WO2020235319A1 - 軟骨又は骨の前駆細胞の拡大培養方法 - Google Patents
軟骨又は骨の前駆細胞の拡大培養方法 Download PDFInfo
- Publication number
- WO2020235319A1 WO2020235319A1 PCT/JP2020/018130 JP2020018130W WO2020235319A1 WO 2020235319 A1 WO2020235319 A1 WO 2020235319A1 JP 2020018130 W JP2020018130 W JP 2020018130W WO 2020235319 A1 WO2020235319 A1 WO 2020235319A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- cell
- cartilage
- bone
- present
- Prior art date
Links
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0655—Chondrocytes; Cartilage
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/32—Bones; Osteocytes; Osteoblasts; Tendons; Tenocytes; Teeth; Odontoblasts; Cartilage; Chondrocytes; Synovial membrane
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0654—Osteocytes, Osteoblasts, Odontocytes; Bones, Teeth
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/066—Tenocytes; Tendons, Ligaments
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/115—Basic fibroblast growth factor (bFGF, FGF-2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/15—Transforming growth factor beta (TGF-β)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/155—Bone morphogenic proteins [BMP]; Osteogenins; Osteogenic factor; Bone inducing factor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/16—Activin; Inhibin; Mullerian inhibiting substance
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/40—Regulators of development
- C12N2501/415—Wnt; Frizzeled
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/70—Enzymes
- C12N2501/72—Transferases (EC 2.)
- C12N2501/727—Kinases (EC 2.7.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/45—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from artificially induced pluripotent stem cells
Definitions
- the present invention relates to a method for expanding and culturing cartilage or bone progenitor cells, and a kit for the expanding culture.
- Human cartilage is usually not regenerated if it is congenitally deficient or acquired or damaged or deficient. It is estimated that there are about 25 million people with osteoarthritis of the knee, mainly due to aging of articular cartilage, in Japan, and it is expected that the number will increase further in the future as the population ages.
- As a conventional treatment for such human cartilage disease there is a method of collecting cartilage tissue from another site of its own and transplanting it to a defective site, but the collection site and the amount are limited.
- chondrocytes have low proliferative potential and poor regenerative ability, it is difficult to proliferate chondrocytes derived from living organisms.
- chondrocytes for example, pluripotent stem cells
- a method of inducing chondrocyte differentiation from pluripotent stem cells has been reported (for example, Non-Patent Documents 1 and 2, Patent Document 1 and the like).
- these methods require a period of at least 12 days to differentiate pluripotent stem cells into chondrocytes, and a sufficient amount of chondrocytes to be used for the treatment of cartilage disease or bone disease. I can't get it.
- an object of the present invention is to provide a method capable of producing a chondrocyte or the like in a sufficient amount for use in the treatment of a disease while the number of days for differentiation into the chondrocyte or the like is shorter than that of the conventional method.
- the present inventors improve the differentiation efficiency of chondrocytes from progenitor cells such as pluripotent stem cells by improving the type of cytokine or transcription factor and the timing of addition to the medium. If the progenitor cells of chondrocytes with high proliferative ability can be identified by changing the conventional idea of, the induction time can be shortened by proliferating the progenitor cells and inducing differentiation into chondrocytes. I got the idea that a large amount of chondrocytes could be obtained.
- chondrocyte or bone progenitor cells with near-infinite proliferative capacity (the present inventors referred to these cells as "iCOP". (Named iPS-derived chondro / osteogenic progenitor) ”) was assumed to exist, and an attempt was made to actually identify iCOP and establish an expanded culture method for the cells.
- the present inventors first assumed that among the cells contained in the cell population containing the germ layer cells, the cells exhibiting proliferative ability and capable of differentiating into chondrocytes in a short period of time were scleromes (hard nodule cells). ..
- Non-Patent Document 1 when the method described in Non-Patent Document 1 was improved to induce differentiation of induced pluripotent stem cells into clerotome via mesoderm cells, the cells highly expressed the markers described in Non-Patent Document 1 such as PDGFRA or SOX9. I found that I was doing it. After that, as a result of diligent studies on the culture conditions under which sclerome can grow, by culturing in a medium containing a TGF- ⁇ signal inhibitor and FGF, sclerome can grow at least 5 passages while maintaining the expression of cell markers. It was found to be possible (ie, capable of self-renewal). The above findings were surprising because sclerotome was not known to have self-renewal ability.
- sclerotome that was expanded and cultured for such a long period of time still had the ability to differentiate into chondrocytes. Therefore, it was concluded that sclerome is the iCOP. As a result of further research based on the above findings, the present inventors have completed the present invention.
- a method for expanding cartilage or bone progenitor cells which comprises a step of culturing cartilage or bone progenitor cells in a medium containing a TGF- ⁇ signal inhibitor and FGF.
- the method according to [1], wherein the cartilage or bone progenitor cells are cells expressing PDGFRA, SOX9, or PAX1.
- the method according to [1] or [2], wherein L-ascorbic acid or a derivative thereof is further contained in the medium.
- the method according to any one of [1] to [3], wherein the TGF- ⁇ signal inhibitor is SB431542.
- [5] The method according to any one of [1] to [4], wherein the FGF is bFGF.
- the concentration of the TGF- ⁇ signal inhibitor in the medium is 1 ⁇ M to 50 ⁇ M.
- the concentration of the FGF in the medium is 1 ng / mL to 500 ng / mL.
- the cartilage or bone progenitor cells are cells derived from pluripotent stem cells.
- cartilage or bone progenitor cells are cells obtained by suspension-culturing pluripotent stem cells.
- expansion culture is performed by suspension culture.
- a kit for expanding culture of cartilage or bone progenitor cells which comprises a basal medium, a TGF- ⁇ signal inhibitor, and FGF.
- the kit according to [11] further comprising L-ascorbic acid or a derivative thereof.
- the kit according to [11] or [12], wherein the TGF- ⁇ signal inhibitor is SB431542.
- a method for producing chondrocytes or bone cells which comprises a step of inducing differentiation of cartilage or bone progenitor cells obtained by the method according to any one of [1] to [10] into chondrocytes or bone cells. ..
- the differentiation-inducing step comprises culturing cartilage or bone progenitor cells in a medium containing a BMP signal activator and / or a TGF- ⁇ signal activator.
- the TGF- ⁇ signal activator is TGF- ⁇ 3.
- a method for producing ligament node cells which comprises a step of inducing differentiation of cartilage or bone progenitor cells obtained by the method according to any one of [1] to [10] into ligament node cells.
- a method for producing a tendon cell or a ligament cell which comprises a step of inducing differentiation of the ligament node cell obtained by the method according to [18] into a tendon cell or a ligament cell.
- a method for identifying cartilage or bone progenitor cells which comprises the step of detecting or measuring the expression of at least one gene selected from the group consisting of the following genes.
- a reagent for isolating cartilage or bone progenitor cells which comprises an antibody against each of the proteins encoded by at least one gene selected from the group consisting of the following genes.
- cartilage or bone progenitor cells such as iCOP. Since the differentiation induction period from chondrocyte or bone progenitor cells to chondrocytes or osteoocytes is less than half of the differentiation induction period from conventional iPS cells to chondrocytes or osteoocytes, iCOP is expanded and cultured in advance and stocked. If this is done, it is possible to significantly shorten the period for inducing differentiation into chondrocytes or osteoocytes.
- chondrocytes or bone cells can be produced in an extremely large amount by expanding and culturing chondrocytes or bone progenitor cells during the process of differentiation into chondrocytes. According to the present invention, a sufficient amount of chondrocytes can be rapidly produced for use in the treatment of chondrocytes or bone diseases.
- FIG. 1 shows the results of inducing iCOP differentiation from iPS cells and measuring the positive rate of SOX9 expressed by iCOP with a flow cytometer. It was shown that iPS cells are differentiated into iCOP with high efficiency of about 90%.
- FIG. 2 shows the growth curve when iCOP was subcultured every 7 days for 5 passages. The growth curve showed that the daily population doubling (PD) was 0.46.
- FIG. 3 shows the results of verifying the effect of cryopreservation on the cell proliferation ability of iCOP. It was shown that cryopreservation did not reduce daily PD.
- FIG. 1 shows the results of inducing iCOP differentiation from iPS cells and measuring the positive rate of SOX9 expressed by iCOP with a flow cytometer. It was shown that iPS cells are differentiated into iCOP with high efficiency of about 90%.
- FIG. 2 shows the growth curve when iCOP was subcultured every 7 days for 5 passages. The growth curve showed that the daily population doubling (PD) was
- FIG. 4 shows the results of verifying the relative expression level (mRNA amount) of the iCOP marker gene in the cryopreserved iCOP or the non-cryopreserved iCOP. It was shown that cryopreservation did not reduce the relative expression of the iCOP marker gene.
- FIG. 5 shows the results of inducing differentiation of iCOP into osteoocytes and evaluating the relative expression level (mRNA amount) of the bone cell marker gene. It was shown that iCOP retains the ability to differentiate into bone cells.
- FIG. 6 shows the results of inducing differentiation from iCOP into ligament node cells (syndetome) and evaluating the relative expression level (mRNA amount) of the ligament node marker gene. It was shown that iCOP retains the ability to differentiate into ligament cells.
- FIG. 7 shows a growth curve when iCOP was subcultured every 5 days by suspension culture. The growth curve showed that the daily PD was 0.38.
- the present invention provides a method for expanding and culturing cartilage or bone progenitor cells (hereinafter, may be referred to as “the culture method of the present invention”).
- the culturing method of the present invention includes culturing cartilage or bone progenitor cells in a medium containing a TGF- ⁇ signal inhibitor and FGF.
- expansion culture means a culture for the purpose of proliferating cartilage or bone progenitor cells contained in a cell population and increasing the number of cells.
- the increase in cell number may be achieved by increasing the number of cells by proliferation exceeding the decrease by death, and it is not necessary for all cells in the cell population to proliferate.
- cartilage or bone progenitor cell means a cell capable of differentiating into chondrocytes and / or bone cells.
- cartilage or bone progenitor cells express PDGFRA, SOX9, or PAX1, and more preferably PDGFRA, PAX1, and SOX9.
- a "cartilage or bone progenitor cell” is a somitor cell (sclerotome) that expresses at least one of PDGFRA, SOX9, and PAX1.
- the cells When differentiating into chondrocytes and / or osteoocytes by at least one of the methods for inducing differentiation into chondrocytes and / or osteoocytes described below, the cells are said to have the ability to differentiate into chondrocytes and / or osteoocytes. Can be evaluated.
- the expression of PDGFRA, PAX1 and SOX9 in cells can be confirmed by at least one of real-time PCR and flow cytometry.
- cartilage or bone progenitor cells may have the ability to induce differentiation into cells other than cartilage or bone, such as ligament node cells (syndetome).
- "expression” is used in the sense of including at least "production of functional protein” unless otherwise specified, but is preferably used in the sense of further including "production of mRNA”. ..
- “cartilage or bone progenitor cells” are at least one of the following marker genes, preferably two or more (eg, three, four, five, six, seven, eight, nine). , 10 or more genes) may be expressed, but at least one expressing the PDGFRA gene is preferable.
- the TGF- ⁇ signal inhibitor used in the present invention is not particularly limited as long as it is a substance that inhibits the signal transduction from the binding of TGF- ⁇ to the receptor to SMAD, but for example, the TGF- ⁇ receptor.
- examples include substances that inhibit binding to a certain ALK family and substances that inhibit the phosphorylation of SMAD by the ALK family.
- the TGF- ⁇ signal inhibitor is, for example, Lefty-1 (for example, NCBI Accession No., mouse: NM_010094, human: NM_020997), SB431542, SB202190 (above, RKLindemann et al., Mol.
- SB431542 is preferable.
- the concentration of the TGF- ⁇ signal inhibitor in the medium is not particularly limited, but is preferably 1 ⁇ M to 50 ⁇ M, for example, when SB431542 is used, for example, 1 ⁇ M, 2 ⁇ M, 3 ⁇ M, 4 ⁇ M, 5 ⁇ M, 6 ⁇ M, 7 ⁇ M, 8 ⁇ M, 9 ⁇ M, 10 ⁇ M, 11 ⁇ M, 12 ⁇ M, 13 ⁇ M, 14 ⁇ M, 15 ⁇ M, 16 ⁇ M, 17 ⁇ M, 18 ⁇ M, 19 ⁇ M, 20 ⁇ M, 25 ⁇ M, 30 ⁇ M, 35 ⁇ M, 40 ⁇ M, 45 ⁇ M, 50 ⁇ M, but not limited to these. More preferably, it is 2 ⁇ M to 20 ⁇ M, and particularly preferably 5 to 10 ⁇ M. Even when a TGF- ⁇ signal inhibitor other than SB431542 is used, the concentration can be appropriately set based on common general knowledge.
- the FGF used in the present invention is not particularly limited as long as it promotes cell proliferation, but is, for example, FGF-1, bFGF (FGF-2), FGF-3, FGF-4, FGF-5, FGF-6, FGF. -7, FGF-8 (eg FGF-8b), FGF-9, FGF-10, FGF-11, FGF-12, FGF-13, FGF-14, FGF-15, FGF-16, FGF-17, Examples thereof include FGF-18, FGF-19, FGF-20, FGF-21, FGF-22, and FGF-23. Of these, bFGF is preferable.
- the above FGF may be derived from any animal (eg, rodents such as mice, rats, hamsters, guinea pigs, primates such as humans, monkeys, orangutans, orangutans), but depending on the type of cells to be cultured. It can be appropriately selected accordingly.
- human-derived FGF include, for example, human bFGF (eg Endocrine Rev., 8, 95, 1987), bovine bFGF (eg Proc. Natl. Acad. Sci. USA, 81, 6963, 1984), mouse bFGF (eg, Proc. Natl. Acad. Sci. USA, 81, 6963, 1984). Examples include, but are not limited to, Dev. Biol., 138, 454-463, 1990), rat bFGF (eg, Biochem. Biophys. Res. Communi., 157, 256-263, 1988).
- the FGF used in the present invention is not only a natural type but also a modified type thereof.
- a specific region of FGF1 (partial sequence at positions 41 to 83 or partial sequence at positions 62 to 83 of the amino acid sequence of human FGF1 protein) is added to the region of bFGF corresponding to the region. Since the substituted chimeric protein is also known to have the same cell proliferation activity as bFGF (Japanese Patent Laid-Open No. 2012-143234, JP-A-2014-100141), the variant of FGF containing a partial region of bFGF is also described in this article. It can be suitably used for the invention.
