CN102344459A - 一种头孢中间体gcle的制备方法 - Google Patents
一种头孢中间体gcle的制备方法 Download PDFInfo
- Publication number
- CN102344459A CN102344459A CN2011102107546A CN201110210754A CN102344459A CN 102344459 A CN102344459 A CN 102344459A CN 2011102107546 A CN2011102107546 A CN 2011102107546A CN 201110210754 A CN201110210754 A CN 201110210754A CN 102344459 A CN102344459 A CN 102344459A
- Authority
- CN
- China
- Prior art keywords
- cyclization
- gcle
- reaction
- reagent
- preparation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images




Landscapes
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
一种头孢中间体GCLE的制备方法,是将反应物4-苯磺酰基硫代-3-苯乙酰胺基-1-[1-(4-甲氧基苄氧基)羰基-2-甲基-2-丙烯基]氮杂环丁-2-酮用有机溶媒稀释后,加入有机碱作为缚酸-环合试剂,通入氯气进行氯代-环合一步反应得到GCLE。本发明的头孢中间体GCLE制备方法中,采用了氯代-环合一步法制备工艺,且氯代试剂选择价廉易得的氯气,缚酸试剂与环合试剂为同一试剂,该工艺的反应收率可以达到80%以上,产品含量达98%以上。此外,以氯代-环合一步法制备头孢中间体GCLE还具有工艺简单、条件温和、操作简便、污染小、能耗低等特点,达到了本发明的预期效果,具备工业化前景。
Description
技术领域
本发明涉及一种头孢中间体GCLE的制备方法,属于有机合成技术领域,本发明采用氯代-环合一步法制备头孢中间体GCLE。
背景技术
GCLE的化学名称为7-苯乙酰氨基-3-氯甲基-3-头孢烯烷酸对甲氧基苄酯,是一种重要的头孢中间体,其与7-ACA、7-ADCA并称为三大头孢母核。
GCLE的合成方法主要分为两大类:扩环前卤代法(JP615084、JP1156984、JP1308287、JP59172493、JP5872591、JP60255796、WO9910352、WO9858932,Tetrahedron Letters,1973,(32):3001、Tetrahedron Letters,1982,23:2187、NIPPON KAGAKUKAISHI,1995, (8):577-587,等)和扩环后卤代法(EP494118、GB1326531、US4950753、US3637678、US3705897、US5109132、US3658799、US4048163、US337525、US4985554,J.Am.Chem. Soc.,1973,98:2403、J.Am.Chem.Soc.,1976,98(16):5040、J.Am.Chem.Soc.,1977,99(8):2822、Tetrahedron Letters,1974,3991、Tetrahedron Letters,1982,23,2187、Tetrahedron,1983,39 (2):337、Tetrahedron,1983,39(3):461、J.Org.Chem.,1973,38(17):2994、J.Chem.Pharm.Bull.1988, 36(2):528-591、J.Synth.Comm.1986,16(6):649-652,等)。
目前以扩环前卤代法使用最为广泛。该法以青霉素G钾盐为起始原料,依次经酯化、氧化、开环、转化、氯代和环合六步操作制备得到GCLE,其反应简式如下:
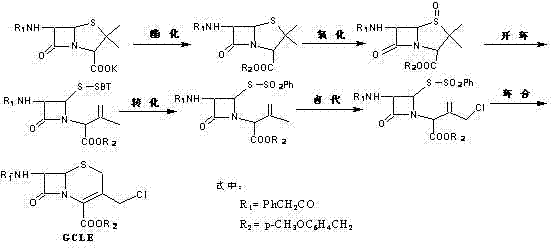
其中,氯代、环合两步反应是制约GCLE合成收率的关键所在,为此,前人进行了相关大量研究。
