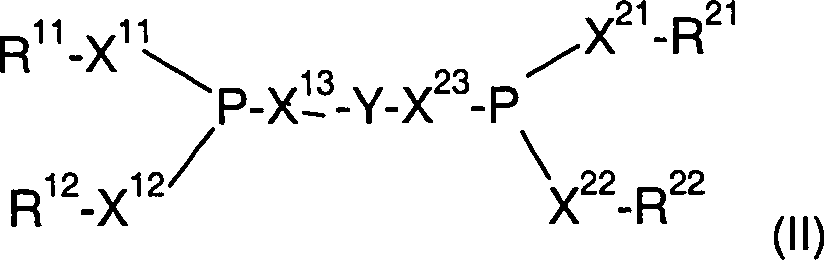-
Die
vorliegende Erfindung betrifft ein Verfahren zur Herstellung von
Nickel(0)-Phosphorligand-Komplexen. Weiterer Gegenstand der vorliegenden
Erfindung sind die durch dieses Verfahren erhältlichen Nickel(0)-Phosphorligand-Komplexe
enthaltenden Mischungen sowie deren Verwendung in der Hydrocyanierung
von Alkenen bzw. Isomerisierung von ungesättigten Nitrilen.
-
Für Hydrocyanierungen
von Alkenen sind Nickelkomplexe von Phosphorliganden geeignete Katalysatoren.
So sind beispielsweise Nickelkomplexe mit einzähnigen Phosphiten bekannt,
welche die Hydrocyanierung von Butadien zur Herstellung einer Mischung
aus isomeren Pentennitrilen katalysieren. Diese Katalysatoren eignen
sich auch in einer sich anschließenden Isomerisierung des verzweigten 2-Methyl-3-butennitrils
zu linearem 3-Pentennitril und der Hydrocyanierung des 3-Pentennitrils
zu Adiponitril, einem wichtigen Zwischenstoff in der Herstellung von
Nylon.
-
US 3,903,120 beschreibt
die Herstellung von nullwertigen Nickelkomplexen mit einzähnigen Phosphitliganden
ausgehend von Nickelpulver. Die phosphorhaltigen Liganden haben
dabei die allgemeine Formel PZ
3, worin Z
einer Alkyl-, Alkoxy- oder Aryloxygruppe entspricht. Bei diesem
Verfahren wird feinverteiltes elementares Nickel verwendet. Darüber hinaus
wird die Umsetzung bevorzugt in der Gegenwart eines nitrilhaltigen
Lösemittels
und in Gegenwart eines Überschusses
an Ligand durchgeführt.
-
US 3,846,461 beschreibt
ein Verfahren zur Herstellung von nullwertigen Nickelkomplexen mit Triorganophosphit-Liganden
durch Reaktion von Triorganophosphit-Verbindungen mit Nickelchlorid
in der Gegenwart eines feinverteilten Reduktionsmetalls, das elektropositiver
als Nickel ist. Die Umsetzung gemäß
US 3,846,461 findet in Gegenwart eines Promotors
statt, der ausgewählt
ist aus der Gruppe bestehend aus NH
3, NH
4X, Zn(NH
3)
2X
2 und Mischungen
von NH
4X und ZnX
2,
worin X einem Halogenid entspricht.
-
Neue
Entwicklungen haben gezeigt, dass es vorteilhaft ist, bei der Hydrocyanierung
von Alkenen Nickelkomplexe mit Chelatliganden (mehrzähnige Liganden)
einzusetzen, da mit diesen bei erhöhter Standzeit sowohl höhere Aktivitäten als
auch höhere Selektivitäten erzielt
werden können.
Die oben beschriebenen Verfahren des Standes der Technik eignen
sich nicht zur Herstellung von Nickelkomplexen mit Chelatliganden.
Aus dem Stand der Technik sind allerdings auch Verfahren bekannt,
welche die Herstellung von Nickelkomplexen mit Chelatliganden ermöglichen.
-
US 5,523,453 beschreibt
ein Verfahren zur Herstellung von nickelhaltigen Hydrocyanierungskatalysatoren,
die zweizähnige
Phosphor-Liganden enthalten. Die Herstellung dieser Komplexe erfolgt ausgehend
von löslichen
Nickel(0)-Komplexen durch Umkomplexierung mit Chelatliganden. Als
Ausgangsverbindungen werden Ni(COD)
2 oder (oTTP)
2Ni(C
2H
4)
verwendet (COD = 1,5-Cyclooctadien; oTTP = P(O-ortho-C
6H
4CH
3)
3).
Dieses Verfahren ist aufgrund der aufwändigen Herstellung der Nickel-Ausgangsverbindungen
kostenintensiv.
-
Alternativ
besteht die Möglichkeit,
Nickel(0)-Komplexe ausgehend von zweiwertigen Nickelverbindungen
und Chelatliganden durch Reduktion herzustellen. Bei dieser Methode
muss im Allgemeinen bei hohen Temperaturen gearbeitet werden, so
dass sich thermisch labile Liganden im Komplex gegebenenfalls zersetzen.
-
US
2003/0100442 A1 beschreibt ein Verfahren zur Herstellung eines Nickel(0)-Chelatkomplexes,
bei dem in Gegenwart eines Chelatliganden und eines nitrilhaltigen
Lösemittels
Nickelchlorid mit einem elektropositiveren Metall als Nickel, insbesondere
Zink oder Eisen, reduziert wird. Um eine hohe Raum-Zeit-Ausbeute
zu erreichen, wird ein Überschuss
an Nickelsalz verwendet, der im Anschluss an die Komplexierung wieder
abgetrennt werden muss. Das Verfahren wird in der Regel mit wasserhaltigem Nickelchlorid
durchgeführt,
was insbesondere bei der Verwendung von hydrolyselabilen Liganden
zu deren Zersetzung führen
kann. Wenn man, insbesondere bei der Verwendung von hydrolyselabilen
Liganden, mit wasserfreiem Nickelchlorid arbeitet, ist es gemäß US 2003/0100442
A1 wesentlich, dass das Nickelchlorid zunächst nach einem speziellen
Verfahren getrocknet wird, bei dem sehr kleine Teilchen mit großer Oberfläche und
damit hoher Reaktivität
erhalten werden. Ein Nachteil des Verfahrens liegt insbesondere
darin, dass dieser durch Sprühtrocknung
hergestellte Feinstaub von Nickelchlorid krebserregend ist. Ein
weiterer Nachteil dieses Verfahrens ist, dass im Allgemeinen bei
erhöhten
Reaktionstemperaturen gearbeitet wird, was insbesondere bei temperaturlabilen
Liganden zur Zersetzung des Liganden oder des Komplexes führen kann.
Weiterhin ist nachteilig, dass mit einem Überschuss an Reagenzien gearbeitet
werden muss, um wirtschaftliche Umsätze zu erzielen. Diese Überschüsse müssen nach
Beendigung der Reaktion aufwändig
entfernt und gegebenenfalls rückgeführt werden.
-
GB 1 000 477 und
BE 621 207 betreffen Verfahren
zur Herstellung von Nickel(0)-Komplexen durch
Reduktion von Nickel(II)-Verbindungen unter Verwendung von phosphorhaltigen
Liganden.
-
Aufgabe
der vorliegenden Erfindung war es somit, ein Verfahren zur Herstellung
von Nickel-Phosphorligand-Komplexen bereitzustellen, das die zuvor
beschriebenen Nachteile des Standes der Technik im Wesentlichen
vermeidet. Dabei soll insbesondere eine wasserfreie Nickelquelle
verwendet werden, damit hydrolyselabile Liganden während der Komplexierung
nicht zersetzt werden. Die Reaktionsbedingungen sollen darüber hinaus
vorzugsweise schonend sein, damit sich temperaturlabile Liganden und
die entstehenden Komplexe nicht zersetzen. Darüber hinaus sollte das erfindungsgemäße Verfahren
vorzugsweise ermöglichen,
dass kein oder nur ein geringer Überschuss
der Reagenzien eingesetzt wird, damit eine Abtrennung dieser Stoffe – nach der Herstellung
des Komplexes – möglichst
nicht nötig ist.
Auch soll sich das Verfahren zur Herstellung von Nickel(0)-Komplexen
mit Chelatliganden eignen.
