reportertest ergibt.
- Wie hier verwendet, ist ein "PPAR-delta-Ligand" eine Verbindung, die an humanes PPAR-alpha mit einem pKi von mehr als 5 bei Untersuchung im nachfolgend beschriebenen Bindungstest bindet. Wie hier verwendet, ist ein "PPAR-delta-Antagonist" ein PPAR-delta-Ligand, der eine mehr als 50%ige Inhibierung der Transaktivierung durch 1000 nM 2-(4-(2-(1-Heptyl-3-(4-fluorphenyl)ureido)ethyl)phenoxy)-2-methylpropionsäure bei Untersuchung im nachfolgend beschriebenen zellbasierten Reportertest ergibt.
- Wie hier verwendet, ist ein "PPAR-Antagonist" eine Verbindung, die ein Antagonist von jedem einzelnen oder mehr als einem PPAR ist. Wie hier verwendet, ist ein "PPAR-Agonist" eine Verbindung, die ein Agonist von jedem einzelnen oder mehr als einem PPAR ist.
- Kurz gesagt offenbart die vorliegende Erfindung in einem Aspekt Verbindungen der Formel (I) oder (II) oder pharmazeutisch akzeptable Salze oder Solvate davon,
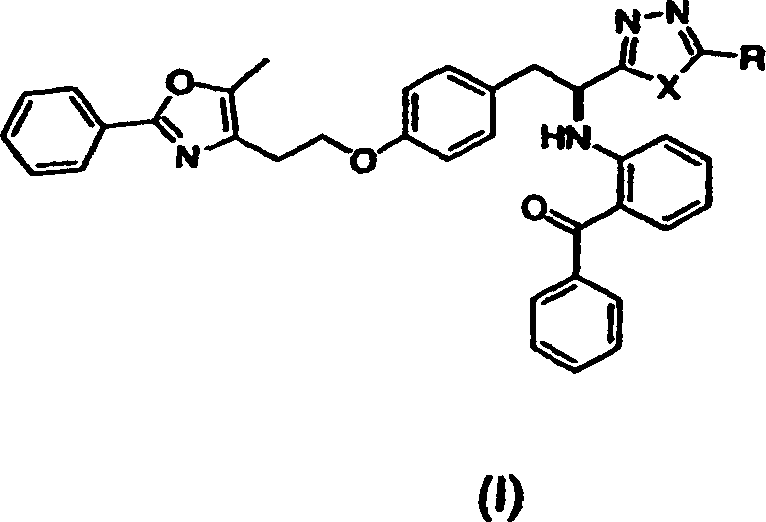
worin in Formel (I) X O, S oder NH ist;
und R Methyl, Ethyl, n-Propyl, i-Propyl, Cyclopropyl, n-Butyl, Phenyl oder -CH2OCH3 ist,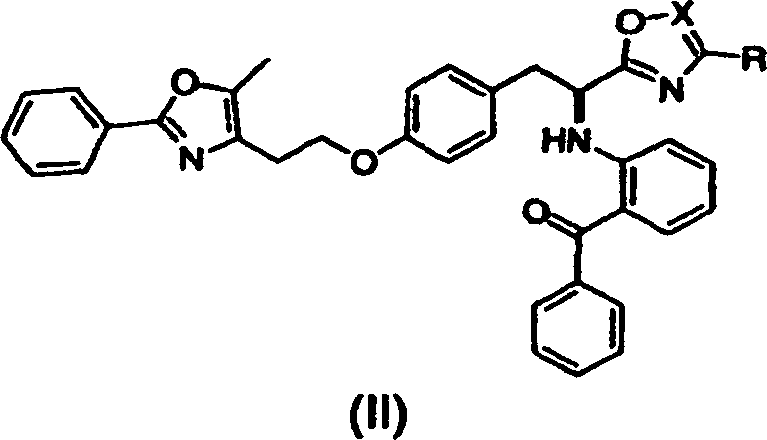
worin in Formel (II) X C oder N ist;
und R Methyl, Ethyl, n-Propyl, i-Propyl, -CH2OCH3 oder -CO2CH3 ist. Diese Verbindungen sind PPAR-gamma-Antagonisten und sind nahe Analoga von PPAR-gamma-Agonisten. - In einem anderen Aspekt offenbart die vorliegende Erfindung ein Verfahren zur Prävention oder Behandlung einer PPAR-gamma-vermittelten Erkrankung oder eines solchen Zustandes, umfassend die Verabreichung einer therapeutisch wirksamen Menge einer Verbindung dieser Erfindung. Wie hier verwendet, bezeichnet "eine Verbindung der Erfindung" eine Verbindung der Formel (I) oder (II) oder ein pharmazeutisch akzeptables Salz oder Solvat davon.
- Geeignete Verbindungen der vorliegenden Erfindung schließen ein:
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-propyl-1,3,4-oxadiazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-ethyl-1,3,4-oxadiazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-phenyl-1,3,4-oxadiazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-butyl-1,3,4-oxadiazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-methyl-1,3,4-oxadiazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-methoxymethyl-1,3,4-oxadiazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-cyclopropyl-1,3,4-oxadiazol,
(S)-{{5-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-3-methyl-1,2,4-oxadiazol,
(S)-{{5-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-3-propyl-1,2,4-oxadiazol,
(S)-{{5-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-3-methoxymethyl-1,2,4-oxadiazol,
(S)-{{5-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-3-ethyl-1,2,4-oxadiazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-isopropyl-1,3,4-thiadiazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-propyl-1,3,4-thiadiazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-methyl-1,3,4-thiadiazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-methyl-1,3,4-triazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-propyl-1,3,4-triazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-4-ethyl-1,3-oxazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-4-isopropyl-1,3-oxazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-4-propyl-1,3-oxazol,
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-4-methoxycarbonyl-l,3-oxazol,
und pharmazeutisch akzeptable Salze und Solvate davon. - Bevorzugte Verbindungen der vorliegenden Erfindung schließen ein:
(S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-propyl-1,3,4-oxadiazol und pharmazeutisch akzeptable Salze und Solvate davon. - Die Fachleute werden anerkennen, daß die Verbindungen oder Antagonisten der vorliegenden Erfindung in Form eines pharmazeutisch akzeptablen Salzes oder Solvats davon verwendet werden können. Physiologisch akzeptable Salze schließen herkömmliche Salze ein, die aus pharmazeutisch akzeptablen anorganischen oder organischen Säuren oder Basen gebildet werden, sowie quaternäre Ammoniumsäureadditionssalze. Besonders spezifische Beispiele für geeignete Säuresalze schließen diejenigen von Chlorwasserstoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Salpetersäure, Perchlorsäure, Fumarsäure, Essigsäure, Propionsäure, Bernsteinsäure, Glycolsäure, Ameisensäure, Milchsäure, Maleinsäure, Weinsäure, Zitronensäure, Pamoasäure, Malonsäuren, Hydroxymaleinsäure, Phenylessigsäure, Glutaminsäure, Benzoesäure, Salicylsäure, Fumarsäure, Toluolsulfonsäure, Methansulfonsäure, Naphthalin-2-sulfonsäure, Benzolsulfonsäure, Hydroxynaphthoesäure, Iodwasserstoffsäure, Äpfelsäure, Stearinsäure, Gerbsäure und dgl. ein. Andere Säuren wie Oxalsäure, obwohl sie selbst nicht pharmazeutisch akzeptabel sind, können nützlich in der Herstellung von Salzen sein, die nützlich als Zwischenstufen zum Erhalt der Verbindungen der Erfindung und ihrer pharmazeutisch akzeptablen Salze sind. Besonders spezifische Beispiele für geeignete basische Salze schließen Natrium-, Lithium-, Kalium-, Magnesium-, Aluminium-, Calcium-, Zink-, N,N'-Dibenzylethylendiamin-, Chlorprocain-, Cholin-, Diethanolamin-, Ethylendiamin-, N-Methylglucamin- und Procainsalze ein.
- Die Fachleute werden anerkennen, daß sich hier Verweise zur Behandlung auf die Prophylaxe sowie auf die Behandlung etablierter Erkrankungen oder Symptome erstrecken. Außerdem wird man anerkennen, daß die Menge einer Verbindung oder eines Antagonisten der Erfindung, die zur Verwendung in der Behandlung erforderlich ist, mit der Natur des behandelten Zustandes und dem Alter und Zustand des Patienten variieren wird und letztlich in der Verantwortung des behandelnden Arztes oder Tierarztes liegen wird. Allgemein werden jedoch Dosen, die für die Behandlung eines erwachsenen Menschen eingesetzt werden, typischerweise im Bereich von 0,02–5000 mg pro Tag, bevorzugt 1–5000 mg pro Tag sein. Die gewünschte Dosis kann zweckmäßig in einer Einzeldosis oder als verteilte Dosen angeboten werden, die in geeigneten Intervallen verabreicht werden, z. B. als zwei, drei, vier oder mehr Unterdosen pro Tag.
- Obwohl es möglich ist, daß Verbindungen oder Antagonisten der vorliegenden Erfindung therapeutisch als Rohchemikalie verabreicht werden können, ist es bevorzugt, den Wirkbestandteil als Teil einer pharmazeutischen Formulierung anzubieten.
- Formulierungen der vorliegenden Erfindung schließen diejenigen ein, die speziell zur oralen, bukkalen, parenteralen, transdermalen, Inhalations-, intranasalen, transmukosalen, Implantations- oder rektalen Verabreichung formuliert sind, jedoch ist die orale Verabreichung bevorzugt. Zur bukkalen Verabreichung kann die Formulierung die Form von Tabletten oder Lutschtabletten annehmen, die in herkömmlicher Weise formuliert werden. Tabletten und Kapseln zur oralen Verabreichung können herkömmliche Exzipienten enthalten, wie z. B. Bindemittel (z. B. Sirup, Gummi arabicum, Gelatine, Sorbit, Tragacanthharz, Schleim aus Stärke oder Polyvinylpyrrolidon), Füllstoffe (z. B. Lactose, Zucker, mikrokristalline Cellulose, Maisstärke, Calciumphosphat oder Sorbit), Schmiermittel (z. B. Magnesiumstearat, Stearinsäure, Talkum, Polyethylenglykol oder Kieselerde), Tablettensprengmittel (z. B. Kartoffelstärke oder Natriumstärkeglykolat) oder Benetzungsmittel, wie z. B. Natriumlaurylsulfat. Die Tabletten können gemäß fachbekannten Verfahren umhüllt werden.