在氯代反应步骤中采用不同的氯代方法,如电解氯代法(JP58222969、JP6154864、JP6154865、JP6169688、JP61174330、SER865651、SER364405、SER71664、SER524689、US4609438、US4058521、US4461897、US4853468等)或化学氯代法,目前主要采用的是化学氯代法,选用不同的氯代试剂如氯气(Cl2)、一氧化二氯(Cl2O)、次氯酸钠(NaClO)、次氯酸钙(Ca(ClO)2)、次氯酸叔丁酯(t-BuClO)、烷氧基氯(ROCl)、五氯化磷(PCl5)、二氯亚砜(SOCl2)、N-氯代丁二酰亚胺(NCS)、N-溴代丁二酰亚胺(NBS)、液溴(Br2)等(US4042585、US4689411、SER524689、SER861626、US4789740、US4048163、WO2004 039813、GB1407348、US4853468、US20070027313A1、WO9855485A、EP0623622A、EP 0500081A2、CN1315841C、CN1006480A、CN1010393A、CN1142908C,Tetrahedron Letters, 1980,21:351-354、Tetrahedron Letters,1980, 32:781-784、Tetrahedron Letters,1984,24:877-880、Tetrahedron Letters,2003,44:6691-6693、J.Org.Chem.,2003, 68:3323、J.Org.Chem.,1982,47:3148等),氯代收率75%~92%;选用不同的氯代溶媒如氯苯、1,2-烷氧基乙烷、二氧六环、四氢呋喃、二甲基亚砜、二氯甲烷、三氯甲烷、甲基吡咯烷酮、醋酸乙酯、醋酸甲酯、水及其混合液等(US4789740、US20070027313A1、WO2004039813、WO2004039813、WO 9855485A、US4042585、US4689411、CN1315841C等);选用不同的缚酸剂如氧化钙、碳酸钙、碳酸氢钙、碳酸钠、碳酸氢钠等(US4789740、US20070027313A1、WO2004039813、CN1315841C等)。
在环合反应步骤中,选用不同的环合试剂如氢氧化钠(钾)(环合收率17%)、氨水或氨气(环合收率74%)、甲醇钠(环合收率84.6%)、乙酸钠(环合收率29%)、碘化钾(环合收率14%)等(US20070027313A1、JP5874689、WO2004039813、CN1315841C等);选用不同的环合溶媒如甲醇、乙醇、DMF、THF、甲基吡咯烷酮及其混合液等(CN1315841C、US20070027313A1、WO2004039813等)。
尽管在已有资料报道中,氯代反应与环合反应步骤均有较高收率者,但实际操作时两步反应的总收率并不高(<78%),且操作过程相对较为麻烦,主要体表现在:
1)因氯代、环合反应步骤的溶媒不同,所以环合反应前应先蒸除氯代溶媒,此过程必须严格控制温度,否则氯代物色泽变深,含量下降;
2)环合反应中要求严格控制体系pH值与温度,稍有出入会大大降低环合收率。
发明内容
本发明的目的是提供一种采用氯代-环合一步法制备头孢中间体GCLE的方法,以简化工艺、提高GCLE的收率和降低生产成本。
本发明提出的氯代-环合一步法制备头孢中间体GCLE的方法是将反应物4-苯磺酰基硫代-3-苯乙酰胺基-1-[1-(4-甲氧基苄氧基)羰基-2-甲基-2-丙烯基]氮杂环丁-2-酮用有机溶媒稀释后,加入有机碱作为缚酸-环合试剂,通入氯气进行氯代-环合一步反应得到GCLE。其反应简式如下。
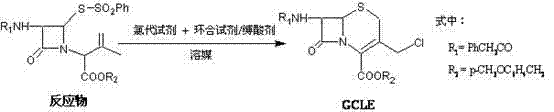
本发明氯代-环合一步法制备头孢中间体GCLE的方法具体是按以下步骤进行:
1)将反应物(4-苯磺酰基硫代-3-苯乙酰胺基-1-[1-(4-甲氧基苄氧基)羰基-2-甲基-2-丙烯基]氮杂环丁-2-酮)用适当的有机溶媒稀释待用,所述的有机溶媒包括但不限于二氧六环、乙酸乙酯、四氢呋喃、二甲基亚砜、四氯化碳、二氯甲烷、氯仿、甲苯或者其混合液,反应物与有机溶媒的质量体积比为1︰5~20;
2)向上述反应物的稀释液中加入一定量的缚酸-环合试剂,所述的缚酸-环合试剂包括但不限于有机碱,如三乙醇胺、三乙胺、吡啶、二甲胺基吡啶等;同时,缚酸-环合试剂的用量在于调节维持反应体系的pH值在8.0~10.0,在反应过程中可根据要求适当补加;
3)控温在-40~80℃,通入氯气进行氯代-环合一步反应,其中,反应物与氯气的摩尔比为1︰2.5~5.0,反应时间3~6h;
4)反应完成后滤除不溶物,将滤液浓缩,加入析晶溶媒进行析晶,即得白色目标产物GCLE。其中,所用析晶溶媒包括但不限于低级脂肪醇,如甲醇、乙醇等。
进一步地,上述制备方法中,步骤3)中控温优选-20~30℃。
本发明制备方法中,步骤4)中析晶后的滤液可回收循环套用于下一批析晶处理。
本发明提出的头孢中间体GCLE制备方法中,采用了氯代-环合一步法制备工艺,且氯代试剂选择价廉易得的氯气,缚酸试剂与环合试剂为同一试剂,该工艺的反应收率可以达到80%以上,产品含量达98%以上。此外,以氯代-环合一步法制备头孢中间体GCLE还具有工艺简单、条件温和、操作简便、污染小、能耗低等特点,达到了本发明的预期效果,具备工业化前景。
