WO2018003694A1 - プリプレグおよびその製造方法 - Google Patents
プリプレグおよびその製造方法 Download PDFInfo
- Publication number
- WO2018003694A1 WO2018003694A1 PCT/JP2017/023199 JP2017023199W WO2018003694A1 WO 2018003694 A1 WO2018003694 A1 WO 2018003694A1 JP 2017023199 W JP2017023199 W JP 2017023199W WO 2018003694 A1 WO2018003694 A1 WO 2018003694A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- prepreg
- resin
- fiber
- thermosetting resin
- layer
- Prior art date
Links
Images


Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/02—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by structural features of a fibrous or filamentary layer
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C70/00—Shaping composites, i.e. plastics material comprising reinforcements, fillers or preformed parts, e.g. inserts
- B29C70/04—Shaping composites, i.e. plastics material comprising reinforcements, fillers or preformed parts, e.g. inserts comprising reinforcements only, e.g. self-reinforcing plastics
- B29C70/06—Fibrous reinforcements only
- B29C70/10—Fibrous reinforcements only characterised by the structure of fibrous reinforcements, e.g. hollow fibres
- B29C70/12—Fibrous reinforcements only characterised by the structure of fibrous reinforcements, e.g. hollow fibres using fibres of short length, e.g. in the form of a mat
- B29C70/14—Fibrous reinforcements only characterised by the structure of fibrous reinforcements, e.g. hollow fibres using fibres of short length, e.g. in the form of a mat oriented
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C70/00—Shaping composites, i.e. plastics material comprising reinforcements, fillers or preformed parts, e.g. inserts
- B29C70/04—Shaping composites, i.e. plastics material comprising reinforcements, fillers or preformed parts, e.g. inserts comprising reinforcements only, e.g. self-reinforcing plastics
- B29C70/06—Fibrous reinforcements only
- B29C70/10—Fibrous reinforcements only characterised by the structure of fibrous reinforcements, e.g. hollow fibres
- B29C70/16—Fibrous reinforcements only characterised by the structure of fibrous reinforcements, e.g. hollow fibres using fibres of substantial or continuous length
- B29C70/20—Fibrous reinforcements only characterised by the structure of fibrous reinforcements, e.g. hollow fibres using fibres of substantial or continuous length oriented in a single direction, e.g. roofing or other parallel fibres
- B29C70/205—Fibrous reinforcements only characterised by the structure of fibrous reinforcements, e.g. hollow fibres using fibres of substantial or continuous length oriented in a single direction, e.g. roofing or other parallel fibres the structure being shaped to form a three-dimensional configuration
- B29C70/207—Fibrous reinforcements only characterised by the structure of fibrous reinforcements, e.g. hollow fibres using fibres of substantial or continuous length oriented in a single direction, e.g. roofing or other parallel fibres the structure being shaped to form a three-dimensional configuration arranged in parallel planes of fibres crossing at substantial angles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C70/00—Shaping composites, i.e. plastics material comprising reinforcements, fillers or preformed parts, e.g. inserts
- B29C70/04—Shaping composites, i.e. plastics material comprising reinforcements, fillers or preformed parts, e.g. inserts comprising reinforcements only, e.g. self-reinforcing plastics
- B29C70/28—Shaping operations therefor
- B29C70/30—Shaping by lay-up, i.e. applying fibres, tape or broadsheet on a mould, former or core; Shaping by spray-up, i.e. spraying of fibres on a mould, former or core
- B29C70/304—In-plane lamination by juxtaposing or interleaving of plies, e.g. scarf joining
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C70/00—Shaping composites, i.e. plastics material comprising reinforcements, fillers or preformed parts, e.g. inserts
- B29C70/04—Shaping composites, i.e. plastics material comprising reinforcements, fillers or preformed parts, e.g. inserts comprising reinforcements only, e.g. self-reinforcing plastics
- B29C70/28—Shaping operations therefor
- B29C70/54—Component parts, details or accessories; Auxiliary operations, e.g. feeding or storage of prepregs or SMC after impregnation or during ageing
- B29C70/545—Perforating, cutting or machining during or after moulding
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/04—Layered products comprising a layer of synthetic resin as impregnant, bonding, or embedding substance
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/12—Layered products comprising a layer of synthetic resin next to a fibrous or filamentary layer
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/38—Layered products comprising a layer of synthetic resin comprising epoxy resins
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/42—Layered products comprising a layer of synthetic resin comprising condensation resins of aldehydes, e.g. with phenols, ureas or melamines
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/02—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by structural features of a fibrous or filamentary layer
- B32B5/12—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by structural features of a fibrous or filamentary layer characterised by the relative arrangement of fibres or filaments of different layers, e.g. the fibres or filaments being parallel or perpendicular to each other
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/22—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by the presence of two or more layers which are next to each other and are fibrous, filamentary, formed of particles or foamed
- B32B5/24—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by the presence of two or more layers which are next to each other and are fibrous, filamentary, formed of particles or foamed one layer being a fibrous or filamentary layer
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/22—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by the presence of two or more layers which are next to each other and are fibrous, filamentary, formed of particles or foamed
- B32B5/24—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by the presence of two or more layers which are next to each other and are fibrous, filamentary, formed of particles or foamed one layer being a fibrous or filamentary layer
- B32B5/26—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by the presence of two or more layers which are next to each other and are fibrous, filamentary, formed of particles or foamed one layer being a fibrous or filamentary layer another layer next to it also being fibrous or filamentary
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/04—Reinforcing macromolecular compounds with loose or coherent fibrous material
- C08J5/0405—Reinforcing macromolecular compounds with loose or coherent fibrous material with inorganic fibres
- C08J5/042—Reinforcing macromolecular compounds with loose or coherent fibrous material with inorganic fibres with carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/241—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres
- C08J5/243—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres using carbon fibres
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C2793/00—Shaping techniques involving a cutting or machining operation
- B29C2793/0081—Shaping techniques involving a cutting or machining operation before shaping
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2063/00—Use of EP, i.e. epoxy resins or derivatives thereof, as moulding material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2105/00—Condition, form or state of moulded material or of the material to be shaped
- B29K2105/06—Condition, form or state of moulded material or of the material to be shaped containing reinforcements, fillers or inserts
- B29K2105/08—Condition, form or state of moulded material or of the material to be shaped containing reinforcements, fillers or inserts of continuous length, e.g. cords, rovings, mats, fabrics, strands or yarns
- B29K2105/0872—Prepregs
- B29K2105/0881—Prepregs unidirectional
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2105/00—Condition, form or state of moulded material or of the material to be shaped
- B29K2105/25—Solid
- B29K2105/251—Particles, powder or granules
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2277/00—Use of PA, i.e. polyamides, e.g. polyesteramides or derivatives thereof, as reinforcement
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2307/00—Use of elements other than metals as reinforcement
- B29K2307/04—Carbon
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2250/00—Layers arrangement
- B32B2250/20—All layers being fibrous or filamentary
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2255/00—Coating on the layer surface
- B32B2255/02—Coating on the layer surface on fibrous or filamentary layer
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2255/00—Coating on the layer surface
- B32B2255/26—Polymeric coating
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2260/00—Layered product comprising an impregnated, embedded, or bonded layer wherein the layer comprises an impregnation, embedding, or binder material
- B32B2260/02—Composition of the impregnated, bonded or embedded layer
- B32B2260/021—Fibrous or filamentary layer
- B32B2260/023—Two or more layers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2260/00—Layered product comprising an impregnated, embedded, or bonded layer wherein the layer comprises an impregnation, embedding, or binder material
- B32B2260/04—Impregnation, embedding, or binder material
- B32B2260/046—Synthetic resin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2262/00—Composition or structural features of fibres which form a fibrous or filamentary layer or are present as additives
- B32B2262/02—Synthetic macromolecular fibres
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2262/00—Composition or structural features of fibres which form a fibrous or filamentary layer or are present as additives
- B32B2262/10—Inorganic fibres
- B32B2262/106—Carbon fibres, e.g. graphite fibres
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2270/00—Resin or rubber layer containing a blend of at least two different polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/50—Properties of the layers or laminate having particular mechanical properties
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/50—Properties of the layers or laminate having particular mechanical properties
- B32B2307/558—Impact strength, toughness
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/70—Other properties
- B32B2307/746—Slipping, anti-blocking, low friction
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2605/00—Vehicles
- B32B2605/18—Aircraft
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2300/00—Characterised by the use of unspecified polymers
- C08J2300/22—Thermoplastic resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2300/00—Characterised by the use of unspecified polymers
- C08J2300/24—Thermosetting resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2363/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
- C08J2363/04—Epoxynovolacs
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2377/00—Characterised by the use of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2381/00—Characterised by the use of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing sulfur with or without nitrogen, oxygen, or carbon only; Polysulfones; Derivatives of such polymers
- C08J2381/06—Polysulfones; Polyethersulfones
Definitions
- the present invention relates to a prepreg for obtaining a carbon fiber reinforced plastic and a method for producing the same.
- Carbon fiber reinforced plastics are attracting attention in industrial applications because of their high specific strength and specific rigidity, excellent mechanical properties, and high functional properties such as weather resistance and chemical resistance.
- it is deployed in applications such as aircraft, spacecraft, automobiles, railways, ships, and sports, and the demand is increasing year by year.
- a cured prepreg laminate is often used for structural members that require mechanical properties.
- prepregs in which carbon fibers are aligned in one direction have a high fiber volume content, and the high fiber elastic modulus and strength of carbon fibers can be utilized to the maximum.
- impregnating the prepreg with a high-functional resin so as to reduce the variation in weight per unit area, the quality of the obtained carbon fiber reinforced plastic is stabilized, so that the material has high mechanical characteristics and reliability.
- the shaping process is the key to determine the quality and productivity of the parts.
- the shaping step is a step of making a prepreg follow a three-dimensional shape to form a preform before the molding and hardening step using an autoclave or the like. If the prepreg is shaped one layer at a time in the shaping step, a high-quality preform can be obtained, but the process time is long and the productivity is poor. Therefore, in order to increase productivity, hot forming is performed in which a prepreg is laminated on a flat plate at high speed in advance using an automatic machine to form a prepreg laminate, and then the prepreg laminate is shaped into a three-dimensional shape while heat is applied.
- the shaping method called is being developed. In the shaping method of patent document 1, shape tracking is enabled by sliding between layers together with bending deformation of each layer of the prepreg laminate.
- the object of the present invention is to provide a prepreg having excellent mechanical properties when the prepreg laminate is made to follow a three-dimensional shape and has excellent formability and a carbon fiber reinforced plastic. Is to provide.
- the present invention provides the following prepreg. That is, a prepreg in which a resin layer containing a thermosetting resin and a thermoplastic resin is present on at least one surface of a fiber layer containing discontinuous carbon fibers oriented in one direction and a thermosetting resin, the prepreg
- the fiber mass of the carbon fiber contained is 120 to 300 g / m 2
- the mass content of the resin with respect to the total mass of the prepreg is 25 to 50%
- the interlayer friction coefficient of the contact surface was measured in increments of 10 ° C. in a temperature range of 40 to 80 ° C.
- the prepreg has a temperature at which the interlaminar friction coefficient is 0.05 or less within a temperature range of 40 to 80 ° C.
- a wrinkle-free preform can be produced in a hot forming process in which a flat prepreg laminate follows a three-dimensional shape, and good mechanical properties are exhibited when a carbon fiber reinforced plastic is used.
- a prepreg can be obtained.
