Campo Técnico
A presente invenção se refere a um novo derivado de aminopiridina 5 substituído usado no campo da medicina que inibe a proliferação de células tumorais baseado em um efeito inibitório contra PLK1, pelo qual exibe um efeito antitumoral e para um inibidor PLK1 e um agente antitumoral contendo o derivado.
Fundamento da Técnica
A proliferação é conhecida como sendo geralmente ativa em células cancerígenas comparado com células normais e, em muitos casos, acredita-se que desordens da proliferação devido a uma anormalidade no mecanismo de controle do ciclo celular é a causa do câncer. Uma fase mitótica (fase M) do ciclo celular é a etapa de divisão em partes iguais de um cromossomo em células-filhas e um controle rigoroso no processo é essencial para a proliferação e sobrevivência da célula. Portanto, acredita-se que a inibição da progressão da fase M é um meio eficiente para inibição da proliferação celular e, praticamente, agentes antitumorais que têm como alvo a fase M como o taxol, a vincristina ou semelhantes têm atingido resultados clinicamente eficientes.
Sabe-se que várias etapas na progressão da fase M são controladas por proteínas quinases que fosforilam proteínas. Uma família PLK (quinases tipo polo) é uma quinase serina-treonina que desempenha um papel importante no controle do ciclo celular incluindo a fase M, e esta família inclui quatro proteínas semelhantes de PLK1, PLK2, PLK3 e SAK (Nature. Review. Molecular. Cell 25 Biology (Nat. Rev. Mol. Cell Biol.), Vol. 5, 429, (2004)). Destas, a PLK1 é conhecida por participar em uma infinidade de estágios importantes na fase M em células de mamíferos: tem sido relatado que a PLK1 participa em cada etapa na entrada da fase M, controle de centrossomo, separação de cromossomo e citocinese, ao fosforilar vários substratos (Nature, review. Molecular. Cell Biology.) 30 (Nat. Rev. Mol. Cell Biol.), Vol. 5, 429, (2004)).
Além disso, há muitos relatos que sugerem que a PLK1 é superexpressa em vários tecidos cancerosos em humanos. Por exemplo, a PLK1 é aprovada como sendo superexpressa em câncer de pulmão de pequenas células ((Oncogene, Vol. 14, 543, (1997)) e câncer de cabeça e pescoço (Cancer 35 Research, Vol. 15, 2794, (1999)) e há dados mostrando que a superexpressão de PLK1 está relacionada com um prognóstico de pacientes com estas doenças.
Também está relatado que a expressão de PLK1 é aumentada em outros tipos de cânceres como no câncer de cólon, câncer de esôfago, câncer ovariano e melanoma. Tais relatos sugerem que a superexpressão de PLK1 está relacionada à alteração maligna de células de uma maneira ou de outra e também que a 5 função de PLK1 é particularmente importante na progressão da fase M em células cancerígenas.
A partir destes fatos, acredita-se que a PLK1 é um possível alvo para a abordagem antitumoral. De fato, há muitos relatos sobre experimentos para analisar o efeito inibitório sobre a função da PLK1 contra células cancerígenas ao 10 usar várias técnicas experimentais. Por exemplo, foi relatado que no experimento de expressão de uma PLK1 mutante com função inibida em células ao usar um vetor viral, a inibição da PLK1 promove a apoptose seletiva de célula cancerígena (Cell growth &Differentiation (Cell growth & Diff.), Vol. 11, 615, (2000)). Há também um relato mostrando que a PLK1 siRNA induz a inibição do crescimento 15 e apoptose de células cancerígenas (Journal of National Cancer Institute (J. Natl. Cancer Inst.), Vol. 94, 1863 (2002)). Além disso, foi relatado que a PLK1 shRNA (Journal of National Cancer Institute (J. Natl. Cancer Inst.), Vol. 96, 862, (2004)) ou um oligonucleotídeo anti-sentido (Oncogene, Vol. 21, 3162 (2002)) apresenta um efeito antitumoral em um modelo de xenoenxerto em camundongos. Estes 20 resultados experimentais mostram que a inibição da atividade de PLK1 causa a promoção da inibição do crescimento e apoptose das células cancerígenas e sugere fortemente que o inibidor da PLK1 é um agente antitumoral eficiente.
Os presentes inventores depositaram um pedido de patente sobre um derivado imidazol substituído contendo um efeito PLK inibitório (Publicação 25 Internacional W02006/025567).
Descrição da Invenção
É um objeto da presente invenção fornecer um novo derivado aminopirimidina que exibe um efeito PLK1 inibitório e uma excelente atividade citostática (inibição do crescimento celular) baseada no efeito inibitório, desta 30 forma desenvolver um agente antitumoral baseado em tal efeito PLK1 inibitório.
Com o objetivo de atingir tal objetivo, os inventores da presente invenção sintetizaram uma ampla variação de derivados de aminopiridinas e descobriram que uma atividade citostática baseada no efeito inibitório, completando, portanto, a invenção.
Ou seja, a presente invenção se refere a um composto representado pela Fórmula [1]:

onde R1 e R2, que podem ser o mesmo ou diferentes, são cada, um átomo de hidr gênio; um átomo de halogênio; um grupo alquila baixo contendo um a dois átomos de carbono, que pode ser substituído por um ou três átomos de flúor;ou um grupo ciclopropila; um de R3 e R4 é um átomo de hidrogênio, enquanto o outro de R3 ou R4 é: a) um grupo alquila baixo substituído com NRaRb, onde Ra e Rb, que podem ser 0 mesmo ou diferentes, são cada, um átomo de hidrogênio, um grupo alquila baixo, um grupo benzila, um grupo cicloalquila contendo três a seis átomos de carbono, onde um grupo cicloalquila pode ser substituído por umsubstituintes, que podem ser o mesmo ou diferente, selecionado a seguintes 1) a 3): 1) um grupo alquila baixo; 2) um substituinte selecionado a partir de <Grupo Substituinte β>; a) um grupo alquila substituído com um ou mais substituintes selecionados a partir de <Grupo Substituinte β>; e 0 grupo cicloalquila pode incluir uma ligação insaturada; b) um grupo heterociclico alifático com 4- a 6- membros selecionados a partir de um grupo azetidinila, um grupo pirrolidinila, um grupo piperidinila e um grupo piperazinila; c) um grupo alquila baixo substituído com um grupo heterociclico alifático com 4- a 6- membros selecionado a partir de um grupo azetidinila, um grupo pirrolidinila, um grupo piperidinila e um grupo piperazinila; d) um grupo heterociclico aromático com 5- a 6- membros selecionados a partir de um grupo pirrolila, um grupo imidazolila, um grupo purazolula, um grupo piridila, um grupo pirazinila e um grupo pirimidinila; ou ou mais partir dose e) um grupo alquila baixo substituícom com um grupo heterocíclico aromático com 5- ou 6- membros a partir de um grupo pirrolila, um grupo imidazolila, um grupo pirazolila, um grupo piridila, um grupo pirazinila e um grupo pirimidinila, onde o grupo heterocíclico alifático e o grupo heterocíclico aromático 5 cada um independentemente pode ser substituído por um ou mais substituintes que podem ser o mesmo ou diferentes, selecionados a partir dos seguintes 1) a 4): 1) um grupo alquila baixo; 2) um substituinte selecionado a partir do <Grupo Substituinte β>; 3) um grupo alquila substituído com um ou mais substituintes selecionados a partir de <Substituinte Grupo β>; e 4) um grupo cicloalquila contendo 3 a 6 átomos de carbono que podem ser substituídos por um ou mais substituintes selecionados a partir de <Grupo Substituinte β>; R5 é um átomo de hidrogênio, um grupo ciano, um átomo de halogênio ou um grupo alquila baixo; e <Grupo Substituinte β>definido como abaixo: <Grupo Substituinte β>: um átomo de halogênio, um grupo hidroxila, um grupo nitro, um grupo ciano, um grupo amino, um grupo carbamoila, um grupo aminosulfonila, um grupo imino, um grupo alquilsulfonila baixo, um grupo 20 alquilsulfonilamina, um grupo alcóxi baixo, um grupo alcoxicarbonila baixo, um grupo alcoxicarbonilamino baixo, um grupo alcanoila baixo, um grupo alcanoilóxi baixo, um grupo alquitio baixo, um grupo carboxila e um grupo benzila; ou um sal ou éster farmaceuticamente aceitável dos mesmos. O composto representado pela Fórmula (1) acima inclui todos os enantiômeros e diastereoisômeros em 25 adição aos racematos do composto.
A invenção também se refere à preparação combinada para administração simultânea, separada ou seqüencial no tratamento do câncer, onde a preparação combinada inclui duas preparações separadas de: * uma preparação incluindo, junto um carreador ou diluente 30 farmaceuticamente aceitável, o composto representado pela Fórmula (1) ou um sal ou éster farmaceuticamente aceitável do mesmo; e * uma preparação incluindo, junto com um carreador ou diluente farmaceuticamente aceitável, um agente antitumoral selecionado a partir do grupo que consiste de agentes alquilantes antitumorias, antimetabólitos antitumorais, 35 antibióticos antitumorais, agentes antitumorais derivados de plantas, compostos complexos antitumorais de platina, derivados antitumorais de camptotecina, * inibidores antitumorais de tirosina quinase anticorpos monoclonais, interferons, modificadores de resposta biológica, e outros agentes antitumorais ou um sal ou éster farmaceuticamente aceitável do mesmo, onde: os agentes alquilantes antitumorais são N-óxido de mostarda nitrogenada, 5 ciclofosfamida, isofosfamida, melfalan, busulfano, mitobronitol, carboquona, tiotepa, ranimustina, nimustina, temozolomida e carmustina; os antimetabólitos antitumorais são o metotrexato, ribosídeo 6- mercaptopurina, mercaptopurina, 5-fluorouracula, tegafur, doxifluridina, carmofur, citarabina, ocfosfato de citarabina, enocitabina, S-l, gemcitabina, fludarabina e 10 pemetrexede dissódico; os antibióticos antitumorais são actinomicida D, doxorrubicina, daunorrubicina, neocarzinostatina, bleomicina, peplomicina, mitomicina C, aclarubicina, pirarubicina, epirubicina, zinostatina estimalamer, idarubicina, sirolimus e valrubicina; os agentes antitumorais derivados de planta são vincristina, vinblastina, vindesina, etoposídeo, sobuzoxano, docetaxel, paclitaxel e vinorelbina; os compostos de complexo de platina são cisplatina, carboplatina, nedaplatina e oxaliplatina; os derivados antitumorais de camptotecina são irinotecano, topotecano e 20 camptotecina; os inibidores antitumorais de tirosina quinase são gefitinibe, imatinibe e erlotinibe; os anticorpos monoclonais são cetuximabe, bevacizumabe, rituximabe, bevacizumabe, alemtuzumabe e trastuzumabe; os interferons são a-interferon, a2a-interferon, a2b-interferon, β-interferon, 25 yla-interferon e yn1-interferon; os modificadores de resposta biológica são Krestin, lentinano, sizofirano, picibanil e ubenimex; e os outros agentes antitumorais são mitoxantrona, L-asparaginase, procarbazina, dacarbazina, hidroxicarbamida, pentostatina, tretinoína, alefacept, 30 darbepoetina alfa, anastrazol, exemestano, bicalutamida, leuprorelina, flutamida, fulvestrant, pegaptanibe octassódico, denileucina, diftitox, aldesleucina, alfa tirotropina, trióxido de arsênico, bortezomibe, capecitabina e goserelina.
Além disso, a invenção se refere a uma composição farmacêutica incluindo, junto com um carreador ou diluente farmaceuticamente aceitável, o 35 composto representado pela Fórmula (1) ou um sal ou éster farmaceuticamente aceitável do mesmo; e um agente antitumoral selecionado a partir do grupo que consiste de agente alquilante antitumoral, antimetabólitos antitumorais, antibióticos antitumorais, agentes antitumorais derivados de planta, compostos antitumorais de complexto de platina, derivados antitumorais de camptotecina, inibidores antitumorais de tirosina quinase, anticorpos monoclonais, modificadores de resposta biológica e outros agentes antitumorais (onde, a definição de cada agente antitumoral tem o mesmo significado que a definida acima), ou um sal ou éster farmaceuticamente aceitável do mesmo.
A invenção também se refere a um método para tratamento de cânceres, compreendendo a administração simultânea, separada ou seqüencial de uma quantidade terapeuticamente eficiente do composto representado pela Fórmula (1) acima ou um sal ou éster farmaceuticamente aceitável do mesmo, em combinação com uma quantidade terapeuticamente eficiente de um agente antitumoral selecionado a partir do grupo de agentes antitumorais alquilantes, antimetabólitos antitumorais, antibióticos antitumorais, agentes antitumorais derivados de planta, compostos antitumorais de complexo de platina, derivados antitumorais de camptotecina, inibidores antitumorais de tirosina quinase, anticorpos monoclonais, interferons, modificadores da resposta biológica, e outros agentes antitumorias (onde, a definição de cada agente antitumoral tem o mesmo significado que o definido acima), ou um sal ou éster farmaceuticamente ativo do mesmo.
A invenção também se refere ao uso de um inibidor de PLK1 para a fabricação de um medicamento para o tratamento de câncer e o uso de um inibidor de PLK1 em combinação com um agente antitumoral para a produção de um medicamento para o tratamento de câncer. Além disso, a invenção se refere a um método para o tratamento de cânceres em mamíferos (particularmente em humanos) que compreende a administração de uma quantidade terapeuticamente eficiente de um inibidor de PLK1 aos mamíferos em combinação com uma quantidade terapeuticamente eficiente de uma agente antitumoral.
Além disso, a invenção se refere a um agente para tratamento de cânceres incluindo um inibidor de PLK1 como um princípio ativo e a um agente para tratamento de cânceres que compreende, junto com um agente antitumoral, um inibidor de PLK1 como um princípio ativo. Deste ponto em diante, os símbolos e termos descritos na presente especificação serão explicados.
O “grupo alquila baixo” na Fórmula (1) se refere a um grupo alquila de cadeia reta ou ramificada contendo 1 a 6 átomo(s) de carbono e exemplos do mesmo incluem um grupo metila, um grupo etila, um grupo propila, um grupo isopropila, um grupo butila, um grupo isobutila, um grupo séc-butila, um grupo tert-butila, um grupo pentila, um grupo hexila, e semelhantes.
Um “grupo cicloalquila” na Fórmula (1) acima se refere a um grupo alicíclico com 3- a 8- membros e exemplos do mesmo incluem um grupo ciclopropila, um grupo ciclobutila, um grupo ciclopentila, um grupo ciclohexila, um grupo cicloheptila, um grupo ciclooctila e semelhantes, e preferencialmente se refere a grupos alicíclicos de 3- a 6- membros. Exemplos preferenciais incluem um grupo ciclopropila, um grupo ciclobutila, um grupo ciclopentila, e um grupo ciclohexila.
O “grupo heterocíclico alifátido” na Fórmula (1) se refere a uma grupo heterocíclico alifático insaturado ou saturado geralmente tendo pelo menos um átomo selecionado a partir de um átomo de nitrogênio, um átomo de oxigênio e um átomo de enxofre, em adição aos átomos de carbono, que seja um anel de um, dois ou três anéis fundidos. Exemplos incluem um grupo azetidila, um grupo pirrolidinila, um grupo piperidinila, um grupo piperazinila, um grupo morfolino, um grupo tetrahidrofuranila, um grupo imidazolidinila, um grupo trimorfolino, um grupo tetrahidroquinolila, um grupo tetrahidroisoquinolila e semelhantes. O “grupo heterocíclico alifático de 4- a 6- membros” na Fórmula (1) se refere a um grupo heterocíclico alifático saturado ou insaturado que é um anel monocíclico de 4- a 6- membros e exemplos do mesmo incluem um grupo azetidila, um grupo pirrolidinila, um grupo piperidinila, um grupo piperazinila, e semelhantes. O “grupo heterocíclico aromático” na Fórmula (1) acima geralmente se refere a um grupo heterocíclico com propriedades aromáticas contendo pelo menos um heteroátomo como um átomo de nitrogênio, um átomo de oxigênio, ou semelhante, e exemplos do mesmo incluem grupos heterocíclicos monocíclicos com 5- a 7- membros e grupos heterocíclicos de anéis fundidos formados pela fusão de um anel de 3- a 8- membros a um grupo heterocíclico monocíclico e semelhantes.
Especificamente, um grupo tienila, um grupo pirrolila, um grupo furila, um grupo tiazolila, um grupo imidazolila, um grupo pirazolila, um grupo oxazolila, um grupo piridila, um grupo pirazinila, um grupo pirimidinila, um grupo piridazinila, um grupo isoxazolila, um grupo isoquinolila, um grupo isoindolila, um grupo indazolila, um grupo indolilda, um grupo quinoxalinila, um grupo quinolila, um grupo benzoimidazolila, um grupo benzofuranila, e semelhantes podem ser mencionados. O “grupo heterocíclico aromático de 5- ou 6- membros” na Fórmula (1) acima se refere a um grupo heterocíclico monocíclico de 5- ou 6- membros com propriedades aromáticas e exemplos do mesmo incluem um grupo pirrolila, um grupo imidazolila, um grupo pirazolila, um grupo piridila, um grupo pirazinila, um grupo pirimidinila, e semelhantes.
O “átomo de halogênio” na Fórmula (1) acima pode ser exemplificado por 5 um átomo de flúor, um átomo de clore, um átomo de bromo, um átomo de iodo ou semelhantes e, entre estes, por exemplo, um átomo de flúor, um átomo de cloro ou um átomo de bromo são preferenciais.
O “grupo alquilsulfonila baixo” na Fórmula (1) acima se refere a um substituinte formado pela ligação do “grupo alquila baixo” a um átomo de enxofre 10 de um grupo sulfonila e exemplos do mesmo incluem um grupo metilsulfonila, um grupo etilsulfonila, um grupo butilsulfonila e semelhantes.
O “grupo alquilsulfonila baixo” na Fórmula (1) acima se refere a um substituinte formado por N-substituição do “grupo alquilsulfonila baixo” por um grupo amino, e exemplos do mesmo incluem um grupo metilsulfonilamina, um 15 grupo etilsulfonilamino, um grupo butilsulfonilamino e semelhantes.
O “grupo alquoxi baixo” na Fórmula (1) acima se refere a um grupo formado pela ligação do “grupo alquila baixo” a um átomo de oxigênio e exemplos do mesmo incluem um grupo metóxi, um gruo etóxi, um grupo propóxi, um grupo isopropóxi, um grupo butóxi, um grupo isobutóxi, um grupo séc-butóxi, um grupo 20 tert-butóxi, um grupo pentióxi, um grupo neopentilóxi, um grupo hexilóxi, um grupo isohexilóxi, e semelhantes;
O “grupo alcoxicarbonila baixo” na Fórmula (1) acima se refere a um grupo formado pela ligação do “grupo alcóxi baixo” a um grupo carbonila e exemplos específicos do mesmo incluem um grupo metóxicarbonila, um grupo 25 etoxicarbonila, um grupo propoxicarbonila, um grupo isopropoxicarbonila, um grupo butoxicarbonila, um grupo isobutoxicarbonila, um grupo sec-butoxicarbonila, um grupo tert-butoxicarbonila, um grupo pentiloxicarbonila, um grupo neopentiloxicarbonila, um grupo hexiloxicarbonila, um grupo isohexiloxicarbonila e semelhantes. 30 O “grupo alcoxicarbonilamino” na Fórmula (1) acima se refere a um gripo formado por N-substituição do “grupo alcoxicarbonila” a um grupo amino e exemplos específicos do mesmo incluem um grupo metoxicarbonilamino, um grupo etoxicarbonilamino, um grupo propoxicarbonilamino, um grupo isso propoxicarbonilamino, um grupo butoxicarbonilamino, um grupo 35 isobutoxicarbonilamino, um grupo sec-butoxicarbonilamino, um grupo tert- butoxicarbonilamino, um grupo pentoxilcarbonilamino, um grupo isohexiloxicarbonilamino e semelhantes.
O grupo “alcanoil baixo” na Fórmula (1) se refere a um grupo formado pela ligação do “grupo alquila baixo” a um grupo carbonila, e é preferencialmente um grupo onde o grupo alquila contendo 1 a 5 átomos de carbono se liga a um grupo carbonila. Por exemplo, um grupo acetila, um grupo propionila, um grupo butirila, um grupo isobutirila, um grupo valerila, um grupo isovalerila, um grupo pivalolila, um grupo pentanoila, e semelhantes podem ser mencionados.
O “grupo alcanoilóxi" na Fórmula (1) acima se refere a um grupo formado pela ligação do “grupo alcanoila baixo” a um átomo de oxigênio, e exemplos do mesmo incluem um grupo acetilóxi, um grupo propionilóxi, um grupo butirilóxi, um grupo isobutirilóxi, um grupo valerilóxi, um grupo isovalerilóxi, um grupo pivaloilóxi, um grupo pentanoilóxi e semelhantes.
O “grupo” alquitio baixo” na Fórmula (1) acima se refere a um substituinte formado pela ligação do “grupo alquil baixo” a um átomo de enxofre e exemplos do mesmo incluem um grupo metiltio, um grupo etiltio, um grupo butiltio e semelhantes.
O termo “PLK” represente uma quinase tipo polo.
O termo “PLK1” é um dos membros da família PLK (quinase tipo polo) constituída por PLK1, PLK2, PLK3 e SAK.
O termo “inibidor de PLK1" é um medicamento para inibição da quinase tipo polo-1. Os termos “sal ou éster farmaceuticamente aceitáveis” e “carreador ou diluente farmaceuticamente aceitáveis” serão explicados adiante.
O termo “tratamento de câncer” (“câncer em tratamento”) como usado na presente especificação significa a inibição do crescimento da célula do câncer ao administrar um agente antitumoral aos pacientes com câncer. O tratamento preferencialmente regressa o crescimento da célula do câncer, ou seja, reduz o tamanho do tumor que pode ser medido. O tratamento mais preferencialmente erradica completamente o câncer. O termo “câncer” como usado na presente especificação inclui tumores sólidos e cânceres hematopoiéticos. Exemplos de tumores sólidos incluem câncer cerebral, câncer de cabeça e pescoço, câncer de esôfago, câncer de tireóide, câncer de pulmão de pequenas células, câncer de pulmão de células não pequenas, câncer de mama, câncer gástrico, câncer de vesícula biliar/ducto hepático, câncer hepático, câncer pancreático, câncer de cólon, câncer retal, câncer ovariano, corioepitelioma, câncer de útero, câncer cervical, câncer pélvico renal/uretral, câncer de bexiga urinária, câncer de próstata, câncer de pênis, câncer testicular, câncer embrionário, tumor de Wilms, câncer de pele, melanoma maligno, neuroblastoma, osteosarcoma, sarcoma de Ewing, sarcoma de tecido molde e semelhantes. Exemplos de cânceres hematopoiéticos incluem leucemia, leucemia linfática crônica, leucemia 5 mielogênica crônica, policitemia verdadeira, linfoma maligno, mieloma múltiplo, linfoma não-Hodgkins e semelhantes.
O termo “preparação” como usado na presente especificação inclui preparações orais e preparações parenterais. A preparação oral está exemplificada por um comprimido, cápsula, pó, granulo ou semelhante, 10 preferencialmente um comprimido, cápsula ou semelhante. A preparação parenteral está exemplificada por uma preparação líquida esterilizada como uma solução, suspensão e semelhante, que é especificamente uma preparação injetável, infusão de gotejamento e semelhantes, e preferencialmente é uma injeção intravenosa e um gotejamento intravenoso.
O termo “preparação combinada” como usado na presente especificação se refere à preparação incluindo dois ou mais tipos para administração simultânea, separada ou seqüencial no tratamento, que pode ser fornecido como uma preparação tipo kit ou uma composição farmacêutica. O termo “preparação combinada” também inclui preparações preparadas por outras combinações de 20 uma ou mais preparações à preparação combinada acima incluindo duas preparações separadas que são úteis no tratamento de câncer.
Em adição às duas preparações separadas, uma ou mais preparações incluindo pelo menos um agente antitumoral selecionado a partir do grupo que consiste de agentes alquilantes antitumorais, anti meta bólitos antitumorais, 25 anticorpos antitumorais, agentes antitumorais derivados de planta, compostos antitumorais de complexo de platina, derivados antitumorais de camptotecina, inibidores antitumorais de tirosina quinase, anticorpos monoclonais, interferons, modificadores da resposta biológica e outros agentes antitumorais (onde a definição de agente antitumoral tem o mesmo significado como definido acima) ou 30 um sal ou éster farmaceuticamente aceitável do mesmo, pode ser ainda combinado junto com um carreador ou diluente farmaceuticamente aceitável. Neste caso, a combinação adicional de pelo menos uma preparação pode ser administrada simultaneamente, separadamente ou sequencialmente com as duas preparações separadas. A preparação combinada incluindo três preparações 35 pode ser exemplificada por: uma preparação compreendendo o composto representado pela Fórmula (1) acima; uma preparação compreendendo 5-fluorouracils; e uma preparação compreendendo leucovorina. Para a preparação combinada, ambas das duas preparações separadas podem ser uma preparação ou uma preparação oral ou uma preparação parenteral, ou de forma alternativa, uma das duas preparações separadas pode ser uma preparação oral enquanto a outra pode ser uma preparação parenteral (preparação injetável ou infusão por gotejamento).
Para a “preparação” de acordo com a invenção, uma quantidade terapeuticamente eficiente do composto de acordo com a invenção pode ser incluída, junta com um carreador ou diluente farmaceuticamente estável. A técnica de formulação acredita-se ser a técnica comum conhecida pelos especialistas no campo técnico, que é, portanto, bem conhecida.
Preferencialmente, tal preparação pode ser formada em, junto com um carreador ou diluente farmaceuticamente aceitável, uma preparação para administração oral, infusão intravenosa ou injeção usando um número de métodos geralmente conhecidos para os especialistas no assunto.
O termo “administração” como usado na presente especificação inclui administração parenteral e/ou administração oral no caso do uso de preparação combinada de acordo com a invenção. Ou seja, quando a preparação combinada é administrada, ambos podem ser administradas via parenteral ou uma pode ser administrada via parenteral enquanto a outro pode ser administrada via oral, ou ainda, ambas podem ser administradas via oral. Aqui o termo “administração parenteral” inclui, por exemplo, administração intravenosa, administração subcutânea, administração intramuscular e semelhantes e preferencialmente inclui administração intravenosa. Quando administrar três ou mais preparações em combinações, pelo menos uma preparação pode ser administrada via parenteral, preferencialmente via intravenosa, e mais preferencialmente administrada por gotejamento intravenosa ou injeção intravenosa.
Adicionalmente, para o caso de administração de três ou mais preparações em combinação, qualquer uma das preparações pode ser uma preparação oral ou uma preparação parenteral.
Ao realizar a invenção, o composto representado pela Fórmula (1) acima pode ser administrado simultaneamente com um outro agente antitumoral. Além disso, o composto representado pela Fórmula (1) acima pode ser administrado ou de forma alternativa, o outro agente antitumoral pode ser administrado primeiro e subsequentemente o composto representado pela Fórmula (1) acima pode ser administrado. Ainda, o composto representado pela Fórmula (1) acima pode ser administrado e em seguida o outro agente antitumoral pode ser administrado separadamente em intervalo de tempo ou de forma alternativa, o outro agente antitumoral pode ser administrado e o composto representado pela Fórmula (1) acima pode ser administrado separadamente no intervalo de tempo. A ordem de administração e intervalo de administração podem ser adequadamente selecionados pelos especialistas na técnica, dependendo da preparação usada incluindo o composto representado pela Fórmula (1) acima, preparação incluindo um agente antitumoral que pode ser usado em combinação, o tipo de células de câncer a serem tratadas e as condições do paciente.
O termo “simultaneamente” como usado na presente especificação significa administração quase ao mesmo tempo para um tratamento. O termo “separadamente” significa em um momento diferente para um tratamento e, por exemplo, se refere ao caso onde uma medicação é usada no primeiro dia e outra medicação é usada no dia seguinte para um tratamento. O termo “sequencialmente” significa administração em ordem, por exemplo, se refere ao caso onde uma medicação é usada primeiro, em seguida a outra medicação é usada após um intervalo de tempo pré-determinado para um tratamento.
O termo “agentes alquilantes antitumorais” como usado na presente especificação significa um agente alquilante com atividade antitumoral, e o “agente alquilante” aqui em geral se refere a um agente que fornece um grupo alquila na reação de alquilação de compostos orgânicos onde o átomo de hidrogênio é substituído por um grupo alquila. Exemplos de “agentes alquilantes antitumorais” incluem N-óxido de mostarda nitrogenada, ciclofosfamida, ifosfamida, melfalan, busulfan, mitobronitol, carboquone, tiotepa, ranimustina, nimustina, temozolomida, carmustina e semelhantes.
