CN1052002C - 四氢吡喃和四氢呋喃的醚衍生物,其制备方法和含有这些化合物的药物组合物 - Google Patents
四氢吡喃和四氢呋喃的醚衍生物,其制备方法和含有这些化合物的药物组合物 Download PDFInfo
- Publication number
- CN1052002C CN1052002C CN94191925A CN94191925A CN1052002C CN 1052002 C CN1052002 C CN 1052002C CN 94191925 A CN94191925 A CN 94191925A CN 94191925 A CN94191925 A CN 94191925A CN 1052002 C CN1052002 C CN 1052002C
- Authority
- CN
- China
- Prior art keywords
- bases
- methyl
- oxos
- tetrahydroquinoline
- thio
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/08—Bridged systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen- Or Sulfur-Containing Heterocyclic Ring Compounds With Rings Of Six Or More Members (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
本发明涉及式(Ⅰ)的醚衍生物或其可药用的盐;Q1-X-Ar-Q2,其中Q1是任意取代或不取代的9-、10-或11-元双环杂环部分,该杂环部分含有一个或两个氮杂原子,并且可以任选地含有另一个选自氮、氧和硫的杂原子;X是氧、硫、亚磺酰基或磺酰基;Ar是任意取代或不取代的亚苯基、吡啶二基、嘧啶二基、噻吩二基、呋喃二基、噻唑二基、唑二基、噻二唑二基或恶二唑二基;并且Q2是选自式(Ⅱ)和(Ⅲ)的基团,其中R1是氢、(2-5C)链烷酰基或任意取代或不取代的苯甲酰基;R2是(1-4C)烷基;和R3是氢或(1-4C)烷基;或者R2与R3连结在一起形成亚甲基、1,2-亚乙烯基、亚乙基或三亚甲基;其制备方法:含有这些化合物的药物组合物及其用作5-脂氧合酶抑制剂的用途。
Description
本发明涉及醚衍生物,特别是作为5-脂氧合酶抑制剂(下文称作5-LO)的醚衍生物。本发明还涉及制备所述醚衍生物的方法和含有这些醚衍生物的新药物组合物。本发明还包括所述醚衍生物在治疗各种疾病例如炎性疾病和/或变应性疾病(其中涉及了5-LO催化的花生四烯酸的氧化作用的直接或间接产物)中的应用和制备用于这些应用的新的药物。
如上所述,下文所述的醚衍生物是5-LO抑制剂,已知5-LO参与花生四烯酸的催化氧化作用,从而通过阶式方法升高具有药理学活性的白三烯如白三烯B4(LTB4)和肽基-类脂白三烯如白三烯C4(LTC4)和白三烯D4(LTD4)和各种代谢物。
G.W.Taylor和S.R.Clarke在Trends in PharmacologicalSciences,1986,7,100-103中总结了白三烯的生物合成关系和药理学性质。白三烯及其代谢物与多种疾病[例如各种炎症和变应性疾病如关节炎(特别是类风湿关节炎、骨关节炎和痛风)、胃肠道炎症(特别是肠炎、溃疡性结肠炎和胃炎)、皮肤病(特别是牛皮癣、湿疹和皮炎)、眼科疾病(特别是变应性结膜炎和眼色素层炎)以及呼吸系统疾病(特别是气喘、支气管炎和变应性鼻炎)]的产生和发展有关,例如与心血管和脑血管疾病(如心肌梗塞、动脉粥样硬化斑的形成、高血压、血小板凝集、咽峡炎、中风、再灌注损伤、血管损伤包括再狭窄和外周血管疾病)的产生和发展有关,例如与休克和损伤(如烧伤、毒血症或外科手术后产生的)疾病的形成有关,例如与各种骨代谢疾病[如骨质疏松(包括老年性骨质疏松和后黄体期骨质疏松)、佩吉特病、骨转移、高钙血、甲状旁腺机能亢进、骨硬化、骨硬化病和牙周炎]有关,以及与伴随有类风湿关节炎和骨关节炎的骨代谢异常变化有关。另外,因白三烯能够调节淋巴细胞和白细胞的功能,所以白三烯是炎性疾病的介体。通过环氧合酶对花生四烯酸的作用,花生四烯酸的其它药理活性代谢物如前列腺素和thromboxanes升高。
在EP 0385662中公开了某些杂环衍生物对5-LO具有抑制活性。EP 0420511、0462812和0462813也涉及了对5-LO具有抑制活性的杂环衍生物。我们发现,某些结构特征与上述申请中所公开的化合物类似、而其它特征(特别是醇基)不同于早期申请的一些醚衍生物是5-LO酶的有效抑制剂,因此也是白三烯生物合成的有效抑制剂。因此,这些化合物在例如单独或者部分由一种或多种白三烯介导的变应性疾病、牛皮癣、气喘、心血管与脑血管疾病、和/或炎性疾病和关节炎、和/或骨代谢疾病的治疗方面是有价值的治疗剂。
已发现,在EP 0462812中公开的某些化合物具有不希望的自诱导性质,即由于动物肝脏代谢该化合物,需要给温血动物重复施用该化合物以增加其功效。结果是降低了在动物血液中存在的重复施用的化合物的量,例如通过降低所达到的最大浓度(Cmax)或者,例如减少了动物与该化合物的接触(根据以血液中化合物的浓度对给药后的时间绘制的曲线下面积(AUC)测量)。化合物4-甲氧基-4-[5-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-2-基]四氢吡喃具有不希望的自诱导性质。
已发现,EP 0462812中所公开的某些化合物是非晶态的,例如它们是油状的或胶态的,或者它们以泡沫的形式分离。当考虑到制备、纯化、分析、加工和大量制备该化合物时,这些非晶形的化合物是不希望的。化合物(2S,4R)-4-甲氧基-2-甲基-4-[5-(1-甲基-2-硫代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-2-基]四氢吡喃具有不希望的粘稠油状特性。
本发明一方面提供了式I的醚衍生物或其可药用的盐
Q1-X-Ar-Q2 I其中Q1是9-、10-或11-元双环杂环部分,该杂环部分含有一个或两个氮杂原子,并且可以任选地含有另一个选自氮、氧和硫的杂原子,并且Q1可以任意地带有至多4个取代基,这些取代基选自卤素、羟基、氰基、甲酰基、氧代、硫代、(1-4C)烷基、(3-4C)链烯基、(3-4C)链炔基、(1-4C)烷氧基、氟-(1-4C)烷基、羟基-(1-4C)烷基、(2-5C)链烷酰基、苯基、苯甲酰基和苄基,其中所述苯基、苯甲酰基和苄基取代基可以任意地带有一个或两个选自卤素、(1-4C)烷基和(1-4C)烷氧基的取代基;X是氧、硫、亚磺酰基或磺酰基;Ar是亚苯基、吡啶二基、嘧啶二基、噻吩二基、呋喃二基、噻唑二基、噁唑二基、噻二唑二基或噁二唑二基,它们可以任意地带有1或2个选自卤素、氰基、三氟甲基、羟基、氨基、(1-4C)烷基、(1-4C)烷氧基、(1-4C)烷氨基和二-(1-4C)烷氨基的取代基;并且Q2是选自式II和III的基团:- 其中R1是氢、(2-5C)链烷酰基或苯甲酰基,并且其中所述苯甲酰基可以任意地带有1或2个选自卤素、(1-4C)烷基和(1-4C)烷氧基的取代基;R2是(1-4C)烷基;和R3是氢或(1-4C)烷基;或者R2与R3连结在一起形成亚甲基、1,2-亚乙烯基、亚乙基或三亚甲基。
其中R1是氢、(2-5C)链烷酰基或苯甲酰基,并且其中所述苯甲酰基可以任意地带有1或2个选自卤素、(1-4C)烷基和(1-4C)烷氧基的取代基;R2是(1-4C)烷基;和R3是氢或(1-4C)烷基;或者R2与R3连结在一起形成亚甲基、1,2-亚乙烯基、亚乙基或三亚甲基。
本发明另一方面提供了式I的醚衍生物或其可药用的盐
Q1-X-Ar-Q2 I其中Q1是10-元双环杂环部分,该杂环部分含有一个或两个氮杂原子,并且可以任选地含有另一个选自氮、氧和硫的杂原子,并且Q1可以任意地带有至多4个取代基,这些取代基选自卤素、羟基、氰基、甲酰基、氧代、硫代、(1-4C)烷基、(1-4C)烷氧基、氟-(1-4C)烷基、羟基-(1-4C)烷基、(2-5C)链烷酰基、苯基、苯甲酰基和苄基,其中所述苯基、苯甲酰基和苄基取代基可以任意地带有一个或两个选自卤素、(1-4C)烷基和(1-4C)烷氧基的取代基;X是氧、硫、亚磺酰基或磺酰基;Ar是亚苯基、吡啶二基、嘧啶二基、噻吩二基、呋喃二基、噻唑二基、噁唑二基、噻二唑二基或噁二唑二基,它们可以任意地带有1或2个选自卤素、氰基、三氟甲基、羟基、氨基、(1-4C)烷基、(1-4C)烷氧基、(1-4C)烷氨基和二-(1-4C)烷氨基的取代基;并且Q2是选自式II和III的基团:- 其中R1是氢、(2-5C)链烷酰基或苯甲酰基,并且其中所述苯甲酰基可以任意地带有1或2个选自卤素、(1-4C)烷基和(1-4C)烷氧基的取代基;R2是(1-4C)烷基;和R3是氢或(1-4C)烷基。
其中R1是氢、(2-5C)链烷酰基或苯甲酰基,并且其中所述苯甲酰基可以任意地带有1或2个选自卤素、(1-4C)烷基和(1-4C)烷氧基的取代基;R2是(1-4C)烷基;和R3是氢或(1-4C)烷基。
在本说明书中,一般性术语“烷基”包括直链和支链烷基。但是对于单个的烷基如“丙基”则仅仅是特指直链丙基,对于单个的支链烷基如“异丙基”则仅仅是特指支链异丙基。类似的规定适用于其它的一般性术语。
应该理解,在上述定义的某些式I化合物中可以存在互变异构现象,并且本文图示的任何结构式都仅仅表示了一种可能的互变异构形式,本发明的定义中包括所有具有抑制5-LO性质的式I化合物的任何互变异构形式,而不仅仅限于任何一种图示结构式中所用的互变异构形式。
还应该理解,在上述定义中的某些式I化合物可以通过一个或多个不对称碳原子以旋光活性形式或外消旋形式存在,本发明的定义中包括具有抑制5-LO性质的任何旋光活性形式或外消旋形式。旋光活性形式的合成可以通过本领域众所周知的有机化学技术进行,例如由旋光活性的原料合成或者拆分外消旋体。
上述一般性术语的合适基团包括下面所列的那些。
当Q1为含有一或两个氮杂原子、并且任选地含有另一个选自氮、氧和硫的杂原子的9元双环杂环部分时,其合适的基团是例如苯环稠合的杂环部分或其氢化衍生物,例如吲哚基、二氢吲哚基、异吲哚基、异二氢吲哚基、中氮茚基、苯并咪唑基、2,3-二氢苯并咪唑基、1H-吲唑基、2,3-二氢-1H-吲唑基、苯并噁唑基、2,3-二氢苯并噁唑基、苯并[c]异噁唑基、苯并[d]异噁唑基、2,3-二氢苯并[d]异噁唑基、苯并噻唑基、2,3-二氢苯并噻唑基、苯并[c]异噻唑基、苯并[d]异噻唑基和2,3-二氢苯并[d]异噻唑基,或者例如吡啶稠合的杂环部分或其氢化衍生物,例如1H-吡咯并[2,3-b]吡啶基、2,3-二氢-1H-吡咯并[2,3-b]吡啶基、1H-吡咯并[2,3-c]吡啶基、2,3-二氢-1H-吡咯并[2,3-c]吡啶基、1H-咪唑并[4,5-b]吡啶基、2,3-二氢-1H-咪唑并[4,5-b]吡啶基、1H-咪唑并[4,5-c]吡啶基和2,3-二氢-1H-咪唑并[4,5-c]吡啶基。
当Q1为含有一或两个氮杂原子、并且任选地含有另一个选自氮、氧和硫的杂原子的10元双环杂环部分时,其合适的基团是例如10元苯环稠合的杂环部分,例如喹啉基、异喹啉基、噌啉基、喹唑啉基、喹喔啉基、4H-1,4-苯并噁嗪基或4H-1,4-苯并噻嗪基,或者其氢化衍生物,例如1,2-二氢喹啉基、1,2,3,4-四氢喹啉基、1,2-二氢异喹啉基、1,2,3,4-四氢喹唑啉基、2,3-二氢-4H-1,4-苯并噁嗪基或2,3-二氢-4H-1,4-苯并噻嗪基;或者例如10元吡啶稠合的杂环部分,例如1,7-二氮杂萘基、1,8-二氮杂萘基、吡啶并[2,3-d]嘧啶基、吡啶并[2,3-b]吡嗪基、4H-吡啶并[3,2-b][1,4]噁嗪基和4H-吡啶并[3,2-b][1,4]噻嗪基,或者其氢化衍生物。
当Q1为含有一或两个氮杂原子、并且任选地含有另一个选自氮、氧和硫的杂原子的11元双环杂环部分时,其合适的基团是例如11元苯环稠合的杂环部分例如1H-苯并[b]氮杂,或者其氢化衍生物如2,3,4,5-四氢-1H-苯并[b]氮杂。
杂环部分可以通过双环杂环的两个环上任何有效的位置连接,包括通过有效的氮原子连接。杂环部分在有效氮原子上可以带有合适的取代基,例如(1-4C)烷基、(3-4C)链烯基、(3-4C)链炔基、氟-(1-4C)烷基、苯基、苯甲酰基或苄基取代基。
在Q1或Ar上、在Q1的苯基取代基上、在含有苯基的Q1的任何取代基上或者在R1(当R1是苯甲酰基时)上可以存在的合适的取代基包括,例如:-卤素: 氟、氯、溴和碘;(1-4C)烷基: 甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基和叔丁基;(3-4C)链烯基: 烯丙基、2-丁烯基和3-丁烯基;(3-4C)链炔基: 2-丙炔基和2-丁炔基;(1-4C)烷氧基: 甲氧基、乙氧基、丙氧基、异丙氧基和丁氧基;氟-(1-4C)烷基: 氟甲基、二氟甲基、三氟甲基、2-氟乙基、2,2,2-三氟乙基和五氟乙基;羟基-(1-4C)烷基: 羟甲基、2-羟乙基和3-羟丙基;(2-5C)链烷酰基: 乙酰基、丙酰基和丁酰基;(1-4C)烷氨基: 甲氨基、乙氨基和丙氨基;和二-(1-4C)烷氨基:二甲氨基、二乙氨基和N-乙基-N-甲氨基。
当Ar是亚苯基时,合适的Ar是例如1,3-亚苯基或1,4-亚苯基。
当Ar是吡啶二基、嘧啶二基、噻吩二基、呋喃二基、噻唑二基、噁唑二基、噻二唑二基或噁二唑二基时,合适的Ar是例如2,4-、2,5-或3,5-吡啶二基、4,6-嘧啶二基、2,4-或2,5-噻吩二基、2,4-或2,5-呋喃二基、2,4-或2,5-噻唑二基、2,4-或2,5-噁唑二基、2,5-噻二唑二基或2,5-噁二唑二基。
当R1是(2-5C)链烷酰基时,合适的R1是例如乙酰基、丙酰基或丁酰基。
当R2或R3是(1-4C)烷基时,合适的R2或R3是例如甲基、乙基、丙基或异丙基。
取代基R3可以与形成基团Q2的环的任何有效碳原子连接,包括与带有取代基R2的碳原子连接。当R2与R3连接在一起形成亚甲基、亚乙烯基、亚乙基或三亚甲基时,如果取代基R2位于形成基团Q2的环的氧原子的α碳原子上,则取代基R3优选位于另一个α碳原子上。
本发明化合物的合适的可药用盐是例如:具有足够碱性的本发明化合物的酸加成盐,例如与例如无机酸或有机酸的酸加成盐,所述酸是例如盐酸、氢溴酸、硫酸、磷酸、三氟乙酸、柠檬酸或马来酸。另外,合适的具有足够酸性的本发明化合物的可药用盐是碱金属盐如钠或钾盐、碱土金属盐如钙或镁盐、铵盐或者与能够提供生理可接受阳离子的有机碱的盐,例如与甲胺、二甲胺、三甲胺、哌啶、吗啉或三-(2-羟乙基)胺的盐。
本发明的具体化合物包括例如式I的醚衍生物或其可药用的盐,其中:-(a)Q1是含有一或两个氮杂原子、并且任选地含有另一个选自氧和硫的杂原子的10元苯环稠合的杂环部分,该杂环部分可以任选地带有1或2个氧代或硫代取代基以及最多2个其它的取代基,这些其它的取代基选自除氧代或硫代之外的上述对Q1所定义的任何取代基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(b)Q1是喹啉基、1,2-二氢喹啉基、1,2,3,4-四氢喹啉基或2,3-二氢-4H-1,4-苯并噁嗪基,它可以任选地带有1个氧代或硫代取代基以及最多3个其它的取代基,这些其它的取代基选自氟、氯、甲基、乙基、烯丙基、2-丙炔基、甲氧基、乙氧基、三氟甲基、乙酰基、丙酰基、苯基、苯甲酰基和苄基,并且其中每个苯基、苯甲酰基或苄基取代基可以任意地带有一个选自氟、氯、甲基和甲氧基的取代基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(c)Q1是2-氧代-1,2-二氢喹啉基、2-硫代-1,2-二氢喹啉基、2-氧代-1,2,3,4-四氢喹啉基、2-硫代-1,2,3,4-四氢喹啉基或3-氧代-2,3-二氢-4H-1,4-苯并噁嗪基,它可以任意地带有最多3个取代基,这些取代基选自氟、氯、甲基、乙基、甲氧基、乙氧基、三氟甲基、乙酰基、丙酰基、苯基、苯甲酰基和苄基,并且其中每个苯基、苯甲酰基或苄基取代基可以任意地带有一个选自氟、氯、甲基和甲氧基的取代基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(d)Q1是2-氧代-1,2-二氢喹啉-6-基、2-氧代-1,2,3,4-四氢喹啉-6-基或3-氧代-2,3-二氢-4H-1,4-苯并噁嗪-7-基,它可以任意地带有最多3个取代基,这些取代基选自氟、氯、甲基、乙基、甲氧基、乙氧基和三氟甲基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(e)Q1是2-氧代-1,2-二氢喹啉-6-基或2-氧代-1,2,3,4-四氢喹啉-6-基,它在1-位带有1个选自甲基、乙基、丙基、烯丙基和2-丙炔基的取代基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(f)Q1是含有一或两个氮杂原子、并且任选地含有另一个选自氧和硫的杂原子的9元苯环稠合的杂环部分,该杂环部分可以任选地带有1个氧代或硫代取代基以及最多3个其它的取代基,这些其它的取代基选自除氧代或硫代之外的上述对Q1所定义的任何取代基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(g)Q1是吲哚基、二氢吲哚基、苯并咪唑基、2,3-二氢苯并咪唑基、苯并噁唑基、2,3-二氢苯并噁唑基、苯并噻唑基或2,3-二氢苯并噻唑基,它可以任选地带有1个氧代或硫代取代基以及最多3个其它的取代基,这些其它的取代基选自氟、氯、甲基、乙基、烯丙基、2-丙炔基、甲氧基、乙氧基、三氟甲基、乙酰基、丙酰基、苯基、苯甲酰基和苄基,并且其中每个苯基、苯甲酰基或苄基取代基可以任意地带有一个选自氟、氯、甲基和甲氧基的取代基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(h)Q1是2-氧代二氢吲哚基、2-氧代-2,3-二氢苯并咪唑基、2-氧代-2,3-二氢苯并噁唑基或2-氧代-2,3-二氢苯并噻唑基,它可以任选地带有最多3个取代基,这些取代基选自氟、氯、甲基、乙基、甲氧基、乙氧基、三氟甲基、乙酰基、丙酰基、苯基、苯甲酰基和苄基,并且其中每个苯基、苯甲酰基或苄基取代基可以任意地带有一个选自氟、氯、甲基和甲氧基的取代基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(i)Q1是2-氧代二氢吲哚-5-基或2-氧代二氢吲哚-6-基,它在1-位带有1个选自甲基、乙基、丙基、烯丙基和2-丙炔基的取代基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(j)Q1是含有一或两个氮杂原子的11元苯环稠合的杂环部分,其中杂环部分可以任选地带有1个氧代或硫代取代基以及最多3个其它的取代基,这些其它的取代基选自除氧代或硫代之外的上述对Q1所定义的任何取代基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(k)Q1是1H-苯并[b]氮杂卓或2,3,4,5-四氢-1H-苯并[b]氮杂卓,它可以任选地带有1个氧代或硫代取代基以及最多3个其它的取代基,这些其它的取代基选自氟、氯、甲基、乙基、烯丙基、2-丙炔基、甲氧基、乙氧基、三氟甲基、乙酰基、丙酰基、苯基、苯甲酰基和苄基,并且其中每个苯基、苯甲酰基或苄基取代基可以任意地带有一个选自氟、氯、甲基和甲氧基的取代基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(1)Q1是2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂,它可以任选地带有最多3个其它的取代基,这些其它的取代基选自氟、氯、甲基、乙基、甲氧基、乙氧基、三氟甲基、乙酰基、丙酰基、苯基、苯甲酰基和苄基,并且其中每个苯基、苯甲酰基或苄基取代基可以任意地带有一个选自氟、氯、甲基和甲氧基的取代基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(m)Q1是2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂卓-7-基,它在1-位带有1个选自甲基、乙基、丙基、烯丙基和2-丙炔基的取代基;并且X、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(n)X是硫、亚磺酰基或磺酰基;并且Q1、Ar和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(o)Ar是亚苯基,它可以任意地带有1或2个选自卤素、三氟甲基、(1-4C)烷基和(1-4C)烷氧基的取代基,Ar是吡啶二基或嘧啶二基,它可以任意地带有一个选自卤素、三氟甲基和氨基的取代基,或者Ar是噻吩二基或噻唑二基;并且Q1、X和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(p)Ar是1,3-亚苯基或5-氟-1,3-亚苯基;并且Q1、X和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(q)Ar是2,4-噻吩二基(X基团位于2-位)或2,5-噻吩二基;并且Q1、X和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(r)Ar是2,4-噻唑二基(X基团位于2-位)或2,5-噻唑二基(X基团位于2-位);并且Q1、X和Q2的含义同前或者同有关本发明具体化合物的本节中的含义;(s)Q2是选自式IV和V的基团:-其中R2是甲基、乙基或丙基;并且Q1、X和Ar的含义同前或者同有关本发明具体化合物的本节中的含义;(t)Q2是式IV基团,其中R2是甲基、乙基或丙基;并且Q1、X和Ar的含义同前或者同有关本发明具体化合物的本节中的含义。