- the present inventors can produce a wrinkle-free preform in a hot forming process in which a prepreg laminate in which a plurality of prepregs composed of a thermosetting resin and carbon fiber are laminated follows a three-dimensional shape, and carbon fiber.
- a prepreg laminate in which a plurality of prepregs composed of a thermosetting resin and carbon fiber are laminated follows a three-dimensional shape, and carbon fiber.
- the carbon fibers are discontinuous carbon fibers oriented in one direction, and the thermosetting resin and the thermoplastic resin for increasing toughness are provided on at least one side of the fiber layer containing the carbon fibers and the thermosetting resin.
- interlayer friction coefficient the friction coefficient (hereinafter referred to as interlayer friction coefficient) of the surfaces where the prepregs are in contact with each other is reduced. Investigated that the problem can be solved.
- the fiber layer in the prepreg of the present invention is a layer containing discontinuous carbon fibers arranged in one direction and a thermosetting resin.
- carbon fibers aligned in one direction are continuous in the prepreg, when the prepreg is bent, the prepreg stretches on the tension side of the neutral axis of the bending, causing wrinkles on the compression side of the neutral axis of the bending. It becomes easy to do.
- the discontinuous carbon fibers it is possible to suppress such a tension, thereby suppressing the generation of wrinkles when the prepreg is bent and shaped.
- the mass ratio of the discontinuous carbon fibers in the carbon fibers constituting the fiber layer is not particularly limited, but the proportion of the mass of the discontinuous carbon fibers in the mass of the entire carbon fibers constituting the fiber layer is 50% or more. If so, it is preferable because the tension of the base material can be effectively suppressed. More preferably, it is 70 mass% or more, More preferably, it is 100 mass%. Different types of carbon fibers may be used for discontinuous carbon fibers and continuous fibers.
- “arranged in one direction” means that the number of carbon fibers within the range of ⁇ 10 ° in the direction with an angle formed in the prepreg surface is 90% or more among the carbon fibers present in the prepreg. Indicates that there is a certain direction. More preferably, it indicates that there is a direction in which the number of carbon fibers having the angle within a range of ⁇ 5 ° is 90% or more. Such a direction is called a fiber direction.
- the longitudinal direction of the prepreg is the fiber direction unless otherwise specified.
- the discontinuous carbon fiber refers to a carbon fiber having a finite fiber length in the prepreg, that is, a carbon fiber having a fiber length shorter than the total length of the prepreg in the fiber direction.
- the fiber length of the discontinuous carbon fiber is not particularly limited, and is preferably determined from the balance of mechanical properties and shape complexity required for a carbon fiber reinforced plastic produced using a prepreg.
- the fiber length is short, the tension of the fiber on the tension side of bending can be suppressed even at the corners with a small radius of curvature, and while the shapeability is improved, the mechanical properties of carbon fiber reinforced plastics are reduced.
- the fiber length is long, the bending property of the fiber on the tension side of bending is reduced even at the corners with a small radius of curvature, but the formability decreases, but the mechanical properties of carbon fiber reinforced plastics are improved. .
- the preferred range of the fiber length is 5 to 100 mm, more preferably 10 to 50 mm.
- carbon fibers having different fiber lengths may be mixed, it is preferable that all the carbon fibers have substantially the same length in consideration of the stability of the quality of the prepreg.
- substantially the same length means that 90% or more of the number of carbon fibers is a fiber length within a range of an average length of all carbon fibers ⁇ 10%.
- Substantially all of the carbon fibers contained in the fiber layer may be discontinuous, or cuts may be inserted only in the carbon fibers in the region used for shaping in the prepreg. From the viewpoint of formability, it is particularly preferred that substantially all the carbon fibers in the fiber layer are discontinuous.
- substantially all the carbon fibers in the fiber layer being discontinuous means that the number of carbon fibers that are not discontinuous among the carbon fibers constituting the fiber layer is 5% or less.
- the method for producing a fiber layer containing discontinuous carbon fibers oriented in one direction is not particularly limited. After producing discontinuous carbon fiber first, it may be produced by compounding with a thermosetting resin, or after producing a fiber layer containing continuous carbon fiber, the carbon fiber is discontinuously produced. You may manufacture by performing the process to do.
- a method of producing a discontinuous carbon fiber first a method of winding a carbon fiber with a roll having a different speed, cutting a part of the carbon fiber by the speed difference, a method of arranging short tows in one direction, An example is a method of arranging discontinuous carbon fibers and arranging them in one direction.
- An example is a method of discontinuously processing carbon fibers.
- a method of discontinuously processing carbon fibers by inserting cuts into a fiber layer containing continuous carbon fibers, a fiber layer with excellent surface smoothness can be obtained, and the effect of the barrier layer described later This is preferable because the interlayer slip of the prepreg becomes good due to the synergistic effect.
- a rotary blade, a laser, a punching die, or the like can be used for cutting the carbon fiber.
- Discontinuous carbon with controlled orientation of carbon fibers and distance between discontinuous carbon fibers by inserting cuts into continuous fibers in a fiber layer containing continuous carbon fibers oriented in one direction.
- a fiber layer containing fibers can be obtained. Thereby, the strength reduction due to the deviation of the fiber bundle can be suppressed.
- ⁇ A cut may or may not be inserted in the resin layer.
- an effect of easily discharging the air inside the laminated body can be expected by evacuating the laminated body when producing a laminated body in which a plurality of prepregs are laminated.
- the formability is not inferior to the case where the notch penetrating the resin layer is inserted.
- the length of the cut there is no particular limitation on the length of the cut, but intermittent cut is preferable.
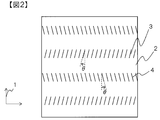
- the intermittent cut is, for example, as shown in FIG. 1, that the cut length l is finite in the prepreg 2, that is, the cut depth is larger than the total length of the prepreg in the fiber direction. It means that the length l is short.
- the cut is intermittent as shown in FIG. Even if it exists, substantially all the carbon fibers can be made discontinuous.
- the cutting angle is not particularly limited, but it is preferable that the cutting is inserted obliquely with respect to the fiber direction. Thereby, the three-dimensional shape followability of the prepreg and the mechanical properties of the carbon fiber reinforced plastic can be further improved.
- the absolute value of ⁇ is preferably 2 to 60 °.
- the absolute value of ⁇ is preferably 25 ° or less, since the mechanical properties, particularly the tensile strength, is remarkably improved.
- the absolute value of ⁇ is smaller than 2 °, it becomes difficult to insert the cut stably.
- the absolute value of ⁇ is preferably 2 ° or more.
- the absolute value of ⁇ is substantially the same, and further includes both a positive cut in which ⁇ is a positive cut and a negative cut in which ⁇ is a negative cut.
- FIG. 2 A conceptual diagram of such a cutting pattern is shown in FIG.
- the carbon fibers are oriented in the fiber direction 1 direction of the prepreg 2.
- the carbon fiber is divided by the positive cut 3 and the negative cut 4 and is discontinuous.
- the normal cut 3 here refers to a cut in which the fiber direction 1 is 0 ° and the cut angle ⁇ is clockwise within the range of 0 ° ⁇ ⁇ 90 °.
- the negative cut 4 indicates a cut in which the fiber direction 1 is 0 ° and the cut angle ⁇ is in the range of 0 ° ⁇ ⁇ 90 ° counterclockwise as shown in FIG. That the absolute value of ⁇ is substantially the same means that the absolute value of ⁇ is within an average value ⁇ 1 ° of the absolute value of ⁇ in all the cuts.
- substantially the same number of positive cuts and negative cuts are included.
- the inclusion of substantially the same number of positive cuts and negative cuts means that the number of cuts for which ⁇ is positive and the number of cuts for which ⁇ is negative are both 45% or more and 55 when expressed as a percentage based on the number. % Or less.
- the prepreg has a cutting pattern in which the absolute value of ⁇ formed by the cutting and the orientation direction of the carbon fiber is substantially the same, and the positive cutting and the negative cutting are substantially the same number, the cutting direction It becomes possible to laminate without worrying about the above.
- thermosetting resin used in the fiber layer is not particularly limited as long as the resin causes a crosslinking reaction by heat to form at least a partial three-dimensional crosslinked structure.
- thermosetting resins include unsaturated polyester resins, vinyl ester resins, epoxy resins, benzoxazine resins, phenol resins, urea resins, melamine resins, and polyimide resins. Modified resins of these resins and resins of two or more types can also be used. Further, these thermosetting resins may be resins that are self-curing by heat, or may be blended with a curing agent, a curing accelerator, or the like. The filler for improving electroconductivity and heat resistance may be blended.
- thermosetting resins epoxy resins are preferably used because of their excellent balance of heat resistance, mechanical properties, and adhesion to carbon fibers.
- an epoxy resin having an amino group or phenol-derived structure is preferably used.
- an aminophenol type epoxy resin a glycidyl aniline type epoxy resin and a tetraglycidyl amine type epoxy resin are preferably used.
- the glycidylamine type epoxy resin include tetraglycidyldiaminodiphenyl, triglycidyl-p-aminophenol, and triglycidylaminocreosol.
- High-purity tetraglycidylamine-type epoxy resin average epoxide equivalent (EEW) in the range of 100-115
- tetraglycidylamine-type epoxy resin high-purity aminophenol-type epoxy resin, in the range of 90-104 amino
- a phenol type epoxy resin is preferably used for suppressing volatile components that may cause voids in the resulting carbon fiber reinforced plastic.
- Tetraglycidyldiaminodiphenylmethane has excellent heat resistance and is preferably used as a composite material resin for aircraft structural members.
- a glycidyl ether type epoxy resin having a phenol-derived structure is also preferably used as the thermosetting resin.
- epoxy resins include bisphenol A type epoxy resins, bisphenol F type epoxy resins, bisphenol S type epoxy resins, phenol novolac type epoxy resins, creosole novolac type epoxy resins, and resorcinol type epoxy resins.
- Liquid bisphenol A type epoxy resin, bisphenol F type epoxy resin and resorcinol type epoxy resin are preferably used in combination with other epoxy resins because of low viscosity.
- a bisphenol A type epoxy resin that is solid at room temperature has a structure in which the crosslinking density in the cured resin is lower than that of a liquid bisphenol A type epoxy resin that is liquid at room temperature (about 25 ° C.).
- the heat resistance is lower, but the toughness is higher. Therefore, it is preferable to use in combination with a glycidylamine type epoxy resin, a liquid bisphenol A type epoxy resin or a bisphenol F type epoxy resin.
- an epoxy resin having a naphthalene skeleton is a cured resin having low absorbency and high heat resistance.
- biphenyl type epoxy resins, dicyclopentadiene type epoxy resins, phenol aralkyl type epoxy resins, and phenyl fluorine type epoxy resins can be preferably used because they are cured resins having low absorbability.
- urethane-modified epoxy resins and isocyanate-modified epoxy resins can be preferably used because they become cured resins having high fracture toughness and elongation.
- epoxy resins may be used alone or may be blended as appropriate.
- an epoxy resin having a bifunctional, trifunctional or higher functional group is added to the resin composition, the resulting prepreg satisfies both workability and processability and heat resistance under wet conditions as a fiber-reinforced composite.
- This is preferable because it is possible.
- the combination of a glycidylamine type epoxy resin and a glycidyl ether type epoxy resin can achieve processability, heat resistance and water resistance.
- blending at least one epoxy resin that is liquid at room temperature and at least one epoxy resin that is solid at room temperature is effective in providing both tackiness and draping properties suitable for the prepreg.
