-
Diese Erfindung betrifft Substanzen,
die eine GewebePlasminogenaktivator-artige (t-PA) Aktivität aufweisen.
Insbesondere betrifft diese Erfindung "rekombinante" thrombolytische Proteine, ein Verfahren
zur Gewinnung der Proteine aus gentechnisch behandelten Zellen und
die therapeutische Verwendung der Substanzen als thrombolytische
Wirkstoffe.
-
Diese Proteine sind aktive thrombolytische
Wirkstoffe, die verbesserte fibrinolytische Profile im Vergleich
zu nativem menschlichem t-PA aufweisen sollten. Dies kann an einer
erhöhten
Fibrin-Affinität,
einer geringeren Reaktivität
mit t-PAInhibitoren, einer beschleunigten Thrombolyse, einer erhöhten fibrinolytischen
Aktivität
und/oder einer verlängerten
biologischen Halbwertszeit erkennbar sein. Die erfindungsgemäßen Proteine
sollten außerdem
leichter in einer homogeneren Form als natives menschliches t-PA
hergestellt werden können.
Diese Proteine sollten ein verbessertes pharmakokinetisches Gesamtprofil
aufweisen.
-
Die Struktur von nativem menschlichem
t-PA umfaßt
einen Amino-(N-)-Terminus von etwa 91 Aminosäureresten, zwei als "Kringel" bezeichnete Bereiche
und am Carboxy-Terminus eine Serin-Protease-artige Domäne. Es wurde
festgestellt, daß der
NTerminus mehrere Subdomänen
enthält,
die unter anderem bei der Bindung an Fibrin und bei der Ausscheidung
des Proteins in vivo eine funktionelle Rolle spielen. Kürzlich wurde die
Isolierung einer weiteren t-PA-Form beschrieben, der der native
N-Terminus und der erste Kringel-Bereich fehlen, vgl. die veröffentlichte
Europäische
Patentanmeldung Nr. 0 196 920 (am B. Oktober 1986 veröffentlicht).
Gemäß dieser
Veröffentlichung
ist die verkürzte
Form von t-PA, die mit Ala-160 von nativem menschlichem t-PA beginnt,
fibrinolytisch aktiv.
-
Wie hier nachstehend genauer beschrieben,
stellt diese Erfindung neue Proteinanaloge von menschlichem t-PA
bereit, die beide Kringelbereiche von nativem menschlichem t-PA
behalten, jedoch innerhalb des N-Terminus modifiziert sind. Während bei
bestimmten Ausführungsformen
die Modifikationen Deletionen am N-Terminus umfassen, bleibt der
erste Kringelbereich intakt, und am N-Terminus sind niemals mehr
als 94 Aminosäuren
deletiert. Die meisten Ausführungsformen
sind in weitaus geringerem Umfang deletiert und/oder durch Aminosäuresubstitution(en)
verändert.
Durch Beibehaltung eines größeren Teils
der Struktur von nativem menschlichem t-PA sollten die erfindungsgemäßen Proteine
entsprechend einen größeren Teil
der gewünschten
biologischen Aktivitäten
von nativem menschlichem t-PA behalten und weniger immunogen als stärker modifizierte
t-PA-Analoge sein. Die erfindungsgemäßen Proteine sollten somit
verglichen sowohl mit nativem menschlichem t-PA als auch mit dem
verkürzten
Ala-160t-PA sowie weiteren modifizierten t-PA-Formen verbesserte
fibrinolytische und pharmakokinetische Profile aufweisen.
-
Das Polypeptidrückgrat von natürlichem
menschlichem t-PA enthält
außerdem
vier Konsensus-Asn-Glykosylierungsstellen.
Es wurde gezeigt, daß zwei
dieser Stellen im t-PA von Säugerzellen,
die von Melanomzellen stammen, typischerweise glykosyliert sind,
d. h., am Asn117 und Asn448 Asn184 ist gelegentlich glykosyliert
und Asn218 ist typischerweise nicht glykosyliert.
t-PA aus Säugerzellen,
die von Melanomzellen stammen, z. B. Bowes- Zellen, wird hier nachstehend auch als "natives" oder "natürliches" menschliches t-PA bezeichnet.
-
Diese Erfindung betrifft, wie vorstehend
beschrieben, die neuen Proteinanaloge von menschlichem t-PA, die
in den Ansprüchen
gekennzeichnet sind. Die Merkmale der erfindungsgemäßen Proteine
sind nachstehend genauer beschrieben. Ungeachtet der verschiedenen
Modifikationen wird die Nummerierung von Aminosäuren, deren Sequenz im Ein-Buchstabencode in
Tabelle 1 dargestellt ist, beibehalten.
-
A. Modifikationen am N-Terminus
-
In einer Ausführungsform der Erfindung sind
Proteine durch eine Deletion von Cys-51 bis Asp-87 im Vergleich
zu nativem menschlichem t-PA gekennzeichnet. In einer weiteren besonderen
Ausführungsform
sind Cys-6 bis Ile-86 deletiert. In einer anderen Ausführungsform
sind Cys-6 bis Cys-51 deletiert. In anderen Ausführungsformen sind im Nterminalen
Bereich der Proteine mehr konservative Modifikationen vorhanden.
Bestimmte erfindungsgemäße Proteine
enthalten beispielsweise eine oder mehrere Aminosäuredeletionen
oder -substitutionen innerhalb der Unterbereiche, auf die in den
Ansprüchen
Bezug genommen wird. Die Unterbereiche von menschlichem t-PA lauten
wie folgt:
-
Die beanspruchten Modifikationen
innerhalb des N-Terminus
sind hier nachstehend genauer beschrieben.
-
B. Modifikationen an N-Glykosylierungsstellen
-
Die erfindungsgemäßen Proteinvarianten enthalten
außerdem
im Vergleich zu natürlichem
menschlichem t-PA keine N-gebundenen Kohlenhydrateinheiten oder
sind nur partiell glykosyliert. Ein hier nachstehend als "partiell glykosyliert" bezeichnetes Protein
ist ein Protein, das weniger N-gebundene Kohlenhydrateinheiten als
vollständig
glykosylierter nativer menschlicher t-PA enthält. Diese fehlende oder nur
partielle Glykosylierung wird durch eine Aminosäuresubstitution oder -deletion
an einer oder mehreren der Konsensus-N-Glykosylierungserkennungsstellen
im nativen t-PA-Molekül
verursacht. Es wurde festgestellt, daß erfindungsgemäße Proteinvarianten,
die eine solche Modifikation an einer oder mehreren N-Glykosylierungsstellen
aufweisen, eine t-PA-artige thrombolytische Aktivität mit einer
gelegentlich sogar größeren fibrinolytischen
Aktivität
aufweisen, wobei sie leichter in homogenerer Form als natives t-PA
produziert werden können
und häufig
längere
in vivo-Halbwertszeiten als natives t-PA aufweisen.
-
Es wird gegenwärtig angenommen, daß N-Glykosylierungserkennungsstellen
Tripeptidsequenzen umfassen, die durch die geeigneten zellulären Glykosylierungsenzyme
spezifisch erkannt werden. Diese Tripeptidsequenzen sind entweder
Asparagin-X-Threonin
oder Asparagin-X-Serin, wobei X meistens jede Aminosäure sein
kann. Ihre Lage innerhalb der t-PA-Peptidsequenz ist in Tabelle 1 dargestellt.
Durch einige Austausche oder Deletionen von Aminosäuren an
einer oder mehreren der drei Stellen einer Glykosylierungserkennungsstelle
wird die Glykosylierung der modifizierten Sequenz verhindert. Asn117 und Asn184 im
t-PA sind beispielsweise in einer Ausführungsform beide durch Thr
und in einer weiteren Ausführungsform
durch Gln ausgetauscht worden. Zumindest wenn Gln zweimal eingetauscht
wird, sollte das erhaltene Glykoprotein (Gln117Gln184) auf jeden Fall nur eine N-gebundene
Kohlenhydrateinheit (an Asn448) und nicht
zwei oder drei solcher Einheiten wie im nativen t-PA enthalten.
Der Fachmann wird erkennen, daß analoge
Glykoproteine mit der gleichen Asn448-Monoglykosylierung
hergestellt werden können
durch Deletion von Aminosäuren
oder Austausch gegen andere Aminosäuren an den Positionen 117
und 184 und/oder durch Deletion oder Substitution von einer oder
mehreren Aminosäuren
an anderen Positionen innerhalb der jeweiligen Glykosylierungserkennungsstellen,
z. B. an Ser119 und Ser186,
vorstehend beschrieben, und/oder durch Substitution oder stärker bevorzugt
durch Deletion an einer oder mehreren "X"-Positionen
der Tripeptidstellen. In einer anderen Ausführungsform ist Asn an den Positionen
117, 184 und 448 durch Gln ersetzt. Die erhaltenen Varianten sollten im
Unterschied zu nativem t-PA mit zwei oder drei solcher Einheiten
keine N-gebundenen Kohlenhydrateinheiten enthalten.
-
In anderen Ausführungsformen sind potentielle
Glykosylierungsstellen individuell modifiziert worden, beispielsweise
durch Austausch von Asn, z. B. gegen Gln an Position 117 in einer
gegenwärtig
bevorzugten Ausführungsform,
an Position 184 in einer anderen Ausführungsform und an Position
448 in einer weiteren Ausführungsform.
Die Erfindung umfaßt
solche nicht-glykosylierten,
monoglykosylierten, diglykosylierten und triglykosylierten t-PA-Varianten.
-
Beispiele für Modifikationen an einer oder
mehreren der drei Konsensus-N-Glykosylierungssequenzen, R1, R2 und R3, wie in mehreren erfindungsgemäßen Ausführungsformen
festgestellt, sind nachstehend dargestellt:
-
Beispiele
für Modifikationen
an n-Glykosylierungsstellen
-
C. Modifikation an der
Arg-275/Ile-276-Spaltstelle
-
Ein erfindungsgemäßer Aspekt betrifft die Modifikation
von Varianten gegebenenfalls an der proteolytischen Spaltstelle
im Bereich von Arg-275 bis Ile-276, wobei Arg-275 deletiert oder
gegen eine weitere Aminosäure,
vorzugsweise eine Aminosäure
außer
Lys oder His, ausgetauscht ist. Thr ist gegenwärtig eine besonders bevorzugte
Aminosäure
zum Austausch von Arg-275 in den verschiedenen erfindungsgemäßen Ausführungsformen.
Eine proteolytische Spaltung am Arg-275 von nativem t-PA liefert
das so genannte "Zwei-Ketten"-Molekül, wie es im Stand der Technik
bekannt ist. Die durch eine Modifikation dieser Spaltstelle gekennzeichneten
erfindungsgemäßen Proteine
können
auf eine einfachere Weise mit größerer Homogenität als das entsprechende,
an der Spaltstelle nicht modifizierte Protein hergestellt werden
und, vielleicht wichtiger noch, dürften ein verbessertes fibrinolytisches
Profil und verbesserte pharmakokinetische Eigenschaften aufweisen.
-
Diese Erfindung stellt daher eine
Familie von neuen, mit menschlichem t-PA verwandten, thrombolytischen
Proteinen bereit. Diese Familie umfaßt mehrere Gattungen von Proteinen.
-
In einer Ausführungsform sind die Proteine
durch eine in den Ansprüchen
gekennzeichnete Peptidsequenz charakterisiert, in der Arg-275 deletiert
oder durch eine andere Aminosäure,
vorzugsweise durch eine andere als Lysin oder Histidin, ersetzt
ist, und mindestens eine der Konsensus-Asn-Glykosylierungsstellen deletiert oder
zu einer von der Konsensus-Asn-Glykosylierungssequenz abweichenden
Sequenz modifiziert ist. Beispiele für Proteine dieser Ausführungsform
sind nachstehend in Tabelle 1 dargestellt. Die erfindungsgemäßen Proteine
sind t-PA-Analoge, die durch die verschiedenen Modifikationen oder
Kombinationen von Modifikationen, wie hier offenbart, gekennzeichnet
sind, die auch noch weitere Variationen mit einer noch vorhandenen
thrombolytischen Aktivität,
z. B. allele Variationen, oder (eine) weitere Deletion(en), Substitution(en)
oder Insertion(en) von Aminosäuren
enthalten können,
sofern die diese Proteine codierende DNA (vor der erfindungsgemäßen Modifikation)
mit einer menschlichen t-PA codierenden DNA-Sequenz unter stringenten
Bedingungen hybridisieren kann.