- the FGF used in the present invention can be produced by a method known per se, or can be obtained by purchasing a commercially available product.
- the medium containing FGF can be prepared by adding soluble FGF to the medium, or a carrier such as beads on which the FGF is immobilized on the surface (eg, StemBeads FGF2, etc.) may be added to the medium. It can also be produced by.
- the concentration of FGF in the medium is not particularly limited, but for example, when bFGF is used, it is preferably 1 ng / mL to 500 ng / mL, for example, 1 ng / mL, 2 ng / mL, 3 ng / mL, 4 ng.
- ng / mL 5 ng / mL, 6 ng / mL, 7 ng / mL, 8 ng / mL, 9 ng / mL, 10 ng / mL, 11 ng / mL, 12 ng / mL, 13 ng / mL, 14 ng / mL, 15 ng / mL, 16 ng / mL, 17 ng / mL, 18 ng / mL, 19 ng / mL, 20 ng / mL, 25 ng / mL, 30 ng / mL, 35 ng / mL, 40 ng / mL, 45 ng / mL, 50 ng / mL, 100 ng / mL, but not limited to these.
- the concentration can be appropriately set based on the common general technical knowledge.
- basal medium used in the present invention examples include DMEM, EMEM, IMDM (Iscove's Modified Dulbecco's Medium), GMEM (Glasgow's MEM), RPMI-1640, ⁇ -MEM, Ham's Medium F-12, Ham's Medium F-10, Ham's.
- the medium used in the present invention may contain additives known per se.
- additives include, for example, growth factors (eg PDGF, insulin, etc.), iron sources (eg, transferrin, etc.), hedgehog signal activators, polyamines (eg, putresin, etc.), minerals (eg, selenic acid).
- each additive is contained within a concentration range known per se.
- vitamin Cs mean L-ascorbic acid and its derivatives
- L-ascorbic acid derivatives mean those that become vitamin C by an enzymatic reaction in the living body.
- vitamin C phosphate glucoside ascorbic acid, ascorbyl ethyl, vitamin C ester, ascorbyl tetrahexyl decanoate, ascorbyl stearate, -2-phosphate-6 palmitic acid and the like can be used. Illustrated.
- Vitamin C phosphate eg, Ascorbic acid 2-phosphate
- Vitamin C phosphate is preferred and is, for example, phosphorus such as sodium phosphate-L-ascorbic acid, Mg phosphate-L-ascorbic acid. Acid-L-ascorbate can be mentioned.
- the concentration of vitamin Cs in the medium is not particularly limited, but the concentration in the medium when ascorbic acid-2-phosphate is used is typically 20 ⁇ M to 2 mM (eg, 50 ⁇ M, 100 ⁇ M, 150 ⁇ M, 200 ⁇ M, 250 ⁇ M, 300 ⁇ M, 500 ⁇ M, 1 mM, etc.). Even when vitamin Cs other than ascorbic acid-2-phosphoric acid are used, the concentration can be appropriately set based on common general technical knowledge.
- the PDGF used in the present invention may be PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC, or PDGF-DD that binds to the PDGF receptor, but PDGF-BB is preferable.
- the concentration of PDGF in the medium is not particularly limited, but the concentration in the medium when PDGF-BB is used is typically 10 ng / mL to 1 ⁇ g / mL (eg, 50 ng / mL, 100 ng / mL, 150 ng / mL, 300 ng / mL, etc.). Even when PDGF other than PDGF-BB is used, the concentration can be appropriately set based on common general technical knowledge.
- the hedgehog signal activator used in the present invention is not particularly limited as long as it is a substance that activates a signal mediated by a 12-transmembrane type Patched (Ptc) and a 7-transmembrane type Smoothened (Smo).
- hedgehog proteins eg Hh, Shh, Ihh, Dhh, etc.
- Smoothened agonists eg SAG (Hh-Ag 1.3), SAG21k (3-chloro-4,7-difluoro-N- (4-) methoxy-3- (pyridin-4-yl) benzyl) -N-((1r, 4r) -4- (methylamino) cyclohexyl) benzo [b] thiophene-2-carboxamide), Hh-Ag 1.1, Hh-Ag 1.5 , Purmorphamine, etc., but a Smoothened agonist is preferable, and SAG is more preferable.
- the concentration of the hedgehog signal activator in the medium is not particularly limited, but the concentration in the medium when SAG is used is typical. 30 nM to 3 ⁇ M (eg 100 nM, 200 nM, 300 nM, 400 nM, 500 nM, 1 ⁇ M, etc.). Even when using a hedgehog signal activator other than SAG, the technique Based on common wisdom, the concentration can be set as appropriate.
- the medium used in the present invention may contain serum.
- the serum is not particularly limited as long as it is animal-derived serum, but is preferably mammalian-derived serum (eg, fetal bovine serum, human serum, etc.).
- the serum concentration may be within the concentration range known per se.
- chondrocytes induced to differentiate from the cells, etc. are used for medical purposes, heterologous components may become infection sources or heterologous antigens of blood-borne pathogens. Therefore, it is preferable that it does not contain serum.
- alternative additives for serum for example, Knockout Serum Replacement (KSR) (Invitrogen), Chemically-defined Lipid concentrated (Gibco), B-27 Supplement (Gibco), etc.
- KSR Knockout Serum Replacement
- incubators examples include flasks, tissue culture flasks, dishes, petri dishes, tissue culture dishes, multi-dish, microplates, microwell plates, multi-plates, multi-well plates, microslides, and chamber slides. , Petri dishes, tubes, trays, culture bags, roller bottles, etc.
- the incubator may be a cell adhesion incubator used for adhesive culture or a cell non-adhesive incubator used for suspension culture, and can be appropriately selected according to the purpose.
- Cell-adhesive incubators are coated with any cell-supporting substrate such as extracellular matrix (ECM, also called extracellular matrix) for the purpose of improving the adhesion of the surface of the incubator to cells.
- ECM extracellular matrix
- the cell-supporting substrate can be any substance intended for cell adhesion.
- the feeder cells are not used in the culturing method of the present invention, it is preferable to cultivate using an extracellular matrix or an active fragment thereof, or an artificial substance that mimics the functions thereof.
- the extracellular matrix is not particularly limited as long as it is usually used for culturing cells for the purpose of improving the adhesion between the surface of the incubator and the cells, and for example, laminin (laminin 511, laminin 332, etc.), fibronectin, etc.
- laminin laminin 511, laminin 332, etc.
- fibronectin Known substances such as vitronectin, collagen, elastin, and adhesamine can be used.
- the active fragment of the extracellular matrix may be any fragment thereof having cell adhesion activity equivalent to that of the extracellular matrix, and known ones can also be used.
- the E8 fragment of laminin 511 eg, iMatrix-511 (Nippi), etc.
- the E8 fragment of laminin 332 disclosed in Japanese Patent Application Laid-Open No.
- the extracellular matrix and its active fragment may be commercially available, and are available from, for example, Life Technologies, BD Falcon, Biolamina, Nippi and the like. Two or more kinds of these extracellular matrices and active fragments thereof may be used in combination.
- Matrigel (trade name) or Geltrex Matrix (trade name), which is a mixture of complex basement membrane components containing proteins and polysaccharides extracted and purified from mouse EHS sarcoma that overproduces the basement membrane, may be used.
- the extracellular matrix and its active fragment may be suspended in a suitable solution and applied to a container suitable for culturing cells. Artificial substances that mimic the function of extracellular matrix are also not particularly limited as long as they are usually used for cell culture. For example, Corning's Synthemax (registered trademark), Ultraweb (registered trademark), and Sigma-Aldrich Known products such as Hy-STEM series, polylysine, polyornithine, etc. can be used.
- the extracellular matrix or active fragment thereof used in the present invention, or an artificial substance that mimics the functions thereof is preferably Matrigel, or an active fragment of laminin 511 or laminin 511, and more preferably an active fragment of laminin 511 (. That is, the E8 fragment of laminin 511).
- cartilage or bone progenitor cells can be proliferated endlessly (proliferation is possible for at least 5 weeks or more). Can be selected as appropriate.
- the culture temperature is not particularly limited, but is 30 to 40 ° C., preferably 37 ° C., and the culture is carried out in the presence of CO 2 containing air, and the CO 2 concentration is preferably 2 to 5%.
- cartilage or bone progenitor cells used in the present invention for example, cells isolated from a biological sample by cell sorting using PDGFRA as an index may be used, or from stem cells such as pluripotent stem cells and mesenchymal stem cells. Differentiation-induced cells may be used, but cells derived from pluripotent stem cells are preferable.
- pluripotent stem cell refers to embryonic stem cells (ES cells) and similar differentiated pluripotent cells, that is, various tissues of the living body (endoderm, mesodermal, etc.). It means a cell that has the potential to differentiate into (all of the ectoderm).
- pluripotent stem cells used in the present invention include embryonic stem cells (embryonic stem cells), induced pluripotent stem cells (iPS cells), pluripotent germ stem cells, and embryonic germ stem cells. (EG cells) and the like, preferably ES cells or iPS cells, and among them, iPS cells are preferable.
- the pluripotent stem cell is an ES cell or an arbitrary cell derived from a human embryo
- the cell is a cell produced by destroying an embryo, even if the cell is produced by destroying the embryo. It may be, but preferably cells produced without destroying the embryo.
- ES cell means a pluripotent stem cell established from the inner cell mass of an early embryo (for example, blastocyst) of a mammal such as a human or a mouse. ES cells were discovered in mice in 1981 (MJ Evans and MH Kaufman (1981), Nature 292: 154-156), and ES cell lines were subsequently established in primates such as humans and monkeys (JA Thomson et). al. (1998), Science 282: 1145-1147; JA Thomson et al. (1995), Proc. Natl. Acad. Sci. USA, 92: 7844-7848; JA Thomson et al. (1996), Biol. Reprod ., 55: 254-259; JA Thomson and VS Marshall (1998), Curr. Top. Dev. Biol., 38: 133-165).
- induced pluripotent stem cell is a pluripotent stem cell derived from a somatic cell, and by reprogramming the somatic cell, pluripotency similar to that of an embryonic stem cell is artificially created. It means a cell that has been given.
- iPS cells induced pluripotent stem cells
- differentiated cells such as fibroblasts by expressing genes such as Oct3 / 4, Sox2, Klf4, and Myc can be mentioned. ..
- Yamanaka et al Established induced pluripotent stem cells from mouse fibroblasts (Cell, 2006, 126 (4), p663-676).
- induced pluripotent stem cells with pluripotency similar to embryonic stem cells were established from human fibroblasts (Cell, 2007, 131 (5), p861-872; Science, 2007, 318 ( 5858), p1917-1920; Nat Biotechnol., 2008, 26 (1), p101-106).
- pluripotent stem cells examples include, but are limited to, rodent-derived cells such as mice, rats, hamsters, and guinea pigs, and primate-derived cells such as humans, monkeys, orangutans, and orangutans. Not done. Human-derived cells are preferred.
- the pluripotent stem cell used in the present invention may be a pre-established and stocked cell, or a newly established cell. Therefore, a step of establishing pluripotent stem cells may be performed prior to the culture method of the present invention.
- the step of establishing the pluripotent stem cell is not particularly limited as long as it includes a step of introducing a specific reprogramming factor into the somatic cell. For example, a step of collecting somatic cells, introducing reprogramming factors such as Oct3 / 4, Sox2, Klf4, and Myc to artificially express them, and then selecting and expanding the cells that have acquired pluripotency.
- Step of introducing reprogramming factors (Oct3 / 4, Sox2, Klf4, and c-Myc) into human peripheral blood lymphocytes using a retroviral vector or Sendai viral vector and culturing them can be mentioned.
- somatic cells from which the induced pluripotent stem cells used in the present invention are derived are not particularly limited.
- somatic cells include lymphocytes in peripheral blood, fibroblasts such as skin, skin cells, visual cells, brain cells, hairy cells, oral mucosa, lung cells, hepatocytes, gastric mucosal cells, and intestinal cells.
- Genes included in the reprogramming factors include, for example, Oct3 / 4, Sox2, Sox1, Sox3, Sox15, Sox17, Klf4, Klf2, c-Myc, N-Myc, L-Myc, Nanog, Lin28, Fbx15, ERas, ECAT15-2, Tcl1, beta-catenin, Lin28b, Sall1, Sall4, Esrrb, Nr5a2, Tbx3, Glis1, etc. are exemplified, and these reprogramming factors may be used alone or in combination.
- the combinations of reprogramming factors include WO2007 / 069666, WO2008 / 118820, WO2009 / 007852, WO2009 / 032194, WO2009 / 058413, WO2009 / 057831, WO2009 / 075119, WO2009 / 079007, WO2009 / 091659, WO2009 / 101084, WO2009 / 101407, WO2009 / 102983, WO2009 / 114949, WO2009 / 117439, WO2009 / 126250, WO2009 / 126251, WO2009 / 126655, WO2009 / 157593, WO2010 / 009015, WO2010 / 033906, WO2010 / 033920, WO2010 / 042800, WO2010 / 050626, WO2010 / 056831, WO2010 / 068955, WO2010 / 098419, WO2010/102267
- mesenchymal stem cell is derived from bone marrow or bone membrane, peripheral blood, umbilical cord blood, or adipose tissue, and is a tissue of the mesenchymal tissue system (adipose tissue, cartilage tissue, bone tissue). Etc.) means stem cells that can differentiate into.
- mesenchymal stem cells bone marrow mesenchymal stem cells are preferable because it is easy to collect cells from living tissue and a culture method after collection has been established, and they are collected as surplus tissue from living body.
- Adipose tissue-derived mesenchymal stem cells are preferable because they are easy to collect and are less invasive at the time of collection.
- the method for inducing differentiation of stem cells such as pluripotent stem cells and mesenchymal stem cells into cartilage or bone progenitor cells can be appropriately performed according to a method known per se.
- pluripotent stem cells can be obtained by the methods described in Patent Document 1, Non-Patent Documents 1 and 2, JP-A-2005-511083 and the like.
- a cell population in the middle of the process of differentiating from chondrocytes (for example, 4 to 8 days after induction of differentiation) can be used by selecting cells expressing PDGFRA or the like, if necessary.
- the method described in JP-A-2004-254655 can be appropriately referred to.