- the phenol novolac type epoxy resin and the creosole novolac type epoxy resin have high heat resistance and low absorbability, and thus become a cured resin having high heat resistance and water resistance.
- the tackiness and draping properties of the prepreg can be adjusted while improving the heat and water resistance.
- the curing agent for the epoxy resin may be any compound having an active group capable of reacting with an epoxy group.
- compounds having an amino group, an acid anhydride group or an azide group are suitable as the curing agent.
- More specific examples of curing agents include isomers of dicyandiamide, diaminodiphenylmethane and diaminodiphenylsulfone; aminobenzoic acid esters, various acid anhydrides, phenol novolac resins, cresol novolac resins, polyphenol compounds, imidazole derivatives, fats Group amine, tetramethylguanidine, thiourea addition amine, methylhexahydrophthalic anhydride, other carboxylic anhydride, carboxylic hydrazide, carboxylic amide, polymercaptan, boron trifluoride ethylamine complex and other Lewis acid complexes Etc.
- These curing agents can be used alone or in combination.
- an aromatic diamine as a curing agent, a cured resin having good heat resistance can be obtained.
- various isomers of diaminodiphenylsulfone are most preferable because a cured resin having good heat resistance can be obtained.
- the addition amount of the aromatic diamine curing agent is preferably stoichiometrically equivalent, but in some cases, by using about 0.7 to 0.9 equivalent to the epoxy resin, a high elastic modulus can be obtained. A cured resin can be obtained.
- imidazole or dicyandiamide and a urea compound for example, 3-phenol-1,1-dimethylurea, 3- (3-chlorophenyl) -1,1-dimethylurea, 3- (3,4-dichlorophenyl) -1, 1-dimethylurea, 2,4-toluenebisdimethylurea, and 2,6-toluenebisdimethylurea
- a urea compound for example, 3-phenol-1,1-dimethylurea, 3- (3-chlorophenyl) -1,1-dimethylurea, 3- (3,4-dichlorophenyl) -1, 1-dimethylurea, 2,4-toluenebisdimethylurea, and 2,6-toluenebisdimethylurea
- the storage stability of the prepreg can be increased by using a substance having the possibility of forming one of these curing agents, for example, a microencapsulated substance.
- a substance having the possibility of forming one of these curing agents for example, a microencapsulated substance.
- tackiness and draping properties hardly change even when left at room temperature.
- composition products obtained by partially prereacting these epoxy resins or curing agents, or both are also possible to add to the composition products obtained by partially prereacting these epoxy resins or curing agents, or both. In some cases, this method is effective for viscosity adjustment and storage stability improvement.
- Thermoplastic resin may be blended with thermosetting resin and dissolved.
- a thermoplastic resin is usually a heat having a bond selected from a carbon-carbon bond, an amide bond, an imide bond, an ester bond, an ether bond, a carbonate bond, a urethane bond, a thioether bond, a sulfone bond and a carbonyl bond.
- a plastic resin it may have a partially crosslinked structure.
- thermoplastic resin may or may not have crystallinity.
- at least one resin selected from the group consisting of polyaramid, polyether nitrile, and polybenzimidazole is blended and dissolved in the thermosetting resin.
- thermoplastic resins may be commercially available polymers or so-called oligomers having a molecular weight lower than that of commercially available polymers.
- oligomer an oligomer having a functional group capable of reacting with a thermosetting resin at a terminal or in a molecular chain is preferable.
- thermosetting resin When using a blend of a thermosetting resin and a thermoplastic resin, the brittleness of the thermosetting resin can be covered with the toughness of the thermoplastic resin compared to the case of using only one of them. Since the difficulty of molding can be covered with a thermosetting resin, a balanced main agent can be obtained.
- the mass ratio of the thermosetting resin to the thermoplastic resin is preferably in the range of 100: 2 to 100: 50, and more preferably in the range of 100: 5 to 100: 35 in terms of balance.
- any type of carbon fiber can be used regardless of whether it is a polyacrylonitrile-based carbon fiber or a pitch-based carbon fiber, depending on the application.
- carbon fibers having a tensile elastic modulus of 230 to 400 GPa are preferred.
- a carbon fiber reinforced plastic having high rigidity and mechanical strength can be obtained, and therefore carbon fibers having a tensile strength of 4.4 to 7.0 GPa are preferably used.
- the tensile elongation is an important factor, and carbon fibers having a tensile elongation of 1.7 to 2.3% are preferable. Accordingly, carbon fibers having the characteristics that the tensile modulus is at least 230 GPa, the tensile strength is at least 4.4 GPa, and the tensile elongation is at least 1.7% are most suitable.
- Examples of commercially available carbon fibers that can be preferably used include “Torayca (registered trademark)" T1100G-24K, “Torayca (registered trademark)” T1100G-12K, “Torayca (registered trademark)” T800S-24K, and “Torayca (registered trademark)” “T800S-12K", “Torayca (registered trademark)” T300-3K, and “Torayca (registered trademark)” T700S-12K (manufactured by Toray Industries, Inc.).
- the fiber mass of the carbon fiber contained in the prepreg of the present invention is 120 to 300 g / m 2 , more preferably 140 to 280 g / m 2 .
- the fiber mass is the mass of carbon fibers contained per unit area of the prepreg.
- the fiber mass is smaller than 120 g / m 2 , there is a problem that the number of prepreg layers required for obtaining a desired carbon fiber reinforced plastic thickness increases, and the number of man-hours for production increases.
- the fiber mass is larger than 300 g / m 2 , the resin is hardly impregnated in the fiber, and when the carbon fiber reinforced plastic is used, the non-impregnated portion remains as a void, which may lead to a decrease in physical properties.
- the prepreg of the present invention has a resin mass content of 25 to 50%, more preferably 30 to 40%, based on the total mass of the prepreg.
- the mass content of the resin is the ratio of the mass of all resin components excluding carbon fibers to the total mass of the prepreg.
- the mass content of the resin is larger than 50%, the carbon fiber content decreases, and when the carbon fiber reinforced plastic is used, the strength and elastic modulus are decreased.
- the resin mass content is less than 25%, particularly in the configuration of the present invention in which the resin layer is provided on the surface of the prepreg, the amount of the resin in the fiber layer decreases, and the fiber surface cannot be completely covered with the resin. , Cracks are likely to occur between the fibers, causing unexpected breakage and increasing quality variations.
- the resin layer includes a thermosetting resin and a thermoplastic resin.
- a thermosetting resin the resin seed
- the thermosetting resin of the resin layer may be the same as the thermosetting resin of the fiber layer or may be different from the thermosetting resin of the fiber layer.
- the resin layer contains a thermoplastic resin from the viewpoint of mechanical properties of the obtained carbon fiber reinforced plastic.
- a thermoplastic resin from the viewpoint of mechanical properties of the obtained carbon fiber reinforced plastic.
- Carbon fiber reinforced plastics that have cured prepreg laminates are prone to breakage between layers when impact is applied, so the thermoplastic resin is included in the resin layer and the toughness between the layers is improved. Excellent carbon fiber reinforced plastic can be obtained.
- the type of the thermoplastic resin is not particularly limited, and may be the same type as the various thermoplastic resins exemplified above.
- the resin layer may be disposed only on one side of the fiber layer or on both sides.
- the arrangement on both sides is preferable because the mechanical properties are particularly improved.
- a resin layer is arrange
- the resin layer is a resin layer containing a solid thermoplastic resin soluble in a thermosetting resin.
- the solid thermoplastic resin soluble in the thermosetting resin has a clear boundary with the thermosetting resin at a temperature of 40 to 80 ° C., which is the temperature of the shaping process, and is a heat that disperses the resin.
- thermosetting It means a thermoplastic resin having the property of being dissolved in the resin.
- the clear boundary means that when the cross section of the prepreg is observed with an optical microscope, the interface between the solid thermoplastic resin and the surrounding thermosetting resin can be clearly seen.
- the solid thermoplastic resin does not dissolve, so that a large amount of thermoplastic resin can be supplied to the resin layer. Therefore, the toughness of the resin layer existing between the layers can be further improved after molding. Is possible.
- the solid thermoplastic resin soluble in the thermosetting resin may be the same as the various thermoplastic resins exemplified above.
- polyethersulfone is preferable because it greatly improves impact resistance due to excellent toughness.
- the form of the solid thermoplastic resin soluble in the thermosetting resin may be a non-woven fabric or fiber, but particles are preferred in order to obtain better moldability. Since the solid thermoplastic resin is in the form of particles, when the layers slide, the positional relationship between the particles can be changed, so that the interlayer friction coefficient can be reduced as compared with the non-woven fabric or fiber form. .
- the shape of the particles may be any of spherical, non-spherical, porous, needle-like, whisker-like, and flake-like, but a spherical shape having a small contact area between the particles is particularly preferable. In the case of a spherical shape, the sphericity is preferably 90-100.
- thermoplastic resin insoluble in the thermosetting resin means that the temperature of the thermosetting resin in which the thermoplastic resin is dispersed is increased to 180 ° C. at a temperature increase rate of 1.5 ° C./min in an autoclave. It means a thermoplastic resin that does not dissolve in the thermosetting resin when cured by heating and pressing at 180 ° C. and a pressure of 7 kg / cm 2 for 2 hours.
- thermoplastic resin insoluble in the thermosetting resin a thermoplastic resin having a glass transition temperature in the range of 80 ° C. to 180 ° C. is preferable.
- thermoplastic resin having such a relatively high glass transition temperature does not deform in the form of heat curing, so a stable interlayer thickness is formed in a carbon fiber reinforced plastic obtained by curing a prepreg laminate.
- a carbon fiber reinforced plastic having excellent interlaminar toughness and high compressive strength during wet heat can be obtained.
- the glass transition temperature of the thermoplastic resin is less than 80 ° C.
- the carbon fiber reinforced plastic is inferior in the balance between interlayer toughness and compressive strength during wet heat.
- the glass transition temperature of the thermoplastic resin exceeds 180 ° C., the toughness of the thermoplastic resin itself tends to decrease, and the interfacial adhesiveness between the thermoplastic resin and the matrix resin becomes low, and the carbon fiber having low interlayer toughness. It becomes reinforced plastic.
- thermoplastic resin insoluble in the thermosetting resin may be the same type as the various thermoplastic resins exemplified above. Of these, polyamide is most preferred because it greatly improves impact resistance due to excellent toughness.
- polyamides polyamide 12, polyamide 6, polyamide 66, polyamide 11, polyamide 6/12 copolymer and epoxy compound described in Example 1 of JP-A-1-104624 and semi-IPN (polymer interpenetrating network)
- the (structured) polyamide (semi-IPN polyamide) has particularly good adhesive strength with the thermosetting resin. Accordingly, the delamination strength when the carbon fiber reinforced plastic is used is increased, and the impact resistance is increased, which is preferable.
- the resin layer containing a thermoplastic resin that is insoluble in the thermosetting resin may further contain a thermoplastic resin that is soluble in the thermosetting resin.
- the toughness between layers is further improved by the presence of a thermoplastic resin that is insoluble in the thermosetting resin in the resin layer in which the thermoplastic resin soluble in the thermosetting resin and the thermosetting resin are dissolved. be able to.
- the form of the thermoplastic resin insoluble in the thermosetting resin may be a nonwoven fabric or a fiber, but particles are preferable in order to obtain better moldability. Since the thermoplastic resin is in the form of particles, when the interlayer of the prepreg slides, the positional relationship between the particles can be changed, so that the interlayer friction coefficient can be reduced as compared with the non-woven fabric or fiber form. it can.