附图说明
图1是实施例1制备得到的头孢中间体GCLE的质谱图。
图2是头孢中间体GCLE对照品的质谱图。
图3是实施例1制备得到的头孢中间体GCLE的红外光谱图。
图4是头孢中间体GCLE对照品的红外光谱图。
具体实施方式
实施例1。
1)取4-苯磺酰基硫代-3-苯乙酰胺基-1-[1-(4-甲氧基苄氧基)羰基-2-甲基-2-丙烯基]氮杂环丁-2-酮40.0g(0.0687mol)于反应瓶中,以有机溶媒二氧六环650ml稀释,搅拌下加入缚酸-环合试剂三乙胺,至调节体系的pH=9.0~10.0,然后控温在25~30℃。
2)快速搅拌下,采取先快后慢的方式通入干燥氯气0.1924mol,约3.5~4h通完,继续控温在25~30℃,计时搅拌反应30min。其间,随着通氯气量的增加,反应混合液逐渐变得浑浊,且有白色烟雾生成,为控制体系的pH值,在通氯气反应过程中可适当补加缚酸-环合试剂。
3)反应完成后,滤除不溶物,将滤液浓缩得到粘稠状剩余物,加入甲醇150ml,搅拌降温,即有白色细小颗粒物析出,于-10~0℃养晶1h,过滤,洗涤,干燥,即得目标产物GCLE白色固体粉末28.78g,收率86.21%,含量98.72%。
回收甲醇析晶滤液,循环套用于下批氯代-环合反应完成后的析晶后处理操作。
目标产物的质谱图如图1,红外光谱谱图如图3,与GCLE对照品的质谱图和红外光谱图对比一致,证明得到的产物为GCLE。
实施例2。
1)取4-苯磺酰基硫代-3-苯乙酰胺基-1-[1-(4-甲氧基苄氧基)羰基-2-甲基-2-丙烯基]氮杂环丁-2-酮40.0g(0.0687mol)于反应瓶中,以有机溶媒乙酸乙酯700ml稀释,搅拌下加入缚酸-环合试剂三乙醇胺,至调节体系的pH=8.0~9.0,然后控温在-20~-10℃。
2)快速搅拌下,采取先快后慢的方式通入干燥氯气0.2016mol,约3~4h通完,继续控温在-20~-10℃,计时搅拌反应30min。其间,随着通氯气量的增加,反应混合液逐渐变得浑浊,且有白色烟雾生成,为控制体系的pH值,在通氯气反应过程中可适当补加缚酸-环合试剂。
3)反应完成后,滤除不溶物,将滤液浓缩得到粘稠状物,加入乙醇200ml,搅拌降温即有白色细小颗粒物析出,于-10~0℃养晶1h,过滤,洗涤,干燥,即得目标产物GCLE白色固体粉末26.87g,收率80.47%,含量98.31%。
回收乙醇析晶滤液,循环套用于下批氯代-环合反应完成后的析晶后处理操作。
实施例3。
1)取4-苯磺酰基硫代-3-苯乙酰胺基-1-[1-(4-甲氧基苄氧基)羰基-2-甲基-2-丙烯基]氮杂环丁-2-酮35.0g(0.0601mol)于反应瓶中,以有机溶媒四氢呋喃600ml稀释,搅拌下加入缚酸-环合试剂吡啶,至调节体系的pH=9.0~10.0,然后控温在-15~-5℃。
2)快速搅拌下,采取先快后慢的方式通入干燥氯气0.2404mol,约5~6h通完,继续控温在-15~-5℃,计时搅拌反应30min。其间,随着通氯气量的增加,反应混合液逐渐变得浑浊,且有白色烟雾生成,为控制体系的pH值,在通氯气反应过程中可适当补加缚酸-环合试剂。
3)反应完成后,滤除不溶物,将滤液浓缩得到粘稠状物,加入回收甲醇160ml,搅拌降温即有白色细小颗粒物析出,于-10~0℃养晶1h,过滤,洗涤,干燥,即得目标产物GCLE白色固体粉末25.10g,收率85.93%,含量99.13%。
回收甲醇析晶滤液,继续循环套用于下批氯代-环合反应完成后的析晶后处理操作。
实施例4。
1)取4-苯磺酰基硫代-3-苯乙酰胺基-1-[1-(4-甲氧基苄氧基)羰基-2-甲基-2-丙烯基]氮杂环丁-2-酮40.0g(0.0687mol)于反应瓶中,以有机溶媒二氯甲烷750ml稀释,搅拌下加入缚酸-环合试剂三乙胺,至调节体系的pH=9.0~10.0,然后控温在-10~0℃。
2)快速搅拌下,采取先快后慢的方式通入干燥氯气0.3092mol,约4~5h通完,继续控温在-10~0℃,计时搅拌反应30min。其间,随着通氯气量的增加,反应混合液逐渐变得浑浊,且有白色烟雾生成,为控制体系的pH值,在通氯气反应过程中可适当补加缚酸-环合试剂。
3)反应完成后,滤除不溶物,将滤液浓缩得到粘稠状物,加入回收乙醇180ml,搅拌降温即有白色细小颗粒物析出,于-10~0℃养晶1h,过滤,洗涤,干燥,即得目标产物GCLE白色固体粉末27.16g,收率81.35%,含量99.57%。
回收乙醇析晶滤液,继续循环套用于下批氯代-环合反应完成后的析晶后处理操作。
实施例5。
1)取4-苯磺酰基硫代-3-苯乙酰胺基-1-[1-(4-甲氧基苄氧基)羰基-2-甲基-2-丙烯基]氮杂环丁-2-酮40.0g(0.0687mol)于反应瓶中,以有机溶媒四氯化碳600ml稀释,搅拌下加入缚酸-环合试剂4-二甲氨基吡啶,至调节体系的pH=8.0~9.0,然后控温在0~10℃。