- pluripotent stem cells are cultured in a medium containing a Wnt signal activator, a BMP inhibitor, FGF and / or a TGF- ⁇ signal activator for about 1 to 2 days (preferably).
- a medium containing TGF- ⁇ signal inhibitor, Wnt signal activator, BMP inhibitor and / or FGF for about 1 to 2 days (preferably 1 day).
- the cells are cultured in a medium containing a TGF- ⁇ signal inhibitor, a BMP inhibitor, a Wnt signal inhibitor and / or a MAPK / ERK kinase (MEK) inhibitor for about 1 to 2 days (preferably 1 day).
- MPK MAPK / ERK kinase
- Wnt signal activator examples include CHIR99021 (6-[[2-[[4- (2,4-Dichlorophenyl) -5- (5-methyl-1H-imidazol-2-yl) -2-pyrimidinyl). ] amino] ethyl] amino] -3-pyridinecarbonitrile), WNT proteins (eg Wnt-1, Wnt-2, Wnt-2b, Wnt-3a, Wnt-4, Wnt-5a, Wnt-5b, Wnt-6, Wnt-7a, Wnt-7a / b, Wnt-7b, Wnt-8a, Wnt-8b, Wnt-9a, Wnt-9b, Wnt-10a, Wnt-10b, Wnt-11, Wnt-16b, etc.), RSPO proteins (Example: RSP02), lithium chloride, TDZD8 (4-Benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione), BIO-ace
- BMP inhibitor examples include NOGGIN, CHORDIN, LDN193189 (4- [6- (4-Piperazin-1-yl-phenyl) -pyrazolo [1,5-a] pyrimidin-3-yl] -quinoline hydrochloride).
- LDN193189 When used, its concentration in the medium is typically 0.03 ⁇ M to 3 ⁇ M (eg 0.3 ⁇ M, etc.).
- FGF examples include the same ones used in the above-mentioned culture method of the present invention, but bFGF is preferable.
- bFGF concentration in the medium is typically 10 ng / mL to 1000 ng / mL (eg, 100 ng / mL, etc.).
- TGF- ⁇ signal activator examples include TGF- ⁇ (eg, TGF- ⁇ 1, TGF- ⁇ 2, TGF- ⁇ 3), activin A, IDE1 (1- [2-[(2-Carboxyphenyl) methylene]]. hydrazide] heptanoic acid), IDE2 (1- (2-cyclopentylidenehydrazide) -heptanedioic acid), Nodal, etc., but activin A is preferable. When activin A is used, its concentration in the medium is typically between 3 ng / mL and 300 ng / mL (eg, 30 ng / mL, etc.).
- TGF- ⁇ signal inhibitor examples include the above 1.
- the same ones listed in (1) can be mentioned, but SB431542 is preferable.
- SB431542 When SB431542 is used, its concentration in the medium is typically 1 ⁇ M to 100 ⁇ M (eg, 10 ⁇ M, etc.).
- Wnt signal inhibitor examples include C59 (4- (2-methyl-4-pyridinyl) -N- [4- (3-pyridinyl) phenyl] benzeneacetamide), DKK1, and IWP-2 (N- (6- (6-).
- Examples of the above MEK inhibitor include PD184352 (2- (2-chloro-4-iodophenylamino) -N-cyclopropylmethoxy-3,4-difluorobenzamide) and PD98059 (2- (2-amino-3-methoxyphenyl) -4H- 1-benzopyran-4-one), U0126, SL327, PD0325901 (N-[(2R) -2,3-Dihydroxypropoxy] -3,4-difluoro-2-[(2-fluoro-4-iodophenyl) amino]- Benzamide), trametinib, cobimetinib, binimetinib and the like, with PD0325901 being preferred.
- PD0325901 When PD0325901 is used, its concentration in the medium is typically 0.03 ⁇ M to 3 ⁇ M (eg 0.3 ⁇ M, etc.).
- hedgehog signal activator examples include hedgehog proteins (eg, Hh, Shh, Ihh, Dhh, etc.), Smoothened agonists (eg, SAG (Hh-Ag 1.3), SAG21k (3-chloro-4,7)).
- -difluoro-N- (4-methoxy-3- (pyridin-4-yl) benzyl) -N-((1r, 4r) -4- (methylamino) cyclohexyl) benzo [b] thiophene-2-carboxamide), Hh -Ag 1.1, Hh-Ag 1.5, purmorphamine, etc. are mentioned, but Smoothened agonists are preferable, and SAG is more preferable.
- SAG When SAG is used, its concentration in the medium is typically 0.03 ⁇ M to 3 ⁇ M. (Example: 0.3 ⁇ M, etc.).
- the medium used in step (i) contains CHIR99021, LDN193189, bFGF and activin A
- the medium used in step (ii) contains SB431542, CHIR99021, LDN193189 and bFGF.
- SB431542, LDN193189, IWR-1 and PD0325901 are included, and the medium used in the above step (iv) contains IWR-1 and SAG.
- the cartilage or bone progenitor cells induced to differentiate in this way may be used as they are in the culture method of the present invention, or may be cryopreserved (also referred to as cryopreservation) and thawed before use before the present invention. It may be used as a culture method. In addition, differentiation-induced cartilage or bone progenitor cells may be expanded and cultured before cryopreservation. Therefore, in another aspect of the present invention, a method for cryopreserving the cells, or a frozen stock produced by the method, comprising the step of cryopreserving the progenitor cells of cartilage or bone obtained by the culturing method of the present invention. A manufacturing method is provided.
- a culture solution in which cells are immersed, a physiological buffer solution, or the like is used as a cryopreservation solution, and a cryoprotectant is added thereto, or the culture solution is replaced with a cryopreservation solution containing a cryoprotectant. You may do it after performing processing such as.
- the cryopreservation solution may be added after substantially all the culture solution has been removed, or the cryopreservation solution may be added while a part of the culture solution remains.
- a commercially available solution may be used, and examples thereof include STEM-CELLBANKER (registered trademark) (ZENOAQ).
- the cells can be thawed by any known cell thawing method.
- cryopreserved cells are subjected to solid, liquid or gas having a temperature higher than the freezing temperature by using a water bath, an incubator, a thermostat or the like. Achieved by contact with a medium (eg, water, culture).
- a medium eg, water, culture
- the basal medium used for inducing differentiation of stem cells into cartilage or bone progenitor cells is not particularly limited, and examples thereof include the same basal medium used in the culture method of the present invention described above.
- the medium may contain additives, serum and / or serum substitutes, for example, the same as the additives, serum and serum substitutes that can be used in the culture method of the present invention described above.
- the incubator used for inducing differentiation is not particularly limited, and examples thereof include the same incubator used in the above-mentioned culturing method of the present invention.
- the incubator may be cell-adherent or non-cell-adhesive, and is appropriately selected according to the purpose.
- a cell-adhesive incubator is coated with any cell-supporting substrate such as extracellular matrix (ECM, also called extracellular matrix) for the purpose of improving the adhesion of the surface of the incubator to cells.
- ECM extracellular matrix
- the cell-supporting matrix may be the same as the cell-supporting substrate that can be used in the above-mentioned culture method of the present invention.
- the cell culturing method may be adhesive culturing or suspension culturing.
- "suspension culture” means culturing a target cell or cell mass without adhering it to the bottom surface of the incubator, and even if the cell or cell mass touches the bottom surface, the culture solution is used. Suspension culture also includes culturing in a state in which cells and cell clusters float in the culture solution when shaken lightly.
- the suspension culture may be a static culture, but cell seeding, medium exchange, cell image acquisition, and cultured cell recovery are automatically performed under mechanical control in a closed environment, and pH, temperature, and oxygen are used.
- a bioreactor eg, single-use bioreactor, etc.
- an automatic culturing device capable of culturing at high density while controlling the concentration and the like.
- Fed-batch culture, continuous culture, and perfusion culture are methods for supplying a new medium in the middle of culture using these devices and supplying the required substances to cells and / or tissues in just proportion.
- the culture temperature is not particularly limited, but is 30 to 40 ° C., preferably 37 ° C., and the culture is carried out in the presence of CO 2 containing air, and the CO 2 concentration is preferably 2 to 5%.
- the present invention also provides a cartilage or bone progenitor cell expansion kit (hereinafter sometimes referred to as the "kit of the present invention").
- the kit of the present invention contains a basal medium, a TGF- ⁇ signal inhibitor and FGF.
- the basal medium, TGF- ⁇ signal inhibitor and FGF include the above 1.
- the same ones listed in (1) can be mentioned, but among them, DMEM / F-12 mixed medium is preferable as the basal medium, SB431542 is preferable as the TGF- ⁇ signal inhibitor, and bFGF is preferable as the FGF.
- the kit of the present invention may also include cartilage or bone precursor cells, media additives, serum, serum substitutes, incubators and / or cell support substrates, and cartilage or bone precursors.
- Examples of cells, additives to media, serum, serum substitutes, incubators and substrates for supporting cells include the above 1. The same ones listed in.
- the kit of the present invention may further include a document or instruction manual describing the procedure for expansion culture.
- kits containing a medium containing a TGF- ⁇ signal inhibitor and / or FGF, or a medium for expanding culture of cartilage or bone progenitor cells containing a TGF- ⁇ signal inhibitor and FGF is provided.
- the present invention also provides a method for producing chondrocytes or osteoocytes (hereinafter, may be referred to as “method for producing the present invention”).
- the production method of the present invention comprises a step of inducing differentiation of cartilage or bone progenitor cells (hereinafter sometimes referred to as “progenitor cells of the present invention”) obtained by the culture method of the present invention into chondrocytes or bone cells. Including.
- the progenitor cells of the present invention are induced to differentiate into chondrocytes by appropriately referring to the methods described in Patent Document 1, Non-Patent Documents 1 and 2, JP-A-2005-511083, and JP-A-2004-254655. can do. More specifically, for example, by culturing the progenitor cells of the present invention in a medium containing a BMP signal activator and / or a TGF- ⁇ signal activator, differentiation can be induced into chondrocytes.
- the progenitor cells of the present invention may be expanded and cultured again by the method of the present invention before the induction of differentiation is started (for example, after the cryopreserved cells are put to sleep).
- the medium, TGF- ⁇ signal inhibitor, FGF, medium additive, serum and its substitute, incubator, etc. are described in 1. above. As described in.
- BMP signal activator examples include bone morphogenetic proteins (BMP) (eg, BMP-2, BMP-4, BMP-7, etc.), Alantolactone, FK506, isoliquiritigenin, 4'-hydroxychalcone and the like. However, it is preferably a bone morphogenetic protein, and BMP-4 is more preferable.
- BMP-4 bone morphogenetic protein
- its concentration in the medium is typically between 2 ng / mL and 200 ng / mL (eg, 20 ng / mL, etc.).
- TGF- ⁇ signal activator examples include the above 1. The same as those listed in the above can be mentioned, but TGF- ⁇ is preferable, and TGF- ⁇ 3 is more preferable.
- the concentration in the medium is typically 1 ng / mL to 100 ng / mL (eg, 10 ng / mL, etc.).
- the medium used in the culture contains BMP-4 and TGF- ⁇ 3.
- chondrocytes induced to differentiate from cartilage or bone progenitor cells express COL2A1, ACAN and EPIPHYCAN at at least mRNA levels. Expression of such markers can be detected by methods known per se, and detection of marker protein expression can be detected by immunological assays using antibodies such as ELISA, immunostaining, Western blotting, flow cytometry. Can be done using. Further, the expression of the marker gene can be detected by using, for example, a nucleic acid amplification method and / or a nucleic acid detection method such as real-time PCR, microarray, biochip and RNAseq.
- a method for inducing differentiation of the progenitor cells of the present invention into bone cells a method known per se can be used.
- progenitor cells of the present invention by culturing the progenitor cells of the present invention in a medium containing, for example, dexamethasone, ⁇ -glycerophosphate and vitamin Cs, a Rho kinase inhibitor and a BMP signal activator, and / or a calcium antagonist.
- each of the additives to the medium is contained within a concentration range known per se.
- a method of inducing differentiation of progenitor cells into chondrocytes and then differentiating from chondrocytes into bone cells can also be used.
- Examples of the above-mentioned vitamin Cs include the same ones listed in 1 above.
- Examples of the Rho kinase inhibitor include (+)-trans-4- (1-aminoethyl) -1- (4-puridylcarbamoyl) cyclohexane and (+)-trans-N- (1H-pyrrolo [2,3-].
- Examples of the calcium channel blocker include dihydropyridine calcium channel blockers (eg, benidipine, diphedipine, amlodipine, synolenidipine, etc.), phenylalkylamine calcium channel blockers (eg, verapamil, galopamil, bepridil, etc.), and benzothiazem.
- Calcium channel blockers eg, diltiazem
- zonisamide fasdil, romelysin, pregavalin, cyclanderate, idebenon, buflomedil, atociban and the like.
- Confirmation of bone cells can be performed by confirming alkaline phosphatase activity and / or expression of one or more bone markers.
- the bone marker include RUNX2, COL1A1, Osteopontin (OPN), Osterix, ALP, Osteocalcin and the like.
- bone cells induced to differentiate from cartilage or bone progenitor cells express COL2A1 and OPN at at least mRNA levels.
- Alkaline phosphatase activity can be confirmed by methods known per se (eg Kind-King method, Bessey-Lowry method, GSCC method, SSCC method, JSCC method, etc.) and commercially available kits (eg TRACP & ALP Assay Kit (Takara Bio). ) Etc.) can be used.
- the expression of the bone marker can be detected by the same method as the detection of the expression of the cartilage marker described above.
- the basal medium used for inducing differentiation of the progenitor cells of the present invention into chondrocytes or osteoocytes is not particularly limited, and examples thereof include the same basal medium used in the above-mentioned culturing method of the present invention.
- the medium may also contain additives, serum and / or serum substitutes, for example the same additives, serum and serum substitutes that can be used in the culture method of the present invention described above. Can be mentioned.
- the incubator used for inducing differentiation is not particularly limited, and examples thereof include the same incubator used in the above-mentioned culturing method of the present invention.
- the incubator may be cell-adherent or non-cell-adhesive, and is appropriately selected according to the purpose.
- the cell-adhesive incubator is coated with any cell-supporting substrate such as extracellular matrix (ECM, also called extracellular matrix) for the purpose of improving the adhesion of the surface of the incubator to cells.