-
Die
Aufgabe wird erfindungsgemäß gelöst durch
ein Verfahren zur Herstellung von Nickel(0)-Phosphorligand-Komplexen,
enthaltend mindestens ein Nickel(0)-Zentralatom und mindestens einen
phosphorhaltigen Liganden.
-
Das
erfindungsgemäße Verfahren
ist dadurch gekennzeichnet, dass ein durch Azeotropdestillation
getrocknetes wasserhaltiges Nickel(II)-halogenid in Gegenwart mindestens
eines phosphorhaltigen Liganden reduziert wird.
-
Azeotropdestillation
-
In
der Azeotropdestillation wird ein wasserhaltiges Nickel(II)-halogenid
verwendet. Wasserhaltiges Nickel(II)-halogenid ist ein Nickelhalogenid,
welches ausgewählt
ist aus der Gruppe von Nickelchlorid, Nickelbromid und Nickeliodid,
das mindestens 2 Gew.-% Wasser enthält. Beispiele hierfür sind Nickelchlorid-Dihydrat,
Nickelchlorid-Hexahydrat, eine wässrige
Lösung
von Nickelchlorid, Nickelbromid-Trihydrat, eine wässrige Lösung von
Nickelbromid, Nickeliodid-Hydrate oder eine wässrige Lösung von Nickeliodid. Im Fall
von Nickelchlorid werden bevorzugt Nickelchlorid-Hexahydrat oder
eine wässrige
Lösung von
Nickelchlorid eingesetzt. Im Fall von Nickelbromid und Nickeliodid
werden bevorzugt die wässrigen Lösungen eingesetzt.
Besonders bevorzugt ist eine wässrige
Lösung
von Nickelchlorid.
-
Im
Falle einer wässrigen
Lösung
ist die Konzentration des Nickel(II)-halogenids in Wasser an sich
nicht kritisch. Als vorteilhaft hat sich ein Anteil des Nickel(II)-halogenids
an der Gewichtssumme aus Nickel(II)-halogenid und Wasser von mindestens 0,01
Gew.-%, vorzugsweise mindestens 0,1 Gew.-%, besonders bevorzugt
mindestens 0,25 Gew.%, insbesondere bevorzugt mindestens 0,5 Gew.-%
erwiesen. Als vorteilhaft hat sich ein Anteil des Nickel(II)-halogenids
an der Gewichtssumme aus Nickel(II)-halogenid und Wasser im Bereich von
höchstens
80 Gew.-%, vorzugsweise höchstens
60 Gew.-%, besonders bevorzugt höchstens
40 Gew.-% erwiesen. Aus praktischen Gründen ist es von Vorteil, einen
Anteil von Nickelhalogenid in der Mischung aus Nickelhalogenid und
Wasser nicht zu überschreiten, der
unter den gegebenen Temperatur- und Druckbedingungen eine Lösung ergibt.
Im Falle einer wässrigen
Lösung
von Nickelchlorid ist es daher aus praktischen Gründen von
Vorteil, bei Raumtemperatur einen Anteil von Nickelhalogenid an
der Gewichtssumme aus Nickelchlorid und Was sers von höchstens
31 % Gew.-% zu wählen.
Bei höheren
Temperaturen können
entsprechend höhere
Konzentrationen gewählt
werden, die sich aus der Löslichkeit
von Nickelchlorid in Wasser ergeben.
-
Das
wasserhaltige Nickel(II)-halogenid wird vor der Reduktion durch
eine Azeotropdestillation getrocknet. In einer bevorzugten Ausführungsform
der vorliegenden Erfindung ist die Azeotropdestillation ein Verfahren
zur Entfernung von Wasser aus dem entsprechenden wasserhaltigen
Nickel(II)-halogenid, wobei dieses mit einem Verdünnungsmittel
versetzt wird, dessen Siedepunkt im Falle der Nichtazeotrop-Bildung
des Verdünnungsmittels
mit Wasser unter den Druckbedingungen der nachfolgend genannten
Destillation höher
ist als der Siedepunkt von Wasser und das an diesem Siedepunkt des
Wassers flüssig
vorliegt oder das ein Azeotrop oder Heteroazeotrop mit Wasser unter
den Druck- und Temperaturbedingungen der nachfolgend genannten Destillation bildet,
und die Mischung, enthaltend das wasserhaltige Nickel(II)-halogenid
und das Verdünnungsmittel, unter
Abtrennung von Wasser oder des genannten Azeotrops oder des genannten
Heteroazeotrops von dieser Mischung und unter Erhalt einer wasserfreien Mischung,
enthaltend Nickel(II)-halogenid und das besagte Verdünnungsmittel,
destilliert wird.
-
Die
Ausgangsmischung kann neben dem wasserhaltigen Nickel(II)-halogenid
weitere Bestandteile enthalten, wie ionische oder nichtionische,
organische oder anorganische Verbindungen, insbesondere solche,
die mit der Ausgangsmischung homogen einphasig mischbar oder in
der Ausgangsmischung löslich
sind.
-
Erfindungsgemäß versetzt
man das wasserhaltige Nickel(II)-halogenid mit einem Verdünnungsmittel,
dessen Siedepunkt unter den Druckbedingungen der Destillation höher ist
als der Siedepunkt von Wasser und das an diesem Siedepunkt des Wassers flüssig vorliegt.
-
Die
Druckbedingungen für
die nachfolgende Destillation sind an sich nicht kritisch. Als vorteilhaft haben
sich Drücke
von mindestens 10–4 MPa, vorzugsweise
mindestens 10–3 MPa,
insbesondere mindestens 5·10–3 MPa
erwiesen. Als vorteilhaft haben sich Drücke von höchstens 1 MPa, vorzugsweise höchstens
5·10–1 MPa,
insbesondere höchstens 1,5·10–1 MPa
erwiesen.
-
In
Abhängigkeit
von den Druckbedingungen und der Zusammensetzung des zu destillierenden Gemischs
stellt sich dann die Destillationstemperatur ein. Bei dieser Temperatur
liegt das Verdünnungsmittel
vorzugsweise flüssig
vor. Im Sinne der vorliegenden Erfindung wird unter dem Begriff
Verdünnungsmittel
sowohl ein einzelnes Verdünnungsmittel
wie auch ein Gemisch solcher Verdünnungsmittel verstanden, wobei
sich im Falle eines solchen Gemischs die in der vorliegenden Erfindung
genannten physikalischen Eigenschaften auf dieses Gemisch beziehen.
-
Weiterhin
weist das Verdünnungsmittel
unter diesen Druck- und Temperaturbedingungen vorzugsweise einen
Siedepunkt auf, der im Falle der Nichtazeotrop-Bildung des Verdünnungsmittels
mit Wasser höher
als der von Wasser liegt, vorzugsweise um mindestens 5 °C, insbesondere
mindestens 20 °C, und
vorzugsweise höchstens
200 °C,
insbesondere höchstens
100 °C.
-
In
einer bevorzugten Ausführungsform
kann man Verdünnungsmittel
einsetzen, die mit Wasser ein Azeotrop oder Heteroazeotrop bilden.
Die Menge an Verdünnungsmittel
gegenüber
der Menge an Wasser in dem Gemisch ist an sich nicht kritisch. Vorteilhaft
sollte man mehr flüssiges
Verdünnungsmittel einsetzen
als den durch die Azeotrope abzudestillierenden Mengen entspricht,
so dass überschüssiges Verdünnungsmittel
als Sumpfprodukt verbleibt.
-
Setzt
man ein Verdünnungsmittel
ein, das mit Wasser kein Azeotrop bildet, so ist die Menge an Verdünnungsmittel
gegenüber
der Menge an Wasser in dem Gemisch an sich nicht kritisch.
-
Das
eingesetzte Verdünnungsmittel
ist dabei insbesondere ausgewählt
aus der Gruppe bestehend aus organischen Nitrilen, aromatischen
Kohlenwasserstoffen, aliphatischen Kohlenwasserstoffen und Mischungen
der zuvor genannten Lösemittel.