- the shape of the particles may be any of spherical, non-spherical, porous, needle-like, whisker-like, and flake-like, but a spherical shape having a small contact area between the particles is particularly preferable. In the case of a spherical shape, the sphericity is preferably 90-100. In the case where a soluble thermoplastic resin and an insoluble thermoplastic resin are present, it is preferable that both particles are particles because they contribute to a reduction in frictional resistance.
- thermoplastic resin whether soluble or insoluble in the thermosetting resin
- sphericity of the thermoplastic resin is measured by the following procedure. First, particles are photographed with a scanning electron microscope at a magnification of 1000 times, and the minor axis and major axis of 30 particles arbitrarily selected on the photographed image are measured. Next, the minor axis / major axis of each particle is calculated, and the average value ⁇ 100 of the minor axis / major axis of 30 particles is defined as sphericity (%).
- the inter-layer friction coefficient of the contact surface between the prepregs when the center prepreg is drawn out the drawing speed 0.2 mm / min, the vertical stress 0.08 MPa, the drawing length 1 mm.
- the temperature at which the interlayer friction coefficient is 0.05 or less exists within the temperature range of 40 to 80 ° C.
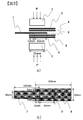
- Interlayer friction coefficient refers to the friction coefficient generated between prepreg layers in a prepreg laminate in which prepregs are laminated. As shown in FIG.
- one prepreg 7 is sandwiched between two prepregs 8, and a predetermined load P (vertical load) is applied to the prepreg vertically from the outside of the prepreg surface using the pressure plate 5.
- a predetermined load P vertical load
- a value obtained by dividing the load obtained when pulling out the sandwiched prepreg 7 by twice the vertical load applied to the overlap portion among the vertical loads is defined as an interlayer friction coefficient.
- the reason for dividing by twice the vertical load is that there are two prepreg surfaces subject to frictional resistance.
- the prepreg is cut out so as to be long in the fiber direction, and three prepregs are laminated in the same fiber direction so that the prepreg 7 and the prepreg 8 overlap in a range of 30 mm in width and 15 mm in length.
- the spacer 9 which cut the prepreg of the same fiber direction of width 30mm so that it may touch the overlap part of the center prepreg 7 is installed. As the prepreg is pulled out, the area of the overlap portion is reduced, and the area where pressure is applied by the pressure plate 1 is biased. Therefore, there is a possibility that the pressure plate 1 hits one side and a high load is locally applied. 9 is arranged so that the pressure plate 5 does not tilt. During the test, a constant vertical load of 168 N was applied to the range where the overlap part and spacer are pressed by the pressure plate 5 (range of width 30 mm, length 70 mm) to a predetermined temperature with the pressure plate 5 having a heating source. to continue.
- this vertical load When this vertical load is converted into a vertical stress, it becomes 0.08 MPa.
- the pulling load is measured while pulling out the central prepreg layer 7 in the fiber direction at a pulling rate of 0.2 mm / min.
- a value obtained by dividing the pulling load by twice the vertical load (36 N at the start of the test) applied to the overlap portion (the range of 30 mm width and 15 mm at the start of the test) is calculated as the interlayer friction coefficient.
- the area of the overlap portion where the central prepreg layer receives a vertical load is reduced along with the drawing, the area of the overlap portion (width 30 mm, length 15 mm ⁇ drawing length range) appropriately converted to the drawing length
- the vertical load applied to the overlap part is calculated proportionally assuming that the area received by the spacer (the range of 30 mm in width and 55 mm in length) is received as 168 N, and it is pulled out at twice the vertical load.
- the load divided is the inter-layer friction coefficient.
- the interlayer friction coefficient changes not only with temperature but also with drawing speed, normal stress, and time.
- the drawing rate is 0.2 mm / min
- the vertical stress is 0.08 MPa
- the interlayer friction coefficient at a drawing length of 1 mm is measured. The measurement is performed 5 times, and the average value is taken as the interlayer friction coefficient.
- the temperature at which the interlayer friction coefficient is 0.05 or less exists in the temperature range of 40 to 80 ° C. in the measurement of the interlayer friction coefficient.
- a temperature at which the interlayer friction coefficient is preferably 0.04 or less, more preferably 0.03 or less, and particularly preferably 0.02 or less is present at 40 to 80 ° C. More preferably, in the measurement of the interlayer friction coefficient, the temperature at which the interlayer friction coefficient falls within the above range is in the temperature range of 50 to 80 ° C.
- the prepreg laminate is made to follow the three-dimensional shape in the temperature range where the curing reaction does not start, that is, about 80 ° C or less. Even if the shaping is performed at a temperature at which the interlayer friction coefficient is minimum, the interlayer is difficult to slip and wrinkles may occur.
- the temperature range where the interlayer friction coefficient is 0.05 or less exists in the temperature range of 20 ° C. or more in the temperature range of 40 to 80 ° C.
- temperature distribution often occurs in the prepreg laminate depending on the temperature control conditions. Since the temperature region where the interlayer friction coefficient is 0.05 or less exists in the temperature region having a width of 20 ° C. or more, even if there is uneven temperature of the prepreg, the amount of interlayer slip of the prepreg can be easily increased. It becomes a prepreg suitable for larger-scale shaping.
- the temperature at which the interlaminar friction coefficient is 0.04 or less, more preferably 0.03 or less, and particularly preferably 0.02 or less is present in a temperature range having a width of 20 ° C. or more.
- the interlayer friction coefficient is set in a temperature range of 40 to 80 ° C. under conditions of a drawing speed of 0.2 mm / min, a normal stress of 0.08 MPa, a drawing length of 1 mm, and a drawing length of 2 mm.
- the rate of increase of the interlayer friction coefficient at the drawing length of 2 mm with respect to the interlayer friction coefficient at the drawing length of 1 mm within 10 ° C. above and below the temperature at which the interlayer friction coefficient becomes the minimum at the drawing length of 1 mm when measured in increments of 10 ° C. Is a prepreg having a temperature within 40%.
- there is a temperature at which the rate of increase is within 20%.
- the temperature region in which the rate of increase is within 40% should be present in a range of 20 ° C. or more, and more preferably, the temperature region in which the rate of increase is within 20% is present in a range of 20 ° C. or more. It is good.
- the rate of increase (%) is calculated as ⁇ (interlayer friction coefficient at 2 mm pulling length) ⁇ (interlayer friction coefficient at 1 mm pulling length) ⁇ / (interlayer friction coefficient at 1 mm pulling length) ⁇ 100. Is the value to be
- a laminate obtained by laminating and forming a prepreg in a pseudo isotropic manner and curing is processed into a flat test piece defined by ASTM D7137 / 7137M-07.
- the prepreg has a compression strength after impact (CAI) of 250 MPa or more of the laminate measured according to ASTM D7137 / 7137M-07.
- CAI compression strength after impact
- the compressive strength after impact is preferably 300 MPa or more, and more preferably 350 MPa or more. However, the compressive strength after impact that is practically realizable is 450 MPa or less.
- the falling weight impact process for causing delamination on the test piece is performed according to ASTM D7136 / 7136M-07.
- the term “pseudo isotropic lamination” means that the fiber orientation of the laminated prepreg is slightly shifted so that the fiber orientation becomes isotropic as a whole laminate. In the present invention, it refers to laminating four layers of prepregs by shifting the fiber directions of adjacent prepregs by 45 °.
- the method for realizing the prepreg having a low interlayer friction coefficient of the present invention is not particularly limited, but at a boundary between the resin layer and the fiber layer at 40 to 80 ° C., the viscosity is higher than that of the thermosetting resin in the resin layer. It is preferable that a barrier layer made of a resin having a temperature range exists. The barrier layer has an effect of preventing the thermosetting resin in the resin layer from moving to the fiber layer. When the prepreg laminate is heated and pressurized for shaping, the thermosetting resin in the resin layer may move into the fiber layer. In such a case, the proportion of the thermoplastic resin present as a solid in the resin layer or the hardener added as a solid in the resin layer at a molding process temperature of 40 to 80 ° C.
- thermosetting resin in the resin layer increases. Interfering with the fibers of the fiber layer increases the interlayer friction coefficient. Therefore, an increase in the interlayer friction coefficient can be suppressed by providing a barrier layer that prevents the thermosetting resin in the resin layer from shifting to the fiber layer. Even when the prepreg is stored for a long period of time, since the thermosetting resin in the resin layer may migrate into the fiber layer, the resin constituting the barrier layer is included in the resin layer even at room temperature of 10 to 30 ° C. It is preferable to have a higher viscosity than the thermosetting resin.
- the barrier layer does not need to form a layer in the obtained carbon fiber reinforced plastic by being dispersed in the thermosetting resin at a molding temperature, for example, around 180 ° C.
- the barrier layer may act as a lubricant at a shaping process temperature of 40 to 80 ° C. Since the barrier layer itself slides as a lubricant, the interlayer friction coefficient can be further reduced.
- the resin that acts as the lubricant is not particularly limited, and specifically, a thermoplastic resin, a thermosetting resin that is solid at room temperature, or a film, nonwoven fabric, or particle made of a mixture thereof is preferable.
- a barrier layer having an effect of a lubricant can be formed by arranging a resin that is solid at 25 ° C. and has a viscosity of 10,000 Pa ⁇ s or less at 80 ° C. at the boundary between the resin layer and the fiber layer.
- a resin that is solid at 40 ° C. and has a viscosity of 10,000 Pa ⁇ s or less at 80 ° C. is preferable.
- the effect of preventing migration is enhanced.
- the viscosity is 10000 Pa ⁇ s or less at 80 ° C., and more preferably, the viscosity is 1000 Pa ⁇ s or less at 80 ° C., the effect as a lubricant is enhanced.
- the resin constituting the barrier layer are not particularly limited, and examples thereof include epoxy resins, particularly bisphenol A type epoxy resins, bisphenol F type epoxy resins, biphenyl type epoxy resins, and phenoxy resins.
- a fiber layer is formed by impregnating carbon fibers arranged in one direction with a thermosetting resin, and then a resin constituting the barrier layer is disposed on at least one side of the fiber layer. Then, there is a method of arranging a resin layer on the surface on which the resin is arranged. That is, by arranging the resin on the carbon fiber in three stages, a barrier layer can be provided between the fiber layer and the resin layer.
- the method of disposing the resin constituting the barrier layer is not particularly limited. For example, a method of dispersing the resin powder on the fiber layer, and a method of laminating the resin film on the fiber layer. Etc.
- the followability to a three-dimensional shape is good, but it may be used not only for hot forming but also for press molding.
- the manufacturing process of the preform may be omitted, but when the preform is manufactured, it is preferable to press in the range of 40 to 80 ° C. using a press machine.
- the production methods and evaluation methods of resin raw materials, prepregs, and carbon fiber reinforced plastics used in the examples are shown below.
- the production environment and evaluation of the prepregs of the examples are performed in an atmosphere at a temperature of 25 ° C. ⁇ 2 ° C. and a relative humidity of 50% unless otherwise specified.
- CAI compressive strength after impact
- D According to the test method prescribed in ASTM D7136 / 7136M-07, a falling weight impact process and ultrasonic flaw detection were performed, and the damaged area was measured. The energy of impact given to the panel was calculated from the average of 9 molded plate thicknesses, and was uniformly 28.4 J.
- CAI was measured using an “Instron (registered trademark)” universal tester model 4208 according to the test method defined in ASTM D7137 / 7137M-07. The number of test pieces measured was 5, and the average value was CAI.
- the interlayer friction coefficient was measured by the following operations (a) to (c).