优选的本发明化合物包括式I的醚衍生物或其可药用的盐其中Q1是2-氧代-1,2-二氢喹啉基、2-氧代-1,2,3,4-四氢喹啉基或3-氧代-2,3-二氢-4H-1,4-苯并噁嗪基,它可以任意地带有1、2或3个选自甲基和乙基的取代基;X是硫、亚磺酰基或磺酰基;Ar是1,3-亚苯基,它可以任意地带有1或2个氟取代基,或者Ar是3,5-吡啶二基、2-氨基-4,6-嘧啶二基、2,4-或2,5-噻吩二基或者2,4-或2,5-噻唑二基;并且Q2是选自式IV和V基团,其中R2是甲基或乙基。
进一步优选的本发明化合物包括式I的醚衍生物或其可药用的盐其中Q1是2-氧代-二氢吲哚基、2-氧代-1,2-二氢喹啉基、2-氧代-1,2,3,4-四氢喹啉基或2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂基,它可以任意地带有1、2或3个选自氟、氯、甲基、乙基、烯丙基和2-丙炔基的取代基;X是硫、亚磺酰基或磺酰基;Ar是1,3-亚苯基,它可以任意地带有1或2个氟取代基,或者Ar是2,4-或2,5-噻吩二基或者2,4-或2,5-噻唑二基;并且Q2是式II的基团,其中R1是氢;R2是甲基或乙基;和R3是氢或甲基;或者R2和R3连接在一起形成亚乙基。
进一步优选的本发明化合物包括式I的醚衍生物或其可药用的盐其中Q1是2-氧代二氢吲哚基,它可以任意地带有1、2或3个选自氟、氯、甲基、乙基、烯丙基和2-丙炔基的取代基;X是硫、亚磺酰基或磺酰基;Ar是1,3-亚苯基,它可以任意地带有1或2个氟取代基,或者Ar是2,4-或2,5-噻吩二基或者2,4-或2,5-噻唑二基;并且Q2是式II的基团,其中R1是氢;R2是甲基或乙基;和R3是氢或甲基。
进一步优选的本发明化合物包括式I的醚衍生物或其可药用的盐其中Q1是2-氧代-1,2-二氢喹啉-6-基或2-氧代-1,2,3,4-四氢喹啉-6-基,它在1-位带有一个选自甲基、乙基和丙基的取代基;X是硫、亚磺酰基或磺酰基;Ar是1,3-亚苯基或5-氟-1,3-亚苯基,或者Ar是2,4-噻吩二基(X基团在2-位)、2,5-噻吩二基、2,4-噻唑二基(X基团在2-位)或2,5-噻唑二基(X基团在2-位);并且Q2是式IV的基团其中R2是甲基。
进一步优选的本发明化合物包括式I的醚衍生物或其可药用的盐其中Q1是2-氧代二氢吲哚-5-基、2-氧代-1,2-二氢喹啉-6-基、2-氧代-1,2,3,4-四氢喹啉-6-基或2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂卓-7-基,它在1-位带有一个选自甲基、乙基、烯丙基和2-丙炔基的取代基,并且它还任意地带有另一个选自氟、氯和甲基的取代基;X是硫、亚磺酰基或磺酰基;Ar是1,3-亚苯基或5-氟-1,3-亚苯基,或者Ar是2,4-噻吩二基(X基团在2-位)、2,5-噻吩二基、2,4-噻唑二基(X基团在2-位)或2,5-噻唑二基(X基团在2-位);并且Q2是式IV的基团,其中R2是甲基。
进一步优选的本发明化合物包括式I的醚衍生物或其可药用的盐其中Q1是1-甲基-2-氧代-1,2-二氢喹啉-6-基或1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基;X是硫、亚磺酰基或磺酰基;Ar是1,3-亚苯基或5-氟-1,3-亚苯基,或者Ar是2,4-噻吩二基(X基团在2-位)、2,5-噻吩二基、2,4-噻唑二基(X基团在2-位)或2,5-噻唑二基(X基团在2-位);并且Q2是式IV的基团,其中R2是甲基。
进一步优选的本发明化合物包括式I的醚衍生物或其可药用的盐其中Q1是1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基或1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基;X是硫、亚磺酰基或磺酰基;Ar是2,4-噻唑二基(X基团在2-位)或2,5-噻唑二基(X基团在2-位);并且Q2是式IV的基团,其中R2是甲基。
进一步优选的本发明化合物包括式I的醚衍生物或其可药用的盐其中Q1是1-甲基-2-氧代二氢吲哚-5-基、1-甲基-2-氧代-1,2-二氢喹啉-6-基、1-烯丙基-2-氧代-1,2,3,4-四氢喹啉-6-基、1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基、1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、1-(2-丙炔基)-2-氧代-1,2,3,4-四氢喹啉-6-基、8-氯-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、8-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、1,8-二甲基-2-氧代-1,2,3,4-四氢喹啉-6-基或1-甲基-2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂-7-基;X是硫、亚磺酰基或磺酰基;Ar是2,4-噻吩二基(X基团在2-位)、2,5-噻吩二基或2,5-噻唑二基(X基团在2-位);并且Q2是式IV的基团,其中R2是甲基。
进一步优选的本发明化合物包括式I的醚衍生物或其可药用的盐其中Q1是1-甲基-2-氧代-1,2-二氢喹啉-6-基或1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基;X是硫、亚磺酰基或磺酰基;Ar是1,3-亚苯基或5-氟-1,3-亚苯基;并且Q2是式IV的基团,其中R2是甲基。
进一步优选的本发明化合物包括式I的醚衍生物或其可药用的盐其中Q1是1-甲基-2-氧代二氢吲哚-5-基、1-甲基-2-氧代-1,2-二氢喹啉-6-基、1-烯丙基-2-氧代-1,2,3,4-四氢喹啉-6-基、1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基、1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、1-(2-丙炔基)-2-氧代-1,2,3,4-四氢喹啉-6-基、8-氯-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、8-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、1,8-二甲基-2-氧代-1,2,3,4-四氢喹啉-6-基或1-甲基-2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂-7-基;X是硫、亚磺酰基或磺酰基;Ar是1,3-亚苯基或5-氟-1,3-亚苯基;并且Q2是式IV的基团,其中R2是甲基。
特别优选的本发明化合物是下列式I化合物或其可药用的盐:-(2S,4R)-4-[5-氟-3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃或(2S,4R)-4-[5-氟-3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)苯基]-4-羟基-2-甲基四氢吡喃。
其它特别优选的本发明化合物是下列式I化合物或其可药用的盐:-(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻唑-5-基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)噻唑-5-基]四氢吡喃,(2S,4R)-4-[2-(7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻唑-5-基]-4-羟基-2-甲基四氢吡喃或(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代二氢吲哚-5-基硫代)噻唑-5-基]四氢吡喃。
其它特别优选的本发明化合物是下列式I化合物或其可药用的盐:-(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)噻吩-4-基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-5-基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2-二氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1,8-二甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃,4-[2-(8-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]-4-羟基-2-甲基四氢吡喃,4-[2-(7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]-4-羟基-2-甲基四氢吡喃或(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代二氢吲哚-5-基硫代)噻吩-4-基]四氢吡喃。
其它特别优选的本发明化合物是下列式I化合物或其可药用的盐:-(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)苯基]四氢吡喃,(2S,4R)-4-[3-(1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃,(2S,4R)-4-[3-(7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2-二氢喹啉-6-基硫代)苯基]四氢吡喃,(2S,4R)-4-[3-(8-氯-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃或(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代二氢吲哚-5-基硫代)苯基]四氢吡喃。
在本发明的另一方面,我们发现本发明的某些化合物不存在基本程度的不希望的自诱导性质。这对于例如实施例3、9和28中所公开的那些化合物是确定的。这些化合物在各种白三烯依赖性疾病如温血动物的炎性疾病和/或变应性疾病的治疗方面特别有价值,因为它们不具有产生自诱导作用的缺点。因此,例如,如果试验化合物显示出具有明显程度的自诱导作用,则对药理学和毒理学数据的评估更为复杂。另外,自诱导作用预示了一般性的酶诱导作用,这对于测定任何联合施用药物的代谢速度的增加具有不利的影响。
在本发明的另一方面,我们发现本发明的某些化合物是晶形的。本发明的结晶化合物是在下文的实施例中给出了熔点的那些,例如实施例27和28的化合物。因此,当需要大规模制备时,这些化合物是有价值的。如果它形成结晶,则易于纯化、分析和加工。已知,例如从非结晶的油状物质中除去残留的溶剂是成问题的。另外,制备含有结晶物质的药物组合物是常规方法。该组合物可以是例如适于口服的形式,如片剂或胶囊剂;或者是例如适于吸入法给药的形式,如细磨的粉末或微晶形式。如果这些物质是油状的,那么如此配制这些物质是不可行的。
包括式I的醚衍生物或其可药用盐的本发明化合物可以通过任何已知可用于制备结构相关化合物的方法进行制备。这些方法作为本发明的另一个特征,并通过下列典型的实例进行说明,例如其中除非另外指明,Q1、X、Ar和Q2的含义如前所定义,条件是当Q1、Ar和Q2中存在氨基、烷氨基或羟基时,这些基团可以任意地被常规的保护基保护,当需要时,这些保护基可以通过常规方法除去。(a)通常在合适的碱存在下,将式Q1-X-H化合物与式Z-Ar-Q2化合物偶合,其中Z是可置换的基团。
合适的可置换基团Z是例如卤素或磺酰氧基,例如氟、氯、溴、碘、甲磺酰氧基或甲苯-4-磺酰氧基。
用于偶合反应的合适的碱是例如,碱金属或碱土金属碳酸盐、(1-4C)醇盐、氢氧化物或氢化物,例如碳酸钠、碳酸钾、乙醇钠、丁醇钾、氢氧化锂、氢氧化钠、氢氧化钾、氢化钠或氢化钾,或者有机金属碱如(1-4C)烷基锂,例如正丁基锂。偶合反应通常在合适的惰性溶剂或稀释剂(例如N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基吡咯烷-2-酮、二甲基亚砜、丙酮、1,2-二甲氧基乙烷或四氢呋喃)中,在例如10-150℃、通常在100℃或接近100℃的温度下进行。
通常该反应可以在合适的催化剂例如金属催化剂(例如钯(O)或铜(I)如四(三苯膦)钯、氯化亚铜或溴化亚铜)存在下进行。
氨基或烷氨基的合适的保护基是例如酰基,如(2-4C)链烷酰基(特别是乙酰基)、(1-4C)烷氧羰基(特别是甲氧羰基、乙氧羰基或叔丁氧羰基)、芳甲氧羰基(特别是苄氧羰基)或芳酰基(特别是苯甲酰基)。上述保护基的脱保护条件需要根据所选择的保护基而变化。因此,例如酰基如链烷酰基或烷氧羰基或芳酰基可以通过例如用合适的碱如碱金属氢氧化物(如氢氧化锂或氢氧化钠)水解除去。或者可以通过例如用合适的酸如盐酸、硫酸或磷酸或三氟乙酸进行处理,除去酰基如叔丁氧羰基,以及例如通过在催化剂如披钯碳上进行氢化,可以除去芳甲氧羰基如苄氧羰基。
合适的羟基保护基是例如酰基如(2-4C)链烷酰基(特别是乙酰基)、芳酰基(特别是苯甲酰基)、芳甲基(特别是苄基)、三-(1-4C)烷基甲硅烷基(特别是三甲基甲硅烷基或叔丁基二甲基甲硅烷基)或芳基二(1-4C)烷基甲硅烷基(特别是二甲基苯基甲硅烷基)。上述保护基的脱保护条件需要根据所选择的保护基而变化。因此,例如酰基如链烷酰基或芳酰基可以通过例如用合适的碱如碱金属氢氧化物(如氢氧化锂或氢氧化钠)水解除去。或者可以例如在催化剂如披钯碳上进行氢化,除去芳甲基如苄基。或者可以通过例如用合适的酸如盐酸、硫酸、磷酸或三氟乙酸进行处理,或者用碱金属氟化物或氟化铵如氟化钠、或者优选用氟化四丁基铵进行处理,除去三烷基甲硅烷基或芳基二烷基甲硅烷基如叔丁基二甲基甲硅烷基或二甲基苯基甲硅烷基。
式Q1-X-H与式Z-Ar-Q2的原料可以通过标准有机化学方法得到。原料的制备描述于所附的非限制性实施例中。或者可以按照与有机化学领域普通技术人员所述的类似方法得到所需的原料。EP 0385662、0420551、0462812和0462813的公开内容具体涉及到合适原料的制备。(b)通常在如上定义的合适的碱存在下,将式Q1-Z化合物(其中Z是如上定义的可置换的基团)与式H-X-Ar-Q2化合物偶合。
偶合反应通常在如上定义的合适的惰性溶剂中,在例如10-150℃、通常在100℃或接近100℃的温度下进行。通常该反应可以在如上定义的合适的催化剂存在下进行。
式Q1-Z与式H-X-Ar-Q2的原料可以通过标准有机化学方法得到。原料的制备描述于所附的非限制性实施例中。或者可以按照与有机化学领域普通技术人员所述的类似方法得到所需的原料。EP 0385662、0420551、0462812和0462813的公开内容具体涉及到合适原料的制备。(c)将式Q1-X-Z化合物(其中Z是如上定义的可置换基团,或者当X是硫代基团时,Z可以是式Q1-X-基团)与式M-Ar-Q2的有机金属试剂偶合,其中M是碱金属或碱土金属如锂或钙,或者M表示常规格利雅试剂的卤化镁部分。
偶合反应通常在如上定义的合适的惰性溶剂或稀释剂中,在例如-80至+50℃、通常在-80℃至室温的温度下进行。
式Q1-X-Z与式M-Ar-Q2的原料可以通过标准有机化学方法制备。原料的制备描述于所附的非限制性实施例中。或者可以按照与有机化学领域普通技术人员所述的类似方法得到所需的原料。上述欧洲专利申请的公开内容具体涉及到合适原料的制备。(d)为了制备其中X是亚磺酰基或磺酰基的式I化合物,将其中X是硫的式I化合物氧化。
合适的氧化剂是例如将硫氧化成亚磺酰基和/或磺酰基领域中任何已知的试剂,例如过氧化氢、过酸(如3-氯过苯甲酸或过乙酸)、碱金属过硫酸盐(如过一硫酸钾)、三氧化铬或在铂存在下的氧气。氧化通常尽可能在中等条件下、在所需的化学计量量的氧化剂存在下进行,以降低氧化过程中的危险和对其它官能基的损伤。通常该反应在合适的溶剂或稀释剂(如二氯甲烷、氯仿、丙酮、四氢呋喃或叔丁基甲基醚)中,在例如接近室温、即15-35℃的温度下进行。当需要带有亚磺酰基的化合物时,也可以使用通常在极性溶剂如乙酸或乙醇中的较温和的氧化剂,例如偏高碘酸钠或偏高碘酸钾。显然,当需要含有磺酰基的式I化合物时,可以通过氧化相应的亚磺酰基化合物和相应的硫代化合物得到。(e)为了制备其中Q2中的R1是(2-5C)链烷酰基或苯甲酰基的式I化合物,将其中Q2中的R1是氢的式I化合物酰化。