-
Tabelle
1
Beispiele für
Proteine, die eine Modifikation am Arg-275 enthalten und an mindestens einer
N-Glykosylierungs-stelle
-
In einer zweiten Ausführungsform
sind die Proteine durch eine im wesentlichen der Peptidsequenz von menschlichem
t-PA entsprechenden Peptidsequenz gekennzeichnet, in der eine oder
mehrere Aminosäuren innerhalb
des N-Terminus im Bereich von Val-4 bis Val-72 deletiert sind und
in der (a) eine oder mehrere Asn-Glykosylierungsstellen deletiert
oder auf andere Weise zu einer anderen als einer Konsensus-Asn-Glykosylierungsstelle
modifiziert sind und/oder (b) Arg-275 gegebenenfalls durch eine
andere Aminosäure,
vorzugsweise nicht durch Lysin oder Histidin, deletiert oder ersetzt
ist. Beispiele für
Proteine dieser Ausführungsform sind
nachstehend dargestellt:
-
Beispiele für Proteine
mit N-terminalen Deletionen
-
Die nachstehend dargestellten Proteine
weisen die in Tabelle 1 dargestellte Peptidsequenz auf, in der R1, R2 und R3 die wt-Tripeptidsequenzen sind, in der
jedoch die N-Termini (Gly-(-3)
bis Thr-91) wie folgt ausgetauscht sind:
-
-
Diese Ausführungsform schließt eine
Untergattung von Proteinen ein, in der 1 bis etwa 69 Aminosäuren des
Bereichs Val-4 bis Val-72 deletiert sind, und eine oder mehrere
Asn-Glykosylierungsstellen
deletiert oder auf andere Weise zu einer anderen als einer Konsensus-Asn-Glykosylierungssequenz,
wie vorstehend beschrieben, modifiziert sind. Dazu gehört außerdem eine
Untergattung von Verbindungen, in der 1 bis etwa 69 Aminosäuren im
Bereich Val-4 bis Val-72 deletiert sind und Arg-275 deletiert oder
durch eine andere Aminosäure, vorzugsweise
nicht durch Lysin oder Histidin, ersetzt ist. Eine weitere Untergattung
dieser Ausführungsform
ist durch eine Deletion von 1 bis etwa 69 Aminosäuren innerhalb des Bereichs
Val-4 bis Val-72, durch eine Deletion oder Modifikation einer oder
mehrerer Asn-Glykosylierungsstellen (vgl. z. B. die Tabelle auf
Seite 6) und durch eine Deletion von Arg-275 oder sein Austausch
gegen eine andere Aminosäure
gekennzeichnet. Beispiele für
die Proteine dieser Untergattungen sind nachstehend in den Tabellen
2 und 2.5 dargestellt.
-
Diese Ausführungsform schließt außerdem eine
Untergattung von Proteinen ein, wobei die Deletion am N-Terminus eine Deletion
von einer oder mehrerer Aminosäuren
im Bereich Val-4 bis Ser-50 umfasst. Außerdem ist eine Untergattung
von Proteinen eingeschlossen, wobei eine oder mehrere Aminosäuren im
Bereich Val-4 bis Ser-50 deletiert und eine oder mehrere Glykosylierungsstellen,
wie vorstehend beschrieben, modifiziert sind. Eine weitere Untergattung
umfaßt
Proteine, in denen Aminosäuren
im Bereich Val-4 bis Ser-50 deletiert sind, wobei Arg-275 deletiert
oder durch eine andere Aminosäure
ersetzt ist. Außerdem
ist eine Untergattung eingeschlossen, in der eine oder mehrere Aminosäuren im
Bereich Val-4 bis Ser-50 deletiert sind und in der gegebenenfalls
sowohl (a) eine oder mehrere Glykosylierungsstellen als auch (b)
Arg-275, wie vorstehend beschrieben, modifiziert sind. Beispiele
für die
Proteine dieser Untergattungen sind nachstehend in Tabelle 3 sowie
in den Tabellen 2 und 2.5 dargestellt.
-
Tabelle
2
Beispiele für
Proteine, die eine Deletion von einer oder mehrerer Aminosäuren am
N-Terminus und eine Modifikation an mindestens einer N-Glykosylierungsstelle
und gegebenenfalls am Arg-275 enthalten (die allgemeine Sequenz
ist in Tabelle 1 dargestellt)
-
-
Tabelle
2.5
Beispiele für
N-Termini, die eine Deletion von einer oder mehrerer Aminosäuren enthalten
-
-
Die spezifischen erfindungsgemäßen Proteine
erhalten eine 3-teilige
Bezeichnung, die eine Verbindungsnummer von Tabelle 2 mit einer
anschließenden
Bezeichnung des N-Terminus und danach eine Angabe zu Position 275
umfaßt.
Verbindung Nr. 2-11/N-6/Arg bezeichnet beispielsweise ein Protein,
in dem die 3 Glykosylierungsstellen deletiert sind ("2–11", vgl. Tabelle 2), C-36 bis C-43 deletiert
sind (N-Terminus #N-6) und Arg-275 erhalten bleibt.
-
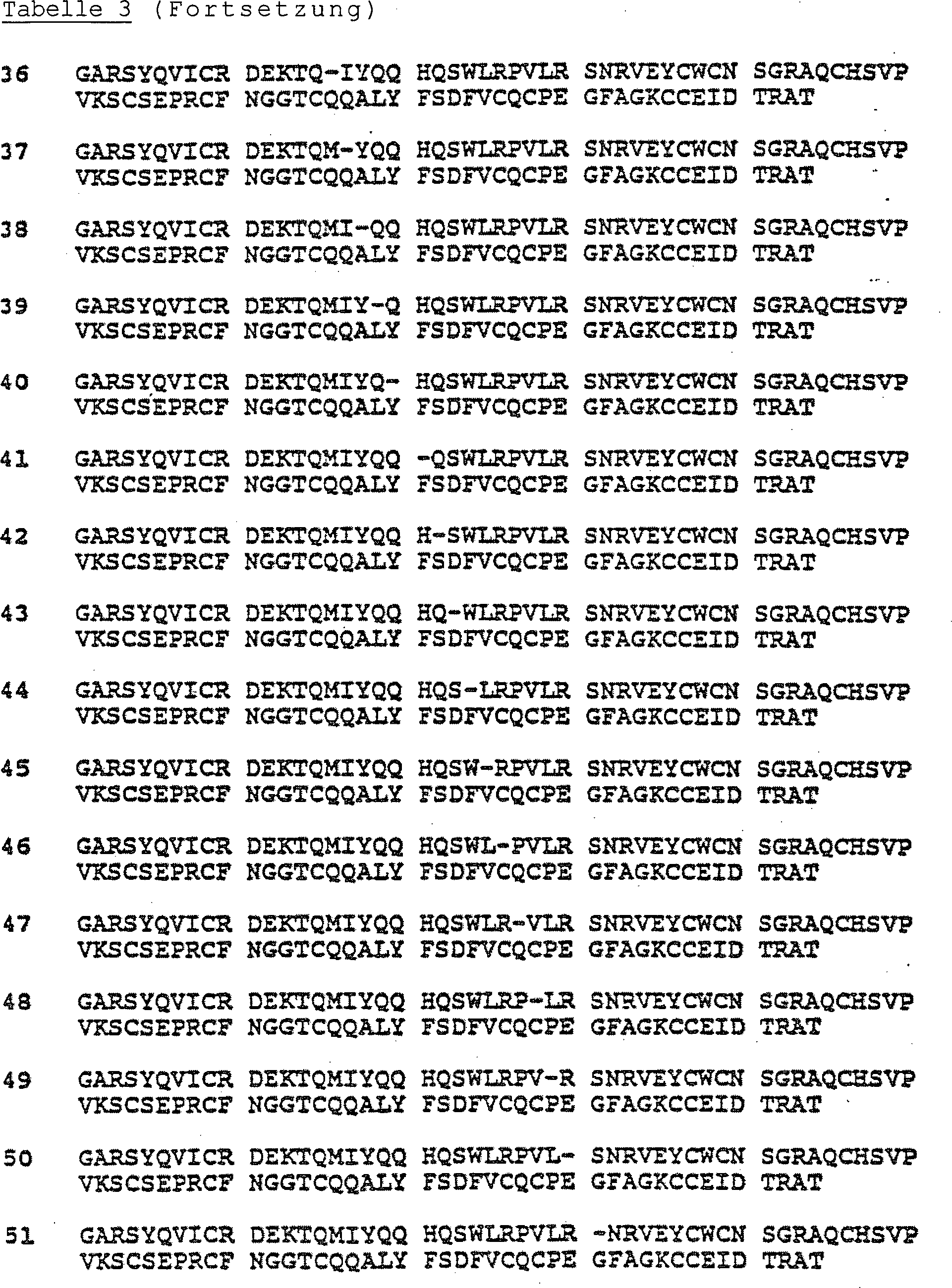
Tabelle 3
-
Beispiele für Proteine,
die eine Deletion von 1–45
Aminosäuren
im Bereich Val-4 bis Ser-50 und eine Modifikation entweder nur (a)
am Arg-275 oder (b) an mindestens einer N-Glykosylierungsstelle oder sowohl (a)
am Arg-275 als auch (b) an mindestens einer N-Glykosylie-rungsstelle
aufweisen (die allgemeine Sequenz ist in Tabelle 1 dargestellt)
-
Beispiele für Proteine entsprechen den
in Tabelle 2 definierten, wobei allerdings die Wildtyp (wt)-Sequenz
von Gly-(-3) bis Thr-91 gegen die nachstehend dargestellten N-Termini
ausgetauscht ist:
-
Die in Tabelle 3 dargestellten modifizierten
N-Termini zeigen, daß mehr
als eine Aminosäure
deletiert sein kann. Wenn mehrere Aminosäuren deletiert sind, können sie
einander benachbart oder durch eine oder mehrere Aminosäuren getrennt
sein. Bei der Bezeichnung der Verbindungen mit solchen N-Termini wird die Größe der Deletion
durch "Δn" angezeigt, wobei "n" die Zahl der deletierten Aminosäuren ist.
Beispielsweise kann der N-Terminus #27, in dem eine Aminosäure deletiert
ist, wie in Tabelle 3 dargestellt, als "N-27Δ1" bezeichnet werden.
Bei einer Deletion von zwei Aminosäuren, z. B. bei Y-4 und I-5,
wird der N-Terminus als "N-27Δ2" etc. bezeichnet.
Bei einer Kombination von Deletionen, wobei z. B. Y-4, I-5 und D-8
deletiert sind, kann der N-Terminus als "N-27Δ2", "N-31Δ1" bezeichnet werden.
Wie im Text im Anschluß an
Tabelle 2.5 beschrieben ist, besteht die Bezeichnung bestimmter
Verbindungen aus einem dreiteiligen Code, der eine Verbindungsnummer
aus Tabelle 2, anschließend
eine Bezeichnung des N-Terminus #, z. B. von Tabelle 2.5, und danach
eine Statusangabe der Position 275 umfaßt. Die Verbindung 2-26/N-N-27Δ2/N-31Δ1 bezeichnet somit das
Protein, in dem alle drei Glykosylierungsstellen sowie R-275, S-1,
Y-2 und I-5 deletiert sind.
-
Diese Ausführungsform schließt ferner
eine Untergattung von Proteinen ein, in denen eine oder mehrere
Aminosäuren
im Bereich Val-4 bis Val-27 deletiert sind. Proteine dieser Untergattung
können
gegebenenfalls, so modifiziert werden, daß Arg-275 deletiert oder gegen
eine andere Aminosäure
ausgetauscht ist, vorzugsweise nicht gegen Lysin oder Histidin.
Proteine dieser Untergattung sind alternativ beispielsweise durch Entfernung
einer oder mehrerer N-Glykosylierungsstellen, wie vorstehend beschrieben,
modifiziert.
-
Beispiele von Proteinen dieser Untergattung
sind in den Tabellen 2 und 3 dargestellt, sie enthalten jedoch N-Termini, wie sie
beispielsweise nachstehend in Tabelle 4 angegeben sind.