- Alternativ können die Verbindungen und Antagonisten der vorliegenden Erfindung in orale flüssige Zubereitungen eingeführt werden, wie z. B. wäßrige oder ölige Suspensionen, Lösungen, Emulsionen, Sirupe oder Elixiere. Außerdem können Formulierungen, die diese Verbindungen oder Antagonisten enthalten, als ein Trockenprodukt zur Herrichtung mit Wasser oder einem anderen geeigneten Träger vor der Verwendung angeboten werden. Solche flüssigen Zubereitungen können herkömmliche Additive enthalten, wie z. B. Suspendiermittel, wie Sorbitsirup, Methylcellulose, Glucose/Zuckersirup, Gelatine, Hydroxyethylcellulose, Carboxymethylcellulose, Aluminiumstearatgel oder hydrierte eßbare Fette; Emulgatoren wie Lecithin, Sorbitanmonooleat oder Gummi arabicum; nicht-wäßrige Träger (die eßbare Öle einschließen können), wie Mandelöl, fraktioniertes Kokosöl, Ölsäureester, Propylenglykol oder Ethylalkohol; und Konservierungsstoffe wie Methyl- oder Propyl-p-hydroxybenzoate oder Sorbinsäure. Solche Zubereitungen können ebenfalls als Suppositorien formuliert werden, die z. B. herkömmliche Suppositorienbasen wie Kakaobutter oder andere Glyceride enthalten.
- Zusätzlich können Formulierungen der vorliegenden Erfindung zur parenteralen Verabreichung durch Injektion oder kontinuierliche Infusion formuliert werden. Formulierungen zur Injektion können solche Formen wie Suspensionen, Lösungen oder Emulsionen in öligen oder wäßrigen Trägern annehmen und können Formulierungsmittel wie Suspendiermittel, Stabilisatoren und/oder Dispergiermittel enthalten. Alternativ kann der Wirkbestandteil in Pulverform zur Herrichtung mit einem geeigneten Träger (z. B. sterilem pyrogenfreiem Wasser) vor der Verwendung sein.
- Die erfindungsgemäßen Formulierungen können ebenfalls als Depotzubereitung formuliert werden. Solche langwirkenden Formulierungen können durch Implantation (z. B. subkutan oder intramuskulär) oder durch intramuskuläre Injektion verabreicht werden. Entsprechend können die Verbindungen und Antagonisten der Erfindung mit geeigneten polymeren oder hydrophoben Materialien (z. B. als eine Emulsion in einem akzeptablen Öl), Ionenaustauscherharzen oder als schwachlösliche Derivate, z. B. als schwachlösliches Salz, formuliert werden.
- Die erfindungsgemäßen Formulierungen können zwischen 0,1 und 99% des Wirkbestandteils enthalten, zweckmäßig 30 bis 95% für Tabletten und Kapseln und 3 bis 50% flüssige Zubereitungen.
- Die Verbindungen und Antagonisten dieser Erfindung können durch organische Standardchemie hergestellt werden, wie es durch die begleitenden Ausführungsbeispiele erläutert wird. Die folgenden Beispiele werden zur Erläuterung der Synthese einiger besonderer Verbindungen der vorliegenden Erfindung und zur exemplarischen Darstellung allgemeiner Verfahren ange geben. Entsprechend ist der nachfolgende Abschnitt über Beispiele keineswegs zur Beschränkung des hier erwogenen Erfindungsumfangs gedacht.
- Beispiele
- Wie hier verwendet, sind die in diesen Verfahren, Schemata und Beispielen verwendeten Symbole und Konventionen übereinstimmend mit den in der zeitgenössischen wissenschaftlichen Literatur verwendeten, z. B. im Journal of American Chemical Society. Wenn nichts anderes angegeben wird, wurden alle Ausgangsstoffe von gewerblichen Lieferanten erhalten und ohne weitere Reinigung verwendet. Spezifisch können die folgenden Abkürzungen in den Beispielen und durchgehend in der Beschreibung verwendet werden: g (Gramm); mg (Milligramm); 1 (Liter); ml (Milliliter); μl (Mikroliter); N (normal); mM (millimolar); mmol (Millimol); i.v. (intravenös); Hz (Hertz); MHz (Megahertz); mol (Mole); RT (Raumtemperatur); min (Minuten); h (Stunden); Smp. (Schmelzpunkt); DC (Dünnschichtchromatographie); HPLC (Hochdruck-Flüssigchromatographie); MS (Massenspektrum); ES+ (Elektrospray); Rf (Retentionsfraktion); tr (Retentionszeit); RP (Umkehrphase); McOH (Methanol); TFA (Trifluoressigsäure); HCl (Salzsäure); HCO2H (Ameisensäure); THF (Tetrahydrofuran); CH3CN (Acetonitril); EtOH (Ethanol); CDCl3 (deuteriertes Chloroform); DMSO (Dimethylsulfoxid); DMSO-d6 (deuteriertes Dimethylsulfoxid); EtOAc (Ethylacetat); DCM oder CH2Cl2 (Dichlormethan); DMF (Dimethylformamid); Et3N (Triethylamin); MgSO4 (Magnesiumsulfat); H2O (Wasser); LAH (Lithiumaluminiumhydrid); NaH (Natriumhydrid), Na2CO3 (Natriumcarbonat); Na2SO4 (Natriumsulfat); MnO2 (Mangandioxid); NaOH (Natriumhydroxid); LiOH (Lithiumhydroxid); DIEA (Diisopropylethylamin); Et2O (Diethylether); Diethylazodicarboxylat (DEAD); tert-Butyloxycarbonyl (BOC); NaHCO3 (gesättigtes wäßriges Natriumbicarbonat). Kochsalzlösung bezeichnet eine gesättigte wäßrige Lösung von NaCl. Wenn nichts anderes angegeben wird, sind alle Temperaturen in °C (Grad Celsius) ausgedrückt. Alle Reaktionen wurden bei Raumtemperatur durchgeführt, wenn nichts anderes angegeben wird.
- Die 1H-NMR-Spektren wurden an einem Varian VXR-300-, einem Varian Unity-300- oder einem Varian Unity-400-Instrument aufgezeichnet. Chemische Verschiebungen sind in Teilen pro Million (ppm, δ-Einheiten) ausgedrückt. Kopplungskonstanten sind in Einheiten von Hertz (Hz). Aufspaltungsmuster sind als s, Singulett; d, Dublett; t, Triplett; q, Quartett; m, Multiplett; br, breit; hept, Heptuplett bezeichnet.
- Niedrig auflösende Massenspektren (MS) wurden an einem JOEL JMS-AX505HA, JOEL SX-102 oder SCIEX-APIiii-Spektrometer aufgezeichnet. Alle Massenspektren wurden unter Elektrospray-Ionisation (ES, entweder im positiven Ionenmodus oder negativen Ionenmodus) oder durch Fast Atom Bombardment-Verfahren (FAB) aufgenommen. Infrarotspektren (IR) wurden an einem Nicolet 510 FT-IR-Spektrometer unter Verwendung einer 1 mm-NaCl-Zelle erhalten. Alle Reaktionen wurden durch Dünnschichtchromatographie an 0,25 mm E. Merck-Kieselgel-Platten (60F-254) überwacht, visualisiert mit UV-Licht, Iodanfärbung oder 7%iger ethanolischer Phosphomolybdänsäure-Lösung oder p-Anisaldehyd-Lösung. Flash-Säulenchromatographie wurde an Kieselgel (230–300 mesh, Merck) durchgeführt.
- Die analytische Reinheit wurde an einem System Hewlett Packard Reihe 1050 oder 1100, ausgerüstet mit einem Dioden-Array-Spektrometer, gemessen. Die stationäre Phase war entweder eine Dynamax-C8-Säule (25 cm × 4,4 mm), eine Dynamax 60A C18-Säule (25 cm × 4,6 mm), eine Vydac C18-Säule (5 m, 4,6 mm × 250 mm), eine Supelco C18-Säule (5 m, 4,6 mm × 150 mm), oder eine Rainin C18-Säule (5 m, 4,6 mm × 250 mm). Die Fließgeschwindigkeit betrug 1,0 bis 1,5 ml/min (t0 = 2,8 oder 3,0 min), und die Lösungsmittelsysteme waren wie nachfolgend beschrieben. Die Enantiomerenreinheit wurde unter Verwendung entweder einer Chiralpak AD-Säule (25 cm × 4,6 mm) oder einer Chiralpak OD-Säule (25 cm × 4,6 mm) an entweder einem HPLC-System Hewlett Packard Reihe 1050, ausgerüstet mit einem Diodenarrayspektrometer, oder an einem Supercritical Fluid-System (SFC) unter Verwendung von CO2/Methanol als mobile Phase.
- Die folgenden Ausführungsbeispiele sind alles Verbindungen dieser Erfindung. Diese Verbindungen wurden entwickelt und hergestellt unter Verwendung der Entwicklungs- und Herstellungsverfahren dieser Erfindung. Daher sind die folgenden Ausführungsbeispiele erläuternd für die Verbindungen dieser Erfindung, die Antagonisten dieser Erfindung und das Verfahren der Entwicklung oder Herstellung dieser Erfindung.
- Entwicklungsverfahren
- Verbindungen wurden entwickelt, um die saure Gruppe eines PPAR-gamma-Agonisten durch eine Anordnung von Atomen zu ersetzen, die a) nicht mehr eine starke Wasserstoffbindung mit Tyrosin 473 oder Histidin 323 der Liganden-Bindungsdomäne bilden konnten und/oder b) Tyrosin 473 oder Histidin 323 der Liganden-Bindungsdomäne von ihren Agonisten-gebundenen Positionen verdrängten. Die in B.R. Henke et al., J. Med. Chem. (1998), Bd. 41, 5020–5036 beschriebene Verbindung 20 ist ein PPAR-gamma-Agonist und enthält eine Carbonsäure-Gruppe, die eine Wasserstoffbindung mit Tyrosin 473 und/oder Histidin 323 der PPAR-gamma-Liganden-Bindungsdomäne bilden kann. In den folgenden Beispielen wurde die saure Gruppe durch andere Gruppen ersetzt, und keine Modifikationen wurden an der Struktur von Verbindung 20 vorgenommen.