合适的酰化剂是例如将醇酰化成酯领域中任何已知的试剂,例如酰卤(如在上述合适的碱存在下的(2-5C)链烷酰氯或链烷酰溴或者苯甲酰氯或苯甲酰溴),链烷酸酐例如(2-5C)链烷酸酐,或者链烷酸的混合酸酐,例如在合适的碱存在下、通过链烷酸与(1-4C)烷氧羰基卤(如(1-4C)烷氧羰基氯)反应形成的混合酸酐。通常该反应在合适的溶剂或稀释剂(如二氯甲烷、丙酮、四氢呋喃、叔丁基甲基醚或冰醋酸)中,在例如接近室温、即15-35℃的温度下进行。如果需要时,合适的碱是例如吡啶、4-二甲氨基吡啶、三乙胺、乙基二异丙基胺、N-甲基吗啉、碱金属碳酸盐如碳酸钾或碱金属羧酸盐如乙酸钠。(f)为了制备其中Q1在有效氮原子上带有一个烷基或取代烷基取代基的式I化合物,将其中Q1在所述有效氮原子上带有氢原子的式I化合物烷基化。
合适的烷基化试剂是例如有效氮原子烷基化领域中已知的任何试剂,例如烷基卤或取代烷基卤,如在上述的合适碱存在下的(1-4C)烷基氯、(1-4C)烷基溴或(1-4C)烷基碘或者取代的(1-4C)烷基氯、取代的(1-4C)烷基溴或取代的(1-4C)烷基碘。烷基化反应优选在合适的惰性溶剂或稀释剂(如N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲基亚砜、丙酮、1,2-二甲氧基乙烷或四氢呋喃)中,在例如10-150℃、通常在室温或接近室温的温度下进行。(g)为了制备其中Q1带有1或2个硫代取代基的式I化合物,将其中Q1带有1或2个氧代取代基的式I化合物与硫化剂进行反应,由此将各个氧代取代基转化为硫代取代基。
合适的硫化剂是例如将氧代基团转化为硫代基团领域中任何已知的试剂,例如2,4-二(4-甲氧基苯基)-1,3-二硫杂-2,4-二膦烷-2,4-二硫化物(Lawesson′s试剂)或五硫化磷。硫杂化反应通常用所需化学计量量的硫化剂进行,以降低对其它官能团损伤的危险。该反应通常在合适的溶剂或稀释剂(如甲苯、二甲苯或四氢呋喃)中,在例如溶剂或稀释剂的回流温度或接近该温度、即65-150℃的温度下进行。
当需要新的式I化合物的可药用盐时,可以采用常规方法、例如通过所述化合物与合适的酸或碱反应获得。当需要旋光活性的式I化合物时,可以使用旋光活性原料、通过进行上述一种方法获得,或者采用常规方法、通过拆分所述化合物的外消旋体获得。
如上所述,式I化合物是5-LO抑制剂。这种抑制作用可以采用下列一种或多种标准方法证明:-
a)体外分析系统包括用肝素化的人血液(预先用钙离子载体A23187激发)培养试验化合物,然后采用Carey和Forder所述的特异性放射免疫法、通过测定LTB4的量间接测量对5-LO的抑制作用(F.Carey和R.A.Forder,Prostaglandins,Leukotrienes Med.,1986,22,57; Prostaglandins,1984,28,666;Brit.J.Pharmacol.1985,84,34P),该方法包括使用利用Young等的方法制备的蛋白-LTB4共轭物(Prostaglandins,1983,26(4),605-613)。同时可以采用Carey和Forder所述的对thromboxane B2(TxB2)的特异性放射免疫分析(如上)测量试验化合物对环氧合酶的作用(包括在花生四烯酸的替代代谢途径中的作用,以及造成前列腺素、thromboxane和有关代谢物的升高)。该试验表明了在血细胞和蛋白质存在下试验化合物对5-LO和环氧合酶的作用。这样就可以评估对5-LO或环氧合酶的选择性抑制作用。
(b)体内分析系统是上述试验(a)的改变,包括给一组试验大鼠施用试验化合物(通常以混悬液的形式口服,是将试验化合物的二甲基亚砜溶液加入羧甲基纤维素中制成的),收集血液,肝素化,用A23187激发并进行LTB4和TxB2的放射免疫分析。该试验表明了试验化合物作为5-LO或环氧合酶抑制剂的生物利用度。
(c)体内分析系统包括测量给雄性大鼠口服试验化合物对由大鼠背部皮下组织产生的气囊中的酵母多糖引起的LTB4释放的影响。将大鼠麻醉,通过注射无菌空气(20ml)形成气囊,3天后同样地注射空气(10ml)。在开始注射空气6天后施用试验化合物(通常以混悬液的形式口服,是将试验化合物的二甲基亚砜溶液加入羧甲基纤维素中制成的),然后在气囊内注射酵母多糖(1ml,在生理盐水中的1%混悬液)。3小时后处死大鼠,用生理盐水灌洗气囊,采用上述特异性放射免疫分析测定洗出物中的LTB4。该试验表明了对炎性环境中5-LO的抑制作用。
尽管如预料的那样,式I化合物的药理学性质随结构的变化而改变,但是总的来说,在至少上述a)-c)的一个试验中下列浓度或剂量的式I化合物具有5-LO抑制作用:-
试验a):IC50(LTB4)在例如0.01-40μM的范围内
IC50(TxB2)在例如40-200μM的范围内;
试验b):口服ED50(LTB4)在例如0.1-100mg/kg的范围内;
试验c):口服ED50(LTB4)在例如0.1-100mg/kg的范围内。
当以最低抑制剂量或浓度的几倍量施用试验化合物时,在试验b)和/或c)中没有出现明显的毒性和其它不适当的作用。
因此,例如,化合物(2S,4R)-4-[5-氟-3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)苯基]-4-羟基-2-甲基四氢吡喃在试验a)中对LTB4的IC50值为0.03μM,在试验c)中对LTB4的ED50值约为0.15mg/kg;化合物(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃在试验a)中对LTB4的IC50值为0.03μM,在试验c)中对LTB4的ED50值约为0.02mg/kg;化合物(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代二氢吲哚-5-基硫代)噻吩-4-基]四氢吡喃在试验a)中对LTB4的IC50值为0.02μM,在试验c)中对LTB4的ED50值约为0.05mg/kg;和化合物(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻唑-5-基]四氢吡喃在试验a)中对LTB4的IC50值为0.05μM,在试验c)中对LTB4的ED50值约为0.2mg/kg。
这些化合物是本发明化合物的实例,相对于环氧合酶来说,它们对5-LO具有选择性抑制性质,预期这种选择性可以改善治疗性质,例如降低或免除通常与环氧合酶抑制剂有关的胃肠道副作用。
本发明的另一个特征是提供了一种药物组合物,该组合物含有式I的醚衍生物或其可药用盐以及可药用的稀释剂或载体。
该组合物可以是适于口服的形式,例如片剂、胶囊、水溶液或油溶液、混悬液或乳液;局部施用的形式,例如乳膏、油膏、凝胶或水溶液或油溶液或混悬液;鼻用的形式,例如嗅剂、鼻喷雾剂或鼻滴剂;阴道或直肠施用的形式,例如栓剂;吸入施用的形式,例如细磨的粉末如干粉剂、微晶形式或液体气雾剂;舌下或颊下施用的形式,例如片剂或胶囊;或者肠道施用的形式(包括静脉内、皮下、肌内、血管内或输注),例如无菌水溶液或油溶液或混悬液。一般来说,上述组合物可以采用常规赋形剂以常规方法制备。
与一种或多种赋形剂结合以制备单一剂型的活性成分(即式I的醚衍生物或其可药用的盐)的用量需要根据接受治疗的对象和具体的给药途径变化。例如,给人口服的制剂一般含有例如0.5mg至2g与适当和常规用量的赋形剂结合的活性成分,赋形剂的含量可以占组合物总重量的约5%至约98%。单位剂量形式一般含有约1mg至约500mg的活性成分。
本发明的另一个特征是提供了用于治疗人或动物体的方法的式I醚衍生物或其可药用的盐。
本发明还包括单独或者部分由一种或多种白三烯介导的疾病的治疗方法,该方法包括给需要这种治疗的温血动物施用有效量的上述活性成分。本发明还提供了这种活性成分在制备用于白三烯介导的疾病或医学疾病的新药物的用途。
治疗或预防用的式I化合物的剂量大小自然是根据疾病的性质和严重程度、动物或患者的年龄和性别以及给药途径、根据已知的药学原理而变化。如上所述,式I化合物适用于治疗例如变应性疾病和炎性疾病,以及单独或者部分由于通过线性(5-LO催化的)途径(特别是由5-LO介导产生的白三烯)升高的花生四烯酸的代谢作用造成的骨代谢疾病。如前所述,这些疾病包括例如气喘、变应性反应、变应性鼻炎、变应性休克、牛皮癣、变应性皮炎、心血管疾病和脑血管疾病、关节炎和炎性关节疾病、肠炎、结膜炎、休克或损伤疾病以及各种骨代谢疾病。
在使用式I化合物进行治疗或预防时,一般是以例如每公斤体重0.5mg-75mg的日剂量施用,如果需要可以以分次剂量给药。当经非肠道途径使用时,一般以较低的剂量施用。因此,例如对于静脉注射来说,一般使用的剂量范围是例如每公斤体重0.5mg-30mg。同样地,对于吸入给药来说,使用的剂量范围是例如每公斤体重0.5mg-25mg。
尽管式I化合物主要是温血动物(包括人)用的有价值的治疗剂,但是无论何时需要,它们也适用于抑制5-LO酶。因此,它们适合于作为药学标准,用于发展新的生物学试验以及用于研究新的药学试剂。
由于其对白三烯的作用,式I化合物具有某些细胞保护作用,例如它们适用于降低或抑制某些环氧合酶抑制非甾族抗炎剂(NSAIA)(如消炎痛、乙酰水杨酸、布洛芬、苏灵大、甲苯酰吡酸和炎痛喜康)的胃肠道副作用。再者,式I的5-LO抑制剂与NSAIA联合施用可以降低产生治疗作用所需的后一试剂的用量,因而降低了产生副作用的可能性。本发明的另一特征是提供了一种药物组合物,该组合物含有与环氧合酶抑制非甾族抗炎剂(如上述那些药物)结合或混合的式I的醚衍生物或其可药用的盐,以及可药用的稀释剂或载体。
式I化合物的细胞保护作用可以例如采用标准实验模型进行证明,该模型用于评估对在大鼠胃肠道中由消炎痛引起的或由乙醇引起的溃疡形成的保护作用。
另外,本发明的组合物可以含有一种或多种已知治疗这种疾病有价值的治疗剂或预防剂。因此,例如已知的血小板凝集抑制剂、降脂血剂、抗高血压剂、β肾上腺素能阻滞剂或血管舒张药也可以包括在本发明的药物组合物中,用于治疗心脏或血管疾病。同样地,例如抗组胺药、类固醇(例如二丙酸氯地米松)、色甘酸钠、磷酸二酯酶抑制剂或β肾上腺素能兴奋药也可以包括在本发明的药物组合物中,用于治疗肺病。
本发明将在下列非限制性实施例中进行说明,其中,除非另外指明:-
(i)蒸发通过真空旋转蒸发进行,并且在过滤除去固体残余物后进行处理过程;
(ii)在室温、即18-25℃以及在惰性气氛如氩气氛下进行操作;
(iii)在由E.Merck,Darmstadt,Germany得到的MerckKieselgel硅胶(Art.9385)或Merck lichroprep RP-18(Art.9303)反相硅胶上进行柱色谱(闪式方法)和中压液相色谱;
(iv)产率仅仅是说明性的,而不一定是可以得到的最大产率;
(v)式I的终产物的微量分析是令人满意的,并且其结构通过核磁共振(NMR)和质谱技术得到证实;除非另外指明,采用式I终产物的CDCl3溶液进行NMR谱数据的测定,化学迁移值以δ标度测量;采用下列缩写:s,单峰;d,双峰;t,三峰;q,四峰;m,多峰;
(vi)中间体的特征一般没有完全测定,并且纯度通过薄层色谱、红外(IR)或NMR分析进行评估;
(vii)熔点没有校准,采用Mettler SP62自动熔点装置或油浴装置测定;式I终产物的熔点是在从单一的常规有机溶剂如乙醇、甲醇、丙酮、乙醚或己烷中或其混合物中结晶后测定的;和
(viii)采用下列缩写:-
NMP N-甲基吡咯烷-2-酮;
DMF N,N-二甲基甲酰胺;
THF 四氢呋喃;
DMSO 二甲基亚砜。实施例1
将6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮(0.19g)、(2S,4R)-4-(3,5-二氟苯基)-4-羟基-2-甲基四氢吡喃(0.23g)、一水合氢氧化锂(0.05g)和NMP(1ml)的混合物搅拌并加热至130℃,保持6小时。将混合物冷却至室温,在乙醚和水之间进行分配。有机相用水洗涤,用硫酸镁干燥并蒸发。用持续增加极性的己烷和乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到(2S,4R)-4-[5-氟-3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃(0.065g,16%),熔点181-183℃(从甲醇中重结晶)。NMR Spectrum:1.2(d,3H),1.5-1.75(m,3H),2.05(m,1H),2.65(m,2H),2.9(m,2H),3.38(s,3H),3.85-4.0(m,3H),6.7(m,1H),7.0(m,2H),7.2(m,1H),7.28(m,1H),7.36(m,1H).
如下制备用作原料的6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮:-
向搅拌着的二-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基)二硫化物(欧洲专利申请0462812,其中的实施例7;38.4g)、三苯膦(29g)和1,4-二噁烷(300ml)的混合物中加入浓盐酸(5滴)和水(50ml)的混合物。在室温搅拌该混合物30分钟。将该混合物蒸发浓缩,使体积减少约一半。残余物在乙酸乙酯和0.5N氢氧化钠水溶液之间进行分配。水相用乙醚洗涤,然后加入稀盐酸水溶液酸化至pH2。用乙酸乙酯萃取酸性混合物。用硫酸镁干燥有机相并蒸发。将残留的油溶于乙醚中并加入己烷。由此得到固体的6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮(35.5g,92%),该产物不需进一步纯化即可使用。
如下制备用作原料的(2S,4R)-4-(3,5-二氟苯基)-4-羟基-2-甲基四氢吡喃:-
采用与EP 0462813实施例9有关原料制备部分的第一段所述类似的方法,将由3,5-二氟溴苯得到的格利雅试剂与(2S)-2-甲基四氢吡喃-4-酮(EP 0385662,其中的实施例20)进行反应,得到油状的(2S,4R)-4-(3,5-二氟苯基)-4-羟基-2-甲基四氢吡喃,产率25%。实施例2
在室温将(2S,4R)-4-[5-氟-3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃(0.095g)、过一硫酸钾(0.22g)、乙醇(2ml)和水(2ml)的混合物搅拌18小时。将混合物在乙酸乙酯和水之间进行分配。有机相用硫酸镁干燥并蒸发。用持续增加极性的二氯甲烷和乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到(2S,4R)-4-[5-氟-3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)苯基]-4-羟基-2-甲基四氢吡喃(0.06g,58%),熔点151-154℃(从己烷和乙酸乙酯的混合物中重结晶)。NMR Spectrum:1.22(d,3H),1.55-1.80(m,3H),2.08(m,1H),2.69(m,2H),2.97(m,2H),3.36(s,3H),3.85-4.0(m,3H),7.07(m,1H),7.4(m,1H),7.5(m,1H),7.72(m,1H),7.8-7.9(m,2H).实施例3
将6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮(0.052g)加入(2S,4R)-4-(2-氯噻唑-5-基)-4-羟基-2-甲基四氢吡喃(0.062g)、碳酸钾(0.04g)和NMP(2ml)的混合物中。将该混合物搅拌并加热至100℃,保持90分钟。将混合物冷却至室温,在乙酸乙酯和水之间进行分配。有机相用盐水洗涤、用硫酸镁干燥并蒸发。用石油醚(沸点40-60℃)和乙酸乙酯的1∶3混合物作洗脱剂,经柱色谱纯化残余物。由此得到泡沫状的(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻唑-5-基]四氢吡喃(0.064g,62%);NMR Spectrum:1.17(d,3H),1.6-1.9(m,4H),2.05(m,1H),2.7(m,2H),2.95(m,2H),3.4(s,3H),3.9(m,3H),7.05(d,1H),7.45(d,1H),7.5(s,1H),7.55(m,1H).
如下制备用作原料的(2S,4R)-4-(2-氯噻唑-5-基)-4-羟基-2-甲基四氢吡喃:-
向冷却至0℃并搅拌着的2-氨基噻唑(10g)在浓盐酸(50ml)中的溶液中滴加饱和亚硝酸钠(6.9g)水溶液。在0℃搅拌该混合物75分钟。分批加入氯化亚铜(9.9g),保持反应温度为0℃,并搅拌该混合物2.5小时。加入10N氢氧化钠水溶液中和该混合物。将该混合物在乙醚和水之间进行分配。有机相用盐水洗涤、用硫酸镁干燥并蒸发。通过蒸馏纯化残余物。由此得到2-氯噻唑(3.95g,在68mmHg的沸点为68℃)。
用10分钟,向冷却至-78℃的乙醚(5ml)中同时加入2-氯噻唑(0.5g)的乙醚(4ml)溶液和正丁基锂(2.5M的己烷溶液,1.8ml)。搅拌该混合物3.5小时并使其温热至-20℃。将混合物再冷却至-78℃,加入(2S)-2-甲基四氢吡喃-4-酮[EP0385662(其中的实施例20);0.43g]的乙醚(4ml)溶液。加入5%氯化铵水溶液,用乙醚萃取该混合物。有机相用盐水洗涤、用硫酸镁干燥并蒸发。用石油醚(沸点40-60℃)和乙酸乙酯的1∶1混合物作洗脱剂,经柱色谱纯化残余物。由此得到油状(2S,4R)-4-(2-氯噻唑-5-基)-4-羟基-2-甲基四氢吡喃(0.11g,13%)。实施例4
向冷却至0℃并搅拌着的(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻唑-5-基]四氢吡喃(0.28g)的二氯甲烷(5ml)溶液中加入3-氯过苯甲酸(0.493g)。将该混合物温热至室温并搅拌16小时。加入饱和碳酸氢钠水溶液,用乙醚萃取该混合物。有机相用盐水洗涤、用硫酸镁干燥并蒸发。用石油醚(沸点40-60℃)和乙酸乙酯的1∶4混合物作洗脱剂,经柱色谱纯化残余物。由此得到油状(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)噻唑-5-基]四氢吡喃(0.22g,72%);NMR Spectrum:1.20-1.22(d,3H),1.69-1.75(m,1H),1.80-1.84(m,1H),1.88-1.94(m,1H),2.06-2.09(m,1H),2.31(s,1H),2.67-2.71(m,2H),2.97-3.01(m,2H),3.37(s,3H),3.87-3.95(m,3H),7.10-7.12(d,1H),7.75(s,1H),7.86-7.88(m,1H),7.96-7.99(m,1H).实施例5
将6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮(1.15g)、(2S,4R)-4-羟基-4-(2-巯基噻吩-4-基)-2-甲基四氢吡喃(0.92g)、氯化亚铜(0.12g)、碳酸钾(1.66g)和DMF(40ml)的混合物搅拌并加热至130℃,保持2小时。将该混合物冷却至室温并在乙酸乙酯和水之间分配。有机相用水和盐水洗涤、用硫酸镁干燥并蒸发。用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到泡沫状(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃(0.6g);NMR Spectrum:1.2(d,3H),1.7(m,3H),2.1(m,1H),2.6(m,2H),2.9(m,2H),3.3(s,3H),3.9(m,3H),6.9(d,1H),7.2(m,4H).