-
Tabelle 4
-
Beispiele für Proteine,
die eine Deletion von einer oder mehrerer Aminosäuren im Bereich Val-4 bis Val-72
aufweisen (die allgemeine Sequenz ist in Tabelle 1 dargestellt)
-
Beispiele für Proteine sind, wie in Tabelle
2 definiert, wobei jedoch die Wildtyp (wt)-Sequenz von Gly-(-3)
bis Thr-91 gegen die nachstehend dargestellten N-Termini ausgetauscht
ist:
-
Im Hinblick auf die vorstehende Tabelle
4 muß angemerkt
werden, daß darin
mehrere Untergruppen von Proteinen offenbart sind. Beispielsweise
sind Proteine dargestellt, die eine Deletion von 1 bis 47 Aminosäuren (einzeln,
aufeinander folgend oder kombiniert) von Val-4 bis Val-72 enthalten,
sowie Proteine, die eine Deletion von 1 bis 22 Aminosäuren von
Cys-51 bis Val-72 und eine Deletion von einer oder mehreren Aminosäuren im
Bereich von Val-4 bis Cys-51 (vgl. N-76) enthalten. Bei Verbindungen,
die einen N-Terminus enthalten, der aus den N-Termini von N-76 bis
N-97 ausgewählt
sind, ist es selbstverständlich,
daß mehr
als eine Aminosäure
deletiert sein kann. Beispielsweise kann N-77, in dem, wie in Tabelle
4 dargestellt, eine Aminosäure
deletiert ist, als "N-77Δ1" bezeichnet werden.
Bei einer Deletion von sechs Aminosäuren, z. B. S-52 bis F-57,
wird der N-Terminus als "N-77Δ6"etc. bezeichnet.
Bestimmte Proteine werden wie in den nachstehenden Tabellen 2 und
3 bezeichnet. Beispiele, in denen die drei Glykosylierungsstellen
und Arg-275 deletiert sind, sind nachstehend dargestellt:

-
Beispiele für Verbindungen, die an der
ersten N-Glykosylierungsstelle
modifiziert sind, wobei Asn-117 durch Gln ausgetauscht ist, sind
nachstehend aufgeführt:
2-1/N-424/Arg
2-1/N-425/Arg
2-1/N-426/Arg
2-1/N-427/Arg
2-1/N-428/Arg
2-1/N-63Δ2,N-92Δ3/Arg
2-1/N-63Δ1,N-92Δ2/Arg
2-1/N-63Δ1,N-92Δ1/Arg
-
Beispiele für Verbindungen mit den gleichen
vorstehend beschriebenen Deletionen, die zudem an allen drei Glykosylierungsstellen
modifiziert sind, wobei Asn jeweils durch Gln ausgetauscht ist,
sind nachstehend aufgeführt:
2-7/N-424/Arg
2-7/N-425/Arg
2-7/N-426/Arg
2-7/N-427/Arg
2-7/N-428/Arg
2-7/N-63Δ2,N-92Δ3/Arg
2-7/N-63Δ1,N-92Δ2/Arg
2-7/N-63Δ1,N-92Δ1/Arg
-
Somit schließt diese Ausführungsform
außerdem
eine Untergattung von C-Proteinen ein, in denen eine oder mehrere
Deletionen von weniger als etwa 20 Aminosäuren im Bereich Val-4 bis Val-72
vorhanden sind. Proteine dieser Untergattung sind an einer oder
mehreren Asn-Glykosylierungsstellen und gegebenenfalls am Arg-275
modifiziert. Beispiele für
Proteine dieser Untergattung entsprechen den in Tabellen 2 bis 4 dargestellten,
sie enthalten jedoch anstelle des Wildtyp-N-Terminus einen N-Terminus, wie er beispielsweise nachstehend
in Tabelle 5 dargestellt ist. Weitere Beispiele für Verbindungen
dieser Untergattung sind ebenfalls vorstehend mit ihren aus einem
dreiteiligen Code bestehenden Bezeichnungen aufgeführt.
-
In einer dritten Ausführungsform
sind die Proteine durch eine Peptidsequenz gekennzeichnet, die im wesentlichen
der Peptidsequenz von menschlichem t-PA entspricht, in der jedoch
Aminosäuren
im Bereich Arg-23 bis Val-72 durch verschiedene Aminosäuren ausgetauscht
sind. Diese Ausführungsform
schließt
eine Untergattung von Verbindungen ein, die durch den Austausch
von einer oder mehreren Aminosäuren
innerhalb des vorstehend beschriebenen N-Terminus und durch eine Modifikation
am Arg-275, wie vorstehend beschrieben, gekennzeichnet ist. Diese
Ausführungsform
von Verbindungen ist ebenfalls gekennzeichnet durch die Modifikation,
wie vorstehend beschrieben, an einer oder mehreren der Konsensus-Asn-Glykosylierungsstellen.
Eine weitere Untergattung dieser Ausführungsformen ist durch Austausch
einer oder mehrerer Aminosäuren
innerhalb des N-Terminus und durch Modifikationen, wie vorstehend
beschrieben, sowohl am Arg-275 als auch an einer oder mehreren der
N-Glykosylierungsstellen gekennzeichnet. In einer weiteren Ausführungsform
werden ein bis etwa elf, vorzugsweise ein bis etwa 6, Aminosäuren innerhalb
eines oder mehrerer der nachstehend aufgeführten Bereiche ausgetauscht.
-
-
Ein weiterer Aspekt dieser Ausführungsform
betrifft die Anwesenheit der Substitution(en) in einem oder in mehreren
der nachstehend aufgeführten
Bereiche: N-3' bis
C-42 und H-44 bis
S-50. Ein weiterer Gesichtspunkt dieser Ausführungsform betrifft die Modifikation
des N-Terminus wiederum durch Austausch von einer bis etwa elf,
vorzugsweise einer bis etwa sechs, Aminosäuren in einem oder in mehreren
der vorstehend definierten Bereiche und außerdem durch Deletion von einer
bis 50, vorzugsweise 1 bis etwa 45, und stärker bevorzugt 1 bis etwa 15,
Aminosäuren.
-
Beispiele für Aminosäuresubstitutionen sind nachstehend
in Tabelle 6-A und Beispiele für
Proteine in Tabelle 6-B
dargestellt. Von den in Tabelle 6-A dargestellten, anstelle , von
R-40, A-41 und Q-42 eingesetzten Aminosäuren werden S bevorzugt anstelle
von R-40 und V und L bevorzugt anstelle von A-41 bzw. Q-42 eingesetzt.
Es soll darauf hingewiesen werden, daß die erfindungsgemäßen Ausführungsformen
der Proteine, in denen R-40, A-41 und Q-42 entsprechend der Tabelle
6A ausgetauscht sind, kombiniert werden mit Modifikationen an mindestens
einer Glykosylierungsstelle und gegebenenfalls mit (einer) weiteren
Substitution en) und/oder Deletionen im NTerminus und/oder Modifikationen an
R-275. Je mehr die Proteine durch Substitution statt durch Deletion
modifiziert sind, desto mehr sollten die Proteine die native t-PA-Konformation
und selektiv mehr der gewünschten
biologischen Aktivitäten
von nativem t-PA beibehalten.
-
Tabelle 5
-
Beispiele für Proteine
mit einer oder mehreren Deletionen von weniger als –20 Aminosäuren innerhalb
des Bereichs Val-4 bis Val-72
-
(Tabelle 1 zeigt die allgemeine
Sequenz)
-
Die Beispiele für Proteine entsprechen den
in Tabelle 2 definierten, sie weisen jedoch die nachstehend aufgeführten N-Termini
auf, die die Wildtyp (wt)-Sequenz im Bereich Gly-(-3) bis Thr-91
ersetzen:
-
Tabelle
6-A
Beispiele für
Aminosäure-Substitutionen
-
Tabelle 6-B
-
Beispiele für Proteine,
die Substitutionen für
eine oder mehrere Aminosäuren
im Bereich von Arg-23 bis Val-72 enthalten (die allgemeine Sequenz
ist in Tabelle 1 dargestellt)
-
Beispiele für Proteine entsprechen den
in Tabelle 2 definierten, wobei allerdings die Wildtyp (wt)-Sequenz
von Gly-(-3) bis Thr-91 gegen die nachstehend dargestellten N-Termini
ausgetauscht ist:
-
Erfindungsgemäße Ausführungsformen der Proteine,
die (eine) Aminosäuresubstitution(en)
enthalten, können,
wie vorstehend beschrieben, mit einem dreiteiligen Code bezeichnet
werden. Es kann selbstverständlich
mehr als eine wt-Aminosäure
ausgetauscht sein. Bei der Bezeichnung von Verbindungen mit solchen N-Termini
wird die Zahl der Substitutionen durch "sn" angegeben,
wobei "n" die Zahl der ausgetauschten
Aminosäuren
ist, wobei z. B. für
den Austausch die in Tabelle 6-A aufgeführten Aminosäuren (jedoch
nicht ausschließlich)
verwendet werden können.
Beispielsweise bezeichnet der N-Terminus #N-14451 den N-Terminus 144,
wie in Tabelle 6-B dargestellt, während der N-Terminus #N-14454
den N-Terminus bezeichnet, in dem R-23 durch G ersetzt und die folgenden
drei wt-Aminosäuren
durch andere Aminosäuren
ausgetauscht sind. Die Proteine dieser Ausführungsform, die mehrere Aminosäuresubstitutionen
enthalten, können
durch eine Aneinanderreihung von N-Terminus-Bezeichnungen, die spezifische
Austausche anzeigen, wie nachstehend dargestellt, bezeichnet werden:
-
Beispiele von Verbindungen mit mehreren
Substitutionen am N-Terminus,
wobei Asn-117 durch Gln und Arg-275 durch Thr ersetzt ist:
-
Eine Untergattung von besonderem
Interesse ist durch den Austausch einer oder mehrerer Aminosäuren im
Bereich von Y-67 bis S-69 gegebenenfalls mit Deletion und/oder Austausch
einer oder mehrerer Aminosäuren
im Bereich von Arg-23 bis L-66 mit einer Modifikation, wie vorstehend
beschrieben, an einer oder mehreren Glykosylierungsstellen und gegebenenfalls
am Arg-275 gekennzeichnet.
-
In einem erfindungsgemäßen Gesichtspunkt
enthalten die Proteine mindestens eine als "komplexes Kohlenhydrat" bezeichnete, für Säuger-Glykoproteine
charakteristische Zuckereinheit. Wie nachstehend genauer beschrieben,
können
derartige, ein "komplexes
Kohlenhydrat" enthaltende
Glykoproteine durch Expression eines DNA-Moleküls, das die gewünschte Polypeptidsequenz
codiert, in Wirtszellen von Säugern
produziert werden. Geeignete Wirtszellen von Säugern und Verfahren zur Transformation,
Kultur, Vermehrung, zum Absuchen, zur Herstellung und Reinigung
des Produkts sind im Stand der Technik bekannt. Vgl. z. B. Gething und
Sambrook, Nature 293 (1981), 620–625, oder auch Kaufman et
al., Molecular and Cellular Biology 5 (7) (1985), 1750–1759, oder
Howley et al., US-Patent Nr. 4,419,446.
-
Ein weiterer erfindungsgemäßer Gesichtspunkt
betrifft tPA-Varianten,
wie vorstehend beschrieben, in denen jede Kohlenhydrateinheit eine
prozessierte Form des anfänglichen
Dolicolgebundenen Oligosaccharids ist, das für in Insektenzellen hergestellte
Glykoproteine charakteristisch ist, im Gegensatz zu einem "komplexen Kohlenhydrat"-Substituenten, der für Säugerglykoproteine, einschließlich des
Säuger-t-PAs,
kennzeichnend ist. Eine derartige Glykosylierung wie bei Insektenzellen
wird hier zur Vereinfachung als Kohlenhydrat mit "viel Mannose" bezeichnet. In dieser
Beschreibung entsprechen die komplexen und viel Mannose enthaltenden
Kohlehydrate der Definition von Kornfeld et al., Ann. Rev. Biochem.
54 (1985), 631–64.
Erfindungsgemäße Varianten
mit "viel Mannose" sind, wie vorstehend
beschrieben, durch ein variables Polypeptidrückgrat charakterisiert, das
mindestens eine besetzte N-Glykosylierungsstelle enthält. Derartige
Varianten können
durch Expression einer die Variante codierenden DNA-Sequenz in Insekten-Wirtszellen
produziert werden. Geeignete Insekten-Wirtszellen sowie zur Durchführung dieses
Gesichtspunktes der Erfindung nützliche
Verfahren und Materialien zur Transformation/Transfektion, zur Züchtung von
Insektenzellen, zum Absuchen und zur Herstellung und Reinigung des
Produkts sind im Stand der Technik bekannt. So hergestellte Glykoproteine
unterscheiden sich auch dadurch von natürlichem t-PA und von t-PA,
das bisher durch Rekombinationsverfahren in Säugerzellen hergestellt wurde,
daß die
Varianten dieses erfindungsgemäßen Gesichtspunkts
keine terminalen Sialinsäuren
oder Galaktose-Substituenten auf den Kohlenhydrateinheiten oder
andere Proteinmodifizierungen, die für von Säugern stammende Glykoproteine
charakteristisch sind, enthalten.