O termo “antimetabólitos antitumorais” como usado na presente especificação se refere a um antagonista metabólico que contém uma atividade antitumoral e o “antagonista metabólico” aqui em senso amplo inclui substancias que interferem uma mudança metabólica normal para ocorrer devido à sua estrutura ou função semelhantes aos metabólitos (vitaminas, co-enzimas, aminoácidos, açúcares, etc.) que são fatores importantes nos organismos e substâncias que previnem a produção de intermediários de alta energia ao inibirem o sistema de transporte de elétrons. Exemplos de “antimetabólitos antitumorais” incluem metotrexato, 6-mercaptopurina, ribosídeo, mercaptopurina, 5-fluorouracila, tegafur, doxifluridina, carmofur, citarabina, octofosfato de citarabina, enocitabina, S-1, gemcitabina, fludarabina, pemetrexede dissódico e semelhantes.
O termo “antibióticos antitumorais” como usado na presente especificação se refere a um antibiótico com atividade antitumoral e os “antibióticos” aqui é 5 preparado por microorganismos e inclui substâncias que inibem o crescimento ou outras funções nas células de microorganismos ou outros organismos. Exemplos de “antibióticos antitumorais” incluem actinomicina-D, doxorubicina, daunorubicina, neocarzinostatina, bleomicina, peplomicina, mitomicina C, aclarubicina, pirarubicina, epirubicina, zinostatina estimalamer, idarubicina, 10 sirolimus, valrubicina e semelhantes.
O termo “agentes antitumorais derivados de planta” como usado na presente especificação inclui compostos contendo atividade antitumoral, originados de plantas, e estes compostos que estão sujeitos a modificações químicas. Exemplos de “agentes antitumorais derivados de plantas” incluem 15 vincristina, vinblastina, vindesina, etoposídeo, sobuzoxano, docetaxel, paclitaxel, vinorelbina e semelhantes.
O termo “derivados antitumorais de camptotecina” como usado na presente especificação inclui a camptotecina per se e se refere a um composto inibitório contra a proliferação de células cancerígenas, que é estruturalmente relacionado 20 à camptotecina. Os “derivados antitumorais da camptotecina” não são particularmente limitados a, mas podem ser exemplificados por, camptotecina, 10- hidróxi camptotecina, topotecano, irinotecano, 9-aminocaptotecina, ou semelhantes. O irinotecano é metabolizado in vivo e exibe atividade antitumoral como SN-38. Acredita-se que o derivado de camptotecina tem um mecanismo de 25 ação e atividade quase semelhantes à camptotecina (Nitta et al, Cancer and Chemotherapeutics, 14, 850-857 (1987), etc.).
O termo “compostos antitumorais de complexo de platina” como usado na presente especificação se refere a um composto de complexo de platina contendo atividade antitumoral e o “composto de complexo de platina” aqui significa um 30 composto de complexo de platina que fornece a platina na forma de um íon. Exemplos preferenciais de compostos de platina incluem cisplatina; íon de platina (II) cis-diamina diaquo; cloreto de cloro(dietilenotriamino)-platina(ll); dicloro(etilenodiamino)-platina(H); diamina(1,1 -ciclobutano-dicarboxilato)platina(ll)(carboplatina); espiroplatina; iproplatina; diamino(2- etilmalonato)-platina(ll); etilenodiamino malonato de platina(ll); aqua (1,2-diamino- diciclohexano) sulfato de platina (II); aqua(1,2-diamino-diciclohexano) malonato de platina (II); (1,2-diamino-ciclohexano) de platina (II); (1,2-diaminociclohexano)- (isocitrato) de platina (II); (1,2-diaminociclohexano) oxalato de platina (H); ormaplatina; tetraplatina; carboplatina; nedaplatina; e oxalaplatina. Além disso, outros compostos antitumorais de complexo de platina exemplificados na 5 presente especificação são geralmente conhecidos e comercialmente disponíveis e/ou podem ser para os especialistas no assunto em conformidade com as técnicas convencionais.
O termo “inibidores antitumorais da tirosina quinase” como usado na presente especificação se refere a um inibidor da tirosina quinase contendo 10 atividade antitumoral e o “inibidor da tirosina quinase” aqui se refere a uma substância química para inibir uma “tirosina quinase” que envolve a transferência de um grupo y-fosfato de um ATP para um grupo hidroxila de uma tirosina específica em proteínas. Exemplos de “inibidores antitumorais de tirosina quinase” incluem gefitinibe, imatinibe, erlotinibe e semelhantes.
O termo “anticorpos monoclonais” como usado na presente especificação se refere a um anticorpo produzido a partir de células monoclonais formadores de anticorpos e exemplos incluem cetuximabe, bevacizumabe, rituximabe, alemtuzumabe, trastuzumabe e semelhantes.
O termo “interferons” como usado na presente especificação se refere a um 20 interferon contendo atividade antitumoral e geralmente no caso de infecção virai, é uma glicoproteína com peso molecular de cerca de 20.000 que é produzido/secretado a partir de maioria das células animais. Bem como inibir a proliferação virai, o interferon inibe a proliferação de células (células tumorais em particular) e exibe varias atividades executoras da imunidade incluindo aumento 25 da atividade de natural killer,e é conhecida como uma das citocinas. Exemplos de “interferon” incluem a-interferon. a2a-interferon, a2b-interferon, β-interferon, y1a- interferon, y1 n-interferon e semelhantes.
O termo “modificadores da resposta biológica” como usado na presente especificação também é abreviado como BRM e geralmente é um termo genérico 30 de substâncias ou medicamentos que levam a atingir benefícios individuais contra tumores, infecções ou outras doenças ao regular reações biológicas como o mecanismo de defesa controlado pelos organismos e sobrevivência, proliferação ou diferenciação de células de tecido. Exemplos de “modificadores de resposta biológica” incluem krestin, lentinano, esquizofilano, picibanil, ubenimex e 35 semelhantes.
O termo “outros agentes antitumorais"como usado na presente especificação se refere a um agente antitumoral contendo atividade que não está incluída em nenhuma das acima. Exemplos de “outros agentes antitumorais” incluem mitoxantrona, L-asparaginase, procarbazina, dacarbazina, hidroxicarbamida, pentostaina, tretinoína, alefacept, alfa darbepoetina, anastrazol, exemestano, bicalutamida, leuprorelina, flutamida, fulvestrano, pegaptanibe, denileucina diftitox octassódica, aldesleucina, alfa tirotropina, trióxido de arsênico, bortezomibe, capecitabina, goserelina e semelhantes. Todos os “agentes antitumorais alquilantes”, “antimetabólitos antitumorais”, “antibióticos antitumorais”, “agentes antitumorais derivados de plantas”, “compostos antitumorais de complexo de platina”, “derivados antitumorais de camptotecina”, “inibidores antitumorais de tirosina quinase”, “anticorpos monoclonais”, “interferons”, “modificadores da resposta biológica” e “outros agentes antitumorais” acima são geralmente conhecidos e disponíveis comercialmente ou podem ser daqueles especialistas no assunto em conformidade com os métodos conhecidos per se ou métodos comumente conhecidos/usados. É descrito um processo para a produção de gefitinibe, por exemplo, na especificação de Patente U.S. No. 5.770.599; um processo para produção de cetuximabe, por exemplo, na Publicação Internacional W096/10210; um processo para produção de oxaliplatina, por exemplo, nas especificações da Patente U.S. Nos. 5.420.319 e 5.959.133; um processo para produção de gemcitabina, por exemplo, nas especificações da Patente U.S. Nos. 5.434.254 e 5.223.608; um processo para produção de camptotecina nas especificações da Patete U.S. Nos. 5.162.532, 5.247.089, 5.191.082, 5.200.524, 5.243.050 e 5.321.140; um processo para produção de irinotecano, por exemplo, na especificação da Patente U.S. No. 4.604.463; um processo para produção de topotecano, por exemplo, na especificação da Patente U.S. No. 5.734.056; um processo para produção de temozolomida, por exemplo na especificação da Publicação de Patente Japonesa Não Avaliada No. H4-5029; e um processo para produção de rituximabe na especificação de Publicação de Patente Japonesa Não Avaliada No. H2-503143.
Para os agentes antitumorais alquilantes, por exemplo, o N-óxido de mostarda nitrogenada está comercialmente disponível sob o nome de marca Nitromin da Mitsubishi Pharma Corporation; a ciclofosfamida está comercialmente disponível sob o nome de marca Endoxan da Shionogi &Co., Ltd.; ifosfamida está comercialmente disponível sob o nome de marca ifomide de Shionogi &Co., Ltd.;melfalan está comercialmente disponível sob o nome de marca Alkeran da GlaxoSmithKline; busulfano está comercialmente disponível sob o nome de marca Myebrol da Kyorin Pharmaceutical Co., Ltd.; carboquone está comercialmente disponível sob o nome de marca Esquinon da Sankyo Co., Ltd; tiotepa está comercialmente disponível sob o nome de marca Tespamin sobre Sumitomo Pharmaceuticals Co., Ltd; ranimustina está comercialmente disponível sob o nome Cymerin da Mitsubishi Pharma Corporation; nimustina está comercialmente disponível sob o nome de marca Nidran da Sankyo Co., Ltd.; temozolomida está comercialmente disponível sob o nome de marca Temodal da Schering-Plough Co., Ltd.; e carmustina está comercialmente disponível sob o nome de marca Gliadel Wafer da Guilford Pharmaceuticals. Para os antimetabólitos antitumorais, por exemplo, metotrexato está comercialmente disponível sob o nome de marca Metotrexaro da Takeda pharmaceutical; ribosídeo de 6-mercaptopurina está comercialmente disponível sob o nome de marca Thioinosine da Aventos Co., Ltd.; mercaptopurina está comercialmente disponível sob o nome de marca Leukrein da Takeda pharmaceutical; 5-fluorouracila está comercialmente disponível sob o nome de marca 5-FU da Kyowa Hakko Co., Ltd.; tegafur está comercialmente disponível sob o nome de marca Futraful da Taiho Pharmaceutical Co., Ltd.; doxifluridina está comercialmente disponível sob o nome de marca Yamafur da Yamanouchi Pharmaceutical; citarabina está comercialmente disponível sob o nome de marca Cylocide da Nippon Shinyaku Co., Ltd.; octofosfato de citarabina está comercialmente disponível sob o nome de marca Starasid da Nippon Kayaku Co., Ltd.; enocitabina está comercialmente disponível sob o nome de marca Sunrabin da Asahi Kasei Corporation; S-1 está comercialmente disponível sob o nome de marca TS-1 da Taiho Pharmaceutical Co., Ltd.; gemcitabina está comercialmente disponível sob o nome de marca Gemzar da Eli Lily Co., Ltd.; fludarabina está comercialmente disponível sob o nome de marca Fludara da Japan Schering- Plough K.K.; e pemetrexede dissódico está comercialmente disponível sob o nome de marca Alimta da Eli Lily Co., Ltd.
Para os antibióticos antitumorais, por exemplo, actinomicina D está comercialmente disponível sob o nome de marca Cosmegen da Banyu Pharmaceutical Co., Ltd.; doxorubicina está comercialmente disponível sob o nome de marca Daunomycin da Meiji Seika, Ltd.; neocarzinostatina está comercialmente disponível sob o nome de marca Neocarzinastatin da Yamanouchi Pharmaceutical; bleomicina está comercialmente disponível sob o nome de marca Bleo da Nippon Kayaku Co., Ltd.; peplomicina está comercialmente disponível sob o nome de marca Peplo da Nippon Kayaku Co., Ltd.; mitomicina C está comercialmente disponível sob o nome de marca Mitomycin da Kyowa Hakko Co., Ltd.; aclarubicina está comercialmente disponível sob o nome de marca Aclacinon da Yamanouchi Pharmaceutical; pirarubicina está comercialmente disponível sob o nome de marca Pinorubin da Nippon Kayaku Co., Ltd.; epirubicina está comercialmente disponível sob o nome de marca Farmorubicina da Pharmacia corporation; zinostatina estimalamer está comercialmente disponível sob o nome de marca Smancs da Yamanouchi Pharmaceutical; idarubicina está comercialmente disponível sob o nome de marca Idamycin da Pharmacia corporation; sirolimus está comercialmente disponível sob o nome de marca Rapamune da Wyeth; e valrubicina está comercialmente disponível sob o nome de marca Valstar da Anthra pharmaceutical. Para os agentes antitumorais derivados de plantas, por exemplo, vincristina está comercialmente disponível sob o nome de marca Oncovin da Shionogu & Co., Ltd.; vinblastina está comercialmente disponível sob o nome de marca Vinblastin da Kyorin Pharmaceutica Co., Ltd.; vindesina está comercialmente disponível sob o nome de marca Fildesin da Shionogi &Co., Ltd.; etoposídeo está comercialmente disponível sob o nome de marca Lastet da Nippon Kayaku & Co., Ltd.; sobzoxano está comercialmente disponível sob o nome de marca Perazolin da Zenyaku Kogyo; docetaxel está comercialmente disponível sob o nome de marca Taxotere da Aventis Co., Ltd.; paclitaxel está comercialmente disponível sob o nome de marca Taxol da Bristol-Myers K.K.; e vinorelbina está comercialmente disponível sob o nome de marca Navelbine da Kyowa Hakko Co., Ltd.
Para os compostos antitumorais de complexo de platina, por exemplo, cisplatina está comercialmente disponível sob o nome de marca Randa da Nippon Kayaku Co., Ltd.; carboplatina está comercialmente disponível sob o nome de marca Paraplatin da Bristol-Myers K.K.; nedaplatina está comercialmente disponível sob o nome de marca Eloxatin da Sanofi K.K.
Para os derivados antitumorais de camptotecina, por exemplo, irinotecano está comercialmente disponível sob o nome de marca Campto da Yakult Co., Ltd.; topotecano está comercialmente disponível sob o nome de marca Hycamtin da GlaxoSmithKline; e captotecina está comercialmente disponível da Aldrich Chemical Company, U.S.A., etc.
Para os inibidores de tirosina quinase, por exemplo, gefitinibe está comercialmente disponível sob o nome de marca Iressa da AstraZeneca; imitinabe está comercialmente disponível sob o nome de marca Gleevec da Novartis pharma K.K.; e erlotinibe está comercialmente disponível sob o nome de 5 marca Tarceva da OSI pharmaceutical, Inc. Para os anticorpos monoclonais, por exemplo, cetuximabe está comercialmente disponível sob o nome de marca Erbitux da Bristol-Myers Squibb Company; bevacizumabe está comercialmente disponível sob o nome de marca Avastin da Genetech Inc.; rituximabe está comercialmente disponível sob o nome de marca Rituxan da Biogen Idee Inc.; 10 alemtuzumabe está comercialmente disponível sob o nome de marca Campath da Berlex, Inc.; e trastuzumabe está comercialmente disponível sob o nome de marca Herceptin da Chugai Pharmaceutical Co., Ltd.
Para os interferons, por exemplo, a-interferon está comercialmente disponível sob o nome de marca Sumiferon da Sumitomo Pharma Co., Ltd; α2a- 15 interferon está comercialmente disponível sob o nome de marca Canferon-A da Takeda pharmaceutical; α2b-interferon está comercialmente disponível sob o nome de marca Intron A da Schering-Plough Co., Ltd.; β-interferon está comercialmente disponível sob o nome de marca IFN β da Mochida Pharmaceutical Co., Ltd.; y1 a-interferon está comercialmente disponível sob o 20 nome de marca Imunomax-y da Shionogi &Co., Ltd.; e yn1 -interferon está comercialmente disponível sob o nome de marca Ogamma da Otsuka Pharmaceutical Co., Ltd.
Para os modificadores de resposta biológica, por exemplo krestin está comercialmente disponível sob o nome de marca Krestin da Sankyo Co., Ltd.; 25 lentinano está comercialmente disponível sob o nome de marca Lentinan da Aventis Co., Ltd.; ezquizofilano está comercialmente disponível sob o nome de marca Sonifilan da Kaken Pharmaceutical Co., Ltd.; picibanil está comercialmente disponível sob o nome de marcaPicibanil da Chugai Pharmaceutical Co., Ltd.; e ubenimex está comercialmente disponível sob o nome de marca Bestatin da 30 Nippon Kayaku Co., Ltd.
Para os outros agentes antitumorais, por exemplo, mitoxantrona está comercialmente disponível sob o nome de marca Novantron da Wyeth-Lederie Japan; L-asparaginase está comercialmente disponível sob o nome de marca Leunase da Kyowa Hakko Co., Ltd.; procarbazina está comercialmente disponível 35 sob o nome de marca Natula da Roche Japan; dacarbazina está comercialmente disponível sob o nome de marca Dacarbazine da Kyowa Hakko Co., Ltd.; hidroxicarbamida está comercialmente disponível sob o nome de marca sob o nome Hydrea da Bristol K.K.; pentostatina está comercialmente disponível sob o nome de marca Coforin da Chemo-Sero-Therapeutic Research Institute; tretinoína está comercialmente disponível sob o nome de marca Vesanoid da Roche Japan; alefacept está comercialmente disponível sob o nome de marca Amevive da Biogen Idee Inc.; alfa darbepoetina está comercialmente disponível sob o nome de marca Aranesp da Amgem Inc., anastrazol está comercialmente disponível sob o nome de marca Arimidex da AstraZeneca; exemestano está comercialmente disponível sob o nome de marca Aromasin da Pfizer Inc.; bicalutamida está comercialmente disponível sob o nome de marca Casodex da AstraZeneca; leuprorelina está comercialmente disponível sob o nome de marca Lupulin da Takeda Pharmaceutical; flutamida está comercialmente disponível sob o nome de marca Eulexin da Schering-Plough Co., Ltd.; fulvestrano está comercialmente disponível sob o nome de marca Faslodex da AstraZeneca; pegaptanibe octassódico está comercialmente disponível sob o nome de marca Macugen da Guilead Sciences Inc.; denileucina diftitox está comercialmente disponível sob o nome de marca Ontak da Ligan Pharmaceuticals Inc.; aldesleucina está comercialmente disponível sob o nome de marca Proleukin da Chiron Corporation; alfa tirotropina está comercialmente disponível sob o nome de marca Thyrogen da Genzyme; trióxido de arsênico está comercialmente disponível sob o nome de marca Trisenox da Cell Therapeutics, Inc.; bortezomibe é produzido sob o nome de marca Velcade da Millenium; capecitabina é produzida sob o nome de marca Xeloda da Roche; e goserelina é produzida sob o nome de marca Zoladex da AstraZeneca. O termo “agentes antitumorais” como usado na presente especificação inclui agentes antitumorais selecionados a partir de “agentes antitumorais alquilantes”, “antimetabólitos antitumorais”, “antibióticos antitumorais”, “agentes antitumorais derivados de planta”, “compostos antitumorais de complexo de platina”, “antitumorias derivados de camptotecina”, “inibidores antitumorais de tirosina quinase”, “anticorpos monoclonais”, “interferons”, “modificadores de resposta biológica” e “outros agentes antitumorais”. Realizações do composto representado pela Fórmula (1) acima serão descritas em maiores detalhes. Ri e R2, que podem ser o mesmo ou diferente, são cada, um átomo de hidrogênio, um átomo de halogênio, um grupo alquila baixo que pode ser substituído por um ou dois átomos de flúor; ou um grupo ciclopropila; R-i é preferencialmente um grupo alquila baixo contendo 1 a 2 átomos de carbono que podem ser substituídos por 1 a 3 átomos de flúor; um grupo ciclopropila; ou um átomo de cloro. R1 é mais preferível que seja um grupo metila, um grupo etila, um grupo difluorometila, um grupo trifluorometila, um grupo ciclopropila, ou um átomo de cloro. RT é ainda mais preferível que seja um grupo etila, um grupo difluorometila, um grupo trifluorometila, um grupo ciclopropila, ou um átomo de cloro. R2 é preferencialmente um átomo de hidrogênio. Para R3 e R4, um de R3 e R4 é um átomo de hidrogênio e o outro de R3 θ R4 é: a) um grupo alquila baixo substituído com NR3R4, onde R3 e R4, que podem ser o mesmo ou diferentes são, cada um, um átomo de hidrogênio, um grupo alquila baixo, um grupo benzila ou um grupo cicloalquila contendo três a seis átomos de carbono, onde o grupo cicloalquila pode ser substituído por um ou mais substituintes, que pode ser 0 mesmo ou diferente, selecionado a partir do seguinte 1) a 3): 1) um grupo alquila baixo; 2) um substituinte selecionado do <Grupo Substituinte β>; e 3) um grupo alquila baixo substituído por um ou mais substituintes selecionados a partir do <Grupo Substituinte β>; e o grupo cicloalquila pode incluir uma ligação insaturada; b) um grupo heterocíclico alifático com 4- a 6- membros selecionado a partido do grupo azetidinila, um grupo pirrolidinila, um grupo piperidinila, e um grupo piperazinila; ou c) um grupo alquila baixo substituído com um grupo heterocíclico alifático com 4- a 6- membros selecionados a partir do grupo azetidinila, um grupo pirrolidinila, um grupo piperidinila e um grupo pirrolila, um grupo imidazolila, um grupo pirazolila, um grupo piridila, um grupo pirazinila, e um grupo e um grupo pirimidinila; ou e) um grupo alquila baixo substituído com um grupo heterocíclico aromático com 5- ou 6- membros selecionado a partir de um grupo pirrolila, um grupo imidazolila, um grupo pirazolila, um grupo piridila, um grupo pirazinila e um grupo pirimidinila, onde o grupo heterocíclico alifático e o grupo heterocíclico aromático cada um independentemente pode ser substituído por um ou mais substituintes, que podem ser 0 mesmo ou diferentes, selecionados a partir de 1) a 4): 1) um grupo alquila baixo; 2) um substituinte selecionado do <Grupo Substituinte β>; 3) um grupo alquila substituído com um ou mais substituintes selecionados a partir do <Grupo Substituinte β>; e 4) um grupo cicloalquila contendo 3 a 6 átomos de carbono, que podem ser substituídos com um ou mais substituintes selecionados a partir do <Grupo Substituinte β>. Para R3 e R4, é preferencial que um de R3 e R4 seja um átomo de hidrogênio e o outro de R3 e R4 seja: a) um grupo alquila substituído com NR3R4 onde R3 e R4, que pode ser o mesmo ou diferente, cada um, um átomo de hidrogênio, onde um grupo alquila baixo ou um grupo cicloalquila contendo cinco a seis átomos de carbono, onde o grupo cicloalquila pode ser substituído por um ou mais substituintes, que podem ser o mesmo ou mais substituintes que podem ser o mesmo ou diferentes, selecionados a partir do seguinte 1) a 3): 1) um grupo alquila baixo; 2) um substituinte selecionado do <Grupo Substituinte β>; e 3) um grupo alquila substituído com um ou mais substituintes selecionados a partir do <Grupo Substituinte β>; ou b) um grupo heterociclico alifático selecionado a partir de um grupo azetidinila, um grupo pirrolidinila e um grupo piperidinila, onde o grupo heterociclico alifático pode ser substituído com um ou mais substituintes, que podem ser o mesmo ou diferentes, selecionados a partir de 1) a 3): 1) um grupo alquila baixo; 2) um substituinte selecionado a partir do <Grupo Substituinte β>; e 3) um grupo alquila baixo substituído com um ou mais substituintes selecionados a partir do <Grupo Substituinte β>. O <Grupo Substituinte β>para R3 e R4 é preferencialmente um grupo que consiste de um átomo de halogênio, um grupo amino, um grupo alquilsulfonila baixo e um grupo alcóxi baixo.
Para R3 e R4, é mais preferível que um de R3 e R4 seja um átomo de hidrogênio e ou outro de R3 e R4 seja um grupo alquila baixo (onde o dito alquila baixo é um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono que é N-substituído ou N,N-di-substuiído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo piperidinila que é N- substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo pirrolidinila que é N-substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo azetidinila que é N- substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; ou um grupo cicloalquila contendo cinco a seis átomos de carbono, onde 5 um grupo piperidinila, o grupo pirrolidina e o grupo azetidinila cada um independentemente pode ser ainda substituído por um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono, e o grupo cicloalquila pode ser substituído por um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono opcionalmente tendo um grupo hidróxi. Aqui, o grupo piperidinila é 10 preferencialmente piperidin-3-ila N-substituída, piperidin-4-ila N-substituída ou semelhantes. O grupo pirrolidinila é preferencialmente pirrolidin-2-ila N- substituída, pirrolidin-3-ila N-substituída, ou semelhantes e mais preferencialmente pirrolidin-2-ila N-substituída, pirrolidin-3-ila N-substituída, ou semelhantes, e mais preferencialmente pirrolidin-2-ila N-substituída. O grupo 15 azetidinila é preferencialmente azetitidin-3-ila ou semelhante. O grupo cicloalquila é preferencialmente ciclopentila, ciclohexila ou semelhante, mais preferencialmente ciclopentila. Para R3 e R4, é ainda mais preferencial que um de R3 e R4 seja um átomo de hidrogênio, e o outro de R3 e R4 seja um grupo alquila linear ou ramificado 20 contendo 1 a 3 átomos de carbono que é substituído por um grupo dimetilamino, um grupo isopropilamino, um grupo 1,1-dimetilpropilamino, ou um grupo t- butilamino; um grupo piperidinila que é N-substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo pirrolidinila que é N- substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de 25 carbono; um grupo azetidinila que é N-substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; ou um grupo ciclopentila que pode ser substituído com um grupo metila ou um grupo hidroximetila, onde 0 grupo piperidinila, o grupo pirrolidinila e o grupo azetidinila podem ainda ser substituídos por um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono. R5 é um átomo de hidrogênio, um grupo ciano, um átomo de halogênio, ou um grupo alquila baixo. R5é preferencialmente um átomo de hidrogênio, um grupo ciano, um átomo de halogênio, ou um grupo metila, mais preferencialmente um grupo ciano, um átomo de halogênio, ou um grupo metila, particularmente preferencial um grupo ciano, um átomo de flúor ou um grupo metila.
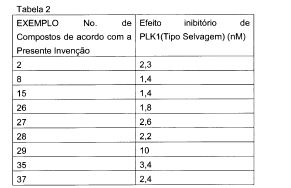
Para R1 a R5, do ponto de vista da exibição do efeito inibitório de proliferação celular com base no efeito inibitório de PLK1, o seguinte caso (A) é preferencial, o caso seguinte (B) é mais preferencial e o caso seguinte (C) é ainda mais preferencial. (A) Caso onde: Ri é um grupo alquila contendo 1 a 2 átomos de carbono que pode ser substituído por 1 a 3 átomos de flúor, um grupo ciclopropila ou um átomo de halogênio; R2 é um átomo de hidrogênio; um de R3 e R4 é um átomo de hidrogênio, enquanto o outro de R3 e R4 é um grupo alquila amino baixo (onde o dito alquila é um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono) que é N-substituído ou N,N-di-substituído com um grupo alquila N- substituído linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo piperidinila que é N-susbtituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo pirrolidinina que é N-substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo azetidinila que é N-substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; ou um grupo cicloalquila contendo cinco a seis átomos de carbono, onde um grupo piperidinila, o grupo pirrolidinila e o grupo azetidinila cada um independentemente pode ser ainda substituído por um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono, e 0 grupo cicloalquila pode ser substituído por um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono opcionalmente contendo um grupo hidróxi; e R5 é um grupo ciano, um átomo de halogênio ou um grupo metila. (B) Caso onde: R-i é um grupo metila, um grupo etila, um grupo dilfuorometila, um grupo trifluorometila, um grupo ciclopropila ou um átomo de cloro; R2 é um átomo de hidrogênio; um de R3 e R4 é um átomo de hidrogênio, enquanto o outro de R3 e R4 é um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono) que é substituído com um grupo dimetilamino, um grupo isopropilamino, um grupo 1,1- dimetilpropilamino ou um grupo t-butilamino; um grupo piperidinila que é N- susbtituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo pirrolidinina que é N-substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo azetidinila que é N- substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; ou um grupo ciclopentila que pode ser substituído com um grupo metila ou um grupo hidroximetila, onde o grupo piperidinila, o grupo pirrolidinila e o grupo azetidinila cada um independentemente pode ser ainda substituído por um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono; e R5 é um grupo ciano, um átomo de flúor ou um grupo metila. (C) Caso onde: R1 é um grupo etila, um grupo difluorometila, um grupo trifluorometila, um grupo ciclopropila ou um átomo de cloro. 5 R2 é um átomo de hidrogênio; um de R3 e R4 é um átomo de hidrogênio, enquanto o outro de R3 e R4 é um grupo t-butilaminometila; um grupo 1-metil-dimetilaminoetila; um grupo piperidin-3-ila que é N-substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo piperidin-4-ila que é N-substituído 10 com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo pirrolidin-2-ila que é N-substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo azetidin-3-ila que é N- substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; ou um grupo ciclopentila que pode ser substituído por um grupo metila 15 ou um grupo hidroximetila, onde o grupo piperidin-3-ila, o grupo piperidin-4-ila, o grupo pirrolidin-2-ila e o grupo azetidin-3-ila podem ainda ser substituídos com um grupo metila; e R5 é um grupo ciano ou um átomo de flúor.