- ECM extracellular matrix
- the cell-supporting matrix may be the same as the cell-supporting substrate that can be used in the above-mentioned culture method of the present invention.
- the culture period is not particularly limited as long as the chondrocytes can differentiate, but is preferably 3 days or more (eg, 4 days, 5 days or more).
- the upper limit is not particularly limited, but is preferably 20 days or less (eg, 15 days, 10 days, 9 days, 8 days, 7 days or less). In a preferred embodiment, it is 6 days.
- the culture period is not particularly limited as long as the bone cells can differentiate, but is 7 days or more (eg, 8 days, 9 days, 10 days, 11 days, 12 days, 13 days or more) is preferable.
- the upper limit is not particularly limited, but is preferably 30 days or less (eg, 25 days, 20 days, 19 days, 18 days, 17 days, 16 days, 15 days or less, and examples thereof include 14 days.
- the temperature is not particularly limited, but is 30 to 40 ° C., preferably 37 ° C., and the culture is carried out in the presence of CO 2 containing air, and the CO 2 concentration is preferably 2 to 5%.
- the culture of the progenitor cells of the present invention may be adhesive culture or suspension culture.
- the suspension culture may be a static culture, but it can also be performed by the above-mentioned bioreactor or automatic culture device. When culturing using these devices, any of the methods of fed-batch culture, continuous culture and perfusion culture can be used in the present invention.
- the culture temperature is not particularly limited, but is 30 to 40 ° C., preferably 37 ° C., and the culture is carried out in the presence of CO 2 containing air, and the CO 2 concentration is preferably 2 to 5%.
- the present invention provides a method for producing ligamentous node cell (hereinafter, may be referred to as "method for producing ligamentous node cell of the present invention”).
- the method for producing a ligament node cell of the present invention includes a step of inducing differentiation of the progenitor cell of the present invention into a ligament node cell.
- the ligament node cells produced in this manner may be referred to as "the ligament node cells of the present invention”. It is also possible to produce tendon cells or ligament cells by inducing differentiation of the ligament node cells of the present invention into tendon cells or ligament cells.
- chondrocytes bone cells, ligament node cells, tendon cells, ligament cells and progenitor cells of the present invention produced as described above are defined as "sclerotome of the present invention”. -lineage cells) "may be used.
- the progenitor cells of the present invention can be induced to differentiate into ligament node cells by appropriately referring to the method described in Nakajima T. et al., Development, 145 (16): dev165431 (2016). More specifically, for example, (A) a step of culturing the precursor cells of the present invention in a medium containing FGF and / or TGF- ⁇ signal activator, and (B) cells cultured by the step (A).
- the medium used in steps (A) and (B) preferably contains vitamin Cs, and specific vitamin Cs include, for example, the above 1. The same ones listed in.
- the concentration of vitamin C in the medium is also the above 1. It is preferable that the concentration is the same as that described in 1.
- the above FGF includes the above 1. The same as those listed in the above can be mentioned, but FGF-8 (eg, FGF-8b) is preferable.
- FGF-8 eg, FGF-8b
- its concentration in the medium is typically 1 ng / mL to 100 ng / mL (eg, 10 ng / mL, etc.).
- TGF- ⁇ signal activator examples include the above 1. The same as those listed in the above can be mentioned, but TGF- ⁇ is preferable, and TGF- ⁇ 3 is more preferable. When TGF- ⁇ 3 is used, its concentration in the medium is typically 1 ng / mL to 100 ng / mL (eg, 10 ng / mL, etc.).
- BMP-4 is preferable.
- its concentration in the medium is typically 1 ng / mL to 100 ng / mL (eg, 10 ng / mL, etc.).
- TGF- ⁇ 3 and FGF-8 are included when used in step (A) and / or medium used in step (B) contains BMP-4 and TGF- ⁇ 3. Is done.
- Confirmation that it is a ligament node cell can be performed by confirming the expression of two or more types of ligament node markers.
- the cells may be cultured while applying mechanical stress.
- the ligament node marker include two or more types of markers selected from COL1A1, TNMD and SCX.
- ligament node cells induced to differentiate from cartilage or bone progenitor cells express COL2A1 and TNMD at least at the mRNA level. Expression of such markers can be detected by methods known per se, and detection of marker protein expression can be detected by immunological assays using antibodies such as ELISA, immunostaining, Western blotting, flow cytometry. Can be done using. Further, the expression of the marker gene can be detected by using, for example, a nucleic acid amplification method and / or a nucleic acid detection method such as real-time PCR, microarray, biochip and RNAseq.
- ligament node cells of the present invention As a method for inducing differentiation of ligament node cells of the present invention into tendon cells or ligament cells, a method known per se can be used.
- the ligament node cells of the present invention can be induced to differentiate into tendon cells or ligament cells by appropriately referring to the methods described in JP-A-2011-205964 and the like.
- the basal medium used for inducing differentiation into ligament node cells, tendon cells or ligament cells is not particularly limited, and examples thereof include the same basal medium used in the above-mentioned culture method of the present invention.
- the medium may also contain additives, serum and / or serum substitutes, for example the same additives, serum and serum substitutes that can be used in the culture method of the present invention described above. Can be mentioned.
- the incubator used for inducing differentiation is not particularly limited, and examples thereof include the same incubator used in the above-mentioned culturing method of the present invention.
- the incubator may be cell-adherent or non-cell-adhesive, and is appropriately selected according to the purpose.
- the cell-adhesive incubator is coated with any cell-supporting substrate such as extracellular matrix (ECM, also called extracellular matrix) for the purpose of improving the adhesion of the surface of the incubator to cells.
- ECM extracellular matrix
- the cell-supporting matrix may be the same as the cell-supporting substrate that can be used in the above-mentioned culture method of the present invention.
- the culture period for differentiating ligament node cells from the progenitor cells of the present invention is not particularly limited as long as the ligament node cells can be differentiated, but is preferably 3 days or more (eg, 4 days, 5 days or more).
- the upper limit is not particularly limited, but is preferably 20 days or less (eg, 15 days, 10 days, 9 days, 8 days, 7 days or less). In a preferred embodiment, it is 8 days.
- the culture period of step (A) is preferably 1 to 3 days (example: 2 days), and the period of step (B) is preferably 4 to 8 days (example: 6 days).
- the culture period for differentiating tendon cells or ligament cells from ligament node cells is also not particularly limited as long as the tendon cells or ligament cells can be differentiated.
- the culture temperature is not particularly limited, but is 30 to 40 ° C., preferably 37 ° C., and the culture is carried out in the presence of CO 2 containing air, and the CO 2 concentration is preferably 2 to 5%.
- the culture may be an adhesive culture or a suspension culture.
- the suspension culture is described in 1. above. It can be carried out in the same manner as described in.
- the present invention also provides a cell transplantation therapy agent (hereinafter, also referred to as “cell transplantation therapy agent of the present invention”) containing the hardline lineage cells of the present invention and a method for producing the agent. ..
- the culture method of the present invention can obtain a sufficient amount of chondrocyte or bone precursor cells for use in the treatment or prevention of cartilage disease or bone disease, and by inducing differentiation of the cells.
- a sufficient amount of chondrocytes or bone cells can be provided for use in the treatment or prevention of cartilage or bone disease.
- ligament node cells, tendon cells or ligament cells induced to differentiate from the progenitor cells of the present invention can be used for the treatment or prevention of tendon or ligament-related diseases.
- the culture method of the present invention can obtain a cell population in which the residual risk of undifferentiated cells causing tumors and the like is significantly reduced. Therefore, the hardline lineage cells of the present invention are suitable for use as a raw material for a cell transplantation therapy agent, and the cell transplantation therapy agent is a cartilage disease or bone disease such as osteoarthritis or rheumatoid arthritis, or a ligament injury. It is useful for the treatment or prevention of tendon or ligament-related diseases such as Eras Dunlos's symptoms.
- the gangular lineage cells of the present invention also include a cell population containing the cells.
- a step of providing progenitor cells of cartilage or bone by the culture method of the present invention and (2) an effective amount of the present invention provided by the step (1).
- a method for producing a cell transplantation therapeutic agent of the present invention which comprises the step of preparing a preparation containing progenitor cells, is provided.
- a method for producing a cell transplantation therapeutic agent of the present invention which comprises a step of preparing a preparation containing cells and the like.
- a preparation containing ligament node cells, tendon cells or ligament cells can also be prepared in the same manner as described above.
- a mammal eg, human, mouse, rat, monkey, bovine, horse
- an effective amount of the gangular lineage cell of the present invention or the cell transplantation therapeutic agent of the present invention is treated or prevented.
- Pigs, dogs, etc. to treat or prevent cartilage or bone disease.
- iPS cells established from somatic cells having the same or substantially the same HLA genotype of the transplanted individual from the viewpoint of not causing rejection. It is desirable to use cells derived from.
- substantially the same means that the transplanted cells have the same HLA genotype to the extent that the immune response can be suppressed by the immunosuppressant, and for example, HLA-A and HLA-B.
- HLA-A and HLA-B It is a somatic cell having an HLA type in which 3 loci of HLA-DR or 4 loci with HLA-C are matched.
- the hardline lineage cells of the present invention have gene mutations that cause cartilage disease, bone disease, etc., for example, using a technique such as genome editing (eg, CRISPR system, TALEN, ZFN, etc.), It is preferable to repair the mutation of the gene that causes the disease in advance. If sufficient cells cannot be obtained due to age or constitution, it is possible to implant the cells in a capsule such as polyethylene glycol or silicon, a porous container, etc. to avoid rejection. is there.
- the hardline lineage cells of the present invention are produced as parenteral preparations such as injections, suspensions, and infusions by mixing with a pharmaceutically acceptable carrier according to conventional means.
- Pharmaceutically acceptable carriers that can be included in the parenteral preparation include, for example, isotonic solutions containing saline, glucose and other adjuvants (eg, D-sorbitol, D-mannitol, sodium chloride, etc.).
- An aqueous solution for injection can be mentioned.
- the cell transplantation therapeutic agent of the present invention is, for example, a buffer (for example, phosphate buffer, sodium acetate buffer), a soothing agent (for example, benzalkonium chloride, procaine hydrochloride, etc.), and a stabilizer (for example, human).
- the transplant therapeutic agent of the present invention may be blended with serum albumin, polyethylene glycol, etc.), preservatives, antioxidants, etc.
- the transplant therapeutic agent of the present invention is formulated as an aqueous suspension
- the ganglion lineage cells of the present invention are suspended in the above aqueous solution so as to have a concentration of about 1 ⁇ 10 6 to about 1 ⁇ 10 8 cells / mL. Just let me do it.
- the dose or the amount of transplantation and the number of administrations or the number of transplants of the nodular lineage cell or the cell transplantation therapeutic agent of the present invention can be appropriately determined depending on the age, body weight, symptoms and the like of the mammal to be administered.
- the cell transplantation therapeutic agent of the present invention is provided in a cryopreserved state under conditions normally used for cryopreservation of cells, and can be thawed before use.
- serum or a substitute thereof an organic solvent (eg, DMSO) and the like may be further contained.
- the concentration of serum or a substitute thereof is not particularly limited, but may be about 1 to about 30% (v / v), preferably about 5 to about 20% (v / v).
- the concentration of the organic solvent is not particularly limited, but may be 0 to about 50% (v / v), preferably about 5 to about 20% (v / v).
- the hardline lineage cells of the present invention are useful for screening compounds for the treatment or prevention of cartilage or bone diseases, or tendon or ligament-related diseases.
- the test compound alone or in combination with other agents may be contacted with the gangular lineage cells of the invention to promote differentiation, proliferation, maturation of the cells, or cartilage or bone matrix by methods known per se.
- the production amount of the above-mentioned test compound is increased, it can be evaluated that the test compound is useful as a therapeutic compound for the above-mentioned diseases.
- the cells used for the screening are preferably cells that exhibit the same phenotype as the disease to be treated, and are particularly preferably produced by inducing differentiation of induced pluripotent stem cells prepared from somatic cells derived from disease patients. It is a cell.
- Examples of the cartilage disease or bone disease include the above 4. The same diseases listed in (1) can be mentioned.
- test compound examples include peptides, proteins, antibodies, non-peptide compounds, synthetic compounds, fermentation products, cell extracts, plant extracts, animal tissue extracts, plasma and the like.
- the test compound may form a salt.
- salts with physiologically acceptable acids eg, inorganic acids, organic acids
- bases eg, alkali metal salts, alkaline earth metal salts, aluminum salts
- the salt include salts with inorganic acids (eg, hydrochloric acid, phosphoric acid, hydrobromic acid, sulfuric acid), or organic acids (eg, acetic acid, formic acid, propionic acid, fumaric acid, maleic acid, succinic acid, tartrate acid).
- Citric acid malic acid, oxalic acid, benzoic acid, methanesulfonic acid, benzenesulfonic acid
- sodium salt potassium salt, calcium salt, magnesium salt, barium salt, aluminum salt can be used.
- the hard node lineage cells of the present invention can also be used for verification of drug discovery targets and analysis of disease mechanisms.
- cartilage or bone progenitor cells can be identified by detecting or measuring the expression of at least one gene selected from the group consisting of the following genes.
- at least one of the following genes preferably two or more (eg, three, four, five, six, seven, eight, nine, ten or more)
- Those in which gene expression is detected can be identified as cartilage or bone progenitor cells, but it is preferable that at least PDGFRA gene expression is detected.
- the detection or measurement of gene expression can be performed by the same method as the above-mentioned detection or measurement of cartilage marker expression.
- cell surface antigen genes of the present invention the following cell surface antigen genes (hereinafter sometimes referred to as “cell surface antigen genes of the present invention"), and the antigen encoded by the genes is referred to as "the present At least one, preferably two or more (eg, three, four, five, six, seven, eight, nine, ten) selected from (sometimes referred to as "cell surface antigens of the invention”).
- the present antigen encoded by one or more genes as an index
- cartilage or bone progenitor cells can be isolated from a cell population containing cartilage or bone progenitor cells.
- PDGFRA is preferable as one of the cell surface antigens of the present invention.
- Cell isolation can be performed by methods known per se (eg, FACS, MACS, etc.).
- the present invention also comprises at least an antibody against each of one or more of the cell surface antigens of the present invention for isolating cartilage or bone progenitor cells.
- a reagent (hereinafter sometimes referred to as “reagent of the present invention”) is provided.
- the reagents of the present invention contain antibodies against two or more antigens, the reagents can be provided as a reagent kit containing each antibody in a separate reagent.