- release paper 6 having a width of 40 mm and a length of 150 mm was pasted so as to overlap the first and third layers.
- C Thirty seconds after starting to apply the vertical load, the pulling load was measured while pulling the second layer of the prepreg in the fiber direction at a pulling rate of 0.2 mm / min.
- the vertical load received by the area of the overlap part converted by the drawing displacement is double, that is, 168 N ⁇ (15 mm ⁇ drawing displacement) ⁇ (70 mm-pulling displacement) x 2 divided by the pulling load is defined as the interlaminar friction coefficient, and after 5 min and 10 min from the start of pulling, that is, the interlaminar friction coefficient at the pulling displacement of 1 mm and 2 mm is measured 5 times.
- the interlaminar friction coefficient at the pulling displacement of 1 mm and 2 mm is measured 5 times.
- Hot forming shaping test A hot forming shaping test and wrinkle determination were performed by the following operations (a) to (d).
- a prepreg laminate having a width of 15 cm and a length of 15 cm was produced by laminating 16 prepregs with a [45 / ⁇ 45 / 0/90] 2S laminate structure with 0 ° as the length direction.
- the prepreg laminate was set on the frame 14 having the shape mold so that the longitudinal direction of the shaping mold was 0 °, and the temperature was adjusted in an oven set at 60 ° C. for 30 minutes.
- C After placing the prepreg laminate 10 on the shaping mold 12 and adjusting the temperature in the oven for 10 minutes, the inside of the frame 14 was evacuated 11 over 150 seconds. As a result, a post-molding prepreg laminate 16 in which both ends of the laminate were bent by 90 ° was obtained.
- D Wrinkles generated inside the bent portion of the prepreg laminate 16 after shaping were determined in two stages, “wrinkle generation” and “no wrinkles”.
- thermoplastic resin particles 16 ply of prepreg was laminated with the fiber direction aligned in the same direction.
- the laminated prepreg is covered with a polyamide film so that there are no gaps, heated to 180 ° C. at a heating rate of 1.5 ° C./min in an autoclave, and then heated and pressed at a temperature of 180 ° C. and a pressure of 7 kg / cm 2 for 2 hours. And cured to obtain a unidirectional reinforcing material (carbon fiber reinforced plastic). Polishing the 0 ° cut cross-section of this unidirectional reinforcement material until the interface between the carbon fiber and the thermosetting resin is clearly seen, and observing the surface with an optical microscope.
- thermoplastic resin particles in the resin layer to be observed were observed. At this time, if the interface between the granular thermoplastic resin particles and the surrounding thermosetting resin was clearly visible, it was insoluble. On the other hand, when the thermoplastic resin particles were indistinguishable from the surrounding thermosetting resin, it was considered soluble.
- thermosetting resin Transparent polyamide product name: “Griramid (registered trademark)”-TR55, EMSER Weke
- epoxy Resin product name: “Epicoat (registered trademark)” 828, manufactured by Shell Petrochemical Co., Ltd.
- curing agent product name: “Tomide (registered trademark)” # 296, manufactured by Fuji Chemical Industry Co., Ltd.” 2 0.5 parts by mass was added to a solvent mixture containing 300 parts by mass of chloroform and 100 parts by mass of methanol to obtain a uniform solution.
- the obtained uniform solution was atomized with a spray gun for coating and sprayed toward a liquid surface of 3000 parts by mass of n-hexane.
- the precipitated solid was separated by filtration, washed thoroughly with n-hexane, and then vacuum-dried at 100 ° C. for 24 hours to obtain spherical epoxy-modified polyamide particles insoluble in the thermosetting resin.
- the obtained epoxy-modified polyamide particles were classified with a CCE classifier manufactured by CCE Technologies.
- the obtained particles had a 90% by volume particle size of 28 ⁇ m and a CV value of 60%. Further, when the obtained powder was observed with a scanning electron microscope as described in the specification, the sphericity was 96 fine particles and the average particle size was 14 ⁇ m.
- thermosetting resin composition (B) Preparation of thermosetting resin composition
- the materials used for the preparation of the thermosetting resin composition are as follows.
- thermosetting resin compositions (A) to (D) were prepared by the following procedure.
- thermosetting resin composition (A) 13 parts by weight of PES5003P was added to and dissolved in 60 parts by weight of “Araldite®” MY9655 and 12.6 parts by weight of “Epon®” 825 in a kneader. Next, 45 parts by mass of “Aradur (registered trademark)” 9664-1 as a curing agent was added and further kneaded to prepare a thermosetting resin composition (A).
- thermosetting resin composition (B) 16 parts by weight of PES5003P was added to 60 parts by weight of “Araldite (registered trademark)” MY9655 and 40 parts by weight of “EPON (registered trademark)” 825 in a kneader, and further dissolved. 80 parts by mass of the thermoplastic resin particles prepared in “Preparation of resin particles” were added and kneaded. Next, 45 parts by mass of “Aradur (registered trademark)” 9664-1 as a curing agent was added and further kneaded to prepare a thermosetting resin composition (B).
- thermosetting resin composition (C) 16 parts by weight of PES5003P was added to and dissolved in 60 parts by weight of “Araldite®” MY9655 and 40 parts by weight of “EPON®” 825 in a kneader. Next, 45 parts by mass of “Aradur (registered trademark)” 9664-1 as a curing agent was added and further kneaded to prepare a thermosetting resin composition (C).
- thermosetting resin composition (D) 13 parts by weight of PES5003P was added to and dissolved in 60 parts by weight of “Araldite®” MY9655 and 40 parts by weight of “EPON®” 825 in a kneader. Next, 45 parts by mass of “Aradur (registered trademark)” 9664-1 was added as a curing agent and further kneaded to prepare a thermosetting resin composition (D).
- thermosetting resin composition (A) was apply
- Teka registered trademark
- solid epoxy resin “jER (registered trademark)” 1001 bisphenol A type epoxy resin, manufactured by Mitsubishi Chemical Corporation
- jER registered trademark
- 1001 is solid at 25 ° C., and uses a viscoelasticity measuring device “ARES-G2” (manufactured by TA Instruments), with a heating rate of 2 ° C./min, a vibration frequency of 0.5 Hz, The viscosity measured under the condition of a parallel plate (diameter 40 mm) was 120 Pa ⁇ s at 80 ° C. Next, both sides were sandwiched between release papers, sealed with bag film, and then evacuated for 5 minutes while controlling the temperature at 60 ° C.
- thermosetting resin composition (B) is applied to a release paper with a knife coater, two resin films having a resin amount of 30 g / m 2 are prepared, and both sides of the fiber layer on which the barrier layer is placed are formed. After laminating and sealing with a bag film, the resin layer containing thermoplastic resin particles insoluble in the thermosetting resin was laminated on the barrier layer by vacuuming for 5 minutes while adjusting the temperature to 50 ° C.
- a prepreg having a barrier layer and a resin layer disposed on both sides of the fiber layer, a fiber mass of 270 g / m 2 , and a matrix resin mass content of 34% by mass was produced. Thereafter, the prepreg was pressed against a rotary blade roller having a blade disposed at a predetermined position, a notch penetrating the prepreg was inserted, and the carbon fiber was discontinuous.
- the cutting area was the entire prepreg.
- the cutting pattern is the pattern shown in FIG. 1, the length L of the cut carbon fiber is 30 mm, the cutting length l is 1 mm, and the angle ⁇ between the cutting and the orientation direction of the carbon fiber is 14 °. did.
- thermosetting resin composition (A) was apply
- Teka registered trademark
- solid epoxy resin “jER (registered trademark)” 1001 is pulverized into a powder in a mortar, and using a 32 ⁇ m sieve, on both sides of the fiber layer prepared earlier, Each side was loaded with 10 g / m 2 .
- both sides were sandwiched between release papers, sealed with bag film, and then evacuated for 5 minutes while controlling the temperature at 60 ° C.
- the above thermosetting resin composition (C) is applied to a release paper with a knife coater, two resin films having a resin amount of 23 g / m 2 are prepared, and both sides of the fiber layer on which the barrier layer is placed are formed.
- thermosetting resin As solid thermoplastic resin particles soluble in thermosetting resin, particulate PES5003P is placed on both sides of the prepreg by 7 g / m 2 on one side so that it is soluble in the thermosetting resin on the barrier layer. A resin layer containing thermoplastic resin particles was laminated. In this manner, a prepreg having a barrier layer and a resin layer disposed on both surfaces of the fiber layer, a fiber mass of 270 g / m 2 , and a matrix resin mass content of 34% by mass was produced.
- the cutting area was the entire prepreg.
- the cutting pattern is the pattern shown in FIG. 1, the length L of the cut carbon fiber is 30 mm, the cutting length l is 1 mm, and the angle ⁇ between the cutting and the orientation direction of the carbon fiber is 14 °. did.
- the obtained prepreg was used for interlayer friction coefficient measurement, insolubility evaluation test, and shaping test. Moreover, the carbon fiber reinforced plastic was produced using the obtained prepreg, and CAI was measured. The results are shown in Tables 1 and 2.
- thermosetting resin composition (D) was apply
- Teka registered trademark
- solid epoxy resin “jER (registered trademark)” 1001 is pulverized into a powder in a mortar, and using a 32 ⁇ m sieve, on both sides of the fiber layer prepared earlier, Each side was loaded with 10 g / m 2 .
- both sides were sandwiched between release papers, sealed with bag film, and then evacuated for 5 minutes while controlling the temperature at 60 ° C.
- the above thermosetting resin composition (B) is applied to a release paper with a knife coater, two resin films having a resin amount of 30 g / m 2 are prepared, and both sides of the fiber layer on which the barrier layer is placed are formed.
- the resin layer containing thermoplastic resin particles insoluble in the thermosetting resin was laminated on the barrier layer by vacuuming for 5 minutes while adjusting the temperature to 50 ° C. In this manner, a prepreg having a barrier layer and a resin layer disposed on both surfaces of the fiber layer, a fiber mass of 190 g / m 2 , and a matrix resin mass content of 39% by mass was produced.
- the cutting area was the entire prepreg.
- the cutting pattern is the pattern shown in FIG. 1, the length L of the cut carbon fiber is 30 mm, the cutting length l is 1 mm, and the angle ⁇ between the cutting and the orientation direction of the carbon fiber is 14 °. did.
- the obtained prepreg was used for interlayer friction coefficient measurement, insolubility evaluation test, and shaping test. Moreover, the carbon fiber reinforced plastic was produced using the obtained prepreg, and CAI was measured. The results are shown in Tables 1 and 2.
- thermosetting resin composition (D) was apply
- the two resin films thus prepared were laminated on both sides of a sheet-like carbon fiber ("Treka (registered trademark)" T800S-12K) arranged in one direction, and the same conditions as in Example 1 were obtained.
- a fiber layer was prepared by impregnating the carbon fiber sheet with resin by heating and pressing.
- the thermosetting resin composition (B) is applied to a release paper with a knife coater to produce two resin films having a resin amount of 20 g / m 2 and laminated on both sides of the fiber layer prepared earlier.
- thermosetting resin By heating and pressurizing, a resin layer containing thermoplastic resin particles insoluble in the thermosetting resin was laminated on the fiber layer.
- the resin layer was arrange
- the cutting area was the entire prepreg.
- the cutting pattern is the pattern shown in FIG. 1, the length L of the cut carbon fiber is 30 mm, the cutting length l is 1 mm, and the angle ⁇ between the cutting and the orientation direction of the carbon fiber is 14 °. did.