- Beispiel 1
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-propyl-1,3,4-oxadiazol
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-propyl-1,3,4-oxadiazol wurde als ein PPARgamma-Antagonist durch Austausch der Carbonsäure in Verbindung 20 gegen einen substituierten 5-gliedrigen heterocyclischen Ring entwickelt.
- Zu einer gerührten, gekühlten (°C) Lösung aus (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure [Verbindung 20, beschrieben in B.R. Henke et al., J. Med. Chem. (1998), Bd. 41, 5020–5036] (5,00 g, 9,15 mmol) in THF (120 ml) wurde 4-Methylmorpholin (1,20 g, 11,8 mmol) gefolgt von Isobutylchlorformiat (1,79 g, 13,1 mmol) gegeben. Die resultierende Suspension wurde für 30 min gerührt und dann in eine gerührte, gekühlte (0°C) Lösung aus Hydrazin (1,43 g, 44,6 mmol) in THF (80 ml) filtriert. Nach Rühren für 45 min wurde die Mischung in EtOAc gegossen und mit Wasser, gesättigtem wäßrigem Ammoniumchlorid und Kochsalzlösung gewaschen und über MgSO4 getrocknet. Entfernung des Lösungsmittels unter reduziertem Druck lieferte das gewünschte Monoacylhydrazid (5,35 g) als gelben Feststoff.
- Zu einer gerührten Lösung aus dem obigen Monoacylhydrazid (5,34 g) und Trimethylorthobutyrat (4,07 g, 27,5 mmol) in Dioxan (100 ml) wurde Methansulfonsäure (178 mg, 1,85 mmol) gegeben. Die Mischung wurde in ein auf 105°C vorgeheiztes Ölbad gegeben und für 15 min gerührt. Nach Abkühlen auf Raumtemperatur wurde die Mischung in EtOAc gegossen, mit gesättigtem wäßrigem NaHCO3 und Kochsalzlösung gewaschen und über MgSO4 getrocknet. Das Lösungsmittel wurde unter reduziertem Druck unter Erhalt eines gelben Öls entfernt. Dieses Material wurde mit drei anderen Ansätzen vereinigt, die in einer ähnlichen weise aus der oben angegebenen Carbonsäure hergestellt worden waren (5,54 g, 5,99 g und 5,06 g), und an Kieselgel unter Elution mit 2 : 3 EtOAc in Hexan chromatographiert, um 20,8 g eines klebrigen gelben Glases zu liefern. Dieses Material wurde in Et2O gelöst, langsam eindampfen gelassen und im Vakuum getrocknet, um das gewünschte 1,3,4-Oxadiazol (19,7 g, 84%) als amorphen gelben Feststoff zu liefern. Smp. = 78–82°C;
300 MHz 1H-NMR (CDCl3) δ 8,97 (d, 1H), 7,97 (m, 2H), 7,59–7,30 (m, 10H), 7,12 (d, 2H), 6,86 (d, 1H), 6,79 (d, 2H), 6,59 (t, 1H), 5,09 (m, 1H), 4,18 (t, 2H), 3,30 (d, 2H), 2,94 (t, 2H), 2,75 (t, 2H), 2,34 (s, 3H), 1,73 (m, 2H), 0,95 (t, 3H); niedrigauflösendes MS (ES+) m/e 613 (MH+).
Analyse berechnet für C38H36N4O4·0,5 H2O: C, 73,41; H, 6,00; N, 9,01.
Gefunden: C, 73,51; H, 6,04; N, 9,01. - Beispiel 2
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-ethyl-1,3,4-oxadiazol
- Hergestellt aus der in Beispiel 1 beschriebenen Zwischenstufe (2S)[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäurehydrazid und Triethylorthopropionat, um das gewünschte 1,3,4-Oxadiazol (82%) als gelben Schaum bereitzustellen. Niedrigauflösendes MS (ES+) m/e 599 (MH+).
400 MHz 1H-NMR (CDCl3) δ 8,97 (d, 1H), 7,97 (m, 2H), 7,59–7,31 (m, 10H), 7,13 (d, 2H), 6,87 (d, 1H), 6,80 (d, 2H), 6,59 (t, 1H), 5,09 (m, 1H), 4,18 (t, 2H), 3,30 (d, 2H), 2,94 (t, 2H), 2,81 (m, 2H), 2,34 (s, 3H), 1,31 (t, 3H).
Analyse berechnet für C37H34N4O4·0,5 H2O: C, 73,13; H, 5,81; N, 9,22.
Gefunden: C, 73,06; H, 5,74; N, 8,82. - Beispiel 3
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-phenyl-1,3,4-oxadiazol
- Hergestellt aus der in Beispiel 1 beschriebenen Zwischenstufe (2S)[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäurehydrazid und Trimethylorthobenzoat, um das gewünschte 1,3,4-Oxadiazol (85%) als gelben Schaum bereitzustellen. Niedrigauflösendes MS (ES+) m/e 647 (MH+).
300 MHz 1H-NMR (CDCl3) δ 9,05 (d, 1H), 7,98 (m, 4H), 7,61 – 7,31 (m, 13H), 7,19 (d, 2H), 6,94 (d, 1H), 6,81 (d, 2H), 6,60 (t, 1H), 5,19 (m, 1H), 4,17 (t, 2H), 3,38 (d, 2H), 2,94 (t, 2H), 2,34 (s, 3H). - Beispiel 4
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-butyl-1,3,4-oxadiazol
- Zu einer gerührten Lösung aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure (200 mg, 0,37 mmol) in CH2Cl2 (4 ml) wurden 1-Hydroxybenzotriazol (56 mg, 0,41 mmol) und 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid (773 mg, 0,40 mmol) gefolgt von 4-Methylmorpholin (92 mg, 0,91 mmol) gegeben. Die Mischung wurde für 5 Minuten gerührt, und dann wurde Valeriansäurehydrazid (50 mg, 0,43 mmol) hinzugegeben. Nach Rühren über Nacht wurde die resultierende dicke Suspension filtriert, und der aufgefangene Feststoff wurde mit CH2Cl2 gewaschen und im Vakuum getrocknet, um das gewünscht Diacylhydrazid (157 mg, 66%) als gelben Feststoff zu liefern.
- Eine Suspension aus P2O5 (137 mg, 0,97 mmol) in Methansulfonsäure (1,04 g, 10,8 mmol) wurde für 1 h gerührt. Zu dieser Mischung wurde das obige Diacylhydrazid (121 mg, 0,19 mmol) gegeben, und die resultierende Mischung wurde in ein auf 75°C vorgeheiztes Ölbad gegeben. Die Mischung wurde langsam homogen und wurde über Nacht stehengelassen. Nach Abkühlen auf Raumtemperatur wurde die Mischung mit CH2Cl2 verdünnt und vorsichtig zu einer gerührten Lösung aus wäßrigem 2 M K2CO3 gegeben. Die zwei Schichten wurden getrennt, und die wäßrige Schicht wurde weiter mit CH2Cl2 extrahiert. Die vereinigten organischen Schichten wurden mit Kochsalzlösung gewaschen, über MgSO4 getrocknet und zur Trockene unter reduziertem Druck eingedampft. Der zurückbleibende Rückstand wurde an Kieselgel unter Elution mit 1 : 3 EtOAc in Hexan chromatographiert, um das gewünschte 1,3,4-Oxadiazol (68 mg, 57%) als gelben Schaum zu liefern. Niedrigauflösendes MS (ES+) m/e 627 (MH+).
400 MHz 1H-NMR (CDCl3) δ 8,97 (d, 1H), 7,97 (m, 2H), 7,59–7,30 (m, 10H), 7,12 (d, 2H), 6,86 (d, 1H), 6,79 (d, 2H), 6,60 (t, 1H), 5,09 (m, 1H), 4,18 (t, 2H), 3,30 (d, 2H), 2,94 (t, 2H), 2,77 (m, 2H), 2,36 (s, 3H), 1,68 (m, 2H), 1,33 (m, 2H), 0,88 (t, 3H).
Analyse berechnet für C39H38N4O4·0,25 H2O: C, 74,21; H, 6,15; N, Gefunden: C, 74,20; H, 6,39; N, 8,48. - Beispiel 5
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-methyl-1,3,4-oxadiazol
- Hergestellt durch das in Beispiel 4 beschriebene Verfahren aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure und Acetylhydrazid. Cyclisierung lieferte das gewünschte 1,3,4-Oxadiazol (52%) als gelben Schaum. Niedrigauflösendes MS (ES+) m/e 585 (MH+).
400 MHz 1H-NMR (CDCl3) δ 8,98 (d, 1H), 7,96 (m, 2H), 7,59–7,30 (m, 10H), 7,14 (d, 2H), 6,86 (d, 1H), 6,80 (d, 2H), 6,61 (t, 1H), 5,08 (m, 1H), 4,18 (t, 2H), 3,29 (d, 2H), 2,94 (t, 2H), 2,50 (s, 3H), 2,35 (s, 3H). - Beispiel 6
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-methoxymethyl-1,3,4-oxadiazol
- Zu einer gerührten Lösung aus der in Beispiel 1 beschriebenen Zwischenstufe (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäurehydrazid (242 mg, 0,43 mmol) in CH2Cl2 (5 ml) wurde Triethylamin (109 mg, 1,08 mmol) gefolgt von Methoxy acetylchlorid (53 mg, 0,49 mmol) gegeben. Nach Rühren über Nacht wurde die Mischung mit CH2Cl2 verdünnt, mit wäßrigem 1 N HCl und Kochsalzlösung gewaschen und über MgSO4 getrocknet. Das Lösungsmittel wurde unter reduziertem Druck entfernt. Der verbleibende Rückstand wurde an Kieselgel unter Elution mit 1 : 1 EtOAc in Hexan gefolgt von EtOAc chromatographiert, um das gewünschte Diacylhydrazid (161 mg, 59%) als gelben Feststoff zu liefern.