Bezüglich
der organischen Nitrile werden vorzugsweise Acetonitril, Propionitril,
n-Butyronitril, n-Valeronitril, Cyanocyclopropan,
Acrylnitril, Crotonitril, Allylcyanid, cis-2-Pentennitril, trans-2-Pentennitril,
cis-3-Pentennitril, trans-3-Pentennitril, 4-Pentennitril, 2-Methyl-3-butennitril, Z-2-Methyl-2-butennitril,
E-2-Methyl-2-butennitril, Ethylsuccinnitril, Adipodinitril, Methylglutarnitril
oder Mischungen davon verwendet. Bezüglich der aromatischen Kohlenwasserstoffe können vorzugsweise
Benzol, Toluol, o-Xylol, m-Xylol, p-Xylol oder Mischungen davon
verwendet werden. Aliphatische Kohlenwasserstoffe können vorzugsweise
aus der Gruppe der linearen oder verzweigten aliphatischen Kohlenwasserstoffe,
besonders bevorzugt aus der Gruppe der Cycloaliphaten, wie Cyclohexan
oder Methylcyclohexan, oder Mischungen davon gewählt werden. Besonders bevorzugt
werden cis-3-Pentennitril, trans-3-Pentennitril, Adipodinitril,
Methylglutarnitril oder Mischungen daraus als Lösemittel verwendet.
-
Setzt
man als Verdünnungsmittel
ein organisches Nitril bzw. Mischungen, enthaltend mindestens ein
organisches Nitril ein, so hat es sich als vorteilhaft erwiesen,
die Menge an Verdünnungsmittel
so zu wählen,
dass in der fertigen Mischung der Anteil des Nickel(II)-halogenids
an der Gewichtssumme aus Nickel(II)-halogenid und Verdünnungsmittel
mindestes 0,05 Gew.-%, vorzugsweise mindestens 0,5 Gew.-%, besonders
bevorzugt mindestens 1 Gew.-% beträgt.
-
Setzt
man als Verdünnungsmittel
ein organisches Nitril bzw. Mischungen, enthaltend mindestens ein
organisches Nitril ein, so hat es sich als vorteilhaft erwiesen,
die Menge an Verdünnungsmittel
so zu wählen,
dass in der fertigen Mischung der Anteil des Nickel(II)-halogenids
an der Gewichtssumme aus Nickel(II)-halogenid und Verdünnungsmittel
höchstens 50
Gew.-%, vorzugsweise höchstens
30 Gew.-%, besonders bevorzugt höchstens
20 Gew.-% beträgt.
-
Erfindungsgemäß destilliert
man die Mischung, enthaltend das wasserhaltige Nickel(II)-halogenid und das
Verdünnungsmittel,
unter Abtrennung von Wasser von dieser Mischung und unter Erhalt
einer wasserfreien Mischung, enthaltend Nickel(II)-halogenid und
das besagte Verdünnungsmittel.
In einer bevorzugten Ausführungsform
wird zunächst
die Mischung hergestellt und anschließend destilliert. In einer
anderen bevorzugten Ausführungsform
wird das wasserhaltige Nickelhalogenid, besonders bevorzugt die
wässrige
Lösung
des Nickelahlogenids, während der
Destillation nach und nach zu dem siedenden Verdünnungsmittel zugegeben. Dadurch
kann die Bildung eines verfahrenstechnisch schwer zu handhabenden,
schmierigen Feststoffs im wesentlichen vermieden werden.
-
Im
Falle von Pentennitril als Verdünnungsmittel
kann man die Destillation vorteilhaft bei einem Druck von höchstens
200 kPa, vorzugsweise höchstens
100 kPa, insbesondere höchstens
50 kPa, besonders bevorzugt höchstens
20 kPa, durchführen.
-
Im
Falle von Pentennitril als Verdünnungsmittel
kann man die Destillation vorzugsweise bei einem Druck von mindestens
1 kPa, vorzugsweise mindestens 5 kPa, besonders bevorzugt 10 kPa, durchführen.
-
Die
Destillation kann vorteilhaft durch einstufige Verdampfung, bevorzugt
durch fraktionierende Destillation in einer oder mehreren, wie 2
oder 3 Destillationsapparaturen erfolgen. Dabei kommen für die Destillation
hierfür übliche Apparaturen
in Betracht, wie sie beispielsweise in: Kirk-Othmer, Encyclopedia
of Chemical Technology, 3. Ed., Vol. 7, John Wiley & Sons, New York,
1979, Seite 870–881
beschrieben sind, wie Siebbodenkolonnen, Glockenbodenkolonnen, Packungskolonnen,
Füllkörperkolonnen,
Kolonnen mit Seitenabzug oder Trennwandkolonnen.
-
Die
Destillation kann diskontinuierlich erfolgen.
-
Die
Destillation kann kontinuierlich erfolgen.
-
Ligand
-
Das
erfindungsgemäße Verfahren
zur Herstellung von Nickel(0)-Phosphorligand-Komplexen, enthaltend
mindestens ein Nickel(0)-Zentralatom und mindestens einen phosphorhaltigen
Liganden, ist dadurch gekennzeichnet, dass das durch Azeotropdestil lation
getrocknetes Nickel(II)-halogenid in Gegenwart mindestens eines
phosphorhaltigen Liganden reduziert wird.
-
In
dem erfindungsgemäßen Verfahren
sind die phosphorhaltigen Liganden vorzugsweise ausgewählt aus
der Gruppe bestehend aus Phosphinen, Phosphiten, Phosphiniten und
Phosphoniten.
-
Diese
phosphorhaltigen Liganden weisen vorzugsweise die Formel (I) P(X1R1)(X2R2)(X3R3) (I)auf.
-
Unter
Verbindung (I) wird im Sinne der vorliegenden Erfindung eine einzelne
Verbindung oder ein Gemisch verschiedener Verbindungen der vorgenannten
Formel verstanden.
-
Erfindungsgemäß sind X1, X2, X3 unabhängige voneinander
Sauerstoff oder Einzelbindung.
-
Falls
alle der Gruppen X1, X2 und
X3 für
Einzelbindungen stehen, so stellt Verbindung (I) ein Phosphin der
Formel P(R1R2R3) mit den für R1,
R2 und R3 in dieser
Beschreibung genannten Bedeutungen dar.
-
Falls
zwei der Gruppen X1, X2 und
X3 für
Einzelbindungen stehen und eine für Sauerstoff, so stellt Verbindung
(I) ein Phosphinit der Formel P(OR1)(R2)(R3) oder P(R1)(OR2)(R3) oder P(R1)(R2)(OR3) mit den für R1, R2 und R3 in dieser Beschreibung genannten Bedeutungen
dar.
-
Falls
eine der Gruppen X1, X2 und
X3 für
eine Einzelbindung steht und zwei für Sauerstoff, so stellt Verbindung
(I) ein Phosphonit der Formel P(OR1)(OR2)(R3) oder P(R1)(OR2)(OR3) oder P(OR1)(R2)(OR3) mit den für R1, R2 und R3 in dieser Beschreibung genannten Bedeutungen
dar.
-
In
einer bevorzugten Ausführungsform
sollten alle der Gruppen X1, X2 und
X3 für
Sauerstoff stehen, so dass Verbindung (I) vorteilhaft ein Phosphit der
Formel P(OR1)(OR2)(OR3) mit den für R1,
R2 und R3 in dieser
Beschreibung genannten Bedeutungen darstellt.
-
Erfindungsgemäß stehen
R1, R2, R3 unabhängig
voneinander für
gleiche oder unterschiedliche organische Reste.
-
Als
R1, R2 und R3 kommen unabhängig voneinander Alkylreste,
vorzugsweise mit 1 bis 10 Kohlenstoffatomen, wie Methyl, Ethyl,
n-Propyl, i-Propyl, n-Butyl, i-Butyl, s-Butyl, t-Butyl, Aryl-Gruppen, wie Phenyl, o-Tolyl,
m-Tolyl, p-Tolyl, 1-Naphthyl, 2-Naphthyl, oder Hydrocarbyl, vorzugsweise
mit 1 bis 20 Kohlenstoffatomen, wie 1,1'-Biphenol, 1,1'-Binaphthol
in Betracht.
-
Die
Gruppen R1, R2 und
R3 können
miteinander direkt, also nicht allein über das zentrale Phosphor-Atom,
verbunden sein. Vorzugsweise sind die Gruppen R1,
R2 und R3 nicht
miteinander direkt verbunden.
-
In
einer bevorzugten Ausführungsform
kommen als Gruppen R1, R2 und
R3 Reste ausgewählt aus der Gruppe bestehend
aus Phenyl, o-Tolyl, m-Tolyl und p-Tolyl in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
sollten dabei maximal zwei der Gruppen R1,
R2 und R3 Phenyl-Gruppen
sein.