O <Grupo Substituinte β>é um grupo consistindo de um átomo de 20 halogênio, um grupo hidróxi, um grupo nitro, um grupo ciano, um grupo amino, um grupo carbamoila, um grupo aminosulfanila, um grupo imino, um grupo alquilsulfonila baixo, um grupo alquilsulfonilamino baixo, um grupo alcóxi baixo, um grupo alcoxicarbonila baixo, um grupo alcoxicarbonilamino baixo, um grupo alcanoila baixo, um grupo alcanoilóxi baixo, um grupo alquitio baixo, um grupo 25 carboxila baixo e um grupo benzila.
O <Grupo Substituinte β>é preferencialmente um grupo que consiste de um átomo de halogênio, um grupo hidróxi, um grupo alquilsufonila baixo, e um grupo alcóxi baixo.
Em relação ao átomo de carbono assimétrico indicado na estrutura a 30 seguir, o composto representado pela Fórmula (1) é preferencialmente uma forma S.

O composto da Fórmula (1) acima é preferencialmente (a)2-[((1S)-1-{4-[2-(tert-butilamino)-1-hidroxietil]fenil}etil)amino]-4-(8- etilimidazo[1,2-a]piridin-3-il)pirimidina-5-carbonitrila (Exemplos 2 e 3); (b)4-(8-etilimidazo[1,2-a]piridin-3-ila)-2-[(( 1 S)-1 -{4-[hid róxi( 1 - isopropilpiperidin-4-ila)metil]fenil}etill)amino]pirimidina-5-carbonitrila (Exemplos 9 e10); (c)4-(8-ciclopropilimidazo[1,2-a]piridin-3-ila)-2-[((1 S)-1 -{4-[hidróxi(1 - metilpiperidin-4-ila)metil]fenil}etil)amino]pirimidina-5-carbonitrila (Exemplo 12); (d)4-(8-etilimidazo[l,2-a]piridin-3-ila)-2-[((15)-l-{4-[hidróxi(l-metilpiperidin-3- ila)metil]fenil}etil)amino]pirimidina-5-carbonitrila (Exemplos 13, 14, 15 e 16); (e)2- [((1 S)-l-{4-t2-(tert-butilamino)-1 -hidroxietil]fenil}etil)amino]-4-(8-cloroimidazo[1,2- a]piridin-3-ila)pirimidina-5-carbonitrila (Exemplo 22); (f )2-[(( 1 S)-1 -{4-[2-(tert-butilamino)-1 -hid roxietil]fenil}etil)amino]-4-[8- (difluorometil)imidazo[l,2-a]piridin-3-ila]pirimidina-5-carbonitrila (Exemplos 4 e 21); (g) 4-(8-ciclopropilimidazo[1,2-a]pirid in-3-ila)-2-[(( 1 S)-1 -{4-[( 1,2- dimetilpirrolidin-2- ila)(hidróxi)metil]fenil}etil)amino]pirimidina-5-carbonitrila (Exemplos 23, 24, 25, e 26); (h) (1S)-2-(tert-butilamino)-1-[4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3- ila)-5-fluoropirimidin-2-ila]amino}etil)fenil]etanol (Exemplo 27); (i) (1S)-1-[4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-ila)-5-fluoropirimidin- 2- ila]amino}etil)fenil]-2-[(l-metilciclopentil)amino]etanol (Exemplo 28); (j) (IS)-2- (tert-butilamino)-l -[4-((1 S)-1 -{[4-(8-ciclopropilimidazo[1,2-a]piridin-3-ila)-5- metilpirimidin-2-ila]amino}etil)fenil]etanol (Exemplo 29); (1) (1S)-2-(tert-butilamino-1-[4-((1S)-1-{[4-(8-ciclopropilimidazo[1,2-a]piridin- 3-ila)-5- fluoropirimidin-2-ila]amino}etil)fenil]etanol (Exemplo 30); (2) (1 S)-2-(tert-butilamino)-1 -{4-[( 1 S)-1 -({5-fluoro-4-[8- (trifluorometila)imidazo[1,2-a]piridin-3-ila]pirimidin-2-ila}amino)etil]fenil}etanol (Exemplo 31); (m) [4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-ila)-5-fluoropirimidin-2- ila]amino}etil)fenil](1,2-dimetilpirrolidin-2-ila)metanol (Exemples 32 e 33); (n) l-[4-(( 1 S)-]-{[4-(8-cloroimidazo[1,2-a]piridin-3-ila)-5-fluoropirimidin-2- ila]amino}etill)fenil]-2-(dimetilamino)-2-metilpropan-1-ol (Exemplos 34 e 35); (o) [4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-ila)-5-fluoropirimidin-2- ila]amino}etil)fenil](l- isopropilazetidin-3-ila)metanol (Exemplos 36 e 37); ou (p) (1S)-2-(tert-butilamino)-1-{4-[(1S)-1-({4-[8-(difluorometil)imidazo[1,2- a]piridin-3-ila]-5- fluoropirimidin-2-ila}amino)etill]fenil}etanol (Exemplo 38), ou urn sal ou éster farmaceuticamente aceitável do mesmo. Adicionalmente, os aspectos preferenciais da presente invenção também podem ser representados como a seguir: (1) Um composto de Fórmula (1) acima ou um sal ou éster farmaceuticamente aceitável do mesmo onde R5 é um átomo de hidrogênio, um grupo ciano, um átomo de halogênio ou um grupo metila; ou (3) O composto descrito em (1) acima ou um sal ou éster farmaceuticamente ativo do mesmo onde R1 é um grupo alquila baixo contendo um ou dois átomos de carbono que podem ser substituídos por 1 a 3 átomos de flúor; um grupo ciclopropila; ou um átomo e R2 é um átomo de hidrogênio; ou (4) O composto descrito em (1) ou (2) acima ou um sal ou éster farmaceuticamente aceitável do mesmo onde o <Grupo Substituinte β>é um átomo de halogênio, um grupo hidróxi, um grupo amino, um grupo alquil Sulfonila e um grupo alcóxi baixo; ou (5) O composto descrito em um de (1) a (3) acima ou um sal ou éster farmaceuticamente aceitável do mesmo, onde um de R3 e R4 é um átomo de hidrogênio e o outro de R3 e R4: a) o grupo alquila baixo substituído com NRaRb, onde Ra e Rb, que podem ser o mesmo ou diferente, são cada um, átomo de hidrogênio, um grupo alquila baixo ou um grupo cicloalquila contendo cinco a seis átomos de carbono, onde o grupo cicloalquila pode ser substituído com um ou mais substituintes, que pode ser o mesmo ou diferente, selecionados a partir de 1) a 3): 1) um grupo alquila; (6) um substituinte selecionado a partir do <Grupo Substituinte β>; e (7) um grupo alquila baixo substituído com um ou mais substituinte selecionados a partir do <Grupo Substituinte β>; ou b) um grupo heterociclico alifático com 4- ou 6- membros selecionados a partir do grupo azetidinila, um grupo pirrolidinila e um grupo piperidinila, onde o grupo heterociclico alifático pode ser substituído com um ou mais substituintes, que podem ser o mesmo ou diferentes selecionados a partir dos seguintes 1) a 4): 1) um grupo alquila baixo; 2) um substituinte selecionado a partir do <Grupo Substituinte β>; e 3) um grupo alquila baixo substituído por um ou mais substituintes selecionados a partir do <Grupo Substituinte β>; ou (5) O composto descrito em um de (1) a (4) acima ou um sal ou éster farmaceuticamente aceitável do mesmo, onde Ri é um grupo alquila contendo 1 a 2 átomos de carbono que pode ser substituído por 1 a 3 átomos de flúor, um grupo ciclopropila ou um átomo de halogênio; R2 é um átomo de hidrogênio; um de R3 e R4 é um átomo de hidrogênio, enquanto o outro de R3 e R4 é um grupo alquila amino baixo (onde o dito alquila baixo é um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono) que é N-substituído ou N,N-di-substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo piperidinila que é N- susbtituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; ou um grupo cicloalquila contendo cinco a seis átomos de carbono, onde um grupo piperidinila, o grupo pirrolidinila e o grupo azetidinila cada um independentemente pode ser ainda substituído por um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono, e o grupo cicloalquila pode ser substituído por um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono opcionalmente contendo um grupo hidróxi; e R5 é um grupo ciano, um átomo de halogênio ou um grupo metila; ou (6) O composto descrito em um de (1) a (5) acima ou um sal ou éster farmaceuticamente aceitável do mesmo, onde Ri é um grupo metila, um grupo etila, um grupo dilfuorometila, um grupo trifluorometila, um grupo ciclopropila ou um átomo de cloro; R2 é um átomo de hidrogênio; um de R3 e R4 é um átomo de hidrogênio, enquanto 0 outro de R3 e R4 é um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono) que é substituído com um grupo dimetilamino, um grupo isopropilamino, um grupo 1,1- dimetilpropilamino ou um grupo t-butilamino; um grupo piperidinila que é N- susbtituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo pirrolidinina que é N-substituído com um grupo alquila linear ou ramificado contendo 1 a 5 átomos de carbono; um grupo azetidinila que é N- substituído com um grupo aliquila linear ou ramificado contendo 1 a 5 átomos de carbono; ou um grupo ciclopentila que pode ser substituído com um grupo metila ou um grupo hidroximetila, onde o grupo piperidinila, o grupo pirrolidinila e 0 grupo azetidinila cada um independentemente pode ser ainda substituído por um grupo alquila linear ou ramificado contendo 1 a 3 átomos de carbono; e R5 é um grupo ciano, um átomo de flúor ou um grupo metila. Para a preparação combinada de acordo com a invenção, que é formada por duas preparações separadas, é preferível que uma ou duas das preparações separadas seja uma preparação oral ou uma preparação parenteral. Na preparação combinada de acordo com a invenção que é formado por duas preparações separadas, preferencialmente uma preparação junto com um carreador ou diluente farmaceuticamente aceitáveis, seja uma preparação que compreenda: (a)2-[((1 S)-1-{4-[2-(tert-butilamino)-1-hidroxietil]fenil}etil)amino]-4-(8- etilimidazo[1,2-a]piridin-3-ila)pirimidina-5-carbonitrila; (b)4-(8-etilimidazo[1,2-a]piridin-3-ila)-2-[((1 S)-1 -{4-[hidróxi(1 - isopropilpiperidin-4-ila)metil]fenil}etil)amino]pirimidina-5-carbonitrila; (c)4-(8-ciclopropilimidazo[1,2-a]piridin-3-ila)-2-[((1S)-1-{4-[hidróxi(1- metilpiperidin-4-ila)metil]fenil}etil)amino]pirimidina-5-carbonitrila; (d)4-(8-etilimidazo[1,2-a]piridin-3-ila)-2-[((1 S)-1 -{4-[hidróxi(1-metilpiperidin- 3-ila)metil]fenil}etil)amino]pirimidina-5-carbonitrila; (e)2-[((1S)-1-{4-[2-(tert- butilamino)-1-hidroxietil]fenil}etil)amino]-4-(8-cloroimidazo[l,2-a]piridin-3- ila)pirimidina-5-carbonitrila; (f )2-[(( 1S)-1 -{4-[2-(tθrt-butilamino)-1 -hidroxietil]fenil}etil)amino]-4-[8- (difluorometil)imidazo[1,2-a]pi rid i n-3-i la]pi ri mid i na-5-ca rbo n itri la; (f )2-[(( 1S)-1 -{4-[2- (tert-butilamino)-1-hidroxietil]fenil}etil)amino]-4-[8-(difluorometil)imidazo[1,2- a]piridin-3-ila]pirimidina-5-carbonitrila; (g) 4-(8-ciclopropilimidazo[1,2-a]piridin-3- ila)-2-[((1 S)-1 -{4-[(1,2-dimetilpirrolidin-2- ila)(hid róxi)metil]fenil}etil)amino]pirimid ina-5-carbonitrila; (h) (1 S)-2-(tert- butilamino)-1-[4-((1S)- 1 -{[4-(8-cloroimidazo[1,2-a]piridin-3-ila)-5-fluoropirimidin-2- ila]amino}etil)fenil]etanol; (i) (1 S)-1 -[4-((1 S)-1 -{[4-(8-cloroimidazo[1,2-a]piridin-3-ila)-5-fluoropirimidin- 2- ila]amino}etil)fenil]-2-[(1-metilciclopentil)amino]etanol; 0) (1S)-2-(tert-butilamino)-1-[4-((1S)-1-{[4-(8-ciclopropilimidazo[1,2-a]piridin- 3-ila)-5- metilpirimidin-2-ila]amino}etil)fenil]etanol; (k) (1S)-2-(tert-butilamino)-1-[4- ((1 S)-1 -{4-(8-ciclopropilimidazo[1,2-a]pirid in-3-ila)-5-f luoropirimid in-2- ila]amino}etil)fenil]etanol; (I) (1S)-2-(tert-butilamino)-1-{4-[(1S)-1-({5-fluoro-4-[8- (trifluorometila)imidazo[1,2-a]piridin-3-ila]pirimidin-2-ila}amino)etil]fenil}etanol; (m) [4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-ila)-5-fluoropirimidin-2- ila]amino}etil)fenil](1,2-dimetilpirrolidin-2-ila)metanol; (n) 1-[4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-ila)-5-fluoropirimidin-2- ila]amino}etil)fenila]-2-(dimetilamino)-2-metilpropan-1-ol; (o) [4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-ila)-5-fluoropirimidin-2- ila]amino}etil)fenil](1-isopropilazetidin-3-ila)metanol; ou (p) (1S)-2-(tert-butilamino)- 1-{4-[(1S)-1-({4-[8-(difluorometil)imidazo[1,2-a]piridin-3-ila]-5-fluoropirimidin-2- ila}amino)etil]fenil}etanol, ou um sal ou éster farmaceuticamente aceitável do mesmo.
A preparação combinada de acordo com a invenção que compreende duas preparações separadas pode ser ainda combinada com pelo menos uma preparação incluindo, junto com um carreador ou diluente farmaceuticamente aceitável, um agente antitumoral selecionado a partir do grupo derivado de agentes antitumorais, compostos antitumorais de complexo de platina, antitumorais derivados de camptotecina, antitumorais inibidores de tirosina quinase, anticorpos monoclonais, interferons, modificadores de resposta biológica e outros agentes antitumorais (aqui, a definição de cada agente antitumoral tem o mesmo significado conforme definido acima) ou um sal ou éster farmaceuticamente aceitável do mesmo. Uma composição farmacêutica de acordo com a invenção preferencialmente inclui, junto com um carregador ou diluente farmaceuticamente aceitável, (a)2-[((1S)-1-{4-[-2-(tert-butilamino)-1-hidroxietil]fenil}etil)amino]-4-(8- etilimidazo[1,2-a]pirid in-3-ila)pirimidina-5-carbonitrila; (b)4-(8-etilimidazo[1,2- a]piridin-3-ila)-2-[((1 S)-1 -{4-[hidróxi(1 -isopropilpiperidin-4- ila)metil]fenil}etil)amino]pirimidina-5-carbonitrila; (c)4-(8-ciclopropilimidazo[1,2-a]piridin-3-ila)-2-[((1 S)-1 -{4-[hidróxi(1 - metilpiperidin-4-ila)metil]fenil}etil)amino]pirimidina-5-carbonitrila; (d)4-(8-etilimidazo[1,2-a]piridin-3-ila)-2-[((1 S)-1 -{4-[hidróxi(1 -metilpiperidin- 3- ila)metil]fenil}etil)amino]pirimidina-5-carbonitrila; (e)2-[((1 S)-1 -{4-[2-(tert-butilamino)-1 -hidroxietil]fenil}etil)amino]-4-(8- cloroimidazo[1,2- a]piridin-3-ila)pirimidina-5-carbonitrila; (f)2-[((1S)-1-{4-[2-(tert-butilamino)-1-hidroxietil]fenil}etil)amino]-4-[8- (difluorometil)imidazo[1,2-a]piridin-3-ila]pirimidina-5-carbonitrila; (g) 4-(8-ciclopropilimidazo[1,2-a]piridin-3-ila)-2-[((1 S)-1 -{4-[( 1,2- dimetilpirrolidin-2- ila)(hidróxi)metil]fenil}etil)amino]pirimidina-5-carbonitrila; (h) (1S)-2-(tert-butilamino)-1-[4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-ila)-5- fluoropirimidin-2-ila]amino}etil)fenil]etanol; (i) (1S)-1-[4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-ila)-5-fluoropirimidin- 2-ila]amino} etil)fenil]-2-[(1-metilciclopentila)amino]etanol; (j) (1S)-2-(tert-butilamino)-1-[4-((1S)-1-{[4-(8-ciclopropilimidazo[1,2-a]piridin- 3-ila)-5- metilpirimidin-2-ila]amino}etil)fenil]etanol; (k) (1S)-2-(tert-butilamino-1-[4-((1S)-1-{[4-(8-ciclopropilimidazo[1,2-a]piridin- 3-ila)-5- fluoropirimidin-2-ila]amino}etil)fenil]etanol; [4-((1 S)-1 -{[4-(8-cloroimidazo[1,2-a]pirid in-3-ila)-5-f luoropirimid in-2- ila]amino}etil)fenil](1,2-dimetilpirrolidin-2-ila)metanol; (n) 1-[4-((1S)-1-{[4-(8- cloroimidazo[1,2-a]piridin-3-ila)-5-fluoropirimidin-2-ila]amino}etil)fenil]-2- (dimetilamino)-2-metilpropan-1-ol; (o) [4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3- ila)-5-fluoropirimidin-2-ila]amino}etil)fenil]( 1 -isopropilazetidin-3-ila)metanol; ou (p) (1S)-2-(tert-butilamino)-1-{4-[(1S)-1-({4-[8-(difluorometil)imidazo[1,2-a]piridin-3-ila]- 5-fluoropirimidin-2-ila}amino)etil]fenil}etanol, ou um sal ou éster farmaceuticamente aceitável do mesmo. De agora em diante, os processos representativos para a produção do composto da presente invenção serão descritos. Esquema 1a: Processo para Produção do Composto da Fórmula (1) a partir do Composto da Fórmula (lIa)

Esquema 1a
O composto da Fórmula (1) de acordo com a invenção (onde Ri, R2, R3, R4 e R5 são os mesmos conforme definido acima) pode ser sintetizado ao sujeitar primeiramente o composto da fórmula (Ha) (onde R-i, R2, θ R5 são os mesmos conforme definidos acima) a uma reação da oxidação para obter o composto da fórmula (Ha) (onde Ri, R2, e R5 são os mesmos conforme definidos acima, e n é 1 ou 2) e então conduzindo uma reação da substituição entre o composto da fórmula (Ha) e o fenetilamina representado pela Fórmula acima (IV) (onde R3 θ R4 são os mesmos conforme definidos acima). O composto da fórmula (Ha) pode ser sintetizado por oxidação do composto da fórmula (Ha) em um solvente como o diclorometano, clorofórmio, 0 N,N-dimetilformamida, ou semelhantes, com o uso de um agente oxidante como 0 ácido m-cloroperbenzóico (m-CPBA), o peróxido de benzoila, solução peróxido de hidrogênio, periodato do sódio, ou semelhantes, preferencialmente com 0 uso do ácido m-cloroperbenzóico. Na reação, 0 ácido m- cloroperbenzóico (m-CPBA) é usado na quantidade de 2 a 5 moles, preferencialmente 2 moles, para 1 mol do composto da Fórmula (Ha). A temperatura da reação pode ser adequadamente selecionada pelos especialistas no assunto em conformidade com o composto de partida usado ou com o solvente da reação, mas a temperatura da reação é geralmente de 0°C a temperatura ambiente. A reação é geralmente completada em 1 a 24 horas, mas a duração da reação pode ser adequadamente aumentada ou diminuída. Além disso, 0 composto obtido da Fórmula (Ha) acima pode ser sujeito à seguinte reação sem ser separado e purificado. A reação de substituição entre o composto da Fórmula (Ha) acima e fenetilamina da Fórmula (IV) acima é preferencialmente conduzido na presença de uma base (por exemplo, uma base inorgânica como carbonato de potássio e hidrogênio carbonato de sódio, ou uma base orgânica como trietilamina e diisopropiletilamina). O solvente da reação para uso inclui clorofórmio, tetrahidrofurano, N,N-dimetilformamida, dimetilSuIfóxido, e semelhantes e preferencialmente inclui clorofórmio e tetrahidrofurano. Na reação, fenetilamina da Fórmula (IV) acima é usada na quantidade de 0,5 a 3 moles, para 1 mol do composto representado pela Fórmula (Ha). A temperatura da reação pode ser adequadamente selecionada pelos especialistas no assunto em conformidade com o composto de partida usado ou com o solvente da reação, mas a temperatura da reação é geralmente entre temperatura ambiente até o ponto de ebulição do solvente, preferencialmente a temperatura ambiente. A reação é geralmente completada em 1 a 24 horas, mas a duração da reação pode ser adequadamente aumentada ou diminuída. Esquema 1b: Processo para Produção do Composto da Fórmula (1) a partir do Composto da Fórmula (IIb)

Esquema 1b
O composto da Fórmula (1) de acordo com a invenção (onde Ri, R2, R3, R4 e R5 são os mesmos conforme definidos acima) pode ser sintetizado pela condução da reação de substituição entre o composto da Fórmula (lia) (onde R-i, R2, e R5 são os mesmos conforme definidos acima) e fenetilamina representada pela Fórmula (IV) (onde R3 e R4 são os mesmos conforme definidos acima).
A reação de substituição entre o composto da Fórmula (llb) acima e fenetilamina da Fórmula (IV) acima é preferencialmente conduzida na presença de uma base (por exemplo, uma base inorgânica como carbonato de potássio, césio sódico e hidrogênio carbonato de sódio, ou uma base orgânica como trietilamina e diisopropiletilamina). O solvente da reação para uso inclui clorofórmio, tetrahidrofurano, N,N-dimetilformamida, N,N-dimetilacetoamida, N- metil-2-pirrolidinona, dimetilSuIfóxido e semelhantes e preferencialmente inclui N,N-dimetilformamida, N,N-dimetilacetoamida, N-metil-2-pirrolidinona. Na reação, fenetilamina da Fórmula (IV) acima é usada na quantidade de 0,5 a 3 moles, para 1 mol do composto representado pela Fórmula (llb). A temperatura da reação pode ser adequadamente selecionada pelos especialistas no assunto em conformidade com o composto de partida usado ou com o solvente da reação, mas a temperatura da reação é geralmente entre temperatura ambiente até o ponto de ebulição do solvente, preferencialmente 80 a 200°C. A reação é geralmente completada em 1 a 24 horas, mas a duração da reação pode ser adequadamente aumentada ou diminuída. Esquema 2a Processo Representativo para Produção do Composto da Fórmula (lia)
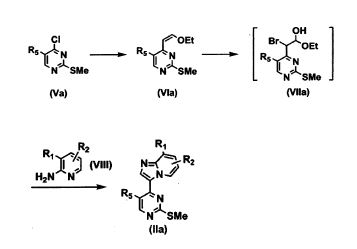
Esquema 2a
O composto da Fórmula (Via) (onde R5 é o mesmo conforme definido acima) pode ser sintetizado pela reação de acoplamento de Still entre cis-1-etóxi- 2-tri-b-butiltin (que pode ser sintetizado de acordo com o método descrito em J. Am. Chem. Soc, 1977, 99, 7365) e o composto da Fórmula (Va) acima (onde R5 é o mesmo conforme definido acima), usando diclorobis (trifenilfosfina) paládio (II) como um catalisador. O solvente da reação é preferencialmente acetonitrila e a temperatura de reação é geralmente entre temperatura ambiente até o ponto de ebulição do solvente, e é preferencialmente o ponto de ebulição do solvente. A reação é geralmente completada em 1 a 24 horas, mas a duração da reação pode ser adequadamente aumentada ou diminuída. O composto da Fórmula (Vila) (onde R5 é o mesmo conforme definido acima) pode ser preparado pela reação do composto da Fórmula (Via) acima com N-bromosuccinimida em 1,4-dioxano. Na reação, 1 a 3 moles, preferencialmente 1 mol de N-bromosuccinimida, para 1 mol do composto da Fórmula (Via) são usados. A temperatura da reação pode ser adequadamente selecionada pelos especialistas no assunto em conformidade com o composto de partida usado ou com o solvente da reação, mas a temperatura da reação é preferencialmente de 0°C até temperatura ambiente. A reação é geralmente completada em 1 a 12 horas, mas a duração da reação pode ser adequadamente aumentada ou diminuída. O composto da Fórmula (Vila) obtido pode ser sujeito à reação subsequente sem além disso ser isolado e purificado.
O composto da Fórmula (Ha) (onde Ri, R2 e R5 são os mesmos conforme definidos acima) podem ser sintetizados a partir do composto da Fórmula (Vila) acima e o composto da Fórmula (VIII) acima (onde RÍ e R2 são os mesmos conforme definidos acima) em 1,4-dioxano. Na reação, 1 a 3 moles, preferencialmente 1 mol do composto da Fórmula (VIII) acima, para 1 mol do composto da Fórmula (Vila) são usados. A temperatura da reação pode ser adequadamente selecionada pelos especialistas no assunto em conformidade com o composto de partida usado ou com o solvente da reação, mas a temperatura da reação é geralmente entre temperatura ambiente até 0 ponto de ebulição do solvente, e preferencialmente de temperatura ambiente até 50°C.
A reação é geralmente completada em 1 a 24 horas, mas a duração da reação pode ser adequadamente aumentada ou diminuída.
O composto da Fórmula (Va), por exemplo, é 3-cloro-2-(meti1Sulfanil)-5- pirimidinecarbonitrila ou semelhante e o composto da Fórmula (VIII) acima, por exemplo, é 2-amino-3-picolina ou semelhante. Estes compostos são comercialmente disponíveis quanto podem ser sintetizados a partir de um composto comercialmente disponível por um método geralmente conhecido pelos especialistas no assunto ou um método análogo a este (Literatura: Publicação Internacional WO 2004/043936, página 32 - 33, etc.).Esquema 2b Processo Representativo para Produção do Composto da Fórmula (IIb)
Esquema 2b
O composto da Fórmula (Hb) (onde Ri, R2 e R5 são os mesmos conforme definidos acima) pode ser sintetizado usando 0 mesmo método como 5 apresentado no Esquema 2a a partir do composto da Fórmula (Vb) (onde R5 é definido conforme acima) como um material de partida no lugar do composto da Fórmula (Va) no Esquema 2a; vide Esquema 2b.
O composto da Fórmula (Vb), por exemplo, é 2,4-dicloro-5-metilpiridina e semelhantes e também o composto da Fórmula (VIII) acima, por exemplo, é 2- 10 amino-picolina e semelhantes, ambos dos quais são tanto comercialmente disponíveis quanto podem ser sintetizados a partir de um composto comercialmente disponível por um método geralmente conhecido pelos especialistas no assunto ou um método análogo a este (Literatura: Publicação lnternacionalW02004/043936, página 32 to 33, etc.). Esquema 3: Processo Representativo para Produção do Composto da Fórmula (IV)
Esquema 3
O composto da Fórmula (IX) acima (onde Boc é um grupo tert- butoxicarbonila) pode ser sintetizado usando (S)-(-)-1-(4-bromofenil)etilamina comercialmente disponível (composto (VIII)) acima em conformidade com o método geralmente conhecido pelos especialistas no assunto (Literatura: Protective Groups in Organic Synthesis, na terceira edição, escrito por T. W. Greene, John Wiley &Sons Publication, página 518 - 524, etc.).Em seguida, o composto da Fórmula (XI) acima (onde Boc, R3 e R4 são os mesmos conforme definidos acima) pode ser sintetizado ao sujeitar primeiramente 0 n-butil-lítio ao composto da Fórmula (IX) em tetrahidrofurano para obter aril-lítio como um intermediário de reação e em seguida reagindo com uma cetona da Fórmula (Xa) acima (onde R3 e R4 são os mesmos conforme definidos acima) ou um aldeído da Fórmula (Xb) (onde R4 é 0 mesmo conforme definido acima) ambos dos quais são eletrófilos. Na reação, 2 a 5 moles, preferencialmente 2 mol de n-butil-lítio, para 1 mol do composto da Fórmula (IX) são usados. Além disso, 1 a 3 moles de cetona da Fórmula (Xa) acima ou 0 aldeído da Fórmula (Xb) acima para 1 mol do composto da Fórmula (IX) são usados. A temperatura da reação é geralmente de -78°C até 0°C e preferencialmente -78°C. A reação é geralmente completada em 1 a 24 horas, mas a duração da reação pode ser adequadamente aumentada ou diminuída.