- Das obige Diacylhydrazid (130 mg, 0,21 mmol) wurde in ein wiederverwendbares dickwandiges Druckrohr gegeben und unter Rühren in Chlorbenzol (0,3 ml) suspendiert. Hexamethyldisilazan (230 mg, 1,42 mmol) und 1 M Tetrabutylammoniumfluorid in THF (15 μl, 0,015 mmol) wurden zur Suspension gegeben. Das Reaktionsgefäß wurde versiegelt und in ein auf 130°C vorgeheiztes Ölbad gegeben. Nach 72 h wurde die Mischung auf Raumtemperatur abgekühlt, auf eine Säule aus Kieselgel übertragen und mit 1 : 9 EtOAc in Hexan, gefolgt von 3 : 7 EtOAc in Hexan und 1 : 1 EtOAc in Hexan eluiert. Weitere Reinigung an Kieselgel unter Verwendung von Radialchromatographie lieferte das gewünschte 1,2,4-Oxadiazol (19 mg, 14%) als gelben Schaum. Niedrigauflösendes MS (ES+) m/e 615 (MH+).
400 MHz 1H-NMR (CDCl3) δ 9,00 (d, 1H), 7,99 (m, 2H), 7,59–7,30 (m, 10H), 7,13 (d, 2H), 6,85 (d, 1H), 6,79 (d, 2H), 6,60 (t, 1H), 5,12 (m, 1H), 4,58 (s, 2H), 4,19 (t, 2H), 3,38 (s, 3H), 3,33 (d, 2H), 2,96 (t, 2H), 2,38 (s, 3H). - Beispiel 7
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-cyclopropyl-1,3,4-oxadiazol
- Zu einer gerührten Lösung aus der in Beispiel 1 beschriebenen Zwischenstufe (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäurehydrazid (158 mg, 0,28 mmol) in CH2Cl2 (5 ml) wurde 4-Methylmorpholin (37 mg, 0,37 mmol) gefolgt von Cyclopropancarbonylchlorid (35 mg, 0,33 mmol) gegeben. Die resultierende gelatinöse Mischung wurde über Nacht gerührt und filtriert. Das aufgefangene feste Material wurde mit Hexan gefolgt von einer minimalen Menge CH2Cl2 gewaschen und im Vakuum getrocknet, um das gewünschte Diacylhydrazid (165 mg, 94%) als gelben Feststoff zu liefern.
- Das obige Diacylhydrazid (140 mg, 0,22 mmol) wurde wie in Beispiel 5 beschrieben cyclisiert, um das gewünschte 1,3,4-Oxadiazol (66 mg, 49%) als gelben Schaum zu liefern. Niedrigauflösendes MS (ES+) m/e 611 (MH+).
400 MHz 1H-NMR (CDCl3) δ 8,94 (d, 1H), 7,97 (m, 2H), 7,59–7,30 (m, 10H), 7,12 (d, 2H), 6,86 (d, 1H), 6,80 (d, 2H), 6,60 (t, 1H), 5,05 (m, 1H), 4,18 (t, 2H), 3,27 (d, 2H), 2,94 (t, 2H), 2,35 (s, 3H), 2,07 (m, 1H), 1,27 (m, 2H), 1,08 (m, 2H). - Beispiel 8
- (S)-{{5-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-3-methyl-1,2,4-oxadiazol
- Zu einer gerührten, gekühlten (0°C) Lösung aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure (254 mg, 0,46 mmol) in CH2Cl2 (5 ml) wurde 1,3-Dicyclohexylcarbodiimid (50 mg, 0,24 mmol) gegeben. Nach 1 h wurde die Mischung filtriert und unter reduziertem Druck zur Trockene eingedampft. Der zurückbleibende Rückstand wurde in Pyridin (2 ml) gelöst, und eine Lösung aus Acetamidoxim [hergestellt wie beschrieben in C.D. Bedford et al.; J. Med. Chem. (1986), Bd. 29, 2174–2183] (14,5 mg, 0,20 mmol) in Pyridin (1 ml) wurde hinzugetropft. Die Mischung wurde zum Rückfluß erwärmt und über Nacht stehengelassen. Nach Abkühlen auf Raumtemperatur wurde die Mischung mit CH2Cl2 verdünnt, mit wäßrigem 1 N HCl und Kochsalzlösung gewaschen und über MgSO4 getrocknet. Das Lösungsmittel wurde unter reduziertem Druck entfernt, und der resultierende Rückstand wurde an Kieselgel unter Elution mit 1 : 9 EtOAc in Hexan gefolgt von 1 : 3 EtOAc in Hexan chromatographiert, um das gewünschte 1,2,4-Oxadiazol (20 mg, 17%) als gelben Schaum zu liefern. Niedrigauflösendes MS (ES+) m/e 585 (MH+).
- Beispiel 9
- (S)-{{5-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-3-propyl-1,2,4-oxadiazol
- Zu einer gerührten Lösung aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure (498 mg, 0,91 mmol) in CH2Cl2 (15 ml) wurden 1-Hydroxybenzotriazol (122 mg, 0,90 mmol) und 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid (173 mg, 0,90 mmol) gefolgt von 4-Methylmorpholin (230 mg, 2,27 mmol) gegeben. Die Mischung wurde für 5 Minuten gerührt, und dann wurde eine Lösung aus Butanamidoxim (124 mg, 1,21 mmol) in CH2Cl2 (10 ml) hinzugegeben. Die Mischung wurde über Nacht gerührt, mit CH2Cl2 verdünnt, mit Wasser und Kochsalzlösung gewaschen und über MgSO4 getrocknet. Das Lösungsmittel wurde unter reduziertem Druck entfernt, und der resultierende Rückstand wurde an Kieselgel unter Elution mit 2 : 1 EtOAc in Hexan chromatographiert, um das gewünschte Acylamidoxim (423 mg, 76%) als gelben Feststoff zu liefern.
- Das obige Acylamidoxim (185 mg, 0,29 mmol) wurde in ein wiederverwendbares dickwandiges Druckrohr gegeben und in p-Xylol (1 ml) unter Rühren suspendiert. Das Reaktionsgefäß wurde versiegelt, in ein auf 125°C vor geheiztes Ölbad gegeben und über Nacht stehengelassen. Nach Abkühlen auf Raumtemperatur wurde die Mischung auf eine Säule aus Kieselgel übertragen und mit 1 : 9 EtOAc in Hexan gefolgt von 15 : 85 EtOAc in Hexan eluiert.
- Weitere Reinigung an Kieselgel unter Verwendung von Radialchromatographie lieferte das gewünscht 1,2,4-Oxadiazol (95 mg, 53%) als gelbes Glas. Niedrigauflösendes MS (ES+) m/e 635 (MH+).
300 MHz 1H-NMR (CDCl3) δ 9,05 (d, 1H), 7,97 (m, 2H), 7,61 – 7,31 (m, 10H), 7,11 (d, 2H), 6,79 (d, 2H), 6,71 (d, 1H), 6,60 (t, 1H), 5,06 (m, 1H), 4,18 (t, 2H), 3,33 (d, 2H), 2,94 (t, 2H), 2,67 (t, 2H), 2,35 (s, 3H), 1,73 (m, 2H), 0,93 (t, 3H).
Analyse berechnet für C38H36N4O4·0,25 H2O: C, 73,95; H, 5,96; N, 9,08.
Gefunden: C, 73,67; H, 5,90; N, 8,97. - Beispiel 10
- (S)-{{5-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-3-methoxymethyl-1,2,4-oxadiazol
- Hergestellt aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure und Methoxyacetamidoxim, um das gewünschte 1,2,4-Oxadiazol (31%) als gelben Schaum zu liefern. Niedrigauflösendes MS (ES+) m/e 615 (MH+). 400 MHz 1H-NMR (CDCl3) δ 9,06 (d, 1H), 7,97 (m, 2H), 7,60–7,29 (m, 10H), 7,13 (d, 2H), 6,80 (d, 2H), 6,69 (d, 1H), 6,60 (t, 1H), 5,10 (m, 1H), 4,55 (s, 2H), 4,18 (t, 2H), 3,49 (s, 3H), 3,35 (m, 2H), 2,95 (t, 2H), 2,35 (s, 3H).
Analyse berechnet für C37H34N4O4·0,50 H2O: C, 71,25; H, 5,66; N, 8,98.
Gefunden: C, 71,00; H, 5,67; N, 8,61. - Beispiel 11
- (S)-{{5-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-3-ethyl-1,2,4-oxadiazol
- Hergestellt aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure und Propanamidoxim, um das gewünschte 1,2,4-Oxadiazol (45%) als gelben Schaum zu liefern. Niedrigauflösendes MS (ES+) m/e 599 (MH+).
300 MHz 1H-NMR (CDCl3) δ 9,05 (d, 1H), 7,97 (m, 2H), 7,60–7,29 (m, 10H), 7,12 (d, 2H), 6,80 (d, 2H), 6,69 (d, 1H), 6,60 (1, 1H), 5,06 (m, 1H), 4,17 (t, 2H), 3,33 (d, 2H), 2,94 (t, 2H), 2,74 (q, 2H), 2,34 (s, 3H), 1,30 (t, 3H). - Beispiel 12
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-isopropyl-1,3,4-thiadiazol
- Zu einer gerührten Lösung aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure (501 mg, 0,92 mmol) in CH2Cl2 (10 ml) wurden Isobuttersäurehydrazid (112 mg, 1,10 mmol), 1-Hydroxybenzotriazol (136 mg, 1,01 mmol) und Triethylamin (233 mg, 2,30 mmol) gegeben. Die Mischung wurde für 5 min gerührt, und dann wurde 1-(3-Dimethylaminopropyl-3-ethylcarbodiimidhydrochlorid (0,211 g, 1,01 mmol) hinzugegeben. Ein gelber Niederschlag bildete sich. Nach Rühren über Nacht wurde die Mischung filtriert, und der aufgefangene Feststoff wurde im Vakuum getrocknet, um das gewünschte Diacylhydrazid (1,29 g, 100%) als gelben Feststoff zu liefern.