-
In
einer anderen bevorzugten Ausführungsform
sollten dabei maximal zwei der Gruppen R1,
R2 und R3 o-Tolyl-Gruppen
sein.
-
Als
besonders bevorzugte Verbindungen (I) können solche der Formel (o-Tolyl-O-)w(m-Tolyl-O-)x(p-Tolyl-O-)y(Phenyl-O-)zP mit
w, x, y, z eine natürliche
Zahl
mit w + x + y + z = 3 und
w, z kleiner gleich 2
eingesetzt
werden, wie (p-Tolyl-O-)(Phenyl-O-)2P, (m-Tolyl-O-)(Phenyl-O-)2P, (o-Tolyl-O-)(Phenyl-O-)2P,
(p-Tolyl-O-)2(Phenyl-O-)P, (m-Tolyl-O-)2(Phenyl-O-)P, (o-Tolyl-O-)2(Phenyl-O-)P, (m-Tolyl-O-)(p-Tolyl-O)(Phenyl-O-)P,
(o-Tolyl-O-)(p-Tolyl-O-)(Phenyl-O-)P,
(o-Tolyl-O-)(m-Tolyl-O-)(Phenyl-O-)P, (p-Tolyl-O-)3P,
(m-Tolyl-O-)(p-Tolyl-O-)2P, (o-Tolyl-O-)(p-Tolyl-O-)2P, (m-Tolyl-O-)2(p-Toluyl-O-)P,
(o-Tolyl-O-)2(p-Tolyl-O-)P, (o-Tolyl-O-)(m-Tolyl-O-)(p-Tolyl-O)P,
(m-Tolyl-O-)3P, (o-Tolyl-O-)(m-Tolyl-O-)2P (o-Tolyl-O-)2(m-Tolyl-O-)P, oder
Gemische solcher Verbindungen eingesetzt werden.
-
So
können
beispielsweise Gemische enthaltend (m-Tolyl-O-)3P,
(m-Tolyl-O-)2(p-Tolyl-O-)P, (m-Tolyl-O-)(p-Tolyl-O-)2P und (p-Tolyl-O-)3P
durch Umsetzung eines Gemisches enthaltend m-Kresol und p-Kresol,
insbesondere im Molverhältnis
2 : 1, wie es bei der destillativen Aufarbeitung von Erdöl anfällt, mit
einem Phosphortrihalogenid, wie Phosphortrichlorid, erhalten werden.
-
Solche
Verbindungen (I) und deren Herstellung sind an sich bekannt.
-
In
dem erfindungsgemäßen Verfahren
ist es allerdings bevorzugt, dass der phosphorhaltige Ligand zweizähnig ist.
Daher weist der in dem erfindungsgemäßen Verfahren verwendete Ligand
vorzugsweise die Formel (II)
mit
X
11,
X
12, X
13, X
21, X
22, X
23 unabhängig
voneinander Sauerstoff oder eine Einzelbindung
R
11,
R
12 unabhängig voneinander gleiche oder
unterschiedliche, einzelne oder verbrückte organische Reste
R
21, R
22 unabhängig voneinander
gleiche oder unterschiedliche, einzelne oder verbrückte organische Reste,
Y
Brückengruppe
auf.
-
Unter
Verbindung (II) wird im Sinne der vorliegenden Erfindung eine einzelne
Verbindung oder ein Gemisch verschiedener Verbindungen der vorgenannten
Formel verstanden.
-
In
einer bevorzugten Ausführungsform
können
X11, X12, X13, X21, X22, X23 Sauerstoff
darstellen. In einem solchen Fall ist die Brückengruppe Y mit Phosphit-Gruppen
verknüpft.
-
In
einer anderen bevorzugten Ausführungsform
können
X11 und X12 Sauerstoff
und X13 eine Einzelbindung oder X11 und X12 Sauerstoff
und X12 eine Einzelbindung darstellen, so
dass das mit X11, X12 und X13 umgebene Phosphoratom Zentralatom eines Phosphonits
ist. In einem solchen Fall können
X21, X22 und X23 Sauerstoff oder X21 und
X22 Sauerstoff und X23 eine
Einzelbindung oder X21 und X23 Sauerstoff
und X22 eine Einzelbindung oder X23 Sauerstoff und X21 und
X22 eine Einzelbindung oder X21 Sauerstoff
und X22 und X23 eine
Einzelbindung oder X21, X22 und
X23 eine Einzelbindung darstellen, so dass
das mit X21, X22 und
X23 umgebene Phosphoratom Zentralatom eines Phosphits,
Phosphonits, Phosphinits oder Phosphins, vorzugsweise eines Phosphonits, sein
kann.
-
In
einer anderen anderen bevorzugten Ausführungsform können X13 Sauerstoff und X11 und
X12 eine Einzelbindung oder X11 Sauerstoff
und X12 und X13 eine
Einzelbindung darstellen, so dass das mit X11,
X12 und X13 umgebene
Phosphoratom Zentralatom eines Phosphonits ist. In einem solchen
Fall können
X21, X22 und X23 Sauerstoff oder X23 Sauerstoff und
X21 und X22 eine
Einzelbindung oder X21 Sauerstoff und X22 und X23 eine Einzelbindung
oder X21, X22 und
X23 eine Einzelbindung darstellen, so dass
das mit X21, X22 und
X23 umgebene Phosphoratom Zentralatom eines
Phosphits, Phosphinits oder Phosphins, vorzugsweise eines Phosphinits,
sein kann.
-
In
einer anderen bevorzugten Ausführungsform
können
X11, X12 und X13 eine Einzelbindung darstellen, so dass
das mit X11, X12 und
X13 umgebene Phosphoratom Zentralatom eines
Phosphins ist. In einem solchen Fall können X21,
X22 und X23 Sauerstoff oder
X21, X22 und X23 eine Einzelbindung darstellen, so dass
das mit X21, X22 und
X23 umgebene Phosphoratom Zentralatom eines
Phosphits oder Phosphins, vorzugsweise eines Phosphins, sein kann.
-
Als
Brückengruppe
Y kommen vorzugsweise substituierte, beispielsweise mit C1-C4-Alkyl, Halogen, wie
Fluor, Chlor, Brom, halogeniertem Alkyl, wie Trifluormethyl, Aryl,
wie Phenyl, oder unsubstituerte Arylgruppen in Betracht, vorzugsweise
solche mit 6 bis 20 Kohlenstoffatomen im aromatischen System, insbesondere
Pyrocatechol, Bis(phenol) oder Bis(naphthol).
-
Die
Reste R11 und R12 können unabhängig voneinander
gleiche oder unterschiedliche organische Reste darstellen. Vorzugsweise
kommen als Reste R11 und R12 Arylreste,
insbesondere solche mit 6 bis 10 Kohlenstoffatomen, in Betracht,
die unsubstituiert oder einfach oder mehrfach substituiert sein können, insbesondere
durch C1-C4-Alkyl,
Halogen, wie Fluor, Chlor, Brom, halogeniertem Alkyl, wie Trifluormethyl,
Aryl, wie Phenyl, oder unsubstituierte Arylgruppen.
-
Die
Reste R21 und R22 können unabhängig voneinander
gleiche oder unterschiedliche organische Reste darstellen. Vorzugsweise
kommen als Reste R21 und R22 Arylreste,
vorzugsweise solche mit 6 bis 10 Kohlenstoffatomen, in Betracht,
die unsubstituiert oder einfach oder mehrfach substituiert sein können, insbesondere
durch C1-C4-Alkyl,
Halogen, wie Fluor, Chlor, Brom, halogeniertem Alkyl, wie Trifluormethyl,
Aryl, wie Phenyl, oder unsubstituierte Arylgruppen.
-
Die
Reste R11 und R12 können einzeln
oder verbrückt
sein.
-
Die
Reste R21 und R22 können einzeln
oder verbrückt
sein.