-
Die erfindungsgemäßen Proteine, die keine N-gebundenen Kohlehydrateinheiten
enthalten, können ebenfalls
durch Expression eines DNA-Moleküls,
das die gewünschte
Variante codiert, z. B. die Verbindungen 1–6 bis 1–11 von Tabelle 1, in Wirtszellen
produziert werden, die Säuger-,
Insekten-, Hefe oder Bakterienzellen sind, wobei eukaryotische Wirtszellen
gegenwärtig
bevorzugt werden. Wie vorstehend angegeben, sind geeignete Säuger- und
Insekten-Wirtszellen
und ferner geeignete Hefe- und bakterielle Wirtszellen sowie bei
der Durchführung
dieses Gesichtspunktes der Erfindung nützliche Verfahren und Materialien
zur Transformation/Transfektion, zur Zellkultur, zum Absuchen und
zur Herstellung und Reinigung des Produkts ebenfalls im Stand der
Technik bekannt.
-
Wie es jedem Durchschnittsfachmann
verständlich
sein sollte; schließt
die Erfindung darüberhinaus weitere
t-PA-Varianten ein,
die anstelle einer Aminosäure-Deletion
im Bereich Val-4 bis Val-72 durch eine oder mehrere Aminosäuresubstitutionen
innerhalb des Bereichs, insbesondere im Bereich Val-4 bis Val-72
oder durch eine Kombination einer Deletion und Substitution gekennzeichnet
sind. Die diese Verbindungen codierenden cDNAs können z. B. durch Verfahren,
die den hier beschriebenen Mutageneseverfahren entsprechen, unter
Verwendung von geeigneten Mutagenese-Oligonucleotiden einfach hergestellt
werden. Die cDNAs werden an einem oder mehreren der Codons für R1, R2 und R3 und gegebenenfalls am Arg-275 mutagenisiert
und können
durch die hier beschriebenen Verfahren in Expressionsvektoren inseriert
und in Wirtszellen exprimiert werden. Diese Proteine sollten die
vorteilhaften pharmakokinetischen Eigenschaften der anderen erfindungsgemäßen Verbindungen
aufweisen, und durch sie sollte eine schädliche Antigenität nach der
Verabreichung in Arzneimitteln, die den hier beschriebenen entsprechen,
möglicherweise
vermieden werden.
-
Wie der vorstehenden Beschreibung
zu entnehmen ist, werden sämtliche
erfindungsgemäßen Varianten
durch Rekombinationsverfahren unter Verwendung von DNA-Sequenzen
hergestellt, die Analoge codieren, die auch weniger oder keine potentiellen
Glykosylierungsstellen im Vergleich zu natürlichem menschlichem t-PA enthalten
und gegebenenfalls eine Deletion oder einen Austausch des Arg-275
enthalten. Derartige DNA-Sequenzen können durch eine übliche ortsspezifische
Mutagenese von t-PA codierenden DNA-Sequenzen hergestellt werden.
-
t-PA codierende DNA-Sequenzen sind
cloniert und charak terisiert worden: Vgl. z. B. D. Pennica et al., Nature
(London) 301 (1983), 214, und R. Kaufman et al., Mol. Cell. Biol.
5(7) (1985), 1750. Clon ATCC 39891, der ein thrombolytisch aktives
t-PA-Analogon codiert, ist insofern einzigartig, als er an Position
245 anstelle der Aminosäure
Val einen Met-Rest enthält.
Typischerweise codiert die DNA-Sequenz eine Leadersequenz, die prozessiert,
d. h., durch die Wirtszelle erkannt und entfernt wird, worauf die
Aminosäurereste
des vollständigen Proteins,
das mit Gly.Ala.Arg.Ser.Tyr.Gln. etc. beginnt, folgen. Je nachdem,
in welchen Medien und welcher Wirtszelle die DNA-Sequenz exprimiert
wird, kann das so produzierte Protein mit dem Aminoterminus G1y.Ala.Arg
beginnen oder weiter. prozessiert werden, so dass die ersten drei
Aminosäurereste
proteolytisch entfernt werden. Im letzten Fall weist das reife Protein
einen Aminoterminus auf, der Ser.Tyr.Gln.Leu. etc. umfaßt. t-PA-Varianten,
die einen der Aminotermini aufweisen, sind thrombolytisch aktiv
und Bestandteil der Erfindung. Zu den erfindungsgemäßen Varianten
gehören
auch Proteine, die entweder Met245 oder
Val 245 enthalten, sowie weitere Varianten, z. B. allele Varianten
oder andere Aminosäuresubstitutionen
oder Deletionen, die thrombolytische Aktivität beibehalten.
-
Wie vorstehend beschrieben, können individuelle
erfindungsgemäße Varianten
codierende DNA-Sequenzen durch übliche
ortsspezifische Mutagenese einer DNA-Sequenz hergestellt werden,
die menschliches t-PA oder Analoge oder ihre Varianten codiert.
Zu derartigen Mutagenese-Verfahren gehört das M13System von Zoller
und Smith, Nucleic Acids Res. 10 (1982), 6487–6500, Methods Enzymol. 100
(1983), 468–500,
und DNA 3 (1984), 479–488,
in dem einzelsträngige
DNA verwendet wird, und das Verfahren von Morinaga et al., Bio/technology
(Juli 1984), 636–639,
in dem DNA als Heteroduplex verwendet wird. Mehrere Beispiele von
Oligonucleotiden, die gemäß derartiger
Verfahren zum Erhalt von Deletionen am N-Terminus oder zur Umwandlung
eines Asparaginrestes in beispielsweise Threonin oder Glutamin verwendet
werden, sind in Tabelle 7 dargestellt. Es ist natürlich selbstverständlich,
daß eine
DNA, die eines der erfindungsgemäßen Glykoproteine
codiert, durch einen Fachmann durch eine ortsspezifische Mutagenese
unter Verwendung von zweckmäßig gewählten Oligonucleotiden
bzw. eines Oligonucleotids analog produziert werden kann. Eine Expression
der DNA durch übliche
Verfahren in einem Wirtszellsystem aus Säuger-, Hefe-, Bakterien- oder
Insektenzellen liefert die gewünschte
Variante. Säuger-Expressionssysteme
und die dadurch erhaltenen Varianten werden gegenwärtig bevorzugt.
-
Die hier beschriebenen Expressionsvektoren
für Säugerzellen
können
durch dem Fachmann vertraute Verfahren synthetisiert werden. Die
Bestandteile der Vektoren, beispielsweise die bakteriellen Replicons,
Selektionsgene, Enhancer, Promotoren etc., können von natürlichen
Quellen oder durch Synthese mit bekannten Verfahren erhalten werden.
Vgl. Kaufman et al., J. Mol. Biol. 159 (1982), 51–521, und
Kaufman, Proc. Natl. Acad. Sci. 82 (1985) 689–693.
-
Etablierte Zellinien, zu denen transformierte
Zellinien gehören,
sind als Wirte geeignet. Normale diploide Zellen, Zellstämme, die
von einer in vitro-Kultur von primärem Gewebe stammen, sowie primäre Explantate (die
relativ undifferenzierte Zellen, wie hämatopoetische Stammzellen,
einschließen)
sind ebenfalls geeignet. Die Kandidatenzellen müssen hinsichtlich des Selektionsgens
nicht genotypisch defizient sein, wenn das Selektionsgen dominant
ist.
-
Die Wirtszellen sind vorzugsweise
etablierte Säugerzellinien.
Zur stabilen Integration der Vektor-DNA in die chromosomale DNA
und zur anschließenden
Vermehrung der integrierten Vektor-DNA, jeweils durch übliche Verfahren,
werden gegenwärtig
CHO-Zellen (Eierstockzellen des Chinesischen Hamsters) bevorzugt.
In einer anderen Ausführungsform
kann die Vektor-DNA das gesamte oder einen Teil des Genoms des Rinder-Papillomavirus
enthalten (Lusky et al., Cell 36 (1984), 391–401) und als ein stabiles
episomales Element in Zellinien, wie C127-Mauszellen, eingeführt werden.
Weitere verwendbare Säuger-Zellinien
schließen
HeLa-Zellen, COS-1-Affenzellen, Maus-L-929-Zellen, 3T3-Linien, die
von "Swiss"-, BALB/c- oder NIH-Mäusen stammen,
BHK- oder HaK-Hamster-Zellinien etc. ein, sie sind jedoch nicht
darauf beschränkt.
-
Stabile Transformanten werden anschließend durch
immunologische oder enzymatische Standardtests abgesucht, um festzustellen,
ob das Produkt exprimiert wird. Die Gegenwart der die Proteinvarianten
codierenden DNA kann durch Standardverfahren, wie dem Southern-Blot-Verfahren,
nachgewiesen werden. Die vorübergehende
Expression der die Varianten codierenden DNA während mehrerer Tage nach der
Einführung der
Expressions-Vektor-DNA in geeignete Wirtszellen, beispielsweise
in COS-1Affenzellen, wird ohne Selektion durch einen Aktivitäts- oder
Immuntest der Proteine im Kulturmedium gemessen.
-
Zur Expression in Bakterien kann
die die Variante codierende DNA weiter modifiziert werden, wobei sie
verschiedene, im Stand der Technik bekannte Codons zur Expression
in Bakterien enthält
und vorzugsweise im Leseraster mit einer Nucleotidsequenz funktionell
verbunden wird, die ein sekretorisches Leader-Polypeptid codiert,
das die Expression, Sekretion und das Prozessieren der reifen Proteinvariante
in Bakterien ermöglicht,
wie dies im Stand der Technik ebenfalls bekannt ist. Die in Säuger-, Insekten-,
Hefe- oder bakteriellen Wirtszellen exprimierten Verbindungen können anschließend gewonnen,
gereinigt und/oder im Hinblick auf physikalisch-chemische, biochemische und/oder klinische
Parameter charakterisiert werden, wobei immer bekannte Verfahren
verwendet werden.
-
Es wurde festgestellt, daß diese
Verbindungen gegen menschliches t-PA gerichtete monoclonale Antikörper binden
und daher durch Immunaffinitätschromatographie
unter Verwendung solcher Antikörper
gewonnen und/oder gereinigt werden können. Ferner weisen diese Verbindungen
eine t-PA-artige Enzymaktivität auf,
d. h., die erfindungsgemäßen Verbindungen
aktivieren Plasminogen in Gegenwart von Fibrin effektiv, wobei eine
Fibrinolyse bewirkt wird, die durch einen im Stand der Technik bekannten
indirekten Test unter Verwendung des chromogenen Substrats S-2251
für Plasmin
gemessen werden kann.
-
Diese Erfindung schließt außerdem Zusammensetzungen
zur thrombolytischen Therapie ein, die eine therapeutisch wirksame
Menge einer vorstehend beschriebenen Variante zusammen mit einem
pharmazeutisch verträglichen
parenteralen Träger
umfassen. Eine derartige Zusammensetzung kann dem menschlichen t-PA
entsprechend verwendet werden und sollte bei Menschen oder niederen
Tieren, beispielsweise Hunden, Katzen und anderen Säugern nützlich sein,
bei denen bekanntermaßen
thrombotische Probleme im Herzkreislaufsystem auftreten. Die Zusammensetzungen
sollen sowohl zur Behandlung als auch, falls gewünscht, zur Verhinderung von
thrombotischen Zuständen
verwendet werden. Die genaue Dosierung und das Verabreichungsverfahren
werden durch den behandelnden Arzt je nach Potenz und pharmakokinetischein
Profil der jeweiligen Verbindung sowie nach verschiedenen Faktoren
festgelegt, die die Wirkungen von Arzneimitteln modifizieren, beispielsweise
das Körpergewicht,
Geschlecht, die Ernährung,
Zeit der Verabreichung, Arzneimittelkombination, Reaktionsempfindlichkeiten
und die Schwere des jeweiligen Falls.
-
Die nachstehenden Beispiele sollen
die erfindungsgemäßen Ausführungsformen
erläutern.
Es ist selbstverständlich,
daß die
Erfindung durch diese Beispiele lediglich erläutert wird und nicht auf sie
beschränkt ist,
es sei denn es ist in den beigefügten
Ansprüchen
entsprechend angegeben.
-
In allen Beispielen, in denen Insektenzellen
zur Expression verwendet wurden, war das verwendete nukleäre Polyedervirus
die L-1-Variante von Autographa Californica und die verwendete Insekten-Zellinie
die Zellinie Spodoptera frugiperda IPLB-SF21 (J. L. Vaughn et al.,
In Vitro 13 (1977), 213–217).
Die Manipulationen an den Zellen und Viren entsprachen den Angaben
in den Veröffentlichungen
(G. D. Pennock et al., a, a. 0., D. W. Miller, P. Safer und L. K.