2)快速搅拌下,采取先快后慢的方式通入干燥氯气0.2405mol,约3~4h通完,继续控温在0~10℃,计时搅拌反应30min。其间,随着通氯气量的增加,反应混合液逐渐变得浑浊,且有白色烟雾生成,为控制体系的pH值,在通氯气反应过程中可适当补加缚酸-环合试剂。
3)反应完成后,滤除不溶物,将滤液浓缩得到粘稠状物,加入甲醇200ml,搅拌降温即有白色细小颗粒物析出,于-10~0℃养晶1h,过滤,洗涤,干燥,即得目标产物GCLE白色固体粉末28.27g,收率84.68%,含量98.96%。
回收甲醇析晶滤液,继续循环套用于下批氯代-环合反应完成后的析晶后处理操作。
Claims (4)
1.一种头孢中间体GCLE的制备方法,是将反应物4-苯磺酰基硫代-3-苯乙酰胺基-1-[1-(4-甲氧基苄氧基)羰基-2-甲基-2-丙烯基]氮杂环丁-2-酮用有机溶媒稀释后,加入有机碱作为缚酸-环合试剂,通入氯气进行氯代-环合一步反应得到GCLE。
2.根据权利要求1所述的头孢中间体GCLE的制备方法,其特征是按以下步骤进行:
1)将反应物4-苯磺酰基硫代-3-苯乙酰胺基-1-[1-(4-甲氧基苄氧基)羰基-2-甲基-2-丙烯基]氮杂环丁-2-酮用适当的有机溶媒稀释待用,所述的有机溶媒包括但不限于二氧六环、乙酸乙酯、四氢呋喃、二甲基亚砜、四氯化碳、二氯甲烷、氯仿、甲苯或者其混合液,反应物与有机溶媒的质量体积比为1︰5~20;
2)向上述反应物的稀释液中加入一定量的缚酸-环合试剂,所述的缚酸-环合试剂包括但不限于三乙醇胺、三乙胺、吡啶、二甲胺基吡啶;缚酸-环合试剂的用量为调节维持反应体系的pH值在8.0~10.0;
3)控温在-40~80℃,通入氯气进行氯代-环合一步反应,其中,反应物与氯气的摩尔比为1︰2.5~5.0,反应时间3~6h;
4)反应完成后滤除不溶物,将滤液浓缩,加入析晶溶媒进行析晶,得到白色目标产物GCLE,其中,所用析晶溶媒包括但不限于甲醇或乙醇。
3.根据权利要求2所述的头孢中间体GCLE的制备方法,其特征是所述步骤3)中控温-20~30℃。
4.根据权利要求2所述的头孢中间体GCLE的制备方法,其特征是步骤4)中析晶后的滤液回收循环套用于下一批析晶处理。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201110210754.6A CN102344459B (zh) | 2011-07-27 | 2011-07-27 | 一种头孢中间体gcle的制备方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201110210754.6A CN102344459B (zh) | 2011-07-27 | 2011-07-27 | 一种头孢中间体gcle的制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN102344459A true CN102344459A (zh) | 2012-02-08 |
| CN102344459B CN102344459B (zh) | 2014-08-06 |
Family
ID=45543566
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN201110210754.6A Active CN102344459B (zh) | 2011-07-27 | 2011-07-27 | 一种头孢中间体gcle的制备方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN102344459B (zh) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102690243A (zh) * | 2012-05-25 | 2012-09-26 | 伊犁川宁生物技术有限公司 | 一种制备7-苯乙酰胺-3-氯甲基头孢烷酸对甲氧基苄酯过程中原料回收的方法 |
| CN102863460A (zh) * | 2012-09-29 | 2013-01-09 | 广东省石油化工研究院 | 一种头孢菌素类抗生素母核gcle的制备方法 |
| CN105440053A (zh) * | 2015-12-24 | 2016-03-30 | 湖北凌晟药业有限公司 | 一种gcle结晶母液的回收方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4689411A (en) * | 1981-04-10 | 1987-08-25 | Otsuka Kagaku Yakuhin Kabushiki Kaisha | 4-thio azetidinone intermediates and process for the preparation of the same |