Além disso, o composto da Fórmula (XI) também pode ser sintetizado pelo seguinte método. Ou seja, o composto da Fórmula (IX) acima é sujeitado a n-butil- lítio para obter aril-lítio como um intermediário de reação, que reage, então, com um eletrófilo como amida de Weinreb (Xc) (onde R3 é o mesmo conforme definido acima). O composto obtido é sujeito à reação de redução com 0 uso de borohidreto de sódio para sintetizar 0 composto da Fórmula (XI) acima.
Na reação, 2 a 5 moles, preferencialmente 2 mol de n-butil-lítio, para 1 mol do composto da Fórmula (IX) são usados. Além disso, 1 a 3 moles da amida da Fórmula (Xc) acima para 1 mol do composto da Fórmula (IX) é usado. A temperatura da reação é geralmente de -78°C até 0°C e preferencialmente -78°C. A reação é geralmente completada em 1 a 24 horas, mas a duração da reação pode ser adequadamente aumentada ou diminuída. Na reação de redução, 1 a 3 moles, preferencialmente 1 mol de borohidreto de sódio, para 1 mol do composto da Fórmula (IX) é usado.
O composto da Fórmula (IV) (onde R3 e R4 são os mesmos em conforme definidos acima) pode ser sintetizado usando o composto da Fórmula (XI) acima em conformidade com o método geralmente conhecido pelos especialistas no assunto (Literatura: Protective Groups in Organic Synthesis, terceira edição, escrito por T. W. Greene, John Wiley &Sons Publication, página 518 - 524, etc.). O composto da Fórmula (Xa) acima, por exemplo, é 1 -metil-4-piperidona ou semelhante, e o composto da Fórmula (Xb) acima, por exemplo, é benzil 2- formilpirrolidina-1-carboxilato ou semelhante. O composto da Fórmula (Xc) acima, por exemplo, é benzil 4-(N-metoxi-N-metilcarbamoil)piperidina-1-carboxilato ou semelhante. Estes compostos são comercialmente disponíveis quanto podem ser sintetizados a partir de um composto comercialmente disponível por um método geralmente conhecido pelos especialistas no assunto ou um método análogo a este (Literatura: Bioorg. Med. Chem., 2003, 11, 3153, Pamphlet of Publicação lntemacionalW003/011285, página 60-61, etc.). Esquema 4: Processo Representativo para Produção do Composto da Fórmula (XVI)

Esquema 4
O composto da Fórmula (XII) acima (onde Boc é um grupo tert- butilóxicarbonila) pode ser obtido ao sujeitar o composto da Fórmula (IX) a uma reação de acoplamento (onde Boc é o mesmo conforme definido acima) e viniltrifluoroborato de potássio ou viniltributiltin em um solvente como N,N- dimetilformamida, 1,4-dioxano, tolueno, tetrahidrofurano, metanol, dimetoxietano ou semelhante na presença de um paládio catalítico como tetrakis (trifenilfosfina) paládio (0) ou diclorobis(trifenilfosfina) paládio (II). Quando viniltrifluoroborato de potássio é usado aqui, a reação é preferencialmente conduzida na presença de uma base inorgânica como o carbonato de sódio, hidrogênio carbonato de sódio ou carbonato de potássio ou uma base orgânica como trietilamina ou diisopropiletilamina.
Na reação, a temperatura da reação pode ser adequadamente selecionada pelos especialistas na técnica em conformidade com o reagente usado ou com o solvente da reação, mas a temperatura da reação é geralmente entre temperatura ambiente e o ponto de ebulição do solvente. A reação é geralmente completada em 1 a 24 horas, mas a duração da reação pode ser adequadamente aumentada ou diminuída.
O composto da Fórmula (XIII) acima (onde Boc é o mesmo conforme definido acima) pode ser sintetizado ao sujeitar o composto da Fórmula (XII) acima a uma reação de dihidroxilação em uma mistura de solvente de acetona e água com o uso de tetróxido de ósmio e N-óxido de N-metil-morfolina. É preferível que a reação seja conduzida fora da temperatura usual de 0°C até temperatura ambiente. A reação é geralmente completada em 1 a 48 horas, mas a duração da reação pode ser adequadamente aumentada ou diminuída.
Uma substância opticamente ativa do composto da Fórmula (XIII) acima pode ser sintetizado ao sujeitar o composto da Fórmula (XII) acima a uma reação de dihidroxilação assimétrica de Sharpless (Literatura: Chem, Rev., 1994, 94, 2483, etc.) com o uso de AD-mix-α ou β (disponível na Aldrich Corporation) ao invés de tetróxido de ósmio, como um agente oxidante.O composto de Fórmula (XIV) acima (onde Boc é o mesmo conforme definido acima) pode ser sintetizado pela introdução seletiva de um grupo de saída a um grupo 1 hidróxi primário do composto da Fórmula (XIII) e em seguida sujeitando o resultado a uma reação de ciclização por aquecimento em um solvente como diclorometano, clorofórmio, tolueno, tetrahidrofurano ou semelhante na presença ou de uma base inorgânica como hidróxido de sódio, hidróxido de potássio, carbonato de potássio ou semelhante ou uma base orgânica como trietilamina, diisopropilamina, piridina ou semelhante. Neste caso, o grupo metanosulfonila, o grupo p-toluenosulfonila ou semelhante podem ser usados como grupos de saída. Na redução de introdução do grupo de saída, 1 a 3 moles, preferencialmente 1,1 moles de cloreto de metanesulfonila ou cloreto de p-toluenosulfonila, para 1 a 3 moles do composto da Fórmula (XIII) são usados. A temperatura de reação é de 0°C até temperatura ambiente e preferencialmente 0°C. A reação é geralmente completada em 1 a 24 horas, mas a duração da reação pode ser adequadamente aumentada ou diminuída.
O composto da Fórmula (XV) acima (onde Nu é um substituinte derivado a de um agente nucleofílico como um grupo tert-butilamino e Boc é o mesmo conforme definido acima) pode ser sintetizado pela reação de um epóxido representado pela Fórmula (XIV) acima com um agente nucleofílico em um solvente como metanol, etanol ou água. Na reação 1, 1 mole de quantidade excessiva preferencialmente cerca de 10 moles do agente nucleofílico para 1 mol do composto da Fórmula (XV) acima são usados. A temperatura da reação pode ser adequadamente selecionada pelos especialistas no assunto em conformidade com o composto de partida usado com o composto de partida ou com o solvente da reação, mas a temperatura da reação é geralmente entre temperatura ambiente até o ponto de ebulição do solvente, preferencialmente de 40°C até o ponto de ebulição do solvente. A reação é geralmente completada em 1 a 24 horas, mas a duração da reação pode ser adequadamente aumentada ou diminuída. O agente nucleofílico na reação, por exemplo, é o tert-butilamina, piperazina ou semelhante. Estes são ou comercialmente disponíveis ou podem ser sintetizados a partir de um composto comercialmente disponível por um método geralmente conhecido pelos especialistas ou um método análogo a este.
O composto da Fórmula (XVI) (onde Nu é o mesmo como definido acima) pode ser sintetizado a partir do composto da Fórmula (XV) acima em conformidade com o método geralmente conhecido pelos especialistas no assunto (Literatura: Protective Groups in Organic Synthesis, terceira edição, escrito por T. W. Greene, John Wiley &Sons Publication, página 518 - 524, etc.).Os processos produtivos descritos nos Esquemas 1 a 4, os compostos desejáveis podem ainda ser obtidos pelo uso do método comumente usado em química orgânica sintética como métodos de proteger ou desproteger os grupos funcionais [por exemplo, ver Protective Groups in Organic Synthesis, terceira edição, escrita por T. W. Greene, John Wiley &Sons Publication] conforme necessário.Em seguida, o efeito inibitório e o efeito inibitório de proliferação celular do composto da Fórmula (1) será explicado.
1. Medida do efeito inibitório contra a atividade de PLK1 (Método A) (1) Preparação de PLK1-T210D
Foi descoberto que o 210° códon da PLK1 humana que originalmente codifica para treonina pode ser alterado para um tipo ativo ao alterar o local dentro do ácido aspártico [Molecular and Cell Biology (Mol. Cell. Biol.), 17th edition, 3408 (1997)]. Com o objetivo de obter a proteína tipo PLK1 humana ativa, o cDNA da PLK1 mutada (PLK1-T210D) da qual os códigos do 210° códon para ácido aspártico pela substituição de uma base no 210° códon da PLK1 cDNA foi preparado. Um baculovírus para o qual o N-terminal do cDNA da PLK1-T210D é fundido com GST (glutationa S-transferase) foi preparado e em seguida a PLK1- T210D transfectada em uma célula do inseto Spodoptera frugiperda (Sf) 9 foi altamente expressa como uma proteína GST fundida. As células foram recuperadas e suspendidas em um tampão de lise (50 mM de tampão de ácido tris-cloroacético (pH 7,4)/150 mM de cloreto de sódio / 1mM de EDTA (ácido tetracético de etilenodiamina)/ 1mM de ditiotreitol / 0,1% de monolaurato sorbitano de polioxietileno) para quebrar as células com sonicador e o sobrenadante foi recuperado após a centrifugação. O sobrenadante foi reagido com pérolas de glutationa sefarase e em seguidas as pérolas foram lavadas com um tampão de lise. Posteriormente, as pérolas foram reagidas em um tampão de lise contendo Protease de Precision(disponível da GE Healthcare Bioscience Company) para recuperar o sobrenadante. (2) Medida da atividade de PLK1-T210D
Para a medida da atividade da PLK1T210, um peptídeo sintético (arginina- arginina-ácido aspártico-ácido glutâmico-metionina-ácido glutâmico-alanina- serina-fenilalanina-alanina-ácido aspártico-glutamina-ácido glutâmico-alanina- lisina-valina) (SEQ. ID. NO.: 1) onde a seqüência de serina que cerca a seqüência de aminoácido No. 198 de CDC25C tendo sido relatado como o local para o substrato de PLK1 [EMBO Report, 3rd edition, 341 (2002)] é alterado, foi usado como substrato.A reação foi conduzida em conformidade com o método de Toyoshima-Morimoto et al. [Nature, Vol, 410, 215 - 220, (2001)]. O volume da solução de reação foi de 21,1 pL e a composição do tampão de reação foi 20 mM de tampão de ácido tris-hidroclórico (pH 7,4)/10 MM cloreto de magnésio / 0,5 mM ditiotreitol / 1 mM de EGTA (etileno glicol-bis(beta-aminoetileter)N,N,N’,N’,-ácido tetraacético). A isto a PLK1 purificada, 50 pM do substrato peptídeo, 50 pM de trifosfato de adenosina não marcada (ATP) e 1 pCi de ATP[y-33P] marcada (2000 a 4000 Ci/mmol) foram adicionados para conduzir a reação a 25°C por 20 minutos. Em seguida, 10 pL de tampão fosfato 350 mM foram adicionados ao sistema da reação para terminar a reação. A solução resultante foi manchada em um filtro de fosfocelulose multi-screen em uma placa de 96 poços. Após lavagem do filtro de fosfocelulose com tampão fosfato 75 mM, o filtro foi seco para medir a radioatividade com um contador de cintilação líquida. O ATP não marcado e o ATP [y-33P] marcado foram adquiridos da Amersham Bioscience Corp., e o filtro de fosfocelulose multi-screen foi adquirido da Millipore Corp. A adição do composto de acordo com a invenção do sistema de reação foi conduzido pela adição de 1,1 pL da solução preparada pela dissolução preliminar em dimetilSuIfóxido a uma concentração 20 vezes da concentração final. Um controle foi fornecido pela adição de 1,1 pL de dimetilSuIfóxido ao sistema de reação.
O valor IC50 do composto de acordo com a invenção para a atividade da PLK1-T210D foi determinado e os resultados são mostrados na Tabela 1 abaixo.
De acordo com a Bula do Produto da Carna Biosciences Inc., a presente enzima é uma enzima obtida pelo preparo do baculovírus para o qual a N- terminação de um cDNA de comprimento total da PLK1 do tipo selvagem é fundida com GST (glutationa S-transferase), seguido por PLK1 transfectada altamente expressa em uma célula de inseto como proteína GST-fundida e em seguida, é conduzida uma purificação com o uso de cromatografia de glutationa sefarose.
(2) Medida da atividade de PLK1 (tipo selvagem)
Para a medida da atividade da PLK1, um peptídeo sintético (arginina- arginina-ácido aspártico-ácido glutâmico-leucina-metionina-ácido glutâmico- alanina-serina-fenilalanina-alanina-ácido aspártico-glutamina-ácido glutâmico- alanina-lisina-valina) (SEQ. ID. NO.: 1) onde a seqüência de serina que cerca a seqüência de aminoácido No. 198 de CDC25C tendo sido relatado como o local para o substrato de PLK1 [EMBO Report, 3rd edition, 341 (2002)] é alterado, foi usado como substrato para a PLK1.
A reação foi conduzida em conformidade com o método como descrito por Toyoshima-Morimoto et al. [Nature, Vol. 410, 215 - 220, (2001)]. O volume da solução de reação foi de 10,5 pL e a composição do tampão de reação foi de tampão Tris-HCI 20 mM (pH 7,4)/10 mM cloreto de magnésio / 0,5 mM ditiotreitol / 1 mM EGTA (etilenoglicol-is (betaOaminoetileter-N,N,N’,N’-ácido tetraacético). A isto, a PLK1 purificada, 20 pM do substrato peptídeo, 10 pM de adenosina trifosfato não marcada (ATP) e 0,3 pCi de ATP marcada [y-33P] (>2500 Ci/mmol) foram adicionados para realizar a reação a uma temperatura de reação de 25°C por 120 minutos. Portanto, 20 pl de tampão fosfato 350 mM foram adicionados ao sistema de reação para terminar a reação, e a solução resultante foi aplicada sobre um filtro multi-tela de fosfocelulose em uma placa de 384 poços. Após o filtro de fosfocelulose ser lavado com um tampão fosfato 75 mM e, em seguida, seco, a radioatividade foi medida com um contador líquido de cintilação. Os ATPs marcados [y-33P] e não marcados e o filtro fosfocelulose multi-tela foram adquiridos da GE Healthcare Bio-Ciências e Millipore Corporation, respectiva mente.
A adição do composto da presente invenção ao sistema de reação foi realizada pela adição de 0,5 pl da solução preparada pela dissolução preliminar do composto em dimetilSuIfóxido a uma concentração 20 vezes da concentração final. Um controle foi fornecido pela adição de 0,5 pl do dimetilSuIfóxido ao sistema de reação.
O valor IC5o valor dos compostos da presente invenção para a atividade de PLK 1 foi determinada, e os resultados são apresentados na Tabela 2 abaixo.
Como mostrado acima, os mesmos resultados sobre a atividade inibitória dos compostos da invenção contra PLK1 podem ser obtidos por qualquer método de A e B do método, e é claro que a atividade inibitória é extremamente elevada.
3. Medida do efeito inibitório contra a proliferação de células: Medida da atividade inibitória contra PLK1 em Nível Celular
(1) O método de Cultura de Células
Para a medida da atividade inibitória de PLK 1 do composto em um nível celular, linhagens de células cancerígenas de cérvice uterino humano HeLaS3 foram utilizadas. A célula HeLaS3 foi obtida da American Type Culture Collection (ATTC), e cultivada em incubadora de CO2 de vapor saturado usando Meio Dulbecco's Modified Eagle modificado contendo soro fetal bovino 10% a 37°C na presença de 5% de CO2.
(2) Medida da atividade inibitória do composto de acordo com a invenção
Tem sido relatado que PLK 1 desempenha um papel importante em vários estágios da fase de mitose (fase M) em células de mamíferos (Nature Review Molecular Cell Biology (Nat. Rev. Mol. Cell. Biol.), Vol. 5, 429, (2004)). Na verdade, quando as células de mamíferos são tratados com PLK 1 SiRNA para controlar o nível de expressão, a progressão do ciclo celular é inibida e, portanto, as células são presas na fase M. Neste momento, quando o nível de fosforilação do 10° resíduo de serina na histona H3, que é considerado necessário para uma condensação de cromossomos na fase M, é examinada, em seguida, observa-se que o nível é aumentado para um nível elevado. Assim, após o tratamento das células com o composto de acordo com a invenção, o nível de fosforilação da histona H3 foi examinado pela técnica de anticorpo de imunofluorescência indireta, as células da fase M foram identificadas usando o nível do mesmo como um indicador para analisar a proporção de células presas na fase M; e ainda o valor EC5O de cada composto foi calculado para avaliar a atividade inibitória de PLK 1 em um nível celular.
Primeiro, as células HeLaS3 foram semeadas em placa de 96 poços tratadas com lisina (Falcon Cocp). na proporção de 3000 por poço, e deixadas em repouso ainda na incubadora de CO2 acima mencionados. 24 horas após a semeadura, 0 composto de acordo com a invenção, que é diluído em série, foi adicionado em cada poço da placa, e ainda deixado em repouso na incubadora de CO2. 18 horas após a adição do composto de acordo com a invenção, o meio de cultura contendo os compostos de acordo com a invenção em cada poço da placa foi removido e, em seguida 100 pL de metanol gelado 100% (Wako Pure Chemical Industries, Ltd.) foi adicionado, para realizar a fixação das células por 10 minutos e tratamento do aumento da permeabilidade da membrana. Posteriormente, para os poços em que o metanol foi removido, 50 mL de BSA 1%/PBS foi adicionado, e então o bloqueio foi realizado por 30 minutos. Posteriormente, para a reação do anticorpo primário, 50 pL de BSA 1%/PBS contendo 2,5 mg/ml anticorpo Anti- fosdo histona H3 (SerlO) (Upstate Corp) foi adicionado aos poços e a placa foi deixada em temperatura ambiente por 90 minutos. Após encerramento da reação, cada poço foi lavado uma vez com PBS, e para a segunda reação de anticorpos, 50 pL de BSA 1%/PBS contendo 1,5 mg/mL de anticorpo IgG ant—coelho marcado com Cy5 (H+L) (Chemicon) e 10 pg/mL de DAPI (Sigma), que é um regente de coloração de núcleo, foi adicionado, e ainda deixado em temperatura ambiente por 90 minutos. Após o término da reação, a solução de reação em poços foi removida e substituída com 100 pL de PBS e, em seguida imagens de fluorescência foram capturados usando Analisador de Célula IN 1000 (fabricados pela GE Amersham) para analisar a proporção de células em fase M (índice mitótico) em cada vista. Quando o valor máximo da proporção de células presas na fase M, que podem ser induzidas por cada droga é assumida como 100%, a concentração da droga necessária para induzir 50% de que 100% é definida como EC50.Valores de EC5Q obtidos pelo método acima mencionados são apresentados na Tabela 3 abaixo.

Como mostrado na Tabela 3, uma vez que o composto da invenção apresenta um forte efeito inibidor contra a proliferação de células, é considerado extremamente útil como um agente antitumoral. Quando comparado com os compostos revelados na Publicação Internacional WQ2006/025567, é evidente que o composto da invenção apresenta atividade inibitória contra a proliferação celular significativamente excelente, devido a conter a seguinte estrutura parcial:
Como mencionado acima, o composto de acordo com a invenção tem uma atividade inibitória de PLK 1 excelente e também tem um forte efeito inibidor contra a proliferação celular. Portanto, acredita-se ser útil como um agente antitumoral for inibir fortemente a proliferação de células cancerosas. Ou seja, uma composição farmacêutica contendo o novo derivado de aminopiridina substituído de acordo com a invenção ou um sal farmaceuticamente aceitável ou éster do mesmo, ou um agente antitumor contendo o novo derivado aminopiridina substituído de acordo com a invenção ou um sal farmaceuticamente aceitável ou éster do mesmo, acredita-se que seja eficaz no tratamento de pacientes com câncer. Além disso, a composição farmacêutica ou o agente antitumoral podem conter transportadores farmaceuticamente aceitáveis ou diluentes. Aqui, o termo "transportadores ou diluentes farmaceuticamente aceitáveis"refere-se aos excipientes [por exemplo, gorduras, cera de abelha, polióis semi-sólidos ou líquidos, óleo natural ou hidrogenado, etc]; água (por exemplo, água destilada, água especialmente destilada para injeção, etc) salmoura fisiológica, álcool (por exemplo, etanol), glicerol, poliol, glicose aquosa, manitol, óleo vegetal, ou semelhantes, aditivos [por exemplo, agente de volume, desintegrante, aglutinante, lubrificante, agente umectante, estabilizante, emulSificante, dispersante, conservante, edulcorante, corante, agente flavorizante ou substância aromática, agente espessante, diluente, substância tampão, solvente ou solubilizante, droga para alcançar efeito do armazenamento, sal para ajuste de pressão osmótica, o agente de revestimento, ou antioxidante] e semelhantes.
O composto de acordo com a invenção pode também ser usado como uma pró-droga contendo ésteres. Aqui, o termo pró-droga refere-se geralmente a um derivado onde certa molécula de droga é quimicamente modificada, que se mostra sem atividade fisiológica, mas depois de ter sido administrada in vivo, transforma de volta à sua molécula da droga original para apresentar a eficácia da droga. Como a pró-droga do composto de acordo com a invenção, um composto acima de Fórmula (I) em que o grupo hidróxi 1 é acilado com um grupo fosfato ou como pode ser exemplificado. A pró-droga e éster podem ser produzidos de acordo com o método comumente conhecido ou utilizado por aqueles especialistas na técnica.
Além disso, para os tumores adequado para esperar um efeito terapêutico do composto de acordo com a invenção, por exemplo, tumores sólidos humanos e outros podem ser mencionados. Exemplos de tumores humanos sólidos incluem o câncer cerebral, câncer de cabeça e pescoço, câncer do esôfago, câncer de tireóide, câncer de células pequenas, câncer de células não-pequenas, câncer da mama, câncer gástrico, câncer do dueto biliar e/ou vesícula biliar, o câncer hepático, câncer pancreático, câncer de cólon, câncer do reto, câncer ovariano, corioepitelioma, câncer uterino, câncer cervical, câncer renal pélvico/ureteral, câncer de bexiga, câncer de próstata, câncer de pênis, câncer testicular, câncer embrionário, tumor de Wilson, câncer de pele, melanoma maligno, neuroblastoma, osteosarcoma, sarcoma de Ewing, sarcoma de tecido mole e semelhantes.
Em seguida, o "sal farmaceuticamente aceitável ou éster do mesmo descrito acima será explicado. Quando o composto de acordo com a invenção é usado como um agente antitumoral ou semelhantes, o composto pode ser usado sob a forma de um sal farmaceuticamente aceitável. Exemplos típicos de sal farmaceuticamente aceitáveis incluem sais ácidos inorgânicos, como sais de metais alcalinos, por exemplo, sódio, potássio, e semelhantes, cloridrato, sulfato, nitrato, fosfato, carbonato, carbonato de hidrogênio, e hiperclorato, sais de ácidos orgânicos, tais como acetato, propionato, lactato, maleato, fumarato, tartarato, malato, citrato e ascorbato, sulfonatos como metanosulfonato, isotionato, benzenosulfonato e toluenosulfonato; sais ácidos de aminoácidos como aspartato e glutamato, e semelhantes. Um sal preferencial do composto de acordo com a invenção é um sal de cloridrato, e semelhantes.
A preparação dos sais farmaceuticamente aceitáveis do composto de acordo com a invenção podem ser realizados por uma combinação apropriada de métodos que são convencionalmente usados no campo da química orgânica sintética. Especificamente, um método de neutralizar uma solução por titulação do composto de acordo com a invenção de uma forma livre com uma solução alcalina ou com uma solução ácida, ou semelhantes, podem ser mencionadas.
Exemplos de éster do composto de acordo com a invenção incluem éster metílico, éster etílico, e semelhantes. Estes ésteres podem ser preparados por esterificação de um grupo carboxila livre de acordo com métodos padronizados.
Para a forma de administração utilizada na administração do composto de acordo com a invenção como um agente antitumoral ou semelhantes, várias formas podem ser selecionados. Por exemplo, preparações orais, como comprimidos, cápsulas, pó, granulado e líquido; e preparações parenterais de líquidos esterilizados, como solução e suspensão, e semelhantes podem ser mencionadas. Aqui, preparações sólidas podem ser preparadas, sem modificações, na forma de comprimidos, cápsulas, grânulos ou pó de acordo com métodos padronizados, mas também pode ser preparado usando aditivos. Exemplos de aditivos incluem açúcares como a lactose e glicose, amidos como as de milho, trigo e arroz; ácidos graxos como o ácido esteárico, sais inorgânicos, como metassilicato de sódio, aluminato de magnésio, e fosfato de cálcio anidro; polímeros sintéticos, como polivinilpirrolidona e polialquilenoglicol, sais de ácidos graxos como o estearato de cálcio e estearato de magnésio, álcoois, como o álcool estearílico e álcool benzílico e derivados sintéticos de celulose, tais como a metilcelulose, carboximetilcelulose, etilcelulose e hidroxipropilmetilcelulose. Além destes, aditivos geralmente usados como água, gelatina, talco, óleos vegetais, e goma arábica, e semelhantes, também pode ser mencionados. Estas preparações sólidas, como comprimidos, cápsulas, grânulos e pó podem geralmente contêm 0,1 a 100%, em peso, de preferência 5 a 100%, em peso, do ingrediente ativo.
Preparações líquidas podem ser preparadas na forma de suspensão, xarope, preparação injetável, ou semelhantes, usando de aditivos apropriados, que são geralmente utilizados para a preparação de líquidos, como água, álcoois, óleos derivados de plantas, por exemplo, óleo de soja, óleo de amendoim, óleo de gergelim, e semelhantes. Em particular, os exemplos solvente ou diluente apropriados usados no caso de administração parenteral através de injeção intramuscular, injeção intravenosa ou injeção subcutânea incluem água destilada para injeção, solução aquosa de cloridrato de lidocaína (por injeção intramuscular), salmoura fisiológica, solução aquosa de glicose, etanol, fluido para injeção intravenosa (por exemplo, solução aquosa de ácido cítrico, citrato de sódio, e semelhantes), solução de eletrólitos (por exemplo, o fluido para infusão ou para injeção intravenosa) e semelhantes e soluções misturadas das mesmas.
Estas preparações injetáveis podem estar na forma de pó ou composto com aditivos apropriados, que deverão ser dissolvidas no momento do uso, além da forma que preliminarmente dissolveu. Tais líquidos injetáveis podem geralmente conter 0,1 a 10% em peso do ingrediente ativo.
O líquido para administração oral, tais como a suspensão, xarope ou semelhante, pode conter 0,5 a 10% em peso do ingrediente ativo. Para o tratamento de acordo com a invenção, o curso preferencial da medicação pode ser variada de acordo com a forma de administração do composto representado pela Fórmula (I), o tipo de composto representado pela Fórmula (I) a ser utilizado, a formulação compreendendo o compost representado pela Fórmula (I) a ser utilizado, o tipo, forma de administração, e formulação de outro(s) agente(s) antitumoral(is) para serem utilizados em combinação, e as células cancerígenas a serem tratadas e as condições do paciente. O tratamento mais apropriado em uma condição pré-determinado pode ser determinada pelos especialistas na técnica com base no curso de uma terapêutica comumente usada e/ou considerando as especificações presentes.
A quantidade preferencial do composto de acordo com a invenção deve ser administrada na prática, pode ser apropriadamente aumentada ou diminui de acordo com o tipo de composto a ser utilizado, o tipo de composição misturada, a frequência de administração, o local específico a ser tratado, e as condições do paciente. Por exemplo, a dose diária para um adulto é, no caso de administração oral, 10 a 500 mg, e no caso de administração parenteral, preferencialmente injeção intravenosa, 10 a 100 mg, por dia. Além disso, a freqüência da dose pode variar dependendo do modo de administração e os sintomas, mas a administração pode ser realizada uma vez, ou dividida em 2 a 5 porções.