- the obtained prepreg was used for interlayer friction coefficient measurement, insolubility evaluation test, and shaping test. Moreover, the carbon fiber reinforced plastic was produced using the obtained prepreg, and CAI was measured. The results are shown in Tables 1 and 2.
- thermosetting resin composition (A) was apply
- Teka registered trademark
- solid epoxy resin “jER (registered trademark)” 1001 is pulverized into a powder in a mortar, and using a 32 ⁇ m sieve, on both sides of the fiber layer prepared earlier, Each side was loaded with 10 g / m 2 .
- both sides were sandwiched between release papers, sealed with bag film, and then evacuated for 5 minutes while controlling the temperature at 60 ° C.
- the above thermosetting resin composition (B) is applied to a release paper with a knife coater, two resin films having a resin amount of 30 g / m 2 are prepared, and both sides of the fiber layer on which the barrier layer is placed are formed.
- the resin layer containing thermoplastic resin particles insoluble in the thermosetting resin was laminated on the barrier layer by vacuuming for 5 minutes while adjusting the temperature to 50 ° C.
- a prepreg having a barrier layer and a resin layer disposed on both surfaces of the fiber layer, a fiber mass of 270 g / m 2 , and a matrix resin mass content of 34% by mass was produced. Since the cutting insertion process was not performed, all the carbon fibers contained in the prepreg were continuous carbon fibers and did not contain discontinuous carbon fibers.
- the obtained prepreg was used for interlayer friction coefficient measurement, insolubility evaluation test, and shaping test. Moreover, the carbon fiber reinforced plastic was produced using the obtained prepreg, and CAI was measured. The results are shown in Tables 1 and 2.
- thermosetting resin composition (D) was apply
- the produced two resin films are laminated on both sides of a sheet-like carbon fiber ("Treka (registered trademark)" T800S-12K) arranged in one direction, and the resin is carbonized by heating and pressing.
- a fiber sheet was produced by impregnating the fiber sheet.
- the thermosetting resin composition (C) containing no thermoplastic particles is applied to a release paper with a knife coater to produce two resin films having a resin amount of 30 g / m 2 , and the fiber layer produced earlier.
- grains of a thermoplastic resin was laminated
- a prepreg having resin layers disposed on both sides of the fiber layer, a fiber mass of 270 g / m 2 and a matrix resin mass content of 34% by mass was produced. Since the cutting insertion process was not performed, all the carbon fibers contained in the prepreg were continuous carbon fibers and did not contain discontinuous carbon fibers.
- the prepreg of the present invention can be shaped into a wrinkle-free preform and is suitable for producing a high-quality fiber-reinforced plastic. Since the prepreg of the present invention exhibits high mechanical properties when it is made of fiber reinforced plastic, it can be developed for structural uses such as aircraft, spacecraft, automobiles, railways, ships, electrical appliances, and sports.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Composite Materials (AREA)
- Mechanical Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Textile Engineering (AREA)
- Inorganic Chemistry (AREA)
- Reinforced Plastic Materials (AREA)
Abstract
Description
次の(a)~(e)の操作によりCAIを測定した。
(a)0°を長さ方向として、プリプレグを[45/0/-45/90]2Sの積層構成で16ply積層した。
(b)積層したプリプレグをポリアミドフィルムで隙間のないように覆い、オートクレーブ中で昇温速度1.5℃/分で180℃まで昇温した後、温度180℃、圧力7kg/cm2で2時間加熱加圧して硬化し、平板状の擬似等方材(炭素繊維強化プラスチック)を得た。
(c)平板状の炭素繊維強化プラスチックより、0°を長さ方向とし、長さ150±0.25mm、幅100±0.25mmのCAI試験片を切り出した。
(d)ASTM D7136/7136M-07に規定する試験方法に従い、落錘衝撃工程および超音波探傷を行い、損傷面積を測定した。パネルに与えたインパクトのエネルギーは、成形板厚さ9点の平均から算出し、一律28.4Jとした。
(e)ASTM D7137/7137M-07に規定する試験方法に従い、“インストロン(登録商標)”万能試験機4208型を用い、CAIを測定した。測定した試験片の数は5とし、平均値をCAIとした。
次の(a)~(c)の操作により、層間摩擦係数を測定した。
(a)図3に示すように、0°を長さ方向として、幅40mm、長さ150mmに裁断した1層目のプリプレグ8に、幅30mm、長さ105mmに裁断した2層目のプリプレグ7を幅30mm、長さ15mmの範囲でオーバーラップするように積層し、さらに2層目のオーバーラップ部に接するように幅30mm、長さ65mmのスペーサー9用プリプレグを積層した後、幅40mm、長さ150mmの3層目のプリプレグ8を1層目と重なるように積層した。その後、幅40mm×長さ150mmの離型紙6を1層目および3層目の外側に重なるよう貼り付けた。
(b)オーバーラップ部とスペーサーの長さ10mmの範囲(幅30mm、長さ70mmの範囲)を、加熱源を有した圧板5を用いて、所定の温度に温調しながら168Nの一定垂直荷重を加えた。
(c)垂直荷重を加え始めて30秒後に、2層目のプリプレグを繊維方向に引抜速度0.2mm/minで引抜きながら、引抜荷重を測定した。引抜きとともに2層目のプリプレグが垂直荷重を受けるオーバーラップ部の面積が減少するため、引抜き変位で換算したオーバーラップ部の面積で受ける垂直荷重の2倍、すなわち168N×(15mm-引抜き変位)÷(70mm-引抜き変位)×2で引抜き荷重を割ったものを層間摩擦係数とし、引抜開始から5分後および10分後、すなわち引抜き変位1mmおよび2mmにおける層間摩擦係数をそれぞれ5回測定し、それぞれの平均値を層間摩擦係数の値とした。
次の(a)~(d)の操作により、ホットフォーミング賦形試験およびしわ判定を行った。
(a)0°を長さ方向として[45/-45/0/90]2Sの積層構成で、プリプレグを16枚積層し、幅15cm、長さ15cmのプリプレグ積層体を作製した。
(b)図4に示すような、幅5cm、高さ10cm、X=6cm、Y=0.8cmの段差が設けられ、全辺がR=5mmの賦形型12をシリコンラバー13、シール15が具備されたフレーム14に賦形型の長手方向が0°となるようにプリプレグ積層体をセットし、60℃に設定したオーブンで30分温調した。
(c)プリプレグ積層体10を賦形型12の上に配置し、オーブン内で10分温調した後、フレーム14内を150秒かけて真空引き11した。これによって、積層体の両端部が90°曲げられた賦型後プリプレグ積層体16が得られた。
(d)賦型後プリプレグ積層体16の曲げられた部分の内側に生成するしわを、「しわ発生」および「しわなし」の2段階で判定した。
プリプレグを、繊維方向を同方向に揃えて16ply積層した。積層したプリプレグをポリアミドフィルムで隙間のないように覆い、オートクレーブ中で昇温速度1.5℃/分で180℃まで昇温した後、温度180℃、圧力7kg/cm2で2時間加熱加圧して硬化し、一方向強化材(炭素繊維強化プラスチック)を得た。繊維方向を0°として、この一方向強化材の0°カット断面を、炭素繊維と熱硬化性樹脂との界面が明確に見えるまで研磨し、その表面を光学顕微鏡で観察し、繊維層間に存在する樹脂層中の熱可塑性樹脂粒子を観察した。この際に、粒状の熱可塑性樹脂粒子と周囲の熱硬化性樹脂との界面が明確に見える場合は不溶とした。一方、熱可塑性樹脂粒子が周囲の熱硬化性樹脂との区別がつかない場合は可溶とした。
(a)熱硬化性樹脂に不溶な熱可塑性樹脂の粒子の調製
透明ポリアミド(製品名:“Grilamid(登録商標)”-TR55、EMSER Werke社)90質量部、エポキシ樹脂(製品名:“エピコート(登録商標)”828、シェル石油化学社製)7.5質量部および硬化剤(製品名:“トーマイド(登録商標)”#296、フジ化成工業株式会社製)2.5質量部を、クロロホルム300質量部およびメタノール100質量部を含有する溶媒混合物に加えて均一な溶液とした。次に、得られた均一な溶液を塗装用スプレーガンで霧化し、n-ヘキサン3000質量部の液体表面に向けて噴霧した。沈殿した固体を濾過により分離し、n-ヘキサンで十分に洗浄し、次いで100℃で24時間真空乾燥させて、熱硬化性樹脂に不溶な球状エポキシ変性ポリアミド粒子を得た。得られたエポキシ変性ポリアミド粒子をCCEテクノロジーズ社製のCCE分級機で分級した。得られた粒子の90体積%粒子径は28μm、CV値が60%であった。また、得られた粉体を、明細書中に記したように走査型電子顕微鏡にて観察したところ、真球度は96の微粒子形状、平均粒子径は14μmであった。
熱硬化性樹脂組成物の調製に用いた材料は次の通りである。
・“Araldite(登録商標)”MY9655(テトラグリシジルジアミノジフェノルメタン、Huntsman社製)
・“EPON(登録商標)”825(液状ビスフェノールA型エポキシ樹脂、Hexion社製)
(熱可塑性樹脂)
・“スミカエクセル(登録商標)”PES5003P(ポリエーテルスルホン、住友化学(株)社製)
(硬化剤)
・“Aradur(登録商標)”9664-1(4,4’―ジアミノジフェニルスルホン、Huntsman社製)
これらを用いて、熱硬化性樹脂組成物(A)~(D)を次の手順で作製した。
13質量部のPES5003Pを、混練機中で60質量部の“Araldite(登録商標)”MY9655および12.6質量部の“Epon(登録商標)”825に加えて溶解させた。次いで硬化剤として、“Aradur(登録商標)”9664-1を45質量部加えてさらに混練して熱硬化性樹脂組成物(A)を作製した。
16質量部のPES5003Pを、混練機中で60質量部の“Araldite(登録商標)”MY9655および40質量部の“EPON(登録商標)”825に加えて溶解させ、さらに上記「(a)熱可塑性樹脂粒子の調製」で調製した熱可塑性樹脂粒子を80質量部加えて混練した。次いで硬化剤として“Aradur(登録商標)”9664-1を45質量部加えてさらに混練して熱硬化性樹脂組成物(B)を作製した。
16質量部のPES5003Pを、混練機中の60質量部の“Araldite(登録商標)”MY9655および40質量部の“EPON(登録商標)”825に加えて溶解させた。次いで硬化剤として“Aradur(登録商標)”9664-1を45質量部加えてさらに混練して熱硬化性樹脂組成物(C)を作製した。
13質量部のPES5003Pを、混練機中で60質量部の“Araldite(登録商標)”MY9655および40質量部の“EPON(登録商標)”825に加えて溶解させた。次いで硬化剤として、“Aradur(登録商標)”9664-1を45質量部加えてさらに混練して熱硬化性樹脂組成物(D)を作製した。