- Antibodies contained in the reagents of the present invention can be provided, for example, in the form bound to fluorescent dyes, metal isotopes or beads (eg, magnetic beads), depending on the isolation means.
- the antibody shall also include an antibody fragment or a variant having the ability to bind to a cell surface antigen (eg, Fab fragment, scFab fragment, ScFv fragment, etc.).
- Example 1 Preparation of iCOP having proliferative ability and chondrogenic ability
- iCOP iPS cell-induced chondro / osteogenic progenitor
- iPS cells induced pluripotent stem cells
- the proliferative potential The ability to differentiate into chondrocytes was evaluated.
- the iPS cells the 1210B2 strain purchased from iPS Academia Japan was used.
- the medium On the first day of differentiation, the medium was replaced with 5 mL of 2 differentiation medium containing RPMI 1640, 2% B27, 10 ⁇ M SB431542 (ReproCell), 5 ⁇ M CHIR99021, 100 ng / mL bFGF, and 0.3 ⁇ M LDN 193189.
- the medium On the second day of the following day, the medium was replaced with 5 mL of 3 differentiation medium containing RPMI1640, 2% B27, 10 ⁇ M SB431542, 3 ⁇ M IWR-1 (Sigma), 0.3 ⁇ M PD0325901 (Cayman Chemical), and 0.3 ⁇ M LDN 193189.
- the medium was replaced with 5 mL of 4 differentiation medium containing RPMI1640, 2% B27, 3 ⁇ M IWR-1, and 0.3 ⁇ M SAG (Cayman Chemical).
- 4 differentiation medium containing RPMI1640, 2% B27, 3 ⁇ M IWR-1, and 0.3 ⁇ M SAG (Cayman Chemical).
- 70% of the culture supernatant of the single-use bioreactor was exchanged with 4 differentiation media, and the cells on the 6th day were designated as iCOP.
- SOX9 expressing iCOP was labeled using Alexa Fluor® 647 Mouse Anti-Sox9 (BD Biosciences), and its positive rate was measured by Attune NxT Flow Cytometer (Thermo Fisher Scientific). As a result, it was confirmed that iCOP can be induced with high efficiency of about 90% (Fig. 1).
- iCOP was harvested from a single-use bioreactor, centrifuged, suspended in 1 mL of Accumax cell detachment solution (Merck), and allowed to stand for 10 minutes. Suspend and centrifuge again until the cell mass collapses, DMEM / F12 (Thermo Fisher Scientific), 2% B27, 5 ⁇ M SB431542, 10 ng / mL bFGF, 250 ⁇ M L-ascorbic acid-2-phosphate sesquimagnesium salt ( The cells were suspended in iCOP growth medium containing Sigma) and the number of cells was counted.
- Example 2 Verification of cartilage differentiation ability of iCOP
- iCOP of each passage number was washed with PBS (-), immersed in Accutase (Nacalai Tesque), and incubated at 37 ° C. for 5 minutes. ..
- the cells are detached, they are collected in a tube, centrifuged, suspended in 5 differentiation media containing DMEM / F12, 2% B27, 10 ng / mL TGF- ⁇ 3 (PeproTech), and 20 ng / mL BMP4 (R & D systems), and the cells are suspended. The number was measured.
- Example 3 Verification of cryopreservability of iCOP
- iCOP prepared from iPS cells was cryopreserved and then thawed to evaluate proliferative ability and differentiation ability into chondrocytes.
- the iPS cells the 1231A3 strain purchased from iPS Academia Japan was used.
- iPS cells were seeded on a 10 cm dish (BD FALCON) pre-coated with 0.5 ⁇ g / cm 2 iMatrix-511 (Nippi), and StemFit containing 10 ⁇ M Y-27632 (StemFit). Registered trademark) AK03N was cultured in an incubator at 37 ° C and 5% CO 2 .
- the medium was replaced with 8 mL of differentiation 1 medium containing RPMI1640, 20% StemFit® For Differentiation®, 30 ng / mL Activin A, 10 ⁇ M CHIR99021, 100 ng / mL bFGF, and 0.3 ⁇ M LDN 193189.
- the medium was replaced with 8 mL of differentiated 2 medium containing RPMI1640, 20% StemFit® For Differentiation, 10 ⁇ M SB431542, 5 ⁇ M CHIR99021, 100 ng / mL bFGF, 0.3 ⁇ M LDN 193189.
- the medium was replaced with 8 mL of 3 differentiation medium containing RPMI1640, 20% StemFit® For Differentiation, 10 ⁇ M SB431542, 3 ⁇ M IWR-1, 0.3 ⁇ M PD0325901, 0.3 ⁇ M LDN 193189.
- the cells were replaced with 8 mL of 4 differentiation medium containing RPMI1640, 20% StemFit® For Differentiation, 3 ⁇ M IWR-1, and 0.3 ⁇ M SAG.
- the medium was exchanged with 4 differentiation media, and the cells on the 6th day were designated as iCOP.
- the number of viable cells of iCOP was measured, and 2 ⁇ 10 5 cells of iCOP were seeded in a 10 cm dish pre-coated with 0.5 ⁇ g / cm 2 iMatrix-511, DMEM / F12, 20% StemFit® For Differentiation, Incubated in iCOP growth medium containing 250 ⁇ M L-Ascorbic Acid 2-phosphate, 10 ⁇ M SB431542, 100 ng / mL bFGF, and 0.3 ⁇ M SAG at 37 ° C. and 5% CO 2 .
- the number of iCOP cells was counted, and the cells were divided into a subculture group and a frozen stock preparation group.
- 1 ⁇ 10 6 cells were suspended in 1 mL STEM-CELLBANKER® GMP grade (Zenoaq) and dispensed into serum tubes (IWAKI). After that, the serum tube was put into BICELL (Japan Freezer) that had been cooled to 4 ° C in advance, frozen in the -80 ° C deep freezer, and transferred to the -150 ° C deep freezer the next day.
- Example 4 Verification of bone differentiation ability of iCOP
- differentiation was induced from iCOP to osteoocyte (Osteocyte), and the gene expression level of the bone cell marker was evaluated.
- iPS cells 1231A3 strain, 1210B2 strain, and 201B7 strain purchased from iPS Academia Japan were used.
- the iCOP cryopreserved in the first passage was put to sleep, DMEM / F12, 20% StemFit® For Differentiation, 250 ⁇ M L-Ascorbic Acid 2-phosphate, 10 ⁇ M SB431542, 100 ng / mL bFGF, 0.3.
- the cells were expanded to the third passage in iCOP growth medium containing ⁇ M SAG and 10 ng / mL PDGF-BB (Peprotech).
- iCOP When iCOP reaches 90-100% confluence, count the number of viable cells, DMEM containing 4.5 g / L Glucose (Nakarai Tesk), 20% StemFit® For Differentiation, 250 ⁇ M L-Ascorbic Acid 2-phosphate, 100 Suspended in a bone differentiation medium containing nM dexamethasone (Sigma) and 10 mM ⁇ -glycero phosphate (Nakaraitesku). 2 ⁇ 10 5 cells of iCOP were seeded on a 24-well plate (BD FALCON) pre-coated with 0.5 ⁇ g / cm 2 iMatrix-511 and allowed to stand in an incubator at 37 ° C. and 5% CO 2 .
- BD FALCON BD FALCON
- Example 5 Verification of ligament node cell differentiation ability of iCOP
- differentiation was induced from iCOP to ligament node cells (syndetome), and the gene expression level of the ligament node marker was evaluated.
- iPS cells 1231A3 strain, 1210B2 strain, and 201B7 strain purchased from iPS Academia Japan were used.
- the iCOP cryopreserved in the first passage was put to sleep, DMEM / F12, 20% StemFit® For Differentiation, 250 ⁇ M L-Ascorbic Acid 2-phosphate, 10 ⁇ M SB431542, 100 ng / mL bFGF, 0.3. It was expanded to the 5th passage in iCOP growth medium containing ⁇ M SAG.
- iCOP When iCOP reaches 90-100% confluence, count viable cells, DMEM / F12, 20% StemFit® For Differentiation, 250 ⁇ M L-Ascorbic Acid 2-phosphate, 10 ng / mL TGF- ⁇ 3 (Peprotech) ), Suspended in ligament node cell differentiation medium 1 containing 20 ng / mL FGF8b (Peprotech). 2.5 ⁇ 10 5 cells iCOP was seeded on a 24-well plate pre-coated with 0.5 ⁇ g / cm 2 iMatrix-511 and allowed to stand in an incubator at 37 ° C. and 5% CO 2 .
- Example 6 Expanded culture by suspension culture of iCOP
- iCOP was grown by suspension culture, and its proliferative potential and ability to differentiate into chondrocytes were evaluated.
- the iPS cells the 1231A3 strain purchased from iPS Academia Japan was used.
- the number of viable cells of iCOP prepared from iPS cells was measured, and DMEM / F12, 20% StemFit® For Differentiation, 250 ⁇ M L-Ascorbic Acid 2-phosphate, 10 ⁇ M SB431542, 100 ng / mL bFGF, 0.3 ⁇ M Suspended in iCOP growth medium containing SAG, 10 ng / mL PDGF-BB.
- the cells were seeded in a 5 mL single-use bioreactor (Biott) at a cell density of 2 ⁇ 10 5 cells / mL and cultured with stirring in an incubator at 37 ° C. and 5% CO 2 at a stirring rate of 80 rpm. 70% medium was exchanged every other day.
- iCOP was harvested from a single-use bioreactor, centrifuged, suspended in 1 mL of Accumax cell detachment solution (Merck), and allowed to stand for 10 minutes. The cells were suspended again until the cell mass collapsed, centrifuged, suspended in iCOP growth medium, and the number of cells was counted. The same operation was repeated every 5 days, and the growth curve up to the 3rd generation was obtained. As a result, the daily PD was 0.38 (Fig. 7).
- the gene expression of iCOP markers PAX1, SOX9 and PDGFRA
- the expression was retained even after 3 passages (Table 3).
- chondrocyte genes were eventually expressed (Table 4). From the above, it was shown that iCOP can be expanded and cultured in a floating state.
- Example 7 Identification of iCOP-specific markers
- iCOP-specific markers prepared from iPS cells were analyzed by the next-generation sequencer (NGS) to identify iCOP-specific markers.
- NGS next-generation sequencer
- As iPS cells the 1231A3 strain purchased from iPS Academia Japan was used.
- iPS cells were seeded in 4 wells of a 24-well plate (BD FALCON) coated with 0.5 ⁇ g / cm 2 iMatrix-511 (Nippi) in advance, and 10 ⁇ M Y-27632 was used. Incubated in a 5% CO 2 incubator at 37 ° C. in StemFit® AK03N containing.
- the medium was replaced with 500 ⁇ L of differentiation 3 medium containing RPMI1640, 20% StemFit® For Differentiation, 10 ⁇ M SB431542, 3 ⁇ M IWR-1, 0.3 ⁇ M PD0325901, 0.3 ⁇ M LDN 193189.
- the cells were replaced with 500 ⁇ L of differentiated 4 medium containing RPMI1640, 20% StemFit® For Differentiation, 3 ⁇ M IWR-1, and 0.3 ⁇ M SAG.
- the medium was exchanged with 4 differentiation media, and the number of viable cells of iCOP was measured using the cells on the 6th day as iCOP.
- the iCOP at this point was designated as the 0th passage iCOP, and a cell suspension containing 1 ⁇ 10 6 cells of the acquired cells was collected in a 1.5 mL Eppendorf tube, and the supernatant was removed after centrifugation for NGS analysis. As a sample, it was cryopreserved in the form of pellets.
- iCOP 3 ⁇ 10 5 cells of iCOP were sown in a 10 cm dish coated with 0.5 ⁇ g / cm 2 iMatrix-511 in advance, and DMEM / F12, 20% StemFit® For Differentiation, 250 ⁇ M L-Ascorbic Acid 2 Incubated in iCOP growth medium containing -phosphate, 10 ⁇ M SB431542, 100 ng / mL bFGF, 0.3 ⁇ M SAG at 37 ° C. and 5% CO 2 .
- the medium When the medium is changed once every two days and 90 to 100% confluence is reached, the number of iCOP cells is counted, and the iCOP at this point is used as the first passage iCOP, and some of the acquired cells are passaged to 1 ⁇ 10 6 cells were cryopreserved in the form of pellets as a sample for NGS analysis.
- iCOP frozen samples 0th, 1st, 3rd, 5th, 8th, and 10th passages were obtained. The obtained frozen sample was thawed, and RNA was extracted using a PureLink TM RNA Mini kit (Thermo Fischer Scientific).
- RNA Integrity Number was measured using a bioanalyzer (Agilent RNA6000 Nano kit, Agilent).
- the KAPA Stranded mRNA-Seq Kit KAPA Biosystems
- KAPA Unique Dual-Indexed Adapter Kit KAPA Biosystems
- a bioanalyzer (Agilent DNA1000 kit, Agilent) was used to check the quality of the prepared library, and Qubit® 2.0 Fluorometer (Thermo Fisher Scientific) and Qubit® dsDNA HS assay kit (registered trademark) were used to measure the concentration. Thermo Fisher Scientific) was used.
- the prepared library was used for sequencing with the NextSeq 500 System (illumina).
- FASTQ files were obtained from the BaseSpace® Onsite (illumina) system and trimmed at the 3'end with the Trimmomatic tool on Bio-Linux.
- On the Jupyter notebook confirm the quality improvement by trimmomatic by FastQC, execute the RNA-seq pipeline consisting of mapping to the human genome, count of reads, and normalization, and use the output read count file.
- the expression variation gene (DEG) was analyzed using the TCC package. As a result of the analysis, 110 genes extracted as iCOP-specific marker genes are shown below.
- the gene group that is the cell surface antigen is shown below.
- the present invention it is possible to expand and culture chondrocytes or bone progenitor cells, and if the progenitor cells are expanded and cultured in advance and stocked, the period for inducing differentiation into chondrocytes or osteoocytes can be significantly shortened.
- the present invention can rapidly produce a sufficient amount of chondrocytes for use in the treatment of chondrocytes or bone diseases.