如下制备用作原料的6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮:-
在3.5个大气压的氢气氛下将1-甲基-2-氧代-1,2-二氢喹啉-2-酮(EP 0420511,其中的实施例1;8g)、10%披钯炭催化剂(2g)和乙醇(60ml)的混合物搅拌24小时。过滤并蒸发该混合物。用二氯甲烷和乙醚的9∶1混合物作洗脱剂,经柱色谱纯化残余物。由此得到油状的1-甲基-2-氧代-1,2,3,4-四氢喹啉-2-酮(7.88g,98%)。
将由此得到的一部分产物(1.2g)、一氯化碘(1.9g)和冰醋酸(25ml)的混合物搅拌并加热至80℃,保持2小时。将混合物冷却至室温并倒入稀硫代硫酸钠水溶液中。加入碳酸氢钠中和该混合物并用乙酸乙酯萃取。有机相用盐水洗涤、用硫酸镁干燥并蒸发。残余物从己烷和乙酸乙酯的混合物中重结晶。由此得到6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮(1.09g,51%);NMR Spectrum:(CD3SOCD3)2.65(t,2H),2.85(t,2H),3.2(s,3H),6.9(d,1H),7.6(m,2H).
如下制备用作原料的(2S,4R)-4-羟基-4-(2-巯基噻吩-4-基)-2-甲基四氢吡喃:-
向冷却至-75℃并搅拌着的2,4-二溴噻吩(J.Org.Chem.,1988,53,417;10g)和乙醚(150ml)的混合物中加入正丁基锂(1.6M的己烷溶液,28.2ml)。在-75℃搅拌该混合物1小时。加入二甲二硫(3.1g)的乙醚(10ml)溶液,用1小时使该混合物温热至-20℃。将混合物倒入水中。有机相用盐水洗涤、用硫酸镁干燥并蒸发。由此得到4-溴-2-甲基硫代噻吩(8.9g)。
向冷却至-70℃的、如此得到的一部分物质(4.9g)的乙醚(70ml)溶液中加入正丁基锂(1.6M的己烷溶液,14.7ml)。在-70℃搅拌该混合物30分钟。加入(2S)-2-甲基四氢吡喃-4-酮(2g)的乙醚(5ml)溶液。在-70℃搅拌该混合物30分钟,然后使其温热至室温。将混合物倒入水中。有机相用水和盐水洗涤、用硫酸镁干燥并蒸发。用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到(2S,4R)-4-羟基-2-甲基-4-(2-甲基硫代噻吩-4-基)四氢吡喃(0.65g);NMR Spectrum:1.2(d,3H),1.7(m,3H),2.0(m,2H),2.5(s,3H),3.9(m,3H),7.1(m,2H),和(2S,4S)-4-羟基-2-甲基-4-(2-甲基硫代噻吩-4-基)四氢吡喃(2.45g);NMR Spectrum:(CDCl3+D2O)1.2(d,3H),1.65(m,1H),1.95(m,1H),2.2(m,2H),2.5(s,3H),3.4(m,2H),3.95(m,1H),7.1(m,2H).
重复上述反应后,将所得产物(1.1g)、甲硫醇(methanethiolate)钠(0.42g)和DMF(20ml)的混合物搅拌并加热至130℃,保持90分钟。将混合物冷却至室温,并在乙酸乙酯和水之间分配。有机相用水和盐水洗涤、用硫酸镁干燥并蒸发。由此得到液体的(2S,4R)-4-羟基-4-(2-巯基噻吩-4-基)-2-甲基四氢吡喃(0.92g),该产物不需进一步纯化即可使用。实施例6
将6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮(0.23g)、(2S,4R)-4-羟基-4-(3-碘苯基)-2-甲基四氢吡喃(0.35g)、碳酸钾(0.2g)、氯化亚铜(0.05g)和DMF(3ml)的混合物搅拌并加热至120℃,保持2小时。将混合物冷却至室温,并在乙酸乙酯和水之间分配。有机相用硫酸钠干燥并蒸发。用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃(0.33g),熔点135-137℃(从己烷和乙酸乙酯的混合物中重结晶);NMR Spectrum:1.21(d,3H),1.55-1.8(m,4H),2.1(m,1H),2.66(m,2H),2.86(m,2H),3.35(s,3H),3.85-4.05(m,3H),6.93(d,1H),7.13(m,1H),7.2-7.4(m,4H),7.47(m,1H).
如下制备用作原料的(2S,4R)-4-羟基-4-(3-碘苯基)-2-甲基四氢吡喃:-
向冷却至-70℃并搅拌着的1,3-二碘苯(19.8g)的THF(200ml)溶液中滴加正丁基锂(1.5M的己烷溶液,40ml)。在-70℃搅拌该混合物12分钟。加入(2S)-2-甲基四氢吡喃-4-酮(5.7g)。搅拌该混合物,使其温热至室温并在室温搅拌1小时。通过加入冰醋酸使该混合物酸化,并将其在乙醚和水之间分配。有机相用盐水洗涤、用硫酸钠干燥并蒸发。将在残余物的乙醚(50ml)溶液加到冷却至0℃的浓硫酸(35%v/v,200ml)中。搅拌混合物并使其温热至室温。在室温搅拌该混合物3小时。将混合物倒入碎冰中,并用乙醚萃取。有机相用水、饱和碳酸氢钠水溶液和盐水洗涤,用硫酸钠干燥并蒸发。用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到(2S,4R)-4-羟基-4-(3-碘苯基)-2-甲基四氢吡喃(12g,75%)。实施例7
采用与实施例2所述类似的方法,将(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃氧化,得到(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)苯基]四氢吡喃,产率82%;NMR Spectrum:1.22(d,3H),1.5-1.95(m,4H),2.1(m,1H),2.66(m,2H),2.96(m,2H),3.35(s,3H),3.85-4.05(m,3H),7.06(m,1H),7.50(m,1H),7.65-7.75(m,2H),7.8-7.9(m,2H).实施例8
采用与实施例6所述类似的方法,将1-乙基-6-巯基-1,2,3,4-四氢喹啉-2-酮与(2S,4R)-4-羟基-4-(3-碘苯基)-2-甲基四氢吡喃进行反应,得到(2S,4R)-4-[3-(1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃,产率55%,熔点148-149℃(从乙醚中重结晶);NMR Spectrum:1.15-1.3(m,6H),1.55-1.8(m,4H),2.1(m,1H),2.65(m,2H),2.85(m,2H),3.85-4.05(m,5H),6.96(d,1H),7.12(m,1H),7.2-7.4(m,4H),7.48(m,1H).如下制备用作原料的1-乙基-6-巯基-1,2,3,4-四氢喹啉-2-酮:-
在5个大气压的氢气氛下将2-羟基喹啉(12g)、10%披钯炭催化剂(5g)和乙醇(150ml)的混合物搅拌36小时。过滤并蒸发该混合物。由此得到1,2,3,4-四氢喹啉-2-酮(9.0g),熔点156-158℃。
向冷却至0℃并搅拌着的1,2,3,4-四氢喹啉-2-酮(0.64g)、碘乙烷(1.7ml)和DMF(5ml)的混合物中加入氢化钠(在矿物油中的50%分散液,0.23g)。将该混合物温热至室温并搅拌90分钟。通过加入冰醋酸使混合物酸化。将混合物在乙醚和水之间分配。有机相用盐水洗涤、用硫酸钠干燥并蒸发。用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到油状的1-乙基-1,2,3,4-四氢喹啉-2-酮(0.7g,91%)。
将如此得到的物质与氯磺酸(1.2ml)的混合物搅拌并加热至60℃,保持1小时。将混合物倒入碎冰中,用二氯甲烷萃取。有机相用硫酸钠干燥并蒸发。由此得到1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰氯(0.7g,70%),该产物不需进一步纯化即可使用。
将如此得到的物质、三甲基甲硅烷基氯(2.27ml)、碘化钾(3.4g)和乙腈(20ml)的混合物在室温搅拌72小时。加入水,并通过加入碳酸钾使该混合物碱化至pH8。滴加焦亚硫酸钠以除去因碘的存在所导致的棕色颜色反应。混合物用乙酸乙酯萃取。有机相用硫酸钠干燥并蒸发。由此得到胶状的二-(1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基)二硫化物(0.52g),该产物不需进一步纯化即可使用。
采用与实施例1第一段有关原料制备部分所述类似的方法,将二-(1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基)二硫化物还原,得到胶状的1-乙基-6-巯基-1,2,3,4-四氢喹啉-2-酮,产率70%。实施例9
采用与实施例4所述类似的方法,将(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃氧化,得到(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)噻吩-4-基]四氢吡喃,产率52%,熔点64-66℃,从乙醚和二氯甲烷的50∶1混合物中重结晶后熔点为110℃;NMR Spectrum:1.2(d,3H),1.6-1.82(m,3H),1.96-2.05(m,1H),2.68(m,2H),2.96(m,2H),3.36(s,3H),3.83-3.91(m,3H),7.07(d,1H),7.51(s,1H),7.70(s,1H),7.76(m,1H),7.88(m,1H).实施例10
采用与实施例6所述类似的方法,将6-巯基-1,2,3,4-四氢喹啉-2-酮与(2S,4R)-4-羟基-4-(3-碘苯基)-2-甲基四氢吡喃进行反应,得到(2S,4R)-4-羟基-2-甲基-4-[3-(2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃,产率25%,熔点180-181℃(从乙酸乙酯中重结晶);NMR Spectrum:1.21(d,3H),1.55-1.8(m,4H),2.1(m,1H),2.64(m,2H),2.90(m,2H),3.85-4.05(m,3H),6.72(d,1H),7.1(m,1H),7.2-7.4(m,4H),7.45(m,1H),7.9(broad s,1H).
如下制备用作原料的6-巯基-1,2,3,4-四氢喹啉-2-酮:-
采用与实施例8有关原料制备的第3、4和5段所述类似的方法,将1,2,3,4-四氢喹啉-2-酮依次转化为:-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰氯,产率82%,熔点205-208℃;二-(2-氧代-1,2,3,4-四氢喹啉-6-基)二硫化物,产率95%,熔点264-265℃;和6-巯基-1,2,3,4-四氢喹啉-2-酮,产率70%,熔点154-156℃。实施例11
采用与实施例2所述类似的方法,将(2S,4R)-4-羟基-2-甲基-4-[3-(2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃氧化,得到(2S,4R)-4-羟基-2-甲基-4-[3-(2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)苯基]四氢吡喃,产率51%,熔点178-180℃(从乙酸乙酯中重结晶);NMR Spectrum:1.2(d,3H),1.55-1.8(m,3H),1.9(s,1H),2.1(m,1H),2.65(m,2H),3.0(m,2H),3.85-4.05(m,3H),6.85(d,1H),7.5(t,1H),7.68(m,1H),7.7-7.9(m,3H),8.1(m,1H),8.4(broad s,1H).实施例12
采用与实施例2所述类似的方法,将(2S,4R)-4-[3-(1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃氧化,得到(2S,4R)-4-[3-(1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)苯基]-4-羟基-2-甲基四氢吡喃氧化,产率97%,熔点122-124℃(从乙醚中重结晶);NMR Spectrum:1.15-1.3(m,6H),1.55-1.8(m,4H),2.1(m,1H),2.65(m,2H),2.95(m,2H),3.85-4.05(m,5H),7.1(d,1H),7.5(t,1H),7.6-7.75(m,2H),7.9(m,2H),8.12(m,1H).实施例13
采用与实施例6所述类似的方法,将7-氟-6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮与(2S,4R)-4-羟基-4-(3-碘苯基)-2-甲基四氢吡喃进行反应,得到(2S,4R)-4-[3-(7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃,产率55%,熔点90-92℃(从乙醚中重结晶);NMR Spectrum:1.2(d,3H),1.55-1.8(m,4H),2.1(m,1H),2.65(m,2H),2.85(m,2H),3.35(s,3H),3.85-4.05(m,3H),6.78(d,1H),7.1(m,1H),7.2-7.35(m,3H),7.45(m,1H).
如下制备用作原料的7-氟-6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮:-
在室温将2,4-二氟苯甲醛(2.84g)、膦酰乙酸三甲酯(4.5g)、碳酸钾(3.1g)和水(2ml)的混合物剧烈搅拌18小时。加入水(50ml),分离沉淀并干燥。由此得到2,4-二氟肉桂酸甲酯(3.2g,80%),熔点38-40℃。
在室温,将如此得到的一部分物质(3g)与甲胺(16.5g)的乙醇(50ml)溶液搅拌3小时。蒸发该混合物,用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到2,4-二氟-N-甲基肉桂酰胺(1.57g,45%),熔点142-143℃。
将如此得到的物质、10%披钯碳催化剂(0.2g)和乙醇(50ml)的混合物在氢气氛下搅拌2小时。将该混合物过滤,蒸发滤液,得到3-(2,4-二氟苯基)-N-甲基丙酰胺(1.46g,95%),熔点90-91℃。
向搅拌着的部分N-甲基丙酰胺(1.3g)的NMP(10ml)溶液中分批加入氢化钠(在矿物油中的60%分散液,0.3g),在室温搅拌该混合物3小时,在60℃搅拌30分钟。将该混合物冷却至室温,在乙酸乙酯和水之间分配。有机相用硫酸钠干燥并蒸发。用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到胶状的7-氟-1-甲基-1,2,3,4-四氢喹啉-2-酮(0.38g,32%)。
采用与实施例8有关原料制备的第3、4和5段所述类似的方法,将7-氟-1-甲基-1,2,3,4-四氢喹啉-2-酮依次转化为:-7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰氯,产率96%,熔点122-124℃;二-(7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基)二硫化物,产率95%,熔点163-165℃;和7-氟-6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮,产率83%,熔点114-115℃。实施例14
采用与实施例2所述类似的方法,将(2S,4R)-4-[3-(7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃氧化,得到(2S,4R)-4-[3-(7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)苯基]-4-羟基-2-甲基四氢吡喃,产率64%,熔点135-138℃(从乙醚中重结晶);NMR Spectrum:1.2(d,3H),1.55-1.8(m,4H),2.1(m,1H),2.65(m,2H),2.95(m,2H),3.32(s,3H),3.85-4.05(m,3H),6.7(d,1H),7.52(t,1H),7.75(m,1H),7.8-8.0(m,2H),8.15(m,1H).实施例15
采用与实施例6所述类似的方法,将5-氟-6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮与(2S,4R)-4-羟基-4-(3-碘苯基)-2-甲基四氢吡喃进行反应,得到(2S,4R)-4-[3-(5-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃,产率82%,熔点141-142℃(从己烷和乙酸乙酯的混合物中重结晶);NMR Spectrum:1.2(d,3H),1.55-1.8(m,4H),2.1(m,1H),2.65(m,2H),2.95(m,2H),3.35(s,3H),3.85-4.05(m,3H),6.78(d,1H),7.1(m,1H).
如下制备用作原料的5-氟-6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮:-
将2,6-二氟苯甲醛(2.84g)、膦酰乙酸三甲酯(4.5g)、碳酸氢钾(4g)和水(2ml)的混合物剧烈搅拌并加热至100℃,保持1小时。将该混合物冷却至室温。加入水(50ml)和己烷(5ml),搅拌该混合物形成不溶的胶,沉淀形成结晶。由此得到2,6-二氟肉桂酸甲酯(3.2g,81%),熔点48-50℃。
采用与实施例13有关原料制备部分的第2-5段所述类似的方法,将2,6-二氟肉桂酸甲酯依次转化为:-2,6-二氟-N-甲基肉桂酰胺,产率67%,熔点143-145℃;3-(2,6-二氟苯基)-N-甲基丙酰胺,产率95%,熔点115-117℃;5-氟-1-甲基-1,2,3,4-四氢喹啉-2-酮,产率74%,熔点55-57℃;5-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰氯,产率79%,熔点129-130℃;二-(5-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基)二硫化物,产率91%,熔点173-175℃;和5-氟-6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮,产率97%,熔点120-123℃。实施例16
采用与实施例2所述类似的方法,将(2S,4R)-4-[3-(5-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃氧化,得到(2S,4R)-4-[3-(5-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)苯基]-4-羟基-2-甲基四氢吡喃,产率63%,熔点149-151℃(从乙醚和乙酸乙酯的混合物中重结晶);NMR Spectrum:1.2(d,3H),1.55-1.9(m,4H),2.1(m,1H),2.65(m,2H),2.9(m,2H),3.35(s,3H),3.85-4.05(m,3H),6.9(d,1H),7.55(t,1H),7.75(m,1H),7.9-8.05(m,2H),8.15(m,1H).实施例17
采用与实施例6所述类似的方法,将8-氯-6-巯基-1,2,3,4-四氢喹啉-2-酮与(2S,4R)-4-羟基-4-(3-碘苯基)-2-甲基四氢吡喃进行反应,得到胶状的(2S,4R)-4-[3-(8-氯-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃,产率69%;NMR Spectrum:1.21(d,3H),1.55-1.8(m,4H),2.1(m,1H),2.64(m,2H),2.96(m,2H),3.85-4.05(m,3H),7.1-7.2(m,2H),7.25-7.45(m,3H),7.5(m,1H),7.8(broad s,1H).
如下制备用作原料的8-氯-6-巯基-1,2,3,4-四氢喹啉-2-酮:-
用45分钟,向冷却至0℃并搅拌着的2-氯苯胺(43ml)的二氯甲烷(20ml)溶液中滴加3-氯丙酰氯(19.1ml)的二氯甲烷(80ml)溶液。在0℃搅拌该混合物2小时。加入二氯甲烷(100ml),该混合物用水洗涤、用硫酸钠干燥并蒸发。残余物用己烷研制。由此得到3,2′-二氯丙酰苯胺(39.1g,90%),熔点79-80℃。
将如此得到的物质分批加入氯化铝(71.4g)中,搅拌该混合物并加热至120℃。添加完成后,将混合物加热至120℃,保持4小时。将该混合物冷却至80℃并倒入碎冰中。所得混合物用二氯甲烷萃取。有机溶液用饱和碳酸氢钠水溶液洗涤、用硫酸钠干燥并蒸发。由此得到8-氯-1,2,3,4-四氢喹啉-2-酮(28.1g,86%),熔点107-109℃。
采用与实施例8有关原料制备部分的第3、4和5段所述类似的方法,将8-氯-1,2,3,4-四氢喹啉-2-酮依次转化为:-8-氯-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰氯,产率84%,熔点185-189℃;二-(8-氯-2-氧代-1,2,3,4-四氢喹啉-6-基)二硫化物,产率100%,熔点156-160℃;和8-氯-6-巯基-1,2,3,4-四氢喹啉-2-酮,产率74%,熔点163-165℃(从己烷和乙醚的混合物中重结晶)。实施例18
向搅拌着的(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-4-[3-(8-氯-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-2-甲基四氢吡喃(0.25g)、甲基碘(0.015g)和DMF(3ml)的混合物中分批加入氢化钠(在矿物油中的50%分散液,0.05g)。在室温搅拌该混合物30分钟。将混合物在稀氯化铵水溶液和乙醚之间分配。有机相用盐水洗涤、用硫酸钠干燥并蒸发。将所得残余物与氟化四丁铵(0.2M的THF溶液,5ml)的混合物搅拌并加热至70℃,保持2小时。将该混合物蒸发,用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到胶状的(2S,4R)-4-[3-(8-氯-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃(0.18g,89%);NMR Spectrum:1.21(d,3H),1.55-1.8(m,4H),2.1(m,1H),2.58(m,2H),2.8(m,2H),3.45(s,3H),3.85-4.05(m,3H),7.04(m,1H),7.2-7.45(m,4H),7.55(m,1H).