上記の熱硬化性樹脂組成物(A)をナイフコーターで離型紙に塗布して、樹脂量30g/m2の樹脂フィルムを2枚作製した。次に、作製したこの2枚の樹脂フィルムを、一方向に配列されたシート状の炭素繊維(“トレカ(登録商標)”T800S-12K)の両面に積層し、加熱・加圧して樹脂を炭素繊維シートに含浸させ、繊維層を作製した。その後、バリア層を構成する樹脂として、固形エポキシ樹脂“jER(登録商標)”1001(ビスフェノールA型エポキシ樹脂、三菱化学(株)製)を乳鉢で粉体となるよう粉砕し、32μmの目の篩を用いて、先ほど作製した繊維層の両面に、片面10g/m2ずつ乗せた。なお、“jER(登録商標)”1001は25℃では固体であり、粘弾性測定装置“ARES―G2”(TA Instruments社製)を用い、昇温速度2℃/分、振動周波数0.5Hz、パラレルプレート(直径40mm)の条件下で測定した粘度は、80℃で120Pa・sであった。次いで、両面を離型紙で挟み、バグフィルムで密封した後、60℃に温調しながら5分間真空引きした。さらに、上記の熱硬化性樹脂組成物(B)をナイフコーターで離型紙に塗布して、樹脂量30g/m2の樹脂フィルムを2枚作製して、バリア層を乗せた繊維層の両面に積層し、バグフィルムで密封した後、50℃に温調しながら5分間真空引きすることにより、バリア層の上に熱硬化性樹脂に不溶な熱可塑性樹脂の粒子を含む樹脂層を積層した。
上記の熱硬化性樹脂組成物(A)をナイフコーターで離型紙に塗布して、樹脂量30g/m2の樹脂フィルムを2枚作製した。次に、作製したこの2枚の樹脂フィルムを、一方向に配列されたシート状の炭素繊維(“トレカ(登録商標)”T800S-12K)の両面に積層し、加熱・加圧して樹脂を炭素繊維シートに含浸させ、繊維層を作製した。その後、バリア層を構成する樹脂として、固形エポキシ樹脂“jER(登録商標)”1001を乳鉢で粉体となるよう粉砕し、32μmの目の篩を用いて、先ほど作製した繊維層の両面に、片面10g/m2ずつ乗せた。次いで、両面を離型紙で挟み、バグフィルムで密封した後、60℃に温調しながら5分間真空引きした。さらに、上記の熱硬化性樹脂組成物(C)をナイフコーターで離型紙に塗布して、樹脂量23g/m2の樹脂フィルムを2枚作製して、バリア層を乗せた繊維層の両面に積層し、バグフィルムで密封した後、50℃に温調しながら5分間真空引きした。さらに、熱硬化性樹脂に可溶な固形熱可塑性樹脂の粒子として、粒子状のPES5003Pを、プリプレグ両面に片面7g/m2ずつ乗せることにより、バリア層の上に熱硬化性樹脂に可溶な熱可塑性樹脂の粒子を含む樹脂層を積層した。このようにして、繊維層の両面に、バリア層および樹脂層が配置され、繊維質量が270g/m2でマトリックス樹脂の質量含有率が34質量%のプリプレグを作製した。
上記の熱硬化性樹脂組成物(D)をナイフコーターで離型紙に塗布して、樹脂量20g/m2の樹脂フィルムを2枚作製した。次に、作製したこの2枚の樹脂フィルムを、一方向に配列されたシート状の炭素繊維(“トレカ(登録商標)”T800S-12K)の両面に積層し、加熱・加圧して樹脂を炭素繊維シートに含浸させ、繊維層を作製した。その後、バリア層を構成する樹脂として、固形エポキシ樹脂“jER(登録商標)”1001を乳鉢で粉体となるよう粉砕し、32μmの目の篩を用いて、先ほど作製した繊維層の両面に、片面10g/m2ずつ乗せた。次いで、両面を離型紙で挟み、バグフィルムで密封した後、60℃に温調しながら5分間真空引きした。さらに、上記の熱硬化性樹脂組成物(B)をナイフコーターで離型紙に塗布して、樹脂量30g/m2の樹脂フィルムを2枚作製して、バリア層を乗せた繊維層の両面に積層し、バグフィルムで密封した後、50℃に温調しながら5分間真空引きすることにより、バリア層の上に熱硬化性樹脂に不溶な熱可塑性樹脂の粒子を含む樹脂層を積層した。このようにして、繊維層の両面に、バリア層および樹脂層が配置され、繊維質量が190g/m2でマトリックス樹脂の質量含有率が39質量%のプリプレグを作製した。
上記の熱硬化性樹脂組成物(D)をナイフコーターで離型紙に塗布して、樹脂量30g/m2の樹脂フィルムを2枚作製した。次に、作製したこの2枚の樹脂フィルムを、一方向に配列されたシート状の炭素繊維(“トレカ(登録商標)”T800S-12K)の両面に積層し、実施例1と同様の条件、加熱・加圧して樹脂を炭素繊維シートに含浸させ、繊維層を作製した。さらに、上記の熱硬化性樹脂組成物(B)をナイフコーターで離型紙に塗布して、樹脂量20g/m2の樹脂フィルムを2枚作製して、先ほど作製した繊維層の両面に積層し、加熱・加圧してすることにより、繊維層の上に熱硬化性樹脂に不溶な熱可塑性樹脂の粒子を含む樹脂層を積層した。このようにして、繊維層の両面に、樹脂層が配置され、繊維質量が190g/m2でマトリックス樹脂の質量含有率が34.5質量%のプリプレグを作製した。
上記の熱硬化性樹脂組成物(A)をナイフコーターで離型紙に塗布して、樹脂量30g/m2の樹脂フィルムを2枚作製した。次に、作製したこの2枚の樹脂フィルムを、一方向に配列されたシート状の炭素繊維(“トレカ(登録商標)”T800S-12K)の両面に積層し、加熱・加圧して樹脂を炭素繊維シートに含浸させ、繊維層を作製した。その後、バリア層を構成する樹脂として、固形エポキシ樹脂“jER(登録商標)”1001を乳鉢で粉体となるよう粉砕し、32μmの目の篩を用いて、先ほど作製した繊維層の両面に、片面10g/m2ずつ乗せた。次いで、両面を離型紙で挟み、バグフィルムで密封した後、60℃に温調しながら5分間真空引きした。さらに、上記の熱硬化性樹脂組成物(B)をナイフコーターで離型紙に塗布して、樹脂量30g/m2の樹脂フィルムを2枚作製して、バリア層を乗せた繊維層の両面に積層し、バグフィルムで密封した後、50℃に温調しながら5分間真空引きすることにより、バリア層の上に熱硬化性樹脂に不溶な熱可塑性樹脂の粒子を含む樹脂層を積層した。このようにして、繊維層の両面に、バリア層および樹脂層が配置され、繊維質量が270g/m2でマトリックス樹脂の質量含有率が34質量%のプリプレグを作製した。切込挿入工程は行わなかったので、プリプレグに含まれる炭素繊維は全て連続の炭素繊維であり、不連続の炭素繊維を含まなかった。
上記の熱硬化性樹脂組成物(D)をナイフコーターで離型紙に塗布して、樹脂量40g/m2の樹脂フィルムを2枚作製した。次に、作製したこの2枚の樹脂フィルムを、一方向に配列されたシート状の炭素繊維(“トレカ(登録商標)”T800S-12K)の両面に積層し、加熱・加圧して樹脂を炭素繊維シートに含浸させ、繊維層を作製した。さらに上記の熱可塑粒子を含まない熱硬化性樹脂組成物(C)をナイフコーターで離型紙に塗布して、樹脂量30g/m2の樹脂フィルムを2枚作製して、先ほど作製した繊維層の両面に積層し、加熱・加圧することにより、繊維層の上に熱可塑性樹脂の粒子を含まない樹脂層を積層した。このようにして、繊維層の両面に樹脂層が配置され、繊維質量が270g/m2でマトリックス樹脂の質量含有率が34質量%のプリプレグを作製した。切込挿入工程は行わなかったので、プリプレグに含まれる炭素繊維は全て連続の炭素繊維であり、不連続の炭素繊維を含まなかった。


2:プリプレグ
3:正切込
4:負切込
5:圧板
6:離型紙
7:2層目のプリプレグ
8:1層目、3層目のプリプレグ
9:スペーサー用プリプレグ
10:プリプレグ積層体
11:真空引き
12:賦形型
13:シリコンラバー
14:フレーム
15:シール
16:賦型後プリプレグ積層体
θ:切込角度
L:分断された炭素繊維の長さ
l:切込の長さ
Claims (10)
- 一方向に配向した不連続の炭素繊維と熱硬化性樹脂とを含む繊維層の少なくとも片面に、熱硬化性樹脂と熱可塑性樹脂を含む樹脂層が存在するプリプレグであって、該プリプレグに含まれる炭素繊維の繊維質量が120~300g/m2、プリプレグ全質量に対する樹脂の質量含有率が25~50%であり、前記プリプレグを3層積層し、中央のプリプレグを引き抜いた際のプリプレグ同士の接触面の層間摩擦係数を、引抜速度0.2mm/min、垂直応力0.08MPa、引抜長さ1mmの条件下において、40~80℃の温度範囲で10℃刻みに測定した場合に、該層間摩擦係数が0.05以下となる温度が40~80℃の温度範囲内に存在する、プリプレグ。
- 樹脂層が、熱硬化性樹脂に可溶な固形熱可塑性樹脂を含む、請求項1に記載のプリプレグ。
- 熱硬化性樹脂に可溶な固形熱可塑性樹脂の形態が粒子である、請求項2に記載のプリプレグ。
- 樹脂層が、熱硬化性樹脂に不溶な熱可塑性樹脂を含む、請求項1~3のいずれかに記載のプリプレグ。
- 熱硬化性樹脂に不溶な熱可塑性樹脂の形態が粒子である、請求項4に記載のプリプレグ。
- 前記層間摩擦係数の測定において層間摩擦係数が0.05以下となる温度領域が20℃以上の幅の温度領域において存在する、請求項1~5のいずれかに記載のプリプレグ。
- 前記層間摩擦係数の測定において、引抜長さ1mmにおける層間摩擦係数に対する、引抜長さ2mmにおける層間摩擦係数の上昇率が40%以内となる温度が、引抜長さ1mmにおける層間摩擦係数が最低となる温度の上下10℃以内に存在する、請求項1~6のいずれかに記載のプリプレグ。
- プリプレグを擬似等方に積層および成形し、ASTM D7137/7137M-07に準拠して測定した衝撃後圧縮強度が250MPa以上である、請求項1~7のいずれかに記載のプリプレグ。
- 樹脂層と繊維層との境界に、40~80℃において、樹脂層中の熱硬化性樹脂よりも高粘度な温度域が存在する樹脂からなるバリア層が存在する、請求項1~8のいずれかに記載のプリプレグ。
- 請求項1~9のいずれかに記載のプリプレグの製造方法であって、一方向に配向した連続の炭素繊維と熱硬化性樹脂とを含む繊維層における連続繊維に切込を挿入することによって、一方向に配向した不連続の炭素繊維と熱硬化性樹脂とを含む繊維層を形成する工程を含む、プリプレグの製造方法。
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2019100991A RU2019100991A (ru) | 2016-06-28 | 2017-06-23 | Препрег и способ его производства |
| CA3020078A CA3020078C (en) | 2016-06-28 | 2017-06-23 | Prepreg and production method therefor |
| US16/099,342 US20190210301A1 (en) | 2016-06-28 | 2017-06-23 | Prepreg and production method therefor |
| AU2017287341A AU2017287341A1 (en) | 2016-06-28 | 2017-06-23 | Prepreg and production method therefor |
| BR112018071500-0A BR112018071500A2 (pt) | 2016-06-28 | 2017-06-23 | pré-impregnado e método de produção do mesmo |
| CN201780032655.9A CN109196026B (zh) | 2016-06-28 | 2017-06-23 | 预浸料坯及其制造方法 |
| EP17820051.5A EP3476886A4 (en) | 2016-06-28 | 2017-06-23 | PREPREG AND MANUFACTURING METHOD THEREFOR |
| KR1020187031195A KR102309912B1 (ko) | 2016-06-28 | 2017-06-23 | 프리프레그 및 그 제조 방법 |
| JP2017538737A JP7003662B2 (ja) | 2016-06-28 | 2017-06-23 | プリプレグおよびその製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016127270 | 2016-06-28 | ||
| JP2016-127270 | 2016-06-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018003694A1 true WO2018003694A1 (ja) | 2018-01-04 |
Family
ID=60786428
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/023199 WO2018003694A1 (ja) | 2016-06-28 | 2017-06-23 | プリプレグおよびその製造方法 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US20190210301A1 (ja) |
| EP (1) | EP3476886A4 (ja) |
| JP (1) | JP7003662B2 (ja) |
| KR (1) | KR102309912B1 (ja) |
| CN (1) | CN109196026B (ja) |
| AU (1) | AU2017287341A1 (ja) |
| BR (1) | BR112018071500A2 (ja) |
| CA (1) | CA3020078C (ja) |
| RU (1) | RU2019100991A (ja) |
| TW (1) | TWI723188B (ja) |
| WO (1) | WO2018003694A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2021052466A (ja) * | 2019-09-24 | 2021-04-01 | 株式会社明電舎 | 炭素繊維強化樹脂成形品 |
| WO2021117465A1 (ja) * | 2019-12-11 | 2021-06-17 | 東レ株式会社 | プリプレグ、積層体および一体化成形品 |
| EP3835343A4 (en) * | 2018-09-18 | 2022-05-25 | Toray Industries, Inc. | PREPREG, PREPREG LAMINATE AND FIBER REINFORCED COMPOSITE |
| WO2024204522A1 (ja) * | 2023-03-30 | 2024-10-03 | 東レ株式会社 | 離型シ-ト付き樹脂フィルム |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3390506A4 (en) * | 2015-12-16 | 2019-08-07 | Toray Industries, Inc. | PREPREG, LAMINATE BODY, FIBER-REINFORCED COMPOSITE MATERIAL AND MANUFACTURING METHOD FOR FIBER-REINFORCED COMPOSITE MATERIAL |
| US10921859B2 (en) * | 2017-04-10 | 2021-02-16 | Securaplane Technologies, Inc. | Composite electronics cases and methods of making and using the same |
| CN113840723A (zh) * | 2019-05-23 | 2021-12-24 | 东丽株式会社 | 预浸料坯、层叠体及成型品 |
| CN111453523A (zh) * | 2020-04-28 | 2020-07-28 | 山东省东平县华东纸业有限责任公司 | 韧性强度优良的三层抽纸及其制备方法 |