Miller, Genetic Engineering Bd. 8, Seite 277–298, J. K. Setlow und A. Hollaender
(Hrsg.) Plenum Press (1986)). Die RF-m13-Vektoren, mp18 und mpll,
sind von New England Biolabs erhältlich.
Der Durchschnittsfachmann, den diese Erfindung betrifft, wird jedoch
wissen, daß weitere
Viren, Stämme,
Wirtszellen, Promotoren und Vektoren, die, wie vorstehend beschrieben,
die betreffende cDNA enthalten, zur Durchführung jeder erfindungsgemäßen Ausführungsform
ebenfalls verwendet werden können. Die
verwendeten DNA-Manipulationen entsprechen, sofern hier nicht gesondert
angegeben, denen von Maniatis et al., Molecular Cloning: A Laboratory
Manual (Cold Spring Harbor, NY 1982).
-
Tabelle
7
Beispiele für
Oligonucleotide zur Mutagenese
-
Plasmidderivate
-
Eine Mutagenese von cDNAs an den
verschiedenen Aminosäurencodons
wurde durch das Verfahren von Zoller und Smith unter Verwendung
eines geeigneten Restriktionsfragments der cDNA in M13-Plasmiden durchgeführt. Deletionen
innerhalb der cDNA wurden durch eine "loopout"-Mutagenese unter Verwendung eines geeigneten
Restriktionsfragments, z. B. des SacI-Fragments, der cDNA entweder in M13-Vektoren
oder durch Heteroduplex-"loopout" im Plasmid pSVPA4
durchgeführt.
-
Das Plasmid pSVPA4 wurde konstruiert,
damit das t-PAGlycoprotein
in Säugerzellen
exprimiert werden kann. Das Plasmid wurde hergestellt, indem zunächst die
DNA, die das große
SV40-T-Polypeptid codiert, vom Plasmid pspLT5 (Z. Zhu et al., J.
Virology 51 (1984), 170–180)
entfernt wurde. Dies wurde durch eine vollständige Spaltung mit XhoI erreicht,
wonach anschließend
mit der Restriktionsendonuclease BamHI partiell gespalten wurde.
Der das große
T codierende Bereich von SV40 in pspLT5 wurde durch die menschlichen
t-PA codierende Sequenz ausgetauscht, indem ein t-PA codierendes
SalI/BamHI-Restriktionsfragment
mit überstehenden
Enden, das durch Spaltung des Plasmids J205 (ATCC Nr. 39568) mit
SalI und BamHI isoliert worden war, mit dem parentalen, mit XhoI/BamHI
gespaltenen Vektor pspLT5, der wie vorstehend beschrieben hergestellt
worden war, ligiert wurde. Daher wird das t-PA nach der Einführung in
Säugerzellen
in diesem Vektor unter der Kontrolle des späten Promotors von SV40 transkribiert.
Diese schließlich
erhaltene Konstruktion erhält die
Bezeichnung pSVPA4.
-
Das Plasmid pLDSG ist ein amplifizierbarer
Vektor, mit dem t-PA in Säugerzellen,
beispielsweise in CHO-Zellen, exprimiert werden kann. pLDSG enthält eine
Maus-DHFR-cDNA- Transkriptionseinheit,
die den späten
Hauptpromotor (MLP) des Typ 2-Adenovirus, den Affenvirus 40-(SV40)-Enhancer
und -Replikationsursprung, den späten Promotor von SV40 (in der
gleichen Orientierung wie der MLP von Adenovirus) verwendet, ein
Gen, das eine Tetracyclinresistenz codiert, und eine cDNA, die menschliches
t-PA (Met-245) in der richtigen Orientierung im Hinblick auf den
MLP des Typ 2-Adenovirus.
Die Herstellung von pLDSG aus pCVSVL2 (ATCC Nr. 39813) und einer
t-PA codierenden cDNA ist genauer beschrieben worden, wie auch die
gleichzeitige Transformation mit und Amplifikation von pLDSG in
CHO-Zellen. Kaufman et al., Mol. and Cell. Bio. 5(7) (1985), 1750–1759.
-
Das Plasmid pWGSM ist mit der Ausnahme,
daß die
cDNAInsertion menschliches Val-245-t-PA codiert, mit pLDSG identisch.
pWSGM kann unter Verwendung von cDNA vom Plasmid J205 (ATCC Nr.
39568) oder pIVPA/1 (ATCC Nr. 39891) konstruiert werden. In dieser
Beschreibung können
pWGSM und pLDSG austauschbar verwendet werden, obwohl, wie vorstehend
beschrieben, der erste Vektor Val-245-Proteine und der letzte Met-245Proteine
produziert.
-
pIVPA/I (ATCC Nr. 39891) ist ein
baculoviraler Transplacement-Vektor, der eine t-PA codierende cDNA enthält. pIVPA/I
und mutagenisierte Derivate davon werden zur Insertion einer gewünschten
cDNA in ein baculovirales Genom verwendet, so dass die Transkription
der cDNA vom baculoviralen PolyhedrinPromotor kontrolliert wird.
-
Heteroduplex-Mutagenese
-
Die Mutagenese mittels einer Heteroduplex-DNA
in bestimmten Bereichen vom t-PA-Expressionsplasmid, pSVPA4, schließt die nachstehend
beschriebenen Schritte ein:
-
Herstellung der Ampicillin-sensitiven
pSVPA4-DNA
-
- 1. Plasmid pSVPA4 (15 μg)
wurde mit PvuI vollständig
linearisiert. Das Gemisch wurde mit Phenol/Chloroform extrahiert
und die DNA unter Verwendung von zwei Volumina Ethanol sowie 0,1
M NaCl ausgefällt.
- 2. Die DNA wurde in 21 μ1
Wasser, 1 μ1
dNTP-Lösung
(die 2 mM dATP, dGTP, dTTP und dCTP enthielt), 2,5 μ1 10 × NickTranslationspuffer
(0,5 M Tris-Cl, pH 7,5, 0,1 M MgSO4, 10
mM DTT, 500 μg/ml)
und 0,5 μ1
(2 Einheiten) des großen
Fragments der DNA-Polymerase I (New England Biolabs) resuspendiert.
Dieses Gemisch wurde dreißig
Minuten bei Raumtemperatur inkubiert und anschließend mit
Phenol/Chloroform extrahiert, woran sich, wie vorstehend beschrieben,
eine Ethanolausfällung
anschloss.
- 3. Die ausgefällte
DNA wurde durch Zugabe von 75 μl
Wasser zu 0,2 μg/μl resuspendiert.
-
Herstellung von Ampicillin-resistenter
pSVPA4-DNA
-
- 1. Das Plasmid pSVPA4 (15 μg) wurde mit SacI gespalten,
die dieses Plasmid zweimal innerhalb der t-PAcodierenden Sequenz
spaltet, wobei zwei Restriktionsfragmente erzeugt werden, ein 1,4
kbp t-PAcodierendes Restriktionsfragment sowie der parentale Vektor.
Nach der Restriktion wurde 1 μl
(28 Einheiten) alkalische Phosphatase des Kälberdarms (Boehringer Mannheim)
zugesetzt und anschließend
5 Minuten bei 37°C
inkubiert. Die beiden Banden wurden getrennt, indem dieses Gemisch
auf ein 0,7 % Agarosegel aufgetragen wurde. Das Restriktionsfragment
des parentalen Vektors wurde aus dem Gel ausgeschnitten und bei
4°C durch Adsorption
an Siliziumdioxid extrahiert, wonach 30 Minuten bei 37°C mit 50
mM Tris/1 mM EDTA eluiert wurde. Die eluierte DNA wurde auf eine
Endkonzentration von 0,2 μg/μl eingestellt.
-
Aneinanderlagerung zur
Heteroduplex
-
- 1. Herstellung eines Gemisches aus 6 μl (1,2 μl) Ampicillin-sensitiver pSVPA4-DNA
und 6 μl
(1,2 μg)
Ampicillin-resistenter pSVPA4-DNA.
- 2. Zugabe eines gleichen Volumens (12 μl) 0,4 M NaOH. Inkubation für 10 Minuten
bei Raumtemperatur.
- 3. Langsame Zugabe von 4,5 Volumina (108 μl) 0,1 M Tris-HCl bei einem
pH-Wert von 7,5/20 mM HCl.
- 4. Zugabe von 50 picomol (5 μl)
phosphoryliertes mutagenes Oligonucleotid zu 45 μl Heteroduplexgemisch.
- 5. Dieses Gemisch wurde zwei Stunden bei 68°C inkubiert und anschließend bei
Raumtemperatur langsam abgekühlt.
-
Mutagenese
-
- 1. Zur Mutagenese wurde jede Reaktionslösung auf die nachstehend angegebenen
Konzentrationen eingestellt, indem 7 μl 2 mM MgCl2/0,2
mM ATP/60 μM
dATP, dTTP, dGTP, dCTP/4 mM DTT/40 Einheiten/ml Klenowfragment der
E. coli-DNA-Polymerase
I (B. R. L.), 2000 Einheiten/ml T4-DNA-Ligase (N. E. B.) zu den
Heteroduplexgemischen zugesetzt wurden. Dieses Gemisch wurde 2 Stunden
bei Raumtemperatur inkubiert.
- 2. Die Reaktionslösung
wurde anschließend
mit Phenol/Chloroform extrahiert, wonach mit Ethanol ausgefällt wurde.
Die ausgefällte
DNA wurde in 12 μl
50 mM Tris-Cl/1 mM EDTA resuspendiert. 4 μl wurden zur Transformation
kompetenter HB101-Bakterien verwendet.
- 3. Ampicillin-resistente Kolonien wurden mit 1 × 106 cpm/ml eines 32P-markierten
Oligonucleotids in 5 × SSC, 0,1
% SDS, 5 × Denhardt-Reagens
und 100 μg/ml
denaturierter Lachssperma-DNA abgesucht.
- 4. Die Filter wurden mit 5 × SSC
und 0,1 % SDS bei einer Temperatur gewaschen, die 5°C unterhalb
der berechneten Schmelztemperatur der Oligonucleotidsonde lag.
- 5. Die DNA wurde aus positiv hybridisierenden Clonen hergestellt
und zu Beginn durch Spaltung mit verschiedenen Restriktionsenzymen
und einer Agarose-Gelelektrophorese
analysiert. Die DNA wurde auf Nitrozellulose transferiert, die Filter
wurden präpariert
und mit den absuchenden Sonden hybridisiert, um sicherzustellen, dass
das mutagene Oligonucleotid in das richtige Fragment eingeführt worden
war.
- 6. Die DNA wurde anschließend
erneut in E. coli transformiert und Ampicillin-resistente Kolonien
wurden auf eine Hybridisierung mit der Oligonucleotidsonde getestet.
- 7. Mutationen wurden schließlich
durch eine DNA-Sequenzierung
(Sauger) überprüft.
-
Herstellung von mutagenisierten
cDNAs: M13-Verfahren
-
Die nachstehende schematische Restriktionskarte
stellt eine menschlichen t-PA codierende cDNA (oberhalb) dar, wobei
die Spaltstellen bestimmter Endonucleasen (unterhalb) angegeben
sind:
-
Das
Initiationscodon ATG und die (a) codierenden cDNABereiche
-
R1, R2 und R3 sind angegeben.
Somit kann eine Mutagenese am N-Terminus beispielsweise unter Verwendung
des SacI-Fragments
oder des BglII/NarI-Fragments durchgeführt werden. Eine Mutagenese
am Arg-275 und/oder R1 und/oder R2 kann
beispielsweise unter Verwendung des SacI-Fragments öder des BglII/SacI-Fragments
durchgeführt
werden. Eine Mutagenese am R3 kann unter
Verwendung eines EcoRI/XmaI- oder EcoRI/ApaI-Fragments durchgeführt werden.
Die Wahl des Restriktionsfragments kann davon abhängen, welche
bestimmten Vektoren zur Mutagenese und/oder zur Konstruktion von
Expressionsvektoren geeignet sind.
-
Üblicherweise
kann das zu mutagenisierende cDNA-Restriktionsfragment aus der vollständigen cDNA,
die z. B. in pWGSM, pIVPA/I oder pSVPA4 vorhanden ist, unter Verwendung
der/des angegebenen Endonucleaseenzyms(e) ausgeschnitten und anschließend mutagenisiert
werden, z. B. mit den in Tabelle 7 dargestellten Oligonucleotiden
oder anderen, für
die gewünschte
Mutagenese konstruierten Oligonucleotiden.
-
Beispiele für mutagenisierte cDNA-Fragmente,
die so hergestellt werden können,
sind nachstehend in Tabelle 8 dargestellt.