如下制备用作原料的(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-4-[3-(8-氯-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-2-甲基四氢吡喃:-
向搅拌着的(2S,4R)-4-羟基-4-(3-碘苯基)-2-甲基四氢吡喃(1.64g)、1,4,7,10,13-五氧杂环十五烷(下文称作15-冠-5,0.05g)和THF(30ml)的混合物中分批加入氢化钠(在矿物油中的50%分散液,0.3g),在室温搅拌该混合物30分钟。加入叔丁基二甲基甲硅烷基氯(0.9g),将该混合物搅拌并加热至60℃,保持6小时。将混合物冷却至室温并在乙醚与稀氯化铵水溶液之间分配。有机溶液用盐水洗涤、用硫酸钠干燥并蒸发。用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到油状的(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-4-(3-碘苯基)-2-甲基四氢吡喃(1.9g,88%)。
采用与实施例6所述类似的方法,将如此得到的物质与8-氯-6-巯基-1,2,3,4-四氢喹啉-2-酮进行反应。由此得到胶状的(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-4-[3-(8-氯-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-2-甲基四氢吡喃,产率77%。实施例19
采用与实施例18所述类似的方法,将(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-2-甲基-4-[3-(2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃与烯丙基氯进行反应,并用氟化四丁基铵处理所得产物,得到(2S,4R)-4-[3-(1-烯丙基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃,产率86%,熔点107-109℃(从乙醚中重结晶);NMR Spectrum:1.2(d,3H),1.55-1.8(m,4H),2.1(m,1H),2.7(m,2H),2.9(m,2H),3.85-4.05(m,3H),4.55(m,2H),5.1-5.3(m,2H),5.75-6.0(m,1H),6.95(d,1H),7.12(d,1H),7.2-7.4(m,4H),7.5(m,1H).
采用与实施例6所述类似的方法,通过6-巯基-1,2,3,4-四氢喹啉-2-酮与(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-4-(3-碘苯基)-2-甲基四氢吡喃进行反应,得到用作原料的(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-2-甲基-4-[3-(2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃。由此得到胶状的所需原料,产率36%。实施例20
重复实施例19所述的方法,只是用3-三甲基甲硅烷基丙-2-炔基溴代替烯丙基氯。由此得到(2S,4R)-4-羟基-2-甲基-4-[3-(2-氧代-1-(2-丙炔基)-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃,产率22%,熔点110-113℃(从乙醚中重结晶);NMR Spectrum:1.21(d,3H),1.55-1.8(m,4H),2.1(m,1H),2.24(m,1H),2.7(m,2H),2.88(m,2H),3.85-4.05(m,3H),4.7(d,1H),7.1-7.4(m,6H),7.5(m,1H).实施例21
在室温将(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃(0.15g)、过一硫酸钾(0.12g)、水(1ml)和乙醇(2ml)的混合物剧烈搅拌30分钟。加入水(10ml),用乙酸乙酯萃取该混合物。有机溶液用硫酸钠干燥并蒸发。用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到胶状的(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基亚磺酰)苯基]四氢吡喃(0.06g,38%);NMR Spectrum:(CD3SOCD3+CD3CO2D)1.1(d,3H),1.45-1.7(m,4H),2.5(m,2H),2.9(m,2H),3.22(s,3H),3.7-4.0(m,3H),7.18(m,1H),7.4-7.65(m,5H),7.88(m,1H).实施例22
将(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃(0.1g)、乙酸酐(1ml)和冰醋酸(1ml)的混合物搅拌并加热至回流4小时。将混合物冷却至室温并在乙酸乙酯与水之间分配。有机相用硫酸钠干燥并蒸发。用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到胶状的(2S,4R)-4-乙酸基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃(0.05g,45%);NMR Spectrum:1.21(d,3H),1.65(m,1H),1.9-2.1(m,4H),2.4(m,2H),2.65(m,2H),2.87(m,2H),3.35(s,3H),3.7-4.05(m,3H),6.93(d,1H),7.1-7.4(m,6H).实施例23
将(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃(0.05g)、硫代甲醇钠(0.05g)和NMP(0.5ml)的混合物搅拌并加热至120℃,保持18小时。加入第二份硫代甲醇钠(0.05g),在120℃再搅拌该混合物24小时。将混合物冷却至室温并在乙酸乙酯与水之间分配。有机相用硫酸钠干燥并蒸发。用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2-二氢喹啉-6-基硫代)苯基]四氢吡喃(0.02g,40%),熔点134-137℃(从乙醚中重结晶);NMR Spectrum:1.2(d,3H),1.55-1.8(m,4H),2.1(m,1H),3.72(s,3H),3.85-4.05(m,3H),6.72(d,1H),7.12(m,1H),7.25-7.4(m,3H),7.5(m,1H),7.56-7.64(m,3H).实施例24
采用与实施例6所述类似的方法,将5-巯基-1-甲基二氢吲哚-2-酮与(2S,4R)-4-羟基-4-(3-碘苯基)-2-甲基四氢吡喃进行反应,得到(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代二氢吲哚-5-基硫代)苯基]四氢吡喃,产率55%,熔点110-111℃(从乙醚和乙酸乙酯的混合物中重结晶);NMR Spectrum:1.2(d,3H),1.55-1.8(m,4H),2.1(m,1H),3.22(s,3H),3.50(m,2H),3.85-4.05(m,3H),6.8(d,1H),7.05(m,1H),7.2-7.35(m,3H),7.4-7.5(m,2H).
如下制备用作原料的5-巯基-1-甲基二氢吲哚-2-酮:-
向在冰浴中冷却并搅拌着的2-氧代二氢吲哚(21.8g)和甲醇(40ml)的混合物中加入在水(5ml)和甲醇(55ml)的混合物中的氢氧化钾(46g)溶液。搅拌该混合物20分钟。向在冰浴中冷却并搅拌着的该混合物中滴加硫酸二甲酯(77ml)。使该混合物温热至室温并搅拌3小时。将该混合物过滤并蒸发滤液。残余物在乙酸乙酯与水之间分配。有机相用硫酸镁干燥并蒸发。用石油醚(沸点40-60℃)和乙酸乙酯的7∶3混合物作洗脱剂,经柱色谱纯化残余物。由此得到1-甲基-2-氧代二氢吲哚(18g,75%),熔点83-85℃。
向冷却至0℃并搅拌着的氯磺酸(6.2ml)中分批加入如此得到的一部分物质(1g)。将该混合物在0℃搅拌30分钟,然后加热至60℃,保持2小时。将混合物冷却至室温,倒入冰水混合物中,并用乙醚萃取。有机溶液用盐水洗涤、用硫酸镁干燥并蒸发。用石油醚(沸点40-60℃)和乙酸乙酯的33∶17混合物作洗脱剂,经柱色谱纯化残余物。由此得到1-甲基-2-氧代二氢吲哚-5-基磺酰氯(1.6g,87%),熔点159℃。
将一部分如此得到的物质(0.95g)加入氢碘酸水溶液(57%,2.7ml)中,搅拌该混合物并加热至100℃,保持90分钟。将该混合物冷却至室温,并分批加入焦亚硫酸氢钠以除去因碘的存在所导致的棕色颜色反应。将混合物在乙酸乙酯与饱和焦亚硫酸氢钠水溶液之间分配。分离沉淀物并进行干燥。由此得到二-(1-甲基-2-氧代二氢吲哚-5-基)二硫化物(0.6g,44%),熔点155℃。
向二-(1-甲基-2-氧代二氢吲哚-5-基)二硫化物(1g)、水(1.25ml)和1,4-二噁烷(15ml)的混合物中加入三苯膦(0.865g),搅拌该混合物并加热至100℃,保持16小时。蒸发掉大量的有机溶剂,通过加入2N氢氧化钠水溶液使该混合物碱化至pH12。碱性溶液用二氯甲烷洗涤,并通过加入2N盐酸水溶液使其酸化。用二氯甲烷萃取该含水混合物。所得有机溶液用盐水洗涤、用硫酸镁干燥并蒸发。由此得到5-巯基-1-甲基二氢吲哚-2-酮(0.83g,82%),熔点100-101℃。实施例25
采用与实施例5所述类似的方法,将7-氟-6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮与(2S,4R)-4-羟基-4-(2-巯基噻吩-4-基)-2-甲基四氢吡喃进行反应,得到胶状的4-[2-(7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]-4-羟基-2-甲基四氢吡喃,产率85%;NMR Spectrum:1.2(d,3H),1.55-1.85(m,4H),2.1(m,1H),2.65(m,2H),2.8(m,2H),3.3(s,3H),3.85-4.05(m,3H),6.7(d,1H),7.07(d,1H),7.25(m,2H).
如下制备用作原料的7-氟-6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮:-
将7-氟-1-甲基-1,2,3,4-四氢喹啉-2-酮(0.4g)、一氯化碘(0.6g)和冰醋酸(3ml)的混合物搅拌并加热至80℃,保持1小时。将该混合物冷却至室温,倒入稀焦亚硫酸氢钠水溶液中,并用乙酸乙酯萃取。有机溶液用硫酸钠干燥并蒸发。用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到7-氟-6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮(0.45g,66%),熔点125-126℃(从己烷与乙醚的混合物中重结晶)。实施例26
将7-氟-6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮(0.25g)、(2S,4R)-4-(2-氯噻唑-5-基)-4-羟基-2-甲基四氢吡喃(0.22g)、碳酸钾(0.2g)和DMF(2ml)的混合物搅拌并加热至100℃,保持2小时。将该混合物冷却至室温,在乙酸乙酯与水之间分配。有机溶液用硫酸钠干燥并蒸发。用持续增加极性的己烷与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。由此得到胶状的(2S,4R)-4-[2-(7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻唑-5-基]-4-羟基-2-甲基四氢吡喃(0.25g,61%);NMR Spectrum:1.2(d,3H),1.55-2.1(m,4H),2.2(s,1H),2.65(m,2H),2.9(m,2H),3.35(s,3H),3.8-4.0(m,3H),6.85(d,1H),7.42(d,1H),7.45(s,1H).实施例27
采用与实施例26所述类似的方法,只是用NMP代替DMF作为反应溶剂,将5-巯基-1-甲基二氢吲哚-2-酮与(2S,4R)-4-(2-氯噻唑-5-基)-4-羟基-2-甲基四氢吡喃进行反应,得到(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代二氢吲哚-5-基硫代)噻唑-5-基]四氢吡喃,产率27%,熔点140℃;NMR Spectrum:1.17-1.19(d,3H),1.63-1.69(m,1H),1.73-1.78(m,1H),1.82-1.87(m,1H),1.91(s,1H),1.98-2.04(m,1H),3.24(s,3H),3.56(s,2H),3.84-3.89(m,3H),6.86-6.88(d,1H),7.46(s,1H),7.52-7.53(m,1H),7.61-7.63(m,1H).实施例28
向搅拌着的(2S,4R)-4-羟基-4-(2-巯基噻吩-4-基)-2-甲基四氢吡喃(3.15g)的DMSO(25ml)溶液中分批加入叔丁醇钾(1.66g),在室温搅拌该混合物5分钟。加入5-溴-1-甲基二氢吲哚-2-酮(3.71g)和四(三苯膦)钯(O)(1.59g),将该混合物加热至100℃,保持75分钟。将混合物冷却至室温并在乙酸乙酯与冰水混合物之间进行分配。有机相用盐水洗涤、用硫酸镁干燥并蒸发。用持续增加极性的石油醚(沸点40-60℃)与乙酸乙酯的混合物作洗脱剂,经柱色谱纯化残余物。用二氯甲烷与丙酮的5∶1混合物作洗脱剂,经柱色谱进一步纯化如此得到的物质。由此得到(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代二氢吲哚-5-基硫代)噻吩-4-基]四氢吡喃(2.45g,48%),熔点125-127℃(从含有几滴二氯甲烷的乙醚中重结晶),从乙酸乙酯中进一步重结晶后的熔点为129.5-130.5℃;NMR Spectrum:1.15(d,3H),1.70(m,4H),1.95(m,1H),3.12(s,3H),3.40(s,2H),3.85(m,3H),6.68(d,1H),7.18(m,3H),7.27(d,1H).
如下制备用作原料的5-溴-1-甲基二氢吲哚-2-酮:-
向溴化钾(15.55g)的水(57ml)溶液中加入溴(3.36ml)。向加热至80℃并搅拌着的1-甲基-2-氧代二氢吲哚(9.58g)的水(285ml)溶液中滴加上述混合物。当添加完成式,将混合物冷却至室温,并分离沉淀。将如此得到的物质溶于乙酸乙酯中。有机溶液用水和盐水洗涤、用硫酸镁干燥并蒸发。用石油醚(沸点40-60℃)和乙醚的1∶1混合物作洗脱剂,经柱色谱纯化残余物。由此得到5-溴-1-甲基二氢吲哚-2-酮(11.3g,77%),熔点134-135℃。实施例29
向冷却至-50℃的(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-2-甲基-4-(2-噻吩基)四氢吡喃(0.624g)的THF(20ml)溶液中滴加正丁基锂(1.6M的己烷溶液,1.4ml)。在-25℃搅拌反应混合物90分钟。将混合物再冷却至-70℃,加入二-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基)二硫化物(0.8g)的THF(20ml)溶液。在-70℃搅拌该混合物1小时,然后使其温热至0℃。将混合物在乙醚与饱和氯化铵水溶液之间分配。有机相用盐水洗涤、用硫酸镁干燥并蒸发。用己烷和乙酸乙酯的5∶1混合物作洗脱剂,经柱色谱纯化残余物。将所得残余物(0.55g)的THF(20ml)溶液冷却至0℃,加入氟化四丁基铵(1M的THF溶液,1.6ml)。将混合物温热至室温并搅拌2小时。将混合物在乙醚与饱和碳酸氢钠水溶液之间分配。有机相用盐水洗涤、用硫酸镁干燥并蒸发。用己烷和乙酸乙酯的3∶7混合物作洗脱剂,经柱色谱纯化残余物。由此得到(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-5-基]四氢吡喃(0.393g,48%),熔点50-52℃。
如下制备用作原料的(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-2-甲基-4-(2-噻吩基)四氢吡喃:-
向在乙醚(7ml)中由2-溴噻吩(0.82g)和镁屑(0.15g)制备的格利雅试剂中加入(2S)-2-甲基四氢吡喃-4-酮(0.627g)的甲苯(1ml)溶液。在室温搅拌该混合物90分钟。将混合物在乙醚与饱和氯化铵水溶液之间分配。有机溶液用盐水洗涤、用硫酸镁干燥并蒸发。用己烷和乙酸乙酯的5∶1混合物作洗脱剂,经柱色谱纯化残余物。由此得到:-(2S,4R)-4-羟基-2-甲基-4-(2-噻吩基)四氢吡喃(0.48g,48%)和相应的(2S,4S)-异构体(0.35g,35%)。
重复前述步骤,向在冰浴中冷却并搅拌着的氢化钾(在矿物油中的35%分散液,0.56g)、1,4,7,10,13,16-六氧杂环十八烷(下文称作18-冠-6,0.012g)和THF(4ml)的混合物中加入(2S,4R)-异构体(0.81g)的THF(22ml)溶液。在0℃搅拌该混合物10分钟。加入叔丁基二甲基甲硅烷基氯(0.685g)的THF(4ml)溶液,在室温搅拌该混合物16小时。将混合物在乙醚和饱和碳酸氢钠水溶液之间分配。有机溶液用盐水洗涤、用硫酸镁干燥并蒸发。用己烷和乙酸乙酯的10∶1混合物作洗脱剂,经柱色谱纯化残余物。由此得到油状的(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-2-甲基-4-(2-噻吩基)四氢吡喃(0.63g,50%);NMR Spectrum:0.3(s,6H),0.85(s,9H),1.18(d,3H),1.6-1.72(m,1H),1.96-2.13(m,3H),3.61-3.95(m,3H),6.7-6.98(m,2H),7.20(m,1H).实施例30
采用与实施例4所述类似的方法,将(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-5-基]四氢吡喃氧化,得到(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)噻吩-5-基]四氢吡喃,产率59%,熔点80-82℃。实施例31
采用与实施例5所述类似的方法,将6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮与4-羟基-4-(2-巯基噻吩-4-基)-2,6-二甲基四氢吡喃进行反应,得到泡沫状的4-羟基-2,6-二甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃,产率62%;NMR Spectrum:1.2(d,6H),1.6-1.9(m,4H),2.6(m,2H),2.9(m,2H),3.3(s,3H),4.0(m,2H),6.9-7.3(m,5H);羟基和每个甲基都是顺式关系。
如下制备用作原料的4-羟基-4-(2-巯基噻吩-4-基)-2,6-二甲基四氢吡喃:-
重复实施例5有关原料制备部分的最后两段所述方法,只是用2,6-二甲基四氢吡喃-4-酮代替(2S)-2-甲基四氢吡喃-4-酮。由此得到所需的液体原料,该原料应立即使用,因为它会氧化成相应的二硫化物。实施例32
采用与实施例5所述类似的方法,将6-碘-1-甲基-1,2-二氢喹啉-2-酮(EP 0420511,其中的实施例1)与(2S,4R)-4-羟基-4-(2-巯基噻吩-4-基)-2-甲基四氢吡喃进行反应,得到泡沫状的(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2-二氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃,产率65%;NMR Spectrum:1.2(d,3H),1.6(s,1H),1.7-1.9(m,3H),2.1(m,1H),3.7(s,3H),4.0(m,3H),6.7(d,1H),7.3(d,3H),7.5(m,3H).实施例33
采用与实施例5所述类似的方法,将6-碘-1,8-二甲基-1,2,3,4-四氢喹啉-2-酮与(2S,4R)-4-羟基-4-(2-巯基噻吩-4-基)-2-甲基四氢吡喃进行反应,得到泡沫状的(2S,4R)-4-羟基-2-甲基-4-[2-(1,8-二甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃,产率60%;NMR Spectrum:1.2(d,3H),1.6(s,1H),1.65-1.9(m,4H),2.1(m,1H),2.3(s,3H),2.5(q,2H),2.8(q,2H),3.3(s,3H),3.9(m,3H),7.0(d,2H),7.3(d,2H).