Abstract
Description
[1] 軟骨又は骨の前駆細胞を、TGF-βシグナル阻害剤及びFGFを含む培地中で培養する工程を含む、軟骨又は骨の前駆細胞の拡大培養方法。
[2] 前記軟骨又は骨の前駆細胞がPDGFRA、SOX9、若しくはPAX1を発現する細胞である、[1]に記載の方法。
[3] 前記培地中にL-アスコルビン酸又はその誘導体がさらに含まれる、[1]又は[2]に記載の方法。
[4] 前記TGF-βシグナル阻害剤がSB431542である、[1]~[3]のいずれかに記載の方法。
[5] 前記FGFがbFGFである、[1]~[4]のいずれかに記載の方法。
[6] 前記TGF-βシグナル阻害剤の培地中での濃度が1μM~50μMである、[1]~[5]のいずれかに記載の方法。
[7] 前記FGFの培地中での濃度が1ng/mL~500ng/mLである、[1]~[6]のいずれかに記載の方法。
[8] 前記軟骨又は骨の前駆細胞が多能性幹細胞に由来する細胞である、[1]~[7]のいずれかに記載の方法。
[9] 前記軟骨又は骨の前駆細胞が多能性幹細胞を浮遊培養することで得られる細胞である、[1]~[8]のいずれかに記載の方法。
[10] 拡大培養が浮遊培養により行われる、[1]~[9]のいずれかに記載の方法。
[11] 基礎培地、TGF-βシグナル阻害剤及びFGFを含む、軟骨又は骨の前駆細胞の拡大培養用キット。
[12] L-アスコルビン酸又はその誘導体をさらに含む、[11]に記載のキット。
[13] 前記TGF-βシグナル阻害剤がSB431542である、[11]又は[12]に記載のキット。
[14] 前記FGFがbFGFである、[11]~[13]のいずれかに記載のキット。
[15] [1]~[10]のいずれかに記載の方法で得られた軟骨又は骨の前駆細胞を、軟骨細胞又は骨細胞へ分化誘導させる工程を含む、軟骨細胞又は骨細胞の製造方法。
[16] 前記分化誘導工程が、軟骨又は骨の前駆細胞をBMPシグナル活性化剤及び/又はTGF-βシグナル活性化剤を含む培地で培養する工程を含む、[15]に記載の方法。
[17] 前記TGF-βシグナル活性化剤がTGF-β3である、[15]又は[16]に記載の方法。
[18] [1]~[10]のいずれかに記載の方法で得られた軟骨又は骨の前駆細胞を、靭帯節細胞へ分化誘導させる工程を含む、靭帯節細胞の製造方法。
[19] [18]に記載の方法で得られた靭帯節細胞を腱細胞又は靭帯細胞へ分化誘導させる工程を含む、腱細胞又は靭帯細胞の製造方法。
[20] [15]~[19]のいずれかに記載の方法で得られた細胞を含有してなる、細胞移植療法剤。
[21] 以下の遺伝子からなる群から選択される少なくとも1つの遺伝子の発現を検出又は測定する工程を含む、軟骨又は骨の前駆細胞を同定する方法。
A2M, ABCA9, ACAT2, ADAMTS9, ADGRF5, AGTR2, AMER1, AMOT, ANGPT1, ARHGAP5-AS1, ARHGEF26, ATP8A1, BCHE, BEGAIN, BOC, C3ORF52, CACNA1G, CELF2, CLSPN, COL25A1, COL26A1, CORO1A, CRISPLD1, CTTNBP2, CYB5B, DLGAP1, DNAJC12, DUSP9, EBF1, EBF2, EFEMP1, ESCO2, EYA1, FAM35A, FAM78A, FAR2, FGFBP2, FGFR4, FKBP4, FRZB, GCNT4, GNG11, HAAO, HMGCS1, HS3ST5, ID4, IGDCC3, IRF8, KCNA6, KCNB2, KITLG, LGR5, LHCGR, LHFP, LPPR5, LUM, MAPK8IP2, MECOM, MEF2C, MEOX2, METTL7A, NAB1, NCALD, NGF, NGFR, NKAIN2, NPR1, NR2F1, OLFML1, PCDH18, PCDH19, PCSK9, PDE4D, PDGFRA, PEG10, PLVAP, PRELP, PRRT4, RARRES2, RBMS3, RBPMS2, RSPO3, RUNX1T1, SAMD5, SCARA5, SKIDA1, SLC12A2, SLC27A3, SLC39A8, SLC7A2, SLC8A3, SLCO1C1, SOX8, SPRED2, STARD8, STOM, SYNPO, TBX18, TMC6, TNRC6C-AS1, TRIM2, TRIM9, TRIP13, TSPAN15, UBE2N, UHRF1, UNC5C, VIT, ZBTB46及びZNF488
[22] 軟骨又は骨の前駆細胞を含む細胞集団から、以下の遺伝子からなる群から選択される少なくとも1つの遺伝子を発現する細胞を単離する工程を含む、軟骨又は骨の前駆細胞の単離方法。
ABCA9, ATP8A1, BOC, KITLG, NKAIN2, ADGRF5, AGTR2, CACNA1G, C3ORF52, COL25A1, CYB5B, FAR2, FGFR4, GCNT4, HS3ST5, IGDCC3, LGR5, LHFP, LHCGR, NPR1, NGFR, PLVAP, PDGFRA, KCNA6, KCNB2, PCDH18, PCDH19, SCARA5, SLC12A2, SLC27A3, SLC39A8, SLC7A2, SLC8A3, SLCO1C1, STOM, TSPAN15, TMC6及びUNC5C
[23] 以下の遺伝子からなる群から選択される少なくとも1つの遺伝子にコードされるタンパク質の各々に対する抗体を含んでなる、軟骨又は骨の前駆細胞の単離用試薬。
ABCA9, ATP8A1, BOC, KITLG, NKAIN2, ADGRF5, AGTR2, CACNA1G, C3ORF52, COL25A1, CYB5B, FAR2, FGFR4, GCNT4, HS3ST5, IGDCC3, LGR5, LHFP, LHCGR, NPR1, NGFR, PLVAP, PDGFRA, KCNA6, KCNB2, PCDH18, PCDH19, SCARA5, SLC12A2, SLC27A3, SLC39A8, SLC7A2, SLC8A3, SLCO1C1, STOM, TSPAN15, TMC6及びUNC5C
本発明は、軟骨又は骨の前駆細胞を拡大培養する方法(以下「本発明の培養方法」と称することがある)を提供する。本発明の培養方法は、軟骨又は骨の前駆細胞を、TGF-βシグナル阻害剤及びFGFを含む培地中で培養する工程を含む。
本発明はまた、軟骨又は骨の前駆細胞の拡大培養用キット(以下「本発明のキット」と称することがある)を提供する。本発明のキットには、基礎培地、TGF-βシグナル阻害剤及びFGFが含まれる。かかる基礎培地、TGF-βシグナル阻害剤及びFGFとしては、例えば、上記1.で列挙したものと同一のものが挙げられるが、中でも、基礎培地としてはDMEM/F-12混合培地が好ましく、TGF-βシグナル阻害剤としてはSB431542が好ましく、FGFとしてはbFGFが好ましい。
本発明はまた、軟骨細胞又は骨細胞の製造方法(以下「本発明の製造方法」と称することがある)を提供する。本発明の製造方法は、本発明の培養方法で得られた軟骨又は骨の前駆細胞(以下「本発明の前駆細胞」と称することがある)を、軟骨細胞又は骨細胞へ分化誘導させる工程を含む。
さらに、本発明は、靭帯節細胞の製造方法(以下「本発明の靭帯節細胞の製造方法」と称することがある)を提供する。本発明の靭帯節細胞の製造方法は、本発明の前駆細胞を、靭帯節細胞へ分化誘導させる工程を含む。以下では、このようにして製造された靭帯節細胞を、「本発明の靭帯節細胞」と称することがある。本発明の靭帯節細胞を、腱細胞(tendon cell)又は靭帯細胞(ligament cell)に分化誘導させることで、腱細胞又は靭帯細胞を製造することもできる。本明細書において、上記のようにして製造された軟骨細胞、骨細胞、靭帯節細胞、腱細胞、靭帯細胞及び本発明の前駆細胞を包含するものとして、「本発明の硬節系譜細胞(sclerotome-lineage cells)」との用語を用いることがある。
本発明はまた、本発明の硬節系譜細胞を含有してなる、細胞移植療法剤(以下、「本発明の細胞移植療法剤」ともいう)及び該剤の製造方法を提供する。上述の通り、本発明の培養方法により、軟骨疾患又は骨疾患の治療又は予防に用いるのに十分な量の軟骨又は骨の前駆細胞を得ることができ、また、該細胞を分化誘導することにより、軟骨疾患又は骨疾患の治療又は予防に用いるのに十分な量の軟骨細胞又は骨細胞を提供することができる。同様に、本発明の前駆細胞から分化誘導した靭帯節細胞、腱細胞又は靭帯細胞は、腱又は靭帯関連疾患の治療又は予防に用いることができる。また、本発明の培養方法により、腫瘍などの原因となる未分化細胞の残存リスクが顕著に低下された細胞集団が得られる。よって、本発明の硬節系譜細胞は、細胞移植療法剤の原料として用いることに適しており、該細胞移植療法剤は、変形性関節症や関節リウマチなどの軟骨疾患又は骨疾患、あるいは靭帯損傷やエーラスダンロス症候などの腱又は靭帯関連疾患の治療又は予防に有用である。本明細書において、本発明の硬節系譜細胞には、該細胞を含む細胞集団も包含される。
さらに、本発明の硬節系譜細胞は、軟骨疾患若しくは骨疾患、あるいは腱若しくは靭帯関連疾患の治療又は予防用化合物のスクリーニングに有用である。例えば、試験化合物を単独で又は他の薬剤と組み合わせて、本発明の硬節系譜細胞と接触させ、自体公知の方法により、当該細胞の分化、増殖、成熟を促進する、あるいは軟骨基質又は骨基質の産生量を増加する場合には、当該試験化合物が上記疾患の治療用化合物として有用であると評価することができる。上記スクリーニングに用いる細胞は、治療対象となる疾患と同様の表現型を呈する細胞が好ましく、特に好ましくは、疾患患者由来の体細胞から作製された人工多能性幹細胞を分化誘導することにより製造された細胞である。上記軟骨疾患又は骨疾患としては、上記4.で列挙した疾患と同一のものが挙げられる。
以下の実施例で示す通り、軟骨又は骨の前駆細胞であるiCOPが、以下の遺伝子を発現することが実証された。従って、以下の遺伝子からなる群から選択される少なくとも1つの遺伝子の発現を検出又は測定することで、軟骨又は骨の前駆細胞を同定することができる。該方法では、以下の遺伝子のうちの少なくとも1つ、好ましくは2つ以上(例:3つ、4つ、5つ、6つ、7つ、8つ、9つ、10つ又はそれ以上)の遺伝子の発現が検出されたものを軟骨又は骨の前駆細胞として同定することができるが、少なくともPDGFRA遺伝子の発現が検出されることが好ましい。遺伝子の発現の検出又は測定は、上述の軟骨マーカーの発現の検出又は測定と同様の方法により行うことができる。
また、上記タンパク質のうち、下記の細胞表面抗原遺伝子(以下「本発明の細胞表面抗原遺伝子」と称することがあり、該遺伝子にコードされる抗原を「本発明の細胞表面抗原」と称することがある)から選択される少なくとも1つ、好ましくは2つ以上(例:3つ、4つ、5つ、6つ、7つ、8つ、9つ、10つ又はそれ以上)の遺伝子にコードされる抗原を指標として、軟骨又は骨の前駆細胞を含む細胞集団から、軟骨又は骨の前駆細胞を単離することができる。本発明の細胞表面抗原の1つとしては、PDGFRAが好ましい。細胞の単離は、自体公知の方法(例:FACS、MACS等)により行うことができる。
本発明はまた、本発明の細胞表面抗原のうちの1つ以上の抗原の各々に対する抗体を少なくとも含んでなる、軟骨又は骨の前駆細胞の単離用試薬(以下「本発明の試薬」と称することがある)を提供する。本発明の試薬が2つ以上の抗原に対する抗体を含む場合、該試薬は、各抗体を別個の試薬中に含む試薬キットとして提供され得る。本発明の試薬に含まれる抗体は、単離手段に応じて、例えば、蛍光色素、金属同位体又はビーズ(例:磁気ビーズ)に結合した形態で提供され得る。本明細書において、該抗体には、細胞表面抗原への結合能を有する抗体断片や改変体(例:Fab断片、scFab断片、ScFv断片等)も含まれるものとする。
以下の実施例では、人工多能性幹細胞(iPS細胞)からiCOP(iPS cell-induced chondro/osteogenic progenitor)を作製し、その増殖性と軟骨細胞への分化能を評価した。iPS細胞として、iPSアカデミアジャパン社より購入した1210B2株を用いた。
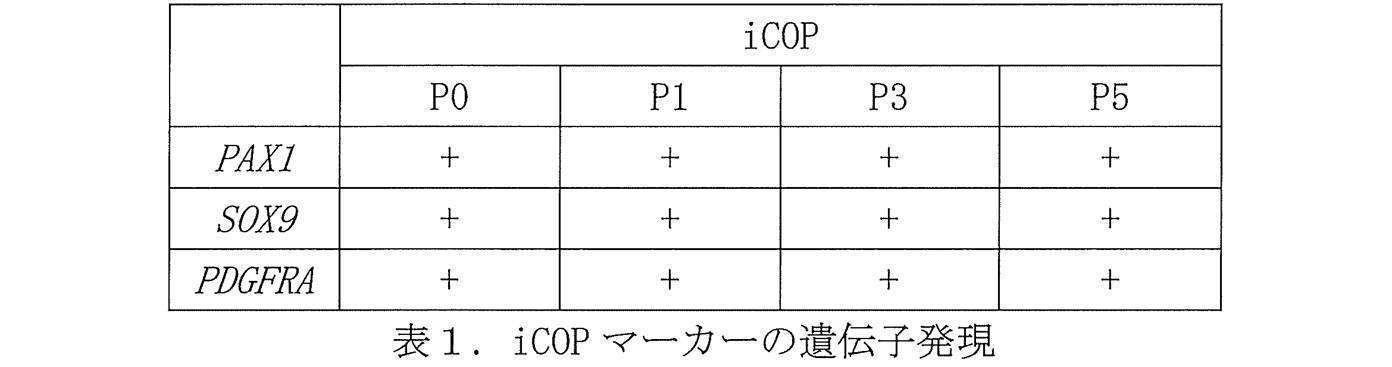
iCOPの軟骨分化能を検証するため、各継代数のiCOPをPBS(-)で洗浄後、Accutase(ナカライテスク)に浸して37℃、5分間インキュベーションした。細胞が剥離したらチューブに回収して遠心し、DMEM/F12、2% B27、10 ng/mL TGF-β3(PeproTech)、20 ng/mL BMP4(R&D systems)を含む分化5培地に懸濁し、細胞数を計測した。1×106cellsのiCOPを1 mLの分化5培地を含むElplasiaTM Non-adherent surface 24 well plate(Kuraray)に播種し、37℃、5% CO2条件下でインキュベーションした。培養上清を毎日半量交換し、6日目に細胞を回収した。培養後の軟骨細胞の遺伝子発現をreal-time PCR法により評価した結果、5継代したiCOPは軟骨細胞への分化能が保持していた(表2)。

以下の実験例では、iPS細胞から作製したiCOPを凍結保存し、その後解凍して増殖能と軟骨細胞への分化能を評価した。iPS細胞として、iPSアカデミアジャパン社より購入した1231A3株を用いた。
以下の実験例では、iCOPから骨細胞(Osteocyte)へ分化誘導し、骨細胞マーカーの遺伝子発現量を評価した。iPS細胞として、iPSアカデミアジャパン社より購入した1231A3株、1210B2株、及び201B7株を用いた。
以下の実験例では、iCOPから靱帯節細胞(syndetome)へ分化誘導し、靱帯節マーカーの遺伝子発現量を評価した。iPS細胞として、iPSアカデミアジャパン社より購入した1231A3株、1210B2株、及び201B7株を用いた。
以下の実験例では、iCOPを浮遊培養により増殖させ、その増殖性と軟骨細胞への分化能を評価した。iPS細胞として、iPSアカデミアジャパン社より購入した1231A3株を用いた。


以下の実験例では、iPS細胞から作製したiCOPの次世代シーケンサー(NGS)による解析を実施し、iCOP特異的マーカーの同定を行った。iPS細胞として、iPSアカデミアジャパン社より購入した1231A3株を使用した。


Claims (12)
- 軟骨又は骨の前駆細胞を、TGF-βシグナル阻害剤及びFGFを含む培地中で培養する工程を含む、軟骨又は骨の前駆細胞の拡大培養方法。
- 前記軟骨又は骨の前駆細胞がPDGFRA、SOX9、若しくはPAX1を発現する細胞である、請求項1に記載の方法。
- 前記TGF-βシグナル阻害剤がSB431542である、請求項1又は2に記載の方法。
- 前記FGFがbFGFである、請求項1~3のいずれか1項に記載の方法。
- 前記軟骨又は骨の前駆細胞が多能性幹細胞に由来する細胞である、請求項1~4のいずれか1項に記載の方法。
- 前記軟骨又は骨の前駆細胞が多能性幹細胞を浮遊培養することで得られる細胞である、請求項1~5のいずれか1項に記載の方法。
- 拡大培養が浮遊培養により行われる、請求項1~6のいずれか1項に記載の方法。
- 基礎培地、TGF-βシグナル阻害剤及びFGFを含む、軟骨又は骨の前駆細胞の拡大培養用キット。
- 前記TGF-βシグナル阻害剤がSB431542である、請求項8に記載のキット。
- 前記FGFがbFGFである、請求項8又は9に記載のキット。
- 請求項1~7のいずれか1項に記載の方法で得られた軟骨又は骨の前駆細胞を、軟骨細胞又は骨細胞へ分化誘導させる工程を含む、軟骨細胞又は骨細胞の製造方法。
- 請求項1~7のいずれか1項に記載の方法で得られた軟骨又は骨の前駆細胞を、靭帯節細胞へ分化誘導させる工程を含む、靭帯節細胞の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA3141455A CA3141455A1 (en) | 2019-05-20 | 2020-04-28 | Expansion culture method for cartilage or bone precursor cells |
| JP2021520684A JPWO2020235319A1 (ja) | 2019-05-20 | 2020-04-28 | |
| EP20809188.4A EP3974519A4 (en) | 2019-05-20 | 2020-04-28 | EXPANSION CULTURE METHOD FOR CARTILAGE OR BONE PRECURSOR CELLS |
| US17/455,763 US20220073882A1 (en) | 2019-05-20 | 2021-11-19 | Expansion culture method for cartilage or bone precursor cells |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019094878 | 2019-05-20 | ||
| JP2019-094878 | 2019-05-20 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US17/455,763 Continuation US20220073882A1 (en) | 2019-05-20 | 2021-11-19 | Expansion culture method for cartilage or bone precursor cells |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020235319A1 true WO2020235319A1 (ja) | 2020-11-26 |
Family
ID=73458414
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/018130 WO2020235319A1 (ja) | 2019-05-20 | 2020-04-28 | 軟骨又は骨の前駆細胞の拡大培養方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20220073882A1 (ja) |
| EP (1) | EP3974519A4 (ja) |
| JP (1) | JPWO2020235319A1 (ja) |
| CA (1) | CA3141455A1 (ja) |
| WO (1) | WO2020235319A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20240052317A1 (en) * | 2022-07-19 | 2024-02-15 | Lung Biotechnology Pbc | Methods for Differentiating Stromal Cells or Pericytes |
Citations (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001017562A1 (en) | 1999-09-02 | 2001-03-15 | Yamanouchi Pharmaceutical Co., Ltd. | Osteogenesis promoting agents |
| JP2004254655A (ja) | 2003-02-27 | 2004-09-16 | Kanegafuchi Chem Ind Co Ltd | 軟骨の分化誘導方法 |
| JP2005511083A (ja) | 2001-12-07 | 2005-04-28 | ジェロン コーポレイション | ヒト胚幹細胞由来の軟骨細胞前駆細胞 |