-
Die
Reste R11, R12,
R21 und R22 können alle einzeln,
zwei verbrückt
und zwei einzeln oder alle vier verbrückt sein in der beschriebenen
Art.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in
US 5,723,641 genannten Verbindungen
der Formel I, II, III, IV und V in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in
US 5,512,696 genannten Verbindungen
der Formel I, II, III IV, V, VI und VII, insbesondere die dort in
den Beispielen 1 bis 31 eingesetzten Verbindungen, in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in
US 5,821,378 genannten Verbindungen
der Formel I, II, III, IV, V, VI, VII, VIII, IX, X, XI, XII, XIII,
XIV und XV, insbesondere die dort in den Beispielen 1 bis 73 eingesetzten
Verbindungen, in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in
US 5,512,695 genannten Verbindungen
der Formel I, II, III, IV, V und VI, insbesondere die dort in den
Beispielen 1 bis 6 eingesetzten Verbindungen, in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in
US 5,981,772 genannten Verbindungen
der Formel I, II, III, IV, V, VI, VII, VIII, IX, X, XI, XII, XIII
und XIV, insbesondere die dort in den Beispielen 1 bis 66 eingesetzten
Verbindungen, in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in
US 6,127,567 genannten Verbindungen
und dort in den Beispielen 1 bis 29 eingesetzten Verbindungen in
Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in
US 6,020,516 genannten Verbindungen
der Formel I, II, III, IV, V, VI, VII, VIII, IX und X, insbesondere
die dort in den Beispielen 1 bis 33 eingesetzten Verbindungen, in
Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in
US 5,959,135 genannten Verbindungen
und dort in den Beispielen 1 bis 13 eingesetzten Verbindungen in
Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in
US 5,847,191 genannten Verbindungen
der Formel I, II und III in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in
US 5,523,453 genannten Verbindungen,
insbesondere die dort in Formel 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,
12, 13, 14, 15, 16, 17, 18, 19, 20 und 21 dargestellten Verbindungen,
in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in WO 01/14392 genannten Verbindungen, vorzugsweise die
dort in Formel V, VI, VII, VIII, IX, X, XI, XII, XIII, XIV, XV,
XVI, XVII, XXI, XXII, XXIII dargestellten Verbindungen, in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in WO 98/27054 genannten Verbindungen in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in WO 99/13983 genannten Verbindungen in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in WO 99/64155 genannten Verbindungen in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in der deutschen Patentanmeldung
DE 100 380 37 genannten Verbindungen
in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in der deutschen Patentanmeldung
DE 100 460 25 genannten Verbindungen
in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in der deutschen Patentanmeldung
DE 101 502 85 genannten Verbindungen
in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in der deutschen Patentanmeldung
DE 101 502 86 genannten Verbindungen
in Betracht.
-
In
einer besonders bevorzugten Ausführungsform
kommen die in der deutschen Patentanmeldung
DE 102 071 65 genannten Verbindungen
in Betracht.
-
In
einer weiteren besonders bevorzugten Ausführungsform der vorliegenden
Erfindung kommen die in der US 2003/0100442 A1 genannten phosphorhaltigen
Chelatliganden in Betracht.
-
In
einer weiteren besonders bevorzugten Ausführungsform der vorliegenden
Erfindung kommen die in der prioritätsgleichen deutschen Patentanmeldung
mit dem Titel „Phosphinitphosphite" der BASF AG genannten
phosphorhaltigen Chelatliganden in Betracht.
-
Solche
Verbindungen (I) und (II) und deren Herstellung sind an sich bekannt.
-
Als
phosphorhaltiger Ligand können
auch Mischungen, enthaltend die Verbindungen I und II, eingesetzt
werden.
-
Reduktion
-
Das
erfindungsgemäße Verfahren
zur Herstellung von Nickel(0)-Phosphorligand-Komplexen, enthaltend mindestens ein
Nickel(0)-Zentralatom und mindestens einen phosphorhaltigen Liganden,
durch Reduktion wird vorzugsweise in Gegenwart eines Lösemittels
durchgeführt.
Das Lösemittel
ist dabei insbesondere ausgewählt
aus der Gruppe bestehend aus organischen Nitrilen, aromatischen
Kohlenwasserstoffen, aliphatischen Kohlenwasserstoffen und Mischungen
der zuvor genannten Lösemittel.
Bezüglich
der organischen Nitrile werden vorzugsweise Acetonitril, Propionitril,
n-Butyronitril,
n-Valeronitril, Cyanocyclopropan, Acrylnitril, Crotonitril, Allylcyanid, cis-2-Pentennitril, trans-2-Pentennitril,
cis-3-Pentennitril, trans-3-Pentennitril, 4-Pentennitril, 2-Methyl-3-butennitril,
Z-2-Methyl-2-butennitril, E-2-Methyl-2-butennitril, Ethylsuccinnitril,
Adipodinitril, Methylglutarnitril oder Mischungen davon verwendet. Bezüglich der
aromatischen Kohlenwasserstoffe können vorzugsweise Benzol, Toluol,
o-Xylol, m-Xylol,
p-Xylol oder Mischungen davon verwendet werden. Aliphatische Kohlenwasserstoffe
können
vorzugsweise aus der Gruppe der linearen oder verzweigten aliphatischen
Kohlenwasserstoffe, besonders bevorzugt aus der Gruppe der Cycloaliphaten, wie
Cyclohexan oder Methylcyclohexan, oder Mischungen davon gewählt werden.
Besonders bevorzugt werden cis-3-Pentennitril, trans-3-Pentennitril, Adipodinitril,
Methylglutarnitril oder Mischungen daraus als Lösemittel verwendet.
-
Vorzugsweise
wird ein inertes Lösemittel verwendet.
-
Die
Konzentration des Lösemittels
beträgt vorzugsweise
10 bis 90 Massen-%, besonders bevorzugt 20 bis 70 Massen-%, insbesondere
30 bis 60 Massen-%, jeweils bezogen auf die fertige Reaktionsmischung.
-
In
einer besonderen Ausführungsform
der vorliegenden Erfindung ist das Lösemittel identisch zu dem Verdünnungsmittel,
das in dem oben beschriebenen erfindungsgemäßen Verfahren zur Herstellung
der wasserfreien Mischung, enthaltend das Nickel(II)-Halogenid und
das Verdünnungsmittel,
verwendet wird.
-
In
dem erfindungsgemäßen Verfahren
beträgt
die Konzentration des Liganden in dem Lösemittel vorzugsweise 1 bis
90 Gew.-%, besonders bevorzugt 5 bis 80 Gew.-%, insbesondere 50
bis 80 Gew.-%.
-
Das
in dem erfindungsgemäßen Verfahren verwendete
Reduktionsmittel ist vorzugsweise ausgewählt aus der Gruppe bestehend
aus Metallen, die elektropositiver als Nickel sind, Metallalkylen,
elektrischem Strom, komplexen Hydriden und Wasserstoff.
-
Wenn
in dem erfindungsgemäßen Verfahren als
Reduktionsmittel ein Metall, das elektropositiver als Nickel ist,
verwendet wird, so ist dieses Metall vorzugsweise ausgewählt aus
der Gruppe bestehend aus Natrium, Lithium, Kalium, Magnesium, Calcium, Barium,
Strontium, Titan, Vanadium, Eisen, Kobalt, Kupfer, Zink, Cadmium,
Aluminium, Gallium, Indium, Zinn, Blei und Thorium. Besonders bevorzugt
sind hierbei Eisen und Zink. Wird Aluminium als Reduktionsmittel
verwendet, so ist es von Vorteil, wenn dieses durch Reaktion mit
einer katalytischen Menge Quecksilber(II)-Salz oder Metallalkyl
voraktiviert wird. Bevorzugt wird für die Voraktivierung Triethylaluminium
in einer Menge von vorzugsweise 0,05 bis 50 Mol.-%, besonders bevorzugt
0,5 bis 10 Mol-%, verwendet. Das Reduktionsmetall ist vorzugsweise
fein verteilt, wobei der Ausdruck „fein verteilt" bedeutet, dass das
Metall in einer Partikelgröße von weniger als
10 mesh, besonders bevorzugt weniger als 20 mesh, verwendet wird.
-
Wenn
in dem erfindungsgemäßen Verfahren als
Reduktionsmittel ein Metall verwendet wird, das elektropositiver
ist als Nickel, so beträgt
die Menge an Metall vorzugsweise 0,1 bis 50 Gew.-%, bezogen auf
die Reaktionsmasse.