如下制备用作原料的6-碘-1,8-二甲基-1,2,3,4-四氢喹啉-2-酮:-
将8-甲基喹啉(7g)、碘化钾(17ml)和乙腈(30ml)的混合物搅拌并加热至60℃,保持8天。将混合物冷却至室温,加入乙醚(100ml)。分离沉淀,用乙醚洗涤。由此得到碘化1,8-二甲基喹啉鎓(12g)。
向冷却至5℃并搅拌着的铁氰化钾(51g)在氢氧化钠水溶液(10%重量/体积,120ml)中的溶液中分批加入上述得到的物质。在室温搅拌该混合物4小时。将混合物在乙酸乙酯和水之间分配。有机溶液用水和盐水洗涤、用硫酸镁干燥并蒸发。由此得到1,8-二甲基-1,2-二氢喹啉-2-酮(6g,79%)。NMR Spectrum:(CD3SOCD3)2.7(s,3H),3.8(s,3H),6.6(d,1H),7.1(t,1H),7.4(d,1H),7.5(d,1H),7.8(d,1H).
在5个大气压的氢气氛下,将如此得到的物质、10%披钯碳催化剂(2g)和乙醇(200ml)的混合物搅拌36小时。由此得到1,8-二甲基-1,2,3,4-四氢喹啉-2-酮(5.4g,88%)。
将一部分如此得到的物质(0.28g)、一氯化碘(0.27g)和冰醋酸(3ml)的混合物搅拌并加热至80℃,保持3小时。加入另一份一氯化碘(0.27g),并将混合物加热至80℃,保持16小时。将混合物冷却至室温,倒入饱和碳酸氢钠水溶液中。所得混合物用乙酸乙酯萃取。有机溶液用水和盐水洗涤、用硫酸镁干燥并蒸发。由此得到6-碘-1,8-二甲基-1,2,3,4-四氢喹啉-2-酮(0.38g,79%)。NMR Spectrum:2.3(s,3H),2.5(m,2H),2.8(m,2H),3.3(s,3H),7.3(s,1H),7.4(s,1H).实施例34
采用与实施例4所述类似的方法,将(2S,4R)-4-羟基-2-甲基-4-[2-(1,8-二甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃氧化,得到(2S,4R)-4-羟基-2-甲基-4-[2-(1,8-二甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)噻吩-4-基]四氢吡喃,产率77%;NMR Spectrum:1.2(m,3H),1.6-1.9(m,4H),2.0(m,1H),2.4(s,3H),2.6(q,2H),2.9(q,2H),3.4(s,3H),3.9(m,3H),7.5(d,1H),7.6(s,1H),7.7(d,2H).实施例35
采用与实施例5所述类似的方法,将8-氟-6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮与(2S,4R)-4-羟基-4-(2-巯基噻吩-4-基)-2-甲基四氢吡喃进行反应,得到泡沫状的(2S,4R)-4-[2-(8-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]-4-羟基-2-甲基四氢吡喃,产率40%;NMR Spectrum:1.2(d,3H),1.7(m,6H),2.1(m,2H),2.6(m,2H),2.8(m,2H),3.4(d,3H),3.9(m,3H),6.8(d,2H),7.4(d,2H).
如下制备用作原料的8-氟-6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮:-
向冷却至0℃的乙酸酐(5.4g)中滴加甲酸(3g)。在0℃搅拌该混合物15分钟,然后加热至55℃,保持2小时。将混合物冷却至室温。加入THF(5ml)和2-氟苯胺(2.22g)的THF(15ml)溶液,并在室温搅拌该混合物2小时。将混合物蒸发,得到2′-氟-N-甲酰苯胺(2.8g),该产物不需进一步纯化即可使用。
向冷却至0℃并搅拌着的2′-氟-N-甲酰苯胺(2.8g)的THF(12ml)溶液中滴加甲硼烷:二甲二硫复合物(5ml)。将该混合物在0℃搅拌15分钟,然后加热至回流2.5小时。将混合物再冷却至0℃,并滴加甲醇(8ml)。在室温搅拌该混合物1小时。通过引入氯化氢气体将混合物酸化至pH 2,将所得混合物加热至回流1小时。蒸发该混合物,残余物在乙醚和稀氢氧化钠水溶液之间分配。有机溶液用水和盐水洗涤、用硫酸镁干燥并蒸发。由此得到液体的2-氟-N-甲基苯胺(1.59g,59%)。
完成上述步骤后,向冷却至0℃并搅拌着的3-氯丙酰氯(1.93g)的二氯甲烷(30ml)溶液中滴加2-氟-N-甲基苯胺(3.8g)。将该混合物在0℃搅拌1小时,并在3℃贮存16小时。该混合物用冷的1N盐酸溶液、水和盐水洗涤,用硫酸镁干燥并蒸发。由此得到3-氯-2′-氟-N-甲基丙酰苯胺(3.74g,57%)。
将如此得到的物质分批加入氯化铝(7.46g)中,将该混合物搅拌并加热至100℃,保持1小时。该混合物冷却至约80℃,倒入碎冰中。所得混合物用乙醚萃取。有机溶液用水和盐水洗涤、用硫酸镁干燥并蒸发。用己烷和乙酸乙酯的10∶3混合物作洗脱剂,经柱色谱纯化残余物。由此得到8-氟-1-甲基-1,2,3,4-四氢喹啉-2-酮(1.84g,60%);NMR-Spectrum:2.6(m,2H),2.9(m,2H),3.5(d,3H),7.0(m,3H).
将如此得到的四氢喹啉-2-酮、一氯化碘(2.15g)和冰醋酸(30ml)的混合物搅拌并加热至80℃,保持16小时。加入另一份一氯化碘(1.65g),将混合物加热至80℃,保持3小时。将混合物冷却至室温,缓缓倒入饱和碳酸氢钠水溶液中。用乙酸乙酯萃取该混合物。有机溶液用水和盐水洗涤、用硫酸镁干燥并蒸发。用己烷和乙酸乙酯的10∶3混合物作洗脱剂,经柱色谱纯化残余物。由此得到8-氟-6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮(1.1g,36%);NMR Spectrum:2.6(m,2H),2.9(m,2H),3.4(s,3H),7.2(m,2H).实施例36
采用与实施例5所述类似的方法,将6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮与4-羟基-4-(2-巯基噻吩-4-基)-2,2-二甲基四氢吡喃进行反应,得到泡沫状的4-羟基-2,2-二甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃,产率72%;NMR Spectrum:1.2(s,3H),1.47(s,3H),1.7-1.8(m,1H),1.85(s,2H),2.0-2.2(m,1H),2.6-2.68(m,2H),2.8-2.9(m,2H),3.3(s,3H),3.7-3.8(m,1H),4.02-4.18(m,1H),6.85-7.2(m,3H),7.28(m,2H).
如下制备用作原料的4-羟基-4-(2-巯基噻吩-4-基)-2,2-二甲基四氢吡喃:-
向冷却至-85℃并搅拌着的4-溴-2-甲基硫代噻吩(1.24g)的乙醚(35ml)溶液中滴加正丁基锂(1.4M的己烷溶液,4.2ml)。在-70℃搅拌该混合物1小时。加入2,2-二甲基四氢吡喃-4-酮(EP 0375404,其中的实施例48;0.75g)的乙醚(5ml)溶液。将该混合物在-70℃搅拌1小时,然后使其温热至-30℃。将该混合物倒入冰和饱和氯化铵水溶液的混合物中。有机溶液用盐水洗涤、用硫酸镁干燥并蒸发。用己烷和乙酸乙酯的10∶3混合物作洗脱剂,经柱色谱纯化残余物。由此得到4-羟基-2,2-二甲基-4-(2-甲基硫代噻吩-4-基)四氢吡喃(1.0g,66%)。NMR Spectrum:1.2(s,3H),1.46(s,3H),1.57-1.85(m,3H),2.0-2.2(m,1H),2.5(s,3H),3.7-3.8(m,1H),4.0-4.2(m,1H),7.08(d,1H),7.12(d,1H).
将一部分所得物质(0.27g)、甲硫醇钠(0.28g)和DMF(3ml)的混合物搅拌并加热至130℃,保持40分钟。将该混合物冷却至室温,并在乙酸乙酯和稀柠檬酸水溶液之间分配。有机溶液用盐水洗涤、用硫酸镁干燥并蒸发。由此得到4-羟基-4-(2-巯基噻吩-4-基)-2,2-二甲基四氢吡喃,该产物不需进一步纯化即可使用。实施例37
采用与实施例4所述类似的方法,将4-羟基-2,2-二甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃氧化,得到4-羟基-2,2-二甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)噻吩-4-基]四氢吡喃,产率95%;NMR Spectrum:1.2(s,3H),1.45(s,3H),1.65(m,1H),1.8(s,2H),2.02(m,1H),2.65(m,2H),2.95(m,2H),3.75(m,1H),4.05(m,1H),7.07(d,1H),7.5(d,1H),7.7(d,1H),7.76(d,1H),7.88(m,1H).实施例38
采用与实施例5所述类似的方法,将6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮与4-羟基-4-(2-巯基噻吩-4-基)-2,6-二甲基四氢吡喃进行反应,得到4-羟基-2,6-二甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃,产率28%;NMR Spectrum:1.1(s,3H),1.43(s,3H),1.5(broad s,1H),1.7(m,3H),2.09(m,1H),2.55(m,2H),2.79(m,2H),3.25(s,3H),4.15(m,2H),6.8(m,1H),7.05(m,1H),7.15(m,1H),7.2(m,2H).
如下制备用作原料的4-羟基-4-(2-巯基噻吩-4-基)-2,6-二甲基四氢吡喃:-
重复实施例36有关原料制备部分所述的方法,只是用2,6-二甲基四氢吡喃-4-酮代替2,2-二甲基四氢吡喃-4-酮。于是得到所需的原料,该原料不需进一步纯化即可使用。实施例39
采用与实施例4所述类似的方法,将4-羟基-2,6-二甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃氧化,得到4-羟基-2,6-二甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)噻吩-4-基]四氢吡喃,产率83%;NMR Spectrum:1.2(s,3H),1.5(s,3H),1.5-2.0(m,5H),2.12(m,1H),2.70(m,2H),2.96(m,2H),3.36(s,3H),4.2(m,2H),7.08(m,1H),7.5(d,1H),7.7(d,1H),7.75(m,1H),7.88(m,1H).实施例40
采用与实施例5所述类似的方法,将6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮与3-羟基-3-(2-巯基噻吩-4-基)-8-氧杂双环[3,2,1]辛烷进行反应,得到3-羟基-3-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]-8-氧杂双环[3,2,1]辛烷,产率28%;NMR Spectrum:1.7-1.8(m,4H),2.05(m,2H),2.25(m,2H),2.55(m,2H),2.85(m,2H),3.2(s,3H),4.35(m,2H),7.0-7.2(m,3H),7.25(d,1H),7.4(d,1H).
如下制备用作原料的3-羟基-3-(2-巯基噻吩-4-基)-8-氧杂双环[3,2,1]辛烷:-
在1个大气压的氢气氛下,将8-氧杂双环[3,2,1]辛-6-烯-3-酮(J.Chem.Res.(S),1981,246;3.2g)、10%披钯碳催化剂(0.45g)、水(2ml)和乙醇(25ml)的混合物搅拌5小时。将该混合物过滤并蒸发滤液。残余物在乙醚和盐水之间分配。有机相用硫酸镁干燥并蒸发。由此得到8-氧杂双环[3,2,1]辛-3-酮(2.84g)。
重复实施例36有关原料制备部分所述的方法,只是用8-氧杂双环[3,2,1]辛-3-酮代替2,2-二甲基四氢吡喃-4-酮。由此得到3-羟基-3-(2-巯基噻吩-4-基)-8-氧杂双环[3,2,1]辛烷,产率35%。实施例41
采用与实施例26所述类似的方法,只是向反应混合物中加入催化量(0.01g)的碘化钾,将6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮与4-(2-氯噻唑-5-基)-4-羟基-2,2-二甲基四氢吡喃进行反应,得到泡沫状的4-羟基-2,2-二甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻唑-5-基]四氢吡喃,产率92%;NMR Spectrum:1.05(s,3H),1.3(s,3H),1.55-1.9(m,4H),2.55(m,2H),2.9(m,2H),3.3(s,3H),3.55(m,1H),3.85(m,1H),5.6(broads,1H),7.0-7.5(m,4H).
如下制备用作原料的4-(2-氯噻唑-5-基)-4-羟基-2,2-二甲基四氢吡喃:-
将2-氯噻唑(0.75g)的乙醚(8ml)溶液和正丁基锂(1.4M的己烷溶液,4.5ml)同时、但是分别加入冷却至-80℃的乙醚(8ml)中。在-75℃搅拌该混合物10分钟,并使其温热至-30℃。将该混合物再冷却至-80℃,加入2,2-二甲基四氢吡喃-4-酮(0.76g)的乙醚(5ml)溶液。搅拌该混合物,并使其温热至-30℃。将该混合物倒入冰和饱和氯化铵水溶液的混合物中。有机溶液用盐水洗涤、用硫酸镁干燥并蒸发。用己烷和乙酸乙酯的10∶3混合物作洗脱剂,经柱色谱纯化残余物。由此得到油状的4-(2-氯噻唑-5-基)-4-羟基-2,2-二甲基四氢吡喃(0.67g,46%);NMR Spectrum:1.2(s,3H),1.45(s,3H),1.8-2.15(m,4H),3.75(m,1H),4.1(m,1H),7.38(s,1H).实施例42
采用与实施例26所述类似的方法,只是向反应混合物中加入催化量(0.01g)的碘化钾,将6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮与4-(2-氯噻唑-5-基)-4-羟基-2,6-二甲基四氢吡喃进行反应,得到泡沫状的4-羟基-2,6-二甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻唑-5-基]四氢吡喃,产率53%;NMR Spectrum:1.2(s,3H),1.5(s,3H),1.7-1.9(m,3H),2.15(m,1H),2.66 (m,2H),2.95(m,2H),3.39(s,3H),4.18(m,2H),7.0(d,1H),7.5(m,2H),7.55(m,1H).
如下制备用作原料的4-(2-氯噻唑-5-基)-4-羟基-2,6-二甲基四氢吡喃:-
重复实施例41有关原料制备部分所述的方法,只是用2,6-二甲基四氢吡喃-4-酮代替2,2-二甲基四氢吡喃-4-酮。由此得到所需的原料,产率77%。NMR Spectrum:1.2(s,3H),1.5(s,3H),1.7-2.0(m,3H),2.16(m,2H),4.2(m,2H),7.38(s,1H).实施例43
采用与实施例5所述类似的方法,将7-碘-1-甲基-2,3,4,5-四氢-1H-苯并[b]氮杂卓-2-酮与3-羟基-3-(2-巯基噻吩-4-基)-8-氧杂双环[3,2,1]辛烷进行反应,得到油状的3-羟基-3-[2-(1-甲基-2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂卓-7-基硫代)噻吩-4-基]-8-氧杂双环[3,2,1]辛烷,产率85%;NMR Spectrum:1.75-1.9(m,3H),1.95(m,2H),2.15(m,2H),2.25-2.4(m,6H),2.65(t,2H),3.3(s,3H),4.5(m,2H),7.08(m,3H),7.27(m,2H).
如下制备用作原料的7-碘-1-甲基-2,3,4,5-四氢-1H-苯并[b]氮杂卓-2-酮:-
用1小时向搅拌着的1,2,3,4-四氢萘-1-酮(8g)和多磷酸(110ml)的混合物中分批加入叠氮化钠(3.9g)。将该混合物缓缓温热至50℃并搅拌5小时。将混合物冷却至室温,倒入冰水混合物中。该混合物通过加入浓氢氧化钠水溶液(40%重量/体积)进行中和,并用二氯甲烷萃取。有机溶液用水洗涤、用硫酸镁干燥并蒸发。残余物用乙醚研制。由此得到2,3,4,5-四氢-1H-苯并[b]氮杂卓-2-酮(5.4g),熔点137-139℃。
向冷却至0℃并搅拌着的2,3,4,5-四氢-1H-苯并[b]氮杂卓-2-酮(3g)的DMF(100ml)溶液中分批加入氢化钠(在矿物油中的60%分散液,0.82g)。在10℃搅拌该混合物1小时。滴加碘化钾(2.64g)的THF(8ml)溶液,在室温搅拌该混合物2小时。蒸发混合物,残余物在乙酸乙酯和水之间分配。有机溶液用水洗涤、用硫酸钠干燥并蒸发。用甲苯和乙酸乙酯的1∶1混合物作洗脱剂,经柱色谱纯化残余物。由此得到液体的1-甲基-2,3,4,5-四氢-1H-苯并[b]氮杂卓-2-酮(2.25g,69%)。
将一部分如此得到的物质(2g)、碘(1.27g)、碘酸(1.86g)、浓硫酸(1.5ml)和乙酸(8.5ml)的混合物搅拌并加热至95℃,保持2小时。将混合物冷却至室温,并在乙酸乙酯和饱和碳酸氢钠水溶液之间分配。有机溶液用饱和硫代硫酸钠水溶液洗涤、用硫酸钠干燥并蒸发。用己烷和乙酸乙酯的3∶2混合物作洗脱剂,经柱色谱纯化残余物。由此得到7-碘-1-甲基-2,3,4,5-四氢-1H-苯并[b]氮杂卓-2-酮(2.1g),熔点124-126℃。实施例44
采用与实施例4所述类似的方法,将3-羟基-3-[2-(1-甲基-2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂卓-7-基硫代)噻吩-4-基]-8-氧杂双环[3,2,1]辛烷氧化,得到3-羟基-3-[2-(1-甲基-2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂卓-7-基磺酰)噻吩-4-基]-8-氧杂双环[3,2,1]辛烷,产率73%;NMR Spectrum:(CDCl3+CD3CO2D)1.8-2.0(m,4H),2.2-2.4(m,8H),2.75-2.86(t,2H),3.4(s,3H),4.5-4.6(m,2H),7.35(d,1H),7.55(d,1H),7.7(d,1H),7.85(d,1H),7.95(m,1H).实施例45
采用与实施例6所述类似的方法,将6-巯基-1-甲基-1,2,3,4-四氢喹啉-2-酮与(2SR,4RS)-2-乙基-4-羟基-4-(3-碘苯基)四氢吡喃进行反应,得到胶状的(2SR,4RS)-2-乙基-4-羟基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃,产率63%;NMR Spectrum:0.95(t,3H),1.4-1.8(m,6H),2.1(m,1H),2.65(m,2H),2.85(m,2H),3.35(s,3H),3.7(m,1H),3.95(m,2H),6.85(d,1H),7.12(m,1H),7.2-7.4(m,4H),7.5(m,1H).