| WO2006123699A1 (ja) | 2005-05-17 | 2006-11-23 | Kyowa Hakko Kogyo Co., Ltd. | 幹細胞に対する骨芽細胞への分化促進剤 |
| WO2007069666A1 (ja) | 2005-12-13 | 2007-06-21 | Kyoto University | 核初期化因子 |
| JP2008514188A (ja) * | 2004-09-24 | 2008-05-08 | アンジオブラスト システムズ,インコーポレーテッド | 間葉系前駆細胞(mpc)の増殖および/または生存を増強させる方法 |
| WO2008118820A2 (en) | 2007-03-23 | 2008-10-02 | Wisconsin Alumni Research Foundation | Somatic cell reprogramming |
| WO2009007852A2 (en) | 2007-06-15 | 2009-01-15 | Izumi Bio, Inc | Multipotent/pluripotent cells and methods |
| WO2009032194A1 (en) | 2007-08-31 | 2009-03-12 | Whitehead Institute For Biomedical Research | Wnt pathway stimulation in reprogramming somatic cells |
| WO2009057831A1 (ja) | 2007-10-31 | 2009-05-07 | Kyoto University | 核初期化方法 |
| WO2009058413A1 (en) | 2007-10-29 | 2009-05-07 | Shi-Lung Lin | Generation of human embryonic stem-like cells using intronic rna |
| WO2009075119A1 (ja) | 2007-12-10 | 2009-06-18 | Kyoto University | 効率的な核初期化方法 |
| WO2009079007A1 (en) | 2007-12-17 | 2009-06-25 | Gliamed, Inc. | Stem-like cells and method for reprogramming adult mammalian somatic cells |
| WO2009091659A2 (en) | 2008-01-16 | 2009-07-23 | Shi-Lung Lin | Generation of tumor-free embryonic stem-like pluripotent cells using inducible recombinant rna agents |
| WO2009102983A2 (en) | 2008-02-15 | 2009-08-20 | President And Fellows Of Harvard College | Efficient induction of pluripotent stem cells using small molecule compounds |
| WO2009101084A1 (en) | 2008-02-13 | 2009-08-20 | Fondazione Telethon | Method for reprogramming differentiated cells |
| WO2009101407A2 (en) | 2008-02-11 | 2009-08-20 | Cambridge Enterprise Limited | Improved reprogramming of mammalian cells, and the cells obtained |
| WO2009117439A2 (en) | 2008-03-17 | 2009-09-24 | The Scripps Research Institute | Combined chemical and genetic approaches for generation of induced pluripotent stem cells |
| WO2009114949A1 (en) | 2008-03-20 | 2009-09-24 | UNIVERSITé LAVAL | Methods for deprogramming somatic cells and uses thereof |
| WO2009126251A2 (en) | 2008-04-07 | 2009-10-15 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through use of an hdac modulator |
| WO2009146408A1 (en) | 2008-05-30 | 2009-12-03 | Summa Health Systems Llc | Methods for using tgf-b receptor inhibitors or activin-like kinase (alk) 5 inhibitors a-83-01 and sb-431542 to treat eye disease and wound healing conditions |
| WO2009157593A1 (en) | 2008-06-27 | 2009-12-30 | Kyoto University | Method of efficiently establishing induced pluripotent stem cells |
| WO2010009015A2 (en) | 2008-07-14 | 2010-01-21 | Oklahoma Medical Research Foundation | Production of pluripotent cells through inhibition of bright/arid3a function |
| WO2010033906A2 (en) | 2008-09-19 | 2010-03-25 | President And Fellows Of Harvard College | Efficient induction of pluripotent stem cells using small molecule compounds |
| WO2010033920A2 (en) | 2008-09-19 | 2010-03-25 | Whitehead Institute For Biomedical Research | Compositions and methods for enhancing cell reprogramming |
| WO2010042800A1 (en) | 2008-10-10 | 2010-04-15 | Nevada Cancer Institute | Methods of reprogramming somatic cells and methods of use for such cells |
| WO2010050626A1 (en) | 2008-10-30 | 2010-05-06 | Kyoto University | Method for producing induced pluripotent stem cells |
| WO2010056831A2 (en) | 2008-11-12 | 2010-05-20 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through use of an hdac modulator |
| WO2010068955A2 (en) | 2008-12-13 | 2010-06-17 | Dna Microarray | MICROENVIRONMENT NICHE ASSAY FOR CiPS SCREENING |
| WO2010098419A1 (en) | 2009-02-27 | 2010-09-02 | Kyoto University | Novel nuclear reprogramming substance |
| WO2010102267A2 (en) | 2009-03-06 | 2010-09-10 | Ipierian, Inc. | Tgf-beta pathway inhibitors for enhancement of cellular reprogramming of human cells |
| WO2010111422A2 (en) | 2009-03-25 | 2010-09-30 | The Salk Institute For Biological Studies | Induced pluripotent stem cell generation using two factors and p53 inactivation |
| WO2010111409A2 (en) | 2009-03-25 | 2010-09-30 | The Salk Institute For Biological Studies | Pluripotent stem cells |
| WO2010115050A2 (en) | 2009-04-01 | 2010-10-07 | The Regents Of The University Of California | Embryonic stem cell specific micrornas promote induced pluripotency |
| WO2010124290A2 (en) | 2009-04-24 | 2010-10-28 | Whitehead Institute For Biomedical Research | Compositions and methods for deriving or culturing pluripotent cells |
| WO2010147612A1 (en) | 2009-06-18 | 2010-12-23 | Lixte Biotechnology, Inc. | Methods of modulating cell regulation by inhibiting p53 |
| WO2010147395A2 (en) | 2009-06-16 | 2010-12-23 | Korea Research Institute Of Bioscience And Biotechnology | Medium composition comprising neuropeptide y for the generation, maintenance, prologned undifferentiated growth of pluripotent stem cells and method of culturing pluripotent stem cell using the same |
| JP2011078370A (ja) | 2009-10-08 | 2011-04-21 | Osaka Univ | ヒト多能性幹細胞用培養基材およびその利用 |
| JP2011205964A (ja) | 2010-03-30 | 2011-10-20 | Japan Health Science Foundation | 腱及び/又は靭帯細胞の分化誘導剤 |
| JP2012143234A (ja) | 2007-10-12 | 2012-08-02 | National Institute Of Advanced Industrial Science & Technology | 高機能化キメラ蛋白質を含有する細胞増殖促進用培地 |
| JP2013231082A (ja) * | 2008-09-30 | 2013-11-14 | Oriental Yeast Co Ltd | 新しい軟骨細胞増殖及び分化誘導剤 |
| JP2014100141A (ja) | 2012-10-23 | 2014-06-05 | National Institute Of Advanced Industrial & Technology | 多能性幹細胞培養方法 |
| JP2015171345A (ja) * | 2014-03-12 | 2015-10-01 | 俊文 東 | 骨前駆細胞 |
| WO2016141084A1 (en) | 2015-03-03 | 2016-09-09 | The Board Of Trustees Of The Leland Stanford Junior University | Producing mesodermal cell types and methods of using the same |
| JP2017517259A (ja) * | 2014-05-22 | 2017-06-29 | フレッド ハッチンソン キャンサー リサーチ センター | Lilrb2およびnotchを介した造血前駆細胞の拡大培養 |
| JP2018029522A (ja) * | 2016-08-24 | 2018-03-01 | 国立大学法人 東京大学 | 軟骨細胞の製造方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3064577B1 (en) * | 2013-11-01 | 2020-09-09 | Kyoto University | Novel chondrocyte induction method |
-
2020
- 2020-04-28 JP JP2021520684A patent/JPWO2020235319A1/ja active Pending
- 2020-04-28 CA CA3141455A patent/CA3141455A1/en active Pending
- 2020-04-28 WO PCT/JP2020/018130 patent/WO2020235319A1/ja unknown
- 2020-04-28 EP EP20809188.4A patent/EP3974519A4/en active Pending
-
2021
- 2021-11-19 US US17/455,763 patent/US20220073882A1/en active Pending
Patent Citations (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001017562A1 (en) | 1999-09-02 | 2001-03-15 | Yamanouchi Pharmaceutical Co., Ltd. | Osteogenesis promoting agents |
| JP2005511083A (ja) | 2001-12-07 | 2005-04-28 | ジェロン コーポレイション | ヒト胚幹細胞由来の軟骨細胞前駆細胞 |
| JP2004254655A (ja) | 2003-02-27 | 2004-09-16 | Kanegafuchi Chem Ind Co Ltd | 軟骨の分化誘導方法 |
| JP2008514188A (ja) * | 2004-09-24 | 2008-05-08 | アンジオブラスト システムズ,インコーポレーテッド | 間葉系前駆細胞(mpc)の増殖および/または生存を増強させる方法 |
| WO2006123699A1 (ja) | 2005-05-17 | 2006-11-23 | Kyowa Hakko Kogyo Co., Ltd. | 幹細胞に対する骨芽細胞への分化促進剤 |
| WO2007069666A1 (ja) | 2005-12-13 | 2007-06-21 | Kyoto University | 核初期化因子 |
| WO2008118820A2 (en) | 2007-03-23 | 2008-10-02 | Wisconsin Alumni Research Foundation | Somatic cell reprogramming |
| WO2009007852A2 (en) | 2007-06-15 | 2009-01-15 | Izumi Bio, Inc | Multipotent/pluripotent cells and methods |
| WO2009032194A1 (en) | 2007-08-31 | 2009-03-12 | Whitehead Institute For Biomedical Research | Wnt pathway stimulation in reprogramming somatic cells |
| JP2012143234A (ja) | 2007-10-12 | 2012-08-02 | National Institute Of Advanced Industrial Science & Technology | 高機能化キメラ蛋白質を含有する細胞増殖促進用培地 |
| WO2009058413A1 (en) | 2007-10-29 | 2009-05-07 | Shi-Lung Lin | Generation of human embryonic stem-like cells using intronic rna |
| WO2009057831A1 (ja) | 2007-10-31 | 2009-05-07 | Kyoto University | 核初期化方法 |
| WO2009075119A1 (ja) | 2007-12-10 | 2009-06-18 | Kyoto University | 効率的な核初期化方法 |
| WO2009079007A1 (en) | 2007-12-17 | 2009-06-25 | Gliamed, Inc. | Stem-like cells and method for reprogramming adult mammalian somatic cells |
| WO2009091659A2 (en) | 2008-01-16 | 2009-07-23 | Shi-Lung Lin | Generation of tumor-free embryonic stem-like pluripotent cells using inducible recombinant rna agents |
| WO2009101407A2 (en) | 2008-02-11 | 2009-08-20 | Cambridge Enterprise Limited | Improved reprogramming of mammalian cells, and the cells obtained |
| WO2009101084A1 (en) | 2008-02-13 | 2009-08-20 | Fondazione Telethon | Method for reprogramming differentiated cells |
| WO2009102983A2 (en) | 2008-02-15 | 2009-08-20 | President And Fellows Of Harvard College | Efficient induction of pluripotent stem cells using small molecule compounds |
| WO2009117439A2 (en) | 2008-03-17 | 2009-09-24 | The Scripps Research Institute | Combined chemical and genetic approaches for generation of induced pluripotent stem cells |
| WO2009114949A1 (en) | 2008-03-20 | 2009-09-24 | UNIVERSITé LAVAL | Methods for deprogramming somatic cells and uses thereof |