| US20050215782A1 (en) * | 2004-03-25 | 2005-09-29 | Nobuo Matsumoto | Process for preparing crystalline 3-chloromethyl-3-cephem derivatives |
| CN1708501A (zh) * | 2002-11-01 | 2005-12-14 | 幽兰化学医药有限公司 | 制备氯甲基头孢烯衍生物的改进方法 |
| CN1849324A (zh) * | 2003-09-09 | 2006-10-18 | 日本化学工业株式会社 | 3-氯甲基-3-头孢烯衍生物的制造方法 |
| CN101186614A (zh) * | 2007-12-13 | 2008-05-28 | 吴江市方霞企业信息咨询有限公司 | 一种甲氧基苄酯的制备方法 |
| CN101260116A (zh) * | 2008-04-21 | 2008-09-10 | 湖南有色凯铂生物药业有限公司 | 7-苯乙酰胺-3-氯甲基-4-头孢烷酸对甲氧苄酯的合成方法 |
| CN101429208A (zh) * | 2007-11-07 | 2009-05-13 | 上海五洲药业股份有限公司 | 7-苯乙酰胺-3-氯甲基头孢烷酸对甲氧基苄酯的合成方法 |
| CN101525340A (zh) * | 2008-03-04 | 2009-09-09 | 山东方兴科技开发有限公司 | 7-苯乙酰氨-3-氯甲基头孢烷烯酸对甲氧基苄酯的合成方法 |
-
2011
- 2011-07-27 CN CN201110210754.6A patent/CN102344459B/zh active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4689411A (en) * | 1981-04-10 | 1987-08-25 | Otsuka Kagaku Yakuhin Kabushiki Kaisha | 4-thio azetidinone intermediates and process for the preparation of the same |
| CN1708501A (zh) * | 2002-11-01 | 2005-12-14 | 幽兰化学医药有限公司 | 制备氯甲基头孢烯衍生物的改进方法 |
| CN1849324A (zh) * | 2003-09-09 | 2006-10-18 | 日本化学工业株式会社 | 3-氯甲基-3-头孢烯衍生物的制造方法 |
| US20050215782A1 (en) * | 2004-03-25 | 2005-09-29 | Nobuo Matsumoto | Process for preparing crystalline 3-chloromethyl-3-cephem derivatives |
| CN101429208A (zh) * | 2007-11-07 | 2009-05-13 | 上海五洲药业股份有限公司 | 7-苯乙酰胺-3-氯甲基头孢烷酸对甲氧基苄酯的合成方法 |
| CN101186614A (zh) * | 2007-12-13 | 2008-05-28 | 吴江市方霞企业信息咨询有限公司 | 一种甲氧基苄酯的制备方法 |
| CN101525340A (zh) * | 2008-03-04 | 2009-09-09 | 山东方兴科技开发有限公司 | 7-苯乙酰氨-3-氯甲基头孢烷烯酸对甲氧基苄酯的合成方法 |
| CN101260116A (zh) * | 2008-04-21 | 2008-09-10 | 湖南有色凯铂生物药业有限公司 | 7-苯乙酰胺-3-氯甲基-4-头孢烷酸对甲氧苄酯的合成方法 |
Non-Patent Citations (1)
| Title |
|---|
| 魏文珑,等: "头孢类中间体GCLE的合成研究", 《中国抗生素杂志》 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102690243A (zh) * | 2012-05-25 | 2012-09-26 | 伊犁川宁生物技术有限公司 | 一种制备7-苯乙酰胺-3-氯甲基头孢烷酸对甲氧基苄酯过程中原料回收的方法 |
| CN102690243B (zh) * | 2012-05-25 | 2014-07-16 | 伊犁川宁生物技术有限公司 | 一种制备7-苯乙酰胺-3-氯甲基头孢烷酸对甲氧基苄酯过程中原料回收的方法 |