O curso do composto representado pela Fórmula (I) e outros agentes antitumorais para serem usados em combinação não é particularmente limitado, mas pode ser apropriadamente determinada em conformidade com a literatura pública, se necessário, pelos especialistas na técnica.
Exemplos
De agora em diante, a presente invenção será descrita em maiores detalhes com referência a Exemplos, mas a invenção não se destina a ser limitada pelos exemplos, por qualquer meio. Nos exemplos, cromatografia em camada delgada foi realizada utilizando sílica gel 60F254 (Merck &Co., Inc.) ou NH (Fuji Silysia Chemical, Ltd.) para a placa, e um detector de UV para a detecção. Wakogel ™ C-300 ou C-200 (Wako Pure Chemical Industries, Ltd.), NH (Fuji Silysia Química, Ltd.), Biotage Si ou Biotage NH (Biotage), ou Purif-Pack (MORITEX) foram utilizados como a sílica gel para a coluna. Em uma cromatografia líquida de fase reversa preparativa, CombiPrep Pro C18 (YMC) foi usada para uma coluna, e 0,1% de ácido trifluoroacético solução aquosa e 0,1% de ácido trifluoroacético acetonitrila foram utilizados para uma fase móvel. Para o espectro de MS, JMS SX-102A (JEOL, Co. Ltd.), ou QUATTRO II (Micromass) foram utilizados, ou para a LC-MS, ZMD (Micromass) foi utilizado para a medição. Para o espectro de RMN, dimetilSuIfóxido foi utilizado como padrão interno no processo no caso de medição em uma solução de dimetilSuIfóxido deuterada, tetrametilSilano foi utilizado como padrão interno no caso de medição em uma solução de clorofórmio deuterado, e metanol foi utilizado como padrão interno no caso de medição em uma solução de metanol deuterada. Além disso, um espectrômetro, como Mercury 400 (400 MHz; Varian, Inc.), Inova 400 (400 MHz; Varian, Inc.), ou JNM AL400 (400 MHz; JEOL, Co. Ltd.) foi utilizado para a medição. Todos os valores δ foram expressos ppm. Os significados das siglas usadas em Exemplos são dados abaixo. s: Singleto d: Dubleto; dd: Duplo dubleto t: Tripleto dt: Duplo tripleto q: Quarteto dq: Duplo quarteto quint: Quinteto m: Multipleto br: Largo brs: Singleto largo J: Constante de acoplamento Hz: Hertz DMSOdθ: dimetilSuIfóxido deuterado CDCI3: Clorofórmio deuterado CD3OD: Metanol deuterado RT: Tempo de retenção. Deste ponto em diante, a fórmula estrutural dos compostos dos Exemplos 1 a 38 serão mostradas nas Tabelas 4 a 8. Nas tabelas, os compostos marcados com * próximo ao átomo de carbono assimétrico para o qual R3, R4 e OH estão ligados, indica que um ou mais átomos de carbono assimétricos estão em uma forma R e outro está em uma forma S. No entanto, Exemplos 13 a 16 são mostrados a seguir. Na Tabela 5, compostos marcados com ** perto de um átomo de carbono assimétrico para o qual R3, R4, θ OH estão ligados, indicam que os átomos de carbono assimétricos são os mesmos isômeros de cada de uma forma R ou uma forma S. Enquanto isso, os compostos marcados com *** indicam que seus átomos de carbono assimétricos são os mesmos isômeros de uma forma R ou uma forma S, mas eles são isômeros diferentes dos compostos marcados com **. Tabela 4

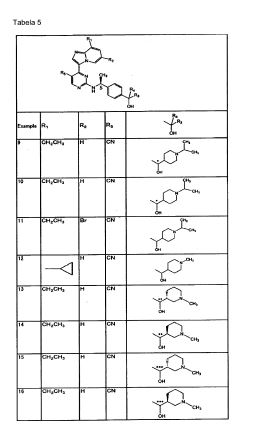


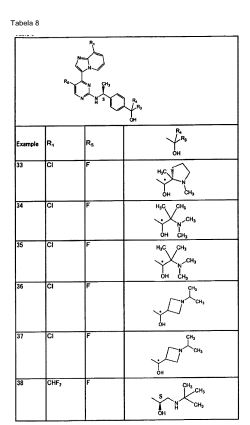
Síntese de 2-[(( 1 S)-1 -{4-[2-(dimetilamino)-1 -hidroxietila]fenil}etil)amino]-4- (8-metilimidazo1,2-a]piridin-3-il)pirimidina-5-carbonitrila [1] (doravante, referida como o composto [I]) (1), 2 g de S-(-)-1-(4-bromofenil)etilamina foi dissolvido em 20 mL de clorofórmio e 4,18 mL de trietilamina foi adicionado. A mesma, 2,62 g de dicarbonato di-tert-butila foi adicionada em uma condição gelada, e a mistura foi agitada por 30 minutos em temperatura ambiente. A solução de reação foi diluída em 300 mL de acetato de etila e lavada com água e salmoura saturada nessa ordem, e na camada orgânica resultante foi seca sobre sulfato de magnésio anidro. Os insolúveis foram filtrados, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi dissolvido em uma pequena quantidade de clorofórmio e solidificado com hexano, para obter 2,6 g de [tert-butil(1S)-1-(4- bromofenil)] carbamato de etila [1-1] (doravante referido como o composto [1-1]) como um sólido branco. (2) Uma mistura de 600 mg do composto [1-1], 75 mg de diclorobis(trifenilfosfina)paládio (II), 743 pL de tributil(1 -etoxivinl) estanho e 3 mL de 1,4-dioxano, foram agitados durante a noite a 100°C. Após deixar resfriar, os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. O resíduo resultante foi purificado em coluna de sílica gel (eluente: Hexano/acetato de etila = 8/2) para obter 367 mg de [tert-butil(1 S)-1 -(4-acetilfenil)] carbamato de etila [1-2] (doravante referido como o composto [1-2]) como um sólido incolor. (3) 984 mg de brometo de benzil tributil amónio foi dissolvido em uma mistura de solventes de 3 mL de clorofórmio e 2 mL de metanol, e depois de 141 pL de bromo foi adicionado. A mesma, uma solução preparada dissolvendo 363 mg do composto [1-2] em uma mistura de solvente de 4 mL de clorofórmio e 0,8 mL de metanol foi adicionado. A mistura de reação foi agitada por 30 minutos a 40°C. Após deixar para resfriar, a água foi adicionada à solução de reação, que foi extraída com clorofórmio. A camada orgânica foi lavada com salmoura saturada e seca com sulfato de sódio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado em coluna de sílica gel (eluente: hexano / acetato de etila = 7/3), para obter 170 mg de tert-butil{(1S)-1-[4- (bromoacetil)fenil]etil}carbamato[1-3] (de agora em diante, chamado como o composto [1-3]) como produto oleoso incolor. (4) 50 mg do composto [1-3] foi dissolvida em 0,5 mL de N, N- dimetilformamida, e 365 pL de dimetilamina (2,0 M solução tetrahidrofurano) foi adicionado à mesma. A mistura foi agitada durante a noite em temperatura ambiente, e, em seguida, para a solução de reação foi adicionada uma solução aquosa saturada de bicarbonato de sódio, e a solução foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada e seca com sulfato de sódio anidro. Os insolúveis foram filtrados, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado por cromatografia em camada delgada preparativa, para obter 26,3 mg de tert-butil{(1S)-1-[4-(N,N- dimetilglicil)fenil]etil}carbamato[1 -4] (de agora em diante, chamado de composto [1-4]) como um produto oleoso incolor. (5), 26,3 mg do composto [1-4] foi dissolvido em 1,5 mL de metanol, 16,2 mg de sódio boronhidrido foi adicionado ao mesmo, e mistura foi agitada por 30 minutos em temperatura ambiente. Para a solução de reação foi adicionada uma solução aquosa saturada de bicarbonato de sódio, e a solução foi extraída com uma mistura de solvente de clorofórmio e metanol (proporção de mistura: 9/1). A camada orgânica foi lavada com salmoura saturada e seca com sulfato de sódio anidro. Os insolúveis foram filtradas, e o filtrado foi concentrado sob pressão reduzida, para obter tert- butil((1S)-1-{4-[2-(dimetilamino)-1-hidroxietil]fenil}etil)carbamato[1-5] (de agora em diante, chamado como o composto [1-5]). O composto [1-5] foi usado na reação subsequente sem outra purificação. (6) 10 g de 4-[(Z)-2-etoxivinil]-2-(metiltio)-5-pirimidinacarbonitrila (sintetizado de acordo com o método divulgado na Publicação Internacional W02006/025567, páginas 90-91) foi dissolvido em uma mistura de solvente de 150 mL de 1,4-dioxano e 30 mL de água e, em seguida, 8,04 g de N- Bromosuccinimida foi adicionada em uma condição gelada, e a mistura foi agitada por 1,5 horas em temperatura ambiente. Para a solução da reação, 4,89 g de 2- amino-3-picolina foi adicionado, e a mistura foi novamente agitada por 2,5 horas. 200 mL de água foi adicionado à solução de reação para extrair com 2 L de uma mistura de solventes de clorofórmio e metanol (proporção de mistura: 9 /1). A camada orgânica foi lavada com água e seca com sulfato de magnésio anidro. Posteriormente, os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. O resíduo resultante foi dissolvido em uma pequena quantidade de clorofórmio e solidificado com hexano para obter 4,02 g de 4-(8- metilimidazo[1,2-a] piridina-3-il)-2-(metiltio) pirimidina-5-carbonitrila (de agora em diante, chamado de composto [1-6] como um sólido marrom claro. (7), 20,1 mg do composto [1-6] foi dissolvido em 1,5 mL de clorofórmio e em seguida 24,7 mg de ácido m-cloroperbenzóico foi adicionado em uma condição gelada, e a mistura foi agitada por 30 minutos a 0°C. À solução de reação foi adicionada uma solução aquosa saturada de bicarbonato de sódio, e a solução foi extraída com de clorofórmio. A camada orgânica resultante foi lavada com salmoura saturada e em seguida seca com sulfato de sódio anidro. Os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida para obter a forma oxidada do composto [1-6]. Em um frasco separado, o composto [1- 5] foi dissolvido em 1,5 mL de clorofórmio e 1,5 mL de ácido trifluoroacético foi adicionado à mesma. A mistura foi agitada por 1 hora à temperatura ambiente. A solução de reação foi concentrada sob pressão reduzida, e o resíduo resultante foi dissolvido em metanol, que foi passado por uma resina de troca aniônica fraca (Bond Elut Regular tipo PSA, GL Sciences Inc.) para remover o ácido trifluoroacético, e o solvente foi destilado sob pressão reduzida. O resíduo resultante foi dissolvido em 0,5 mL de tetrahidrofurano, e para a solução resultante foi adicionada uma solução preparada misturando 9,9 pL de trietilamina e a forma oxidada obtida anteriormente em 1 mL de clorofórmio, e a solução foi agitada por 3 horas à temperatura ambiente. À solução de reação foi adicionada uma solução aquosa saturada de bicarbonato de sódio, e a solução foi extraída com de clorofórmio. A camada orgânica resultante foi lavada com salmoura saturada e em seguida seca com sulfato de sódio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e em seguida purificada por cromatografia em camada delgada preparativa (eluente de desenvolvimento: Clorofórmio/metanol = 20/1), para obter 14,5 mg do composto de título [1] como um sólido amarelo. Os dados espectrais do composto [1] são apresentados abaixo. 1H-NMR (CDCI3) δ: 9,64 - 9,62 (m, 1Hx1/5), 8,95 - 8,74 (m, 1H+1HX4/5), 8,56 (s, 1Hx 1/5), 8,53 (s, 1HX4/5), 7,43 - 7,15 (m, 5H), 6,64 - 6,63 (m, 1H), 6,02 - 6,01 (m, 1HX4/5), 5,85 - 5,76 (m, 1Hx1/5), 5,42 - 5,33 (m, 1H+1Hx1/5), 5,16 - 5,12 (m, 1HX4/5), 4,74 - 4,69 (m, 1H), 2,69 (s, 1HX3/5), 2,64 (s, 2H+1Hx2/5), 2,36 (s, 3H), 2,03 - 1,80 (m, 2H), 1,63 (d, J = 6,8Hz, 3H) massa: 442 (M+1)+.
EXEMPLO 2
Síntese de 2-[((1S)-1-{4-[(1S)-2-(tert-butilamino)-1- hidroxietil]fenil}etil)amino]-4-(8-etilimidazo[1,2-a]piridin-3-il)pirimidina-5-carbonitrila [2] (de agora em diante, chamado de composto [2]) (1) uma mistura de 1,0 g do composto [1-1], 892 mg de viniltrifluoroborato de potássio, 468 mg de dicloreto (trifenilfosfina) de paládio (II), 10 mL de solução 2,0M de solução aquosa de carbonato de sódio e 20 mL de N , N- dimetilformamida, foi agitada durante a noite em 90°C. Após deixar para resfriar, a água foi adicionada à solução de reação, que foi extraída com acetato de etila. A camada orgânica resultante foi lavada com salmoura saturada e em seguida seca com sulfato de sódio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado em coluna de sílica gel (eluente: hexano / acetato de etila = 100/0-75/25), para obter 485 mg [tert-butílico(1 S)-1 -(4-vinilfenil)] carbamato de etila [2-1] (doravante referido como o composto [2-1]) como um sólido branco. (2) 25,3 g de AD-mix-alfa (disponível a partir de Aldrich Co.) foi adicionado a uma mistura de solvente de 110 mL de tert-butanol e 110 mL de água para preparar uma suspensão. À mesma, 4,34 g do composto [2-1] foi adicionada sob uma condição gelada, e a mistura foi agitada por 24 horas na mesma temperatura. 25 g de sulfito de sódio foi adicionada à solução de reação, que foi agitado durante 30 minutos a 0°C e em seguida extraídos com clorofórmio. A camada orgânica resultante foi seca sobre sulfato de sódio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado em coluna de sílica gel (eluente: clorofórmio/metanol = 100/0 - 90/10), para obter 4,32 g de tert-butil((1S)-1-{4-[(1S)-1,2- dihidroxietil]fenil}etil)carbamato [2-2] (de agora em diante chamado de composto [2-2]) como um sólido branco. (3), 4,32 g do composto [2-2] foi dissolvido em 50 mL de piridina e 3,66 g de cloreto de p-toluenessulfonila foi adicionado em uma mesma condição gelada. A mistura de reação foi agitada durante a noite a 0°C, a água foi adicionada à mesma, e, em seguida, a mistura foi extraída com acetato de etila, e na camada orgânica resultante foi seca sobre sulfato de sódio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado em coluna de sílica gel (eluente: hexano/acetato de etila = 50/50 - 0/100) para obter 6,52 g de (2S)-2-(4-{(1S)-1-[(tert-butoxicarbonil)amino]etil)fenil)- 2-hidroxietil 4-metilbenzenesulfonato [2-3] (doravante referido como o composto [2-3]) como um amorfo incolor. (4) 6,52 g do composto [2-3] foi dissolvida em 44,4 mL de clorofórmio, uma solução preparada pela dissolução de 2,4 g de hidróxido de sódio em 5,54 mL de água foi adicionado, e 203 mg de sulfato de hidrogênio tetrabutilamônio foi adicionado à mesma. A mistura de reação foi agitada durante a noite em temperatura ambiente, a água foi adicionada à mesma, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada e seca com sulfato de sódio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado em coluna de sílica gel (eluente: 50% de clorofórmio-hexano/acetato de etila = 100/0 - 75/25), para obter 4.4. g de tert-butil((1S)-1-{4-[(2S)-oxiran-2-il]fenil}etil)carbamato [2-4] (doravante denominado como composto [2-4] como um sólido branco. (5), 4,4 g do composto [2-4] foi dissolvido em 40 mL de etanol, 20 mL de tert- butilamina foi adicionado à mesma, e a mistura foi agitada por 3 dias a 50°C. A solução de reação foi concentrada, e o resíduo resultante foi purificado em cromatografia de coluna de sílica gel (eluente: hexano / acetato de etila = 100/0 - 0/100), para obter 3,1 g tert-butil((1S)-1-{4-[(1S)-2-(tert-butilamino)-1- hidroxietil]fenil}etil)carbamato [2-5] (doravante denominado como composto [2-5]) como um sólido branco. (6) 4,89 g de 4-[(Z)-2-etoxivinil]-2-(metiltio)-5-pirimidinacarbonitrila (sintetizado de acordo com o método divulgado na Publicação Internacional W02006/025567, páginas 90 a 91) foi dissolvido em uma mistura de solvente de 50 mL de 1,4-dioxano e 5 mL de água e, em seguida, 3,93 g de N- bromosuccinimida foi adicionada em uma condição gelada, e a mistura foi agitada por 1,5 horas em temperatura ambiente. À solução de reação foi adicionado 2,7 g de 3-etilpiridina-2-amina (sintetizado de acordo com o método revelado na Publicação Internacional W02006/025567, página 142), e a solução foi agitada durante a noite em temperatura ambiente. À solução de reação foi adicionada uma solução aquosa saturada de bicarbonato de sódio, a solução foi extraída com clorofórmio, e a camada orgânica resultante foi seca sobre sulfato de sódio anidro. Os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. Éter dietílico foi adicionado ao resíduo resultante, e a solução resultante foi agitada por 3 horas. Assim sólidos produzidos foram tomados por filtração e secos sob pressão reduzida, para obter 4,46 g de 4 - (8-etilimidazo,[1,2-a]piridina- 3-il)-2- (metiltio)pirimidina-5-carbonitrila [2 - 6] (doravante referido como o composto [2-6]) como um sólido amarelo pálido. (7) 2 g do composto [2-6] foi dissolvida em 150 mL de clorofórmio e 1,95 g de ácido m-cloroperbenzóico foi adicionado em uma mesma condição gelada. A mistura de reação foi agitada por 30 minutos a 0°C. À solução de reação foi adicionada uma solução aquosa saturada de bicarbonato de sódio, e a solução foi extraída com de clorofórmio. A camada orgânica foi lavada com salmoura saturada e seca com sulfato de sódio anidro. Os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida para obter a forma oxidada do composto [2-6]. Em um frasco separado, o composto [2-5] foi dissolvido em 20 mL de clorofórmio e 10 mL de ácido trifluoroacético foi adicionado à mesma sob condição gelada. A mistura foi agitada por 1 hora à temperatura ambiente. A solução de reação foi concentrada sob pressão reduzida, o resíduo resultante foi dissolvido em 20 mL de tetrahidrofurano e uma solução preparada pela mistura de 25 mL de trietilamina e a anteriormente obtida na forma oxidada em 20 mL de clorofórmio foi adicionado ao mesmo, e a mistura foi agitada por 1 hora à temperatura ambiente. Para a solução de reação foi adicionada uma solução aquosa saturada de bicarbonato de sódio, e a solução foi extraída com uma mistura de solvente de clorofórmio e metanol (proporção de mistura: 9/1). A camada orgânica foi lavada com salmoura saturada e seca com sulfato de sódio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado em coluna de sílica gel (eluente: clorofórmio/metanol = 100/0 - 90/10), para obter 1,2 mg do composto [2] como um sólido amarelo pálido. 803 mg do composto [2] foi suspenso em 20 mL de etanol, e 1,66 mL de uma solução aquosa de ácido clorídrico 1N foi adicionado ao mesmo, para gerar uma solução homogênea. A mistura foi concentrada sob pressão reduzida, e 20 mL de etanol foi adicionado ao resíduo resultante, e a solução resultante foi novamente concentrada sob pressão reduzida. Assim o sólido produzido foi seco sob pressão reduzida a 40°C e 835 mg de sal cloridrato do composto de título [2] foi obtida como um sólido amarelo pálido. Os dados espectrais do composto [2] são apresentados abaixo. 1H-NMR (DMSOd6) δ: 9,99 (d, J = 6,8Hz, 1H x 1/2), 9,10 (d, J = 6,3hz, 1H x 1/2), 9,04 (d,J = 7,3Hz, 1H x 1/2), 8,99 (d, J = 7,8Hz, 1H x 1/2), 8,89 (brs, 1H), 8,76 (s, 1H), 8,71 (s, 1H x 1/2), 8,64 (s, 1H x 1/2), 8,42 - 8,40 (m, 1H), 7,46 - 7,38 (m, 4H), 7,19 (t, J = 7,1 Hz, 1H x 1/2), 7,07 (t, J = 7,1 Hz, 1H x 1/2), 6,10 (m, 1H), 5,24 (t, J = 7,3Hz, 1H x 1/2), 5,15 (t, J = 7,1Hz, 1H x 1/2), 4,87 (m, 1H), 3,07 - 2,97 (m, 3H), 2,90 - 2,85 (m, 1H), 1,53 (d, J = 7,3Hz, 3Hx 1/2), 1,50 (d, J = 6,8Hz, 3Hx1/2), 1,33 (t, J = 7,6Hz, 3Hx1/2), 1,30 (t, J = 7,3Hz, 3Hx1/2), 1,29 (s, 9Hx1/2), 1,26 (s, 9Hx1/2) massa: 484 (M+1)+.
EXEMPLO 3
Síntese de 2-[((1S)-1-{4-[(1R)-2-(tert-butilamino)-1- hidroxietil]fenil}etil)amino]-4-(8-etilimidazo[1,2-a]piridin-3-il)pirimidina-5-carbonitrila [3] (doravante chamado de composto [3]) (1) 2,5 g de tert-butil((1S)-1-{4-[(1R)-2-(tert-butilamino)-1- hidroxietil]fenil}etil)carbamato [3-1] (doravante denominado composto [3-1]) foi obtido como um sólido branco de 4,14 g do composto [2-1] e 24 g de AD-mix-β (disponível de Aldrich Corporation) de acordo com os métodos do Exemplo 2-(2) a (5). (2) 1,17 g de sal de cloridrato de composto [3] foi obtido como um produto espumoso amarelo pálido de 1,5 g do composto [2-6] e 1,56 g do composto [3-1] de acordo com o método do exemplo 2 - (7). Os dados espectrais do composto [3] são apresentados abaixo. 1H-NMR (DMSOd6) δ: 9,98 (d, J = 6,8Hz, 1H x 1/2), 9,04 (d, J = 8,3hz, 1H x 1/2), 9,01 (d, J = 8.3Hz, 1Hx1/2), 8,97-8,92 (m, 1H), 8,76 (s, 1Hx1/2), 8,76 (s, 1Hx1/2), 8,71 (s, 1Hx1/2), 7,49 (d, J = 6.3hz, 1Hx1/2), 7,44-7,37 (m, 5H), 7,21 (t, J = 7.3Hz, 1Hx1/2), 7,08 (t, J = 7.3Hz, 1Hx1/2), 6,11 (BRS, 1Hx1/2), 5,27-5,20 (m, 1Hx1/2), 5,17-5,10 (m, 1Hx1/2), 4,89-4,85 (m, IH), 3,05-2,95 (m, 3H+1Hx1/2), 2,91 - 2,80 (m, 1Hx1/2), 1,53 - 1,49 (m, 3H), 1,34 - 1,30 (m, 3H), 1,28 (s, 9Hx1/2), 1,25 (s, 9Hx 1/2) massa: 484 (M+ 1)+.
EXEMPLO 4
Síntese de 2-[((1S)-1-{4-[(1R)-2-(tert-butilamino)-1- hidroxietil]fenil}etil)amino]-4-[8-(difluorometil)imidazo[1,2-a]piridin-3-il]pirimidine-5- carbonitrila [4] (doravante denominado como composto [4]) (1) 7,15 g de 4-(8-formilimidazo[1,2-a]piridina-3-il)-2-(metiltio)[pirimidina-5- carbonitrila [4-1] (doravante denominado composto [4-1]) foi obtido como um sólido marrom de 5,69 g de 2-amino-3-formilpiridina e 10 g de 4-[(Z)-2-etoxivinil]- 2-(metiltio)-5-pirimidinecarbonitrila de acordo com Exemplo do método de 1-(6). (2) 5 g do composto [4-1] foi suspenso em 200 mL de clorofórmio, em seguida, 7,8 mL de trifluoreto de bis(2-metoxietilico)aminosulfur foi adicionado em uma condição gelada, e a mistura foi agitada no mesma temperatura. Após cerca de 30 minutos, a mistura de reação que se transformou em uma solução marrom clara foi agitada ainda durante 30 minutos, e então 300 mL de uma solução aquosa saturada de bicarbonato de sódio foi adicionado à mesma. A camada orgânica e a fase aquosa foram separadas, e a fase aquosa foi extraída com um solvente misto de clorofórmio e metanol (proporção de mistura: 9 /1). A camada orgânica foi combinada com a camada orgânica anterior que foi lavada com água e seca com sulfato de magnésio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado em coluna de sílica gel (eluente: clorofórmio/acetato de etila = 100/0 - 30/70). O solvente foi destilado, em seguida, dissolvido em uma pequena quantidade de clorofórmio e hexano foi adicionado a mesma para ser solidificado, por meio disso, obtendo-se 2,03 g de 4-[8-(difluorometila)imidazo[1,2-a] piridina-3-il]-2-(metiltio)pirimidina-5- carbonitrila [4-2] (doravante denominado o composto [4-2]) como um sólido marrom claro.(3) 12 mg do composto de título [4] foi obtida como um produto espumoso branco de 20 mg do composto [4-2] e 21 mg do composto [3-1] acordo com o método de exemplo 1-(7). Os dados espectrais do composto [4] são apresentados abaixo. 1H-NMR (DMSOd6) δ: 10,20 (d, J = 7,3Hz, 1Hx1/2), 9,09 (d, J = 7,3Hz, 1 Hx1/2), 9,05 (d, J = 7,3Hz, 1 Hx1/2), 8,96 (d, J = 7,3Hz, 1 Hx1/2), 8,78 (s, 1 Hx1/2), 8,77 (s, 1Hx1/2), 8,74 (s, 1Hx1/2), 8,63 (s, 1Hx1/2), 7,86 (d, J = 7,3Hz, 1Hx1/2), 7,78 (d, J = 7,3Hz, 1Hx1/2), 7,51 (t, J = 54,6Hz, 1Hx1/2), 7,45 (t, J = 54,1 Hz, 1Hx1/2), 7,37 - 7,29 (m, 5H+1Hx1/2), 7,12 (t, J = 7,3Hz, 1Hx1/2), 5,30 - 5,04 (m, 2H), 4,51 - 4,45 (m, IH), 2,60 - 2,51 (m, 2H), 1,54 - 1,49 (m, 3H), 1,00 (s, 9Hx1/2), 0,94 (s, 9Hx 1/2) massa: 506 (M+ 1)+.
EXEMPLO 5
Síntese de 2-[((1 S)-1 -{4-[hidroxi(piridim-2-il)metil]fenil}etil)amino]-4-(8- metilimidazo[1,2-a]piridin-3-il)pirimidina-5-carbonitrila [5] (doravante chamado de composto [5]) (1) 500 mg do composto [1-1] foi dissolvido em 10 mL de tetrahidrofurano e resfriado a -78°C. Em seguida, 2,33 mL de n-butil lítio (1,57M solução de hexano) foi adicionado à mesma. Após 1 hora de agitação na mesma temperatura, 193 uL de picolinaldeído foi adicionado à suspensão branca resultante, e a suspensão foi agitada durante a noite, enquanto se aquece volta à temperatura ambiente. 50 mL de água foi adicionada à mistura de reação, e a mistura foi extraída com 200 mL de acetato de etila. A camada orgânica foi lavada com salmoura saturada e seca com sulfato de magnésio anidro. Posteriormente, os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. O resíduo resultante foi purificado em coluna de silica gel (eluente: hexano /acetato de etila = 100/0 - 30/70) para obter 212 mg de tert-butil((1S)-1-{4-[hidroxi(piridin-2-il)metil]fenil}etil)carbamato [5- 1] (doravante denominado como composto [5-1] como produto oleoso marrom claro. (2) 24 mg do composto de título [5] foi obtida como um produto espumoso cinza de 58 mg do composto [5-1] e 41 mg do composto [1-6] acordo com o método de exemplo 1-(7). Os dados espectrais do composto [5] são apresentados abaixo. 1H-NMR (DMSOd6) δ: 9,95 (d, J = 6,8Hz, 1Hx1/2), 8,94 (d, J = 7,1 Hz, 1 Hx1/2), 8,88 (d, J = 7,8 Hz, 1Hx1/2), 8,75-8,70 (m, 1H+1Hx1/2), 8,67 (s, 1Hx1/2), 8,56-8,55 (m, 1Hx 1/2), 8,42-8,35 (m, 1H), 7,77-7,67 (m, 1H), 7,56-7,51 (m, t 1H), 7,42-7,25 (m, 5H), 7,21-7,15 (m, 1H), 7,11 (J = 6,8Hz, 1Hx1/2), 6,68-6,61 (m, 1 Hx1/2), 6,03-6,00 ( m, 1H), 5,66-5,65 (m, 1H), 5,19 (Quint, J = 7,1Hz, 1Hx1/2), 5,02 (Quint, J = 6,8Hz, 1Hx1/2), 2,58 (s, 3Hx1/2) , 2,52 (s, 3Hx1/2), 1,49 (d, J = 7,1 Hz, 3HX 1/2), 1,45 (d, J = 6,8Hz, 3Hx 1/2) massa: 462 (M+l)+.