-
Tabelle
8
Beispiele für
mutagenisierte cDNA-Fragmente
Im Anschluss an die Mutagenese kann das Fragment,
mit oder ohne weitere Mutagenese, aus dem M13-Vektor ausgeschnitten
und in einen Expressionsvektor zurück ligiert werden, der die
gesamte oder einen Teil der cDNA enthält und zuvor mit den (dem)
gleichen zum Ausschneiden des mutagenisierten Fragments aus dem M13-Vektor
verwendeten Enzym en) gespalten worden ist. Durch dieses Verfahren
kann die vollständige,
nach Wunsch mutagenisierte cDNA erneut unter Verwendung eines oder
mehrerer mutagenisierter Fragmente als Restriktionsfragment-Kassetten
erneut zusammengesetzt werden.
-
cDNAs, die die nachstehenden Verbindungsbeispiele
codieren (vgl. die Tabelle auf Seite 9 und die Tabellen 2.0, 2.5 & 3) können, wie
nachstehend beschrieben, aus den mutagenisierten Fragmenten von
Tabelle 8 hergestellt werden:
-
Die Plasmide pIVPA oder pSVPA4 können neben
ihrer Nützlichkeit
als Expressionsvektoren auch als ein "Depot" zur Konstruktion von cDNAs, die jede
gewünschte
Permutation von mutagenisierten Stellen aufweisen, verwendet werden.
Daher können
die (mittels M13 oder Heteroduplexbildung) mutagenisierten Plasmide "pIVPA/Δ" oder "pSVPA4/Δ", die eine gewünschte Modifikation
des den N-terminalen Bereich codierenden cDNA-Bereichs enthalten,
mit NarI (partiell) und XmaI (SmaI) (vollständig) gespalten werden, um
den cDNA-Bereich zu entfernen, der die R1,
R2 und R3 umfassende
Proteindomäne
codiert.
-
Ein zweites Plasmid pIVPA oder pSVPA4,
mutagenisiert, falls erwünscht,
(mittels M13 oder Heteroduplexbildung) in jeder Kombination von
Arg-275, R1, R2 und
R3 codierenden Bereichen, kann anschließend mit NarI
(vollständig)
und XmaI (SmaI) (vollständig)
gespalten und das NarI/XmaI (SmaI)-Fragment identifiziert, isoliert
und mit dem NarI/XmaI (SmaI)-gespaltenen pIVPA/Δ oder pSVPA4/Δ ligiert
werden. Unter Verwendung dieser NarI/XmaI (SmaI)-Restriktionsfragment-Kassette können beispielsweise
die gewünschten
mutagenisierten cDNAs in pIVPA oder pSVPA4 konstruiert werden. Die
mutagenisierte cDNA kann anschließend, z. B. als eine BglII/XmaI-Restriktionsfragment-Kassette
in das BglII/XmaI-gespaltene pWGSM zur Expression in Säugern, falls
erwünscht,
transferiert werden.
-
Beispiele
-
Beispiel 1
-
Herstellung von G1n117-Deletionsvarianten
-
A. Herstellung von am
Gln-117 verkürzter
cDNA
-
cDNA-Moleküle, die die Polypeptidsequenz
der Verbindungen 2-1/N-21/Arg codieren, wurden unter Verwendung
des Oligonucleotid-gerichteten Mutageneseverfahrens von Zoller und
Smith hergestellt. Insbesondere wurde der das t-PA-Gen enthaltende
Mutagenesevektor RF-M13/t-PA aus dem Säuger-t-PA-Expressionsplasmid pSVPA4 konstruiert.
RF M13/t-PA wurde konstruiert, indem zunächst pSVPA4 mit der Restriktionsendonuclease
SacI vollständig
gespalten wurde. Das etwa 1436 Basenpaar (bp)-SacI-Fragment codiert
einen grossen Teil der Polypeptidsequenz von t-PA und schließt die Nucleotidsequenzen
ein, die die Konsensus-N-Glykosylierungsstellen
codieren, die die Asparaginreste 117, 184 und 218 enthalten. Dieses
1436 by (hier nachstehend als 1,4 kbp bezeichnet) Fragment wurde
durch präparative
Agarose-Gelelektrophorese gereinigt.
-
Das SacI-Fragment der t-PA-cDNA,
das vorstehend als ein SacI-Fragment erhalten worden war, wurde
mit einem linearisierten doppelsträngigen RF-M13mp18-DNA-Vektor
ligiert, der zuvor mit SacI gespalten worden war. Das Ligierungsgemisch
wurde verwendet, um Transformationskompetente bakterielle JM101-Zellen
zu transformieren. M13-Plaques, die die von transformierten Zellen
produzierte, von t-PA stammende DNA enthielten, wurden durch analytische
DNA-Restriktionsanalyse und/oder Plaquehybridisierung identifiziert
und isoliert. Radioaktiv-markierte Oligonucleotide (~17-mere, mit
einer positiven Polarität),
die einem Bereich zwischen den SacIRestriktionsstellen der t-PA
codierenden, in Tabelle 1 dargestellten Nucleotidsequenz entsprachen,
wurden als Sonden verwendet, wenn mittels Filterhybridisierung t-PA-DNA
enthaltende virale Plaques nachgewiesen werden sollten. Sämtliche
Oligonucleotide wurden nach Herstellerangaben durch automatisierte
Synthese mit einem DNA-Synthesizer (Applied Biosystems) hergestellt.
-
Einige der durch Restriktions- oder
Hybridisierungsanalyse nachgewiesenen positiven Plaques wurden anschließend durch
eine übliche
Plaquereinigung weiter geclont. Der durch das Plaque-Reinigungsverfahren
erhaltene, gereinigte M13/t-PA-Bakteriophage
wurde zur Infektion von JM101-Zellen verwendet. Diese infizierten
Zellen produzieren eine cytoplasmatische doppelsträngige "RF"-M13/t-PA-Plasmid-DNA.
Die infizierten Zellen produzieren im Kulturmedium auch Bakteriophagen,
die einzelsträngige,
zu dem 1,4 kbp-SacI-Fragment
von t-PA und der M13-DNA komplementäre DNA enthalten. Die einzelsträngige DNA
wurde aus dem M13/t-PA enthaltenden, aus dem Kulturmedium isolierten
Phagen gereinigt. Diese einzelsträngige M13/t-PA-DNA wurde als
Matrize in einer Mutagenesereaktion gemäß dem Verfahren von Zoller
und Smith unter Verwendung des Oligonucleotids #3 von Tabelle 7
verwendet. Dieses Mutageneseereignis verursacht durch Umwandlung
der DNA-Sequenz "AAC" in "CAG" einen Austausch
des Asn-Codons zu einem Gln-Codon an Position 117 des dadurch erhaltenen
codierenden DNA-Strangs. Nach der Mutagenesereaktion wurde die DNA
in den bakteriellen Stamm JM101 transformiert. Zur Identifizierung
von mutagenisierten cDNAs wurden die Plaques der Transformanten
durch DNA-Hybridisierung
unter Verwendung des radioaktiv-markierten Oligonucleotids #4 von
Tabelle 7 abgesucht. Sämtliche
Oligonucleotidbeispiele in Tabelle 7 weisen eine positive Polarität auf, d.
h., sie stellen Teile eines codierenden und nicht eines nicht-codierenden
DNA-Stranges dar. Sämtliche
positiv hybridisierenden Plaques wurden durch anschließende sekundäre Infektionen
von JM101-Zellen mit M13-Phagen, die die mutagenisierte DNA enthielten,
weiter gereinigt.
-
RF-M13/t-PA-Plasmid-DNA wurde aus
JM101-Zellen, die mit dem gereinigten, die mutagenisierte t-PA-cDNA
enthaltenden M13Phagen infiziert worden waren, gereinigt. Das so
erhaltene RFM13/t-PA-Plasmid enthält das am Gln117 mutagenisierte
SacIRestriktionsfragment der t-PA-DNA. Dieses mutagenisierte Restriktionsfragment
kann anschließend
weiter mutagenisiert werden, wiederum nach dem Verfahren von Zoller
und Smith, allerdings unter Verwendung der nachstehend beschriebenen
Oligonucleotide. Die nachstehend beschriebenen Oligonucleotide wurden
konstruiert, um eine Deletion (Schleifenbildung) innerhalb des cDNA-Bereichs
der N-terminalen Domäne
zu induzieren.
-
Deletionsmutagenese 1: Das Oligonucleotid
#8 in Tabelle 7 bewirkte eine Deletion der cDNA, die von Cys-6 bis
einschließlich
Ser-50 reichte. Im Anschluss an diese zweite Mutagenesereaktion
wurde die DNA in JM101-Zellen transformiert. Zur Identifizierung
von mutagenisierten cDNAs wurden die Plaques der Transformanten,
wie vorstehend beschrieben, abgesucht, allerdings unter Verwendung
des radioaktiv-markierten Oligonucleotids #9 von Tabelle 7. Die
positiv hybridisierenden Plaques können durch anschließende erneute
Infektionen vonJM101-Zellen mit dem M13-Phagen, der die zweifach
mutagenisierte t-PAcDNA enthält,
gereinigt werden. Die wie nachstehend beschrieben hergestellte cDNA,
die dieses mutagenisierte Restriktionsfragment enthält, codiert
Verbindung 2-1/N-21/Arg,
in der Ile-5 über
eine Peptidbindung mit Cys-51 kovalent verbunden ist.
-
Dieses mutagenisierte Restriktionsfragment
kann als eine SacI-Kassette durch Verfahren, die den in Beispiel
#3B beschriebenen entsprechen, erneut mit dem Säuger-Expressionsvektor pSVPA4
ligiert werden oder zur Insertion in den Insektenzellen-Expressionsvektor
pIVPA/I (ATCC Nr. 39891) als eine BglII/SacI-Kassette, die von der
modifizierten RF-M13/tPA-DNA stammt, verwendet werden.
-
B. Herstellung von Vektoren,
die zur Expression von Gln117-Deletionsvarianten
mit viel Mannose verwendet werden
-
Das gereinigte RF-M13/tPA, das die
modifizierte und verkürzte
t-PA-cDNA enthält,
die wie vorstehend beschrieben hergestellt wird, kann mit den Restriktionsendonucleasen
BglII und SacI gespalten werden. Das etwa 1,2 kbp-BglII/SacIRestriktionsfragment
wurde durch eine übliche
präparative
Gelelektrophorese gereinigt. Das so erhaltene BglII/SacIFragment
stellt eine mutagenisierte Kassette dar, der ein 5'- und 3'-Bereich der DNA
fehlt, die den Amino- und Carboxy-Terminus des translatierten Proteins
codiert.
-
Der Insekten-Expressionsvektor pIVPA/I
(ATCC Nr. 39891) enthält
eine Wildtyp-t-PA-cDNA-Insertion, die mit einem Polyhedrin-Promotor
sowie flankierenden DNA-Sequenzen des Baculovirus funktionell verbunden
ist. pIVPA/I wurde mit BglII und SacI gespalten, wobei ein t-PA-codierender
Bereich ausgeschnitten wird, der den N-Terminus und R1 und R2 umfasst.
Die BglII/SacI-Kassette, die die mutagenisierten, am N-Terminus modifizierten
t-PA-cDNA-Fragmente enthält,
kann anschließend
jeweils mit pIVPA/I-Expressionsvektor-DNA ligiert werden, die zuvor
nach einer Spaltung mit BglII und SacI gereinigt wurde. Das erhaltene
Plasmid pIVPA/Δ FBR,
Gln117 sollte die mutagenisierten cDNAs
enthalten, die die Verbindung 2-1/N-21/Arg codieren, die nun mit dem
Polyhedrin-Promotor funktionell verbunden ist. Die Nucleotidsequenz
der mutagenisierten cDNA-Insertion kann durch Sequenzierung des
Supercoils mit dem Plasmid als Substrat überprüft werden. Vgl. z. B. E. Y. Chen
t al., DNA 4(2) (1985), 165–170.
-
B. Einführung der
mutagenisierten cDNA in das Insektenvirus
-
Das pIVPA-Plasmid, das die mutagenisierte
cDNA enthält,
kann in das Insektenvirus durch gemeinsame Transfektion mit Wildtyp-AcNPV
in Spodopterazellen eingeführt
werden. 1 μg
gereinigte Autographa californica NPV-DNA und 10 μg der gewünschten
pIVPA-DNA werden durch ein CalciumphosphatTransfektionsverfahren
(K. N. Potter und L. K. Miller, J. Invertebr. Path. 36 (1980), 431–432) in
auf Gewebekulturgefäßen wachsende
Spodoptera-Zellen eingeführt.