-
Wenn
in dem erfindungsgemäßen Verfahren als
Reduktionsmittel Metallalkyle verwendet werden, so handelt es sich
bevorzugt um Lithiumalkyle, Natriumalkyle, Magnesiumalkyle, insbesondere
Grignard-Reagenzien, Zinkalkyle oder Aluminiumalkyle. Besonders
bevorzugt sind Aluminiumalkyle, wie Trimethylaluminium, Triethylaluminium,
Triisopropylaluminium oder Mischungen hiervon, insbesondere Triethylaluminium.
Die Metallalkyle können
in Substanz oder gelöst
in einem inerten organischen Lösesmittel,
wie Hexan, Heptan oder Toluol, eingesetzt werden.
-
Wenn
in dem erfindungsgemäßen Verfahren komplexe
Hydride als Reduktionsmittel verwendet werden, so werden bevorzugt
Metallaluminiumhydride, wie Lithiumaluminiumhydrid, oder Metallborhydride,
wie Natriumborhydrid, eingesetzt.
-
Das
molare Verhältnis
der Redoxäquivalente zwischen
der Nickel(II)-Quelle und dem Reduktionsmittel beträgt vorzugsweise
1 : 1 bis 1 : 100, besonders bevorzugt 1 : 1 bis 1 : 50, insbesondere
1 : 1 bis 1 : 5.
-
In
dem erfindungsgemäßen Verfahren
kann der zu verwendende Ligand auch in einer Ligandlösung vorliegen,
die bereits als Katalysatorlösung
in Hydrocyanierungsreaktionen eingesetzt wurde und an Nickel(0)
abgereichert ist. Diese „Rück-Katalysatorlösung" hat im Allgemeinen
die folgende Zusammensetzung:
- – 2 bis
60 Gew.-%, insbesondere 10 bis 40 Gew.-% Pentennitrile,
- – 0
bis 60 Gew.-%, insbesondere 0 bis 40 Gew.-% Adipodinitril,
- – 0
bis 10 Gew.-%, insbesondere 0 bis 5 Gew.-% andere Nitrile,
- – 10
bis 90 Gew.-%, insbesondere 50 bis 90 Gew.-% phosphorhaltiger Ligand
und
- – 0
bis 2 Gew.-%, insbesondere 0 bis 1 Gew.-% Nickel(0).
-
Der
in der Rück-Katalysatorlösung enthaltene
freie Ligand kann nach dem erfindungsgemäßen Verfahren somit wieder
zu einem Nickel(0)-Komplex umgesetzt werden.
-
In
einer besonderen Ausführungsform
der vorliegenden Erfindung ist das Verhältnis der Nickel(II)-Quelle
zu phosphorhaltigem Ligand 1 : 1 bis 1 : 100. Weitere bevorzugte
Verhältnisse
von Nickel(II)-Quelle zu phosphorhaltigem Ligand sind 1 : 1 bis
1 : 3, insbesondere 1 : 1 bis 1 : 2.
-
Das
erfindungsgemäße Verfahren
kann bei beliebigem Druck durchgeführt werden. Aus praktischen
Gründen
sind Drücke
zwischen 0.1 bara und 5 bara, vorzugsweise 0.5 bara und 1.5 bara,
bevorzugt.
-
Das
erfindungsgemäße Verfahren
kann in Batchfahrweise oder kontinuierlich durchgeführt werden.
-
Es
ist möglich,
das Nickel(II)-Ether-Addukt dirket in der so erhaltenen Lösung bzw.
Suspension zur Herstellung der Nickel(0)-Phosphorligand-Komplexe
zu verwenden. Alternativ kann das Addukt auch zunächst isoliert
und gegebenenfalls getrocknet werden und zur Herstellung des Nickel(0)-Phosphorligand-Komplexes
wieder gelöst
bzw. resuspendiert werden. Eine Isolierung des Adduktes aus der
Suspension kann durch dem Fachmann an sich bekannte Verfahren erfolgen,
wie Filtration, Zentrifugation, Sedimentation oder durch Hydrocyclone,
wie beispielsweise in Ullmann's
Encyclopedia of Industrial Chemistry, Unit Operation I, Vol. B2,
VCH, Weinheim, 1988, in Kapitel 10, Seiten 10-1 bis 10-59, Kapitel
11, Seiten 11-1 bis 11–27
und Kapitel 12, Seiten 12-1 bis 12-61, beschrieben.
-
In
dem erfindungsgemäßen Verfahren
ist es möglich,
ohne Überschuss
an Nickel(II)-halogenid oder
Reduktionsmittel, beispielsweise Zink, zu arbeiten, so dass deren
Abtrennung nach der Nickel(0)-Komplexbildung nicht notwendig ist.
-
In
einer besonderen Ausführungsform
der vorliegenden Erfindung umfasst das erfindungsgemäße Verfahren
die folgenden Verfahrensschritte:
- (1) Trocknung
eines wasserhaltigen Nickel(II)-halogenids durch Azeotropdestillation,
- (2) Vorkomplexierung des azeotrop getrockneten Nickel(II)-halogenids
in einem Lösemittel
in Gegenwart eines phosphorhaltigen Liganden,
- (3) Zugabe mindestens eines Reduktionsmittels zu der aus Verfahrensschritt
(2) stammenden Lösung
oder Suspension bei einer Zugabetemperatur von 20 bis 120°C,
- (4) Rühren
der aus Verfahrensschritt (3) stammenden Suspension oder Lösung für bei einer Umsetzungstemperatur
von 20 bis 120 °C.
-
Die
Vorkomplexierungstemperaturen, Zugabetemperaturen und Umsetzungstemperaturen
können,
jeweils unabhängig
voneinander, 20 °C
bis 120 °C
betragen. Besonders bevorzugt sind bei der Vorkomplexierung, Zugabe
und Umsetzung Temperaturen von 30 °C bis 80 °C.
-
Die
Vorkomplexierungszeiträume,
Zugabezeiträume
und Umsetzungszeiträume
können,
jeweils unabhängig
voneinander, 1 Minute bis 24 Stunden betragen. Der Vorkomplexierungszeitraum
beträgt
insbesondere 1 Minute bis 3 Stunden. Der Zugabezeitraum beträgt vorzugsweise
1 Minute bis 30 Minuten. Der Umsetzungszeitraum beträgt vorzugsweise
20 Minuten bis 5 Stunden.
-
Weiterer
Gegenstand der vorliegenden Erfindung sind die durch das erfindungsgemäße Verfahren
erhältlichen
Nickel(0)-Phosphorligand-Komplexe enthaltenden Lösungen sowie deren Verwendung
in der Hydrocyanierung von Alkenen und von ungesättigten Nitrilen, insbesondere
in der Hydrocyanierung von Butadien zur Herstellung einer Mischung
von Pentennitrilen und der Hydrocyanierung von Pentennitrilen zu
Adiponitril. Die vorliegende Erfindung betrifft auch deren Verwendung
in der Isomerisierung von Alkenen und von ungesättigten Nitrilen, insbesondere
von 2-Methyl-3-butennitril zu 3-Pentennitril.
-
Weiterer
Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Herstellung
von einem durch Azeotropdestillation getrocknetem Nickel(II)-halogenid
durch Entfernung von Wasser aus Mischungen, enthaltend mindestens
ein wasserhaltiges Nickel(II)-halogenid,
wobei die Mischung mit einem Verdünnungsmittel versetzt wird,
dessen Siedepunkt im Falle der Nichtazeotrop-Bildung des genannten
Verdünnungsmittels
mit Wasser unter den Druckbedingungen der nachfolgend genannten
Destillation höher
ist als der Siedepunkt von Wasser und das an diesem Siedepunkt des
Wassers flüssig
vorliegt oder das ein Azeotrop oder Heteroazeotrop mit Wasser unter
den Druck- und Temperaturbedingungen der nachfolgend genannten Destillation
bildet, und die Mi schung, enthaltend das wasserhaltige Nickel(II)-halogenid
und das Verdünnungsmittel,
unter Abtrennung von Wasser oder des genannten Azetrops oder des
genannten Heteroazeotrops von dieser Mischung und unter Erhalt einer
wasserfreien Mischung, enthaltend Nickel(II)-halogenid und das besagte
Verdünnungsmittel
destilliert wird. Weitere Ausführungen
und Ausgestaltungen dieses Verfahren sind bereits oben beschrieben.
-
Die
vorliegenden Erfindungen werden anhand der folgenden Ausführungsbeispiele
näher erläutert.