EXEMPLO 6
Síntese de 2-[((1S)-1-{4-[1-hidroxi-2-(1-metilpiperidin-4- il)etil]fenil}etil)amino]-4-(8-metilimidazo[1,2-a]piridin-3-il)pirimidine-5-carbonitrila [6] (doravante denominado composto [6]) (1) 43 mg de benzil4-{2-[4-((1S)-1-{[5-ciano-4-(8-metilimidazo[1,2-a]piridin- 3-il)pirimidin-2-il]amino}etil)fenil]-2-hidroxietil}piperidina-1 -carboxilato [6-1] (doravante denominado [6-1]) foi obtido como um sólido amarelo pálido a partir de 300 mg do composto [1-1], 516 mg de 1-benziloxicarbonil-4-(formilmetil)piperidina (sintetizado de acordo com o método divulgado na especificação da Publicação de Patente Européia No. 0367110, páginas 108 a 109), e 41 mg do composto [1- 6], de acordo com o método do Exemplo 5. (2) 21 mg do composto [6-1], foi dissolvido em uma mistura de solventes, de 3 mL de tetrahidrofurano e 3 mL de metanol, em seguida, 20 mg de um catalisador de carbono de 20% de hidróxido de paládio foi adicionado ao mesmo, e a mistura foi agitada por 2 horas em temperatura ambiente sob atmosfera de hidrogênio. O catalisador foi filtrado através de celite e o filtrado foi concentrado sob pressão reduzida. Éter dietílico foi adicionado ao resíduo resultante, e assim o sólido produzido foi tomado por filtração para a obtenção de 6 mg de 2-({(1S)-1-[4-(1- hidroxi-2-piperidin-4-il-etil)fenil]etil}amino)-4-(8-metilimidazo[1,2-a]piridin-3- il)pirimidina-5-carbonitrila [6-2] (doravante denominado composto [6-2]). (3) Em uma mistura de 2 mg do composto [6-2] e 100 pL de uma solução de formol a 37%, 200 mL de solução metanol contendo 4 mg de cloreto de zinco e 4 mg de sódio cianotrIHidroborato foi adicionado, e a mistura foi agitada durante a noite em temperatura ambiente. À mistura de reação foi adicionada água e clorofórmio, e se a camada orgânica foi separada da camada aquosa. A camada obtida orgânica foi lavada com uma solução aquosa saturada de bicarbonato de sódio e gorduras saturadas salmoura nessa ordem, e seca com sulfato de sódio anidro. Os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. Éter dietílico foi adicionado ao resíduo resultante, e o sólido resultante foi tomado por filtração, para obter 2,2 mg do composto título [6] como um sólido cinza. Os dados espectrais do composto [6] são apresentados abaixo. 1H-NMR (CDCI3) ó: 9,62 (d, J = 7,2Hz, 1Hx1/5), 8,92 - 8,64 (m, 1H+1HX4/5), 8,55 (s, 1Hx1/5), 8,41 (s, 1HX4/5), 7,45 - 6,55 (m, 6H), 6,23 - 6,20 (m, 1HX4/5), 5,95 - 5,90 (m, 1Hx1/5), 5,40 - 4,70 (m, 2H), 3,00 - 2,70 (m, 4H), 2,58 (s, 3H), 2,30 (s, 3H), 2,10 -1,40 (m, 7H), 1,62 (d, J = 6,8Hz, 3H) massa: 496 (M+ 1)+.
EXEMPLOS 7 e 8
Síntese de 4-(8-etilimidazo[1,2-a]piridin-3-il)-2-[((1 S)-1 -{4-[hidroxi(1 - metilpiperidin-4-il)metil]fenil}etil)amino]pirimidina-5-carbonitrilas [7] (doravante denominados como o composto [7]) e [8] (doravante denominado composto [8]), (aqui, 0 composto [7] e o composto [8] são diastereômeros. Por favor, vide Tabela 4) (1) À uma mistura de 975 mg de magnésio, 100 pL de Bromoetano e 20 mL de tetrahidrofurano, uma quantidade de catalisador de iodo foi adicionado, e a mistura foi agitada por cerca de 10 minutos até que a cor marrom do iodo desaparece. Ao mesmo, 20 mL de uma solução contendo 5,9 g de tetrahidrofurano de 4-cloro-1 metilpiperidina foi adicionado e aquecido por 2,5 horas sob refluxo. Assim a suspensão branca produzida foi resfriada a 0°C e 10 mL de uma solução contendo 2 g do composto [39] foi adicionada à mesma (vide REFERÊNCIA 1). Após a agitação durante a noite sob aquecimento gradualmente de volta à temperatura ambiente, uma solução aquosa saturada de cloreto de amónio foi adicionado ao mesmo e extraído com acetato de etila. A camada orgânica obtida foi lavada com salmoura saturada e seca com sulfato de magnésio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado em coluna de sílica gel (eluente: clorofórmio / metanol = 100/0 - 90/10), para obter 2,27 g de tert- butil((1S)-1-{4-[hidroxi(1-metilpiperidin-4-il)metil]fenil}etil)carbamato [7-1] (doravante denominado composto [7-1]). (2) 1,2 mg dos compostos de título [7] e [8] foram obtidas de 1,42 g do composto [7-1] e 1 mg do composto [2-6] de acordo com o método de Exemplo 1- (7). (3) 700 mg da mistura dos compostos [7] e [8] foi resolvido usando Chiralcel OD-H. As condições de resolução óptica são os seguintes, coluna: Chiralcel OD-H (Daicel Chemical Industries, Ltd.), diâmetro de 20 mm, comprimento de 250 mm; eluente: Hexano/etanol/dietilamina = 85/15/0,1; taxa de fluxo: 15 mL/min. A solução obtida foi concentrada sob pressão reduzida, e o resíduo foi dissolvido em uma pequena quantidade de clorofórmio para ser solidificado com éter dietilico, obtendo-se 217 mg do composto título [7] (TR = 18,3 minutos) como um sólido branco e 230 mg do composto título [8] (TR = 24,3 minutos) como um sólido amarelo pálido. Os dados espectrais do composto [7] e do composto [8] são apresentados abaixo. Composto [7] 1H-NMR (DMSO-dβ) δ: 9,98 (d, J = 6,3Hz, 1HX2/5), 8,97 (d, J = 7,1 Hz, 1HX3/5), 8,90 (d, J = 7,3Hz, 1H), 8,73 (s, 1H), 8,70 (s, 1HX2/5), 8,60 (s, 1 HX3/5), 7,42 (d, J = 6,1 Hz, 1HX2/5), 7,35 - 7,33 (m, 2H+1Hx3/5), 7,26 - 7,21 (m, 2H), 7,15 (t, J = 7,1 Hz, 1HX2/5), 6,89 (t, J = 7,1 Hz, 1HX3/5), 5,24 (quint, J = 7,1 Hz, 1HX2/5), 5,17 - 5,05 (m, 1H+1HX3/5), 4,20 - 4,13 (m, 1H), 3,02 (q, J = 7,6Hz, 2Hx2/5), 2,96 (q, J = 7,6Hz, 2Hx3/5), 2,74 - 2,63 (m, 1H+1HX2/5), 2,50 - 2,48 (m, 1HX3/5), 2,06 (s, 3HX2/5), 2,02 (s, 3HX3/5), 1,73 - 1,62 (m, 2H+1HX2/5), 1,52 (d, J = 7,1 Hz, 3Hx2/5), 1,49 (d, J = 7,1 Hz, 3Hx3/5), 1,32 (t, J = 7,6Hz, 3Hx2/5), 1,28 (t, J = 7,6Hz, 3Hx3/5), 1,22 -1,04 (m, 4H), 0,97 - 0,94 (m, 1 HX3/5) massa: 496 (M+ 1)+. Composto [8] 1H-NMR (DMSO-dβ) δ: 9,98 (d, J = 6,1 Hz, 1Hx1/2), 8,97 (d, J = 7,1 Hz, 1Hx1/2), 8,90 - 8,89 (m, 1H), 8,73 (s, 1Hx1/2), 8,73 (s, 1Hx1/2), 8,70 (s, 1Hx1/2), 8,60 (s, 1HX1/2), 7,43 (d, J = 6,1Hz, 1HX1/2), 7,35 - 7,33 (m, 2H+1HX1/2), 7,25 (d, J = 8,3Hz, 1Hx1/2), 7,22 (d, J = 8,0Hz, 1HX1/2), 7,15 (t, J = 7,1 Hz, 1Hx1/2), 6,92 (t, J = 7,1 Hz, 1HX1/2), 5,24 (quint, J = 7,1 Hz, 1Hx1/2), 5,08 - 5,05 (m, 1H+1 Hx1/2), 4,20 - 4,16 (m, 1H), 3,01 (q, J = 7,6Hz, 2Hx 1/2), 2,96 (q, J = 7,6Hz, 2Hx1/2), 2,74 - 2,64 (m, 1H+1Hx1/2), 2,55 - 2,51 (m, 1Hx1/2), 2,06 (s, 3Hx 1/2), 2,02 (s, 3Hx1/2), 1,72 - 1,55 (m, 2H+1 Hx1/2), 1,52 (d, J = 7,1 Hz, 3Hx1/2), 1,49 (d, J = 7,1 Hz, 3Hx1/2), 1,32 (t, J = 7,6Hz, 3Hx1/2), 1,28 (t, J = 7,6Hz, 3Hx 1/2), 1,22 - 1,11 (m, 4H), 1,03 -1,00 (m, 1 Hx1/2)massa: 496 (M+ 1)+.
EXEMPLOS 9 e 10
Síntese de 4-(8-etilimidazo[1,2-a]piridin-3-il)-2-[((1 S)-1 -{4-[hidroxi(1 - isopropilpiperidin-4-il)metil]fenil}etil)amino]pirimidina-5-carbonitrilas [9] (doravante denominados como o composto [9]) e [10] (doravante denominado composto [10]), (aqui, o composto [9] e o composto [10] são diastereômeros. Por favor, vide Tabela 5) (1) 786 mg do composto [25-3] foi dissolvido em 15 mL de clorofórmio, em seguida, 2,4 mL de acetona e 728 mg de sódio triacetoxiborohidrido foram adicionados aos mesmos, e a mistura foi agitada a noite de 50°C. Depois de deixada resfriar, à solução de reação foi adicionada uma solução aquosa saturada de bicarbonato de sódio, e a solução foi extraída com clorofórmio. A camada orgânica foi lavada com água e seca com sulfato de magnésio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado em coluna de sílica gel (eluente: clorofórmio / metanol = 100/0 - 93/7), para obter 738 mg de uma mistura de compostos de título [9] e [10]. A mistura foi resolvido usando Chiralcel OD-H. As condições de resolução óptica são os seguintes, coluna: Chiralcel OD-H (Daicel Chemical Industries, Ltd.), diâmetro de 20 mm, comprimento de 250 mm; eluente: hexano/etanol/dietilamina = 85/15/0,1; taxa de fluxo: 15 mL/min. A solução obtida foi concentrada sob pressão reduzida, e o resíduo foi dissolvido em uma pequena quantidade de clorofórmio para ser solidificado com éter dietílico, obtendo-se 175 mg do composto título [9] (TR = 17 minutos) como um sólido branco e 390 mg do composto título [10] (TR = 21 minutos) como um sólido cinza. Os dados espectrais do composto [9] e do composto [10] são apresentados abaixo. Composto [9] RMN-H1(de DMSO-d6) δ: 9,98 (d, J = 6,6Hz, 1Hx1/2), 8,95 (d, J = 7,1 Hz, 1Hx1/2), 8,90 - 8,88 (m, 1H), 8,73 (s, 1Hx1/2), 8,72 (s, 1Hx1/2), 8,70 (s, 1Hx1/2), 8,61 (s, 1Hx1/2), 7,42 (d, J = 6,8Hz, 1Hx1/2), 7,35 - 7,32 (m, 2H+1HX1/2), 7,26 - 7,20 (m, 2H), 7,14 (t, J = 7,1 Hz, 1Hx 1/2), 6,89 (t, J = 7,1 Hz, 1 Hx1/2), 5,24 (quint, J = 7,3Hz, 1Hx1/2), 5,06 (quint, J = 7,1 Hz, 1Hx1/2), 5,03 - 5,01 (m, 1H), 4,20 - 4,18 (m, 1Hx1/2), 4,15 - 4,12 (m, 1Hx1/2), 3,02 (q, J = 7,6Hz, 2Hx1/2), 2,96 (q, J = 7,6Hz, 2Hx1/2), 2,75 - 2,60 (m, 1H+1Hx1/2), 2,58 - 2,48 (m, 1Hx1/2), 1,95 - 1,88 (m, 1H+1Hx1/2), 1,76 - 1,73 (m, 1H+1Hx1/2), 1,52 (d, J = 7,3Hz, 3Hx 1/2), 1,50 (d, J = 7, 1Hz, 3Hx 1/2), 1,38 - 1,25 (m, 11H), 1,32 (t, J = 7,6Hz, 3Hx1/2), 1,27 (t, J = 7,6Hz, 3Hx1/2), 1,18 - 1,09 (m, 2H), 1,00 - 0,96 (m, 1H), 0,89-0,83 (m, 6H) massa: 524 (M+ 1)+. Composto [10] RMN-H1 (de DMSO-d6) δ: 9.98 (d, J = 6,8Hz, 1 Hx1/2), 8,96 (d, J = 7,1 Hz, 1Hx1/2), 8,91 - 8,88 (m, 1H), 8,73 (s, 1H), 8,71 (s, 1Hx1/2), 8,60 (s, 1Hx1/2), 7,43 (d, J = 7,8Hz, 1Hx1/2), 7,35 - 7,33 (m, 2H+1Hx1/2), 7,25 - 7,20 (m, 2H), 7,15 (t, J = 6,8Hz, 1Hx1/2), 6,92 (t, J = 6,8Hz, 1Hx1/2), 5,26 - 5,22 (m, 1Hx1/2), 5,09 - 5,06 (m, 1Hx1/2), 5,04 - 5,00 (m, 1H), 4,21 - 4,16 (m, 1H), 3,02 (q, J = 7,3Hz, 2Hx1/2), 2,96 (q, J = 7,3Hz, 2Hx1/2), 2,74 - 2,71 (m, 1Hx1/2), 2,67 - 2,63 (m, 1H), 2,55 - 2,48 (m, 1Hx1/2), 1,98 - 1,65 (m, 3H), 1,52 (d, J = 7,1 Hz, 3Hx1/2), 1,50 (d, J = 7,3Hz, 3Hx1/2), 1,35 - 1,26 (m, 1H), 1,32 (t, J = 7,6Hz, 3Hx1/2), 1,28 (t, J = 7,6Hz, 3Hx1/2), 1,14-1,03 (m, 3H), 0,88 (d, J = 6,3Hz, 6Hx1/2), 0,85 (d, J = 6,6Hz, 6Hx 1/2) massa: 524 (M+1)+.
EXEMPLO 11
Sintese de 4-(6-bromo-8-etilimidazo[1,2-a]piridin-3-il)-2-[((1S)-1-{4- [hidroxi(1 - isopropilpiperidin-4-il)metil]fenil}etil)amino]pirimidina-5-carbonitrila [11] (doravante denominado composto [H]) (1) Para uma solução preparada pela dissolução de 360 mg de 3- etilpiridina-2-amina (sintetizado de acordo com o método divulgado na Publicação Internacional W02006/025567, página 142) em uma mistura de solventes, de 12 mL de 1,4-dioxano e 4 mL de água, 553 mg de N-bromosuccinimida foi adicionado em 0°C, e a mistura foi agitada por 1,5 horas na mesma temperatura. A água foi adicionada à solução de reação, que foi extraída com acetato de etila. A camada orgânica obtida foi lavada com água e salmoura saturada nesta ordem e seca com sulfato de sódio anidro. Posteriormente, os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. O resíduo obtido foi purificado em coluna de sílica gel (eluente: hexano / acetato de etila = 75/25), para obter 394 mg de 5-[3-bromo-etilpiridina-2-amina 11-1] (doravante referido como o composto [11- 1]). (2) 530 mg de 4-([6-bromo-8-etilimidazo[1 ,2-a]piridina-3-il)-2-(metiltio)- pirimidina-5-carbonitrila [11-2] (doravante referido como o composto [11-2]) foi obtido a partir de 388 mg do composto [11-1], de acordo com o método de exemplo 1-(6). (3) Para uma solução preparada pela dissolução de 2,98 g do composto [40] (vide REFERÊNCIA 2) em uma mistura de solventes, de 30 mL de tetrahidrofurano e 10 mL de 2-propanol, 750 mg de um catalisador de carbono de hidróxido de paládio 20% foi adicionado, e a mistura foi agitada durante a noite em temperatura ambiente sob atmosfera de hidrogênio. O catalisador foi filtrado através de celite e o filtrado foi concentrado sob pressão reduzida, para obter 2,3 g de tert-butil((1S)-1-{4-[hidroxi(piperidin-4- il)metil]fenil}etil)carbamato [11-3]) foi usado na reação subsequente sem outra purificação. (4) 2,14 g de tert-butil butil((1 S)-1 -{4-[hidroxi(1 -isopropilpiperidin-4- il)metil]fenil}etil)carbamato [11-4] (doravante denominado composto [11-4] foi obtido de 2,3 g do composto [11-3] e 15 mL de acetona de acordo com o método do Exemplo 6-(3). (2) 41 mg do composto de título [11] foi obtida como um produto amarelo amorfo de 70 mg do composto [11-4] e 63 mg do composto [11-2] acordo com o método de exemplo 1-(7). Os dados espectrais do composto [11] são apresentados abaixo. 1H-NMR (CDCI3) δ: 9,89 (s, 1HX5/6), 9,74 (s, 1 Hx1/6), 8,93 (s, 1HX5/6), 8,86 (s, 1Hx1/6), 8,55 (s, 1 Hx1/6), 8,16 (s, 1HX5/6), 7,41 - 7,27 (m, 5H), 6,76 (d, J = 8,0Hz, 1 HX5/6), 6,29 (d, J = 8,0Hz, 1Hx1/6), 5,35 (brs, 1Hx1/6), 5,27 (quint, J = 7,5Hz, 1HX5/6), 4,38 (d, J = 7,5Hz, 1H), 3,10 (q, J = 7,5Hz, 2H), 2,98 - 2,72 (m, 5H), 2,14 - 1,92 (m, 3H), 1,69 (d, J = 7,5Hz, 3H), 1,59 - 1,50 (m, 1H), 1,43 - 1,25 (m, 2H), 1,41 (t, J = 7,5Hz, 3H), 1,02 (d, J = 7,5Hz, 6H) massa: 602, 604 (M+ 1)+.
EXEMPLO 12
Síntese de 4-(8-ciclopropilimidazo[1,2-a]piridin-3-il)-2-[((1S)-1-{4-[hidroxi(1- metilpiperidin-4-il)metil]fenil}etil)amino]pirimidine-5-carbonitrila [12] (doravante denominado composto [12]) (1) 750 mg de 4-(8-ciclopropilimidazo[1,2-a]piridina-3-il)-2- (metiltio)[pirimidina-5-carbonitrila [12-1] (doravante denominado composto [12-1]) foi obtido como um sólido de laranja a partir de 390 mg de 3-ciclopropilpiridina-2-amina (sintetizado de acordo com o método divulgado na Publicação Internacional W02006/025567, página 142) de acordo com o método de exemplo 1-(6). (2) 66 mg do composto de título [12] foi obtida como um produto amarelo sólido de 100 mg do composto [12-1] e 110 mg do composto [7-1] de acordo com o método de exemplo 1-(7). Os dados espectrais do composto [12] são apresentados abaixo. 5 1H-NMR (CD3OD) δ: 9,85 (d, J = 8,0Hz, 1Hx1/3), 8,78 - 8,73 (m, 1HX2/3), 8,68 (s, 1Hx1/3), 8,61 (s, 1HX2/3), 8,43 (s, 1HX2/3), 8,36 (s, 1Hx1/3), 7,40 - 7,25 (m, 4H), 6,93 (t, J = 8,0Hz, 1HX2/3), 6,87 (d, J = 8,0Hz, 1HX2/3), 6,71 (d, J = 8,0Hz, 1Hx1/3), 6,67 (t, J = 8,0Hz, 1Hx1/3), 5,23 (q, J = 7,5Hz, 1Hx1/3), 5,01 (quint, J = 7,5Hz, 1HX2/3), 4,28 - 4,22 (m, 1H), 2,87 - 2,71 (m, 2Hx2/3), 2,59 - 10 2,51 (m, 2Hx1/3), 2,39 - 2,36 (m, 1H), 2,16 (s, 3Hx1/3), 2,12 (s, 3Hx2/3), 1,99 - 1,79 (m, 2H), 1,71 - 1,04 (m, 7H), 1,56 (d, J = 7,5Hz, 3Hx2/3), 1,06 (d, J = 7,5Hz, 3Hx 1/3), 0,82 - 0,73 (m, 2H) massa: 508 (M+ 1)+.
EXEMPLOS 13e14
Síntese de 4-(8-etilimidazo[1,2-a]piridina-3-il)-2-([(1S)-1-(4- ([hidroxi[(3R)-1 - 15 metilpiperidin-3-il]metil]}fenil)etil]amino}pirimidina-5-carbonitrila [13] (doravante referido como o composto [13]) e 4-(8-etilimidazo[1,2-a]piridina-3-il)-2-([(1S)-1-(4- ([hidroxi(3S)-1 metil]-metilpiperidin-3-il]fenil)etil]amino}pirimidina-5-carbonitrila [14] (doravante denominado composto [14]) (aqui, o composto [13] e os compostos [14] são diastereoisômeros. Por favor, vide Tabela 5) 20 (1) 11.8 g de benzil-3-[(4-{(1S)-1-[(tert- butoxicarbonil)amino]etil}fenil)(hidroxi)metil]piperidina-1-carboxilato [13-1] (doravante denominado composto [13-1]) foi obtido como uma mistura de quatro isômeros, a partir de 15 g de benzil(3R)-3-formilpiperidina-1-carboxilato (sintetizado de acordo com o método divulgado na Publicação Internacional 25 WO02/46157) e 16,6 g do composto [1-1] de acordo com 0 método do Exemplo 5- (1). (2) 6,81 g de benzil-3[[4-((1S)-1-{[5-ciano-4-(8-etilimidazo[1,2-a]piridina-3- i I )pi ri m id i n-2-i I amino}etil)fenil](hidroxi)metil]piperidina-1 -carboxilato [13-2] (doravante denominado composto [13-2]) foi obtido a partir de 7,56 g do composto 30 [2-6] e 11,8 g do composto [13-1], de acordo com o método de exemplo 1-(7). (3), 5 g de 4-(8-etilimidazo[1,2-a]piridin-3-il)-2-[((1S)-1-{4-[hidroxi(piperidin-3- il)metil]fenil}etil)amino]pirimidina-5-carbonitrila [13-3] foi obtida a partir de 6,81 g do composto [13-2] de acordo com o método do Exemplo 6-(2). O composto obtido [13-3] que é uma mistura de quatro isômeros foi purificada por 35 cromatografia de coluna de silica gel (eluente: metanol/clorofórmio = 100/0 - 90/10), obtendo-se, por meio disso, 2,21 g de um composto de baixa polaridade (composto que primeiro eluiu, na condição acima) [13-3a] (doravante referido como o composto [13-3]) e 1,88 g de um composto de alta polaridade (composto que eluiu depois sob a condição acima) [13-3b] (doravante referido como o composto [13-3b)J cada um como uma mistura de dois isômeros. Aqui, o composto [13-3a] é uma mistura de precursores dos compostos [13] e [14], respectivamente, e o composto [13-3b], é uma mistura de precursores de compostos [15] e [16] descritos depois, respectivamente. (2) 1,89 mg de uma mistura dos compostos de título [13] e [14] foram obtidas de 2,21 g do composto [13-3a] de acordo com o método de Exemplo 6- (3). A mistura do resolvida usando Chiralpack AD. As condições de resolução óptica são os seguintes, coluna: Chiralpack AD (Daicel Chemical Industries Ltd.), diâmetro de 50 mm, comprimento de 5.000 mm; eluente: hexano/2- propanol/dietilamina = 65/35/0.1; taxa de fluxo: 100 mL/min. A solução obtida foi concentrada sob pressão reduzida, e o resíduo foi dissolvido em uma pequena quantidade de clorofórmio para ser solidificado com éter dietilico, obtendo-se 892 mg do composto título [13] (TR = 15,5 minutos) como um sólido amarelo pálido e 298 mg do composto título [14] (TR = 35 minutos) como um sólido amarelo pálido. Os dados espectrais do composto [13] e do composto [14] são apresentados abaixo. Composto [13] 1H-NMR (DMSO-d6) δ: 9,98 (d, J = 6,8Hz, 1H/1/3), 8,96 (d, J = 6,8Hz, 1HX2/3), 8,89 - 8,86 (m, 1H), 8,73 (s, 1H), 8,70 (s, 1 Hx1/3), 8,61 (s, 1HX2/3), 7,41 - 7,16 (m, 5H+1Hx1/3), 6,89 (t, J = 7,1Hz, 1Hx2/3), 5,30 - 5,00 (m, 2H), 4,29 - 4,19 (m, 1H), 3,32 (s, 3H), 3,02 - 2,93 (m, 2H), 2,65 - 2,50 (m, 1H), 2,32 - 2,28 (m, 1Hx1/3), 2,12 - 2,06 (m, 1HX2/3), 1,80 - 1,40 (m, 9H), 1,32 - 1,20 (m, 3H) massa: 496 (M+ 1)+. Composto [14] 1H-NMR (DMSOd6) δ: 9,90 (m, 1HX2/5), 8,98 - 8,60 (m, 3H+1Hx3/5), 7,60 - 6,85 (m, 6H), 5,18 - 5,05 (m, 2H), 4,30 - 4,20 (m, 1H), 3,34 (s, 3H), 3,20 - 2,80 (m, 2H), 2,70 - 2,50 (m, 1H), 2,18 -1,45 (m, 10H), 1,40 - 1,20 (m, 3H) massa: 496 (M+ 1)+-
EXEMPLOS 15e 16
Síntese de 4-(8-etilimidazo[1,2-a]piridin-3-il)-2-{[(1S)-1-(4-{hidroxi[(3R)-1- metilpiperidin-3-il]metil}fenil)etil]amino}pirimidina-5-carbonitrila [15] (doravante denominado composto [15]) e 4-(8-etilimidazo[1,2-a]piridin-3-il)-2-{[(1S)-1-(4- {hidroxi[(3S)-1 -metilpiperidin-3-il]metil}fenil)etil]amino}pirimidina-5-carbonitrila [16] (doravante denominado composto [16]) (aqui, o composto [15] e o composto [16] são diasteroisômeros. Por favor, vide Tabela 5) 921 mg de uma mistura dos compostos de título [15] e [16] foram obtidas de 917 mg do composto [13-3b] de acordo com o método de Exemplo 6-(3). A mistura do resolvida usando Chiralcel OD-H. As condições de resolução óptica são os seguintes, coluna: Chiralcel OD-H (Daicel Chemical Industries, Ltd.), diâmetro de 20 mm, comprimento de 250 mm; eluente: hexano/etanol/dietilamina = 85/15/0,1; taxa de fluxo: 20 mL/min. A solução obtida foi concentrada sob pressão reduzida, e o resíduo foi dissolvido em uma pequena quantidade de clorofórmio para ser solidificado com éter dietílico, obtendo-se 510 mg do composto título [15] (TR = 16,8 minutos) como um sólido amarelo pálido e 253 mg do composto título [16] (TR = 23,1 minutos) como um sólido amarelo. Os dados espectrais do composto [15] e do composto [16] são apresentados abaixo. Composto [15] 1H-NMR (DMSO-dθ) δ: 10,00 (d, J = 6,8Hz, 1HX2/5), 8,98 (d, J = 6,8Hz, 1HX3/5), 8,92 - 8,87 (m, 1H), 8,76 (s, 1H), 8,73 (s, 1HX2/5), 8,63 (s, !Hx3/5), 7,46 - 7,15 (m, 5H+1Hx2/5), 6,90 (t ,J = 7,1 Hz, 1Hx3/5), 5,25 - 5,05 (m, 2H), 4,22 - 4,20 (m, 1H), 3,34 (s, 3H), 3,10 - 2,80 (m, 2H), 2,70 - 2,50 (m, 1H), 2,15 - 1,60 (m, 10H), 1,39- 1,22 (m, 3H) massa: 496 (M+l)+. Composto [16] 1H-NMR (DMSO-d6) δ: 9,90 (m, 1HX2/5), 9,00 - 8,60 (m, 3H+1 Hx3/5), 7,40 - 6,80 (m, 6H), 5,20 - 5,00 (m, 2H), 4,20 - 4,00 (m, 1H), 3,30 (s, 3H), 3,40 - 3,20 (m, 2H), 2,70 -1,40 (m, 8H), 1,40 -1,20 (m, 5H) massa: 496 (M+ 1)+.