- Das obige Diacylhydrazid (261 mg, 0,36 mmol) und Lawesson-Reagens (290 mg, 0,72 mmol) in Toluol (5 ml) wurden für 4 h refluxiert. Nach Abkühlen auf Raumtemperatur wurde die Mischung auf konzentriert und an Kieselgel unter Elution mit EtOAc in Hexan (1 : 10 zu 1 : 1) chromatographiert. Das isolierte halbgereinigte Material wurde dann auf eine basische Aluminiumoxidsäule aufgetragen und mit 100 : 1 CH2Cl2-MeOH eluiert, um das gewünschte 1,3,4-Thiadiazol (62 mg, 27%) als rotbraunen Schaum zu liefern. Niedrigauflösendes MS (ES+) m/e 628 (MH+).
400 MHz 1H-NMR (CDCl3) δ 9,04 (d, 1H), 7,90 (dd, 2H), 7,52 (d, 2H), 7,48 (d, 1H), 7,40 – 7,30 (m, 7H), 7,25 (d, 2H), 6,75 (d, 2H), 6,65 (d, 1H), 6,50 (t, 1H), 5,15 (m, 1H), 4,10 (t, 2H), 3,35 – 3,10 (m, 3H), 2,9 (t, 2H), 2,35 (s, 3H), 1,32 (t, 6H). - Beispiel 13
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-propyl-1,3,4-thiadiazol
- Hergestellt aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure und Buttersäurehydrazid. Cyclisierung lieferte das gewünschte 1,3,4-Thiadiazol. Niedrigauflösendes MS (ES+) m/e 629 (MH+).
400 MHz 1H-NMR (CDCl3) δ 9,50 und 8,88 (d, 1H, Rotamere), 7,80 (bd, 2H), 7,60 – 7,40 (m, 10H), 7,20 – 7,05 (m, 2H), 6,95–6,75 (m, 3H), 6,65–6,55 (m, 1H), 5,50 und 5,40 (q, 1H, Rotamere), 4,25 (m, 2H), 3,30 – 3,15 (m, 2H), 2,90 (t, 2H), 2,90–2,80 (m, 2H), 2,3 (s, 3H), 1,65 (p, 2H), 0,85 (t, 3H). - Beispiel 14
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-methyl-1,3,4-thiadiazol
- Hergestellt aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure und Propionsäurehydrazid. Cyclisierung lieferte das gewünschte 1,3,4-Thiadiazol. Niedrigauflösendes MS (ES+) m/e 601 (MH+).
400 MHz 1H-NMR (CDCl3) δ 9,06 (d, 1H), 7,90 (dd, 2H), 7,62–7,38 (m, 10H), 7,20 (d, 2H), 6,82 (d, 2H), 6,70 (d, 1H), 6,58 (t, 1H), 5,2 (m, 1H), 4,18 (t, 2H), 3,42–3,25 (m, 2H), 2,9 (t, 2H), 2,7 (s, 3H), 2,35 (s, 3H). - Beispiel 15
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-methyl-1,3,4-triazol
- Zu einer gerührten, gekühlten (0°C) Lösung aus der in Beispiel 1 beschriebenen Zwischenstufe (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäurehydrazid (197 mg, 0,35 mmol) in THF (5 ml) wurde Ethylacetimidathydrochlorid (51 mg, 0,41 mmol), gefolgt von Triethylamin (47 mg, 0,47 mmol) gegeben. Nach 1,5 h wurde die Mischung auf Raumtemperatur erwärmen gelassen und über Nacht gerührt. Entfernung des Lösungsmittels unter reduziertem Druck ergab einen Rückstand, der in EtOAc gelöst, mit gesättigtem wäßrigem NaHCO3 und Kochsalzlösung gewaschen und über MgSO4 getrocknet wurde. Das Lösungsmittel wurde unter reduziertem Druck entfernt, und der verbleibende Rückstand wurde an Kieselgel unter Elution mit EtOAc gefolgt von 5 : 95 McOH in CH2Cl2 chromatographiert, um das gewünschte Acylamidrazon (153 mg, 73%) als gelben Schaum zu liefern.
- Das obige Acylamidrazon (150 mg, 0,25 mmol) wurde in ein wiederverwendbares dickwandiges Druckrohr gegeben und in p-Xylol (1,5 ml) unter Rühren suspendiert. Das Reaktionsgefäß wurde versiegelt, in ein auf 120°C erwärmtes Ölbad gegeben und über Nacht stehengelassen. Nach Abkühlen auf Raumtemperatur wurde die Mischung auf eine Säule aus Kieselgel überführt und mit 1 : 1 EtOAc in Hexan, gefolgt von 3 : 2 EtOAc in Hexan eluiert. Weitere Reinigung an Kieselgel unter Verwendung von Radialchromatographie lieferte das gewünschte 1,2,4-Triazol (103 mg, 71%) als gelben Feststoff. Niedrigauflösendes MS (ES+) m/e 584 (MH+).
400 MHz 1H-NMR (CDCl3) δ 8,92 (d, 1H), 7,96 (m, 2H), 7,58–7,31 (m, 10H), 7,10 (d, 2H), 6,78 (d, 2H), 6,63 (d, 1H), 6,55 (t, 1H), 4,95 (m, 1H), 4,13 (m, 2H), 3,33 (m, 1H), 3,20 (m, 1H), 2,93 (m, 2H), 2,50 (s, 3H), 2,34 (s, 3H). - Beispiel 16
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-propyl-1,3,4-triazol
- Hergestellt durch das in Beispiel 15 beschriebene Verfahren aus der in Beispiel 1 beschriebenen Zwischenstufe (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäurehydrazid (207 mg, 0,37 mmol) und Ethylbutyrimidathydrochlorid (68 mg, 0,45 mmol). Cyclisierung lieferte das gewünschte 1,3,4-Triazol (78%) als gelben Schaum. Niedrigauflösendes MS (ES+) m/e 612 (MH+).
300 MHz 1H-NMR (CDCl3) δ 8,92 (d, 1H), 7,96 (m, 2H), 7,59–7,39 (m, 10H), 7,09 (d, 2H), 6,77 (d, 1H), 6,65 (d, 1H), 6,56 (t, 1H), 4,97 (m, 1H), 4,16 (t, 2H), 3,28 (m, 2H), 2,93 (t, 2H), 2,74 (t, 2H), 2,34 (s, 3H), 1,80 (m, 2H), 0,98 (s, 3H).
Analyse berechnet für C39H37N5O3·0,5 H2O: C, 73,53; H, 6,17; N, 11,28.
Gefunden: C, 73,88; H, 6,35; N, 10,91. - Beispiel 17
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-4-ethyl-1,3-oxazol
- Zu einer gerührten Lösung aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure (501 mg, 0,92 mmol) in CH2Cl2 (30 ml) wurden 2-Aminobutanol (273 mg, 3,06 mmol), 1-Hydroxybenzotriazol (414 mg, 3,06 mmol) und Triethylamin (511 mg, 5,05 mmol) gegeben. Die Mischung wurde für 5 min gerührt, und dann wurde 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid (640 mg, 3,34 mmol) hinzugegeben. Nach Rühren über Nacht wurde die Mischung mit CH2Cl2 verdünnt, mit Wasser gewaschen, über MgSO4 getrocknet, filtriert und aufkonzentriert. Der Rohmaterial wurde an Kieselgel unter Elution mit 40 : 1 CH2Cl2 in McOH chromatographiert, um das gewünschte Amid (1,32 g, 77%) als gelben Feststoff zu liefern.
- Eine Lösung aus dem obigen Amid (1,32 g, 2,14 mmol) und 4-Methylmorpholin (1,00 g, 8,56 mmol) in CH2Cl2 wurde über MgSO4 getrocknet und filtriert. Celite (0,50 g) wurde zur Lösung hinzugegeben, und nach Rühren für 5 Minuten wurde die Mischung mit TPAP (0,188 g, 0,535 mmol) behandelt. Die Reaktion wurde für 5 min gerührt und dann durch ein Kissen, das aus 3" Celite auf 3" Kieselerde bestand, unter Elution mit CH2Cl2 filtriert. Entfernung des Lösungsmittels unter reduziertem Druck lieferte den gewünschten Aldehyd (247 mg, 19%).
- Eine Lösung aus Iod (203 mg, 0,80 mmol), Triphenylphosphin (210 mg, 0,80 mmol) und Triethylamin (162 mg, 1,60 mmol) in CH2Cl2 (5 ml) wurde für 5 min gerührt. Zu dieser Mischung wurde eine Lösung aus dem obigen Aldehyd (247 mg, 0,40 mmol) in CH2Cl2 (5 ml) gegeben. Nach Rühren für 1,5 h wurde die Mischung in eine gesättigte Lösung von Na2S3O3 gegossen. Die Schichten wurden getrennt, und die wäßrige Schicht wurde weiter mit Diethylether extrahiert. Die vereinigten organischen Schichten wurden über MgSO4 getrocknet, filtriert und unter reduziertem Druck aufkonzentriert. Reinigung des Rohmaterials durch Chromatographie an Kieselgel unter Elution mit 3 : 1 EtOAc in Hexan lieferte das gewünschte Oxazol (40 mg, 17%). Niedrigauflösendes MS (ES+) m/e 620 (M + Na).
400 MHz 1H-NMR (CDCl3) δ 8,97 (d, 1H), 7,92 (m, 2H), 7,59 (d, 2H), 7,5 (t, 1H), 7,44–7,39 (m, 7H), 7,29 (t, 1H), 7,02 (d, 2H), 6,71 (d, 3H), 6,48 (t, 1H), 4,84 (q, 1H), 4,12 (t, 2H), 3,21 (t, 2H), 2,90 (t, 2H), 2,46 (q, 2H), 2,29 (s, 3H), 1,98 (t, 3H). - Beispiel 18
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-4-isopropyl-1,3-oxazol
- Hergestellt aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure und Valinol. TPAP-Oxidation und Cyclisierung lieferte das gewünschte Oxazol. Niedrigauflösendes MS (ES+) m/e 612 (MH+).
400 MHz 1H-NMR (CDCl3) δ 9,02 (d, 1H), 7,96 (dd, 2H), 7,59 (d, 2H), 7,49 (d, 1H), 7,44–7,39 (m, 6H), 7,29 (t, 1H), 7,2 (s, 1H), 7,07 (d, 2H), 6,79 (dd, 3H), 6,54 (t, 1H), 4,91 (q, 1H), 4,18 (t, 2H), 3,26 (t, 2H), 2,93 (g, 2H), 2,79 (p, 1H), 2,35 (s, 3H), 1,98 (dd, 6H).