Die gemeinsame Einführung
dieser DNAs in die Zellen verursacht ein doppeltes Rekombinationsereignis
zwischen dem pIVPA-Plasmid (das die mutagenisierten cDNAs enthält) und
der viralen DNA zwischen den gemeinsamen Homologiebereichen, das
heißt,
der PolyhedrinGenbereich des von dem Rekombinationsereignis abstammenden
Virus enthält
die mutagenisierte cDNA-Insertion des pIVPA-Plasmids.
-
Isolierung des Virus, das die die
erfindungsgemäßen Proteine
codierende Nucleotidsequenz enthält.
-
Die in den Medien über den
transfizierten Zellen vorhandenen Virusnachkommen werden auf frische einlagige
Schichten von Zellen in mehreren verschiedenen Verdünnungen
zur Plaquebildung transferiert. Die Plaques werden getestet und
die Rekombinanten mit einem PIB-minus-Phänotyp, wie nachstehend beschrieben;
ausgewählt:
Ein Virus, das sein Polyhedrin-Gen verloren hat, wie beispielsweise
ein Virus, das eine mutagenisierte cDNA enthält, produziert kein PIB. Scheinbar
PIB-defiziente Plaques werden selektiert, ausgeschnitten und auf
frischen Zellen vermehrt. Die Zellüberstände werden anschließend auf
eine dem t-PA-artige Enzymaktivität getestet. Positive Testergebnisse
zeigen an, dass das Glycoprotein tatsächlich produziert wird.
-
Ein alternatives Reinigungsverfahren
für Viren
mittels des Plaque-Lifting-Protokolls weicht nur geringfügig von
der vorstehend beschriebenen Vorgehensweise ab und ist nachstehend
beschrieben. Die Nachkommen des transfizierten Virus werden in einer
geeigneten Verdünnung
auf Zellkulturen enthaltende Schalen zur Plaquebildung transferiert.
Eine Kopie der einlagigen Schicht von Zellen und der Virus-Plaques
werden auf Nitrozellulose hergestellt. Die Agaroseschicht über der
Platte wird als Virusquelle aufbewahrt, nachdem die Ergebnisse der
nachstehend beschriebenen Schritte erhalten worden sind.
-
Der Nitrozellulosefilter wird mit
radioaktiven DNA-Fragmenten,
die dem in das Virus-Genom eingebauten Gen entsprechen, abgesucht.
Als positiv werden die gewertet, die das Fremdgen enthalten. Die
hybridisierte Sonde wird entfernt. Der Filter wird erneut mit radioaktiver
DNA abgesucht, die einem durch Fremd-DNA ersetzten Teil des Virus-Chromosoms
entspricht. Als positiv werden die gewertet, die noch ein Polyhedrin-Gen enthalten.
-
Die hybridisierte Sonde wird entfernt.
Der Filter wird mit einem radioaktiven DNA-Fragment, das Virus-Plaques
ungeachtet des Status des Polyhedrin-Gens identifizieren kann, erneut
abgesucht. Ein geeignetes Fragment kann das EcoRI-Fragment sein.
Diese werden als Virus-Nachkommen bewertet. Die Plaques, die für die Fremdgen-DNA-Sonde
positiv, für
die Polyhedrin-Gen-Sonde negativ und für die Virus-DNA-Sonde positiv
sind, werden ausgewählt.
Diese weisen, mit großer
Wahrscheinlichkeit den gewünschten
Genotyp auf.
-
C. Herstellung und Charakterisierung
von Glycoproteinen mit viel Mannose
-
Antikörper sind verwendet worden,
um die Gegenwart der Proteinvarianten in den extrazellulären Medien
von infizierten Zellen anzuzeigen. Das wie vorstehend beschrieben
produzierte rekombinante Virus wird zur Infektion von Zellen verwendet,
die in der üblichen
TC-100 (Gibco)-Nährsalzlösung gezüchtet worden
sind, die anstelle von 10% fötalem
Kälberserum,
wie in Standardmedien, mit 50% Eigelb angereichert ist (auf 1% Gesamtvolumen)(ScottBiologicals).
Zuvor durchgeführte
Experimente hatten gezeigt, daß unter
diesen Bedingungen ein unversehrteres Protein erhalten wird. Der Überstand
der infizierten Zellen wird auf einer Affinitätssäule fraktioniert, die einen
gebundenen monoclonalen Antikörper
gegen natürliches
menschliches t-PA trägt. Das
in dieser Säule
spezifisch zurückgehaltene
Protein wird eluiert und auf eine t-PA-Enzymaktivität getestet. Eine
festgelegte Menge aktiver Einheiten davon und von t-PA-Präparaten
als Kontrolle wird auf einem Acrylamidgel getrennt. Dieses Gel wird
anschließend
mit einem auf Silber basierenden Reagens gefärbt, um das Proteinmuster zu
erhalten. Dieses zeigt, daß das
Virus nach der Infektion von Insektenzellen ein Protein mit einer t-PAähnlichen
Aktivität
extrazellulär
produziert.
-
Radioaktiv markiertes Protein wird
zur. weiteren Charakterisierung produziert, indem zunächst mit dem
Virus (m. o. i=1) infizierte Spodoptera frugiperda-Zellen 48 Stunden.
inkubiert werden. Die Kulturplatten werden anschließend mit
Methionin-defizienten Medien gewaschen. Mit 35S-Methionin
versetzte, Methionin-defiziente Medien werden danach den Kulturplatten
zugesetzt. Die Zellkulturen werden 4 Stunden inkubiert. Der das
radioaktiv-markierte Glykoprotein enthaltende Überstand kann durch SDS-PAGE
(7,5 %) gegen in Insektenzellen analog produziertes Wildtyp-t-PA
(d. h., mit vollständiger
Länge und
vollständig
glykosyliert) und gegen Säuger-t-PA,
das z. B. durch das Verfahren von R. Kaufman et al., Mol. Cell.
Biol. 5 (7) (1985), 1750, produziert wird, allerdings in Gegenwart
von Tunicamycin (nicht-glykosyliert), analysiert werden. Die in
Beispiel 1 produzierten, partiell glykosylierten, verkürzten Proteine
sollten im Vergleich zum vollständig
glykosylierten Analogon und dem nicht-glykosylierten Analogon mit
vollständiger
Länge eine
erhöhte
Gelmobilität
aufweisen.
-
Beispiel 2
-
Herstellung weiterer erfindungsgemäßer Proteine
-
A. Herstellung weiterer
cDNAs
-
Die Mutageneseverfahren von Beispiel
1 können
auch mit anderen, in üblicher
Weise hergestellten, synthetischen Oligonucleotiden durchgeführt werden,
die die ursprüngliche
t-PA-DNA-Sequenz modifizieren, wobei Proteine produziert werden,
die am N-Terminus und/oder gegebenenfalls an N-Glykosylierungsstellen und/oder am Arg-275
mit den(m) vorstehend beschriebenen, geeigneten Codonaustausch(en)
modifiziert sind. Vgl. z. B. "Preparation
of Mutagenized cDNAs: M13 Method" und
die vorstehend beschriebenen Wege (a) bis (h).
-
Beispielsweise kann die cDNA, die
die Verbindungen D- 6,
D-1 und D-3 codiert, unter Verwendung des SacI-Restriktionsfragments in M13/t-PA und
durch Mutagenisierung mit den Oligonucleotiden #8, 10 bzw. 12, jedoch
ohne das Oligonucleotid #3, hergestellt werden. Arg-275 kann deletiert
oder z. B. durch Thr unter Verwendung der Oligonucleotide 14 bzw.
15 ersetzt werden. Die Vektorkonstruktion, Transfektion und Expression kann,
wie in Beispiel 1 für
Insektenzellen oder, wie nachstehend in Beispiel 3 beschrieben,
für Säugerzellen durchgeführt werden.
-
Eine einzelsträngige DNA, die aus dem wie
in Beispiel 1 produzierten M13-Mutagenesevektor (RF-M13/t-PA) erzeugt
wird, kann auch als Matrize verwendet werden, um ortsspezifisch
Arg-275 und/oder die Glykosylierungsstelle(n) R1 oder
R2 oder beide zu mutagenisieren. Der Bereich,
der das Asn218 enthaltende Konsensus-Tripeptid
codiert, kann in ähnlicher
Weise mutagenisiert werden. Zur Herstellung mehrerer Modifikationen
des Proteins an diesen Stellen kann ein sich wiederholendes Verfahren
verwendet werden. Beispielsweise kann im Anschluss an die Identifizierung
und Reinigung des M13-Phagen, der eine modifizierte R1-Stelle enthält, eine
einzelsträngige
DNA, die diese modifizierte Stelle enthält, von diesem Phagen gereinigt
und als Matrize verwendet werden, um eine zweite Runde der Mutagenese
innerhalb der R2-Stelle und/oder am Arg-275
zu initiieren. Dieses Verfahren kann wiederholt werden, bis alle
gewünschten
Modifikationen erhalten werden. Die cDNR, die die Verbindung 2-2/N-21/Arg
codiert, kann somit durch das Verfahren von Beispiel 1 hergestellt
werden, wobei jedoch das Mutagenese-Oligonucleotid #5 für das Oligonucleotid #3 substituiert
ist und das Screening-Oligonucleotid #6 für Oligonucleotid #4. Die cDNA,
die die Verbindung 2-6/N-21/Arg codiert, kann durch zweifache Mutagenisierung
des SacI-Fragments, wie in Beispiel 1 beschrieben, und eine weitere
Mutagenese und Absuchen mit den Oligonucleotiden #5 und #6 hergestellt
werden. Die Vektorkonstruktion, Transfektion und Expression werden,
wie in Beispiel 1 für
Insektenzellen oder, wie nachstehend für Säugerzellen beschrieben, durchgeführt. Vgl.
die vorstehend beschriebenen Wege (a) bis (h).
-
Der RF-M13/t-PA-Mutagenesevektor
enthält
keine DNA-Sequenz,
die R3 codiert, wobei die N-Glykosylierungsstelle
von t-PA dem Carboxyterminus des Proteins am nächsten liegt. Daher wurde zur
Durchführung
von DNA-Modifikationen an dieser Stelle ein neuer M13/t-PA-Mutagenese-RF-Vektor
mit der Bezeichnung M13/t-PA:Rl-XmaI hergestellt. Dieser Vektor
wurde durch vollständige
Spaltung des M13-Vektors M13mpll mit EcoRI und XmaI konstruiert.
Der RI/XmaI-gespaltene M13-Vektor
wurde mit einem gereinigten EcoRI/XmaI-t-PA-Restriktionsfragment (etwa 439 bp, hier
nachstehend als 0,4 kbp bezeichnet) ligiert, das einen die Glykosylierungsstelle
R3 umfassenden Polypeptidbereich codiert.
Dieses 0,4 kbp-Restriktionsfragment wurde
nach der Spaltung des Plasmids pWGSM mit EcoRI und XmaI gereinigt.
Das das t-PA-Gen codierende Säuger-Expressionsplasmid
pWGSM entspricht innerhalb des 439 bp-EcoRI/XmaI-Fragments dem Plasmid pLDSG,
das von Kaufman et al., Mol. Cell Biol. 5 (1985), 1750–1759, beschrieben
ist.
-
Das Ligierungsgemisch wurde zur Transformation
kompetenter JM101-Bakterienzellen verwendet. Mehrere Plaques wurden
ausgewählt
und durch eine übliche
DNA-Restriktionsfragmentanalyse
auf die Gegenwart des 0,4 kbp-t-PA-EcoRI/XmaI-Fragments
getestet. Doppelsträngige
RF-M13-DNA wurde aus Zellen, die das 0,4 kbp-t-PA-Fragment enthielten,
gereinigt. Diese DNA erhielt die Bezeichnung RF-M13/t-PA RI--XmaI-Mutagenese-Vektor.
Wie zuvor in Beispiel 1A beschrieben, kann dieser in kompetente
JM101-Zellen transformierte Vektor zur Herstellung eines M13/t-PA
RI-XmaI-Phagen verwendet werden, aus dem die einzelsträngige M13/t-PA
RI-XmaI-DNA gereinigt werden kann. Diese einzelsträngige DNA
kann als Matrize in der ortsspezifischen Mutagenesereaktion verwendet
werden, um die t-PA-DNA an der N-Glykosylierungsstelle
R3 zu modifizieren.