-
Ausführungsbeispiele
-
Bei
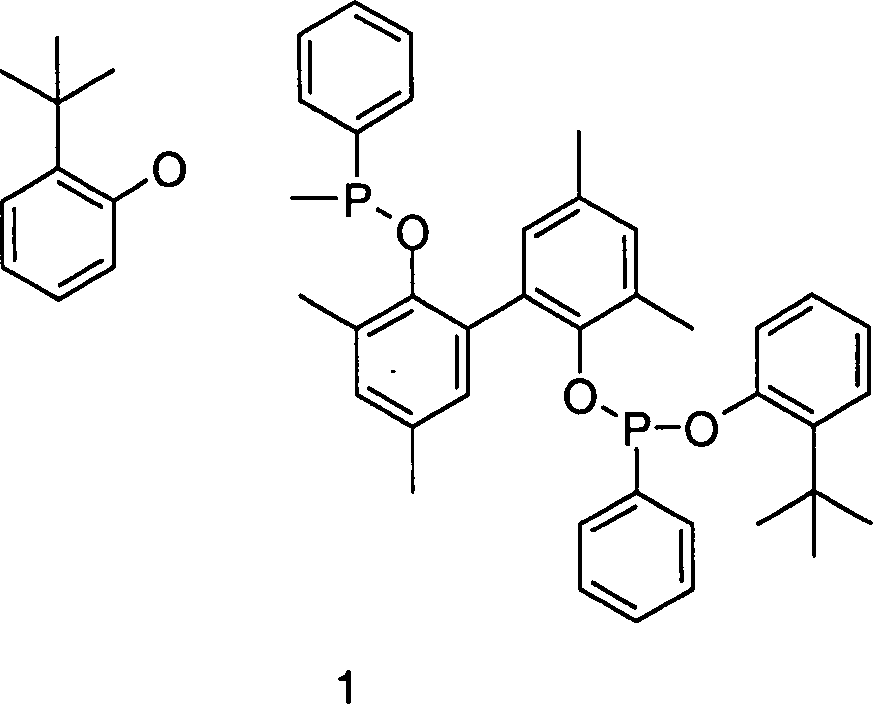
den Beispielen zur Komplexsynthese wurde als Chelatligand-Lösung eine
Lösung
des Chelatphosphonits 1
in 3-Pentennitril (65 Gew.-%
Chelat, 35 Gew.-% 3-Pentennitril) eingesetzt.
-
Zur
Bestimmung des Umsatzes wurden die hergestellten Komlplexlösungen auf
ihren Gehalt an aktivem, komplexierten Ni(0) untersucht. Hierzu
wurden die Lösungen
mit Tri(m/p-tolyl)phosphit (typischerweise 1 g Phosphit pro 1 g
Lösung)
versetzt und ca. 30 Min. bei 80 °C
gehalten, um eine vollständige Umkomplexierung
zu erzielen. Anschließend
wurde für
die elektrochemische Oxidation in einer cyclovoltammetrischen Messapparatur
die Strom-Spannungs-Kurve in ruhender Lösung gegen eine Referenzelektrode
gemessen, der der Konzentration proportionale Peakstrom ermittelt
und über
eine Kalibrierung mit Lösungen
bekannter Ni(0)-Konzentrationen der Ni(0)-Gehalt der Testlösung – korrigiert
um die nachträgliche
Verdünnung
mit Tri(m/p-tolyl)phosphit – bestimmt.
Die in den Beispielen genannten Ni(0)-Werte geben den nach dieser
Methode bestimmten Gehalt an Ni(0) in Gew.-% bezogen auf die gesamte
Reaktionslösung
an.
-
Die
Beispiele 1–4
beschreiben die Herstellung der Suspensionen mit direkt anschließender Komplexbildung:
-
Beispiel 1:
-
In
einem 250-ml-Kolben mit Rührer
und Wasserauskreiser wurden von 9.7 g NiCl2·6H2O (41 mmol) in 100 ml 3-Pentennitril suspendiert.
Die Mischung wurde unter Rückfluss
zum Sieden erhitzt und das Wasser dabei ausgekreist. Es wurde eine feine
Suspension in 3-Pentennitril erhalten. Die Suspension wurde fast
bis zur Trockene eingeengt, in 13 g 3-Pentenntril resuspendiert
und mit 100 g Chelatlösung
(86 mmol Ligand) versetzt. Bei 50 °C wurden 4 g Zn-Pulver (61 mmol,
1.4 Äq.)
zugegeben, der Ansatz auf 60 °C
erwärmt
und 3 h gerührt.
Da kein Umsatz beobachtet wurde, wurde noch 3 h bei 80 °C nachgerührt. Es
wurde ein Ni(0)-Wert von 1.1 % (56 % Umsatz) gemessen.
-
Beispiel 2:
-
In
einem 500-ml-Kolben mit Rührer
und Wasserauskreiser wurde eine Lösung von 9.7 g NiCl2·6H2O (41 mmol) in 10 g Wasser mit 150 g 3-Pentennitril
versetzt. Die zweiphasige Mischung wurde unter Rückfluss zum Sieden erhitzt
und das Wasser dabei ausgekreist. Es wurde eine feine Suspension
in 3-Pentennitril erhalten. Die Suspension wurde fast bis zur Trockene
eingeengt, in 13 g 3-Pentennitril resuspendiert und mit 100 g Chelatlösung (86 mmol
Ligand) versetzt. Bei 80 °C
wurden 4 g Zn-Pulver (61 mmol, 1.4 Äq.) zugegeben und der Ansatz
6 h gerührt.
Es wurde ein Ni(0)-Wert von 1.4 % (71 % Umsatz) gemessen.
-
Beispiel 3:
-
Eine
analog zu Beispiel 2 hergestellte Suspension wurde fast bis zur
Trockene eingeengt, in 3 g 3-Pentennitril resuspendiert und mit
50 g Chelatlösung
(43 mmol Ligand) versetzt. Bei 80 °C wurden 4 g Zn-Pulver (61 mmol,
1.4 Äq.)
zugegeben und der Ansatz 4 h gerührt.
Es wurde ein Ni(0)-Wert von 2.3 % (60 % Umsatz) gemessen.
-
Beispiel 4:
-
In
einem 250-ml-Kolben mit Rührer
und Wasserauskreiser wurde eine Lösung von 19.7 g NiCl2·6H2O (83 mmol) in 20 g Wasser mit 61 g 3-Pentennitril
versetzt. Die zweiphasige Mischung wurde unter Rückfluss zum Sieden erhitzt
und das Wasser dabei ausgekreist. Es wurde eine dicke, gerade noch rührfähige Suspension
in 3-Pentennitril erhalten. Sie Suspension wurde nach Abkühlen auf
80 °C mit
100 g Chelatlösung
(86 mmol Ligand) versetzt. Anschließend wurden bei 80 °C 8 g Zn-Pulver (122 mmol, 1.4 Äq.) zugegeben
und der Ansatz 4 h gerührt.
Es wurde ein Ni(0)-Wert von 1.2 (45 % Umsatz) gemessen.
-
Die
Beispiele 5 und 6 beschreiben die separate Herstellung einer NiCl2-Suspension.
-
Beispiel 5:
-
In
einem 2-l-Kolben mit Rührer
und Wasserauskreiser wurde eine Lösung von 194 g NiCl2·6H2O (816 mmol) in 100 g Wasser mit 300 g 3-Pentennitril versetzt.
Die zweiphasige Mischung wurde unter Rückfluss zum Sieden erhitzt
und das Wasser dabei ausgekreist. Nachdem 161 g Wasser (86 % der
theoretischen Menge) abgetrennt waren, war die Suspension im Kolben
teils so zähflüssig und
teils zu großen Feststoffagglomeraten
erstarrt, dass der Versuch abgebrochen werden musste.
-
Beispiel 6:
-
In
einem 2-l-Kolben mit Rührer,
Wasserauskreiser und Tropftrichter wurden 700 g 3-Pentennitril unter
Rückfluss
zum Sieden erhitzt. Zu diesem siedenden Pentennitril wurde eine
Lösung
von 194 g NiCl2·6H2O
(816 mmol) in 105 g Wasser gerade so schnell zugetropft wie das
Wasser im Wasserauskreiser wieder abgetrennt wurde. Es wurde eine
feine, fast homogene Suspension in 3-Pentennitril erhalten.