通过重复实施例6有关原料制备部分的方法,只是用2-乙基四氢吡喃-4-酮(Chem.Ber.,1955,88,1053)代替(2S)-2-甲基四氢吡喃-4-酮,制备用作原料的(2SR,4RS)-2-乙基-4-羟基-4-(3-碘苯基)四氢吡喃。由此得到油状的所需原料,产率63%。实施例46
采用与实施例5所述类似的方法,将6-碘-1-甲基-2-氧代-1,2,3,4-四氢喹唑啉与(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-4-(3-巯基苯基)-2-甲基四氢吡喃进行反应,得到泡沫状的(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹唑啉-6-基硫代)苯基]四氢吡喃,产率41%。将如此得到的物质(0.12g)和氟化四丁基铵(1M的THF溶液,5ml)的混合物搅拌并加热至回流2小时。将混合物冷却至室温,在乙酸乙酯和水之间分配。有机溶液用硫酸钠干燥并蒸发。用持续增加极性的二氯甲烷和甲醇的混合物作洗脱剂,经柱色谱纯化残余物。由此得到胶状的(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹唑啉-6-基硫代)苯基]四氢吡喃(0.07g);NMR Spectrum:1.2(d,3H),1.5-1.8(m,4H),2.05(m,1H),3.3(s,3H),3.8-4.05(m,3H),4.38(s,2H),5.0(broad s,1H),6.82(d,1H),7.05(m,1H),7.12(m,1H),7.2-7.37(m,3H),7.42(m,1H).
如下制备用作原料的6-碘-1-甲基-2-氧代-1,2,3,4-四氢喹唑啉:-
在室温将5-碘邻氨基苯甲酸(10.5g)、N-羟基琥珀酰亚胺(4.92g)、N,N′-二环己基碳二亚胺(9g)和乙酸乙酯(200ml)的混合物搅拌2小时。将混合物过滤并蒸发滤液。加入饱和氨的甲醇(200ml)溶液,在室温搅拌该混合物18小时。将混合物蒸发,并在乙酸乙酯和1N氢氧化钠水溶液之间分配。有机溶液用盐水洗涤、用硫酸镁干燥并蒸发。由此得到5-碘邻氨基苯甲酰胺(6.7g),该产物不需进一步纯化即可使用。
在室温将如此得到的一部分物质(1.4g)与甲硼烷:THF复合物(1M的THF溶液,16ml)的混合物搅拌18小时。蒸发该混合物。向残余物中加入甲醇,将该混合物再蒸发。向残余物中加入2N盐酸水溶液(30ml),在室温搅拌该混合物30分钟,通过加入2N氢氧化钠水溶液使该混合物碱化至pH 9,用二氯甲烷萃取。有机溶液用盐水洗涤、用硫酸钠干燥并蒸发。残余物用乙醚研制。由此得到2-氨基-5-碘苯甲胺(0.95g,73%)。
重复上述反应后,将2-氨基-5-碘苯甲胺(2.1g)、1,1′-羰基二咪唑(1.64g)和THF(20ml)的混合物搅拌并加热至回流30小时。将混合物冷却至室温,分离沉淀。由此得到6-碘-2-氧代-1,2,3,4-四氢喹唑啉(1.88g,91%),熔点242-244℃。
在室温将一部分如此得到的物质(0.5g)、氢化钠(在矿物油中的60%分散液,0.07g)和DMF(5ml)的混合物搅拌30分钟。加入碘化钾(0.24ml),在室温搅拌该混合物2小时。将混合物在乙酸乙酯和饱和氯化铵水溶液之间分配。有机溶液用盐水洗涤、用硫酸钠干燥并蒸发。由此得到6-碘-1-甲基-2-氧代-1,2,3,4-四氢喹唑啉(0.3g,57%),熔点175-178℃。
如下制备用作原料的(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-4-(3-巯基苯基)-2-甲基四氢吡喃:-
采用与实施例6有关原料制备部分所述类似的方法,将1,3-二溴苯与(2S)-2-甲基四氢吡喃-4-酮进行反应,得到油状的(2S,4R)-4-羟基-4-(3-溴苯基)-2-甲基四氢吡喃,产率66%。
采用与实施例18有关原料制备部分所述类似的方法,将如此得到的4-(3-溴苯基)-2-甲基四氢吡喃与叔丁基二甲基甲硅烷基氯进行反应,得到油状的(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-4-(3-溴苯基)-2-甲基四氢吡喃,产率78%。
将如此得到的物质(0.96g)的THF(5ml)溶液冷却到-80℃,滴加正丁基锂(1.6M的己烷溶液,1.6ml)。在-80℃搅拌该混合物30分钟。加入硫(0.08g),并在-80℃搅拌该混合物30分钟。加入2N氢氧化钠水溶液(20ml),将混合物温热至室温。该混合物用乙醚洗涤,通过加入2N盐酸水溶液将其酸化至pH 3,用乙酸乙酯萃取。有机溶液用盐水洗涤、用硫酸钠干燥并蒸发。由此得到油状的(2S,4R)-4-(叔丁基二甲基甲硅烷氧基)-4-(3-巯基苯基)-2-甲基四氢吡喃(0.22g,26%),该产物不需进一步纯化即可使用。实施例47
采用与实施例5所述类似的方法,将6-碘-1,3-二甲基-2-氧代-1,2,3,4-四氢喹唑啉与(2S,4R)-4-羟基-4-(2-巯基噻吩-4-基)-2-甲基四氢吡喃进行反应,得到胶状的(2S,4R)-4-羟基-2-甲基-4-[2-(1,3-二甲基-2-氧代-1,2,3,4-四氢喹唑啉-6-基硫代)噻吩-4-基]四氢吡喃,产率74%;NMR Spectrum:1.2(d,3H),1.5-1.85(m,4H),2.05(m,1H),3.02(s,3H),3.3(s,3H),3.8-4.05(m,3H),4.32(s,2H),6.75(d,1H),7.05(d,1H),7.2-7.3(m,3H).
如下制备用作原料的6-碘-1,3-二甲基-2-氧代-1,2,3,4-四氢喹唑啉:-
向搅拌着的6-碘-2-氧代-1,2,3,4-四氢喹唑啉(0.55g)、碘化钾(0.71ml)和DMF(5ml)的混合物分批加入氢化钠(在矿物油中的60%分散液,0.24g),在室温搅拌所得混合物30分钟。将该混合物在乙酸乙酯和饱和氯化铵水溶液之间分配。有机溶液用盐水洗涤、用硫酸钠干燥发病蒸发。残余物用乙醚研制,由此得到6-碘-1,3-二甲基-2-氧代-1,2,3,4-四氢喹唑啉(0.38g,63%),该产物不需进一步纯化即可使用。实施例48
向搅拌着的(2S,4R)-4-羟基-4-(2-巯基噻吩-4-基)-2-甲基四氢吡喃(0.4g)的DMSO(5ml)溶液中加入叔丁醇钾(0.212g),在室温搅拌该混合物5分钟。依次加入6-碘-1-甲基-2-硫代-1,2,3,4-四氢喹啉(0.526g)和四(三苯膦)钯(O)(0.21g),将该混合物搅拌并加热至90℃,保持2.5小时。加入另一份钯催化剂(0.1g),将该混合物加热至90℃,再保持1小时。将混合物冷却至室温,在乙酸乙酯和水之间分配。有机相用盐水洗涤、用硫酸镁干燥并蒸发。用二氯甲烷和乙醚的1∶1混合物作洗脱剂,经柱色谱纯化残余物。由此得到固体的(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-硫代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃(0.09g,13%);NMR Spectrum:1.2(d,3H),1.7(m,3H),2.05(m,1H),2.72(m,2H),3.16(m,2H),3.87(s,3H),3.92(m,3H),7.02(m,2H),7.13(m,1H),7.30(m,2H).
如下制备用作原料的6-碘-1-甲基-2-硫代-1,2,3,4-四氢喹啉:-
将6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮(0.6g)、2,4-二(4-甲氧基苯基)-1,3-二硫杂-2,4-二膦(diphosphetane)-2,4-二硫化物(Lawesson′试剂,0.49g)和甲苯(7ml)的混合物搅拌并加热至100℃,保持1小时。将混合物冷却至室温并蒸发。用石油醚(沸点40-60℃)和二氯甲烷的4∶1混合物作洗脱剂,经柱色谱纯化残余物。由此得到6-碘-1-甲基-2-硫代-1,2,3,4-四氢喹啉(0.42g),熔点202-204℃;NMR Spectrum:2.76(m,2H),3.17(m,2H),3.86(s,3H),6.88(d,1H),7.50(d,1H),7.61(m,1H).实施例49
采用与实施例3所述类似的方法,将6-巯基-1-甲基-2-硫代-1,2,3,4-四氢喹啉与(2S,4R)-4-(2-氯噻唑-5-基)-4-羟基-2-甲基四氢吡喃进行反应,得到泡沫状的(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-硫代-1,2,3,4-四氢喹啉-6-基硫代)噻唑-5-基]四氢吡喃(0.365g,45%);NMR Spectrum:1.19(d,3H),1.6-2.1(m,5H),2.83(m,2H),3.22(m,2H),3.86(m,3H),3.90(s,3H),7.17(d,1H),7.44(d,1H),7.52(d,1H),7.55(m,1H).
如下制备用作原料的6-巯基-1-甲基-2-硫代-1,2,3,4-四氢喹啉:-
将二-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基)二硫化物(1g)、2,4-二(4-甲氧基苯基)-1,3-二硫杂-2,4-二膦-2,4-二硫化物(0.65g)和甲苯(10ml)的混合物搅拌并加热至回流1小时。将该混合物蒸发,用二氯甲烷作洗脱剂,经柱色谱纯化残余物。由此得到二-(1-甲基-2-硫代-1,2,3,4-四氢喹啉-6-基)二硫化物(0.86g,79%),熔点180-182℃。
采用与实施例1有关原料制备部分所述类似的方法,在浓盐酸存在下将如此得到的物质与三苯膦进行反应。由此得到固体的6-巯基-1-甲基-2-硫代-1,2,3,4-四氢喹啉,产率85%,该产物不需进一步纯化即可使用。实施例50
采用与实施例5所述类似的方法,将6-碘-1-甲基-1,2,3,4-四氢喹啉-2-酮与(2RS,3SR)-3-羟基-3-(2-巯基噻吩-4-基)-2-甲基四氢呋喃进行反应,得到泡沫状的(2RS,3SR)-3-羟基-2-甲基-3-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基)四氢呋喃,产率42%;NMR Spectrum:1.17(d,3H),2.0(broad s,1H),2.2-2.55(m,2H),2.64(m,2H),2.86(m,2H),3.32(s,3H),3.85-4.2(m,3H),6.9(d,1H),7.11(d,1H),7.19(m,1H),7.22(d,1H),7.37(d,1H).
如下制备用作原料的(2RS,3SR)-3-羟基-3-(2-巯基噻吩-4-基)-2-甲基四氢呋喃:-
将(2RS,3SR)-3-羟基-2-甲基-3-(2-甲基硫代噻吩-4-基)四氢呋喃(EP 0555068,其中的实施例80;0.44g)、甲硫醇钠(0.21g)和DMF(6.3ml)的混合物搅拌并加热至130℃,保持90分钟。将混合物冷却至室温,在乙酸乙酯和水之间分配。通过加入1M柠檬酸水溶液将混合物酸化。有机溶液用水和盐水洗涤、用硫酸镁干燥并蒸发,由此得到(2RS,3SR)-3-羟基-3-(2-巯基噻吩-4-基)-2-甲基四氢呋喃(0.36g,86%),该产物不需纯化即可使用。实施例51
下面对用于人的治疗或预防的、含有式I化合物或其可药用盐(下文称为化合物X)的典型药物剂型进行说明:(a)片剂I mg/片化合物X 100乳糖(Ph.Eur) 182.75Croscarmellose sodium 12.0玉米淀粉浆(5% w/v浆) 2.25硬脂酸镁 3.0(b)片剂II mg/片化合物X 50乳糖(Ph.Eur) 223.75Croscarmellose sodium 6.0玉米淀粉 15.0聚乙烯吡咯烷酮(5% w/v浆) 2.25硬脂酸镁 3.0(c)片剂III mg/片化合物X 1.0乳糖(Ph.Eur) 93.25Croscarmellose sodium 4.0玉米淀粉浆(5% w/v浆) 0.75硬脂酸镁 1.0(d)胶囊 mg/胶囊化合物X 10乳糖(Ph.Eur) 488.5硬脂酸镁 1.5(e)注射液I (50mg/ml)化合物X 5.0% w/v1M氢氧化钠溶液 15.0% v/v 0.1M盐酸(调节pH至7.6)聚乙二醇 4.5% w/v注射用水 至 100%(f)注射液II (10mg/ml)化合物X 1.0% w/v磷酸钠BP 3.6% w/v1M氢氧化钠溶液 15.0% v/v注射用水 至 100%(g)注射液III (1mg/ml,缓冲至pH6)化合物X 0.1% w/v磷酸钠BP 2.26% w/v柠檬酸 0.38% w/v聚乙二醇400 3.5% v/v注射用水 至 100%(h)气雾剂I mg/ml化合物X 10.0脱水山梨醇三油酸酯 13.5三氯氟甲烷 910.0二氯二氟甲烷 490.0(i)气雾剂II mg/ml 化合物X 0.2脱水山梨醇三油酸酯 0.27三氯氟甲烷 7.0二氯二氟甲烷 280.0二氯四氟乙烷 1094.0(j)气雾剂III mg/ml化合物X 2.5脱水山梨醇三油酸酯 3.38三氯氟甲烷 67.5二氯二氟甲烷 1086.0二氯四氟乙烷 191.6(k)气雾剂IV mg/ml化合物X 2.5大豆卵磷脂 2.7三氯氟甲烷 67.5二氯二氟甲烷 1086.0二氯四氟乙烷 191.6注:
上述制剂可以通过药物领域公知的方法得到。片剂(a)-(c)可以按常规方法进行肠包衣,例如包上乙酸邻苯二甲酸纤维素包衣。气雾剂(h)-(k)可以与标准的计量给药的气雾剂供给器结合使用,并且可以用替代的悬浮剂如脱水山梨醇一油酸酯、脱水山梨醇倍半油酸酯、多乙氧基醚、聚甘油油酸酯或油酸代替悬浮剂脱水山梨醇三油酸酯和大豆卵磷脂。
Claims (12)
1.式I的醚衍生物或其可药用的盐
Q1-X-Ar-Q2 I其中Q1是9-、10-或11-元双环杂环部分,该杂环部分含有一个或两个氮杂原子,并且Q1可以任意地带有至多4个取代基,这些取代基选自卤素、羟基、氰基、甲酰基、氧代、硫代、(1-4C)烷基、(3-4C)链烯基、(3-4C)链炔基、(1-4C)烷氧基、氟-(1-4C)烷基、羟基-(1-4C)烷基、(2-5C)链烷酰基、苯基、苯甲酰基和苄基,其中所述苯基、苯甲酰基和苄基取代基可以任意地带有一个或两个选自卤素、(1-4C)烷基和(1-4C)烷氧基的取代基;X是氧、硫、亚磺酰基或磺酰基;Ar是亚苯基、噻吩二基、噻唑二基、它们可以任意地带有1或2个选自卤素、氰基、三氟甲基、羟基、氨基、(1-4C)烷基、(1-4C)烷氧基、(1-4C)烷氨基和二-(1-4C)烷氨基的取代基;并且Q2是选自式II和III的基团: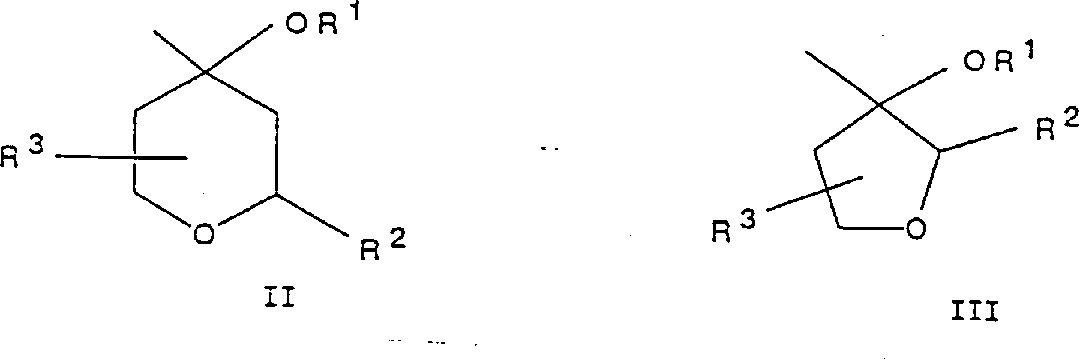 其中R1是氢、(2-5C)链烷酰基或苯甲酰基,并且其中所述苯甲酰基可以任意地带有1或2个选自卤素、(1-4C)烷基和(1-4C)烷氧基的取代基;R2是(1-4C)烷基;和R3是氢或(1-4C)烷基;或者R2与R3连结在一起形成亚甲基、1,2-亚乙烯基、亚乙基或三亚甲基。
其中R1是氢、(2-5C)链烷酰基或苯甲酰基,并且其中所述苯甲酰基可以任意地带有1或2个选自卤素、(1-4C)烷基和(1-4C)烷氧基的取代基;R2是(1-4C)烷基;和R3是氢或(1-4C)烷基;或者R2与R3连结在一起形成亚甲基、1,2-亚乙烯基、亚乙基或三亚甲基。
2.权利要求1的式I醚衍生物或其可药用的盐其中Q1是2-氧代-二氢吲哚基、2-氧代-1,2-二氢喹啉基、2-氧代-1,2,3,4-四氢喹啉基或2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂基,它可以任意地带有1、2或3个选自氟、氯、甲基、乙基、烯丙基和2-丙炔基的取代基;X是硫、亚磺酰基或磺酰基;Ar是1,3-亚苯基,它可以任意地带有1或2个氟取代基,或者Ar是2,4-或2,5-噻吩二基或者2,4-或2,5-噻唑二基;并且Q2是式II的基团,其中R1是氢;R2是甲基或乙基;和R3是氢或甲基;或者R2和R3连接在一起形成亚乙基。
3.权利要求1的式I醚衍生物或其可药用的盐其中Q1是2-氧代二氢吲哚基,它可以任意地带有1、2或3个选自氟、氯、甲基、乙基、烯丙基和2-丙炔基的取代基;X是硫、亚磺酰基或磺酰基;Ar是1,3-亚苯基,它可以任意地带有1或2个氟取代基,或者Ar是2,4-或2,5-噻吩二基或者2,4-或2,5-噻唑二基;并且Q2是式II的基团,其中R1是氢;R2是甲基或乙基;和R3是氢或甲基。
4.权利要求1的式I醚衍生物或其可药用的盐其中Q1是2-氧代二氢吲哚-5-基、2-氧代-1,2-二氢喹啉-6-基、2-氧代-1,2,3,4-四氢喹啉-6-基或2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂-7-基,它在1-位带有一个选自甲基、乙基、烯丙基和2-丙炔基的取代基,并且它还任意地带有另一个选自氟、氯和甲基的取代基;X是硫、亚磺酰基或磺酰基;Ar是1,3-亚苯基或5-氟-1,3-亚苯基,或者Ar是X基团在2-位的2,4-噻吩二基、2,5-噻吩二基、X基团在2-位的2,4-噻唑二基或X基团在2-位的2,5-噻唑二基;并且Q2是式IV的基团 其中R2是甲基。
其中R2是甲基。
5.权利要求4的式I醚衍生物或其可药用的盐其中Q1是1-甲基-2-氧代二氢吲哚-5-基、1-甲基-2-氧代-1,2-二氢喹啉-6-基、1-烯丙基-2-氧代-1,2,3,4-四氢喹啉-6-基、1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基、1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、1-(2-丙炔基)-2-氧代-1,2,3,4-四氢喹啉-6-基、8-氯-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、8-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、1,8-二甲基-2-氧代-1,2,3,4-四氢喹啉-6-基或1-甲基-2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂-7-基;X是硫、亚磺酰基或磺酰基;Ar是X基团在2-位的2,4-噻吩二基、2,5-噻吩二基、或X基团在2-位的2,5-噻唑二基;并且Q2是式IV的基团,其中R2是甲基。
6.权利要求4的式I醚衍生物或其可药用的盐其中Q1是1-甲基-2-氧代二氢吲哚-5-基、1-甲基-2-氧代-1,2-二氢喹啉-6-基、1-烯丙基-2-氧代-1,2,3,4-四氢喹啉-6-基、1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基、1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、1-(2-丙炔基)-2-氧代-1,2,3,4-四氢喹啉-6-基、8-氯-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、8-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基、1,8-二甲基-2-氧代-1,2,3,4-四氢喹啉-6-基或1-甲基-2-氧代-2,3,4,5-四氢-1H-苯并[b]氮杂卓-7-基;X是硫、亚磺酰基或磺酰基;Ar是1,3-亚苯基或5-氟-1,3-亚苯基;并且Q2是式IV的基团,其中R2是甲基。
7.权利要求1的式I醚衍生物或其可药用的盐,选自:-(2S,4R)-4-[5-氟-3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃和(2S,4R)-4-[5-氟-3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)苯基]-4-羟基-2-甲基四氢吡喃。
8.权利要求1的式I醚衍生物或其可药用的盐,选自:-(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻唑-5-基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)噻唑-5-基]四氢吡喃,(2S,4R)-4-[2-(7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻唑-5-基]-4-羟基-2-甲基四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代二氢吲哚-5-基硫代)噻唑-5-基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)噻吩-4-基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-5-基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代-1,2-二氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1,8-二甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]四氢吡喃,4-[2-(8-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]-4-羟基-2-甲基四氢吡喃,4-[2-(7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)噻吩-4-基]-4-羟基-2-甲基四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代二氢吲哚-5-基硫代)噻吩-4-基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基磺酰)苯基]四氢吡喃,(2S,4R)-4-[3-(1-乙基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃,(2S,4R)-4-[3-(7-氟-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃,(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代-1,2-二氢喹啉-6-基硫代)苯基]四氢吡喃,(2S,4R)-4-[3-(8-氯-1-甲基-2-氧代-1,2,3,4-四氢喹啉-6-基硫代)苯基]-4-羟基-2-甲基四氢吡喃或(2S,4R)-4-羟基-2-甲基-4-[3-(1-甲基-2-氧代二氢吲哚-5-基硫代)苯基]四氢吡喃。
9.权利要求1的式I醚衍生物是:-(2S,4R)-4-羟基-2-甲基-4-[2-(1-甲基-2-氧代二氢吲哚-5-基硫代)噻吩-4-基]四氢吡喃或其可药用的盐。