| CN113561519B (zh) * | 2021-09-26 | 2021-12-28 | 昌亚新材料科技有限公司 | 纤维增韧的多层塑料型材及其制备方法、可降解食物餐盒 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01104624A (ja) * | 1987-10-16 | 1989-04-21 | Toray Ind Inc | 樹脂微粒子を用いたプリプレグ |
| JP2003026768A (ja) * | 2001-07-13 | 2003-01-29 | Toray Ind Inc | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| JP2006169541A (ja) * | 1996-12-18 | 2006-06-29 | Toray Ind Inc | プリプレグ |
| JP2007217665A (ja) * | 2006-01-19 | 2007-08-30 | Toray Ind Inc | プリプレグおよび炭素繊維強化複合材料 |
| WO2008038591A1 (en) * | 2006-09-28 | 2008-04-03 | Toray Industries, Inc. | Process for producing composite prepreg base, layered base, and fiber-reinforced plastic |
| WO2016111190A1 (ja) * | 2015-01-07 | 2016-07-14 | 東レ株式会社 | プリプレグ |
| WO2017110991A1 (ja) * | 2015-12-25 | 2017-06-29 | 東レ株式会社 | プリプレグおよびその製造方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5772950A (en) | 1994-08-31 | 1998-06-30 | The Boeing Company | Method of vacuum forming a composite |
| BRPI0809295B1 (pt) * | 2007-04-17 | 2018-10-16 | Hexcel Corp | material compósito pré-impregnado para curar em uma temperatura de cura entre 140 ºc e 200 ºc, parte do compósito que compreende um material compósito pré-impregnado e métodos para produzir os mesmos |
| US8686069B2 (en) * | 2010-10-12 | 2014-04-01 | Hexcel Corporation | Solvent resistance of epoxy resins toughened with polyethersulfone |
| JP5900327B2 (ja) * | 2011-03-17 | 2016-04-06 | 東レ株式会社 | プリプレグ、プリプレグの製造方法および炭素繊維強化複合材料の製造方法 |
| US9200125B2 (en) * | 2012-08-26 | 2015-12-01 | Hexcel Corporation | Composite material with polyamide particles |
| US9187636B2 (en) * | 2012-08-26 | 2015-11-17 | Hexcel Corporation | Composite material with polyamide particle mixtures |
-
2017
- 2017-06-23 EP EP17820051.5A patent/EP3476886A4/en not_active Withdrawn
- 2017-06-23 BR BR112018071500-0A patent/BR112018071500A2/pt not_active Application Discontinuation
- 2017-06-23 US US16/099,342 patent/US20190210301A1/en not_active Abandoned
- 2017-06-23 AU AU2017287341A patent/AU2017287341A1/en not_active Abandoned
- 2017-06-23 RU RU2019100991A patent/RU2019100991A/ru not_active Application Discontinuation
- 2017-06-23 WO PCT/JP2017/023199 patent/WO2018003694A1/ja unknown
- 2017-06-23 CN CN201780032655.9A patent/CN109196026B/zh active Active
- 2017-06-23 CA CA3020078A patent/CA3020078C/en active Active
- 2017-06-23 KR KR1020187031195A patent/KR102309912B1/ko active IP Right Grant
- 2017-06-23 JP JP2017538737A patent/JP7003662B2/ja active Active
- 2017-06-26 TW TW106121213A patent/TWI723188B/zh active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01104624A (ja) * | 1987-10-16 | 1989-04-21 | Toray Ind Inc | 樹脂微粒子を用いたプリプレグ |
| JP2006169541A (ja) * | 1996-12-18 | 2006-06-29 | Toray Ind Inc | プリプレグ |
| JP2003026768A (ja) * | 2001-07-13 | 2003-01-29 | Toray Ind Inc | エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料 |
| JP2007217665A (ja) * | 2006-01-19 | 2007-08-30 | Toray Ind Inc | プリプレグおよび炭素繊維強化複合材料 |
| WO2008038591A1 (en) * | 2006-09-28 | 2008-04-03 | Toray Industries, Inc. | Process for producing composite prepreg base, layered base, and fiber-reinforced plastic |
| WO2016111190A1 (ja) * | 2015-01-07 | 2016-07-14 | 東レ株式会社 | プリプレグ |
| WO2017110991A1 (ja) * | 2015-12-25 | 2017-06-29 | 東レ株式会社 | プリプレグおよびその製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3476886A4 * |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3835343A4 (en) * | 2018-09-18 | 2022-05-25 | Toray Industries, Inc. | PREPREG, PREPREG LAMINATE AND FIBER REINFORCED COMPOSITE |
| US11760053B2 (en) | 2018-09-18 | 2023-09-19 | Toray Industries, Inc. | Prepreg, prepreg laminate, and fiber-reinforced composite material |
| JP2021052466A (ja) * | 2019-09-24 | 2021-04-01 | 株式会社明電舎 | 炭素繊維強化樹脂成形品 |
| JP7521182B2 (ja) | 2019-09-24 | 2024-07-24 | 株式会社明電舎 | 覆い部材 |
| WO2021117465A1 (ja) * | 2019-12-11 | 2021-06-17 | 東レ株式会社 | プリプレグ、積層体および一体化成形品 |
| JPWO2021117465A1 (ja) * | 2019-12-11 | 2021-06-17 | ||
| JP7088320B2 (ja) | 2019-12-11 | 2022-06-21 | 東レ株式会社 | プリプレグ、積層体および一体化成形品 |
| CN114746491A (zh) * | 2019-12-11 | 2022-07-12 | 东丽株式会社 | 预浸料坯、层叠体及一体化成型品 |
| WO2024204522A1 (ja) * | 2023-03-30 | 2024-10-03 | 東レ株式会社 | 離型シ-ト付き樹脂フィルム |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3476886A4 (en) | 2020-01-08 |
| US20190210301A1 (en) | 2019-07-11 |
| TWI723188B (zh) | 2021-04-01 |
| EP3476886A1 (en) | 2019-05-01 |
| CN109196026B (zh) | 2021-04-23 |
| KR20190022461A (ko) | 2019-03-06 |
| CA3020078C (en) | 2024-01-16 |
| JP7003662B2 (ja) | 2022-02-10 |
| CA3020078A1 (en) | 2018-01-04 |
| TW201819489A (zh) | 2018-06-01 |
| KR102309912B1 (ko) | 2021-10-07 |
| JPWO2018003694A1 (ja) | 2019-04-18 |
| CN109196026A (zh) | 2019-01-11 |
| RU2019100991A (ru) | 2020-07-28 |
| BR112018071500A2 (pt) | 2019-02-19 |
| AU2017287341A1 (en) | 2018-10-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2018003694A1 (ja) | プリプレグおよびその製造方法 | |
| US10723087B2 (en) | Prepreg and method for manufacturing same | |
| EP2691199B1 (en) | Prepreg, fiber reinforced composite material, and manufacturing method for fiber reinforced composite material | |
| JP6007914B2 (ja) | 真空成形用プリプレグ、繊維強化複合材料およびその製造方法 | |
| JP5723505B2 (ja) | 樹脂組成物、硬化物、プリプレグ、および繊維強化複合材料 | |
| JP2009286895A (ja) | プリプレグおよび繊維強化複合材料の成形方法 | |
| US20160289405A1 (en) | Prepreg, fibre-reinforced composite material, and particle-containing resin composition | |
| KR20140127867A (ko) | 섬유강화 복합 재료 | |
| US20160289404A1 (en) | Prepreg, fibre-reinforced composite material, and particle-containing resin composition | |
| JP2021172694A (ja) | プリプレグ | |
| JPWO2018181254A1 (ja) | プリプレグおよび繊維強化複合材料 | |
| KR20150033691A (ko) | 섬유 강화 복합 재료 | |
| US11565497B2 (en) | Prepreg, laminate body, fiber-reinforced composite material, and manufacturing method for fiber-reinforced composite material | |
| US20160280872A1 (en) | Prepreg, fibre-reinforced composite material, and particle-containing resin composition | |
| US20160289403A1 (en) | Prepreg, fibre-reinforced composite material, and particle-containing resin composition | |
| WO2023182432A1 (ja) | 熱硬化性プリプレグ及びその製造方法 | |
| JP7143473B2 (ja) | プリプレグ及びその製造方法、並びに繊維強化複合材料 | |
| JP2021178902A (ja) | プリプレグ | |
| JP2021195473A (ja) | プリプレグ | |
| JP2020045481A (ja) | プリプレグ、離型シート付プリプレグおよび繊維強化複合材料 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2017538737 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3020078 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2017287341 Country of ref document: AU Date of ref document: 20170623 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20187031195 Country of ref document: KR Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112018071500 Country of ref document: BR |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17820051 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2017820051 Country of ref document: EP Effective date: 20190128 |
|
| ENP | Entry into the national phase |
Ref document number: 112018071500 Country of ref document: BR Kind code of ref document: A2 Effective date: 20181018 |