| WO2009126251A2 (en) | 2008-04-07 | 2009-10-15 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through use of an hdac modulator |
| WO2009126655A2 (en) | 2008-04-07 | 2009-10-15 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through use of a small molecule modulator |
| WO2009126250A2 (en) | 2008-04-07 | 2009-10-15 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through rna interference |
| WO2009146408A1 (en) | 2008-05-30 | 2009-12-03 | Summa Health Systems Llc | Methods for using tgf-b receptor inhibitors or activin-like kinase (alk) 5 inhibitors a-83-01 and sb-431542 to treat eye disease and wound healing conditions |
| WO2009157593A1 (en) | 2008-06-27 | 2009-12-30 | Kyoto University | Method of efficiently establishing induced pluripotent stem cells |
| WO2010009015A2 (en) | 2008-07-14 | 2010-01-21 | Oklahoma Medical Research Foundation | Production of pluripotent cells through inhibition of bright/arid3a function |
| WO2010033906A2 (en) | 2008-09-19 | 2010-03-25 | President And Fellows Of Harvard College | Efficient induction of pluripotent stem cells using small molecule compounds |
| WO2010033920A2 (en) | 2008-09-19 | 2010-03-25 | Whitehead Institute For Biomedical Research | Compositions and methods for enhancing cell reprogramming |
| JP2013231082A (ja) * | 2008-09-30 | 2013-11-14 | Oriental Yeast Co Ltd | 新しい軟骨細胞増殖及び分化誘導剤 |
| WO2010042800A1 (en) | 2008-10-10 | 2010-04-15 | Nevada Cancer Institute | Methods of reprogramming somatic cells and methods of use for such cells |
| WO2010050626A1 (en) | 2008-10-30 | 2010-05-06 | Kyoto University | Method for producing induced pluripotent stem cells |
| WO2010056831A2 (en) | 2008-11-12 | 2010-05-20 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through use of an hdac modulator |
| WO2010068955A2 (en) | 2008-12-13 | 2010-06-17 | Dna Microarray | MICROENVIRONMENT NICHE ASSAY FOR CiPS SCREENING |
| WO2010098419A1 (en) | 2009-02-27 | 2010-09-02 | Kyoto University | Novel nuclear reprogramming substance |
| WO2010102267A2 (en) | 2009-03-06 | 2010-09-10 | Ipierian, Inc. | Tgf-beta pathway inhibitors for enhancement of cellular reprogramming of human cells |
| WO2010111409A2 (en) | 2009-03-25 | 2010-09-30 | The Salk Institute For Biological Studies | Pluripotent stem cells |
| WO2010111422A2 (en) | 2009-03-25 | 2010-09-30 | The Salk Institute For Biological Studies | Induced pluripotent stem cell generation using two factors and p53 inactivation |
| WO2010115050A2 (en) | 2009-04-01 | 2010-10-07 | The Regents Of The University Of California | Embryonic stem cell specific micrornas promote induced pluripotency |
| WO2010124290A2 (en) | 2009-04-24 | 2010-10-28 | Whitehead Institute For Biomedical Research | Compositions and methods for deriving or culturing pluripotent cells |
| WO2010147395A2 (en) | 2009-06-16 | 2010-12-23 | Korea Research Institute Of Bioscience And Biotechnology | Medium composition comprising neuropeptide y for the generation, maintenance, prologned undifferentiated growth of pluripotent stem cells and method of culturing pluripotent stem cell using the same |
| WO2010147612A1 (en) | 2009-06-18 | 2010-12-23 | Lixte Biotechnology, Inc. | Methods of modulating cell regulation by inhibiting p53 |
| JP2011078370A (ja) | 2009-10-08 | 2011-04-21 | Osaka Univ | ヒト多能性幹細胞用培養基材およびその利用 |
| JP2011205964A (ja) | 2010-03-30 | 2011-10-20 | Japan Health Science Foundation | 腱及び/又は靭帯細胞の分化誘導剤 |
| JP2014100141A (ja) | 2012-10-23 | 2014-06-05 | National Institute Of Advanced Industrial & Technology | 多能性幹細胞培養方法 |
| JP2015171345A (ja) * | 2014-03-12 | 2015-10-01 | 俊文 東 | 骨前駆細胞 |
| JP2017517259A (ja) * | 2014-05-22 | 2017-06-29 | フレッド ハッチンソン キャンサー リサーチ センター | Lilrb2およびnotchを介した造血前駆細胞の拡大培養 |
| WO2016141084A1 (en) | 2015-03-03 | 2016-09-09 | The Board Of Trustees Of The Leland Stanford Junior University | Producing mesodermal cell types and methods of using the same |
| JP2018029522A (ja) * | 2016-08-24 | 2018-03-01 | 国立大学法人 東京大学 | 軟骨細胞の製造方法 |
Non-Patent Citations (38)
| Title |
|---|
| "NCBI", Database accession no. NM_020997 |
| BIOCHEM. BIOPHYS. RES. COMMUN., vol. 157, 1988, pages 256 - 263 |
| CELL, vol. 131, no. 5, 2007, pages 861 - 872 |
| DEV. BIOL., vol. 138, 1990, pages 454 - 463 |
| EMINLI S ET AL., STEM CELLS., vol. 26, 2008, pages 2467 - 2474 |
| ENDOCRINE REV., vol. 8, 1987, pages 95 |
| FENG B ET AL., NAT CELL BIOL., vol. 11, 2009, pages 197 - 203 |
| HAN J ET AL., NATURE, vol. 463, 2010, pages 1096 - 100 |
| HENG JC ET AL., CELL STEM CELL., vol. 6, 2010, pages 167 - 74 |
| HU J. ET AL., TISSUE ENG. PART A, vol. 16, 2010, pages 3507 - 3514 |
| HUANGFU D ET AL., NAT BIOTECHNOL., vol. 26, no. 1, 2008, pages 1269 - 1275 |
| HUANGFU D ET AL., NAT. BIOTECHNOL., vol. 26, 2008, pages 795 - 797 |
| ICHIDA JK ET AL., CELL STEM CELL, vol. 5, 2009, pages 491 - 503 |
| J.A. THOMSON ET AL., BIOL. REPROD., vol. 55, 1996, pages 254 - 259 |
| J.A. THOMSON ET AL., PROC. NATL. ACAD. SCI. USA, vol. 92, 1995, pages 7844 - 7848 |
| J.A. THOMSON ET AL., SCIENCE, vol. 282, 1998, pages 1145 - 1147 |
| J.A. THOMSONV.S. MARSHALL, CURR. TOP. DEV. BIOL., vol. 38, 1998, pages 133 - 165 |
| JUDSON R.L. ET AL., NAT. BIOTECH., vol. 27, 2009, pages 459 - 461 |
| KIM JB ET AL., NATURE, vol. 461, 2009, pages 649 - 643 |
| LEE K.W. ET AL., STEM CELLS DEV., vol. 19, 2010, pages 557 - 568 |
| LOH K.M. ET AL., CELL, vol. 166, no. 2, 2016, pages 451 - 467 |
| LYSSIOTIS CA ET AL., PROC NATL ACAD SCI U S A., vol. 106, 2009, pages 8912 - 8917 |
| M.J. EVANSM.H. KAUFMAN, NATURE, vol. 292, 1981, pages 154 - 156 |
| MAEKAWA M ET AL., NATURE, vol. 474, 2011, pages 225 - 229 |
| MAHMOOD A. ET AL., J. BONE MINER. RES., vol. 25, 2010, pages 1216 - 1233 |
| MALI P ET AL., STEM CELLS, vol. 28, 2010, pages 713 - 720 |
| NAKAJIMA T. ET AL., DEVELOPMENT, vol. 145, no. 16, 2018, pages dev165431 |
| NAKAJIMA, TAIKI ET AL.: "Modeling human somite development and fibrodysplasia ossificans progressiva with induced pluripotent stem cells", DEVELOPMENT, vol. 145, no. dev165431, 2018, pages 1 - 14, XP055641586, DOI: 10.1242/dev.165431 * |
| NATURE METHODS, vol. 8, 2011, pages 424 - 429 |
| NISHIMURA, T. ET AL., CELL STEM CELL, vol. 12, 2013, pages 114 - 126 |
| PROC. NATL. ACAD. SCI. USA, vol. 81, 1984, pages 6963 |
| R.K. LINDEMANN ET AL., MOL. CANCER, vol. 2, 2003, pages 20 |
| SCIENCE, vol. 318, no. 5858, 2007, pages 1917 - 1920 |
| See also references of EP3974519A4 |
| YAMANAKA, CELL, vol. 126, no. 4, 2006, pages 663 - 676 |
| YAMASHITA A. ET AL., STEM CELL REPORTS, vol. 4, no. 3, 2015, pages 404 - 418 |
| ZHAO Y ET AL., CELL STEM CELL, vol. 3, 2008, pages 132 - 135 |
| ZOU L. ET AL., SCI. REP., vol. 3, 2013, pages 2243 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3974519A4 (en) | 2023-07-12 |
| EP3974519A1 (en) | 2022-03-30 |
| US20220073882A1 (en) | 2022-03-10 |
| JPWO2020235319A1 (ja) | 2020-11-26 |
| CA3141455A1 (en) | 2020-11-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2021104021A (ja) | 軟骨細胞系統細胞及び/又は軟骨様組織を作製するための方法及び組成物 | |
| CN110662832B (zh) | 由间介中胚层细胞分化诱导肾祖细胞的方法和由多能干细胞分化诱导肾祖细胞的方法 | |
| US10787640B2 (en) | Producing mesodermal cell types and methods of using the same | |
| EP3223831B1 (en) | Methods for generation of podocytes from pluripotent stem cells and cells produced by the same | |
| KR20180135482A (ko) | 망막 조직의 제조 방법 | |
| US10208287B2 (en) | CD82-positive cardiac progenitor cells | |
| AU2016320991A1 (en) | Method for producing retinal pigment epithelial cells | |
| JP7341433B2 (ja) | 腸管神経前駆細胞の製造方法 | |
| CN106536718B (zh) | 胰芽细胞的制造方法及含有胰芽细胞的胰疾病治疗剂 | |
| WO2017188458A1 (ja) | 骨格筋前駆細胞及び骨格筋細胞の製造方法 | |
| WO2023106122A1 (ja) | 間葉系譜への分化に特化した神経堤細胞の製造方法 | |
| US20210340498A1 (en) | Medium for direct differentiation of pluripotent stem cell-derived mesenchymal stem cell, method for preparing mesenchymal stem cell by using same, and mesenchymal stem cell prepared thereby | |
| Kumar et al. | Generation of an expandable intermediate mesoderm restricted progenitor cell line from human pluripotent stem cells | |
| US20220073882A1 (en) | Expansion culture method for cartilage or bone precursor cells | |
| KR20200091885A (ko) | 세포의 배양 방법 | |
| JP2017108705A (ja) | 心筋細胞の製造方法 | |
| JP2023165901A (ja) | 多能性幹細胞から各種細胞への段階的製造方法 | |
| WO2020100481A1 (ja) | 脳オルガノイドの製造方法 | |
| TW202113062A (zh) | 腎間質細胞的製造方法 | |
| WO2021187601A1 (ja) | 心筋細胞の精製方法 | |
| KR100937456B1 (ko) | 인간배아줄기세포를 조골세포 직계열로 분화시키는 방법 | |
| CN111918961B (zh) | 心肌细胞成熟促进剂 | |
| RU2813532C1 (ru) | Способ очистки кардиомиоцитов | |
| WO2022196613A1 (ja) | 椎板細胞の製造方法および該椎板細胞の利用 | |
| WO2023060322A1 (en) | Methods for producing cartilage and bones |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20809188 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021520684 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3141455 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020809188 Country of ref document: EP Effective date: 20211220 |