Analyse berechnet für C39H37N3O4: C, 76,57; H, 6,10; N, 6,87.
Gefunden: C, 76,82; H, 6,24; N, 6,66. - Beispiel 19
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-4-propyl-1,3-oxazol
- Hergestellt aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}propionsäure und 2-Aminopentanol. TPAP-Oxidation und Cyclisierung lieferte das gewünschte Oxazol. Niedrigauflösendes MS (ES+) m/e 612 (MH+).
400 MHz 1H-NMR (CDCl3) δ 9,02 (d, 1H), 7,97 (dd, 2H), 7,59 (d, 2H), 7,52–7,39 (m, 9H), 7,29 (t, 1H), 7,07 (d, 2H), 6,78 (dd, 3H), 6,54 (t, 1H), 4,91 (q, 1H), 4,18 (t, 2H), 3,26 (dd, 2H), 2,93 (t, 2H), 2,45 (t, 2H), 2,42 (s, 3H), 0,90 (t, 3H). - Beispiel 20
- (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-4-propyl-1,3-oxazol
- Hergestellt aus der in Beispiel 1 angegebenen (2S)-[(2-Benzoylphenyl)amino]-3-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy] phenyl}propionsäure und Serinmethylester. Oxidation durch das Verfahren von Swern [A.J. Mancuso; S.L. Huang; D. Swern, J. Org. Chem. (1978), Bd. 43, 2480–2482] und Cyclisierung lieferte das gewünschte Oxazol (50%). Niedrigauflösendes MS (ES+) m/e 628 (MH+).
400 MHz 1H-NMR (CDCl3) δ 9,04 (d, 1H), 7,96 (dd, 2H), 7,60–7,27 (m, 13H), 7,11 (d, 2H), 6,81–6,75 (m, 4H), 6,57 (t, 1H), 5,03 (q, 1H), 4,19 (t, 2H), 3,91 (s, 3H), 3,31 (t, 2H), 2,94 (t, 2H). - Bindungstest
- Testverbindungen wurden zur Bindung an die humane PPAR-gamma-Rezeptor-Ligandenbindungsdomäne getestet wie beschrieben in J.S. Nichols, D.J. Parks, T.G. Consler und S.G. Blanchard, Anal. Biochem. (1998), Bd. 257, S. 112–119. Jedes der obigen Beispiele 1–20 hatte ein pKi >5 in diesem Bindungstest.
- Repräsentative Bindungstests für PPAR-alpha und PPAR-delta werden in Xu et al., Mol. Cell (1999), Bd. 3, S. 397–403 beschrieben.
- Zellbasierter Reportertest
- CV-1-Zellen wurden in DME High Glucose-Medium (Irvine Scientific), ergänzt mit 10% fötalem Rinderserum und 2 mM Glutamin, gehalten. Die Zellen wurden in D-MEM/F-12-Medium (Gibco), das mit 10% an Aktivkohle gestripptem fötalem Rinderserum ergänzt war, für 3 d vor dem Ernten aufgespalten. Die Zellen wurden in D-MEM/F-12-Medium (Gibco), das mit 10% an Aktivkohle gestripptem fötalem Rinderserum ergänzt war, geerntet und gezählt. Die Zellen wurden mit einer Dichte von 24 000 Zellen pro Vertiefung in Platten mit 96 Vertiefungen geimpft und über Nacht bei 5% CO2 und 37°C inkubiert. Die Zellen wurden für 6 bis 20 Stunden auf der Basis des Lipofectaminprotokolls (Gibco) mit den folgenden Mengen von DNA pro Vertiefung transfiziert: 2 ng PSGS GAL4-humanes PPAR-gamma, 8 ng UAS-tk-SPAP, 25 ng beta-gal, 45 ng pBluescript. Siehe J.M. Lehmann et al., J. Biol. Chem. (1995), Bd. 270, S. 12953–12956 und P.J. Brown et al., Chem. Biol (1997), Bd. 4, S. 909–918. Die Zellen wurden über Nacht bei 5% CO2 und 37°C inkubiert. Testverbindungen wurden auf 10 mM in DMSO solubilisiert. Die Testverbindungen wurden dann seriell von 1e-5 M bis 1e-10 M in D-MEM/F-12-Medium (Gibco) verdünnt, das mit 10% delipidiertem und an Aktivkohle gestripptem Kälberserum (Sigma), hitzeinaktiviert bei 60°C für 30 Minuten, 2 mM Glutamin und Pen-Strep ergänzt war. Diese Medium, in dem die Testverbindungen verdünnt wurden, enthielt ebenfalls 100 nM Rosiglitazon. Diese Testverbindungsverdünnungen wurden mit 100 μl/Vertiefung in die transfizierten Zellplatten gegeben, nachdem die Transfektionsmedien abgesaugt waren. DMSO-Kontrollen und Kontrollen mit 1 μM Rosiglitazon wurden zu jeder Zellplatte gegeben. Die Zellen wurden über Nacht bei 5% CO2 und 37°C inkubiert. Die Zellen wurden mit 25 μl 0,5% Triton X-100 lysiert. Zwei Tochterplatten wurden aus jeder Mutterplatte hergestellt. Eine Tochter erhielt 200 μl/Vertiefung SPAP-Substrat (Sigma 104), und die andere Tochter erhielt 200 μl/Vertiefung beta-gal-Substrat (Sigma N-1127). Nach Entwicklung wurden die Zellplatten bei 405 nm ausgelesen. SPAP-Daten wurden auf beta-gal normalisiert, und die prozentuale maximale Inhibierung der Transaktivierung wurde relativ zur Positivkontrolle mit 1 μm Rosiglitazon berechnet. Jedes der obigen Beispiele 1–20 besaß >50% Inhibierung der Transaktivierung durch 100 nm Rosiglitazon in diesem PPAR-gamma-zellbasierten Reportergentest.
- Der PPAR-alpha-zellbasierte Reportertest war wie für PPAR-gamma beschrieben, außer daß er GAL4-humanes PPAR-alpha anstelle von GAL-4-humanes PPAR-gamma und 2-(4-(2-(1-Heptyl-3-(4-fluorphenyl)ureido)ethyl)phenoxy)-2-methylpropionsäure anstelle von Rosiglitazon einsetzte.
- Der PPAR-delta-zellbasierte Reportertest war wie für PPAR-alpha beschrieben, außer daß er GAL4-humanes PPAR-delta anstelle von GAL4-humanes PPAR-alpha und eine 10-fach höhere Konzentration von 2-(4-(2-(1-Heptyl-3-(4-fluorphenyl)ureido)ethyl)phenoxy)-2-methylpropionsäure als Agonist einsetzte.
- Adipozytendifferenzierungstest
- C3H10T1/2 Klon 8 murine Fibroblasten (American Type Culture Collection) unter Passage 22 wurden in Dulbeccos modifiziertem Eagle-Medium (Life Technologies, Inc.), ergänzt mit 10% fötalem Kälberserum und 100 Einheiten/ml Penicillin G und 100 μg/ml Streptomycin, gehalten. Einen Tag nach Passage in Mikrotiterplatten mit 96 Vertiefungen (12,5 × 103 Zellen/cm2) wurden die Zellen mit 150 nM Rosiglitazon plus 1 μM Insulin und 1 μM 9-cis-Retinolsäure (Sigmal, St. Louis, Mo) behandelt. Träger oder Testverbindungen, die auf 10 mM in DMSO solubilisiert und dann seriell von 1e-5 M bis 1e-10 M in das Medium verdünnt worden waren, wurden hinzugegeben. Nach 7 Tagen wurden die Zellen in 0,01% Digitonin (Sigma, St. Louis, Mo) lysiert und die lipogene Aktivität durch Messung der Gesamttriglyceride unter Verwendung eines Glycerol-Triglycerid-Kits (GPO-Trinder) (337-B, Sigma, St. Louis, Mo) bestimmt. Die Mischung wurde bei 37°C für 2 h inkubiert und die Extinktion bei 550 nm ausgelesen. Die prozentuale maximale Inhibierung der Lipogenese wurde relativ zu den Träger-behandelten Zellen berechnet. Jedes der obigen Beispiele 1–20 besaß >50% Inhibierung der Lipogenese, induziert durch 150 nM Rosiglitazon, in diesem Adipozytendifferenzierungstest.
- Diese Daten lassen vermuten, daß PPAR-gamma-Antagonisten dieser Erfindung keine Gewichtszunahme oder erhöhte Fettsucht verursachen werden.
- Experimenteller Tierversuch
- In Alter und Gewicht übereinstimmende männliche ZDF/GMI fa/fa-Ratten (Genetic Models) wurden bei 72° F und 50% relativer Feuchtigkeit mit einem 12 h Hell/Dunkelzyklus gehalten. Beginnend mit einem Alter von 8 Wochen wurden die Tiere zweimal täglich mit einer Magensonde mit 150 mg/kg von Beispiel 2 dosiert. Nach 2 Wochen wurden die Tiere mit Isofluoran anästhesiert, Blut wurde durch Herzpunktur abgenommen, und nicht-nüchterne Messungen von Glucose, Triglyceriden und nicht-veresterten freien Fettsäuren (NEFAs) wurden erhalten. Die Daten wurden als Mittelwert und Standardfehler (SEM) aus Experimenten berechnet, die an 10 Tieren pro Behandlungsgruppe durchgeführt wurden. Zweiseitige Tests wurden durchgeführt, um P-Werte unter Verwendung von Excel auf einem Personal-Computer zu berechnen. Diese Forschungsstudie entsprach den Prinzipien der Versuchstierfürsorge (NIH-Veröffentlichung Nr. 86–23, überarbeitet 1985) und der Firmenpolitik für die Pflege und Verwendung von Tieren und verwandten Praxiskodizes.
-

- Diese Daten legen nahe, daß PPAR-gamma-Antagonisten dieser Erfindung nützlich zur Behandlung von Diabetes, Fettsucht, metabolischem Syndrom, pathologischer Glucosetoleranz, Syndrom X und Kreislauferkrankungen einschließlich Dyslipidämie sein können.