-
Die
Beispiele 7–12
beschreiben die Herstellung der Nickelkomplexe aus einer separat
hergestellten Suspension.
-
Beispiel 7:
-
In
einem 500-ml-Kolben mit Rührer
wurden unter Argon 74 g einer nach Beispiel 6 hergestellten Suspension
(83 mmol NiCl2) mit 100 g Chelatlösung (86
mmol Ligand) versetzt und 15 Min. bei 80 °C gerührt. Anschließend wurden
bei 80 °C
8 g Zn-Pulver (122 mmol, 1.5 Äq.)
zugegeben und 5 h bei 80 °C
gerührt.
Es wurde ein Ni(0)-Wert von 1.7 % (64 % Umsatz) gemessen.
-
Beispiel 8:
-
In
einem 250-ml-Kolben mit Rührer
wurden unter Argon 37 g einer nach Beispiel 6 hergestellten Suspension
(42 mmol NiCl2) mit 50 g Chelatlösung (43
mmol Ligand) versetzt und 15 Min. bei 50 °C gerührt. Anschließend wurden
bei 50 °C
3 g Zn-Pulver (46 mmol, 1.1 Äq.)
zugegeben und 5 h bei 50 °C
gerührt.
Es wurde ein Ni(0)-Wert von 1.2 % (43 % Umsatz) gemessen.
-
Beispiel 9:
-
Es
wurde eine Reaktion analog Beispiel 8 durchgeführt, jedoch wurde vor Zugabe
des Zn-Pulvers auf 80 °C
erwärmt.
Nach 5 h wurde ein Ni(0)-Wert von 1.4 % (50 % Umsatz) gemessen.
-
Beispiel 10:
-
Es
wurde eine Reaktion analog Beispiel 8 durchgeführt, jedoch wurden alle Schritte
bei 80 °C durchgeführt. Nach
5 h wurde ein Ni(0)-Wert von 1.8 % (61 % Umsatz) gemessen.
-
Beispiel 11:
-
Es
wurde eine Reaktion analog Beispiel 7 durchgeführt, jedoch wurden statt Zn-Pulver
6.8 g Fe-Pulver (122 mmol, 1.5 Äq.)
zugegeben. Nach 5 h wurde ein Ni(0)-Wert von 1.2 % (53 % Umsatz)
gemessen.
-
Beispiel 12:
-
In
einem 250-ml-Kolben mit Rührer
wurden unter Argon 47.6 g einer nach Beispiel 6 hergestellten Suspension
(53 mmol NiCl2) in 67.3 g Chelatlösung (58
mmol Ligand) suspendiert und auf 0 °C abgekühlt. Anschließend wurden
26.5 g einer 25%igen Lösung
von Triethylaluminium in Toluol (58 mmol) langsam zudosiert. Nach
Aufwärmen
der Lösung
auf Raumtemperatur wurde noch 10 h gerührt. Es wurde ein Ni(0)-Wert
von 0.64 % (28 % Umsatz) gemessen.
-
Im
Beispiel 13 wurde als Ligandlösung
eine „Rück-Katalysatorlösung" eingesetzt, die
bereits als Katalysatorlösung
in Hydrocyanierungsreaktionen eingesetzt und stark an Ni(0) abgereichert
worden war. Die Zusammensetzung der Lösung beträgt ca.
-
20
Gew.-% Pentennitrile, ca. 6 Gew.-% Adipodinitril, ca. 3 Gew.-% andere
Nitrile, ca. 70 Gew.-% Ligand (bestehend aus einer Mischung von
40 Mol-% Chelatphosphonit 1 und 60 Mol-% Tri(m/p-tolylphosphit)
und einem Nickel(O)-Gehalt von nur noch 0,8 %.
-
Beispiel 13:
-
In
einem 250-ml-Kolben mit Rührer
wurden unter Argon 37 g einer nach Beispiel 6 hergestellten Suspension
(42 mmol NiCl2) mit 50 g Rück-Katalysatorlösung versetzt
und 15 Min. bei 80 °C
gerührt.
Anschließend
wurden bei 80 °C
3 g Zn-Pulver (46 mmol, 1.1 Äq.)
zugegeben und 5 h bei 80 °C
gerührt.
Es wurde ein Ni(0)-Wert von 1.64 % (entsprechend einem Verhältnis von
P : Ni von 4 : 1) gemessen.
-
In
den Beispielen 14 bis 19 wurde Tri(m/p-tolylphosphit) als Ligand
eingesetzt.
-
Beispiel 14:
-
In
einem 250-ml-Kolben mit Rührer
wurden unter Argon 100 g einer analog zu Beispiel 6 hergestellten
Suspension (25 mmol NiCl2) mit 36 g (100 mmol)
Tri(m/p-Tolyl)phosphit versetzt und 5 Min. bei 80 °C gerührt. Anschließend wurden
bei 80 °C
1.8 g Zn-Pulver (28 mmol, 1.1 Äq.)
zugegeben und 4 h bei 80 °C
gerührt.
Es wurde ein Ni(0)-Wert von 0.75 % (72 % Umsatz) gemessen.
-
Beispiel 15:
-
Es
wurde eine Reaktion analog Beispiel 14 durchgeführt, jedoch wurden 53.8 g (152
mmol) Tri(m/p-tolylphosphit) eingesetzt. Es wurde ein Ni(0)-Wert
von 0.8 % (85 Umsatz) gemessen.
-
Beispiel 16:
-
Es
wurde eine Reaktion analog Beispiel 15 durchgeführt, jedoch wurden alle Verfahrensstufen bei
40 °C durchgeführt. Es
wurde ein Ni(0)-Wert von 0.6 % (65 % Umsatz) gemessen.
-
Beispiel 17:
-
Es
wurde eine Reaktion analog Beispiel 15 durchgeführt, jedoch wurden alle Verfahrensstufen bei
60 °C durchgeführt. Es
wurde ein Ni(0)-Wert von 0.95 % (99 % Umsatz) gemessen.
-
Beispiel 18:
-
Es
wurde eine Reaktion analog Beispiel 14 durchgeführt, jedoch wurden 71.8 g (203
mmol) Tri(m/p-tolylphosphit) eingesetzt. Es wurde ein Ni(0)-Wert
von 0.5 % (85 Umsatz) gemessen.
-
In
den Vergleichsbeispielen wurde kommerziell erhältliches, wasserfreies Nickelchlorid
als Nickelquelle eingesetzt.
-
Vergleichsbeispiel 1:
-
In
einem 500-ml-Kolben mit Rührer
wurden unter Argon 11 g (85 mmol) NiCl2 in
13 g 3-Pentennitril
suspendiert, mit 100 g Chelatlösung
(86 mmol Ligand) versetzt und 15 Min. bei 80 °C gerührt. Nach Abkühlen auf
40 °C wurden
8 g Zn-Pulver (122 mmol, 1.4 Äq.)
zugegeben und 4 h bei 40 °C
gerührt.
Es wurde ein Ni(0)-Wert von 0.05 % (1 % Umsatz) gemessen.
-
Vergleichsbeispiel 2:
-
Es
wurde eine Reaktion analog Vergleichsbeispiel 1 durchgeführt, jedoch
wurde die Temperatur bei der Zugabe des Zn-Pulvers bei 80 °C gehalten. Nach
5 h wurde ein Ni(0)-Wert von 0.4 % (10 % Umsatz) gemessen.
-
Vergleichsbeispiel 3:
-
In
einem 500-ml-Kolben mit Rührer
wurden unter Argon 11 g (85 mmol) NiCl2 in
13 g 3-Pentennitril
suspendiert, mit 100 g Chelatlösung
(86 mmol Ligand) versetzt und 15 Min. bei 80 °C gerührt. Nach Abkühlen auf
60 °C wurden
5.3 g Zn-Pulver (95 mmol, 1.1 Äq.)
zugegeben und 10 h bei 60–65 °C gerührt. Es
wurde ein Ni(0)-Wert von 0.16 % (4 Umsatz) gemessen.
-
Vergleichsbeispiel 4:
-
Es
wurde eine Reaktion analog Vergleichsbeispiel 3 durchgeführt, jedoch
wurde die Temperatur bei der Zugabe des Fe-Pulvers bei 80 °C gehalten. Nach
10 h wurde ein Ni(0)-Wert von 0.4 % (10 % Umsatz) gemessen.