10.权利要求1-9的式I醚衍生物或其可药用盐的制备方法,包括:-(a)将式Q1-X-H化合物与式Z-Ar-Q2化合物偶合,其中Z是可置换的基团;(b)将其中Z是可置换的基团的式Q1-Z化合物与式H-X-Ar-Q2化合物偶合;(c)将其中Z是可置换基团、或者当X是硫代基团时,Z可以是式Q1-X-基团的式Q1-X-Z化合物与式M-Ar-Q2的有机金属试剂偶合,其中M是碱金属或碱土金属或者M表示常规格利雅试剂的卤化镁部分;(d)为了制备其中X是亚磺酰基或磺酰基的式I化合物,将其中X是硫的式I化合物氧化;(e)为了制备其中Q2中的R1是(2-5C)链烷酰基或苯甲酰基的式I化合物,将其中Q2中的R1是氢的式I化合物酰化;(f)为了制备其中Q1在有效氮原子上带有一个烷基或取代烷基取代基的式I化合物,将其中Q1在所述有效氮原子上带有氢原子的式I化合物烷基化;(g)为了制备其中Q1带有1或2个硫代取代基的式I化合物,将其中Q1带有1或2个氧代取代基的式I化合物与硫化剂进行反应,由此将各个氧代取代基转化为硫代取代基;
并且当需要新的式I化合物的可药用盐时,可以采用常规方法、通过所述化合物与合适的酸或碱反应获得,而且当需要旋光活性的式I化合物时,可以使用旋光活性原料、通过进行上述一种方法获得,或者采用常规方法、通过拆分所述化合物的外消旋体获得。
11.药物组合物,该组合物含有权利要求1-9的式I醚衍生物或其可药用盐以及可药用的稀释剂或载体。
12.权利要求1-9的式I醚衍生物或其可药用盐在制备用于白三烯介导的疾病或医学疾病的药物中的用途。
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP93401120 | 1993-04-29 | ||
| EP93401120.6 | 1993-04-29 | ||
| EP93401991 | 1993-08-02 | ||
| EP93401991.0 | 1993-08-02 | ||
| EP94400190.8 | 1994-01-28 | ||
| EP94400190 | 1994-01-28 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1121715A CN1121715A (zh) | 1996-05-01 |
| CN1052002C true CN1052002C (zh) | 2000-05-03 |
Family
ID=27235558
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN94191925A Expired - Fee Related CN1052002C (zh) | 1993-04-29 | 1994-04-25 | 四氢吡喃和四氢呋喃的醚衍生物,其制备方法和含有这些化合物的药物组合物 |
Country Status (24)
| Country | Link |
|---|---|
| US (2) | US5478842A (zh) |
| EP (1) | EP0623614B1 (zh) |
| JP (1) | JP3553127B2 (zh) |
| KR (1) | KR100358464B1 (zh) |
| CN (1) | CN1052002C (zh) |
| AP (1) | AP9400632A0 (zh) |
| AT (1) | ATE163417T1 (zh) |
| AU (1) | AU681886B2 (zh) |
| CA (1) | CA2121199C (zh) |
| CZ (1) | CZ280695A3 (zh) |
| DE (1) | DE69408593T2 (zh) |
| DK (1) | DK0623614T3 (zh) |
| ES (1) | ES2112483T3 (zh) |
| FI (2) | FI109123B (zh) |
| GB (1) | GB9408001D0 (zh) |
| HK (1) | HK1004777A1 (zh) |
| HU (1) | HU9502968D0 (zh) |
| IL (1) | IL109254A (zh) |
| NO (1) | NO309191B1 (zh) |
| NZ (1) | NZ266139A (zh) |
| PL (1) | PL311300A1 (zh) |
| SK (1) | SK134995A3 (zh) |
| TW (1) | TW252978B (zh) |
| WO (1) | WO1994025453A1 (zh) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3990972B2 (ja) * | 2001-11-20 | 2007-10-17 | 有限会社 キック | 血管再狭窄防止薬及び該防止薬がコーティングされた血管内埋め込み器具 |
| SE0104274D0 (sv) * | 2001-12-17 | 2001-12-17 | Astrazeneca Ab | Novel process |
| DE102004002557A1 (de) * | 2004-01-17 | 2005-08-04 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Verwendung von substituierten Pyrimido(5,4-d)pyrimidinen zur Behandlung von Atemwegserkrankungen |
| US8158362B2 (en) | 2005-03-30 | 2012-04-17 | Decode Genetics Ehf. | Methods of diagnosing susceptibility to myocardial infarction and screening for an LTA4H haplotype |
| JP2009523820A (ja) | 2006-01-23 | 2009-06-25 | アミラ ファーマシューティカルス,インコーポレーテッド | 5−リポキシゲナーゼの三環系抑制剤 |
| JP2010511632A (ja) | 2006-11-30 | 2010-04-15 | アミラ ファーマシューティカルス,インコーポレーテッド | 5−リポキシゲナーゼ活性化タンパク質インヒビターおよび一酸化窒素モジュレーターを含んでいる組成物および治療法 |
| US10793515B2 (en) * | 2008-03-19 | 2020-10-06 | Aurimmed Pharma, Inc. | Compounds advantageous in the treatment of central nervous system diseases and disorders |
| NZ624963A (en) | 2009-04-29 | 2016-07-29 | Amarin Pharmaceuticals Ie Ltd | Pharmaceutical compositions comprising epa and a cardiovascular agent and methods of using the same |
| WO2015120038A1 (en) | 2014-02-04 | 2015-08-13 | Bioscience Pharma Partners, Llc | Use of flap inhibitors to reduce neuroinflammation mediated injury in the central nervous system |
| ME03663B (me) | 2015-05-04 | 2020-07-20 | Astrazeneca Ab | Derivati rirazola korisni kao inhibitori proteina koji aktivira 5-lipoksigenazu (flap) |
| JP7041146B2 (ja) | 2016-10-28 | 2022-03-23 | アストラゼネカ・アクチエボラーグ | (1R,2R)-2-[4-(3-メチル-1H-ピラゾール-5-イル)ベンゾイル]-N-(4-オキソ-4,5,6,7-テトラヒドロピラゾロ[1,5-a]ピラジン-3-イル)シクロヘキサンカルボキサミド |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1046903A (zh) * | 1989-02-28 | 1990-11-14 | 帝国化学工业公司 | 制备杂环醚衍生物的方法 |
| EP0420511A2 (en) * | 1989-09-29 | 1991-04-03 | Imperial Chemical Industries Plc | Heterocyclic derivatives |
| EP0462812A2 (en) * | 1990-06-21 | 1991-12-27 | Zeneca Limited | Pyran derivatives as inhibitors of 5-lipoxygenase |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4794188A (en) * | 1982-12-01 | 1988-12-27 | Usv Pharmaceutical Corporation | Certain unsymmetrical quinolinyl ethers having anti-inflammatory and anti-allergic activity |
| US4567184A (en) * | 1982-12-01 | 1986-01-28 | Usv Pharmaceutical Corporation | Certain aryl or hetero-aryl derivatives of 1-hydroxy-pentane or 1-hydroxy-hexane which are useful for treating inflammation and allergies |
| US4625034A (en) * | 1985-02-04 | 1986-11-25 | Usv Pharmaceutical Corp. | 1,2-Dihydro; 1,2,3,4-tetrahydro; 5,8 dihydro; and 5,6,7,8-tetrahydroquinoline derivatives |
| US4839369A (en) * | 1985-04-16 | 1989-06-13 | Rorer Pharmaceutical Corporation | Aryl and heteroaryl ethers as agents for the treatment of hypersensitive ailments |
| US4631287A (en) * | 1985-04-16 | 1986-12-23 | Usv Pharmaceutical Corp. | Aryl and heteroaryl ethers as agents for the treatment of hypersensitive ailments |
| US4725619A (en) * | 1985-04-16 | 1988-02-16 | Usv Pharmaceutical Corporation | Aryl and heteroaryl ethers as agents for the treatment of hypersensitive ailments |
| US4728668A (en) * | 1985-04-16 | 1988-03-01 | Usv Pharmaceutical Corporation | Aryl and heteroaryl ethers as agents for the treatment of hypersensitive ailments |
| US4868193A (en) * | 1986-09-24 | 1989-09-19 | Rorer Pharmaceutical Corporation | Styryl tetrazoles and anti-allergic use thereof |
| GB8926981D0 (en) * | 1988-12-23 | 1990-01-17 | Ici Plc | Heterocyclic derivatives |
| US5234950A (en) * | 1988-12-23 | 1993-08-10 | Imperial Chemical Industries Plc | Tetrahydrofuran derivatives |
| US5202326A (en) * | 1989-02-28 | 1993-04-13 | Imperial Chemical Industries Plc | Heterocyclic ethers |
| US5236919A (en) * | 1989-02-28 | 1993-08-17 | Imperial Chemical Industries Plc | Quinoxalinyl derivatives suitable for use in leukotriene mediated disease |
| US5134148A (en) * | 1989-02-28 | 1992-07-28 | Imperial Chemical Industries Plc | Heterocycles for use as inhibitors of leukotrienes |
| IE64358B1 (en) * | 1989-07-18 | 1995-07-26 | Ici Plc | Diaryl ether heterocycles |
| US5088927A (en) * | 1990-06-18 | 1992-02-18 | Lee Howard G | Radio opaque plastics and process of making |
| AU637500B2 (en) * | 1990-06-21 | 1993-05-27 | Zeneca Limited | Bicyclic heterocyclic compounds |
| GB9113137D0 (en) * | 1990-07-13 | 1991-08-07 | Ici Plc | Thioxo heterocycles |
| US5272173A (en) * | 1990-11-28 | 1993-12-21 | Imperial Chemical Industries Plc | 5-lipoxygenase inhibitors |
| IE913866A1 (en) * | 1990-11-28 | 1992-06-03 | Ici Plc | Aryl derivatives |
| EP0501578A1 (en) * | 1991-02-28 | 1992-09-02 | Merck Frosst Canada Inc. | Pyranylphenyl hydroxyalkylnaphthoic acids as inhibitors of leukotriene biosynthesis |
| EP0501579A1 (en) * | 1991-02-28 | 1992-09-02 | Merck Frosst Canada Inc. | Naphthalene lactones as inhibitors of leukotriene biosynthesis |
| US5308852A (en) * | 1992-06-29 | 1994-05-03 | Merck Frosst Canada, Inc. | Heteroarylnaphthalenes as inhibitors of leukotriene biosynthesis |
-
1994
- 1994-04-07 AP APAP/P/1994/000632A patent/AP9400632A0/en unknown
- 1994-04-07 IL IL10925494A patent/IL109254A/en not_active IP Right Cessation
- 1994-04-13 CA CA002121199A patent/CA2121199C/en not_active Expired - Fee Related
- 1994-04-22 GB GB9408001A patent/GB9408001D0/en active Pending
- 1994-04-25 WO PCT/EP1994/001277 patent/WO1994025453A1/en active IP Right Grant
- 1994-04-25 CN CN94191925A patent/CN1052002C/zh not_active Expired - Fee Related
- 1994-04-25 CZ CZ952806A patent/CZ280695A3/cs unknown
- 1994-04-25 HU HU9502968A patent/HU9502968D0/hu unknown
- 1994-04-25 SK SK1349-95A patent/SK134995A3/sk unknown
- 1994-04-25 PL PL94311300A patent/PL311300A1/xx unknown
- 1994-04-25 AU AU67212/94A patent/AU681886B2/en not_active Ceased
- 1994-04-25 KR KR1019950704723A patent/KR100358464B1/ko not_active IP Right Cessation
- 1994-04-25 NZ NZ266139A patent/NZ266139A/xx not_active IP Right Cessation
- 1994-04-26 ES ES94303012T patent/ES2112483T3/es not_active Expired - Lifetime
- 1994-04-26 DK DK94303012T patent/DK0623614T3/da active
- 1994-04-26 JP JP08882694A patent/JP3553127B2/ja not_active Expired - Fee Related
- 1994-04-26 AT AT94303012T patent/ATE163417T1/de not_active IP Right Cessation
- 1994-04-26 EP EP94303012A patent/EP0623614B1/en not_active Expired - Lifetime
- 1994-04-26 DE DE69408593T patent/DE69408593T2/de not_active Expired - Fee Related
- 1994-04-28 US US08/234,148 patent/US5478842A/en not_active Expired - Lifetime
- 1994-04-30 TW TW083103922A patent/TW252978B/zh active
-
1995
- 1995-05-12 US US08/440,132 patent/US5512594A/en not_active Expired - Lifetime
- 1995-10-27 FI FI955165A patent/FI109123B/fi active
- 1995-10-27 NO NO954321A patent/NO309191B1/no not_active IP Right Cessation
-
1998
- 1998-05-05 HK HK98103836A patent/HK1004777A1/xx not_active IP Right Cessation
-
2002
- 2002-02-11 FI FI20020270A patent/FI20020270A/fi unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1046903A (zh) * | 1989-02-28 | 1990-11-14 | 帝国化学工业公司 | 制备杂环醚衍生物的方法 |
| EP0420511A2 (en) * | 1989-09-29 | 1991-04-03 | Imperial Chemical Industries Plc | Heterocyclic derivatives |
| EP0462812A2 (en) * | 1990-06-21 | 1991-12-27 | Zeneca Limited | Pyran derivatives as inhibitors of 5-lipoxygenase |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1028174C (zh) | 制备具有类视黄酸生物活性的带有二取代的乙炔部分的化合物的方法 | |
| CN1036920C (zh) | 含杂环碳酸衍生物 | |
| CN1052002C (zh) | 四氢吡喃和四氢呋喃的醚衍生物,其制备方法和含有这些化合物的药物组合物 | |
| CN1221417A (zh) | 具有抗糖尿病、降低血脂、抗高血压性能的噻唑烷二酮化合物,其制备方法及其药物组合物 | |
| CN1264382A (zh) | 新的化合物 | |
| CN1036393C (zh) | 7-(2-氨乙基)-苯并噻唑酮 | |
| CN1030757A (zh) | 苯并噻唑衍生物 | |
| CN1040025A (zh) | 新的四氢苯并吲哚丙酸磺酰胺的制备 | |
| CN1264378A (zh) | 作为nk-2和nk-3受体拮抗剂的喹啉-4-甲酰胺衍生物 | |
| WO1996011916A1 (fr) | Derive d'azole | |
| CN1145627C (zh) | 吡唑并[4,3-d]嘧啶类 | |
| CN1070488C (zh) | 杀灭螺旋杆菌用的取代芳烷基硫代烷基硫代吡啶 | |
| CN1437605A (zh) | 作为肿瘤坏死因子抑制剂的噻吩并二苯并薁化合物 | |
| CN1031262C (zh) | 环取代的2-氨基-1,2,3,4-四氢化萘和3-氨基苯并二氢吡喃 | |
| CN1323306A (zh) | 萘啶衍生物 | |
| CN1249063C (zh) | 新颖的免疫调制性化合物 | |
| CN101048392A (zh) | 新用途 | |
| CN1015627B (zh) | 芳香基呱嗪基-亚烷基苯基-杂环化合物的制备方法 | |
| CN1461301A (zh) | 苯并[b]噻吩衍生物及其制备方法 | |
| CN1195739C (zh) | 2-氨磺酰苯甲酸衍生物 | |
| CN1027066C (zh) | 吲哚衍生物的制备方法 | |
| CN1025027C (zh) | 噻吩-2-羧酰胺类的制备方法 | |
| CN1128029A (zh) | 作为脂氧合酶抑制剂的噻唑衍生物 | |
| CN1146545C (zh) | 苯甲酰基哒嗪 | |
| CN1084166A (zh) | 取代的(苯并噻唑基-和喹喔啉基-甲氧基)苯基-乙酸衍生物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20000503 Termination date: 20100425 |