Claims (7)
- Verbindung der Formel (I) oder (II) oder pharmazeutisch akzeptable Salze oder Solvate davon,worin in Formel (2) X 0, S oder NH ist; und R Methyl, Ethyl, n-Propyl, i-Propyl, Cyclopropyl, n-Butyl, Phenyl oder -CH2OCH3 ist,
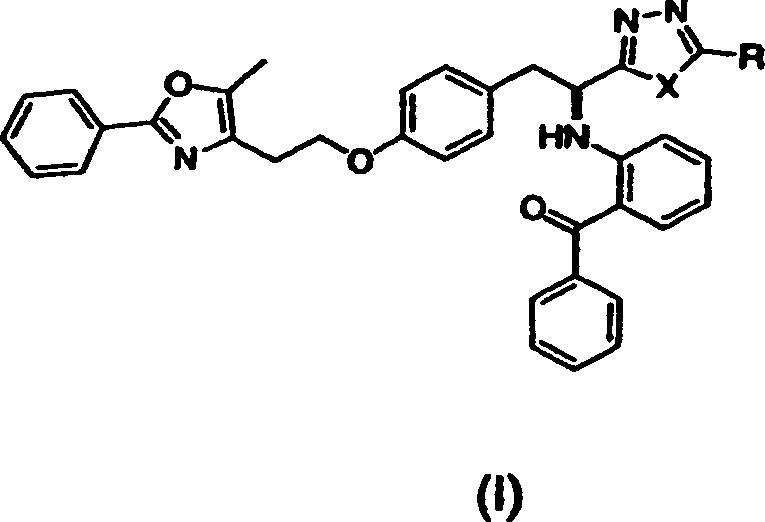 und worin in Formel (II) X C oder N ist; und R Methyl, Ethyl, n-Propyl, i-Propyl, -CH2OCH3 oder -CO2CH3 ist.
und worin in Formel (II) X C oder N ist; und R Methyl, Ethyl, n-Propyl, i-Propyl, -CH2OCH3 oder -CO2CH3 ist.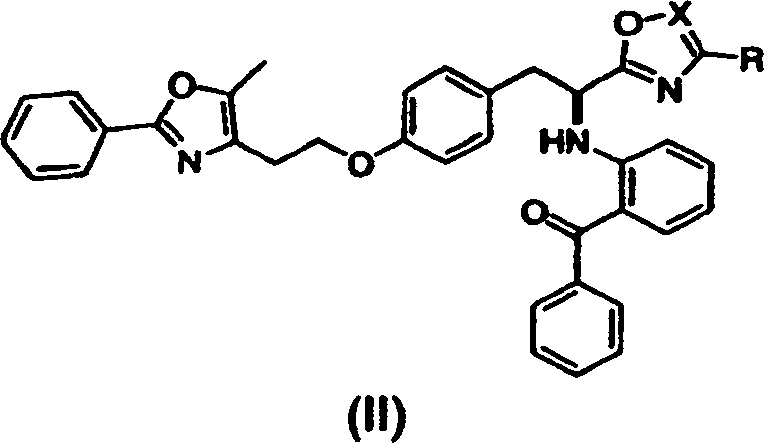
- Verbindung gemäß Anspruch 1, worin die Verbindung ein PPARgamma-Antagonist ist.
- Verbindung gemäß Anspruch 2, worin die Verbindung aus der Gruppe ausgewählt ist, die aus: (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-propyl-1,3,4-oxadiazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4- yl)ethoxy]phenyl}ethyl}}-5-ethyl-1,3,4-oxadiazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-phenyl-1,3,4-oxadiazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-butyl-1,3,4-oxadiazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-methyl-1,3,4-oxadiazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-(4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-methoxymethyl-1,3,4-oxadiazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-cyclopropyl-1,3,4-oxadiazol, (S)-{{5-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-3-methyl-1,2,4-oxadiazol, (S)-{{5-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-3-propyl-1,2,4-oxadiazol, (S)-{{5-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4- yl)ethoxy]phenyl}ethyl}}-3-methoxymethyl-1,2,4-oxadiazol, (S)-{{5-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-3-ethyl-1,2,4-oxadiazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-isopropyl-1,3,4-thiadiazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-propyl-1,3,4-thiadiazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-methyl-1,3,4-thiadiazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-methyl-1,3,4-triazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-propyl-1,3,4-triazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-4-ethyl-1,3-oxazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-(2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-4-isopropyl-1,3-oxazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-4-propyl-1,3-oxazol, (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-4-methoxycarbonyl-1,3-oxazol, und pharmazeutisch akzeptablen Salzen und Solvaten davon besteht.
- Verbindung gemäß Anspruch 2, worin die Verbindung aus der Gruppe ausgewählt ist, die aus (S)-{{2-[1-(2-Benzoylphenyl)amino]-2-{4-[2-(5-methyl-2-phenyloxazol-4-yl)ethoxy]phenyl}ethyl}}-5-propyl-1,3,4-oxadiazol und pharmazeutisch akzeptablen Salzen und Solvaten davon besteht.
- Verwendung einer Verbindung gemäß Anspruch 2 zur Herstellung eines Medikaments zur Vorbeugung oder Behandlung einer PPAR-gamma-vermittelten Erkrankung oder eines PPAR-vermittelten Zustandes.
- Verwendung gemäß Anspruch 5, worin die Erkrankung oder der Zustand aus der Gruppe ausgewählt ist, die aus Diabetes, Fettsucht, metabolischem Syndrom, pathologischer Glucosetoleranz, Syndrom X und Kreislauferkrankungen besteht.
- Verwendung gemäß Anspruch 6, worin die Erkrankung oder der Zustand aus der Gruppe ausgewählt ist, die aus Diabetes und Kreislauferkrankungen besteht.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15276199P | 1999-09-08 | 1999-09-08 | |
| US152761P | 1999-09-08 | ||
| US19996900P | 2000-04-27 | 2000-04-27 | |
| US199969P | 2000-04-27 | ||
| PCT/US2000/024364 WO2001017994A1 (en) | 1999-09-08 | 2000-09-01 | Oxazole ppar antagonists |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE60008763D1 DE60008763D1 (de) | 2004-04-08 |
| DE60008763T2 true DE60008763T2 (de) | 2005-02-10 |
Family
ID=26849844
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE60008763T Expired - Fee Related DE60008763T2 (de) | 1999-09-08 | 2000-09-01 | Oxazol ppar antagonisten |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US6506781B1 (de) |
| EP (1) | EP1210345B1 (de) |
| JP (1) | JP2003508529A (de) |
| AT (1) | ATE260912T1 (de) |
| AU (1) | AU7115300A (de) |
| DE (1) | DE60008763T2 (de) |
| ES (1) | ES2215719T3 (de) |
| WO (1) | WO2001017994A1 (de) |
Families Citing this family (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6982251B2 (en) | 2000-12-20 | 2006-01-03 | Schering Corporation | Substituted 2-azetidinones useful as hypocholesterolemic agents |
| GB0101933D0 (en) * | 2001-01-25 | 2001-03-07 | Astrazeneca Ab | Therapy |
| ES2290562T3 (es) | 2001-01-26 | 2008-02-16 | Schering Corporation | Combinaciones del activador del receptor activado por el proliferador de los peroxisomas (ppar) fenofibrato con el inhibidor de la absorcion de esteroles ezetimiba para indicaciones vasculares. |
| US7071181B2 (en) | 2001-01-26 | 2006-07-04 | Schering Corporation | Methods and therapeutic combinations for the treatment of diabetes using sterol absorption inhibitors |
| KR20070120617A (ko) | 2001-01-26 | 2007-12-24 | 쉐링 코포레이션 | 치환된 아제티딘온 화합물을 포함하는 약제학적 조성물 |
| EP1911462A3 (de) | 2001-01-26 | 2011-11-30 | Schering Corporation | Zusammensetzungen enthaltend einen Sterolabsorptionshemmer |
| WO2002076959A1 (en) * | 2001-03-23 | 2002-10-03 | Takeda Chemical Industries, Ltd. | Five-membered heterocyclic alkanoic acid derivative |
| ATE480236T1 (de) * | 2001-04-25 | 2010-09-15 | Takeda Pharmaceutical | Verwendung des abc-expressionspromotors pioglitazon zur behandlung von arteriosklerose obliterans |
| US6967212B2 (en) * | 2001-05-30 | 2005-11-22 | Bristol-Myers Squibb Company | Substituted azole acid derivatives useful as antidiabetic and antiobesity agents and method |
| US20030092736A1 (en) * | 2001-05-30 | 2003-05-15 | Cheng Peter T. | Substituted azole acid derivatives useful as antidiabetic and antiobesity agents and method |
| CA2458621A1 (en) * | 2001-08-29 | 2003-03-06 | Warner-Lambert Company Llc | Oral antidiabetic agents |
| AU2002336532C1 (en) | 2001-09-14 | 2008-10-16 | Amgen, Inc | Linked biaryl compounds |
| US7056906B2 (en) | 2001-09-21 | 2006-06-06 | Schering Corporation | Combinations of hormone replacement therapy composition(s) and sterol absorption inhibitor(s) and treatments for vascular conditions in post-menopausal women |
| US7132415B2 (en) | 2001-09-21 | 2006-11-07 | Schering Corporation | Methods and therapeutic combinations for the treatment of xanthoma using sterol absorption inhibitors |
| US7053080B2 (en) | 2001-09-21 | 2006-05-30 | Schering Corporation | Methods and therapeutic combinations for the treatment of obesity using sterol absorption inhibitors |
| GB2381866A (en) * | 2001-11-12 | 2003-05-14 | Karobio Ab | Assays for liver X receptor (LXR) modulators |
| US7205321B2 (en) | 2001-11-15 | 2007-04-17 | Eli Lilly And Company | Peroxisome proliferator activated receptor alpha agonists |
| EP1465882B1 (de) | 2001-12-21 | 2011-08-24 | X-Ceptor Therapeutics, Inc. | Heterocyclische modulatoren von nukleären rezeptoren |
| US6870466B2 (en) * | 2002-04-03 | 2005-03-22 | Hewlett-Packard Development Company, L.P. | Data display system and method for an object traversing a circuit |
| US6716842B2 (en) | 2002-04-05 | 2004-04-06 | Warner-Lambert Company, Llc | Antidiabetic agents |
| US6790929B2 (en) | 2002-06-12 | 2004-09-14 | General Electric Company | Method for making an aromatic polycarbonate |
| GEP20084475B (en) * | 2002-07-09 | 2008-09-10 | Bristol Myers Squibb Co | Substituted heterocyclic derivatives useful as antidiabetic and antiobesity agents and method for their production |