EXEMPLOS 17e18
Síntese de 2-{[(1S)-1-(4-{hidroxi[(2S)-1-metilpirrolidin-2- il]metil}fenil)etil]amino}-4-(8-metilimidazo[1,2-a]piridin-3-il)pirimidina-5-carbonitrilas [17] (doravante denominado composto [17]) e [18] (doravante denominado composto 18]) (aqui o composto [17] e o composto [18] são diasteroisômeros. Por favor, vide Tabela 6) (1) 95 mg de benzil(2S)-2-[(4-{(1S)-1-[(tert- butoxicarbonil)amino]etil}fenil)(hidroxi)metil]pirrolidina-1 -carboxilato[17-1 ] (doravante denominado composto [17-1]) foi obtido a partir de 467 mg de benzil(2S)-2-formilpirrolidina-1- carboxilato (sintetizado de acordo com o método divulgado em Bioorg. Med. Chem., 2003.11. 3153-3164) e 500 mg do composto [1-1] de acordo com o método do Exemplo 5-(1). (2) 68 mg de benzil(2S)-2-[[4-((1S)-1-{[5-ciano-4-(8-metilimidazo[1,2- a]piridin-3- il)pirimidin-2-il]amino}etil)fenil](hidroxi)metil]pirrolidina-1 -carboxilato [17-2] (doravante, referido como o composto [17-2]) foi obtido a partir de 93 mg do composto [17-1], e 48 mg do composto [1-6] de acordo com o método do Exemplo 1-(7). (3) 10 mg de 2-{[(1S)-1-(4-([hidroxi(2S)-pirrolidin-2-il]metil}fenil)etil]amino]}- 4-(8- metilimidazo[1,2-a]piridina-3-il)pirimidina-5-carbonitrila [17-3] (doravante referido como o composto [17-3]) foi obtido a partir de 30 mg do composto [17-2], de acordo com o método de Exemplo 6 - (2). (4) 7 mg do composto [17-3] foi dissolvido em 1 mL de clorofórmio, em seguida, 4 mg de solução de formaldeído a 37% e 10 mg de sódio triacetoxiborohidrido foram adicionados aos mesmos, e a mistura foi agitada por 2,5 horas a temperatura ambiente. À solução de reação foi adicionada uma solução aquosa saturada de bicarbonato de sódio, e a solução foi extraída com clorofórmio. A camada orgânica foi lavada com salmoura saturada e seca com sulfato de sódio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado por cromatografia em camada delgada preparativa (eluente de desenvolvimento: metanol/clorofórmio = 20/1), para resolver o composto [17] e os compostos [18], que são diasterômeros. De acordo com a condição de purificação acima, 3 mg do composto título [17], que é um composto de baixa polaridade, e 2 mg do composto título [18], que é um composto de alta polaridade, foram obtidos ambos como um sólido branco. Os dados espectrais do composto [17] e do composto [18] são apresentados abaixo. Composto [17] 1H-NMR (CDCI3) δ: 9,63 (d, J = 6,3Hz, 1Hx 1/5), 8,94 (s, 1Hx 1/5), 8,89 (s, 1HX4/5), 8,76 (d, J = 6,3Hz, 1HX4/5), 8,57 (s, 1/5), 8,53 (s, 1HX4/5), 7,42 - 7,15 (m, 5H), 6,64 - 6,60 (m, 1H), 6,07 - 6,05 (m, 1Hx4/5), 5,88 - 5,80 (m, 1Hx1/5), 5,36 - 5,25 (m, 1H+1Hx1/5), 5,15 - 5,11 (m, 1HX4/5), 4,94 (s, 1H), 2,65 - 2,40 (m, 3H), 2,69 (s, 1HX3/5), 2,64 (s, 2H+1Hx2/5), 2,15 - 1,70 (m, 6H), 2,53 (s, 3H), 1,82 - 1,64 (m, 4H), 1,63 (d, J = 7,2Hz, 3H) massa: 468 (M+ 1)+. Composto [18] 1H-NMR (CDCh) δ: 9,64 - 9,63 (m, 1Hx1/5), 8,94 - 8,88 (m, 1 H+1 HX4/5), 8,53 (s, 1 Hx1/5), 8,52 (s, 1HX4/5), 7,43 - 7,16 (m, 5H), 6,66 - 6,62 (m, 1H), 6,04 - 6,02 (m, 1HX4/5), 5,83 - 5,82 (m, 1 Hx1/5), 5,18 - 5,13 (m, 1HX4/5), 4,55 - 4,54 (m, 1H), 3,40 - 3,20 (m, 1H), 2,69 (s, 1HX3/5), 2,65 (s, 2H+1Hx2/5), 2,57 - 2,45 (m, 6H), 2,38 (s, 3H), 1,96 - 1,55 (m, 4H), 1,63 (d, J = 7,2Hz, 3H) massa: 468 (M+ 1)+.
EXEMPLOS 19 e 20
Síntese de 4-(8-etilimidazo[1,2-a]piridin-3-il)-2-{[( 1 S)-1 -(4-{hidroxi[(2R)-1 - metilpirrolid in-2-il]metil}fenil)etil]amino}pirimidina-5-carbonitri las [19] (doravante denominado composto [19]) e [20] (doravante referido como composto [20]) (aqui os composto [19] e o composto [20] são diasteroisômeros. Por favor, vide Tabela 6) (1) 300 mg do composto [41] (Vide REFERÊNCIA 3) foi dissolvido em uma mistura de solvente de 6 mL de tetrahidrofurano e 6 mL de metanol, em seguida 100 mg de um catalisador de carbono de 20% de hidróxido de paládio foram adicionados e a mistura foi agitada por 2,5 horas em temperatura ambiente sob atmosfera de hidrogênio. O catalisador foi filtrado através de celite e o filtrado foi concentrado sob pressão reduzida. O resíduo resultante foi dissolvido em 6 mL de tetrahidrofurano, em seguida, 147 pL de solução de formaldeído a 37% e 420 mg de triacetoxiborohidrido de sódio foram adicionados aos mesmos, e a mistura foi agitada por 1,5 horas a temperatura ambiente. À solução de reação foi adicionada uma solução aquosa saturada de bicarbonato de sódio, e a solução foi extraída com uma mistura de solvente de clorofórmio e metanol (proporção de mistura: 9/1), e a camada orgânica resultante foi lavada com salmoura saturada e seca com sulfato de sódio anidro. Posteriormente, os insolúveis foram filtradas, e o filtrado foi concentrado sob pressão reduzida, para a obtenção de 220 mg [tert- butil( 1 S)-1 -(4-([hidroxi(2R)-1 ]metilpirrolidin-2-il]metil}fenil)etil]carbamato [19-1 ] (doravante referido como o composto [19-1]). (2) 15 mg do composto de título [19] e 13 mg do composto de título [20] foram obtidos ambos como um sólido incolor de 73 mg do composto [19-1] e 65 mg do [compostos 2-6 ] de acordo com o método de exemplo 1-(7). De acordo com as condições de purificação mencionadas no Exemplo 1-(7), o composto [19] foi um composto de baixa polaridade e do composto [20] foi um composto de alta polaridade. Os dados espectrais do composto [19] e do composto [20] são apresentados abaixo. Composto [19] 1H-NMR (CDCI3) δ: 9,67 - 9,60 (m, 1Hx1/5), 8,95 - 8,89 (m, 1H+1HX4/5), 8,57 (s, 1Hx1/5), 8,52 (s, 1HX4/5), 7,45 - 7,17 (m, 5H), 6,70 - 6,67 (m, 1H), 6,03 - 6,02 (m, 1HX4/5), 5,85 - 5,76 (m, 1Hx1/5), 5,35 - 5,34 (m, 1Hx1/5), 5,16 - 5,13 (m, 1HX4/5), 4,88 - 4,87 (m, 1H), 3,14 - 3,05 (m, 3H), 2,46 (s, 3H), 2,53 - 2,24 (m, 3H), 1,75-1,51 (m, 4H), 1,63 (d, J = 6,8Hz, 3H), 1,37 (t, J = 7,8Hz, 3H) massa: 482 (M+1)+. Composto [20] 1H-NMR (CDCI3) δ: 9,64 (d, J = 6,8Hz, 1 Hx1/5), 8,94 (s, 1Hx1/5), 8,89 (s, 1 HX4/5), 8,80 (d, J = 6,8Hz, 1HX4/5), 8,56 (s, 1Hx1/5), 8,52 (s, 1HX4/5), 7,43 - 7,16 (m, 5H), 6,70 - 6,66 (m, 1H), 6,04 - 6,03 (m,1 Hx4/5), 5,85 - 5,78 (m, 1Hx1/5), 5,36 - 5,26 (m, 1Hx1/5), 5,16 - 5,11 (m, 1HX4/5), 4,34 - 4,33 (m, 1H), 3,10 - 3,04 (m, 3H), 2,74 - 2,71 (m, 1H), 2,44 - 2,17 (m, 2H), 2,26 (s, 1 HX3/5), 2,17 (s, 2H+1 Hx2/5), 1,90 - 1,68 (m, 4H), 1,64 (d, J = 6,8Hz, 3H), 1,37 (t, J = 7,8Hz, 3H) massa: 482 (MH-1)+
EXEMPLO 21
Síntese de 2-[((1S)-1-{4-[(1S)-2-(tert-butilamino)-1- hidroxietil]fenil}etil)amino]-4-[8-(difluorometil)imidazo[1,2-a]piridin-3-il]pirimidina-5- carbonitrila [21] (doravante denominado como o composto [21]) 29 mg de sal cloridrato do título de composto [21] foi obtido como um sólido branco a partir de 101 mg do composto [2-5] e 95 mg do composto [4-2] de acordo com o método do Exemplo 2-(7). Os dados espectrais do composto [21] são apresentados abaixo. 1H-NMR (DMSOd6) δ: 10,20 (d, J = 6,8Hz, 1Hx1/2), 9,11 (d, J = 6,4Hz, 1Hx1/2), 9,06 (d, J = 7,2Hz, 1Hx1/2), 8,96 (d, J = 8,0Hz, 1Hx1/2), 8,78 (s, 1Hx1/2), 8,77 (s, 1Hx1/2), 8,74 (s, 1Hx1/2), 8,64 (s, 1Hx1/2), 7,86 (d, J = 8,0Hz, 1Hx1/2), 7,79 (d, J = 7,2Hz, 1Hx1/2), 7,51 (t, J = 54,4Hz, 1Hx1/2), 7,45 (t, J = 54,4Hz, 1Hx1/2), 7,37 - 7,29 (m, 5H+1Hx1/2), 7,13 (t, J = 7,2Hz, 1Hx1/2), 5,30 - 5,04 (m, 2H), 4,51 -4,45 (m, 1H), 2,60-2,50 (m, 2H), 1,54 -1,48 (m, 3H), 1,00 (s, 9Hx1/2), 0,93 (s, 9Hx1/2), massa: 506 (M+l)+.
EXEMPLO 22
Síntese de 2-[((1S)-1-{4-[(1S)-2-(tert-butilamino)-1- hidroxietil]fenil}etil)amino]-4-(8-cloroimidazo[1,2-a]piridin-3-il)pirimidina-5- carbonitrila [22] (doravante denominado como o composto [22]) 1,2 g de sal de cloridrato de composto [22] foi obtido como um sólido branco a partir de 2,23 g do composto [2-5] e 2 g do composto [9-1] de acordo com o método do exemplo 2 - (7). Os dados espectrais do composto [22] são apresentados abaixo. 1H-NMR (DMSOd6) õ: 10,06 (dd, J = 6,8Hz, 0,8Hz, 1Hx 1/2), 9,06 (d, J = 6,8Hz, 1HX1/2), 8,99 (dd, J = 7,2Hz, 1,2Hz, 1 Hx1/2), 8,97 (d, J = 8,4Hz, 1Hx1/2), 8,78 (s, 1Hx1/2), 8,75 (d, J = 5,2Hz, 1Hx1/2), 8,62 (s, 1Hx 1/2), 7,79 (dd, J = 7,6Hz, 1,2Hz, 1Hx1/2), 7,72 (dd, J = 7,6Hz, 0,8Hz, 1Hx1/2), 7,37 - 7,28 (m, 5H+1Hx1/2), 7,19 (t, J = 7,2Hz, 1Hx1/2), 7,00 (t, J = 7,2Hz, 1Hx1/2), 5,28 - 5,05 (m, 2H), 4,49 - 4,44 (m, 1H), 2,57 - 2,51 (m, 2H), 1,54 - 1,48 (m, 3H), 1,00 (s, 9Hx 1/2), 0,93 (s, 9Hx1/2) massa: 490, 492 (M+1)+.
EXEMPLOS 23 e 24
Síntese de 4-(8-ciclopropilimidazo[1,2-a]piridin-3-il)-2-[((1S)-1-(4-[[(2S)-1,2- dimetilpirrolidin-2-il](hidroxi)metil]fenil}etil)amino]pirimidina-5-carbonitrila [23] (doravante denominado como o composto [23]) e [24] (doravante denominado como o composto [24]) (aqui o composto [23] e o composto [24] são diastereisômeros. Por favor, vide Tabela 6) (1) Para a solução preparada pela dissolução de 500 mg de H-a-Me-OH- Pro (comercialmente disponível de Inc. Chem-lmpex) e 693 mg de cloroformato benzílico em 10 mL de 1,4-dioxano e 5 mL de água, uma solução 1 M de hidróxido de sódio aquosa foi adicionado à temperatura ambiente, de modo a manter o pH em 10, e a solução foi agitada a temperatura ambiente por 30 minutos. À solução da reação, HCI 5 M foi adicionado de forma a ajustar o pH a 2, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada e seca com sulfato de magnésio anidro. Os insolúveis foram filtradas, e o filtrado foi concentrado sob pressão reduzida, para a obtenção de 842 mg de 1-[(benziloxi)carbonil]-2-metil-1-prolina [23-1] (doravante referido como o composto [23-1]). (2) Para uma solução obtida pela dissolução de 840 mg do composto [23-1] em 16 mL de tetrahidrofurano, 4,31 mL de um complexo de sulfeto de metila borano (solução de tetrahidrofurano 2M) foi adicionada à temperatura ambiente, e a mistura foi aquecida sob refluxo por 4 horas. À solução de reação foi adicionada uma solução aquosa saturada de bicarbonato de sódio, e a solução foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada e seca com sulfato de magnésio anidro. Os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. O resíduo obtido foi purificado em coluna de sílica gel (eluente: hexano / acetato de etila = 100/0 a 60/40) para obtenção de 498 mg de benzil (2S)-2- (hidroximetil)-2-metilpirrolidina-1-carboxilato [23-2] (doravante referida como a [composto 23-2]). (3) Para uma solução obtida pela dissolução de 377 mg do composto [23-2] e 147 mg de piridina em 7,5 mL de clorofórmio, 673 mg de periodinano Dess- Martin foi adicionado à temperatura ambiente, e a mistura foi aquecida sob refluxo por 1 hora. À solução de reação, uma solução saturada aquosa de tiossulfato de sódio foi adicionada, e a mistura foi extraída com clorofórmio. A camada obtida orgânica foi lavada com uma solução aquosa saturada de bicarbonato de sódio e salmoura saturada, em seqüência, e seca com sulfato de sódio anidro. Os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. O resíduo obtido foi purificado em coluna de sílica gel (eluente: hexano / acetato de etila = 100/0 a 60/40) para obtenção de 346 mg de benzil (2S)-2- formil-2- metilpirrolidina-1-carboxilato [23-3] (doravante referida como a [composto 23-3]). (4) 423 mg do composto [1-1] e 508 mg de N,N,N',N'-diamina tetrametiletileno foram dissolvidos em 22 mL de tetrahidrofurano, a temperatura da solução foi mantida a -78°C, e, em seguida, 1,09 mL de n-butil lítio (solução de hexano 2,66 M) foi adicionado à mesma. Depois de ter agitado à mesma temperatura por 1 hora para obter a suspensão branca resultante, 2,2 mL de uma solução tetrahidrofurano preparada pela dissolução de 384 mg do composto [23- 3], foi adicionado à mesma, e a mistura foi agitada na mesma temperatura de 1,5 horas. À mistura de reação, uma solução saturada de cloreto de amónio aquoso foi adicionada, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada e seca com sulfato de sódio anidro. Posteriormente, os insolúveis foram filtradas, e o filtrado foi concentrado sob pressão reduzida para obter benzil-(2S)-2-[(4-{(1S)-1-[(tert- butoxicarbonil)amino]etil}fenil)(hidroxi)metil]-2-metilpirrolidina-1-carboxilato [23-4]. O composto [23-4] obtido foi uma mistura de dois isômeros, que foi então purificado por cromatografia de coluna de sílica gel (eluente: Hetaxo/acetato de etila = 100/0 - 40/60), para obter 199 mg de um composto de baixa polaridade (composto que primeiro eluiu, na condição acima) [23-4a] (doravante referido como o composto [23-4a]) e 117 mg de um composto de alta polaridade (composto que eluiu depois sob a condição acima) [23-4b] (doravante referido como o composto [23-4b]). Aqui, o composto [23-4a] é um precursor de um composto [23], e o composto [23-4b] é um precursor de um composto [24] descrito adiante. (5) 23,3 mg do composto de título [23] foi obtido como um sólido branco de 38 mg do composto [12-1] e 57,5 mg do composto [23-4a] de acordo com os métodos dos EXEMPLOS 17, 18-(2) a (4). Os dados espectrais do composto [23] são apresentados abaixo. 1H-NMR (CDCI3) δ: 9,70-9,62 (m, 1H 1/5), 8,98 (d, J = 6,8 Hz, 1H 4/5), 8,92 (s, 1H 1/5), 8,88 (s, 1H 4/5), 8,57 (s, 1H 1/5), 8,52 (s, 1H 4/5), 7,36-7,28 (m, 4H), 6,96-6,88 (m, 1H+1H 1/5), 6,71- 6,68 (m, 1H 4/5), 5,38-5,27 (m, 1H 1/5), 5,15-5,13 (m, 1H 4/5), 4,39 (s, 1H), 3,10-3,07 (m, 1H), 2,59- 2,45 (m, 2H), 2,19-2,03 (m, 2H), 2,10 (s, 3H), 1,73-1,51 (m, 2H), 1,62 (d, J = 6,8 Hz, 3H), 1,31-1,26 (m, 1 H), 1,17- 1,15 (m,2H), 0,92-0,86 (m, 5H) massa: 508 (M+ 1)+. (6) 10,6 mg do composto de título [24] foi obtida como um sólido branco de 38 mg do composto [12-1] e 57,5 mg do composto [23-4b] de acordo com os métodos dos EXEMPLOS 17, e 18-(2) a (4). Os dados espectrais do composto [24] são apresentados abaixo. 1H-NMR (CDCI3) δ: 9,63-9,56 (m, 1H 1/5), 8,97 (s, 1H 1/5), 8,90 (s, 1H 4/5), 8,74 (d, J = 6,8 Hz, 1H 4/5), 8,58 (s, 1H 1/5), 8,53 (s, 1H 4/5), 7,44-7,28 (m, 4H), 6,93-6,85 (m, 1H+1H 1/5), 6,68- 6,64 (m, 1H 4/5), 6,04-6,02 (m, 1H 4/5), 5,85-5,80 (m, 1H 1/5), 5,40-5,28 (m, 1H 1/5), 5,14-5,11 (m, 1H 4/5), 4,43 (s, 1H), 3,15-3,10 (m, 1H), 2,62-2,49 (m, 2H), 2,28 (s, 3H), 2,13-2,05 (m, 1H), 1,78-1,50 (m, 2H), 1,62 (d, J = 6,8 Hz, 3H), 1,38-1,25 (m, 1H), 1,15-1,12 (m, 2H), 0,94-0,85 (m, 5H) massa: 508 (M+l)+.
EXEMPLOS 25 e 26
Síntese de 4-(8-ciclopropilimidazo[1,2-a]piridin-3-il)-2-[((1S)-1-{4-[(R)-[(2R)- 1,2- dimetipirrolidin-2-il](hidroxi)metil]fenil}etil)amino]pirimidina-5-carbonitrila [25] (doravante denominado composto [25]) e 4-(8-ciclopropilimidazo[1,2-a]piridin-3-il)- 2-[((1 S)-1 -{4-[(S)-[(2R)-1,2-d imetilpirrolidin-2- il](hidroxi)metil]fenil}etil)amino]pirimidina-5-carbonitrila [26] (doravante denominado composto [26]) (aqui, o composto [25] e o composto [26] são diasteroisômeros. Por favor, vide a Tabela 12) (1) Benzil (2R)-2-[(4-{(1S)-1-[(tert- butoxicarbonil)amino]etil}fenil)(hidroxi)metil]-2-metilpirrolidina-1 -carboxilato [25-1 ] foi obtido de 1 g de H-a-Me-D-Pro-OH (comercialmente disponível de Chem- Impex International Inc.) de acordo com os métodos dos EXEMPLOS 23 e 24-(1) a (4). O composto [25-1] obtido foi uma mistura de dois isômeros, e 195 mg de [25- 1a] (doravante denominado composto [25-1 a]) e 134 mg de [25-1 b] (doravante denominado composto [25-1 b]) foram obtidos sob condição de purificação descrita nos EXEMPLOS 23 e 24-(4). Nos EXEMPLOS, o composto de baixa polaridade (composto que eluiu primeiro sob a condição acima) é um composto [25-1 a], e o composto de alta polaridade (composto que eluiu depois sob a condição acima) é um composto [25-1 b], (2) 140 mg de tert-butil((1S)-1-{4- [(R)-[(2R)-1,2-d imetilpirrolid in-2- il](hidroxi)metil]fenil}etil)carbamato [25-2] (doravante denominado composto [25-2]) foi obtido de 193 mg do composto [25- 1a] de acordo com os métodos dos EXEMPLOS 19 e 20-(1). (3) 21,4 mg do composto título [25] (doravante denominado composto [25]) foi obtido como um sólido amarelo pálido de 45,6 mg do composto [12-1] e 49 mg do composto [25-2] de acordo com o método do EXEMPLO 1-(7). Os dados espectrais do composto [25] são apresentados abaixo. 1H-NMR (CDCh) δ: 9,63-9,55 (m, 1H 1/5), 8,97 (s, 1H 1/5), 8,89 (s, 1H 4/5), 8,73 (d, J = 6,8 Hz, 1H 4/5), 8,58 (s, 1H 1/5), 8,53 (s, 1H 4/5), 7,37-7,29 (m, 4H), 6,93-6,84 (m, 1H+1H 1/5), 6,68- 6,65 (m, 1H 4/5), 6,02-6,00 (m, 1H 4/5), 5,84-5,77 (m, 1H 1/5), 5,38-5,26 (m, 1H 1/5), 5,13-5,09 (m, 1H 4/5), 4,67-4,50 (m, 1H), 4,34 (s, 1H), 3,11-3,07 (m, 1H), 2,62-2,45 (m, 2H), 2,17-2,11 (m, 2H), 2,04 (s, 3H), 1,74-1,61 (m, 2H), 1,62 (d, J = 6,8 Hz, 3H), 1,32-1,20 (m, 2H), 1,15-1,12 (m, 1H), 0,90-0,85 (m, 5H) massa: 508 (M+ 1)+. (4) A configuração absoluta do composto título [25] foi determinada pelo método seguinte. À 9,2 mg do composto [25-1 a], 1 mL de uma solução 2 M de hidróxido de potássio aquoso foi adicionada à temperatura ambiente, e a mistura foi agitada na mesma temperatura por 2 horas. A solução de reação foi concentrado sob pressão reduzida. Para o resíduo obtido, foi adicionada água, e a mistura foi extraída com clorofórmio. A camada orgânica foi lavada com salmoura saturada e seca com sulfato de sódio anidro. Os insolúveis foram filtrados e 0 filtrado foi concentrado sob pressão reduzida. O resíduo obtido foi purificado por cromatografia de camada delgada preparativa para obter 6,1 mg de tert-butil((1S)-1-{4-[(1 R,7aR)-7a-metil-3-oxotetrahidro-1 H-pirrolo[1,2-c][1,3]oxazol- 1-il]fenil}etil)carbamato [25-4] (doravante denominado composto [25-4]). Por medida de NOE (Nucleus Overhauser Effect) do composto [25-4], as configurações absolutas do composto [25-4] e o composto título [25] foram determinadas. (3) 15,7 mg do composto título [26] (doravante denominado composto [26]) foi obtido como um sólido amarelo pálido de 45,6 mg do composto [12-1] e 132 mg do composto [25-1 a] de acordo com o método dos EXEMPLOS 25 e 26-(2) e (3). Os dados espectrais do composto [26] são mostrados abaixo. 1H-NMR (CDCI3) δ: 9,63-9,55 (m, 1H 1/5), 8,97-8,90 (m, 1H+1H 4/5), 8,58 (s, 1H 1/5), 8,53 (s, 1H 4/5), 7,43-7,32 (m, 4H), 6,93-6,85 (m, 1H+1H 1/5), 6,66- 6,62 (m, 1H 4/5), 6,04-6,03 (m, 1H 4/5), 5,85-5,78 (m, 1H 1/5), 5,38-5,28 (m, 1H 1/5), 5,16-5,13 (m, 1H 4/5), 4,80-4,67 (m, 1H), 4,43 (s, 1H), 3,15-3,07 (m, 1H), 2,63-2,49 (m, 2H), 2,27 (s, 3H), 2,09-2,05 (m, 1H), 1,85-1,50 (m, 2H), 1,62 (d, J = 6,8 Hz, 3H), 1,36-1,26 (m, 2H), 1,16-1,14 (m, 1H), 0,90-0,80 (m, 5H) massa: 508 (M+l)+. (6) A configuração absoluta do composto título [26] foi determinada pelo método seguinte. 11,6 mg de tert-butil((1S)-1-{4-[(1S,7aR)-7a-metil-3- oxotetrahidro-1H-pirrolo[1,2-c][1,3]oxazol-1-il]fenil}etil)carbamato [25-6] (doravante denominado composto [25-6]) foi obtida de 30 mg do composto [25-1 b] de acordo com os métodos dos EXEMPLOS 65 e 66-(4). Por determinação de NOE (Nucleus Overhauser Effect) do composto [25-6], as configurações absolutas do composto [25-6] e o composto título [26] foram determinadas.