| CN102863460A (zh) * | 2012-09-29 | 2013-01-09 | 广东省石油化工研究院 | 一种头孢菌素类抗生素母核gcle的制备方法 |
| CN105440053A (zh) * | 2015-12-24 | 2016-03-30 | 湖北凌晟药业有限公司 | 一种gcle结晶母液的回收方法 |
| CN105440053B (zh) * | 2015-12-24 | 2017-11-10 | 湖北凌晟药业有限公司 | 一种gcle结晶母液的回收方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102344459B (zh) | 2014-08-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102311392B (zh) | 嘧菌酯及其合成中专用中间体的合成方法 | |
| CN107365275B (zh) | 高纯度的赛乐西帕 | |
| CN109912624A (zh) | 一种巴洛沙韦酯关键母核中间体的合成方法 | |
| CA2664052C (en) | An improved process for the preparation of temozolomide and analogs | |
| CN102344459B (zh) | 一种头孢中间体gcle的制备方法 | |
| CN101357911B (zh) | 合成(z)-2-(α-甲氧亚胺)呋喃乙酸铵的方法 | |
| CN101570543B (zh) | 一种美洛西林钠溶媒结晶制备方法 | |
| CN107445917A (zh) | 一种mica活性酯环保生产方法 | |
| CN102351802B (zh) | N-单取代哌嗪-2,3-二酮的合成方法 | |
| CN106749259B (zh) | 一种环戊基嘧啶并吡咯类化合物的合成方法 | |
| CN103724258A (zh) | 一种索拉非尼的制备方法 | |
| CN107963994A (zh) | 一种制备5-氟尿嘧啶的绿色方法 | |
| CN103936759B (zh) | (3aS,6aR)-1,3-二苄基-四氢-4H-噻吩并[3,4-d]咪唑-2,4-(1H)-二酮的简便制备方法 | |
| CN105111103B (zh) | 邻羟基苯甲腈及其衍生物的制备方法 | |
| CN107056720A (zh) | 一种缬沙坦的制备和纯化方法 | |
| CN103709164B (zh) | 一种腺嘌呤的合成方法 | |
| CN104098502B (zh) | 一种(S)-α-羟基-γ-N-邻苯二甲酰氨基丁酸的合成方法 | |
| CN101302198B (zh) | 一种氮杂卓衍生物的化学合成方法 | |
| CN103012266B (zh) | 一种7-氯-5-氧代-2,3,4,5-四氢-1h-1-苯并氮杂卓的制备方法 | |
| CN108623602B (zh) | 一种制备和纯化依鲁替尼的方法 | |
| CN109761871A (zh) | 一种氨曲南单环母核的合成方法 | |
| CN101463039A (zh) | 3-羟甲基头孢烷酸的制备方法及以该中间体合成7β-氨基-3-无头孢环-4-羧酸的新工艺 | |
| CN105399738A (zh) | 一种阿齐沙坦酯的制备方法 | |
| CN102249970A (zh) | 一种多尼培南侧链的合成工艺 | |
| CN101328146B (zh) | 一种5-氰基亚氨基芪的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CP01 | Change in the name or title of a patent holder |
Address after: 030500 Shanxi city of Lvliang province Jiaocheng Cross Ridge Road from the 307 National Road 1000 meters Patentee after: Shanxi xintianyuan Pharmaceutical Co. Ltd. Address before: 030500 Shanxi city of Lvliang province Jiaocheng Cross Ridge Road from the 307 National Road 1000 meters Patentee before: Shanxi Xintianyuan Pharm. & Chem. Co., Ltd. |