| US7137963B2 (en) * | 2002-08-26 | 2006-11-21 | Flowcardia, Inc. | Ultrasound catheter for disrupting blood vessel obstructions |
| AU2003262023A1 (en) | 2002-09-10 | 2004-04-30 | Takeda Pharmaceutical Company Limited | Five-membered heterocyclic compounds |
| MXPA05004810A (es) | 2002-11-06 | 2005-07-22 | Schering Corp | Inhibidores de la absorcion del colesterol para el tratamiento de la desmielinizacion. |
| CA2512879A1 (en) * | 2003-01-17 | 2004-08-12 | Soumya P. Sahoo | N-cyclohexylaminocarbonyl benzenesulfonamide derivatives |
| CA2513157A1 (en) * | 2003-01-22 | 2004-08-12 | Merck & Co., Inc. | Peroxisome proliferator-activated receptor |
| DE10308353A1 (de) * | 2003-02-27 | 2004-12-02 | Aventis Pharma Deutschland Gmbh | Diarylcycloalkylderivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| WO2004081004A1 (en) | 2003-03-07 | 2004-09-23 | Schering Corporation | Substituted azetidinone compounds, formulations and uses thereof for the treatment of hypercholesterolemia |
| US7459442B2 (en) | 2003-03-07 | 2008-12-02 | Schering Corporation | Substituted azetidinone compounds, processes for preparing the same, formulations and uses thereof |
| EP1601669B1 (de) | 2003-03-07 | 2008-12-24 | Schering Corporation | Substituierte azetidinon-derivate, deren pharmazeutische formulierungen und deren verwendung zur behandlung von hypercholesterolemia |
| ATE406364T1 (de) | 2003-03-07 | 2008-09-15 | Schering Corp | Substituierte azetidinon-derivate, deren pharmazeutische formulierungen und deren verwendung zur behandlung von hypercholesterolemia |
| US6987118B2 (en) | 2003-05-21 | 2006-01-17 | Pfizer Inc. | Tetrahydroisoquinoline derivatives as PPAR-alpha activators |
| NZ550857A (en) * | 2004-04-01 | 2009-12-24 | Aventis Pharma Inc | 1,3,4-oxadiazol-2-ones as PPAR delta modulators |
| CA2561230A1 (en) * | 2004-04-01 | 2005-10-20 | Aventis Pharmaceuticals Inc. | 1,3,4-oxadiazol-2-ones as ppar delta modulators and their use thereof |
| CN100467456C (zh) * | 2004-05-24 | 2009-03-11 | 北京摩力克科技有限公司 | 作为hPPARα和/或hPPARγ激活剂的α-哌嗪取代的苯丙酸衍生物 |
| CN100436430C (zh) * | 2004-05-24 | 2008-11-26 | 北京摩力克科技有限公司 | 作为hPPARα和hPPARγ激动剂的烷酰基取代的酪氨酸衍生物 |
| AU2005272786B2 (en) * | 2004-08-12 | 2011-12-22 | Amgen Inc. | Bisaryl-sulfonamides |
| US20070142326A1 (en) * | 2004-09-30 | 2007-06-21 | Youe-Kong Shue | Treatment of a condition in a mammal with administration of aminosugar and uses thereof |
| WO2006041150A1 (ja) * | 2004-10-15 | 2006-04-20 | Mitsubishi Pharma Corporation | 糖尿病の予防および/または治療薬 |
| WO2006044961A1 (en) * | 2004-10-20 | 2006-04-27 | Smithkline Beecham Corporation | Ppar-gamma modulatores |
| PL371841A1 (pl) * | 2004-12-20 | 2006-06-26 | ADAMED Sp.z o.o. | Nowe związki pochodne kwasu 3-fenylopropionowego |
| PL372332A1 (pl) | 2005-01-19 | 2006-07-24 | ADAMED Sp.z o.o. | Nowe związki, pochodne kwasu 3-fenylopropionowego |
| PL372356A1 (pl) | 2005-01-20 | 2006-07-24 | ADAMED Sp.z o.o. | Nowe związki, pochodne kwasu 3-fenylopropionowego |
| WO2006094014A2 (en) | 2005-02-28 | 2006-09-08 | The Regents Of The University Of California | Methods for diagnosis and treatment of endometrial cancer |
| WO2006113526A2 (en) | 2005-04-15 | 2006-10-26 | The Regents Of The University Of California | Prevention of chlamydia infection using a protective antibody |
| US8648052B2 (en) * | 2005-04-15 | 2014-02-11 | The Regents Of The University Of California | Prevention of chlamydia infection using SIRNA |
| US8318906B2 (en) | 2005-04-15 | 2012-11-27 | The Regents Of The University Of California | EMP2 antibodies and their therapeutic uses |
| JP5911805B2 (ja) | 2009-11-20 | 2016-04-27 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 上皮膜タンパク質−2(emp2)および増殖性硝子体網膜症(pvr) |
| US20130156720A1 (en) | 2010-08-27 | 2013-06-20 | Ironwood Pharmaceuticals, Inc. | Compositions and methods for treating or preventing metabolic syndrome and related diseases and disorders |
| MX377523B (es) | 2013-09-11 | 2025-03-10 | Inst Nat Sante Rech Med | Metodos y composiciones farmaceuticas para el tratamiento de la infeccion por el virus de la hepatitis b. |
| US20160199364A1 (en) * | 2015-01-13 | 2016-07-14 | Creighton University | Seizure treatment compositions and methods |
| CN106146483A (zh) * | 2015-04-23 | 2016-11-23 | 上海迪诺医药科技有限公司 | 杂环类法尼酯衍生物x受体调节剂 |
| CN110944635A (zh) | 2017-03-30 | 2020-03-31 | 国家医疗保健研究所 | 用于减少附加体病毒的持久性和表达的方法和药物组合物 |
-
2000
- 2000-09-01 US US10/070,474 patent/US6506781B1/en not_active Expired - Fee Related
- 2000-09-01 EP EP00959913A patent/EP1210345B1/de not_active Expired - Lifetime
- 2000-09-01 AU AU71153/00A patent/AU7115300A/en not_active Abandoned
- 2000-09-01 AT AT00959913T patent/ATE260912T1/de not_active IP Right Cessation
- 2000-09-01 WO PCT/US2000/024364 patent/WO2001017994A1/en not_active Ceased
- 2000-09-01 DE DE60008763T patent/DE60008763T2/de not_active Expired - Fee Related
- 2000-09-01 JP JP2001522217A patent/JP2003508529A/ja not_active Withdrawn
- 2000-09-01 ES ES00959913T patent/ES2215719T3/es not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| EP1210345A1 (de) | 2002-06-05 |
| WO2001017994A1 (en) | 2001-03-15 |
| ATE260912T1 (de) | 2004-03-15 |
| JP2003508529A (ja) | 2003-03-04 |
| EP1210345B1 (de) | 2004-03-03 |
| ES2215719T3 (es) | 2004-10-16 |
| DE60008763D1 (de) | 2004-04-08 |
| US6506781B1 (en) | 2003-01-14 |
| AU7115300A (en) | 2001-04-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE60008763T2 (de) | Oxazol ppar antagonisten | |
| DE69706658T2 (de) | Substituierte 4-hydroxyphenylalkansaure-derivate mit agonistischer wirkung auf ppar-gamma | |
| DE60010333T2 (de) | Substituierte oxazole und thiazole derivate als hppar-alpha aktivatore | |
| DE69916457T2 (de) | Substituierte oxazole und thiazole derivate als hppar-gamma und hppar-alpha aktivatore | |
| DE60223245T2 (de) | Thiazol- und oxazolderivate, die sich für die behandlung von herzkreislauferkrankungen und ähnlichen erkrankungen eignen | |
| US11420934B2 (en) | PPAR agonists | |
| DE60020701T2 (de) | Thiazol- und oxazol-derivate und ihre pharmazeutische verwendung | |
| US10550149B2 (en) | PPAR agonists and methods of use thereof | |
| DE69922703T2 (de) | Ureido-thiobuttersäurederivate als ppar-agonisten | |
| JP4658073B2 (ja) | グアニジン化合物および5−ht5受容体への結合相手としてのそれの使用 | |
| DE60107449T2 (de) | Substituierte aminopropoxyarylderivate als lxr agonisten | |
| DE60315603T2 (de) | Modulatoren von peroxisome proliferator-aktivierten rezeptoren | |
| DE60319080T2 (de) | Phenyloxyalkansäure-derivate als hppar aktivatore | |
| DE60025621T2 (de) | O-anisamid-derivate | |
| JP2003503486A (ja) | 選択的npy(y5)アンタゴニスト | |
| DE102006014688A1 (de) | Azolopyridin-3-on-derivate als Inhibitoren von Lipasen und Phospholipasen | |
| JP7310071B2 (ja) | 神経筋障害の治療のための化合物 | |
| DE69922528T2 (de) | N-imidazolyl-alkyl substituierte cyklische amine als histamin-h3 agonisten oder antagonisten | |
| DE69923995T2 (de) | Thiazol-derivate als ppar-gamma-ligande | |
| DE3621775A1 (de) | Neue substituierte thiazole und oxazole, diese verbindungen enthaltende arzneimittel und verfahren zu ihrer herstellung | |
| JP2001261674A (ja) | ベンゾチオフェン誘導体およびそれを有効成分として含有する核内レセプター作動薬 | |
| DE60223805T2 (de) | Oxazol/thiazol-derivate als aktivatoren des hppar-alpha-rezeptors | |
| Pingali et al. | Design and synthesis of novel oxazole containing 1, 3-Dioxane-2-carboxylic acid derivatives as PPAR α/γ dual agonists | |
| US20040077659A1 (en) | Method for treating ppar gamma mediated diseases or conditions | |
| DE69107874T2 (de) | Imidazolderivate und diese Imidazolderivate als Wirkstoffe enthaltende Antiepileptika. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8339 | Ceased/non-payment of the annual fee |