-
Sequenzen, die ein modifiziertes
R3 codieren, können verwendet werden, um die
Wildtyp-R3-Sequenzen zu ersetzen, die entweder
im modifizierten pIVPA/I, wie in Beispiel 1 hergestellt (am R1 und/oder R2 verkürzt und/oder
modifiziert), oder in der Wildtyp-pIVPA/I-Plasmid-DNA vorhanden
sind. Dies kann erreicht werden, indem der am R3 modifizierte
M13/t-PA:RI/XmaIMutagenese-Plasmid-Vektor zunächst mit SacI/ApaI vollständig gespalten
und das so produzierte am R3 modifizierte
165 bp-t-PA-Restriktionsfragment isoliert wird. Der Insekten-Expressionsvektor
pIVPA/I oder die pIVPA/I-Plasmid-DNA, die z. B, wie in Beispiel
1 modifiziert ist, können
in ähnlicher
Weise mit SacI und ApaI vollständig
gespalten werden, um das 165 bp-Wildtyp-t-PA-Restriktionsfragment,
das die nicht modifizierte R3-Stelle codiert, auszuschneiden. Durch
Ligierung des gereinigten Insekten-Expressionsvektors, dem das 165
bp-Fragment fehlt, mit dem modifizierten 165 bp-R3-Fragment wird ein
neuer Insekten-Expressionsvektor erhalten. Durch Expression des
Vektors wird ein verkürztes
Protein erhalten, das an der R3-Stelle sowie
an irgendeiner oder sämtlichen
anderen Konsensus-N-Glykosylierungsstellen, die in natürlichem
t-PA und/oder am Arg-275 vorhanden sind, modifiziert ist.
-
Das pIVPA-Plasmid, das die modifizierte
cDNA enthält,
kann ebenfalls verwendet werden, um das BglII/ApaI-Fragment der modifizierten
t-PA-cDNA zu erzeugen, die den Deletionsbereich in der N-terminalen Domäne sowie
den R1, R2 und R3 codierenden Bereich umfasst, oder das NarI/XmaI-Fragment, das R1, R2 und R3 umfasst. Jedes dieser Fragmente kann in
Säuger-Expressionsvektoren,
beispielsweise in pSVPA4 oder pWGSM, wie in Beispiel 3 beschrieben,
inseriert werden.
-
Beispiel 3
-
Produktion der Verbindungen
D-6, D-1 und D-3 in Säugerzellen
-
A. Herstellung von cDNA
-
cDNA-Moleküle, die die Polypeptidsequenzen
der Verbindungen D-6, D-1 und D-3 codieren, wurden unter Verwendung
der Mutagenese-Oligonucleotide #8, 10 bzw. 12 und des SacI-Fragments
der t-PA-cDNA als Matrize durch das M13-Verfahren von Beispiel 1 oder durch
Heteroduplex-Mutagenese
(Moranaga-Heteroduplex-Mutagenesis-Protokoll) (beide, a.a.O.) hergestellt.
Durch eine DNA-Sequenzanalyse wurde der Nachweis erbracht, dass
Mutanten, die durch DNA-Hybridisierung
unter Verwendung der Oligonucleotide 9, 11 bzw. 13 als Sonden selektiert
worden waren, eine korrekt modifizierte DNA-Sequenz aufwiesen.
-
B. Herstellung des modifizierten
t-PA-Vektors
-
Alle in Beispiel 1A hergestellten
modifizierten cDNAs (ΔGln117)
oder 3A (Δ)
wurden zunächst
aus dem M13-Mutagenese Vektor RF-M13/t-PA durch vollständige Spaltung
des Vektors mit SacI entfernt. Das etwa 1,4 kbp-Restriktionsfragment
jeder mutagenisierten cDNA wurde durch Gelelektrophorese gereinigt
und anschließend,
wie nachstehend beschrieben, mit pSVPA4 ligiert. Zunächst wurde
pSVPA4 zur Entfernung des 1,4 kbp-Wildtyp-t-PA-Restriktionsfragments
mit SacI gespalten. Der verbliebene Teil von SacI-gespaltenem pSVPA4
wurde anschließend
mit dem 1,4 kbp-Restriktionsfragment der mutagenisierten cDNA ligiert.
Durch dieses Ligierungsereignis können zwei Orientierungen des
inserierten Fragments erhalten werden. Die geeignete Orientierung
kann für
jeden Fall unter Verwendung von EcoRI und PvuII als Enzyme in einer üblichen
analytischen Restriktionsenzym-Analyse ermittelt werden. Durch diesen
Austausch kann das SacI-Fragment als Kassettenfragment zwischen
dem RF-M13/t-PA-Mutagenesevektor und dem pSVPA4-Säuger-Expressionsvektor
verwendet werden. Modifizierte M13-SacI-Fragmente (verkürzt und gegebenenfalls am R1 und/oder R2 modifiziert)
können
in die SacI-gespaltene pSVPA4-DNA inseriert werden, die, falls erwünscht, zuvor
am R3 modifiziert worden ist oder anschließend modifiziert
wird. Alternativ kann die zuvor am R1, R2 und/oder R3 modifizierte
DNA aus den Vektoren, beispielsweise aus pIVPA oder pSVPA4, als
ein NarI/ApaI- oder NarI/XmaI-Fragment ausgeschnitten werden. Das
so erhaltene Fragment kann anschließend in Vektoren inseriert werden,
beispielsweise in pSVPA4 oder pWGSM, die zuvor mit NarI (partiell)
und ApaI oder XmaI (vollständig) gespalten
wurden. Durch dieses Verfahren kann jede Kombination von Nterminaler
Deletion und/oder Substitution und/oder Mutagenese von Glykosylierungsstellen
und/oder einer Mutagenese am Arg-275 erhalten werden.
-
C. Transfektion von COS-Zellen
(SV40-transformierte Nierenzellen der afrikanischen grünen Meerkatze)
-
Die COS-1-Zellen (ATCC CRL 1650)
wurden durch das Verfahren von M. A. Lopata et al., Nucl. Acids Res.
12 (1984), 5707–5717,
mit den in Beispiel 3B hergestellten Vektoren, d: h. mit modifiziertem
pSVPA4, transfiziert.
-
Serum-enthaltendes Medium wurde 24
Stunden nach der Transfektion durch Serum freies Medium ausgetauscht
und 48 und 72 Stunden nach der Transfektion das konditionierte Medium
sowohl auf die Gegenwart der Plasminogenstimulierenden Aktivität unter
Verwendung des chromogenen Substrats S-2251 als auch auf die Gegenwart
des tPA-Antigens durch einen ELISA-Test untersucht.
-
D. Virus-Vermehrung in
CV1-Zellen (Nierenzellen der afrikanischen grünen Meerkatze)
-
Ein modifiziertes komplexes Kohlenhydratprotein
kann durch Infektion von CV1-Zellen (ATCC CCL 70) mit SV40-Virus-Stammlösungen,
die wie von Gething und Sambrook (Nature 293 (1981), 620–625) beschrieben
vermehrt wurden, produziert werden. Dies wurde zunächst durch
vollständige
Spaltung des modifizierten pSVPA4 mit der Restriktionsendonuclease
BamHI durchgeführt,
um den bakteriellen Shuttle-Vektor pXf3 von der viralen SV40-DNA
zu entfernen. Vor der Transfektion dieser DNA zusammen mit der Helfervirus-SV40-rINS-pBR322-DNA
(nachstehend beschrieben) in CVI-Zellen wird die mit BamHI linearisierte SV40/t-PA-DNA durch Ligierung
mit verdünnten
DNA-Konzentrationen (1 μg/ml)
zirkularisiert. Dieses Verfahren wurde mit dem Insulin enthaltenden
SV40-Vektor SV40-rINS-pBR322 (M. Horowitz et al., Eukaryotic Viral Vectors
(1982), 47–53,
Cold Spring Harbor Laboratory) wiederholt. Der bakterielle Shuttle-Vektor
pBR322 in SV40-rINS-pBR322 wurde durch vollständige Spaltung mit EcoRI entfernt.
Die linearisierte virale Insulin/SV40-DNA wurde anschließend durch
Ligierung bei einer DNAKonzentration von 1 μg/ml zirkularisiert. Die transfizierten
CV-1-Zellen müssen
mit zirkulären
ligierten pSVPA4- und SV40rINS-DNAs in äquimolaren Mengen ligiert werden,
um einen Virus-Vorrat zu erzeugen. SV40-rINS wird zur Produktion
der "späten" SV40-Proteine verwendet,
während
pSVPA4 die für
die Virus-DNA-Produktion erforderlichen, "frühen" SV40-Proteine bereitstellt
sowie die erfindungsgemäßen Proteine
codiert. Wenn demnach Zellen mit beiden DNAs transfiziert werden,
wie von Gething und Sambrook beschrieben, wird, das SV40-Virus produziert,
das DNA von jedem Virus-Vektor enthält. Durch eine anschließende Infektion
von CVI-Zellen mit dem vermehrten Virus wird ein Protein mit t-PA-artiger
Aktivität
erhalten, das 72 Stunden nach der Infektion, wie in Beispiel 3C
beschrieben, getestet werden kann.
-
Beispiel 4
-
Herstellung von weiteren
Proteinen
-
cDNAs, die verschiedene erfindungsgemäße Proteine
codieren, sind durch die Verfahren der Beispiele 1, 2 und 3 hergestellt
worden. Die BglII/XmaI-Restriktionsfragment-Kassette kann anschließend entweder aus
dem pIVPA- oder pSVPA4-Vektor ausgeschnitten werden, der die cDNA
enthält,
die das verkürzte
Protein mit oder ohne Modifikation an einer oder mehreren Glykosylierungsstellen
codiert. Das ausgeschnittene BglII/XmaI-Fragment kann anschließend zur
Einführung
in Säugerzellen
mit dem BglII/XmaI-gespaltenen pSVPA4 oder pWGSM ligiert werden.
Die Expression derartiger cDNAs in Säuger-Wirtszellen, z. B., durch
das Verfahren von Beispiel 3, durch das Verfahren von Kaufman et
al., a.a.O., (CHO-Wirtszellen) oder durch das Verfahren von Howley
et al., US-Patent Nr. 4,419,446 (1983) (BPV-Expressionssysteme),
liefert die entsprechenden, von Säugern stammenden, verkürzten Proteine.
So wurde cDNA, die die Verbindung 2-1/N-21/Arg (Δ FBR, G1n117)
codiert, wie vorstehend beschrieben, hergestellt und in pSVPA4 inseriert.
In ähnlicher
Weise wurden die cDNAs, die die Verbindungen D-1 (Δ FBR) und
D-3 (Δ EGF/FBR)
codieren, durch M13-Mutagenese, wie vorstehend beschrieben, hergestellt
und als SacI-Fragment in das SacI-gespaltene pSVPA4 inseriert. Die cDNA,
die die Verbindung D-6 (Δ EGF)
codiert, wurde durch das vorstehend beschriebene Heteroduplexverfahren
unter Verwendung von pSVPA4 als Matrize und des MutageneseOligonucleotids
#12 und durch Absuchen mit dem Oligonucleotid # 13 hergestellt.
Zur Vermehrung und Expression der die Proteine codierenden cDNAs
in Säugerzellen
wird die in pSVPA4 oder pIVPA enthaltene cDNA als ein BglII/XmaI-Fragment
ausgeschnitten und mit dem gereinigten, mit BglII/XmaI-gespaltenen
pWGSM ligiert. In jedem Fall wird der erhaltene pWGSM-Vektor in
CHO-Zellen eingeführt
und durch das Verfahren von Kaufman, a.a.O., vermehrt. Die transformierten
und vermehrten CHO-Zellen
produzieren die Verbindungen D-6, D-1, D-3 bzw. 2-1/N-21/Arg, die durch
menschliche, t-PA-spezifische Antikörper, im Kulturmedium nachgewiesen
wurden. Die Verbindungen können
anschließend
durch Immunaffinitätschromatographie
gewonnen und gereinigt werden.
-
Beispiel 5
-
Beispiel 4 kann unter Verwendung
der cDNA wiederholt werden, die die Proteine codiert, die innerhalb des
N-Terminus und/oder am R-275 mit oder ohne Modifikation am R1, R2 und/oder R3 verändert
sind, um das gewünschte
Protein in CHO-Zellen zu produzieren. Die mutagenisierten cDNAs
können,
wie vorstehend beschrieben, hergestellt werden. Somit werden die
cDNAs, die die Verbindung 2-7/N-21/Arg
codieren, wie in Beispiel 2 beschrieben, durch pIVPR hergestellt.
Die cDNAs können
anschließend
als BglII/XmaI-Fragment ausgeschnitten und mit gereinigtem, BglII/XmaI-gespaltenem
pWGSM ligiert und der erhaltene Vektor in CHO-Zellen, wie in Beispiel
4, transformiert und vermehrt werden, um die Verbindung 2-7/N-21/Arg
herzustellen.