EXEMPLO 27
Síntese de (1S)-2-(tert-butilamino)-1-[4-((1S)-1-{[4-(8-cloroimidazo[1,2- a]piridin-3-il)-5-fluoropirimidin-2-il]amino}etil)fenil]etanol [27] (doravante denominado composto [27]) (1) Uma mistura de 18,9 g de 2,4-dicloro-5-fluoropirimidina (disponível comercialmente de Fluorochem Co. Ltd), 42,9 g de L-cis-etoxi-2-tri-n- butilStaniletileno (sintetizada pelo método descrito em J. Am. Chem. Soc, 1977, 99, 7365), 3,97 g de diclorobis(trifenilfosfino) de paládio (II), e 380 mL de acetonitrila foram agitados a 80°C por 3 horas. Após o resfriamento da mistura de reação, 113 mL de água e 53 g de fluoreto de potássio foram adicionados aos mesmos, e a mistura foi agitada durante a noite em temperatura ambiente. As insolúveis foram filtrados através de Celite, e o filtrado foi extraído com acetato de etila. A camada orgânica foi lavada com salmoura saturada, em sequência, e seca com sulfato de sódio anidro. Os insolúveis foram filtrados e 0 filtrado foi concentrado sob pressão reduzida. O resíduo obtido foi purificado em cromatografia de coluna de sílica gel (eluente: hexano / acetato de etila = 100/0 a 40/60) para obter 14,0 g de 2-cloro-4-[(Z)-2-etoxivinil] -5-fluoropirimidina [27-1] (doravante denominado composto [27-1]) como um sólido amarelo. (2) 25,2 g do composto [27-1], foi dissolvido em uma mistura de solvente de 320 mL de 1,4-dioxano e 32 mL de água. Em seguida, 22,2 g de imida de ácido N-bromosuccínico foi adicionada em uma condição gelada, e a mistura foi agitada a temperatura ambiente por 2 horas. À solução da reação, 16,0 g de 2-amino-3- cloropiridina foi adicionado, e a mistura foi agitada durante a noite em temperatura ambiente. Uma solução aquosa saturada de bicarbonato de sódio foi adicionada, e a mistura foi extraída com uma mistura de solvente de clorofórmio e metanol (proporção de mistura: 9/1). A camada orgânica resultante foi seca sobre sulfato de sódio anidro. Os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. Para o resíduo obtido, foi adicionado hexano, e a mistura foi agitada por 30 minutos. O sólido foi tirado por filtração e seco sob pressão reduzida, para obter 20,0 g de 8-(cloro-3-(2-cloro-5-fluoropirimidin-4- il)imidazol[1,2-a]piridina [27-2] (doravante referido como o composto [27-2]) como um sólido amarelo pálido. (3) 15,8 g do composto [2-5] foi dissolvido em 50 mL de clorofórmio, 45 mL de ácido trifluoroacético foi adicionado em uma condição gelada, e a mistura foi agitada em temperatura ambiente por 1 hora. A solução de reação foi concentrada sob pressão reduzida, e o resíduo obtido foi purificado por cromatografia em coluna de sílica gel (eluente: clorofórmio /metanol = 100/0 a 90/10) para a obtenção 10,7 g (1S)-1-{4-[(1S)aminoetil-1-fenil}-2-(tert- butilamino)etanol [27-3] (doravante denominado composto [27-3]) como um sólido branco. (4) Uma mistura de 3,00 g do composto [27-2], 3,01 g do composto [27-3] 1,12 g de carbonato de sódio e 10 mL de N-metil-2-pirrolidinona foi agitada para a 170°C por 2 horas. À solução de reação, água foi adicionada e a mistura foi extraída com uma mistura de solventes de clorofórmio e acetato de etila (proporção de mistura: 1/10). A camada orgânica foi lavada com salmoura saturada e seca com sulfato de sódio anidro. Os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. O resíduo obtido foi purificado em coluna de sílica gel (eluente: clorofórmio/metanol = 100/0 a 98/2). A solução obtida foi concentrada sob pressão reduzida, e o resíduo foi dissolvido acetato de etila e solidificado para obter 3,79 g do composto título [27] como um sólido amarelo pálido. Os dados espectrais do composto [27] são apresentados abaixo. 1H-NMR (CDCI3) δ: 9,22 (brs, 1H), 8,41 (d, J = 3,9 Hz, 1H), 8,24 (d, J = 3,9 Hz, 1H), 7,42-7,37 (m, 5H), 6,68 (brs, 1H), 5,50 (d, J = 5,9 Hz, 1H), 5,03 (quint, J = 6,8 Hz, 1H), 4,57 (dd, J = 9,0 Hz, 3,7 Hz, 1H), 2,87 (dd, 12,0 Hz, 3,7 Hz, 1H), 2,53 (dd, J = 12,0 Hz, 9,0 Hz, 1H), 1,60 (d, J = 6,8 Hz, 3H), 1,07 (s, 9H) massa: 483 (M+l)+.
EXEMPLO 28
Síntese de (1S)-1-[4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-il)-5- fluoropirimidin-2-il]amino}etil)fenil]-2-[(1-metilciclopentil)amino]etanol [28] (doravante denominado composto [28]) (1) Uma mistura de 500 mg do composto [2-4], 639 mg de monocloridrato de 1- metilciclopentilamina (sintetizada pelo método descrito em J. Med. Chem., 2006, 49, 3068), 613 mg de N,N-diisopropil etilamina, 5 mL of etanol, e 5 mL de água foi aquecida à noite sob refluxo. A solução de reação foi concentrada. Para o resíduo obtido, foi adicionada água, e a mistura foi extraída com acetato de etila. A camada orgânica resultante foi seca sobre sulfato de magnésio anidro. Os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. O resíduo obtido foi purificado em coluna de sílica gel (eluente: clorofórmio / metanol = 100/0 a 95/5) para obter 365 mg de [tert-butil(1S)-1-(4- {(1S)-1-hidroxi-2-[(1-metilciclopetil)amino]etil}fenil)etil]carbamato [28-1] (doravante denominado composto [28-1]) como um sólido branco. (2) 25,9 mg do composto título [28] (doravante denominado composto [28]) foi obtido como um sólido amarelo pálido de 45 mg do composto [27-2] e 150 mg do composto [28-1] de acordo com o método do EXEMPLO 27-(3) e (4). Os dados espectrais do composto [28] são apresentados abaixo. 1H-NMR (CDCI3) δ: 9,21 (br, 1H), 8,41 (d, J = 3,9 Hz, 1H), 8,23 (d, J = 3,4 Hz, 1H), 7,42-7,37 (m, 5H), 7,26 (s, 1H), 6,68 (brs, 1H), 5,54 (brs, 1H), 5,07-4,99 (m, 1H), 4,58 (dd, J = 3,9 Hz, 9,3 Hz, 1H), 2,88 (dd, J = 3,9 Hz, 12,2 Hz, 1H), 2,53 (dd, J = 9,3 Hz, 12,2 Hz, 1H), 1,65-1,52 (m, 6H), 1,60 (d, J = 6,8 Hz, 3H), 1,47- 1,39 (m, 2H), 1,30-1,27 (m, 1H), 1,12 (s, 3H) massa: 509, 511 (M+l)+.
EXEMPLO 29
Síntese de (1S)-2-(tert-butilamino)-1-[4-((1S)-1- {[4-(8- ciclopropilimidazo[1,2-a]piridin-3-il)-5-metilpirimidin-2-il]amino}etil)fenil]etanol [29] (doravante denominado composto [29]) (1) 23,1 mg de 3-(2-cloro-5-metilprimidin-4-il)-8-ciclopropilimidazo[1,2- a]piridina [29-1] (doravante denominado composto [29-1]) foi obtido a partir de 139 mg de 2,4-dicloro-5-metilpirimidina (disponível comercialmente de Aldrich Corporation) e 50,2 mg de 3-ciclopropilpiridina-2-amina (sintetizada pelo método descrito na página 142 da Publicação International WO 2006/025567) de acordo com os métodos de EXEMPLO 27-( 1) e (2). (2) 10,9 mg do composto título [29] (doravante denominado composto [29]) foi obtido como um sólido branco a partir de 45 mg do composto [27-3] e 21,3 mg do composto [29-1] de acordo com o método do EXEMPLO 27-(4). Os dados espectrais do composto [29] são apresentados abaixo. 1H-NMR (CDCh) δ: 8,77 (brs, 1H), 8,21 (s, 1H), 8,06 (s, 1H), 7,38 (s, 4H), 6,77 (d, J = 6,8 Hz, 1H), 6,54 (brs, 1H), 5,38 (d, J = 5,9 Hz, 1H), 5,11 (quint, J = 7,0 Hz, 1H), 4,60 (dd, J = 9,0 Hz, 3,7 Hz, 1H), 2,89 (dd, J = 11,7 Hz, 3,4 Hz, 1H), 2,64-2,56 (m, 2H), 2,35 (s, 3H), 1,56 (d, J = 6,8 Hz, 3H), 1,15-1,10 (m, 2H), 1,08 (s, 9H), 0,92-0,82 (m, 2H) massa: 485 (M+l)+.
EXEMPLO 30
Síntese de (1 S)-2-(tert-butilamino)-1 -[4-((1 S)-1 -{[4-(8-ciclopropilimidazo[1,2- a]piridin-3-il)-5-fluoropirimidin-2-il]amino}etil)fenil]etanol [30] (doravante denominado composto [30]) (1) 13,4 g de 3-(2-cloro-5-fluoropirimidin-4-il)-8-ciclopropilimidazo[1,2- a]piridina [30-1] (doravante denominado composto [30-1]) foi obtido a partir de 11,5 g de 2,4-dicloro-5-fluoropirimidina (disponível comercialmente de Fluorochem Co. Ltd.) e 9,21 g de 3-ciclopropilpiridina-2-amina (sintetizada pelo método descrito na página 142 da Publicação Internacional do WO 2006/025567) de acordo com os métodos de EXEMPLO 27-(1) e (2). (2) 673 mg do composto título [30] (doravante denominado composto [30]) foi obtido como um sólido laranja a partir de 1,17 mg do composto [27-3] e 1,1 mg do composto [30-1] de acordo com o método do EXEMPLO 27-(4). Os dados espectrais do composto [30] são apresentados abaixo. 1H-NMR (CDCh) δ: 9,20 (brs, 1H), 8,41 (d, J = 3,9 Hz, 1H), 8,19 (d, J = 3,9 Hz, 1H), 7,39 (q, 4H), 6,83 (d, J = 6,8 Hz, 1H), 6,69 (m, 1H), 5,44 (d, J = 5,9 Hz, 1H), 5,06 (quint, J = 7,3 Hz, 1H), 4,57 (dd, J = 8,8 Hz, 3,4 Hz, 1H), 2,85 (dd, 12,0 Hz, 3,7 Hz, 1H), 2,62 (dd, J = 11,7 Hz, 8,8 Hz, 1H), 1,59 (d, J = 6,8 Hz, 3H), 1,13 (d, J = 8,8 Hz, 2H), 1,06 (s, 9H), 0,87 (m, 2H) massa: 489 (M+ 1)+.
EXEMPLO 31
Síntese de (1 S)-2-(tert-butilamino)-1 -{4-[( 1 S)-1 -({5-fluoro-4-[8- (trifluorometil)imidazo[1,2-a] pi rid in-3-i I] pi ri m id i n-2-i l}am i no )etil]fen i l}eta no I [31 ] (doravante denominado composto [31]) (1) 143 mg de 3-(2-cloro-5-fluoropirimidin-4-il)-8- (trifluorometil)imidazo[1,2a]piridina [31-1] (doravante denominado composto [31- 1]) foi obtido a partir de 113 mg de 2,4 - dicloro-5-fluoropirimidina (disponível comercialmente a partir de Fluorochem Co. Ltd.) e 104 mg de 3- trifluorometilpiridina-2-amina (sintetizada pelo método descrito na página 142 da Publicação Internacional do WO 2006/025567) de acordo com os métodos de EXEMPLO 27-(1)e(2). (2) 30,6 mg do composto título [31] (doravante denominado composto [31]) foi obtido como um sólido amarelo pálido de 45,6 mg do composto [27-3] e 47 mg do composto [31-1], de acordo com o método de EXEMPLO 27-(4). Os dados espectrais do composto [31] são apresentados abaixo. (3) NMR (CDCI3) õ: 9,55-9,25 (m, 1H), 8,47 (d, J = 3,9 Hz, 1H), 8,26 (d, J = 3,9 Hz, 1H), 7,64 (d, J = 7,3 Hz, 1H), 7,43-7,37 (m, 4H), 6,85-6,73 (m, 1H), 5,55- 5,53 (m, 1H), 5,04-5,01 (m, 1H), 4,59-4,56 (m, 1H), 2,87 (dd, 12,0 Hz, 3,7 Hz, 1H), 2,53 (dd, J = 12,0 Hz, 8,8 Hz, 1H), 1,61 (d, J = 6,8 Hz, 3H), 1,07 (s, 9H) massa: 517 (M+l)+.
EXEMPLO 32
Síntese de (R)-[4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-il)-5- fluoropirimidin-2-il]amino}etil)fenil][(2R)-1,2-dimetilpirrolidin-il]metanol [32] (doravante denominado composto [32]). 15.6 mg do composto título [32] (doravante denominado composto [32]) foi obtida como um sólido amarelo a partir de 34,2 mg do composto [25-4] e 30 mg do composto [27-2] de acordo com os métodos do EXEMPLO 27-(3) e (4). Os dados espectrais do composto [32] são mostrados abaixo. 1H-NMR (CDCI3) δ: 9,27-9,05 (m, 1H), 8,39 (d, J = 3,9 Hz, 1H), 8,24 (d, J = 3,9 Hz, 1H), 7,42-7,30 (m, 5H), 6,78-6,70 (m, 1H), 5,01-4,99 (m, 1H), 4,36 (s, 1H), 3,11-3,07 (m, 1H), 2,52-2,46 (m, 1H), 2,26 (s, 3H), 2,09 (s, 3H), 1,75-1,53 (m, 2H), 1,60 (d, J = 6,8 Hz, 3H), 1,25-1,21 (m, 1H), 0,86 (s, 3H) massa: 495, 497 (M+l)+.
EXEMPLO 33
Síntese de [4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-il)-5-fluoropirimidin- 2-il]amino}eti l)fenil][(2S)-1,2-dimetilpirrolidin-2-il]metanol [33] (doravante denominado composto [33]) (1) 1,69 g of tert-butil((1S)-1-(4-[[(2S)-1,2- d imeti I pi rro I id i n-2-i l]( h id roxi )metil]fen i l}eti I )carbamato [33-1 ] (doravante denominado composto [33-1]) foi obtido a partir de 2,55 g do composto [23-4a] de acordo com os métodos dos EXEMPLOS EXAMPLES 19 e 20-(1). (2) 10,5 mg do composto título [33] (doravante denominado composto [33]) foi obtido a partir de 30 mg do composto [33-1] e 18,4 mg do composto [27-2] de acordo com os métodos do EXEMPLO 27-(3) e (4). Os dados espectrais do composto [33] são mostrados abaixo. 1H-NMR (CDCh) δ: 9,48-9,35 (m, 1H), 8,42 (d, J = 3,4 Hz, 1H), 8,24 (d, J = 3,9 Hz, 1H), 7,42-7,28 (m, 5H), 6,78-6,70 (m, 1H), 5,05-5,03 (m, 1H), 4,37 (s, 1H), 3,12-3,07 (m, 1H), 2,51-2,47 (m, 1H), 2,08 (s, 3H), 1,85 (s, 3H), 1,85-1,71 (m, 3H), 1,59 (d, J = 6,8 Hz, 3H), 0,85 (s, 3H) massa: 495, 497 (M+l)+.
EXEMPLOS 34 e 35
Síntese de 1-[4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-il)-5- fluoropirimidin-2-il]amino}etil)fenil]-2-(dimetilamino)-2-metilpropan-1-ol [34] (doravante denominado composto [34]) (doravante referido como composto [34] e [35]) (doravante referido como composto [34]) (aqui, o composto [34] e o composto [35] são diasteroisômeros. Por favor, vide a Tabela 14) (1) 51 g de dimetilSuIfóxido foi dissolvido em 70 mL de diclorometano, e a temperatura da solução foi mantida a -78°C. Então, 160 mL de uma solução de diclorometano em que 59,1 g de dicloreto de oxila é dissolvido foi adicionado. A mistura foi agitada na mesma temperatura por 30 minutos e, posteriormente, 120 mL de uma solução de diclorometano em que 18,2 g de 2-(dimetilamino)-2- metilpropan-1-ol é dissolvido foi adicionado à mesma. A mistura foi agitada na mesma temperatura por 20 minutos, 107 g de trietilamina foi adicionado ao mesmo, e a mistura foi agitada na mesma temperatura por 40 minutos. A mistura de reação foi aquecida à temperatura ambiente e, em seguida diluídos com diclorometano. A camada obtida orgânica foi lavada com uma solução aquosa saturada de bicarbonato de sódio e salmoura saturada, em seqüência, e seca com sulfato de magnésio anidro. Posteriormente, os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. O resíduo obtido foi purificado por destilação sob pressão reduzida para obter 11,1 g de 2-(dimetilamino)-2- metilpropanal [34-1] (doravante referido como composto [34-1]). (2) 12,8 g de tert-butil ((1S)-1-{4-[2-(dimetilamino)-1-hidroxi-2- metilpropil]fenil}etil)carbamato [34-2] (doravante referido como composto [34-2]) foi obtido a partir de 24,1 g do composto [1-1] e 11,1 g do composto [34-1] de acordo com os métodos dos EXEMPLOS 23 e 24-(4). (3) 2,31 g de uma mistura dos compostos de título [34] e [35] foi obtida a partir de 6,24 g do composto [34-2] e 2,34 g do composto [27-2] de acordo com os métodos dos EXEMPLOS 27-(3) e (4). (4) 70,2 mg de uma mistura de compostos [34] e [35] foi separada em Chiralcel AD-H. Condições para separação óptica são os seguintes. Coluna: Chiralcel AD-H (Chiralcel AD-H, fabricado pela Daicel Chemical Industries, LTD.), diâmetro de 20 mm e comprimento de 250 mm Eluente: hexano/2- propanol/dietilamina = 65/35/0,1; Taxa de fluxo: ,100 mL/min. A solução obtida foi concentrada sob pressão reduzida, e o resíduo foi dissolvido em uma pequena quantidade de clorofórmio para ser solidificado com éter dietílico, obtendo-se 892 mg do composto título [13] (TR = 15,5 minutos) como um sólido amarelo pálido e 298 mg do composto título [14] (TR = 35 minutos) como um sólido amarelo pálido. Os dados espectrais do composto [34] são apresentados abaixo. 1H-NMR (CD3OD) δ: 8,37 (s, 1H), 8,29 (s, 1H), 7,75 (d, J = 7,0 Hz, 1H), 7,51-7,42 (m, 5H), 7,21-7,05 (m, 1H), 5,12-5,01 (m, 1H), 4,95 (s, 1H), 2,94 (s, 3H), 2,80 (s, 3H), 1,58 (d, J = 7,0 Hz, 3H), 1,16 (s, 3H), 1,13 (s, 3H) massa: 483, 485 (M+l)+ Os dados espectrais do composto [35] são apresentados abaixo. 1H-NMR ((CD3OD) δ: 8,38 (s, 1H), 8,29 (s, 1H), 7,75 (d, J = 7,0 Hz, 1H), 7,51-7,43 (m, 5H), 7,19-7,04 (m, 1H), 5,10-5,01 (m, 1H), 4,96 (s, 1H), 2,94 (s, 3H), 2,80 (s, 3H), 1,58 (d, J = 7,0 Hz, 3H), 1,17 (s, 3H), 1,12 (s, 3H) massa: 483, 485 (M+l)+
EXEMPLOS 36 e 37
Síntese de [4-((1S)-1-{[4-(8-cloroimidazo[1,2-a]piridin-3-il)-5-fluoropirimidin- 2- il]amino}etil)fenil](1 -isopropilazetidin-3-il) metanol [36] (doravante denominado composto [36]) e [37] (doravante denominado composto [37]) (aqui, o composto [36] e o composto [37] são diastereoisômeros. Por favor, vide Tabela 15) (5) 9,94 g de [tert-butil(1 S)-1 -(4-{[1 -(difenilmetil)] azetidin-3- il]carbonil}fenil)etil]carbamato [36-1] (doravante referido como o composto [36-1]) foi obtido a partir do ácido 1-benzidrilazetidina-3-carboxílico de acordo com os métodos de EXEMPLO 1 - (1) e (2). (6) 9,94 g do composto [36-1], foi dissolvido em 100 mL de tetrahidrofurano e 20 mL de metanol, 799 mg de borohidreto de sódio foi adicionado ao mesmo à temperatura ambiente, e a mistura foi agitada na mesma temperatura por 2,5 horas Para a mistura de reação, a água foi adicionada, e a mistura foi extraída com clorofórmio. A camada orgânica resultante foi seca sobre sulfato de magnésio anidro. Posteriormente, os insolúveis foram filtrados e o filtrado foi concentrado sob pressão reduzida. O resíduo obtido foi purificado em coluna de sílica gel (eluente: hexano / acetato de etila = 100/0 a 30/70) a obtenção de 632 mg de tert-butil((1 S)-1 -{4-[[1 -(difenilmetil)azetidin-3- il](hidroxi)metil]fenil}etil)carbamato [36-2] (doravante denominado composto [36- 2]). (7) 6,26 g of tert-butil((1 S)-1-{4-[hidroxi(1-isopropilazetidina3-il) metil]fenil}etil)carbamato [36-3] (doravante denominado como composto [36-3]) foi obtido a partir de 9,79 g do composto [36-2] de acordo com os métodos do EXEMPLO 11-(3) e (4). (8) 135 mg de uma mistura de compostos de título [36] e [37] foram obtidas a partir de 720 mg do composto [36-3] e 200 mg do composto [27-2] de acordo com os métodos do EXEMPLO 23-(3) e (4). (9) 130 mg da mistura dos compostos [36] e [37] foi separado em Chiralcel AD-H. As condições de separação óptica são os seguintes. Coluna: Chiralcel AD-H (Chiralcel AD-H, fabricado pela DAICEL CHEMICAL INDUSTRIES, LTD.), diâmetro de 20 mm e comprimento de 250 mm Eluente: Hexano/etanol/dietilamina = 70/30/1; Taxa de fluxo: 12 mL/min. A solução obtida foi concentrada sob pressão reduzida para obter 36 mg do composto título [36] (TR = 11,4 minutos) e 63 mg do composto título [37] (TR = 21,8 minutos).
Os dados espectrais do composto [36] são apresentados abaixo. 1H-NMR (CDCI3) δ: 8,90 (br, 1H), 8,22-8,16 (m, 2H), 7,43-7,38 (m, 4H), 7,32 (d, J = 7,3 Hz, 1H), 7,27 (s, 1H), 6,60 (brs, 1H), 5,58 (brs, 1H), 4,95 (brs, 1H), 4,89 (d, J = 6,8 Hz, 1H), 3,22 (d, J = 5,9 Hz, 2H), 3,15-3,12 (m, 1H), 3,05-3,04 (m, 1H), 2,68-2,65 (m, 1H), 2,30-2,25 (m, 1H) massa: 495, 497 (M+1)+.
Os dados espectrais do composto [37] são apresentados abaixo. 1H-NMR (CDCI3) δ: 8,99 (br, 1H), 8,29 (brs, 1H), 8,18-8,17 (m, 1H), 7,41- 7,37 (m, 4H), 7,32 (d, J = 6,8 Hz, 1H), 7,27 (s, 1H), 6,57 (br, 1H), 5,61 (br, 1H), 4,99-4,95 (m, 1H), 4,87 (d, J = 6,3 Hz, 1H), 3,21 (d, J = 6,3 Hz, 2H), 3,14-3,10 (m, 1H), 3,05-3,02 (m, 1H), 2,66-2,62 (m, 1H), 2,27 (quint, J = 6,3 Hz, 1H), 1,58 (d, J = 6,8 Hz, 3H), 0,89 (d, J = 5,9 Hz, 6H)massa: 495, 497 (M+l)+.
EXEMPLO 38
Síntese de (1S)-2-(tert-butilamino)-1-{4-[(1S)-1-({4-[8- (difluorometil)imidazo[1,2-a]piridin-3-il]-5-fluoropirimidin-2-il}amino)etil]fenil}etanol [38] (doravante referido como o composto [38]) (10) 180 mg de 3-(2-cloro-5-fluoropirimidin-4-il)imidazo[1,2-a]piridin-8- carboaldeído [38-1] (doravante referenciado como o composto [38-1]) foi obtido a partir de 568 mg de 2,4-dicloro-5-fluoropirimidina (comercialmente disponível de Fluorochem Co. Ltd) e 415 mg de 2-aminonicotinaldeído (disponível comercialmente de Aldrich Corporation), de acordo com os métodos de EXEMPLO 27-(1)e(2). (11) 97 mg de 3-(2-cloro-5-fluoropirimidin-4-il)-8-(difluorometil)imidazo[1,2a] [38-2] (doravante referida como o composto [38-2]) foi obtido a partir de 180 mg do composto [38-1], de acordo com o método de EXEMPLO 4-(2). (12) 12,9 mg do composto título [38] (doravante denominado composto [38]) foi obtido a partir de 33,5 mg do composto [27-3] e 30 mg do composto [38-2] de acordo com o método do EXEMPLO 27-(4).
Os dados espectrais do composto [38] são apresentados abaixo. 1H-NMR (CDCh) δ: 9,36 (brs, 1H), 8,43-8,40 (m, 1H 4/5), 8,25-8,22 (m, 1H 4/5), 7,97- 7,93 (m, 1H 1/5+1H 1/5), 7,63-7,58 (m, 1H 4/5), 7,50-7,37 (m, 4H+1H 1/5+1H 1/5), 7,32 (t, J = 55,1 Hz, 1H), 6,87-6,79 (m, 1H 4/5), 5,57-5,51 (m, 1H 4/5), 5,06-5,00 (m, 1H 4/5), 4,67-4,57 (m, 1H 4/5+1H 1/5+1H 1/5), 4,32-4,26 (m, 1H 1/5), 2,98-2,91 (m, 1H 1/5), 2,90-2,84 (m, 1H 4/5), 2,59-2,51 (m, 1H), 1,62-1,58 (m, 3H), 1,12-1,10 (m, 9H 1/5), 1,09-1,06 (m, 9H 4/5) massa: 499 (M+1)+.
REFERÊNCIA 1
Síntese de tert-butil(1S)-1-(4-formilfenil)etil]carbamato [39] 5 g do composto [1-1] foi dissolvida em 100 mL de tetrahidrofurano e resfriada a - 78°C. Em seguida, 31,2 mL de n-butil lítio (solução de hexano 1,6 M) foi adicionado ao mesmo, e a mistura foi agitada por 1 hora à mesma temperatura. 774 pL de N,N- dimetilformamida foi adicionada à suspensão branca produzida, que foi agitada por 5 horas enquanto gradualmente aquecida de volta à temperatura ambiente. À mistura de reação, uma solução saturada de cloreto de amónio aquoso foi adicionada, e a mistura foi extraída com acetato de etila. A camada orgânica foi lavada com salmoura saturada e seca com sulfato de magnésio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado em coluna de sílica gel (eluente: hexano / acetato de etila = 100/0 - 50/50), a obtenção de 514 mg de [tert-butil[(1S)-1-(4- formilfenil)etil]carbamato [39] como um amorfo amarelo-pálido.
REFERÊNCIA 2
Síntese de benzi 14-[(4-{(1 S)-1-[(tert- butoxicarbonil)amino]etil}fenil)(hidroxi)metil]piperidina-1-carboxilato [40] 267 mg do composto [42] (Vide REFERÊNCIA 4), foi dissolvido em um solvente misturado de 10 mL de tetrahidrofurano e 1 mL de metanol, em seguida, 22 mg de borohidreto de sódio foi adicionado, e a mistura foi agitada durante a noite em temperatura ambiente. A água foi adicionada à solução de reação, que foi extraída com acetato de etila. A camada orgânica obtida foi lavada com salmoura saturada e seca com sulfato de magnésio anidro. Os insolúveis foram filtradas, o filtrado foi concentrado sob pressão reduzida, e o resíduo resultante foi purificado em coluna de sílica gel (eluente: hexano / acetato de etila = 100/0 - 30/70), para obter 256 mg de benzil4-[(4-{(1S)-1-[(tert- butoxicarbonil)amino]etil}fenil)(hidroxi)metil]piperidina-1-carboxilato [40].
REFERÊNCIA 3
Síntese de benzyl (2R)-2-[(4-{(1S)-1-[(tert- butoxicarbonil)amino]etil}fenil)(hidroxi)metil]pirrolidina-1 -carboxilato [41 ] 337 mg de benzil(2R)-2-[(4-{(1S)-1-[(tert- butoxicarbonil)amino]etil}fenil)(hidroxi)metil]pirrolidina-1-carboxilato [41] foi obtido a partir de 1,06 g de benzil(2R)-2-formilpirrolidina-1-carboxilato (sintetizado de acordo com o método divulgado em Bioorg. Med. Chem., 2003.11. 3153-3164) e 1,14 g do composto [1-1] de acordo com o método do Exemplo 2-(1).
Referência 4 Síntese de benzil4-(4-{(1S)-1-[(tert-butoxicarbonil)amino]etil} benzeu)piperidina-1 -carboxilato [42] 1,26 g of benzil4-(4-{(1S)-1-[(tert- butoxicarbonil)amino]etil}benzeu)piperidina-1- carboxilato [42] foi obtido a partir de 2,39 g de benzil4-(N-metoxi-N-metilcarbamoil)piperidina-1- carboxilato e 2,13 g do composto [1-1] de acordo com o método do Exemplo 2-(1).
Aplicabilidade Industrial
O composto da presente invenção tem um excelente efeito inibitório contra PLK1 e proliferação celular e, portanto, espera-se para servir como um agente antitumoral útil no campo da medicina.